ES2774517T3 - Moduladores del receptor nuclear (ROR) para el tratamiento de enfermedades inflamatorias y autoinmunes - Google Patents
Moduladores del receptor nuclear (ROR) para el tratamiento de enfermedades inflamatorias y autoinmunes Download PDFInfo
- Publication number
- ES2774517T3 ES2774517T3 ES16731721T ES16731721T ES2774517T3 ES 2774517 T3 ES2774517 T3 ES 2774517T3 ES 16731721 T ES16731721 T ES 16731721T ES 16731721 T ES16731721 T ES 16731721T ES 2774517 T3 ES2774517 T3 ES 2774517T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- mmol
- optionally substituted
- methyl
- reaction mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000023275 Autoimmune disease Diseases 0.000 title description 8
- 208000027866 inflammatory disease Diseases 0.000 title description 4
- 230000002757 inflammatory effect Effects 0.000 title description 4
- 108020005497 Nuclear hormone receptor Proteins 0.000 title description 3
- 108020004017 nuclear receptors Proteins 0.000 title description 2
- 102000006255 nuclear receptors Human genes 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 173
- -1 -OCHF2 Chemical group 0.000 claims abstract description 134
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 81
- 150000003839 salts Chemical class 0.000 claims abstract description 38
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 37
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 35
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 26
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 24
- 125000003386 piperidinyl group Chemical group 0.000 claims abstract description 20
- 125000001424 substituent group Chemical group 0.000 claims abstract description 18
- 125000002757 morpholinyl group Chemical group 0.000 claims abstract description 17
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 14
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 11
- 125000004193 piperazinyl group Chemical group 0.000 claims abstract description 11
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 10
- 229910052721 tungsten Inorganic materials 0.000 claims abstract description 10
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 9
- 150000002367 halogens Chemical class 0.000 claims abstract description 9
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 8
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims abstract description 8
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 7
- 125000003566 oxetanyl group Chemical group 0.000 claims abstract description 7
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 7
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 7
- 125000001475 halogen functional group Chemical group 0.000 claims abstract description 6
- 229910052727 yttrium Inorganic materials 0.000 claims abstract description 6
- 125000002393 azetidinyl group Chemical group 0.000 claims abstract description 5
- 125000005960 1,4-diazepanyl group Chemical group 0.000 claims abstract description 4
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims abstract description 3
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 claims abstract description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract 3
- 125000000217 alkyl group Chemical group 0.000 claims description 83
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 19
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 2
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 claims 1
- 125000002947 alkylene group Chemical group 0.000 abstract description 6
- 201000006417 multiple sclerosis Diseases 0.000 description 396
- 238000000034 method Methods 0.000 description 311
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 305
- 239000011541 reaction mixture Substances 0.000 description 203
- 239000000243 solution Substances 0.000 description 201
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 185
- 238000002360 preparation method Methods 0.000 description 182
- 238000005160 1H NMR spectroscopy Methods 0.000 description 177
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 170
- 235000019439 ethyl acetate Nutrition 0.000 description 151
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 105
- 239000000203 mixture Substances 0.000 description 99
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 92
- 235000019341 magnesium sulphate Nutrition 0.000 description 92
- 239000012044 organic layer Substances 0.000 description 91
- 230000002829 reductive effect Effects 0.000 description 89
- 239000007787 solid Substances 0.000 description 74
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 60
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 56
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 54
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 50
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 50
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 50
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 45
- 239000000741 silica gel Substances 0.000 description 43
- 229910002027 silica gel Inorganic materials 0.000 description 43
- 238000006243 chemical reaction Methods 0.000 description 42
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 38
- 238000004440 column chromatography Methods 0.000 description 38
- 239000010410 layer Substances 0.000 description 38
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 36
- 229920006395 saturated elastomer Polymers 0.000 description 36
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 35
- 239000012267 brine Substances 0.000 description 34
- 239000003814 drug Substances 0.000 description 34
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 34
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 30
- 238000010898 silica gel chromatography Methods 0.000 description 29
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 27
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 27
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 27
- 201000010099 disease Diseases 0.000 description 25
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 24
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 24
- 150000004820 halides Chemical class 0.000 description 24
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 23
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 22
- 125000001309 chloro group Chemical group Cl* 0.000 description 22
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 21
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 21
- 150000001408 amides Chemical class 0.000 description 21
- 239000003795 chemical substances by application Substances 0.000 description 21
- 238000007429 general method Methods 0.000 description 20
- 239000003112 inhibitor Substances 0.000 description 20
- 238000010992 reflux Methods 0.000 description 20
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 20
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 19
- 208000035475 disorder Diseases 0.000 description 19
- LCEDQNDDFOCWGG-UHFFFAOYSA-N morpholine-4-carbaldehyde Chemical class O=CN1CCOCC1 LCEDQNDDFOCWGG-UHFFFAOYSA-N 0.000 description 19
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 19
- 239000011734 sodium Substances 0.000 description 19
- 239000000725 suspension Substances 0.000 description 19
- 102000004127 Cytokines Human genes 0.000 description 18
- 108090000695 Cytokines Proteins 0.000 description 18
- 229910052796 boron Inorganic materials 0.000 description 18
- 239000000706 filtrate Substances 0.000 description 18
- 239000003921 oil Substances 0.000 description 18
- 235000019198 oils Nutrition 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- 235000017557 sodium bicarbonate Nutrition 0.000 description 17
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 17
- 239000002253 acid Substances 0.000 description 15
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 15
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 15
- 229940079593 drug Drugs 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 150000003141 primary amines Chemical class 0.000 description 14
- 101100322888 Escherichia coli (strain K12) metL gene Proteins 0.000 description 13
- 239000007864 aqueous solution Substances 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 13
- 150000003335 secondary amines Chemical class 0.000 description 13
- 229940124597 therapeutic agent Drugs 0.000 description 13
- 210000001744 T-lymphocyte Anatomy 0.000 description 12
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 12
- 239000002244 precipitate Substances 0.000 description 12
- 229940002612 prodrug Drugs 0.000 description 12
- 239000000651 prodrug Substances 0.000 description 12
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 12
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 11
- 102000000589 Interleukin-1 Human genes 0.000 description 11
- 108010002352 Interleukin-1 Proteins 0.000 description 11
- 102000013691 Interleukin-17 Human genes 0.000 description 11
- 108050003558 Interleukin-17 Proteins 0.000 description 11
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 11
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 11
- 239000005557 antagonist Substances 0.000 description 11
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 11
- 229960000485 methotrexate Drugs 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 229960001940 sulfasalazine Drugs 0.000 description 11
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 11
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 11
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 10
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 10
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 10
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 10
- 150000003936 benzamides Chemical class 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 229910052739 hydrogen Inorganic materials 0.000 description 10
- 239000003446 ligand Substances 0.000 description 10
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 9
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 9
- 201000004681 Psoriasis Diseases 0.000 description 9
- 229960002170 azathioprine Drugs 0.000 description 9
- 239000003246 corticosteroid Substances 0.000 description 9
- 239000013058 crude material Substances 0.000 description 9
- 239000001257 hydrogen Substances 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 9
- 235000019345 sodium thiosulphate Nutrition 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 8
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 8
- 108010036949 Cyclosporine Proteins 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 229960001265 ciclosporin Drugs 0.000 description 8
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 8
- 229960001334 corticosteroids Drugs 0.000 description 8
- 229930182912 cyclosporin Natural products 0.000 description 8
- PXBYFCRSEFPAKW-SFYZADRCSA-N ethyl (3s,4s)-3-methylpiperidine-4-carboxylate Chemical compound CCOC(=O)[C@H]1CCNC[C@H]1C PXBYFCRSEFPAKW-SFYZADRCSA-N 0.000 description 8
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 8
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 8
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 8
- 239000005541 ACE inhibitor Substances 0.000 description 7
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 7
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 7
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 7
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 7
- 210000000068 Th17 cell Anatomy 0.000 description 7
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 7
- 239000000556 agonist Substances 0.000 description 7
- 125000003118 aryl group Chemical group 0.000 description 7
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000006184 cosolvent Substances 0.000 description 7
- 235000019152 folic acid Nutrition 0.000 description 7
- 239000011724 folic acid Substances 0.000 description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 7
- 229960001680 ibuprofen Drugs 0.000 description 7
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 7
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 7
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 7
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 229960005205 prednisolone Drugs 0.000 description 7
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 7
- 206010039073 rheumatoid arthritis Diseases 0.000 description 7
- 230000011664 signaling Effects 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 6
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 6
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 6
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- 108010065805 Interleukin-12 Proteins 0.000 description 6
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 6
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 6
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 6
- 230000027455 binding Effects 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 6
- 229960000590 celecoxib Drugs 0.000 description 6
- 125000000753 cycloalkyl group Chemical group 0.000 description 6
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 6
- 125000000623 heterocyclic group Chemical group 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- 239000008101 lactose Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- DSYBYAOEKDFWGJ-UHFFFAOYSA-N methyl 3-(bromomethyl)-2,4-dichlorobenzoate Chemical compound COC(=O)C1=CC=C(Cl)C(CBr)=C1Cl DSYBYAOEKDFWGJ-UHFFFAOYSA-N 0.000 description 6
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 230000000770 proinflammatory effect Effects 0.000 description 6
- 239000011347 resin Substances 0.000 description 6
- 229920005989 resin Polymers 0.000 description 6
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 6
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 6
- AZPWJMHMHRSWGK-UHFFFAOYSA-N 2-iodo-6-methyl-4-(trifluoromethoxy)aniline Chemical compound IC1=C(N)C(=CC(=C1)OC(F)(F)F)C AZPWJMHMHRSWGK-UHFFFAOYSA-N 0.000 description 5
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- 206010009900 Colitis ulcerative Diseases 0.000 description 5
- 208000011231 Crohn disease Diseases 0.000 description 5
- 206010012438 Dermatitis atopic Diseases 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 5
- 108010050904 Interferons Proteins 0.000 description 5
- 102000014150 Interferons Human genes 0.000 description 5
- 108090000978 Interleukin-4 Proteins 0.000 description 5
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 5
- 235000019502 Orange oil Nutrition 0.000 description 5
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 5
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 5
- 201000006704 Ulcerative Colitis Diseases 0.000 description 5
- 229940022663 acetate Drugs 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 201000008937 atopic dermatitis Diseases 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- PXBYFCRSEFPAKW-JGVFFNPUSA-N ethyl (3r,4r)-3-methylpiperidine-4-carboxylate Chemical compound CCOC(=O)[C@@H]1CCNC[C@@H]1C PXBYFCRSEFPAKW-JGVFFNPUSA-N 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 229940014259 gelatin Drugs 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 239000011630 iodine Substances 0.000 description 5
- 229960000681 leflunomide Drugs 0.000 description 5
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 5
- KBOPZPXVLCULAV-UHFFFAOYSA-N mesalamine Chemical compound NC1=CC=C(O)C(C(O)=O)=C1 KBOPZPXVLCULAV-UHFFFAOYSA-N 0.000 description 5
- 229960004963 mesalazine Drugs 0.000 description 5
- MSDQAKYIAGOKSJ-UHFFFAOYSA-N methyl 2,4-dichloro-3-(2-oxoethyl)benzoate Chemical compound COC(=O)C1=CC=C(Cl)C(CC=O)=C1Cl MSDQAKYIAGOKSJ-UHFFFAOYSA-N 0.000 description 5
- 235000001968 nicotinic acid Nutrition 0.000 description 5
- 239000011664 nicotinic acid Substances 0.000 description 5
- 239000010502 orange oil Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 229960005489 paracetamol Drugs 0.000 description 5
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 229960004618 prednisone Drugs 0.000 description 5
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 5
- TVDSBUOJIPERQY-UHFFFAOYSA-N prop-2-yn-1-ol Chemical compound OCC#C TVDSBUOJIPERQY-UHFFFAOYSA-N 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 150000003431 steroids Chemical class 0.000 description 5
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 5
- MDNKYIORSOLYFR-UHFFFAOYSA-N 2-fluoro-3-methyl-5-(trifluoromethyl)benzaldehyde Chemical compound FC1=C(C=O)C=C(C=C1C)C(F)(F)F MDNKYIORSOLYFR-UHFFFAOYSA-N 0.000 description 4
- WGUMRJHAMGWMMB-UHFFFAOYSA-N 2-iodo-3-methyl-5-(trifluoromethyl)aniline Chemical compound IC1=C(N)C=C(C=C1C)C(F)(F)F WGUMRJHAMGWMMB-UHFFFAOYSA-N 0.000 description 4
- OWIAZVCCDRLAAA-UHFFFAOYSA-N 2-iodo-6-methyl-4-(trifluoromethyl)aniline Chemical compound CC1=CC(C(F)(F)F)=CC(I)=C1N OWIAZVCCDRLAAA-UHFFFAOYSA-N 0.000 description 4
- XWOPESLRBNIPLR-UHFFFAOYSA-N 3-formyl-2,4-dimethylbenzoic acid Chemical compound C(=O)C=1C(=C(C(=O)O)C=CC=1C)C XWOPESLRBNIPLR-UHFFFAOYSA-N 0.000 description 4
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 4
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 4
- 101150013553 CD40 gene Proteins 0.000 description 4
- WJOHZNCJWYWUJD-IUGZLZTKSA-N Fluocinonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)COC(=O)C)[C@@]2(C)C[C@@H]1O WJOHZNCJWYWUJD-IUGZLZTKSA-N 0.000 description 4
- 208000009329 Graft vs Host Disease Diseases 0.000 description 4
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 102000003815 Interleukin-11 Human genes 0.000 description 4
- 108090000177 Interleukin-11 Proteins 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 4
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 4
- 208000008589 Obesity Diseases 0.000 description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 102000001253 Protein Kinase Human genes 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 208000026935 allergic disease Diseases 0.000 description 4
- 206010003246 arthritis Diseases 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 208000006673 asthma Diseases 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- RMRCNWBMXRMIRW-BYFNXCQMSA-M cyanocobalamin Chemical compound N#C[Co+]N([C@]1([H])[C@H](CC(N)=O)[C@]\2(CCC(=O)NC[C@H](C)OP(O)(=O)OC3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)C)C/2=C(C)\C([C@H](C/2(C)C)CCC(N)=O)=N\C\2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O RMRCNWBMXRMIRW-BYFNXCQMSA-M 0.000 description 4
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 4
- 229960001259 diclofenac Drugs 0.000 description 4
- 239000008298 dragée Substances 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 229960000785 fluocinonide Drugs 0.000 description 4
- 229960000304 folic acid Drugs 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 208000024908 graft versus host disease Diseases 0.000 description 4
- 239000003102 growth factor Substances 0.000 description 4
- 125000001072 heteroaryl group Chemical group 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 4
- 229960000905 indomethacin Drugs 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 231100000682 maximum tolerated dose Toxicity 0.000 description 4
- 229960001428 mercaptopurine Drugs 0.000 description 4
- 208000030159 metabolic disease Diseases 0.000 description 4
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 229960002009 naproxen Drugs 0.000 description 4
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 4
- 235000020824 obesity Nutrition 0.000 description 4
- 229960004110 olsalazine Drugs 0.000 description 4
- QQBDLJCYGRGAKP-FOCLMDBBSA-N olsalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=C(C(O)=CC=2)C(O)=O)=C1 QQBDLJCYGRGAKP-FOCLMDBBSA-N 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 229920000728 polyester Polymers 0.000 description 4
- 108060006633 protein kinase Proteins 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 4
- 229960000371 rofecoxib Drugs 0.000 description 4
- 239000012047 saturated solution Substances 0.000 description 4
- 229940083542 sodium Drugs 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 235000000346 sugar Nutrition 0.000 description 4
- 238000004808 supercritical fluid chromatography Methods 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- RNPVDZKNTIPWAH-UHFFFAOYSA-N 1,4-dimethyl-6-(trifluoromethyl)indole-3-carbaldehyde Chemical compound CN1C=C(C2=C(C=C(C=C12)C(F)(F)F)C)C=O RNPVDZKNTIPWAH-UHFFFAOYSA-N 0.000 description 3
- WUOYHGRVXDXONQ-UHFFFAOYSA-N 1-(benzenesulfonyl)-4-methyl-6-(trifluoromethyl)indole Chemical compound CC1=C2C=CN(C2=CC(=C1)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1 WUOYHGRVXDXONQ-UHFFFAOYSA-N 0.000 description 3
- WTXXASKXVSSFDI-UHFFFAOYSA-N 1-(benzenesulfonyl)-5-bromo-7-methylindole Chemical compound BrC=1C=C2C=CN(C2=C(C=1)C)S(=O)(=O)C1=CC=CC=C1 WTXXASKXVSSFDI-UHFFFAOYSA-N 0.000 description 3
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 3
- ZXWRJAVZDOLKFN-UHFFFAOYSA-N 1-[2-fluoro-3-methyl-5-(trifluoromethyl)phenyl]ethanol Chemical compound FC1=C(C=C(C=C1C)C(F)(F)F)C(C)O ZXWRJAVZDOLKFN-UHFFFAOYSA-N 0.000 description 3
- UKDCDBZJHUOORZ-UHFFFAOYSA-N 1-fluoro-4-iodo-3-methyl-2-nitrobenzene Chemical compound FC1=C(C(=C(C=C1)I)C)[N+](=O)[O-] UKDCDBZJHUOORZ-UHFFFAOYSA-N 0.000 description 3
- SQUCWHYKYDDSKP-UHFFFAOYSA-N 2,3,5-trichloro-4-(diethoxymethyl)pyridine Chemical compound ClC1=NC=C(C(=C1Cl)C(OCC)OCC)Cl SQUCWHYKYDDSKP-UHFFFAOYSA-N 0.000 description 3
- HSKQBSLVSASUSQ-UHFFFAOYSA-N 2,4-dichloro-3-(2-hydroxyethyl)benzoic acid Chemical compound ClC1=C(C(=O)O)C=CC(=C1CCO)Cl HSKQBSLVSASUSQ-UHFFFAOYSA-N 0.000 description 3
- LHAKBNBGEDNPIC-UHFFFAOYSA-N 2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]benzoic acid Chemical compound ClC1=C(C(=O)O)C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl LHAKBNBGEDNPIC-UHFFFAOYSA-N 0.000 description 3
- VYUHXPMAUNKWGW-UHFFFAOYSA-N 2,4-dichloro-3-formylbenzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C(C=O)=C1Cl VYUHXPMAUNKWGW-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- JZNQNKXUCYQJIT-UHFFFAOYSA-N 2-(2,6-dichloro-3-methoxycarbonylphenyl)acetic acid Chemical compound ClC1=C(C(=CC=C1C(=O)OC)Cl)CC(=O)O JZNQNKXUCYQJIT-UHFFFAOYSA-N 0.000 description 3
- QQVIMQAJDZWEFD-UHFFFAOYSA-N 2-(3-bromo-2,6-dimethylphenyl)acetic acid Chemical compound CC1=CC=C(Br)C(C)=C1CC(O)=O QQVIMQAJDZWEFD-UHFFFAOYSA-N 0.000 description 3
- OPXHHVDZUDAMES-UHFFFAOYSA-N 2-(3-methoxycarbonyl-2,6-dimethylphenyl)acetic acid Chemical compound COC(=O)C=1C(=C(C(=CC=1)C)CC(=O)O)C OPXHHVDZUDAMES-UHFFFAOYSA-N 0.000 description 3
- HGMXNZOHBYBJBW-UHFFFAOYSA-N 2-(3-methylpiperidin-4-yl)acetic acid hydrochloride Chemical compound Cl.CC1CNCCC1CC(O)=O HGMXNZOHBYBJBW-UHFFFAOYSA-N 0.000 description 3
- LLGFQMFRYYNXAS-UHFFFAOYSA-N 2-bromo-4-methyl-6-(trifluoromethyl)pyridin-3-amine Chemical compound BrC1=NC(=CC(=C1N)C)C(F)(F)F LLGFQMFRYYNXAS-UHFFFAOYSA-N 0.000 description 3
- FJDWPMVYWMIGKO-UHFFFAOYSA-N 2-bromo-5-(trifluoromethyl)pyridin-3-amine Chemical compound NC1=CC(C(F)(F)F)=CN=C1Br FJDWPMVYWMIGKO-UHFFFAOYSA-N 0.000 description 3
- IIDBMILLZRYZCH-UHFFFAOYSA-N 2-methyl-4-(trifluoromethoxy)aniline Chemical compound CC1=CC(OC(F)(F)F)=CC=C1N IIDBMILLZRYZCH-UHFFFAOYSA-N 0.000 description 3
- QHUHCKNDRFYKBY-UHFFFAOYSA-N 2-methyl-4-nitro-6-(trifluoromethyl)pyridine Chemical compound CC1=NC(=CC(=C1)[N+](=O)[O-])C(F)(F)F QHUHCKNDRFYKBY-UHFFFAOYSA-N 0.000 description 3
- RKOREZRSQWTGLZ-UHFFFAOYSA-N 2-methyl-6-nitro-4-(trifluoromethoxy)aniline Chemical compound CC1=CC(OC(F)(F)F)=CC([N+]([O-])=O)=C1N RKOREZRSQWTGLZ-UHFFFAOYSA-N 0.000 description 3
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- CEHZMNAKUGZOHI-UHFFFAOYSA-N 3,5-dichloro-2-methoxypyridine-4-carbaldehyde Chemical compound ClC1=C(C=O)C(=CN=C1OC)Cl CEHZMNAKUGZOHI-UHFFFAOYSA-N 0.000 description 3
- LMZAZXGOOGERDK-UHFFFAOYSA-N 3-iodo-4-methyl-6-(trifluoromethyl)-2H-indazole Chemical compound IC1=NNC2=CC(=CC(=C12)C)C(F)(F)F LMZAZXGOOGERDK-UHFFFAOYSA-N 0.000 description 3
- WGNMVCRTUYDGKV-UHFFFAOYSA-N 3-methyl-5-(methylamino)-4-nitrobenzonitrile Chemical compound CC=1C=C(C#N)C=C(C=1[N+](=O)[O-])NC WGNMVCRTUYDGKV-UHFFFAOYSA-N 0.000 description 3
- LXECQKKRYRWEKN-UHFFFAOYSA-N 4-amino-3-iodo-2-methylbenzonitrile Chemical compound NC1=C(C(=C(C#N)C=C1)C)I LXECQKKRYRWEKN-UHFFFAOYSA-N 0.000 description 3
- UNNAJGSGZMFAPL-UHFFFAOYSA-N 4-amino-3-methyl-5-(methylamino)benzonitrile Chemical compound NC1=C(C=C(C#N)C=C1NC)C UNNAJGSGZMFAPL-UHFFFAOYSA-N 0.000 description 3
- VMNUNODKDDCZKW-UHFFFAOYSA-N 4-amino-3-methyl-5-nitrobenzonitrile Chemical compound CC1=CC(C#N)=CC([N+]([O-])=O)=C1N VMNUNODKDDCZKW-UHFFFAOYSA-N 0.000 description 3
- PTTJGVZKKZYQDO-UHFFFAOYSA-N 4-amino-5-iodo-6-methylpyridine-2-carbonitrile Chemical compound NC1=CC(=NC(=C1I)C)C#N PTTJGVZKKZYQDO-UHFFFAOYSA-N 0.000 description 3
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 description 3
- PIZSHCRWJOXLFZ-UHFFFAOYSA-N 4-bromo-6-(trifluoromethyl)pyridazin-3-amine Chemical compound BrC1=C(N=NC(=C1)C(F)(F)F)N PIZSHCRWJOXLFZ-UHFFFAOYSA-N 0.000 description 3
- QVZIMQZFEGYKJU-UHFFFAOYSA-N 4-iodo-2-methyl-6-(trifluoromethyl)pyridin-3-amine Chemical compound IC1=C(C(=NC(=C1)C(F)(F)F)C)N QVZIMQZFEGYKJU-UHFFFAOYSA-N 0.000 description 3
- GVWDELRRDIOTNF-UHFFFAOYSA-N 4-iodo-3-methyl-5-nitrobenzonitrile Chemical compound IC1=C(C=C(C#N)C=C1[N+](=O)[O-])C GVWDELRRDIOTNF-UHFFFAOYSA-N 0.000 description 3
- VOOSBHAMKUIWNZ-UHFFFAOYSA-N 4-methyl-6-(trifluoromethyl)-1H-indazole Chemical compound CC1=C2C=NNC2=CC(=C1)C(F)(F)F VOOSBHAMKUIWNZ-UHFFFAOYSA-N 0.000 description 3
- WBUIUDXMWUNWMT-UHFFFAOYSA-N 4-methyl-6-(trifluoromethyl)pyridazin-3-amine Chemical compound Cc1cc(nnc1N)C(F)(F)F WBUIUDXMWUNWMT-UHFFFAOYSA-N 0.000 description 3
- JHXYLCHHNINYTI-UHFFFAOYSA-N 5-bromo-1-chloro-2-nitroso-3-(trifluoromethyl)benzene Chemical compound BrC=1C=C(C(=C(C=1)Cl)N=O)C(F)(F)F JHXYLCHHNINYTI-UHFFFAOYSA-N 0.000 description 3
- PZGNEGICJCQNAW-UHFFFAOYSA-N 5-bromo-2-iodo-1-methyl-3-nitrobenzene Chemical compound CC1=CC(Br)=CC([N+]([O-])=O)=C1I PZGNEGICJCQNAW-UHFFFAOYSA-N 0.000 description 3
- KEQIFLHCBPPNJG-UHFFFAOYSA-N 5-bromo-2-iodo-3-methylaniline Chemical compound BrC=1C=C(C(=C(N)C=1)I)C KEQIFLHCBPPNJG-UHFFFAOYSA-N 0.000 description 3
- DFGKQCOFARYQOA-UHFFFAOYSA-N 5-bromo-4,6-dimethyl-2-oxo-1h-pyridine-3-carbonitrile Chemical compound CC=1NC(=O)C(C#N)=C(C)C=1Br DFGKQCOFARYQOA-UHFFFAOYSA-N 0.000 description 3
- QJIDZLYWLXOVCJ-UHFFFAOYSA-N 5-bromo-4,6-dimethylpyridine-3-carbonitrile Chemical compound CC1=NC=C(C#N)C(C)=C1Br QJIDZLYWLXOVCJ-UHFFFAOYSA-N 0.000 description 3
- VFEVDRLQIJPUOP-UHFFFAOYSA-N 7-chloro-5-(trifluoromethyl)-1H-indole Chemical compound ClC=1C=C(C=C2C=CNC=12)C(F)(F)F VFEVDRLQIJPUOP-UHFFFAOYSA-N 0.000 description 3
- WMSGBDJYUJELMH-UHFFFAOYSA-N 7-methyl-5-(trifluoromethoxy)-1H-indole Chemical compound CC=1C=C(C=C2C=CNC=12)OC(F)(F)F WMSGBDJYUJELMH-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 201000001320 Atherosclerosis Diseases 0.000 description 3
- KSPPSZRTTSKLHW-DHZHZOJOSA-N C(C)OC(\C=C/1\C(CN(CC\1)C(=O)OCC)C)=O Chemical compound C(C)OC(\C=C/1\C(CN(CC\1)C(=O)OCC)C)=O KSPPSZRTTSKLHW-DHZHZOJOSA-N 0.000 description 3
- DQTGNXNXHZFXKU-UHFFFAOYSA-N CC1=C(C(=CC(=C1)C(F)(F)F)C#C[Si](C)(C)C)NS(=O)(=O)C1=CC=CC=C1 Chemical compound CC1=C(C(=CC(=C1)C(F)(F)F)C#C[Si](C)(C)C)NS(=O)(=O)C1=CC=CC=C1 DQTGNXNXHZFXKU-UHFFFAOYSA-N 0.000 description 3
- FGGNDLCLTQLJBS-UHFFFAOYSA-N CC=1C(=C(C=C(C=1)C(F)(F)F)NS(=O)(=O)C1=CC=CC=C1)C#C[Si](C)(C)C Chemical compound CC=1C(=C(C=C(C=1)C(F)(F)F)NS(=O)(=O)C1=CC=CC=C1)C#C[Si](C)(C)C FGGNDLCLTQLJBS-UHFFFAOYSA-N 0.000 description 3
- VUQOGAVMPYVWLO-UHFFFAOYSA-N CC=1C=C(C=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)B1OC(C(O1)(C)C)(C)C Chemical compound CC=1C=C(C=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)B1OC(C(O1)(C)C)(C)C VUQOGAVMPYVWLO-UHFFFAOYSA-N 0.000 description 3
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 3
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 3
- 102100032937 CD40 ligand Human genes 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 3
- 102400000792 Endothelial monocyte-activating polypeptide 2 Human genes 0.000 description 3
- IOLOADBJLBCMMF-UHFFFAOYSA-N FC1=C(C(=C(C=C1)C(F)(F)F)C)[N+](=O)[O-] Chemical compound FC1=C(C(=C(C=C1)C(F)(F)F)C)[N+](=O)[O-] IOLOADBJLBCMMF-UHFFFAOYSA-N 0.000 description 3
- 101150021185 FGF gene Proteins 0.000 description 3
- 108010072051 Glatiramer Acetate Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 3
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 3
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 3
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 3
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 3
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 3
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 3
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 3
- 101000800116 Homo sapiens Thy-1 membrane glycoprotein Proteins 0.000 description 3
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 3
- 206010022489 Insulin Resistance Diseases 0.000 description 3
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 3
- 101800001003 Interleukin-1 receptor type 2, soluble form Proteins 0.000 description 3
- 102400000027 Interleukin-1 receptor type 2, soluble form Human genes 0.000 description 3
- 108090000174 Interleukin-10 Proteins 0.000 description 3
- 108090000176 Interleukin-13 Proteins 0.000 description 3
- 101800003050 Interleukin-16 Proteins 0.000 description 3
- 102000049772 Interleukin-16 Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 108010065637 Interleukin-23 Proteins 0.000 description 3
- 102000013264 Interleukin-23 Human genes 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- 108010002586 Interleukin-7 Proteins 0.000 description 3
- 108090001007 Interleukin-8 Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- GNEHJQPWRLKFOL-UHFFFAOYSA-N N-[2-ethynyl-6-methyl-4-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound C(#C)C1=C(C(=CC(=C1)C(F)(F)F)C)NS(=O)(=O)C1=CC=CC=C1 GNEHJQPWRLKFOL-UHFFFAOYSA-N 0.000 description 3
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 102000016978 Orphan receptors Human genes 0.000 description 3
- 108070000031 Orphan receptors Proteins 0.000 description 3
- 239000012826 P38 inhibitor Substances 0.000 description 3
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 108091008773 RAR-related orphan receptors γ Proteins 0.000 description 3
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 3
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 3
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 3
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 3
- 102100033523 Thy-1 membrane glycoprotein Human genes 0.000 description 3
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 3
- 206010046851 Uveitis Diseases 0.000 description 3
- IGEWFMNCMDNSTA-UHFFFAOYSA-N [2,4-dichloro-3-(1-hydroxyprop-2-ynyl)phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1C(C#C)O)Cl)C(=O)N1CCOCC1 IGEWFMNCMDNSTA-UHFFFAOYSA-N 0.000 description 3
- MGVGMXLGOKTYKP-ZFOBEOMCSA-N acetic acid;(6s,8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-17-(2-hydroxyacetyl)-6,10,13-trimethyl-7,8,9,11,12,14,15,16-octahydro-6h-cyclopenta[a]phenanthren-3-one Chemical compound CC(O)=O.C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)CO)CC[C@H]21 MGVGMXLGOKTYKP-ZFOBEOMCSA-N 0.000 description 3
- 239000000464 adrenergic agent Substances 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 239000003146 anticoagulant agent Substances 0.000 description 3
- 229960004676 antithrombotic agent Drugs 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000001363 autoimmune Effects 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 229910000019 calcium carbonate Inorganic materials 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 3
- 239000004074 complement inhibitor Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 108010057085 cytokine receptors Proteins 0.000 description 3
- 102000003675 cytokine receptors Human genes 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 3
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- JOLZZOLPFCVDBL-UHFFFAOYSA-N ethyl 4,6-dichloro-5-formylpyridine-3-carboxylate Chemical compound ClC1=C(C(=NC=C1C(=O)OCC)Cl)C=O JOLZZOLPFCVDBL-UHFFFAOYSA-N 0.000 description 3
- SIWKZRPTLRLLKS-UHFFFAOYSA-N ethyl 4-(2-ethoxy-2-oxoethyl)-3-methylpiperidine-1-carboxylate Chemical compound C(C)OC(CC1C(CN(CC1)C(=O)OCC)C)=O SIWKZRPTLRLLKS-UHFFFAOYSA-N 0.000 description 3
- 229940014144 folate Drugs 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 3
- 229960004171 hydroxychloroquine Drugs 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 229960000598 infliximab Drugs 0.000 description 3
- 229940079322 interferon Drugs 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 229940043355 kinase inhibitor Drugs 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 206010025135 lupus erythematosus Diseases 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 108010000525 member 1 small inducible cytokine subfamily E Proteins 0.000 description 3
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- IOUUGYLLLHFVEL-UHFFFAOYSA-N methyl 2,4-dichloro-3-(2-hydroxyethyl)benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CCO)Cl IOUUGYLLLHFVEL-UHFFFAOYSA-N 0.000 description 3
- MSFSNIXPBYILBF-UHFFFAOYSA-N methyl 2,4-dichloro-3-(2-oxobutyl)benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CC(CC)=O)Cl MSFSNIXPBYILBF-UHFFFAOYSA-N 0.000 description 3
- OILFPGVEUFVNER-UHFFFAOYSA-N methyl 2,4-dichloro-3-methylbenzoate Chemical compound COC(=O)C1=CC=C(Cl)C(C)=C1Cl OILFPGVEUFVNER-UHFFFAOYSA-N 0.000 description 3
- HDEBJVHBOSDRQX-UHFFFAOYSA-N methyl 2-(3-methylpiperidin-4-yl)acetate hydrochloride Chemical compound Cl.COC(=O)CC1CCNCC1C HDEBJVHBOSDRQX-UHFFFAOYSA-N 0.000 description 3
- MYPGNKNDYKVDPA-UHFFFAOYSA-N methyl 3-(3-bromo-2-oxobutyl)-2,4-dichlorobenzoate Chemical compound BrC(C(CC=1C(=C(C(=O)OC)C=CC=1Cl)Cl)=O)C MYPGNKNDYKVDPA-UHFFFAOYSA-N 0.000 description 3
- WWFDAHPJYNPSLC-UHFFFAOYSA-N methyl 3-(chloromethyl)-2,4-dimethylbenzoate Chemical compound ClCC=1C(=C(C(=O)OC)C=CC=1C)C WWFDAHPJYNPSLC-UHFFFAOYSA-N 0.000 description 3
- YLFRZCFECDQHLA-UHFFFAOYSA-N methyl 3-(hydroxymethyl)-2,4-dimethylbenzoate Chemical compound OCC=1C(=C(C(=O)OC)C=CC=1C)C YLFRZCFECDQHLA-UHFFFAOYSA-N 0.000 description 3
- SSFJOKUOIUWZNZ-UHFFFAOYSA-N methyl 3-formyl-2,4-dimethylbenzoate Chemical compound C(=O)C=1C(=C(C(=O)OC)C=CC=1C)C SSFJOKUOIUWZNZ-UHFFFAOYSA-N 0.000 description 3
- OACWRRPQDLJKOA-UHFFFAOYSA-N methyl 6-(trifluoromethoxy)-1h-indole-2-carboxylate Chemical compound C1=C(OC(F)(F)F)C=C2NC(C(=O)OC)=CC2=C1 OACWRRPQDLJKOA-UHFFFAOYSA-N 0.000 description 3
- 229960004584 methylprednisolone Drugs 0.000 description 3
- 229960001293 methylprednisolone acetate Drugs 0.000 description 3
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 3
- 229960004866 mycophenolate mofetil Drugs 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 3
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000008177 pharmaceutical agent Substances 0.000 description 3
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 3
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 3
- 229930002330 retinoic acid Natural products 0.000 description 3
- 229960004889 salicylic acid Drugs 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 229960002930 sirolimus Drugs 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 description 3
- 235000017550 sodium carbonate Nutrition 0.000 description 3
- 235000010288 sodium nitrite Nutrition 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- PXDDCUVFKNTKET-UHFFFAOYSA-N tert-butyl 2,4-dichlorobenzoate Chemical compound CC(C)(C)OC(=O)C1=CC=C(Cl)C=C1Cl PXDDCUVFKNTKET-UHFFFAOYSA-N 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 229960002117 triamcinolone acetonide Drugs 0.000 description 3
- YNDXUCZADRHECN-JNQJZLCISA-N triamcinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O YNDXUCZADRHECN-JNQJZLCISA-N 0.000 description 3
- LNPDTQAFDNKSHK-UHFFFAOYSA-N valdecoxib Chemical compound CC=1ON=C(C=2C=CC=CC=2)C=1C1=CC=C(S(N)(=O)=O)C=C1 LNPDTQAFDNKSHK-UHFFFAOYSA-N 0.000 description 3
- 229960002004 valdecoxib Drugs 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- VDPLLINNMXFNQX-UHFFFAOYSA-N (1-aminocyclohexyl)methanol Chemical compound OCC1(N)CCCCC1 VDPLLINNMXFNQX-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- MTDHILKWIRSIHB-UHFFFAOYSA-N (5-azaniumyl-3,4,6-trihydroxyoxan-2-yl)methyl sulfate Chemical compound NC1C(O)OC(COS(O)(=O)=O)C(O)C1O MTDHILKWIRSIHB-UHFFFAOYSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- XCRYTCRFIXRLES-UHFFFAOYSA-N 1,4-dimethyl-6-(trifluoromethyl)indazole-3-carbaldehyde Chemical compound CN1N=C(C2=C(C=C(C=C12)C(F)(F)F)C)C=O XCRYTCRFIXRLES-UHFFFAOYSA-N 0.000 description 2
- QIAWEAHTCAGLQV-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-bromo-4-chloroindole Chemical compound BrC1=CC(=C2C=CN(C2=C1)S(=O)(=O)C1=CC=CC=C1)Cl QIAWEAHTCAGLQV-UHFFFAOYSA-N 0.000 description 2
- XRSUHCJGWFWVIX-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-bromo-4-fluoroindole Chemical compound BrC1=CC(=C2C=CN(C2=C1)S(=O)(=O)C1=CC=CC=C1)F XRSUHCJGWFWVIX-UHFFFAOYSA-N 0.000 description 2
- NWVJVFWGWBWXHG-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-methyl-5-(trifluoromethyl)indole Chemical compound CC=1C=C(C=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)C(F)(F)F NWVJVFWGWBWXHG-UHFFFAOYSA-N 0.000 description 2
- ICPNYFORWGNPAH-UHFFFAOYSA-N 1-(benzenesulfonyl)-7-methylindol-5-ol Chemical compound CC=1C=C(C=C2C=CN(C=12)S(=O)(=O)C1=CC=CC=C1)O ICPNYFORWGNPAH-UHFFFAOYSA-N 0.000 description 2
- NJIFZIQNTQHZLU-UHFFFAOYSA-N 1-methyl-6-(trifluoromethoxy)indole-2-carbaldehyde Chemical compound CN1C(=CC2=CC=C(C=C12)OC(F)(F)F)C=O NJIFZIQNTQHZLU-UHFFFAOYSA-N 0.000 description 2
- MVXVYAKCVDQRLW-UHFFFAOYSA-N 1h-pyrrolo[2,3-b]pyridine Chemical compound C1=CN=C2NC=CC2=C1 MVXVYAKCVDQRLW-UHFFFAOYSA-N 0.000 description 2
- INLOKTSZAJODQA-UHFFFAOYSA-N 2,2,2-trifluoro-N-[2-methyl-6-nitro-4-(trifluoromethoxy)phenyl]acetamide Chemical compound FC(C(=O)NC1=C(C=C(C=C1[N+](=O)[O-])OC(F)(F)F)C)(F)F INLOKTSZAJODQA-UHFFFAOYSA-N 0.000 description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- JCRFPKOMSMKMHW-UHFFFAOYSA-N 2,4-dichloro-3-(hydroxymethyl)benzoic acid Chemical compound ClC1=C(C(=O)O)C=CC(=C1CO)Cl JCRFPKOMSMKMHW-UHFFFAOYSA-N 0.000 description 2
- MZLWADLOQKKXFN-UHFFFAOYSA-N 2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-3-yl]methyl]benzoic acid Chemical compound ClC1=C(C(=O)O)C=CC(=C1CC1=CN(C2=CC(=CC(=C12)C)C(F)(F)F)C)Cl MZLWADLOQKKXFN-UHFFFAOYSA-N 0.000 description 2
- ATCRIUVQKHMXSH-UHFFFAOYSA-N 2,4-dichlorobenzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1Cl ATCRIUVQKHMXSH-UHFFFAOYSA-N 0.000 description 2
- WAMNAZKASOMLLM-UHFFFAOYSA-N 2,6-dichloro-3-(morpholine-4-carbonyl)benzaldehyde Chemical compound ClC1=C(C=O)C(=CC=C1C(=O)N1CCOCC1)Cl WAMNAZKASOMLLM-UHFFFAOYSA-N 0.000 description 2
- RXDIRAKBQLATCC-UHFFFAOYSA-N 2,6-dichloro-3-[(2-methylpropan-2-yl)oxycarbonyl]benzoic acid Chemical compound C(C)(C)(C)OC(=O)C=1C(=C(C(=O)O)C(=CC=1)Cl)Cl RXDIRAKBQLATCC-UHFFFAOYSA-N 0.000 description 2
- CMATWTQNJDZMBV-UHFFFAOYSA-N 2,6-dimethyl-3-(morpholine-4-carbonyl)benzaldehyde Chemical compound CC1=C(C=O)C(=CC=C1C(=O)N1CCOCC1)C CMATWTQNJDZMBV-UHFFFAOYSA-N 0.000 description 2
- WWGJUEBXGNZEFS-UHFFFAOYSA-N 2-(2,6-dichloro-4-nitrophenyl)acetic acid Chemical compound OC(=O)CC1=C(Cl)C=C([N+]([O-])=O)C=C1Cl WWGJUEBXGNZEFS-UHFFFAOYSA-N 0.000 description 2
- BKFJNJSLBBPQQA-UHFFFAOYSA-N 2-[2,6-dichloro-3-(morpholine-4-carbonyl)phenyl]acetic acid Chemical compound ClC1=C(C(=CC=C1C(=O)N1CCOCC1)Cl)CC(=O)O BKFJNJSLBBPQQA-UHFFFAOYSA-N 0.000 description 2
- NOEMTXYEHPVRRH-UHFFFAOYSA-N 2-iodo-1-methyl-3-nitro-5-(trifluoromethoxy)benzene Chemical compound IC1=C(C=C(C=C1[N+](=O)[O-])OC(F)(F)F)C NOEMTXYEHPVRRH-UHFFFAOYSA-N 0.000 description 2
- ZWLIZCSTDUPRKB-UHFFFAOYSA-N 2-iodo-3-methyl-5-(trifluoromethyl)phenol Chemical compound IC1=C(C=C(C=C1C)C(F)(F)F)O ZWLIZCSTDUPRKB-UHFFFAOYSA-N 0.000 description 2
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- PAXQXJDYVORMOO-UHFFFAOYSA-N 2-methyl-4-(trifluoromethyl)aniline Chemical compound CC1=CC(C(F)(F)F)=CC=C1N PAXQXJDYVORMOO-UHFFFAOYSA-N 0.000 description 2
- BWDBLIAZUCVCKA-UHFFFAOYSA-N 2-methyl-6-(trifluoromethyl)pyridin-4-amine Chemical compound CC1=CC(N)=CC(C(F)(F)F)=N1 BWDBLIAZUCVCKA-UHFFFAOYSA-N 0.000 description 2
- HRSOGFVRYLPHKR-UHFFFAOYSA-N 2-methyl-6-nitro-4-(trifluoromethyl)aniline Chemical compound CC1=CC(C(F)(F)F)=CC([N+]([O-])=O)=C1N HRSOGFVRYLPHKR-UHFFFAOYSA-N 0.000 description 2
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 2
- RARCPPUDWWMXRG-UHFFFAOYSA-N 3,7-dimethyl-1H-indole-5-carbonitrile Chemical compound CC1=CNC2=C(C=C(C=C12)C#N)C RARCPPUDWWMXRG-UHFFFAOYSA-N 0.000 description 2
- PSDIKPIIWIBLFV-UHFFFAOYSA-N 3,7-dimethyl-5-(trifluoromethyl)-1H-indole Chemical compound CC1=CNC2=C(C=C(C=C12)C(F)(F)F)C PSDIKPIIWIBLFV-UHFFFAOYSA-N 0.000 description 2
- JVAQCRXOWYZRLD-UHFFFAOYSA-N 3,7-dimethyl-5-(trifluoromethyl)-1H-pyrrolo[2,3-c]pyridine Chemical compound CC1=CNC2=C(N=C(C=C21)C(F)(F)F)C JVAQCRXOWYZRLD-UHFFFAOYSA-N 0.000 description 2
- YERHWVXGFDTLQF-UHFFFAOYSA-N 3,7-dimethyl-5-(trifluoromethyl)-2H-indazole Chemical compound CC1=NNC2=C(C=C(C=C12)C(F)(F)F)C YERHWVXGFDTLQF-UHFFFAOYSA-N 0.000 description 2
- QSJNNXVRLVUQND-UHFFFAOYSA-N 3-amino-4-iodo-5-methylbenzonitrile Chemical compound NC=1C=C(C#N)C=C(C=1I)C QSJNNXVRLVUQND-UHFFFAOYSA-N 0.000 description 2
- GTAIJORGXRXVOP-UHFFFAOYSA-N 3-bromo-2,6-dimethylbenzaldehyde Chemical compound CC1=CC=C(Br)C(C)=C1C=O GTAIJORGXRXVOP-UHFFFAOYSA-N 0.000 description 2
- GEHGMGPAUZLWTD-UHFFFAOYSA-N 3-iodo-1,4-dimethyl-6-(trifluoromethyl)indazole Chemical compound IC1=NN(C2=CC(=CC(=C12)C)C(F)(F)F)C GEHGMGPAUZLWTD-UHFFFAOYSA-N 0.000 description 2
- TWZNMXNBADSZKE-UHFFFAOYSA-N 3-iodo-2-methyl-4-nitrobenzonitrile Chemical compound IC=1C(=C(C#N)C=CC=1[N+](=O)[O-])C TWZNMXNBADSZKE-UHFFFAOYSA-N 0.000 description 2
- LIPCGCXRTKXDEE-UHFFFAOYSA-N 3-iodo-2-methyl-6-(trifluoromethyl)pyridin-4-amine Chemical compound IC=1C(=NC(=CC=1N)C(F)(F)F)C LIPCGCXRTKXDEE-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- ZXFVKFUXKFPUQJ-UHFFFAOYSA-N 4-bromo-2-methyl-6-nitroaniline Chemical compound CC1=CC(Br)=CC([N+]([O-])=O)=C1N ZXFVKFUXKFPUQJ-UHFFFAOYSA-N 0.000 description 2
- RCTDVYQFHWTQEL-UHFFFAOYSA-N 4-methyl-6-(trifluoromethoxy)-1h-indole Chemical compound CC1=CC(OC(F)(F)F)=CC2=C1C=CN2 RCTDVYQFHWTQEL-UHFFFAOYSA-N 0.000 description 2
- SAVVDZDBLITBHR-UHFFFAOYSA-N 4-methyl-6-(trifluoromethyl)-1h-indole Chemical compound CC1=CC(C(F)(F)F)=CC2=C1C=CN2 SAVVDZDBLITBHR-UHFFFAOYSA-N 0.000 description 2
- UUEREACRVVTQKF-UHFFFAOYSA-N 5-bromo-2-chloro-4,6-dimethylpyridine-3-carbonitrile Chemical compound CC1=NC(Cl)=C(C#N)C(C)=C1Br UUEREACRVVTQKF-UHFFFAOYSA-N 0.000 description 2
- QQGXFWWAZULHIZ-UHFFFAOYSA-N 5-bromo-4,6-dimethylpyridine-3-carboxylic acid Chemical compound CC1=NC=C(C(O)=O)C(C)=C1Br QQGXFWWAZULHIZ-UHFFFAOYSA-N 0.000 description 2
- OWKONBYDKAWKAK-UHFFFAOYSA-N 5-cyclopropyl-7-fluoro-1H-indole Chemical compound C1(CC1)C=1C=C2C=CNC2=C(C=1)F OWKONBYDKAWKAK-UHFFFAOYSA-N 0.000 description 2
- VTODDAKQRGKCGF-UHFFFAOYSA-N 6-bromo-4-fluoro-1h-indole Chemical compound FC1=CC(Br)=CC2=C1C=CN2 VTODDAKQRGKCGF-UHFFFAOYSA-N 0.000 description 2
- OFLGMWPMVSXEIN-UHFFFAOYSA-N 6-chloro-n,4-dimethyl-3-nitropyridin-2-amine Chemical compound CNC1=NC(Cl)=CC(C)=C1[N+]([O-])=O OFLGMWPMVSXEIN-UHFFFAOYSA-N 0.000 description 2
- BXCGZPXDHAMVET-UHFFFAOYSA-N 7-ethenyl-5-(trifluoromethyl)-1H-indole Chemical compound FC(C=1C=C2C=CNC2=C(C=1)C=C)(F)F BXCGZPXDHAMVET-UHFFFAOYSA-N 0.000 description 2
- MQMGGZVBFKKCSZ-UHFFFAOYSA-N 7-ethyl-5-(trifluoromethyl)-1H-indole Chemical compound C(C)C=1C=C(C=C2C=CNC=12)C(F)(F)F MQMGGZVBFKKCSZ-UHFFFAOYSA-N 0.000 description 2
- SZSMQVXYMOXQCJ-UHFFFAOYSA-N 7-methyl-5-(trifluoromethyl)-1H-indole Chemical compound CC=1C=C(C=C2C=CNC=12)C(F)(F)F SZSMQVXYMOXQCJ-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 206010003267 Arthritis reactive Diseases 0.000 description 2
- 208000025705 Axial Spondyloarthritis Diseases 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 108090000426 Caspase-1 Proteins 0.000 description 2
- 108010076667 Caspases Proteins 0.000 description 2
- 102000011727 Caspases Human genes 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 108010008165 Etanercept Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- 101000868215 Homo sapiens CD40 ligand Proteins 0.000 description 2
- 101000713602 Homo sapiens T-box transcription factor TBX21 Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 239000003458 I kappa b kinase inhibitor Substances 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 108090000172 Interleukin-15 Proteins 0.000 description 2
- 102000003812 Interleukin-15 Human genes 0.000 description 2
- 102100030703 Interleukin-22 Human genes 0.000 description 2
- 208000003456 Juvenile Arthritis Diseases 0.000 description 2
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 2
- 208000005777 Lupus Nephritis Diseases 0.000 description 2
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- UTZFSVNXLWTNCF-UHFFFAOYSA-N N-[2-iodo-5-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound IC1=C(C=C(C=C1)C(F)(F)F)NS(=O)(=O)C1=CC=CC=C1 UTZFSVNXLWTNCF-UHFFFAOYSA-N 0.000 description 2
- YWMXUNQCLOAACV-UHFFFAOYSA-N N-[2-iodo-6-methyl-4-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound IC1=C(C(=CC(=C1)C(F)(F)F)C)NS(=O)(=O)C1=CC=CC=C1 YWMXUNQCLOAACV-UHFFFAOYSA-N 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 2
- 102100023421 Nuclear receptor ROR-gamma Human genes 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- 108091008680 RAR-related orphan receptors Proteins 0.000 description 2
- 206010039085 Rhinitis allergic Diseases 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 201000002661 Spondylitis Diseases 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- CYQFCXCEBYINGO-UHFFFAOYSA-N THC Natural products C1=C(C)CCC2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3C21 CYQFCXCEBYINGO-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- VDNDOUHEEWHDGB-UHFFFAOYSA-N [2,4-dichloro-3-(chloromethyl)phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CCl)Cl)C(=O)N1CCOCC1 VDNDOUHEEWHDGB-UHFFFAOYSA-N 0.000 description 2
- BIMYKUROHMJBQR-UHFFFAOYSA-N [2,4-dichloro-3-(hydroxymethyl)phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CO)Cl)C(=O)N1CCOCC1 BIMYKUROHMJBQR-UHFFFAOYSA-N 0.000 description 2
- RPZVCPIAHVCCRS-UHFFFAOYSA-N [2,4-dichloro-3-[(3-methylimidazo[4,5-b]pyridin-2-yl)methyl]phenyl]-morpholin-4-ylmethanone Chemical class ClC1=C(C=CC(=C1CC1=NC=2C(=NC=CC=2)N1C)Cl)C(=O)N1CCOCC1 RPZVCPIAHVCCRS-UHFFFAOYSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 125000005042 acyloxymethyl group Chemical group 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 229960002459 alefacept Drugs 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 125000005206 alkoxycarbonyloxymethyl group Chemical group 0.000 description 2
- 201000009961 allergic asthma Diseases 0.000 description 2
- 201000010105 allergic rhinitis Diseases 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 201000004982 autoimmune uveitis Diseases 0.000 description 2
- 230000005784 autoimmunity Effects 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- UTZLYIFQYGQUPA-UHFFFAOYSA-N benzyl 2,5-dioxopyrrolidine-1-carboxylate Chemical compound O=C1CCC(=O)N1C(=O)OCC1=CC=CC=C1 UTZLYIFQYGQUPA-UHFFFAOYSA-N 0.000 description 2
- 229960001102 betamethasone dipropionate Drugs 0.000 description 2
- CIWBQSYVNNPZIQ-XYWKZLDCSA-N betamethasone dipropionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CIWBQSYVNNPZIQ-XYWKZLDCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 2
- 239000004327 boric acid Substances 0.000 description 2
- 229960002645 boric acid Drugs 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 125000001589 carboacyl group Chemical group 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000005754 cellular signaling Effects 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 238000004296 chiral HPLC Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- 208000019069 chronic childhood arthritis Diseases 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 239000003245 coal Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 229940038717 copaxone Drugs 0.000 description 2
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(i) oxide Chemical compound [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 229940111134 coxibs Drugs 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 229960002104 cyanocobalamin Drugs 0.000 description 2
- 235000000639 cyanocobalamin Nutrition 0.000 description 2
- 239000011666 cyanocobalamin Substances 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 2
- JBDSSBMEKXHSJF-UHFFFAOYSA-M cyclopentanecarboxylate Chemical compound [O-]C(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-M 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- PCCPERGCFKIYIS-AWEZNQCLSA-N daxalipram Chemical compound C1=C(OC)C(OCCC)=CC([C@@]2(C)OC(=O)NC2)=C1 PCCPERGCFKIYIS-AWEZNQCLSA-N 0.000 description 2
- CYQFCXCEBYINGO-IAGOWNOFSA-N delta1-THC Chemical compound C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@@H]21 CYQFCXCEBYINGO-IAGOWNOFSA-N 0.000 description 2
- 230000000779 depleting effect Effects 0.000 description 2
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960004193 dextropropoxyphene Drugs 0.000 description 2
- XLMALTXPSGQGBX-GCJKJVERSA-N dextropropoxyphene Chemical compound C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 XLMALTXPSGQGBX-GCJKJVERSA-N 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- FLKPEMZONWLCSK-UHFFFAOYSA-N diethyl phthalate Chemical compound CCOC(=O)C1=CC=CC=C1C(=O)OCC FLKPEMZONWLCSK-UHFFFAOYSA-N 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 208000016097 disease of metabolism Diseases 0.000 description 2
- 229960004242 dronabinol Drugs 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 239000003974 emollient agent Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 229960000403 etanercept Drugs 0.000 description 2
- PIHFWZTVGIJVBU-UHFFFAOYSA-N ethyl 1-benzyl-5-methyl-3,6-dihydro-2h-pyridine-4-carboxylate Chemical compound C1CC(C(=O)OCC)=C(C)CN1CC1=CC=CC=C1 PIHFWZTVGIJVBU-UHFFFAOYSA-N 0.000 description 2
- AAUBVINEXCCXOK-UHFFFAOYSA-N ethyl 4,6-dichloropyridine-3-carboxylate Chemical compound CCOC(=O)C1=CN=C(Cl)C=C1Cl AAUBVINEXCCXOK-UHFFFAOYSA-N 0.000 description 2
- QRSPXEKUBUYLMW-UHFFFAOYSA-N ethyl 4-fluoro-1-methyl-6-(trifluoromethyl)indole-2-carboxylate Chemical compound FC1=C2C=C(N(C2=CC(=C1)C(F)(F)F)C)C(=O)OCC QRSPXEKUBUYLMW-UHFFFAOYSA-N 0.000 description 2
- KVIIMCWOHLXJQU-UHFFFAOYSA-N ethyl 4-fluoro-6-(trifluoromethyl)-1H-indole-2-carboxylate Chemical compound FC1=C2C=C(NC2=CC(=C1)C(F)(F)F)C(=O)OCC KVIIMCWOHLXJQU-UHFFFAOYSA-N 0.000 description 2
- 229960004979 fampridine Drugs 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 2
- 238000001030 gas--liquid chromatography Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 229960002849 glucosamine sulfate Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 239000007902 hard capsule Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 208000002557 hidradenitis Diseases 0.000 description 2
- 201000007162 hidradenitis suppurativa Diseases 0.000 description 2
- 229960002764 hydrocodone bitartrate Drugs 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 102000009634 interleukin-1 receptor antagonist activity proteins Human genes 0.000 description 2
- 108040001669 interleukin-1 receptor antagonist activity proteins Proteins 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 229960000448 lactic acid Drugs 0.000 description 2
- 108020001756 ligand binding domains Proteins 0.000 description 2
- 238000004460 liquid liquid chromatography Methods 0.000 description 2
- 231100000053 low toxicity Toxicity 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 229960001929 meloxicam Drugs 0.000 description 2
- GQJCAQADCPTHKN-UHFFFAOYSA-N methyl 2,2-difluoro-2-fluorosulfonylacetate Chemical compound COC(=O)C(F)(F)S(F)(=O)=O GQJCAQADCPTHKN-UHFFFAOYSA-N 0.000 description 2
- GDAVHDNJGMASIM-UHFFFAOYSA-N methyl 2,4-dichloro-3-(2-hydroxybutyl)benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CC(CC)O)Cl GDAVHDNJGMASIM-UHFFFAOYSA-N 0.000 description 2
- LEDJIWCFZBHHGO-UHFFFAOYSA-N methyl 2,4-dichloro-3-(cyanomethyl)benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CC#N)Cl LEDJIWCFZBHHGO-UHFFFAOYSA-N 0.000 description 2
- YYNDTSNUQWSZEY-UHFFFAOYSA-N methyl 2,4-dichloro-3-formylbenzoate Chemical compound COC(=O)C1=CC=C(Cl)C(C=O)=C1Cl YYNDTSNUQWSZEY-UHFFFAOYSA-N 0.000 description 2
- PQTXGZGFXOHGLN-UHFFFAOYSA-N methyl 2-azaspiro[3.3]heptane-6-carboxylate 2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.COC(=O)C1CC2(CNC2)C1 PQTXGZGFXOHGLN-UHFFFAOYSA-N 0.000 description 2
- IHKYCPIKNFXPSI-UHFFFAOYSA-N methyl 5-ethenyl-4,6-dimethylpyridine-3-carboxylate Chemical compound CC1=C(C(=NC=C1C(=O)OC)C)C=C IHKYCPIKNFXPSI-UHFFFAOYSA-N 0.000 description 2
- SLXBHPRLDZZGKV-UHFFFAOYSA-N methyl 5-formyl-4,6-dimethylpyridine-3-carboxylate Chemical compound C(=O)C=1C(=NC=C(C(=O)OC)C=1C)C SLXBHPRLDZZGKV-UHFFFAOYSA-N 0.000 description 2
- OJLOPKGSLYJEMD-URPKTTJQSA-N methyl 7-[(1r,2r,3r)-3-hydroxy-2-[(1e)-4-hydroxy-4-methyloct-1-en-1-yl]-5-oxocyclopentyl]heptanoate Chemical compound CCCCC(C)(O)C\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1CCCCCCC(=O)OC OJLOPKGSLYJEMD-URPKTTJQSA-N 0.000 description 2
- 229960000282 metronidazole Drugs 0.000 description 2
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 229960005249 misoprostol Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- 229910052750 molybdenum Inorganic materials 0.000 description 2
- 239000011733 molybdenum Substances 0.000 description 2
- QACIMZNAAXTLTN-UHFFFAOYSA-N n-methoxy-n-methyl-6-(trifluoromethoxy)-1h-indole-2-carboxamide Chemical compound C1=C(OC(F)(F)F)C=C2NC(C(=O)N(C)OC)=CC2=C1 QACIMZNAAXTLTN-UHFFFAOYSA-N 0.000 description 2
- 229960005027 natalizumab Drugs 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- MUZQPDBAOYKNLO-RKXJKUSZSA-N oxycodone hydrochloride Chemical compound [H+].[Cl-].O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C MUZQPDBAOYKNLO-RKXJKUSZSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- XUYJLQHKOGNDPB-UHFFFAOYSA-N phosphonoacetic acid Chemical compound OC(=O)CP(O)(O)=O XUYJLQHKOGNDPB-UHFFFAOYSA-N 0.000 description 2
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 2
- 230000035479 physiological effects, processes and functions Effects 0.000 description 2
- SIOXPEMLGUPBBT-UHFFFAOYSA-M picolinate Chemical compound [O-]C(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-M 0.000 description 2
- 229960005330 pimecrolimus Drugs 0.000 description 2
- KASDHRXLYQOAKZ-ZPSXYTITSA-N pimecrolimus Chemical compound C/C([C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@]2(O)O[C@@H]([C@H](C[C@H]2C)OC)[C@@H](OC)C[C@@H](C)C/C(C)=C/[C@H](C(C[C@H](O)[C@H]1C)=O)CC)=C\[C@@H]1CC[C@@H](Cl)[C@H](OC)C1 KASDHRXLYQOAKZ-ZPSXYTITSA-N 0.000 description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 2
- 229960002702 piroxicam Drugs 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 229940068968 polysorbate 80 Drugs 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- LXNHXLLTXMVWPM-UHFFFAOYSA-N pyridoxine Chemical compound CC1=NC=C(CO)C(CO)=C1O LXNHXLLTXMVWPM-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 208000002574 reactive arthritis Diseases 0.000 description 2
- 210000003289 regulatory T cell Anatomy 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 108090000064 retinoic acid receptors Proteins 0.000 description 2
- 102000003702 retinoic acid receptors Human genes 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- YPNVIBVEFVRZPJ-UHFFFAOYSA-L silver sulfate Chemical compound [Ag+].[Ag+].[O-]S([O-])(=O)=O YPNVIBVEFVRZPJ-UHFFFAOYSA-L 0.000 description 2
- 229910000367 silver sulfate Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- RSIJVJUOQBWMIM-UHFFFAOYSA-L sodium sulfate decahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].[O-]S([O-])(=O)=O RSIJVJUOQBWMIM-UHFFFAOYSA-L 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- AGHLUVOCTHWMJV-UHFFFAOYSA-J sodium;gold(3+);2-sulfanylbutanedioate Chemical compound [Na+].[Au+3].[O-]C(=O)CC(S)C([O-])=O.[O-]C(=O)CC(S)C([O-])=O AGHLUVOCTHWMJV-UHFFFAOYSA-J 0.000 description 2
- 201000005671 spondyloarthropathy Diseases 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- SEEPANYCNGTZFQ-UHFFFAOYSA-N sulfadiazine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)NC1=NC=CC=N1 SEEPANYCNGTZFQ-UHFFFAOYSA-N 0.000 description 2
- 229960004306 sulfadiazine Drugs 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 229960005349 sulfur Drugs 0.000 description 2
- 239000002511 suppository base Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 229960001967 tacrolimus Drugs 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- YTGBPZMHQRYUGQ-UHFFFAOYSA-N tert-butyl 2,4-dichloro-3-formylbenzoate Chemical compound ClC1=C(C(=O)OC(C)(C)C)C=CC(=C1C=O)Cl YTGBPZMHQRYUGQ-UHFFFAOYSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 2
- 229940033663 thimerosal Drugs 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- 210000001541 thymus gland Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 229960001727 tretinoin Drugs 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 2
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- NITUEMISTORFON-PPFXTMJRSA-N (2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2R)-2-aminopropanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-3-methylbutanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-phenylpropanoyl]amino]propanoyl]amino]-4-oxobutanoyl]amino]-3-methylpentanoyl]amino]-3-methylbutanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carboxylic acid Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@@H](C)N)C(C)C)C(C)C)C1=CC=CC=C1 NITUEMISTORFON-PPFXTMJRSA-N 0.000 description 1
- SFGFYNXPJMOUHK-PKAFTLKUSA-N (2r)-2-[[(2r)-2-amino-5-(diaminomethylideneamino)pentanoyl]amino]-n-[(2r)-1-[[(2r)-1-[[(2r)-1-[[(2r)-1-[[(2r)-1-[[(2r)-1-[[2-[[(2r)-1-amino-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohe Chemical compound NC(N)=NCCC[C@@H](N)C(=O)N[C@H](CCCC)C(=O)N[C@H](CCCC)C(=O)N[C@H](CCCC)C(=O)N[C@H](CCCN=C(N)N)C(=O)N[C@H](CCCC)C(=O)N[C@H](CCCC)C(=O)N[C@H](CCCC)C(=O)NCC(=O)N[C@@H](C(N)=O)CC1=CC=C(O)C=C1 SFGFYNXPJMOUHK-PKAFTLKUSA-N 0.000 description 1
- ZZMYWYZPNZRYPX-SANMLTNESA-N (2r)-2-amino-2-[5-[4-[2-(4-phenylphenyl)ethoxy]-3-(trifluoromethyl)phenyl]-1h-imidazol-2-yl]propan-1-ol Chemical compound N1C([C@@](N)(CO)C)=NC=C1C(C=C1C(F)(F)F)=CC=C1OCCC1=CC=C(C=2C=CC=CC=2)C=C1 ZZMYWYZPNZRYPX-SANMLTNESA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- SPFMQWBKVUQXJV-BTVCFUMJSA-N (2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal;hydrate Chemical compound O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O SPFMQWBKVUQXJV-BTVCFUMJSA-N 0.000 description 1
- DRSJLVGDSNWQBI-SFHVURJKSA-N (2s)-2-[(2,6-dichlorobenzoyl)amino]-3-[4-(2,6-dimethoxyphenyl)phenyl]propanoic acid Chemical compound COC1=CC=CC(OC)=C1C(C=C1)=CC=C1C[C@@H](C(O)=O)NC(=O)C1=C(Cl)C=CC=C1Cl DRSJLVGDSNWQBI-SFHVURJKSA-N 0.000 description 1
- JEQDSBVHLKBEIZ-REOHCLBHSA-N (2s)-2-chloropropanoyl chloride Chemical compound C[C@H](Cl)C(Cl)=O JEQDSBVHLKBEIZ-REOHCLBHSA-N 0.000 description 1
- OTHYPAMNTUGKDK-UHFFFAOYSA-N (3-acetylphenyl) acetate Chemical compound CC(=O)OC1=CC=CC(C(C)=O)=C1 OTHYPAMNTUGKDK-UHFFFAOYSA-N 0.000 description 1
- OSLRVFJRYRWWCB-WMLDXEAASA-N (3R,4R)-1-[2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]benzoyl]-3-methylpiperidine-4-carboxylic acid Chemical compound ClC1=C(C(=O)N2C[C@@H]([C@@H](CC2)C(=O)O)C)C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl OSLRVFJRYRWWCB-WMLDXEAASA-N 0.000 description 1
- OSLRVFJRYRWWCB-PBHICJAKSA-N (3S,4S)-1-[2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]benzoyl]-3-methylpiperidine-4-carboxylic acid Chemical compound ClC1=C(C(=O)N2C[C@H]([C@H](CC2)C(=O)O)C)C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl OSLRVFJRYRWWCB-PBHICJAKSA-N 0.000 description 1
- DKSZLDSPXIWGFO-BLOJGBSASA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;phosphoric acid;hydrate Chemical compound O.OP(O)(O)=O.OP(O)(O)=O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC DKSZLDSPXIWGFO-BLOJGBSASA-N 0.000 description 1
- CXAGHAZMQSCAKJ-WAHHBDPQSA-N (4s,7s)-n-[(2r,3s)-2-ethoxy-5-oxooxolan-3-yl]-7-(isoquinoline-1-carbonylamino)-6,10-dioxo-2,3,4,7,8,9-hexahydro-1h-pyridazino[1,2-a]diazepine-4-carboxamide Chemical compound CCO[C@@H]1OC(=O)C[C@@H]1NC(=O)[C@H]1N(C(=O)[C@H](CCC2=O)NC(=O)C=3C4=CC=CC=C4C=CN=3)N2CCC1 CXAGHAZMQSCAKJ-WAHHBDPQSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- IRELROQHIPLASX-SEYXRHQNSA-N (z)-2-cyano-3-hydroxy-n-[4-(trifluoromethyl)phenyl]hept-2-en-6-ynamide Chemical compound C#CCCC(/O)=C(\C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 IRELROQHIPLASX-SEYXRHQNSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- HHLCSFGOTLUREE-UHFFFAOYSA-N 1,2,3-trichloro-5-nitrobenzene Chemical compound [O-][N+](=O)C1=CC(Cl)=C(Cl)C(Cl)=C1 HHLCSFGOTLUREE-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- JZFSLMDLIPKTAO-UHFFFAOYSA-N 1,3-dihydroazepin-2-one Chemical compound O=C1CC=CC=CN1 JZFSLMDLIPKTAO-UHFFFAOYSA-N 0.000 description 1
- AFIRLJILFZZZIA-UHFFFAOYSA-N 1,4-dimethyl-6-(trifluoromethyl)indole Chemical compound CN1C=CC2=C(C=C(C=C12)C(F)(F)F)C AFIRLJILFZZZIA-UHFFFAOYSA-N 0.000 description 1
- BMEMBBFDTYHTLH-UHFFFAOYSA-N 1-(2-methoxyethyl)piperazine Chemical compound COCCN1CCNCC1 BMEMBBFDTYHTLH-UHFFFAOYSA-N 0.000 description 1
- 125000005846 1-(alkanoyloxy)ethyl group Chemical group 0.000 description 1
- 125000005848 1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- FZRDGLKRGKUTME-UHFFFAOYSA-N 1-(benzenesulfonyl)-4,6-dichloroindole Chemical compound ClC1=C2C=CN(C2=CC(=C1)Cl)S(=O)(=O)C1=CC=CC=C1 FZRDGLKRGKUTME-UHFFFAOYSA-N 0.000 description 1
- VJNFLJOAJQAMMN-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-bromo-4-methylindole Chemical compound BrC1=CC(=C2C=CN(C2=C1)S(=O)(=O)C1=CC=CC=C1)C VJNFLJOAJQAMMN-UHFFFAOYSA-N 0.000 description 1
- YNLCQDBPLOYFFA-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-cyclopropyl-4-fluoroindole Chemical compound C1(CC1)C1=CC(=C2C=CN(C2=C1)S(=O)(=O)C1=CC=CC=C1)F YNLCQDBPLOYFFA-UHFFFAOYSA-N 0.000 description 1
- QOQNWFSCVSEJOD-UHFFFAOYSA-N 1-(benzenesulfonyl)-6-methoxy-4-methylindole Chemical compound COC1=CC(=C2C=CN(C2=C1)S(=O)(=O)C1=CC=CC=C1)C QOQNWFSCVSEJOD-UHFFFAOYSA-N 0.000 description 1
- AOYSLJHKVBSXRU-UHFFFAOYSA-N 1-(oxetan-3-yl)piperazine Chemical compound C1OCC1N1CCNCC1 AOYSLJHKVBSXRU-UHFFFAOYSA-N 0.000 description 1
- QHTSZBPSLDEABZ-DZGCQCFKSA-N 1-O-benzyl 4-O-ethyl (3R,4R)-3-methylpiperidine-1,4-dicarboxylate Chemical compound C[C@H]1CN(CC[C@H]1C(=O)OCC)C(=O)OCC1=CC=CC=C1 QHTSZBPSLDEABZ-DZGCQCFKSA-N 0.000 description 1
- QHTSZBPSLDEABZ-HIFRSBDPSA-N 1-O-benzyl 4-O-ethyl (3S,4S)-3-methylpiperidine-1,4-dicarboxylate Chemical compound C[C@@H]1CN(CC[C@@H]1C(=O)OCC)C(=O)OCC1=CC=CC=C1 QHTSZBPSLDEABZ-HIFRSBDPSA-N 0.000 description 1
- OSLRVFJRYRWWCB-UHFFFAOYSA-N 1-[2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]benzoyl]-3-methylpiperidine-4-carboxylic acid Chemical compound ClC1=C(C(=O)N2CC(C(CC2)C(=O)O)C)C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl OSLRVFJRYRWWCB-UHFFFAOYSA-N 0.000 description 1
- MDBVGPLRLOTVBU-UHFFFAOYSA-N 1-[2-fluoro-3-methyl-5-(trifluoromethyl)phenyl]ethanone Chemical compound FC1=C(C=C(C=C1C)C(F)(F)F)C(C)=O MDBVGPLRLOTVBU-UHFFFAOYSA-N 0.000 description 1
- AIBRJIYOSLKVFL-UHFFFAOYSA-N 1-bromo-4-fluoro-2-methyl-3-nitrobenzene Chemical compound CC1=C(Br)C=CC(F)=C1[N+]([O-])=O AIBRJIYOSLKVFL-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- SPICZPCIWHHXED-UHFFFAOYSA-N 1-fluoro-2-methyl-4-(trifluoromethyl)benzene Chemical compound CC1=CC(C(F)(F)F)=CC=C1F SPICZPCIWHHXED-UHFFFAOYSA-N 0.000 description 1
- JSXAJLMUBSEJFF-UHFFFAOYSA-N 1-fluoro-3-methyl-2-nitrobenzene Chemical compound CC1=CC=CC(F)=C1[N+]([O-])=O JSXAJLMUBSEJFF-UHFFFAOYSA-N 0.000 description 1
- 125000005847 1-methyl-1-(alkanoyloxy)-ethyl group Chemical group 0.000 description 1
- 125000005849 1-methyl-1-(alkoxycarbonyloxy)ethyl group Chemical group 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- BLRHMMGNCXNXJL-UHFFFAOYSA-N 1-methylindole Chemical compound C1=CC=C2N(C)C=CC2=C1 BLRHMMGNCXNXJL-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- CNLIIAKAAMFCJG-UHFFFAOYSA-N 2,3,5-trichloropyridine Chemical compound ClC1=CN=C(Cl)C(Cl)=C1 CNLIIAKAAMFCJG-UHFFFAOYSA-N 0.000 description 1
- RZNIINCSHCFUNN-UHFFFAOYSA-N 2,3,5-trichloropyridine-4-carbaldehyde Chemical compound ClC1=CN=C(Cl)C(Cl)=C1C=O RZNIINCSHCFUNN-UHFFFAOYSA-N 0.000 description 1
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 1
- BCDSOFBPNSSRIZ-UHFFFAOYSA-N 2,4-dichloro-3-(methylsulfonyloxymethyl)benzoic acid Chemical compound ClC1=C(C(=O)O)C=CC(=C1COS(=O)(=O)C)Cl BCDSOFBPNSSRIZ-UHFFFAOYSA-N 0.000 description 1
- SICGCSFQCNBUDT-UHFFFAOYSA-N 2,4-dichloro-3-[(3-methylimidazo[1,2-a]pyridin-2-yl)methyl]benzamide Chemical class ClC1=C(C(=O)N)C=CC(=C1CC=1N=C2N(C=CC=C2)C=1C)Cl SICGCSFQCNBUDT-UHFFFAOYSA-N 0.000 description 1
- BOEYOJGUYQCWTR-UHFFFAOYSA-N 2,6-dichloro-3-methoxybenzaldehyde Chemical compound COC1=CC=C(Cl)C(C=O)=C1Cl BOEYOJGUYQCWTR-UHFFFAOYSA-N 0.000 description 1
- CMJNIYUZFQWYCK-UHFFFAOYSA-N 2,6-dichloro-4-methyl-3-nitropyridine Chemical compound CC1=CC(Cl)=NC(Cl)=C1[N+]([O-])=O CMJNIYUZFQWYCK-UHFFFAOYSA-N 0.000 description 1
- QOJQBWSZHCKOLL-UHFFFAOYSA-N 2,6-dimethylbenzaldehyde Chemical compound CC1=CC=CC(C)=C1C=O QOJQBWSZHCKOLL-UHFFFAOYSA-N 0.000 description 1
- SZXUTTGMFUSMCE-UHFFFAOYSA-N 2-(1h-imidazol-2-yl)pyridine Chemical class C1=CNC(C=2N=CC=CC=2)=N1 SZXUTTGMFUSMCE-UHFFFAOYSA-N 0.000 description 1
- ZAVJTSLIGAGALR-UHFFFAOYSA-N 2-(2,2,2-trifluoroacetyl)cyclooctan-1-one Chemical compound FC(F)(F)C(=O)C1CCCCCCC1=O ZAVJTSLIGAGALR-UHFFFAOYSA-N 0.000 description 1
- MMEXIIGRXCODNK-UHFFFAOYSA-N 2-(2,6-dimethylphenyl)acetic acid Chemical compound CC1=CC=CC(C)=C1CC(O)=O MMEXIIGRXCODNK-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 1
- VTAKZNRDSPNOAU-UHFFFAOYSA-M 2-(chloromethyl)oxirane;hydron;prop-2-en-1-amine;n-prop-2-enyldecan-1-amine;trimethyl-[6-(prop-2-enylamino)hexyl]azanium;dichloride Chemical compound Cl.[Cl-].NCC=C.ClCC1CO1.CCCCCCCCCCNCC=C.C[N+](C)(C)CCCCCCNCC=C VTAKZNRDSPNOAU-UHFFFAOYSA-M 0.000 description 1
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 1
- RQVKVJIRFKVPBF-VWLOTQADSA-N 2-[[(2s)-2-amino-3-phenylpropyl]amino]-3-methyl-5-naphthalen-2-yl-6-pyridin-4-ylpyrimidin-4-one Chemical compound C([C@H](N)CNC=1N(C(C(C=2C=C3C=CC=CC3=CC=2)=C(C=2C=CN=CC=2)N=1)=O)C)C1=CC=CC=C1 RQVKVJIRFKVPBF-VWLOTQADSA-N 0.000 description 1
- NBGAYCYFNGPNPV-UHFFFAOYSA-N 2-aminooxybenzoic acid Chemical class NOC1=CC=CC=C1C(O)=O NBGAYCYFNGPNPV-UHFFFAOYSA-N 0.000 description 1
- KOFZWAGTGPUOCM-UHFFFAOYSA-N 2-bromo-3-methyl-5-nitroaniline Chemical compound CC1=CC([N+]([O-])=O)=CC(N)=C1Br KOFZWAGTGPUOCM-UHFFFAOYSA-N 0.000 description 1
- MJTPNVWLMZFFIU-UHFFFAOYSA-N 2-bromo-3-nitro-5-(trifluoromethyl)pyridine Chemical compound [O-][N+](=O)C1=CC(C(F)(F)F)=CN=C1Br MJTPNVWLMZFFIU-UHFFFAOYSA-N 0.000 description 1
- UKDQALCDSBOTRX-UHFFFAOYSA-N 2-bromo-6-methyl-4-(trifluoromethyl)aniline Chemical compound CC1=CC(C(F)(F)F)=CC(Br)=C1N UKDQALCDSBOTRX-UHFFFAOYSA-N 0.000 description 1
- BVBRVPXLHHOEMP-UHFFFAOYSA-N 2-bromo-6-methyl-4-nitropyridine Chemical compound CC1=CC([N+]([O-])=O)=CC(Br)=N1 BVBRVPXLHHOEMP-UHFFFAOYSA-N 0.000 description 1
- GCTGNIQJWWFHGZ-UHFFFAOYSA-N 2-chloro-6-iodo-4-(trifluoromethyl)aniline Chemical compound NC1=C(Cl)C=C(C(F)(F)F)C=C1I GCTGNIQJWWFHGZ-UHFFFAOYSA-N 0.000 description 1
- KFEHNXLFIGPWNB-UHFFFAOYSA-N 2-fluoro-4-(trifluoromethyl)benzaldehyde Chemical compound FC1=CC(C(F)(F)F)=CC=C1C=O KFEHNXLFIGPWNB-UHFFFAOYSA-N 0.000 description 1
- UZCLUOWLQYOFSZ-UHFFFAOYSA-N 2-fluoro-6-iodo-4-(trifluoromethyl)aniline Chemical compound NC1=C(F)C=C(C(F)(F)F)C=C1I UZCLUOWLQYOFSZ-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- ZLAKLXWPUUUYNN-UHFFFAOYSA-N 2-iodo-3-methyl-5-(trifluoromethoxy)aniline Chemical compound IC1=C(N)C=C(C=C1C)OC(F)(F)F ZLAKLXWPUUUYNN-UHFFFAOYSA-N 0.000 description 1
- OEHDGSONCOREHH-UHFFFAOYSA-N 2-iodo-4-methyl-5-(trifluoromethyl)aniline Chemical compound CC1=CC(I)=C(N)C=C1C(F)(F)F OEHDGSONCOREHH-UHFFFAOYSA-N 0.000 description 1
- SCFZBZGJXHIKEP-UHFFFAOYSA-N 2-iodo-5-(trifluoromethoxy)aniline Chemical compound NC1=CC(OC(F)(F)F)=CC=C1I SCFZBZGJXHIKEP-UHFFFAOYSA-N 0.000 description 1
- KJTQOUZTJKNWGK-UHFFFAOYSA-N 2-iodo-5-(trifluoromethyl)aniline Chemical compound NC1=CC(C(F)(F)F)=CC=C1I KJTQOUZTJKNWGK-UHFFFAOYSA-N 0.000 description 1
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 1
- FUJKUIAVFVGTGS-UHFFFAOYSA-N 2-methyl-6-(trifluoromethyl)pyridin-3-amine Chemical compound CC1=NC(C(F)(F)F)=CC=C1N FUJKUIAVFVGTGS-UHFFFAOYSA-N 0.000 description 1
- FZDJDEXABKVIDA-UHFFFAOYSA-N 2-o-tert-butyl 6-o-methyl 2-azaspiro[3.3]heptane-2,6-dicarboxylate Chemical compound C1C(C(=O)OC)CC21CN(C(=O)OC(C)(C)C)C2 FZDJDEXABKVIDA-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- TZOYXRMEFDYWDQ-UHFFFAOYSA-N 3,4-dihydro-1h-quinolin-2-one Chemical compound C1=CC=C2NC(=O)CCC2=C1 TZOYXRMEFDYWDQ-UHFFFAOYSA-N 0.000 description 1
- XMASGFYUZZRWOE-UHFFFAOYSA-N 3,5-dichloro-2-methoxypyridine Chemical compound COC1=NC=C(Cl)C=C1Cl XMASGFYUZZRWOE-UHFFFAOYSA-N 0.000 description 1
- UOTYNGKRLCSNSU-UHFFFAOYSA-N 3,7-dimethyl-5-(trifluoromethyl)-1H-pyrrolo[3,2-b]pyridine Chemical compound CC1=CNC=2C1=NC(=CC=2C)C(F)(F)F UOTYNGKRLCSNSU-UHFFFAOYSA-N 0.000 description 1
- DDYUBCCTNHWSQM-UHFFFAOYSA-N 3-(3-cyclopentyloxy-4-methoxyphenyl)-3-(1,3-dioxoisoindol-2-yl)propanamide Chemical compound COC1=CC=C(C(CC(N)=O)N2C(C3=CC=CC=C3C2=O)=O)C=C1OC1CCCC1 DDYUBCCTNHWSQM-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- REVKQTVWHUOMRX-UHFFFAOYSA-N 3-(morpholine-4-carbonyl)benzaldehyde Chemical compound O=CC1=CC=CC(C(=O)N2CCOCC2)=C1 REVKQTVWHUOMRX-UHFFFAOYSA-N 0.000 description 1
- TVIDQWBXASWURT-UHFFFAOYSA-N 3-amino-2-methyl-4-nitrobenzonitrile Chemical compound CC1=C(N)C([N+]([O-])=O)=CC=C1C#N TVIDQWBXASWURT-UHFFFAOYSA-N 0.000 description 1
- HXHUNOWRVCNLIF-UHFFFAOYSA-N 3-amino-4-bromo-5-methylbenzonitrile Chemical compound CC1=CC(C#N)=CC(N)=C1Br HXHUNOWRVCNLIF-UHFFFAOYSA-N 0.000 description 1
- JURDBYHADODTPF-UHFFFAOYSA-N 3-amino-4-iodobenzonitrile Chemical compound NC1=CC(C#N)=CC=C1I JURDBYHADODTPF-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- WYDJMBLMXGCHOL-UHFFFAOYSA-N 3-methyl-5-(trifluoromethyl)aniline Chemical compound CC1=CC(N)=CC(C(F)(F)F)=C1 WYDJMBLMXGCHOL-UHFFFAOYSA-N 0.000 description 1
- WHZRGBVRNHCWLM-UHFFFAOYSA-N 3-methyl-5-(trifluoromethyl)benzene-1,2-diamine Chemical compound CC1=CC(C(F)(F)F)=CC(N)=C1N WHZRGBVRNHCWLM-UHFFFAOYSA-N 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- NIXGYRHZQFZCCV-UHFFFAOYSA-N 4,6-dichloro-1h-indole Chemical compound ClC1=CC(Cl)=C2C=CNC2=C1 NIXGYRHZQFZCCV-UHFFFAOYSA-N 0.000 description 1
- ILMIEWNDXAKVNI-UHFFFAOYSA-N 4,6-dichloropyridine-3-carboxylic acid Chemical compound OC(=O)C1=CN=C(Cl)C=C1Cl ILMIEWNDXAKVNI-UHFFFAOYSA-N 0.000 description 1
- OCYMJCILWYHKAU-UHFFFAOYSA-N 4,6-dimethyl-2-oxo-1h-pyridine-3-carbonitrile Chemical compound CC1=CC(C)=C(C#N)C(O)=N1 OCYMJCILWYHKAU-UHFFFAOYSA-N 0.000 description 1
- NBUHTTJGQKIBMR-UHFFFAOYSA-N 4,6-dimethylpyrimidin-5-amine Chemical compound CC1=NC=NC(C)=C1N NBUHTTJGQKIBMR-UHFFFAOYSA-N 0.000 description 1
- XQNVDQZWOBPLQZ-UHFFFAOYSA-N 4-(trifluoromethoxy)benzaldehyde Chemical compound FC(F)(F)OC1=CC=C(C=O)C=C1 XQNVDQZWOBPLQZ-UHFFFAOYSA-N 0.000 description 1
- ZZLCFHIKESPLTH-UHFFFAOYSA-N 4-Methylbiphenyl Chemical compound C1=CC(C)=CC=C1C1=CC=CC=C1 ZZLCFHIKESPLTH-UHFFFAOYSA-N 0.000 description 1
- DJKNRCWSXSZACF-UHFFFAOYSA-N 4-acetamido-n-tert-butylbenzamide Chemical compound CC(=O)NC1=CC=C(C(=O)NC(C)(C)C)C=C1 DJKNRCWSXSZACF-UHFFFAOYSA-N 0.000 description 1
- UOWVTQFTEAYDLM-UHFFFAOYSA-N 4-amino-3-iodobenzonitrile Chemical compound NC1=CC=C(C#N)C=C1I UOWVTQFTEAYDLM-UHFFFAOYSA-N 0.000 description 1
- WZRJTRPJURQBRM-UHFFFAOYSA-N 4-amino-n-(5-methyl-1,2-oxazol-3-yl)benzenesulfonamide;5-[(3,4,5-trimethoxyphenyl)methyl]pyrimidine-2,4-diamine Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1.COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 WZRJTRPJURQBRM-UHFFFAOYSA-N 0.000 description 1
- JRUCLQINUHVRJW-UHFFFAOYSA-N 4-bromo-6-(trifluoromethyl)-1h-indazole Chemical compound FC(F)(F)C1=CC(Br)=C2C=NNC2=C1 JRUCLQINUHVRJW-UHFFFAOYSA-N 0.000 description 1
- AAEDEYUFAPYBPJ-UHFFFAOYSA-N 4-fluoro-1-methyl-6-(trifluoromethyl)indole-2-carbaldehyde Chemical compound FC1=C2C=C(N(C2=CC(=C1)C(F)(F)F)C)C=O AAEDEYUFAPYBPJ-UHFFFAOYSA-N 0.000 description 1
- WCWWRSIRPRSDDH-UHFFFAOYSA-N 4-methyl-6-(trifluoromethyl)-1H-indole-3-carbaldehyde Chemical compound CC1=C2C(=CNC2=CC(=C1)C(F)(F)F)C=O WCWWRSIRPRSDDH-UHFFFAOYSA-N 0.000 description 1
- GLJGKHHMJXKHPG-UHFFFAOYSA-N 4-methyl-6-(trifluoromethyl)pyridin-3-amine Chemical compound CC1=CC(C(F)(F)F)=NC=C1N GLJGKHHMJXKHPG-UHFFFAOYSA-N 0.000 description 1
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical compound O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 description 1
- OHXXDADUZZSIDO-UHFFFAOYSA-N 5-(difluoromethoxy)-7-methyl-1H-indole Chemical compound FC(OC=1C=C2C=CNC2=C(C=1)C)F OHXXDADUZZSIDO-UHFFFAOYSA-N 0.000 description 1
- KWSLGOVYXMQPPX-UHFFFAOYSA-N 5-[3-(trifluoromethyl)phenyl]-2h-tetrazole Chemical compound FC(F)(F)C1=CC=CC(C2=NNN=N2)=C1 KWSLGOVYXMQPPX-UHFFFAOYSA-N 0.000 description 1
- DKBQOOXGPIRVPC-UHFFFAOYSA-N 5-bromo-1-fluoro-3-methyl-2-nitrobenzene Chemical compound CC1=CC(Br)=CC(F)=C1[N+]([O-])=O DKBQOOXGPIRVPC-UHFFFAOYSA-N 0.000 description 1
- TZRRRWOVYVGRSL-UHFFFAOYSA-N 5-bromo-7-fluoro-1h-indole Chemical compound FC1=CC(Br)=CC2=C1NC=C2 TZRRRWOVYVGRSL-UHFFFAOYSA-N 0.000 description 1
- HOUJSOAXJSBNRH-UHFFFAOYSA-N 5-bromo-7-methyl-1h-indole Chemical compound CC1=CC(Br)=CC2=C1NC=C2 HOUJSOAXJSBNRH-UHFFFAOYSA-N 0.000 description 1
- UZHXMAXLRQCDQP-UHFFFAOYSA-N 5-bromo-N,3-dimethyl-2-nitroaniline Chemical compound CNc1cc(Br)cc(C)c1[N+]([O-])=O UZHXMAXLRQCDQP-UHFFFAOYSA-N 0.000 description 1
- BOCNOBUKLSOYER-UHFFFAOYSA-N 5-iodo-2-(trifluoromethyl)pyridin-4-amine Chemical compound NC1=CC(C(F)(F)F)=NC=C1I BOCNOBUKLSOYER-UHFFFAOYSA-N 0.000 description 1
- SVAGFBGXEWPNJC-SPIKMXEPSA-N 6,9-bis(2-aminoethylamino)benzo[g]isoquinoline-5,10-dione;(z)-but-2-enedioic acid Chemical compound OC(=O)\C=C/C(O)=O.OC(=O)\C=C/C(O)=O.O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN SVAGFBGXEWPNJC-SPIKMXEPSA-N 0.000 description 1
- FYSRKRZDBHOFAY-UHFFFAOYSA-N 6-(N-carbamoyl-2,6-difluoroanilino)-2-(2,4-difluorophenyl)-3-pyridinecarboxamide Chemical compound FC=1C=CC=C(F)C=1N(C(=O)N)C(N=1)=CC=C(C(N)=O)C=1C1=CC=C(F)C=C1F FYSRKRZDBHOFAY-UHFFFAOYSA-N 0.000 description 1
- WPZXOYUWWTZHFO-UHFFFAOYSA-N 6-(trifluoromethyl)pyridazin-3-amine Chemical compound NC1=CC=C(C(F)(F)F)N=N1 WPZXOYUWWTZHFO-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- JSFRDTNSPFYYTQ-UHFFFAOYSA-N 6-bromo-4-chloro-1h-indole Chemical compound ClC1=CC(Br)=CC2=C1C=CN2 JSFRDTNSPFYYTQ-UHFFFAOYSA-N 0.000 description 1
- OJTPNPQPCYJAIN-UHFFFAOYSA-N 6-chloro-2-N,4-dimethylpyridine-2,3-diamine Chemical compound CNc1nc(Cl)cc(C)c1N OJTPNPQPCYJAIN-UHFFFAOYSA-N 0.000 description 1
- HSAHCMOZFNSMLH-UHFFFAOYSA-N 6-chloro-4-methylpyridazin-3-amine Chemical compound CC1=CC(Cl)=NN=C1N HSAHCMOZFNSMLH-UHFFFAOYSA-N 0.000 description 1
- UWDLNRUFHRYMSE-UHFFFAOYSA-N 6-chloro-5-methylpyridazin-3-amine Chemical compound CC1=CC(N)=NN=C1Cl UWDLNRUFHRYMSE-UHFFFAOYSA-N 0.000 description 1
- IDDIKXCCZQGDAD-UHFFFAOYSA-N 6-methoxy-4-methyl-1h-indole Chemical compound COC1=CC(C)=C2C=CNC2=C1 IDDIKXCCZQGDAD-UHFFFAOYSA-N 0.000 description 1
- MWPUOIRDBPZGDP-UHFFFAOYSA-N 7-fluoro-5-(trifluoromethyl)-1H-indole Chemical compound Fc1cc(cc2cc[nH]c12)C(F)(F)F MWPUOIRDBPZGDP-UHFFFAOYSA-N 0.000 description 1
- DMSQROQHQXQMNU-UHFFFAOYSA-N 7-methyl-5-(trifluoromethyl)-1H-indazole Chemical compound CC=1C=C(C=C2C=NNC=12)C(F)(F)F DMSQROQHQXQMNU-UHFFFAOYSA-N 0.000 description 1
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical class O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 102100039705 Beta-2 adrenergic receptor Human genes 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 125000004399 C1-C4 alkenyl group Chemical group 0.000 description 1
- VAUBWHIQMRKGBN-BHHHYXKXSA-N C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)CC2=O Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)CC2=O VAUBWHIQMRKGBN-BHHHYXKXSA-N 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102100035904 Caspase-1 Human genes 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 229940122444 Chemokine receptor antagonist Drugs 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 229920002567 Chondroitin Polymers 0.000 description 1
- YTTSOPMMGFGSKL-UHFFFAOYSA-N ClC=1C=C(C=C2C=C(NC=12)[Si](C)(C)C)C(F)(F)F Chemical compound ClC=1C=C(C=C2C=C(NC=12)[Si](C)(C)C)C(F)(F)F YTTSOPMMGFGSKL-UHFFFAOYSA-N 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 102000008169 Co-Repressor Proteins Human genes 0.000 description 1
- 108010060434 Co-Repressor Proteins Proteins 0.000 description 1
- 108050005238 Collagenase 3 Proteins 0.000 description 1
- 102100027995 Collagenase 3 Human genes 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 206010058675 Dermatitis psoriasiform Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- CYQFCXCEBYINGO-DLBZAZTESA-N Dronabinol Natural products C1=C(C)CC[C@H]2C(C)(C)OC3=CC(CCCCC)=CC(O)=C3[C@H]21 CYQFCXCEBYINGO-DLBZAZTESA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 101000759376 Escherichia phage Mu Tail sheath protein Proteins 0.000 description 1
- 241000400611 Eucalyptus deanei Species 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- POPFMWWJOGLOIF-XWCQMRHXSA-N Flurandrenolide Chemical compound C1([C@@H](F)C2)=CC(=O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O POPFMWWJOGLOIF-XWCQMRHXSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 238000010268 HPLC based assay Methods 0.000 description 1
- MUQNGPZZQDCDFT-JNQJZLCISA-N Halcinonide Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CCl)[C@@]1(C)C[C@@H]2O MUQNGPZZQDCDFT-JNQJZLCISA-N 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 108010008212 Integrin alpha4beta1 Proteins 0.000 description 1
- 108010005716 Interferon beta-1a Proteins 0.000 description 1
- 108010005714 Interferon beta-1b Proteins 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- 101800000542 Interleukin-1 receptor type 1, soluble form Proteins 0.000 description 1
- 102400000025 Interleukin-1 receptor type 1, soluble form Human genes 0.000 description 1
- 102000004560 Interleukin-12 Receptors Human genes 0.000 description 1
- 108010017515 Interleukin-12 Receptors Proteins 0.000 description 1
- 102100035018 Interleukin-17 receptor A Human genes 0.000 description 1
- 101710186083 Interleukin-17 receptor A Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000000585 Interleukin-9 Human genes 0.000 description 1
- ZCVMWBYGMWKGHF-UHFFFAOYSA-N Ketotifene Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2CC(=O)C2=C1C=CS2 ZCVMWBYGMWKGHF-UHFFFAOYSA-N 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical class NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 108010016230 MBP-8298 Proteins 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- FEWJPZIEWOKRBE-XIXRPRMCSA-N Mesotartaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-XIXRPRMCSA-N 0.000 description 1
- QXKHYNVANLEOEG-UHFFFAOYSA-N Methoxsalen Chemical compound C1=CC(=O)OC2=C1C=C1C=COC1=C2OC QXKHYNVANLEOEG-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- NXGRJILTXLIIJH-UHFFFAOYSA-N N-(6-cyano-3-iodo-2-methylpyridin-4-yl)benzenesulfonamide Chemical compound C(#N)C1=CC(=C(C(=N1)C)I)NS(=O)(=O)C1=CC=CC=C1 NXGRJILTXLIIJH-UHFFFAOYSA-N 0.000 description 1
- ATTZBAWRZYAHGB-UHFFFAOYSA-N N-[2-bromo-5-(trifluoromethyl)pyridin-3-yl]benzenesulfonamide Chemical compound BrC1=NC=C(C=C1NS(=O)(=O)C1=CC=CC=C1)C(F)(F)F ATTZBAWRZYAHGB-UHFFFAOYSA-N 0.000 description 1
- GCTPQRNXVDSRSV-UHFFFAOYSA-N N-[2-iodo-3-methyl-5-(trifluoromethoxy)phenyl]benzenesulfonamide Chemical compound IC1=C(C=C(C=C1C)OC(F)(F)F)NS(=O)(=O)C1=CC=CC=C1 GCTPQRNXVDSRSV-UHFFFAOYSA-N 0.000 description 1
- CHIMYWFBDUJNTD-UHFFFAOYSA-N N-[2-iodo-3-methyl-5-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound IC1=C(C=C(C=C1C)C(F)(F)F)NS(=O)(=O)C1=CC=CC=C1 CHIMYWFBDUJNTD-UHFFFAOYSA-N 0.000 description 1
- UQQPAXCGXDREEC-UHFFFAOYSA-N N-[2-iodo-5-(trifluoromethoxy)phenyl]benzenesulfonamide Chemical compound IC1=C(C=C(C=C1)OC(F)(F)F)NS(=O)(=O)C1=CC=CC=C1 UQQPAXCGXDREEC-UHFFFAOYSA-N 0.000 description 1
- QVIYATZHLCJAFK-UHFFFAOYSA-N N-methoxy-N,1-dimethyl-6-(trifluoromethoxy)indole-2-carboxamide Chemical compound CON(C(=O)C=1N(C2=CC(=CC=C2C=1)OC(F)(F)F)C)C QVIYATZHLCJAFK-UHFFFAOYSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- JOCBASBOOFNAJA-UHFFFAOYSA-N N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid Chemical compound OCC(CO)(CO)NCCS(O)(=O)=O JOCBASBOOFNAJA-UHFFFAOYSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 208000036110 Neuroinflammatory disease Diseases 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 102000008299 Nitric Oxide Synthase Human genes 0.000 description 1
- 108010021487 Nitric Oxide Synthase Proteins 0.000 description 1
- 102000042846 PKC family Human genes 0.000 description 1
- 108091082203 PKC family Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920000148 Polycarbophil calcium Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920002642 Polysorbate 65 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 108010005366 RDP 1258 Proteins 0.000 description 1
- 102000042839 ROR family Human genes 0.000 description 1
- 108091082331 ROR family Proteins 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical class [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229940078123 Ras inhibitor Drugs 0.000 description 1
- DRFDPXKCEWYIAW-UHFFFAOYSA-M Risedronate sodium Chemical compound [Na+].OP(=O)(O)C(P(O)([O-])=O)(O)CC1=CC=CN=C1 DRFDPXKCEWYIAW-UHFFFAOYSA-M 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 101100485158 Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) wzzE gene Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 102100025750 Sphingosine 1-phosphate receptor 1 Human genes 0.000 description 1
- 101710155454 Sphingosine 1-phosphate receptor 1 Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 102100036840 T-box transcription factor TBX21 Human genes 0.000 description 1
- JACAAXNEHGBPOQ-LLVKDONJSA-N Talampanel Chemical compound C([C@H](N(N=1)C(C)=O)C)C2=CC=3OCOC=3C=C2C=1C1=CC=C(N)C=C1 JACAAXNEHGBPOQ-LLVKDONJSA-N 0.000 description 1
- 239000004012 Tofacitinib Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102000011117 Transforming Growth Factor beta2 Human genes 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 208000035896 Twin-reversed arterial perfusion sequence Diseases 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 1
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- WVHBEIJGAINUBW-UHFFFAOYSA-N Xaliproden hydrochloride Chemical compound Cl.FC(F)(F)C1=CC=CC(C=2CCN(CCC=3C=C4C=CC=CC4=CC=3)CC=2)=C1 WVHBEIJGAINUBW-UHFFFAOYSA-N 0.000 description 1
- PCWZKQSKUXXDDJ-UHFFFAOYSA-N Xanthotoxin Natural products COCc1c2OC(=O)C=Cc2cc3ccoc13 PCWZKQSKUXXDDJ-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- OGQICQVSFDPSEI-UHFFFAOYSA-N Zorac Chemical compound N1=CC(C(=O)OCC)=CC=C1C#CC1=CC=C(SCCC2(C)C)C2=C1 OGQICQVSFDPSEI-UHFFFAOYSA-N 0.000 description 1
- JPKKQJKQTPNWTR-BRYCGAMXSA-N [(1r,5s)-8-methyl-8-azabicyclo[3.2.1]octan-3-yl] 3-hydroxy-2-phenylpropanoate;sulfuric acid;hydrate Chemical compound O.OS(O)(=O)=O.C([C@H]1CC[C@@H](C2)N1C)C2OC(=O)C(CO)C1=CC=CC=C1.C([C@H]1CC[C@@H](C2)N1C)C2OC(=O)C(CO)C1=CC=CC=C1 JPKKQJKQTPNWTR-BRYCGAMXSA-N 0.000 description 1
- QWRYEDTUBAMBON-UHFFFAOYSA-N [2,4-dichloro-3-(2-hydroxyethyl)phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CCO)Cl)C(=O)N1CCOCC1 QWRYEDTUBAMBON-UHFFFAOYSA-N 0.000 description 1
- NVYIMYFBPUMFNN-UHFFFAOYSA-N [2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]phenyl]-[4-(2-methoxyethyl)piperazin-1-yl]methanone Chemical compound ClC1=C(C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl)C(=O)N1CCN(CC1)CCOC NVYIMYFBPUMFNN-UHFFFAOYSA-N 0.000 description 1
- YOLONXDHSMBQTJ-UHFFFAOYSA-N [2,4-dichloro-3-[[1,4-dimethyl-6-(trifluoromethyl)indol-2-yl]methyl]phenyl]-[4-(oxetan-3-yl)piperazin-1-yl]methanone Chemical compound ClC1=C(C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)C)Cl)C(=O)N1CCN(CC1)C1COC1 YOLONXDHSMBQTJ-UHFFFAOYSA-N 0.000 description 1
- CWBSCPVTABVSEI-UHFFFAOYSA-N [2,4-dichloro-3-[[1-methyl-6-(trifluoromethyl)indol-2-yl]methyl]phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CC=1N(C2=CC(=CC=C2C=1)C(F)(F)F)C)Cl)C(=O)N1CCOCC1 CWBSCPVTABVSEI-UHFFFAOYSA-N 0.000 description 1
- SKCIUTDOKGNNGB-UHFFFAOYSA-N [2,4-dichloro-3-[[4-methyl-6-(trifluoromethyl)-1H-indol-2-yl]methyl]phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CC=1NC2=CC(=CC(=C2C=1)C)C(F)(F)F)Cl)C(=O)N1CCOCC1 SKCIUTDOKGNNGB-UHFFFAOYSA-N 0.000 description 1
- ACHDVHJAGAJRRG-UHFFFAOYSA-N [2,4-dichloro-3-[[6-(trifluoromethyl)-1H-indol-2-yl]methyl]phenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CC=1NC2=CC(=CC=C2C=1)C(F)(F)F)Cl)C(=O)N1CCOCC1 ACHDVHJAGAJRRG-UHFFFAOYSA-N 0.000 description 1
- DRVGMTIOBCTOKB-UHFFFAOYSA-N [3-(1-hydroxyprop-2-ynyl)-2,4-dimethylphenyl]-morpholin-4-ylmethanone Chemical compound OC(C#C)C=1C(=C(C=CC=1C)C(=O)N1CCOCC1)C DRVGMTIOBCTOKB-UHFFFAOYSA-N 0.000 description 1
- AMJLVIVQNZBFMF-UHFFFAOYSA-N [3-[[1-(benzenesulfonyl)-4-methyl-6-(trifluoromethyl)indol-2-yl]-hydroxymethyl]-2,4-dichlorophenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1C(C=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1)O)Cl)C(=O)N1CCOCC1 AMJLVIVQNZBFMF-UHFFFAOYSA-N 0.000 description 1
- FCBWCBDLTOMAQS-UHFFFAOYSA-N [3-[[1-(benzenesulfonyl)-4-methyl-6-(trifluoromethyl)indol-2-yl]-hydroxymethyl]-2,4-dimethylphenyl]-morpholin-4-ylmethanone Chemical compound OC(C=1C(=C(C=CC=1C)C(=O)N1CCOCC1)C)C=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1 FCBWCBDLTOMAQS-UHFFFAOYSA-N 0.000 description 1
- WJUKQHUVHJKRFS-UHFFFAOYSA-N [3-[[1-(benzenesulfonyl)-4-methyl-6-(trifluoromethyl)indol-2-yl]methyl]-2,4-dichlorophenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CC=1N(C2=CC(=CC(=C2C=1)C)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1)Cl)C(=O)N1CCOCC1 WJUKQHUVHJKRFS-UHFFFAOYSA-N 0.000 description 1
- IYEKNMYRESTRNT-UHFFFAOYSA-N [3-[[1-(benzenesulfonyl)-6-(trifluoromethyl)indol-2-yl]-hydroxymethyl]-2,4-dichlorophenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1C(C=1N(C2=CC(=CC=C2C=1)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1)O)Cl)C(=O)N1CCOCC1 IYEKNMYRESTRNT-UHFFFAOYSA-N 0.000 description 1
- FGBRYKWKOGSVPJ-UHFFFAOYSA-N [3-[[1-(benzenesulfonyl)-6-(trifluoromethyl)indol-2-yl]methyl]-2,4-dichlorophenyl]-morpholin-4-ylmethanone Chemical compound ClC1=C(C=CC(=C1CC=1N(C2=CC(=CC=C2C=1)C(F)(F)F)S(=O)(=O)C1=CC=CC=C1)Cl)C(=O)N1CCOCC1 FGBRYKWKOGSVPJ-UHFFFAOYSA-N 0.000 description 1
- YKIYEKRLGCHTNK-UHFFFAOYSA-N [4-fluoro-1-methyl-6-(trifluoromethyl)indol-2-yl]methanol Chemical compound FC1=C2C=C(N(C2=CC(=C1)C(F)(F)F)C)CO YKIYEKRLGCHTNK-UHFFFAOYSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 229960001683 abetimus Drugs 0.000 description 1
- RSWGJHLUYNHPMX-ONCXSQPRSA-N abietic acid Chemical compound C([C@@H]12)CC(C(C)C)=CC1=CC[C@@H]1[C@]2(C)CCC[C@@]1(C)C(O)=O RSWGJHLUYNHPMX-ONCXSQPRSA-N 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 229960005339 acitretin Drugs 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000012042 active reagent Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 230000011759 adipose tissue development Effects 0.000 description 1
- 239000000048 adrenergic agonist Substances 0.000 description 1
- 229940126157 adrenergic receptor agonist Drugs 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- DCSBSVSZJRSITC-UHFFFAOYSA-M alendronate sodium trihydrate Chemical compound O.O.O.[Na+].NCCCC(O)(P(O)(O)=O)P(O)([O-])=O DCSBSVSZJRSITC-UHFFFAOYSA-M 0.000 description 1
- 229960004343 alendronic acid Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- IHUNBGSDBOWDMA-AQFIFDHZSA-N all-trans-acitretin Chemical compound COC1=CC(C)=C(\C=C\C(\C)=C\C=C\C(\C)=C\C(O)=O)C(C)=C1C IHUNBGSDBOWDMA-AQFIFDHZSA-N 0.000 description 1
- 208000002029 allergic contact dermatitis Diseases 0.000 description 1
- 208000028004 allergic respiratory disease Diseases 0.000 description 1
- 108010004614 allotrap Proteins 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- PECIYKGSSMCNHN-UHFFFAOYSA-N aminophylline Chemical compound NCCN.O=C1N(C)C(=O)N(C)C2=NC=N[C]21.O=C1N(C)C(=O)N(C)C2=NC=N[C]21 PECIYKGSSMCNHN-UHFFFAOYSA-N 0.000 description 1
- 229960003556 aminophylline Drugs 0.000 description 1
- 229960000836 amitriptyline Drugs 0.000 description 1
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- XYXNTHIYBIDHGM-UHFFFAOYSA-N ammonium thiosulfate Chemical compound [NH4+].[NH4+].[O-]S([O-])(=O)=S XYXNTHIYBIDHGM-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- NUZWLKWWNNJHPT-UHFFFAOYSA-N anthralin Chemical compound C1C2=CC=CC(O)=C2C(=O)C2=C1C=CC=C2O NUZWLKWWNNJHPT-UHFFFAOYSA-N 0.000 description 1
- 230000003092 anti-cytokine Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000003430 antimalarial agent Substances 0.000 description 1
- 229940033495 antimalarials Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000001483 arginine derivatives Chemical class 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- METKIMKYRPQLGS-UHFFFAOYSA-N atenolol Chemical class CC(C)NCC(O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-UHFFFAOYSA-N 0.000 description 1
- 229960002028 atropine sulfate Drugs 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 229940009100 aurothiomalate Drugs 0.000 description 1
- XJHSMFDIQHVMCY-UHFFFAOYSA-M aurothiomalic acid Chemical compound OC(=O)CC(S[Au])C(O)=O XJHSMFDIQHVMCY-UHFFFAOYSA-M 0.000 description 1
- 208000029618 autoimmune pulmonary alveolar proteinosis Diseases 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 229940003504 avonex Drugs 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- IPOKCKJONYRRHP-FMQUCBEESA-N balsalazide Chemical compound C1=CC(C(=O)NCCC(=O)O)=CC=C1\N=N\C1=CC=C(O)C(C(O)=O)=C1 IPOKCKJONYRRHP-FMQUCBEESA-N 0.000 description 1
- 229960004168 balsalazide Drugs 0.000 description 1
- 229960000560 balsalazide disodium Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- PPDJNZTUDFPAHX-UHFFFAOYSA-N benzyltrimethylammonium dichloroiodate Chemical compound Cl[I-]Cl.C[N+](C)(C)CC1=CC=CC=C1 PPDJNZTUDFPAHX-UHFFFAOYSA-N 0.000 description 1
- 108010014499 beta-2 Adrenergic Receptors Proteins 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- SNHRLVCMMWUAJD-SUYDQAKGSA-N betamethasone valerate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]1(C)C[C@@H]2O SNHRLVCMMWUAJD-SUYDQAKGSA-N 0.000 description 1
- 229960004311 betamethasone valerate Drugs 0.000 description 1
- 229940021459 betaseron Drugs 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940046731 calcineurin inhibitors Drugs 0.000 description 1
- 229960002882 calcipotriol Drugs 0.000 description 1
- LWQQLNNNIPYSNX-UROSTWAQSA-N calcipotriol Chemical compound C1([C@H](O)/C=C/[C@@H](C)[C@@H]2[C@]3(CCCC(/[C@@H]3CC2)=C\C=C\2C([C@@H](O)C[C@H](O)C/2)=C)C)CC1 LWQQLNNNIPYSNX-UROSTWAQSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229930003827 cannabinoid Natural products 0.000 description 1
- 239000003557 cannabinoid Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 229940107810 cellcept Drugs 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- CKMOQBVBEGCJGW-UHFFFAOYSA-L chembl1200760 Chemical compound [Na+].[Na+].C1=C(C([O-])=O)C(O)=CC=C1N=NC1=CC=C(C(=O)NCCC([O-])=O)C=C1 CKMOQBVBEGCJGW-UHFFFAOYSA-L 0.000 description 1
- RCTCWZRPYFBGLQ-KVBIMOIYSA-N chembl2105639 Chemical compound C([C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 RCTCWZRPYFBGLQ-KVBIMOIYSA-N 0.000 description 1
- XMEVHPAGJVLHIG-FMZCEJRJSA-N chembl454950 Chemical compound [Cl-].C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H]([NH+](C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O XMEVHPAGJVLHIG-FMZCEJRJSA-N 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000002559 chemokine receptor antagonist Substances 0.000 description 1
- DLGJWSVWTWEWBJ-HGGSSLSASA-N chondroitin Chemical compound CC(O)=N[C@@H]1[C@H](O)O[C@H](CO)[C@H](O)[C@@H]1OC1[C@H](O)[C@H](O)C=C(C(O)=O)O1 DLGJWSVWTWEWBJ-HGGSSLSASA-N 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- DIOIOSKKIYDRIQ-UHFFFAOYSA-N ciprofloxacin hydrochloride Chemical compound Cl.C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 DIOIOSKKIYDRIQ-UHFFFAOYSA-N 0.000 description 1
- 229960001229 ciprofloxacin hydrochloride Drugs 0.000 description 1
- 230000027288 circadian rhythm Effects 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- CBGUOGMQLZIXBE-XGQKBEPLSA-N clobetasol propionate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]1(C)C[C@@H]2O CBGUOGMQLZIXBE-XGQKBEPLSA-N 0.000 description 1
- 229960004703 clobetasol propionate Drugs 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 229940047766 co-trimoxazole Drugs 0.000 description 1
- 239000011280 coal tar Substances 0.000 description 1
- 229940053219 coal tar / salicylic acid Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229960004415 codeine phosphate Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- FZCHYNWYXKICIO-FZNHGJLXSA-N cortisol 17-valerate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)CO)(OC(=O)CCCC)[C@@]1(C)C[C@@H]2O FZCHYNWYXKICIO-FZNHGJLXSA-N 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 229940109248 cromoglycate Drugs 0.000 description 1
- IMZMKUWMOSJXDT-UHFFFAOYSA-N cromoglycic acid Chemical compound O1C(C(O)=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C(O)=O)O2 IMZMKUWMOSJXDT-UHFFFAOYSA-N 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000002993 cycloalkylene group Chemical group 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 239000003260 cyclooxygenase 1 inhibitor Substances 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 229960003662 desonide Drugs 0.000 description 1
- WBGKWQHBNHJJPZ-LECWWXJVSA-N desonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O WBGKWQHBNHJJPZ-LECWWXJVSA-N 0.000 description 1
- 229960002593 desoximetasone Drugs 0.000 description 1
- VWVSBHGCDBMOOT-IIEHVVJPSA-N desoximetasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@H](C(=O)CO)[C@@]1(C)C[C@@H]2O VWVSBHGCDBMOOT-IIEHVVJPSA-N 0.000 description 1
- SSQJFGMEZBFMNV-PMACEKPBSA-N dexanabinol Chemical compound C1C(CO)=CC[C@@H]2C(C)(C)OC3=CC(C(C)(C)CCCCCC)=CC(O)=C3[C@H]21 SSQJFGMEZBFMNV-PMACEKPBSA-N 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- ASWXNYNXAOQCCD-UHFFFAOYSA-N dichloro(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Cl)(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 ASWXNYNXAOQCCD-UHFFFAOYSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- KPHWPUGNDIVLNH-UHFFFAOYSA-M diclofenac sodium Chemical compound [Na+].[O-]C(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl KPHWPUGNDIVLNH-UHFFFAOYSA-M 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229960002124 diflorasone diacetate Drugs 0.000 description 1
- BOBLHFUVNSFZPJ-JOYXJVLSSA-N diflorasone diacetate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H](C)[C@@](C(=O)COC(C)=O)(OC(C)=O)[C@@]2(C)C[C@@H]1O BOBLHFUVNSFZPJ-JOYXJVLSSA-N 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- HYPPXZBJBPSRLK-UHFFFAOYSA-N diphenoxylate Chemical compound C1CC(C(=O)OCC)(C=2C=CC=CC=2)CCN1CCC(C#N)(C=1C=CC=CC=1)C1=CC=CC=C1 HYPPXZBJBPSRLK-UHFFFAOYSA-N 0.000 description 1
- 229960004192 diphenoxylate Drugs 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 229960002311 dithranol Drugs 0.000 description 1
- MVCOAUNKQVWQHZ-UHFFFAOYSA-N doramapimod Chemical compound C1=CC(C)=CC=C1N1C(NC(=O)NC=2C3=CC=CC=C3C(OCCN3CCOCC3)=CC=2)=CC(C(C)(C)C)=N1 MVCOAUNKQVWQHZ-UHFFFAOYSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 230000002497 edematous effect Effects 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 239000003602 elastase inhibitor Substances 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 201000002491 encephalomyelitis Diseases 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 239000002662 enteric coated tablet Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HVJJYOAPXBPQQV-UHFFFAOYSA-N ethyl 2-azidoacetate Chemical compound CCOC(=O)CN=[N+]=[N-] HVJJYOAPXBPQQV-UHFFFAOYSA-N 0.000 description 1
- ROFKLBRCQTUZGH-UHFFFAOYSA-N ethyl 3,5-dichloro-4-formylpyridine-2-carboxylate Chemical compound ClC=1C(=NC=C(C=1C=O)Cl)C(=O)OCC ROFKLBRCQTUZGH-UHFFFAOYSA-N 0.000 description 1
- YQONNJPKBHDJJM-UHFFFAOYSA-N ethyl 3-methyl-4-oxopiperidine-1-carboxylate Chemical compound CCOC(=O)N1CCC(=O)C(C)C1 YQONNJPKBHDJJM-UHFFFAOYSA-N 0.000 description 1
- PXBYFCRSEFPAKW-UHFFFAOYSA-N ethyl 3-methylpiperidine-4-carboxylate Chemical compound CCOC(=O)C1CCNCC1C PXBYFCRSEFPAKW-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 229960004511 fludroxycortide Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229940043075 fluocinolone Drugs 0.000 description 1
- 229960001347 fluocinolone acetonide Drugs 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 125000005643 gamma-butyrolacton-4-yl group Chemical group 0.000 description 1
- 229940044627 gamma-interferon Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- 229940015045 gold sodium thiomalate Drugs 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 229960002383 halcinonide Drugs 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 230000000887 hydrating effect Effects 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229960000631 hydrocortisone valerate Drugs 0.000 description 1
- USZLCYNVCCDPLQ-UHFFFAOYSA-N hydron;n-methoxymethanamine;chloride Chemical compound Cl.CNOC USZLCYNVCCDPLQ-UHFFFAOYSA-N 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 229960002927 hydroxychloroquine sulfate Drugs 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 229960001550 hyoscyamine sulfate Drugs 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- JXXWJMUPNZDILL-UHFFFAOYSA-N imidazo[4,5-b]pyridin-2-one Chemical class C1=CC=NC2=NC(=O)N=C21 JXXWJMUPNZDILL-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 229940073062 imuran Drugs 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 230000003960 inflammatory cascade Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 229940030980 inova Drugs 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940074383 interleukin-11 Drugs 0.000 description 1
- 230000024949 interleukin-17 production Effects 0.000 description 1
- 108010074108 interleukin-21 Proteins 0.000 description 1
- 108010074109 interleukin-22 Proteins 0.000 description 1
- 229940118526 interleukin-9 Drugs 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 229960004958 ketotifen Drugs 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 229960004393 lidocaine hydrochloride Drugs 0.000 description 1
- YECIFGHRMFEPJK-UHFFFAOYSA-N lidocaine hydrochloride monohydrate Chemical compound O.[Cl-].CC[NH+](CC)CC(=O)NC1=C(C)C=CC=C1C YECIFGHRMFEPJK-UHFFFAOYSA-N 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- PGYPOBZJRVSMDS-UHFFFAOYSA-N loperamide hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)N(C)C)CCN(CC1)CCC1(O)C1=CC=C(Cl)C=C1 PGYPOBZJRVSMDS-UHFFFAOYSA-N 0.000 description 1
- 229960002983 loperamide hydrochloride Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 210000005210 lymphoid organ Anatomy 0.000 description 1
- 238000012792 lyophilization process Methods 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- FRIJBUGBVQZNTB-UHFFFAOYSA-M magnesium;ethane;bromide Chemical compound [Mg+2].[Br-].[CH2-]C FRIJBUGBVQZNTB-UHFFFAOYSA-M 0.000 description 1
- LROBJRRFCPYLIT-UHFFFAOYSA-M magnesium;ethyne;bromide Chemical compound [Mg+2].[Br-].[C-]#C LROBJRRFCPYLIT-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000010907 mechanical stirring Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229940051129 meperidine hydrochloride Drugs 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 101150089110 metN gene Proteins 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229960004469 methoxsalen Drugs 0.000 description 1
- SIDXTUGPBDOKID-UHFFFAOYSA-N methyl 2,4-dichloro-3-(3-chloro-2-oxobutyl)benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CC(C(C)Cl)=O)Cl SIDXTUGPBDOKID-UHFFFAOYSA-N 0.000 description 1
- NABKPKYIJXQGKY-UHFFFAOYSA-N methyl 2,4-dichloro-3-[[4-methyl-6-(trifluoromethyl)-1H-indol-2-yl]methyl]benzoate Chemical compound ClC1=C(C(=O)OC)C=CC(=C1CC=1NC2=CC(=CC(=C2C=1)C)C(F)(F)F)Cl NABKPKYIJXQGKY-UHFFFAOYSA-N 0.000 description 1
- XBTVTQJQOYOXGH-UHFFFAOYSA-N methyl 2-(2-azaspiro[3.3]heptan-6-yl)acetate Chemical compound COC(=O)CC1CC2(CNC2)C1 XBTVTQJQOYOXGH-UHFFFAOYSA-N 0.000 description 1
- RXYCUKSOYJWGPE-UHFFFAOYSA-N methyl 2-azidoacetate Chemical compound COC(=O)CN=[N+]=[N-] RXYCUKSOYJWGPE-UHFFFAOYSA-N 0.000 description 1
- VOUIHMBRJVKANW-UHFFFAOYSA-N methyl 2-piperidin-4-ylacetate Chemical compound COC(=O)CC1CCNCC1 VOUIHMBRJVKANW-UHFFFAOYSA-N 0.000 description 1
- ADBDFGZYGJGDNJ-UHFFFAOYSA-N methyl 2-piperidin-4-ylacetate;hydrochloride Chemical compound Cl.COC(=O)CC1CCNCC1 ADBDFGZYGJGDNJ-UHFFFAOYSA-N 0.000 description 1
- PZMMESDUAFUZSK-UHFFFAOYSA-N methyl 3,5-dichloro-4-formylbenzoate Chemical compound COC(=O)C1=CC(Cl)=C(C=O)C(Cl)=C1 PZMMESDUAFUZSK-UHFFFAOYSA-N 0.000 description 1
- WZOYBQZGQLXQME-UHFFFAOYSA-N methyl 5-(1-hydroxyprop-2-ynyl)-4,6-dimethylpyridine-3-carboxylate Chemical compound OC(C#C)C=1C(=NC=C(C(=O)OC)C=1C)C WZOYBQZGQLXQME-UHFFFAOYSA-N 0.000 description 1
- RHMACOFOSAMNHB-UHFFFAOYSA-N methyl 5-bromo-4,6-dimethylpyridine-3-carboxylate Chemical compound COC(=O)C1=CN=C(C)C(Br)=C1C RHMACOFOSAMNHB-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229960000334 methylprednisolone sodium succinate Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 208000008275 microscopic colitis Diseases 0.000 description 1
- 229960002853 midazolam hydrochloride Drugs 0.000 description 1
- PLYSCVSCYOQVRP-UHFFFAOYSA-N midazolam hydrochloride Chemical compound Cl.C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F PLYSCVSCYOQVRP-UHFFFAOYSA-N 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004023 minocycline Drugs 0.000 description 1
- DYKFCLLONBREIL-KVUCHLLUSA-N minocycline Chemical compound C([C@H]1C2)C3=C(N(C)C)C=CC(O)=C3C(=O)C1=C(O)[C@@]1(O)[C@@H]2[C@H](N(C)C)C(O)=C(C(N)=O)C1=O DYKFCLLONBREIL-KVUCHLLUSA-N 0.000 description 1
- 239000010446 mirabilite Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 229960002744 mometasone furoate Drugs 0.000 description 1
- WOFMFGQZHJDGCX-ZULDAHANSA-N mometasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(Cl)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)C(=O)CCl)C(=O)C1=CC=CO1 WOFMFGQZHJDGCX-ZULDAHANSA-N 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 229960004715 morphine sulfate Drugs 0.000 description 1
- GRVOTVYEFDAHCL-RTSZDRIGSA-N morphine sulfate pentahydrate Chemical compound O.O.O.O.O.OS(O)(=O)=O.O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O.O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O GRVOTVYEFDAHCL-RTSZDRIGSA-N 0.000 description 1
- 125000005858 morpholino(C2-C3)alkyl group Chemical group 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- WDBIZWZHDIQGBT-UHFFFAOYSA-N n-[2-methyl-6-nitro-4-(trifluoromethyl)phenyl]acetamide Chemical compound CC(=O)NC1=C(C)C=C(C(F)(F)F)C=C1[N+]([O-])=O WDBIZWZHDIQGBT-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229950006327 napsilate Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- RQTOOFIXOKYGAN-UHFFFAOYSA-N nedocromil Chemical compound CCN1C(C(O)=O)=CC(=O)C2=C1C(CCC)=C1OC(C(O)=O)=CC(=O)C1=C2 RQTOOFIXOKYGAN-UHFFFAOYSA-N 0.000 description 1
- 229960004398 nedocromil Drugs 0.000 description 1
- 230000003959 neuroinflammation Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- SBQLYHNEIUGQKH-UHFFFAOYSA-N omeprazole Chemical compound N1=C2[CH]C(OC)=CC=C2N=C1S(=O)CC1=NC=C(C)C(OC)=C1C SBQLYHNEIUGQKH-UHFFFAOYSA-N 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 230000000771 oncological effect Effects 0.000 description 1
- 230000005305 organ development Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 108090000629 orphan nuclear receptors Proteins 0.000 description 1
- 102000004164 orphan nuclear receptors Human genes 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229960000797 oxitropium Drugs 0.000 description 1
- NVOYVOBDTVTBDX-PMEUIYRNSA-N oxitropium Chemical compound CC[N+]1(C)[C@H]2C[C@@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)[C@H](CO)C1=CC=CC=C1 NVOYVOBDTVTBDX-PMEUIYRNSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 210000001986 peyer's patch Anatomy 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 239000008196 pharmacological composition Substances 0.000 description 1
- 239000002935 phosphatidylinositol 3 kinase inhibitor Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000005856 piperidino(C2-C3)alkyl group Chemical group 0.000 description 1
- ISWRGOKTTBVCFA-UHFFFAOYSA-N pirfenidone Chemical compound C1=C(C)C=CC(=O)N1C1=CC=CC=C1 ISWRGOKTTBVCFA-UHFFFAOYSA-N 0.000 description 1
- 229960003073 pirfenidone Drugs 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 206010035485 plasmacytosis Diseases 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229950005134 polycarbophil Drugs 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229940099511 polysorbate 65 Drugs 0.000 description 1
- 239000013312 porous aromatic framework Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 239000000276 potassium ferrocyanide Substances 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 229940116317 potato starch Drugs 0.000 description 1
- DQKXQSGTHWVTAD-UHFFFAOYSA-N pramocaine Chemical compound C1=CC(OCCCC)=CC=C1OCCCN1CCOCC1 DQKXQSGTHWVTAD-UHFFFAOYSA-N 0.000 description 1
- 229960001896 pramocaine Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- XXPDBLUZJRXNNZ-UHFFFAOYSA-N promethazine hydrochloride Chemical compound Cl.C1=CC=C2N(CC(C)N(C)C)C3=CC=CC=C3SC2=C1 XXPDBLUZJRXNNZ-UHFFFAOYSA-N 0.000 description 1
- 229960002244 promethazine hydrochloride Drugs 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 230000006825 purine synthesis Effects 0.000 description 1
- 239000003379 purinergic P1 receptor agonist Substances 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 235000008160 pyridoxine Nutrition 0.000 description 1
- 239000011677 pyridoxine Substances 0.000 description 1
- 125000005857 pyrrolidino(C2-C3)alkyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000000754 repressing effect Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960000759 risedronic acid Drugs 0.000 description 1
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 1
- 229960002586 roflumilast Drugs 0.000 description 1
- ZCBUQCWBWNUWSU-SFHVURJKSA-N ruboxistaurin Chemical compound O=C1NC(=O)C2=C1C(C1=CC=CC=C11)=CN1CCO[C@H](CN(C)C)CCN1C3=CC=CC=C3C2=C1 ZCBUQCWBWNUWSU-SFHVURJKSA-N 0.000 description 1
- 229950000261 ruboxistaurin Drugs 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 229940023036 salicylic acid / sulfur Drugs 0.000 description 1
- 235000015067 sauces Nutrition 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000002453 shampoo Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- BBMHARZCALWXSL-UHFFFAOYSA-M sodium dihydrogenphosphate monohydrate Chemical compound O.[Na+].OP(O)([O-])=O BBMHARZCALWXSL-UHFFFAOYSA-M 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 229910001379 sodium hypophosphite Inorganic materials 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 229940050865 sodium phosphate,monobasic,monohydrate Drugs 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- VIDRYROWYFWGSY-UHFFFAOYSA-N sotalol hydrochloride Chemical compound Cl.CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 VIDRYROWYFWGSY-UHFFFAOYSA-N 0.000 description 1
- OAVGBZOFDPFGPJ-UHFFFAOYSA-N sotrastaurin Chemical compound C1CN(C)CCN1C1=NC(C=2C(NC(=O)C=2C=2C3=CC=CC=C3NC=2)=O)=C(C=CC=C2)C2=N1 OAVGBZOFDPFGPJ-UHFFFAOYSA-N 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 1
- 229960000894 sulindac Drugs 0.000 description 1
- 230000000475 sunscreen effect Effects 0.000 description 1
- 239000000516 sunscreening agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940065721 systemic for obstructive airway disease xanthines Drugs 0.000 description 1
- 229950004608 talampanel Drugs 0.000 description 1
- ZMELOYOKMZBMRB-DLBZAZTESA-N talmapimod Chemical compound C([C@@H](C)N(C[C@@H]1C)C(=O)C=2C(=CC=3N(C)C=C(C=3C=2)C(=O)C(=O)N(C)C)Cl)N1CC1=CC=C(F)C=C1 ZMELOYOKMZBMRB-DLBZAZTESA-N 0.000 description 1
- 239000011269 tar Substances 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960000565 tazarotene Drugs 0.000 description 1
- 229960000195 terbutaline Drugs 0.000 description 1
- 229960000331 teriflunomide Drugs 0.000 description 1
- UTNUDOFZCWSZMS-YFHOEESVSA-N teriflunomide Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC=C(C(F)(F)F)C=C1 UTNUDOFZCWSZMS-YFHOEESVSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229960004989 tetracycline hydrochloride Drugs 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- XOGGUFAVLNCTRS-UHFFFAOYSA-N tetrapotassium;iron(2+);hexacyanide Chemical compound [K+].[K+].[K+].[K+].[Fe+2].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-] XOGGUFAVLNCTRS-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 229950010980 tiplimotide Drugs 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- XFYDIVBRZNQMJC-UHFFFAOYSA-N tizanidine Chemical compound ClC=1C=CC2=NSN=C2C=1NC1=NCCN1 XFYDIVBRZNQMJC-UHFFFAOYSA-N 0.000 description 1
- 229960000488 tizanidine Drugs 0.000 description 1
- 229960001350 tofacitinib Drugs 0.000 description 1
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 229960004380 tramadol Drugs 0.000 description 1
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940062627 tribasic potassium phosphate Drugs 0.000 description 1
- KVSKGMLNBAPGKH-UHFFFAOYSA-N tribromosalicylanilide Chemical compound OC1=C(Br)C=C(Br)C=C1C(=O)NC1=CC=C(Br)C=C1 KVSKGMLNBAPGKH-UHFFFAOYSA-N 0.000 description 1
- 229950001807 tribromsalan Drugs 0.000 description 1
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 1
- DCGLONGLPGISNX-UHFFFAOYSA-N trimethyl(prop-1-ynyl)silane Chemical compound CC#C[Si](C)(C)C DCGLONGLPGISNX-UHFFFAOYSA-N 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 239000002447 tumor necrosis factor alpha converting enzyme inhibitor Substances 0.000 description 1
- 229950008396 ulobetasol propionate Drugs 0.000 description 1
- BDSYKGHYMJNPAB-LICBFIPMSA-N ulobetasol propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]2(C)C[C@@H]1O BDSYKGHYMJNPAB-LICBFIPMSA-N 0.000 description 1
- 229940045136 urea Drugs 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 229940011671 vitamin b6 Drugs 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 1
- CGTADGCBEXYWNE-JUKNQOCSSA-N zotarolimus Chemical compound N1([C@H]2CC[C@@H](C[C@@H](C)[C@H]3OC(=O)[C@@H]4CCCCN4C(=O)C(=O)[C@@]4(O)[C@H](C)CC[C@H](O4)C[C@@H](/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C3)OC)C[C@H]2OC)C=NN=N1 CGTADGCBEXYWNE-JUKNQOCSSA-N 0.000 description 1
- 229950009819 zotarolimus Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/16—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Epidemiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Ophthalmology & Optometry (AREA)
- Urology & Nephrology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Indole Compounds (AREA)
- Steroid Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un compuesto de Formula (I-a): **(Ver fórmula)** o una sal farmaceuticamente aceptable del mismo, en el que: G y J son independientemente CH o N; W es C o CRa, L1 esta conectado a W o Y; y A y E son independientemente C o N, siempre que ambos no sean N; V es CR3 o N; X es CRa, NRa, N u O; Y es C, CRa, NRa, N, O o S; Z es CR3 o N; o W es N o NRa, L1 esta conectado a W o Y; y A y E son independientemente C o N, siempre que ambos no sean N; V es CR3 o N; X es CRa, NRa o N; Y es C, CRa o N; Z es CR3 o N; o L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 y R2 son independientemente halogeno, -O-(C1-C3)alquilo, ciclopropilo, o (C1-C3)alquilo; cada R3 es independientemente H, -CF3, -CN, halogeno, -OCHF2, -OCF3, -C(O)N(Re)2, -NReCORe, -(C1-C3)alquilo, -O-(C1-C3)alquilo, -S-(C1-C3)alquilo, (C3-C6)cicloalquilo, o morfolinilo; R4 es (C1-C6)alquilo opcionalmente sustituido con uno o mas sustituyentes seleccionados independientemente de halo, -CO2H, y CO2(C1-C4)alquilo; o R4 es -NH-CH2-tetrahidro-2H-piranilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de -OH y O(C1-C4)alquilo; o R4 es -N(H)-(C3-C6)alquilo opcionalmente sustituido con uno o mas sustituyentes seleccionados independientemente de F, -OH, -COOH, y -CO2(C1-C4)alquilo; o R4 es (C3-C6)cicloalquilo opcionalmente sustituido con uno o mas sustituyentes seleccionados independientemente de F, -OH, -COOH y -CO2(C1-C4)alquilo; o R4 es azabiciclo[2,2,1]heptanilo o azabiciclo[3,1,0]hexanilo, cada uno opcionalmente sustituido con -CO2H o - CO2(C1-C4)alquilo; o R4 es azaspiro[3,3]heptanilo opcionalmente sustituido con -CH2CO2H, -CH2CO2CH3, -CO2H, -CO2 CH3; o R4 es 2-oxa-6-azaspiro[3,3]heptanilo, 2-oxa-7-azaspiro[3,5]nonanilo, o 2-oxa-8-azaspiro[4,5]decanilo, cada uno opcionalmente sustituido con -CH2CO2H; o R4 es azetidinilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de - CH3, -OH, -OCH3, -(C1-C4)alquileno-OH, -CH2OCH3, -(C1-C4) alquilenoO(C1-C4)alquilo, -CO2(C1-C4) alquilo,-CO2H, -(C1-C4)alquileno-CO2H, -(C1-C4) alquileno-CO2-(C1-C4)alquilo, -N(CH3)2, y grupos pirrolidinilo; o R4 es 1,2-diazepanilo o 1,4-diazepanilo, cada uno opcionalmente sustituido con -(C1-C4)alquileno-OH; o R4 es morfolinilo opcionalmente sustituido con =O; o R4 es piperazinilo, piperidinilo o pirrolidinilo, cada uno opcionalmente sustituido con uno o mas sustituyentes seleccionados independientemente de -halo, -CN, -(C1-C4)alquilo, -CH2-ciclopropil, -(C1-C4)alquileno-F, -CF3, - CH2CF3, -COOH, -(C1-C4)alquilenoCOOH, -CH(OH)CO2H, -COCH3, -CO2(C1-C4)alquilo,-CH(OH)CO2CH3, -(C1- C4)alquileno-C(=O)O(C1-C4)alquilo, -(C1-C4)alquileno-OH, -OH, -O(C1-C4)alquilo,-(C1-C4)alquilenoO(C1-C4)alquilo, -O(C1-C4)alquilenoCO2H, -O(C1-C4)alquilenoC(=O)O(C1-C4)alquilo, -CONHCH3, -SO2-(C1-C4)alquilo, -NH2, - NH(C1-C4)alquilo, -N((C1-C4)alquilo)2, acido ciclobutanocarboxilico, y oxetanilo; cada Ra es independientemente H, -C(O)CH3, (C1-C6)alquilo opcionalmente sustituido, (C3-C6)cicloalquilo opcionalmente sustituido o -S(O)2-fenilo; y Re es independientemente H o -(C1-C3)alquilo; siempre que no mas de dos de A, E, W, X y Y sean N.
Description
DESCRIPCIÓN
Moduladores del receptor nuclear (ROR) para el tratamiento de enfermedades inflamatorias y autoinmunes
Campo técnico
La presente invención se refiere a nuevos compuestos que modulan la actividad de Rorc y el receptor RORYt, y su uso como medicamentos.
Antecedentes de la invención
Las isoformas del receptor huérfano relacionado con el ácido retinoico (ROR) RORa, RORp y RORy son miembros de la superfamilia de receptores de hormonas nucleares esteroideas, y desempeñan papeles prominentes en una variedad de procesos biológicos que incluyen el desarrollo de órganos, inmunidad, homeostasis y metabolismo de los lípidos, y ritmos circadianos (Jetten y otros. NURSA 2009, 7, 1). Los miembros de la familia ROR están compuestos por un dominio de unión a ligando (LBD) y un dominio de unión a ADN (DBD). La unión a ligando provoca un cambio conformacional que modula la unión de proteínas correguladoras: los agonistas reclutan coactivadores; los antagonistas y los agonistas inversos interrumpen la unión de los coactivadores o aumentan la unión de los correpresores reprimiendo así la transcripción de genes diana (Fauber y otros. J. Med. Chem. 2014, 57, 5871).
El timo y del receptor huérfano relacionado con el ácido retinoico (RORy, también denominado RORc y NR1F3) es codificado por Rorc; RORy humano y de ratón comparten una alta homología de secuencia y una homología de sitio de unión casi idéntica (Jin, L. y otros. Mol. Endocrinol. 2010, 24, 923). El RORy de mamíferos existe en dos isoformas distintas, RORy y RORYt, que poseen LBD idénticos y difieren solo en sus secuencias W-terminales (Medvedev y otros. Gene 1996, 181, 199). La expresión de la isoforma RORYt está restringida a los órganos linfoides, incluido el timo, mientras que RORy se expresa más ampliamente (hígado, músculo, riñón), similar al RORa que también se encuentra en el cerebro y el tejido adiposo (Kurebayashi, S. y otros. Proc. Natl. Acad. Sci., USA 2000, 97, 10132). RORp se localiza en la corteza cerebral (Hirose, T. y otros. Biochem. Biophys Res. Commun. 1993, 194, 1371). RORYt es crítico para el desarrollo de ganglios linfáticos y parches de Peyer y para la diferenciación normal de células T helper-17 (Th17), células T y5 y células LTi (Sun y otros. Science 2000, 288, 2369).
RORYt es un factor de transcripción obligatorio que controla la diferenciación de las células T CD4+ vírgenes en el linaje Th17, y regula la transcripción de la citocina efectora IL-17 en las células Th17 y las células de la respuesta inmune innata tanto en roedores como en humanos (Ivanov, I. y otros. Cell 2006, 126, 1121). Las citocinas proinflamatorias que incluyen IL-17A, IL-17F e IL-22 producidas por células Th17 y otros linfocitos RORYt+ activan y dirigen la respuesta inmune a los patógenos extracelulares (Eberl, G. y otros. Nat. Immunol. 2004, 5, 64). La interrupción de RORy por la ablación genética de Rorc en ratones atenúa la gravedad de la enfermedad en modelos murinos de autoinmunidad e inflamación, incluida la encefalomielitis autoinmune experimental (EAE) inducida por péptido antigénico, dermatitis psoriasiforme inducida por imiquimod y enfermedad alérgica de las vías respiratorias. La desregulación de la transcripción y secreción de IL-17 ha implicado múltiples trastornos autoinmunes humanos, que incluyen psoriasis, artritis reumatoide, enfermedad inflamatoria intestinal (EII), asma y esclerosis múltiple (EM) (por ejemplo, ver: Yang, X. y otros. Inmunidad 2008, 28, 29; Pantelyushin, S. y otros, J. Clin. Invest. 2012, 122, 2252; Leppkes, M. y otros. Gastroenterología 2009, 136, 257; y Tilley, J. y otros, J. Immunol. 2007, 178, 3208). El resultado de ensayos clínicos recientes con anticuerpos neutralizantes contra IL-17A y su receptor IL-17RA sirve para resaltar el papel de esta citocina en la patogénesis de la enfermedad de psoriasis (Papp, K. y otros. New. Engl. J. Med. 2012, 366, 1181; Leonardi, C. y otros. New. Engl. J. Med. 2012, 366, 1190;. La atenuación de la producción de IL-17 a partir de células T activadas y Th17, por ejemplo, mediante inhibición o RORYt, puede ofrecer un beneficio terapéutico similar.
El documento WO 2015036411 A1 (Hoffman La Roche) describe una serie de derivados de ceto-imidazopiridina como moduladores de RORc y establece que estos compuestos pueden usarse para el tratamiento de enfermedades inflamatorias tales como la artritis.
Existen agentes terapéuticos para tratar una variedad de enfermedades inflamatorias y autoinmunes, pero aún existe una importante necesidad médica no satisfecha en estas áreas terapéuticas. Dado el papel de IL-17 en la enfermedad humana y la validación de IL-17 y RORy como objetivos en modelos murinos de enfermedad, se contemplan compuestos capaces de modular la actividad de RORYt para proporcionar un beneficio terapéutico en el tratamiento de múltiples trastornos inmunes e inflamatorios.
Resumen de la invención
En una primera realización, la invención proporciona un compuesto de Fórmula (I-a)

Fórmula (la),
o una sal farmacéuticamente aceptable del mismo, en el que
G y J son independientemente CH o N;
W es C o CRa , L1 está conectado a W o Y; y
A y E son independientemente C o N, siempre que ambos no sean N;
V es CR3 o N;
X es CRa , NRa, N u O;
Y es C, CRa, NRa , N, O o S;
Z es CR3 o N; o
W es N o NRa , L1 está conectado a W o Y; y
A y E son independientemente C o N, siempre que ambos no sean N;
V es CR3 o N;
X es CRa , NRa o N;
Y es C, CRa o N;
Z es CR3 o N; o
L1 es -CH2- o C(O);
L2 es -C(O)-;
R1 y R2 son independientemente halógeno, -O-(Cr C3)alquilo, ciclopropilo, o (Cr C3)alquilo;
cada R3 es independientemente -H, -CF3 , -CN, halógeno, -OCHF2 , -OCF3 , -C(O)N(Re)2 , NReCORe , -(Cr C3)alquilo, -O-(C1-C3)alquilo, -S-(C1-C3)alquilo, (C3-C6)cicloalquilo o morfolinilo;
R4 es (C1-C6)alquilo opcionalmente sustituido con uno o más halo, -CO2 H, o -CO2(C1-C4)alquilo; o
R4 es -NH-CH2-tetrahidro-2H-piranilo opcionalmente sustituido con uno o dos -OH o -O(Cr C4)alquilo; o
R4 es -N(H)-(C3-C6)cicloalquilo opcionalmente sustituido con uno o más F, -OH, -COOH o -CO2(C1-C4)alquilo; o R4 es (C3-C6)cicloalquilo opcionalmente sustituido con uno o más F, -OH, -COOH, o -CO2(C1-C4)alquilo; o
R4 es azabiciclo[2,2,1]heptanilo o azabiciclo[3,1,0]hexanilo, cada uno opcionalmente sustituido con -CO2H o -CO2(C1-C4)alquilo; o
R4 es azaspiro[3,3]heptanilo opcionalmente sustituido con -CH2CO2 H, -CH2CO2CH3 , -CO2 H, -CO2 CH3 ; o
R4 es 2-oxa-6-azaspiro[3,3]heptanilo, 2-oxa-7-azaspiro[3,5]nonanilo, o 2-oxa-8-azaspiro[4,5]decanilo, cada uno opcionalmente sustituido con -CH2CO2 H; o
R4 es azetidinilo opcionalmente sustituido con uno o dos grupos -CH3 , -OH, -OCH3 , -(C 1-C4)alquileno-OH, -CH2OCH3 , -(C1-C4)alquilenoO(C1 -C4)alquilo, -CO2(C1-C4)alquilo, -CO2 H, -(C1-C4)alquileno-CO2 H, -(Cr C4)alquileno-CO2-(Cr C4)alquilo, -N(CH3)2 , o pirrolidinilo; o
R4 es 1,2-diazepanilo o 1,4-diazepanilo, cada uno opcionalmente sustituido con -(C 1-C4)alquileno-OH; o
R4 es morfolinilo opcionalmente sustituido con =O; o
R4 es piperazinilo, piperidinilo o pirrolidinilo, cada uno opcionalmente sustituido con uno o más -halo, -CN, -(C 1-C4)alquilo, -CH2-ciclopropilo, -(C1-C4)alquileno-F, -CF3 , -CH2CF3 , -COOH, -(C1-C4)alquileno COOH, -CH(OH)CO2 H, -COCH3 , -CO2(C1-C4)alquilo, -CH(OH)CO2CH3 , -(C1-C4)alquileno -C(=O)O(C1-C4)alquilo, -(C1-C4)alquileno -OH, -OH, -O(Cr C4)alquilo, -(C1-C4)alquileno O(C1-C4)alquilo, -O(C1-C4)alquilenoCO2 H, -O(C1-C4)alquileno C(=O)O(C1-C4)aquilo, -CONHCH3, -SO2-(C1-C4)alquilo -NH2 , -NH(C1-C4)alquilo, -N((C1-C4)alquilo)2 , ácido ciclobutanocarboxílico, u oxetanilo cada Ra es independientemente H, -C(O)CH3 , (C1-C6)alquilo opcionalmente sustituido, (C3-C6)cicloalquilo opcionalmente sustituido o -S(O)2-fenilo; y
Re es independientemente H o (C 1-C3)alquilo;
siempre que no más de dos de A, E, W, X y Y sean N.
En una segunda realización, la invención proporciona un compuesto de acuerdo con cualquiera de las realizaciones anteriores, en el que R1 y R2 son ambos halo o -CH3.
En una tercera realización, la invención proporciona un compuesto de acuerdo con cualquiera de las realizaciones anteriores, en el que W es C o CH, A es C y E es C.
En una cuarta realización, la invención proporciona un compuesto de acuerdo con cualquiera de las realizaciones anteriores, en el que G es CH.
En una quinta realización, la invención proporciona un compuesto de acuerdo con cualquiera de las realizaciones anteriores, en el que J es CH.
En una sexta realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es CH; X es CRa , en el que Ra es H o -CH3 ; Y es N; A es C; E es C; V es CR3 , en el que R3 es H; Z es N o CR3 , en el que R3 es H, -CH3 , o -CF3 ; L1 está conectado a Y; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl o -CH3 ; R2 es -Cl o -CH3 ; cada R3 es independientemente H, -F, -Cl, -CN, -CH3 , -CH(CH3)2 , -CF3 , -OCHF2 , -OCF3 , -SCH3 , o ciclopropilo; y R4 es 2-oxa-6-azaspiro[3,3]heptanilo opcionalmente sustituido, piperidinilo opcionalmente sustituido.
En una séptima realización, la invención proporciona un compuesto de acuerdo con la sexta realización en el que el R42-oxa-6-azaspiro[3,3]heptanilo opcionalmente sustituido está opcionalmente sustituido por -CH2CO2 H.
En una octava realización, la invención proporciona un compuesto de acuerdo con la sexta realización en el que R4 piperidinilo opcionalmente sustituido está opcionalmente sustituido por -CH3 , -COOH, -CH2CO2 H, -CH2CH2CO2 H, -CO2CH3 , -CO2CH2CH3 , o -OH.
En una novena realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es CH; X es N; Y es N; Z es cR3; A es C; E es C; V es CR3; y L1 está conectado a Y.
En una décima realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es C; X es NRa , en el que Ra es H o -CH3; Y es CRa, en el que Ra es H; Z es N; A es C; E es C; V es CR3 , en el que R3 es H; L1 está conectado a W; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CH3 , o -CF3 ; y R4 es morfolinilo, opcionalmente sustituido, piperazinilo opcionalmente sustituido o piperidinilo opcionalmente sustituido.
En una undécima realización, la invención proporciona un compuesto de acuerdo con la novena realización, en el que el R4 piperazinilo opcionalmente sustituido está opcionalmente sustituido con -SO2CH3 ; el piperidinilo opcionalmente sustituido está opcionalmente sustituido por -CN, -CH3 , -OH, -COOH, -CH2COOH, -CH2CH2OCH3 , o -SO2CH3,
En una duodécima realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es C; X es CR, en el que Ra es H o -CH3 ; Y es N; A es N; E es C; Z es N; V es CR3 , en el que R3 es H o -CH3 ; L1 está conectado a W; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -Cl, -CN, -CH3 , o -CF3 ; y R4 es morfolinilo, opcionalmente sustituido, piperazinilo opcionalmente sustituido (por ejemplo, sustituido con -CH3, -SO2CH3, o oxetanilo), o piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH2COOH, oxetanilo).
En una decimotercera realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es C; X es CRa , en el que Ra es H o -CH3 ; Y es N; Z es CR3 , en el que R3 es H; A es N; E es C; V es Cr3, en el que R3 es H; L1 es conectado a W; L1 es -CH2-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CN, -CH3 , o -CF3 ; R4 es morfolinilo opcionalmente sustituido, piperazinilo opcionalmente sustituido (por ejemplo, sustituido con oxetanilo), piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -OH, -OCH3 , -CH2OH, -CH2CH2OH, -CH2CH2OCH3 , -CH(OH)CH3 , -C(OH)(CH3)2 , -CH2CO2 H, -CH2CO2 Me, -N(CH3)2 , oxetanilo), o pirrolinilo opcionalmente sustituido (por ejemplo, sustituido con -Oh).
En una decimocuarta realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es N; X es CRa , en el que Ra es H o -CH3 ; Y es N; Z es CR3 , en el que R3 es H; A es C; E es C; V es Cr3, en el que R3 es H; L1 está connextado a W; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CH3 , o -CF3 ; y R4 es morfolinilo opcionalmente sustituido o piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH2COOH).
En una decimoquinta, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es N; X es CRa , en el que Ra es H o -CH3; Y es N; Z es CR3 , en el que R3 es H; E es C; V es CR3 , en el que R3 es H; L1 está conectado a Y; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CH3 , o -CF3; y R4 es morfolinilo opcionalmente sustituido o piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH2COOH, oxetanilo).
En una decimosexta realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es N; X es NRa , en el que Ra es H o -CH3 ; Y es C; Z es CR3 , en el que R3 es H; A es C; E es C; V es CR3 , en el que R3 es H; L1 está conectado a Y; L1 es -CH2-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl;
cada R3 es independientemente H, -CH3, o -CF3 ; y R4 es piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH2COOH).
En una decimoséptima realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es C; X es NRa, en el que Ra es H o CH3 ; Y es CRa , en el que Ra es H; Z es CR3 , en el que R3 es H; A es C; E es C; V es N; L1 está conectado a W; L1 es -CH2-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CN, -CF3 , o CH3 ; y R4 es morfolinilo opcionalmente sustituido o piperazinilo opcionalmente sustituido (por ejemplo, sustituido con oxetanilo).
En una decimoctava realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es C; X es CRa, en el que Ra es H o CH3 ; Y es N; Z es CR3, en el que R3 es H; A es C; E es C; V es N; L1 está conectado a Y; L1 es -CH2- o -C(O)-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CF3 , o -CH3; y R4 es morfolinilo opcionalmente sustituido o piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH2COOH).
En una decimonovena realización, la invención proporciona un compuesto de acuerdo con una cualquiera de las realizaciones primera a quinta, en el que W es CRa , en el que Ra es H; X es O o NRa , en el que Ra es H o -CH3; Y es C; Z es CR3, en el que R3 es H o -CF3 ; A es C; E es C; V es Cr3, en el que R3 es H; L1 está conextado a Y; L1 es -CH2-; L2 es -C(O)-; R1 es -Cl; R2 es -Cl; cada R3 es independientemente H, -CH3 , -CF3, o -OCF3 ; y R4 es piperidinilo opcionalmente sustituido (por ejemplo, sustituido con -CH3 , -COOH, -CH2COOH, -CH2CO2CH3 , -CO2CH2CH3).
En una vigésima realización, la invención proporciona un compuesto de acuerdo con cualquiera de las realizaciones anteriores, en el que el compuesto es:

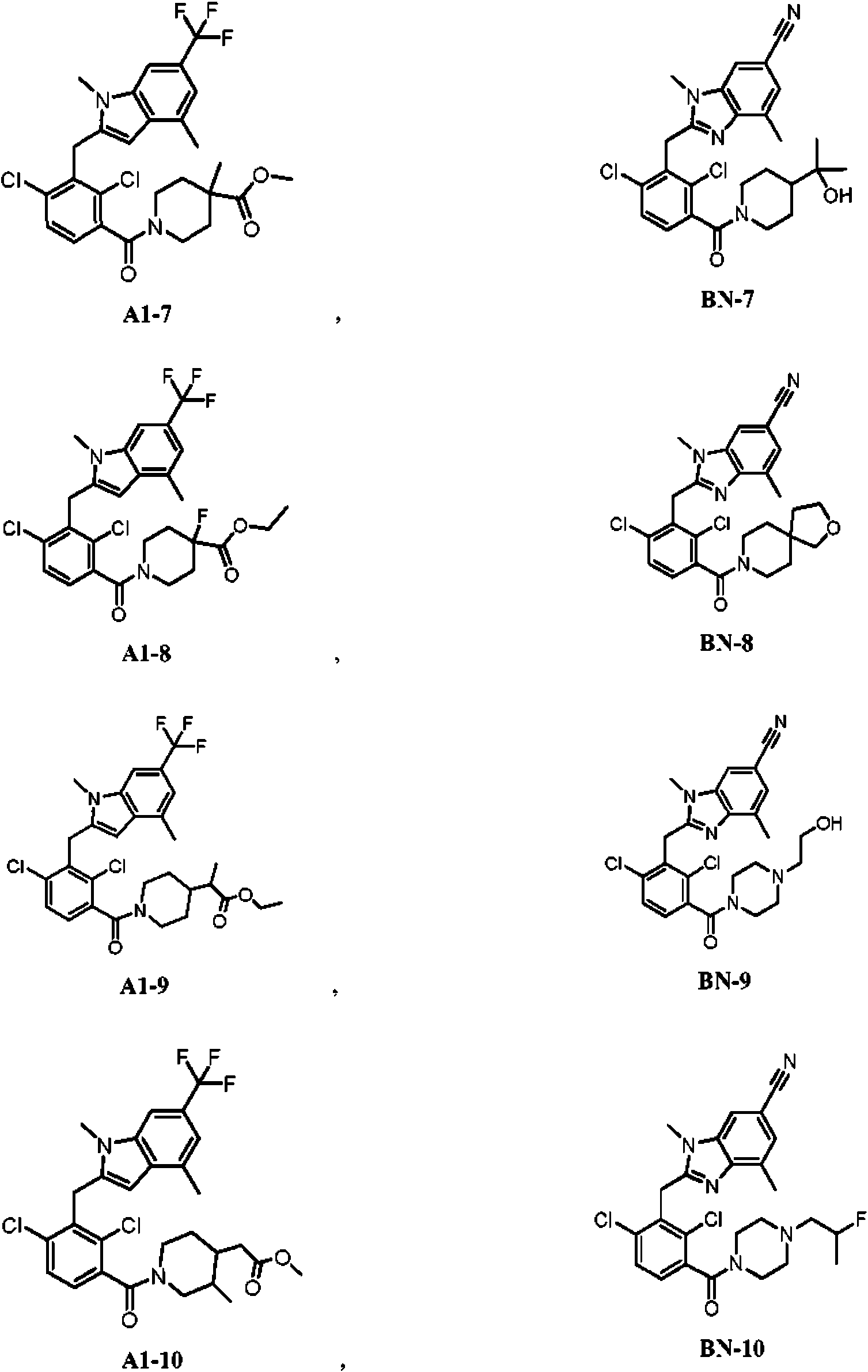



























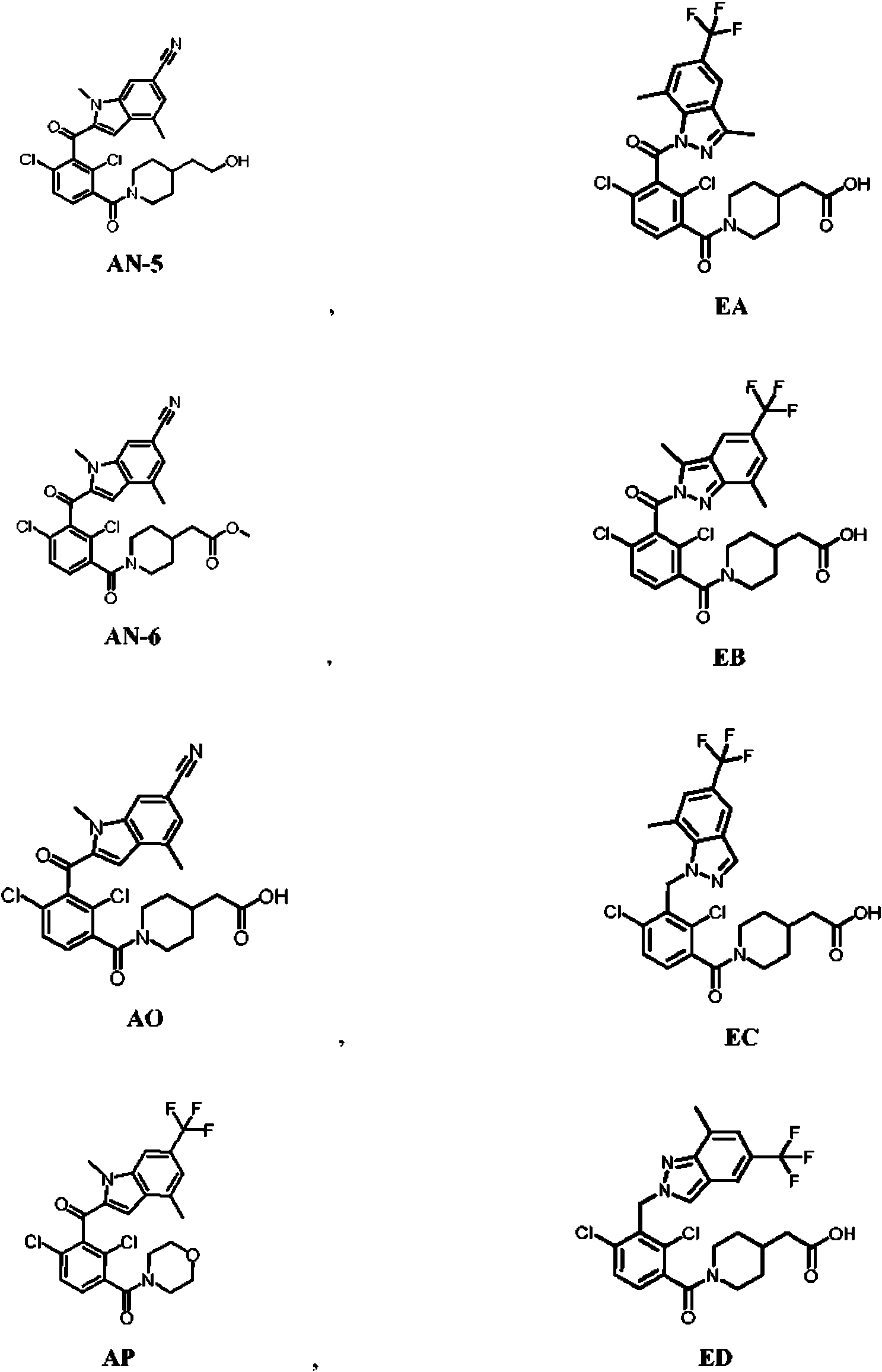







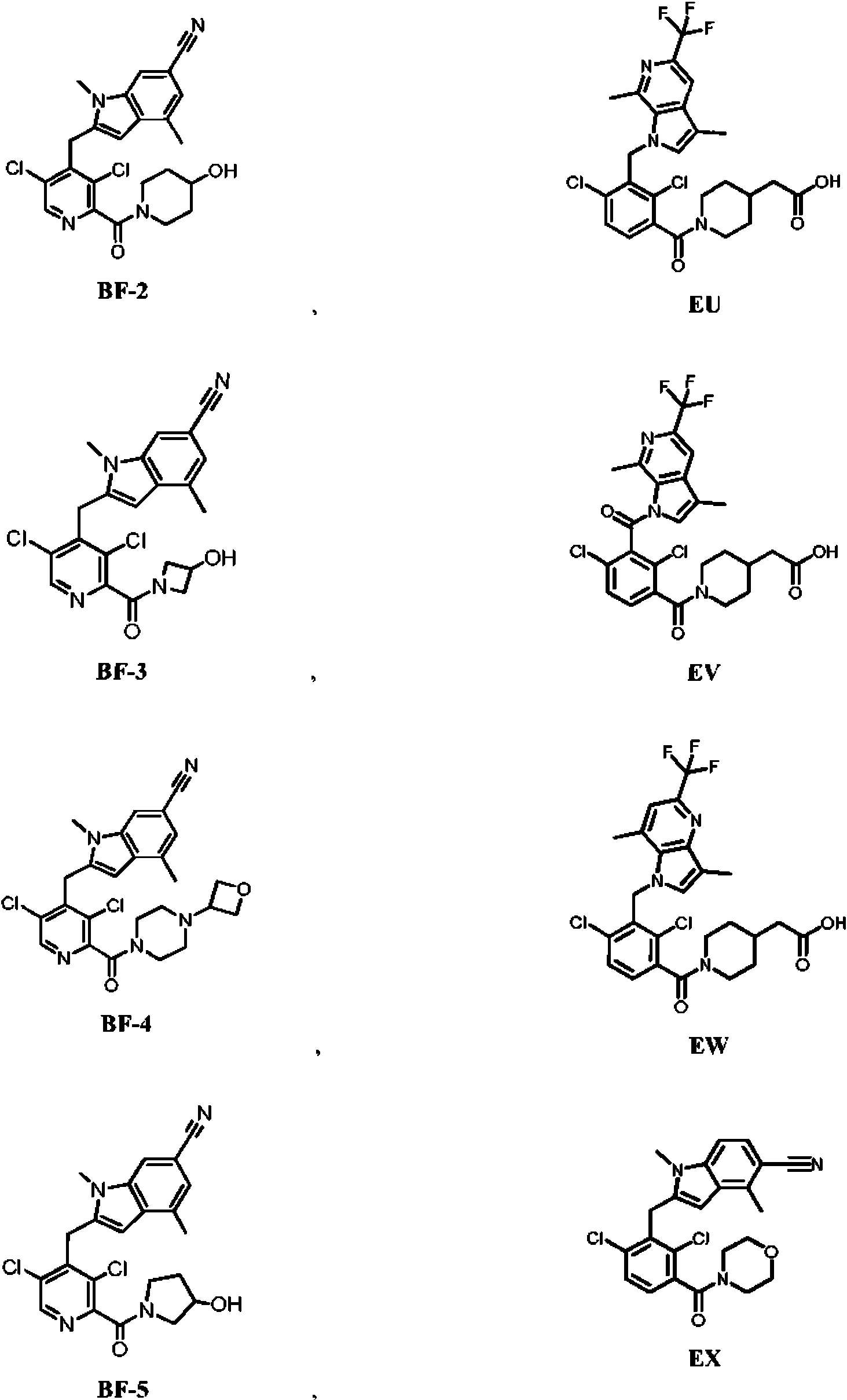





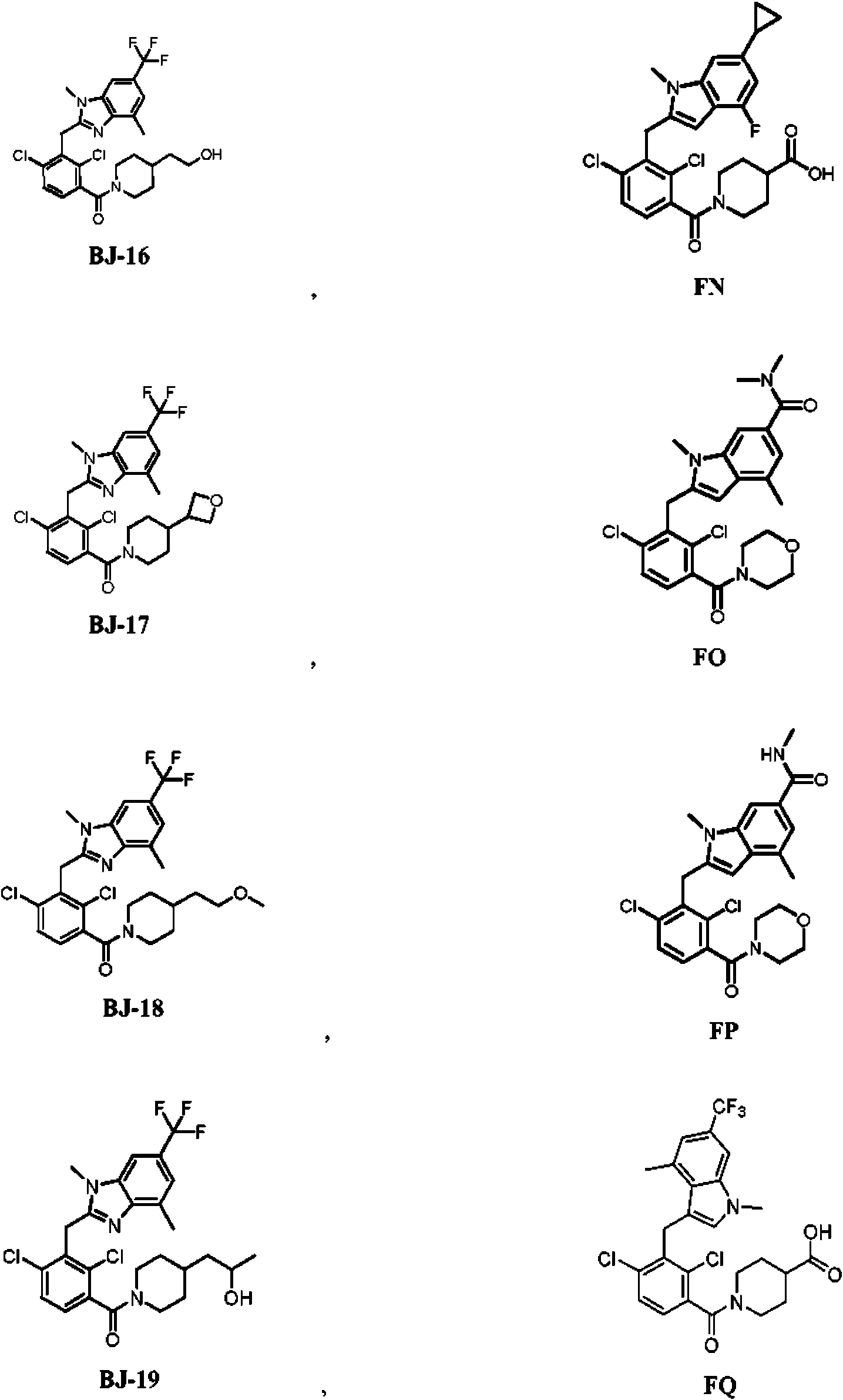


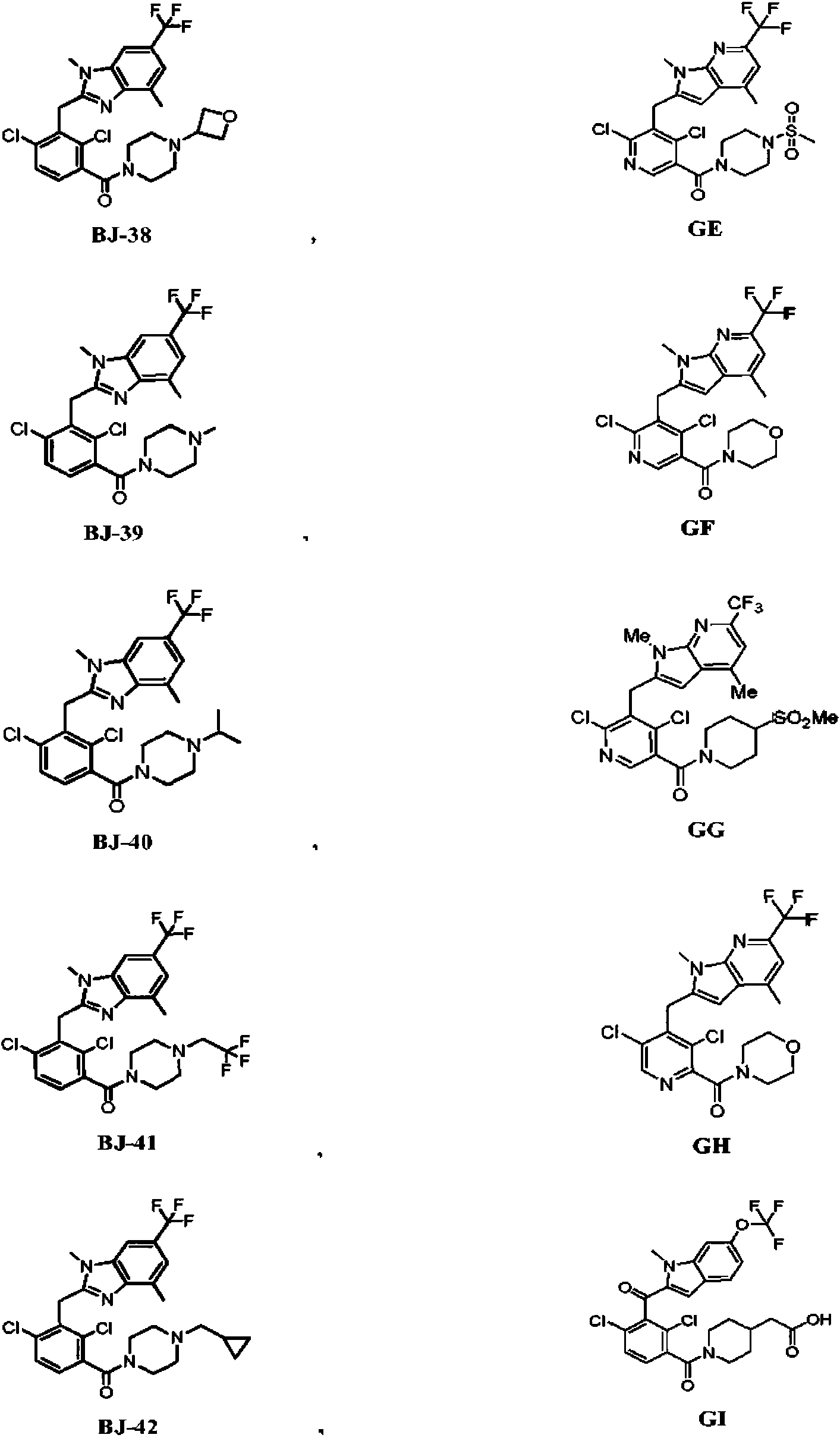

o una sal farmacéuticamente aceptable del mismo.
En una vigésimo primera realización, la invención proporciona una composición farmacéutica que comprende un compuesto de acuerdo con cualquiera de las realizaciones anteriores y uno o más excipientes farmacéuticamente aceptables.
En una vigésimo segunda realización, la invención proporciona un compuesto de cualquiera de las realizaciones anteriores para usar en un método de tratamiento de una enfermedad, en el que la enfermedad es artritis reumatoide, artritis reumatoide juvenil, psoriasis, artritis psoriásica, enfermedad de Crohn, enfermedad inflamatoria intestinal, colitis ulcerosa, espondilitis anquilosante, artritis reactiva, artritis asociada con enfermedad inflamatoria intestinal, espondiloartritis indiferenciada, lupus eritematoso sistémico, nefritis lúpica, uveítis, dermatitis atópica, esclerosis múltiple, espondiloartritis axial o hidradenitis supurativa.
En una vigésimo tercera realización, la invención proporciona un kit que comprende un producto empacado que comprende componentes con los que administrar un compuesto de cualquiera de las realizaciones anteriores para el tratamiento de un trastorno autoinmune.
En una vigésimo cuarta realización, la invención proporciona el kit de acuerdo con la trigésimo séptima realización en el que el producto empacado comprende un compuesto de acuerdo con cualquiera de las realizaciones e instrucciones de uso anteriores.
Descripción detallada
Los compuestos de la presente invención son útiles para prevenir, diagnosticar y tratar diversos trastornos médicos en humanos o animales. Los compuestos de la presente invención se usan para inhibir o reducir una o más actividades asociadas con los receptores RORYt, en relación con los receptores RORYt en ausencia de los mismos compuestos. Por lo tanto, en la presente se describe un compuesto de acuerdo con la Fórmula (I-a), forma estereoisomérica, N-óxido, sal, solvato, hidrato o composición farmacéutica farmacéuticamente aceptable como se describe en la presente descripción para su uso en el tratamiento de una enfermedad o trastorno seleccionado de una enfermedad o trastorno autoinmune, psoriasis, artritis psoriásica, asma, una enfermedad o trastorno alérgico, una enfermedad o trastorno metabólico y cáncer. En ciertas realizaciones, la enfermedad o trastorno autoinmune se selecciona de psoriasis, artritis psoriásica, espondilitis anquilosante, artritis reumatoide, esclerosis múltiple, enfermedad inflamatoria intestinal, enfermedad de Crohn, colitis ulcerosa, uveítis, enfermedad de injerto contra huésped y lupus. En ciertas realizaciones, la enfermedad o trastorno alérgico se selecciona de dermatitis atópica, rinitis alérgica, asma o enfermedad pulmonar obstructiva crónica (EPOC). En ciertas realizaciones, la enfermedad o trastorno metabólico se selecciona de obesidad, resistencia a la insulina inducida por la obesidad, aterosclerosis y diabetes tipo II. En ciertas realizaciones, la enfermedad o trastorno oncológico es el melanoma.
En una realización, la enfermedad o trastorno es psoriasis o artritis psoriásica. Ver, por ejemplo: Pantelyushin, S. y otros. "RORYt+ innate lymphocytes and y5 T cells initiate psoriasiform plaque formation in mice" J. Clin. Invest. 2012, 122: 2252; y Raychaudhuri, S. y otros. "Role of IL-17 in psoriasis and psoriatic arthritis," Clin. Rev. Allergy Immunol. 2013, 44: 183.
En una realización, la enfermedad o trastorno es la dermatitis atópica. Ver, por ejemplo: Ma, L. y otros. "The imbalance of Th17 cells and CD4+CD25highFoxp3+ Treg cells in patients with atopic dermatitis" J. Eur. Acad. Dermatol. Venereol.
2014, 28: 1079; y Peiser, M. "Role of Th17 cells in skin inflammation of allergic contact dermatitis" Clin. Dev. Immunol.
2013, 261037.
En otra realización, la enfermedad o trastorno es la artritis reumatoide. Ver, por ejemplo: Park, T.-Y. y otros. "RORYtspecific transcriptional interactomic inhibition suppresses autoimmunity associated with TH17 cells" Proc. Natl. Acad. Sci. USA 2014, 111:18673; y Solt, L. y otros. "Action of RORs and their ligands in (patho)\physiology" Trends Endocrinol. Metab. 2012, 23: 619; y Chang, M. y otros. "Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor y es therapeutic in the collagen-induced arthritis experimental model" Arthritis Rheumatol., 2014, 66:579.
En otra realización, la enfermedad o trastorno es la espondilitis anquilosante. Ver, por ejemplo: Bidad, K. y otros "Effect of all-transretinoic acid on Th17 and T regulatory cell subsets in patients with ankylosing spondylitis" J. Rheumatol. 2013, 40: 476; y Toussirot, E. y otros. "The IL-23/Thl7 pathway as a therapeutic target in chronic inflammatory diseases," Inflamm. Allergy Drug Targets 2012, 1:159.
En otra realización, la enfermedad o trastorno es la esclerosis múltiple. Ver, por ejemplo: Martinez, N. y otros. "RORYt, but not T-bet, overexpression exacerbates an autoimmune model for multiple sclerosis" J. Neuroimmunol. 2014, 276: (1/2) 142-149; y Codarri, L. y otros., "RORYt drives production of the cytokine GM-CSF in helper T cells, which es essential for the effector phase of autoimmune neuroinflammation" Nat. Immunol. 2011, 12: 560.
En otra realización, la enfermedad o trastorno es la enfermedad inflamatoria intestinal. Ver, por ejemplo: Troncone, E. y otros. "Th17 cytokines in inflammatory bowel diseases: discerning the good from the bad" Intl. Rev. Immunol. 2013, 32:526; y Leppkes, M. y otros. "RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F" Gastroenterology 2009, 136:257-267.
En otra realización, las enfermedades o trastornos son colitis ulcerosa o enfermedad de Crohn. Ver, por ejemplo: Dong, Z. y otros. "Aberrant expression of circulating Th17, Th1 and Tc1 cells in patients with active and inactive ulcerative colitis" Intl. J. Mol. Med. 2013, 31:989; and Kumawat, A. y otros. "Microscopic colitis patients demonstrate a mixed Th17/Tc17 and Th1/Tc1 mucosal cytokine profile" Mol Immunol. 2013, 55:355.
En otra realización, la enfermedad o trastorno es la uveítis autoinmune. Ver, por ejemplo: Horai, R. y otros. "Cytokines in autoimmune uveitis" J. Interferon Cytokine Res. 2011, 31:733.
En otra realización, la enfermedad o trastorno es lupus. Ver, por ejemplo: Yoh, K. y otros. "Overexpression of RORYt under control of the CD2 promoter induces polyclonal plasmacytosis and autoantibody production in transgenic mice" Eur. J. Immunol. 2012, 42:1999.
En otra realización, la enfermedad o trastorno es la enfermedad de injerto contra huésped (EICH). Ver, por ejemplo: Yu, Y. y otros. "Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORYt in mice" Blood 2011, 118:5011.
En otra realización, la enfermedad o trastorno es la aterosclerosis. Ver, por ejemplo: Erbel, C. y otros. "IL-17A influences essential functions of the monocyte/macrophage lineage and es involved in advanced murine and human atherosclerosis" J. Immunol. 2014, 193, 4344.
En otra realización, las enfermedades o trastornos son rinitis alérgica, asma o enfermedad pulmonar obstructiva crónica (EPOC). Ver, por ejemplo: Loubaki, L. y otros. "Co-culture of human bronchial fibroblasts and CD4+ T cells increases Th17 cytokine signature" PLoS One 2013, 8:e81938/1; and Solt, L. y otros. "Action of RORs and their ligands in (patho)\physiology" Trends Endocrinol. Metab. 2012, 23:619.
En otra realización, las enfermedades o trastornos son obesidad, y/o resistencia a la insulina, y/o diabetes tipo II. Ver, por ejemplo: Solt, L. y otros. "ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes" Endocrinology 2015, 156:869; y Meissburger, B. y otros. "Adipogenesis and insulin sensitivity in obesity are regulated by retinoid-related orphan receptor gamma" EMBO Mol. Med. 2011,3:637; y Solt, L. "Ligand regulation of retinoic acid receptor-related orphan receptors: implications for development of novel therapeutics" Curr. Opin. Lipidology 2010, 21:204.
En otra realización, la enfermedad o trastorno es melanoma. Ver, por ejemplo: Purwar, R. y otros. "Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells" Nat. Med. 2012, 18:1248.
Debe entenderse además que las combinaciones que se incluirán dentro de esta invención son aquellas combinaciones útiles para su propósito pretendido. Los agentes expuestos a continuación tienen propósitos ilustrativos y no están destinados a ser limitados. Las combinaciones, que son parte de esta invención, pueden ser los compuestos de la presente invención y al menos un agente adicional seleccionado de las listas a continuación. La combinación también puede incluir más de un agente adicional, por ejemplo, dos o tres agentes adicionales si la combinación es tal que la composición formada puede realizar su función prevista.
Las combinaciones preferidas son fármacos antiinflamatorios no esteroideos, también conocidos como AINE, que incluyen fármacos como el ibuprofeno. Otras combinaciones preferidas son los corticosteroides, incluida la prednisolona; los efectos secundarios bien conocidos del uso de esteroides pueden reducirse o incluso eliminarse disminuyendo la dosis de esteroides requerida cuando se trata a pacientes en combinación con los compuestos de esta invención. Los ejemplos no limitantes de agentes terapéuticos para la artritis reumatoide con los que se puede combinar un compuesto de Fórmula (I-a) de la invención incluyen los siguientes: fármacos antiinflamatorios supresores de citocinas (CSAID); anticuerpos o antagonistas de otras citocinas humanas o factores de crecimiento, por ejemplo, LT, IL-1, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-21, IL-23, interferones, EMAP-II, GM-CSF, FGF, MMP-13 y PDGF. Los compuestos de la invención pueden combinarse con anticuerpos contra moléculas de la superficie celular tales como CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD80 (B7,1), CD86 (B7,2), CD90, CTLA o sus ligandos incluyendo CD154 (gp39 o CD40L).
Las combinaciones preferidas de agentes terapéuticos pueden interferir en diferentes puntos de la cascada autoinmune e inflamatoria posterior; los ejemplos preferidos incluyen inhibidores de IL-1 (inhibidores de la enzima convertidora de interleucina-1, IL-1RA, etc.) pueden ser efectivos por la misma razón. Otras combinaciones preferidas incluyen interleucina 11. Aún otras combinaciones preferidas son los otros actores clave de la respuesta autoinmune que pueden actuar paralelamente, dependiendo de o en sintonía con la función IL-18; se prefieren especialmente los antagonistas de IL-12 que incluyen anticuerpos de IL-12 o receptores de IL-12 solubles, o proteínas de unión a IL-12. Se ha demostrado que IL-12 e IL-18 tienen funciones superpuestas pero distintas y una combinación de antagonistas para ambos puede ser más efectiva. Otra combinación preferida son los inhibidores anti-CD4 no depletores. Aún otras combinaciones preferidas incluyen antagonistas de la ruta coestimuladora CD80 (B7.1) o CD86 (B7.2) que incluyen anticuerpos, receptores solubles o ligandos antagonistas.
Un compuesto de Fórmula (I-a) de la invención también puede combinarse con agentes, tales como metotrexato, 6-mercaptopurina, azatioprina, sulfasalazina, mesalazina, olsalazina, cloroquinina/hidroxicloroquina, pencilamina, aurotiomalato (intramuscular y oral), azatioprina, cochicina corticosteroides (oral, inhalada e inyección local), agonistas de los receptores adrenérgicos beta-2 (salbutamol, terbutalina, salmeteral), xantinas (teofilina, aminofilina), cromoglicato, nedocromilo, ketotifeno, ipratropio y oxitropio, ciclosporina, FK506, rapamicina, micofenolato de mofetilo, leflunomida, AINE, por ejemplo, ibuprofeno, corticosteroides como prednisolona, inhibidores de la fosfodiesterasa, agonistas de adensosina, agentes antitrombóticos, inhibidores del complemento, agentes adrenérgicos, agentes que interfieren con la señalización de las citocinas proinflamatorias tales como IL-1 (por ejemplo, inhibidores de la quinasa NIK, IKK, JAK1, JAK2, JAK3, p38 o MAP), inhibidores de la enzima convertidora de IL-1P, inhibidores de señalización de células T tales como el inhibidor de la quinasa, inhibidores de metaloproteinasas, sulfasalazina, 6-mercaptopurinas, inhibidores de la enzima convertidora de angiotensina, receptores de citocinas solubles y derivados de los mismos (por ejemplo, sI L-1RI, sIL-1RII, sIL-6R), citocinas antiinflamatorias (por ejemplo, IL-4, IL-10, IL -11, IL-13), celecoxib, ácido fólico, sulfato de
hidroxicloroquina, rofecoxib, naproxeno, valdecoxib, sulfasalazina, metilprednisolona, meloxicam, acetato de metilprednisolona, tiomalato sódico de oro, aspirina, acetónido de triamcinolona, napsilato de propoxifeno/APAP, diclofenaco sódico/misoprostol, fentanilo, anakinra, tramadol HCl, salsalato, sulindac, cianocobalamina/fa/piridoxina, paracetamol, alendronato sódico, prednisolona, sulfato de morfina, clorhidrato de lidocaína, indometacina, sulfato de glucosamina/condroitina, amitriptilina HCl, sulfadiazina, oxicodona HCl/paracetamol, olopatadina HCl misoprostol, naproxeno sódico, omeprazol, ciclofosfamida, rituximab, tofacitinib, TRAP IL-1, MRA, CTLA4-IG, IL-18 BP, anti-IL-12, Anti-IL15, BIRB-796, SCIO-469, VX-702, AMG-548, VX- 740, roflumilast, IC-485, CDC-801, agonistas de S1P1 (tal como FTY720), inhibidores de la familia PKC (tales como ruboxistaurina o AEB-071) y mesopram. Las combinaciones preferidas incluyen metotrexato o leflunomida y en casos de artritis reumatoide moderada o grave, y ciclosporina.
Los ejemplos no limitantes de agentes terapéuticos para la enfermedad inflamatoria intestinal con los que se puede combinar un compuesto de Fórmula (I-a) de la invención incluyen los siguientes: budenósida; factor de crecimiento epidérmico; corticosteroides ciclosporina, sulfasalazina; aminosalicilatos; 6-mercaptopurina; azatioprina; metronidazol; inhibidores de lipoxigenasa; mesalamina; olsalazina; balsalazida; antioxidantes; inhibidores de tromboxano; antagonistas del receptor de IL-1; anticuerpos monoclonales anti-IL-1p; anticuerpos monoclonales anti-IL-6; factores de crecimiento; inhibidores de elastasa; compuestos de piridinil-imidazol; anticuerpos o antagonistas de otras citocinas humanas o factores de crecimiento, por ejemplo, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, IL-23, EMAP-II, GM-CSF, FGF, y PDGF; moléculas de la superficie celular tales como CD2, CD3, CD4, CD8, CD25, CD28, CD30, CD40, CD45, CD69, CD90 o sus ligandos; metotrexato; ciclosporina; FK506; rapamicina; micofenolato de mofetilo; leflunomida; AINE, por ejemplo, ibuprofeno; corticosteroides como la prednisolona; inhibidores de fosfodiesterasa; agonistas de adenosina; agentes antitrombóticos; inhibidores del complemento; agentes adrenérgicos; agentes que interfieren con la señalización por citocinas proinflamatorias tales como IL-1 (por ejemplo, inhibidores de la quinasa NIK, IKK, p38 o MAP); inhibidores de la enzima convertidora de IL-ip; inhibidores de señalización de células T tales como inhibidores de quinasas; inhibidores de metaloproteinasas; sulfasalazina; azatioprina; 6-mercaptopurinas; inhibidores de la enzima convertidora de angiotensina; receptores de citocinas solubles y derivados de los mismos (por ejemplo, sIL-IRI, sIL-1RII, sIL-6R) y citocinas antiinflamatorias (por ejemplo, IL-4, IL-10, IL-11, IL-13). Los ejemplos preferidos de agentes terapéuticos para la enfermedad de Crohn con los que se puede combinar un compuesto de Fórmula (I-a) incluyen inhibidores de PDE4. Un compuesto de Fórmula (Ia) se puede combinar con corticosteroides, por ejemplo, budenósida y dexametasona; sulfasalazina, ácido 5-aminosalicílico; olsalazina; y agentes que interfieren con la síntesis o acción de las citocinas proinflamatorias tales como IL-1, por ejemplo, inhibidores de la enzima convertidora de IL-1p e IL-1ra; inhibidores de la señalización de células T, por ejemplo, inhibidores de tirosina quinasa; 6-mercaptopurina; IL-11; mesalamina; prednisona azatioprina; mercaptopurina; infliximab; succinato de metilprednisolona sódica; difenoxilato/sulfato de atropina; clorhidrato de loperamida; metotrexato; omeprazol; folato ciprofloxacina/dextrosa-agua; bitartrato de hidrocodona/apap; clorhidrato de tetraciclina; fluocinonida; metronidazol; timerosal/ácido bórico; colestiramina/sacarosa; clorhidrato de ciprofloxacina; sulfato de hiosciamina; clorhidrato de meperidina; clorhidrato de midazolam; oxicodona HCl/paracetamol; clorhidrato de prometazina; fosfato de sodio; sulfametoxazol/trimetoprima; celecoxib; policarbofilo; napsilato de propoxifeno; hidrocortisona; multivitaminas; balsalazida disódica; fosfato de codeína/apap; colesevelam HCl; cianocobalamina; ácido fólico; levofloxacina; metilprednisolona; natalizumab e interferón gamma.
Los ejemplos no limitantes de agentes terapéuticos para la esclerosis múltiple con los que se puede combinar un compuesto de Fórmula (I-a) incluyen los siguientes: corticosteroides; prednisolona; metilprednisolona; azatioprina; ciclofosfamida; ciclosporina; metotrexato; 4-aminopiridina; tizanidina; interferón-p1a (AVONEX®; Biogen); interferón-p1b (BETASERON®; Chiron/Berlex); interferón a-n3) (Interferon Sciences/Fujimoto), interferón-a (Alfa Wassermann/J & J), interferón p1 A-IF (Serono/Inhale Therapeutics), Peginterferon a 2b (Enzon/Schering-Plough), Copolímero 1 (Cop-1; COPAXONE®; Teva Pharmaceutical Industries, Inc.); oxígeno hiperbárico; inmunoglobulina intravenosa; cladribina; anticuerpos o antagonistas de otras citocinas humanas o factores de crecimiento y sus receptores, por ejemplo, LT, IL-1, IL-2, IL-6, IL-7, IL-8, IL-12, IL-23, IL-15, IL-16, EMAP-II, GM-CSF, FGF y PDGF. Un compuesto de Fórmula (I-a) puede combinarse con anticuerpos contra moléculas de la superficie celular tales como CD2, CD3, CD4, CD8, CD19, CD20, CD25, CD28, CD30, CD40, CD45, CD69, CD80, CD86, CD90 o sus ligandos. Un compuesto de Fórmula (I-a) también se puede combinar con agentes tales como metotrexato, ciclosporina, FK506, rapamicina, micofenolato de mofetilo, leflunomida, un agonista de S1P1, AINE, por ejemplo, ibuprofeno, corticosteroides como prednisolona, inhibidores de la fosfodiesterasa, agonistas de la adensosina, agentes antitrombóticos, inhibidores del complemento, agentes adrenérgicos, agentes que interfieren con la señalización por citocinas proinflamatorias como IL-1 (por ejemplo, NIK, IKK, p38 o inhibidores de la quinasa MAP), inhibidores de la enzima convertidora de IL-1p, inhibidores de TACE, inhibidores de la señalización de células T tales como inhibidores de quinasas, inhibidores de metaloproteinasas, sulfasalazina, azatioprina, 6-mercaptopurinas, inhibidores de la enzima convertidora de angiotensina, receptores de citocinas solubles y derivados de los mismos (por ejemplo, sIL-1RI, sIL-1RII, sIL-6R) y citocinas antiinflamatorias (por ejemplo, IL-4, IL-10, IL-13 y TGFp).
Los ejemplos preferidos de agentes terapéuticos para la esclerosis múltiple en los que un compuesto de Fórmula (I-a) se puede combinar para incluir interferón-p, por ejemplo, IFNp1a e IFNp1b; copaxona, corticosteroides, inhibidores de caspasa, por ejemplo, inhibidores de caspasa-1, inhibidores de IL-1 y anticuerpos contra el ligando CD40 y CD80.
Un compuesto de Fórmula (I-a) también se puede combinar con agentes, como alemtuzumab, dronabinol, daclizumab, mitoxantrona, clorhidrato de xaliproden, fampridina, acetato de glatiramer, natalizumab, sinnabidol, a-inmunocina NNSO3, ABR-215062, AnergiX.MS, antagonistas del receptor de quimioquinas, BBR-2778, calagualina, CPI-1189, LEM
(mitoxantrona encapsulada en liposomas), THC.CBD (agonista de cannabinoides), MBP-8298, mesopram (inhibidor de PDE4), MNA-715, anticuerpo anti-receptor de IL-6, neurovax, pirfenidona, allotrap 1258 (RDP-1258), sTNF-R1, anticuerpos o inhibidores de TNF de molécula pequeña, talampanel, teriflunomida, TGF-beta2, tiplimotida, antagonistas de VLA-4 (por ejemplo, TR-14035, VLA4 Ultrahaler, Antegran-ELAN/Biogen), antagonistas de interferón gamma y agonistas de IL-4.
Los ejemplos no limitantes de agentes terapéuticos para la espondilitis anquilosante con los que se puede combinar un compuesto de Fórmula (I-a) incluyen los siguientes: ibuprofeno, diclofenaco, misoprostol, naproxeno, meloxicam, indometacina, diclofenaco, celecoxib, rofecoxib, sulfasalazina, metotrexato, azatioprina, minociclina y prednisona
Los ejemplos no limitantes de agentes terapéuticos para la psoriasis con los que se puede combinar un compuesto de Fórmula (I-a) incluyen los siguientes: calcipotrieno, propionato de clobetasol, acetónido de triamcinolona, propionato de halobetasol, tazaroteno, metotrexato, fluocinonida, dipropionato de betametasona aumentada, acetónido de fluocinolona, acitretina, champú de alquitrá, valerato de betametasona, furoato de mometasona, ketoconazol, pramoxina/fluocinolona, valerato de hidrocortisona, flurandrenólido, urea, betametasona, propionato de clobetasol/emoliente, propionato de fluticasona, azitromicina, hidrocortisona, fórmula hidratante, ácido fólico, desonida, pimecrolimus, alquitrán de hulla, diacetato de diflorasona, folato de etanercept, ácido láctico, metoxsalen, hc/bismuto subgal/znox/resor, acetato de metilprednisolona, prednisona, protector solar, halcinonida, ácido salicílico, antralina, pivalato de clocortolona, extracto de carbón, alquitrán de hulla/ácido salicílico, alquitrán de hulla/ ácido salicílico/azufre, desoximetasona, diazepam, emoliente, fluocinonida/emoliente, aceite mineral/aceite de ricino/na lact, aceite mineral/aceite de maní, petróleo/miristato de isopropilo, psoraleno, ácido salicílico, jabón/tribromsalan, timerosal/ácido bórico, celecoxib, infliximab, ciclosporina, alefacept, efalizumab, tacrolimus, pimecrolimus, PUVA, UVB, sulfasalazina, ABT-874 y ustekinamab.
Los ejemplos no limitantes de agentes terapéuticos para la artritis psoriásica con los que se puede combinar un compuesto de fórmula (I-a) incluyen los siguientes: metotrexato, etanercept, rofecoxib, celecoxib, ácido fólico, sulfasalazina, naproxeno, leflunomida, acetato de metilprednisolona, indometacina, sulfato de hidroxicloroquina, prednisona, sulindac, dipropionato de betametasona aumentada, infliximab, metotrexato, folato, acetónido de triamcinolona, diclofenaco, dimetilsulfóxido, piroxicam, diclofenaco sódico, ketoprofeno, meloxicam, metilprednisolona, nabumetona, tolmetina sódica, calcipotrieno, ciclosporina, diclofenaco sódico/misoprostol, fluocinonida, sulfato de glucosamina, tiomalato sódico de oro, bitartrato de hidrocodona/apap, ibuprofeno, risedronato sódico, sulfadiazina, tioguanina, valdecoxib y alefacept
Los ejemplos no limitantes de agentes terapéuticos para la restenosis con los que se puede combinar un compuesto de Fórmula (I-a) incluyen los siguientes: sirolimus, paclitaxel, everolimus, tacrolimus, ABT-578 y paracetamol.
Los ejemplos preferidos de agentes terapéuticos para LES (Lupus) con los que se puede combinar un compuesto de Fórmula (I-a) incluyen los siguientes: AINe , por ejemplo, diclofenaco, naproxeno, ibuprofeno, piroxicam, indometacina; inhibidores de COX2, por ejemplo, celecoxib, rofecoxib, valdecoxib; antipalúdicos, por ejemplo, hidroxicloroquina; esteroides, por ejemplo, prednisona, prednisolona, budenósido, dexametasona; citotóxicos, por ejemplo, azatioprina, ciclofosfamida, micofenolato de mofetilo, metotrexato; inhibidores de PDE4 o inhibidor de síntesis de purina, por ejemplo Cellcept®. Un compuesto de Fórmula (I-a) también puede combinarse con agentes tales como sulfasalazina, ácido 5-aminosalicílico, olsalazina, Imuran® y agentes que interfieren con la síntesis, producción o acción de citocinas proinflamatorias tal como IL-1, por ejemplo, inhibidores de caspasa como los inhibidores de la enzima convertidora de IL-ip y la IL-1ra. También se puede usar un compuesto de Fórmula (I-a) con inhibidores de señalización de células T, por ejemplo, inhibidores de tirosina quinasa; o moléculas que se dirigen a moléculas de activación de células T, por ejemplo, CTLA-4-IgG o anticuerpos de la familia anti-B7, anticuerpos de la familia anti-PD-1. Un compuesto de Fórmula (I-a) (puede combinarse con IL-11 o anticuerpos anti-citoquinas, por ejemplo, fonotolizumab (anticuerpo anti-IFNg), o anticuerpos antirreceptores, por ejemplo, anticuerpo del antirreceptor de IL-6 y anticuerpos contra moléculas de la superficie de las células B. Un compuesto de Fórmula (I-a) también se puede usar con LJp 394 (abetimus), agentes depletores o que inactivan las células B, por ejemplo, rituximab (anticuerpo anti-CD20) y linfostato-B (anticuerpo anti-BlyS).
En esta invención, son aplicables las siguientes definiciones:
Una "cantidad terapéuticamente efectiva" es una cantidad de un compuesto de Fórmula (I-a) o una combinación de dos o más de tales compuestos, que inhibe, total o parcialmente, la progresión de la afección o alivia, al menos parcialmente, uno o más síntomas de la afección. Una cantidad terapéuticamente efectiva también puede ser una cantidad que es profilácticamente efectiva. La cantidad terapéuticamente efectiva dependerá del tamaño y sexo del paciente, la afección a tratar, la gravedad de la afección y el resultado buscado. Para un paciente dado, se puede determinar una cantidad terapéuticamente efectiva mediante métodos conocidos por los expertos en la técnica.
"Sales farmacéuticamente aceptables" se refiere a aquellas sales que retienen la efectividad biológica y las propiedades de las bases libres y que se obtienen por reacción con ácidos inorgánicos, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico y ácido fosfórico o ácidos orgánicos como ácido sulfónico, ácido carboxílico, ácido fosfórico orgánico, ácido metanosulfónico, ácido etanosulfónico, ácido p- toluenosulfónico, ácido cítrico, ácido fumárico, ácido maleico, ácido succínico, ácido benzoico, ácido salicílico, ácido láctico, ácido tartárico (por ejemplo, ácido (+) o (-)-tartárico o mezclas de los mismos), aminoácidos (por ejemplo (+) o (-)-aminoácidos o mezclas de los mismos) y similares. Estas sales se pueden preparar por métodos conocidos por los expertos en la técnica.
Ciertos compuestos de Fórmula (I-a) que tienen sustituyentes ácidos pueden existir como sales con bases farmacéuticamente aceptables. La presente invención incluye tales sales. Los ejemplos de tales sales incluyen sales de sodio, sales de potasio, sales de lisina y sales de arginina. Estas sales pueden prepararse por métodos conocidos por los expertos en la técnica.
Ciertos compuestos de Fórmula (I-a) y sus sales pueden existir en más de una forma cristalina y la presente invención incluye cada forma cristalina y sus mezclas.
Ciertos compuestos de Fórmula (I-a) y sus sales también pueden existir en forma de solvatos, por ejemplo, hidratos, y la presente invención incluye cada solvato y mezclas de los mismos.
Ciertos compuestos de Fórmula (I-a) pueden contener uno o más centros quirales y existir en diferentes formas ópticamente activas. Cuando los compuestos de Fórmula (I-a) contienen un centro quiral, los compuestos existen en dos formas enantioméricas y la presente invención incluye tanto enantiómeros como mezclas de enantiómeros, tales como mezclas racémicas. Los enantiómeros pueden resolverse mediante métodos conocidos por los expertos en la técnica, por ejemplo mediante la formación de sales diastereoisoméricas que pueden separarse, por ejemplo, mediante cristalización; formación de derivados o complejos diastereoisoméricos que pueden separarse, por ejemplo, por cristalización, cromatografía de líquidos o de gas-líquido; reacción selectiva de un enantiómero con un reactivo específico de enantiómero, por ejemplo esterificación enzimática; o cromatografía de líquidos o de gas-líquido en un entorno quiral, por ejemplo en un soporte quiral, por ejemplo, sílice con un ligando quiral unido o en presencia de un disolvente quiral. Se apreciará que cuando el enantiómero deseado se convierte en otra entidad química mediante uno de los procedimientos de separación descritos anteriormente, se requiere una etapa adicional para liberar la forma enantiomérica deseada. Alternativamente, se pueden sintetizar enantiómeros específicos mediante síntesis asimétrica usando reactivos, sustratos, catalizadores o disolventes ópticamente activos, o convirtiendo un enantiómero en el otro mediante transformación asimétrica.
Cuando un compuesto de Fórmula (I-a) contiene más de un centro quiral, éste puede existir en formas diastereoisoméricas. Los compuestos diastereoisoméricos pueden separarse por métodos conocidos por los expertos en la técnica, por ejemplo, cromatografía o cristalización y los enantiómeros individuales pueden separarse como se describió anteriormente. La presente invención incluye cada diastereoisómero de los compuestos de Fórmula (I-a) (y mezclas de los mismos).
Ciertos compuestos de Fórmula (I-a) pueden existir en diferentes formas tautoméricas o como diferentes isómeros geométricos, y la presente invención incluye cada tautómero y/o isómero geométrico de los compuestos de Fórmula (I-a) y mezclas de los mismos.
Ciertos compuestos de Fórmula (I-a) pueden existir en diferentes formas conformacionales estables que pueden ser separables. La asimetría torsional debido a la rotación restringida alrededor de un enlace simple asimétrico, por ejemplo, debido a impedimento estérico o tensión del anillo, puede permitir la separación de diferentes conformadores. La presente invención incluye cada isómero conformacional de los compuestos de Fórmula (I-a) y mezclas de los mismos.
Ciertos compuestos de fórmula (I-a) pueden existir en forma de ion híbrido y la presente invención incluye cada forma de compuesto de ion híbrido de fórmula (I-a) y sus mezclas.
Como se usa en la presente descripción, el término "profármaco" se refiere a un agente que se convierte en el fármaco original in vivo por algún proceso químico fisiológico (por ejemplo, un profármaco al alcanzar el pH fisiológico se convierte en la forma de fármaco deseada). Los profármacos a menudo son útiles porque, en algunas situaciones, pueden ser más fáciles de administrar que el fármaco original. Por ejemplo, pueden estar biodisponibles para administración oral mientras que el fármaco original no lo está. El profármaco también puede tener una solubilidad mejorada en composiciones farmacológicas sobre el fármaco original. Un ejemplo, sin limitación, de un profármaco sería un compuesto de la presente invención en el que se administra como un éster (el "profármaco") para facilitar la transmisión a través de una membrana celular donde la solubilidad en agua no es beneficiosa, pero luego se hidroliza metabólicamente al ácido carboxílico una vez dentro de la célula, donde la solubilidad en agua es beneficiosa.
Los profármacos tienen muchas propiedades útiles. Por ejemplo, un profármaco puede ser más soluble en agua que el fármaco final, lo que facilita la administración intravenosa del fármaco. Un profármaco también puede tener un mayor nivel de biodisponibilidad oral que el fármaco definitivo. Después de la administración, el profármaco se escinde enzimática o químicamente para administrar el fármaco definitivo en la sangre o el tejido.
Los profármacos ilustrativos tras la escisión liberan el ácido libre correspondiente, y tales residuos formadores de ésteres hidrolizables de los compuestos de esta invención incluyen, pero no se limitan a sustituyentes de ácido carboxílico en los que el hidrógeno libre se sustituye por (C1-C4)alquilo, (C1-C 12 )alcanoiloximetilo, (C4-Cg)1-(alcanoiloxi)etilo, 1 -metil-1-(alcanoiloxi)-etilo que tiene de 5 a 10 átomos de carbono, alcoxicarboniloximetilo que tiene de 3 a 6 átomos de carbono, 1-(alcoxicarboniloxi)etilo que tiene de 4 a 7 átomos de carbono, 1-metil-1-(alcoxicarboniloxi)etilo que tiene de 5 a 8 átomos de carbono, W-(alcoxicarbonil)aminometilo que tiene de 3 a 9 átomos de carbono, 1-(W-(alcoxicarbonil)amino)etilo que tiene de 4 a 10 átomos de carbono, 3-ftalidilo, 4-crotonolactonilo, gamma-butirolacton-4-ilo, di-W,W-(Cr C2)alquilamino(C2
C3)alquilo (tal como p-dimetilaminoetilo), carbamoil-(C i -C2)alquilo, W,W-di(Ci -C2)-alquilcarbamoil-(C i -C2)alquilo y piperidino-, pirrolidino- o morfolino(C2-C3)alquilo.
Otros profármacos ilustrativos liberan un alcohol de Fórmula (I-a) en el que el hidrógeno libre del sustituyente hidroxilo (por ejemplo, R1 contiene hidroxilo) se reemplaza por (Cr C6)alcanoiloximetilo, 1-((Cr C6)alcanoiloxi)etilo, i-m etil-i-((C i -Ca)alcanoiloxi)etilo, (C i -C i 2 )alcoxicarboniloximetilo, N-(C i -C6)alcoxicarbonilamino-metilo, succinoilo, (C i -C6)alcanoilo, aamino(C i -C4)alcanoilo, arilactilo y a-aminoacilo, o a-aminoacil-a-aminoacilo en el que dichos restos a-aminoacilo son independientemente cualquiera de los L-aminoácidos que se encuentran naturalmente en las proteínas, P(O)(OH)2 , -P(O)(O(Ci -C6)alquilo)2 o glicosilo (el radical resultante de la separación del hidroxilo del hemiacetal de un carbohidrato).
El término "heterocíclico", "heterociclilo" o "heterociclileno", como se usa en la presente descripción, incluye sistemas de anillos no aromáticos, que incluyen, pero no se limitan a, anillos monocíclicos, bicíclicos, tricíclicos y espirocíclicos, que pueden estar completamente saturados o que pueden contener una o más unidades de insaturación, para evitar dudas, el grado de insaturación no da como resultado un sistema de anillo aromático) y tienen de 5 a i2 átomos que incluyen al menos un heteroátomo, tal como nitrógeno, oxígeno o azufre. Para fines de ejemplificación, que no deben interpretarse como limitantes del alcance de esta invención, los siguientes son ejemplos de anillos heterocíclicos: azepinilo, azetidinilo, indolinilo, isoindolinilo, morfolinilo, piperazinilo, piperidinilo, pirrolidinilo, quinucludinilo, tiomorfolinilo, tetrahidropiranilo, tetrahidrofuranilo, tetrahidroindolilo, tiomorfolinilo y tropanilo.
El término "heteroarilo" o "heteroarileno", como se usa en la presente descripción, incluye sistemas de anillos aromáticos, que incluyen, pero no se limitan a, anillos monocíclicos, bicíclicos y tricíclicos, y tienen de 5 a i2 átomos que incluyen al menos un heteroátomo, tal como nitrógeno, oxígeno, o azufre. Para fines de ejemplificación, que no deben interpretarse como limitantes del alcance de esta invención: azaindolilo, benzo(6)tienilo, bencimidazolilo, benzofuranilo, benzoxazolilo, benzotiazolilo, benzotiadiazolilo, benzoxadi-azolilo, furanilo, imidazolilo, imidazopiridinilo, indolilo, indazolilo, isoxazolilo, isotiazolilo, oxadiazolilo, oxazolilo, purinilo, piranilo, pirazinilo, pirazolilo, piridinilo, pirimidinilo, pirrolilo, pirrolo[2,3-d]pirimidinilo, pirazolo[3,4-C]pirimidinilo, quinolinilo, quinazolinilo, triazolilo, tiazolilo, tiofenilo, tetrazolilo, tiadiazolilo, tienilo, 6H-pirrolo[2,3-e][i ,2,4]triazolo[4,3-a]pirazinilo, 6H-imidazo[i ,5-a]pirrolo[2,3-e]pirazinilo, i ,6-dihidropirazolo[3,4-C]pirrolo[2,3-í)]piridina, 3H-3,4,6,8a-tetraaza-asindacenilo, 3H-imidazo[i ,2-a]pirrolo[2,3-e]pirazinilo, pirazolo[3,4-C]pirrolo[2,3-í)]piridinilo, i ,6-dihidro-i,2,5,6-tetraza-as-indacenilo, 3H-3,4,8a-triaza-as-indacenilo, 6H-3-oxa-2,5,6-triazaas-indacenilo, 3,6-dihidro-2,3,6-tetraaza-as-indacenilo, i ,6-dihidro-dipirrolo[2,3-fo;2'3'-C]piridinilo, 6H-3-thia-2,5,6-triazaas-indacenilo, 4,5-dihidro-i H-benzo[b]azepin-2(3H)-ona, 3,4-dihidroquinolin-2(i H)-ona, 2H-benzo[6][i ,4]oxazin-3(4H)-ona, o 6,7-dihidro-4H-pirazolo[5,i-c][i,4]oxazinilo o i,6-dihidroimidazo[4,5-c(]pirrolo[2,3-b]piridina.
Como se usa en la presente descripción, "alquilo", "alquileno" o notaciones tales como "(C i -Cs )" incluyen hidrocarburos de cadena lineal o ramificada que están completamente saturados. Los ejemplos de alquilos son metilo, etilo, propilo, isopropilo, butilo, pentilo, hexilo e isómeros de los mismos. Como se usa en la presente descripción, "alquenilo", "alquenileno", "alquinileno" y "alquinilo" significa C2-C8 e incluye hidrocarburos de cadena lineal o ramificada que contienen una o más unidades de insaturación, uno o más dobles enlaces para alquenilo y uno o más enlaces triples para alquinilo.
Como se usa en la presente descripción, los grupos "aromáticos" (o grupos "arilo" o "arileno") incluyen sistemas de anillos aromáticos carbocíclicos (por ejemplo, fenilo) y sistemas de anillos aromáticos policíclicos fusionados (por ejemplo, naftilo, bifenilo y i,2,3,4-tetrahidronaftilo).
Como se usa en la presente descripción, "cicloalquilo" o "cicloalquileno" significa hidrocarburos de C3-C i 2 monocíclicos o multicíclicos (por ejemplo, bicíclico, tricíclico, espirocíclicos, etc.) que están completamente saturados. Los ejemplos de un grupo cicloalquilo son ciclopropilo, ciclobutilo, ciclopentilo, b ic iclo[i,i,i]pentilo y ciclohexilo.
Como se usa en la presente descripción, "cicloalquenilo" significa hidrocarburos de C3-C i 2 monocíclicos o multicíclicos (por ejemplo, bicíclico, tricíclico, espirocíclicos, etc.) que tienen uno o más enlaces insaturados, pero no equivale a un grupo aromático. Los ejemplos de un grupo cicloalquenilo son ciclopentenilo y ciclohexenilo.
Como se usa en la presente descripción, muchos restos o sustituyentes se denominan "sustituidos" u "opcionalmente sustituidos". Cuando un resto se modifica por uno de estos términos, a menos que se indique lo contrario, denota que cualquier porción del resto que un experto en la técnica conozca que está disponible para la sustitución puede sustituirse, lo que incluye uno o más sustituyentes, donde si hay más de un sustituyente, entonces cada sustituyente se selecciona independientemente. Tales medios para la sustitución son bien conocidos en la técnica y/o enseñados por la presente divulgación. Para los propósitos de ejemplificación, que no deben interpretarse como limitantes del alcance de esta invención, algunos ejemplos de grupos que son sustituyentes son: grupos (C i -Cs )alquilo, grupos (C2-Cs )alquenilo, grupos (C2-Cs )alquinilo, grupos (C3-C io )cicloalquilo, halógeno (F, Cl, Br o I), grupos (C i -Cs )alquilo halogenados (por ejemplo, pero sin limitarse a -CF3), grupos -(C i -Cs )alquilo, =O, =CH2 , -OH, -CH2OH, -CH2 NH2 , (C i -C4)alquilo-OH, -CH2CH2OCH2CH3 , grupos -S-(C i -Cs )alquilo, -SH, grupos -NH(C i -Cs )alquilo, grupos -N((C i -Cs )alquilo)2 , -NH2 , -C(O)NH2 , -CH2 NHC(O)(Ci -C4)alquilo, -CH2 NHC(O)CH2Cl, -CH2NHC(O)CH2CN, -CH2NHC(O)CH2CH2N(CH3)2 , -CH2NHC(O)C(=CH2)CH3 , -CH2 NHC(O)(C2-C4)quinilo -CH2 NHC(O)CH2CH2-piperidinilo, -(C i -C4)alquilo-morfolinilo, -CH2 NHC(O)CH2O-fenilo en el que el fenilo está opcionalmente sustituido con halógeno, (C i -C4)alcoxi, -C(O)(C i -C4)alquilo, -C(O)(C i -C4)alcoxi, -C(O)N(H)2 , -C(O)N(CH3)2 , -C(O)(C i -C6)heteroarilo, -N(CH3)2 , -NHC(O)(C i -C4)alquilo, -NHC(O)(C2-C4)alquenilo, -NHC(O)CH2CN, -S(O)2(C i -C4)alquilo, -S(O)2(C i -C6)heteroarilo, -S(O)2(C i -C6) (C i -C6)heterociclilo, 4metilpiperazinacarbonilo, -(C i -C4)alquiloC(O)NH2 , grupos -C(O)NH(C i -C8)alquilo, -C(O)N((C i -C8)alquilo)2 , grupos -C(O)N(H)(C3-C8)cidoalquilo, -C(O)(C1-C4)alcoxi, -NHC(O)H, grupos -NHC(O)(Cr C8)alquilo, grupos -NHC(O)(C3-C8)cicloalquilo, -N((C i -C8)alquilo)C(O)H, grupos -N((C i -C8)alquilo)C(O)(Ci -C8)alquilo, -NHC(O)NH2 , grupos -NHC(O)NH(Ci -C8)alquilo, grupos -N((C i -C8)alquilo)C(O)NH2 , grupos -NHC(O)N((C i -C8)alquilo)2 , grupos -N((C i -C8)alquilo)C(O)N((C1-C8)alquilo)2 , -N((Cr C8)alquilo)C(O)NH((Cr C8)alquilo), -NHCH2-heteroarilo, bencilo, -OCH2-heteroarilo, grupos -C(O)H, -C(O)(C1-C8)alquilo, -CN, -NO2 , grupos -S(O)(C1-C8)alquilo, grupos -S(O)2(C1-C8)alquilo, grupos -S(O)2 N((C1-C8)alquilo)2 , grupos -S(O)2 NH(C1-C8)alquilo, grupos -S(O)2 NH(C3-C8)cicloalquilo, grupos -S(O)2 NH2 , grupos -NHS(O)2(C1-C8)alquilo, grupos -N((Cr C8)alquilo)S(O)2(Cr C8)alquilo, grupos -(C1-C8)alquilo-O-(C1-C8)alquilo, grupos -O-(C1-C8)alquilo-O-(C1-C8)alquilo, grupos -C(O)OH, -C(O)O(C1-C8)alquilo, NHOH, grupos NHO(Cr C8)alquilo, grupos -O-(C1-C8)alquilo halogenados (por ejemplo, pero sin limitarse a -OCF3), grupos (Cr C8)alquilo -S(O)2-halogenados (por ejemplo, pero sin limitarse a -S(O)2CF3), grupos (C1-C8)alquilo -S-halogenados (por ejemplo, pero sin limitarse a -SCF3), -(C r C6)heterociclilo (por ejemplo, pero sin limitarse a pirrolidina, tetrahidrofurano, pirano o morfolina), -(C1-C6)heteroarilo (por ejemplo, pero sin limitarse a tetrazol, imidazol, furano, pirazina o pirazol), -fenilo, bencilo opcionalmente sustituido, grupos -NHC(O)O-(C1-C6)alquilo, grupos -N((C1-C6)alquilo)C(O)O-(C1-C6)alquilo, grupos -C(=NH)-(Cr C6)alquilo, grupos -C(=NOH)-(C1-C6)alquilo, o grupos -C(=N-O-(C1-C6)alquilo)-(C1-C6)alquilo.
El término "kit", como se usa en la presente descripción, se refiere a un producto empacado que comprende componentes con los que administrar un compuesto de Fórmula (I-a) de la invención para el tratamiento de un trastorno autoinmune. El kit comprende preferiblemente una caja o contenedor que contiene los componentes del kit. La caja o el contenedor tiene adherida una etiqueta o un protocolo aprobado por la Administración de Fármacos y Alimentos. La caja o contenedor contiene componentes de la invención que están contenidos preferiblemente dentro de recipientes de plástico, polietileno, polipropileno, etileno o propileno. Los recipientes pueden ser tubos tapados o botellas. El kit también puede incluir instrucciones para administrar un compuesto de Fórmula (I-a).
Uno o más compuestos de esta invención pueden administrarse a un paciente humano por sí mismos o en composiciones farmacéuticas donde se mezclan con vehículos o excipientes biológicamente adecuados a dosis para tratar o mejorar una enfermedad o afección como se describe en la presente descripción. Las mezclas de estos compuestos también se pueden administrar al paciente como una mezcla simple o en composiciones farmacéuticas formuladas adecuadas. Una dosis terapéuticamente efectiva se refiere a esa cantidad del compuesto o compuestos suficiente para dar como resultado la prevención o atenuación de una enfermedad o afección como se describe en la presente descripción. Las técnicas para la formulación y administración de los compuestos de la presente solicitud se pueden encontrar en referencias bien conocidas por un experto en la técnica, tales como "Remington's Pharmaceutical Sciences", Mack Publishing Co., Easton, PA, última edición.
Las vías de administración adecuadas pueden incluir, por ejemplo, administración oral, gota para ojos, rectal, transmucosa, tópica o intestinal; administración parenteral, incluidas inyecciones intramusculares, subcutáneas, intramedulares, así como inyecciones intratecales, intraventriculares directas, intravenosas, intraperitoneales, intranasales o intraoculares.
Alternativamente, el compuesto se puede administrar de manera local en lugar de sistémica, por ejemplo, mediante inyección del compuesto directamente en un sitio edematoso, a menudo en un depósito o formulación de liberación sostenida.
Además, el fármaco se puede administrar en un sistema de administración de fármacos dirigido, por ejemplo, en un liposoma recubierto con anticuerpo específico de células endoteliales.
Las composiciones farmacéuticas de la presente invención pueden fabricarse de una manera que es conocida por sí misma, por ejemplo, mediante procesos convencionales de mezcla, disolución, granulación, fabricación de grageas, levigación, emulsión, encapsulación, atrapamiento o liofilización.
Las composiciones farmacéuticas para usar de acuerdo con la presente invención, por lo tanto, pueden formularse de manera convencional usando uno o más vehículos fisiológicamente aceptables que comprenden excipientes y auxiliares que facilitan el procesamiento de los compuestos activos en preparaciones que pueden usarse farmacéuticamente. La formulación adecuada depende de la ruta de administración elegida.
Para la inyección, los agentes de la invención pueden formularse en soluciones acuosas, preferiblemente en tampones fisiológicamente compatibles tales como la solución de Hank, la solución de Ringer o el tampón salino fisiológico. Para la administración transmucosa, en la formulación se usan penetrantes apropiados para la barrera a atravesar. Tales penetrantes son generalmente conocidos en la técnica.
Para la administración oral, los compuestos pueden formularse fácilmente combinando los compuestos activos con vehículos farmacéuticamente aceptables bien conocidos en la técnica. Tales vehículos permiten que los compuestos de la invención se formulen como comprimidos, píldoras, grageas, cápsulas, líquidos, geles, jarabes, lechadas, suspensiones y similares, para la ingestión oral de un paciente a tratar. Las preparaciones farmacéuticas para uso oral se pueden obtener combinando el compuesto activo con un excipiente sólido, opcionalmente moliendo una mezcla resultante y procesando la mezcla de gránulos, después de agregar auxiliares adecuados, si se desea, para obtener comprimidos o
núcleos de grageas. Los excipientes adecuados son, en particular, cargas tales como azúcares, que incluyen lactosa, sacarosa, manitol o sorbitol; preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona (PVP). Si se desea, se pueden agregar agentes desintegrantes, tales como la polivinilpirrolidona reticulada, agar o ácido algínico o una sal de los mismos, tal como alginato de sodio.
Los núcleos de las grageas están provistos de recubrimientos adecuados. Para este propósito, se pueden usar soluciones de azúcar concentradas, que opcionalmente pueden contener goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y solventes orgánicos adecuados o mezclas de solventes. Se pueden agregar colorantes o pigmentos a los comprimidos o a los recubrimientos de las grageas para identificación o para caracterizar diferentes combinaciones de dosis de compuestos activos.
Las preparaciones farmacéuticas que se pueden usar por vía oral incluyen cápsulas duras hechas de gelatina, así como cápsulas selladas blandas hechas de gelatina y un plastificante, como glicerol o sorbitol. Las cápsulas duras pueden contener los ingredientes activos mezclados con relleno tales como lactosa, aglutinantes como almidones y/o lubricantes como talco o estearato de magnesio y, opcionalmente, estabilizadores. En cápsulas las blandas, los compuestos activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, se pueden agregar estabilizadores. Todas las formulaciones para administración oral deben estar en dosis adecuadas para dicha administración.
Para la administración bucal, las composiciones pueden tomar la forma de comprimidos o pastillas formuladas de manera convencional.
Para la administración por inhalación, los compuestos para usar de acuerdo con la presente invención se administran convenientemente en forma de presentación en aerosol de paquetes presurizados o un nebulizador, con el uso de un propulsor adecuado, por ejemplo, diclorodifluorometano, triclorofluorometano, diclorotetrafluoroetano, dióxido de carbono u otro gas adecuado. En el caso de aerosol presurizado, la unidad de dosificación se puede determinar proporcionando una válvula para administrar una cantidad medida. Se pueden formular cápsulas y cartuchos de, por ejemplo, gelatina para usar en un inhalador o insuflador que contiene una mezcla en polvo del compuesto y una base en polvo adecuada tal como lactosa o almidón.
Los compuestos pueden formularse para administración parenteral mediante inyección, por ejemplo, inyección en bolo o infusión continua. Las formulaciones para inyección pueden presentarse en forma de dosificación unitaria, por ejemplo, en ampollas o en envases multidosis, con un conservante añadido. Las composiciones pueden tomar formas tales como suspensiones, soluciones o emulsiones en vehículos aceitosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilizantes y/o dispersantes.
Las formulaciones farmacéuticas para administración parenteral incluyen soluciones acuosas de los compuestos activos en forma soluble en agua. Además, las suspensiones de los compuestos activos pueden prepararse como suspensiones de inyección oleosas apropiadas. Los disolventes o vehículos lipofílicos adecuados incluyen aceites grasos tales como aceite de sésamo o ésteres de ácidos grasos sintéticos, tales como oleato o triglicéridos de etilo, o liposomas. Las suspensiones de inyección acuosa pueden contener sustancias que aumentan la viscosidad de la suspensión, como carboximetilcelulosa de sodio, sorbitol o dextrano. Opcionalmente, la suspensión también puede contener estabilizadores o agentes adecuados que aumentan la solubilidad de los compuestos para permitir la preparación de soluciones altamente concentradas.
Alternativamente, el ingrediente activo puede estar en forma de polvo para constitución con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso.
Los compuestos también pueden formularse en composiciones rectales tales como supositorios o enemas de retención, por ejemplo, que contienen bases de supositorios convencionales tales como manteca de cacao u otros glicéridos.
Además de las formulaciones descritas anteriormente, los compuestos también pueden formularse como una preparación de depósito. Dichas formulaciones de acción prolongada pueden administrarse por implantación (por ejemplo, por vía subcutánea o intramuscular o por inyección intramuscular). Así, por ejemplo, los compuestos pueden formularse con materiales poliméricos o hidrófobos adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico, o como derivados escasamente solubles, por ejemplo, como una sal escasamente soluble.
Un ejemplo de un vehículo farmacéutico para los compuestos hidrófobos de la invención es un sistema codisolvente que comprende alcohol bencílico, un tensioactivo no polar, un polímero orgánico miscible en agua y una fase acuosa. El sistema codisolvente puede ser el sistema codisolvente VPD. VPD es una solución de 3% p/v de alcohol bencílico, 8% p/v del polisorbato tensioactivo no polar 80 y 65% p/v de polietilenglicol 300, hasta el volumen en etanol absoluto. El sistema codisolvente VPD (VPD:5W) consiste en VPD diluido 1:1 con una solución de dextrosa al 5% en agua. Este sistema codisolvente disuelve bien los compuestos hidrófobos, y en sí mismo produce una baja toxicidad tras la administración sistémica. Naturalmente, las proporciones de un sistema codisolvente pueden variar considerablemente sin destruir sus características de solubilidad y toxicidad. Además, la identidad de los componentes codisolventes puede
variar: por ejemplo, se pueden usar otros tensioactivos no polares de baja toxicidad en lugar de polisorbato 80; el tamaño de la fracción de polietilenglicol puede variar; otros polímeros biocompatibles pueden reemplazar al polietilenglicol, por ejemplo, polivinilpirrolidona; y otros azúcares o polisacáridos pueden sustituir a la dextrosa.
Alternativamente, se pueden emplear otros sistemas de suministro para compuestos farmacéuticos hidrófobos. Los liposomas y las emulsiones son ejemplos bien conocidos de vehículos de entrega o vehículos para fármacos hidrófobos. Ciertos disolventes orgánicos como el dimetilsulfóxido también pueden emplearse, aunque generalmente a costa de una mayor toxicidad. Además, los compuestos pueden administrarse usando un sistema de liberación sostenida, tal como matrices semipermeables de polímeros hidrófobos sólidos que contienen el agente terapéutico. Se han establecido diversos materiales de liberación sostenida y son bien conocidos por los expertos en la técnica. Las cápsulas de liberación sostenida pueden, dependiendo de su naturaleza química, liberar los compuestos durante algunas semanas hasta más de 100 días. Dependiendo de la naturaleza química y la estabilidad biológica del reactivo terapéutico, se pueden emplear estrategias adicionales para la estabilización de proteínas.
Las composiciones farmacéuticas también pueden comprender vehículos o excipientes en fase sólida o de gel adecuados. Los ejemplos de tales vehículos o excipientes incluyen, pero no se limitan a, carbonato de calcio, fosfato de calcio, diversos azúcares, almidones, derivados de celulosa, gelatina y polímeros tales como polietilenglicoles.
Muchos de los compuestos de la invención pueden proporcionarse como sales con contra iones farmacéuticamente compatibles. Las sales farmacéuticamente compatibles pueden formarse con muchos ácidos, incluidos, entre otros, clorhídrico, sulfúrico, acético, láctico, tartárico, málico, succínico, etc. Las sales tienden a ser más solubles en solventes acuosos u otros solventes protónicos que las formas de base libre correspondientes.
Las composiciones farmacéuticas adecuadas para su uso en la presente invención incluyen composiciones en las que los ingredientes activos están contenidos en una cantidad efectiva para lograr su propósito pretendido. Más específicamente, una cantidad terapéuticamente efectiva significa una cantidad efectiva para prevenir el desarrollo o para aliviar los síntomas existentes del sujeto que está siendo tratado. La determinación de las cantidades efectivas está dentro de la capacidad de los expertos en la técnica.
Para cualquier compuesto para usar en el tratamiento de una enfermedad o trastorno descrito aquí, la dosis terapéuticamente efectiva puede estimarse inicialmente a partir de ensayos celulares. Por ejemplo, se puede formular una dosis en modelos celulares y animales para lograr un rango de concentración circulante que incluya el IC50 como se determina en los ensayos celulares (por ejemplo, la concentración del compuesto de prueba que logra una inhibición semi máxima de una proteína quinasa dada actividad). En algunos casos es apropiado determinar el IC50 en presencia de 3 a 5% de albúmina de suero ya que una determinación tal aproxima los efectos de unión de la proteína plasmática en el compuesto. Dicha información se puede utilizar para determinar con mayor precisión las dosis útiles en humanos. Además, los compuestos más preferidos para la administración sistémica inhiben eficazmente la señalización de la proteína quinasa en células intactas a niveles que se pueden lograr de forma segura en plasma.
Una dosis terapéuticamente efectiva se refiere a la cantidad del compuesto que produce una mejoría de los síntomas en un paciente. La toxicidad y la eficacia terapéutica de dichos compuestos puede determinarse mediante procedimientos farmacéuticos estándar en cultivos celulares o animales experimentales, por ejemplo, para determinar la dosis máxima tolerada (MTD) y la ED50 (dosis efectiva para una respuesta máxima del 50%). La relación de dosis entre los efectos tóxicos y terapéuticos es el índice terapéutico y puede expresarse como la relación entre MTD y ED50. Se prefieren los compuestos que exhiben altos índices terapéuticos. Los datos obtenidos de estos ensayos de cultivo celular y estudios en animales pueden usarse para formular un rango de dosis para uso en humanos. La dosificación de tales mentiras compuestos preferiblemente dentro de un intervalo de concentraciones circulantes que incluyen la DE50 con poca o ninguna toxicidad. La dosificación puede variar dentro de este rango dependiendo de la forma de dosificación empleada y la vía de administración utilizada. La formulación exacta, la vía de administración y la dosificación pueden ser elegidas por el médico individual en vista de la condición del paciente (véase, por ejemplo, Fingl y otros, 1975, en The Pharmacological Basis of Therapeutics, cap. 1, pág. 1). En el tratamiento de crisis, puede ser necesaria la administración de un bolo agudo o una infusión que se aproxime al MTD para obtener una respuesta rápida.
La cantidad de dosis y el intervalo pueden ajustarse individualmente para proporcionar niveles plasmáticos del resto activo que sean suficientes para mantener los efectos moduladores de la quinasa, o una concentración efectiva mínima (MEC). El MEC variará para cada compuesto, pero puede estimarse a partir de datos in vitro; por ejemplo, la concentración necesaria para lograr una inhibición del 50-90% de la proteína quinasa usando los ensayos descritos en este documento. Las dosis necesarias para lograr el MEC dependerán de las características individuales y la vía de administración. Sin embargo, los ensayos o bioensayos de HPLC pueden usarse para determinar las concentraciones plasmáticas.
Los intervalos de dosificación también se pueden determinar usando el valor MEC. Los compuestos deben administrarse usando un régimen que mantenga niveles en plasma por encima del MEC durante 10-90% del tiempo, preferiblemente entre 30-90% y lo más preferiblemente entre 50-90% hasta que se logre la mejoría deseada de los síntomas. En casos de administración local o absorción selectiva, la concentración local efectiva del fármaco puede no estar relacionada con la concentración plasmática.
La cantidad de composición administrada dependerá, por supuesto, del sujeto a tratar, del peso del sujeto, la gravedad de la aflicción, la forma de administración y el juicio del médico que prescribe.
Las composiciones pueden, si se desea, presentarse en un paquete o dispositivo dispensador que puede contener una o más formas de dosificación unitarias que contienen el ingrediente activo. El paquete puede comprender, por ejemplo, una lámina de metal o plástico, tal como un paquete de blíster. El paquete o dispositivo dispensador puede ir acompañado de instrucciones de administración. Las composiciones que comprenden un compuesto de la invención formulado en un vehículo farmacéutico compatible también se pueden preparar, colocar en un recipiente apropiado y etiquetar para el tratamiento de una afección indicada.
En algunas formulaciones puede ser beneficioso usar los compuestos de la presente invención en forma de partículas de tamaño muy pequeño, por ejemplo, obtenidas por molienda de energía fluida.
El uso de los compuestos de la presente invención en la fabricación de composiciones farmacéuticas se ilustra mediante la siguiente descripción. En esta descripción, el término "compuesto activo" denota cualquier compuesto de la invención, pero particularmente cualquier compuesto que sea el producto final de uno de los siguientes Ejemplos:
a) Cápsulas
En la preparación de cápsulas, se pueden desagregar y mezclar 10 partes en peso de compuesto activo y 240 partes en peso de lactosa. La mezcla se puede rellenar en cápsulas de gelatina dura, cada cápsula contiene una dosis unitaria o parte de una dosis unitaria de compuesto activo.
b) Comprimidos
Los comprimidos se pueden preparar, por ejemplo, a partir de los siguientes ingredientes.
Partes en peso
Compuesto activo 10
Lactosa 190
Almidón de maíz 22
Polivinilpirrolidona 10
Estearato de magnesio 3
El compuesto activo, la lactosa y algo del almidón se pueden desagregar, mezclar y la mezcla resultante se puede granular con una solución de polivinilpirrolidona en EtOH. El granulado seco se puede mezclar con el estearato de magnesio y el resto del almidón. La mezcla se comprime luego en una máquina de fabricación de comprimidos para dar comprimidos que contienen cada uno una dosis unitaria o una parte de una dosis unitaria de compuesto activo.
c) Comprimidos con cubierta entérica
Los comprimidos se pueden preparar por el método descrito en (b) arriba. Los comprimidos pueden recubrirse entéricamente de manera convencional usando una solución de 20% de acetato ftalato de celulosa y 3% de ftalato de dietilo en EtOH:DCM (1:1).
d) Supositorios
En la preparación de supositorios, por ejemplo, se pueden incorporar 100 partes en peso de compuesto activo en 1300 partes en peso de base de supositorio de triglicéridos y la mezcla se forma en supositorios que contienen cada uno una cantidad terapéuticamente efectiva de ingrediente activo.
En las composiciones de la presente invención, si se desea, el compuesto activo puede asociarse con otros ingredientes farmacológicamente activos compatibles. Por ejemplo, los compuestos de esta invención pueden administrarse en combinación con otro agente terapéutico que se sabe que trata una enfermedad o afección descrita en la presente descripción. Los compuestos de la invención pueden administrarse antes, después o simultáneamente con el agente farmacéutico adicional, cualquiera que sea el curso de administración apropiado. Los agentes farmacéuticos adicionales incluyen, pero no se limitan a, esteroides antidémicos, AINE, inhibidores de ras, agentes anti-IL1, antihistamínicos, antagonistas de PAF, inhibidores de COX-1, inhibidores de COX-2, inhibidores de NO sintasa, inhibidores de Akt/PTB, inhibidores de IGF-1R, inhibidores de PKC, inhibidores de PI3 quinasa, inhibidores de calcineurina e inmunosupresores. Los compuestos de la invención y los agentes farmacéuticos adicionales actúan de forma aditiva o sinérgica.
La presente invención también comprende el uso de un compuesto de Fórmula (I-a) como un medicamento.
La presente invención también proporciona un compuesto de Fórmula (I-a) para usar en un método para el tratamiento de artritis reumatoide, artritis reumatoide juvenil, psoriasis, artritis psoriásica, enfermedad de Crohn, enfermedad inflamatoria intestinal, colitis ulcerosa, espondilitis anquilosante, artritis reactiva, artritis asociada con la enfermedad inflamatoria intestinal, espondiloartritis indiferenciada, lupus eritematoso sistémico, nefritis lúpica, uveítis, dermatitis atópica, esclerosis múltiple, espondiloartritis axial o hidradenitis supurativa y/u otros trastornos del sistema inmunitario.
ABREVIATURAS

Esquemas sintéticos generales
Los compuestos de la invención pueden prepararse usando las transformaciones sintéticas ilustradas en los Esquemas IXXIII. Los materiales de partida están disponibles comercialmente, pueden prepararse mediante los procedimientos descritos en la presente descripción, mediante procedimientos de literatura o mediante procedimientos que serían bien conocidos por un experto en la técnica de la química orgánica.
Esquema I. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((1-metil-1H-indol-2-il)metil) benzamida de la invención se ilustran en el Esquema I, y se describen adicionalmente en el Ejemplo A, Al, L, M, V, AA, AC, AX: (R= Br, CN, CF3 , Me, H; R1 = CN, CF3 , Me, H; R2= CN, CF3 , Me, H; R3=amina primaria y secundaria).

Esquema II. Los métodos generales para preparar los compuestos (2,4-dicloro-3-((1-alquil-1H-indol-2-il)metil)fenil)(morfolino)metanona de la invención se ilustran en el Esquema II, y se describen adicionalmente en el Ejemplo
C, D, E, P, AB, AE: (R= CN, CF3, Me, H; R-,= CN, CF3, Me, H; R2= CN, CF3, Me, H; R3= C1, Me, H; R4= H, sustituyente acilo o alquilo).

Esquema III. Los métodos generales para preparar los compuestos 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzamida)) de la invención se ilustran en el Esquema III, y se describen adicionalmente en el Ejemplo F:
(R= CN, CF3 ,Me, H; Ri= cN, CF3 ,Me, H; R2= amina primaria y secundaria).
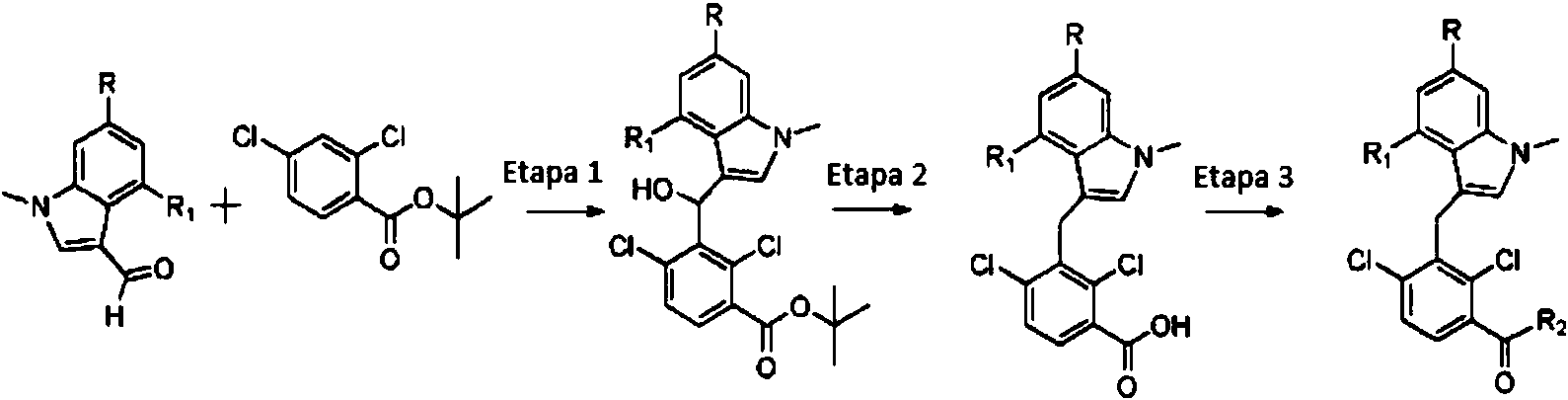
Esquema IV. Los métodos generales para preparar los compuestos 2-(2,6-dicloro-3-alcoxibencil)-1-metil-1H-indol de la
invención se ilustran en el Esquema IV, y se describen adicionalmente en los Ejemplos W, X, Y, Z, AF, AG, AH, AI, AJ,
AK, AL, BI: (R= CN, CF3, Me, H; R1= CN, CF3, Me, H; R2= CN, CF3, Me, H; R3= H, alquilo, cicloalquilo, CH2-heterociclo;
X=C o N).

Esquema V. Los métodos generales para preparar compuestos 3,5-dicloro-4-((1-metil-1H-indol-2-il)metil)benzamida de la invención se ilustran en el Esquema V, y se describen adicionalmente en el Ejemplo R: (R= CN, CF3 , Me, C1, H; Ri = CN, CF3 , Me, C1, H; R2= CN, CF3 , Me, C1, H; R3= amina primaria y secundaria).

Esquema VI. Los métodos generales para preparar los compuestos 2,4-disustituido-3-((1-metil-1H-benzo[C]imidazol-2-il)metil)benzamida de la invención se ilustran en el Esquema Vi, y se describen adicionalmente en el Ejemplo BJ, BK, BL, BM, bN, BQ, BR, BU, BV, BY, BZ, CA, CB: (R= CN, CF3 , alquilo, haluro, amida, H; R1 = CN, CF3 , alquilo, haluro, amida, H; R2= CN, CF3 , alquilo, haluro, amida, H; R3= CN, CF3, alquilo, haluro, amida, H; R4 =C1, alquilo; R5 amina primaria y secundaria).

Esquema VII. Los métodos generales para preparar los compuestos (2,4-dicloro-3-((3-metil-3H-imidazo[4,5-b]piridin-2-il)metil)fenil)(morfolino)metanona de la invención se ilustran en el Esquema VII, y se describen adicionalmente en el Ejemplo CI, DA: (R= CN, CF3 , alquilo, haluro, amida, H).

Esquema VIII. Los métodos generales para preparar los compuestos W-(3,5-dicloro-4-((1-metil-1H-benzo[C]imidazol-2-il)metil)fenil)acetamida de la invención se ilustran en el Esquema VIII, y se describen adicionalmente en el Ejemplo CJ: (R= cN, CF3, alquilo, H; R1= CN, CF3, alquilo, H; R2= alquilo o cicloalquilo).

Esquema IX. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((1-metil-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzamida de la invención se ilustran en el Esquema IX, y se describen adicionalmente en el Ejemplo CL : (R= CN, CF3, alquilo, H; R1= CN, CF3, alquilo, H, halógeno; R2= amina primaria y secundaria).

Esquema X. Los métodos generales para preparar los compuestos (2,4-dicloro-3-((1-metil-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona de la invención se ilustran en el Esquema X, y se describen adicionalmente en el Ejemplo CO : (R= CN, CF3, alquilo, H; R1= CN, CF3, alquilo, H; R2= amina primaria y secundaria).
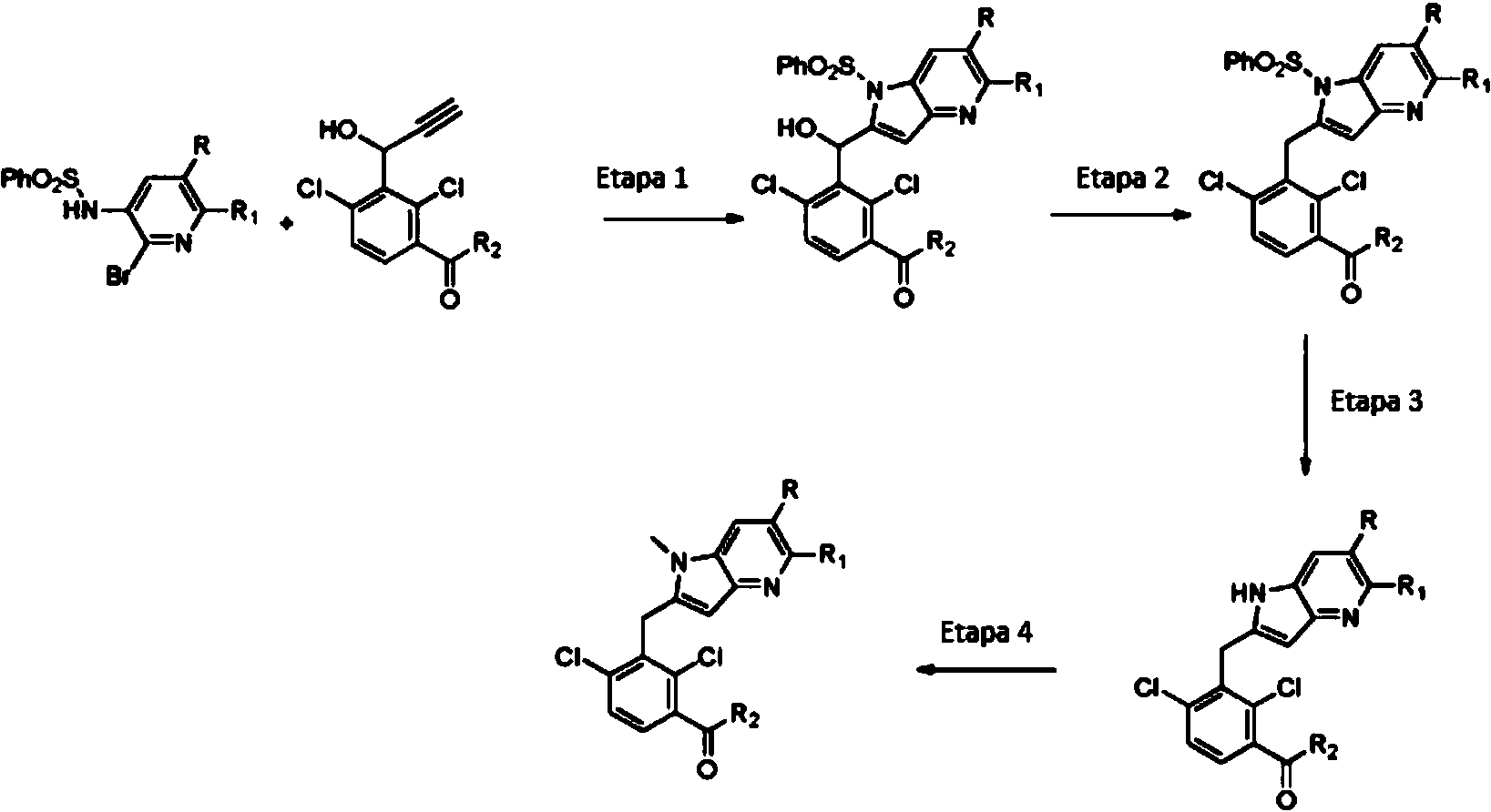
Esquema XI. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((1-metiMH-pirrolo[3,2-c]piridin-2-il)metil)benzamida de la invención se ilustran en el Esquema XI, y se describen adicionalmente en el Ejemplo CQ, CR y CS : (R= CN, CF3 , alquilo, H; R1 = CN, CF3 , alquilo, H; R2= amina primaria y secundaria).

Esquema XII. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((3-metilimidazo[l,2-a]piridin-2-il)metil)benzamida de la invención se ilustran en el Esquema XII, y se describen adicionalmente en el Ejemplo CT, CV, CW y CZ : (R= CN, CF3 , alquilo, haluro, H; R1 = CN, CF3 , alquilo, haluro, H; R2= CN, CF3 , alquilo, haluro, H; R3= amina primaria y secundaria; X=C o N).

Esquema XIII. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((1-metil-3,4-dihidroisoquinolin-2(1H)-il)metil)benzamida de la invención se ilustran en el Esquema XIII, y se describen adicionalmente en el Ejemplo DO:

Esquema XIV. Los métodos generales para preparar los compuestos 2,4-disustituido-3-((1-alquil-1H-benzo[C]imidazol-2-il)metil)benzamida de la invención se ilustran en el Esquema XIV, y se describen adicionalmente en los Ejemplos CG y CH : (R= H; R1 = CN, CF3, alquilo, haluro, amida, H; R2= CN, CF3 , alquilo, haluro, amida, H; R3= CN, CF3 , alquilo, haluro, amida, H; R4 =C1, alquilo; R5 amina primaria y secundaria; R6 = alquilo).

Esquema XV. Los métodos generales para preparar los compuestos 2,4-disustituido-3-((1-metil-1H-benzo[C]imidazol-2-il)metil)bencilamina de la invención se ilustran en el Esquema XV, y se describen adicionalmente en los Ejemplos BW y BX : (R= CN, CF3 , alquilo, haluro, amida, H; R1 = CN, CF3 , alquilo, haluro, amida, H; R2= CN, CF3 , alquilo, haluro, amida, H; R3= CN, CF3 , alquilo, haluro, amida, H; R4 =C1, alquilo; R5 amina primaria y secundaria).
A partir del Esquema XII

Esquema XVI. Los métodos generales para preparar los compuestos (2,4-dicloro-3-((3-metil-1H-indazoM-il)metil)benzamida o (2,4-dicloro-3-((3-metil-2H-indazol-2-il)metil)benzamida de la invención se ilustran en el Esquema XVI, y se describen adicionalmente en los Ejemplos DD, DD1, DE, DF y DG : (R 1 = CN, CF3 , alquilo, haluro, amida, H; R2= CN, CF3, alquilo, haluro, amida, H; R3= CN, CF3, alquilo, haluro, amida, H; R*= CN, CF3 , alquilo, haluro, amida, H; R5=metilo; R6 = amina primaria y secundaria; R7=cloro).

Esquema XVII. L o s m é t o d o s g e n e r a l e s p a r a p r e p a r a r l o s c o m p u e s t o s (2 ,4 - d i c l o r o - 3 - ( ( 3 - m e t i l - 3 H - i m i d a z o [ 4 , 5 - b ] p i r i d i n - 2 -i l ) m e t i l ) f e n i l ) ( m o r f o l i n o ) m e t a n o n a d e la i n v e n c i ó n s e i l u s t r a n e n e l E s q u e m a X V I I , y s e d e s c r i b e n a d i c i o n a l m e n t e e n e l E j e m p l o D C : ( R = C N , C F 3 , a lq u i l o , h a l u r o , a m i d a , H ) .

Esquema XVIII. L o s m é t o d o s g e n e r a l e s p a r a p r e p a r a r l o s c o m p u e s t o s 2 ,4 - d i c l o r o - 3 - ( ( 1 - m e t i l - 1 H - i n d o l - 2 -

Esquema XIX. d e l a i n v e n c i ó n C N , C F 3 , M e , H

Esquema XX. Los métodos generales para preparar los compuestos 2,4-dicloro-3-(1 -metil-1 H-indol-2-carbonil)benzamida de la invención se ilustran en el Esquema XX, y se describen adicionalmente en el Ejemplos AN, AO, AP y AQ: (A= C, N; R= CN, CF3 , Me, H; R1 = CN, CF3 , Me, H; R2= Cn, CF3 , Me, H; R3= amina primaria y secundaria).
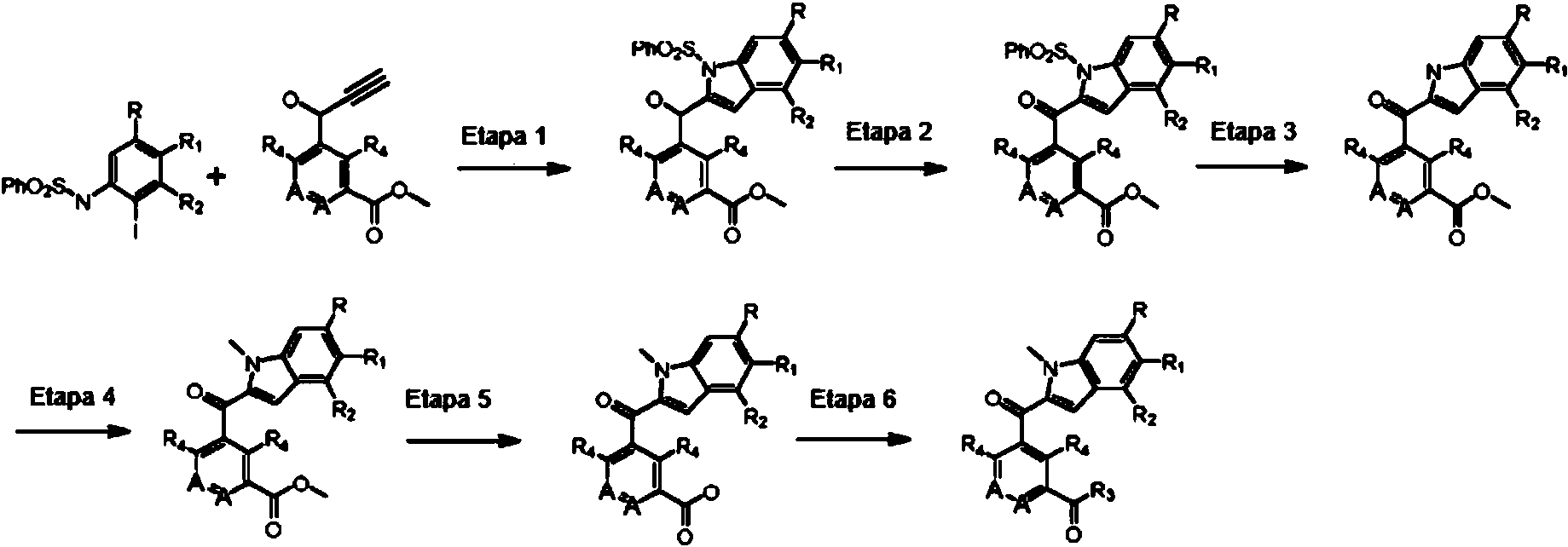
Esquema XXI. Los métodos generales para preparar los compuestos 2,4-disustituido-3-((1-metil-1H-indol-2-il)metil)bencilamina de la invención se ilustran en el Esquema XXI, y se describen adicionalmente en el Ejemplo AU : (R= CN, CF3 , alquilo, haluro, amida, H; R1 = CN, CF3 , alquilo, haluro, amida, H; R2= CN, CF3 , alquilo, haluro, amida, H; R3 = CN, CF3 , alquilo, haluro, amida, H; R4 =C1, alquilo; R5 amina primaria y secundaria).
A partir del Esquema I

Esquema XXII. Los métodos generales para preparar los compuestos 2,4-dicloro-3-((1-metil-6-morfolino-1H-indol-2 il)metil) benzamida de la invención se ilustran en el Esquema XXII, y se describen adicionalmente en el Ejemplo AY : (R1 = CN, CF3 , Me, H; R2= CN, CF3 , Me, H; R3= amina primaria y secundaria).

Esquema XXIII. Los métodos generales para preparar los compuestos 2,4-dicloro-3-(1H-indol-1-il)metil) benzamida de la invención se ilustran en el Esquema XXIII, y se describen adicionalmente en el Ejemplos J, DH, DI, DJ, DK, DL, DM y DN: (R i = CN, CF3, Me, H; R2= CN, CF3 , Me, H; R3= CN, CF3 , Me, H; R4= CN, CF3 , Me, H; Rs = Me, H; R6= amina primaria y secundaria; X= H, O, Rz= Me o terc-Bu).

Métodos analíticos
Los datos analíticos se incluyeron dentro de los procedimientos a continuación, en las ilustraciones de los procedimientos generales, o en las tablas de ejemplos. A menos que se indique lo contrario, todos los datos de 1H NMR se recolectaron en un Varian Mercury Plus 400 MHz o un instrumento Varian Inova 600 MHz y los cambios químicos se mencionan en partes por millón (ppm). Los datos de LC/MS y HPLC se mencionan en la tabla de condiciones de LC/MS y HPLC usando la letra del método en minúscula proporcionada en la Tabla 1.
Tabla 1. Métodos de LC/MS y HPLC.


Tabla 2. Métodos de HPLC uiral

Preparaciones y ejemplos
L o s m é t o d o s s i n t é t i c o s g e n e r a l e s u s a d o s e n c a d a p r e p a r a c i ó n s i g u e n e i n c l u y e n u n a i l u s t r a c ió n d e u n c o m p u e s t o q u e s e s in t e t iz ó . N i n g u n a d e l a s c o n d i c i o n e s y r e a c t i v o s e s p e c í f i c o s in d i c a d o s e n la p r e s e n t e d e s c r i p c i ó n d e b e n in t e r p r e t a r s e c o m o l i m it a n t e s d e l a l c a n c e d e la i n v e n c i ó n y s e p r o p o r c io n a n ú n i c a m e n t e c o n f i n e s i l u s t r a t iv o s . T o d o s l o s m a t e r i a l e s d e p a r t id a e s t á n d i s p o n i b l e s c o m e r c i a l m e n t e d e S i g m a - A l d r i c h ( i n c l u i d o s F l u k a y D i s c o v e r y C P R ) a m e n o s q u e s e i n d iq u e lo c o n t r a r io d e s p u é s d e l n o m b r e q u í m i c o . L o s n o m b r e s d e r e a c t i v o s / r e a c t i v o s d a d o s s o n l o s q u e f ig u r a n e n l a b o t e l la c o m e r c i a l o l o s g e n e r a d o s p o r l a s c o n v e n c i o n e s I U P A C , C a m b r i d g e S o f t ® C h e m D r a w U lt r a 9 .0 .7 , C a m b r i d g e S o f t ® C h e m i s t r y E - N o t e b o o k 9 .0.127 o A u t o N o m 2000 . L o s c o m p u e s t o s d e s i g n a d o s c o m o s a l e s ( p o r e je m p l o , h id r o c lo r u r o , a c e t a t o ) p u e d e n c o n t e n e r m á s d e u n e q u i v a l e n t e m o l a r d e la s a l . L o s c o m p u e s t o s d e la i n v e n c i ó n e n l o s q u e la e s t e r e o q u í m i c a a b s o l u t a s e h a d e t e r m in a d o m e d ia n t e e l u s o d e u n m a t e r ia l d e p a r t id a e n a n t i o m é r i c a m e n t e p u r o c o m e r c i a l m e n t e d i s p o n i b l e o u n p r o d u c t o in t e r m e d io d e f in i d o e s t e r e o q u í m i c a m e n t e , o m e d i a n t e d i f r a c c i ó n d e r a y o s X s e d e n o t a n c o n u n a s t e r i s c o d e s p u é s d e l n ú m e r o d e e je m p l o . L o s e j e m p l o s q u e n o e n t r a n d e n t r o d e l a l c a n c e d e l a s r e i v i n d i c a c i o n e s a d j u n t a s s e p r o p o r c io n a n c o m o e j e m p l o s c o m p a r a t i v o s .
Preparación #1. Metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato:

Etapa A: metil 2,4-dicloro-3-metilbenzoato

A una solución de ácido 2,4-didoro-3-metilbenzoico (250 g, 1.219 mmol) en MeOH (1,5 L) se añadió ácido sulfúrico (78 mL, 1.463 mmol) y la reacción se agitó a reflujo durante 20 horas. La mezcla de reacción se concentró al vacío. El producto crudo se disolvió en diclorometano. La capa orgánica se lavó con agua, luego con una solución acuosa saturada de NaHCO3 y salmuera, se secó sobre sulfato de magnesio y se concentró a vacío para dar metil 2,4-dicloro-3-metilbenzoato (196 g, 73,4%) como un aceite incoloro que precipita lentamente. LC/MS (Método h) Rt = 2,78 min.; MS m/z: 217 [M-H]-.
1H NMR (DMSO-cfe, 300 MHz): 57,61 (d, J= 8,4 Hz, 1H), 7,55 (d, J= 8,4 Hz, 1H), 3,87 (s, 3H), 2,37 (s, 3H).
Etapa B: metil 3-(bromometil)-2,4-diclorobenzoato:

A una solución de metil 2,4-dicloro-3-metilbenzoato (19,6 g, 89 mmol) en ACN (100 mL) se añadió N -bromosuccinimida (20,70 g, 116 mmol) y 2,2'-azobis(2-metilpropionitrilo) (0,735 g, 4,47 mmol) en pequeñas fracciones a temperatura ambiente. La mezcla de reacción se agitó a reflujo 18 horas y luego se concentró al vacío. El producto crudo se diluyó con EtOAc y el sólido se filtró y se lavó con EtOAc. La capa orgánica se lavó sucesivamente con una solución acuosa saturada de NaHCO3, solución acuosa de Na2S2O310%, se secó sobre sulfato de magnesio, se filtró y evaporó para dar metil 3-(bromometil)-2,4-diclorobenzoato (24,9 g, 93% de rendimiento) como un sólido de color blanco roto. No LC/MS 1H NMR (DMSO-cfe, 300 MHz): 57,78 (d, J= 9 Hz, 1H), 7,67 (d, J= 9 Hz, 1H), 4,84 (s, 2H), 3,88 (s, 3H).
Etapa C: metil 2,4-dicloro-3-formilbenzoato:

A una solución de metil 3-(bromometil)-2,4-diclorobenzoato (51,6 g, 173 mmol) en ACN (400 mL) se añadió 4-metilmorfolina N-óxido (81 g, 693 mmol) en solución en agua (160 mL). La mezcla de reacción se agitó a reflujo por 1,5 horas, se enfrió a temperatura ambiente y se diluyó con agua hasta la precipitation del compuesto esperado. El sólido se filtró, se lavó con agua y se secó al vacío para dar metil 2,4-dicloro-3-formilbenzoato (25,48 g, 63,1%) como un sólido de color blanco.
LC/MS (Método h) Rt = 2,21 min.; sin ionización 1H NMR (DMSO-cfe, 300 MHz): 5 10,33 (s, 1H), 7,95 (d, J = 8,4 Hz, 1H), 7,72 (d, J = 8,4 Hz, 1H), 3,89 (s, 3H).
Etapa D: metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato

A una suspensión de metil 2,4-dicloro-3-formilbenzoato (19,36 g, 83 mmol) en THF (80 mL) se añadió gota a gota bromuro de etinilmagnesio (0,5M en THF, 199 mL, 100 mmol) y la mezcla de reacción se agitó a temperatura ambiente por 1 hora. Luego se diluyó con una solución HCl 1N y se extrajo EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 0-30% EtOAc en ciclohexano) para dar metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (19,6 g, 91%) como un sólido de color blanco. LC/MS (Método h) Rt = 1,95 min.; MS m/z: 259 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,67 (d, J = 9 Hz, 1H), 7,59 (d, J = 9 Hz, 1H), 6,29 (s, 1H), 6,15 (d, J=2,48Hz, 1H), 3,87 (s, 3H), 3,51 (d, J=2,48Hz, 1H).
Preparación #2. Ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético

Etapa A: metil 2,4-didoro-3-(cianometil)benzoato

A una solución calentada (40°C) de KCN (8,52 g, 131 mmol) en DMSO (150 mL) se añadió gota a gota una solución de metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (30 g, 101 mmol) en DMSO (90 mL). La mezcla de reacción se agitó a 40 °C por 2h30. Luego se enfrió a temperatura ambiente y se añadió una solución acuosa saturada de NaHCO3. El producto se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-50% EtOAc en ciclohexano) para dar metil 2,4-dicloro-3-(cianometil)benzoato (19,1 g, 78%) como un sólido de color blanco. LC/MS (Método h) Rt = 2,19 min.; MS m/z: 242 [M-H]‘.1H NMR (DMSO-cfe, 300 MHz): 57,83 (d, J = 9 Hz, 1H), 7,72 (d, J = 9 Hz, 1H), 4,26 (s, 2H), 3,89 (s, 3H).
Etapa B: metil 2,4-dicloro-3-(2-oxoetil)benzoato

A una solución de metil 2,4-dicloro-3-(cianometil)benzoato (11 g, 45,1 mmol) en una mezcla de piridina (171 mL), ácido acético (85 mL) y agua (85 mL), se añadió hipofosfito de sodio (31,7 g, 361 mmol). La mezcla de reacción se agitó 15 min a temperatura ambiente. Se añadió níquel Raney activado (13,13 mL, 77 mmol) y la mezcla de reacción se agitó a 80 °C durante 1,5 horas. La mezcla de reacción se filtró, se lavó con EtOAc y agua. Las capas se separaron y la capa orgánica se lavó con una solución de HCl 1M, y luego se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 10-25% EtOAc en ciclohexano) para dar metil 2,4-dicloro-3-(2-oxoetil)benzoato (10,24 g, 65,3 % de rendimiento). LC/MS (Método h) Rt = 2,19 min., 1,59 min. (hidrato); MS m/z: 323 [M-H]- +CH3COOH 1H NMR (DMSO-cfe, 300 MHz): 59,76 (s, 1H), 7,72 (d, J = 9 Hz, 1H), 7,64 (d, J = 9 Hz, 1H), 4,27 (s, 2H), 3,87 (s, 3H).
Etapa C: ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético

A una solución de metil 2,4-dicloro-3-(2-oxoetil)benzoato (3g, 12,14 mmol) en THF (50 mL) y agua (50,0 mL) se añadieron sucesivamente 2-metil-2-buteno (60,7 mL, 121 mmol), clorito de sodio (8,79 g, 97 mmol) y fosfato de sodio, monobásico, monohidratado (15,08 g, 109 mmol). La mezcla se agitó a temperatura ambiente por 1,5 horas y luego se diluyó con diclorometano y una solución acuosa saturada de NaHCO3. Las capas se separaron y la capa acuosa se volvió a acidificar con solución de HCl 1N y se extrajo con diclorometano. La capa orgánica se secó sobre sulfato de magnesio y se concentró a presión reducida. El residuo se trituró en pentano para dar ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (3 g, 94%). LC/MS (Método h) Rt = 1,91 min.; MS m/z: 263 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 5 12,78 (ancho, 1H), 7,70 (d, J= 9 Hz, 1H), 7,62 (d, J = 9 Hz, 1H), 3,97 (s, 2H), 3,87 (s, 3H).
Preparación #3. (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona

Etapa A: ácido 2,4-didoro-3-formilbenzoico

A una solución de metil 2,4-didoro-3-formilbenzoato (Preparación #1, Etapa C) (20 g, 86 mmol) en THF (240 mL) y agua (120 mL) se añadió hidróxido de litio (3.083 mg, 129 mmol) y la mezcla de reacción se a temperatura ambiente durante la noche. La mezcla de reacción se evaporó parcialmente. La capa acuosa resultante se lavó con diclorometano y luego se acidificó con una solución de HCl 1 M a (pH = 2-3) y el compuesto se extrajo sucesivamente con diclorometano y EtOAc. Las capas orgánicas se secaron sobre sulfato de magnesio y se concentraron a presión reducida para dar ácido 2,4-dicloro-3-formilbenzoico (17,6 g, 94%). LC/MS (Método h) Rt = 1,02 min.; MS m/z: 217 [M-H]-1H NMR (DMSO-cfe, 300 MHz): 513,88 (ancho, 1H), 10,36 (s, 1H), 7,92 (d, J= 9 Hz, 1H), 7,67 (d, J = 9 Hz, 1H).
Etapa B: 2,6-didoro-3-(morfolina-4-carbonil)benzaldehído

A una suspensión de ácido 2,4-dicloro-3-formilbenzoico (17,2 g, 79 mmol) en diclorometano (340 mL) se añadió HATU (32,8 g, 86 mmol) y 4-metilmorfolina (9,51 mL, 86 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 20 minutos, y luego se añadió morfolina (10,19 mL, 118 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche. La mezcla de reacción se diluyó con diclorometano y se lavó con una solución acuosa saturada de NaHCO3. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-40% EtOAc en ciclohexano) para dar 2,6-dicloro-3-(morfolina-4-carbonil)benzaldehído (23,8g, 100%) como un sólido de color blanco.
LC/MS (Método h) Rt = 1,58 min.; MS m/z: 288 [M+H]+ 1H NMR (DMSO-de, 300 MHz): 510,36(s, 1H), 7,69 (d, J=9Hz, 1H), 7,64 (d, J=9Hz, 1H), 3,66 (m, 4H), 3,54 (m, 2H), 3,16 (m, 2H).
Etapa C: (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona:

Usando un procedimiento similar a la Preparación #1, se preparó (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (14,1 g, 99%) a partir de 2,6-dicloro-3-(morfolina-4-carbonil)benzaldehído (13 g, 45,1 mmol). LC/MS (Método h) Rt = 1,43 min.; MS m/z: 314 [M+H]+ 1H NMR (DMSO-de , 300 MHz): 57,55 (d, J=9Hz, 1H), 7,38 (d, J=9Hz, 1H), 6,26 (s, 1H), 6,09 (d d, J=3Hz, 1H), 3,65 (m, 4H), 3,54 (m, 2H), 3,51 (d, J=3Hz, 1H), 3,12 (m, 2H).
Preparación #4. (3-(1 -hidroxiprop-2-in-1 -il)-2,4-dimetilfenil)(morfolino)metanona

Etapa A: 3-bromo-2,6-dimetilbenzaldehído

A una suspensión de tricloruro de aluminio (14,91 g, 112 mmol) en diclorometano (80 mL) se añadieron gota a gota a 0 °C 2,6-dimetilbenzaldehído (10 g, 74,5 mmol) en solución en diclorometano (80 mL) y dibromina (3,72 mL, 72,3 mmol). La mezcla se agitó 4 horas a 0 °C. La mezcla de reacción se vertió en hielo y se extrajo dos veces con diclorometano. Las capas orgánicas se lavaron con una solución de HCl 1N, una solución acuosa saturada de NaHCO3, se secaron sobre sulfato de magnesio y se concentraron a presión reducida para dar 3-bromo-2,6-dimetilbenzaldehído (14,4g, 91% de rendimiento) como un aceite amarillo.
LC/MS (Método h) Rt = 2,62 min.; sin ionización. 1H NMR (DMSO-cfe, 300 MHz): 510,47 (s, 1H), 7,72 (d, J=9Hz, 1H), 7,12 (d, J=9Hz, 1H), 2,57 (s, 3H), 2,47 (s, 3H).
Etapa B: ácido 3-formil-2,4-dimetilbenzoico

En un reactor de microondas se añadieron acetato de paladio(II) (13,70 mg, 0,061 mmol), tetrafluoroborato de tri-tercbutilfosfonio (17,70 mg, 0,061 mmol) y hexacarbonilo de molibdeno (354 mg, 1,342 mmol), 3-bromo-2,6-dimetilbenzaldehído (260 mg, 1,220 mmol) en solución en dimetoxietano (2 mL) y carbonato de sodio (194 mg, 1,830 mmol) en solución en agua (0,5 mL). La mezcla de reacción se agitó una hora a 120 °C bajo irradiación de microondas. El dimetoxietano se evaporó y el residuo se diluyó con agua y se lavó con EtOAc. La capa acuosa se acidificó con solución de HCl 1N (pH 1) y se extrajo con diclorometano, se secó sobre sulfato de magnesio y se concentró a presión reducida para dar ácido 3-formil-2,4-dimetilbenzoico (150 mg, 69%) como un polvo blanco. LC/MS (Método h) Rt = 1,50 min.; MS m/z: 177 [M-H]-.1H NMR (DMSO-cfe, 300 MHz): 5 13,06 (ancho, 1H), 10,56 (s, 1H), 7,78 (d, J=9Hz, 1H), 7,24 (d, J=9Hz, 1H), 2,65 (s, 3H), 2,52 (s, 3H).
Etapa C: 2,6-dimetil-3-(morfolina-4-carbonil)benzaldehído

A una solución de ácido 3-formil-2,4-dimetilbenzoico (1 g, 5,61 mmol) en DMF (5 mL) se añadió 1,1'-carbonildiimidazol (1,183 g, 7,30 mmol) y la mezcla de reacción se agitó a temperatura ambiente por 4 horas. Morfolina (0,971 mL, 11,22 mmol) y trietilamina (1,564 mL, 11,22 mmol) se añadieron y la mezcla de reacción se agitó a temperatura ambiente por una noche. La mezcla de reacción se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa obtenida se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH al 0,1-5% en diclorometano) para dar 2,6-dimetil-3-(morfolina-4-carbonil) benzaldehído (780 mg, 56%).
LC/MS (Método h) Rt = 1,46 min.; MS m/z: 248 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 5 10,53 (s, 1H), 7,32 (d, J=9Hz, 1H), 7,24 (d, J=9Hz, 1H), 3,65(m, 4H), 3,48 (m, 2H), 3,11 (m, 2H), 2,54 (s, 3H), 2,42 (s, 3H).
Etapa D: (3-(1-hidroxiprop-2-in-1-il)-2,4-dimetilfenil)(morfolino)metanona:

Usando un procedimiento similar a la Preparación #1, (3-(1-hidroxiprop-2-in-1-il)-2,4-dimetilfenil)(morfolino)metanona (680 mg, 64%) se preparó a partir de 2,6-dimetil-3-(morfolina-4-carbonil)benzaldehído (953 mg, 3,85 mmol). LC/MS (Método h) Rt = 1,39 min.; MS m/z: 274 [M+H]+ . 1H NMR (DMSO-cfe, 300 MHz): 5 7,06 (d, J=7,6Hz, 1H), 6,98 (d, J=7,6Hz, 1H), 5,83 (m, 1H), 5,76 (m, 1H), 3,64 (m, 4H), 3,47 (m, 2H), 3,41 (d, J=2,3Hz, 1H), 3,10 (m, 2H), 2,44 (s, 3H), 2,34 (s, 3H).
Preparación #5. (3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona:

Usando un procedimiento similar a la Preparación #1, (3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (2 g, 73%) se preparó a partir de 3-(morfolina-4-carbonil)benzaldehído (descrito en WO2004/058762) (2,54 g, 11,59 mmol). LC/MS (Método h) Rt = 1,09 min.; MS m/z: 246 [M+H]+. 1H NMR (DMSO d-, 300 MHz): 57,62 (m, 2H), 7,47-7,34 (m, 2H), 5,47 (d, J=2Hz, 1H), 3,90-3,30 (m ancho, 9H), 2,67 (d, J=2Hz, 1H).
Preparación #6: metil 3,5-didoro-4-(1-hidroxiprop-2-in-1-il)benzoato:

Usando un procedimiento similar a la Preparación #1, metil 3,5-dicloro-4-(1-hidroxiprop-2-in-1-il)benzoato (1 g, 60%) se preparó a partir de metil 3,5-dicloro-4-formilbenzoato (descrito en WO2013/149997) (1,5 g, 6,44 mmol). LC/MS (Método h) Rt = 2,14 min.; sin ionización 1H NMR (DMSO-d6, 300 MHz): 57,91 (s, 2H), 6,37 (s, 1H), 6,09 (d, J=2Hz, 1H), 3,88 (s, 3H), 3,54 (d, J=2Hz, 1H).
Preparación #7. 1-(2,6-dicloro-3-metoxifenil)prop-2-in-1-ol:

Usando un procedimiento similar a la Preparación #1, 1-(2,6-dicloro-3-metoxifenil)prop-2-in-1-ol (1,68 g, 93%) se preparó a partir de 2,6-dicloro-3-metoxibenzaldehído (descrito en WO2006/049952) (1,6 g, 7,80 mmol). LC/MS (Método h) Rt = 1,95 min.; sin ionización 1H NMR (DMSO-cfe, 300 MHz): 57,42 (d, J=9Hz, 1H), 7,14 (d, J=9Hz, 1H), 6,12 (s, 1H), 6,05 (d, J=2,4Hz, 1H), 3,86 (s, 3H), 3,41 (d, J=2,4Hz, 1H).
Preparación #8. Ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)fenil)acético
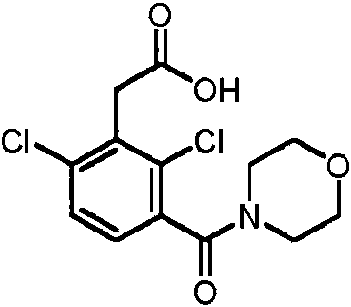
Etapa A: metil 2,4-dicloro-3-(2-hidroxietil)benzoato

A una solución de metil 2,4-dicloro-3-(2-oxoetil)benzoato (Preparación #2, Etapa B) (1,88 g, 7,61 mmol) en MeOH (28 mL) y se enfrió a 0 °C se añadió borohidruro de sodio (0,288 g, 7,61 mmol) y mezcla de reacción se agitó a 0 °C por 30
minutos. Se añadió agua y el MeOH se evaporó. La capa acuosa resultante se extrajo con diclorometano. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar metil 2,4-dicloro-3-(2-hidroxietil)benzoato (1,6 g, 84%). LC/MS (Método h) Rt = 1,93 min.; MS m/z: 249 [M+H]+. 1H NMR (DMSO-cf6, 300 MHz): 5 7,61 (d, J=8,4Hz, 1H), 7,56 (d, J=8,4Hz, 1H), 4,92 (ancho, 1H), 3,85 (s, 3H), 3,56 (m, 2H), 3,13 (t, J=7,8Hz, 2H)
Etapa B: ácido 2,4-dicloro-3-(2-hidroxietil)benzoico

Usando un procedimiento similar a la Preparación #3, Etapa A, se preparó ácido 2,4-dicloro-3-(2-hidroxietil)benzoico (1,41 g, 93%) a partir de metil 2,4-dicloro-3-(2-hidroxietil)benzoato (1,60 g, 6,42 mmol). LC/MS (Método h) Rt = 1,20 min.; MS m/z: 233 [M-H]-. 1H NMR (DMSO-cf6, 300 MHz): 5 13,56 (ancho, 1H), 7,56 (d, J=8,4Hz, 1H), 7,51 (d, J=8,4Hz, 1H), 4,92 (ancho, 1H), 3,55 (t, J=2,7Hz, 2H), 3,12 (t, J=7,8Hz, 2H)
Etapa C: (2,4-dicloro-3-(2-hidroxietil)fenil)(morfoIino)metanona

A una suspensión de ácido 2,4-dicloro-3-(2-hidroxietil)benzoico (1,4 g, 5,96 mmol) en diclorometano (75 mL) se añadió clorhidrato de 1-(3-dimetilaminopropil)-3-etilcarbodiimida (1,484 g, 7,74 mmol) y 1-hidroxibenzotriazol (1,046 g, 7,74 mmol) morfolina (1,031 mL, 11,91 mmol) y trietilamina (1,660 mL, 11,91 mmol). La mezcla de reacción se agitó a temperatura ambiente por una noche. Luego se diluyó con diclorometano. Las capas se separaron y la capa orgánica obtenida se lavó con agua, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-40% EtOAc en ciclohexano) para dar (2,4-dicloro-3-(2-hidroxietil)fenil)(morfolino)metanona (1,2 g, 66%) como un sólido de color blanco.
LC/MS (Método h) Rt = 1,41 min.; MS m/z: 304 [M+H]+. 1H NMR (DMSO-cf6, 300 MHz): 5 7,53 (d, J=9Hz, 1H), 7,27 (d, J=9Hz, 1H), 4,91 (t, J=6Hz, 1H), 3,65-3,50 (m, 8H), 3,10 (m, 4H).
Etapa D: ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)fenil)acético

A una solución de óxido de cromo(vi) (182 mg, 1,816 mmol) en agua (325 pl) y enfriada a 0 °C se añadió ácido sulfúrico (162 pL, 3,05 mmol) y la mezcla de reacción se agitó a 0 °C por 10 min. Esta solución se añadió lentamente a una solución de (2,4-dicloro-3-(2-hidroxietil)fenil)(morfolino)metanona (325 mg, 1,068 mmol) en acetona (3898 pL) enfriada a 0 °C. La mezcla de reacción se agitó a 0 °C por 15min, luego a temperatura ambiente por 30 minutos. La mezcla de reacción se filtró y se lavó con acetona. El filtrado se concentró. El residuo se diluyó en EtOAc y se lavó con una solución de HCl 1N. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró para dar ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)fenil)acético (283 mg, 83%). LC/MS (Método h) Rt = 1,42 min.; MS m/z: 318 [M+H]+ 1H NMR (DMSO-d6, 300 MHz): 5 12,74 (ancho, 1H), 7,59 (d, J=9Hz, 1H), 7,36 (d, J=9Hz, 1H), 3,93 (s, 2H), 3,66 (m, 4H), 3,52 (m, 2H), 3,11 (m, 2H).
Preparación #9. Ácido 2-(3-(metoxicarbonil)-2,6-dimetilfenil)acético

Etapa A: ácido 2-(3-bromo-2,6-dimetilfenil)acético

A una solución calentada (40 °C) de ácido 2-(2,6-dimetilfenil) acético (2 g, 12,18 mmol) en ácido acético (12 mL), se añadió gota a gota durante 1 hora una solución de dibromina (0,845 mL, 16,44 mmol) en ácido acético (5 mL). La mezcla de reacción se agitó a 45 °C por 18 horas y luego se concentró hasta la sequedad. El residuo se trituró en ciclohexano y el sólido se filtró y se lavó con ciclohexano para dar ácido 2-(3-bromo-2,6-dimetilfenil) acético (2,4 g, 71%) como un sólido de color blanco. LC/MS (Método h) Rt = 2,26 min.; MS m/z: 241 [M-H]-. 1H NMR (DMSO-cfe, 300 MHz): 5 12,45 (ancho, 1H), 7,38 (d, J=8,1Hz, 1H), 6,97 (d, J=8,1Hz, 1H), 3,69 (s, 2H), 2,33 (s, 3H), 2,24 (s, 3H)
Etapa B: ácido 2-(3-(metoxicarbonil)-2,6-dimetilfenil)acético

En un vial de microondas se añadió ácido 2-(3-bromo-2,6-dimetilfenil)acético (1,5 g, 6,17 mmol), trans-bis(acetato)bis[o-(di-o-tolilfosfino)bencil]dipaladio(M) (0,289 g, 0,309 mmol), tetrafluoroborato de tri-terc-butilfosfonio (0,358 g, 1,234 mmol) y hexacarbonilo de molibdeno (1,629 g, 6,17 mmol) en MeOH (30 mL). Se añadió gota a gota 1,8-diazabiciclo[5,4,0] undec-7-eno (2,82 g, 18,51 mmol) y la mezcla se agitó vigorosamente a temperatura ambiente durante 5 minutos. Luego, el vial se calentó en microondas a 110 °C durante 30 minutos y a 140 °C durante 15 minutos. Después de enfriar a temperatura ambiente, la mezcla de reacción se filtró y el disolvente se eliminó a presión reducida. El residuo se diluyó con agua y EtOAc. Las fases se separaron y la acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron y descartaron. La capa de agua básica se acidificó a pH 2-3 con solución de HCl 1N y se extrajo completamente con EtOAc. Las fases orgánicas se combinaron, se lavaron con salmuera, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida para dar ácido 2-(3-(metoxicarbonil)-2,6-dimetilfenil)acético (1,17 g, 79%) como un sólido verde. LC/MS (Método h) Rt = 1,86 min.; MS m/z: 223 [M+H]+1H NMR (CDCla, 300 MHz): 57,66 (d, J=9Hz, 1H), 7,11 (d, J=9Hz, 1H), 3,89 (s, 3H), 3,81 (s, 2H), 2,54 (s, 3H), 2,39 (s, 3H).
Preparación #10. Ácido 2-(2,6-dicloro-4-nitrofenil)acético

A una suspensión de malonato de dietilo (1,006 mL, 6,62 mmol) y carbonato de cesio (3,60 g, 11,04 mmol) en DMF (5 mL) calentada a 70°C se añadió 3,4,5-tricloronitrobenceno (1 g, 4,42 mmol) y la mezcla se agitó a 70 °C por 3 horas. La mezcla se enfrió a temperatura ambiente. Se añadió una solución de HCl 2 M y la mezcla de reacción se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio y se concentraron. El residuo se suspendió en ácido clorhídrico (7,36 mL 44,2 mmol) y se agitó a 100 °C durante 24 horas. La mezcla se enfrió a temperatura ambiente, se introdujo en agua fría y la mezcla de reacción se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio y se concentraron. El residuo se disolvió en dioxano (15 mL). Se añadió hidróxido de sodio (11,99 mL, 11,99 mmol) y la mezcla se agitó a temperatura ambiente durante 48 horas. Se añadió un exceso de hidróxido de sodio (11,99 mL, 11,99 mmol) y
la agitación continuó durante 24 horas. Se añadió otro exceso de hidróxido de sodio (11,99 mL, 11,99 mmol) y la agitación continuó durante 48 horas. La mezcla se diluyó con agua y se lavó dos veces con EtOAc. Las capas acuosas se acidificaron con solución de HCl 10 M (pH=2) y se extrajeron con diclorometano. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio y se concentraron para dar ácido 2-(2,6-dicloro-4-nitrofenil)acético (465 mg, 42%) como un sólido de color blanco. lC/MS (Método h) Rt = 1,98 min.; MS miz: 497 [2M-H]-1H NMR (DMSO-cfe 300 MHz): 512,96 (ancho, 1H), 8,32 (s, 2H), 4,03 (s, 2H)
Preparación #11. (2,4-didoro-3-(dorometM)feml)(morfolmo)metanona

Etapa A: ácido 2,4-dicloro-3-(hidroximetil)benzoico

A una solución de metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (10 g, 33,6 mmol) en dioxano (100 mL) se añadió hidróxido de sodio (84 mL, 84 mmol) y la mezcla de reacción se agitó a reflujo por 5 horas. La mezcla de reacción se concentró hasta la sequedad. El residuo se disolvió en agua y se acidificó a pH1. El sólido de color blanco se filtró, se lavó con agua y se secó al vacío para dar ácido 2,4-dicloro-3-(hidroximetil)benzoico (5,9 g, 80%). LC/MS (Método h) Rt = 0,70 min.; MS m/z: 219 [M-H]-1H NMR (DMSO-cfe, 300 MHz): 5 13,58 (ancho, 1H), 7,67 (d, J=9Hz, 1H), 7,52 (d, J=9Hz, 1H), 5,28 (m, 1H), 4,74 (s, 2H).
Etapa B: (2,4-didoro-3-(hidroximetil)fenil)(morfolino)metanona

Usando un procedimiento similar a la Preparación #8, (2,4-dicloro-3-(hidroximetil)fenil)(morfolino)metanona (2,6 g, 41%) se preparó a partir de ácido 2,4-dicloro-3-(hidroximetil)benzoico (4,8 g, 21,72 mmol). LC/MS (Método h) Rt = 1,19 min.; MS m/z: 290 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,53 (d, J=9Hz, 1H), 7,34 (d, J=9Hz, 1H), 5,28 (s, 1H), 4,70 (s, 2H), 3,64 (m, 4H), 3,52 (m, 2H), 3,12 (m, 2H).
Etapa C: (2,4-dicloro-3-(clorometil)fenil)(morfolino)metanona

A una solución de (2,4-dicloro-3-(hidroximetil)fenil)(morfolino)metanona (653 mg, 2,251 mmol) en DCM (18 mL) se añadió cloruro de metanosulfonilo (0,349 mL, 4,50 mmol) y trietilamina (0,627 mL, 4,50 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se diluyó con DCM. La capa orgánica obtenida se lavó con agua y salmuera. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 7-60% EtOAc en DCM) para dar (2,4-dicloro-3-(clorometil)fenil)(morfolino)metanona (548 mg, 79%). LC/mS (Método h) Rt = 2,12 min.; MS miz: 308 [M+H]+.
1H NMR (DMSO-de, 300 MHz): 57,62 (d, J=9Hz, 1H), 7,44 (d, J=9Hz, 1H), 4,93 (s, 2H), 3,65 (m, 4H), 3,52 (m, 2H), 3,12 (m, 2H).
Preparación #12. etil 3-((metilsulfonil)oxi)ciclopentanocarboxilato

A una solución de etil 3-hidroxicidopentanocarboxilato (0,8 g, 5,06 mmol) y trietilamina (1,762 mL, 12,64 mmol) en DCM (8 mL) y enfriada a 0 °C se añadió una solución de cloruro de metanosulfonilo (0,588 mL, 7,59 mmol) en DCM (3 mL). La reacción se agitó a temperatura ambiente durante 1 hora. Se añadió agua y la mezcla de reacción se extrajo con DCM. La capa orgánica se lavó con salmuera y se secó sobre MgSO4 , se filtró y evaporó para dar etil 3-((metilsulfonil)oxi)ciclopentanocarboxilato (1,1 g, 92%) como un líquido incoloro: No LC/MS. 1H NMR (DMSO-Ce, 300 MHz): 55,08 (m, 1H), 4,05 (m, 2H), 3,60 (m, 4H), 2,86 (m, 1H), 2,30 (m, 1H), 2,05 (m, 1H), 1,80 (m, 3H), 1,18 (m, 3H).
Preparación #13. metil 2,4-dicloro-3-(3-doro-2-oxobutil)benzoato

Se calentó a reflujo una suspensión de zinc (0,241 g, 3,69 mmol) en THF (1 mL) y se añadieron 1,2-dibromoetano (0,025 g, 0,134 mmol) y clorotrimetilsilano (0,015 g, 0,134 mmol). La mezcla de reacción se agitó 30 minutos. Después, se añadió gota a gota una solución de metil 3-(bromometil)-2,4-diclorobenzoato (1 g, 3,36 mmol) (Preparación #1, Etapa B) en THF (3 mL) durante 5 minutos y la mezcla se agitó a reflujo por 2 horas. Esta solución se añadió a temperatura ambiente a una mezcla del complejo de di(cloruro de litio) cianuro de cobre (I) (3,36 mL, 3,36 mmol) y N,N-dimetilformamida (246 mg, 3,36 mmol) y la solución se agitó 30 minutos a temperatura ambiente. Luego, se añadió una solución de cloruro de 2-cloropropionilo (427 mg, 3,36 mmol) en tolueno (4 mL) y tetrakis(trifenilfosfina)paladio(0) (116 mg, 0,101 mmol). La reacción se agitó una hora a temperatura ambiente, se diluyó con 10 mL de hexano y 10 mL de EtOAc y se añadió gel de sílice sólido. La mezcla se agitó 5 minutos y el sólido se filtró y se lavó con EtOAc. La capa orgánica se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar metil 2,4-dicloro-3-(3-cloro-2-oxobutil)benzoato (560 mg, 54%) como un aceite incoloro. LC/MS (Método h) Rt = 2,75 min.; MS m/z: 309 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 57,71 (d, J=9Hz, 1H), 7,62 (d, J=9Hz, 1H), 5,05 (q, J=7Hz, 1H), 4,56 (d, J=15Hz, 1H), 4,43 (d, J=15Hz, 1H), 3,87 (s, 3H), 1,63 (d, J=7Hz, 3H).
Preparación #14, metil 3-(3-bromo-2-oxobutil)-2,4-diclorobenzoato

Etapa A: metil 2,4-dicloro-3-(2-hidroxibutil)benzoato

A una solución de metil 2,4-dicloro-3-(2-oxoetil)benzoato (Preparación #2, Etapa B) (100 mg, 0,405 mmol) en THF (1 mL) se añadió bromuro de etilmagnesio (1 M en THF, 0,486 mL, 0,486 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas. La mezcla de reacción se diluyó con una solución de HCl 1N acuoso y la capa acuosa obtenida se extrajo con EtOAc. La capa orgánica se secó sobre MgSO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-20% EtOAc en ciclohexano) para dar metil 2,4-dicloro-3-(2-hidroxibutil)benzoato (100 mg, 89%) como un aceite incoloro. LC/MS (Método h) Rt = 2,39 min.; MS m/z: 277 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 57,58 (m, 2H), 4,65 (d, J=6Hz, 1H), 3,85 (s, 3H), 3,72 (m, 1H), 3,05 (m, 2H), 1,42 (m, 2H), 0,89 (t, J=6Hz, 3H).
Etapa B: metil 2,4-dicloro-3-(2-oxobutil)benzoato

A una solución de metil 2,4-didoro-3-(2-hidroxibutil)benzoato (100 mg, 0,361 mmol) en DCM (1,5 mL) se añadió periodinano Dess-Martin (0,750 mL, 0,361 mmol). La mezcla de reacción se agitó a temperatura ambiente por 18 horas. La mezcla de reacción se diluyó con DCM. La capa orgánica se lavó con una solución acuosa saturada de Na2S2O3, luego se secó sobre MgSO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-20% EtOAc en ciclohexano) para dar metil 2,4-dicloro-3-(2-oxobutil)benzoato (63 mg, 63%) como un aceite incoloro. LC/MS (Método h) Rt = 2,53 min.; MS m/z: 275 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5 7,69 (d, J=9Hz, 1H), 7,60 (d, J=9Hz, 1H), 4,21 (s, 2H), 3,86 (s, 3H), 2,64 (q, J=7,2Hz, 2H), 0,99 (t, J=7,2, 3H).
Etapa C: metil 3-(3-bromo-2-oxobutil)-2,4-diclorobenzoato

Una suspensión de bromuro cúprico (81 mg, 0,363 mmol) en cloroformo (300 |_iL) y EtOAc (300 mL) se calentó a reflujo. Metil 2,4-dicloro-3-(2-oxobutil)benzoato (100 mg, 0,363 mmol) en solución en cloroformo (80 pL) y EtOAc (80 pL) se añadió y la mezcla de reacción se agitó a reflujo por 72 horas. La mezcla de reacción se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-20% EtOAc en ciclohexano) para dar metil 3-(3-bromo-2-oxobutil)-2,4-diclorobenzoato (105 mg, 47%) como un aceite incoloro. LC/MS (Método h) Rt = 2,82 min.; MS m/z: 353 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,70 (d, J=9Hz, 1H), 7,63 (d, J=9Hz, 1H), 5,10 (q, J=6 ,6 Hz, 1H), 4,63 (d, J=18Hz, 1H), 4,46 (d, J=18Hz, 1H), 3,86 (s, 3H), 1,72 (d, J=6,65Hz, 3H).
Preparación # 15.1-(3,5-dicloro-2-metoxipiridin-4-il)prop-2-in-1-ol

Etapa A: 3,5-dicloro-2-metoxiisonicotinaldehído
Una solución de 3,5-dicloro-2-metoxipiridina (16 g, 90 mmol) en THF (100 mL) se añadió lentamente a una solución de LDA (53,9 mL, 108 mmol) (2M en THF) a -78 °C. DMF (14 mL, 180 mmol) se añadió a la mezcla de reacción y la solución resultante se agitó a -78 °C durante aproximadamente 1h. La mezcla de reacción se vertió después en una solución acuosa saturada de NH4CL La capa acuosa se extrajo con EtOAc (3 x 20 mL). La porción orgánica combinada se secó sobre Na2SO4 anhidro, se filtró, y se concentró a presión reducida para dar 3,5-dicloro-2-metoxiisonicotinaldehído (17,54 g, 90 %). LC/MS (Tabla 1, Método d) Rt = 1,86 min.; MS m/z: 205, 207 [M+H]+.
Etapa B: 1-(3,5-dicloro-2-metoxipiridin-4-il)prop-2-in-1-ol

Usando un procedimiento similar a la Preparación #1, 1-(3,5-dicloro-2-metoxipiridin-4-il)prop-2-in-1-ol (8,74 g, 76 %) se preparó a partir de 3,5-dicloro-2-metoxiisonicotinaldehído (10 g, 48,5 mmol). Lc /MS (Tabla 1, Método b) Rt = 1,90 min.; MS(M+1)= 231,233
1H NMR (DMSO-de, 400 MHz): 58,24 (s,1H), 6,42 (s, 1H), 6,00 (s, 1H), 3,95 (s, 3H), 3,56 (s, 1H).
Preparación #16. W-(2-yodo-3-metil-5-(tnfluorometil)feml)bencenosulfonamida

Etapa A: 2-yodo-3-metil-5-(trifluorometil)anilina

A una solución de 3-amino-5-metilbenzotrifluoruro (10 g, 57,1 mmol) en DCM (200 mL) y MeOH (50 mL) se añadió dicloroyodato de benciltrimetilamonio (39,7 g, 114 mmol) y carbonateo de calcio (14,29 g, 143 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche, se filtró y el sólido se lavó con DCM. El filtrado se lavó con una solución acuosa saturada de Na2S2O3, y la capa orgánica obtenida se lavó con agua y después se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-100% en ciclohexano) para dar 2-yodo-3-metil-5-(trifluorometil)anilina (9,1 g, 53%) como un líquido de color naranja. LC/MS (Método h) Rt = 2,83 min.; MS m/z: 302 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 86,87 (d, J = 1,8 Hz, 1H), 6,81 (d, J = 1,8 Hz, 1H), 5,68 (s, 2H), 2,38 (s, 3H).
Etapa B: W-(2-yodo-3-metil-5-(trifluorometil)feml)bencenosulfonamida

A una solución de 2-yodo-3-metil-5-(trifluorometil)anilina (9,1 g, 30,2 mmol) en piridina (50 mL) se añadió cloruro de bencenosulfonilo (7,75 mL, 60,5 mmol). La reacción se agitó a temperatura ambiente durante la noche, luego se repartió entre agua y EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se disolvió en 1,4-dioxano (66,7 mL) y se añadió hidróxido de potasio 3 N (40,3 mL, 121 mmol) a la mezcla de reacción. La reacción se calentó a reflujo durante dos horas, luego se diluyó con agua y se acidificó con HCl concentrado. La capa acuosa se extrajo con DCM y la capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar W-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfon-amida (13,1 g, 98%) como un sólido de color marrón. LC/MS (Método h) Rt = 2,96 min.; MS m/z: 440 [M-H]-.
1H NMR (DMSO-Ce, 300 MHz): 510,9 (s, 1H), 7,71 (m, 3H), 7,59 (m, 3H), 6,92 (s, 1H), 2,46 (s, 3H).
Preparación #17. A/-(2-yodo-4-metil-5-(trifluorometil)fenil)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, W-(2-yodo-4-metil-5-(trifluorometil)fenil)bencenosulfonamida (3 g, 100%) se preparó a partir de 2-yodo-4-metil-5-(trifluorometil)anilina (descrito en WO2006/002342) (2 g, 6,64 mmol). LC/MS (Método h) Rt = 2,95 min.; MS m/z: 440 [M-H]‘1H NMR (DMSO-cfe, 300 MHz): 5 10,02 (s, 1H), 7,97 (s, 1H), 7,70 (m, 3H), 7,66 (m, 2H), 7,07 (s, 1h ), 2,33 (s, 3H).
Preparación #18. W-(5-ciano-2-yodo-3-metilfeml)bencenosulfonamida

Etapa A: 4-amino-3-metil-5-nitrobenzonitrilo

A una suspensión de 4-bromo-2-metil-6-nitroanilina (25 g, 108 mmol) en DMF (300 mL) se añadió yoduro de cobre(I) (1,030 g, 5,41 mmol) y cianuro de zinc (38,1 g, 325 mmol). La mezcla se agitó a temperatura ambiente durante 15 minutos, luego se añadió tetrakis paladio (12,50 g, 10,82 mmol) y la mezcla se agitó a 100°C durante 24 horas. Se añadió más tetrakis paladio (12,50 g, 10,82 mmol) y la mezcla se agitó a 130°C durante 6 horas. La mezcla de reacción se filtró y se lavó con EtOAc. El filtrado se lavó sucesivamente con solución de NaHCO3 acuoso y salmuera después se secó sobre sulfato de magnesio y se concentró a reducir la presión saturada. El residuo se recristalizó en 600 ml de EtOH y se obtuvo 4-amino-3-metil-5-nitrobenzonitrilo (12 g, 63%) como un sólido amarillo. LC/MS (Método h) Rt = 1,77 min.; MS m/z: 176 [M-H]-.1H NMR (DMSO-cfe, 300 MHz): 58,35 (s, 1H), 7,65 (ancho, 2H), 7,68 (s, 1H), 2,22 (s, 3H).
Etapa B: 4-yodo-3-metil-5-nitrobenzonitrilo

A una solución de 4-amino-3-metil-5-nitrobenzonitrilo (12 g, 67,7 mmol) en DMSO (176 mL) y enfriada a 0 °C se añadió gota a gota ácido sulfúrico (240 mL, 67,7 mmol). La temperatura se mantuvo por debajo de 10 ° C sin congelar el DMSO. Luego a la mezcla de reacción se añadió gota a gota nitrito de sodio (7,01 g, 102 mmol) en agua (25 mL) para mantener la temperatura a 0 °C y la mezcla de reacción se agitó durante 1 hora entre 0-5 °C. Se añadió gota a gota una solución de yoduro de potasio (33,7 g, 203 mmol) y yodo (12,89 g, 50,8 mmol) en agua (100 mL) manteniendo la temperatura entre 0-5 °C y la mezcla de reacción se agitó a 0 °C durante 1 hora. Se añadió una solución acuosa de Na2S2Os al 20% a la mezcla y el sólido se filtró y se lavó con agua. El sólido se recristalizó en EtOH puro (150 mL) y todos los licores madres se purificaron por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-100% en ciclohexano). Se obtuvo 4-yodo-3-metil-5-nitrobenzonitrilo (23,5 g, 70%) como un sólido amarillo.
LC/MS (Método h) Rt = 1,77 min.; sin ionización 1H NMR (CDCla, 300 MHz): 57,69 (m, 2H), 2,64 (s, 3H).
Etapa C: 3-amino-4-yodo-5-metilbenzonitrilo

A una solución de 4-yodo-3-metil-5-nitrobenzonitrilo (3,11 g, 10,80 mmol) en EtOH (100 mL) se añadió hierro (6,03 g, 108 mmol), cloruro de amonio (1,733 g, 32,4 mmol) y agua (30 mL). La mezcla de reacción se agitó a reflujo por 1 hora. La mezcla de reacción se filtró a través de celita y se lavó con EtOH y EtOAc. El filtrado se concentró, el residuo se disolvió en EtOAc y se lavó sucesivamente con solución acuosa de NH4Cl 2M y salmuera, se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 10% EtOAc en ciclohexano) para dar 3-amino-4-yodo-5-metilbenzonitrilo (2,55g, 80%) como un sólido de color marrón. LC/MS (Método h) Rt = 2,29 min.; MS m/z: 259 [M+H]+. 1H NMR (CDCl3, 300 MHz): 56,87 (s, 1H), 6,77 (s, 1H), 4,41 (ancho, 2H), 2,24 (s, 3H).
Etapa D: A/-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, W-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (9,43 g, 91%) como un sólido de color naranja a partir de 3-amino-4-yodo-5-metilbenzonitrilo (6,7 g, 26 mmol). LC/MS (Método k) Rt = 2,68 min.; MS m/z: 397 [M-H]‘ . 1H NMR (CDCl3 , 300 MHz): 57,72 (m, 2H), 7,64 (m, 1H), 7,50 (m, 1H), 7,41 (m, 2H), 7,16 (m, 1H), 7,09 (s, 1H), 2,34 (s, 3H).
Preparación #19. W-(4-ciano-2-yodofenil)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, A/-(4-ciano-2-yodofenil)bencenosulfonamida (3,7 g, 100%) se obtuvo como un sólido de color naranja a partir de 4-amino-3-yodobenzonitrilo (2 g, 8,20 mmol). LC/MS (Método h) Rt = 2,41 min.; MS m/z: 383 [M-H]-1H NMR (CDCla, 300 MHz): 57,96 (m, 1H), 7,83 (m, 2H), 7,71 (m, 1H), 7,60 (m, 2H), 7,51 (m, 2H), 7,17 (m, 1H).
Preparación #20: W-(5-ciano-2-yodofenil)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, W-(5-ciano-2-yodofenil)bencenosulfonamida (1,6 g, 51%) se obtuvo como un sólido de color naranja a partir de 3-amino-4-yodobenzonitrilo (2 g, 8,20 mmol). LC/MS (Método h) Rt = 2,38 min.; MS m/z: 383 [M-H]-1H NMR (DMSO-cfe, 300 MHz): 5 10,14 (ancho, 1H), 8,05 (d, J = 6Hz, 1H), 7,70 (m, 3H), 7,61 (m, 2H), 7,42 (d, J = 9Hz, 1H), 7,36 (d, J = 3Hz, 1H).
Preparación #21: N-1,3-dimetil-5-(trifluorometil)benceno-1,2-diamina
En un reactor Parr se introdujeron bajo nitrógeno, 2-bromo-6-metil-4-(trifluorometil)anilina (descrito en WO2009/009411) (14,5 g, 57,1 mmol) en THF (14,5 mL). Se añadieron metilamina (40% en agua) (72,5 mL, 2094 mmol) y óxido de cobre (I) (0,817 g, 5,71 mmol). El reactor se cerró bajo nitrógeno y se calentó a 110 °C, durante 16 horas. Se añadió agua a la mezcla y se extrajo con EtOAc. La capa orgánica se lavó con agua, se secó sobre sulfato de magnesio, se filtró y se evaporó. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 5-40% EtOAc en ciclohexano) para dar W-1,3-dimetil-5-(trifluorometil)benceno-1,2-diamina (2,26 g, 19%). LC/MS (Método h) Rt = 2,07 min.; MS m/z: 205 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 56,68 (s, 1H), 6,42 (s, 1H), 4,99 (d, J = 5Hz, 1H), 4,85 (s, 2H), 2,73 (d, J = 5Hz, 3H).
Preparación #22: 4-amino-3-metil-5-(metilamino)benzonitrilo

Etapa A: 5-bromo-N,3-dimetil-2-nitroanilina

A una solución de 5-bromo-1-fluoro-3-metil-2-nitrobenceno (5 g, 21,37 mmol) en EtOH, (50 mL) se añadió metilamina (2M en THF) (32,0 mL, 64,1 mmol) y la mezcla de reacción se agitó a 70 °C por 1,5 horas y durante la noche a temperatura ambiente. Se añadió 0 (2 M en THF) (32,0 mL, 64,1 mmol) y la mezcla de reacción se agitó a 70°C durante 1 hora. La mezcla de reacción se concentró a presión reducida. El residuo se diluyó con EtOAc y se lavó con salmuera, se secó sobre sulfato de magnesio, y se evaporó para dar 5-bromo-W,3-dimetil-2-nitroanilina (5,13 g, 98%) como un sólido de color naranja. LC/MS (Método h) Rt = 2,77 min.; MS m/z: 245 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 5. 6,86 (s, 1H), 6,77 (s, 1H), 6,72 (m, 1H), 2,77 (d, J = 3Hz, 3H), 2,28 (s, 3H)
Etapa B: 3-metil-5-(metilamino)-4-nitrobenzonitrilo

En un reactor de microondas se añadieron ferrocianuro de potasio (3006 mg, 8,16 mmol), Pd(dppf)Cl2 (179 mg, 0,245 mmol), carbonato de sodio (865 mg, 8,16 mmol) y 5-bromo-N, 3-dimetil- 2-nitroanilina (2000 mg, 8,16 mmol) en NMP (15 mL). La mezcla de reacción se agitó en microondas 2 horas a 130 °C. La mezcla de reacción se filtró y se lavó con EtOAc. El filtrado se lavó con agua y salmuera. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-25% en ciclohexano) para dar 3-metil-5-(metilamino)-4-nitrobenzonitrilo (1,21 g, 64%). LC/mS (Método h) Rt = 2,28 min.; MS m/z: 192 [M+H]+ 1H NMR (DMSO-Ce, 300 MHz): 5.7,13 (s, 1H), 7,01(s, 1H), 6,60 (q, J = 4,8Hz, 1H), 2,76 (d, J = 4,8Hz, 3H), 2,25 (s, 3H)
Etapa C: 4-amino-3-metil-5-(metilamino)benzonitrilo

A una suspensión de 3-metil-5-(metilamino)-4-nitrobenzonitrilo (1,795 g, 9,39 mmol) en EtOAc (84 mL) y MeOH (84 mL) se añadió Pd/C (0,400 g, 1,878 mmol) y la mezcla de reacción se agitó a temperatura ambiente bajo H2 (1 bar) durante 1 hora. La mezcla de reacción se filtró y se lavó con EtOAc. El filtrado se concentró a presión reducida para dar 4-amino-3-metil-5-(metilamino)benzonitrilo (1,51 g, 100%) como un sólido de color rojo. LC/m S (Método h) Rt = 1,40 min.; MS m/z: 162 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 5.6,79 (s, 1H), 6,50(s, 1H), 5,18 (s, 2H), 5,02(m, 1h ), 2,72 (d, J = 5,1Hz, 3H), 2,06 (s, 3H).
Preparación #23: A/-1,3,5-trimetilbenceno-1,2-diamina

Etapa A: N-(2-metil-4-(tnfluorometil)feml)acetamida

Se añadió anhídrido acético (8,08 mL, 86 mmol) a 2-metil-4-(trifluorometil)anilina (5 g, 28,5 mmol) a 0 °C, y la mezcla de reacción se agitó a 0 °C durante 20 minutos. El sólido formado se trituró con mortero, se puso en suspensión en agua y se neutralizó con amoníaco. El precipitado se filtró y se secó al vacío para dar W-(2-metil-4-(trifluorometil)fenil)acetamida (5,91 g, 95%) como un sólido de color beige.
LC/MS (Método h) Rt = 1,99 min.; MS m/z: 218 [M+H]+ 1H NMR (DMSO-Ce, 300 MHz): 5. 9,45 (s, 1H), 7,76 (d, J = 8,4Hz, 1H), 7,57 (s, 1H), 7,50 (d, J = 8,4Hz, 1H), 2,30 (s, 3h ), 2,11 (s, 3H)
Etapa B: N -(2-metil-6-nitro-4-(trifluorometil)fenil) acetamida
A una solución de W-(2-metil-4-(trifluorometil)fenil)acetamida (5,9 g, 27,2 mmol) en ácido sulfúrico (20 mL, 375 mmol) a 0 °C se añadió ácido nítrico gota a gota (20 mL, 448 mmol). La mezcla de reacción se agitó a 0 °C durante 2 horas y a temperatura ambiente durante la noche. La mezcla de reacción se diluyó en agua y se extrajo con EtOAc. Las capas se separaron y la capa orgánica se lavó con solución de NaHCO3 acuoso saturado y salmuera. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró para dar W-(2-metil-6-nitro-4-(trifluorometil)fenil) acetamida (6,6 g, 93%). LC/MS (Método h) Rt = 2,00 min.; MS m/z: 263 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 5. 10,14 (ancho, 1H), 8,08 (s, 1h ), 8,04 (s, 1H), 2,39 (s, 3H), 2,07 (s, 3H).
Etapa C: 2-metil-6-nitro-4-(trifluorometil)anilina
A una solución de W-(2-metil-6-nitro-4-(trifluorometil)fenil)acetamida (2,95 g, 11,25 mmol) en MeOH (2,5 mL) y agua (1 mL) se añadió hidróxido de potasio (0,189 g, 3,38 mmol) y la mezcla de reacción se agitó a 60 °C durante 18 horas. Se vertió hielo en la mezcla de reacción y se formó un precipitado, se filtró y se lavó con agua fría. El sólido se secó al vacío para dar 2-metil-6-nitro-4-(trifluorometil)anilina (1,65 g, 67%). LC/MS (Método h) Rt = 2,51 min.; MS m/z: 219 [MH]-. 1H NMR (DMSO-Ce, 300 MHz): 5. 8,16 (s, 1H), 7,66 (s, 1H), 7,62 (s, 2H), 2,27 (s, 3H).
Etapa D: 3-metil-5-(trifluorometil)benceno-1,2-diamina

A una solución de 2-metil-6-nitro-4-(trifluorometil)anilina (1,33 g, 6,04 mmol) en EtOH (22 mL) y agua (11 mL) se añadieron hierro (1,687 g, 30,2 mmol) y cloruro de calcio (1,341 g, 12,08 mmol) y la mezcla de reacción se agitó a reflujo durante 5 horas. La mezcla de reacción se filtró y el filtrado se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH al 0-5% en DCM) para dar 3-metil-5-(trifluorometil)benceno-1,2-diamina (862 mg, 75%). LC/MS (Método h) Rt = 1,86 min.; MS m/z: 191 [M+H]+.
1H NMR (DMSO-Ce, 300 MHz): 5. 6,68 (s, 1H), 6,61 (s, 1H), 4,81 (m, 4H), 2,07 (s, 3H).
Etapa E: etil (2-amino-3-metil-5-(trifluorometil)fenil)carbamato

A una solución de 3-metil-5-(trifluorometil)benceno-1,2-diamina (860 mg, 4,52 mmol) en DMF (9 mL) y se enfrió a 0 °C se añadió hidruro de sodio (109 mg, 4,52 mmol) y la mezcla de reacción se agitó a 0 °C durante 1 hora. Se añadió cloroformiato de etilo (0,432 mL, 4,52 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas. La mezcla de reacción se hidrolizó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 0-40% de EtOAc en DCM) para dar etil (2-amino-3-metil-5-(trifluorometil)fenil)carbamato (512 mg, 43%) LC/MS (Método h) Rt = 2,28 min.; MS m/z: 263 [M+H]+. 1H NMR (DMSO-Ce, 300 MHz): 5. 8,69 (s, 1H), 7,44 (s, 1H), 7,10 (s, 1H), 5,30 (s, 2H), 4,11 (q, J = 7,2Hz, 2H), 2,13 (s, 3H), 1,24 (t, J = 7,2Hz, 3H).
Etapa F: N -1,3,5-trimetilbenceno-1,2-diamina

A una solución de etil (2-amino-3-metil-5-(trifluorometil)fenil)carbamato (510 mg, 1,945 mmol) en THF (12 mL) enfriada a 0 °C se añadió una suspensión de LiAlH4 (9,72 ml, 9,72 mmol) en THF (5 mL) y la mezcla de reacción se calentó a reflujo durante 18 horas. La mezcla de reacción se trató a temperatura ambiente con sales de Glauber durante 7 horas y se filtró. El filtrado se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 0-40% de EtOAc en DCM) para dar W1,3,5-trimetilbenceno-1,2-diamina (197 mg, 67%). LC/MS (Método h) Rt = 1,05 min.; MS miz: 151 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5. 6,16 (s, 1H), 6,10 (s, 1H), 4,53 (m, 1H), 3,95 (s, 2H), 2,67 (d, J = 5,1Hz, 3H), 2,10 (s, 3H), 2,00 (s, 3H).
Preparación #24: 5-bromo-A/ -1,3-dimetilbenceno-1,2-diamina

Usando un procedimiento similar a la Preparación #23, Etapa D, se preparó 5-bromo-W-1,3-dimetilbenceno-1,2-diamina (712 mg, 100%) a partir de 5-bromo-W,3-dimetil-2-nitroanilina (Preparación #22, Etapa A) (815 mg, 3,33 mmol). LC/MS (Método h) Rt = 1,93 min.; MS miz: 215 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5. 6,49 (s, 1H), 6,32 (s, 1H), 4,91 (m, 1H), 4,34 (ancho, 2H), 2,67 (d, J = 4,8Hz, 3H), 2,03 (s, 3H).
Preparación #25: 6-cloro-A/-2,4-dimetilpiridina-2,3-diamina

Etapa A: 6-cloro-N, 4-dimetil-3-nitropiridin-2-amina

A una solución de 2,6-dicloro-4-metil-3-nitropiridina (2,2 g, 10,63 mmol) en ACN (60 mL) se añadieron trietilamina (4,44 mL, 31,9 mmol) y metilamina (2 M en THF) (6,38 mL, 12,75 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 horas. La mezcla de reacción se inactivó con solución acuosa saturada de NaHCO3 y se diluyó con agua y EtOAc. Las capas se separaron y la acuosa se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-30% en ciclohexano) para dar 6-cloro-W,4-dimetil-3-nitropiridin-2-amina (2,05 g, 43%) como un amarillo sólido. LC/MS (Método h) Rt = 2,45 min.; MS miz: 202 [M+H]+. 1H NMR (DMSO-cfe 400 MHz): 57,93 (ancho, 1H), 6,72 (s, 1H), 2,89 (s, 3H), 2,38 (s, 3H).
Etapa B: 6-doro-W2,4-dimetilpindma-2,3-diamma

Usando un procedimiento similar al ejemplo Preparación #23, Etapa D, se preparó 6-cloro-N2,4-dimetilpiridina-2,3-diamina (90 mg, 51%) a partir de 6-cloro-N,4-dimetil-3-nitropiridin-2-amina (200 mg, 0,992 mmol). LC/MS (Método h) Rt = 1,40 min.; MS m/z: 172 [M+H]+. 1H NMR (DMSO-d6, 300 MHz): 56,30 (s, 1H), 5,93 (ancho, 1H), 4,48 (ancho, 2H), 2,78 (d, J = 3 Hz, 3H), 2,00 (s, 3H).
Preparación #26: W-(2-bromo-5-(trifluorometil)piridm-3-il)bencenosulfonamida

Etapa A: 2-bromo-5-(trifluorometil)piridin-3-amina

A una solución de 2-bromo-3-nitro-5-(trifluorometil)piridina (descrita en el documento WO2013/038381) (2,6 g, 9,59 mmol) es EtOH (40 mL) y agua (16 mL) se añadió hierro (4,02 g, 72,0 mmol) y ácido acético (3,7 mL). La mezcla de reacción se agitó a 110 °C durante 3 horas, luego se filtró y se evaporó el disolvente. El residuo se diluyó con EtOAc, y la solución acuosa saturada de NaHCO3 se extrajo, se secó sobre sulfato de magnesio, se filtró y se evaporó para dar 2-bromo-5-(trifluorometil)piridin-3-amina (2,35 g, 100%) como un sólido de color blanco. LC/MS (Método h) Rt = 1,91 min.; MS m/z: 241 [M+H]+. 1H NMR (DMSO-ak, 300 MHz): 5. 7,98 (s, 1H), 7,34 (s, 1H), 6,06 (s, 2H).
Etapa B: M-(2-bromo-5-(trifluorometil)piridm-3-il)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se preparó N-(2-bromo-5-(trifluorometil)piridin-3-il)bencenosulfonamida (954 mg, 50%) a partir de 2-bromo-5-(trifluorometil)piridina-3-amina (1,2 g, 4,98 mmol) LC/MS (Método h) Rt = 2,52 min.; MS m/z: 381 [M+H]+. 1H NMR (DMSO-d6 , 300 MHz,) 5 10,67 (ancho, 1H), 8,66 (s, 1H), 7,85 (d, J = 2,3 Hz, 1H), 7,73 (m, 3H), 7,59 (m, 2H).
Preparación #27: M-(5-yodo-2-(trifluorometil)piridm-4-il)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se preparó W-f5-yodo-2-(trifluorometil)piridin-4-il)bencenosulfonamida (13,32 g, 52%) a partir de 5-yodo-2-(trifluorometil)piridina-4-amina (descrita en WO2010/091310) (17,3 g, 60,1 mmol) LC/MS (Método h) Rt = 2,57 min.; MS m/z: 429 [M+H]+ .1H NMR (DMSO-d6 300 MHz,) 58,99 (s, 1H), 7,93 (m, 2H), 7,66 (m, 3H), 7,48 (s, 1H).
Preparación #28: 6-doro-4-metilpiridazin-3-amina y 6-doro-5-metilpiridazin-3-amina

Una suspensión de 3,6-didoro-4-metilpiridazina (3,38 g, 20,74 mmol) en hidróxido de amonio (55 mL, 1412 mmol) se calentó en un reactor a 130 °C durante aproximadamente 16 h. La mezcla de reacción se enfrió a temperatura ambiente. Apareció un precipitado que se filtró, se lavó con agua y se secó al vacío para dar una mezcla inseparable de 6-cloro-4-metilpiridazin-3-amina y 6-cloro-5-metilpiridazin-3-amina (relación 57/43) (2,25 g, 76%). 1H NMR (DMsO-d6 300 MHz,) 5 7,30 (s, 1H), 6,45 (ancho, 2H), 2,08 (s, 3H) y 6,74 (s, 1H), 6,47 (ancho, 2H), 2,19 (s, 3H).
Preparación #29: W-(2-yodo-5-(trifluorometil)feml)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se preparó N-[2-yodo-5-(trifluorometil)fenil]bencenosulfonamida (136 g, 100%) a partir de 2-yodo-5-(trifluorometil)anilina (85 g, 296 mmol) (descrito en US20070129334).
Preparación #30: N1,3-dimetil-4-(trifluorometil)benceno-1,2-diamma

Etapa A: 1-fluoro-4-yodo-3-metil-2-nitrobenceno

A una solución de 3-fluoro-2-nitrotolueno (1 g, 6,45 mmol) en ácido trifluorometanosulfónico (2,83 mL, 32,2 mmol) y enfriado a 0 °C se añadió en porciones N-yodosuccinimida (1,740 g, 7,74 mmol) y la mezcla se mezcló agitado a temperatura ambiente durante 1 hora. La mezcla se vertió en agua y se extrajo dos veces con éter dietílico. La capa orgánica se lavó sucesivamente con una solución acuosa de Na2S2O3 al 10%, después con salmuera y se concentró para dar 1-fluoro-4-yodo-3-metil-2-nitrobenceno como un sólido de color beige (1,86 g, 74%).
LC/MS (Método i) Rt = 2,28 min.; sin ionización 1H NMR (DMSO-d6 , 300MHz): 58,16 (dd, J = 6 Hz, 9Hz, 1H), 7,31 (t, J = 9 Hz, 1H), 2,39 ppm (s, 3H).
Etapa B: 1-fluoro-3-metil-2-nitro-4-(trifluorometil)benceno

A una suspensión de 1-fluoro-4-yodo-3-metil-2-nitrobenceno (2,39 g, 8,50 mmol), yoduro de cobre(I) (2,430 g, 12,76 mmol) y N,N-diisopropiletilamina (4,41 mL, 25,5 mmol) en DMF (8 mL) se añadió metil 2,2-difluoro-2-(fluorosulfonil)acetato (3,25 mL, 25,5 mmol) y la mezcla se calentó a 80 °C durante 20 horas en un tubo sellado. La mezcla de reacción se vertió en agua y se extrajo dos veces con EtOAc. La capa orgánica se lavó sucesivamente con una solución acuosa de NaHCO3 al 10% y salmuera, se secó sobre sulfato de magnesio y se concentró para dar 1-fluoro-3-metil-2-nitro-4
(trifluorometil)benceno (2,37 g, 62%). LC/MS (Método i) Rt = 2,25 min.; sin ionización 1H NMR (DMSO-d6, 300MHz): 5 8,09 (dd, J = 6 Hz, 9Hz, 1H), 7,71 (t, J = 9 Hz, 1H), 2,43 (m, 1H).
Etapa C: A/,3-dimetil-2-nitro-4-(trifluorometil)anilina

A una solución de 1-fluoro-3-metil-2-nitro-4-(trifluorometil)benceno (2,3 g, 10,31 mmol) en MeOH (15 mL) se añadió metilamina (15,46 mL, 30,9 mmol) (2M en THF) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró a presión reducida y el residuo se diluyó con EtoAc y se lavó con salmuera. La fase orgánica se secó sobre sulfato de magnesio y se evaporó. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5% en ciclohexano) para dar W,3-dimetil-2-nitro-4-(trifluorometil)anilina (1 g, 43%) como un sólido de color amarillo. LC/MS (Método i) Rt = 2,29 min.; MS m/z: 235 [M+H]+ .
1H NMR (DMSO-d6, 300MHz): 57,63 (d, J = 8,9 Hz, 1H), 6,77 (d, J = 8,9 Hz, 1H), 6,59 (ancho, 1H), 2,77 (d, J = 4,5 Hz, 3H), 2,24 (m, 3H).
Etapa D: W1,3-dimetil-4-(trifluorometil)benceno-1,2-diamma

A una suspensión de W,3-dimetil-2-nitro-4-(trifluorometil)anilina (1 g, 4,27 mmol) en EtOH (48 mL) se añadió hierro (2,385 g, 42,7 mmol), cloruro de amonio (0,685 g, 12,81 mmol) y agua (16 mL) y la mezcla de reacción se agitó a reflujo durante 1 hora. La mezcla de reacción se filtró a través de Celite y el filtrado se concentró. El residuo se disolvió en DCM y se lavó sucesivamente con una solución acuosa de NH4Cl 2M y salmuera; luego la fase orgánica se secó sobre sulfato de magnesio y se concentró para dar W1,3-dimetil-4-(trifluorometil)benceno-1,2-diamina como un sólido de color amarillo (884 mg, 77%). LC/MS (Método i) Rt = 1,96 min.; MS m/z: 205 [M+H]+ .
1H NMR (DMSO-d6 , 300MHz): 56,87 (d, J = 8,4 Hz, 1H), 6,33 (d, J = 8,4 Hz, 1H), 5,28 (d ancho, J = 5,0 Hz, 1H), 4,57 (s, 2H), 2,75 (d, J = 5,0 Hz, 3H), 2,12 ppm (m, 3H)
Preparación #31: 4-bromo-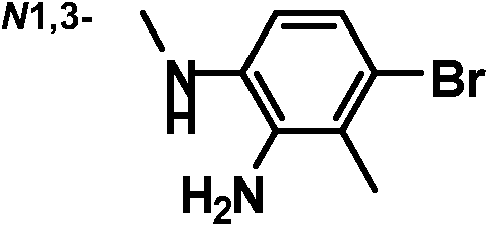 dimetilbenceno-1,2-diamina
dimetilbenceno-1,2-diamina
Etapa A: 4-bromo-W,3-dimetil-2-nitroamlma

Usando un procedimiento similar a la Preparación #30 Etapa C, se preparó 4-bromo-W,3-dimetil-2-nitroanilina (428 mg, 82%) a partir de 1-bromo-4-fluoro-2-metil-3-nitrobenceno (0,5 g, 2,14 mmol). LC/MS (Método h) Rt = 2,73 min.; MS m/z: 245 [M+H]+ .1H NMR (DMSO-d6 , 300MHz)57,55 (d, J = 9,1 Hz, 1h ), 6,65 (d, J = 9,1 Hz, 1H), 6,23 (m, 1H), 2,71 (d, J = 4,8 Hz, 3H), 2,24 (s, 3H).
Etapa B: 4-bromo-W1,3-dimetilbenceno-1,2-diamma

Usando un procedimiento similar a la Preparación #30 Etapa D, se preparó 4-bromo-W1,3-dimetilbenceno-1,2-diamina (360 mg, 82%) a partir de 4-bromo-W,3-dimetil-2-nitroanilina (425 mg, 1,73 mmol). LC/MS (Método h) Rt = 1,73 min.; MS
m/z: 215 [M+H]+. 1H NMR (DMSO-da, 300MHz): 56,73 (d, J=8,4 Hz, 1H), 6,21 (d, J=8,4 Hz, 1H), 4,60 (ancho, 3H), 2,67 (s, 3H), 2,15 (s, 3H).
Preparación #32: 5-bromo-W1-metil-3-(tnfluorometil)benceno-1,2-diamma

Etapa A: 5-bromo-1-cloro-2-nitroso-3-(trifluorometil)benceno

A una solución de 4-bromo-2-doro-6-(trifluorometil)anilina (1,396 mL, 9,00 mmol) en 1,2-dicloroetano (60 mL) se añadió ácido 3-cloroperoxibenzoico (4,66 g, 27,0 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 20 minutos y a 50 °C durante la noche. Se añadió DCM y la capa orgánica se lavó sucesivamente con una solución acuosa saturada de tiosulfato de amonio y una solución saturada de bicarbonato de sodio, y luego la fase orgánica se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc del 5% al 20% en ciclohexano) para dar 5-bromo-1-cloro-2-nitroso-3-(trifluorometil)benceno como un sólido de color beige (1,89 g, 69%). LC/MS (Método h) Rt = 2,98 min.; no ionization
1H NMR (DMSO-d6, 300MHz): 58,48 (d, J=1,8 Hz, 1H), 8,33 (d, J=1,8 Hz, 1H).
Etapa B: 5-bromo-W-meí/7-2-mtroso-3-(trifluorometil)amlma

A una solución de 5-bromo-1-cloro-2-nitroso-3-(trifluorometil)benceno (2,7 g, 9,36 mmol) en THF (25 mL) se añadió metilamina (10,30 mL, 20,59 mmol) (2M en THF) y la mezcla de reacción se agitó a temperatura ambiente durante 5 minutos. La mezcla de reacción se inactivó con agua y se extrajo con EtOAc. La capa orgánica se secó con sulfato de magnesio, se filtró y se concentró a presión reducida para dar 5-bromo-W-metil-2-nitroso-3-(trifluorometil)anilina como un líquido de color marrón (2,65 g, 89%).
LC/MS (Método h) Rt = 2,60 min.; MS m/z: 283 [M+H]+ .1H NMR (DMSO-d6 , 300MHz): 511,56 (ancho, 1H), 7,69 (d, J=1,8 Hz, 1H), 7,39 (d, J=1,8 Hz, 1H), 2,92 (d, J=5,3 Hz, 3H).
Etapa C: 5-bromo-W1-metil-3-(triftuorometil)benceno-1,2-diamma

Usando un procedimiento similar a la Preparación #23 Etapa D, se preparó 5-bromo-W1-metil-3-(trifluorometil)benceno-1,2-diamina (1,33 g, 51%) a partir de 5-bromo-W-metil-2-nitroso-3-(trifluorometil)anilina (2,65 g, 9,36 mmol). LC/MS (Método h) Rt = 2,67 min.; MS m/z: 269 [M+H]+ .
1H NMR (DMSO-d6 , 300MHz): 56,77 (d, J=2,1 Hz, 1H), 6,60 (d, J=2,1 Hz, 1H), 5,48 (m, 1H), 5,10 (s, 2H), 2,73 (d, J=4,8 Hz, 3H).
Preparación #33: terc-butil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato

Etapa A: 2,4-diclorobenzoato de tere-butilo

A una solución de ácido 2,4-diclorobenzoico (13,15 g, 68,8 mmol) en THF (68,5 mL) se añadió dicarbonato de di-terc-butilo (15,98 mL, 68,8 mmol) y N,N-dimetilpiridin-4-amina (0,841 g, 6,88 mmol) y la reacción se agitó a 40 °C durante 24 horas y luego se evaporó a sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-10% en ciclohexano) para dar terc-butil 2,4-diclorobenzoato (13,1 g, 72%) como un aceite incoloro. LC/MS (Método i) Rt = 2,60 min.; MS m/z: 247 [M+H]+ 1H NMR (DMSO-d6 , 300MHz): 57,75 (m, 2H), 7,53 (dd, J=8,3, 2,1 Hz, 1H), 1,55 ppm (s, 9H).
Etapa B: tere- butil 2,4-dicloro-3-formilbenzoato

A una solución de terc-butil 2,4-diclorobenzoato (13,1 g, 53,0 mmol) en THF (130 mL) y enfriada a -78 °C, se añadió gota a gota durante 10 minutos diisopropilamida de litio (31,8 mL, 63,6 mmol) diluida en THF (25 mL) y ya enfriada a 0 °C. La mezcla de reacción se agitó durante 90 minutos a -75 °C, luego se añadió DMF (12,31 mL, 159 mmol) y la mezcla de reacción se agitó durante 10 minutos entre -65 °C y -75 °C. La mezcla de reacción se inactivó con ácido acético (6 mL) y se dejó calentar a temperatura ambiente. La mezcla de reacción se repartió entre éter dietílico y solución de HCl 1N. La capa acuosa se extrajo con éter dietílico, la capa orgánica se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-10% en ciclohexano) para dar tercbutil 2,4-dicloro-3-formilbenzoato (10,6 g, 71%) como un aceite amarillo.
LC/MS (Método h) Rt = 2,88 min.; MS m/z: 275 [M+H]+ .1H NMR (DMSO-d6 , 300MHz): 5 10,34 (s, 1H), 7,86 (d, J=8,4 Hz, 1H), 7,69 (d, J=8,4 Hz, 1H), 1,56 (s, 9H).
Etapa C: tere- butil 2,4-didoro-3-(1-hidroxiprop-2-in-1-il)benzoato

Usando un procedimiento similar a la Preparación #1 Etapa D, terc-butil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (4 g, 100%) se preparó a partir de terc-butil 2,4-dicloro-3-formilbenzoato (3,39 g, 12,3 mmol). LC/mS (Método i) Rt = 2,14 min.; MS m/z: 301 [M+H]+ .
1H NMR (DMSO-d6 , 300MHz): 57,56 (s, 2H), 6,26 (d, J=5,0 Hz, 1H), 6,10 (dd, d, J=5,0 Hz y 2,3 Hz, 1H), 3,51 (d, J=2,3 Hz, 1H), 1,55 (s, 9H).
Preparación #34: W-(2-yodo-6-metil-4-(tnfluorometil)feml)bencenosulfonamida

Etapa A: 2-yodo-6-metil-4-(trifluorometil)anilina

A una solución de 2-metil-4-(trifluorometil)anilina (3,5 g, 19,98 mmol) en EtOH (80 mL) se añadió yodo (5,07 g, 19,98 mmol) y sulfato de plata (6,23 g, 19,98 mmol), y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se diluyó con una solución acuosa de Na2S2O3 al 10%. La capa acuosa obtenida se extrajo con EtOAc y la capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 2-yodo-6-metil-4-(trifluorometil)anilina como una resina de color naranja (6,3 g, 90%). LC/MS (Método i) Rt = 2,32 min.; MS m/z: 302 [M+H]+. 1H NMR (DMSO-de, 300MHz): 57,70 (s, 1H), 7,31 (s, 1H), 5,55 (s, 2H), 2,20 (s, 3H)
Etapa B: N-(2-yodo-6-metM-4-(tnfluorometil)feml)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, se preparó N-(2-yodo-6-metil-4-(trifluorometil)fenil)bencenosulfonamida (2,73 g, 32%) a partir de 2-yodo-6-metil-4-(trifluorometil)anilina (5,8 g, 19,3 mmol). LC/MS (Método i) Rt = 2,35 min.; MS m/z: 442 [M+H]+. 1 H NMR (DMSO-de, 300MHz): 5 10,03 (s, 1H), 8,01 (d, J=2,1 Hz, 1H), 7,60 (m, 6H), 2,04 (s, 3H).
Preparación #35: W-(5-bromo-2-yodo-3-metilfeml)bencenosulfonamida

Etapa A: 5-bromo-2-yodo-1-metil-3-nitrobenceno

A una solución de 4-bromo-2-metil-6-nitroanilina (250 mg, 1,082 mmol) en cloroformo (1,6 mL) se añadió nitrito de isoamilo (190 mg, 1,623 mmol) y yodo (549 mg, 2,164 mmol) y la mezcla de reacción se agitó a reflujo durante 3,5 horas. La mezcla de reacción se diluyó con una solución acuosa de Na2S2O3 al 50%. La capa acuosa obtenida se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 5-bromo-2-yodo-1-metil-3-nitrobenceno (350 mg, 30%) como una resina marrón. Se usó cruda en la siguiente etapa. LC/MS (Método h) Rt = 2,90 min.; sin ionización
Etapa B: 5-bromo-2-yodo-3-metilanilina

Usando un procedimiento similar a la Preparación #23 Etapa D, se preparó 5-bromo-2-yodo-3-metilanilina (1,6 g, 56%) a partir de 5-bromo-2-yodo-1-metil-3-nitrobenceno (7,25 g 8,69 mmol). Se usó crudo en la siguiente etapa. LC/MS (Método h) Rt = 2,74 min.; MS m/z: 312 [M+H]+. 1H NMR (DMSO-da, 300MHz): 56,76 (d, J=2,5 Hz, 1H), 6,70 (m, 1H), 5,49 (s, 2H), 2,29 (s, 3H).
Etapa C: N-(5-bromo-2-yodo-3-metilfeml)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, se preparó W-(5-bromo-2-yodo-3-metilfenil)bencenosulfonamida (2,21 g, 95%) a partir de 5-bromo-2-yodo-3-metilanilina (1, 6 g, 5,13 mmol). LC/MS (Método h) Rt = 2,94 min.; MS m/z: 450 [M-H]-.
1H NMR (DMSO-d6, 300MHz): 59,95 (s, 1H), 7,65 (m, 5H), 7,44 (d, J=2,1 Hz, 1H), 6,87 (d, J=2,1 Hz, 1H), 2,36 (s, 3H).
Preparación #36: Preparación de metil 5-(1-hidroxiprop-2-in-1-il)-4,6-dimetilnicotinato

Etapa A: 5-bromo-4,6-dimetil-2-oxo-1,2-dihdropiridina-3-carbonitrilo

A una solución de 4,6-dimetil-2-oxo-1,2-dihidropiridin-3-carbonitrilo (35 g, 236 mmol) en ácido acético (200 mL) se añadió bromo (41,5 g, 260 mmol). La mezcla de reacción se agitó durante aproximadamente 30 minutos a temperatura ambiente. La mezcla de reacción se concentró luego a sequedad a presión reducida. La resina resultante precipitó en EtOH acuoso para obtener 5-bromo-4,6-dimetil-2-oxo-1,2-dihidropiridina-3-carbonitrilo (70 g, 95%). LCMS (Tabla 1, Método c) RT= 1,63 min.; MS m/z = 226, 228 (M+H).
Etapa B: 5-bromo-2-cloro-4,6-dimetilnicotinonitrilo

A una solución de 5-bromo-4,6-dimetil-2-oxo-1,2-dihidropiridina-3-carbonitrilo (14 g, 61,7 mmol) en POCl3 (10 mL) se añadió pentaclorofosforano (12,84 g, 61,7 mmol). La reacción se calentó a reflujo durante aproximadamente 8 h. El POCh se concentró a presión reducida. El residuo resultante se repartió entre DCM (100 mL) y NaOH acuoso al 5% (100 mL). Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron al vacío. El residuo resultante se disolvió en POCh (20 mL) y se trató con pentaclorofosforano (13 g, 62,4 mmol). La reacción se calentó a reflujo durante aproximadamente 12 h. El POCl3 se concentró a presión reducida. El residuo resultante se repartió entre DCM (100 mL) y NaOH acuoso al 5% (100 mL). Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron al vacío para proporcionar 5-bmmo-2-dom-4,6-dimetilnicotinonitrilo (12 g, 79%). LCMS (Tabla 1, Método d) RT = 2,12 min.; MS m/z = 246, 248 (M+H).
Etapa C: 5-bromo-4,6-dimetilnicotinonitrilo

A una mezcla de 5-bromo-2-cloro-4,6-dimetilnicotinonitrilo (10 g, 40,7 mmol) y fósforo rojo (12,62 g, 407 mmol) se añadió HI (44,7 mL, 489 mmol). La reacción se agitó a aproximadamente 120 °C durante aproximadamente 4 h. La mezcla se enfrió a temperatura ambiente y después se vertió en NaHCO3 acuoso saturado (500 mL) y Na2 SO3 (10 g). Los sólidos se filtraron y el filtrado restante se concentró al vacío. El sólido resultante se extrajo con MeOH, luego se concentró a
presión reducida para proporcionar 5-bromo-4,6-dimetilnicotinonitrilo (7 g, 44,0%). LCMS (Tabla 1, Método f) RT = 1,63 min.; MS m/z = 211,213 (M+H).
Etapa D: ácido 5-bromo-4,6-dimetilnicotínico

A una mezcla de 5-bromo-4,6-dimetilnicotinonitrilo (10 g, 47,4 mmol) en agua (20 mL) se añadió KOH (26,6 g, 47,4 mmol). La reacción se agitó a aproximadamente 110 °C durante aproximadamente 16 h. La mezcla de reacción se enfrió luego a temperatura ambiente y se neutralizó mediante la adición de HCl concentrado. El sólido resultante se filtró para proporcionar ácido 5-bromo-4,6-dimetilnicotínico (9 g, 79%). LCMS (Tabla 1, Método e) RT = 1,61 min.; MS m/z = 232, 233 (M+H).
Etapa E: metil 5-bromo-4,6-dimetilnicotinato

A una solución de ácido 5-bromo-4,6-dimetilnicoínico (10 g, 43,5 mmol) en MeOH (10 mL) se añadió cloruro de tionilo (5,17 g, 43,5 mmol) a aproximadamente 0 °C. La mezcla de reacción se calentó a aproximadamente 80 °C. Después de aproximadamente 12 h, la mezcla de reacción se concentró al vacío. El residuo resultante se diluyó con DCM, se secó sobre Na2 SO4 anhidro, se filtró y se concentró a presión reducida para proporcionar 5-bromo-4,6-dimetinicotinato (9,6 g, 81%). LCMS (Tabla 1,’ Método e) RT = 1,99 min.; MS m/z = 245, 246 (M+H).
Etapa F: metil 4,6-dimetil-5-vinilnicotinato

Metil 5-bromo-4,6-dimetilnicotinato (15 g, 61,5 mmol), 4,4,5,5-tetrametil-2-vinil-1,3,2-dioxaborolano (14,2 g, 92 mmol), Pd(PPh3 ) 4 (7,1 g, 6,1 mmol), y KF (5,4 g, 92 mmol) se combinaron en DMF (20 mL). La mezcla se calentó a aproximadamente 100 °C durante aproximadamente 16 h. La mezcla se enfrió a temperatura ambiente y el disolvente se concentró al vacío. El residuo restante se purificó por cromatografía sobre gel de sílice (10:1 éter de petróleo/EtOAc) para proporcionar metil4,6-dimetil-5-vinilnicotinato (9 g, 56%). LC/MS (Tabla 1, Método e) RT = 1,23 min.; MS m/z = 192 (M+H).
Etapa G: metil 5-formil-4,6-dimetilnicotinato

A una solución de metil 4,6-dimetil-5-vinilnicotinato (12 g, 62,8 mmol) y 2,6-dimetilpiridina ( 8 mL, 69 mmol) en una mezcla 3:1 de 1,4-dioxano:agua (50,0 mL) se añadió OsO4 (12 mL, 0,94 mmol, 0,08 M en t-BuOH) y peryodato de sodio (40 g, 188 mmol). La mezcla se agitó a temperatura ambiente durante aproximadamente 36 h. La mezcla de reacción se filtró, y el filtrado se lavó con solución acuosa saturada de Na2 S2 O3. La mezcla se extrajo con EtOAc, se secó sobre Na2 SO4 anhidro, se filtró, y se concentró al vacío. El material crudo se purificó por cromatografía en gel de sílice (5:1 a 2:1 éter de petróleo: EtOAc) para proporcionar metil 5 formil-4,6-dimetilnicotinato ( 8 g, 59%). LCMS (Tabla 1, Método d) RT = 1,55 min.; MS m/z = 194 (M+H).
Etapa H: metil 5-(1-hidroxiprop-2-in-1-il)-4,6-dimetilnicotinato
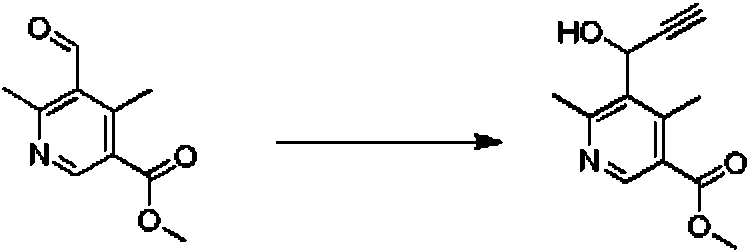
A una solución de metil 5-formil-4,6-dimetilnicotinato ( 8 g, 37,3 mmol) en THF anhidro (100 mL) a aproximadamente 0 °C se añadió bromuro de etinilmagnesio (82 mL, 41 mmol). La reacción resultante se agitó a aproximadamente 0 °C durante aproximadamente 1 h. La reacción se diluyó con cloruro de metileno, se lavó con una solución acuosa saturada de NH4 Cl (3 x 100 mL). La capa orgánica se secó sobre Na2 SO4 anhidro, se filtró, y se concentró al vacío. El material crudo se purificó por cromatografía sobre gel de sílice (2:1 hexanos:EtOAc) para proporcionar metil 5-(1-hidroxiprop-2-in-1-il)-4,6-dimetilnicotinato ( 6 g, 73%). LCMS (Tabla 1, Método c) RT = 1,73 min.; Ms m/z = 220 (M+H).
Preparación #37: etil 3,5-didoro-4-(1-hidroxiprop-2-in-1-N) picolinato

Etapa A: 2,3,5-tridoroisonicotinaldehído

A una solución de LDA (99 mL, 197 mmol) en THF (100 mL), se añadió una solución de 2,3,5-tricloropiridina (30 g, 164 mmol) en THF (200 mL) a aproximadamente -78 °C. La reacción resultante se agitó a aproximadamente -78 °C durante aproximadamente 1 h. Se añadió cuidadosamente formiato de metilo (20 mL, 329 mmol) a la reacción, luego la mezcla se agitó a aproximadamente -78 °C durante aproximadamente 1 h, luego se calentó a temperatura ambiente y se agitó durante 16 h. La reacción se vertió en solución acuosa saturada de NH4 CL La capa acuosa se extrajo con EtOAc (3 x 20 mL). Las capas orgánicas combinadas se secaron sobre Na2 SO4 , anhidro, se filtraron a través de un embudo de vidrio sinterizado, y se concentraron a presión reducida para dar 2,3,5-trid oroisonicotinaldehído (26,2 g, 53% 70% de pureza). LCMS (Tabla 1, Método c) RT = 1,83 min.; MS m/z = 209, 211 (M+H).
Etapa B: 2,3,5-tricloro-4-(dietoximetil) piridina

Una mezcla de 2,3,5-tricloroisonicotinaldehído (26,2 g, 87 mmol), ácido p-toluenosulfónico monohidratado (18,23 g, 96 mmol), MgSO4 (11,54 g, 96 mmol) y ortoformiato de trietilo (21,77 mL, 131 mmol) en DCE (300 mL) se calentó a aproximadamente 50 °C durante aproximadamente 5 h. La solución se diluyó con agua, se extrajo con DCM (3 x 50 mL). Las capas orgánicas combinadas se secaron sobre Na2 SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar 2,3,5-tricloro-4-(dietoximetil)piridina (20,67 g, 75%). LCMS (Tabla 1, Método d) RT = 2,12 min.; MS m/z = 284, 286 (M+H).
Etapa C: etil 3,5-dicloro-4-(dietoximetil)picolinato

Una solución de 2,3,5-tricloro-4-(dietoximetil)piridina (20 g, 70,3 mmol). PdCl2 (PPh3 ) 2 (4,93 g, 7,03 mmol), y TEA (29 mL, 211 mmol) en EtOH (200 mL) se trató con CO a una presión de 30 bares y se calentó a aproximadamente 100 °C en tubo
sellado durante aproximadamente 24 h. La reacción se filtró a través de una almohadilla de celita, y el filtrado se concentró a presión reducida. El material crudo se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 5%/heptanos para dar etil 3,5-dicloro-4-(dietoximetil)picolinato (14 g, 61,8%). LCMS (Tabla 1, Método d) RT = 2,02 min.; MS m/z = 322, 324 (M+H).
Etapa D: etil 3,5-didoro-4-formilpicolinato

Un matraz de fondo redondo se cargó con una mezcla de etil 3,5-dicloro-4-(dietoximetil)picolinato (14 g, 43,5 mmol) y HCl (100 mL, 3291 mmol). La reacción se agitó durante aproximadamente 1 hora a temperatura ambiente. La reacción se diluyó con EtOAc, se lavó con NaOH ac.1N (3 x 50 mL). La porción orgánica se secó sobre Na2 SO4 anhidro, se filtró a través de embudo de vidrio sinterizado y se concentró a presión reducida para dar etil 3,5-dicloro-4-formilpicolinato (10 g, 93%). LCMS (Tabla 1, Método d) RT = 1,92 min.; MS m/z = 247, 249 (M+H).
Etapa E: etil 3,5-didoro-4-(1-hidroxiprop-2-in-1-il)picolinato

A una solución de etil 3,5-dicloro-4-formilpicolinato (9,6 g, 32,9 mmol) en THF anhidro (100 mL) a aproximadamente 0 °C se añadió bromuro de etinil magnesio (72 mL, 36,2 mmol). La reacción resultante se agitó a aproximadamente 0 °C durante aproximadamente 1 h. La reacción se diluyó con cloruro de metileno, se lavó con una solución acuosa saturada de NH4 Cl (3 x 100 mL). La capa orgánica se secó sobre Na2 SO4 anhidro, se filtró, y se concentró al vacío. El material crudo se purificó por cromatografía sobre gel de sílice (10% EtOAc/heptanos) para proporcionar etil 3,5-dicloro-4-(1-hidroxiprop-2in-1il)picolinato (7,9 g, 8 8 %). LC/MS (Tabla 1, Método d) RT = 1,65 min.; Ms m/z = 274, 276 (M+H).
Preparación #38: Preparación de etil 4,6-dicloro-5-(1-hidroxiprop-2-in-1-il)nicotinato

Etapa A: etil 4,6-dicloronicotinato

Una mezcla de ácido 4,6-dicloronicotínico (20 g, 104 mmol) en DCM (200 mL) se enfrió a 0 °C antes de la adición de DMF (0,807 mL, 10,42 mmol) y cloruro de oxalilo (10,94 mL, 125 mmol). La reacción resultante se agitó a aproximadamente 0 °C-25 °C. Después de aproximadamente 2 h, se añadió lentamente EtOH (48,7 mL, 833 mmol) y se agitó a aproximadamente 25 °C durante 2 h. La mezcla de reacción se diluyó con éter (50 mL) y la solución resultante se lavó con NaHCO3 acuoso saturado (3 x 50 mL). Los extractos se combinaron y se secaron sobre Na2 SO4 anhidro, se filtraron y el disolvente se eliminó a vacío para proporcionar etil 4,6-dicloronicotinato (18 g.79%): LC/MS (Tabla 1, Método d) Rt = 1,91 min.; MS m/z = 220 (M+H).
Etapa B: etil 4,6-dicloro-5-formilnicotinato

A un matraz que contenía LDA (27,3 mL, 54,5 mmol) en THF (100 mL) se añadió una solución de etil 4,6-didoronicotinato (10 g, 45,4 mmol) en THF (200 mL) de modo que la temperatura interna se mantuvo a aproximadamente -78 °C. Después de aproximadamente 30 minutos, se añadió formiato de metilo (5,57 mL, 91 mmol) gota a gota a la mezcla de reacción y se agitó a -78 °C durante 1 hora. La mezcla de reacción se vertió después en Na2 SO4 acuoso saturado, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice (20:1 hexanos:EtOAc) para proporcionar etil 4,6-dicloro-5formilnicotinato (5,19 g, 46%). LCMS (Tabla 1, Método c) RT = 1,95 min.; MS m/z = 248 (M+H).
Etapa C: etil 4,6-didoro-5-(1-hidroxiprop-2-in-1-il)nicotinato

A una solución de etil 4,6-dicloro-5-formilnicotinato (12,9 g, 52,0 mmol) en THF anhidro (120 mL) a aproximadamente 0 °C se añadió bromuro de etinilmagnesio (114 mL, 57,2 mmol). La reacción resultante se agitó a aproximadamente 0 °C durante aproximadamente 1 h. La reacción se diluyó con cloruro de metileno, se lavó con una solución acuosa saturada de NH4 Cl (3 x 100 mL). La capa orgánica se secó sobre Na2 SO4 anhidro, se filtró, y se concentró al vacío. El material crudo se purificó por cromatografía en gel de sílice (10:1 hexanos/EtOAc) para proporcionar etil 4,6-dicloro-5-(1-hidroxiprop-2-in-1-il)nicotinato (11,1 g, 78%). LCMS (Tabla 1, Método d) Rt= 1,70 min.; MS m/z = 274 (M+H).
Preparación #39: A/-(3-yodo-2-metil-6-(trifluorometil)piridin-4-il)bencenosulfonamida

Etapa A: 2-metil-4-nitro-6-(trifluorometil)piridina

A una solución de 2-bromo-6-metil-4-nitropiridina (WO2013/059587) (1,84 g, 8,48 mmol) y yoduro de cobre (I) (1,938 g, 10,17 mmol) en NMP (15,73 mL) se añadió metil 2,2-difluoro-2-(fluorosulfonil)acetato (3,24 mL, 25,4 mmol) y la reacción se irradió en microondas a 100 °C durante 5 horas. La mezcla de reacción se lavó con agua y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 2-metil-4-nitro-6-(trifluorometil)piridina (1,48 g, 80%). LC/MS (Método h) Rt = 2,16 min; sin ionización 1 H NMR (DMSO-d6 , 300MHz): 58,41 (d, J=1, 8 Hz, 1H), 8,32 (d, J=1,8 Hz, 1H), 2,74 (s, 3H)
Etapa B: 2-metil-6-(trifluorometil)piridin-4-amina
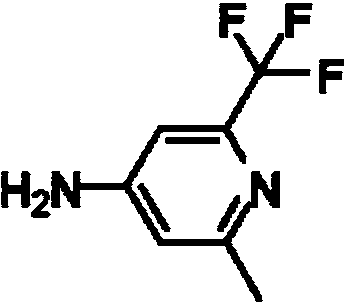
Usando un procedimiento similar al descrito en la Preparación #18, Etapa C, se preparó metil-6-(trifluorometM)piridin-4-amina (409 mg, 42%) a partir de 2-metil-4-nitro-6-(trifluorometil)piridina (1,05 g, 5,09 mmol). LC/mS (Método k) Rt = 2,15 min.; MS m/z: 177 [M+H]+. 1H NMR (DMSO-de, 300MHz): 56,73 (d, J=1,7 Hz, 1H), 6,49 (d, J=1,7 Hz, 1H), 6,42 (s, 2H), 2,29 (s, 3H).
Etapa C: 3-yodo-2-metil-6-(trifluorometil)piridin-4-amina

A una solución de 2-metil-6-(trifluorometil)piridin-4-amina (405 mg, 2,299 mmol) en EtOH (8 mL) se añadió sulfato de plata (717 mg, 2,299 mmol) y yodo (584 mg, 2,299 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 4 horas. La mezcla de reacción se inactivó con una solución acuosa saturada de Na2 S2 O3 y se extrajo dos veces con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-40% en ciclohexano) para dar 3-yodo-2-metil-6-(trifluorometil)piridin-4-amina (608 mg, 88%) como un sólido amorfo. LC/MS (Método i) Rt = 1,78 min.; MS m/z: 303 [M+H]+.
1 H NMR (DMSO-de, 300MHz): 5 6,86 (s, 1H), 6,63 (ancho, 2H), 2,59 (s, 3H).
Etapa D: W-(3-yodo-2-metil-6-(trifluorometil)piridm-4-il)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16 Etapa B, se preparó W-(3-yodo-2-metil-6-(trifluorometil)piridin-4-il)bencenosulfonamida (414 mg, 33%) a partir de 3-yodo-2- metil-6-(trifluorometil)piridin-4-amina (740 mg, 2,45 mmol). LC/MS (Método i) Rt = 2,24 min.; MS m/z: 443 [M+H]+. 1 H NMR (DMSO-d6 , 300MHz): 5 7,90 (m, 2H), 7,64 (m, 3H), 7,22 (s, 1H), 2,70 (s, 3H).
Preparación #40: N-(6-ciano-3-yodo-2-metilpiridm-4-il)bencenosulfonamida

Etapa A: 4-amino-5-yodo-6-metilpicolinonitrilo

A una solución de 4-amino-6-met¡lp¡col¡non¡tr¡lo (1,756 g, 13,19 mmol) (descrito en W02005/030213) en ACN (44,0 mL) se añadió W-yodosuccinimida (3,26 g, 14,51 mmol) y la mezcla de reacción se agitó a reflujo por 24 horas. La mezcla de reacción se hidrolizó con una solución acuosa saturada de Na2 S2 O3 y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-60% en ciclohexano) para dar 4-amino-5-yodo-6-metilpicolinonitrilo (1,18 g, 34%) como un sólido de color marrón. LC/MS (Método h) Rt = 1,55 min.; MS m/z: 260 [M+H]+.
1 H NMR (DMSO-d6 , 500MHz): 56,88 (s, 1H), 6,66 (ancho, 2H), 2,55 (s, 3H)
Etapa B: W-(6-ciano-3-yodo-2-metilpiridm-4-il)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se preparó N-(6-ciano-3-yodo-2-metilpiridin-4-il)bencenosulfonamida (400 mg, 23%) a partir de 4-amino-5-yodo-6-metilpicolinonitrilo (1,15 g, 4,44 mmol). El compuesto se usó directamente en la siguiente etapa.
LC/MS (Método h) Rt = 2,28 min.; MS m/z: 400 [M+H]+.
Preparación #41: 4-metil-6-(trifluorometil)piridazin-3-amina

Etapa A: 4-bromo-6-(trifluorometil)piridazin-3-amina

A una solución de 6-(trifluorometil)piridazin-3-amina (730 mg, 4,48 mmol) y bicarbonato de sodio (752 mg, 8,95 mmol) en MeOH (15 mL) se añadió bromo (0,231 mL, 4,48 mmol) gota a gota. La mezcla de reacción se agitó a temperatura ambiente durante una noche, después se hidroliza con una solución acuosa saturada de Na2S2O3. El MeOH se evaporó y la solución resultante se diluyó con EtOAc y las dos fases se separaron. La capa orgánica se lavó con una solución acuosa saturada de Na2S2O3, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 7-60% en ciclohexano) para dar 4-bromo-6-(trifluorometil)piridazin-3-amina (513 mg, 47%) como un sólido de color beige. LC/MS (Método i) Rt = 1,33 min.; MS m/z: 242 [M+H]+ 1H NMR (DMSO-de, 300MHz): 58,23 (s, 1H), 7,55 (ancho, 2H).
Etapa B: 4-metil-6-(trifluorometil)piridazin-3-amina

A una solución de 4-bromo-6-(trifluorometil)piridazin-3-amina (460 mg, 1,901 mmol), tris (dibencilideneacetona) dipaladio(0) (17,41 mg, 0,019 mmol), 2-(diciclohexilfosfino)- 2',4',6'-triisopropilbifenilo (36,2 mg, 0,076 mmol) y fosfato de potasio tribásico (807 mg, 3,80 mmol) en 1,4-dioxano (3,7 mL) se añadió trimetilboroxina (0,398 mL, 2,85 mmol). La mezcla de reacción se agitó a 100 °C durante una noche, después se diluyó con H2 O. La capa acuosa obtenida se extrajo con EtOAc y la capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El mismo procedimiento se realizó tres veces con el residuo obtenido para aumentar la conversión. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 7-60% en ciclohexano) para dar 4-metil-6-(trifluorometil)piridazin-3-amina (318 mg, 94%) como un sólido de color beige. LC/MS (Método i) Rt = 0,88 min.; MS m/z: 178 [M+H]+.
1 H NMR (DMSO-de, 300MHz): 5 7,54 (s, 1H), 6,94 (br. s., 2H), 2,09 (s, 3H)
Preparación #42: 3,7-dimetil-5-(trifluorometil)-1H-indazol
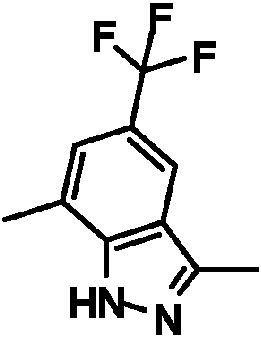
Etapa A: 2-fluoro-3-metil-5-(trifluorometil)benzaldehído
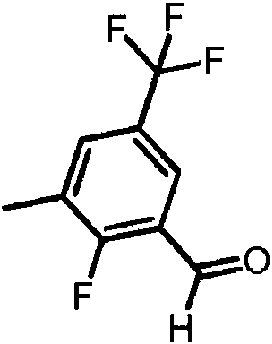
A una solución de 4-fluoro-3-metilbenzotrifluoruro (1,5 g, 8,42 mmol) en THF (11 mL) y enfriado a -78 °C se añadió butil litio (5,42 mL, 8,67 mmol). La mezcla se agitó durante 30 minutos a -78 °C, luego se añadió gota a gota una solución de DMF (0,724 mL, 9,35 mmol) en THF (3 mL). La reacción se agitó a -78 °C durante 1 hora y luego se calentó a 0 °C. Se añadió una solución al 10% de ácido cítrico a la mezcla de reacción a 0 °C y la mezcla se extrajo con EtOAc. La capa orgánica se lavó con salmuera y se secó sobre sulfato de magnesio, se filtró y se evaporó para dar 2-fluoro-3-metil-5-(trifluorometil)benzaldehído (2 g, 100%) como un líquido incoloro. LC/MS (Método h) Rt = 2,52 min.; sin ionización 1H NMR (DMSO-da, 300MHz): 5 10,23 (s, 1H), 8,09 (m, 1H), 7,97 (m, 1H), 2,39 ppm (m, 3H)
Etapa B: 1-(2-fluoro-3-metil-5-(trifluorometil)fenil)etanol

A una solución de 2-fluoro-3-metil-5-(trifluorometil)benzaldehído (2 g, 9,70 mmol) en THF (42 mL) y enfriada a -50 °C se añadió bromuro de metilmagnesio (3,88 mL, 11,64 mmol) y la mezcla se agitó a temperatura ambiente durante 2 horas. La mezcla se inactivó con una solución acuosa saturada de NH4 G y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se evaporó para dar 1-(2-fluoro-3-metil-5-(trifluorometil)fenil)etanol (1,21 g, 41%) como un líquido incoloro: LC/MS (Método h) Rt = 2,42 min.; MS m/z: 281 [M-H]- +CH3 COOH
1 H NMR (DMSO-da, 300MHz): 5 7,65 (m, 1H), 7,59 (m, 1H), 5,53 (d, J=4,6 Hz, 1H), 5,00 (m, 1H), 2,30 (d, J=2,3 Hz, 3H), 1,34 (d, J=6,6 Hz, 3H).
Etapa C: 1-(2-fluoro-3-metil-5-(trifluorometil)fenil)etenona

Usando un procedimiento similar a la Preparación #14 Etapa B, se preparó 1-(2-fluoro-3-metil-5-(trifluorometil)fenil)etanona (854 mg, 75%) a partir de 1-(2-fluoro-3-metilo -5-(trifluorometil)fenil)etanol (1,1 g, 4,95 mmol). LC/MS (Método h) Rt = 2,63 min.; sin ionización
1 H NMR (DMSO-de, 300MHz): 5 7,99 (m, 1H), 7,91 (m, 1H), 2,63 (d, J=4,3 Hz, 3H), 2,37 (d, J=2,5 Hz, 3H).
Etapa D: 3,7-dimetil-5-(trifluorometil)-1H-indazol

A una solución de 1-(2-fluoro-3-metil-5-(trifluorometil)fenil)etanona (200 mg, 0,908 mmol) en etano-1,2-diol (700 pl, 12,55 mmol) se añadió hidrato de hidrazina (53,0 pL, 1,090 mmol) y la mezcla se agitó a temperatura ambiente durante 20 horas. Se añadió más hidrato de hidrazina (53,0 pL, 1,090 mmol) y la mezcla se calentó a 50 °C durante 1 hora. El calentamiento se aumentó a 170 °C y continuó durante 2 horas. La mezcla se enfrió a temperatura ambiente: el indazol final precipitó. Se diluyó con agua, se filtró; y se enjuagó con agua y se secó a 50 °C al vacío para dar 3,7-dimetil-5-(trifluorometil)-1H-indazol (130 mg, 61%) como un sólido de color blanco. LC/MS (Método i) Rt = 1,99 min.; MS m/z: 215 [M+H]+1H NMR (DMSO-de, 300MHz): 513,14 (ancho, 1H), 7,97 (s, 1H), 7,39 (s, 1H), 2,55 (s, 3H), 2,54 (s, 3H).
Preparación #43: 3,7-dimetil-5-(trifluorometil)-1H-mdol

Etapa A: W-alil-2-yodo-6-metil-4-(trifluorometil)amlma

A una solución de 2-yodo-6-metil-4-(trifluorometil)anilina (Preparación #34, Etapa A) (11,1 g, 36,9 mmol) en tolueno (56 mL) se añadió carbonato de potasio (7,64 g, 55,3 mmol), bromuro de tetrabutilamonio (1,189 g, 3,69 mmol), hidróxido de potasio (2,276 g, 40,6 mmol) y bromuro de alilo (5,80 g, 47,9 mmol) La mezcla de reacción se agitó a 65 °C durante 18 horas. Luego se añadieron más carbonato de potasio (0,8 eq), bromuro de tetrabutilamonio (0,05 eq), hidróxido de potasio (0,5 eq) y bromuro de alilo (0,6 eq). La mezcla de reacción se agitó a 65 °C durante 18 horas. Luego se añadieron más carbonato de potasio (0,8 eq), bromuro de tetrabutilamonio (0,05 eq), hidróxido de potasio (0,5 eq) y bromuro de alilo (0,6 eq). La mezcla de reacción se agitó a 65 °C durante 18 horas. Luego se añadieron más carbonato de potasio (0,8 eq), bromuro de tetrabutilamonio (0,05 eq), hidróxido de potasio (0,5 eq) y bromuro de alilo (0,6 eq). La mezcla de reacción se agitó a 65 °C durante 18 horas. La mezcla de reacción se enfrió a 5 °C. Se diluyó con agua y se acidificó con una solución de HCl 5N hasta pH = 3-4. La capa acuosa se extrajo con EtOAc. La capa orgánica se lavó con agua, salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó dos veces por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-30% en ciclohexano) y luego (eluyendo con EtOAc al 5-10% en ciclohexano) para dar N-alil-2-yodo-6-metil-4-(trifluorometil)anilina (7,25 g, 57%) como un aceite de color naranja. LC/MS (Método i) Rt = 2,69 min.; MS m/z: 342 [M+H]+1H NMR (CLOROFORMO-d, 300MHz): 57,82 (s, 1H), 7,32 (s, 1H), 5,96 (dd, J = 17,2, 10,2 Hz, 1H), 5,30 (dd, J = 17,2, 1,4 Hz, 1,4H, 1H), 5,16 (dd, J = 10,2, 1,4 Hz, 1H), 3,84 (ancho, 1H), 3,73 (d, J = 5,8 Hz, 2H ), 2,38 (s, 3H),
Etapa B: 3,7-dimetil-5-(trifluorometil)- 1H-indol

A una solución de W-alil-2-yodo-6-metil-4-(trifluorometil)anilina (1 g, 2,93 mmol) en ACN (5 mL) se añadió acetato de paladio(N) (0,013 g, 0,059 mmol) y tri-o-tolilfosfina (0,036 g, 0,117 mmol). La mezcla de reacción se calentó a 45 °C y se añadió trietilamina (1,5 mL). La mezcla de reacción se agitó a 75 °C durante 15 horas. Se añadió más acetato de paladio (II) (0,05 eq) y tri-o-tolilfosfina (0,1 eq) y la agitación continuó durante 2 horas. La mezcla de reacción se enfrió a temperatura ambiente y el disolvente se eliminó a presión reducida. Se añadió EtOAc al residuo y el precipitado obtenido se filtró y se lavó con EtOAc. El filtrado se concentró a presión reducida. Este residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con 0-15% de EtOAc en ciclohexano) para dar 3,7-dimetil-5-(trifluorometil)-1 H-indol (460 mg, 68%) como un aceite de color naranja. LC/MS (Método i) Rt = 2,32 min.; MS miz: 214 [M+H]+1H Nm R (DMSO-da, 300MHz): 511,18 (ancho, 1H), 7,67 (s, 1H), 7,28 (s, 1H), 7,16 (s, 1H), 2,51 (s, 3H), 2,29 (s, 3H)
Preparación #44: tere- butil 2,4-didoro-3-(clorocarbonil)benzoato

Etapa A: ácido 3-(terc-butoxicarbonil)-2,6-didorobenzoico

A una solución de tere -butil 2,4-diclorobenzoato (Preparación #33, Etapa A) (15 g, 60,7 mmol) en THF (100 mL) y enfriada a -70 °C se añadió diisopropilamida de litio (36,4 mL, 72,8 mmol). La mezcla de reacción se agitó durante 90 minutos a -70 °C y luego se añadió dióxido de carbono triturado (30 g, 682 mmol) secado sobre sulfato de magnesio. La mezcla de reacción se agitó durante 15 minutos entre -65 °C y - 70 °C, luego se calentó a temperatura ambiente. La mezcla de reacción se diluyó con agua y el THF se evaporó. La capa acuosa básica se lavó con éter dietílico y luego se acidificó a pH = 1 mediante la adición de solución de HCl 1N y se extrajo con EtOAc. La capa orgánica se lavó con una solución saturada de NaCl, se secó sobre sulfato de magnesio y se concentró para dar ácido 3-(terc-butoxicarbonil)-2,6-diclorobenzoico (13 g, 74%) de un sólido de color beige. LC/MS (Método i) Rt = 1,53 min.; sin ionización 1H NMR (DMSO-d6, 300MHz): 57,76 (d, J=8,4 Hz, 1H), 7,65 (d, J=8,4 Hz, 1H), 1,55 (s, 9H).
Etapa B: terc-butil 2,4-didoro-3-(dorocarbom l)benzoato

A una solución de ácido 3-(terc-butoxicarbonil)-2,6-diclorobenzoico (455 mg, 1,563 mmol) en DCM (23 mL) y enfriada a 0 °C se añadió cloruro de oxalilo (0,205 mL, 2,344 mmol) diluido en DCM (2 mL) con 2 gotas de DMF. La mezcla de reacción se agitó durante 45 minutos a 0 °C. La mezcla de reacción se concentró a vacío para dar el terc-butil 2,4-dicloro-3-(clorocarbonil)benzoato que se usó directamente crudo en la siguiente etapa.
Preparación #45: 3,7-dimetil-1H -indol-5-carbonitrilo

4-amino-3-doro-5-metilbenzonitrilo (250 mg, 1,501 mmol), carbonato de potasio (622 mg, 4,50 mmol), 3-bromoprop-1-eno (0,130 mL, 1,501 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (71,5 mg, 0,150 mmol) se disolvieron en DME (7,5 mL) y se añadió acetato de paladio(II) (16,84 mg, 0,075 mmol). La mezcla de reacción se agitó 95 minutos a 150 °C bajo irradiación de microondas. La mezcla de reacción se concentró hasta la sequedad y el residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 15-20% en ciclohexano) para dar 3,7-dimetil-1H -indol-5-carbonitrilo (107 mg, 25 %) como un sólido de color amarillo pálido. LC/MS (Método i) Rt = 1,91 min.; MS m/z: 171 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 5 11,34 (ancho, 1H), 7,86 (s, 1H), 7,30 (s, 1H), 7,22 (s, 1H), 2,47 (s, 3H), 2,27 (s, 3H)
Preparación #46: W-(2-bromo-5-ciano-3-metilfeml)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se obtuvo W-(2-bromo-5-ciano-3-metilfenil)bencenosulfonamida (2,03 g, 67%) como un sólido de color naranja a partir de 3-amino-4-bromo-5-metilbenzonitrilo (descrito en Journal of Medicinal Chemistry, 2007, 50, 6519-6534) (1,5 g, 7,11 mmol). LC/MS (Método h) Rt = 2,48 min.; MS m/z: 349 [M-H]- 1 H NMR (CDCla, 300 MHz): 5 7,81 (m, 3H), 7,59 (m, 1H), 7,49 (m, 2H), 7,24 (m, 2H), 2,36 (s, 3H).
Preparación #47: N-(2-yodo-3-metil-5-(trifluorometoxi)fenil)bencenosulfonamida

A una solución de 2-metil-4-(trifluorometoxi)anilina (1 g, 5,23 mmol) en DCM (4 mL) y enfriada a 0 °C se añadió anhídrido 2,2,2-trifluoroacético (1,921 mL, 13,60 mmol) y la mezcla se agitó durante una hora a 0 °C. Se añadió lentamente nitrato de potasio (0,661 g, 6,54 mmol) a 0 °C y la mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla se diluyó con agua y se extrajo tres veces con DCM. Las fases orgánicas se lavaron con una solución saturada de NaCl, se secaron sobre sulfato de magnesio, se filtraron y se evaporaron para dar 2,2,2-trifluoro-N-(2-metil-6-nitro-4-(trifluorometoxi)fenil)acetamida (1,7 g, 100%) como un sólido amarillo. LC/MS (Método i) Rt = 2,17 min.; MS m/z: 331 [M-H]- 1 H NMR (DMSO-d6 , 300MHz): 5 = 11,59 (s, 1H), 8,05 (m, 1H), 7,88 (m, 1H), 2,35 (s, 3H).
Etapa B: 2-metil-6-nitro-4-(trifluorometoxi)anilina

A una solución de 2,2,2-trifluoro-N-(2-metil-6-nitro-4-(trifluorometoxi)fenil)acetamida (4 g, 12,04 mmol) en MeOH (80 mL) se añadió carbonato de potasio (6,66 g, 48,2 mmol) y la mezcla de reacción se agitó a 85 °C durante 36 horas. El disolvente se evaporó, el residuo se diluyó con acetato de etilo y agua y se neutralizó mediante la adición de una solución de HCl 1N. Después de dos extracciones con acetato de etilo, las capas orgánicas se lavaron con salmuera, se secaron sobre sulfato de magnesio y se evaporaron para dar 2-metil-6-nitro-4-(trifluorometoxi)anilina (3,24 g, 100%). LC/MS (Método i) Rt = 2,16 min.; MS m/z: 235 [M-H]-1 H NMR (DMSO-d6 , 300MHz): d = 7,83 (d, J=2,8 Hz, 1H), 7,47 (d, J=2,8 Hz, 1H), 7,33 (br, 2H), 2,25 (s, 3H)
Etapa C: 2-yodo-1-metil-3-nitro-5-(trifluorometoxi)benceno

A una solución de 2-metil-6-nitro-4-(trifluorometoxi)anilina (2,5 g, 10,59 mmol) en agua (10,6 mL) se añadió gota a gota ácido clorhídrico concentrado (10,61 mL, 349 mmol). La solución insoluble se enfrió a 0 °C, luego se añadió gota a gota una solución de nitrito de sodio (1,169 g, 16,94 mmol) en agua (15 mL) para mantener la temperatura por debajo de 10 °C. La mezcla de reacción se agitó 1 hora entre 0 °C y 10 °C. Se añadió gota a gota una solución de yoduro de potasio (5,27 g, 31,8 mmol) en agua (15 mL) para mantener la temperatura por debajo de 10 °C y la mezcla de reacción se agitó una hora a 5-10 °C. Se añadió una solución saturada de tiosulfato de sodio y la reacción se extrajo tres veces con EtOAc. Las capas orgánicas se lavaron con una solución saturada de NaCl, se secaron sobre sulfato de magnesio, se filtraron y se evaporaron. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar 2-yodo-1-metil-3-nitro-5-(trifluorometoxi)benceno (2,81 g, 76%) como un aceite amarillo. LC/MS (Método i) Rt = 2,43 min.; sin ionización
1 H NMR (DMSO-d6 , 300MHz): d = 7,89 (d, J=2,0 Hz, 1H), 7,72 (d, J=2,0 Hz, 1H), 2,55 (s, 3H).
Etapa D: 2-yodo-3-metil-5-(trifluorometoxi)anilina

A una solución de 2-yodo-1-metil-3-nitro-5-(trifluorometoxi)benceno (2 g, 5,76 mmol) en EtOH (50 mL) se añadió hierro (0,805 g, 14,41 mmol), agua (10,42 ml) y ácido clorhídrico (0,154 mL, 5,07 mmol) y la mezcla de reacción se agitó a 80 °C durante una noche. La mezcla de reacción se filtró y el sólido se lavó con EtOH. El filtrado se concentró a presión reducida y el residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar 2-yodo-3-metil-5-(trifluorometoxi)anilina (1,36 g, 75 %) como un líquido de color marrón. LC/MS (Método i) Rt = 2,38 min.; MS m/z: 318 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 5 6,56 (s, 1H), 6,52 (s, 1H), 5,62 (s, 2H), 2,33 (s, 3H)
Etapa E: N-(2-yodo-3-metil-5-(trifluorometoxi)fenil)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, se preparó N-(2-yodo-3-metil-5-(trifluorometoxi)fenil)bencenosulfonamida (1,98 g, 100%) a partir de 2-yodo-3-metil-5-(trifluorometoxi)anilina (1,36 g, 4,29 mmol). LC/MS (Método i) Rt = 2,47 min.; MS m/z: 456 [M-H]-1H NMR (DMSO-da, 300MHz): 510,02 (s, 1H), 7,71 (m, 3H), 7,59 (m, 2H), 7,28 (m, 1H), 6,62-6,70 (m, 1H), 2,41 (s, 3H).
Preparación #48: 6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol

A una solución de 6-bromo-4-cloro-1H-indol (1 g, 4,34 mmol) en N,N-dimetilformamida (10 mL) y enfriada a 0 °C se añadió hidruro de sodio (0,208 g, 5,21 mmol) y la mezcla de reacción se agitó a 0 °C durante 1 hora. Se añadió cloruro de bencenosulfonilo (0,668 mL, 5,21 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se diluyó con una solución saturada de NH4 Cl y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-40% en ciclohexano) para dar 6-bromo-4-cloro-1-(fenil-sulfonil)-1H-indol (1,57 g, 98%) como un sólido de color beige. LC/MS (Método i) Rt = 2,71 min.; sin ionización 1 H NMR (DMSO-d6 , 300MHz): 5 8,05 (m, 4H), 7,75 (m, 1H), 7,65 (m, 3H), 6,90 (dd, J=3,8, 0,8 Hz, 1H)
Preparación #49: 7-metil-5-(trifluorometil)-1H-indol

Etapa A: N-(2-metil-4-(trifluorometil)-6-((trimetilsilil) etinil)fenil)bencenosulfonamida

A una solución de N-(2-yodo-6-metil-4-(trifluorometil)fenil)bencenosulfonamida (Preparación #34) (400 mg, 0,907 mmol) en THF (4,5 mL) se añadió dicloruro de bis(trifenilfosfina)paladio (31,8 mg, 0,045 mmol), yoduro de cobre(I) (17,27 mg, 0,091 mmol), trimetilsililacetileno (351 |_il, 2,54 mmol) y trietilamina (4,5 mL). La mezcla de reacción se agitó a 65 °C en microondas durante 1,5 horas. La mezcla de reacción se filtró, se enjuagó con EtOAc y el filtrado se evaporó para dar N-(2-metil-4-(trifluorometil)-6-((trimetilsilil)etinil)fenil)bencenosulfonamida (495 mg, 100%) como un marrón sólido. LC/MS (Método i) Rt = 2,76 min.; MS m/z: 412 [M+H]+
1 H NMR (DMSO-d6 , 300MHz): 59,65 (br, 1H), 7,70 (m, 2H), 7,55 (m, 5H), 2,07 (s, 3H), 0,15 (m, 9H).
Etapa B: N-(2-etinil-6-metil-4-(trifluorometil)fenil)bencenosulfonamida

A una solución de N-(2-metil-4-(trifluorometil)-6-((trimetilsilil) etinil)fenil)bencenosulfonamida (373 mg, 0,906 mmol) en THF (3,2 mL) y e nfriada a 0 °C se añadió fluoruro de tetrabutilamonio (4,08 mL, 4,08 mmol) y la mezcla de reacción se agitó a 0 °C durante 15 minutos. La mezcla de reacción se inactivó con ácido cítrico y se diluyó con agua. Luego se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida para dar N-(2-etinil-6-metil-4-(trifluorometil)fenil)bencenosulfonamida (413 mg, 100%). El compuesto se usó crudo en la siguiente etapa. LC/MS (Método i) Rt = 2,28 min.; MS m/z: 338 [M-H]
Etapa C: 7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-indol

A una solución de N-(2-etinil-6-metil-4-(trifluorometil)fenil)bencenosulfonamida (308 mg, 0,908 mmol) en dicloroetano (4,5 mL) se añadió acetato de cobre(II) (165 mg, 0,908 mmol) y la reacción se calentó a 150 °C bajo microondas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-15% en ciclohexano) para dar 7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-indol (231 mg, 75%) como un aceite rosa. LC/MS (Método i) Rt = 2,59 min.; MS miz: 340 [M+H]+
1H NMR (DMSO-da, 300MHz): 58,06 (d, J=3,8 Hz, 1H), 7,93 (s, 1H), 7,76 (m, 3H), 7,64 (m, 2H), 7,41 (m, 1H), 7,05 (d, J=3,8 Hz, 1H), 2,53 (s, 3H).
Etapa D: 7-metil-5-(trifluorometil)-1H-indol

A una solución de 7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-indol (230 mg, 0,68 mmol) en THF (8,4 mL) se añadió fluoruro de tetrabutilamonio (813 pL, 0,813 mmol) y la mezcla de reacción se agitó a 65 °C durante 2,5 horas. La mezcla de reacción se concentró a presión reducida. El residuo se recogió en DCM y se lavó con una solución saturada de NaHCO3. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar 7-metil-5-(trifluorometil)-1H-indol (83 mg, 61%). LC/MS (Método i) Rt = 2,20 min.; MS miz: 198 [M-H]- 1 H NMR (DMSO-d6 , 300MHz): 511,52 (br, 1H), 7,77 (m, 1H), 7,50 (m, 1H), 7,17 (s, 1H), 6,60 (m, 1H), 2,54 (s, 3H).
Preparación #50: clorhidrato de metil 2-(3-metilpiperidin-4-il)acetato

Etapa A: (E)-etil 4-(2-etoxi-2-oxoetilideno)-3-metilpiperidina-1-carboxilato

A una suspensión de hidruro de sodio (0,972 g, 24,30 mmol) en 1,2-dimetoxietano (30 mL) y enfriada a 0 °C se añadió fosfonacetato de trietilo (4,52 mL, 22,68 mmol) en solución en 1,2-dimetoxietano (30,0 mL) y la mezcla de reacción se agitó a 0 °C durante 30 minutos y 1 hora a temperatura ambiente. La mezcla de reacción se enfrió a 10 °C y a la mezcla de reacción se añadió gota a gota 3-metil-4-oxopiperidina-1-carboxilato de etilo (3 g, 16,20 mmol) en 1,2-dimetoxietano (30,0 mL). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se inactivó con agua y se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar (E)-etil 4-(2-etoxi-2-oxoetilideno)-3-metilpiperidina-1-carboxilato (3,6 g, 87%) como un líquido amarillo pálido.
LC/MS (Método i) Rt = 2,05 min.; MS miz: 256 [M+H]+
Etapa B: etil 4-(2-etoxi-2-oxoetil)-3-metilpiperidina-1-carboxilato

A una solución de (E)-etil 4-(2-etoxi-2-oxoetilideno)-3-metilpiperidina-1-carboxilato (3,60 g, 14,10 mmol) en EtOH (100 mL) se añadió paladio sobre carbono (0,36 g, 3,38 mmol) y la reacción se agitó bajo hidrógeno (1 atm) durante 3 horas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida para dar etil 4-(2-etoxi-2-oxoetil)-3-metilpiperidina-1-carboxilato (3,6 g, 100%).
Etapa C: clorhidrato de ácido 2-(3-metilpiperidin-4-il) acético

Una solución de etil 4-(2-etoxi-2-oxoetil)-3-metilpiperidina-1-carboxilato (200 mg, 0,777 mmol) en ácido clorhídrico (1 mL, 3,29 mmol) se agitó a reflujo durante una noche. La mezcla de reacción se diluyó con H2O y se liofilizó para dar clorhidrato de ácido 2-(3-metilpiperidin-4-il) acético (151 mg, 100%).
Etapa D: clorhidrato de metil 2-(3-metilpiperidin-4-il) acetato

A una solución de clorhidrato de ácido 2-(3-metilpiperidin-4-il) acético (151 mg, 0,780 mmol en MeOH (2 mL) y enfriada a 0 °C se añadió cloruro de tionilo (0,566 mL, 7,80 mmol) y la mezcla de reacción se agitó a reflujo durante 4 horas. La mezcla de reacción se concentró al vacío para dar clorhidrato de metil 2-(3-metilpiperidin-4-il)acetato (151 mg, 93%) como un aceite amarillo. (70% de cis, 30% de trans).
Preparación #51: 1,4-dimetil-6-(trifluorometil)-1H-indol-3-carbaldehído

Etapa A: N-(3-metil-5-(trifluorometil)-2-((trimetilsilil) etinil)fenil)bencenosulfonamida

A una solución de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (5 g, 11,33 mmol) en THF (40 mL) se añadió dicloruro de bis(trifenilfosfina)paladio(II) (0,398 g, 0,567 mmol), yoduro cuproso (0,216 g, 1,133 mmol), trimetilsililacetileno (3,12 g, 31,7 mmol) y trietilamina (40,0 mL) y la mezcla de reacción se agitó a 65 °C en microondas durante 1,5 horas. La mezcla de reacción se filtró y se concentró a presión reducida para dar N-(3-metil-5-(trifluorometil)-2-((trimetilsilil) etinil)fenil)bencenosulfonamida (4,5 g, 85%) como una resina naranja. El producto se usa crudo en la siguiente etapa.
LC/MS (Método j) Rt = 2,85 min.; MS miz: 412 [M+H]+
Etapa B: 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol

A una solución de N-(3-metil-5-(trifluorometil)-2-((trimetilsilil) etinil)fenil)bencenosulfonamida (4,5 g, 10,94 mmol) en THF (40 mL) y se enfrió a 0 °C se añadió fluoruro de tetrabutilamonio (12,87 g, 49,2 mmol) y la mezcla de reacción se agitó a 0 °C durante 1 hora y a temperatura ambiente durante 1 hora. La mezcla de reacción se inactivó con ácido cítrico y se diluyó con agua. Se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida para dar 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol (3,5 g, 84%) como un marrón sólido. LC/MS (Método i) Rt = 2,58 min.; MS m/z: 338 [M-H]- 1H NMR (CLORO-FORMO-d, 300MHz): 5 8,12 (s, 1H), 7,90 (m, 2H), 7,70 (d, J=3,3 Hz, 1h ), 7,56 (m, 1H), 7,48 (m, 2H), 7,28 (m, 1H), 6,74 (d, J=3,3 Hz, 1H), 2,52 (s, 3H)
Etapa C: 4-metil-6-(trifluorometil)-1H-indol

A una solución de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol (3,1 g, 9,14 mmol) en THF (10 mL) se añadió fluoruro de tetrabutilamonio (10,96 mL, 10,96 mmol) y la mezcla de reacción se agitó a 60 °C durante 1,5 horas. Se añadió más fluoruro de tetrabutilamonio (0,6 eq) y la agitación continuó durante 3 horas. Se añadió más fluoruro de tetrabutilamonio (0,9 eq) y la agitación continuó durante 18 horas. Se añadió más fluoruro de tetrabutilamonio (0,9 eq) y la agitación continuó durante 3 horas. La mezcla de reacción se concentró a presión reducida. El residuo se recogió en diclorometano y se lavó con una solución saturada de NaHCO3. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar 4-metil-6-(trifluorometil)-1H-indol (1,18 g, 62%) como un aceite de color marrón pálido. LC/MS (Método i) Rt = 2,24 min.; MS m/z: 198 [M-H]- 1H NMR (CLORO-FORMO-d, 300MHz): 58,30 (br, 1H), 7,53 (s, 1H), 7,34 (t, J=2,9 Hz, 1H), 7,15 (s, 1H), 6,62 (td, J=2,9, 1,1 Hz, 1H), 2,60 (s, 3H)
Etapa D: 4-metil-6-(trifluorometil)-1H-indol-3-carbaldehído

Como solución de N, N-dimetilformamida (1,431 mL, 18,48 mmol) en DCM (15 mL) y enfriada a 0 °C, se añadió cloruro de oxalilo (1,617 mL, 18,48 mmol) diluido en DCM (15 mL) y la mezcla de reacción se agitó durante 30 minutos. Luego se añadió gota a gota a una solución de 4-metil-6-(trifluorometil)-1H-indol (920 mg, 4,62 mmol) en DCM (15 mL) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Se añadió una solución de NaOH 5N hasta pH 10. La capa acuosa se extrajo con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 4-metil-6-(trifluorometil)-1H-indol-3-carbaldehído (1 g, 78%) como un sólido de color marrón. El compuesto se usa crudo en la siguiente etapa.
LC/MS (Método i) Rt = 1,96 min.; MS m/z: 228 [M+H]+
Etapa E: 1,4-dimetil-6-(trifluorometil)-1H-indol-3-carbaldehído

A una solución de 4-metil-6-(trifluorometil)-1H-indol-3-carbaldehído (1,09 g, 4,80 mmol) en DMF, (60 mL) se añadió hidruro de sodio (0,192 g, 4,80 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 30 minutos. Se añadió yodometano (0,300 mL, 4,80 mmol) y la mezcla de reacción se agitó una hora a temperatura ambiente. La mezcla de reacción se diluyó con agua. El precipitado obtenido se filtró, se lavó con agua y se concentró hasta la sequedad para dar 1,4-dimetil-6-(trifluorometil)-1H-indol-3-carbaldehído (815 mg, 70%) como un sólido rosa. LC/MS (Método i) Rt = 2,09 min.;
MS m/z: 242 [M+H]+ 1H NMR (DMSO-da, 300MHz): 59,98 (s, 1H), 8,46 (s, 1H), 7,83 (s, 1H), 7,35 (s, 1H), 3,96 (s, 3H), 2,84 (s, 3H)
Preparación #52: 1-metil-6-(trifluorometoxi)-1H-indol-2-carbaldehído

Etapa A: metil (Z)-2-azido-3-(4-(trifluorometoxi)fenil) acrilato

En un matraz de fondo redondo equipado con agitación mecánica, se añadió metóxido de sodio al 25% en MeOH (107 mL, 480 mmol) a MeOH (MeOH) (80 mL) para dar una solución incolora y se enfrió a aprox. -20 °C. Se añadió gota a gota una mezcla de 4-(trifluorometoxi)benzaldehído (38,8 g, 204 mmol) y metil-2-azidoacetato (55,22 g, 480 mmol) durante 15 minutos, manteniendo la temperatura a aprox. -20 °C. Después de completar la adición, la reacción se convirtió en un aceite de color amarillo espeso. Calentado a 0-5 °C. Después de 1 h, la mezcla se vertió en una mezcla acuosa de HCl 2M (400 mL)/MTBE (400 mL). Las capas se separaron y se extrajo la fase acuosa con MTBE (2 x 200 mL). Los compuestos orgánicos se lavaron sobre salmuera, se secaron sobre sulfato de sodio y se eliminó el disolvente al vacío. Dio 80,43 g de material crudo como un aceite de naranja. El material crudo se cromatografió en el sistema Intelliflash 280 usando una columna de gel de sílice Silicycle SiliaSep de 330 g usando el siguiente gradiente: A: heptano; B: 10% de EtOAc en heptano; 0% B a 100% B durante 30 min. Las fracciones combinadas del producto se concentraron al vacío para dar 37,85 g (65% de rendimiento) de metil (Z)-2-azido-3-(4-(trifluorometoxi)fenil)acrilato. 1H NMR (400 MHz, DMSO-d6) 58,00 (d, J = 9,0 Hz, 1H), 7,40 (d, J = 8,0 Hz, 1H), 6,97 (s, 1H), 3,87 (s, 3H).
Etapa B: metil 6-(trifluorometoxi)-1H-indol-2-carboxilato

En un matraz de fondo redondo, Xileno (500 mL) que se había secado durante el fin de semana sobre tamices moleculares, y luego purgado con nitrógeno durante 1 hora, se calentó a 140 °C. Una solución de (Z)-metil 2-azido-3-(4-(trifluorometoxi)fenil)acrilato (37,85 g, 132 mmol) en xileno (75 mL) se añadió gota a gota a la solución durante 45 minutos. La LC/MS del material después de completar la adición muestra el consumo completo del material de partida. Se dejó enfriar a temperatura ambiente. Se eliminó el disolvente al vacío, se trituró con heptanos (100 mL) durante el fin de semana. Se filtró, lavado con heptano. Se secó al vacío. Dio 25,08 g (73% de rendimiento) de metil 6-(trifluorometoxi)-1H-indol-2-carboxilato como un sólido cristalino. 1H NMR (400 MHz, DMSO-d6) 512,15 (s, 1H), 7,78 (d, J = 8,8 Hz, 1H), 7,42 7,32 (m, 1H), 7,23 (dd, J = 2,2, 0,9 Hz, 1H), 7,07 (ddt, J = 8,7, 2,1, 1,0 Hz, 1H), 3,89 (s, 3H); LC/MS (Método i) R, = 1,59 min.; MS m/z: 258,17 (M-H)-.
Etapa C: N-metoxi-N-metil-6-(trifluorometoxi)-1H-indol-2-carboxamida

En un matraz de fondo redondo, se añadió metil 6-(trifluorometoxi)-1H-indol-2-carboxilato (25,08 g, 97 mmol) en THF (150 mL) para dar una solución amarilla. Se añadió clorhidrato de N, O-dimetilhidroxilamina (10,38 g, 106 mmol) y la mezcla se enfrió a 0 °C. Se añadió gota a gota bis (trimetilsilil) amida de litio 1,0 M en THF (300 mL, 300 mmol), manteniendo la temperatura < 5 °C. La LC/MS 10 minutos después de completar la adición se indica casi completa. Se añadió HCl acuoso 2 N (275 mL), manteniendo la temperatura < 25 °C. Se extrajo la capa acuosa con MTBE (275 mL) y se lavó con salmuera orgánica (75 mL). Se secó sobre sulfato de magnesio y se eliminó el disolvente al vacío. Los residuos se suspendieron en heptano (100 mL) durante 20 minutos, luego se filtraron, lavando con heptano mínimo. El sólido de color amarillo se secó al vacío para dar 25,57 g (92% de rendimiento) de N-metoxi-N-metil-6-(trifluorometoxi)-1H-indol-2-carboxamida que se llevó directamente a la siguiente etapa sin purificación adicional. 1H NMR (400 MHz, DMSO-d6) 5 11,77 (s, 1H), 7,78 (d, J = 8,7 Hz, 1H), 7,42-7,38 (m, 1H), 7,22 (dd, J = 2,2, 0,9 Hz, 1H), 7,03 (ddt, J = 8,8, 2,1, 1,0 Hz, 1H), 3,81 (s, 3h ), 3,34 (s, 3H); LC/MS (Método i) R, = 1,48 min.; MS m/z: 289,26 (M+H)+; MS m/z: 287,17 (M-h )-.
Etapa D: N-metoxi-N, 1-dimetil-6-(trifluorometoxi)-1H-indol-2-carboxamida

En un matraz de fondo redondo, se añadió N-metoxi-N-metil-6-(trifluorometoxi)-1H-indol-2-carboxamida (25,57 g, 89 mmol) en dimetilformamida (DMF) (250 mL) para dar una solución amarilla. Se enfrió a 0 °C y se añadió terc-butóxido de sodio (8,95 g, 93 mmol), manteniendo la temperatura < 5 °C. La solución se agitó 30 minutos a 0 °C, luego se añadió gota a gota una solución de yodometano 2,0 M en MTBE (46,6 mL, 93 mmol), manteniendo la temperatura < 5 °C. Se agitó a ~ 0 °C. Después de 2 horas, se consumió casi todo el material de partida. Se añadieron MTBE (600 mL) y ácido clorhídrico 2 N (250 mL). Las capas se separaron y la capa acuosa se extrajo con MTBE (200 mL) y luego las capas orgánicas combinadas se lavaron con salmuera (2 x 150 mL). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio y el disolvente se eliminó al vacío. El material crudo se cromatografió en el sistema Intelliflash 280 usando una columna de gel de sílice Silicycle SiliaSep de 220 g usando el siguiente gradiente: A: heptano; B: EtOAc; 20% B a 100% B durante 30 min. para dar 21,67 g (80% de rendimiento) de N-metoxi-N, 1-dimetil-6-(trifluorometoxi)-1H-indol-2-carboxamida. 1H NMR (400 MHz, DMSO-d6) 57,75 (dd, J = 8,6, 0,5 Hz, 1H), 7,61 (tt, J = 1,4, 0,8 Hz, 1H), 7,08 (ddq, J = 8,6, 2,1, 1,0 Hz, 1H), 7,05 (d, J = 0,9 Hz, 1H), 3,85 (s, 3H), 3,65 (s, 3H), 3,33 (s, 3h ); LC/MS (Método i) R, = 1,57 min.; MS m/z: 303,14(M+H)+.
Etapa E: 1-metil-6-(trifluorometoxi)-1H-indol-2-carbaldehído

En un matraz de fondo redondo, se añadió N-metoxi-N,1-dimetil-6-(trifluorometoxi)-1H-indol-2-carboxamida (21,67 g, 71,7 mmol) en THF (200 mL) para dar una solución amarilla. La solución se enfrió a -78 °C y se añadió gota a gota hidruro de litio y aluminio 2,0 M en THF (50,2 mL, 100 mmol), manteniendo la temperatura <-65 °C. Después de completar la adición, LC/Ms indicó el consumo completo de material de partida. Se añadió sulfato de sodio decahidratado (23,10 g, 71,7 mmol) en porciones, manteniendo la temperatura <-60 °C seguido de MTBE (300 mL). La solución se calentó a temperatura ambiente. A aproximadamente -10 °C se formó un precipitado. Se añadió MTBE adicional hasta que se agitó libremente. Se filtró a través de una almohadilla de Celite® 545, lavando con MTBE (2 x 120 mL). El disolvente se eliminó al vacío, lavando con heptano (2 x 120 mL), dando 14,29 g de una mezcla de aceite amarillo. El material crudo se cromatografió en el sistema Intelliflash 280 usando una columna de gel de sílice Grace de 220 g usando el siguiente gradiente: A:
heptano; B: EtOAc; 0% B a 60% B durante 30 minutos, dando 11,21 g (64% de rendimiento) de 1-metil-6-(trifluorometoxi)-1H-indol-2-carbaldehído. 1H NMR (400 MHz, DMSO-d6) 59,93 (s, 1H), 7,89 (dd, J = 8,7, 0,5 Hz, 1H), 7,68 (dp, J = 2,5, 0,9 Hz, 1H), 7,51 (d, J = 0,9 Hz, 1H), 7,14 (ddq, J = 8,7, 2,1, 1,0 Hz, 1H), 4,04 (s, 3H). LC/MS (Método i) R, = 1,62 min.; MS m/z: 244,19 (M+H)+.
Preparación #53: metil 2-(1-(2,4-didoro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il)acetato

Etapa A: ferc-butil 2,4-didoro-3-(hidroximetil)benzoato

Usando un procedimiento similar a la Preparación #8, Etapa A, ferc-butil 2,4-dicloro-3-(hidroximetil)benzoato (11,8 g, 97%) se preparó a partir de ferc-butil 2,4-dicloro-3-formilbenzoato (13,5 g, 44 mmol) (Preparación #33, Etapa B). LC/MS (Método i): Rt = 2,02 min; MS m/z: 277 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,55 (m, 2H), 5,28 (s, 1H), 4,72 (s, 2H), 1,54 (s, 9H).
Etapa B: ferc-butil 2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoato

A una solución de ferc- butil 2,4-dicloro-3-(hidroximetil)benzoato (11,9 g, 42,9 mmol) en DCM (472 mL) y enfriada a -10 °C se añadió trietilamina (8,98 mL, 64,4 mmol) y cloruro de metanosulfonilo (5,33 mL, 68,7 mmol). La mezcla de reacción se agitó a -10 °C durante 1 hora, luego se diluyó con DCM y se lavó con agua. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío para dar ferc -butil 2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoato (15,04 g, 99%) como un aceite amarillo. LC/MS (Método i): Rt = 2,25 min; MS m/z: 355 [M+H]+; 1H NMR (DMSO-de, 300MHz): 5 7,76 (d, J=8,9 Hz, 1H), 7,68 (d, J=8,9 Hz, 1H), 5,48 (s, 2H) 3,32 (s, 3H), 1,55 (s, 9H).
Etapa C: metil 2-(1-(2,4-didoro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il)acetato

A una solución de ácido 2,4-dicloro-3-(((metilsulfonil)oxi)metil) benzoico (7,56 g, 25,3 mmol) en DCM (145 mL) y enfriada a 0 °C se añadieron 1-(3-dimetilaminopropilo)-3-etilcarbodiimida hidrocloruro (4,84 g, 25,3 mmol) y 1-hidroxibenzotriazol (3,42 g, 25,3 mmol). La mezcla de reacción se agitó durante 10 minutos a 0 °C y luego se añadieron clorhidrato de metil 2-(piperidin-4-il)acetato (4,89 g, 25,3 mmol) y trietilamina (3,52 mL, 25,3 mmol). La mezcla de reacción se agitó a 0 °C durante 50 minutos y se diluyó con agua y DCM. Las capas se separaron y la capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-5% en ciclohexano) para dar metil 2-(1-(2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il) acetato (6,74 g, 61%). LC/mS (Método i): Rt = 1,80 min; MS m/z: 438 [M+H]+;
1H NMR (DMSO-Ce, 300MHz): 5 7,66 y 7,64 (d, J=8,3 Hz, 1H), 7,53 y 7,45 (m, 1H), 5,46 y 5,45 (s, 2H), 4,47 (m, 1H), 3,58 (s, 3H), 3,33 (ancho, 3H), 3,20 (m, 1H), 3,03 (m, 1 h ), 2,81 (m, 1H), 2,27 (m, 2H), 1,96 (m, 1H), 1,75 (m, 1H), 1,58 (m, 1H), 1,16 (m, 2H).
Preparación #54: metil 3-(clorometil)-2,4-dimetilbenzoato

Etapa A: metil 3-formil-2,4-dimetilbenzoato

Usando un procedimiento similar a la Preparación #1, Etapa A, se preparó metil 3-formil-2,4-dimetilbenzoato (7 g, 93%) a partir de ácido 3-formil-2,4-dimetilbenzoico (7 g, 39,3 mmol) (Preparación #4, Etapa B). LC/MS (Método h): Rt = 2,15 min; 1 H NMR (DMSO-Ce, 300 MHz): 5 10,55 (s, 1H), 7,78 (d, J=7,5 Hz, 1H), 7,27 (d, J=7,5 Hz, 1H), 3,83 (s, 3H), 2,51 (s, 3H), 2,46 (s, 3H).
Etapa B: metil 3-(hidroximetil)-2,4-dimetilbenzoato

Usando un procedimiento similar a la Preparación #8, Etapa A, se preparó metil 3-(hidroximetil)-2,4-dimetilbenzoato (6,08 g, 86%) a partir de metil 3-formil-2,4-dimetilbenzoato (7 g, 36,4 mmol). LC/MS (Método h): Rt = 1,65 min; MS m/z: 195 [M+H]+; 1H NMR (DMSO-Ce, 300 MHz): 5 7,52 (d, J=7,5 Hz, 1H), 7,09 (d, J=7,5 Hz, 1H), 4,83 (t, J=5,4 Hz, 1H), 4,53 (d, J=5,4 Hz, 2H), 3,80 (s, 3H), 2,49 (s, 3H), 2,39 (s, 3H).
Etapa C: metil 3-(clorometil)-2,4-dimetilbenzoato

Usando un procedimiento similar a la Preparación #11, Etapa C, se preparó metil 3-(clorometil)-2,4-dimetilbenzoato (2,6 g, 88%) a partir de metil 3-(hidroximetil)-2,4-dimetilbenzoato (2,5 g, 12,87 mmol). lC/MS (Método h): Rt = 2,66 min; MS m/z: 213 [M+H]+; 1H NMR (DMSO-Ce, 300 MHz): 5 7,60 (d, J=7,9 Hz, 1H), 7,19 (d, J=7,9 Hz, 1H), 4,84 (s, 2H), 3,82 (s, 3H), 2,52 (s, 3H), 2,43 (s, 3H).
Preparación #55: terc-butil 2,4-dicloro-3-(fluorocarbonil)benzoato

A una solución de ácido 3-(íerc-butoxicarbonil)-2,6-diclorobenzoico (0,95 g, 3,26 mmol) (Preparación #44, Etapa A) en DCM (32,6 mL) se añadieron diisopropiletilamina (2,26 mL, 13,05 mmol) seguido por trifluoruro de bis(2-metoxietil)aminoazufre (2,65 mL, 7,18 mmol) gota a gota. La reacción es ligeramente exotérmica y se agitó a temperatura ambiente durante 22 horas. La mezcla de reacción se lavó con ácido cítrico acuoso al 10%, una solución saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre sulfato de magnesio y se concentró a presión reducida para dar íerc-butil 2,4-dicloro-3-(fluorocarbonil)benzoato (1 g, 100%) como un aceite naranja. LC/MS (Método j): Rt = 1,98 min; MS miz: 291 [MH]-; H NMR (CDCla, 300MHz): 57,83 (d, J=8,4 Hz, 1H), 7,44 (d, J=8,4 Hz, 1H), 1,60 (s, 9H).
Preparación #56: íerc-butil 4,6-didoro-5-(dorocarbom l)m cotinato
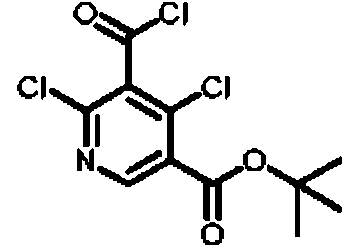
Etapa A: ácido 5-(íerc-butoxicarbonil)-2,4-didoromcotmico

Usando un procedimiento similar a la Preparación #44, Etapa A, se preparó ácido 5-(íerc-butoxicarbonil)-2,4-dicloronicotínico (1,96 g, 43%) a partir de íerc-butil 4,6-dicloronicotinato (3,85 g, 15,5 mmol) ((Ejemplo DO, Etapa 11). LC/MS (Método i): Rt = 1,31 min; MS miz: 292 [M+H]+1H NMR (DMSO-cfe, 300MHz): 58,78 (s, 1H), 1,56 (s, 9H).
Etapa 2: íerc-butil 4,6-dicloro-5-(clorocarbonil)nicotinato
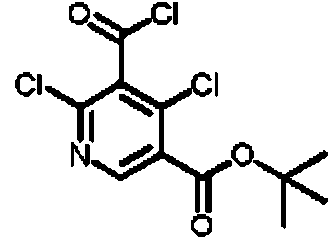
Usando un procedimiento similar a la Preparación #44, Etapa B, se preparó íerc-butil 4,6-dicloro-5-(clorocarbonil)nicotinato (2 g, 100%) a partir de ácido 5-(íerc-butoxicarbonil)-2,4-dicloronicotínico (1,96 g, 6,71 mmol). El residuo se usó en la siguiente etapa sin purificación adicional.
Preparación #57:. íerc-butil 4,6-dicloro-5-(((metilsulfonil)oxi)metil)nicotinato

Etapa A: íerc-butil 4,6-dicloro-5-(hidroximetil)nicotinato

Usando un procedimiento similar a la Preparación #8, Etapa A, íerc-butil 4,6-dicloro-5-(hidroximetil)nicotinato (503 mg, 97%) se preparó a partir de íerc-butil 4,6-dicloro-5-formilnicotinato (500 mg, 1,8 mmol) (Ejemplo DO, Etapa 12). LC/MS
(Método i): Rt = 1,83 min; MS m/z: 278 [M+H]+1H NMR (DMSO-de, 300MHz): 5 8,63 (s, 1H), 5,45 (t, J=5,4 Hz, 1H), 4,72 (d, J=5,4 Hz, 2H), 1,56 (s, 9H).
Etapa B: terc-butil 4,6-didoro-5-(((metilsulfonil)oxi)metil)nicotinato
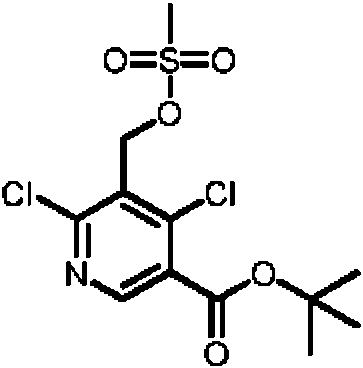
Usando un procedimiento similar a la Preparación #11, Etapa C, íerc-butil 4,6-didoro-5-(((metilsulfonil)oxi)metil)nicotinato y íerc-butil 4,6-dicloro-5-(clorometil)nicotinato (160 mg) se prepararon a partir de íerc-butil 4,6-dicloro-5-(hidroximetil)nicotinato (130 mg, 0,47 mmol). El residuo se usó crudo en la siguiente etapa. LC/MS (Método j): Rt = 1,37 min; MS m/z: 356 [M+H]+; 1H NMR (CDCla, 300MHz): 58,73 (s, 1H), 5,55 (s, 2H), 3,67 (s, 3H), 161 (s, 9H).
Preparación #58: íerc-butil 2-doro-4-cidopropil-3-(1-hidroxiprop-2-in-1-il)benzoato

Etapa A: íerc-butil 4-bromo-2-cloro-3-formilbenzoato

Usando un procedimiento similar a la Preparación #33, Etapa B, íerc-butil 4-bromo-2-cloro-3-formilbenzoato (2,66 g, 32%) se preparó a partir de íerc-butil 4-bromo-2-clorobenzoato (5 g, 17,15 mmol). LC/MS (Método i): Rt = 2,38 min; MS m/z: 319 [M+H]+; 1 H NMR (DMSO-de, 300MHz): 5 10,23 (s, 1H), 7,85 (d, J=7,8 Hz, 1H), 7,75 (d, J=7,8 Hz, 1H), 1,55 (s, 9H).
Etapa B: íerc-butil 2-cloro-4-ciclopropil-3-formilbenzoato

Usando un procedimiento similar a la Preparación #66, íerc-butil 2-cloro-4-ciclopropil-3-formilbenzoato (1,18 g, 88%) se preparó a partir de íerc-butil 4-ciclopropil-2-clorobenzoato (2 g, 4,17 mmol). LC/MS (Método i): Rt = 2,49 min; MS m/z: 281 [M+H]+; 1H NMR (DMSO-de, 300MHz): 5 10,56 (s, 1H), 7,71 (d, J=8,3 Hz, 1H), 7,12 (d, J=8,3 Hz, 1H), 2,45 (m, 1H), 1,53 (m, 9h ), 1,03 (m, 2H), 0,77 (m, 2H).
Etapa C: íerc-butil 2-doro-4-ddopropil-3-(1-hidroxiprop-2-m-1-N)benzoato

Usando un procedimiento similar a la Preparación #1, Etapa D, se preparó íerc-butil 2-cloro-4-ciclopropil-3-(1-hidroxiprop-2-in-1-il)benzoato (1,11 g, 86%) a partir de íerc-butil-2-cloro-4-ciclopropil-3-formilbenzoato (1,18 g, 4,2 mmol).
LC/MS (Método j) : Rt = 1,28 min; MS m/z: 365 [M-H]-+ CH3 COOH; 1 H NMR (DMSO-Ce, 300MHz): 5 7,41 (d, J=8,1 Hz, 1H), 6,91 (d, J=8,1 Hz, 1H), 6,26 (m, 1H), 6,19 (s, 1H), 3,49 (d, J=2,3 Hz, 1H), 2,73 (m, 1H), 1,53 (s, 9H), 1,03 (m, 2H), 0,83 (m, 1H), 0,70 (m, 1H).
Preparación #59: (E)-íerc-butil 2,4-didoro-3-(3-((metilsulfonil)oxi)prop-1-en-1-il)benzoato

Etapa A: (E)-íerc-butil 2,4-didoro-3-(3-etoxi-3-oxoprop-1-en-1-il)benzoato

A una suspensión de hidruro de sodio (0,407 g, 10,18 mmol) en THF (6,2 mL) y enfriada a 0 °C se añadió gota a gota fosfonacetato de trietilo (2,03 mL, 10,18 mmol) y la mezcla de reacción se dejó calentar a temperatura ambiente y se agitó hasta que fue homogénea. La mezcla de reacción se enfrió a 0 °C y una solución de íerc-butil 2,4-dicloro-3-formilbenzoato (2,8 g, 10,18 mmol) (Preparación #33, Etapa B) en THF (16 mL) se añadió gota a gota a la mezcla de reacción, apareció una goma marrón y fue difícil de agitar. La mezcla de reacción se dejó calentar a temperatura ambiente y la mezcla de reacción se inactivó gota a gota con una solución saturada de NaHCO3. La capa acuosa obtenida se extrajo tres veces con EtOAc y las capas orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en DCM) para dar (E)-íerc- butil 2,4-dicloro-3-(3-etoxi-3-oxoprop-1-en-1-il)benzoato (2,6 g, 77%) como un aceite amarillo. LC/MS (Método i): Rt = 2,68 min; MS m/z: 345 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,66 (s, 2H), 7,62 (d, J=16,3 Hz, 1H), 6,48 (d, J=16,3 Hz, 1H), 4,24 (q, J=7,1 Hz, 2H), 1,55 (s, 9H), 1,27 (t, J=7,1 Hz, 3H).
Etapa B: (E)-ferc-butil 2,4-dicloro-3-(3-hidroxiprop-1-en-1-il)benzoato

A una solución de (E)-íerc-butil 2,4-dicloro-3-(3-etoxi-3-oxoprop-1-en-1-il)benzoato (2,59 g, 7,50 mmol) en THF (68 mL) y enfriada a -78 °C se añadió gota a gota hidruro de diisobutilaluminio (24,38 mL, 29,3 mmol) y la mezcla de reacción se agitó durante 30 minutos a -78 °C. Se añadió EtOAc lentamente, seguido de 68 mL de una solución al 30% de tartrato de sodio y potasio. La mezcla se agitó hasta que hubo dos capas. La mezcla se extrajo dos veces con EtOAc y la capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 7-60% en DCM) para dar (E)-íerc-butil 2,4
didoro-3-(3-hidroxiprop-1-en-1-ilo)benzoato (2,19 g, 96%). LC/MS (Método i): Rt = 2,19 min; MS miz: 303 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 5 = 7,56 (d, J=7,9 Hz, 1H), 7,51 (d, J=7,9 Hz, 1H), 6,53 (dd, J=15, 3,3 Hz, 1H), 6,26 (dd, J=15, 4,3 Hz, 1H), 5,08 (t, J=5,6Hz, 1H), 4,19 (m, 2H), 1,54 (s, 9H).
Etapa C: (E)-íerc-butM 2,4-didoro-3-(3-((metilsulfonil)oxi)prop-1-en-1-il)benzoato

Usando un procedimiento similar a la Preparación 53, Etapa B, (E)-ferc-butil 2,4-dicloro-3-(3-((metilsulfonil)oxi)prop-1-en-1-il)benzoato (2,95 g, 86%) se preparó a partir de (E)-íerc-butil 2,4-didoro-3-(3-hidroxiprop-1-en-1-il)benzoato (2,18 g, 7,19 mmol). LC/MS (Método i): Rt = 2,39 min; MS miz: 381 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 57,60 (m, 2H), 6,77 (d, J=15 Hz, 1H), 6,26 (dt, J=15, 5,9 Hz, 1H), 4,98 (dd, J=5,9, 1,6 Hz, 2H), 3,26 (s, 3H), 1,54 (s, 9H).
Preparación #60: íerc-butil 4,6-didoro-5-(1-hidroxiprop-2-m-1-il)mcotmato

Usando un procedimiento similar a la Preparación #1, Etapa D, íerc-butil 4,6-dicloro-5-(1-hidroxiprop-2-in-1-il)nicotinato (5,8 g, 79%) se preparó a partir de íerc-butil 4,6-dicloro-5-formilnicotinato (6,75 g, 24,45 mmol) (Ejemplo DO, Etapa 12). LC/MS (Método i): Rt = 2,00 min; MS miz: 302 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 58,65 (s, 1H), 6,43 (d, J=4,8 Hz, 1H), 6,10 (dd, J=4,8, 2,5 Hz, 1H), 3,60 (d, J=2,5 Hz, 1H), 1,56 (s, 9H).
Preparación #61: 3,7-dimetil-5-(trifluorometoxi)-'ÍH-mdol

Etapa A: 2-yodo-6-metil-4-(trifluorometoxi)anilina

Usando un procedimiento similar a la Preparación #34, Etapa A, se preparó 2-yodo-6-metil-4-(trifluorometoxi)anilina (5,25 g, 79%) a partir de 2-metil-4-(trifluorometoxi)anilina (4 g, 20,9 mmol). LC/MS (Método i): Rt = 2,32 min; MS miz: 318 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,43 (s, 1H), 7,05 (s, 1H), 5,07 (s, 2H), 2,17 (s, 3H).
Etapa B: 3,7-dimetil-5-(trifluorometoxi)-1H-mdol

A una solución de 2-yodo-6-metil-4-(trifluorometoxi)anilina (500 mg, 1,57 mmol) en W,W-dimetilformamida (se añadieron 3 ml de acetato de paladio(II) (35,4 mg, 0,16 mmol), cloruro de litio (66,9 mg, 1,58 mmol), trifenilfosfina (41,4 mg, 0,16 mmol), acetato de sodio (259 mg, 3,15 mmol) y 1 -(trimetilsilil)-1 -propino (0,470 mL, 3,15 mmol) y la mezcla de reacción se calentó a 140 °C durante 1 hora bajo irradiación de microondas. La mezcla de reacción se lavó con agua y se extrajo dos veces con EtOAc. La capa orgánica se lavó sucesivamente con salmuera y agua, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se disolvió en THF (8 mL) y se añadió fluoruro de tetrabutilamonio (0,718 mL, 2,481 mmol). La mezcla de reacción se agitó a reflujo durante 4 horas y luego se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-60% en ciclohexano) para dar 3,7-dimetil-5-(trifluorometoxi)- 1H-indol (285 mg, 79%) como un líquido de color marrón. LC/MS (Método i): Rt = 2,34 min; MS miz: 230 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 511,00 (s, 1H), 7,23 (s, 2H), 6,85 (s, 1H), 2,46 (s, 3H), 2,24 (s, 3H).
Preparación #62: 7-cloro-5-(trifluorometil)-1H-indol

A una solución de 2-cloro-6-yodo-4-(trifluorometil)anilina (3,2 g, 9,95 mmol) en DMF (64 mL) se añadió trimetilsililacetileno (1,075 g, 10,95 mmol), acetato de paladio(II) (0,335 g, 1,493 mmol) y carbonato de sodio (3,17 g, 29,9 mmol) y la mezcla de reacción se agitó a 120 °C en microondas durante 20 minutos. El residuo se suspendió en salmuera y se extrajo dos veces con DMC. La capa orgánica se secó luego sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-5% en ciclohexano) para dar 7-cloro-5-(trifluorometil)-2-(trimetilsilil)-1H-indol (1 g, 20%). El residuo se disolvió en THF (11 mL), se añadió fluoruro de tetrabutilamonio (3,65 mL, 3,65 mmol) y la mezcla de reacción se agitó a 50 °C durante 30 minutos. La mezcla de reacción se concentró a presión reducida. El residuo se recogió en DCM y se lavó con una solución acuosa de NaHCO3. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-10% en ciclohexano) para dar 7-cloro-5-(trifluorometil)-1 H-indol (470 mg, 46%). LC/MS (Método i): Rt = 2,23 min; MS m/z: 218 [M-H]-; 1H NMR (CDCla, 300MHz): 5 8,54 (ancho, 1H), 7,86 (s, 1H), 7,45 (m, 1H), 7,36 (m, 1H), 6,70 (m, 1H).
Preparación #63: 7-etM-5-(tnfluorometil)-1 H-indol

Etapa A: 5-(trifluorometN)-7-vimM H-indol
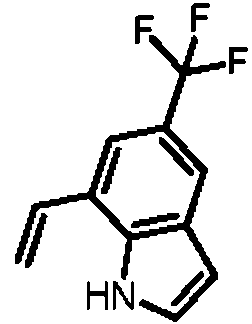
A una solución de 7-cloro-5-(trifluorometil)-1 H-indol (115 mg, 0,52 mmol) (Preparación #62) en 1,4-dioxano (3,4 mL) se añadió fluoruro de cesio (159 mg, 1,05 mmol) y bis(tri-íerc-butilfosfina)paladio(0) (53,5 mg, 0,10 mmol) y tributil(vinil)estaño (830 mg, 2,62 mmol) y la mezcla de reacción se agitó a 120 °C durante 45 minutos. La mezcla de reacción se dejó enfriar a temperatura ambiente y el disolvente se eliminó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-5% en ciclohexano) para dar 5-(trifluorometil)-7-vinil-1 H-indol (110 mg, 81%) como un aceite de color marrón. LC/MS (Método i): Rt = 2,23 min; MS m/z: 212 [M+H]+; 1H NMR (CDCl3, 300MHz): 58,56
(ancho, 1H), 7,86 (s, 1H), 7,46 (s, 1H), 7,34 (s, 1H), 7,00 (dd, J=17,7, 11,2 Hz, 1H), 6,67 (m, 1H), 5,87 (d, J=17,7 Hz, 1H), 5,55 (d, J=11,2 Hz, 1H).
Etapa B: 7-etil-5-(trifluorometil)-1H-indol

A una solución de 5-(trifluorometil)-7-vinil-1 H- indol (110 mg, 0,52 mmol) en EtOH (2 mL) bajo argón, se añadió paladio sobre carbono (11,09 mg, 10,42 mmol). La mezcla de reacción se agitó a temperatura ambiente bajo presión atmosférica de hidrógeno durante 5 horas. La mezcla de reacción se filtró y se lavó con EtOH. El disolvente se eliminó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-1% en ciclohexano) para dar 7-etil-5-(trifluorometil)-1H-indol (30 mg, 18%) como una resina de color beige. LC/MS (Método i): Rt = 2,22 min; MS m/z: 214 [M+H]+
Preparación #64: 7-metil-5-(trifluorometoxi)-1H-indol

Usando un procedimiento similar a la Preparación #34, Etapa A, se preparó 2-yodo-6-metil-4-(trifluorometoxi)anilina (6,8 g, 82%) a partir de 2-metil-4-(trifluorometoxi)anilina (5 g, 26,2 mmol). Lc/MS (Método i): Rt = 2,33 min; MS m/z: 318 [M+H]+; 1H NMR (DMSO-cfe 300MHz): 57,43 (s, 1H), 7,05 (s, 1H), 5,07 (s, 2H), 2,17 (s, 3H).
Etapa B: 7-metil-5-(trifluorometoxi)-1H-indol

A una solución de 2-yodo-6-metil-4-(trifluorometoxi)anilina (3,3 g, 10,41 mmol) en W,W-dimetilformamida, (24 mL) se añadieron Pd(PPh3)2Cl2 (0,14 g, 0,21 mmol), yoduro de cobre(I) (0,099 g, 0,520 mmol), etiniltrimetilsilano (1,584 mL, 11,45 mmol) y dietilamina (2,392 mL, 22,90 mmol). La mezcla de reacción se agitó a 120 °C bajo irradiación de microondas durante 40 minutos. La mezcla de reacción se filtró y se lavó con EtOAc. La mezcla se diluyó con agua y se extrajo dos veces con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio y se concentró. El residuo se disolvió en DMF (30 mL) y se añadieron carbonato de calcio (1,045 g, 10,44 mmol) y yoduro de cobre(I) (1,988 g, 10,44 mmol), y la mezcla de reacción se agitó durante 20 minutos a 150 °C bajo irradiación de microondas. La mezcla de
reacción se diluyó con agua. La capa acuosa se extrajo dos veces con EtOAc, y las capas orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se evaporaron. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-20% en ciclohexano) para dar 7-metil-5-(trifluorometoxi)-1H-indol (1 g, 49%) como un sólido amarillo. LC/MS (Método j) : Rt = 1,49 min; MS m/z: 216 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 11,34 (s, 1H), 7,46 (t, J=2,9 Hz, 1H), 7,34 (s, 1H), 6,87 (m, 1H), 6,50 (m, 1H), 2,50 (s, 3H).
Preparación #65: 5-(difluorometoxi)-7-metil-1 H - indol

Etapa A: 5-bromo-7-metil-1-(fenilsulfonil)-1 H - indol

Usando un procedimiento similar a la Preparación #48, 5-bromo-7-metil-1-(fenilsulfonil)-1H-indol (1,6 g, 63%) se preparó a partir de 5-bromo-7-metil-1H-indol (1 g, 4,76 mmol). LC/MS (Método i): Rt = 2,58 min; MS m/z: 350 [M+H]+; 1H NMR (CDCls, 300MHz): 5 = 7,79 (d, J=3,8 Hz, 1h ), 7,40-7,69 (m, 6H), 7,14 (m, 1h ), 6,64 (d, J=3,8 Hz, 1H), 2,50 (s, 3H).
Etapa B: 7-metil-1-(fenilsulfonil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-indol

A una solución de 5-bromo-7-metil-1-(fenilsulfonil)-1H-indol (1,6 g, 4,57 mmol) en THF (55 mL) se añadió bis(pinacolato)diboro (2,32 g, 9,14 mmol), [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (0,83 g, 1,14 mmol) y acetato de potasio (1,12 g, 11,42 mmol). El tubo se selló y se calentó a 80 °C durante la noche. La mezcla de reacción se dejó enfriar a temperatura ambiente y se inactivó con agua. La capa acuosa se extrajo dos veces con DCM. La capa orgánica se lavó con agua y salmuera, se secó sobre sulfato de magnesio y se concentró hasta sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar 7-metil-1-(fenilsulfonil)-5-(4,4,5,5-tetrametil-1,3, 2-dioxaborolan-2-il)-1H-indol (1,45 g, 74%) como una resina de color amarillo. LC/MS (Método i): Rt = 2,73 min; MS m/z: 398 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 = 7,88 (d, J=3,8 Hz, 1H), 7,81 (s, 1H), 7,70 (m, 3H), 7,61 (m, 2H), 7,34 (s, 1H), 6,94 (d, J=3,8 Hz, 1H), 2,47 (s, 3H), 1,28 (s, 12H).
Etapa C: 7-metil-1-(fenilsulfonil)-1H-indol-5-ol

A una solución de 7-metil-1-(fenilsulfonil)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-indol (1,6 g, 4,6 mmol) en THF (40 mL) y enfriada a 5 °C se añadieron peróxido de hidrógeno (0,59 g, 8,68 mmol) e hidróxido de sodio (0,27 g, 6,85 mmol) y la mezcla de reacción se agitó a 5 °C durante 7 horas. La mezcla de reacción se inactivó con agua y se extrajo dos veces con DCM. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró hasta la sequedad para dar 7-metil-1-(fenilsulfonil)-1H-indol-5-ol (1,1 g, 73%) como una resina marrón. LC/MS (Método i): Rt = 1,96 min; MS m/z: 288 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 = 9,27 (s, 1H), 7,68 (m, 4H), 7,57 (m, 2H), 6,72 (m, 2H), 6,52 (m, 1H), 2,42 (s, 3H).
Etapa D: 5-(difluorometoxi)-7-metiM-(femlsulfonM)-1H-mdol

A una solución de 7-metil-1-(fenilsulfonil)-1H-indol-5-ol (1 g, 3,48 mmol) en ACN (11 ml y agua (11 mL) y enfriada a 0 °C se añadió hidróxido de potasio (3,91 g, 69,6 mmol) y la mezcla de reacción se agitó a 0 °C durante 15 minutos. Se añadió dietil(bromodifluorometil)fosfonato (1,67 g, 6,26 mmol) y la mezcla de reacción se agitó a 0 °C durante 5 minutos antes de dejar que se calentara a temperatura ambiente. La agitación continuó durante 1,5 horas. La mezcla de reacción se inactivó con agua y se acidificó con HCl (1N) hasta pH = 1. Luego se extrajo dos veces con DCM. La capa orgánica se lavó con agua y salmuera, se secó sobre sulfato de magnesio y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-10% en ciclohexano) para dar 5-(difluorometoxi)-7-metil-1-(fenilsulfonil)-1H-indol (550 mg, 43%) como una resina de color amarillo. LC/MS (Método i): Rt = 2,42 min; MS m/z: 338 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,93 (d, J=3,8 Hz, 1H), 7,73 (m, 3H), 7,61 (m, 2H), 7,29 (d, J=2,3 Hz, 1H), 7,17 (t, J=74 Hz, 1H), 6,92 (d, J=2,3 Hz, 2H), 2,48 (s, 3H).
Etapa E: 5-(difluorometoxi)-7-metil-1 H- indol

Usando un procedimiento similar a la Preparación #51, Etapa C, se preparó 5-(difluorometoxi)-7-metil-1H-indol (280 mg, 73%) a partir de 5-(difluorometoxi)-7-metil-1-(fenilsulfonil)-1H-indol (550 mg, 1,63 mmol). LC/MS (Método i): Rt = 1,98 min; MS m/z: 198 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 511,19 (s, 1H), 7,39 (m, 1H), 7,15 (s, 1H), 7,05 (t, J=74 Hz, 1H), 6,74 (s, 1H), 6,43 (m, 1H), 2,47 (s, 3H).
Preparación #66: 5-ciclopropil-7-fluoro-1 H- indol (10467249-0995

5-bromo-7-fluoroindol (1 g, 4,67 mmol), ácido ciclopropilborónico (0,602 g, 7,01 mmol), [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (0,308 g, 0,420 mmol), y carbonato de cesio se mezclaron (4,57 g, 14,02 mmol) y se añadieron 1,4-dioxano (12,6 mL) y agua (1,3 mL). La mezcla de reacción se agitó a 110 °C bajo irradiación de microondas durante 30 minutos. Se añadieron ácido ciclopropilborónico (0,201 g, 2,336 mmol) y [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio(II) (0,154 g, 0,210 mmol) y la mezcla de reacción se agitó a 110 °C bajo irradiación de microondas durante 30 minutos. La mezcla de reacción se diluyó con agua. La capa acuosa se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar 5-ciclopropil-7-fluoro-1H-indol (1,19 g, 73%) como un líquido amarillo. LC/MS (Método i): Rt = 2,12 min; MS m/z: 174
[M-H]-; 1H NMR (DMSO-ó6, 300MHz): 511,39 (ancho, 1H), 7,33 (d, J=3,0 Hz, 1H), 7,10 (s, 1H), 6,63 (d, J=12,9, 1H), 6,39 (t, J=3,0 Hz, 1H), 1,97 (m, 1H), 0,89 (m, 2H), 0,63 (m, 2H).
Preparación #67: 7-fluoro-5-(trifluorometil)-1H-mdol

Usando un procedimiento similar a la Preparación 64, Etapa B, se preparó 7-fluoro-5-(trifluorometil)-1H-indol (230 mg, 22%) a partir de 2-fluoro-6-yodo-4-(trifluorometil)anilina (1 g, 3,61 mmol). LC/MS (Método i): Rt = 2,12 min; MS m/z: 202 [M-H]-; 1H NMR (DMSO-ó6, 300MHz): 512,10 (ancho, 1H), 7,83 (s, 1H), 7,61 (d, J=3,1 Hz, 1H), 7,27 (d, J=11,5, 1H), 6,71 (t, J=3,1 Hz, 1H).
Preparación #68: 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridma

Etapa A: 4-yodo-2-metil-6-(trifluorometil)piridin-3-amina

Usando un procedimiento similar a la Preparación #34, Etapa A, se preparó 4-yodo-2-metil-6-(trifluorometil)piridin-3-amina (3,4 g, 37%) a partir de 2-metil-6-(trifluorometil)piridin-3-amina (4,9 g, 27,8 mmol). LC/MS (Método i): Rt = 1,92 min; MS m/z: 303 [M+H]+; 1H NMR (CDCh, 300MHz): 57,79 (s, 1H), 4,43 (ancho, 2H), 2,53 (s, 3H).
Etapa B: W-alil-4-yodo-2-metil-6-(trifluorometil)piridm-3-amma

Usando un procedimiento similar a la Preparación #43, Etapa A, se preparó W-alil-4-yodo-2-metil-6-(trifluorometil)piridin-3-amina (3,7 g, 87%) a partir de 4-yodo-2- metil-6-(trifluorometil)piridin-3-amina (3,4 g, 11,26 mmol). Lc/MS (Método i): Rt = 2,35 min; MS m/z: 343 [M+H]+; 1H NMR (CDCh, 300MHz): 57,85 (s, 1H), 5,93 (m, 1H), 5,34 (d, J=17 Hz, 1H), 5,20 (d, J=9 Hz, 1H), 3,83 (ancho, 3H), 2,62 (s, 3H).
Etapa C: 3,7-dimetil-5-(trifluorometil)- 1H-pirrolo[2,3-c]piridina

Usando un procedimiento similar a la Preparación #43, Etapa B, 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina (2 g, 85%) se preparó a partir de W-alil-4-yodo-2-metil-6-(trifluorometil)piridin-3-amina (3,7 g, 10,82 mmol). LC/m S (Método i): Rt = 1,81 min; MS m/z: 215 [M+H]+; 1H NMR (DMSO-cfe 300MHz): 5 = 11,78 (ancho, 1H), 7,84 (s, 1H), 7,52 (s, 1H), 2,70 (s, 3H), 2,30 ppm (s, 3H)
Preparación #69: 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-b]pmdma

Etapa A: 2-bromo-4-metil-6-(trifluorometil)piridin-3-amina

A una solución de 4-metil-6-(trifluorometil)piridin-3-amina (3,4 g, 19,30 mmol) en dimetilsulfóxido (83 mL) y agua (4,5 mL) se añadió 1-bromopirrolidina-2,5-diona (8,59 g, 48,3 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 24 horas. La mezcla de reacción se inactivó con agua, y el sólido formado se filtró. El filtrado se extrajo dos veces con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. Los dos residuos se purificaron por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 10-30% en ciclohexano) para dar 2-bromo-4-metil-6-(trifluorometil)piridin-3-amina (4,3 g, 85%). Lc/Ms (Método i): Rt = 1,86 min; MS m/z: 255 [M+H]+; 1 H NMR (DMSO-Ce, 300MHz): 5 7,52 (s, 1H), 5,97 (s, 2H), 2,23 (s, 3H).
Etapa B: W-(2-bromo-4-metil-6-(tnfluorometil)piridm-3-il)-2,2,2-tnfluoroacetamida

A una solución de 2-bromo-4-metil-6-(trifluorometil)piridin-3-amina (522 mg, 2,05 mmol en DCM (6 mL) se añadió piridina (0,33 mL, 4,09 mmol) y anhídrido trifluoroacético (0,572 mL, 4,09 mmol) lentamente. La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se lavó con agua y se extrajo dos veces con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar W-(2-bromo-4-metil-6-(trifluorometil)piridin-3-il)-2,2,2-trifluoroacetamida (764 mg, 100%) como un sólido de color beige. LC/MS (Método i): Rt = 2,01 min; MS m/z: 351 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 11,80 (ancho, 1H), 8,09 (s, 1H), 2,37 (s, 3H).
Etapa C: W-alil-W-(2-bromo-4-metil-6-(tnfluorometil)pindm-3-il)-2,2,2-tnfluoroacetamida

A una solución de W-(2-bromo-4-metil-6-(trifluorometil)piridin-3-il)-2,2,2-trifluoroacetamida (719 mg, 2,048 mmol) en ACN (5 mL) se añadió carbonato de sodio (651 mg, 6,14 mmol) y bromuro de alilo (0,71 mL, 8,19 mmol) y la mezcla de reacción se agitó a 70 °C durante 2 horas. La mezcla de reacción se concentró a presión reducida, y el residuo se diluyó en EtOAc y se lavó con agua. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar W-alil-W-(2-bromo-4-metil-6-(trifluorometil)piridin-3-il)-2,2,2-trifluoroacetamida (795 mg, 99%). LC/MS (Método i): Rt = 2,40 min; MS m/z: 392 [M+H]+; 1H NMR (DMSO-de, 300MHz): 58,15 (s, 1H), 5,94 (m, 1H), 5,32 (dq, J=17, 1,5 Hz, 1H), 5,23 (m, 1H), 4,53 (dd, J=14,2, 6,3 Hz, 1H), 4,21 (dd, J=14,2, 7,6 Hz, 1H), 2,44 (s, 3H).
Etapa D: 3,7-dimetil-5-(tnfmorometil)-7H-pirrolo[3,2-b]pmdma
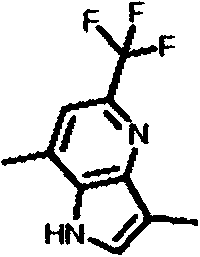
A una solución de W-alil-W-(2-bromo-4-metil-6-(trifluorometil)piridin-3-il)-2,2,2-trifluoroacetamida (200 mg, 0,256 mmol) en W,W-dimetilformamida (1 mL) se añadió acetato de paladio(II) (2,296 mg, 10,23 |_imol), cloruro de tetrabutilamonio (78 mg, 0,281 mmol) y trietilamina (0,078 mL, 0,563 mmol) y la mezcla de reacción se agitó a 110 °C durante 1,5 horas. La mezcla de reacción se lavó con agua y se extrajo con EtOAc. La capa orgánica se lavó con agua, luego se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 10-60% en ciclohexano) para dar 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-b]piridina (29 mg, 53%). LC/MS (Método i): Rt = 1,86 min; MS m/z: 215 [M+H]+; 1H NMR (DMSO-de, 300MHz): 511,53 (ancho, 1H), 7,61 (s, 1H), 7,39 (s, 1H), 2,57 (s, 3H), 2,30 (s, 3H).
Preparación #70: W-(4-ciano-2-yodo-3-metilfeml)bencenosulfonamida

Etapa A: 3-yodo-2-metil-4-mtrobenzomtnlo
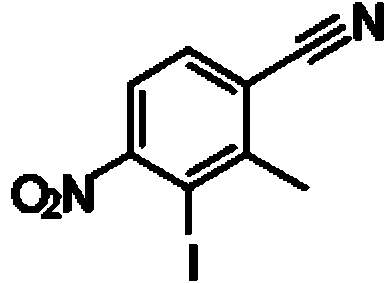
A una solución de 3-amino-2-metil-4-nitrobenzonitrilo (870 mg, 4,91 mmol) en THF (2,5 mL) y enfriada a -10 °C se añadió gota a gota dietil eterato de trifluoruro de boro (1,079 mL, 8,59 mmol) y la reacción se agitó durante 15 minutos a -10 °C. Luego se añadió gota a gota nitrito de isoamilo (0,986 mL, 7,37 mmol) y la mezcla de reacción se agitó durante 2 horas a 0 °C. A la mezcla de reacción se añadió pentano (2 mL) y se formó un precipitado beige, se filtró y se lavó varias veces con éter dietílico. Este residuo se añadió a una mezcla de yodo (935 mg, 3,68 mmol) y yoduro de potasio (1,6 g, 9,82 mmol) en acetona (6 mL) a 0 °C y la mezcla de reacción se agitó hasta que cesó el desprendimiento de N2 (15 minutos a 0 °C y luego 1 hora a temperatura ambiente. Una solución acuosa de tiosulfato de sodio al 10% se añadió a la mezcla de reacción y se agitó durante 15 minutos. Luego se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio y se concentraron para dar 3-yodo-2-metil-4-nitrobenzonitrilo (1,3 g, 88%) como un sólido amarillo. LC/MS (Método i): Rt = 2,06 min; MS m/z: 287 [M-H]-; 1H NMR (DMSO-de, 300MHz): 58,08 (d, J=8,3 Hz, 1H), 7,84 (d, J=8,3 Hz, 1H), 2,73 (s, 3H).
Etapa B: 4-amino-3-yodo-2-metilbenzonitrilo

Usando un procedimiento similar a la Preparación #18, Etapa C, se preparó 4-amino-3-yodo-2-metilbenzonitrilo (990 mg, 85%) a partir de 3-yodo-2-metil-4-nitrobenzonitrilo (1,3 g, 4,51 mmol). LC/MS (Método i): Rt = 1,84 min; MS miz: 259 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,39 (d, J=8,4 Hz, 1H), 6,64 (d, J=8,4 Hz, 1H), 6,23 (s, 2H), 2,51 (s, 3H).
Etapa C: W-(4-ciano-2-yodo-3-metilfeml)bencenosulfonamida

Usando un procedimiento similar a la Preparación #16, Etapa B, W-(4-ciano-2-yodo-3-metilfenil)bencenosulfonamida (1,8 g, 97%) se preparó a partir de 4-amino-3-yodo-2-metilbenzonitrilo (1,2 g, 4,65 mmol). LC/MS (Método i): Rt = 2,15 min; MS m/z: 397 [M-H]-; 1H NMR (DMSO-Ce, 300MHz): 5 10,11 (m, 1H), 7,81 (m, 2H), 7,70 (m, 2H), 7,59 (m, 2H), 7,01 (d, J=8,4 Hz, 1H), 2,59 (s, 3H).
Preparación #71: 4,6-dicloro-1-(femlsulfoml)-1H-mdol

Usando un procedimiento similar a la Preparación #48, se preparó 4,6-dicloro-1-(fenilsulfonil)-1H-indol (1,3 g, 74%) a partir de 4,6-dicloro-1H-indol (1 g, 5,38 mmol). LC/MS (Método i): Rt = 2,67 min; MS m/z: 326 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,10 (m, 1H), 8,06 (m, 1H), 8,03 (d, J=3,6 Hz, 1H), 7,94 (m, 1H), 7,75 (m, 1H), 7,65 (m, 2H), 7,54 (s, 1H), 6,90 (m, 1H).
Preparación #72: 6-metoxi-4-metiM-(femlsulfoml)-1H-indol

Usando un procedimiento similar a la Preparación #48, 6-metoxi-4-metil-1-(fenilsulfonil)-1H-indol (1,3 g, 81%) se preparó a partir de 6-metoxi-4-metil-1H-indol (900 mg, 5,58 mmol). LC/MS (Método i): Rt = 2,36 min; MS m/z: 302 [M+H]+; 1H Nm R (DMSO-Ce, 300MHz): 57,96 (m, 2H), 7,63 (m, 4H), 7,25 (m, 1H), 6,80 (dd, J=3,6, 0,8 Hz, 1H), 6,72 (m, 1H), 3,80 (s, 3H), 2,37 (s, 3H).
Preparación #73: 4-fluoro-1-metil-6-(trifluorometil)-1H-mdol-2-carbaldehído

Etapa A: (Z)-etil 2-azido-3-(2-fluoro-4-(trifluorometil)fenil)acrilato

A una solución de 2-fluoro-4-(trifluorometil)benzaldehído (2,128 mL, 15,62 mmol) en EtOH (40 mL) la mezcla de reacción se enfrió a -5 °C con un baño de hielo salino fresco y se añadió azidoacetato de etilo (7,14 mL, 62,5 mmol) y etanolato de sodio (5,31 g, 78 mmol). La mezcla de reacción se agitó a 0 °C durante 1 hora y una noche a temperatura ambiente. La mezcla de reacción se lavó con agua y se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar (Z)-etil 2-azido-3-(2-fluoro-4-(trifluorometil)fenil)acrilato (1,75 g, 34%) como un líquido amarillo. LC/MS (Método i): Rt = 2,69 min; MS m/z: 276 [M-N2+H]+; 1H NMR (DMSO-cfe 300MHz): 5 8,41 (dd, J=8,9, 7,8 Hz, 1H), 7,77 (dd, J=10,7, 1 Hz, 1H), 7,67 (dd, J=8,9, 1 Hz, 1H), 6,95 (s, 1H), 4,35 (q, J=7,1 Hz, 2H), 1,33 ppm (t, J=7,1 Hz, 3h )
Etapa B: etil acetato de 4-fluoro-6-(trifluorometM)-1H-mdol-2-carboxMato

Una solución de (Z)-etil 2-azido-3-(2-fluoro-4-(trifluorometil)fenil)acrilato (1,75 g, 5,77 mmol) en xileno (5 mL) se agitó a 140 °C durante 5 horas. El residuo se purificó directamente por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-40% en ciclohexano) para dar etil 4-fluoro-6-(trifluorometil)-1H-indol-2-carboxilato (1,2 g, 64%) como un sólido amarillo. LC/MS (Método i): Rt = 2,41 min; MS m/z: 274 [M-H]-; 1H NMR (DMSO-Ce, 300MHz): 512,74 (ancho, 1H), 7,64 (s, 1H), 7,29 (m, 2H), 4,39 (q, J=7,1 Hz, 2H), 1,36 ppm (t, J=7,1 Hz, 3H).
Etapa C: etil 4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-carboxilato

A una solución de etil 4-fluoro-6-(trifluorometil)-1H-indol-2-carboxilato (1 g, 3,63 mmol) en ACN (35 mL) se añadió sulfato de dimetilo (1,7 mL, 18,17 mmol) y carbonato de cesio (5,92 g, 18,17 mmol) y la mezcla de reacción se agitó a 50 °C durante la noche. La mezcla de reacción se diluyó con H2O y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-20% en ciclohexano) para dar etil 4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-carboxilato (240 mg, 23%). LC/MS (Método i): Rt = 2,61 min; MS m/z: 290 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,99 (s, 1H), 7,36 (s, 1H), 7,30 (d, J=10 Hz, 1H), 4,37 (q, J=7,1 Hz, 2H), 4,13 (s, 3H), 1,36 (t, J=7,1 Hz, 3H).
Etapa D: (4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metanol

A una solución de etil 4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-carboxilato (232 mg, 0,802 mmol) en THF (5 mL) y se enfrió a 0 °C se añadió lentamente hidruro de aluminio y litio (91 mg, 2,406 mmol). La mezcla de reacción se agitó a 0 °C durante 30 minutos. La sal de Glauber se añadió en porciones a la mezcla de reacción y se agitó durante una noche. La sal se filtró y se lavó con DCM. El filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-40% en DCM) para dar (4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metanol (166 mg, 84%) como un sólido de color blanco. LC/MS (Método i): Rt = 2,02 min; MS m/z: 248 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 = 7,78 (s, 1H), 7,13 (d, J=10,6 Hz, 1H), 6,56 (s, 1H), 5,43 (t, J=5,5 Hz, 1H), 4,69 (d, J=5,5 Hz, 2H), 3,84 (s, 3H).
Etapa E: 4-fluoro-1-metM-6-(tnfluorometM)-1H-mdol-2-carbaldehído

A una solución de (4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metanol (162 mg, 0,65 mmol) en DCM (2 mL) se añadió dióxido de manganeso (342 mg, 3,93 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 días. La mezcla de reacción se filtró y el sólido se lavó con DCM. El filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-20% en DCM) para dar 4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-carbaldehído (117 mg, 73 %) como un sólido de color beige. LC/MS (Método i): Rt = 2,28 min; MS m/z: 246 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 10,02 (s, 1H), 8,01 (s, 1H), 7,63 (s, 1H), 7,32 (d, J=10,3 Hz, 1H), 4,14 (s, 3H).
Preparación #74: 6-(difluorometoxi)-4-metiM-(femlsulfoml)-1H-mdol

Etapa A: 6-bromo-4-metiM-(fenilsulfoml)-1H-mdol

Usando un procedimiento similar al descrito en la Preparación #48, 6-bromo-4-metil-1-(fenilsulfonil)-1H-indol (3,49 g, 84%) se preparó a partir de 6-bromo-4-metil-1W-indol (2,5 g, 11,9 mmol). LC/MS (Método i): Rt = 2,41 min; MS miz: 350 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 57,99 (m, 2H), 7,89 (m, 1H), 7,85 (d, J=3,8 Hz, 1H), 7,72 (m, 1H), 7,62 (m, 2H), 7,28 (m, 1H), 6,92 (d, J=3,6 Hz, 1H), 2,43 (s, 3H).
Etapa B: 4-metiM-(femlsulfoml)-1H-indol-
Usando un procedimiento similar al descrito en la Preparación #65, Etapa B y Etapa C, se preparó 4-metil-1-(fenilsulfonil)-1W-indol-6-ol (2,35 g, 82%) a partir de 6-bromo-4 metil-1-(fenilsulfonil)-1 W-indol (3,49 g, 9,96 mmol). LC/MS (Método i): Rt = 2,03 min; MS miz: 288 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 59,48 (s, 1H), 7,87 (m, 2H), 7,69 (m, 1H), 7,61 (m, 2H), 7,54 (d, J=3,8 Hz, 1H), 7,14-7,19 (m, 1H), 6,74 (d, J=3,8 Hz, 1H), 6,55 (m, 1H), 2,32 (s, 3H).
Etapa C: 6-(difluorometoxi)-4-metiM-(femlsulfonil)-1H-mdol

Usando un procedimiento similar a la Preparación 65, Etapa D, 6-(difluorometoxi)-4-metil-1-(fenilsulfonil)-1W-indol (1,73 g, 63%) se preparó a partir de 4-metil-1 -(fenilsulfonil)-1 W-indol-6-ol (2,35 g, 8,18 mmol). LC/MS (Método i): Rt = 2,44 min; MS miz: 338 [M+H]+; 1H NMR (DMSO-C& 300MHz): 5 = 7,99 (m, 2H), 7,83 (d, J=3,6 Hz, 1H), 7,71 (m, 1H), 7,60 (m, 2H), 7,54 (m, 1H), 7,24 (t, J=74,0 Hz, 1H), 6,95 (m, 1H), 6,91 (d, J=3,8 Hz, 1H), 2,43 (s, 3H).
Preparación #75: W-(2-yodo-5-(trifluorometoxi)feml)bencenosulfonamida

Usando un procedimiento similar al descrito en la Preparación #16, Etapa B, se preparó N-(2-yodo-5-(trifluorometoxi)fenil)bencenosulfonamida (4,28 g, 86%) a partir de 2-yodo-5-(trifluorometoxi)anilina (3,2 g, 10,56 mmol).
LC/MS (Método i): Rt = 2,28 min; MS miz: 444 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 10,06 (s, 1H), 7,95 (d, J=8,8 Hz, 1H), 7,70 (m, 3H), 759 (m, 2H), 7,04 (m, 1H), 6,88 (m, 1H).
Preparación #76: W-(2-bromo-3-metil-5-nitrofenil)bencenosulfonamida
Usando un procedimiento similar al descrito en la Preparación #16, Etapa B, se preparó W-(2-bromo-3-metil-5-nitrofenil)bencenosulfonamida (3,24 g, 38%) a partir de 2-bromo-3-metil-5-nitroanilina (5,3 g, 22,94 mmol). LC/MS (Método i): Rt = 2,16 min; MS miz: 369 [M-H]-; 1H NMR (DMSO-Ce, 300MHz): 510,42 (ancho, 1H), 8,10 (d, J=2,5 Hz, 1H), 7,77 (m, 3H), 7,99 (m, 1H), 7,59 (m, 2H), 2,42 (s, 3H).
Preparación #77: 6-cidopropM-4-fluoro-1-(femlsulfoml)-1H-mdol

Etapa A: 6-bromo-4-fluoro-1-(fenilsulfonil)-1H-indol

Usando un procedimiento similar al descrito en la Preparación #48, se preparó 6-bromo-4-fluoro-1-(fenilsulfonil)-1H-indol (251 mg, 76%) a partir de 6-bromo-4-fluoro-1H-indol (200 mg, 0,93 mmol). LC/MS (Método j): Rt = 1,93 min; MS m/z: 354 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 = 8,06 (m, 2H), 7,95 (m, 2H), 7,76 (m, 1H), 7,65 (m, 2H), 7,44 (d, J=9,4 Hz, 1H), 6,96 ppm (d, J=3,8 Hz, 1H).
Etapa B: 6-ciclopropil-4-fluoro-1-(fenilsulfonil)-1H-indol

Usando un procedimiento similar al descrito en la Preparación #12, se preparó 6-cidopropil-4-fluoro-1-(fenilsulfonil)-1H-indol (100 mg, 80%) a partir de 6-bromo-4-fluoro-1H-indol (140 mg, 0,39 mmol). 1H Nm R (DMSO-Ce, 300MHz): 58,01 (m, 2H), 7,78 (d, J=3,8 Hz, 1H), 7,73 (m, 1H), 7,63 (m, 2H), 7,52 (s, 1H), 6,84 (dd, J=3,6, 0,7 Hz, 1H), 6,78 (dd, J=11,5, 1,2 Hz, 1H), 2,11 (m, 1H), 1,02 (m, 2H), 0,72 (m, 2H).
Preparación #78: 7-metil-5-(trifluorometM)-1H-mdazol

Usando un procedimiento similar al descrito en la Preparación #42, Etapa D, se preparó 7-metil-5-(trifluorometil)-1 H-indazol (550 mg, 46%) a partir de 2-fluoro-3-metil-5-(trifluorometil) benzaldehído (1 g, 4,85 mmol) (Preparación #42, Etapa A). LC/MS (Método i): Rt = 1,87 min; MS m/z: 201 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 = 13,58 (m, 1H), 8,25 (s, 1H), 8,04 (s, 1H), 7,41 (s, 1h ), 2,59 (s, 3H).
Preparación #79: 1,4-dimetil-6-(trifluorometil)-1H-indazol-3-carbaldehído

Etapa A: 4-metil-6-(trifluorometil)-1H-indazol
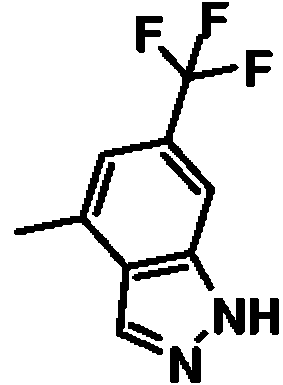
Usando un procedimiento similar al descrito en la Preparación #41, Etapa B, se preparó 4-metil-6-(trifluorometil)-1 H-indazol (4,91 g, 72%) a partir de 4-bromo-6-(trifluorometil)-1H-indazol (9 g, 34 mmol). Lc /MS (Método i): Rt = 1,89 min; MS m/z: 201 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 13,49 (ancho, 1H), 8,30 (s, 1h ), 7,73 (s, 1H), 7,18 (s, 1H), 2,64 (s, 3H).
Etapa B: 3-yodo-4-metil-6-(trifluorometil)-1H-indazol

A una solución de 4-metil-6-(trifluorometil)-1H-indazol (3 g, 14,99 mmol) en 1,4-dioxano (89,7 mL) se añadió 2 N de solución de hidróxido de sodio (11,24 mL, 22,48 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Luego se añadió diyodo (4,5 g, 17,99 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 20 horas. Se añadió una S solución de Na2S2O3 al 10%, y la mezcla se agitó 15 min a temperatura ambiente, después se extrajo con EtOAc. Se añadió HCl 1M en la capa acuosa y se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-15% en ciclohexano) para dar 3-yodo-4-metil-6-(trifluorometil)-1H-indazol (4,33 g, 89%) como un polvo blanco. LC/MS (Método i): Rt = 2,22 min; MS m/z: 327 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 513,89 (ancho, 1H), 7,80 (s, 1H), 7,20 (s, 1H), 2,85 (s, 3H).
Etapa C: 3-yodo-1,4-dimetil-6-(trifluorometil)-1H-indazol

Usando un procedimiento similar al descrito en la Preparación #51, Etapa E, 3-yodo-1,4-dimetil-6-(trifluorometil)-1 H-indazol (3,5 g, 67%) se preparó a partir de 3-yodo-4- metil-6-(trifluorometil)-1 H-indazol (4,3 g, 13,3 mmol). LC/MS (Método i): Rt = 2,44 min; MS m/z: 341 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 8,06 (s, 1H), 7,22 (s, 1H), 4,12 (s, 3H), 2,83 (s, 3H).
Etapa D: 1,4-dimetM-6-(tnfluorometM)-3-vimM H-indazol

A una solución de 3-yodo-1,4-dimetil-6-(trifluorometil)-1 H-indazol (3,3 g, 9,70 mmol) en tolueno (105 mL) se añadió cloruro de bis(trifenilfosfina)paladio(II) (681 mg, 0,970 mmol) y tributil(vinil)estaño (3,97 mL, 13,58 mmol), y la mezcla de reacción se agitó 2 horas a 90 °C. Se añadió SO 2, y la mezcla de reacción se evaporó a sequedad y se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar 1,4-dimetil-6-(trifluorometil)-3-vinil-1 H -indazol (2,33 g, 90%) como un polvo blanco. LC/MS (Método i): Rt = 2,33 min; MS m/z: 241 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,95 (s, 1H), 7,19 (s, 1H), 7,22 (dd, C=17,3, 11,1 Hz, 1H), 6,02 (dd, C=17,3, 2,1 Hz, 1H), 5,40 (dd, C=11,1,2,1 Hz, 1H), 4,12 (s, 3H), 2,71 (s, 3H).
Etapa E: 1,4-dimetM-6-(tnfluorometM)-1H-mdazol-3-carbaldehído

A una solución de 1,4-dimetil-6-(trifluorometil)-3-vinil-1 H-indazol (2,3 g, 9,70 mmol) en ACN (66 mL) y agua (14,51 mL) se añadió una solución de cloruro de rutenio(III) hidratado (9,70 mL, 0,388 mmol) en agua (9,70 mL), seguido de peryodato de sodio (4,1 g, 19,40 mmol). La mezcla de reacción se agitó durante la noche a temperatura ambiente. Se añadió una solución saturada de tiosulfato de sodio, y la mezcla se extrajo dos veces con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar 1,4-dimetil-6-(trifluorometil)-1 H-indazol-3-carbaldehído (1,1 g, 47%) como un polvo blanco. LC/MS (Método i): Rt = 2,15 min; MS m/z: 243 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 10,14 (s, 1H), 8,15 (s, 1H), 7,45 (s, 1H), 4,29 (s, 3H), 2,87 (s, 3H).
Preparación #80: 4-metil-6-(trifluorometoxi)-1 H -indol

Usando un procedimiento similar al descrito en la Preparación 64, Etapa B, se preparó 4-metil-6-(trifluorometoxi)-1 H-indol (373 mg, 57%) a partir de 2-yodo-3-metil-5-(trifluorometoxi)anilina (1,1 g, 3,47 mmol) (Preparación #47, Etapa D).
LC/MS (Método i): Rt = 2,24 min; MS m/z: 214 [M-H]-; 1H NMR (DMSO-Ce, 300MHz): 5 = 11,27 (ancho, 1H), 7,42 (m, 1H), 7,17 (m, 1H), 6,79 (m, 1H), 6,50 (m, 1H), 2,49 (s, 3h ).
Preparación #81: metil 2-azaspiro[3,3]heptano-6-carboxilato 2,2,2-trifluoroacetato

A una solución de metil 2-boc-2-aza-spiro[3,3]heptano-6-carboxilato (400 mg, 1,57 mmol) en DCM (6 mL) se añadió TFA (3,02 mL, 39,2 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 18 horas. La mezcla de reacción se concentró a presión reducida para dar metil 2-azaspiro[3,3]heptano-6-carboxilato 2,2,2-trifluoro-acetato (422 mg, 100%). 1H NMR (DMSO-Ce, 300MHz): 53,98 (t, J=6,2 Hz, 2H), 3,90 (t, J=6,2 Hz, 2H), 3,59 (s, 3H), 3,01 (m, 1H), 2,50 (m, 4H).
Preparación #82: clorhidrato de metil 2-(2-azaspiro[3,3]heptan-6-il)acetato

A una solución de ácido 2-(2-(íerc-butoxicarbonil)-2-azaspiro [3,3]heptan-6-il)acético (309 mg, 1,21 mmol) en EtO-HMeOH (2,3 mL) se añadió gota a gota cloruro de oxalilo (0,21 mL, 2,42 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 20 horas, luego se concentró a presión reducida para dar clorhidrato de metil 2-(2-azaspiro[3,3]heptan-6-il)acetato (249 mg, 100%) como un sólido de color blanco. 1H NMR (DMSO-Ce, 300MHz): 53,94 (m, 2H), 3,81 (m, 2H), 3,76 (s, 1H), 3,56 (s, 3H), 2,38 (m, 4H), 1,88 (m, 2H).
Preparación #83: (3S,4S)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Etapa 1: (cis)-etil 3-metilpiperidina-4-carboxilato

Se cargó un matraz con Pd(OH)2 (2,71 g, 3,86 mmol) y se disolvió en EtOH (100 mL) bajo una manta de nitrógeno. Una solución de etil 1-bencil-5-metil-1,2,3,6-tetrahidropiridin-4-carboxilato (10 g, 38,6 mmol) (Journal of Med Chem, 2010, vol.
53, # 21 p. 7682-7698) en EtOH (50 mL) se añadió y la reacción se purgó con globo de hidrógeno y se agitó a temperatura ambiente. El catalizador se filtró y el disolvente se concentró a presión reducida para proporcionar un aceite (cis)-etil 3-metilpiperidin-4-carboxilato (29 g, 169 mmol, 97% de rendimiento). LC/MS (Método i) Rt = 0,1 min.; MS m/z: 130 (M+H)+.
Etapa 2: (3R,4R)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico y (3S,4S)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Se añadió N-etil-N-isopropilpropan-2-amina (5,35 mL, 30,7 mmol) a una solución de rac-(3R,4R)-etil 3-metil-ilpiperidina-4-carboxilato (5,00 g, 29,2 mmol) (mezcla de isómeros cis/trans 10:1) se disolvió en DCM (97 mL) a temperatura ambiente en un matraz de 250 mL. Bencil 2,5-dioxopirrolidina-1-carboxilato (6,81 g, 28,6 mmol) se añadió por lotes a temperatura ambiente. Después de 3 horas, la reacción se concentró hasta la sequedad y luego se redisolvió en éter. La solución se lavó con agua, solución de HCl al 10% (2x 20 mL), seguido de una solución de salmuera (20 mL), y luego se secó sobre sulfato de magnesio. Los isómeros cis se separaron por SFC prep: isopropanol al 10% en CO2 (60 mL/min, 140 bar, 40 °C). El tiempo de ciclo fue de 4,40 min, con un tiempo de ejecución simple de 22 min. Se usó isopropanol de grado HPLC con CO2 grado SFC. La cromatografía usó una columna YMC-SC de 30 x 150 mm (partículas de 5 |_im). LC/MS (Método i) Rt = 1,60 min.; MS m/z: 306 (M+H)+.
Etapa 3: (3S,4S)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Se añadió paladio sobre carbono (4,18 g, 3,93 mmol) a un matraz de fondo redondo de 50 mL bajo una manta de nitrógeno. (3S,4S)-1-bencil 4-etil 3-metilpiperidina-1,4-dicarboxilato (12 g, 39,3 mmol) se disolvió en EtOH (7 mL) y se añadió al matraz de reacción. El matraz se purgó con una atmósfera de hidrógeno y luego se agitó durante aproximadamente 18 h. Al finalizar, se eliminó el hidrógeno y la mezcla se filtró a través de una almohadilla de Celite®. La solución se concentró hasta la sequedad. Se añadió éter (10 mL) seguido de HCl en éter (58,9 mL, 58,9 mmol), que precipitó un sólido de color blanco. El sólido se filtró bajo una corriente de nitrógeno y luego se secó en un horno de vacío durante aproximadamente dos horas. El material se recogió como un sólido de color blanco puro (3S,4S)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico (5,3 g, 25,5 mmol, rendimiento del 64,9%). LC/MS (Método i) Rt = 0,1 min.; MS m/z: 130 (M+H)+.
Preparación #84: (3R,4R)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Etapa 1: (cis)-etil 3-metilpiperidina-4-carboxilato

Se cargó un matraz con Pd(OH) 2 (2,71 g, 3,86 mmol) y se disolvió en EtOH (100 mL) bajo una manta de nitrógeno. Una solución de etil 1-bencil-5-metil-1,2,3,6-tetrahidropiridin-4-carboxilato (10 g, 38,6 mmol) (Journal of Med Chem, 2010, vol.
53, # 21 p.7682-7698) en EtOH (50 mL) se añadió y la reacción se purgó con globo de hidrógeno y se agitó a temperatura ambiente durante aproximadamente 18 horas. El catalizador se filtró y el disolvente se concentró a presión reducida para proporcionar (cis)-etil 3-metilpiperidina-4-carboxilato (29 g, 169 mmol, 97%) como un aceite. LC/MS (Método i) Rt = 0,1 min.; MS m/z: 130 (M+H)+.
Etapa 2: (3R,4R)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Se añadió N-etil-N-isopropilpropan-2-amina (5,35 mL, 30,7 mmol) a una solución de rac-(3R,4R)-etil 3-metilpiperidina-4-carboxilato (5,00 g, 29,2 mmol) (mezcla de isómeros cis/trans 10:1) disueltos en DCM (97 mL) a temperatura ambiente en un matraz de 250 mL. Se añadió por lotes bencil 2,5-dioxopirrolidina-1-carboxilato (6,81 g, 28,6 mmol) a temperatura ambiente. Después de 3 horas, la reacción se concentró hasta la sequedad y luego se redisolvió en éter. La solución se lavó con agua, solución de HCl al 10% (2x 20 mL), seguido de una solución de salmuera (20 mL), y luego se secó sobre sulfato de magnesio. Los isómeros cis se separaron por SFC prep: isopropanol al 10% en CO2 (60 mL/min, 140 bar, 40 °C). El tiempo de ciclo fue de 4,40 min, con un tiempo de ejecución simple de 22 min. Se usó isopropanol de grado HPLC con CO2 grado SFC. La cromatografía usó una columna YMC-SC de 30 x 150 mm (partículas de 5 mm). LC/MS (Método i) Rt = 1,60 min.; MS m/z: 306 (M+H)+.
Etapa 3: (3R,4R)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico

Se añadió paladio sobre carbono (2,67 g, 2,505 mmol) a un matraz seco de 500 mL de 2 bocas purgado con nitrógeno. Se añadió una cantidad mínima de EtOAc para humedecer el catalizador y luego se añadió EtOH (200 mL) cuidadosamente al matraz mientras se agitaba. Se añadió (3R,4R)-1-bencil 4-etil 3-metilpiperidina-1,4-dicarboxilato (15,3 g, 50,1 mmol) en 50 mL de EtOH y luego el matraz se purgó tres veces con una atmósfera de hidrógeno. La reacción se dejó agitar durante la noche. La mezcla se evacuó luego con nitrógeno y luego se filtró a través de una almohadilla de Celite® y el filtrado se concentró hasta un aceite crudo. El aceite se disolvió en éter (50 mL) y luego se añadió HCl 4 M en éter. La solución se filtró en un embudo de vidrio fritado bajo una corriente de nitrógeno y luego se secó en un horno de vacío durante un par de horas para proporcionar (3R,4R)-etil 3-metilpiperidina-4-carboxilato, ácido clorhídrico (6,88 g, 66%). LC/MS (Método i) Rt = 0,1 min.; MS m/z: 130 (M+H)+.
Preparación #85: 2-yodo-3-metil-5-(trifluorometil)fenol

A una solución de 2-yodo-3-metil-5-(trifluorometil)anilina (3 g, 9,97 mmol) (Preparación #16, Etapa A) en ácido acético (30 mL, 9,97 mmol) y enfriada a 10 °C se añadió gota a gota nitrito de sodio (1,031 g, 14,95 mmol) en solución en ácido sulfúrico (18 mL, 9,97 mmol). La mezcla de reacción se agitó 30 minutos entre 10 y 15 °C. Esta solución se añadió gota a gota a una solución de agua a reflujo (54 mL) y la mezcla de reacción se agitó 30 minutos a reflujo. La reacción se enfrió a temperatura ambiente y la mezcla se extrajo dos veces con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 2-yodo-3-metil-5-(trifluorometil)fenol (3 g, 100%) como un aceite naranja. LC/MS (Método i): Rt = 2,19 min; MS m/z: 301 [M-H]-; 1H NMR (DMSO-cfe, 300MHz): 510,96 (s, 1H), 7,13 (s, 1H), 6,93 (s, 1H), 2,44 (s, 3H).
Ejemplo A: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(2-metoxietil)piperazin-1-il)metanona

Etapa 1: metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)- 1H-indol-2-il)metil)benzoato

A una solución de W-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (300 mg, 0,680 mmol) en DMF (2 mL) se añadió Pd(PPh3 )2 Cl2 (23,86 mg, 0,034 mmol), yoduro de cobre(I) (6,47 mg, 0,034 mmol), trietilamina (2 mL) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-ilo)benzoato (229 mg, 0,884 mmol) (Preparación #1). La mezcla de reacción se calentó a aproximadamente 110 °C bajo microondas durante 20 minutos y luego se diluyó con agua. La capa acuosa se extrajo con EtOAc, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-60% en ciclohexano) para dar metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (400 mg, 99%) como un aceite de color marrón. LC/MS (Método h) Rt = 3,33 min.; MS m/z: 630 [M-H]-+ CH3 COOH.1H NMR (DMSO-C6, 300 MHz): 5 8,08 (s, 1H) 7,80 (m, 1H), 7,78 (m, 1H), 7,68 (m, 2H), 5,56 (m, 3H), 7,43 (s, 1H), 7,02 (d, 1H), 6,88 (s, 1H), 6,75 (d, 1H), 3,86 (s, 3H), 2,47 (s, 3H).
Etapa 2: metil 2,4-didoro-3-((4-metiM-(femlsulfonM)-6-(tnfluorometM)- 1H-mdol-2-M)metil)benzoato

A una solución de metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (440 mg, 0,769 mmol) en DCM (4,6 mL) enfriado a 0 °C se añadió trietilsilano (0,613 mL, 3,84 mmol), dietil eterato de trifluoruro de boro (0,483 mL, 3,84 mmol) y TFA (0,046 mL). La mezcla de reacción se agitó a temperatura ambiente durante una hora, luego se diluyó con agua. La capa acuosa obtenida se extrajo con DCM y la capa orgánica se lavó con solución acuosa saturada de NaHCO3, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)- 1H-indol-2-il)metil)benzoato (413 mg, 94%) como un sólido negro. LC/MS (Método h) Rt = 3,79 min.; MS m/z: 556 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,24 (s, 1H), 7,92 (m, 2H), 7,81 (m, 2H), 7,70 (m, 3H), 7,42 (s, 1H), 5,98 (s, 1H), 4,61 (s, 2H), 3,86 (s, 3H), 2,35 (s, 3H)
Etapa 3: metil 2,4-didoro-3-((4-metil-6-(trifluorometil)- 1H-mdol-2-M)metM)benzoato

A una solución de metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (6,16 g, 11,07 mmol) en THF (200 mL) se añadió gota a gota fluoruro de tetrabutilamonio (13,29 mL, 13,29 mmol). La mezcla de reacción se agitó a reflujo durante una hora y se añadió fluoruro de tetrabutilamonio (3,3 mL, 3,3 mmol) y se calentó a reflujo durante una hora más. La mezcla de reacción se concentró a presión reducida y el residuo se recogió en EtOAc y se lavó con solución acuosa saturada de NaHCO3. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se evaporó a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-20% en ciclohexano) para dar metil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (3,8 g, 66%) como un sólido amarillo. LC/MS (Método h) Rt = 3,39 min.; MS m/z: 416 [M+H]+. 1H NMR (CDCfe, 300 MHz): 58,36 (s, 1H), 7,64 (d, J = 9,0 Hz, 1H), 7,43 (m, 2H), 7,09 (s, 1H), 6,39 (s, 1H), 4,56 (s, 2H), 3,94 (s, 3H), 2,52 (s, 3H)
Etapa 4: metil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato

A una solución de metil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (3,80 g, 9,13 mmol) en DMF (100 mL) se añadió en porciones hidruro de sodio (0,402 g, 10,04 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos, luego se añadió yodometano (0,628 mL, 10,04 mmol) y la mezcla de reacción se agitó durante una hora. La mezcla de reacción se diluyó con agua. El precipitado resultante se filtró, se lavó con agua y se secó para obtener metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (3,7 g, 94%) como sólido de color beige.
LC/MS (Método h) Rt = 3,51 min.; MS m/z: 430 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 57,80 (d, J = 9,0 Hz, 1H), 7,70 (m, 2H), 7,06 (s, 1H), 5,67 (s, 1H), 4,50 (s, 2H), 3,92 (s, 3H), 3,88 (s, 3H), 2,36 (s, 3H)
Etapa 5: 2,4-didoro-3-(((trifluorometM)-1H-mdol-2-M)metM-6- 1,4-dimetil)benzoico

A una solución de metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (3,7 g, 8,60 mmol) en THF (100 mL) se añadió hidróxido de litio (0,722 g, 17,20 mmol) en agua (50 mL). La mezcla de reacción se agitó a temperatura ambiente durante la noche y luego se concentró a presión reducida. El residuo se tomó en agua y se acidificó a pH = 3-4 mediante la adición de una solución de HCl 1N. El precipitado obtenido se filtró, se lavó con agua y se secó para dar ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)benzoico (3,5 g, 95% de rendimiento) como un polvo blanco. LC/MS (Método h) Rt = 3,08 min.; MS m/z: 416 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5 13,55 (ancho, 1H), 7,76 (d, J = 9,0 Hz, 1H), 7,68 (m, 2H), 7,06 (s, 1H), 5,67 (s, 1H), 4,84 (s, 2H), 3,92 (s, 3H), 2,49 (s, 3H)
Etapa 6: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(2-metoxietil)piperazin-1-il)metanona

A una solución de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)- 1H-indol-2-il)metil)benzoico (102 mg, 0,245 mmol) en DCM (2 mL) se añadió trietilamina (0,044 mL, 0,319 mmol), 1-(3-dimetilaminopropil)-3-etilcarbodiimida (49,5 mg, 0,319 mmol) y 1-hidroxibenzotriazol (43,0 mg, 0,319 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 30 minutos. Se añadió 1-(2-metoxietil)piperazina (0,044 mL, 0,294 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante la noche, luego se diluyó con agua y se extrajo con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH 0-5% en DCM) para dar (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)fenil)(4-(2-metoxietil)piperazin-1-il)metanona (59 mg, 43,6%) como un sólido de color blanco. LC/MS (Método g) Rt = 1,34 min.; MS m/z: 542 [M+H]+.
1H NMR (DMSO-C6, 400 MHz): 5 7,70 (s, 1H), 7,67 (d, J = 8,1Hz, 1H), 7,41 (d, J = 8,1Hz, 1H), 7,07 (s, 1H), 5,67 (s,
Tabla A. Se prepararon los siguientes intermedios a partir de 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-ilmetilbenzoico Eemlo A Etaa 5 usando el mismo rocedimiento con la amina adecuada.




Ejemplo A1: (2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)feml)(4-(oxetan-3-M) piperazin-1-il)metanona

A una solución de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)benzoico (Ejemplo A, Etapa 5) (100 mg, 0,240 mmol) en DMF (519 |_iL) se añadió HATU (110 mg, 0,288 mmol) y 4-metilmorfolina (106 pL, 0,961 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 15 minutos y luego se añadió 1-(oxetan-3-il)piperazina (41,0 mg, 0,288 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. Se añadió agua y el precipitado formado se filtró y se lavó con agua y se trituró en éter dietílico para dar (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(oxetan-3-il)piperazin-1-il)metanona (81 mg, 62%) como un sólido de color blanco. LC/MS (Método g) Rt = 1,73 min.; MS m/z: 540 [M+H]+ .1H NMR (DMSO-cfe, 400 MHz): 57,70 (s, 1H), 7,68 (d, J = 8,1Hz, 1H), 7,43 (d, J = 8,1Hz, 1H), 7,06 (s, 1H), 5,68 (s, 1H), 4,52 (m, 2H), 4,43 (m, 4H), 3,92 (s, 3H), 3,67 (m; 2H), 3,43 (m, 1H), 3,20 (m, 2H), 2,36 (s, 3H), 2,38-2,18 (m, 4h ).
Tabla A1. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-in l-2-ilm il nz i E m l A E n l mi m r imi n n l min r i .


Ejemplo B: ácido 1-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-M)metN)benzoN)piperidma-4-carboxNico

Usando un procedimiento similar al Ejemplo A, Etapa 5, acido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxílico (1,465 g, 95% de rendimiento) se preparó a partir de etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato (Tabla A. A-1) (1,6 g, 2,88 mmol).
LC/MS (Método g) Rt = 1,79 min.; MS m/z: 527 [M+H]+. 1H NMR (DMSO-óe, 500 MHz): 513,36 (ancho, 1H), 7,87 (s, 1H), 7,67 (m, 1H), 7,42 (m, 1H), 6,87 (s, 1H), 5,67 (m, 1H), 4,48 (m, 2H), 4,35 (m, 1H), 3,92 (s, 3H), 3,31 (m, 1H), 3,08 (m, 1H), 2,97 (m, 1H), 2,58 (m, 1H), 2,35 (s, 3H), 1,93 (m, 1H), 1,75 (m, 1H), 1,50 (m, 2H)
Tabla B. Los siguientes Ejemplos se prepararon usando el mismo procedimiento a partir de los ésteres apropiados (como se describe en las Tablas A y A1, BH).




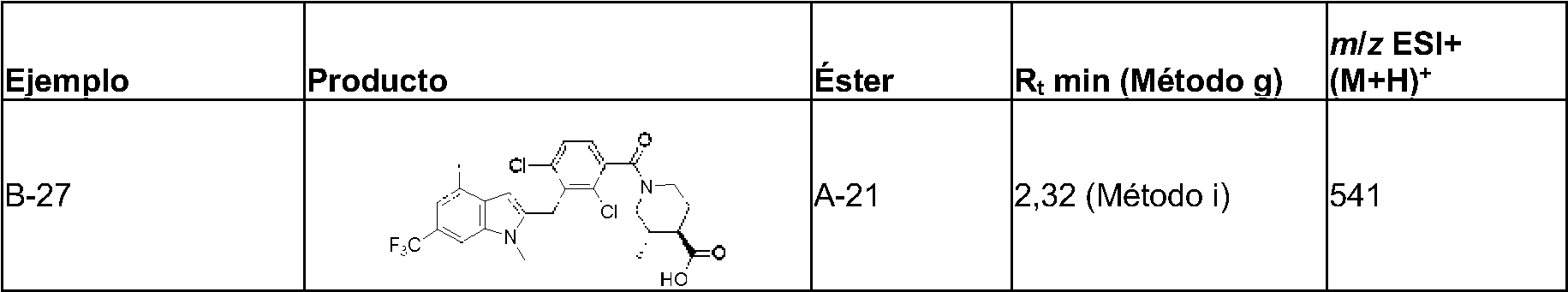
Ejemplo B.2: ácido (3S,4S)-1-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-M)metM)benzoN)-3-metilpiperidina-4-carboxílico y ácido (3R,4R)-1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico

Ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico (Ejemplo B1, Tabla B, 400 mg) se disolvió en cloruro de metileno y se purificó usando el Método o de los métodos de purificación quiral (Tabla 2). Las fracciones del primer isómero eluyente se agruparon y se analizaron.
Ácido (3S,4S)-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico (0,123 g, 0,227 mmol, 28,7% de rendimiento) LC/MS (Método 1) Rt = 2,5 min.; MS m/z: 539 [M-H]-, 541 [M+H]+.1H NMR (CDCla, 400 MHz): 5 12,33 (bs, 1H), 7,73-7,64 (m, 2H), 7,47-7,37 (m, 1H), 7,07 (s, 1H), 4,15-4,55 (m, 3H), 3,91-3,94 (m, 3H), 3,28-3,22 (m, 1H), 3,18-2,92 (m, 2H), 2,72-2,63 (m, 1H), 2,38-2,34 (m, 3H), 2,34-2,06 (m, 1H), 1,74-1,50 (m, 2H), 0,95-0,70 (m,4H)
HPLC quiral 2,9 min (Método n, >99% ee).
Las fracciones del segundo pico de elución se agruparon y analizaron.
Ácido (3R,4R)-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil) benzoil)-3-metilpiperidina-4-carboxílico (0,138 g, 0,255 mmol, 32,2% de rendimiento) LC/MS (Método 1) Rt = 2,5 min.; MS m/z: 539 [M-H]-, 541 [M+H]+.1H NMR (CDCla, 400 MHz): 5 12,33 (bs, 1H), 7,73-7,64 (m, 2H), 7,47-7,37 (m, 1H), 7,07 (s, 1H), 4,15-4,55 (m, 3H), 3,91-3,94 (m, 3H), 3,28-3,22 (m, 1H), 3,18-2,92 (m, 2H), 2,72-2,63 (m, 1H), 2,38-2,34 (m, 3H), 2,34-2,06 (m, 1H), 1,74-1,50 (m, 2H), 0,95-0,70 (m,4H).
HPLC quiral 3,7 min (Método n, >99% ee).
Ejemplo C: (2,4-dicloro-3-((4-metil-6-(trifluorometil)- 1H-mdol-2-M)metM)feml)(morfolmo)metanona

Etapa 1: (2,4-didoro-3-(hidroxi(4-metiM-(femlsulfonM)-6-(trifluorometM)-1H-mdol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-didoro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifiuorometil)-1 H-indol-2-il)metil)fenil)(morfolino)metanona (3,7 g, 87%) se preparó a partir de W-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (3 g, 6,80 mmol) y (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (2,78 g, 8,84 mmol) (Preparación #3). LC/MS (Método k) Rt = 3,10 min.; MS miz: 627[M+H]+.
1H NMR (CDCla, 300 MHz): 58,25 (s, 1H), 7,90 (m, 2H), 7,52 (m, 1H), 7,45 (m, 3H), 7,30 (m, 2H), 7,12 (m, 1H), 6,35 (m, 1H), 4,02 (m, 1H), 3,82 (m, 4H), 3,79 (m, 2H), 3,30 (m, 2H), 2,41 (s, 3H).
Etapa 2: (2,4-didoro-3-((4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 2, (2,4-didoro-3-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (3 g, 86%) se preparó a partir de (2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (3,67 g, 5,85 mmol).
LC/MS (Método k) Rt = 3,56 min.; MS miz: 611[M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,24 (s, 1H), 7,92 (m, 2H), 7,68 (m, 1H), 7,65 (m, 3H), 7,46 (m, 1H), 7,42 (s, 1H), 6,00 (s, 1H), 4,57 (m, 2H), 3,62 (m, 4H), 3,47 (m, 2H), 3,18 (m, 2H), 2,35 (s, 3H).
Etapa 3: (2,4-didoro-3-((4-metM-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona
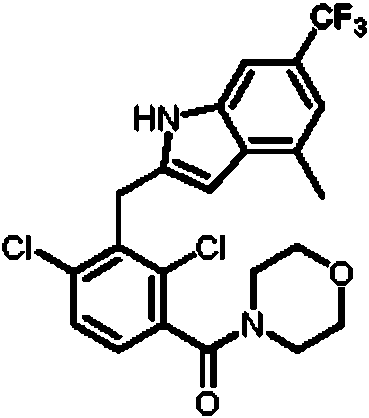
Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (130 mg, 59%) se preparó a partir de (2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (286 mg, 0,468 mmol). LC/MS (Método g) Rt = 1,36min.; MS miz: 471 [M+H]+. 1H NMR (DMSO-C6, 400 MHz): 5 11,55 (s, 1H), 7,66 (m, 1H), 7,48 (m, 1H), 7,42 (m, 1H), 7,02 (s, 1H), 5,97 (s, 1H), 4,48 (m, 2H), 3,65 (m, 4H), 3,52 (m, 2H), 3,18 (m, 2H), 2,40 (s, 3H).
Ejemplo D: (2,4-didoro-3-((1,4-dimetM-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (26 mg, 50%) se preparó a partir de (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (Ejemplo C) (50 mg, 0,106 mmol). LC/MS (Método g) Rt = 1,91 min.; MS m/z: 485[M+H]+.
1H NMR (DMSO-de, 400 MHz): 57,75 (s, 1H), 7,70 (d, J = 9,0Hz, 1H), 7,49 (d, J = 9,0Hz, 1H), 7,06 (s, 1H), 5,67 (s, 1H), 4,48 (m, 2H), 3,92 (s, 3H), 3,62 (m, 4H), 3,51 (m, 2H), 3,20 (m, 2H), 2,36 (s, 3H).
Tabla D. Los siguientes análogos se prepararon a partir de (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1 H-indol-2-il)metil)fenil)(morfolino)metanona (Ejemplo C) usando el mismo procedimiento y usando el electrófilo apropiado.

Ejemplo E: (2,4-dicloro-3-((1-metM-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona
Etapa 1: (2,4-didoro-3-(hidroxi(1-(femlsulfoml)-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-didoro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (491 mg, 81%) se preparó a partir de N-(2-yodo-5-(trifluorometil)fenil)bencenosulfonamida (420 mg, 0,983 mmol) (Preparación #29) y (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (402 mg, 1,28 mmol) (Preparación #3). LC/MS (Método g) Rt = 1,77 min.; MS miz: 612[M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 58,21 (s, 1H), 7,91 (m, 1H), 7,85 (m, 2H), 7,72 (m, 1H), 7,61 (m, 3H), 7,53 (m, 1H), 7,38 (m, 1H), 6,98 (m, 1H), 6,82 (m, 1H), 6,65 (m, 1H), 3,68 (m, 4h ), 3,57 (m, 2h ), 3,15 (m, 2H).
Etapa 2: (2,4-didoro-3-((1-(femlsulfoml)-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo) metanona

Usando un procedimiento similar al Ejemplo A, Etapa 2, (2,4-didoro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (196 mg, 47%) se preparó a partir de (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (430 mg, 0,701 mmol). LC/MS (Método g) Rt = 2,00 min.; MS miz: 597 [M+H]+. 1H NMR (DMSO-C6, 400 MHz): 58,41 (s, 1H), 7,94 (m, 2H), 7,80 (m, 1H), 7,68 (m, 4H), 7,60 (m, 1H), 6,48 (m, 1H), 6,06 (s, 1H), 4,55 (m, 2H), 3,65 (m, 4H), 3,51 (m, 2H), 3,21 (m, 2H).
Etapa 3: (2,4-didoro-3-((6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-didoro-3-((6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (103 mg, 69 %) se preparó a partir de (2,4-dicloro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (182 mg, 0,305 mmol). LC/MS (Método g) Rt = 1,76 min.; Ms miz:
457 [M+H]+. 1H NMR (DMSO-C6, 400 MHz): 511,53 (s, 1H),7,65 (m, 2H), 7,57 (d, J = 8,0Hz, 1H), 7,41 (d, J = 8,0Hz, 1H), 7,21 (dd, J = 8,0Hz, J = 4,0Hz, 1H), 5,97 (s, 1H), 4,46 (s, 2H), 3,64 (m, 4H), 3,53 (m, 2H), 3,18 (m, 2H).
Etapa 4: (2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (68 mg, 77%) se preparó a partir de (2,4-dicloro-3-((6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (86 mg, 0,188 mmol). LC/MS (Método g) Rt = 1,83 min.; Ms miz: 471 [M+H]+. 1H NMR (DMSO-cfe, 400 MHz): 57,87 (s, 1H), 7,67 (d, J = 8,0Hz, 1H), 7,56 (d, J = 8,0Hz, 1H), 7,45 (d, J = 8,0Hz, 1H), 7,25 (d, J = 8,0Hz, 1H), 5,72 (s, 1H), 4,45 (s, 2h ), 3,93 (s, 3H), 4,65 (m, 4H), 3,52 (m, 2H), 3,18 (m, 2H).
Ejemplo F: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: terc-butil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)(hidroxi)metil)benzoato

A una solución de terc-butil 2,4-diclorobenzoato (Preparación #33, Etapa A) (748 mg, 3,03 mmol) en THF (20 mL) y enfriada a -78 °C se añadió diisopropilamida de litio (1,816 mL, 3,63 mmol) diluido en THF (10 mL) y ya enfriado a 0 °C. La mezcla de reacción se agitó a -78 °C durante 1 hora. A -78 °C, se añadió 1,4-dimetil-6-(trifluorometil)-1H-indol-3-carbaldehído (Preparación #51) (730 mg, 3,03 mmol) y la mezcla de reacción se agitó durante 15 minutos. Luego se dejó calentar a temperatura ambiente y la agitación continuó durante 30 minutos. La mezcla de reacción se inactivó con una solución saturada de NH4 CL La capa acuosa se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró hasta la sequedad a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-50% en ciclohexano) para dar terc-butil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)(hidroxi)metil)benzoato (355 mg, 23%) como un sólido naranja. LC/MS (Método i) Rt = 2,79 min.; MS miz: 546 [M-H]- + CH3 COOH 1H NMR (DMSO-d6, 300MHz): 5 7,64 (s, 1H), 7,58 (s, 2H), 7,13 (s, 1H), 6,98 (s, 1h ), 6,87 (d, J=5,6 Hz, 1H), 5,93 (d, J=5,6 Hz, 1H), 3,75 (s, 3H), 2,85 (s, 3H), 1,54 (s, 9H)
Etapa 2: ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoico (20 mg, 31 %) se preparó a partir de terc-butil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)(hidroxi)metil)benzoato (50 mg, 0,10 mmol ). Mezcla con la indolina correspondiente (30%) LC/MS (Método i) Rt = 2,56 min.; MS m/z: 416 [M+H]+
Etapa 3: metil 2-(1-(2,4-did oro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acetato (65 mg, 77%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(tri-fluorometil)-1H-indol-3-il)metil)benzoico (50 mg, 0,12 mmol) y clorhidrato de metil (4-piperidil)acetato (26 mg, 0,13 mmol). Mezcla con la indolina correspondiente (22%). LC/MS (Método i) Rt = 2,72 min.; MS m/z: 555 [M+H]+
Etapa 4: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acético
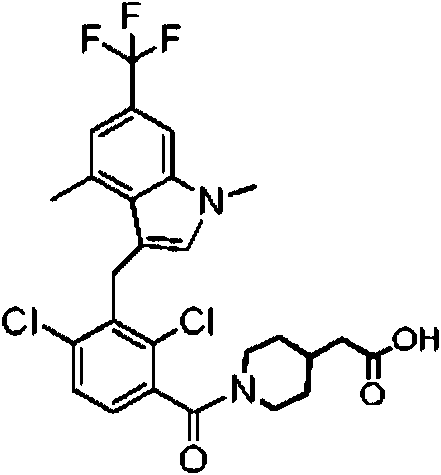
A una solución de metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acetato (65 mg, 0,117 mmol) en dioxano (1,25 mL) y agua (0,5 mL) se añadió hidróxido de sodio (11,70 mg, 0,293 mmol) y la mezcla de reacción se agitó a 45 °C durante 1 hora. La mezcla de reacción se concentró a presión reducida. El residuo se tomó en agua y se acidificó hasta pH = 3-4 mediante la adición de una solución de HCl 1N. El precipitado obtenido se lavó con agua y se secó a presión reducida. El residuo se purificó por LC/MS preparativa (Método 12) para dar ácido 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidin-4-il)acético (18 mg, 28%) como un sólido de color blanco. LC/MS (Método g) Rt = 1,92 min.; MS m/z: 541 [M+H]+.1H NMR (DMSO-d6, 400MHz): 5 12,12 (br, 1H), 7,63 (m, 2H), 7,39 y 7,32 (d, J= 8,1 Hz, 1H), 7,09 (s, 1H), 6,53 (m, 1H), 4,60 (m, 2H), 4,47 (m, 1H), 3,70 (m, 3H), 3,30 (m, 1H), 3,03 (m, 1H), 2,78 (m, 4H), 2,17 (m, 2H), 1,91 (m, 1H), 1,77 (m, 1H), 1,62 (m, 1H), 1,16 (m, 2H).
Tabla F. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-3-ilmetilbenzoico Eemlo F Etaa 2 usando el mismo rocedimiento con la amina aroiada.
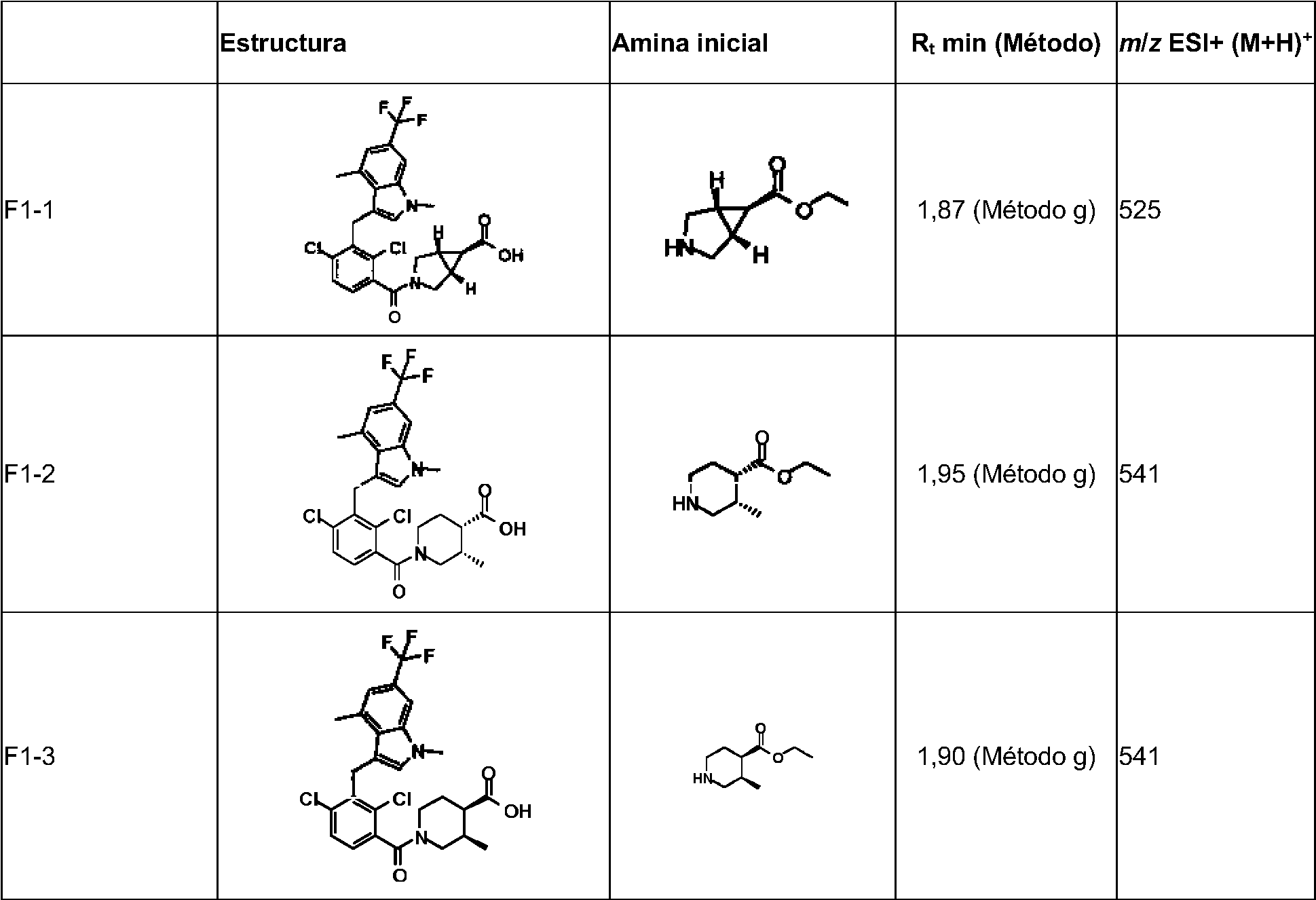
Ejemplo G: (3-((1,4-dimetM-6-(trifluorometM)-1H-mdol-2-M)metM)-2,4-dimetM-feml)(morfolmo)metanona

Ejemplo G1: (2,6-dimetM-3-(morfolma-4-carboml)feml)(1,4-dimetM-6-(trifluorometN)-1H-mdol-2-M)metanona

Etapa 1: (3-(hidroxi(4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-mdol-2-M)metM)-2,4-dimetMfeml) (morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (374 mg, 94%) se preparó a partir de W-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (300 mg, 0,680 mmol) y (3-(1 -hidroxiprop-2-in-1-il)-2,4-dimetilfenil)(morfolino)metanona (223 mg, 0,816 mmol) (Preparación #4). LC/MS (Método h) Rt = 3,05 min.; MS miz: 587 [M+H]+. 1H NMR (DMSO-da, 300 MHz): 58,08 (m, 1H), 7,98 (m, 2H), 7,85 (m, 1H), 7,68 (m, 1H), 7,60 (m, 2H), 7,48 (s, 1H), 7,06 (m, 2H), 6,75 (m, 1H), 6,25 (m, 1H), 3,63 (m, 4H), 3,46 (m, 2H), 3,12 (m, 2H), 2,89 (s, 3H), 2,73 (s, 3H), 2,45 (m, 3H).
Etapa 2: (2,4-dimetM-3-((4-metiM-(fenMsulfoml)-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 2, (2,4-dimetil-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (346 mg, 95%) se preparó a partir de (3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (374 mg, 0,638 mmol). lC/MS (Método h) Rt = 3,30 min.; MS miz: 571 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,29 (s, 1H), 7,96 (m, 2H), 7,76 (m, 1H), 7,68 (m, 2H), 7,41 (s, 1H), 7,17 (m, 1H), 7,08 (m, 1H), 5,73 (s, 1H), 4,25 (m, 2H), 3,62 (m, 4H), 3,44 (m, 2H), 3,10 (m, 2H), 2,30 (s, 3h ), 2,05 (s, 3h ), 1,90 (s, 3H).
Etapa 3: (2,4-dimetM-3-((4-metM-6-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-dimetil-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (198 mg, 76%) se preparó a partir de (2,4-dimetil-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (346 mg, 0,606 mmol). LC/MS (Método g) Rt = 1,80min.; MS m/z: 431 [M+H]+ 1H NMR (D M S O C 400 MHz): 5 11,39 (s, 1H), 7,46 (s, 1H), 7,17 (d, J = 8,0Hz, 1H), 7,02 (m, 2H), 5,81 (s, 1H), 4,19 (s, 2H), 3,64 (m, 4H), 3,48 (m, 2H), 3,13 (m, 2H), 2,38 (s, 3H), 2,30 (s, 3H), 2,15 (s, 3H).
Etapa 4: (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona y (2,6-dimetil-3-(morfolma-4-carboml)feml)(1,4-dimetM-6-(trifluorometM)-1H-mdol-2-M)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 4, (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil) (morfolino)metanona (30 mg, 19%) se preparó a partir de (2,4-dimetil-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (154 mg, 0,358 mmol). LC/MS (Método g) Rt = 1,87 min.; MS miz: 445[M+H]+.1H NMR (DMSO-cfe, 400 MHz): 57,69 (s, 1H), 7,19 (d, J = 8Hz, 1H), 7,06 (m, 2H), 5,51 (s, 1H), 4,17 (m, 2H), 3,91 (s, 3H), 3,65 (m, 4H), 3,48 (m, 2H), 3,13 (m, 2H), 2,33 (s, 3H), 2,25 (s, 3H), 2,11 (s, 3H).
Esta reacción también proporcionó un producto de oxidación que se aisló y caracterizó: (2,6-dimetil-3-(morfolina-4-carbonil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metanona (32 mg, 19%). LC/MS (Método g) Rt = 1,84 min.; MS m/z: 459[M+H]+.
1H NMR (DMSO-C6, 400 MHz): 5 7,95 (s, 1H), 7,26 (s, 2H), 7,22 (s, 1H), 6,82 (s, 1H), 4,24 (s, 3H), 3,62 (m, 4H), 3,48 (m, 2H), 3,16 (m, 2H), 2,44 (s, 3H), 2,17 (s, 3H), 2,00 (s, 3H).
Ejemplo H: (2,4-didoro-3-((4-metM-6-(trifluorometN)-1H-mdol-2-M)metM)feml)(4-(dimetNammo)pipe-ridm-1-il)metanona

Etapa 1: ácido 2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (280 mg, 82%) se preparó a partir de metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (Ejemplo A, Etapa 2) (350 mg, 0,629 mmol). LC/MS (Método h) Rt = 3,33 min.; MS m/z: 542 [M+H]+.1H NMR (DMSO-C6, 300 MHz): 5 8,25 (s, 1H), 7,92 (m, 2H), 7,75 (m, 1H), 7,67 (m, 3H), 7,58 (m, 1H), 7,41 (s, 1H), 5,93 (s, 1H), 4,58 (s, 2H), 2,35 (s, 3H).
Etapa 2: (2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino) piperidin-1-il)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifiuorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino)piperidin-1-il)metanona (305 mg, 72%) se preparó a partir de ácido 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H -indol-2-il)metil)benzoico (350 mg, 0,645 mmol) y N,N-dimetilpiperidin-4-amina (165 mg, 1,29 mmol). LC/MS (Método h) Rt = 3,34 min.; MS m/z: 652 [M+H]+.
1H NMR (DMSO-C6, 300 MHz): 58,25 (s, 1H), 7,92 (m, 2H), 7,75 (m, 1H), 7,66 (m, 3H), 7,48 (m, 2H), 5,98 (m, 1H), 4,50 (m, 2H), 4,41 (m, 1H), 2,98 (m, 1H), 2,80 (m, 1h ), 2,36 (s, 3H), 2,30 (m, 2H), 2,20 (s, 3H), 2,09 (s, 3H), 1,82 (m, 1h ), 1,64 (m, 1h ), 1,27 (m, 2h ).
Etapa 3: (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino)piperidin-1-il)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1 H-indol-2-il)metil)fenil)(4-(dimetilamino)piperidin-1-il)metanona (190 mg, 86%) se preparó a partir de (2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino) piperidin-1-il)metanona (280 mg, 0,429 mmol).
LC/MS (Método g) Rt = 1,22 min.; MS m/z: 512 [M+H]+. 1H NMR (DMSO-cfe, 400 MHz): 511,40 (s, 1H), 7,64 (m, 1H), 7,48 (s, 1H), 7,38 (m, 1H), 7,03 (s, 1H), 5,95 (s, 1H), 4,46 (m, 3H), 3,30 (m, 1H), 3,00 (m, 1H), 2,85 (m, 1h ), 2,41 (s, 3h ), 2,32 (m, 1h ), 2,16 (s, 3h ), 2,12 (s, 3h ), 1,82 (m, 1h ), 1,65 (m, 1h ), 1,32 (m, 2h ).
Ejemplo I: (2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)feml)(4-(dimetMami-no)pipendm-1 -iljmetanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino) piperidin-1-il)metanona (10 mg, 13%) se preparó a partir de (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-(dimetilamino) piperidin-1-il)metanona (Ejemplo H, Etapa 3) (75 mg, 0,146 mmol). LC/MS (Método g) Rt = 1,29 min.; MS m/z: 526 [M+H]+.1H NMR (DMSO-C6, 300 MHz): 5 7,70 (m, 1H), 7,65 (m, 1H), 7,43
(m, 1H), 7,07 (s, 1H), 5,68 (m, 1H), 4,46 (m, 3H), 3,92 (m, 3H), 2,99 (m, 1H), 2,85 (m, 1H), 2,36 (s, 3H), 2,32 (m, 1H), 2,17 (s, 3H), 2,11 (s, 3H), 1,83 (m, 1H), 1,68 (m, 1H), 1,32 (m, 2h ).
Ejemplo J: ácido 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: metil 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato

A una solución de 7-metil-5-(trifluorometil)-1H-indol (Preparación #49) (0,850 g, 4,27 mmol) en N,N-dimetil-formamida (10,67 mL) y se enfrió a 0 °C se añadió hidruro de sodio (0,188 g, 4,69 mmol) y la reacción se agitó 30 minutos. Se añadió metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (1,272 g, 4,27 mmol) y la mezcla de reacción se agitó 1 hora a 0 °C. La mezcla de reacción se diluyó con agua y EtOAc. Las capas se separaron y la acuosa se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua y luego con una solución saturada de NaCl, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar metil 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1- il)metil)benzoato (1,55 g, 80%). LC/MS (Método j) Rt = 2,32 min.; MS miz: 416 [M+H]+1H NMR (DMSO-d6, 300MHz): 5 7,89 (d, J=8,4 Hz, 1H), 7,79 (m, 1H), 7,76 (d, J=8,4 Hz, 1H), 7,23 (m, 1H), 6,74 (d, J=3,3 Hz, 1H), 6,56 (d, J=3,3 Hz, 1H), 5,98 (s, 2H), 3,88 (s, 3H), 2,94 (s, 3H).
Etapa 2: ácido 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico
Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil) benzoico (1,44 g, 96%) se preparó a partir de 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (1,55 g, 3,72 mmol). LC/MS (Método j) Rt = 1,86 min.; MS miz: 402 [M+H]+1H NMR (DMSO-de, 300MHz): 5 13,8 (br, 1H), 7,82 (d, J=8,3 Hz, 1H), 7,79 (s, 1H), 7,69 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,73 (d, J=3,3 Hz, 1H), 6,56 (d, J=3,3 Hz, 1H), 5,97 (s, 2H), 2,95 (s, 3H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil) benzoil)piperidin-4-il) acetato (118 mg, 88%) se preparó a partir de ácido 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (100 mg, 0,25 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (62,6, 0,32 mmol).
LC/MS (Método j) Rt = 2,10 min.; MS miz: 541 [M+H]+1H NMR (DMSO-d6, 300MHz): 5 7,79 (s, 1H), 7,70 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,76 (m, 1H), 6,57 (m, 1H), 5,94 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,21 (m, 1H), 3,03 (m, 1H), 2,94 (s, 3H), 2,80 (m, 1H), 2,28 (m, 2H), 1,96 (m, 1H), 1,75 (m, 1H), 1,59 (m, 1H), 1,17 (m, 2H)
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (99 mg, 89%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (114 mg, 0,21 mmol). LC/MS (Método g) Rt = 1,85 min.; MS miz: 527 [M+H]+
1H NMR (DMSO-d6, 400MHz): 5 12,10 (br, 1H), 7,79 (s, 1H), 7,70 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,76 (m, 1H), 6,58 (m, 1H), 5,94 (m, 2H), 4,46 (m, 1H), 3,27 (m, 1H), 3,03 (m, 1H), 2,94 (s, 3H), 2,80 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,14 (m, 2H).
Ejemplo K: (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(hidroxi)metil)-2,4-dimetilfenil)(morfolino)metanona

Ejemplo K1: (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(metoxi)metil)-2,4-dimetilfenil)(morfolino)metanona

Etapa 1: (3-(hidroxi(4-metM-6-(trifluorometM)-1H-mdol-2-M)metM)-2,4-dimetMfeml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (3-(hidroxi(4-metil-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (189 mg, 39%) se preparó a partir de (3-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (Ejemplo G, Etapa 1) (629 mg, 1,072 mmol). LC/MS (Método h) Rt = 2,66 min.; MS m/z: 447 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 511,32 (s, 1H), 7,52 (d, J = 4,8Hz, 1H), 7,12 (m, 1H), 7,03 (m, 2H) 6,38 (s, 1H), 6,24 (d, J = 4Hz, 1H), 5,94 (s, 1H), 3,62 (m, 4H), 3,49 (m, 2H), 3,136 (m, 2h ), 2,41 (s, 3H), 2,40 (s, 3H), 2,12 (s, 3H).
Etapa 2: (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(hidroxi)metil)-2,4-dimetilfenil)(morfolino)metanona y (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(metoxi)metil)-2,4-dimetilfenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(hidroxi)metil)-2,4-dimetilfenil)(morfolino)metanona (14 mg, 7%) se preparó a partir de (3-(hidroxi(4-metil-6-(trifluorometil)-1H-indol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (189 mg, 0,423 mmol). LC/m S (Método g) Rt = 1,70 min.; MS miz: 461 [M+H]+. 1H NMR (DMSO-cfe, 500 MHz): 57,68 (s, 1H), 7,14 (m, 1H), 7,07 (m, 2H), 6,39 (s, 1H), 6,10 (s, 1h ), 5,96 y 5,92 (s, 1H), 3,86 y 3,82 (s, 3H), 3,63 (m, 4H), 3,48 (m, 2H), 3,16 (m, 2H), 2,39 y 2,38 (s, 3H), 2,34 y 2,31 (s, 3H), 2,21 y 2,19 (s, 3H). Durante esta reacción un producto de doble metilación también se aisló y se caracterizó como (3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)(metoxi)metil)-2,4-dimetilfenil)(morfolino)metanona (0,5 mg, 0,2%). LC/MS (Método g) Rt = 1,85 min.; MS miz: 475 [M+H]+. 1H NMR (DMSOCs, 500 MHz): 57,72 (s, 1H), 7,20 (m, 1H), 7,14 (m, 1H), 7,08 (s, 1H), 6,05 (s, 1H), 5,80 (s, 1H), 3,91 (s, 3H), 3,65-3,48 (m, 6H), 3,33 (s, 3H), 3,12 (m, 2H), 2,36 (s, 3H), 2,32 (s, 3H), 2,19 (s, 3H).
Ejemplo L: (2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-hidroxipiperidin-1-il)metanona

Etapa 1: metil 2,4-didoro-3-(hidroxi(1-(femlsulfoml)-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (7,8 g, 78%) se preparó a partir de W-(2-yodo-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #29) (7,00 g, 16,4 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (4,7 g, 18,1 mmol).
LC/MS (Método h) Rt = 3,27 min.; MS m/z: 540 [M-H]"+CHsCOOH.1H NMR (DMSO-C6, 300 MHz): 5 8,24 (s, 1H), 7,85 (m, 3H), 7,69 (m, 2H), 7,58 (m, 4H), 7,03 (m, 1H), 6,84 (s, 1H), 6,73 (m, 1H), 3,87 (s, 3H).
Etapa 2: metil 2,4-didoro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-dicloro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (6,3 g, 90%) se preparó a partir de metil 2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(tri-fluorometil)-1H-indol-2-il)metil)benzoato (7,15 g, 12,8 mmol). LC/MS (Método h) Rt = 3,73 min.; MS m/z: 540 [M-H]-. 1H NMR (CDCla, 300 MHz): 5 8,45 (s, 1H), 7,80 (m, 2H), 7,63 (d, J = 9Hz, 1H), 7,51 (m, 1H), 7,38 (m, 5H), 5,68 (s, 1H), 4,63 (m, 2H), 3,86 (s, 3H).
Etapa 3: metil 2,4-didoro-3-((6-(trifluorometN)-1H-mdol-2-N)metN)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-dicloro-3-((6-(trifluorometil)-1H-indol-2-il)metil)benzoato (4,3 g, 93%) se preparó a partir de metil 2,4-dicloro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (6,2 g, 11,4 mmol).
LC/MS (Método h) Rt = 3,23 min.; MS m/z: 402 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 5 11,5 (ancho, 1H), 7,75 (d, J = 9Hz, 1H), 7,69 (d, J = 9Hz, 1H), 7,65 (s, 1H), 7,57 (d, J = 6Hz, 1H), 7,23 (dd, J = 6Hz, J = 1,5Hz, 1H), 5,96 (s, 1H), 4,50 (s, 2H), 2,87 (s, 3H).
Etapa 4: metil 2,4-didoro-3-((1-metN-6-(trifluorometM)-1H-mdol-2-N)metN)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 4, metil 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-1-il)metil)benzoato (3,86 g, 89%) se preparó a partir de metil 2,4-dicloro-3-((6-(trifluorometil)-1H-indol-2-il)metil)benzoato (4,17 g, 10,37 mmol). LC/MS (Método g) Rt = 2,06 min.; MS m/z: 416 [M+H]+.1H NMR (DMSO-C6, 300 MHz): 5 7,87 (s, 1H), 7,79 (d, J = 9Hz, 1H), 7,71 (d, J = 9Hz, 1H), 7,55 (d, J = 9Hz, 1H), 7,24 (d, J = 9Hz, 1H), 5,68 (s, 1H), 4,50 (s, 2H), 3,94 (s, 3H), 3,88 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((1-metM-6-(trifluorometN)-1H-mdol-2-M)metM)benzoico
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (1,53 g, 87%) se preparó a partir de metil 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (1,81 g, 4,35 mmol).
LC/MS (Método h) Rt = 3,08 min.; MS m/z: 402 [M+H]+.
1H NMR (D M S O C 300 MHz): 57,87 (s, 1H), 7,74 (d, J = 9Hz, 1H), 7,67 (d, J = 9Hz, 1H), 7,56 (d, J = 9Hz, 1H), 7,24 (dd, J = 9Hz, J = 1,5Hz, 1H), 5,68 (s, 1H), 4,49 (s, 2H), 3,94 (s, 3h ).
Etapa 6: (2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(4-hidroxipiperidin-1-il)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-didoro-3-((1-metil-6-(trifluorometil)-1 H-indol-2-il)metil)fenil)(4-hidroxipiperidin-1-il)metanona (65 mg, 53%) se preparó a partir de ácido 2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (100 mg, 0,249 mmol) y piperidin-4-ol (37,7 mg, 0,37 mmol). LC/MS (Método g) Rt = 1,70 min.; MS m/z: 485 [M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 5 7,87 (s, 1H), 7,65 (dd, J = 6,3Hz, J = 1,8Hz, 1H), 7,56 (d, J = 6,3Hz, 1H), 7,41 (m, 1H), 7,25 (d, J = 6Hz, 1H), 5,70 (m, 1H), 4,80 (m, 1h ), 4,38 (m, 2h ), 4,05 (m, 1H), 3,96 (s, 3H), 3,73 (m, 1H), 3,20 (m, 2H), 3,07 (m, 1 h ), 1,81 (m, 1H), 1,75 (m, 1H), 1,39 (m, 2H).
Tabla L. Los siguientes análogos se prepararon a partir de ácido 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-ilm il nz i E m l L E n l mi m r imi n n l min r i .

Ejemplo M: ácido 1-(2,4-dicloro-3-((1-metN-6-(trifluorometM)-1H-mdol-2-N)metN)benzoN)piperidma-4-carboxíNco

Etapa 1: metil 1-(2,4-didoro-3-((1-metM-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoM)pipendma-4-carboxMato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 1-(2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato (172 mg, 73%) se preparó a partir de ácido 2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (Ejemplo L, Etapa 5) (180 mg, 0,448 mmol) y metil 4-piperidinacarboxilato (96 mg, 0,67 mmol). LC/MS (Método h) Rt = 3,17 min.; MS m/z: 527 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,81 (s, 1H), 7,65 (d, J = 8,4Hz, 1H), 7,56 (d, J = 7,8Hz, 1H), 7,46 (d, J = 8,4Hz, 0,5H), 7,39 (d, J = 8,4Hz, 0,5H), 7,26 (d, J = 7,8Hz, 1H), 5,72 y 5,69 (s, 1h ), 4,46 (m, 2H), 4,38 (m, 1h ), 3,93 (s, 3H), 3,62 y 3,58 (s, 3H), 3,27 (m, 1H), 3,13-2,94 (m, 2H), 2,67 (m, 1H), 1,82 (m, 2h ), 1,52 (m, 2h ).
Etapa 2: ácido 1-(2,4-didoro-3-((1-metN-6-(trifluorometM)-1H-mdol-2-M)metM)benzoN)piperidma-4-carboxNico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxílico (154 mg, 93%) se preparó a partir de metil 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato (170 mg, 0,32 mmol). LC/MS (Método g) Rt = 1,73 min.; MS m/z: 513 [M+H]+. 1H NMR (D M S O C 400 MHz): 512,32 (ancho, 1H), 7,87 (s, 1H), 7,65 (d, J = 8,4Hz, 1H), 7,56 (d, J = 8,4Hz, 1H), 7,46 (d, J = 8,4Hz, 0,5H), 7,39 (d, J = 8,4Hz, 0,5H), 7,24 (d, J = 8,4Hz, 1H), 5,71 (m, 1H), 4,51 (m, 2H), 4,45 (m, 1H), 3,93 (s, 3H), 3,29 (m, 1H), 3,12 (m, 1H), 2,95 (m, 1H), 2,54 (m, 1H), 1,95 (m, 1H), 1,76 (m, 1H), 1,50 (m, 2H).
Ejemplo N: ácido 1-(2,4-didoro-3-((1-metM-6-(trifluorometN)-1H-mdol-2-M)metM)benzoN)-3-hidroxipiperidma-4-carboxílico
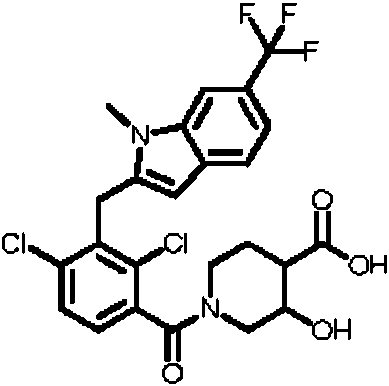
Etapa 1: etil 1-(2,4-didoro-3-((1-metM-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoM)-3-oxopipendma-4-carboxMato

Usando un procedimiento similar al Ejemplo A, Etapa 6, etil 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-oxopiperidina-4-carboxilato (221 mg, 59%) se preparó a partir de 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (Ejemplo L, Etapa 5) (250 mg, 0,62 mmol) y clorhidrato de etil 3-oxopiperidina-4-carboxilato (194 mg, 0,93 mmol). LC/MS (Método h) Rt = 3,52 min.; MS m/z: 555 [M+H]+. 1H NMR (DMSO-ds, 300 MHz): 7,87 (s, 1H), 7,69 (m, 1H), 7,65 (m, 1H), 7,46 (m, 1H), 7,26 (m, 1H), 5,74 (s, 1H), 5,73 y 5,69 (s, 1H), 4,48 (s, 2H), 4,30 (m, 1H), 4,17 (m, 2H), 3,92 (m, 3H), 3,51 (m,2H), 3,32 (m, 1H), 3,30 (m, 2H), 1,20 (m, 3H).
Etapa 2: etil 1-(2,4-didoro-3-((1-metM-6-(trífluorometN)-1H-mdol-2-N)metM)benzoM)-3-hidroxipiperidma-4-carboxilato:

A una suspensión de etil 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-oxo-piperidina-4-carboxilato (218 mg, 0,393 mmol) en EtOHMeoH (4 mL) enfriada a 0 °C se añadió NaBH4 (7,43 mg, 0,196 mmol) y la mezcla se agitó a 5 °C durante 4 horas. Se añadió nuevamente NaBH4 (7,43 mg, 0,196 mmol) y la reacción se agitó durante 5 horas a 5 °C. La reacción se hidrolizó con solución acuosa saturada de NH4Cl y se extrajo con DCM. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-70% en ciclohexano) para dar etil 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil) benzoil)-3-hidroxipiperidina-4-carboxilato (150 mg, 69%). LC/MS (Método g) Rt = 1,85 min.; MS m/z: 557 [M+H]+. 1H NMR (DMSO-ds, 400 MHz): 57,87 (s, 1H), 7,69 (m, 1H), 7,51 (m, 1H), 7,38 (m, 1H), 7,25 (m, 1H), 5,72 (m, 1H), 5,40 y 5,22 y 5,13 y 5,00 y 4,82 (m, 1H), 4,52-4,25 (m, 3H), 4,22-4,00 (m, 3H), 3,94 (s, 3H), 3,60 (m, 0,5H), 3,38-3,18 (m, 0,5H), 3,10-2,55 (m, 2,5H), 2,45 (m, 0,5H), 2,00-1,40 (m, 2H), 1,18 (m, 3H).
Etapa 3: ácido 1-(2,4-didoro-3-((1-metM-6-(trífluorometN)-1H-mdol-2-N)metM)benzoM)-3-hidroxipiperidma-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-hidroxipiperidina-4-carboxílico (73 mg, 66%) se preparó a partir de etil 1-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-hidroxipiperidina-4-carboxilato (Ejemplo M, Etapa 2) (116 mg, 0,208 mmol). LC/MS (Método g) Rt = 2,60 min.; MS m/z: 527 [M-H]-.1H NMR (DMSO-ds, 400 MHz): 57,87 (s, 1H), 7,68 (m, 1H), 7,58 (m, 1H), 7,38 (m, 1H), 7,23 (m, 1H), 5,70 (m, 1H), 4,47 (m, 3H), 4,19 y 4,02 (m, 1H), 3,93 (s, 3H), 3,20 (m, 1H), 3,09 (m, 1H), 2,70 (m, 2H), 1,85 (m, 1H), 1,63 y 1,50 (m, 1H).
Ejemplo O: ácido 3-(4-(2,4-didoro-3-((1-metM-6-(trifluorometM)-1H-mdol-2-M)metN)benzoN)piperazm-1-il)ciclobutanocarboxílico

Etapa 1: íerc-butil 4-(2,4-didoro-3-((1-metM-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoM)piperazma-1-carboxMato

Usando un procedimiento similar al Ejemplo A, Etapa 6, íerc-butil 4-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil) benzoil)piperazina-1-carboxilato (840 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (Ejemplo L, Etapa 5) (500 mg, 1,243 mmol) y 1-boc-piperazina (347 mg, 1,86 mmol). LC /MS (Método h) Rt = 3,42 min.; MS m/z: 570 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,88 (s, 1H), 7,68 (d, J = 9Hz, 1H), 7,56 (d, J = 9Hz, 1H), 7,46 (d, J = 9Hz, 1H), 7,26 (J = 9Hz, 1H), 5,74 (s, 1H), 4,46 (s, 2H), 3,94 (s, 3H), 3,60 (m, 2H), 3,42 (m, 2H), 3,20 (m, 4H), 1,40 (s, 9H).
Etapa 2: (2,4-didoro-3-((1-metM-6-(tnfluorometM)-1H-mdol-2-M)metM)feml)(piperazm-1-M)metanona

A una solución de íerc-butil 4-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)pipera-zina-1-carboxilato (840 mg, 1,473 mmol) en DCM (32 mL) y enfriada a 0 °C se añadió TFA (8 mL, 104 mmol). El baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 1,5 horas. La mezcla de reacción se concentró a presión reducida. El producto crudo se diluyó con DCM y se lavó con solución acuosa saturada de NaHCO3. La capa orgánica se lavó con agua, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (850 mg, 100%) como un polvo naranja. LC/MS (Método h) Rt = 2,07 min.; MS m/z: 470 [M+H]+. 1H NMR (CDCla, 300 MHz): 57,51 (s, 1H), 7,40 (m, 2H), 7,19 (m, 2H), 5,73 (s, 1H), 4,36 (s, 2H), 3,81 (s, 3H), 3,72 (m, 2H), 3,16 (m, 2H), 2,89 (m, 2H), 2,73 (m, 2H).
Etapa 3: metil 3-(4-(2,4-didoro-3-((1-metM-6-(tnfluorometM)-1-H-mdol-2-M)metM)benzoM)piperazm-1-il)ciclobutanocarboxilato

A una solución de (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (50 mg, 0,106 mmol) y metil 3-oxociclobutanocarboxilato (31,8 mg, 0,149 mmol) en 1,2-dicloroetano (1,4 mL) se añadió ácido acético
(50 |_il, 0,873 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 45 minutos. Luego se añadió triacetoxiborohidruro de sodio (33,8 mg, 0,159 mmol) y la agitación continuó durante 2,5 horas. La mezcla de reacción se concentró a presión reducida para dar 3-(4-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxilato (80 mg, 100%). LC/MS (Método h) Rt = 2,46 min.; MS m/z: 582 [M+H]+. 1H NMR (CDCla, 300 MHz): 57,51 (s, 1H), 7,41 (m, 2H), 7,18 (m, 2H), 5,72 (s, 1H), 4,36 (s, 2H), 3,81 (s, 3H), 3,75 (m, 2H), 3,60 (s, 3H), 3,20 (m, 2H), 2,68 (m, 2H), 2,36-2,05 (m, 8H).
Etapa 4: ácido 3-(4-(2,4-didoro-3-((1-metM-6-(trifluorometN)-1H-mdol-2-N)metN)benzoM)piperazm-1-il)ciclobutanocarboxílico

A una solución de metil 3-(4-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piper-azin-1-il) ciclobutanocarboxilato (200 mg, 0,343 mmol) en dioxano (3,75 mL) y agua (1,5 mL) se añadió hidróxido de sodio (54,9 mg, 1,374 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora y se concentró a presión reducida. El residuo se tomó en agua y se acidificó hasta pH = 3-4 mediante la adición de una solución de HCl 1N. Después de la extracción con EtOAc, la capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH al 0-10% en DCM) para dar ácido 3-(4-(2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H -indol-2-il)metil) benzoil)piperazin-1-il)ciclobutanocarboxílico (115 mg, 57%). LC/MS (Método g) Rt = 1,30 min., 1,33min; MS m/z: 568 [M+H]+. 1H n Mr (DMSO-da, 400 MHz): 57,87 (s, 1H), 7,65 (d, J = 8Hz, 1H), 7,56 (d, J = 8,4Hz, 1H), 7,41 (d, J = 8Hz, 1H), 7,25 (d, J = 8,4Hz, 1H), 5,71 (s, 1H), 4,49 (s, 2H), 3,93 (s, 3H), 3,62 (m, 2H), 3,17 (m, 2h ), 2,69 (m, 2H), 2,31 (m, 2H), 2,21 (m, 4H), 1,90 (m, 2h ).
Ejemplo P: (2,4-didoro-3-((1-metM-5-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Etapa 1: (2,4-didoro-3-(hidroxi(1-(femlsulfoml)-5-(trifluorometM)-1H-mdol-2-M)metM)feml) (morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (850 mg, 74%) se preparó a partir de N-(2-yodo-4-(trifluorometil)fenil)bencenosulfonamida (descrito en la patente FR2890071) (800 mg, 1,873 mmol) y (2,4-dicloro -3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (647 mg, 2,060 mmol) (Preparación #3). LC/MS (Método h) Rt = 2,92 min.; MS m/z: 613 [M+H]+.1H NMR (DMSO-d6, 300 MHz): 5 8,01 (m, 1H), 7,95 (m, 2H), 7,91 (m, 1H), 7,65 (m, 4H), 7,53 (m, 1H), 7,40 (m, 1H), 6,95 (m, 1H), 6,77 y 6,71 (s, 1H), 6,69 (m, 1H), 3,64 (m, 4h ), 3,52 (m, 2H), 3,16 (m, 2H).
Etapa 2: (2,4-dicloro-3-((1-(fenilsulfonil)-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 2, (2,4-dicloro-3-((1-(fenilsulfonil)-5-(trifluorometil)-1H -indol-2-il)metil)fenil)(morfolino)metanona (540 mg, 58%) se preparó a partir de (2,4-dicloro-3-(hidroxi(1-(fenil-sulfonil)-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (840 mg, 1,369 mmol). LC/MS (Método h) Rt = 3,33 min.; MS miz: 597 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,33 (d, J = 8,4Hz, 1H), 7,98 (m, 2H), 7,90 (s, 1H), 7,77 (m, 1H), 7,67 (m, 4H), 7,49 (d, J = 8,1Hz, 1H), 6,03 (s, 1H), 4,59 (m, 2H), 3,63 (m, 4H), 3,51 (m, 2H), 3,17 (m, 2H).
Etapa 3: (2,4-didoro-3-((5-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3 (2,4-dicloro-3-((5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (230 mg, 69%) se preparó a partir de (2,4-dicloro-3-((1-(fenilsulfonil)-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (500 mg, 0,837 mmol). LC/MS (Método h) Rt = 2,86 min.; Ms miz:
457 [M+H]+.1H NMR (DMSO-C6, 300 MHz): 5 11,55 (ancho, 1H), 7,78 (s, 1H), 7,65 (d, J = 8,1Hz, 1H), 7,50 (d, J = 8,7Hz, 1H), 7,41 (d, J = 8,7Hz, 1H), 7,30 (d, J = 8,1Hz, 1H), 5,97 (s, 1H), 4,44 (s, 2H), 3,65 (m, 4H), 3,52 (m, 2H), 3,17 (m, 2H).
Etapa 4: (2,4-didoro-3-((1-metM-5-(trifluorometM)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

A una solución de (2,4-dicloro-3-((5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (100 mg, 0,219 mmol) en ACNACN (2 mL) se añadió sulfato de dimetilo (0,031 mL, 0,328 mmol) y carbonato de cesio (143 mg, 0,437 mmol). La mezcla se agitó a temperatura ambiente durante 24 horas, luego se diluyó con agua y se extrajo con DCM. La capa orgánica se lavó con salmuera y se secó sobre sulfato de magnesio. El residuo se purificó por LC/MS preparativa para dar (2,4-dicloro-3-((1 -metil-5-(trifluorometil)-1 H-indol-2-il)metil)fenil)(morfolino)metanona (50 mg, 45%). lC/MS (Método g) Rt = 1,78 min.; MS m/z: 471 [M+H]+.1H NMR (D M S O C 400 MHz): 5 7,78 (s, 1H), 7,66 (m, 2H), 7,45 (d, J = 8,4Hz, 1H), 7,38 (d, J = 8,8Hz, 1H), 5,75 (s, 1H), 4,44 (s, 2H), 3,91 (s, 3H), 3,66 (m, 4H), 3,55 (m, 2H), 3,17 (m, 2h ).
Ejemplo Q: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)indolin-2-il)metil)fenil)(morfolino)metanona

Etapa 1: (2,4-didoro-3-((4-metil-6-(trifluorometil)indolin-2-il)metil)fenil)(morfolino)metanona

A una solución de (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (Ejemplo C) (256 mg, 0,543 mmol) en TFA (4 mL) se añadió cianoborohidruro de sodio (170 mg, 2,71 mmol) por fracción. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas y se añadió una solución acuosa de NaOH 1N (pH 7). La capa acuosa se extrajo con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por LC/MS preparativa para dar (2,4-dicloro-3-((4-metil-6-(trifluorometil)indolin-2-il)metil)fenil)(morfolino)metanona (82 mg, 43 %) como un sólido amarillo. LC/MS (Método h) Rt = 3,08 min.; MS m/z: 473 [M+H]+. 1H NMR (DMSO-de, 300 MHz): 57,58 (d, J = 9Hz, 1H), 7,32 (J = 9Hz, 1H), 6,67 (s, 1H), 6,49 (dm, 1H), 6,14 (m, 1H), 4,22 (m, 1H), 3,65 (m, 4H), 3,54 (m, 2H), 3,16 (m, 4H), 2,90 (m, 1H), 2,72 (m, 1H), 2,17 (s, 3H).
Etapa 2: (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil) indolin-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)indolin-2-il)metil)fenil)(morfolino)metanona (5 mg, 20%) se preparó a partir de (2,4-dicloro-3-((4-metil-6-(trifluorometil)indolin-2-il)metil)fenil)(morfolino)metanona (24 mg, 0,051 mmol). LC/MS (Método g) Rt = 1,97 min.; MS m/z: 487 [M+H]+. 1H NMR (DMSO-d6, 400 MHz): 57,61 (d, J = 8,4Hz, 1H), 7,35 (d, J = 8,4Hz, 1H), 6,76 (s, 1H), 6,57 (s, 1H), 3,90 (m, 1H), 3,69 (m, 4H), 3,54 (m, 2H), 3,40 (m, 1H), 3,13 (m, 3H), 2,93 (m, 1H), 2,82 (m, 3H), 2,72 (m, 1H), 2,14 (s, 3H).
Ejemplo R: ácido 2-(1-(2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((4-doro-6-dano-1-metiMH-indol-2-M)metM)benzoM)piperidin-4-M)acetato

Usando un procedimiento similar al Ejemplo A1, se preparó metil 2-(1-(2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (101 mg, 83%) a partir de ácido 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoico (Ejemplo DQ, Etapa 6) (90 mg, 0,23 mmol) y clorhidrato de metil (4-piperidil)acetato (66,4 mg, 0,34 mmol).
LC/MS (Método i) Rt = 2,43 min.; MS m/z: 532 [M+H]+ 1H NMR (DMSO-cfe, 300MHz): 5 8,17 (m, 1H), 7,68 y 7,67 (d, J=8,4 Hz, 1H), 7,51 y 7,50 (s, 1H), 7,47 y 7,40 (d, J=8,4 Hz, 1H), 5,68 y 5,65 (m, 1H), 4,47 (m, 3H), 3,96 y 3,95 (s, 3H), 3,59 y 3,56 (s, 3H), 3,25 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,25 (m, 2h ), 1,95 (m, 1H), 1,75 (m, 1H), 1,59 (m, 1H), 1,17 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, se preparó ácido 2-(1-(2,4-dicloro-3-((4-doro-6-dano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (93 mg, 95%) a partir de metil 2-(1-(2,4-dicloro-3-((4-doro-6-dano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (101 mg, 0,19 mmol). LC/MS (Método g) Rt = 1,63 min.; MS m/z: 518 [M+H]+ 1H NMR (DMSO-cfe, 400MHz): 5 12,09 (m, 1H), 8,17 (s, 1H), 7,68 y 7,67 (d, J=8,4 Hz, 1H), 7,50 (m, 1H), 7,46 y 7,39 (d, J=8,4 Hz, 1H), 5,68 y 5,66 (m, 1H), 4,49 (m, 3H), 3,96 y 3,95 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,15 (m, 2H), 193 (m, 1H), 1,77 (m, 1h ), 1,61 (m, 1H), 1,15 (m, 2H).
Ejemplo S: ácido 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(tnfluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxílico
Etapa 1: ferc-butil 4-(2,4-dicloro-3-((1,4-dimetil-6-(tnfluorometil)-1H-indol-2-il)metil)benzoil)piperazina-1-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, ferc-butil 4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazina-1-carboxilato (700 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (Ejemplo A, Etapa 5) (500 mg, 1,201 mmol) y 1-boc-piperazina (336 mg, 1,8 mmol). LC/MS (Método h) Rt = 3,49 min.; MS m/z: 584 [M+H]+ 1H NMR (CDCl3, 300 MHz): 57,49 (d, J = 9Hz, 1H), 7,43 (s, 1H), 7,26 (d, J = 9Hz, 1H), 7,09 (s, 1H), 5,76 (s, 1H), 4,44 (m, 2H), 3,87 (s, 3H), 3,75 (m, 2H), 3,53 (m, 2H), 3,39 (m, 2H), 3,22 (m, 2H), 2,43 (s, 3H), 1,46 (m, 9H).
Etapa 2: (2,4-dicloro-3-((1,4-dimetil-6-(tnfluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona

Usando un procedimiento similar al del Ejemplo O, Etapa 2, (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (535 mg, 92%) se preparó a partir de íerc-butil 4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazina-1-carboxilato (700 mg, 1,198 mmol). LC/Ms (Método h) Rt = 2,73 min.; MS m/z: 483 [M+H]+ 1H NMR (CDCla, 300 MHz): 57,49 (d, J = 9Hz, 1H), 7,43 (s, 1H), 7,26 (d, J = 9Hz, 1H), 7,08 (s, 1H), 5,78 (s, 1H), 4,43 (m, 2H), 3,87 (s, 3H), 3,80 (m, 2H), 3,23 (m, 2H), 2,95 (m, 2H), 2,88 (m, 1H), 2,80 (m, 1h ), 2,42 (s, 3h ).
Etapa 3: íerc-butil 3-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometN)-1H-mdol-2-N)metN)benzoM)piperazm-1-il)cidobutanocarboxilato

Usando un procedimiento similar al del Ejemplo O, Etapa 3, íerc-butil 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxilato (105 mg, 80%) se preparó a partir de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2 -il)metil)fenil)(piperazin-1-il)metanona (100 mg, 0,206 mmol) y íerc-butil 3-oxociclobutanocarboxilato (52,7 mg, 0,310 mmol). lC/MS (Método h) Rt = 3,39 min.; MS m/z: 638 [M+H]+1H NMR (DMSO-da, 300 MHz): 57,70 (s, 1H), 7,67 (d, J = 9Hz, 1H), 7,42 (d, J = 9Hz, 1H), 7,06 (s, 1H), 5,67 (s, 1H), 4,46 (m, 2H), 3,92 (s, 3H), 3,60 (m, 2H), 3,15 (m, 2h ), 2,70 (m, 2H), 2,28 (s, 3H), 2,20 (m, 6H), 2,17 (m, 2H), 1,39 (s, 9H).
Etapa 4: ácido 3-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometN)-1H-mdol-2-N)metN)benzoM)piperazm-1-il)ddobutanocarboxNico:
A una solución de íerc-butil 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxilato (105 mg, 0,164 mmol) en DCM (3,5 mL) y enfriada a 0 °C se añadió TFA (0,8 mL, 10,38 mmol). El baño de hielo se retiró y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se concentró a presión reducida. El producto crudo se diluyó con DCM y se lavó con solución acuosa saturada de NaHCO3. La capa orgánica se lavó con agua, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El crudo se tomó en agua y el pH se ajustó a 7. La capa acuosa se extrajo con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar ácido 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxílico (45 mg, 45,8%). LC/MS (Método g) Rt = 1,38 min.; MS m/z: 582 [M+H]+1H NMR (Piridina-d5, 500 MHz): 57,66 (s, 1H), 7,53 (d, J = 8Hz, 1H), 7,44 (d, J = 8Hz, 1H), 7,18 (s, 1H), 6,10 (s, 1H), 4,48 (m, 2H), 3,90 (m, 2H), 3,76 (s, 3H), 3,32 (m, 2H), 2,98 (m, 1H), 2,56 (m, 1h ), 2,32 (m, 8H), 2,26 (s, 3H).
Ejemplo T: ácido 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)-2-metilpropanoico

Etapa 1: etil 2-(4-(2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoM)piperazm-1-M)-2-metilpropanoato

A una solución de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (Ejemplo S, Etapa 2) (50 mg, 0,103 mmol) y 2-bromoisobutirato de etilo (60,4 mg, 0,310 mmol) en DMF (0,3 mL) se añadió carbonato de potasio (42,8 mg, 0,310 mmol) y la mezcla de reacción se agitó a 60 °C por la noche. Se añadieron carbonato de potasio (3 eq) y 2-bromoisobutirato de etilo (3 eq) y la agitación continuó durante 24 horas. La mezcla de reacción se diluyó con agua y EtOAc. La capa acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 20-80% en ciclohexano) para dar etil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)-2-metilpropanoato (30 mg, 49%).
LC/MS (Método h) Rt = 3,35 min.; MS miz: 598 [M+H]+
Etapa 2: ácido 2-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometN)-1H-mdol-2-N)metN)benzoN)piperazm-1-M)-2-metilpropanoico

Usando un procedimiento similar al del Ejemplo O, Etapa 4, ácido 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil) benzoil)piperazin-1-il)-2-metilpropanoico (60 mg, 96%) se preparó a partir de etil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)-2-metilpropanoato (60 mg, 0,100 mmol). LC/MS (Método g) Rt = 1,59 min.; MS miz. 570 [M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 5 7,70 (s, 1H), 7,66 (d, J = 8Hz, 1H), 7,47 (d, J = 8Hz, 1H), 7,06 (s, 1H), 5,68 (s, 1H), 4,49 (m, 2H), 3,90 (s, 3H), 3,65 (m, 2H), 3,18 (m, 2H), 2,62 (m, 2H), 2,58 (m, 2H), 2,36 (s, 3H), 1,19 (s, 6H).
Ejemplo U: ácido 2-(4-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-N)metN)benzoM)piperazm-1-il)propanoico

Etapa 1: etil 2-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometN)-1H-mdol-2-N)metN)benzoN)piperazm-1-M)propanoato

Usando un procedimiento similar al del Ejemplo T, Etapa 1, etil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)propanoato (43 mg, 66%) se preparó a partir de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (Ejemplo S, Etapa 2) (50 mg, 0,103 mmol) y 2-bromopropionato de etilo (28 mg, 0,155 mmol). LC/MS (Método k) Rt = 3,05 min.; MS miz: 584 [M+H]+ 1H NMR (cDch, 300 MHz): 57,48 (d, J = 9Hz, 1H), 7,43 (s, 1H), 7,25 (d, J = 9Hz, 1H), 7,08 (s, 1H), 5,78 (s, 1H), 4,42 (m, 2H), 4,14 (m, 2H), 3,86 (s, 3H), 3,84 (m, 2H), 3,28 (m, 3H), 2,71 (m, 4H), 2,43 (s, 3h ), 1,28 (m, 6H).
Etapa 2: ácido 2-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-M)metM)benzoM)piperazm-1-il)propanoico

Usando un procedimiento similar al del Ejemplo O, Etapa 4, ácido 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)propanoico (45 mg, 53%) se preparó a partir de etil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H -indol-2-il)metil)benzoil)piperazin-1-il) propanoato (85 mg, 0,145 mmol). LC/MS (Método g) Rt = 1,52 min.; MS miz: 556 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 57,70 (s, 1H), 7,66 (d, J = 8Hz, 1H), 7,40 (d, J = 8Hz, 1H), 7,06 (s, 1H), 5,67 (s, 1H), 4,46 (m, 2H), 3,92 (s, 3H), 3,63 (m, 3H), 3,16 (m, 4h ), 2,57 (m, 2H), 2,36 (s, 3H), 1,73 (m, 3H).
Ejemplo V: ácido 1-(2,4-didoro-3-((1,5-dimetM-6-(trifluorometN)-1H-mdol-2-N)metN)benzoN)piperidma-4-carboxNico

Etapa 1: metil 2,4-dicloro-3-(hidroxi(5-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-(hidroxi(5-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (1 g, 96%) se preparó a partir de W-(2-yodo-4-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #17) (800 mg, 1,813 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (564 mg, 2,176 mmol). LC/MS (Método h) Rt = 3,36 min.; MS miz: 630 [M-H]- +CH3 COOH 1H NMR (DMSO-ds, 300 MHz): 5 8,29 (s, 1H), 7,79 (m, 2H), 7,68-7,53 (m, 6H), 6,97 (d, J = 3Hz, 1H), 6,77 (s, 1H), 6,72 (d, J = 8Hz, 1H), 3,86 (s, 3H), 2,46 (s, 3H).
Etapa 2: metil 2,4-did oro-3-((5-metiM-(femlsulfonM)-6-(trifluorometM)-1H-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-didoro-3-((5-metil-1-(fenilsulfonil)-6-(trifluorometil)-1 H-indol-2-il)metil)benzoato (841 mg, 87%) se preparó a partir de metil 2,4-dicloro-3-(hidroxi(5-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil) (1 g, 1,747 mmol). LC/MS (Método h) Rt = 3,80 min.; MS miz: 556 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 58,38 (s, 1H), 7,90 (d, J = 8Hz, 1H), 7,79 (m, 3H), 7,69 (m, 3H), 7,50 (s, 1H), 5,88 (s, 1H), 4,58 (s, 2H), 3,87 (s, 3H), 2,44 (s, 3H).
Etapa 3: metil 2,4-did oro-3-((5-metN-6-(trifluorometM)-1H-mdol-2-N)metN)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, 179enzo 2,4-didoro-3-((5-metil-6-(trifluorometil)-1H-indol-1-il)179enzo)179enzoate (420 mg, 67%) se 179enzoat a partir de 179enzo 2,4-dicloro-3-((5-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)179enzo)179enzoate (841 mg, 1,512 mmol) LC/MS (Método h) Rt = 3,43 min.; MS miz: 416 [M+H]+ 1H NMR (DMSO-C6, 300 MHz): 5 11,35 (s, 1H), 7,75 (d, J = 9Hz, 1H), 7,68 (d, J = 9Hz, 1H), 7,62 (s, 1H), 7,37 (s, 1H), 5,87 (s, 1H), 4,47 (s, 2H), 3,87 (s, 3H), 2,42 (s, 3H).
Etapa 4: metil 2,4-did oro-3-((1,5-dimetM-6-(trifluorometN)-1H-mdol-2-M)metM)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-didoro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (530 mg, 84%) se preparó a partir de metil 2,4-didoro-3-((5-metil-6-(trifluorometil)-1H -indol-2-il)metil)benzoato (592 mg, 1,422 mmol) LC/MS (Método h) Rt = 3,57 min.; MS m/z: 430 [M+H]+ 1H NMR (DMSO-de, 300 MHz): 57,79 (s, 1H), 7,78 (d, J = 8Hz, 1H), 7,71 (d, J = 8Hz, 1H), 7,36 (s, 1H), 5,56 (s, 1H), 4,47 (s, 2H), 3,90 (s, 3H), 3,86 (s, 3H), 2,42 (s, 3H).
Etapa 5: ácido 2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico
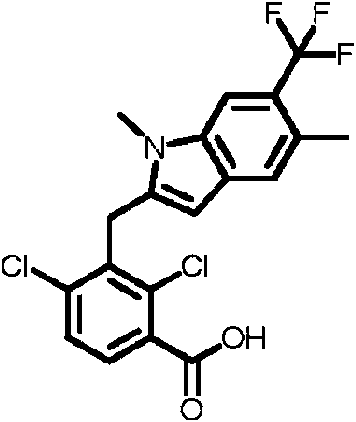
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (424 mg, 100%) se preparó a partir de metil 2,4-didoro-3-((1,5-dimetil-6-(trifluorometil)-1H -indol-2-il)metil)benzoato (438 mg, 1,018 mmol) LC/MS (Método h) Rt = 3,13 min.; MS m/z: 416 [M+H]+ 1H NMR (DMSO-de, 300 MHz): 5 13,67 (ancho, 1H), 7,79 (s, 1H), 7,75 (d, J = 9Hz, 1H), 7,67 (d, J = 9Hz, 1H), 7,36 (s, 1H), 5,57 (s, 1H), 4,47 (d, 2H), 3,90 (s, 3H), 2,43 (s, 3H).
Etapa 6: etil 1-(2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato

Usando un procedimiento similar al Ejemplo A1, etil 1-(2,4-didoro-3-((1,5-dimetil-6-(trifluorometil)-1H -indol-2-il)metil)benzoil)piperidina-4-carboxilato (133 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (100 mg, 0,240 mmol) y etil piperidina-4-carboxilato (37,8 mg, 0,24 mmol). LC/MS (Método h) Rt = 3,43 min.; MS m/z: 555 [M+H]+ 1H NMR (DMSO-de, 300 MHz): 5 7,79 (s, 1H), 7,65 (d, J = 6Hz, 1H), 7,46 (d, J = 6Hz, 0,5H), 7,38 (m, 1,5H), 5,60 y 5,58 (s, 1H), 4,43 (m, 2H), 4,37 (m, 1H), 4,07 (m, 2H), 3,89 (s, 3H), 3,27 (m, 1H), 3,13 (m, 2H), 2,65 (m, 1H), 2,43 (s, 3H), 1,93 (m, 1H), 1,81 (m, 1H), 1,51 (m, 2H), 1,18 (m, 3H).
Etapa 7: ácido 1-(2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxílico (102 mg, 77%) se preparó a partir de etil 1-(2,4-dicloro-3-((1,5-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato (133 mg, 0,239 mmol). LC/MS (Método g) Rt = 1,82 min.; MS m/z: 527 [M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 512,40 (ancho, 1H), 7,79 (s, 1H), 7,67 (d, J = 10Hz, 1H), 7,45 (d, J = 10Hz, 0,5H), 7,37 (m, 1,5H), 5,60 y 5,58 (s, 1H), 4,46 (s, 2H), 4,36 (m, 1H), 3,96 (s, 3H), 3,29 (m, 1H), 3,12 (m, 1H), 3,01 (m, 1H), 2,56 (m, 1H), 2,49 (s, 3H), 1,92 (m, 1h ), 1,78 (m, 1H), 1,53 (m, 2H).
Ejemplo W: 2-(2,6-did oro-3-metoxibend l)-1,4-dimetM-6-(tnfluorometM)-1H-mdol

Etapa 1: (2,6-did oro-3-metoxifeml)(4-metiM-(femlsulfoml)-6-(tnfluorometM)-1H-mdol-2-M)metanol

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,6-dicloro-3-metoxifenil)(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metanol (2,51 g, 90%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (2,262 g, 5,13 mmol) y 1-(2,6-dicloro- 3-metoxifenil)prop-2-in-1 -ol (Preparación #7 (1,54 g, 6,66 mmol). LC/MS (Método h) Rt = 3,39 min.; MS m/z: 602 [M-H]"+CHsCOOH 1H NMR (DMSO-C6, 300 MHz): 5 8,08 (m, 1H), 7,82 (m, 2H), 7,67 (m, 1H), 7,56 (m, 2H), 7,40 (m, 2H), 7,14 (d, J = 9Hz, 1H), 6,98 (d, 6Hz, 1H), 6,79 (s, 1H), 6,57 (d, J = 6Hz, 1H), 3,87 (s, 3H), 2,45 (s, 3H).
Etapa 2: 2-(2,6-did oro-3-metoxibend l)-4-metiM-(femlsulfoml)-6-(tnfluorometM)-1H-mdol

Usando un procedimiento similar al Ejemplo A, Etapa 2, 2-(2,6-didoro-3-metoxibendl)-4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol (2,12 g, 87 %) se preparó a partir de (2,6-didoro-3-metoxifenil)(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metanol (2,5 g, 4,59 mmol). LC/MS (Método h) Rt = 3,85 min.; m S m/z: 528 [m H]+1H NMR (DMSO-da, 300 MHz): 58,24 (s, 1H), 7,93 (m, 2H), 7,76 (m, 1H), 7,68 (m, 2H), 7,53 (d, J = 9Hz, 1H), 7,41 (s, 1H), 7,23 (d, J = 9Hz, 1H), 5,90 (s, 1H), 4,54 (s, 2H), 3,90 (s, 3H), 2,34 (s, 3H).
Etapa 3: 2-(2,6-d¡cloro-3-metox¡benc¡l)-4-met¡l-6-(tr¡fluoromet¡l)-1H-mdol

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-didoro-3-metoxibendl)-4-metil-6-(trifluorometil)-1H-indol (1,52 g, 98%) se preparó a partir 2-(2,6-didoro-3-metoxibendl)-4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol (2,12 g, 4,01 mmol). LC/MS (Método h) Rt = 3,47 min.; MS m/z: 388 [M+H]+ 1H NMR (DMSO-da, 300 MHz): 5 11,50 (ancho, 1H), 7,52 (d, J = 9Hz, 1H), 7,48 (s, 1H), 7,17 (d, J = 9Hz, 1H), 7,02 (s, 1H), 5,95 (s, 1H), 4,42 (s, 2H), 3,89 (s, 3H), 2,40 (s, 3h ).
Etapa 4: 2-(2,6-dicloro-3-metox¡benc¡l)-1,4-d¡met¡l-6-(tr¡fluorometil)-1H-mdol

Usando un procedimiento similar al del Ejemplo P, Etapa 4, 2-(2,6-dicloro-3-metoxibencil)-1,4-dimetil-6-(trifluorometil)-1 H-indol (517 mg, 93%) se preparó a partir de 2-(2,6-dicloro-3-metoxibencil)-4-metil-6-(trifluorometil)-1H-indol (1,36 g, 3,50 mmol) LC/EM (procedimiento g) Rt 2,17 min.; MS m/z: 402 [M+H]+1H NMR (DMSO-cía, 300 MHz): 5 7,69 (s, 1H), 7,54 (d, J = 9Hz, 1H), 7,22 (d, J = 9Hz, 1H), 7,05 (s, 1H), 5,65 (s, 1H), 4,41 (s, 2H), 3,91 (s, 6H), 2,35 (s, 3H).
Ejemplo X: 2,4-d¡cloro-3-((1,4-d¡met¡l-6-(tr¡fluoromet¡l)-1H-mdol-2-¡l)met¡l)fenol

A una solución de 2-(2,6-dicloro-3-metoxibencil)-1,4-dimetil-6-(trifluorometil)-1H-indol (Ejemplo W) (0,517 g, 1,285 mmol) en DCM (51,4 mL) y enfriada a -10 °C se añadió BBr3 (3,86 mL, 3,86 mmol). La reacción se agitó a -10 °C durante 1 hora y se calentó a temperatura ambiente durante 1,5 horas. La reacción se hidrolizó por adición de solución acuosa saturada de NaHCO3, y la mezcla básica se extrajo con DCM. La capa orgánica se lavó sucesivamente con agua y solución saturada de NaCl, se secó sobre sulfato de magnesio, se filtró y se evaporó. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-30% en ciclohexano) para dar 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil) fenol (389 mg, 78%). LC/MS (Método g) Rt 1,97 min.; MS miz: 388 [M+H]+1H NMR (DMSO-cfe, 400 MHz): 510,58 (ancho, 1H), 7,68 (s, 1 h ), 7,35 (d, J = 8,8Hz, 1H), 7,05 (s, 1H), 7,01 (d, J = 8,8Hz, 1H), 5,66 (s, 1H), 4,37 (s, 2H), 3,90 (s, 3H), 2,36 (s, 3H).
Ejemplo Y: ácido 2-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-N)metN)fenoxi)acético

Etapa 1: metil 2-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1-H-indol-2-il)metil)fenoxi)acetato

A una solución de 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)- 1H-indol-2-il)metil)fenol (Ejemplo X) (100 mg, 0,258 mmol) en ACN (1 mL) se añadió Cs2CO3 (92 mg, 0,283 mmol) y la mezcla se agitó a temperatura ambiente durante 10 minutos. Se añadió bromoacetato de metilo (0,036 mL, 0,386 mmol) y la mezcla se agitó a temperatura ambiente durante 7 horas. La mezcla de reacción se diluyó con agua y se extrajo con DCM. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-30% en ciclohexano) para dar metil 2-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)acetato (98 mg, 83%). LC/MS (Método h) Rt 3,46 min.; MS miz: 460 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 57,70 (s, 1H), 7,52 (d, J = 9Hz, 1H), 7,16 (d, J = 9Hz, 1H), 7,06 (s, 1H), 5,65 (s, 1h ), 5,00 (s, 2H), 4,43 (s, 2H), 3,92 (s, 3H), 3,72 (s, 3H), 2,36 (s, 3H).
Etapa 2: ácido 2-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)fenoxi)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)acético (88 mg, 93%) se preparó a partir de 2-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)acetato (98 mg, 0,213 mmol) LC/MS (Método g) Rt 1,98 min.; MS miz: 446 [M+H]+ 1H NMR (DMSO-de, 400 MHz): 5 13,30 (ancho, 1H), 7,70 (s, 1H), 7,50 (d, J = 8,8Hz, 1H), 7,09 (d, J = 8,8Hz, 1H), 7,06 (s, 1H), 5,66 (s, 1H), 4,83 (s, 2H), 4,42 (s, 2H), 3,91 (s, 3H), 2,34 (s, 3H).
Ejemplo Y1: ácido 3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)propanoico

A una solución de 2,4-didoro-3-((1,4-dimetN-6-(trifluorometil)-1H-indol-2-il)metil)fenol (Ejemplo X) (150 mg, 0,386 mmol) en DMF (2 mL) se añadió NaH (23,18 mg, 0,580 mmol) y la mezcla se agitó a temperatura ambiente durante 10 minutos. Se añadió beta-propiolactona (0,036 mL, 0,580 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió agua a la mezcla, y la mezcla se acidificó a pH = 3-4 con solución de HCl 1N. El sólido se filtró, se lavó con agua y éter dietílico y se secó al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH 0-20 en DCM) para dar ácido 3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)propanoico (101 mg, 37%) como un sólido de color blanco. LC/MS (Método g) Rt 1,97 min.; MS m/z: 460 [M+H]+ 1H NMR (DMSO-CÍ6, 400 MHz): 5 12,56 (ancho, 1H), 7,67 (s, 1H), 7,52 (d, J = 9,2Hz, 1H), 7,24 (d, J = 9,2Hz, 1H), 7,05 (s, 1H), 5,65 (s, 1H), 4,40 (s, 2H), 4,30 (t, J = 6Hz, 2H), 3,90 (s, 3H), 2,74 (t, J = 6Hz, 2H), 2,36 (s, 3H).
Ejemplo Z: 2-((3,5-dicloro-2-metoxipiridin-4-il)metil)-1,4-dimetil-6-(trifluorometil)-1H-indol

Etapa 1: (3,5-dicloro-2-metoxipindin-4-il)(4-metiM-(fenilsulfonil)-6-(tnfluorometil)-1H-indol-2-il)metanol

Usando un procedimiento similar al Ejemplo A, Etapa 1, (3,5-dicloro-2-metoxipiridin-4-il)(4-metil-1-(fenilsul-fonil)-6-(trifluorometil)-1H-indol-2-il)metanol (3,47 g, 94%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (3 g, 6,80 mmol) y 1-(3,5-dicloro-2-metoxipiridin-4-il)prop-2-in-1-ol (Preparación #15) (1,894 g, 8,16 mmol). LC/MS (Método k) Rt 3,59 min.; MS miz: 545 [M+H]+ 1H NMR (DMSO-cfe, 300 MHz): 58,21 (s, 1H), 8,09 (s, 1H), 7,82 (m, 2H), 7,71 (m, 1H), 7,58 (m, 1H), 7,44 (s, 1H), 6,94 (s, 1H), 6,87 (m, 2H), 3,95 (s, 3H), 2,48 (s, 3H).
Etapa 2: 2-((3,5-did oro-2-metoxipindm-4-M)metM)-4-metiM-(femlsulfoml)-6-(tnfluorometM)-1H-mdol

A una solución de (3,5-dicloro-2-metoxipiridin-4-il)(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metanol (50 mg, 0,092 mmol) en cloroformo (420 pL) se añadió cloruro de tionilo (19,95 pL, 0,275 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante una noche. La mezcla de reacción se basificó (pH 8) con solución acuosa saturada de NaHCO3. La capa acuosa obtenida se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se disolvió en ácido acético (420 pL) y se añadió zinc (30,0 mg, 0,458 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas, luego se filtró, se lavó con DCM y el filtrado se concentró a presión reducida. El residuo se diluyó con EtOAc y se lavó con agua. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 2-((3,5-dicloro-2-metoxipiridin-4-il)metil)-4-metil-1-(fenilsulfo-nil)-6-(trifluorometil)-1H-indol (50 mg, 94%) como un sólido de color beige. LC/MS (Método k) Rt 4,19 min.; MS miz: 529 [M+H]+ 1H NMR (DMSO-C6, 300 MHz): 5 8,32 (s, 1H), 8,22 (s, 1H), 7,92 (m, 2H), 7,77 (m, 1H), 7,67 (m, 2h ), 7,41 (s, 1H), 6,21 (s, 1H), 4,54 (s, 2H), 3,39 (s, 3H), 2,37 (s, 3H).
Etapa 3: 2-((3,5-did oro-2-metoxipindm-4-M)metM)-4-metM-6-(tnfluorometM)-1H-mdol

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-((3,5-didoro-2-metoxipiridin-4-il)metil)-4-metil-6-(trifluorometil)-1H-indol (1,2 g, 86%) se preparó a partir 2-((3,5-didoro-2-metoxipiridin-4-il)metil)-4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol (1,9 g, 3,59 mmol) LC/MS (Método h) LC/MS (Método h) Rt 3,45 min.; MS m/z: 389 [M+H]+ 1H NMR (DMSO-óe, 300 MHz): 511,49 (s, 1H), 8,31 (s, 1H), 7,48 (s, 1H), 7,03 (s, 1H), 6,06 (s, 1H), 4,42 (s, 2H), 3,98 (s, 3H), 2,42 (s, 3H).
Etapa 4: 2-((3,5-dicloro-2-metoxipmdm-4-M)metil)-1,4-dimetM-6-(trifluorometM)-1H-mdol

Usando un procedimiento similar al Ejemplo A, Etapa 4, 2-((3,5-didoro-2-metoxipiridin-4-il)metil)-1,4-dimetil-6-(trifluorometil)-1H-indol (963 mg, 77%) se preparó a partir de 2-((3,5-didoro-2-metoxipiridin-4-il)metil)-4-metil-6-(trifluorometil)-1 H-indol (1,2 g, 3,08 mmol). LC/MS (Método h) Rt 3,60 min.; MS m/z: 403 [M+H]+1H NMR (DMSO-óe, 300 MHz): 58,35 (s, 1H), 7,70 (s, 1H), 7,07 (s, 1H), 5,80 (s, 1H), 4,43 (s, 2H), 4,00 (s, 3H), 3,92 (s, 3H), 2,38 (s, 3H).
Ejemplo AA: 2-(2,6-dicloro-3-(4-(oxetan-3-M)piperazma-1-carboml)bencil)-1,4-dimetiMH-mdol-6-carbomtrMo

Etapa 1: metil 2,4-didoro-3-((6-ciano-4-metiM-(femlsulfoml)-1H-mdol-2-M)(hidroxi)metM)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (1,32 g, 71%) se obtuvo como un sólido de color marrón a partir de W-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #18) (1,4 g, 3,52 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (1,093 g, 4,22 mmol). LC/MS (Método h) Rt = 2,97 min.; MS m/z: 587[M-H]- + CH3 COOH. 1H NMR (D M S O C 300 MHz): 58,24 (s, 1H), 7,90 (m, 2H), 7,67 (m, 2H), 7,53 (m, 4H), 7,02 (m, 1H), 6,89 (s, 1H), 6,77 (d, J = 6Hz, 1H), 3,87 (s, 3H), 2,43 (s, 3H).
Etapa 2: metil 2,4-didoro-3-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-dicloro-3-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (1,6 g, 100%) se obtuvo como un sólido de color amarillo a partir de metil 2,4-dicloro-3-(6-ciano-4-metil-1-((fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (1,52 g, 2,87 mmol). LC/MS (Método h) Rt = 3,45 min.; MS m/z: 513 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,36 (s, 1H), 8,07 (d, J = 9Hz, 2H), 7,81 (m, 2H), 7,69 (m, 3H), 7,48 (s, 1H), 5,98 (s, 1H), 4,63 (s, 2H), 3,86 (s, 3H), 2,31 (s, 3H).
Etapa 3: metil 2,4-didoro-3-((6-dano-4-metiMH-mdol-2-N)metN)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, se obtuvo metil 2,4-dicloro-3-((6-dano-4-metil-1H-indol-2-il)metil)benzoato (850 mg, 67%) como un sólido de color marrón a partir de metil 2,4-dicloro-3-((6-dano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (1,6 g, 3,12 mmol).
LC/MS (Método h) Rt = 3,03 min.; MS m/z: 373 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5 11,66 (ancho, 1H), 7,76 (d, J = 6Hz, 1H), 7,68 (d, J = 6Hz, 1H), 7,64 (s, 1H), 7,08 (s, 1H), 5,99 (s, 1H), 4,50 (s, 2H), 3,87 (s, 3H), 2,37 (s, 3H).
Etapa 4: metil 2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 4, metil 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)benzoato (660 mg, 75%) se obtuvo como un sólido de color beige a partir de metil 2,4-dicloro-3-((6-ciano-4-metil-1H-indol-2-il)metil)benzoato (847 mg, 2,27 mmol).
LC/MS (Método k) Rt = 3,29 min.; MS m/z: 387 [M+H]+. 1H NMR (DMSO-C6, 300 MHz): 57,92 (s, 1H), 7,80 (d, J = 9Hz, 1H), 7,72 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,70 (s, 1H), 4,50 (s, 2H), 3,91 (s, 3H), 3,88 (s, 3H), 2,33 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-N)metil)benzoico
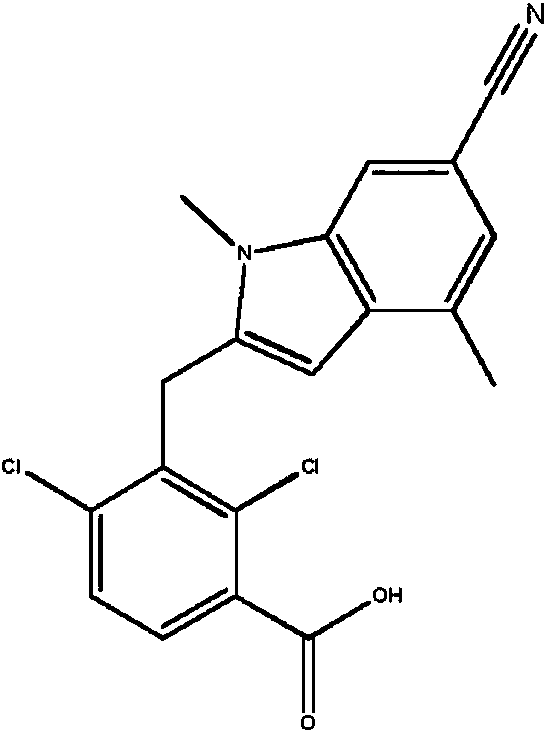
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoico (660 mg, 100%) se obtuvo como un sólido de color blanco a partir de metil 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoato (655 mg, 1,69 mmol). LC/MS (Método k) Rt = 2,82 min.; MS miz: 373 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 57,92 (s, 1h ), 7,77 (d, J = 9Hz, 1H), 7,68 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,70 (s, 1H), 4,49 (s, 2H), 3,91 (s, 3H), 2,33 (s, 3H).
Etapa 6: 2-(2,6-didoro-3-(4-(oxetan-3-M)piperazma-1-carboml)bencM)-1,4-dimetiMH-mdol-6-carbomtnlo

Usando un procedimiento similar al Ejemplo A.1,2-(2,6-dicloro-3-(4-(oxetan-3-il)piperazina-1-carbonil)bencil)-1,4-dimetil-1H- indol-6-carbonitrilo (700 mg, 88%) se obtuvo como un sólido de color blanco a partir de ácido 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoico (600 mg, 1,608 mmol) y 1-(oxetan-3-il)piperazina (260 mg, 1,83 mmol). LC/MS (Método g) Rt = 1,47 min.; MS miz: 497 [M+H]+ 1H NMR (DMSOCs, 400 MHz): 57,92 (s, 1H), 7,67 (d, J = 8Hz, 1H), 7,43 (d, J = 8Hz, 1H), 7,11 (s, 1H), 5,71 (s, 1H), 4,53 (m, 2H), 4,47 (m, 4H), 3,90 (s, 3H), 3,69 (m, 2H), 3,48 (m, 1H), 3,20 (m, 2H), 2,38 (m, 2H), 2,23 (s, 3H), 2,18 (m, 2H).
Tabla AA. Los siguientes ejemplos se prepararon a partir de 2,4-dicloro-3-((6-ciano-1,4-dimetiMH-indol-2-ilm il nz i E m l AA E n ' l mi m r imi n n l min r i .


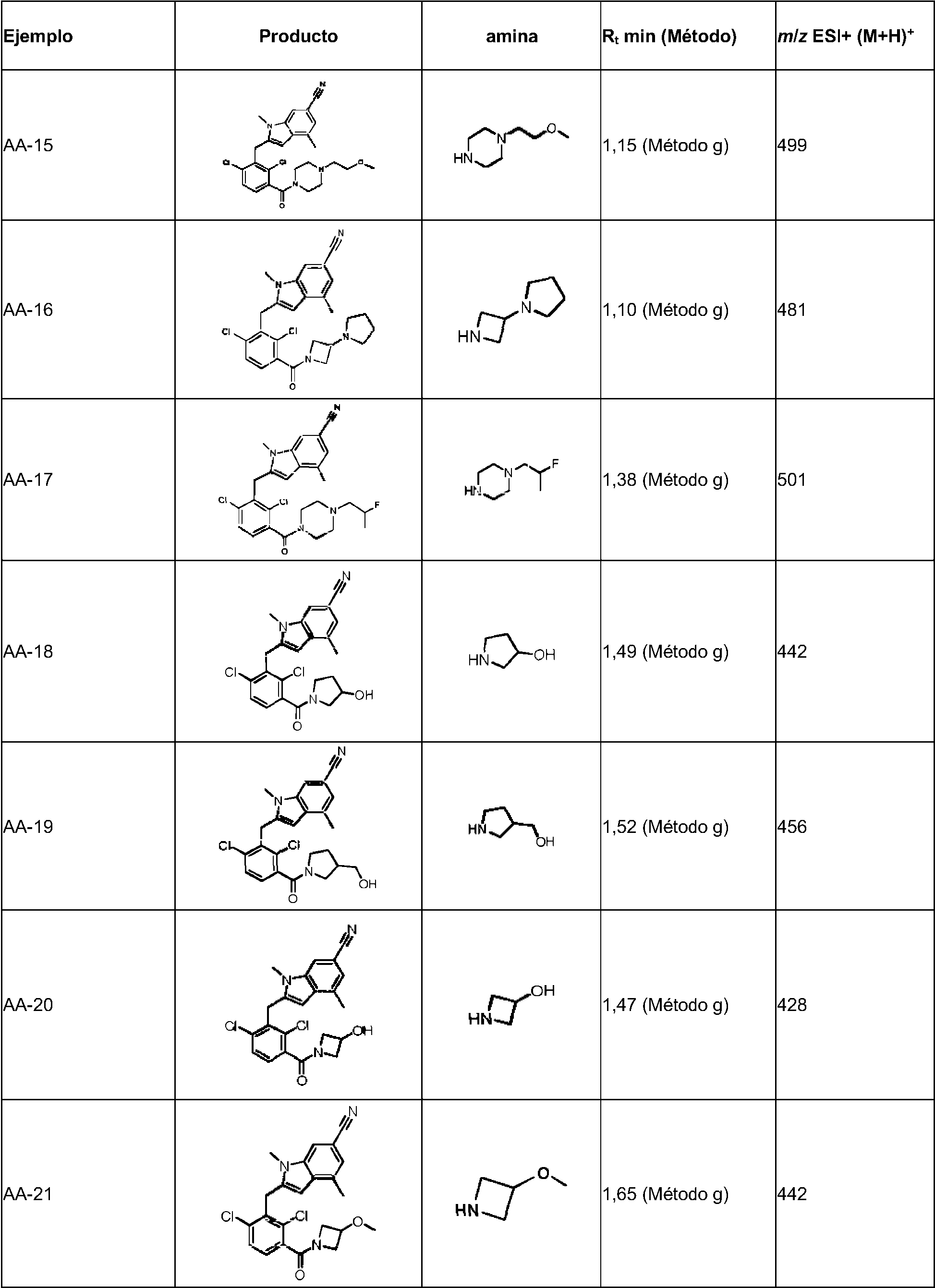



Ejemplo AB: 2-(2,6-dicloro-3-(morfolma-4-carbonN)bencN)-1-metiMH-mdol-5-carbomtnlo

Etapa 1: 2-((2,6-dicloro-3-(morfolma-4-carbonN)fenM)(hidroxi)metN)-1-(fenNsulfonN)-1H-mdol-5-carbomtnlo

Usando un procedimiento similar al Ejemplo A, Etapa 1, 2-((2,6-didoro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-1-(fenilsulfonil)-1H-indol-5-carbonitrilo (500 mg, 100%) se obtuvo como un sólido de color marrón a partir de (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (Preparación #3) (319 mg, 1,015 mmol) y N-(4-ciano-2-yodofenil)bencenosulfonamida (Preparación #19) (300 mg, 0,781 mmol). LC/MS (Método h) Rt = 2,52 min.; MS m/z: 570 [M+H]+
1H NMR (DMSO-cfe, 300 MHz): 58,14 (m, 2H), 7,94 (m, 2H), 7,70 (m, 1H), 7,55 (m, 4H), 7,52 (m, 2H), 7,01 (m, 1H), 6,71 (m, 1H), 3,65 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H).
Etapa 2: 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-(fenilsulfonil)-1H-indol-5-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 2, 2-(2,6-didoro-3-(morfolina-4-carbonil)bencil)-1-(fenil- sulfonil)-1H-indol-5-carbonitrilo (20 mg, 29%) se obtuvo como un sólido de color blanco a partir de 2-((2,6-dicloro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-1-(fenilsulfonil)-1H-indol-5-carbonitrilo (70 mg, 0,123 mmol). LC/mS (Método g) Rt = 1,78 min.; MS m/z: 554 [M+H]+
1H NMR (DMSO-cfe, 300 MHz): 5 8,30 (d, J = 9Hz, 1H), 7,98 (m, 3H), 7,77 (m, 2H), 7,68 (m, 3H), 7,48 (d, J = 9Hz, 1H), 5,99 (s, 1H), 4,58 (m, 2H), 3,65 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H).
Etapa 3: 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1H-indol-5-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1H-indol-5-carbonitrilo (465 mg, 85%) se obtuvo como de un sólido de color amarillo a partir de 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-(fenilsulfonil)-1H-indol-5-carbonitrilo (670 mg, 1,208 mmol). LC/MS (Método h) Rt = 2,40 min.; MS m/z: 414 [M+H]+ 1H NMR (DMSO-cía, 300 MHz): 511,70 (s, 1H), 7,90 (s, 1H), 7,64 (d, J = 9Hz, 1H), 7,47 (m, 1H), 7,39 (m, 2H), 5,96 (s, 1H), 4,44 (s, 2H), 3,65 (m, 4H), 3,52 (m, 2H), 3,17 (m, 2H).
Etapa 4: 2-(2,6-d¡cloro-3-(morfolma-4-carboml)benc¡l)-1-met¡l-ÍH-mdol-5-carbomtr¡lo:

Usando un procedimiento similar al Ejemplo P, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-indol-5-carbonitrilo (115 mg, 56%) se obtuvo como un sólido de color blanco a partir de 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1H-indol-5-carbonitrilo (200 mg, 0,483 mmol).
LC/MS (Método g) Rt = 1,49 min.; MS m/z: 428 [M+H]+
1 H NMR (DMSO-CÍ6 , 400 MHz): 5 7,90 (m, 1H), 7,68 (d, J = 8Hz, 1H), 7,65 (d, J = 8Hz, 1H), 7,46 (m, 2H), 5,73 (s, 1H), 4,44 (s, 2H), 3,90 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,19 (m, 2H).
Ejemplo AC: 2-(2,6-d¡cloro-3-(morfol¡na-4-carboml)benc¡l)-1-met¡MH-mdol-6-carbomtr¡lo

Etapa 1: met¡l 2,4-d¡cloro-3-((6-c¡ano-1-(femlsulfoml)-1H-mdol-2-¡l)(h¡drox¡)met¡l)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-((6-ciano-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (715 mg, 89%) se obtuvo como un sólido de color blanco a partir de N-(5-ciano-2-yodofenil)bencenosulfonamida (Preparación #20) (600 mg, 1,562 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (445 mg, 1,718 mmol). LC/MS (Método h) Rt = 2,87 min.; MS miz: 573 [M-H]- +CH 3 COOH 1 H NMR (DMSO-óe, 300 MHz): 5 8,40 (s, 1H), 7,98 (dd, J = 3Hz, 9Hz, 2H), 7,79 (d, J = 6Hz, 1H), 7,68 (m, 3H), 7,58 (m, 3H), 7,03 (dd, J = 3Hz, 6Hz, 1H), 6,82 (s, 1H), 6,76 (d, J = 6Hz, 1H), 3,86 (s, 3H).
Etapa 2: metil 2,4-dicloro-3-((6-ciano-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-dicloro-3-((6-ciano-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (400 mg, 100%) se obtuvo como un sólido de color amarillo a partir de metil 2,4-dicloro-3-((6-ciano-1-(fenil-sulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (400 mg, 0,776 mmol). LC/MS (Método h) Rt = 3,39 min.; MS miz: 497 [M-H]-1H NMR (DMSO-óe, 300 MHz): 58,51 (s, 1H), 8,07 (d, J = 9Hz, 2H), 7,80 (m, 2H), 7,68 (m, 5H), 6,00 (s, 1H), 4,62 (s, 2H), 3,86 (s, 3H).
Etapa 3: metil 2,4-didoro-3-((6-ciano-1H -indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, se obtuvo metil 2,4-dicloro-3-((6-ciano-1H-indol-2-il)metil)benzoato (213 mg, 74%) como un sólido de color amarillo a partir de metil 2,4-dicloro-3-((6-ciano-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (400 mg, 0,801 mmol). LC/MS (Método h) Rt = 2,93 min.; MS miz: 359 [M+H]+1H NMR (DMSO-
da, 300 MHz): 511,65 (ancho, 1H), 7,79 (d, J = 9Hz, 1H), 7,74 (s, 1H), 7,68 (d, J = 9Hz, 1H), 7,54 (d, J = 9Hz, 1H), 7,27 (dd, J = 3Hz, 9Hz, 1H), 7,98 (s, 1h), 4,50 (s, 2H), 3,87 (s, 3H).
Etapa 4: metil 2,4-didoro-3-((6-ciano-1-metiMH-mdol-2-il)metM)benzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, se obtuvo metil 2,4-dicloro-3-((6-ciano-1-metil-'/H-indol-2-il)metil)benzoato (210 mg, 93%) como un sólido de color blanco a partir de metil 2,4-dicloro-3-((6-ciano-1H -indol-2-il)metil)benzoato (210 mg, 0,585 mmol). LC/MS (Método h) Rt = 3,04 min.; MS miz: 373 [M+H]+1H NMR (DMSOd 300 MHz): 58,08 (s, 1H), 7,80 (d, J = 9Hz, 1H), 7,72 (d, J = 9Hz, 1H), 7,53 (d, J = 9Hz, 1H), 7,30 (dd, J = 3Hz, 9Hz, 1H), 5,71 (s, 1H), 4,50 (s, 2H), 3,92 (s, 3H), 3,88 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((6-dano-1-metiMH-mdol-2-N)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, se obtuvo ácido 2,4-dicloro-3-((6-ciano-1-metil-1H-indol-2-il)metil)benzoico (170 mg, 8 8 %) como un sólido de color blanco a partir de metil 2,4-dicloro-3-((6-ciano-1-metil-1H-indol-2-il)metil)benzoato (200 mg, 0,536 mmol). LC/MS (Método h) Rt = 2,61 min.; MS miz: 359 [M+H]+
1 H NMR (DMSO-d6 , 300 MHz): 58,08 (d, J = 3Hz, 1H), 7,75 (d, J = 9Hz, 1h), 7,66 (d, J = 9Hz, 1H), 7,54 (d, J = 9Hz, 1H), 7,30 (dd, J = 3Hz, 9Hz, 1H), 5,71 (s, 1H), 4,50 (s, 2H), 3,92 (s, 3H).
Etapa 6: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1-metiMH-mdol-6-carbomtrMo:

Usando un procedimiento similar al Ejemplo A, Etapa 6 , 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-indol-6-carbonitrilo (100 mg, 50%) se obtuvo como un sólido de color blanco a partir de ácido 2,4-dicloro-3-((6-ciano-1-metil-1Hindol-2-il)metil)benzoico (165 mg, 0,459 mmol) y morfolina (80 mg, 0,92 mmol). LC/MS (Método g) Rt = 1,53 min.; MS miz: 428 [M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 58,08 (s, 1H), 7,67 (d, J = 8Hz, 1H), 7,54 (d, J = 8Hz, 1H), 7,45 (d, J = 8Hz, 1H), 7,30 (dd, J = 3Hz, 8Hz, 1H), 5,75 (s, 1H), 4,46 (s, 2H), 3,92 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,18 (m, 2H).
Ejemplo AD: 2-(2,6-didoro-3-(morfolma-4-carboml)benzoM)-1,4-dimetiMH-mdol-6-carbomtrNo

Etapa 1: 2-((2,6-didoro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1 H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 1,2-((2,6-dicloro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (3 g, 41%) se obtuvo como un sólido de color naranja a partir de W-(2-bromo-5-ciano-3-metilfenil)bencenosulfonamida (Preparación #46) (3,6 g, 10,25 mmol) y (2,4-dicloro-3-(1-hidroxi-prop-2-in-1-il)fenil)(morfolino)metanona (Preparación #3) (4,83 g, 15,38 mmol). LC/MS (Método h) Rt = 2,60 min.; MS miz: 584 [M+H]+ 1H NMR (CDCla, 300 MHz): 58,30 (s, 1H), 7,92 (m, 2H), 7,54 (m, 2H), 7,47 (m, 3H), 7,28 (m, 3H), 6,43 y 6,31 (s, 1H), 3,81 (m, 4H), 3,67 (m, 2H), 3,29 (m, 2H), 2,40 (s, 3H).
Etapa 2: 2-(2,6-didoro-3-(morfolma-4-carboml) benzoil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo

A una solución de 2-((2,6-dicloro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metiM-(fenilsulfonil)-1H-indol-6-carbonitrilo (321 mg, 0,549 mmol) en DCM (5 mL) se añadió periodinano Dess-Martin (2,284 mL, 1,098 mmol). La reacción se agitó a temperatura ambiente durante 2 horas, luego se filtró y se lavó con ACN. El filtrado se lavó con solución acuosa saturada de NaHCO3 , salmuera y se secó sobre sulfato de magnesio; después se concentró para dar 2-(2,6-dicloro-3-(morfolina-4-carbonil)benzoil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo como un sólido de color amarillo (252 mg, 72%). LC/MS (Método h) Rt = 2,79 min.; MS m/z: 582 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 58,53 (s, 1H), 8,28 (m, 2h), 7,80 (m, 1H), 7,73 (m, 4H), 7,65 (m, 2h), 3,63 (m, 4H), 3,52 (m, 2H), 3,23 (m, 2H), 2,48 (s, 3H).

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-dicloro-3-(morfolina-4-carbonil)benzoil)-4-metil-1/-/-indol-6-carbonitrilo (165 mg, 73%) se obtuvo como un sólido de color amarillo a partir de 2-(2,6-dicloro-3-(morfolina-4-carbonoil)benzoil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (250 mg, 0,429 mmol). LC/MS (Método h) Rt = 2,34 min.; MS m/z:
442 [M+H]+
1H NMR (DMSO-cfe, 300 MHz): 512,72 (ancho, 1H), 7,79 (m, 2H), 7,66 (d, J = 6 Hz, 1H), 7,33 y 7,12 (s, 1H), 7,24 (s, 1H), 3,66 (m, 4H), 3,56 (m, 2H), 3,18 (m, 2H), 2,48 (s, 3H).
Etapa 4: 2-(2,6-didoro-3-(morfolma-4-carboml)benzoM)-1,4-dimetiMH-mdol-6-carbomtrMo:

Usando un procedimiento similar al Ejemplo A, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)benzoil)-1,4-dimetil-1H-indol-6 -carbonitrilo (49 mg, 29%) se obtuvo como un sólido de color amarillo a partir de 2-(2,6-dicloro-3-(morfolina-4
carbonil)benzoil)-4-metil-1H-indol-6-carbonitrilo (160 mg, 0,362 mmol). LC/MS (Método g) Rt = 1,58 min.; MS miz: 456 [M+H]+
1H NMR (DMSO-de, 400 MHz): 58,21 (s, 1H), 7,75 (d, J = 8Hz, 1H), 7,65 (d, J = 8Hz, 1H), 7,30 y 7,10 (ancho, 1H), 7,29 (s, 1H), 4,20 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,19 (m, 2H), 2,48 (s, 3H).
Ejemplo AE: 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6-carbonitrilo

Etapa 1: 2-((2,6-dimetM-3-(morfolma-4-carbonM)fenM)(hidroxi)metM)-4-metiM-(fenMsulfonM)-1H-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 1,2-((2,6-dimetil-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (730 mg, 71%) se obtuvo como un aceite de color amarillo a partir de N-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #18) (730 mg, 1,833 mmol) y (3-(1-hidroxi-prop-2-in-1-il)-2,4-dimetilfenil)(morfolino)metanona (Preparación #4) (601 mg, 2,2 mmol).
LC/MS (Método h) Rt = 2,66min.; MS miz: 544 [M+H]+1H NMR (DMSO-d6, 300 MHz): 58,24 (d, J = 9Hz, 1H), 8,08 (d, J = 9Hz, 1H), 7,97 (d, J = 9Hz, 1H), 7,95 (s, 1H), 7,71 (t, J = 9Hz, 1H), 7,59 (t, J = 9Hz, 2H), 7,48 (s, 1H), 7,06 (m, 1H), 6,78 y 6,72 (s, 1H), 6,80 y 6,42 (s, 1H), 6,30 (ancho, 1H), 3,64 (m, 4H), 3,47 (m, 2H), 3,12 (m, 2H), 2,89 (s, 3H), 2,73 (s, 3H), 2,36 y 2,39 (s, 3H).
Etapa 2: 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 2, 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1-(fenilsulfonil)-l H-indol-6-carbonitrilo (700 mg, 100%) se obtuvo como un sólido de color amarillo a partir de 2-((2,6-dimetil-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonilo)indolina-6-carbonitrilo (725 mg, 1,329 mmol). LC/MS (Método h) Rt = 2,94 min.; MS m/z: 528 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 58,39 (s, 1H), 8,08 (m, 2H), 7,81 (m, 1H), 7,67 (m, 2H), 7,47 (s, 1H), 7,18 (d, J = 9Hz, 1H), 7,07 (d, J = 9Hz, 1H), 5,73 (s, 1H), 4,28 (m, 2H), 3,62 (m, 4H), 3,44 (m, 2H), 3,09 (m, 2H), 2,26 (s, 3H), 2,06 (s, 3H), 1,89 (s, 3H).
Etapa 3: 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1H-indol-6-carbonitrilo (380 mg, 64%) se obtuvo como un sólido de color amarillo a partir de 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (700 mg, 1,327 mmol). LC/MS (Método h) Rt = 2,52min.; MS m/z: 388 [M+H]+ 1H NMR (DMSO-C6, 300 MHz): 5 11,56 (ancho, 1H), 7,61 (s, 1H), 7,17 (d, J = 6Hz, 1H), 7,06 (m, 1H), 7,03 (d, J = 6Hz, 1H), 5,83 (s,1H), 4,18 (s, 2H), 3,64 (m, 4H), 3,47 (m, 2H), 3,12 (m, 2H), 2,35 (s, 3H), 2,30 (s, 3H), 2,13 (s, 3H).
Etapa 4: 2-(2,6-dimetM-3-(morfolma-4-carboml)bencM)-1,4-dimetiMH-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo P, Etapa 4, 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6 -carbonitrilo (70 mg, 33%) se obtuvo como un sólido de color rosa a partir de 2-(2,6-dimetil-3-(morfolina-4-carbonil)bencil)-4-metil-1H-indol-6-carbonitrilo (200 mg, 0,516 mmol). LC/MS (Método g) Rt = 1,56min.; Ms miz: 402 [M+H]+1H NMR (DMSO-cfe, 400 MHz): 57,90 (s, 1H), 7,19 (d, J = 8 Hz, 1H), 7,09 (m, 1H), 7,07 (d, J = 8 Hz, 1H), 5,53 (s, 1H), 4,18 (m, 2H), 3,90 (s, 3H), 3,65 (m, 4H), 3,50 (m, 2H), 3,13 (m, 2H), 2,29 (s, 3H), 2,25 (s, 3H), 2,10 (s, 3H).
Ejemplo AF: ácido frans-4-(2,4-didoro-3-((6-dano-1,4-dimetiMH-m dol-2-il)metil)fenoxi)ddohexanocarboxNico
Ejemplo AF1: ácido c/s-4-(2,4-didoro-3-((6-dano-1,4-dim etiMH-m dol-2-il)m etil)fenoxi)ddohexanocarboxNico

Etapa 1: 2-((2,6-didoro-3-metoxifenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 1,2-((2,6-dicloro-3-metoxifenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (2,25 g, 67%) se obtuvo como un sólido de color beige a partir de 1-(2,6-dicloro-3-metoxifenil)prop-2-in-1-ol (Preparación #7) (1,810 g, 7,83 mmol) y W-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #18) (2,4 g, 6,03 mmol). LC/MS (Método h) Rt = 2,99min.; MS miz: 559 [M-H]- CH3 COOH.
1 H NMR (DMSO-C6 , 300 MHz): 58,24 (s, 1H), 7,90 (m, 2H), 7,68 (m, 1h), 7,57 (m, 2H), 7,49 (s, 1H), 7,40 (d, J = 9Hz, 1H), 7,14 (d, J = 9Hz, 1H), 6,98 (dd, J = 3Hz, 6 Hz, 1H), 6,79 (s, 1H), 6,62 (d, J = 6 Hz, 1H), 3,86 (s, 3H), 2,41 (s, 3H).
Etapa 2: 2-(2,6-dicloro-3-metoxibencil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 2, 2-(2,6-dicloro-3-metoxibencil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (1 ,99 g, 84%) se obtuvo como un sólido de color amarillo a partir de 2-((2,6-dicloro-3-metoxifenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-indol-6-carbonitrilo (2,2 g, 4,39 mmol). LC/MS (Método h) Rt = 3,50min.; MS miz: 485 [M+H]+
1H NMR (DMSO-cfe, 300 MHz): 58,36 (s, 1H), 8,06 (m, 2H), 7,79 (t, J = 9Hz, 1H), 7,67 (t, J = 9Hz, 2H), 7,52 (d, J = 9Hz, 1H), 7,48 (s, 1H), 7,28 (d, J = 9H, 1H), 7,89 (s, 1H), 4,56 (s, 2H), 3,89 (s, 3h), 2,30 (s, 3H).
Etapa 3: 2-(2,6-dicloro-3-metoxibencil)-4-metil-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-dicloro-3-metoxibencil)-4-metiMH-indol-6-carbonitrilo (1,22 g, 78%) se obtuvo como un sólido de color beige a partir de 2-(2,6-didoro-3-metoxibendl)-4-metiM-(fenilsulfonil)-1 H-indol-6 -carbonitrilo (1,99 g, 4,10 mmol).
LC/MS (Método h) Rt = 3,10min.; MS m/z: 345 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 511,61 (ancho, 1H), 7,63 (s, 1H), 7,52 (d, J = 9Hz, 1H), 7,18 (d, J = 9Hz, 1H), 7,07 (s, 1H), 5,96 (s, 1H), 4,42 (s, 2H), 3,89 (s, 3H), 2,36 (s, 3H).
Etapa 4: 2-(2,6-didoro-3-metoxibendl)-1,4-dimetiMH-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo P, Etapa 4, 2-(2,6-dicloro-3-metoxibencil)-1,4-dimetiMH-indol-6-carbonitrilo (1,20 g, 80%) se obtuvo como un sólido de color rosa a partir de 2-(2,6-dicloro-3-metoxibencil)-4-metil-1H-indol-6-carbonitrilo (1,2 g, 3,48 mmol) LC/MS (Método g) Rt = 1,96 min.; MS m/z: 359 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 5 7,91 (s, 1H), 7,55 (d, J = 9Hz, 1H), 7,21 (d, J = 9Hz, 1H), 7,10 (s, 1H), 5,68 (s, 1H), 4,41 (s, 2H), 3,91 (s, 3H), 3,90 (s, 3H), 2,32 (s, 3H).
Etapa 5: 2-(2,6-didoro-3-hidroxibendl)-1,4-dimetiMH-mdol-6-carbom trilo

Usando un procedimiento similar al Ejemplo X, se obtuvo 2-(2,6-dicloro-3-hidroxibencil)-1,4-dimetil-1H -indol-6 -carbonitrilo (63 mg, 67%) como un sólido de color blanco a partir de 2-(2,6-dicloro-3-metoxibencil)-1,4-dimetil-1H-indol-6-carbonitrilo (200 mg, 0,557 mmol). LC/MS (Método g) Rt = 1,71 min.; MS m/z: 345 [M+H]+1H NMR (DMSO-cfe, 400 MHz): 510,58 (ancho, 1H), 7,90 (s, 1H), 7,36 (d, J = 8 Hz, 1H), 7,10 (s, 1H), 7,02 (d, J = 8 Hz, 1H), 5,69 (s, 1H), 4,38 (s, 2H), 3,89 (s, 3H), 2,33 (s, 3H).
Etapa 6: etil trans -4-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-il)metM) fenoxi) ciclohexanocarboxilato y etil c/s-4-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-M)metM)fenoxi) ciclohexanocarboxilato

A una solución de etil 4-((metilsulfonil)oxi)ddohexanocarboxilato (descrito en WO2011/035159) (75 mg, 0,300 mmol) en DMSO (0,5 mL) se añadió 2-(2,6-dicloro-3-hidroxibencilo)-1,4-dimetiMH-indol-6-carbonitrilo (100 mg, 0,290 mmol) y carbonato de cesio (123 mg, 0,377 mmol) en DMSO (0,5 mL). La reacción se agitó a 90 °C durante 26 horas. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se lavó con salmuera y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 10% en ciclohexano) para dar etil trans-4-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1 H-indol-2-il)metil) fenoxi)ciclohexanocarboxilato (15 mg, 10%) LC/MS (Método k) Rt = 4,04 min.; MS m/z: 499 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 57,91 (s, 1H), 7,48 (d, J = 9Hz, 1H), 7,31 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,68 (s, 1H), 4,47 (m, 1H), 4,40 (s, 2H), 4,08 (m, 2h), 3,89 (s, 3h), 2,37 (m, 1H), 2,32 (s, 3H), 2,06 (m, 2h), 1,94 (m, 2H), 1,51 (m, 4H), 1 , 1 6 (m, 3H) y etil c/s-4-(2,4-didoro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxilato (26 mg, 18%) como sólidos de color blanco. LC/MS (Método k) Rt = 4,04 min.; MS m/z: 499 [M+H]+ 1H NMR (DMSOC 300 MHz): 57,91 (s, 1H), 7,48 (d, J = 9Hz, 1H), 7,31 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,68 (s, 1H), 4,47 (m, 1H), 4,40 (s, 2H), 4,08 (m, 2H), 3,89 (s, 3H), 2,37 (m, 1H), 2,32 (s, 3H), 2,06 (m, 2H), 1,94 (m, 2h), 1,51 (m, 4h), 1,16 (m, 3h).
Etapa 7: ácido frans-4-(2,4-didoro-3-((6-dano-1,4-dimetiMH-m dol-2-il)metil)fenoxi)ddohexanocarboxNico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido trans-4-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi) ciclohexanocarboxílico (16,5 mg, 45%) se obtuvo como un sólido de color blanco a partir de etil trans-4- (2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-ilo)metil)fenoxi)ciclohexanocarboxilato (37 mg, 0,074 mmol). LC/MS (Método g) Rt = 1,87 min.; MS m/z: 471 [M+H]+ 1H NMR (DMSOC 500 MHz): 512,12 (ancho, 1H), 7,90 (s, 1H), 7,48 (d, J = 9Hz, 1H), 7,30 (d, J = 9Hz, 1H), 7,10 (s, 1H), 5,69 (s, 1H), 4,45 (m, 1H), 4,40 (s, 2H), 3,89 (s, 3h), 2,32 (s, 3h), 2,28 (m, 1H), 2,08 (m, 2H), 1,95 (m, 2H), 1,49 (m, 4H).
Etapa 7a: ácido c/s-4-(2,4-didoro-3-((6-ciano-1,4-dimetiM H-mdol-2-il)metil)fenoxi)ddohexanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido c/s-4-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico (14,5 mg, 54%) se obtuvo como un sólido de color blanco a partir de etil c/s-4-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi) ciclohexanocarboxilato (26 mg, 0,052 mmol). LC/MS (Método g)
Rt = 1,89 min.; MS m/z: 471 [M+H]+1H NMR (DMSO-cfe, 500 MHz): 512,10 (ancho, 1H), 7,91 (s, 1H), 7,49 (d, J = 9Hz, 1H), 7,26 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,69 (s, 1H), 4,72 (m, 1H), 4,41 (s, 2H), 3,90 (s, 3H), 2,39 (m, 1H), 2,32 (s, 3H), 1,87 (m, 2H), 1,77 (m, 2H), 1,69 (m, 4h).
Ejemplo AG: ácido trans -3-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-M)metM)fenoxi)ddopentanocarboxíMco
Ejemplo AG1: ácido c/s-3-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-N)metN)fenoxi)ddopentanocarboxNico

Etapa 1: etil trans -3-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-il)metM)fenoxi)ddopentanocarboxMato y etil c/s-3-(2,4-didoro-3-((6-dano-1,4-dimetiMH-mdol-2-M)metM)fenoxi)ddopentanocarboxMato

Usando un procedimiento similar al Ejemplo AF, Etapa 6 , etil írans-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil) fenoxi)ddopentanocarboxilato (52 mg, 32%) LC/mS (Método h) Rt = 3,62 min.; mS m/z: 485 [M+H]+ 1H NMR (DmSo-C6 , 300 MHz): 57,90 (s, 1H), 7,51 (d, J = 9Hz, 1H), 7,21 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,68 (s, 1H), 5,05 (m, 1H), 4,40 (s, 2H), 4,07 (q, J = 6 Hz, 2H), 3,89 (s, 3H), 3,01 (m, 1H), 2,32 (s, 3H), 2,09 (m, 4H), 1,81 (m, 2h), 1,18 (t, J = 6 Hz, 3H); y etil c/'s-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ddopentanocarboxilato (15 mg, 9%) se obtuvieron a partir de 2-(2,6-didoro-3-hidroxibendl)-1,4-dimetil-1H-indol-6-carbonitrilo (Ejemplo AF, Etapa 5) (100 mg, 0,290 mmol) y etil 3-((metilsulfonilo)oxi)ciclopentanocarboxilato (Preparación #1 2 ) (90 mg, 0,381 mmol) LC/MS (Método h) Rt = 3,54 min.; MS m/z: 485 [M+H]+1H NMR (DMSO-C6 , 300 MHz): 57,90 (s, 1h), 7,50 (d, J = 9Hz, 1H), 7,20 (d, J = 9Hz, 1H), 7,11 (s, 1H), 5,70 (s, 1H), 4,97 (m, 1H), 4,40 (s, 2H), 4,01 (q, J = 6 Hz, 2H), 3,89 (s, 3H), 2,92 (m, 1H), 2,32 (s, 3H), 2,28 (m, 1H), 2,09 (m, 2h), 1,94 (m, 3h), 1,10 (t, J = 6 Hz, 3H).
Etapa 2: ácido trans-3-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclopentanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido frans-3-(2,4-didoro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)cidopentanocarboxílico (35 mg, 48%) se obtuvo a partir de etil fraas-3-(2,4-dicloro-3-((6-dano-1,4-dimetil-1H-ndol-2-il)metil)fenoxi)ciclopentanocarboxilato (73 mg, 0,150 mmol). LC/MS (Método g) Rt = 1,85 min.; MS miz: 457 [M+H]+ 1H NMR (DMSO-cfe, 500 MHz): 512,21 (ancho, 1H), 7,90 (s, 1H), 7,50 (d, J = 9Hz, 1H), 7,22 (d, J = 9Hz, 1H), 7,10 (s, 1H), 5,69 (s, 1H), 5,04 (m, 1H), 4,40 (s, 2H), 3,89 (s, 3H), 2,94 (m, 1H), 2,32 (s, 3H), 2,11 (m, 2H), 2,05 (m, 2h), 1;80 (m, 2H).
Etapa 2a: ácido c/s-3-(2,4-dicloro-3-((6-dano-1,4-dimetiM H-mdol-2-il)metil)fenoxi)cidopentanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido c/s-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclopentanocarboxílico (5 mg, 21%) se obtuvo a partir de etil c/s-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclopentanocarboxilato (24 mg, 0,049 mmol). LC/MS (Método g) Rt = 1,83 min.; MS miz: 457 [M+H]+ 1H NMR (DMSO-Ca, 500 MHz): 512,15 (ancho, 1H), 7,90 (s, 1H), 7,50 (d, J = 9Hz, 1h), 7,19 (d, J = 9Hz, 1H), 7,10 (s, 1H), 5,69 (s, 1H), 4,94 (m, 1H), 4,40 (s, 2H), 3,89 (s, 3h), 2,81 (s, 1H), 2,36 (m, 1H), 2,32 (s, 3H), 1,94 (m, 5H).
Ejemplo AH: ácido frans-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-mdol-2-il)metil)fenoxi)ddobutanocarboxNico Ejemplo AH1: ácido c/s-3-(2,4-didoro-3-((6-dano-1,4-dim etiMH-m dol-2-il)m etil)fenoxi)ddobutanocarboxNico

Etapa 1: etil frans-3-(2,4-didoro-3-((6-dano-1,4-dimetiMH-m dol-2-il)metil)fenoxi)ddobutanocarboxilato y etil c/s-3-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxilato

Usando un procedimiento similar al Ejemplo AF, Etapa 6 , metil frans-3-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1/-/-indol-2-il)metil)fenoxi)cidobutanocarboxilato (200 mg, 29%) LC/MS (Método h) Rt = 3,37 min.; MS miz: 457 [M+H]+1H NMR (DMSO-cfe, 300MHz): 5 7,91 (s, 1H), 7,49 (d, J=8,9 Hz, 1H), 7,11 (s, 1H), 6,99 (d, J=8,9 Hz, 1H), 5,68 (s, 1H), 4,96 (m, 1H), 4,41 (s, 2H), 3,90 (s, 3H), 3,67 (s, 3H), 3,23 (m, 1H), 2,73 (m, 2H), 2,43 (m, 2H), 2,32 (s, 3H) y metil cis- 3-(2,4-didoro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)cidobutanocarboxilato (75 mg, 11 %) LC/MS (Método h) Rt = 3,30 min.; MS miz: 457 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 57,91 (s, 1H), 7,50 (d, J=8,9 Hz, 1H), 7,11 (s, 1H), 7,05 (d, J=9,1 Hz, 1H), 5,68 (m, 1H), 4,79 (m, 1H), 4,41 (s, 2H), 3,90 (s, 3H), 3,63 (s, 3H), 2,80 (m, 3H), 2,32 (s, 3H), 2,25 (m, 2H) se obtuvieron a partir de 2-(2,6-didoro-3-hidroxibendl)-1,4-dimetil-1H-indol-6-carbonitrilo (Ejemplo AF, Etapa 5) (400 mg, 1,16 mmol) y metil 3-((metilsulfonil)oxi)ciclobutanocarboxilato (descrito en W O2011/035159) (314 mg, 1,50 mmol). Etapa 2: ácido frans-3-(2,4-dicloro-3-((6-dano-1,4-dimetil-1H-mdol-2-il)metil)fenoxi)dclobutanocarboxNico

Usando un procedimiento similar al Ejemplo A, Etapa 5 ácido fraas-3-(2,4-didoro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxílico (130 mg, 71%) se preparó a partir de metil fraas-3-(2,4-dicloro-3-((6-dano-1,4-dimetil-fH-indol-2-il)metil)fenoxi)cidobutanocarboxilato ( 180 mg, 0,38 mmol). LC/MS (Método g) Rt = 1,79 min.; MS miz: 443 [M+H]+ 1H NMR (DMSO-d6, 500MHz): 5 12,40 (ancho, 1H), 7,91 (s, 1H), 7,49 (d, J=8,9 Hz, 1H), 7,11 (s, 1H), 7,00 (d, J=9,1 Hz, 1H), 5,70 (s, 1H), 4,96 (m, 1H), 4,42 (s, 2H), 3,91 (s, 3H), 3,11 (m, 1H), 2,72 (m, 2H), 2,41 (m, 2H), 2,33 (s, 3H). Etapa 2a: ácido c/s-3-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido cis -3-(2,4-dicloro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxílico (35 mg, 44%) se preparó a partir de metil c/s-3-(2,4-dicloro-3-((6-dano-1,4dimetil-1 H-indol-2-il)metil)fenoxi)ciclobutanocarboxilato (70 mg, 0,15 mmol). LC/MS (Método g) Rt = 1,76 min.; MS m/z: 443 [M+H]+ 1H NMR (DMSO-d6, 500MHz): 5 12,34 (ancho, 1H), 7,91 (s, 1H), 7,50 (d, J=8,9 Hz, 1H), 7,10-7,15 (m, 1H), 7,06 (d, J=8,9 Hz, 1H), 5,69 (s, 1H), 4,78 (m, 1H), 4,42 (s, 2H), 3,91 (s, 3H), 2,77 (m, 3H), 2,33 (s, 3H), 2,24 (m, 2H).
Ejemplo AI: ácido írans-4-(2,4-dicloro-3-((1,4-dimetil-6- (trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico
Ejemplo AI1: ácido cis -4-(2,4-dicloro-3-((1,4-dimetil-6- (trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico

Etapa 1: etil íraas-4-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-mdol-2-il)metil)fenoxi) ciclohexanocarboxilato y etil cis -4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil) fenoxi) ciclohexanocarboxilato

Usando un procedimiento similar al del Ejemplo AF, Etapa 6 etil fraas-4-(2,4-didoro-3-((1,4-dimetil-6-(trifiuorometil)-1H-indol-2-il)metil)fenoxi)cidohexanocarboxilato (48 mg, 14%) LC/MS (Método h) Rt = 3,99 min.; MS miz: 542 [M+H]+ 1 H NMR (DMSO-da, 300MHz): 57,69 (s, 1H), 7,49 (d, J=9,1 Hz, 1H), 7,30 (d, J=9,1 Hz, 1h), 7,05 (s, 1H), 5,66 (s, 1H), 4,47 (m, 2H), 4,40 (q, J=7,1 Hz, 2H), 4,07 (m, 2H), 3,91 (s, 3H), 3,30 (s, 1H), 2,32 (m, 4H), 2,08 (m, 1H), 1,95 (m, 1h), 1,52 (m, 4h), 1,19 (t, J=7,1 Hz, 3h) y etil c/s-4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxilato (70 mg, 25%) LC/MS (Método h) Rt = 3,96 min.; MS miz: 542 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 57,69 (s, 1H), 7,49 (d, J=9,1 Hz, 1H), 7,26 (d, J=9,1 Hz, 1H), 7,06 (s, 1H), 5,66 (s, 1H), 4,74 (m, 1H), 4,41 (m, 2H), 4,05 (q, J=7,0 Hz, 2H), 3,91 (m, 4H), 3,30 (s, 1H), 2,36 (m, 4H), 1,87 (m, 2H), 1,65 (m, 4H), 1,16 (t, J=7,1 Hz, 3H) se preparó a partir de 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenol (200 mg, 0,51 mmol) (Ejemplo X) y etil 4-((metilsulfonil)oxi)ciclohexanocarboxilato (193 mg, 0,77 mmol).
Etapa 7: ácido frans-4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi)ddohexanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido fraas-4-(2,4-didoro-3-((1,4-dimetil-6-(trifiuorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico (13 mg, 28%) se preparó a partir de etil íraas-4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2 -il)metil)fenoxi)ddohexanocarboxilato (48 mg, 0,088 mmol). LC/mS (Método g) Rt = 2 , 1 2 min.; MS miz: 514 [M+H]+ 1 H NMR (DMSO-d6 , 400MHz): 512,15 (broad 1H), 7,69 (s, 1H), 7,49 (d, J=9,0 Hz, 1H), 7,30 (d, J=9,0 Hz, 1H), 7,05 (s, 1H), 5,66 (s, 1H), 4,46 (m, 1H), 4,40 (s, 2H), 3,91 (s, 3H), 2,32 (s, 3H), 2,30 (m, 1H), 2,08 (m, 2H), 1,97 (m, 2h), 149 (m, 4H).
Etapa 7a: ácido crs-4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido c/s-4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)cidohexanocarboxílico (23 mg, 46%) se preparó a partir de etil c/s-4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2 -il)metil)fenoxi)cidohexanocarboxilato (52 mg, 0,096 mmol). LC/mS (Método g) Rt = 2 , 1 1 min.; MS m/z: 514 [M+H]+1H NMR (DMSO-d6 , 400MHz): 512,08 (ancho, 1H), 7,69 (s, 1H), 7,49 (d, J=9,0 Hz, 1H), 7,26 (d, J=9,0 Hz, 1H), 7,05 (s, 1H), 5,66 (s, 1H), 4,72 (m, 1H), 4,41 (s, 2H), 3,91 (s, 3H), 2,36 (s, 4H), 1,87 (ancho, 2H), 1,78 (t, J=10,2 Hz, 2h), 1,61-1,74 (m, 4H).
Ejemplo AJ: ácido trans-4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi)ddohexanocarboxílico
Ejemplo AJ1: ácido cis -4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi)ddohexanocarboxílico
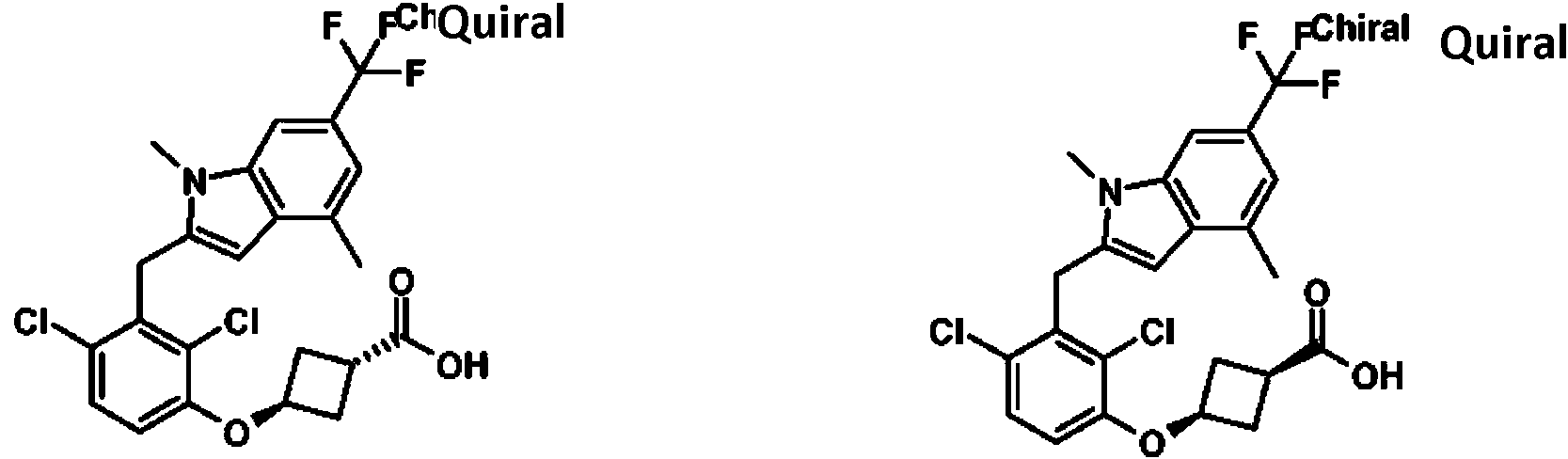
Etapa 1: metil trans -3-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi) ciclobutanocarboxilato y metil cis -3-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi)ciclobutanocarboxilato

Usando un procedimiento similar al del Ejemplo AF, Etapa 6 , metil trans -3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxilato (200 mg, 63%) LC/MS (Método h) Rt = 3,72 min.; MS m/z: 500 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 57,69 (s, 1H), 7,49 (d, J=8,9 Hz, 1H), 7,06 (s, 1H), 6,99 (d, J=8,9 Hz, 1H), 5,66 (s, 1H), 4,97 (m, 1H), 4,41 (s, 2H), 3,91 (s, 3H), 3,67 (s, 3H), 3,23 (m, 1H), 2,72 (m, 2H), 2,44 (m, 2H), 2,36 (s, 3h) y metil c/s-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)fenoxi) ciclobutanocarboxilato
LC/MS (Método h) Rt = 3,65 min.; MS m/z: 500 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 57,69 (s, 1H), 7,50 (d, J=8,9 Hz, 1H), 7,02-7,09 (m, 2H), 5,65 (s, 1H), 4,80 (m, 1H), 4,40 (s, 2H), 3,91 (s, 3H), 3,63 (s, 3H), 2,86 (m, 3H), 2,36 (s, 3H), 2,26 (m, 2H) se prepararon a partir de 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)fenol (Ejemplo IA, Etapa 5) (200 mg, 0,51 mmol) y metil 3-((metilsulfonil)oxi)ciclobutanocarboxilato (161 mg, 0,77 mmol).
Etapa 2: ácido trans -4-(2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)fenoxi)ddohexanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido trans -4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)cidohexanocarboxílico (125 mg, 98%) se preparó a partir de metil trans -3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)fenoxi)ciclobutanocarboxilato (130 mg, 0,26 mmol). LC/MS (Método g) Rt = 2,05 min.; MS m/z: 486 [M+H]+1H NMR (DMSO-da, 400MHz): 512,36 (ancho, 1H), 7,69 (s, 1H), 7,48 (d, J=9,0 Hz, 1H), 7,05 (s, 1H), 6,99 (d, J=9,0 Hz, 1H), 5,66 (s, 1H), 4,95 (m, 1H), 4,41 (s, 2H), 3,91 (s, 3h), 3,12 (m, 1H), 2,70 (m, 2H), 2,41 (m, 2H), 2,36 (s, 3H).
Etapa 2a: ácido cis -4-(2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)fenoxi) ciclohexanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, c/s-4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclohexanocarboxílico (25 mg, 70%) se preparó a partir de metil c/s-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclobutanocarboxilato (33 mg, 0,066 mmol). LC/MS (Método g) Rt = 2,02 min.; MS m/z: 486 [M+H]+ 1 H NMR (DMSO-d6 , 400MHz): 512,35 (ancho, 1H), 7,69 (s, 1H), 7,49 (d, J=8 , 8 Hz, 1H), 7,03-7,08 (m, 2H), 5,66 (s, 1H), 4,78 (m, 1H), 4,40 (s, 2H), 3,91 (s, 3H), 2,76 (m, 3H), 2,36 (s, 3H), 2,22 (m, 2H).
Ejemplo AK: ácido trans-3-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-il)metil)fenoxi)ciclopentanocarboxílico
Ejemplo AK1: ácido cis-3-(2,4-dicloro-3-((1,4-dimetil-6- (trifluorometil)-1H-indol-2-il)metil)fenoxi)ciclopentanocarboxílico

Etapa 1: etil fraas-3-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-mdol-2-il)metil)fenoxi) ciclopentanocarboxilato y etil c/s-3-(2,4-didoro-3-((l,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi) ciclopentanocarboxilato

Usando un procedimiento similar al del Ejemplo AF, Etapa 6 , etil trans-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)cidopentanocarboxilato (125 mg, 46%) LC/MS (Método h) Rt = 3,94 min.; MS m/z: 528 [M+H]+1H NMR (DMSO-da, 300MHz): 57,69 (s, 1H), 7,51 (d, J=9,1 Hz, 1H), 7,22 (d, J=9,1 Hz, 1H), 7,06 (s, 1H), 5,66 (s, 1H), 5,06 (m, 1H), 4,40 (s, 2H), 4,03 (q, J= 7,1Hz, 2H), 3,91 (s, 3H), 3,02 (m, 1H), 2,36 (s, 3h), 2,10 (m, 4H), 1,80 (m, 2H), 1,20 (t, J= 7,1Hz, 3H) y etil c/s-3-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenoxi)cidopentanocarboxilato (55 mg, 20%) (que contiene 20% de isómero trans) LC/MS (Método h) Rt = 3,86 min.; MS m/z: 528 [M+H]+1H NMR (DMSO-d6 , 300MHz): 57,69 (s, 1H), 7,50 (d, J=9,1 Hz, 1H), 7,19 (d, J=9,1 Hz, 1H), 7,05 (s, 1H), 5,67 (s, 1H), 4,97 (m, 1H), 4,39 (s, 3H), 3,02 (q, J=7,1 Hz, 2H), 3,91 (s, 3H), 2,92 (m, 1H), 2,32 (m, 4H), 1,95 (m, 4H), 1,11 (t, J=7,1 Hz, 3H)
se prepararon a partir de 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenol (Ejemplo AI, Etapa 5) (200 mg, 0,51 mmol) y etil 3-((metilsulfonil)oxi)ciclopentanocarboxilato (Preparación #12) (183 mg, 0,77 mmol).
Etapa 2: ácido frans-3-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)fenoxi)ddopentanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido trans-3-(2,4-dicloro-3-(((trifluorometil)-1H-indol-2-il)metil) fenoxi-6-1,4-dimetil) ciclopentanocarboxílico (80 mg, 6 8 %) se preparó a partir de acetato de trans-3-(2,4-dicloro-3-((1,4-dimetil-6 -(trifluorometil)-1 H-indol-2 -il)metil)fenoxi) ciclopentanocarboxilato ( 1 2 2 mg, 0,23 mmol). LC/Ms (Método g) Rt = 2,09 min.; MS m/z: 500 [M+H]+ 1H NMR (DMSO-d6 , 400MHz): 512,21 (ancho, 1H), 7,69 (s, 1H), 7,51 (d, J=9,0 Hz, 1H), 7,22 (d, J=9,0 Hz, 1H), 7,05 (s, 1H), 5,66 (s, 1H), 5,04 (m, 1H), 4,40 (s, 2H), 3,91 (s, 3H), 2,95 (m, 1H), 2,36 (s, 3H), 2,08 (m, 4H), 1,81 (m, 2H).
Etapa 2a: ácido c/s -3-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-mdol-2-il)metil)fenoxi)cidopentanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido cis -3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2- il)metil)fenoxi)cidopentanocarboxílico (18 mg, 34%) (que contiene 20% de isómero trans) se preparó a partir de etil cis- 3- (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil) fenoxi)ciclopentanocarboxilato (52 mg, 0,098 mmol). LC/MS (Método g) Rt = 2,07 min.; MS m/z: 500 [M+H]+1H NMR (DMSO-da, 400MHz): 512,14 (ancho, 1H), 7,69 (s, 1h), 7,50 (d, J=9.2 Hz, 1H), 7,19 (d, J=9.2 Hz, 1H), 7,05 (s, 1H), 5,66 (s, 1H), 4,95 (m, 1h), 4,39 (s, 2H), 3,91 (s, 3H), 2,80 (m, 1H), 2,36 (m, 4H), 1,92 (m, 5H).
Ejemplo AL: 2-((3,5-didoro-2-((tetrahidro-2H-piran-4-N) metoxi)piridin-4-il)metil)-1,4-dimetil-6-(trifluorometil)-1 H-indol

Etapa 1: 3,5-didoro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-2-ol

Usando un procedimiento similar al Ejemplo X, 3,5-didoro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-2-ol (716 mg, 100%) se preparó a partir 2-((3,5-didoro-2-metoxipiridin-4-il)metil)-1,4-dimetil-6-(trifluorometil)-1H-indol (Ejemplo Z, Etapa 4) (647 mg, 1,6 mmol). LC/MS (Método h) Rt = 2,68 min.; MS m/z: 389 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 5 12,60 (ancho, 1H), 7,81 (s, 1H), 7,70 (s, 1H), 7,07 (s, 1H), 5,97 (s, 1H), 4,31 (s, 2H), 3,89 (s, 3H), 2,41 (s, 3H).
Etapa 2: 2-((3,5-didoro-2-((214etal214ydro-2H-piran-4-il)metoxi)piridin-4-il)214etal)-1,4-dimetil-6-(trifluorometil)-1H-indol y 3,5-didoro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)214etal)-1-((214etal214ydro-2H-piran-4-il)214etal)piridin-2(1H)-ona

A una solución de 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-2-ol (200 mg, 0,514 mmol) en DMF (2,6 mL) se añadió carbonato de potasio (284 mg, 2,056 mmol) y 4-(bromometil)tetrahidropirano (0,135 mL, 1,028 mmol) y la mezcla de reacción se agitó a 100 °C durante 2 horas. La mezcla de reacción se diluyó con agua y la capa acuosa obtenida se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 5-100% en DCM) y luego por LCMS preparativa para dar 2-((3,5-dicloro-2-((tetrahidro-2H-piran-4-il) metoxi)piridin-4-il)metil)-1,4-dimetil-6-(trifluorometil)-1 H-indol (108 mg, 40%) como un sólido de color blanco LC/MS (Método g) Rt = 2,26 min.; MS m/z: 487 [M+H]+ 1 H NMR (DMSO-d6 , 400MHz): 5 8,32 (m, 1H), 7,70 (s, 1H), 7,07 (s, 1H), 5,80 (s, 1H), 4,42 (s, 2H), 4,24 (d, J=6,3 Hz, 2H), 3,91 (s, 3H), 3,88 (m, 2H), 3,34 (m, 2H), 2,38 (s, 3H), 2,07 (m, 1H), 1,68 (m, 2H), 1,39 (m, 2H) y 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)- 1H-indol-2-il)metil)-1-((tetrahidro-2H-piran-4-il)metil)piridin-2(1H)-ona (46 mg, 18%) como un sólido de color blanco. LC/MS (Método g) Rt = 1,84 min.; MS m/z: 487 [M+H] + 1 H NMR (DMSO-d6 , 400MHz): 5 8,14 (s, 1H), 7,69 (s, 1H), 7,07 (s, 1H), 6,00 (s, 1H), 4,30 (s, 2H), 3,82-3,95 (m, 7H), 3,20-3,30 (m, 2H), 2,41 (s, 3H), 2,09 (m, 1H), 1,48 (m, 2H), 1,30 (m, 2H).
Ejemplo AM: ácido 2-((1-(2,4-dicloro-3-((6-ciano-1,4-dimetiMH -indol-2-il)metil)benzoil)piperidin-4-il)oxi)acético

Etapa 1: etil 2-((1-(2,4-dicloro-3-((6-ciano-1,4-dimetil- 1H-indol-2-il)metil)benzoil)piperidin-4-il)oxi)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, etil 2-((1-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)oxi)acetato (337 mg, 88%) se obtuvo como un aceite de color naranja a partir de ácido 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoico (Ejemplo AA, Etapa 5) (150 mg, 0,402 mmol) y clorhidrato de
W-1-((etilimino)metileno)-W3,W3-dimetilpropano-1,3-diamina (100 mg, 0,52 mmol) y se usó crudo en la siguiente etapa. LC/MS (Método h) Rt = 2,90 min.; MS m/z: 542 [M+H]+
Etapa 2: ácido 2-((1-(2,4-didoro-3-((6-ciano-1,4-dimetil- 1H-mdol-2-M)metM)benzoM)piperidm-4-M)oxi)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-((1-(2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)oxi)acético (33 mg, 18%) se obtuvo como un sólido de color blanco a partir de etil 2-((1-(2,4-dicloro-3-((6-dano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)oxi)acetato (336 mg, 0,353 mmol). LC/MS (Método g) Rt = 1,52 min.; MS m/z: 514 [M+H]+ 1H NMR (DMSO-da, 400 MHz): 5 12,57 (ancho, 1H), 7,92 (s, 1H), 7,66 (d, J = 9Hz, 1H), 7,46 y 7,42 (d, J = 9Hz, 1H), 7,12 (s, 1H), 5,70 (m, 1H), 4,47 (m, 2H), 4,05 y 4,03 (s, 2H), 3,96 (m, 1H), 3,90 (s, 3H), 3,63 (m, 1H), 3,37 (m, 2H), 3,05 (m, 1H), 2,33 (s, 3H), 1,91 (m, 1H), 1,78 (m, 1H), 1,60-1,40 (m, 2H).
Tabla AM. Los siguientes Ejemplos se prepararon usando el mismo procedimiento a partir de los ésteres a ro iados de la Tabla AA.

Ejemplo AN: 2-(2,6-didoro-3-(4-hidroxipiperidma-1-carbonM)benzoM)-1,4-dimetiMH-mdol-6-carbomtrMo

Etapa 1: metil 2,4-dicloro-3-(6-ciano-4-metiM-(femlsulfoml)-1H-mdol-2-carboml)benzoato

Usando un procedimiento similar al del Ejemplo AD, Etapa 2, metil 2,4-didoro-3-(6-tiano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)benzoato (5,98 g, 100 %) se obtuvo como una espuma de color beige a partir de metil 2,4-dicloro-3-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (Ejemplo AA, Etapa 1) (6 g, 11,33 mmol). LC/MS (Método i) Rt = 2,57 min.; MS m/z: 527 [M+H]+ 1H NMR (DMSO-de, 300MHz): 58,54 (s, 1H), 8,29 (m, 2H), 8,05 (m, 2H), 7,68-7,91 (m, 4H), 7,63 (m, 1H), 3,88 (s, 3H), 2,26 (s, 3H)
Etapa 2: metil 2,4-dicloro-3-(6-ciano-4-metiM-(femlsulfoml)-1H-mdol-2-carboml)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-dicloro-3-(6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)benzoato (3,32 g, 76 %) se obtuvo como una espuma de color beige a partir de metil 2,4-dicloro-3-(6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)benzoato (5,98 g, 11,34 mmol). LC/MS (Método i) Rt = 2,33 min.; MS m/z: 387 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 512,72 (s, 1H), 8,03 (d, J=8,4 Hz, 1H), 7,80 (m, 2H), 7,15 (m, 2H), 3,90 (s, 3H), 2,50 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-(6-ciano-1,4-dimetiMH -indol-2-carbonil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 4, metil-2,4-dicloro-3-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)benzoato (3,34 g, 97%) se obtuvo como un sólido de color blanco a partir de metil 2,4-dicloro-3-(6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)benzoato (3,32 g, 8,57 mmol). LC/MS (Método i) Rt = 2,49 min.; MS miz: 401 [M+H]+ 1H NMR (DMSO-da, 300MHz): 58,20 (s, 1H), 8,03 (d, J=8,1 Hz, 1H), 7,79 (d, J=8,1 Hz, 1H), 7,26-7,34 (m, 1H), 7,20 (s, 1H), 4,20 (s, 3H), 3,90 (s, 3H), 2,47 (s, 3H)
Etapa 4: ácido 2,4-didoro-3-(6-dano-1,4-dimetiMH-indol-2-carbom l)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, se obtuvo ácido 2,4-dicloro-3-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)benzoico (3,22 g, 100%) como un sólido de color blanco a partir de 2,4-dicloro-3-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)benzoato (3,34 g, 8,32 mmol).
LC/MS (Método i) Rt = 2,12 min.; MS miz: 387 [M+H]+ 1 H NMR (DMSO-da, 300MHz): 5 8,20 (s, 1H), 8,00 (d, J=8,6 Hz, 1H), 7,75 (d, J=8,6 Hz, 1H), 7,29 (m, 1H), 7,17 (m, 1H), 4,20 (s, 3H), 2,47 (s, 3H).
Etapa 5: 2-(2,6-didoro-3-(4-hidroxipiperidma-1-carbonM)benzoN)-1,4-dimetiMH-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-(2,6-dicloro-3-(4-hidroxipiperidina-1-carbonil)benzoil)-1,4-dimetil-1H-indol-6-carbonitrilo (101 mg, 83%) se obtuvo como un sólido de color amarillo a partir de ácido 2,4-dicloro-3-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)benzoico (100 mg, 0,26 mmol) y piperidin-4-ol (39 mg, 0,38 mmol). LC/MS (Método g) Rt = 1,47 min.; MS miz: 470 [M+H] + 1 H NMR (DMSO-d6 , 400MHz): 5 8,20 (s, 1H), 7,74 (d, J=8,4 Hz, 1H), 7,61 (m, 1H), 7,29 (s, 1H), 7,20 y 7,07 (m, 1H), 4,81 y 4,76 (m, 1H), 4,20 y 4,19 (s, 3H), 4,02 (m, 1H), 3,75 (m, 1H), 3,25 (m, 1H), 3,08 (m, 1H), 2,48 (s, 3H), 1,70 (m, 2H), 1,35 (m, 2H).

Ejemplo AO: ácido 2-(1-(2,4-dicloro-3-(6-ciano-1,4-dimetiMH-mdol-2-carboml)benzoil)piperidm-4-il) acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, 2-(1-(2,4-dicloro-3-(6-dano-1,4-dimetil-1H-indol-2-carbonil)benzoil)piperidin-4-il)acético (107 mg, 94%) se obtuvo como un sólido de color blanco a partir de metil 2-(1-(2,4-didoro-3-(6-dano-1,4-dimetil-1H-indol-2-carbonil)benzoil)piperidin-4-il) acetato (Tabla AN, Ejemplo AN-6 ) (116 mg, 0,22 mmol). LC/MS (Método g) Rt = 1,54 min.; MS m/z: 512 [M+H]+ 1H NMR (DMSO-da, 400MHz): 512,12 (broad., 1H), 8,20 (s, 1H), 7,75 (m, 1H), 7,63 y 7,57 (d, J=8,4 Hz, 1H), 7,29 (s, 1H), 7,01-7,26 (m, 1H), 4,47 (m, 1H), 4,20 y 4,19 (s, 3H), 3,55 (m, 1H), 3,06 (m, 1H), 2,81 (m, 1H), 2,48 (m, 3H), 2,19 (m, 1h), 2,13 (m, 1H), 1,95 (m, 1H), 1,77 (m, 1H), 1,64 (m, 1H), 1,14 (m, 2H).
Ejemplo AP: (2,4-didoro-3-(1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-carboml)feml)(morfolmo)metanona
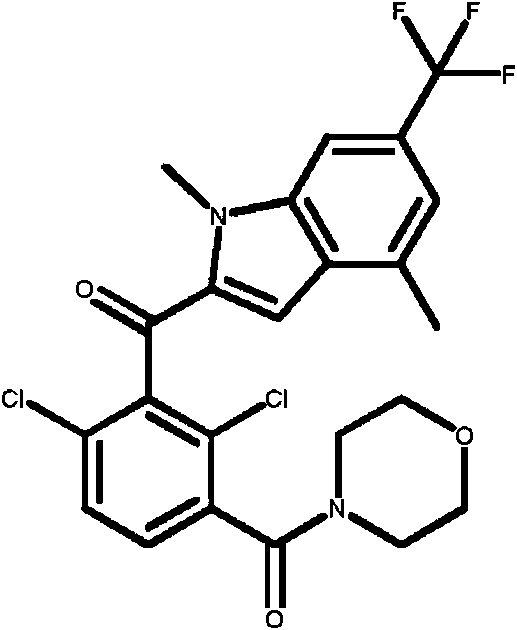
Etapa 1: metil 2,4-didoro-3-(4-metiM-(femlsulfoml)-6-(trifluorometN)-1H-mdol-2-carboml)benzoato

Usando un procedimiento similar al del Ejemplo AD, Etapa 2, metil 2,4-didoro-3-(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-carbonil)benzoato (5,88 g, 100%) se obtuvo como una espuma de color beige a partir de 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)benzoato (Ejemplo A, Etapa 1) (5,9 g, 10,3 mmol). LC/MS (Método i) Rt = 2,80 min.; MS m/z: 570 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 58,37 (s, 1H), 8,12 (m, 2H), 8,01 (d, J=8,4 Hz, 1H), 7,70 (m, 5H), 7,58 (s, 1H), 3,88 (s, 3H), 2,56 (s, 3H).
Etapa 2: metil 2,4-didoro-3-(4-metiM-(femlsulfoml)-6-(trifluorometN)-1H-mdol-2-carboml)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-didoro-3-(4-metil-1-(fenilsulfonil)-6-(trifiuorometil)-1H-indol-2-carbonil)benzoato (4 g, 79%) se obtuvo como una espuma incolora a partir de metil 2,4-dicloro-3-(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-carbonil)benzoato (5,88 g, 10,3 mmol). LC/MS (Método i) Rt = 2,59min.; MS miz:
430 [M+H]+ 1H NMR (DMSO-da, 300MHz): 512,64 (s, 1H), 8,03 (d, J=8,4 Hz, 1H), 7,80 (d, J=8,4 Hz, 1H), 7,62 (s, 1H), 7,18 (s, 2H), 3,90 (s, 3H), 2,56 (s, 3H).
Etapa 3: metil 2,4-didoro-3-(1,4-dimetN-6-(trifluorometM)-1H-mdol-2-carboml)benzoato
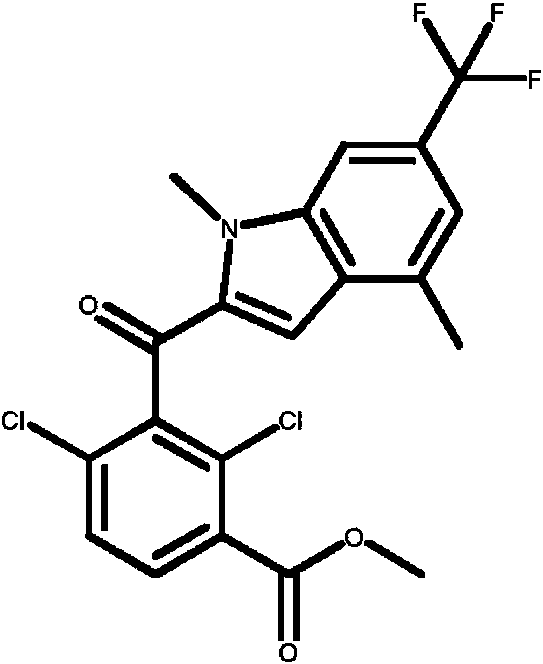
Usando un procedimiento similar al Ejemplo A, Etapa 4, metil 2,4-didoro-3-(1,4-dimetil-6-(trifluorometil)-1H -indol-2-carbonil)benzoato (2,45 g, 92 %) se obtuvo como un sólido de color blanco a partir de metil 2,4-dicloro-3-(4-metil-6-(trifluorometil)-1H-indol-2-carbonil)benzoato (2,58 g, 6 mmol). LC/MS (Método i) Rt = 2,75 min.; MS miz: 444 [M+H]+ 1 H NMR (DMSO-d6 , 300MHz): 58,02 (d, J=8,5 Hz, 1h), 7,96 (s, 1H), 7,79 (d, J=8,5 Hz, 1H), 7,24 (s, 1H), 7,18 (s, 1H), 4,24 (s, 3H), 3,32 (s, 3H), 2,50 (s, 3H).
Etapa 4: ácido 2,4-didoro-3-(1,4-dimetM-6-(trifluorometN)-1H-mdol-2-carbonM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil) benzoico (2,37 g, 100%) se obtuvo como un sólido de color blanco a partir de metil 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)benzoato (2,45 g, 5,52 mmol). LC/MS (Método i) Rt = 2,42 min.; MS miz: 430 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 13,84 (ancho, 1H), 7,99 (d, J=8,4 Hz, 1H), 7,96 (s, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,24 (s, 1H), 7,15 (s, 1H), 4,24 (s, 3H), 2,50 (s, 3H).
Etapa 5(2,4-didoro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)fenil)(morfolino)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)fenil)(morfolino)metanona (101 mg, 87 %) se obtuvo como un sólido de color blanco a partir de ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)benzoico (100 mg, 0,23 mmol) y morfolina (26,3 mg, 0,30 mmol). LC/MS (Método g) Rt = 1,87 min.; MS miz: 499 [M+H]+1H NMR (DMSO-da, 400MHz): 5 7,96 (s, 1H), 7,76 (d, J=8,1 Hz, 1H), 7,65 (d, J=8,1 Hz, 1H), 7,30 y 7,09 (m, 1H), 7,24 (s, 1H), 4,23 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,36 (m, 1H), 3,15 (m, 1H), 2,50 (s, 3H).
Tabla AP. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1 H-indol-2-carbonil) benzoico (Ejemplo Ap, Etapa 4) usando el mismo procedimiento con la amina apropiada.


Ejemplo AQ: ácido 2-(1-(2,4-didoro-3-(1,4-dimetN-6-(trifluorometM)-1H-mdol-2-carboml)benzoN)piperidm-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)benzoil)piperidin -4-il)acético (108 mg, 82%) se obtuvo como un sólido de color blanco a partir de ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)benzoico (Ejemplo AP, Etapa 4) (100 mg, 0,23 mmol). LC/MS (Método g) Rt = 1,80 min.; MS m/z: 555 [M+H]+
1H NMR (DMSO-da, 400MHz): 5 12,10 (ancho, 1H), 7,96 (s, 1H), 7,74 (m, 1H), 7,63 y 7,57 (d, J=8 Hz, 1H), 7,24-7,00 (m, 2H), 4,47 (m, 1H), 4,23 y 4,22 (s, 3H), 3,20 (m, 1H), 3,06 (m, 1H), 2,81 (m, 1H), 2,50 (s, 3H), 2,20 (m, 2H), 1,94 (m, 1H), 1,78 (m, 1H), 1,65 (m, 1H), 1,15 (m, 2H).
Tabla AQ. Los siguientes ejemplos se prepararon usando el mismo procedimiento a partir de los ésteres apropiados (como se describe en la Tabla AP).

Ejemplo AR: (2,6-didoro-3-(hidroximetil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metanona

A una solución de metil 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)benzoato (Ejemplo AP, Etapa 3) (250 mg, 0,563 mmol) en EtOH MeOH (10 mL) se añadió a temperatura ambiente tetrahidroborato de sodio (63,9 mg, 1,688 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Se añadió más tetrahidroborato de sodio (63,9 mg, 1,688 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 24 horas. La reacción se inactivó con solución acuosa saturada de NH4CL La mezcla de reacción se extrajo con EtOAc y la capa orgánica se secó sobre sulfato de magnesio y se concentró hasta la sequedad. El producto crudo se purificó por LCMs preparativa para dar (2,6-dicloro-3-(hidroximetil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metanona (71 mg, 29%). lC/MS (Método g) Rt = 1,92 min.; MS m/z: 416 [M+H]+
1H NMR (DMSO-d6, 400MHz): 57,95 (s, 1H), 7,73 (d, J=8 Hz, 1H), 7,66 (d, J=8 Hz, 1H), 7,23 (s, 1H), 7,02 (s, 1H), 5,62 (t, J=5,4 Hz, 1H), 4,61 (d, J=5,4 Hz, 2H), 4,23 (s, 3H), 2,50 (s, 3H).
Ejemplo AS: (2,4-didoro-3-((7-metil-5-(trifluorometil)- 1H-mdol-2-M)metM)feml)(morfolmo)metanona

Etapa 1: íerc-butil 2,4-didoro-3-(hidroxi(7-metiM-(femlsulfoml)-5-(tnfluorometM)-1 H- indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, íerc-butil 2,4-dicloro-3-(hidroxi(7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1 H-indol-2-il)metil)benzoato (1,75 g, 49%) se obtuvo como una resina naranja a partir de W-(2-yodo-6-metil-4-(trifluorometil)fenil)bencenosulfonamida (Preparación #34) (2 g, 4,53 mmol) y íerc-butil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #33) (1,23 g, 4,08 mmol). LC/MS (Método i) Rt = 2,84 min.; MS m/z: 672 [M-H]-+ CH3COOH 1H NMR (DMSO-d6, 300MHz): 57,60 (m, 8H), 7,44 (s, 1H), 6,97 (dd, J=5,9 y 1,5Hz, 1H), 6,74 (d, J=1,5 Hz, 1H), 6,66 (d, J=5,9 Hz, 1H), 2,45 (s, 3h), 1,49 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-((7-metiM-(femlsulfoml)-5-(trifluorometN)-1H-mdol-2-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-indol-2-il)metil)benzoico (800 mg, 52%) se obtuvo como un polvo de color amarillo a partir de íerc -butil 2,4-dicloro-3-(hidroxi(7-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-indol-2 -il)metil)benzoato (1,7 g, 2,77 mmol). LC/MS (Método i) Rt =
2,67 min.; MS miz: 542 [M+H]+1H NMR (DMSO-da, 300MHz): 57,51-7,80 (m, 6H), 7,40 (m, 3H), 5,98 (s, 1H), 4,49 (s, 2H), 2,66 (s, 3H).
Etapa 3: ácido 2,4-didoro-3-((-7-metN-5-(trifluorometM)-1H-mdol-2-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, ácido 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-2-il)metil)benzoico (620 mg, 99%) se obtuvo como un polvo de color naranja a partir de 2,4-dicloro-3-((7-metil-1-(fenilsulfonil)-5-(trifluorometil)- 1H-indol-2-il)metil)benzoico (800 mg, 1,47 mmol). Lc/mS (Método i) Rt = 2,37 min.; MS miz:
402 [M+H]+
1H NMR (DMSO-d6, 300MHz): 511,57 (s, 1H), 7,55 (s, 1H), 7,41 (m, 1H), 7,29 (m, 2H), 7,10 (s, 1H), 5,77 (s, 1H), 4,40 (s, 2H), 2,54 (s, 3H).
Etapa 4: (2,4-didoro-3-((7-metM-5-(trifluorometN)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (se obtuvo 95 mg, 20%) como un polvo de color naranja a partir de ácido 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-2-il)metil)benzoico (400 mg, 0,99 mmol). LCiMS (Método g) Rt = 1,79 min.; MS miz: 471 [M+H]+
1H NMR (DMSO-d6, 500MHz): 511,56 (s, 1H), 7,65 (d, J=8,4 Hz, 1H), 7,58 (s, 1H), 7,42 (d, J=8,4 Hz, 1H), 7,12 (s, 1H), 5,80 (m, 1H), 4,45 (s, 2H), 3,65 (m, 4H), 3,55 (m, 2H), 3,17 (m, 2H), 2,54 (s, 3H).
Ejemplo AT: (2,4-didoro-3-((1,7-dimetil-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-dicloro-3-((1,7-dimetil-5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (48 mg, 63%) se obtuvo como un polvo de color beige a partir de (2,4-dicloro-3-((7-metil5-(trifluorometil)-1H-indol-2-il)metil)fenil)(morfolino)metanona (Ejemplo AS, Etapa 4) (70 mg, 0,149 mmol). LC/MS (Método g) Rt = 1,88 min.; MS m/z: 485 [M+H]+
1H NMR (DMSO-d6, 400MHz): 57,67 (d, J=8,1 Hz, 1H), 7,59 (s, 1H), 7,45 (d, J=8,1 Hz, 1H), 7,09 (s, 1H), 5,67 (s, 1H), 4,39 (m, 2H), 4,12 (s, 3H), 3,65 (m, 4h), 3,55 (m, 2H), 3,20 (m, 2H), 2,83 (s, 3H).
Ejemplo AU: 2-(2,6-dicloro-3-((3-oxomorfolino)metil) bencN)-1,4-dimetiMH-mdol-6-carbomtrilo
Ejemplo A U1: 2-(2,6-dicloro-3-((3-oxomorfolino)metil) benzoil)-1,4-dim etiMH -indol-6-carbonitrilo

Etapa 1: 2-(2,6-dicloro-3-(hidroximetil)bencil)-1,4-dimetil-1H -indol-6-carbonitrilo

A una solución de metil 2,4-dicloro-3-((6-ciano-1,4-dimetil-7 H-indol-2-il)metil)benzoato (Ejemplo AA, Etapa 4) (300 mg, 0,775 mmol) en THF (10 mL) se añadió borohidruro de litio (1,162 mL, 2,32 mmol) (2M en THF) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió más borohidruro de litio (0,5 mL, 1,17 mmol) (2M en THF) y la mezcla se agitó a 50 °C durante una hora, luego se enfrió a 0 °C y se inactivó lentamente mediante la adición de solución acuosa de HCl 1M. El THF se concentró, el precipitado se filtró, se lavó con agua y se secó al vacío. El residuo se disolvió en DCM, se secó con sulfato de magnesio y se concentró para dar 2-(2,6-dicloro-3-(hidroximetil)bencil)-1,4-dimetil-1 H-indol-6-carbonitrilo (290 mg, 89%) como un sólido de color blanco. LC/mS (Método h) Rt = 2,77 min.; MS m/z: 359 [M+H]+ 1H NMR (DMSO-de, 300MHz): 57,91 (s, 1H), 7,60 (m, 2H), 7,11 (s, 1H), 5,67 (s, 1H), 5,55 (t, J=5,5 Hz, 1H), 4,59 (d, J=5,5 Hz, 2H), 4,44 (s, 2H), 3,91 (s, 3H), 2,31 (s, 3H).
Etapa 2: 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)bencil metanosulfonato

Usando un procedimiento similar a la Preparación #12, 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)bencil metanosulfonato (327 mg, 54%) se obtuvo como un sólido de color amarillo a partir de 2-(2,6-dicloro-3-(hidroximetil)bencil)-1,4-dimetil-1H-indol-6-carbonitrilo (290 mg, 0,80 mmol). LC/MS (Método h) Rt = 2,95 min.; MS m/z: 437 [M+H]+ 1H NMR (DMSO-de, 300MHz): 57,92 (s, 1H), 7,67 (m, 2H), 7,10 (m, 1H), 5,70 (m, 1H), 5,39 (s, 2H), 4,48 (s, 2H), 3,92 (s, 3H), 3,30 (s, 3H), 2,32 (m, 3H).
Etapa 3: 2-(2,6-didoro-3-((3-oxomorfolino)metil) bencil)-1,4-dim etiMH -indol-6-carbonitrilo
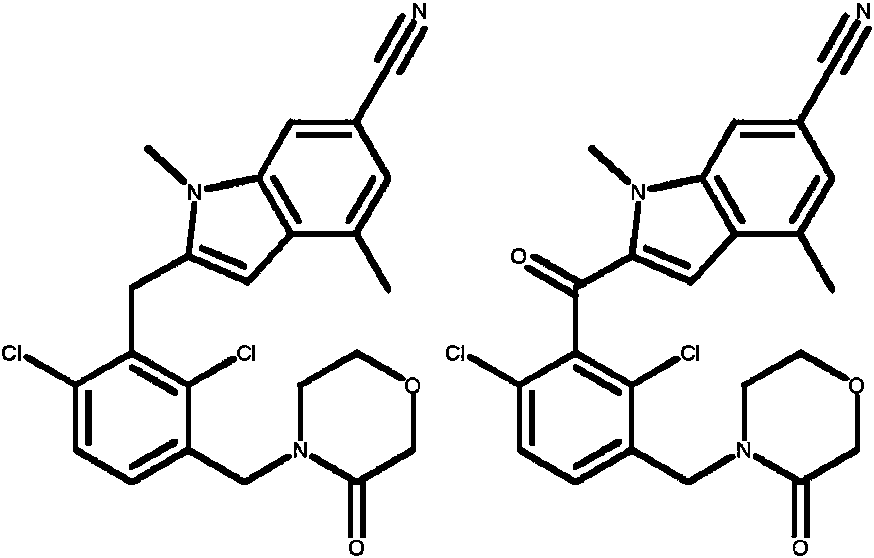
A una solución de morfolin-3-ona (139 mg, 1,372 mmol) en DMF (9 mL) se añadió hidruro de sodio (63,1 mg, 1,578 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 minutos. A la mezcla de reacción se añadió 2,4-dicloro-3-((6-ciano-1,4-dimetiMH-indol-2-il)metil)bencil metanosulfonato (300 mg, 0,686 mmol) y se agitó a temperatura ambiente durante 20 horas. La mezcla de reacción se vertió en una solución de agua y EtOAc; la capa orgánica se extrajo, se lavó con salmuera; se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 30-70% en ciclohexano) y luego se purificó por LCMS preparativa para dar 2-(2,6-didoro-3-((3-oxomorfolino)metil)bencil)-1,4-dimetil-1H-indol-6-carbonitrilo (35 mg, 11%) como un sólido amarillo. LC/MS (Método g) Rt = 1,65 min.; MS m/z: 443 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 57,91 (s, 1H), 7,60 (d, J=8,4 Hz, 1H), 7,30 (d, J=8,4 Hz, 1H), 7,04-7,14 (m, 1H), 5,70 (s, 1H), 4,66 (s, 2H), 4,46 (s, 2H), 4,16 (s, 2h), 3,91 (s, 3H), 3,89 (m, 2H), 3,35 (m, 2H), 2,32 (s, 3H).
Durante esta reacción, se aisló un producto secundario de oxidación y se caracterizó como 2-(2,6-dicloro-3-((3-oxomorfolino)metil)benzoil)-1,4-dimetil-1H-indol-6-carbonitrilo (30 mg, 9%) como un sólido de color blanco. LC/MS (Método g) Rt = 1,62 min.; MS m/z: 456 [M+H]+1H NMR (DMSO-d6, 400MHz): 58,20 (s, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,46 (d, J=8,4 Hz, 1H), 7,29 (m, 1H), 7,09 (s, 1H), 4,67 (m, 2H), 4,20 (s, 3H), 4,17 (s, 2H), 3,91 (m, 2H), 3,40 (m, 2H), 2,47 (s, 3H).
Ejemplo AV: etil 2-(4-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)benzoM)piper-azm-1-N)acetato

Usando un procedimiento similar al del Ejemplo T, Etapa 1, etil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)acetato (51 mg, 49%) se preparó a partir de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2- il)metil)fenil)(piperazin-1-il)metanona (Ejemplo S, Etapa 2) (85 mg, 0,17 mmol) y bromoacetato de etilo (30 mg, 0,18 mmol). LC/MS (Método g) Rt = 1,90 min.; MS m/z: 570 [M+H]+1H NMR (DMSO-d6, 400MHz): 57,70 (s, 1H), 7,67 (d, J=8,1 Hz, 1H), 7,41 (d, J=8,1 Hz, 1H), 7,06 (s, 1H), 5,68 (s, 1H), 4,48 (d, J=16 Hz, 1H), 4,43 (d, J=16 Hz, 1H), 4,06 (q, J=7,2 Hz, 2H), 3,92 (s, 3H), 3,68 (m, 1H), 3,61 (m, 1H), 3,27 (s, 2H), 3,16 (m, 2H), 2,60 (m, 2H), 2,50 (m, 2h), 2,36 (s, 3H), 1,16 (t, J=7,2 Hz, 3H).
Ejemplo AW: ácido 2-(4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)acético

Etapa 1: metil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)acetato

Usando un procedimiento similar al del Ejemplo T, Etapa 1,metil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)acetato (115 mg, 100%) se preparó a partir de (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)fenil)(piperazin-1-il)metanona (Ejemplo S, Etapa 2) (100 mg, 0,206 mmol) y bromoacetato de metilo (42,8 mg, 0,31 mmol). LC/MS (Método h) Rt = 3,11 min.; MS miz: 556 [M+H]+1H NMR (CLOROFORM-d, 300MHz): 57,41 (d, J=8,1 Hz, 1H), 7,36 (s, 1H), 7,19 (d, J=8,1 Hz, 1H), 7,01 (s, 1H), 5,70 (s, 1H), 4,36 (m, 2H), 3,80 (m, 5H), 3,65 (s, 3H), 3,25 (m, 2H), 3,20 (s, 2H), 2,62 (m, 2H), 2,50 (m, 2H), 2,35 (s, 3H).
Etapa 2: ácido 2-(4-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-N)metN)benzoM)piperazm-1-N)acético

Usando un procedimiento similar al del Ejemplo O, Etapa 4, ácido 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperazin-1-il)acético (25 mg, 15%) se preparó a partir de metil 2-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1-H-indol-2-il)metil)benzoil)piperazin-1-il)acetato (170 mg, 0,30 mmol). LC/mS (Método g) Rt = 1,50 min.; MS m/z: 542 [M+H]+ 1H NMR (DMSO-d6, 500MHz): 57,70 (s, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,41 (d, J=8,4 Hz, 1H), 7,06 (s, 1H), 5,68 (s, 1H), 4,46 (m, 2h), 3,91 (s, 3H), 3,65 (m, 2H), 3,19 (m, 4H), 3,06 (m, 2H), 2,58 (m, 1H), 2,48 (m, 1h), 2,36 (s, 3H).
Ejemplo AX: (3-((6-bromo-1,4-dimetil-1H-indol-2-il)metil)-2,4-didorofenil)(morfolino)metanona

Etapa 1: metil 3-((6-bromo-4-metiM-(fenMsulfoml)-1H-mdol-2-M)(hidroxi)metM)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 3-((6-bromo-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-didorobenzoato (1,9 g, 66%) se preparó a partir de W-(5-bromo-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #35) (2,21 g, 4,89 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (1,39 g, 5,38 mmol). LC/MS (Método h) Rt = 3,33 min.; MS m/z:640 [M-H]-+ CH3COOH 1H NMR (DMSO-da, 300MHz): 57,94 (s, 1H), 7,85 (m, 2H), 7,60 (m, 5H), 7,28 (s, 1H), 6,98 (dd, J=5,8, 1,2 Hz, 1H), 6,71 (s, 1H), 6,64 (d, J=5,8 Hz, 1H, 3,86 (s, 3H), 2,37 (s, 3H).
Etapa 2: metil 3-((6-bromo-4-metiM-(fenMsulfoml)-1H-mdol-2-M)metM)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 3-((6-bromo-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoato (1,73 g, 94%) se preparó a partir de metil 3-((6-bromo-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-diclorobenzoato (1,9 g, 3,26 mmol). LC/MS (Método h) Rt = 3,77 min.; MS m/z: 566 [M+H]+1H NMR (DMSO-d6, 300MHz): 5 8,10 (s, 1H), 7,93 (m, 2H), 7,79 (m, 2H), 7,69 (m, 3H), 7,27 (s, 1H), 5,84 (s, 1H), 4,55 (s, 2H), 3,86 (s, 3H), 2,26 (s, 3H).
Etapa 3: metil 3-((6-brom o-4-m etiM H -indol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3 metil 3-((6-bromo-4-metil-1H-indol-2-il)metil)-2,4-diclorobenzoato (1,05 g, 81%) se preparó a partir de metil 3-((6-bromo-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoato (1,73 g, 3,05 mmol). LC/MS (Método h) Rt = 3,36 min.; MS m/z: 426 [M+H]+1H NMR (DMSO-da, 300MHz): 511,19 (s, 1H), 7,75 (m, 1H), 7,65 (m, 1H), 7,32 (s, 1H), 6,88 (s, 1H), 5,84 (s, 1H), 4,43 (s, 2H), 3,87 (s, 3H), 2,32 (s, 3H).
Etapa 4: metil 3-((6-bromo-1,4-dimetiMH-mdol-2-N)metil)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 3-((6-bromo-1,4-dimetil-1/-/-indol-2-il)metil)-2,4-diclorobenzoato (916 mg, 89%) se preparó a partir de metil 3-((6-bromo-4-metil-1H-indol-2-il)metil)-2,4-diclorobenzoato (1 g, 2,34 mmol). LC/MS (Método h) Rt = 3,52 min.; MS m/z: 440 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 57,80 (m, 1H), 7,70 (m, 1H), 7,54 (s, 1H), 6,91 (s, 1H), 5,54 (m, 1H), 4,43 (s, 2H), 3,88 (s, 3H), 3,82 (s, 3H), 2,27 (s, 3h).
Etapa 5: ácido 3-((6-bromo-1,4-dimetiMH-mdol-2-M)metM)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3-((6-bromo-1,4-dimetil-1H-indol-2-il)metil)-2,4-diclorobenzoico (850 mg, 96%) se preparó a partir de metil 3-((6-bromo-1,4-dimetil-1 H-indol-2-il)metil)-2,4-diclorobenzoato (916 mg, 2,07 mmol). El compuesto se usó directamente en la siguiente etapa. LC/MS (Método h) Rt = 3,06 min.; MS m/z: 426 [M+H]+
Etapa 6: (3-((6-bromo-1,4-dimetiMH-mdol-2-M)metM)-2,4-didorofeml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A1, (3-((6-bromo-1,4-dimetil-1H-indol-2-il)metil)-2,4-diclorofenil) (morfolino)metanona (970 mg, 97 %) se preparó a partir de ácido 3-((6-bromo-1,4-dimetil-1H-indol-2-il)metil)-2,4-diclorobenzoico (850 mg, 1,99 mmol) y morfolina (260 mg, 2,99 mmol). LC/MS (Método g) Rt = 1,90 min.; mS miz: 495 [M+H]+ 1H NMR (DMSO-da, 400MHz): 57,67 (d, J=8,4 Hz, 1h), 7,53 (m, 1H), 7,44 (d, J=8,4 Hz, 1h), 6,92 (s, 1H), 5,57 (s, 1H), 4,39 (m, 2H), 3,81 (s, 3H), 3,65 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,28 (s, 3H).
Ejemplo AY: (2,4-didoro-3-((1,4-dimetN-6-morfolmo-1H -indol-2-il)metil)fenil)(morfolino)metanona

A una suspensión de (3-((6-bromo-1,4-dimetil-1H-indol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (Ejemplo AW, Etapa 6), (200 mg, 0,403 mmol) y fosfato de potasio tribásico (171 mg, 0,806 mmol) en dioxano (2,5 mL) se añadió 2-diciclohexilfosfino-2',6'-di-isopropoxi-1,1'-bifenilo (11,28 mg, 0,024 mmol), cloro(2-diciclohexilfosfino-2',6'-di-isopropoxi-1,1'-bifenil)[2-(2-aminoetilfenil)]paladio(II), aducto de metil-t-butiléter (9,88 mg, 0,012 mmol) y morfolina (38,6 mg, 0,443 mmol). La mezcla se agitó a reflujo durante 8 horas. Se añadió más fosfato de potasio tribásico (171 mg, 0,806 mmol), 2-diciclohexilfosfino-2',6'-di-isopropoxi-1,1'-bifenilo (11,28 mg, 0,024 mmol), cloro(2-diciclohexilfosfino-2',6'-di-isopropoxi-1,1'-bifenil)[2-(2-aminoetilfenil)]paladio(II), aducto de metil-íerc-butiléter (9,88 mg, 0,012 mmol) y morfolina (38,6 mg, 0,443 mmol) y la mezcla se agitó a reflujo durante una noche. La mezcla se diluyó con EtOAc y la fase orgánica se lavó sucesivamente con agua y una solución acuosa saturada de NaCl, se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por LCMS preparativa para dar (2,4-dicloro-3-((1,4-dimetil-6-morfolino-1H-indol-2-il)metil)fenil)(morfolino)metanona (25 mg, 12%) como un sólido de color beige. LC/MS (Método g) Rt = 1,38 min.; MS miz:
502 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 57,65 (d, J=8,1 Hz, 1H), 7,42 (d, J=8,1 Hz, 1H), 6,73 (s, 1H), 6,55 s, 1H), 5,40 (s, 1H), 4,36 (m, 2H), 3,76 (m, 7H), 3,65 (m, 4H), 3,54 (m, 2H), 3,17 (m, 2H), 3,07 (m, 4H), 2,23 (s, 3H).
Ejemplo AZ: ácido 1 -(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)benzoil)-3,3-difluoropiperidina-4-carboxílico

A una solución de acetato de 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3,3-difluoropiperidina-4-carboxilato (Tabla A, Ejemplo A-8) (100 mg, 0,169 mmol) en ácido acético (6,5 mL) se añadió a temperatura ambiente ácido clorhídrico (323 |_il, 3,34 mmol) y la mezcla de reacción se agitó a reflujo por 18 horas. Se añadió más cloruro de (140 |_il, 1,449 mmol) a temperatura ambiente y la mezcla de reacción se agitó a reflujo durante 24 horas. La mezcla de reacción se concentró hasta la sequedad y el residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con MeOH al 7-10% en DCM) para dar ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3,3-difluoro-4-carboxílico (16 mg, 16%) como un sólido de color blanco. LC/mS (Método g) Rt = 1,86 min.; MS m/z: 563 [M+H]+ 1H NMR (DMSO-d6, 500MHz): 57,70 (m, 2H), 7,38 (m, 1H), 7,06 (s, 1H), 5,67 (m, 1H), 4,47 (m, 2H), 4,28 (m, 1H), 4,10 (m, 1H), 3,92 (s, 3H), 3,39 (m, 1h), 3,17 (m, 1H), 2,82 (m, 1H), 2,36 y 2,34 (s, 3H), 1,90 (m, 2H).
Ejemplo BA: ácido 1-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)benzoN)-3-fluoropiperidma-4-carboxílico
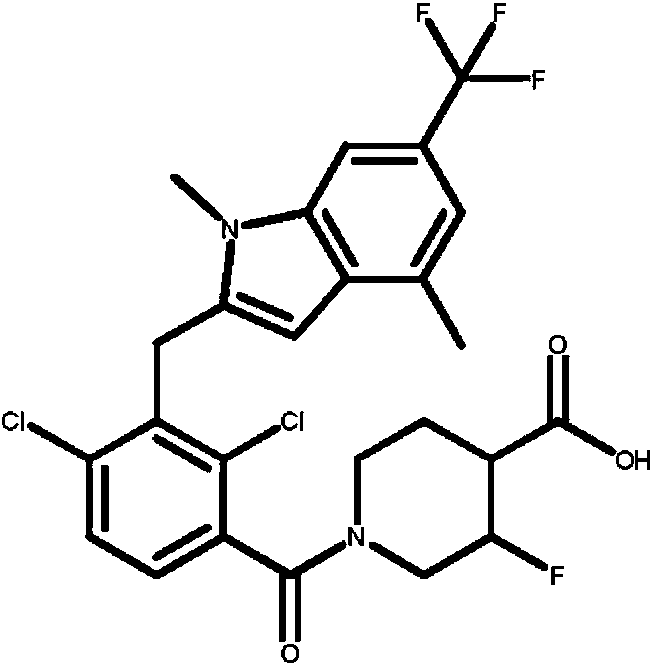
Usando un procedimiento similar al del Ejemplo AZ, ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-fluoropiperidin-4-carboxílico (11 mg, 13%) se preparó a partir de etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)-3-fluoropiperidina-4-carboxilato (Tabla A, Ejemplo A-9) (80 mg, 0,14 mmol). LC/MS (Método i) Rt = 2,42 min.; MS m/z: 545 [M+H]+1H NMR (DMSO-d6, 500MHz): 512,81 (ancho, 1H), 7,70 (m, 2h), 7,50 y 7,40 (m, 1H), 7,06 (s, 1h), 5,68 (m, 1H), 4,89-4,70 (m, 1H), 4,39-4,58 (m, 2H), 4,31-3,90 (m, 4H), 3,40-3,58 (m, 2H), 3,10-3,26 (m, 2H), 2,38 (m, 3H), 1,85-2,08 (m, 1H), 1,56-1,76 (m, 1H)
Ejemplo BB: (5-((1,4-dimetM-6-(trifluorometM)-1H-mdol-2-M)metM)-4,6-dimetMpiridm-3-M)(morfolmo)metanona

Etapa 1: metil 5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifiuorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinato (1,4 g, 69%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (1,8 g, 4,16 mmol) y metil 5-(1-hidroxiprop- 2-in-1-il)-4,6-dimetilnicotinato (Preparación #36) (830 mg, 3,79 mmol). El compuesto se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,51 min.; MS m/z: 533 [M+H]+
Etapa 2: metil 4,6-dimetil-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, metilo 4,6-dimetil-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (1,28 g, 94%) se preparó a partir de metil 5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinato (1,4 g, 2,63 mmol). El producto crudo se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,74 min.; MS m/z: 517 [M+H]+
Etapa 3: metil 4,6-dimetil-5-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 4,6-dimetil-5-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (700 mg, 75%) se preparó a partir de metil 4,6-dimetil-5-((4-metil-1-(fenilsulfonil)-6-(trifiuorometil)-1H-indol-2-il)metil)nicotinato (1,28 g, 2,48 mmol). LC/MS (Método i) Rt = 2,45 min.; MS miz: 377 [M+H]+ 1H NMR (DMSO-da, 300MHz): 511,42 (s, 1H), 8,72 (s, 1H), 7,46 (s, 1H), 7,02 (s, 1H), 5,91 (s, 1H), 4,26 (s, 2H), 3,32 (s, 3H), 2,54 (s, 3H), 2,47 (s, 3H), 2,40 (s, 3H).
Etapa 4: metil 5-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-M)metM)-4,6-dimetNmcotmato

Usando un procedimiento similar al Ejemplo A, Etapa 4, metil 5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinato (540 mg, 75 %) se preparó a partir de metil 4,6-dimetil-5 metil-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (700 mg, 1,86 mmol). LC/MS (Método i) Rt = 2,53 min.; MS miz: 391 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,76 (s, 1H), 7,70 (s, 1H), 7,05 (s, 1H), 5,57 (s, 1H), 4,27 (s, 2H), 3,93 (s, 3H), 3,32 (s, 3H), 2,50 (s, 3H), 2,43 (s, 3H), 2,34 (s, 3h)

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotínico (420 mg, 81 %) se preparó a partir de metil 5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinato (540 mg, 1,38 mmol). LC/MS (Método i) Rt = 1,89 min.; MS miz: 377 [M+H]+1H NMR (DMSO-da, 300MHz): 5 13,21 (ancho, 1H), 8,76 (s, 1H), 7,70 (s, 1H), 7,05 (s, 1H), 5,57 (s, 1H), 4,25 (s, 2H), 3,92 (s, 3H), 2,48 (s., 3H), 2,45 (s, 3H), 2,34 (s, 3H).
Etapa  metanona
metanona
Usando un procedimiento similar al del Ejemplo A1, (5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilpiridin-3-il)(morfolino)metanona (19 mg, 26%) se preparó a partir ácido 5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotínico (59 mg, 0,16 mmol) y morfolina (25 mg, 0,29 mmol). LC/MS (Método g) Rt = 1,50 min.; mS miz: 446 [M+H]+1H NMR (DMSO-d6, 400MHz): 5 8,26 (s, 1H), 7,70 (s, 1H), 7,05 (s, 1H), 5,58 (s, 1H), 4,23 (s, 2H), 3,92 (s, 3H), 3,67 (m, 4H), 3,51 (m, 2H), 3,18 (m, 2H), 2,46 (s, 3H), 2,35 (s, 3H), 2,14 (s, 3H).
Ejemplo BC: ác¡do 2-(1-(5-((1,4-d¡met¡l-6-(tr¡fluoromet¡l)-1H-¡ndol-2-¡l)met¡l)-4,6-d¡met¡ln¡cot¡no¡l)p¡per¡d¡n-4-¡l)acét¡co

Etapa 1: met¡l 2-(1-(5-((1,4-d¡met¡l-6-(tr¡fluoromet¡l)-1H-¡ndol-2-¡l)met¡l)-4,6-d¡met¡ln¡cot¡no¡l) p¡per¡d¡n-4-¡l) acetato

Usando un procedimiento similar al Ejemplo A1, metil 2-(1-(5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinoil)piperidina-4-il) acetato (137 mg, 100%) se preparó a partir ácido 5-((1,4-dimetil-6-(trifluorometil)-1H-indol-
2-il)metil)-4,6-dimetilnicotínico (Ejemplo BB, Etapa 5) (100 mg, 0,26 mmol) y clorhidrato de metil (4-piperidil)acetato (66,9 mg, 0,34 mmol). El compuesto se usó crudo en la siguiente etapa. LC/MS (Método i) Rt = 2,41 min.; Ms miz: 516 [M+H]+ Etapa 2: ácido 2-(1-(5-((1,4-dimetM-6-(trifluorometN)-1H-mdol-2-M)metM)-4,6-dimetNmcotmoN)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinoil)piperidin-4-il)acético (116 mg, 87%) se preparó a partir de 2-(1-(5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)-4,6-dimetilnicotinoil)piperidin-4-il)acetato (137 mg, 0,26 mmol).
LC/MS (Método g) Rt = 1,43 min.; MS miz: 502 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 512,09 (ancho, 1H), 8,30 y 8,24 (s, 1H), 7,70 (s, 1H), 7,05 (s, 1H), 5,57 (m, 1H), 4,52 (m, 1H), 4,25 (m, 2H), 3,92 (s, 3H), 3,34 (m, 1H), 3,05 (m, 1H), 2,81 (m, 1h), 2,48 (s, 3h), 2,35 y 2,34 (s, 3H), 2,06-2,25 (m, 5H), 1,92 (m, 1H), 1,78 (m, 1H), 1,63 (m, 1H), 1,17 (m, 2H).
Ejemplo BD: 2-[2,4-dimetil-5-[4-(oxetan-3-il)piperidina-1-carbonil]piridina-3-carbonil]-1,4-dimetil-indol-6-carbonitrilo

Etapa 1: metil 5-((6-ciano-4-metiM-(femlsulfoml)-1H-mdol-2-N)(hidroxi)metN)-4,6-dimetMmcotmato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 5-((6-ciano-4-metil-1 -(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-4,6-dimetilnicotinato (14 g, 81%) se preparó a partir de W-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #18) (14 g, 35,2 mmol) y metil 5-(1-hidroxiprop-2-in-1-il)-4,6-dimetilnicotinato (Preparación #36) (8 ,86 g, 40,4 mmol). LC/MS (Método i) Rt = 2,22 min.; MS miz: 490 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 8,65 (s, 1H), 8,25 (s, 1H), 7,93 (m, 2H), 7,69 (m, 1H), 7,52-7,64 (m, 2H), 7,50 (s, 1H), 6,75 (s, 2H), 6,54 (s, 1H), 3,85 (s, 3H), 2,46 (s, 3H), 2,42 (s, 3H), 2,41 (s, 3H).
Etapa 2: 5-(6-dano-4-metiM-(femlsulfoml)-1H-mdol-2-carboml)-4,6-dimetMmcotmato

Usando un procedimiento similar al del Ejemplo AD, Etapa 2, metil 5-(6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)-4,6-dimetilnicotinato (7,5 g, 100%) se preparó a partir de metil 5-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-4,6-dimetilnicotinato (6,9 g, 14 mmol). LC/MS (Método i) Rt = 2,40 min.; MS miz: 488 [M+H]+1H NMR (DMSO-d6, 300MHz): 58,96 (s, 1H), 8,51 (s, 1H), 8,37 (m, 2H), 7,88 (m, 1H), 7,75 (m, 2H), 7,62 (m 1H), 7,55 (s, 1H), 3,88 (s, 3H), 2,50 (s, 3H), 2,37 (s, 3H), 2,36 (s, 3H).
Etapa 3: metil 5-(6-dano-4-metM-1H-mdol-2-carboml)-4,6-dimetMmcotmato

Usando un procedimiento similar al Ejemplo A, Etapa 3, se preparó metil 5-(6-ciano-4-metil-1H-indol-2-carbonil)-4,6-dimetilnicotinato (5 g, 100%) a partir de metil 5-(6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-carbonil)-4,6-dimetilnicotinato (6,87 g, 14 mmol). LC/MS (Método i) Rt = 2,05 min.; MS miz: 348 [M+H]+1H NMR (DMSO-d6, 300MHz): 5 12,64 (ancho, 1H), 8,97 (s, 1H), 7,78 (s, 1H), 7,23 (s, 1H), 7,09 (s, 1H), 3,89 (s, 3H), 2,47 (s, 3H), 2,36 (s, 6 H).
Etapa 4: metil 5-(6-ciano-1,4-dimetiMH-mdol-2-carboml)-4,6-dimetNmcotmato

Usando un procedimiento similar al Ejemplo A, Etapa 4, se preparó 5-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)-4,6-dimetilnicotinato (2,6 g, 51%) a partir de metil 5-(6-ciano-4-metil-1H-indol-2-carbonil)-4,6-dimetilnicotinato (4,89 g, 14 mmol). LC/MS (Método i) Rt = 2,20 min.; MS m/z: 362 [M+H]+1H NMR (DMSO-da, 300MHz): 5 8,96 (s, 1H), 8,21 (s, 1H), 7,27 (s, 1H), 7,06 (s, 1H), 4,23 (s, 3H), 3,32 (s, 3H), 2,45 (s, 3H), 2,38 (s, 3H), 2,37 (s, 3H).
Etapa 5: ácido 5-(6-ciano-1,4-dimetiMH-mdol-2-carboml)-4,6-dimetNmcotmico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 5-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)-4,6-dimetilnicotínico (2 g, 95%) se preparó a partir de metil 5-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)-4,6-dimetilnicotinato (2,19 g, 6,06 mmol). LC/MS (Método i) Rt = 1,80 min.; MS m/z: 348 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 13,42 (ancho, 1H), 8,96 (s, 1H), 8,21 (s, 1H), 7,27 (s, 1H), 7,05 (s, 1H), 4,23 (s, 3H), 2,45 (s, 3H), 2,39 (s, 3H), 2,36 (s, 3H).
Etapa 6: 2-[2,4-dimetil-5-[4-(oxetan-3-il)piperidina-1-carbonil]piridina-3-carbonil]-1,4-dimetil-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A1, 2-[2,4-dimetil-5-[4-(oxetan-3-il)piperidina-1-carbonil]piridina-3-carbonil]-1,4-dimetil-indol-6-carbonitrilo (73 mg, 54%) se preparó partir de ácido 5-(6-ciano-1,4-dimetil-1H-indol-2-carbonil)-4,6-dimetilnicotínico (100 mg, 0,28 mmol) y 4-(oxetan-3-il) piperidina (53 mg, 0,374 mmol). LC/MS (Método g) Rt = 1,42 min.; MS m/z: 471 [M+H]+1H NMR (DMSO-d6, 400MHz): 58,44 (ancho, 1H), 8,20 (s, 1H), 7,27 (s, 1H), 7,02 (ancho, 1H), 4,58
(m, 3H), 4,34 (m, 2H), 4,21 (s, 3H), 3,44 (m, 1H), 3,08 (m, 1H), 2,78 (m, 2H), 2,46 (m, 3H), 2,35 (s, 3H), 2,04 (m, 3H), 1,90 (m, 1H), 1,69 (m, 1H), 1,56 (m, 1H), 1,06 (m, 2H).
Ejemplo BE: (3,5-dicloro-4-((1,4-dimetM-6-(tnfluorometM)-1 H-mdol-2-M)metM)pindm-2-M)(morfoMno)metanona

Etapa 1: etil 3,5-dicloro-4-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)picolinato
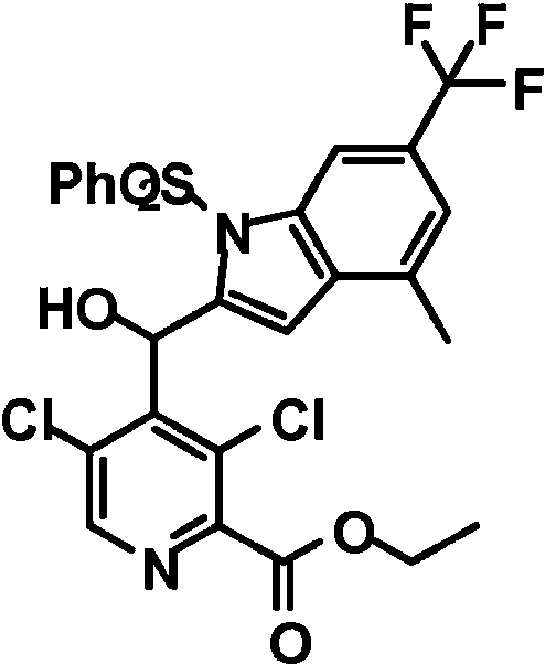
Usando un procedimiento similar al Ejemplo A, Etapa 1, etil 3,5-didoro-4-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)picolinato (702 mg, 53%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (1 g, 0,267 mmol) y etil 3,5-dicloro-4-(1-hidroxiprop-2-in-1-il)picolinato (Preparación #37) (0,87 mg, 3,17 mmol). LC/MS (Método h) Rt = 3,30 min.; MS miz: 587 [M+H]+1H NMR (DMSO-de, 300MHz): 5 8,65 (s, 1H), 8,08 (s, 1H), 7,84 (m, 2H), 7,70 (m, 1H), 7,61 (m, 2H), 7,45 (s, 1H), 7,03 (m, 2H), 6,94 (s, 1H), 4,40 (q, J=7,1 Hz, 2H), 2,50 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 2: etil 3,5-didoro-4-((4-m etiM -(fenilsulfonil)-6-(trifluorometil)-1 H-mdol-2-il)metN)picolmato

Usando un procedimiento similar al Ejemplo Z, Etapa 2, etil 3,5-didoro-4-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)picolinato (474 mg, 69%) se preparó a partir de etil 3,5-didoro-4-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)- 1H-indol-2-il)metil)picolinato (702 mg, 1,19 mmol). LC/MS (Método h) Rt = 3,65 min.; MS miz: 571 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,77 (s, 1h), 8,23 (s, 1H), 7,91 (m, 2H), 7,77 (m, 1H), 7,69 (m, 2H), 7,42 (s, 1H), 6,31 (s, 1H), 4,61 (s, 2H), 4,39 (q, J=7,1 Hz, 2H), 2,38 (s, 3H), 1,32 (t, J=7,1 Hz, 3H).
Etapa 3: etil 3,5-didoro-4-((4-metM-6-(trifluorometN)-1H-mdol-2-N)metN)picolmato

Usando un procedimiento similar al Ejemplo A, Etapa 3, etil 3,5-dicloro-4-((4-metil-6-(trifluorometil)- 1H-indol-2-il)metil)picolinato (221 mg, 51%) se preparó a partir de etil 3,5-dicloro-4-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)- 1H-indol-2-il)metil)picolinato (427 mg, 0,82 mmol). LC/MS (Método h) Rt = 3,27 min.; MS miz: 431 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 511,53 (s, 1H), 8,77 (s, 1H), 7,48 (s, 1H), 7,04 (s, 1h), 6,10 (m, 1H), 4,50 (s, 2H), 4,40 (q, J=7,1 Hz, 2H), 2,43 (s, 3H), 1,32 (t, J=7,1 Hz, 3H).
Etapa 4: etil 3,5-didoro-4-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-M)metM)picolmato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, etil 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolinato (191 mg, 69%) se preparó a partir de etil 3,5-dicloro-4-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)picolinato (220 mg, 0,51 mmol). LC/MS (Método h) Rt = 3,37 min.; MS miz : 445 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,80 (s, 1H), 7,72 (s, 1H), 7,07 (s, 1H), 5,83 (s, 1H), 4,51 (s, 2H), 4,41 (d, J=7,1 Hz, 2H), 3,92 (s, 3H), 2,38 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 5: acido 3,5-didoro-4-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)picolmico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolínico (154 mg, 97%) se preparó a partir de etil 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolinato (170 mg, 0,38 mmol). LC/MS (Método h) Rt = 2,61 min.; MS m/z: 417 [M+H]+ 1H NMR (DMSO-da, 300MHz): 58,74 (s, 1H), 7,72 (s, 1H), 7,07 (s, 1h), 5,79 (s, 1H), 4,50 (s, 2H), 3,93 (s, 3H), 2,38 (s, 3H).
Etapa 6: (3,5-didoro-4-((1,4-dimetM-6-(trifluorometM)-1H-mdol-2-M)metM)pmdm-2-M)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-2-il)(morfolino)metanona (108 mg, 72%) se preparó a partir de ácido 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolínico (129 mg, 0,30 mmol). LC/MS (Método g) Rt = 1,79 min.; MS m/z: 486 [M+H]+1H NMR (DMSO-da, 300MHz): 58,78 (s, 1H), 7,72 (s, 1H), 7,07 (s, 1H), 5,83 (s, 1H), 4,48 (s, 2H), 3,92 (s, 3H), 3,54 (s, 2H), 3,31 (m, 4H), 3,21 (m, 2H), 2,38 (s, 3H).
Ejemplo BF: 2-[[3,5-didoro-2-(morfolina-4-carbonil)-4-piridil] metil]-1,4-dimetil-indol-6-carbonitrilo


Usando un procedimiento similar al Ejemplo A, Etapa 1, etil 3,5-didoro-4-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)picolinato (7,6 mg, 39%) se preparó a partir de W-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (Preparación #18) (12 g, 30, mmol) y etil 3,5-dicloro-4-(1-hidroxiprop-2-in-1-il)picolinato (Preparación #37) (9,42 g, 34,4 mmol). LC/MS (Método i) Rt = 2,39 min.; MS m/z: 544 [M+H]+ 1H NMR (DMSO-da, 300MHz): 58,64 (s, 1h), 8,25 (s, 1H), 7,95 (m, 2H), 7,71 (m, 1H), 7,60 (m, 2H), 7,52 (m, 1H), 7,06 (s, 1H), 7,02 (m, 1H), 6,92 (m, 1H), 4,40 (q, J=7,1 Hz, 2h), 2,44 (s, 3H), 1,32 (t, J=7,1 Hz, 3H).
Etapa 2: etil 3,5-didoro-4-((6-dano-4-metiM-(femlsulfoml)-1H-mdol-2-il)metil)picolmato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, etil 3,5-didoro-4-((6-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)picolinato (5,3 g, 56%) se preparó a partir de etil 3,5-dicloro-4-((6-ciano-4-metil-1-(fenilsulfonil)- 1H-indol-2-il)(hidroxi)metil)picolinato (7,3 g, 13,4 mmol). LC/MS (Método i) Rt = 2,67 min.; MS m/z: 528 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,77 (s, 1H), 8,35 (s, 1H), 8,05 (m, 2H), 7,78 (m, 1H), 7,65 (m, 2H), 7,48 (s, 1H), 6,32 (s, 1H), 4,63 (s, 2H), 4,39 (q, J=7,1 Hz, 2H), 2,33 (s, 3H), 1,32 (t, J=7,1 Hz, 3H).
Etapa 3: etil 3,5-didoro-4-((6-dano-4-m etil-íH -indol-2-il)metil)picolinato

Usando un procedimiento similar al Ejemplo A, Etapa 3, etil 3,5-dicloro-4-((6-ciano-4-metiMH-indol-2-il)metil)picolinato (4,9 g, 100%) se preparó a partir de etil 3,5-didoro-4-((6-dano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)picolinato (5,3 g, 10 mmol). El producto se usa crudo en la siguiente etapa. LC/MS (Método i) Rt = 2,36 min.; MS miz: 388 [M+H]+
Etapa 4: etil 3,5-didoro-4-((6-dano-1,4-dimetiMH-mdol-2-N)metN)picolmato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, etil 3,5-dicloro-4-((6-dano-1,4-dimetil-1H-indol-2-il)metil)picolinato (1,25 g, 31%) se preparó a partir de etil 3,5-dicloro-4-((6-ciano-4-metiMH-indol-2-il)metil)picolinato (3,89 g, 10,02 mmol). El producto se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,41 min.; MS miz: 402 [M+H]+
Etapa 5: ácido 3,5-didoro-4-((6-dano-1,4-dimetiMH-mdol-2-N)metN)picolímco

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3,5-dicloro-4-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)picolínico (1,06 g, 88%) se preparó a partir de etil 3,5-dicloro-4-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)picolinato (1,25 g, 3,11 mmol). LC/MS (Método i) Rt = 1,80 min.; MS m/z: 374 [M+H]+1H NMR (DMSO-da, 300MHz): 5 14,15 (ancho, 1H), 8,76 (s, 1H), 7,93 (s, 1H), 7,13 (s, 1H), 5,83 (s, 1H), 4,51 (s, 2H), 3,91 (s, 3H), 2,35 (s, 3H).
Etapa 6: 2-[[3,5-didoro-2-(morfolina-4-carbonil)-4-piridil]metil]-1,4-dimetil-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A1, 2-[[3,5-dicloro-2-(morfolina-4-carbonil)-4-piridil]metil]-1,4-dimetil-indol-6-carbonitrilo (49 mg, 41%) se preparó a partir de ácido 3,5-dicloro-4-((6-ciano-1,4-dimetil-1H-indol-2-il)metil)picolínico (100 mg, 0,26 mmol) y morfolina (30 mg, 0,347 mmol). LC/MS (Método g) Rt = 1,53 min.; MS m/z: 443 [M+H]+1H NMR (DMSO-da, 400MHz): 58,78 (s, 1H), 7,93 (s, 1H), 7,12 (m, 1H), 5,87 (s, 1H), 4,49 (s, 2H), 3,91 (s, 3H), 3,67 (s, 4H), 3,53 (m, 2H), 3,22 (m, 2H), 2,35 (s, 3H).
Tabla BF. Los siguientes ejemplos se prepararon a partir de ácido 3,5-dicloro-4-((6-ciano-1,4-dimetil-1 H-indol-2-il)metil)picolínico (Ejemplo BA, Etapa 5) usando el mismo procedimiento con la amina apropiada.


Ejemplo BG: ácido 2-[1-[3,5-didoro-4-[(6-ciano-1,4-dimetil-indol-2-il)metil]piridina-2-carbonil]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-[1-[3,5-didoro-4-[(6-ciano-1,4-dimetiMndol-2-il)metil]piridina-2-carbonil]-4-piperidil]acético (49 mg, 37%) se preparó a partir de metil 2-[1-[3,5-didoro-4-[(6-ciano-1,4-dimetil-indol-2-il)metil]piridina-2-carbonil]-4-piperidil]acetatoo(Tabla BF, Ejemplo BF-6 ) (0,27 mmol). LC/MS (Método g) Rt = 1,48 min.; MS m/z: 499 [M+H]+1H NMR (DMSO-da, 400MHz): 5 12,11 (ancho, 1H), 8,76 (s, 1H), 7,93 (s, 1H), 7,10 (m,
1H), 5,83 (s, 1H), 4,49 (m, 2H), 4,44 (m, 1H), 3,91 (s, 3H), 3,25 (m, 1H), 3,07 (m, 1H), 2,85 (m, 1H), 2,34 (s, 3H), 2,17 (m, 2H), 1,96 (m, 1H), 1,79 (m, 1H), 1,63 (m, 1H), 1,14 (m, 2H).
Ejemplo BH: (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-3-il)(morfolino)metanona

Etapa 1: (4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-3-il)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)piridin-3-il)(morfolino)metanona (4,3 mg, 32%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (Preparación #16) (8,72 g, 19,7 mmol) y etil 4,6-dicloro-5-(1-hidroxiprop-2-in-1-il)nicotinato (Preparación #38) (6,5 g, 23,7 mmol). LC/MS (Método h) Rt = 2,65 min.; MS miz: 587 [M+H]+ 1H nMr (DMSO-d6, 300MHz): 58,69 (s, 1H), 8,08 (s, 1H), 7,80 (m, 2H), 7,69 (m, 1H), 7,60 (m, 2H), 7,45 (s, 1H), 7,07 (s, 1H), 6,91 (m, 2H), 4,37 (q, J=7,1 Hz, 2H), 2,50 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 2: etil 4,6-did oro-5-((4-m etiM -(femlsulfom l)-6-(tNfluorometM)-1H-mdol-2-N)metN)mcotmato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, etil 4,6-didoro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (3,35 g, 80%) se preparó a partir de (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-3-il)(morfolino)metanona (4,3 g, 7,32 mmol). LC/MS (Método h) Rt = 2,57 min.; MS miz: 571 [M+H]+1H NMR (DMSO-d6, 300MHz): 58,84 (s, 1H), 8,23 (s, 1H), 7,93 (m, 2H), 7,77 (m, 1H), 7,69 (m, 2H), 7,42 (s, 1H), 6,33 (s, 1H), 4,60 (s, 2H), 4,38 (q, J=7,1 Hz, 2H), 2,38 (s, 3H), 1,34 (t, J=7,1 Hz, 3H).
Etapa 3: ácido 4,6-didoro-5-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotínico

A una solución de etil 4,6-dicloro-5-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (3,29 g, 5,76 mmol) en THF (30 mL) y MeOH (30 mL) se añadió hidróxido de sodio (28 mL) y la mezcla se agitó a temperatura ambiente durante la noche. Los disolventes se concentraron al vacío y el residuo se diluyó con agua. El pH se ajustó a 5 con la adición de solución acuosa de HCl 1N y el precipitado obtenido se filtró, se lavó con agua y se secó para dar ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotínico (2,27 g, 98%) como un polvo beige. El producto se usó directamente en la siguiente etapa. LC/MS (Método h) Rt = 1,46 min.; MS miz: 403 [M+H]+
Etapa 4: metil 4,6-didoro-5-((1,4-dimetM-6-(tnfluorometM)-1H-mdol-2-M)metM)mcotmato

A una solución de ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-indol-2-il)metil)nicotínico (2 g, 4,96 mmol) en ACN (50 mL) se añadió carbonato de cesio (4,85 g, 14,88 mmol) y sulfato de dimetilo (1,176 mL, 12,40 mmol) y la mezcla se agitó durante una noche a 45 °C. Se añadió más sulfato de dimetilo (624 mg, 4,9 mmol) y la mezcla se agitó durante 6 horas. Se añadió agua a la mezcla y la capa acuosa se extrajo con EtOAc. La fase orgánica se lavó con una solución acuosa saturada de NaCl, se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-80% en ciclohexano) para dar metil 4,6-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (1 g, 47%) como un polvo rosa. lC/MS (Método h) Rt = 2,13 min.; MS miz: 431 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,84 (s, 1H), 7,71 (s, 1H), 7,07 (s, 1H), 5,82 (s, 1H), 4,50 (s, 2H), 3,92 (s, 6 H), 2,38 (s, 3H). Etapa 5: ácido 4,6-didoro-5-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)mcotmico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)nicotínico (88 mg, 100%) se preparó a partir de metil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (92 mg, 0,21 mmol). LC/MS (Método h) Rt = 2,70 min.; MS miz: 417 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 8,79 (s, 1H), 7,71 (s, 1H), 7,07 (s, 1H), 5,81 (s, 1H), 4,49 (s, 2H), 3,92 (s, 3H), 2,38 (s, 3H).
Etapa 6: (4,6-didoro-5-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)piridm-3-N)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6 , (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-3-il)(morfolino)metanona (81 mg, 78%) se preparó a partir de ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)meti)nicotínico (85 mg, 0,20 mmol). lC/MS (Método g) Rt = 1,78 min.; MS miz: 486 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 58,51 (s, 1H), 7,71 (s, 1H), 7,07 (s, 1H), 5,85 (s, 1H), 4,46 (m, 2H), 3,92 (s, 3H), 3,70(m, 4H), 3,55 (m, 2H), 3,25 (m, 2H), 2,39 (s, 3H).
Tabla BH. Los siguientes ejemplos se prepararon a partir de ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-indol-2 -il)metil) nicotínico (Ejemplo BH, Etapa 5) usando el mismo procedimiento con la amina apropiada.

Ejemplo BI: ácido 3-(3,5-didoro-4-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)-2-oxopiridm-1(2H)-il)propanoico

A una suspensión de 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)piridin-2-ol (Ejemplo AL, Etapa 1) (50 mg, 0,128 mmol) en DMF (700 pL) se añadió hidruro de sodio (4,62 mg, 0,193 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 15 minutos. Se añadió beta-propiolactona (12,12 pL, 0,193 mmol) a la mezcla de reacción y la mezcla de reacción se agitó a 50 °C durante 24 horas. La mezcla de reacción se diluyó con agua. La capa acuosa obtenida se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por LCMS preparativa para dar ácido 3-(3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-H-indol-2 -il)metil)-2 -oxopiridin-1 (2 H)-il)propanoico (7 mg, 12 %) como un sólido de color blanco. LC/MS (Método g) Rt = 1,68 min.; MS m/z: 461 [M+H]+
1H NMR (DMSO-da, 500MHz): 58,15 (s, 1H), 7,69 (s, 1H), 7,07 (s, 1H), 5,98 (s, 1H), 4,30 (s, 2H), 4,17 (t, J=6,9 Hz, 2H), 3,89 (s, 3H), 2,66 (m, 2H), 2,41 ppm (s, 3H)
Ejemplo BJ: (2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)feml)(morfolmo)metanona

Etapa 1: metil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoato

Una mezcla de W-1,3-dimetil-5-(trifluorometil)benceno-1,2-diamina (Preparación #21) (100 mg, 0,490 mmol), ácido 2-(2,6-didoro-3-(metoxicarbonil)fenil)acético (Preparación #2) (129 mg, 0,490 mmol), HATU (186 mg, 0,490 mmol) y 4-metilmorfolina (216 pL, 1,959 mmol) se calentó a 100 °C bajo agitación rápida durante 18 horas. Se añadió ácido acético (245 pl) y la mezcla se agitó a 100 °C durante 1 hora adicional. La mezcla de reacción se diluyó con agua, se extrajo con EtOAc. La capa orgánica se lavó con solución acuosa saturada de NaHCO3, agua y salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró para dar metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoato (199 mg, 94%) como un sólido de color beige. LC/ MS (Método k) Rt = 2,98 min.; MS miz: 431 [M+H]+ 1H NMR (DMSO-Ca, 300 MHz): 57,83 (s, 1H), 7,76 (d, J = 9Hz, 1H), 7,67 (d, J = 9Hz, 1H), 7,27 (s, 1H), 4,63 (s, 2H), 3,98 (s, 3H), 3,87 (s, 3H), 2,41 (s, 3H).
Etapa 2: ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[rf]imidazol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoico (7,4 g, 98%) se preparó a partir de metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoato (7,81 g, 18,16 mmol). LC/MS (Método h) Rt = 2,41 min.; MS miz: 417 [M+H]+.
1H NMR (DMSO-cfe, 300 MHz): 513,67 (ancho, 1H), 7,82 (s, 1H), 7,73 (d, J = 9Hz, 1H), 7,62 (d, J = 9Hz, 1H), 7,27 (s, 1H), 4,63 (s, 2H), 3,97 (s, 3H), 2,41 (s, 3H).
Etapa 3: (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)- 1H-benzo[d]imidazol-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)- 1 H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona (191 mg, 56%) se preparó a partir de ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoico (290 mg, 0,695 mmol) y morfolina (79 mg, 0,90 mmol). LC/MS (Método h) Rt = 2,59 min.; MS m/z: 486 [M+H]+.1H NMR (DMSOd 400 MHz): 57,82 (s, 1 h), 7,65 (d, J = 8Hz, 1H), 7,41 (d, J = 8Hz, 1H), 7,26 (s, 1H), 4,63 (d, J = 16Hz, 1H), 4,56 (d, J = 16Hz, 1H), 3,97 (s, 3H), 3,63 (m, 4H), 3,52 (m, 2h), 3,17 (m, 2H), 2,39 (s, 3H).
Tabla BJ. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[ d ]imidazol-2-il)metil)benzoico usando el mismo procedimiento con la amina apropiada.




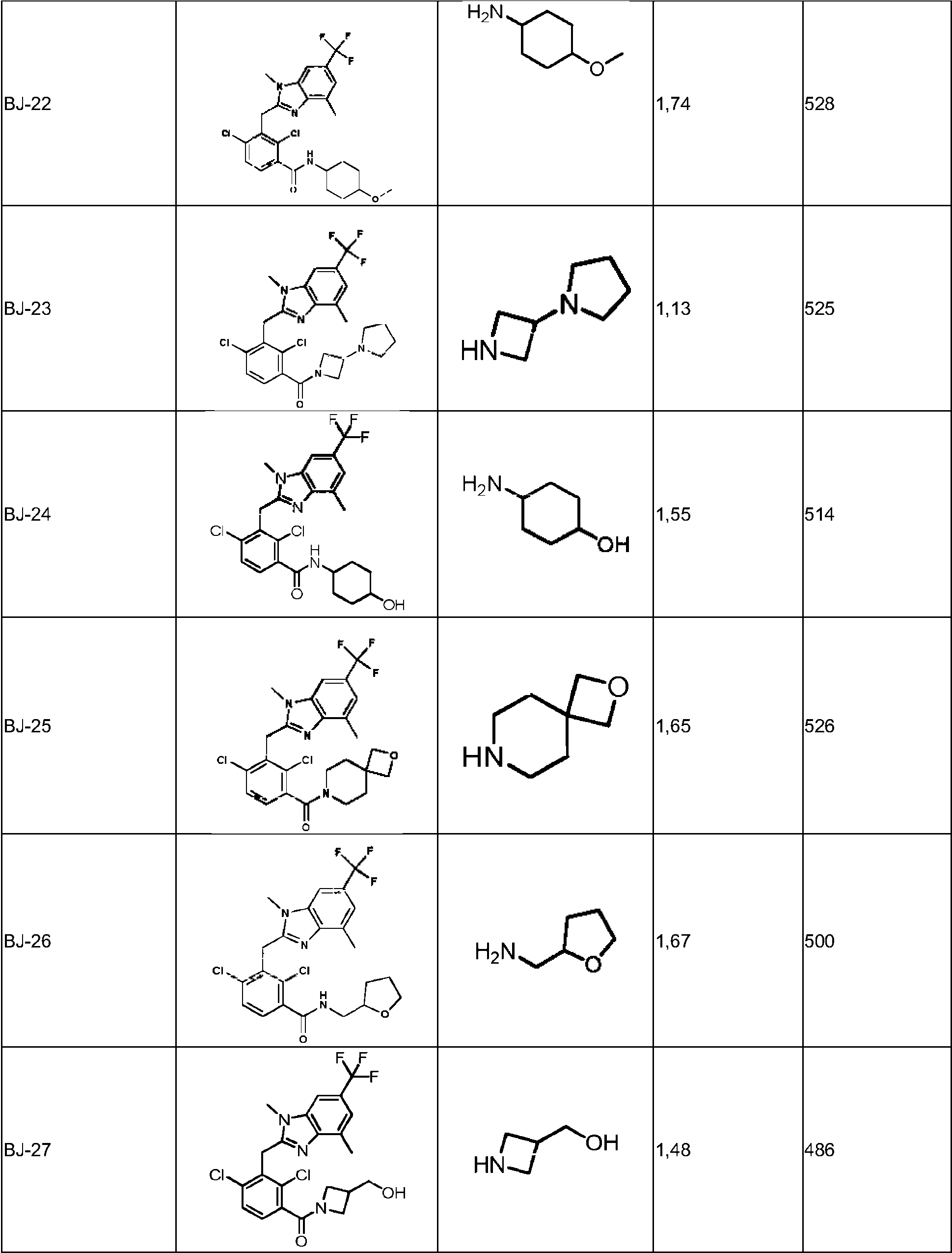
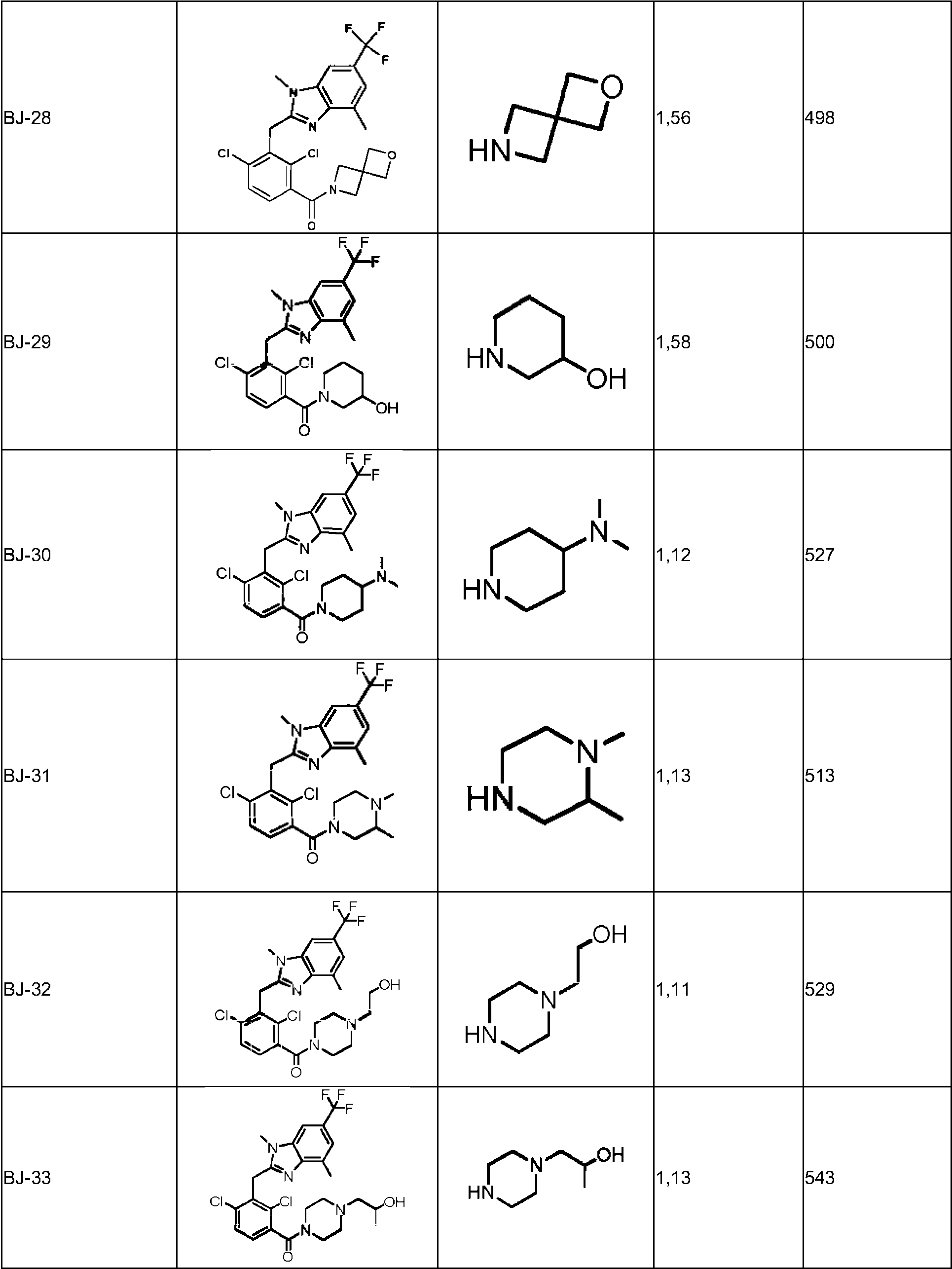


Ejemplo BK: ácido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)pipendma-4-carboxílico

Etapa 1: etil 1-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)pipe-ndma-4-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidina-4-carboxilato (110 mg, 63%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (130 mg, 0,312 mmol) y etil piperidina-4-carboxilato (73,5 mg, 0,467 mmol). LC/MS (Método h) Rt = 2,97 min.; Ms miz: 556 [M+H]+.1H NMR (dMsO- d6, 300 MHz): 57,82 (s, 1H), 7,62 (d, J = 9Hz, 1H), 7,42 y 7,35 (d, J = 9Hz, 1H), 7,26 (s, 1H), 4,65 (m, 1H), 4,56 (d, J = 16Hz, 1H), 4,38 y 4,34 (m, 1H), 4,04 (m, 2H), 3,97 (s, 3H), 3,27 (m, 1H), 3,09 (m, 1H), 2,97 (m, 1H), 2,64 (m, 1H), 2,40 (s, 3H), 1,93 (m, 1H), 1,76 (m, 1h), 1,50 (m, 2h), 1,16 (m, 3H).
Etapa 2: ácido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidma-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidina-4-carboxílico (88 mg, 83%) se preparó a partir de etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidina-4-carboxilato (110 mg, 0,198 mmol). LC/MS (Método g) Rt = 1,53 min.; MS miz: 528 [M+H]+.1H NMR (DMSO-d6, 400 MHz): 57,83 (s, 1H), 7,62 (d, J = 8Hz, 1H), 7,42 y 7,35 (d, J = 8Hz, 1H), 7,27 (s, 1H), 4,65 (dd, J = 6Hz, 12Hz, 1H), 4,56 (d, J = 12Hz, 1H), 4,36 y 4,33 (m, 1H), 3,97 (s, 3h), 3,28 (m, 1H), 3,07 (m, 1H), 2,96 (m, 1H), 2,54 (m, 1H), 2,40 (s, 3H), 1,92 (m, 1H), 1,76 (m, 1H), 1,60-1,40 (m, 2H).
Ejemplo BL: ácido (1R, 5S, 6r)-3-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)-3-azabiddo[3,1,0]hexano-6-carboxílico

Etapa 1: (1R, 5S)-etil-3-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)-3-azabicido[3,1,0]hexano-6-carboxilato
Quiral

Usando un procedimiento similar al Ejemplo A, Etapa 6, (1R,5S)-etil-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1/-/-benzo[c(]imidazol-2-il)metil)benzoil)-3-azabiddo[3,1,0]hexano-6-carboxilato (110 mg, 52%) se preparó a partir de ácido 2,4-did oro-3-((1,4-dimetil-6-(trifluorometil)-1R-benzo[C(]imidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (160 mg, 0,384 mmol) y clorhidrato de (1R,5S)-etil 3-azabiciclo[3,1,0]hexano-6-carboxilato (89 mg, 0,464 mmol). LC/MS (Método g) Rt = 1,77 min.; MS m/z: 554 [M+H]+.1H NMR (DMSO-cfe, 300 MHz): 57,82 (s, 1H), 7,63 (d, J = 9Hz, 1H), 7,42 (d, J = 9Hz, 1H), 7,26 (s, 1H), 4,60 (m, 2H), 4,05 (m, 2H), 3,97 (s, 3H), 3,91 (m, 1H), 3,50 (m, 2h), 3,29 (m, 1H), 3,20 (m, 1h), 2,40 (s, 3h), 2,13 (m, 1H), 2,05 (m, 1H), 1,15 (m, 3H).
Etapa 2: ácido (1R, 5S, 6r)-3-(2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)-3-azabiciclo[3,1,0]hexano-6-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5 ácido (1R, 5S, 6r)-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)-3-azabicido[3,1,0]hexano-6-carboxflico (74 mg, 62%) se preparó a partir de etil (1 R, 5S, 6r)-3-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-benzo[c(]imidazol-2-il)metil)benzoil)-3-azabiciclo[3,1,0]hexano-6-carboxilato (122 mg, 0,220 mmol). LC/MS (Método g) Rt = 1,50 min.; MS miz: 526 [M+H]+.1H NMR (DMSO-cfe, 400 MHz): 57,82 (s, 1H), 7,62 (d, J = 9Hz, 1H), 7,41 (d, J = 9Hz, 1 h), 7,26 (s, 1H), 4,60 (m, 2H), 3,97 (s, 3H), 3,88 (m, 1H), 3,48 (m, 2H), 3,20 (m, 1H), 2,40 (s, 3H), 2,07 (m, 1H), 1,99 (m, 1H), 1,33 (m, 1H).
Ejemplo BM: ácido 1-[2,4-didoro-3-[[1,4-dimetil-6-(trifluorometil)bendmidazol-2-il]metil]benzoil]-3-metilpiperidina-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 6 y 5, ácido 1-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometil)bencimidazol-2-il]metil]benzoil]-3-metil-piperidina-4-carboxílico se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-4-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (60 mg, 0,144 mmol) y clorhidrato de metil 3-metilpiperidina-4-carboxilato (0,216 mmol). LC/MS (Método g) Rt = 1,61 min.; MS miz: 542 [M+H]+.
1H NMR (DMSO-cfe, 400 MHz): 512,26 (ancho, 1H), 7,83 (s, 1H), 7,61 (m, 1H), 7,37 (m, 1H), 7,27 (s, 1H), 4,62 (m, 2H), 4,38 y 4,23 (m, 1H), 3,97 (m, 3H), 3,28-2,95 (m, 3h), 2,69 (m, 1H), 2,40 (m, 3H), 2,29 y 2,10 (m, 1H), 1,75-1,48 (m, 2h), 0,89 and 0,74 (m, 3H).
Ejemplo BM-1: ácido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-2-il)metil)benzoil)-4-fluoropipe-ridma-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 6 y 5, ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifiuorometil)-1H-indol-2-il)metil)benzoil)-4-fluoropiperidina-4-carboxílico (91 mg, 99%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (95 mg, 0,166 mmol) y clorhidrato de etil 4-fluoropiperidina-4-carboxilato (76 mg, 0,36 mmol). LC/Ms (Método g) Rt = 1,92 min.; MS miz: 545 [M+H]+.
1H NMR (DMSO-d6, 400MHz): 57,69 (m, 2H), 7,54 y 7,44 (d, J=, 8Hz 1H), 7,06 (s, 1H), 5,69 (m, 1H), 4,48 (m, 3H), 3,92 (s, 3H), 3,26 (m, 1H), 3,08 (m, 1H), 2,37 y 2,36 (s, 3H), 2,00 (m, 2H), 1,88 (m, 2H)
Ejemplo BN: 2-(2,6-didoro-3-(4-(2-hydroxietil)piperidma-1-carboml)bendl)-1,4-dimetiMH-benzo[d]imidazol-6-carbonitrilo

Etapa 1: metil 2,4-didoro-3-((6-ciano-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)benzoato (1,65 g, 44%) se preparó a partir de 4-amino-3-metil-5-(metilamino)benzonitrilo (Preparación #22) (1,55 g, 9,62 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (2,53 g, 9,62 mmol). LC/MS (Método k) Rt = 2,69 min.; MS m/z: 388 [M+H]+. 1H NMR (DMSO-C6, 300 MHz): 5. 8,02 (s, 1H), 7,76 (d, J = 9Hz, 1H), 7,68 (d, J = 9Hz, 1H), 7,34 (s, 1H), 4,64 (s, 2H), 3,95 (s, 3H), 3,87 (s, 3H), 2,37 (s, 3H).
Etapa 2: ácido 2,4-dicloro-3-((6-ciano-1,4-dimetil-1H-benzo[d]imidazol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((6-dano-1,4-dimetil-1H-benzo[C]imidazol-2-il)metil)benzoico (1,7 g, 87%) se preparó a partir de 2,4-didoro-3-((6-dano-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)benzoato (2,028 g, 5,22 mmol).
LC/MS (Método h) Rt = 2,04 min.; MS m/z: 374 [M+H]+. 1H NMR (D M S O C 300 MHz): 5. 13,63 (ancho, 1H), 8,02 (s, 1H), 7,73 (d, J = 9Hz, 1H), 7,63 (d, J = 9Hz, 1H), 7,34 (s, 1H), 4,63 (s, 2H), 3,95 (s, 3H), 2,37 (s, 3H).
Etapa 3: 2-(2,6-dicloro-3-(4-(2-hidroxietil)piperidma-1-carboml)bencil)-1,4-dimetiMH-benzo[d]imidazol-6-carbonitrilo:

Usando un procedimiento similar al Ejemplo A1, 2-(2,6-didoro-3-(4-(2-hidroxietil)piperidina-1-carbonil)bencil)-1,4-dimetil-1H-benzo[c(]imidazol-6-carbonitrilo (55 mg, 65%) se preparó a partir de ácido 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-benzo[C]imidazol-2-il)metil)benzoico (65 mg, 0,174 mmol) y 4-piperidinaetanol (0,26 mmol). LC/MS (Método h) Rt = 2,04 min.; MS m/z: 485 [M+H]+.1H NMR (DMSO-cfe, 300 MHz): 5.8,02 (s, 1H), 7,61 y 7,60 (d, J=8,1Hz, 1H), 7,40 y 7,32 (d, J=8,1Hz, 1H), 7,34 (s, 1H), 4,66 y 6,64 (d, J=17,1, 1H), 4,56 (d, J=17,1, 1h), 4,46 (m, 1H), 3,95 (s, 3H), 3,42 (m, 3H), 3,27 (m, 1H), 2,99 (m, 1H), 2,75 (m, 1H), 2,36 (m, 3H), 1,75 (m, 1 h), 1,86 (m, 1H), 1,57 (m, 1H), 1,35 (m, 2H), 1,06 (m, 2H)
Tabla BN. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((6-ciano-1,4-dimetil-1 H- benzo[C]imidazol-2-il)metil)benzoico (Ejemplo BN, Etapa 2) usando el mismo procedimiento con la amina apropiada.



Ejemplo BO: (2,4-dicloro-3-((1,4,6-trimetil-1H-benzo[tf]imidazol-2-il)metil)fenil)(morfolino)metanona

Etapa 1: (2,4-didoro-3-((1,4,6-tnmetiMH-benzo[d]imidazol-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, (2,4-didoro-3-((1,4,6-trimetil-1H-benzo[c(]imidazol-2-il)metil)fenil)(morfolino)metanona (17 mg, 12%) se preparó a partir de W1,3,5-trimetilbenceno-1,2-diamina (Preparación #23) (50 mg, 0,333 mmol) y ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)fenil)acético (Preparación #8) (100 mg, 0,314 mmol).
LC/MS (Método g) Rt = 1,05 min.; MS miz: 432 [M+H]+. 1H NMR (DMSO-cfe, 300 MHz): 5. 7,62 (d, J = 9Hz, 1H), 7,38 (d, J = 9Hz, 1H), 7,11 (s, 1H), 6,75 (s, 1H), 4,55 (d, J = 15Hz, 1H), 4,46 (d, J = 15Hz, 1H), 3,82 (s, 3H), 3,64 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,38 (s, 3H), 2,27 (s, 3H).
Ejemplo BP: (2,4-didoro-3-((1,7-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona

Etapa 1: (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, (2,4-didoro-3-((4-metil-6-(trifluorometil)-1 H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona (70 mg, 11%) se preparó a partir de 3-metil-5-(trifluorometil)benceno-1,2-diamina (Preparación #23, Etapa 4) (251 mg, 1,320 mmol) y ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)fenil)acético (Preparación #8) (420 mg, 1,320 mmol) LC/MS (Método h) Rt = 2,32 min.; MS m/z: 472 [M+H]+.
1H NMR (DMSO-cfe, 300 MHz): 5.12,92 and 12,62 (ancho, 1H), 7,63 (m, 2H), 7,41 (m, 1H), 7,24 y 7,16 (m, 1H), 4,56 (m, 2H), 3,64 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,56 y 2,50 (s, 3H).
Etapa 2: (2,4-didoro-3-((1,7-dimetil-5-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-dicloro-3-((1,7-dimetil-5-(trifluorometil)-1 H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona (8 mg, 11%) se preparó a partir de (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona (70 mg, 0,148 mmol). LC/mS (Método g) Rt = 1,54 min.; MS m/z: 486 [M+H]+.1H NMR (DMSO-cfe, 400 MHz): 5.7,69 (s, 1h), 7,62 (d, J = 9Hz, 1H), 7,41 (d, J = 9Hz, 1H), 7,25 (s, 1H), 4,56 (m, 2H), 4,15 (s, 3H), 3,65 (m, 4H), 3,53 (m, 2h), 3,16 (m, 2H), 2,81 (s, 3H).
Ejemplo BQ: (3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorofeml)(morfolmo)metanona

Etapa 1: metil 3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 3-((6-bromo-1,4-dimetil-1H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoato (451 mg, 42%) se preparó a partir de 5-bromo-W-1,3-dimetilbenceno-1,2-diamina (Preparación #24) (440 mg, 2,046 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil) fenil)acético (Preparación #2) (538 mg, 2,046 mmol). LC/MS (Método h) Rt = 2,89 min.; MS m/z: 441 [M+H]+.1H NMR (DMSO-Ca, 300 MHz): 5.7,75 (d, J = 9Hz, 1H), 7,67 (d, J = 9Hz, 1H), 7,63 (m, 1H), 7,10 (m, 1H), 4,57 (s, 2H), 3,88 (s, 3H), 3,87 (s, 3H), 2,32 (s, 3H).
Etapa 2: ácido 3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorobenzoico (248 mg, 57%) se preparó a partir de 3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorobenzoato (445 mg, 1,006 mmol).
LC/MS (Método h) Rt = 2,16 min.; MS m/z: 427 [M+H]+.1H NMR (DMSOC 300 MHz): 5.7,72 (d, J = 9Hz, 1H), 7,65 (s, 1H), 7,62 (d, J = 9Hz, 1 h), 7,11 (s, 1H), 4,57 (s, 2H), 3,88 (s, 3H), 2,33 (s, 3H).
Etapa3: (3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorofeml)(morfolmo)metanona:

Usando un procedimiento similar al Ejemplo A1, (3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (223 mg, 78%) se preparó a partir de ácido 3-((6-bromo-1,4-dimetil-1 H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoico (245 mg, 0,572 mmol) y morfolina (64,8 mg, 0,744 mmol). LC/MS (Método g) Rt = 1,53 min.; MS m/z: 496 [M+H]+.1H NMR (DMSO-C6, 300 MHz): 5.7,63 (s, 1H), 7,62 (d, J = 9Hz, 1H), 7,40 (d, J = 9Hz, 1H), 7,10 (s, 1H), 4,56 (d, J = 15Hz, 1H), 4,48 (d, J = 15Hz, 1H), 3,87 (s, 3H), 3,64 (m, 4H), 3,52 (m, 2H), 3,17 (m, 2H), 2,31 (s, 3H).
Ejemplo BR: (3-((6-bromo-1,4-dimetil-1H-benzo[tf]imidazol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona

Etapa 1: metil 3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-dimetilbenzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-dimetilbenzoato (620 mg, 70%) se preparó a partir de ácido 2-(3-(metoxicarbonil)-2,6-dimetilfenil)acético (Preparación #9) (500 mg, 2,250 mmol) y 5-bromo-W1,3-dimetilbenceno-1,2-diamina (Preparación #24) (484 mg, 2,250 mmol) LC/MS (Método h) Rt = 2,62 min.; MS m/z: 401 [M+H]+. 1H NMR (DMSO-cf6, 300 MHz): 57,61 (m, 1H), 7,55 (d, J = 9Hz, 1H), 7,18 (d, J = 9Hz, 1H), 7,08 (m, 1H), 4,28 (s, 2H), 3,85 (s, 3H), 3,81 (s, 3H), 2,36 (s, 3H), 2,33 (s, 3H), 2,28 (s, 3H).
Etapa 2: ácido 3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-dimetilbenzoico

Usando un procedimiento similar al Ejemplo E, Etapa 1, ácido 3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-dimetilbenzoico (580 mg, 94%) se preparó a partir de metil 3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-dimetilbenzoato 620 mg, 1,545 mmol) LC/MS (Método h) Rt = 2,09 min.; MS m/z: 387 [M+H]+. 1H NMR (DMSO-cfe, 400 MHz): 5 12,72 (ancho, 1H), 7,63 (s, 1H), 7,57 (d, J = 9Hz, 1H), 7,14 (d, J = 9Hz, 1H), 7,09 (s, 1H), 4,27 (s, 2H), 3,33 (s, 3H), 2,38 (s, 3H), 2,33 (s, 3H), 2,28 (s, 3H).
Etapa 3: (3-((6-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-dimetilfeml)(morfolmo)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 6, (3-((6-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-dimetilfenil)(morfolino)metanona (580 mg, 97%) se preparó a partir de ácido 3-((6-bromo-1,4-dimetil-1 H-benzo[C]imidazol-2-il)metil)-2,4-dimetilbenzoico (465 mg, 1,201 mmol) y morfolina (157 mg, 1,80 mmol). LC/MS (Método g) Rt = 1,99 min.; MS m/z: 456 [M+H]+. 1H NMR (D M S O C 300 MHz): 57,61 (m, 1H), 7,12 (d, J = 9Hz, 1H), 7,08 (m, 1H), 7,01 (d, J = 9Hz, 1H), 4,26 (m, 2H), 3,83 (s, 3H), 3,64 (m, 4H), 3,48 (m, 2H), 3,14 (m, 2H), 2,32 (s, 3H), 2,25 (s, 3H), 2,12 (s, 3H).
Ejemplo BS: (2,4-didoro-3-(1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-carbonil)fenil)(morfolino)metanona

A una solución de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)fenil)(morfolino)metanona (Ejemplo BJ, Etapa 3) (35 mg, 0,072 mmol) en dioxano (500 pL) se añadió dióxido de manganeso (67 mg, 0,771 mmol). La mezcla de reacción se agitó a 65 °C durante 24 horas. Se añadió dióxido de manganeso (55 mg, 0,633 mmol) y la mezcla de reacción se agitó a 65 °C durante 24 horas. Se añadió dióxido de manganeso (55 mg, 0,633 mmol) y la mezcla de reacción se agitó a 80 °C durante 24 horas. Se añadió dióxido de manganeso (55 mg, 0,633 mmol) y la mezcla de reacción se agitó a 80 °C durante 36 horas. La mezcla de reacción se filtró y se lavó con EtOAc. El filtrado se concentró a presión reducida. El residuo se purificó por LCMS preparativa para dar (2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-carbonil)fenil)(morfolino)metanona (20 mg, 55%). LC/Ms (Método g) Rt = 1,77 min.; MS m/z: 500 [M+H]+. 1H NMR (DMSO-C6, 400 MHz): 5 8,16 (s, 1H), 7,76 (d, J = 9Hz, 1H), 7,66 (d, J = 9Hz, 1H), 7,51 (s, 1H), 4,31 (s, 3H), 3,65 (m, 4H), 3,56 (m, 2H), 3,20 (m, 2H), 2,51 (s, 3H).
Ejemplo BT: 2-(2,6-didoro-3-(morfolma-4-carboml)benzoil)-1,4-dimetiMH-benzo[d]imidazol-6-carboxamida

Usando un procedimiento similar al Ejemplo BS, 2-(2,6-dicloro-3-(morfolina-4-carbonil)benzoil)-1,4-dimetil-1H-benzo[c(]imidazol-6-carboxamida (15 mg, 18%) se preparó a partir de 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-benzo[c(]imidazol-6-carbonitrilo (Tabla BN, Ejemplo BN-1) (180 mg, 0,406 mmol). LC/MS (Método g) Rt = 1,12 min.; MS m/z: 475 [M+H]+.1H NMR (DMSOC 300 MHz): 58,18 (s, 1H), 8,06 (ancho, 1H), 7,75 (d, J = 9Hz, 1H), 7,71 (m, 1H), 7,64 (d, J = 9Hz, 1H), 7,46 (ancho, 1H), 4,27 (s, 3h), 3,65 (m, 4H), 3,55 (m, 2H), 3,20 (m, 2h), 2,46 (s, 3h).
Ejemplo BU: ácido 2-(1-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidin-4-il)acetato (163 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (120 mg, 0,288 mmol) y clorhidrato de metil (4-piperidil)acetato (67 mg, 0,345 mmol). LC/Ms (Método i) Rt = 2,37 min.; mS m/z: 556 [M+H]+.1H NmR (DMSO-d6, 300MHz): 57,85 (s, 1H), 7,62 (m, 1H), 7,40 y 7,33 (d, J=8,3 Hz, 1H), 7,29 (s, 1H), 4,64 (m, 2H), 4,47 (m, 1h), 3,98 y 3,97 (s, 3H), 3,59 y 3,56 (s, 3H), 3,28 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1H), 2,40 (s, 3H), 2,26 (m, 2H), 1,95 (m, 1H), 1,74 (m, 1h), 1,56 (m, 1H), 1,13 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperid-in-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[cflimidazol-2-il)metil)benzoil)piperidin-4-il)acético (130 mg, 81%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[cflimidazol-2-il)metil)benzoil)piperidin-4-il)acetato (163 mg, 0,0,293 mmol). LC/MS (Método g) Rt = 1,56 min.; MS m/z : 542 [M+H]+.1H NMR (DMSO-de, 400MHz): 512,01 (s, 1H), 7,85 (s, 1H), 7,61 (m, 1H), 7,40 y 7,33 (d, J=8,4 Hz, 1H), 7,29 (s, 1H), 4,65(m, 2H), 4,47 (m, 1H), 3,98 y 3,97 (s, 3H), 3,29 (m, 1H), 3,04 (m, 1H), 2,79 (m, 1H), 2,40 (s, 3H), 2,18 (m, 1H), 2,13 (m, 1H), 1,91 (m, 1H), 1,62(m, 1H), 1,59 (m, 1H), 1,17 (m, 2H).
Ejemplo BV: ácido 3-[4-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometil)bencimidazol-2-il]metil]benzoil]piperazin-1-MjciclobutanocarboxíMco

Etapa 1: terc-butil 4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometM)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperazina-1-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, tere- butil 4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[cflimidazol-2-il)metil)benzoil)piperazina-1-carboxilato (480 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[cflimidazol-2-il)metil)benzoico (Ejemplo BJ, Etapa 2) (300 mg, 0,719 mmol) y tercbutil piperazina-1-carboxilato (201 mg, 1,079 mmol). LC/MS (Método h) Rt = 3,11 min.; MS m/z: 585 [m+H]+.
1H NMR (DMSO-d6, 300MHz): 57,83 (s, 1H), 7,64 (d, J=8,3 Hz, 1H), 7,41 (d, J=8,3 Hz, 1H), 7,27 (s, 1H), 4,65 (d, J=16 Hz, 1H), 4,53 (d, J=16 Hz, 1H), 3,97 (s, 3H), 3,61 (m, 2H), 3,21 (m, 2H), 3,15 (m, 2H), 2,58 (m, 2H), 2,41 (s, 3H), 1,30 (s, 9H).
Etapa 2: (2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)feml)(piperazm-1-il)metanona

Usando un procedimiento similar al del Ejemplo O, Etapa 2, (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)fenilXpiperazin-1-il)metanona (340 mg, 98%) se preparó a partir de terc-butil 4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperazine-1-carboxilato (420 mg, 0,717 mmol). LC/MS (Método h) Rt = 2,46 min.; MS m/z: 485 [M+H]+.1H NMR (DMSO-da, 300MHz): 57,82 (s, 1H), 7,61 (d, J=8,3 Hz, 1H), 7,37 (d, J=8,3 Hz, 1H), 7,26 (s, 1H), 4,66 (d, J=16 Hz, 1H), 4,53 (d, J=16 Hz, 1H), 3,97 (s, 3H), 3,53 (m, 2H), 3,07 (m, 2H), 2,6770 (m, 2H), 2,60 (m, 2H), 2,39 (s, 3H).
Etapa 3: metil 3-(4-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperazm-1-il)ciclobutanocarboxilato

Usando un procedimiento similar al del Ejemplo O, Etapa 3, metil 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperazin-1-il)cidobutanocarboxilato (105 mg, 65%) se preparó a partir de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)fenil)(piperazin-1-il)metanona (130 mg, 0,268 mmol) y metil 3-oxociclobutanocarboxilato (85,8mg, 0,67 mmol). LC/MS (Método h) Rt = 2,89 min.; MS m/z: 597 [M+H]+.1H NMR (DMSO-d6, 300MHz): 57,82 (s, 1H), 7,62 (d, J=8,1 Hz, 1H), 7,37 (d, J=8,1 Hz, 1H), 7,27 (s, 1H), 4,65 (d, J=16 Hz, 1H), 4,53 (d, J=16 Hz, 1H), 3,97 (s, 3H), 3,65 (m, 1H), 3,58 (s, 3H), 3,14 (m, 2H), 2,82 (m, 1H), 2,65 (m, 1H), 2,39 (s, 3H), 2,32 (m, 1h), 2,20 (m, 6H), 1,93 (m, 2H).
Etapa 4: ácido 3-[4-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometil)bencimidazol-2-il]metil]benzoil]piperazin-1-il]ciclobutanocarboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3-[4-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometil)bencimidazol-2-il]metil]benzoil]piperazin-1 -il]cidobutanocarboxílico (65 mg, 63%) se preparó a partir de metil 3-(4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoil)piperazin-1-il)ciclobutanocarboxilato (105 mg, 0,176 mmol). LC/MS (Método g) Rt = 1,16 min.; MS m/z: 583 [M+H]+. 1H NMR (DMSO-da, 300MHz,): 57,82 (s, 1H), 7,61 (d, J =8,3 Hz, 1H), 7,37 (d, J =8,3 Hz, 1H), 7,27 (s, 1H), 4,65 (d, J =16 Hz, 1H), 4,53 (d, J=16 Hz, 1H), 3,96 (s, 3H), 3,61 (m, 2H), 3,13 (m, 2H), 2,40 (s, 3H), 2,18 (m, 6H), 1,97 (m, 2H), 1,81 (m, 2H)
Ejemplo BW: 2-[[2,6-didoro-3-[[4-(2-metoxietil)-1-pipendil]metil]feml]metil]-1,4-dimetil-6-(trifluorometil)bencimidazol

Etapa 1: (2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)feml)metanol

A una solución de metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoato (Ejemplo BJ, Etapa 1) (2,45 g, 5,68 mmol) en THF (100 mL) se añadió en porciones borohidruro de litio (0,371 g, 17,04 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Se añadió más borohidruro de litio (0,185 g, 8,5 mmol) y la reacción se agitó a 50 °C durante 3 horas. La mezcla se enfrió a 0 °C y se inactivó lentamente mediante la adición de una solución acuosa de HCl 1N. Se obtuvo un precipitado blanco. El THF se concentró y el precipitado se filtró, se lavó con
agua y se secó bajo vacío para dar (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)fenil)metanol (1,83 g, 80%) como un polvo blanco.
LC/MS (Método h) Rt = 2,66 min.; MS m/z: 403 [M+H]+.1H NMR (DMSO-da, 300MHz): 57,82 (s, 1H), 7,55 (s, 2H), 7,26 (s, 1H), 5,54 (t, J=5,6 Hz, 1H), 4,53-4,60 (m, 4H), 3,97 (s, 3H), 2,41 (s, 3H).
Etapa 2: 2,4-didoro-3-((1,4-dimetil-6-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)bencil metanosulfonato

A una suspensión de (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)fenil)metanol (1,820 g, 4,51 mmol) en DCM (150 mL) se añadió trietilamina (0,913 g, 9,03 mmol) y cloruro de metanosulfonilo (0,776 g, 6,77 mmol) y la mezcla se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se diluyó con DCM. La capa orgánica obtenida se lavó sucesivamente con agua y una solución acuosa de HCl 1N. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)bencil metanosulfonato (2,1 g, 97%) como un polvo blanco. Lc/MS (Método h) Rt = 2,92 min.; MS m/z: 481 [M+H]+.1H NMR (DMSO-d6, 300MHz): 57,83 (s, 1H), 7,64 (m, 2H), 7,26 (s, 1H), 5,37 (s, 2H), 4,62 (s, 2H), 3,98 (s, 3H), 3,28 (s, 3H), 2,40 ppm (s, 3H).
Etapa 3: 2-[[2,6-dicloro-3-[[4-(2-metoxietil)-1-piperidil]metil]fenil]metil]-1,4-dimetil-6-(trifluorometil)bencimidazol

A una solución de clorhidrato de 4-(2-metoxietil)piperidina (52 mg, 0,29 mmol) en DMF (0,5 mL) se añadió carbonato de potasio (40 mg, 0,29 mmol) y 2,4-dicloro-3-((1, 4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)bencil metanosulfonato (70 mg, 0,145 mmol) en solución en DMF (1,5 mL). La mezcla se agitó a 60 °C durante 18 horas. Se añadió agua y la mezcla se extrajo con DCM. La fase orgánica se secó por filtración a través de una membrana hidrófoba y se concentró. El residuo se purificó por LCMS preparativa para dar 2-[[2,6-dicloro-3-[[4-(2-metoxietil)-1-piperidil]metil]fenil]metil]-1,4-dimetil-6-(trifluorometil)bencimidazol (52 mg, 68%). LC/MS (HPLC) Rt = 1,24 min.; MS m/z:
528 [M+H]+.1H NMR (DMSO-d6, 400MHz): 57,79 (s, 1H), 7,50 (d, J=8 Hz, 1H), 7,45 (d, J=8 Hz, 1H), 7,23 (s, 1H), 4,58 (s, 2H), 3,96 (s, 3H), 3,54 (s, 2H), 3,32 (t, J= 6Hz, 2H), 3,21 (s, 3H), 2,80 (m, 2h), 2,40 (s, 3H), 2,00 (m, 2H), 1,60 (m, 2H), 1,42 (m, 2H), 1,32 (m, 1H), 1,10 (m, 2H)
Tabla BW. Los siguientes ejemplos se prepararon a partir de 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)bendl metanosulfonato usando el mismo procedimiento con la amina apropiada.



Ejemplo 4-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-benzo[d]imidazol-2-N)metN)bendl)morfolm-3-ona

A una solución de morfolin-3-ona (42,0 mg, 0,416 mmol) en DMF (3 mL) se añadió hidruro de sodio (19,11 mg, 0,478 mmol) y la reacción se agitó a temperatura ambiente durante 15 minutos. 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)bendl metanosulfonato (Ejemplo BW, Etapa 2) (100 mg, 0,208 mmol) se añadió y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora, luego se añadió agua y EtOAc. La capa orgánica se decantó, se lavó con salmuera; se secó sobre sulfato de magnesio y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 40-70% en ciclohexano) para dar 4-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)bencil)morfolin-3-ona (20 mg, 19%) como un sólido de color blanco. LC/MS (HPLC) Rt = 1,62 min.; MS m/z: 486 [M+H]+.
1H NMR (DMSO-d6, 400MHz): 57,82 (s, 1H), 7,55 (d, J=8,4 Hz, 1H), 7,26 (m, 2H), 4,64 (s, 2H), 4,60 (s, 2H), 4,16 (s, 2H), 3,97 (s, 3H), 3,89 (m, 2H), 3,34 (m, 2H), 2,41 (s, 3H).
Ejemplo BY: ácido 1-(2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)- 1H-benzo[d]imidazol-2-il)metil)benzoil)piperidina-4-carboxílico

Etapa 1: metil 2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoato (1,2 g, 61%) se preparó a partir de W1,3-dimetil-4-(trifluorometil)benceno-1,2-diamina (Preparación #30) (880 mg, 4,31 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (1,13 g, 4,31 mmol). LC/MS (Método i) Rt = 2,50 min.; MS m/z: 431 [M+H]+.
1H NMR (DMSO-da, 300MHz): 57,6 (d, J=9 Hz, 1H), 7,68 (d, J=9 Hz, 1H), 7,54 (d, J=9 Hz, 1H), 7,50 (d, J=9 Hz, 1H), 4,63 (s, 2H), 3,95 (s, 3H), 3,87 (s, 3H), 2,49 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoico (1,09 g, 98%) se preparó a partir de metil 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)benzoato (1,1 g, 2,55 mmol). LC/Ms (Método i) Rt = 2,08 min.; MS m/z: 417 [M+H]+.
1H NMR (DMSO-d6, 300MHz): 513,63 (ancho, 1H), 7,73 (d, J=9 Hz, 1H), 7,63 (d, J=9 Hz, 1H), 7,54 (d, J=9 Hz, 1H),7,50 (d, J=9 Hz, 1H), 4,62 (s, 2H), 3,95 (s, 3H), 2,50 (s, 3H).
Etapa 3: metil 1-(2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidma-4-carboxilato

Usando un procedimiento similar al del Ejemplo A1, metil 1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidina-4-carboxilato (255 mg, 70%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (200 mg, 0,479 mmol) y clorhidrato de metil piperidina-4-carboxilato (103 mg, 0,575 mmol). LC/MS (Método i) Rt = 2,34 min.; MS m/z: 542 [M+H]+.1H NMR (DMSO-da, 300MHz): 57,62 (d, J=8,3 Hz, 1H), 7,56 (d, J=9 Hz, 1H), 7,50 (d, J=9 Hz, 1H), 7,43 y 7,35 (d, J=8,3 Hz, 1H), 4,60 (m, 2H), 4,36 (m, 1H), 3,94 (m, 3H), 3,62 y 3,57 (s, 3H), 3,30 (m., 1H), 3,10 (m, 1H), 2,94 (m, 1H), 2,69 (m, 1H), 2,50 (m, 3H), 1,93 (m, 1H), 1,77 (m, 1H), 1,38-1,66 (m, 2H)
Etapa 4: ácido 1-(2,4-didoro-3-((1,4-dimetM-5-(trifluorometN)-1H-benzo[d]imidazol-2-M)metM)benzoN)piperidma-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidina-4-carboxílico (80 mg, 32%) se preparó a partir de metil 1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidina-4-carboxilato (254 mg, 0,468 mmol). LC/MS (Método g) Rt = 1,55 min.; MS m/z: 528 [M+H]+.1H NMR (DMSO-d6, 400MHz): 512,31 (ancho, 1H), 7,61 (m, 1H), 7,52 (d, J=9 Hz, 1H), 7,50 (d, J=9 Hz, 1H), 7,42 y 7,35 (d, J=8 Hz, 1H), 4,59 (m, 2H), 4,35 (m, 1H), 3,94 (m, 3H), 3,21 (m, 1H), 3,09 (m, 1H), 2,96 (m, 1H), 2,54 (m, 1H), 2,50 (m, 3H), 1,91 (m, 1H), 1,74 (m, 1H), 1,50 (m, 2h).
Ejemplo BZ: ácido 2-(1-(2,4-didoro-3-((1,4-dimetM-5-(trifluorometN)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidin-4-il)acetato (275 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (Ejemplo BY, Etapa 2) (200 mg, 0,479 mmol) y clorhidrato de metil (4-piperidil)acetato (111 mg, 0,57 mmol). LC/MS (Método i) Rt = 2,39 min.; MS miz: 556 [M+H]+.1H NmR (DMSO-da, 300MHz): 57,61 (m, 1H), 7,55 (d, J=9 Hz, 1H), 7,51 (d, J=9 Hz, 1H), 7,40 y 7,33 (d, J=8,3 Hz, 1H), 4,62 (m, 2H), 4,46 (m, 1H), 3,94 (m, 3h), 3,59 y 3,55 (s, 3H), 3,28 (m, 1H), 3,06 (m, 1H), 2,78 (m, 1H), 2,47 (m, 3h), 2,25 (m, 2H), 1,94 (m, 1H), 1,75 (m, 1H), 1,57 (m, 1H), 1,14 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperid-m-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidin-4-il)acético (210 mg, 76%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoil)piperidin-4-il)acetato (272 mg, 0,46 mmol). LC/MS (Método g) Rt = 1,57 min.; MS miz: 542 [M+H]+.1H NMR (DMSO<k 400MHz): 512,07 (ancho, 1H), 7,61 (m, 1h), 7,54 (d, J=9 Hz, 1H), 7,50(d, J=9 Hz, 1H), 7,40 y 7,32 (d, J=8,1 Hz, 1H), 4,4,62 (m, 2H), 4,47 (m, 1H), 3,94 (m, 3H), 3,30 (m, 1H), 3,04 (m, 1h), 2,79 (m, 1H), 2,48, 2,47 (s, 3H), 2,10 (m, 2h), 1,93 (m, 1H), 1,77 (m, 1H), 1,57 (m, 1H), 1,10 (m, 2H).
Ejemplo CA: (2,4-didoro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)fenil)(morfolino)metanona (90 mg, 76%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-5-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)benzoico (Ejemplo bY, Etapa 2) (200 mg, 0,479 mmol) y morfolina (31 mg, 0,360 mmol). LC/MS (Método g) Rt = 1,63 min.; MS miz: 486 [M+H]+.1H NMR (DMSO-d6, 400MHz): 5 = 7,63 (d, J=8,1 Hz, 1H), 7,55 (d, J=8 Hz, 1H), 7,50 (d, J=8 Hz, 1h), 7,41 (d, J=8,1 Hz, 1H), 4,64 (d, J=16 Hz, 1H), 4,56 (d, J=16 Hz, 1H), 3,94 (s, 3h), 3,64 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,48 (s, 3H).
Ejemplo CB: (3-((5-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorofeml)(morfolmo)metanona

Etapa 1: metil 3-((5-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 3-((5-bromo-1,4-dimetil-1H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoato (508 mg, 67%) se preparó a partir de 4-bromo-W1,3-dimetilbenceno-1,2-diamina (Preparación #31) (350 mg, 1,63 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (428 mg, 1,63 mmol). LC/MS (Método h) Rt = 2,98 min.; MS m/z: 441 [M+H]+.1H NMR (DMSO-da, 300MHz): 57,75 (d, J=9 Hz, 1h), 7,66 (d, J=9 Hz, 1H), 7,38 (d, J=9 Hz, 1H), 7,34 (d, J=9 Hz, 1H), 4,58 (s, 2H), 3,88 (s, 3H), 3,86 (s, 3H), 2,37 (s, 3h).
Etapa 2: ácido 3-((5-bromo-1,4-dimetiMH-benzo[cf]imidazol-2-il)metil)-2,4-diclorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3-((5-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorobenzoico (450 mg, 93%) se preparó a partir de 3-((5-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorobenzoato (500 mg, 1,13 mmol).
LC/MS (Método h) Rt = 2,14 min.; MS m/z: 427 [M+H]+.1H NMR (DMSO-d6, 300MHz): 57,71 (d, J=9 Hz, 1H), 7,63 (d, J=9 Hz, 1H), 7,37 (d, J=9 Hz, 1H), 7,34 (d, J=9 Hz, 1H), 4,57 (s, 2H), 3,88 (s, 3H), 2,37 (s, 3H).
Etapa 3: (3-((5-bromo-1,4-dimetiMH-benzo[d]imidazol-2-il)metil)-2,4-diclorofeml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A1, (3-((5-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (465 mg, 84%) se preparó a partir de ácido 3-((5-bromo-1,4-dimetil-1H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoico (436 mg, 4,02 mmol) y morfolina (133 mg, 1,53 mmol). LC/MS (Método g) Rt = 1,53 min.; MS m/z: 496 [M+H]+. 1H NMR (DMSO-da, 400MHz): 5 7,63 (d, J=8,1 Hz, 1H), 7,40 (m, 3H), 4,63 (d, J=16 Hz, 1H), 4,55 (d, J=16 Hz, 1H), 3,89 (s, 3H), 3,65 (m, 4H), 3,52 (m, 2H), 3,17 (m, 2H), 2,38 (s, 3H).
Ejemplo CC: 2-(2,6-didoro-3-(morfolma-4-carboml)bencil)-1,4-dimetiMH-benzo[d]imidazol-5-carbomtrilo

En un vial de microondas se puso (3-((5-bromo-1,4-dimetil-1H-benzo[c(]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (Ejemplo CB, Etapa 3) (140 mg, 0,28 mmol), cianuro de zinc (24,8 mg, 0,211 mmol) Pd(Ph3P)4 (32,5 mg, 0,028mmol) en DMF (800 mL) y la mezcla de reacción se agitó a 120 °C durante 60 minutos bajo irradiación de microondas. La mezcla de reacción se diluyó con EtOAc y se filtró. Se añadió agua al filtrado. Las capas se separaron y la acuosa se lavó con EtOAc. Las capas orgánicas se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 50-60% en ciclohexano) para dar 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-benzo[C]imidazol-5-carbonitrilo (30 mg, 23%). LC/MS (Método g) Rt = 1,32 min.; MS m/z: 443 [M+H]+. 1H NMR (DMSO-d6, 400MHz): 57,63 (d, J=8,1 Hz, 1H), 7,56 (m, 2H), 7,41 (d, J=8,1 Hz, 1H), 4,64 (d, J=16 Hz, 1H), 4,56 (d, J=16 Hz, 1H), 3,94 (s, 3H), 3,60 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,50 (s, 3H).
Ejemplo CD: 2-(2,6-didoro-3-(morfolma-4-carboml)bencil)-1-metiMH-benzo[d]imidazol-5-carbomtrilo

Etapa 1: metil 2,4-didoro-3-((5-dano-1-metiMH-benzo[d]imidazol-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 2,4-dicloro-3-((5-ciano-1-metil-1H-benzo[C]imi-dazol-2-il)metil)benzoato (460 mg, 82%) se preparó a partir de 3-amino-4-(metilamino)benzonitrilo (WO 2003060078) (220 mg, 1,49 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (393 mg, 1,49 mmol). LC/MS (Método h) Rt = 2,39 min.; MS m/z: 374 [M+H]+. 1H NMR (DMSO-d6, 300MHz): 58,04 (d, J=1,5 Hz, 1H), 7,76 (m, 2H), 7,66 (d, J=9 Hz, 1H), 7,62 (dd, J=9, 1,5 Hz, 1H), 4,64 (s, 2H), 3,97 (s, 3H), 3,87 (s, 3H).
Etapa 2: ácido 2,4-dicloro-3-((5-ciano-1-metiMH-benzo[rf]imidazol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5 ácido 2,4-dicloro-3-((5-ciano-1-metil-1H-benzo[C]imidazol-2-il)metil) benzoico (400 mg, 92%) se preparó a partir de metil 2,4-dicloro-3-((5-ciano-1-metil-1H-benzo[c(]imidazol-2-il)metil)benzoato (450 mg, 1,20 mmol).
LC/MS (Método h) Rt = 1,80 min.; MS m/z: 360 [M+H]+. 1H NMR (DMSO-d6, 300MHz): 58,04 (d, J=1,3 Hz, 1H), 7,77 (d, J=8,4 Hz, 1H), 7,72 (d, J=9 Hz, 1H), 7,62 (m, 2H), 4,63 (s, 2H), 3,97 (s, 3H).
Etapa 3: 2-(2,6-dicloro-3-(morfolma-4-carboml)bencil)-1-metiMH-benzo[d]imidazol-5-carbomtrilo

Usando un procedimiento similar al Ejemplo A1, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-benzo[C]imidazol-5-carbonitrilo (178 mg, 38%) se preparó a partir de ácido 2,4-dicloro-3-((5-ciano-1-metil-1H-benzo[c]imidazol-1-il)metil)benzoico (387 mg, 1,07 mmol) y morfolina (140 mg, 1,61 mmol). LC/MS (HPLC) Rt = 1,19 min.; MS m/z: 429 [M+H]+.
1H NMR (DMSO-d6, 400MHz): 58,05 (s, 1H), 7,77 (d, J=8,4 Hz, 1H), 7,62 (m, 2H), 7,41 (d, J=8,4 Hz, 1H), 4,61 (m, 2H), 3,97 (s, 3H), 3,64 (m, 4H), 3,54 (m, 2H), 3,16 (m, 2H).
Ejemplo CE: (3-((6-bromo-1-metil-4-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona

Etapa 1: metil 3-((6-bromo-1-metil-4-(tnfluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[c(]imidazol-2-il)metil)-2,4-didorobenzoato (827 mg) se preparó a partir de 5-bromo-W1-metil-3-(trifluorometil)benceno-1,2-diamina (Preparación #32) (450 mg, 1,67 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (440 mg, 1,67 mmol). Se usó crudo en la siguiente etapa. LC/MS (Método h) Rt = 3,19 min.; MS m/z: 494 [M+H]+.
Etapa 2: ácido 3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-diclorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoico (487 mg, 88%) se preparó a partir de metil 3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[C]imidazol-2-il)metil)-2,4-diclorobenzoato (830 mg, 1,67 mmol). LC/MS (Método h) Rt = 2,77 min.; MS m/z: 481 [M+H]+.
1H NMR (DMSO-d6, 300MHz): 58,24 (d, J=1,0 Hz, 1H), 7,74 (d, J=9 Hz, 1H), 7,63 (d, J=9 Hz, 1H), 7,59 (d, J=1,0 Hz, 1H), 4,64 (s, 2H), 3,97 (s, 3H).
Etapa 3: (3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A1, (3-((6-bromo-1 -metil-4-(trifluorometil)-1 H-benzo[d]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (442 mg, 79%) se preparó a partir de ácido 3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-diclorobenzoico (478 mg, 0,99 mmol) y morfolina (86 mg, 0,99 mmol). Lc/MS (Método g) Rt = 1,76 min.; MS m/z: 550 [M+H]+.1H NMR (DMSO-d6, 400MHz): 58,23 (s, 1H), 7,63 (d, J=8,4 Hz, 1H), 7,58 (s, 1H), 7,40 (d, J=8,4 Hz, 1H), 4,66 (d, J=16 Hz, 1H), 4,56 (d, J=16 Hz, 1H), 3,96 (s, 3H), 3,63 (m, 4H), 3,40-3,54 (m, 2H), 3,12 (m, 2H).
Ejemplo CF: 2-(2,6-didoro-3-(morfolma-4-carboml)bencM)-1-metM-4-(tnfluorometM)-1H-benzo[d]imidazol-6-carbonitrilo

Usando un procedimiento similar al del Ejemplo CC, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1 -metil-4-(trifluorometil)-1H-benzo[d]imidazol-6-carbonitrilo (57 mg, 42%) se preparó a partir de (3-((6-bromo-1-metil-4-(trifluorometil)-1H-benzo[d]imidazol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (Ejemplo CE, Etapa 3) (151 mg, 0,0,27 mmol). LC/MS (Método g) Rt = 1,50 min.; MS m/z: 497 [M+H]+.1H NMR (DMSO-d6, 400MHz): 58,58 (s, 1H), 7,94 (m, 1H), 7,64 (d, J=8,4 Hz, 1H), 7,41 (d, J=8,4 Hz, 1H), 4,74 (d, J=16 Hz, 1H), 4,64 (d, J=16 Hz, 1H), 4,03 (s, 3H), 3,63 (m, 4H), 3,48 (m, 2H), 3,12 (m, 2H).
Ejemplo CG: 2-(2,6-didoro-3-(morfolma-4-carboml)bencM)-1-etM-4-metiMH-benzo[d]imidazol-6-car-bomtrMo

Etapa 1: metil 2,4-didoro-3-((6-ciano-4-metiMH-benzo[d]imidazol-2-N)metN)benzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 2,4-dicloro-3-((6-ciano-4-metil-1H-benzo[d]imi-dazol-2-il)metil)benzoato (1,16 g, 76%) se preparó a partir de 3,4-diamino-5-metilbenzonitrilo (WO2005021510) (600 mg, 4,08 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2 ) (440 mg, 1,67 mmol). LC/MS (Método h) Rt = 2,35 min.; MS m/z: 374 [M+H]+. 1H NMR (DMSO-d6, 300MHz): 5 13,07 and 12,78 (ancho, 1H), 7,77 (m, 2H), 7,66 (d, J=9 Hz, 1H), 7,36 (s, 1H), 4,61 (s, 2H), 3,87 (s, 3H), 2,48 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((6-dano-4-metiMH-benzo[d]imidazol-2-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-ciano-4-metil-1H-benzo[d]imidazol-2-il)metil)benzoico (462 mg, 76%) se preparó a partir de 2,4-dicloro-3-((6-ciano-4-metil-1H-benzo[d]imidazol-2-il)metil)benzoato (600 mg, 1,60 mmol).
LC/MS (Método h) Rt = 1,72 min.; MS m/z: 360 [M+H]+. 1H NMR (DMSO-d6, 300MHz): 57,85 (d, J=8,4 Hz, 1H), 7,78 (s, 1H), 7,52 (d, J=8,4 Hz, 1H), 7,47 (s, 1H), 5,03 (s, 2H), 2,74 (s, 3H).
Etapa 3: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-4-metiMH-benzo[d]imidazol-6-carbomtrNo

Usando un procedimiento similar al Ejemplo A1, 2-(2,6-didoro-3-(morfolina-4-carbonil)bendl)-4-metil-1H-benzo[c(]imidazol-6-carbonitrilo (129 mg, 71%) se preparó a partir de ácido 2,4-dicloro-3-((6-dano-4-metil-1H-benzo[c(]imidazol-2-il)metil)benzoico (150 mg, 0,416 mmol) y morfolina (43,5 mg, 0,5 mmol). LC/MS (Método h) Rt = 1,97 min.; MS m/z: 429 [M+H]+.
1H NMR (DMSO-da, 300MHz): 513,04 and 12,76 (s, 1H), 7,85 y 7,76 (s, 1H), 7,63 (m, 1H), 7,39 (m, 2H), 4,57 (m, 2H), 3,65 (m, 4H), 3,53 (m, 2H), 3,16 (m, 2H), 2,50 y 2,45 (s, 3H).

Usando un procedimiento similar al Ejemplo A, Etapa 4, 2-(2,6-didoro-3-(morfolina-4-carbonil)bendl)-1-etil-4-metil-1H-benzo[c(]imidazol-6-carbonitrilo (63 mg, 47%) se preparó a partir de 2-(2,6-dicloro-3-(morfolina-4-carbon-il)bendl)-4-metil-1H-benzo[c(]imidazol-6-carbonitrilo (125 mg, 0,29 mmol) y bromoetano (56 mg, 0,52 mmol). LC/MS (Método g) Rt = 1,43 min.; MS m/z: 457 [M+H]+.1H NMR (DMSO-d6, 500MHz): 58,05 (s, 1H), 7,64 (d, J=8,2 Hz, 1H), 7,42 (d, J=8,4 Hz, 1H), 7,34 (s, 1H), 4,65 (d, J=16 Hz, 1H), 4,57 (d, J=8,2 Hz, 1H), 4,45 (q, J=7,1 Hz, 2H), 3,65 (m, 4h), 3,53 (m, 2H), 3,16 (m, 2H), 2,34 (s, 3H), 1,39 (t, J=7,1 Hz, 3H).

Usando un procedimiento similar al Ejemplo A1, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-isopropil-4-metil-1H-benzo[c(]imidazol-6-carbonitrilo (20 mg, 14%) se preparó a partir de 2-(2,6-dicloro-3-(morfolina-4-carbo-nil)bencil)-4-metil-1H-benzo[d]imidazol-6-carbonitrilo (Ejemplo CG, Etapa 3) (130 mg, 0,30 mmol) y 2-yodopropano (61 mg, 0,36 mmol). LC/MS (Método g) Rt = 1,52 min.; MS m/z: 471 [M+H]+.1H NMR (DMSO-d6, 500MHz): 58,15 (s, 1H), 7,63 (d, J=8,2 Hz, 1H), 7,41 (d, J=8,2 Hz, 1H), 7,33 (s, 1H), 5,05 (sept, J=6,9 Hz, 1H), 4,66 (d, J=16 Hz, 1H), 4,58 (d, J=16 Hz, 1H), 3,65 (m, 4h), 3,52 (m, 2h), 3,16 (m, 2H), 2,31 (s, 3H), 1,64 (d, J=6,9 Hz, 6H).
Ejemplo CI: 2-(2,6-didoro-3-(morfolma-4-carboml)bend l)-3,7-dimetil-3H-imidazo[4,5-to]pmdma-5-car-bomtnlo

Etapa 1: metil 2,4-didoro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-fo]piridm-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo BJ, Etapa 1, metil 2,4-dicloro-3-((5-cloro-3,7-dimetil-3H-imidazo[4,5-6]piridin-2-il)metil)benzoato (350 mg, 50%) se preparó a partir de 6-cloro-W2,4-dimetilpiridina-2,3-diamina (Preparación #25) (150 mg, 0,874 mmol) y ácido 2-(2,6-dicloro-3-(metoxicarbonil)fenil)acético (Preparación #2) (230 mg, 0,874 mmol) y se usó crudo en la siguiente etapa. LC/MS (Método h) Rt = 2,78 min.; Ms miz: 398 [M+H]+.
Etapa 2: ácido 2,4-didoro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-fo]piridm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2,4-dicloro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoico (250 mg, 86%) se preparó a partir de metil 2,4-dicloro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoato (300 mg, 0,753 mmol). LC/MS (Método h) Rt = 2,21 min.; MS miz: 384 [M+H]+.1H NMR (DMSO-cfe, 300 MHz): 513,64 (ancho, 1H), 7,72 (d, J = 9Hz, 1H), 7,62 (d, J = 9Hz, 1H), 7,14 (s, 1H), 4,62 (s, 2H), 3,88 (s, 3H), 2,39 (s, 3H).
Etapa 3: (2,4-didoro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-to]pmdm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-dicloro-3-((5-doro-3,7-dimetil-3H-imidazo[4,5-ib]pi-ridin-2-il)metil)fenil)(morfolino)metanona (160 mg, 87%) se preparó a partir de ácido 2,4-dicloro-3-((5-cloro-3,7-dimetil-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoico (150 mg, 0,390 mmol) y morfolina (51 mg, 0,585 mmol). Lc/MS (Método g) Rt = 1,43 min.; MS m/z: 453 [M+H]+.1H NMR (DMSO-cfe, 400 MHz): 57,63 (d, J = 9Hz, 1H), 7,41 (d, J = 9Hz, 1H), 7,13 (s,1H), 4,63 (d, J = 12Hz, 1H), 4,56 (d, J = 12Hz, 1H), 3,87 (s, 3H), 3,64 (m, 4H), 3,52 (m, 2H), 3,15 (m, 2H), 2,38 (s, 3H).
Etapa 4: 2-(2,6-dicloro-3-(morfolma-4-carboml)bencil)-3,7-dimetil-3H-imidazo[4,5-to]piridma-5-carbomtrilo

En un vial de microondas se pusieron (2,4-dicloro-3-((5-cloro-3,7-dimetil-3H-imidazo[4,5-ib]piridin-2-il)metil)fenil)(morfolino)metanona (70 mg, 0,154 mmol), cianuro de zinc (15,40 mg, 0,131 mmol) y Pd(Ph3P)4 (17,83 mg, 0,015 mmol) en DMF (250 pL) y la mezcla de reacción se agitó a 110 °C durante 30 minutos bajo irradiación de microondas. La mezcla de reacción se diluyó con EtOAc y se filtró. El filtrado se lavó luego con agua. Las capas se separaron y la acuosa se lavó con EtOAc. Las capas orgánicas se combinaron, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-100% en ciclohexano) para dar 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-3,7-dimetil-3H-imidazo[4,5-¿]piridina-5-carbonitrilo (18 mg, 26%). LC/MS (Método g) Rt = 1,32 min.; MS m/z: 444 [M+H]+.1H NMR (DMSO-C6, 400 MHz): 57,70 (s, 1H), 7,64 (d, J = 9Hz, 1H), 7,43 (d, J = 9Hz, 1H), 4,72 (d, J = 16Hz, 1H), 4,64 (d, J = 16Hz, 1H), 3,94 (s, 3H), 3,65 (m, 4H), 3,53 (m, 2H), 3,15 (m, 2H), 2,43 (s, 3H).
Ejemplo CJ: 4-cloro-2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-indol-6-carbonitrilo

Etapa 1: terc-butil 3-((6-bromo-4-doro-l-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-didorobenzoato

A una solución de 6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol (Preparación #48) (200 mg, 0,54 mmol) en THF (1,5 mL) y enfriada a -78 °C se añadió diisopropilamida de litio (0,30 mL, 0,59 mmol) y la mezcla de reacción se agitó durante 1 hora a -78 °C. A la mezcla de reacción se añadió terc-butil 2,4-didoro-3-formilbenzoato (Preparación #33, Etapa B) (178 mg, 0,65 mmol) en solución en THF (1,5 mL) y la mezcla de reacción se agitó durante 2 horas a -78 °C. La mezcla de reacción se inactivó con una solución saturada de NH4 Cl y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar terc-butilo 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il) (hidroxi)metil)-2,4-diclorobenzoato (262 mg, 75%). LC/MS (Método j) Rt = 2,75 min.; MS miz: 702 [M-H]-+ CH3 COOH 1H NMR (DMSO-da, 300MHz): 58,11 (m, 1h), 7,85 (m, 2H), 7,72 (m, 1H), 7,66 (d, J= 6 Hz, 1H), 7,55 (m, 4H), 6,95 (m, 1H), 6,75 (m, 1H), 6 , 6 6 (m, 1H), 1,54 ppm (s, 9H)
Etapa 2: ácido 3-((6-bromo-4-doro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-1,4-diclorobenzoico (1,17 g, 93%) se preparó a partir de terc-butil 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-didorobenzoato (1,42 g, 2,2 mmol). LC/MS (Método i) Rt = 2,81 min.; MS miz: 570 [M-H]-1H NMR (DMSO-da, 300MHz): 58,27 (m, 1H), 8,00 (m, 2H), 7,80 (m, 2H), 7,69 (m, 4H), 5,70 (m, 1h), 4,55 (s, 2H).
Etapa 3: ácido 3-((6-bromo-4-doro-1H-indol-2-il)metil)-2,4-didorobenzoico
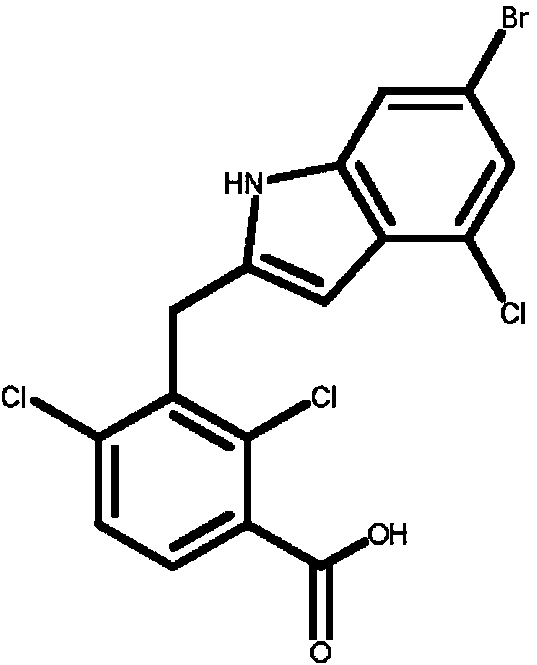
[1101] Usando un procedimiento similar al Ejemplo A, Etapa 3, se preparó ácido 3-((6-bromo-4-cloro-1H-indol-2-il)metil)-2,4-diclorobenzoico (657 mg, 100%) a partir de ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoico (870 mg, 1,52 mmol). El compuesto se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,48 min.; MS miz: 432 [M+H]+
Etapa 4: metil 3-((6-bromo-4-doro-1-metil-1H-indol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 3-((6-bromo-4-cloro-1-metil-1H-indol-2-il)metil)-2,4-diclorobenzoato (568 mg, 81%) se preparó a partir de ácido 3-((6-bromo-4-cloro-1H-indol-2-il)metil)-2,4-diclorobenzoico
(657 mg, 1,52 mmol). LC/MS (Método j) Rt = 2,46 min.; MS m/z: 460 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,80 (m, 2H), 7,72 (m, 1H), 7,23 (d, J=1,5 Hz, 1H), 5,52 (m, 1H), 4,46 (s, 2H), 3,89 (s, 3H), 3,88 (s, 3H).
Etapa 5: metil 2,4-didoro-3-((4-doro-6-dano-1-metiMH-mdol-2-M)metM)benzoato

A una solución de metil 3-((6-bromo-4-doro-1-metil-1H-indol-2-il)metil)-2,4-didorobenzoato (559 mg, 1,211 mmol) en N,N-dimetilformamida (4,3 mL) se añadió cianuro de zinc (107 mg, 0,908 mmol) y tetrakis(trifenilfosfina)paladio (140 mg, 0,121 mmol) y la mezcla de reacción se calentó a 120 °C en microondas durante 1 hora. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-40% en ciclohexano) para dar metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (200 mg, 40%). LC/MS (Método i) Rt = 2,59 min.; MS m/z: 405 [M-H]-1H NMR (DMSO-d6, 300MHz): 5 = 8,18 (m, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,73 (d, J=8,4 Hz, 1H), 7,50 (d, J=1,2 Hz, 1H), 5,67 (m, 1H), 4,54 (s, 2H), 3,96 (s, 3H), 3,88 (s, 3H).
Etapa 6: ácido 2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil) benzoico (180 mg, 94%) se preparó a partir de metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (198 mg, 0,48 mmol). LC/MS (Método i) Rt = 2,25 min.; MS m/z: 393 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 13,60 (br, 1H), 8,18 (m, 1H), 7,79 (d, J=8,4 Hz,, 1H), 7,69 (d, J=8,4 Hz,, 1H), 7,50 (d, J =1,2 Hz, 1H), 5,67 (m, 1H), 4,53 (s, 2H), 3,96 (s, 3H).
Etapa 7: 4-doro-2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1-metiMH-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A1, se preparó 4-cloro-2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-indol-6-carbonitrilo (89 mg, 82%) a partir de ácido 2,4-didoro-3-((4-doro-6-dano-1-metiMH-indol-2-il)metil)benzoico (90 mg, 0,23 mmol) y morfolina (23,9 mg, 0,27 mmol). LC/MS (Método g) Rt = 1,69 min.; MS miz: 462 [M+H]+
1H NMR (DMSO-d6, 400MHz): 58,17 (s, 1H), 7,69 (d, J=8,1 Hz, 1H), 7,50 (d, J=1,3 Hz, 1H), 7,47 (d, J=8,1 Hz, 1H), 5,70 (m, 1H), 4,50 (m, 2H), 3,96 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,15 (m, 2H).
Ejemplo CK: (2,4-didoro-3-((1-metiMH-pirrolo[2,3-to]pmdm-2-il)metil)feml)(morfolmo)metanona

Etapa 1: metil 2,4-dicloro-3-((1-(fenilsulfonil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato

A una solución de 1-(fenilsulfonil)-1H-pirrolo[2,3-ib]piridina (descrita en W 02006/050076) 1,5 g, 5,81 mmol) en THF (45 mL) y enfriada a -78 °C se añadió gota a gota butil litio (2,439 mL, 6,10 mmol) y la mezcla de reacción se agitó durante 45 minutos a -78 °C. La mezcla de reacción se calentó a -20 °C y se agitó 5 minutos a esta temperatura. A -78 °C, se añadió gota a gota metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (2,249 g, 7,55 mmol) previamente disuelto en THF, (20 mL) y la mezcla de reacción se mezcló se agitó durante 10 minutos a -78 °C y durante 18 horas a temperatura ambiente. La mezcla de reacción se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró hasta la sequedad al vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 10-20% en ciclohexano) para dar metil 2,4-dicloro-3-((1-(fenilsulfonil)-1H-pirrolo[2,3-¿]piridin-2-il)metil)benzoato (1 g, 22%). LC/MS
(Método h) Rt = 3,09 min.; MS m/z: 475 [M+H]+.1H NMR (DMSO-da, 300 MHz): 58,32 (dd, J = 4,8Hz, 1,6Hz, 1H), 8,15 (m, 2H), 7,87-7,62 (m, 6H), 7,23 (dd, J = 4,8Hz, 7,7Hz, 1H), 5,81 (m, 1H), 4,79 (s, 2H), 3,88 (s, 3h).
Etapa 2: metil 3-((1H-pirrolo[2,3-to]pindm-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 3-((1H-pirrolo[2,3-ib]piridin-2-il)metil)-2,4-didorobenzoato (125 mg, 71%) se obtuvo como un sólido de color blanco a partir metil 2,4-dicloro-3-((1-(fenilsulfonil)-1H-pirrolo[2,3-¿>]piridin-2-il)metil)benzoato (250 mg, 0,526 mmol). LC/MS (Método h) Rt = 2,34 min.; MS m/z: 335 [M+H]+ 1H NMR (DMSO-da, 300 MHz): 511,71 (ancho, 1H), 8,12 (dd, J = 5Hz, 1,6Hz, 1H), 7,75 (d, J = 8,4Hz, 1H), 7,74 (dd, J = 7,7Hz, 1,6Hz, 1H), 7,68 (d, J = 8,4Hz, 1H), 6,97 (dd, J = 7,7Hz, 5Hz, 1H), 5,68 (m, 1H), 4,46 (s, 2H), 3,87 (s, 3H).
Etapa 3: metil 2,4-didoro-3-((1-metiMH-pin'olo[2,3-fo]piridm-2-il)metil)benzoato
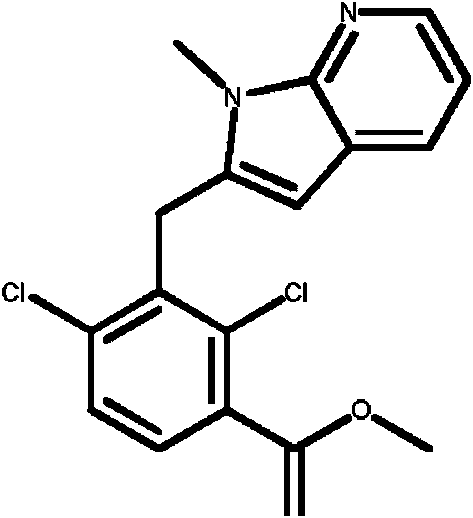
Usando un procedimiento similar al Ejemplo A, Etapa 4, metil 2,4-dicloro-3-((1-metil-1/-/-pirrolo[2,3-b]piridin-2-il)metil)benzoato (97 mg, 76%) se obtuvo como un sólido de color amarillo a partir de metil 3-((1H-pirrolo[2,3-d]piridin-2-il)metil)-2,4-diclorobenzoato (123 mg, 0,367 mmol). LC/MS (Método h) Rt = 2,70 min.; MS m/z: 349 [M+H]+ 1H NMR (DMSO-d6, 300 MHz): 58,19 (dd, J = 4,6Hz y 1,5Hz, 1H), 7,80 (d, J = 8,4Hz, 1H), 7,76 (dd, J = 7,1Hz y 1,5Hz, 1H), 7,71 (d, J = 8,4Hz, 1H), 7,01 (dd, J = 4,6Hz y 7,1Hz, 1H), 5,56 (m, 1H), 4,50 (s, 2H), 3,90 (s, 3H), 3,88 (s, 3H).
Etapa 4: ácido 2,4-didoro-3-((1-metiMH-pirrolo[2,3-fo]piridm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1-metil-1H-pirrolo[2,3-d]piridin-2-il)metil)benzoico (87 mg, 96%) se obtuvo a partir de metil 2,4-dicloro-3-((1-metil-1H-pirrolo[2,3-d]piridin-2-il)metil)benzoato (94 mg, 0,269 mmol).
LC/MS (Método h) Rt = 2,44 min.; MS m/z: 335 [M+H]+
Etapa 5: (2,4-didoro-3-((1-metiMH-pirrolo[2,3-to]pmdm-2-il)metil)feml)(morfolmo)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-dicloro-3-((1-metil-1H-pin"olo[2,3-ib]piridin-2-il)metil)fenil)(morfolino)metanona (82 mg, 79%) como un sólido de color amarillo a partir de ácido 2,4-dicloro-3-((1-metil-1H-pir-rolo[2,3-ib]piridin-2-il)metil)benzoico (86 mg, 0,257 mmol) y morfolina (33,5 mg, 0,385 mmol). LC/MS (Método g) Rt = 1,34 min.; MS m/z: 404 [M+H]+1H NMR (DMSO-cfe, 400 MHz): 58,19 (dd, J = 4,6Hz, y 1,5Hz, 1H), 7,77 (dd, J = 7,9Hz, y 1,5Hz, 1H), 7,67(d, J = 8,4Hz, 1H), 7,45 (d, J = 8,4Hz, 1H), 7,02 (dd, J = 4,6Hz, y 7,9Hz, 1H), 5,60 (m, 1H), 4,46 (s, 2H), 3,90 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,18 (m, 2H).
Ejemplo CL: 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-pirrolo[2,3-b]piridina-6-car-bonitrilo

Etapa 1: metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato

A una solución de diisopropilamida de litio (8,81 mL, 17,63 mmol) en THF (27 mL) enfriada a -25 °C se añadió gota a gota durante 10 minutos 4-metil-1-(fenilsulfonil)-1H-pirrolo[2,3-¿]piridina (descrita en WO2013/015984) (2,40 g, 8,81 mmol) en solución en THF (44 mL) y la mezcla de reacción se agitó durante 20-25 minutos a -25 °C. Se añadió gota a gota metil 2,4-dicloro-3-formilbenzoato (Preparación #1, Etapa C) (2,157 g, 9,25 mmol) previamente disuelto en THF (22 mL) durante 15 minutos a -25 °C. La mezcla de reacción se agitó durante 1,5 horas a -25 °C. La mezcla de reacción se inactivó con solución acuosa saturada de NH4Cl, y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre sulfato de magnesio y se concentró hasta sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice
(eluyendo con EtOAc al 10-20% en ciclohexano) para dar metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoato (1,32 g, 27%) como una espuma naranja. LC/MS (Método h) Rt = 2,69 min.; MS miz: 505 [M+H]+ 1H NMR (DMSO-da, 300 MHz): 58,20 (d, J = 4,9Hz, 1H), 8,00 (m, 2H), 7,68 (m, 2H), 7,56 (m, 3H), 7,13 (dd, J = 1,1Hz, 6,1Hz, 1H), 7,10 (d, J = 4,9Hz, 1H), 6,70 (d, J = 1,1Hz, 1H), 6,68 (d, J = 6,1Hz, 1H), 3,86 (s, 3H), 2,42 (s, 3H).
Etapa 2: metil 2,4-dicloro-3-((4-metiM-(femlsulfonil)-1H-pirrolo[2,3-fo]piridm-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-1H-pirrolo[2,3-6]piridin-2-il)metil)benzoato (1,85 g, 70%) (mezcla con doble enlace exocíclico) se obtuvo a partir de metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-1H-pirrolo[2,3-¿]piridin-2-il)metil)benzoato (2,12 g, 4,19 mmol). LC/MS (Método h) Rt = 3,00 min. y 3,11 min; MS miz: 489 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 58,15 (m, 3H), 7,85 (d, J = 8,4Hz, 1H), 7,76-7,60 (m, 4H), 7,06 (d, J = 5,2Hz, 1H), 5,85 (m, 1H), 4,79 (s, 2H), 3,88 (s, 3H), 2,29 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-((4-metiMH-pin'olo[2,3-fo]piridm-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-dicloro-3-((4-metil-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoato (86 mg, 93%) se obtuvo como un sólido de color amarillo a partir de metil 2,4-dicloro-3-((4-metil-1-(fenilsulfo-nil)-1H-pirrolo[2,3-¿]piridin-2-il)metil)benzoato (130 mg, 0,266 mmol). lC/MS (Método h) Rt = 2,27 min; MS miz:
349 [M+H]+
1H NMR (DMSO-C6, 300 MHz): 511,55 (ancho, 1H), 8,00 (d, J = 4,9Hz, 1H), 7,75 (d, J = 8,4Hz, 1H), 7,68 (d, J = 8,4Hz, 1H), 6,80 (d, J = 4,9Hz, 1H), 5,72 (m, 1H), 4,45 (s, 2H), 3,87 (s, 3h), 2,34 (s, 3H).
Etapa 4: 2-(2,6-didoro-3-(metoxicarboml)bencil)-4-metiMH-pirrolo[2,3-to]piridma 7-óxido

A una solución de metil 2,4-dicloro-3-((4-metil-1H-pirrolo[2,3-í)]piridin-2-il)metil)benzoato (160 mg, 0,458 mmol) en DME (6 mL) y enfriada a 0 °C se añadió ácido 3-clorobenzoperoxoico (138 mg, 0,802 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró hasta la sequedad, el residuo se solubilizó en EtOAc y se lavó con solución acuosa saturada de NaHCO3 y salmuera. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró hasta la sequedad para dar 2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metil-1 H-pirrolo[2,3-ib]piridina 7-óxido (140 mg, 84%). LC/MS (Método h) Rt = 2,14 min; MS m/z: 365 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 512,60 (ancho, 1H), 7,95 (d, J = 6,2Hz, 1H), 7,77 (d, J = 8,4Hz, 1H), 7,69 (d, J = 8,4Hz, 1H), 6,82 (d, J = 6,4Hz, 1H), 5,78 (s, 1H), 4,46 (s, 2h), 3,87 (s, 3H), 2,33 (s, 3H).

A una solución de 2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metil-1H-pirrolo[2,3-ib]piridina 7-óxido (70 mg, 0,192 mmol) en ACN (4 mL) se añadió trietilamina (0,067 mL, 0,479 mmol) y luego cianuro de trimetilsililo (0,154 mL, 1,150 mmol). La mezcla de reacción se agitó a reflujo durante la noche. Se añadió más trietilamina (0,067 mL, 0,479 mmol) y cianuro de trimetilsililo (0,154 mL, 1,150 mmol). La mezcla de reacción se agitó a reflujo durante 24 horas. La mezcla de reacción se inactivó con solución acuosa saturada de NaHCO3 y se extrajo con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró hasta sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar metil 2,4-dicloro-3-((6-ciano-4-metil-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoato (22 mg, 30%).
LC/MS (Método k) Rt = 2,93 min; MS m/z: 374 [M+H]+ 1H NMR (DMSO-C6, 300 MHz): 512,29 (ancho, 1H), 7,78 (d, J = 8,4Hz, 1H), 7,69 (d, J = 8,4Hz, 1H), 7,41 (s, 1H), 5,95 (s, 1H), 4,51 (s, 2H), 3,87 (s, 3H), 2,43 (s, 3H).
Etapa 6: metil 2,4-didoro-3-((6-ciano-1,4-dimetiMH-pirrolo[2,3-fo]piridm-2-M)metil)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-pirrolo[2,3-6]piridin-2-il)metil)benzoato (63 mg, 95%) se obtuvo a partir de metil 2,4-dicloro-3-((6-ciano-4-metil-1 H-pirrolo[2,3-ib]piridin-2-il)metil)benzoato (64 mg, 0,171 mmol)
LC/MS (Método h) Rt = 3,06 min; MS miz: 388 [M+H]+1H NMR (DMSO-cfe, 300 MHz): 57,83 (d, J = 8,4Hz, 1H), 7,73 (d, J = 8,4Hz, 1H), 7,45 (s, 1H), 5,82 (s, 1H), 4,56 (s, 2H), 3,93 (s, 3H), 3,88 (s, 3H), 2,41 (s, 3H).
Etapa 7: ácido 2,4-didoro-3-((6-ciano-1,4-dimetiMH-pin'olo[2,3-fo]pmdm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((6-dano-1,4-dimetil-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoico (59 mg, 99%) se obtuvo a partir de metil 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoato (62 mg, 0,160 mmol). LC/MS (Método h) Rt = 2,56 min; mS miz : 374 [M+H]+
1H NMR (DMSO-C6, 300 MHz): 513,66 (ancho, 1H), 7,76 (d, J = 8,4Hz, 1H), 7,67 (d, J = 8,4Hz, 1H), 7,45 (s, 1H), 5,81 (s, 1H), 4,55 (s, 2H), 3,93 (s, 3H), 2,42 (s, 3H).
Etapa 8: 2-(2,6-didoro-3-(morfolma-4-carboml)bencil)-1,4-dimetiMH-pirrolo[2,3-to]pindma-6-carbomtrilo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-(2,6-didoro-3-(morfolina-4-carbonil)bendl)-1,4-dimetil-1H-pirrolo[2,3-ib]piridina-6-carbonitrilo (55 mg, 79%) se obtuvo a partir de ácido 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-pir
rolo[2,3-í)]piridin-2-il)metil)benzoico (58 mg, 0,155 mmol) y morfolina (23,5 mg, 0,23 mmol). LC/MS (Método g) Rt = 1,57 min; MS m/z: 443 [M+H]+ 1H NMR (DMSO-cfe, 400 MHz): 57,69 (d, J = 8,4Hz, 1H), 7,47 (d, J = 8,4Hz, 1H), 7,46 (m, 1H), 5,76 (s, 1H), 4,53 (m 2H), 3,92 (s, 3H), 3,65 (m, 4H), 3,53 (m, 2H), 3,18 (m, 2H), 2,42 (s, 3H).
Tabla CL. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-((6-dano-1,4-dimetil-1H-pirrolo[2,3-6]piridin-2-il)metil)benzoico (Ejemplo CL, Etapa 7) usando el mismo procedimiento con la amina apropiada.

Ejemplo CM: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona
Ejemplo CM1: (2,4-didoro-3-((6-doro-1,4-dimetiMH-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona

Etapa 1: metil 2-(2,6-didoro-3-(metoxicarboml)bendl)-6-yodo-4-metiMH-pirrolo[2,3-b]piridma-1-carboxilato

A una solución de 2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metiMH-pirrolo[2,3-ib]piridina 7-óxido (preparado de manera similar al Ejemplo cL, Etapa 4) (60 mg, 0,164 mmol) en THF (3 mL) se añadió bis(trimetilsilil)amina (0,034 mL, 0,164 mmol), yodotrimetilsilano (0,067 mL, 0,493 mmol) y luego cloroformiato de metilo (0,025 mL, 0,33 mmol) gota a gota. La reacción se agitó 20 horas a temperatura ambiente. La mezcla de reacción se concentró a presión reducida y el residuo se disolvió en EtOAc. La capa orgánica se lavó sucesivamente con solución acuosa saturada de NaHCO3 , salmuera y se secó sobre sulfato de magnesio, se filtró y luego se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-5% en ciclohexano) para dar metil 2-(2,6-dicloro-3-(metoxicarbo-nil)bencil)-6-yodo-4-metil-1H-pirrolo[2,3-ib]piridina-1-carboxilato (33 mg, 14%) como un sólido de color amarillo pálido. El compuesto se usó directamente en la siguiente etapa. LC/MS (Método h) Rt = 3,30 min; MS m/z: 533 [M+H]+
Etapa 2: metil 2-(2,6-didoro-3-(metoxicarboml)bendl)-4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]pirid-ma-1-carboxilato y metil 6-doro-2-(2,6-didoro-3-(metoxicarboml)bendl)-4-metiMH-pirrolo[2,3-b]piridma-1-carboxilato

A una solución de metil 2-(2,6-dicloro-3-(metoxicarbonil)bendl)-6-yodo-4-metil-1H-pirrolo[2,3-ib]piridina-1-carboxilato (134 mg, 0,251 mmol) en DMF (3 mL) se añadió a temperatura ambiente metil 2,2-difluoro-2-(fluorosulfonil)acetato (0,127 mL, 1,005 mmol) y yoduro de cobre(I) (47,9 mg, 0,251 mmol), y la mezcla de reacción se agitó a 115 °C durante 24 horas en un tubo sellado. El residuo se diluyó en EtOAc. La capa orgánica se lavó sucesivamente con agua, con solución acuosa de LiCl al 10% y salmuera y se secó sobre sulfato de magnesio, se filtró y luego se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 10-20% en ciclohexano) para dar una mezcla de metil 2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridina-1-carboxilato LC/MS (Método h) Rt = 3,30 min; MS m/z: 475 [M+H]+ y metil 6-cloro-2-(2,6-dicloro-3-(metoxicar-bonyl)bencil)-4-metiMH-pirrolo[2,3-íb]piridina-1-carboxilato (120 mg) con una relación de 43/57. LC/MS (Método h) Rt = 3,12 min; Ms m/z: 441 [M+H]+
Etapa 3: ácido 2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridm-2-il)metil)benzoico y ácido 2,4-didoro-3-((6-doro-4-metiMH-pirrolo[2,3-b]piridm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-6]piridin-2-il)metil)benzoico LC/MS (Método h) Rt = 2,65 min; MS m/z: 403 [M+H]+ y ácido 2,4-dicloro-3-((6-cloro-4-metil-1H-pirrolo[2,3-¿]piridin-2-il)metil)benzoico (85 mg) LC/MS (Método h) Rt = 2,48 min; MS m/z: 369 [M+H]+ se obtuvieron con una relación de 35/65 a partir de una mezcla de metil 2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metil-6-(trifluorometil)-1H-pirrolo[2,3-¿]piridina-1-carboxilato y metil 6-cloro-2-(2,6-dicloro-3-(metoxicarbonil)bencil)-4-metil-1H-pirrolo[2,3-6]piridina-1-carboxilato (120 mg) con una relación de 43/57.
Etapa 4: (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona y (2,4-didoro-3-((6-doro-4-metiMH-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-didoro-3-((4-metil-6-(trifluorometil)-1 H-pirrolo[2,3-ib]piridin-2-il)metil)fenil)(morfolino)metanona LC/MS (Método h) Rt = 2,68 min; MS miz: 472 [M+H]+ y (2,4-dicloro-3-((6-cloro-4-metil-1H-pirrolo[2,3-ib]piridin-2-il)metil)fenil)(morfolino)metanona (102 mg) LC/MS (Método h) Rt = 2,51 min; MS miz: 438 [M+H]+ se obtuvieron con una relación de 39/61 a partir de una mezcla de ácido 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)benzoico y ácido 2,4-dicloro-3-((6-cloro-4-metil-1 H-pirrolo[2,3-ib]piridin-2-il)metil)benzoico (102 mg) con una relación de 35/65.
Etapa 5: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona y (2,4-didoro-3-((6-doro-1,4-dimetiMH-pirrolo[2,3-to]piridm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo P, Etapa 4, (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-¿]piridin-2-il)metil)fenil)(morfolino)metanona (24 mg, 23%)
LC/MS (Método g) Rt = 1,83 min; MS m/z: 486 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 5 7,69 (d, J=8,4 Hz, 1H), 7,47 (d, J=8,4 Hz, 1H), 7,34 (d, J=0,9 Hz, 1H), 5,81 (m, 1H), 4,51 (m, 2H), 3,93 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,20 (m, 2H), 2,46 (s, 3H) y (2,4-dicloro-3-((6-cloro-1,4-dimetil-1H-pirrolo[2,3-¿]piridin-2-il)metil)fenil)(morfolino)metanona (30 mg, 31%) LC/MS (Método g) Rt = 1,72 min; MS m/z: 452 [M+H]+1H NMR (DMSO-d6, 400MHz): 57,67 (d, J =8,4 Hz, 1H), 7,45 (d, J =8,4 Hz, 1H), 6,94 (d, J =0,9 Hz, 1H), 5,68 (m, 1H), 4,44 (m, 2H), 3,84 (s, 3H), 3,65 (m, 4H), 3,52 (m, 2H), 3,19 (m, 2H), 2,35 (s, 3H) se obtuvieron a partir de una mezcla de (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-¿]piridin-2-il)metil)fenil)(morfolino)metanona y (2,4-dicloro-3-((6-cloro-4-metil-1H-pirrolo[2,3-¿]piridin-2-il)metil)fenil)(morfolino)metanona (100 mg) con una relación de 39/61.
Ejemplo CN: (2,4-didoro-3-(hidroxi(1-metil-5-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)fenil)(morfolino)metanona

Etapa 1: metil 3-((1-((3-(ferc-butil)feml)sulfoml)-5-(tnfluorometil)-1H-pirrolo[3,2-to]pmdm-2-il)(hidroxi)metil)-2,4-diclorobenzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 3-((1-((3-(terf-butil)fenil)sulfonil)-5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)(hidroxi)metil)-2,4-diclorobenzoato (540 mg, 7 6 %) a partir de 3-(íerí-butil)-W-(2-yodo-6-(trifluorometil)piridin-3-il)bencenosulfonamida (descrita en WO2007/026104) (500 mg, 1,032 mmol) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (Preparación #1) (348 mg, 1,342 mmol) LC/MS (Método h) Rt = 3,43 min.; MS m/z: 615 [M+H]+.1H NMR (DMSO-da, 300MHz,) 58,70 (d, J = 8,7Hz, 1H), 7,94 (m, 1H), 7,87 (d, J = 8,7Hz, 1H), 7,76 (d, J = 7,9Hz, 1H), 7,70 (d, J = 7,9Hz, 1H), 7,68 (d, J = 8,7Hz, 1H), 7,53 (m, 2H), 7,02 (ancho, 1H), 6,95 (ancho, 1H), 6,92 (s, 1H), 3,86 (s, 3h), 1,21 (s, 9H).
Etapa 2: ácido 2,4-dicloro-3-(hidroxi(5-(trifluorometil)-1H-pirrolo[3,2-b]piridin-2-il)metil)benzoico

A una solución de metil 3-((1-((3-(ferc-butil)fenil)sulfonil)-5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)(hidroxi)metil)-2,4-diclorobenzoato (500 mg, 0,812 mmol) en THF (5 mL) y agua (2 mL) se añadió hidróxido de litio (97 mg, 4,06 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. Se añadió hidróxido de litio (97 mg, 4,06 mmol) (10 mg, 1 eq) y la reacción se calentó a reflujo durante 5 horas y se agitó durante la noche a temperatura ambiente. La mezcla de reacción se diluyó con agua y se acidificó con solución acuosa de HCl 1M. La capa acuosa obtenida se extrajo con DCM y la capa orgánica obtenida se lavó con salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida para dar ácido 2,4-dicloro-3-(hidroxi(5-(trifluorometil)-1H-pyrro-lo[3,2-¿]piridin-2-il)metil)benzoico (374
mg, 45%) como un sólido de color marrón. LC/MS (Método h) Rt = 1,85 min.; MS m iz: 405 [M+H]+. 1H NMR (DMSO-d6, 300MHz,) 5 11,76 (s, 1H), 7,90 (d, J = 9Hz, 1H), 7,68 (d, J = 9Hz, 1H), 7,56 (d, J = 9Hz, 1H), 7,50 (d, J = 9Hz, 1H), 6,82 (ancho, 1H), 6,20 (s, 1H).
Etapa 3: (2,4-dicloro-3-(hidroxi(5-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-didoro-3-(hidroxi(5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (87 mg, 15%) se preparó a partir de ácido 2,4-didoro-3-(hidroxi(5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)benzoico (370 mg, 0,913 mmol) y se usó crudo en la siguiente etapa y morfolina (239 mg, 2,74 mmol). LC/MS (Método h) Rt = 2,07 min.; MS miz: 474 [M+H]+.
Etapa 4: (2,4-dicloro-3-(hidroxi(1-metil-5-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al del Ejemplo P, Etapa 4 (2,4-didoro-3-(hidroxi(1-metil-5-(trifluorometil)-1H-pir-rolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (28 mg, 34%) se preparó a partir de (2,4-dicloro-3-(hidroxi(5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (80 mg, 0,169 mmol). Lc/MS (Método g) Rt = 1,36 min.; MS miz: 488 [M+H]+.1H NMR (DMSO-d6, 400MHz,) 5 8,13 (m, 1H), 7,62 (m, 2H), 7,46 (d, J = 8Hz, 1H), 6,73 (m, 1H), 6,65 (m, 1H), 6,32 y 6,29 (s, 1H), 3,88 y 3,86 (s, 3H), 3,66 (m, 3H), 3,55 (m, 3H), 3,21 (m, 2H).
Ejemplo CO: (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-pirrolo[3,2-to]piridin-2-il)metil)fenil)(morfolino)metanona

Etapa 1: (2,4-dicloro-3-(hidroxi(1-(femlsulfoml)-6-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-didoro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (520 mg, 87%) se preparó a partir de W-(2-bromo-4-(trifluorometil)piridin-3-il)bencenosulfonamida (Preparación #26) (370 mg, 0,971 mmol) y (2,4-dicloro-3-(1-hy-droxiprop-2-in-1-il)fenil)(morfolino)metanona (Preparación #3) (396 mg, 1,262 mmol). LC/MS (Método g) Rt = 1,62 min.; MS miz: 614 [M+H]+.1H NMR (DMSO-d6, 400MHz,) 58,92 (s, 1H), 8,56 (m, 1H), 7,89 (m, 2H), 7,74 (m, 1H), 7,60 (m, 2H), 7,52 y 7,45 (d, J = 8Hz, 1H), 7,39 (m, 1H), 7,03 y 6,92 (s, 1H), 6,98 y 6,88 (m, 1H), 6,87 y 6,83 (d, J = 8Hz, 1H), 3,64 (m, 4H), 3,54 (m, 2H), 3,17 (m, 2H).
Etapa 2: (2,4-dicloro-3-((1-(femlsulfoml)-6-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo Z, Etapa 2, (2,4-didoro-3-((1-(fenilsulfonil)-6-(trifluorometil)-1H-pir-rolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (314 mg, 96%) se preparó a partir de (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (336 mg, 0,547 mmol). LC/MS (Método h) Rt = 3,05 min.; MS miz: 598 [M+H]+.
1H NMR (DMSO-d6, 300MHz,) 58,89 (m, 1H), 8,70 (m, 1H), 8,05 (m, 2H), 7,83 (m, 1H), 7,67 (m, 3H), 7,46 (d, J = 9Hz, 1H), 6,18 (s, 1H), 4,58 (m, 2h), 3,64 (m, 4H), 3,49 (m, 2H), 3,21 (m, 2H).
Etapa 3: (2,4-dicloro-3-((6-(trifluorometil)-1H-pirrolo[3,2-to]piridm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-dicloro-3-((6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (237 mg, 100%) se preparó a partir de (2,4-didoro-3-((1-(fenilsulfonil)-4-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (310 mg, 0,518 mmol) LC/mS (Método h) Rt = 2,10 min.; MS miz: 456 [M-H]-.1H NMR (DMSO-da, 300MHz,) 511,83 (ancho, 1H), 8,56 (m, 1H), 8,02 (m, 1H), 7,66 (d, J = 9Hz, 1H), 7,46 (d, J = 9Hz, 1H), 6,13 (s, 1H), 4,53 (s, 2h), 3,65 (m, 4H), 3,54 (m, 2H), 3,19 (m, 2H).
Etapa 4: (2,4-didoro-3-((1-metil-6-(tnfluorometil)-1H-pirrolo[3,2-to]pmdm-2-il)metil)feml)(morfolmo)metanona:

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-didoro-3-((1-metil-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (16 mg, 31%) se preparó a partir de (2,4-didoro-3-((6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (50 mg, 0,109 mmol) LC/MS (Método g) Rt = 1,46 min.; MS miz: 472 [M+H]+.
1H NMR (DMSO-d6, 400MHz,) 58,57 (m, 1H), 8,37 (m, 1H), 7,69 (d, J = 8Hz, 1H), 7,47 (d, J = 8Hz, 1H), 5,85 (s, 1H), 4,52 (s, 2H), 3,98 (s, 3h), 3,56 (m, 4H), 3,55 (m, 2H), 3,21 (m, 2H).
Ejemplo CP: (2,4-dicloro-3-(1-metil-6-(trifluorometil)-1H-pirrolo[3,2-b]piridina-2-carbonil)fenil)(morfolino)metanona

Etapa 1: (2,4-didoro-3-(1-(femlsulfoml)-6-(trifluorometil)-1H-pirrolo[3,2-to]piridma-2-carbonil)fenil)(morfolino)metanona

A una solución de (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-2-il)metil)fenil)(morfolino)metanona (Ejemplo CO, Etapa 1) (580 mg, 0,944 mmol) en dicloroetano (10 mL) se añadió dióxido de manganeso (821 mg, 9,44 mmol) y la mezcla se agitó a 85 °C durante 24 horas. La mezcla se filtró y se lavó con DCM y ACN. El filtrado se concentró para dar (2,4-dicloro-3-(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridina-2-carbonil)fenil)(morfolino)metanona (540 mg, 80%) como un polvo beige. LC/MS (Método h) Rt = 2,92 min.; mS m/z: 612 [M+H]+.
1H NMR (DMSO-da, 300MHz,) 59,13 (s, 1H), 8,93 (s, 1H), 8,26 (d, J = 9Hz, 2H), 7,85 (ancho, 1H), 7,84 (m, 1H), 7,71 (m, 3H), 7,64 (d, J = 9Hz, 1H), 3,66 (m, 4H), 3,53 (m, 2h), 3,25 (m, 2H).
Etapa 2: (2,4-didoro-3-(6-(trifluorometil)-1H-pirrolo[3,2-to]piridma-2-carboml)feml)(morfolmo)met-anona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-didoro-3-(6-(trifluorometil)-1H-pirrolo[3,2-ib]piridina-2-carbonil)fenil)(morfolino)metanona (363 mg, 86%) se preparó a partir de (2,4-dicloro-3-(1-(fenilsulfonil)-6-(tri-fluorometil)-1H-pirrolo[3,2-ib]piridina-2-carbonil)fenil)(morfolino)metanona (530 mg, 0,865 mmol). LC/MS (Método h) Rt = 2,31 min.; MS m/z: 472 [M+H]+.1H NMR (DMSO-d6, 300MHz,) 513,00 (ancho, 1H), 8,84 (m, 1H), 8,20 (m, 1H), 7,77 (d, J = 9Hz, 1H), 7,68 (d, J = 9Hz, 1H), 7,50 y 7,20 (ancho, 1H), 3,66 (m, 4H), 3,57 (m, 2H), 3,40 (m, 1H), 3,19 (m, 1H).
Etapa 3: (2,4-dicloro-3-(1-metil-6-(trifluorometil)-1H-pirrolo[3,2-to]piridma-2-carboml)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-didoro-3-(1-metil-6-(trifluorometil)-1H-pirrolo[3,2-ib]piridina-2-carbonil)fenil)(morfolino)metanona (28 mg, 27%) se preparó a partir de (2,4-didoro-3-(6-(trifluorometil)-1H-pirrolo[3,2-¿]piridina-2-carbonil)fenil)(morfolino)metanona (100 mg, 0,212 mmol). LC/MS (Método g) Rt = 1,56 min.; mS m/z: 486 [M+H]+.1H NMR (DMSO-d6, 400MHz,) 58,87 (m, 1H), 8,75 (m, 1H), 7,77 (d, J = 8Hz, 1H), 7,66 (d, J = 8Hz, 1H), 7,45 (ancho, 1H), 4,27 (s, 3H), 3,66 (m, 4H), 3,55 (m, 2H), 3,26 (m, 2H).
Ejemplo CQ: (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridm-2-il)metil)feml)(morfolmo)metanona
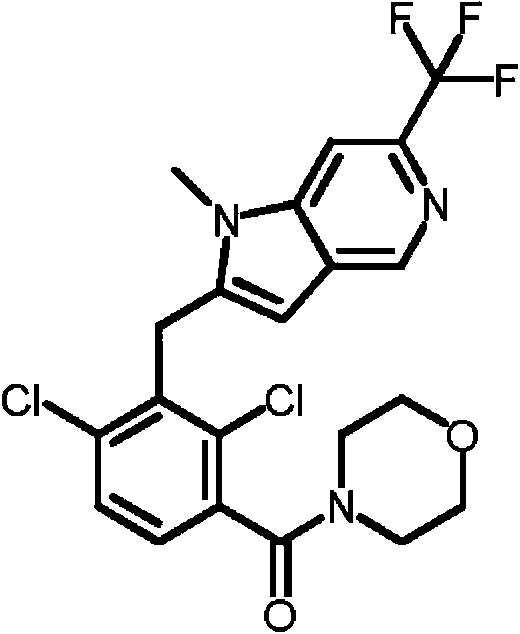
Etapa 1: (2,4-didoro-3-(hidroxi(1-(femlsulfoml)-6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (1,94 g, 68%) se preparó a partir de N-(5-yodo-2-(trifluorometil)piridin-4-il)bencenosulfonamida (Preparación #27) (2 g, 4,67 mmol) y (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(tetrahidro-2H-piran-4-il)metanona (Preparación #3) (1,902 g, 6,07 mmol). LC/MS (Método h) Rt = 2,63 min.; MS miz: 614 [M+H]+.1H NMR (DMSO-d6, 300MHz) 59,07 and 9,05 (s, 1H), 8,32 y8,29 (s, 1H), 7,95 (m, 2H), 7,75 (m, 1H), 7,63 (m, 2H), 7,51 (m, 1H), 7,40 (m, 1H), 6,97 (m, 2H), 6,79 y 6,74 (d, J = 8Hz, 1H), 3,65 (m, 4H), 3,54 (m, 2H), 3,16 (m, 2H).
Etapa 2: (2,4-didoro-3-((1-(femlsulfoml)-6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-il)metil)fenil)(morfolino)metanona
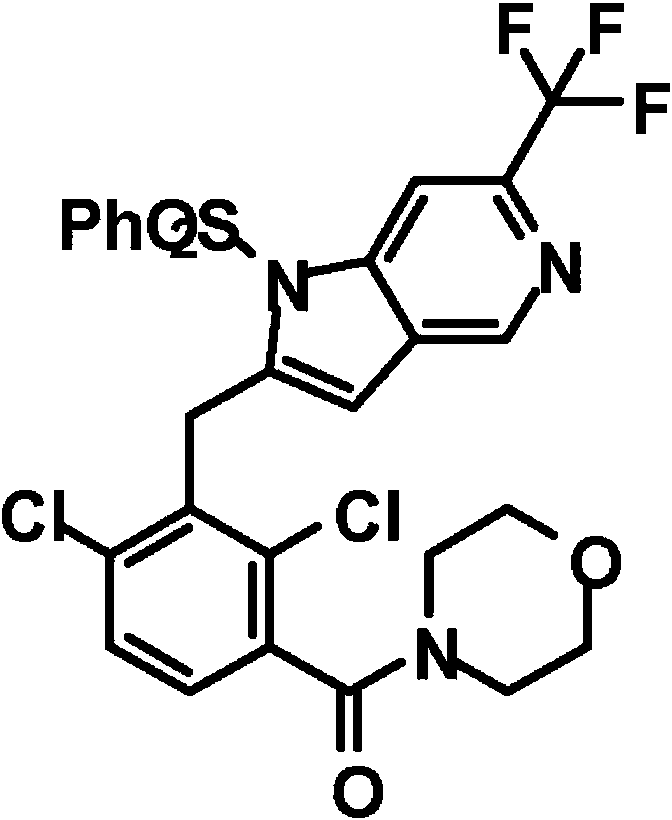
Usando un procedimiento similar al del Ejemplo Z, Etapa 2(2,4-dicloro-3-((1-(fenilsulfonil)-6-(trifiuorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (2,1 g, 100%) se preparó a partir de (2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (1,93 g, 3,14 mmol). lC/MS (Método k) Rt = 3,14 min.; MS miz: 598 [M+H]+.1H NMR (DMSO-d6, 300MHz,) 58,94 (s, 1H), 8,48 (s, 1H), 8,05 (m, 1H), 7,96 (m, 1h), 7,81 (m, 1H), 7,66 (m, 2H), 7,47 (m, 1H), 7,41 (m, 1H), 6,21 (s, 1H), 4,52 (m, 2h), 3,64 (m, 4H), 3,49 (m, 2H), 3,16 (m, 2H).
Etapa 3: (2,4-didoro-3-((6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-didoro-3-((6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (1,05 g, 65%) se preparó a partir de (2,4-didoro-3-((1-(fenilsulfonil)-6-(tri-fluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfblino)metanona (2,1 g, 3,51 mmol). LC/MS (Método k) Rt = 2,35 min.; MS miz: 458 [M+H]+.1H NMR (DMSO-da, 300MHz,) 512,05(ancho, 1H), 8,80 (s, 1H), 7,78 (s, 1H), 7,66 (d, J = 9Hz, 1H), 7,43 (d, J = 9Hz, 1H), 6,14 (s, 1H), 4,49 (s, 2H), 3,66 (m, 4H), 3,54 (m, 2H), 3,18 (m, 2H).
Etapa 4: (2,4-didoro-3-((1-metM-6-(trifluorometN)-1H-pirrolo[3,2-c]pmdm-2-N)metN)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4 (2,4-dicloro-3-((1-metil-6-(trifluorometil)-1/-/-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (78mg, 50%) se preparó a partir de (2,4-didoro-3-((6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (150 mg, 0,327 mmol) LC/MS (Método g) Rt = 2,36 min.; MS m iz : 472 [M+H]+.
1H NMR (DMSO-d6, 400MHz,) 58,76 (s, 1H), 8,09 (s, 1H), 7,69 (d, J = 8Hz, 1H), 7,47 (d, J = 8Hz, 1H), 5,91 (s, 1H), 4,49 (s, 2H), 3,98 (s, 3h), 3,65 (m, 4H), 3,55 (m, 2H), 3,19 (m, 2H).
Ejemplo CR: (2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-pirrolo[3,2-c]pindm-2-il)metil)fenil)(morfolino)metanona
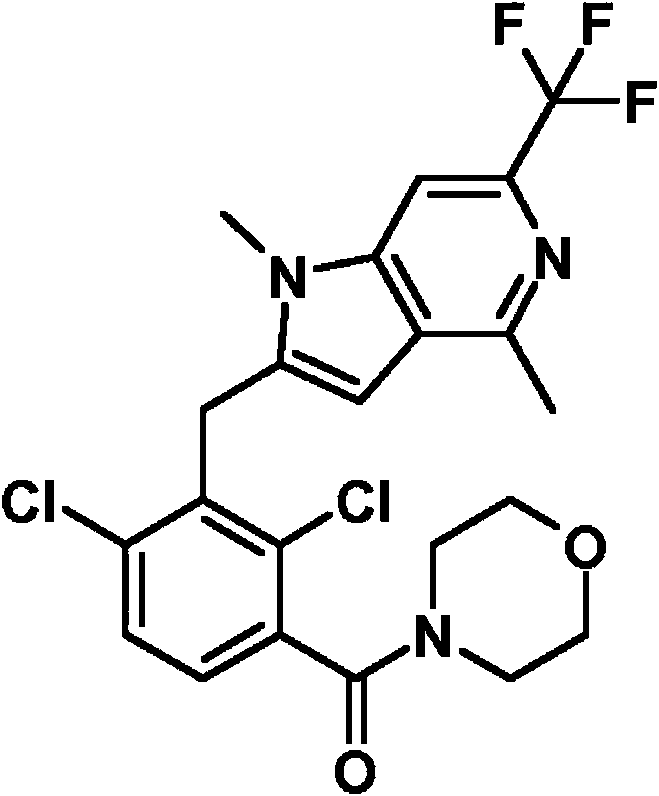
Etapa 1: (2,4-didoro-3-(hidroxi(4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 1, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pyrro-lo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (180 mg, 50%) se preparó a partir de W-(3-iodo-2-metil-6-(trifluorometil)piridin-4-il)bencenosulfonamida (Preparación #39) (233 mg, 0,53 mmol) y (2,4-didoro-3-(1-hidroxi-prop-2-in-1-il)fenil)(morfolino)metanona (199 mg, 0,63 mmol) (Preparación #3). LC/MS (Método i) Rt = 2,21 min; Ms miz: 628 [M+H]+1H NMR (DMSO-da, 300MHz): 58,14 (m, 1H), 7,94 (d, J=7,8 Hz, 1H), 7,86 (d, J=7,8 Hz, 1H), 7,75 (m, 1H), 7,62 (m, 2H), 7,50 (m, 1H), 7,40 (m, 1H), 7,00 (m, 1H), 6,91 (m, 1H), 6,74 y 6,81 (d, J=6 Hz, 1H), 3,65 (m, 4H), 3,55 (m, 2H), 3,18 (m, 2H), 2,68 y 2,69 (s, 3H).
Etapa 2: (2,4-didoro-3-((4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo Z, Etapa 2, (2,4-dicloro-3-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (202 mg, 100%) se preparó a partir de (2,4-dicloro-3-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (175 mg, 0,28mmol) como una resina de color amarillo. El compuesto se usa directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,48 min; MS m/z: 612 [M+H]+
Etapa 3: (2,4-didoro-3-((4-metM-6-(trifluorometM)-1H-pirrolo[3,2-c] piridin-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 3, (2,4-dicloro-3-((4-metil-6-(trifluorometil)-1 H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (115 mg, 87%) se preparó a partir de (2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (171 mg, 0,28 mmol). LC/MS (Método i) Rt = 1,98 min; MS m/z: 472 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 11,96 (s, 1H), 7,65 (m, 2H), 7,43 (d, J=8,3 Hz, 1H), 6,13 (s, 1H), 4,48 (s, 2H), 3,65 (dm, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,58 (s, 3H).
Etapa 4: (2,4-didoro-3-((1,4-dimetM-6-(trifluorometM)-1H-pirrolo[3,2-c]piridm-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 4, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (25 mg, 21%) se preparó a partir de (2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(morfolino)metanona (112 mg, 0,24 mmol). LC/MS (Método g) Rt = 1,53 min; MS miz: 486 [M+H]+1H NMR (DMSO-da, 400MHz): 57,92 (s, 1H), 7,69 (d, J=8,4 Hz, 1H), 7,47 (d, J=8,4 Hz, 1H), 5,88 (s, 1H), 4,48 (m, 2H), 3,96 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,21 (m, 2H), 2,54 (s, 3H).
Ejemplo CS: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1,4-dimetiMH-pirrolo[3,2-c]pindma-6-carbomtrMo

Etapa 1: 2-((2,6-didoro-3-(morfolma-4-carboml)fenM)(hidroxi)metM)-4-metiM-(femlsulfoml)-1H-pirrolo[3,2-c]piridina-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 1,2-((2,6-dicloro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (204 mg, 42%) se preparó a partir de W-(6-ciano-3-yodo-2-metilpiridin-4-il)bencenosulfonamida (Preparación #40 ) (300 mg, 0,75 mmol) y (2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)fenil)(morfolino)metanona (283 mg, 0,90 mmol) (Preparación #3). LC/MS (Método h) Rt = 2,37 min; MS miz: 585 [M+H]+ 1H NMR (DMSO-da, 300MHz): 58,44 y 8,42 (s, 1H), 8,07 (m, 1h), 7,99 (m, 1H), 7,75 (m, 1H), 7,63 (m, 2H), 7,52 y 7,49 (d, J= 8,1 Hz, 1H), 7,40 y 7,39 (d, J= 8,1 Hz, 1H), 7,06 y 6,89 (s, 1H), 7,03 y 6,93 (d, J= 5,5 Hz, 1H), 6,83 y 6,77 (d, J= 5,5 Hz, 1H), 3,64 (m, 4H), 3,55 (m, 2H), 3,16 (m, 2H), 2,65 y 2,63 (s, 3H).
Etapa 2: 2-(2,6-didoro-3-(morfolma-4-carbonM)bencM)-4-metiM-(femlsulfoml)-1H-pirrolo [3,2-c] piridina-6-carbonitrilo

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-4-metil-1-(fenilsulfonil)-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (248 mg, 100%) se preparó a partir de 2-((2,6-dicloro-3-(morfolina-4-carbonil)fenil)(hidroxi)metil)-4-metil-1-(fenilsulfonil)-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (200 mg, 0,34 mmol) como una resina de color amarillo. El compuesto se usa directamente en la siguiente etapa. LC/mS (Método h) Rt = 2,77 min; MS m / z : 569 [M+H]+
Etapa 3: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-4-metiMH-pirrolo[3,2-c]piridma-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-4-metil-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (100 mg, 47%) se preparó a partir de 2-(2,6-dicloro-3-(morfolina-4-carbon-il)bencil)-4-metil-1-(fenilsulfonil)-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (248 mg, 0,43 mmol). LC/MS (Método h) Rt = 2,07 min; MS m / z : 429 [M+H]+
1H NMR (DMSO-d6, 300MHz): 512,14 (ancho, 1H), 7,85 (s, 1H), 7,66 (d, J=8,3 Hz, 1H), 7,43 (d, J=8,3 Hz, 1H), 6,15 (s, 1H), 4,48 (s, 2H), 3,65 (m, 4H), 3,53 (m, 2H), 3,17 (m, 2H), 2,56 (s, 3H).
Etapa 4: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1,4-dimetiMH-pirrolo[3,2-c]piridma-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (18 mg, 16%) se preparó a partir de 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-4-metil-1H-pirrolo[3,2-c]piridina-6-carbonitrilo (100 mg, 0,23 mmol). LC/MS (Método g) Rt = 1,31 min; MS miz: 443 [M+H]+ 1H NMR (DMSO-da, 400MHz): 5 8,17 (s, 1H), 7,69 (d, J=8,1 Hz, 1H), 7,47 (d, J=8,1 Hz, 1H), 5,92 (s, 1H), 4,48 (m, 2H), 3,94 (s, 3H), 3,64 (m, 4H), 3,54 (m, 2H), 3,20 (m, 2H), 2,52 (s, 3H).
Ejemplo CT: (2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)fenil)(morfolino)metanona

Etapa 1: metil 2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoato

A una solución de metil 2,4-dicloro-3-(3-cloro-2-oxobutil)benzoato (Preparación #13) (176 mg, 0,568 mmol) en W-metil-2-pirrolidinona (300 pL) se añadió 3-metil-5-(trifluorometil)piridin-2-amina (descrita en WO2007/089034) (100 mg, 0,568 mmol). La mezcla de reacción se agitó a 120 °C durante 48 horas y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar metil 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoate (55 mg, 22%) LC/MS (Método h) Rt = 2,96 min.; MS miz: 431 [M+H]+. 1H NMR (DMSO-d6, 300MHz) 58,59 (s, 1H), 7,67 (d, J = 8Hz, 1H), 7,62 (d,J= 8Hz, 1H), 7,22 (s, 1H), 4,43 (s, 2H), 3,85 (s, 3H), 2,48 (s, 3H), 2,42 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoico (18 mg, 35%) se preparó a partir de metil 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoato (53 mg, 0,123 mmol) LC/MS (Método h) Rt = 2,03 min.; MS miz: 417 [M+H]+. 1H NMR (DMSO-da, 300MHz,) 513,57 (ancho, 1H), 8,59 (s, 1H), 7,61 (d, J = 9Hz, 1H), 7,55 (d, J = 9Hz, 1H), 7,22 (s, 1H), 4,42 (s, 2H), 2,47 (s, 3H), 2,43 (s, 3H).
Etapa 3: (2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo A1, (2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)fenil)(morfolino)metanona (14 mg, 19%) se preparó a partir de ácido 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoico (62 mg, 0,149 mmol) y morfolina (19,4 mg, 0,22 mmol). LC/MS (Método g) Rt = 1,47 min.; MS miz: 486 [M+H]+. 1H NMR (DMSO-da, 400MHz,) 58,59 (s, 1H), 7,56 (d, J = 8Hz, 1H), 7,32 (d, J = 8Hz, 1H), 7,22 (s, 1H), 4,40 (m, 2H), 3,66 (m, 4H), 3,55 (m, 2H), 3,14 (m, 2H), 2,48 (s, 3H), 2,41 (s, 3H).
Tabla CT. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoico (Ejemplo CT, Etapa 2) usando el mismo procedimiento con la amina apropiada.



Ejemplo CU: ácido 2-(1-(2,4-didoro-3-((3,8-dimetN-6-(trifluorometM)imidazo[1,2-a]piridm-2-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5 ácido 2-(1-(2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoil)piperidin-4-il)acético (73 mg, 48%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-a]piridin-2-il)metil)benzoil)piperidin-4-il)acetato (Tabla CT, CT-12) (155 mg, 0,28 mmol). LC/MS (Método g) Rt = 1,43 min; MS m/z: 542 [M+H]+1H NMR (DMSO-da, 400MHz): 512,08 (ancho, 1h), 8,60 (s, 1h), 7,54 (m, 1H), 7,32 y 7,24 (d, J=8,1 Hz, 1H), 7,22 (s, 1H), 4,43 (m, 3H), 3,23 (m, 1H), 3,02 (m, 1H), 2,79 (m, 1H), 2,47 (s, 3H), 2,41 (s, 3H), 2,15 (m, 2H), 1,96 (m, 1H), 1,75 (m, 1H), 1,61 (m, 1H), 1,15 (m, 2H).
Ejemplo CV: 2-[[2,6-didoro-3-[4-(2-hidroxietil)piperidina-1-carbonil]fenil]metil]-3,8-dimetil-imidazo[1,2-a]piridina-6-carbonitrilo

Etapa 1: ácido 2,4-didoro-3-((6-ciano-3,8-dimetilimidazo[1,2-a]piridin-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo CT, Etapa 1, seguido del Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-ciano-3,8-dimetilimidazo[1,2-a]piridin-2-il)metil)benzoico (1,17 g, 34%) se preparó a partir de metil 2,4-dicloro-3-(3-cloro-2-oxobutil)benzoato (Preparación #13) (2,67 g, 8,64 mmol) y 6-amino-5-metilnicotinonitrilo (1,15 g, 8,64 mmol). LC/MS (Método g) Rt = 1,74 min; MS m/z: 374 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 13,55 (ancho, 1H), 8,95 (s, 1H), 7,63 (d, J=9 Hz, 1H), 7,57 (d, J=9 Hz, 1H), 7,24 (s, 1H), 4,42 (s, 2H), 2,44 (s, 3H), 2,38 (s, 3H).
Etapa 2: 2-[[2,6-didoro-3-[4-(2-hidroxietil)piperidina-1-carbonil]fenil]metil]-3,8-dimetil-imidazo[1,2-a]piridina-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A1, 2-[[2,6-dicloro-3-[4-(2-hidroxietil)piperidina-1-carbonil]fenil]metil]-3,8-dimetil-imidazo[1,2-a]piridina-6-carbonitrilo (40 mg, 52%) se preparó a partir de ácido 2,4-dicloro-3-((6-ciano-3,8-dimetilimidazo[1,2-a]piridin-2-il)metil)benzoico (60 mg, 0,16 mmol). Lc/MS (Método g) Rt = 1,33 min; MS m/z: 485 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 58,94 (s, 1H), 7,54 y 7,53 (d, J=8,1, 1H), 7,31 y 7,24 (d, J=8,4 Hz, 1H), 7,22 (m, 1H), 4,45 (m, 1H), 4,38 (m, 3H), 3,42 (m, 2H), 3,24 (m, 1H), 2,98 (m, 1H), 2,75 (m, 1H), 2,44 (s, 3h), 2,35 (m, 3H), 1,75 (m, 1H), 1,66 (m, 1H), 1,57 (m, 1H), 1,36 (m, 2H), 0,94-1,20 (m, 2H).
Tabla CV. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-((6-ciano-3,8-dimetilimidazo[1,2-a]piridin-2-il)metil)benzoico (Ejemplo CV, Etapa 1) usando el mismo procedimiento con la amina apropiada.

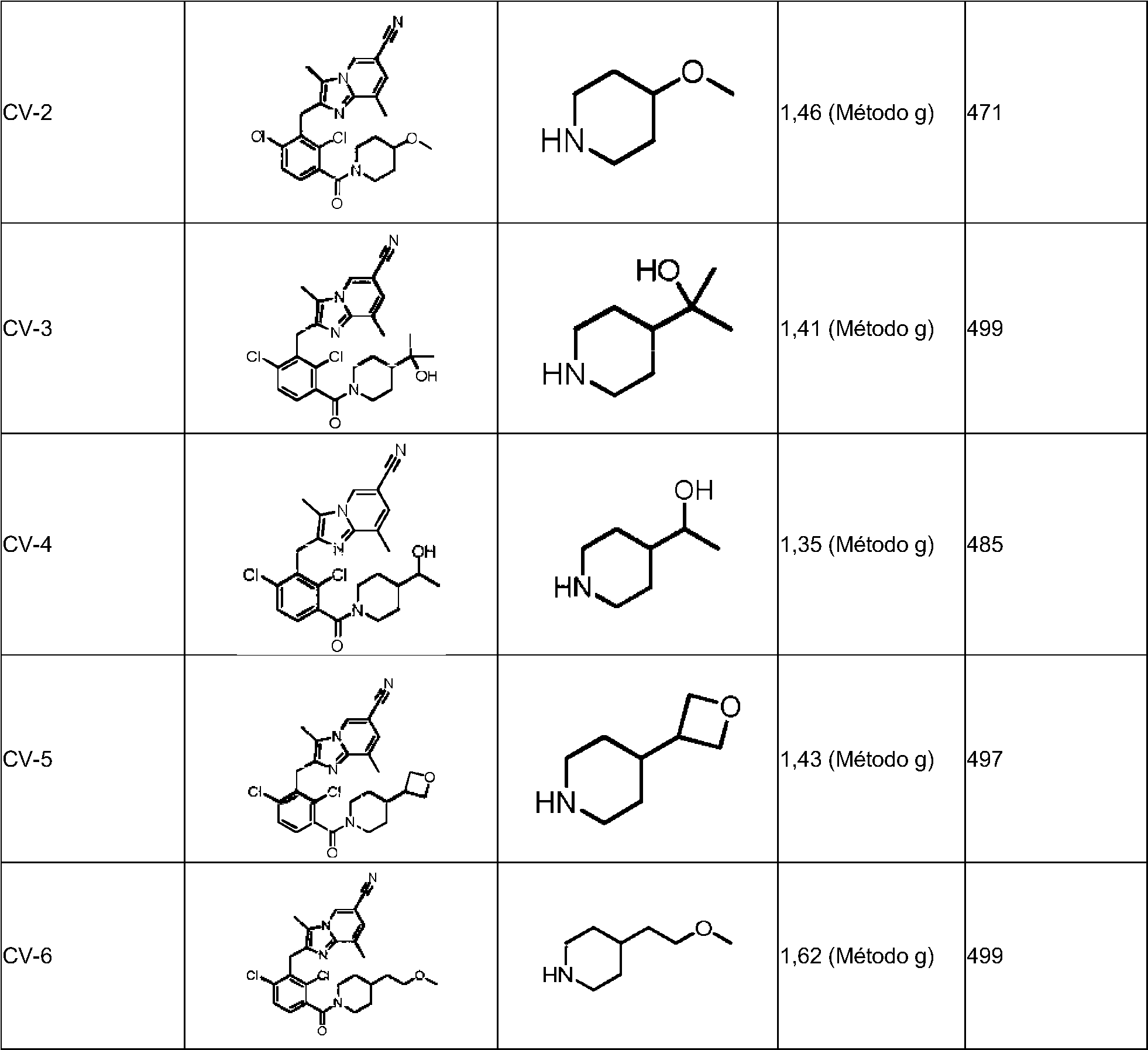
Ejemplo CW: (2,4-didoro-3-((6-doro-3,8-dimetilimidazo[1,2-b]piridazm-2-il)metil)feml)(morfolmo)metanona

Etapa 1: metil 2,4-didoro-3-((6-doro-3,8-dimetilimidazo[1,2-to]pindazm-2-il)metil)benzoato y metil-2,4-dicloro-3-((6-doro-3,7-dimetilimidazo[1,2-b]piridazm-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo CT, Etapa 1, metil 2,4-dicloro-3-((6-cloro-3,8-dimetilimidazo[1,2-ib]piridazin-2-il)metil)benzoato (47 mg, 34%)
LC/MS (Método h) Rt = 3,10 min.; MS m/z: 398 [M+H]+.1H NMR (DMSO-da, 400MHz) 57,68 (d, J = 8Hz, 1H), 7,63 (d, J = 8Hz, 1H), 7,15 (s, 1H), 4,45 (s, 2H), 3,87 (s, 3H), 2,44 (s, 6H); y metil 2,4-dicloro-3-((6-cloro-3,7-dimetilimidazo[1,2-6]piridazin-2-il)metil)benzoato (38 mg, 28%) se prepararon a partir de una mezcla de 6-cloro-5-metilpiridazin-3-amina y 6-cloro-4-metilpiridazin-3-amina (Preparación #28) (23,12 mg, 0,161 mmol) y metil 3-(3-bromo-2-oxobutil)-2,4-diclorobenzoato (Preparación #14) (57 mg, 0,161 mmol). LC/MS (Método h) Rt = 2,98 min.; MS m/z: 398 [M+H]+.1H NMR (DMSO-d6, 400MHz,) 57,98 (s, 1 h), 7,66 (d, J = 8Hz, 1H), 7,60 (d, J = 8Hz, 1H), 4,43 (s, 2H), 3,85 (s, 3H), 2,49 (s, 3H), 2,33 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((6-doro-3,8-dimetilimidazo[1,2-fo]piridazm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-cloro-3,8-dimetilimidazo[1,2-ib]piridazin-2-il)metil)benzoico (330 mg, 93%) se preparó a partir de metil 2,4-dicloro-3-((6-cloro-3,8-dimetilimidazo[1,2-ib]piridazin-2-il)metil)benzoato (366 mg, 0,918 mmol) LC/MS (Método h) Rt = 2,47 min.; MS m/z: 384 [M+H]+.1H NMR (DMSO-d6, 300MHz,) 513,57 (ancho, 1H), 7,65 (d, J = 9Hz, 1H), 7,57 (d, J = 9Hz, 1H), 7,15 (s, 1H), 4,45 (s, 2H), 2,45 (s, 3H), 2,43 (s, 3H).
Etapa 3: (2,4-didoro-3-((322-doro-3,8-dimetilimidazo[1,2-to]pindazm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A1, (2,4-dicloro-3-((6-cloro-3,8-dimetilimidazo[1,2-ib]piridazin-2-il)metil)fenil)(morfolino)metanona (356 mg, 92%) se preparó a partir de ácido 2,4-dicloro-3-((6-cloro-3,8-dimetilimidazo[1,2-¿]piridazin-2-il)metil)benzoico (327 mg, 0,850 mmol) y morfolina (89 mg, 1,02 mmol). lC/MS (Método g) Rt =
1,62 min.; MS miz. 453 [M+H]+.1H NMR (DMSO-da, 300MHz,) 57,57 (d, J = 8Hz, 1H), 7,33 (d, J = 8Hz, 1H), 7,15 (m, 1H), 4,42 (m, 2H), 3,65 (m, 4H), 3,54 (m, 2H), 3,14 (m, 2H), 2,44 (s, 6H).
Ejemplo CY: (2,4-didoro-3-((6-doro-3,7-dimetilimidazo[1,2-to]piridazm-2-il)metil)feml)(morfolmo)metanona

Etapa 1: ácido 2,4-didoro-3-((6-doro-3,7-dimetilimidazo[1,2-fo]piridazm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-cloro-3,7-dimetilimidazo[1,2-ib]piridazin-2-il)metil)benzoico (21 mg, 66%) se preparó a partir de metil 2,4-dicloro-3-((6-cloro-3,7-dimetilimidazo[1,2-ib]piridazin-2-il)metil)benzoato (Ejemplo CW, Etapa 1) (33 mg, 0,083 mmol). LCiMS (Método h) Rt = 2,30 min.; MS miz: 384 [M+H]+.1H NMR (DMSO-d6, 300MHz,) 513,55 (ancho, 1H), 8,01 (s, 1 h), 7,64 (d, J = 9Hz, 1H), 7,56 (d, J = 9Hz, 1H), 4,45 (s, 2H), 2,48 (s, 3H), 2,36 (s, 3H).
Etapa 2: (2,4-didoro-3-((6-doro-3,323-dimetilimidazo[1,2-to]pindazm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A1, (2,4-dicloro-3-((6-cloro-3,7-dimetilimidazo[1,2-ib]piridazin-2-il)metil)fenil)(morfolino)metanona (15 mg, 70%) se preparó a partir de ácido 2,4-dicloro-3-((6-cloro-3,7-dimetilimi-dazo[1,2-6]piridazin-2-il)metil)benzoico (18 mg, 0,047 mmol) y morfolina (6,12 mg, 0,07 mmol). lC/MS (Método g) Rt = 1,56 min.; MS miz: 453 [M+H]+.1H NMR (DMSO-d6, 400MHz,) 57,99 (s, 1h), 7,55 (d, J = 8Hz, 1H), 7,33 (d, J = 8Hz, 1H), 4,40 (m, 2H), 3,63 (m, 4H), 3,52 (m, 2H), 3,12 (m, 2H), 2,48 (s, 3H), 2,34 (s, 3H).
Ejemplo CZ: (2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-to]piridazm-2-il)metil)fenil)(morfolino)metanona

Etapa 1: metil 2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-b]piridazin-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo CT, Etapa 1 metil 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imi-dazo[1,2-6]piridazin-2-il)metil)benzoato (135 mg, 37%) se preparó a partir de metil 2,4-dicloro-3-(3-cloro-2-oxobutil)benzoato (Preparación #13) (262 mg, 0,847 mmol) y 4-metil-6-(trifluorometil)piridazin-3-amina (Preparación #41) (150 mg, 0,847 mmol). LC/MS (Método i) Rt = 2,65 min; MS m/z: 432 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,69 (d, J=9 Hz, 1H), 7,64 (, J=9 Hz, 1H), 7,47 (d, J=1,0 Hz, 1H), 4,52 (s, 2H), 3,86 (s, 3H), 2,55 (d, J=1 Hz, 3H), 2,51 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-b]piridazm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-6]piridazin-2-il)metil)benzoico (100 mg, 80%) se preparó a partir de metil 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazin-2-il)metil)benzoato (129 mg, 0,29 mmol). LC/MS (Método i) Rt = 2,27 min; MS m/z: 418 [M+H]+
1H NMR (DMSO-d6, 300MHz): 5 13,61 (ancho, 1H), 7,65 (d, J=9 Hz, 1H), 7,61 (d, J=9 Hz, 1H), 7,47 (d, J=1,0 Hz, 1H), 4,52 (s, 2H), 2,55 (d, J=0,8 Hz, 3H), 2,50 (s, 3H).
Etapa 3: (2,4-didoro-3-((3,8-dimetil-6-(tnfluorometil)imidazo[1,2-to]pindazm-2-il)metil)feml)(morfolmo)metanona

Usando un procedimiento similar al Ejemplo A1, (2,4-didoro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazin-2-il)metil)fenil)(morfolino)metanona (22 mg, 38%) se preparó a partir de metil 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazin-2-il)metil)benzoato (48 mg, 0,11 mmol). LC/MS (Método g) Rt = 1,75 min; MS miz: 487 [M+H]+ 1H NMR (DMSO-da, 400MHz): 5 7,58 (d, J=8,4 Hz, 1H), 7,46 (d, J=1,1 Hz, 1H), 7,35 (d, J=8,4 Hz, 1H), 4,49 (m, 2H), 3,64 (m, 4H), 3,54 (m, 2H), 3,15 (m, 2H), 2,54 (d, J=0,9 Hz, 3H) 2,50 (s, 3H).
Tabla CZ. Los siguientes ejemplos se prepararon usando el mismo procedimiento que en el Ejemplo CZ, Etapa 3 con las aminas apropiadas.

Ejemplo CZ-1A: ácido 2-(1-(2,4-didoro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-b]pmdazma-2-carbonil)benzoyl)piperidin-4-il)acético

Etapa 1: ácido 2,4-didoro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-b]piridazma-2-carboml)benzoico

Usando un procedimiento similar al Ejemplo AD, Etapa 2, ácido 2,4-dicloro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-6]piridazina-2-carbonil)benzoico (347 mg, 43%) se preparó a partir de ácido 2,4-dicloro-3-((3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazin-2-il)metil)benzoico (420 mg, 1 mmol) (Ejemplo CZ, Etapa 2). LC/MS (Método g): Rt = 1,78 min; MS m/z: 432 [M+H]+; 1H NMR (DMSO-C6, 500MHz): 513,67 (ancho,1H), 7,92 (d, J=8,5 Hz, 1H), 7,70 (d, J=8,5 Hz, 1H), 7,68 (s, 1H), 2,87 (s, 3H), 2,56 (s, 3H)
Etapa 2: metil 2-(1-(2,4-didoro-3-(3,8-dimetil-6-(tnfluorometil)imidazo[1,2-to]pmdazma-2-carbonil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,8-dimetil-6-(trifluorometil)im-idazo[1,2-ib]piridazina-2-carbonil)benzoil)piperidin-4-il)acetato (45 mg, 57%) se preparó a partir de ácido 2,4-dicloro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazina-2-carbonil)benzoico (60 mg, 0,14 mmol) y clorhidrato de metil (4-piperidil)acetato (40 mg, 0,21 mmol). LC/MS (Método i): Rt = 2,33 min; MS m/z: 571 [M+H]+; 1H NMR (DMSO-C6, 300MHz): 57,95 (s, 1H), 7,60 (m, 2H), 4,47 (m, 1H), 3,60 (m, 3H), 3,27 (m, 1H), 3,09 (m, 1H), 2,87 (s, 3H), 2,80 (m, 1H), 2,55 (m, 3H), 2,27 (m, 2H), 1,97 (m, 1H), 1,75 (m, 1H), 1,62 (m, 1H), 1,15 (m, 2H).
Etapa 3: ácido 2-(1-(2,4-didoro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-b]piridazma-2-carbonil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-ib]piridazina-2-carbonil)benzoil)piperidin-4-il)acético (31 mg, 70%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-(3,8-dimetil-6-(trifluorometil)imidazo[1,2-í)]piridazina-2-carbonil)benzoil)piperidin-4-il)acetato (45 mg, 0,08 mmol). LC/MS (Método g): Rt = 1,66 min; MS m/z: 557 [M+H]+; 1H NMR (DMSO-C6, 400MHz): 5 = 12,11 (ancho, 1H), 7,69 (m, 2H), 7,59 y 7,52 (d, J=8,6 Hz, 1H), 4,46 (m, 1H), 3,24 (m, 1H), 3,10 (m, 1H), 2,87 (s, 3H), 2,81 (m, 1H), 2,57 (m, 3H), 2,16 (m, 2H), 1,95 (m, 1H), 1,77 (m, 1H), 1,64 (m, 1H), 1,18 (m, 2H).
Ejemplo DA: 2-(2,6-didoro-3-(morfolma-4-carboml)bencil)-3,8-dimetilimidazo[1,2-b]piridazma-6-carbomtrilo

Usando un procedimiento similar al Ejemplo CI, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bendl)-3,8-dimetilimidazo[1,2-í)]piridazina-6-carbonitrilo (41 mg, 79%) a partir de (2,4-dicloro-3-((6-doro-3,8-dimetilimidazo[1,2-6]piridazin-2-il)metil)fenil)(morfolino)metanona (Ejemplo CW, Etapa 3) (50 mg, 0,11 mmol). LC/MS (Método g) Rt = 1,52min; MS m/z: 444 [M+H]+1H NMR (DMSO-d6, 400MHz): 57,58 (d, J=8,4 Hz, 1H), 7,53 (d, J=1,1 Hz, 1H), 7,35 (d, J=8,4 Hz, 1H), 4,49 (m, 2H), 3,64 (m, 4H), 3,54 (m, 2H), 3,14 (m, 2H), 2,54 m (s, 3H), 2,50 (s, 3H).
Ejemplo DB: 2-(2,6-didoro-3-(morfolma-4-carboml)bencil)-3,7-dimetilimidazo[1,2-to]piridazma-6-carbomtrilo

Usando un procedimiento similar al Ejemplo CI, Etapa 4, 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-3,7-dimetillimidazo[1,2-ib]piridazina-6-carbonitrilo (30 mg, 13%) se preparó a partir de (2,4-dicloro-3-((6-cloro-3,7-dimetilimidazo[1,2-í)]piridazin-2-il)metil)fenil)(morfolino)metanona (Ejemplo CY, Etapa 2) (100 mg, 0,22 mmol). LC/MS (Método g) Rt = 1,48 min; MS m/z: 444 [M+H]+ 1H NMR (DMSO-da, 400MHz): 58,10 (d, J=1,1 Hz, 1H), 7,56 (d, J=8,4 Hz, 1H), 7,34 (d, J=8,4 Hz, 1H), 4,47 (m, 2H), 3,65 (m, 4H), 3,53 (m, 2H), 3,13 (m, 2H), 2,53 (s, 3H), 2,47 (m, 3H).
Ejemplo DC: (2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-to]piridm-2-il)metil)fenil)(morfolino)metanona

Etapa 1: metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-b]piridin-2-il)metil)benzoato

A una solución de metil 2,4-dicloro-3-((5-cloro-3,7-dimetil-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoato (Ejemplo CI, Etapa 1) (350 mg, 0,878 mmol) en DMF (2,5 mL) se añadieron metil 2,2-difluoro-2-(fluorosulfonil)acetato (0,333 mL, 2,63 mmol) y yoduro de cobre(I) (167 mg, 0,878 mmol) y la mezcla de reacción se agitó a 115 °C durante 24 horas. La mezcla de reacción se diluyó con EtOAc. La capa orgánica se lavó con agua, con solución acuosa de LiCl al 10%, con salmuera y se secó sobre sulfato de magnesio, se filtró, se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-5% en ciclohexano) para dar metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-¿]piridin-2-il)metil)benzoato (165 mg, 17%) como un polvo amarillo pálido. El producto se usó directamente en la siguiente etapa.
LC/MS (Método h) Rt = 2,86 min; MS m/z: 432 [M+H]+
Etapa 2: ácido 2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-fo]piridm-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-6]piridin-2-il)metil) benzoico (210 mg, 94%) se preparó a partir de metil 2,4-didoro-3-((3,7-dimetil-5-(tri-fluorometil)-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoato (230 mg, 0,53 mmol). LC/MS (Método h) Rt = 2,15 min; MS miz: 418 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,84 y 7,66 (d, J=8,4 Hz, 1H), 7,50 (m, 1H), 7,13 (m, 1H), 4,61 (s, 1H), 4,53 (s, 1h), 3,87 (m, 3H), 2,39 y 2,37 (s, 3H).

Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-ib]piridin-2-il)metil)fenil)(morfolino)metanona (33 mg, 13%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-3H-imidazo[4,5-ib]piridin-2-il)metil)benzoico (210 mg, 0,50 mmol) y morfolina (125 mg, 0,65 mmol).
LC/MS (Método g) Rt = 1,49 min; MS miz: 487 [M+H]+
1H NMR (DMSO-d6, 500MHz): 57,93 (d, J=8,4 Hz, 1H), 7,49 (d, J=8,4 Hz, 1H), 7,13 (d, J=0,7 Hz, 1H), 4,55 (s, 2H), 3,86 (m, 3H), 3,53-3,73 (m, 5H), 3,47 (m, 1H), 3,20 (m, 1H), 3,10 (m, 1H), 2,37 (s, 3H).
Ejemplo DD: (2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-mdazoM-il)metil)feml)(morfolmo)metanona

Etapa 1: metil 2,4-didoro-3-((3,7-dimetM-5-(tnfluorometM)-1H-mdazoM-M)metM)benzoato y metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoato

A una solución de 3,7-dimetil-5-(trifluorometil)-1H-indazol (Preparación #42) (140 mg, 0,654 mmol) en DMF (5 mL) se añadió carbonato de potasio (117 mg, 0,850 mmol) y la mezcla de reacción se agitó 30 minutos a temperatura ambiente. Se añadió metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (214 mg, 0,719 mmol) y la reacción se calentó a 50 °C durante 2 horas. La mezcla de reacción se vertió en agua (100 mL) y se extrajo con EtOAc. La capa orgánica combinada se lavó con salmuera y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 10-20% en ciclohexano) para dar metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1 -il)metil)benzoato (82 mg, 17%) LC/MS (Método h) Rt = 3,39 min; MS miz. 431 [M+H]+1H NMR (DMSO-da, 500MHz): 5 7,98 (s, 1H), 7,78 (d, J=8,5 Hz, 1H), 7,67 (d, J=8,5 Hz, 1H), 7,47 (s, 1H), 6,02 (s, 2H), 3,86 (s, 3H), 2,94 (s, 3H), 2,39 (s, 3H) y metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoato (215 mg, 49%). LC/Ms (Método h) Rt = 3,28 min; MS miz. 431 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 58,05 (s, 1H), 7,82 (d, J=8,5 Hz, 1H), 7,70 (d, J=8,5 Hz, 1H), 7,15 (s, 1H), 5,85 (s, 2H), 3,86 (s, 3H), 2,83 (s, 3H), 2,36 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdazoM-N)metN)benzoico
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (75 mg, 90%) se preparó a partir de metil 2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-in-dazol-1-il)metil)benzoato (80 mg, 0,18 mmol).
LC/MS (Método h) Rt = 2,93 min; MS m/z: 417 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,98 (s, 1H), 7,74 (d, J=8,5 Hz, 1H), 7,62 (d, J=8,5 Hz, 1H), 7,48 (s, 1H), 6,01 (s, 2H), 2,94 (s, 3H), 2,39 (s, 3H).
Etapa 3: (2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdazoM-N)metN)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)fenil)(morfolino)metanona (25 mg, 30%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (70 mg, 0,17 mmol) y morfolina (29 mg, 0,33 mmol). LC/MS (Método g) Rt = 1,83 min; MS m / z : 486 [M+H]+
1H NMR (DMSO-da, 400MHz): 57,98 (s, 1H), 7,62 (d, J=8,4 Hz, 1H), 7,47 (s, 1H), 7,44 (d, J=8,4 Hz, 1H), 6,00 (d, J=12 Hz, 1H), 5,97 (d, J=12 Hz, 1H), 3,60 (m, 4H), 3,52 (m, 2H), 3,13 (m, 2H), 2,93 (s, 3H), 2,39 (s, 3H)
Ejemplo DD-1: (2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdazoM-N)metN)feml)(4-(oxetan-3-N)piperidm-1-il)metanona
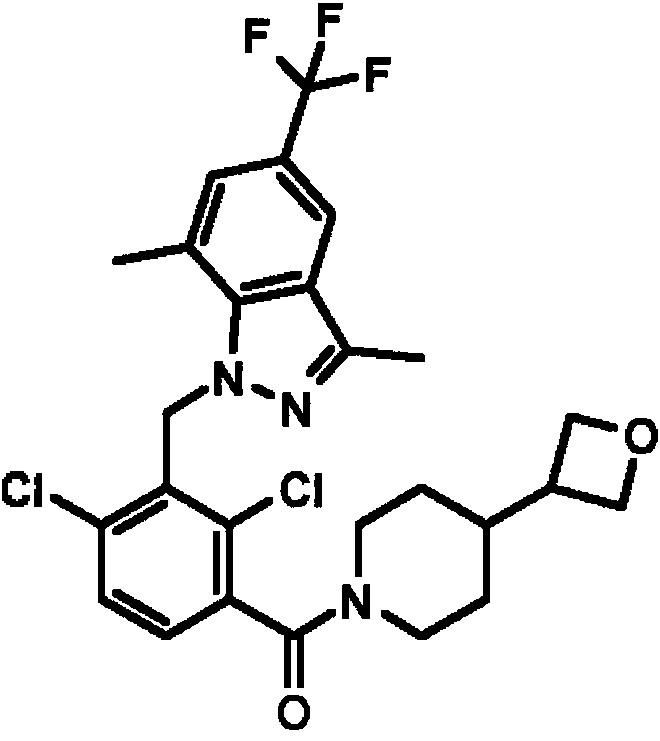
Usando un procedimiento similar al del Ejemplo A1, (2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)fenil)(4-(oxetan-3-il)piperidin-1-il)metanona (128 mg, 72%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (133 mg, 0,32 mmol) (Ejemplo dD, Etapa 1) y 4-(oxetan-3-il) piperidina (58 mg, 0,41 mmol). LC/MS (Método g): Rt = 1,91 min; MS m/z: 540 [M+H]+ 1H NMR (DMSO-ck, 400MHz): 57,98 (s, 1H), 7,61 y 7,60 (d, J =8,3 Hz, 1H), 7,47 (s, 1H), 7,44 y 7,37 (d, J =8,3 Hz, 1H), 5,99 (m, 2H), 4,58 (m, 2H), 4,49 (m, 1H), 4,33 (m, 2h), 3,25 (m, 1H), 3,02 (m, 1H), 2,93 (s, 3H), 2,76 (m, 2H), 2,39 y 2,38 (s, 3H), 1,89 (m, 1H), 1,67 (m, 1H), 1,51 (m, 1H), 0,96 (m, 2H).
Ejemplo DE: ácido 2-(1-(2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-1H-mdazoM-N)metN)benzoN)piperidm-4-il)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdazoM-N)metN)benzoM)piperidm-4-N)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-in-dazol-1-il)metil)benzoil)piperidin-4-il)acetato (90 mg, 73%) se preparó a partir de ácido 2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (Ejemplo DD, Etapa 2) (92 mg, 0,22 mmol) y clorhidrato de metil (4-piperidil)acetato (51 mg, 0,26 mmol). LC/MS (Método i) Rt = 2,59 min; MS m/z: 556 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,98 (s, 1H), 7,60 y 7,59 (d, J=8,3 Hz, 1H), 7,47 (s, 1H), 7,43 y 7,36 (d, J=8,3 Hz, 1H), 5,90 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,22 (m, 1h), 3,02 (m, 1H), 2,93 (s, 3H), 2,79 (m, 1H), 2,39 (m, 3H), 2,27 (m, 2H), 1,97 (m, 1H), 1,74 (m, 1H), 1,57 (m, 1H), 1,15 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-1H-mdazoM-M)metM)benzoN)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoil)piperidin-4-il)acético (64 mg, 71%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifiuorometil)-1w-indazol-1-il)metil)benzoil)piperidin-4-il)acetato (90 mg, 0,16 mmol). LC/MS (Método g) Rt = 1,77 min; MS
m/z: 542 [M+H]+1H NMR (DMSO-da, 500MHz): 5 12,09 (ancho, 1H), 7,98 (s, 1H), 7,60 (m, 1H), 7,47 (s, 1H), 7,43 y 7,36 (d, J=8,3 Hz, 1H), 5,96 (m, 2H), 4,46 (m, 1H), 3,23 (m, 1H), 2,02 (m, 1H), 2,93 (s, 3H), 2,79 (m, 1H), 2,39 (m, 3H), 2,15 (m, 2H), 1,92 (m, 1H), 1,76 (m, 1H), 1,60 (m, 1H), 1,10 (m, 2H).
Ejemplo DF: (2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-2H-mdazol-2-N)metN)feml)(morfolmo)metanona

Etapa 1: ácido 2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-2H-mdazol-2-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoico (160 mg, 76%) se preparó a partir de metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoato (Ejemplo DD, Etapa 1) (200 mg, 0,46 mmol). LC/MS (Método h) Rt = 2,81 min; MS m/z: 417 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 13,68 (s, 1H), 8,05 (s, 1H), 7,78 (d, J=9 Hz, 1H), 7,65 (d, J=9 Hz, 1H), 7,15 (s, 1H), 5,84 (s, 2H), 2,83 (s, 3H), 2,36 (s, 3H).
Etapa 2: (2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)fenil)(morfolino)metanona (153 mg, 82%) se preparó a partir de ácido 2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoico (156 mg, 0,37 mmol) y morfolina (65 mg, 0,75 mmol). LC/MS (Método g) Rt = 1,78 min; MS miz: 486 [M+H]+
1H NMR (DMSO-da, 400MHz): 5 8,05 (s, 1H), 7,64 (d, J=8,4 Hz, 1H), 7,47 (d, J=8,4 Hz, 1H), 7,15 (s, 1H), 5,81 (m, 2H), 3,60 (m, 4H), 3,53 (m, 2H), 3,14 (m, 2H), 2,83 (s, 3H), 2,34 (s, 3H).
Ejemplo DG: ácido 2-(1-(2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-2H-mdazol-2-N)metN)benzoN)piperidm-4-il)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-in-dazol-2-il)metil)benzoil)piperidin-4-il)acetato (192 mg, 56%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoico (Ejemplo DF, Etapa 1) (237 mg, 0,56 mmol) y clorhidrato de metil (4-piperidil)acetato (132 mg, 0,68 mmol). LC/MS (Método i): Rt = 2,52 min; MS miz: 556 [M+H]+
1H NMR (DMSO-d6, 300MHz): 58,05 (s, 1H), 7,63 y 7,60 (d, J=8,3 Hz, 1H), 7,46 y 7,38 (d, J=8,3 Hz, 1H), 7,15 (s, 1H), 5,82 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,22 (m, 1H), 3,03 (m, 1H), 2,83 (m, 3H), 2,79 (m,1H), 2,34 (s, 3H), 2,20 (m, 2H), 1,99 (m, 1H), 1,75 (m, 1H), 1,56 (m, 1H), 1,15 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-2H-mdazol-2-M)metM)benzoN)piperidm-4-M)acético
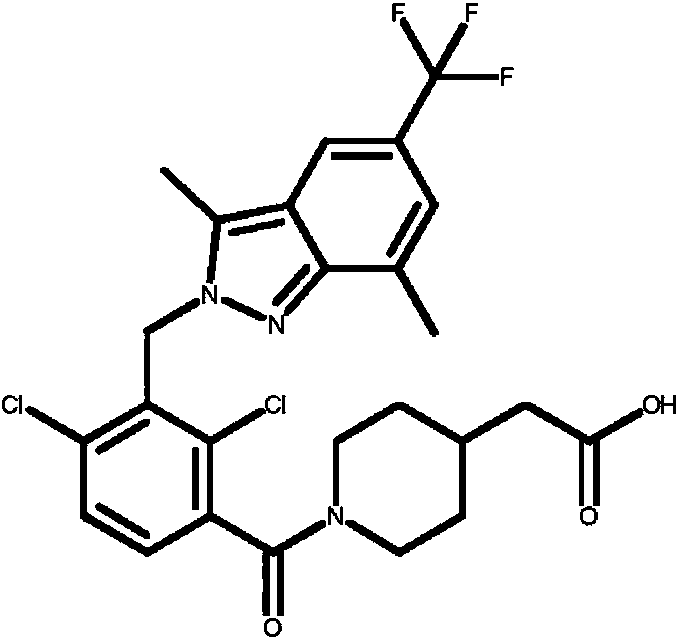
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoil)piperidin-4-il)acético (170 mg, 89%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoil)piperidin-4-il)acetato (190 mg, 0,34 mmol). LC/MS (Método g): Rt = 1,71 min; MS m/z: 542 [M+H]+1H NMR (DMSO-da, 400MHz): 512,15 (s, 1H), 8,04 (s, 1H), 7,61 (m, 1H), 7,46 y 7,38 (d, J=8,3 Hz, 1H), 7,14 (s, 1H), 5,73-5,91 (m, 2H), 4,47 (m, 1H), 3,21 (m, 1H), 3,03 (m, 1h), 2,83 (s, 3H), 2,78 (m, 1H), 2,34 (s, 3H), 2,10 (m, 2H), 1,93 (m, 1H), 1,77 (m, 1H), 1,58 (m, 1H), 1,10 (m, 2H).
Ejemplo DH: ácido 1-(2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdoM-M)metM)benzoN)piperidma-4-carboxílico

Etapa 1: metil 2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-1H-mdoM-M)metM)benzoato

A una solución de 3,7-dimetil-5-(trifluorometil)-1H-indol (Preparación #43) (90 mg, 0,422 mmol) en DMF (1,1 mL) y enfriada a 0 °C se añadió hidruro de sodio (18,57 mg, 0,464 mmol). Después de 30 minutos de agitación, se añadió metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (126 mg, 0,422 mmol) y la agitación continuó durante 1 hora a 0 °C. La mezcla de reacción se diluyó con agua y EtOAc. Las capas se separaron y la acuosa se extrajo con EtOAc. Las capas orgánicas se combinaron, se secaron sobre sulfato de magnesio, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc 0-5% en ciclohexano) para dar 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (100 mg, 53%) como un polvo blanco. LC/MS (Método i): Rt = 2,86 min; MS m/z: 430 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 57,88 (d, J=8,4 Hz, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,68 (s, 1H), 7,23 (s, 1H), 6,55 (d, J=1,2 Hz, 1H), 5,92 (s, 2H), 3,88 (s, 3H), 2,93 (s, 3H), 2,17 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdoM-N)metN)benzoico

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (90 mg, 93%) se preparó a partir de metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (100 mg, 0,23 mmol).
LC/MS (Método i): Rt = 2,57 min; MS m/z: 416 [M+H]+1H NMR (DMSO-d6, 300MHz): 57,67 (s, 1H), 7,55 (s, 2H), 7,22 (s, 1H), 6,52 (s, 1H), 5,87 (s, 2H), 2,93 (s, 3H), 2,16 (s, 3H).
Etapa 3: metil 1-(2,4-didoro-3-((3,7-dimetM-5-(trifluorometN)-1H-mdoM-M)metM)benzoN)piperidma-4-carboxMato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidina-4-carboxilato (84 mg, 72%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-4-(trifluorometil)-1H-indol-1-il)metil)benzoico (90 mg, 0,12 mmol) y clorhidrato de metil piperidina-4-carboxilato (43 mg, 0,24 mmol). LC/MS (Método i): Rt = 2,69 min; MS m/z: 541 [M+H]+1H NMR (DMSO-d6, 300MHz): 57,69 (m, 2H), 7,55 y 7,48 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,56 (m, 1H), 5,90 (m, 2H), 4,35 (m, 1H), 3,62 y 3,60 (s, 3h), 3,31 (m, 1H), 2,94-3,15 (m, 2H), 2,92 (s, 3H), 2,67 (m, 1h), 2,18 (m, 3h), 1,90 (m, 1H), 1,77 (m, 1H), 1,50 (m, 2H)
Etapa 4: ácido 1-(2,4-didoro-3-((3,7-dimetN-5-(trifluorometM)-1H-mdoM-N)metN)benzoM)piperidma-4-carboxNico

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidina-4-carboxílico (67 mg, 82%) se preparó a partir de metil 1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indoM-il)metil)benzoil)piperidina-4-carboxilato (84 mg, 0,15 mmol). LC/MS (Método g): Rt = 1,87 min; MS m/z: 527 [M+H]+1H NMR (DMSO-de, 400MHz): 512,30 (ancho, 1H), 7,70 (m, 2h), 7,55 y 7,48 (d, J=8,1 Hz, 1H), 7,22 (s, 1H), 6,57 y 6,55 (d, 1H), 5,90 (m, 2H), 4,34 (m, 1H), 3,29 (m, 1H), 3,00 (m, 2H), 2,92 (s, 3H), 2,54 (m, 1H), 2,18 (s, 3H), 1,92 (m, 1H), 1,76 (m, 1h), 1,49 (m, 2H).
Tabla DH. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (Ejemplo DH, Etapa 2) usando el mismo procedimiento que en el Ejemplo DH, Etapa 3, con la amina apropiada.

Tabla DH1. Los siguientes ejemplos se prepararon usando el mismo procedimiento que el Ejemplo DH, Etapa 4 con los ésteres apropiados descritos en la Tabla DH.


Ejemplo DI: ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(tnfluorometil)-1H-indoM-il)metil)benzoil)pipendin-4-il)acético

Etapa 1: metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-didoro-3-((3,7-dimetil-5-(trifluorometN)-1H-indoM-il)metil)benzoil)piperidin-4-il)acetato (120 mg, 84%) se preparó a partir de ácido 2,4-didoro-3-((3,7-dimetil-4-(trifluorometM)-1H-indoM-N)metil)benzoico (Ejemplo DH, Etapa 2) (105 mg, 0,25 mmol) y clorhidrato de metil (4-piperidil)acetato (54 mg, 0,27 mmol). LC/MS (Método i): Rt = 2,73 min; MS m/z: 555 [M+H]+
1H NMR (DMSO-d6, 300MHz): 57,69 (m, 2H), 7,52 y 7,45 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,55 (m, 1H), 5,88 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,25 (m, 1H), 3,04 (m, 1H), 2,92 (s, 3H), 2,80 (m, 1h), 2,27 (m, 2H), 2,17 (s, 3h), 1,95 (m, 1H), 1,75 (m, 1H), 1,58 (m, 1h), 1,10 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(tnfluorometil)-1H-indoM-il)metil)benzoil)pipendin-4-il)acético

Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (70 mg, 58%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-indoM-il)metil)benzoil)piperidin-4-il)acetato (120 mg, 0,21 mmol). LC/MS (Método g): Rt = 1,89 min; MS miz: 541 [M+H]+ 1H NMR (DMSO-da, 400MHz): 5 12,10 (ancho, 1H), 7,71 (m, 2H), 7,52 y 7,45 (d, J=8,4 Hz, 1H), 7,22 (s, 1H), 6,55 y 6,53 (s, 1H), 5,85 (m, 2H), 4,46 (m, 1H), 3,26 (m, 1H), 3,03 (m, 1H), 2,92 (s, 3H), 2,80 (m, 1H), 2,19 (m, 5H), 1,90 (m, 1H), 1,77 (m, 1H), 1,60 (m, 1H), 1,10 (m, 2H).
Ejemplo DJ: ácido 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-1H-mdoM-carboml)benzoM)piperidm-4-N) acético

Etapa 1: terc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoato

A una solución de 3,7-dimetil-5-(trifluorometil)- 1 H-indol (Preparación #43) (170 mg, 0,797 mmol) en DMF (5 mL) y enfriada a °C se añadió hidruro de sodio (57,4 mg, 1,435 mmol) y la mezcla de reacción se agitó a 0 °C durante 20 minutos. Luego se añadió terc-butil 2,4-dicloro-3-(clorocarbonil)benzoato (Preparación #44) (443 mg, 1,435 mmol) diluido en DMF (4 mL) y la mezcla de reacción se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se inactivó con una solución acuosa saturada de NH4 Cl y se extrajo con EtOAc. La capa orgánica se lavó con agua y salmuera, se secó sobre sulfato de magnesio, se filtró y se concentró hasta sequedad. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar íerc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1 H-indol-1 -carbonil)benzoato (300 mg, 76%) como una resina de color amarillo. LC/MS (Método j): Rt = 2,87 min; MS m/z: 486 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,99 (d, J=8,4 Hz, 1H), 7,82 (m, 2H), 7,61 (s, 1H), 7,27 (s, 1H), 2,73 (s, 3H), 2,21 (s, 3H), 1,57 (s, 9H)
Etapa 2: ácido 2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-1H-mdoM-carbonM)benzoico

A una solución de íerc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoato (300mg, 0,617 mmol) en DCM (5 mL) se añadió TFA (2,5 mL, 32,4 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró con tolueno a presión reducida para dar ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoico (260 mg, 96%) como un sólido de color beige.
LC/MS (Método i): Rt = 2,50 min; MS m/z: 430 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 513,87 (ancho, 1H), 8,05 (d, J=8,4 Hz, 1H), 7,81 (m, 2H), 7,61 (s, 1H), 7,25 (s, 1H), 2,74 (s, 3H), 2,21 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(tnfluorometM)-1H-mdoM-carboml)benzoM)pipendm-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (150 mg, 81%) se preparó a partir de ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoico (105 mg, 0,25 mmol) y clorhidrato de metil (4-piperidil)acetato (64 mg, 0,33 mmol). LC/MS (Método i): Rt = 2,71 min; MS m/z: 569 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 57,76-81 (m, 2H), 7,65 (m, 2H), 7,43, 7,37 y 7,17 (m, 1H), 4,47 (m, 1H), 3,60 (m, 3H), 3,23 (m, 1H), 3,07 (m, 1H), 2,85 (m, 1H), 2,72 (s, 3H), 2,30 (m, 2h), 2,23 (m, 3H), 1,92 (m, 1H), 1,77 (m, 1H), 1,62 (m, 1H), 1,06-1,27 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-(3,7-dimetN-5-(trifluorometM)-1H-mdoM-carboml)benzoN)piperidm-4-M)acético
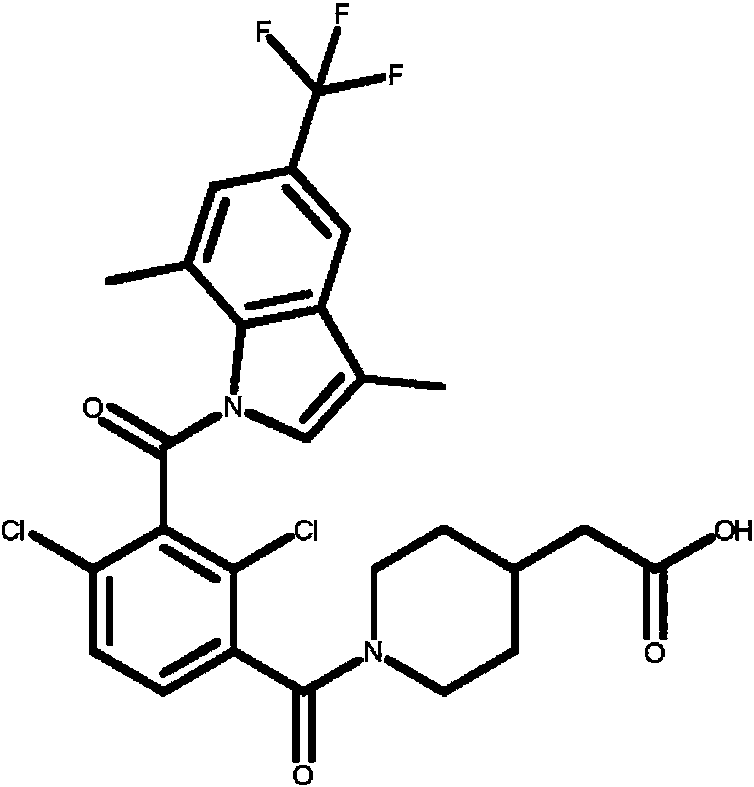
Usando un procedimiento similar al Ejemplo F, Etapa 4, ácido 2-(1-(2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acético (96 mg, 65%) se preparó a partir de 2-(1-(2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (150 mg, 0,26 mmol). LC/MS (Método g): Rt = 1,89 min; MS miz: 555 [M+H]+1H NMR (DMSO-da, 400MHz): 5 12,10 (s, 1H), 7,82 (m, 2H), 7,57-7,72 (m, 2H), 7,42, 7,37 y 7,16 (m, 1H), 4,48 (m, 1H), 3,57 y 3,26 (m, 1H), 3,08 (m, 1H), 2,82 (m, 1H), 2,72 (s, 3H), 2,23 (m, 3H), 2,19 (m, 2H), 1,95 (m, 1H), 1,77 (m, 1H), 1,63 (m, 1H), 1,16 (m, 2H).
Tabla DJ. Los siguientes intermedios se prepararon a partir de (Ejemplo DJ, Etapa 2) usando el mismo procedimiento que en el Ejemplo DJ, Etapa 3, con la amina apropiada.

Tabla DJ1. Los siguientes ejemplos se prepararon usando el mismo procedimiento que el ejemplo DJ, Etapa 4, con los ésteres apropiados descritos en la Tabla DJ.

Ejemplo DK: 1-(2,6-didoro-3-(4-hidroxipipendma-1-carbonM)bendl)-3,7-dimetiMH-mdol-5-carbomtrMo

Etapa 1: metil 2,4-didoro-3-((5-dano-3,7-dimetiMH-mdoM-M)metM)benzoato

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-didoro-3-((5-ciano-3,7-dimetil-1H-indoM-il)metil)benzoato (262 mg, 56%) se preparó a partir de 3,7-dimetil-1 H-indol-5-carbonitrilo (Preparación #45) (242 mg, 0,87 mmol) y 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (466 mg, 1,56 mmol). El compuesto se usa directamente en el siguiente Etapa LC/MS (Método i): Rt = 2,58 min; MS m/z: 387 [M+H]+
Etapa 2: ácido 2,4-didoro-3-((5-ciano-3,7-dimetiMH-mdoM-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((5-ciano-3,7-dimetil-1H-indoM-il)metil)benzoico (100 mg, 90%) se preparó a partir de metil 2,4-didoro-3-((5-dano-3,7-dimetil-1H-indol-1-il)metil)benzoato (113 mg, 0,29 mmol). LC/MS (Método i): Rt = 2,22 min; MS m/z: 373 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 57,88 (s, 1H), 7,82 (d, J=8,4 Hz, 1H), 7,69 (d, J =8,4 Hz, 1H), 7,29 (s, 1H), 6,56 (d, J=1 Hz, 1H), 5,90 (s, 2H), 2,89 (s, 3H), 2,15 (d, J=1 Hz, 3H).
Etapa 3: 1-(2,6-didoro-3-(4-hidroxipiperidma-1-carbonM)bendl)-3,7-dimetiMH-mdol-5-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 1-(2,6-dicloro-3-(4-hidroxipiperidina-1-carbonil)bendl)-3,7-dimetil-1H-indol-5-carbonitrilo (48 mg, 65%) se preparó a partir de ácido 2,4-didoro-3-((5-dano-3,7-dimetil-1H-indol-1-il)metil)benzoico (60 mg, 0,16 mmol) y 4-hidroxipiperidina (19,5 mg, 0,19 mmol). LC/MS (Método g): Rt = 1,54 min; MS miz: 456 [M+H]+1H NMR (DMSO-d6, 400MHz): 57,89 (s, 1H), 7,69 y 7,68 (d, J=8,4 Hz, 1H), 7,49 y 7,48 (d, J= 8,4 Hz, 1H), 7,29 (d, J=1 Hz, 1H), 6,57 y 7,56 (d, J=1 Hz, 1H), 5,88 (m, 2H), 4,79 (m, 1H), 4,01 (m, 1H), 3,73 (m, 1H), 3,26 (m, 2h), 3,01 (m, 1H), 2,88 (m, 3H), 2,16 (s, 3H), 1,78 (m, 1H), 1,66 (m, 1h), 137 (m, 2H).
Ejemplo DL: ácido 2-(1-(2,4-didoro-3-((5-ciano-3,7-dimetiMH-mdoM-M)metM)benzoN)piperidm-4-M)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((5-ciano-3,7-dimetiMH-mdoM-M)metM)benzoM)pipendm-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-didoro-3-((5-dano-3,7-dimetil-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (111 mg, 99%) se preparó a partir de ácido 2,4-didoro-3-((5-dano-3,7-dimetil-fH -indol-1-il)metil)benzoico (Ejemplo DK, Etapa 2) (80 mg, 0,21 mmol) y clorhidrato de metil (4-piperidil)acetato (49,8 mg, 0,27 mmol). El compuesto se usa directamente en la siguiente etapa. LC/MS (Método i): Rt = 2,43 min; MS miz: 512 [M+H]+
Etapa 2: ácido 2-(1-(2,4-didoro-3-((5-dano-3,7-dimetiMH-mdoM-M)metM)benzoN)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((5-dano-3,7-dimetil-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (51 mg, 45%) se preparó a partir de metil 2-(1-(2,4-didoro-3-((5-ciano-3,7-dimetil-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (110 mg, 0,21 mmol). LC/MS (Método g): Rt = 1,62 min; MS m/z: 498 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 5 12,09 (ancho, 1H), 7,88 (m, 1H), 7,69 y 7,68 (d, J=8,4 Hz, 1H), 7,52 y 7,46 (d, J=8,4 Hz, 1H), 7,29 (s, 1H), 6,58 y 6,55 (s, 1H), 5,87 (m, 2H), 4,46 (m, 1H), 3,27 (m, 1H), 3,03 (m, 1H), 2,88 (s, 3H), 2,80 (m, 1H), 2,16 (m, 5H), 1,93 (m, 1H), 1,77 (m, 1H), 1,60 (m, 1H), 1,15 (m, 2H).
Ejemplo DM: 1-(2,6-didoro-3-(4-hidroxipipeNdma-1-carboml)benzoM)-3,7-dimetiMH-mdol-5-carbomtrMo

Etapa 1: terc-butil 2,4-didoro-3-(5-dano-3,7-dimetN-1H-mdoM-carboml)benzoato

Usando un procedimiento similar al Ejemplo DJ, Etapa 1, tere- butil 2,4-didoro-3-(5-dano-3,7-dimetil-1H-indol-1-carbonil)benzoato (290 mg, 75%) se preparó a partir de 3,7-dimetil-1H-indol-5-carbonitrilo (Preparación #45) (242 mg,
0,87 mmol) y terc-butil 2,4-didoro-3-(clorocarbonil)benzoato (Preparación #44) (483 mg, 1,56 mmol). El producto se usa directamente en la siguiente etapa. LC/MS (Método i): Rt = 2,87 min; MS miz: 443 [M+H]+
Etapa 2: ácido 2,4-didoro-3-(5-dano-3,7-dim etiMH-indoM-carbom l)benzoico

Usando un procedimiento similar al Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-(5-dano-3,7-dimetil-1H-indoM-carbonil)benzoico (232 mg, 82%) se preparó a partir de tere- butil 2,4-dicloro-3-(5-dano-3,7-dimetil-1H-indoM-carbon-il)benzoato (290 mg, 0,65 mmol). LC/MS (Método i): Rt = 2,17 min; MS miz: 387 [M+H]+1 H NMR (DMSO-da, 300MHz): 513,91 (ancho, 1H), 8,05 (d, J =8,6 Hz, 1H), 8,02 (m, 1H), 7,81 (d, J =8,6 Hz, 1H), 7,71 (s, 1H), 7,27 (d, J =1,3 Hz, 1H), 2,69 (s, 3H), 2,19 (d, J=1,3 Hz, 3H).
Etapa 3: 1-(2,6-didoro-3-(4-hidroxipiperidma-1-carbonM)benzoN)-3,7-dimetiMH-mdol-5-carbomtrMo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 1-(2,6-dicloro-3-(4-hidroxipiperidina-1-carbonil)benzoil)-3,7-dimetil-1H-indol-5-carbonitrilo (65 mg, 45%) se preparó a partir de ácido 2,4-didoro-3-(5-dano-3,7-dimetil-1H-indol-1-carbonil)benzoico (113 mg, 0,3 mmol) y 4-hidroxipiperidina (36,4 mg, 0,36 mmol). LC/MS (Método g): Rt = 1,54 min; MS miz: 470 [M+H]+1H NMR (DMSO-d6, 400MHz): 58,03 (s, 1H), 7,80 (m, 1H), 7,70 (m, 1H), 7,67 (m, 1H), 7,45, 7,41 y 7,19 (m, 1H), 4,80 (m, 1H), 4,02 (m, 1H), 3,75 (m, 1H), 3,50 (m, 1H), 3,27 (m, 1H), 3,12 (m, 1h), 2,67 (s, 3H), 2,20 (s, 3H), 1,80 (m, 1h), 1,70 (m, 1H), 1,35 (m, 2H).
Ejemplo DN: ácido 2-(1-(2,4-didoro-3-(5-dano-3,7-dimetiMH-mdoM-carboml)benzoN)piperídm-4-N)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-(5-ciano-3,7-dimetiMH-mdoM-carboml)benzoN)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(5-ciano-3,7-dimetiMH-indoM-carbonil)benzoil)piperidin-4-il)acetato (140 mg, 72%) se preparó a partir de ácido 2,4-dicloro-3-(5-ciano-3,7-dimetiMH-indol-1-carbonil)benzoico (Ejemplo DM, Etapa 2) (115 mg, 0,29 mmol) y clorhidrato de metil (4-piperidil)acetato (69 mg, 0,35 mmol). El producto se usa directamente en la siguiente etapa. LC/Ms (Método i): Rt = 2,42 min; MS m/z: 526 [M+H]+
Etapa 2: ácido 2-(1-(2,4-didoro-3-(5-dano-3,7-dimetiMH-mdoM-carboml)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(5-dano-3,7-dimetil-1H-indoM-carbonil)benzoil)piperidin-4-il)acético (123 mg, 85%) se preparó a partir de 2-(1-(2,4-dicloro-3-(5-ciano-3,7-dimetil-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (138 mg, 0,26 mmol). LC/MS (Método i): Rt = 2,18 min; MS miz: 512 [M+H]+ 1H NMR (DMSO-da, 400MHz): 5 12,10 (ancho, 1H), 8,02 (s, 1H), 7,80 (m, 1H), 7,65 (m, 2H), 7,45 y 7,39 y 7,19 (m, 1H), 4,48 (m, 1H), 3,57 (m, 1H), 3,24 (m, 1H), 3,07 (m, 1H), 2,82 (m, 1H), 2,67 (s, 3H), 2,18 (m, 4H), 1,95 (m, 1H), 1,78 (m, 1H), 1,63 (m, 1H), 1,17 (m, 2H).
Ejemplo DO: (4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-3-il)(4-hidroxipiperidin-1-il)metanona

Etapa 1: 4-metM-6-(trifluorometN)pmdm-2(1H)-ona

Se enfrió una solución de CHCl3 (1,25 L) a aproximadamente 0 °C y se añadió anhídrido 2,2,2-trifluoroacético (0,141 L, 1012 mmol) seguido de la adición de cloruro de 3-metilbut-2-enoilo (0,094 L, 843 mmol). Se añadió gota a gota trietilamina (0,259 L, 1856 mmol) de modo que la temperatura interna se mantuvo por debajo de aproximadamente 10 °C (exotérmica). Después de la adición, la mezcla se agitó a aproximadamente 0 °C durante aproximadamente 1 hora y luego se calentó a temperatura ambiente lentamente y se agitó durante la noche. La mezcla de reacción se lavó con agua (1 L), NaHCO3 saturado (1 L), agua/salmuera (1:1,500 mL), se secó sobre MgSO4, se filtró y se concentró al vacío. El residuo se disolvió en ácido acético (1,250 L) y se añadió ácido acético/sal de amoniaco (130 g, 1687 mmol). El matraz de reacción se tapó con un globo y la mezcla se calentó a aproximadamente 115 °C durante la noche. Después de aproximadamente 20 h, la mezcla de reacción se enfrió a aproximadamente 40 °C y se concentró al vacío. El jarabe se vertió en agua (~ 2 L) con agitación. Después de aproximadamente 1 h, los sólidos se recogieron por filtración para proporcionar 4-metil-6-(trifluorometil)piridin-2(1H)-ona (101 g, 68%) como un sólido color canela. LC/MS (Método a) Rt = 1,59 min.; MS miz: 178 [M+H]+. 1H NMR (CDCla, 400 MHz): 56,79 (s, 1H), 6,70 (s, 1H), 2,34 (s, 3H).
Etapa 2: 2-cloro-4-metil-6-(trifluorometil)piridina

Un matraz de 50 mL en forma de pera equipado con un cabezal de destilación de recorrido corto se cargó con 4-metil-6-(trifluorometil)piridin-2(1H)-ona (20 g, 113 mmol). Al matraz se añadió dicloruro fenilfosfónico (19,2 mL, 135 mmol). La mezcla de reacción se calentó a aproximadamente 160 °C. Después de aproximadamente 3 h, la presión sobre la mezcla de reacción se redujo lentamente a 100 mBar y la destilación comienza con una temperatura del cabezal de aproximadamente 120-130 °C. Periódicamente, cuando la destilación había disminuido significativamente, la presión sobre la mezcla de reacción se redujo a 80 mBar y la temperatura del baño se elevó a aproximadamente 170 °C. Después
de aproximadamente 3 h, la destilación había dejado de proporcionar 2-doro-4-metil-6-(trifluorometil)piridina (12 g, 54 %). LC/MS (Método a) Rt = 2,4 min.; MS miz: 196 [M+H]+.1H NMR (DMSOC 400 MHz): 7,79 (s, 1 H), 7,71 (s, 1H), 2,43 (s, 3 H).
Etapa 3: íerc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato

Un matraz cargado con 2-cloro-4-metil-6-(trifluorometil)piridina (35,1 g, 180 mmol), carbamato de íerc-butilo (42,1 g, 359 mmol), Pd2 (dba) 3 (4,11 g, 4,49 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (X-Phos) (4,28 g, 8,98 mmol) y carbonato de cesio (205 g, 629 mmol) se evacuó y se llenó con N2 (repetido 3 veces) antes de la adición de 1,4-dioxano desgasificado (350 mL). La mezcla se calentó luego a aproximadamente 80 °C durante aproximadamente 2 h. La mezcla de reacción se enfrió a temperatura ambiente y la mezcla de reacción se repartió entre agua y EtOAc. La capa acuosa se extrajo adicionalmente con EtOAc (2 x 100 mL). La capa orgánica se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad para dar un aceite de color rojo oscuro. El material se usó sin purificación adicional, suponiendo un rendimiento del 100%. LC/MS (Método a) Rt = 2,71 min.; MS miz: 275 [M+H]+.1H NMR (400 MHz, DMSO-C6) 510,13 (s, 1H), 7,90 (dt, J = 1,3, 0,7 Hz, 1H), 7,34 (CC, J= 1,3, 0,7 Hz, 1H), 2,38 (t, J = 0,6 Hz, 3H), 1,45 (s, 9H).
Etapa 4: 4-metil-6-(trifluorometil)piridin-2-amina

Una mezcla de íerc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato (49,6 g, 180 mmol) y cloruro de hidrógeno (4M en dioxano) (597 mL, 2388 mmol) se agitó a temperatura ambiente durante aproximadamente 2 h. El disolvente se eliminó a presión reducida y el residuo se filtró. El insoluble se lavó con EtOAc y el filtrado se diluyó con EtOAc y se extrajo con HCl 6N (3x). La capa acuosa se lavó con DCM (4 x 100 mL) y después se ajustó a pH 8 con la adición de Na2 CO3 lentamente. El sólido de color amarillo brillante se recogió por filtración y se lavó con agua. Después, el sólido se disolvió en DCM, se lavó con salmuera, se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con éter de petróleo y se recogió por filtración, se lavó con éter de petróleo y se secó al aire para dar 4-metil-4-(trifluorometil)piridin-2-amina (27,7 g, 88%) como un sólido de color amarillo pálido. LC/MS (Método a) Rt = 1,92 min.; MS miz: 177 [M+H]+.1H NMR (400 MHz, DMSO-C6) 56,72 (s, 1 H), 6,45 (s, 1 h), 6,35 (s, 2 H), 2,20 (s, 3 H).
Etapa 5: 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina

Una mezcla de 4-metil-6-(trifluorometil)piridin-2-amina (31,4 g, 179 mmol) y NBS (66,7 g, 375 mmol) en ACN (250 mL) se calentó a aproximadamente 70 °C durante aproximadamente 2 h. Al matraz de reacción se añadió agua (~500 mL). El sólido resultante se recogió por filtración, se lavó con agua y se secó al aire durante aproximadamente 5 minutos. El sólido se solubilizó en DCM, se lavó con agua, se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con heptano y se recogió por filtración, se lavó con heptano y se secó para dar 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,9 g, 96%) como un sólido de color amarillo claro. LC/MS (Método a) Rt = 2,55 min.; MS miz: 333 [M+H]+.1H NMR (400 MHz, DMSO-C6) 56,90 (s, 2H), 2,53 (s, 3H).
Etapa 6: 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina

A una solución de color naranja clara de 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,98 g, 171 mmol) en THF (570 mL) a aproximadamente -78 °C se añadió n-butil litio (2,5 M en hexano) (68,3 mL, 171 mmol) gota a gota (convertido en una solución marrón). La mezcla se agitó a aproximadamente -78 °C durante aproximadamente 45 minutos. LCMS indicó solo conversión parcial y se añadió gota a gota una segunda porción de BuLi (2,5 M en hexano) (20,48 mL, 51,2 mmol). Se añadió gota a gota una tercera porción de butil litio (2,5 M en hexano) (13,65 mL, 34,1 mmol) para completar la conversión al producto. El matraz de reacción se transfirió a un baño de agua con hielo y se añadieron rápidamente 20 mL de agua. La mezcla de reacción se calentó luego a temperatura ambiente. La mezcla de reacción se repartió entre NH4 Cl saturado y EtOAc. La capa orgánica se lavó con NaHCO3 , se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con heptano y se recogió por filtración, se lavó con hepano para dar la primera cosecha de producto como un sólido de color blanquecino (38,9 g, 85%). El filtrado se concentró hasta la sequedad y se añadió heptano. El sólido se recogió por filtración al vacío y se lavó con heptano y se secó para dar la segunda cosecha de producto como un sólido de color amarillo pálido (1,9 g, 3%). LC/MS (Método a) Rt = 2,29 min.; MS miz: 255 [M+H]+.1H NMR (400 MHz, DMSO-C6 ) 56,96 (s, 1 H), 6,74 (s, 1 H), 2,32 (s, 3 H).
Etapa 7: (£)-2-(2-Etoxivinil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano

Bajo una atmósfera de nitrógeno en un matraz de fondo redondo, etoxietileno, solución al 50% en hexanos (100 g, 710 mmol) y DCM (996 mL) se agitó a aproximadamente 0-5 °C. A la solución agitada se añadió 4,4,5,5-tetrametil-1,3,2-dioxaborolano (113 mL, 781 mmol) en una porción seguido de la adición de bis(ciclopentadienil)circonio hidrido-cloruro (9,16 g, 35,5 mmol) a aproximadamente 0-5 °C. La suspensión (naranja) se dejó calentar gradualmente a temperatura ambiente durante aproximadamente 30 minutos. La disolución ocurrió dentro de aproximadamente 10 min. La mezcla de reacción se agitó a temperatura ambiente durante la noche (rojo muy oscuro). A la solución de reacción se añadió éter (2L) y la solución se lavó con solución saturada acuosa de NH4 CL Los disolventes se eliminaron a presión reducida, se añadió DCM mínimo (100 mL) y la solución se filtró a través de una almohadilla de alúmina, cubierta con Celita. La alúmina se lavó con DCM y el disolvente del filtrado se eliminó al vacío para producir (E)-2-(2-etoxivinil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (114,9 g, 82 %) como un aceite rojo muy oscuro.1H nMr (400 Mhz, DmSO-C6 ) 57,05 (d, 1 H), 4,45 (d, 1 H), 3,83 (q, 2 H), 1,30 (t, 3 H), 1,28 (s, 12 H).
Etapa 8: (£)-3-(2-etoxivmN)-4-metM-6-(trifluorometN)pmdm-2-amma

Un matraz cargado con 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina (38,3 g, 143 mmol), (E)-2-(2-etoxivinil)-4,4,5, 5-tetrametil-1,3,2-dioxaborolano (56,5 g, 285 mmol), diacetoxipaladio (0,960 g, 4,28 mmol), diciclohexil(2',4',6'-triisopropil-[1,1'-bifenil]-2-il)fosfina (Xphos) (4,28 g, 8,98 mmol) y carbonato de cesio (116 g, 356 mmol) se desgasificó con N2 durante 15 min antes de la adición de 320 mL 1,4-dioxano desgasificado/H2O (4:1). La mezcla se calentó a aproximadamente 80 °C durante aproximadamente 2 h. La mezcla se enfrió a temperatura ambiente. La mezcla de reacción se repartió entre EtOAc y agua y la capa acuosa se extrajo adicionalmente con EtOAc (2 x 50 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Al residuo se añadió heptano y el sólido resultante se recogió por filtración y se lavó con heptano para dar un sólido de color blanquecino (1,45 g). El filtrado se concentró hasta la sequedad nuevamente para dar un aceite negro espeso que se dejó a temperatura ambiente durante la noche. Se observó una formación significativa de sólidos y el sólido se diluyó con heptano, se sonicó y se filtró, se lavó con heptano para dar un sólido marrón claro (15,2 g, 42%). El filtrado se concentró hasta la sequedad para dar un aceite negro que se purificó por cromatografía ultrarrápida (EtOAc 0-25%/heptano durante 30 minutos). El producto que contenía fracciones se concentró casi hasta la sequedad. El sólido se recogió por filtración, se lavó con una pequeña cantidad de heptano y se secó para dar la segunda cosecha de producto como un sólido de color amarillo claro (12,7 g, 35%). LC/MS (Método 1) Rt = 1,48
min.; MS m/z: 247 [M+H]+. 1H NMR (400 MHz, DMSO-C6) 56,82 (s, 1H), 6,73 (d, J = 13,1 Hz, 1H), 6,12 (s, 2H), 5,45 (d, J = 13,1 Hz, 1H), 3,93 (q, J = 7,0 Hz, 2H), 2,23 (s, 3H), 1,25 (t, J = 7,0 Hz, 3H).
Etapa 9: 4-metil-6-(tnfluorometil)-1H-pirrolo[2,3-to]pmdma

Una mezcla de (E)-3-(2-etoxivinil)-4-metil-6-(trifluorometil)piridin-2-amina (27,9 g, 113 mmol) y ácido acético (130 mL) se calentó a aproximadamente 100 °C durante la noche. La mezcla se enfrió a temperatura ambiente y el precipitado se recogió por filtración, se lavó con ACN y se secó para dar la primera cosecha de producto.
El filtrado se concentró hasta aproximadamente 50 mL y el sólido se recogió por filtración, se lavó con ACN y se secó para dar la segunda cosecha de producto. Ambos parecían de una pureza similar y, por lo tanto, se combinaron. El filtrado todavía tenía algo de producto y se dejó a un lado. El sólido se suspendió en aproximadamente 250 mL de EtOAc y se calentó a reflujo para disolverse. La mayor parte del disolvente se eliminó a presión reducida y se añadió heptano. El sólido se recogió por filtración al vacío, se secó para dar el producto como un sólido de color blanquecino (19,8 g, 87%). LC/MS (Método 1-3) Rt = 1,30 min.; MS m/z: 201 [M+H]+. 1H NMR (400 MHz, DMSO-C6) 5 12,13 -11,98 (br, 1H), 7,67 (t, J = 3,1 Hz, 1H), 7,34 (d, J = 1,0 Hz, 1H), 6,63 (dd, J = 3,5, 1,9 Hz, 1H), 2,59 (s, 3H).
Etapa 10: 4-metiM-(femlsulfoml)-6-(trifluorometil)-1H-pirrolo[2,3-b]pmdma

A una suspensión de hidruro de sodio (60% en aceite mineral) (4,35 g, 109 mmol) en DMF (40 mL) a aproximadamente 0 °C se añadió una solución de 4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (19,8 g, 99 mmol) bajo N2. Después de la adición, se retiró el baño de agua con hielo y la mezcla se agitó a temperatura ambiente durante aproximadamente 30 minutos. Se enfrió nuevamente a aproximadamente 0 °C, se añadió gota a gota cloruro de bencenosulfonilo (13,27 mL, 104 mmol). Después de la adición, la reacción se dejó calentar a temperatura ambiente y se agitó a temperatura ambiente durante aproximadamente 1 hora.
La reacción se inactivó con la adición de 100 mL de solución saturada de NH4Cl seguido de la adición de 350 mL de agua. El sólido se recogió por filtración, se lavó con agua y se secó en un horno de vacío a aproximadamente 70 °C durante aproximadamente 2 días para dar un sólido de color blanquecino. El sólido se disolvió en EtOAc, se filtró a través de 70 g de gel de sílice y la almohadilla se lavó con EtOAc. El filtrado se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. El sólido se trituró con EtOAc/heptano para dar la primera cosecha de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (28,5 g, 85 %) como sólido blanco.
El filtrado se concentró hasta la sequedad y el sólido se trituró con éter/hepatano. El sólido resultante se recogió por filtración, se lavó con heptano y se secó para dar la segunda cosecha de 4-metil-1-(fenilsulfonil)-6-(tri-fluorometil)-ÍH-pirrolo[2,3-¿]piridina (3,65 g, 11%) como un sólido de color blanquecino. LC/MS (Método 1-3) Rt = 1,84 min.; MS m/z: 341 [M+H]+. 1H NMR (400 MHz, DMSO-C6) 5 8,15 -8,07 (m, 3H), 7,75 -7,67 (m, 1H), 7,64 -7,56 (m, 3H), 7,03 (d, J = 4,1 Hz, 1H), 2,57 (s, 3H).
Etapa 11: íerc-butil 4,6-dicloronicotinato

Una mezcla de ácido 4,6-didoronicotínico (8,2 g, 42,7 mmol) y BOC2 O (19,83 mL, 85 mmol) en THF (100 mL) se agitó a temperatura ambiente, después se añadió DMAP (1,044 g, 8,54 mmol) a la solución. La mezcla resultante se agitó a aproximadamente 70 °C durante aproximadamente 1 h. La muestra se depositó sobre gel de sílice y se cargó en una columna de gel de sílice y se eluyó con EtOAc al 5%/heptano. Las siguientes fracciones se recogieron para dar íerc-butil 4,6-dicloronicotinato (10 g, 94 %) como un aceite incoloro.1H NMR (400 MHz, CDCh) 58,76 (s, 1 H), 7,43 (s, 1 H), 1,61 (s, 9 H).
Etapa 12: íerc-butil 4,6-didoro-5-formilnicotinato

El íerc-butil 4,6-dicloronicotinato (10 g, 40,3 mmol) se disolvió en THF (100 mL), se agitó y se enfrió a aproximadamente -78 °C. A la solución se añadió LDA (22,2 mL, 44,3 mmol) a una velocidad para mantener la temperatura por debajo de aproximadamente -70 °C. La solución resultante se agitó a aproximadamente -78 °C durante aproximadamente 10 minutos. Luego se añadió formiato de metilo (4,94 mL, 81 mmol) a la solución y la mezcla se agitó a aproximadamente -78 °C durante aproximadamente 30 minutos. La mezcla se vertió en NH4 Cl saturado, se extrajo con EtOAc (3x) y las capas orgánicas combinadas se secaron con Na2 SO4 , se filtraron y se concentraron para proporcionar un aceite de color marrón. La muestra se depositó sobre gel de sílice y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 2%/heptano. Se recogieron las siguientes fracciones para dar íerc-butil 4,6-dicloro-5-formilnicotinato (5,0 g, 45% de rendimiento) como un sólido amarillo.1H NMR (400 MHz, CDCh) 510,47 (s, 1 H), 8,7 (s, 1 H), 1,65 (s, 9 H).
Etapa 13: íerc-butil 4,6-didoro-5-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]pi-ridin-2-il)metil)nicotinato

Usando un procedimiento similar al descrito en el Ejemplo CL, Etapa 1, íerí-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato se preparó a partir de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (6,0 g, 17,6 mmol) y íerc-butil 4,6-dicloro-5-formilnicotinato (5,35 g, 19,4 mmol). LC/MS (Método 1-3) Rt = 2,15 min.; MS m/z: 616 [M+H]+.1H NMR (400 MHz, DMSO-C6) 58,67 (s, 1 H), 7,95 (d, 2 h), 7,69 (m, 1 H), 7,61 (s, 1 H), 7,55 (m, 2 H), 7,05 (s, 1 H), 7,04 (d, 1 H), 6,92 (d, 1 H), 2,54 (s, 3 H), 1,54 (s, 9 H).
Etapa 14: terc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo [2,3-b] piridin-2-il)metil)nicotinato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, íerc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato se preparó a partir de íerc-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato (1,51 g, 2,44 mmol). LC/MS (Método 1-3) Rt = 2,33 min.; MS m/z: 600 [M+H]+.
Etapa 15: ácido 4,6-didoro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico

A una solución de íerc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato (334,9 mg, 0,558 mmol) en 1,4-dioxano (7339 mL) se añadió hidróxido de sodio (2M) (1394 pL, 2,79 mmol). La mezcla se calentó a aproximadamente 50 °C durante la noche. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con agua y se acidificó con HCl (1M). La mezcla se extrajo dos veces con EtOAc y los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se evaporaron para dar ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico (0,23 g, 100%) como un sólido de color marrón pálido. LC/MS (Método 1-3) Rt = 1,02 min.; MS m/z: 404 [M+H]+. 1H NMR (400 MHz, DMSO-d6 ) 5 12,16 (s, 1H), 8,75 (s, 1H), 7,29 (d, J = 0,8 Hz, 1H), 6,09 (dd, J = 2,1, 1,0 Hz, 1H), 4,48 (s, 3H).
Etapa 16: metil 4,6-didoro-5-((1,4-dimetil-6-(tnfluorometil)-1H-pirrolo[2,3-to]pmdm-2-il)metil)mcotmato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato se preparó a partir de ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico (0,23 g, 0,567 mmol) LC/MS (Método 1-3) Rt = 2,01 min.; MS m/z: 432 [M+H]+.
Etapa 17: ácido 4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pin'olo[2,3-fo]piridm-2-il)metil) nicotínico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-tó]piridin-2-il)metil)nicotínico se preparó a partir de metil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (0,25 g, 0,578 mmol) LC/MS (Método 1-3) Rt = 1,14 min.; MS miz: 418 [M+H]+.
Etapa 18: (4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pin'olo[2,3-fo]piridm-2-il)metil)pmdm-3-il)(4-hidroxipiperidin-1-il)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6 , (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-tó]piridin-2-il)metil)piridin-3-il)(4-hidroxipiperidin-1-il)metanona se preparó a partir de ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotínico (0,116 g, 0,278 mmol) y piperidin-4-ol (0,042 g, 0,42 mmol). LC/MS (Método a) Rt = 2,28 min.; MS miz: 501 [M+H]+. 1H NMR (400 MHz, DMSO-d6 ) 58,31 (s, 1 H), 7,26 (s, 1 H), 7,22 (s, 1 H), 5,81 (s, 1 H), 4,46 (m, 2 H), 4,15 (m, 1 H), 4,04 (m, 1 H), 3,98 (s, 3 H), 3,65-3,45 (m, 3 H), 3,13 (m, 2 H), 2,00 (m, 1 H), 1,87 (m, 2 H), 1,70 (m, 2 H).
Ejemplo DP: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-mdol-2-il)metil)benzoil)piperidm-4-il)acético

Etapa 1: metil 2,4-didoro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-didoro-3-(hidroxi(4-metil-1-(fenilsulfonil)-4-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (814 mg, 79%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometoxi)fenil)bencenosulfonamida (Preparación #47) (800 mg, 1,75 mmol) y metil 2,4-dicloro-3-(1-hidroxi-prop-2-in-1-il)benzoato (Preparación #1) (544 mg, 2,1 mmol). LC/MS (Método i) Rt = 2,69 min.; MS miz: 646 [M-H]-+ CH3C0 OH 1H NMR (DMSO-d6, 300MHz): 57,89 (m, 2H), 7,74 (s, 1H), 7,69 (m, 2H), 7,58 (m, 3H), 7,12 (m, 1H), 7,03 (dd, J=5,9, 1,2 Hz, 1H), 6,74 (m, 1H), 6,67 (d, J=5,9 Hz, 1H), 3,87 (s, 3H), 2,40 (s, 3H).
Etapa 2: metil 2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (540 mg, 68%) a partir de metil 2,4-didoro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifiuorometoxi)-1H-indol-2-il)metil)benzoato (811 mg, 1,38 mmol). LC/MS (Método j) Rt = 2,65 min.; MS m/z: 572 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,96 (m, 2H), 7,88 (s, 1H), 7,83 (m, 1H), 7,76 (d, J=7,6 Hz, 1H), 7,68 (m, 3H), 7,12 (m, 1H), 5,89 (m, 1H), 4,61 (m, 2H), 3,87 (s, 3H), 2,29 (s, 3H).
Etapa 3: metil 2,4-didoro-3-((4-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-didoro-3-((4-metil-6-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (408 mg, 100%) se preparó a partir de metil 2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (540 mg, 0,94 mmol). LC/MS (Método j) Rt = 2,25 min.; MS m/z: 432 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 11,26 (s, 1H), 7,74 (d, J= 8,4 Hz, 1H), 7,68 (d, J= 8,4 Hz, 1H), 7,11 (s, 1H), 6,73 (s, 1H), 5,90 (s, 1H), 4,45 (s, 2H), 3,87 (s, 3H), 2,36 (s, 3H)
Etapa 4: metil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (178 mg, 42%) se obtuvo a partir de metil 2,4-dicloro-3-((4-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (408 mg, 0,944 mmol). LC/MS (Método j) Rt = 2,42 min.; MS m/z: 446 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 7,80 (d, J=8,4 Hz, 1H), 7,71 (d, J=8,4 Hz, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,59 (s, 1H), 4,46 (s, 2H), 3,88 (s, 3H), 3,85 (s, 3H), 2,31 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (170 mg, 97 %) se preparó a partir de metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (178 mg, 0,4 mmol).
LC/MS (Método i) Rt = 2,53 min.; MS m/z: 432 [M+H]+ 1H NMR (DMSO-da, 300MHz): 5 13,60 (br, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,59 (s, 1H), 4,45 (s, 2H), 3,85 (s, 3H), 2,32 (s, 3H)
Etapa 6: metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (102 mg, 96%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (80 mg, 0,18 mmol) y clorhidrato de metil (4-piperidil)acetato (54 mg, 0,28 mmol).
LC/MS (Método i) Rt = 2,68 min.; MS m/z: 571 [M+H]+ 1H NMR (DMSO-da, 300MHz): 57,66 y 7,65 (d, J=8,4 Hz, 1H), 7,42 y 7,37 (d, J=8,4 Hz, 1H), 7,34 (m, 1H), 6,77 (m, 1H), 5,59 y 5,56 (m, 1H), 4,44 (m, 3H), 3,85 y 3,84 (s, 3H), 3,59 y 3,55 (s, 3h), 3,25 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,31 y 2,30 (s, 3H), 2,24 (m, 2H), 1,95 (m, 1h), 1,74 (m, 1H), 1,58 (m, 1H), 1,17 (m, 2H).
Etapa 7: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (90 mg, 89%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (102 mg, 0,18 mmol).
LC/MS (Método g) Rt = 1,88 min.; MS m/z: 557 [M+H]+ 1H NMR (DMSO-d6, 400MHz): 512,10 (m, 1H), 7,66 y 7,65 (d, J=8,3 Hz, 1H), 7,43 y 7,36 (d, J=8,3 Hz, 1H), 7,34 (s, 1H), 6,77 (m, 1H), 5,59 y 5,56 (s, 1H), 4,42 (m, 3H), 3,84 (m, 3H), 3,26 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,31 y 2,30 (s, 3H), 2,15 (m, 2H), 1,95 (m, 1H), 1,77 (m, 1H), 1,62 (m, 1H), 1,15 (m, 2H).
Ejemplo DP-1: (2,4-didoro-3-((1,4-dimetM-6-(trifluorometoxi)-1H-mdol-2-N)metN)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)fenil)(morfolino)metanona (79 mg, 83%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (80 mg, 0,18 mmol) (Ejemplo DP, Etapa 5) y morfolina (19 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,96 min; MS m/z: 501 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 57,67 (d, J=8,4 Hz, 1H), 7,44 (d, J=8,4 Hz, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,61 (s, 1H), 4,42 (m, 2H), 3,84 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,17 (m, 2H), 2,32 (s, 3H).
Ejemplo DP-2: ácido (3S,4S)-1-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoN)-3-metilpiperidina-4-carboxílico

Etapa 1: (3S,4S)-etil 1-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoN)-3-metNpiperidma-4-carboxilato
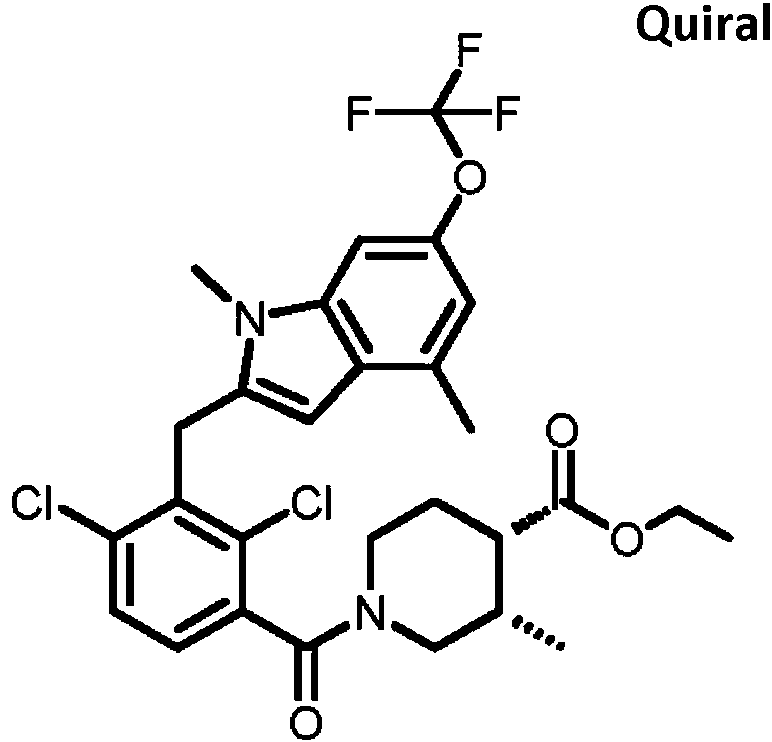
Usando un procedimiento similar al Ejemplo A, Etapa 6, (3S,4S)-etil 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxilato (166 mg, 82%) a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (80 mg, 0,18 mmol) (ejemplo DP, Etapa 5) y clorhidrato de (3S,4S)-etil 3-metilpiperidina-4-carboxilato (108 mg, 0,52 mmol) (Preparación 83). LC/MS (Método i): Rt = 2,80 min; MS miz: 585 [M+H]+1H NMR (DMSO-C6, 300MHz): 57,66 (d, J=8,3 Hz, 1H), 7,42 (m, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,59 (m, 1H), 4,41 (m, 2H), 4,08 (m, 2H), 3,85 y 3,84 (s, 3h), 3,16 (m, 3H), 2,79 (m, 1H), 2,32 (m, 3H), 2,27 (m, 1h), 2,12 (m, 1H), 1,72 (m, 2H), 1,16 (m, 3H), 0,82 (m, 3H).
Etapa 2: ácido (3S,4S)-1-(2,4-didoro-3-((1,4-dimetN-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoN)-3-metN-piperidina-4-carboxílico
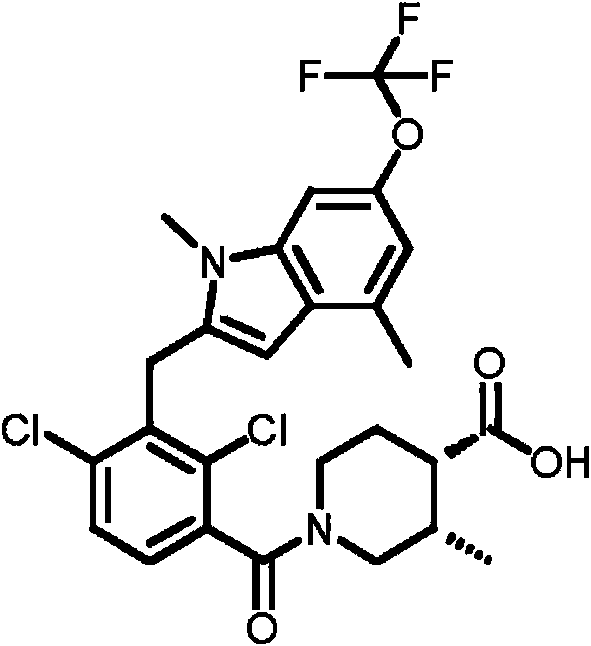
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido (3S,4S)-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico (72 mg, 46%) se preparó a partir de (3S,4S)-etil-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxilato (160 mg, 0,27 mmol). LC/MS (Método g): Rt = 1,90 min; MS m/z: 557 [M+H]+; 1H NMR (DMSOC 400MHz): 512,25 (ancho, 1H), 7,65 (m, 1H), 7,41 (m, 1H), 7,34 (s, 1H), 6,77 (m, 1H), 5,58 (m, 1H), 4,30 (m, 3H), 3,85 y 3,84 (s, 3H), 3,08 (m, 3H), 2,67 (m, 1H), 2,32 (m, 3H), 2,28 y 2,11 (m, 1h), 1,65 (m, 2H), 0,84 (m, 3H).
Ejemplo DQ: 4-doro-2-(2,6-didoro-3-(morfolma-4-carboml)bend l)-1-m etiM H-mdol-6-carbomtrilo

A una solución de 6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol (Preparación #48) (200 mg, 0,54 mmol) en THF (1,5 mL) y enfriado a -78 °C se añadió diisopropilamida de litio (0,30 mL, 0,59 mmol) y la mezcla de reacción se agitó durante 1 hora a -78 °C. Se añadió íerc-butil 2,4-dicloro-3-formilbenzoato (Preparación #33, Etapa B) (178 mg, 0,65 mmol) en solución en THF (1,5 mL) a la mezcla de reacción y la mezcla de reacción se agitó durante 2 horas a -78 °C. La mezcla de reacción se inactivó con una solución saturada de NH4Cl y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar íerc-butil 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il) (hidroxi)metil)-2,4-diclorobenzoato (262 mg, 75%). LC/MS (Método j) Rt = 2,75 min.; MS m/z: 702 [M-H]-+ CH3COOH 1H NMR (DMSO-d6, 300MHz): 58,11 (m, 1H), 7,85 (m, 2H), 7,72 (m, 1H), 7,66 (d, J=6 Hz, 1H), 7,55 (m, 4H), 6,95 (m, 1H), 6,75 (m, 1H), 6,66 (m, 1H), 1,54 ppm (s, 9H)
Etapa 2: ácido 3-((6-bromo-4-doro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoico (1,17 g, 93%) se preparó a partir de terc-butil 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-1-il)(hidroxi)metil)-2,4-didorobenzoato (1,42 g, 2,2 mmol). LC/MS (Método i) Rt = 2,81 min.; MS miz: 570 [M-H]-1H NMR (DMSO-da, 300MHz): 58,27 (m, 1H), 8,00 (m, 2H), 7,80 (m, 2H), 7,69 (m, 4H), 5,70 (m, 1h), 4,55 (s, 2H).
Etapa 3: ácido 3-((6-bromo-4-doro-1H-indol-2-il)metil)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, se preparó ácido 3-((6-bromo-4-cloro-1H-indol-2-il)metil)-2,4-diclorobenzoico (657 mg, 100%) a partir de ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoico (870 mg, 1,52 mmol). El compuesto se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,48 min.; MS miz: 432 [M+H]+
Etapa 4: metil 3-((6-bromo-4-doro-1-metil-1H-indol-2-il)metil)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 3-((6-bromo-4-cloro-1-metil-1H-indol-2-il)metil)-1,4-diclorobenzoato (568 mg, 81%) se preparó a partir de ácido 3-((6-bromo-4-doro-1H-indol-2-il)metil)-2,4-didorobenzoico (657 mg, 1,52 mmol).
LC/MS (Método j) Rt = 2,46 min.; MS m/z: 460 [M+H]+1H NMR (DMSO-d6, 300MHz): 57,80 (m, 2H), 7,72 (m, 1H), 7,23 (d, J=1,5 Hz, 1H), 5,52 (m, 1H), 4,46 (s, 2H), 3,89 (s, 3H), 3,88 (s, 3H).
Etapa 5: metil 2,4-didoro-3-((4-doro-6-dano-1-metiMH-mdol-2-M)metil)benzoato

A una solución de metil 3-((6-bromo-4-doro-1-metiMH-indol-2-il)metil)-2,4-didorobenzoato (559 mg, 1,211 mmol) en N,N-dimetilformamida (4,3 mL) se añadió cianuro de zinc (107 mg, 0,908 mmol) y tetrakis(trifenilfosfina)paladio (140 mg, 0,121 mmol) y la mezcla de reacción se calentó a 120 °C en microondas durante 1 hora. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-40% en ciclohexano) para dar metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (200 mg, 40%). LC/MS (Método i) Rt = 2,59 min.; MS m/z: 405 [M-H]-1H NMR (DMSO-d6, 300MHz): 5 = 8,18 (m, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,73 (d, J=8,4 Hz, 1H), 7,50 (d, J=1,2 Hz, 1H), 5,67 (m, 1H), 4,54 (s, 2H), 3,96 (s, 3H), 3,88 (s, 3H).
Etapa 6: ácido 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil) benzoico (180 mg, 94%) se preparó a partir de metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (198 mg, 0,48 mmol). LC/MS (Método i) Rt = 2,25 min.; MS m/z: 393 [M+H]+
1H NMR (DMSO-d6, 300MHz): 5 13,60 (br, 1H), 8,18 (m, 1H), 7,79 (d, J=8,4 Hz,, 1H), 7,69 (d, J=8,4 Hz, 1H), 7,50 (d, J=1,2 Hz, 1H), 5,67 (m, 1H), 4,53 (s, 2H), 3,96 (s, 3H).
Etapa 7: 4-doro-2-(2,6-didoro-3-(morfolina-4-carbonil)bencil)-1-metil-1H-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A1, se preparó 4-cloro-2-(2,6-didoro-3-(morfolina-4-carbonil)bendl)-1-metil-1H-indol-6-carbonitrilo (89 mg, 82%) a partir de ácido 2,4-didoro-3-((4-doro-6-dano-1-metiMH-indol-2-il)metil)benzoico (90 mg, 0,23 mmol) y morfolina (23,9 mg, 0,27 mmol). LC/MS (Método g) Rt = 1,69 min.; MS miz: 462 [m+H]+1H NMR (DMSO-da, 400MHz): 58,17 (s, 1H), 7,69 (d, J=8,1 Hz, 1H), 7,50 (d, J=1,3 Hz, 1h), 7,47 (d, J=8,1 Hz, 1H), 5,70 (m, 1H), 4,50 (m, 2H), 3,96 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,15 (m, 2H).
Ejemplo DP-1: (2,4-didoro-3-((1,4-dimetM-6-(tnfluorometoxi)-1H-mdol-2-M)metM)feml)(morfolmo)metanona

Usando un procedimiento similar al del Ejemplo A1, (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)fenil)(morfolino)metanona (79 mg, 83%) se preparó a partir de ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (80 mg, 0,18 mmol) (Ejemplo DP, Etapa 5) y morfolina (19 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,96 min; MS miz: 501 [M+H]+; 1H NMR (DMSO-C6, 400MHz): 57,67 (d, J=8,4 Hz, 1H), 7,44 (d, J=8,4 Hz, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,61 (s, 1H), 4,42 (m, 2H), 3,84 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,17 (m, 2H), 2,32 (s, 3H).
Ejemplo DP-2: ácido (3S,4S)-1-(2,4-didoro-3-((1,4-dimetM-6-(tnfluorometoxi)-1H-mdol-2-M)metM)benzoM)-3-metilpiperidina-4-carboxílico 
Etapa 1: (3S,4S)-etil 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoil)-3-metilpiperidma-4-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, (3S,4S)-etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxilato (166 mg, 82%) se preparó a partir de ácido 2,4-dicloro-1-((1,4-dimetil-6-(trifluorometoxi)-1H -indol-2-il)metil)benzoico (80 mg, 0,18 mmol) (Ejemplo DP, Etapa 5) y clorhidrato de (3S,4S)-etil 3-metilpiperidina-4-carboxilato (108 mg, 0,52 mmol) (Preparación 83). LC/MS (Método i): Rt = 2,80 min; MS miz: 585 [M+H]+1H NMR (DMSO-C6, 300MHz): 57,66 (d, J=8,3 Hz, 1H), 7,42 (m, 1H), 7,34 (s, 1H), 6,77 (s, 1H), 5,59 (m, 1H), 4,41 (m, 2H), 4,08 (m, 2H), 3,85 y 3,84 (s, 3h), 3,16 (m, 3H), 2,79 (m, 1H), 2,32 (m, 3H), 2,27 (m, 1h), 2,12 (m, 1H), 1,72 (m, 2H), 1,16 (m, 3H), 0,82 (m, 3H).
Etapa 2: ácido (3S,4S)-1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoil)-3-metilpiperidina-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido (3S,4S)-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico (72 mg, 46%) se preparó a partir de (3S,4S)-etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxilato (160 mg, 0,27 mmol). LC/MS (Método g): Rt= 1,90 min; MS miz: 557 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 512,25 (ancho, 1H), 7,65 (m, 1H), 7,41 (m, 1H), 7,34 (s, 1H), 6,77 (m, 1H), 5,58 (m, 1H), 4,30 (m, 3H), 3,85 y 3,84 (s, 3H), 3,08 (m, 3H), 2,67 (m, 1H), 2,32 (m, 3H), 2,28 y 2,11 (m, 1H), 1,65 (m, 2H), 0,84 (m, 3H).
Ejemplo DP-3: ácido (3R,4R)-1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoil)-3-metilpiperidina-4-carboxílico

Ácido (3R,4R)-1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)-3-metilpiperidina-4-carboxílico se preparó de una manera similar al Ejemplo DP-2 usando clorhidrato de (3R,4R)-etil 3-metilpiperidina-4-carboxilato (Preparación 84) en la Etapa 1. LC/MS (Método g): Rt = 1,90 min; MS miz: 557 [M+H]+
Ejemplo DQ: 4-doro-2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1-metiMH-mdol-6-carbomtrMo
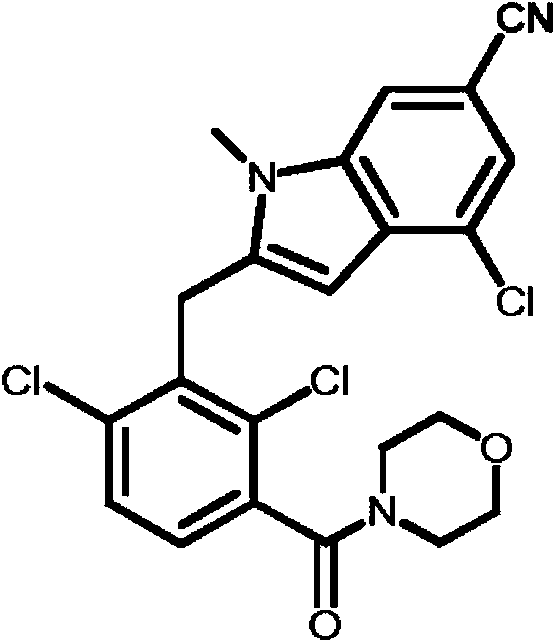
Etapa 1: íerc-butil 3-((6-bromo-4-doro-1-(femlsulfoml)-1H-mdol-2-M)(hidroxi)metM)-2,4-didorobenzoato

A una solución de 6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol (Preparación #48) (200 mg, 0,54 mmol) en THF (1,5 mL) y enfriada a -78 °C se añadió diisopropilamida de litio (0,30 mL, 0,59 mmol) y la mezcla de reacción se agitó durante 1 hora a -78 °C. Se añadió íerc-butil 2,4-dicloro-3-formilbenzoato (Preparación #33, Etapa B) (178 mg, 0,65 mmol) en solución en THF (1,5 mL) a la mezcla de reacción y la mezcla de reacción se agitó durante 2 horas a -78 °C. La mezcla de reacción se inactivó con una solución saturada de NH4Cl y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar terc-butil 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-diclorobenzoato (262 mg, 75%). LC/MS (Método j) Rt = 2,75 min.; MS miz: 702 [M-H]-+ CH3COOH; 1H NMR (DMSO-d6, 300MHz): 5 8,11 (m, 1H), 7,85 (m, 2H), 7,72 (m, 1H), 7,66 (d, J=6 Hz, 1H), 7,55 (m, 4H), 6,95 (m, 1H), 6,75 (m, 1H), 6,66 (m, 1H), 1,54 ppm (s, 9H)
Etapa 2: ácido 3-((6-bromo-4-doro-1-(femlsulfoml)-1H-mdol-2-N)metN)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoico (1,17 g, 93%) se preparó a partir de terc-butil 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)-2,4-didorobenzoato (1,42 g, 2,2 mmol). LC/MS (Método i) Rt = 2,81 min.; MS miz: 570 [M-H]-; 1H NMR (DMSO-da, 300MHz): 58,27 (m, 1H), 8,00 (m, 2H), 7,80 (m, 2H), 7,69 (m, 4h), 5,70 (m, 1H), 4,55 (s, 2H).
Etapa 3: ácido 3-((6-bromo-4-doro-1H-indol-2-il)metil)-2,4-didorobenzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, se preparó ácido 3-((6-bromo-4-cloro-1H-indol-2-il)metil)-2,4-diclorobenzoico (657 mg, 100%) a partir de ácido 3-((6-bromo-4-cloro-1-(fenilsulfonil)-1H-indol-2-il)metil)-2,4-diclorobenzoico (870 mg, 1,52 mmol). El compuesto se usó directamente en la siguiente etapa. LC/MS (Método i) Rt = 2,48 min.; MS miz: 432 [M+H]+
Etapa 4: metil 3-((6-bromo-4-doro-1-metiMH-mdol-2-N)metN)-2,4-didorobenzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 3-((6-bromo-4-cloro-1-metil-1H-indol-2-il)metil)-2,4-diclorobenzoato (568 mg, 81%) se preparó a partir de ácido 3-((6-bromo-4-doro-1H-indol-2-il)metil)-2,4-didorobenzoico (657 mg, 1,52 mmol). LC/MS (Método j) Rt = 2,46 min.; MS m/z: 460 [M+H]+ ; 1H NMR (DMSO-da, 300MHz): 5 7,80 (m, 2H), 7,72 (m, 1H), 7,23 (d, J=1,5 Hz, 1H), 5,52 (m, 1H), 4,46 (s, 2H), 3,89 (s, 3H), 3,88 (s, 3H).
Etapa 5: metil 2,4-didoro-3-((4-doro-6-dano-1-metiMH-mdol-2-M)metil)benzoato

A una solución de metil 3-((6-bromo-4-doro-1-metiMH-indol-2-il)metil)-2,4-didorobenzoato (559 mg, 1,211 mmol) en N,N-dimetilformamida (4,3 mL) se añadió cianuro de zinc (107 mg, 0,908 mmol) y tetrakis(trifenilfosfina)paladio (140 mg, 0,121 mmol) y la mezcla de reacción se calentó a 120 °C en microondas durante 1 hora. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-40% en ciclohexano) para dar metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (200 mg, 40%). LC/MS (Método i) Rt = 2,59 min.; MS m/z: 405 [M-H]-; 1H NMR (DMSO-d6, 300MHz): 5 = 8,18 (m, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,73 (d, J=8,4 Hz, 1H), 7,50 (d, J=1,2 Hz, 1H), 5,67 (m, 1H), 4,54 (s, 2H), 3,96 (s, 3H), 3,88 (s, 3H).
Etapa 6: ácido 2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil) benzoico (180 mg, 94 %) se preparó a partir de metil 2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoato (198 mg, 0,48 mmol). LC/MS (Método i) Rt = 2,25 min.; MS m/z: 393 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 13,60 (br, 1H), 8,18 (m, 1H), 7,79 (d, J=8,4 Hz,, 1H), 7,69 (d, J=8,4 Hz,, 1H), 7,50 (d, J =1,2 Hz, 1H), 5,67 (m, 1H), 4,53 (s, 2H), 3,96 (s, 3H).
Etapa 7: 4-doro-2-(2,6-didoro-3-(morfolma-4-carboml)bencM)-1-metiMH-mdol-6-carbomtrMo

Usando un procedimiento similar al Ejemplo A1,4-doro-2-(2,6-didoro-3-(morfolina-4-carbonil)bencil)-1-metiMH-indol-6-carbonitrilo (89 mg, 82%) se preparó a partir de ácido 2,4-didoro-3-((4-doro-6-dano-1-metiMH-indol-2-il)metil)benzoico (90 mg, 0,23 mmol) y morfolina (23,9 mg, 0,27 mmol). LC/MS (Método g) Rt = 1,69 min.; MS miz: 462 [M+H]+ ; 1H NMR (DMSO-da, 400MHz): 58,17 (s, 1H), 7,69 (d, J=8,1 Hz, 1H), 7,50 (d, J=1,3 Hz, 1H), 7,47 (d, J=8,1 Hz, 1H), 5,70 (m, 1H), 4,50 (m, 2H), 3,96 (s, 3H), 3,65 (m, 4H), 3,55 (m, 2H), 3,15 (m, 2H).
Ejemplo DR: ácido2-(1-(2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il) acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato
Usando un procedimiento similar al Ejemplo A1, se preparó metil 2-(1-(2,4-dicloro-3-((4-doro-6-dano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (101 mg, 83%) a partir de ácido 2,4-dicloro-3-((4-doro-6-dano-1-metil-1H-indol-2-il)metil)benzoico (Ejemplo DQ, Etapa 6 ) (90 mg, 0,23 mmol) y clorhidrato de metil (4-piperidil)acetato (66,4 mg, 0,34 mmol). LC/MS (Método i) Rt = 2,43 min.; MS m/z: 532 [M+H]+ 1H NMR (DMSO-d6 , 300MHz): ' 8,17 (m, 1H), 7,68 y 7,67 (d, J=8,4 Hz, 1H), 7,51 y 7,50 (s, 1H), 7,47 y 7,40 (d, J=8,4 Hz, 1H), 5,68 y 5,65 (m, 1H), 4,47 (m, 3H), 3,96 y 3,95 (s, 3H), 3,59 y 3,56 (s, 3H), 3,25 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,25 (m, 2H), 1,95 (m, 1H), 1,75 (m, 1H), 1,59 (m, 1H), 1,17 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-dicloro-3-((4-cloro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, se preparó ácido 2-(1-(2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (93 mg, 95%) a partir de metil 2-(1-(2,4-didoro-3-((4-doro-6-ciano-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (101 mg, 0,19 mmol). LC/MS (Método g) Rt = 1,63 min.; MS m/z: 518 [M+H]+1H NMR (DMSO-d6, 400MHz): 5 12,09 (m, 1H), 8,17 (s, 1H), 7,68 y 7,67 (d, J=8,4 Hz, 1H), 7,50 (m, 1H), 7,46 y 7,39 (d, J=8,4 Hz, 1H), 5,68 y 5,66 (m, 1H), 4,49 (m, 3H), 3,96 y 3,95 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,15 (m, 2H), 193 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,15 (m, 2H).
Ejemplo DS: ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: metil 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato

Usando un procedimiento similar al Ejemplo DH, Etapa 1, metil 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (1,55 g, 80%) se preparó a partir de 7-metil-5-(trifluorometil)-1H-indol (Preparación #49) (0,85 mg, 4,27 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (Preparación #1, Etapa B) (1,27 g, 0,27 mmol).
LC/MS (Método j) Rt = 2,32 min.; MS m/z: 416 [M+H]+ 1H NMR (DMSO-d6 , 300MHz): ' 7,89 (d, J=8,4 Hz, 1H), 7,79 (m, 1H), 7,76 (d, J=8,4 Hz, 1H), 7,23 (m, 1H), 6,74 (d, J=3,3 Hz, 1H), 6,56 (d, J=3,3 Hz, 1H), 5,98 (s, 2H), 3,88 (s, 3H), 2,94 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico

Usando un procedimiento similar al Ejemplo F, Etapa 3, ácido 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (1,44 g, 96 %) se preparó a partir de metil 2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (1,55 g, 3,72 mmol). LC/MS (Método j) Rt = 1,86 min.; MS m/z: 402 [M+H]+ 1H NMR (DMSO-d6, 300MHz): 5 13,8 (br, 1H), 7,82 (d, J=8,3 Hz, 1H), 7,79 (s, 1H), 7,69 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,73 (d, J=3,3 Hz, 1H), 6,56 (d, J=3,3 Hz, 1H), 5,97 (s, 2H), 2,95 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6 , metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (118 mg, 88%) se preparó a partir de ácido 2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (100 mg, 0,25 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (62 ,6 , 0,32 mmol). LC/MS (Método j) Rt = 2,10 min.; MS m/z: 541 [M+H]+
1H NMR (DMSO-d6, 300MHz): 5 7,79 (s, 1H), 7,70 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,76 (m, 1H), 6,57 (m, 1H), 5,94 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,21 (m, 1H), 3,03 (m, 1H), 2,94 (s, 3H), 2,80 (m, 1H), 2,28 (m, 2H), 1,96 (m, 1H), 1,75 (m, 1H), 1,59 (m, 1H), 1,17 (m, 2H)
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (99 mg, 89%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (114 mg, 0,21 mmol).
LC/MS (Método g) Rt = 1,85 min.; MS m/z: 527 [M+H]+1H NMR (DMSO-da, 400MHz): 512,10 (br, 1H), 7,79 (s, 1H), 7,70 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,76 (m, 1H), 6,58 (m, 1H), 5,94 (m, 2H), 4,46 (m, 1H), 3,27 (m, 1H), 3,03 (m, 1h), 2,94 (s, 3H), 2,80 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,14 (m, 2H).
Ejemplo DT: (4,6-didoro-5-((1,4-dimetil-6-(tnfluorometil)-1H-pirrolo[2,3-to]pindm-2-il)metil)pmdm-3-il)(4-hidroxipiperidin-1-il)metanona

Etapa 1: 4-metil-6-(trifluorometil)piridin-2(1H)-ona

Una solución de CHCl3 (1,25 L) se enfrió a aproximadamente 0 °C y se añadió anhídrido 2,2,2-trifluoroacético (0,141 L, 1012 mmol) seguido de la adición de cloruro de 3-metilbut-2-enoilo (0,094 L, 843 mmol). Se añadió gota a gota trietilamina (0,259 L, 1856 mmol) de modo que la temperatura interna se mantuvo por debajo de aproximadamente 10 °C (exotérmica). Después de la adición, la mezcla se agitó a aproximadamente 0 °C durante aproximadamente 1 hora y luego se calentó lentamente a temperatura ambiente y se agitó durante la noche. La mezcla de reacción se lavó con agua (1 L), NaHCO3 saturado (1 L), agua/salmuera (1:1,500 mL), se secó sobre MgSO4 , se filtró y se concentró al vacío. El residuo se disolvió en ácido acético (1,250 L) y se añadió ácido acético/sal de amoniaco (130 g, 1687 mmol). El matraz de reacción se tapó con un globo y la mezcla se calentó a aproximadamente 115 °C durante la noche. Después de aproximadamente 20 h, la mezcla de reacción se enfrió a aproximadamente 40 °C y se concentró al vacío. El jarabe se vertió en agua (~ 2 L) con agitación. Después de aproximadamente 1 h, los sólidos se recogieron por filtración para proporcionar 4-metil-6(trifluorometil)piridin-2(1H)-ona (101 g, 68%) como un sólido color canela. LC/MS (Método a) Rt = 1,59 min.; MS miz: 178 [M+H]+.1H NMR (CDCla, 400 MHz): 56,79 (s, 1H), 6,70 (s, 1H), 2,34 (s, 3H).
Etapa 2: 2-doro-4-metil-6-(trifluorometil)piridina

Un matraz de 50 mL en forma de pera equipado con un cabezal de destilación de recorrido corto se cargó con 4-metil-6-(trifluorometil)piridin-2(1H)-ona (20 g, 113 mmol). Al matraz se añadió dicloruro fenilfosfónico (19,2 mL, 135 mmol). La mezcla de reacción se calentó a aproximadamente 160 °C. Después de aproximadamente 3 horas, la presión sobre la mezcla de reacción se redujo lentamente a 100 mBar y la destilación comienza con una temperatura del cabezal de aproximadamente 120-130 °C. Periódicamente, cuando la destilación había disminuido significativamente, la presión sobre la mezcla de reacción se redujo a 80 mBar y la temperatura del baño se elevó a aproximadamente 170 °C. Después de aproximadamente 3 horas, la destilación había cesado, proporcionando 2-cloro-4-metil-6-(trifluorometil)piridina (12 g, 54%). LC/MS (Método a) Rt = 2,4 min.; MS miz: 196 [M+H]+.1H NMR (DMSO-Ce, 400 MHz): 7,79 (s, 1 h), 7,71 (s, 1H), 2,43 (s, 3 H).
Etapa 3: íerc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato

Un matraz cargado con 2-cloro-4-metil-6-(trifluorometil)piridina (35,1 g, 180 mmol), carbamato de íerc-butilo (42,1 g, 359 mmol), Pd2 (dba) 3 (4,11 g, 4,49 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (X-Phos) (4,28 g, 8,98 mmol) y carbonato de cesio (205 g, 629 mmol) se evacuó y se llenó con N2 (repetido 3 veces) antes de la adición de 1,4-dioxano desgasificado (350 mL). La mezcla se calentó luego a aproximadamente 80 °C durante aproximadamente 2 h. La mezcla de reacción se enfrió a temperatura ambiente y la mezcla de reacción se repartió entre agua y EtOAc. La capa acuosa se extrajo adicionalmente con EtOAc (2 x 100 mL). La capa orgánica se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad para dar un aceite de color rojo oscuro. El material se usó sin purificación adicional, 100% de rendimiento. LC/MS (Método a) Rt = 2,71 min.; MS miz: 275 [M+H]+.1H NMR (400 MHz, DMSO-C6) 510,13 (s, 1H), 7,90 (dt, J = 1,3, 0,7 Hz, 1H), 7,34 (dd, J = 1,3, 0,7 Hz, 1H), 2,38 (t, J = 0,6 Hz, 3H), 1,45 (s, 9H).
Etapa 4: 4-metil-6-(trifluorometil)piridin-2-amina
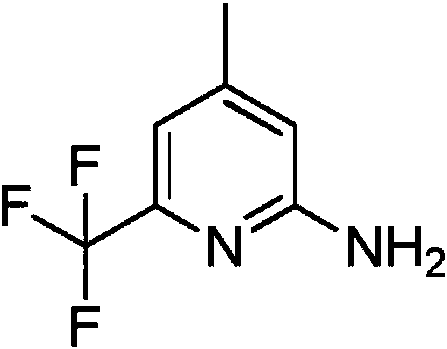
Una mezcla de íerc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato (49,6 g, 180 mmol) y cloruro de hidrógeno (4M en 1,4-dioxano) (597 mL, 2388 mmol) se agitó a temperatura ambiente durante aproximadamente 2 h. El disolvente se eliminó a presión reducida y el residuo se filtró. El material insoluble se lavó con EtOAc y el filtrado se diluyó con EtOAc y se extrajo con HCl 6 N (3x). La capa acuosa se lavó con DCM (4 x 100 mL) y después se ajustó a pH 8 con la adición de Na2 CO3 lentamente. El sólido de color amarillo brillante se recogió por filtración y se lavó con agua. Después, el sólido se disolvió en DCM, se lavó con salmuera, se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con éter de petróleo y se recogió por filtración, se lavó con éter de petróleo y se secó al aire para dar 4-metil-6-(trifluorometil)piridin-2-amina (27,7 g, 88%) como un sólido de color amarillo pálido. LC/MS (Método a) Rt = 1,92 min.; MS miz: 177 [M+H]+.1H NMR (400 MHz, DMSO-C6) 56,72 (s, 1 H), 6,45 (s, 1 h), 6,35 (s, 2 H), 2,20 (s, 3 H).
Etapa 5: 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina

Una mezcla de 4-metil-6-(trifluorometil)piridin-2-amina (31,4 g, 179 mmol) y W-bromosuccinimida (66,7 g, 375 mmol) en ACN (250 mL) se calentó a aproximadamente 70 °C durante aproximadamente 2 h. Al matraz de reacción se añadió agua (~500 mL). El sólido resultante se recogió por filtración, se lavó con agua y se secó al aire durante aproximadamente 5 minutos. El sólido se solubilizó en DCM, se lavó con agua, se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con heptano y se recogió por filtración, se lavó con heptano y se secó para dar 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,9 g, 96%) como un sólido de color amarillo claro. LC/MS (Método a) Rt = 2,55 min.; MS m/z: 333 [M+H]+.1H NMR (400 MHz, DMSO-C6 ) 56,90 (s, 2H), 2,53 (s, 3H).
Etapa 6: 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina

A una solución de color naranja clara de 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,98 g, 171 mmol) en THF (570 mL) a aproximadamente -78 °C se añadió n-butil litio (2,5 M en hexano) (68,3 mL, 171 mmol) gota a gota (convertido en una solución marrón). La mezcla se agitó a aproximadamente -78 °C durante aproximadamente 45 minutos. LC/MS indicó solo una conversión parcial y se añadió gota a gota una segunda porción de BuLi (2,5 M en hexano) (20,48 mL, 51,2 mmol). Se añadió gota a gota una tercera porción de BuLi (2,5 M en hexano) (13,65 mL, 34,1 mmol) para completar la conversión al producto. El matraz de reacción se transfirió a un baño de agua con hielo y se añadieron rápidamente 20 mL de agua. La mezcla de reacción se calentó luego a temperatura ambiente. La mezcla de reacción se repartió entre NH4 Cl saturado y EtOAc. La capa orgánica se lavó con NaHCO3 , se secó sobre Na2 SO4 , se filtró y se concentró casi hasta la sequedad. El sólido se trituró con heptano y se recogió por filtración, se lavó con hepano para dar la primera cosecha de producto como un sólido de color blanquecino (38,9 g, 85%). El filtrado se concentró hasta la sequedad y se añadió heptano. El sólido se recogió por filtración al vacío y se lavó con heptano y se secó para dar la segunda cosecha de producto como un sólido de color amarillo pálido (1,9 g, 3%). LC/MS (Método a) Rt = 2,29 min.; MS m/z: 255 [M+H]+.1H NMR (400 MHz, DMSO-C6 ) 56,96 (s, 1 H), 6,74 (s, 1 H), 2,32 (s, 3 H).
Etapa 7: (E)-2-(2-EtoxivmM)-4,4,5,5-tetrametiM,3,2-dioxaborolano

Bajo una atmósfera de nitrógeno en un matraz de fondo redondo, etoxietileno, una solución al 50% en hexanos (100 g, 710 mmol) y DCM (996 mL) se agitaron a aproximadamente 0-5 °C. A la solución agitada se añadió 4,4,5,5-tetrametil-1,3,2-dioxaborolano (113 mL, 781 mmol) en una porción seguido de la adición de clorhidrato de bis(ciclopentadienil)circonio (9,16 g, 35,5 mmol) a aproximadamente 0-5 °C. La suspensión (naranja) se dejó calentar gradualmente a temperatura ambiente durante aproximadamente 30 minutos. La disolución ocurrió dentro de aproximadamente 10 min. La mezcla de reacción se agitó a temperatura ambiente durante la noche (rojo muy oscuro). A la solución de reacción se añadió éter (2L) y la solución se lavó con solución saturada acuosa de NH4 CL Los disolventes se eliminaron a presión reducida, se añadió DCM (100 mL) y la solución se filtró a través de una almohadilla de alúmina, cubierta con Celite®. La alúmina se lavó con diclorometano y el disolvente del filtrado se eliminó al vacío para producir (E)-2-(2-etoxivinil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (114,9 g, 82 %) como un aceite rojo muy oscuro.1H nMr (400 MHz, DMSO-C6 ) 57,05 (d, 1 H), 4,45 (d, 1 H), 3,83 (q, 2 H), 1,30 (t, 3 H), 1,28 (s, 12 H).
Etapa 8: (E)-3-(2-etoxivmM)-4-metM-6-(tnfluorometM)pmdm-2-amma

Un matraz cargado con 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina (38,3 g, 143 mmol), (E)-2-(2-etoxivinil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (56,5 g, 285 mmol), diacetoxipaladio (0,960 g, 4,28 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (XPhos) (4,28 g, 8,98 mmol) y carbonato de cesio (116 g, 356 mmol) se desgasificó con N2 durante 15 min antes de la adición de 320 mL de 1,4-dioxano desgasificado/H2O (4:1). La mezcla se calentó a aproximadamente 80 °C durante aproximadamente 2 horas. La mezcla se enfrió a temperatura ambiente. La mezcla de reacción se repartió entre EtOAc y agua y la capa acuosa se extrajo adicionalmente con EtOAc (2 x 50 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Al residuo se añadió heptano y el sólido resultante se recogió por filtración y se lavó con heptano para dar un sólido de color blanquecino (1,45 g). El filtrado se concentró hasta la sequedad nuevamente para dar un aceite negro espeso que se dejó a temperatura ambiente durante la noche. Se observó una formación significativa de sólidos y el sólido se diluyó con heptano, se sonicó y se filtró, se lavó con heptano para dar un sólido marrón claro (15,2 g, 42%). El filtrado se concentró hasta la sequedad para dar un aceite negro que se purificó por cromatografía ultrarrápida (EtOAc 0-25%/heptano durante 30 minutos). El producto que contenía fracciones se concentró casi hasta la sequedad. El sólido se recogió por filtración, se lavó con una pequeña cantidad de heptano y se secó para dar la segunda cosecha de producto como un sólido de color amarillo claro (12,7 g, 35%). LC/MS (Método l) Rt = 1,48 min.; MS m/z: 247 [M+H]+. 1H NMR (400 MHz, DMSO-d6) 56,82 (s, 1H), 6,73 (d, J = 13,1 Hz, 1H), 6,12 (s, 2H), 5,45 (d, J = 13,1 Hz, 1H), 3,93 (q, J = 7,0 Hz, 2H), 2,23 (s, 3H), 1,25 (t, J = 7,0 Hz, 3H).
Etapa 9: 4-metil-6-(tnfluorometil)-1H-pirrolo[2,3-to]pmdma

Una mezcla de (E)-3-(2-etoxivinil)-4-metil-6-(trifluorometil)piridin-2-amina (27,9 g, 113 mmol) y ácido acético (130 mL) se calentó a aproximadamente 100 °C durante la noche. La mezcla se enfrió a temperatura ambiente y el precipitado se recogió por filtración, se lavó con ACN y se secó para dar la primera cosecha de producto. El filtrado se concentró hasta ~ 50 mL y el sólido se recogió por filtración, se lavó con ACN y se secó para dar la segunda cosecha de producto. Ambos cultivos se combinaron. El filtrado todavía tenía algo de producto y se dejó a un lado. El sólido se suspendió en ~ 250 ml de EtOAc y se calentó a reflujo para disolverse. La mayor parte del disolvente se eliminó a presión reducida y se añadió heptano. El sólido se recogió por filtración al vacío, se secó para dar el producto como un sólido de color blanquecino (19,8 g, 87%). LC/MS (Método l) Rt = 1,30 min.; MS m/z: 201 [M+H]+. 1H NMR (400 MHz, DMSO-d6) 5 12,13-11,98 (br, 1H), 7,67 (t, J = 3,1 Hz, 1H), 7,34 (d, J = 1,0 Hz, 1H), 6,63 (dd, J = 3,5, 1,9 Hz, 1H), 2,59 (s, 3H).
Etapa 10: 4-metiM-(femlsulfoml)-6-(trifluorometil)-1H-pirrolo[2,3-to]pmdma

A una suspensión de hidruro de sodio (60% en aceite mineral) (4,35 g, 109 mmol) en N ,N - dimetilformamida (40 mL) a aproximadamente 0 °C se añadió una solución de 4-metil-6-(trifluorometil)-1H-pirrolo[2,3-í)]piridina (19,8g, 99 mmol) bajo N2. Después de la adición, se retiró el baño de agua con hielo y la mezcla se agitó a temperatura ambiente durante aproximadamente 30 minutos. Se enfrió nuevamente a aproximadamente 0 °C, se añadió gota a gota cloruro de bencenosulfonilo (13,27 mL, 104 mmol). Después de la adición, la reacción se dejó calentar a temperatura ambiente y se agitó durante aproximadamente 1 h. La reacción se inactivó con la adición de 100 mL de solución saturada de NH4Cl seguido de la adición de 350 mL de agua. El sólido se recogió por filtración, se lavó con agua y se secó en un horno de vacío a aproximadamente 70 °C durante aproximadamente 2 días para dar un sólido de color blanquecino. El sólido se
disolvió en EtOAc, se filtró a través de 70 g de gel de sílice y la almohadilla se lavó con EtOAc. El filtrado se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. El sólido se trituró con EtOAc/heptano para dar la primera cosecha de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridina (28,5 g, 85 %) como un sólido de color blanco. El filtrado se concentró hasta la sequedad y el sólido se trituró con éter/hepatano. El sólido resultante se recogió por filtración, se lavó con heptano y se secó para dar la segunda cosecha de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-6 ]piridina (3,65 g, 11 %) como un sólido de color blanquecino. LC/MS (Método l) Rt = 1,84 min.; MS m/z: 341 [M+H]+. 1H NMR (400 MHz, DMSO-C6) 5 8,15-8,07 (m, 3H), 7,75-7,67 (m, 1H), 7,64-7,56 (m, 3H), 7,03 (d, J = 4,1 Hz, 1H), 2,57 (s, 3H).
Etapa 11: íerc-butil 4,6-didoronicotinato

Una mezcla de ácido 4,6-dicloronicotínico (8,2 g, 42,7 mmol) y di-íerc-butilo dicarbonato (19,83 mL, 85 mmol) en THF (100 mL) se agitó a temperatura ambiente, luego a la solución se añadió 4-(W,W-dimetilamino)piridina (1,044 g, 8,54 mmol). La mezcla resultante se agitó a aproximadamente 70 °C durante aproximadamente 1 h. La muestra se depositó sobre gel de sílice y se cargó en una columna de gel de sílice y se eluyó con EtOAc al 5%/heptano. Las siguientes fracciones se recogieron para dar íerc-butil 4,6-dicloronicotinato (10 g, 94 %) como un aceite incoloro. 1H NMR (400 MHz, CDCh) 58,76 (s, 1 H), 7,43 (s, 1 H), 1,61 (s, 9 H).
Etapa 12: íerc-butil 4,6-didoro-5-formilnicotinato

El íerc-butil 4,6-dicloromcotinato (10 g, 40,3 mmol) se disolvió en THF (100 mL), se agitó y se enfrió a aproximadamente -78 °C. A la solución se añadió diisopropilamida de litio (22,2 mL, 44,3 mmol) a una velocidad para mantener la temperatura por debajo de aproximadamente -70 °C. La solución resultante se agitó a aproximadamente -78 °C durante aproximadamente 10 minutos. Luego se añadió formiato de metilo (4,94 mL, 81 mmol) a la solución y la mezcla se agitó a aproximadamente -78 °C durante aproximadamente 30 minutos. La mezcla se vertió en NH4Cl saturado, se extrajo con EtOAc (3x) y las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron para proporcionar un aceite de color marrón. La muestra se depositó sobre gel de sílice y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 2%/heptano. Se recogieron las siguientes fracciones para dar íerc-butil 4,6-dicloro-5-formilnicotinato (5,0 g, 45% de rendimiento) como un sólido amarillo. 1H NMR (400 MHz, CDC 13 ) 5 10,47 (s, 1 H), 8,7 (s, 1 H), 1,65 (s, 9 H).
Etapa 13: tere- butil 4,6-didoro-5-(hidroxi(4-metiM-(fenilsulfoml)-6-(trifluorometil)-1H-pirrolo[2,3-fo]pi- ridin-2-il)metil)nicotinato

Usando un procedimiento similar al del Ejemplo CL, Etapa 1, íerc-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinatoe se preparó a partir de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridina (6,0 g, 17,6 mmol) y íerc-butil 4,6-dicloro-5-formilnicotinato (5,35 g, 19,4 mmol). LC/MS (Método l) Rt = 2,15 min.; MS m/z: 616 [M+H]+.1H NMR (400 MHz, DMSO-C6) 58,67 (s, 1 H), 7,95 (d, 2 H), 7,69 (m, 1 H), 7,61 (s, 1 H), 7,55 (m, 2 H), 7,05 (s, 1 H), 7,04 (d, 1 H), 6,92 (d, 1 H), 2,54 (s, 3 h), 1,54 (s, 9 H).
Etapa 14: tere- butil 4,6-didoro-5-((4-metiM-(femlsulfoml)-6-(tnfluorometil)-1H-pirrolo[2,3-to]piridm-2-il)metil)nicotinato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, íerc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato se preparó a partir de íerc-butil 4,6-dicloro-5-(hidroxi(4-me-til-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotinato (1,51 g, 2,44 mmol). LC/MS (Método l) Rt = 2,33 min.; MS m/z: 600 [M+H]+.
Etapa 15: ácido 4,6-didoro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridm-2-il)metil)mcotmico

A una solución de íerc-butil 4,6-dicloro-5-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3Hb]pi-ridin-2-il)metil)nicotinato (334,9 mg, 0,558 mmol) en 1,4-dioxano (7339 pL) se añadió hidróxido de sodio (2M) (1394 pL, 2,79 mmol). La mezcla se calentó a aproximadamente 50 °C durante la noche. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con agua y se acidificó con HCl (1M). La mezcla se extrajo dos veces con EtOAc y los extractos orgánicos combinados se secaron sobre Na2 SO4 , se filtraron y se evaporaron para dar ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico (0,23 g, 100%) como un sólido de color marrón pálido. LC/MS (Método l) Rt = 1,02 min.; MS m/z: 404 [M+H]+.1H NMR (400 MHz, DMSO-d6) 512,16 (s, 1H), 8,75 (s, 1H), 7,29 (d, J = 0,8 Hz, 1H), 6,09 (dd, J = 2,1, 1,0 Hz, 1H), 4,48 (s, 3H).
Etapa 16: metil 4,6-didoro-5-((1,4-dimetil-6-(tnfluorometil)-1H-pirrolo[2,3-to]pmdm-2-il)metil)mcotmato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pir-rolo[2,3-b]piridin-2-il)metil)nicotinato se preparó a partir de ácido 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotínico (0,23 g, 0,567 mmol) LC/MS (Método l) Rt = 2,01 min.; MS miz: 432 [M+H]+.
Etapa 17: ácido 4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pin'olo[2,3-fo]piridm-2-il)metil) nicotínico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-6]piridin-2-il)metil)nicotmico se preparó a partir de metil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1 H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (0,25 g, 0,578 mmol) LC/MS (Método l) Rt = 1,14 min.; MS miz: 418 [M+H]+.
Etapa 18: (4,6-didoro-5-((1,4-dimetil-6-(trifluorometM)-1H-pin'olo[2,3-fo]piridm-2-M)metil)pmdm-3-il)(4-hidroxipiperidin-1-il)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6 , (4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)piridin-3-il)(4-hidroxipiperidin-1-il)metanona se preparó a partir de ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-ib]piridin-2-il)metil)nicotínico (0,116 g, 0,278 mmol) y piperidin-4-ol (0,042 g, 0,42 mmol). LC/MS (Método a) Rt = 2,28 min.; MS miz: 501 [M+H]+. 1H NMR (400 MHz, DMSO-d6 ) 58,31 (s, 1 H), 7,26 (s, 1 H), 7,22 (s, 1 H), 5,81 (s, 1 H), 4,46 (m, 2 H), 4,15 (m, 1 H), 4,04 (m, 1 H), 3,98 (s, 3 H), 3,65-3,45 (m, 3 H), 3,13 (m, 2 H), 2,00 (m, 1 H), 1,87 (m, 2 H), 1,70 (m, 2 H).
Ejemplo DU: ácido 1-[2,4-didoro-3-[[1,4-dimetN-6-(trifluorometoxi)-1H-mdol-3-M]metM]benzoN]piperidma-4-carboxílico

Etapa 1: íerc-butil 2,4-didoro-3-[[4-metN-6-(trifluorometoxi)-1H-mdol-3-M]metM]benzoato

Una solución de 4-metil-6-(trifluorometoxi)-1H-indol (200 mg, 0,93 mmol) (Preparación 80) y íerc-butil 2,4-dicloro-3-formilbenzoato (281 mg, 1,02 mmol) (Preparación #33, Etapa B) en DCM (3 mL) se añadió gota a gota a una solución fría (baño de hielo) de DCM (1,25 mL) de 2,2,2-TFA (0,11 mL, 1,39 mmol) y trietilsilano (0,44 mL, 2,79 mmol) en un matraz de fondo redondo de 5 mL bajo nitrógeno, manteniendo la temperatura por debajo de 5 °C (coloración amarilla y luego marrón). Después de 20 minutos 0 °C, la mezcla de reacción se agitó 2 horas a temperatura ambiente. La mezcla de reacción se diluyó con DCM, se lavó con agua, luego con una solución acuosa saturada de NaHCO3 y finalmente con salmuera. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. La resina marrón obtenida se cromatografió sobre gel de sílice (columna de 15 g, eluyente ciclohexano/EtOAc 95: 5 a 80/20) para dar hidruro de íerc-butil 2,4-dicloro sodio (42,1 mg, 1,054 mmol), se añadió en porciones -6-(trifluorometoxi)-1H-indol-3-il)metil)benzoato (266 mg, 56%) como una resina incolora. LC/MS (Método j): Rt = 2,69 min; MS m/z: 474 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 5 10,96 (m, 1H), 7,63 (s, 2H), 7,13 (s, 1H), 6,77 (s, 1H), 6,32 (m, 1H), 4,59 (s, 2H), 2,77 (s, 3H), 1,55 (s, 9H).
Etapa 2: íerc-butil 2,4-didoro-3-[[1,4-dimetM-6-(trifluorometoxi)-1H-mdol-3-M]metM]benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 4, íerc-butil 2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoato (383 mg, 74%) se preparó a partir de íerc-butil 2,4-dicloro-3-((4-metil-6-(trifluoromet-oxi)-1H-indol-3-il)metil)benzoato (476 mg, 1,0 mmol). LC/MS (Método j): Rt = 2,90 min; MS miz: 488 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,64 (s, 2H), 7,23 (s, 1H), 6,81 (s, 1H), 6,39 (s, 1H), 4,59 (s, 2H), 3,62 (s, 3H), 2,78 (s, 3H), 1,55 (s, 9H).
Etapa 3: ácido 2,4-didoro-3-[[1,4-dimetN-6-(trifluorometoxi)-1H-mdol-3-M]metM]benzoico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1 H-indol-3-il)metil)benzoico como un sólido de color marrón (335 mg, 99%) se preparó a partir de íerc-butil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il)metil)benzoato (381mg, 0,78 mmol). Lc/MS (Método j): Rt = 2,01 min; MS miz: 432 [M+H]+; 1H NMR ((DMSO-de, 300MHz): 513,58 (ancho, 1H), 7,71 (d, J=8,4 Hz, 1h), 7,64 (d, J=8,4 Hz, 1H), 7,23 (s, 1H), 6,81 (s, 1H), 6,37 (s, 1H), 4,60 (s, 2H), 3,61 (s, 3H), 2,78 (s, 3H).
Etapa 4: metil 1-[2,4-didoro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoil]piperidina-4-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, 1-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoil]piperidina-4-carboxilato (104 mg, 63%) se preparó a partir de ácido 2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoico (125 mg) y clorhidrato de metil piperidina-4-carboxilato (67 mg, 0,38 mmol). LC/MS (Método i): Rt = 2,74 min; MS miz: 557 [M+H]+; 1H NMR ((DMSO-de, 300MHz): 57,62 (d, J=8,3 Hz, 1H), 7,41 y
7,34 (d, J=8,3 Hz, 1H), 7,23 (s, 1H), 6,80 (s, 1H), 6,40 y 6,38 (s, 1H), 4,57 (m, 2H), 4,35 (m, 1H), 3,60 (m, 6H), 3,33 (m, 1H), 3,03 (m, 2H), 2,77 (s, 3H), 2,68 (m, 1H), 1,99 (m, 1H), 1,78 (m, 1H), 1,53 (m, 2H).
Etapa 5: ácido 1-[2,4-didoro-3-[[1,4-dimetN-6-(trifluorometoxi)-1H-mdol-3-M]metM]benzoN]piperidma-4-carboxNico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoil]piperidina-4-carboxílico (85 mg, 83%) se preparó a partir de metil 1-[2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]benzoil]piperidina-4-carboxilato (103 mg, 0,18 mmol). LC/MS (Método g): Rt = 1,93 min; MS m/z: 543 [M+H]+; 1H NMR ((DMSO-cfe 400MHz): 5 = 7,63 (d, J=8,4 Hz, 1H), 7,40 y 7,34 (d, J=8,4 Hz, 1H), 7,23 (s, 1H), 6,81 (s, 1H), 6,39 y 6,36 (s, 1H), 4,57 (m, 2H), 4,35 (m, 1H), 3,62 y 3,61 (s, 3H), 3,32 (m, 1H), 3,08 (m, 1H), 2,95 (m, 1h), 2,77 (s, 3h), 2,55 (m, 1h), 1,94 (m, 1H), 1,79 (m, 1H), 1,50 (m, 2H).
Tabla DU. Los siguientes intermedios se prepararon a partir de ácido 2,4-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1 H-indol-3-il]metil] benzoico (Ejemplo Du, Etapa 5), usando el mismo procedimiento con la amina apropiada.

Tabla DU1. Los siguientes Ejemplos se prepararon usando el mismo procedimiento que en el Ejemplo A, Etapa 5, partiendo de los ésteres apropiados (de la Tabla DU).

Ejemplo DV: ácido 2-[1-[4,6-didoro-5-[[1,4-dimetM-6-(trifluorometN)-1H-mdol-3-N]metM]piridma-3-carboml]-4-piperidil]acético
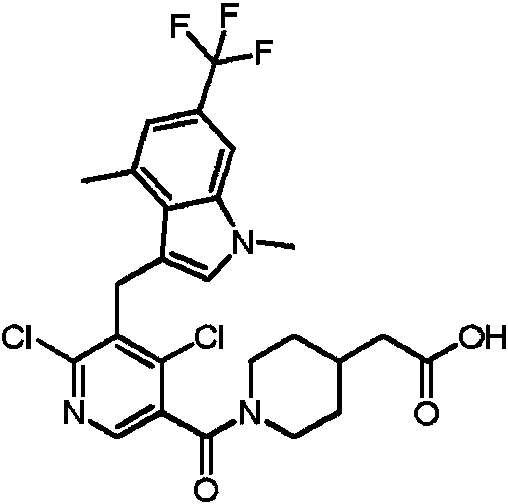
Etapa 1: ferc-butil 4,6-didoro-5-[[4-metM-6-(trifluorometN)-1H-mdol-3-N]metM] piridina-3-carboxilato

Usando un procedimiento similar al del Ejemplo DU Etapa 1, se obtuvo ferc-butil 4,6-didoro-5-[[4-metil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxilato (544 mg, 79%) a partir de 4-metil-6-(trifluorometil)-1H-indol (250 mg, 1,255 mmol) (Preparación #51, Etapa C) y ferc-butil 4,6-dicloro-5-formyl-piridina-3-carboxilato (381 mg, 1,38 mmol) (Ejemplo DO, Etapa
12). LC/MS (Método i): Rt = 2,89 min; MS m/z: 459 [M+H]+; 1H NMR ((DMSO-de, 300MHz): 5 = 11,26 (s, 1H), 8,71 (s, 1H), 7,51 (s, 1H), 7,05 (s, 1H), 6,68 (m, 1H), 4,62 (s, 2h), 2,82 (s, 3H), 1,57 (s, 9H).
Etapa 2: íerc-butil 4,6-didoro-5-[[1,4-dimetM-6-(trifluorometN)-1H-mdol-3-M]metM]piridma-3-carboxNato

Usando un procedimiento similar al Ejemplo A, Etapa 4" se obtuvo íerc-butil 4,6-didoro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxilato (353 mg, 70%) a partir de íerc-butil 4,6-didoro-5-[[4-metil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxilato (490 mg, 1,07 mmol) y yoduro de metilo (80 |_il, 1,28 mmol).
LC/MS (Método j): Rt = 2,67 min; MS m/z: 473 [M+H]+; 1H NMR ((DMSO-de, 300MHz): 58,73 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,74 (s, 1H), 4,61 (s, 2H), 3,70 (s, 3H), 2,82 (s, 3H), 1,57 (s, 9H).
Etapa 3: ácido 4,6-didoro-5-[[1,4-dimetN-6-(trifluorometM)-1H-mdol-3-N]metN]pmdma-3-carboxNico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, se obtuvo ácido 4,6-didoro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxílico (281 mg, 91%) a partir de íerc-butil 4,6-didoro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxilato (350 mg, 0,739 mmol). LC/MS (Método j): Rt = 1,63 min; MS m/z: 417 [M+H]+; 1H NMR ((DMSO-de, 300MHz): 513,99 (ancho, 1H), 8,76 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,71 (s, 1H), 4,62 (s, 2H), 3,70 (s, 3H), 2,83 (s, 3H).
Etapa 4: metil
piperidil]acetato 
Usando un procedimiento similar al Ejemplo A, Etapa 6, se obtuvo metil 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acetato (87 mg, 73%) a partir de ácido 4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carboxflico (90 mg, 0,216 mmol) y clorhidrato metil 2-(4-piperidil)acetato (46 mg, 0,237 mmol). LC/MS (Método j): Rt=2,01 min; MS m/z: 556 [M+H]+; 1H NMR ((DMSO-Ce, 300MHz): 58,46 y 8,39 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,76 y 6,71 (s, 1h), 4,50 (m, 3H), 3,71 (s, 3H), 3,60 y 3,57 (s, 3H), 3,44 (m, 1H), 3,07 (m, 1h), 2,82 (m, 4h), 2,29 (m, 2H), 1,98 (m, 1H), 1,77 (m, 1H), 1,62 (br s, 1H), 1,17 (m, 2H).
Etapa 5: ácido 2-[1-[4,6-didoro-5-[[1,4-dimetM-6-(trifluorometN)-1H-mdol-3-N]metN]piridma-3-carboml]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, se obtuvo ácido 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acético (71 mg, 83%) a partir de metil 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acetato (85 mg, 0,15 mmol). Lc/MS (Método g): Rt= 1,82 min; MS m/z: 542 [M+H]+ 1H NMR ((DMSO-Ce, 400MHz): 58,45 y 8,39 (s, 1h), 7,61 (s, 1H), 7,09 (s, 1H), 6,75 y 7,71 (s, 1H), 4,54 (m, 3H), 3,71 (s, 3H), 3,40 (m, 1H), 3,06 (m, 1H), 2,82 (s, 4H), 2,15 (m, 2H), 1,93 (m, 1H), 1,79 (m, 1H), 1,64 (m, 1h), 1,14 (m, 2H).
Ejemplo DW: ácido 2-[1-[3,5-didoro-4-[[1,4-dimetM-6-(trifluorometN)-1H-mdol-3-N]metN]piridma-2-carboml]-piperidil]acético

Etapa 1: etil 3,5-dicloro-4-[[4-metil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxilato

Usando un procedimiento similar al del Ejemplo DU Etapa 1, se obtuvo etil 3,5-didoro-4-[[4-metil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxilato (444 mg, 68%) a partir de 4-metil-6-(trifluorometil)-1H-indol (250 mg, 1,255 mmol) (Preparación #51, Etapa C) y etil 3,5-dicloro-4-formil-piridina-2-carboxilato (343 mg, 1,381 mmol) (Preparación #37, Etapa D). LC/MS (Método i): Rt = 2,67 min; MS m/z: 431 [M+H]+; 1H NMR ((DMSO-Ce, 300MHz): 511,28 (s, 1H), 8,75 (s, 1H), 7,52 (s, 1H), 7,06 (s, 1H), 6,66 (m, 1H), 4,64 (s, 2H), 4,40 (q, J=7,1 Hz, 2H), 2,82 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 2: etil 3,5-didoro-4-[[1,4-dimetM-6-(tnfluorometM)-1H-mdol-3-M]metM]pmdma-2-carboxMato

Usando un procedimiento similar al Ejemplo A, Etapa 4, se obtuvo etil 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1 H-indol-3-il]metil]piridina-2-carboxilato (190 mg, 47%) a partir de etil 3,5-dicloro-4-[[4-metil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxilato (390 mg, 0,90 mmol) y yoduro de metilo (79 |_il, 1,27 mmol). LC/MS (Método j): Rt=2,32 min; MS m/z: 445 [M+H]+; 1H NMR ((DMSO-Ce, 300MHz): 58,76 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,72 (s, 1H), 4,63 (s, 2H), 4,40 (q, J=7,1 Hz, 2H), 3,70 (s, 3H), 2,82 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 3: ácido 3,5-didoro-4-[[1,4-dimetN-6-(trifluorometM)-1H-mdol-3-N]metN]pmdma-2-carboxNico
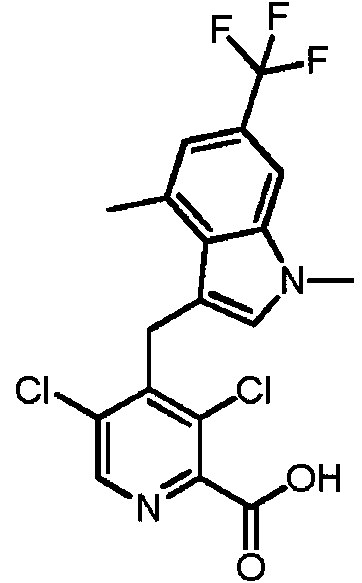
Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxílico (195 mg, 100% de rendimiento, sólido de color naranja) se preparó a partir de etil 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxilato (190 mg, 0,427 mmol). LC/Ms (Método j): Rt = 1,62 min; MS m/z: 417 [M+H]+; 1H NMR ((DMSO-Ce, 300MHz): 58,60 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,64 (s, 1H), 4,59 (s, 2H), 3,70 (s, 3H), 2,82 (s, 3H).
Etapa 4: metil 2-[1-[3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil] acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, se obtuvo metil 2-[1-[3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acetato (72 mg, 67%) a partir de ácido 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carboxílico (81 mg, 0,194 mmol) y clorhidrato de metil 2-(4-piperidil)acetato (53 mg, 0,272 mmol). LC/MS (Método j): Rt = 2,05 min; MS m/z: 556 [M+H]+; 1H NMR ((DMSO-Ce, 300MHz): 5 = 8,72 (s, 1H), 7,63 (s, 1H), 7,10 (s, 1H), 6,69 (s, 1H), 4,60 (s, 2H), 4,46 (m, 1H), 3,70 (s, 3H), 3,58 (s, 3H), 3,28 (m, 1H), 3,05 (m, 1h), 2,81 (m, 4h), 2,29 (m, 2H), 1,99 (m, 1H), 1,78 (m, 1H), 1,61 (m, 1H), 1,16 (m, 2H).
Etapa 5: ácido 2-[1-[3,5-didoro-4-[[1,4-dimetM-6-(trifluorometN)-1H-mdol-3-N]metN]piridma-2-carboml]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, se obtuvo ácido 2-[1-[3,5-didoro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acético (62 mg, 89%) a partir de metil 2-[1-[3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometil)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acetato (70 mg, 0,126 mmol). Lc/MS (Método g): Rt = 1,83 min; MS m/z: 542 [M+H]+1H NMR ((DMSO-Ce, 400MHz): 58,72 (s, 1H), 7,62 (s, 1H), 7,10 (s, 1H), 6,69 (s, 1H), 4,60 (s, 2H), 4,46 (m, 1H), 3,70 (s, 3H), 3,28 (m, 1H), 3,04 (m, 1H), 2,82 (s, 4H), 2,16 (m, 2H), 1,95 (m, 1H), 1,80 (m, 1H), 1,62 (m, 1h), 1,14 (m, 2H).
Ejemplo DX: ácido 2-[1-[3,5-didoro-4-[[1,4-dimetN-6-(trifluorometoxi)-1H-mdol-3-M]metM]piridma-2-carboml]-3-piperidil]acético
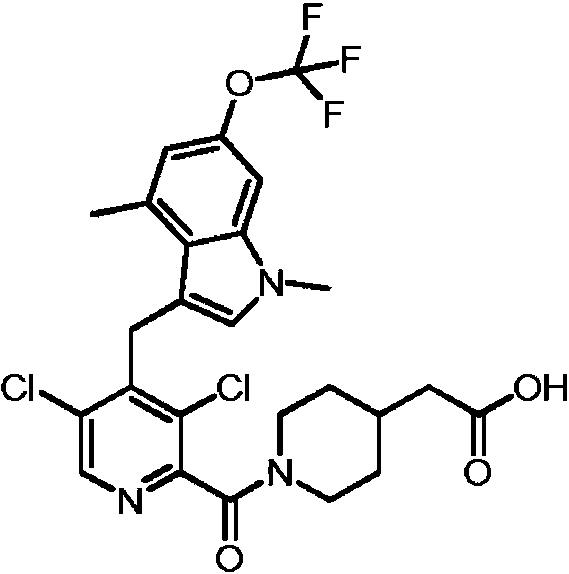
Etapa 1: etil 3,5-dicloro-4-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilato

Usando un procedimiento similar al del Ejemplo DU, Etapa 1, se obtuvo etil 3,5-dicloro-4-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilato (185 mg, 59%) a partir de 4-metil-6-(trifluorometoxi)-1H-indol (150 mg, 0,697 mmol, (Preparación 80) y etil 3,5-dicloro-4-formil-piridina-2-carboxilato (190 mg, 0,767 mmol) (Preparación #37, Etapa D). LC/MS (Método i): Rt = 2,68 min; MS m/z: 447 [M+H]+; 1H NMR ((DMSO-cfe 300MHz): 511,05 (s, 1H), 8,75 (s, 1H), 7,14 (s, 1H), 6,78 (s, 1H), 6,49 (s, 1H), 4,60 (s, 2H), 4,40 (q, J=7,1 Hz, 2H), 2,77 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 2: etil 3,5-didoro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 4, se obtuvo etil 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilato (210 mg, 79%) a partir de etil 3,5-dicloro-4-[[4-metil-6-(trifluoromet-oxi)-1H-indol-3-il]metil]piridina-2-carboxilato (185 mg, 0,41 mmol) y yoduro de metilo (26 pL, 0,41 mmol). LC/MS (Método i): Rt = 2,82 min; MS m/z: 461 [M+H]+; 1H NMR ((DMSO-cfe 300MHz): 5 = 8,76 (s, 1H), 7,25 (s, 1H), 6,82 (s, 1H), 6,56 (s, 1H), 4,60 (s, 2H), 4,40 (q, J=6,9 Hz, 2H), 3,62 (s, 3H), 2,73 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 3: ácido 3,5-dicloro-4-[[1,4-dimetN-6-(trífluorometoxi)-1H-mdol-3-N]metN]pirídma-2-carboxíNco

Usando un procedimiento similar al Ejemplo A, Etapa 5, sepreparó 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilic acid (180 mg, 84%) a partir de etil 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxilato (210 mg, 0,45 mmol). LC/MS (Método i): Rt = 2,26 min; MS m/z: 433 [M+H]+; 1H nMr ((DMSO- d6, 300MHz): 58,70 (s, 1H), 7,25 (s, 1H), 6,82 (m, 1H), 6,52 (s, 1H), 4,58 (s, 2H), 3,62 (s, 3H), 2,77 (s, 3H).
Etapa 4: metil 2-[1-[3,5-dicloro-4-[[1,4-dimetN-6-(trífluorometoxi)-1H-mdol-3-N]metN]pirídma-2-carbon-N]-4-piperidil]acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, se obtuvo metil 2-[1-[3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acetato (110 mg, 41%) a partir de ácido 3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carboxílico (180 mg, 0,416 mmol) y clorhidrato de metil 2-(4-piperidil)acetato (89 mg, 0,457 mmol).
LC/MS (Método i): Rt=2,65 min; MS m/z: 572 [M+H]+; 1H NMR ((DMSO-d6, 300MHz): 5 = 8,72 (s, 1H), 7,25 (s, 1H), 6,82 (s, 1H), 6,53 (s, 1H), 4,57 (s, 2H), 4,46 (m, 1H), 3,62 (s, 3H), 3,58 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,80 (m, 4H), 2,29 (m, 2H), 1,99 (m, 1H), 1,76 (m, 1H), 1,60 (m, 1H), 1,15 (m, 2H).
Etapa 5: ácido 2-[1-[3,5-didoro-4-[[1,4-dimetM-6-(tNfluorometoxi)-1H-mdol-3-N]metN]pindma-2-carboml]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, se obtuvo ácido 2-[1-[3,5-dicloro-4-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acético (44 mg, 80%) a partir de metil 2-[1-[3,5-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-2-carbonil]-4-piperidil]acetato (55 mg, 0,096 mmol). LC/MS (Método g): Rt= 1,86 min; MS m/z: 558 [M+H]+; 1H NMR ((DMSO-d6, 400MHz): 5 12,11 (s, 1H), 8,72 (s, 1H), 7,25 (s, 1H), 6,82 (s, 1H), 6,53 (s, 1H), 4,57 (s, 2H), 4,46 (m, 1H), 3,62 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,84 (m, 1H), 2,77 (m, 3H), 2,18 (m, 2H), 1,96 (m, 1H), 1,80 (m 1H), 1,63 (m, 1H), 1,15 (m, 2H).
Ejemplo DY: ácido 2-[1 -[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1 H-mdol-3-N]metN]pindma-3-carboml]-4-piperidil]acético

Etapa 1: ferc-butil 4,6-didoro-5-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato

Usando un procedimiento similar al del Ejemplo DU Etapa 1, ferc-butil 4,6-dicloro-5-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato (100 mg, 45%) se obtuvo a partir de 4-metil-6-(trifluorometoxi)-1H-indol (100 mg, 0,465 mmol) (Preparación 80) y ferc-butil 4,6-dicloro-5-formil-piridina-3-carboxilato (141 mg, 0,511 mmol) (Ejemplo DO, Etapa 12). LC/MS (Método i): Rt = 2,89 min; MS m/z: 475 [M+H]+; 1H NMR ((DMSO-d6, 300MHz): 5 11,03 (s, 1H), 8,70 (s, 1H), 7,13 (s, 1H), 6,77 (m, 1H), 6,80 (s, 1H), 4,58 (s, 2H), 2,77 (s, 3H), 1,57 (s, 9H).
Etapa 2: ferc-butil 4,6-didoro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 4, terc-butil 4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato (145 mg, 92% de rendimiento) se obtuvo a partir de terc-butil 4,6-dicloro-5-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato (135 mg, 0,28 mmol) y yoduro de metilo (18 pL, 0,284 mmol). LC/MS (Método j): Rt = 2,68 min; MS m/z: 489 [M+H]+; 1H NMR ((DMSO-ofe, 300MHz): 5 = 8,72 (s, 1H), 7,24 (s, 1H), 6,81 (s, 1H), 6,57 (s, 1H), 4,58 (s, 2h), 3,62 (s, 3H), 2,77 (s, 3H), 1,57 (s, 9H).
Etapa 3: ácido 4,6-dicloro-5-[[1,4-dimetN-6-(trifluorometoxi)-1H-indol-3-N]metN]piridina-3-carboxíNco

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, se obtuvo ácido 4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxílico (135 mg, 74 % de rendimiento) a partir de terc-butil 4,6-dicloro-5-[[4-metil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxilato (140 mg, 0,286 mmol). LC/MS (Método i): Rt = 2,32 min; MS m/z: 433 [M+H]+
Etapa 4: metil 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il] metil]piridina-3-carbon-il] -4-piperidil] acetato

Usando un procedimiento similar al del Ejemplo A Etapa 6, 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acetato (60 mg, 32% de rendimiento) se obtuvo a partir de ácido 4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carboxNico (135 mg, 0,312 mmol) y clorhidrato de metil 2-(4-piperidil)acetato (66,4 mg, 0,343 mmol). LC/MS (Método i): Rt = 2,63 min; MS m/z: 572 [M+H]+; 1H NMR ((DMSO-ofe, 300MHz): 5 = 8,45 y 8,38 (s, 1H), 7,24 (s, 1H), 6,81 (s, 1H), 6,59 y 6,54 (s, 1H), 4,54 (m, 3H), 3,62 (s, 3H), 3,60 y 3,57 (s, 3H), 3,41 (m, 1H), 3,07 (m, 1H), 2,81 (m, 4H), 2,29 (m, 2H), 1,99 (m, 1H), 1,78 (m, 1H), 1,61 (m, 1H), 1,17 (m, 2H). Etapa 5: ácido 2-[1 -[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1 H-mdol-3-N]metN]piridma-3-carboml]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-[1-[4,6-dicloro-5-[[1,4-dimetil-6-(trifluorometoxi)-1H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acético (43 mg, 78%) se obtuvo a partir de metil 2-[1-[4,6-dicloro-3-[[1,4-dimetil-6-(trifluorometoxi)-1 H-indol-3-il]metil]piridina-3-carbonil]-4-piperidil]acetato (55 mg, 0,09 mmol). LC/MS (Método g): Rt = 1,85 min; MS m/z: 558 [M+H]+; 1H NMR ((DMSO-Ce, 400MHz): 5 = 12,11 (ancho, 1H), 8,45 y 8,38 (s, 1H), 7,24 (s, 1H), 6,81 (s, 1H), 6,59 y 6,54 (s, 1H), 4,55 (m, 3H), 3,63 (s, 3H), 3,38 (m, 1H), 3,07 (m, 1H), 2,79 (m, 4H), 2,18 (m, 2h), 1,96 (m, 1H), 1,79 (m, 1H), 1,64 (m, 1H), 1,16 (m, 2H).
Ejemplo DZ: ácido 2-(1-(2,4-didoro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: ferc-butil 2,4-didoro-3-(3-(2-yodo-3-metil-5-(trifluorometil)fenoxi)prop-1-en-1-il)benzoato

A una solución de ferc-butil 2,4-dicloro-3-(3-((metilsulfonil)oxi)prop-1-en-1-il)benzoato (2,95 g, 7,74 mmol) (Preparación 59) en ACN (78 mL) se añadió carbonato de potasio (2,139 g, 15,47 mmol), yoduro de potasio (1,2 g, 7,74 mmol) y 2-yodo-3-metil-5-(trifluorometil)fenol (2,3 g, 7,74 mmol) (Preparación 85). La mezcla de reacción se agitó a 50 °C durante 45 minutos. La mezcla de reacción se diluyó con H2 O y se extrajo dos veces con EtOAc. La capa orgánica obtenida se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó para dar 3,7-dimetil-5-(trifluorometoxi)-fH-indol ferc-butil 2,4-dicloro-3-(3-(2-yodo-3-metil-5-(trifluorometil)fenoxi)prop-1-en-1-il)benzoato (3,6 g, 80%) como un sólido de color amarillo pálido. lC/MS (Método i): Rt=3,07 min.1H NMR (DMSO-Ce, 300MHz): 57,59 (d, J=8,6 Hz, 1H), 7,54 (d, J=8,6 Hz, 1H), 7,34 (s, 1H), 7,17 (s, 1H), 6,81 (d, J=16,3 Hz, 1H), 6,33 (dt, J=16,3, 4,8 Hz, 1H), 5,03 (m, 2H), 2,6 (s, 3H), 1,54 (s, 9H).
Etapa 2: ferc-butil 2,4-didoro-3-((4-metN-6-(trifluorometM)benzofuran-3(2H)-iNdeno)metM)benzoato

A una solución de íerc-butil 2,4-did oro-3-(3-(2-yodo-3-metil-5-(trifluorometil) fenoxi) prop-1-en-1-il)benzoato (500 mg, 0,85 mmol) en DMF (2,5 mL) se añadió cloruro de tetrabutilamonio (260 mg, 0,937 mmol), carbonato de sodio (226 mg, 2,129 mmol), formiato de sodio (57,9 mg, 0,852 mmol) y bis(acetonitrilo)dicloropaladio(II) (17,67 mg, 0,068 mmol). La mezcla de reacción se desgasificó con argón y se agitó a 80 °C durante 18 horas. La mezcla de reacción se filtró y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con EtOAc al 0-10% en ciclohexano) para dar íerc-butil 2,4-dicloro-3-((4-metil-6-(trifluorometil)benzofuran-3(2H)-ilideno)metil)benzoato (240 mg, 45%) como un aceite amarillo. Se usó crudo en la siguiente etapa. lC/MS (Método j): Rt = 2,88 min.1H NMR (DMSO-Ce, 300MHz): 57,67 (m, 2H), 7,21 (s, 1H), 7,15 (s, 1H), 6,92 (m, 1H), 5,01 (m, 2H), 2,62 (s, 3H), 1,56 (s, 9H).
Etapa 3: ácido 2,4-didoro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoico

Usando un procedimiento similar al Ejemplo DJ, Etapa 2, se preparó ácido 2,4-dicloro-3-((4-metil-6-(trifluorometil) benzofuran-3-il)metil)benzoico (100 mg, 61%) a partir de íerc-butil 2,4-dicloro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoato (115 mg, 0,25 mmol) crudo. Se usó crudo en la siguiente etapa. LC/MS (Método i): Rt = 2,55 min; MS m/z: 401 [M-H]-; 1H NMR (DMSO-Ce, 500MHz): 513,59 (ancho, 1H), 7,83 (s, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,42 (s, 1H), 7,26 (s, 1H), 4,55 (s, 2h), 2,83 (s, 3H).
Etapa 4: metil 2-(1-(2,4-didoro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acetato (54 mg, 50%) se preparó a partir de ácido 2,4-dicloro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoico crudo (66 mg, 0,16 mmol) y clorhidrato de metil (4-piperidil)acetato (48 mg, 0,25 mmol). LC/MS (Método i): Rt = 2,73 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,82 (s, 1H), 7,65 y
7,63 (d, J=8,3 Hz, 1H), 7,35 (m, 3H), 4,45 (m, 3H), 3,59 y 3,57 (s, 3H), 3,36 (m, 1H), 3,02 (m, 1H), 2,82 (s, 3H), 2,75 (m, 1H), 2,27 (m, 2H), 1,90 (m, 1H), 1,73 (m, 1H), 1,58 (m, 1H), 1,14 (m, 2H).
Etapa 5: ácido 2-(1-(2,4-didoro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((4-metil-6-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acético (25 mg, 50%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((4-metil-3-(trifluorometil)benzofuran-3-il)metil)benzoil)piperidin-4-il)acetato (52 mg, 0,09 mmol). LC/MS (Método g): Rt = 1,93 min; MS m/z: 528 [M+H]+; 1H NMR (DMSO-cfe, 500MHz): 512,11 (m, 1H), 7,82 (s, 1H), 7,64 y 7,63 (d, J=8,3 Hz, 1H), 7,35 (m, 3H), 4,51 (m, 3H), 3,28 (m, 1H), 3,02 (m, 1H), 2,79 (m, 4H), 2,17 (m, 2H), 1,92 (m, 1H), 1,77 (m, 1H), 1,62 (m, 1H), 1,13 (m, 2H).
Ejemplo EA: ácido 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-1H-mdazoM-carboml)benzoN)piperidm-4-il)acético

Etapa 1: íerc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoato y íerc-butil 2,4-didoro-3-(3,7-dimetN-5-(trifluorometM)-2H-mdazol-2-carboml)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoato (120 mg, 4%) y íerc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-2H-inda-zole-2-carbonil)benzoato (870 mg, 43%) se prepararon a partir de 3,7-dimetil-5-(trifluorometil)-1H-indazol (600 mg, 2,8 mmol) (Preparación #42) y íerc-butil 2,4-dicloro-3-(clorocarbonil)benzoato (1,3 g, 4,2 mmol) (Preparación #44). íerc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoato. LC/MS (Método j): Rt = 2,55 min; Ms m/z: 487 [M+H]+; 1H NmR (DMSO de, 300MHz): 58,20 (s, 1H), 7,91 (d, J=8,4 Hz, 1H), 7,90 (s, 1H), 7,75 (d, J=8,4 Hz, 1H), 2,88 (s, 3H), 2,51 (s, 3H), 1,55 (s, 9H).
íerí-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoato. LC/MS (Método j): Rt=2,61 min; MS miz: 487 [M+H]+; 1H NMR (DMSO-de, 300MHz): 58,24 (s, 1H), 7,97 (d, J=8,4 Hz, 1H), 7,79 (d, J=8,4 Hz, 1H), 7,33 (s, 1H), 3,07 (s, 3H), 2,31 (s, 3H), 1,55 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-1H-mdazoM-carboml)benzoico
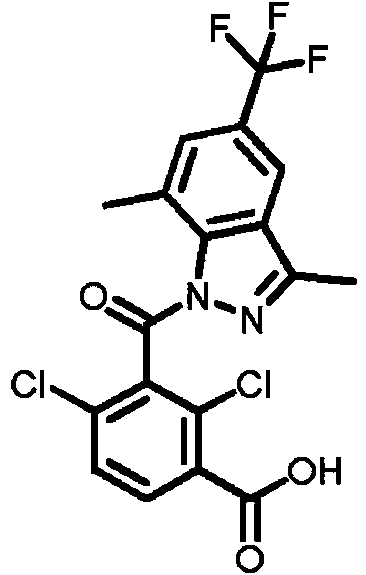
Usando un procedimiento similar al Ejemplo DJ, Etapa 2, ácido 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoico (100 mg, 83%) se preparó a partir de íerc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoato (110 mg, 0,22 mmol). LC/MS (Método j): Rt = 1,76 min; MS miz: 431 [M+H]+; 1H nMr (DMSO-de, 300MHz): 58,19 (m, 1H), 7,96 (d, J=8,4 Hz, 1H), 7,90 (s, 1H), 7,74 (d, J=8,4 Hz, 1H), 2,88 (s, 3H), 2,49 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(tnfluorometM)-1H-mdazoM-carboml)benzoM)pipendm-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoil)piperidin-4-il)acetato (64 mg, 42%) se preparó a partir de ácido 2,4-dicloro-3-(3,7-dimetil-3-(trifluorometil)-1H-indazol-1-carbonil)benzoico (115 mg, 0,27 mmol) y clorhidrato metil 2-(piperidin-4-il)acetato (67 mg, 0,35 mmol). LC/MS (Método i): Rt = 2,58 min; MS miz: 570 [M+H]+; 1H NMR (DMSO-de, 300MHz): 58,21 (s, 1H), 7,91 (s, 1H), 7,72 (m, 1H), 7,56 (m, 1H), 4,7 (m, 1H), 3,59 (m, 4H), 3,21 (m, 1H), 3,09 (m, 2H), 2,89 (m, 3H), 2,82 (m, 1H), 2,48 (m, 1H), 2,29 (m, 2H), 1,99 (m, 1h), 1,74 (m, 1H), 1,64 (m, 1H), 1,15 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-(3,7-dimetN-5-(trifluorometM)-1H-mdazoM-carboml)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazole-1-carbonil)benzoil)piperidin-4-il)acético (53 mg, 86%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-indazol-1-carbonil)benzoil)piperidin-4-il)acetato (63 mg, 0,11 mmol). LC/MS (Método g): Rt = 1,86 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 5 = 12,11 (ancho, 1H), 8,21 (m, 1H), 7,91 (s, 1H), 7,73 (m, 1H), 7,63 y 7,55 (m, 1H), 4,47 (m, 1H), 3,27 (m, 1H), 3,09 (m, 1H), 2,85 (m, 4H), 2,47 (m, 3H), 2,18 (m, 2H), 1,95 (m, 1H), 1,78 (m, 1h), 1,65 (m, 1h), 1,18 (m, 2H).
Ejemplo EB: ácido 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-2H-mdazol-2-carboml)benzoN)piperidm-4-il)acético

Etapa 1: ácido 2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-2H-mdazol-2-carboml)benzoico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoico (145 mg, 86%) se preparó a partir de terc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoato (190 mg, 0,39 mmol) (Ejemplo EA, Etapa 1). LC/MS (Método i): Rt = 2,42 min; MS m/z: 431 [M+H]+;
1H NMR (DMSO-Ce, 300MHz): 5 = 8,26 (s, 1H), 8,04 (d, J=8,4 Hz, 1H), 7,79 (d, J=8,4 Hz, 1H), 7,33 (s, 1H), 3,09 (s, 4H), 2,32 (s, 3H).
Etapa 2: metil 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-2H-mdazol-2-carboml)benzoN)piperidm-3-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoil)piperidin-4-il)acetato (125 mg, 63%) se preparó a partir de ácido 2,4-dicloro-3-(3,7-dimetil-3-(trifluorometil)-2H-indazol-2-carbonil)benzoico (140 mg, 0,32 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (82 mg, 0,42 mmol). LC/MS (Método i): Rt = 2,62 min; MS m/z: 570 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,26 (s, 1H), 7,70 (m, 2H), 7,33 (s, 1H), 4,46 (m, 1H), 3,56 (m, 3H), 3,29 (m, 1H), 3,08 (s, 4H), 2,81 (m, 1H), 2,28 (m, 5H), 1,99 (m, 1 h), 1,75 (m, 1h), 1,60 (m, 1H), 1,17 (m, 2H).
Etapa 3: ácido 2-(1-(2,4-didoro-3-(3,7-dimetN-5-(trifluorometM)-2H-mdazol-2-carboml)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoil)piperidin-4-il)acético (102 mg, 87%) se preparó a partir de 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-2H-indazol-2-carbonil)benzoil)piperidin-4-il)acetato (120 mg, 0,21 mmol). LC/MS (Método g): Rt = 1,89 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 5 = 12,12 (ancho, 1H), 8,26 (s, 1H), 7,70 (m, 2H), 7,33 (s, 1H), 4,46 (m, 1H), 3,28 (m, 1H), 3,08 (m, 4H), 2,80 (m, 1H), 2,31 (m, 4H), 2,11 (m, 1h), 1,94 (m, 1H), 1,79 (m, 1H), 1,62 (m, 1H), 0,15 (m, 2H).
Ejemplo EC: ácido 2-(1-(2,4-didoro-3-((7-metN-5-(trifluorometM)-1H-mdazoM-N)metN)benzoM)piperidm-4-N)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoil)piperidin-4-il)acetato y metil 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-2H-indazol-2-il)metil)benzoil)piperi-din-4-il)acetato

Usando un procedimiento similar al Ejemplo DD, Etapa 1, metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoil)piperidin-4-il)acetato (115 mg, 8,5%) y metil 2-[1-[2,4-dicloro-3-[[7-metil-5-(trifluorometil)-2H-indazol-2-il]metil]benzoil]-4-piperidil]acetato (350 mg, 26%) se prepararon a partir de 7-metil-5-(trifluorometil)-1H-indazol (500 mg, 2,5 mmol) (Preparación 78) y metil 2-(1-(2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il)acetato (1,4 g, 3,2 mmol) (Preparación 53). metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoil)piperidin-4-il)acetato. LC/MS (Método i): Rt = 2,51 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 58,14 (s, 1h), 8,06 (s, 1H), 7,61 y 7,60 (d, J=8,3 Hz, 1H), 7,50 (s, 1H), 7,45 y 7,37 (d, J=8,3 Hz, 1H), 6,06 y 6,07 (s, 2H), 4,44 (m, 1H), 3,59 y 3.57 (s, 3H), 3,20 (m, 1H), 3,03 (m, 1H), 2,97 (s, 3H), 2,78 (m, 1H), 2,50 (s, 3H), 2,27 (m, 2H), 1,95 (m, 1h), 1,74 (m, 1H), 1.57 (m 1H), 1,13 (m, 2H).
metil 2-[1-[2,4-didoro-3-[[7-metil-5-(trifluorometil)-2H-indazol-2-il]metil]benzoil]-4-piperidil]acetato. LC/MS (Método i): Rt = 2,47 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,55 y 8,53 (s, 1H), 8,01 (s, 1H), 7,66 (m, 1H), 7,51 y 7,43 (d, J=8,26 Hz, 1H), 7,22 (s, 1H), 5,97 (s, 2H), 4,45 (m, 1H), 3,59 y 3,58 (s, 3H), 3,28 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,50 (m, 3H), 2,27 (m, 2H), 1,96 (m, 1H), 1,76 (m, 1H), 1,57 (m, 1H), 1,17 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((7-metN-5-(trifluorometM)-1H-idazoM-N)metN)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometil)- 1H-indazol-1-il)metil)benzoil)piperidin-4-il)acético (113 mg, 97%) se preparó a partir de 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoil)piperidin-4-il)acetato (119 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,73 min; mS m/z: 528 [M+H]+;
1H NMR (DMSO-Ce, 400MHz): 512,10 (s, 1H), 8,14 (s, 1H), 8,06 (s, 1H), 7,61 y 8,60 (d, J=8,3 Hz, 1H), 7,50 (s, 1H), 7,45 y 7,37 (d, J=8,3 Hz, 1H), 6,08 y 7,07 (s, 2H), 4,44 (m, 1H), 3,21 (m, 1H), 3,02 (m, 1H), 2,97 (s, 3H), 2,78 (m, 1H), 2,16 (m, 2h), 1,92 (m, 1H), 1,76 (m, 1H), 1,59 (m, 1H), 1,12 (m, 2H).
Ejemplo ED: ácido 2-(1-(2,4-didoro-3-((7-metN-5-(trifluorometM)-2H-mdazol-2-N)metN)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometil)-2H-indazol-1-il)metil)benzoil)piperidin-4-il)acético (76 mg, 64%) se preparó a partir de metil 2-[1-[2,4-dicloro-3-[[7-metil-5-(trifluorometil)indazol-2-il]metil]benzoil]-4-piperidil]acetato (121 mg, 0,22 mmol) (Ejemplo EC, etapa 1). LC/MS (Método g): Rt=1,70 min; MS m/z: 528 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 512,13 (ancho, 1H), 8,55 y 8,53 (s, 1H), 8,01 (s, 1h), 7,67 y 7,65 (d, J=8,4 Hz, 1H), 7,51 y 7,43 (d, J=8,4 Hz, 1H), 7,22 (s, 1H), 597 (m, 2H), 4,47 (m, 1H), 3,31 (m, 4H), 3,03 (m, 1H), 2,80 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1H), 1,76 (m, 1H), 1,60 (m, 1H), 1,17 (m, 2h).
Ejemplo EE: ácido 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: íerc-butil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)- 1H-mdazol-3-M)(hidroxi)metM)benzoato

Usando un procedimiento similar al del Ejemplo FE, Etapa 1, íerc-butil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)(hidroxi)metil)benzoato (149 mg, 63%) se preparó a partir de 1,4-dimetil-6-(trifluorometil)-1H -inda-zol-3-carbaldehído (100 mg, 0,41 mmol) (Preparación 79) y íerc-butil 2,4-diclorobenzoato (153 mg, 0,62 mmol) ((Preparación #33, Etapa A). LC/MS (Método i): Rt=2,72 min; MS miz: 489 [M+H]+; 1H NMR (DMSO-de, 400MHz): 5 7,93 (s, 1H), 7,56 (m, 2H), 7,24 (s, 1H), 6,95 (d, J=5,7 Hz, 1H), 6,35 (d, J=5,7 Hz, 1H), 3,96 (s, 3H), 2,88 (s, 3H), 1,55 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-((1,4-dimetN-6-(trifluorometM)-1H-mdazol-3-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2 y calentamiento a 50 °C, ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)metil)benzoico (66 mg, 61%) se preparó a partir de íerc-butil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)(hidroxi)metil)benzoato (149 mg, 0,26 mmol). LC/MS (Método i): Rt = 2,39 min; MS miz: 417 [M+H]+; 1H (DMSO-de, 300MHz): 5 = 13,50 (ancho, 1H), 7,89 (s, 1H), 7,65 (m, 2H), 7,19 (s, 1H), 4,80 (s, 2H), 3,92 (s, 3H), 2,85 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)metil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-in-dazol-3-il)metil)benzoil)piperidin-4-il)acetato (110 mg, 82%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)metil)benzoico (100 mg, 0,24 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (69,6 mmol). LC/MS (Método i): Rt = 2,62 min; MS miz: 556 [M+H]+; 1H NMR (DMSO-de, 300MHz): 5 7,89 (s, 1H), 7,59 y 7,56 (d, J=8,3 Hz,
1H), 7,35 y 7,27 (d, J=8,3 Hz, 1H), 7,19 (s, 1H), 4,77 (m, 2H), 4,47 (m, 1H), 3,91 y 3,90 (s, 3H), 3,59 y 3,57 (s, 3H), 3,28 (m, 1H), 3,05 (m, 1H), 2,85 (s, 3H), 2,78 (m, 1H), 2,27 (m, 2h), 1,97 (m, 1H), 1,77 (m, 1H), 1,60 (m, 1H), 1,14 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-((1,4-dimetM-6-(trifluorometN)-1H-mdazol-3-M)metM)benzoN)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5 ácido 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-in-dazol-3-il)metil)benzoil)piperidin-4-il)acético (71 mg, 67%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indazol-3-il)metil)benzoil)piperidin-4-il)acetato (105 mg, 0,19 mmol). LC/MS (Método g): Rt = 1,78 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-Ce, 500MHz): 512,10 (ancho, 1H), 7,88 (s, 1H), 7,58 y 7,57 (d, J=8,3 Hz, 1H), 7,36 y 7,28 (d, J=8,3 Hz, 1H), 7,19 (s, 1H), 4,78 (m, 2H), 4,48 (m, 1H), 3,91 y 3,90 (s, 3H), 3,25 (m, 1H), 3,04 (m, 1H), 2,85 (s, 3h), 2,79 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1H), 1,76 (m, 1H), 1,62 (m, 1H), 1,13 (m, 2H).
Ejemplo EF: ácido 2-(1-(2,4-didoro-3-((3-metN-5-(trifluorometM)-1H-mdazoM-M)metM)benzoN)piperidm-4-N)acético

Etapa 1: metil 2,4-didoro-3-((3-metN-5-(trifluorometM)-1H-mdazoM-M)metM)benzoato

Usando un procedimiento similar al Ejemplo DD, Etapa 1 metil 2,4-didoro-3-((3-metil-5-(trifluorometil)-1H-inda-zol-1-il)metil)benzoato (1,44 g, 86%) se preparó a partir 3-metil-5-(trifluorometil)-1H-indazol (800 mg, 4 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (1,5 g, 5,2 mmol) (Preparación #1, Etapa b). LC/MS (Método i): Rt = 2,55 min; MS m/z:
417 [M+H]+; 1H NMR (DMSO-cfe, 500MHz): 58,17 (m, 1H), 7,94 (d, J=8,9 Hz, 1H), 7,77 (d, J=8,4 Hz, 1H), 7,71 (d, J=8,9 Hz, 1H), 7,66 (d, J=8,4 Hz, 1H), 5,82 (s, 2H), 3,84 (s, 3H), 2,45 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3-metM-5-(trifluorometN)-1H-mdazoM-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (1,22 g, 88%) se preparó a partir de metil 2,4-dicloro-3-((3-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoato (1,44 g, 3,45 mmol). LC/MS (Método i): Rt = 2,23 min; MS m/z: 403 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 5 13,69 (br., 1H), 8,19 (m, 1H), 7,96 (d, J=8,8 Hz, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,73 (d, J=8,8 Hz, 1H), 7,63 (d, J=8,4 Hz, 1H), 5,83 (s, 2H), 2,47 (s, 3H).
Etapa 3: ácido 2-(1-(2,4-didoro-3-((3-metN-5-(trifluorometM)-1H-mdazoM-N)metN)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 6 y el Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((3-metil-5-(trifluorometil)-1H-indazoM-il)metil)benzoil)piperidin-4-il)acético (104 mg, 79%) se preparó a partir de ácido 2,4-dicloro-3-((3-metil-5-(trifluorometil)-1H-indazol-1-il)metil)benzoico (100 mg, 0,25 mmol). LC/MS (Método g): Rt = 1,71 min; MS m/z: 528 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 512,10 (ancho, 1H), 8,19 (s 1H), 94 (d, J=9,0 Hz, 1H), 7,72 (d, J=9,0 Hz, 1H), 7,61 y 7,60 (d, J=8,3 Hz, 1H), 7,33-7,44 y 7,37 (d, J=8,3 Hz, 1H), 5,80 (m, 2H), 4,45 (m, 1H), 3,20 (m, 1H), 3,02 (m, 1 h), 2,78 (m, 1H), 2,47 y 2,46 (ds, 3H), 2,16 (m, 2H), 1,92 (m, 1H), 1,76 (m, 1H), 1,59 (m, 1H), 1,14 (m, 2H).
Ejemplo EG: ácido 2-[1-[2,4-didoro-3-[[3,7-dimetN-5-(trifluorometoxi)-1H-mdoM-M]metM]benzoN]-4-piperidil]acético

Etapa 1: metil 2,4-didoro-3-((3,7-dimetM-5-(trifluorometoxi)-1H-mdoM-N)metN)benzoato

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-didoro-3-((3,7-dimetil-5-(trifluorometoxi)-1 H-indol-1-il)metil)benzoato (336 mg, 62%) se preparó a partir de 3,7-dimetil-5-(trifluorometoxi)-1H-indol (Preparación #61) (280 mg, 1,22 mmol) y íerc-butil 3-(bromometil)-2,4-diclorobenzoato (498 mg, 1,46 mmol) (Preparación # 1, Etapa B). LC/MS (Método i): Rt = 2,82 min; MS m/z: 446 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 57,87 (d, J=8,4 Hz, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,26 (s, 1H), 6,93 (s, 1H), 6,48 (s, 1H), 5,86 (s, 2H), 3,88 (s, 3H), 2,88 (s, 3H), 2,11 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((3,7-dimetN-5-(trifluorometoxi)-1H-mdoM-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (272 mg, 85%) se preparó a partir de metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (332 mg, 0,7 mmol). LC/MS (Método i): Rt = 2,53 min; MS m/z: 432 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 513,65 (ancho, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,71 (d, J=8,4 Hz, 1H), 7,26 (s, 1H), 6,93 (m, 1H), 6,47 (s, 1H), 5,86 (s, 2h), 2,88 (s, 3H), 2,11 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-((3,7-dimetN-5-(trifluorometoxi)-1H-mdoM-M)metM)benzoM)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (100 mg, 76%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-4-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (100 mg, 0,2 mmol) y clorhidrato de metil (4-piperidil)acetato (90 mg, 0,4
mmol). LC/MS (Método i): Rt = 2,68 min; MS miz: 571 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,69 y 7,67 (d, J=8,3 Hz, 1H), 7,52 (d, J=8,3 Hz, 0,5H), 7,45 (d, J=8,3 Hz, 0,5H), 7,26 (s, 1H), 6,93 (s, 1H), 6,50 y 6,47 (s, 1H), 5,80 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,28 (m, 1H), 2,08 (m, 1H), 2,87 (s, 3H), 2,80 (m, 1H), 2,26 (m, 2H), 2,12 (s, 3H), 1,97 (m, 1H) 1,75 (m, 1H), 1,58 (m, 1H), 1,17 (m, 2H).
Etapa 4: ácido 2-(1 -(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1 H-indoM -il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al del Ejemplo 1, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (78 mg, 83%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (96 mg, 0,16 mmol). LC/MS (Método g): Rt = 1,96 min; MS m iz : 557 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 12,10 (ancho, 1H), 7,69 y 7,68 (d, J=8,3Hz, 1H), 7,52 (d, J=8,3 Hz, 0,5H), 7,46 (d, J=8,3 Hz, 0,5H), 7,26 (s, 1H), 6,93 (s, 1H), 6,50 y 6,46 (s, 1H), 5,82 (m, 2H), 4,46 (m, 1H), 3,27 (m, 1H), 3,04 (m, 1H), 2,87 (s, 3H), 2,82 (m, 1H), 2,18 (m, 2H), 2,12 (s, 3H), 1,94 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,17 (m, 2H). Tabla EG. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (Ejemplo EG Etapa 2) usando el mismo procedimiento como se describe en el ejemplo EG Etapa 3 y 4 con las aminas apropiadas.

Ejemplo EH: ácido 2-[1-[2,4-didoro-3-[3,7-dimetil-5-(trifluorometoxi)indol-1-carbonil]benzoil]-4-piperi-dil]acético

Etapa 1: terc-butil 2,4-dieloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbón il)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, terc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoato (330 mg, 40%) se preparó a partir de 3,7-dimetil-5-(trifluorometoxi)-1H-indol (Preparación 61) (415 mg, 1,81 mmol) y terc-butil 2,4-dicloro-3-(clorocarbonil)benzoato (513 mg, 1,65 mmol) (Preparación #44). LC/MS (Método j): Rt = 2,71 min; MS m/z: 502 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,98 (d, J=8,4 Hz, 1H), 7,81 (d, J=8,4 Hz, 1H), 7,43 (s, 1H), 7,27 (m, 1H), 7,21 (s, 1H), 2,69 (s, 3H), 2,16 (d, J=1,3 Hz, 3H), 1,57 (s, 9H).
Etapa 2: terc-butil 2,4-didoro-3-(3,7-dimetM-5-(trifluorometoxi)-1H-indoM-carbonil)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoico (280 mg, 96%) se preparó a partir de terc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoato (330 mg, 0,65 mmol). LC/MS (Método k): Rt = 2,56 min; MS m/z: 446 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 13,9 (ancho, 1H), 8,04 (d, J=8,4 Hz, 1H), 7,80 (d, J=8,4 Hz, 1H), 7,43 (s, 1H), 7,28 (s, 1H), 7,18 (d, J=1,2 Hz, 1H), 2,69 (s, 3H), 2,16 (d, J=1,2 Hz, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-(3,7-dimetM-5-(tnfluorometoxi)-1H-indoM-carbonM)benzoM)piperidin-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (93 mg, 71%) se preparó a partir de ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoico (100 mg, 0,22 mmol) y clorhidrato de metil (4-piperidil)acetato (87 mg, 0,44 mmol). LC/MS (Método k): Rt = 2,70 min; MS m/z: 585 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,79 (m, 1H), 7,85 (m, 1H), 7,45 (s, 1H), 7,25 (s, 1H), 7,35 y 7,31 y 7,09 (s, 1H), 4,47 (m, 1H), 3,59 (m, 3H), 3,24 (m, 1H), 3,07 (m, 1H), 2,83 (m, 1H), 2,68 (s, 3H), 2,25 (m, 2H), 2,18 (m, 3H), 1,99 (m, 1H), 1,76 (m, 1H), 1,59 (m, 1H), 1,12 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-mdoM-carboml)benzoil)piperidm-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 5, 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1 -carbonil)benzoil)piperidin-4-M ) acético (48 mg, 55%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (90 mg, 0,15 mmol). LC/MS (Método g): Rt = 1,95 min; MS m/z: 571 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 12,11 (ancho, 1H), 7,79 (m, 1H), 7,65 (m, 1H), 7,43 (ancho, 1H), 7,37 y 7,31 y 7,10 (m, 1H), 7,27 (ancho, 1H), 4,47 (m, 1H), 3,57 (m, 1H), 3,24 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,68 (s, 3H), 2,15 (m, 5H), 1,95 (m, 1H), 1,78 (m, 1H), 1,65 (m, 1H), 1,15 (m, 2H).
Tabla EH. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometoxi)-1H-indol-1-carbonil) benzoico (Ejemplo Eh, Etapa 2) usando el mismo procedimiento como se describió en el Ejemplo EH, Etapa 3 y 4 con las aminas apropiadas.

Ejemplo EI: ácido 2-(1-(2,4-didoro-3-((5-(trifluorometoxi)-1H-mdoM-N)metN)benzoN)piperidm-4-N)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((5-(tnfluorometoxi)-1H-mdoM-M)metM)benzoM)pipendm-4-M)acetato
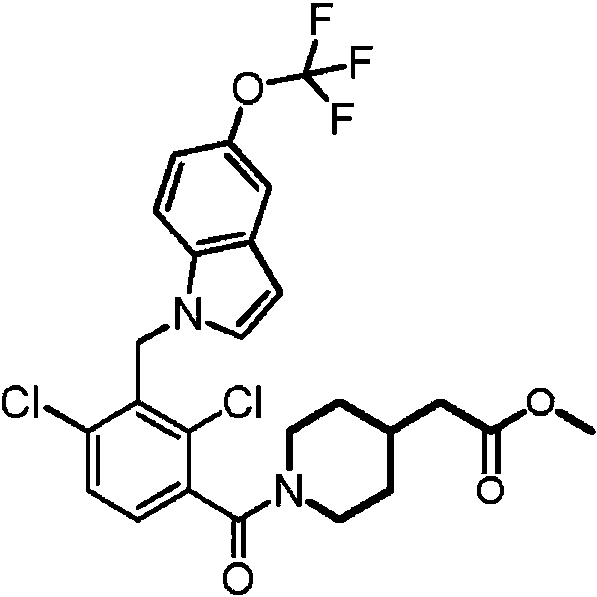
Usando un procedimiento similar al Ejemplo DH, Etapa 1, metil 2-(1-(2,4-didoro-3-((5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (141 mg, 77%) se preparó a partir de 5-(trifluorometoxi)-1H-indol (68 mg, 0,34 mmol) y metil 2-(1-(2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il)acetato (Preparation 53) (192 mg, 0,44 mmol). LC/MS (Método i): Rt = 2,60 min; MS m/z: 543 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,69 (m, 2H), 7,56 (m, 1H), 7,50 y 7,42 (d, J=8,4Hz, 1H), 7,15 (m, 2H), 6,54 (m, 1H), 5,62 (m, 2H), 4,45 (m, 1H), 3,59 y 3,58 (s, 3H), 3,20 (m, 1H), 3,01 (m, 1h), 2,79 (m, 1H), 2,27 (m, 2H), 1,97 (m, 1H), 1,75 (m, 1H), 1,57 (m, 1H), 1,13 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((5-(triftuorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (75 mg, 58%) se preparó a partir de metil 2-(1-(2,4-didoro-3-((5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (132 mg, 0,24 mmol). LC/MS (Método g): Rt = 1,77 min; Ms m/z: 529 [M+H]+;
1H NMR (DMSO-Ce, 400MHz): 512,10 (ancho, 1H), 7,66 (m, 2H), 7,56 (s, 1H), 7,50 y 7,42 (d, J=8,4 Hz, 1H), 7,14 (m, 2H), 6,54 (m, 1H), 5,62 (m, 2H), 4,45 (m, 1H), 3,23 (m, 1H), 3,02 (m, 1H), 2,78 (m, 1H), 2,16 (m, 2H), 1,93 (m, 1H), 1,77 (m, 1H), 1,59 (m, 1H), 1,13 (m, 2H).
Tabla EI. Los siguientes ejemplos se prepararon a partir de metil 2-(1-(2,4-dicloro-3-(((metilsulfonil)oxi)metil)benzoil)piperidin-4-il) acetato (Preparación 53) usando el mismo procedimiento que se describe en ejemplo EI, Etapa 1 y 2 con el indol apropiado.


Ejemplo EJ: ácido 2-(1-(2,4-didoro-3-((7-metN-5-(trifluorometoxi)-1H-mdoM-N)metN)benzoN)piperidm-4-M)acético

Etapa 1: metil 2,4-didoro-3-((7-metM-5-(tnfluorometoxi)-1H-mdoM-M)metMo)

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (910 mg, 69%) se preparó a partir 7-metil-5-(trifluorometoxi)-1H-indol (Preparación #64) (600 mg, 2,8 mmol) y íerc-butil 3-(bromometil)-2,4-diclorobenzoato (914mg, 3,07 mmol) (Preparación #1, Etapa B). LC/MS (Método j): Rt = 2,34 min; MS m/z: 432 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,88 (d, J=8,4 Hz, 1H), 7,76 (d, J=8,4 Hz, 1H), 7,37 (s, 1H), 6,94 (s, 1H), 6,67 (d, J=3,3 Hz, 1H), 6,45 (d, J=3,3 Hz, 1H), 5,93 (s, 2H), 3,88 (s, 3H), 2,90 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((7-metM-5-(trifluorometoxi)-1H-mdoM-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (840 mg, 91%) se preparó a partir de metil 2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (900 mg, 2,08 mmol). LC/MS (Método j): Rt = 1,85 min; MS m/z: 418 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 13,78 (ancho, 1H), 7,82 (d, J=8,4 Hz, 1H), 7,70 (d, J=8,4 Hz, 1H), 7,37 (s, 1H), 6,94 (s, 1H), 6,66 (d, J=3,3 Hz, 1H), 6,45 (d, J=3,3 Hz, 1H), 5,92 (s, 2H), 2,90 (s, 3H)
Etapa 3: metil 2-(1-(2,4-didoro-3-((7-metM-5-(trifluorometoxi)-1H-mdoM-N)metN)benzoN)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, se preparó metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (240 mg, 82%) a partir de ácido 2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (200 mg, 0,48 mmol) y clorhidrato de metil (4-piperidil)acetato (111 mg, 0,57 mmol). LC/MS (Método j): Rt = 2,14 min; MS m/z: 557 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,70 y 7,71 (d, J=8,3 Hz, 1H), 7,53 y 7,45 (d, J=8,3 Hz, 1H), 7,37 (s, 1H), 6,94 (s, 1H), 6,69 (m, 1H), 6,46 (m, 1H), 5,89 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,26 (m, 1H), 3,03 (m, 1H), 2,99 (s, 3H), 2,79 (m, 1H), 2,27 (m, 2H), 1,97 (m, 1H), 1,75 (m, 1H), 1,57 (m, 1H), 1,05-1,27 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il) acético A-1668537.0

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (150 mg, 67%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((7-metil-4-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (230 mg, 0,41 mmol). LC/MS (Método g): Rt = 1,88 min; MS m/z: 543 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 512,08 (m, 1H), 7,69 (m, 1H), 7,53 y 7,45 (d, J=8,1Hz, 1H), 7,37 (s, 1H), 6,93 (s, 1H), 6,69 y 6,68 (d, J=8,1Hz 1H), 6,46 (m, 1H), 5,89 (m, 2H), 4,46 (m, 1H), 3,29 (m, 1H), 3,03 (m, 1h), 2,89 (s, 3h), 2,80 (m, 1H), 2,15(m, 2H), 1,93(m, 1H), 1,77 (m, 1H), 1,60 (m, 1H), 1,12(m, 2H).
Tabla EJ. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil) benzoico (Ejemplo Ej, Etapa 2) usando el mismo procedimiento como se describe en el ejemplo EJ, Etapa 3 y 4 con las aminas apropiadas.

Ejemplo EK: ácido 2-(1-(2,4-dimetN-3-((7-metN-5-(trifluorometoxi)-1H-mdoM-M)metM)benzoN)piperidm-4-M)acético

Etapa 1: metil 2,4-dimetM-3-((7-metM-5-(tnfluorometoxi)-1H-mdoM-M)metM)benzoato
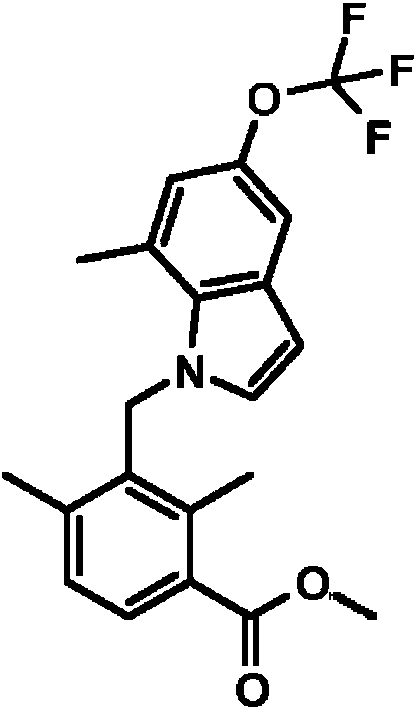
A una solución de 7-metil-5-(trifluorometoxi)-1 H-indol (Preparación 64) (200 mg, 0,93 mmol) en ACN (4 mL) se añadió carbonato de cesio (454 mg, 1,4 mmol) y 3-(clorometil)-2,4-dimetilbenzoato (227 mg, 1,1 mmol) (Preparación #54) y la mezcla de reacción se agitó a 70 °C durante la noche, luego se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con acetato de etilo 10% a 20% en ciclohexano) para dar 2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (300 mg, 64%). LC/MS (Método j): Rt = 2,37 min; MS m/z: 392 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,71 (d, J=7,9 Hz, 1H), 7,36 (s, 1H), 7,29 (d, J=7,9 Hz, 1H), 6,92 (s, 1H), 6,46 (d, J=3,3 Hz, 1H), 6,39 (d, J=3,3 Hz, 1H), 5,73 (m, 2H), 3,82 (s, 3H), 2,93 (s, 3H), 2,36 (s, 3H), 2,29 (s, 3H). Etapa 2: ácido 2,4-dimetM-3-((7-metN-5-(trifluorometoxi)-1H-mdoM-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil) benzoico (245 mg, 84%) se preparó a partir de metil 2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoato (292 mg, 0,75 mmol). LC/MS (Método j): Rt = 1,89 min; MS m/z: 378 [M+H]+; 1H NMR (DMSO-de, 300MHz): 513,01 (ancho, 1H), 7,70 (d, J=7,9 Hz, 1H), 7,35 (s, 1H), 7,25 (d, J=7,9 Hz, 1H), 6,92 (s, 1H), 6,47 (d, J=3,3 Hz, 1H), 6,38 (d, J=3,3 Hz, 1h), 5,71 (s, 2H), 2,93 (s, 3H), 2,38 (s, 3H), 2,28 (s, 3H).
Etapa 3: metil 2-(1-(2,4-dimetN-3-((7-metM-5-(trifluorometoxi)-1H-mdoM-N)metN)benzoM)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1 H -indol-1-il)metil)benzoil)piperidin-4-il)acetato (135 mg, 90%) se preparó a partir de ácido 2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (110 mg, 0,29 mmol) y clorhidrato de metil (4-piperidil)acetato (68 mg, 0,35 mmol). LC/MS
(Método j): Rt = 2,08 min; MS miz: 517 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,35 (s, 1H), 7,20 (m, 1H), 7,16 y 7,08 (d, J=9 Hz, 1H), 6,91 (s, 1H), 6,49 (m, 1H), 6,40 (m, 1H), 5,68 (s, 2H), 4,52 (m, 1H), 3,59 y 3,57 (s, 3H), 3,27 (m, 1H), 2,97 (m, 1H), 2,92 (s, 3h), 2,76 (m, 1h), 2,30 (m, 2h), 2,24 (s, 3h), 2,10 y 2,05 (s, 3H), 1,92 (m, 1H), 1,75 (m, 1H), 1,55 (m, 1H), 1,12 (m, 2H).
Etapa 4: ácido 2-(1 -(2,4-dimetN-3-((7-metN-5-(tnfluorometoxi)-1 H-indol-1 -il)metil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dimetil-3-((7-metil-5-(trifluorometoxi)-1H-indoM-il)metil)benzoil)piperidin-4-il)acético (115 mg, 85%) se preparó a partir de metil 2-(1-(2,4-dimetil-3-((7-metil-4-(trifluorometoxi)-1H-indoM-il)metil)benzoil)piperidin-4-il)acetato (130 mg, 0,25 mmol). LCiMS (Método g): Rt = 1,84 min; MS miz: 503 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 511,90 (br, 1H), 7,35 (s, 1H), 7,22 y 7,21 (d, J=7,7 Hz, 1h), 7,16 y 7,09 (d, J=7,7 Hz, 1H), 6,91 (s, 1H), 6,49 (m, 1H), 6,40 (m, 1H), 5,68 (m, 2H), 4,51 (m, 1H), 3,27 (m, 1H), 2,98 (m, 1H), 2,92 (s, 3H), 2,75 (m, 1H), 2,25 (s, 3h), 2,16 (m, 2h), 2,10 y 2,05 (s, 3H), 1,92 (m, 1H), 1,74 (m, 1H), 1,53 (m, 1H), 1,05 (m, 2H).
Ejemplo EL: ácido 2-(1-(2,4-dicloro-3-(7-metN-5-(trifluorometoxi)-1H-indoM-carbonN)benzoN)piperidm-4-N)acético

Etapa 1: terc-butil 2,4-didoro-3-(7-metil-5-(trifluorometoxi)-1H-mdoM-carboml)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, terc-butil 2,4-dicloro-3-(7-metil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoato (840 mg, 88%) se preparó a partir 7-metil-5-(trifluorometoxi)-1H-indol (Preparación 64) (600 mg, 2,8 mmol) y terc-butil 2,4-dicloro-3-(fluorocarbonil)benzoato (631 mg, 2 mmol) (Preparación 55). LCiMS (Método j): Rt = 2,74 min; MS miz: 488 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 57,99 (d, J=8,4 Hz, 1H), 7,82 (d, J=8,4 Hz, 1H), 7,54 (s, 1H), 7,41 (d, J=3,8 Hz, 1H), 7,27 (m, 1H), 6,84 (d, J=3,8 Hz, 1H), 2,69 (s, 3H), 1,57 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-(7-metN-5-(tnfluorometoxi)-1H-mdol-1-carbonN)benzoico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-(7-metil-5-(trifluorometoxi)-'/H-/ncfo/-f-carbonil) benzoico (705 mg, 97%) se preparó de íerc-butil 2,4-dicloro-3-(7-metil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoato (807 mg, 1,65 mmol). LC/MS (Método j): Rt = 1,77 min; MS m/z: 432 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 5 13,90 (ancho, 1H), 8,03 (d, J=8,6 Hz, 1H), 7,81 (d, J=8,6 Hz, 1H), 7,54 (s, 1H), 7,38 (d, J=3,8 Hz, 1H), 7,27 (m, 1H), 6,84 (d, J=3,8 Hz, 1H), 2,70 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-(7-metil-5-(trifluorometoxi)-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato
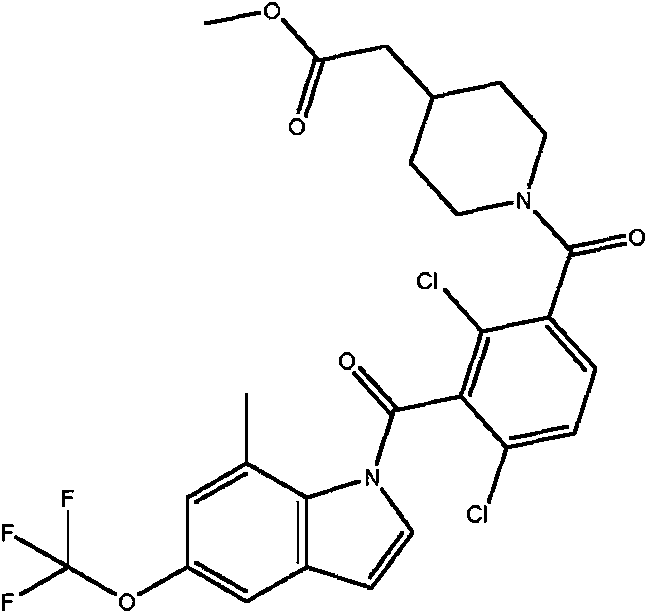
Usando un procedimiento similar al Ejemplo A, Etapa 6, se preparó metil 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (240 mg, 82%) a partir de ácido 2,4-didoro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoico (200 mg, 0,48 mmol) y clorhidrato de metil (4-piperidil)acetato (111 mg, 0,57 mmol). LC/MS (Método j): Rt = 2,14 min; MS m/z: 557 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,70 y 7,71 (d, J=8,3 Hz, 1H), 7,53 y 7,45 (d, J=8,3 Hz, 1H), 7,37 (s, 1H), 6,94 (s, 1H), 6,69 (m, 1H), 6,46 (m, 1H), 5,89 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,26 (m, 1H), 3,03 (m, 1H), 2,99 (s, 3H), 2,79 (m, 1H), 2,27 (m, 2H), 1,97 (m, 1H), 1,75 (m, 1H), 1,57 (m, 1H), 1,05-1,27 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-(7-metM-5-(trifluorometoxi)-1H-mdoM-carboml)benzoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (150 mg, 67%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((7-metil-5-(trifluorometoxi)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (230 mg, 0,41 mmol). LC/MS (Método g): Rt = 1,88 min; MS m/z: 543 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 5 12,08 (m, 1H), 7,69 (m, 1H), 7,53 y 7,45 (d, J=8,1Hz, 1H), 7,37 (s,
1H), 6,93 (s, 1H), 6,69 y 6,68 (d, J=8,1Hz 1H), 6,46 (m, 1H), 5,89 (m, 2H), 4,46 (m, 1H), 3,29 (m, 1H), 3,03 (m, 1H), 2,89 (s, 3h), 2,80 (m, 1H), 2,15(m, 2H), 1,93(m, 1H), 1,77 (m, 1H), 1,60 (m, 1H), 1,12(m, 2H).
Tabla EL. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-(7-metil-5-(trifluorometoxi)-1H-indol-1-carbonil) benzoico (Ejemplo EL, Etapa 2) usando el mismo procedimiento con las aminas apropiadas como se describe en el ejemplo EL, Etapa 3 y 4.

Ejemplo EM: ácido 2-(1-(2,4-didoro-3-((5-(difluorometoxi)-7-metiMH-mdoM-il)metil)benzoil)piperidm-4-il)acético

Etapa 1: metil 2,4-didoro-3-((5-(difluorom etoxi)-7-metiMH-mdoM-il)metil)benzoato

Usando un procedimiento similar al Ejemplo DH, Etapa 1, metil 2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indoM-il)metil)benzoato (300 mg, 47%) se preparó a partir de 5-(difluorometoxi)-7-metil-1 H-indol (Preparación 65) (280 mg, 1,42 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (423 mg, 1,42 mmol) (Preparación #1, Etapa B).
LC/MS (Método i): Rt = 2,63 min; MS miz: 414 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,87 (d, J=8,4 Hz, 1H), 7,75 (d, J=8,4 Hz, 1H), 7,17 (s, 1H), 7,10 (t, J=75 Hz, 1H), 6,81(m, 1H), 6,61 (d, J=3,1Hz, 1H), 6,39 (d, J=3,1Hz, 1H), 5,90 (s, 2H), 3,88 (s, 3H), 2,87 (s, 3H).
Etapa 2: ácido 2,4-didoro-3-((5-(difluorom etoxi)-7-metil-íH -indol-1-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indol-1-il)metil) benzoico (270 mg, 93%) se preparó a partir de metil 2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indol-1-il)metil)benzoato (295 mg, 0,7 mmol). LC/MS (Método i): Rt = 2,29 min; MS m/z: 400 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 513,74 (ancho, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,70 (d, J=8,4 Hz, 1H), 7,17 (m, 1H), 7,10 (t, J=75 Hz, 1H), 6,80 (m, 1H), 6,60 (m, 1H), 6,39 (m, 1H), 5,89 (s, 2H), 2,87 (s, 3H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-((5-(difluorometoxi)-7-metiMH-mdoM-il)metil)benzoil)piperidm-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1Hindol-1-il)metil)benzoil)piperidin-4-il)acetato (120 mg, 83%) se preparó a partir de ácido 2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indol-1-il)metil)benzoico (100 mg, 0,25 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (53 mg, 0,27 mmol). LC/MS (Método i): Rt = 2,50 min; MS m/z: 539 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,69 y 7,68 (dd, J=8,3 Hz, 1H), 7,52 y 7,44 (d, J=8,3 Hz, 1H), 7,17 (m, 1H), 7,10 (t, J=75 Hz, 1H), 6,80 (m, 1H), 6,63 (m, 1H), 6,40 (m, 1H), 5,87 (m, 2H), 4,46 (m, 1H), 3,59 y 3,57 (s, 3H), 3,26 (m, 1H), 3,03 (m, 1H), 2,88 (s, 3H), 2,81 (m, 1H), 2,28 (m, 2H), 1,97 (m, 1H), 1,75 (m, 1H), 1,56 (m, 1H), 1,13 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((5-(difluorometoxi)-7-metiMH-mdoM-il)metil)benzoil)piperidm-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (105 mg, 85%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((5-(difluorometoxi)-7-metil-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (120 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,71 min; MS m/z: 525 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 11,13 (ancho, 1H), 7,69 y 7,68 (d, J=8,4 Hz, 1H), 7,52 y 7,45 (d, J=8,4 Hz, 1H), 7,17 (m, 1H), 7,10 (t, J=75 Hz 1H), 6,80 (m, 1H), 6,63 (m, 1H), 6,40 (m, 1H), 5,87 (m, 2H), 4,47 (m, 1H), 3,29 (m, 1H), 3,06 (m, 1H), 2,86 (s, 3H), 2,81 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1H), 1,78 (m, 1H), 1,61 (m, 1 H), 1,14 (m, 2H).
Ejemplo EN: ácido 2-(1-(2,4-dicloro-3-((5-ciclopropil-7-fluoro-1H-mdoM-il)metil)benzoil)piperidm-4-il)acético
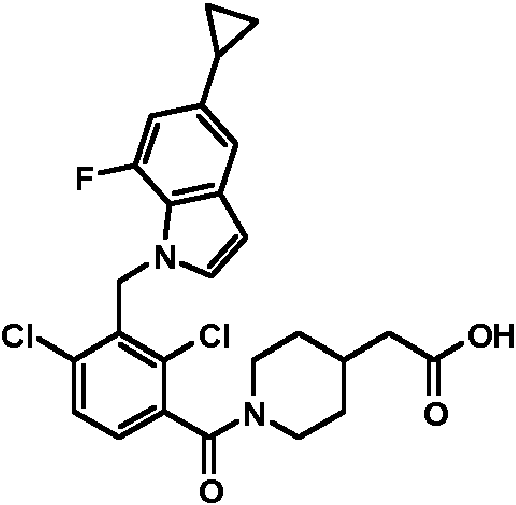
Etapa 1: metil 2,4-dicloro-3-((5-cidopropil-7-fluoro-1H -indol-1-il)metil)benzoato

Usando un procedimiento similar al Ejemplo DH, Etapa 1, metil 2,4-dicloro-3-((5-ddopropil-7-fluoro-1H-indol-1-il)metil)benzoato (610 mg, 68%) se preparó a partir de 5-ddopropil-7-fluoro-1 H- indol (Preparación 66) (400 mg, 2,28 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (680 mg, 2,28 mmol) (Preparación #1, Etapa B). LC/MS (Método i): Rt = 2,84 min; MS m/z: 392 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,83 (d, J=8,4 Hz, 1H), 7,72 (d, J=8,4 Hz, 1H), 7,10 (m, 1H), 6,75 (m, 2H), 6,38 (m, 1H), 5,76 (m, 2H), 3,87 (s, 3H), 1,98 (m, 1H), 0,92 (m, 2H), 0,66 (m, 2H).
Etapa 2: ácido 2,4-dicloro-3-((5-ciclopropN-7-fluoro-1H-mdoM-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, el ácido 2,4-didoro-3-((5-ddopropil-7-fluoro- 1H -indol-1-il)metil) benzoico (602 mg, 97%) preparado a partir de metil 2,4-didoro-3-((5-cidopropil-7-fluoro- 1H -indol-1-il)metil)benzoato (610 mg, 1,55 mmol). LC/MS (Método i): Rt = 2,49 min; MS m/z: 378 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,77 (d, J=8,4 Hz, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,10 (m, 1H), 6,76 (m, 2H), 6,38 (m, 1H), 5,75 (s, 2H), 1,98 (m, 1H), 0,93 (m, 2H), 0,67 (m, 2H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-((5-ciclopropM-7-fluoro-1H-mdoM-M)metM)benzoN)piperídm-4-M)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-didoro-3-((5-cidopropil-7-fluoro-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (115 mg, 84%) se preparó a partir de ácido 2,4-dicloro-3-((5-dclopropil-7-fluoro-1H-indol-1-il)metil)benzoico (100 mg, 0,26 mmol) y clorhidrato de metil (4-piperidil)acetato (102 mg, 0,52 mmol). LC/MS (Método i): Rt = 2,69 min; MS m/z: 517 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,67 y 7,65 (d, J=8,3 Hz, 1H), 7,48 y 7,40 (d, J=8,3 Hz, 1H), 7,10 (m, 1H), 6,80 (m, 1H), 6,74 y 6,69 (s, 1H), 6,39 (m, 1H), 5,69-5,73 (m, 2H), 4,45 (m, 1H), 3,59 y 3,57 (s, 3H), 3,28 (m, 1H), 3,02 (m, 1H), 2,77 (m, 1H), 2,27 (m, 2H), 1,98 (m, 2H), 1,75 (m, 1H), 1,57 (m, 1H), 1,17 (m, 2H), 0,90 (m, 2H), 0,66 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((5-ciclopropil-7-fluoro-1H-mdoM-il)metil)benzoil)piperidm-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((5-cidopropil-7-fluoro-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (114 mg, 99%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((5-dclopropil-7-fluoro-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (115 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,88 min; MS m/z: 503 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 5 12,10 (ancho, 1H), 7,66 y 7,64 (d, J=8,4 Hz, 1H), 7,49 y 7,41 (d, J=8,4 Hz, 1H), 7,10 (m, 1H), 6,80 (m, 1H), 6,73 y 6,70 (s, 1H), 6,40 (m, 1H), 5,73 (m, 2H), 4,46 (m, 1H), 3,25 (m, 1H), 3,02 (m, 1H), 2,78 (m, 1H), 2,17 (m, 2H), 1,94 (m, 2H), 1,76 (m, 1H), 1,59 (m, 1H), 1,13 (m, 2H), 0,90 (m, 2H), 0,66 (m, 2H).
Ejemplo EO: ácido 2-(1-(2,4-dicloro-3-(5-ciclopropil-7-fluoro-1H-mdoM-carboml)benzoil)piperidm-4-il)acético

Etapa 1: ferc-butil 2,4-dicloro-3-(5-ciclopropil-7-fluoro- 1H-indol-1-carbonil)benzoato

Usando un procedimiento similar al Ejemplo DJ, Etapa 1, se preparó terc-butil 2,4-dicloro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoato (1,05 g, 94%) a partir de 5-cidopropil-7-fluoro-1H-indol (Preparación 66) (400 mg, 2,28 mmol) y terc-butil 2,4-dicloro-3-(fluorocarbonil)benzoato (1 g, 3,4 mmol) (Preparación 55). LC/MS (Método i): Rt = 2,89min; MS miz: 448 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,98 (d, J=9 Hz, 1H), 7,79 (d, J=9 Hz, 1H), 7,28 (d, J=3,8 Hz, 1H), 7,21 (s, 1H), 6,99 (m, 1H), 5,76 (s, 1H), 2,05 (m, 1H), 1,56 (m, 9H), 0,99 (m, 2H), 0,75 (m, 2H).
Etapa 2: ácido 2,4-dicloro-3-(5-ciclopropil-7-fluoro-1H-indoM-carboml)benzoico

Usando un procedimiento similar al Ejemplo DJ, Etapa 2, se preparó ácido 2,4-dicloro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoico (834 mg, 91%) a partir de terc-butil 2,4-didoro-3-(5-ddopropil-7-fluoro-1H-indol-1-carbonil)benzoato (1 g, 2,3 mmol). LC/MS (Método i): Rt = 2,23 min; MS miz: 392 [M+H]+; 1H NMR (DMSO-dí, 300MHz): 5 13,86 (ancho, 1H), 8,03 (d, J=9Hz, 1H), 7,79 (d, J=9Hz, 1H), 7,25 (m, 2H), 6,97 (m, 1H), 6,76 (m, 1H), 2,05 (m, 1H), 0,99 (m, 2H), 0,75 (m, 2H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-(5-ciclopropil-7-fluoro-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-(5-ddopropil-7-fluoro-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (107 mg, 79%) se preparó a partir de ácido 2,4-dicloro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoico (100 mg, 0,25 mmol) y clorhidrato de metil (4-piperidil)acetato (74 mg, 0,38 mmol). LC/MS (Método i): Rt = 2,50 min; MS miz: 531 [M+H]+1H NMR (DMSO-dí, 300MHz): 5 = 8,13-7,14 (m, 4H), 7,02-6,74 (m, 2H), 4,45 (m, 1H), 3,62-3,56 (m, 3H), 3,26-3,15 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,28 (m, 2H), 1,99 (m, 2H), 1,76 (m, 1H), 1,60 (m, 1H), 1,15 (m, 2H), 0,97 (m, 2H), 0,73 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoil)piperidin-4-il)acético (80 mg, 77%) se preparó a partir de metil 2-(1-(2,4-didoro-3-(5-cidopropil-7-fluoro-1H-indol-1-carbonil)benzoil)piperidin-4-il)acetato (107 mg, 0,2 mmol). LC/MS (Método g): Rt = 1,72 min; MS m/z: 517 [M+H]+; 1H NMR (DMSO-d6, 500MHz): 5 12,04 (ancho, 1H), 8,12-7,16 (m, 4H), 7,00-6,73 (m, 2H), 4,45 (m, 1H), 3,56 (m, 1H), 3,22 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,19 (m, 1H), 2,05 (m, 1H), 1,95 (m, 1H), 1,78 (m, 1H), 1,63 (m, 1H), 1,13 (m, 2H), 0,99 (m, 2H), 0,75 (m, 2H).
Ejemplo EP: ácido 2-(1-(2,4-didoro-3-((7-fluoro-5-(trilluorometil)-1H-mdoM-il)metil)benzoil)piperidm-4-il)ace-tico
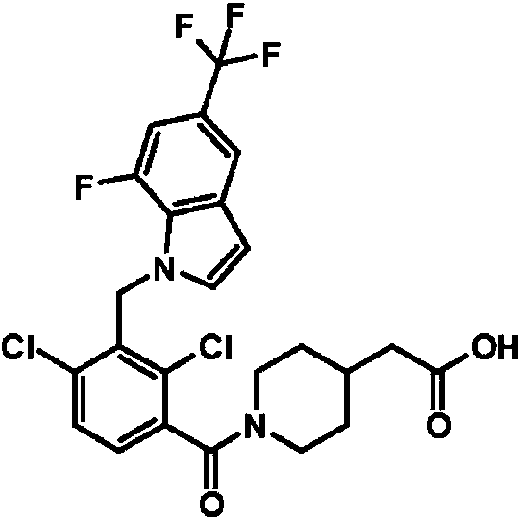
Etapa 1: metil 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-mdoM-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (324 mg, 69%) se preparó a partir de 7-fluoro-5-(trifluorometil)-1H-/ndo/ (Preparación 67) (225 mg, 1,1 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (350 mg, 1,1 mmol) (Preparación #1, Etapa B). LC/MS (Método i): Rt = 2,81 min; MS m/z: 420 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,85 (m, 2H), 7,72 (d, J=8,4Hz, 1H), 7,38 (m, 1H), 7,07 (d, J=3,3 Hz, 1H), 6,71 (m, 1H), 5,88 (s, 2H), 3,87 (s, 3H)
Etapa 2: ácido 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indoM-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (299 mg, 97%) se preparó a partir de metil 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoato (320 mg, 0,76 mmol). LC/MS (Método i): Rt = 2,47 min; MS miz: 406 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 13,73 (ancho, 1H), 7,85 (s, 1H), 7,82 (d, J=8,3 Hz, 1H), 7,70 (J=8,3 Hz, 1H), 7,39 (m, 1H), 7,06 (d, J=3,3 Hz, 1H), 6,71 (m, 1H), 5,88 (s, 2H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-((7-fluoro-5-(trífluorometM)-1H-indoM-N)metN)benzoN)piperídin-4-M)ac-etato

Usando un procedimiento similar al del Ejemplo A1 metil 2-(1-(2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (103 mg, 77%) se preparó a partir de ácido 2,4-didoro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoico (100 mg, 0,26 mmol) y clorhidrato de metil (4-piperidil)acetato (71 mg, 0,36 mmol). LC/MS (Método i): Rt = 2,67 min; MS miz: 545 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,86 (s, 1H), 7,68 y 7,66 (d, J=8,3 Hz, 1H), 7,50 y 7,43 (d, J=8,3 Hz, 1H), 7,38 (m, 1H), 7,08 (m, 1H), 6,72 (m, 1H), 5,84 (m, 2H), 4,45 (m, 1H), 3,59 y 3,57 (s, 3H), 3,28 (m, 1H), 3,02 (m, 1H), 2,77 (m, 1H), 2,27 (m, 2H), 1,96 (m, 1H), 1,76 (m, 1H), 1,56 (m, 1 H), 1,14 (m, 2H). Etapa 4: ácido 2-(1-(2,4-dicloro-3-((7-fluoro-5-(trífluorometN)-1H-indoM-M)metM)benzoM)piperídin-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acético (84 mg, 83%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((7-fluoro-5-(trifluorometil)-1H-indol-1-il)metil)benzoil)piperidin-4-il)acetato (103 mg, 0,17 mmol). LC/MS (Método g): Rt = 1,87 min; MS miz: 531 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 12,11 (br, 1H), 7,86 (s, 1H), 7,68 y 6,66 (d, J=8,3 Hz, 1H), 7,51 y 7,43 (d, J=8,3 Hz, 1H), 7,38 (m, 1H), 7,08 (m, 1H), 6,72 (m, 1H), 5,84 (m, 2H), 4,45 (m, 1H), 3,27 (m, 1H), 3,02 (m, 1H), 2,80 (m, 1H), 2,17 (m, 2H), 1,92 (m, 1H), 1,76 (m, 1H), 1,59 (m, 1H), 1,14 (m, 2H).
Ejemplo EQ: ácido 2-(1-(4,6-didoro-5-(3,7-dimetM-5-(trífluorometM)-1H-mdoM-carboml)nicotmoM)piperídm-4-il)acético

Etapa 1: íerc-butil 4,6-didoro-5-(3,7-dimetM-5-(trífluorometM)-1H-indoM-carbonM)mcotinato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, cbutil 4,6-didoro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinato (1,36 g, 63%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-indol (Preparación #43) (0,95 g, 4,46 mmol) y íerc-butil 4,6-dicloro-5-(clorocarbonil)nicotinato (2,07 g, 6,68 mmol) (Preparación 56). LC/MS (Método i): Rt = 2,81 min; MS m/z: 487 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 59,01 (s, 1H), 7,82 (s, 1H), 7,62 (s, 1H), 7,56 (s, 1H), 2,73 (s, 3H), 2,22 (s, 3H), 1,59 (s, 9H).
Etapa 2: ácido 4,6-didoro-5-(3,7-dimetM-5-(trífluorometN)-1H-indoM-carboml)mcotímco

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 4,6-didoro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotínico (1,48 g, 100%) se preparó a partir de íerc- butil 4,6-didoro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinate (1,36 g, 2,79 mmol). LC/MS (Método i): Rt = 2,30 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 14,20 (ancho, 1H), 9,04 (s, 1H), 7,82 (s, 1H), 7,62 (s, 1H), 7,54 (s, 1H), 2,73 (s, 3H), 2,22 (s, 3H).
Etapa 3: metil 2-(1-(4,6-dicloro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(4,6-didoro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinoil)piperidin-4-il)acetato (280 mg, 42%) se preparó a partir de ácido 4,6-dicloro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotmico (500 mg, 1,16 mmol) y clorhidrato de metil (4-piperidil)acetato (337 mg, 1,74 mmol). LC/MS (Método i): Rt = 2,48 min; MS m/z: 570 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 58,70 (m, 1H), 7,83 (s, 1H), 7,72 y 7,50 (m, 1H), 7,61 (s, 1H), 4,47 (m, 1H), 3,72 (m, 1H), 3,60 (m, 3H), 3,10 (m, 1H), 2,86 (m, 1H), 2,71 (s, 3H), 2,31 (m, 2H), 2,23 (s, 3H), 2,00 (m, 1H), 1,77 (m, 1H), 1,64 (m, 1H), 1,18 (m, 2H).
Etapa 4: ácido 2-(1-(4,6-didoro-5-(3,7-dimetN-5-(trifluorometM)-1H-mdoM-carboml)mcotmoM)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(4,6-didoro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinoil)piperidin-4-il)acético (176 mg, 64%) se preparó a partir de metil 2-(1-(4,6-dicloro-5-(3,7-dimetil-5-(trifluorometil)-1H-indol-1-carbonil)nicotinoil)piperidin-4-il)acetato (278 mg, 0,48 mmol). LC/mS (Método g): Rt = 1,86 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 512,11 (ancho, 1H), 8,70 (m, 1H), 7,83 (s, 1H), 7,72 y 7,50 (m, 1H), 7,62 (s, 1H), 4,47 (m, 1H), 3,73 y 3,45 (m, 1H), 3,10 (m, 1H), 2,85 (m, 1H), 2,71 (s, 3H), 2,23 (s, 3H), 2,17 (m, 2H), 1,96 (m, 1h), 1,75 (m, 1H), 1,66 (m, 1H), 1,15 (m, 2H).
Ejemplo ER: ácido 2-(1-(4,6-didoro-5-((3,7-dimetM-5-(trifluorometN)-1H-mdoM-M)metM)mcotmoN)piperidm-4-il)acético

Etapa 1: íerc-butil 4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinato

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, terc-butil 4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinato (85 mg, 28%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-indol (Preparación #43) (100 mg, 0,47 mmol) y terc-butil 4,6-dicloro-5-(((metilsulfonil)oxi)metil)nicotinato (167 mg, 0,47 mmol) (Preparación 57). LC/MS (Método j): Rt = 2,66 min; MS m/z: 473 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 58,82 (s, 1H), 7,69 (s, 1H), 7,23 (s, 1H), 6,74 (s, 1H), 5,90 (s, 2H), 2,91 (s, 3H), 2,18 (s, 3H), 1,57 (s, 9H).
Etapa 2: ácido 4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotínico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotínico (80 mg, 70%) se preparó a partir de terc-butil 4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinato (85 mg, 0,18 mmol). LC/MS (Método i): Rt = 2,30 min; MS m/z: 417 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 14,14 (ancho, 1H), 8,85 (s, 1H), 7,69 (s, 1H), 7,23 (s, 1H), 6,72 (s, 1H), 5,91 (s, 2H), 2,91 (s, 3H), 2,18 (s, 3H).
Etapa 3: metil 2-(1-(4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinoil)piperidin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A, etapa 6, metil 2-(1-(4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinoil)piperidin-4-il)acetato (60 mg, 60%) se preparó a partir de ácido 4,6-dicloro-5-((3,7-dimetil-4-(trifluorometil)-1H-indol-1-il)metil)nicotínico (70 mg, 0,17 mmol) y clorhidrato de metil (4-piperidil)acetato (41,8 mg, 0,21 mmol). LC/MS (Método i): Rt = 2,58 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 58,56 y 8,49 (s, 1H), 7,69 (s, 1H), 7,23 (s, 1H), 6,76 y 6,73 (s, 1H), 5,88 (m, 2H), 4,46 (m, 1H), 3,50 y 3,57 (s, 3H), 3,40 (m, 1H), 3,07 (m, 1H), 2,90 (s, 3H), 2,84 (m, 1H), 2,29 (s, 2H), 2,19 (s, 3H), 1,99 (m, 1H), 1,76 (m, 1H), 1,60 (m, 1H), 1,17 (m, 2H).
Etapa 4: ácido 2-(1-(4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinoil)piperidin-4-il)acético

Usando un procedimiento similar al del Ejemplo O, Etapa 4, ácido 2-(1-(4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1 -il)metil)nicotinoil)piperidin-4-il)acético (36 mg, 70%) a partir de metil 2-(1-(4,6-dicloro-5-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)nicotinoil)piperidin-4-il)acetato (50 mg, 0,09 mmol). LC/MS (Método g): Rt = 1,80 min; MS miz: 542 [M+H]+; 1H NMR (dMSO-C6, 400MHz): 512,10 (ancho, 1H), 8,56 y 8,49 (s, 1H), 7,69 (s, 1H), 7,23 (s, 1H), 6,75 y 6,73 (s, 1H), 5,87 (m, 2H), 4,48 (m, 1H), 3,40 (m, 1H), 3,07 (m, 1H), 2,90 (s, 3H), 2,83 (m, 1H), 2,19 (m, 5H), 1,95 (m, 1h), 1,80 (m, 1H), 1,63 (m, 1H), 1,17 (m, 2H).
Ejemplo ES: ácido 2-(1-(2,4-didoro-3-(7-metM-5-(trifluorometN)-1H-mdoM-carbonM)benzoN)piperidm-4-N)acético

Etapa 1: íerc-butil 2,4-didoro-3-(7-metM-5-(trifluorometN)-1H-mdoM-carboml)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, íerc-butil 2,4-dicloro-3-(7-metil-5-(trifluorometil)-1H-indol-1-carbonil)benzoato (684 mg, 72%) se preparó a partir 7-metil-5-(trifluorometil)-1 H-indol (Preparación #49) (400 mg, 2 mmol) y íerc-butil 2,4-dicloro-3-(clorocarbonil)benzoato (684 mg, 2,2 mmol) (Preparación #44). LC/MS (Método j): Rt = 2,61 min; MS miz: 472 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,00 (d, J=8,4 Hz, 1H), 7,93 (s, 1H), 7,83 (d, J=8,4 Hz, 1H), 7,60 (s, 1H), 7,48 (d, J=4,0 Hz, 1H), 6,92 (d, J=4,0 Hz, 1H), 2,74 (s, 3H), 1,57 (s, 9h).
Etapa 2: ácido 2,4-didoro-3-(7-metN-5-(trifluorometM)-1H-mdoM-carboml)benzoico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-dicloro-3-(7-met¡l-5-(triflüoromet¡l)-1H-¡ndoN-carbonil)benzoico (595 mg, 99%) se preparó de ferc-butil 2,4-didoro-3-(7-metil-5-(trifluorometil)-1H-indoM-carbonil)benzoato (684 mg, 1,5 mmol). LC/MS (Método j): Rt = 1,68 min; MS m/z: 416 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 5 13,91 (ancho, 1H), 8,06 (d, J=8,4 Hz, 1H), 7,93 (s, 1H), 7,82 (d, J=8,4 Hz, 1H), 7,60 (s, 1H), 7,45 (d, J=3,8 Hz, 1H), 6,92 (d, J=3,8 Hz, 1H), 2,74 (s, 3H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-(7-metil-5-(tnfluorometil)-1H-indoM-carbonil)benzoil)pipendin-4-il)acetato

Usando un procedimiento similar al del Ejemplo A, Etapa 6, metil 2-(1-(2,4-didoro-3-(7-metil-5-(trifluorometil)-1H-indoM-carbonil)benzoil)piperidin-4-il)acetato (118 mg, 88%) se preparó a partir de ácido 2,4-didoro-3-(7-metil-5-(trifluorometil)-1H-indol-1-carbonil)benzoico (100 mg, 0,24 mmol) y clorhidrato de metil (4-piperidil)acetato (93 mg, 0,48 mmol). LC/MS (Método i): Rt = 2,58 min; MS m/z: 555 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 57,93 (m, 1H), 7,82 y 7,80 (d, J=8,4 Hz, 1H), 7,72-7,35 (m, 3H), 6,93 (m, 1H), 4,45 (m, 1H), 3,56 (m, 4H), 3,06 (m, 1H), 2,81 (m, 1H), 2,72 (s, 3H), 2,28 (m, 2H), 1,99 (m, 1H), 1,69-1,77 (m, 1H), 1,61 (m, 1H), 1,18 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-(7-metil-5-(tnfluorometil)-1H-indoM-carbonil)benzoil)pipendin-4-il)acético

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-(7-metil-5-(trifluorometil)-1H-indoM-carbonil)benzoil)piperidin-4-il)acético (99 mg, 84%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-(7-metil-5-(trifluorometil)-1H-indoM-carbonil)benzoil)piperidin-4-il)acetato (118 mg, 0,21 mmol). LC/MS (Método g): Rt = 1,86 min; MS m/z: 541 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 5 12,16 (ancho, 1H), 7,93 (m, 1H), 7,81 y 7,80 (d, J=8,4 Hz, 1H), 7,72 7,36 (m, 3H), 6,93 (m, 1H), 4,47 (m, 1H), 3,55 y 3,24 (m, 1H), 3,06 (m, 1H), 2,81 (m, 1H), 2,72 (s, 3H), 2,18 (m, 2H), 1,95 (m, 1H), 1,77 (m, 1H), 1,63 (m, 1H), 1,16 (m, 2H).
Ejemplo ET: (2,4-dicloro-3-((3,7-dimetil-5-(tnfluorometil)-1H-pin*olo[2,3-c]pindin-1-il)metil)fenil)(morfolino)metanona

Usando un procedimiento similar al Ejemplo DH, Etapa 1, (2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)fenil)(morfolino)metanona (101 mg, 67%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina (Preparación 68) (66 mg, 0,31 mmol) y (2,4-dicloro-3-(clorometil)fenil)(morfolino)metanona (105 mg, 0,34 mmol) (Preparación #11). LC/MS (Método g): Rt = 1,68 min; MS m/z: 486 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 7,87 (s, 1H), 7,73 (d, J=8,4 Hz, 1H), 7,55 (d, J=8,4 Hz, 1H), 6,83 (m, 1H), 5,96 (d, J=15 Hz, 1H), 5,89 (d, J=15 Hz, 1H), 3,64 (m, 4H), 3,54 (m, 2H), 3,19 (m, 2H), 3,08 (s, 3H), 2,20 (m, 3H).
Ejemplo EU: ácido 2-(1-(2)4-dicloro-3-((3)7-dimetM-5-(trifluorometN)-1H-pirrolo[2,3-c]piridin-1-M)metM)benzoM)piperidin-4-M)acético

Etapa 1: metil 2,4-dicloro-3-((3,7-dimetM-5-(tnfluorometM)-1H-pirrolo[2,3-c]piridin-1-M)metM)benzoato

Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoato (600 mg, 74%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina (Preparación 68) (300 mg, 1,4 mmol) y metil 3-(bromometil)-2,4-diclorobenzoato (417 mg, 1,4 mmol) ((Preparación #1, Etapa B). LC/MS (Método i): Rt = 2,60 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,90 (d, J=8,4 Hz, 1H), 7,87 (s, 1H), 7,76 (d, J=8,4 Hz, 1H), 6,82 (s, 1H), 5,96 (s, 2H), 3,88 (s, 3H), 3,09 (s, 3H), 2,19 (s, 3H).
Etapa 2: ácido 2,4-dicloro-3-((3,7-dimetM-5-(trífluorometN)-1H-pirrolo[2,3-c]pirídin-1-M)metM)benzoico

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoico (610 mg, 99%) se preparó a partir de metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoato (600 mg, 1,4 mmol). LC/MS (Método i): Rt = 2,23 min; MS m/z: 417 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,86 (s, 1H), 7,70 (d, J=8,4 Hz, 1H), 7,63 (d, J=8,4 Hz, 1H), 6,78 (s, 1H), 5,93 (s, 2H), 3,09 (s, 3H), 2,18 (s, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-((3,7-dimetN-5-(trífluorometN)-1H-pirrolo[2,3-c]pirídm-1-N)metN)benzoN)piperídm-4-il)acetato
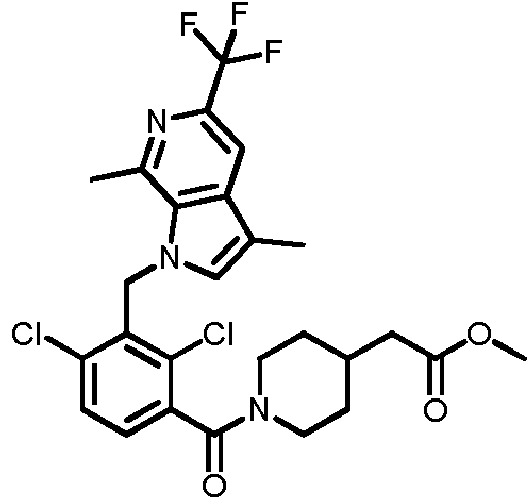
Usando un procedimiento similar al del Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoil)piperidin-4-il)acetato (225 mg, 73%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1 H-pirrolo[2,3-c]piridin-1 -il)metil)benzoico (230 mg, 0,55 mmol) y clorhidrato de metil (4-piperidil)acetato (117 mg, 0,60 mmol). LC/MS (Método i): Rt = 2,46 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-Ó6, 300MHz): 57,87 (s, 1H), 7,72 y 7,71 (d, J=8,3 Hz, 1H), 7,54 y 7,47 (d, J=8,3 Hz, 1H), 6,82 y 6,80 (s, 1H), 5,92 (m, 2H), 4,44 (m, 1H), 3,59 y 3,57 (s, 3H), 3,27 (m, 1H), 3,08 (s, 3H), 3,02 (m, 1H), 2,81 (m, 1H), 2,28 (m, 2H), 2,20 (s, 3H), 1,99 (m, 1H), 1,72 (m, 1H), 1,57 (m, 1H), 1,14 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-didoro-3-((3,7-dimetN-5-(trífluorometN)-1H-pirrolo[2,3-c]pirídm-1-N)metN)benzoN)piperidin-4-il)acético

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoil)piperidin-4-il)acético (130 mg, 59%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridin-1-il)metil)benzoil)piperidin-4-il)acetato (225 mg, 0,40 mmol). LC/MS (Método g): Rt = 1,63 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 5 12,09 (ancho, 1H), 7,87 (s, 1H), 7,71 y
7,70 (d, J=8,3 Hz, 1H), 7,55 y 7,53 (d, J=8,3 Hz, 1H), 6,82 y 6,79 (s, 1H), 5,92 (m, 2H), 4,46 (m, 1H), 3,25 (m, 1H), 3,08 (s, 3H), 3,03 (m, 1H), 2,80 (m, 1H), 2,20 (s, 3H), 2,15 (m, 2H), 1,94 (m, 1H), 1,76 (m, 1H), 1,61 (m, 1H), 1,12 (m, 2H). Ejemplo EV: ácido 2-(1-(2,4-didoro-3-(3,7-dimetN-5-(trifluorometM)-1H-pirrolo[2,3-c]piridma-1-carbonil)benzoil)piperidin-4-il)acético

Etapa 1: íerc-butil 2,4-didoro-3-(3,7-dimetM-5-(tnfluorometM)-1H-pirrolo[2,3-c]piridma-1-carboml)benzoato

Usando un procedimiento similar al del Ejemplo DJ, Etapa 1, íerc-butil 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoato (690 mg, 68%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina (Preparación 68) (350 mg, 1,6 mmol) y íerc-butil 2,4-dicloro-3-(clorocarbonil)benzoato (911 mg, 2,94 mmol) (Preparación #44). LC/MS (Método i): Rt = 2,80 min; MS m/z: 487 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 5 8,05 (s, 1H), 8,03 (d, J=8,4 Hz, 1H), 7,84 (d, J=8,4 Hz, 1H), 7,57 (m, 1H), 2,92 (s, 3H), 2,24 (s, 3H), 1,57 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-(3,7-dimetM-5-(trifluorometN)-1H-pirrolo[2,3-c]pmdma-1-carbonM)benzoico

Usando un procedimiento similar al Ejemplo DJ, Etapa 2, ácido 2,4-didoro-3-(3,7-dimetil-5-(trifluorometil)-1H-pyrro-lo[2,3-c]piridina-1-carbonil)benzoico (370 mg, 52%) se preparó a partir de íerc-butil 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoato (690 mg, 1,4 mmol). LC/MS (Método i): Rt = 2,19 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-^ 6, 300MHz): 58,08 (d, J=8,4 Hz, 1H), 8,05 (s, 1H), 7,84 (d, J=8,4 Hz, 1h), 7,55 (d, J=1,3 Hz, 1H), 2,93 (s, 3H), 2,24 (d, J=1,3 Hz, 3H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(tnfluorometil)-1H-pirrolo[2,3-c]pindina-1-carbonil)benzoil)piperidin-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoil)piperidin-4-il)acetato (85 mg, 58%) se preparó a partir de ácido 2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoico (100 mg, 0,23 mmol) y clorhidrato de metil (4-piperidil)acetato (49 mg, 0,25 mmol). LC/MS (Método i): Rt = 2,45 min; MS m/z: 570 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 58,05 (s, 1H), 7,83 (m, 1H), 7,67 (m, 1,5H), 7,48 (m, 0,5H), 4,46 (m, 1H), 3,60 (m, 3H), 3,28 (m, 1H), 3,06 (m, 1H), 2,91 (s, 3H), 2,82 (m, 1H), 2,30 (m, 2H), 2,25 y 2,24 (s, 3H), 1,98 (m, 1H), 1,77 (m, 1H), 1,62 (m, 1H), 1,17 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(tnfluorometil)-1H-pirrolo[2,3-c]pindina-1-carbonil)benzoil)piperidin-4-il)acético

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoil)piperidin-4-il)acético (48 mg, 57%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-(3,7-dimetil-5-(trifluorometil)-1H-pirrolo[2,3-c]piridina-1-carbonil)benzoil)piperidin-4-il)acetato (85 mg, 0,15 mmol). LC/MS (Método g): Rt = 1,73 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 58,05 (s, 1H), 7,82 (m, 1H), 7,70 (m, 1,5H), 7,47 (m, 0,5H), 3,60 (m, 1H), 3,25 (m, 1H), 3,09 (m, 1H), 2,92 (s, 3H), 2,84 (m, 1H), 2,25 (s, 3H), 2,17 (m, 2H), 1,94 (m, 1H), 1,77 (m, 1H), 1,65 (m, 1H), 1,14 (m, 2H).
Ejemplo EW: ácido2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(tnfluorometil)-1H-pirrolo[3,2-b]pindin-1-il)metil)benzoyl)piperidin-4-il)acético


Usando un procedimiento similar al del Ejemplo DH, Etapa 1, metil 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-1-il)metil)benzoato (579 mg, 96%) se preparó a partir de 3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-6]piridina (300 mg, 1,4 mmol) (Preparación 69) y metil 3-(bromometil)-2,4-diclorobenzoato (417 mg, 1,4mmol) (Preparación n# 1, Etapa B). LC/MS (Método i): Rt = 2,56 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,89 (d, J=8,0 Hz, 1H), 7,76 (d, J=8,0 Hz, 1H), 7,46 (s, 1H), 6,92 (m, 1H), 5,91 (s, 2H), 3,88 (s, 3h), 2,96 (s, 3H), 2,19 (m, 3H).

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-1-il)metil)benzoico (545 mg, 97%) se preparó a partir de 2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-ib]piridin-1-il)metil)benzoato (579 mg, 1,34 mmol). LC/MS (Método i): Rt = 2,23 min; MS m/z: 417 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 513,70 (ancho, 1H), 7,85 (d, J=8,4 Hz, 1H), 7,72 (d, J=8,4 Hz, 1H), 7,46 (s, 1H), 6,92 (m, 1H), 5,90 (s, 2H), 2,96 (s, 3H), 2,19 (m, 3H).
Etapa 3: metil 2-(1-(2,4-didoro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-fo]piridm-1-il)metil)benzoil)piperidm-4-il)acetato

Usando un procedimiento similar al del Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-¿]piridin-1-il)metil)benzoil)piperidin-4-il)acetato (86 mg, 64%) se preparó a partir de ácido 2,4-dicloro-3-((3,7-dimetil-5-(triíluorometil)-1H-pirrolo[3,2-i)]piridin-1-il)metil)benzoico (100 mg, 0,24 mmol) y clorhidrato de metil (4
piperidil)acetato (93 mg, 0,48 mmol). LC/MS (Método i): Rt = 2,44 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 5 = 7,71 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,45 (s, 1H), 6,93 y 6,91 (m, 1H), 5,87 (m, 2H), 4,44 (m, 1H), 3,59 y 3,57 (s, 3H), 3,31 (m, 1H), 3,05 (m, 1H), 2,95 (s, 3H), 2,79 (m, 1H), 2,28 (m, 2H), 2,20 (m, 3H), 1,97 (m, 1H), 1,75 (m, 1H), 1,57 (m, 1H), 1,15 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(tnfluorometil)-1H-pin*olo[3,2-b]pindin-1-il)metil)benzoil)pipendin-4-il)acético

Usando un procedimiento similar al del Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-b]piridin-1-il)metil)benzoil)piperidin-4-il)acético (64 mg, 80%) se preparó a partir de metil 2-(1-(2,4-dichlo-ro-3-((3,7-dimetil-5-(trifluorometil)-1H-pirrolo[3,2-b]piridin-1-il)metil)benzoil)piperidin-4-il)acetato (81 mg, 0,15 mmol). LC/MS (Método i): Rt = 2,20 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 512,18 (ancho, 1H), 7,70 y 7,69 (d, J=8,3 Hz, 1H), 7,53 y 7,46 (d, J=8,3 Hz, 1H), 7,45 (s, 1H), 6,94 y 6,91 (s, 1H), 5,87 (m, 2h), 4,46 (m, 1H), 3,28 (m, 1H), 3,03 (m, 1H), 2,94 (s, 3H), 2,79 (m, 1H), 2,20 (m, 3H), 2,13 (m, 2H), 1,92 (m, 1H), 1,76 (m, 1H), 1,61 (m, 1H), 1,14 (m, 2H). Ejemplo EX: 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetiMH-indol-5-carbonitnlo

Etapa 1: metil 2,4-dicloro-3-((5-ciano-4-metiM-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-((5-ciano-4-metiM-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (1,92 g, 76%) se preparó a partir de N-(4-ciano-2-yodo-3-metilfenil)bencenosulfonamida (1,7 g, 4,3 mmol) (Preparación 70) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (1,2 g, 4,7 mmol) (Preparación #1). LC/MS (Método i): Rt = 2,41 min; MS m/z: 587 [M-H]- + CH3COOH; 1H NMR (DMSO-cf6, 300MHz): 58,01 (d, J=8,8 Hz, 1H), 7,90 (m, 2H), 7,69 (m, 3H), 7,58 (m, 3H), 7,04 (d, J=5,8 Hz, 1H), 6,88 (s, 1H), 6,73 (d, J=5,8 Hz, 1H), 3,86 (s, 3H), 2,57 (s, 3H).
Etapa 2: metil 2,4-dicloro-3-((5-ciano-4-metiM-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (10459453-791-001)

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-dicloro~3~((5-ciano-4-metil-1~(fen¡lsulfon¡l)~1H~¡ndol-2~ il)metil)benzoato (1,74 g, 85%) se preparó a partir de metil 2,4-didoro-3-((5-dano-4-metil-1-(fenil-sulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (1,9 g, 3,6 mmol).
LC/MS (Método i): Rt = 2,74 min; MS m/z: 513 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 58,14 (d, J=8,9 Hz, 1H), 7,99 (m, 2H), 7,82 (m, 2H), 7,86 (m, 4H), 6,05 (s, 1H), 4,62 (s, 2H), 3,87 (s, 3H), 2,46 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-((5-ciano-4-metiMH-indol-2-il)metil)benzoato
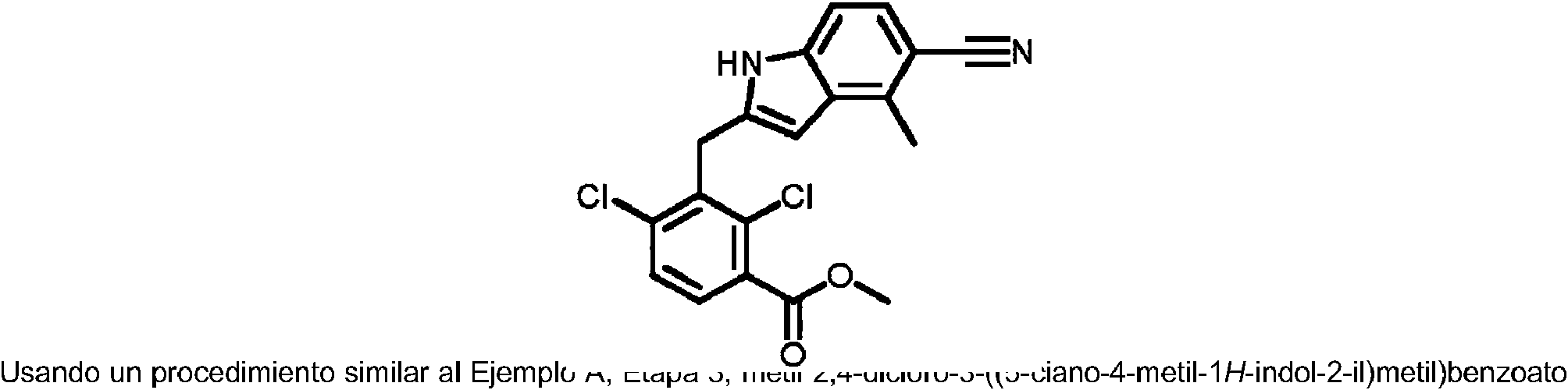
(1,13 g, 83%) se preparó a partir de metil 2,4-didoro-3-((5-ciano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (1,72 g, 3,3 mmol). LC/MS (Método i): Rt = 2,38 min; MS m/z: 373 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 5 11,63 (s, 1H), 7,77 (d, J=8,4 Hz, 1H), 7,69 (d, J=8,4 Hz, 1H), 7,31 (s, 2H), 6,03 (m, 1H), 4,48 (s, 2H), 3,87 (s, 3H), 2,53 (s, 3H).
Etapa 4: metil 2,4-dicloro-3-((5-ciano-1,4-dimetiMH-indol-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((5-dano-1,4-dimetil-1H-indol-2-il)metil)benzoato (1,1 g, 94%) se preparó a partir de metil 2,4-dicloro-3-((5-ciano-4-metil-1H-indol-2-il)metil)benzoato (1,1 g, 2,95 mmol). LC/MS (Método i): Rt = 2,48 min; MS m/z: 387 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 57,80 (d, J=8,1 Hz, 1H), 7,72 (d, J=8,1 Hz, 1H), 7,49 (d, J=8,6 Hz, 1H), 7,40 (d, J=8,6 Hz, 1H), 5,75 (s, 1H), 4,48 (s, 2H), 3,90 (s, 3H), 3,88 (s, 3H), 2,48 (s, 3H).
Etapa 5: ácido 2,4-dicloro-3-((5-ciano-1,4-dimetiMH-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoico (1,01 g, 93%) se preparó a partir de metil 2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoato (1,1 g, 2,81 mmol). LC/MS (Método i): Rt = 2,15 min; MS m/z: 373 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 5 13,71 (ancho,
1H), 7,75 (d, J=8,4 Hz, 1H), 7,67 (d, J=8,4 Hz, 1H), 7,49 (d, J=8,8 Hz, 1H), 7,40 (d, J=8,8 Hz, 1H), 5,75 (s, 1H), 4,47 (s, 2H), 3,89 (s, 3H), 2,49 (s, 3H).
Etapa 6: 2-(2,6-didoro-3-(morfolma-4-carboml)bendl)-1,4-dimetiMH-mdol-5-carbonitrMo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-(2,6-didoro-3-(morfolina-4-carbonil)bendl)-1,4-dimetiM H-indol-5-carbonitrilo (80 mg, 66%) se preparó a partir de ácido 2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoico (100 mg, 0,27 mmol) y morfolina (28 mg, 0,32 mmol). LC/MS (Método g): Rt = 1,60 min; MS miz: 442 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 57,68 (d, J=8,1 Hz, 1H), 7,49 (d, J=8,4 Hz, 1H), 7,46 (d, J=8,4 Hz, 1H), 7,40 (d, J=8,1 Hz, 1H), 5,77 (s, 1H), 4,44 (m, 2H), 3,89 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,19 (m, 2H), 2,49 (s, 3H).
Ejemplo EY: ácido 2-(1-(2,4-didoro-3-((5-dano-1,4-dimetiMH-mdol-2-M)metN)benzoM)piperidm-4-N)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((5-dano-1,4-dimetiMH-mdol-2-M)metM)benzoM)piperidm-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (150 mg, 84%) se preparó a partir de ácido 2,4-dicloro-3-((5-dano-1,4-dimetil-1H-indol-2-il)metil)benzoico (130 mg, 0,35 mmol) (Ejemplo EX, Etapa 5) y clorhidrato de metil (4-piperidil)acetato (81 mg, 0,42 mmol). LC/MS (Método i): Rt = 2,33 min; MS miz: 512 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,66 y 7,64 (d, J=8,3 Hz, 1H), 7,43 (m, 3H), 5,76 y 7,72 (s, 1H), 4,46 (m, 3H), 3,89 (s, 3H), 3,59 y 3,56 (s, 3H), 3,30 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1h), 2,49 y 2,47 (s, 3H), 2,27 (m, 2h), 1,96 (m, 1h), 1,75 (m, 1H), 1,59 (m, 1H), 1,14 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((5-dano-1,4-dimetiMH-mdol-2-N)metM)benzoN)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (122 mg, 88%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((5-ciano-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (140 mg, 0,27 mmol). LC/MS (Método g): Rt = 1,56 min; MS miz: 498 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 512,10 (ancho, 1h), 7,66 y 7,64 (d, J=8,4 Hz, 1H), 7,42 (m, 3H), 5,76 y 5,72 (s, 1H), 4,47 (m, 3H), 3,89 y 3,88 (s, 3H), 3,28 (m, 1H), 3,05 (m, 1H), 2,78 (m, 1H), 2,49 y 2,47 (s, 3H), 2,14 (m, 2H), 1,93 (m, 1h), 1,77 (m, 1h), 1,62 (m, 1H), 1,14 (m, 2H).
Ejemplo EZ: (2,4-didoro-3-((4,6-didoro-1-metiMH-mdol-2-M)metM)feml)(morfolmo)metanona

Etapa 1: íerc-butil 2,4-didoro-3-((4,6-didoro-1-(femlsulfoml)-1H-mdol-2-M)(hidroxi)metM)benzoato

Usando un procedimiento similar al del Ejemplo CJ, Etapa 1, íerc-butil 2,4-dicloro-3-((4,6-didoro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (870 mg, 44%) se preparó a partir de 4,6-dicloro-1-(fenilsulfonil)-1H-indol (1,08 g, 3,31 mmol) (Preparación 71) y íerc-butil 2,4-dicloro-3-formilbenzoato (900 mg, 3,27 mmol) (Preparación #33, Etapa B). LC/MS (Método j): Rt = 2,70 min; MS m/z: 658 [M-H]- CH3COOH; 1H NMR (DMSO-de, 300MHz): 5 7,99 (s, 1H), 7,87 (m, 2H), 7,73 (m, 1H), 7,54 (m, 5H), 6,97 (s, 1H), 6,76 (s, 1H), 6,67 (s, 1H), 1,54 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-((4,6-didoro-1-(femlsulfoml)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((4,6-didoro-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoico (565 mg, 60%) se preparó a partir de íerc-butil 2,4-dicloro-3-((4,6-didoro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (1 g, 1,76 mmol). LC/Ms (Método j): Rt = 2,30 min; mS m/z: 526 [M-H]-; 1H NmR (DMSO-d e, 300MHz): 5 13,68 (ancho, 1H), 8,14 (m, 1H), 8,02 (m, 2H), 7,80 (m, 2H), 7,69 (m, 3H), 7,55 (m, 1H), 5,71 (m, 1H), 4,57 (m, 2H).
Etapa 3: ácido 2,4-didoro-3-((4,6-didoro-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A1, (2,4-dicloro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)fenil)(morfolino)metanona (39 mg, 52%) se preparó a partir de ácido 2,4-dicloro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)benzoico (80 mg, 0,16 mmol) y morfolina (27,7 mg, 0,32 mmol). LC/MS (Método g): Rt = 1,96 min; MS miz: 471 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 57,68 (d, J=8,4 Hz, 1H), 7,66 (m, 1H), 7,46 (d, J=8,4 Hz, 1H), 7,13 (d, J=1,5 Hz, 1H), 5,55 (m, 1H), 4,43 (m, 2H), 3,87 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,20 (m, 2H).
Ejemplo FA: ácido 2-(1-(2,4-didoro-3-((4,6-didoro-1-metiMH-mdol-2-N)metN)benzoN)piperidm-4-N)acético

Etapa 1: metil 2-(1-(2,4-didoro-3-((4,6-didoro-1-metiMH-mdol-2-N)metN)benzoM)piperidm-4-N)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-dicloro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (32 mg, 47%) se preparó a partir de ácido 2,4-dicloro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)benzoico (63 mg, 0,12 mmol) (Ejemplo EZ, Etapa 5) y clorhidrato de metil (4-piperidil)acetato (96 mg, 0,5 mmol). LC/MS (Método i): Rt = 2,69 min; MS miz: 541 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,66 (m, 2H), 7,44 y 7,37 (d, J=8,3 Hz, 1H), 7,13 (m, 1H), 5,52 (m, 1H), 4,44 (m, 3H), 3,87 y 3,86 (s, 3H), 3,59 y 3,56 (s, 3H), 3,23 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1h), 2,25 (m, 2H), 1,86 (m, 1H), 1,77 (m, 1H), 1,59 (m, 1H), 1,14 (m, 2H).
Etapa 2: ácido 2-(1-(2,4-didoro-3-((4,6-didoro-1-metiMH-mdol-2-N)metN)benzoN)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (17 mg, 57%) se preparó a partir de metil 2-(1-(2,4-didoro-3-((4,6-didoro-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (30 mg, 0,055 mmol). LC/MS (Método g): Rt = 1,87 min; MS miz: 527 [M+H]+;
1H NMR (DMSO-Ce, 400MHz): 512,13 (ancho, 1H), 7,66 (m, 2H), 7,44 y 7,37 (d, J=8,4 Hz, 1H), 7,13 (m, 1H), 5,53 (m, 1H), 4,44 (m, 3H), 3,87 y 3,86 (s, 3H), 3,24 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,15 (m, 2h), 1,93 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,14 (m, 2H).
Ejemplo FB: 2-((2,4-didoro-5-(2-oxa-8-azaspiro[4,5]decano-8-carboml)piridm-3-N)metN)-1,4-dimetiMH-mdol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 1, etil 4,6-didoro-5-((6-dano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)nicotinato (4,46 g, 41%) se preparó a partir de N-(5-ciano-2-yodo-3-metilfenil)bencenosulfonamida (7,91 g, 19,8mmol) (Preparación #18) y etil 4,6-dicloro-5-(1-hidroxiprop-2-in-1-il)nicotinato (6,53g, 23,82 mmol) (Preparación #38). LC/MS (Método i): Rt = 2,40 min; MS m/z: 544 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 58,70 (s, 1H), 8,24 (s, 1H), 7,89 (m, 2H), 7,71 (m, 1H), 7,56 (m, 3H), 7,07 (s, 1H), 6,93 (s, 2H), 4,37 (q, J=7,1 Hz, 2H), 2,45 (s, 3H), 1,33 (t, J=7,1 Hz, 3H).
Etapa 2: etil 4,6-dicloro-5-((6-ciano-4-metiM-(fenMsulfonN)-1H-indol-2-N)metM)nicotinato

Usando un procedimiento similar al del Ejemplo Z, Etapa 2, etil 4,6-didoro-5-((6-dano-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)nicotinato (3,6 g, 83%) se preparó a partir de etil 4,6-didoro-5-((6-dano-4-metil-1-(fenilsulfo-nil)-1H-indol-2-il)(hidroxi)metil)nicotinato (4,46 g, 8,19 mmol). LC/MS (Método i): Rt = 2,71 min; MS m/z: 528 [M+H]+;
1H NMR (DMSO-de, 300MHz): 58,84 (s, 1H), 8,35 (s, 1H), 8,05 (m, 2H), 7,78 (m, 1H), 7,68 (m, 2H), 7,48 (m, 1H), 6,34 (s, 1H), 4,62 (s, 2H), 4,37 (q, J=7,1 Hz, 2H), 2,33 (s, 3H), 1,34 (t, J=7,1 Hz, 3H).
Etapa 3: ácido 4,6-dicloro-5-((6-ciano-4-metiMH-indol-2-N)metM)nicotmico

Usando un procedimiento similar al Ejemplo A, Etapa 3, 4,6-did oro-5-((6-d ano-4-metiMH -indol-2-il)metil)nicotínico (3,1 g, 100%) se preparó a partir de etil 4,6-did oro-5-((6-d ano-4-metil-1-(fenilsulfonil)-1H -indol-2-il)metil)nicotinato (3,55 g, 6,73 mmol). LC/MS (Método i): Rt = 1,85 min; MS miz: 360 [M+H]+; 1H NMR (DMSO-C e, 300MHz): 5 11,67 (s, 1H), 8,63 (s, 1H), 7,63 (s, 1H), 7,09 (s, 1H), 6,08 (s, 1H), 4,47 (s, 2H), 2,39 (s, 3H).
Etapa 4: metil 4,6-didoro-5-((6-dano-1,4-dimetiMH-mdol-2-M)metM)nicotmato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 4,6-dicloro-5-((6-d ano-1,4-dimetil-1H-indol-2-il)metil)nicotinato (1,05 g, 31%) se preparó a partir de ácido 4,6-did oro-5-((6-d ano-4-metil-1H-indol-2-il)metil)nicotínico (3,1 g, 8,61 mmol). LC/MS (Método i): Rt = 2,35 min; MS miz: 388 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,84 (s, 1H), 7,93 (s, 1H), 7,12 (s, 1H), 5,87 (s, 1H), 4,50 (s, 2H), 3,92 (s, 3H), 3,91 (ds, 3H), 2,34 (s, 3H).
Etapa 5: ácido 4,6-didoro-5-((6-dano-1,4-dimetiMH-mdol-2-N)metil)mcotmico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-((6-d ano-1,4-dimetil-1H-indol-2-il)metil)nicotínico (870 mg, 86%) se preparó a partir metil 4,6-dicloro-5-((6-d ano-1,4-dimetil-1H-indol-2-il)metil)nicotinato (1,05 g, 2,7 mmol). LC/MS (Método i): Rt = 1,87 min; MS miz: 374 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 58,74 (s, 1H), 7,92 (s, 1H), 7,12 (s, 1H), 5,84 (s, 1H), 4,49 (s, 2H), 3,91 (s, 3H), 2,34 (s, 3H).
Etapa 6: 2-((2,4-didoro-5-(2-oxa-8-azaspiro[4,5]decano-8-carboml)piridm-3-M)metM)-1,4-dimetiMH-mdol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-((2,4-dicloro-5-(2-oxa-8-azaspiro[4,5]decane-8-carbonil)piridin-3-il)metil)-1,4-dimetiMH-indol-6-carbonitrilo (20 mg, 15%) se preparó a partir de ácido 4,6-dicloro-5-((6-dano-1,4-dimetil-1H-indol-2-il)metil)nicotínico (100 mg, 0,27 mmol) y 2-oxa-8-azaspiro[4,5]decano (49 mg, 0,35 mmol). LC/MS (Método g): Rt = 1,60 min; MS m/z: 497 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 58,48 (s, 1H), 7,92 (s, 1H), 7,12 (s, 1H), 5,86 (m, 1h), 4,47 (m, 2H), 3,90 (s, 3H), 3,75 (m, 4H), 3,48 (m, 2H), 3,24 (m, 2H), 2,35 (s, 3H), 1,74 (m, 2H), 1,56 (m, 4H).
Ejemplo FC: 2-((2,4-dicloro-5-(4-(oxetan-3-il)pipendina-1-carbonil)pindin-3-il)metil)-1,4-dimetiMH-indol-6-carbonitrilo

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-((2,4-dicloro-5-(4-(oxetan-3-il)piperidina-1-carbonil)piridin-3-il)metil)-1,4-dimetiMH-indol-6-carbonitrilo (54 mg, 38%) se preparó a partir de ácido 4,6-dicloro-5-((6-dano-1,4-dimetil-1H-indol-2-il)metil)nicotínico (100 mg, 0,27 mmol) y 4-(oxetan-3-il)piperidina (49 mg, 0,35 mmol) (Ejemplo FB, Etapa 5). LC/MS (Método g): Rt = 1,56 min; MS m/z: 497 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 58,51 y 8,44 (s, 1H), 7,92 (s, 1h), 7,12 (s, 1H), 5,88 y 5,83 (s, 1H), 4,46 (m, 7H), 3,90 (s, 3H), 3,43 (m, 1H), 3,07 (m, 1H), 2,71 (m, 2H), 2,35 y 2,33 (s, 3h), 1,91 (m, 1h), 1,69 (m, 1H), 1,54 (m, 1H), 1,02 (m, 2H).
Ejemplo FD: ácido 2-(1-(2,4-dicloro-3-((6-metoxM,4-dimetiMH-indol-2-il)metil)benzoil)pipendin-4-il)acético

Etapa 1: ferc-butil 2,4-dicloro-3-(hidroxi(6-metoxi-4-metiM-(fenilsulfonil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo CJ, Etapa 1, ferc-butil 2,4-dicloro-3-(hidroxi(6-metoxi-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (732 mg, 45%) se preparó a partir de 6-metoxi-4-metil-1-(fenilsulfonil)-1H-indol (857 mg, 2,84 mmol) (preparación 72) y ferc-butil 2,4-dicloro-3-formilbenzoato (939 mg, 3,41 mmol) (Preparación #33, Etapa B). LC/MS (Método i): Rt = 2,71 min; MS m/z: 634 [M-H]- + CH3COOH; 1H NMR (DMSO-cf6, 300MHz): 57,90 (m, 2H), 7,67 (m, 1H), 7,57 (m, 4H), 7,31 (m, 1H), 7,01 (m, 1H), 6,71 (m, 1H), 6,53 (s, 1H), 6,46 (m, 1H), 3,78 (s, 3H), 2,30 (s, 3h), 1,54 (s, 9H).
Etapa 2: ácido 2,4-dicloro-3-((6-metoxi-4-metiM-(femlsulfoml)-1H-mdol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((6-metoxi-4-metiM-(fenilsulfonil)-1H-indol-2-il)metil)benzoico (638 mg, 100%) se preparó a partir de ferc-butil 2,4-dicloro-3-(hidroxi(6-metoxi-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (730 mg, 1,27 mmol). LC/MS (Método i): Rt = 2,44 min; MS m/z: 504 [M+H]+; 1H NMR (DMSO-de, 300MHz): 5 13,65 (ancho, 1H), 7,92 (m, 2H), 7,75 (m, 2H), 7,66 (m, 3H), 7,47 (m 1H), 6,71 (m, 1H), 5,67 (s, 1H), 4,56 (s, 2H), 3,81 (s, 3H), 2,20 (s, 3H).
Etapa 3: ácido 2,4-dicloro-3-((6-metoxi-4-metiMH-mdol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, se preparó ácido 2,4-dicloro-3-((6-metoxi-4-metiMH-indol-2-il)metil)benzoico a partir de ácido 2,4-dicloro-3-((6-metoxi-4-metiM-(fenilsulfonil)-1H-indol-2-il)metil)benzoico (55 mg, 1,1 mmol) y se usó crudo en la siguiente etapa. LC/MS (Método i): Rt = 2,11 min; MS m/z: 364 [M+H]+
Etapa 4: metil 2,4-dicloro-3-((6-metox¡-1,4-d¡met¡MH-mdol-2-il)met¡l)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((6-metoxi-1,4-dimetiMH-indol-2-il)metil)benzoato (500 mg, 100%)se preparó a partir de ácido 2,4-dicloro-3-((6-metoxi-4-metiM-(fenilsulfonil)-1H-indol-2-il)metil)benzoico crudo. LC/MS (Método i): Rt = 2,53 min; MS m/z: 392 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,77 (d, J=7,8 Hz, 1H), 7,70 (d, J=7,8 Hz, 1H), 6,81 (m, 1H), 6,43 (m, 1H), 5,41 (m, 1H), 4,40 (s, 2H), 3,88 (s, 3H), 3,77 (s, 3H), 3,76 (s, 3H), 2,22 (s, 3H).
Etapa 5: ácido 2,4-dicloro-3-((6-metoxM,4-dimetiMH-mdol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoico (305 mg, 62%) se preparó a partir de metil 2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoato (509 mg, 1,3 mmol). LC/MS (Método i): Rt = 2,20 min; MS m iz: 378 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 13,59 (ancho, 1H), 7,73 (d, J=8,3 Hz, 1H), 7,65 (d, J=8,3 Hz, 1H), 6,81 (d, J=2,0 Hz, 1H), 6,43 (m, 1H), 5,42 (m, 1H), 4,39 (s, 2H), 3,77 (s, 3H), 3,76 (s, 3H), 2,23 (s, 3H).
Etapa 6: metil 2-(1-(2,4-dicloro-3-((6-metoxM,4-dimetiMH-indol-2-N)metN)benzoM)piperídin-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (83 mg, 61%) se preparó a partir de ácido 2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoico (100 mg, 0,26 mmol) y clorhidrato de metil (4-piperidil)acetato (102 mg, 0,53 mmol). LC/MS (Método i): Rt = 2,37 min; MS miz: 517 [M+H]+1H NMR (DMSO-d6, 300MHz): 57,64 y 7,62 (d, J=8,3 Hz, 1H), 7,41 y 7,34 (d, J=8,3 Hz, 1H), 6,81 (s, 1H), 6,43 (m, 1H), 5,42 y 5,39 (s, 1H), 4,41 (m, 3H), 3,77 (s, 6H), 3,59 y 3,57 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,28 (m, 2H), 2,22 y 2,21 (s, 3H), 1,86-2,05 (m, 1H), 1,75 (m, 1H), 1,58 (m, 1H), 1,17 (m, 2H).
Etapa 7: ácido 2-(1-(2,4-dicloro-3-((6-metoxi-1,4-dimetiMH-indol-2-M)metM)benzoN)piperídin-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (55 mg, 68%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((6-metoxi-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (80 mg, 0,15 mmol). LC/MS (Método i): Rt = 2,13 min; MS miz: 503 [M+H]+; 1H NMR (DMSO-de, 400MHz): 5 = 12,08 (s, 1H), 7,64 y 7,62 (d, J=8,3 Hz, 1H), 7,41 y 7,34 (d, J=8,3 Hz, 1H), 6,80 (s, 1H), 6,43 (s, 1H), 5,43 y 5,40 (s, 1H), 4,36 (m, 3H), 3,77 (s, 6H), 3,27 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1H), 2,21 y 2,21 (s, 3H), 2,14 (m, 2H), 1,95 (m, 1H), 1,79 (m, 1H), 1,61 (m, 1H), 1,16 (m, 2H).
Ejemplo FE: ácido 2-(1-(2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético

Etapa 1: terc-butil 2,4-dicloro-3-((4-fluoro-1 -metil-6-(trifluorometil)-1 H-mdol-2-M)(hidroxi)metM)benzoato

A una solución de íerc-butil 2,4-diclorobenzoato (125 mg, 0,507 mmol) (Preparación #33, Etapa A) en THF (1,5 mL) bajo argón y enfriado a -78 °C se añadió gota a gota diisopropilamida de litio (0,230 ml, 0,461 mmol). La mezcla de reacción se agitó durante 1 hora a -78 °C y luego se añadió 4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-carbaldehyde (113 mg, 0,461 mmol) (Preparación #73) en solución en THF (1,5 mL). La mezcla de reacción se agitó durante 30 minutos a -78 °C. La mezcla de reacción se inactivó con una solución saturada de NaHCO3 y se extrajo dos veces con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (eluyendo con EtOAc al 5-20% en ciclohexano) para dar tere- butil 2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)(hidroxi)metil)benzoato (156 mg, 69%). LC/MS (Método i): Rt = 2,76 min; MS m iz : 492 [M+H]+; 1H NMR (DMSO-ck, 300MHz): 57,83 (s, 1H), 7,66 (d, J=8,3 Hz, 1H), 7,61 (d, J=8,3 Hz, 1H), 7,15 (dd, J=10,7, 0,7 Hz, 1H), 6,73 (d, J=5,4 Hz, 1H), 6,59 (d, J=5,6 Hz, 1H), 6,13 (s, 1H), 3,90 (s, 3H), 1,55 (s, 9H).
Etapa 2: ácido 2,4-didoro-3-((4-fluoro-1-metM-6-(tnfluorometM)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (43 mg, 100%) se preparó a partir de terc-butil 2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)(hidroxi)metil)benzoato (50 mg, 0,1 mmol). LC/MS (Método i): Rt = 2,43 min; MS miz: 420 [M+H]+; 1H NMR (DMSO-ck, 300MHz): 5 13,50 (ancho, 1H), 7,83 (s, 1H), 7,77 (d, J=8,9 Hz, 1H), 7,68 (d, J=8,9 Hz, 1H), 7,12 (dd, J=10,4, 0,7 Hz, 1H), 5,69 (d, J=0,7 Hz, 1H), 4,51 (s, 2H), 3,97 (s, 3H).
Etapa 3: metil 2-(1-(2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperid-in-4-il)acetato

Usando un procedimiento similar al del Ejemplo A1, metil 2-(1-(2,4-didoro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (48 mg, 60%) se preparó a partir de ácido 2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoico (60 mg, 0,14 mmol) y clorhidrato de metil (4-piperidil)acetato (41 mg, 0,21 mmol). LC/MS (Método i): Rt = 2,58 min; MS miz: 559 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 5 7,83 (s, 1H), 7,66 y 7,65 (d, J=8,2 Hz, 1H), 7,45 y 7,38 (d, J=8,2 Hz, 1H), 7,12 (d, J=10,2 Hz, 1H), 5,72 y 5,68 (s, 1H), 4,47 (m, 3H), 3,97 (s, 3H), 3,59 y 3,56 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,81 (m, 1H), 2,26 (m, 2H), 1,95 (m, 1H), 1,75 (m, 1H), 1,56 (m, 1H), 1,15 (m, 2H).
Etapa 4: ácido 2-(1-(2,4-dicloro-3-((4-fluoro-1-metM-6-(trifluorometM)-1H-mdol-2-N)metM)benzoM)piperidm-4-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (40 mg, 86%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((4-fluoro-1-metil-6-(trifluorometil)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (46 mg, 0,08 mmol). LC/MS (Método g): Rt = 1,83 min; MS miz: 545 [M+H]+; 1H NMR (DMSO-ó6, 400MHz): 5 = 12,14 (ancho, 1H), 7,82 (s, 1H), 7,66 y 7,65 (d, J=8,2 Hz, 1H), 7,45 y 7,38 (d, J=8,2 Hz, 1H), 7,12 (d, J=10,3 Hz, 1H), 5,72 y 5,68 (s, 1H), 4,47 (m, 3H), 3,97 y 3,96 (s, 3H), 3,24 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1H), 2,15 (m, 2H), 1,93 (m, 1H), 1,79 (m, 1H), 1,60 (m, 1H), 1,14 (m, 2H).
Los siguientes ejemplos se prepararon a partir de acido 2.4-dicloro-3-((4-fluoro-1-metil-6-<trltluorometil>-1H-indol-2-il)metil>benzoico (ejemplo FE, etapa el mismo procedimiento con las aminas apropiadas como se describe en el ejemplo FE. etapas 3 y 4.

Ejemplo FF: ácido 2-(1-(2,4-dieloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético
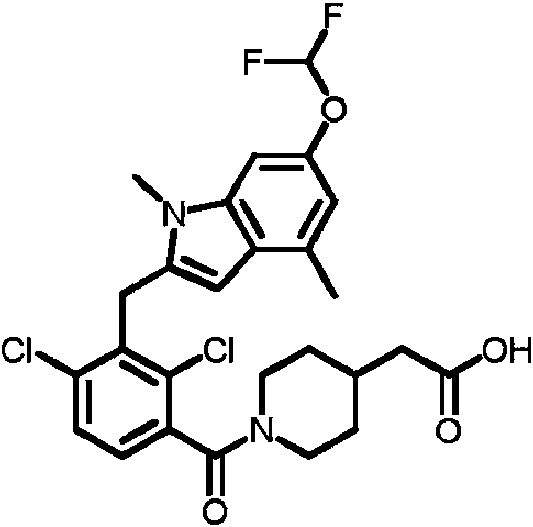
Etapa 1: terc-butil 2,4-dicloro-3-((6-(-difluorometoxi)-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato

Usando un procedimiento similar al Ejemplo CJ, Etapa 1, terc-butil 2,4-didoro-3-((6-(difluorometoxi)-4-metil-1-(fenilsulfonil)-1/7-indol-2-il)(hidroxi)metil)benzoato (807 mg, 36%) se preparó a partir de 6-(difluorometoxi)-4-metil-1-(fenilsulfonil)-1/7-indol (1,2 g, 3,6 mmol) (Preparación #ZZ-24) y terc-butil 2,4-dicloro-3-formilbenzoato (1 g, 3,97 mmol)
(Preparación #33, Etapa B). LC/MS (Método i): Rt = 2,77 min; MS m/z: 670 [M-H]- CH3COOH; 1H NMR (DMSO-d6, 300MHz): 57,88 (m, 2H), 7,68 (m, 1H), 7,56 (m, 5H), 7,23 (t, J=73 Hz, 1H), 6,98 (m, 2H), 6,69 (s, 1H), 6,58 (s, 1H), 2,37 (s, 3H), 1,54 (s, 9H).
Etapa 2: ácido 2,4-dicloro-3-((6-(-difluorometoxi)-4-metil-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-dicloro-3-((6-(difluorometoxi)-4-metil-1-(fenilsulfo-nyl)-1H-indol-2-il)metil)benzoico (666 mg, 100%) se preparó a partir de ferc-butil 2,4-dicloro-3-((6-(difluorometoxi)-4-metil-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (655 mg, 1 mmol). LC/MS (Método j): Rt = 1,92 min; mS m/z: 540 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 513,6 (ancho, 1H), 7,94 (m, 2H), 7,76 (m, 3H), 7,66 (m, 3H), 7,24 (t, J=74,3 Hz, 1H), 6,94 (s, 1H), 5,80 (s, 1H), 4,58 (s, 2H), 2,26 (s, 3H).
Etapa 3: ácido 2,4-dicloro-3-((6-(difluorometoxi)-4-metiMH-indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, 2,4-dicloro-3-((6-(difluorometoxi)-4-metil-1H-indol-2-il)metil)benzoico (456 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((6-(difluorometoxi)-4-metiM-(fenilsulfonil)-1 H-indol-2-il)metil)benzoico (616 mg, 1,1 mmol). El crudo se usó en el siguiente etapa sin purificación. LC/MS (Método j): Rt = 1,53 min; MS m/z: 400 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 = 11,05 (s, 1H), 7,45 (s, 2H), 7,21 (m, 1H), 6,99 (t, J=75,1 Hz, 1H), 6,85 (s, 1H), 6,51 (s, 1H), 5,73 (s, 1H), 4,31 (s, 2H), 2,24 (s, 3H).
Etapa 4: metil 2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoato (350 mg, 72%) se preparó a partir de ácido 2,4-dicloro-3-((6-(difluorometoxi)-4-metil-1H-indol-2-il)metil)benzoico (456 mg, 1,1 mmol). LC/MS (Método j): Rt = 2,10 min; MS m/z: 428 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,78 (d, J=8,2 Hz, 1H), 7,70 (d, J=8,2 Hz, 1H), 7,14 (s, 1H), 7,11 (t, J=75 Hz, 1H), 6,64 (s, 1H), 5,53 (s, 1H), 4,44 (s, 2h), 3,88 (s, 3H), 3,81 (s, 3H), 2,28 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((6-(difluorometoxi)-1,4-dimetiMH-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoico (319 mg, 94%) se preparó a partir de metil 2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoato (349 mg, 0,8 mmol). LC/MS (Método j): Rt = 1,62 min; MS miz. 414 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,70 (d, J=8,2 Hz, 1H), 7,64 (d, J=8,2 Hz, 1H), 7,14 (s, 1H), 7,11 (t, J=75 Hz, 1H), 6,64 (s, 1H), 5,54 (s, 1H), 4,42 (s, 2h), 3,81 (s, 3H), 2,29 (s, 3H).
Etapa 6: metil 2-(1-(2,4-didoro-3-((6-(difluorometoxi)-1,4-dimetiMH-mdol-2-N)metM)benzoN)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (123 mg, 92%) a partir de ácido 2,4-dicloro-3-((6-(difluorometh-oxi)-1,4-dimetil-1H-indol-2-il)metil)benzoico (100 mg, 0,24 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (51,4 mg, 0,26 mmol). LC/MS (Método j): Rt = 1,89 min; MS miz. 553 [M+H]+; 1H NMR (DMSO-Ce, 300MHz): 57,65 (m, 1H), 7,74 y 7,36 (d, J=7,7 Hz, 1H), 7,13 (s, 1H), 7,11 (t, J=75 Hz, 1H), 6,64 (s, 1H), 5,54 y 5,51 (s, 1H), 4,42 (m, 3H), 3,80 (s, 3H), 3,59 y 3,56 (s, 3H), 3,24 (m, 1h), 3,05 (m, 1H), 2,80 (m, 1h), 2,27 (m, 5H), 1,95 (m, 1H), 1,75 (m, 1H), 1,58 (m, 1H), 1,15 (m, 2H).
Etapa 7: ácido 2-(1-(2,4-didoro-3-((6-(difluorometoxi)-1,4-dimetiMH-mdol-2-M)metN)benzoM)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (98 mg, 83%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (120 mg, 0,22 mmol). LC/MS (Método g): Rt = 1,72 min; MS miz. 539 [M+H]+; 1H NMR (DMSO-Ce, 400MHz): 512,07 (ancho, 1H), 7,65 y 7,64 (d, J=8,3 Hz, 1H), 7,42 y 7,72 (d, J=8,3 Hz, 1H), 7,13 (s, 1H), 7,11 (t, J=75 Hz, 1H), 6,64 (s, 1H), 5,54 y 5,51 (s, 1H), 4,43 (m, 3H), 3,80 (s, 3H), 3,25 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,29 y 2,28 (s, 3H), 2,14 (m, 2H), 1,93 (m, 1H), 1,77 (m, 1H), 1,61 (m, 1H), 1,15 (m, 2H).
Tabla FF. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((6-(difluorometoxi)-1,4-dimetil-1H-indol-2-il)metil)benzoico (Ejemplo FF, Etapa 5) usando el mismo procedimiento con las aminas apropiadas como se describe en el Ejemplo FF, Etapas 6 y 7.

Ejemplo FG: ácido 2-(1-(4,6-dicloro-5-(1,4-dimetil-6-(tnfluorometil)-1H-indol-2-carboml)nicotinoil)pipendin-4-il)acético

Etapa 1: ferc-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato

Usando un procedimiento similar al Ejemplo A, Etapa 1, ferc-butil 4,6-dicloro-5-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (5,45 g, 55%) se preparó a partir de N-(2-yodo-3-metil-5-(trifluorometil)fenil)bencenosulfonamida (7,06 g, 16 mmol) (Preparación #16) y ferc-butil 4,6-dicloro-5-(1-hidroxiprop-2-yn-1-il)nicotinate (5,8 g, 19,2 mmol) (Preparación 60). LC/MS (Método i): Rt = 2,77 min; MS m/z: 615 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 58,63 (s, 1H), 8,08 (s, 1H), 7,78 (m, 2H), 7,69 (m, 1H), 7,58 (m, 2H), 7,45 (s, 1H), 7,07 (s, 1H), 6,90 (m, 2H), 2,49 (s, 3H), 1,56 (s, 9H).
Etapa 2: ferc-butil 4,6-didoro-5-(4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-mdol-2-carboml)mcoti-nato

Usando un procedimiento similar al del Ejemplo AD, Etapa 2, ferc-butil 4,6-dicloro-5-(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-carbonil)nicotinato (3,84 g, 71%) se preparó a partir de ferc-butil 4,6-dicloro-5-(hidroxi(4-me-til-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-il)metil)nicotinato (5,4 g, 8,77 mmol). LC/MS (Método i): Rt = 2,91 min; MS m/z: 613 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 58,97 (s, 1H), 8,33 (m, 1H), 8,12 (m, 3H), 7,80 (m, 1H), 7,71 (m, 2H), 7,59 (s, 1H), 2,58 (s, 3H), 1,57 (s, 9H).
Etapa 3: ácido 4,6-didoro-5-(4-metM-6-(trifluorometM)-1H-mdol-2-carboml)mcotmico

A una solución de ferc-butil 4,6-dicloro-5-(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-indol-2-carbon-il)nicotinato (3,74 g, 6,1 mmol) en THF (75 mL) se añadió hidróxido de sodio (30,5 mL, 30,5 mmol) y la mezcla se agitó 20 horas a temperatura ambiente y 6 horas a 50 °C. La mezcla de reacción se evaporó parcialmente y luego se añadió HCl 1M. El precipitado formado se filtró para dar ácido 4,6-dicloro-5-(4-metil-6-(trifluorometil)-1 H-indol-2-carbonil)nicotínico (3,25 g, 59%). Se usa crudo en la siguiente etapa. LC/MS (Método i): Rt = 201 min; MS m/z: 417 [M+H]+
Etapa 4: metil 4,6-didoro-5-(1,4-dimetM-6-(trifluorometM)-1H-mdol-2-carboml)mcotmato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 4,6-dicloro-5-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)nicotinato (850 mg, 24%) se preparó a partir de ácido 4,6-dicloro-5-(4-metil-6-(trifluorometil)-1H-indol-2-carbonil)nicotínico (3,25 g, 7,79 mmol). Lc/MS (Método j): Rt = 2,06 min; MS m/z: 445 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 59,03 (s, 1H), 7,96 (s, 1H), 7,53 (s, 1H), 7,25 (s, 1H), 4,24 (s, 3H), 3,94 (s, 3H), 2,52 (s, 3H).
Etapa 5: ácido 4,6-didoro-5-(1,4-dimetN-6-(trifluorometM)-1H-mdol-2-carboml)mcotmico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)nicotínico (780 mg, 95%) se preparó a partir de metil 4,6-dicloro-5-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)nicotinato (0,85 g, 1,9 mmol). LC/MS (Método i): Rt = 2,20 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 5 = 14,22 (ancho, 1H), 9,00 (s, 1H), 7,96 (s, 1H), 7,51 (s, 1H), 7,24 (s, 1H), 4,24 (s, 3H), 2,53 (s, 3H).
Etapa 6: ácido 2-(1-(4,6-didoro-5-(1,4-dimetM-6-(trifluorometM)-1H-mdol-2-carboml)mcotmoM)piperídm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 6, seguido del Ejemplo A, Etapa 5, ácido 2-(1-(4,6-dicloro-5-(1,4-dimetil-6-(trifluorometil)-1 H-indol-2-carbonil)nicotinoil)piperidin-4-il) acético (59 mg, 46%) se preparó a partir de ácido 4,6-dicloro-5-(1,4-dimetil-6-(trifluorometil)-1H-indol-2-carbonil)nicotínico (100 mg, 0,23 mmol). LC/Ms (Método g): Rt = 1,81 min; MS m/z: 556 [M+H]+; 1H NMR (DMSO-ó6, 400MHz): 58,66 (m, 1H), 7,96 (s, 1H), 7,55 (m, 1H), 7,24 (s, 1H), 4,46 (m, 1H), 4,23 (s, 3H), 3,74 (m, 1H), 3,11 (m, 1H), 2,85 (m, 1H), 2,53 (s, 3H), 2,19 (m, 1H), 2,13 (m, 1H), 1,96 (m, 1H), 1,80 (m, 1H), 1,66 (m, 1H), 0,18 (m, 2H).
Ejemplo FH: ácido 2-(1-(3,5-didoro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolinoyl)piperidin-4-il)acético

Etapa 1: metil 2-(1-(3,5-didoro-4-((1,4-dimetM-6-(trífluorometM)-1H-mdol-2-M)metM)picolmoN)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato (89 mg, 74%) se preparó a partir de ácido 3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolínico (90 mg, 0,21 mmol) (Ejemplo BE, Etapa 5). LC/MS (Método j): Rt = 1,85 min; MS m /z : 556 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 58,76 (s, 1H), 7,71 (s, 1H), 7,07 (s, 1H), 5,79 (s, 1H), 4,48 (s, 3H), 3,92 (s, 3H), 3,58 (s, 3H), 3,25 (m, 1H), 3,07 (m, 1H), 2,85 (m, 1H), 2,37 (s, 3H), 2,28 (m, 2H), 1,99 (m, 1H), 1,76 (m, 1H), 1,63 (m, 1H), 1,17 (m, 2H).
Etapa 2: ácido 2-(1-(3,5-didoro-4-((1,4-dimetN-6-(trifluorometM)-1H-mdol-2-N)metN)picolmoM)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-indol-2-il)metil)picolinoyl)piperidin-4-il)acético (60 mg, 70%) se preparó a partir de metil 2-(1-(3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1 H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato acid (85 mg, 0,15 mmol). LC/MS (Método g): Rt = 1,76 min; MS m/z: 542 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 512,10 (ancho, 1H), 8,76 (s, 1H), 7,72 (s, 1H), 7,07 (s, 1H), 5,79 (s, 1H), 4,48 (m, 3H), 3,92 (s, 3H), 3,26 (m, 1H), 3,08 (m, 1H), 2,84 (m, 1H), 2,37 (s, 3H), 2,17 (m, 2H), 1,95 (m, 1H), 1,78 (m, 1H), 1,62 (m, 1H), 1,14 (m, 2H).
Ejemplo FI: ácido 2-(1-(2,4-didoro-3-((1-metil-6-(trifluorometoxi)-1H-mdol-2-il)metil)benzoil)piperidm-4-il)acético

Etapa 1: metil 2,4-didoro-3-(hidroxi(1-(femlsulfoml)-6-(trifluorometoxi)-1H-mdol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (2,32 g, 80%) se preparó a partir de N-(2-yodo-5-(trifluorometoxi)fenil)bencenosulfonamida (2,2 g, 4,96 mmol) (Preparación 75) y metil 2,4-dicloro-3-(1-hidroxiprop-2- in-1-il)benzoato (1,54 g, 5,96 mmol) (Preparación #1). LC/MS (Método i): Rt = 2,39 min; MS m/z: 632 [M-H]- CH3COOH; 1H NMR (DMSO-cfe, 300MHz): 5 7,93 (m, 3H), 7,68 (m, 3H), 7,60 (m, 3H), 7,29 (m, 1H), 7,05 (m, 1H), 6,67 (m, 2H), 3,87 (s, 3H).
Etapa 2: metil 2,4-didoro-3-((1-(femlsulfoml)-6-(trifluorometoxi)-1H-mdol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-dicloro-3-((1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (2,25 g, 98%) se preparó a partir de metil 2,4-dicloro-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (2,3 g, 4 mmol). LC/MS (Método i): R = 2,68 min; MS m/z: 558 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 = 8,04 (s, 1H), 7,96 (m, 2H), 7,82 (m, 1H), 7,76 (m, 1H), 7,69 (m, 3H), 7,57 (m, 1H), 7,28 (m, 1H), 5,91 (m, 1H), 4,61 (m 2H), 3,87 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-((6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-didoro-3-((6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (1,4 g, 60%) se preparó a partir metil 2,4-didoro-3-((1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (2,6 g, 4,6 mmol). LC/MS (Método i): Rt = 2,38 min; MS m/z: 418 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 11,30 (s, 1H), 7,74 (d, J=8,3 Hz, 1H), 7,68 (d, J=8,3 Hz, 1H), 7,44 (d, J=8,6 Hz, 1H), 7,28 (m, 1H), 6,92 (m, 1H), 5,89 (s, 1H), 4,45 (s, 2H), 3,87 (s, 3H).
Etapa 4: metil 2,4-didoro-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-N)metN)benzoato
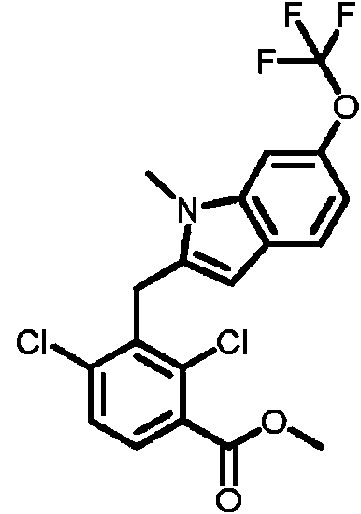
Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-didoro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (1,1 g, 54%) se preparó a partir de metil 2,4-didoro-3-((6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (1,35 g, 3,23 mmol). LC/MS (Método j): Rt = 1,79 min; MS m/z: 432 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 7,80 (d, J =8,4 Hz, 1H), 7,71 (d, J =8,4 Hz, 1H), 7,52 (s, 1H), 7,44 (d, J =8,6 Hz, 1H), 6,94 (m, 1H), 5,59 (s, 1H), 4,46 (s, 2H), 3,88 (s, 3H), 3,86 (s, 3H).
Etapa 5: ácido 2,4-didoro-3-((1-metM-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (990 mg, 100%) se preparó a partir de metil 2,4-didoro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (1 g, 3,3 mmol). LC/MS (Método i): Rt = 2,47 min; MS m/z: 418 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 = 13,64 (ancho, 1H), 7,74 (d, J=8,4 Hz, 1H), 7,66 (d, J=8,4 Hz, 1H), 7,52 (s, 1H), 7,44 (d, J=8,6 Hz, 1H), 6,94 (m, 1H), 5,60 (m, 1H), 4,45 (s, 2H), 3,86 (s, 3H).
Etapa 6: metil 2-(1-(2,4-didoro-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoN)piperidm-4-N)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (180 mg, 88%) se preparó a partir de ácido 2,4-dicloro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (150 mg, 0,36 mmol) y clorhidrato de metil 2-(piperidin-4-il)acetato (83 mg, 0,43 mmol). LC/MS (Método i): Rt = 2,64 min; MS m/z: 557 [M+H]+; 1H NMR (DMSO-cfe, 300MHz): 57,64 y 7,63 (d, J=8,3 Hz, 1H), 7,51 (s, 1H), 7,43 (m, 1,5H), 7,36 (d, J=8,3 Hz, 0,5H), 6,94 (m, 1H), 5,63 y 6,60 (s, 1H), 4,42 (m, 3H), 3,85 (s, 3H), 3,59 y 3,56 (s, 3h), 3,28 (m, 1H), 3,05 (m, 1H), 2,80 (m, 1H), 2,29 (m, 2H), 1,97 (m, 1H), 1,73 (m, 1H), 1,59 (m, 1H), 1,16 (m, 2H).
Etapa 7: ácido 2-(1-(2,4-dicloro-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-N)metil)benzoN)piperidm-4-N)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-dicloro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (150 mg, 88%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (170 mg, 0,30 mmol). LC/MS (Método g): Rt = 1,83 min; MS m/z: 543 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 57,64 y 7,63 (d, J=8,1 Hz, 1H), 7,51 (s, 1H), 7,44 (m, 1,5H), 7,36 (d, J=8,4 Hz, 0,5H), 6,4 (m, 1H), 5,63 y 5,61 (s, 1H), 4,44 (m, 3H), 3,85 (s, 3H), 3,27 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1H), 2,17 (m, 2H), 1,93 (m, 1h), 1,78 (m, 1H), 1,62 (m, 1H), 1,14 (m, 2H).
Tabla FI. Los siguientes ejemplos se prepararon a partir de ácido 2,4-dicloro-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (Ejemplo FI, Etapa 5) usando el mismo procedimiento con las aminas apropiadas como se describe en el Ejemplo FI, Etapa 6 y 7.

Ejemplo FJ: (2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(4-(oxetan-3-il)piperazin-1 -il)metanona

Etapa 1: metil 2,4-didoro-3-(hidroxi(4-metiM-(fenMsulfonM)-6-(tnfluorometM)-1H-pirrolo[3,2-c]pind-in-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-dicloro-3-(hidroxi(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (1,16 g, 60%) a partir de N-(3-yodo-2-metil-6-(trifluorometil)piridin-4-il)bencenosulfonamida (1,5 g, 3,4 mmol) (Preparación #39) y metil 2,4-dicloro-3-(1-hidroxiprop-2-in-1-il)benzoato (1,0 g, 4 mmol) (Preparación #1). LC/MS (Método i): Rt = 2,46 min; MS m/z: 573 [M+H]+; 1H NMR (DMSO- d6, 300MHz): 58,17 (s, 1H), 7,85 (m, 2H), 7,72 (m, 1H), 7,67 (d, J=8,4 Hz, 1H), 7,60 (m, 2H), 7,52 (d, J=8,4 Hz, 1H), 7,01 (s, 2H), 6,62 (m, 1H), 3,86 (s, 3H), 2,69 (s, 3H).
Etapa 2: metil 2,4-dicloro-3-((4-metiM-(fenilsulfonil)-6-(tnfluorometil)-1H-pin*olo[3,2-c]pindin-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo Z, Etapa 2, metil 2,4-dicloro-3-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (644 mg, 58%) se preparó a partir de metil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (946 mg, 1,6 mmol). LC/MS (Método i): Rt = 2,79 min; MS m/z: 557 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 58,32 (s, 1H), 8,05 (m 2H), 7,80 (m, 2H), 7,70 (m, 3H), 6,20 (s, 1H), 4,57 (s, 2H), 3,86 (s, 3H), 2,57 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-((4-metil-6-(tnfluorometil)-1H-pin*olo[3,2-c]pindin-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (471 mg, 98%) se preparó a partir de metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (644 mg, 1,15 mmol). LC/MS (Método i): Rt = 2,30 min; MS m/z: 417 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 5 = 11,95 (ancho, 1H), 7,77 (d, J=8,1 Hz, 1H), 7,70 (d, J=8,1 Hz, 1h), 7,61 (s, 1H), 6,13 (m, 1H), 4,52 (s, 2H), 3,88 (s, 3H), 2,58 (s, 3H).
Etapa 4: metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1 H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (500 mg, 100%) se preparó a partir de metil 2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (468 mg, 1,12 mmol). LC/MS (Método i): Rt = 2,42 min; MS m/z: 431 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 57,92 (s, 1H), 7,82 (d, J=8,3 Hz, 1H), 7,73 (d, J=8,3 Hz, 1H), 5,86 (s, 1H), 4,52 (s, 2H), 3,96 (s, 3H), 3,88 (s, 3H), 2,54 (s, 3H).
Etapa 5: ácido 2,4-dicloro-3-((1,4-dimetN-6-(tnfluorometN)-1H-pirrolo[3,2-c]pmdin-2-N)metN)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoico (350 mg, 75%) se preparó a partir de metil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoato (484 mg, 1,12 mmol). LC/MS (Método i): Rt = 2,03 min; MS m/z: 417 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 57,92 (s, 1H), 7,65 (m, 2H), 5,84 (s, 1H), 4,49 (s, 2H), 3,96 (s, 3H), 2,53 (s, 3H).
Etapa 6: (2,4-dicloro-3-((1,4-dimetN-6-(tnfluorometN)-1H-pirrolo[3,2-c]pindin-2-N)metN)feml)(4-(oxe-tan-3-il)piperazin-1-il)metanona

Usando un procedimiento similar al Ejemplo A, Etapa 6 , (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)fenil)(4-(oxetan-3-il)piperazin-1-il)metanona (64 mg, 59%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[3,2-c]piridin-2-il)metil)benzoico (80 mg, 0,19 mmol) y 1-(oxetan-3-il)piperazina (41 mg, 0,29 mmol). LC/MS (Método g): Rt = 1,39 min; MS m/z: 541 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 5 7,92 (s, 1H), 7,68 (d, J=8,4 Hz, 1H), 7,44 (d, J=8,4 Hz, 1H), 5,86 (s, 1H), 4,48 (m, 6 H), 3,96 (s, 3H), 3,67 (m, 2H), 3,43 (m, 1H), 3,21 (m, 2H), 2,54 (s, 3H), 2,33 (m, 2H), 2,23 (m, 2H).
Ejemplo FK: W-(2-(2,6-dicloro-3-(morfolina-4-carboml)bencN)-1,4-dimetiMH-mdol-6-N)acetamida

Etapa 1: metil 2,4-dicloro-3-(hidroxi(4-metil-6-nitro-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, metil 2,4-didoro-3-(hidroxi(4-metil-6-nitro-1-(fenilsul-fonyl)-1H-indol-2-il)metil)benzoato (559 mg, 63%) se preparó a partir de N-(2-bromo-3-metil-5-nitrofenil)bence-nosulfonamida (600 mg, 1,6 mmol) (Preparación 76) y metil 2,4-didoro-3-(1-hidroxiprop-2-in-1-il)benzoato (544 mg, 2,1 mmol) (Preparación #1). LC/MS (Método i): Rt = 2,49 min; MS m/z: 531 [M+H]+ -H2 O; 1H NMR (DMSO-cf6, 300MHz): 58,71 (m, 1H), 8,01 (m, 1H), 7,81 (m, 2H), 7,70 (m, 2H), 7,50 (m, 3H), 6,99 (m, 2H), 6,82 (m, 1H), 3,86 (s, 3h), 2,50 (s, 3H).
Etapa 2: metil 2,4-dicloro-3-(hidroxi(4-metil-6-nitro-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 3, metil 2,4-dicloro-3-(hidroxi(4-metil-6-nitro-1H-indol-2-il)metil)benzoato (506 mg, 61%) se preparó a partir de metil 2,4-didoro-3-(hidroxi(4-metil-6-nitro-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoato (1,1 g, 2,0 mmol). LC/MS (Método i): Rt = 2,26 min; MS m/z: 409 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 511,88 (m, 1H), 8,16 (m, 1H), 7,73 (d, J=8,3 Hz, 1H), 7,67 (m, 1H), 7,63 (d, J=8,3 Hz, 1h), 6,82 (m, 2H), 6,14 (s, 1H), 3,86 (s, 3H), 2,45 (s, 3H).
Etapa 3: metil 2,4-dicloro-3-((4-metil-6-nitro-1H-indol-2-il)metil)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 2, metil 2,4-didoro-3-((4-metil-6-nitro-1H-indol-2-il)metil)benzoato (130 mg, 27%) se preparó a partir de metil 2,4-didoro-3-(hidroxi(4-metil-6-nitro-1H-indol-2-il)metil)benzoato (493 mg, 1,2
mmol). LC/MS (Método i): Rt = 2,52 min; MS m/z: 393 [M+H]+; 1H NMR (DMSO-cíe, 300MHz): 511,84 (s, 1H), 8,11 (m, 1H), 7,77 (d, J =8,8 Hz, 1H), 7,70 (d, J =8,8 Hz, 1H), 7,67 (m, 1H), 6,08 (m, 1H), 4,54 (s, 2H), 3,87 (s, 3H), 2,44 (s, 3h).
Etapa 4: metil 2,4-d¡cloro-3-((1,4-dimet¡l-6-mtro-1H-mdol-2-¡l)met¡l)benzoato

Usando un procedimiento similar al del Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)benzoato (161 mg, 100%) se preparó a partir de metil 2,4-didoro-3-((4-metil-6-nitro-1H-indol-2-il)metil)benzoato (152 mg, 0,4 mmol). LC/MS (Método i): Rt = 2,59 min; MS m/z: 407 [M+H]+; 1H NMR (DMSO-cíe, 300MHz): 58,35 (s, 1H), 7,81(d, J=8,4 Hz, 1H), 7,72 (d, J=8,4 Hz, 1H), 7,70 (s, 1H), 5,76 (s, 1H), 4,54 (s, 2h), 3,98 (s, 3H), 3,88 (s, 3H), 2,39 (s, 3H).
Etapa 5: ácido 2,4-dicloro-3-((1,4-dimetil-6-mtro-1H-mdol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)benzoico (145 mg, 93%) se preparó a partir de 2,4-didoro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)benzoato (161 mg, 0,39 mmol). LC/MS (Método i): Rt = 2,24 min; MS m/z: 393 [M+H]+; 1H NMR (DMSO-cíe, 300MHz): 513,68 (br., 1H), 8,35 (m, 1H), 7,77 (d, J=8,2 Hz, 1H), 7,71(m, 1H), 7,68 (d, J=8,2 Hz, 1H), 5,76 (s, 1H), 4,53 (s, 2H), 3,98 (s, 3H), 2,39 (s, 3H).
Etapa 6: ^^ -d ic lo ro ^ -^ l^ -d im etil^ -rn tro -I
Usando un procedimiento similar al Ejemplo A, Etapa 6, (2,4-dicloro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)fenil)(morfolino)metanona (217 mg, 100%) se preparó a partir de ácido 2,4-dicloro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)benzoico (145 mg, 0,36 mmol) y morfolina (38,6 mg, 0,44 mmol). LC/MS (Método i): Rt = 2,27 min; MS m/z: 462 [M+H]+; 1H NMR (DMSO-de, 300MHz): 58,35 (s, 1H), 7,70 (m, 2H), 7,47 (d, J=8,3 Hz, 1H), 5,79 (m, 1H), 4,50 (m, 2H), 3,98 (s, 3H), 3,65 (s, 4H), 3,55 (m, 2H), 3,18 (m, 2H), 2,39 (s, 3H).
Etapa 7: (3-((6-am¡no-1,4-d¡met¡l-1H-¡ndol-2-¡l)met¡l)-2,4-d¡clorofen¡l)(morfol¡no)metanona

Usando un procedimiento similar a la Preparación #18, Etapa C, (3-((6-amino-1,4-dimetil-1H-indol-2-il)metil)-2,4-didorofenil)(morfolino)metanona (91 mg, 45%) se preparó a partir de (2,4-dicloro-3-((1,4-dimetil-6-nitro-1H-indol-2-il)metil)fenii)(morfolino)metanona (217 mg, 0,5 mmol). lC/MS (Método i): Rt = 1,51 min; Ms m/z: 432 [M+H]+; 1H NMR (DMSO-da, 300MHz): 57,65 (d, J=8,2 Hz, 1H), 7,40 (d, J=8,2 Hz, 1H), 6,32 (s, 1H), 6,15 (s, 1H), 5,30 (s, 1H), 4,64 (m, 2H), 4,31 (m, 2H), 3,65 (m, 4H), 3,63 (s, 3H), 3,54 (m, 2H), 3,20 (m, 2H), 2,13 (s, 3H).
Etapa 8: W-(2-(2,6-didoro-3-(morfolma-4-carboml)bencM)-1,4-dimetiMH-mdol-6-M)acetamida

A una solución de (3-((6-amino-1,4-dimetil-1H-indol-2-il)metil)-2,4-diclorofenil)(morfolino)metanona (91 mg, 0,21 mmol) en DCM (2 mL) se añadió cloruro de acetilo (0,016 mL, 0,232 mmol) y trietilamina (0,053 mL, 0,379 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. Se añadió agua y la mezcla se extrajo dos veces con DCM. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyendo con DCM al 10% en EtOAc) para dar N-(2-(2,6-dicloro-2-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6-il)acetamida (57 mg, 54%). LC/MS (Método g): Rt = 1,34 min; MS m/z: 474 [M+H]+; 1H NMR (DMSO-da, 500MHz): 59,76 (s, 1H), 7,69 (s, 1h), 7,66 (d, J=8,3 Hz, 1H), 7,43 (d, J=8,3 Hz, 1H), 6,80 (s, 1H), 5,46 (s, 1H), 4,41 (d, J=16,3 Hz, 1H), 4,36 (d, J=16,3 Hz, 1H), 3,74 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,17 (m, 2H), 2,24 (s, 3H), 2,03 (s, 3H).
Ejemplo FL: ácido 2-(1-(2-doro-4-ddopropN-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-N)metN)benzoN)pipe-ridm-4-il)acético

Etapa 1: ferc-butil 2-doro-4-ddopropN-3-(hidroxi(1-(femlsulfoml)-6-(trifluorometoxi)-1H-mdol-2-N)metN)benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 1, ferc-butil 2-cloro-4-ciclopropil-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (786 mg, 38%) se preparó a partir de N-(2-yodo-5-(trifluorometoxi)fenil)bencenosulfonamida (1,26 g, 2,8 mmol) (Preparación 75) y ferc-butil 2-cloro-4-ciclopropil-3-(1-hidrox
iprop-2-in-1-il)benzoato (1,05 g, 3,42 mmol) (Preparación 58). LC/MS (Método j): Rt = 2,65 min; MS miz: 680 [M-H]-+CH3COOH; 1H NMR (DMSO-de, 300MHz): 58,04 (m, 2H), 7,92 (s, 1H), 7,65 (m, 4H), 7,48 (d, J=8,3 Hz, 1H), 7,26 (m, 2H), 6,96 (d, J=8,3 Hz, 1H), 6,60 ppm (d, J=5,6 Hz, 1H), 6,52 (s, 1H), 2,68 (m, 1H), 1,53 (s, 9H), 0,80 (m, 4H).
Etapa 2: ácido 2-doro-4-ddopropM-3-((1-(femlsulfoml)-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2-doro-4-ddopropil-3-((1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (223 mg, 81%) se preparó a partir de íerc-butil 2-cloro-4-cidopropil-3-(hidroxi(1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (250 mg, 0,4 mmol). LC/MS (Método j): Rt = 2,17 min; MS miz: 548 [M-H]-;
1H NMR (DMSO-de, 300MHz): 513,29 (m, 1H), 8,06 (s, 1H), 7,96 (m, 2H), 7,77 (m, 1H), 7,66 (m, 3H), 7,58 (d, J=8,6 Hz, 1H), 7,25 (m, 1H), 7,14 (d, J=8,6 Hz, 1H), 5,83 (s, 1h), 4,62 (s, 2H), 1,69 (m, 1H), 0,68 (m, 4H).
Etapa 3: ácido 2-doro-4-ddopropM-3-((6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, ácido 2-cloro-4-ddopropil-3-((6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (164 mg, 100%) se preparó a partir de ácido 2-doro-4-ddopropil-3-((1-(fenilsulfonil)-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (220 mg, 0,4 mmol). LC/MS (Método j): Rt = 1,77 min; MS miz: 410 [M+H]+; 1H NMR (DMSO-de, 300MHz): 511,29 (s, 1H), 7,53 (d, J=8,3 Hz, 1H), 7,42 (d, J=8,6 Hz, 1H), 7,27 (s, 1H), 7,08 (d, J=8,1 Hz, 1H), 6,88 (m, 1H), 5,83 (s, 1H), 4,50 (s, 2H), 1,99 (s, 1H), 0,90 (m, 2H), 0,69 (m, 2H).
Etapa 4: metil 2-doro-4-ddopropM-3-((1-metM-6-(tnfluorometoxi)-1H-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 2-doro-4-ddopropil-3-((1-metil-6-(trifluoromet-oxi)-1H-indol-2-il)metil)benzoato (100 mg, 57%) se preparó a partir de ácido 2-cloro-4-ddopropil-3-((6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (131 mg, 0,32 mmol). LC/MS (Método j): Rt = 2,37 min; MS miz: 438 [M+H]+; 1H NMR (DMSO-de, 300MHz): 57,65 (d, J=8,1 Hz, 1H), 7,51 (s, 1H), 7,42 (d, J=8,4 Hz, 1H), 7,15 (d, J=8,1 Hz, 1H), 6,93 (m, 1H), 5,56 (s, 1H), 4,50 (s, 2H), 3,87 (s, 3h), 3,84 (s, 3H), 1,94 (m, 1H), 0,91 (m, 2H), 0,71 (m, 2h).
Etapa 5: ácido 2-doro-4-ddopropM-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-cloro-4-ciclopropil-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (77 mg, 81%) se preparó a partir de metil 2-cloro-4-ciclopropil-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (98 mg, 0,22 mmol). LC/MS (Método j): Rt = 1,90 min; MS m/z: 424 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 513,34 (ancho, 1H), 7,58 (d, J=8,1 Hz, 1H), 7,51 (s, 1H), 7,42 (d, J=8,6 Hz, 1H), 7,10 (d, J=8,1 Hz, 1H), 6,91 (m, 1H), 5,57 (s, 1H), 4,48 (s, 2H), 3,87 (s, 3H), 1,92 (s, 1H), 0,89 (m, 2H), 0,69 (m, 2H).
Etapa 6: metil 2-(1-(2-doro-4-ddopropM-3-((1-metN-6-(trifluorometoxi)-1H-mdol-2-N)metN)benzoM)pip-eridm-4-il)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2-cloro-4-ciclopropil-3-((1-metil-6-(trifluor-ometoxi)-1H-indol-2-il)metM)benzoil)piperidin-4-il)acetato (80 mg, 80%) se preparó a partir ácido 2-cloro-4-ciclopropil-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoico (75 mg, 0,18 mmol) y clorhidrato de metil (4-piperidil)acetato (38 mg, 0,19 mmol). LC/MS (Método j): Rt = 2,15 min; MS m/z: 563 [M+H]+; 1H NMR (DMSO-d6, 300MHz): 57,51 (s, 1H), 7,43 y 7,42 (d, J =8,5 Hz, 1H), 7,17 (m, 2H), 6,92 (m, 1H), 5,59 y 5,56 (s, 1H), 4,46 (m, 3H), 3,86 (s, 3H), 3,59 y 3,56 (s, 3h), 3,24 (m, 1H), 3,01 (m, 1h), 2,79 (m, 1H), 2,25 (m, 2H), 1,90 (m, 2H), 174 (m, 1H), 1,58 (m, 1H), 1,14 (m, 2H), 0,87 (m, 2H), 0,67 (m, 2H).
Etapa 7: ácido 2-(1 -(2-doro-4-ddopropil-3-((1 -metil-6-(trifluorometoxi)-1 H-mdol-2-M)metM)benzoM)piperidm-2-il)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2-cloro-4-ciclopropil-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (66 mg, 84%) se preparó a partir de metil 2-(1-(2-cloro-4-ciclo-propil-3-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (80 mg, 0,14 mmol). lC/MS (Método g): Rt = 1,83 min; MS m/z: 549 [M+H]+; 1H NMR (DMSO-d6, 400MHz): 512,21 (m, 1H), 7,51 (s, 1H), 7,43 y 7,42 (d, J=8,6 Hz, 1H), 7,25 y 7,18 (d, J=8,6 Hz, 1H), 7,11 (m, 1H), 6,92 (m, 1H), 5,59 y 5,57 (s, 1H), 4,46 (m, 3H), 3,86 (s, 3H), 3,26 (m, 1H), 3,01 (m, 1h), 2,77 (m, 1H), 2,17 (m, 1H), 2,11 (m, 1H), 1,90 (m, 2H), 1,76 (m, 1H), 1,62 (m, 1H), 1,12 (m, 2H), 0,88 (m, 2H), 0,67 (m, 2H).
Ejemplo FM: ácido 2-(1-(2,4-didoro-3-((6-cidopropN-4-fluoro-1-metiMH-mdol-2-N)metM)benzoN)piperidm-4-il)acético

Etapa 1: ferc-butil 2,4-didoro-3-((6-ddopropil-4-fluoro-1-(femlsulfoml)-1H-mdol-2-il)(hidroxi)metil)benzoato

Usando un procedimiento similar al del Ejemplo CJ, Etapa 1, ferc-butil 2,4-didoro-3-((6-ddopropil-4-fluoro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (180 mg, 71%) se preparó a partir de 6-ddopropil-4-fluoro-1-(fenil-sulfonil)-1H-indol (135 mg, 0,43 mmol) (Preparación 77) y ferc-butil 2,4-dicloro-3-formilbenzoato (177 mg, 0,64 mmol) (Preparación #33, Etapa B). LC/MS (Método j): Rt = 2,48 min; MS m/z: 572 [M+H]+-H2O; 1H NMR (DMSO-óe, 300MHz): 57,89 (m, 2H), 7,69 (m, 1H), 7,56 (m, 5H), 6,99 (m, 1H), 6,79 (m, 1H), 6,61 (m, 1H), 6,56 (s, 1H), 2,09 (m, 1H), 1,54 (s, 9H), 1,01 (m, 2H), 0,70 (m, 2H).
Etapa 2: ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-(femlsulfoml)-1H-mdol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 2,4-didoro-3-((6-ddopropil-4-fluoro-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoico (700 mg, 100%) se preparó a partir de terc-butil 2,4-didoro-3-((6-dclopropil-4-fluoro-1-(fenilsulfonil)-1H-indol-2-il)(hidroxi)metil)benzoato (920 mg, 1,2 mmol). LC/MS (Método j): Rt = 2,09 min; MS m/z: 516 [M-H]-; 1H NMR (DMSO-óe, 300MHz): 5 = 13,64 (ancho, 1H), 7,96 (m, 2H), 7,78 (m, 2H), 7,68 (m, 4H), 6,78 (m, 1H), 5,68 (s, 1H), 4,57 (s, 2H), 2,12 (m, 1H), 1,03 (m, 2H), 0,73 (m, 2H).
Etapa 3: ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-fH -indol-2-il)metil)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 3, ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1H-indol-2-il)metil)benzoico (620 mg, 69%) se preparó a partir de ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-(fenilsulfonil)-1H-indol-2-il)metil)benzoico (697 mg, 1,3 mmol). LC/MS (Método j): Rt = 1,59 min; MS m/z: 378 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 5 = 13,60 (ancho, 1H), 11,17 (s, 1H), 7,70 (d, J=8,6 Hz, 1H), 7,63 (d, J=8,6 Hz, 1H), 6,89 (s, 1H), 6,44 (m, 1H), 5,73 (m, 1H), 4,42 (m, 2H), 1,97 (m, 1H), 0,91 (m, 2H), 0,63 (m, 2H).
Etapa 4: metil 2,4-didoro-3-((6-ddopropil-4-fluoro-1-metiMH-mdol-2-M)metM)benzoato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoato (219 mg, 33%) se preparó a partir de ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1H-indol-2-il)metil)benzoico (618 mg, 1,6 mmol). LC/MS (Método j): Rt = 2,26 min; MS m/z: 406 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 5 7,78 (d, J=8,8 Hz, 1H), 7,70 (d, J=8,8 Hz, 1H), 7,04 (s, 1H), 6,50 (m, 1H), 5,42 (s, 1H), 4,42 (s, 2H), 3,88 (s, 3H), 3,81 (s, 3H), 2,01 (m, 1H), 0,94 (m, 2H), 0,71 (m, 2H).
Etapa 5: ácido 2,4-didoro-3-((6-cidopropM-4-fluoro-1-metiMH-mdol-2-M)metM)benzoico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoico (145 mg, 57%) se preparó a partir de metil 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoato (263 mg, 0,64 mmol). LC/MS (Método j): Rt = 1,78 min; MS m/z: 392 [M+H]+
Etapa 6: metil 2-(1-(2,4-didoro-3-((6-cidopropN-4-fluoro-1-metiMH-mdol-2-N)metN)benzoN)piperidm-4-M)acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 2-(1-(2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acetato (81 mg, 84%) se preparó a partir de ácido 2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoico (71 mg, 0,18 mmol) y clorhidrato de metil (4-piperidil)acetato (70 mg, 0,36 mmol). LC/MS (Método i): Rt = 2,63 min; MS m/z: 531 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 57,64 y 7,63 (d, J=8,3 Hz, 1H), 7,42 y 7,35 (d, J=8,3 Hz, 1H), 7,04 (s, 1H), 6,49 (m, 1H), 5,44 (m, 1H), 4,41 (m, 3H), 3,81 (s, 3H), 3,59 y 3,56 (s, 3H), 3,27 (m, 1H), 3,05 (m, 1H), 2,78 (m, 1H), 2,26 (m, 2H), 1,99 (m, 2H), 1,75 (m, 1H), 1,59 (m, 1H), 1,17 (m, 2H), 0,94 (m, 2H), 0,70 (m, 2H).
Etapa 7: ácido 2-(1-(2,4-didoro-3-((6-cidopropM-4-fluoro-1-metiMH-mdol-2-M)metN)benzoM)piperidm-4-M)acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-(1-(2,4-didoro-3-((6-ddopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoil)piperidin-4-il)acético (41 mg, 53%) se preparó a partir de metil 2-(1-(2,4-dicloro-3-((6-cidopro-pyl-4-fluoro-1-metiMH-indol-2-il)metil)benzoil)piperidin-4-il)acetato (79 mg, 0,15 mmol). LC/MS (Método g): Rt = 1,83 min; MS miz : 517 [M+H]+; 1H NMR (DMSO-cf6, 400MHz): 5 = 12,10 (s, 1H), 7,64 y 7,63 (d, J=8,1 Hz, 1H), 7,42 y 7,35 (d, J=8,1 Hz, 1H), 7,04 (s, 1H), 6,49 (s, 1H), 5,45 (m, 1H), 4,42 (m, 3H), 3,81 (s, 3H), 3,26 (m, 1H), 3,04 (m, 1H), 2,80 (m, 1H), 2,15 (m, 2h), 1,97 (m, 2H), 1,77 (m, 1H), 1,61 (m, 1H), 1,14 (m, 2H), 0,93 (m, 2H), 0,70 (m, 2H).
Ejemplo FN: ácido carboxílico 
Etapa 1: metil 1-(2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metiMH-indol-2-il)metil)benzoil)pipendina-4-carboxilato

Usando un procedimiento similar al Ejemplo A, Etapa 6, metil 1-(2,4-didoro-3-((6-ddopropil-4-fluoro-1-metiMH-indol-2-il)metil)benzoil)piperidina-4-carboxilato (74 mg, 80%) se preparó a partir de ácido 2,4-dicloro-3-((6-ddopropil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoico (70 mg, 0,18 mmol) (Ejemplo FM, Etapa 5) y clorhidrato de metil piperidina-4-carboxilato (64 mg, 0,36 mmol). LC/MS (Método j): Rt = 1,98 min; MS m/z: 517 [M+H]+; 1H NMR (DMSO-cf6, 300MHz): 57,64 (d, J=8,3 Hz, 1H), 7,45 y 7,38 (d, J=8,3 Hz, 1H), 7,04 (s, 1H), 6,50 (m, 1H), 5,45 (m, 1H), 4,38 (m, 3H), 3,81 (m, 3H), 3,62 y 3,59 (s, 3H), 3,28 (m, 1H), 3,13 (m, 1H), 2,97 (m, 1H), 2,68 (m, 1H), 1,98 (m 2H), 1,77 (m, 1H), 1,52 (m, 2H), 0,93 (m, 2H), 0,71 (m, 2H).
Etapa 2: ácido 1-(2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metiMH-indol-2-il)metil)benzoil)pipendina-4-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 1-(2,4-dicloro-3-((6-ciclopropil-4-fluoro-1-metiMH-indol-2-il)metil)benzoil)piperidina-4-carboxílico (34 mg, 48%) se preparó a partir de metil 1-(2,4-dicloro-3-((6-ciclo-propil-4-fluoro-1-metil-1H-indol-2-il)metil)benzoil)piperidina-4-carboxilato (72 mg, 0,13 mmol). LC/MS (Método g): Rt = 1,81 min; MS miz: 503 [M+H]+; 1H NMR (DMSO-ó6, 400MHz): 5 12,33 (ancho, 1H), 7,64 (d, J=8,1 Hz, 1H), 7,44 y 7,38 (d, J=8,1 Hz, 1H), 7,04 (s, 1H), 6,50 (m, 1H), 5,45 (m, 1H), 4,38 (m, 3H), 3,81 (s, 3H), 3,25 (m, 1H), 3,08 (m, 1H), 2,98 (m, 1H), 2,54 (m, 1H), 2,01 (m, 1H), 1,92 (m, 1H), 1,78 (m, 1H), 1,49 (m, 2H), 0,94 (m, 2H), 0,71 (m, 2H).
Ejemplo FO: 2-[[2,6-dicloro-3-(morfolina-4-carbonil)fenil]metil]-N,N,1,4-tetrametil-indol-6-carboxamida

Etapa 1: ácido 2-(2,6-didoro-3-(morfolma-4-carboml)bencM)-1,4-dimetiMH-mdol-6-carboxNico

A una suspensión de 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetiMH-indol-6-carbonitrilo (Ejemplo AA-35) en un etilenglicol (600 |_il, 10,76 mmol) se añadió a una solución 3M de hidróxido de potasio (400 |_iL, 1,20 mmol) y la mezcla de reacción se agitó 1,5 horas a 130 °C bajo irradiación de microondas. La mezcla de reacción se diluyó con agua y se lavó dos veces con DCM. La capa acuosa se acidificó con una solución de HCl 1M, se extrajo con DCM, se secó sobre sulfato de magnesio, se filtró y se evaporó. El ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6-carboxílico (16 mg, 53,4%) se aisló como un polvo de color blanco roto. LC/MS (Método i): Rt = 1,90 min; MS m/z: 461 [M+H]+; 1H NMR (DMSO-ó6, 300MHz): 5 12,47 (ancho, 1H), 7,92 (s, 1H), 7,68 (d, J=8,4 Hz, 1H), 7,45 (d, J=8,4 Hz, 1H), 7,40 (s, 1H), 5,64 (m, 1H), 4,46 (m, 2H), 3,89 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,18 (m, 2H), 2,33 (m, 3H).
Etapa 2: 2-[[2,6-didoro-3-(morfolina-4-carbonil)fenil]metil]-N,N,1,4-tetrametil-indol-6-carboxamida

Usando un procedimiento similar al Ejemplo A, Etapa 6, 2-[[2,6-dicloro-3-(morfolina-4-carbonil)fenil]metil]-N,N,1,4-tetrametil-indol-6-carboxamida (29 mg, 55%) se preparó a partir de ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6-carboxílico (50 mg, 0,11 mmol). LC/MS (Método g): Rt = 1,42 min; MS m/z: 488 [M+H]+; 1H NMR (DMSO-óe, 400MHz): 5 = 7,68 (d, J=8,1 Hz, 1H), 7,45 (d, J=8,1 Hz, 1H), 7,35 (s, 1H), 6,81 (s, 1H), 5,58 (s, 1H), 4,43 (m, 2H), 3,85 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,12-3,26 (m, 2H), 2,98 (s, 6H), 2,30 (s, 3H).
Ejemplo FP: 2-[[2,6-dicloro-3-(morfolina-4-carbonil)fenil]metil]-N,1,4-trimetil-indol-6-carboxamida
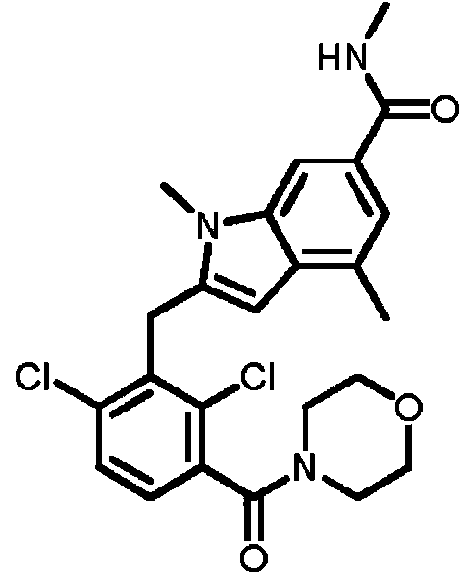
Usando un procedimiento similar al Ejemplo A1, 2-[[2,6-dicloro-3-(morfolina-4-carbonil)fenil]metil]-N,1,4-tri-metil-indol-6-carboxamida (35 mg, 68%) se preparó a partir de ácido 2-(2,6-dicloro-3-(morfolina-4-carbonil)bencil)-1,4-dimetil-1H-indol-6-carboxílico (50 mg, 0,11 mmol) (Ejemplo FO, Etapa 1) y metilamina (0,217 pL, 0,43 mmol). Lc/MS (Método g): Rt = 1,32 min; MS m/z: 474 [M+H]+; 1H NMR (DMSO-cfe, 400MHz): 5 8,26 (m, 1H), 7,82 (s, 1H), 7,68 (d, J=8,4 Hz, 1H), 7,45 (d, J=8,4 Hz, 1H), 7,31 (m, 1H), 5,59 (s, 1H), 4,44 (m, 2H), 3,87 (s, 3H), 3,65 (m, 4H), 3,54 (m, 2H), 3,19 (m, 2H), 2,80 (d, J=4,4 Hz, 3H), 2,32 (s, 3H).
Ejemplo FQ: ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-mdol-3-il)metil)benzoil)piperidma-4-carboxílico

Etapa 1: 2-bromo-1-metil-3-nitro-5-(trifluorometil)benceno

Una solución bifásica amarilla de 1-bromo-2-metil-4-(trifluorometil)benceno (40,65 g, 170 mmol) (Combi-blocks) y H2SO4 (203 mL) se enfrió en un baño de hielo y se añadió nitrato y potasio (20,63 g, 204 mmol) en porciones, manteniendo la temperatura <25 °C. El baño de hielo se retiró y la temperatura se mantuvo entre 20 y 25 °C. Después de 1,5 horas, LC/MS mostró reacción completa. La solución se enfrió en un baño de hielo y se añadió agua (400 mL) durante 10 minutos, manteniendo la temperatura <30 °C. Se extrajo la capa acuosa con MTBE (2x100 mL), la capa orgánica se lavó con salmuera (2x50 mL). Los extractos se combinaron y se secaron sobre Na2SO4, se filtraron y el disolvente se eliminó para producir 2-bromo-1-metil-3-nitro-5-(trifluorometil)benceno (45,1 g, 159 mmol, 93% de rendimiento), que se usa sin purificación adicional. LC/MS (Método i) Rf = 1,73 min.; MS m/z: 282,13 & 283,95 (M-H)-; 1H NMR cruda muestra una relación de isómeros de 5:1
Etapa 2: bencil 2-ciano-2-(2-metil-6-nitro-4-(trifluorometil)fenil)acetato

Una mezcla de 2-bromo-1-metil-3-nitro-5-(trifluorometil)benceno (86,6 g, 305 mmol), DMF (178 mL), cianoacetato de bencilo (46,7 mL, 305 mmol) y carbonato de potasio (93 g, 671 mmol) se calentó a 80 °C. Después de 90 minutos, la solución se enfrió a <20 °C mediante la adición de agua/hielo (800 mL), seguido de HCl 2N (534 mL, 1067 mmol) cuidadosamente a <25 °C. El producto crudo se dividió entre la fase acuosa ácida y EtOAc (4x150 mL). Los extractos orgánicos se combinaron, se lavaron con agua (3x200 mL) y salmuera (200 mL), se secaron sobre Na2SO4, se filtraron y
se eliminó el disolvente al vacío para producir una goma oscura que solidificó al reposar. El sólido se trituró con MTBE (100 mL) durante 15 minutos antes de la adición gota a gota de heptano (200 mL). El sólido se recogió y se lavó con 1:2 MTBE/heptanos (2x50 mL), finalmente Et2 O (2x20 mL) y se secó para dar un polvo sólido de color beige de bencil 2-ciano-2-(2-metil-6-nitro-4-(trifluorometil)fenil)acetato (71 g, 184 mmol, 60,3%). LC/Ms (Método i) Rf = 1,73 min.; MS m/z: 377,31 (M-H)-.
Etapa 3: 4-metil-6-(trifluorometil)-1H-indol

Bencil 2-ciano-2-(2-metil-6-nitro-4-(trifluorometil)fenil)acetato (63,5 g, 168 mmol), EtOH (490 mL), agua (49 mL), ácido acético (49 mL) se añadieron a Pd/C al 5% (JM#9 húmedo) (21,51 g, 101 mmol) en un hidrogenador Parr de 1,7 L y se agitó durante 23 horas a 70 psi y 50 °C. La mezcla se enfrió a temperatura ambiente y se filtró, se concentró al vacío a 50 °C, se diluyó con EtOAc (200 mL) y agua (200 mL). La capa acuosa se separó y se extrajo con EtOAc (2x50 mL). EtOAc original y los extractos se combinaron, se lavaron con HCl 2N (2x80 mL) seguido por Na2 S2 O3 acuoso 10 % (2x100 mL), NaHCO3 saturado acuoso (3 x 150 mL) y salmuera (2x50mL), se secó (Na2 SO4 ), se filtró y se destiló el disolvente para producir un líquido púrpura que solidificó al dejar en reposo 4-metil-6-(trifluorometil)-1H-indol (38 g, 191 mmol, 100% de rendimiento). Se uso sin purificación adicional. LC/MS (Método i) Rf = 1,58 min.; MS m/z: 198,23 (M-H)-.
Etapa 4: terc-butil 2,4-didoro-3-((4-metil-6-(trifluorometil)-1H-indol-3-il)metil)benzoato

Una solución de 4-metil-6-(trifluorometil)-1H-indol (14,4 g, 72,3 mmol) en DCM (75 mL) se enfrió a <5 °C y se añadió tertbutil 2,4-dicloro-3-formilbenzoato (21,88 g, 80 mmol) (Preparación #33, Etapa B), trietilsilano (34,6 mL, 217 mmol) y 2,2,2-TFA (8,36 mL, 108 mmol), manteniendo la temperatura <5 °C. Después de 2 horas la mezcla de reacción se vertió sobre una solución agitada de MTBE (200 mL) y NaHCO3 saturado (200 mL). Después de 30 minutos, la capa orgánica se separó, se lavó con salmuera y se concentró al vacío para proporcionar terc-butil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-3-il)metil)benzoato como un aceite. El producto crudo se usó sin purificación adicional.
Etapa 5: ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil) benzoico
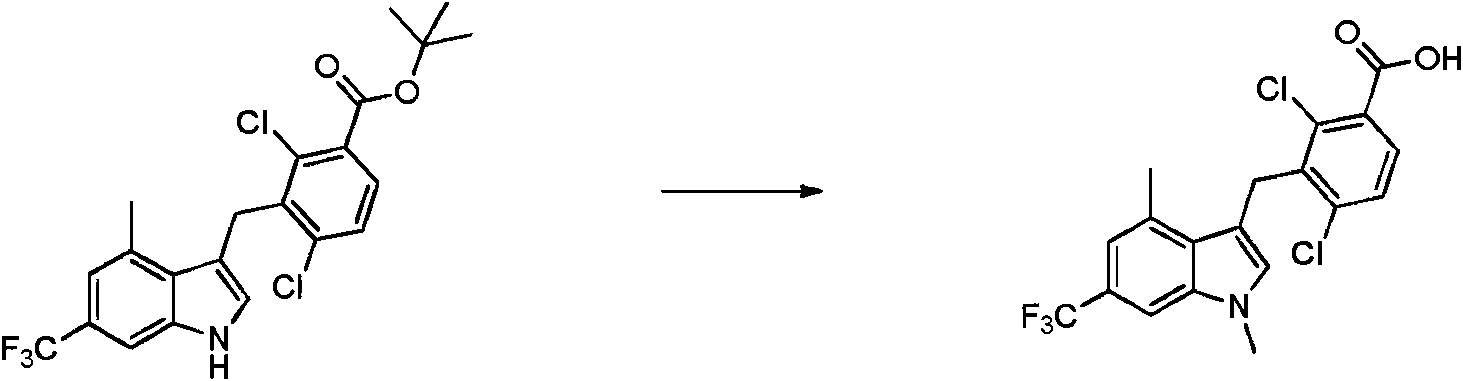
A una solución del terc-butil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-indol-3-il)metil)benzoato crudo (33 g, 72,0 mmol) en DMF (100 mL) se añadió yodometano (4,95 mL, 79 mmol) seguido de una solución de 2-metilpropan-2-olato de sodio (7,61 g, 79 mmol) en DMF (50 mL) mientras se mantenía una temperatura de aproximadamente 25-30 °C . Después de 4 h, la mezcla de reacción se diluyó con MTBE (200 mL) y NH4 Cl (100 mL). La capa orgánica se separó, se lavó con agua (100 mL) y salmuera (100 mL), se secó (Na2 SO4 ), se filtró y se concentró al vacío. El éster crudo se disolvió en 1,4-dioxano (240 mL) y una solución 6N de cloruro de hidrógeno (120 mL, 720 mmol). La mezcla de reacción se calentó a 85 °C. Después de 48 horas, la mezcla de reacción se enfrió a temperatura ambiente y se añadió MTBE (200 mL). La capa orgánica se separó, se lavó con salmuera, se secó sobre Na2 SO4 , se filtró y se concentró al vacío. El material crudo se suspendió en ACN (100 mL), se calentó brevemente a 70 °C, se enfrió a temperatura ambiente y se filtró. Los sólidos contenían producto y sales inorgánicas (no solubles en DMSO o MeOH). Los sólidos se disolvieron en EtOAc (200 mL) y se lavaron con agua (100 mL) y salmuera (100 mL), se secaron sobre Na2 SO4 , se filtraron y se concentraron al vacío. Los sólidos se suspendieron en aCn (100 mL) calentado a 70 °C con agitación. Después de 2 horas, la mezcla se enfrió a temperatura ambiente y se filtró para proporcionar ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3il)metil)benzoico (20 g, 48,1 mmol, 66,7% como un sólido color canela. LC/MS (Método i) Rf = 2,18 min.; MS miz: 414 (M-H)-.
Etapa 6: ácido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidina-4-carboxílico

A una suspensión de ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoico (20 g, 48,1 mmol) en THF (200 mL) se añadió hexafluorofosfato (V) de 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (20,10 g, 52,9 mmol) seguido de trietilamina (26,8 mL, 192 mmol). Después de 10 minutos, se añadió etil piperidina-4-carboxilato, ácido clorhídrico (10,24 g, 52,9 mmol) en una porción. Después de 2 horas, LC/MS mostró reacción completa (solución amarilla casi homogénea). La mezcla se diluyó con MTBE (300 mL) y se lavó con HCl 1 N (50 mL x 3), NaOH 1N (50 mL x 3), NH4Cl saturado (50 mL) y salmuera (50 mL), se secó sobre Na2SO4, se filtró y se concentró al vacío hasta un aceite amarillo. El aceite se disolvió en MTBE (200 mL) y se añadió carbono decolorante (15 g). Después de 30 minutos de agitación, la mezcla se filtró y se concentró al vacío hasta obtener una espuma. A una solución del éster en 1,4-dioxano (200 mL) se añadió hidróxido de sodio 2 N (96 mL, 192 mmol). La mezcla de reacción se calentó a 75 °C. Se añadió MeOH hasta que fue homogénea. Después de 1 hora se completó la hidrólisis. La solución se enfrió a temperatura ambiente. Se añadió MTBE (500 mL) y el pH se ajustó a 1 con HCl 5N. La capa orgánica se separó, se lavó con agua (2 x 100 mL), salmuera (100 mL), se secó sobre Na2SO4, se filtró y se concentró al vacío hasta un sólido amarillo. Los sólidos se suspendieron en ACN y se agitaron durante aproximadamente 18 horas. Después de 15 horas, los sólidos se recogieron por filtración enjuagando con ACN (2 x 10 mL). Después de secar durante 30 minutos, los sólidos se transfirieron a un vial y se secaron en un horno de vacío a 55 °C durante aproximadamente 18 horas para proporcionar ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-indol-3-il)metil)benzoil)piperidina-4-carboxílico (24,5 g, 46,5 mmol, 97% de rendimiento) como un sólido de color blanco. LC/MS (Método i) Rf = 2,52 min.; MS miz: 525 (M-H)-.
Ejemplo FR: ácido 2-(1-(3,5-didoro-4-((3,7-dimetM-5-(trifluorometN)-1H-mdoM-N)metN)picolmoN)piperidm-4-il)acético

Etapa 1: 1-óxido de 3,5-dicloro-4-(dietoximetil)piridina

A una solución de 3,5-dicloroisonicotinaldehído (50 g, 284 mmol) en EtOH (500 mL) se añadió trietoximetano (47,3 mL, 284 mmol) seguido de ácido sulfúrico (1,514 mL, 28,4 mmol). La mezcla de reacción se calentó a 75 °C. Después de 4 horas, la mezcla de reacción se enfrió a temperatura ambiente, se concentró parcialmente al vacío y los sólidos comenzaron a precipitar. Los sólidos se recogieron por filtración y se secaron al vacío para proporcionar 3,5-dicloro-4-(dietoximetil)piridina (41 g, 164 mmol, 57,7%) como un sólido de color blanquecino. El filtrado se concentró parcialmente al vacío hasta aproximadamente 100 mL y se enfrió en un baño de hielo. Después de 30 minutos, se recogieron sólidos adicionales por filtración, se enjuagaron con EtOH helado y se secaron al vacío para proporcionar 3,5-dicloro-4-(dietoximetil)piridina (20,2 g, 81 mmol, 28,4% de rendimiento) como un sólido de color blanquecino. Los dos cultivos se combinaron. 1H NMR (400 MHz, DMSO-d6) 58,60 (s, 1H), 5,89 (s, 1H), 3,73 (dq, J = 9,7, 7,1 Hz, 2H), 3,50 (dq, J = 9,7, 7,0 Hz, 2H), 1,13 (t, J = 7,0 Hz, 6H).
Etapa 2: 3,5-didoro-4-(dietoximetil)piridina 1-óxido

A una solución de 3,5-didoro-4-(dietoximetil)piridina (41 g, 164 mmol) se añadió ácido 3-clorobenzoperoxoico (44,1 g, 197 mmol). Después de 2 días, la mezcla de rea 2O (aproximadamente 300 mL) y se lavó con NaOH 2N (2 x 300 mL), salmuera (100 mL), se secó so se concentró al vacío. Los sólidos resultantes se suspendieron en heptano (aproximadamente 40 m utos de agitación, los sólidos se recogieron por filtración y se secaron al vacío para proporcionar 3,5-dicloro-4-(dietoximetil)piridina 1-óxido (34,5 g, 130 mmol, 79%) como un sólido de color canela. LC/MS (Método i) Rf = 1,70 min.; MS miz: 266 (M+H)+.
utos de agitación, los sólidos se recogieron por filtración y se secaron al vacío para proporcionar 3,5-dicloro-4-(dietoximetil)piridina 1-óxido (34,5 g, 130 mmol, 79%) como un sólido de color canela. LC/MS (Método i) Rf = 1,70 min.; MS miz: 266 (M+H)+.
Etapa 3: 3,5-didoro-4-(hidroximetil)picolinato
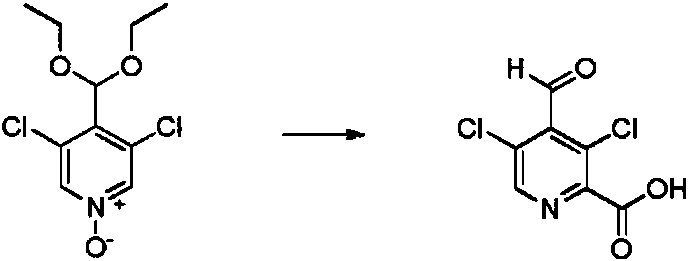
A una solución de 3,5-dicloro-4-(dietoximetil) piridina 1-óxido (67 g, 252 mmol) en ACN (140 mL) se añadió trietilamina (70,2 mL, 504 mmol) seguido de trimetilsilanocarbonitrilo (101 mL, 755 mmol). La mezcla de reacción se purgó con nitrógeno. Después de 10 minutos, la mezcla de reacción se equipó con un condensador de reflujo y un globo. La mezcla de reacción se calentó a 80 °C. Después de 15 horas, la mezcla de reacción se enfrió a temperatura ambiente y se concentró al vacío. La mezcla se diluyó con hidróxido de sodio 2N (629 mL, 1259 mmol) y dioxano (~ 60 mL). La mezcla de reacción se calentó a 80 °C. Después de 15 horas, la mezcla de reacción se enfrió a temperatura ambiente, se diluyó con EtOAc (aproximadamente 600 mL) y el pH de la mezcla de reacción se ajustó a 1 con HCl concentrado. La capa orgánica se separó, se secó sobre Na2SO4, se filtró y se concentró al vacío. Los sólidos se suspendieron en heptano (aproximadamente 100 mL) con agitación. Después de 2 horas, los sólidos se recogieron por filtración para proporcionar ácido 3,5-dicloro-4-(dietoximetil)picolínico. El acetal se suspendió en HCl 6N (500 mL) y 1,4-dioxano (aproximadamente 50 mL). Después de 2 horas, LC/MS mostró que se habían formado el aldehido y los sólidos significativos. Los sólidos se disolvieron en EtOAc (600 mL). La capa acuosa ácida se extrajo con EtOAc (3 x 200 mL). Todas las fases orgánicas se combinaron, se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron al vacío usando intercambio de disolvente con heptanos durante la concentración. Los sólidos resultantes se recogieron por filtración para proporcionar ácido 3,5-dicloro-4-formilpicolínico (47 g, 214 mmol, 85% de rendimiento) como un sólido de color canela. LC/MS (Método i) R = 0,40 min.; MS miz: 218 (M-H)-.
Etapa 4: 3,5-dicloro-4-(hidroximetil)picolinato

A una solución de ácido 3,5-dicloro-4-formilpicolínico (51,6 g, 235 mmol) en EtOH (500 mL) se añadió cuidadosamente tetrahidroborato de sodio (18,63 g, 493 mmol) para mantener una temperatura de alrededor de 35 °C durante la adición. El precipitado se formó poco después de la adición. Después de 2 horas a temperatura ambiente, se añadió lentamente MTBE (150 mL). Después de 30 minutos, los sólidos se recogieron por filtración enjuagando con MTBE y se secaron al vacío a 50 °C para obtener 3,5-dicloro-4-(hidroximetil)picolinato de sodio crudo (70 g) que se usó en el siguiente etapa sin purificación adicional. 1H NMR (DMSO, d6): 58,30 (s, 1H); 4,64 (s, 2H).
Etapa 5: ácido 3,5-didoro-4-(dorometil)picolínico

A una suspensión de ácido 3,5-didoro-4-(hidroximetil)picolínico (70 g, 236 mmol) en DCM (150 mL) se añadió dicloruro sulfuroso (69,0 mL, 946 mmol). Después de 15 horas se añadió cuidadosamente agua (5 mL) y después EtOAc (200 mL) para controlar el burbujeo vigoroso. Las capas orgánicas se separaron, se secaron sobre Na2SO4, se filtraron y se concentraron al vacío hasta un sólido de color blanquecino, ácido 3,5-dicloro-4-(clorometil)picolínico (50,7 g, 211 mmol, 89%). LC/MS (Método i) R = 0,30 min.; MS m/z: 240 (M-H)-.
Etapa 6: 3,5-didoro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinico

A una solución de 3,7-dimetil-5-(trifluorometil)-1H-indol (38 g, 178 mmol) (Preparación #43) en THF (400 mL) se añadió hidruro de sodio (15,68 g, 392 mmol). Después de 15 minutos, se añadió ácido 3,5-dicloro-4-(clorometil)picolínico (42,9 g, 178 mmol) en DMF (100 mL). Después de 15 horas, la mezcla de reacción se había solidificado. La mezcla de reacción se diluyó con MTBE (600 mL) y agua (600 mL). El pH de la capa acuosa se ajustó a 1 con HCl 5N. Las capas orgánicas se separaron, se lavaron con salmuera (300 mL) se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. El producto se concentró de ACN varias veces, luego se suspendió en ACN (200 mL) agitado durante la noche. Los sólidos se recogieron, se enjuagaron con ACN (20 mL) y se secaron al vacío para proporcionar ácido 3,5-dicloro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1 -il)metil)picolínico (35,2 g, 84 mmol, 47,3% de rendimiento) como un sólido color canela. LC/MS (Método i) R = 1,20 min.; MS m/z: 417 (M-H)-.
Etapa 7: metil 2-(1-(3,5-didoro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinoil)piperidin-4-il)acetato

A una suspensión de ácido 3,5-dicloro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolínico (30 g, 71,9 mmol) en EtOAc (200 ml), DMF (20 mL) se añadió metil 2-(piperidin-4-il) acetato, ácido clorhídrico (15,32 g, 79 mmol) seguido de hexafluorofosfato(V) de 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (30,1 g, 79 mmol). Se añadió lentamente trietilamina (35,1 mL, 252 mmol) manteniendo la temperatura a 20-25 °C. Después de 30 minutos, se añadieron MTBE (100 mL) y agua (200 mL). La capa orgánica se separó, se lavó con HCl 1N (100 mL x 3), Na2CO3 saturado (100 mL x 2), NH4Cl saturado (100 mL) y salmuera (50 mL) y se concentró al vacío. El producto crudo se purificó por cromatografía en columna ultrarrápida usando DCM/MTBE (5-10%) como eluyente en una columna de SiO2330 g x 2 para proporcionar metil 2-(1-(3,5-dicloro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinoil)piperid-in-4-il)acetato (35 g, 62,9 mmol, 87% de rendimiento) como un aceite amarillo. LC/MS (Método i) Rf = 2,00 min.; MS m/z: 556 (M+H)+.
Etapa 8: ácido 2-(1-(3,5-didoro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinoil)piperidin-4-il) acético

A una solución de metil 2-(1-(3,5-didoro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinoil)piperidin-4-il)acetato (40 g, 71,9 mmol) en THF (300 mL) se añadió una solución 1M de hidróxido de sodio (71,9 mL, 144 mmol). La mezcla de reacción se calentó a 45-50 °C y se añadió MeOH hasta que resultó una mezcla de reacción homogénea. Después de 3 horas, la mezcla de reacción se enfrió a temperatura ambiente y se concentró parcialmente al vacío, eliminando aproximadamente 200 mL de disolvente. Se añadieron MTBE (300 mL) y HCl 2N (100 mL). La capa orgánica se separó, se lavó con agua (100 mL x 2), salmuera (100 mL), se secó sobre Na2 SO4 , se filtró y se concentró al vacío, intercambio de disolventes con ACN. Después de tres ciclos de concentración, se añadió ACN (400 mL) y la suspensión resultante se calentó a 50 °C. Después de 1 hora, la suspensión se enfrió lentamente a temperatura ambiente con agitación. Después de 15 horas, los sólidos se recogieron por filtración, se secaron en horno de vacío a 55 °C para proporcionar ácido 2-(1-(3,5-dicloro-4-((3,7-dimetil-5-(trifluorometil)-1H-indol-1-il)metil)picolinoil)piperidin-4-il)acético (30,5 g, 56,2 mmol, 78%) como un sólido de color blanco. LC/MS (Método i) Rf = 1,68 min.; MS miz: 540 (M-H)-.
Ejemplo FS: ácido 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxílico

Etapa 1: 4-metil-6-(trifluorometil)piridin-2(1H)-ona

Un matraz cargado con CHCh (1,25 L) se enfrió a 0 °C. Se añadió anhídrido 2,2,2-trifluoroacético (0,141 L, 1012 mmol) seguido de cloruro de 3-metilbut-2-enoilo (0,094 L, 843 mmol). Se añadió gota a gota trietilamina (0,259 L, 1856 mmol) de modo que la temperatura interna se mantuvo por debajo de 10 °C. Después de la adición, la mezcla se agitó a 0 °C durante 1 hora y luego se calentó a temperatura ambiente lentamente y se agitó durante la noche. La mezcla de reacción se lavó con agua (1 L), NaHCO3 saturado (1 L), agua/salmuera (1:1,500 mL), se secó sobre MgSO4 , se filtró y se concentró al vacío. El residuo se disolvió en ácido acético (1,25 L). Se añadió NH4 OAc (130 g, 1687 mmol). La reacción preparada se tapó con un globo y la mezcla se calentó a 115 °C durante la noche. Después de 20 horas, la mezcla de reacción se enfrió a 40 °C y se concentró al vacío. El jarabe se vertió en agua (aproximadamente 2 L) con agitación. Después de 1 hora, los sólidos se recogieron por filtración para proporcionar 4-metil-6-(trifluorometil)piridin-2(1H)-ona (101 g, 570 mmol, 67,6%) como un sólido color canela. LC/MS (Método i) Rf = 1,59 min.; MS miz: 178 (M+H)+; 1H NMR (400 MHz, Cloroformd) 56,79 (s, 1H), 6,70 (s, 1H), 2,34 (s, 3H).
Etapa 2: 2-cloro-4-metil-6-(trifluorometil)piridina

Un matraz de 50 mL en forma de pera equipado con un cabezal de destilación de recorrido corto se cargó con 4-metil-6-(trifluorometil)piridin-2(1H)-ona (20 g, 113 mmol). Al matraz se añadió dicloruro fenilfosfónico (19,21 mL, 135 mmol). La mezcla de reacción se calentó a 160 °C. Después de 3 horas, la presión sobre la mezcla de reacción se redujo lentamente a 100 mBar. La destilación comenzó a una temperatura de aproximadamente 120-130 °C. Cuando la destilación se desaceleró significativamente, la presión sobre la mezcla de reacción se redujo a 80 mBar y la temperatura del baño se elevó a 170 °C. Después de 3 horas, la destilación había cesado proporcionando 2-cloro-4-metil-6-(trifluorometil)piridina (12 g, 61,4 mmol, 54,3% de rendimiento).1H NMR (400 MHz, DMSO-d6) 57,81 (s, 1H), 7,73 (s, 1h), 2,43 (s, 3h).
Etapa 3: terc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato

Un matraz cargado con 2-doro-4-metil-6-(trifluorometil)piridina (35,12 g, 180 mmol), carbamato de terc-butilo (42,1 g, 359 mmol), Pd2(dba)3 (4,11 g, 4,49 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (X-phos) (4,28 g, 8,98 mmol) y carbonato de cesio (205 g, 629 mmol) se evacuó y se llenó con N2 (repetido 3 veces) antes de la adición de 1,4-dioxano desgasificado (350 mL). La mezcla se calentó luego a 80 °C durante 2 horas y luego se enfrió a temperatura ambiente. La mezcla de reacción se repartió entre agua (300 mL) y EtOAc (300 mL). La capa acuosa se extrajo adicionalmente con EtOAc (2 x 100 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad para dar un aceite de color rojo oscuro. Se usó crudo en la siguiente reacción.
Etapa 4: 4-metil-6-(trifluorometil)piridin-2-amina

Una mezcla de terc-butil (4-metil-6-(trifluorometil)piridin-2-il) carbamato (49,6 g, 180 mmol) y cloruro de hidrógeno (4M en 1,4-dioxano) (597 mL, 2388 mmol) se agitó a temperatura ambiente durante 2 horas. El disolvente se eliminó a presión reducida. El residuo se suspendió en EtOAc y se filtró. El filtrado se diluyó con EtOAc. La capa orgánica se extrajo con HCl 6N (3 x 50 mL). La capa acuosa se lavó con DCM (4 x 100 mL). El pH de la capa acuosa se ajustó a pH 8 con la adición de Na2CO3. El sólido de color amarillo brillante se recogió por filtración, se lavó con agua. Después, el sólido se disolvió en DCM, se lavó con salmuera, se secó sobre Na2SO4, se filtró y se concentró casi hasta la sequedad. Los sólidos se trituraron con éter de petróleo. El sólido se recogió por filtración, se lavó con éter de petróleo y se secó al aire para dar 4-metil-6-(trifluorometil)piridin-2-amina (27,73 g, 157 mmol, 88%) como un sólido de color amarillo pálido. LC/MS (Método i) R = 2,55 min.; MS m/z: 177 (M+H)+.
Etapa 5: 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina

Una mezcla de 4-metil-6-(trifluorometil)piridin-2-amina (31,45 g, 179 mmol)
se calentó a 70 °C durante 2 horas. Se añadieron aproximadamente 500 mL de agua. El sólido se recogió por filtración, se lavó con agua y se secó al aire durante 5 minutos. El sólido se solubilizó en DCM, se lavó con agua, se secó sobre Na2SO4, se filtró y se concentró casi hasta la sequedad. Los sólidos se trituraron con heptano. El sólido se recogió por filtración, se lavó con heptano y se secó para dar 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,99 g, 171 mmol, 96%) como un sólido de color amarillo claro. LC/MS (Método i) Rf = 2,55 min.; MS m/z: 333 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 56,90 (s, 2H), 2,53 (s, 3H).
Etapa 6: 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina

A una solución de color naranja clara de 3,5-dibromo-4-metil-6-(trifluorometil)piridin-2-amina (56,98 g, 171 mmol) en THF (570 mL) a -78 °C se añadió n-butil litio (2,5 M en hexano) (68,3 mL, 171 mmol) gota a gota. La mezcla se agitó a -78 °C durante 15 minutos. La agitación continuó a -78 °C durante otros 30 minutos. Una segunda porción de butil litio (2,5 M en hexano) (20,48 mL, 51,2 mmol) se añadió gota a gota. Después de 30 minutos, una tercera porción de butil litio (2,5 M en hexano) (13,65 mL, 34,1 mmol) se añadió gota a gota. LC/MS después de 15 minutos mostró que la reacción se había completado. El matraz de reacción se transfirió a un baño de agua con hielo. Se añadieron rápidamente 20 mL de agua y
la reacción se dejó calentar a temperatura ambiente. La mezcla de reacción se repartió entre NH4Cl saturado (300 mL) y EtOAc (300 mL). La capa orgánica se lavó con NaHCO3 (300 mL), se secó sobre Na2SO4, se filtró y se concentró casi hasta la sequedad. Los sólidos se trituraron con heptano. El sólido se recogió por filtración, se lavó con hepano para dar 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina (38,87 g, 145 mmol, 85%), como un sólido de color blanquecino. LC/MS (Método i) R, = 2,29 min.; MS m /z : 255 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 56,96 (s, 1H), 6,74 (s, 2H), 2,32 (s, 3H).
Etapa 7: (E)-3-(2-etoxivinil)-4-metil-6-(trifluorometil)piridin-2-amina

Un matraz cargado con 3-bromo-4-metil-6-(trifluorometil)piridin-2-amina (38,26 g, 143 mmol), (E)-2-(2-etoxivinil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (56,5 g, 285 mmol), diacetoxipaladio (0,960 g, 4,28 mmol), diciclohexil(2',4',6'-triisopropil-[1,1 '-bifenil]-2-il)fosfina (XPhos) (4,28 g, 8,98 mmol) y carbonato de cesio (116 g, 356 mmol) se desgasificó con N2 durante 15 minutos antes de la adición de 320 mL de 1,4-dioxano desgasificado/H2O (4:1). La mezcla se calentó a 80 °C durante 2 horas y luego se enfrió a temperatura ambiente. La mezcla de reacción se repartió entre EtOAc (200 mL) y agua (200 mL). La capa acuosa se extrajo adicionalmente con EtOAc (2 x 50 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Los sólidos se diluyeron con heptano, se filtraron, se lavaron con heptano para dar un sólido de color blanquecino (1,45 g) que se desechó. El filtrado se concentró hasta la sequedad nuevamente para dar un aceite negro espeso, que se dejó a temperatura ambiente durante la noche. Se observó una formación significativa de sólidos. Los sólidos se diluyeron nuevamente con heptano, se sonicaron y filtraron, se lavaron con heptano para dar un sólido marrón claro. 15,20 g 95% de pureza por LC/MS, 1H NMR. El filtrado se concentró hasta la sequedad para dar un aceite negro que se purificó por cromatografía ultrarrápida (EtOAc 0-25%/heptano durante 30 minutos; columna Redi-Sep, 330 g, 309 nm). Las fracciones se concentraron casi hasta la sequedad. El sólido se recogió por filtración, se lavó con una pequeña cantidad de heptano y se secó para dar la segunda cosecha de producto como un sólido de color amarillo claro. 12,69 g. Dos cultivos se combinaron para dar (E)-3-(2-etoxivinil)-4-metil-6-(trifluorometil)piridin-2-amina (27,89 g, 113 mmol, 79%). LC/MS (Método i) R, = 1,48 min.; MS m/z: 247 (M+H)+.
Etapa 8: 4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina

Una mezcla de (E)-3-(2-etoxivinil)-4-metil-6-(trifluorometil)piridin-2-amina (27,89 g, 113 mmol) y ácido acético (130 mL) se calentó a 100 °C durante la noche. Se enfrió a temperatura ambiente. El precipitado se recogió por filtración, se lavó con ACN y se secó para dar la primera cosecha de producto. El filtrado se concentró hasta aproximadamente 50 mL. El sólido se recogió por filtración, se lavó con ACN y se secó para dar la segunda cosecha de producto. El filtrado todavía tenía algún producto. El sólido se suspendió en aproximadamente 250 mL de EtOAc, se calentó a reflujo para disolverlo y luego se evaporó mediante rotación para eliminar la mayor parte del disolvente. Se añadió heptano y la suspensión se filtró y se secó para dar el producto como un sólido de color blanquecino. Los tres cultivos del producto se combinaron para dar 4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (19,77 g, 99 mmol, 88%). LC/MS (Método i) R, = 1,30 min.; MS m/z: 201 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 5 12,13 -11,98 (br, 1H), 7,67 (,, J= 3,1 Hz, 1H), 7,34 (d, J = 1,0 Hz, 1H), 6,63 (dd, J = 3,5, 1,9 Hz, 1H), 2,59 (s, 3H).
Etapa 9: 4-m etiM -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina

A una suspensión de hidruro de sodio (60% en aceite mineral) (4,35 g, 109 mmol) en DMF (40 mL) a 0 °C se añadió una solución de 4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (19,77 g, 99 mmol) bajo N2. Después de la adición, se retiró el baño de agua helada y la mezcla se agitó a temperatura ambiente durante 30 minutos y luego se enfrió a 0 °C. Cloruro de bencenosulfonilo (13,27 mL, 104 mmol) se añadió gota a gota. Después de la adición, la reacción se dejó calentar a temperatura ambiente y se agitó durante 1 hora. La reacción se inactivó con la adición de 100 mL de solución saturada
de NH4 CI seguido de la adición de 350 mL de agua. El sólido se recogió por filtración, se lavó con agua y se secó en un horno de vacío a 70 °C durante el fin de semana para dar un sólido de color blanquecino. Los sólidos se disolvieron en EtOAc, se filtraron a través de 70 g de gel de sílice y se lavaron con EtOAc. El filtrado se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad. Los sólidos se trituraron con EtOAc/heptano para dar 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (28,52 g, 84 mmol, 85%) como un sólido de color blanco. LC/MS (Método i): Rt = 1,84 min.; MS m/z: 341 (M+H)+; 1H NMR (400 MHz, DMSO-d6 ) 58,15-8,07 (m, 3H), 7,75-7,67 (m, 1H), 7,64-7,56 (m, 3H), 7,03 (d, J = 4,1 Hz, 1H), 2,57 (s, 3H).
Etapa 10: terc-butil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b] piridin-2-il)metil)benzoato

Una solución incolora de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (3 g, 8,82 mmol) en THF (45 mL) se enfrió a -78 °C bajo N2. Se añadió gota a gota LDA (1,0 M en THF/hexano) (10,58 mL, 10,58 mmol) (temperatura interna < -70 °C) (la solución se tornó naranja oscura al final de la adición). La mezcla se agitó a -78 °C durante 0,5 h. Una solución de tert-butil 2,4-dicloro-3-formilbenzoato (2,91 g, 10,58 mmol) en THF ( 8 mL) se añadió gota a gota (temperatura interna mantenida por debajo de -70 °C) (la solución permaneció rojo naranja). La mezcla se agitó a -78 °C durante 1 h. La reacción se inactivó con NH4Cl saturado (50 mL) y se extrajo con EtOAc (2x 50 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Los sólidos se purificaron por cromatografía ultrarrápida (EtOAc 0-25%/heptano durante 30 minutos y luego se mantuvo EtOAc 25%/heptano; columna Silicycle, 200 g, 278 nm) para dar terc-butil 2,4-dicloro-3-(hidroxi(4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (4,59 g, 7,46 mmol, 85%) como un sólido de color blanco. LC/MS (Método i) Rt = 2,18 min.; MS m/z: 615 (M+h)+.
Etapa 11: ácido 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoico

A una solución de terc-butil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (2,0571 g, 3,34 mmol) en CHCl3 (20 mL) se añadió SOCl2 (2,440 mL, 33,4 mmol). La mezcla se calentó a 60 °C durante la noche. SOCl2 adicional (2,440 mL, 33,4 mmol) se añadió, la mezcla se calentó a 60 °C durante otras 24 horas. El disolvente se eliminó a presión reducida. El residuo se repartió entre EtOAc (100 mL) y NaHCO3 saturado (100 mL). La capa orgánica se concentró hasta la sequedad para dar una espuma blanca. La TLC (25% EtOAc/heptano) indicó un pico principal. El intermedio se disolvió en ácido acético (45 mL). Se añadió zinc (1,093 g, 16,71 mmol). La mezcla se agitó a temperatura ambiente durante 0,5 horas, se filtró y se lavó con EtOAc. El filtrado se concentró hasta sequedad. El residuo se repartió entre EtOAc (20 mL) y HCl 1N (20 mL). La capa orgánica se lavó con NaHCO3 saturado (20 mL), se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad para dar una espuma blanca. A la espuma anterior se añadió DCM (12 mL) seguido de TFA (4 mL). La solución amarilla se agitó a temperatura ambiente durante 4 horas. Los volátiles se eliminaron a presión reducida. El residuo se repartió entre EtOAc (50 mL) y NH4Cl saturado (50 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad para dar una espuma blanca, que se usó en la siguiente etapa sin purificación adicional.
Etapa 12: 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b] piridin-2-il)metil)benzoato

A una solución de ácido 2,4-didoro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoico (1,816 g, 3,34 mmol) en ACN (54 mL) se añadió carbonato de cesio (3,27 g, 10,03 mmol) seguido de sulfato de dimetilo (0,634 mL, 6,68 mmol). La mezcla se agitó a temperatura ambiente durante 4 horas y luego se calentó a 40 °C durante la noche. La mezcla de reacción se repartió entre EtOAc (20 mL) y agua (20 mL). La capa acuosa se extrajo adicionalmente con EtOAc (20 mL). La capa orgánica se lavó con salmuera (10 mL), se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. El producto crudo se purificó por cromatografía ultrarrápida (EtOAc 0-50%/heptano durante 30 min; columna Redi-Sep, 120 g, 254 nm). El pico principal se concentró hasta la sequedad para dar metil 2,4-dicloro-3-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (1,15 g, 2,063 mmol, 61,8%) como una goma de color amarillo claro. LC/MS (Método i) Rt = 1,49 min.; mS m/z: 557 (M+H)+.
Etapa 13: 2,4-dicloro-3-((4-metN-6-(tnfluorometN)-1H-pirrolo[2,3-b]pmdm-2-N)metN)benzoato

Un matraz cargado con metil 2,4-dicloro-3-((4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (1,15 g, 2,063 mmol) se purgó con N2 durante 15 min. Se añadió THF (23 mL) para formar una solución amarilla. Se añadió TBAF (3,09 mL, 3,09 mmol). La mezcla se calentó a 50 °C durante la noche. El disolvente se eliminó a presión reducida. El residuo se disolvió en EtOAc (20 mL), se lavó con NaHCO3 saturado (2x 10 mL), salmuera (10 mL), se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. El producto crudo se purificó por cromatografía ultrarrápida (EtOAc 0-25%/heptano durante 30 minutos; columna Redi-Sep, 80 g, 284 nm) para dar metil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (662,7 mg, 1,588 mmol, 77%) como un sólido de color blanco. LC/MS (Método i) Rt = 2,01 min.; MS m/z: 417 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 512,16 (s, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,67 (d, J = 8,4 Hz, 1H), 7,28 (d, J = 0,8 Hz, 1H), 5,93 (dd, J = 2,1, 1,1 Hz, 1H), 4,49 (s, 2H), 3,86 (s, 3H), 2,45 (s, 3H).
Etapa 14: metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo [2,3-b] piridin-2-il)metil)benzoato

A una solución de metil 2,4-dicloro-3-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (662 mg, 1,587 mmol) en ACN (50 mL) se añadieron de carbonato de cesio (1551 mg, 4,76 mmol) seguido de sulfato de dimetilo (0,301 mL, 3,17 mmol). La mezcla se agitó a temperatura ambiente durante el fin de semana (se convirtió en una suspensión espesa después de aproximadamente 30 minutos de agitación). La reacción se inactivó con agua (20 mL) y se extrajo con EtOAc (20 mL). La capa orgánica se lavó con salmuera (3 x 10 mL), se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Los sólidos se trituraron con Et2O/heptano. El sólido se recogió por filtración, se lavó con heptano, se secó para dar metil 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (632,8 mg, 1,467 mmol, 92%) como un sólido de color blanco. lC/MS (Método i) Rt = 2,19 min.; MS m/z: 431 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 57,80 (d, J = 8,4 Hz, 1H), 7,71 (d, J = 8,4 Hz, 1H), 7,32 (d, J = 0,8 Hz, 1H), 5,80 -5,75 (m, 1H), 4,54 (s, 2H), 3,92 (s, 3H), 3,87 (s, 3H), 2,44 (d, J = 0,7 Hz, 3H).
Etapa 15: ácido 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoico

A una solución amarilla de metil 2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (632,8 mg, 1,467 mmol) en THF (12 mL) se añadió hidróxido de litio hidratado (123 mg, 2,93 mmol). La mezcla se agitó a temperatura ambiente durante la noche. El disolvente se eliminó a presión reducida. La solución se diluyó con agua (10 mL) y el pH se ajustó a 2-3 con HCl 1N. Se formó un precipitado similar a un gel. El sólido se recogió por filtración, se lavó con agua y se secó en un horno de vacío a 60 °C durante la noche para dar ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoico (389 mg, 64% de rendimiento). LC/MS (Método i) Rf = 1,42 min.; MS m /z : 417 (M+H)+. 1H NMR (400 MHz, DMSO-d6) 5 13,63 (s, 1H), 7,74 (d, J = 8,4 Hz, 1H), 7,65 (d, J = 8,4 Hz, 1H), 7,34-7,30 (m, 1H), 5,77 (s, 1H), 4,52 (s, 2H), 3,92 (s, 3H), 2,44 (s, 3H).
Etapa 16: (2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)fenil)(4-hidroxipiperidin-1-il)metanona
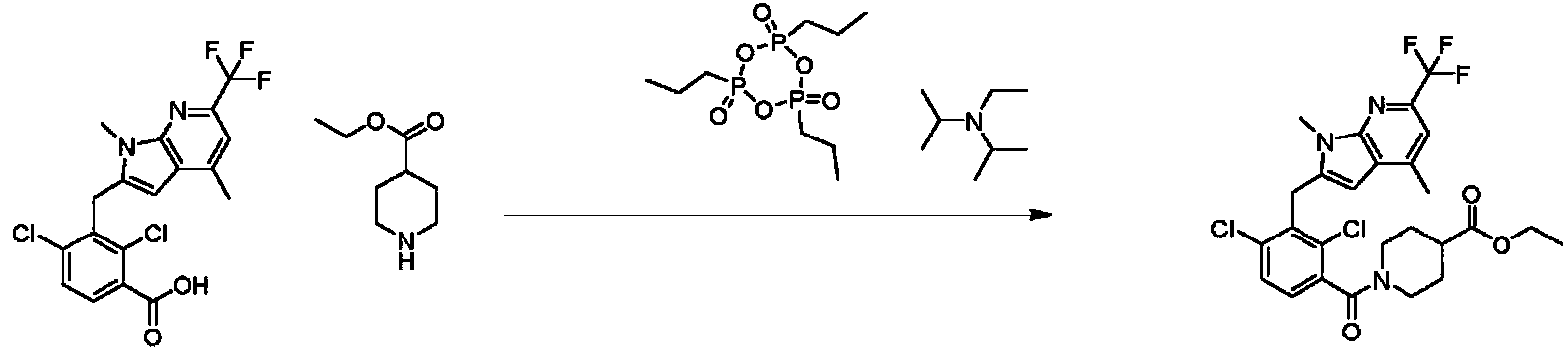
El ácido 2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo [2,3-b] piridin-2-il)metil)benzoico (120 mg, 0,288 mmol) se suspendió en EtOAc (2,4 mL). A la suspensión se añadieron etil piperidina-4-carboxilato (66,5 pL, 0,431 mmol) y N-etil-N-isopropilpropan-2-amina (95 pL, 0,546 mmol). La suspensión se enfrió a 0-5 °C en un baño de hielo y se añadió 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano 2,4,6-trióxido (308 pL, 0,518 mmol) gota a gota (se convirtió en una solución casi transparente con la adición de T3P). La mezcla de reacción se agitó durante 2 horas a 0-5 °C. La mezcla se vertió en un embudo separador, se lavó con HCl 1M (3x20 mL), NaOH 1M (3x20 mL), luego se secó sobre sulfato de sodio, se filtró a través de una almohadilla de gel de sílice, enjuagando con EtOAc. El filtrado se concentró hasta la sequedad para dar 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxi-lato (160 mg, 100%) como una espuma. lC/MS (Método i) Rf = 2,11 min.; MS m/z: 556 (M+H)+.
Etapa 17: ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxílico

Una mezcla de etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxilato (160 mg, 0,288 mmol) e hidróxido de litio hidratado (12,07 mg, 0,288 mmol) en THF (3 mL) y agua (1.000 mL) se agitó a temperatura ambiente durante la noche. Las capas se repartieron entre EtOAc (10 mL), aCN (2 mL) y HCl 1N (10 mL). La capa orgánica se lavó con salmuera (3x 5 mL)) después se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad. Los sólidos se trituraron con ACN. El sólido se recogió por filtración, se lavó con H2O y se secó para dar ácido 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoyl)piperidina-4-carboxílico (118,7 mg, 0,220 mmol, 77% de rendimiento) como un sólido de color blanco. LC/Ms (Método i) Rt = 2,41 min.; MS m/z: 528 (M+H)+.
Ejemplo FT: (2,4-didoro-3-((1,4-dimetM-6-(tnfluorometM)-1H-pirrolo[2,3-b]pindin-2-M)metM)fenM)(4-(2-metoxietil)piperidin-1-il)metanona

(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b] piridin-2-il)metil)fenil)(4-(2-metoxietil)piperidin-1-il)metanona se preparó de manera similar al Ejemplo FS, Etapa l6. LC/MS (Método i) Rt = 3,01 min.; MS m/z: 542 (M+H)+.
Ejemplo FT-1: ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b] piridin-2-il)metil)benzoil)piperidin-4-il)acético

ácido 2-(1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidin-4-il)acético se preparó de manera similar al Ejemplo FS, Etapa 16. lC/MS (Método i) Rt = 2,48 min.; MS m/z: 542 (M H) .
Ejemplo FT-3: etil 1-(2,4-didoro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxilato

Etil 1-(2,4-dicloro-3-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoil)piperidina-4-carboxilato se preparó de manera similar al Ejemplo FS Etapa 16. LC/MS (Método i) Rt = 2,41 min.; MS m/z: 528 (M+H)+.
Ejemplo FU: ácido 2-(1 -(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidin-4-il)acético

Etapa 1: terc-butil 2,4-dicloro-3-(4-metiM-(femlsulfoml)-6-(trifluorometM)-1H-pirrolo[2,3-b]piridma-2-carbonil)benzoato

A una solución transparente de terc-butil 2,4-dicloro-3-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)benzoato (4,73 g, 7,69 mmol) (Ejemplo FS, Etapa 10) en CH2 O2 (50 mL) a 0 °C se añadió peryodinano de Dess-Martin (3,59 g, 8,45 mmol) en una porción. La mezcla se agitó a 0 °C durante 2 horas, después se diluyó con DCM (50 mL), se lavó con 5% NaS2 O3 (30 mL) y NaHCO3 saturado (30 mL), salmuera (20 mL), se secó sobre Na2 SO4 , se filtró y se concentró a sequedad para dar terc-butil 2,4-dicloro-3-(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (4,7 g, 100%) como una espuma blanca. LC/MS (Método i) Rt = 2,28 min.; mS miz : 613 (M+H)+.
Etapa 2: terc-butil 2,4-dicloro-3-(4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato

Un matraz cargado con terc-butil 2,4-dicloro-3-(4-metiM-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (4,71 g, 7,68 mmol) se desgasificó con N2 durante 15 minutos. Se añadió THF (25 mL) para formar una solución amarilla clara. Se añadió fluoruro de tetrabutilamonio (1,0 M en THF) (7,68 mL, 7,68 mmol) para formar una solución marrón. La mezcla se calentó a 50 °C durante la noche. Se añadió fluoruro de tetrabutilamonio adicional (1,0 M en THF) (7,68 mL, 7,68 mmol) y la solución se calentó a 50 °C durante 6 horas. El disolvente se eliminó a presión reducida. El residuo se disolvió en EtOAc (100 mL), se filtró para eliminar los sólidos insolubles, después se lavó con NaHCO3 saturado (2x 50 mL), salmuera (2x 50 mL), se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad. Los sólidos se redisolvieron en THF (25 mL). Se añadió fluoruro de tetrabutilamonio (4,99 mL, 4,99 mmol). La mezcla se calentó a 50 °C durante 4 h. El disolvente se eliminó a presión reducida. El residuo se disolvió en EtOAc (100 mL), se lavó con NaHCO3 saturado (2x 50 mL), salmuera (2x 50 mL), se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad. Los sólidos se purificaron a través de una almohadilla de gel de sílice, eluyendo con EtOAc 40%/heptano. Las fracciones que contenían el producto se concentraron hasta la sequedad para dar terc-butil 2,4-dicloro-3-(4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (3,7127 g, 7,84 mmol, 102 %). LC/MS (Método i) Rt = 2,16 min.; MS m/z: 473 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 513,17 (s, 1H), 7,93 (d, J = 8,5 Hz, 1H), 7,74 (d, J = 8,4 Hz, 1H), 7,48 (s, 1H), 7,30 (s, 1H), 2,61 (s, 3H), 1,55 (s, 9H).
Etapa 3: terc-butil 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato

A una solución amarilla de terc-butil 2,4-dicloro-3-(4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (3,71 g, 7,84 mmol) en ACN (148 mL) se añadió carbonato de cesio (7,66 g, 23,52 mmol) seguido de sulfato de dimetilo (1,487 mL, 15,68 mmol). La mezcla se agitó a temperatura ambiente durante aproximadamente 60 horas. La solución se vertió en 300 mL de agua con agitación. El sólido se recogió por filtración, se lavó con agua y se secó para dar terc-butil 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (3,4 g, 89%) como un sólido de color marrón claro. LC/MS (Método i) Rt = 2,39 min.; MS m/z: 487 (M+H)+. 1H NMR (400 MHz, DMSO-d6) 57,94 (d, J = 8,4 Hz, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,54 (d, J = 0,9 Hz, 1H), 7,35 (s, 1H), 4,18 (s, 3H), 2,61 (d, J = 0,8 Hz, 3H), 1,55 (s, 9H).
Etapa 4: ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoico

A una solución amarilla de terc-butil 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoato (3,4 g, 6,98 mmol) en CH2Cl2 (17 mL) se añadió TFA (16,13 mL, 209 mmol). La mezcla se agitó a temperatura ambiente durante 2 horas. El disolvente se eliminó a presión reducida. Al residuo se añadió éter. La solución se sonicó hasta que comenzó a formarse un sólido y la suspensión resultante se agitó a temperatura ambiente durante 10 minutos. El sólido se recogió por filtración, se lavó con éter y se secó para dar ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoico (2,1816 g, 72%) como un sólido de color blanco. LC/MS (Método i) Rt = 1,39 min.; MS m/z: 431 (M+H)+; 1H NMR (400 MHz, DMSO-d6) 5 13,76 (s, 1H), 8,00 (d, J = 8,5 Hz, 1H), 7,75 (d, J = 8,4 Hz, 1H), 7,54 (d, J = 1,1 Hz, 1H), 7,31 (s, 1H), 4,18 (s, 3H), 2,61 (s, 3H).
Etapa 5: etil 2-(1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidin-4-il)acetato

El ácido 2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoico (150 mg, 0,348 mmol) se suspendió en EtOAc (2,319 mL). A la suspensión se añadieron etil 2-(piperidin-4-il)acetato (89 mg, 0,522 mmol) y N-etil-N-isopropilpropan-2-amina (0,115 mL, 0,661 mmol). La suspensión se enfrió a 0-5 °C en un baño de hielo y se añadió 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano 2,4,6-trióxido (0,372 mL, 0,626 mmol) gota a gota. La mezcla de reacción se agitó durante 2 horas a 0-5 °C. Se añadió HCl 1M (4 mL) y la mezcla acuosa se extrajo con EtOAc (3 x 4 mL). Las capas orgánicas se concentraron hasta la sequedad y se purificaron por cromatografía ultrarrápida (EtOAc 0-75%/heptano durante 30 minutos; Columna Redi-Sep, 24 g, 311 nm). Las fracciones se concentraron hasta la sequedad para dar etil 2-(1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidin-4-il)acetato (0,196 g, 0,335 mmol, 96%) como un aceite transparente. LC/MS (Método i) Rt = 2,16 min.; MS m/z: 584 (M+H)+.
Etapa 6: ácido 2-(1-(2,4-didoro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidin-4-il)acético

Una mezcla de 2-(1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbon-il)benzoil)piperidin-4-il)acetato (0,196 g, 0,335 mmol) e hidróxido de litio hidratado (0,014 g, 0,335 mmol) en THF (3 mL) y agua (1000 mL) se agitó a temperatura ambiente durante la noche. Se eliminó el solvente. Se añadieron agua (10 mL) y HCl (1N, 2 mL). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La suspensión se filtró y se secó para dar ácido 2-(1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidin-4-il)acético (0,162 g, 0,291 mmol, 87%) como un sólido de color blanquecino. LC/MS (Método i) R = 2,52 min.; MS m/z: 556 (M+H)+.
Ejemplo FV: 1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidina-4-carbonitrilo

Se preparó 1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)piperidina-4-carbonitrilo en una manera similar al Ejemplo FU, Etapa 5. LC/MS (Método i) Rt = 2,71 min.; MS m/z: 523 (M+H)+.
Ejemplo FW: (2,6-dicloro-3-((3S,4S)-4-hidroxi-3-metilpiperidina-1-carbonil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metanona y (2,6-dicloro-3-((3R,4R)-4-hidroxi-3-metilpiperidina-1-carbonil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metanona

(2,6-dicloro-3-((3S,4S)-4-hidroxi-3-metilpiperidina-1-carbonil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metanona y (2,6-dicloro-3-((3R,4R)-4-hidroxi-3-metilpiperidina-1-carbonil)fenil)(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metanona se prepararon de una manera similar al Ejemplo FU, Etapa 5, LC/MS (Método i) Rt = 2,71 min.; MS m/z: 523 (M+H)+.
Ejemplo FX: ácido (3R,4R)-1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperid i na-4-carboxíl ico

Ácido (3R,4R)-1-(2,4-Dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperidina-4-carboxílico se preparó de manera similar al Ejemplo FU, Etapa 6. LC/MS (Método i) Rt = 2,49 min.; mS m/z: 556 (M+H)+.
Ejemplo FY: (2,4-dicloro-3-(1,4-dimetN-6-(trifluorometN)-1H-pirrolo[2,3-b]piridma-2-carboml)feml)(4-(2-metoxietil)piperidin-1-il)metanona

(2,4-Dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonM)fenM)(4-(2-metoxietil)piperidin-1 -il)metanona se preparó de manera similar al Ejemplo FU, Etapa 5. LC/MS (Método i) Rt = 2,99 min.; MS m/z: 556 (M+H)+.
Ejemplo FZ: (2,4-dicloro-3-(1,4-dimetM-6-(trifluorometM)-1H-pirrolo[2,3-b]piridma-2-carboml)feml)(4-hidroxipiperidin-1 -il)metanona

(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)fenil)(4-hidroxipipe-ridin-1-il)metanona se preparó de una manera similar al Ejemplo FU, Etapa 5, LC/MS (Método i) Rt = 2,49 min.; MS m/z: 514 (M+H)+.
Ejemplo GA: ácido 2-((3R,4S)-1 -(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometM)-1H-pirrolo[2,3-b]piridma-2-carbonil)benzoil)-3-metilpiperidin-4-il)acético; ácido 2-((3S,4R)-1 -(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperidin-4-il)acético; ácido 2-((3S,4S)-1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperidin-4-il)acético; ácido 2-((3R,4R)-1-(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperidin-4-il)acético

ácido 2-((3R,4S)-1 -(2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)benzoil)-3-metilpiperidin-4-il)acético se preparó de manera similar al Ejemplo FU, Etapa 6. LC/mS (Método i) Rt = 2,53 min.; MS m/z: 570 (M+H)+. Los cuatro isómeros del producto final pueden aislarse por cromatografía quiral (Método quiral p) o prepararse individualmente a partir de materiales de partida quirales como se ejemplifica en la Preparación 83-84.
Ejemplo GB: (2,4-dicloro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)fenil)(4-(metilsulfonil)piperidin-1-il)metanona

(2,4-didoro-3-(1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina-2-carbonil)fenil)(4-(metilsulfo-nyl)piperidin-1-il)metanona se preparó de manera similar al Ejemplo FU, Etapa 5. LC/MS (Método i) Rt = 2,53 min.; mS m/z: 576 (M+H)+.
Ejemplo GC: ácido 2-((3S,4R)-1 -(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acético

Etapa 1: terc-butil 4,6-dicloro-5-(hidroxi(4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato

A una solución de 4-metil-1-(fenilsulfonM)-6-(trifluorometM)-1H-pirrolo[2,3-b]piridina (29 g, 85 mmol) (Ejemplo FS, Etapa 9) en THF (290 mL) se añadió diisopropilamida de litio 2M en THF (42,6 mL, 85 mmol) gota a gota a -78 °C bajo nitrógeno (convertido en una solución de color naranja rojiza, temperatura interna mantenida por debajo de -70 °C). La mezcla se agitó a -78 °C durante 30 minutos. Una solución de terc-butil 4,6-dicloro-5-forminicotinato (25,9 g, 94 mmol) se añadió gota a gota (Ejemplo DO, Etapa 12) en THF (145 mL) (temperatura interna mantenida por debajo de -75 °C). La mezcla se agitó a -78 °C durante 1 h. La reacción se inactivó con la adición de NH4Cl saturado (120 mL). El disolvente THF se eliminó al vacío y la capa acuosa se diluyó con agua (200 mL) y el producto se repartió entre EtOAc (3x100 mL) y la capa acuosa. Los extractos se combinaron y se lavaron con agua (3x70 mL), salmuera (100 mL) y luego se secaron sobre Na2 SO4, se filtraron y el disolvente se eliminó al vacío hasta la sequedad para dar una goma/espuma color canela 68 gdisolvente húmedo. La espuma se trituró con pentano (3x150 mL), gradualmente resultó una goma/sólido. La suspensión se sonicó para producir un polvo fino, se enfrió a aproximadamente 15 °C y el sólido se recogió y se lavó con pentano (3x80 mL) y se secó al vacío para dar un sólido de color blanquecino de producto crudo 42 g. El sólido se agitó con pentano (200 mL), se filtró y se secó para producir 21,5 g. El sólido se agitó con éter (20 mL) durante 10 minutos y se añadieron éter de petróleo (20 mL) gota a gota 30-60 °C con agitación. El sólido se recogió y se lavó con éter de petróleo (2x30 mL) 30-60 °C y se secó para producir un sólido de color blanco de terc-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo [2,3-b]piridin-2-il)metil)nicotinato (17,7 g, 28,7 mmol, 33,7%). LC/MS (Método i) Rf = 2,09 min.; MS m/z: 616 (m+H)+.
Etapa 2: terc-butil 4,6-dicloro-5-((4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato

A una solución de terc-butil 4,6-dicloro-5-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (7,9 g, 12,82 mmol) en CHCL (49,4 mL) se añadió dicloruro de azufre (4,68 mL, 64,1 mmol). La mezcla se calentó a 60 °C durante la noche. La mezcla de reacción se concentró a presión reducida para dar una espuma de color amarillo pálido. Se disolvió en ácido acético (26,0 mL). El zinc (4,19 g, 64,1 mmol) se agitó con ácido acético durante 10 minutos y se filtró, luego se añadió a la mezcla de reacción. Después de 1,5 horas, la reacción se filtró sobre una almohadilla de Celite® y se lavó con EtOAc (50 mL). El filtrado se concentró y se purificó mediante Analogix (columna de 120 g; EtOAc 0-20%/heptano en 10 volúmenes de columna). Las fracciones limpias se recogieron y se concentraron para dar terc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (3,1 g, 5,16 mmol, 40,3%). LC/MS (Método i) R = 2,29 min.; MS m/z: 600,601,602 (M+H)+.
Etapa 3: terc-butil 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato

A una solución de terc-butil 4,6-dicloro-5-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]pi-ridin-2-il)metil)nicotinato (3 g, 5,00 mmol) en THF (50,0 mL) se añadió fluoruro de tetrabutilamonio (5,50 mL, 5,50 mmol). La mezcla de reacción se agitó a temperatura ambiente durante aproximadamente 4 horas. La mezcla de reacción se concentró y se purificó mediante Analogix (columna de 80 g; EtOAC 0-25%/heptano en 7 volúmenes de columna) para dar terc-butil 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (1,83 g, 3,98 mmol, 80%). LC/MS (Método i) Rt = 2,10 min.; MS m/z: 460,461,462(M+H)+.
Etapa 4: terc-butil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato

Se cargó un vial con terc-butil 4,6-dicloro-5-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (1,48 g, 3,22 mmol) y ACN (1,00 mL) seguido de la adición de carbonato de cesio (3,14 g, 9,65 mmol) y sulfato de dimetilo (0,768 mL, 8,04 mmol). La mezcla se agitó a temperatura ambiente durante aproximadamente 60 horas. La mezcla se concentró a presión reducida. El residuo se repartió entre agua (50 mL) y EtOAC (60 mL). La capa orgánica se secó sobre MgSO4, se filtró y se concentró. El material crudo se purificó mediante Analogix (0-30% EtOAC/DCM; columna de gel de sílice Silicycle SiliaSep 40 g). Las fracciones recogidas se concentraron y se suspendieron en éter y se filtraron para dar un sólido de color blanco. El material se secó en un horno de vacío a aproximadamente 60 °C para dar 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (1,35 g, 2,85 mmol, 89%). LC/MS (Método i) Rt = 2,25 min.; MS m/z: 474,475,476 (M+H)+.
Etapa 5: ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico

A una solución amarilla de terc-butil 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinato (1,127 g, 2,376 mmol) en CH2 Cl2 (3 mL) se añadió TFA (6,04 mL, 78 mmol). La mezcla se agitó a temperatura ambiente durante aproximadamente 5 horas. La mezcla se concentró a presión reducida y se disolvió en dCm. Al añadir una solución saturada de bicarbonato de sodio, se formó un precipitado de color blanco que se recogió por filtración y se lavó con agua. El precipitado se secó en un horno de vacío a aproximadamente 60 °C durante la noche para dar ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico (820 mg, 1,961 mmol, 83% de rendimiento). LC/MS (Método i) Rt = 1,15 min.; MS m/z: 418,419,420 (M+H)+.
Etapa 6: etil 2-((3R,4S)-1-(4,6-dicloro- 5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acetato

Se cargó un vial con ácido 4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotínico (162 mg, 0,387 mmol) y etil 2-((3R,4S)-3-metilpiperidin-4-il)acetato, ácido clorhídrico (150 mg, 0,581 mmol) en EtOAc (2 mL). Se añadió N-etil-N-isopropilpropan-2-amina (0,203 mL, 1,162 mmol), seguido de la adición gota a gota de 2,4,6-tripropil-1.3.5.2.4.6- trioxatrifosfinano 2,4,6-trióxido (T3P) (0,530 mL, 0,891 mmol). La reacción se agitó durante la noche. Se añadió 2.4.6- tripropil-1,3,5,2,4,6-trioxatrifosfinano 2,4,6-trióxido (0,230 mL, 0,387 mmol) y N-etil-N-isopropilpropan-2-amina (0,101 mL, 0,581 mmol) y se agitó 3 horas. La reacción se calentó a aproximadamente 40 °C. Después de 2 horas, se añadió 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano 2,4,6-trióxido (0,230 mL, 0,387 mmol) y la reacción se agitó durante la noche a aproximadamente 40 °C. La mezcla de reacción se diluyó con EtOAc (10 mL) y se lavó con HCl 1N (10 mL), después con una solución de NaHCO3 (10 mL). La capa orgánica se secó sobre MgSO4 , se filtró y se concentró y se purificó mediante Analogix (columna de 12 g, EtOAc 10-80% /Hept) para dar etil 2-((3R,4S)-1-(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acetato (152 mg, 0,260 mmol, 67,0% de rendimiento). LC/MS (Método i) Rt = 2,01min.; MS m/z: 585,586,587(M+H)+.
Etapa 7: ácido 2-((3R,4S)-1-(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acético

A una solución de etil 2-((3R,4S)-1-(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acetato (151mg, 0,258 mmol) en THF (4 mL):agua (0,800 mL) se añadió hidróxido de litio hidratado (16,24 mg, 0,387 mmol) y la reacción se agitó a aproximadamente 35 °C durante la noche. La mezcla se concentró a presión reducida. El residuo se disolvió en agua (5 mL) y la capa acuosa se extrajo con DCM (5 mL). Se formó una emulsión. La mezcla completa se acidificó con HCl 1N a un pH de aproximadamente 1-2. Se añadió más DCM, se agitó y se dejó reposar durante aproximadamente 1 h. La capa orgánica se separó. La capa acuosa se lavó con 5% MeOH/DCM. Las capas orgánicas se combinaron, se secaron sobre MgSO4, se filtraron y se concentraron. El producto crudo se purificó
por RP-HPLC. Las fracciones obtenidas se concentraron al 60% del volumen original y los sólidos resultantes se recogieron por filtración y se secaron al vacío a aproximadamente 60 °C durante la noche para dar ácido 2-((3R,4S)-1-(4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicotinoil)-3-metilpiperidin-4-il)acético (102 mg, 0,183 mmol, 70,9% de rendimiento). Lc/MS (Método i) Rf = 2,36 min.; MS miz: 557,558,559 (M+H)+.
Ejemplo GD: (2,4-didoro-3-(1,4-dimetM-6-(tnfluorometM)-1H-pirrolo[2,3-b]pindma-2-carboml)feml)(4-(metilsulfonil)piperidin-1-il)metanona

Ácido 2-((3S,4R)-1-(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)nicoti-noyl)-3-metilpiperidin-4-il)acético (130 mg, 0,233 mmol, 59,6% de rendimiento) se preparó de manera similar al Ejemplo GC, Etapa 7. LC/MS (Método i) R = 2,36 min.; MS miz: 557,558,559 (M+H)+
Ejemplo GE: (2,4-didoro-3-(1,4-dimetM-6-(tnfluorometM)-1H-pirrolo[2,3-b]pindma-2-carboml)feml)(4-(metilsulfonil)piperidin-1-il)metanona

(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-3-il)(4-(metil-sulfonil)piperazin-1-il)metanona ( 110 mg, 0,195 mmol, 78% de rendimiento) de manera similar al Ejemplo GC, Etapa 6. LC/MS (Método i).) Rf = 2,47 min.; MS miz: 564,565, 566 (M+H)+.
Ejemplo GF: (4,6-didoro-5-((1,4-dimetM-6-(trifluorometN)-1H-pirrolo[2,3-b]piridm-2-M)metN)piridm-3-il)(morfolino)metanona

(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-3-il)(morfolino)metanona (102,7 mg, 0,207 mmol, 75% de rendimiento) se preparó de manera similar al Ejemplo GC, Etapa 6. LC/MS (Método i).) Rt = 2,52 min.; MS m/z: 487 (M+H)+.
Ejemplo GG: (4,6-didoro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-3-il)(4-(metilsulfonil)piperidin-1-il)metanona

(4,6-dicloro-5-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-3-il)(4-(metil-sulfonil)piperidin-1 -il)metanona (35 mg, 10% de rendimiento) se preparó de manera similar al Ejemplo GC, Etapa 6. LC/mS (Método i).) Rt = 1,59 min.; MS m/z: 563, 565, 567 (M+H)+.
Ejemplo GH: (3,5-didoro-4-((1,4-dimetN-6-(trifluorometM)-1H-pirrolo[2,3-b]piridm-2-M)metM)piridm-2-il)(morfolino)metanona
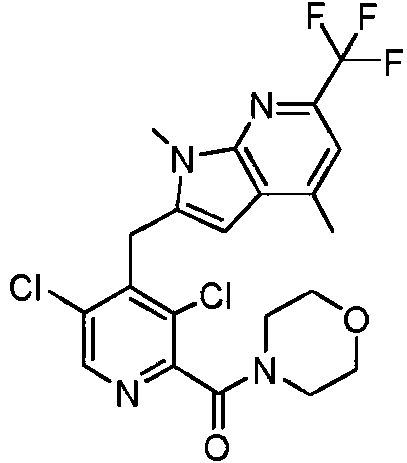
Etapa 1: 3,5-dicloro-2-(morfolina-4-carbonil) isonicotinaldehído

A una solución de ácido 3,5-dicloro-4-formilpicolínico (1,2 g, 5,45 mmol) (Eejemplo FR, Etapa 4) y hexafluorofosfato(V) de 2-(3H-[1,2,3]tri-azolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (2,281 g, 6,00 mmol) en THF (5 mL) se añadió morfolina (1,045 mL, 12,00 mmol). Después de 15 horas la mezcla de reacción se repartió entre EtOAc (10 mL) y NaHCO3 (10 mL). La capa orgánica se separó, se concentró al vacío y se purificó por cromatografía sobre gel de sílice (EtOAc/DCM) para dar 3,5-dicloro-2-(morfolina-4-carbonil)isonicotinaldehído (900 mg, 3,11 mmol, 57,1% de rendimiento) como un aceite espeso.1H NMR (400 MHz, DMSO-d6) 510,29 (s, 1H), 8,81 (s, 1H), 3,51 (t, J = 4,8 Hz, 4H), 3,16 (t, J = 4,8 Hz, 4H). Etapa 2: (3,5-didoro-4-(hidroxi(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona

Una solución incolora de 4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridina (900 mg, 2,64 mmol) (Ejemplo DO, Etapa 10) en THF (15 ml) se enfrió a -78 °C bajo N2. Se añadió gota a gota LDA (1,0 M en THF/hexano) (3,17 mL, 3,17 mmol) (manteniendo la temperatura interna < -75 °C). La mezcla se agitó a -78 °C durante 1 hora. Una solución de 3,5-dicloro-2-(morfolina-4-carbonil)isonicotinaldehído (918 mg, 3,17 mmol) en THF (8 mL) se añadió gota a gota (temperatura interna mantenida por debajo de -75 °C). La mezcla se agitó a -78 °C durante 1 hora. La solución se inactivó con NH4 Cl saturado (20 mL). Se extrajo la capa acuosa con EtOAc (2x 30 mL). La capa orgánica se secó sobre Na2 SO4 , se filtró y se concentró hasta la sequedad. El sólido se purificó por cromatografía ultrarrápida (EtOAc 25-75%/heptano durante 30 minutos; columna de Silicycle, 40 g, 282 nm) para dar (3,5-dicloro-4-(hidroxi(4-metil-1-(fenilsulfonil)-5-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (1,18 g, 1,875 mmol, 70,9% de rendimiento) como un sólido de color blanco. LC/MS (Método i) Rf = 2,63 min.; MS m/z: 573 (M+H)+.
Etapa 3: (3,5-dicloro-4-(cloro(4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona

A una solución transparente de (3,5-dicloro-4-(hidroxi(4-metil-1 -(fenilsulfonM )-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (300 mg, 0,477 mmol) en CHCl3 (3 mL) se añadió SOCl2 (0,174 mL, 2,383 mmol). La mezcla se calentó a 60 °C durante la noche. El disolvente se eliminó a presión reducida. El residuo se repartió entre EtOAc (20 mL) y NaHCO3 saturado (20 mL). La capa orgánica se concentró hasta la sequedad y se purificó por cromatografía ultrarrápida (EtOAc 0-75%/heptano durante 30 minutos; columna Redi-Sep, 24 g, 280 nm) para dar (3,5-dicloro-4-(cloro(4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (200,3 mg, 0,309 mmol, 64,9% de rendimiento) como un sólido de color blanco. lC/MS (Método i) Rf = 1,99 min.; MS m/z: 647 (m+h)+.
Etapa 4: (3,5-dicloro-4-((4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona

A una solución de (3,5-dicloro-4-(cloro(4-metil-1 -(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]pirid-in-2-il)metil)piridin-2-il)(morfolino)metanona (200 mg, 0,309 mmol) en AcOH (2 mL) se añadió zinc (101 mg, 1,543 mmol). La mezcla se agitó a temperatura ambiente durante 2 horas, luego se filtró y se lavó con EtOAc. El filtrado se concentró hasta sequedad. El residuo se repartió entre EtOAc (20 mL) y NaHCO3 saturado (20 mL). La capa acuosa se extrajo con EtOAc (1x10 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró hasta la sequedad para dar (3,5-dicloro-4-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (176,6 mg, 0,248 mmol, 80% de rendimiento) como una espuma blanca. LC/MS (Método i) Rt = 1,90 min.; MS m/z: 613 (M+H)+.
Etapa 5: (3,5-dicloro-4-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona

A una solución amarilla clara de (3,5-dicloro-4-((4-metil-1-(fenilsulfonil)-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (176 mg, 0,247 mmol) en THF (3 mL) bajo N2 se añadió TBAF (1,0 M en THF) (0,370 mL, 0,370 mmol). La mezcla se calentó a 50 °C durante 4 horas. El disolvente se eliminó a presión reducida. Se purificó por cromatografía ultrarrápida (EtOAc 0-75%/heptano durante 30 min; columna Redi-Sep, 24 g, 285 nm) para dar (3,5-dicloro-4-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (139,1 mg, 0,294 mmol, 119% de rendimiento) como un sólido de color blanco. LC/MS (Método i) Rt = 1,48 min.; MS m/z: 473 (M+H)+.
Etapa 6: (3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona

Un vial cargado con (3,5-dicloro-4-((4-metil-6-(trifluorometil)-1H-pirrolo[2,3-b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (139 mg, 0,294 mmol) y carbonato de cesio (239 mg, 0,734 mmol) y después se purgó con N2 durante 10 min antes de la adición de ACN (2 mL). La solución se purgó con N2 durante otros 5 minutos. Se añadió sulfato de dimetilo (0,042 mL, 0,441 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas. La solución se filtró y se lavó con EtOAc. El filtrado se concentró hasta la sequedad y se purificó por cromatografía ultrarrápida (EtOAc 0-75%/heptano durante 30 min; columna Redi-Sep, 24 g, 283 nm). Se purificó adicionalmente en el sistema Interchim RP con 20-95% 0,1% HCO2H en agua/ACN. Las fracciones que contenían el producto se concentraron para eliminar los volátiles y se liofilizaron hasta la sequedad para dar (3,5-dicloro-4-((1,4-dimetil-6-(trifluorometil)-1H-pirrolo [2,3 -b]piridin-2-il)metil)piridin-2-il)(morfolino)metanona (103 mg, 0,207 mmol, 84% de rendimiento) como un polvo blanquecino. LC/MS (Método i) Rt = 2,56 min.; MS m/z: 487 (M+H)+.
Ejemplo GI: ácido 2-(1-(2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoil)piperidin-4-il)acético

Etapa 1: terc-butil 2,4-dicloro-3-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato
En un vial d
THF (THF) (20 mL) para dar una solución incolora. La solución se enfrió a aproximadamente -65 °C en un baño de hielo seco/acetona. Se añadió una solución de terc-butil 2,4-diclorobenzoato (2,56 g, 10,36 mmol) (Preparación #33, Etapa A) en THF (THF) (15 mL) y la mezcla se agitó durante 2 horas. La solución se oscureció con la adición. En un vial de reacción separado, se añadió 1-metil-5-(trifluorometoxi)-1H-indol-2-carbaldehído (2,10 g, 8,64 mmol) (Preparación #52, Etapa N) en THF (THF) (20 mL) dar una solución amarilla y la mezcla se enfrió a aproximadamente -78 °C. La solución del anión se añadió gota a gota a través de una jeringa, manteniendo la temperatura < -65 °C. Se agitó a aproximadamente -65 °C durante 5 minutos. LC/MS muestra aproximadamente 30% de conversión. Se formó un anión adicional (1,2 eq) y se agregó. Después de 5 minutos, se consumió casi todo el material de partida. Se añadió ácido cítrico 1,0 M (100 mL) y se extrajo con MTBE (100 mL). Se lavó con salmuera (50 mL) y se secó sobre sulfato de magnesio. El producto crudto se trituró con heptano (50 mL). Los sólidos se eliminaron por filtración y la capa de heptano se dejó reposar. Se filtró el precipitado adicional que se había formado durante el fin de semana. El disolvente de heptano se eliminó al vacío para dar 5,27 g de un aceite crudo. El material crudo se cromatografió en el sistema Intelliflash 280 usando una columna de gel de sílice Redi-Sep de 120 g usando el siguiente gradiente: A: heptano; B: EtOAc; 0% B a 60% B durante 30 min. Dio terc-butil 2,4-dicloro-3-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (2,072 g, 49% de rendimiento). LC/MS (Método i) LC/MS (Método i) Rt = 2,17 min.; MS m/z: 490,0 (M+H)+.
Etapa 2: terc-butil 2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoato

En un matraz de fondo redondo, terc-butil 2,4-didoro-3-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)benzoato (1,03 g, 2,101 mmol) en DCM (10 mL) y THF (10,00 mL) se añadió para dar una solución amarilla. Se añadió dióxido de manganeso (1,826 g, 21,01 mmol) y la mezcla se agitó a temperatura ambiente. La mezcla se dejó reaccionar durante el fin de semana. La capa inorgánica se filtró y se añadió dióxido de manganeso adicional (1,826 g, 21,01 mmol). Después de 6 horas, se consumió todo el material de partida. La solución se filtró a través de una almohadilla de Celite®, y se lavó con DCM. El disolvente se eliminó a vacío para dar 1,020 g de producto crudo que se usó directamente en la siguiente etapa sin purificación adicional. LC/MS (Método i) Rt = 2,33 min.; MS miz: 488,18 (M+H)+.
Etapa 3: ácido 2,4-didoro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoico

En un matraz de fondo redondo, terc-butil 2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoato (1,020 g, 2,089 mmol) en dioxano (20 mL) se añadió para dar una solución incolora. Se añadió ácido clorhídrico 5 N (2,089 mL, 10,44 mmol) y la mezcla se calentó a 85 °C durante la noche. Se demostró que todo el material de partida se convirtió en producto por LC/MS. La solución se enfrió a temperatura ambiente. Se añadieron MBTE (40 mL) y agua (20 mL). Las capas se separaron y la capa orgánica se lavó con agua (2 x 10 mL), se secó sobre sulfato de magnesio y el disolvente se eliminó al vacío para dar ácido 2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoico (0,908 g, 95% de rendimiento) como un sólido que se usó en la siguiente etapa sin purificación adicional. LC/MS (Método i) LC/MS (Método i) Rt = 1,32 min.; MS m/z: 432,12 (M+H)+; MS m/z: 430,22 (M-H)-.
Etapa 4: metil 2-(1-(2,4-didoro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoil)piperidin-4-il)acetato

En un matraz de fondo redondo, ácido 2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoico (1,411 g, 3,26 mmol) en EtOAc (15 mL) se añadió para dar una solución amarilla. Se añadieron metil 2-(piperidin-4-il)acetato y ácido clorhídrico (0,696 g, 3,59 mmol) y N,N-dimetilformamida (3 mL). La solución se agitó durante 15 minutos. Se añadió HATU (1,366 g, 3,59 mmol) seguido de la adición gota a gota de trietilamina (1,593 mL, 11,43 mmol), manteniendo la temperatura < 25 °C. Después de 1 hora, se añadieron MBTE (40 mL) y agua (15 mL). Las capas se separaron y la capa orgánica se lavó con HCl 1N (3 x 15 mL), bicarbonato de sodio acuoso saturado (2 x 15 mL), cloruro de amonio acuoso saturado (15 mL) y salmuera (15 mL) y luego se secó sobre sulfato de magnesio y el disolvente se eliminó al vacío para dar 1,72 g de material crudo. El material crudo se cromatografió en el sistema Intelliflash 280 usando una columna de gel de sílice Silicycle SiliaSep de 120 g usando el siguiente gradiente: A: heptano; B: EtOAc; 10% B a 100% B durante 30 min. El disolvente se eliminó al vacío a partir de las fracciones 3-8 para dar metil 2-(1-(2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoil)piperidin-4-il)acetato (1,724 g, 91 % de rendimiento). Lc/MS (Método i) Rt = 1,99 min.; MS m/z: 571,17(M+H)+.
Etapa 5: ácido 2-(1-(2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoil)piperidin-4-il)acético

En un matraz de fondo redondo, metil 2-(1-(2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbon-il)benzoil)piperidin-4-il)acetato (1,59 g, 2,78 mmol) en THF (THF) (30 mL) y agua (10,0 mL) se añadió para dar una solución incolora. Se añadió hidróxido de litio (0,133 g, 5,57 mmol) y la mezcla se agitó a temperatura ambiente durante la noche. Mezcla de reacción se concentró y se añadió agua (10 mL). Se acidificó a pH ~ 3 con HCl 1N. Dio un precipitado aglomerado. Se filtró. Se lavó con agua. El residuo se disolvió en DCM. Se separó el agua y se secó sobre sulfato de magnesio. Se eliminó el disolvente al vacío, se secó a 60 °C. Se liofilizó durante el fin de semana (del agua/ACN). Dio ácido 2-(1-(2,4-dicloro-3-(1-metil-6-(trifluorometoxi)-1H-indol-2-carbonil)benzoil)piperidin-4-il)acético (0,993 g, 63% rendimiento). LC/MS (Método i) Rt = 1,69 min.; MS m/z: 557,20, 559,19 (M+H)+; MS m/z: 555,20, 557,22 (M-H)-.
Ejemplo GJ: ácido 2-(1-(4,6-dicloro-5-((1-metN-6-(tnfluorometoxi)-1H-indol-2-N)metN)nicotinoM)pipendin-4-il)acético

Etapa 1: ácido 4,6-dicloro-5-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotínico

A una solución de diisopropilamina (2,461 mL, 17,27 mmol) en
litio (6,91 mL, 17,27 mmol) en hexanos. Después de 20 minutos, la mezcla se enfrió a -75 °C y se añadió lentamente una solución de ácido 4,6-dicloronicotínico (1,658 g, 8,64 mmol) en THF (25 mL), temperatura <65 °C. Después de 30 minutos, se añadió lentamente una solución de 1-metil-6-(trifluorometoxi)-1H-indol-2-carbaldehído (1,05 g, 4,32 mmol) (Preparación #52, Etapa n) en THF (5 mL), temperatura <70 °C. Después de 30 minutos, se añadió HCl 1N (30 mL) y la mezcla de reacción se calentó a temperatura ambiente. Se añadieron MTBE/EtOAc (1:1, 50 mL) y agua (30 mL). La capa orgánica se separó, se lavó con salmuera (30 mL), se secó (Na2SO4), se filtró, se añadió TEA (10 mL) y la mezcla se concentró al vacío para proporcionar ácido 4,6-dicloro-5-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotínico crudo (1,8 g) que se usó sin purificación adicional.
Etapa 2: metil 2-(1-(4,6-dicloro-5-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)pip-eridin-4-il)acetato

A una solución de ácido 4,6-didoro-5-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotínico (1,8 g, 4,14 mmol) en EtOAc (EtOAc) (9 mL) y dimetilformamida (DMF) (9,00 mL) se añadió metil 2-(piperidin-4-il)acetato, ácido clorhídrico (1,682 g, 8,69 mmol), hexafluorofosfato(V) de 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouro-nio (3,30 g, 8,69 mmol) y trietilamina (2,88 mL, 20,68 mmol). Después de 2 h se añadió HCl 1N (20 mL) y MTBE (20 mL). La capa orgánica se separó, se lavó con NaHCO3 (20 mL). Se añadió NH4O (40 mL), lo orgánico se separó, se lavó con salmuera (20 mL), se secó (Na2SO4), se filtró y se concentró al vacío. El producto crudo se purificó por FCC en SiO2 (40 g) (EtOAc/Hep) para proporcionar metil 2-(1-(4,6-dicloro-5-(hidroxi(1-metil-6-(trifluor-ometoxi)-1H-indol-2-il)metil)nicotinoil)piperidin-4-il)acetato (1,4 g, 2,437 mmol, 58,9% de rendimiento) como una espuma. LC/MS (Método i) Rt = 1,55 min.; MS m/z: 574 (M+H)+.
Etapa 3: metil 2-(1-(4,6-dicloro-5-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)piperidin-4-il)acetato

A una solución de metil 2-(1-(4,6-dicloro-5-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)piperidin-4-il)acetato (1,4 g, 2,437 mmol) en DCM (DCM) (50 mL) a 0 °C se añadió trietilsilano (11,68 mL, 73,1 mmol) y 2,2,2-TFA (1,878 mL, 24,37 mmol). Después de 2 h, se calentó a temperatura ambiente. Después de 8 h se diluyó con MTBE (100 mL) y NaOH 2N (50 mL). Orgánico separada, se lavó con salmuera (50 mL), se secó (Na2SO4), se filtró y se concentró al vacío. FCC SiO2 (40 g), EtOAc/Hep para proporcionar metil 2-(1-(4,6-dicloro-5-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicoti-noyl)piperidin-4-il)acetato (800 mg, 1,433 mmol, 58,8% de rendimiento) como una espuma. LC/MS (Método i) Rt = 1,95 min.; MS m/z: 556 (M+H)+.
Etapa 4: ácido 2-(1-(4,6-didoro-5-((1-metN-6-(trifluorometoxi)-1H-mdol-2-N)metN)mcotmoN)piperidm-4-N)acético

A una solución de metil 2-(1-(4,6-dicloro-5-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)pip-eridin-4-il)acetato (1,2 g, 2,149 mmol) en THF (12 mL) se añadió una solución 1N de hidróxido de sodio (4,30 mL, 4,30 mmol). La mezcla de reacción se calentó a 45 °C y se añadió MeOH lentamente hasta que resultó una solución homogénea. Después de 3 horas, la mezcla de reacción se enfrió a temperatura ambiente y se añadieron MTBE:EtOAc (1:1, 30 mL) y agua (20 mL). La capa orgánica se separó, se lavó con salmuera (20 mL), se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se concentró a partir de ACN varias veces (forma de sólidos). El residuo se suspendió en ACN (3 mL), se calentó a 60 °C, se agitó durante 2 horas y se enfrió lentamente a temperatura ambiente y se filtró. El filtrado se concentró parcialmente bajo una corriente de aire y los sólidos resultantes se recogieron por filtración y se combinaron con la primera cosecha. Los sólidos se secaron al vacío a 50 °C para obtener ácido 2-(1-(4,6-dicloro-5-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)piperidin-4-il)acético (350 mg, 0,643 mmol, 29,9% de rendimiento) como un sólido de color blanco. LC/MS (Método i) Rt = 1,61 min.; MS m/z: 544 (M+H)+.
Ejemplo GK: ácido 2-(1-(4,6-dicloro-5-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)nicotinoil)piperidin-4-il)acético

Etapa 1: metil 2-(1-(3,5-didoro-4-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato

En un matraz de fondo redondo de 500 mL, se añadió diisopropilamina (9,12 mL, 64,0 mmol) en THF (53,3 mL) para dar una solución incolora. La solución se enfrió a aproximadamente 0 °C en un baño de hielo. Se añadió gota a gota butil litio (2,5 M) (24 mL, 60,0 mmol) y butil litio (2,0 M en ciclohexano) (2,00 mL, 4,00 mmol) y la solución se agitó a 0 °C durante aproximadamente 7 minutos. La solución se enfrió a aproximadamente -78 °C en un baño de hielo seco/acetona, luego se agitó a -78 °C durante aproximadamente 15 minutos. Una solución de ácido 3,5-dicloropicolínico (6,14 g, 32,0 mmol) (Ark Pharm) en THF (53,3 mL) se añadió gota a gota mediante una jeringa (uso de 10 ml adicionales de THF para enjuagar) durante aproximadamente 32 minutos, y la reacción se agitó durante unos 10 minutos. Una solución de 1-metil-6-(trifluorometoxi)-1H-indol-2-carbaldehído (3,89 g, 16,00 mmol) (Preparación #52, Etapa n) en THF (53,3 mL) se añadió gota a gota con una jeringa (uso de 10 mL adicionales de THF para enjuagar) durante aproximadamente 9 minutos, y la reacción se agitó durante aproximadamente 10 minutos. La mezcla de reacción se vertió en NH4Cl saturado en agitación (200 mL) más hielo (200 g), se añadió EtOAc (400 mL), HCl (1N, 20 mL). Las dos capas se repartieron, la capa orgánica se lavó con agua saturada. NH4Cl (2x100 mL), salmuera (2x100 mL), se secó sobre MgSO4, se filtró y se concentró para dar ácido 3,5-dicloro-4-(hidroxi(1-metil-6-(trifluorometh-oxi)-1H-indol-2-il)metil)picolínico (11 g) como aceite de naranja. El producto crudo se usó como está en la siguiente etapa. LC/MS (Método i) Rf = 1,03 min.; MS m/z: 436 (M+H)+.
Etapa 2: metil 2-(1-(3,5-didoro-4-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato

A una solución de color naranja de ácido 3,5-dicloro-4-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolínico (6,96 g, 16 mmol) en dimetilformamida (DMF) (45,0 mL) se añadió metil 2-(piperidin-4-il)acetato, ácido clorhídrico (6,51 g, 33,6 mmol), la solución se agitó durante aproximadamente 10 minutos y luego se enfrió por baño de hielo. Se añadió hexafluorofosfato(V) 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio (12,78 g, 33,6 mmol) y la solución se agitó durante aproximadamente 10 minutos. Luego se añadió lentamente trietilamina (11,15 mL, 80 mmol). Se eliminó el baño de hielo. La solución se agitó a temperatura ambiente durante 90 minutos. Se añadió EtOAc (400 mL) y NaHCO3 saturado (250 mL). La capa orgánica se lavó con NaHCO3 saturado (2x150 mL) y salmuera (125 mL) se secó (sobre Na2SO4 y MgSO4), se filtró y se concentró al vacío para dar 12,7 g de crudo. FCC SiO2 (220 g) EtOAc/Hep (45-85%) para proporcionar metil 2-(1-(3,5-dicloro-4-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoyl)piperidin-4-il)acetato (4,748 g, 8,27 mmol, 49,1% de rendimiento). LC/MS (Método i) Rf = 1,71 min.; MS m/z: 575 (M+H)+.
Etapa 3: metil 2-(1-(3,5-dicloro-4-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoyl)piperidin-4-il)acetato

En un matraz de fondo redondo de 2 L, metil 2-(1-(3,5-didoro-4-(hidroxi(1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil) picolinoil) piperidin-4-il)acetato (4,74 g, 8,25 mmol) en DCM (450 mL) se añadió para dar una solución amarilla pálida. La solución se enfrió a aproximadamente 0 °C en un baño de hielo. Se añadió trietilsilano (52,7 mL, 330 mmol). Se añadió 2,2,2-TFA (6,36 mL, 83 mmol) gota a gota. Después de 1 hora se retiró el baño de hielo y se agitó a temperatura ambiente. Después de 15 horas se añadió trietilsilano (25 mL, 157 mmol), se agitó a temperatura ambiente durante la noche. La mezcla de reacción se concentró y se purificó por medio de Isco (35-90% EtOAc/heptano) en gel de sílice para proporcionar metil 2-(1-(3,5-dicloro-4-((1-metil-6-(trifluoromet-oxi)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato (1,25 g, 2,194 mmol, 26,6% de rendimiento). LC/MS (Método i) Rf = 1,89 min.; MS m/z: 559 (M+H)+.
Etapa 4: ácido 2-(1-(3,5-didoro-4-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acético

En un vial de reacción de 40 mL, se añadió metil 2-(1-(3,5-dicloro-4-((1-metil-6-(trifluorometoxi)-1H-indol-2-il)metil)picolinoil)piperidin-4-il)acetato (1,25 g, 2,239 mmol) en MeOH (4,33 mL) y agua (2,163 mL) para dar una suspensión blanca. Se añadió hidróxido de sodio (2N) (2,239 mL, 4,48 mmol), se añadió THF (2,2 mL), se calentó a 70 °C durante 30 minutos, se convirtió en una solución roja transparente. La solución se enfrió a aproximadamente 0 °C en un baño de hielo. Cloruro de hidrógeno (0,895 mL, 4,48 mmol) se añadió gota a gota hasta pH = 1, se formó una suspensión gomosa blanca, se filtró y se lavó con agua (5x10 mL) y pentano (5x10 mL) para dar un sólido de color amarillo pálido. Al sólido se añadieron 12 mL de MeOH (10 ml/g), se calentó hasta 100 °C en un vial con tapa de 40 mL durante 10 minutos, todavía una suspensión de color amarillo pálido. La solución se enfrió lentamente a temperatura ambiente, se filtró, se lavó con MeOH (3x2 mL), se secó en embudo durante 2 horas para dar 970 mg de sólido de color amarillo pálido. El sólido se secó en un horno de vacío durante la noche para dar ácido 2-(1-(3,5-dicloro-4-((1-metil-6-(trifluorometoxi)-1 H-indol-2-il)metil)picolinoil)piperidin-4-il)acético (0,97 g, 1,764 mmol, 79% rendimiento). LC/mS (Método i) R = 1,62 min.; MS m/z: 545 (M+H)+.
Ejemplo GL: ácido 2-[1-[2,4-didoro-3-[[1-metN-7-(trifluorometM)-1H-mdol-3-N]metN]benzoN]-4-piperidN]acético

Etapa 1: íerc-butil 2,4-dicloro-3-[[7-(trifluorometil)-1H-indol-3-il]metil]benzoato

Usando un procedimiento similar al del Ejemplo DU Etapa 1, ferc-butil 2,4-didoro-3-[[7-(trifluorometil)-1H-indol-3-il]metil]benzoato (190 mg, 71%) se obtuvo a partir de from 7-(trifluorometil)-1H-indol (100 mg, 0,540 mmol) y ferc-butil 2,4-dicloro-3-formilbenzoato (182 mg, 0,594 mmol) (Preparación #33, Etapa B). LC/MS (Método i): Rt = 2,92 min; MS miz: 444 [M+H]+; 1H NMR ((DMSO-da , 300MHz): 5 = 5 11,28 (s, 1H); 7,96 (d, J = 8,1 Hz, 1H); 7,60 (m, 2H); 7,46 (d, J = 8,1 Hz, 1H); 7,20 (m, 1H); 6,80 (s, 1H); 4,40 (s, 2H); 1,53 (s, 9H)
Etapa 2: ferc-butil 2,4-dicloro-3-[[1-metN-7-(trifluorometN)-1H-mdol-3-N]metN]benzoato

Usando un procedimiento similar al Ejemplo A, Etapa 4, ferc-butil 2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoato (180 mg, 92%) se obtuvo a partir de ferc-butil 2,4-didoro-3-[[7-(trifluorometil)-1H-indol-3-il]metil]benzoato (180 mg, 0,405 mmol) y yoduro de metilo (27,9 L, 0,446 mmol). LC/MS (Método i): Rt = 2,79 min; MS miz: 458 [M+H]+; 1H NMR ((DMSO-da, 300MHz): 58,05 (d, J = 8,1 Hz, 1H); 7,60 (s, 2H); 7,59 (d, J = 8,1 Hz, 1H); 7,25 (m, 1H); 6,89 (s, 1H); 4,38 (s, 2H); 3,78 (s, 3H); 1,54 (s, 9H).
Etapa 3: ácido 2,4-dicloro-3-[[1-metN-7-(trifluorometN)-1H-mdol-3-N]metN]benzoico

Usando un procedimiento similar al del Ejemplo DJ, Etapa 2, ácido 2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoico se obtuvo (150 mg, 84%) a partir de ferf-butil 2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoato (170 mg, 0,371 mmol). LC/MS (Método j): Rt = 1,85 min; MS miz: 402 [M+H]+; 1H NMR ((DMSO-da, 300MHz): 5 13,5 (m broad, 1H); 8,04 (d, J = 8,4 Hz, 1H); 7,60 (m, 3H); 7,32 (m, 1H); 6,87 (s, 1H); 4,39 (s, 2H); 3,78 (s, 3H).
Etapa 4: metil 2-[1-[2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoil]-4-piperidil]acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6 , metil 2-[1-[2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoil]-4-piperidil]acetato (138 mg, 71%) se obtuvo a partir de ácido 2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoico (140 mg, 0,348 mmol) y clorhidrato de metil 2-(4-piperidil)acetato (81 mg, 0,418 mmol). lC/MS (Método j): Rt = 2,15 min; MS m/z: 541 [M+H]+; 1H NMR ((DMSO-da, 300MHz): 58,04 (m, 1H); 7,59 (m, 2H); 7,30 y 7,25 (d, J = 8,4 Hz, 1H); 7,20 (m, 1H); 6,87 (m, 1H); 4,48 (m, 1H); 4,35 (m, 2H); 3,79 (s, 3H); 3,57 y 3,59 (s, 3H); 3,35 (m, 1H); 3,02 (m, 1H); 2,80 (m, 1H); 2,27 (m, 2H); 1,99 (m, 1H); 1,75 (m, 1H); 1,62 (m, 1H); 1,15 (m, 2H).
Etapa 5: ácido 2-[1 -[2,4-dicloro-3-[[1 -metil-7-(trifluorometil)-1 H-indol-3-il]metil]benzoil]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-[1-[2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoil]-4-piperidil]acético (115 mg, 84%) se obtuvo a partir de metil 2-[1-[2,4-didoro-3-[[1-metil-7-(trifluorometil)-1H-indol-3-il]metil]benzoil]-4-piperidil]acetato (135 mg, 0,249 mmol). LC/MS (Método g): Rt = 1,90 min; MS m/z: 527 [M+H]+; 1H NMR ((DMSO-cf6, 300MHz): 512,11 (m broad, 1H); 8,02 (m, 1H); 7,63 (m, 2H); 7,38 y 7,32 (d, J = 8,4 Hz, 1H); 7,22 (m, 1H); 6,88 (m, 1H); 4,50 (m, 1H); 4,38 (m, 2H); 3,78 (s, 3h); 3,27 (m, 1H); 3,00 (m, 1H); 2,80 (m, 1H); 2,16 (m, 2H); 1,95 (m, 1H); 1,76 (m, 1H); 1,60 (m, 1H); 1,13 (m, 2H).
Ejemplo GN: ácido 2-[1-[4,6-dicloro-5-[(6-ciclopropil-4-fluoro-1-metiMH-indol-2-il)metil]piridina-3-carbonil]-4-piperidil]acético

Etapa 1: ferc-butil 5-[[1-(bencenosulfonil)-6-ciclopropil-4-fluoro-1H-indol-2-il]-hidroximetil]-4,6-dicloro-pi-ridina-3-carboxilato

Usando un procedimiento similar al Ejemplo CJ, Etapa 1, terc-butil 5-[[1-(bencenosulfonil)-6-ddopropil-4-fluoro-1H-indol-2-il]-hidroxi-metil]-4,6-didoro-piridina-3-carboxilato (105 mg, 18%) se preparó a partir de 6-ciclopropil-4-fluoro-1-(fenilsulfonil)-1H-indol (170 mg, 0,539 mmol) (Preparación 77) y terc-butil 4,6-dicloro-5-formil-piridina-3-carboxilato (223 mg, 0,809 mmol) (Ejemplo DO, Etapa 12). LC/MS (Método j): Rt = 2,31 min; MS m/z: 591 [M+H]+; 1H NMR ((DMSO-cf6, 300MHz): 5 = 8,63 (s, 1H); 7,84 (d, J=7,79 Hz, 2H); 7,66 (m, 1H); 7,59 (m, 2H); 6,89 (m, 1h); 6,78 (m, 2H); 2,10 (m, 1H): 1,6 (s, 9H); 1,52 (m, 1H); 1,39 (s, 1H); 1,00 (m, 2H); 0,71 (m, 2H).
Etapa 2: ácido 5-[[1-(bencenosulfonil)-6-ciclopropil-4-fluoro-1H-indol-2-il]metil]-4,6-dicloro-piridina-3-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 2, ácido 5-[[1-(bencenosulfonil)-6-ddopropil-4-fluoro-1H-indol-2-il]metil]-4,6-didoro-piridina-3-carboxílico (176 mg, 100%) se preparó a partir de ferc-butil 5-[[1-(bencenosulfonil)-6-ddopropil-4-fluoro-1H-indol-2-il]-hidroxi-metil]-4,6-didoro-piridina-3-carboxilato (200 mg, 0,338 mmol). LC/MS (Método j): Rt = 1,73 min; MS m/z: 519 [M+H]+
Etapa 3: ácido 4,6-dicloro-5-[(6-cidopropil-4-fluoro-1H-indol-2-il)metil]piridina-3-carboxílico

Usando un procedimiento similar al Ejemplo A, Etapa 3, ácido 4,6-didoro-5-[(6-ddopropil-4-fluoro-1H-indol-2-il)metil]piridina-3-carboxílico (165 mg, 81%) se preparó a partir de ácido 5-[[1-(bencenosulfonil)-6-ddopropil-4-fluoro-1H-indol-2-il]metil]-4,6-dicloro-piridina-3-carboxílico (270 mg, 0,390 mmol). LC/MS (Método i): Rt = 2,04 min; sin ionización.
Etapa 4: metil 4,6-d¡cloro-5-[(6-dcloprop¡l-4-fluoro-1-met¡MH-mdol-2-¡l)met¡l]pir¡dma-3-carbox¡lato

Usando un procedimiento similar al Ejemplo P, Etapa 4, metil 4,6-dicloro-5-[(6-ddopropil-4-fluoro-1-metiMH-indol-2-il)metil]piridina-3-carboxilato (172 mg, 100%) se preparó a partir de ácido 4,6-dicloro-5-[(6-ddopropil-4-fluoro-1H-indol-2-il)metil]piridina-3-carboxílico (160 mg, 0,422 mmol). LC/MS (Método i): Rt = 2,60 min; mS m/z: 407 [M+H]+.
Etapa 5: ác¡do 4,6-d¡cloro-5-[(6-c¡cloprop¡l-4-fluoro-1-met¡MH-mdol-2-¡l)met¡l]p¡r¡dma-3-carboxíl¡co

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 4,6-dicloro-5-[(6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carboxíMco (125 mg, 46%) se preparó a partir de metil 4,6-dicioro-5-[(6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carboxilato (200 mg, 0,491 mmol). LC/MS (Método i): Rt = 2,17 min; sin ionización.
Etapa 6: metil 2-[1-[4,6-dicloro-5-[(6-ciclopropN-4-fluoro-1-metiMH-mdol-2-N)metN]piridma-3-carboml]-4-piperidil]acetato

Usando un procedimiento similar al Ejemplo A, Etapa 6 , metil 2-[1-[4,6-dicloro-5-[(6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carbonil]-4-piperidil]acetato (80 mg, 38%) se preparó a partir de ácido 4,6-dicloro-5-[(6-ciclo-propil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carboxílico (120 mg, 0,305 mmol) y clorhidrato de metil (4-piperi-dil)acetato (76 mg, 0,397 mmol).
LC/MS (Método i): Rt = 2,49 min; MS m/z: 532 [M+H]+; 1H NMR ((DMSO-d6, 300MHz): 5 = 8,44 y 8,40 (s, 1H); 7,05 (s, 1H); 6,50 (d, J= 11,7 Hz, 1H); 5,59 (d, J= 17 Hz, 1H); 4,48 (m, 1H); 4,38 (m, 2H); 3,81 (s, 3H); 3,59 (s, 3H); 3,39 (m, 1H); 3,08 (m, 1H); 2,83 (m, 1H); 2,27 (m, 1H); 2,03 (m, 3H); 1,75 (m, 1H); 1,40 (m, 1H); 1,15 (m, 2H); 0,93 (m, 2H); 0,70 (m, 2H).
Etapa 7: ácido 2-[1-[4,6-didoro-5-[(6-ciclopropN-4-fluoro-1-metiMH-mdol-2-N)metN]piridma-3-carboml]-4-piperidil]acético

Usando un procedimiento similar al Ejemplo A, Etapa 5, ácido 2-[1-[4,6-dicloro-5-[(6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carbonil]-4-piperidil]acético (65 mg, 83%) se preparó a partir de metil 2-[1-[4,6-dicloro-5-[(6-ciclopropil-4-fluoro-1-metil-1H-indol-2-il)metil]piridina-3-carbonil]-4-piperidil]acetato (80 mg, 0,15 mmol). Lc/MS (Método g): Rt = 1,72 min; MS m/z: 518 [M+H]+; 1H NMR ((DMSO-d6, 300MHz): 5 = 12,1 (m broad, 1H); 8,47 y 8,40(s, 1H); 7,05 (s, 1H); 6,50 (d, J = 11,5 Hz, 1H); 5,61 y 5,56 (s, 1H); 6,38 (m, 1H); 4,35 (s, 2H); 3,80 (s, 3H); 3,39 (m, 1H); 3,05 (m, 1H); 2,84 (m, 1H); 2,15 (m, 2H); 1,99 (m, 2H); 1,78 (m, 1H); 1,60 (m, 1H); 1,18 (m, 2H); 0,90 (m, 2H); 0,65 (m, 2H).
Actividad biológica
La exploración primaria se realizó mediante ensayos de transactivación transitoria. Estos ensayos basados en células se llevaron a cabo usando células Cos-7 transfectadas con un plásmido de expresión del receptor RORy-Gal4 humano quimérico y un plásmido reportero 5Gal4 pGL3 TK Luc. Las transfecciones se realizaron por un agente químico (Jet PEI). Las células transfectadas se distribuyeron en placas de 384 pocillos y se les permitió recuperarse durante 24 h. Luego se eliminó el medio de cultivo y se añadió medio fresco que contenía los compuestos a ser probados (la concentración final varía de 10-4 M a 310-10 M). Después de una incubación durante la noche, se midió la expresión de luciferasa mediante adición de SteadyGlo de acuerdo con las instrucciones del fabricante (Promega). T0901317 a 10-5 M se usó como referencia. Los resultados se expresaron como veces de inducción en comparación con el nivel basal o como porcentaje de actividad en comparación con las referencias tomadas como 100% (basal) o 0% (T0901317). Las curvas de dosisefecto y la CI50 se calcularon usando el programa Assay Explorer (MDL).
La IC50 para los diferentes ejemplos se informa en la tabla usando las siguientes categorías:
A: IC50 < 0,1 |jM; B: 0,1 pM < IC50 < 0,5 pM; C: 0,5 pM < IC50 < 1 pM;
D: 1 pM < IC50 < 10 pM; E : IC50 > 10 pM.
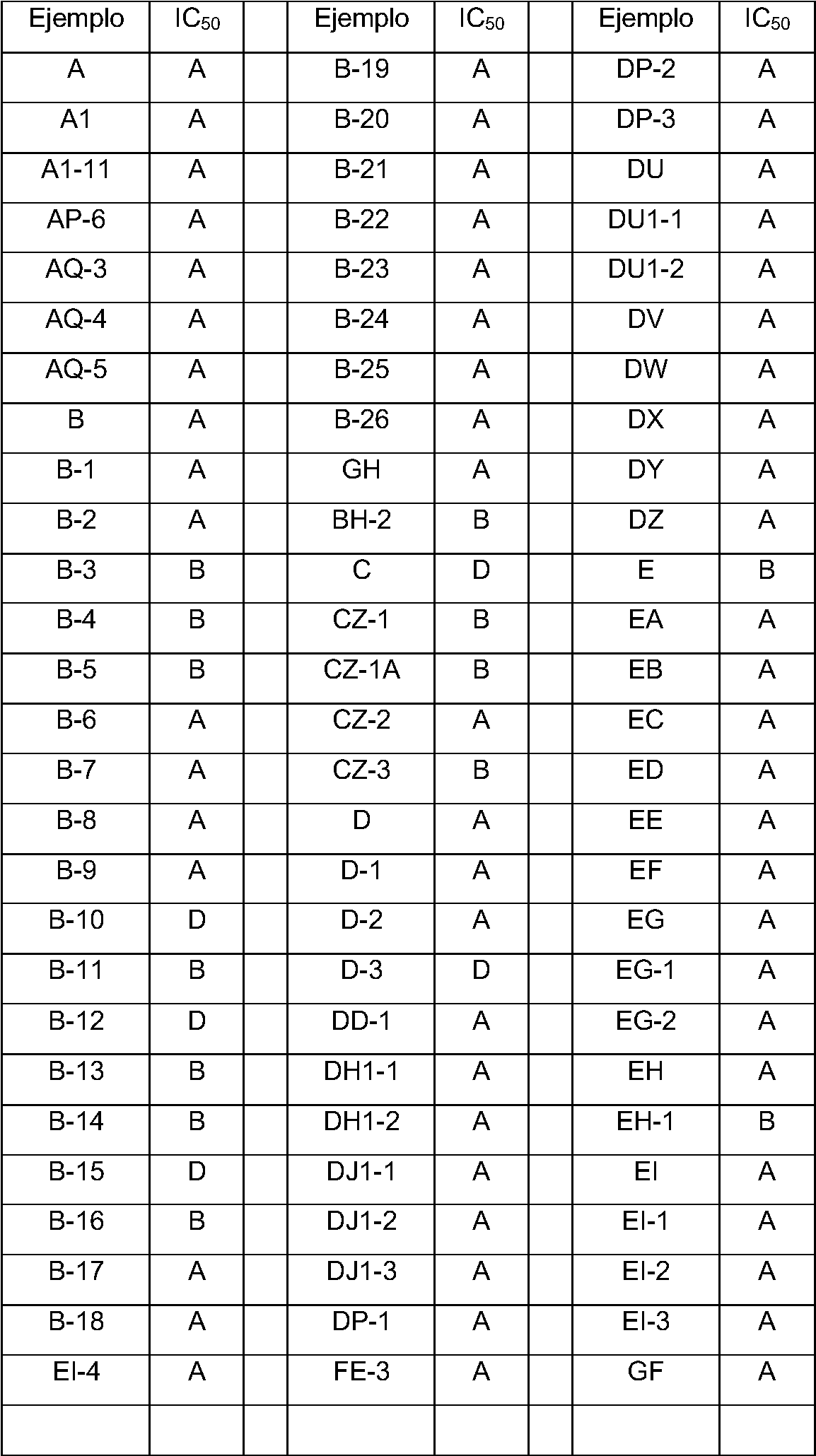



Los compuestos de la invención también se probaron en el modelo IL-23 de
Dermatitis psoriasiforme: Se alojaron ratones hembra C57BL/6" de 18-22 g (Charles River Labs) en una instalación aprobada por AAALAC en condiciones específicas libres de patógenos y se les proporcionó comida y agua a voluntad. Los animales se inyectaron (oreja; intradérmica) con 1 pg de IL-23 murina recombinante en los días 0, 1, 2 y 3 bajo anestesia (isoflurano). En el día 4, los animales se sacrificaron humanamente y se recogieron muestras de plasma y orejas para criterios de valoración de modelo y/o exposiciones a fármacos. Los animales también recibieron vehículo (hidroxipropilmetilcelulosa al 0,5%/Tween 80 al 0,02% en agua) o compuesto de prueba suspendido en el vehículo diariamente por sonda oral, hasta que se terminó el estudio. Los compuestos de esta invención dieron > 50% de reducción en la inflamación de la oreja en relación con el control del vehículo dosificado por vía oral a > 1 mg/kg.
Claims (21)
- REIVINDICACIONESUn compuesto de Fórmula (I-a):
 o una sal farmacéuticamente aceptable del mismo,en el que:G y J son independientemente CH o N;W es C o CRa, L1 está conectado a W o Y; yA y E son independientemente C o N, siempre que ambos no sean N;V es CR3 o N;X es CRa, NRa, N u O;Y es C, CRa, NRa, N, O o S;Z es CR3 o N; oW es N o NRa, L1 está conectado a W o Y; yA y E son independientemente C o N, siempre que ambos no sean N;V es CR3 o N;X es CRa, NRa o N;Y es C, CRa o N;Z es CR3 o N; oL1 es -CH2- o -C(O)-;L2 es -C(O)-;R1 y R2 son independientemente halógeno, -O -(C rC 3)alquilo, ciclopropilo, o (C 1-C3)alquilo;cada R3 es independientemente H, -CF3, -CN, halógeno, -OCHF2, -OCF3, -C(O)N(Re)2, -NReCORe, -(C rC 3)alquilo, -O-(C 1-C3)alquilo, -S-(C 1-C3)alquilo, (C3-C6)cicloalquilo, o morfolinilo;R4 es (C 1-C6)alquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de halo, -CO2H, y CO2(C 1-C4)alquilo; oR4 es -NH-CH2-tetrahidro-2 H-piranilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de -OH y O (C rC 4)alquilo; oR4 es -N(H)-(C3-C6)alquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de F, -OH, -COOH, y -CO2(C rC 4)alquilo; oR4 es (C3-C6)cicloalquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de F, -OH, -COOH y -CO2(C 1-C4)alquilo; oR4 es azabiciclo[2,2,1]heptanilo o azabiciclo[3,1,0]hexanilo, cada uno opcionalmente sustituido con -CO2H o -CO2(C 1-C4)alquilo; oR4 es azaspiro[3,3]heptanilo opcionalmente sustituido con -CH2CO2H, -CH2CO2CH3, -CO2H, -CO2 CH3; o R4 es 2-oxa-6-azaspiro[3,3]heptanilo, 2-oxa-7-azaspiro[3,5]nonanilo, o 2-oxa-8-azaspiro[4,5]decanilo, cada uno opcionalmente sustituido con -CH2CO2H; oR4 es azetidinilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de -CH3, -OH, -OCH3, -(C 1-C4)alquileno-OH, -CH2OCH3, -(C 1-C4) alquilenoO(C1-C4)alquilo, -CO2(C 1-C4) alquilo,-CO2H, -(C 1-C4)alquileno-CO2H, -(C 1-C4) alquileno-CO2-(C 1-C4)alquilo, -N(CH3)2, y grupos pirrolidinilo; oR4 es 1 ,2 -diazepanilo o 1,4-diazepanilo, cada uno opcionalmente sustituido con -(C rC 4)alquileno-OH; o R4 es morfolinilo opcionalmente sustituido con =O; oR4 es piperazinilo, piperidinilo o pirrolidinilo, cada uno opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de -halo, -CN, -(C 1-C4)alquilo, -CH2-ciclopropil, -(C 1-C4)alquileno-F, -CF3, -CH2CF3, -COOH, -(C 1-C4)alquilenoCOOH, -CH(OH)CO2H, -COCH3, -CO2(C 1-C4)alquilo,-CH(OH)CO2CH3, -(C 1-C4)alquileno-C(=O)O(C1-C4)alquilo, -(C 1-C4)alquileno-OH, -OH, -O(C 1-C4)alquilo,-(C1-C4)alquilenoO(C1-C4)alquilo, -O(C 1-C4)alquilenoCO2H, -O(C 1-C4)alquilenoC(=O)O(C1-C4)alquilo, -CONHCH3, -SO2-(C rC 4)alquilo, -NH2, -NH(C1-C4)alquilo, -N((C 1-C4)alquilo)2, ácido ciclobutanocarboxílico, y oxetanilo;cada Ra es independientemente H, -C(O)CH3, (C 1-C6)alquilo opcionalmente sustituido, (C3-C6)cidoalquilo opcionalmente sustituido o -S(O)2-fenilo; yRe es independientemente H o -(Ci-C3)alquilo;siempre que no más de dos de A, E, W, X y Y sean N.
o una sal farmacéuticamente aceptable del mismo,en el que:G y J son independientemente CH o N;W es C o CRa, L1 está conectado a W o Y; yA y E son independientemente C o N, siempre que ambos no sean N;V es CR3 o N;X es CRa, NRa, N u O;Y es C, CRa, NRa, N, O o S;Z es CR3 o N; oW es N o NRa, L1 está conectado a W o Y; yA y E son independientemente C o N, siempre que ambos no sean N;V es CR3 o N;X es CRa, NRa o N;Y es C, CRa o N;Z es CR3 o N; oL1 es -CH2- o -C(O)-;L2 es -C(O)-;R1 y R2 son independientemente halógeno, -O -(C rC 3)alquilo, ciclopropilo, o (C 1-C3)alquilo;cada R3 es independientemente H, -CF3, -CN, halógeno, -OCHF2, -OCF3, -C(O)N(Re)2, -NReCORe, -(C rC 3)alquilo, -O-(C 1-C3)alquilo, -S-(C 1-C3)alquilo, (C3-C6)cicloalquilo, o morfolinilo;R4 es (C 1-C6)alquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de halo, -CO2H, y CO2(C 1-C4)alquilo; oR4 es -NH-CH2-tetrahidro-2 H-piranilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de -OH y O (C rC 4)alquilo; oR4 es -N(H)-(C3-C6)alquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de F, -OH, -COOH, y -CO2(C rC 4)alquilo; oR4 es (C3-C6)cicloalquilo opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de F, -OH, -COOH y -CO2(C 1-C4)alquilo; oR4 es azabiciclo[2,2,1]heptanilo o azabiciclo[3,1,0]hexanilo, cada uno opcionalmente sustituido con -CO2H o -CO2(C 1-C4)alquilo; oR4 es azaspiro[3,3]heptanilo opcionalmente sustituido con -CH2CO2H, -CH2CO2CH3, -CO2H, -CO2 CH3; o R4 es 2-oxa-6-azaspiro[3,3]heptanilo, 2-oxa-7-azaspiro[3,5]nonanilo, o 2-oxa-8-azaspiro[4,5]decanilo, cada uno opcionalmente sustituido con -CH2CO2H; oR4 es azetidinilo opcionalmente sustituido con uno o dos sustituyentes seleccionados independientemente de -CH3, -OH, -OCH3, -(C 1-C4)alquileno-OH, -CH2OCH3, -(C 1-C4) alquilenoO(C1-C4)alquilo, -CO2(C 1-C4) alquilo,-CO2H, -(C 1-C4)alquileno-CO2H, -(C 1-C4) alquileno-CO2-(C 1-C4)alquilo, -N(CH3)2, y grupos pirrolidinilo; oR4 es 1 ,2 -diazepanilo o 1,4-diazepanilo, cada uno opcionalmente sustituido con -(C rC 4)alquileno-OH; o R4 es morfolinilo opcionalmente sustituido con =O; oR4 es piperazinilo, piperidinilo o pirrolidinilo, cada uno opcionalmente sustituido con uno o más sustituyentes seleccionados independientemente de -halo, -CN, -(C 1-C4)alquilo, -CH2-ciclopropil, -(C 1-C4)alquileno-F, -CF3, -CH2CF3, -COOH, -(C 1-C4)alquilenoCOOH, -CH(OH)CO2H, -COCH3, -CO2(C 1-C4)alquilo,-CH(OH)CO2CH3, -(C 1-C4)alquileno-C(=O)O(C1-C4)alquilo, -(C 1-C4)alquileno-OH, -OH, -O(C 1-C4)alquilo,-(C1-C4)alquilenoO(C1-C4)alquilo, -O(C 1-C4)alquilenoCO2H, -O(C 1-C4)alquilenoC(=O)O(C1-C4)alquilo, -CONHCH3, -SO2-(C rC 4)alquilo, -NH2, -NH(C1-C4)alquilo, -N((C 1-C4)alquilo)2, ácido ciclobutanocarboxílico, y oxetanilo;cada Ra es independientemente H, -C(O)CH3, (C 1-C6)alquilo opcionalmente sustituido, (C3-C6)cidoalquilo opcionalmente sustituido o -S(O)2-fenilo; yRe es independientemente H o -(Ci-C3)alquilo;siempre que no más de dos de A, E, W, X y Y sean N. - 2. El compuesto de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que R1 y R2 son ambos halo o -CH3.
- 3. El compuesto de acuerdo con la reivindicación reivindicaciones 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que W es C o CH, A es C y E es C.
- 4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3, o una sal farmacéuticamente aceptable del mismo, en el que G es CH.
- 5. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 4, o una sal farmacéuticamente aceptable del mismo, en el que J es CH.
- 6. El compuesto de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que:W es CH;X es CRa, en el que Ra es H o -CH3;Y es N;A es C;E es C;Y es CR3, en el que R3 es H;Z es N o CR3, en el que R3 es H, -CH3, o -CF3;L1 está conectado a Y;L1 es -CH2- o -C(O)-;L2 es -C(O)-;R1 es -Cl o -CH3;R2 es -Cl o -CH3;cada R3 es independientemente H, -F, -Cl, -CN, -CH3, -CH(CH3)2, -CF3, -OCHF2, -OCF3, -SCH3, o ciclopropilo; y R4 es 2-oxa-6-azaspiro[3,3]heptanilo opcionalmente sustituido o piperidinilo opcionalmente sustituido.
- 7. El compuesto de acuerdo con la reivindicación 6 , o una sal farmacéuticamente aceptable del mismo, en el que el piperidinilo opcionalmente sustituido está opcionalmente sustituido por -CH3, -COOH, -CH2CO2K -C H 2CH2CO2H, -CO2CH3, -CO2CH2CH3, o -OH.
- 8. El compuesto de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que:W es C;X es NRa, en el que Ra es H, -CH3, -CH2CH3, -COCH3, o -CH2CH(CH3)2;Y es CRa, en el que Ra es H;Z es CR3, en el que R3 es H o -CH3;A es C;E es C;V es CR3, en el que R3 es H, -CN, -CH3, o -CF3;L1 está conectado a W;L1 es -CH2- o -C(O)-;L2 es -C(O)-;R1 es -Cl, -CH3, o ciclopropilo;R2 es -Cl o -CH3;cada R3 es independientemente H, -F, -Br, -Cl, -CN, -CH3, -CF3, -OCH3, -OCHF2, -OCF3, -CONHCH3, -CON(CH3)2, -NHCOCH3, ciclopropilo, o morfolinilo;R4 es (C 1-C6)alquilo opcionalmente sustituido, tetrahidro-2H-piranilo opcionalmente sustituido con -NH-CH2-, ciclohexilo opcionalmente sustituido con -NH-, azabiciclo[2,2,1]heptanilo opcionalmente sustituido, 3 azabiciclo[3,1,0]hexanilo opcionalmente sustituido, azaspiro[3,3]heptanilo opcionalmente sustituido, azetidinilo opcionalmente sustituido, ciclobutilo opcionalmente sustituido, ciclohexilo opcionalmente sustituido, ciclopentilo opcionalmente sustituido, 1,4-diazepanilo opcionalmente sustituido, morfolinilo opcionalmente sustituido, 2-oxa-6-azaspiro[3,3]heptanilo opcionalmente sustituido, 2-oxa-7-azaspiro[3,5]nonanilo opcionalmente sustituido, 2-oxa-8-azaspiro[4,5]decanilo opcionalmente sustituido, piperazinilo opcionalmente sustituido, piperidinilo opcionalmente sustituido o pirrolidinilo opcionalmente sustituido.
- 9. El compuesto de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que:W es C;X es NRa, en el que Ra es H o -CH3;Y es CRa, en el que Ra es H;Z es N;A es C;E es C;V es CR3, en el que R3 es H;L1 está conectado a W;L1 es -CH2- o -C(O)-;L2 es -C(O)-;R1 es -Cl;R2 es -Cl;cada R3 es independientemente H, -CH3, o -CF3; yR4 es morfolinilo opcionalmente sustituido, piperazinilo opcionalmente sustituido o piperidinilo opcionalmente sustituido.
- 10. El compuesto de acuerdo con la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, en el que:W es CRa, en el que Ra es H;X es O o NRa, en el que Ra es H o -CH3;V es C;Z es CR3, en el que R3 es H o -CF3;A es C;E es C;V es CR3, en el que R3 es H;L1 está conectado a Y;L1 es -CH2-;L2 es -C(O)-;R1 es -Cl;R2 es -Cl;cada R3 es independientemente H, -CH3, -CF3, o -OCF3; yR4 es piperidinilo opcionalmente sustituido.
- 11. El compuesto de acuerdo con la reivindicación 10 o una sal farmacéuticamente aceptable del mismo, en el que el piperidinilo opcionalmente sustituido está opcionalmente sustituido por -CH3, -COOH, -CH2CO O K -C H 2CO2CH3, o -CO2CH2CH3.
- 12. El compuesto de acuerdo con la reivindicación 1, en el que el compuesto es:





 Ċ
Ċ



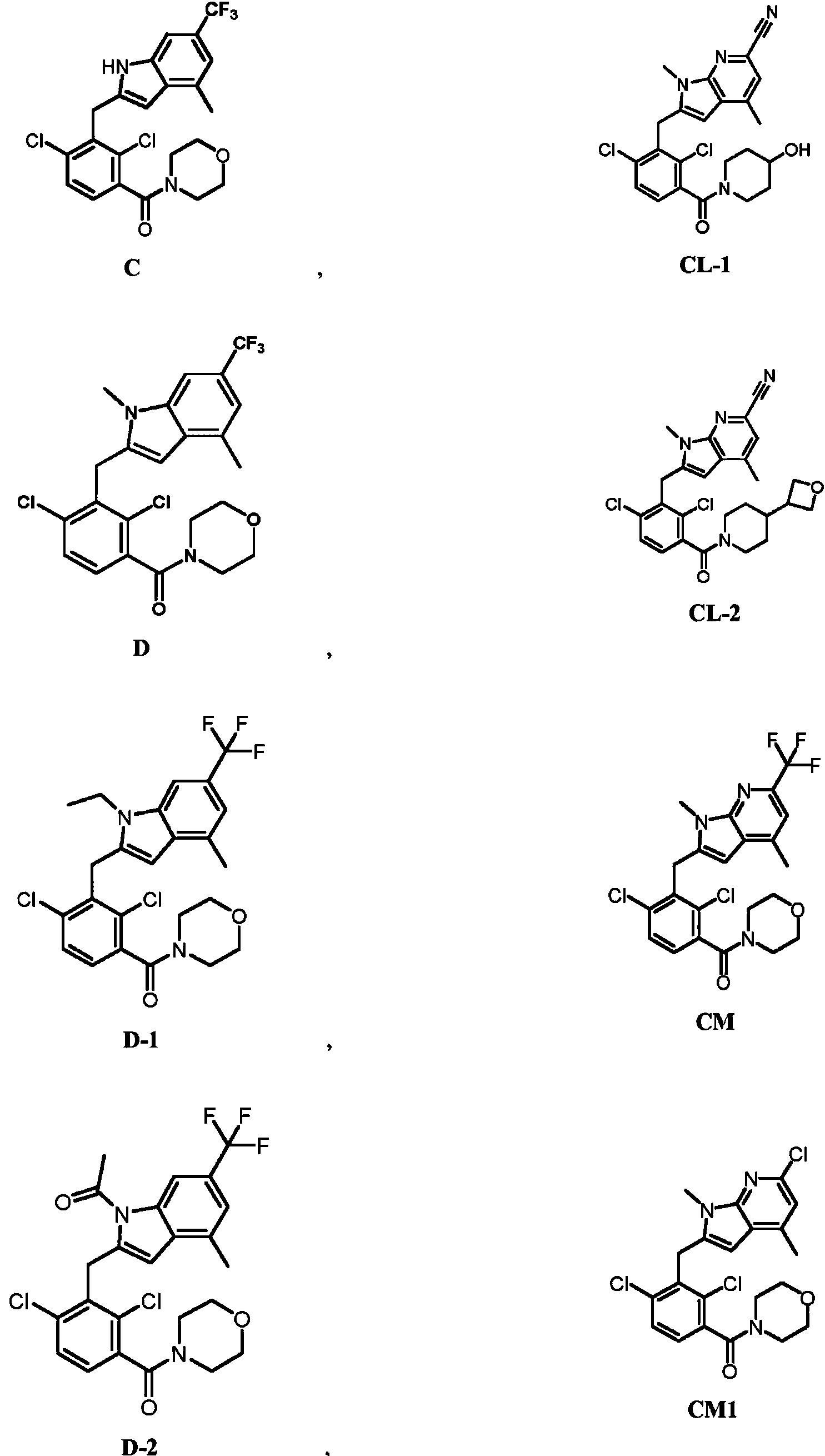



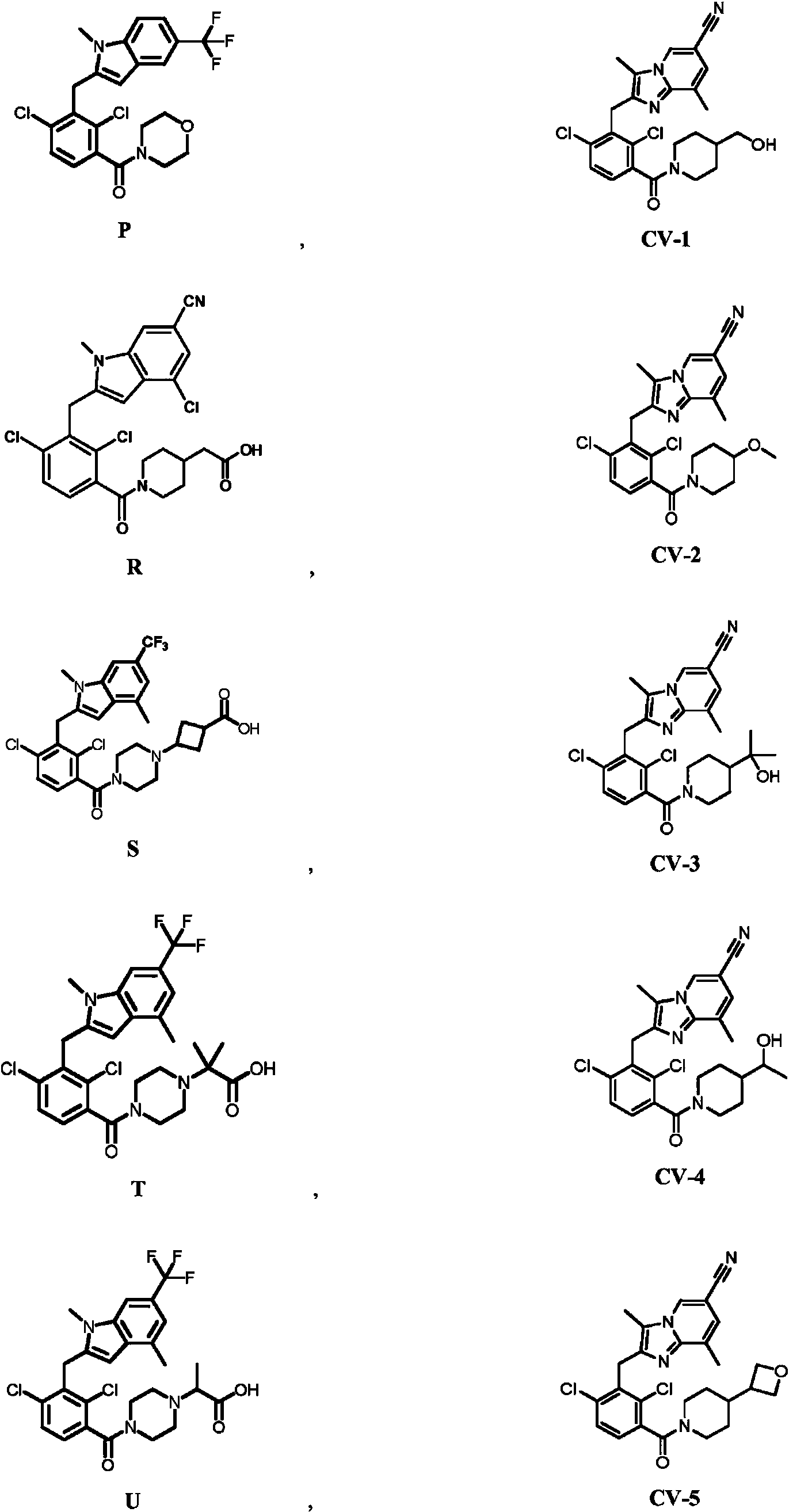 Ċ
Ċ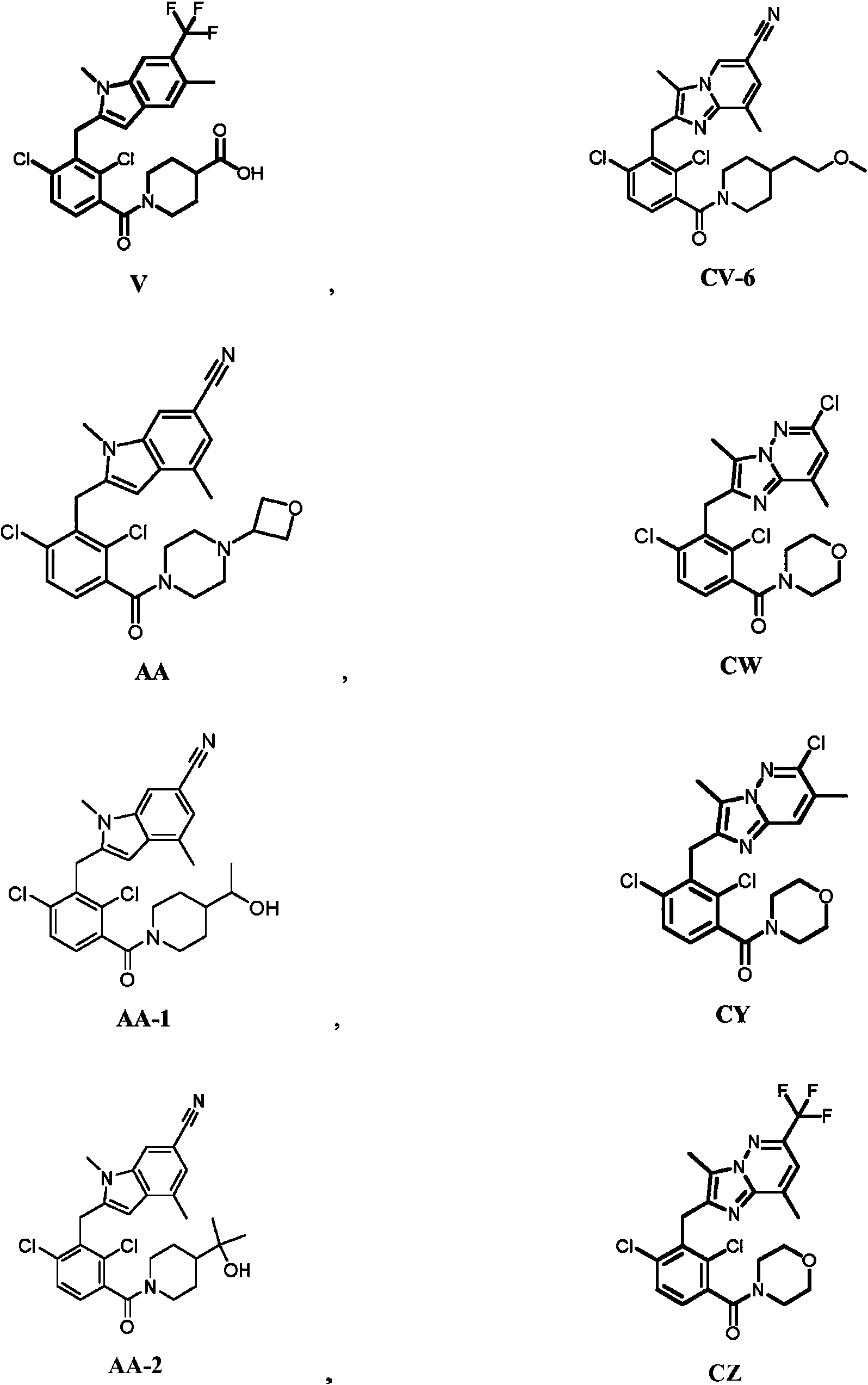



 Ċ
Ċ




 Ċ
Ċ











 o una sal farmacéuticamente aceptable del mismo.
o una sal farmacéuticamente aceptable del mismo. - 13. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es:
 B.
B. - 14. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es una sal farmacéuticamente aceptable de:

- 15. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es:

- 16. El compuesto de acuerdo con la reivindicación 12

- 17. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es:

- 18. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es una sal farmacéuticamente aceptable de:

- 19. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es:

- 20. El compuesto de acuerdo con la reivindicación 12, en el que el compuesto es una sal farmacéuticamente aceptable de:

- 21. Una composición farmacéutica que comprende:(i) un compuesto de una cualquiera de las reivindicaciones 1 a 11, o una sal farmacéuticamente aceptable del mismo, o(ii) un compuesto de la reivindicación 12 o una sal farmacéuticamente aceptable del mismo, o (iii) un compuesto de una cualquiera de las reivindicaciones 13, 15, 17 o 19, o(iv) una sal farmacéuticamente aceptable de un compuesto de una cualquiera de las reivindicaciones 14, 16, 18 o 20,y uno o más excipientes farmacéuticamente aceptables.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2015/001693 WO2016198908A1 (en) | 2015-06-09 | 2015-06-09 | Ror nuclear receptor modulators |
| US201562257806P | 2015-11-20 | 2015-11-20 | |
| US201662343905P | 2016-06-01 | 2016-06-01 | |
| PCT/US2016/036283 WO2016200851A1 (en) | 2015-06-09 | 2016-06-08 | Nuclear receptor modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2774517T3 true ES2774517T3 (es) | 2020-07-21 |
Family
ID=56194583
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16731721T Active ES2774517T3 (es) | 2015-06-09 | 2016-06-08 | Moduladores del receptor nuclear (ROR) para el tratamiento de enfermedades inflamatorias y autoinmunes |
Country Status (36)
| Country | Link |
|---|---|
| US (4) | US10106501B2 (es) |
| EP (2) | EP3636643A1 (es) |
| JP (4) | JP6549735B2 (es) |
| KR (1) | KR20180014433A (es) |
| CN (1) | CN107820494B (es) |
| AU (3) | AU2016276316B2 (es) |
| BR (1) | BR112017026452A2 (es) |
| CA (1) | CA2988502A1 (es) |
| CL (1) | CL2017003147A1 (es) |
| CO (1) | CO2017012594A2 (es) |
| CR (1) | CR20170594A (es) |
| CY (1) | CY1122495T1 (es) |
| DK (1) | DK3307734T3 (es) |
| DO (1) | DOP2017000289A (es) |
| EC (1) | ECSP18001225A (es) |
| ES (1) | ES2774517T3 (es) |
| HR (1) | HRP20200064T1 (es) |
| HU (1) | HUE047988T2 (es) |
| IL (2) | IL256043B (es) |
| LT (1) | LT3307734T (es) |
| MA (1) | MA51556A (es) |
| MX (2) | MX372543B (es) |
| MY (1) | MY193715A (es) |
| PE (1) | PE20230683A1 (es) |
| PH (1) | PH12017502237B1 (es) |
| PL (1) | PL3307734T3 (es) |
| PT (1) | PT3307734T (es) |
| RS (1) | RS59934B1 (es) |
| RU (1) | RU2715897C2 (es) |
| SG (1) | SG10201911849SA (es) |
| SI (1) | SI3307734T1 (es) |
| TW (2) | TW202128619A (es) |
| UA (1) | UA120651C2 (es) |
| UY (1) | UY36716A (es) |
| WO (1) | WO2016200851A1 (es) |
| ZA (1) | ZA201708285B (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3692991A1 (de) | 2015-06-09 | 2020-08-12 | Bayer Pharma Aktiengesellschaft | Positiv allosterische modulatoren des muskarinergen m2 rezeptors |
| DK3307734T3 (da) * | 2015-06-09 | 2020-01-27 | Abbvie Inc | Nukleære receptormodulatorer (ror) til behandling af inflammatoriske og autoimmune sygdomme |
| GB201609601D0 (en) | 2016-06-01 | 2016-07-13 | Nucana Biomed Ltd | Phosphoramidate compounds |
| CN109689654A (zh) | 2016-07-11 | 2019-04-26 | 拜耳医药股份有限公司 | 7-取代的1-吡啶基-萘啶-3-甲酰胺类及其用途 |
| JOP20190045A1 (ar) | 2016-09-14 | 2019-03-14 | Bayer Ag | مركبات أميد حمض 1- أريل-نفثيريدين-3-كربوكسيليك مستبدلة في الموضع 7 واستخدامها. |
| CN108689942B (zh) * | 2017-04-11 | 2023-06-09 | 广东东阳光药业有限公司 | 含氮双环化合物及其制备方法和用途 |
| US11427596B2 (en) * | 2019-07-19 | 2022-08-30 | Indian Institute of Technology Indore | Metal-free solvent-free synthesis of fused-pyrido heterocycles and biomedical applications |
| CN110483561B (zh) * | 2019-08-24 | 2021-07-23 | 中国科学院兰州化学物理研究所 | 一种烯基硼酸酯类化合物的合成方法 |
| CN110407863B (zh) * | 2019-08-24 | 2021-08-17 | 中国科学院兰州化学物理研究所 | 一种通过转移硼化反应合成烯基硼酸酯类化合物的方法 |
| CN114436833B (zh) * | 2021-12-27 | 2024-12-24 | 南京红太阳医药研究院有限公司 | 一种替米沙坦关键中间体4′-溴甲基联苯-2-羧酸酯的制备方法 |
| WO2023232870A1 (en) | 2022-05-31 | 2023-12-07 | Immunic Ag | Rorg/rorgt modulators for the treatment of virus infections like covid-19 |
| AU2024267864A1 (en) * | 2023-05-09 | 2025-11-06 | 11949098 Canada Inc. | Inverse agonists of rar related orphan receptors (rors) |
| EP4719396A2 (en) * | 2023-06-05 | 2026-04-08 | The Board Of Regents Of The University Of Texas System | Non-peptide small molecule fgf14:nav1.6 channel complex protein-protein interaction modulators |
| CN117263810B (zh) * | 2023-09-19 | 2026-01-27 | 上海凌凯科技股份有限公司 | 还原硝基苯制备苯胺类化合物以及选择性碘代苯胺类化合物的方法 |
| CN119241538A (zh) * | 2024-09-30 | 2025-01-03 | 陕西科技大学 | 一类三唑并吡啶结构的化合物、合成方法及其应用 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992004321A1 (en) | 1990-09-10 | 1992-03-19 | Rhone-Poulenc Rorer International (Holdings) Inc. | Substituted bicyclic aryl compounds exhibiting selective leukotriene b4 antagonist activity |
| US5290798A (en) | 1991-09-30 | 1994-03-01 | Merck Frosst Canada, Inc. | (hetero-arylmethoxy)indoles as inhibitors of leukotriene biosynthesis |
| ZA928276B (en) | 1991-10-31 | 1993-05-06 | Daiichi Seiyaku Co | Aromatic amidine derivates and salts thereof. |
| JP3457694B2 (ja) | 1993-02-04 | 2003-10-20 | 第一製薬株式会社 | インフルエンザ感染予防・治療薬 |
| US5589482A (en) | 1994-12-14 | 1996-12-31 | Pfizer Inc. | Benzo-thiophene estrogen agonists to treat prostatic hyperplasia |
| US6124463A (en) | 1998-07-02 | 2000-09-26 | Dupont Pharmaceuticals | Benzimidazoles as corticotropin release factor antagonists |
| US7157487B2 (en) | 2000-12-28 | 2007-01-02 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| US20070129334A1 (en) | 2001-10-30 | 2007-06-07 | Conforma Therapeutics Corporation | Orally Active Purine-Based Inhibitors of Heat Shock Protein 90 |
| WO2003091211A1 (en) | 2002-03-28 | 2003-11-06 | Sumitomo Pharmaceuticals Co., Ltd. | Novel heteroaryl compounds |
| CN1747949A (zh) | 2002-12-20 | 2006-03-15 | 法马西亚公司 | 无环吡唑化合物 |
| MXPA05011539A (es) | 2003-04-30 | 2006-01-23 | Inst For Pharm Discovery Inc | Acidos carboxilicos sustituidos con heterociclo como inhibidores de la proteina tirosina-fosfatasa-1b. |
| CN1856490A (zh) | 2003-07-28 | 2006-11-01 | 詹森药业有限公司 | 苯并咪唑、苯并噻唑和苯并噁唑衍生物及其作为lta4h调节剂的应用 |
| UY28526A1 (es) | 2003-09-24 | 2005-04-29 | Boehringer Ingelheim Pharma | Miméticos de glucocorticoides, métodos de preparación composiciones farmacéuticas y usos de los mismos |
| US7417063B2 (en) | 2004-04-13 | 2008-08-26 | Bristol-Myers Squibb Company | Bicyclic heterocycles useful as serine protease inhibitors |
| AU2005267436A1 (en) | 2004-06-24 | 2006-02-02 | Eli Lilly And Company | Compounds and methods for treating dyslipidemia |
| US7713979B2 (en) | 2004-10-29 | 2010-05-11 | Eli Lilly And Company | Cycloalkyl lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| US7855205B2 (en) | 2004-10-29 | 2010-12-21 | Janssen Pharmaceutica Nv | Pyrimidinyl substituted fused-pyrrolyl compounds useful in treating kinase disorders |
| CN102206216B (zh) * | 2005-06-22 | 2014-11-12 | 普莱希科公司 | 作为蛋白质激酶抑制剂的吡咯并[2,3-b]吡啶衍生物 |
| FR2890071B1 (fr) | 2005-08-30 | 2007-11-09 | Fournier Sa Sa Lab | Nouveaux composes de l'indole |
| BRPI0709612A2 (pt) | 2006-03-15 | 2011-07-19 | Wyeth Corp | composto de fórmula i; método para o tratamento de um distúrbio no sistema nervoso central relacionado ou afetado pelo receptor histamina-3 em um paciente que necessita deste tratamento; método para a inibição do receptor h3; composição farmacêutica; e processo para a preparação de um composto de fórmula i |
| US8026257B2 (en) | 2007-07-11 | 2011-09-27 | Bristol-Myers Squibb Company | Substituted heterocyclic ethers and their use in CNS disorders |
| ES2524787T3 (es) * | 2007-11-15 | 2014-12-12 | Msd Italia S.R.L. | Derivados de piridazinona como inhibidores de PARP |
| MX2011008276A (es) | 2009-02-06 | 2011-12-14 | Elan Pharm Inc | Inhibidores de quinasa jun n-terminal. |
| GB0910003D0 (en) | 2009-06-11 | 2009-07-22 | Univ Leuven Kath | Novel compounds for the treatment of neurodegenerative diseases |
| CA2771710A1 (en) | 2009-09-18 | 2011-03-24 | Zalicus Pharmaceuticals Ltd. | Aryl sulphone derivatives as calcium channel blockers |
| US20130310379A1 (en) | 2010-11-19 | 2013-11-21 | Constellation Pharmaceuticals | Modulators of methyl modifying enzymes, compositions and uses thereof |
| US8466186B2 (en) * | 2010-12-10 | 2013-06-18 | Boehringer Ingelheim International Gmbh | Compounds |
| EP2487159A1 (en) * | 2011-02-11 | 2012-08-15 | MSD Oss B.V. | RorgammaT inhibitors |
| MX336578B (es) | 2011-07-25 | 2016-01-25 | Allergan Inc | Derivados de n-(imidazolidin-2-ilideno)-heterociclopenta[b] pirididina como moduladores de receptores de alfa 2 adrenergicos. |
| WO2013038381A1 (en) | 2011-09-16 | 2013-03-21 | Novartis Ag | Pyridine/pyrazine amide derivatives |
| UA111754C2 (uk) * | 2011-10-06 | 2016-06-10 | Байєр Фарма Акцієнгезелльшафт | Заміщені бензиліндазоли для застосування як інгібіторів bub1-кінази для лікування гіперпроліферативних захворювань |
| RU2014120166A (ru) | 2011-10-20 | 2015-11-27 | ГЛЭКСОСМИТКЛАЙН ЭлЭлСи | Замещенные бициклические аза-гетероциклы и их аналоги в качестве модуляторов сиртуина |
| EP2647628A1 (en) | 2012-04-02 | 2013-10-09 | Almirall, S.A. | Substituted tricyclic compounds with activity towards ep1 receptors |
| WO2014026328A1 (en) * | 2012-08-15 | 2014-02-20 | Merck Sharp & Dohme Corp. | 3-cyclohexenyl substituted indole and indazole compounds as rorgammat inhibitors and uses thereof |
| EP2991994B1 (en) | 2013-05-01 | 2018-08-15 | Vitae Pharmaceuticals, Inc. | Thiazolopyrrolidine inhibitors of ror-gamma |
| LT3027603T (lt) * | 2013-08-02 | 2018-08-10 | Pfizer Inc. | Heterobicikloarilo rorc2 inhibitoriai ir jų panaudojimo būdai |
| CN105531274A (zh) * | 2013-09-11 | 2016-04-27 | 豪夫迈·罗氏有限公司 | 作为RORc调节剂的酮-咪唑并吡啶衍生物 |
| EP3102576B8 (en) | 2014-02-03 | 2019-06-19 | Vitae Pharmaceuticals, LLC | Dihydropyrrolopyridine inhibitors of ror-gamma |
| PT3207043T (pt) | 2014-10-14 | 2019-03-25 | Vitae Pharmaceuticals Llc | Inibidores de di-hidropirrolopiridina de ror-gama |
| DK3307734T3 (da) * | 2015-06-09 | 2020-01-27 | Abbvie Inc | Nukleære receptormodulatorer (ror) til behandling af inflammatoriske og autoimmune sygdomme |
-
2016
- 2016-06-08 DK DK16731721.3T patent/DK3307734T3/da active
- 2016-06-08 CR CR20170594A patent/CR20170594A/es unknown
- 2016-06-08 TW TW110113121A patent/TW202128619A/zh unknown
- 2016-06-08 PH PH1/2017/502237A patent/PH12017502237B1/en unknown
- 2016-06-08 CA CA2988502A patent/CA2988502A1/en not_active Abandoned
- 2016-06-08 MA MA051556A patent/MA51556A/fr unknown
- 2016-06-08 BR BR112017026452-8A patent/BR112017026452A2/pt active Search and Examination
- 2016-06-08 LT LTEP16731721.3T patent/LT3307734T/lt unknown
- 2016-06-08 MX MX2017015980A patent/MX372543B/es active IP Right Grant
- 2016-06-08 CN CN201680036750.1A patent/CN107820494B/zh not_active Expired - Fee Related
- 2016-06-08 PL PL16731721T patent/PL3307734T3/pl unknown
- 2016-06-08 MY MYPI2017704687A patent/MY193715A/en unknown
- 2016-06-08 UY UY0001036716A patent/UY36716A/es unknown
- 2016-06-08 HU HUE16731721A patent/HUE047988T2/hu unknown
- 2016-06-08 US US15/176,309 patent/US10106501B2/en active Active
- 2016-06-08 TW TW105118235A patent/TWI726888B/zh not_active IP Right Cessation
- 2016-06-08 JP JP2017563311A patent/JP6549735B2/ja not_active Expired - Fee Related
- 2016-06-08 HR HRP20200064TT patent/HRP20200064T1/hr unknown
- 2016-06-08 ES ES16731721T patent/ES2774517T3/es active Active
- 2016-06-08 SI SI201630618T patent/SI3307734T1/sl unknown
- 2016-06-08 EP EP19203418.9A patent/EP3636643A1/en not_active Withdrawn
- 2016-06-08 EP EP16731721.3A patent/EP3307734B1/en active Active
- 2016-06-08 SG SG10201911849SA patent/SG10201911849SA/en unknown
- 2016-06-08 RS RS20200097A patent/RS59934B1/sr unknown
- 2016-06-08 RU RU2017146835A patent/RU2715897C2/ru active
- 2016-06-08 KR KR1020187000451A patent/KR20180014433A/ko not_active Ceased
- 2016-06-08 PT PT167317213T patent/PT3307734T/pt unknown
- 2016-06-08 UA UAA201713051A patent/UA120651C2/uk unknown
- 2016-06-08 PE PE2022000685A patent/PE20230683A1/es unknown
- 2016-06-08 AU AU2016276316A patent/AU2016276316B2/en not_active Ceased
- 2016-06-08 WO PCT/US2016/036283 patent/WO2016200851A1/en not_active Ceased
-
2017
- 2017-12-01 IL IL256043A patent/IL256043B/en active IP Right Grant
- 2017-12-06 ZA ZA2017/08285A patent/ZA201708285B/en unknown
- 2017-12-06 CO CONC2017/0012594A patent/CO2017012594A2/es unknown
- 2017-12-07 MX MX2020003224A patent/MX2020003224A/es unknown
- 2017-12-07 CL CL2017003147A patent/CL2017003147A1/es unknown
- 2017-12-07 DO DO2017000289A patent/DOP2017000289A/es unknown
-
2018
- 2018-01-08 EC ECIEPI20181225A patent/ECSP18001225A/es unknown
- 2018-08-22 US US16/108,230 patent/US20190055196A1/en not_active Abandoned
-
2019
- 2019-06-26 JP JP2019118167A patent/JP2019178159A/ja not_active Ceased
- 2019-10-16 US US16/654,141 patent/US20210130293A1/en not_active Abandoned
-
2020
- 2020-01-21 CY CY20201100043T patent/CY1122495T1/el unknown
- 2020-04-16 AU AU2020202584A patent/AU2020202584A1/en not_active Abandoned
- 2020-05-08 IL IL274537A patent/IL274537A/en unknown
- 2020-08-11 JP JP2020135558A patent/JP6938737B2/ja not_active Expired - Fee Related
-
2021
- 2021-07-23 US US17/443,264 patent/US20230066771A1/en not_active Abandoned
- 2021-08-31 JP JP2021140781A patent/JP7235816B2/ja active Active
- 2021-09-15 AU AU2021232727A patent/AU2021232727A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2774517T3 (es) | Moduladores del receptor nuclear (ROR) para el tratamiento de enfermedades inflamatorias y autoinmunes | |
| JP6770127B2 (ja) | Btk阻害薬としての一級カルボキサミド類 | |
| CN114945366A (zh) | Hpk1拮抗剂和其用途 | |
| JP2022527216A (ja) | Ikaros及びAiolosの三環式分解誘導薬 | |
| EP4572755A2 (en) | Cdk2 degraders and uses thereof | |
| WO2016168633A1 (en) | Indazolones as modulators of tnf signaling | |
| WO2016198908A1 (en) | Ror nuclear receptor modulators | |
| CA3185649A1 (en) | Indole compounds as androgen receptor modulators | |
| EP4573094A2 (en) | Cdk2 inhibitors and uses thereof | |
| KR20250175338A (ko) | Tyk2 분해제 및 이의 용도 | |
| TWI915393B (zh) | 作為雄激素受體調節劑的吲哚化合物 | |
| HK1248678B (zh) | 核受体调节剂 |