ES2800311T3 - Nuevos enfoques terapéuticos para tratar la enfermedad de Alzheimer - Google Patents
Nuevos enfoques terapéuticos para tratar la enfermedad de Alzheimer Download PDFInfo
- Publication number
- ES2800311T3 ES2800311T3 ES10773302T ES10773302T ES2800311T3 ES 2800311 T3 ES2800311 T3 ES 2800311T3 ES 10773302 T ES10773302 T ES 10773302T ES 10773302 T ES10773302 T ES 10773302T ES 2800311 T3 ES2800311 T3 ES 2800311T3
- Authority
- ES
- Spain
- Prior art keywords
- levosimendan
- disease
- alzheimer
- composition
- modulator
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 208000024827 Alzheimer disease Diseases 0.000 title claims abstract description 123
- 230000001225 therapeutic effect Effects 0.000 title description 18
- 238000013459 approach Methods 0.000 title description 12
- 239000000203 mixture Substances 0.000 claims abstract description 121
- 229960000692 levosimendan Drugs 0.000 claims abstract description 89
- WHXMKTBCFHIYNQ-SECBINFHSA-N levosimendan Chemical compound C[C@@H]1CC(=O)NN=C1C1=CC=C(NN=C(C#N)C#N)C=C1 WHXMKTBCFHIYNQ-SECBINFHSA-N 0.000 claims abstract description 71
- 150000003839 salts Chemical class 0.000 claims abstract description 50
- 238000009472 formulation Methods 0.000 claims abstract description 47
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 42
- 238000011282 treatment Methods 0.000 claims abstract description 38
- 238000013268 sustained release Methods 0.000 claims abstract description 31
- 239000012730 sustained-release form Substances 0.000 claims abstract description 31
- 239000003814 drug Substances 0.000 claims description 128
- 229940079593 drug Drugs 0.000 claims description 127
- 229960002722 terbinafine Drugs 0.000 claims description 64
- 229960000654 sulfafurazole Drugs 0.000 claims description 61
- NHUHCSRWZMLRLA-UHFFFAOYSA-N Sulfisoxazole Chemical compound CC1=NOC(NS(=O)(=O)C=2C=CC(N)=CC=2)=C1C NHUHCSRWZMLRLA-UHFFFAOYSA-N 0.000 claims description 57
- DOMXUEMWDBAQBQ-WEVVVXLNSA-N terbinafine Chemical compound C1=CC=C2C(CN(C\C=C\C#CC(C)(C)C)C)=CC=CC2=C1 DOMXUEMWDBAQBQ-WEVVVXLNSA-N 0.000 claims description 56
- 229960002684 aminocaproic acid Drugs 0.000 claims description 49
- 229960000794 baclofen Drugs 0.000 claims description 47
- 150000001875 compounds Chemical class 0.000 claims description 47
- 229960002911 zonisamide Drugs 0.000 claims description 39
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 claims description 35
- 210000002569 neuron Anatomy 0.000 claims description 34
- KPYSYYIEGFHWSV-UHFFFAOYSA-N Baclofen Chemical compound OC(=O)CC(CN)C1=CC=C(Cl)C=C1 KPYSYYIEGFHWSV-UHFFFAOYSA-N 0.000 claims description 30
- 239000000890 drug combination Substances 0.000 claims description 29
- UBQNRHZMVUUOMG-UHFFFAOYSA-N zonisamide Chemical compound C1=CC=C2C(CS(=O)(=O)N)=NOC2=C1 UBQNRHZMVUUOMG-UHFFFAOYSA-N 0.000 claims description 28
- 230000001988 toxicity Effects 0.000 claims description 24
- 231100000419 toxicity Toxicity 0.000 claims description 24
- 229960004047 acamprosate Drugs 0.000 claims description 23
- 229960001208 eplerenone Drugs 0.000 claims description 23
- 210000004556 brain Anatomy 0.000 claims description 22
- 229960003243 phenformin Drugs 0.000 claims description 22
- 229920000439 Sulodexide Polymers 0.000 claims description 20
- 229960003491 sulodexide Drugs 0.000 claims description 20
- AFCGFAGUEYAMAO-UHFFFAOYSA-N acamprosate Chemical compound CC(=O)NCCCS(O)(=O)=O AFCGFAGUEYAMAO-UHFFFAOYSA-N 0.000 claims description 19
- DERZBLKQOCDDDZ-JLHYYAGUSA-N cinnarizine Chemical compound C1CN(C(C=2C=CC=CC=2)C=2C=CC=CC=2)CCN1C\C=C\C1=CC=CC=C1 DERZBLKQOCDDDZ-JLHYYAGUSA-N 0.000 claims description 19
- 229960000876 cinnarizine Drugs 0.000 claims description 19
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 claims description 18
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 claims description 18
- 229960003009 clopidogrel Drugs 0.000 claims description 18
- 229960005450 eritrityl tetranitrate Drugs 0.000 claims description 18
- SNFOERUNNSHUGP-ZXZARUISSA-N erythrityl tetranitrate Chemical compound [O-][N+](=O)OC[C@@H](O[N+]([O-])=O)[C@@H](O[N+]([O-])=O)CO[N+]([O-])=O SNFOERUNNSHUGP-ZXZARUISSA-N 0.000 claims description 18
- ICFJFFQQTFMIBG-UHFFFAOYSA-N phenformin Chemical compound NC(=N)NC(=N)NCCC1=CC=CC=C1 ICFJFFQQTFMIBG-UHFFFAOYSA-N 0.000 claims description 18
- 229960000528 amlodipine Drugs 0.000 claims description 17
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 claims description 16
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 claims description 16
- 229960000681 leflunomide Drugs 0.000 claims description 16
- GRYSXUXXBDSYRT-WOUKDFQISA-N (2r,3r,4r,5r)-2-(hydroxymethyl)-4-methoxy-5-[6-(methylamino)purin-9-yl]oxolan-3-ol Chemical compound C1=NC=2C(NC)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1OC GRYSXUXXBDSYRT-WOUKDFQISA-N 0.000 claims description 15
- BQENDLAVTKRQMS-SBBGFIFASA-L Carbenoxolone sodium Chemical compound [Na+].[Na+].C([C@H]1C2=CC(=O)[C@H]34)[C@@](C)(C([O-])=O)CC[C@]1(C)CC[C@@]2(C)[C@]4(C)CC[C@@H]1[C@]3(C)CC[C@H](OC(=O)CCC([O-])=O)C1(C)C BQENDLAVTKRQMS-SBBGFIFASA-L 0.000 claims description 15
- KXNPVXPOPUZYGB-XYVMCAHJSA-N argatroban Chemical compound OC(=O)[C@H]1C[C@H](C)CCN1C(=O)[C@H](CCCN=C(N)N)NS(=O)(=O)C1=CC=CC2=C1NC[C@H](C)C2 KXNPVXPOPUZYGB-XYVMCAHJSA-N 0.000 claims description 15
- 229960003856 argatroban Drugs 0.000 claims description 15
- 229960000530 carbenoxolone Drugs 0.000 claims description 15
- 229960003315 cinacalcet Drugs 0.000 claims description 15
- RCKMWOKWVGPNJF-UHFFFAOYSA-N diethylcarbamazine Chemical compound CCN(CC)C(=O)N1CCN(C)CC1 RCKMWOKWVGPNJF-UHFFFAOYSA-N 0.000 claims description 15
- 229960003974 diethylcarbamazine Drugs 0.000 claims description 15
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 claims description 15
- 229960003365 mitiglinide Drugs 0.000 claims description 15
- MVFGUOIZUNYYSO-UHFFFAOYSA-N prilocaine Chemical compound CCCNC(C)C(=O)NC1=CC=CC=C1C MVFGUOIZUNYYSO-UHFFFAOYSA-N 0.000 claims description 15
- 229960001807 prilocaine Drugs 0.000 claims description 15
- VDHAWDNDOKGFTD-MRXNPFEDSA-N cinacalcet Chemical compound N([C@H](C)C=1C2=CC=CC=C2C=CC=1)CCCC1=CC=CC(C(F)(F)F)=C1 VDHAWDNDOKGFTD-MRXNPFEDSA-N 0.000 claims description 14
- 229960001690 etomidate Drugs 0.000 claims description 14
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 claims description 13
- 229960000901 mepacrine Drugs 0.000 claims description 13
- PMRYVIKBURPHAH-UHFFFAOYSA-N methimazole Chemical compound CN1C=CNC1=S PMRYVIKBURPHAH-UHFFFAOYSA-N 0.000 claims description 13
- 229960002178 thiamazole Drugs 0.000 claims description 13
- 229960004916 benidipine Drugs 0.000 claims description 12
- QZVNQOLPLYWLHQ-ZEQKJWHPSA-N benidipine Chemical compound C1([C@H]2C(=C(C)NC(C)=C2C(=O)OC)C(=O)O[C@H]2CN(CC=3C=CC=CC=3)CCC2)=CC=CC([N+]([O-])=O)=C1 QZVNQOLPLYWLHQ-ZEQKJWHPSA-N 0.000 claims description 12
- SCKYRAXSEDYPSA-UHFFFAOYSA-N ciclopirox Chemical compound ON1C(=O)C=C(C)C=C1C1CCCCC1 SCKYRAXSEDYPSA-UHFFFAOYSA-N 0.000 claims description 12
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 claims description 12
- 229960004588 cilostazol Drugs 0.000 claims description 12
- GPKJTRJOBQGKQK-UHFFFAOYSA-N quinacrine Chemical compound C1=C(OC)C=C2C(NC(C)CCCN(CC)CC)=C(C=CC(Cl)=C3)C3=NC2=C1 GPKJTRJOBQGKQK-UHFFFAOYSA-N 0.000 claims description 12
- 229960000885 rifabutin Drugs 0.000 claims description 12
- 229960000835 tadalafil Drugs 0.000 claims description 12
- ZWBTYMGEBZUQTK-PVLSIAFMSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,32-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-1'-(2-methylpropyl)-6,23-dioxospiro[8,33-dioxa-24,27,29-triazapentacyclo[23.6.1.14,7.05,31.026,30]tritriaconta-1(32),2,4,9,19,21,24,26,30-nonaene-28,4'-piperidine]-13-yl] acetate Chemical compound CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c4NC5(CCN(CC(C)C)CC5)N=c4c(=NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C ZWBTYMGEBZUQTK-PVLSIAFMSA-N 0.000 claims description 11
- NZLBHDRPUJLHCE-UHFFFAOYSA-N aprindine Chemical compound C1C2=CC=CC=C2CC1N(CCCN(CC)CC)C1=CC=CC=C1 NZLBHDRPUJLHCE-UHFFFAOYSA-N 0.000 claims description 11
- 229960004957 aprindine Drugs 0.000 claims description 11
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 claims description 11
- WPGGHFDDFPHPOB-BBWFWOEESA-N mitiglinide Chemical compound C([C@@H](CC(=O)N1C[C@@H]2CCCC[C@@H]2C1)C(=O)O)C1=CC=CC=C1 WPGGHFDDFPHPOB-BBWFWOEESA-N 0.000 claims description 10
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 10
- 229960003791 cefmenoxime Drugs 0.000 claims description 9
- HJJDBAOLQAWBMH-YCRCPZNHSA-N cefmenoxime Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NN=NN1C HJJDBAOLQAWBMH-YCRCPZNHSA-N 0.000 claims description 9
- VQASKUSHBVDKGU-UHFFFAOYSA-N Paramethadione Chemical compound CCC1(C)OC(=O)N(C)C1=O VQASKUSHBVDKGU-UHFFFAOYSA-N 0.000 claims description 8
- 229960003274 paramethadione Drugs 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 5
- 210000002889 endothelial cell Anatomy 0.000 claims description 5
- 239000002253 acid Substances 0.000 claims description 3
- -1 difilin Chemical compound 0.000 description 72
- 230000000694 effects Effects 0.000 description 60
- 210000004027 cell Anatomy 0.000 description 58
- 108090000623 proteins and genes Proteins 0.000 description 56
- 230000035987 intoxication Effects 0.000 description 53
- 231100000566 intoxication Toxicity 0.000 description 53
- 238000012360 testing method Methods 0.000 description 45
- 239000003112 inhibitor Substances 0.000 description 40
- 102000004169 proteins and genes Human genes 0.000 description 39
- 239000003981 vehicle Substances 0.000 description 37
- 238000002648 combination therapy Methods 0.000 description 31
- VIROVYVQCGLCII-UHFFFAOYSA-N amobarbital Chemical compound CC(C)CCC1(CC)C(=O)NC(=O)NC1=O VIROVYVQCGLCII-UHFFFAOYSA-N 0.000 description 30
- 108010092555 Large-Conductance Calcium-Activated Potassium Channels Proteins 0.000 description 28
- 108090000765 processed proteins & peptides Proteins 0.000 description 28
- 102000016469 Large-Conductance Calcium-Activated Potassium Channels Human genes 0.000 description 26
- 208000035475 disorder Diseases 0.000 description 26
- 239000002609 medium Substances 0.000 description 22
- 238000000034 method Methods 0.000 description 22
- 229940089617 risedronate Drugs 0.000 description 22
- 241000699670 Mus sp. Species 0.000 description 21
- 230000033115 angiogenesis Effects 0.000 description 21
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 20
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 20
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 16
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 16
- 241000700159 Rattus Species 0.000 description 16
- 210000003618 cortical neuron Anatomy 0.000 description 16
- 102000005962 receptors Human genes 0.000 description 16
- 108020003175 receptors Proteins 0.000 description 16
- 230000003977 synaptic function Effects 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 229960001301 amobarbital Drugs 0.000 description 15
- 239000012190 activator Substances 0.000 description 14
- SRZNHPXWXCNNDU-RHBCBLIFSA-N cefotetan Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CS[C@@H]21)C(O)=O)=O)C(=O)C1SC(=C(C(N)=O)C(O)=O)S1 SRZNHPXWXCNNDU-RHBCBLIFSA-N 0.000 description 14
- 229960005495 cefotetan Drugs 0.000 description 14
- 239000008194 pharmaceutical composition Substances 0.000 description 14
- 230000011664 signaling Effects 0.000 description 14
- 101000631760 Homo sapiens Sodium channel protein type 1 subunit alpha Proteins 0.000 description 13
- 238000000540 analysis of variance Methods 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 13
- 239000000499 gel Substances 0.000 description 13
- 238000011534 incubation Methods 0.000 description 13
- 239000000243 solution Substances 0.000 description 13
- 239000003826 tablet Substances 0.000 description 13
- 101150111584 RHOA gene Proteins 0.000 description 12
- 239000001963 growth medium Substances 0.000 description 12
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 11
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 11
- 102000017914 EDNRA Human genes 0.000 description 11
- 101000967336 Homo sapiens Endothelin-1 receptor Proteins 0.000 description 11
- 238000004113 cell culture Methods 0.000 description 11
- 230000007960 cellular response to stress Effects 0.000 description 11
- 238000013270 controlled release Methods 0.000 description 11
- 238000002474 experimental method Methods 0.000 description 11
- TVURRHSHRRELCG-UHFFFAOYSA-N fenoldopam Chemical compound C1=CC(O)=CC=C1C1C2=CC(O)=C(O)C(Cl)=C2CCNC1 TVURRHSHRRELCG-UHFFFAOYSA-N 0.000 description 11
- 229960002724 fenoldopam Drugs 0.000 description 11
- 231100000331 toxic Toxicity 0.000 description 11
- 230000002588 toxic effect Effects 0.000 description 11
- 208000037259 Amyloid Plaque Diseases 0.000 description 10
- 102100022387 Transforming protein RhoA Human genes 0.000 description 10
- 239000004480 active ingredient Substances 0.000 description 10
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 10
- 201000010099 disease Diseases 0.000 description 10
- 230000002880 effect on intoxication Effects 0.000 description 10
- 239000002953 phosphate buffered saline Substances 0.000 description 10
- 238000010149 post-hoc-test Methods 0.000 description 10
- 239000000651 prodrug Substances 0.000 description 10
- 229940002612 prodrug Drugs 0.000 description 10
- 238000011002 quantification Methods 0.000 description 10
- 230000004044 response Effects 0.000 description 10
- 239000006228 supernatant Substances 0.000 description 10
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 9
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 9
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 9
- 238000000692 Student's t-test Methods 0.000 description 9
- 230000006907 apoptotic process Effects 0.000 description 9
- 239000011575 calcium Substances 0.000 description 9
- 229910052791 calcium Inorganic materials 0.000 description 9
- 238000000576 coating method Methods 0.000 description 9
- 210000002241 neurite Anatomy 0.000 description 9
- 230000037361 pathway Effects 0.000 description 9
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- 108010025020 Nerve Growth Factor Proteins 0.000 description 8
- 102000009524 Vascular Endothelial Growth Factor A Human genes 0.000 description 8
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 8
- 239000005557 antagonist Substances 0.000 description 8
- 239000011248 coating agent Substances 0.000 description 8
- 238000001514 detection method Methods 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 210000004925 microvascular endothelial cell Anatomy 0.000 description 8
- 231100000572 poisoning Toxicity 0.000 description 8
- 230000000607 poisoning effect Effects 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- 102100036009 5'-AMP-activated protein kinase catalytic subunit alpha-2 Human genes 0.000 description 7
- 101000783681 Homo sapiens 5'-AMP-activated protein kinase catalytic subunit alpha-2 Proteins 0.000 description 7
- 102000015336 Nerve Growth Factor Human genes 0.000 description 7
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 230000004094 calcium homeostasis Effects 0.000 description 7
- 230000004637 cellular stress Effects 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 230000003511 endothelial effect Effects 0.000 description 7
- 230000003371 gabaergic effect Effects 0.000 description 7
- 230000000848 glutamatergic effect Effects 0.000 description 7
- 230000007170 pathology Effects 0.000 description 7
- 230000001681 protective effect Effects 0.000 description 7
- 229940075993 receptor modulator Drugs 0.000 description 7
- 210000002966 serum Anatomy 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- WIHBNMPFWRHGDF-SLVFWPMISA-N (2s)-2-[[(2s)-2-[[2-[[(2s,3s)-2-[[(2s,3s)-2-[[(2s)-2-[[2-[[(2s)-6-amino-2-[[(2s)-4-amino-2-[[(2s)-2-[(2-aminoacetyl)amino]-3-hydroxypropanoyl]amino]-4-oxobutanoyl]amino]hexanoyl]amino]acetyl]amino]propanoyl]amino]-3-methylpentanoyl]amino]-3-methylpentanoy Chemical compound CSCC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](C)NC(=O)CNC(=O)[C@H](CCCCN)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)CN WIHBNMPFWRHGDF-SLVFWPMISA-N 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 6
- 101001046426 Homo sapiens cGMP-dependent protein kinase 1 Proteins 0.000 description 6
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 108090000631 Trypsin Proteins 0.000 description 6
- 102000004142 Trypsin Human genes 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- MBMBGCFOFBJSGT-KUBAVDMBSA-N all-cis-docosa-4,7,10,13,16,19-hexaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCC(O)=O MBMBGCFOFBJSGT-KUBAVDMBSA-N 0.000 description 6
- 108010061598 amyloid beta-protein (25-35) Proteins 0.000 description 6
- 230000002491 angiogenic effect Effects 0.000 description 6
- 230000002146 bilateral effect Effects 0.000 description 6
- 102100022422 cGMP-dependent protein kinase 1 Human genes 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 230000030833 cell death Effects 0.000 description 6
- 230000001419 dependent effect Effects 0.000 description 6
- 230000008482 dysregulation Effects 0.000 description 6
- 239000008187 granular material Substances 0.000 description 6
- 210000001320 hippocampus Anatomy 0.000 description 6
- 239000004005 microsphere Substances 0.000 description 6
- 230000004224 protection Effects 0.000 description 6
- 229960005322 streptomycin Drugs 0.000 description 6
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000012588 trypsin Substances 0.000 description 6
- WRGQSWVCFNIUNZ-GDCKJWNLSA-N 1-oleoyl-sn-glycerol 3-phosphate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)COP(O)(O)=O WRGQSWVCFNIUNZ-GDCKJWNLSA-N 0.000 description 5
- 102100023073 Calcium-activated potassium channel subunit alpha-1 Human genes 0.000 description 5
- 206010012289 Dementia Diseases 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 102100035924 Gamma-aminobutyric acid type B receptor subunit 2 Human genes 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 101001000703 Homo sapiens Gamma-aminobutyric acid type B receptor subunit 2 Proteins 0.000 description 5
- 101000808011 Homo sapiens Vascular endothelial growth factor A Proteins 0.000 description 5
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 102000009065 Netrin-1 Human genes 0.000 description 5
- 108010074223 Netrin-1 Proteins 0.000 description 5
- 102000015499 Presenilins Human genes 0.000 description 5
- 108010050254 Presenilins Proteins 0.000 description 5
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 5
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 5
- 108010064539 amyloid beta-protein (1-42) Proteins 0.000 description 5
- 210000004958 brain cell Anatomy 0.000 description 5
- 210000001638 cerebellum Anatomy 0.000 description 5
- 230000002354 daily effect Effects 0.000 description 5
- 238000013075 data extraction Methods 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 108010082117 matrigel Proteins 0.000 description 5
- 239000012528 membrane Substances 0.000 description 5
- 230000015654 memory Effects 0.000 description 5
- 239000002858 neurotransmitter agent Substances 0.000 description 5
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 210000000225 synapse Anatomy 0.000 description 5
- 230000000946 synaptic effect Effects 0.000 description 5
- 108010026424 tau Proteins Proteins 0.000 description 5
- 102000013498 tau Proteins Human genes 0.000 description 5
- 238000011830 transgenic mouse model Methods 0.000 description 5
- 230000035899 viability Effects 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- 102100026882 Alpha-synuclein Human genes 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 102100021977 Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 Human genes 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 4
- 101000614701 Homo sapiens ATP-sensitive inward rectifier potassium channel 11 Proteins 0.000 description 4
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 4
- 101001046870 Homo sapiens Hypoxia-inducible factor 1-alpha Proteins 0.000 description 4
- 101001098232 Homo sapiens P2Y purinoceptor 1 Proteins 0.000 description 4
- 101001120086 Homo sapiens P2Y purinoceptor 12 Proteins 0.000 description 4
- 102100022875 Hypoxia-inducible factor 1-alpha Human genes 0.000 description 4
- 102000017792 KCNJ11 Human genes 0.000 description 4
- 229930182816 L-glutamine Natural products 0.000 description 4
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 4
- 241000699660 Mus musculus Species 0.000 description 4
- 102100026171 P2Y purinoceptor 12 Human genes 0.000 description 4
- 108010036639 WW Domain-Containing Oxidoreductase Proteins 0.000 description 4
- 102100027534 WW domain-containing oxidoreductase Human genes 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 210000003050 axon Anatomy 0.000 description 4
- 208000010877 cognitive disease Diseases 0.000 description 4
- 230000006735 deficit Effects 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 102000058223 human VEGFA Human genes 0.000 description 4
- 239000007943 implant Substances 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 230000002045 lasting effect Effects 0.000 description 4
- 230000003902 lesion Effects 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 206010027175 memory impairment Diseases 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 239000003094 microcapsule Substances 0.000 description 4
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 4
- 239000008108 microcrystalline cellulose Substances 0.000 description 4
- 229940016286 microcrystalline cellulose Drugs 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 230000004770 neurodegeneration Effects 0.000 description 4
- 208000015122 neurodegenerative disease Diseases 0.000 description 4
- 210000002682 neurofibrillary tangle Anatomy 0.000 description 4
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 4
- 230000008520 organization Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000000007 visual effect Effects 0.000 description 4
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102100033346 Adenosine receptor A1 Human genes 0.000 description 3
- 208000006888 Agnosia Diseases 0.000 description 3
- 102100036514 Amyloid-beta A4 precursor protein-binding family A member 1 Human genes 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 102100032912 CD44 antigen Human genes 0.000 description 3
- 108010050543 Calcium-Sensing Receptors Proteins 0.000 description 3
- 108091006146 Channels Proteins 0.000 description 3
- 102100020756 D(2) dopamine receptor Human genes 0.000 description 3
- 108010014066 DCC Receptor Proteins 0.000 description 3
- 229940118365 Endothelin receptor antagonist Drugs 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 239000001856 Ethyl cellulose Substances 0.000 description 3
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 3
- 102100035650 Extracellular calcium-sensing receptor Human genes 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- 102100032827 Homeodomain-interacting protein kinase 2 Human genes 0.000 description 3
- 101000799712 Homo sapiens Adenosine receptor A1 Proteins 0.000 description 3
- 101000928690 Homo sapiens Amyloid-beta A4 precursor protein-binding family A member 1 Proteins 0.000 description 3
- 101001049859 Homo sapiens Calcium-activated potassium channel subunit alpha-1 Proteins 0.000 description 3
- 101000931901 Homo sapiens D(2) dopamine receptor Proteins 0.000 description 3
- 101001053980 Homo sapiens Disks large homolog 2 Proteins 0.000 description 3
- 101001072029 Homo sapiens Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11A Proteins 0.000 description 3
- 101000619542 Homo sapiens E3 ubiquitin-protein ligase parkin Proteins 0.000 description 3
- 101000590691 Homo sapiens MAGUK p55 subfamily member 2 Proteins 0.000 description 3
- 101000628796 Homo sapiens Microsomal glutathione S-transferase 2 Proteins 0.000 description 3
- 101001098529 Homo sapiens Proteinase-activated receptor 1 Proteins 0.000 description 3
- 101000835995 Homo sapiens Slit homolog 1 protein Proteins 0.000 description 3
- 101000651893 Homo sapiens Slit homolog 3 protein Proteins 0.000 description 3
- 101001098805 Homo sapiens cAMP-specific 3',5'-cyclic phosphodiesterase 4A Proteins 0.000 description 3
- 101000988412 Homo sapiens cGMP-specific 3',5'-cyclic phosphodiesterase Proteins 0.000 description 3
- 102100036721 Insulin receptor Human genes 0.000 description 3
- 102100032498 MAGUK p55 subfamily member 2 Human genes 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 102100026723 Microsomal glutathione S-transferase 2 Human genes 0.000 description 3
- 238000012347 Morris Water Maze Methods 0.000 description 3
- 229930040373 Paraformaldehyde Natural products 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- 102000004257 Potassium Channel Human genes 0.000 description 3
- 102100022033 Presenilin-1 Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 102100037136 Proteinase-activated receptor 1 Human genes 0.000 description 3
- 102000004914 RYR3 Human genes 0.000 description 3
- 108060007242 RYR3 Proteins 0.000 description 3
- 102100027733 Sarcoplasmic/endoplasmic reticulum calcium ATPase 3 Human genes 0.000 description 3
- 102100025490 Slit homolog 1 protein Human genes 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 108700019146 Transgenes Proteins 0.000 description 3
- 229930003448 Vitamin K Natural products 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 230000002776 aggregation Effects 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 108090000185 alpha-Synuclein Proteins 0.000 description 3
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000003376 axonal effect Effects 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- 238000009227 behaviour therapy Methods 0.000 description 3
- 102100037092 cAMP-specific 3',5'-cyclic phosphodiesterase 4A Human genes 0.000 description 3
- 102100029175 cGMP-specific 3',5'-cyclic phosphodiesterase Human genes 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 229940125400 channel inhibitor Drugs 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000001149 cognitive effect Effects 0.000 description 3
- 238000007398 colorimetric assay Methods 0.000 description 3
- 230000001054 cortical effect Effects 0.000 description 3
- 210000000172 cytosol Anatomy 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 235000020669 docosahexaenoic acid Nutrition 0.000 description 3
- 229940090949 docosahexaenoic acid Drugs 0.000 description 3
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 3
- 239000002308 endothelin receptor antagonist Substances 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 235000019325 ethyl cellulose Nutrition 0.000 description 3
- 229920001249 ethyl cellulose Polymers 0.000 description 3
- 230000007717 exclusion Effects 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 239000012894 fetal calf serum Substances 0.000 description 3
- 238000007667 floating Methods 0.000 description 3
- 230000008014 freezing Effects 0.000 description 3
- 238000007710 freezing Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- 235000020686 ginkgo biloba extract Nutrition 0.000 description 3
- 239000003906 humectant Substances 0.000 description 3
- 239000000017 hydrogel Substances 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- CDAISMWEOUEBRE-UHFFFAOYSA-N inositol Chemical compound OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- AGBQKNBQESQNJD-UHFFFAOYSA-N lipoic acid Chemical compound OC(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-N 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 238000011418 maintenance treatment Methods 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 3
- 239000002105 nanoparticle Substances 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000010899 nucleation Methods 0.000 description 3
- 230000010627 oxidative phosphorylation Effects 0.000 description 3
- 239000012188 paraffin wax Substances 0.000 description 3
- 229920002866 paraformaldehyde Polymers 0.000 description 3
- 230000008506 pathogenesis Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 108020001213 potassium channel Proteins 0.000 description 3
- 229920001592 potato starch Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 210000004129 prosencephalon Anatomy 0.000 description 3
- 230000004845 protein aggregation Effects 0.000 description 3
- 230000012846 protein folding Effects 0.000 description 3
- 238000003118 sandwich ELISA Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 230000006886 spatial memory Effects 0.000 description 3
- DUYSYHSSBDVJSM-KRWOKUGFSA-N sphingosine 1-phosphate Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)COP(O)(O)=O DUYSYHSSBDVJSM-KRWOKUGFSA-N 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 239000007916 tablet composition Substances 0.000 description 3
- 238000011287 therapeutic dose Methods 0.000 description 3
- 230000009261 transgenic effect Effects 0.000 description 3
- 230000002792 vascular Effects 0.000 description 3
- 235000019168 vitamin K Nutrition 0.000 description 3
- 239000011712 vitamin K Substances 0.000 description 3
- 150000003721 vitamin K derivatives Chemical class 0.000 description 3
- 229940046010 vitamin k Drugs 0.000 description 3
- 230000003936 working memory Effects 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 2
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 2
- JVJUWEFOGFCHKR-UHFFFAOYSA-N 2-(diethylamino)ethyl 1-(3,4-dimethylphenyl)cyclopentane-1-carboxylate;hydrochloride Chemical class Cl.C=1C=C(C)C(C)=CC=1C1(C(=O)OCCN(CC)CC)CCCC1 JVJUWEFOGFCHKR-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- HSTOKWSFWGCZMH-UHFFFAOYSA-N 3,3'-diaminobenzidine Chemical compound C1=C(N)C(N)=CC=C1C1=CC=C(N)C(N)=C1 HSTOKWSFWGCZMH-UHFFFAOYSA-N 0.000 description 2
- 102100034254 3-oxo-5-alpha-steroid 4-dehydrogenase 1 Human genes 0.000 description 2
- 102100035923 4-aminobutyrate aminotransferase, mitochondrial Human genes 0.000 description 2
- 102100026802 72 kDa type IV collagenase Human genes 0.000 description 2
- 102100032296 A disintegrin and metalloproteinase with thrombospondin motifs 12 Human genes 0.000 description 2
- 108091005671 ADAMTS12 Proteins 0.000 description 2
- 108010011376 AMP-Activated Protein Kinases Proteins 0.000 description 2
- 102000014156 AMP-Activated Protein Kinases Human genes 0.000 description 2
- 102100022714 Acyl-coenzyme A thioesterase 13 Human genes 0.000 description 2
- 101710169767 Acyl-coenzyme A thioesterase 13 Proteins 0.000 description 2
- 102100039675 Adenylate cyclase type 2 Human genes 0.000 description 2
- 108060003345 Adrenergic Receptor Proteins 0.000 description 2
- 102000017910 Adrenergic receptor Human genes 0.000 description 2
- 241001047040 Agnosia Species 0.000 description 2
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 2
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 2
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 2
- 102400000574 Amyloid-beta protein 42 Human genes 0.000 description 2
- 102100034608 Angiopoietin-2 Human genes 0.000 description 2
- 206010003062 Apraxia Diseases 0.000 description 2
- 102100035080 BDNF/NT-3 growth factors receptor Human genes 0.000 description 2
- TUSUWHFYKZZRIG-JQWMYKLHSA-N C([C@@H](NC(=O)[C@@H](C(C)C)NC(=O)[C@@H](CC(C)C)NC)C(=O)N[C@H](CC=1C=CC=CC=1)C(=O)N[C@H](CC(C)C)C(N)=O)C1=CC=CC=C1 Chemical compound C([C@@H](NC(=O)[C@@H](C(C)C)NC(=O)[C@@H](CC(C)C)NC)C(=O)N[C@H](CC=1C=CC=CC=1)C(=O)N[C@H](CC(C)C)C(N)=O)C1=CC=CC=C1 TUSUWHFYKZZRIG-JQWMYKLHSA-N 0.000 description 2
- ANDGGVOPIJEHOF-UHFFFAOYSA-N CX-516 Chemical compound C=1C=C2N=CC=NC2=CC=1C(=O)N1CCCCC1 ANDGGVOPIJEHOF-UHFFFAOYSA-N 0.000 description 2
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 2
- 229940127291 Calcium channel antagonist Drugs 0.000 description 2
- 102000000584 Calmodulin Human genes 0.000 description 2
- 108010041952 Calmodulin Proteins 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 102100032566 Carbonic anhydrase-related protein 10 Human genes 0.000 description 2
- 102000011068 Cdc42 Human genes 0.000 description 2
- 208000005145 Cerebral amyloid angiopathy Diseases 0.000 description 2
- 101000845237 Cereibacter sphaeroides Tryptophan-rich sensory protein Proteins 0.000 description 2
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 2
- 102100021809 Chorionic somatomammotropin hormone 1 Human genes 0.000 description 2
- 240000004307 Citrus medica Species 0.000 description 2
- 102100033781 Collagen alpha-2(IV) chain Human genes 0.000 description 2
- 102100031096 Cubilin Human genes 0.000 description 2
- 102100029140 Cyclic nucleotide-gated cation channel beta-3 Human genes 0.000 description 2
- 101150081028 Cysltr1 gene Proteins 0.000 description 2
- 102100038496 Cysteinyl leukotriene receptor 1 Human genes 0.000 description 2
- 102100036318 Cytoplasmic phosphatidylinositol transfer protein 1 Human genes 0.000 description 2
- 102000016896 DCC Receptor Human genes 0.000 description 2
- 102100033587 DNA topoisomerase 2-alpha Human genes 0.000 description 2
- 208000031124 Dementia Alzheimer type Diseases 0.000 description 2
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 2
- 102100030012 Deoxyribonuclease-1 Human genes 0.000 description 2
- 102100022732 Diacylglycerol kinase beta Human genes 0.000 description 2
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 2
- 108700019745 Disks Large Homolog 4 Proteins 0.000 description 2
- 102000047174 Disks Large Homolog 4 Human genes 0.000 description 2
- 238000001061 Dunnett's test Methods 0.000 description 2
- 102100022207 E3 ubiquitin-protein ligase parkin Human genes 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 101150016325 EPHA3 gene Proteins 0.000 description 2
- 108050004000 Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 Proteins 0.000 description 2
- 102100032155 Ephexin-1 Human genes 0.000 description 2
- 102100030324 Ephrin type-A receptor 3 Human genes 0.000 description 2
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 2
- 102100035111 Farnesyl pyrophosphate synthase Human genes 0.000 description 2
- 102000017695 GABRA2 Human genes 0.000 description 2
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 206010018341 Gliosis Diseases 0.000 description 2
- 102100033417 Glucocorticoid receptor Human genes 0.000 description 2
- 102100022197 Glutamate receptor ionotropic, kainate 1 Human genes 0.000 description 2
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 2
- 102100038104 Glycogen synthase kinase-3 beta Human genes 0.000 description 2
- 102100028006 Heme oxygenase 1 Human genes 0.000 description 2
- RPTUSVTUFVMDQK-UHFFFAOYSA-N Hidralazin Chemical compound C1=CC=C2C(NN)=NN=CC2=C1 RPTUSVTUFVMDQK-UHFFFAOYSA-N 0.000 description 2
- 101000640855 Homo sapiens 3-oxo-5-alpha-steroid 4-dehydrogenase 1 Proteins 0.000 description 2
- 101001000686 Homo sapiens 4-aminobutyrate aminotransferase, mitochondrial Proteins 0.000 description 2
- 101000627872 Homo sapiens 72 kDa type IV collagenase Proteins 0.000 description 2
- 101000959347 Homo sapiens Adenylate cyclase type 2 Proteins 0.000 description 2
- 101000924533 Homo sapiens Angiopoietin-2 Proteins 0.000 description 2
- 101000596896 Homo sapiens BDNF/NT-3 growth factors receptor Proteins 0.000 description 2
- 101000867836 Homo sapiens Carbonic anhydrase-related protein 10 Proteins 0.000 description 2
- 101000710876 Homo sapiens Collagen alpha-2(IV) chain Proteins 0.000 description 2
- 101000922080 Homo sapiens Cubilin Proteins 0.000 description 2
- 101000771083 Homo sapiens Cyclic nucleotide-gated cation channel beta-3 Proteins 0.000 description 2
- 101001074657 Homo sapiens Cytoplasmic phosphatidylinositol transfer protein 1 Proteins 0.000 description 2
- 101001044814 Homo sapiens Diacylglycerol kinase beta Proteins 0.000 description 2
- 101000897035 Homo sapiens Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 Proteins 0.000 description 2
- 101000637325 Homo sapiens Ephexin-1 Proteins 0.000 description 2
- 101001023007 Homo sapiens Farnesyl pyrophosphate synthase Proteins 0.000 description 2
- 101000893333 Homo sapiens Gamma-aminobutyric acid receptor subunit alpha-2 Proteins 0.000 description 2
- 101000926939 Homo sapiens Glucocorticoid receptor Proteins 0.000 description 2
- 101000900515 Homo sapiens Glutamate receptor ionotropic, kainate 1 Proteins 0.000 description 2
- 101001079623 Homo sapiens Heme oxygenase 1 Proteins 0.000 description 2
- 101000840551 Homo sapiens Hexokinase-2 Proteins 0.000 description 2
- 101001066401 Homo sapiens Homeodomain-interacting protein kinase 2 Proteins 0.000 description 2
- 101001044118 Homo sapiens Inosine-5'-monophosphate dehydrogenase 1 Proteins 0.000 description 2
- 101000975428 Homo sapiens Inositol 1,4,5-trisphosphate receptor type 1 Proteins 0.000 description 2
- 101000992298 Homo sapiens Kappa-type opioid receptor Proteins 0.000 description 2
- 101001050577 Homo sapiens Kinesin-like protein KIF2A Proteins 0.000 description 2
- 101000972489 Homo sapiens Laminin subunit alpha-1 Proteins 0.000 description 2
- 101000990902 Homo sapiens Matrix metalloproteinase-9 Proteins 0.000 description 2
- 101000583944 Homo sapiens Methionine adenosyltransferase 2 subunit beta Proteins 0.000 description 2
- 101001013648 Homo sapiens Methionine synthase Proteins 0.000 description 2
- 101000615613 Homo sapiens Mineralocorticoid receptor Proteins 0.000 description 2
- 101000950695 Homo sapiens Mitogen-activated protein kinase 8 Proteins 0.000 description 2
- 101000979223 Homo sapiens N-terminal EF-hand calcium-binding protein 3 Proteins 0.000 description 2
- 101000841744 Homo sapiens Netrin receptor UNC5C Proteins 0.000 description 2
- 101000615538 Homo sapiens Nuclear protein MDM1 Proteins 0.000 description 2
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 description 2
- 101000741790 Homo sapiens Peroxisome proliferator-activated receptor gamma Proteins 0.000 description 2
- 101000605666 Homo sapiens Phospholipase A1 member A Proteins 0.000 description 2
- 101000730670 Homo sapiens Phospholipase D2 Proteins 0.000 description 2
- 101000905839 Homo sapiens Phospholipid-transporting ATPase VA Proteins 0.000 description 2
- 101000728095 Homo sapiens Plasma membrane calcium-transporting ATPase 1 Proteins 0.000 description 2
- 101000605403 Homo sapiens Plasminogen Proteins 0.000 description 2
- 101001126417 Homo sapiens Platelet-derived growth factor receptor alpha Proteins 0.000 description 2
- 101001064864 Homo sapiens Polyunsaturated fatty acid lipoxygenase ALOX12 Proteins 0.000 description 2
- 101001026214 Homo sapiens Potassium voltage-gated channel subfamily A member 5 Proteins 0.000 description 2
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 2
- 101001073427 Homo sapiens Prostaglandin E2 receptor EP1 subtype Proteins 0.000 description 2
- 101000579300 Homo sapiens Prostaglandin F2-alpha receptor Proteins 0.000 description 2
- 101001048456 Homo sapiens Protein Hook homolog 2 Proteins 0.000 description 2
- 101000573199 Homo sapiens Protein PML Proteins 0.000 description 2
- 101000768465 Homo sapiens Protein unc-13 homolog C Proteins 0.000 description 2
- 101000845206 Homo sapiens Putative peripheral benzodiazepine receptor-related protein Proteins 0.000 description 2
- 101001104100 Homo sapiens Rab effector Noc2 Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101001074528 Homo sapiens Regulating synaptic membrane exocytosis protein 1 Proteins 0.000 description 2
- 101000936917 Homo sapiens Sarcoplasmic/endoplasmic reticulum calcium ATPase 3 Proteins 0.000 description 2
- 101000654418 Homo sapiens Schwannomin-interacting protein 1 Proteins 0.000 description 2
- 101000836994 Homo sapiens Sigma non-opioid intracellular receptor 1 Proteins 0.000 description 2
- 101000701334 Homo sapiens Sodium/potassium-transporting ATPase subunit alpha-1 Proteins 0.000 description 2
- 101000703460 Homo sapiens Sphingosine-1-phosphate phosphatase 2 Proteins 0.000 description 2
- 101000600903 Homo sapiens Substance-P receptor Proteins 0.000 description 2
- 101000715050 Homo sapiens Thromboxane A2 receptor Proteins 0.000 description 2
- 101000801481 Homo sapiens Tissue-type plasminogen activator Proteins 0.000 description 2
- 101000637850 Homo sapiens Tolloid-like protein 2 Proteins 0.000 description 2
- 101000845233 Homo sapiens Translocator protein Proteins 0.000 description 2
- 101000823316 Homo sapiens Tyrosine-protein kinase ABL1 Proteins 0.000 description 2
- 101001103033 Homo sapiens Tyrosine-protein kinase transmembrane receptor ROR2 Proteins 0.000 description 2
- 101001087416 Homo sapiens Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 description 2
- 101000955962 Homo sapiens Vacuolar protein sorting-associated protein 51 homolog Proteins 0.000 description 2
- 101001098818 Homo sapiens cGMP-inhibited 3',5'-cyclic phosphodiesterase A Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- 102100021602 Inosine-5'-monophosphate dehydrogenase 1 Human genes 0.000 description 2
- 102100024039 Inositol 1,4,5-trisphosphate receptor type 1 Human genes 0.000 description 2
- 102100023915 Insulin Human genes 0.000 description 2
- 108010001127 Insulin Receptor Proteins 0.000 description 2
- 102100031819 Kappa-type opioid receptor Human genes 0.000 description 2
- 102100023426 Kinesin-like protein KIF2A Human genes 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- 102100022746 Laminin subunit alpha-1 Human genes 0.000 description 2
- 101000761444 Loxosceles laeta Dermonecrotic toxin Proteins 0.000 description 2
- 102100030412 Matrix metalloproteinase-9 Human genes 0.000 description 2
- 102000056430 Member 1 Solute Carrier Family 12 Human genes 0.000 description 2
- 208000026139 Memory disease Diseases 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 102100030932 Methionine adenosyltransferase 2 subunit beta Human genes 0.000 description 2
- 102100031551 Methionine synthase Human genes 0.000 description 2
- 102100021316 Mineralocorticoid receptor Human genes 0.000 description 2
- 102100037808 Mitogen-activated protein kinase 8 Human genes 0.000 description 2
- 108010006519 Molecular Chaperones Proteins 0.000 description 2
- 102100023213 N-terminal EF-hand calcium-binding protein 3 Human genes 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 102000010803 Netrins Human genes 0.000 description 2
- 108010063605 Netrins Proteins 0.000 description 2
- 108090000772 Neuropilin-1 Proteins 0.000 description 2
- 108090000770 Neuropilin-2 Proteins 0.000 description 2
- 108010008858 Nitric Oxide Synthase Type I Proteins 0.000 description 2
- 102100022397 Nitric oxide synthase, brain Human genes 0.000 description 2
- 102100021278 Nuclear protein MDM1 Human genes 0.000 description 2
- 102100026742 Opioid-binding protein/cell adhesion molecule Human genes 0.000 description 2
- 101710096745 Opioid-binding protein/cell adhesion molecule Proteins 0.000 description 2
- 102100040557 Osteopontin Human genes 0.000 description 2
- 108010015181 PPAR delta Proteins 0.000 description 2
- 206010033885 Paraparesis Diseases 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 102100038824 Peroxisome proliferator-activated receptor delta Human genes 0.000 description 2
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 2
- 102100026918 Phospholipase A2 Human genes 0.000 description 2
- 101710096328 Phospholipase A2 Proteins 0.000 description 2
- 102100032983 Phospholipase D2 Human genes 0.000 description 2
- 102100023496 Phospholipid-transporting ATPase VA Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- 102100029751 Plasma membrane calcium-transporting ATPase 1 Human genes 0.000 description 2
- 102100038124 Plasminogen Human genes 0.000 description 2
- 102100030485 Platelet-derived growth factor receptor alpha Human genes 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 102100031949 Polyunsaturated fatty acid lipoxygenase ALOX12 Human genes 0.000 description 2
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 2
- 108010036933 Presenilin-1 Proteins 0.000 description 2
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 2
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 2
- 102100035842 Prostaglandin E2 receptor EP1 subtype Human genes 0.000 description 2
- 102100028248 Prostaglandin F2-alpha receptor Human genes 0.000 description 2
- 102100026375 Protein PML Human genes 0.000 description 2
- 102100024924 Protein kinase C alpha type Human genes 0.000 description 2
- 102100023068 Protein kinase C-binding protein NELL1 Human genes 0.000 description 2
- 102100034433 Protein kinase C-binding protein NELL2 Human genes 0.000 description 2
- 102100027900 Protein unc-13 homolog C Human genes 0.000 description 2
- 102100031269 Putative peripheral benzodiazepine receptor-related protein Human genes 0.000 description 2
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 description 2
- 102100040095 Rab effector Noc2 Human genes 0.000 description 2
- 102100022127 Radixin Human genes 0.000 description 2
- 102100022122 Ras-related C3 botulinum toxin substrate 1 Human genes 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- 102100036240 Regulating synaptic membrane exocytosis protein 1 Human genes 0.000 description 2
- 102000034527 Retinoid X Receptors Human genes 0.000 description 2
- 108010038912 Retinoid X Receptors Proteins 0.000 description 2
- 101001030849 Rhinella marina Mesotocin receptor Proteins 0.000 description 2
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 108091005487 SCARB1 Proteins 0.000 description 2
- 108091006621 SLC12A1 Proteins 0.000 description 2
- 108060007759 SLC6A1 Proteins 0.000 description 2
- 102000005028 SLC6A1 Human genes 0.000 description 2
- 108060006706 SRC Proteins 0.000 description 2
- 102000001332 SRC Human genes 0.000 description 2
- 102100037118 Scavenger receptor class B member 1 Human genes 0.000 description 2
- 102100031396 Schwannomin-interacting protein 1 Human genes 0.000 description 2
- 102100026715 Serine/threonine-protein kinase STK11 Human genes 0.000 description 2
- 101710181599 Serine/threonine-protein kinase STK11 Proteins 0.000 description 2
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 2
- 229920001800 Shellac Polymers 0.000 description 2
- 102100028656 Sigma non-opioid intracellular receptor 1 Human genes 0.000 description 2
- 102100030458 Sodium/potassium-transporting ATPase subunit alpha-1 Human genes 0.000 description 2
- 208000002548 Spastic Paraparesis Diseases 0.000 description 2
- 102100030677 Sphingosine-1-phosphate phosphatase 2 Human genes 0.000 description 2
- 102100037346 Substance-P receptor Human genes 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 102100036704 Thromboxane A2 receptor Human genes 0.000 description 2
- 102100033571 Tissue-type plasminogen activator Human genes 0.000 description 2
- 102100031997 Tolloid-like protein 2 Human genes 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 2
- 102100022596 Tyrosine-protein kinase ABL1 Human genes 0.000 description 2
- 102100039616 Tyrosine-protein kinase transmembrane receptor ROR2 Human genes 0.000 description 2
- 102100033019 Tyrosine-protein phosphatase non-receptor type 11 Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 2
- 108010022133 Voltage-Dependent Anion Channel 1 Proteins 0.000 description 2
- 102100037820 Voltage-dependent anion-selective channel protein 1 Human genes 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000011149 active material Substances 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 230000028600 axonogenesis Effects 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000000480 calcium channel blocker Substances 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 230000028956 calcium-mediated signaling Effects 0.000 description 2
- 239000004203 carnauba wax Substances 0.000 description 2
- 235000013869 carnauba wax Nutrition 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 230000006727 cell loss Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 210000003710 cerebral cortex Anatomy 0.000 description 2
- 210000004720 cerebrum Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229960003749 ciclopirox Drugs 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 230000007278 cognition impairment Effects 0.000 description 2
- 230000003920 cognitive function Effects 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- VFLDPWHFBUODDF-FCXRPNKRSA-N curcumin Chemical compound C1=C(O)C(OC)=CC(\C=C\C(=O)CC(=O)\C=C\C=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-FCXRPNKRSA-N 0.000 description 2
- VEVZSMAEJFVWIL-UHFFFAOYSA-O cyanidin cation Chemical compound [O+]=1C2=CC(O)=CC(O)=C2C=C(O)C=1C1=CC=C(O)C(O)=C1 VEVZSMAEJFVWIL-UHFFFAOYSA-O 0.000 description 2
- 230000003436 cytoskeletal effect Effects 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 231100000517 death Toxicity 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 108020001096 dihydrofolate reductase Proteins 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 2
- 208000025688 early-onset autosomal dominant Alzheimer disease Diseases 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 229960005309 estradiol Drugs 0.000 description 2
- 229930182833 estradiol Natural products 0.000 description 2
- 210000003754 fetus Anatomy 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- 210000004051 gastric juice Anatomy 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 229940014259 gelatin Drugs 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000007387 gliosis Effects 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 2
- 229960001587 hesperetin Drugs 0.000 description 2
- AIONOLUJZLIMTK-AWEZNQCLSA-N hesperetin Chemical compound C1=C(O)C(OC)=CC=C1[C@H]1OC2=CC(O)=CC(O)=C2C(=O)C1 AIONOLUJZLIMTK-AWEZNQCLSA-N 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- JNODQFNWMXFMEV-UHFFFAOYSA-N latrepirdine Chemical compound C1N(C)CCC2=C1C1=CC(C)=CC=C1N2CCC1=CC=C(C)N=C1 JNODQFNWMXFMEV-UHFFFAOYSA-N 0.000 description 2
- 235000019136 lipoic acid Nutrition 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 2
- 229960000907 methylthioninium chloride Drugs 0.000 description 2
- 208000027061 mild cognitive impairment Diseases 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 2
- 230000000324 neuroprotective effect Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 210000000956 olfactory bulb Anatomy 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 229960005489 paracetamol Drugs 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 102000013415 peroxidase activity proteins Human genes 0.000 description 2
- 108040007629 peroxidase activity proteins Proteins 0.000 description 2
- 208000028591 pheochromocytoma Diseases 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 210000002381 plasma Anatomy 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 239000004800 polyvinyl chloride Substances 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 210000003538 post-synaptic density Anatomy 0.000 description 2
- 230000004481 post-translational protein modification Effects 0.000 description 2
- 108010092804 postsynaptic density proteins Proteins 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- 230000003518 presynaptic effect Effects 0.000 description 2
- LXNHXLLTXMVWPM-UHFFFAOYSA-N pyridoxine Chemical compound CC1=NC=C(CO)C(CO)=C1O LXNHXLLTXMVWPM-UHFFFAOYSA-N 0.000 description 2
- 238000010814 radioimmunoprecipitation assay Methods 0.000 description 2
- 108010048484 radixin Proteins 0.000 description 2
- 230000026267 regulation of growth Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 102000003702 retinoic acid receptors Human genes 0.000 description 2
- 108090000064 retinoic acid receptors Proteins 0.000 description 2
- 235000002020 sage Nutrition 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- CDAISMWEOUEBRE-CDRYSYESSA-N scyllo-inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O CDAISMWEOUEBRE-CDRYSYESSA-N 0.000 description 2
- 239000004208 shellac Substances 0.000 description 2
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 2
- 229940113147 shellac Drugs 0.000 description 2
- 235000013874 shellac Nutrition 0.000 description 2
- 235000010413 sodium alginate Nutrition 0.000 description 2
- 239000000661 sodium alginate Substances 0.000 description 2
- 229940005550 sodium alginate Drugs 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 230000009182 swimming Effects 0.000 description 2
- 230000007470 synaptic degeneration Effects 0.000 description 2
- 230000003956 synaptic plasticity Effects 0.000 description 2
- WWJZWCUNLNYYAU-UHFFFAOYSA-N temephos Chemical compound C1=CC(OP(=S)(OC)OC)=CC=C1SC1=CC=C(OP(=S)(OC)OC)C=C1 WWJZWCUNLNYYAU-UHFFFAOYSA-N 0.000 description 2
- 229960002663 thioctic acid Drugs 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- KBPHJBAIARWVSC-XQIHNALSSA-N trans-lutein Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2C(=CC(O)CC2(C)C)C KBPHJBAIARWVSC-XQIHNALSSA-N 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 210000004291 uterus Anatomy 0.000 description 2
- 238000010200 validation analysis Methods 0.000 description 2
- 102000038650 voltage-gated calcium channel activity Human genes 0.000 description 2
- 108091023044 voltage-gated calcium channel activity Proteins 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- 108020001612 μ-opioid receptors Proteins 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- JWZZKOKVBUJMES-UHFFFAOYSA-N (+-)-Isoprenaline Chemical compound CC(C)NCC(O)C1=CC=C(O)C(O)=C1 JWZZKOKVBUJMES-UHFFFAOYSA-N 0.000 description 1
- WMBWREPUVVBILR-WIYYLYMNSA-N (-)-Epigallocatechin-3-o-gallate Chemical compound O([C@@H]1CC2=C(O)C=C(C=C2O[C@@H]1C=1C=C(O)C(O)=C(O)C=1)O)C(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-WIYYLYMNSA-N 0.000 description 1
- NGGMYCMLYOUNGM-UHFFFAOYSA-N (-)-fumagillin Natural products O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)C=CC=CC=CC=CC(O)=O)CCC21CO2 NGGMYCMLYOUNGM-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- 239000001100 (2S)-5,7-dihydroxy-2-(3-hydroxy-4-methoxyphenyl)chroman-4-one Substances 0.000 description 1
- YCYMCMYLORLIJX-SNVBAGLBSA-N (2r)-2-propyloctanoic acid Chemical compound CCCCCC[C@H](C(O)=O)CCC YCYMCMYLORLIJX-SNVBAGLBSA-N 0.000 description 1
- JKQXZKUSFCKOGQ-JLGXGRJMSA-N (3R,3'R)-beta,beta-carotene-3,3'-diol Chemical compound C([C@H](O)CC=1C)C(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)C[C@@H](O)CC1(C)C JKQXZKUSFCKOGQ-JLGXGRJMSA-N 0.000 description 1
- MKJIEFSOBYUXJB-HOCLYGCPSA-N (3S,11bS)-9,10-dimethoxy-3-isobutyl-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-one Chemical compound C1CN2C[C@H](CC(C)C)C(=O)C[C@H]2C2=C1C=C(OC)C(OC)=C2 MKJIEFSOBYUXJB-HOCLYGCPSA-N 0.000 description 1
- NPDLTEZXGWRMLQ-IBGZPJMESA-N (3r)-3-[6-(4-methylphenyl)pyridin-3-yl]oxy-1-azabicyclo[2.2.2]octane Chemical compound C1=CC(C)=CC=C1C(N=C1)=CC=C1O[C@@H]1C(CC2)CCN2C1 NPDLTEZXGWRMLQ-IBGZPJMESA-N 0.000 description 1
- OCKIPDMKGPYYJS-ZDUSSCGKSA-N (3r)-spiro[1-azabicyclo[2.2.2]octane-3,2'-3h-furo[2,3-b]pyridine] Chemical compound C1N(CC2)CCC2[C@]21OC1=NC=CC=C1C2 OCKIPDMKGPYYJS-ZDUSSCGKSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- GQIVTWIJJVAWQR-DANDVKJOSA-N (4r,4ar,7ar,12bs)-9-methoxy-3-methyl-1,2,4,4a,5,6,7a,13-octahydro-4,12-methanobenzofuro[3,2-e]isoquinoline-7-one;(2r,3r)-2,3-dihydroxybutanedioic acid;n-(4-hydroxyphenyl)acetamide Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.CC(=O)NC1=CC=C(O)C=C1.C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC GQIVTWIJJVAWQR-DANDVKJOSA-N 0.000 description 1
- SWXOGPJRIDTIRL-DOUNNPEJSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s)-1-amino-3-(1h-indol-3-yl)-1-oxopropan-2-yl]-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5,8,11,14,17-pent Chemical compound C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 SWXOGPJRIDTIRL-DOUNNPEJSA-N 0.000 description 1
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 description 1
- KWTSXDURSIMDCE-QMMMGPOBSA-N (S)-amphetamine Chemical compound C[C@H](N)CC1=CC=CC=C1 KWTSXDURSIMDCE-QMMMGPOBSA-N 0.000 description 1
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- FCERNNLMTCOKHX-UHFFFAOYSA-O 1,5-diphenyl-2H-tetrazol-1-ium Chemical compound C1=CC=CC=C1C1=[NH+]N=NN1C1=CC=CC=C1 FCERNNLMTCOKHX-UHFFFAOYSA-O 0.000 description 1
- CDKIEBFIMCSCBB-UHFFFAOYSA-N 1-(6,7-dimethoxy-3,4-dihydro-1h-isoquinolin-2-yl)-3-(1-methyl-2-phenylpyrrolo[2,3-b]pyridin-3-yl)prop-2-en-1-one;hydrochloride Chemical compound Cl.C1C=2C=C(OC)C(OC)=CC=2CCN1C(=O)C=CC(C1=CC=CN=C1N1C)=C1C1=CC=CC=C1 CDKIEBFIMCSCBB-UHFFFAOYSA-N 0.000 description 1
- MAYQIFVKVAUMPD-UHFFFAOYSA-N 1-(benzenesulfonyl)-2-methyl-4-piperazin-1-ylbenzimidazole Chemical compound CC1=NC2=C(N3CCNCC3)C=CC=C2N1S(=O)(=O)C1=CC=CC=C1 MAYQIFVKVAUMPD-UHFFFAOYSA-N 0.000 description 1
- CNEWKIDCGDXBDE-UHFFFAOYSA-N 1-[2-(4-phenylphenyl)ethyl]-4-[3-(trifluoromethyl)phenyl]-3,6-dihydro-2h-pyridine Chemical compound FC(F)(F)C1=CC=CC(C=2CCN(CCC=3C=CC(=CC=3)C=3C=CC=CC=3)CC=2)=C1 CNEWKIDCGDXBDE-UHFFFAOYSA-N 0.000 description 1
- RLUCYBFCLXANSO-BTJKTKAUSA-N 1-[3-[2-(1-benzothiophen-5-yl)ethoxy]propyl]azetidin-3-ol;(z)-but-2-enedioic acid Chemical compound OC(=O)\C=C/C(O)=O.C1C(O)CN1CCCOCCC1=CC=C(SC=C2)C2=C1 RLUCYBFCLXANSO-BTJKTKAUSA-N 0.000 description 1
- LIYLTQQDABRNRX-UHFFFAOYSA-N 1-[4-(3,4-dichlorophenyl)-3-fluorophenyl]cyclopropane-1-carboxylic acid Chemical compound C=1C=C(C=2C=C(Cl)C(Cl)=CC=2)C(F)=CC=1C1(C(=O)O)CC1 LIYLTQQDABRNRX-UHFFFAOYSA-N 0.000 description 1
- YFRBKEVUUCQYOW-UHFFFAOYSA-N 1-[6-[(3-cyclobutyl-1,2,4,5-tetrahydro-3-benzazepin-7-yl)oxy]pyridin-3-yl]pyrrolidin-2-one Chemical compound O=C1CCCN1C(C=N1)=CC=C1OC1=CC=C(CCN(CC2)C3CCC3)C2=C1 YFRBKEVUUCQYOW-UHFFFAOYSA-N 0.000 description 1
- OCAPBUJLXMYKEJ-UHFFFAOYSA-N 1-[biphenyl-4-yl(phenyl)methyl]imidazole Chemical compound C1=NC=CN1C(C=1C=CC(=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 OCAPBUJLXMYKEJ-UHFFFAOYSA-N 0.000 description 1
- JQSAYKKFZOSZGJ-UHFFFAOYSA-N 1-[bis(4-fluorophenyl)methyl]-4-[(2,3,4-trimethoxyphenyl)methyl]piperazine Chemical compound COC1=C(OC)C(OC)=CC=C1CN1CCN(C(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)CC1 JQSAYKKFZOSZGJ-UHFFFAOYSA-N 0.000 description 1
- 102100026152 1-acyl-sn-glycerol-3-phosphate acyltransferase epsilon Human genes 0.000 description 1
- 102100038028 1-phosphatidylinositol 3-phosphate 5-kinase Human genes 0.000 description 1
- 102100030390 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase beta-1 Human genes 0.000 description 1
- 102000010400 1-phosphatidylinositol-3-kinase activity proteins Human genes 0.000 description 1
- 108040005121 1-phosphatidylinositol-4-phosphate 5-kinase activity proteins Proteins 0.000 description 1
- 102100036506 11-beta-hydroxysteroid dehydrogenase 1 Human genes 0.000 description 1
- GVEZIHKRYBHEFX-MNOVXSKESA-N 13C-Cerulenin Natural products CC=CCC=CCCC(=O)[C@H]1O[C@@H]1C(N)=O GVEZIHKRYBHEFX-MNOVXSKESA-N 0.000 description 1
- LLMLGZUZTFMXSA-UHFFFAOYSA-N 2,3,4,5,6-pentachlorobenzenethiol Chemical compound SC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl LLMLGZUZTFMXSA-UHFFFAOYSA-N 0.000 description 1
- WCOXQTXVACYMLM-UHFFFAOYSA-N 2,3-bis(12-hydroxyoctadecanoyloxy)propyl 12-hydroxyoctadecanoate Chemical compound CCCCCCC(O)CCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCC(O)CCCCCC)COC(=O)CCCCCCCCCCC(O)CCCCCC WCOXQTXVACYMLM-UHFFFAOYSA-N 0.000 description 1
- YFGHCGITMMYXAQ-UHFFFAOYSA-N 2-[(diphenylmethyl)sulfinyl]acetamide Chemical compound C=1C=CC=CC=1C(S(=O)CC(=O)N)C1=CC=CC=C1 YFGHCGITMMYXAQ-UHFFFAOYSA-N 0.000 description 1
- HSXLMAFNWCSZGP-UHFFFAOYSA-N 2-[1-[(4-tert-butylphenyl)methyl]-5-(3-methylphenyl)indol-3-yl]-2-oxoacetic acid Chemical compound CC1=CC=CC(C=2C=C3C(C(=O)C(O)=O)=CN(CC=4C=CC(=CC=4)C(C)(C)C)C3=CC=2)=C1 HSXLMAFNWCSZGP-UHFFFAOYSA-N 0.000 description 1
- ZBIAKUMOEKILTF-UHFFFAOYSA-N 2-[4-[4,4-bis(4-fluorophenyl)butyl]-1-piperazinyl]-N-(2,6-dimethylphenyl)acetamide Chemical compound CC1=CC=CC(C)=C1NC(=O)CN1CCN(CCCC(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)CC1 ZBIAKUMOEKILTF-UHFFFAOYSA-N 0.000 description 1
- NSVFSAJIGAJDMR-UHFFFAOYSA-N 2-[benzyl(phenyl)amino]ethyl 5-(5,5-dimethyl-2-oxido-1,3,2-dioxaphosphinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylate Chemical compound CC=1NC(C)=C(C(=O)OCCN(CC=2C=CC=CC=2)C=2C=CC=CC=2)C(C=2C=C(C=CC=2)[N+]([O-])=O)C=1P1(=O)OCC(C)(C)CO1 NSVFSAJIGAJDMR-UHFFFAOYSA-N 0.000 description 1
- DUGMCDWNXXFHDE-UHFFFAOYSA-N 2-amino-2-methyl-n-[1-(1-methylsulfonylspiro[2h-indole-3,4'-piperidine]-1'-yl)-1-oxo-3-phenylmethoxypropan-2-yl]propanamide;methanesulfonic acid Chemical compound CS(O)(=O)=O.C1CC2(C3=CC=CC=C3N(C2)S(C)(=O)=O)CCN1C(=O)C(NC(=O)C(C)(N)C)COCC1=CC=CC=C1 DUGMCDWNXXFHDE-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- DDDZBLNULGDPGA-UHFFFAOYSA-N 2-methyl-3-[4-(3-pyrrolidin-1-ylpropoxy)phenyl]-5-(trifluoromethyl)quinazolin-4-one Chemical compound CC1=NC2=CC=CC(C(F)(F)F)=C2C(=O)N1C(C=C1)=CC=C1OCCCN1CCCC1 DDDZBLNULGDPGA-UHFFFAOYSA-N 0.000 description 1
- WLAMNBDJUVNPJU-UHFFFAOYSA-N 2-methylbutyric acid Chemical compound CCC(C)C(O)=O WLAMNBDJUVNPJU-UHFFFAOYSA-N 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- JWUBBDSIWDLEOM-UHFFFAOYSA-N 25-Hydroxycholecalciferol Natural products C1CCC2(C)C(C(CCCC(C)(C)O)C)CCC2C1=CC=C1CC(O)CCC1=C JWUBBDSIWDLEOM-UHFFFAOYSA-N 0.000 description 1
- 102100039377 28 kDa heat- and acid-stable phosphoprotein Human genes 0.000 description 1
- 101710176122 28 kDa heat- and acid-stable phosphoprotein Proteins 0.000 description 1
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 1
- JJZFWROHYSMCMU-UHFFFAOYSA-N 3-(benzenesulfonyl)-8-piperazin-1-ylquinoline Chemical compound C=1N=C2C(N3CCNCC3)=CC=CC2=CC=1S(=O)(=O)C1=CC=CC=C1 JJZFWROHYSMCMU-UHFFFAOYSA-N 0.000 description 1
- SNKZJIOFVMKAOJ-UHFFFAOYSA-N 3-Aminopropanesulfonate Chemical compound NCCCS(O)(=O)=O SNKZJIOFVMKAOJ-UHFFFAOYSA-N 0.000 description 1
- BMUDPLZKKRQECS-UHFFFAOYSA-K 3-[18-(2-carboxyethyl)-8,13-bis(ethenyl)-3,7,12,17-tetramethylporphyrin-21,24-diid-2-yl]propanoic acid iron(3+) hydroxide Chemical compound [OH-].[Fe+3].[N-]1C2=C(C)C(CCC(O)=O)=C1C=C([N-]1)C(CCC(O)=O)=C(C)C1=CC(C(C)=C1C=C)=NC1=CC(C(C)=C1C=C)=NC1=C2 BMUDPLZKKRQECS-UHFFFAOYSA-K 0.000 description 1
- KJNNWYBAOPXVJY-UHFFFAOYSA-N 3-[4-[2-butyl-1-[4-(4-chlorophenoxy)phenyl]imidazol-4-yl]phenoxy]-n,n-diethylpropan-1-amine Chemical compound CCCCC1=NC(C=2C=CC(OCCCN(CC)CC)=CC=2)=CN1C(C=C1)=CC=C1OC1=CC=C(Cl)C=C1 KJNNWYBAOPXVJY-UHFFFAOYSA-N 0.000 description 1
- 102100021834 3-hydroxyacyl-CoA dehydrogenase Human genes 0.000 description 1
- UIAGMCDKSXEBJQ-IBGZPJMESA-N 3-o-(2-methoxyethyl) 5-o-propan-2-yl (4s)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COCCOC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)[C@H]1C1=CC=CC([N+]([O-])=O)=C1 UIAGMCDKSXEBJQ-IBGZPJMESA-N 0.000 description 1
- ZGRIPYHIFXGCHR-UHFFFAOYSA-N 3-o-[2-[(4-fluorophenyl)methyl-methylamino]ethyl] 5-o-propan-2-yl 4-(1,3-benzodioxol-4-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound C=1C=CC=2OCOC=2C=1C1C(C(=O)OC(C)C)=C(C)NC(C)=C1C(=O)OCCN(C)CC1=CC=C(F)C=C1 ZGRIPYHIFXGCHR-UHFFFAOYSA-N 0.000 description 1
- UWSONZCNXUSTKW-UHFFFAOYSA-N 4,5-Dimethylthiazole Chemical compound CC=1N=CSC=1C UWSONZCNXUSTKW-UHFFFAOYSA-N 0.000 description 1
- YKGYIDJEEQRWQH-UHFFFAOYSA-N 4-[6-(diaminomethylideneamino)-1-oxohexoxy]benzoic acid ethyl ester Chemical compound CCOC(=O)C1=CC=C(OC(=O)CCCCCN=C(N)N)C=C1 YKGYIDJEEQRWQH-UHFFFAOYSA-N 0.000 description 1
- NRPQELCNMADTOZ-OAQYLSRUSA-N 4-cyano-n-[(2r)-2-[4-(2,3-dihydro-1,4-benzodioxin-5-yl)piperazin-1-yl]propyl]-n-pyridin-2-ylbenzamide Chemical compound C([C@@H](C)N1CCN(CC1)C=1C=2OCCOC=2C=CC=1)N(C=1N=CC=CC=1)C(=O)C1=CC=C(C#N)C=C1 NRPQELCNMADTOZ-OAQYLSRUSA-N 0.000 description 1
- GXYZREDEYDFJPT-ZMBIFBSDSA-N 4-cyano-n-[(2r)-2-[4-(2,3-dihydro-1,4-benzodioxin-5-yl)piperazin-1-yl]propyl]-n-pyridin-2-ylbenzamide;hydrochloride Chemical compound Cl.C([C@@H](C)N1CCN(CC1)C=1C=2OCCOC=2C=CC=1)N(C=1N=CC=CC=1)C(=O)C1=CC=C(C#N)C=C1 GXYZREDEYDFJPT-ZMBIFBSDSA-N 0.000 description 1
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- CHWVDGYLKPLBES-UHFFFAOYSA-N 5-(2-methoxyphenyl)-2-furoic acid Chemical compound COC1=CC=CC=C1C1=CC=C(C(O)=O)O1 CHWVDGYLKPLBES-UHFFFAOYSA-N 0.000 description 1
- OJBLXSPBJMGZDN-UHFFFAOYSA-N 5-[3-(difluoromethyl)-4-fluorophenyl]-3-[(2-methylimidazol-1-yl)methyl]pyridazine;dihydrochloride Chemical compound Cl.Cl.CC1=NC=CN1CC1=CC(C=2C=C(C(F)=CC=2)C(F)F)=CN=N1 OJBLXSPBJMGZDN-UHFFFAOYSA-N 0.000 description 1
- PYZFRRVBPNGCBX-SECBINFHSA-N 5-chloro-n-[(2s)-3-ethyl-1-hydroxypentan-2-yl]thiophene-2-sulfonamide Chemical compound CCC(CC)[C@@H](CO)NS(=O)(=O)C1=CC=C(Cl)S1 PYZFRRVBPNGCBX-SECBINFHSA-N 0.000 description 1
- PSXOKXJMVRSARX-SCSAIBSYSA-N 5-chloro-n-[(2s)-4,4,4-trifluoro-1-hydroxy-3-(trifluoromethyl)butan-2-yl]thiophene-2-sulfonamide Chemical compound FC(F)(F)C(C(F)(F)F)[C@@H](CO)NS(=O)(=O)C1=CC=C(Cl)S1 PSXOKXJMVRSARX-SCSAIBSYSA-N 0.000 description 1
- 102100027499 5-hydroxytryptamine receptor 1B Human genes 0.000 description 1
- SUBDBMMJDZJVOS-UHFFFAOYSA-N 5-methoxy-2-{[(4-methoxy-3,5-dimethylpyridin-2-yl)methyl]sulfinyl}-1H-benzimidazole Chemical compound N=1C2=CC(OC)=CC=C2NC=1S(=O)CC1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- CUCUKLJLRRAKFN-UHFFFAOYSA-N 7-Hydroxy-(S)-usnate Chemical compound CC12C(=O)C(C(=O)C)C(=O)C=C1OC1=C2C(O)=C(C)C(O)=C1C(C)=O CUCUKLJLRRAKFN-UHFFFAOYSA-N 0.000 description 1
- 102100036512 7-dehydrocholesterol reductase Human genes 0.000 description 1
- VFPMCLQMAUVEHD-UCPSWNCLSA-N 7beta-hydroxyepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3[C@@H](O)C[C@H]21 VFPMCLQMAUVEHD-UCPSWNCLSA-N 0.000 description 1
- 102100031901 A-kinase anchor protein 2 Human genes 0.000 description 1
- 101150092476 ABCA1 gene Proteins 0.000 description 1
- 108091007504 ADAM10 Proteins 0.000 description 1
- 102000017920 ADRB1 Human genes 0.000 description 1
- 108010067409 AN-1792 Proteins 0.000 description 1
- 108700005241 ATP Binding Cassette Transporter 1 Proteins 0.000 description 1
- 108091006112 ATPases Proteins 0.000 description 1
- 102100028247 Abl interactor 1 Human genes 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- 229940121683 Acetylcholine receptor antagonist Drugs 0.000 description 1
- 102100030913 Acetylcholine receptor subunit alpha Human genes 0.000 description 1
- 102100022094 Acid-sensing ion channel 2 Human genes 0.000 description 1
- 239000004925 Acrylic resin Substances 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 1
- 102100022089 Acyl-[acyl-carrier-protein] hydrolase Human genes 0.000 description 1
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 1
- 102000009346 Adenosine receptors Human genes 0.000 description 1
- 108050000203 Adenosine receptors Proteins 0.000 description 1
- 102100032599 Adhesion G protein-coupled receptor B3 Human genes 0.000 description 1
- 102100031786 Adiponectin Human genes 0.000 description 1
- 102100033497 Adiponectin receptor protein 1 Human genes 0.000 description 1
- 102100036775 Afadin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 102100033816 Aldehyde dehydrogenase, mitochondrial Human genes 0.000 description 1
- 108010003133 Aldo-Keto Reductase Family 1 Member C2 Proteins 0.000 description 1
- 102100024089 Aldo-keto reductase family 1 member C2 Human genes 0.000 description 1
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 1
- 239000012103 Alexa Fluor 488 Substances 0.000 description 1
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical compound COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 1
- 102100024401 Alpha-1D adrenergic receptor Human genes 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 102100022531 Amiloride-sensitive sodium channel subunit delta Human genes 0.000 description 1
- 102100028661 Amine oxidase [flavin-containing] A Human genes 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- APKFDSVGJQXUKY-KKGHZKTASA-N Amphotericin-B Natural products O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1C=CC=CC=CC=CC=CC=CC=C[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-KKGHZKTASA-N 0.000 description 1
- 102000002659 Amyloid Precursor Protein Secretases Human genes 0.000 description 1
- 108010043324 Amyloid Precursor Protein Secretases Proteins 0.000 description 1
- 102100031329 Ankyrin repeat family A protein 2 Human genes 0.000 description 1
- 102100040006 Annexin A1 Human genes 0.000 description 1
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 1
- 102100033715 Apolipoprotein A-I Human genes 0.000 description 1
- 102000013918 Apolipoproteins E Human genes 0.000 description 1
- 108010025628 Apolipoproteins E Proteins 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 101100236700 Arabidopsis thaliana MCC1 gene Proteins 0.000 description 1
- 101100256253 Arabidopsis thaliana SCAR2 gene Proteins 0.000 description 1
- NCUCGYYHUFIYNU-UHFFFAOYSA-N Aranidipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCC(C)=O)C1C1=CC=CC=C1[N+]([O-])=O NCUCGYYHUFIYNU-UHFFFAOYSA-N 0.000 description 1
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 102000002804 Ataxia Telangiectasia Mutated Proteins Human genes 0.000 description 1
- 108010004586 Ataxia Telangiectasia Mutated Proteins Proteins 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- YXSLJKQTIDHPOT-UHFFFAOYSA-N Atracurium Dibesylate Chemical compound C1=C(OC)C(OC)=CC=C1CC1[N+](CCC(=O)OCCCCCOC(=O)CC[N+]2(C)C(C3=CC(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=CC=2)(C)CCC2=CC(OC)=C(OC)C=C21 YXSLJKQTIDHPOT-UHFFFAOYSA-N 0.000 description 1
- 108010092776 Autophagy-Related Protein 5 Proteins 0.000 description 1
- 102000016614 Autophagy-Related Protein 5 Human genes 0.000 description 1
- XEAOPVUAMONVLA-QGZVFWFLSA-N Avagacestat Chemical compound C=1C=C(Cl)C=CC=1S(=O)(=O)N([C@H](CCC(F)(F)F)C(=O)N)CC(C(=C1)F)=CC=C1C=1N=CON=1 XEAOPVUAMONVLA-QGZVFWFLSA-N 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 238000000035 BCA protein assay Methods 0.000 description 1
- 108091012583 BCL2 Proteins 0.000 description 1
- 108700020463 BRCA1 Proteins 0.000 description 1
- 101150072950 BRCA1 gene Proteins 0.000 description 1
- 108010001478 Bacitracin Proteins 0.000 description 1
- 102100021334 Bcl-2-related protein A1 Human genes 0.000 description 1
- 108010081589 Becaplermin Proteins 0.000 description 1
- 102100021251 Beclin-1 Human genes 0.000 description 1
- 108090000524 Beclin-1 Proteins 0.000 description 1
- 102100021738 Beta-adrenergic receptor kinase 1 Human genes 0.000 description 1
- CYGODHVAJQTCBG-UHFFFAOYSA-N Bifeprunox Chemical compound C=12OC(=O)NC2=CC=CC=1N(CC1)CCN1CC(C=1)=CC=CC=1C1=CC=CC=C1 CYGODHVAJQTCBG-UHFFFAOYSA-N 0.000 description 1
- 102100024504 Bone morphogenetic protein 3 Human genes 0.000 description 1
- 101000964894 Bos taurus 14-3-3 protein zeta/delta Proteins 0.000 description 1
- 102100025401 Breast cancer type 1 susceptibility protein Human genes 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- QZDCYUCETTWCMO-CDFKWJNJSA-N C1C2C[C@H]3C[N@](C2)CC1[C@H]3Oc1nnc(s1)-c1ccccc1 Chemical compound C1C2C[C@H]3C[N@](C2)CC1[C@H]3Oc1nnc(s1)-c1ccccc1 QZDCYUCETTWCMO-CDFKWJNJSA-N 0.000 description 1
- 102100032986 CCR4-NOT transcription complex subunit 8 Human genes 0.000 description 1
- 102000049320 CD36 Human genes 0.000 description 1
- 108010045374 CD36 Antigens Proteins 0.000 description 1
- KORNTPPJEAJQIU-KJXAQDMKSA-N Cabaser Chemical compound C1=CC([C@H]2C[C@H](CN(CC=C)[C@@H]2C2)C(=O)N(CCCN(C)C)C(=O)NCC)=C3C2=CNC3=C1 KORNTPPJEAJQIU-KJXAQDMKSA-N 0.000 description 1
- 102000000905 Cadherin Human genes 0.000 description 1
- 108050007957 Cadherin Proteins 0.000 description 1
- 102100025805 Cadherin-1 Human genes 0.000 description 1
- 101000975407 Caenorhabditis elegans Inositol 1,4,5-trisphosphate receptor itr-1 Proteins 0.000 description 1
- 235000021318 Calcifediol Nutrition 0.000 description 1
- 108010042955 Calcineurin Proteins 0.000 description 1
- 102000004631 Calcineurin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- 102400000113 Calcitonin Human genes 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- 102100023074 Calcium-activated potassium channel subunit beta-1 Human genes 0.000 description 1
- 102100032583 Calcium-dependent secretion activator 2 Human genes 0.000 description 1
- 102100024316 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Human genes 0.000 description 1
- 102100021534 Calcium/calmodulin-dependent protein kinase kinase 2 Human genes 0.000 description 1
- 102100033088 Calcium/calmodulin-dependent protein kinase type 1D Human genes 0.000 description 1
- 102100025580 Calmodulin-1 Human genes 0.000 description 1
- 101710167800 Capsid assembly scaffolding protein Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102100022344 Cardiac phospholamban Human genes 0.000 description 1
- 101710118321 Casein kinase I isoform alpha Proteins 0.000 description 1
- 102100034356 Casein kinase I isoform alpha-like Human genes 0.000 description 1
- 102100028002 Catenin alpha-2 Human genes 0.000 description 1
- 102100028914 Catenin beta-1 Human genes 0.000 description 1
- 108050001278 Cdc42 Proteins 0.000 description 1
- 102100024490 Cdc42 effector protein 3 Human genes 0.000 description 1
- 108091007854 Cdh1/Fizzy-related Proteins 0.000 description 1
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 101150054987 ChAT gene Proteins 0.000 description 1
- PCLITLDOTJTVDJ-UHFFFAOYSA-N Chlormethiazole Chemical compound CC=1N=CSC=1CCCl PCLITLDOTJTVDJ-UHFFFAOYSA-N 0.000 description 1
- 102100023460 Choline O-acetyltransferase Human genes 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 108010000063 Ciliary Neurotrophic Factor Receptor Proteins 0.000 description 1
- 102000016989 Ciliary Neurotrophic Factor Receptor Human genes 0.000 description 1
- 102100031615 Ciliary neurotrophic factor receptor subunit alpha Human genes 0.000 description 1
- 235000001258 Cinchona calisaya Nutrition 0.000 description 1
- 102000012422 Collagen Type I Human genes 0.000 description 1
- 108010022452 Collagen Type I Proteins 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 108010088874 Cullin 1 Proteins 0.000 description 1
- 102100039195 Cullin-1 Human genes 0.000 description 1
- 108010045171 Cyclic AMP Response Element-Binding Protein Proteins 0.000 description 1
- 102000005636 Cyclic AMP Response Element-Binding Protein Human genes 0.000 description 1
- 108010025454 Cyclin-Dependent Kinase 5 Proteins 0.000 description 1
- 108010016788 Cyclin-Dependent Kinase Inhibitor p21 Proteins 0.000 description 1
- 102100033270 Cyclin-dependent kinase inhibitor 1 Human genes 0.000 description 1
- 102100026805 Cyclin-dependent-like kinase 5 Human genes 0.000 description 1
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 1
- 108010076010 Cystathionine beta-lyase Proteins 0.000 description 1
- 102100038698 Cytochrome P450 7B1 Human genes 0.000 description 1
- 102100036943 Cytoplasmic protein NCK1 Human genes 0.000 description 1
- 102100029813 D(1B) dopamine receptor Human genes 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- HEBKCHPVOIAQTA-QWWZWVQMSA-N D-arabinitol Chemical compound OC[C@@H](O)C(O)[C@H](O)CO HEBKCHPVOIAQTA-QWWZWVQMSA-N 0.000 description 1
- OQEBIHBLFRADNM-UHFFFAOYSA-N D-iminoxylitol Natural products OCC1NCC(O)C1O OQEBIHBLFRADNM-UHFFFAOYSA-N 0.000 description 1
- DSCFFEYYQKSRSV-KLJZZCKASA-N D-pinitol Chemical compound CO[C@@H]1[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@H]1O DSCFFEYYQKSRSV-KLJZZCKASA-N 0.000 description 1
- 101150065997 DLG2 gene Proteins 0.000 description 1
- RPYWXZCFYPVCNQ-RVDMUPIBSA-N DMXB-A Chemical compound COC1=CC(OC)=CC=C1\C=C/1C(C=2C=NC=CC=2)=NCCC\1 RPYWXZCFYPVCNQ-RVDMUPIBSA-N 0.000 description 1
- 102100024829 DNA polymerase delta catalytic subunit Human genes 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- 102100031604 Dedicator of cytokinesis protein 3 Human genes 0.000 description 1
- GCPYCNBGGPHOBD-UHFFFAOYSA-N Delphinidin Natural products OC1=Cc2c(O)cc(O)cc2OC1=C3C=C(O)C(=O)C(=C3)O GCPYCNBGGPHOBD-UHFFFAOYSA-N 0.000 description 1
- CVBMAZKKCSYWQR-BPJCFPRXSA-N Deserpidine Natural products O=C(OC)[C@@H]1[C@H](OC)[C@H](OC(=O)c2cc(OC)c(OC)c(OC)c2)C[C@H]2[C@@H]1C[C@H]1N(C2)CCc2c3c([nH]c12)cccc3 CVBMAZKKCSYWQR-BPJCFPRXSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 102100030215 Diacylglycerol kinase eta Human genes 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- 102100028561 Disabled homolog 1 Human genes 0.000 description 1
- 102100039673 Disintegrin and metalloproteinase domain-containing protein 10 Human genes 0.000 description 1
- 102100031111 Disintegrin and metalloproteinase domain-containing protein 17 Human genes 0.000 description 1
- JWCSIUVGFCSJCK-CAVRMKNVSA-N Disodium Moxalactam Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CO[C@@H]21)C(O)=O)=O)C(=O)C(C(O)=O)C1=CC=C(O)C=C1 JWCSIUVGFCSJCK-CAVRMKNVSA-N 0.000 description 1
- 102100035419 DnaJ homolog subfamily B member 9 Human genes 0.000 description 1
- 229940098778 Dopamine receptor agonist Drugs 0.000 description 1
- 201000010374 Down Syndrome Diseases 0.000 description 1
- 101000975393 Drosophila melanogaster Inositol 1,4,5-trisphosphate receptor Proteins 0.000 description 1
- 102100036367 Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11A Human genes 0.000 description 1
- 206010013976 Dyspraxia Diseases 0.000 description 1
- 102100035989 E3 SUMO-protein ligase PIAS1 Human genes 0.000 description 1
- 102100035813 E3 ubiquitin-protein ligase CBL Human genes 0.000 description 1
- 102100033334 E3 ubiquitin-protein ligase Itchy homolog Human genes 0.000 description 1
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 1
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 1
- 102000007303 ELAV-Like Protein 2 Human genes 0.000 description 1
- 108010008795 ELAV-Like Protein 2 Proteins 0.000 description 1
- 102100030695 Electron transfer flavoprotein subunit alpha, mitochondrial Human genes 0.000 description 1
- 108700041152 Endoplasmic Reticulum Chaperone BiP Proteins 0.000 description 1
- 102100021451 Endoplasmic reticulum chaperone BiP Human genes 0.000 description 1
- 102100039328 Endoplasmin Human genes 0.000 description 1
- 102100033902 Endothelin-1 Human genes 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102100021579 Enhancer of filamentation 1 Human genes 0.000 description 1
- 102000050554 Eph Family Receptors Human genes 0.000 description 1
- 108091008815 Eph receptors Proteins 0.000 description 1
- 102000020086 Ephrin-A1 Human genes 0.000 description 1
- 108010043945 Ephrin-A1 Proteins 0.000 description 1
- 102000006397 Ephrin-B1 Human genes 0.000 description 1
- 108010044099 Ephrin-B1 Proteins 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- LMHIPJMTZHDKEW-XQYLJSSYSA-M Epoprostenol sodium Chemical compound [Na+].O1\C(=C/CCCC([O-])=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 LMHIPJMTZHDKEW-XQYLJSSYSA-M 0.000 description 1
- 102100031855 Estrogen-related receptor gamma Human genes 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- 229920000064 Ethyl eicosapentaenoic acid Polymers 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102100020903 Ezrin Human genes 0.000 description 1
- 241000272186 Falco columbarius Species 0.000 description 1
- TZXKOCQBRNJULO-UHFFFAOYSA-N Ferriprox Chemical compound CC1=C(O)C(=O)C=CN1C TZXKOCQBRNJULO-UHFFFAOYSA-N 0.000 description 1
- 102000003971 Fibroblast Growth Factor 1 Human genes 0.000 description 1
- 108090000386 Fibroblast Growth Factor 1 Proteins 0.000 description 1
- 108090000368 Fibroblast growth factor 8 Proteins 0.000 description 1
- 102100026561 Filamin-A Human genes 0.000 description 1
- 108010009306 Forkhead Box Protein O1 Proteins 0.000 description 1
- 102100035427 Forkhead box protein O1 Human genes 0.000 description 1
- XWLUWCNOOVRFPX-UHFFFAOYSA-N Fosphenytoin Chemical compound O=C1N(COP(O)(=O)O)C(=O)NC1(C=1C=CC=CC=1)C1=CC=CC=C1 XWLUWCNOOVRFPX-UHFFFAOYSA-N 0.000 description 1
- 102100033063 G protein-activated inward rectifier potassium channel 1 Human genes 0.000 description 1
- 102000054184 GADD45 Human genes 0.000 description 1
- WMBWREPUVVBILR-UHFFFAOYSA-N GCG Natural products C=1C(O)=C(O)C(O)=CC=1C1OC2=CC(O)=CC(O)=C2CC1OC(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-UHFFFAOYSA-N 0.000 description 1
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 1
- 102100027541 GTP-binding protein Rheb Human genes 0.000 description 1
- 108091006109 GTPases Proteins 0.000 description 1
- 102100035212 Gamma-aminobutyric acid type B receptor subunit 1 Human genes 0.000 description 1
- HEMJJKBWTPKOJG-UHFFFAOYSA-N Gemfibrozil Chemical compound CC1=CC=C(C)C(OCCCC(C)(C)C(O)=O)=C1 HEMJJKBWTPKOJG-UHFFFAOYSA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010027915 Glutamate Receptors Proteins 0.000 description 1
- 102000018899 Glutamate Receptors Human genes 0.000 description 1
- 102100034009 Glutamate dehydrogenase 1, mitochondrial Human genes 0.000 description 1
- 102100030651 Glutamate receptor 2 Human genes 0.000 description 1
- 102100022630 Glutamate receptor ionotropic, NMDA 2B Human genes 0.000 description 1
- 102100022193 Glutamate receptor ionotropic, delta-1 Human genes 0.000 description 1
- 102100039770 Glutamate receptor-interacting protein 1 Human genes 0.000 description 1
- 102100036076 Glycerophosphocholine cholinephosphodiesterase ENPP6 Human genes 0.000 description 1
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 1
- 102100033945 Glycine receptor subunit alpha-1 Human genes 0.000 description 1
- 102100021196 Glypican-5 Human genes 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- 102100036733 Guanine nucleotide-binding protein subunit alpha-12 Human genes 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101150112743 HSPA5 gene Proteins 0.000 description 1
- 102000017911 HTR1A Human genes 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 102000005548 Hexokinase Human genes 0.000 description 1
- 102100022846 Histone acetyltransferase KAT2B Human genes 0.000 description 1
- 101710110793 Homeodomain-interacting protein kinase 2 Proteins 0.000 description 1
- 102100023607 Homer protein homolog 1 Human genes 0.000 description 1
- 101000691569 Homo sapiens 1-acyl-sn-glycerol-3-phosphate acyltransferase epsilon Proteins 0.000 description 1
- 101000583063 Homo sapiens 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase beta-1 Proteins 0.000 description 1
- 101000928753 Homo sapiens 11-beta-hydroxysteroid dehydrogenase 1 Proteins 0.000 description 1
- 101000896020 Homo sapiens 3-hydroxyacyl-CoA dehydrogenase Proteins 0.000 description 1
- 101000822895 Homo sapiens 5-hydroxytryptamine receptor 1A Proteins 0.000 description 1
- 101000724725 Homo sapiens 5-hydroxytryptamine receptor 1B Proteins 0.000 description 1
- 101000928720 Homo sapiens 7-dehydrocholesterol reductase Proteins 0.000 description 1
- 101000774738 Homo sapiens A-kinase anchor protein 2 Proteins 0.000 description 1
- 101100162766 Homo sapiens ANKRA2 gene Proteins 0.000 description 1
- 101000724225 Homo sapiens Abl interactor 1 Proteins 0.000 description 1
- 101000677540 Homo sapiens Acetyl-CoA carboxylase 2 Proteins 0.000 description 1
- 101000726895 Homo sapiens Acetylcholine receptor subunit alpha Proteins 0.000 description 1
- 101000901079 Homo sapiens Acid-sensing ion channel 2 Proteins 0.000 description 1
- 101000824278 Homo sapiens Acyl-[acyl-carrier-protein] hydrolase Proteins 0.000 description 1
- 101000796801 Homo sapiens Adhesion G protein-coupled receptor B3 Proteins 0.000 description 1
- 101000775469 Homo sapiens Adiponectin Proteins 0.000 description 1
- 101001135206 Homo sapiens Adiponectin receptor protein 1 Proteins 0.000 description 1
- 101000928246 Homo sapiens Afadin Proteins 0.000 description 1
- 101000689685 Homo sapiens Alpha-1A adrenergic receptor Proteins 0.000 description 1
- 101000689696 Homo sapiens Alpha-1D adrenergic receptor Proteins 0.000 description 1
- 101000834898 Homo sapiens Alpha-synuclein Proteins 0.000 description 1
- 101000822355 Homo sapiens Amiloride-sensitive sodium channel subunit delta Proteins 0.000 description 1
- 101000694718 Homo sapiens Amine oxidase [flavin-containing] A Proteins 0.000 description 1
- 101000959738 Homo sapiens Annexin A1 Proteins 0.000 description 1
- 101000733802 Homo sapiens Apolipoprotein A-I Proteins 0.000 description 1
- 101100218947 Homo sapiens BMP3 gene Proteins 0.000 description 1
- 101000894929 Homo sapiens Bcl-2-related protein A1 Proteins 0.000 description 1
- 101000892264 Homo sapiens Beta-1 adrenergic receptor Proteins 0.000 description 1
- 101000751445 Homo sapiens Beta-adrenergic receptor kinase 1 Proteins 0.000 description 1
- 101000942586 Homo sapiens CCR4-NOT transcription complex subunit 8 Proteins 0.000 description 1
- 101001049849 Homo sapiens Calcium-activated potassium channel subunit beta-1 Proteins 0.000 description 1
- 101000867778 Homo sapiens Calcium-dependent secretion activator 2 Proteins 0.000 description 1
- 101001117044 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Proteins 0.000 description 1
- 101000971617 Homo sapiens Calcium/calmodulin-dependent protein kinase kinase 2 Proteins 0.000 description 1
- 101000944258 Homo sapiens Calcium/calmodulin-dependent protein kinase type 1D Proteins 0.000 description 1
- 101000984164 Homo sapiens Calmodulin-1 Proteins 0.000 description 1
- 101000620629 Homo sapiens Cardiac phospholamban Proteins 0.000 description 1
- 101000859073 Homo sapiens Catenin alpha-2 Proteins 0.000 description 1
- 101000916173 Homo sapiens Catenin beta-1 Proteins 0.000 description 1
- 101000762414 Homo sapiens Cdc42 effector protein 3 Proteins 0.000 description 1
- 101000969553 Homo sapiens Cell surface glycoprotein CD200 receptor 1 Proteins 0.000 description 1
- 101000895818 Homo sapiens Chorionic somatomammotropin hormone 1 Proteins 0.000 description 1
- 101000993348 Homo sapiens Ciliary neurotrophic factor receptor subunit alpha Proteins 0.000 description 1
- 101000868333 Homo sapiens Cyclin-dependent kinase 1 Proteins 0.000 description 1
- 101000957674 Homo sapiens Cytochrome P450 7B1 Proteins 0.000 description 1
- 101001024707 Homo sapiens Cytoplasmic protein NCK1 Proteins 0.000 description 1
- 101000865210 Homo sapiens D(1B) dopamine receptor Proteins 0.000 description 1
- 101000909198 Homo sapiens DNA polymerase delta catalytic subunit Proteins 0.000 description 1
- 101000866238 Homo sapiens Dedicator of cytokinesis protein 3 Proteins 0.000 description 1
- 101000864599 Homo sapiens Diacylglycerol kinase eta Proteins 0.000 description 1
- 101001053479 Homo sapiens Dihydropyrimidinase-related protein 5 Proteins 0.000 description 1
- 101000915416 Homo sapiens Disabled homolog 1 Proteins 0.000 description 1
- 101000777461 Homo sapiens Disintegrin and metalloproteinase domain-containing protein 17 Proteins 0.000 description 1
- 101000804119 Homo sapiens DnaJ homolog subfamily B member 9 Proteins 0.000 description 1
- 101000997630 Homo sapiens E3 ubiquitin-protein ligase Itchy homolog Proteins 0.000 description 1
- 101001010541 Homo sapiens Electron transfer flavoprotein subunit alpha, mitochondrial Proteins 0.000 description 1
- 101000812663 Homo sapiens Endoplasmin Proteins 0.000 description 1
- 101000925493 Homo sapiens Endothelin-1 Proteins 0.000 description 1
- 101000898310 Homo sapiens Enhancer of filamentation 1 Proteins 0.000 description 1
- 101000920831 Homo sapiens Estrogen-related receptor gamma Proteins 0.000 description 1
- 101000866287 Homo sapiens Excitatory amino acid transporter 2 Proteins 0.000 description 1
- 101000854648 Homo sapiens Ezrin Proteins 0.000 description 1
- 101000913549 Homo sapiens Filamin-A Proteins 0.000 description 1
- 101000944266 Homo sapiens G protein-activated inward rectifier potassium channel 1 Proteins 0.000 description 1
- 101001051083 Homo sapiens Galectin-12 Proteins 0.000 description 1
- 101001040875 Homo sapiens Glucosidase 2 subunit beta Proteins 0.000 description 1
- 101000870042 Homo sapiens Glutamate dehydrogenase 1, mitochondrial Proteins 0.000 description 1
- 101001010449 Homo sapiens Glutamate receptor 2 Proteins 0.000 description 1
- 101000972850 Homo sapiens Glutamate receptor ionotropic, NMDA 2B Proteins 0.000 description 1
- 101000900493 Homo sapiens Glutamate receptor ionotropic, delta-1 Proteins 0.000 description 1
- 101001034009 Homo sapiens Glutamate receptor-interacting protein 1 Proteins 0.000 description 1
- 101000876254 Homo sapiens Glycerophosphocholine cholinephosphodiesterase ENPP6 Proteins 0.000 description 1
- 101000996297 Homo sapiens Glycine receptor subunit alpha-1 Proteins 0.000 description 1
- 101001040711 Homo sapiens Glypican-5 Proteins 0.000 description 1
- 101001066158 Homo sapiens Growth arrest and DNA damage-inducible protein GADD45 alpha Proteins 0.000 description 1
- 101001072398 Homo sapiens Guanine nucleotide-binding protein subunit alpha-12 Proteins 0.000 description 1
- 101001047006 Homo sapiens Histone acetyltransferase KAT2B Proteins 0.000 description 1
- 101001048469 Homo sapiens Homer protein homolog 1 Proteins 0.000 description 1
- 101001079904 Homo sapiens Hyaluronan and proteoglycan link protein 1 Proteins 0.000 description 1
- 101000962530 Homo sapiens Hyaluronidase-1 Proteins 0.000 description 1
- 101001076642 Homo sapiens Inosine-5'-monophosphate dehydrogenase 2 Proteins 0.000 description 1
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 1
- 101000852815 Homo sapiens Insulin receptor Proteins 0.000 description 1
- 101001078158 Homo sapiens Integrin alpha-1 Proteins 0.000 description 1
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 description 1
- 101000598002 Homo sapiens Interferon regulatory factor 1 Proteins 0.000 description 1
- 101001044893 Homo sapiens Interleukin-20 receptor subunit alpha Proteins 0.000 description 1
- 101000599056 Homo sapiens Interleukin-6 receptor subunit beta Proteins 0.000 description 1
- 101001090172 Homo sapiens Kinectin Proteins 0.000 description 1
- 101001135499 Homo sapiens Kv channel-interacting protein 1 Proteins 0.000 description 1
- 101001021858 Homo sapiens Kynureninase Proteins 0.000 description 1
- 101001054649 Homo sapiens Latent-transforming growth factor beta-binding protein 2 Proteins 0.000 description 1
- 101001054646 Homo sapiens Latent-transforming growth factor beta-binding protein 3 Proteins 0.000 description 1
- 101001064301 Homo sapiens Lipase member K Proteins 0.000 description 1
- 101001064542 Homo sapiens Liprin-beta-1 Proteins 0.000 description 1
- 101001051093 Homo sapiens Low-density lipoprotein receptor Proteins 0.000 description 1
- 101001043562 Homo sapiens Low-density lipoprotein receptor-related protein 2 Proteins 0.000 description 1
- 101001039207 Homo sapiens Low-density lipoprotein receptor-related protein 8 Proteins 0.000 description 1
- 101001005668 Homo sapiens Mastermind-like protein 3 Proteins 0.000 description 1
- 101001032848 Homo sapiens Metabotropic glutamate receptor 3 Proteins 0.000 description 1
- 101000645296 Homo sapiens Metalloproteinase inhibitor 2 Proteins 0.000 description 1
- 101001074975 Homo sapiens Molybdopterin molybdenumtransferase Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101000970561 Homo sapiens Myc box-dependent-interacting protein 1 Proteins 0.000 description 1
- 101001072470 Homo sapiens N-acetylglucosamine-1-phosphotransferase subunits alpha/beta Proteins 0.000 description 1
- 101000978949 Homo sapiens NADP-dependent malic enzyme Proteins 0.000 description 1
- 101000998158 Homo sapiens NF-kappa-B inhibitor beta Proteins 0.000 description 1
- 101000621420 Homo sapiens Neural Wiskott-Aldrich syndrome protein Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101000962041 Homo sapiens Neurobeachin Proteins 0.000 description 1
- 101000603239 Homo sapiens Neuroligin-1 Proteins 0.000 description 1
- 101001128969 Homo sapiens Neuron navigator 1 Proteins 0.000 description 1
- 101000634196 Homo sapiens Neurotrophin-3 Proteins 0.000 description 1
- 101000979342 Homo sapiens Nuclear factor NF-kappa-B p105 subunit Proteins 0.000 description 1
- 101000896414 Homo sapiens Nuclear nucleic acid-binding protein C1D Proteins 0.000 description 1
- 101000602930 Homo sapiens Nuclear receptor coactivator 2 Proteins 0.000 description 1
- 101000836873 Homo sapiens Nucleotide exchange factor SIL1 Proteins 0.000 description 1
- 101001098357 Homo sapiens Orexin receptor type 2 Proteins 0.000 description 1
- 101000886818 Homo sapiens PDZ domain-containing protein GIPC1 Proteins 0.000 description 1
- 101000604540 Homo sapiens PRKCA-binding protein Proteins 0.000 description 1
- 101000735213 Homo sapiens Palladin Proteins 0.000 description 1
- 101000922137 Homo sapiens Peripheral plasma membrane protein CASK Proteins 0.000 description 1
- 101000619805 Homo sapiens Peroxiredoxin-5, mitochondrial Proteins 0.000 description 1
- 101000741788 Homo sapiens Peroxisome proliferator-activated receptor alpha Proteins 0.000 description 1
- 101001123325 Homo sapiens Peroxisome proliferator-activated receptor gamma coactivator 1-beta Proteins 0.000 description 1
- 101000620897 Homo sapiens Phosphatidylcholine transfer protein Proteins 0.000 description 1
- 101000741978 Homo sapiens Phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 2 protein Proteins 0.000 description 1
- 101000605630 Homo sapiens Phosphatidylinositol 3-kinase catalytic subunit type 3 Proteins 0.000 description 1
- 101000869517 Homo sapiens Phosphatidylinositol-3-phosphatase SAC1 Proteins 0.000 description 1
- 101000583474 Homo sapiens Phosphatidylinositol-binding clathrin assembly protein Proteins 0.000 description 1
- 101000730665 Homo sapiens Phospholipase D1 Proteins 0.000 description 1
- 101000609255 Homo sapiens Plasminogen activator inhibitor 1 Proteins 0.000 description 1
- 101001064779 Homo sapiens Plexin domain-containing protein 2 Proteins 0.000 description 1
- 101000620009 Homo sapiens Polyunsaturated fatty acid 5-lipoxygenase Proteins 0.000 description 1
- 101001094629 Homo sapiens Popeye domain-containing protein 2 Proteins 0.000 description 1
- 101000994632 Homo sapiens Potassium voltage-gated channel subfamily A member 2 Proteins 0.000 description 1
- 101001135486 Homo sapiens Potassium voltage-gated channel subfamily D member 2 Proteins 0.000 description 1
- 101001043564 Homo sapiens Prolow-density lipoprotein receptor-related protein 1 Proteins 0.000 description 1
- 101001009547 Homo sapiens Prosaposin receptor GPR37 Proteins 0.000 description 1
- 101001051777 Homo sapiens Protein kinase C alpha type Proteins 0.000 description 1
- 101000942742 Homo sapiens Protein lin-7 homolog A Proteins 0.000 description 1
- 101000822339 Homo sapiens Protein transport protein Sec24D Proteins 0.000 description 1
- 101000878540 Homo sapiens Protein-tyrosine kinase 2-beta Proteins 0.000 description 1
- 101000613391 Homo sapiens Protocadherin beta-16 Proteins 0.000 description 1
- 101000608230 Homo sapiens Pyrin domain-containing protein 2 Proteins 0.000 description 1
- 101001104083 Homo sapiens Rabphilin-3A Proteins 0.000 description 1
- 101000620559 Homo sapiens Ras-related protein Rab-3B Proteins 0.000 description 1
- 101000580043 Homo sapiens Ras-specific guanine nucleotide-releasing factor 2 Proteins 0.000 description 1
- 101000606548 Homo sapiens Receptor-type tyrosine-protein phosphatase gamma Proteins 0.000 description 1
- 101000584743 Homo sapiens Recombining binding protein suppressor of hairless Proteins 0.000 description 1
- 101001074548 Homo sapiens Regulating synaptic membrane exocytosis protein 2 Proteins 0.000 description 1
- 101000823237 Homo sapiens Reticulon-1 Proteins 0.000 description 1
- 101000602187 Homo sapiens Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit delta Proteins 0.000 description 1
- 101000581173 Homo sapiens Rho GTPase-activating protein 17 Proteins 0.000 description 1
- 101001091984 Homo sapiens Rho GTPase-activating protein 26 Proteins 0.000 description 1
- 101000733257 Homo sapiens Rho guanine nucleotide exchange factor 28 Proteins 0.000 description 1
- 101000669917 Homo sapiens Rho-associated protein kinase 1 Proteins 0.000 description 1
- 101000581128 Homo sapiens Rho-related GTP-binding protein RhoG Proteins 0.000 description 1
- 101000944909 Homo sapiens Ribosomal protein S6 kinase alpha-1 Proteins 0.000 description 1
- 101001051714 Homo sapiens Ribosomal protein S6 kinase beta-2 Proteins 0.000 description 1
- 101000650697 Homo sapiens Roundabout homolog 2 Proteins 0.000 description 1
- 101000963987 Homo sapiens SH3 domain-binding protein 5 Proteins 0.000 description 1
- 101000777293 Homo sapiens Serine/threonine-protein kinase Chk1 Proteins 0.000 description 1
- 101000576904 Homo sapiens Serine/threonine-protein kinase MRCK beta Proteins 0.000 description 1
- 101000809308 Homo sapiens Serine/threonine-protein kinase ULK4 Proteins 0.000 description 1
- 101000597662 Homo sapiens Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform Proteins 0.000 description 1
- 101000595252 Homo sapiens Serine/threonine-protein phosphatase PP1-alpha catalytic subunit Proteins 0.000 description 1
- 101000639970 Homo sapiens Sodium- and chloride-dependent GABA transporter 1 Proteins 0.000 description 1
- 101000629638 Homo sapiens Sorbin and SH3 domain-containing protein 2 Proteins 0.000 description 1
- 101000629597 Homo sapiens Sterol regulatory element-binding protein 1 Proteins 0.000 description 1
- 101000640315 Homo sapiens Synaptojanin-1 Proteins 0.000 description 1
- 101000626386 Homo sapiens Synaptotagmin-12 Proteins 0.000 description 1
- 101000658114 Homo sapiens Synaptotagmin-like protein 4 Proteins 0.000 description 1
- 101000617808 Homo sapiens Synphilin-1 Proteins 0.000 description 1
- 101000585070 Homo sapiens Syntaxin-1A Proteins 0.000 description 1
- 101000820490 Homo sapiens Syntaxin-binding protein 6 Proteins 0.000 description 1
- 101000595526 Homo sapiens T-box brain protein 1 Proteins 0.000 description 1
- 101000800055 Homo sapiens Testican-1 Proteins 0.000 description 1
- 101000659879 Homo sapiens Thrombospondin-1 Proteins 0.000 description 1
- 101000633605 Homo sapiens Thrombospondin-2 Proteins 0.000 description 1
- 101000837626 Homo sapiens Thyroid hormone receptor alpha Proteins 0.000 description 1
- 101000819111 Homo sapiens Trans-acting T-cell-specific transcription factor GATA-3 Proteins 0.000 description 1
- 101000635938 Homo sapiens Transforming growth factor beta-1 proprotein Proteins 0.000 description 1
- 101000649014 Homo sapiens Triple functional domain protein Proteins 0.000 description 1
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 1
- 101001026790 Homo sapiens Tyrosine-protein kinase Fes/Fps Proteins 0.000 description 1
- 101000820294 Homo sapiens Tyrosine-protein kinase Yes Proteins 0.000 description 1
- 101001087394 Homo sapiens Tyrosine-protein phosphatase non-receptor type 1 Proteins 0.000 description 1
- 101000807344 Homo sapiens Ubiquitin-conjugating enzyme E2 A Proteins 0.000 description 1
- 101000638886 Homo sapiens Urokinase-type plasminogen activator Proteins 0.000 description 1
- 101000775709 Homo sapiens V-type proton ATPase subunit C 1 Proteins 0.000 description 1
- 101000617921 Homo sapiens VPS10 domain-containing receptor SorCS2 Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 101000650141 Homo sapiens WAS/WASL-interacting protein family member 1 Proteins 0.000 description 1
- 101000759453 Homo sapiens YY1-associated protein 1 Proteins 0.000 description 1
- 101000994496 Homo sapiens cAMP-dependent protein kinase catalytic subunit alpha Proteins 0.000 description 1
- ZRJBHWIHUMBLCN-SEQYCRGISA-N Huperzine A Natural products N1C(=O)C=CC2=C1C[C@H]1/C(=C/C)[C@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-SEQYCRGISA-N 0.000 description 1
- 102000003918 Hyaluronan Synthases Human genes 0.000 description 1
- 108090000320 Hyaluronan Synthases Proteins 0.000 description 1
- 102100028084 Hyaluronan and proteoglycan link protein 1 Human genes 0.000 description 1
- 102100039283 Hyaluronidase-1 Human genes 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 1
- 229920001479 Hydroxyethyl methyl cellulose Polymers 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 102100020755 Hypoxia up-regulated protein 1 Human genes 0.000 description 1
- 108091058560 IL8 Proteins 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- ALOBUEHUHMBRLE-UHFFFAOYSA-N Ibutilide Chemical compound CCCCCCCN(CC)CCCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ALOBUEHUHMBRLE-UHFFFAOYSA-N 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 102100025891 Inosine-5'-monophosphate dehydrogenase 2 Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102100025323 Integrin alpha-1 Human genes 0.000 description 1
- 102100025304 Integrin beta-1 Human genes 0.000 description 1
- 102100036981 Interferon regulatory factor 1 Human genes 0.000 description 1
- 102100022706 Interleukin-20 receptor subunit alpha Human genes 0.000 description 1
- 102100037795 Interleukin-6 receptor subunit beta Human genes 0.000 description 1
- 102000004890 Interleukin-8 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- SFBODOKJTYAUCM-UHFFFAOYSA-N Ipriflavone Chemical compound C=1C(OC(C)C)=CC=C(C2=O)C=1OC=C2C1=CC=CC=C1 SFBODOKJTYAUCM-UHFFFAOYSA-N 0.000 description 1
- 102000017791 KCNJ12 Human genes 0.000 description 1
- 101150037139 KCNJ12 gene Proteins 0.000 description 1
- ZCVMWBYGMWKGHF-UHFFFAOYSA-N Ketotifene Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2CC(=O)C2=C1C=CS2 ZCVMWBYGMWKGHF-UHFFFAOYSA-N 0.000 description 1
- 102100034751 Kinectin Human genes 0.000 description 1
- 238000012313 Kruskal-Wallis test Methods 0.000 description 1
- 102100033173 Kv channel-interacting protein 1 Human genes 0.000 description 1
- 102100036091 Kynureninase Human genes 0.000 description 1
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 description 1
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 description 1
- WXFIGDLSSYIKKV-RCOVLWMOSA-N L-Metaraminol Chemical compound C[C@H](N)[C@H](O)C1=CC=CC(O)=C1 WXFIGDLSSYIKKV-RCOVLWMOSA-N 0.000 description 1
- WZNJWVWKTVETCG-YFKPBYRVSA-N L-mimosine Chemical compound OC(=O)[C@@H](N)CN1C=CC(=O)C(O)=C1 WZNJWVWKTVETCG-YFKPBYRVSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 102100027017 Latent-transforming growth factor beta-binding protein 2 Human genes 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 102100030653 Lipase member K Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 102100031961 Liprin-beta-1 Human genes 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 1
- 102100021922 Low-density lipoprotein receptor-related protein 2 Human genes 0.000 description 1
- 102100040705 Low-density lipoprotein receptor-related protein 8 Human genes 0.000 description 1
- 101000964266 Loxosceles laeta Dermonecrotic toxin Proteins 0.000 description 1
- 101710097496 Lysophospholipid acyltransferase Proteins 0.000 description 1
- 102000042189 MAGUK family Human genes 0.000 description 1
- 108091077533 MAGUK family Proteins 0.000 description 1
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 1
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 1
- 231100000002 MTT assay Toxicity 0.000 description 1
- 238000000134 MTT assay Methods 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101150003567 Mapk12 gene Proteins 0.000 description 1
- 102100025134 Mastermind-like protein 3 Human genes 0.000 description 1
- YJPIGAIKUZMOQA-UHFFFAOYSA-N Melatonin Natural products COC1=CC=C2N(C(C)=O)C=C(CCN)C2=C1 YJPIGAIKUZMOQA-UHFFFAOYSA-N 0.000 description 1
- ZRVUJXDFFKFLMG-UHFFFAOYSA-N Meloxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=NC=C(C)S1 ZRVUJXDFFKFLMG-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102100038352 Metabotropic glutamate receptor 3 Human genes 0.000 description 1
- 102100026262 Metalloproteinase inhibitor 2 Human genes 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 108090000192 Methionyl aminopeptidases Proteins 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- DUGOZIWVEXMGBE-UHFFFAOYSA-N Methylphenidate Chemical compound C=1C=CC=CC=1C(C(=O)OC)C1CCCCN1 DUGOZIWVEXMGBE-UHFFFAOYSA-N 0.000 description 1
- NZXKDOXHBHYTKP-UHFFFAOYSA-N Metohexital Chemical compound CCC#CC(C)C1(CC=C)C(=O)NC(=O)N(C)C1=O NZXKDOXHBHYTKP-UHFFFAOYSA-N 0.000 description 1
- 102000009664 Microtubule-Associated Proteins Human genes 0.000 description 1
- 108010020004 Microtubule-Associated Proteins Proteins 0.000 description 1
- 102100021118 Microtubule-associated protein 2 Human genes 0.000 description 1
- 102100030129 Mitochondrial 2-oxodicarboxylate carrier Human genes 0.000 description 1
- 108010009513 Mitochondrial Aldehyde Dehydrogenase Proteins 0.000 description 1
- 102000056243 Mitogen-activated protein kinase 12 Human genes 0.000 description 1
- 108700015929 Mitogen-activated protein kinase 12 Proteins 0.000 description 1
- WMSYWJSZGVOIJW-ONUALHDOSA-L Mivacurium chloride Chemical compound [Cl-].[Cl-].C([C@@H]1C2=CC(OC)=C(OC)C=C2CC[N+]1(C)CCCOC(=O)CC/C=C/CCC(=O)OCCC[N+]1(CCC=2C=C(C(=CC=2[C@H]1CC=1C=C(OC)C(OC)=C(OC)C=1)OC)OC)C)C1=CC(OC)=C(OC)C(OC)=C1 WMSYWJSZGVOIJW-ONUALHDOSA-L 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 102100027869 Moesin Human genes 0.000 description 1
- 102100035971 Molybdopterin molybdenumtransferase Human genes 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 102100025748 Mothers against decapentaplegic homolog 3 Human genes 0.000 description 1
- 101710143111 Mothers against decapentaplegic homolog 3 Proteins 0.000 description 1
- 101150097381 Mtor gene Proteins 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 101000836750 Mus musculus E3 ubiquitin-protein ligase SIAH1A Proteins 0.000 description 1
- 101100298534 Mus musculus Prnp gene Proteins 0.000 description 1
- 101600076428 Mus musculus WW domain-containing oxidoreductase (isoform 1) Proteins 0.000 description 1
- 102000014415 Muscarinic acetylcholine receptor Human genes 0.000 description 1
- 108050003473 Muscarinic acetylcholine receptor Proteins 0.000 description 1
- 102100021970 Myc box-dependent-interacting protein 1 Human genes 0.000 description 1
- 101001033610 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Inosine-5'-monophosphate dehydrogenase Proteins 0.000 description 1
- IDBPHNDTYPBSNI-UHFFFAOYSA-N N-(1-(2-(4-Ethyl-5-oxo-2-tetrazolin-1-yl)ethyl)-4-(methoxymethyl)-4-piperidyl)propionanilide Chemical compound C1CN(CCN2C(N(CC)N=N2)=O)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 IDBPHNDTYPBSNI-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 102100036710 N-acetylglucosamine-1-phosphotransferase subunits alpha/beta Human genes 0.000 description 1
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 1
- 108010002998 NADPH Oxidases Proteins 0.000 description 1
- 102000004722 NADPH Oxidases Human genes 0.000 description 1
- 102100033457 NF-kappa-B inhibitor beta Human genes 0.000 description 1
- 102100029447 Na(+)/H(+) exchange regulatory cofactor NHE-RF1 Human genes 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 108010032605 Nerve Growth Factor Receptors Proteins 0.000 description 1
- 102000007339 Nerve Growth Factor Receptors Human genes 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- 108010030865 Netrin Receptors Proteins 0.000 description 1
- 102000005951 Netrin Receptors Human genes 0.000 description 1
- 102100021153 Netrin receptor DCC Human genes 0.000 description 1
- 102100029514 Netrin receptor UNC5C Human genes 0.000 description 1
- 102100023031 Neural Wiskott-Aldrich syndrome protein Human genes 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 102000014413 Neuregulin Human genes 0.000 description 1
- 108050003475 Neuregulin Proteins 0.000 description 1
- 102000048238 Neuregulin-1 Human genes 0.000 description 1
- 108090000556 Neuregulin-1 Proteins 0.000 description 1
- 102100039234 Neurobeachin Human genes 0.000 description 1
- 102100038992 Neuroligin-1 Human genes 0.000 description 1
- 102100031225 Neuron navigator 1 Human genes 0.000 description 1
- 101100426589 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) trp-3 gene Proteins 0.000 description 1
- 102100029268 Neurotrophin-3 Human genes 0.000 description 1
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 1
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 1
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 1
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 1
- JZFPYUNJRRFVQU-UHFFFAOYSA-N Niflumic acid Chemical compound OC(=O)C1=CC=CN=C1NC1=CC=CC(C(F)(F)F)=C1 JZFPYUNJRRFVQU-UHFFFAOYSA-N 0.000 description 1
- FAIIFDPAEUKBEP-UHFFFAOYSA-N Nilvadipine Chemical compound COC(=O)C1=C(C#N)NC(C)=C(C(=O)OC(C)C)C1C1=CC=CC([N+]([O-])=O)=C1 FAIIFDPAEUKBEP-UHFFFAOYSA-N 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- 102100023050 Nuclear factor NF-kappa-B p105 subunit Human genes 0.000 description 1
- 102100021713 Nuclear nucleic acid-binding protein C1D Human genes 0.000 description 1
- 102100038494 Nuclear receptor subfamily 1 group I member 2 Human genes 0.000 description 1
- 102100027096 Nucleotide exchange factor SIL1 Human genes 0.000 description 1
- HDVDLQFPDLTOSI-UHFFFAOYSA-L O[AlH]O Chemical compound O[AlH]O HDVDLQFPDLTOSI-UHFFFAOYSA-L 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102100037588 Orexin receptor type 2 Human genes 0.000 description 1
- 101000840556 Oryza sativa subsp. japonica Hexokinase-4, chloroplastic Proteins 0.000 description 1
- 108010081689 Osteopontin Proteins 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- DUDKAZCAISNGQN-UHFFFAOYSA-N Oxyphencyclimine Chemical compound CN1CCCN=C1COC(=O)C(O)(C=1C=CC=CC=1)C1CCCCC1 DUDKAZCAISNGQN-UHFFFAOYSA-N 0.000 description 1
- 102100037600 P2Y purinoceptor 1 Human genes 0.000 description 1
- 101700056750 PAK1 Proteins 0.000 description 1
- 102100039983 PDZ domain-containing protein GIPC1 Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 102100038730 PRKCA-binding protein Human genes 0.000 description 1
- 101150073266 PRKCD gene Proteins 0.000 description 1
- 102100035031 Palladin Human genes 0.000 description 1
- 240000004371 Panax ginseng Species 0.000 description 1
- 235000002789 Panax ginseng Nutrition 0.000 description 1
- BYPFEZZEUUWMEJ-UHFFFAOYSA-N Pentoxifylline Chemical compound O=C1N(CCCCC(=O)C)C(=O)N(C)C2=C1N(C)C=N2 BYPFEZZEUUWMEJ-UHFFFAOYSA-N 0.000 description 1
- 102100027913 Peptidyl-prolyl cis-trans isomerase FKBP1A Human genes 0.000 description 1
- 102100031166 Peripheral plasma membrane protein CASK Human genes 0.000 description 1
- 102100022078 Peroxiredoxin-5, mitochondrial Human genes 0.000 description 1
- 102100038831 Peroxisome proliferator-activated receptor alpha Human genes 0.000 description 1
- 102100028961 Peroxisome proliferator-activated receptor gamma coactivator 1-beta Human genes 0.000 description 1
- 102100022906 Phosphatidylcholine transfer protein Human genes 0.000 description 1
- 102100038633 Phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 2 protein Human genes 0.000 description 1
- 102100038329 Phosphatidylinositol 3-kinase catalytic subunit type 3 Human genes 0.000 description 1
- 102100032286 Phosphatidylinositol-3-phosphatase SAC1 Human genes 0.000 description 1
- 102100031014 Phosphatidylinositol-binding clathrin assembly protein Human genes 0.000 description 1
- 102100026481 Phosphoinositide 3-kinase regulatory subunit 4 Human genes 0.000 description 1
- 102100038331 Phospholipase A1 member A Human genes 0.000 description 1
- 102100032967 Phospholipase D1 Human genes 0.000 description 1
- 102100033616 Phospholipid-transporting ATPase ABCA1 Human genes 0.000 description 1
- OWWLUIWOFHMHOQ-XGHATYIMSA-N Pipecuronium Chemical compound N1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)N2CC[N+](C)(C)CC2)CC[N+](C)(C)CC1 OWWLUIWOFHMHOQ-XGHATYIMSA-N 0.000 description 1
- 108010003044 Placental Lactogen Proteins 0.000 description 1
- 239000000381 Placental Lactogen Substances 0.000 description 1
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 1
- 102100037596 Platelet-derived growth factor subunit A Human genes 0.000 description 1
- 102100040990 Platelet-derived growth factor subunit B Human genes 0.000 description 1
- 102100039277 Pleiotrophin Human genes 0.000 description 1
- 102100031889 Plexin domain-containing protein 2 Human genes 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 102100022364 Polyunsaturated fatty acid 5-lipoxygenase Human genes 0.000 description 1
- 102100034369 Potassium voltage-gated channel subfamily A member 2 Human genes 0.000 description 1
- 102100033170 Potassium voltage-gated channel subfamily D member 2 Human genes 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 108010001511 Pregnane X Receptor Proteins 0.000 description 1
- 101710130420 Probable capsid assembly scaffolding protein Proteins 0.000 description 1
- 102100024819 Prolactin Human genes 0.000 description 1
- 108010057464 Prolactin Proteins 0.000 description 1
- KNAHARQHSZJURB-UHFFFAOYSA-N Propylthiouracile Chemical compound CCCC1=CC(=O)NC(=S)N1 KNAHARQHSZJURB-UHFFFAOYSA-N 0.000 description 1
- 102100030284 Prosaposin receptor GPR37 Human genes 0.000 description 1
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108010038241 Protein Inhibitors of Activated STAT Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- 102100037340 Protein kinase C delta type Human genes 0.000 description 1
- 102100032928 Protein lin-7 homolog A Human genes 0.000 description 1
- 102100022542 Protein transport protein Sec24D Human genes 0.000 description 1
- 102100027897 Protein unc-13 homolog B Human genes 0.000 description 1
- 101710141054 Protein unc-13 homolog B Proteins 0.000 description 1
- 102100037787 Protein-tyrosine kinase 2-beta Human genes 0.000 description 1
- 101000947436 Psychotria longipes Cyclopsychotride-A Proteins 0.000 description 1
- 102000015799 Qa-SNARE Proteins Human genes 0.000 description 1
- 108010010469 Qa-SNARE Proteins Proteins 0.000 description 1
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 1
- 101150058540 RAC1 gene Proteins 0.000 description 1
- 101150020518 RHEB gene Proteins 0.000 description 1
- 102100022851 Rab5 GDP/GTP exchange factor Human genes 0.000 description 1
- 102100040040 Rabphilin-3A Human genes 0.000 description 1
- 102100022306 Ras-related protein Rab-3B Human genes 0.000 description 1
- 102100027555 Ras-specific guanine nucleotide-releasing factor 2 Human genes 0.000 description 1
- 102100025234 Receptor of activated protein C kinase 1 Human genes 0.000 description 1
- 102100039661 Receptor-type tyrosine-protein phosphatase gamma Human genes 0.000 description 1
- 108010044157 Receptors for Activated C Kinase Proteins 0.000 description 1
- 102100030000 Recombining binding protein suppressor of hairless Human genes 0.000 description 1
- 102000043322 Reelin Human genes 0.000 description 1
- 108700038365 Reelin Proteins 0.000 description 1
- 102100036266 Regulating synaptic membrane exocytosis protein 2 Human genes 0.000 description 1
- 101150057388 Reln gene Proteins 0.000 description 1
- 101710203837 Replication-associated protein Proteins 0.000 description 1
- QNVSXXGDAPORNA-UHFFFAOYSA-N Resveratrol Natural products OC1=CC=CC(C=CC=2C=C(O)C(O)=CC=2)=C1 QNVSXXGDAPORNA-UHFFFAOYSA-N 0.000 description 1
- 102100022647 Reticulon-1 Human genes 0.000 description 1
- 102100037593 Retinal rod rhodopsin-sensitive cGMP 3',5'-cyclic phosphodiesterase subunit delta Human genes 0.000 description 1
- 102100027656 Rho GTPase-activating protein 17 Human genes 0.000 description 1
- 102100035744 Rho GTPase-activating protein 26 Human genes 0.000 description 1
- 102100033204 Rho guanine nucleotide exchange factor 28 Human genes 0.000 description 1
- 102100039313 Rho-associated protein kinase 1 Human genes 0.000 description 1
- 102100027605 Rho-related GTP-binding protein RhoG Human genes 0.000 description 1
- 102100033536 Ribosomal protein S6 kinase alpha-1 Human genes 0.000 description 1
- 102100024917 Ribosomal protein S6 kinase beta-2 Human genes 0.000 description 1
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 description 1
- 102100027739 Roundabout homolog 2 Human genes 0.000 description 1
- 229930001406 Ryanodine Natural products 0.000 description 1
- 108010064582 SAP90-PSD95 Associated Proteins Proteins 0.000 description 1
- 102000015266 SAP90-PSD95 Associated Proteins Human genes 0.000 description 1
- 101150105578 SAPK3 gene Proteins 0.000 description 1
- 102100040119 SH3 domain-binding protein 5 Human genes 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 102000012980 SLC1A2 Human genes 0.000 description 1
- 108091006427 SLC25A21 Proteins 0.000 description 1
- 108091006253 SLC8A1 Proteins 0.000 description 1
- 108091006647 SLC9A1 Proteins 0.000 description 1
- 102100037375 SLIT-ROBO Rho GTPase-activating protein 3 Human genes 0.000 description 1
- 101150083405 SRGAP3 gene Proteins 0.000 description 1
- 108010017324 STAT3 Transcription Factor Proteins 0.000 description 1
- 101150112492 SUM-1 gene Proteins 0.000 description 1
- 101150096255 SUMO1 gene Proteins 0.000 description 1
- 240000007164 Salvia officinalis Species 0.000 description 1
- 235000002912 Salvia officinalis Nutrition 0.000 description 1
- 101710109118 Sarcoplasmic/endoplasmic reticulum calcium ATPase 3 Proteins 0.000 description 1
- GMBQZIIUCVWOCD-WWASVFFGSA-N Sarsapogenine Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)CC[C@H](O)C[C@H]4CC[C@H]3[C@@H]2C1)C)[C@@H]1C)[C@]11CC[C@H](C)CO1 GMBQZIIUCVWOCD-WWASVFFGSA-N 0.000 description 1
- 101710204410 Scaffold protein Proteins 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 102000014105 Semaphorin Human genes 0.000 description 1
- 108050003978 Semaphorin Proteins 0.000 description 1
- 102100027974 Semaphorin-3A Human genes 0.000 description 1
- 108010090319 Semaphorin-3A Proteins 0.000 description 1
- 206010039966 Senile dementia Diseases 0.000 description 1
- 102100031081 Serine/threonine-protein kinase Chk1 Human genes 0.000 description 1
- 102100025347 Serine/threonine-protein kinase MRCK beta Human genes 0.000 description 1
- 102100027910 Serine/threonine-protein kinase PAK 1 Human genes 0.000 description 1
- 102100038455 Serine/threonine-protein kinase ULK4 Human genes 0.000 description 1
- 102100035348 Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform Human genes 0.000 description 1
- 102100036033 Serine/threonine-protein phosphatase PP1-alpha catalytic subunit Human genes 0.000 description 1
- 108010090763 Shiga Toxin 2 Proteins 0.000 description 1
- ZRJBHWIHUMBLCN-UHFFFAOYSA-N Shuangyiping Natural products N1C(=O)C=CC2=C1CC1C(=CC)C2(N)CC(C)=C1 ZRJBHWIHUMBLCN-UHFFFAOYSA-N 0.000 description 1
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 1
- 108010052164 Sodium Channels Proteins 0.000 description 1
- 102000018674 Sodium Channels Human genes 0.000 description 1
- 102100028910 Sodium channel protein type 1 subunit alpha Human genes 0.000 description 1
- 102100033927 Sodium- and chloride-dependent GABA transporter 1 Human genes 0.000 description 1
- 102100035088 Sodium/calcium exchanger 1 Human genes 0.000 description 1
- 102100030980 Sodium/hydrogen exchanger 1 Human genes 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 102100026901 Sorbin and SH3 domain-containing protein 2 Human genes 0.000 description 1
- 208000014604 Specific Language disease Diseases 0.000 description 1
- 101710168942 Sphingosine-1-phosphate phosphatase 1 Proteins 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 102100026839 Sterol regulatory element-binding protein 1 Human genes 0.000 description 1
- 101000880156 Streptomyces cacaoi Subtilisin inhibitor-like protein 1 Proteins 0.000 description 1
- 101000745670 Streptomyces venezuelae (strain ATCC 10712 / CBS 650.69 / DSM 40230 / JCM 4526 / NBRC 13096 / PD 04745) Chloramphenicol 3-O phosphotransferase Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 102100033916 Synaptojanin-1 Human genes 0.000 description 1
- 108010057722 Synaptosomal-Associated Protein 25 Proteins 0.000 description 1
- 102100030552 Synaptosomal-associated protein 25 Human genes 0.000 description 1
- 102100024619 Synaptotagmin-12 Human genes 0.000 description 1
- 102100035002 Synaptotagmin-like protein 4 Human genes 0.000 description 1
- 102100021997 Synphilin-1 Human genes 0.000 description 1
- 102100029932 Syntaxin-1A Human genes 0.000 description 1
- 102100035936 Syntaxin-2 Human genes 0.000 description 1
- 102100021681 Syntaxin-binding protein 6 Human genes 0.000 description 1
- 102000002154 T-Lymphoma Invasion and Metastasis-inducing Protein 1 Human genes 0.000 description 1
- 108010001288 T-Lymphoma Invasion and Metastasis-inducing Protein 1 Proteins 0.000 description 1
- 102100036083 T-box brain protein 1 Human genes 0.000 description 1
- 102100033456 TGF-beta receptor type-1 Human genes 0.000 description 1
- 102000003629 TRPC3 Human genes 0.000 description 1
- 108010006877 Tacrolimus Binding Protein 1A Proteins 0.000 description 1
- SEQDDYPDSLOBDC-UHFFFAOYSA-N Temazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 SEQDDYPDSLOBDC-UHFFFAOYSA-N 0.000 description 1
- 108010049264 Teriparatide Proteins 0.000 description 1
- 102100033390 Testican-1 Human genes 0.000 description 1
- IUJDSEJGGMCXSG-UHFFFAOYSA-N Thiopental Chemical compound CCCC(C)C1(CC)C(=O)NC(=S)NC1=O IUJDSEJGGMCXSG-UHFFFAOYSA-N 0.000 description 1
- 102100036034 Thrombospondin-1 Human genes 0.000 description 1
- 102100029529 Thrombospondin-2 Human genes 0.000 description 1
- 102100028702 Thyroid hormone receptor alpha Human genes 0.000 description 1
- DKJJVAGXPKPDRL-UHFFFAOYSA-N Tiludronic acid Chemical compound OP(O)(=O)C(P(O)(O)=O)SC1=CC=C(Cl)C=C1 DKJJVAGXPKPDRL-UHFFFAOYSA-N 0.000 description 1
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 1
- 102100021386 Trans-acting T-cell-specific transcription factor GATA-3 Human genes 0.000 description 1
- LUKBXSAWLPMMSZ-OWOJBTEDSA-N Trans-resveratrol Chemical compound C1=CC(O)=CC=C1\C=C\C1=CC(O)=CC(O)=C1 LUKBXSAWLPMMSZ-OWOJBTEDSA-N 0.000 description 1
- 108010011702 Transforming Growth Factor-beta Type I Receptor Proteins 0.000 description 1
- 102100030742 Transforming growth factor beta-1 proprotein Human genes 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- 102100028101 Triple functional domain protein Human genes 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 101150037542 Trpc3 gene Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 1
- 102100027881 Tumor protein 63 Human genes 0.000 description 1
- 101710140697 Tumor protein 63 Proteins 0.000 description 1
- 102100037333 Tyrosine-protein kinase Fes/Fps Human genes 0.000 description 1
- 102100021788 Tyrosine-protein kinase Yes Human genes 0.000 description 1
- 102100033001 Tyrosine-protein phosphatase non-receptor type 1 Human genes 0.000 description 1
- 101150032479 UNC-5 gene Proteins 0.000 description 1
- 102100037261 Ubiquitin-conjugating enzyme E2 A Human genes 0.000 description 1
- 208000003443 Unconsciousness Diseases 0.000 description 1
- 102100031834 Unconventional myosin-VI Human genes 0.000 description 1
- 102100031358 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 102100032189 V-type proton ATPase subunit C 1 Human genes 0.000 description 1
- 102100021938 VPS10 domain-containing receptor SorCS2 Human genes 0.000 description 1
- 108010062352 Vacuolar Sorting Protein VPS15 Proteins 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 208000032594 Vascular Remodeling Diseases 0.000 description 1
- 201000004810 Vascular dementia Diseases 0.000 description 1
- 102100039037 Vascular endothelial growth factor A Human genes 0.000 description 1
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 1
- 102000003786 Vesicle-associated membrane protein 2 Human genes 0.000 description 1
- 108090000169 Vesicle-associated membrane protein 2 Proteins 0.000 description 1
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 102100027538 WAS/WASL-interacting protein family member 1 Human genes 0.000 description 1
- 102100023267 YY1-associated protein 1 Human genes 0.000 description 1
- JKQXZKUSFCKOGQ-LQFQNGICSA-N Z-zeaxanthin Natural products C([C@H](O)CC=1C)C(C)(C)C=1C=CC(C)=CC=CC(C)=CC=CC=C(C)C=CC=C(C)C=CC1=C(C)C[C@@H](O)CC1(C)C JKQXZKUSFCKOGQ-LQFQNGICSA-N 0.000 description 1
- QOPRSMDTRDMBNK-RNUUUQFGSA-N Zeaxanthin Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCC(O)C1(C)C)C=CC=C(/C)C=CC2=C(C)CC(O)CC2(C)C QOPRSMDTRDMBNK-RNUUUQFGSA-N 0.000 description 1
- OGQICQVSFDPSEI-UHFFFAOYSA-N Zorac Chemical compound N1=CC(C(=O)OCC)=CC=C1C#CC1=CC=C(SCCC2(C)C)C2=C1 OGQICQVSFDPSEI-UHFFFAOYSA-N 0.000 description 1
- GYKFWCDBQAFCLJ-RTWAWAEBSA-N [(2s,3s)-8-chloro-5-[2-(dimethylamino)ethyl]-2-(4-methoxyphenyl)-4-oxo-2,3-dihydro-1,5-benzothiazepin-3-yl] acetate Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=C(Cl)C=C2S1 GYKFWCDBQAFCLJ-RTWAWAEBSA-N 0.000 description 1
- PBHFNBQPZCRWQP-QUCCMNQESA-N [(3ar,8bs)-3,4,8b-trimethyl-2,3a-dihydro-1h-pyrrolo[2,3-b]indol-7-yl] n-phenylcarbamate Chemical compound CN([C@@H]1[C@@](C2=C3)(C)CCN1C)C2=CC=C3OC(=O)NC1=CC=CC=C1 PBHFNBQPZCRWQP-QUCCMNQESA-N 0.000 description 1
- JNWFIPVDEINBAI-UHFFFAOYSA-N [5-hydroxy-4-[4-(1-methylindol-5-yl)-5-oxo-1H-1,2,4-triazol-3-yl]-2-propan-2-ylphenyl] dihydrogen phosphate Chemical compound C1=C(OP(O)(O)=O)C(C(C)C)=CC(C=2N(C(=O)NN=2)C=2C=C3C=CN(C)C3=CC=2)=C1O JNWFIPVDEINBAI-UHFFFAOYSA-N 0.000 description 1
- ZUPXXZAVUHFCNV-UHFFFAOYSA-N [[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [5-(3-carbamoyl-4h-pyridin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl hydrogen phosphate;potassium Chemical compound [K].C1=CCC(C(=O)N)=CN1C1C(O)C(O)C(COP(O)(=O)OP(O)(=O)OCC2C(C(O)C(O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ZUPXXZAVUHFCNV-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000008649 adaptation response Effects 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940062527 alendronate Drugs 0.000 description 1
- 229960001391 alfentanil Drugs 0.000 description 1
- 229960004601 aliskiren Drugs 0.000 description 1
- JAZBEHYOTPTENJ-JLNKQSITSA-N all-cis-5,8,11,14,17-icosapentaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O JAZBEHYOTPTENJ-JLNKQSITSA-N 0.000 description 1
- JKQXZKUSFCKOGQ-LOFNIBRQSA-N all-trans-Zeaxanthin Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2=C(C)CC(O)CC2(C)C JKQXZKUSFCKOGQ-LOFNIBRQSA-N 0.000 description 1
- 229960005039 almitrine Drugs 0.000 description 1
- OBDOVFRMEYHSQB-UHFFFAOYSA-N almitrine Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)N1CCN(C=2N=C(NCC=C)N=C(NCC=C)N=2)CC1 OBDOVFRMEYHSQB-UHFFFAOYSA-N 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- OUJTZYPIHDYQMC-LJQANCHMSA-N ambrisentan Chemical compound O([C@@H](C(OC)(C=1C=CC=CC=1)C=1C=CC=CC=1)C(O)=O)C1=NC(C)=CC(C)=N1 OUJTZYPIHDYQMC-LJQANCHMSA-N 0.000 description 1
- 229960002414 ambrisentan Drugs 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960003731 amlexanox Drugs 0.000 description 1
- SGRYPYWGNKJSDL-UHFFFAOYSA-N amlexanox Chemical compound NC1=C(C(O)=O)C=C2C(=O)C3=CC(C(C)C)=CC=C3OC2=N1 SGRYPYWGNKJSDL-UHFFFAOYSA-N 0.000 description 1
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 1
- 229960003942 amphotericin b Drugs 0.000 description 1
- 230000006933 amyloid-beta aggregation Effects 0.000 description 1
- 229960001694 anagrelide Drugs 0.000 description 1
- OTBXOEAOVRKTNQ-UHFFFAOYSA-N anagrelide Chemical compound N1=C2NC(=O)CN2CC2=C(Cl)C(Cl)=CC=C21 OTBXOEAOVRKTNQ-UHFFFAOYSA-N 0.000 description 1
- 230000019552 anatomical structure morphogenesis Effects 0.000 description 1
- 230000006481 angiogenic pathway Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000181 anti-adherent effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 201000007201 aphasia Diseases 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- ATALOFNDEOCMKK-OITMNORJSA-N aprepitant Chemical compound O([C@@H]([C@@H]1C=2C=CC(F)=CC=2)O[C@H](C)C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)CCN1CC1=NNC(=O)N1 ATALOFNDEOCMKK-OITMNORJSA-N 0.000 description 1
- 229960001372 aprepitant Drugs 0.000 description 1
- 239000013011 aqueous formulation Substances 0.000 description 1
- 229950007556 aranidipine Drugs 0.000 description 1
- 229940090880 ardeparin Drugs 0.000 description 1
- 229960004372 aripiprazole Drugs 0.000 description 1
- 229960002430 atomoxetine Drugs 0.000 description 1
- VHGCDTVCOLNTBX-QGZVFWFLSA-N atomoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C VHGCDTVCOLNTBX-QGZVFWFLSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 229960001862 atracurium Drugs 0.000 description 1
- AUJRCFUBUPVWSZ-XTZHGVARSA-M auranofin Chemical compound CCP(CC)(CC)=[Au]S[C@@H]1O[C@H](COC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O AUJRCFUBUPVWSZ-XTZHGVARSA-M 0.000 description 1
- 229960005207 auranofin Drugs 0.000 description 1
- 230000004900 autophagic degradation Effects 0.000 description 1
- XNEFYCZVKIDDMS-UHFFFAOYSA-N avobenzone Chemical compound C1=CC(OC)=CC=C1C(=O)CC(=O)C1=CC=C(C(C)(C)C)C=C1 XNEFYCZVKIDDMS-UHFFFAOYSA-N 0.000 description 1
- 229960005193 avobenzone Drugs 0.000 description 1
- YEESUBCSWGVPCE-UHFFFAOYSA-N azanylidyneoxidanium iron(2+) pentacyanide Chemical compound [Fe++].[C-]#N.[C-]#N.[C-]#N.[C-]#N.[C-]#N.N#[O+] YEESUBCSWGVPCE-UHFFFAOYSA-N 0.000 description 1
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 1
- 229960003644 aztreonam Drugs 0.000 description 1
- 229960003071 bacitracin Drugs 0.000 description 1
- 229930184125 bacitracin Natural products 0.000 description 1
- CLKOFPXJLQSYAH-ABRJDSQDSA-N bacitracin A Chemical compound C1SC([C@@H](N)[C@@H](C)CC)=N[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(=O)N[C@H](CCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2N=CNC=2)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCCCC1 CLKOFPXJLQSYAH-ABRJDSQDSA-N 0.000 description 1
- IPOKCKJONYRRHP-FMQUCBEESA-N balsalazide Chemical compound C1=CC(C(=O)NCCC(=O)O)=CC=C1\N=N\C1=CC=C(O)C(C(O)=O)=C1 IPOKCKJONYRRHP-FMQUCBEESA-N 0.000 description 1
- 229960004168 balsalazide Drugs 0.000 description 1
- 229950001863 bapineuzumab Drugs 0.000 description 1
- HYNPZTKLUNHGPM-KKERQHFVSA-N becaplermin Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](Cc2cnc[nH]2)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N3CCC[C@H]3C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)[C@@H]4CCCN4C(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](C(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](C(C)C)NC(=O)[C@@H]5CCCN5C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H]6CCCN6C(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CS)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](CS)NC(=O)[C@H](CS)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CS)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@@H]7CCCN7C(=O)[C@H](Cc8c[nH]c9c8cccc9)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H](C)NC(=O)[C@H](CC(=O)N)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H](CO)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCNC(=N)N)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CS)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCSC)NC(=O)[C@H](C)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CCC(=O)O)NC(=O)[C@H](C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)N HYNPZTKLUNHGPM-KKERQHFVSA-N 0.000 description 1
- 229960004787 becaplermin Drugs 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 229960003515 bendroflumethiazide Drugs 0.000 description 1
- HDWIHXWEUNVBIY-UHFFFAOYSA-N bendroflumethiazidum Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1=CC=CC=C1 HDWIHXWEUNVBIY-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- WHQCHUCQKNIQEC-UHFFFAOYSA-N benzbromarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(Br)=C(O)C(Br)=C1 WHQCHUCQKNIQEC-UHFFFAOYSA-N 0.000 description 1
- 229960002529 benzbromarone Drugs 0.000 description 1
- NDTSRXAMMQDVSW-UHFFFAOYSA-N benzthiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1N=C2CSCC1=CC=CC=C1 NDTSRXAMMQDVSW-UHFFFAOYSA-N 0.000 description 1
- 229960001541 benzthiazide Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- UIEATEWHFDRYRU-UHFFFAOYSA-N bepridil Chemical compound C1CCCN1C(COCC(C)C)CN(C=1C=CC=CC=1)CC1=CC=CC=C1 UIEATEWHFDRYRU-UHFFFAOYSA-N 0.000 description 1
- 229960003665 bepridil Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960004980 betanidine Drugs 0.000 description 1
- NIVZHWNOUVJHKV-UHFFFAOYSA-N bethanidine Chemical compound CN\C(=N/C)NCC1=CC=CC=C1 NIVZHWNOUVJHKV-UHFFFAOYSA-N 0.000 description 1
- 229950009087 bifeprunox Drugs 0.000 description 1
- 229960002206 bifonazole Drugs 0.000 description 1
- 229960002470 bimatoprost Drugs 0.000 description 1
- AQOKCDNYWBIDND-FTOWTWDKSA-N bimatoprost Chemical compound CCNC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)CCC1=CC=CC=C1 AQOKCDNYWBIDND-FTOWTWDKSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 229920013641 bioerodible polymer Polymers 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- FZGVEKPRDOIXJY-UHFFFAOYSA-N bitolterol Chemical compound C1=CC(C)=CC=C1C(=O)OC1=CC=C(C(O)CNC(C)(C)C)C=C1OC(=O)C1=CC=C(C)C=C1 FZGVEKPRDOIXJY-UHFFFAOYSA-N 0.000 description 1
- 229960004620 bitolterol Drugs 0.000 description 1
- OHJMTUPIZMNBFR-UHFFFAOYSA-N biuret Chemical compound NC(=O)NC(N)=O OHJMTUPIZMNBFR-UHFFFAOYSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 1
- 229960005520 bryostatin Drugs 0.000 description 1
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 1
- MUIWQCKLQMOUAT-AKUNNTHJSA-N bryostatin 20 Natural products COC(=O)C=C1C[C@@]2(C)C[C@]3(O)O[C@](C)(C[C@@H](O)CC(=O)O[C@](C)(C[C@@]4(C)O[C@](O)(CC5=CC(=O)O[C@]45C)C(C)(C)C=C[C@@](C)(C1)O2)[C@@H](C)O)C[C@H](OC(=O)C(C)(C)C)C3(C)C MUIWQCKLQMOUAT-AKUNNTHJSA-N 0.000 description 1
- MOYGZHXDRJNJEP-UHFFFAOYSA-N buclizine Chemical compound C1=CC(C(C)(C)C)=CC=C1CN1CCN(C(C=2C=CC=CC=2)C=2C=CC(Cl)=CC=2)CC1 MOYGZHXDRJNJEP-UHFFFAOYSA-N 0.000 description 1
- 229960001705 buclizine Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001736 buprenorphine Drugs 0.000 description 1
- 102220352928 c.35C>A Human genes 0.000 description 1
- 102100037093 cGMP-inhibited 3',5'-cyclic phosphodiesterase A Human genes 0.000 description 1
- 229960004596 cabergoline Drugs 0.000 description 1
- GVEZIHKRYBHEFX-UHFFFAOYSA-N caerulein A Natural products CC=CCC=CCCC(=O)C1OC1C(N)=O GVEZIHKRYBHEFX-UHFFFAOYSA-N 0.000 description 1
- JWUBBDSIWDLEOM-DTOXIADCSA-N calcidiol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)CCC1=C JWUBBDSIWDLEOM-DTOXIADCSA-N 0.000 description 1
- 229960004361 calcifediol Drugs 0.000 description 1
- 230000002308 calcification Effects 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229960005084 calcitriol Drugs 0.000 description 1
- GMRQFYUYWCNGIN-NKMMMXOESA-N calcitriol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](CCCC(C)(C)O)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C GMRQFYUYWCNGIN-NKMMMXOESA-N 0.000 description 1
- 235000020964 calcitriol Nutrition 0.000 description 1
- 239000011612 calcitriol Substances 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- ZTWZVMIYIIVABD-OEMFJLHTSA-N candoxatril Chemical compound C([C@@H](COCCOC)C(=O)OC=1C=C2CCCC2=CC=1)C1(C(=O)N[C@@H]2CC[C@@H](CC2)C(O)=O)CCCC1 ZTWZVMIYIIVABD-OEMFJLHTSA-N 0.000 description 1
- 229950004548 candoxatril Drugs 0.000 description 1
- 230000008777 canonical pathway Effects 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229960005296 carboprost tromethamine Drugs 0.000 description 1
- UMMADZJLZAPZAW-OVXHCKHTSA-N carboprost tromethamine Chemical compound OCC([NH3+])(CO)CO.CCCCC[C@](C)(O)\C=C\[C@H]1[C@@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC([O-])=O UMMADZJLZAPZAW-OVXHCKHTSA-N 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 108010051348 cdc42 GTP-Binding Protein Proteins 0.000 description 1
- CZTQZXZIADLWOZ-CRAIPNDOSA-N cefaloridine Chemical compound O=C([C@@H](NC(=O)CC=1SC=CC=1)[C@H]1SC2)N1C(C(=O)[O-])=C2C[N+]1=CC=CC=C1 CZTQZXZIADLWOZ-CRAIPNDOSA-N 0.000 description 1
- 229960004682 cefoperazone Drugs 0.000 description 1
- GCFBRXLSHGKWDP-XCGNWRKASA-N cefoperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC(O)=CC=1)C(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 GCFBRXLSHGKWDP-XCGNWRKASA-N 0.000 description 1
- 229960001668 cefuroxime Drugs 0.000 description 1
- JFPVXVDWJQMJEE-IZRZKJBUSA-N cefuroxime Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 JFPVXVDWJQMJEE-IZRZKJBUSA-N 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 210000005056 cell body Anatomy 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 230000007541 cellular toxicity Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920006217 cellulose acetate butyrate Polymers 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229940106164 cephalexin Drugs 0.000 description 1
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 description 1
- 210000003198 cerebellar cortex Anatomy 0.000 description 1
- 210000000782 cerebellar granule cell Anatomy 0.000 description 1
- 229950005984 cerulenin Drugs 0.000 description 1
- GVEZIHKRYBHEFX-NQQPLRFYSA-N cerulenin Chemical compound C\C=C\C\C=C\CCC(=O)[C@H]1O[C@H]1C(N)=O GVEZIHKRYBHEFX-NQQPLRFYSA-N 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- DDPFHDCZUJFNAT-PZPWKVFESA-N chembl2104402 Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CCCCCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 DDPFHDCZUJFNAT-PZPWKVFESA-N 0.000 description 1
- IWXUVYOOUMLUTQ-CZUORRHYSA-N chembl2179105 Chemical compound C([C@H]([C@@H](C1)C=2NC(=O)C=3C=NN(C=3N=2)C2CCOCC2)C)N1CC1=NC=CC=N1 IWXUVYOOUMLUTQ-CZUORRHYSA-N 0.000 description 1
- 238000002144 chemical decomposition reaction Methods 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 229960002155 chlorothiazide Drugs 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- TZFWDZFKRBELIQ-UHFFFAOYSA-N chlorzoxazone Chemical compound ClC1=CC=C2OC(O)=NC2=C1 TZFWDZFKRBELIQ-UHFFFAOYSA-N 0.000 description 1
- 229960003633 chlorzoxazone Drugs 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- BZMKNPGKXJAIDV-VAWYXSNFSA-N cinalukast Chemical compound CCC(CC)(C(O)=O)CC(=O)NC1=CC=CC(\C=C\C=2SC=C(N=2)C2CCC2)=C1 BZMKNPGKXJAIDV-VAWYXSNFSA-N 0.000 description 1
- 229950006262 cinalukast Drugs 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 229960000358 cisatracurium Drugs 0.000 description 1
- YXSLJKQTIDHPOT-LJCJQEJUSA-N cisatracurium Chemical compound C1=C(OC)C(OC)=CC=C1C[C@H]1[N@+](CCC(=O)OCCCCCOC(=O)CC[N@+]2(C)[C@@H](C3=CC(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=CC=2)(C)CCC2=CC(OC)=C(OC)C=C21 YXSLJKQTIDHPOT-LJCJQEJUSA-N 0.000 description 1
- 229960001653 citalopram Drugs 0.000 description 1
- 229950000308 clentiazem Drugs 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960002286 clodronic acid Drugs 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 229960004414 clomethiazole Drugs 0.000 description 1
- 229960004094 clomocycline Drugs 0.000 description 1
- BXVOHUQQUBSHLD-XCTBDMBQSA-N clomocycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(=C(/O)NCO)/C(=O)[C@@]4(O)C(=O)C3=C(O)C2=C1O BXVOHUQQUBSHLD-XCTBDMBQSA-N 0.000 description 1
- 229960004022 clotrimazole Drugs 0.000 description 1
- VNFPBHJOKIVQEB-UHFFFAOYSA-N clotrimazole Chemical compound ClC1=CC=CC=C1C(N1C=NC=C1)(C=1C=CC=CC=1)C1=CC=CC=C1 VNFPBHJOKIVQEB-UHFFFAOYSA-N 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 235000017471 coenzyme Q10 Nutrition 0.000 description 1
- ACTIUHUUMQJHFO-UPTCCGCDSA-N coenzyme Q10 Chemical compound COC1=C(OC)C(=O)C(C\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CC\C=C(/C)CCC=C(C)C)=C(C)C1=O ACTIUHUUMQJHFO-UPTCCGCDSA-N 0.000 description 1
- 230000006999 cognitive decline Effects 0.000 description 1
- 231100000876 cognitive deterioration Toxicity 0.000 description 1
- 229940096422 collagen type i Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 201000010989 colorectal carcinoma Diseases 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 230000002079 cooperative effect Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229940109248 cromoglycate Drugs 0.000 description 1
- IMZMKUWMOSJXDT-UHFFFAOYSA-N cromoglycic acid Chemical compound O1C(C(O)=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C(O)=O)O2 IMZMKUWMOSJXDT-UHFFFAOYSA-N 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 235000012754 curcumin Nutrition 0.000 description 1
- 239000004148 curcumin Substances 0.000 description 1
- 229940109262 curcumin Drugs 0.000 description 1
- 235000007336 cyanidin Nutrition 0.000 description 1
- SKYSRIRYMSLOIN-UHFFFAOYSA-N cyclopentolate Chemical compound C1CCCC1(O)C(C(=O)OCCN(C)C)C1=CC=CC=C1 SKYSRIRYMSLOIN-UHFFFAOYSA-N 0.000 description 1
- 229960001815 cyclopentolate Drugs 0.000 description 1
- 229960003176 cyclothiazide Drugs 0.000 description 1
- BOCUKUHCLICSIY-QJWLJZLASA-N cyclothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2C1[C@H](C=C2)C[C@H]2C1 BOCUKUHCLICSIY-QJWLJZLASA-N 0.000 description 1
- 230000000120 cytopathologic effect Effects 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- OBATZBGFDSVCJD-UHFFFAOYSA-N de-O-acetyl-lanatoside C Natural products CC1OC(OC2CC3C(C4C(C5(CCC(C5(C)C(O)C4)C=4COC(=O)C=4)O)CC3)(C)CC2)CC(O)C1OC(OC1C)CC(O)C1OC(OC1C)CC(O)C1OC1OC(CO)C(O)C(O)C1O OBATZBGFDSVCJD-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229960003266 deferiprone Drugs 0.000 description 1
- 229960000958 deferoxamine Drugs 0.000 description 1
- 229960004120 defibrotide Drugs 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 235000007242 delphinidin Nutrition 0.000 description 1
- FFNDMZIBVDSQFI-UHFFFAOYSA-N delphinidin chloride Chemical compound [Cl-].[O+]=1C2=CC(O)=CC(O)=C2C=C(O)C=1C1=CC(O)=C(O)C(O)=C1 FFNDMZIBVDSQFI-UHFFFAOYSA-N 0.000 description 1
- ISMCNVNDWFIXLM-WCGOZPBSSA-N deserpidine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C([C]5C=CC=CC5=N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 ISMCNVNDWFIXLM-WCGOZPBSSA-N 0.000 description 1
- 229960001993 deserpidine Drugs 0.000 description 1
- 229960001324 deslanoside Drugs 0.000 description 1
- OBATZBGFDSVCJD-LALPQLPRSA-N deslanoside Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@H]1[C@@H](O)C[C@@H](O[C@@H]1C)O[C@@H]1C[C@@H]2[C@]([C@@H]3[C@H]([C@]4(CC[C@@H]([C@@]4(C)[C@H](O)C3)C=3COC(=O)C=3)O)CC2)(C)CC1)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O OBATZBGFDSVCJD-LALPQLPRSA-N 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 230000028315 developmental maturation Effects 0.000 description 1
- 229960000632 dexamfetamine Drugs 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229960004042 diazoxide Drugs 0.000 description 1
- NZZIMKJIVMHWJC-UHFFFAOYSA-N dibenzoylmethane Chemical compound C=1C=CC=CC=1C(=O)CC(=O)C1=CC=CC=C1 NZZIMKJIVMHWJC-UHFFFAOYSA-N 0.000 description 1
- 229960001585 dicloxacillin Drugs 0.000 description 1
- YFAGHNZHGGCZAX-JKIFEVAISA-N dicloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(Cl)C=CC=C1Cl YFAGHNZHGGCZAX-JKIFEVAISA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- VFLDPWHFBUODDF-UHFFFAOYSA-N diferuloylmethane Natural products C1=C(O)C(OC)=CC(C=CC(=O)CC(=O)C=CC=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-UHFFFAOYSA-N 0.000 description 1
- 229960005156 digoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 229960004704 dihydroergotamine Drugs 0.000 description 1
- HESHRHUZIWVEAJ-JGRZULCMSA-N dihydroergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2[C@@H](C3=CC=CC4=NC=C([C]34)C2)C1)C)C1=CC=CC=C1 HESHRHUZIWVEAJ-JGRZULCMSA-N 0.000 description 1
- ILYCWAKSDCYMBB-OPCMSESCSA-N dihydrotachysterol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1/C[C@@H](O)CC[C@@H]1C ILYCWAKSDCYMBB-OPCMSESCSA-N 0.000 description 1
- 229960000465 dihydrotachysterol Drugs 0.000 description 1
- MUCZHBLJLSDCSD-UHFFFAOYSA-N diisopropyl fluorophosphate Chemical compound CC(C)OP(F)(=O)OC(C)C MUCZHBLJLSDCSD-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960003530 donepezil Drugs 0.000 description 1
- 239000003136 dopamine receptor stimulating agent Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 229960004428 doxacurium Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- IQQBRKLVEALROM-UHFFFAOYSA-N drinabant Chemical compound C=1C(F)=CC(F)=CC=1N(S(=O)(=O)C)C(C1)CN1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 IQQBRKLVEALROM-UHFFFAOYSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- JWJOTENAMICLJG-QWBYCMEYSA-N dutasteride Chemical compound O=C([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)N[C@@H]4CC3)C)CC[C@@]21C)NC1=CC(C(F)(F)F)=CC=C1C(F)(F)F JWJOTENAMICLJG-QWBYCMEYSA-N 0.000 description 1
- 229960004199 dutasteride Drugs 0.000 description 1
- 101150093683 dyn-1 gene Proteins 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000003937 effect on alzheimer disease Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 229960002759 eflornithine Drugs 0.000 description 1
- 229950003102 efonidipine Drugs 0.000 description 1
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 1
- JAZBEHYOTPTENJ-UHFFFAOYSA-N eicosapentaenoic acid Natural products CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O JAZBEHYOTPTENJ-UHFFFAOYSA-N 0.000 description 1
- 229960000756 elcatonin Drugs 0.000 description 1
- 108700032313 elcatonin Proteins 0.000 description 1
- OTLDLQZJRFYOJR-LJQANCHMSA-N eletriptan Chemical compound CN1CCC[C@@H]1CC1=CN=C2[C]1C=C(CCS(=O)(=O)C=1C=CC=CC=1)C=C2 OTLDLQZJRFYOJR-LJQANCHMSA-N 0.000 description 1
- 229960002472 eletriptan Drugs 0.000 description 1
- 229950010020 elgodipine Drugs 0.000 description 1
- 230000026721 endothelial cell chemotaxis Effects 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229960000610 enoxaparin Drugs 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 229940030275 epigallocatechin gallate Drugs 0.000 description 1
- 230000001073 episodic memory Effects 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- 229960004943 ergotamine Drugs 0.000 description 1
- OFKDAAIKGIBASY-VFGNJEKYSA-N ergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2C(C3=CC=CC4=NC=C([C]34)C2)=C1)C)C1=CC=CC=C1 OFKDAAIKGIBASY-VFGNJEKYSA-N 0.000 description 1
- XCGSFFUVFURLIX-UHFFFAOYSA-N ergotaminine Natural products C1=C(C=2C=CC=C3NC=C(C=23)C2)C2N(C)CC1C(=O)NC(C(N12)=O)(C)OC1(O)C1CCCN1C(=O)C2CC1=CC=CC=C1 XCGSFFUVFURLIX-UHFFFAOYSA-N 0.000 description 1
- HCZKYJDFEPMADG-UHFFFAOYSA-N erythro-nordihydroguaiaretic acid Natural products C=1C=C(O)C(O)=CC=1CC(C)C(C)CC1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-UHFFFAOYSA-N 0.000 description 1
- 229960003276 erythromycin Drugs 0.000 description 1
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 description 1
- 229960004341 escitalopram Drugs 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- GXRZIMHKGDIBEW-UHFFFAOYSA-N ethinamate Chemical compound NC(=O)OC1(C#C)CCCCC1 GXRZIMHKGDIBEW-UHFFFAOYSA-N 0.000 description 1
- 229960002209 ethinamate Drugs 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 229960004585 etidronic acid Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000002964 excitative effect Effects 0.000 description 1
- 229940108366 exelon Drugs 0.000 description 1
- 238000010195 expression analysis Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 239000011536 extraction buffer Substances 0.000 description 1
- 208000015756 familial Alzheimer disease Diseases 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 239000007888 film coating Substances 0.000 description 1
- 235000021323 fish oil Nutrition 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- NCWZOASIUQVOFA-FWZJPQCDSA-N florbetaben ((18)F) Chemical compound C1=CC(NC)=CC=C1\C=C\C1=CC=C(OCCOCCOCC[18F])C=C1 NCWZOASIUQVOFA-FWZJPQCDSA-N 0.000 description 1
- AAXVEMMRQDVLJB-BULBTXNYSA-N fludrocortisone Chemical compound O=C1CC[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 AAXVEMMRQDVLJB-BULBTXNYSA-N 0.000 description 1
- 229960002011 fludrocortisone Drugs 0.000 description 1
- SMANXXCATUTDDT-QPJJXVBHSA-N flunarizine Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)N1CCN(C\C=C\C=2C=CC=CC=2)CC1 SMANXXCATUTDDT-QPJJXVBHSA-N 0.000 description 1
- 229960000326 flunarizine Drugs 0.000 description 1
- 229960002200 flunitrazepam Drugs 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 238000002376 fluorescence recovery after photobleaching Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical class FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 229960005051 fluostigmine Drugs 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 1
- 229960001318 fondaparinux Drugs 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 229960005102 foscarnet Drugs 0.000 description 1
- 229960000693 fosphenytoin Drugs 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 229960000936 fumagillin Drugs 0.000 description 1
- NGGMYCMLYOUNGM-CSDLUJIJSA-N fumagillin Chemical compound C([C@H]([C@H]([C@@H]1[C@]2(C)[C@H](O2)CC=C(C)C)OC)OC(=O)\C=C\C=C\C=C\C=C\C(O)=O)C[C@@]21CO2 NGGMYCMLYOUNGM-CSDLUJIJSA-N 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229960002870 gabapentin Drugs 0.000 description 1
- 229950000501 gabexate Drugs 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960003627 gemfibrozil Drugs 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 108010008714 glucose-regulated protein 170 Proteins 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 210000000020 growth cone Anatomy 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- ACGDKVXYNVEAGU-UHFFFAOYSA-N guanethidine Chemical compound NC(N)=NCCN1CCCCCCC1 ACGDKVXYNVEAGU-UHFFFAOYSA-N 0.000 description 1
- 229960003602 guanethidine Drugs 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 229960003878 haloperidol Drugs 0.000 description 1
- 229940109738 hematin Drugs 0.000 description 1
- 229940059489 heme arginate Drugs 0.000 description 1
- 229940025294 hemin Drugs 0.000 description 1
- BTIJJDXEELBZFS-QDUVMHSLSA-K hemin Chemical compound CC1=C(CCC(O)=O)C(C=C2C(CCC(O)=O)=C(C)\C(N2[Fe](Cl)N23)=C\4)=N\C1=C/C2=C(C)C(C=C)=C3\C=C/1C(C)=C(C=C)C/4=N\1 BTIJJDXEELBZFS-QDUVMHSLSA-K 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- AIONOLUJZLIMTK-UHFFFAOYSA-N hesperetin Natural products C1=C(O)C(OC)=CC=C1C1OC2=CC(O)=CC(O)=C2C(=O)C1 AIONOLUJZLIMTK-UHFFFAOYSA-N 0.000 description 1
- 235000010209 hesperetin Nutrition 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- FTODBIPDTXRIGS-UHFFFAOYSA-N homoeriodictyol Natural products C1=C(O)C(OC)=CC(C2OC3=CC(O)=CC(O)=C3C(=O)C2)=C1 FTODBIPDTXRIGS-UHFFFAOYSA-N 0.000 description 1
- ZRJBHWIHUMBLCN-YQEJDHNASA-N huperzine A Chemical compound N1C(=O)C=CC2=C1C[C@H]1\C(=C/C)[C@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-YQEJDHNASA-N 0.000 description 1
- 229960002474 hydralazine Drugs 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 239000000416 hydrocolloid Substances 0.000 description 1
- 229950000208 hydrocortamate Drugs 0.000 description 1
- FWFVLWGEFDIZMJ-FOMYWIRZSA-N hydrocortamate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)COC(=O)CN(CC)CC)(O)[C@@]1(C)C[C@@H]2O FWFVLWGEFDIZMJ-FOMYWIRZSA-N 0.000 description 1
- DMDGGSIALPNSEE-UHFFFAOYSA-N hydroflumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O DMDGGSIALPNSEE-UHFFFAOYSA-N 0.000 description 1
- 229960003313 hydroflumethiazide Drugs 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 1
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 1
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960004053 ibutilide Drugs 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 229960005431 ipriflavone Drugs 0.000 description 1
- QRWOVIRDHQJFDB-UHFFFAOYSA-N isobutyl cyanoacrylate Chemical compound CC(C)COC(=O)C(=C)C#N QRWOVIRDHQJFDB-UHFFFAOYSA-N 0.000 description 1
- RPCVIAXDAUMJJP-PZBABLGHSA-N ispronicline Chemical compound CN[C@@H](C)C\C=C\C1=CN=CC(OC(C)C)=C1 RPCVIAXDAUMJJP-PZBABLGHSA-N 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 229960004958 ketotifen Drugs 0.000 description 1
- WZNJWVWKTVETCG-UHFFFAOYSA-N kojic acid Natural products OC(=O)C(N)CN1C=CC(=O)C(O)=C1 WZNJWVWKTVETCG-UHFFFAOYSA-N 0.000 description 1
- 229940106134 krill oil Drugs 0.000 description 1
- 229960004340 lacidipine Drugs 0.000 description 1
- GKQPCPXONLDCMU-CCEZHUSRSA-N lacidipine Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OCC)C1C1=CC=CC=C1\C=C\C(=O)OC(C)(C)C GKQPCPXONLDCMU-CCEZHUSRSA-N 0.000 description 1
- 229960000448 lactic acid Drugs 0.000 description 1
- 208000011977 language disease Diseases 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 229960000433 latamoxef Drugs 0.000 description 1
- 229950003465 latrepirdine Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 229950007396 lecozotan Drugs 0.000 description 1
- 210000002414 leg Anatomy 0.000 description 1
- 229940039781 leptin Drugs 0.000 description 1
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 description 1
- 102000005861 leptin receptors Human genes 0.000 description 1
- 108010019813 leptin receptors Proteins 0.000 description 1
- 229960004294 lercanidipine Drugs 0.000 description 1
- ZDXUKAKRHYTAKV-UHFFFAOYSA-N lercanidipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)(C)CN(C)CCC(C=2C=CC=CC=2)C=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZDXUKAKRHYTAKV-UHFFFAOYSA-N 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 229960004502 levodopa Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 229960001941 lidoflazine Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960001078 lithium Drugs 0.000 description 1
- 229950007692 lomerizine Drugs 0.000 description 1
- 230000007787 long-term memory Effects 0.000 description 1
- 230000027928 long-term synaptic potentiation Effects 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 210000001699 lower leg Anatomy 0.000 description 1
- WGFOBBZOWHGYQH-MXHNKVEKSA-N lubiprostone Chemical compound O1[C@](C(F)(F)CCCC)(O)CC[C@@H]2[C@@H](CCCCCCC(O)=O)C(=O)C[C@H]21 WGFOBBZOWHGYQH-MXHNKVEKSA-N 0.000 description 1
- 229960000345 lubiprostone Drugs 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- FBQPGGIHOFZRGH-UHFFFAOYSA-N lucanthone Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(C)=CC=C2NCCN(CC)CC FBQPGGIHOFZRGH-UHFFFAOYSA-N 0.000 description 1
- 229950005239 lucanthone Drugs 0.000 description 1
- 235000012680 lutein Nutrition 0.000 description 1
- 229960005375 lutein Drugs 0.000 description 1
- 239000001656 lutein Substances 0.000 description 1
- KBPHJBAIARWVSC-RGZFRNHPSA-N lutein Chemical compound C([C@H](O)CC=1C)C(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\[C@H]1C(C)=C[C@H](O)CC1(C)C KBPHJBAIARWVSC-RGZFRNHPSA-N 0.000 description 1
- ORAKUVXRZWMARG-WZLJTJAWSA-N lutein Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2C(=CC(O)CC2(C)C)C ORAKUVXRZWMARG-WZLJTJAWSA-N 0.000 description 1
- 229960004196 lymecycline Drugs 0.000 description 1
- AHEVKYYGXVEWNO-UEPZRUIBSA-N lymecycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCNCCCC[C@H](N)C(O)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O AHEVKYYGXVEWNO-UEPZRUIBSA-N 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960003963 manidipine Drugs 0.000 description 1
- ANEBWFXPVPTEET-UHFFFAOYSA-N manidipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN2CCN(CC2)C(C=2C=CC=CC=2)C=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ANEBWFXPVPTEET-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229960003951 masoprocol Drugs 0.000 description 1
- HCZKYJDFEPMADG-TXEJJXNPSA-N masoprocol Chemical compound C([C@H](C)[C@H](C)CC=1C=C(O)C(O)=CC=1)C1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-TXEJJXNPSA-N 0.000 description 1
- IMYZQPCYWPFTAG-IQJOONFLSA-N mecamylamine Chemical compound C1C[C@@H]2C(C)(C)[C@@](NC)(C)[C@H]1C2 IMYZQPCYWPFTAG-IQJOONFLSA-N 0.000 description 1
- 229960002525 mecamylamine Drugs 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- FRQMUZJSZHZSGN-HBNHAYAOSA-N medroxyprogesterone Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FRQMUZJSZHZSGN-HBNHAYAOSA-N 0.000 description 1
- 229960003987 melatonin Drugs 0.000 description 1
- DRLFMBDRBRZALE-UHFFFAOYSA-N melatonin Chemical compound COC1=CC=C2NC=C(CCNC(C)=O)C2=C1 DRLFMBDRBRZALE-UHFFFAOYSA-N 0.000 description 1
- 229960001929 meloxicam Drugs 0.000 description 1
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 230000004630 mental health Effects 0.000 description 1
- 229960002342 mephentermine Drugs 0.000 description 1
- RXQCGGRTAILOIN-UHFFFAOYSA-N mephentermine Chemical compound CNC(C)(C)CC1=CC=CC=C1 RXQCGGRTAILOIN-UHFFFAOYSA-N 0.000 description 1
- DAHIQPJTGIHDGO-IAGOWNOFSA-N mesembrine Chemical compound C1=C(OC)C(OC)=CC=C1[C@]1(CCC(=O)C2)[C@@H]2N(C)CC1 DAHIQPJTGIHDGO-IAGOWNOFSA-N 0.000 description 1
- DAHIQPJTGIHDGO-UHFFFAOYSA-N mesembrine Natural products C1=C(OC)C(OC)=CC=C1C1(CCC(=O)C2)C2N(C)CC1 DAHIQPJTGIHDGO-UHFFFAOYSA-N 0.000 description 1
- 210000001259 mesencephalon Anatomy 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- GBLRQXKSCRCLBZ-IYQFLEDGSA-N meso-doxacurium Chemical compound COC1=C(OC)C(OC)=CC(C[C@@H]2[N@@+](CCC3=C2C(=C(OC)C(OC)=C3)OC)(C)CCCOC(=O)CCC(=O)OCCC[N@@+]2(C)[C@@H](C3=C(OC)C(OC)=C(OC)C=C3CC2)CC=2C=C(OC)C(OC)=C(OC)C=2)=C1 GBLRQXKSCRCLBZ-IYQFLEDGSA-N 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229960003663 metaraminol Drugs 0.000 description 1
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 1
- FLOSMHQXBMRNHR-DAXSKMNVSA-N methazolamide Chemical compound CC(=O)\N=C1/SC(S(N)(=O)=O)=NN1C FLOSMHQXBMRNHR-DAXSKMNVSA-N 0.000 description 1
- 229960004083 methazolamide Drugs 0.000 description 1
- 229960002683 methohexital Drugs 0.000 description 1
- NXMXPVQZFYYPGD-UHFFFAOYSA-N methyl 2-methylprop-2-enoate;methyl prop-2-enoate Chemical compound COC(=O)C=C.COC(=O)C(C)=C NXMXPVQZFYYPGD-UHFFFAOYSA-N 0.000 description 1
- VKQFCGNPDRICFG-UHFFFAOYSA-N methyl 2-methylpropyl 2,6-dimethyl-4-(2-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCC(C)C)C1C1=CC=CC=C1[N+]([O-])=O VKQFCGNPDRICFG-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960001344 methylphenidate Drugs 0.000 description 1
- 229960004465 metyrapone Drugs 0.000 description 1
- FJLBFSROUSIWMA-UHFFFAOYSA-N metyrapone Chemical compound C=1C=CN=CC=1C(C)(C)C(=O)C1=CC=CN=C1 FJLBFSROUSIWMA-UHFFFAOYSA-N 0.000 description 1
- VKHAHZOOUSRJNA-GCNJZUOMSA-N mifepristone Chemical compound C1([C@@H]2C3=C4CCC(=O)C=C4CC[C@H]3[C@@H]3CC[C@@]([C@]3(C2)C)(O)C#CC)=CC=C(N(C)C)C=C1 VKHAHZOOUSRJNA-GCNJZUOMSA-N 0.000 description 1
- 229960003248 mifepristone Drugs 0.000 description 1
- PZRHRDRVRGEVNW-UHFFFAOYSA-N milrinone Chemical compound N1C(=O)C(C#N)=CC(C=2C=CN=CC=2)=C1C PZRHRDRVRGEVNW-UHFFFAOYSA-N 0.000 description 1
- 229960003574 milrinone Drugs 0.000 description 1
- 229950002289 mimosine Drugs 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 229960002540 mivacurium Drugs 0.000 description 1
- 229960001165 modafinil Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 108010071525 moesin Proteins 0.000 description 1
- 239000002052 molecular layer Substances 0.000 description 1
- 230000004784 molecular pathogenesis Effects 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 229960001664 mometasone Drugs 0.000 description 1
- QLIIKPVHVRXHRI-CXSFZGCWSA-N mometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CCl)(O)[C@@]1(C)C[C@@H]2O QLIIKPVHVRXHRI-CXSFZGCWSA-N 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 108010049787 myosin VI Proteins 0.000 description 1
- SSRDSYXGYPJKRR-ZDUSSCGKSA-N n-[(3r)-1-azabicyclo[2.2.2]octan-3-yl]-7-chloro-1-benzothiophene-2-carboxamide Chemical compound C1N(CC2)CCC2[C@H]1NC(=O)C1=CC(C=CC=C2Cl)=C2S1 SSRDSYXGYPJKRR-ZDUSSCGKSA-N 0.000 description 1
- CMRLNEYJEPELSM-BTQNPOSSSA-N n-[(3s)-1-azabicyclo[2.2.2]octan-3-yl]-1h-indazole-3-carboxamide;hydrochloride Chemical compound Cl.C1=CC=C2C(C(N[C@H]3C4CCN(CC4)C3)=O)=NNC2=C1 CMRLNEYJEPELSM-BTQNPOSSSA-N 0.000 description 1
- JTSLALYXYSRPGW-UHFFFAOYSA-N n-[5-(4-cyanophenyl)-1h-pyrrolo[2,3-b]pyridin-3-yl]pyridine-3-carboxamide Chemical compound C=1C=CN=CC=1C(=O)NC(C1=C2)=CNC1=NC=C2C1=CC=C(C#N)C=C1 JTSLALYXYSRPGW-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960003255 natamycin Drugs 0.000 description 1
- 239000004311 natamycin Substances 0.000 description 1
- 235000010298 natamycin Nutrition 0.000 description 1
- NCXMLFZGDNKEPB-FFPOYIOWSA-N natamycin Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C[C@@H](C)OC(=O)/C=C/[C@H]2O[C@@H]2C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 NCXMLFZGDNKEPB-FFPOYIOWSA-N 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- OGZQTTHDGQBLBT-UHFFFAOYSA-N neramexane Chemical compound CC1(C)CC(C)(C)CC(C)(N)C1 OGZQTTHDGQBLBT-UHFFFAOYSA-N 0.000 description 1
- 229950004543 neramexane Drugs 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 229960000808 netilmicin Drugs 0.000 description 1
- ZBGPYVZLYBDXKO-HILBYHGXSA-N netilmycin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@]([C@H](NC)[C@@H](O)CO1)(C)O)NCC)[C@H]1OC(CN)=CC[C@H]1N ZBGPYVZLYBDXKO-HILBYHGXSA-N 0.000 description 1
- 230000000626 neurodegenerative effect Effects 0.000 description 1
- 230000016273 neuron death Effects 0.000 description 1
- 230000014511 neuron projection development Effects 0.000 description 1
- 230000006764 neuronal dysfunction Effects 0.000 description 1
- 108010082365 neuronal interleukin-16 Proteins 0.000 description 1
- 230000002981 neuropathic effect Effects 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- 239000003900 neurotrophic factor Substances 0.000 description 1
- PGSADBUBUOPOJS-UHFFFAOYSA-N neutral red Chemical compound Cl.C1=C(C)C(N)=CC2=NC3=CC(N(C)C)=CC=C3N=C21 PGSADBUBUOPOJS-UHFFFAOYSA-N 0.000 description 1
- 229960001783 nicardipine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Chemical compound CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 229960000916 niflumic acid Drugs 0.000 description 1
- 229960005366 nilvadipine Drugs 0.000 description 1
- 229960000715 nimodipine Drugs 0.000 description 1
- 229960000227 nisoldipine Drugs 0.000 description 1
- 229960001721 nitisinone Drugs 0.000 description 1
- OUBCNLGXQFSTLU-UHFFFAOYSA-N nitisinone Chemical compound [O-][N+](=O)C1=CC(C(F)(F)F)=CC=C1C(=O)C1C(=O)CCCC1=O OUBCNLGXQFSTLU-UHFFFAOYSA-N 0.000 description 1
- 229960002460 nitroprusside Drugs 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 229960005017 olanzapine Drugs 0.000 description 1
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229960002369 oxyphencyclimine Drugs 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- GVEAYVLWDAFXET-XGHATYIMSA-N pancuronium Chemical compound C[N+]1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 GVEAYVLWDAFXET-XGHATYIMSA-N 0.000 description 1
- 229960005457 pancuronium Drugs 0.000 description 1
- BUUKFBVDKSFMHN-LKMAISLMSA-N parathar acetate Chemical compound CC(O)=O.C([C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)C(C)C)[C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CN=CN1 BUUKFBVDKSFMHN-LKMAISLMSA-N 0.000 description 1
- 229960000987 paricalcitol Drugs 0.000 description 1
- BPKAHTKRCLCHEA-UBFJEZKGSA-N paricalcitol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@@H](\C=C\[C@H](C)C(C)(C)O)C)=C\C=C1C[C@@H](O)C[C@H](O)C1 BPKAHTKRCLCHEA-UBFJEZKGSA-N 0.000 description 1
- 230000001936 parietal effect Effects 0.000 description 1
- 102000045222 parkin Human genes 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000008807 pathological lesion Effects 0.000 description 1
- YZPOQCQXOSEMAZ-UHFFFAOYSA-N pbt2 Chemical compound ClC1=CC(Cl)=C(O)C2=NC(CN(C)C)=CC=C21 YZPOQCQXOSEMAZ-UHFFFAOYSA-N 0.000 description 1
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 description 1
- 229960005301 pentazocine Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000011458 pharmacological treatment Methods 0.000 description 1
- JTJMJGYZQZDUJJ-UHFFFAOYSA-N phencyclidine Chemical compound C1CCCCN1C1(C=2C=CC=CC=2)CCCCC1 JTJMJGYZQZDUJJ-UHFFFAOYSA-N 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- 229960002695 phenobarbital Drugs 0.000 description 1
- DQDAYGNAKTZFIW-UHFFFAOYSA-N phenprocoumon Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC)C1=CC=CC=C1 DQDAYGNAKTZFIW-UHFFFAOYSA-N 0.000 description 1
- 229960004923 phenprocoumon Drugs 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960001260 pipecuronium Drugs 0.000 description 1
- MXXWOMGUGJBKIW-YPCIICBESA-N piperine Chemical compound C=1C=C2OCOC2=CC=1/C=C/C=C/C(=O)N1CCCCC1 MXXWOMGUGJBKIW-YPCIICBESA-N 0.000 description 1
- WVWHRXVVAYXKDE-UHFFFAOYSA-N piperine Natural products O=C(C=CC=Cc1ccc2OCOc2c1)C3CCCCN3 WVWHRXVVAYXKDE-UHFFFAOYSA-N 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 230000007505 plaque formation Effects 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 108010017843 platelet-derived growth factor A Proteins 0.000 description 1
- 230000036178 pleiotropy Effects 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 1
- 238000011240 pooled analysis Methods 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 150000003109 potassium Chemical class 0.000 description 1
- YRVIKLBSVVNSHF-JTQLQIEISA-N pozanicline Chemical compound CC1=NC=CC=C1OC[C@H]1NCCC1 YRVIKLBSVVNSHF-JTQLQIEISA-N 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960001233 pregabalin Drugs 0.000 description 1
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 description 1
- 210000000063 presynaptic terminal Anatomy 0.000 description 1
- 230000001023 pro-angiogenic effect Effects 0.000 description 1
- 230000000861 pro-apoptotic effect Effects 0.000 description 1
- 230000009219 proapoptotic pathway Effects 0.000 description 1
- 229960002288 procaterol Drugs 0.000 description 1
- FKNXQNWAXFXVNW-BLLLJJGKSA-N procaterol Chemical compound N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)[C@@H](NC(C)C)CC FKNXQNWAXFXVNW-BLLLJJGKSA-N 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 239000000186 progesterone Substances 0.000 description 1
- 229960003387 progesterone Drugs 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 229940097325 prolactin Drugs 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 229960002662 propylthiouracil Drugs 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 239000003801 protein tyrosine phosphatase 1B inhibitor Substances 0.000 description 1
- SCHKZZSVELPJKU-UHFFFAOYSA-N prx-03140 Chemical compound O=C1N(C(C)C)C=2SC=CC=2C(O)=C1C(=O)NCCCN1CCCCC1 SCHKZZSVELPJKU-UHFFFAOYSA-N 0.000 description 1
- RADKZDMFGJYCBB-UHFFFAOYSA-N pyridoxal hydrochloride Natural products CC1=NC=C(CO)C(C=O)=C1O RADKZDMFGJYCBB-UHFFFAOYSA-N 0.000 description 1
- WKSAUQYGYAYLPV-UHFFFAOYSA-N pyrimethamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C=C1 WKSAUQYGYAYLPV-UHFFFAOYSA-N 0.000 description 1
- 229960000611 pyrimethamine Drugs 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- 229960000948 quinine Drugs 0.000 description 1
- 229960004157 rabeprazole Drugs 0.000 description 1
- YREYEVIYCVEVJK-UHFFFAOYSA-N rabeprazole Chemical compound COCCCOC1=CC=NC(CS(=O)C=2NC3=CC=CC=C3N=2)=C1C YREYEVIYCVEVJK-UHFFFAOYSA-N 0.000 description 1
- ZRJBHWIHUMBLCN-BMIGLBTASA-N rac-huperzine A Natural products N1C(=O)C=CC2=C1C[C@@H]1C(=CC)[C@@]2(N)CC(C)=C1 ZRJBHWIHUMBLCN-BMIGLBTASA-N 0.000 description 1
- 108010062302 rac1 GTP Binding Protein Proteins 0.000 description 1
- JQOFKKWHXGQABB-UHFFFAOYSA-N radequinil Chemical compound COC1=CC=CC(C=2C=3C=C(C(=O)NC=3C=CN=2)C=2N=C(C)ON=2)=C1 JQOFKKWHXGQABB-UHFFFAOYSA-N 0.000 description 1
- LVQTUXZKLGXYIU-GWSNJHLMSA-M rapacuronium Chemical compound [Br-].N1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)CC)[N+]2(CC=C)CCCCC2)CCCCC1 LVQTUXZKLGXYIU-GWSNJHLMSA-M 0.000 description 1
- 229960004804 rapacuronium Drugs 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- RUOKEQAAGRXIBM-GFCCVEGCSA-N rasagiline Chemical compound C1=CC=C2[C@H](NCC#C)CCC2=C1 RUOKEQAAGRXIBM-GFCCVEGCSA-N 0.000 description 1
- 229960000245 rasagiline Drugs 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 102000037983 regulatory factors Human genes 0.000 description 1
- 108091008025 regulatory factors Proteins 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 235000021283 resveratrol Nutrition 0.000 description 1
- 229940016667 resveratrol Drugs 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 210000001202 rhombencephalon Anatomy 0.000 description 1
- GLLPUTYLZIKEGF-HAVVHWLPSA-N ridogrel Chemical compound C=1C=CC(C(F)(F)F)=CC=1C(=N/OCCCCC(=O)O)\C1=CC=CN=C1 GLLPUTYLZIKEGF-HAVVHWLPSA-N 0.000 description 1
- 229950006674 ridogrel Drugs 0.000 description 1
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 description 1
- 229960001534 risperidone Drugs 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- 239000002760 rocket fuel Substances 0.000 description 1
- YXRDKMPIGHSVRX-OOJCLDBCSA-N rocuronium Chemical compound N1([C@@H]2[C@@H](O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(CC=C)CCCC2)CCOCC1 YXRDKMPIGHSVRX-OOJCLDBCSA-N 0.000 description 1
- 229960000491 rocuronium Drugs 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- JJSYXNQGLHBRRK-SFEDZAPPSA-N ryanodine Chemical compound O([C@@H]1[C@]([C@@]2([C@]3(O)[C@]45O[C@@]2(O)C[C@]([C@]4(CC[C@H](C)[C@H]5O)O)(C)[C@@]31O)C)(O)C(C)C)C(=O)C1=CC=CN1 JJSYXNQGLHBRRK-SFEDZAPPSA-N 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 230000001953 sensory effect Effects 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- 230000006403 short-term memory Effects 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 239000003009 skin protective agent Substances 0.000 description 1
- 102000030938 small GTPase Human genes 0.000 description 1
- 108060007624 small GTPase Proteins 0.000 description 1
- 230000035943 smell Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 229960002232 sodium phenylbutyrate Drugs 0.000 description 1
- VPZRWNZGLKXFOE-UHFFFAOYSA-M sodium phenylbutyrate Chemical compound [Na+].[O-]C(=O)CCCC1=CC=CC=C1 VPZRWNZGLKXFOE-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- AEQFSUDEHCCHBT-UHFFFAOYSA-M sodium valproate Chemical compound [Na+].CCCC(C([O-])=O)CCC AEQFSUDEHCCHBT-UHFFFAOYSA-M 0.000 description 1
- 108010046571 sodium-hydrogen exchanger regulatory factor Proteins 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- QZWYXEBIQWJXAR-UHFFFAOYSA-N spiro[1,3-dihydroindene-2,3'-imidazo[1,2-a]pyridine]-2'-one Chemical compound C1C2=CC=CC=C2CC21N1C=CC=CC1=NC2=O QZWYXEBIQWJXAR-UHFFFAOYSA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- RXLOZRCLQMJJLC-UHFFFAOYSA-N ssr-180,711 Chemical compound C1=CC(Br)=CC=C1OC(=O)N1C(CC2)CCN2CC1 RXLOZRCLQMJJLC-UHFFFAOYSA-N 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 108010090953 subunit 1 GABA type B receptor Proteins 0.000 description 1
- AXOIZCJOOAYSMI-UHFFFAOYSA-N succinylcholine Chemical compound C[N+](C)(C)CCOC(=O)CCC(=O)OCC[N+](C)(C)C AXOIZCJOOAYSMI-UHFFFAOYSA-N 0.000 description 1
- 229940032712 succinylcholine Drugs 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 229960005404 sulfamethoxazole Drugs 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 229960005349 sulfur Drugs 0.000 description 1
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000002889 sympathetic effect Effects 0.000 description 1
- 230000033504 synapse organization Effects 0.000 description 1
- 230000003976 synaptic dysfunction Effects 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 210000002504 synaptic vesicle Anatomy 0.000 description 1
- 108010016910 synaptojanin Proteins 0.000 description 1
- 102000000580 synaptojanin Human genes 0.000 description 1
- 229960001685 tacrine Drugs 0.000 description 1
- YLJREFDVOIBQDA-UHFFFAOYSA-N tacrine Chemical compound C1=CC=C2C(N)=C(CCCC3)C3=NC2=C1 YLJREFDVOIBQDA-UHFFFAOYSA-N 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229960000565 tazarotene Drugs 0.000 description 1
- 229960003188 temazepam Drugs 0.000 description 1
- 229960000338 teriparatide acetate Drugs 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 229960005333 tetrabenazine Drugs 0.000 description 1
- MPLHNVLQVRSVEE-UHFFFAOYSA-N texas red Chemical compound [O-]S(=O)(=O)C1=CC(S(Cl)(=O)=O)=CC=C1C(C1=CC=2CCCN3CCCC(C=23)=C1O1)=C2C1=C(CCC1)C3=[N+]1CCCC3=C2 MPLHNVLQVRSVEE-UHFFFAOYSA-N 0.000 description 1
- 210000001103 thalamus Anatomy 0.000 description 1
- XCTYLCDETUVOIP-UHFFFAOYSA-N thiethylperazine Chemical compound C12=CC(SCC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 XCTYLCDETUVOIP-UHFFFAOYSA-N 0.000 description 1
- 229960004869 thiethylperazine Drugs 0.000 description 1
- 229960003279 thiopental Drugs 0.000 description 1
- 229960001918 tiagabine Drugs 0.000 description 1
- PBJUNZJWGZTSKL-MRXNPFEDSA-N tiagabine Chemical compound C1=CSC(C(=CCCN2C[C@@H](CCC2)C(O)=O)C2=C(C=CS2)C)=C1C PBJUNZJWGZTSKL-MRXNPFEDSA-N 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- 229940019375 tiludronate Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229960003425 tirofiban Drugs 0.000 description 1
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 1
- 230000030968 tissue homeostasis Effects 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 229960004394 topiramate Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 231100000820 toxicity test Toxicity 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 229960003570 tramiprosate Drugs 0.000 description 1
- 238000013271 transdermal drug delivery Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 229960002368 travoprost Drugs 0.000 description 1
- MKPLKVHSHYCHOC-AHTXBMBWSA-N travoprost Chemical compound CC(C)OC(=O)CCC\C=C/C[C@H]1[C@@H](O)C[C@@H](O)[C@@H]1\C=C\[C@@H](O)COC1=CC=CC(C(F)(F)F)=C1 MKPLKVHSHYCHOC-AHTXBMBWSA-N 0.000 description 1
- PHLBKPHSAVXXEF-UHFFFAOYSA-N trazodone Chemical compound ClC1=CC=CC(N2CCN(CCCN3C(N4C=CC=CC4=N3)=O)CC2)=C1 PHLBKPHSAVXXEF-UHFFFAOYSA-N 0.000 description 1
- 229960003991 trazodone Drugs 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000003656 tris buffered saline Substances 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- OYYDSUSKLWTMMQ-JKHIJQBDSA-N trospium Chemical compound [N+]12([C@@H]3CC[C@H]2C[C@H](C3)OC(=O)C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CCCC1 OYYDSUSKLWTMMQ-JKHIJQBDSA-N 0.000 description 1
- 229960001491 trospium Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960004317 unoprostone Drugs 0.000 description 1
- TVHAZVBUYQMHBC-SNHXEXRGSA-N unoprostone Chemical compound CCCCCCCC(=O)CC[C@H]1[C@H](O)C[C@H](O)[C@@H]1C\C=C/CCCC(O)=O TVHAZVBUYQMHBC-SNHXEXRGSA-N 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 229940004858 usnic acid Drugs 0.000 description 1
- ICTZCAHDGHPRQR-UHFFFAOYSA-N usnic acid Natural products OC1=C(C)C(O)=C(C(C)=O)C2=C1C1(C)C(O)=C(C(=O)C)C(=O)C=C1O2 ICTZCAHDGHPRQR-UHFFFAOYSA-N 0.000 description 1
- WEYVVCKOOFYHRW-UHFFFAOYSA-N usninic acid Natural products CC12C(=O)C(C(=O)C)=C(O)C=C1OC1=C2C(O)=C(C)C(O)=C1C(C)=O WEYVVCKOOFYHRW-UHFFFAOYSA-N 0.000 description 1
- 229940102566 valproate Drugs 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 108010084171 vanutide cridificar Proteins 0.000 description 1
- 229960002730 vapreotide Drugs 0.000 description 1
- 108700029852 vapreotide Proteins 0.000 description 1
- JQSHBVHOMNKWFT-DTORHVGOSA-N varenicline Chemical compound C12=CC3=NC=CN=C3C=C2[C@H]2C[C@@H]1CNC2 JQSHBVHOMNKWFT-DTORHVGOSA-N 0.000 description 1
- 229960004751 varenicline Drugs 0.000 description 1
- 230000007556 vascular defect Effects 0.000 description 1
- 230000006439 vascular pathology Effects 0.000 description 1
- BGSZAXLLHYERSY-XQIGCQGXSA-N vecuronium Chemical compound N1([C@@H]2[C@@H](OC(C)=O)C[C@@H]3CC[C@H]4[C@@H]5C[C@@H]([C@@H]([C@]5(CC[C@@H]4[C@@]3(C)C2)C)OC(=O)C)[N+]2(C)CCCCC2)CCCCC1 BGSZAXLLHYERSY-XQIGCQGXSA-N 0.000 description 1
- 229960003819 vecuronium Drugs 0.000 description 1
- 229960003636 vidarabine Drugs 0.000 description 1
- 229960005318 vigabatrin Drugs 0.000 description 1
- PJDFLNIOAUIZSL-UHFFFAOYSA-N vigabatrin Chemical compound C=CC(N)CCC(O)=O PJDFLNIOAUIZSL-UHFFFAOYSA-N 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000019158 vitamin B6 Nutrition 0.000 description 1
- 239000011726 vitamin B6 Substances 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940011671 vitamin b6 Drugs 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
- 230000001755 vocal effect Effects 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- FJHBOVDFOQMZRV-XQIHNALSSA-N xanthophyll Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2C=C(C)C(O)CC2(C)C FJHBOVDFOQMZRV-XQIHNALSSA-N 0.000 description 1
- ZXIBCJHYVWYIKI-PZJWPPBQSA-N ximelagatran Chemical compound C1([C@@H](NCC(=O)OCC)C(=O)N2[C@@H](CC2)C(=O)NCC=2C=CC(=CC=2)C(\N)=N\O)CCCCC1 ZXIBCJHYVWYIKI-PZJWPPBQSA-N 0.000 description 1
- 229960001522 ximelagatran Drugs 0.000 description 1
- 235000010930 zeaxanthin Nutrition 0.000 description 1
- 239000001775 zeaxanthin Substances 0.000 description 1
- 229940043269 zeaxanthin Drugs 0.000 description 1
- UZBODILCSLUHQR-JLMRSGIVSA-N zenvia Chemical compound C([C@@H]12)CCC[C@]11CCN(C)[C@H]2CC2=CC=C(OC)C=C21.C1C([C@H](C2)C=C)CCN2[C@H]1[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 UZBODILCSLUHQR-JLMRSGIVSA-N 0.000 description 1
- ZAFYATHCZYHLPB-UHFFFAOYSA-N zolpidem Chemical compound N1=C2C=CC(C)=CN2C(CC(=O)N(C)C)=C1C1=CC=C(C)C=C1 ZAFYATHCZYHLPB-UHFFFAOYSA-N 0.000 description 1
- 229960001475 zolpidem Drugs 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/155—Amidines (), e.g. guanidine (H2N—C(=NH)—NH2), isourea (N=C(OH)—NH2), isothiourea (—N=C(SH)—NH2)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/343—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide condensed with a carbocyclic ring, e.g. coumaran, bufuralol, befunolol, clobenfurol, amiodarone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
- A61K31/635—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide having a heterocyclic ring, e.g. sulfadiazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Landscapes
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Emergency Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychiatry (AREA)
- Cardiology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Una composición para uso en el tratamiento de la enfermedad de Alzheimer (EA) o un trastorno relacionado, que comprende al menos levosimendán, o una sal o una formulación de liberación sostenida del mismo.
Description
DESCRIPCIÓN
Nuevos enfoques terapéuticos para tratar la enfermedad de Alzheimer
Campo de la invención
La presente invención se refiere a composiciones para uso en el tratamiento de la enfermedad de Alzheimer (EA) y de trastornos relacionados. Más específicamente, la presente invención se refiere a nuevas terapias combinatorias de la enfermedad de Alzheimer y de trastornos relacionados. En particular, la invención atañe a compuestos que, solos o en combinación(es), pueden modular eficazmente la función sináptica y/o la angiogénesis y/o la respuesta al estrés celular.
Antecedentes de la invención
La EA es la demencia cortical prototípica caracterizada por un déficit de la memoria junto con disfasia (trastorno del lenguaje en el que hay un deterioro del habla y de la comprensión del habla), dispraxia (incapacidad para coordinar y realizar ciertos movimientos y gestos intencionales en ausencia de impedimentos motores o sensoriales) y agnosia (capacidad de reconocer objetos, personas, sonidos, formas u olores) atribuibles a una participación de las áreas de asociación corticales. También pueden estar implicados síntomas especiales tales como la paraparesia espástica (debilidad que afecta a las extremidades inferiores) (1-4).
La incidencia de la enfermedad de Alzheimer aumenta drásticamente con la edad. La EA es actualmente la causa más común de demencia. Se caracteriza clínicamente por una disminución global de la función cognitiva que pro gresa lentamente y deja a los pacientes en su estadio final ligados a la cama, incontinentes y dependientes de la atención de una persona a su cuidado. La muerte tiene lugar, en promedio, 9 años después del diagnóstico (5). La tasa de incidencia de la EA aumenta espectacularmente con la edad. Las predicciones demográficas de las Na ciones Unidas estiman que el número de personas mayores de 80 años se acercará a los 370 millones para el año 2050. Actualmente, se estima que el 50% de las personas mayores de 85 años sufren de EA. Por lo tanto, más de 100 millones de personas en todo el mundo padecerán demencia en 50 años. La enorme cantidad de personas que requerirán cuidados constantes y otros servicios afectará gravemente a los recursos médicos, monetarios y huma nos (6).
El deterioro de la memoria es la primera característica de la enfermedad y afecta a la memoria episódica (memoria para acontecimientos cotidianos). La memoria semántica (memoria para significado el verbal y visual) se ve afectada más tarde en la enfermedad. Por el contrario, la memoria de trabajo (memoria a corto plazo que implica estructuras y procesos utilizados para almacenar y manipular información temporalmente) y la memoria procedural (memoria inconsciente que es una memoria a largo plazo de habilidades y procedimientos) se conservan hasta tarde. A medi da que avanza la enfermedad, surgen las características adicionales de deterioro del lenguaje, las deficiencias vi suales perceptivas y espaciales, las agnosias y las apraxias.
El cuadro clásico de la enfermedad de Alzheimer es suficientemente característico para permitir una identificación en aproximadamente el 80% de los casos (7). Sin embargo, se produce una heterogeneidad clínica y no solo es impor tante para el manejo clínico, sino que proporciona una mayor implicación de los tratamientos con una medicación específica para las formas funcionalmente diferentes (8).
La característica distintiva patológica de la EA incluye placas amiloides que contienen beta-amiloide (Abeta), ovillos neurofibrilares (ONFs) que contienen Tau y disfunción y pérdida neuronal y sináptica (9-11). En la última década se han propuesto dos hipótesis principales sobre la causa de la EA: la "hipótesis de la cascada amiloide", que expone que el proceso neurodegenerativo es una serie de acontecimientos desencadenados por el procesamiento anormal de la proteína precursora amiloidea (APP, por sus siglas en inglés) (12), y la "hipótesis de la degeneración citoesquelética neuronal" (13), que propone que los cambios citoesqueléticos son los acontecimientos desencadenantes. La teoría más ampliamente aceptada que explica la evolución de la EA sigue siendo la hipótesis de la cascada amiloide (14-16) y los investigadores de la EA se han centrado principalmente en determinar los mecanismos subyacen tes a la toxicidad asociada con las proteínas Abeta. En cambio, la proteína Tau ha recibido mucha menos atención por parte de la industria farmacéutica que la amiloide, por intereses tanto fundamentales como prácticos. Además, el cambio en la densidad sináptica es la lesión patológica que mejor correlación tiene con el deterioro cognitivo en relación con las otras dos. Algunos estudios han revelado que la patología amiloide parece desarrollarse en una manera específica de los neurotransmisores, en la que los terminales colinérgicos parecen ser los más vulnerables, seguidos de los terminales glutamatérgicos y finalmente de los terminales GABAérgicos (11).
Compendio de la invención
El propósito de la presente invención es proporcionar nuevos enfoques terapéuticos para tratar la EA y los trastornos relacionados.
Los inventores han identificado diversos fármacos que, solos o en combinación(es), pueden afectar de forma eficaz a las rutas involucradas en la EA y representan terapias nuevas y efectivas para el tratamiento de la EA y los tras
tornos relacionados.
Por lo tanto, la invención proporciona nuevas composiciones para tratar la enfermedad EA y trastornos relacionados, tal y como se definen en las reivindicaciones.
En esta memoria también se describe una composición que comprende una combinación de al menos dos compues tos elegidos a partir del grupo que consiste en ácido aminocaproico, acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina, levosimendán y zonisamida, o sales o formula ciones de liberación sostenida de los mismos, para uso en el tratamiento de la enfermedad de Alzheimer o un tras torno relacionado.
También se describe en esta memoria una composición que comprende una combinación de al menos dos com puestos elegidos a partir del grupo que consiste en ácido aminocaproico, acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexi da, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina, levosimendán y zonisamida, o sales o formulaciones de liberación sostenida de los mismos, para una administración simultánea, por separado o secuencial.
Las combinaciones de fármacos más preferidas comprenden 2, 3, 4 o 5 fármacos distintos, incluso más preferible mente 2 o 3. Además, las combinaciones de fármacos anteriores también pueden usarse en combinación adicional con fármacos o tratamientos adicionales usados actualmente para la EA.
También se describe en esta memoria un método para producir fármaco(s) para tratar la enfermedad de Alzheimer o un trastorno relacionado, en donde el método comprende una etapa de someter a ensayo el o los fármacos candida tos para estudiar la actividad sobre la función sináptica y la angiogénesis y la respuesta al estrés celular y seleccio nar el o los fármacos candidatos que mejoran la función sináptica, atenúan la desregulación angiogénica y modulan la respuesta al estrés celular.
También se describe en esta memoria un método para producir una composición para tratar la enfermedad de Alz heimer o un trastorno relacionado, en donde el método comprende preparar una combinación de un fármaco que modula la función sináptica y/o un fármaco que atenúa la desregulación angiogénica y/o un fármaco que modula la respuesta al estrés celular, para una administración simultánea, por separado o secuencial a un sujeto que lo nece site.
Breve descripción de las Figuras
Figura 1: Efecto de fármacos seleccionados sobre la extensión de neuritas en un cultivo de neuronas corticales pri marias de rata, intoxicadas con beta-amiloide. QQ: p <0,01; QQQQ: p <0,00001: significativamente diferente del vehículo. *: p <0,05; ****: p <0,0001: significativamente diferente de Abeta25-35. Prueba t de Student bilateral. Ap25-35 20 pM produce una intoxicación significativa, superior al 25%, en comparación con las neuronas tratadas con vehículo. Esta intoxicación se evita significativamente con Acamprosato (Fig. 1A) o Zonisamida (Fig. 1B).
Figura 2: Efecto de Fenformina sobre la extensión de neuritas en un cultivo de neuronas corticales primarias de rata intoxicada con beta-amiloide. QQ: p <0,01: significativamente diferente del vehículo. **: p <0,001: significativamente diferente de AP25-35. Prueba t de Student bilateral. AP25-35 20 pM produce una intoxicación significativa, superior al 25%, en comparación con las neuronas tratadas con vehículo. Esta intoxicación se evita significativamente con Fen formina.
Figura 3: Efecto protector de fármacos seleccionados contra la toxicidad del péptido beta-amiloide sobre la liberación de LDH desde células cerebrales endoteliales de rata. ^ : p <0,05: significativamente diferente del vehículo. **: p <0,01; ***: p <0,0001; ****: p <0,00001: significativamente diferente de Ap25-35. Prueba t de Student bilateral. AP25-35 30 pM produce una intoxicación moderada pero significativa (Fig. 3A a D). Esta intoxicación se evita significativa mente con Leflunomida (Fig. 3A), Terbinafina (Fig. 3B), Sulfisoxazol (Fig. 3C) o Baclofeno (-) (Fig. 3D). Además, Leflunomida y Terbinafina no solo evitan el efecto nocivo amiloide, sino que también disminuyen la muerte celular espontánea en el medio de cultivo.
Figura 4: Efecto de fármacos seleccionados sobre la viabilidad de PC12 diferenciadas con NGF después de una intoxicación con beta-amiloide. QQQQ: p <0,00001: significativamente diferente del vehículo. **: p <0,01; ***: p <0,0001: significativamente diferente de Abeta25-35. Prueba t de Student bilateral. Abeta25-35 10 pM produce una intoxicación significativa, superior al 25%, en comparación con las neuronas tratadas con vehículo (Fig. 4A y 4B). Esta intoxicación se evita significativamente con Prilocaína (Fig. 4A) o Amlodipina (Fig. 4B).
Figura 5: Efecto de fármacos seleccionados sobre la liberación de LDH en un cultivo de neuronas corticales prima rias de rata, intoxicadas con beta-amiloide. é é é é : p <0,000001: significativamente diferente del vehículo. *: p <0,05; ***: p <0,001: significativamente diferente de AP25-35. Prueba t de Student bilateral. AP25-3520 pM produce una intoxi cación significativa, superior al 25%, en comparación con las neuronas tratadas con vehículo (Fig. 5A y B). Esta intoxicación se evita significativamente con Zonisamida (Fig. 5A) o Sulfisoxazol (Fig. 5B) o Leflunomida (Fig. 5C).
Figura 6: Efecto de un pretratamiento con fármacos seleccionados contra una lesión con AP1-42 humano en HBMEC. A) Validación del modelo experimental utilizado para la detección de fármacos: 1 hora de pretratamiento con VEGF 10 nM protegía significativamente la red capilar de esa lesión amiloide (+78% de la red capilar en comparación con una intoxicación con amiloide). *: p <0,05: significativamente diferente del control (sin intoxicación) é: p <0,05: signi ficativamente diferente de una intoxicación con amiloide (ANOVA prueba de Dunett Post-Hoc). La intoxicación se evita significativamente con Sulfisoxazol, Levosimendán, Terbinafina, Baclofeno, Ácido aminocaproico, Sulodexida o Fenoldopam, tal y como se muestra en los experimentos de dosis-respuesta, respectivamente en la Fig. 6B, Fig. 6C, Fig. 6D, Fig. 6E, Fig. 6F, Fig. 6G, Fig. 6H. é: p <0,05: significativamente diferente de la siguiente dosis *: p <0,05: significativamente diferente de la intoxicación con amiloide (ANOVA prueba de Dunett Post-Hoc).
Figura 7: Efecto de un pretratamiento con fármacos seleccionados sobre la liberación de LDH en la toxicidad de Ap1-42 humano en células corticales primarias de rata. A) Validación del modelo experimental utilizado para la detección de fármacos: 1 hora de pretratamiento con BDNF (50 ng/ml) protegía significativamente las neuronas de esa lesión amiloide (-62%), lo que se considera un control positivo para la neuroprotección. *: p <0,05: significativamente dife rente del control (sin intoxicación) é: p <0,05: significativamente diferente de la intoxicación con amiloide (ANOVA prueba de Dunett Post-Hoc). Para todos los experimentos, AP1-42 produce una intoxicación significativa en compara ción con las neuronas tratadas con vehículo. La intoxicación se evita significativamente con Baclofeno (-86%) (B), Sulfisoxazol (-42%) (C), Levosimendán (-133%) (D), Etomidato (-50%) (E), Carbenoxolona (-39%) (F), y con Cinarizina (-50%) (G). Para todos los experimentos, é: p <0,05: significativamente diferente de una intoxicación con AP1-42 (ANOVA prueba de Dunett Post-Hoc).
Figura 8: Efecto de una terapia de combinación con Sulfisoxazol y Levosimendán sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. é: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agregado (AP1-42 2,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Sulfisoxazol y Levosimendán (A), mientras que, con esas concentraciones, Levosimendán (B) y Sulfisoxazol (C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
Figura 9: Efecto de una terapia de combinación con Sulfisoxazol y Terbinafina sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. é: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agrega do (AP1-422,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Sulfisoxazol y Levosimendán (A) mientras que, con esas concentraciones, Sulfisoxazol (B) y Terbinafina (C) por sí solos, no tienen un efecto signi ficativo sobre la intoxicación.
Figura 10: Efecto de una terapia de combinación con Baclofeno y Levosimendán sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. é: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agregado (AP1-42 2,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Baclofeno y Levosi mendán (A) mientras que, con esas concentraciones, Levosimendán (B) y Baclofeno (C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
Figura 11: Efecto de una terapia de combinación con Terbinafina y Ácido aminocaproico sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. é: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agregado (AP1-42 2,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Terbinafina y Ácido aminocaproico (A) mientras que, con esas concentraciones, Ácido aminocaproico (B) y Terbinafina (C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
Figura 12: Efecto de la combinación de Ácido aminocaproico y Levosimendán sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. é: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agrega do (AP1-422,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Levosimendán y Ácido amino caproico (A) mientras que, con esas concentraciones, Ácido aminocaproico (B) y Levosimendán (C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
Figura 13: Efecto de una terapia de combinación con Terbinafina y Levosimendán sobre la longitud total de la red capilar en cultivos de HBMEC intoxicados con beta-amiloide. ^: p <0,05, significativamente diferente de AP1-42. *: p <0,05, significativamente diferente del vehículo. ANOVA prueba de Bunett Post-Hoc. El péptido amiloide humano agregado (AP1-42 2,5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de Terbinafina y Levo simendán (A) mientras que, con esas concentraciones, Terbinafina (B) y Levosimendán (C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
Descripción detallada de la invención
La presente invención proporciona nuevos enfoques terapéuticos para tratar la EA o trastornos relacionados. La invención describe el uso novedoso de fármacos o combinaciones de fármacos que permiten una corrección efectiva de tales enfermedades y que se pueden usar para el tratamiento de un paciente.
La expresión "trastorno relacionado con EA" incluye demencia senil de tipo EA (DSTA), demencia con cuerpos de Lewis, demencia vascular, deterioro cognitivo leve (DCL), deterioro de la memoria asociado a la edad (DMAE) y problemas asociados con el envejecimiento, Parkinson post-encefalítico, esclerosis lateral amiotrófica (ELA), escle rosis múltiple (EM) y síndrome de Down.
Tal y como se usa en esta memoria, el "tratamiento" de un trastorno incluye la terapia, prevención, profilaxis, retraso o reducción de los síntomas provocados por el trastorno. El término tratamiento incluye en particular el control de la progresión de una enfermedad y los síntomas asociados.
El término "mejora", cuando se refiere a la función sináptica, incluye cualquier aumento en la función sináptica en comparación con la función existente en el sujeto. Una mejora de ese tipo puede incluir una restauración, es decir, a los niveles normales, o un aumento menor, que todavía son suficientes para mejorar el estado del paciente. Esa mejora puede evaluarse o verificarse utilizando pruebas biológicas conocidas, como las descritas en la sección ex perimental.
El término "aumento", cuando se refiere a la angiogénesis, incluye cualquier aumento de la angiogénesis en compa ración con el nivel existente en el sujeto. Una mejora de ese tipo puede incluir una restauración, es decir, a niveles normales, o un aumento menor, que todavía son suficientes para mejorar el estado del paciente. Ese aumento pue de evaluarse o verificarse usando pruebas biológicas conocidas, como las descritas en la sección experimental. El término "inhibir", cuando se refiere a la respuesta frente al estrés celular ("REC"), incluye cualquier reducción en la REC en comparación con la actividad existente en el sujeto. Esa reducción puede incluir una disminución parcial, por ejemplo, del 5-20%, que es suficiente para mejorar el estado del paciente, así como reducciones más sustanciales, por ejemplo, del 20-50% o más de la inhibición completa, por ejemplo, superior al 50%. La inhibición se puede eva luar o verificar utilizando pruebas biológicas conocidas, como las descritas en la sección experimental.
Además, la designación de compuestos específicos dentro del contexto de esta invención se entiende que incluye no solo las moléculas mencionadas específicamente, sino también cualquier sal farmacéuticamente aceptable de las mismas.
La expresión "tratamiento/terapia combinada o combinatoria" designa un tratamiento en el que al menos dos o más fármacos se administran conjuntamente a un sujeto para causar un efecto biológico. En una terapia combinada de acuerdo con esta invención, los al menos dos fármacos pueden administrarse juntos o por separado, al mismo tiem po o secuencialmente. Además, los al menos dos fármacos pueden administrarse a través de diferentes vías y pro tocolos. Como resultado, aunque se pueden formular juntos, los fármacos de una combinación también se pueden formular por separado.
Como se ha indicado anteriormente, en esta memoria se describen composiciones para tratar la enfermedad de Alzheimer o un trastorno relacionado, en un sujeto que lo necesita, usando fármacos particulares o combinaciones de fármacos que mejoran la función sináptica y/o aumentan la angiogénesis y/o inhiben la respuesta al estrés celu lar.
Mediante una integración integral de datos experimentales que incluyen los resultados de estudios de biología celu lar, experimentos del perfil de expresión y estudios de asociación genética, que describen diferentes aspectos de la enfermedad de Alzheimer y los vínculos existentes con la señalización celular y las rutas funcionales, los inventores han descubierto que la función sináptica, la angiogénesis y la respuesta al estrés celular representan mecanismos importantes que se alteran en sujetos que tienen EA. Mediante investigaciones experimentales adicionales, los in ventores han seleccionado fármacos o combinaciones de fármacos que alteran eficazmente esas rutas y que mejo ran eficazmente la EA, como se ilustra en los ejemplos. Esos fármacos y combinaciones, por lo tanto, representan enfoques novedosos para tratar la EA y trastornos relacionados.
Los genes ubicados en dichas redes funcionales e implicados en la enfermedad de Alzheimer se seleccionaron mediante los siguientes criterios:
(1) - interacción directa con los genes responsables de causar los casos familiares de enfermedad de Alzheimer (APP, ApoE, presenilinas, proteína tau),
(2) - parejas funcionales de los genes seleccionados por el criterio (1),
(3) - parejas funcionales más cercanas de los genes seleccionados por el criterio (2).
A través de este procedimiento, los inventores han establecido que las redes responsables de la función sináptica, la angiogénesis y la respuesta al estrés celular son las principales redes funcionales afectadas en la enfermedad de Alzheimer.
Los inventores han establecido más específicamente que la pérdida sináptica es una característica distintiva, funcio nalmente relevante de la enfermedad de Alzheimer, que finalmente conduce a un deterioro cognitivo progresivo, pérdida de memoria y demencia. Es importante destacar que la pérdida sináptica se correlaciona mejor con el déficit cognitivo caracterizado por la patología de Alzheimer, en comparación con otros marcadores de lesiones celulares específicos de la EA que se manifiestan con un desarrollo de ovillos neurofibrilares o el depósito de placas amiloides. En consecuencia, la organización de la sinapsis y la plasticidad sináptica representan un objetivo importante para las intervenciones terapéuticas en el contexto de la enfermedad de Alzheimer.
La proteína APP se transporta y procesa axonalmente en terminales presinápticas, lo que conduce a una acumula ción elevada de Abeta en las sinapsis. Los oligómeros de Abeta42, así como las propias placas amiloides, son im portantes para inhibir la potenciación a largo plazo y son los principales responsables del deterioro de la memoria en pacientes con EA.
Nuestro procedimiento de integración de datos reveló un grupo de genes, que están implicados en una distorsión sináptica en la EA y que se pueden separar formalmente en tres grupos funcionales principales: proteínas que parti cipan en la organización de la densidad postsináptica ("PSD") y la transmisión correcta de la señal nerviosa a la membrana post-sináptica; proteínas que aseguran la liberación de neurotransmisores; y proteínas involucradas en el crecimiento del axón y la maduración del desarrollo de la maquinaria sináptica.
La presente solicitud describe, por lo tanto, que es importante, para un tratamiento eficaz de la EA, mejorar la activi dad de las proteínas implicadas en la densidad postsináptica.
Entre los genes identificados a través de nuestro análisis, el gen DLG2, que codifica la proteína de la familia MAGUK y crea una interfaz entre los receptores agrupados unidos a la membrana, las moléculas de adhesión celular y el citoesqueleto basado en actina, tiene un interés particular (17-18). Los inventores han identificado un gran grupo de receptores del glutamato ionotróficos/metabotróficos y de factores de crecimiento, que interaccionan directamente con la proteína DLG2 o el complejo de proteínas DLG2/PSD95 en las sinapsis excitadoras y que, por lo tanto, se pueden reconocer como dianas terapéuticas para tratar la enfermedad de Alzheimer.
La presente solicitud también describe que es importante, para un tratamiento eficaz de la EA, mejorar la actividad de las proteínas involucradas en la regulación de la liberación de neurotransmisores en la membrana presináptica. La liberación de neurotransmisores en una zona activa restringida y altamente especializada de la membrana plas mática presináptica se desencadena a través del potencial de acción y se controla mediante acciones combinadas de canales Cav de calcio dependientes del voltaje, canales MaxiK/BK (canales de potasio activados por calcio de amplia conductancia) y proteínas cinasas PRKG dependientes de GMPc, todas las cuales están estrechamente asociadas, como lo demuestra nuestro análisis, con el desarrollo de la enfermedad de Alzheimer. Además de estos módulos funcionales implicados en la liberación de neurotransmisores, los inventores han definido otro grupo de proteínas, ligadas a la desregulación de la neurotransmisión sináptica en el curso de la enfermedad de Alzheimer, que son responsables de la maduración, el acoplamiento y la fusión de las vesículas sinápticas (por ejemplo, las proteínas de andamiaje STX2, STXBP6, BiN1, rAb3B, UNC13C y RIMS1/2). Por lo tanto, estas rutas funcionales se priorizaron como dianas terapéuticas apropiadas para el tratamiento de la enfermedad de Alzheimer.
La presente solicitud describe además que es importante, para un tratamiento eficaz de la EA, mejorar la actividad de las proteínas implicadas en la regulación del crecimiento y la orientación del axón.
Las proteínas que participan en la regulación del crecimiento y la orientación del axón permiten que las células pre cursoras neuronales y los axones migren hacia destinos adecuados para garantizar una ubicación y conectividad correctas; también están implicadas en la maduración del desarrollo de sinapsis establecidas recientemente, así como en la degradación de axones y sinopsis en la enfermedad EA. Estos procesos tienen un papel fundamental para la ejecución de funciones cognitivas y parecen ser extremadamente vulnerables al efecto tóxico de los depósi tos de Abeta.
Las etapas consecutivas del crecimiento y orientación de los axones están estrechamente controladas por acciones combinadas de moléculas Netrinas, Semaforinas, Efrinas, DLL y Slits extracelulares o ligadas a la membrana y sus respectivos receptores funcionales, la mayoría de los cuales han sido reveladas por nuestro enfoque de extracción de datos. Los resultados funcionales de la activación de la mayoría de los receptores del crecimiento axónico están
estrechamente relacionados con su capacidad para modular de forma diferencial la actividad de las pequeñas GTPasas RhoA, Rac1 y Cdc42, siendo RhoA la GTPasa la principal responsable de la retracción de las neuritas y el colapso del cono de crecimiento (19). Estas rutas de señalización se han reconocido como dianas terapéuticas per tinentes para el tratamiento de la enfermedad de Alzheimer.
Por lo tanto, la presente solicitud reconoce que es importante, para un tratamiento eficaz de la EA, mejorar la función sináptica alterada en la enfermedad de Alzheimer y otros trastornos neurodegenerativos, mediante la modulación de los genes y proteínas diana descritos anteriormente.
A través del procedimiento de extracción de datos, los inventores también han establecido que la red responsable de la angiogénesis representa otra red funcional importante afectada por la enfermedad de Alzheimer.
La angiogénesis desempeña un papel fundamental para garantizar una homeostasis tisular y en respuestas adaptativas a desafíos ambientales y fisiológicos, tales como la hipoxia o la cicatrización de heridas; su disfunción contribu ye a la patogénesis de numerosas y heterogéneas patologías que varían desde complicaciones cardiovasculares hasta el crecimiento de un tumor y la metástasis.
Aunque la enfermedad de Alzheimer se considera tradicionalmente como una afección neurodegenerativa acompa ñada de una patología vascular colateral, nuestro análisis permite reevaluar el impacto patógeno de la desregulación vascular y atribuye un papel importante y probablemente causal a las rutas angiogénicas en la etiología de esta enfermedad. Los inventores han descubierto que los genes que regulan la angiogénesis están extremadamente enriquecidos en las redes de señalización implicadas en la enfermedad de Alzheimer. Esta conclusión tiene profun das consecuencias para la prevención y la curación de la enfermedad de Alzheimer y proporciona nuevas pautas para el tratamiento combinatorio de este complejo trastorno neurodegenerativo.
Entre las rutas de señalización estrechamente implicadas en la remodelación vascular asociada con la enfermedad de Alzheimer, se han identificado varios módulos funcionales mediados por VEGFR1, ErbB4, Notch, DCC, CD44, receptores de efrina y cadherinas.
Como se revela a través de nuestro enfoque de extracción de datos, otras proteínas diana, potencialmente involu cradas en el desarrollo de defectos vasculares manifestados en el curso de la enfermedad de Alzheimer, incluyen IL20Ra, LEPTR, NRP1 y NRP2, y receptores de endotelina EDNRA, proteínas que participan en la organización y la remodelación de la matriz extracelular (THBS2, LAMA1, COL4A2, ADAMTS12 y ADAM10) o proteínas (por ejemplo, TLL2) que tienen un papel importante en el procesamiento funcional de moduladores angiogénicos bien conocidos, tales como la prolactina, la hormona del crecimiento y el lactógeno placentario (20).
Además, también hemos descubierto que varios genes, asociados con la enfermedad de Alzheimer, representan moduladores aguas arriba y efectores aguas abajo de las cinasas activadas por AMP, reguladores importantes del sistema vascular (por ejemplo, receptores de leptina y CNTF, ruta de señalización de trombina, cinasas CAMKK2p y LKB1) (21-24). Este hallazgo nos ha permitido definir la red de señalización mediada por AMPK como una diana terapéutica razonable para el tratamiento de la enfermedad de Alzheimer.
El ácido fosfatídico (PA), el ácido lisofosfatídico (LPA) y la esfingosina 1-fosfato (S1P) son fosfolípidos naturales que poseen potentes propiedades de señalización. En particular, estos factores de crecimiento fosfolipídico muestran efectos divergentes sobre el potencial angiogénico de las células endoteliales (25). Utilizando nuestro enfoque de extracción de datos, identificamos una gran cantidad de genes, involucrados en el metabolismo de LPA o modulados por la señalización de LPA y potencialmente vinculados a la progresión de la enfermedad de Alzheimer (MTR, MAT2B, CUBN, ATP10A, THEM2, PITPNC1, ENPPG, SGPP2, AGPAT, DGKH, DGKB, MGST2, PLD2 y DRD2). Por lo tanto, llegamos a la conclusión de que esta red de señalización representa una diana terapéutica adecuada para el tratamiento de la enfermedad de Alzheimer.
La presente solicitud también enfatiza en la importancia de aumentar la angiogénesis alterada en la enfermedad de Alzheimer y otros trastornos neurodegenerativos, mediante una modulación de los genes y las proteínas diana des critos anteriormente.
Finalmente, hemos establecido que la red responsable de la respuesta frente al estrés celular es la tercera red fun cional principal, afectada por la enfermedad de Alzheimer.
Hemos establecido más específicamente que la respuesta al estrés celular es una característica distintiva, funcio nalmente relevante de la enfermedad de Alzheimer. Como se analiza a continuación, los inventores han identificado tres familias de proteínas, dentro de la red de respuesta al estrés celular, que son funcionalmente relevantes para la génesis y el control de la enfermedad de Alzheimer, y representan dianas valiosas para las terapias combinadas. Estos grupos de proteínas son, más específicamente, proteínas que participan en la homeostasis del calcio, en el plegamiento de proteínas y en la ejecución de la apoptosis.
La presente solicitud describe más específicamente composiciones que usan una combinación de fármacos que modula la actividad de una proteína implicada en la homeostasis del calcio.
El calcio, uno de los mensajeros intracelulares más importantes, media una pleiotropía de procesos celulares en las células neuronales y endoteliales, incluida la plasticidad sináptica, la angiogénesis y la apoptosis.
El nivel de calcio intracelular se regula con precisión mediante la acción cooperativa de una serie de canales permeables al calcio, bombas de calcio e intercambiadores de calcio en la membrana plasmática y el retículo endoplasmático (26-27). Hemos identificado una red de genes implicados en la ruta de la homeostasis del calcio, cuya función podría estar modificada por proteínas de presenilina mutantes o por p-amiloide tóxico en el curso de la en fermedad de Alzheimer. Entre ellos, los receptores IP3R (ITPR1) y RYR3, ATP2A3 (SERCA3 Ca2 ATPasa) que regulan la homeostasis del calcio a nivel de RE, la ATPasa ATP2B1 de la membrana plasmática, que extruye los iones de calcio desde las células eucariotas frente a gradientes de concentración y los canales de Na+ activados por voltaje, tienen un interés particular como posibles dianas terapéuticas para el tratamiento de la enfermedad de Alzheimer.
También se describen en esta memoria composiciones que usan una combinación de fármacos que modula la acti vidad de una proteína implicada en el plegamiento o la agregación de proteínas.
La agregación de proteínas es un fenómeno citopatológico central en la EA. Dos características distintivas celulares principales de la enfermedad de Alzheimer se manifiestan en el desarrollo de ovillos neurofibrilares (ONFs) y el de pósito de placas amiloides, compuestas de proteína tau hiperfosforilada agregada y fragmentos Ap de proteína APP, respectivamente. Sin embargo, otra proteína propensa a la agregación, la a-sinucleína, reconocida como una carac terística distintiva bastante específica de la enfermedad de Parkinson, se puede detectar en las placas amiloides en la mayoría de los casos de formas esporádicas y familiares de la enfermedad de Alzheimer.
Hemos determinado varios genes implicados en la modulación del plegamiento, la modificación postraduccional y el procesamiento de cada componente principal de las agregaciones de proteínas asociadas a la enfermedad de Alzheimer, como dianas terapéuticas pertinentes para el tratamiento de la enfermedad de Alzheimer, por ejemplo, las proteínas APBA1 y APBA2BP que interaccionan con APP y regulan su estabilidad y funciones, o la ubicuitinaproteína ligasa PARK2 que está implicada en el aclaramiento de la a-sinucleína (28). Además, la cinasa GSK-3p podría desempeñar un papel particularmente importante en la patogenia del plegamiento defectuoso de proteínas en el curso de la enfermedad de Alzheimer. Esta conclusión se ve reforzada por nuestro hallazgo de que unos pocos módulos de señalización que regulan la actividad de la cinasa GSK-3p y su interacción con la proteína tau - WWOX (29), el receptor de hialuronano CD44, los receptores de Wnt Fz2/ROR2 y el complejo de receptor de insulina/fosfatasa PTPRG (30) - están asociados con la progresión de la enfermedad de Alzheimer.
La presente solicitud también describe composiciones que usan una combinación de fármacos que inhibe la apopto sis que se reconoce como un mecanismo celular principal responsable de la pérdida celular en la enfermedad de Alzheimer.
Tal y como se ha identificado con nuestro análisis, la apoptosis en el caso de la enfermedad de Alzheimer se ejecu ta, muy probablemente, a través de rutas canónicas dependientes de p53.
La proteína p53 se puede regular mediante modificaciones postraduccionales y mediante interacciones con factores reguladores positivos y negativos. Hemos identificado varias de esas proteínas reguladoras (WWOX, MDM1, HIPK2 y PML), confirmando la propuesta sobre el papel fundamental de la proteína p53 en la ejecución de la muerte celular en la enfermedad de Alzheimer (31 -33).
Entre los sistemas receptores que podrían estar directa y específicamente implicados en la inducción de la apoptosis en el contexto de la enfermedad de Alzheimer, los receptores de netrina UNC5C (Homólogo C de Unc-5) y DCC (suprimido en el carcinoma colorrectal, del inglés “Deleted in Colorectal Carcinoma”), que participan en la guía axonal y la angiogénesis, tienen un interés particular. Estos receptores se denominan supresores tumorales condi cionales putativos, ya que se comportan como receptores dependientes de netrina que inducen la apoptosis en au sencia de su ligando (34). La unión de netrina-1 a esos receptores inhibe la apoptosis dependiente de p53, mientras que p53 está implicado directamente en la regulación transcripcional de netrina-1 y sus receptores (33). Además, se sabe que el receptor de DCC está procesado por la presenilina, lo que indica su importante papel en el desarrollo de la enfermedad de Alzheimer (35). Por lo tanto, nuestra extracción de datos sugiere que la muerte celular programada dependiente de los receptores de netrina y mediada por p53, podría ser una de las rutas específicas proapoptóticas implicadas en la pérdida celular patológica en el contexto de la enfermedad de Alzheimer, además de programas proapoptóticos bastante inespecíficos, estimulados por la homeostasis de calcio interrumpida y la producción exce siva de ROS.
La presente solicitud también describe composiciones que emplean una combinación de fármacos que inhibe la actividad de al menos dos proteínas distintas, implicadas en la homeostasis del calcio, en el plegamiento de las proteínas y en la ejecución de la apoptosis.
La presente solicitud propone composiciones novedosas, que se pueden usar para inhibir la respuesta al estrés celular inducida en la enfermedad de Alzheimer y otros trastornos neurodegenerativos, modulando genes y proteí nas diana descritos anteriormente.
Como se ha indicado anteriormente, también se describen en esta memoria composiciones para tratar la enferme dad de Alzheimer o un trastorno relacionado en un sujeto que lo necesita, usando una combinación de fármacos que mejoran la función sináptica y/o aumentan la angiogénesis y/o inhiben la respuesta al estrés celular.
Más específicamente, los inventores han seleccionado y sometido a ensayo una serie de fármacos o combinaciones de fármacos que alteran una o, preferiblemente, todas las rutas descritas anteriormente. Como se describe en los ejemplos, estas combinaciones de fármacos tienen un fuerte efecto sobre la enfermedad de Alzheimer y representan nuevos enfoques terapéuticos de la patología. Estas combinaciones de fármacos son particularmente ventajosas porque afectan a diferentes rutas y, por lo tanto, son más eficaces. Además, debido a su eficacia y modo de acción, las combinaciones de fármacos se pueden usar en dosificaciones bajas, lo cual es una ventaja adicional muy sustancial.
Los fármacos más preferidos se enumeran en la Tabla 1 a continuación.
Tabla 1


A este respecto, se describen en esta memoria composiciones que comprenden una combinación de al menos dos compuestos elegidos a partir del grupo que consiste en ácido aminocaproico, acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina, levosimendán y zonisamida, o sales o formulaciones de liberación sostenida de los mismos, para una administración simultánea, por separado o secuencial.
También se describen en esta memoria composiciones que comprenden al menos un compuesto elegido a partir del grupo que consiste en ácido aminocaproico, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbama zina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina y levosimendán, o sal(es) o formulación(es) de liberación sostenida de los mismos, en combinación con al menos un compuesto elegido a partir del grupo que consiste en acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina y zonisamida, o sal(es) o formulación(es) de liberación sostenida, para una administración simultánea, por separado o secuencial.
Tal y como se describe en los ejemplos, las terapias combinadas que usan al menos 2 de los fármacos menciona dos anteriormente, conducen a una corrección eficaz de la enfermedad de Alzheimer.
La terapia según la invención se puede llevar a cabo sola o como una combinación de fármacos.
En una realización preferida, los fármacos de la invención se usan en combinación(es) para una administración combinada, por separado o secuencial, con el fin de proporcionar el efecto más efectivo. A este respecto, las com posiciones para tratar la enfermedad de Alzheimer emplean fármaco(s) que mejora(n) la función sináptica y fármaco(s) que atenúa(n) la angiogénesis y/o fármaco(s) que inhibe(n) la respuesta al estrés celular.
También se describen en esta memoria las composiciones para uso en el tratamiento de la enfermedad de Alzhei mer o un trastorno relacionado que comprenden al menos una de las siguientes combinaciones de fármacos:
- un modulador de AMPK (preferiblemente, fenformina) y un inhibidor del canal de sodio SCN1A y un activa dor de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferible mente, meticlotiazida),
- un modulador de AMPK (preferiblemente, fenformina) y un modulador de la actividad de los receptores GABAérgicos y glutamatérgicos (seleccionado preferiblementes a partir de acamprosato, etomidato y aprindina),
- un modulador de AMPK (preferiblemente, fenformina) y un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador del receptor de rianodina RYR3 (preferiblemente, prilocaína),
- un modulador del receptor GABBR2 (preferiblemente, baclofeno) y un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato),
- un modulador del receptor GABBR2 (preferiblemente, baclofeno) y un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol),
- un modulador del receptor GABBR2 (preferiblemente, baclofeno) y un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (pre feriblemente, meticlotiazida),
- un modulador del receptor GABBR2 (preferiblemente, baclofeno) y un modulador de las sintasas de hialuronano HAS1-3 (preferiblemente, leflunomida),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador de los receptores de adenosina ADORA1/2/3 (preferiblemente, difilina),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y un antagonis ta del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y un inhibidor de las fosfolipasas PLA1A y PLA2 (preferiblemente, mepacrina),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y un modulador de la actividad de los receptores GABAérgicos y glutamatérgicos (seleccionado preferiblemente a partir de acamprosato, etomidato y aprindina),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y una chaperona química (preferiblemente, rifabutina),
- un modulador de AMPK (preferiblemente, fenformina) y un inhibidor de las fosfodiesterasas PDE11A y PDE4A, PDE5A (seleccionado preferiblemente a partir de tadalafilo, enprofilina y oxtrifilina),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador de la señalización del receptor de trombina F2R (preferiblemente, argatrobán y cefmenoxima),
- un modulador de AMPK (preferiblemente, fenformina) y un modulador de los receptores purinérgicos P2RY1 y P2RY12 (preferiblemente, clopidogrel),
- un modulador de la actividad de los receptores GABAérgicos y glutamatérgicos (seleccionado preferible mente a partir de acamprosato, etomidato y aprindina) y un modulador de CASR (preferiblemente, cinacalcet),
- un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol) y un modulador de CASR (preferiblemente, cinacalcet),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y un modulador de la señalización del receptor de trombina F2R (seleccionado preferiblemente a partir de argatrobán y cefmenoxima),
- un modulador del receptor GABBR2 (preferiblemente, baclofeno) y un modulador de los receptores purinér gicos P2RY1 y P2RY12 (preferiblemente, clopidogrel),
- un modulador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato) y un modulador de los receptores purinérgicos P2RY1 y P2RY12 (preferiblemente, clopidogrel),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un antagonista de los canales de calcio CACNA dependientes de voltaje (seleccionado preferiblemente a partir de cinarizina, benidipina, pa
rametadiona y amlodipina),
- un modulador de la actividad de los receptores GABAérgicos y glutamatérgicos (seleccionado preferible mente a partir de acamprosato, etomidato y aprindina) y un antagonista de los canales de calcio CACNA dependientes de voltaje (seleccionado preferiblemente a partir de cinarizina, benidipina, parametadiona y amlodipina),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador de la señalización de HIF1A (preferiblemente ciclopirox),
- un modulador de los receptores GABAérgicos y glutamatérgicos (seleccionado preferiblemente a partir de acamprosato, etomidato y aprindina) y un modulador de la señalización de HIF1A (preferiblemente, ciclopirox),
- un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol) y un modulador de la fos forilación oxidativa (seleccionado preferiblemente a partir de amobarbital y metimazol),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador de la fosforilación oxidativa (seleccionado preferiblemente a partir de amobarbital y metimazol),
- un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol) y un modulador del meta bolismo de la vitamina K (preferiblemente, cefotetán),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador del metabolismo de la vitamina K (preferiblemente, cefotetán),
- un modulador de la actividad de los receptores GABAérgicos y glutamatérgicos (seleccionado preferible mente a partir de acamprosato, etomidato y aprindina) y un modulador de PRKG1 (preferiblemente, tetranitrato de eritritilo),
- un inhibidor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un modulador de los canales de BK (preferiblemente, meticlotiazida) y un modulador de PRKG1 (preferi blemente, tetranitrato de eritritilo),
- un antagonista del receptor de endotelina EDNRA (preferiblemente, sulfisoxazol) y un modulador de PRKG1 (preferiblemente, tetranitrato de eritrilo),
- un modulador de KCNJ11 (seleccionado preferiblemente a partir de mitiglinida y levosimendán) y un modu lador de PRKG1 (preferiblemente, tetranitrato de eritrilo),
- un modulador de KCNJ11 (seleccionado preferiblemente a partir de mitiglinida y levosimendán) y un inhibi dor del canal de sodio SCN1A y un activador de los canales de BK (preferiblemente, zonisamida) o un mo dulador de los canales de BK (preferiblemente, meticlotiazida),
- un modulador de KCNJ11 (seleccionado preferiblemente a partir de mitiglinida y levosimendán) y un modu lador de RHOA (seleccionado preferiblemente a partir de terbinafina y risedronato).
También se describe en esta memoria cualquier combinación de compuestos seleccionados a partir de ácido aminocaproico, acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina, levosimendán y zonisamida o sales o formulaciones de liberación sostenida de los mismos, para uso en el tratamiento de la enfermedad de Alzheimer o un trastorno relacionado.
También se describe en esta memoria un uso de una composición en el tratamiento de la enfermedad de Alzheimer o un trastorno relacionado, que comprende al menos un compuesto elegido a partir del grupo que consiste en ácido aminocaproico, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina y levosimendán, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de libe ración sostenida de los mismos, en combinación con al menos un compuesto elegido a partir del grupo que consiste en acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, lefluno mida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina y zonisamida, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de liberación sostenida de los mismos.
También se describe en esta memoria una composición para tratar la enfermedad de Alzheimer (EA) o un trastorno relacionado en un sujeto que lo necesita, que comprende al menos ácido aminocaproico, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de liberación sostenida de los mismos. Por ejemplo, el ácido aminocaproico se usa en combinación con al menos un compuesto adicional, seleccionado preferiblemente a partir de acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina, levosimendán y zonisamida o sales o profármacos o derivados o formulaciones de liberación sostenida de los mismos, para una administración combinada, por separado o secuencial.
También se describen en esta memoria composiciones que comprenden ácido aminocaproico, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de liberación sostenida del mismo, y al menos un compuesto adicional selec cionado a partir de baclofeno, sulfisoxazol, terbinafina y levosimendán, o sales o profármacos o derivados o formula ciones de liberación sostenida de los mismos, para una administración combinada, por separado o secuencial. La presente invención se refiere a una composición para tratar la enfermedad de Alzheimer (EA) o un trastorno rela cionado en un sujeto que la necesita, que comprende al menos levosimendán, o sal(es) o formulación(es) de libera ción sostenida del mismo. En una realización particular, levosimendán se usa en combinación con al menos un compuesto adicional seleccionado preferiblemente a partir de ácido aminocaproico, acamprosato, amlodipina, arga trobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenfor mina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina y zonisamida o sales o formula ciones de liberación sostenida de los mismos, para una administración combinada, por separado o secuencial. Una composición preferida de la invención comprende levosimendán, o sal(es) o formulación(es) de liberación sos tenida del mismo, y al menos un compuesto adicional seleccionado a partir de ácido aminocaproico, baclofeno, sulfi soxazol y terbinafina, o sales o formulaciones de liberación sostenida de los mismos, para una administración com binada, por separado o secuencial. Tal composición per se también representa un objeto particular de la invención. También se describe en esta memoria una composición para tratar la enfermedad de Alzheimer (EA) o un trastorno relacionado en un sujeto que lo necesita, en donde la composición comprende al menos Eplerenona, Carbenoxolo na, Sulodexida, Cinarizina o carbamazina, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de liberación sostenida de las mismas.
Por ejemplo, Eplerenona, Carbenoxolona, Sulodexida, Cinarizina o carbamazina, se usan en combinación con al menos un compuesto adicional seleccionado preferiblemente a partir de levosimendán, ácido aminocaproico, acam prosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, ciclopirox, eplerenona, carbenoxolona, sulodexida, carbamazina, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina y zo nisamida, o sales o profármacos o derivados o formulaciones de liberación sostenida de los mismos, para una admi nistración combinada, por separado o secuencial.
También se describe en esta memoria una composición que comprende Eplerenona, Carbenoxolona, Sulodexida, Cinarizina o carbamazina, o sal(es) o profármaco(s) o derivado(s) o formulación(es) de liberación sostenida de las mismas, y al menos un compuesto adicional seleccionado a partir de levosimendán, ácido aminocaproico, baclofeno, sulfisoxazol y terbinafina, o sales o profármacos o derivados o formulaciones de liberación sostenida de los mismos, para una administración combinada, por separado o secuencial. Una composición de ese tipo per se también repre senta un objeto particular de la invención.
También se describe en esta memoria una composición para un tratamiento combinatorio de la enfermedad de Alz heimer (EA) o un trastorno relacionado en un sujeto que lo necesita, que comprende al menos una de las siguientes combinaciones de fármacos para una administración combinada, por separado o secuencial:
- fenformina y zonisamida,
- fenformina y meticlotiazida,
- fenformina y acamprosato,
- fenformina y sulfisoxazol,
- baclofeno y ácido aminocaproico,
- baclofeno y levosimendán,
- baclofeno y terbinafina,
- baclofeno y risedronato,
- baclofeno y sulfisoxazol,
- baclofeno y zonisamida,
- baclofeno y meticlotiazida,
- baclofeno y sulfisoxazol,
- baclofeno y leflunomida,
- ácido aminocaproico y sulfisoxazol,
- ácido aminocaproico y terbinafina,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol,
- levosimendán y terbinafina,
- zonisamida y difilina,
- meticlotiazida y difilina,
- zonisamida y prilocaína,
- meticlotiazida y prilocaína,
- zonisamida y sulfisoxazol,
- fenformina y clopidogrel,
- acamprosato y cinacalcet,
- sulfisoxazol y cinacalcet,
- terbinafina y argatrobán,
- terbinafina y cefmenoxima,
- baclofeno y clopidogrel,
- terbinafina y clopidogrel,
- risedronato y clopidogrel,
- zonisamida y cinarizina,
- acamprosato y tetranitrato de eritritilo,
- sulfisoxazol y tetranitrato de eritritilo,
- mitiglinida o levosimendán y tetranitrato de eritritilo, - mitiglinida o levosimendán y zonisamida,
- mitiglinida o levosimendán y terbinafina,
- mitiglinida o levosimendán y risedronato.
- mitiglinida o levosimendán y meticlotiazida,
- meticlotiazida o zonisamida y sulfisoxazol,
- terbinafina o risedronato y sulfisoxazol,
- terbinafina o risedronato y mepacrina,
- terbinafina o risedronato y acamprosato,
- terbinafina o risedronato y rifabutina,
- tadalafilo o enprofilina u oxtrifilina y fenformina,
- zonisamida o meticlotiazida y argatrobán o cefmenoxima,
- risedronato y argatrobán o cefmenoxima,
- zonisamida o meticlotiazida y cinarizina o benidipina o parametadiona o amlodipina,
- acamprosato y cinarizina o benidipina o parametadiona o amlodipina,
- zonisamida o meticlotiazida y ciclopirox,
- sulfisoxazol y amobarbital,
- zonisamida o meticlotiazida y amobarbital,
- sulfisoxazol y cefotetán,
- zonisamida o meticlotiazida y cefotetán,
- zonisamida o meticlotiazida y tetranitrato de eritritilo.
Los ejemplos específicos de tales composiciones comprenden una de las siguientes combinaciones de fármacos para una administración combinada, por separado o secuencial:
- baclofeno y ácido aminocaproico,
- baclofeno y levosimendán,
- ácido aminocaproico y sulfisoxazol,
- ácido aminocaproico y terbinafina,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol,
- levosimendán y terbinafina,
- eplerenona y levosimendán,
- eplerenona y sulfisoxazol,
- eplerenona y fenoldopam,
- sulodexida and levosimendán,
- sulodexida y sulfisoxazol,
- sulodexida y fenoldopam o
- eplerenona y sulodexida.
Tal y como se ilustra en la sección experimental, las composiciones que comprenden al menos ácido aminocaproico o levosimendán, proporcionan un efecto terapéutico y biológico sustancial para mejorar la enfermedad de Alzheimer en sujetos humanos. Esas composiciones evitan eficazmente los efectos tóxicos de la proteína o péptido amiloide b sobre las células humanas y representan métodos novedosos y potentes para tratar ese trastorno.
En otra realización preferida, las composiciones según la invención comprenden una combinación de al menos tres compuestos, o sales o formulaciones de liberación sostenida de los mismos, para una administración simultánea, por separado o secuencial para un tratamiento combinatorio de la enfermedad de Alzheimer (EA) o un trastorno relacionado en un sujeto que lo necesite.
Los enfoques terapéuticos de acuerdo con la invención pueden usar fármacos solos o combinaciones de fármacos, junto con cualquier otra terapia dirigida a la misma ruta o que tenga modos de acción distintos.
También se describen en esta memoria composiciones que podrían comprender además un fármaco o fármacos, que ya existen o que se podrían desarrollar, que se unen o modulan la actividad de una proteína codificada por un gen seleccionado a partir de ABAT, ABCA1, ABI1, ABL1, ACAT, ACC2, ACCN1, ADAMTS12, ADCY2, ADIPOQ, ADIPOR1/R2, ADORA1/2A/2B, ADRA1A/2, ADRB1/2, AGPAT5, AIP4, AKAP2, AKR1C2, AKT, ALDH2, ALOX12, ALOX5, ANG2, ANK1, ANKRA, ANXA1, APBA1, APBA2BP, APOA1, APOER2, ARHGAP17, ARHGAP26, ATG5/7/12, ATM, ATP10A, ATP1A1, ATP2A3, ATP2B1, ATP6V1C1, ATR, AUH, BACE1, BAD, BAI3, BASSOON, BAX, BCAR1, BCL2, BDNF, BECLIN1, BIN1, canales de BK (KCNMA1, KCNMB1), BMP3A, BRCA1, CA10, CAC-NA1C/2D3/2D4, CADPS2, CALM1-5 (calmodulina), CAMK1D, CAMKK2, CASK, CASR, CAST, CBL, CD36, CD44, CDC2, CDC42, CDC42BPB, CDC42EP3, CDH1/2/13, CDK5, CDKN1A, CHAT, CHK1, CHRM1-5, CHRNA1-7/9/10, CIT (citron), CK1, CNGB3, CNTFR, COL4A2, CPT, CRAM, CREB, CRMP, CSH1, CTNNA2, CTNNB1, CTTN (cortactina), CUBN, CULLIN1, CYP7B1, CYSLTR1/R2, DAB1, DCC, DEPDC2, DGKB/H/Z, DHCR7, DHFR, DLG2/4, DNAJB9, DOCK3, DRD2/5, DYN1/3, EDG1-8, EDN1/2, EDNRA/B, EFNA1/2/4/5/7 (efrina A), EFNB1/2/3 (efrina B), EHHADH, ELAVL2, ENPP2 (autotaxina), ENPP6, EPHA3, EPHBR1/2/3/4/6, ERBB2/4, ERK1/2, ESRRG, ETFA, EZR, F2, F2R, FAS, FDPS, FES, FGF1/2, FKBP12/12.6, FLNA, FLT1 (VEGFR1), FLT4, FOXO1/3A, FRAP (MTOR), FTO, FYN, FZ2, GABBR1/2, GABRA2/G2, GADD45, GAT1, GATA3, GH1, GIPC1/2, GLRA1, GLUD1, GNA12/13, GNPTAB, GPC5, GPHN (gefrina), GRIA2/3, GRID1/2, GRIK1/2, GRIN2B/3A, GRIP1/2, GRK2/5, GRM3/5/6/7/8, GRP170, GSK3B, HAPLN1, HAS1-3, HCRTR2, HIF1A, HIPK2, HK2, HMOX1, HOMER1/2/3, HSD11B1, HSP90B1, HSPA5, HTR1A/1B/1 D, HYAL1/2/3, IDE, IL20RA/B, IL6ST, IL8, IMPDH1/2, INS, INSR, IRF1, ITB1, ITGA1/6, ITGB1, ITPR1, JNK1, KALRN (kalirina), KCNA2/D2, KCNH2, KCNIP1/2, KCNJ11, KCNJ12, KCNJ3, KCNMA1, KCNMB1-4, KDR (VEGFR2), KTN1, KYNU, LAMA1, LDLR, LEP (LEPTINA), LEPR, LIFR, LIN7A/B/C (VELI1/2/3), LIPL2, LKB1, LRP1, LRP2 (megalina), LTBP2, LYN, MAD1L1, MAML3, MAOA/B, MAT2B, MCC1, MDM1, ME1, MET, MGST2, MINT1, MLLT4 (afadina), MMP2, MMP9, MOESIN, MTR, MUC1, MUNC13/18A, MYO6, MYOL, NADPH oxidasa, NAV1, NBEA, NCAM1, NCK1/2, NEDD9, NF2 (merlina), NFKB1, NFKBIB, NGEF (efexina), NGF, NGFR, NHERF, NIL16, NLGN1, NOC2, NOS1/2A/3, NOTCH1/2/3, NPC1/2, NPIST, NR1I2, NR3C1, NR3C2, NRG1/3, NRP1/2, NRX3, NTF3/5, NTN1 (netrina 1), NTRK2 (TRKB), NWASP, OPCML, OPRK1, OPRM, OPRS1, OSBPL3/10, P2RY1, P2RY 12, PAELR, PAI1/2, PAK1/6/7, PALLD, PAP1, PARK2, PC, PCAF, PCTP, PDE11A, PDE1A, PDE3A/3B, PDE4A/4B/4D, PDE5A, PDE6D, PDGFA/B, PDGFRA/B, PI3K, PIAS1, PICALM, PICK1, PIK3C3, PIP5K, PITPNC1, PKCA, PKCD, PLA1A/2, PLAT, PLAU, PLCB1, PLD1/2, PLEXA1, PLG, PLN, PLXDC2, PML, POP2, PPARA, PPARD, PPARG, PPARGC1B, PPFIBP1, PPP1CA, PPP3CA (calcineurina), PRDX5/6, PRKAA (AMPK), PRKACA, PRKG1, PRL, PTGER1, PTGFR, PTGS2, PTN, PTP1B, PTPN11, PTPRF, PTPRG, PTPRM, PVRL1, PXN (paxilina), PYK2, RAB3B, RAC1, RACK1, RAP1, RASGRF2, RBPJ, RDX (radixina), RELN, RGNEF, RHEB, RHOA, RHOG, RIM2, RIMS1/2, ROBO2, ROCK1/2, ROR2, RPH3A (rabfilina), RPH3AL, RPS6KA1, RPS6KB2, RTN1, RXR/RAR, RYR3, SACM1L, SAPAP, SAPK3, SCARB1, SCHIP1, SCN1A/1B, SCNN1D/1G, SEC24D, SEMA3A/3C/3E/4C, SGPP2, SH3BP5, SIAH1A, SIL1, SLC12A1/2/5, SLC1A2, SLC25A21, SLC6A1/A18, SLC8A1/A2/A3, SLC9A1, SLIT1, SLN, SMAD3/4, SNAP25, SNCA, SNCAIP, SORBS2, SORCS2, SPLA2, SPOCK1, SPP1 (osteopontina), SRC, SRD5A1, SREBF1/F2, SRGAP3, STAT3, STX1A/2 (sintaxinas), STXBP6, SUM1, SV2C, SYN1, SYNJ1/2 (sinaptojanina), SYT12, SYTL4 (granufilina), TACE, TACR1, TBR1, TBXA2R, TGFBR1/R2/R3, THBS1/2, THEM2, THRA/B, TIAM1, TIMP2, TLL2, TOP2A, TP53, TP63, TRIO, TRPC3/4/5, TSC1/2, TSPO, UBE2A, ULK4, UNC13C, UNC5C, VAMP2/5, VCL (VINCULINA), VDAC1, VEGFA/C, VEGFR1, VMAT, VPS15, WASPIP, WAVE, WNT1A/5A, WWOX, XANTINA OXIDASA, YAP y YES1.
Las secuencias de todos los genes y proteínas enumerados anteriormente están disponibles en genotecas y se pueden aislar mediante métodos conocidos en la técnica. La actividad de esos genes y proteínas también se puede evaluar mediante métodos conocidos en la técnica.
Hemos identificado fármacos particulares que, ya sea solos o en combinación(es), modulan las rutas descritas ante riormente y se pueden emplear para tratar la enfermedad de Alzheimer o trastornos relacionados.
También se describen en esta memoria composiciones que pueden comprender además al menos un fármaco se leccionado a partir de un inhibidor de ABAT, (preferiblemente, vigabatrina), y/o un inhibidor de ABL1 (preferiblemente imatinib), y/o un inhibidor de ACAT (preferiblemente, hesperetina), y/o un modulador de ADCY2 (preferiblemente, vidarabina), y/o un modulador de los receptores de adenosina ADORA1/2A/3 (seleccionado preferiblemente a partir de clofarabina y defibrotida), y/o un modulador de los receptores adrenérgicos ADRA (seleccionado preferiblemente a partir de propericiazina, metotrimeprazina, mefentermina y dipivefrina), y/o un modulador de los receptores adre nérgicos ADRB (seleccionado preferiblemente a partir de guanetidina, betanidina, bitolterol y procaterol), y/o un inhibidor de ALOX5/12 (seleccionado preferiblemente a partir de dietilcarbamazina y masoprocol), y/o un inhibidor de ATP1A1 (preferiblemente, deslanosida y omeprazol), y/o un activador de la autofagia (preferiblemente, trehalosa), y/o un inhibidor de CA10 (preferiblemente, metazolamida), y/o un modulador de la calcificación (seleccionado prefe riblemente a partir de foscarnet, nitrato de galio, calcifediol, calcitonina, calcitriol, ácido clodrónico, dihidrotaquisterol, elcatonina, ácido etidrónico, ipriflavona y acetato de teriparatida), y/o un modulador de CALM1 (calmodulina) (prefe riblemente, aprindina), y/o un modulador de CD44 (seleccionado preferiblemente a partir de eflornitina y benzbromarona), y/o una chaperona química (preferiblemente seleccionada a partir de arabitol y manitol), y/o un modulador de los receptores muscarínicos CHRM (seleccionado preferiblemente a partir de ciclopentolato, oxifenciclimina, trospio e isoflurofato), y/o un antagonista de los receptores nicotínicos de acetilcolina CHRNA, que no puede atravesar la
barrera hematoencefálica (seleccionado preferiblemente a partir de pancuronio, pipecuronio, rapacuronio, rocuronio, succinilcolina, vecuronio, atracurio, cisatracurio, doxacurio, mecamilamina, metocurina, mivacurio y neomicina), y/o un inhibidor de CNGB3 (preferiblemente, amilorida), y/o un modulador de los receptores eicosanoides CYSLTR1/2, PTGER1, PTGFR y TBXA2R (seleccionado preferiblemente a partir de travoprost, montelukast, cinalukast, amlexanox, carboprost trometamina, bimatoprost y ridogrel), y/o un inhibidor de DHFR (preferiblemente, pirimetamina y triamtereno), y/o un modulador del receptor de dopamina DRD2 (seleccionado preferiblemente a partir de dihidroergotamina y cabergolina), y/o un agonista del receptor de dopamina DRD5 (preferiblemente, fenoldopam), y/o un inhibidor de EDNRA (seleccionado preferiblemente a partir de sulfametoxazol y gentamicina), y/o un modulador de ENPP2 (autotaxina) (preferiblemente, L-histidina) y/o un inhibidor de ERBB2 (preferiblemente, lapatinib), y/o un mo dulador de la trombina F2 (seleccionado preferiblemente a partir de sulodexida, ximelagatrán, warfarina, fenprocumón, enoxaparina, ardeparina, fondaparinux, latamoxef, bacitracina, ticlopidina y erdosteina), y/o un inhibidor de FDPS (preferiblemente, alendronato), y/o un modulador de GABRA2 (seleccionado preferiblemente a partir de fenobarbital, metohexital, cefotiam, clometiazol, tiopental, lubiprostona y aztreonam), y/o un antagonista de GRIK1 (prefe riblemente, topiramato), y/o un modulador de la actividad de GSK3B (seleccionado preferiblemente a partir de albuterol y metaraminol), y/o un modulador de la señalización de HIF1A (seleccionado preferiblemente a partir de meloxicam, topotecán, deferoxamina, ácido úsnico, hidralazina, deferiprona, dibenzoilmetano, avobenzona, dinoprostona, epoprostenol, 2-oxoglutarato y mimosina), y/o un inhibidor de HK2 (hexocinasa II) (seleccionado preferiblemente a partir de quinina, gabexato, bifonazol y clotrimazol), y/o un modulador de HMOX1 (seleccionado preferiblemente a partir de auranofina, hematina/hemina y hemo arginato), y/o un modulador de los receptores HTR1B/1D (selecciona do preferiblemente a partir de ergotamina y eletriptán), y/o un inhibidor de IMPDH1 e IMPDH2 (preferiblemente, tioguanina), y/o un modulador de las integrinas ITGA/B (preferiblemente, rabeprazol), y/o un inhibidor del canal de potasio KCND2 (preferiblemente, lidocaína), y/o un inhibidor del canal de potasio KCNH2 (preferiblemente, ibutilida), y/o un modulador de KCNMA1 (seleccionado preferiblemente a partir de cromoglicato, etinamato, ketoconazol, clorzoxazona, unoprostona, hesperitina, bendroflumetiazida, benzotiazida, clorotiazida, ciclotiazida, diazoxida, hidroflumetiazida, quinetazona y triclorometiazida), y/o un modulador de MGST2 (preferiblemente, balsalazida), y/o un mo dulador de MMP2 y MMP9 (preferiblemente, candoxatril), y/o un modulador de la formación de poros de transición para la permeabilidad mitocondrial (seleccionado preferiblemente a partir de carbenoxolona y ciprofloxacina), y/o un inhibidor de MTOR (preferiblemente, rapamicina), y/o un modulador de NOS1/2A/3 (seleccionado preferiblemente a partir de propiltiouracilo, tietilperazina y ketotifeno), y/o un modulador de la señalización del receptor NR3C1 (selec cionado preferiblemente a partir de metirapona y mometasona), y/o un modulador del receptor NR3C2 (seleccionado preferiblemente a partir de eplerenona y fludrocortisona), y/o un inhibidor de NRP2 (preferiblemente, pegaptanib), y/o un modulador de OPCML (preferiblemente, alfentanilo), y/o un modulador de OPRK1 y OPRS1(seleccionado prefe riblemente a partir de buprenorfina y pentazocina), y/o OPRM (preferiblemente, levalorfano), y/o un modulador de la fosforilación oxidativa (seleccionado preferiblemente a partir de almitrina, eritromicina, kanamicina y cerulenina), y/o un inhibidor de los receptores P2RY1 y/o P2RY12 (preferiblemente, tirofibán), y/o un inhibidor de las fosfodiesterasas PDE11A, PDE4A y PDE5A (seleccionado preferiblemente a partir de mesembrina, milrinona y anagrelida), y/o un inhibidor de las fosfodiesterasas PDE3A/3B y PDE4A/4B y un activador de los canales de BK (preferiblemente, cilostazol), y/o un modulador de los receptores PDGFRA/B (seleccionado preferiblemente a partir de becaplermina, estreptomicina, delfinidina, cianidina y fumagilina), y/o un modulador de PLA2 (seleccionado preferiblemente a partir de ácido niflúmico, hidrocortamato y netilmicina), y/o un modulador de PLAT (preferiblemente, fenilbutirato de sodio), y/o un modulador de PLD2 (preferiblemente, ambrisentán), y/o un inhibidor de PLG (preferiblemente, ácido aminocaproico), y/o un modulador de PPARD (preferiblemente, icosapent), y/o un modulador de PPARG (preferiblemente, fenilbutirato), y/o un modulador de PRKG1 (seleccionado preferiblemente a partir de nitroprusiato, nitroglicerina y paricalcitol), y/o un inhibidor de PTP1B (preferiblemente, tiludronato), y/o un modulador de RHOA/RAC (selecciona do preferiblemente a partir de clortalidona, hidroclorotiazida, clomociclina, limeciclina, natamicina, anfotericina B, cefalexina, cefaloridina, cefuroxima, dicloxacilina), y/o un modulador de RXR/RAR (preferiblemente, tazaroteno), y/o un antagonista de los canales de sodio SCN1A/B (preferiblemente, fosfenitoína), y/o un inhibidor de SLC12A1 (pre feriblemente, bumetanida), y/o un inhibidor de SLC6A1 (preferiblemente, tiagabina), y/o un modulador de SLC9A1 (preferiblemente, buclizina), y/o un inhibidor de SRD5A1 (preferiblemente, dutasterida), y/o un antagonista de TACR1 (seleccionado preferiblemente a partir de aprepitant y vapreotida), y/o un modulador de la señalización de TGFB (preferiblemente, aliskireno), y/o un modulador de THRa /B (seleccionado preferiblemente a partir de liotioronina), y/o un inhibidor de TOP2A (preferiblemente, lucantona), y/o un modulador de TSPO (seleccionado preferible mente a partir de flunitrazepam y temazepam), y/o un modulador de VDAC1 (preferiblemente, dihidroxialuminio), y/o un inhibidor de VEGFR1 (preferiblemente, sunitinib), y/o un modulador del metabolismo de la vitamina K (seleccio nado preferiblemente a partir de cefmetazol, cefamandol y cefoperazona), y/o un inhibidor de VMAT (seleccionado preferiblemente a partir de tetrabenazina, deserpidina y nitisinona), y/o un inhibidor de los canales de calcio depen dientes de voltaje (CACNA) (seleccionado preferiblemente a partir de lercanidipina, pregabalina, mibefradilo, aranidipina, bamidipina, benciclano, bepridilo, clentiazem, efonidipina, elgodipina, etafenona, fendilina, flunarizina, galopamilo, isadipina, lacidipina, lidoflazina, lomerizina, manidipina, nicardipina, nilvadipina, nimodipina, nisoldipina, nitrendipina, perhexilina, prenilamina, semotiadilo y terodilina), y/o un inhibidor de YES1, SRC y EPHA3 (preferiblemente, dasatinib).
Otras terapias utilizadas junto con el o los fármacos o combinaciones de fármacos descritos en esta memoria, pue den comprender uno o varios fármacos que mejoran los síntomas de la enfermedad de Alzheimer o fármacos que se podrían usar para un tratamiento paliativo de la enfermedad de Alzheimer. Preferiblemente, dicho uno o varios fár macos se seleccionan a partir de 3APS, AAB-001, ABT-089, ABT-126, AC-3933, ACC-001, Acetaminofeno, AFFITOPE AD01, AFFITOPE AD02, ácido alfa lipoico, alfa-tocoferol, AN1792, anti-Abeta, AQW051, Aripiprazol, Atomoxetina, Atorvastatina, AVE1625, AVP-923, AZD0328, AZD3480, Bapineuzumab, BAY94-9172 (Zk 6013443), Bifeprunox, Bioperina, BMS-708163, BRL-049653, Briostatina, CAD106, Celecoxib, CERE-110, Cerebrolisina, CHF 5074, Colina, Circadina, Citalopram, Coenzima Q, Cobre, CTS21166, Curcumina, CX516 (Ampalex), CX717, Ciclofosfamato, DCB-AD1, Dextroamfetamina, DHA (Ácido Docosahexaenoico), Digoxina, Dimebon (Latrepirdina), Divalproex, DMXB-A, Donepezilo, Doxiciclina, Egb 761, EHT 0202 tazolato, ELND005 (escilo-inositol), EpAx 1050TG, mesilato de Ergoloid, Epigalocatequina-Galato, Escitalopram, Estradiol, Estrógeno, Etanercept, EVP-6124, EVT101, Exelon, aceite de pescado, FK962, florpiramina F 18, Folato Vitamina B6 Vitamina B21, Gabapentina, Galantamina, Gemfibrozilo, extractos de Ginkgo biloba (por ejemplo, EGb 761 o CP401), extractos mejorados de Ginkgo biloba (por ejemplo, enriquecidos en ingredientes activos o con menos contaminantes) o fármaco que contiene ex tractos de Ginkgo biloba (por ejemplo, Tanakan o Gingkor fort), Glucosa, Ácido L-glutámico, GSI 136, GSI-953, GSK239512, GSK933776A, Haloperidol, HF0220, Huperzina A, Hidrocodona/APAP, Ibuprofeno, IFN-alfa2A, Indometacina, Insulina, Inmunoglobulina intravenosa, Ketasina, Lecozotán, Leuprolida, Levodopa, Ácido lipoico, Litio, Lorazepam, Lovostatina, Luteína, LY2062430 (solanezumab), LY2811376, LY450139, LY451395, MABT5102A, Malato, Masitinib (AB1010), Medroxiprogesterona, Melatonina, MEM 1003, MEM 3454, Memantina, Azul de metileno, Metilfenidato, Mifepristona, MK0249, MK0677, MK0952, MK0952, MK3328, Modafinilo, MPC-7869, NADH, Naproxeno, Nefiracetam, Aceite de krill Neptuno, Neramexano, NIC5-15, parche de Nicoderm, Nicotinamida (vitami na B3), Novasoy, NP031112, NS 2330, NSA-789, AINEs, Olanzapina, Ácidos grasos poliinsaturados omega-3 (EPA+DHA), ONO-2506PO, Oxibato, Panax Ginseng, PAZ-417, PBT2, Perfenazina, PF-04360365, PF-04447943, PF-04494700, Fenserina, Fosfatidilserina, Pitavastatina, Posifeno, PPI-1019 (APAN), Pravastatina, Prazosina, Prednisona, Progesterona, PRX-03140, PYM50028, Quetiapina, R1450, Raloxifeno, Ramiprilo, Rasagilina, Razadina, resveratrol, rifampicina, risperidona, Rivastigmina, RN1219, RO5313534, Rofecoxib, Rosiglitazona, Salvia officinalis (salvia), sAm -315, Sa M-531, SAM-760, SB-742457, Selenio, Sertralina, SGS-742, Simvastatina SK-PC-B70M, So lanezumab, SR57667B, SRA-333, SRA-444, SSR180711C, ST101, T-817MA, Tacrina, Tarenflurbilo, Testosterona, Tramiprosato (3APS), Trazodona, TRx0014 (cloruro de metiltioninio), Triptófano, V950, Valproato, Vareniclina, Vita mina C, Vitamina E, VP4896, Xaliprodeno, Zeaxantina, Zolpidem y ZT-1 (DEBIO-9902 SR).
También se describe en esta memoria un método para seleccionar un fármaco para el tratamiento combinatorio de la enfermedad de Alzheimer o un trastorno relacionado, en donde el método comprende una etapa de someter a ensayo un fármaco candidato para estudiar una actividad en la función sináptica y/o la angiogénesis y/o la respuesta al estrés celular y seleccionar un fármaco candidato que mejora la función sináptica, atenúa la desregulación angiogénica y modula la respuesta al estrés celular.
También se describe en esta memoria un método para seleccionar una composición para tratar la enfermedad de Alzheimer o un trastorno relacionado, en donde el método comprende preparar una combinación de un fármaco que modula la función sináptica y/o un fármaco que atenúa la desregulación angiogénica y/o un fármaco que modula la respuesta al estrés celular, para una administración simultánea, por separado o secuencial a un sujeto que lo nece site.
La composición de la invención se puede administrar repetidamente al sujeto.
Las composiciones de la invención comprenden típicamente uno o varios vehículos o excipientes farmacéuticamente aceptables. La duración de la terapia depende del estadio de la enfermedad que se está tratando, la combinación utilizada, la edad y el estado del paciente, y cómo responde el paciente al tratamiento. La dosificación, la frecuencia y el modo de administración de cada componente de la combinación se pueden controlar de forma independiente. Por ejemplo, un fármaco se puede administrar por vía oral mientras que el segundo fármaco se puede administrar por vía intramuscular. La terapia combinada se puede administrar en ciclos a intervalos que incluyen períodos de descanso de modo que el cuerpo del paciente tiene la oportunidad de recuperarse de cualquier efecto secundario aún no previsto. Los fármacos también se pueden formular juntos de manera que una administración suministre todos los fármacos.
La administración de cada fármaco de la combinación puede ser a través de cualquier medio adecuado que dé lugar a una concentración del fármaco que, combinado con el otro componente, sea capaz de corregir el funcionamiento de las rutas implicadas en la EA.
Si bien es posible que los ingredientes activos de la combinación se administren como el producto químico puro, es preferible presentarlos como una composición farmacéutica, también denominada en este contexto formulación farmacéutica. Las posibles composiciones incluyen aquellas adecuadas para una administración oral, rectal, tópica (incluyendo transdérmica, bucal y sublingual) o parenteral (incluyendo administración subcutánea, intramuscular, intravenosa e intradérmica).
Más comúnmente, esas formulaciones farmacéuticas se prescriben al paciente en "paquetes de paciente" que con tienen una cantidad de unidades de dosificación u otros medios para la administración de dosis unitarias medidas para uso durante un período de tratamiento distinto en un solo envase, generalmente un blíster. En relación con las prescripciones tradicionales, en las que un farmacéutico divide el suministro para un paciente de un producto farma céutico a partir de un suministro a granel, los paquetes de paciente tienen la ventaja de que el paciente tiene siem pre acceso al prospecto contenido en el paquete de paciente, normalmente ausente en las prescripciones tradiciona
les. Se ha comprobado que la inclusión de un prospecto mejora la adaptabilidad del paciente a las instrucciones del médico. Así pues, la invención incluye además una formulación farmacéutica, como se ha descrito anteriormente en la presente memoria, en combinación con un material de envasado adecuado para dichas formulaciones. En un paquete de paciente de ese tipo, el uso previsto de una formulación para el tratamiento combinado puede inferirse mediante instrucciones, recursos, previsiones, adaptaciones y/u otros medios que ayuden a utilizar la formulación del modo más adecuado para el tratamiento. Tales medidas hacen que un paquete de paciente sea específicamente adecuado y esté específicamente adaptado para el uso en un tratamiento con la combinación de la presente inven ción.
El fármaco puede estar contenido, en cualquier cantidad apropiada, en cualquier sustancia de soporte adecuada y puede estar presente en una cantidad de 1-99% en peso del peso total de la composición. La composición puede estar prevista en una forma de dosificación que sea adecuada para la vía de administración oral, parenteral (por ejemplo, intravenosa, intramuscular), rectal, cutánea, nasal, vaginal, mediante inhalación, sobre la piel (parche) u ocular. Así pues, la composición puede estar en forma, por ejemplo, de comprimidos, cápsulas, píldoras, polvos, granulados, suspensiones, emulsiones, soluciones, geles incluyendo hidrogeles, pastas, pomadas, cremas, emplas tos, pociones, dispositivos de liberación osmótica, supositorios, enemas, inyectables, implantes, nebulizadores o aerosoles.
Las composiciones farmacéuticas se pueden formular según la práctica farmacéutica convencional (véase, por ejemplo, Remington: The Science and Practice of Pharmacy (20a ed.), compilador. A. R. Gennaro, Lippincott Wil liams & Wilkins, 2000 y Encycopledia of Pharmaceutical Technology, compiladores J. Swarbrick y J. C. Boylan, 1988-1999, Marcel Dekker, Nueva York).
Las composiciones farmacéuticas según la invención se pueden formular para liberar el fármaco activo sustancial mente de manera inmediata a la administración, o después de la administración en cualquier momento o periodo de tiempo predeterminado.
Las formulaciones de liberación controlada incluyen (i) formulaciones que crean una concentración sustancialmente constante del fármaco dentro del cuerpo durante un periodo de tiempo prolongado; (ii) formulaciones que, después de un periodo de retraso predeterminado, crean una concentración sustancialmente constante del fármaco dentro del cuerpo durante un periodo de tiempo prolongado; (iii) formulaciones que mantienen la acción del fármaco duran te un periodo de tiempo predeterminado manteniendo un nivel eficaz y relativamente constante del fármaco en el cuerpo con una minimización concomitante de efectos secundarios no deseables, asociados a fluctuaciones en el nivel plasmático de la sustancia activa del fármaco; (iv) formulaciones que localizan la acción del fármaco mediante, por ejemplo, una colocación espacial de una composición de liberación controlada adyacente al tejido u órgano en fermo o en el mismo; y (v) formulaciones que guían la acción del fármaco utilizando vehículos o derivados químicos para entregar el fármaco a un tipo concreto de célula diana.
La administración de fármacos en forma de una formulación de liberación controlada se prefiere especialmente en los casos en los que el fármaco, bien solo, o en combinación, tiene (i) un índice terapéutico reducido (es decir la diferencia entre la concentración plasmática que produce efectos secundarios nocivos o reacciones tóxicas y la concentración plasmática que produce un efecto terapéutico es pequeña; en general, el índice terapéutico, IT, se define como la relación entre la dosis letal media (DL50) y la dosis eficaz media (DE50); (ii) una ventana de absor ción estrecha en el tracto gastrointestinal; o (iii) una semivida biológica muy corta, de manera que se requiere una dosificación frecuente durante un día para mantener el nivel plasmático a un nivel terapéutico.
Se puede aplicar cualquiera de varias estrategias con el fin de conseguir una liberación controlada en la que la tasa de liberación sea mayor que la tasa de metabolismo del fármaco en cuestión. Se puede conseguir una liberación controlada mediante una selección apropiada de diversos ingredientes y parámetros de formulación, incluyendo, por ejemplo, diversos tipos de revestimientos y composiciones de liberación controlada. Así pues, el fármaco se formula con excipientes apropiados para obtener una composición farmacéutica que, al administrarla, libera el fármaco de una manera controlada (composiciones de cápsula o comprimido unitario simple o múltiple, soluciones en aceite, suspensiones, emulsiones, microcápsulas, microesferas, nanopartículas, parches y liposomas).
Formas de dosificación sólidas para uso oral
Las formulaciones para uso oral incluyen comprimidos que contienen el o los ingredientes activos en una mezcla con excipientes no tóxicos farmacéuticamente aceptables. Estos excipientes pueden ser, por ejemplo, diluyentes inertes o cargas (por ejemplo, sacarosa, celulosa microcristalina, almidones incluyendo almidón de patata, carbonato de calcio, cloruro de sodio, fosfato de calcio, sulfato de calcio o fosfato de sodio); agentes de granulación y disgregan tes (por ejemplo, derivados de la celulosa incluyendo celulosa microcristalina, almidones incluyendo almidón de patata, croscarmelosa sódica, alginatos o ácido algínico); aglutinantes (por ejemplo, goma arábiga, ácido algínico, alginato de sodio, gelatina, almidón, almidón pregelatinizado, celulosa microcristalina, carboximetilcelulosa sódica, metilcelulosa, hidroxipropil metilcelulosa, etilcelulosa, polivinilpirrolidona o polietilenglicol); y agentes lubricantes, deslizantes y antiadhesivos (por ejemplo, ácido esteárico, sílices o talco). Otros excipientes farmacéuticamente aceptables pueden ser colorantes, aromatizantes, plastificantes, humectantes, agentes tamponadores y similares.
Los comprimidos pueden no estar revestidos o pueden estar revestidos mediante técnicas conocidas, opcionalmente para retrasar la disgregación y la absorción en el tracto gastrointestinal y de este modo proporcionar una acción mantenida durante un periodo más largo. El revestimiento puede estar adaptado para liberar la sustancia activa del fármaco en un patrón predeterminado (por ejemplo, con el fin de lograr una formulación de liberación controlada) o puede estar adaptado para no liberar la sustancia activa del fármaco hasta haber pasado el estómago (revestimiento entérico). El revestimiento puede ser un revestimiento de azúcar, un revestimiento de película (por ejemplo, basado en hidroxipropil metilcelulosa, metilcelulosa, metil hidroxietilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa, copolímeros de acrilato, polietilenglicoles y/o polivinilpirrolidona) o un revestimiento entérico (por ejemplo, basado en un copolímero de ácido metacrílico, ftalato de acetato de celulosa, ftalato de hidroxipropil metilcelulosa, succinato acetato de hidroxipropil metilcelulosa, ftalato acetato de polivinilo, goma laca y/o etilcelulosa). Puede emplearse un material de retardo tal como, por ejemplo, monoestearato de glicerilo o diestearato de glicerilo.
Las composiciones sólidas de comprimido pueden incluir un revestimiento adaptado para proteger la composición contra cambios químicos no deseados (por ejemplo, degradación química antes de la liberación de la sustancia activa del fármaco). El revestimiento puede aplicarse sobre la forma de dosificación sólida de manera similar a como se describe en la Encyclopedia of Pharmaceutical Technology.
En el comprimido pueden estar mezclados entre sí diversos fármacos, o éstos pueden estar divididos. Por ejemplo, el primer fármaco está contenido en el interior del comprimido, y el segundo fármaco se halla en el exterior, de tal manera que se libere una parte considerable del segundo fármaco antes de la liberación del primer fármaco.
Las formulaciones para uso oral pueden presentarse también como comprimidos masticables, o como cápsulas de gelatina dura en las que el ingrediente activo está mezclado con un diluyente sólido inerte (por ejemplo, almidón de patata, celulosa microcristalina, carbonato de calcio, fosfato de calcio o caolín), o como cápsulas de gelatina blanda en las que el ingrediente activo esté mezclado con agua o un medio aceitoso, por ejemplo, parafina líquida o aceite de oliva. Pueden prepararse polvos y granulados utilizando los ingredientes mencionados anteriormente en forma de comprimidos y cápsulas de una manera convencional.
Las composiciones de liberación controlada para uso oral se pueden elaborar, por ejemplo, para que liberen el fár maco activo controlando la disolución y/o la difusión de la sustancia del fármaco activo.
La liberación controlada por disolución o difusión puede lograrse mediante un revestimiento apropiado de una formu lación en forma de comprimido, cápsula, gránulo o granulado de fármacos, o incorporando el fármaco en una matriz apropiada. Un revestimiento de liberación controlada puede incluir una o varias de las sustancias de revestimiento mencionadas anteriormente y/o, por ejemplo, goma laca, cera de abeja, glycowax, cera de ricino, cera de carnauba, alcohol estearílico, monoestearato de glicerilo, diestearato de glicerilo, palmitoestearato de glicerol, etilcelulosa, resinas acrílicas, poli(ácido dl-láctico), acetato butirato de celulosa, poli(cloruro de vinilo), poli(acetato de vinilo), vinilpirrolidona, polietileno, polimetacrilato, metilmetacrilato, 2-hidroximetacrilato, hidrogeles de metacrilato, 1,3 butilenglicol, metacrilato de etilenglicol y/o polietilenglicoles. En una formulación de matriz de liberación controlada, el material de la matriz puede incluir también, por ejemplo, metilcelulosa hidratada, cera de carnauba y alcohol estearílico, carbopol 934, silicona, triestearato de glicerilo, acrilato de metilo-metacrilato de metilo, poli(cloruro de vinilo), polietileno y/o fluorocarburo halogenado.
Una composición de liberación controlada que contiene uno o varios de los fármacos de las combinaciones reivindi cadas puede presentarse también en forma de un comprimido o cápsula flotante (es decir, un comprimido o una cápsula que, después de la administración oral, flota encima del contenido gástrico durante un cierto periodo de tiempo). Una formulación de comprimido flotante del o de los fármacos puede prepararse granulando una mezcla del o de los fármacos con excipientes y un 20-75% p/p de hidrocoloides, tales como hidroxietilcelulosa, hidroxipropilcelu losa o hidroxipropilmetilcelulosa. Los gránulos obtenidos se pueden comprimir a continuación para obtener compri midos. Al ponerse en contacto con el jugo gástrico, el comprimido forma una barrera de gel sustancialmente im permeable al agua alrededor de su superficie. Esta barrera de gel participa a la hora de mantener una densidad de menos de uno, permitiendo de este modo que el comprimido permanezca flotando en el jugo gástrico.
Líquidos para administración por vía oral
Los polvos, polvos dispersables o gránulos adecuados para la preparación de una suspensión acuosa mediante la adición de agua, son formas de dosificación convenientes para la administración por vía oral. La formulación en forma de suspensión proporciona el ingrediente activo en una mezcla con un agente dispersante o humectante, un agente de suspensión y uno o más agentes conservantes. Como agentes de suspensión son adecuados, por ejem plo, la carboximetilcelulosa sódica, la metilcelulosa, el alginato de sodio y similares.
Composiciones parenterales
La composición farmacéutica puede administrarse también por vía parenteral mediante inyección, infusión o implan tación (intravenosa, intramuscular, subcutánea o similares) en formas de dosificación, formulaciones, o a través de dispositivos de liberación adecuados o implantes que contienen vehículos y coadyuvantes convencionales no tóxi cos, farmacéuticamente aceptables. La formulación y la preparación de tales composiciones son bien conocidas por los expertos en la técnica de la formulación farmacéutica.
Las composiciones para uso parenteral pueden estar previstas en formas de dosificación unitarias (por ejemplo, en ampollas monodosis), o en frascos que contienen varias dosis y a los que se puede añadir un agente conservante adecuado (véase posteriormente). La composición se puede presentar en forma de una solución, una suspensión, una emulsión, un dispositivo de infusión o un dispositivo de liberación para la implantación o se puede presentar en forma de un polvo seco que se reconstituye con agua u otro vehículo adecuado antes del uso. Aparte del o de los fármacos activos, la composición puede incluir vehículos y/o excipientes adecuados, aceptables desde el punto de vista parenteral. El o los fármacos activos pueden estar incorporados en microesferas, microcápsulas, nanopartículas, liposomas o similares para una liberación controlada. La composición puede incluir agentes de suspensión, solubilización, estabilización, ajuste del pH y/o dispersantes.
Las composiciones farmacéuticas según la invención se pueden presentar en la forma adecuada para una inyección estéril. Para preparar tal composición, el o los fármacos activos adecuados se disuelven o suspenden en un vehículo líquido aceptable desde el punto de vista parenteral. Entre los vehículos y disolventes aceptables que pueden em plearse, se incluyen agua, agua ajustada a un pH adecuado mediante la adición de una cantidad apropiada de ácido clorhídrico, hidróxido de sodio o un tampón adecuado, 1,3-butanodiol, solución de Ringer y solución de cloruro de sodio isotónica. La formulación acuosa puede contener también uno o varios agentes conservantes (por ejemplo, phidroxibenzoato de n-propilo, etilo o metilo). En los casos en los que uno de los fármacos sea solo moderadamente o ligeramente soluble en agua, se puede añadir un agente que mejora la disolución o la solubilización, o el disolvente puede incluir un 10-60% p/p de propilenglicol o similares.
Las composiciones parenterales de liberación controlada se pueden presentar en forma de suspensiones acuosas, microesferas, microcápsulas, microesferas magnéticas, soluciones en aceite, suspensiones en aceite o emulsiones. Como alternativa, el o los fármacos activos pueden estar incorporados en vehículos biocompatibles, liposomas, nanopartículas, implantes o dispositivos de infusión. Los materiales para l uso en la preparación de microesferas y/o microcápsulas son, por ejemplo, polímeros biodegradables/bioerosionables tales como poligalactina, poli-(cianoacrilato de isobutilo), poli(2-hidroxietil-L-glutamina). Los vehículos biocompatibles que pueden utilizarse a la hora de formular una formulación parenteral de liberación controlada, son carbohidratos (por ejemplo, dextranos), proteínas (por ejemplo, albúmina), lipoproteínas o anticuerpos. Los materiales para el uso en implantes pueden ser no biodegradables (por ejemplo, polidimetil siloxano) o biodegradables (por ejemplo, poli(caprolactona), poli(ácido glicólico) o poli(orto ésteres)).
Composiciones rectales
Para una aplicación rectal, las formas de dosificación adecuadas para una composición incluyen supositorios (de tipo emulsión o suspensión) y cápsulas de gelatina rectales (soluciones o suspensiones). En una formulación típica de supositorio, el o los fármacos activos se combinan con una base de supositorio apropiada, farmacéuticamente aceptable, tal como manteca de cacao, ácidos grasos esterificados, gelatina glicerinada y diversas bases solubles en agua o dispersables como los polietilenglicoles. Pueden incorporarse diversos aditivos, potenciadores o tensioactivos.
Composiciones percutáneas y tópicas
Las composiciones farmacéuticas también se pueden administrar por vía tópica sobre la piel para una absorción percutánea en formas de dosificación o formulaciones que contienen vehículos y excipientes convencionalmente no tóxicos, farmacéuticamente aceptables, incluyendo microesferas y liposomas. Las formulaciones incluyen cremas, pomadas, lociones, linimentos, geles, hidrogeles, soluciones, suspensiones, barras, nebulizadores, pastas, emplas tos y otros tipos de sistemas de liberación transdérmica de fármacos. Los vehículos o excipientes farmacéuticamen te aceptables pueden incluir agentes emulsionantes, antioxidantes, agentes de tamponación, agentes conservantes, humectantes, potenciadores de la penetración, agentes quelantes, agentes formadores de gel, bases de pomadas, perfumes y agentes protectores de la piel.
Los agentes emulsionantes pueden ser gomas naturales (por ejemplo, goma de acacia o goma de tragacanto) Los agentes conservantes, los humectantes y los potenciadores de la penetración pueden ser parabenos, tales co mo p-hidroxibenzoato de propilo o metilo, y cloruro de benzalconio, glicerina, propilenglicol, urea, etc.
Las composiciones farmacéuticas descritas anteriormente para la administración tópica sobre la piel pueden utilizar se también en conexión con una administración tópica sobre la parte del cuerpo que se ha de tratar o cerca de la misma. Las composiciones pueden estar adaptadas para una aplicación directa o para una aplicación por medio de dispositivos especiales de liberación de fármacos tales como vendajes o, como alternativa, emplastos, almohadillas, esponjas, bandas u otras formas de material flexible adecuado.
Dosificaciones y duración del tratamiento
Se apreciará que los fármacos de la combinación pueden administrarse de manera concomitante, en la misma for mulación farmacéutica o en una formulación farmacéutica diferente o secuencialmente. Si se realiza una administra ción secuencial, la demora en administrar el segundo ingrediente activo (o el ingrediente activo adicional) no debería ser tal que se pierda el beneficio del efecto eficaz de la combinación de los ingredientes activos. Un requisito mínimo
para una combinación según esta descripción es que la combinación debería estar destinada a un uso combinado con el beneficio del efecto eficaz de la combinación de los ingredientes activos. El uso previsto de una combinación puede inferirse mediante recursos, previsiones, adaptaciones y/u otros medios que ayuden a utilizar la combinación según la invención.
Aunque los fármacos activos de la presente invención pueden administrarse en dosis divididas, por ejemplo, dos o tres veces al día, se prefiere una sola dosis diaria de cada fármaco en la combinación, prefiriéndose en máxima medida una sola dosis diaria de todos los fármacos en una sola composición farmacéutica (forma de dosificación unitaria).
La expresión "forma de dosificación unitaria" se refiere a unidades físicamente discretas (tales como cápsulas, com primidos o cilindros de jeringa cargados) adecuadas como dosificaciones unitarias para sujetos humanos, conte niendo cada unidad una cantidad predeterminada de material o materiales activos, calculada para producir el efecto terapéutico deseado, en asociación con el vehículo farmacéutico necesario.
La administración puede ser una o varias veces al día durante un tiempo de varios días a varios años y puede inclu so ser durante toda la vida del paciente. En la mayoría de los casos está indicada una administración crónica o al menos repetida periódicamente a largo plazo.
Adicionalmente, la información farmacogenómica (el efecto del genotipo sobre la farmacocinética, la farmacodinámica o el perfil de eficacia de un producto terapéutico) sobre un paciente concreto puede influir en la dosificación utili zada.
Excepto cuando responden a casos especialmente perjudiciales de enfermedad EA, en los que puedan requerirse dosificaciones mayores, la dosificación preferida de cada fármaco en la combinación se encuentra normalmente dentro del intervalo de dosis no superiores a las normalmente prescritas para un tratamiento de mantenimiento a largo plazo o que hayan demostrado ser seguras en estudios clínicos de fase 3.
Una ventaja notable de la invención es que cada compuesto puede utilizarse con dosis bajas en una terapia combi nada, produciendo al mismo tiempo, en combinación, un beneficio clínico considerable para el paciente. La terapia combinada puede, de hecho, ser eficaz en dosis en las que los compuestos no tienen individualmente ningún efecto importante. Por consiguiente, una ventaja concreta de la invención reside en la capacidad de utilizar dosis subópti mas de cada compuesto, es decir, dosis que son menores que las dosis terapéuticas normalmente prescritas, prefe riblemente 1/2 de las dosis terapéuticas, más preferiblemente 1/3, 1/4, 1/5 o incluso más preferiblemente 1/10 a 1/100 de las dosis terapéuticas. En tales dosificaciones subóptimas, los compuestos solos serían sustancialmente inactivos, mientras que la o las combinaciones según la invención son totalmente eficaces.
Una dosificación preferida corresponde a cantidades desde un 1% hasta un 50% de las normalmente prescritas para un tratamiento de mantenimiento a largo plazo.
La dosificación más preferida puede corresponder a cantidades desde un 1% hasta un 10% de las normalmente prescritas para un tratamiento de mantenimiento a largo plazo.
A continuación se ofrecen ejemplos específicos de dosificaciones de fármacos para uso en la invención:
- Ácido aminocaproico desde aproximadamente 0,05 a 15 g por día
- Levosimendán de 0,05 a 4 mg por día
- amlodipina por vía oral desde aproximadamente 0,05 a 1 mg por día
- clopidogrel por vía oral desde aproximadamente 0,75 a 7,5 mg por día,
- tadalafilo por vía oral desde aproximadamente 0,05 a 0,5 mg por día,
- cilostazol por vía oral desde aproximadamente 1 a 10 mg por día,
- terbinafina por vía oral desde aproximadamente 2,5 a 25 mg una o dos veces al día,
- leflunomida por vía oral desde aproximadamente 0,25 a 2,5 mg por día,
- cinacalcet por vía oral desde aproximadamente 0,3 a 3 mg por día,
- acamprosato por vía oral desde aproximadamente 7 a 70 mg tres veces al día,
- metimazol por vía oral desde aproximadamente 0,05 a 1,5 mg por día
- mepacrina por vía oral desde aproximadamente 3 a 30 mg por día,
- fenformina por vía oral desde aproximadamente 0,5 a 5 mg por día,
- baclofeno por vía oral desde aproximadamente 0,4 a 8 mg por día administrados en dos o tres dosis dividi das,
- rifabutina por vía oral desde aproximadamente 6 a 60 mg por día,
- amobarbital por vía oral desde aproximadamente 0,06 a 15 mg por día,
- cefotetán por vía oral desde aproximadamente 0,01 a 0,4 mg por día,
- difilina por vía oral desde aproximadamente 6 a 60 mg por día en dos o tres dosis divididas,
- meticlotiazida por vía oral desde aproximadamente 0,025 a 1 mg por día,
- risedronato por vía oral desde aproximadamente 0,05 a 3 mg por día,
- etomidato por vía oral desde aproximadamente 0,6 a 6 mg por día,
- zonisamida por vía oral desde aproximadamente 1 a 40 mg por día.
Se entenderá que la cantidad del fármaco realmente administrada será determinada por un médico, a la luz de las circunstancias relevantes incluyendo el estado o los estados que se hayan de tratar, la composición exacta que se ha de administrar, la edad, el peso y la respuesta del paciente individual, la gravedad de los síntomas del paciente y la vía de administración elegida. Por lo tanto, los intervalos de dosificación anteriormente indicados están destinados a proporcionar una orientación general y ayuda para las enseñanzas de la presente memoria, pero no están desti nados a limitar el alcance de la invención.
Los siguientes ejemplos se proporcionan con fines ilustrativos y no a modo de limitación.
Ejemplos
I. Los compuestos y las combinaciones de los mismos evitan la toxicidad del péptido AP25-35
En esta primera serie de experimentos se han sometido a ensayo los compuestos candidatos en cuanto a su capa cidad para prevenir o reducir los efectos tóxicos del péptido AP25-35. Los fármacos se someten a ensayo en primer lugar individualmente, a lo que siguen ensayos de su acción combinada. El efecto se determina sobre diversos tipos de células, para documentar ulteriormente la actividad de los compuestos.
En la EA, la proteína APP forma agregados de láminas p insolubles plegadas de proteína Abeta fibrilar (amiloide). El cambio conformacional desde las formas solubles a fibrilares parece ser un evento espontáneo que se incrementa con concentraciones más altas de Abeta, por lo que cualquier producción de cantidades más altas de lo normal de Abeta (o la producción de formas más grandes y menos solubles de Abeta) tenderá a aumentar la formación de placa. Una vez que la placa Abeta ha comenzado a formarse, otras moléculas pueden interaccionar con la placa naciente para producir eventualmente la placa madura con sus áreas asociadas de muerte celular neuronal. Consi derando esto, hemos dado prioridad a someter a ensayo los efectos de los fármacos sobre la viabilidad de las célu las expuestas a la proteína p amiloide.
1.1 Protección frente a la toxicidad del péptido AP25-35 en neuronas corticales
Cultivo celular
Se cultivaron neuronas corticales de rata como describen Singer et al. 1999. Brevemente, se dio muerte a ratas hembra preñadas en el día 15 de gestación mediante luxación cervical (Ratas Wistar; Janvier) y se extrajeron los fetos del útero. Se extrajo el córtex y se colocó en medio helado de Leibovitz (L15; Invitrogen) que contenía un 1% de penicilina-estreptomicina (PS; Invitrogen) y un 1% de albúmina de suero bovino (BSA; Sigma). Los córtex se disociaron mediante tripsina durante 20 min a 37°C (Tripsina EDTA 1X; Invitrogen) diluida en PBS sin calcio ni mag nesio. La reacción se detiene mediante la adición de medio Eagle modificado con Dulbecco (DMEM; Invitrogen) que contiene ADNasa I de grado II (0,1 mg/ml; Roche Diagnostic) y 10% de suero de ternera fetal (FCS; Invitrogen). Las células se disocian mecánicamente mediante 3 pases a través de una pipeta de 10 ml. Las células se centrifugan después a 180 x g durante 10 minutos a 10°C. El material sobrenadante se desecha y las células del sedimento se resuspenden en un medio de cultivo definido que consiste en Neurobasal (Invitrogen) complementado con B27 (2%; Invitrogen), L-glutamina (0,2 mM; Invitrogen), 1% de solución PS y 10 ng/ml de factor neurotrófico derivado del cere bro (BDNF, Pan Biotech). Las células viables se cuentan en un citómetro de Neubauer utilizando la prueba de exclu sión con azul de tripano. Las células se siembran con una densidad de 30.000 células/pocillo en placas de 96 pocillos (los pocillos están recubiertos previamente con poli-L-lisina (10 pg/ml; Sigma)) y se cultivan a 37°C en una at mósfera con aire humidificado (95%)/CO2 (5%).
Después de 6 días de cultivo, las células se incuban con los fármacos (5 concentraciones). Después de 1 hora, las células se intoxican con 20 pM de beta-amiloide (25-35; Sigma) en medio definido sin BDNF pero junto con los fár macos. Las neuronas corticales se intoxican durante 2 días. Se realizan dos cultivos independientes por condición, 6 pocillos por condición.
Cuantificación de la longitud de las neuritas
Las células se fijan con una solución fría de etanol (95%) y ácido acético (5%) durante 10 minutos. Después de la permeabilización con 0,1% de saponina, las células se bloquean durante 2 h con PBS que contiene 10% de suero de cabra. A continuación, las células se incuban con anticuerpo monoclonal dirigido contra la proteína 2 asociada a los microtúbulos (MAP-2; Sigma). Este anticuerpo indica específicamente los cuerpos celulares y las neuritas. El anticuerpo secundario utilizado es una IgG (sonda Molecular) de cabra anti-ratón Alexa Fluor 488. Los núcleos de las neuronas se indican por un tinte fluorescente (solución de Hoechst, SIGMA). Se toman veinte imágenes por pocillo, usando InCell AnalyzerTM 1000 (GE Healthcare) con un aumento de 20x. Todas las imágenes se toman en las mismas condiciones. La longitud de las neuritas se cuantifica utilizando el programa informático Developer (GE Healthcare).
Resultados
Los resultados presentados en la Fig. 1 se extraen a partir de dos cultivos independientes, 6 pocillos por condición. Todos los valores se expresan como media ± e.e. de la media. Se realiza una prueba t de Student bilateral con datos sin procesar. Los resultados se expresan en porcentaje de longitud de las neuritas, en comparación con el control (vehículo).
Los fármacos se incubaron con neuronas corticales primarias de rata una hora antes de la intoxicación con Abeta25-3520 pM que dura 2 días (36).
Dos días después de esta incubación, se cuantificó la longitud de la red de neuritas, reflejando el crecimiento celular axonal. Los resultados muestran que los fármacos sometidos a ensayo ejercen claramente un efecto neuroprotector frente a una intoxicación con Abeta25-35 (Fig. 1 y Fig. 2).
I.2 Protección frente a la toxicidad del péptido AP25-35 en células cerebrales endoteliales
Cultivo celular
El primer cultivo de células cerebrales endoteliales de rata (Vect-Horus SAS, Marsella) se cultiva en el pase 0. Al llegar a confluencia, las células endoteliales se disocian con tripsina EDTA (Pan Biotech Ref: P10-023100). Las células se siembran con una densidad de 25.000 células/pocillo en placas de 96 pocillos (los pocillos están recubier tos con 30 pl de colágeno de rata de tipo I, a 1,5 mg/ml, Vect-Horus SAS, Marsella) y se cultivan en medio MCBD 131 (M-131-500, Invitrogen) complementado con 1% de complemento de crecimiento microvascular (MVGS, S-005-25, Invitrogen). Las células se cultivan a 37°C en una atmósfera de aire humidificado (95%)/CO2 (5%). La mitad del medio se cambia cada dos días por medio fresco.
Después de 4 días, se añaden los fármacos al medio de cultivo celular, con diferentes concentraciones, se disuelven en DMSO al 0,1% o agua. Se realiza una incubación previa de 1 hora, en un medio de cultivo que contiene medio Eagle modificado con Dulbecco (DMEM, Pan Biotech Ref: P04-03600), complementado con 2% de suero fetal bo vino (FBS; Invitrogen ref: 16000-036), 1% de L-glutamina (Pan Biotech ref: P04-80100), 1% de penicilinaestreptomicina (PS; Pan Biotech ref: P06-07100), 0,1 mg/ml de heparina (Sigma), 10 ng/ml de factor de crecimiento epidérmico (EGF, Invitrogen) y 10 ng/ml de factor de crecimiento endotelial vascular (VEGF, PHG0146, Invitrogen). A continuación, las células se intoxican con 30 pM de p-amiloide (25-35; Sigma) junto con los fármacos en el mismo medio de cultivo. Las células se intoxican durante 3 días.
Ensayo de la actividad lactato deshidrogenasa (LDH)
Para cada cultivo, después de 3 días de intoxicación, se recoge el material sobrenadante y se analiza con un kit de detección de citotoxicidad (LDH, Roche Applied Sciences). Este ensayo colorimétrico para la cuantificación de la muerte celular se basa en la medición de la actividad lactato deshidrogenasa (LDH) liberada desde el citosol de las células dañadas en el material sobrenadante. La densidad óptica (DO) se determina mediante un espectrofotómetro a una longitud de onda de 492 nm mediante un aparato de barrido múltiple (Thermo, Ref Ascent).
Resultados
Los resultados presentados en la Figura 3 se extraen a partir de dos cultivos independientes, 6 pocillos por condi ción. Todos los valores se expresan como media ± e.e.m. Se realiza una prueba t de Student bilateral con datos sin procesar. Los resultados se expresan en porcentaje de viabilidad celular, en comparación con el control (vehículo). Las fármacos se incuban con células endoteliales cerebrales primarias de rata una hora antes de la intoxicación con Ap25-3530 pM que dura 3 días.
Tres días después de esta incubación, se cuantifica la liberación de LDH en el medio de cultivo, lo que refleja el nivel de muerte celular.
Los resultados presentados muestran claramente que los compuestos sometidos a ensayo ejercen un potente efecto protector contra esa intoxicación con AP25-35 (Fig. 3).
I.3 Protección contra la toxicidad del péptido AP25-35 en células de feocromocitoma
Cultivo celular de PC12
Las células PC12 (Feocromocitoma de rata, ATCC ref: CRL-1721) procedentes de ATCC (ATCC CRL-1721) se descongelaron rápidamente en agua a 37°C. El material sobrenadante se puso inmediatamente en 9 ml de medio de proliferación de PC12 que contenía medio Eagle modificado con Dulbecco DMEM-F12 (Pan Biotech ref: P04-41450) con 15% de suero de caballo inactivado por calor (Invitrogen ref: 16050-130), 2,5% de suero fetal bovino (FBS; Invitrogen ref: 16000-036), 1% de penicilina 10.000 U/ml y estreptomicina 10 mg/ml (PS; Pan Biotech ref: P06-07100) y 1% de L-glutamina 200 mM (Pan Biotech ref: P04-80100).
Las células se centrifugaron (800 ciclos/min, 4°C durante 5 min) y se añadieron a 5 ml de medio de proliferación de PC12, las células viables se contaron con una cámara Malassez utilizando la prueba de exclusión de rojo neutro (Sigma).
A continuación, las células se sembraron a 3.104 células por cm2 en medio de proliferación de PC12 en matraces de plástico de 75 cm2 (Greiner Ref: 658175) recubiertos previamente con poli-L-lisina (10 pg/ml, Sigma Ref: P2636). El medio se cambió cada dos días. Después de 3 días de cultivo, cuando las células alcanzaron un 80% de con fluencia, se lavaron en HBSS sin calcio ni magnesio (Pan Biotech Ref: P06-33500) y se incubaron en tripsina EDTA, (0,05%, Pan Biotech Ref: P10-023100). La reacción enzimática se detuvo con medio de proliferación de PC12 al que se añadió 0,5 mg/ml de ADNAsa 1 de grado 2 (Pan Biotech Ref: T60-37780100). A continuación, las PC12 se centri fugaron (800 ciclos/min a 4°C durante 10 min) y las células se sembraron con una densidad de 2,9 104 por cm2 en un matraz de cultivo de 175 cm2 (Greiner Ref: 6 6 1195) recubierto previamente con poli-L-lisina.
Intoxicación y ensayo de viabilidad de MTT
Las células PC12 (pase n° 2) se siembran con una base de 3300 células por cm2 en placas de 96 pocillos (Greiner Ref: 655 180) recubiertas previamente con medio neurobasal de poli-L-lisina (Sigma) (Invitrogen, Ref: 21103049) que contenía B27 (2%, Invitrogen, Ref: 21103049), penicilina (50 U/ml)-estreptomicina (50 pg/ml) y glutamina (1%) y 50 ng/ml de NGF (Sigma Ref: N1408). NGF permite que las PC12 se diferencien en células similares a las neuronas simpáticas.
Después de 5 días de cultivo, el medio se cambia por neurobasal al que se añade NGF (50 ng/ml), B27 sin antioxi dante, glutamina y antibióticos. Después de 24 h, las células se incuban durante 1 hora con fármacos con 5 concen traciones, 6 pocillos por condición. Después de 1 hora de preincubación, las células se intoxican con 10 pM de betaamiloide (25-35; Sigma) junto con fármacos en el medio de cultivo celular. Después de 24 h, las células se lavan una vez con PBS (Pan Biotech, Ref: P04-36100) y se evalúa la supervivencia de las células PC12 mediante la prueba de viabilidad con MTT (bromuro de 3,[4,5-dimetiltiazol-2-il]-2,5 difeniltetrazolio).
Cultivo celular de neuronas corticales
Se cultivaron neuronas corticales primarias de rata como describen Singer et al. 1999. Brevemente, se dio muerte a ratas hembras preñadas en el día 15 de gestación mediante luxación cervical (Ratas Wistar; Janvier) y se extrajeron los fetos del útero. Se extrajo el córtex y se colocó en medio helado de Leibovitz (L15; Invitrogen) que contenía un 1% de penicilina-estreptomicina (PS; Invitrogen) y 1% de albúmina de suero bovino (BSA; Sigma). Los córtex se disociaron mediante tripsina durante 20 min a 37°C (Tripsina EDTA 1X; Invitrogen) diluida en PBS sin calcio ni mag nesio. La reacción se detiene mediante la adición de medio Eagle modificado con Dulbecco (DMEM; Invitrogen) que contiene ADNasa I de grado II (0,1 mg/ml; Roche Diagnostic) y 10% de suero de ternera fetal (FCS; Invitrogen). Las células se disocian mecánicamente mediante 3 pases a través de una pipeta de 10 ml. Las células se centrifugan después a 180 x g durante 10 minutos a 10°C. El material sobrenadante se desecha y las células del sedimento se resuspenden en un medio de cultivo definido que consiste en Neurobasal (Invitrogen) complementado con B27 (2%; Invitrogen), L-glutamina (0,2 mM; Invitrogen), 1% de solución PS y 10 ng/ml de factor neurotrófico obtenido del cere bro (BDNF, Pan Biotech). Las células viables se cuentan en un citómetro de Neubauer utilizando la prueba de exclu sión con azul de tripano. Las células se siembran con una densidad de 30.000 células/pocillo en placas de 96 poci llos (los pocillos están recubiertos previamente con poli-L-lisina (10 pg/ml; Sigma)) y se cultivan a 37°C en una at mósfera con aire humidificado (95%)/CO2 (5%).
Después de 6 días de cultivo, las células se incuban con los fármacos (5 concentraciones). Después de 1 hora, las células se intoxican con 20 pM de p-amiloide (25-35; Sigma) en medio definido sin BDNF pero junto con los fárma cos. Las neuronas corticales se intoxican durante 2 días.
Ensayo de la actividad lactato deshidrogenasa (LDH)
Después de 2 días de cultivo, se recoge el material sobrenadante y se analiza con el kit de detección de citotoxicidad (LDH, Roche Applied Sciences). Este ensayo colorimétrico para la cuantificación de la muerte celular se basa en la medición de la actividad lactato deshidrogenasa (LDH) liberada desde el citosol de las células dañadas en el material sobrenadante. La densidad óptica (DO) se determina mediante un espectrofotómetro a una longitud de onda de 492 nm mediante un aparato de barrido múltiple (Thermo, Ref Ascent). Los resultados se expresan en porcentaje de viabilidad celular, en comparación con el control negativo (vehículo).
Resultados
Los resultados presentados en las Figuras 4 y 5 se extraen a partir de dos cultivos independientes, 6 pocillos por condición. Todos los valores se expresan como media ± e.e. de la media. Se realiza una prueba t de Student bilate ral con datos sin procesar. Los resultados se expresan en porcentaje de longitud de neuritas, en comparación con el control (vehículo).
Las células PC12 diferenciadas con NGF se incuban con los fármacos una hora antes de la intoxicación con Abeta25-35 10 gM que dura 24 horas.
Un día después de esa incubación, se cuantifica la viabilidad de las PC12 diferenciadas con NGF, usando el ensayo de MTT. Los resultados muestran claramente que la prilocaína y la amlodipina ejercen un fuerte efecto neuroprotector contra esa intoxicación con Abeta25-35 (Fig. 4).
Las neuronas corticales primarias de rata también se incubaron con compuestos de la invención una hora antes de la intoxicación con Abeta25-3520 gM que dura 2 días. Dos días después de esa incubación, se cuantifica la liberación de LDH en el medio de cultivo, lo que refleja el nivel de muerte celular. Los resultados presentados demuestran que los compuestos para uso en la presente invención ejercen un efecto protector sustancial contra esa intoxicación con Abeta25-35 (Fig. 5).
I.4 Actividad de las combinaciones de fármacos
Los ensayos in vitro también se llevan a cabo con varias combinaciones de fármacos que modulan la función sináptica y/o la angiogénesis y/o la respuesta al estrés celular.
Los fármacos se incuban con las mismas condiciones experimentales que las descritas anteriormente (véanse las secciones I.1-1.3). Las combinaciones de fármacos más eficaces que actúan sobre las dianas se resumen en la Tabla 2.
Tabla 2



II. Los compuestos evitan la toxicidad de Afii-42 humano
En esta serie adicional de experimentos, los compuestos candidatos se han sometido a ensayo para estudiar su capacidad para prevenir o reducir los efectos tóxicos de AP1-42 humano. Api-42 es el péptido de longitud completa que constituye los agregados que se encuentran en las biopsias de pacientes humanos afectados por EA. Los fár macos primero se someten a ensayo individualmente, seguido por ensayos de su acción combinatoria. El efecto se determina sobre varios tipos de células, para documentar adicionalmente la actividad de los compuestos.
II.1. Protección frente a la toxicidad de AP1-42 en un modelo de células endoteliales microvasculares de cerebro hu mano
Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano para estudiar la protección que ofrecen los compuestos candidatos sobre la toxicidad de AP1-42.
Células cerebrales endoteliales microvasculares de cerebro humano (HBMEC, ScienCell Ref: 1000, congeladas en el pase 10) se descongelaron rápidamente en un baño de agua a 37°C. El material sobrenadante se puso inmedia tamente en 9 ml de medio Eagle modificado con Dulbecco (DMEM; Pan Biotech ref: P04-03600) que contenía 10% de suero de ternera fetal (FCS; GIBCO ref. 10270-106). La suspensión celular se centrifugó a 180 x g durante 10 minutos a 4°C y el sedimento se suspendió en medio sin suero CSC (CSC sin suero, Cell System, Ref: SF-4Z0-500-R, Lote 51407-4) con 1,6% de RocketFuel exento de suero (Cell System, Ref: SF-4Z0-500-R, Lote 54102), 2% de 10.000 U/ml de penicilina y 10 mg/ml de estreptomicina (PS; Pan Biotech ref: P06-07100 lote 133080808) y se sembró con una densidad de 20.000 células por pocillo, en placas de 96 pocillos (sistema de angiogénesis de biocapa de capa matrigel, BD, Ref 354150, Lote A8662) con un volumen final de 100 gl. Sobre el soporte matrigel, las células cerebrales endoteliales iniciaron espontáneamente el proceso de morfogénesis de la red capilar (47).
Se realizaron tres cultivos por separado para cada condición, 6 pocillos por condición.
Compuestos candidatos y tratamiento de amiloide-pi-42 humano
Brevemente, el péptido AP1-42 (Bachem, ref: H1368 lote 1010533) se reconstituyó en medio de cultivo definido 20 pM (solución madre) y se agitó lentamente a 37°C durante 3 días en la oscuridad para la agregación. El medio de con trol se preparó en las mismas condiciones.
Después de 3 días, este péptido amiloide humano agregado se usó sobre HBMEC 2,5 pM, diluido en medio de con trol (tiempo de incubación óptimo). El péptido AP1-42 se añadió 2 horas después de la siembra de HBMEC sobre matrigel durante 18 horas de incubación.
Una hora después de la siembra de las HBMEC sobre matrigel, los compuestos del ensayo y VEGF-165 se disolvie ron en medio de cultivo (+0,1% de DMSO) y luego se incubaron previamente con HBMEC durante 1 hora antes de la aplicación de AP1-42 (con un volumen final por pocillo de cultivo de 100 pl). Una hora después, los compuestos del ensayo o de la incubación con VEGF (dos horas después de la siembra de las células sobre matrigel), se añadieron 100 pl de péptido AP1-42 hasta una concentración final de 2,5 pM, diluida en medio de control en presencia de los compuestos del ensayo o VEGF (en un volumen total/pocillo de 200 pl), para evitar diluciones adicionales del fárma co.
Organización de las placas de cultivos
VEGF-165, conocido por ser una isoforma proangiogénica de VEGF-A, se usó para todos los experimentos en este estudio como compuesto de referencia. VEGF-165 es una de las isoformas de VEGF más abundantes implicadas en la angiogénesis. Se usó VEGF como compuesto de referencia del ensayo, 10 nM.
Se establecieron las siguientes condiciones:
- Control negativo: medio solo 0,1 % de DMSO
- Intoxicación: amiloide-p1-42Ü □ (2,5 pM) durante 18 h
- Control positivo: VEGF-165 (10 nM) (1 compuesto de referencia/cultivo) 1 hora antes de la adición de AP1-42 (2,5 pM) durante un tiempo de incubación de 18 h.
- Compuestos del ensayo: Compuesto del ensayo 1 hora antes de la adición de AP1-42 (2,5 pM) durante un tiempo de incubación de 18 h.
Cuantificación de la red capilar
Por pocillo, se tomaron 2 fotos con lente de 4x usando InCell AnalyzerTM 1000 (GE Healthcare) en transmisión de luz. Todas las imágenes se tomaron con las mismas condiciones. El análisis de las redes de angiogénesis se realizó utilizando el programa informático Developer (GE Healthcare). Se evaluó la longitud total de la red capilar.
Procesamiento de datos
Todos los valores se expresan como media ± e.e. de la media de los 3 cultivos (n = 6 por cada condición). Los análi sis estadísticos se realizaron sobre las diferentes condiciones realizando una ANOVA seguida de la prueba de Dunnett cuando se permitía (programa informático Statview versión 5.0). Los valores (como %) insertados en los gráficos muestran la evolución de la toxicidad amiloide. De hecho, la toxicidad amiloide se tomó como el 100% y el efecto del compuesto del ensayo se calculó como un % de esa toxicidad amiloide.
Resultados
Los resultados se muestran en la Figura 6 y la tabla 3. Demuestran que los fármacos, por sí mismos, inducen un efecto protector sustancial contra la toxicidad causada por el péptido Ap 1 -42:
- El ácido aminocaproico solo, con una dosificación baja de, por ejemplo, 160 nM, induce un fuerte efecto protector;
- Levosimendán, con una dosis tan baja como 8 nm, induce un fuerte efecto protector.
II.2 Protección frente a la toxicidad de AP1-42 en células de neuronas corticales primarias
Compuesto del ensayo y tratamiento con amiloide p 1 -42 humano
Las neuronas corticales primarias de rata se cultivan como se ha descrito previamente.
Brevemente, el péptido AP1-42 se reconstituyó en medio de cultivo definido 40 pM (solución madre) y se agitó lenta mente a 37°C durante 3 días en la oscuridad para la agregación. El medio de control se preparó en las mismas condiciones.
Después de 3 días, la solución se usó sobre neuronas corticales primarias de la siguiente manera:
Después de 10 días de cultivo de neuronas, el fármaco se disolvió en el medio de cultivo (+0,1% de DMSO) y luego se incubó previamente con las neuronas durante 1 hora antes de la aplicación de Ap1-42 (con un volumen final por pocillo de cultivo de 100 gl). Una hora después de la incubación del fármaco, se añadieron 100 gl de péptido Ap1-42 hasta una concentración final de 10 gM diluida en presencia de fármaco, para evitar más diluciones del fármaco. Las neuronas corticales se intoxicaron durante 24 horas. Se realizaron tres cultivos por separado con cada condición, 6 pocillos por cada condición.
BDNF (50 ng/ml) y Estradiol-p (100 y 150 nM) se usaron como control positivo y compuestos de referencia, respecti vamente. Se realizarán tres cultivos por separado con cada condición, 12 pocillos por cada condición.
Organización de las placas de cultivo
Se usó estradiol-p 100 y 150 nM como compuesto de ensayo de referencia y BDNF a 50 ng/ml se usó como control positivo.
El estradiol-p y el BDNF se disolvieron en medio de cultivo y se incubaron previamente durante 1 h antes de aplicar el amiloide-p.1-42 agregado.
Se evaluaron las siguientes condiciones:
- 1 PLACA DE CONTROL: 12 pocillos/condición
• Control negativo: medio solo 0,1% de DMSO
• Intoxicación: amiloide-p1-42 (10 gM) durante 24 h
• Control positivo: BDNF (50 ng/ml) 1 hora seguido por amiloide-p1-42 (10 gM) durante 24 h • Compuesto de referencia: Estradiol (150 nM) 1 h seguido de amiloide-p1-42 (10 gM) durante 24 h.
- PLACA CON FÁRMACO: 6 pocillos/condición
• Control negativo: medio solo 0,1% de DMSO
• Intoxicación: amiloide-p1-42 (10 gM) durante 24 h
• Fármaco 1: Fármaco 1- 1 h seguido de p-amiloide1-42 (10 gM) durante 24 h • Fármaco 2: Fármaco 2 - 1 h seguido de p-amiloide1-42 (10 gM) durante 24 h Ensayo de la actividad lactato deshidrogenasa (LDH)
Veinticuatro horas después de la intoxicación, se retiró el material sobrenadante y se analizó con el kit de detección de citotoxicidad (LDH, Roche Applied Science, ref: 11644793001, lote: 11800300). Este ensayo colorimétrico para la cuantificación de la toxicidad celular se basa en la medición de la actividad lactato deshidrogenasa (LDH) liberada desde el citosol de las células moribundas en el material sobrenadante.
Procesamiento de datos
Todos los valores se expresan como media ± e.e. de la media de los 3 cultivos (n = 6 por cada condición). Los análi sis estadísticos se realizaron con las diferentes condiciones (ANOVA seguido de la prueba de Dunnett cuando esta ba permitido, programa informático Statview versión 5.0).
Resultados
Los resultados obtenidos para los fármacos seleccionados individuales en los ensayos de toxicidad sobre células neuronales corticales primarias, se presentan en la tabla 3 y en la figura 7.
Tabla 3


II.3 Efecto de las terapias combinadas sobre la toxicidad delpéptido Afii-42 humano en células HBMEC humanas. La eficacia de las combinaciones de fármacos de la invención se evalúa en células humanas. El protocolo que se utiliza en esos ensayos es el mismo que el que se describe en las secciones II.1 anteriores.
Resultados
Se someten a ensayo las siguientes combinaciones de fármacos sobre células endoteliales microvasculares de cerebro humano:
- baclofeno y ácido aminocaproico,
- baclofeno y levosimendán,
- ácido aminocaproico y sulfisoxazol,
- ácido aminocaproico y terbinafina,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol,
- levosimendán y terbinafina,
- eplerenona y levosimendán,
- eplerenona y sulfisoxazol,
- eplerenona y fenoldopam,
- sulodexida y levosimendán,
- sulodexida y sulfisoxazol,
- sulodexida y fenoldopam o
- eplerenona y sulodexida.
Todas las combinaciones de fármacos sometidas a ensayo proporcionan un efecto protector contra la toxicidad del péptido AP1-42 humano en el modelo de HBMEC, tal y como se muestra en la Tabla 4 a continuación y se ejemplifica en las Figuras 8 a 13.
Tabla 4
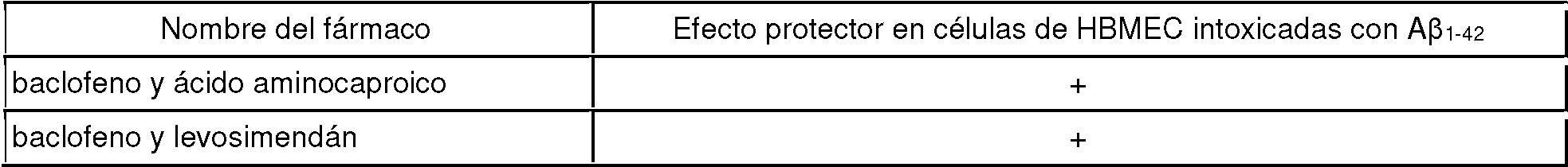

III. La terapia de combinación de levosimendán y sulfisoxazol protege eficazmente las neuronas contra la toxicidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación que emplea levosimendán y sulfisoxazol por su capacidad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 8. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de sulfisoxazol y levosimendán (Fig. 8A) mientras que, a esas concentraciones, levosimendán (Fig. 8B) y sulfisoxazol (Fig. 8C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
IV. La terapia de combinación de terbinafina y sulfisoxazol protege eficazmente las neuronas contra la toxicidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación con terbinafina y sulfisoxazol por su capacidad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 9. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de terbinafina y sulfisoxazol (Fig. 9A) mientras que, a esas concentraciones, el sulfisoxazol (Fig. 9B) y la terbinafina (Fig. 9C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
V. La terapia de combinación de levosimendán y baclofeno protege eficazmente las neuronas contra la toxicidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación que emplea levosimendán y baclofeno por su capacidad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 10. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 pM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de levosimendán y baclofeno (Fig.
10A) mientras que, a esas concentraciones, levosimendán (Fig. 10B) y baclofeno (Fig. 10C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
VI. La terapia de combinación de ácido aminocaproico y terbinafina protege eficazmente las neuronas contra la toxi cidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación que emplea ácido aminocaproico y terbinafina por su capaci dad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 11. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 gM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de ácido aminocaproico y terbinafina (Fig. 11A) mientras que, a esas concentraciones, el ácido aminocaproico (Fig. 11B) y la terbinafina (Fig. 11C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
VII. La terapia de combinación de ácido aminocaproico y levosimendán protege eficazmente las neuronas contra la toxicidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación que emplea ácido aminocaproico y levosimendán por su capa cidad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 12. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 gM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación ácido aminocaproico y levosimendán (Fig. 12A) mientras que, a esas concentraciones, el ácido aminocaproico (Fig. 12B) y el levosimendán (Fig. 12C) por sí solos, no tienen un efecto significativo sobre la intoxicación.
VIII. La terapia de combinación de terbinafina y levosimendán protege eficazmente las neuronas contra la toxicidad de AP1-42 humano
En este ejemplo, se evaluó la terapia de combinación que emplea levosimendán y terbinafina por su capacidad para prevenir o reducir los efectos tóxicos de AP1-42 humano.
La terapia de combinación se sometió a ensayo en las condiciones experimentales descritas en el Ejemplo II.1. Se utilizaron cultivos de células endoteliales microvasculares de cerebro humano, tal y como se describe en II.1., y se incubaron simultánea o secuencialmente con la combinación de fármacos.
Los resultados se presentan en la Figura 13. Muestran claramente que el péptido amiloide humano agregado (AP1-42 2.5 gM) produce una intoxicación significativa, superior al 40%, en comparación con las neuronas tratadas con vehículo. Esa intoxicación se evita significativamente mediante la combinación de terbinafina y levosimendán (Fig. 13A) mientras que, a esas concentraciones, la terbinafina (Fig. 13B) y el levosimendán (Fig. 13C), por sí solos, no tienen un efecto significativo sobre la intoxicación.
IX. Actividad in vivo
Se someten a ensayo compuestos y sus combinaciones de forma activa en pruebas in vitro en un modelo in vivo de enfermedad de Alzheimer. La sobreexpresión de transgenes de precursores de la proteína beta amiloide humana (APP) mutante, ligada a la enfermedad de Alzheimer, ha sido el medio más fiable para promover el depósito de Abeta en los cerebros de ratones transgénicos que servían como modelos de la enfermedad EA en numerosos es tudios. A medida que envejecen, esos ratones con APP mutante desarrollan una patología amiloide fuerte y otras características similares a la EA, que incluyen una disminución de la densidad sináptica, gliosis reactiva y algunos déficits cognitivos. Muchos modelos de ratones con APP mutante muestran poca evidencia de pérdida neuronal manifiesta y patología de ovillo neurofibrilar (ONF). Los ratones hemicigotos para este transgén BRI-Abeta42 son viables y fértiles con una esperanza de vida normal. El ARNm transgénico de BRI-Abeta42 se expresa con un patrón característico del promotor de la proteína priónica de ratón; los niveles más altos de expresión transgénica se detec tan en las células granulares cerebelosas y el hipocampo, seguidos de la corteza, el puente de Varolio, el tálamo y el mesencéfalo. En la proteína de fusión transgénica, Abeta1-42 se fusiona con el extremo C de la proteína BRI en el sitio de escisión similar a la furina, de modo que la escisión da como resultado una secreción eficaz de Abeta1-42 en el lumen o el espacio extracelular. Por lo tanto, esos ratones expresan específicamente la isoforma Abeta1-42. Los ratones hemicigotos BRI-Abeta42 acumulan con la edad beta amiloide insoluble con detergente y desarrollan placas
con núcleo en el cerebelo a los 3 meses de edad. El desarrollo de la patología del prosencéfalo tiene lugar más tarde, las placas de Abeta extracelulares no están presentes de manera consistente en el hipocampo y las cortezas entorrinal/piriforme hasta los 12 meses de edad. Los depósitos beta amiloides (placas con núcleo) pueden observar se ya a los 3 meses en la capa molecular del cerebelo de ratones transgénicos y volverse más pronunciados con la edad; ocasionalmente se observan placas extracelulares en las cortezas entorrinal/piriforme y el hipocampo a los 6 meses de edad, pero no se encuentran de manera constante hasta >12 meses de edad. Los ratones de más edad muestran una patología generalizada con placas con núcleo y difusas en el cerebelo, la corteza, el hipocampo y el bulbo olfatorio. Las placas amiloides extracelulares muestran núcleos amiloides densos con fibrillas radiantes; se observan muchos haces de neuritas distróficas en la periferia de esas placas. Una gliosis reactiva se asocia con las placas.
Tratamientos farmacológicos
Los ratones transgénicos Tg A12E mc (Prnp-ITM2B/APP695*42) (37) se han obtenido en Jackson Laboratory (http://jaxmice.jax.org/strain/007002.html). Los ratones originadores con los niveles plasmáticos más altos de Abeta42, la línea BRI-Abeta42A (12e), se han mantenido en un fondo mixto de B6C3. Los ratones transgénicos machos adultos tienen libre acceso a alimentos y agua. De acuerdo con un protocolo aprobado del Comité Institucional para el Cuidado y Uso de Animales, los ratones se pesan y se les inyecta i.p. o son alimentados a la fuerza, una vez al día durante 10 a 20 semanas consecutivas con una solución de control (placebo) o los fármacos de la presente invención o con combinaciones de fármacos de la Tabla 2, preparadas con dosis diferentes.
Análisis de la supervivencia
Las tasas de supervivencia se han analizado utilizando los métodos de Kaplan-Meier. Los métodos de Holm-Sidak (post hoc) se han utilizado para todas las pruebas de comparación múltiple por parejas. Las muertes externas están censuradas. Todas las comparaciones se han hecho entre los miembros de una camada para limitar cualquier efecto potencialmente confuso por diferencias de la cepa de fondo.
Pruebas conductuales
Las pruebas conductuales fueron diseñadas y realizadas de acuerdo con los métodos publicados por varios autores (38-41).
Aprendizaje y memoria espacial en el laberinto de agua de Morris (MWM, por sus siglas en inglés)
Este experimento se realiza en una piscina circular de 90 cm de diámetro, hecha a base de plástico blanco y rellena de agua de color lechoso. Una plataforma de escape de 8 cm de diámetro, de plástico transparente, se sumergió 0,5 cm por debajo del nivel del agua. Las pistas visuales son proporcionadas por diferentes formas geométricas impre sas en letras de tamaño A4 y colocadas en las cuatro paredes circundantes (la distancia desde la piscina era de 50 a 70 cm). Con cada ratón se han realizado cuatro ensayos diarios (intervalo de 5 a 7 minutos entre ensayos, un total de 16 ensayos) durante 4 días. Cada ensayo se ha realizado a partir de uno de cuatro puntos de partida diferentes. El movimiento de los ratones se controla mediante el programa informático Videotrack (View Point). Se ha determi nado el tiempo necesario para localizar la plataforma de escape (latencia de escape, hasta 60 segundos). Después de localizar la plataforma, al ratón se le ha permitido sentarse sobre ella durante 15 segundos. Los ratones que no pudieron encontrar la plataforma en 60 segundos, fueron guiados hacia ella y se les permitió permanecer en ella durante 15 segundos. Se ingresa una latencia de 60 segundos en el registro para ese caso. Los cuatro ensayos al día se han promediado para un análisis estadístico, excepto el primer ensayo el día 1. El día 9 (5 días después del último entrenamiento) los ratones han sido sometidos a una prueba de ensayo de 60 segundos en la que se elimina la plataforma y se permite que los ratones la busquen. Se registra el tiempo que pasa cada animal en cada cuadran te (tiempo de búsqueda en cuadrante). Se han utilizado varios grupos de ratones macho con 3, 7, 10 y 12 meses. Pocos ratones han mostrado un comportamiento de congelación (por ejemplo, quedándose inmóviles en el agua y negándose a nadar) que interfiere fuertemente con la prueba; esos animales han sido excluidos del análisis de los datos. Todas las pruebas de comportamiento se llevan a cabo en un ambiente tranquilo y con poca luz.
Prueba de memoria de trabajo en el laberinto de agua del brazo radial
Esta medición sensible con base cognitiva de la memoria de trabajo se ha obtenido con la ayuda del aparato que consistía en una piscina de 100 cm de diámetro rellena de agua (también utilizada para el laberinto de agua de Morris y en las tareas de reconocimiento de la plataforma) equipada con un inserto de aluminio para crear seis brazos para nadar, distribuidos radialmente. Las pruebas consisten en cinco ensayos de 1 minuto por sesión diaria durante 9-12 días consecutivos. Al comienzo de cada sesión, una plataforma sumergida transparente se coloca al final de uno de los seis brazos para nadar (seleccionado al azar, cambiado diariamente). Para cada uno de los cuatro prime ros ensayos de adquisición, el animal se coloca en uno de los brazos que no contiene la plataforma (secuencia alea toria) y se le permite buscar la plataforma. Durante el ensayo de 60 s, cada vez que el animal entra en otro brazo que no contiene una plataforma, es devuelto suavemente a su lugar de partida y se registra un error. Después del cuarto ensayo, se permite que el animal repose durante 30 minutos, seguido de un quinto ensayo (de retención) que se origina en el brazo para nadar final que no contiene plataforma. El número de errores (selección de brazos inco
rrectos) y la latencia de escape (tiempo para alcanzar la plataforma, máximo 60 s) se registran para cada ensayo. Aprendizaje de referencia y memoria espacial en la prueba de la plataforma circular
Esta prueba de una tarea con base cognitiva se realiza con la ayuda del aparato que consiste en una plataforma circular de 69 cm de diámetro que tiene 16 orificios de "escape" distribuidos de forma equidistante alrededor de la circunferencia. Un refugio de escape está instalado debajo de uno de los orificios, y una cortina negra, sobre la que se colocan varias señales visuales, rodea la plataforma. El animal se coloca en el centro de la plataforma al inicio de una sola prueba de 5 minutos y se presentan estímulos adversos (luces brillantes, aire de un ventilador). Se registra el número total de errores (toques con la cabeza en los orificios sin escape) y la latencia de escape (tiempo hasta alcanzar el orificio de escape).
Capacidad de reconocimiento en la prueba de reconocimiento de la plataforma
Esta tarea de búsqueda con base cognitiva evalúa la identificación de un objeto y la capacidad de reconocimiento. El objeto diana consiste en una plataforma circular de 9 cm de diámetro provista de un distintivo negro de 10 cm x 40 cm, que se coloca 0,8 cm por encima de la superficie del agua en una piscina circular de 100 cm de diámetro. Las pruebas consisten en cuatro ensayos de 60 s por día, cada uno de cuatro días consecutivos. Cada día, el objeto diana se coloca en un cuadrante diferente de la piscina para cada prueba, y el animal es liberado en la misma ubica ción a lo largo de la circunferencia de la piscina en los cuatro ensayos. Se registra la latencia total (máximo 60 s) para cada ensayo.
Examen de Irwin modificado
Una pantalla completa, modificada de Irwin se utiliza para determinar si alguno de los ratones presentaba impedi mentos fisiológicos, conductuales o sensorimotores relacionados con su genotipo. Para explorar las habilidades motoras, la coordinación y la fuerza muscular, los ratones se colocan sobre un alambre que se había tensado entre dos columnas a 30 cm de altura y se evaluó su capacidad para mantener el equilibrio sobre el cable. Además, se determinó su capacidad para agarrarse y colgarse del cable con las cuatro patas durante al menos 5 segundos y volver a subir al cable.
Cuantificación del depósito amiloide vascular
Para la cuantificación de la angiopatía amiloide cerebral (CAA), se inmunotiñeron secciones de 5 gm incluidas en parafina a intervalos de 30 gm a través de las leptomeninges de la corteza parietal o cerebelar, con anticuerpo Ab9 biotinilado (anti-Ap1 -16, 1:500) durante la noche a 4°C (n = 5-7 ratones por genotipo en cada grupo de edad, n = 6 secciones por ratón). Los vasos sanguíneos teñidos positivamente se evalúan visualmente utilizando el sistema de puntuación de Vonsattel modificado (42). La puntuación de la gravedad de la CAA se calcula multiplicando el núme ro de vasos de CAA por el grado de gravedad de CAA.
Histología: inmunohistoquímica e inmunofluorescencia
Los ratones Tg y WT de 3 a 12 meses se anestesian y se perfunden transcardialmente de forma secuencial con NaCl al 0,9% y paraformaldehído al 4% en 0,1 mol/L de solución salina tamponada con fosfato (PBS) (pH 7,4) o formol al 10% y paraformaldehído al 4% en 0,1 mol/L de PBS (pH 7,4). Los cerebros y las médulas espinales se extirpan y se almacenan en paraformaldehído al 4%. Algunas muestras se incluyen en parafina y se cortan en un microtomo deslizante con un espesor de 10 gm. Las criosecciones (14 gm) se cortan en un criostato y se montan sobre portaobjetos recubiertos con alumbre de cromo. La peroxidasa endógena se inactiva tratando la sección con metanol que contiene un 0,3% de H2O2 durante 30 minutos. Las secciones se bloquean en suero de caballo al 10%. Los anticuerpos primarios se usan y se incuban durante la noche a 4°C en presencia de suero de caballo al 1%. Todos los anticuerpos secundarios biotinilados o acoplados a fluoresceína, rojo de Texas y AMCA, los fluorocromos, el kit ABC y 3,3'-diaminobencidina como cromógeno para la actividad peroxidasa, proceden de Vector Laboratories. La incubación con el anticuerpo secundario se mantiene a temperatura ambiente durante 1 hora. Todas las etapas de lavado (3-10 minutos) y la dilución de los anticuerpos se realizan usando solución salina tamponada con fosfato (0,1 mol/L de PBS, pH 7,4) o solución salina tamponada con Tris (0,01 mol/L de Tris, 0,15 mol/L de NaCl, pH 7,4). La incubación con el complejo ABC y la detección con 3,3'-diaminobencidina se lleva a cabo de acuerdo con el ma nual del fabricante. La contratinción con hematoxilina se realiza de acuerdo con procedimientos convencionales. Para cada determinación se utiliza un mínimo de tres ratones por genotipo, edad y sexo (43).
Preparación de extractos cerebrales
Los cerebros se recogen rápidamente sobre hielo, 90 a 120 minutos después de la inyección mortal y se congelan a -80°C. El hemisferio cerebral derecho de cada ratón se pesa después de la congelación. El análisis de la masa del hemisferio a través de la desviación absoluta media, nos permite excluir las muestras que sobrepasan 4 desviacio nes absolutas medias del resto del conjunto. Los hemisferios cerebrales se homogeneizan y los lisados celulares que contienen proteínas enteras se preparan de acuerdo con las instrucciones del fabricante para los kits de ensayo enzimático (R&D Systems, Inc.). En resumen, las cortezas cerebrales se homogeneizan en 800 gl de tampón de extracción 1x con bajo contenido en sales (kit de R&D) y se incuban sobre hielo durante 10 minutos. Los homogeneizados se centrifugan luego a 13.000 g durante 15 minutos a 4°C. La concentración de proteína en cada muestra se estima de acuerdo con un ensayo derivado de biuret (Pierce). Los niveles de APP, Ap40 y Ap42 se miden me diante técnicas de inmunotransferencia Western y ELISA tipo sándwich. Además, las actividades de las secretasas a, p y y se pueden medir a partir de los mismos extractos.
Ensayo de los niveles de APP total en extractos de corteza cerebral de ratón
Se carga una cantidad igual de proteínas de extractos cerebrales en cada gel, 30 gg por pista para cada muestra. Cada gel contenía ocho tratamientos: control; fármaco 1 dosis de 7,5 mg/kg; y fármaco 2 en varias dosis. Para mini mizar la variación dentro del gel, cada gel contenía tres conjuntos de todos los grupos de tratamiento. Cada transfe rencia se analiza con el anticuerpo 22C11. Cada transferencia también se analiza con el anticuerpo de p-actina para una normalización para transferir la eficiencia. La intensidad de la señal de la banda de APP se normaliza con la de p-actina. Se cargan dos muestras de "control" en cada gel/transferencia para estudiar la variación de transferencia a transferencia. El análisis de las transferencias se realiza de dos maneras: en cuanto a la transferencia (n = 3), para evaluar la variación de gel a gel; y de transferencias combinadas (n = 9 o 10) tal y como se describe (38-39). El análisis en cuanto a la transferencia con n = 3 muestra la misma tendencia que el análisis final con n = 9 o 10. Se presentan los resultados del análisis combinado.
ELISA de tipo sándwich de Ap
Para los ELISA de Ap del cerebro, los niveles de Ap del cerebro anterior y posterior se determinan de forma inde pendiente, y el bulbo olfatorio se excluye del análisis. Para el análisis de Ap en plasma, se extrae sangre en tubos recubiertos con EDTA después de una punción cardíaca. Las muestras de sangre se centrifugan a 3000 rpm durante 10 minutos a 4°C, y el plasma se divide en partes alícuotas y se almacena a -80°C hasta su uso. Los niveles de Ap se determinan mediante ELISAs de tipo sándwich, específicos del final utilizando Ab9 (Ab anti-Ap1-16) como Ab (anticuerpo) de captura para Ap40, 13.1.1-HRP (Ab anti-Ap35-40) como Ab de detección para Ap40, 2.1.3 (Ab anti-Ap35-42) como Ab de captura para Ap42, y Ab9-HRP como Ab de detección para Ap42 (n = 5-7 ratones por genoti po en cada grupo de edad). Los niveles de Ap se normalizan frente a los resultados anteriores, utilizando los mismos conjuntos de ratones como controles internos para minimizar una variabilidad potencial de ELISA, como se describe (46).
Transferencia Western
Las muestras de cerebro anterior congeladas a presión se homogeneizan en tampón de ensayo de radioinmunoprecipitación (RIPA) (Boston BioProducts, Worcester, MA) con una mezcla de inhibidor de proteasa al 1% (Roche). El homogeneizado se centrifuga a 100.000 x g durante 1 ha 4°C. La concentración de proteínas en el material sobre nadante se determina utilizando el ensayo de proteínas BCA (Pierce). Las muestras de proteína (20 gg) se procesan en geles Bis-Tris al 12% XT o geles Bis-Tris al 4-12% XT (Bio-Rad, Hercules, CA) y se transfieren a membranas de nitrocelosa de 0,2 gm. Las transferencias se someten a microondas durante 2 minutos en PBS 0,1 M dos veces y se analizan con Ab 82E1 (anti-Ap1 -16, 1:1000; IBL, Gunma, Japón) y 20 aminoácidos C-terminales anti-APP (1:1000) como se describe (46). Las transferencias se estabilizan y se repite el análisis con anti-p-actina (1:1000; Sigma) como control de carga. La intensidad relativa de las bandas se mide con el programa informático ImageJ.
Cuantificación del depósito de amiloide parenquimatoso
Los medios cerebros se fijan mediante inmersión en formalina al 10% y se procesan para la inclusión en parafina. Secciones de tejido cerebral (5 gm) se inmunotiñeron con anticuerpo (Ab) anti-Ap total. Las secciones se contratiñen con hematoxilina. Se utilizan seis secciones por cerebro a través del hipocampo, la corteza piriforme (bregma, -1,70 a -2,80 mm) o el cerebelo (parafloculo, crus ansiforme y lobulillos simples; bregma, -5,40 a -6,36 mm) para la cuanti ficación (n = 5-7 ratones por genotipo en cada grupo de edad). La carga de la placa de Ap se determina utilizando el programa informático MetaMorph (Molecular Devices, Palo Alto, CA). Para la cuantificación de las placas con nú cleo, se tiñen secciones en serie de las analizadas para la carga de Ap con tioflavina S (ThioS) y se cuenta el número de placas positivas para ThioS en el hipocampo, la corteza entorrinal/piriforme o el cerebelo. Todos los análisis anteriores se realizan de forma ciega.
Análisis estadístico de datos in vivo
Los resultados de todos los experimentos se analizan con STATISTICA 8.0 (Statsoft). Los niveles de Ap, la carga de placa amiloide y la gravedad de la CAA, se analizan utilizando ANOVA con la prueba de comparación múltiple Holm-Sidak post hoc o la prueba t de Student de dos colas. Si el conjunto de datos no cumple con los supuestos de la prueba paramétrica, se realiza la prueba de Kruskal-Wallis seguida de la comparación múltiple de Dunn post hoc o la prueba de suma de rango de Mann-Whitney. Para analizar si los niveles de Ap en los ratones bitransgénicos eran compatibles con una suma aditiva de los niveles de Ap en los miembros individuales transgénicos de una camada, se utiliza una regresión lineal múltiple sin prueba de intercepción. Todas las comparaciones se hacen entre miembros de una camada. La modelización de la respuesta al fármaco se realiza excluyendo las muestras de control (0 mg/kg). La DE50 corresponde a la dosis (mg/kg) requerida para inducir un 50% de respuesta máxima inducida por el fármaco en los experimentos. Se calcula utilizando el modelo de la ecuación de Hill para el logaritmo de DE50.
Se realizan experimentos in vivo para combinaciones de fármacos candidatos. Los resultados positivos en la memo ria de aprendizaje y espacial se enumeran en la tabla 5 a continuación.
Tabla 5

Bibliografía
1. Crook R., Verkkoniemi A., et al. (1998). A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 4(4): 452-5.
2. Houlden H., Baker M., et al. (2000). Variant Alzheimer's disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol.
48(5): 806-8.
3. Kwok J.B., Taddei K., et al. (1997). Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzhei mer's disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport. 8(6): 1537-42.
4. Verkkoniemi A., Kalimo H., et al. (2001). Variant Alzheimer disease with spastic paraparesis: neuropathological phenotype. J Neuropathol Exp Neurol. 60(5): 483-92.
5. Citron M. (2004). Strategies for disease modification in Alzheimer's disease. Nat Rev Neurosci. 5(9): 677-85.
6. Suh Y.H. and Checler F. (2002). Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease. Pharmacol Rev. 54(3): 469-525.
7. Blacker D., Albert M.S., et al. (1994). Reliability and validity of NINCDS-ADRDA criteria for Alzheimer's disease. The National Institute of Mental Health Genetics Initiative. Arch Neurol. 51(12): 1198-204.
8. Rossor M.N., Fox N.C., et al. (1996). Clinical features of sporadic and familial Alzheimer's disease. Neurodegeneration. 5(4): 393-7.
9. Glenner G.G., Wong C.W., et al. (1984). The amyloid deposits in Alzheimer's disease: their nature and pathogenesis. Appl Pathol. 2(6): 357-69.
10. Ballatore C., Lee V.M., et al. (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 8(9): 663-72.
11. Bell K.F. and Claudio Cuello A. (2006). Altered synaptic function in Alzheimer's disease. Eur J Pharmacol.
545(1): 11-21.
12. Hardy J.A. and Higgins G.A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science. 256(5054): 184-5.
13. Braak H. and Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol.
82(4): 239-59.
14. Golde T.E. (2005). The Abeta hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol. 15(1): 84-7.
15. Hardy J. and Selkoe D.J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 297(5580): 353-6.
16. Selkoe D.J. (2000). The genetics and molecular pathology of Alzheimer's disease: roles of amyloid and the pre senilins. Neurol Clin. 18(4): 903-22.
17. Huang Y.Z., Won S., et al. (2000). Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 26(2): 443-55.
18. Naruse S., Thinakaran G., et al. (1998). Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 21 (5):1213-21.
19. Leeuwen F.N., Kain H.E., et al. (1997). The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol. 139(3):797-807.
20. Ge G., Fernández C.A., et al. (2007). Bone morphogenetic protein 1 processes prolactin to a 17-kDa antiangiogenic factor. Proc Natl Acad Sci U S A. 104(24):10010-5.
21. Hardie D.G. (2007). AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 8(10): 774-85.
22. Reihill J.A., Ewart M.A., et al. (2007). AMP-activated protein kinase mediates VEGF-stimulated endothelial NO production. Biochem Biophys Res Commun. 354(4):1084-8.
23. Ouchi N., Kobayashi H., et al. (2004). Adiponectin stimulates angiogenesis by promoting cross-talk between
AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 279(2):1304-9.
24. Hug C., Wang J., et al. (2004). T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 101 (28):10308-13.
25. English D., Kovala A.T., et al. (1999). Induction of endothelial cell chemotaxis by sphingosine 1-phosphate and stabilization of endothelial monolayer barrier function by lysophosphatidic acid, potential mediators of hematopoietic angiogenesis. J Hematother Stem Cell Res. 8(6):627-34.
26. Gorlach A., Klappa P., et al. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 8(9-10): 1391-418.
27. Verkhratsky A. (2004). Endoplasmic reticulum calcium signaling in nerve cells. Biol Res. 37(4): 693-9.
28. Cookson M.R. (2003). Neurodegeneration: how does parkin prevent Parkinson's disease? Curr Biol. 13(13): R522-4.
29. Sze C.I., Su M., et al. (2004). Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer's disease. J Biol Chem. 279(29): 30498-506.
30. Walchli S., Curchod M.L., et al. (2000). Identification of tyrosine phosphatases that dephosphorylate the insulin receptor. A brute force approach based on "substrate-trapping" mutants. J Biol Chem. 275(13):9792-6.
31. Chang N.S., Doherty J., et al. (2003). JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J Biol Chem. 278(11):9195-202.
32. D'Orazi G., Cecchinelli B., et al. (2002). Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 4(1 ):11-9.
33. Zhu H., Wu L., et al. (2003). MDM2 and promyelocytic leukemia antagonize each other through their direct interaction with p53. J Biol Chem. 278(49):49286-92.
34. Rodrigues S., De Wever O., et al. (2007). Opposing roles of netrin-1 and the dependence receptor DCC in cancer cell invasion, tumor growth and metastasis. Oncogene. 26(38):5615-25.
35. Taniguchi Y., Kim S.H., et al. (2003). Presenilin-dependent "gamma-secretase" processing of deleted in colorectal cancer (DCC). J Biol Chem. 278(33):30425-8.
36. Singer C., Figueroa-Masot X., Batchelor R., and Dorsa D. Mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J. Neuroscience, 1999, 19(7):2455-2463.
37. McGowan E., et al. (2005) Ap42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron 47: 191-199.
38. Leighty R.E. et al. (2008) Use of artificial neural networks to determine cognitive impairment and therapeutic effectiveness in Alzheimer's transgenic mice. Journal of Neuroscience Methods 167: 358-366
39. Ashe KH (2001) Learning and memory in transgenic mice modelling Alzheimer's disease. Learning and Memory 8: 301-308.
40. Carlson GA, et al. (1997) Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Human Molecular Genetics 6:1951 -1959.
41. Hsiao K, et al. (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102.
42. Greenberg S.M. and Vonsattel J.P. (1997) Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke 28(7):1418-22
43. Schindowski K. et al. (2006) Alzheimer's Disease-Like Tau Neuropathology Leads to Memory Deficits and Loss of Functional Synapses in a Novel Mutated Tau Transgenic Mouse without Any Motor Deficits. Am J Pathol. 169: 599-616.
44. Lahiri DK, et al. (2004) Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. Journal of Pineal Research 36:224-231.
45. Basha MR, et al. (2005) The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amy loid precursor protein and beta-amyloid in the aging brain. Journal of Neuroscience 25: 823-829.
46. Lahiri D.K. et al. (2007) Experimental Alzheimer's Disease Drug Posiphen[(Phenserine] Lowers AmyloidbetaPeptide Levels in Cell Culture and Mice. Journal of Pharmacology and experimental therapeutics 320: 386-396.
47. Paris D, et al. (2005) Anti-angiogenic activity of the mutant Dutch A(beta) peptide on human brain microvascular endothelial cells. Brain Res Mol Brain Res. 136: 212-30.
Claims (9)
1. Una composición para uso en el tratamiento de la enfermedad de Alzheimer (EA) o un trastorno relacionado, que comprende al menos levosimendán, o una sal o una formulación de liberación sostenida del mismo.
2. Una composición para uso según la reivindicación 1, que comprende levosimendán, o una sal o una formulación de liberación sostenida del mismo y al menos un compuesto adicional seleccionado a partir de ácido aminocaproico, acamprosato, amlodipina, argatrobán, baclofeno, cilostazol, cinacalcet, clopidogrel, difilina, fenoldopam, leflunomida, mepacrina, metimazol, fenformina, prilocaína, rifabutina, sulfisoxazol, tadalafilo, terbinafina, cinarizina, eplerenona, carbenoxolona, sulodexida, carbamazina, ciclopirox, amobarbital, cefotetán, tetranitrato de eritritilo, meticlotiazida, risedronato, enprofilina, oxtrifilina, parametadiona, cefmenoxima, aprindina, etomidato, mitiglinida, benidipina y zonisamida, o sales o formulaciones de liberación sostenida de los mismos, para una administración combinada, por separado o secuencial.
3. Una composición para uso según la reivindicación 2, en donde dicha composición comprende levosimendán, o una sal o una formulación de liberación sostenida del mismo, y al menos un compuesto adicional seleccionado a partir de ácido aminocaproico, baclofeno, sulfisoxazol y terbinafina, o sales o formulaciones de liberación sostenida de los mismos, formulados para una administración combinada, por separado o secuencial.
4. La composición para uso según una cualquiera de las reivindicaciones precedentes, en donde dicha composición comprende al menos una de las siguientes combinaciones de fármacos, formulándose los fármacos en cada combi nación para una administración combinada, por separado o secuencial:
- baclofeno y levosimendán,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol,
- levosimendán y terbinafina,
- levosimendán y tetranitrato de eritritilo, o
- levosimendán y zonisamida,
o sal(es) o formulación(es) de liberación sostenida de los mismos.
5. La composición para uso según la reivindicación 4, que comprende al menos una de las siguientes combinaciones de fármacos, formulándose los fármacos en cada combinación para una administración combinada, por separado o secuencial:
- baclofeno y levosimendán,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol, o
- levosimendán y terbinafina,
o sal(es) o formulación(es) de liberación sostenida de los mismos.
6. La composición para uso según una cualquiera de las reivindicaciones precedentes, que comprende un vehículo o un excipiente farmacéuticamente aceptable.
7. La composición para uso según una cualquiera de las reivindicaciones precedentes, en donde dicha composición se administra repetidamente al sujeto.
8. Una composición que comprende levosimendán, o una sal o una formulación de liberación sostenida del mismo, para uso para proteger neuronas o células cerebrales endoteliales contra la toxicidad de Ap en un sujeto que tiene la enfermedad de Alzheimer.
9. Una composición que comprende al menos una de las siguientes combinaciones de fármacos, para una adminis tración combinada, por separado o secuencial:
- baclofeno y levosimendán,
- ácido aminocaproico y levosimendán,
- levosimendán y sulfisoxazol,
- levosimendán y terbinafina,
- levosimendán y tetranitrato de eritritilo, o
- levosimendán y zonisamida,
o sal(es) o formulación(es) de liberación sostenida de los mismos.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP09306048A EP2322163A1 (en) | 2009-11-03 | 2009-11-03 | New therapeutics approaches for treating alzheimer disease |
| PCT/EP2010/066510 WO2011054759A1 (en) | 2009-11-03 | 2010-10-29 | New therapeutic approaches for treating alzheimer disease |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2800311T3 true ES2800311T3 (es) | 2020-12-29 |
Family
ID=41693027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10773302T Active ES2800311T3 (es) | 2009-11-03 | 2010-10-29 | Nuevos enfoques terapéuticos para tratar la enfermedad de Alzheimer |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8809302B2 (es) |
| EP (4) | EP2322163A1 (es) |
| JP (2) | JP5977172B2 (es) |
| CN (2) | CN102834095A (es) |
| AR (1) | AR078846A1 (es) |
| BR (1) | BR112012010470A2 (es) |
| CA (2) | CA3065614A1 (es) |
| EA (1) | EA024465B1 (es) |
| ES (1) | ES2800311T3 (es) |
| IL (1) | IL219412A (es) |
| MX (1) | MX2012005131A (es) |
| WO (1) | WO2011054759A1 (es) |
Families Citing this family (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10164139A1 (de) | 2001-12-27 | 2003-07-10 | Bayer Ag | 2-Heteroarylcarbonsäureamide |
| EP2065038A1 (en) | 2007-11-30 | 2009-06-03 | Pharnext | New therapeutic approaches for treating Charcot-Marie-Tooth disease |
| WO2009133128A1 (en) * | 2008-04-29 | 2009-11-05 | Pharnext | Combination compositions for treating alzheimer disease and related disorders with zonisamide and acamprosate |
| EP2135607A1 (en) | 2008-06-18 | 2009-12-23 | Pharnext | Combination of pilocarpin and methimazol for treating Charcot-MarieTooth disease and related disorders |
| US9925282B2 (en) | 2009-01-29 | 2018-03-27 | The General Hospital Corporation | Cromolyn derivatives and related methods of imaging and treatment |
| EP2263665A1 (en) | 2009-06-02 | 2010-12-22 | Pharnext | New compositions for treating CMT and related disorders |
| US9393241B2 (en) | 2009-06-02 | 2016-07-19 | Pharnext | Compositions for treating CMT and related disorders |
| EP2322163A1 (en) | 2009-11-03 | 2011-05-18 | Pharnext | New therapeutics approaches for treating alzheimer disease |
| US9387206B2 (en) | 2009-11-03 | 2016-07-12 | Pharnext | Therapeutic approaches for treating Alzheimer's disease |
| US9283211B1 (en) | 2009-11-11 | 2016-03-15 | Rapamycin Holdings, Llc | Oral rapamycin preparation and use for stomatitis |
| PE20170923A1 (es) | 2010-05-17 | 2017-07-12 | Forum Pharmaceuticals Inc | Una forma cristalina de clorhidrato de (r)-7-cloro-n-(quinuclidin-3-il)benzo[b]tiofeno-2-carboxamida monohidratado |
| EP2705842A1 (en) * | 2012-09-05 | 2014-03-12 | Pharnext | Therapeutic approaches for treating parkinson's disease |
| US9248111B2 (en) | 2011-03-01 | 2016-02-02 | Pharnext | Therapeutic approaches for treating parkinson's disease |
| US10010515B2 (en) | 2011-03-01 | 2018-07-03 | Pharnext | Therapeutic approaches for treating Parkinson's disease |
| KR101937782B1 (ko) | 2011-03-01 | 2019-01-11 | 파넥스트 | 바클로펜 및 아캄프로세이트에 기초한 신경 장애의 치료 |
| UA113165C2 (xx) | 2011-03-01 | 2016-12-26 | Застосування комбінації баклофену і акампросату для лікування неврологічних захворювань та композиція, яка містить баклофен і акампросат | |
| US9241933B2 (en) | 2011-03-01 | 2016-01-26 | Pharnext | Compositions for treating amyotrophic lateral sclerosis |
| CN102861017A (zh) * | 2011-07-08 | 2013-01-09 | 辽宁省计划生育科学研究院 | 一种化合物在防治老年痴呆的应用 |
| SMT201700373T1 (it) * | 2012-03-01 | 2017-09-07 | Pharnext | Nuove composizioni per il trattamento della sclerosi laterale amiotrofica |
| AU2013224959B2 (en) * | 2012-03-01 | 2017-12-07 | Pharnext | New compositions for treating amyotrophic lateral sclerosis |
| EP3461481A1 (en) | 2012-05-08 | 2019-04-03 | Forum Pharmaceuticals Inc. | Methods of maintaining, treating or improving cognitive function |
| EP2705841A1 (en) * | 2012-09-05 | 2014-03-12 | Pharnext | Combinations of nootropic agents for treating cognitive dysfunctions |
| EP3563849A3 (en) | 2012-10-25 | 2020-02-12 | The General Hospital Corporation | Combination therapies for the treatment of alzheimer's disease and related disorders |
| US10058530B2 (en) | 2012-10-25 | 2018-08-28 | The General Hospital Corporation | Combination therapies for the treatment of Alzheimer's disease and related disorders |
| US8652527B1 (en) | 2013-03-13 | 2014-02-18 | Upsher-Smith Laboratories, Inc | Extended-release topiramate capsules |
| US9101545B2 (en) | 2013-03-15 | 2015-08-11 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| US10525005B2 (en) | 2013-05-23 | 2020-01-07 | The General Hospital Corporation | Cromolyn compositions and methods thereof |
| DK3003268T3 (en) | 2013-06-05 | 2018-11-05 | Pharnext | STABLE ORAL SOLUTIONS FOR COMBINED API |
| WO2015019125A1 (en) * | 2013-08-05 | 2015-02-12 | Societe De Developpement Et De Recherche Industrielle | Molecular targets for the treatment of wounds, in particular chronic wounds |
| CN110305095A (zh) | 2013-10-22 | 2019-10-08 | 综合医院公司 | 色甘酸衍生物以及成像和治疗的相关方法 |
| KR20150056443A (ko) * | 2013-11-15 | 2015-05-26 | 한미약품 주식회사 | 타다라필 및 암로디핀을 포함하는 복합제제 |
| WO2015072700A1 (en) * | 2013-11-15 | 2015-05-21 | Hanmi Pharm. Co., Ltd. | Composite formulation comprising tadalafil and amlodipine |
| US9700544B2 (en) | 2013-12-31 | 2017-07-11 | Neal K Vail | Oral rapamycin nanoparticle preparations |
| MX2016010409A (es) | 2014-02-11 | 2016-11-30 | Pharnext | Combinacion de baclofeno, acamprosato y trigliceridos de cadena media para el tratamiento de trastornos neurologicos. |
| CN106604747A (zh) * | 2014-08-26 | 2017-04-26 | 基础应用医学研究基金会 | 用于治疗和预防伴随认知缺陷或缺损的神经障碍以及神经变性疾病的产品 |
| WO2016085765A1 (en) | 2014-11-25 | 2016-06-02 | President And Fellows Of Harvard College | Methods for generation of podocytes from pluripotent stem cells and cells produced by the same |
| JP6887952B2 (ja) | 2015-02-25 | 2021-06-16 | プリレニア ニューロセラピューティクス リミテッド | 認知機能を改善するため、およびアルツハイマー病を治療するためのプリドピジンの使用 |
| US11471449B2 (en) | 2015-02-25 | 2022-10-18 | Prilenia Neurotherapeutics Ltd. | Use of pridopidine to improve cognitive function and for treating Alzheimer's disease |
| US10520493B2 (en) * | 2015-03-13 | 2019-12-31 | National Cheng Kung University | Method for assessment of neural function by establishing analysis module |
| CN104888198A (zh) * | 2015-04-21 | 2015-09-09 | 徐志强 | 降钙素在制造延缓脑老化药物的新用途 |
| EP4275750B1 (en) * | 2015-09-10 | 2026-05-06 | Alzheon, Inc. | Treatment of alzheimer's disease in a particular patient population |
| US11026942B2 (en) * | 2015-12-29 | 2021-06-08 | Kyoto University | Agent for preventing and/or treating Alzheimer's disease |
| WO2017201364A1 (en) | 2016-05-20 | 2017-11-23 | Vanda Pharmaceuticals Inc. | Method for improving or enhancing cognition |
| IL263412B (en) | 2016-06-03 | 2022-08-01 | Univ Columbia | Treatment methods for Freder Willi syndrome |
| US10383870B2 (en) | 2016-06-10 | 2019-08-20 | Pharnext | Early treatment of CMT disease |
| JP2019524865A (ja) | 2016-08-31 | 2019-09-05 | ザ ジェネラル ホスピタル コーポレイション | 神経変性疾患と関連する神経炎症におけるマクロファージ/ミクログリア |
| KR101910153B1 (ko) * | 2016-11-04 | 2018-10-19 | 한국과학기술연구원 | 타우 관련 질환의 예방 또는 치료용 레보시멘단 화합물 |
| CN111050761A (zh) | 2017-04-24 | 2020-04-21 | 法奈克斯公司 | 阿尔茨海默病的基于idalopirdine的组合治疗 |
| WO2018213838A1 (en) * | 2017-05-19 | 2018-11-22 | Biscayne Neurotherapeutics, Inc. | Modified release pharmaceutical compositions of huperzine and methods of using the same |
| JP7202376B2 (ja) | 2017-07-20 | 2023-01-11 | エーゼットセラピーズ, インコーポレイテッド | クロモリンナトリウムおよびイブプロフェンの粉末製剤 |
| US9962384B1 (en) * | 2017-09-07 | 2018-05-08 | Korea Institute Of Science And Technology | Levosimendan compound for preventing or treating tau-related diseases |
| US11439608B2 (en) | 2017-09-25 | 2022-09-13 | Qun Lu | Roles of modulators of intersectin-CDC42 signaling in Alzheimer's disease |
| JP7037814B2 (ja) * | 2018-03-28 | 2022-03-17 | 国立大学法人山口大学 | ホルボールエステルを有効成分とする軸索の伸展剤 |
| JP2019182845A (ja) * | 2018-04-02 | 2019-10-24 | 学校法人藤田学園 | キヌレニンアミノトランスフェラーゼ2(kat2)阻害剤 |
| KR102317080B1 (ko) * | 2018-05-09 | 2021-10-25 | 한국과학기술연구원 | 타우 관련 질환의 예방 또는 치료용 조성물 |
| WO2019216589A1 (ko) * | 2018-05-09 | 2019-11-14 | 한국과학기술연구원 | 타우 관련 질환의 예방 또는 치료용 조성물 |
| AU2019299347A1 (en) | 2018-07-02 | 2021-01-21 | Aztherapies, Inc. | Powdered formulations of cromolyn sodium and alpha-lactose |
| CN108707204B (zh) * | 2018-07-04 | 2020-09-15 | 吉林大学 | 过氧化氢响应型靶向载药纳米材料及制备方法 |
| MX2021004586A (es) | 2018-11-19 | 2021-06-15 | Supernus Pharmaceuticals Inc | Uso de dosis mas altas de formulaciones de huperzina de liberacion modificada. |
| MX2021006869A (es) | 2018-12-10 | 2021-07-02 | Massachusetts Gen Hospital | Esteres de cromolin y usos de los mismos. |
| WO2021011421A1 (en) * | 2019-07-12 | 2021-01-21 | University Of South Florida | Compositions and methods for treating alzheimers disease |
| WO2021037027A1 (en) * | 2019-08-29 | 2021-03-04 | The Hong Kong University Of Science And Technology | Genetic variants for diagnosis of alzheimer's disease |
| ES2821599A1 (es) * | 2019-10-24 | 2021-04-26 | Univ Del Pais Vasco / Euskal Herriko Unibertsitatea | Compuestos y metodos para el tratamiento de la enfermedad de alzheimer |
| CN110656170A (zh) * | 2019-11-08 | 2020-01-07 | 新乡医学院 | 一种用于阿尔茨海默病诊断的试剂、诊断产品、治疗组合物,候选药物筛选方法及应用 |
| WO2021142312A1 (en) * | 2020-01-10 | 2021-07-15 | The Board Of Regents Of The University Of Texas | Methods and compositions related to heparinoids |
| WO2021207060A1 (en) | 2020-04-06 | 2021-10-14 | The General Hospital Corporation | Methods of treatment of coronavirus-induced inflammation conditions |
| US20230278965A1 (en) * | 2020-06-19 | 2023-09-07 | Korea Institute Of Science And Technology | Novel carbonohydrazonoyl dicyanide compounds comprising 2 or more aryl or heteroaryl connected via linker and use thereof |
| US12590066B2 (en) | 2020-06-19 | 2026-03-31 | Korea Institute Of Science And Technology | Fused-heterocyclyl-carbonohydrazonoyl dicyanide compounds and use thereof |
| US20230293726A1 (en) * | 2020-08-11 | 2023-09-21 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Method for the treatment of wwox associated diseases |
| CN111983241B (zh) * | 2020-09-04 | 2022-05-20 | 四川大学华西医院 | 诊断脑卒中认知障碍的蛋白标志物以及模型的训练方法 |
| JP7578275B2 (ja) | 2020-12-14 | 2024-11-06 | 日本メナード化粧品株式会社 | Bmpr2産生促進剤 |
| WO2022146914A1 (en) | 2020-12-28 | 2022-07-07 | The General Hospital Corporation | Cromolyn derivatives and uses thereof |
| CN114832116B (zh) * | 2022-05-19 | 2024-01-26 | 沈阳药科大学 | 基于小胶质细胞表型调节和脑内铁清除的ros响应型纳米载体及其制备方法与应用 |
| CN115354048B (zh) * | 2022-06-08 | 2024-02-02 | 暨南大学 | 阿尔茨海默病动物模型的构建方法、用途和Aβ42重组表达载体 |
| CN115851946B (zh) * | 2022-11-24 | 2023-09-01 | 中山大学孙逸仙纪念医院 | CaMK I作为治疗靶点在制备胰腺癌治疗药物中的应用 |
| CN117568466B (zh) * | 2023-11-20 | 2024-07-09 | 南方医科大学南方医院 | 动脉重塑标志物及其应用 |
| CN121513038A (zh) * | 2026-01-16 | 2026-02-13 | 湖北工业大学 | 包含舒洛地特和头孢曲松的组合物在制备治疗细菌性脑膜炎的药物中的应用 |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0572560B1 (en) * | 1991-02-22 | 2003-01-29 | SHAPIRO, Howard, K. | Use of pharmaceutical compounds in the treatment of symptoms of disorders related to neurological diseases and etiologically related symptomology |
| EP1013275A3 (en) * | 1991-11-26 | 2001-01-10 | Sepracor, Inc. | Method and compositions for treating hypertension, angina and other disorders using optically pure (-) amlodipine |
| US5721263A (en) * | 1993-06-07 | 1998-02-24 | Takeda Chemical Industries, Ltd. | Pharmaceutical composition for angiotensin II-mediated diseases |
| US6573251B2 (en) * | 1994-03-30 | 2003-06-03 | Denis Barritault | Drug and pharmaceutical composition for the treatment of lesions of the nervous system and fractions enriched in heparan sulfate |
| JPH101439A (ja) * | 1996-06-11 | 1998-01-06 | Mochida Pharmaceut Co Ltd | 神経変性疾患治療剤 |
| US6391922B1 (en) | 1998-01-13 | 2002-05-21 | Synchroneuron, Llc | Treatment of posttraumatic stress disorder, obsessive-compulsive disorder and related neuropsychiatric disorders |
| AU3173301A (en) * | 2000-02-11 | 2001-08-20 | European Molecular Biology Laboratory | Methods and compositions for treatment of alzheimer's disease by enhancing plasmin or plasmin-like activity |
| US20030149010A1 (en) * | 2001-07-19 | 2003-08-07 | Keller Bradley T. | Combination of an aldosterone receptor antagonist and an HMG CoA reductase inhibitor |
| US20040067918A1 (en) * | 2002-03-18 | 2004-04-08 | Keller Bradley T. | Combination of an aldosterone receptor antagonist and nicotinic acid or a nicotinic acid derivative |
| US20040102525A1 (en) * | 2002-05-22 | 2004-05-27 | Kozachuk Walter E. | Compositions and methods of treating neurological disease and providing neuroprotection |
| EP1380290A1 (en) * | 2002-07-09 | 2004-01-14 | Universitair Medisch Centrum Utrecht | Cross-beta structure pathway and its therapeutic relevance |
| ES2584606T3 (es) * | 2002-10-10 | 2016-09-28 | Ono Pharmaceutical Co., Ltd. | Microesferas que comprenden ONO-1301 |
| TWI323660B (en) * | 2003-02-25 | 2010-04-21 | Otsuka Pharma Co Ltd | Pten inhibitor or maxi-k channels opener |
| FI20040674A0 (fi) * | 2004-05-12 | 2004-05-12 | Orion Corp | Menetelmä tromboembolisten sairauksien estoon |
| US20080025962A1 (en) * | 2004-11-30 | 2008-01-31 | Angesmg,Inc. | Remedy for Alzheimer's Disease |
| US20080188510A1 (en) * | 2005-05-23 | 2008-08-07 | Eisai R & D Management Co., Ltd. | Novel methods using zonisamide |
| CN101309682A (zh) * | 2005-10-21 | 2008-11-19 | 脑细胞股份有限公司 | 通过pde抑制调节神经发生 |
| AU2006308889A1 (en) * | 2005-10-31 | 2007-05-10 | Braincells, Inc. | GABA receptor mediated modulation of neurogenesis |
| JP2009514851A (ja) * | 2005-11-08 | 2009-04-09 | ランバクシー ラボラトリーズ リミテッド | (3r,5r)−7−[2−(4−フルオロフェニル)−5−イソプロピル−3−フェニル−4−[(4−ヒドロキシメチルフェニルアミノ)カルボニル]−ピロール−1−イル]−3,5−ジヒドロキシ−ヘプタン酸ヘミカルシウム塩の製法 |
| JP2009532663A (ja) * | 2006-02-16 | 2009-09-10 | レリアント ファーマスーティカルズ インコーポレイテッド | ω−3脂肪酸を用いた、痴呆、脳血管性痴呆、パーキンソン痴呆、アルツハイマー病及び/又は軽度認知障害の治療により利益を得ると考えられる患者を同定し、治療する方法 |
| WO2007112288A2 (en) * | 2006-03-23 | 2007-10-04 | Mount Sinai School Of Medicine | Cardiovascular composition and use the same for the treatment of alzheimers disease |
| WO2007139178A1 (ja) * | 2006-05-31 | 2007-12-06 | Dnavec Corporation | アルツハイマー病治療薬 |
| KR20090026275A (ko) * | 2006-07-06 | 2009-03-12 | 로스캄프 리서치 엘엘씨 | 베타-아밀로이드 생성을 억제하기 위한 화합물 및 그의 조합물, 및 그의 사용 방법 |
| AR062057A1 (es) * | 2006-07-17 | 2008-10-15 | Boehringer Ingelheim Int | Uso de los inhibidores directos de la trombina |
| KR101441725B1 (ko) * | 2007-05-22 | 2014-09-18 | 오츠카 세이야쿠 가부시키가이샤 | 알츠하이머병 치료를 위한 카르보스티릴 유도체 및 도네페질을 포함하는 치료제 |
| JP5736098B2 (ja) * | 2007-08-21 | 2015-06-17 | アッヴィ・インコーポレイテッド | 中枢神経系障害を治療するための医薬組成物 |
| US20100137442A2 (en) | 2008-02-01 | 2010-06-03 | Xenoport, Inc. | Sustained Release Particulate Oral Dosage Forms of (R)-Baclofen and Methods of Treatment |
| DK2282779T3 (da) * | 2008-04-29 | 2013-05-27 | Pharnext | Nye terapeutiske fremgangsmåder til behandling af alzheimer sygdom og beslægtede lidelser gennem en modulation af cellestress-respons |
| WO2009133128A1 (en) * | 2008-04-29 | 2009-11-05 | Pharnext | Combination compositions for treating alzheimer disease and related disorders with zonisamide and acamprosate |
| PL2282778T3 (pl) * | 2008-04-29 | 2017-08-31 | Pharnext | Nowe podejścia terapeutyczne do leczenia choroby alzheimera i powiązanych zaburzeń poprzez modulację angiogenezy |
| EP2377865B1 (en) * | 2008-11-27 | 2014-12-17 | National University Corporation Kagawa University | Cyclobutyl purine derivative, angiogenesis promoting agent, lumenization promoting agent, neurocyte growth promoting agent, and drug |
| EP2322163A1 (en) | 2009-11-03 | 2011-05-18 | Pharnext | New therapeutics approaches for treating alzheimer disease |
| KR101937782B1 (ko) | 2011-03-01 | 2019-01-11 | 파넥스트 | 바클로펜 및 아캄프로세이트에 기초한 신경 장애의 치료 |
-
2009
- 2009-11-03 EP EP09306048A patent/EP2322163A1/en not_active Withdrawn
-
2010
- 2010-10-29 EP EP10773302.4A patent/EP2496226B1/en active Active
- 2010-10-29 CA CA3065614A patent/CA3065614A1/en not_active Abandoned
- 2010-10-29 CA CA2779070A patent/CA2779070C/en active Active
- 2010-10-29 EP EP20206600.7A patent/EP3797767A1/en not_active Withdrawn
- 2010-10-29 WO PCT/EP2010/066510 patent/WO2011054759A1/en not_active Ceased
- 2010-10-29 MX MX2012005131A patent/MX2012005131A/es active IP Right Grant
- 2010-10-29 CN CN2010800604548A patent/CN102834095A/zh active Pending
- 2010-10-29 BR BR112012010470A patent/BR112012010470A2/pt not_active Application Discontinuation
- 2010-10-29 EP EP19216647.8A patent/EP3650021A1/en not_active Withdrawn
- 2010-10-29 CN CN201710347219.2A patent/CN107261144A/zh active Pending
- 2010-10-29 AR ARP100104015A patent/AR078846A1/es unknown
- 2010-10-29 EA EA201200686A patent/EA024465B1/ru not_active IP Right Cessation
- 2010-10-29 JP JP2012537355A patent/JP5977172B2/ja not_active Expired - Fee Related
- 2010-10-29 ES ES10773302T patent/ES2800311T3/es active Active
-
2012
- 2012-04-25 IL IL219412A patent/IL219412A/en active IP Right Grant
- 2012-05-02 US US13/462,034 patent/US8809302B2/en not_active Expired - Fee Related
-
2015
- 2015-04-27 JP JP2015090340A patent/JP6062995B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CN102834095A (zh) | 2012-12-19 |
| EP2496226B1 (en) | 2020-03-04 |
| JP5977172B2 (ja) | 2016-08-24 |
| US8809302B2 (en) | 2014-08-19 |
| EP3650021A1 (en) | 2020-05-13 |
| IL219412A0 (en) | 2012-06-28 |
| CA3065614A1 (en) | 2011-05-12 |
| US20120270836A1 (en) | 2012-10-25 |
| CN107261144A (zh) | 2017-10-20 |
| CA2779070A1 (en) | 2011-05-12 |
| JP2013510114A (ja) | 2013-03-21 |
| EA201200686A1 (ru) | 2013-05-30 |
| MX2012005131A (es) | 2012-05-29 |
| AR078846A1 (es) | 2011-12-07 |
| EP3797767A1 (en) | 2021-03-31 |
| EP2496226A1 (en) | 2012-09-12 |
| EP2322163A1 (en) | 2011-05-18 |
| JP6062995B2 (ja) | 2017-01-18 |
| CA2779070C (en) | 2020-02-25 |
| JP2015157841A (ja) | 2015-09-03 |
| WO2011054759A1 (en) | 2011-05-12 |
| BR112012010470A2 (pt) | 2016-03-08 |
| EA024465B1 (ru) | 2016-09-30 |
| IL219412A (en) | 2017-05-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2800311T3 (es) | Nuevos enfoques terapéuticos para tratar la enfermedad de Alzheimer | |
| US10874644B2 (en) | Therapeutic approaches for treating Alzheimer disease and related disorders through a modulation of synapse function | |
| US8552041B2 (en) | Therapeutic approaches for treating alzheimer disease and related disorders through a modulation of cell stress response | |
| US9387206B2 (en) | Therapeutic approaches for treating Alzheimer's disease | |
| CN102065898A (zh) | 通过调节血管发生治疗阿茨海默病和相关病症的新治疗手段 | |
| HK1172566A (en) | New therapeutic approaches for treating alzheimer disease | |
| HK1172566B (en) | New therapeutic approaches for treating alzheimer disease |