ES2800433T3 - Derivado de la triazolopirazinona útil como inhibidor de la pde1 humana - Google Patents
Derivado de la triazolopirazinona útil como inhibidor de la pde1 humana Download PDFInfo
- Publication number
- ES2800433T3 ES2800433T3 ES17761381T ES17761381T ES2800433T3 ES 2800433 T3 ES2800433 T3 ES 2800433T3 ES 17761381 T ES17761381 T ES 17761381T ES 17761381 T ES17761381 T ES 17761381T ES 2800433 T3 ES2800433 T3 ES 2800433T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- human
- butyl
- scheme
- refers
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003112 inhibitor Substances 0.000 title description 3
- 101100243084 Dictyostelium discoideum pdsA gene Proteins 0.000 title 1
- 101100243086 Schizosaccharomyces pombe (strain 972 / ATCC 24843) cgs2 gene Proteins 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 67
- 150000003839 salts Chemical class 0.000 claims abstract description 13
- 238000000034 method Methods 0.000 claims description 27
- 238000002360 preparation method Methods 0.000 claims description 13
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 10
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 208000020832 chronic kidney disease Diseases 0.000 claims description 7
- 239000003085 diluting agent Substances 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 5
- 230000008569 process Effects 0.000 claims description 5
- 238000002560 therapeutic procedure Methods 0.000 claims description 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 40
- 239000000047 product Substances 0.000 description 32
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 30
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 29
- 239000007787 solid Substances 0.000 description 27
- 239000000203 mixture Substances 0.000 description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 21
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- 238000000746 purification Methods 0.000 description 19
- 238000001914 filtration Methods 0.000 description 17
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 16
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- 239000010410 layer Substances 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- 239000003960 organic solvent Substances 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- NERXDJUMUSZWOD-UHFFFAOYSA-N 1-butyl-3-hydrazinylpyrido[2,3-b]pyrazin-2-one Chemical compound C(CCC)N1C2=C(N=C(C1=O)NN)N=CC=C2 NERXDJUMUSZWOD-UHFFFAOYSA-N 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- 101001117044 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Proteins 0.000 description 8
- 101001117099 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Proteins 0.000 description 8
- 101001117094 Homo sapiens Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1C Proteins 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 239000000758 substrate Substances 0.000 description 8
- LIVGUKDGBMXQAH-UHFFFAOYSA-N 1-butyl-4H-pyrido[2,3-b]pyrazine-2,3-dione Chemical compound C(CCC)N1C2=C(NC(C1=O)=O)N=CC=C2 LIVGUKDGBMXQAH-UHFFFAOYSA-N 0.000 description 7
- 102100024318 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1B Human genes 0.000 description 7
- 102100024317 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1C Human genes 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 101001117259 Homo sapiens High affinity cGMP-specific 3',5'-cyclic phosphodiesterase 9A Proteins 0.000 description 7
- 101001072037 Homo sapiens cAMP and cAMP-inhibited cGMP 3',5'-cyclic phosphodiesterase 10A Proteins 0.000 description 7
- 101001098858 Homo sapiens cGMP-dependent 3',5'-cyclic phosphodiesterase Proteins 0.000 description 7
- 101000988412 Homo sapiens cGMP-specific 3',5'-cyclic phosphodiesterase Proteins 0.000 description 7
- 239000007832 Na2SO4 Substances 0.000 description 7
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 229910052938 sodium sulfate Inorganic materials 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 101001072029 Homo sapiens Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11A Proteins 0.000 description 6
- 101001117261 Homo sapiens High affinity cAMP-specific and IBMX-insensitive 3',5'-cyclic phosphodiesterase 8A Proteins 0.000 description 6
- 101000609947 Homo sapiens Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit alpha Proteins 0.000 description 6
- 101000988419 Homo sapiens cAMP-specific 3',5'-cyclic phosphodiesterase 4D Proteins 0.000 description 6
- 101001117266 Homo sapiens cAMP-specific 3',5'-cyclic phosphodiesterase 7B Proteins 0.000 description 6
- 101001098818 Homo sapiens cGMP-inhibited 3',5'-cyclic phosphodiesterase A Proteins 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- -1 PDE6AB Proteins 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 5
- TULFFCFYCRQCDI-UHFFFAOYSA-N 3-n-butylpyridine-2,3-diamine Chemical compound CCCCNC1=CC=CN=C1N TULFFCFYCRQCDI-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 102100024232 cAMP-specific 3',5'-cyclic phosphodiesterase 7B Human genes 0.000 description 5
- 102100029175 cGMP-specific 3',5'-cyclic phosphodiesterase Human genes 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 102000055551 human PDE1A Human genes 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- ADFLKSRNLCTWCK-UHFFFAOYSA-N n-butyl-2-nitropyridin-3-amine Chemical compound CCCCNC1=CC=CN=C1[N+]([O-])=O ADFLKSRNLCTWCK-UHFFFAOYSA-N 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 4
- IJVFHCSUEBAAOZ-UHFFFAOYSA-N 3-fluoro-2-nitropyridine Chemical compound [O-][N+](=O)C1=NC=CC=C1F IJVFHCSUEBAAOZ-UHFFFAOYSA-N 0.000 description 4
- ITGKRQZBQHPNEX-UHFFFAOYSA-N C(CCC)N1C2=C(N=C(C1=O)NN=CC1(CC1)C)N=CC=C2 Chemical compound C(CCC)N1C2=C(N=C(C1=O)NN=CC1(CC1)C)N=CC=C2 ITGKRQZBQHPNEX-UHFFFAOYSA-N 0.000 description 4
- 101001117089 Drosophila melanogaster Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1 Proteins 0.000 description 4
- 102100024228 High affinity cAMP-specific and IBMX-insensitive 3',5'-cyclic phosphodiesterase 8A Human genes 0.000 description 4
- 102100024227 High affinity cGMP-specific 3',5'-cyclic phosphodiesterase 9A Human genes 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- 206010020772 Hypertension Diseases 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 102100036377 cAMP and cAMP-inhibited cGMP 3',5'-cyclic phosphodiesterase 10A Human genes 0.000 description 4
- 102100029170 cAMP-specific 3',5'-cyclic phosphodiesterase 4D Human genes 0.000 description 4
- 102100038953 cGMP-dependent 3',5'-cyclic phosphodiesterase Human genes 0.000 description 4
- 102100037093 cGMP-inhibited 3',5'-cyclic phosphodiesterase A Human genes 0.000 description 4
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 4
- 238000001952 enzyme assay Methods 0.000 description 4
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- UVPXLSNLTHNWES-UHFFFAOYSA-N 1-butyl-3-chloropyrido[2,3-b]pyrazin-2-one Chemical compound C(CCC)N1C2=C(N=C(C1=O)Cl)N=CC=C2 UVPXLSNLTHNWES-UHFFFAOYSA-N 0.000 description 3
- DOJZSEYEQKWUSI-UHFFFAOYSA-N 1-methylcyclopropane-1-carbaldehyde Chemical compound O=CC1(C)CC1 DOJZSEYEQKWUSI-UHFFFAOYSA-N 0.000 description 3
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 3
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 229920000936 Agarose Polymers 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- 102100024316 Calcium/calmodulin-dependent 3',5'-cyclic nucleotide phosphodiesterase 1A Human genes 0.000 description 3
- 102100036367 Dual 3',5'-cyclic-AMP and -GMP phosphodiesterase 11A Human genes 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- 239000005909 Kieselgur Substances 0.000 description 3
- 102100039177 Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit alpha Human genes 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 102000054918 human PDE10A Human genes 0.000 description 3
- 102000057579 human PDE11A Human genes 0.000 description 3
- 102000055063 human PDE2A Human genes 0.000 description 3
- 102000048009 human PDE6A Human genes 0.000 description 3
- 102000053871 human PDE9A Human genes 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 238000002821 scintillation proximity assay Methods 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 102000000584 Calmodulin Human genes 0.000 description 2
- 108010041952 Calmodulin Proteins 0.000 description 2
- 101000609949 Homo sapiens Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- RLUOAGRKZGINOM-UHFFFAOYSA-N N'-(1-butyl-2-oxopyrido[2,3-b]pyrazin-3-yl)-1-methylcyclopropane-1-carbohydrazide Chemical compound C(CCC)N1C2=C(N=C(C1=O)NNC(=O)C1(CC1)C)N=CC=C2 RLUOAGRKZGINOM-UHFFFAOYSA-N 0.000 description 2
- 229940124639 Selective inhibitor Drugs 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 230000003276 anti-hypertensive effect Effects 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 102000055047 human PDE3A Human genes 0.000 description 2
- 102000048135 human PDE4D Human genes 0.000 description 2
- 102000048107 human PDE5A Human genes 0.000 description 2
- 102000049886 human PDE8A Human genes 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000003495 polar organic solvent Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000012536 storage buffer Substances 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 238000001665 trituration Methods 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- DIZKLZKLNKQFGB-UHFFFAOYSA-N 1-methylcyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(C)CC1 DIZKLZKLNKQFGB-UHFFFAOYSA-N 0.000 description 1
- HUTNOYOBQPAKIA-UHFFFAOYSA-N 1h-pyrazin-2-one Chemical compound OC1=CN=CC=N1 HUTNOYOBQPAKIA-UHFFFAOYSA-N 0.000 description 1
- DCBOUXKSNFDXER-UHFFFAOYSA-N 1h-pyrido[2,3-b]pyrazin-2-one Chemical compound C1=CC=C2NC(=O)C=NC2=N1 DCBOUXKSNFDXER-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- 102000001707 3',5'-Cyclic-AMP Phosphodiesterases Human genes 0.000 description 1
- 108010054479 3',5'-Cyclic-AMP Phosphodiesterases Proteins 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- NMXJQUZKQRADRK-UHFFFAOYSA-N 8-butyl-3-(1-methylcyclopropyl)-2,4,5,8,13-pentazatricyclo[7.4.0.02,6]trideca-1(9),3,5,10,12-pentaen-7-one Chemical compound C(CCC)N1C(C=2N(C3=C1C=CC=N3)C(=NN=2)C1(CC1)C)=O NMXJQUZKQRADRK-UHFFFAOYSA-N 0.000 description 1
- JKSCIPFPPZANSY-XADNCYFJSA-N CCCCN(/C=C/C=C\N)C(C(CN(C=C)C(C1(C)CC1)=N)=N)=O Chemical compound CCCCN(/C=C/C=C\N)C(C(CN(C=C)C(C1(C)CC1)=N)=N)=O JKSCIPFPPZANSY-XADNCYFJSA-N 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 101100351286 Dictyostelium discoideum pdeE gene Proteins 0.000 description 1
- 101100407341 Drosophila melanogaster Pde9 gene Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-O Htris Chemical compound OCC([NH3+])(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-O 0.000 description 1
- 206010023421 Kidney fibrosis Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 229940121836 Phosphodiesterase 1 inhibitor Drugs 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102100039174 Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta Human genes 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940124301 concurrent medication Drugs 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 206010013990 dysuria Diseases 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 102000055462 human PDE1B Human genes 0.000 description 1
- 102000054507 human PDE6B Human genes 0.000 description 1
- 102000050549 human PDE7B Human genes 0.000 description 1
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- VNXBKJFUJUWOCW-UHFFFAOYSA-N methylcyclopropane Chemical compound CC1CC1 VNXBKJFUJUWOCW-UHFFFAOYSA-N 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 238000007339 nucleophilic aromatic substitution reaction Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 101150037969 pde-6 gene Proteins 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- 230000037081 physical activity Effects 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 125000001567 quinoxalinyl group Chemical class N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 230000008327 renal blood flow Effects 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- JABYJIQOLGWMQW-UHFFFAOYSA-N undec-4-ene Chemical compound CCCCCCC=CCCC JABYJIQOLGWMQW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Urology & Nephrology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
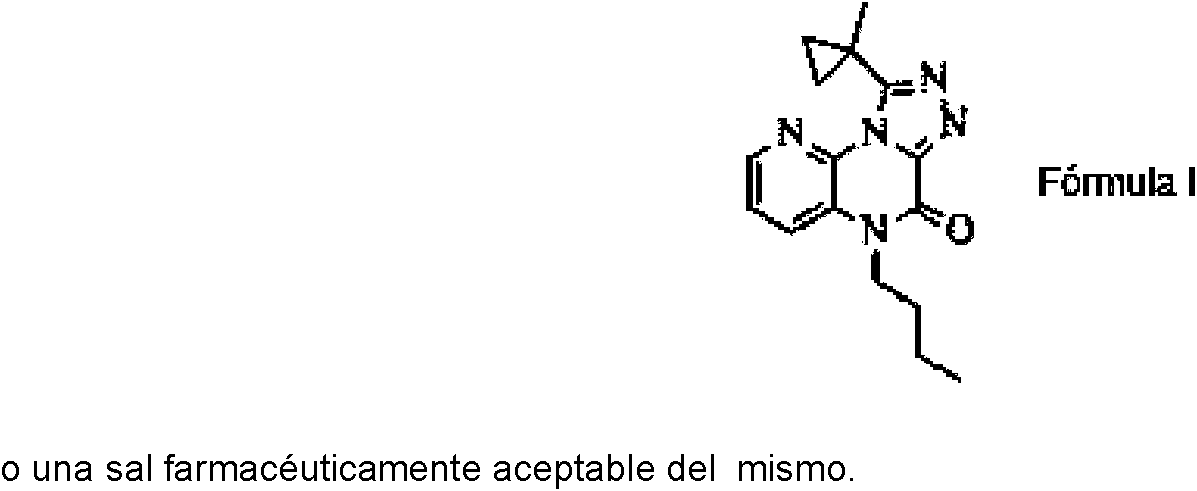
Un compuesto de fórmula: **(Ver fórmula)** o una sal farmacéutica aceptable del mismo.
Description
DESCRIPCIÓN
Derivado de la triazolopirazinona útil como inhibidor de la pdel humana
La presente invención se refiere a un determinado inhibidor de la PDE1 en el ser humano, a las composiciones farmacéuticas que componen el compuesto, a los procedimientos de utilización del compuesto para tratar los trastornos fisiológicos y a los intermediarios y procesos útiles para la síntesis del compuesto.
Las fosfodiesterasas (PDEs) son enzimas que regulan los niveles celulares de cAMP y cGMP controlando la velocidad de hidrolización de estos nucleótidos cíclicos. La PDE1, una PDE dependiente del calcio y la calmodulina, es una de las 11 familias de PDE conocidas. La PDE1 se expresa en muchos tejidos, incluidos el cerebro, el corazón, el pulmón, el riñón y el músculo liso. Además, la PDE1 está compuesta por una familia de tres isoformas conocidas, PDE1A, PDE1B y PDE1C.
Los pacientes que sufren de diabetes a menudo desarrollan una forma de enfermedad renal crónica conocida como enfermedad renal diabética (o nefropatía diabética). Se ha estimado que la enfermedad renal diabética puede afectar hasta el 40 por ciento de los pacientes diabéticos. Las opciones de tratamiento para la enfermedad renal diabética son limitadas e incluyen el uso de medicamentos que reducen la presión arterial, el control de los niveles de glucosa en la sangre, la dieta y el peso, y la realización de actividad física regular. Por lo tanto, es necesario contar con opciones de tratamiento adicionales para los pacientes que sufren de enfermedad renal crónica, en particular la enfermedad renal diabética.
La patente de los Estados Unidos No.8,299,080 revela ciertos derivados de la quinoxalina que tienen una actividad inhibidora de la PDE9, útil para el tratamiento de diversos trastornos, como la disuria y la hipertensión. Además, la Patente Europea No. 0 040 401 revela ciertos sustituidos triazoloquinoxalin-4-onas que poseen actividad antihipertensiva.
La presente invención proporciona un cierto compuesto nuevo que es un inhibidor de la PDE1 humana. Además, la presente invención proporciona un cierto compuesto novedoso que es un inhibidor selectivo de la PDE1A, PDE1B y PDE1C humanas en relación con otras PDE humanas, como la PDE2A, PDE3A, PDE4D, PDE5A, PDE6AB, PDE7B, PDE8A. PDE9A, PDE10A y PDR11A. Además, la presente invención proporciona un cierto compuesto novedoso que puede tener efectos antihipertensivos y también puede mejorar el flujo sanguíneo renal. Además, el compuesto de la presente invención puede reducir la fibrosis renal.
Por consiguiente, la presente invención proporciona un compuesto de Fórmula I:
La presente invención también proporciona un procedimiento para tratar la enfermedad renal crónica en un paciente, que consiste en administrar a un paciente que necesita dicho tratamiento una cantidad efectiva de un compuesto de la Fórmula I.
La presente invención también proporciona un procedimiento para tratar la enfermedad renal diabética en un paciente, que consiste en administrar a un paciente que necesita dicho tratamiento una cantidad efectiva de un compuesto de la Fórmula 1.
La presente invención también proporciona un procedimiento de tratamiento de la hipertensión en un paciente que consiste en administrar a un paciente que necesita dicho tratamiento una cantidad efectiva de un compuesto de la Fórmula 1.
Además, la invención proporciona un compuesto de la Fórmula I para su uso en la terapia. La invención también proporciona un compuesto de la Fórmula I para su uso en el tratamiento de la enfermedad renal crónica. Además, la invención proporciona un compuesto de la Fórmula I para su uso en el tratamiento de la enfermedad renal diabética. Además, la invención proporciona un compuesto de Fórmula I para su uso en el tratamiento de la hipertensión. Además, la invención proporciona el uso de un compuesto de Fórmula I para la fabricación de un medicamento para el tratamiento de la enfermedad renal crónica. Además, la invención proporciona el uso de un compuesto de Fórmula I para la fabricación de un medicamento para el tratamiento de la enfermedad renal diabética. La invención además
proporciona el uso de un compuesto de Fórmula I para la fabricación de un medicamento para el tratamiento de la hipertensión.
La invención proporciona además una composición farmacéutica, que comprende un compuesto de la Fórmula 1 con uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables. La invención además proporciona un proceso para preparar una composición farmacéutica, que comprende la mezcla de un compuesto de Fórmula 1 con uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables. La presente invención también abarca nuevos intermediarios y procesos para la síntesis del compuesto de la Fórmula I.
Tal como se utilizan en el presente documento, los términos "trato", "tratamiento" o "tratar" incluyen prohibir, restringir, ralentizar, detener o invertir la progresión o la gravedad de un síntoma o trastorno existente.
Tal como se usa en el presente documento, el término "paciente" se refiere a un mamífero, como un ratón, un conejillo de indias, una rata, un perro o un humano. Se entiende que el paciente preferido es un humano.
Tal como se utiliza en el presente documento, el término "cantidad efectiva" se refiere a la cantidad o dosis del compuesto de la invención, o de una sal farmacéuticamente aceptable del mismo que, tras la administración de una dosis única o múltiple al paciente, proporciona el efecto deseado en el paciente bajo diagnóstico o tratamiento.
Una cantidad efectiva puede ser fácilmente determinada por un experto en la materia utilizando técnicas conocidas y observando los resultados obtenidos en circunstancias análogas. Para determinar la cantidad efectiva para un paciente, un experto en la materia considera una serie de factores, incluyendo, pero no limitándose a: el tamaño, edad y salud general del paciente; la enfermedad o trastorno específico involucrado; el grado de implicación o la severidad de la enfermedad o trastorno; la respuesta del paciente individual; el compuesto particular administrado; el modo de administración; las características de biodisponibilidad del preparado administrado; el régimen de dosis seleccionado; el uso de medicación concomitante; y otras circunstancias relevantes.
El compuesto de la Fórmula I es generalmente efectivo en un amplio rango de dosis. Por ejemplo, las dosis diarias normalmente están dentro del rango de aproximadamente 0,01 a aproximadamente 20 mg/kg de peso corporal. En algunos casos, los niveles de dosificación por debajo del límite inferior de la gama mencionada pueden ser más que adecuados, mientras que en otros casos pueden emplearse dosis aún mayores con efectos secundarios aceptables y, por lo tanto, la gama de dosificación mencionada no tiene por objeto limitar en modo alguno el ámbito de la invención.
Los compuestos de la invención se formulan preferentemente como composiciones farmacéuticas administradas por cualquier vía que haga que el compuesto sea biodisponible, incluidas las vías oral y parenteral. Preferentemente, tales composiciones son para administración oral. Tales composiciones farmacéuticas y procesos para prepararlas son bien conocidos en la técnica. (Véase, por ejemplo, Remington: La ciencia y la práctica de la farmacia. L.V. Allen, Editor, 22a Edición. Pharmaceutical Press, 2012).
Una sal farmacéuticamente aceptable del compuesto de la invención puede formarse, por ejemplo, mediante la reacción de una base libre apropiada del compuesto de la invención y un ácido apropiado farmacéuticamente aceptable en un disolvente adecuado en condiciones estándar bien conocidas en la técnica. La formación de tales sales es bien conocida y apreciada en la técnica. Véanse, por ejemplo, Gould, P.L., "Selección de sales para fármacos de base", International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin, R.J., y otros, "Selección de sales y procedimientos de optimización de entidades novedosas farmaquímicas", Organic Process Research and Development, 4: 427-435 (2000); y Berge, S.M., y otros, "Sales farmacéuticas", Journal of Pharmaceutical Sciences, 66: 1-19, (1977 ).
Ciertas abreviaturas se definen de la siguiente manera: "ACN" se refiere al acetonitrilo; "AcOH" se refiere al ácido acético glacial; "DBU" se refiere al 1,8-diazabiciclo[5.4].0]undec-7-eno; "DCM" se refiere al diclorometano o al cloruro de metileno; "DIPEA" se refiere a la N,N-diisopropilletina; "DMF" se refiere a la N,N-dimetilformamida; "DMSO" se refiere al dimetilsulfóxido; "EDCI" se refiere a la 1-etil-3-(3-dimetilaminopropil)carbodiimida; "ES/MS" se refiere a la Espectrometría de Masas por Electrospray; "EtOAc" se refiere al acetato de etilo; "Et2O" se refiere al éter dietílico; "EtOH" se refiere al etanol; "HMDS" se refiere al hezametildisilazano; "HOBT" se refiere al hidroxibenzotriazol: "hr" se refiere a la hora u horas; "IC50" se refiere a la concentración de un agente que produce el 50% de la máxima respuesta inhibitoria posible para ese agente; "pmol" se refiere a micromol o micromoles; "min" se refiere al minuto o minutos; "MeOH" se refiere al metanol o al alcohol metílico; "MTBE" se refiere al metil-terc-butil éter; "NiNTA" se refiere a la cromatografía con una fase estacionaria de agarosa funcionalizada con ácido nitrilotriacético como quelante; "POCh" se refiere al oxicloruro de fósforo; "RT" se refiere a la temperatura ambiente; "SNAr" se refiere a la sustitución aromática nucleófila; "TEA" se refiere a la trietilamina; "THF" se refiere al tetrahidrofurano; "Tris" se refiere al 2-Amino-2-hidroximetil-propano-1,3-diol; "U/ml" se refiere a las unidades por mililitro; "wt" se refiere al peso.
Los compuestos de la presente invención pueden prepararse mediante una variedad de procedimientos conocidos por un experto en la técnica, algunos de los cuales se ilustran en los esquemas, preparaciones y ejemplos que figuran a continuación. El experto en la técnica reconocerá que los pasos sintéticos específicos para cada una de las vías descritas pueden combinarse de diferentes maneras, o en conjunción con pasos de diferentes esquemas, para preparar los compuestos de la invención. Los productos de cada paso de los esquemas que figuran a continuación
pueden recuperarse mediante procedimientos convencionales bien conocidos en la técnica, entre ellos la extracción, la evaporación, la precipitación, la cromatografía, la filtración, la trituración y la cristalización. En los esquemas que figuran a continuación, todos los sustitutos, a menos que se indique otra cosa, son los definidos anteriormente. Los reactivos y los materiales de partida son fácilmente accesibles para un experto en la técnica. Sin limitar el ámbito de la invención, se proporcionan los siguientes esquemas, preparaciones y ejemplos para ilustrar mejor la invención:

El esquema 1 describe la síntesis del compuesto de la Fórmula I. En el esquema 1, el paso A. La reacción SNAr de la 3-fluoro-2-nitropiridina, lograda con varios nucleófilos, es muy apreciada en la técnica. Por ejemplo, aproximadamente 1 equivalente de 3-fluoro-2-nitropiridina se reacciona con aproximadamente 3 equivalentes de butano-1-amina en un disolvente polar adecuado como el EtOH. El producto puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la extracción. Por ejemplo, la mezcla de la reacción puede diluirse con agua y extraerse con un disolvente orgánico polar adecuado como el EtOAc. Los extractos orgánicos pueden combinarse, secarse sobre sulfato de sodio anhidro, filtrarse y concentrarse bajo presión reducida para proporcionar N-butil-2-nitro-piridina-3-amina, el producto del paso A, es de suficiente pureza para su uso en el siguiente paso sin purificación adicional.
La posterior reducción del grupo nitro es bien conocida en la técnica. En el Esquema 1, paso B, por ejemplo, aproximadamente 1 equivalente de N-butil-2-nitro-piridina-3-amina, el producto del paso A, puede ser hidrogenado en presencia de un catalizador de metal de transición apropiado, como el paladio sobre el carbono, en una variedad de solventes orgánicos, como el MeOH. El producto reducido puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la filtración y la evaporación. Por ejemplo, la mezcla de reacción cruda puede filtrarse a través de un lecho de tierra de diatomeas, y el filtrado puede concentrarse a presión reducida para obtener N3-butilpiridina-2,3-diamina, el producto del Esquema 1, paso B, es de suficiente pureza para su uso en el siguiente paso sin purificación adicional.
La ciclización al producto dieno del Esquema 1, paso C, puede lograrse en condiciones de acilación térmica con oxalato de dietilo en un disolvente orgánico apropiado como el EtOH. Por ejemplo, aproximadamente 1 equivalente de N3-butilpiridin-2,3-diamina puede tratarse con unos 5 equivalentes de oxalato de dietilo en un disolvente orgánico polar adecuado, como el EtOH, en un tubo sellado a unos 100 °C. El producto ciclado puede entonces aislarse utilizando técnicas bien conocidas en la técnica, como la precipitación y la filtración. Por ejemplo, la mezcla de la reacción puede enfriarse hasta unos -10 a 0 °C, y el precipitado subsiguiente puede recogerse mediante filtración y lavado con éter dietílico para obtener 1-butil-4H-pirido[2,3-b]pirazina-2,3-diona, el producto del Esquema 1, paso C, es de suficiente pureza para su uso en el siguiente paso sin purificación adicional.
La deshidratación de un carbonilo activado con un nucleófilo como la hidracina es muy apreciada en la técnica. Por ejemplo, aproximadamente 1 equivalente de 1-butil-4H-pirido[2,3-b]pirazin-2,3-diona, el producto del Esquema 1, paso C, puede tratarse con aproximadamente 5 equivalentes de hidracina monohidratada a unos 100 °C en un tubo
presurizado. El producto puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la precipitación y la filtración. Por ejemplo, la mezcla de reacción cruda puede enfriarse hasta unos 0 °C, y el precipitado resultante puede recogerse mediante filtración y lavado con éter dietílico para obtener 1 -butil-3-hidracino-pirido[2,3-b]pirazin-2-ona, el producto del Esquema 1, paso D, es de suficiente pureza para su uso en el siguiente paso sin necesidad de purificación adicional.
La subsiguiente alquilación del producto de la hidracina del paso D puede lograrse mediante diversas técnicas de aminación reductora bien conocidas en la técnica. Por ejemplo, aproximadamente 1 equivalente de 1-butil-3-hidracinopirido[2,3-b]pirazin-2-ona, el producto del Esquema 1, paso D, puede ser tratado con aproximadamente 2 equivalentes de un alquilo aldehído sustituido apropiadamente, como 1-metilciclopropanocarbaldehído (CAS # 4515-89-3, Enamine LLC, USA), en un solvente alcohólico apropiado como el MeOH que contiene una cantidad catalítica de un ácido apropiado, como el AcOH, a aproximadamente RT al reflujo. El producto puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la cristalización y la filtración. Por ejemplo, la mezcla de reacción cruda puede concentrarse a presión reducida, y el producto puede obtenerse por cristalización con un disolvente orgánico adecuado, como los hexanos, con filtración posterior para obtener 1-butil-3-[2-[(1-metilciclopropil)metileno]hidracino]pirido[2,3-b]pirazin-2-ona, el producto del Esquema 1, paso E.
La preparación del compuesto de la Fórmula I puede lograrse utilizando un ciclo oxidativo hipervalente mediado por yodo (R. Aggarwal & G. Sumran, Synthetic Communications, 36: 1873-1876, 2006 ) enla imina sustituida 1-butil-3-[2-[(1-metilciclopropil)metileno]hidracino]pirido[2,3-b]pirazin-2-ona en un disolvente orgánico adecuado, como el DCM, a temperaturas que oscilan entre 0 °C y TA. Por ejemplo, aproximadamente 1 equivalente de 1 -butil-3-[2-[(1 -metilciclopropil)metileno]hidracino]pirido[2,3-b]pirazin-2-ona, el producto del Esquema I, paso E, puede disolverse en DCM y tratarse con aproximadamente 2 equivalentes de diacetato de yodosobenceno (CAS # 3240-34-4) a temperaturas que oscilan entre 0 °C y TA. El producto puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la extracción y la cromatografía. Por ejemplo, la mezcla de reacción puede ser diluida con agua y extraída con DCM. Las capas pueden ser separadas y la capa orgánica es lavada secuencialmente con NaHCO3 acuoso saturado, secada sobre Na2SO4, filtrada y concentrada bajo presión reducida. El residuo resultante puede ser purificado por cromatografía sobre sílice, utilizando un gradiente de una mezcla de disolventes apropiada como el EtOAc y hexanos, para obtener el compuesto de la Fórmula I, producto del Esquema I, paso F.

El esquema 2 muestra una síntesis alternativa del compuesto de la Fórmula I. En el Esquema 2, paso A, 1-butil-4H-pirido[2,3-b]pirazin-2,3-diona, el producto del Esquema I, paso C, puede convertirse en el compuesto de cloro, como es bien conocido en la técnica, usando un agente de cloración adecuado, como POCh, SOCh, cloruro de oxalilo, o PCl5, en un solvente orgánico apropiado como DCM o ACN que contiene una cantidad catalítica de DMF a temperaturas que van desde TA hasta el reflujo. Por ejemplo, aproximadamente 1 equivalente de 1 -butil-4H-pirido[2,3-b]pirazin-2,3-diona, el producto del Esquema 1, paso C, puede disolverse en ACN que contiene DMF, y la mezcla de reacción resultante puede tratarse con aproximadamente 3 equivalentes de cloruro de tionilo y calentarse hasta el reflujo durante unas 3 hr. La mezcla de reacción se concentra a presión reducida después de enfriarse a temperatura ambiente para obtener el producto del Esquema 2, paso A, 1-butil-3-cloro-pirido[2,3-b]pirazin-2-ona, adecuado para su uso posterior sin adicional purificación.
En el esquema 2, paso B, el desplazamiento del cloruro puede lograrse tratando aproximadamente 1 equivalente de 1-butil-3-doro-pirido[2,3-b]pirazin-2-ona, el producto del esquema 2, paso A, con una solución de aproximadamente 4 equivalentes de hidracina acuosa monohidratada en un disolvente apto para polos como el THF en TA durante unas 8 a 24 horas. Por ejemplo, la mezcla de reacción puede diluirse con agua y filtrarse, y la torta de filtro puede lavarse con un disolvente orgánico adecuado de alto punto de ebullición como el tolueno o el MTBE, y el agua resultante puede eliminarse de la mezcla bifásica mediante una simple destilación azeotrópica en un evaporador rotatorio a presión reducida con un disolvente adecuado de alto punto de ebullición, como el 2-metil-tetrahidrofurano, para obtener 1-butil-3-hidrazino-pirido[2,3-b]pirazin-2-ona, producto del paso B del esquema 2.
La acilación del producto del paso B puede realizarse con un ácido carboxílico adecuado utilizando una variedad de técnicas de acoplamiento de amida bien conocidas en la técnica. Por ejemplo, aproximadamente yo equivalente de 1-butil-3-hidracino-pirido[2,3-b]pirazin-2-ona, el producto del esquema 2, paso B, puede ser acoplado con aproximadamente 1,5 equivalentes de 1-metilciclopropano de ácido carboxílico en un solvente orgánico adecuado, como THF, DMF, o DMSO, conteniendo aproximadamente 1,5 equivalentes de EDCI y 1,5 equivalentes de HOBT con la adición subsiguiente de aproximadamente 3-5 equivalentes de una base orgánica no nucleófila como DIPEA o TEA. El producto puede entonces ser aislado utilizando técnicas bien conocidas en la técnica, como la extracción. Por ejemplo, la mezcla de la reacción puede ser neutralizada con un ácido mineral adecuado como e1HCl acuoso diluido con agua, y lavada con un solvente orgánico adecuado como el DCM, EtOAc, MTBE o Et2O. Las capas pueden separarse y la capa acuosa resultante puede basificarse a pH ~ 7-8 con un sólido alcalino apropiado, como K2CO3, NaHCO3, o Na2SO3, con extracción posterior con un disolvente orgánico adecuado como DCM, EtOAc, o Et2O. Las capas orgánicas pueden lavarse secuencialmente con agua, NaCl acuoso saturado, secarse sobre Na2SO4, filtrarse, y el filtrado concentrarse bajo presión reducida para dar N'-(1-butil-2-oxo-pirido[2,3-b]pirazin-3-yl)-1 -metilciclopropanecarbohidracida, el producto del Esquema 2, paso C.
En el esquema 2, paso D, el compuesto de la Fórmula 1 puede lograrse mediante ciclización en condiciones térmicas o de microondas bien conocidas en la técnica. Por ejemplo, puede calentarse aproximadamente 1 equivalente de N'-(1-butil-2-oxopirido[2,3-b]pirazin-3-il)-1-metil-ciclopropanecarbohidrato durante unas 2 a 12 horas bajo reflujo en un disolvente adecuado como el hexametildisilazano que contiene aproximadamente 0,2 equivalentes de una base orgánica no nucleófila adecuada como el 1,8-diazabiciclo[5,4,0]undec-7-eno. El producto puede entonces aislarse utilizando técnicas bien conocidas en la técnica, como la dilución, la filtración, la trituración y la cromatografía. Por ejemplo, la mezcla de reacción puede verterse en agua y el precipitado resultante puede recogerse por filtración, con la subsiguiente división del sólido recogido entre una mezcla adecuada no miscible de un disolvente orgánico, como el DCM, y agua. La capa orgánica puede separarse, lavarse secuencialmente con agua y NaCl acuoso saturado, secarse sobre Na2SO4, filtrarse y concentrarse a presión reducida. El residuo resultante puede ser triturado con un solvente orgánico caletar o hirviendo adecuado, como el EtOAc, durante aproximadamente 1 hora, y el sólido resultante puede ser recogido por filtración al enfriarse. El sólido puede purificarse más por cromatografía sobre sílice, utilizando un gradiente de una mezcla de disolventes adecuada como el EtOAc y el DCM, para obtener el compuesto de la Fórmula I, producto del Esquema 2, paso D.
Preparación 1
N-butil-2-nitro-piridin-3-amina

Esquema 1, paso A: Se disolvió 3-fluoro-2-nitropiridina (5,0 g, 35,2 mmol) en EtOH (30 mL) y se enfrió la mezcla a 0 °C en un baño de hielo. Se añadió butan-1-amina (7,7 g, 105,6 mmol) a la mezcla, se dejó calentar la mezcla a TA y se agitó a TA durante 2 hr. Se diluyó la mezcla con agua y se extrajo con EtOAc. Se lavó la capa orgánica con NaCl acuoso saturado, se secó sobre Na2SO4, se filtró y concentró a presión reducida para dar al compuesto del título (6,2 g, 90% de rendimiento) como aceite amarillo, apto para su uso sin purificación adicional. ES/MS m/z 196.1 (M+1).
Procedimiento alternativo de preparación 1
Se añadió 3-fluoro-2-nitropiridina (92 g, 0,65 mol) en EtOH (552 mL) a ~20-25 °C. Se enfrió la mezcla a 0 °C en un baño de hielo. Se añadió butan-1-amina (118,4 g, 1.6187 mol) en ~ 0-5 °C durante 40 min. Se calentó a ~ 20-25 °C y se agitó durante 16 hr. Se añadió agua (800 mL) a la mezcla de reacción, se extrajo con EtOAc (2 x 600 mL), se separaron las capas y se lavaron las capas orgánicas combinadas con agua (2 x 1L), NaCl acuoso saturado (2 x 500 mL), se secó sobre Na2SO4, se filtró y se concentró a presión reducida a 35 °C para obtener el compuesto del título (120,00 g, rendimiento del 95%) como un aceite de color amarillo intenso, adecuado para su uso sin purificación adicional. ES/MS m/z 196.1 (M+1).
Preparación 2
N3-butilpiridin-2,3-diamina

Esquema 1, paso B: Se añadió 5% de Pd/C (3,0 g, 1,4 mmol) a una solución de N-butil-2-nitro-piridin-3-amina (6,0 g, 30,7 mmol) se disolvió en el MeOH (50 mL) bajo N2. Se agitó la mezcla en TA bajo un globo de H2 durante 8 hr. Se filtró la mezcla a través de una almohadilla de tierra de diatomeas, se lavó con el MeOH, y se concentró el filtrado bajo presión reducida para dar al compuesto del título (5,0 g, 98% de rendimiento) como un sólido negro, adecuado para su uso sin purificación adicional. Es /MS m/z 166,1 (M+1).
Procedimiento alternativo de preparación 2
Se añadió N-butil-2-nitro-piridina-3-amina (128,0 g, 0,7 mol) en el MeOH (1024 mL) a -20-25 °C. Se añadió un 5 % de Pd/C húmedo (64 g, 50% de carga) a - 20-25 °C. Se agitó la mezcla resultante bajo 3 atm H2 en ~ 20-25 °C durante 3 hr. Se filtró la mezcla de reacción a través de tierra de diatomeas, se lavó la torta de filtro con MeOH (5 x 500 mL), y se concentró el filtrado a presión reducida para obtener el compuesto del título (101,9 g, rendimiento del 94%) como un sólido negro, adecuado para su uso sin purificación adicional. ES/MS m/z 166.1 (M+1).
Preparación 3
1-butil-4H-pirido[2,3-b]pirazin-2,3-diona

Esquema 1, paso C: Se añadió oxalato de dietilo (20,1 mL, 148,3 mmol) a una mezcla de N3-butilpiridin-2,3-diamina (4,9 g, 29,65 mmol) en EtOH (30 mL). Se calentó la mezcla en un tubo sellado a 100 °C durante 14 horas. Se enfrió la mezcla de reacción a 0 °C y se aisló el sólido resultante por filtración. Se lavó el sólido con Et2O y se secó al vacío a 40 °C para obtener el compuesto del título (3,3 g, rendimiento del 51%) como un sólido de color verde, apto para ser utilizado sin purificación adicional. ES/MS m/z 219,8 (M+1).
Procedimiento alternativo de preparación 3
Se añadió N3-butilpiridina-2,3-diamina (81,4 g, 0,5 mol) en EtOH (550 mL) en ~ 20-25 "C. Se añadió 30 % en peso de NaOEt en EtOH (427,4 g, 1,0 mol) en una porción en ~ 20-25 "C. Se añadió oxalato de dietilo (87,1 g, 0,6 mol) gota a gota en ~ 20-30 °C y se agitó a TA durante 2,5 hr. Se vertió la mezcla de reacción en una mezcla de 0,5 M acuosa HCl/DCM (1600 mL/1200mL) en ~ 0-10 °C con agitación. Se separaron dos capas, se extrajo la capa acuosa con DCM (2 x 800 mL), se lavó con agua (2 x 1600 mL), NaCl acuoso saturado (1600 mL) y se secó sobre Na2SO4. Se filtró y se concentró el filtrado a presión reducida. Se diluyo el residuo sólido resultante en NaCl (200 mL) a ~20-25 °C durante 30 min y se aisló el sólido resultante por filtración para dar al compuesto del título (70.0g, 65% de rendimiento) como un sólido verde, apto para su uso sin purificación adicional. ES/MS m/z 220.1 (M+1).
Preparación 4
1-butil-3-hidracino-pirido[2,3-b]pirazin-2-ona

Esquema 1, paso D: Se añadió monohidrato de hidracina (3,55 mL, 73,0 mmol) a una mezcla de 1-butil-4H-pirido[2,3-b]pirazin-2,3-diona (3,2 g, 14,6 mmol) en EtOH (20 mL). Se calentó la mezcla en un tubo sellado a 100 °C durante 14 horas. Se enfrió la mezcla de reacción a 0 °C y se aisló el sólido por filtración. Se lavó el sólido con Et2O y se secó al vacío a 45 °C para obtener el compuesto del título (2,8 g, rendimiento del 82%) como un sólido de color verde, apto para ser utilizado sin purificación adicional. ES/MS m/z 234,2 (M+1).
Preparación 5
1-butM-3-[2-[(1-metMciclopropM)metMeno]hidracino]pirido[2,3-b]pirazin-2-ona

Esquema 1, paso E: Se añadió 1-metilciclopropanocarbaldehído (1,15 mL, 13,7 mmol) y AcOH (39,3 j L) a una mezcla de 1- butil-3-hidrazino-pirido[2,3-b]pirazin-2-ona (1,6 g, 6,9 mmol) en el MeOH (20 mL). Se agitó la mezcla en TA durante 1 hr. Se concentró la mezcla a presión reducida y recristalizandose el producto a partir del hexano (50 mL). Se aisló el sólido por filtración y se lavó con hexano para obtener el compuesto del título (1,30 g, 63% de rendimiento) como un sólido negro, apto para ser utilizado sin purificación adicional. Es /MS m/z 300,2 (M+1).
Preparación 6
1-butil-3-cloro-pirido[2,3-b]pirazin-2-ona

Esquema 2, paso A: Se disolvió 1-butil-4H-pirido[2,3-b]pirazin-2,3-diona (60,8 g, 0,3 mol) en ACN (10 mL/g, 600 mL) en ~ 20-25 °C. Se añadió DMF (4,2 mL) y se añadió SoCl2 (99,0g, 0,8 mol) en una porción. Se calentó la mezcla resultante al reflujo en ~ 75-80 °C durante 2,5 hr. Se concentró la mezcla de reacción a la sequedad bajo presión reducida para obtener el compuesto del título crudo (93,0g, >99% de rendimiento) como un sólido negro, apto para su uso sin purificación adicional. ES/MS m/z 238,1 (M+1).
Preparación 7
1-butil-3-hidracino-pirido[2,3-b]pirazin-2-ona

Esquema 2, paso B: Se añadió THF (900 mL) a una solución acuosa al 85% (peso/peso) de hidracina monohidratada (48 g. 1,5 mol,) a TA. Se añadió 1-butil-3-cloro-pirido[2,3-b]pirazin-2-ona (90,0 g, 0,4 mol) para formar una pasta y remover a TA durante 16 hr. Se añadió agua (100 mL) a la mezcla de reacción y se agitó durante 20 min. Se filtró y se lavó la torta de filtro con agua (2 x 400 mL) y luego con MTBE (2 x 400 mL). Se eliminó el agua mediante azeotrópico con 2-metil-THF (3 x 600 mL) a presión reducida para obtener el compuesto del título (50.0g, 75% de rendimiento) como un sólido verde, apto para su uso sin purificación adicional. ES/MS m/z 234.1 (M+1).
Preparación 8
N'-(1-butil-2-oxo-pirido[2,3-b]pirazin-3-il)-1-metil-ciclopropanocarbohidrato

Esquema 2, paso C: Se añadió ácido 1-metilciclopropanocarboxílico (30,9 g, 0,3 mol) al DMF (350 mL) a temperatura ambiente y se enfrió la mezcla a 0 °C. En ~ -5-0 °C, se añadió EDCI (61,0 g, 0,3 mol) seguido de HOBT (41,75 g, 0,3 mol). A -10-0 °C, se añadió TEA (62,47 g, 0,6 mol) en gota durante 40 min y se agitó la mezcla resultante a ~ -5-0 °C
durante 20 min. Se añadió 1-butil-3-hidrazino-pirido[2,3-b]pirazin-2-ona (48,0 g, 0,2 mol.) en ~ 0-5 °C en una porción, se calentó a TA, y se agitó la mezcla resultante durante 16 hr. Se vertió la mezcla de reacción en HCl acuoso de 0,6 M (1600 mL) y se lavó con MTBE (3 x 500 mL); se separaron las capas, se desechó la capa de MTBE y se añadió DCM (1000 mL) a la capa acuosa. Se ejustó el pH ~ 7-8 con NaHCO3 sólido (140 g), se separaron las capas, se extrajo la capa acuosa con DCM (3 x 600 mL), y se lavó las capas orgánicas combinadas secuencialmente con agua (3 x 1000 mL) y NaCl acuoso saturado (2 x 1000 mL). Se evaporó a presión reducida para dar al compuesto del título (45,0 g, 69% de rendimiento) como un sólido negro, apto para su uso sin purificación adicional. e S/MS m/z 316.2 (M+1).
Ejemplo 1
5-butil-9-(1-metilciclopropil)pirido[3,2-e][1,2,4]triazolo[4,3-a]pirazin-6(5H)-ona

Esquema 1, paso F: Se añadió 1-butil-3-[2-[(1-metilciclopropil)metilen]-hidracino]pirido[2,3-b]pirazin-2-ona (1,3 g, 4,3 mmol) a DCM (15 mL) y se enfrió la solución a 0 °C en un baño de hielo. Se añadió diacetato de yodosobenceno (2,9 g, 8,7 mmol) a la solución y se agitó la mezcla a TA durante 1 hr. Se enfrió la mezcla de reacción con agua y extrajo con DCM. Se lavó las capas orgánicas con NaHCO3 saturado, se secó sobre Na2SO4, se filtró y se concentró a presión reducida y se purificó el residuo resultante por cromatografía sobre sílice, eluyendo con EtOAc:hexanos (3:1), para obtener el compuesto del título (1,1 g, rendimiento del 85%) como un sólido blanquecino. ES/MS m/z 298.2 (M+1).
Procedimiento alternativo del ejemplo 1
Esquema 2. paso D: Se diluyo N'-(1-butil-2-oxo-pirido[2,3-b]pirazin-3-yl)-1-metil-ciclopropanecarbohidracida (45,0 g, 0,1 mol) en HMDS (360 mL) en TA. Se añadió DBU (4,34 g, 28,5 mmol) y se calentó a 125 °C. Se agitó la solución resultante durante 6 horas bajo reflujo. Se enfrió la mezcla de la reacción a TA, se vertió la mezcla en agua (800 mL), y se filtró y se recogió el sólido resultante. Se disolvió el sólido en DCM (400 mL) / H2O (100 mL), se separaron las capas resultantes, y se lavó la fase orgánica con NaCl acuoso saturado (100 mL), se secó sobre Na2SO4, se filtró, y se concentró el filtrado bajo presión reducida para dar un residuo. Se trituró con EtOAc (200 mL) a 40-50 °C durante 1 h, y se ailsló el sólido resultante (22,0 g, 98% de pureza determinada por LCMS) por filtración. Se combinó el lote de 22,0 g con otro lote de material (10,0 g, 100% de pureza determinada por LCMS) y se purificó adicionalmente por cromatografía sobre gel de sílice, eluyendo con DCM:EtOAc (1:1), para obtener un residuo después de la evaporación del disolvente. Se trituró el residuo resultante con EtOAc caliente durante 30 min y se aisló el sólido resultante por filtración para obtener el compuesto del título (25,70 g. 42% de rendimiento) como un sólido blanco. ES/MS m/z 298.2 (M+1).
Generación de proteínas PDE
Las secuencias de nucleótidos que codifican los PDE1A (NP_001003683.1), PDE1C (NP_005011.1), PDE5A (NP_001074.2), PDE7B (NP_061818.1) y PDE9A (NP_002597.1) humanos de longitud completa se insertan en el vector pFastBac1 (Invitrogen) con una etiqueta HIS de terminal N. Las secuencias de nucleótidos que codifican la PDE4D humana de longitud completa (NP_006194.2) y el dominio catalítico (residuo 641-1141) de la PDE3A (NP_000912.3) se insertan en el vector pFastBac 1 (Invitrogen) con una etiqueta HIS de terminal C. Las secuencias de nucleótidos que codifican la PDE8A (NP_002596.1) y la PDE11A (AAI12394.1) humanas de longitud completa se insertan en el vector pFastBac1 (Invitrogen) con una etiqueta de bandera N-terminal. Las secuencias de nucleótidos que codifican la PDE10A humana de longitud completa (AAD32595.1) se insertan en el vector pFastBac1 (Invitrogen) con una etiqueta de bandera C-terminal. Las secuencias de nucleótidos que codifican la PDE6A (NP_000431.2) y la PDE6B (AAH00249.1) humanas de longitud completa se insertan en el vector pFastBacDual (Invitrogen) con una etiqueta HIS de terminal N y una etiqueta de bandera de terminal N, respectivamente, para la producción de dímeros de PDE6A/6B. La generación de baculovirus y la expresión de proteínas en las células Sf9 se llevan a cabo de acuerdo con el protocolo del sistema de Expresión de Baculovirus (Invitrogen) Bac-to-Bac. Las secuencias de nucleótidos que codifican la PDE1B (NP_000915.1) y la PDE2A (NP_002590.1) humanas de longitud completa se insertan en el pIEX4 (Novagen) con una etiqueta HIS C-terminal, y ambas producciones de proteínas en células Sf9 se llevan a cabo según el protocolo del proveedor (Novagen). Las proteínas PDE marcadas con His se purifican utilizando agarosa Ni-NTA (Qiagen) seguida de cromatografía de exclusión de tamaño en una columna SUPERDEX® 200 (GE Healthcare) en tampón de almacenamiento (20 mM Tris-HCl, pH7,5, 150 mM NaCl, 10% Glicerol). Las proteínas PDE marcadas con la bandera, incluidas las PDE6A/6B, se purifican utilizando agarosa M2 anti-bandera (Sigma), después de la purificación mediante cromatografía en columna de NiNTA y se eluyen en un tampón de almacenamiento (50 mM TrisHCl, pH7,5, 150 mM NaCI, 10% Glicerol 0,1 mg/ml Péptido de la bandera). Todas las proteínas purificadas se almacenan a -80°C en pequeñas alícuotas.
Ensayos de la enzima fosfodiesterasa
Todas las actividades enzimáticas de la fosfodiesterasa nucleotídica cíclica (PDE) de 3', 5' se miden con un ensayo enzimático radiométrico basado en el sistema de detección SPA (ensayo de proximidad de centelleo). Los compuestos que se van a analizar se diluyen en dimetilsulfóxido puro (DMSO) utilizando curvas de respuesta de concentración de diez puntos. La concentración máxima de compuestos en la mezcla de reacción es de 10 o 100 pM. Los compuestos a la concentración apropiada se preincuban con cualquiera de las enzimas PDE durante 30 minutos antes de que se inicie la reacción mediante la adición de sustrato. Las reacciones se dejan continuar durante 60 minutos a temperatura ambiente. A continuación, las reacciones se detienen mediante la adición de perlas de SPA. Las muestras se leen 12 horas más tarde en un MICROBETA™ TRILUX® Counter. "IC50" se refiere a la concentración del compuesto que produce el 50% de la máxima respuesta inhibitoria posible para ese compuesto. Los valores de IC50 se calculan trazando los datos normalizados frente a log [compuesto] y ajustando los datos mediante una ecuación logística de cuatro parámetros.
Ensayos de enzimas PDE dependientes de Ca2+-calmodulina
PDE1B, PDE1A y PDE1C son clonados y purificados siguiendo los procedimientos estándar de generación de proteínas. El tampón de ensayo se prepara para dar una concentración final en el ensayo de 50 mM de Tris-HCl, 50 mM de MgCh, 4 mM de CaCh, 0,1% de albúmina sérica bovina y 6 U/ml de calmodulina en agua, a pH 7,5. La concentración final de la enzima es de 0,25, 0,074 y 0,0012 nM, para PDE1A, PDE1B y PDE1C respectivamente. Las reacciones se inician con la adición del sustrato, [3H]cAMP, para dar una concentración final de 47 nM.
Tabla 1: Potencia in vitro del Ejemplo 1 contra la PDE1A, PDE1B y PDE1C humanas.

Los datos de la Tabla 1 demuestran que el compuesto del Ejemplo 1 inhibe la actividad de las enzimas PDE1A, PDE1B y PDE1C humanas in vitro.
Ensayos de enzimas PDE usando [3H1cAMP como sustrato
Las siguientes actividades de la fosfodiesterasa se miden utilizando [3H]cAMP como sustrato de reacción: PDE3A humana (dominio catalítico), PDE4D humana. PDE7B humana y PDE8A humana. Todas estas enzimas se clonan y purifican siguiendo procedimientos estándar. El tampón de ensayo se prepara para dar una concentración final en el ensayo de 50 mM de Tris-HCl, 8,3 mM de MgCh, 1,7 mM de ácido etilendiaminotetraacético (EDTA) y 0,1% de albúmina sérica bovina a pH 7,5. Las concentraciones finales de las enzimas son de 0,008, 0,021, 0,5 y 0,06 nM para PDE3A, PDE4D, PDE7B y PDE8A respectivamente. Las reacciones se inician con la adición del sustrato, [3H]cAMP, para dar una concentración final de 47 nM.
Tabla 2: Potencia in vitro del ejemplo 1 frente a la PDE3A humana (dominio catalítico). PDE4D, PDE7B y PDE8A.

Ensayos de enzimas PDE usando [3H1cGMP como sustrato
Las siguientes actividades de la fosfodiesterasa se miden utilizando [3H]cGMP como sustrato de reacción: PDE2A humana, PDE5A humana, PDE6A/6B humana, PDE9A humana, PDE10A humana y PDE11A humana. La forma activa catalítica de la PDE6 humana es un dímero compuesto por una subunidad a (PDE6A humana) y p (PDE6B humana). El dímero de la PDE6A/6B humana se produce mediante la estrategia de expresión y purificación, utilizando dos pasos de purificación, a saber, la cromatografía de NiNTA y la cromatografía sefarótica anti-bandera. El resto de las enzimas se clonan y purifican en casa siguiendo procedimientos estándar. El tampón de ensayo se prepara para dar una concentración final en el ensayo de 50 mM de Tris-HCl, 8,3 mM de MgCh, 1,7 mM de EDTA y 0,1 % de albúmina sérica bovina a pH 7,5. Las concentraciones finales de las enzimas son de 0,2, 0,002, 5, 1, 0,03 y 0,03 nM para la PDE2A humana, la PDE5A humana, la PDE6AB humana, la PDE9A humana, la PDE10A humana y la PDE11A humana, respectivamente. Las reacciones se inician mediante la adición del sustrato, [3H]cGMP, para dar una concentración final de 80 nM en el caso de los ensayos de PDE2A, PDE10A, PDE5A, PDe6a B y PDE11A humanas, mientras que para la PDE9A humana se utilizan 20 nM de [3H]cGMP.
Tabla 3: Potencia in vitro del Ejemplo 1 frente a PDE2A, PDE5A, PDE6AB, PDE9A, PDE10A y PDE11A.

Los datos de las Tablas 1, 2 y 3 demuestran que el compuesto del Ejemplo 1 es un inhibidor selectivo de la PDE1A, PDE1B y PDE1C humanas en relación con la PDE2A, PDE3A, PDE4D, PDE5A, PDE6AB, PDE7B, PDE8A, PDE9A, PDE10A y PDE11A humanas in vitro.
Claims (10)
1. Un compuesto de fórmula:

o una sal farmacéutica aceptable del mismo.
2. El compuesto según la reivindicación 1 que es:

3. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 para su uso en terapia.
4. Un compuesto o una sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 para su uso en el tratamiento de enfermedades renales crónicas.
5. Un compuesto o una sal farmacéuticamente aceptable del mismo según la reivindicación 1 para su uso en el tratamiento de la enfermedad renal diabética.
6. Composición farmacéutica que comprende un compuesto o una sal farmacéutica del mismo según la reivindicación 1, con uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables.
7. Un procedimiento de preparación de una composición farmacéutica que comprende la adición de un compuesto según la reivindicación 1, o de una sal del mismo que sea aceptable farmacéuticamente, con uno o más vehículos, diluyentes o excipientes aceptables farmacéuticamente.
8. Un compuesto según la reivindicación 2 para el tratamiento de la enfermedad renal crónica.
9. Un compuesto según la reivindicación 2 para su uso en el tratamiento de la enfermedad renal diabética.
10. Composición farmacéutica que comprende un compuesto según la reivindicación 2, con uno o más vehículos, diluyentes o excipientes farmacéuticamente aceptables.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662379372P | 2016-08-25 | 2016-08-25 | |
| PCT/US2017/047479 WO2018039051A1 (en) | 2016-08-25 | 2017-08-18 | Triazolopyrazinone derivative useful as a human pde1 inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2800433T3 true ES2800433T3 (es) | 2020-12-30 |
Family
ID=59762063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17761381T Active ES2800433T3 (es) | 2016-08-25 | 2017-08-18 | Derivado de la triazolopirazinona útil como inhibidor de la pde1 humana |
Country Status (37)
| Country | Link |
|---|---|
| US (1) | US10138244B2 (es) |
| EP (1) | EP3504208B1 (es) |
| JP (1) | JP6633246B2 (es) |
| KR (1) | KR20190028544A (es) |
| CN (1) | CN109641902B (es) |
| AR (1) | AR109328A1 (es) |
| AU (1) | AU2017317111A1 (es) |
| BR (1) | BR112019000988A2 (es) |
| CA (1) | CA3034828A1 (es) |
| CL (1) | CL2019000487A1 (es) |
| CO (1) | CO2019001644A2 (es) |
| CR (1) | CR20190078A (es) |
| CY (1) | CY1123406T1 (es) |
| DK (1) | DK3504208T3 (es) |
| DO (1) | DOP2019000021A (es) |
| EA (1) | EA201990313A1 (es) |
| EC (1) | ECSP19013196A (es) |
| ES (1) | ES2800433T3 (es) |
| HR (1) | HRP20201187T1 (es) |
| HU (1) | HUE050419T2 (es) |
| JO (1) | JOP20170164A1 (es) |
| LT (1) | LT3504208T (es) |
| MA (1) | MA46039B1 (es) |
| MD (1) | MD3504208T2 (es) |
| ME (1) | ME03791B (es) |
| MX (1) | MX2019002065A (es) |
| PE (1) | PE20190452A1 (es) |
| PH (1) | PH12019500370A1 (es) |
| PL (1) | PL3504208T3 (es) |
| PT (1) | PT3504208T (es) |
| RS (1) | RS60442B1 (es) |
| SG (1) | SG11201901311WA (es) |
| SI (1) | SI3504208T1 (es) |
| SV (1) | SV2019005838A (es) |
| TN (1) | TN2019000038A1 (es) |
| TW (1) | TWI651323B (es) |
| WO (1) | WO2018039051A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR112457A1 (es) * | 2017-08-02 | 2019-10-30 | Lilly Co Eli | Derivados de [1,2,4]triazolo[4,3-a]pirazin-6(5h)-ona |
| AR112346A1 (es) | 2017-08-10 | 2019-10-16 | Lilly Co Eli | Derivados de [1,2,4]triazolo |
| US11453673B2 (en) | 2018-02-06 | 2022-09-27 | Eli Lilly And Company | Substituted [1,2,4]triazolo[4,3-a]pyrazines as phosphodiesterase inhibitors |
| US20220280517A1 (en) * | 2019-08-22 | 2022-09-08 | Intra-Cellular Therapies, Inc. | Organic compounds |
| WO2025059641A1 (en) | 2023-09-14 | 2025-03-20 | Eli Lilly And Company | Pde1 inhibitors |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4354027A (en) | 1980-05-19 | 1982-10-12 | Usv Pharmaceutical Corporation | Triazoloquinoxalin-4-ones |
| US8299080B2 (en) * | 2006-12-13 | 2012-10-30 | Aska Pharmaceutical Co., Ltd. | Substituted imidazo[1,5-A] quinoxalines as a PDE9 inhibitor |
| WO2008103357A1 (en) | 2007-02-21 | 2008-08-28 | E. I. Du Pont De Nemours And Company | Fungicidal tricyclic 1,2,4-triazoles |
| KR20130097178A (ko) | 2010-09-07 | 2013-09-02 | 아스텔라스세이야쿠 가부시키가이샤 | 퀴녹살린 화합물 |
| CA2877146C (en) * | 2012-06-18 | 2020-10-20 | Dart Neuroscience (Cayman) Ltd | Substituted thiophene- and furan-fused azolopyrimidine-5-(6h)-one compounds |
| WO2016022825A1 (en) * | 2014-08-07 | 2016-02-11 | Intra-Cellular Therapies, Inc. | Organic compounds |
| US20160083400A1 (en) * | 2014-09-18 | 2016-03-24 | Sunovion Pharmaceuticals Inc. | Tricyclic guanidine derivative |
| TW201629064A (zh) * | 2014-10-10 | 2016-08-16 | H 朗德貝克公司 | 作爲pde1抑制劑之三唑並吡酮 |
| TWI609870B (zh) | 2016-02-12 | 2018-01-01 | 美國禮來大藥廠 | Pde1抑制劑 |
-
2017
- 2017-08-10 JO JOP/2017/0164A patent/JOP20170164A1/ar unknown
- 2017-08-11 AR ARP170102252A patent/AR109328A1/es unknown
- 2017-08-11 TW TW106127199A patent/TWI651323B/zh not_active IP Right Cessation
- 2017-08-18 KR KR1020197005013A patent/KR20190028544A/ko not_active Withdrawn
- 2017-08-18 MX MX2019002065A patent/MX2019002065A/es unknown
- 2017-08-18 PL PL17761381T patent/PL3504208T3/pl unknown
- 2017-08-18 MD MDE20190732T patent/MD3504208T2/ro unknown
- 2017-08-18 CA CA3034828A patent/CA3034828A1/en not_active Abandoned
- 2017-08-18 SG SG11201901311WA patent/SG11201901311WA/en unknown
- 2017-08-18 AU AU2017317111A patent/AU2017317111A1/en not_active Abandoned
- 2017-08-18 PE PE2019000371A patent/PE20190452A1/es unknown
- 2017-08-18 SI SI201730305T patent/SI3504208T1/sl unknown
- 2017-08-18 RS RS20200738A patent/RS60442B1/sr unknown
- 2017-08-18 EA EA201990313A patent/EA201990313A1/ru unknown
- 2017-08-18 HU HUE17761381A patent/HUE050419T2/hu unknown
- 2017-08-18 US US15/680,250 patent/US10138244B2/en active Active
- 2017-08-18 DK DK17761381.7T patent/DK3504208T3/da active
- 2017-08-18 ME MEP-2020-144A patent/ME03791B/me unknown
- 2017-08-18 EP EP17761381.7A patent/EP3504208B1/en active Active
- 2017-08-18 PT PT177613817T patent/PT3504208T/pt unknown
- 2017-08-18 LT LTEP17761381.7T patent/LT3504208T/lt unknown
- 2017-08-18 CN CN201780050004.2A patent/CN109641902B/zh active Active
- 2017-08-18 HR HRP20201187TT patent/HRP20201187T1/hr unknown
- 2017-08-18 BR BR112019000988-4A patent/BR112019000988A2/pt not_active IP Right Cessation
- 2017-08-18 ES ES17761381T patent/ES2800433T3/es active Active
- 2017-08-18 CR CR20190078A patent/CR20190078A/es unknown
- 2017-08-18 JP JP2019510969A patent/JP6633246B2/ja active Active
- 2017-08-18 TN TNP/2019/000038A patent/TN2019000038A1/en unknown
- 2017-08-18 MA MA46039A patent/MA46039B1/fr unknown
- 2017-08-18 WO PCT/US2017/047479 patent/WO2018039051A1/en not_active Ceased
-
2019
- 2019-01-30 DO DO2019000021A patent/DOP2019000021A/es unknown
- 2019-02-19 SV SV2019005838A patent/SV2019005838A/es unknown
- 2019-02-21 PH PH12019500370A patent/PH12019500370A1/en unknown
- 2019-02-22 CL CL2019000487A patent/CL2019000487A1/es unknown
- 2019-02-22 EC ECSENADI201913196A patent/ECSP19013196A/es unknown
- 2019-02-22 CO CONC2019/0001644A patent/CO2019001644A2/es unknown
-
2020
- 2020-08-14 CY CY20201100760T patent/CY1123406T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2879645T3 (es) | Inhibidor de PDE1 | |
| ES2800433T3 (es) | Derivado de la triazolopirazinona útil como inhibidor de la pde1 humana | |
| CN102482278B (zh) | 作为pi3k抑制剂的嘧啶酮类 | |
| US20100035900A1 (en) | 6-Arylmethyl-substituted pyrazolopyrimidines | |
| US20080221092A1 (en) | Heterobicyclic metalloprotease inhibitors | |
| KR101651933B1 (ko) | 세로토닌 5-ht6 수용체의 2-아미노-3-설포닐-테트라하이드로-피라졸로[1,5-a]피리도-피리미딘 길항제, 이의 제조방법 및 이의 용도 | |
| ZA200605781B (en) | 6-amino-5-cyano-pyrimidine-4-ones used for improving perception, power of concentration, learning efficiency, and/or memory power | |
| ES2904938T3 (es) | Derivados de [1,2,4]triazolo[4,3-a]pirazin-6(5H)-ona | |
| ES2942442T3 (es) | Derivados de [1,2,4]triazolo[4,3-a]pirazin-8-ona | |
| ES2895969T3 (es) | Derivados de [1,2,4]triazolo como inhibidores de PDE1 para el tratamiento de diabetes | |
| WO2025059641A1 (en) | Pde1 inhibitors | |
| CN121986104A (en) | PDE1 inhibitors |