ES2755424T3 - Análogos de salinomicina que contienen nitrógeno, síntesis y utilización contra células troncales cancerosas y paludismo - Google Patents
Análogos de salinomicina que contienen nitrógeno, síntesis y utilización contra células troncales cancerosas y paludismo Download PDFInfo
- Publication number
- ES2755424T3 ES2755424T3 ES15766107T ES15766107T ES2755424T3 ES 2755424 T3 ES2755424 T3 ES 2755424T3 ES 15766107 T ES15766107 T ES 15766107T ES 15766107 T ES15766107 T ES 15766107T ES 2755424 T3 ES2755424 T3 ES 2755424T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- group
- advantageously
- aryl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC([C@@]1OC([C@@](C)[C@](*)[C@](C)C(C([C@](C(C)C[C@]2C)OC2(C=C[C@]2*)O[C@]2(CC2)O[C@]2(C)[C@@](CC2)O[C@@](C)[C@]2(CC)O)=CC)=O)[C@@](C)CC1)C(*)=O Chemical compound CCC([C@@]1OC([C@@](C)[C@](*)[C@](C)C(C([C@](C(C)C[C@]2C)OC2(C=C[C@]2*)O[C@]2(CC2)O[C@]2(C)[C@@](CC2)O[C@@](C)[C@]2(CC)O)=CC)=O)[C@@](C)CC1)C(*)=O 0.000 description 1
- GKLDIEGAYOFYNY-KXOMQEMHSA-N CCCCCCCCCCCCN[C@@H]1C=C[C@]2(O[C@H]([C@@H](CC)C([C@@H](C)[C@H](C(C3O[C@@H](CC(O)=O)CC[C@@H]3C)=C)O)=O)[C@@H](C)C[C@H]2C)O[C@]1(CC1)O[C@]1(C)[C@@H](CC1)O[C@@H](C)[C@]1(CC)O Chemical compound CCCCCCCCCCCCN[C@@H]1C=C[C@]2(O[C@H]([C@@H](CC)C([C@@H](C)[C@H](C(C3O[C@@H](CC(O)=O)CC[C@@H]3C)=C)O)=O)[C@@H](C)C[C@H]2C)O[C@]1(CC1)O[C@]1(C)[C@@H](CC1)O[C@@H](C)[C@]1(CC)O GKLDIEGAYOFYNY-KXOMQEMHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/12—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains three hetero rings
- C07D493/20—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
- A61K31/366—Lactones having six-membered rings, e.g. delta-lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/664—Amides of phosphorus acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/14—Drugs for genital or sexual disorders; Contraceptives for lactation disorders, e.g. galactorrhoea
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/06—Heterocyclic radicals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Pregnancy & Childbirth (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gynecology & Obstetrics (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Compuesto de fórmula (I), enantiómeros, mezcla de enantiómeros, diastereoisómeros y mezcla de diastereoisómeros del mismo:**Fórmula** en el que: - W se selecciona de entre el grupo que consiste en =O; -NR1R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; - NR3-(CH2)n-N+R6R7R8 y -O-(CH2)n-N+R6R7R8; - X se selecciona de entre el grupo que consiste en =O, -OH; -NR1R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8 y -O-(CH2)n-N+R6R7R8, - Y se selecciona de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR4R5; y -NR3-(CH2)n-N+R6R7R8, R1 y R2, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); cicloalquilo C3-C16; arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1- C6)-heteroarilo; o R1 representa H y R2 representa OR9, en el que R9 es H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo; R3 se selecciona de entre el grupo que consiste en H; alquilo C1-C6; alquil (C1-C6)-arilo; R4 y R5, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C1-C6); arilo y alquil (C1-C6)-arilo; R6, R7 y R8, idénticos o diferentes, se seleccionan de entre el grupo que consiste en alquilo (C1-C6); arilo y alquil (C1-C6)-arilo; - Z se selecciona de entre el grupo que consiste en OH; NHNR9R10; NHOC(O)R11; N(OH)-C(O)R11; OOH, SR12; 2-aminopiridina; 3-aminopiridina; -NR3-(CH2)n-NR4R5; y -NR3-(CH2)n-OH, en el que: R9 y R10, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo; R11 se selecciona de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1-C6)-heteroarilo; R12 se selecciona de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1-C6)-heteroarilo, n = 0, 2, 3, 4, 5 o 6.
Description
DESCRIPCIÓN
Análogos de salinomicina que contienen nitrógeno, síntesis y utilización contra células troncales cancerosas y paludismo.
Campo de la invención
La presente invención se refiere a derivados amino de salinomicina, a procedimientos para su preparación y dichos derivados para la utilización como fármaco, en particular en el tratamiento del cáncer y en el tratamiento del paludismo.
Estado de la técnica
La salinomicina es un poliéter monocarboxílico que presenta propiedades inofóricas, de la fórmula siguiente:

Hasta el momento, la salinomicina se ha utilizado ampliamente en medicina veterinaria como antibiótico y antiprotozoario.
Recientemente, el cribado de 16000 compuestos ha permitido identificar un pequeño número de compuestos capaces de eliminar selectivamente las células troncales cancerosas (CSC) y las células iniciadoras de tumor (TIC) sin afectar a las células normales. Dicho estudio demuestra que la utilización de CSC y TIC como diana permite la regresión de la masa tumoral y evita las metástasis.
En dicho estudio, se ha identificado la salinomicina como compuesto potente contra estas células, siendo capaz de reducir la cantidad de TIC con una eficacia 100 veces superior a la del paclitaxel, un fármaco anticanceroso utilizado comúnmente.
Otros estudios asimismo han demostrado que la salinomicina induce la muerte celular en células de leucemia linfocítica crónica mediante la inhibición de la ruta de Wnt; en células de cáncer de próstata, mediante la inducción de especies de oxígeno reactivo y mediante la inducción de una caída del potencial de la membrana mitocondrial. Sin embargo, la salinomicina adolece de la desventaja de que es neurotóxica, conduciendo a neuropatías periféricas. Además, su actividad contra las CSC y TIC sigue siendo moderada.
Se han descrito en la técnica anterior análogos de salinomicina. Las modificaciones de la salinomicina consisten esencialmente en la sustitución de la función ácido 1-carboxílico por un éster, o la acilación del grupo 20-hidroxilo de la salinomicina.
Por lo tanto, existe una necesidad de mejora de la actividad de la salinomicina contra las CSC y las TIC, aunque asimismo de diseño de derivados de dicho compuesto que presenten una neurotoxicidad reducida.
En el contexto de la presente invención se ha descubierto que los derivados 9- y/o 11- y/o 20-amino de la salinomicina presentan una actividad superior contra las CSC y las TIC.
La capacidad de la salinomicina de acumularse en los lisosomas y de estimular la formación de especies de oxígeno reactivo (EOR) y la permeabilidad de la membrana lisosómica sugiere una función de los derivados de salinomicina en la quelación del hierro para catalizar la reacción de Fenton (es decir, la conversión de H2O2 en EOR mediada por complejos de hierro). Por lo tanto, los derivados de salinomicina que porten modificaciones químicas que favorezcan la unión del hierro y la reacción de Fenton resultan de considerable interés.
Por lo tanto, la presente invención se refiere a derivados 9- y/o 11- y/o 20-amino de salinomicina, enantiómeros, mezcla de enantiómeros, diastereoisómeros y mezcla de diastereoisómeros de los mismos, de fórmula (I):

en la que:
- W se selecciona de entre el grupo que consiste en =O; -NRi R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NRs-(CH2)n-N R6R7R8; y -O-(CH2)n-N R6R7R8,
- X se selecciona de entre el grupo que consiste en =O, OH; -NRi R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N R6R7R8, y -O-(CH2)n-N+R6R7R8,
- Y se selecciona de entre el grupo que consiste en -NRi R2; -NR3-(CH2)n-NR4R5, y -NR3-(CH2)n-N+R6R7R8, Ri y R2, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C i-C i6), alquenilo (C3-C16), alquinilo (C3-C16), cicloalquilo (C3-C16), arilo, heteroarilo,
alquil (C1-C6)-arilo, alquil (C1-C6)-heteroarilo, o R1 representa H y R2 representa OR9, en el que R9 es H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo,
R3 se selecciona de entre el grupo que consiste en H, alquilo (C1-C6), alquil (C1-C6)-arilo,
R4 y R5, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (C1-C6), arilo, alquil (C1-C6)-arilo,
R6, R7 y R8, idénticos o diferentes, se seleccionan de entre el grupo que consiste en alquilo (C1-C6), arilo, alquil (C1-C6)-arilo,
Z es un grupo funcional capaz de quelar sales de hierro, tales como OH, NHNR9R10 (hidrazina), NHOC(O)R11 (O-acil hidroxilamina), N(OH)-C(O)R11 (N-acil hidroxilamina), OOH, SR12, 2-aminopiridina, 3-aminopiridina, -NR3-(CH2)n-NR4R5; -NR3-(CH2)n-OH, en los que:
R9 y R10, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo,
R11 se selecciona de entre el grupo que consiste en H, alquilo (C1-C16), alquenilo (C3-C16), alquinilo (C3-C16), arilo, heteroarilo, alquil (C1-C6)-arilo, alquil (C1-C6)-heteroarilo,
R12 se selecciona de entre el grupo que consiste en H, alquilo (C1-C16), alquenilo (C3-C16), alquinilo (C3-C16), arilo, heteroarilo, alquil (C1-C6)-arilo, alquil (C1-C6)-heteroarilo y n = 0, 2, 3, 4, 5 o 6.
Ventajosamente, los derivados 9- y/o 11- y/o 20-amino de salinomicina, enantiómeros, mezcla de enantiómeros, diastereoisómeros y mezcla de diastereoisómeros de la invención son de fórmula (I):

en la que:
- W se selecciona de entre el grupo que consiste en =O; -NR1R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N R6R7R8; y -O-(CH2)n-N R6R7R8,
- X se selecciona de entre el grupo que consiste en =O, OH; -NR1R2 ; -NR3-(CH2)n-NR4R5 ; -O-(CH2)n-NR4 R5 ;
-NRa-(CH2)n-N+R6RyR8, y -O-(CH2)n-N+RaRyR8 ,
- Y se selecciona de entre el grupo que consiste en -NR1R2 ; -NR3-(CH2)n-NR4R5, y -NR3-(CH2)n-N+R6RyR8, R1 y R2 , idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (Ci-Cia), alquenilo (C3-Cia), alquinilo (C3-Cia), arilo, heteroarilo, alquil (Ci-Ca)-arilo, alquil (Ci-Ca)-heteroarilo, o Ri representa H y R2 representa OR9, en los que Rg es H, alquilo (Ci-Ca), arilo y alquil (Ci-Ca)-arilo, R3 se selecciona de entre el grupo que consiste en H, alquilo (Ci-Ca), alquil (Ci-Ca)-arilo,
R4 y R5 , idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (Ci-Ca), arilo, alquil (Ci-Ca)-arilo,
Ra, Ry y R8, idénticos o diferentes, se seleccionan de entre el grupo que consiste en alquilo (Ci-Ca), arilo, alquil (Ci-Ca)-arilo,
n=2, 3, 4, 5 o a.
Z es un grupo funcional capaz de quelar sales de hierro, tales como OH, NHNR9Rio (hidrazina), NHOC(O)Rii (O-acil hidroxilamina), N(OH)-C(O)Rii (N-acil hidroxilamina), OOH, SRi2 , 2-aminopiridina, 3-aminopiridina, -NR3-(CH2)n-NR4R5 ; -NR3-(CH2)n-OH, en los que:
R9 y Rio, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (Ci-Ca), arilo y alquil (Ci-Ca)-arilo,
Rii se selecciona de entre el grupo que consiste en H, alquilo (Ci-Cia), alquenilo (C3-Cia), alquinilo (C3-Cia), arilo, heteroarilo, alquil (Ci-Ca)-arilo, alquil (Ci-Ca)-heteroarilo,
R i2 se selecciona de entre el grupo que consiste en H, alquilo (Ci-Cia), alquenilo (C3-Cia), alquinilo (C3-Cia), arilo, heteroarilo, alquil (Ci-Ca)-arilo, alquil (Ci-Ca)-heteroarilo.
Ventajosamente, n=0, 2, 3 o 4.
Ventajosamente, W y/o X se seleccionan de entre el grupo que consiste en -NRiR2 ; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5 , más ventajosamente -NRiR2, y/o Y se selecciona de entre el grupo que consiste en -NRiR2 ; -NR3-(CH2)n-NR4R5 , más ventajosamente -NRiR2.
Ventajosamente, Ri y R2, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (Ci-Cia), ventajosamente alquilo (C3-C i4), más ventajosamente alquilo (C8-C i4); alquenilo (C3-Cia), ventajosamente alquenilo (C3-C5); alquinilo (C3-Cia), ventajosamente alquinilo (C3-C5); cicloalquilo (C3-Cia), ventajosamente cicloalquilo (C3-Ca), y alquil (Ci-Ca)-heteroarilo, ventajosamente CH2-piridinilo.
Ri y R2 , idénticos o diferentes, asimismo pueden seleccionarse de entre grupo que consiste en H, alquilo (Ci-Cia), ventajosamente alquilo (C8-C i4); alquinilo (C3-Cia), ventajosamente alquinilo (C3-C5), y alquil (Ci-Ca)-heteroarilo, ventajosamente CH2-piridinilo.
Ventajosamente, Ri y R2 no son ambos H.
Más ventajosamente, Ri es H y R2 se selecciona de entre el grupo que consiste en alquilo (Ci-Cia), ventajosamente alquilo (C3-C i4), más ventajosamente alquilo (C8-C i4); alquenilo (C3-Cia), ventajosamente alquenilo (C3-C5); alquinilo (C3-Cia), ventajosamente alquinilo (C3-C5); cicloalquilo (C3-Cia), ventajosamente cicloalquilo (C3-Ca) y alquil (Ci-Ca)-heteroarilo, ventajosamente CH2-piridinilo.
Ri asimismo puede ser H y R2 asimismo puede seleccionarse de entre grupo que consiste en alquilo (Ci-Cia), ventajosamente alquilo (C8-C i4), y alquinilo (C3-Cia), ventajosamente alquinilo (C3-C5), y alquil (Ci-Ca)-heteroarilo, ventajosamente CH2-piridinilo.
Ventajosamente, R3 se selecciona de entre el grupo que consiste en H y alquilo (Ci-Ca). Preferentemente, R3 es H.
Ventajosamente, R4 y R5, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H y alquilo (Ci-Cia). Más ventajosamente, R4 y R5 son H o alquilo (Ci-Ca). Preferentemente, R4 y R5 son idénticos. En una forma de realización ventajosa, el grupo -(CH2)n-NR4R5 se selecciona de entre el grupo que consiste en -(CH2)2-N(CH3)2, -(CH2)3-N(CH3)2, -(CH2)2-NH2 y -(CH2)3-NH2.
Ventajosamente, Ra, Ry y R8, idénticos o diferentes, se seleccionan de entre el grupo que consiste en alquilo (CiC6) y arilo. Más ventajosamente, R5, R7 y R8 son alquilo (Ci-C6). Preferentemente, R6, R7 y R8 son idénticos. En una forma de realización ventajosa, el grupo -(CH2)n-N+R6R7R8 se selecciona de entre el grupo que consiste en -(CH2)2-N+(CHs)s y -(CH2)s-N+(CH3)s.
Ventajosamente, Z es OH, OOH, NHNH
2
, NHOH o NH
2
OH, preferentemente OH. En otra forma de realización particular, Z es SH.
En una primera forma de realización según la presente invención, el compuesto es un derivado 9-, 11-, 20-triamino de salinomicina, un enantiómero, una mezcla de enantiómeros, un diastereoisómero y una mezcla de diastereoisómeros de los mismos de fórmula (Ia):

en la que W, X, Y, Z, R 1 , R 2 , R 3 , R 4 , R 5 , R6, R 7 , R8 y n son tal como se define en la fórmula (I).
En una segunda forma de realización según la presente invención, el compuesto es un derivado diamino de salinomicina, en el que W y X se seleccionan de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8 y -O-(CH2)n-N+R6R7R8, e Y se selecciona de entre el grupo que consiste en -NR 1 R 2 ; -NR3-(CH2)n-NR4R5; y -NR3-(CH2)n-N+R6R7R8, de fórmula (lb),
en la que W, X, Y, Z, R1, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (I).
El compuesto de fórmula (Ib) puede ser un derivado 9-, -20-diamino de salinomicina, un enantiómero, una mezcla de enantiómeros, un diastereoisómero y una mezcla de diastereoisómeros de los mismos de fórmula (Ib1):

en la que X se selecciona de entre el grupo que consiste en -NR 1 R 2 ; -NR 3 -(CH 2 )n-NR 4 R 5 ; -O-(CH 2 )n-NR 4 R 5 ; -NR 3 -(CH 2 )n-N+R 6 R 7 R 8 y -O-(CH 2 )n-N+R 6 R 7 R 8 , e Y se selecciona de entre el grupo que consiste en -NR 1 R 2 ; -NR3-(CH2)n-NR4R5 y -NR3-(CH2)n-N+R6R7R8, y
Z, R1, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (lb).
La descripción da a conocer además un compuesto de fórmula (Ib) puede ser un derivado 9-, -11-diamino de salinomicina, un enantiómero, una mezcla de enantiómeros, un diastereoisómero y una mezcla de diastereoisómeros de los mismos de fórmula (Ib2):

en la que W y X, idénticos o diferentes, se seleccionan de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR 4 R 5 ; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8 y -O-(CH2)n-N+R6R7R8 y
Z, Ri, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (Ib),
El compuesto de fórmula (Ib) puede ser un derivado 11-, -20-diamino de salinomicina, un enantiómero, una mezcla de enantiómeros, un diastereoisómero y una mezcla de diastereoisómeros de los mismos de fórmula (Ib3):

en la que W se selecciona de entre el grupo que consiste en-NR1R2; -NR3-(CH2)n.NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8y -O-(CH2)n-N+R6R7R8, Y se selecciona de entre el grupo que consiste en -NR1R2; -NR3-(CH2VNR4R5; and -NR3-(CH2)n-N+R6R7R8,
X es OH o =O, y
Z, R1, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (lb).
En una forma de realización ventajosa según la presente invención, el compuesto de fórmula (I) es un derivado monoamina de salinomicina de fórmula (Ic) y sólo Y es un grupo -NR1R2; -NR3-(CH2)n-NR4R5 o -NR3-(CH2)n-N+R6R7R8 y
W, X, Y, Z, R1, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (I).
La descripción da a conocer además un compuesto de fórmula (Ic) que puede ser un derivado 9-amino de salinomicina de fórmula (Ic1):
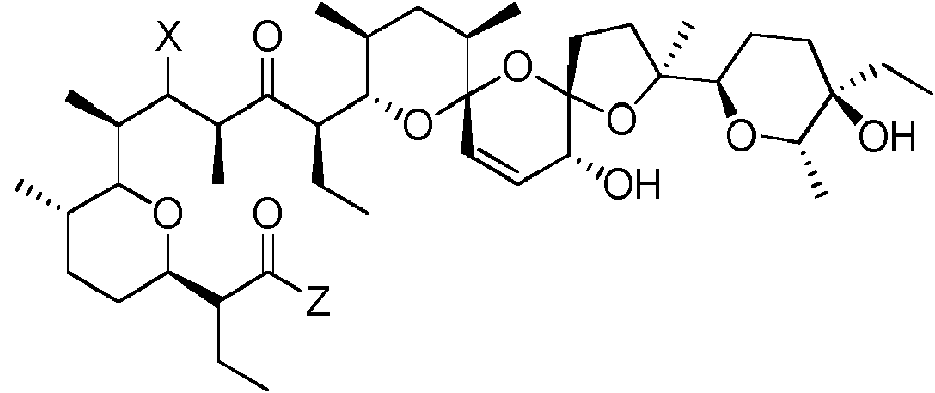
en la que:
X se selecciona de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8 y -O-(CH2)n-N+R6R7R8; y
Z, R1, R2, R3, R4, R5, R6, R7, R8 y n son tal como se define en la fórmula (Ic).
Ventajosamente, Z es OH.
En el contexto de la presente invención en efecto se ha descubierto que la presencia de un grupo amino y el ácido carboxílico en la posición 1 dando como resultado compuestos que presentan una actividad mejorada contra CSC y TIC.
La descripción da a conocer además un compuesto de fórmula (Ic) que puede ser un derivado 11-amino de salinomicina de fórmula (Ic2):

en la que:
W se selecciona de entre el grupo que consiste en -NR i R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8, y -O-(CH2)n-N+R6R7R8, y
Z, Ri, R2, R3, R4, R5, R6, R7, R8 y n son p tal como se define en la fórmula (Ic).
El compuesto de fórmula (Ic) puede ser ventajosamente un derivado 20-amino de salinomicina de fórmula (Ic3):

en la que:
X se selecciona de entre el grupo que consiste en OH y =O,
Y se selecciona de entre el grupo que consiste en -NR1R2 ; -NR3-(CH2)n-NR4R5 y-NR3-(CH2)n-N+R6R7R8 , y Z, Ri, R2, R3 , R4 , R5, R6, R7, R8 y n son tal como se define en la fórmula (I).
En una realización ventajosa, en los compuestos de fórmula (Ic3), X es OH, Z es OH e Y es -NR1R2. Más ventajosamente, R1 es H y R2 se selecciona de entre el grupo que consiste en alquilo (C1-C16), ventajosamente alquilo (C8-C14), alquenilo (C3-C16), ventajosamente alquenilo (C3-C5), alquinilo (C3-C16), ventajosamente alquinilo (C3-C5), cicloalquilo (C3-C16), ventajosamente cicloalquilo (C3-C6) y alquil (C1-C6)-heteroalquilo, ventajosamente (CH2)-piridinilo.
Los inventores en efecto han encontrado que la presencia de un grupo amino y el ácido carboxílico en la posición 1 dan como resultado compuestos que presentan una actividad mejorada contra CSC y TIC.
En otra forma de realización, X es =O, Y es NR1R2 y Z es NHOH. Más ventajosamente, R1 es H y R2 es (CH2)-piridinilo, preferentemente CH2-(2-piridinilo). Alternativamente, R1 es H y R2 es cicloalquilo (C3-C16) y alquinilo (C3-C16).
En una forma de realización particular, en el caso de que Z sea -NHOH, W sea =O y X sea -OH, Y no es un grupo propargilo.
En una forma de realización particular, en el caso de que Z sea -OH, W sea =O y X sea -OH, Y no es NCH2CH2N(CH3)2.
Los compuestos de fórmula (I) pueden seleccionarse ventajosamente del grupo que consiste en:

Ċ


Los compuestos de fórmula (I) pueden seleccionarse de entre grupo que consiste en:


En una forma de realización particular, W es =O, X es -OH, Y es -NR1R2 preferentemente Ri es H y R2 es cicloalquilo C3-C16, preferentemente ciclopropilo y Z es -OH.
En una forma de realización particular, W es =O, X es -OH, Y es -NR1R2 preferentemente R1 es H y R2 es alquinilo C3-C16, preferentemente propargilo y Z es -OH.
La invención se refiere además a una composición farmacéutica que comprende por lo menos un compuesto de fórmula (I) tal como se ha definido anteriormente, una sal, solvato o hidrato farmacéuticamente aceptable del mismo, y por lo menos un excipiente farmacéuticamente aceptable.
La composición farmacéutica puede comprender además por lo menos otro fármaco anticanceroso, tal como adriamicina y ciclofosfamida (AC), docetaxel (Taxol), trastuzumab, degarelix, capecitabina, ifosfamida o cisplatino. Ventajosamente, la composición farmacéutica comprende además adriamicina y ciclofosfamida (AC) o docetaxel (Taxol).
Las composiciones farmacéuticas de la invención pueden estar destinadas a la administración oral, sublingual, subcutánea, intramuscular, intravenosa, transdérmica, tópica o rectal. El principio activo puede administrarse en formas unitarias de administración, mezclado con portadores farmacéuticos convencionales, en seres humanos o en seres humanos.
En el caso de que se prepare una composición sólida en la forma de tabletas, el principio activo principal se mezcla con un vehículo farmacéutico y otros excipientes convencionales conocidos por el experto en la materia.
Los compuestos de la invención pueden utilizarse en una composición farmacéutica en una dosis comprendida entre 0,01 mg y 1000 mg al día, administrados en una única dosis o en varias dosis a lo largo del día, por ejemplo dos veces al día. La dosis administrada diariamente está comprendida ventajosamente entre 5 mg y 500 mg, y más ventajosamente, entre 10 mg y 200 mg. Sin embargo, puede resultar necesario utilizar dosis fuera de estos
intervalos, lo que puede ser advertido por el experto en la materia.
Se ha demostrado que la salinomicina, además de resultar en la inhibición de la proliferación o en la inducción de apoptosis en CSC y TIC en una diversidad de tipos de cáncer, incluyendo los cánceres de mama, sangre, pulmón, páncreas y colon, dificulta la migración de estas células.
Por lo tanto, la presente invención se refiere a un compuesto de fórmula (I) tal como se ha definido anteriormente o una composición farmacéutica tal como se ha definido anteriormente para la utilización como fármaco. La presente invención se refiere además a un compuesto de fórmula (I) tal como se ha definido anteriormente o a una composición farmacéutica tal como se ha definido anteriormente, para la utilización en el tratamiento del cáncer, tal como carcinoma, sarcoma, trastornos metastásicos, cáncer de próstata, colon, pulmón, mama o hígado, y leucemias, ventajosamente cáncer de mama y/o para la prevención de la recaída del cáncer y/o las metástasis.
Otro aspecto de la presente invención se refiere a la utilización de un compuesto de fórmula (I) tal como se ha definido anteriormente o una composición farmacéutica tal como se ha definido anteriormente, para la preparación de un medicamento, ventajosamente para la utilización en el tratamiento del cáncer, tal como carcinoma, sarcoma, trastornos metastásicos, cáncer de próstata, colon, pulmón, mama o hígado, y leucemias, ventajosamente cáncer de mama, y/o en la prevención de la recaída del cáncer y/o las metástasis.
Se da a conocer además un método de tratamiento del cáncer, tal como carcinoma, sarcoma, trastornos metastásicos, cáncer de próstata, colon, pulmón, mama o cáncer, y leucemias, ventajosamente cáncer de mama, y/o la prevención de la recaída del cáncer y/o metástasis, que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) tal como se ha definido anteriormente o una composición farmacéutica tal como se ha definido anteriormente en una persona que lo necesita.
El compuesto de fórmula (I) tal como se ha definido anteriormente puede administrarse solo o en combinación con una terapia contra el cáncer, por ejemplo con otros fármacos anticancerosos. Los fármacos anticancerosos son conocidos de la técnica.
Por lo tanto, un aspecto adicional de la invención se refiere a un producto terapéutico que comprende: a) el compuesto de fórmula (I) tal como se ha definido anteriormente, y b) otro compuesto quimioterapéutico, tal como adriamicina y ciclofosfamida (AC), docetaxel (Taxol), trastuzumab, degarelix, capecitabina, ifosfamida o cisplatino, ventajosamente adriamicina y ciclofosfamida (AC), docetaxel (Taxol), como producto de combinación para la utilización simultánea, separada o escalonada como medicamento, en particular en el tratamiento del cáncer, ventajosamente el cáncer de mama.
La expresión “producto de combinación” se refiere en la presente memoria a que el compuesto de fórmula (I) de la presente invención se administra en el individuo tratado de esta manera anteriormente, durante (incluyendo concurrentemente con; preferentemente coformulado con) y/o después del tratamiento del individuo con el otro fármaco anticanceroso. Las formulaciones pueden presentarse convenientemente en forma de administración unitaria mediante métodos conocidos por el experto en la materia. Preferentemente, el kit de componentes contiene instrucciones que indican la utilización de la forma de administración para conseguir un efecto deseable y la cantidad de forma de administración que debe administrarse durante un periodo de tiempo especificado. Preferentemente, dicho producto de combinación está destinado al tratamiento del cáncer, tal como carcinoma, sarcoma, trastornos metastásicos, cáncer de próstata, colon, pulmón, mama o hígado, y leucemias, ventajosamente cáncer de mama y/o para la prevención de la recaída del cáncer y/o las metástasis.
Debido a que los compuestos de fórmula (I) tal como se ha definido anteriormente son capaces de inhibir la autofagia, la presente invención se refiere además a un compuesto de fórmula (I) para la utilización en el tratamiento de enfermedades que involucran autofagia, en particular el paludismo.
Otro aspecto de la presente invención se refiere a la utilización de un compuesto de fórmula (I) tal como se ha definido anteriormente, para la preparación de un medicamento, ventajosamente para la utilización en el tratamiento de enfermedades que involucran autofagia, en particular el paludismo.
Se da a conocer además un método de tratamiento de enfermedades en las que está involucrada la autofagia, en particular el paludismo, que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de fórmula (I) tal como se ha definido anteriormente en una persona que lo necesita.
Los compuestos de fórmula (I) pueden prepararse siguiendo los métodos ilustrados en el esquema 1 y descritos posteriormente.

Los grupos NR1R2; -NR3-(CH2)n-NR4R5 y -NR3-(CH2)n-N+R6R7R8 en la posición 20 de la salinomicina pueden introducirse mediante el método siguiente:
(a) Oxidar el alcohol alílico en la posición 20 de la salinomicina en la cetona a,p-insaturada.
Los métodos para la oxidación de los alcoholes alílicos son conocidos de la técnica. Ventajosamente, dicha oxidación se lleva a cabo con MnO2.
(b) Proteger el ácido carboxílico en la posición 1 de la cetona a,p-insaturada de la salinomicina; puede utilizarse cualquier grupo protector adecuado para ácidos carboxílicos. Ventajosamente, el ácido carboxílico se protege en forma de un éster, tal como éster metílico o un éster alílico. El ácido carboxílico en la posición 1 asimismo puede protegerse antes de la oxidación en cetona a,p-insaturada.
Los grupos protectores adecuados se dan a conocer, por ejemplo, en Greene, "Protective Groups In Organic synthesis", John Wiley & Sons, New York (1981).
(c) Hacer reaccionar la cetona a,p-insaturada con una amina y simultánea o posteriormente reducir la imina. Los métodos para preparar aminas mediante aminación reductora son conocidos de la técnica. Ventajosamente, la imina se forma haciendo reaccionar la amina en un solvente polar en presencia de un ácido. En una forma de realización particular, la imina se forma en una mezcla de un alcohol, tal como metanol o etanol, y ácido acético. La reducción de la imina en amina se lleva a cabo ventajosamente con un borohidruro, tal como borohidruro sódico o cianoborohidruro sódico, en presencia de una sal de cerio, tal como tricloruro de cerio. CeCh.
(d) Desproteger el éster en la posición 1, proporcionando el ácido carboxílico.
Los métodos para la desprotección de un éster se dan a conocer en, por ejemplo, Greene, "Protective Groups In Organic synthesis", John Wiley & Sons, New York (1981).
Los grupos NR1R2; -NR3-(CH2)n-NR4R5 y -NR3-(CH2)n-N+R6R7R8 en la posición 11 pueden obtenerse haciendo reaccionar el grupo ceto en la posición 11 con una amina y la reducción simultánea o posterior de la imina. Los métodos para preparar aminas mediante aminación reductora son conocidos de la técnica. Ventajosamente, la imina se forma haciendo reaccionar la amina en un solvente polar en presencia de un ácido. En una forma de realización particular, la imina se forma en una mezcla de un alcohol, tal como metanol o etanol, y ácido acético. La reducción de la imina en amina se lleva a cabo ventajosamente con un borohidruro, tal como borohidruro sódico o cianoborohidruro sódico.
El ácido carboxílico y/o los grupos hidroxilo de la salinomicina, en caso de estar presentes, pueden protegerse. Los grupos protectores adecuados se dan a conocer, por ejemplo, en Greene, "Protective Groups In Organic synthesis", John Wiley & Sons, New York (1981). Ventajosamente, el grupo protector es un grupo trietilsililo. Los grupos NR1R2 ; -NR3-(CH2)n-NR4R5 y -NR3-(CH2),-N+R6R7Rs en la posición 9 pueden introducirse mediante las etapas siguientes:
(a) protección de los grupos hidroxilo en las posiciones 11, 20 y 28 de la salinomicina,
(b) transformación del grupo hidroxilo en la posición 9 en un grupo saliente.
El grupo saliente puede ser, por ejemplo, un sulfonato, tal como un mesilato o un trifluorometilsulfonato. Los métodos para la sustitución de un grupo hidroxilo con una amina son conocidos de la técnica:
(c) hacer reaccionar el producto obtenido en la etapa (b) con una amina apropiada,
(e) desproteger los grupos hidroxilo y el ácido carboxílico.
Los grupos -O-(CH2)n-NR4R5 y -O-(CH2)n-N+R6R7Rs pueden introducirse partiendo de los análogos 9, 11 y/o 20 de la salinomicina.
El grupo hidroxilo en la posición 11 de la salinomicina puede obtenerse mediante reducción del grupo ceto en esta posición utilizando métodos conocidos de la técnica. La cetona puede reducirse, por ejemplo, con borohidruro sódico en un alcohol, tal como metanol o etanol.
La reacción se lleva a cabo mediante las etapas siguientes:
(a) protección del ácido carboxílico en la posición 1 y los grupos hidroxilo en las posiciones 9, 11 y/o 20, en caso de hallarse presentes, y el grupo hidroxilo en la posición 28.
Ventajosamente, el ácido carboxílico se protege en forma de un éster alílico.
(b) Transformación del grupo hidroxilo en un grupo saliente.
El grupo saliente puede ser, por ejemplo, un sulfonato, tal como un mesilato o un trifluorometilsulfonato. Los métodos para transformar un grupo hidroxilo en un grupo saliente son conocidos de la técnica. La reacción se lleva a cabo preferentemente en presencia de una base, tal como piridina.
(c) Hacer reaccionar el producto obtenido en la etapa (b) con un compuesto de fórmula HO-(CH2)n-NR4R5 o HO-(CH2)n-N+RaR7R8 en la presencia de una base.
Ventajosamente, la reacción se lleva a cabo con una base fuerte, tal como hidruro sódico. Preferentemente, el alcóxido se prepara por separado, antes de hacerse reaccionar con el producto obtenido en la etapa (b). (d) Desprotección de los grupos hidroxilo y el ácido carboxílico.
Por lo tanto, la presente invención se refiere además a un procedimiento para preparar un derivado 20-amino, 9-, 20-diamino o 9-, 11-, 20-triamino de la salinomicina de fórmula (I), en la que Y se selecciona de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR4R5 y -NR3-(CH2)n-N+R6R7R8 , en los que R1, R2, R3, R4 , R5 , R6, R7, R8 y n son tal como se ha definido para la fórmula (I), que comprende las etapas de:
(a) hacer reaccionar un compuesto de fórmula (II):

en la que:
X es tal como se define en la reivindicación 1, opcionalmente protegido,
GP1 es un grupo protector de ácido carboxílico, ventajosamente metilo,
con una amina de fórmula R2NH2 o NH2-(CH2)n-NR4R5, o NH2-(CH2)n-N+R6R7R8 ,
(b) reducir la imina obtenida en la etapa (a), ventajosamente con un borohidruro, en presencia de una sal de cerio, tal como tricloruro de cerio,
(c) desproteger el ácido carboxílico en la posición 1 ,
(d) opcionalmente alquilar la amina.
La presente descripción da a conocer además un procedimiento para preparar un derivado 9-amino, 9-, 20-diamino o 9-, 11-, 20-triamino de la salinomicina de fórmula (I), en la que Y se selecciona de entre el grupo que consiste en --O-(CH2)n-NR4R5 y -O-(CH2)n-N+RaR7R8, en los que R4, R5, R6, R7 , R8 y n son tal como se ha definido para la fórmula (I), que comprende las etapas de:
(a) hacer reaccionar un compuesto de fórmula (III):

en la que:
X es tal como se define en la fórmula (I), opcionalmente protegido,
OL1 es un grupo saliente, tal como un sulfonato, ventajosamente un mesilato,
GP1 es un grupo protector de ácido carboxílico, tal como metilo o alilo, ventajosamente alilo;
GP2 es un grupo protector de hidroxilo, ventajosamente trietilsililo, con un compuesto de fórmula M-O-(CH2V N R 4R5 o M-O-(CH2)n-N+RaR7R8, en los que:
M es un metal seleccionado del grupo que consiste en Na, K y Li,
(b) desproteger el ácido carboxílico en la posición 1 y los grupos hidroxilo.
Definiciones:
Los compuestos de fórmula (I), en la que X o Y es un grupo -NR3-(CH2)n-N+R6R7R8 o-O-(CH2)n-N+R6R7R8 son betaínas. En ese caso, el ácido carboxílico en la posición 1 de la salinomicina se encuentra en la forma del carboxilato, es decir, Z será O' en lugar de OH.
Dentro de los grupos, radicales o fragmentos definidos en la descripción y en las reivindicaciones, el número de átomos de carbono se especifica entre paréntesis. Por ejemplo, alquilo (C1-C16) designa un grupo o radical alquilo con 1 a 16 átomos de carbono.
Para los grupos que comprenden dos o más subgrupos, la unión se indica con “-”. Por ejemplo “alquil (C1-C6)-arilo” indica un radical alquilo unido a un radical arilo, en el que el alquilo está unido al resto de la molécula.
En el contexto de la presente invención, la expresión “alquilo (C1-C16)” designa una cadena hidrocarburo lineal o ramificada acíclica saturada opcionalmente sustituida que comprende 1 a 16 átomos de carbono. Entre los ejemplos de grupos de alquilo (C1-C16) se incluyen metilo, etilo, propilo, butilo, pentilo, hexilo, heptilo, octilo y dodecilo. A menos que se indique explícitamente, las definiciones propilo, butilo, pentilo, hexilo, dodecilo, etc. incluyen todos los isómeros posibles. Por ejemplo, butilo comprende n-butilo, isobutilo, sec-butilo y terc-butilo. En el contexto de la presente invención, la expresión “alquenilo (C3-C16)” designa una cadena hidrocarburo lineal o ramificada acíclica saturada opcionalmente sustituida que comprende 3 a 16 átomos de carbono, por lo menos dos de los cuales están unidos mediante un doble enlace. Entre los ejemplos de "-alquenilo (C3-C16)" se incluyen propenilo, butenilo, pentenilo o hexenilo. A menos que se indique explícitamente, las definiciones de propenilo, butenilo, pentenilo y hexenilo incluyen todos los isómeros posibles.
En el contexto de la presente invención, la expresión “alquinilo (C3-C16)” designa una cadena hidrocarburo lineal o ramificada acíclica saturada opcionalmente sustituida que comprende 3 a 16 átomos de carbono, por lo menos dos de los cuales están unidos mediante un triple enlace. Entre los ejemplos de "-alquinilo (C3-C16)" se incluyen propinilo, butinilo, pentinilo o hexinilo. A menos que se indique explícitamente, las definiciones de propinilo, butinilo, pentinilo y hexinilo incluyen todos los isómeros posibles.
En el contexto de la presente invención, la expresión “cicloalquilo (C3-C16)” designa una cadena hidrocarburo saturada cíclica saturada opcionalmente sustituida que comprende 1 a 16 átomos de carbono. Entre los ejemplos de grupos cicloalquilo (C3-C16) se incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo y ciclododecilo. Ventajosamente, el grupo cicloalquilo (C3-C16) se selecciona de ciclopropilo, ciclobutilo y ciclopentilo
La expresión “sustituido opcionalmente” tal como se utiliza en la presente memoria se refiere a que cualquiera de los átomos de hidrógeno puede sustituirse por un sustituyente, tal como flúor.
El término “arilo” designa un anillo monocíclico aromático que puede fusionarse con un segundo anillo saturado, insaturado o aromático. El término arilo incluye, aunque sin limitarse a los ejemplos siguientes, fenilo, indanilo, indenilo, naftilo, antracenilo, fenantrenilo, tetrahidronaftilo y dihidronaftilo. Los arilos preferidos son los que comprenden un anillo aromático de seis elementos. El grupo arilo puede sustituirse con uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alcoxi, halógeno, hidroxilo, amino, nitro, ciano, trifluoro, ácido carboxílico o éster carboxílico. Son ejemplos de grupos fenilo sustituidos, metoxifenilo, dimetoxifenilo, trimetoxifenilo, fluorofenilo y trifluorometilfenilo.
El término “alquil (C1-C6)-arilo” designa, en el contexto de la presente invención, un grupo arilo, tal como se ha definido anteriormente, unido al resto de la molécula mediante una cadena alquilo que contiene 1 a 6 átomos de carbono. Ventajosamente, el “alquil (C1-C6)-arilo” es un bencilo sustituido o no sustituido. Entre los ejemplos de grupos bencilo sustituidos se incluyen metoxibencilo, cianobencilo, nitrobencilo o fluorobencilo.
El término heteroarilo designa un arilo monocíclico o policíclico tal como se ha definido anteriormente en el que uno o más átomos de carbono han sido sustituidos por uno o más heteroátomos seleccionados del grupo que consiste en N, O y S. A menos que se indique explícitamente, el término “heteroarilo” incluye todos los isómeros posibles. Entre los ejemplos de grupos heteroarilo se incluyen furilo, tienilo, imidazolilo, piridilo, pirrolilo, N-alquilpirrolilo, pirimidinilo, pirazinilo, tetrazolilo, triazolilo y triazinilo. El grupo heteroarilo puede sustituirse con uno o más grupos seleccionados independientemente del grupo que consiste en alquilo, alcoxi, halógeno, hidroxilo, amino, nitro, ciano, trifluoro, ácido carboxílico o éster carboxílico. Los heteroarilos preferidos son los que presentan 5 o 6 átomos en el anillo, tales como indolilo, pirrolilo, piridinilo, pirazolilo, triazolilo, furanilo o tienilo.
El término “alquil (C1-C6)-arilo” designa, en el contexto de la presente invención, un grupo heteroarilo, tal como se ha definido anteriormente, unido al resto de la molécula mediante una cadena alquilo que contiene 1 a 6 átomos de carbono. Ventajosamente, el “alquil (C1-C6)-heteroarilo” es un heteroarilo sustituido o alquil C1-heteroariloencilo.
En el contexto de la presente invención, el término “halógeno” designa un átomo de flúor, cloro, bromo o yodo.
Para el propósito de la invención, la expresión “farmacéuticamente aceptable” pretende referirse a que resulta útil para la preparación de una composición farmacéutica y que resulta generalmente seguro y no tóxico para un uso farmacéutico.
La expresión “sal, hidrato o solvato farmacéuticamente aceptable” pretende referirse, en el contexto de la presente invención, a una sal de un compuesto que resulta farmacéuticamente aceptable, tal como se ha definido anteriormente, y que posee la actividad farmacológica del compuesto correspondiente. Dichas sales comprenden:
(1) hidratos y solvatos,
(2) sales de adición de ácido formadas con ácidos inorgánicos, tales como los ácidos clorhídrico, bromhídrico, sulfúrico, nítrico y fosfórico, y similares; o formados con ácidos orgánicos, tales como los ácidos acético, bencenosulfónico, fumárico, glucoheptónico, glucónico, glutámico, glicólico, hidroxinaftoico, 2-hidroxietanosulfónico, láctico, maleico, málico, mandélico, metanosulfónico, mucónico, 2-naftalenosulfónico, propiónico, succínico, dibenzoil-L-tartárico, tartárico, p-toluenosulfónico, trimetilacético y trifluoroacético, y similares, y
(3) sales formadas al sustituir un protón ácido presente en el compuesto por un ion metálico, por ejemplo un ion metal alcalino, un ion alcalinotérreo o un ion de aluminio, o coordinados con una base orgánica o inorgánica. Las bases orgánicas aceptables comprenden dietanolamina, etanolamina, N-metilglucamina, trietanolamina, trometamina y similares. Las bases inorgánicas aceptables comprenden hidróxido de aluminio, hidróxido de calcio, hidróxido potásico, carbonato sódico e hidróxido sódico.
Descripción de las figuras
La figura 1 representa la viabilidad de las células HMLER CD24-(línea continua) y de las células HMLER CD24+ (línea de puntos) a diferentes concentraciones de salinomicina (1A), AM5 (1B), AM9 (1C) o AM13 (1D). El eje Y representa la viabilidad celular y se expresa en porcentaje.
El eje X representa la concentración de cada producto, en pM. Las concentraciones utilizadas son, de izquierda a derecha desde la intersección de los ejes X e Y: 0.0001 pM; 0.001; 0.01 pM; 0.1 pM, 1 pM y 10 pM. Cada punto en la línea representa la viabilidad celular medida a la concentración correspondiente.
La figura 2 es una fotomicrografía de contraste de fases representativa de mamosferas formadas tras 11 días en ausencia de ningún compuesto añadido (Control) o en presencia de una cantidad definida de salinomicina, AM5, AM9 o AM13. Los análogos de sal (AM5, AM13) redujeron el número y tamaño de las mamosferas a concentraciones nanomolares bajas. El tamaño de las mamosferas se correlaciona con la proliferación de las células progenitoras, mientras que el número de mamosferas formado tras pases en serie a densidad clonal se correlaciona con la capacidad de autorrenovación de las células troncales cancerosas primitivas. Una masa más pequeña indica muerte celular y regresión de las mamosferas.
La figura 3A es una fotomicrografía de contraste de fases representativa de mamosferas formadas tras 7 o 14 días en ausencia de ningún compuesto añadido (no tratado) o en presencia de una cantidad definida de salinomicina, AM5, taxol o la combinación de AM5 y taxol. Una masa más pequeña indica muerte celular y regresión de las mamosferas.
La figura 3B representa la cuantificación del número y tamaño de las mamosferas. La combinación de AM5 a una concentración de 15 nM y de taxol a 5 nM reduce el número y tamaño de las mamosferas con una eficacia mejorada respecto a AM5 solo a 15 nM o 5 nM.
La figura 4 representa las curvas de crecimiento tumoral de MCF-7 de ratones tratados con compuesto (3 mg por kg de peso corporal al día, inyección intraperitoneal, n=5). Las inyecciones no letales de un análogo de Sal activo (AM5) inhiben el crecimiento tumoral de cáncer de mama en ratones (n=5; barras de error, s.e.m.). Figura 5: análogo de salinomicina - la muerte celular inducida resulta inhibida por el secuestrador de EOR N-acetilcisteína (NAC). Las estirpes celulares se incubaron con o sin 500 nM de análogos de Sal durante 48 h. Se evaluó la apoptosis mediante tinción con anexina V-FITC y PI y análisis de FACS. Todos los datos se expresan como medias ± s.d. De tres experimentos individuales (*; P<0.05).
Figura 6: el hierro lisosómico media en la señalización de muerte celular activada por análogos de Sal. La muerte celular inducida por análogos de Sal resulta inhibida por el quelante de hierro lisosómico mesilato de deferoxamina (DFO). Las células se trataron tal como en (c) con o sin la concentración indicada de DFO durante 48 h. La apoptosis se evaluó tal como en c.
La figura 7 representa la cuantificación del número de mamosferas contadas en los experimentos mostrados en la figura 9. El número de mamosferas formado tras los pases en serie a la densidad clonal se correlaciona con la capacidad de autorrenovación de las células troncales cancerosas primitivas. Una masa más pequeña indica muerte celular y regresión de las mamosferas. En comparación con las células no tratadas o las células tratadas con salinomicina, sólo AM5 y más eficientemente a M23 redujeron el número de mamosferas.
La figura 8 representa la cuantificación del tamaño de las mamosferas contadas en los experimentos mostrados en la figura 9. El tamaño de la mamosfera sólo se redujo con la dosis más alta de AM23, mientras que la salinomicina, AM5 y dosis inferiores de AM23 no alteraron los diámetros tumorales.
La figura 9 representa imágenes de la tercera generación de mamosferas formadas a partir de células HML34 CD24low individuales tratadas durante 7 días con el fármaco indicado. El tamaño de las mamosferas se correlaciona con la capacidad de proliferación de las células progenitoras, mientras que el número de mamosferas formado tras pases en serie a la densidad clonal se correlaciona con la capacidad de autorrenovación de las células troncales cancerosas primitivas. Una masa más pequeña indica muerte celular y regresión de las mamosferas.
La figura 10 es un dibujo ORTEP de la estructura de rayos X de AM23.
La figura 11 representa los análisis de FACS de EOR en células HMLER CD24low tratadas con salinomicina, AM5 y AM23.
La figura 12 es un gráfico que representa la influencia de la salinomicina, AM5, AM13 y AM9 sobre la
concentración intracelular de sodio.
La figura 13 representa la evaluación de la prevención del crecimiento tumoral en ratones portadores de xenoinjerto de Mc F-7.
La figura 14 representa imágenes comparativas de tinción de H+E de tejidos periféricos de ratones tratados tal como en la figura 13, representativos de cinco réplicas biológicas (barra de escala: 100 ^m).
La figura 15 es un gráfico que representa el peso corporal de los ratones durante el tratamiento con AM5. Las barras de error para el peso corporal de los ratones representan s.d. Y corresponden a cinco animales en cada grupo.
La figura 16 es una representación gráfica del porcentaje de células muertas (negativas para DIOC6(3)/positivas o negativas para DAPI).
La figura 17 representa un análisis de FACS de EOR en células tratadas durante 48 h.
Ejemplos
Ejemplo 1: síntesis de compuesto de fórmula (I)
Preparación de salinomicina ácida oxidada (oxo-Sal-H) 2:

Se disolvió salinomicina sodio (2.00 g, 2.587 mmoles) en 250 ml de DCM y se añadió óxido de manganeso (9.00 g, 103.5 mmoles, 40 eq). La suspensión se agitó durante la noche a temperatura ambiente. Tras completar la conversión del material de partida, la mezcla se filtró a través de Celite. Se extrajo el filtrado con solución acuosa 15 mM de H2SO4 , se secó sobre MgSO4 y se concentró, proporcionando el producto 2 (1.71 g, 2.28 mmoles, 96%) en forma pura y de espuma blanca sin ninguna purificación adicional.
RMN 1H (CDCls, 500 MHz, ta): 0.64-0.72 (6H, m), 0.72-0.82 (6H, m), 0.83-0.98 (12H, m), 1.04-1.17 (4H, m), 1.19 1.27 (2H, m), 1.30-1.57 (12H,m), 1.59-2.05 (14H, m), 2.43-2.60 (2H, m), 2.63-2.73 (1H, m) 2.76-2.88 (1H, m), 3.38 3.52 (1H, m), 3.66 (1H, d, J = 9.6 Hz), 3.76 (1H, d, J = 10.2 Hz), 3.88-4.04 (2H, m), 4.11-4.22 (1H, m), 6.20 (1H, d, J = 10.7), 7.12 (1H, d, J = 10.7).
RMN 13C (CDCls, 500 MHz, ta): 6.6, 7.0, 11.3, 12.1, 12.6, 13.1, 14.2, 15.5, 16.0, 17.6, 19.8, 20.7, 22.9, 26.4, 27.2, 28.3, 28.6, 32.1, 32.2, 33.2, 34.2, 35.5, 38.4, 40.2, 50.3, 51.6, 55.7, 67.7, 69.6, 71.0,73.2, 75.8, 76.5, 76.7, 90.0, 98.0, 105.3, 107.1, 142.3, 183.2, 187.9, 217.9.
HRMS (ESI) m/z: calculado para C42H6sNaOn+ [M+Na+] 771.4654, observado: 771.4560.
Metilación de oxo-Sal-H para formar oxo-Sal-Me 3:

En un matraz de Schlenk seco a la llama y enjuagado con Ar, (100 mg, 0.134 mmoles) se introdujo 2 y se disolvió en DMF anhidro. Se añadió carbonato de cesio (56.5 mg, 0.174 mmoles, 1.3 eq.) seguido de yoduro de metilo (11 pl, 0.174 mmoles, 1.3 eq.) y la solución se agitó durante 24 h a temperatura ambiente. Tras completar la reacción, se eliminó el solvente, se introdujo el residuo en DCM y se extrajo la solución con solución acuosa 15 mM de H2SO4, solución saturada de NaHCO3, agua y solución hipersalina, y se secó sobre MgSO4. Se filtró la solución, se concentró y se purificó en gel de sílice con un CombiFlash utilizando DCM/MeOH 10/0.2. El producto puro 3 (96.5 mg, 0.126 mmoles, 95%) se aisló en forma de una espuma blanca.
(El procedimiento de oxidación-metilación puede invertirse; los rendimientos en ambas etapas no cambian mucho).
RMN 1H (CDCl3, 300 MHz, ta): 0.63-0.72 (9H, m), 0.72-0.77 (3H, d, J = 7.0 Hz), 0.80-0.89 (12H, m), 1.06-1.17 (7H, m), 1.17-1.21 (2H, m), 1.21-1.32 (3H, m), 1.32-1.46 (8H, m), 1.46-1.58 (3H, m), 1.60-1.80 (4H, m), 1.80-1.94 (2H, m), 1.98-2.12 (2H, m), 2.48-2.56 (1H, m), 2.58-2.68 (1H, m), 2.81-2.99 (2H, m), 3.26-3.32 (1H, m), 3.51 (1H, dd, J = 9.8Hz, 1.5 Hz), 3.59-3.73 (2H, m), 3.70 (3H, s, OMe), 3.85-3.97 (2H, m), 6.16 (1H, d, J = 10.7 Hz), 7.18 (1H, d, 10.7 Hz).
RMN 13C (CDCls, 300 MHz, ta): 6.6, 7.2, 11.1, 11.9, 12.1, 14.0, 15.0, 17.9, 18.7, 19.8, 20.8, 22.6, 22.7, 26.3, 28.1, 29.2, 29.7, 30.3, 34.26, 34.30, 34.4, 36.6, 39.2, 39.9, 47.7, 49.1,52.7, 57.5, 70.0, 71.2, 71.8, 72.3, 75.1,77.1,77.4, 88.7, 97.6, 105.5, 127.3, 144.2, 176.6, 190.9, 214.3.
HRMS (ESI) m/z: calculado para C43H70NaOn+ [M+Na+] 785.4810, observado: 785.4807.
Procedimiento para las reacciones de aminación reductora en 2 o 3:
Se disolvieron 100 mg de material de partida 2 en 3 ml de MeOH; se añadió la amina primaria (10 eq.) seguido de AcOH (50 pl). La solución se agitó durante una hora a temperatura ambiente antes de añadir CeCl37H2O. Se añadió muy lentamente una solución de NaBH3CN (1.05-1.3 eq.) en 2 ml de MeOH con ayuda de una bomba de jeringa durante un periodo de 8 h a temperatura ambiente. Tras 4 h adicionales de agitación a temperatura ambiente, se extrajo una muestra de la mezcla de reacción y se llevó a cabo un mini-tratamiento final, seguido de cromatografía en capa fina (TLC). En el caso de que no se consumiese por completo el material de partida, se añadía lentamente algo más de NaBH3CN en MeOH hasta que la conversión completa era visible. A continuación, se añadió cuidadosamente una solución acuosa de H2SO4 15 mM (2 a 4 ml), seguido de DCM Se separaron las capas y se reextrajo la capa acuosa 2 veces con DCM. Las capas orgánicas agrupadas se lavaron con solución acuosa 15 mM de H2SO4 , solución acuosa sat. de NaHCO3, agua y solución hipersalina. La solución se secó sobre MgSO4 y se concentró antes de la purificación con un Combi Flash, utilizando gradualmente MeOH 1% a 3% en DCM sobre gel de sílice. La mayoría de productos secundarios pudieron eliminarse mediante esta etapa. Para la purificación final, el producto se purificó mediante HPLC en una columna C18 de fase inversa. Gradiente de elución: 50%/50% ACN/H2O (ambos con ácido fórmico al 0.1%) a 100% ACN en 12 min, 10 a 20 min 100% ACN (dependiendo de la polaridad de los productos y productos secundarios). Las aminas eluyeron en aproximadamente 60-90% de ACN (AM5: 60%, AM9: 70%, AM13: 90-100%). Detección con un detector de UV a una longitud de onda de 217 nm.
Preparación de AM5

Se preparó sal-propargilamina utilizando 103 mg de 2 (0.134 mmoles), 86 pl (1.34 mmoles, 10 eq.) de propargilamina, 11 mg (0.174 mmoles, 1.3 eq.) de NaBH3CN, 50.0 mg (0.134 mmoles, 1 eq.) de CeCl37H2O y 50 pl de ácido acético en 8 ml de MeOH. Tras la purificación con CombiFlash y HPLC, pudieron aislarse 25 mg (0.032 mmoles, 24%) de producto puro en forma de espuma incolora.
RMN 1H (CDCl3, 600 MHz, 5°C): 0.66 (3H, d, J=7.2 Hz, C39H3), 0.71 (3H, d, J=6.6 Hz, C34H3), 0.75 (3H, dd, J=J=7.8 Hz, C37H3), 0.77 (3H, d, J=7.2 Hz, C38H3), 0.85 (3H, d, J=6.6 Hz, C35H3), 0.86 (3H, m, C32H3), 0.88 (3H, m, C40H3), 0.90 (3H, m, C42H3), 1.13 (1H, dd, J=J=13.8 Hz, C15H), 1.21 (3H, d, J=7.2 Hz, C30H3), 1.25 1.38 (8H, m, C41H, C31H2, C36H, C33H3, C4H), 1.39-1.48 (3H, m, C5H, C8H, C41H), 1.49-1.55 (1H, m, C26H), 1.55-1.63 (3H, m, C26H, C27H2), 1.63-1.71 (3H, m, C14H, C15H, C16H), 1.71-1.80 (3H, m, C6H, C23H, C5H), 1.80-1.91 (2H, m, C36H, C4H), 1.91-2.00 (2H, C22H2), 2.09-2.16 (1H, m, C23H), 2.36-2.40 (1H, s, =CH), 2.55 2.64 (2H, m, C12H, C10H), 2.84-2.91 (1H, dt, C2H), 3.53 (1H, m, C13H), 3.57-3.63 (2H, C25H, m, C7H), 3.81-3.85
(1H, m, C29H), 3.91-4.01 (2H, m, C3H, C20H), 4.15 (1H, d, J = 10.2 Hz, C9H), 4.28 (2H, bs, NHCH2), 6.28 (1H, m, C19H), 6.44 (1H, d, J =9.6 Hz, C18H).
RMN 13C (CDCla, 600 MHz, 5°C): 6.5 (C32), 7.1 (C39), 11.2 (C40), 12.2 (C38), 12.5 (C42), 13.3 (C37), 14.5 (C30), 15.6 (C34), 16.7 (C36), 17.6 (C35), 20.0 (C4), 21.8 (C26), 22.9 (C41), 25.0 (C33), 26.4 (C5), 28.1 (C6), 28.8 (C27), 30.7 (C31), 30.8 (C23), 32.3 (C14), 36.1 (C8), 37.1 (NHCH2), 37.6 (C15), 39.0 (C16), 40.0 (C22), 49.2 (C2), 50.0 (C10), 53.0 (C20), 55.2 (C12), 68.9 (C9), 71.1 (C28), 71.5 (C7), 72.9 (C25), 75.5 (C3), 75.8 (C13), 76.3 (=CH), 76.9 (C29), 77.3 (=C-), 88.6 (C24), 98.6 (C17), 105.6 (C21), 125.8 (C19), 132.2 (C18), 180.8 (C1), 216.1 (C11).
HRMS (ESI) m/z: calculado para C42H74NO10+ [M+H+] 788.5307, observado: 788.5304.
Preparación de AM9

Se preparó sal-propargilamina utilizando 106 mg de 3 (0.139 mmoles), 89 ^l (1.39 mmoles, 10 eq.) de propargilamina, 9,6 mg (0.153 mmoles, 1.1 eq.) de NaBHsCN, 51.8 mg (0.138 mmoles, 1 eq.) de C e C h ^^ O y 50 ^l de ácido acético en 8 ml de MeOH. Tras la purificación con CombiFlash y HPLC, pudieron aislarse 25 mg (0.031 mmoles, 22%) de producto puro en forma de espuma incolora.
RMN 1H (CDCla, 500 MHz, 5°C): 0.72 (3H, d, J = 6.9 Hz), 0.75-0.87 (11 H, m), 0.88-0.99 (9H, m), 1.07 (1H, ddd, J = 13.1 Hz, 13.1Hz, 12.1Hz), 1.20-1.68 (21H, m), 1.70-2.05 (8Hz, m), 2.10-2.26 (3H, m), 2.30-2.41 (2H, m), 2.69 2.74 (1H, m), 3.04 (1H, dt, J = 10.8 Hz, 4.1Hz), 3.47-3.72 (4H, m), 3.80-3.88 (1H, m), 3.90 (3H, s), 4.02-4.09 (2H, m), 6.01-6.08 (2H, m).
RMN 13C (500 MHz, CDCh, 5°C): 6.5, 7.4, 11.0, 12.0, 13.2, 13.9, 14.7, 15.7, 17.5, 19.7, 22.2, 22.7, 25.5, 26.2, 28.0, 29.0, 30.6, 30.7, 32.9, 36.4, 37.1 (2C), 38.6, 38.7, 40.3, 48.0, 48.6, 52.6, 55.2, 56.6, 69.2, 71.0, 71.7, 73.9, 75.1, 76.9, 77.3, 80.1, 88.0, 98.6, 102.0, 108.3, 123.2, 130.5, 176.2, 214.0.
HRMS (ESI) m/z: calculado para C46H76NO10+ [M+H+] 802.5464, observado: 802.5465.
Preparación de AM13

Se preparó sal-dodecilamina utilizando 103 mg de 2 (0.138 mmoles), 255,8 mg (1.38 mmoles, 10 eq.) de dodecilamina, 9 mg (0.145 mmoles, 1.05 eq.) de NaBH3CN, 51.4 mg (0.138 mmoles, 1 eq.) de CeCl37H2O y 20 ^l de ácido acético en 8 ml de MeOH. Tras la purificación con CombiFlash y HPLC, pudieron aislarse 14 mg (0.0152 mmoles, 11%) de producto puro en forma de espuma incolora.
RMN 1H (CDCh, 500 MHz, 5°C): 0.67 (3H, d, J=6.9 Hz), 0.66-0.77 (9H, m), 0.78-0.90 (15H, m), 1.10-1.51 (34H, m), 1.51-1.78 (9H, m), 1.80-2.10 (6H, m), 2.50-2.60 (2H, m), 2.77-2.87 (1H, m), 3.30-3.62 (5H, m), 3.68-3.80 (2H, m), 3.97-4.04 (1H, m), 4.20-4.30 (1H, m), 6.32-6.42 (2H, m).
RMN 13C (500 MHz, CDCh, 5°C): 6.5, 7.1, 11.3, 12.3, 12.8, 13.1, 14.3, 14.4, 15.5, 16.5, 17.6, 20.3, 21.8, 22.81, 22.84, 24.5, 26.4, 26.7, 27.3, 28.2, 29.0, 29.5, 29.6, 29.76, 29.81 (4C), 30.5, 31.0, 32.0, 32.2, 35.8, 37.7, 39.0, 40.6, 48.6, 49.3, 50.5, 55.0, 55.1,71.05, 71.14, 73.0, 75.5, 76.4, 77.0, 88.8, 99.0, 106.5, 128.0, 130.9, 204.7, 214.8.
HRMS (ESI) m/z: calculado para C54H96NOio+ [M+H+] 918.7029, observado: 918.7034.
Preparación de AM23

Se preparó sal-cidopropilamina utilizando 100 mg de 2 (0.133 mmoles), 94 pl (1.33 mmoles, 10 eq.) de cidopropilamina, 11 mg (0.17 mmoles, 1.3 eq.) de NaBH3CN, 50.0 mg (0.134 mmoles, 1 eq.) de CeCl37H2O y 50 pl de ácido acético. Se obtuvo AM23 en forma de espuma incolora (44 mg, 42%).
RMN 1H (CDCl3, 500 MHz, 278 K) 50.47-0.58 (2H, m), 0.69 (3H, J = 10.0 Hz), 0.71-0.78 (9H, m), 0.78-0.95 (14H, m), 1.12-1.50 (16H, m), 1.50- 1.75 (5H, m), 1.76-1.91 (4H, m), 2.02-2.20 (2H, m), 2.60 (1H, d, J = 10.5 Hz), 2.62 2.68 (1H, m), 2.70-2.78 (1H, m), 2.78-2.88 (1H, m), 3.37 (1H, s), 3.53-3.80 (3H, m), 3.82-3.89 (1H, m), 4.08 (1H, d, J = 9.5 Hz), 5.15 (2H, br s), 6.13 (2H, s).
RMN 13C (CD3CN, 125 MHz, 278 K) 55.9, 6.4, 6.9, 7.7, 12.0, 12.4, 13.6, 13.7, 15.2, 16.1, 17.4, 17.9, 21.0, 22.7, 24.0, 25.7, 27.3, 29.1,30.1,31.8, 31.9, 33.4, 36.8, 39.0, 39.5, 41.2, 49.0, 50.4, 56.7, 57.6, 69.5, 71.5, 72.4, 74.7, 76.0, 77.1,77.9, 89.7, 99.9, 107.9, 126.0, 130.8, 178.8, 214.7.
HRMS (ESI) m/z: calculado para C45H75NO10+ [M+H+] 789.5385, observado: 789.5381.
La figura 10 muestra la estructura 3D del compuesto AM23, que confirma inequívocamente la estructura cristalina y la estereoquímica de AM23.
Ejemplo 2: evaluación de la IC
50
Se llevó a cabo un ensayo de viabilidad celular mediante la siembra en placa de 1000 células por pocillo en placas de 96 pocillos. Se pretrató con NAC (2 mM, A9165 Sigma) o DFO (1 mM) 2 horas antes del tratamiento con compuesto. Se añadió reactivo CellTiter-Blue® (Promega; G3582) (20pl/pocillo) tras 24, 48 o 72 horas de tratamiento y las células se incubaron durante 1 hora antes de registrar la fluorescencia (560(20)Ex/590(10)Em) utilizando un lector de microplacas Perkin Elmer Wallac 1420 Victor2.
Resultados:




Los resultados anteriores muestran que los compuestos que no contenían una funcionalidad amina en la posición 20 presentan menor potencia que la salinomicina contra las células troncales cancerosas.
La introducción de una función amina en la posición 20 de la salinomicina da como resultado una actividad significativamente mejorada contra las células CD24 (AM 5, AM8, AM11, AM12, AM13 y AM23); una mejora de hasta 18 veces.
La sustitución de la funcionalidad ácido carboxílico en la posición 1 de la salinomicina con un grupo éster da como resultado compuestos con una eficiencia más baja que la salinomicina (AM9 y AM10).
Estos resultados demuestran que tanto la amina como un grupo funcional capaz de quelar el hierro, tal como un ácido carboxílico, resultan necesarios para una actividad mejorada. Se encuentra contemplado que la presencia de estos dos grupos funcionales ayude a la coordinación del hierro, favoreciendo de esta manera la reacción de Fenton en los lisosomas.
Por lo tanto, los compuestos de fórmula (I) resultan útiles para el tratamiento del cáncer y/o para la prevención de las recaídas del cáncer y/o las metástasis.
Ejemplo 3: efecto de AM5, AM9 y AM13 sobre la proliferación de las células HMLER CD24-:
Se evaluaron AM5, AM9, AM13 y salinomicina para su capacidad de inhibir la proliferación celular y la formación de mamosferas.
Los resultados se presentan en la figura 2.
A 30 nM, AM5 y AM13 inhibían la proliferación celular con una eficacia mejorada en 10 veces respecto a la de la salinomicina.
En contraste, AM9 no inhibió la proliferación celular, ni siquiera a una concentración de 500 nM.
De esta manera, estos resultados indican que los compuestos de fórmula (I) según la presente invención son capaces de inhibir la formación de mamosferas más eficientemente que la salinomicina.
Ejemplo 4: efecto de AM5, taxol y combinaciones de los mismos sobre la proliferación de las células HMLER CD24-Se evaluaron AM5, taxol y una combinación de AM5 y taxol para su capacidad de inhibir la proliferación celular y la formación de mamosferas.
Los resultados se presentan en la figura 3.
La combinación de AM5 a una concentración de 15 nM y de taxol a 5 nM inhibe la proliferación celualr y la formación de mamosferas con una eficacia mejorada respecto a AM5 solo a 15 nM o 5 nM.
Ejemplo 5: efecto de AM5 sobre la formación de tumores de xenoinjerto
Se recogieron cultivos de células de la estirpe de cáncer de mama humano MCF-7, se disociaron enzimáticamente, se lavaron en PBS y se resuspendieron en mezcla de PBS/Matrigel (1:1 en volumen). A continuación, se implantaron 0.1 ml de dicha mezcla en la almohadilla grasa mamaria de ratones Fox1nu desnudos atímicos hembra de 5 semanas de edad (Harlan, Francia). Los ratones se mantuvieron en jaulas individualmente ventiladas (Tecniplast, Francia) bajo temperatura y humedad constantes; todos los experimentos se llevaron a cabo bajo flujo laminar (Tecniplast, Francia). Los ratones recibieron suplementación de estradiol (0.4 mg/kg) el mismo día y el 7° día desde la inyección de células, y se observaron y palparon para la aparición de tumores. Los ratones recibieron un análogo de la salinomicina (en la presente memoria, AM5, 3 mg por kg de peso corporal al día, inyección intraperitoneal) cada 5 días laborables de la semana durante 33 días. Se midió semanalmente el crecimiento tumoral utilizando calibradores. Se determinó el volumen tumoral utilizando la fórmula estándar: L*W2*0.52, donde L y W son los diámetros más largo y más corto, respectivamente. Todo el trabajo con los animales se llevó a cabo siguiendo las directrices del Comité coordinador del Reino Unido en la investigación del cáncer.
Los resultados se proporcionan en la figura 4.
Tras el tratamiento de AM5, el volumen tumoral y el peso tumoral eran más bajos.
Estos resultados son consistentes con el ensayo in vitro e indican que los compuestos de fórmula (I) según la presente invención son capaces de inhibir la formación tumoral en ratones desnudos.
Ejemplo 6: la salinomicina y análogos activos inducen la muerte celular a través de la catálisis de Fenton lisosómica.
La muerte celular inducida por análogos de salinomicina resulta inhibida por el secuestrador de EOR N-acetilcisteína (NAC). Las estirpes celulares se incubaron con o sin 500 nM de análogos de Sal durante 48 h. Se evaluó la apoptosis mediante tinción con anexina V-FITC y PI y análisis de FACS.
Los resultados se proporcionan en la figura 5. Todos los datos se expresan como medias ± s.d. De tres experimentos individuales (*; P<0.05).
Los datos indican que la salinomicina y AM5 inducen la muerte celular mediante la producción lisosómica de EOR.
Ejemplo 7: el hierro lisosómico media en la señalización de muerte celular activada por análogos de salinomicina.
La muerte celular inducida por análogos de salinomicina resulta inhibida por el quelante de hierro lisosómico mesilato de deferoxamina (DFO). Las células se trataron tal como en el ejemplo 6, con o sin la concentración indicada de DFO durante 48 h. La apoptosis se evaluó tal como en el ejemplo 6.
Los resultados se proporcionan en la figura 6.
Los datos indican que el hierro lisosómico media en la señalización de muerte celular activada por análogos de salinomicina.
Ejemplo 8: efecto de AM5 y AM23 sobre la proliferación de las células HMLER CD24-:
Se evaluaron AM5 y AM23 y salinomicina para su capacidad de inhibir la proliferación celular y la formación de mamosferas.
Los resultados se proporcionan en las figuras 7 a 9.
A 30 nM, AM5 y AM23 inhibían la proliferación celular con una eficacia mejorada en diez veces en comparación con la salinomicina.
De esta manera, estos resultados indican que los compuestos de fórmula (I) según la presente invención son capaces de inhibir la formación de mamosferas más eficientemente que la salinomicina.
Ejemplo 9: IC50 de la salinomicina, AM5 y AM23 sobre las estirpes de células mamarias:
La tabla 1, a continuación, representa la IC50 de la salinomicina (Sal) y sus derivados AM5 y AM23 para un amplio
abanico de estirpes de células mamarias.
Las células se sembraron en una placa de 6 pocilios a una densidad de 5x105 células/pocillo y se cultivaron durante la noche. A continuación, las células se trataron con diversas concentraciones (15, 30, 100, 500, 1000 y 10000 nM) de salinomicina, AM5 y AM23 durante 72 h, 96 h y 108 h. Tras el tratamiento, se cuantificó la muerte celular utilizando un ensayo de anexina V-FITC/yoduro de propidio (PI) siguiendo el protocolo del fabricante (kit II de detección de apoptosis de FITC-anexina V, 556570, BD PharmingenTM) y se analizaron mediante un citómetro de flujo LSRFortessa™ (BD Bioscience, San Jose, CA). Los datos se procesaron utilizando el software Cell Quest (BD Biosciences). Se determinaron las curvas de dosis-respuesta de muerte celular para el tiempo indicado. Para las células tumorales, las células se clasificaron en función de su sensibilidad a los fármacos. En la presente memoria, las células más sensibles se incubaron durante 72 h; las células de sensibilidad intermedia, durante 96 h, y las células menos sensitivas o células resistentes se incubaron durante 108 h con fármaco a varias concentraciones.
Se utilizaron las concentraciones 30 nM, 500 nM y 1 pM para determinar la IC50 de los fármacos.
Los intervalos [30 - 500 nM] se refieren a que la IC50 está incluida en dicho intervalo con exclusión del valor 30 nM.
Tabla 1

Las estirpes celulares SW620 y SW480 proceden de tumores de colon. La tabla 2 describe las especificidades esenciales de cada estirpe celular:
Tabla 2


Estos resultados indican que AM5 y AM23 presentan una IC50 en comparación con la salinomicina similar o mejor según las células.
Ejemplo 10: influencia de AM23 en la inducción de EOR
Se midieron los niveles de especies de oxígeno reactivo (EOR) mediante citometría de flujo o mediante microscopía de inmunofluorescencia de barrido confocal utilizando CM-H2DCF-DA (C6827, Invitrogen). Brevemente, se trataron células U2OS y HMLER CD24low tal como se indica en la figura 11 (3o nM, 500 nM o 1 pM de salinomicina, AM5 o AM23, o no tratadas, durante 48 h). A continuación, estas células se tripsinizaron y se incubaron con 5 pM de CM-H2DCF-DA a 37°C durante 40 min, se lavaron una vez con PBS y se contratiñeron con DAPI (0,5 pg/ml) para excluir las células no viables. Se determinó la intensidad media de fluorescencia como producción de EOR mediante citometría de flujo con un citómetro LSRFortessa™ (BD Bioscience, San Jose, CA). Para el análisis de microscopía de inmunofluorescencia, las células se sembraron sobre cubreobjetos y se trataron con derivados de salinomicina (inyección en medio de cultivo, seguido de tratamiento durante 24 h, 48 h y 72 h). Para visualizar los lisosomas se utilizó LysoTracker® Red DND-99 (L-7528, Life technologies). A continuación, las células se fijaron con PFA al 4%/PBS. Se utilizó DAPI para visualizar el ADN nuclear. Se obtuvieron imágenes celulares utilizando un microscopio en tiempo real Deltavision (Applied Precision) o un microscopio ApoTome.2 (Zeiss). Se utilizó ImageJ para el procesamiento adicional de las imágenes.
Tal como se muestra en la figura 11, AM23 induce EOR en las células HMLER CD24low.
Ejemplo 11: medición del sodio intracelular y el crecimiento tumoral en ratones portadores de xenoinjerto de MCF-7
Medición del sodio intracelular: se mezclaron tampones de sodio y potasio (HEPES 10 mM, CaCh 1 mM, MgCh 1mM, D-gluconato sódico 130 mM o D-gluconato potásico, NaCl/KCl 30 mM ) en diferentes proporciones para producir cinco tampones con diversas concentraciones de sodio (0, 20, 40, 80, 160 mM). Se utilizó nigericina (N7143, Sigma, 10 pM) y monensina (M5273, Sigma, 10 pM) para equilibrar la concentración intracelular de sodio y establecer una curva de calibración. Se recolectaron las células HMLER CD24low y se resuspendieron en tampón ECS (HEPES 15 mM, KCI 5.4 mM, NaCl 140 mM, glucosa 10 mM, MgCh 1 mM, CaCh 1.8 mM, BSA al 0.1%, pH 7.6) que contenía 10 pM de la sonda específica de sodio (SBFI-AM, S-1263, Molecular Probes®) y Pluronic F-127 al 0.2% (P2443, Sigma), y se incubaron durante 1 h en la oscuridad a 37°C. A continuación, las células se lavaron para eliminar el exceso de pigmento y se incubaron durante 30 min adicionales en tampón de ECS. Las células se introdujeron en una placa de 96 pocillos (1000 células/pocillo) y se trataron con derivados de salinomicina a una concentración comprendida entre 0.03 y 20 pm (AM5: 0.120 pM, AM13: 0.120 pM, AM9: 20 pM; salinomicina: 20 pM y 1 pM) durante 5 min. Cada pocillo se excitó secuencialmente a 340 y 370 nm y se registró la emisión a
500 nm. Se evaluó la respuesta espectral del SBFI tras la unión de sodio, mediante la medición de la proporción de excitación (340/370 nm). Las mediciones se llevaron a cabo en un lector de microplacas Perkin Elmer Wallac 1420 Victor2 a 37°C.
Experimentos de formación de tumores de xenoinjerto: se recolectaron cultivos de células MCF-7, se disociaron enzimáticamente, se lavaron con PBS y se resuspendieron en una mezcla de PBS/Matrigel (1:1 v/v). A continuación, la mezcla (0.1 ml) se implantó en la almohadilla grasa mamaria de ratones Foxlnu desnudos atímicos hembra de 5 semanas de edad (Harlan, Francia). Los ratones se mantuvieron en jaulas individualmente ventiladas (Tecniplast, Francia) bajo temperatura y humedad constantes. Todos los experimentos se llevaron a cabo bajo flujo laminar (Tecniplast, Francia). Los ratones recibieron suplementación de estradiol (0.4 mg/kg) el mismo día y el 7° día desde la inyección de células, y se observaron y palparon para la aparición de tumores. Los ratones se trataron con AM5 o paclitaxel (3 mg/kg peso corporal/día) mediante inyecciones intraperitoneales cada 5 días laborables de la semana. Se midió semanalmente el crecimiento tumoral utilizando calibradores. Se determinó el volumen tumoral utilizando la fórmula estándar: L*W2*0.52, donde L y W son los diámetros más largo y más corto, respectivamente. Todos los estudios con animales fueron aprobados por la Direction des services Vétérinaires, Prefecture de Police, Paris, Francia (número de autorización A75-14-08) y el comité ético (número 34) de la Paris Descartes University. No se utilizó aleatorización y los experimentos fueron ciegos a los tratamientos farmacológicos y análisis de tejidos.
Aunque la salinomicina indujo un rápido incremento del sodio intracelular utilizando una dosis de hasta veinte veces el valor de la IC50, los derivados de salinomicina no presentaron ningún efecto sobre el transporte del sodio a dosis eficaces contra la proliferación de células HMLER CD24low (fig. 12). Estos datos contradicen la idea de que la salinomicina afecta selectivamente al mantenimiento de las CSC mediante la alteración directa de los potenciales de membrana. En contraste, AM9 resultó ineficaz en estos ensayos, validando al carboxilato como motivo requerido para alterar el mantenimiento de las CSC, y el paclitaxel por sí solo presentó una eficacia reducida contra la formación de tumoresferas.
Además, AM5 evitó el crecimiento tumoral en ratones portadores de xenoinjerto de MCF-7 (fig. 13).
Ejemplo 12: evaluación de la toxicidad
Histología. Se extrajeron los órganos de los ratones en el momento del sacrificio. Para los análisis morfológicos, se fijaron los órganos con paraformaldehído al 4%, se incluyeron en parafina y se tiñeron secciones de 4 pm con hematoxilina y eosina (H+E). Las secciones se escanearon a alta resolución utilizando un escáner de portas (NanoZoomer 2.0-HT, Hamamatsu, Massy, Francia). Se muestran imágenes representativas en la figura 14.
No se observó toxicidad genérica tras el tratamiento con una dosis eficaz de AM5, según se observa por la integridad de los tejidos periféricos y el peso corporal constante durante todo el tratamiento (figura 15).
Todas las muestras de pulmón de los grupos no tratados y tratados mostraron agregados de macrófagos multifocales mínimos a moderados en los alveolos. Este resultado es poco significativo y se observa habitualmente en ratones. En 4 ratones (2 no tratados y 2 tratados), se observaron infiltrados de células mononucleares intersticiales extrapulmonares. Muy probablemente poco significativos y no relacionados con los tratamientos. En los ratones no tratados, se observó una densificación pulmonar subpleural focal con fibrosis intersticial y infiltrados celulares atípicos que evocaban células tumorales (metástasis). Un artefacto de grandes dimensiones sobre la lesión (pliegue de tejidos) interfería con el análisis.
No se observaron cambios significativos en riñones.
Ejemplo 13:
Se cultivaron células MDA-MB-231 con o sin inhibidor de catepsina-B (COA74-Me, 30 pM) y/o salinomicina, AM5 o AM23 (500 nM) durante el periodo indicado (48 h, 96 h, 108 h). A partir del tratamiento, se evaluaron las células muertas mediante ensayo de DIOC6(3)/DAPI y se analizaron mediante citometría de flujo. La representación gráfica del porcentaje de células muertas (negativas para DIOC6(3)/positivas o negativas para DAPI) se muestra en la figura 16. La inhibición de catepsina-B y el tratamiento de salinomicina, AM5 o AM23 se combinaron para inducir la muerte de la estirpe celular de cáncer mamario humano MDA-MB-231.
El análisis de FACS de EOR en las células tratadas durante 48 h se representa en la figura 17. La inhibición farmacológica de la catepsina-B evita que AM5 induzca la producción de EOR en células HMLER CD24low.
Claims (16)
1. Compuesto de fórmula (I), enantiómeros, mezcla de enantiómeros, diastereoisómeros y mezcla de diastereoisómeros del mismo:

en el que:
- W se selecciona de entre el grupo que consiste en =O; -NRi R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NRs-(CH2)n-N R6R7R8 y -O-(CH2)n-N+R6R7R8;
- X se selecciona de entre el grupo que consiste en =O, -OH; -NRi R2; -NR3-(CH2)n-NR4R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N R6R7R8 y -O-(CH2)n-N+R6R7R8,
- Y se selecciona de entre el grupo que consiste en -NRi R2; -NR3-(CH2)n-NR4R5; y -NR3-(CH2)n-N+R6R7R8, Ri y R2, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C i-C i6); alquenilo (C3-C16); alquinilo (C3-C16); cicloalquilo C3-C16; arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1-C6)-heteroarilo; o R1 representa H y R2 representa OR9, en el que R9 es H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo;
R3 se selecciona de entre el grupo que consiste en H; alquilo C1-C6 ; alquil (C1-C6)-arilo;
R4 y R5, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C1-C6); arilo y alquil (C1-C6)-arilo;
R6, R7 y R8, idénticos o diferentes, se seleccionan de entre el grupo que consiste en alquilo (C1-C6); arilo y alquil (C1-C6)-arilo;
- Z se selecciona de entre el grupo que consiste en OH; NHNR9R10; NHOC(O)R11; N(OH)-C(O)R11; OOH, SR12; 2-aminopiridina; 3-aminopiridina; -NR3-(CH2)n-NR4R5; y -NR3-(CH2)n-OH, en el que:
R9y R10, idénticos o diferentes, se seleccionan de entre el grupo que consiste en H, alquilo (C1-C6), arilo y alquil (C1-C6)-arilo;
R11 se selecciona de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1-C6)-heteroarilo;
R12 se selecciona de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); arilo; heteroarilo; alquil (C1-C6)-arilo; alquil (C1-C6)-heteroarilo, n = 0, 2, 3, 4, 5 o 6.
2. Compuesto según la reivindicación 1, en el que:
R1 y R2 , idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C1-C16); alquenilo (C3-C16); alquinilo (C3-C16); cicloalquilo (C3-C16); y alquil (C1-C6)-heteroarilo,
R3 se selecciona de entre el grupo que consiste en H; y alquilo (C1-C6);
R4 y R5 , idénticos o diferentes, se seleccionan de entre el grupo que consiste en H; alquilo (C1-C6); y alquil (C1-C6)-arilo.
3. Compuesto según la reivindicación 1 o 2, en el que R1 es H y R2 se selecciona de entre el grupo que consiste en alquilo (C1-C16), ventajosamente alquilo (C3-C14); alquenilo (C3-C16), ventajosamente alquenilo (C3-C5); alquinilo (C3-C16), ventajosamente alquinilo (C3-C5); cicloalquilo (C3-C16), ventajosamente cicloalquilo (C3-C6); y alquil (C1-C6)-heteroalquilo, ventajosamente CH2-piridinilo.
4. Compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 3, en el que Z es OH o NHOH, ventajosamente OH.
5. Compuesto según cualquiera de las reivindicaciones 1 a 4, en el que W y X, idénticos o diferentes, se seleccionan de entre el grupo que consiste en -NR1R2 ; -NR3-(CH2)n-NR4 R5; -O-(CH2)n-NR4R5; -NR3-(CH2)n-N+R6R7R8; y -O-(CH2)n-N+R6R7R8; R1 a R8 y n siendo como se ha definido anteriormente.
6. Compuesto según cualquiera de las reivindicaciones 1 a 4, en el que X o Z, idénticos o diferentes, se seleccionan de entre el grupo que consiste en -NR1R2 ; -NR3-(CH2)n-NR4 R5; -O-(CH2)n-NR4R5; -NR1-(CH2)n-N+R6R7R8 ; y -O-(CH2)n-N+R6R7R8; R1 a R8 y n siendo como se ha definido anteriormente.
7. Compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 4, en el que X se selecciona de entre el grupo que consiste en =O y OH, ventajosamente OH e Y se selecciona de entre el grupo que consiste en -NR1R2; -NR3-(CH2)n-NR4R5; y -NR1-(CH2)n-N+R6R7R8; R1 a R8 y n siendo como se ha definido anteriormente.
8. Compuesto según la reivindicación 7, en el que X es OH, Z es OH e Y es NR1R2 en el que R1 es H y R2 se selecciona de entre el grupo que consiste en alquilo (C1-C16), ventajosamente alquilo (C8-C14); alquenilo (C3-C16), ventajosamente alquenilo (C3-C5); alquinilo (C3-C16), ventajosamente alquinilo (C3-C5) y cicloalquilo (C3-C16), ventajosamente cicloalquilo (C3-C6).
9. Compuesto según la reivindicación 1, seleccionado de entre el grupo que consiste en:



10. Compuesto según cualquiera de las reivindicaciones anteriores para la utilización como un fármaco.
11. Compuesto para la utilización según la reivindicación 10 en el tratamiento del cáncer, ventajosamente el cáncer de mama.
12. Compuesto para la utilización según la reivindicación 10 en la prevención de la recaída de cáncer y/o las metástasis.
13. Compuesto para la utilización según la reivindicación 10 en el tratamiento del paludismo.
14. Composición farmacéutica que comprende por lo menos un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 9, una sal farmacéuticamente aceptable, solvato o hidrato del mismo y por lo menos un excipiente farmacéuticamente aceptable, ventajosamente para la utilización en el tratamiento del cáncer, tal como el cáncer de mama.
15. Composición farmacéutica según la reivindicación 14, que comprende además otro fármaco anticanceroso, ventajosamente adriamicina y ciclofosfamida o docetaxel.
16. Producto farmacéutico que comprende:
a) el compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 9, y
b) otro compuesto de quimioterapia, tal como adriamicina y ciclofosfamida o docetaxel,
como producto de combinación para la utilización simultánea, separada o escalonada como un medicamento, en particular en el tratamiento del cáncer, ventajosamente el cáncer de mama.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14306409 | 2014-09-12 | ||
| PCT/EP2015/070975 WO2016038223A1 (en) | 2014-09-12 | 2015-09-14 | Nitrogen-containing analogs of salinomycin, synthesis and use against cancer stem cells and malaria |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2755424T3 true ES2755424T3 (es) | 2020-04-22 |
Family
ID=51570451
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15766107T Active ES2755424T3 (es) | 2014-09-12 | 2015-09-14 | Análogos de salinomicina que contienen nitrógeno, síntesis y utilización contra células troncales cancerosas y paludismo |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10287298B2 (es) |
| EP (1) | EP3191493B1 (es) |
| JP (1) | JP6599444B2 (es) |
| CN (1) | CN107428772B (es) |
| CA (1) | CA2960910C (es) |
| DK (1) | DK3191493T3 (es) |
| ES (1) | ES2755424T3 (es) |
| PL (1) | PL3191493T3 (es) |
| WO (1) | WO2016038223A1 (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11573241B2 (en) * | 2016-03-09 | 2023-02-07 | Institute National De La Sante Et De La Recherche Medicale (Inserm) | Total cellular iron as a marker of cancer stem cells and uses thereof |
| CN107417699B (zh) * | 2016-05-24 | 2020-07-14 | 中国医学科学院药物研究所 | 盐霉素肟及肟醚衍生物、其制备方法和抗肿瘤用途 |
| US20230220480A1 (en) * | 2019-11-06 | 2023-07-13 | Centre National De La Recherche Scientifique (Cnrs) | Iron-score and in vitro method for identifying high risk dlbcl subjects and therapeutic uses and methods |
| PL242212B1 (pl) | 2020-02-07 | 2023-01-30 | Fileclo Spolka Z Ograniczona Odpowiedzialnoscia | Związki stanowiące C20-modyfikowane pochodne salinomycyny, sposób ich otrzymywania, kompozycja je zawierająca, zastosowanie tych związków oraz sposób otrzymywania produktu pośredniego |
| EP4175628A1 (en) * | 2020-07-03 | 2023-05-10 | Centre National de la Recherche Scientifique (CNRS) | Nitrogen-containing analogs of salinomycin for use in acute myeloid leukemia |
| CN117693337A (zh) | 2021-05-05 | 2024-03-12 | 国家科学研究中心 | 用于在多发性骨髓瘤(mm)中使用的盐霉素的含氮类似物 |
| PL246999B1 (pl) * | 2021-08-05 | 2025-04-22 | Fileclo Spolka Z Ograniczona Odpowiedzialnoscia | Związki stanowiące C20-modyfikowane pochodne salinomycyny, sposób ich otrzymywania, kompozycja je zawierająca, zastosowanie tych związków oraz produkty pośrednie |
| PL4396189T3 (pl) * | 2021-09-03 | 2026-01-19 | Centre National De La Recherche Scientifique | Aktywowane żelazem, zawierające azot pochodne salinomycyny i narazyny, synteza i zastosowanie w leczeniu nowotworów |
| WO2023031402A1 (en) * | 2021-09-03 | 2023-03-09 | Centre National De La Recherche Scientifique | Nitrogen-containing derivatives of salinomycin, synthesis and uses thereof |
| WO2023118165A1 (en) | 2021-12-21 | 2023-06-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and compositions for treating melanoma |
| CN120398907A (zh) * | 2024-01-31 | 2025-08-01 | 南京安吉生物科技有限公司 | 盐霉素衍生物及其制备方法与应用 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU5259801A (en) * | 2000-04-26 | 2001-11-07 | Meiji Seika Kaisha Ltd. | Compositions for treating or preventing malaria and method of treating malaria |
| JP2006347881A (ja) * | 2003-06-17 | 2006-12-28 | Astellas Pharma Inc | トリアミド誘導体及びその生産方法 |
| WO2011085544A1 (zh) * | 2010-01-13 | 2011-07-21 | 安徽辉克药业有限公司 | 有效清除肿瘤细胞的化合物 |
| CN103193841A (zh) * | 2012-01-06 | 2013-07-10 | 维瑞斯特姆有限公司 | 治疗性化合物及相关使用方法 |
-
2015
- 2015-09-14 DK DK15766107T patent/DK3191493T3/da active
- 2015-09-14 JP JP2017513792A patent/JP6599444B2/ja active Active
- 2015-09-14 PL PL15766107T patent/PL3191493T3/pl unknown
- 2015-09-14 CN CN201580061663.7A patent/CN107428772B/zh active Active
- 2015-09-14 EP EP15766107.5A patent/EP3191493B1/en active Active
- 2015-09-14 ES ES15766107T patent/ES2755424T3/es active Active
- 2015-09-14 WO PCT/EP2015/070975 patent/WO2016038223A1/en not_active Ceased
- 2015-09-14 CA CA2960910A patent/CA2960910C/en active Active
- 2015-09-14 US US15/510,427 patent/US10287298B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP6599444B2 (ja) | 2019-10-30 |
| CN107428772B (zh) | 2020-09-18 |
| JP2017526722A (ja) | 2017-09-14 |
| EP3191493B1 (en) | 2019-08-14 |
| WO2016038223A1 (en) | 2016-03-17 |
| CA2960910C (en) | 2022-10-25 |
| CN107428772A (zh) | 2017-12-01 |
| EP3191493A1 (en) | 2017-07-19 |
| US10287298B2 (en) | 2019-05-14 |
| US20170253610A1 (en) | 2017-09-07 |
| DK3191493T3 (da) | 2019-11-18 |
| CA2960910A1 (en) | 2016-03-17 |
| PL3191493T3 (pl) | 2020-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2755424T3 (es) | Análogos de salinomicina que contienen nitrógeno, síntesis y utilización contra células troncales cancerosas y paludismo | |
| JP7398156B2 (ja) | Stat3二機能性リン酸化部位を標的とするトリアロマティック化合物のクラスおよびその応用 | |
| AU2017273726B2 (en) | Use of EZH2 inhibitors for treating cancer | |
| CA3002884A1 (en) | Protein kinase inhibitors, preparation method and medical use thereof | |
| ES2762641T3 (es) | Inhibidores de proteína quinasa de 2-aminopiridina sustituida con piridina | |
| CN116253719B (zh) | 一种雌激素和雄激素受体双重降解剂及其应用 | |
| ES2743701T3 (es) | Conjugados de ligando-fármaco del receptor sigma-2 como compuestos antitumorales, métodos de síntesis y usos de los mismos | |
| JP2024506518A (ja) | アザヘテロアリール化合物、その調製方法及び使用 | |
| CN101220037B (zh) | 10-位取代高喜树碱类化合物及其用途 | |
| KR101693326B1 (ko) | 3,4-디히드로퀴나졸린 유도체 및 그를 포함하는 복합제 | |
| CN102875462B (zh) | 一种抗肿瘤2-氨基烟腈及其应用、制备方法 | |
| JP2017071567A (ja) | 抗がん剤デリバリー分子 | |
| CN116419753B (zh) | 作为bcl-2抑制剂的杂环化合物 | |
| CN119698299A (zh) | 通过放疗的n-氧化物的前药活化 | |
| CA2980084C (en) | Asymmetric bis-acridines with antitumour activity and their uses | |
| CN110698491A (zh) | 2-(喜树碱-10-氧基)乙酰胺类化合物和应用 | |
| CN106349180B (zh) | 4,5-二苯基异噁唑衍生物及其制备方法和应用 | |
| AU2024308508A1 (en) | Heterobifunctional compounds and methods of use thereof | |
| CN108299390A (zh) | 抗肿瘤化合物dcz0415及其制备方法和应用 | |
| WO2025193796A1 (en) | Ros-activatable prodrugs of doxazolidine as novel cancer therapy paradigm | |
| CN120136949A (zh) | 甘草次酸-金(ⅰ)化合物的制备方法及在肝癌中的应用 | |
| CA3209238A1 (en) | Heteroaromatic phosphonium salts and their use treating cancer | |
| CN121378247A (zh) | 一种新型cdk6共价抑制剂及其制备方法和医药用途 | |
| KR20260057135A (ko) | Pkmyt1 표적화용 이작용성 화합물 및 이의 사용 방법 | |
| CA3092149A1 (en) | Indolizine compound, preparation method and use thereof |