ES2774473T3 - Forma de dosificación combinada de un antagonista del receptor opioide mu y un agente opioide - Google Patents
Forma de dosificación combinada de un antagonista del receptor opioide mu y un agente opioide Download PDFInfo
- Publication number
- ES2774473T3 ES2774473T3 ES16718062T ES16718062T ES2774473T3 ES 2774473 T3 ES2774473 T3 ES 2774473T3 ES 16718062 T ES16718062 T ES 16718062T ES 16718062 T ES16718062 T ES 16718062T ES 2774473 T3 ES2774473 T3 ES 2774473T3
- Authority
- ES
- Spain
- Prior art keywords
- dosage form
- axelopran
- weight
- opioid
- solid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002552 dosage form Substances 0.000 title claims description 94
- 239000003795 chemical substances by application Substances 0.000 title description 7
- 239000002623 mu opiate receptor antagonist Substances 0.000 title description 7
- QNFFAKFVKCGLPK-WTKDUOFSSA-N chembl3137319 Chemical compound OS(O)(=O)=O.NC(=O)C1=CC=CC([C@H]2C[C@H]3CC[C@H](N3CCN(CC3CCCCC3)C(=O)[C@@H](O)CO)C2)=C1 QNFFAKFVKCGLPK-WTKDUOFSSA-N 0.000 claims abstract description 47
- 239000000203 mixture Substances 0.000 claims abstract description 42
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims abstract description 40
- 239000004372 Polyvinyl alcohol Substances 0.000 claims abstract description 33
- 229920002451 polyvinyl alcohol Polymers 0.000 claims abstract description 33
- 239000008247 solid mixture Substances 0.000 claims abstract description 29
- 229940050929 polyethylene glycol 3350 Drugs 0.000 claims abstract description 22
- 235000010323 ascorbic acid Nutrition 0.000 claims abstract description 20
- 239000011668 ascorbic acid Substances 0.000 claims abstract description 20
- 229960005070 ascorbic acid Drugs 0.000 claims abstract description 20
- 229950007844 axelopran Drugs 0.000 claims description 105
- ATLYLVPZNWDJBW-KIHHCIJBSA-N chembl3137313 Chemical compound NC(=O)C1=CC=CC([C@H]2C[C@H]3CC[C@H](N3CCN(CC3CCCCC3)C(=O)[C@@H](O)CO)C2)=C1 ATLYLVPZNWDJBW-KIHHCIJBSA-N 0.000 claims description 105
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical group O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 claims description 49
- 229960002085 oxycodone Drugs 0.000 claims description 45
- 239000003814 drug Substances 0.000 claims description 43
- 239000011248 coating agent Substances 0.000 claims description 42
- 238000000576 coating method Methods 0.000 claims description 42
- 229940079593 drug Drugs 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 34
- 239000000243 solution Substances 0.000 claims description 28
- 238000009472 formulation Methods 0.000 claims description 23
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical compound O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 claims description 21
- 239000003612 morphinomimetic agent Substances 0.000 claims description 21
- 239000007787 solid Substances 0.000 claims description 18
- 229960005118 oxymorphone Drugs 0.000 claims description 17
- 208000002193 Pain Diseases 0.000 claims description 16
- 230000036407 pain Effects 0.000 claims description 15
- 239000007909 solid dosage form Substances 0.000 claims description 15
- 239000007864 aqueous solution Substances 0.000 claims description 14
- 239000012729 immediate-release (IR) formulation Substances 0.000 claims description 13
- 239000000014 opioid analgesic Substances 0.000 claims description 13
- 230000000694 effects Effects 0.000 claims description 9
- 229920001223 polyethylene glycol Polymers 0.000 claims description 9
- 239000002202 Polyethylene glycol Substances 0.000 claims description 8
- 241000124008 Mammalia Species 0.000 claims description 7
- 238000000338 in vitro Methods 0.000 claims description 7
- 229920000642 polymer Polymers 0.000 claims description 7
- 230000002496 gastric effect Effects 0.000 claims description 6
- 239000004014 plasticizer Substances 0.000 claims description 6
- 238000002560 therapeutic procedure Methods 0.000 claims description 5
- 239000003826 tablet Substances 0.000 description 49
- BQNSLJQRJAJITR-UHFFFAOYSA-N 1,1,2-trichloro-1,2-difluoroethane Chemical compound FC(Cl)C(F)(Cl)Cl BQNSLJQRJAJITR-UHFFFAOYSA-N 0.000 description 18
- 229960003617 oxycodone hydrochloride Drugs 0.000 description 18
- 239000011247 coating layer Substances 0.000 description 16
- 239000011324 bead Substances 0.000 description 15
- 239000010410 layer Substances 0.000 description 15
- 238000004090 dissolution Methods 0.000 description 13
- 238000013265 extended release Methods 0.000 description 13
- 230000008569 process Effects 0.000 description 13
- BCGJBQBWUGVESK-KCTCKCTRSA-N Oxymorphone hydrochloride Chemical compound Cl.O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BCGJBQBWUGVESK-KCTCKCTRSA-N 0.000 description 12
- 238000013270 controlled release Methods 0.000 description 12
- 229960005374 oxymorphone hydrochloride Drugs 0.000 description 12
- 230000002093 peripheral effect Effects 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 206010010774 Constipation Diseases 0.000 description 10
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 10
- 239000007921 spray Substances 0.000 description 10
- 208000030053 Opioid-Induced Constipation Diseases 0.000 description 9
- 238000002425 crystallisation Methods 0.000 description 9
- 230000008025 crystallization Effects 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 238000011260 co-administration Methods 0.000 description 8
- 239000003401 opiate antagonist Substances 0.000 description 8
- 239000000902 placebo Substances 0.000 description 8
- 229940068196 placebo Drugs 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 241000282472 Canis lupus familiaris Species 0.000 description 7
- 229940005483 opioid analgesics Drugs 0.000 description 7
- 238000012545 processing Methods 0.000 description 7
- 230000004584 weight gain Effects 0.000 description 7
- 235000019786 weight gain Nutrition 0.000 description 7
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 6
- 230000003647 oxidation Effects 0.000 description 6
- 238000007254 oxidation reaction Methods 0.000 description 6
- 239000008213 purified water Substances 0.000 description 6
- 238000005507 spraying Methods 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 5
- 239000012530 fluid Substances 0.000 description 5
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 description 5
- 229960000240 hydrocodone Drugs 0.000 description 5
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 5
- 239000011325 microbead Substances 0.000 description 5
- 229960005181 morphine Drugs 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 description 5
- -1 2 - ((cyclohexylmethyl) ((2S) -2,3-dihydroxypropanoyl) amino) ethyl Chemical group 0.000 description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 238000007726 management method Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 229940068021 opana Drugs 0.000 description 4
- 229940105606 oxycontin Drugs 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 238000000634 powder X-ray diffraction Methods 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- VDPLLINNMXFNQX-UHFFFAOYSA-N (1-aminocyclohexyl)methanol Chemical compound OCC1(N)CCCCC1 VDPLLINNMXFNQX-UHFFFAOYSA-N 0.000 description 3
- IUAXSWPNEQYKDR-GXTZACRKSA-N (4R,4aR,7aR,12bS)-9-methoxy-3-methyl-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinolin-7-one 2,3-dihydroxybutanedioic acid dihydrate Chemical compound O.O.OC(C(O)C(O)=O)C(O)=O.COc1ccc2C[C@@H]3[C@@H]4CCC(=O)[C@@H]5Oc1c2[C@]45CCN3C IUAXSWPNEQYKDR-GXTZACRKSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 229960002764 hydrocodone bitartrate Drugs 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 229960005489 paracetamol Drugs 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 150000003839 salts Chemical group 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 102000003840 Opioid Receptors Human genes 0.000 description 2
- 108090000137 Opioid Receptors Proteins 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- WVLOADHCBXTIJK-YNHQPCIGSA-N hydromorphone Chemical compound O([C@H]1C(CC[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O WVLOADHCBXTIJK-YNHQPCIGSA-N 0.000 description 2
- 229960001410 hydromorphone Drugs 0.000 description 2
- 229960003943 hypromellose Drugs 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000005414 inactive ingredient Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000011859 microparticle Substances 0.000 description 2
- 102000051367 mu Opioid Receptors Human genes 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- CWERGRDVMFNCDR-UHFFFAOYSA-N thioglycolic acid Chemical compound OC(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-N 0.000 description 2
- 239000004408 titanium dioxide Substances 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- 108020001612 μ-opioid receptors Proteins 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- DYUTXEVRMPFGTH-UHFFFAOYSA-N 4-(2,5-dimethylphenyl)-5-methyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C(=CC=C(C)C=2)C)=C1C DYUTXEVRMPFGTH-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 229910002483 Cu Ka Inorganic materials 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010021518 Impaired gastric emptying Diseases 0.000 description 1
- 206010028391 Musculoskeletal Pain Diseases 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 108010079943 Pentagastrin Proteins 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000000889 atomisation Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 230000006652 catabolic pathway Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000005253 cladding Methods 0.000 description 1
- 239000008199 coating composition Substances 0.000 description 1
- 229920001688 coating polymer Polymers 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 229920001531 copovidone Polymers 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 239000008380 degradant Substances 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 101150007166 ensa gene Proteins 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 229940010443 exalgo Drugs 0.000 description 1
- 239000013022 formulation composition Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- XHILEZUETWRSHC-NRGUFEMZSA-N hydromorphone hydrochloride Chemical compound [H+].[Cl-].O([C@H]1C(CC[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O XHILEZUETWRSHC-NRGUFEMZSA-N 0.000 description 1
- 229960002738 hydromorphone hydrochloride Drugs 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 229940065676 hysingla Drugs 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 1
- 229960003086 naltrexone Drugs 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 229940121367 non-opioid analgesics Drugs 0.000 description 1
- 239000003402 opiate agonist Substances 0.000 description 1
- 229940124583 pain medication Drugs 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- ANRIQLNBZQLTFV-DZUOILHNSA-N pentagastrin Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1[C]2C=CC=CC2=NC=1)NC(=O)CCNC(=O)OC(C)(C)C)CCSC)C(N)=O)C1=CC=CC=C1 ANRIQLNBZQLTFV-DZUOILHNSA-N 0.000 description 1
- 229960000444 pentagastrin Drugs 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229940001607 sodium bisulfite Drugs 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229940001482 sodium sulfite Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940035024 thioglycerol Drugs 0.000 description 1
- 235000010215 titanium dioxide Nutrition 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229940116567 xartemis Drugs 0.000 description 1
- 229940056604 zohydro Drugs 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/46—8-Azabicyclo [3.2.1] octane; Derivatives thereof, e.g. atropine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/2853—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, polyethylene oxide, poloxamers, poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2893—Tablet coating processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Una composición sólida en donde la composición comprende: (a) entre 50 % y 95 % en peso de sulfato de axelopran, (b) entre 5 % y 50 % en peso de alcohol polivinílico, (c) entre 0 % y 45 % en peso de polietilenglicol 3350, y (d) entre 0 % y 10 % en peso de ácido ascórbico.
Description
DESCRIPCIÓN
Forma de dosificación combinada de un antagonista del receptor opioide mu y un agente opioide Antecedentes de la invención
Campo de la invención
La invención se refiere a una composición sólida de un antagonista periférico del receptor opioide mu y a una combinación de la composición antagonista del receptor opioide mu y un agente analgésico opioide. En particular, la invención se refiere a una forma de dosificación unitaria en donde el antagonista del receptor opioide mu está en forma de liberación inmediata y el agente analgésico opioide puede estar, por ejemplo, en forma de liberación prolongada, liberación sostenida, liberación modificada o liberación controlada y a métodos de preparación de dicha forma de dosificación unitaria.
Estado de la técnica
Los compuestos que funcionan como agonistas en receptores opioides, de los cuales oxicodona, hidrocodona, morfina y oximorfona son ejemplos comunes, son los pilares de la terapia analgésica para el tratamiento del dolor moderado a severo. Se entiende que los analgésicos opioides ejercen su efecto beneficioso principalmente mediante la activación de los receptores opioides mu en el cerebro y el sistema nervioso central. Sin embargo, Los receptores opioides se expresan en todo el cuerpo, tanto en el sistema nervioso central como en regiones periféricas, incluyendo el tracto gastrointestinal (GI). Desafortunadamente, los efectos secundarios adversos del uso de analgésicos opioides pueden ser causados por la activación de estos receptores periféricos. En particular, los pacientes que reciben analgésicos opioides para tratamiento del dolor a corto o largo plazo frecuentemente experimentan una variedad de síntomas gastrointestinales adversos, en particular estreñimiento inducido por opioides (OIC). Acompañando al estreñimiento, vaciamiento gástrico retrasado, molestias abdominales y náuseas, OIC puede ser extremadamente debilitante y no es propenso a tolerancia. Los síntomas gastrointestinales pueden ser lo suficientemente graves como para comprometer seriamente la gestión del dolor en algunos pacientes.
Se ha mostrado que los compuestos antagonistas opioides de acción periférica que no atraviesan fácilmente la barrera hematoencefálica son beneficiosos para contrarrestar el estreñimiento inducido por opioides. En particular, se ha demostrado clínicamente que axelopran, 3-((1 R,3r,5S)-8-(2-((ciclohexilmetil)((2S)-2,3-dihidroxipropanoil)amino)etil)-8-azabiciclo[3.2.1]octan-3-il)benzamida,
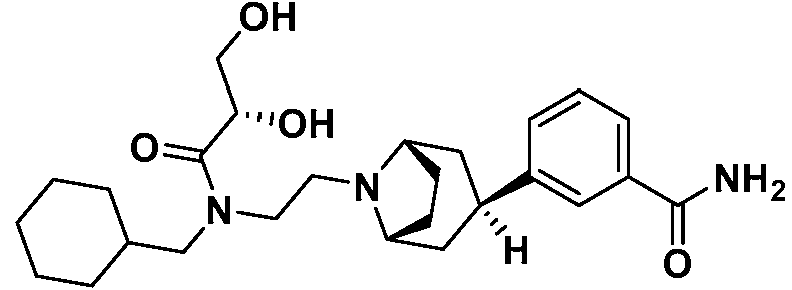
(Patentes de Estados Unidos N.os 7.622.508 y 7.943.772) mejora los síntomas de OIC en pacientes con dolor no canceroso en un régimen de opioides estable sin comprometer la analgesia. (Vickery et al. Pain Week 2012, Las Vegas, NV (P-87).
Los pacientes que toman analgésicos opioides para el tratamiento del dolor también pueden encontrar útil tomar un antagonista opioide periférico para controlar los efectos secundarios adversos de sus medicamentos para el dolor. A tales pacientes, que puede estar sufriendo una variedad de condiciones serias, también se les prescriben frecuentemente fármacos adicionales y, lo tanto, se les carga con la gestión de un régimen farmacéutico complicado. Por tanto, puede ser deseable combinar múltiples fármacos en una forma de dosificación combinada para facilitar la administración y mejora del cumplimiento terapéutico. En particular, el uso de una combinación de un antagonista opioide periférico, como axelopran y un agonista opioide pueden abordar las preocupaciones de OIC antes de desarrollarse.
Según la necesidad clínica, los analgésicos opioides pueden administrarse por las vías oral, transdérmica y parenteral para uso crónico o agudo, de las cuales se prefiere comúnmente la administración oral. Aunque los opioides de liberación inmediata proporcionan una gestión eficaz del dolor, especialmente para dolor agudo y penetrante, los opioides de liberación controlada o prolongada han mostrado un valor clínico significativo. Sin embargo, el efecto profiláctico de un antagonista opioide periférico se logra generalmente mediante la liberación inmediata del agente antagonista. Sería deseable proporcionar una forma de dosificación que combinara un antagonista opioide periférico en una forma de liberación inmediata con un agente analgésico opioide que pueda estar en una liberación inmediata o en una forma de dosificación de liberación modificada (controlada, extendida o sostenida).
Sin embargo, existen varios problemas técnicos que pueden surgir al combinar un antagonista opioide periférico con un agente analgésico opioide. En primer lugar, el antagonista opioide periférico debe ser suficientemente estable durante el almacenamiento de la forma de dosificación. Por ejemplo, se sabe que axelopran se degrada debido a la oxidación en su forma amorfa. Adicionalmente, el antagonista opioide periférico no debería afectar significativamente al perfil de liberación u otras propiedades del agente analgésico opioide.
US2008207676 proporciona una sal de sulfato cristalino de 3-endo-(8-{2-[ciclohexilmetil-((S)-2,3-dihidroxipropionil)amino]etil}-8-aza-biciclo[3.2.1]oct-3-il)benzamida o un solvato de la misma. La invención también proporciona composiciones farmacéuticas que comprenden tales formas de sal cristalina, métodos para usar tales formas de sal cristalina para tratar enfermedades asociadas con la actividad del receptor opioide mu, y procesos útiles para preparar tales formas de sal cristalina.
Sumario de la invención
La presente invención proporciona una composición sólida según la reivindicación 1; una dosis combinada según la reivindicación 5; y una solución acuosa según la reivindicación 12
Se ha mostrado que axelopran es estable como sal de sulfato cristalina, aunque se sabe que axelopran amorfo es insuficientemente estable para su uso en un fármaco. La presente invención es posible gracias al descubrimiento de los requisitos clave para la conversión de axelopran amorfo en axelopran cristalino durante o después de cualquier proceso de preparación del medicamento que inicialmente pueda formar axelopran amorfo. Específicamente, según la elección juiciosa de excipientes y relación relativa de excipientes para axelopran y, opcionalmente, además mediante control de las condiciones del proceso, se puede preparar una composición sólida que comprende sulfato de axelopran en forma cristalina con propiedades físicas deseables.
Específicamente, la composición sólida comprende
(a) entre 50 % y 95 % en peso de sulfato de axelopran,
(b) entre 5 % y 50 % en peso de alcohol polivinílico,
(c) entre 0 % y 45 % en peso de polietilenglicol 3350, y
(d) entre 0 % y 10 % en peso de ácido ascórbico.
En un aspecto, la composición sólida de la invención comprende sulfato de axelopran en forma cristalina. En un aspecto, la composición sólida que comprende sulfato de axelopran cristalino es estable tras almacenamiento en condiciones aceleradas durante aproximadamente 3 meses. La composición sólida puede formar una perla sólida o puede servir como una película o revestimiento en una comprimidos sólido inerte o en una forma de dosificación sólida.
La capacidad de proporcionar formulaciones que puedan convertir controlablemente el axelopran amorfo en una forma cristalina estable tiene una amplia utilidad para el desarrollo de productos de combinación estables. Los productos comerciales opioides, así como los que están en desarrollo, son el resultado de un importante esfuerzo de investigación para proporcionar una liberación modificada, así como propiedades disuasorias de abuso. El enfoque actual permite que axelopran se combine con la amplia gama de productos opioides actualmente en el mercado o en desarrollo sin afectar significativamente al perfil de liberación o propiedades disuasorias de abuso del agente analgésico opioide.
La forma de dosificación combinada comprende sulfato de axelopran como una capa de revestimiento activa, llamada equivalentemente capa superior de fármaco, en una forma de dosificación de analgésico opioide. La capa de revestimiento activa comprende la composición sólida de la invención. En un aspecto, la capa de revestimiento activa comprende sulfato de axelopran en forma cristalina. Opcionalmente, la forma de dosificación combinada comprende además una capa inferior entre la capa de revestimiento activa y la forma de dosificación de opioide. En un aspecto, la forma de dosificación de la invención proporciona el beneficio de un producto combinado que permite la liberación inmediata de un antagonista periférico del receptor opioide mu sin afectar las características de liberación del fármaco de un agente analgésico opioide desde una forma de dosificación de liberación modificada (controlada, extendida o sostenida) y además sin afectar las características farmacocinéticas de cada componente farmacológico.
Se ha demostrado que el sulfato de axelopran es eficaz para mejorar los síntomas del estreñimiento inducido por opioides a dosis bajas. En un aspecto, la invención proporciona una forma de dosificación combinada que comprende entre aproximadamente 2 mg y aproximadamente 30 mg de axelopran en una formulación de liberación inmediata y un agente analgésico opioide en una forma de liberación modificada. En un aspecto, la forma de dosificación analgésica opioide comprende clorhidrato de oxicodona o clorhidrato de oximorfona.
La invención proporciona una composición para uso en un método para tratar dolor en un mamífero, comprendiendo
el método administrar al mamífero una forma de dosificación combinada según la invención.
La invención también proporciona una forma de dosificación combinada según la invención para usar en un método para mejorar un efecto secundario gastrointestinal de la terapia analgésica con opioides, comprendiendo el método administrar la forma de dosificación combinada según la invención.
En un aspecto adicional, la invención proporciona un método para formar la forma de dosificación combinada de la invención, comprendiendo el método proporcionar un agente analgésico opioide en una forma de dosificación sólida y revestir la forma de dosificación sólida con una solución acuosa según la invención.
Breve descripción de los dibujos
FIG. 1A y FIG. 1B muestran el porcentaje de fármaco (axelopran y clorhidrato de oxicodona) disuelto en función del tiempo de una forma de dosificación combinada de axelopran (declarado en etiqueta 10 mg) y comprimido de liberación controlada de clorhidrato de oxicodona (declarado en etiqueta 20 mg) para el revestimiento del fármaco axelopran Formulación A y revestimiento del fármaco axelopran Formulación B, respectivamente.
FIG. 2A y FIG. 2B muestran el porcentaje de fármaco (axelopran y clorhidrato de oximorfona) disuelto en función del tiempo de una forma de dosificación combinada de axelopran (declarado en etiqueta 10 mg) y tableta de liberación prolongada de clorhidrato de oximorfona (declarado en etiqueta 10 mg) para el revestimiento del fármaco axelopran Formulación A y revestimiento del fármaco axelopran Formulación B, respectivamente.
FIG. 3A y FIG. 3B muestran la concentración plasmática media ± desviación estándar (ng/ml) en función del tiempo de clorhidrato de oxicodona y axelopran, respectivamente, de un estudio farmacocinético de dosis única en perros beagle administrados con la forma de dosificación combinada de axelopran y clorhidrato de oxicodona, los dos componentes coadministrados como comprimidos individuales y los componentes administrados individualmente.
FIG. 4A y FIG. 4B muestran la concentración plasmática media ± desviación estándar (ng/ml) en función del tiempo de clorhidrato de oximorfona y axelopran, respectivamente, de un estudio farmacocinético de dosis única en perros beagle administrados con la forma de dosificación combinada de axelopran y clorhidrato de oximorfona, los dos componentes coadministrados como comprimidos individuales y los componentes administrados individualmente.
FIG. 5 muestra la concentración plasmática media de oxicodona ± desviación estándar (ng/ml) en función del tiempo de un estudio farmacocinético de dosis única en sujetos humanos sanos a los que se administró la forma de dosificación combinada de axelopran y clorhidrato de oxicodona, los dos componentes coadministrados como comprimidos individuales y los componentes administrados individualmente.
FIG. 6 muestra la concentración plasmática media de axelopran ± desviación estándar (ng/ml) en función del tiempo de un estudio farmacocinético de dosis única en sujetos humanos sanos a los que se administró la presente forma de dosificación combinada de axelopran y clorhidrato de oxicodona, los dos componentes coadministrados como comprimidos individuales y los componentes administrados individualmente.
FIG. 7 muestra un patrón de difracción de rayos X en polvo de una forma de dosificación combinada de sulfato de axelopran (declarado en la etiqueta 10 mg) y comprimido de liberación controlada de clorhidrato de oxicodona (declarado en la etiqueta 20 mg) Formulación A obtenida con un difractómetro de rayos X Bruker D8-Advance usando Radiación de Cu-Ka (A = 1,54051 Á) con voltaje de salida de 45 kV y corriente de 40 mA.
FIG. 8 muestra el cambio de peso de una película revestida por pulverización de sulfato de axelopran y alcohol polivinílico en una relación 4:1 p/p sometida a una humedad relativa del 70 % a 25 0C.
Descripción detallada de la invención
Definiciones
Como se usa en este documento, la expresión "forma de dosificación sólida" se refiere a una formulación farmacéutica en forma sólida para administración oral a un paciente. La expresión comprende píldoras, comprimidos, perlas, microperlas, micropartículas, y cápsulas. En particular, el término perla como se usa en el presente documento incluye microperlas, micropartículas, y similares.
Como se usa en este documento, la expresión forma de "liberación modificada" se refiere a una formulación de un fármaco que altera el momento y/o velocidad de liberación de la sustancia farmacológica activa. La expresión forma de 'liberación modificada' incluye formas que se describen de diversas maneras como liberación controlada, liberación sostenida, liberación prolongada y acción prolongada.
Como se usa en este documento, la expresión forma de "liberación inmediata" se refiere a una formulación diseñada para liberar el fármaco activo inmediatamente después de administración oral. En formulaciones de liberación inmediata, no se ha intentado modificar la tasa de liberación farmacológica.
Según la presente invención, se proporciona una forma de dosificación combinada que incluye sulfato de axelopran y un agente analgésico opioide. El agente analgésico opioide puede estar presente en una formulación farmacéutica de liberación modificada diseñada para afectar la velocidad de liberación del agente después de la administración a
un paciente, como se define en el presente documento. La forma de dosificación de analgésicos opioides también puede incluir propiedades disuasorias de abuso.
Los agentes analgésicos opioides adecuados para inclusión en la presente invención incluyen oxicodona, hidrocodona, morfina, oximorfona, e hidromorfona. Numerosas formulaciones de liberación controlada o liberación prolongada de estos agentes están disponibles como productos comerciales o en desarrollo inicial o tardío. Los ejemplos de dichos agentes incluyen, pero sin limitación:
oxicodona: OxyContin® (liberación controlada de clorhidrato de oxicodona, Purdue Pharma), Oxicodona DETERx™ (oxicodona, liberación prolongada, Collegium Pharma), Egalet-002 (oxicodona, liberación prolongada, Egalet Corporation),
hidrocodona: Hysingla™ ER (bitartrato de hidrocodona, liberación prolongada, Purdue Pharma), Zohydro™ ER (bitartrato de hidrocodona, liberación prolongada, Zogenix, Inc.), Vantrela™ (bitartrato de hidrocodona, liberación prolongada, Teva Pharmaceutical Industries, Ltd)
morfina: Egalet-001 (Egalet Corporation), ER morfina (Inspirion Delivery Technologies), MS Contin® (Purdue Pharma)
oximorfona: Opana® ER (clorhidrato de oximorfona, liberación prolongada, Endo Pharmaceuticals), Col-172 (Collegium Pharma)
hidromorfona: Exalgo® (clorhidrato de hidromorfona, liberación prolongada, Mallinckrodt Pharmaceuticals) Ejemplos adicionales incluyen productos que también incorporan un agente analgésico no opioide, en particular acetaminofeno. Los ejemplos de tales combinaciones incluyen, pero sin limitación:
Xartemis™ XR (clorhidrato de oxicodona y acetaminofeno, liberación prolongada, Mallinckrodt Pharmaceuticals) MNK-155 (bitartrato de hidrocodona y acetaminofeno, liberación controlada, Mallinckrodt Pharmaceuticals) Como se describe en la Patente de Estados Unidos N.° 7.943.772, el sulfato de axelopran forma una forma cristalina estable, mientras que axelopran amorfo no es suficientemente estable para uso en un fármaco. Como se describe en el presente documento, la forma cristalina se caracteriza por un patrón de difracción de rayos X en polvo (PXRD) que tiene dos o más picos de difracción a valores de 20 seleccionados entre 6,58 ± 0,20, 7,52 ± 0,20, 9,35 ± 0,20, 14,69 ± 0,20, 16,01 ± 0,20, 17,45 ± 0,20, 17,99 ± 0,20, 18,62 ± 0,20, 19,76 ± 0,20, 21,1160,20, 22,07 ± 0,20, 23,18 ± 0,20, 23,74 ± 0,20, 24,56 ± 0,20, 25,63 ± 0,20, 26,45 ± 0,20, 27,86 ± 0,20, 28,31 ± 0,20, 29,54 ± 0,20, 30,59 ± 0,20, 31,58 ± 0,20, 33,89 ± 0,20 y 36,02 ± 0,20. En particular, se ha demostrado la estabilidad de la forma cristalina de sulfato de axelopran en condiciones aceleradas. Así, Es deseable que cualquier fármaco contenga axelopran en forma cristalina.
La provisión de formas de dosificación de combinación estables que incluyen axelopran es posible gracias al descubrimiento por los presentes inventores de los requisitos clave para la conversión de axelopran amorfo en axelopran cristalino durante o después de cualquier proceso que inicialmente pueda formar axelopran amorfo. Se encontró que la elección de los excipientes en la solución de revestimiento es un factor. Como se muestra en el Ejemplo 10 a continuación, los polímeros de revestimiento convencionales no conducen a películas que sean suficientemente estables cuando se prueban en condiciones aceleradas. El ejemplo también muestra que la relación de axelopran a excipientes es otro factor crítico.
Los presentes inventores además han demostrado que un alto contenido de humedad favorece la cristalización como se demuestra en Ejemplo 11 y FIG. 8. Una película revestida por pulverización de sulfato de axelopran y alcohol polivinílico en una relación 4:1 p/p se sometió a 70 % de humedad relativa durante 48 horas a 25 0C. El aumento de peso durante las dos primeras horas puede atribuirse a la absorción de humedad por el sulfato de axelopran amorfo. La pérdida de peso durante el periodo de tiempo posterior puede atribuirse a la cristalización de sulfato de axelopran que libera la humedad absorbida por la forma amorfa.
La oxidación se identificó como la principal vía de degradación del axelopran amorfo. Por tanto, en cualquier proceso de preparación de una capa que contiene axelopran cristalino a partir de una solución en donde el compuesto es amorfo, es útil controlar la oxidación antes de que tenga lugar la cristalización. Aunque se encontró un alto contenido de humedad para favorecer la cristalización, al mismo tiempo, el alto contenido de humedad favorece la oxidación. Una opción viable, entonces, es usar un gas inerte, como nitrógeno o argón, como gas atomizador durante un proceso de revestimiento por pulverización para reducir o eliminar la oxidación. Otra opción es usar un antioxidante en la formulación para controlar la oxidación durante el revestimiento por pulverización.
Basándose en estos descubrimientos, una composición sólida que favorece la cristalización del axelopran comprende sulfato de axelopran y alcohol polivinílico, un polímero formador de película. La composición sólida comprende sulfato de axelopran, polietilenglicol 3530 (un plastificante), y opcionalmente ácido ascórbico, un antioxidante.
La composición sólida puede prepararse a partir de una solución acuosa o una combinación de agua con un alcohol inferior, por ejemplo, hasta aproximadamente 40 % de metanol o etanol en agua, pero preferentemente se prepara a
partir de una solución acuosa. Los ejemplos de polímeros formadores de película conocidos en la técnica incluyen alcohol polivinílico, hidroxipropilmetil celulosa, etilcelulosa, hidroxipropilcelulosa, hidroxietilcelulosa, copovidona, y sus combinaciones. Sin embargo, como se ejemplifica a continuación, se ha determinado que no todos los polímeros formadores de película son igualmente compatibles con fomentar la cristalización del axelopran y/o estabilidad química. En la presente invención, se usa alcohol polivinílico. Los plastificantes hidrófilos conocidos en la técnica incluyen polietilenglicoles, glicerina, polietilenglicol monometil éter, propilenglicol, triacetina y solución de sorbitol sorbitán. Además se determinó, que la elección del plastificante también es importante. En la presente formulación se usa polietilenglicol 3350. El polietilenglicol es un polímero de etilenglicol que tiene un número molecular promedio de 3350. Los antioxidantes incluyen ácido ascórbico, galato de propilo, sulfito sódico, metabisulfito sódico, bisulfito sódico, tioglicerol, ácido tioglicólico, hidroxitolueno butilado, hidroxianisol butilado, y combinaciones de los mismos. En la presente invención se usa ácido ascórbico.
En la composición sólida que favorece la cristalización, el sulfato de axelopran está presente en una cantidad que varía del 50 % al 95 % en peso, incluyendo del 50 % al 85 % en peso, y del 50 % al 70 % en peso. El alcohol polivinílico, está presente en una cantidad entre 5 % y 50 % en peso, incluyendo entre 10 % y 50 % en peso, entre 10 % y 35 % en peso, y entre 12 % y 25 % en peso. El polietilenglicol 3350 está presente en una cantidad entre 0 % y 45 % en peso, incluyendo entre 5 % y 30 % en peso, y entre 12 % y 25 % en peso. El ácido ascórbico está presente en una cantidad entre 0 % y 10 % en peso, incluyendo entre 0,5 % y 10 % en peso, y entre 2 % y 6 % en peso.
En otro aspecto, la invención proporciona una composición sólida que comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 10 % y 50 % en peso de alcohol polivinílico,
(c) entre 5 % y 30 % en peso de polietilenglicol 3350, y
(d) entre 0,5 % y 10 % de ácido ascórbico.
En otro aspecto, la composición sólida en peso comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 12 % y 25 % en peso de alcohol polivinílico,
(c) entre 12 % y 25 % en peso de polietilenglicol 3350, y
(d) entre 2 % y 6 % en peso de ácido ascórbico.
Como se ha descrito anteriormente, la composición sólida de la invención puede prepararse a partir de una solución o suspensión acuosa, en particular una solución o suspensión que comprende sulfato de axelopran, alcohol polivinílico, opcionalmente polietilenglicol 3350 y opcionalmente ácido ascórbico. Típicamente, la composición sólida se prepara a partir de una solución acuosa. En la solución acuosa, sulfato de axelopran, alcohol polivinílico, polietilenglicol 3350 opcional y ácido ascórbico opcional están presentes en la misma proporción que en la composición sólida. Los componentes de la composición sólida, es decir, los componentes no acuosos de la solución, denominados colectivamente "contenido sólido", pueden comprender entre aproximadamente 1 % y aproximadamente 50 %, generalmente entre aproximadamente 5 % y aproximadamente 15 %, por ejemplo, aproximadamente 10 %, de la solución acuosa, el resto es generalmente agua purificada.
En otro aspecto, por tanto, la invención proporciona una solución acuosa que comprende entre 5 % y 15 % de contenido sólido, en donde el contenido sólido comprende sulfato de axelopran, alcohol polivinílico, polietilenglicol 3350 opcional, y ácido ascórbico en las proporciones de la composición sólida descrita anteriormente.
La composición sólida puede constituir una capa, es decir, la capa de revestimiento activa, de una forma de dosificación combinada, o puede formar un revestimiento en un comprimido sólido inerte para proporcionar axelopran como monoterapia. Alternativamente, la composición sólida puede prepararse como una microperla sólida que, por ejemplo, puede combinarse con microperlas de un agente diferente en una forma de dosificación combinada.
En una forma de dosificación combinada que comprende la composición sólida de la invención como una capa de revestimiento activa, llamada equivalentemente capa superior de fármaco, en una forma de dosificación de analgésico opioide, la capa de revestimiento activa que comprende sulfato de axelopran está diseñada para liberar el fármaco en un patrón de liberación inmediata.
En otro aspecto, la invención proporciona una forma de dosificación combinada que comprende una forma de dosificación sólida analgésica opioide y una capa superior de fármaco que comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 10 % y 50 % en peso de alcohol polivinílico,
(c) entre 5 % y 30 % en peso de polietilenglicol 3350, y
(d) entre 0,5 % y 10 % en peso de ácido ascórbico.
En otro aspecto más, la capa superior de fármaco comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 12 % y 25 % en peso de alcohol polivinílico,
(c) entre 12 % y 25 % en peso de polietilenglicol 3350, y
(d) entre 2 % y 6 % en peso de ácido ascórbico.
Como se describe en los ejemplos a continuación, una capa superior de fármaco activo particularmente útil comprende aproximadamente 68 % en peso de sulfato de axelopran, aproximadamente 14 % de alcohol polivinílico en peso, aproximadamente 14 % en peso de polietilenglicol 3350 en peso y aproximadamente 4 % en peso de ácido ascórbico, que corresponde a una relación en peso del equivalente de base libre de axelopran: alcohol polivinílico: polietilenglicol 3350: ácido ascórbico de 12:3:3:1.
Opcionalmente, la forma de dosificación combinada comprende además una capa inferior entre la capa de revestimiento activa y la forma de dosificación de opioide. Es conveniente emplear el mismo polímero formador de película y plastificante en la capa inferior que en el revestimiento activo. En un aspecto particular, la forma de dosificación combinada de la presente invención incluye una capa inferior opcional que comprende alcohol polivinílico y polietilenglicol 3350, por ejemplo en una relación de aproximadamente 1:2 de alcohol polivinílico a polietilenglicol 3350.
El revestimiento activo puede prepararse a partir de una solución de revestimiento acuosa descrita anteriormente como solución acuosa para la preparación de la presente composición sólida. Una solución de revestimiento para aplicar el subrevestimiento opcional comprende entre aproximadamente 5 % y aproximadamente 15 % de componentes de subrevestimiento, es decir, alcohol polivinílico y polietilenglicol 3350, con el resto siendo agua purificada.
La presente forma de dosificación de combinación se puede preparar aplicando la solución de revestimiento acuosa descrita en el presente documento sobre un comprimido opioide opcionalmente subrevestido, píldora, perlas, microperla o cápsula. El proceso de preparación de la capa de revestimiento activa y la capa inferior opcional puede utilizar un revestidor de bandeja o lecho fluido o columna de revestimiento Wurster. Por ejemplo, como se describe en los ejemplos adjuntos, se puede usar una capa superior para pulverizar una solución de revestimiento de subrevestimiento y posteriormente la solución de revestimiento de capa activa acuosa sobre comprimidos opioides. El proceso se realiza generalmente a una temperatura de escape de aproximadamente 40 °C. También se ha demostrado el revestimiento por pulverización sobre perlas con un diámetro característico de aproximadamente 500 pm usando un lecho fluido.
En un aspecto, por tanto, la invención proporciona un método para preparar una forma de dosificación combinada, comprendiendo el método (a) proporcionar un agente analgésico opioide en una forma de dosificación sólida, y (b) revestir la forma de dosificación sólida con una capa superior de fármaco como se describió anteriormente.
El método puede comprender además aplicar una solución acuosa de revestimiento de inferior que comprende alcohol polivinílico y polietilenglicol 3350 sobre la forma de dosificación sólida opioide antes de aplicar la solución de revestimiento de capa activa.
Se ha descubierto que someter una capa que comprende sulfato de axelopran en una forma amorfa o en una forma en donde la cristalización es incompleta a una humedad relativa alta, por ejemplo entre 70 % y 86 % de humedad relativa a una temperatura entre 20 0C y 45 0C da como resultado la conversión del sulfato de axelopran amorfo en la forma cristalina. En particular, se ha descubierto que la capacidad de cristalizar el post-revestimiento de axelopran mejora mediante la formulación específica de la capa sólida desvela en el presente documento.
Los parámetros del proceso de revestimiento por pulverización descritos a continuación en los Ejemplos 2, 3 y 7 en donde se pulveriza una solución acuosa a 40 0C o 45 0C proporcionan las condiciones propicias para la formación de sulfato de axelopran cristalino, como se demuestra por el patrón de difracción de rayos X en polvo de una forma de dosificación combinada preparada mediante el proceso de revestimiento por pulverización ejemplificado mostrado en la FIG. 7.
Además, se ha demostrado que una forma de dosificación combinada preparada mediante un proceso como el descrito en los ejemplos es estable después de almacenamiento. Después de tres meses en condiciones aceleradas de 40 0C y 75 % de humedad relativa, sin cambios significativos en el contenido de axelopran y el perfil de impurezas, así como en el perfil de disolución, se observaron en una dosis combinada de sulfato de axelopran y clorhidrato de oxicodona, lo que indica una buena estabilidad de la forma de dosificación combinada, que además se puede tomar como evidencia de cristalinidad.
Axelopran ha demostrado ser eficaz para mejorar los síntomas del estreñimiento inducido por opioides a dosis bajas. Al ajustar adecuadamente las condiciones de procesamiento, Se puede preparar una forma de dosificación
combinada que contenga entre aproximadamente 2 mg y aproximadamente 30 mg de axelopran por dosis unitaria con buena uniformidad de contenido. Por ejemplo, los inventores han demostrado una forma de dosificación combinada de aproximadamente 10 mg de axelopran de describen comprimidos de liberación controlada de clorhidrato de oxicodona de 20 mg y en comprimidos de liberación prolongada de clorhidrato de oximorfona de 10 mg con una desviación estándar relativa del contenido de axelopran inferior a aproximadamente 5 %.
La presente forma de dosificación combinada que comprende un revestimiento activo de axelopran y un opioide de liberación modificada muestra una liberación inmediata de axelopran y una liberación prolongada del opioide. Por ejemplo, el perfil de disolución del fármaco in vitro de un revestimiento aproximado de 10 mg de capa superior de axelopran en un comprimido de clorhidrato de oxicodona de liberación controlada (FIG. 1A y FIG. 1B) muestra que esencialmente todo el axelopran se liberó en el primer cuarto de hora, mientras que la oxicodona se liberó gradualmente durante 12 horas. Además, el perfil de disolución del fármaco in vitro del comprimido de clorhidrato de oxicodona revestido con axelopran de 10 mg que se había almacenado a 40 0C y 75 % de humedad relativa no cambió desde el perfil de disolución de los comprimidos de tiempo cero. Igualmente, la disolución in vitro de comprimidos de liberación prolongada de clorhidrato de oximorfona revestidos con axelopran demuestra que esencialmente todo el axelopran se liberó en el primer cuarto de hora, mientras que la oximorfona se liberó gradualmente durante 12 horas (FIG. 2A y FIG. 2B).
Además, se ha demostrado la presente forma de dosificación combinada in vivo para proporcionar una exposición sistémica de los dos componentes de manera coherente con las exposiciones observadas en la coadministración de los dos componentes en formas de dosificación separadas. En el Ejemplo 8 se describe un estudio farmacocinético de comprimidos de clorhidrato de oxicodona revestidos con axelopran y comprimidos de clorhidrato de oximorfona revestidos con axelopran en perros. Se observa poca diferencia en la exposición sistémica de oxicodona cuando se administra como la forma de dosificación combinada con axelopran, en comparación con la exposición de la administración de axelopran y oxicodona en formas de dosificación separadas, y con la administración de oxicodona solo, en FIG. 3A. La exposición sistémica de axelopran de la forma de dosificación combinada con oxicodona se muestra coherente con la exposición de la administración en formas de dosificación separadas y de la administración de axelopran solo en la FIG. 3B. También se observaron exposiciones consistentes para la forma de dosificación combinada de axelopran y oximorfona como se ilustra en FIG. 4A y FIG. 4B.
La presente forma de dosificación combinada de axelopran y oxicodona también se estudió en sujetos humanos sanos. Tomando la coadministración de los comprimidos individuales como producto de referencia, La forma de dosificación combinada de 10 mg de axelopran y 20 mg de clorhidrato de oxicodona demostró una exposición total de axelopran dentro de 16 % del valor de referencia y una exposición total de oxicodona dentro de 5 % del valor de referencia en sujetos humanos sanos. Además, La exposición a oxicodona satisfizo las pautas de bioequivalencia de la FDA de Estados Unidos en comparación con el producto de referencia. Además, la exposición total de oxicodona de la combinación fue equivalente a la exposición experimentada cuando la oxicodona se administró sola. Como se ilustra en la FIG. 5, la forma de dosificación combinada de axelopran y oxicodona no alteró la exposición sistémica de oxicodona.
Utilidad
Los agentes terapéuticos opioides como oxicodona, hidrocodona, morfina, y oximorfona, se utilizan ampliamente para el tratamiento del dolor, incluyendo dolor intenso, dolor crónico y dolor no canceroso, como dolor musculoesquelético y dolor postoperatorio. Como se espera que la forma de dosificación combinada de axelopran y un agente analgésico opioide proporcione una exposición equivalente del opioide después de la administración oral, se espera que la presente forma de dosificación combinada sea útil para el tratamiento del dolor.
En un aspecto, por tanto, la invención proporciona una forma de dosificación combinada de la invención para usar en un método de tratamiento del dolor en un mamífero, particularmente un paciente humano, comprendiendo el método administrar al mamífero una forma de dosificación combinada según la invención.
Se ha demostrado que el antagonista del receptor opioide mu axelopran mejora los síntomas del estreñimiento inducido por opioides en pacientes con un régimen de opioides estable que es reducir uno de los efectos secundarios molestos de la terapia con opioides. Como también se espera que la forma de dosificación combinada de axelopran y un analgésico opioide proporcione una exposición equivalente de axelopran en comparación con la administración de axelopran por separado, Se espera que la presente combinación sea útil para la mejora de los efectos secundarios gastrointestinales inducidos por opioides. En otro aspecto, por tanto, la presente invención proporciona una forma de dosificación combinada según la invención para usar en un método para mejorar un efecto secundario gastrointestinal de la terapia con opioides en un mamífero, en particular un paciente humano, comprendiendo el método administrar al mamífero una forma de dosificación combinada según la invención.
Ejemplos
Los siguientes ejemplos se ofrecen para ilustrar la invención, y no deben interpretarse de ninguna manera como limitantes del alcance de la invención.
Para los siguientes ejemplos, el sulfato de axelopran se sintetizó según el proceso descrito en la Patente de Estados Unidos N.° 7.943.772. Se usó una formulación de comprimido clínico de axelopran como artículo de ensayo para estudios in vivo. La oxicodona se proporcionó como comprimidos OxyContin® (liberación controlada de clorhidratos de oxicodona, 20 mg) obtenidos comercialmente. Según el prospecto, los comprimidos contienen 20 mg de oxicodona por comprimido como sal de clorhidrato y los siguientes principios inactivos: hidroxitolueno butilado, hipromelosa, polietilenglicol 400, óxido de polietileno, estearato de magnesio, dióxido de titanio, polisorbato 80, y óxido de hierro rojo. La oximorfona se proporcionó como comprimidos Opana® ER (liberación prolongada de clorhidrato de oximorfona, 10 mg) obtenidos comercialmente. Según el prospecto, los comprimidos de oximorfona contienen 10 mg de clorhidrato de oximorfona por comprimido y los siguientes ingredientes inactivos: hipromelosa, óxido de polietileno, polietilenglicol, a-tocoferol, ácido cítrico, alcohol polivinílico, dióxido de titanio, macrogel, talco, y FD&C amarillo N.° 6.
Ejemplo 1: Proceso de revestimiento por pulverización
Para preparar las formas de dosificación combinadas de los Ejemplos 2 y 3 se usó un revestidor de bandeja Thomas Compu-Lab de 19 pulgadas. Los parámetros de procesamiento optimizados para rendimiento y eficacia del revestimiento se resumen en la Tabla 1.

Ejemplo 2: Preparación de la forma de dosificación combinada de axelopran (10 mg) y clorhidrato de oxicodona (20 mg)
Los núcleos de placebo se fabricaron para que coincidieran con los comprimidos de OxyContin® y se usaron como carga principal del sustrato para el revestimiento.
(a) Preparación de la capa inferior
Para preparar la solución de revestimiento, se añadió alcohol polivinílico (40 g) al agua purificada calentada USP. La mezcla se agitó y calentó hasta su disolución y luego se añadió polietilenglicol 3350 (80 g) y la mezcla se agitó hasta su disolución. Se añadió agua purificada USP con agitación para proporcionar una solución de aproximadamente 10 % de contenido sólido (es decir, contenido no acuoso; el contenido sólido no está en forma sólida).
Un total de aproximadamente 4 kg de núcleos de placebo y un pequeño número de comprimidos de OxyContin® se revistieron por pulverización con la solución de revestimiento inferior según los parámetros de procesamiento de la Tabla I, Conjunto I, para lograr un aumento de peso aproximado del 3 % de la composición de revestimiento inferior indicada en Tabla 2.
Tabla 2. Com osición de formulación de revestimiento inferior

(b) Preparación de la capa de revestimiento activa
Las cantidades de componentes para crear soluciones de revestimiento activas para dos formulaciones se enumeran en la Tabla 3. Para preparar cada solución de revestimiento activa, se añadió alcohol polivinílico al agua purificada calentada USP. La mezcla se agitó y calentó hasta disolución y luego se añadió polietilenglicol, la mezcla se agitó hasta disolución, y se añadió ácido ascórbico con agitación seguido de sulfato de axelopran. Se añadió agua purificada USP con agitación para proporcionar una solución de revestimiento de aproximadamente un 10 % de contenido sólido (por ejemplo, contenido no acuoso; el contenido sólido no está en forma sólida).
Tabla 3. Com osiciones de formulación de ca a de revestimiento activa de fármaco Axelo ran

Al finalizar la capa inferior, los núcleos de placebo sub-revestidos y los comprimidos opioides se retuvieron en la bandeja de revestimiento y se revistieron por pulverización con la solución de revestimiento activa según los parámetros de procesamiento de la Tabla 1, Conjunto I, para lograr el aumento de peso objetivo. Los comprimidos opioides revestidos se separaron de los núcleos revestidos de placebo por la diferencia de color al finalizar el revestimiento.
Ejemplo 3: Preparación de la forma de dosificación combinada de axelopran (10 mg) y clorhidrato de oximorfona (20 mg)
Los núcleos de placebo se fabricaron para que coincidan con los comprimidos Opana® ER y se usaron como carga principal del sustrato para el revestimiento.
(a) Preparación de la capa inferior
Se preparó una solución de revestimiento inferior como en el Ejemplo 2(a). Un total de aproximadamente 4 kg de núcleos de placebo y un pequeño número de comprimidos Opana® ER se revistieron por pulverización según los parámetros de procesamiento de la Tabla I, Conjunto II, dando como resultado un aumento de peso aproximado del 3 % de la composición de revestimiento inferior enumerada en la Tabla 2.
(b) Preparación de la capa de revestimiento activa
Las soluciones de revestimiento para las dos formulaciones de la Tabla 3 se prepararon como se describe en el Ejemplo 2(a). Al finalizar la capa inferior, los núcleos de placebo sub-revestidos y los comprimidos opioides se retuvieron en el revestidor de bandeja y se revistieron por pulverización con las soluciones de revestimiento activo según los parámetros de procesamiento de la Tabla 1, Conjunto I, para lograr el aumento de peso objeto. Los comprimidos opioides revestidos se separaron de los núcleos revestidos de placebo por la diferencia de color al finalizar el revestimiento.
Ejemplo 4: Análisis de uniformidad del contenido farmacológico
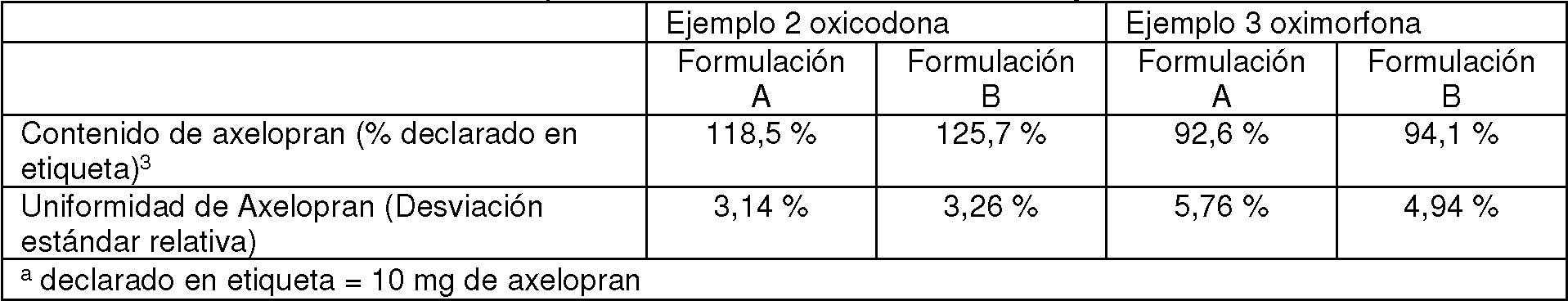
Los comprimidos opioides revestidos con fármaco axelopran (n = 10 comprimidos por formulación) se analizaron para determinar potencia y uniformidad del contenido del fármaco mediante cromatografía líquida de alta presión (HPLC) con detección UV. Los resultados para las formas de dosificación combinadas preparadas en los Ejemplos 2 y 3 se presentan en la Tabla 4.
Tabla 4: Potencia uniformidad del Contenido farmacoló ico Axelo ran
Ejemplo 5: Perfil de disolución farmacológica
Los comprimidos opioides revestidos con axelopran de los Ejemplos 2 y 3 (n = 6 comprimidos por formulación) se analizaron para determinar la velocidad de disolución del fármaco de los componentes individuales según la monografía USP 711 y las metodologías desarrolladas internamente. Se recogieron alícuotas de muestra a los 13, 30, 45, 60, minutos, y en momentos de disolución de 4 y 12 horas y analizados por HPLC de fase inversa con detección UV. Los resultados de la tasa de disolución para los componentes individuales de los comprimidos de oxicodona revestidos con axelopran se muestran en la Tabla 5 y en las FIGs. 1A y 1B, mientras que los resultados de la tasa de disolución análoga para comprimidos de oximorfona revestidos con axelopran se muestran en la Tabla
6 y en las FIGs. 2A y 2B. En todos los casos, esencialmente se observa que todo el axelopran se ha disuelto en el primer momento de 0,25 horas, mientras que el opioide de liberación controlada se disuelve gradualmente durante 12 horas.
Tabla 5: Disolución in vitro de Forma de Dosificación de Combinación de Axelo ran/Oxicodona

Tabla 6: Disolución in vitro de Forma de Dosificación de Combinación de Axelo ran/Oximorfona

Ejemplo 6: Estudio de estabilidad
Las muestras de una forma de dosificación de combinación de sulfato de axelopran/clorhidrato de oxicodona con la formulación de capa de revestimiento activa del Ejemplo 2 Formulación A almacenada en condiciones aceleradas de 40 0C y 75 % de humedad relativa (HR) se analizaron por HPLC para el contenido de axelopran y el perfil de impurezas como se muestra en la Tabla 7 y para la velocidad de disolución como se muestra en la Tabla 8.
T l En Axl r n Im rz n =

T l Di l in in vir Frm ifi in m in Axl r n xi n n =

Ejemplo 7: Preparación de perlas revestidas con axelopran
La viabilidad de preparar una forma de dosificación combinada en una forma de dosificación sólida compuesta de perlas se demostró revistiendo esferas de azúcar que tenían un diámetro característico de 500 ^m.
La composición para preparar un revestimiento de axelopran nominal de 10 mg por 450 mg de perlas se enumera en la Tabla 9.
T l m iin l rl r i n xl rn

Se realizaron tres ejecuciones de revestimiento:
A. Cantidad de lote 200 g con capa inferior
Se prepararon una solución de revestimiento inferior y una solución de capa de revestimiento activo de axelopran según el proceso descrito en el Ejemplo 2. Se cargaron perlas de azúcar en el recipiente de un minilecho fluido Glatt y se calentaron a aproximadamente 450C. La solución de revestimiento inferior se pulverizó sobre las perlas a una velocidad de pulverización de 0,6 g/min hasta que se logró un aumento de peso del 10 %. Al finalizar el
revestimiento inferior, la solución de la capa de revestimiento activa se pulverizó sobre las perlas de azúcar subrevestidas a una velocidad de pulverización de 0,6 g/min. La temperatura del producto se mantuvo a aproximadamente 45 0C en el lecho fluido con una presión de aire de atomización de 0,8 a 1,0 bar. Al finalizar la deposición de la capa de revestimiento activa, las perlas se secaron a 45 °C durante 10 minutos antes de descargarlas.
B. Cantidad de lote 200 g sin revestimiento inferior
Una solución de capa de revestimiento activa de composición equivalente a la descrita en la Tabla 8 se roció directamente sobre perlas de azúcar como se describió anteriormente.
C. Cantidad de lote 2 kg con revestimiento inferior
Se prepararon dos lotes de perlas revestidas con axelopran denominadas C-1 y C-2, a partir de composiciones con cantidades de lote 10 veces las cantidades enumeradas en la Tabla 8 usando un lecho fluido Glatt GPCG-3. Los parámetros de procesamiento habituales se enumeran en la Tabla 10.
Tabl 1 P r m r r mi n l h fl i l 2 kg)

Las perlas resultantes se probaron para determinar la uniformidad del contenido y el degradante principal.
Tabla 11. Contenido de uniformidad ureza

Ejemplo 8: Farmacocinética y biodisponibilidad relativa en perros
Un estudio farmacocinético cruzado, una sola dosis, cuatro periodos, se realizó en perros beagle en ayunas (6 perros machos por grupo) para evaluar las características farmacocinéticas y la biodisponibilidad relativa de axelopran y opioide (oxicodona u oximorfona) después de administración oral de la presente forma de dosificación combinada, coadministración de los componentes individuales y la forma de dosificación individual de cada componente.
Para el estudio de axelopran/oxicodona se usaron forma de dosificación combinada del Ejemplo 2 Formulación A (11,85 mg de axelopran, 19,42 mg de oxicodona por comprimido) y comprimidos individuales de axelopran (10,21 mg), oxicodona (19,42 mg). Para el estudio de axelopran/oximorfona se usaron la forma de dosificación combinada del Ejemplo 3 Formulación A (9,26 mg de axelopran, 10,39 mg de oximorfona) y comprimidos individuales de axelopran (10,21 mg), oximorfona (10,39 mg). Entre cada fase se observó un periodo de lavado de al menos 5 días. Los perros recibieron un pretratamiento intramuscular de pentagastrina (6 pg/kg) para reducir el pH del estómago. Los perfiles plasmáticos de axelopran y opioide de los estudios farmacocinéticos se muestran en FIGs 3A y 3B (oxicodona) y FIGs 4A y 4B (oximorfona), respectivamente.
Ejemplo 9: Farmacocinética y biodisponibilidad relativa en sujetos humanos sanos
Un estudio cruzado de cuatro secuencias aleatorizado, dosis única, sin ocultación, cuatro periodos de tratamiento, de la biodisponibilidad relativa de axelopran y oxicodona después de la administración oral como la forma de dosificación de combinación actual, después de coadministración de los componentes individuales, y después de la administración de la forma de dosificación individual de cada componente se estudió en sujetos humanos sanos. La naltrexona se administró 15 y 3 horas antes y 9 y 21 horas después de la administración de cada tratamiento para bloquear los efectos adversos del opioide. Entre cada periodo se observó un periodo de lavado de siete días. El
número de sujetos que completaron al menos un periodo fue 28 con 26 sujetos que completaron los cuatro periodos de estudio.
Se prepararon comprimidos combinados revestidos (9,66 mg de axelopran, 19,42 mg de oxicodona por comprimido) según el proceso del Ejemplo 2, capa de axelopran en la formulación A de la Tabla 3. Para el estudio se usaron comprimidos individuales de axelopran (10,21 mg), oxicodona (19,42 mg).
Los parámetros farmacocinéticos (PK) se determinaron mediante análisis no compartimental utilizando WinNonlin Versión 6.2.1 (Pharsight, St. Lous, MO) Se informaron los siguientes parámetros PK:
Cmáx: concentración máxima en plasma
AUCo-último: área bajo la curva de concentración-tiempo desde el momento de la dosificación hasta la última concentración medible
AUCo-~: área bajo la curva de concentración-tiempo extrapolada al infinito
La biodisponibilidad relativa de oxicodona y axelopran para dosificación combinada en comparación con la coadministración de los componentes individuales se proporcionan en las Tablas 12 y 14, respectivamente. La biodisponibilidad relativa de oxicodona para la dosificación combinada en comparación con la oxicodona sola se proporcionan en la Tabla 13. Las exposiciones plasmáticas de oxicodona y axelopran se muestran en FIG. 5 y FIG.
6, respectivamente.
El estudio demostró que la forma de dosificación combinada de axelopran y oxicodona proporcionó exposiciones clínicamente equivalentes a oxicodona y axelopran en relación con la coadministración de comprimidos individuales. Las relaciones medias geométricas de los parámetros farmacocinéticos de oxicodona Cmáx, AUC0-último, y AUCo-~ y los intervalos de confianza asociados están dentro de 80 % a 125 % del producto de referencia. Las relaciones medias geométricas Cmáx, AUC0-último, y AUCo-~ para axelopran a partir de la dosificación combinada con respecto a la coadministración de comprimidos individuales son aproximadamente 85 %.
Tabla 12: Biodisponibilidad relativa de oxicodona para la forma de dosificación combinada en comparación con la coadministración de com onentes individuales

Tabla 13: Biodisponibilidad relativa de oxicodona para la forma de dosificación combinada en comparación con oxicodona soloa

Tabla 14: Biodisponibilidad relativa de Axelopran para la forma de dosificación combinada comparado con la coadministración de com onentes individuales3

Ejemplo 10: Estudio de compatibilidad de excipientes
La compatibilidad del axelopran con cuatro composiciones de materiales de revestimiento comerciales se estudió en función de la relación del material de revestimiento a axelopran. Las mezclas de sulfato de axelopran y los materiales de la Tabla 15 se secaron a 40 0C en botes de pesada para formar películas. La HPLC analizó la estabilidad química de las películas almacenadas en condiciones aceleradas de 40 0C y 88 % de humedad relativa durante 2 semanas.
Tabla 15: Materiales de revestimiento

Tabla 16: Ensa o de Axelo ran

Ejemplo 11: Análisis de sorción de humedad de película de Axelopran
Una solución acuosa de sulfato de axelopran y alcohol polivinílico en una relación 4:1 p/p con una carga sólida del 2,5 % se revistió por pulverización sobre una superficie de vidrio con camisa a 60 0C. Se colocó una pequeña pieza de película en un analizador de sorción de humedad (SGA-100, VTI Corp. Hialeah, FL). La humedad se ajustó a aproximadamente 0 % de humedad relativa (HR) durante 10 minutos y luego se elevó al 70 % de HR durante 48 horas. La temperatura se ajustó a 25 0C. Como se muestra en la FIG. 8, la película aumentó de peso durante las primeras dos horas y comenzó a perder peso después del aumento inicial. El aumento de peso durante las dos primeras horas puede atribuirse a la absorción de humedad por el sulfato de axelopran amorfo. La pérdida de peso
durante el periodo de tiempo posterior puede atribuirse a la cristalización de sulfato de axelopran que libera la humedad absorbida por la forma amorfa. El aumento neto del peso de la película puede atribuirse a la absorción de humedad por el alcohol polivinílico.
Claims (16)
1. Una composición sólida en donde la composición comprende:
(a) entre 50 % y 95 % en peso de sulfato de axelopran,
(b) entre 5 % y 50 % en peso de alcohol polivinílico,
(c) entre 0 % y 45 % en peso de polietilenglicol 3350, y
(d) entre 0 % y 10 % en peso de ácido ascórbico.
2. La composición sólida de la reivindicación 1 en donde el sulfato de axelopran está en forma cristalina.
3. La composición sólida de la reivindicación 1 o la reivindicación 2, en donde la composición comprende
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 10 % y 50 % en peso de alcohol polivinílico,
(c) entre 5 % y 30 % en peso de polietilenglicol 3350, y
(d) entre 0,5 % y 10 % en peso de ácido ascórbico.
4. La composición sólida de la reivindicación 1 o la reivindicación 2 en donde la composición comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 12 % y 25 % en peso de alcohol polivinílico,
(c) entre 12 % y 25 % en peso de polietilenglicol 3350, y
(d) entre 2 % y 6 % en peso de ácido ascórbico.
5. Una forma de dosificación combinada que comprende:
una forma de dosificación sólida que comprende un agente analgésico opioide, y
una parte de liberación inmediata en donde la parte de liberación inmediata comprende una composición sólida como se reivindica en una cualquiera de las reivindicaciones 1 a 4 y es una capa superior de fármaco en forma de dosificación sólida.
6. La forma de dosificación combinada de la reivindicación 5 en donde el agente analgésico opioide está en una formulación de liberación modificada.
7. La forma de dosificación combinada de la reivindicación 6 que comprende además una capa inferior entre la capa superior de fármaco y la forma de dosificación sólida que comprende un agente analgésico opioide y en donde la capa superior de fármaco comprende sulfato de axelopran en forma cristalina y comprende además un polímero formador de película y un plastificante.
8. La forma de dosificación combinada de la reivindicación 7 en donde la capa inferior comprende alcohol polivinílico y polietilenglicol.
9. La forma de dosificación combinada de una cualquiera de las reivindicaciones 5 a 8 en donde el agente analgésico opioide es oxicodona u oximorfona.
10. La forma de dosificación combinada de la reivindicación 5, en donde la capa superior del fármaco comprende sulfato de axelopran en forma cristalina y comprende además un polímero formador de película y un plastificante; la forma de dosificación comprende además una capa inferior en donde la capa inferior comprende alcohol polivinílico y polietilenglicol; en donde el agente analgésico opioide es oxicodona u oximorfona; y en donde la capa superior de fármaco comprende:
(a) entre 50 % y 70 % en peso de sulfato de axelopran,
(b) entre 12 % y 25 % en peso de alcohol polivinílico,
(c) entre 12 % y 25 % en peso de polietilenglicol 3350, y
(d) entre 2 % y 6 % en peso de ácido ascórbico.
11. La forma de dosificación combinada de la reivindicación 10 en donde la forma de dosificación proporciona una tasa de liberación in vitro de axelopran durante menos de una hora y una tasa de liberación in vitro del analgésico opioide de hasta 12 horas.
12. Una solución acuosa en donde los componentes no acuosos de la solución comprenden los componentes (a), (b), (c) y (d) como se define en una cualquiera de las reivindicaciones 1, 3 y 4.
13. Un método para preparar una forma de dosificación combinada como se reivindica en una cualquiera de las reivindicaciones 5 a 11, comprendiendo el método:
(a) proporcionar un agente analgésico opioide en una forma de dosificación sólida,, y
(b) revestir la forma de dosificación sólida con una solución acuosa como se reivindica en la reivindicación 12.
14. El método de la reivindicación 13, que comprende además revestir la forma de dosificación sólida de opioide con una capa inferior que comprende alcohol polivinílico y polietilenglicol sobre la forma de dosificación sólida opioide antes de aplicar la capa superior de fármaco que comprende axelopran.
15. El método de la reivindicación 13, en donde el sulfato de axelopran contenido en la forma de dosificación combinada está en forma cristalina, comprendiendo el método además la etapa (c):
(c) exponer la forma de dosificación sólida revestida a una humedad relativa entre 70 % y 86 % a una temperatura entre 20 0C y 45 0C hasta que el sulfato de axelopran se convierta en forma cristalina.
16. Una composición como se reivindica en una cualquiera de las reivindicaciones 5 a 11, para uso en el tratamiento del dolor en un mamífero, o para mejorar un efecto secundario gastrointestinal de la terapia con analgésicos opioides.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562141981P | 2015-04-02 | 2015-04-02 | |
| PCT/US2016/025176 WO2016161069A1 (en) | 2015-04-02 | 2016-03-31 | Combination dosage form of a mu opioid receptor antagonist and an opioid agent |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2774473T3 true ES2774473T3 (es) | 2020-07-21 |
Family
ID=55806767
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16718062T Active ES2774473T3 (es) | 2015-04-02 | 2016-03-31 | Forma de dosificación combinada de un antagonista del receptor opioide mu y un agente opioide |
Country Status (27)
| Country | Link |
|---|---|
| US (4) | US20160287573A1 (es) |
| EP (1) | EP3277278B1 (es) |
| JP (2) | JP6713483B2 (es) |
| KR (1) | KR20170132325A (es) |
| CN (1) | CN107820424B (es) |
| AU (1) | AU2016243691A1 (es) |
| BR (1) | BR112017021120A2 (es) |
| CA (1) | CA2980328A1 (es) |
| CY (1) | CY1122828T1 (es) |
| DK (1) | DK3277278T3 (es) |
| ES (1) | ES2774473T3 (es) |
| HK (1) | HK1247105A1 (es) |
| HR (1) | HRP20200432T1 (es) |
| HU (1) | HUE048785T2 (es) |
| IL (1) | IL254593A0 (es) |
| LT (1) | LT3277278T (es) |
| ME (1) | ME03671B (es) |
| MX (1) | MX2017012474A (es) |
| PH (1) | PH12017501767A1 (es) |
| PL (1) | PL3277278T3 (es) |
| PT (1) | PT3277278T (es) |
| RS (1) | RS60074B1 (es) |
| RU (1) | RU2017134794A (es) |
| SI (1) | SI3277278T1 (es) |
| SM (1) | SMT202000157T1 (es) |
| TW (1) | TW201642856A (es) |
| WO (1) | WO2016161069A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HK1247105A1 (zh) | 2015-04-02 | 2018-09-21 | Theravance Biopharma R&D Ip, Llc | Mu类鸦片受体拮抗剂与类鸦片药剂的组合剂型 |
| JP7222244B2 (ja) * | 2017-08-08 | 2023-02-15 | 三菱ケミカル株式会社 | 医薬錠剤、およびその製造方法 |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001526228A (ja) | 1997-12-22 | 2001-12-18 | ユーロ−セルティーク,エス.エイ. | オピオイド作動薬/拮抗薬の併用 |
| US6419960B1 (en) | 1998-12-17 | 2002-07-16 | Euro-Celtique S.A. | Controlled release formulations having rapid onset and rapid decline of effective plasma drug concentrations |
| US6451806B2 (en) | 1999-09-29 | 2002-09-17 | Adolor Corporation | Methods and compositions involving opioids and antagonists thereof |
| AR029538A1 (es) * | 2000-07-06 | 2003-07-02 | Wyeth Corp | Composiciones farmaceuticas de agentes estrogenicos |
| US7842307B2 (en) | 2001-08-06 | 2010-11-30 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and gelling agent |
| ATE431738T1 (de) * | 2001-08-06 | 2009-06-15 | Euro Celtique Sa | Opioid-agonist-formulierungen mit freisetzbarem und sequestriertem antagonist |
| AU2003301121A1 (en) * | 2002-12-18 | 2004-07-14 | Pain Therapeutics, Inc. | Oral dosage forms with therapeutically active agents in controlled release cores and immediate release gelatin capsule coats |
| US20050013863A1 (en) | 2003-07-18 | 2005-01-20 | Depomed, Inc., A Corporation Of The State Of California | Dual drug dosage forms with improved separation of drugs |
| CA2540308C (en) | 2003-09-26 | 2013-08-06 | Alza Corporation | Drug coating providing high drug loading and methods for providing the same |
| TWI415635B (zh) | 2004-05-28 | 2013-11-21 | 必治妥施貴寶公司 | 加衣錠片調製物及製備彼之方法 |
| NZ560828A (en) * | 2005-03-02 | 2011-01-28 | Theravance Inc | Quinolinone compounds as 5-HT4 receptor agonists |
| ES2366058T3 (es) * | 2006-03-01 | 2011-10-14 | Theravance, Inc. | Compuestos de 8-azabiciclo[3.2.1]octano como antagonistas de los receptores opioides mu. |
| MY145633A (en) | 2006-03-01 | 2012-03-15 | Theravance Inc | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
| CN101500542A (zh) * | 2006-07-11 | 2009-08-05 | 共有药物有限公司 | 控释制剂 |
| TWI409067B (zh) | 2007-02-28 | 2013-09-21 | Theravance Inc | 8-氮雜雙環〔3.2.1〕辛烷化合物之結晶型 |
| TWI415850B (zh) | 2007-07-20 | 2013-11-21 | Theravance Inc | 製備mu類鴉片受體拮抗劑之中間物的方法 |
| WO2009137672A1 (en) * | 2008-05-07 | 2009-11-12 | Salix Pharmaceuticals, Ltd. | Methods of treating bowel disease by administering a bowel cleanser and an antibiotic |
| EP2210595A1 (en) | 2009-01-14 | 2010-07-28 | LEK Pharmaceuticals d.d. | Active coating of pharmaceutical dosage forms |
| CN102421423B (zh) * | 2009-05-12 | 2014-11-05 | Bpsi控股有限责任公司 | 增强型防潮即释薄膜包衣系统和覆有该包衣的基体 |
| IT1398930B1 (it) * | 2010-03-24 | 2013-03-28 | Molteni & C | Formulazioni farmaceutiche bistrato contenenti agonisti ed antagonisti oppioidi. |
| EP2611442B1 (en) * | 2010-09-03 | 2018-07-04 | Bristol-Myers Squibb Company | Drug formulations using water soluble antioxidants |
| BR112013019026A2 (pt) | 2011-02-01 | 2016-10-04 | Astrazeneca Uk Ltd | formulações farmacêuticas incluindo um composto amina |
| US9339489B2 (en) * | 2013-03-15 | 2016-05-17 | Aprecia Pharmaceuticals Company | Rapid disperse dosage form containing levetiracetam |
| HK1247105A1 (zh) | 2015-04-02 | 2018-09-21 | Theravance Biopharma R&D Ip, Llc | Mu类鸦片受体拮抗剂与类鸦片药剂的组合剂型 |
-
2016
- 2016-03-31 HK HK18106740.0A patent/HK1247105A1/zh unknown
- 2016-03-31 PT PT167180629T patent/PT3277278T/pt unknown
- 2016-03-31 ES ES16718062T patent/ES2774473T3/es active Active
- 2016-03-31 MX MX2017012474A patent/MX2017012474A/es unknown
- 2016-03-31 RU RU2017134794A patent/RU2017134794A/ru not_active Application Discontinuation
- 2016-03-31 HR HRP20200432TT patent/HRP20200432T1/hr unknown
- 2016-03-31 ME MEP-2020-48A patent/ME03671B/me unknown
- 2016-03-31 HU HUE16718062A patent/HUE048785T2/hu unknown
- 2016-03-31 LT LTEP16718062.9T patent/LT3277278T/lt unknown
- 2016-03-31 CN CN201680019821.7A patent/CN107820424B/zh active Active
- 2016-03-31 SM SM20200157T patent/SMT202000157T1/it unknown
- 2016-03-31 CA CA2980328A patent/CA2980328A1/en not_active Abandoned
- 2016-03-31 KR KR1020177031800A patent/KR20170132325A/ko not_active Withdrawn
- 2016-03-31 JP JP2017550832A patent/JP6713483B2/ja active Active
- 2016-03-31 BR BR112017021120-3A patent/BR112017021120A2/pt not_active Application Discontinuation
- 2016-03-31 WO PCT/US2016/025176 patent/WO2016161069A1/en not_active Ceased
- 2016-03-31 PL PL16718062T patent/PL3277278T3/pl unknown
- 2016-03-31 RS RS20200315A patent/RS60074B1/sr unknown
- 2016-03-31 EP EP16718062.9A patent/EP3277278B1/en active Active
- 2016-03-31 DK DK16718062.9T patent/DK3277278T3/da active
- 2016-03-31 TW TW105110406A patent/TW201642856A/zh unknown
- 2016-03-31 AU AU2016243691A patent/AU2016243691A1/en not_active Abandoned
- 2016-03-31 US US15/086,816 patent/US20160287573A1/en not_active Abandoned
- 2016-03-31 SI SI201630671T patent/SI3277278T1/sl unknown
-
2017
- 2017-09-19 IL IL254593A patent/IL254593A0/en unknown
- 2017-09-26 PH PH12017501767A patent/PH12017501767A1/en unknown
- 2017-10-31 US US15/798,647 patent/US10369142B2/en active Active
-
2019
- 2019-06-27 US US16/454,353 patent/US10946009B2/en active Active
-
2020
- 2020-02-04 JP JP2020016858A patent/JP2020073583A/ja not_active Withdrawn
- 2020-03-17 CY CY20201100246T patent/CY1122828T1/el unknown
-
2021
- 2021-02-08 US US17/248,787 patent/US11452723B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| SU1706373A3 (ru) | Способ получени средства с регулируемым высвобождением активного соединени | |
| ES2582646T5 (es) | Formulación de comprimido recubierto y método | |
| ES2633496T3 (es) | Composiciones farmacéuticas que comprenden everolimus | |
| CZ284382B6 (cs) | Pevná léková forma s řízeným uvolňováním účinné látky | |
| ES2656412T3 (es) | Nueva combinación | |
| US20250367213A1 (en) | Immediate release multilayer tablet | |
| US11452723B2 (en) | Combination dosage form of a mu opioid receptor antagonist and an opioid agent | |
| JP7735301B2 (ja) | ドキシラミンコハク酸塩およびピリドキシン塩酸塩の調節放出マルチプルユニット型経口剤形、ならびにその調製方法 | |
| RS60355B1 (sr) | Farmaceutski sastavi i njihova upotreba | |
| WO2025111482A1 (en) | Complexing agent salt formulations of pharmaceutical compounds at low stoichiometric ratios | |
| WO2010094996A1 (es) | Composición farmacéutica oral para uso en enfermedades respiratorias | |
| US20080193537A1 (en) | Morphine Sulfate Formulations | |
| CN102920678A (zh) | 一种奥氮平固体制剂及其制备方法 | |
| US20230248673A1 (en) | Novel multiparticulate pharmaceutical composition of tamsulosin and solifenacin | |
| AU2017251803A1 (en) | Choline salt of an anti-inflammatory substituted cyclobutenedione compound |