ES2746105T3 - Profármacos de 5-aza-pirimidina sililada útiles para tratar el cáncer - Google Patents
Profármacos de 5-aza-pirimidina sililada útiles para tratar el cáncer Download PDFInfo
- Publication number
- ES2746105T3 ES2746105T3 ES15849800T ES15849800T ES2746105T3 ES 2746105 T3 ES2746105 T3 ES 2746105T3 ES 15849800 T ES15849800 T ES 15849800T ES 15849800 T ES15849800 T ES 15849800T ES 2746105 T3 ES2746105 T3 ES 2746105T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- silylated
- hydrogen
- pyrimidine
- independently selected
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims description 32
- 201000011510 cancer Diseases 0.000 title claims description 12
- 239000000651 prodrug Substances 0.000 title description 59
- 229940002612 prodrug Drugs 0.000 title description 59
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 title description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 63
- 239000001257 hydrogen Substances 0.000 claims abstract description 58
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 58
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 34
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 29
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 23
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 201000009030 Carcinoma Diseases 0.000 claims description 7
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 201000005787 hematologic cancer Diseases 0.000 claims description 5
- 208000002250 Hematologic Neoplasms Diseases 0.000 claims description 4
- 210000000481 breast Anatomy 0.000 claims description 4
- 210000001072 colon Anatomy 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 210000004072 lung Anatomy 0.000 claims description 3
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 claims 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 37
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- 239000000203 mixture Substances 0.000 description 26
- 150000003230 pyrimidines Chemical class 0.000 description 24
- -1 silyl gemcitabine Chemical compound 0.000 description 24
- 238000000034 method Methods 0.000 description 22
- 238000002360 preparation method Methods 0.000 description 21
- 239000011541 reaction mixture Substances 0.000 description 16
- 230000001225 therapeutic effect Effects 0.000 description 16
- GQGVBSHMRYHBTF-UOWFLXDJSA-N 4-amino-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2-one Chemical compound O=C1N=C(N)N=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 GQGVBSHMRYHBTF-UOWFLXDJSA-N 0.000 description 15
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 12
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 12
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 10
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 10
- YXSLMHOPYBUDME-TZLGMAPXSA-N 4-amino-1-[(2r,4s,5r)-4-benzoyl-3,3-difluoro-4-hydroxy-5-(1-hydroxy-2-oxo-2-phenylethyl)oxolan-2-yl]-1,3,5-triazin-2-one Chemical compound O=C1N=C(N)N=CN1[C@H]1C(F)(F)[C@@](O)(C(=O)C=2C=CC=CC=2)[C@@H](C(O)C(=O)C=2C=CC=CC=2)O1 YXSLMHOPYBUDME-TZLGMAPXSA-N 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 9
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 8
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 8
- VKIGAWAEXPTIOL-UHFFFAOYSA-N 2-hydroxyhexanenitrile Chemical compound CCCCC(O)C#N VKIGAWAEXPTIOL-UHFFFAOYSA-N 0.000 description 7
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 7
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 7
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 238000004007 reversed phase HPLC Methods 0.000 description 7
- 238000011282 treatment Methods 0.000 description 7
- 229960003962 trifluridine Drugs 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- MFEFTTYGMZOIKO-UHFFFAOYSA-N 5-azacytosine Chemical group NC1=NC=NC(=O)N1 MFEFTTYGMZOIKO-UHFFFAOYSA-N 0.000 description 6
- BVSDIUUYQZNNRW-UOWFLXDJSA-N 6-amino-3-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,4-dihydro-1,3,5-triazin-2-one Chemical compound C1NC(N)=NC(=O)N1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 BVSDIUUYQZNNRW-UOWFLXDJSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N acetonitrile Substances CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 229940125904 compound 1 Drugs 0.000 description 6
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 6
- 229960005277 gemcitabine Drugs 0.000 description 6
- 230000002209 hydrophobic effect Effects 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical class O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 239000002777 nucleoside Substances 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical group NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 230000013595 glycosylation Effects 0.000 description 4
- 238000006206 glycosylation reaction Methods 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000007908 nanoemulsion Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 0 *C([C@@](C[N-])N[C@@]1N(C=C(C(F)(F)F)C(N2)=N)C2=[U])C1(F)F Chemical compound *C([C@@](C[N-])N[C@@]1N(C=C(C(F)(F)F)C(N2)=N)C2=[U])C1(F)F 0.000 description 3
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 3
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 3
- 235000011130 ammonium sulphate Nutrition 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 125000004665 trialkylsilyl group Chemical group 0.000 description 3
- VSQQQLOSPVPRAZ-RRKCRQDMSA-N trifluridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(C(F)(F)F)=C1 VSQQQLOSPVPRAZ-RRKCRQDMSA-N 0.000 description 3
- GJJVAFUKOBZPCB-ZGRPYONQSA-N (r)-3,4-dihydro-2-methyl-2-(4,8,12-trimethyl-3,7,11-tridecatrienyl)-2h-1-benzopyran-6-ol Chemical class OC1=CC=C2OC(CC/C=C(C)/CC/C=C(C)/CCC=C(C)C)(C)CCC2=C1 GJJVAFUKOBZPCB-ZGRPYONQSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 2
- DRSHXJFUUPIBHX-UHFFFAOYSA-N COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 Chemical compound COc1ccc(cc1)N1N=CC2C=NC(Nc3cc(OC)c(OC)c(OCCCN4CCN(C)CC4)c3)=NC12 DRSHXJFUUPIBHX-UHFFFAOYSA-N 0.000 description 2
- VCNVMGCFHZBMBU-ZGDXACBUSA-N C[Si]([C@@]1([C@@H](O[C@@H]([C@]1(O)[Si](C)(C)C)C(O)[Si](C)(C)C)N1C(=O)N=C(N)N=C1)O)(C)C Chemical compound C[Si]([C@@]1([C@@H](O[C@@H]([C@]1(O)[Si](C)(C)C)C(O)[Si](C)(C)C)N1C(=O)N=C(N)N=C1)O)(C)C VCNVMGCFHZBMBU-ZGDXACBUSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 2
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- FIWILGQIZHDAQG-UHFFFAOYSA-N NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F Chemical compound NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F FIWILGQIZHDAQG-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 102000028649 Ribonucleoside-diphosphate reductase Human genes 0.000 description 2
- 108010038105 Ribonucleoside-diphosphate reductase Proteins 0.000 description 2
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical class O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 229960002756 azacitidine Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 150000002191 fatty alcohols Chemical class 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 230000003301 hydrolyzing effect Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 229930003799 tocopherol Natural products 0.000 description 2
- 239000011732 tocopherol Substances 0.000 description 2
- 229930003802 tocotrienol Natural products 0.000 description 2
- 239000011731 tocotrienol Substances 0.000 description 2
- 229940068778 tocotrienols Drugs 0.000 description 2
- 235000019148 tocotrienols Nutrition 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical class CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical compound OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 1
- OKMWKBLSFKFYGZ-UHFFFAOYSA-N 1-behenoylglycerol Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(O)CO OKMWKBLSFKFYGZ-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- IAJINJSFYTZPEJ-UHFFFAOYSA-N 1h-pyrimidin-3-ium-2-one;chloride Chemical compound Cl.O=C1N=CC=CN1 IAJINJSFYTZPEJ-UHFFFAOYSA-N 0.000 description 1
- 239000000263 2,3-dihydroxypropyl (Z)-octadec-9-enoate Substances 0.000 description 1
- RZRNAYUHWVFMIP-GDCKJWNLSA-N 3-oleoyl-sn-glycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-GDCKJWNLSA-N 0.000 description 1
- LMNPKIOZMGYQIU-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-pyrimidine-2,4-dione Chemical compound FC(F)(F)C1=CNC(=O)NC1=O LMNPKIOZMGYQIU-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- LAOLDMMWVYDDID-KVQBGUIXSA-N 6-amino-3-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,4-dihydro-1,3,5-triazin-2-one Chemical compound C1NC(N)=NC(=O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 LAOLDMMWVYDDID-KVQBGUIXSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- DYKDDBLOMATDSD-AMXGMDGSSA-N C[Si](C)(C)[C@@]1(C([C@H](O)[C@@H](CO)O1)(F)F)N1C(=O)N=C(N)N=C1 Chemical compound C[Si](C)(C)[C@@]1(C([C@H](O)[C@@H](CO)O1)(F)F)N1C(=O)N=C(N)N=C1 DYKDDBLOMATDSD-AMXGMDGSSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- ZAKOWWREFLAJOT-CEFNRUSXSA-N D-alpha-tocopherylacetate Chemical compound CC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-CEFNRUSXSA-N 0.000 description 1
- 230000007067 DNA methylation Effects 0.000 description 1
- ASMQGLCHMVWBQR-UHFFFAOYSA-N Diphenyl phosphate Chemical class C=1C=CC=CC=1OP(=O)(O)OC1=CC=CC=C1 ASMQGLCHMVWBQR-UHFFFAOYSA-N 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 229920002685 Polyoxyl 35CastorOil Polymers 0.000 description 1
- 102000001853 Pyrimidine Phosphorylases Human genes 0.000 description 1
- 108010054917 Pyrimidine Phosphorylases Proteins 0.000 description 1
- 235000019484 Rapeseed oil Nutrition 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- 229910018557 Si O Inorganic materials 0.000 description 1
- NWGKJDSIEKMTRX-AAZCQSIUSA-N Sorbitan monooleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O NWGKJDSIEKMTRX-AAZCQSIUSA-N 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- 229940122020 Thymidine phosphorylase inhibitor Drugs 0.000 description 1
- 102000005497 Thymidylate Synthase Human genes 0.000 description 1
- AOBORMOPSGHCAX-UHFFFAOYSA-N Tocophersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-UHFFFAOYSA-N 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- GCSPRLPXTPMSTL-IBDNADADSA-N [(2s,3r,4s,5s,6r)-2-[(2s,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[C@@]1([C@]2(CO)[C@H]([C@H](O)[C@@H](CO)O2)O)O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GCSPRLPXTPMSTL-IBDNADADSA-N 0.000 description 1
- BHIIGRBMZRSDRI-UHFFFAOYSA-N [chloro(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(Cl)OC1=CC=CC=C1 BHIIGRBMZRSDRI-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 238000009104 chemotherapy regimen Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- ZAKOWWREFLAJOT-UHFFFAOYSA-N d-alpha-Tocopheryl acetate Natural products CC(=O)OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-UHFFFAOYSA-N 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 238000006481 deamination reaction Methods 0.000 description 1
- STORWMDPIHOSMF-UHFFFAOYSA-N decanoic acid;octanoic acid;propane-1,2,3-triol Chemical compound OCC(O)CO.CCCCCCCC(O)=O.CCCCCCCCCC(O)=O STORWMDPIHOSMF-UHFFFAOYSA-N 0.000 description 1
- 239000005549 deoxyribonucleoside Substances 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 1
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000008344 egg yolk phospholipid Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 150000002194 fatty esters Chemical class 0.000 description 1
- 235000021323 fish oil Nutrition 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229940049654 glyceryl behenate Drugs 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 150000002431 hydrogen Chemical group 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- RZRNAYUHWVFMIP-UHFFFAOYSA-N monoelaidin Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-UHFFFAOYSA-N 0.000 description 1
- 201000000050 myeloid neoplasm Diseases 0.000 description 1
- 229940078812 myristyl myristate Drugs 0.000 description 1
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical group C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- QUANRIQJNFHVEU-UHFFFAOYSA-N oxirane;propane-1,2,3-triol Chemical compound C1CO1.OCC(O)CO QUANRIQJNFHVEU-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical compound CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Inorganic materials [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- DZKXJUASMGQEMA-UHFFFAOYSA-N tetradecyl tetradecanoate Chemical compound CCCCCCCCCCCCCCOC(=O)CCCCCCCCCCCCC DZKXJUASMGQEMA-UHFFFAOYSA-N 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229940042585 tocopherol acetate Drugs 0.000 description 1
- 150000003611 tocopherol derivatives Chemical class 0.000 description 1
- 125000002640 tocopherol group Chemical class 0.000 description 1
- 235000019149 tocopherols Nutrition 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 1
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/695—Silicon compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/12—Triazine radicals
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
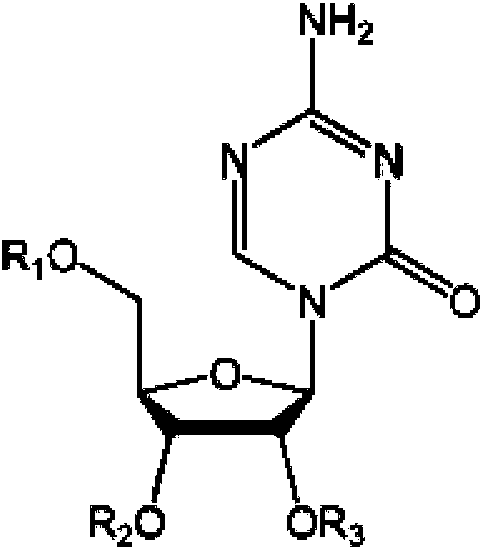
Un compuesto sililado que tiene la fórmula (a), definida como sigue: **Fórmula** en la que R1, R2, R7, y R8 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(R6), en la que R4, R5, y R6 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R1, R2, R7, o R8 sea 5 Si(R4)(R5)(R6).
Description
DESCRIPCIÓN
Profármacos de 5-aza-pirimidina sililada útiles para tratar el cáncer
Campo de la invención
La presente invención proporciona profármacos de 5-aza-pirimidina sililada que son compuestos de 5-aza-2',2'-difluorodeoxicitidina sililada, composiciones que incluyen dichos profármacos de 5-aza-pirimidina sililada, procedimientos para preparar dichos profármacos y composiciones de 5-aza-pirimidina sililada, y dichos profármacos y composiciones de 5-aza-pirimidina sililada para su uso en procedimientos para tratar el cáncer.
Antecedentes de la invención
Los análogos de pirimidina son conocidos por ser agentes quimioterapéuticos efectivos. La gemcitabina, un análogo de la citidina con un azúcar modificado, una 2',2’-difluorodeoxirribosa, es un fármaco quimioterapéutico actualmente aprobado como terapia única para el tratamiento del cáncer de páncreas y como parte de los regímenes combinados de quimioterapia para el tratamiento del cáncer de pulmón de células no pequeñas, cáncer de mama y cáncer de ovario (Gemzar (gemcitabina HCI) para inyección. 2007. Eli Lilly y Company.) Las siguientes propiedades hacen que la gemcitabina sea un agente anticancerígeno efectivo: (1) es un inhibidor de la ribonucleósido difosfato reductasa (RR), la enzima que cataliza la conversión de ribonucleósidos difosforilados en desoxirribonucleósidos difosforilados; (2) actúa como un terminador de cadena de ADN; y (3) no es un sustrato para la pirimidina nucleósido fosforilasa, lo que impide la descomposición de un nucleósido en una base separada y un resto de azúcar (Plunkett, W.,et al., "Gemcitabine: Metabolism, Mechanism of Action and Self-Potentiation," Semin. Oncol. 22(4; Suppl 11):3-1O, 1995; Mini, E., et al., "Cellular Pharmacology of Gemcitabine," Ann. Oncol. 17 (Suppl 5) v7-v12. 2006). Se han propuesto profármacos de gemcitabina sililo para mejorar su penetración a través de la pared intestinal, las membranas celulares y la barrera hematoencefálica (documento WO 2004/050665).
Otros dos análogos de citidina, 5-azacitidina y 5-azadeoxicitidina, están aprobados para el tratamiento de tumores hematológicos y actúan mediante la inhibición de ADN metil transferasa (DNMT), la enzima requerida para la metilación del ADN (US 2006/0205685). Se sabe que los análogos de 5-azacitidina son inestables en agua y se escinden en la posición 6 de la base (Christman, J.K., "5-azacitidina and 5-aza-2'-deoxicitidina as lnhibitors of DNA Metilation: Mechanistic Studies and Their lmplications for Cancer Therapy, Oncogene 21:5483-5495, 2002.)
La trifluridina, 5-trifluorotimidina, también ha demostrado actividad antineoplásica y se cree que actúa mediante la inhibición de la timidilato sintasa. Sin embargo, su desarrollo como agente antineoplásico ha sido obstaculizado por su corta vida media (12 minutos). La coadministración de trifluridina con un inhibidor de la timidina fosforilasa ha dado como resultado una farmacocinética más aceptable con vida media entre 1,37 y 1,57 horas. (Hong DS, Abbruzzese JL, Bogaard K et al. Phase 1 study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer. 2006; 15(107): 1383-1390.)
Un enfoque que se ha propuesto para prolongar la vida media circulatoria de los análogos de pirimidina es generar un profármaco que puede producir una molécula más hidrófoba que se puede formular en una nanoformulación a base de lípidos y liberar rápidamente el fármaco activo tras la exposición a un ambiente acuoso en el sitio del tumor. La conjugación de un fármaco que tiene una vida media corta con un resto lipófilo tal como, por ejemplo, trimetilsililo (TMS) u otros restos sililo, y la formulación del profármaco lipófilo en un medio hidrófobo pueden dar como resultado una vida media circulante prolongada.
Aún existe la necesidad de nuevas terapias contra el cáncer con mejores eficacia, seguridad y/o perfiles farmacocinéticos. La invención proporciona nuevos profármacos de 5-aza-pirimidina sililada que son compuestos y composiciones de 5-aza-2',2'-difluordeoxicitidina sililada para su uso en el tratamiento del cáncer.
Sumario de la invención
La presente invención proporciona compuestos farmacológicos terapéuticos que son profármacos de pirimidina sililada, composiciones que incluyen los profármacos de pirimidina sililada, procedimientos para preparar los profármacos de pirimidina sililada y procedimientos para tratar el cáncer usando los profármacos y composiciones de pirimidina sililada.
En un aspecto de la divulgación, se proporcionan profármacos de pirimidina sililada. Los profármacos de pirimidina sililada de la divulgación son análogos de pirimidina que tienen un grupo sililo unido en una o más posiciones hidroxilo del azúcar ribosa. En una realización, los profármacos de pirimidina sililada son análogos de citidina. En una realización, un profármaco de pirimidina sililada es un análogo de trimetilsilil citidina. En una realización, los profármacos de pirimidina sililada son análogos de timidina. En una realización, un profármaco de pirimidina sililada es un análogo de trimetilsilil timidina. También se proporcionan procedimientos para preparar los profármacos de pirimidina sililada.
En una realización, la invención proporciona un compuesto de 5-aza-2 ’,2 ’-difluorodeoxicitidina sililada, es decir, el llamado compuesto (a) que tiene la fórmula:

en la que R-|, R2, R7, y Rg se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5 y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R-|, R2, R7, o Rg sea Si(R4)(R5)(Rg). En otra realización, la invención proporciona un compuesto que tiene la fórmula:

en la que Rq, R2 y R7 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R i, R2, o R7 sea Si(R4)(R5)(Rg), En una realización adicional, la invención proporciona un compuesto que tiene la fórmula:

en la que R i y R2 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4, R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de Ri o R2 sea Si(R4)(R5)(Rg). En una realización particular, la invención proporciona (a) en la que R i y R2 son Si(R4)(R5)(Rg) y R4 R5 y Rg son metilo.
En una segunda realización, la divulgación proporciona un compuesto de pirimidina sililada (b) que tiene la fórmula:

en la que R j, R2, R3, R7, y Rg se seleccionan de modo independiente de hidrógeno o Si(R4)(Rg)(Rg), en la que R4, R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R j, R2 R3, R7, o Rg sea Si(Rq)(Rg)(Rg). En otra realización, la divulgación proporciona un compuesto que tiene la fórmula:

en la que R j , R2, R3, y R7 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R j, R2, R3, o R7 sea Si(R4)(Rg)(Rg). En una realización adicional, la divulgación proporciona un compuesto que tiene la fórmula:
en la que R j, R2 y R3 se seleccionan de modo independiente de hidrógeno o Si(R4)(Rg)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R j, R2, o R3 sea Si(R4)(R5)(Rg). En una realización particular, la divulgación proporciona un compuesto en la que R j, R2, y R3 son Si(R4)(R5)(Rg) y R4 R5, y Rg son metilo. En una tercera realización, la divulgación proporciona un compuesto de pirimidina sililada (e) que tiene la fórmula:

en la que Rq, R2, R7, y Rg se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5 y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R i, R2, R7, o Rg sea Si(R4)(R5)(Rg). En otra realización, la divulgación proporciona un compuesto que tiene la fórmula:

en la que R i, R2 y R7 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R i, R2, o R7 sea Si(R4)(R5)(Rg). En una realización adicional, la divulgación proporciona un compuesto que tiene la fórmula:

en la que R i y R2 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4, R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de Ri o R2 sea Si(R4)(R5)(Rg). En una realización particular, la divulgación proporciona un compuesto en la que Ri y R2 son Si(R4 )(R5)(Rg) y R4 R5, y Rg son metilo.
En una cuarta realización, la divulgación proporciona un compuesto de pirimidina sililada (d) que tiene la fórmula:

en la que Rq y R2 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4, R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R i o R2 sea Si(R4)(R5)(Rg). En otra realización, la divulgación proporciona un compuesto en la que R i y R2 son Si(R4 )(R5)(Rg) y R4 R5, y Rg son metilo.
En una quinta realización, la divulgación proporciona un compuesto de pirimidina sililada (e) que tiene la fórmula:

en la que R i y R2 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4, R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de Ri o R2 sea Si(R4)(R5)(Rg). En una realización particular, la divulgación proporciona un compuesto en la que Ri y R2 son Si(R4 )(R5)(Rg) y R4 R5, y Rg son metilo.
En otro aspecto, la invención proporciona composiciones que incluyen los compuestos de 5-aza-2',2’-difluorodeoxicitidina sililada, denominado compuesto (a) de la invención. Las composiciones incluyen uno o más del compuesto (a), un vehículo o diluyente farmacéuticamente aceptable, y opcionalmente, uno o más agentes terapéuticos adicionales. Las composiciones son útiles para la administración del compuesto (a) para tratar el cáncer. También se proporcionan procedimientos para preparar las composiciones de compuesto (a).
En otro aspecto de la invención, se proporcionan el compuesto (a) y las composiciones para usar en procedimientos para tratar el cáncer. En la presente se describe un procedimiento para administrar una pirimidina terapéutica a un sitio tumoral mediante permeabilidad y retención mejoradas, que comprende administrar una cantidad efectiva de una composición farmacéutica de la invención al sujeto que lo necesite.
En otra realización, la invención proporciona dicho compuesto (a) para su uso en un procedimiento para tratar un cáncer, que comprende administrar una cantidad efectiva de una composición farmacéutica de la invención al sujeto que lo necesita.
En otra realización, la invención proporciona dicho compuesto (a) para su uso en un procedimiento para tratar un carcinoma de tumor sólido, que comprende administrar una cantidad terapéuticamente efectiva de una composición farmacéutica de la invención a un sujeto que lo necesite. Los carcinomas de tumor sólido representativos incluyen carcinomas de mama, pulmón de células no pequeñas y colon.
En otra realización más, la invención proporciona dicho compuesto (a) para su uso en un procedimiento de tratamiento de una neoplasia maligna hematológica, que comprende administrar una cantidad terapéuticamente efectiva de una composición farmacéutica de la invención a un sujeto que lo necesite. Las neoplasias hematológicas representativas incluyen leucemias.
En los procedimientos indicados anteriormente, en ciertas realizaciones, el sujeto es un ser humano.
g
Breve descripción de los dibujos
La FIGURA 1 ilustra la liberación de 5-azacitidina de un compuesto representativo de silil-pirimidina de fórmula (b) (NUC025) en solución salina tamponada con fosfato (PBS) a 20 °C
Descripción detallada de la invención
La invención se define por las reivindicaciones adjuntas. La descripción que sigue está sujeta a esta limitación. Todos los aspectos y realizaciones no cubiertos por las reivindicaciones son meramente aspectos de la presente divulgación y no forman parte de la invención. Cualquier divulgación que se encuentre fuera del alcance de dichas reivindicaciones solo tiene fines ilustrativos y comparativos.
La presente divulgación proporciona compuestos farmacológicos terapéuticos que son profármacos de pirimidina sililada, composiciones que incluyen los profármacos de pirimidina sililada, procedimientos para preparar los profármacos y composiciones de pirimidina sililada, y procedimientos para tratar el cáncer usando los profármacos y composiciones de pirimidina sililada.
Como se señaló anteriormente, la 5-azacitidina y sus derivados son inestables en agua con la escisión hidrolítica del anillo de citosina. Esto presenta desafíos con la reconstitución de fármacos, y es un factor que afecta adversamente la vida media circulatoria, y quizás la efectividad clínica. El desarrollo de profármacos de 5-azacitidinas se complica por esta inestabilidad porque ciertos motivos pueden aumentar la inestabilidad. Por ejemplo, la adición de un resto del vehículo de ácido graso en el N4-amino, como se hizo para DHADC (5,6-dihidro-5-azadeoxicitidina) para aumentar la biodisponibilidad oral y evitar la desaminación, se ha asumido que es un riesgo para aumentar la inestabilidad de la citosina. En efecto, es posible imaginar un escenario en el que un profármaco produzca en la administración predominante de las especies inactivas al sitio del tumor (ver, por ejemplo, vías de reacción para la apertura del anillo y la hidrólisis del residuo de 5-azacitosina en el ADN en solución y después de la unión covalente al sitio activo de un DNMT descrito en Christman JK, Oncogene 21: 5483-5495, 2002). Un posible remedio es administrar profármacos de 5-aza que se puedan formular en una matriz hidrófoba en el sitio del tumor. Tal matriz adecuadamente formulada no solo evitará hidrólisis, sino que puede proporcionar otros beneficios, tal como mayor permeabilidad y retención (EPR).
EPR es la propiedad por la cual ciertos tamaños de moléculas o formulaciones (tales como las nanogotas de lípidos) tienden a acumularse en el tejido tumoral a una concentración más alta que en los tejidos normales. Los tumores dependen del suministro de sangre de una neovasculatura para su suministro nutricional y de oxígeno. Estos vasos tumorales recién formados son generalmente de forma y arquitectura anormales. Tienen células endoteliales de detección mal alineadas con fenestraciones anchas que permiten la entrada de nanogotas. Además, los tejidos tumorales generalmente carecen de drenaje linfático efectivo. Por lo tanto, las nanogotas pueden penetrar los sitios del tumor preferentemente y se eliminan más lentamente, lo que lleva a la acumulación del fármaco activo en el sitio del tumor. El tamaño del nanogota es un parámetro de diseño crítico que rige su capacidad de penetración tumoral. Debido a que el transporte tumoral es un proceso de difusión limitada, se encuentra que las nanopartículas de menor tamaño (< 20 nm) se difunden casi universalmente de manera más eficiente a través del tejido tumoral que las partículas de mayor tamaño (> 100 nm).
La formulación de un 5 aza nucleósido en una nanogota hidrófoba requiere que el profármaco de 5-aza nucleósido sea hidrófobo y que los restos vehículos se liberen rápidamente en el sitio del tumor, antes de la escisión hidrolítica del anillo de citosina. Tal papel se puede cumplir mediante la modificación de los grupos OH del azúcar o el grupo NH2 de la base con grupos sililo, tal como trimetilsililo (TMS). Se ha demostrado que TMS es relativamente no tóxico, aparte de la sedación, en estudios realizados en varias especies animales para la NASA por Corning. La administración intraperitoneal o intravenosa de 100-200 mg/kg de TMS dio como resultado una rápida absorción de TMS y un nivel de sangre de 1 mg/ml necesario para la anestesia ligera en roedores. Debido a los pesos moleculares de las pirimidinas y TMS, es poco probable que se alcancen los niveles que producen toxicidad después de la administración de una dosis terapéutica de una pirimidina medicinal.
La modificación de un nucleósido con un grupo sililo se describe en a presente. En una realización representativa, la 5-azacitidina se modificó con TMS para proporcionar 2',3',5'-tri-(trimetilsilil)-5-azacitidina (NUC025) como se describe en el Ejemplo 6.
En un aspecto de la invención, se proporcionan profármacos de pirimidina sililada. En una realización, los profármacos de pirimidina sililada son análogos de citidina. En una realización, un profármaco de pirimidina sililada es un análogo de trimetilsilil citidina. En una realización, los profármacos de pirimidina sililada son análogos de timidina. En una realización, un profármaco de pirimidina sililada es un análogo de trimetilsilil timidina.
En una realización, la presente invención proporciona un profármaco sililado de 2',2'-difluoro-5-azadeoxicitidina, es decir, el llamado compuesto (a), que tiene la estructura:

en la que Rq y R2 se seleccionan de modo independiente de hidrógeno o SXR3XR4XR5), en la que R3, R4 y R5 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R j o R2 sea Si(R 3XR4XR5). En una realización, R j es SXR3XR4XR5) y R2 es hidrógeno. En otra realización, R j es hidrógeno y R2 es Si(R3)(Rq)(R5). En una realización adicional, R j y R2 son SXR3XR4XR5). En una realización, R3, R4, y R5 son metilo.
En una realización, la presente invención proporciona un profármaco sililado de 2',2'-difluoro-5-azadeoxicitidina, trimetilsilil-2',2'-difluoro-5-azadeoxicitidina que tiene la estructura:

En una realización, la presente invención proporciona un profármaco sililado de 2',2'-difluoro-5,6-dihidro-5-azadeoxicitidina que tiene la estructura:

en la que R j y R2 se seleccionan de modo independiente de hidrógeno o Si(R3)(R4)(R5), en la que R3, R4 y R5 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de Rj o R2 sea Si(R3)(R4)(R5). En una realización, R j es Si(R3)(R4)(R5) y R2 es hidrógeno. En otra realización, R j es hidrógeno y R2 es Si(R3)(R4)(R5). En una realización adicional,
R j y R2 son Si(R3)(R4)(R5). En una realización, R3, R4, y R5 son metilo.
En una realización, la presente divulgación proporciona un profármaco sililado de 2',2-difluorozebularina que tiene la estructura:

en la que R¡ y R2 se seleccionan de modo independiente de hidrógeno o Si(R3)(R4)(R5), en la que R3, R4 y R5 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R^ o R2 sea Si(R 3XR4XR5). En una realización, R1 es SXR3XR4XR5) y R2 es hidrógeno. En otra realización, R^ es hidrógeno y R2 es SXR3XR4 )(R5). En una realización adicional, R1 y R2 son SXR3XR4XR5). En una realización, R3, R4, y R5 son metilo.
En una realización, la presente divulgación proporciona un profármaco sililado de 2',2'-difluoro-5,6-dihidro-5-azazebularina que tiene la estructura:

en la que R1 y R2 se seleccionan de modo independiente de hidrógeno o SXR3XR4XR5), en la que R3, R4 y R5 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R1 o R2 sea Si(R 3XR4XR5). En una realización, R1 es SXR3XR4XR5) y R2 es hidrógeno. En otra realización, R1 es hidrógeno y R2 es SXR3XR4 )(R5). En una realización adicional, R1 y R2 son Si(R3)(R4)(R5). En una realización, R3, R4 y R5 son metilo.
En una realización, la presente divulgación proporciona un profármaco sililado de 2',2'-difluorodeoxiribosa-trifluorotimidina que tiene la estructura:

en la que Rq y R2 se seleccionan de modo independiente de hidrógeno o SXR3XR4XR5), en la que R3, R4 y R5 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de Rj o R2 sea Si(R3)(R4)(R5). En una realización, R j es Si(R3)(R4)(R5) y R2 es hidrógeno. En otra realización, R j es hidrógeno y R2 es Si(R3)(R4)(R5). En una realización adicional, 5 R j y R2 son SXR3XR4XR5). En una realización, R3, R4 y R5 son metilo.
En una realización, la presente divulgación proporciona un profármaco sililado de azacitidina que tiene la estructura:

en la que R j, R2, y R3, se seleccionan de modo independiente de hidrógeno o Si(R4)(Rg)(Rg), en la que R4 R4 y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R j, R2 o R3, sea Si(R4)(R5)(Rg). En una ¡ 0 realización, R j es SXR3XR4XR5) y R2 y R3 son hidrógeno. En otra realización, R j es hidrógeno, R2 es SXR3XR4XR5), y R3 es hidrógeno. En una realización adicional, R j y R2 son hidrógeno y R3 es SXR3XR4XR5). De modo similar, la divulgación proporciona todas las combinaciones posibles en que dos de R j, R2, o R3 son SXR3XR4 )(Rg). En una realización, cada uno de R j, R2 y R3 es Si(R3)(R4 )(Rg). En una realización, R4 R5, y Rg son metilo.
En otras realizaciones, la divulgación proporciona los profármacos de citidina indicados anteriormente que tienen uno o dos grupos R ¡ 5 (por ejemplo, grupos TMS) acoplados al grupo amino N4.
En una realización representativa, el profármaco pirimidina N-sililada tiene la estructura:
!0

en las que Rq, R2, R3, R7, y Rg se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R1, R2 R3, R7, o Rg sea Si(R4)(R5)(Rg). En una realización, R4 R5, y Rg son metilo.
En los compuestos de pirimidina sililada de la divulgación descripta anteriormente, el término "alquilo" se refiere al grupo alquilo del grupo trialquilsililo y es un grupo alquilo C1-C10 (por ejemplo, metilo, etilo, n-propilo, i-propilo, n -butilo, s-butilo, i-butilo, t-butilo) que puede ser un grupo alquilo de cadena lineal, ramificada o cíclica. En ciertas realizaciones, el grupo alquilo es un grupo alquilo Ci-C4. En ciertas realizaciones específicas, el grupo alquilo es un grupo metilo. En ciertas realizaciones, cada grupo alquilo del grupo trialquilsililo es el mismo (por ejemplo, trimetilsililo). En otras realizaciones, el grupo trialquilsililo incluye dos o tres grupos alquilo diferentes (por ejemplo, t-butildimetilsililo).
La presente divulgación proporciona compuestos de pirimidina sililada terapéuticos representativos. En vista de la enseñanza de la presente divulgación expuesta en la presente, se apreciará que la presente divulgación incluye versiones sililadas de compuestos de pirimidina terapéuticos (es decir, compuestos de pirimidina sililada terapéuticos). Muchos compuestos de pirimidina terapéuticos son conocidos en la técnica, así como su utilidad y limitaciones. La presente divulgación aborda esas limitaciones al proporcionar una nueva clase de compuestos de pirimidina terapéuticos: compuestos de pirimidina sililada terapéuticos. Los compuestos de pirimidina sililada terapéuticos de la invención que son compuestos de 5-aza-2',2'-difluorodesoxicitidina sililada proporcionan propiedades ventajosas y previamente inalcanzables y ventajosas a los compuestos de pirimidina terapéuticos. Las ventajas de los compuestos representativos de pirimidina sililada enumerados y demostrados en la presente acumulan compuestos de pirimidina sililada terapéuticos. El efecto de la sililación es hacer que los compuestos de pirimidina terapéuticos sean hidrófobos y se administren fácilmente usando las composiciones de la invención. En ciertos aspectos, los compuestos de pirimidina sililada de la invención son profármacos: liberan el compuesto de-sililado (es decir, compuesto de pirimidina terapéutico) in vivo (por ejemplo, por hidrólisis del enlace Si-O). Como tales, los compuestos sililados terapéuticos de la invención se administran fácilmente, tienen una biodisponibilidad aumentada y se pueden administrar ventajosamente en el sitio de acción con una efectividad mucho mejor.
En otro aspecto de la invención, se proporcionan procedimientos para preparar los profármacos de pirimidina sililada. Los compuestos de pirimidina sililada de la invención se pueden preparar para tratar un compuesto de pirimidina con un agente sililante, tal como los conocidos en la técnica (por ejemplo, cloruro de trimetilsililo). En ciertas realizaciones, para los compuestos de pirimidina que tienen grupos amina e hidroxilo reactivos con sililo, los compuestos de pirimidina sililada de la invención tienen grupos amina y grupos hidroxi sililados. En otras realizaciones, para los compuestos de pirimidina que tienen grupos amilo e hidroxilo reactivos con sililo, los compuestos de pirimidina sililada de la invención tienen solo grupos hidroxi sililados.
La preparación de 2',2'-difluoro-5-azadeoxicitidina se proporciona en el Ejemplo 1. La preparación de 2',2’-difluoro-5,6-dihidro-5-azadeoxicitidina se proporciona en el Ejemplo 2. La preparación de 2',2'-difluorozebularina se proporciona en el Ejemplo 3. La preparación de 2',2’-difluorodeoxirribosa-trifluorotimidina se proporciona en el Ejemplo 4. Los profármacos de pirimidina sililada de la invención se pueden prepararse mediante el tratamiento con un análogo de pirimidina con un exceso de cloruro de trimetilsililo y trietilamina en tetrahidrofurano. Alternativamente, los profármacos de pirimidina sililada de la invención se pueden preparar mediante el tratamiento de un análogo de pirimidina con un exceso de 1,1,1,3,3,3-hexametildisilazano y cantidades catalíticas de sulfato de amonio a 125 °C. La preparación de un compuesto representativo de pirimidina sililada (a) de la invención, 2 ’,2’-difluoro-5-azadeoxicitidina-trimetilsililo, se proporciona en el Ejemplo 5.
En otros aspectos, la presente invención proporciona composiciones farmacéuticas que comprenden al menos un profármaco de pirimidina sililada de la invención junto con un vehículo farmacéuticamente aceptable adecuado para la administración a un sujeto humano o animal, ya sea solo o junto con otros agentes terapéuticos y/o anticáncer. Los profármacos de pirimidina sililada de la invención se pueden formular en una composición que además comprende vehículos farmacéuticamente aceptables adecuados, que incluyen excipientes y otros compuestos que facilitan la administración de los profármacos de pirimidina sililada a un sujeto mamífero.
En una realización de la invención, el vehículo farmacéuticamente aceptable es un medio lipófilo. El medio lipófilo puede ser cualquiera de una variedad de medios lipófilos que incluyen, por ejemplo, aceites. En una realización, el medio lipófilo incluye un tocoferol (por
ejemplo, a-tocoferol). En una realización, el medio lipófilo incluye aceite de maíz. Los aceites representativos útiles como medio lipófilo incluyen los siguientes:
Ácidos grasos y sus ésteres, que incluyen ácidos carboxílicos de varias longitudes de cadena, en su mayoría de cadena lineal, pero que se pueden ramificar, cuyos ejemplos incluyen ácido cáprico, caprílico, caproico, láurico, mirístico, esteárico, oleico, linoleico, behénico y también ácidos grasos y ésteres saturados o insaturados;
Ácidos grasos esterificados con glicerina para cultivar mono, di o triglicéridos, que pueden ser sintéticos o derivados de fuentes naturales, que incluyen, pero sin limitación, glicéridos tales como aceite de soja, aceite de semilla de algodón, aceite de colza, aceite de pescado, aceite de ricino, Capmul MCM, Captex 300, Miglyol 812, monooleato de glicerilo, triacetina, monoglicérido acetilado, tristearina, behenato de glicerilo y ésteres de ácido diacetil tartárico de monoglicéridos;
Glicéridos conjugados con otros restos, tales como polietilenglicol (por ejemplo, Labrasol, Labrafac, Cremophor EL);
Fosfolípidos, naturales o sintéticos, tales como dimiristilfosfatidilcolina, lecitina de huevo y fosfolípidos pegilados;
Otros ésteres grasos que incluyen alcoholes grasos (miristato de miristilo, palmitato de isopropilo) o azúcares (monooleato de sorbitano, SPAN 80, Tween 80, laurato de sacarosa);
Alcoholes grasos tales como alcohol estearílico, alcohol laurílico, alcohol bencílico, o ésteres o éteres de los mismos, tales como benzoato de bencilo;
Vitaminas y derivados liposolubles, por ejemplo, vitamina E (que incluyen todos los tocoferoles y tocotrienoles, y derivados de tocoferol y tocotrienoles, tales como succinato de vitamina E, acetato de vitamina E y succinato de vitamina E, polietilenglicol (TPGS)).
En ciertas realizaciones, los compuestos de pirimidina sililada de la invención se pueden formular en una emulsión, tal como una nanoemulsión. La concentración del compuesto de pirimidina sililada puede variar de acuerdo con el compuesto, los componentes de la emulsión y la concentración deseada para administrar (por ejemplo, 0,5 a aproximadamente 10 mg/ml). El tamaño de las gotas de aceite en la emulsión también se puede variar según sea necesario (por ejemplo, de 10 a 200 nm o menos de 100 nm). En el Ejemplo 5 se describe una nanoemulsión que comprende un compuesto de pirimidina sililada representativo de la invención (NUC041).
En otro aspecto, en la presente se describen procedimientos para tratar sujetos humanos o animales que padecen una enfermedad celular proliferativa tal como cáncer. Las enfermedades proliferativas de células representativas tratables por los compuestos de la invención incluyen cánceres hematológicos, tales como leucemia, linfoma y mieloma, y cánceres no hematológicos, tales como carcinomas de tumor sólido (por ejemplo, mama, ovario, páncreas, colon, colorrectal, pulmón no pequeño y vejiga), sarcomas y gliomas. La presente divulgación proporciona procedimientos para tratar a un sujeto humano o animal que necesita dicho tratamiento, que comprende administrar al sujeto una cantidad terapéuticamente efectiva de uno o más profármacos de pirimidina sililada de la invención, solos o en combinación con uno o más de otros agentes terapéuticos. y/o anticáncer.
Las composiciones que incluyen uno o más profármacos de pirimidina sililada de la invención se administran para administrar cantidades terapéuticamente efectivas del profármaco de pirimidina sililada. Las cantidades terapéuticamente efectivas de los profármacos de pirimidina sililada generalmente variarán hasta la dosis máxima tolerada, pero las concentraciones no son críticas y pueden variar ampliamente. Las cantidades precisas empleadas por el médico tratante variarán, por supuesto, de acuerdo con el compuesto, vía de administración, afección física del paciente y otros factores. La dosis diaria se puede administrar como una dosis única o se puede dividir en dosis múltiples para la administración. La administración de los profármacos de pirimidina sililada de la invención se realiza por cualquier vía efectiva, por ejemplo, parenteral u oral.
La cantidad de los profármacos de pirimidina sililada de la invención realmente administrada será una cantidad terapéuticamente efectiva, tal término que se usa en la presente para indicar la cantidad necesaria para producir un efecto beneficioso sustancial. Las dosis efectivas se pueden extrapolar a partir de curvas de dosis-respuesta derivadas de sistemas de prueba in vitro o de modelos animales. El modelo animal también se usa típicamente para determinar un intervalo de dosis y una vía de administración deseables. Tal información se puede usar posteriormente para determinar dosis y vías útiles para la administración en seres humanos u otros mamíferos. La determinación de una dosis efectiva está dentro de la capacidad de los expertos en la técnica. Por lo tanto, la cantidad realmente administrada dependerá del individuo al que se aplicará el tratamiento, y preferiblemente será una cantidad optimizada de tal manera que se logre el efecto deseado sin efectos secundarios significativos.
Los siguientes ejemplos se proporcionan con el fin de ilustrar, no limitar, la invención.
Ejemplos
Ejemplo 1
La preparación de 2',2'-Difluoro-5-azadeoxicitidina
Se usaron dos vías alternativas, mostradas en el Esquema 1, para preparar 2',2'-difluoro-5-azadeoxicitidina (Compuesto IV).
A. Preparación de 3'.5'-dibenzoil-2'.2'-difluoro-5-azadeoxicitidina (Compuesto III) por medio de 1-bromo-2-deoxi-2.2-difluoroD-ribofuranosil-3.5-dibenzoato (Compuesto 1).
(1) . El material de partida, 2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato conocido en la técnica (Chou et al., Synthesis 1992, pp. 565-570) se convirtió en análogo 1-bromo por un procedimiento similar al descripto en el documento WO 2006/070985. La ribosa de partida (3,9 g, 10,3 mmol) se disolvió en 31 ml de tolueno y se añadió trietilamina seca (1,43 ml, 10,3 mmol). La solución se enfrió a 0 °C y se añadió cloruro de difenilfosforilo (2,64 ml, 12,3 mmol) en 8 ml de tolueno durante 15 min. La mezcla de reacción se calentó a temperatura ambiente y se incubó durante 3,5 horas. La reacción se inactivó mediante la adición de HCl 1 M (10,2 mil, la capa de tolueno se separó y la capa acuosa se extrajo de nuevo con 10 ml de éter. Las capas orgánicas se combinaron, se extrajeron en consecuencia con agua, NaHCO3 saturado y NaCl saturado (cada una 20 ml) y se secaron sobre MgSO4. Los disolventes se eliminaron y el producto se aisló por cromatografía flash sobre gel de sílice en gradiente de hexano-acetato de etilo. Rendimiento 4,6 g, 73%.
(2) Al análogo intermediario de difenilfosfato (4,6 g, 7,5 mmol) preparado como se describe en la Etapa (1), se añadió HBr en ácido acético (30%, 16,2 ml, 81,3 mmol) y la mezcla de reacción se incubó durante 6,5 ha temperatura ambiente. La mezcla de reacción se diluyó con 80 ml de cloruro de metileno y se extrajo dos veces con agua con hielo, NaHCO3 saturado y NaCl saturado (cada uno100 ml). La capa orgánica se secó con MgSO4, se filtró y se evaporó para producir 1-bromo-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto 1, 3,1 g, 93%), que es una mezcla 9: 1 de isómeros a y p.
(3) Se suspendieron 5-azacitosina (98%, Aldrich, 5,0 g, 44,8 mmol) y sulfato de amonio (25 mg) en hexametildisilazano (25 ml) y se añadió clorotrimetilsilano (0,2 ml). La mezcla de reacción se calentó a reflujo durante 17 h. La solución transparente se enfrió, se evaporó, se co-evaporó dos veces con xileno seco y se secó al vacío para producir sólidos blanquecinos de la 5-azacitosina sililada (7 g), que se usa en gran medida en la siguiente etapa de glicosilación.
(4) Para la etapa de glucosilación 1-bromo-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto 1) (0,78 g, 1,77 mmol), preparado como se describe en la Etapa (2), se disolvió en 4 ml de anisol y se transfirió a los sólidos de 5-azacitosina sililada preparados como en la Etapa (3). La suspensión se calentó a 150 °C y se añadieron 2 ml de anisol para completar la disolución de todos los sólidos. Después de 4 horas, la mezcla de reacción se enfrió y se añadió SnCL4 (0,2 ml, 2 mmol). La mezcla de reacción se volvió a calentar a 150 °C durante 6 horas, se enfrió a temperatura ambiente, se inactivó mediante la adición de cloruro de metileno (30 ml), metanol (10 ml) y gel de sílice (20 g) y se secó para producir 3',5'-dibenzoil-2',2'-difluoro-5-azadeoxi-citidina (Compuesto III). El polvo resultante se aplicó en la parte superior de la columna de gel de sílice empaquetada y los productos se aislaron por cromatografía flash en gradiente de cloroformo-metanol. Primero se eluyó el isómero p seguido de un isómero a (solo se logró una separación parcial, se obtuvo un isómero p del 12%, un isómero a 5%). La separación completa de isómeros se logró mediante RP HPLC en Gemini C185u (columna de 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5) -acetonitrilo.
1H RMN en CDO3 para el compuesto III isómero p: 8,12 ppm (s, 1H, H6), 7,8-8,1 (m, 4H, benzoílo), 7,4-7,6 (m, 2H, benzoílo),
7,2-7,4 (m, 4H, benzoílo), 6,38 (br.t, J=8,0 Hz, 1H, H1'), 5,65 (m, 1H, H3'), 4,70 (m, 2H, H5'), 4,58 (m, 1H, H4'). ESI MS: 473,3 [M+Hj+, 471,1 [M-H]-.
B. Preparación de 2'■2'-difluoro-5-azadeoxicitidina (Compuesto IV). 3',5'-dibenzoil-2',2’-difluoro-5-azadeoxi-citidina (Compuesto III) (15 mg, mezcla 3: 1 de isómeros a y P), preparado como en la Etapa A, se disolvió en 2 ml de MeOH anhidro y NaOMe 1 M en MeOH (0,1 ml). Después de 1 hora a temperatura ambiente, la mezcla de reacción se evaporó y los isómeros desprotegidos se aislaron por RP HPLC en Gemini C18 5u (columna 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5) -acetonitrilo. Inmediatamente después de la separación, las fracciones correspondientes al isómero p se mezclaron y se evaporaron a menos de 10 °C para producir 0,4 mg (19%) de 2 ’,2’-difluoro-5-azadeoxicitidina (Compuesto IV).
1H RMN en DMSO-d6 para el compuesto IV isómero p: 8,48 ppm (s, 1H, H6), 7,79 (br, 2H, NH2), 6,35 (br, 1H, 3'-0H), 6,07 t, J=8,0 Hz, 1H, H1'), 5,30 (br, 1H, 5'-0H), 4,90 (br, 1H, 5'-0H), 4,23 (br. m, 1H, H3'), 3,85 (m, 1H, H4'), 3,78 (m, 1H, H5'), 3,63 (m, 1H, H5'), UV 240 nm (sh) en acetato de trietilamonio 50 mM (pH 7,5)
C. Preparación de 3',5'-dibenzoil-2',2'-difluoro-5-azadeoxicitidina (Compuesto III) por medio de 1-metilsulfonil-2-deoxi-2,2-di-fluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto II).
La 5-azacitosina sililada (2,3 g, 9 mmol) preparada como se describe en la Etapa A (3), se disolvió en 2 ml de anisol a 130 °C. 1-metilsulfonil-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto II) (0,4 g, 0,87 mmol), preparado según lo descrito por Chou et. al., Synthesis 1992, pp. 565-570, se disolvió en 1 ml de anisol y se añadió a la solución caliente de la 5-azacitosina sililada. La mezcla de reacción se incubó a 150 °C durante 7 horas, se enfrió a temperatura ambiente, se inactivó mediante la adición de 15 ml de cloruro de metileno, 15 g de gel de sílice y 5 ml de metanol y la suspensión se secó al vacío. El polvo resultante se aplicó sobre la parte superior de la columna de gel de sílice empaquetada y los productos se aislaron por cromatografía instantánea en gradiente de cloroformo-metanol. Las fracciones apropiadas se mezclaron y se evaporaron para producir 3',5'-dibenzoil-2',2’-difluoro-5-azadeoxicitidina (Compuesto III) como una mezcla 2: 1 de isómeros a y p (100 mg, 25% de rendimiento).
D. Preparación de 2',2'-difluoro-5-azadeoxicitidina (Compuesto IV). 3', 5'-dibenzoil-2',2’-difluoro-5-azadeoxi citidina (Compuesto III)
(80 mg, mezcla 2: 1 de isómeros a y P), preparado como en la Etapa C, se disolvió en 2 ml de MeOH anhidro y se añadió NaOMe 1 M en MeOH (0,1 ml). Después de 1 hora a temperatura ambiente, la mezcla de reacción se evaporó y los isómeros desprotegidos se aislaron por RP HPLC en Gemini C18 5u (columna 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5) -acetonitrilo. Inmediatamente después de la separación, las fracciones correspondientes al isómero P se mezclaron, se evaporaron a menos de 10 °C para producir 2,1 mg (13%) de 2 ’,2’-difluoro-5-azadeoxicitidina (Compuesto IV).
El tiempo de retención de RP HPLC y las características espectrales (es decir, 1H RMN y UV) para el Compuesto IV fueron las mismas que en la Etapa B. ESI MS: 265,2 [M H] .

Ms = metilsulfonilo; TMS = trimetilsililo; Bz = benzoílo; Ph = fenilo; Me = metilo
Esquema 1. Síntesis de 3',5'-dibenzoil-2',2'-difluoro-5-azadeoxicitidina
(Compuesto III) y 2',2'-difluoro-5-azadeoxicitidina (Compuesto IV).
Ejemplo 2
La preparación de 2'.2'-difluoro-5.6-dihidro-5-azadeoxicitidina
La preparación de 2',2'-difluoro-5,6-dihidro-5-azadeoxicitidina (Compuesto VI) se muestra en el Esquema 2.
A. Preparación de 3',5'-dibenzoil-2',2'-difluoro-5-azadeoxicitidina (Compuesto V). El isómero P puro de 3',5'-dibenzoil-2',2’-difluoro-5-azadeoxicitidina (Compuesto III) (144 mg, 0,30 mmol), preparado como en el Ejemplo 1, se disolvió en 3 ml de cloruro de metileno y se añadieron trietilamina (0,12 ml) y clorotrimetilsilano (0,1 ml). Después de 0,5 horas, la mezcla de reacción se diluyó con 5 ml de ácido acético y se añadió borohidruro de sodio (100 mg, 2,6 mmol) como polvo. Después de 1 h a temperatura ambiente, la mezcla de reacción se evaporó y se separó por RP HPLC en Gemini C185u (columna 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5) -acetonitrilo. Las fracciones correspondientes al producto se mezclaron y se evaporaron para producir 83 mg (57%) de 3',5'-dibenzoil-2',2’-difluoro-5-azadeoxicitidina (Compuesto V).
1H RMN en DMSO-d6 para el compuesto V isómero P: 7,93-8,07 (m, 4H, benzoílo), 7,64-7,77 (m, 2H, benzoílo), 7,47-7,62 (m,4H, benzoílo), 6,08 (t, J=7,2 Hz, 1H, H1'), 5,63 (m, 1H, H3'), 4,55-4,70 (m, 3H, H4' y H5'), 4,48 (s, 2H, CH2). ESI MS: 475,4 [M+H]+.
B. Preparación de 2'■2'-difluoro-5■6-dihidro-5-azadeoxicitidina (Compuesto VI). 3',5'-dibenzoil-2',2'-difluoro-5- azadeoxicitidina (Compuesto V) (80 mg, 0,168 mmol), preparado como en la Etapa A, se disolvió en 6 ml de MeOH anhidro y se añadió NaOMe 1 M en MeOH (0,3 ml). Después de 1 h a temperatura ambiente, la mezcla de reacción se inactivó mediante la adición de ácido acético (0,02 ml), se evaporó y el producto desprotegido se aisló mediante RP HPLC en Gemini C185u (columna 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5)-acetonitrilo. Las fracciones apropiadas se mezclaron y se evaporaron para producir 49 mg
(95% de rendimiento) de 2',2'-difluoro-5,6-dihidro-azadeoxicitidina (Compuesto VI).
1H RMN en DMSO-d6 para el compuesto VI: 5,87 (dd, J1=9,6 Hz, J2=10,8 Hz, 1H, H1'), 4,51 y 4,29 (AB, 2H, CH2), 3,98 (ddd, J1=8,4 Hz, J2=12,8 Hz, J3=12,8 Hz, 1H, H3'), 3,67 (m, 1H, H5'), 3,58 (m, 1H, H4'), 3,52 (m, 1H, H5'). ESI MS: 267,0 [M+H]+.

Esquema 2 Síntesis de 2',2'-difluoro-5,6-dihidro-5-azadeoxicitidina (Compuesto VI).
Ejemplo de referencia 3
La preparación de 2',2'-difluorozebularina
La preparación de 2',2'-difluorozebularina (Compuesto VIII) se muestra en el Esquema 3.
(1) El material de partida 1-bromo-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto 1) se preparó como se describe en el Ejemplo 1.
(2) Clorhidrato de 2-hidroxipirimidina (0,57 g, 4,3 mmol) se suspendió en hexametildisilazano (100 ml). La mezcla de reacción se calentó a reflujo durante 1,5 h. La solución transparente se decantó bajo argón, se evaporó, se coevaporó dos veces con xileno seco y se secó al vacío.
(3) Para la etapa de glicosilación, se disolvió 1-bromo-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto 1) (1,20 g, 2,7 mmol), preparado como en la Etapa (1), en 20 ml de dicloroetano y se transfirió al matraz que contiene la 2-hidroxipirimidina sililada. La mezcla de reacción se calentó a reflujo bajo argón durante 16 h, posteriormente se reemplazó el dicloroetano con 10 ml de anisol y la temperatura se elevó a 150 °C. La mezcla de reacción se agitó a 150 °C durante 18 h, se enfrió a temperatura ambiente y el producto, 1-P-(2-desoxi-2',2-difluoro-D-ribofuranosil)-pirimidina-2-ona (Compuesto VII) se aisló como una mezcla (1: 6) de isómeros p y a mediante cromatografía flash sobre gel de sílice en metanol/cloroformo (3:97). Rendimiento: 0,09 a 7%.
(4) 1-P-(2-desoxi-2’,2’-difluoro-D-ribofuranosil)pirimidina-2-ona (Compuesto VII), preparado como en las Etapas (1) a (3), (85 mg, 6: 1 mezcla de isómeros a y P) se disolvió en 2 ml de MeOH anhidro y se añadió NaOMe 1 M en MeOH (0,1 ml). Después de 1 h a 4 °C, la mezcla de reacción se evaporó y los isómeros desprotegidos se aislaron por RP HPLC en Gemini C18 5u (columna 21,2x250 mm) en gradiente de acetato de trietilamonio 50 mM (pH 7,5) -acetonitrilo. Inmediatamente después de la separación, las fracciones correspondientes al isómero p se mezclaron y se evaporaron para producir 3,4 mg (7,4%) de 2 ’,2’-difluorozebularina (Compuesto VIII).
1H RMN en DMSO-d6 compuesto VIII isómero P: 8,66 ppm (dd, J1=4 Hz, J2=2,4 Hz, 1H), 8,57 (dd, J1=6,8 Hz, J2=2,4 Hz, 1H), 6,62 (dd, J1=6,8 Hz, J2=4 Hz, 1H), 6,07 (t, J=7,2 Hz, 1H), 4,42-4,29 (m, 1H), 4,05-3,97 (m, 2H), 3,88-3,82 (m, 1H). ESI MS: 249 [M+H]+.

Ejemplo de referencia 4
Preparación de 2'.2'-Difluorodeoxiribose-trifluorotimidina
La preparación de 2',2'-difluorodeoxiribosa-trifluorotimidina (Compuesto X) se muestra en el Esquema 4. 2', 2 'Difluorodeoxiribosatrifluorotimidina (Compuesto X) se prepara por glicosilación de 5-(trifluorometil)uracilo sililado con 1-bromo-2-desoxi-2,2-difluoro-D-ribofuranosil-3,5-dibenzoato (Compuesto 1), preparado como en el Ejemplo 1, en anisol usando SnCL como catalizador seguido de desprotección con cantidades catalíticas de metóxido de sodio en metanol.

Ejemplo 5
La preparación de un profármaco de pirimidina sililada representativa:
2'■2'-Difluoro-5-azadeoxicitidina-trimetilsililo (NUC041)
Procedimiento l. La preparación de 2',2'-difluoro-5-azadeoxicitidina-trimetilsililo (Compuesto XI) se muestra en el Esquema 5. 2',2'-Difluoro-5-azadeoxicitidina-trimetilsililo (Compuesto XI) se prepara mediante el tratamiento de 2',2'-difluoro-5-azadeoxicitidina (Compuesto IV), preparado como en el Ejemplo 1, con exceso de cloruro de trimetilsililo y trietilamina en tetrahidrofurano.

Esquema 5. 2',2'-Difluoro-5-azadeoxicitidina-trimetilsililo (Compuesto XI)
Procedimiento ll. Un procedimiento alternativo para preparar 2 ’,2’-difluoro-5-azadeoxicitidina-trimetilsililo (Compuesto XI) es el siguiente. La 2 ’,2’-difluoro-5-azadeoxicitidina-trimetilsililo (Compuesto XI) se prepara mediante el tratamiento de la 2',2'-difluoro-5-azadeoxicitidina (Compuesto IV), preparada como en el Ejemplo 1, con un exceso de 1,1,1,3,3,3-hexametildisilazano y cantidades catalíticas de sulfato de amonio a 125 °C. El producto se aísla por cromatografía flash.
El compuesto XI se formuló con éxito en una nanoemulsión a una concentración de 3 mg/ml. La nanoemulsión tiene una fase oleosa que consiste en un aceite inyectable y lecitina y una fase acuosa que comprende un agente de ajuste de tonicidad, un estabilizante y agua. Las emulsiones son de tipo aceite en agua con las gotas de aceite medias de menos de 100 nm. Cada vehículo tiene un pH neutro (5-7) y es aproximadamente isotónico. La toxicidad del Compuesto XI formulado se probó en ratones. Los ratones se inyectaron por vía intravenosa en tres días consecutivos con el Compuesto XI a dosis de hasta 60 mg/kg y se observaron al final de una semana. No se observó evidencia de toxicidad medida por la pérdida de peso.
Ejemplo de referencia 6
2'.3'.5'-Tri(trimetilsilil)-5-azacitidina (NUC025)
En este ejemplo se describen las características de 2',3',5'-tri(trimetilsilil)-5-azacitidina (NUC025). La estructura de NUC025 se muestra continuación.

2',3',5'-Tri(trimetilsilil)-5-azacitidina (NUC025) se prepara por un procedimiento análogo a la preparación de 2',2'-difluoro-5-azadeoxicitidina-trimetilsililo descripto anteriormente en el Ejemplo 5.
La citotoxicidad in vitro de NUC025 se compara con 5-azacitidina para mamas, colon, y luecemia en la Tabla 1.
Tabla 1. Citotoxicidad in vitro de NUC025 y 5-azacitidina (IC50 en pM).

Los datos en la Tabla 1 muestran que NUC025 conserva la actividad de 5-azacitidina in vitro. Tales profármacos de 5-aza nucleósidos pueden demostrar superioridad in vivo, donde es importante una vida media en circulación más larga.
La Figura 1 ilustra la liberación de 5-azacitidina del profármaco 5-azacitidina-TMS (NUC025) por hidrólisis, lo que sugiere que, al menos en PBS a temperatura ambiente, se puede recuperar aproximadamente el 80% del fármaco activo.
Si bien se han ilustrado y descrito realizaciones ilustrativas, se apreciará que se pueden realizar varios cambios en ellas sin apartarse del alcance de las reivindicaciones adjuntas.
Claims (9)
1. Un compuesto sililado que tiene la fórmula (a), definida como sigue:

en la que R1, R2, R7, y R8 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(R6), en la que R4 R5, y R6 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R1, R2, R7, o R8 sea Si(R4)(R5)(R6).
2. El compuesto de la Reivindicación 1 que tiene la fórmula:

en la que R1, R2, y R7 se seleccionan de modo independiente de hidrógeno o Si(Rq)(R5)(R6), en la que R4 R5, y R6 se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R1, R2 o R7 sea Si(R4)(R5)(R6).
3. El compuesto de la Reivindicación 1 que tiene la fórmula:

en la que Rq y R2 se seleccionan de modo independiente de hidrógeno o Si(R4)(R5)(Rg), en la que R4 R5, y Rg se seleccionan de modo independiente de hidrógeno o alquilo, con la condición de que al menos uno de R i o R2 sea Si(R4)(R5)(Rg).
4. El compuesto de una cualquiera de las reivindicaciones 1-3, en el que en (a), Ri y R2 son Si(R4)(R5)(Rg) y R4, R5, y Rg son metilo, y más preferiblemente tiene la fórmula:

en la R i y R2 son Si(CH3)3
5. Una composición farmacéutica, que comprende un compuesto sililado de una cualquiera de las reivindicaciones 1-4 y un vehículo farmacéuticamente aceptable.
6. El compuesto de una cualquiera de las Reivindicaciones 1 a 4 para su uso en el tratamiento de un cáncer y un carcinoma de tumor sólido o neoplasia hematológica, mediante la administración de una cantidad efectiva del compuesto de una cualquiera de las Reivindicaciones 1 a 4 a un sujeto que lo necesite.
7. El compuesto de una cualquiera de las Reivindicaciones 1 a 4 para el uso de la Reivindicación 6, en el que el carcinoma de tumor sólido se selecciona del grupo que consiste en carcinomas de mama, pulmón de células no pequeñas, y colon.
8. El compuesto de una cualquiera de las Reivindicaciones 1 a 4 para el uso de la Reivindicación 6, en el que la neoplasia hematológica es una leucemia.
9. El compuesto de una cualquiera de las Reivindicaciones 1 a 4 para su uso según cualquiera de las Reivindicaciones 6 a 8, en el que el sujeto es un ser humano.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462061526P | 2014-10-08 | 2014-10-08 | |
| PCT/US2015/054755 WO2016057828A1 (en) | 2014-10-08 | 2015-10-08 | Silylated pyrimidine prodrugs and methods of their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2746105T3 true ES2746105T3 (es) | 2020-03-04 |
Family
ID=55653798
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15849800T Active ES2746105T3 (es) | 2014-10-08 | 2015-10-08 | Profármacos de 5-aza-pirimidina sililada útiles para tratar el cáncer |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10479807B2 (es) |
| EP (1) | EP3204121B1 (es) |
| JP (1) | JP6724909B2 (es) |
| CA (1) | CA2963686A1 (es) |
| ES (1) | ES2746105T3 (es) |
| WO (1) | WO2016057828A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109152791A (zh) * | 2016-02-26 | 2019-01-04 | 表观遗传学药业有限责任公司 | 2',2'-二氟-5-氮杂-2'-脱氧胞苷或其前药治疗tp53 野生型肿瘤的方法 |
| WO2017183215A1 (ja) * | 2016-04-21 | 2017-10-26 | 大原薬品工業株式会社 | 5-アザシチジン類の糖部シリルエーテル誘導体 |
| US10227374B2 (en) | 2016-04-21 | 2019-03-12 | Ohara Pharmaceutical Co., Ltd. | Silyl etherified derivatives of 5-azacytidines in carbohydrate moiety |
| JP6162349B1 (ja) * | 2016-04-21 | 2017-07-12 | 大原薬品工業株式会社 | 5−アザシチジン類の糖部シリルエーテル誘導体 |
| JP6956937B2 (ja) * | 2018-02-19 | 2021-11-02 | 大原薬品工業株式会社 | Dnmt阻害剤の用途 |
| US12472196B2 (en) | 2019-09-26 | 2025-11-18 | Ohara Pharmaceutical Co., Ltd. | Use of DNMT inhibitor |
| WO2024196735A2 (en) * | 2023-03-17 | 2024-09-26 | Epigenetics Pharma Llc | Pyrimidine prodrugs formulations and methods of uses thereof |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR920002143B1 (ko) * | 1988-07-11 | 1992-03-12 | 다이호 야꾸힝고오교 가부시끼가이샤 | 5-치환 우리딘 유도체 및 그의 제조 중간체 |
| US5120842A (en) | 1991-04-01 | 1992-06-09 | American Home Products Corporation | Silyl ethers of rapamycin |
| US6413945B1 (en) | 1997-04-04 | 2002-07-02 | Pharmacia Corporation | Gastro-specific prodrugs |
| CA2331386A1 (en) | 1998-06-26 | 2000-01-06 | Incyte Pharmaceuticals, Inc. | Human signal peptide-containing proteins |
| AU2003302624A1 (en) * | 2002-11-29 | 2004-06-23 | Amedis Pharmaceuticals Ltd. | Silicon compounds |
| GB0227906D0 (en) | 2002-11-29 | 2003-01-08 | Amedis Pharm Ltd | Compounds and their use |
| US20080261918A1 (en) | 2004-12-17 | 2008-10-23 | Graham Andrew Showell | Silicon Compounds and Their Use |
| WO2006070985A1 (en) | 2004-12-30 | 2006-07-06 | Hanmi Pharm. Co., Ltd. | METHOD FOR THE PREPARATION OF 2#-DEOXY-2#,2#-DIFLUOROCYTIDINE |
| US7250416B2 (en) * | 2005-03-11 | 2007-07-31 | Supergen, Inc. | Azacytosine analogs and derivatives |
| WO2010014883A2 (en) | 2008-08-01 | 2010-02-04 | Dr. Reddy's Laboratories Ltd. | Azacitidine process and polymorphs |
| UA117095C2 (uk) | 2011-12-22 | 2018-06-25 | Аліос Біофарма, Інк. | Нуклеозидна сполука або її фармацевтично прийнятна сіль |
-
2015
- 2015-10-08 EP EP15849800.6A patent/EP3204121B1/en active Active
- 2015-10-08 CA CA2963686A patent/CA2963686A1/en not_active Abandoned
- 2015-10-08 US US15/516,653 patent/US10479807B2/en active Active
- 2015-10-08 ES ES15849800T patent/ES2746105T3/es active Active
- 2015-10-08 JP JP2017517661A patent/JP6724909B2/ja not_active Expired - Fee Related
- 2015-10-08 WO PCT/US2015/054755 patent/WO2016057828A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017531639A (ja) | 2017-10-26 |
| EP3204121A1 (en) | 2017-08-16 |
| US20170305941A1 (en) | 2017-10-26 |
| JP6724909B2 (ja) | 2020-07-15 |
| EP3204121B1 (en) | 2019-06-19 |
| CA2963686A1 (en) | 2016-04-14 |
| US10479807B2 (en) | 2019-11-19 |
| WO2016057828A1 (en) | 2016-04-14 |
| EP3204121A4 (en) | 2018-05-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2746105T3 (es) | Profármacos de 5-aza-pirimidina sililada útiles para tratar el cáncer | |
| AU2013204313B2 (en) | Combination Therapy | |
| JP6769000B2 (ja) | 4’−チオヌクレオシドの新規な化合物、並びにその調製方法、その医薬組成物及びその用途 | |
| US20130131008A1 (en) | Lipophilic monophosphorylated derivatives and nanoparticles | |
| ES2545340T3 (es) | Terapia de la combinación para el tratamiento de enfermedades proliferativas | |
| CN102369204A (zh) | 3-脱氮瓶菌素衍生物 | |
| ES2310860T3 (es) | Profarmaco amida de gemcitabina, composiciones y uso del mismo. | |
| ES2587381T3 (es) | Uso de sapacitabina para tratar una enfermedad proliferativa | |
| ES2928666T3 (es) | Piridinetionas, sus composiciones farmacéuticas y su uso terapéutico para el tratamiento de enfermedades proliferativas, inflamatorias, neurodegenerativas o inmunomediadas | |
| ES2778524T3 (es) | Profármaco mutuo que comprende ácidos grasos de cadena corta y zebularina o 1'-ciano-citarabina para el tratamiento del cáncer | |
| WO2018074387A1 (ja) | Mdm2阻害剤とdnaメチルトランスフェラーゼ阻害剤との併用治療法 | |
| CA2963819A1 (en) | Vitamin e-nucleoside prodrugs | |
| ES2877103T3 (es) | Análogos de pirimidina fluorados y métodos de uso de los mismos | |
| ES2958817T3 (es) | Compuesto de fosfato cíclico nucleosídico de profármaco de citarabina basado en la administración específica al hígado y uso | |
| RU2471486C2 (ru) | Противоопухолевое средство для непрерывного внутривенного введения, содержащее цитидиновое производное | |
| WO2018230479A1 (ja) | ヌクレオシド系抗がん剤又は抗ウィルス剤の5'位シリルエーテル誘導体 | |
| CN104619325A (zh) | 治疗肿瘤的组合药物及其应用 | |
| RU2482855C2 (ru) | Противоопухолевое средство, включающее производное цитидина и карбоплатин | |
| ES2582339T3 (es) | Composiciones que comprenden oligómeros de gemcitabina para su uso en terapia | |
| HK1232229B (zh) | 4’-硫代核苷的新型化合物及其制备方法、药物组合物和应用 |