ES2713960T3 - Composición que comprende cebranopadol en forma disuelta - Google Patents
Composición que comprende cebranopadol en forma disuelta Download PDFInfo
- Publication number
- ES2713960T3 ES2713960T3 ES16706540T ES16706540T ES2713960T3 ES 2713960 T3 ES2713960 T3 ES 2713960T3 ES 16706540 T ES16706540 T ES 16706540T ES 16706540 T ES16706540 T ES 16706540T ES 2713960 T3 ES2713960 T3 ES 2713960T3
- Authority
- ES
- Spain
- Prior art keywords
- weight
- cebranopadol
- vehicle
- organic solvent
- composition according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/141—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers
- A61K9/143—Intimate drug-carrier mixtures characterised by the carrier, e.g. ordered mixtures, adsorbates, solid solutions, eutectica, co-dried, co-solubilised, co-kneaded, co-milled, co-ground products, co-precipitates, co-evaporates, co-extrudates, co-melts; Drug nanoparticles with adsorbed surface modifiers with inorganic compounds
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Inorganic Chemistry (AREA)
- Medicinal Preparation (AREA)
Abstract
Composición farmacéutica que comprende: (a) cebranopadol disuelto, (b) disolvente orgánico con un punto de ebullición de 110 º a 350 ºC, y (c) vehículo sólido.
Description
DESCRIPCION
Composicion que comprende cebranopadol en forma disuelta
Antecedentes de la invencion
La presente invencion se refiere a una composicion que comprende cebranopadol en forma disuelta y a formas de dosificacion oral que comprenden dicha composicion. La invencion se refiere ademas a un procedimiento de produccion de la composicion que comprende cebranopadol en una forma disuelta y al procedimiento correspondiente de produccion de una forma de dosificacion oral que contiene la composicion de la invencion. El cebranopadol es un analgesico que, de acuerdo con se notifica, tiene un mecanismo de accion unico. Se supone que el cebranopadol se une al receptor nociceptina y activa el mismo (tambien conocido como receptor de orfanina Fq ), asf como a los receptores opioides clasicos, por ejemplo el receptor opioide p. Por este mecanismo de accion, se sugiere que el cebranopadol es un potente analgesico para el dolor cronico medio a fuerte, incluido el dolor neuropatico, como tal en pacientes con tumor(es).
En el contexto de esta invencion, la denominacion IUPAC de "cebranopadol" es (1r,4r)-6'-fluoro-W,W-dimetil-4-fenil-4',9'-dihidro-3'H-espiro[ciclohexano-1,1'-pirano [3,4-6)]indol]-4-amina, que esta representado por la siguiente Formula (I).

Las rutas de smtesis de cebranopadol y las sales de adicion del mismo y el uso como analgesico se han descrito, por ejemplo, en el documento EP 1560835 B1.
Ademas, el documento WO 2013/007361 A1 desvela formas polimorficas cristalinas de cebranopadol y solvatos de cebranopadol que pueden distinguirse por difraccion de rayos X. Ademas, se notifica que los polimorfos cristalinos se podnan convertir entre sf, por ejemplo, en presencia de disolventes o cuando se aplica calor o energfa mecanica. Sin embargo, tal comportamiento (la ocurrencia de cambios morfologicos) puede ser desfavorable, por ejemplo, para formas de dosificacion tales como comprimidos, ya que puede causar cambios en el estado solido en la forma de dosificacion que a menudo dan como resultado diferentes propiedades de disolucion y farmacocineticas. Por ende, dichos cambios pueden requerir un estricto control de la temperatura de las formas de dosificacion, especialmente en verano y/o en las zonas climaticas III y IV. Adicionalmente, dichos cambios en el estado solido pueden llevar a desventajas regulatorias y comerciales.
Es mas, se notifica que el cebranopadol es poco soluble y que se debe aplicar en cantidades muy pequenas (<1 mg). Las composiciones farmaceuticas que comprenden dichos ingredientes farmaceuticos activos con dichas propiedades tienden a tener problemas con la capacidad de solubilidad/biodisponibilidad y especialmente con la uniformidad de contenido del ingrediente farmaceutico activo. Dado que el ingrediente farmaceutico activo se administra en dosis muy bajas, incluso las composiciones farmaceuticas poco homogeneas, por ejemplo debido a una pequena degradacion, pueden reducir los efectos beneficiosos significativamente o incluso pueden ser la base de efectos secundarios indeseables.
El documento WO 2012/016703 A2 describe una forma de dosificacion farmaceutica que comprende cebranopadol. Dicha dosificacion comprende un nucleo lfquido que comprende cebranopadol en forma dispersa o disuelta rodeado por un material de encapsulacion. Dicho sistema podna considerarse como un sistema de administracion de farmacos autoemulsionante (SEDDS). Sin embargo, la produccion de tales SEDDS es compleja y, por lo tanto, consume tiempo y dinero. Adicionalmente, las cantidades de los tensioactivos para producir el nucleo lfquido de la dosificacion podnan producir efectos secundarios no deseados en el tracto gastrointestinal, tal como diarrea.
Por ende, un objeto de la presente invencion era superar los inconvenientes anteriores de la formulacion mencionada anteriormente.
En particular, un objeto de la presente invencion era proporcionar cebranopadol en una forma en la que no requiere un control de temperature espedfico durante el almacenamiento o cuando se usa en las zonas climaticas III y IV. Ademas, debe proporcionarse una forma de cebranopadol que muestre propiedades de disolucion y farmacocineticas ventajosas, en particular cuando se usa despues del almacenamiento o en zonas climaticas III y IV. Ademas, se debe proporcionar una forma de cebranopadol que muestre propiedades mejoradas con respecto a la procesabilidad.
Adicionalmente, era un objeto proporcionar una composicion farmaceutica, en la que la composicion no contiene un nucleo lfquido encapsulado como se describe en el documento WO 2012/016703 y/o polimorfos como se describe en el documento WO 2013/007361. No obstante, la composicion o forma de dosificacion de la presente invencion debe proporcionar propiedades in vitro e in vivo similares a la formulacion descrita en el documento 2012/016703 y/o a los polimorfos descritos en el documento WO 201 3/007361.
Todos los objetivos mencionados anteriormente deben resolverse preferentemente para una forma de dosificacion disenada para liberacion inmediata ("LI") y para liberacion modificada ("LM"). En particular, se desea un rapido alivio del dolor aunque se desea un efecto terapeutico de larga duracion.
Sumario de la invencion
De acuerdo con la presente invencion, los objetivos anteriores se pueden lograr mediante una composicion farmaceutica que comprende cebranopadol disuelto, un disolvente organico con un alto punto de ebullicion y un vehnculo. En la composicion de la presente invencion, cebranopadol esta presente en forma disuelta en un disolvente organico con un alto punto de ebullicion y puede adherirse a dicho vehnculo o preferentemente se adsorbe sobre dicho vehnculo en forma disuelta. La composicion puede procesarse ventajosamente en formas de dosificacion oral.
Por consiguiente, el objeto de la invencion es una composicion farmaceutica, preferentemente una composicion farmaceutica que tiene un aspecto solido, que comprende
(a) cebranopadol disuelto,
(b) disolvente organico con un punto de ebullicion de 110 a 350 °C a 1.013 mbar, y
(c) vehnculo solido.
La presion atmosferica, 1.013 mbar y 760 mm Hg son valores equivalentes y se refieren a la presion estandar. Un objeto adicional de la presente invencion es una forma de dosificacion oral que comprende la composicion de la invencion y opcionalmente otro(s) excipiente(s) farmaceutico(s).
Otro objeto de la presente invencion es un metodo para producir la composicion de acuerdo con la invencion que comprende las etapas que consisten en:
i) disolver cebranopadol (a) en un disolvente organico (b) con un punto de ebullicion de 110 a 350 °C, en el que el punto de ebullicion se mide a 1.013 mbar,
ii) mezclar el vehnculo solido (c) y la solucion de la etapa i),
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii).
Ademas, el objeto de la presente invencion se refiere a un procedimiento de produccion de una forma de dosificacion oral que comprende las etapas que consisten en:
i) disolver cebranopadol (a) en un disolvente organico (b) con un punto de ebullicion de 110 a 350 °C, en el que el punto de ebullicion se mide a 1.013 mbar,
ii) mezclar el vehnculo solido (c) y la solucion de la etapa i),
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii),
iv) opcionalmente, agregar otros excipiente(s) a la mezcla de la etapa ii) o la etapa iii),
v) opcionalmente, granular la mezcla de la etapa ii), etapa iii) o etapa iv)
vi) procesar la mezcla de la etapa ii), etapa iii) o etapa iv) o los granulados del etapa v) en una forma de dosificacion oral.
Los objetos ilustrados anteriormente de la presente invencion son soluciones alternativas a los problemas senalados anteriormente.
Descripcion detallada de la invencion
La presente invencion se refiere a una composicion farmaceutica que es una mezcla de un vehnculo en estado solido
y cebranopadol en estado disuelto. Preferentemente, la apariencia ffsica de toda la composicion es solida a 25 °C y 1.013 mbar. Lo mismo se aplica a la forma de dosificacion de la presente invencion.
En el contexto de esta invencion, cebranopadol puede referirse a cebranopadol en la forma de la base libre. Ademas, cebranopadol puede referirse a sales farmaceuticamente aceptables, hidratos, solvatos, polimorfos, estereoisomeros, como enantiomeros, y mezclas de los mismos. Por ejemplo, la invencion tambien se refiere a enantiomeros o sales farmaceuticamente aceptables de cebranopadol, a solvatos de sales o hidratos de polimorfos o similares.
En una realizacion preferida de la invencion, cebranopadol se refiere a cebranopadol en forma de base libre.
En una realizacion preferida alternativa, cebranopadol se refiere a sales farmaceuticamente aceptables del mismo, preferentemente sales de adicion de acidos farmaceuticamente aceptables. Los acidos que se pueden usar para preparar las sales de adicion de acido farmaceuticamente aceptables son preferentemente aquellos que forman sales de adicion de acido no toxicas. Los acidos no toxicos para formar sales acidas son, por ejemplo, acido clorfndrico, acido bromfndrico, acido yodfndrico, acido mtrico, acido sulfurico, acido fosforico, acido formico, acido acetico, acido oxalico, acido lactico, acido cftrico, acido tartarico, acido succmico, acido maleico, acido fumarico, acido mandelico, acido gluconico, acido sacarico, acido glutamico, acido benzoico, acido 2,4,6-trimetilbenzoico, acido acetilsalidlico, acido metansulfonico, acido etansulfonico, acido bencensulfonico, acido p-toluensulfonico, y acido pamoico, acido [1,1'metilen-bis-(2-hidroxi-3-naftoico), acido hipurico y acido nicotfnico.
En caso de que el peso del cebranopadol se mencione en la presente invencion, esto se refiere a cebranopadol en su forma libre.
En una realizacion particularmente preferida, la composicion de la presente invencion, asf como la forma de dosificacion oral de la presente invencion, comprenden cebranopadol como el unico agente activo farmaceutico. En una realizacion alternativa, la composicion de la presente invencion, asf como la forma de dosificacion oral de la presente invencion puede comprender cebranopadol en combinacion con otro(s) agente(s) farmaceutico(s) activo(s). En la presente invencion, el cebranopadol esta presente en forma disuelta.
El termino "cebranopadol disuelto" se puede usar en el contexto de esta invencion para designar el compuesto anterior en el que los componentes (atomos, iones o moleculas) no exhiben una disposicion periodica en un gran intervalo (= orden de largo alcance), tal como se conoce habitualmente a partir de sustancias cristalinas. En consecuencia, el presente cebranopadol, por ejemplo, no muestra ninguna interferencia clara determinada por medio de difraccion de rayos X. Los presentes componentes de cebranopadol (atomos, iones o moleculas, preferentemente atomos) estan cada uno preferentemente rodeados por una corteza de solvato. Esta corteza de solvato puede estar compuesta de varias capas de moleculas de disolvente en las que cuanto mas interactuen las moleculas de las diversas capas de la corteza de solvato con la molecula central, mas cerca estan de dicha molecula central.
Las moleculas solvatadas pueden considerarse preferentemente como una entidad flexible cuya corteza de solvato esta en interaccion con las moleculas solventes.
En una realizacion preferida, el cebranopadol disuelto se puede considerar como cebranopadol no solido o, en otras palabras, cebranopadol en una forma no solida.
El disolvente organico (b) tiene un punto de ebullicion de 110 °C a 350 °C, en el que el punto de ebullicion se mide a 1.013 mbar. Sin embargo, esto no implica que cebranopadol se deba disolver a 1.013 mbar en el disolvente organico (b). Mas preferido, el disolvente organico puede tener un punto de ebullicion de al menos 120 °C, 130 °C, 140 °C, 150 °C, 160 °C, 170 °C, 180 °C o 190 °C. Mas preferido, el disolvente organico puede tener un punto de ebullicion de hasta 340 °C, 330 °C, 320 °C, 310 °C, 300 °C, 290 °C, 280 °C, 270 °C, 260 °C o 250 °C. Tambien se prefieren todas las combinaciones posibles (p. ej., 170 a 340 °C) de los lfmites inferiores y superiores anteriores. Preferentemente, la temperatura se determina a 1.013 mbar. En esta solicitud, un "punto de ebullicion de 110 a 350 °C" tambien abarca aquellos disolventes organicos (b) que se descomponen en dicho intervalo de temperatura. Ademas, el punto de ebullicion no se relaciona con una temperatura unica, sino que tambien puede referirse a un intervalo de temperaturas, por ejemplo, cuando se utiliza una mezcla de disolventes organicos.
Preferentemente, el punto de ebullicion se determina de acuerdo con Pharm. Eur. 6.0, Capftulo 2.2.12.
En una realizacion preferida de la composicion, el disolvente organico (b) tiene un punto de fusion entre -80 °C y 25 °C, preferentemente entre -75 °C y 10 °C, mas preferentemente entre -72 °C y 5 °C, especialmente entre -70 ° y -5 °C a 1.013 mbar. Por lo tanto, el disolvente organico utilizado en la presente invencion es lfquido a temperatura ambiente (23 °C).
En una realizacion adicional de la invencion, el disolvente organico (b) tiene una densidad de 0,95 a 1,30 g/ml.
Ademas, se prefiere que el disolvente organico (b) tenga una densidad de 0,97 a 1,25 g/ml a 25 °C, incluso mas preferentemente de 1,00 a 1,20 g/ml, especialmente de 1,01 a 1,15 g/ml. La densidad puede determinate mediante la siguiente formula:

m = la masa del disolvente
V = volumen del disolvente
En una realizacion adicional de la invencion, el disolvente organico (b) tiene una presion de vapor de menos de 10 hPa o mbar a 20 °C, preferentemente menos de 1 hPa o mbar a 20 °C.
La presion de vapor es el vapor ejercido por un vapor P en un equilibrio termodinamico con su fase lfquida a una temperatura dada en un sistema cerrado. De acuerdo con la ecuacion de Antoine, se pueden calcular las presiones de vapor estimadas.
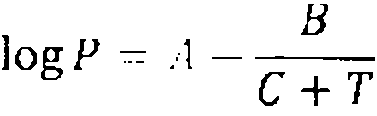
en el que A, B y C son coeficientes espedficos de la sustancia (es decir, constantes o parametros) y
T es la temperatura del Kquido.
Por ejemplo, el disolvente organico (b) puede ser polietilenglicoles tales como tetraetilenglicol y pentaetilenglicol, alcohol, polietilenglicol eter, tal como dietilenglicol monoetil eter, glicerol, propilenglicol, tal como 1,2-propilenglicol, alquil dioles, tales como 2,3-butanodiol, alquil trioles, tales como 1,2,6-hexantriol, isosorbida dimetil eter, glicofurol (tetrahidrofufuril alcohol polietilenglicol eter), polidimetil siloxano, y mezclas de los mismos.
Particularmente preferido como disolvente organico (b) es glicofurol. Alternativamente, se prefiere particularmente como disolvente organico (b) dietilenglicol monoetil eter.
La composicion de la presente invencion puede tener preferentemente una relacion en peso de cebranopadol disuelto (a) a disolvente organico (b) de 1:1 a 1:400, preferentemente de 1:2 a 1:150, mas preferentemente de 1:3 a 1:100, incluso mas preferentemente 1:4 a 1:80, lo mas preferentemente de 1:5 a 1:60.
Resulto que con la relacion anterior se puede asegurar particularmente que la cantidad completa de cebranopadol permanece en estado disuelto y se no precipita como una sustancia solida (cristalina).
La presente composicion comprende ademas un vehfculo (c). El vehfculo es preferentemente solido, en el que "solido" se refiere a la apariencia a 25 °C. El termino "vetnculo (c)" puede referirse a un unico vetnculo (c) o a una mezcla de mas de un vetnculo (c). El vetnculo (c) se puede considerar como una sustancia a la que se puede adherir/adsorber el cebranopadol disuelto (a) y el disolvente organico (b), en el que se asegura que cebranopadol mantiene su estado disuelto. Por lo tanto, el vetnculo (c) se puede considerar como un estabilizador de cebranopadol disuelto (a) en el disolvente organico (b). En general, el vetnculo solido (c) puede ser una sustancia que es capaz de inhibir la transformacion del cebranopadol disuelto en cualquier estado solido (p. ej., amorfo o cristalino) del cebranopadol.
Ademas, debido al vetnculo solido (c), la composicion ffsica se puede proporcionar en un estado adecuado para un procesamiento adicional, tal como relleno de una capsula.
En una realizacion preferida, la composicion de la invencion puede comprender cebranopadol (a) y un vetnculo (c), en el que la relacion en peso de cebranopadol (disuelto) a vetnculo (c) puede ser de 1:1 a 1:150, preferentemente de 1:1 a 1:135, mas preferentemente de 1:1 a 1:120, incluso mas preferentemente de 1:1 a 1:110, y particularmente de 1:1 a 1:100.
Generalmente, el vetnculo (c) puede ser una sustancia no quebradiza o quebradiza.
Los excipientes farmaceuticos, tales como los vetnculos, pueden clasificarse generalmente con respecto al cambio en la forma de las partfculas bajo presion de compresion (compactacion): los excipientes plasticos se caracterizan por la deformacion plastica, mientras que cuando se ejerce una fuerza de compresion sobre sustancias quebradizas, las partfculas tienden a romperse en partfculas mas pequenas. El comportamiento quebradizo en la parte del sustrato se puede cuantificar por el aumento en el area de la superficie en un moldeo. En la tecnica, es habitual clasificar la fragilidad en terminos de la "presion de fluencia". De acuerdo con una clasificacion simple, los valores para la "presion de fluencia" son bajos para sustancias plasticas, pero altos en el caso de sustancias friables
(Duberg, M., Nystrom, C., 1982, "Studies on direct compression of tablets VI. Evaluation of methods for the estimation of particle fragmentation during compaction", Acta Pharm. Suec. 19, 421-436; Humbert-Droz P., Mordier D., Doelker E., "Methode rapide de determination du comportement a la compression pour des etudes de preformulation", Pharm. Acta Helv., 57, 136-143 (1982)). La "presion de fluencia" describe la presion que debe alcanzarse para que el excipiente (es decir, preferentemente el vehnculo) comience a fluir plasticamente.
La "presion de fluencia" se calcula preferentemente utilizando el redproco del gradiente de la grafica de Meckel, como se describe en York, P., Drug Dev. Ind. Pharm. 18, 677 (1992). La medicion en este caso se realiza preferentemente a 25 °C y a una velocidad de deformacion de 0,1 mm/s.
En el contexto de la presente invencion, se considera que un excipiente (especialmente un vetnculo) es un excipiente no quebradizo cuando tiene una "presion de fluencia" de no mas de 120 MPa, preferentemente no mas de 100 MPa, en particular 5 a 80 MPa. Un excipiente se describe generalmente como un excipiente quebradizo cuando tiene una "presion de fluencia" de mas de 80 MPa, preferentemente mas de 100 MPa, particularmente preferentemente mas de 120 MPa, especialmente mas de 150 MPa. Los excipientes quebradizos pueden exhibir una "presion de fluencia" de hasta 300 MPa o hasta 400 MPa o incluso hasta 500 MPa.
Ejemplos de excipientes no quebradizos (vehnculos) son manitol o almidon.
Ejemplos de excipientes quebradizos (vehnculos) son silicatos o aluminosilicatos, preferentemente aluminosilicatos de magnesio.
En una realizacion particularmente preferida, las sustancias quebradizas se utilizan como vehnculo (c) en la forma de dosificacion oral de la presente invencion.
Ademas, se prefiere que el vehnculo (c) sea una sustancia insoluble en agua. Una sustancia insoluble en agua generalmente es un excipiente farmaceutico como se especifica en la Farmacopea Europea con una solubilidad en agua de menos de 33 mg/ml medida a 25 °C. Preferentemente, la sustancia insoluble en agua tiene una solubilidad de 10 mg/ml o menos, mas preferentemente de 5 mg/ml o menos, especialmente de 0,1 mg/ml, lo que significa practicamente insoluble (determinado de acuerdo con el metodo de elucion de la columna en virtud de la Directiva de la UE RL67-548-EWG, Apendice V Cap. A6).
En una realizacion preferida de la invencion, el vehnculo puede ser un poftmero organico o una sustancia inorganica. En una realizacion alternativa preferida de la invencion, el vehnculo (c) puede ser preferentemente un poftmero organico. Ademas, el vehnculo (c) tambien puede incluir sustancias que se comportan como poftmeros. Ejemplos de estas sustancias son grasas y ceras. Ademas, el vehnculo (c) tambien puede incluir compuestos solidos, no polimericos, que preferentemente pueden contener grupos laterales polares. Ejemplos de estos compuestos son alcoholes de azucar o disacaridos.
En una realizacion preferida, el vehnculo (c) puede ser un poftmero. El poftmero a utilizar para la preparacion de la composicion farmaceutica puede tener preferentemente una temperatura de transicion vftrea (Tv) de mas de 45 °C, mas preferentemente de 50 °C a 150 °C, en particular de 55 °C a 120 °C. Una Tv respectiva puede ser importante para lograr las propiedades deseadas de la forma de dosificacion resultante.
En la presente invencion, el termino "temperatura de transicion vftrea" (Tv) describe la temperatura a la que los poftmeros amorfos o parcialmente cristalinos cambian del estado solido al estado ftquido. En el procedimiento, se produce un cambio distinto en los parametros ffsicos, p. ej., dureza y elasticidad. Por debajo de la Tv, un poftmero suele ser vftreo y duro, mientras que por encima de la Tv cambia de un estado similar a una goma al viscoso. La temperatura de transicion vftrea se determina en el contexto de esta invencion por medio de calorimetna de barrido diferencial (CBD).
Para este fin, se puede utilizar un aparato Mettler Toledo® DSC 1. El trabajo se realiza a una velocidad de calentamiento de 1-20 °C/min, preferentemente de 10 °C/min, y a una velocidad de enfriamiento de 5 °C a 50 °C/min, preferentemente de 50 °C/min.
En general, el poftmero organico que se utiliza como vehnculo (c) puede tener preferentemente un peso molecular promedio en peso de 1.000 a 500.000 g/mol, mas preferentemente de 1.500 a 100.000 g/mol y particularmente de 2.000 a 50.000 g/mol. El peso molecular promedio en peso se determina preferentemente mediante cromatograffa de permeacion en gel.
En la presente invencion, los poftmeros hidrofilos se pueden utilizar preferentemente como vehnculo (c). El termino "poftmeros hidrofilos" se refiere generalmente a poftmeros que poseen grupos hidrofilos. Ejemplos de grupos hidrofilos adecuados pueden ser grupos hidroxi, sulfonato, carboxilato y amonio cuaternario.
El vehftculo (c) puede comprender, por ejemplo, los siguientes poftmeros: celulosa microcristalina, polisacaridos,
tales como hidroxipropilmetilcelulosa (HPMC), etilcelulosa, metilcelulosa, hidroxietilcelulosa, etilhidroxietilcelulosa, hidroxipropilcelulosa (HPC), alcohol polivimlico, y mezclas de los mismos.
Alternativamente, tambien puede utilizarse como vehnculo (c) una silicona, preferentemente mezclada adicionalmente con dioxido de silicio, tal como simeticona.
En una realizacion mas preferida de la invencion, el vehnculo (c) puede ser una sustancia inorganica. Una sustancia inorganica puede considerarse preferentemente como un compuesto que no contiene un grupo hidrocarburo. Ademas, se prefiere que el vehnculo (c) pueda ser un fosfato o un silicato, preferentemente un silicato, mas preferentemente un aluminosilicato.
Ejemplos de sustancias inorganicas adecuadas para ser utilizadas como vehnculos son fosfatos, tales como fosfato dicalcico, sflice amorfa tal como aerosol y gel de sflice, minerales de arcilla tales como caolinita, bentonita y montmorillonita, kieselguhr (celite), zeolitas, sflice mesoporosa tal como Aeroperl® 300, MSU-G, MSU-F, A1-MCM-41, MCM-48, SBA-15 y SbA-16, aluminosilicatos de magnesio tales como AhO3 MgO 1.7SiO2 xH2O (Neusilin®), y aluminosilicatos tales como A1-MCM-41 o mezclas de los mismos.
Ademas, el carbon activo (es decir, carbon activado) se puede utilizar preferentemente como vehnculo (c).
En una realizacion preferida, el vehnculo (c), en particular el vehnculo inorganico (c), tiene un area superficial espedfica de 50 a 450 m2/g, mas preferentemente de 75 a 400 m2/g, en particular de 100 a 300 m2/g. El area superficial espedfica se determina preferentemente por adsorcion de gas de acuerdo con Ph. Eur., 6a edicion, Capttulo 2.9.26. Para este fin, se utiliza un ASAP® 2020 (Micrometrics) y una temperatura de "desgasificacion" de 40 °C. Se ha descubierto de manera sorprendente que el area superficial espedfica mencionada anteriormente podna ser beneficiosa para lograr los objetos mencionados anteriormente (p. ej., estabilizacion del estado disuelto del cebranopadol).
En una realizacion preferida, el vehnculo (c) se selecciona entre simeticona, carbon activado, celulosa macrocristalina, almidon, polisacaridos, alcoholes de azucar, fosfatos, dioxidos de silicio, minerales de arcilla, kieselguhr (celite), zeolitas, sflice mesoporosa y aluminosilicatos de magnesio, o mezclas de los mismos.
En una realizacion mas preferida, el vehnculo (c) se selecciona entre simeticona, carbon activo, celulosa microcristalina, fosfatos, dioxidos de silicio, minerales de arcilla, kieselguhr, zeolitas, sflice mesoporosa y aluminosilicatos de magnesio, o mezclas de los mismos.
Los mas preferidos como vehnculos (c) son silicatos, en particular aluminosilicatos de magnesio, especialmente Al2O3 MgO 1.7SiO2 xH2O.
Alternativamente, lo mas preferido como vehnculo (c) es sflice, en particular sflice mesoporosa.
Ademas, se prefiere que la sflice mesoporosa este en forma de granulados mesoporosos similares a perlas. Los granulados pueden tener preferentemente un tamano de partfculas medio D50 de 5 a 250 pm, mas preferentemente de 10 a 100 pm, aun mas preferentemente de 15 a 80 pm, en particular de 20 a 50 pm.
Preferentemente, la sflice mesoporosa tiene un area superficial espedfica de 250 a 350 m2/g, en particular, de 280 a 320 m2/g. Preferentemente, el volumen de la sflice mesoporosa es de 1,0 a 2,5 ml/g, mas preferentemente de 1,4 a 1,8 ml/g.
El tamano de partfculas medio puede referirse al valor D50 de la distribucion del tamano de partfculas. El tamano de partfculas medio puede determinarse mediante difractometna laser. En particular, se puede utilizar un Malvern Instruments Mastersizer 2000 para determinar el tamano (preferentemente medicion en humedo con ultrasonido durante 60 segundos, 2.000 rpm, preferentemente dispersado en aceite de girasol, la evaluacion se realiza de acuerdo con la Partfcula RI establecida en 1.520 y Absorcion de 2).
En una realizacion particularmente preferida, se utiliza Aeroperl® 300.
En una realizacion particularmente preferida, se utiliza Neusilin®.
En una realizacion preferida, la composicion de la presente invencion puede comprender preferentemente las siguientes cantidades de componentes:
0,5 a 1.000 pg de cebranopadol, preferentemente de 0,8 a 800 pg de cebranopadol, mas preferentemente de 5 a 600 pg de cebranopadol, particularmente de 10 a 400 pg, especialmente 200 pg de cebranopadol,
0,01 a 150 mg de disolvente organico, preferentemente de 0,02 a 100 mg de disolvente organico, en particular de 0,1 a 80 mg de disolvente organico,
0,005 a 150 mg de vehnculo, preferentemente de 0,01 a 120 mg de vehnculo, particularmente de 0,05 a 90 mg de
vehnculo.
La composicion de la presente invencion se puede aplicar en forma de una forma de dosificacion oral, en particular en forma de una forma de dosificacion oral solida. Por lo tanto, otro objeto de la presente invencion es una forma de dosificacion oral solida que comprende una composicion de cebranopadol de acuerdo con la presente invencion y otro(s) excipiente(s) farmaceutico(s).
Los excipientes farmaceuticos son excipientes con los que esta familiarizado el experto en la materia, como los que se describen en la Farmacopea Europea (Ph. Eur.) y/o en la Farmacopea de los Estados Unidos (USP).
En una realizacion preferida de la presente invencion, la forma de dosificacion oral puede comprender ademas uno o mas excipientes seleccionados entre tensioactivos (d), cargas (e), aglutinantes (f), desintegrantes (g), lubricantes (h), y deslizantes (j).
Los tensioactivos (d) se pueden considerar como sustancias que disminuyen la tension interfacial entre dos fases, permitiendo o apoyando la formacion de dispersiones o actuando como un solubilizante. Los tensioactivos comunes son alquilsulfatos (por ejemplo, laurilsulfato de sodio), sales de alquiltrimetilamonio, alcohol etoxilados y similares. Los tensioactivos se pueden utilizar en una cantidad de 0 a 2 % en peso, preferentemente de 0,1 a 1,5 % en peso, basado en el peso total de la forma de dosificacion oral.
Se prefiere particularmente que la forma de dosificacion oral de la presente invencion no contenga un tensioactivo. Se pueden utilizar cargas (e) o diluyentes para aumentar el volumen aparente y el peso de un farmaco de dosis baja hasta un lfmite en el que se pueda formar una forma de dosificacion farmaceutica. Las cargas deben cumplir varios requisitos, como ser qmmicamente inertes, no higroscopicas, biocompatibles, facilmente procesables y con buenas propiedades biofarmaceuticas. Ejemplos de cargas son lactosa, sacarosa, glucosa, manitol, carbonato de calcio, celulosa y otros. Las cargas (e) se pueden utilizar en una cantidad de 0 a 75 % en peso, mas preferentemente de 5 a 40 % en peso, basado en el peso total de la forma de dosificacion.
Los aglutinantes (f) se pueden agregar a la formulacion farmaceutica con el fin de asegurar que las formas de dosificacion oral, preferentemente comprimidos, se puedan formar con la resistencia mecanica requerida. El aglutinante puede ser, por ejemplo, almidon, polivinilpirrolidona o derivados de celulosa. El agente aglutinante puede estar presente en una cantidad de 0 a 20 % en peso, preferentemente de 1 a 18 % en peso, mas preferentemente de 2 a 15 % en peso, en particular de 3 a 12 % en peso, basado en el peso total de la formulacion farmaceutica. Los desintegrantes (g) son compuestos que potencian la capacidad de la forma de dosificacion, preferentemente la capacidad del comprimido a romperse en fragmentos mas pequenos cuando estan en contacto con un lfquido, preferentemente agua. Los desintegrantes preferidos son carboximetilalmidon de sodio, polivinilpirrolidona reticulada (crospovidona), carboximetil glicolato de sodio (por ejemplo, Explotab®), polisacarido hinchable, por ejemplo polisacarido de soja, carragenina, agar, pectina, almidon y derivados de los mismos, protema, por ejemplo, formaldehndo-casema, bicarbonato de sodio, o mezclas de los mismos. Son mas preferidos carboximetilcelulosa sodica y la polivinilpirrolidona reticulada (crospovidona). Los desintegrantes se pueden utilizar en una cantidad de 0 a 15 % en peso, preferentemente de 1 a 2 % en peso, mas preferentemente de 3 a 10 % en peso, basado en el peso total de la forma de dosificacion.
Se notifica que la funcion de los lubricantes (h) asegura que la formacion y eyeccion de comprimidos pueda ocurrir con una baja friccion entre el solido y la pared de la matriz. Ademas, los lubricantes generalmente pueden aumentar la fluidez del polvo. El lubricante es preferentemente un estearato o acido graso, mas preferentemente un estearato de metal alcalinoterreo, tal como estearato de magnesio. El lubricante esta presente adecuadamente en una cantidad de 0 a 2 % en peso, preferentemente de aproximadamente 0,1 a 1,0 % en peso, basado en el peso total de la forma de dosificacion.
Los deslizantes (j) tambien se pueden utilizar para mejorar la fluidez. Tradicionalmente, el talco se utilizaba como deslizante, pero hoy en dfa esta casi completamente reemplazado con sflice coloidal (por ejemplo, Aerosil®). Preferentemente, el deslizante puede estar presente en una cantidad de 0 a 3 % en peso, mas preferentemente de 0,1 a 2,5 % en peso, en particular de 0,25 a 2,0 % en peso, basado en el peso total de la forma de dosificacion. La naturaleza de los excipientes farmaceuticos radica en que a veces pueden realizar mas de una funcion en una formulacion farmaceutica. A este respecto, generalmente se senala que, debido a la naturaleza de los excipientes farmaceuticos, no se puede excluir que un determinado compuesto cumpla con los requisitos de mas de uno de los componentes (b) o (c) y (d) a (j). Por lo tanto, la sflice coloidal (Aerosil) puede funcionar como un vehnculo para formar la composicion de acuerdo con la invencion, asf como un deslizante (j), es decir, el hecho de que la sflice coloidal se utilice como componente (c) para formar la composicion de acuerdo con la invencion no significa que no pueda actuar tambien como un deslizante (j).
Sin embargo, para permitir una distincion no ambigua, en la presente solicitud se prefiere que uno y el mismo
compuesto farmaceutico solo puedan funcionar como uno de los compuestos (b) o (c) y (d) a (j). Por ejemplo, si la celulosa microcristalina funciona como un vehnculo (c), no puede funcionar adicionalmente como un desintegrante (g), incluso aunque la celulosa microcristalina tambien exhiba un cierto efecto desintegrador.
La forma de dosificacion oral de la presente invencion puede comprender preferentemente las siguientes cantidades de componentes:
0,5 a 1.000 |jg de cebranopadol, preferentemente de 0,8 a 800 j de cebranopadol, mas preferentemente de 5 a 600 |jg de cebranopadol, particularmente de 10 a 400 jg, especialmente de 200 jg de cebranopadol,
0,01 a 150 mg de disolvente organico, preferentemente de 0,02 a 100 mg de disolvente organico, particularmente de 0,1 a 80 mg de disolvente organico,
0,005 a 150 mg de vehnculo, preferentemente de 0,01 a 120 mg de vehnculo, particularmente de 0,05 a 90 mg de vehnculo,
0 a 20 mg de tensioactivo, preferentemente de 2 a 15 mg de tensioactivo, particularmente de 4 a 10 mg de tensioactivo,
0 a 400 mg de carga, preferentemente de 25 a 300 mg de carga, particularmente de 40 a 200 mg de carga, de 0 a 150 mg de aglutinante, preferentemente de 15 a 100 mg de aglutinante, particularmente de 25 a 75 mg de aglutinante,
0 a 100 mg de desintegrante, preferentemente de 5 a 75 mg de desintegrante, particularmente de 10 a 50 mg de desintegrante,
0 a 15 mg de lubricante, preferentemente de 1 a 10 mg de lubricante, particularmente de 2 a 8 mg de lubricante, 0 a 25 mg de deslizante, preferentemente de 1 a 15 mg de deslizante, particularmente de 2 a 7 mg de deslizante. En una realizacion preferida, la forma de dosificacion oral de la presente invencion puede comprender preferentemente:
0,005 a 2 % en peso de cebranopadol, preferentemente de 0,01 a 1,5 % en peso de cebranopadol, particularmente de 0,02 a 1 % en peso de cebranopadol,
0,1 a 30% en peso de disolvente organico, preferentemente de 2 a 28% en peso de disolvente organico, particularmente de 5 a 25 % en peso de disolvente organico,
0,1 a 28 % en peso de vetnculo, preferentemente de 2 a 26 % en peso de vehnculo, particularmente de 5 a 24 % en peso de vehnculo,
0 a 2 % en peso de tensioactivo, preferentemente de 0,05 a 1,66 % en peso de tensioactivo, particularmente de 0,1 a 1,5 % en peso de tensioactivo,
0 a 75 % en peso de carga, preferentemente de 3 a 65 % en peso de carga, particularmente de 5 a 40 % en peso de carga,
0 a 20 % en peso de aglomerante, preferentemente de 2 a 15 % en peso de aglomerante, particularmente de 3 a 12 % en peso de aglomerante,
0 a 15 % en peso de desintegrante, preferentemente de 1 a 12 % en peso de desintegrante, particularmente de 3 a 10 % en peso de desintegrante,
0 a 2 % en peso de lubricante, preferentemente 0,1 a 1,0 % en peso de lubricante, particularmente de 0,2 a 0,8 % en peso de lubricante,
0 a 3 % en peso de deslizante, preferentemente de 0,1 a 2,5 % en peso de deslizante, particularmente de 0,25 a 2,0 % en peso de deslizante,
basado en el peso total de la forma de dosificacion oral.
En una realizacion adicional de la presente invencion, la forma de dosificacion oral puede ser una capsula o un comprimido, mas preferentemente un comprimido, para uso peroral. Alternativamente, la forma de dosificacion oral solida se puede rellenar como un polvo o granulado en dispositivos similares a sobres o sobres tubulares.
La presente invencion se refiere ademas a un metodo para producir una composicion de acuerdo con la invencion. Por ende, un objeto adicional de la presente invencion es un metodo para producir una composicion que comprende cebranopadol disuelto (a), disolvente organico (b) y vehnculo (c) que comprende las etapas que consisten en i) disolver cebranopadol en un disolvente organico con un punto de ebullicion de 110° a 350 °C (b),
ii) mezclar el vehnculo (c) y la solucion de la etapa que consiste en i), y
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii).
En general, los comentarios realizados anteriormente para cebranopadol, disolvente organico y vehnculo tambien pueden aplicarse al metodo de la presente invencion.
En la etapa i) del metodo de la invencion, el cebranopadol (a) se disuelve en un disolvente organico (b).
El termino "disolucion" significa que una sustancia, tal como cebranopadol, se pone en contacto con el disolvente, preferentemente con un disolvente o mezcla de disolventes como se ha definido anteriormente para el compuesto (b), p. ej., un glicofurol, en el que el disolvente moja la superficie de la sustancia o la sustancia se puede disolver
completamente en el disolvente. Cuando se obtiene una solucion clara y no se puede detectar cebranopadol (cristales) por control visual, esto puede considerarse como una disolucion completa de cebranopadol en un disolvente organico.
En una realizacion preferida, el cebranopadol se anade preferentemente a un disolvente organico. Ademas, se prefiere que el disolvente organico se agite y/o caliente preferentemente, de manera preferente a una temperatura de aproximadamente 80 °C.
En una realizacion preferida, cebranopadol (a) se puede disolver en un disolvente organico (b), preferentemente bajo agitacion durante la etapa de disolucion, preferentemente a una velocidad de agitado de 300 a 450 rpm (rotaciones por minuto). Adicionalmente, se prefiere que el disolvente este a una temperatura elevada, preferentemente a aproximadamente 80 °C, durante la etapa de disolucion. Ademas, el cebranopadol se anade preferentemente en forma cristalina.
Ademas, para apoyar la formacion de la solucion de la etapa i), el cebranopadol en el disolvente organico puede someterse a un tratamiento mecanico, tal como un tratamiento ultrasonico. Generalmente, el tratamiento ultrasonico se puede llevar a cabo sumergiendo cebranopadol y disolvente organico en un dispositivo ultrasonico, por ejemplo, en un bano ultrasonico. Ejemplos de tratamiento ultrasonico son la cavitacion hidrodinamica, la fragmentacion sonora y/o la cavitacion sonora o la molienda simultanea. Por ejemplo, el tratamiento ultrasonico se puede llevar a cabo con el equipo ultrasonico Tesla.
El tratamiento ultrasonico puede realizarse preferentemente utilizando ondas ultrasonicas que tengan una frecuencia de 5 a 100 kHz, mas preferentemente de l0 a 80 kHz. Ademas, el tratamiento ultrasonico se realiza preferentemente mediante el uso de ondas ultrasonicas que tienen una intensidad de 50 a 5.000 W, mas preferentemente de 500 a 1.000 W. Como ejemplo, se pueden utilizar 1.000 W y 20 kHz o 500 W y 58 kHz.
Normalmente, el tratamiento mecanico puede llevarse a cabo durante 1 a 30 minutos, preferentemente durante 5 a 20 minutos.
Una vez que el cebranopadol se disuelve completamente en el disolvente organico (b) en la etapa ii), el vehfculo (c) y la solucion de la etapa i) se pueden mezclar.
En una realizacion preferida, el vehfculo (c) se anade a la solucion de la etapa i). Se prefiere que la solucion de la etapa i) este a temperatura elevada, preferentemente a aproximadamente 80 °C, cuando se lleva a cabo la mezcla con el vehfculo (c). Ademas, la solucion se agita preferentemente, de manera preferente a una velocidad de agitado de 300 a 550 rpm (rotaciones por minuto) durante la etapa de mezcla ii).
En una realizacion preferida, la mezcla de la etapa ii), preferentemente despues de dejar enfriar a 23 °C, se puede obtener como un material similar al polvo.
En una realizacion alternativa, se puede anadir mas agente activo farmaceutico a la mezcla entre la etapa ii) y la etapa opcional iii).
En la etapa opcional iii), la mezcla de la etapa ii) se puede moler y/o tamizar preferentemente.
El molido se puede realizar preferentemente en aparatos de molido convencionales, tales como en un molino de bolas, molino de chorro de aire, molino de puas, molino clasificador, molino batidor de martillos cruzados, molino de disco, molino de mortero o un molino de rotor. Preferentemente se utiliza un molino de bolas planetario.
El tiempo de molienda es preferible de 0,5 minutos a 30 minutos, preferentemente de 1 a 15 minutos, mas preferentemente de 3 a 7 minutos.
Es preferible que el tamizado de la mezcla de la etapa ii) se realice con un tamiz que tenga un tamano de malla de 25 a 1000 pm, preferentemente de 50 a 800 pm, especialmente de 100 a 600 pm.
Ademas, el objeto de la presente invencion se refiere a un metodo para preparar la dosificacion oral de la invencion que comprende las etapas que consisten en:
i) disolver cebranopadol en un disolvente organico con un punto de ebullicion de 110° a 350 °C (b),
ii) mezclar el vehfculo (c) y la solucion de la etapa i),
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii),
iv) opcionalmente, agregar otro(s) excipiente(s) a la mezcla de la etapa ii) o la etapa iii),
v) opcionalmente, granular la mezcla de la etapa ii), la etapa iii) o la etapa iv),
vi) procesar la mezcla de la etapa ii), la etapa iii) o la etapa iv) o los granulados de la etapa v) en una forma de dosificacion oral.
En las etapas i) a iii) se proporciona una composicion de acuerdo con la presente invencion, es dedr, todas las etapas de procedimiento anteriores i), ii) y iii) que conducen a la presente composicion tambien se aplican al procedimiento de preparacion de la presente forma de dosificacion oral.
En la etapa iv) se pueden agregar opcionalmente otros excipientes adicionales y/o mas agentes farmaceuticamente activos a la mezcla de la etapa ii) o etapa iii). Durante o despues de la adicion de los excipientes opcionales y/o el agente farmaceuticamente activo adicional, la mezcla resultante se puede combinar preferentemente. Los excipientes pueden seleccionarse preferentemente entre los excipientes (d), (e), (f), (g), (h) y (j) como se ha descrito anteriormente.
En la etapa v), la mezcla de la etapa ii), la etapa iii) o la etapa iv) se puede granular opcionalmente.
Por "granulacion" se entiende generalmente la formacion de un material de agregado granular o relativamente grueso en forma de polvo mediante el ensamblaje y/o agregacion de partfculas de polvo mas finas (formacion de aglomerados o granulacion acumulada) y/o la formacion de granulos mas finos mediante la separacion de agregados mas gruesos (desintegracion o granulacion por descomposicion).
La granulacion puede significar convencionalmente granulacion en humedo o en seco.
La granulacion en seco, que es preferente, se lleva a cabo generalmente utilizando presion. En una realizacion preferida de la invencion, la granulacion de la mezcla de la etapa iv) se puede realizar, por ejemplo, mediante "agitacion", utilizando una gran prensa rotativa pesada y rompiendo las capsulas en granulados con un molino de martillos o mediante compactacion con rodillos, utilizando, por ejemplo, compactadores de rodillos de Powtec o Alexanderwerk. Los granulados se tamizan opcionalmente.
En la etapa vi), la mezcla de la etapa ii), la etapa iii), la etapa iv) o los granulados de la etapa v) se procesan en una forma de dosificacion oral solida. El procesamiento de la mezcla de la etapa ii), la etapa iii) o la etapa iv) o los granulados de la etapa v) en una forma de dosificacion oral solida puede comprender preferentemente llenar dicha mezcla en capsulas, preferentemente capsulas de gelatina dura. Opcionalmente, el procesamiento de la mezcla de la etapa ii), la etapa iii) o la etapa iv) o los granulados de la etapa v) en comprimidos se puede llevar a cabo comprimiendo dicha formulacion en una prensa rotativa, p. ej., en una Fette® (Fette GmbH, Alemania) o una Riva® piccola (Riva, Argentina). La fuerza de compresion principal puede variar de 1 a 50 kN, preferentemente de 3 a 40 kN. Los comprimidos resultantes pueden tener una dureza de 30 a 400, mas preferentemente de 50 a 250 N, en particular preferentemente de 30 a 180 N, mas preferentemente de 40 a 150 N, en los que la dureza se puede medir de acuerdo con Ph. Eur. 6.0, Capftulo 2.9.8. Para el llenado opcional de la formulacion en capsulas, se pueden utilizar sistemas de dosificacion dependientes (por ejemplo, una barrena) o, preferentemente, sistemas de dosificacion independientes (por ejemplo, MG2, Matic (IMA)).
Ademas, la forma de dosificacion, preferentemente el comprimido, de la invencion tiene preferentemente una uniformidad de contenido, es decir, un contenido de agente(s) activo(s), que se encuentra dentro de la concentracion de 90 a 110 %, preferentemente de 95 a 105 %, especialmente preferido de 98 a 102 % del contenido medio de los agentes activos. La "uniformidad del contenido" se determina con una prueba de acuerdo con Ph. Eur., 6.0, Capftulo 2.9.6. De acuerdo con esa prueba, el contenido de agente(s) activo(s) de cada comprimido individual de 20 comprimidos ha de estar entre 90 y 110 %, preferentemente entre 95 y 105 %, especialmente entre 98 y 102 % del contenido medio de agentes activos. Por lo tanto, el contenido de los farmacos activos en cada comprimido de la invencion difiere del contenido medio del agente activo como maximo en un 10 %, preferentemente como maximo en un 5 % y especialmente como maximo en un 2 %.
Ademas, el comprimido resultante tiene preferentemente una friabilidad inferior al 5 %, en particular preferentemente inferior al 2 %, especialmente inferior al 1 %. La friabilidad se determina de acuerdo con Ph. Eur., 6.0, Capftulo 2.9.7. La friabilidad de los comprimidos se refiere generalmente a comprimidos sin recubrimiento.
La formulacion farmaceutica de la invencion puede ser un comprimido peroral que puede tragarse sin masticar. El comprimido puede estar recubierto preferentemente con una peftcula.
Generalmente, los recubrimientos de peftcula que no afectan la liberacion del(de los) agente(s) activo(s) y los recubrimientos de peftcula que afectan la liberacion del(de los) agente(s) activo(s) pueden emplearse con comprimidos de acuerdo con la invencion. Se prefieren los recubrimientos de peftcula que no afectan la liberacion del(de los) agente(s) activo(s).
Los ejemplos preferidos de recubrimientos de peftcula que no afectan la liberacion del principio activo pueden ser aquellos que incluyen poli(met)acrilato, metilcelulosa (MC), hidroxipropilmetilcelulosa (HPMC), hidroxipropilcelulosa (HPC), hidroxietilcelulosa (HEC), polivinilpirrolidona (PVP) y mezclas de los mismos. Estos poftmeros pueden tener un peso molecular promedio en peso de 10.000 a 150.000 g/mol.
En una realizacion preferida alternativa, el recubrimiento de peftcula puede afectar la liberacion del agente activo.
Ejemplos de recubrimientos de peKcula que afectan la liberacion del agente activo son los recubrimientos de peKcula resistentes a los jugos gastricos y los recubrimientos retardados.
En una realizacion preferida, la pelfcula puede tener un espesor de 2 pm a 150 pm, preferentemente de 10 a 100 pm, mas preferentemente de 20 a 60 pm.
En una realizacion preferida, la forma de dosificacion de la invencion es para liberacion modificada. En ese caso, el perfil de liberacion de la formulacion farmaceutica, preferentemente del comprimido, de acuerdo con el metodo USP (aparato de alabe USP, 900 ml de medio de prueba, en tampon fosfato a pH 6,8 y 37 °C, 100 rpm) despues de 2 horas indica una liberacion de contenido de 0 a 90 %, preferentemente de 10 a 80 %, mas preferentemente de 15 a 75 %, mas preferentemente de 20 a 50 % y particularmente de 25 a 40 %.
En una realizacion alternativa preferida de la invencion, la forma de dosificacion es para liberacion inmediata. En ese caso, el perfil de liberacion de la formulacion farmaceutica, preferentemente del comprimido, de acuerdo con el metodo de la USP (aparato de alabe USP, 900 ml de medio de prueba, en tampon fosfato a pH 6,8 a 37 °C, 100 rpm) despues de 15 minutos indica una liberacion de contenido de al menos 50 %, preferentemente al menos 70 %, especialmente al menos 90 %.
Ademas, la invencion se refiere a un metodo para tratar el dolor cronico que incluye dolor neuropatico, como tal en pacientes con tumor(es), que comprende administrar a un sujeto en necesidad del mismo una cantidad terapeuticamente eficaz de una composicion farmaceutica que comprende (a) cebranopadol disuelto, (b) disolvente organico con un punto de ebullicion de aproximadamente 110 °C a 350 °C, y (c) vehfculo solido. Para la composicion farmaceutica administrada en el metodo mencionado anteriormente, lo mismo se aplica a la composicion como se ha descrito anteriormente en el texto.
Parte experimental
Metodos analiticos:
Las composiciones de acuerdo con la invencion se examinaron mediante difraccion de rayos X en polvo.
Difraccion de rayos X en polvo
Las mediciones se realizaron de la siguiente manera: Las muestras se midieron en un difractometro de rayos X en polvo D8 Advance (Bruker-AXS, Karlsruhe, Alemania) en un soporte de muestra PMMA (diametro: 25 m; profundidad: 1 mm) girando a 20 rpm durante la medicion en modo reflexion (geometna Bragg-Brentano). Las condiciones adicionales para las mediciones se resumen a continuacion. Los datos en bruto se analizaron con el programa EVA (Bruker-AXS, Karlsruhe, Alemania). No se realizaron sustraccion de fondo y separacion de Ka2. radiacion Cu Ka1/a2
fuente 34 kV/40 mA
detector Vantec-1 (ventana electronica: 3°) filtro kp Ni (haz difractado)
medicion del diametro del drculo 435 mm
ranura de ventana de detector 12 mm
ranura anti-dispersion 8 mm
ranura de divergencia v6.00 (variable)
ranura de soller (haz incidente/difractado) 2,5°
intervalo 20 2°<20<55°
tamano de etapa 0,016°
tiempo de etapa 0,2 s
Ejemplos
El cebranopadol se disolvio en disolvente a 80 °C. El material del vehfculo se anadio en porciones (0,05 g) a 80 °C hasta que la mezcla fuera un polvo homogeneo que flrna libremente. La composicion se dejo enfriar a temperatura ambiente (23 °C).
Las cantidades empleadas y el estado solido resultante de las formulaciones se especifican en la Tabla 1.
Tabla 1: Composicion y analisis de estado solido para formulaciones de cebranopadol


RESULTADOS
La Figura A-1 muestra una descripcion general de los difractogramas de rayos X en polvo de todos los vehnculos utilizados.
En la Figura A-2 se muestra el difractograma del Ejemplo 1. El difractograma se parece esencialmente al difractograma de Neusilin® puro (comparese con la Figura A-1).
En la Figura A-3 se muestra el difractograma del Ejemplo 2. Nuevamente, el difractograma se parece esencialmente al difractograma de SBA-16 puro (comparese con la Figura A-1).
En la Figura A-4 se muestra el difractograma del Ejemplo 3. El difractograma se parece esencialmente al difractograma de SBA-15 puro (comparese con la Figura A-1).
En la Figura A-5 se muestra el difractograma del Ejemplo 4. El difractograma se parece esencialmente al difractograma de MSU-F puro (comparese con la Figura A-1).
En la Figura A-6 se muestra el difractograma del Ejemplo 5. El difractograma se parece esencialmente al difractograma de MCM-48 puro (comparese con la Figura A-1).
En la Figura A-7 se muestra el difractograma del Ejemplo 6. El difractograma se parece esencialmente al difractograma de A1-MCM-41 puro (comparese con la Figura A-1).
En la Figura A-8 se muestra el difractograma del Ejemplo 7. El difractograma se parece esencialmente al difractograma de Celite puro (comparese con la Figura A-1).
En la Figura A-9 se muestra el difractograma del Ejemplo 8. El difractograma se parece esencialmente al difractograma de Aeroperl® 300 puro (comparese con la Figura A-1).
El disolvente organico glicofurol (polietilenglicol tetrahidrofurfuril eter) tiene un punto de ebullicion de 100-145 °C a 0,4 Hg. Aunque se obtuvo una alta concentracion posible de cebranopadol en solucion, no hay indicios de que el cebranopadol cristalice en la presente muestra, es decir, el cebranopadol permanece disuelto.
En la Figura A-10 se muestra el difractograma del Ejemplo 9. El difractograma parece esencialmente al difractograma de Neusilin® puro (comparese con la Figura A-1).
Se obtienen resultados similares para los otros ejemplos 10 a 16.
El disolvente organico dietilenglicol monoetil eter tiene un punto de ebullicion de 202 °C a presion atmosferica. Aunque se obtuvo una alta concentracion posible de cebranopadol en solucion, no hay indicios de que el
cebranopadol cristalice en la presente muestra, es dedr, el cebranopadol permanece disuelto.
Ċ
Claims (14)
1. Composicion farmaceutica que comprende:
(a) cebranopadol disuelto,
(b) disolvente organico con un punto de ebullicion de 110° a 350 °C, y
(c) vehnculo solido.
2. Composicion de acuerdo con la reivindicacion 1, en la que el disolvente organico (b) tiene un punto de ebullicion de 170 °C a 345 °C.
3. Composicion de acuerdo con la reivindicacion 1 o 2, en la que el disolvente organico (b) tiene una densidad de 0,95 a 1,30 g/ml, medida a 20 °C.
4. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en la que la relacion en peso de cebranopadol disuelto (a) a disolvente organico (b) es de 1:1 a 1:150.
5. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en la que el vehnculo (c) es una sustancia quebradiza, que tiene una presion de fluencia de 80 MPa a 500 MPa.
6. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 5, en la que el vehnculo (c) es un polfmero organico o una sustancia inorganica.
7. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 6, en la que el vehnculo (c) posee un area superficial espedfica de 75 a 350 mi) 2/g, por lo que el area superficial espedfica se mide de acuerdo con Ph. Eur. 6.0, 2.9.26.
8. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 7, en la que el vehnculo (c) es un silicato, preferentemente un aluminosilicato de magnesio o sflice mesoporosa.
9. Composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 8, en la que la relacion en peso de cebranopadol disuelto (a) a vehnculo (c) es de 1:1 a 1:100.
10. Forma de dosificacion oral que comprende una composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 9 y opcionalmente otro(s) excipiente(s) farmaceutico(s).
11. Forma de dosificacion oral de acuerdo con la reivindicacion 10, en la que la forma de dosificacion comprende: 0,005 a 2 % en peso de cebranopadol, preferentemente de 0,01 a 1,5 % en peso de cebranopadol, particularmente de 0,02 a 1 % en peso de cebranopadol,
0,1 a 30 % en peso de disolvente organico, preferentemente de 2 a 28 % en peso de disolvente organico,
0,1 a 28 % en peso de vehnculo, preferentemente de 2 a 26 % en peso de vehnculo,
0 a 2 % en peso de tensioactivo, preferentemente de 0,05 a 1,6 % en peso de tensioactivo,
0 a 75 % en peso de carga, preferentemente de 3 a 65 % en peso de carga,
0 a 20 % en peso de aglutinante, preferentemente de 2 a 15 % en peso de aglutinante,
0 a 15 % en peso de desintegrante, preferentemente de 1 a 12 % en peso de desintegrante,
0 a 2 % en peso de lubricante, preferentemente de 0,1 a 1,0 % en peso de lubricante,
0 a 3 % en peso de deslizante, preferentemente de 0,1 a 2,5 % en peso de deslizante,
basado en el peso total de la forma de dosificacion oral.
12. Metodo para producir una composicion de acuerdo con una cualquiera de las reivindicaciones 1 a 9, que comprende las etapas que consisten en:
i) disolver cebranopadol (a) en un disolvente organico con un punto de ebullicion de 110 °C a 350 °C (b) ii) mezclar un vehnculo (c) y la solucion de la etapa i)
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii).
13. Metodo para producir una forma de dosificacion oral de acuerdo con la reivindicacion 10 u 11 que comprende las etapas que consisten en:
i) disolver cebranopadol (a) en un disolvente organico con un punto de ebullicion de 110 °C a 350 °C (b) ii) mezclar un vehnculo (c) y la solucion de la etapa i)
iii) opcionalmente, moler y/o tamizar la mezcla de la etapa ii)
iv) opcionalmente, agregar otro(s) excipiente(s) a la mezcla de la etapa ii) o la etapa iii),
v) opcionalmente, granular la mezcla de la etapa ii), la etapa iii) o la etapa iv)
vi) procesar la mezcla de la etapa ii), la etapa iii) o la etapa iv) o los granulados de la etapa v) en una forma de
dosificacion oral.
14. Una composicion de acuerdo con la reivindicacion 1 para su uso en el tratamiento de dolor cronico que incluye dolor neuropatico, preferentemente en pacientes con tumor(es).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15153405 | 2015-02-02 | ||
| PCT/EP2016/052017 WO2016124513A1 (en) | 2015-02-02 | 2016-02-01 | Composition comprising cebranopadol in a dissolved form |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2713960T3 true ES2713960T3 (es) | 2019-05-24 |
Family
ID=52440596
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16706540T Active ES2713960T3 (es) | 2015-02-02 | 2016-02-01 | Composición que comprende cebranopadol en forma disuelta |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20180021304A1 (es) |
| EP (1) | EP3253374B1 (es) |
| ES (1) | ES2713960T3 (es) |
| WO (1) | WO2016124513A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT201700057899A1 (it) | 2017-05-29 | 2018-11-29 | Univ Degli Studi Di Camerino | Co-attivatori dei recettori MOP e NOP nel trattamento della dipendenza da droghe e/o psicostimolanti |
| CN109589418B (zh) * | 2018-12-14 | 2020-08-18 | 华南理工大学 | 一种具有pH响应性的席夫碱共聚物包覆的介孔二氧化硅载药纳米粒子及其制备方法和应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10252667A1 (de) | 2002-11-11 | 2004-05-27 | Grünenthal GmbH | Spirocyclische Cyclohexan-Derivate |
| CA2718255C (en) * | 2008-03-11 | 2016-08-23 | Aska Pharmaceutical Co., Ltd. | Solid dispersion and pharmaceutical composition of the same, and production processes thereof |
| RS54438B1 (sr) | 2010-08-04 | 2016-06-30 | Grünenthal GmbH | Farmaceutski dozni oblik koji sadrži 6`-fluoro-(n-metil-ili n,n-dimetil-)-4-fenil-4`,9`-dihidro-3`h-spiro[cikloheksan-1,1`-pirano[3,4,b]indol]-4-amina |
| CN106939005A (zh) | 2011-07-08 | 2017-07-11 | 格吕伦塔尔有限公司 | 晶态的(1r,4r)‑6’‑氟‑N,N‑二甲基‑4‑苯基‑4’,9’‑二氢‑3’H‑螺[环己烷‑1,1’‑吡喃并[3,4,b]吲哚]‑4‑胺 |
| GB201115079D0 (en) * | 2011-08-31 | 2011-10-19 | Iota Nanosolutions Ltd | Method of preparing carrier liquids |
-
2016
- 2016-02-01 ES ES16706540T patent/ES2713960T3/es active Active
- 2016-02-01 WO PCT/EP2016/052017 patent/WO2016124513A1/en not_active Ceased
- 2016-02-01 US US15/545,738 patent/US20180021304A1/en not_active Abandoned
- 2016-02-01 EP EP16706540.8A patent/EP3253374B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3253374B1 (en) | 2018-11-28 |
| WO2016124513A1 (en) | 2016-08-11 |
| EP3253374A1 (en) | 2017-12-13 |
| US20180021304A1 (en) | 2018-01-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI727949B (zh) | 調節蛋白質激酶之化合物的固態形式 | |
| JP7172997B2 (ja) | エンザルタミドを含有する経口投与用医薬組成物 | |
| JP2000007566A (ja) | ジプラシドン組成物 | |
| EA006777B1 (ru) | Фармацевтические композиции адсорбатов аморфного лекарственного средства | |
| RS56408B1 (sr) | Čvrsti oblici doziranja bendamustina | |
| JP2020514291A (ja) | Rad1901−2hclの多形性形態 | |
| WO2008072534A1 (ja) | マンニトール又は乳糖を含有する固形製剤 | |
| JP2022506062A (ja) | 置換されたインダンを含む固体分散体及び医薬組成物並びにそれの製造方法及び使用 | |
| JP6964195B2 (ja) | 経口生体利用率を改善するための薬物−層状シリケート複合体、これを含む経口用薬学組成物、及び前記複合体の製造方法 | |
| US20220002306A1 (en) | Crystal form of upadacitinib and preparation method and use thereof | |
| ES2971644T3 (es) | Sales de 4-amino-N-(1-((3-cloro-2-fluorofenil)amino)-6-metilisoquinolin-5-il)tieno[3,2-d]pirimidin-7-carboxamida y formas cristalinas de las mismas | |
| EP3697418A1 (en) | Improved bromocriptine formulations | |
| CN111615389A (zh) | 药物调配物 | |
| US20160136112A1 (en) | Composition comprising tapentadol in a dissolved form | |
| CA3159315A1 (en) | Ahr inhibitors and uses thereof | |
| TW201840320A (zh) | 包含5-氯-n4-[2-(二甲基磷醯基)苯基]-n2-{2-甲氧基-4-[4-(4-甲基哌嗪-1-基)哌啶-1-基]苯基}嘧啶-2,4-二胺之醫藥調配物 | |
| ES2713960T3 (es) | Composición que comprende cebranopadol en forma disuelta | |
| WO2016175230A1 (ja) | 経口投与用医薬組成物 | |
| BR112021008732A2 (pt) | ingredientes farmacêuticos amorfos ativos compreendendo carbonato de magnésio mesoporoso substancialmente amorfo | |
| TW202541806A (zh) | 維賽替尼之調配物 | |
| CN109963565B (zh) | 一种药物组合物及其制备方法 | |
| CN101884791B (zh) | 一种来曲唑共研磨物及其制备方法和含其药物组合物 | |
| EP1753400A2 (en) | Ziprasidone dosage form | |
| ES2642162T3 (es) | Composición que comprende vemurafenib y HPMC-AS | |
| WO2019105359A1 (zh) | Acalabrutinib的晶型及其制备方法和用途 |