ES2804101T3 - Compuestos de 4-amino-imidazoquinolina - Google Patents
Compuestos de 4-amino-imidazoquinolina Download PDFInfo
- Publication number
- ES2804101T3 ES2804101T3 ES15716824T ES15716824T ES2804101T3 ES 2804101 T3 ES2804101 T3 ES 2804101T3 ES 15716824 T ES15716824 T ES 15716824T ES 15716824 T ES15716824 T ES 15716824T ES 2804101 T3 ES2804101 T3 ES 2804101T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- imidazo
- ethoxymethyl
- quinolin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(C)(*)C[n]1c2c(ccc(*)c3*)c3nc(N)c2nc1* Chemical compound CC(C)(*)C[n]1c2c(ccc(*)c3*)c3nc(N)c2nc1* 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4188—1,3-Diazoles condensed with other heterocyclic ring systems, e.g. biotin, sorbinil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Virology (AREA)
- Transplantation (AREA)
- Biotechnology (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Un compuesto de fórmula **(Ver fórmula)** en la que R1 es alquilo C1-7 o alcoxi C1-7-alquilo C1-7; R2 se selecciona del grupo que consiste en hidrógeno, halógeno, alcoxicarbonil C1-7-alquilo C1-7 y alcoxicarbonil C1-7-alquenilo C2-7; R3 es hidrógeno; R4 se selecciona del grupo que consiste en -O-(CH2)m-NHR5 y -O-(CO)-(CH2)n-NHR6, en las que m es 2, n es 1, R5 es hidrógeno y R6 es hidrógeno; o sales farmacéuticamente aceptables del mismo.
Description
DESCRIPCIÓN
Compuestos de 4-amino-imidazoquinolina
CAMPO DE LA INVENCIÓN
La presente invención se refiere a derivados de 4-amino-imidazoquinolina novedosos que tienen actividad farmacéutica, su fabricación, composiciones farmacéuticas que los contienen y su uso potencial como medicamentos.
En particular, la presente invención se refiere a compuestos de fórmula

en la que de R1 a R4 y X son como se describe a continuación, o a sales farmacéuticamente aceptables de los mismos.
Los compuestos son agonistas de TLR. En particular, los compuestos son agonistas de TLR7 y/o TLR8 y más en particular, agonistas de ambos receptores TLR7 y TLR8. Por tanto, pueden ser útiles para el tratamiento y prevención del cáncer, enfermedades autoinmunitarias e infecciosas. Por ejemplo, pueden ser útiles en una vacuna frente a enfermedades tales como el cáncer, enfermedades autoinmunitarias o infecciosas.
Los receptores de tipo Toll (TLR) son una familia de receptores que abarcan la membrana que se expresan en las células del sistema inmunitario como células dendríticas, macrófagos, monocitos, linfocitos T, linfocitos B, linfocitos NK y mastocitos, pero también en las células endoteliales y epiteliales. (Kawai et al., Immunity, 2011, 34, 637-650, Kawai et al., Nat. Immunol., 2010, 11, 373-384). Los TLR que reconocen componentes bacterianos y fúngicos se expresan en la superficie celular (es decir, TLR1, 2, 4, 5 y 6), mientras que otros que reconocen ácidos nucleicos víricos o microbianos como TLR3, 7, 8 y 9 se localizan en la membrana endolisosómica/fagosómica. (Henessy et al. Nat. Rev. Drug Discovery 2010, 9, 293-307). La activación de TLR da lugar a la inducción y liberación de citocinas proinflamatorias, dependiendo la secuencia de activación y la respuesta específicas del TLR y el tipo de célula específicos. Ambos TLR7 y TLR8 se expresan en monocitos y macrófagos, con TLR7 también altamente expresado en células dendríticas plasmocitoides y TLR8 en células dendríticas mieloides y mastocitos. Ambos receptores se activan por ARNmc y su activación estimula la producción de citocinas tales como IL-6, IL-12, TNF-a e IFN-y y moléculas coestimuladoras adicionales y receptores de quimiocinas. Dependiendo del tipo de célula, los interferones de tipo I, IFNa (de células dendríticas plasmocitoides) e IFNp, también se producen por las células tras la activación con agonistas de TLR7/8 (Uematsu et al., J. Biol. Chem., 2007, 282, 15319-15323).
Se han identificado los agonistas de moléculas pequeñas para tanto el receptor TLR7 como TLR8, así como los análogos modificados para su uso como coadyuvantes o conjugados de vacunas en muchas patentes (es decir, documentos WO1992015582, US 2003187016, WO 2005076783, WO2007024612, WO2009111337, WO2010093436, WO2011017611, WO2011068233, WO2011139348, WO2012066336, WO2012167081, WO2013033345, WO2013067597, WO2013166110 y US2013202629). Se ha obtenido experiencia clínica usando exclusivamente agonistas de TLR7. Una serie de los primeros compuestos han demostrado propiedades antivíricas y anticancerígenas. Por ejemplo, el agonista de TLR7 imiquimod (ALDARA™) se aprobó por la Administración de Medicamentos y Alimentos de los EE. UU. como agente tópico para el tratamiento de condilomas acuminados, carcinoma basocelular superficial y queratosis actínica. Sin embargo, se ha abandonado la aplicación sistémica de los primeros agonistas de TLR7 como resiquimod debido a la cardiotoxicidad intolerable observada tras la estimulación de quimiocinas global a niveles terapéuticos (Holldack, Drug Discovery Today, 2013, 1-4). El conocimiento sobre los agonistas de TLR8 está menos avanzado y se limita principalmente a datos con los primeros agonistas de TLR7/8 mixtos como resiquimod y más recientemente a compuestos descritos por VentiRX Pharmaceuticals (es decir, documentos WO2010054215, WO2012045090). En la actualidad, todavía existe una necesidad de agonistas de TLR7 y TLR8 de moléculas pequeñas adicionales, específicamente aquellos con potencia mejorada.
La presente invención está dirigida a derivados de 1H-imidazo[4,5-c]quinolin-4-amina-2-metilpropan-2-iloxi con potencia celular mejorada con respecto a agonistas de TLR7 y/o TLR8 conocidos de este tipo para su uso en el
tratamiento del cáncer, preferentemente tumores sólidos y linfomas, y para otros usos que incluyen el tratamiento de determinadas dermopatías o enfermedades cutáneas, tales como la dermatitis atópica, el tratamiento de enfermedades infecciosas, preferentemente enfermedades víricas, y para su uso como coadyuvantes en vacunas formuladas para su uso en el tratamiento del cáncer o desensibilizando los receptores por estimulación continua en el tratamiento de enfermedades autoinmunitarias.
Específicamente, la presente invención divulga derivados de 1H-imidazo[4,5-c]quinolin-4-amina-2-metilpropan-2-iloxi que se derivatizan directamente en el alcohol terciario con un resto aminoetilo o glicina. Debido a la escasa reactividad del alcohol terciario, estos derivados obviamente habían eludido los primeros intentos en la síntesis. De forma sorprendente, estos nuevos compuestos poseen una alta potencia celular en TLR7 que es comparable o incluso mejor que el propio resiquimod, mientras que los análogos cercanos que se describieron inicialmente tales como 1-(2-(2-aminoetoxi)etil)-2-(etoximetil)-7H-imidazo[4,5-c]quinolin-4-amina (ejemplo 69 del documento US20030187016) no muestran la actividad requerida. Además y también de forma sorprendente, los derivados de 1H-imidazo[4,5-c]quinolin-4-amina-2-metilpropan-2-iloxi descritos son fuertes agonistas del receptor TLR8 con una potencia comparable o superior a los agonistas de TLR8 divulgados hasta ahora de otras clases químicas y muy mejorada con respecto al propio resiquimod. Por tanto, los compuestos de la presente invención satisfacen la necesidad de activar ambos receptores TLR7 y TLR8 con potencia mejorada.
SUMARIO DE LA INVENCIÓN
La presente invención se refiere a derivados de 4-amino-imidazoquinolina de fórmula

en la que
R1 es alquilo C1-7 o alcoxi C1-7-alquilo C1-7;
R2 se selecciona del grupo que consiste en hidrógeno, halógeno, alcoxicarbonil C1-7-alquilo C1-7 y alcoxicarbonil C1-7-alquenilo C2-7;
R3 es hidrógeno;
R4 se selecciona del grupo que consiste en
-O-(CH2)m-NHR5 y
-O-(CO)-(CH2)n-NHR6,
en la que
m se selecciona de 2,
n se selecciona de 1,
R5 se selecciona del grupo que consiste en hidrógeno y
R6 se selecciona del grupo que consiste en hidrógeno,
o sales farmacéuticamente aceptables de los mismos.
La invención también se refiere a procedimientos para la fabricación de compuestos de fórmula I.
La invención también se refiere a composiciones farmacéuticas que comprenden un compuesto de fórmula I como se describe anteriormente y un vehículo y/o coadyuvante farmacéuticamente aceptable.
Otro aspecto de la invención es el uso de compuestos de fórmula I como principios activos terapéuticos para el
tratamiento de enfermedades que pueden estar mediadas con agonistas de TLR, en particular agonistas de TLR7 y/o TLR8, más en particular, receptores TLR7 y TLR8. Por tanto, la invención se refiere a un procedimiento para el tratamiento de una enfermedad que se puede mediar con agonistas de TLR tales como, por ejemplo, cáncer y enfermedades autoinmunitarias o infecciosas.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado como se entiende comúnmente por un experto en la técnica a la que pertenece la presente invención. Además, las siguientes definiciones se exponen para ilustrar y definir el significado y alcance de los diversos términos usados para describir la invención.
La nomenclatura usada en esta solicitud se basa en la nomenclatura sistemática de la IUPAC, a menos que se indique de otro modo.
El término "compuesto(s) de esta invención" y "compuesto(s) de la presente invención" se refiere a compuestos de fórmula I y solvatos o sales de los mismos (por ejemplo, sales farmacéuticamente aceptables).
El término "sustituyente" indica un átomo o un grupo de átomos que reemplazan un átomo de hidrógeno en la molécula original.
El término "halógeno" se refiere a flúor, cloro, bromo y yodo, siendo flúor, cloro y bromo de interés particular. Más en particular, halógeno se refiere a flúor y cloro.
El término "alquilo", solo o en combinación con otros grupos, se refiere a un radical hidrocarburo alifático saturado monovalente de cadena lineal o ramificada de uno a veinte átomos de carbono, en particular de uno a dieciséis átomos de carbono, más en particular de uno a diez átomos de carbono. El término "alquilo C1-10" se refiere a un radical hidrocarburo alifático saturado monovalente de cadena lineal o ramificada de uno a diez átomos de carbono, tal como, por ejemplo, metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, tere-butilo, pentilo, 1,1,3,3-tetrametilbutilo. Más en particular, el término "alquilo" también abarca grupos alquilo inferior como se describe a continuación. El término "alquilo inferior" o "alquilo C1-7", solo o en combinación, significa un grupo alquilo de cadena lineal o cadena ramificada con de 1 a 7 átomos de carbono, en particular un grupo alquilo de cadena lineal o ramificada con de 1 a 6 átomos de carbono y más en particular un grupo alquilo de cadena lineal o ramificada con de 1 a 4 átomos de carbono. Los ejemplos de grupos alquilo C1-7 de cadena lineal y ramificada son metilo, etilo, propilo, isopropilo, butilo, isobutilo, tere-butilo, los pentilos isómeros, los hexilos isómeros y los heptilos isómeros, en particular metilo y etilo.
El término "alquenilo inferior" o "alquenilo C2-7" significa un residuo hidrocarburo de cadena lineal o de cadena ramificada que comprende un enlace olefínico y de 2 a 7, en particular de 3 a 6, más en particular de 3 a 4 átomos de carbono. Los ejemplos de grupos alquenilo son etenilo, 1-propenilo, 2-propenilo, isopropenilo, 1 -butenilo, 2-butenilo, 3-butenilo e isobutenilo, en particular etenilo.
El término "alcoxi inferior" o "alcoxi C 1-7" se refiere al grupo R'-O-, en el que R' es alquilo inferior y el término "alquilo inferior" tiene el significado previamente dado. Los ejemplos de grupos alcoxi inferior son metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, sec-butoxi y terc-butoxi, en particular metoxi.
El término "alcoxicarbonilo inferior" o "alcoxicarbonilo C 1-7" se refiere al grupo -COOR, en el que R es alquilo inferior y el término "alquilo inferior" tiene el significado previamente dado. Los grupos alcoxicarbonilo inferior de particular interés son metoxicarbonilo o etoxicarbonilo.
El término "alcoxicarbonilalquilo inferior" o "alcoxicarbonil C1-7-alquilo C1-7" se refiere a grupos alquilo inferior como se define anteriormente en los que al menos uno de los átomos de hidrógeno del grupo alquilo inferior está reemplazado por un grupo alcoxicarbonilo inferior. Entre los grupos alcoxicarbonil-alquilo inferior interesantes particulares está -(CH2)2-Co OC2Hs.
El término "alcoxicarbonilalquenilo inferior" o "alcoxicarbonil C1-7-alquenilo C2-7" se refiere a grupos alquenilo inferior como se define anteriormente en los que al menos uno de los átomos de hidrógeno del grupo alquenilo inferior se reemplaza por un grupo alcoxicarbonilo inferior. Entre los grupos de alcoxicarbonil-alquenilo inferior interesantes particulares está -(CH2)2-COOC2Hs.
El término "farmacéuticamente aceptable" indica un atributo de un material que es útil en la preparación de una composición farmacéutica que en general es segura, no tóxica y ni biológicamente ni de otro modo indeseable y es aceptable para uso farmacéutico veterinario así como humano.
Los compuestos de fórmula I pueden formar sales farmacéuticamente aceptables. El término "sales
farmacéuticamente aceptables" se refiere a las sales que retienen la eficacia biológica y las propiedades de las bases libres o ácidos libres, que no son biológicamente o de otro modo indeseables. Las sales farmacéuticamente aceptables incluyen tanto sales de adición de ácido como de base. Las sales son por ejemplo sales de adición de ácido de compuestos de fórmula I con ácidos minerales fisiológicamente compatibles, tales como ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido carbónico, ácido sulfúrico, ácido sulfuroso o ácido fosfórico; o con ácidos orgánicos, tales como ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido fórmico, ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxílico, ácido láctico, ácido trifluoroacético, ácido cítrico, ácido fumárico, ácido maleico, ácido malónico, ácido tartárico, ácido benzoico, ácido cinámico, ácido mandélico, ácido embónico, ácido succínico o ácido salicílico. Además, se pueden preparar sales farmacéuticamente aceptables a partir de la adición de una base inorgánica o una base orgánica al ácido libre. Las sales derivadas de una base inorgánica incluyen, pero no se limitan a, las sales de sodio, potasio, litio, amonio, calcio, magnesio, cinc, cobre, manganeso y aluminio. Las sales derivadas de bases orgánicas incluyen, pero no se limitan a sales de aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas naturales, aminas cíclicas y resinas de intercambio iónico básicas, tales como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, etanolamina, lisina, arginina, histidina, cafeína, procaína, hidrabamina, colina, betaína, etilendiamina, glucosamina, metilglucamina, teobromina, piperacina, W-etilpiperidina, piperidina y resinas de poliamina. El compuesto de fórmula I también se puede presentar en forma de iones dipolares. Las sales farmacéuticamente aceptables de compuestos de fórmula I de interés particular son las sales de sodio o sales con aminas terciarias.
Los compuestos de fórmula I también pueden estar solvatados, por ejemplo, hidratados. La solvatación se puede efectuar en el transcurso del procedimiento de fabricación o puede tener lugar, por ejemplo, como consecuencia de propiedades higroscópicas de un compuesto inicialmente anhidro de fórmula I (hidratación). El término "sales farmacéuticamente aceptables" también incluye solvatos fisiológicamente aceptables.
El término "agonista" indica un compuesto que potencia la actividad de otro compuesto o sitio receptor como se define por ejemplo, en "The Pharmacological Basis of Therapeutics, de Goodman y Gilman, 7.a ed." en la página 35, Macmillan Publ. Company, Canadá, 1985. Un "agonista completo" efectúa una respuesta completa mientras que un "agonista parcial" efectúa una activación menor que completa incluso cuando ocupa la población de receptores total. Un "agonista inverso" produce un efecto opuesto al de un agonista, aunque se une al mismo sitio de unión al receptor.
El término "concentración eficaz máxima media" (CE50) indica la concentración plasmática de un compuesto particular requerida para obtener un 50 % del máximo de un efecto particular in vivo.
El término "cantidad terapéuticamente eficaz" indica una cantidad de un compuesto de la presente invención que, cuando se administra a un sujeto, (i) trata o previene la enfermedad, afección o trastorno particular, (ii) atenúa, mejora o elimina uno o más síntomas de la enfermedad, afección o trastorno particular, o (iii) previene o retrasa la aparición de uno o más síntomas de la enfermedad, afección o trastorno particular descrito en el presente documento. La cantidad terapéuticamente eficaz variará dependiendo del compuesto, estado de enfermedad que se está tratando, la gravedad o la enfermedad tratada, la edad y salud relativa del sujeto, la vía y forma de administración, el juicio del médico especialista o veterinario, y otros factores.
En detalle, la presente invención se refiere a compuestos de fórmula

en la que
R1 es alquilo C1-7 o alcoxi C i-7-alquilo C1-7;
R2 se selecciona del grupo que consiste en hidrógeno, halógeno, alcoxicarbonil Cwalquilo C1-7 y alcoxicarbonil C1-7-alquenilo C2-7;
R3 es hidrógeno;
R4 se selecciona del grupo que consiste en
-O-(CH2)m-NHR5 y
-O-(CO)-(CH2)n-NHR6,
en la que
m se selecciona de 2,
n se selecciona de 1,
R5 se selecciona del grupo que consiste en hidrógeno y
R6 se selecciona del grupo que consiste en hidrógeno
o sales farmacéuticamente aceptables de los mismos.
En un aspecto, la invención se refiere a compuestos de fórmula I, en la que R1 es alcoxi C1-7-alquilo C1-7. Más en particular, R1 es etoxietilo.
En otro aspecto, la invención también se refiere a compuestos de fórmula I, en la que R4 es -O-(CO)-(CH2)n-NHR6 y en la que n es 1 o 2 y en la que R6 es hidrógeno.
En un aspecto particular, la invención se refiere a compuestos de fórmula I, en la que R2 es hidrógeno.
Los compuestos particulares de fórmula I de acuerdo con la invención son los siguientes:
1- (2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
2- aminoacetato de 1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo,
(E)-3-[4-amino-1-[2-(2-aminoetoxi)-2-metilpropil]-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il]prop-2-enoato de etilo, 3- (4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, 3-(4-amino-1-(2-(2-aminoacetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, 1-(2-(2-aminoetoxi)-2-metilpropil)-2-pentil-1H-imidazo[4,5-c]quinolin-4-amina,
1-(2-(2-aminoetoxi)-2-metilpropil)-7-bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
o sales farmacéuticamente aceptables de los mismos.
En particular, la invención se refiere a los siguientes compuestos de fórmula I:
1- (2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
o sales farmacéuticamente aceptables de los mismos.
Más en particular, la invención se refiere a compuestos de fórmula I seleccionados del grupo que consiste en 2- aminoacetato de 1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo,
3- (4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, y sales farmacéuticamente aceptables de los mismos.
En particular, la invención se refiere a las siguientes sales de compuestos de fórmula I:
clorhidrato de 3-(4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo,
1-(2-(2-aminoetoxi)-2-metilpropil)-7-bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
o sales farmacéuticamente aceptables de los mismos.
Se apreciará que los compuestos de fórmula general I en la presente invención se pueden derivatizar en grupos
funcionales para proporcionar derivados que se pueden volver a convertir en el compuesto original in vivo. Los derivados fisiológicamente aceptables y metabólicamente lábiles, que pueden producir los compuestos originales de fórmula general I in vivo también están dentro del alcance de la presente invención.
Otro aspecto de la presente invención es el procedimiento para la fabricación de compuestos de fórmula I como se define anteriormente, procedimiento que comprende
a) hacer reaccionar un compuesto de fórmula II

en la que R1, R2 y R3 son como se define en el presente documento anteriormente y GP es un grupo protector, con un compuesto de fórmula III

en la que m es como se define en el presente documento anteriormente, en condiciones básicas, y retirar los grupos protectores GP y Boc en condiciones ácidas para obtener un compuesto de fórmula I-a

en la que de R1 a R3 y m son como se define en el presente documento anteriormente, y opcionalmente, acoplar además el compuesto de fórmula I-a con un alcohol o ácido de fórmula R5-OH o un aldehído de fórmula R5=O para obtener un compuesto de fórmula I-c
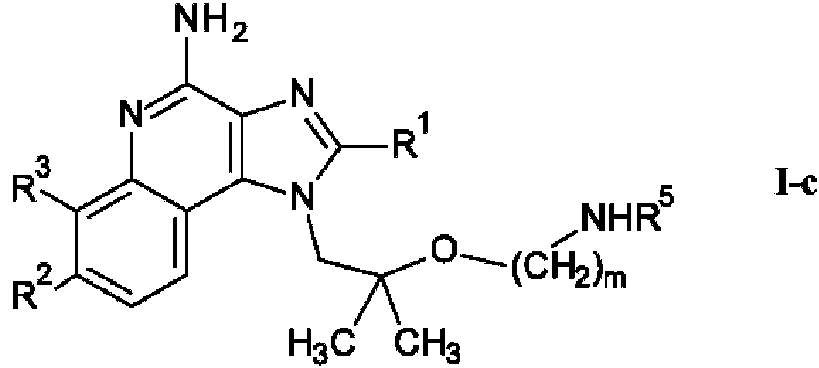
en la que de R1 a R3, m y R5 son como se define en el presente documento anteriormente, y, si se desea, convertir el compuesto obtenido en una sal farmacéuticamente aceptable, o
b) hacer reaccionar un compuesto de fórmula Il-a

en la que R1, R2 y R3 son como se define en el presente documento anteriormente y GP' es un grupo protector, con un ácido carboxílico de fórmula IV

en la que n se define como en el presente documento anteriormente y GP" es un grupo protector, en presencia de un agente de esterificación, y retirar los grupos protectores GP' y GP" con un agente reductor suave para obtener un compuesto de fórmula I-b

en la que de R1 a R3 y n son como se define en el presente documento anteriormente, y opcionalmente, acoplar además el compuesto de fórmula I-a con un alcohol o ácido de fórmula R6-OH o un aldehído de fórmula R6=O para obtener un compuesto de fórmula I-d

en la que de R1 a R3, m y R6 son como se define en el presente documento anteriormente, y, si se desea, convertir el compuesto obtenido en una sal farmacéuticamente aceptable.
En particular, un grupo protector GP adecuado es un grupo protector de amino seleccionado de tritilo (TRT), o de doble protección usando un grupo protector de isoindolina-1,3-diona, bis-bencilo o bis-carboxibencilo (bis-Z).
"En condiciones básicas" significa la presencia de una base tal como hidruro de sodio o ferc-butilato de potasio. La reacción se lleva a cabo en un disolvente adecuado tal como, por ejemplo W,W-dimetilformamida (DMF), dimetilacetamida (DMA), diclorometano o dioxano, a temperaturas entre 0 °C y temperatura ambiente.
"Retirar los grupos protectores GP y Boc en condiciones ácidas" significa tratar el compuesto protegido con ácidos en un disolvente adecuado, por ejemplo, se puede emplear ácido trifluoroacético (TFA) en un disolvente tal como diclorometano (DCM).
Los grupos protectores GP' y GP" adecuados son grupos protectores que forman un anillo cíclico con el átomo de nitrógeno del grupo amino. En particular, GP' o GP" conjuntamente con el átomo de nitrógeno al que están unidos
forman una isoindolina-1,3-diona o representan un grupo protector de bis-bencilo o bis-carboxibencilo.
Un agente de esterificación es un compuesto que facilita una reacción de esterificación. Un agente de esterificación particular es W,W-diisopropil-carbodiimida. La reacción se lleva a cabo, en particular, en un disolvente inerte, tal como DCM.
"Retirar los grupos protectores GP' y GP" con un agente reductor suave", en particular, significa tratar el compuesto protegido con hidracina/agua en un disolvente inerte, tal como THF.
La invención se refiere además a compuestos de fórmula I como se define anteriormente obtenibles de acuerdo con un procedimiento como se define anteriormente.
La síntesis de los compuestos con la estructura general I se puede lograr, por ejemplo, de acuerdo con los esquemas siguientes. A menos que se indique de otro modo, de R1 a R3 y X son como se define anteriormente. Se proporciona una vía de acceso a los materiales de partida de fórmula AG en el esquema 1, y la vía se ha ejemplificado en el documento WO 2013/033345 (Univ. de Minnesota).
Los compuestos AB se pueden obtener a partir de ortoésteres AA adecuadamente sustituidos por condensación con 2-amino-propanodinitrilo y 1-amino-2-metil-propan-2-ol en un disolvente inerte, como por ejemplo, THF en presencia de una base, como por ejemplo, trietilamina. Los ortoésteres AA sustituidos adecuados están disponibles comercialmente, se pueden sintetizar por un experto en la técnica o se han ejemplificado en la parte experimental. Los compuestos AC se pueden obtener a partir de los compuestos AB por diazotización/yodación como es conocido en la técnica; específicamente usando diyodometano como fuente de yoduro e isoamilnitrito como fuente de nitrito en un disolvente inerte como cloroformo a temperaturas de 0 °C a la temperatura de ebullición del disolvente, preferentemente a una temperatura de 80 °C.
Los compuestos de fórmula AC se pueden acoplar con compuestos de fórmula AD donde M indica un grupo saliente de metal y R' indica un hidrógeno y/o un grupo protector adecuado, por procedimientos conocidos en la técnica, para dar compuestos de fórmula AE. Los grupos salientes de metale adecuados pueden ser ácidos borónicos, ésteres de boronato y trifluoroboratos, pero también grupos salientes a base de estaño o cinc. En particular, los ácidos borónicos o ésteres de boronato se pueden usar en acoplamientos de tipo Suzuki-Miyaura usando un catalizador de paladio, como Pd(OAc)2 en presencia de trifenilfosfina, en un disolvente inerte, como DME, conjuntamente con una base adecuada, como carbonato de sodio. La temperatura de reacción puede variar de temperatura ambiente a la temperatura de ebullición del disolvente, siendo la temperatura ambiente una opción adecuada en muchos casos. Si los compuestos AE están protegidos en el grupo amino de anilina (un R' diferente de H), la desprotección para proporcionar compuestos de tipo AF se puede realizar por procedimientos conocidos en la técnica, siendo la escisión ácida del grupo protector usando TFA una opción preferente.
Los compuestos de tipo AG se pueden obtener a partir de compuestos AF por condensación térmica (cierre del anillo) en presencia de un catalizador ácido. Esto se puede lograr simplemente calentando los compuestos AF en un disolvente inerte, como dioxano, en presencia de un ácido, como HCl, durante un tiempo apropiado, por ejemplo, durante 2 horas a 90 °C.

Después del procedimiento de acuerdo con el esquema 2, los compuestos AG se pueden usar como material de partida para la síntesis de compuestos I-a donde R4 es -O-(CH2)m-NH2.
El compuesto BA se puede obtener a partir de AG por reacción con cloruro de tritilo en presencia de una base en un disolvente inerte a temperatura elevada con o sin irradiación de microondas. Una combinación adecuada de basedisolvente es, por ejemplo, trietilamina o DIEA y acetonitrilo, especialmente si la reacción se realiza a temperatura elevada en un reactor de microondas.
Los compuestos de fórmula general BC se pueden obtener a partir de compuestos de fórmula general BA por reacción con Boc-sulfamidato BB en un disolvente adecuado como DMF en presencia de una base adecuada como hidruro de sodio o ferc-butilato de potasio. La reacción se realiza de forma ventajosa a de 0 °C a temperatura ambiente.
Los compuestos de fórmula general I-a se pueden obtener a partir de compuestos de fórmula general BC por retirada de los grupos protectores por tratamiento con ácidos en un disolvente adecuado. Uno de dichos ácidos es TFA con o sin DCM adicional, usado a temperatura ambiente.

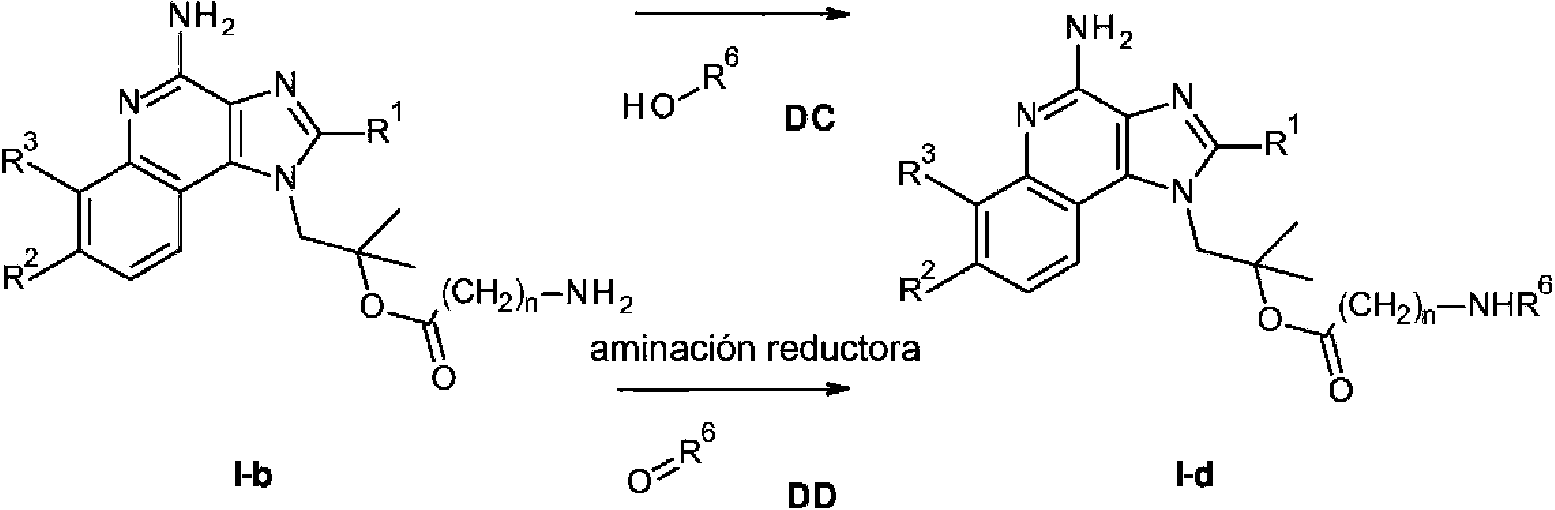
Después del procedimiento de acuerdo con el esquema 3, el compuesto AG se puede usar como material de partida para la síntesis de compuestos I-b donde R4 es -O-(CO)-(CH2)n-NH2.
El compuesto CA se puede obtener a partir de AG por introducción de un grupo protector GP adecuado por procedimientos conocidos en la técnica. Dichos grupos protectores pueden ser, por ejemplo, isoindolina-1,3-diona (ftalilo). Específicamente, el compuesto CA se puede hacer reaccionar con cloruro de ftaloílo en presencia de una base, como 1,4-diazabiciclo[2.2.2]octano, en un disolvente inerte, de alto punto de ebullición, tal como tolueno, a temperaturas elevadas para dar un compuesto de tipo CA con isoindolina-1,3-diona como grupo protector.
El compuesto CC se puede obtener a partir de los compuestos CB y CA por esterificación usando uno de los muchos procedimientos descritos en la técnica. De forma ventajosa, dicha esterificación se puede lograr combinando CB y CA en un disolvente inerte, como DCM, en presencia de W,W-diisopropil-carbodiimida a temperatura elevada. Los ácidos carboxílicos CB adecuados están disponibles comercialmente, se pueden preparar por procedimientos conocidos en la técnica o se han ejemplificado en la parte experimental.
Los compuestos de fórmula general I-b se pueden obtener a partir de compuestos de fórmula general CC por retirada de los grupos protectores con procedimientos conocidos en la técnica. Específicamente, la isoindolina-1,3-diona se puede retirar por tratamiento con hidracina/agua en un disolvente inerte, como THF, a temperatura ambiente.
Después del procedimiento de acuerdo con el esquema 4, los compuestos I-c e I-d se pueden obtener a partir de I-a o I-b por procedimientos conocidos en la técnica, como, por ejemplo, acoplamientos de amida (R5-OH o R6-OH son ácidos) o aminaciones reductoras (R5=O o R6=O son aldehídos) como se explica con más detalle en la parte experimental.
Esquema 4
procedimientos de

procedimientos de
aco lamiento de amida
Si uno de los materiales de partida, los compuestos de fórmula AA, AD, AG, DA, DB, DC o DD, contiene uno o más grupos funcionales que no son estables o son reactivos en las condiciones de reacción de una o más etapas de reacción, se pueden introducir grupos protectores (GP) apropiados (como se describe, por ejemplo, en T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3.a edición) antes de la etapa crítica aplicando procedimientos bien conocidos en la técnica. Dichos grupos protectores se pueden retirar en una fase posterior de la síntesis usando procedimientos estándar conocidos en la técnica.
Si uno o más compuestos de fórmula AA, AD, AG, DA, DB, DC o DD contienen centros quirales, los compuestos de fórmula I se pueden obtener como mezclas de diastereómeros o enantiómeros, que se pueden separar por procedimientos bien conocidos en la técnica, por ejemplo, HPLC (quiral) o cristalización. Se pueden separar los
compuestos racémicos, por ejemplo, en sus antípodas por medio de sales diastereómeras por cristalización o por separación de las antípodas por procedimientos cromatográficos específicos usando un adsorbente quiral o bien un eluyente quiral.
Como se describe en el presente documento anteriormente, los compuestos de fórmula I de la presente invención se pueden usar como medicamentos para el tratamiento de enfermedades que están mediadas por agonistas de TLR, en particular para el tratamiento de enfermedades que están mediadas por agonistas de TLR7 y/o TLR8, más en particular para el tratamiento de enfermedades que están mediadas por agonistas de TLR7 y TL8.
Los compuestos definidos en la presente invención son agonistas de receptores TLR7 y/o TLR8 en ensayos celulares in vitro. Más en particular, los compuestos de la presente invención son agonistas de ambos receptores TLR7 y TLR8. En consecuencia, se espera que los compuestos de la presente invención sean agentes potencialmente útiles en el tratamiento de enfermedades o afecciones médicas que se pueden beneficiar de la activación del sistema inmunitario por medio de agonistas de TLR7 y/o TLR8, más en particular en el tratamiento de enfermedades o afecciones médicas que se pueden beneficiar de la activación del sistema inmunitario por medio de ambos receptores TLR7 y TLR8. Por ejemplo, las siguientes enfermedades y afecciones se pueden tratar con compuestos de la presente invención.
Los compuestos de fórmula I de la presente invención son útiles en oncología, es decir, se pueden usar en el tratamiento de cánceres comunes que incluyen cáncer de vejiga, cáncer de cabeza y cuello, cáncer de próstata, cáncer colorrectal, cáncer de riñón, cáncer de mama, cáncer de pulmón, cáncer de ovario, cáncer de cuello uterino, cáncer de páncreas, cáncer de colon, cáncer de estómago, cáncer de tiroides, melanoma, tumores cutáneos y encefálicos y neoplasias malignas que afectan la médula ósea, tales como leucemias y sistemas linfoproliferativos, tales como linfoma hodgkiniano y linfoma no hodgkiniano; incluyendo la prevención y tratamiento del cáncer metastásico y las recidivas tumorales, y síndromes paraneoplásicos.
Los compuestos de fórmula I de la presente invención también son útiles en el tratamiento de enfermedades autoinmunitarias. Una "enfermedad autoinmunitaria" es una enfermedad o trastorno que surge de y se dirige contra los propios tejidos u órganos de un individuo o una cosegregación o manifestación del mismo o afección resultante del mismo. La "enfermedad autoinmunitaria" puede ser una enfermedad específica de un órgano (es decir, la respuesta inmunitaria se dirige específicamente contra un sistema o aparato tal como el sistema endocrino, el sistema hematopoyético, la piel, el aparato cardiorrespiratorio, el aparato digestivo y el sistema hepático, el aparato urinario, el tiroides, los oídos, el sistema neuromuscular, el sistema nervioso central) o una enfermedad sistémica que puede afectar múltiples sistemas o aparatos (por ejemplo, lupus eritematoso sistémico (LES), artritis reumatoide, polimiositis). En un aspecto particular, la enfermedad autoinmunitaria se asocia con la piel, tejido muscular y/o tejido conjuntivo.
Las enfermedades autoinmunitarias particulares incluyen trastornos reumatológicos autoinmunitarios (tales como, por ejemplo, artritis reumatoide, síndrome de Sjogren, esclerodermia, lupus tales como LES y nefritis lúpica, polimiositis/dermatomiositis, crioglobulinemia, síndrome de anticuerpos antifosfolipídicos y artropatía psoriásica), trastornos gastrointestinales y hepáticos autoinmunitarios (tales como, por ejemplo, enfermedades intestinales inflamatorias, colitis ulcerosa y enfermedad de Crohn), gastritis autoinmunitaria y anemia perniciosa, hepatitis autoinmunitaria, cirrosis biliar primaria, colangitis esclerosante primaria y enfermedad celíaca), vasculitis (tal como, por ejemplo, vasculitis no asociada a ANCA y vasculitis asociada a ANCA, incluyendo vasculitis de Churg-Strauss, granulomatosis de Wegener y poliangitis microscópica), trastornos neurológicos autoinmunitarios (tales como, por ejemplo, esclerosis múltiple, síndrome de opsoclonía-mioclonía, miastenia grave, neuromielitis óptica, enfermedad de Parkinson, enfermedad de Alzheimer y polineuropatías autoinmunitarias), trastornos renales (tales como, por ejemplo, glomerulonefritis, síndrome de Goodpasture y enfermedad de Berger), trastornos dermatológicos autoinmunitarios (tales como, por ejemplo, psoriasis, urticaria, ronchas, pénfigo vulgar, penfigoide ampolloso y lupus eritematoso cutáneo), trastornos hemáticos (tales como, por ejemplo, púrpura trombocitopénica, púrpura trombocitopénica trombótica, púrpura postransfusional y anemia hemolítica autoinmunitaria), ateroesclerosis, uveítis, enfermedades auditivas autoinmunitarias (tales como, por ejemplo, enfermedad del oído interno e hipoacusia), enfermedad de Behpet, síndrome de Raynaud, trasplante de órganos y trastornos endocrinos autoinmunitarios (tales como, por ejemplo, enfermedades autoinmunitarias relacionadas con diabetes tales como diabetes mellitus insulinodependiente (IDDM), enfermedad de Addison y enfermedad tiroidea autoinmunitaria (por ejemplo, enfermedad de Graves y tiroiditis)), afecciones y respuestas alérgicas, alergias alimentarias, alergias a fármacos, alergias a insectos, trastornos alérgicos raros tales como mastocitosis, reacción alérgica, eccema incluyendo eccema alérgico o atópico, asma tal como asma bronquial y asma autoinmunitario, afecciones que implican infiltración de linfocitos T y respuestas inflamatorias crónicas:
Los compuestos de fórmula I de la presente invención también son útiles en el tratamiento de enfermedades infecciosas. Por tanto, pueden ser útiles en el tratamiento de enfermedades víricas, en particular para enfermedades provocadas por infección con virus seleccionados del grupo que consiste en papilomavirus, tales como papilomavirus humano (PVH) y los que provocan condilomas acuminados, verrugas comunes y verrugas plantares, virus del herpes simple, molusco contagioso, virus de la hepatitis B (VHB), virus de la hepatitis C (VHC), virus del dengue, virus de la viruela, virus de inmunodeficiencia humana (VIH), citomegalovirus (CMV), virus del zóster y la
varicela (VZV), rinovirus, enterovirus, adenovirus, coronavirus (por ejemplo, SARS), gripe, paperas y virus paragripal. También pueden ser útiles en el tratamiento de enfermedades bacterianas, en particular para enfermedades provocadas por infección con bacterias seleccionadas del grupo que consiste en Mycobacterium tales como Mycobacterium tuberculosis, Mycobacterium avium y Mycobacterium leprae. Los compuestos de fórmula I de la presente invención pueden ser útiles además en el tratamiento de otras enfermedades infecciosas, tales como clamidia, enfermedades fúngicas, en particular enfermedades fúngicas seleccionadas del grupo que consiste en candidosis, aspergilosis y meningitis criptocócica, y enfermedades parasitarias tales como Pneumocystis carnii, neumonía, criptosporidiosis, histoplasmosis, toxoplasmosis, infección por Trypanosome y leishmaniosis.
Por tanto, la expresión "enfermedades que están mediadas por agonistas de TLR" significa enfermedades que se pueden tratar por la activación del sistema inmunitario con agonistas de TLR7 y/o TLR8 tales como cáncer y enfermedades infecciosas. En particular, la expresión "enfermedades que están mediadas por agonistas de TLR" significa cánceres o enfermedades autoinmunitarias o enfermedades infecciosas seleccionadas del grupo que consiste en enfermedades víricas, enfermedades bacterianas, enfermedades fúngicas y enfermedades parasitarias. En un aspecto particular, la expresión "que están mediadas por agonistas de TLR" se refiere al cáncer seleccionado del grupo que consiste en cáncer de vejiga, cáncer de cabeza y cuello, cáncer de próstata, cáncer colorrectal, cáncer de riñón, cáncer de mama, cáncer de pulmón, cáncer de ovario, cáncer de cuello uterino, cáncer de páncreas, cáncer de colon, cáncer de estómago, cáncer de tiroides, melanoma, tumores cutáneos y encefálicos y neoplasias malignas que afectan la médula ósea, tales como leucemias y sistemas linfoproliferativos, tales como linfoma hodgkiniano y linfoma no hodgkiniano; incluyendo la prevención y tratamiento del cáncer metastásico y las recidivas tumorales, y síndromes paraneoplásicos.
La invención también se refiere a composiciones farmacéuticas que comprenden un compuesto de fórmula I como se define anteriormente y un vehículo y/o coadyuvante farmacéuticamente aceptable. Más específicamente, la invención se refiere a composiciones farmacéuticas útiles para el tratamiento de enfermedades que están mediadas por agonistas de TLR.
Además, la invención se refiere a compuestos de fórmula I como se define anteriormente para su uso como sustancias terapéuticamente activas, en particular como sustancias terapéuticamente activas para el tratamiento de enfermedades que están mediadas por agonistas de TLR. En particular, la invención se refiere a compuestos de fórmula I para su uso en el tratamiento de cánceres o enfermedades autoinmunitarias o enfermedades infecciosas seleccionadas del grupo que consiste en enfermedades víricas, enfermedades bacterianas, enfermedades fúngicas y enfermedades parasitarias.
La invención se refiere además al uso de compuestos de fórmula I como se define anteriormente para el tratamiento de enfermedades que están mediadas por agonistas de TLR.
Además, la invención se refiere al uso de compuestos de fórmula I como se define anteriormente para la preparación de medicamentos para el tratamiento de enfermedades que están mediadas por agonistas de TLR. En particular, la invención se refiere al uso de compuestos de fórmula I como se define anteriormente para la preparación de medicamentos para el tratamiento de cánceres o enfermedades autoinmunitarias o enfermedades infecciosas seleccionadas del grupo que consiste en enfermedades víricas, enfermedades bacterianas, enfermedades fúngicas y enfermedades parasitarias.
En otro aspecto, los compuestos de fórmula I pueden estar en combinación con una o más modalidades de tratamiento adicionales en un régimen para el tratamiento del cáncer.
La politerapia engloba, además de la administración de un compuesto de la invención, el uso complementario de una o más modalidades que sean eficaces en el tratamiento del cáncer. Dichas modalidades incluyen, pero no se limitan a, agentes quimioterápicos, inmunoterápicos, agentes antiangiogénicos, citocinas, hormonas, anticuerpos, polinucleótidos, agentes terapéuticos fotodinámicos y de radiación. En un aspecto específico, se puede usar politerapia para prevenir la recidiva del cáncer, inhibir la metástasis o inhibir el crecimiento y/o diseminación del cáncer o metástasis. Como se usa en el presente documento, "en combinación con" significa que el compuesto de fórmula I se administra como parte de un régimen de tratamiento que comprende una o más modalidades de tratamiento adicionales como se menciona anteriormente. Por tanto, la invención también se refiere a un procedimiento para el tratamiento del cáncer, procedimiento que comprende administrar una cantidad terapéuticamente activa de un compuesto de fórmula I en combinación con uno o más de otros compuestos farmacéuticamente activos a un ser humano o animal.
Los compuestos de fórmula I se pueden usar solos o en combinación con una o más modalidades de tratamiento adicionales en el tratamiento de enfermedades autoinmunitarias.
La politerapia engloba, además de la administración de un compuesto de la invención, el uso complementario de una o más modalidades que ayudan en la prevención o tratamiento de enfermedades autoinmunitarias. Dichas
modalidades incluyen, pero no se limitan a, agentes quimioterápicos, inmunoterápicos, agentes antiangiogénicos, citocinas, hormonas, anticuerpos, polinucleótidos, agentes terapéuticos fotodinámicos y de radiación. Como se usa en el presente documento, "en combinación con" significa que el compuesto de fórmula I se administra como parte de un régimen de tratamiento que comprende una o más modalidades de tratamiento adicionales como se menciona anteriormente. Por tanto, la invención también se refiere a un procedimiento para el tratamiento de enfermedades autoinmunitarias, procedimiento que comprende administrar una cantidad terapéuticamente activa de un compuesto de fórmula I en combinación con uno o más de otros compuestos farmacéuticamente activos a un ser humano o animal.
En otro aspecto, los compuestos de fórmula I se pueden usar solos o en combinación con una o más modalidades de tratamiento adicionales en el tratamiento de enfermedades infecciosas.
La politerapia engloba, además de la administración de un compuesto de la invención, el uso complementario de una o más modalidades que ayudan en la prevención o tratamiento de enfermedades infecciosas. Dichas modalidades incluyen, pero no se limitan a, agentes antivíricos, antibióticos y agentes antifúngicos. Como se usa en el presente documento, "en combinación con" significa que el compuesto de fórmula I se administra como parte de un régimen de tratamiento que comprende una o más modalidades de tratamiento adicionales como se menciona anteriormente. Por tanto, la invención también se refiere a un procedimiento para el tratamiento de enfermedades infecciosas, procedimiento que comprende administrar una cantidad terapéuticamente activa de un compuesto de fórmula I en combinación con uno o más de otros compuestos farmacéuticamente activos a un ser humano o animal. PRUEBA FARMACOLÓGICA
Las siguientes pruebas se llevaron a cabo para determinar la actividad de los compuestos de fórmula I:
Para someter a prueba la actividad de TLR8 y TLR7, se usaron células de TLR8 o TLR7 humano HEK-Blue, respectivamente, (Invivogen, San Diego, CA, Ee . UU.) transfectadas con una construcción de indicador SEAP (fosfatasa alcalina embrionaria secretada), en las que se regula la expresión del indicador por el promotor NF-k B tras la estimulación durante 24 h. La actividad del indicador se determinó usando el kit Quanti Blue (Invivogen, San Diego, Ca, EE. UU.) a una longitud de onda de 640 nm.
Se determinaron los valores de CE50 usando el análisis Activity Base (ID Business Solution, Limited).
Los compuestos de acuerdo con la fórmula I tienen una actividad (valor de CE50) en el ensayo anterior para TLR8 humano en el intervalo de 0,01 nM a 11 pM, más en particular, de 0,01 nM a 3 pM y en el ensayo anterior para TLR7 humano en el intervalo de 0,01 nM a 1 pM, en particular, de 0,01 nM a 0,3 pM y más en particular, de 0,01 nM a 0,1 pM.
Por ejemplo, los siguientes compuestos mostraron los siguientes valores de CE50 en el ensayo descrito anteriormente:

COMPOSICIONES FARMACÉUTICAS
Los compuestos de fórmula I y sus sales farmacéuticamente aceptables se pueden usar como medicamentos, por ejemplo, en forma de preparaciones farmacéuticas para administración entérica, parenteral o tópica. Los compuestos de fórmula I y sus sales farmacéuticamente aceptables se pueden administrar por administración sistémica (por ejemplo, parenteral) o local (por ejemplo, inyección tópica o intralesional). En algunos casos, la formulación farmacéutica se administra por vía tópica, parenteral, oral, vaginal, intrauterina, intranasal o por inhalación. Como se describe en el presente documento, determinados tejidos pueden ser dianas preferentes para el agonista de TLR. Por tanto, la administración del agonista de TLR a ganglios linfáticos, bazo, médula ósea, sangre, así como tejido expuesto a virus, son sitios de administración preferentes.
En un aspecto, la formulación farmacéutica que comprende los compuestos de fórmula I o sus sales farmacéuticamente aceptables se administra por vía parenteral. Las vías de administración parenteral incluyen, pero no se limitan a, inyección transdérmica, transmucosa, nasofaríngea, pulmonar y directa. La administración parenteral por inyección puede ser por cualquier vía de inyección parenteral, incluyendo, pero sin limitarse a, vías intravenosa (i.v.), incluyendo intravenosa rápida e infusión (por ejemplo, rápida o lenta), intraperitoneal (i.p.), intramuscular (i.m.), subcutánea (s.c.) e intradérmica (i.d.). La administración transdérmica y transmucosa se puede lograr, por ejemplo, por inclusión de un vehículo (por ejemplo, dimetilsulfóxido, DMSO), por aplicación de impulsos eléctricos (por ejemplo, iontoforesis) o una combinación de los mismos. Están disponibles una variedad de dispositivos para la administración transdérmica que se pueden usar. Las formulaciones de los compuestos de fórmula I adecuadas para administración parenteral se formulan en general en agua USP o agua para inyectables y pueden comprender además tampones de pH, sales agentes voluminizadores, conservantes y otros excipientes farmacéuticamente aceptables.
La administración transdérmica se logra por aplicación de una crema, enjuague o gel que pueda permitir que el agonista de TLR penetre en la piel y entre en el torrente sanguíneo. Las composiciones adecuadas para administración transdérmica incluyen, pero no se limitan a, suspensiones, aceites, cremas y pomadas farmacéuticamente aceptables aplicados directamente en la piel o incorporados en un vehículo protector tal como un dispositivo transdérmico (denominado "parche"). Se pueden encontrar ejemplos de cremas o pomadas adecuadas, por ejemplo, en el Physician's Desk Reference. La transmisión transdérmica también se puede lograr por iontoforesis, por ejemplo usando parches disponibles comercialmente que administran su producto continuamente a través de la piel intacta durante períodos de varios días o más. El uso de este procedimiento permite la transmisión controlada de composiciones farmacéuticas en concentraciones relativamente grandes, permite la infusión de fármacos en combinación y permite el uso contemporáneo de un promotor de absorción. La administración por medio de las vías transdérmica y transmucosa puede ser continua o pulsátil.
La administración pulmonar se logra por inhalación, e incluye vías de administración tales como vías intranasal, transbronquial y transalveolar. Se proporcionan formulaciones de compuestos de fórmula I adecuadas para administración por inhalación incluyendo, pero sin limitarse a, suspensiones líquidas para formar aerosoles así como formas en polvo para sistemas de administración por inhalación de polvo seco. Los dispositivos adecuados para administración por inhalación incluyen, pero no se limitan a, atomizadores, vaporizadores, nebulizadores y dispositivos de administración por inhalación de polvo seco. Otros procedimientos de administración a la mucosa respiratoria incluyen la administración de formulaciones líquidas, tales como por gotas nasales. La administración por inhalación se logra preferentemente en dosis discretas (por ejemplo, por medio de un inhalador dosificador), aunque se puede lograr una administración similar a una infusión a través del uso de un nebulizador.
Los compuestos de fórmula I y sales farmacéuticamente aceptables de los mismos también se pueden administrar por vía oral, por ejemplo, en forma de comprimidos, comprimidos recubiertos, grageas, cápsulas de gelatina blanda y dura.
La producción de las preparaciones farmacéuticas se puede efectuar de manera que sea consabido para cualquier experto en la técnica llevando los compuestos descritos de fórmula I y sus sales farmacéuticamente aceptables, opcionalmente en combinación con otras sustancias terapéuticamente valiosas, a una forma de administración galénica conjuntamente con materiales de vehículo sólidos o líquidos terapéuticamente compatibles, inertes, no tóxicos, adecuados y, si se desea, coadyuvantes farmacéuticos habituales.
Los materiales de vehículo adecuados no son solo materiales de vehículo inorgánicos, sino también materiales de vehículo orgánicos. Por tanto, se pueden usar, por ejemplo, lactosa, almidón de maíz o derivados del mismo, talco, ácido esteárico o sus sales, como materiales de vehículo para comprimidos, comprimidos recubiertos, grageas y cápsulas de gelatina dura. Los materiales de vehículo adecuados para cápsulas de gelatina blanda son, por ejemplo, aceites vegetales, ceras, grasas y polioles líquidos y semisólidos (dependiendo de la naturaleza del ingrediente activo, sin embargo, puede no requerirse ningún vehículo en el caso de cápsulas de gelatina blanda). Los materiales de vehículo adecuados para la producción de soluciones y jarabes son, por ejemplo, agua, polioles, sacarosa, azúcar invertido. Los materiales de vehículo adecuados para soluciones inyectables son, por ejemplo, agua, alcoholes, polioles, glicerol y aceites vegetales. Los materiales de vehículo adecuados para supositorios son, por ejemplo, aceites naturales o hidrogenados, ceras, grasas y polioles líquidos o semilíquidos. Los materiales de
vehículo adecuados para preparaciones tópicas son glicéridos, glicéridos sintéticos y semisintéticos, aceites hidrogenados, ceras líquidas, parafinas líquidas, alcoholes grasos líquidos, esteroles, polietilenglicoles y derivados de celulosa.
Los estabilizantes, conservantes, agentes humectantes y emulsionantes, agentes de mejora de consistencia, agentes de mejora de sabor, sales para variar la presión osmótica, sustancias tamponadoras, solubilizantes, colorantes y agentes de enmascaramiento y antioxidantes habituales se tienen en cuenta como coadyuvantes farmacéuticos.
La dosificación de los compuestos de fórmula I puede variar dentro de límites amplios dependiendo de la enfermedad que se va a controlar, la edad y la condición individual del paciente y el modo de administración, y por supuesto, se ajustará a los requisitos individuales en cada caso particular. Para pacientes adultos, se tiene en cuenta una dosificación diaria de aproximadamente 1 a 1000 mg, en especial de aproximadamente 1 a 300 mg. Dependiendo de la gravedad de la enfermedad y el perfil farmacocinético preciso, el compuesto se podría administrar con una o varias unidades de dosificación diaria, por ejemplo, en de 1 a 3 unidades de dosificación.
Las preparaciones farmacéuticas contienen de forma práctica aproximadamente 1-500 mg, preferentemente 1 100 mg, de un compuesto de fórmula I.
Los siguientes ejemplos C1 a C3 ilustran composiciones típicas de la presente invención, pero sirven simplemente como representativos de las mismas.
Ejemplo C1
Se pueden fabricar comprimidos recubiertos con película que contienen los siguientes ingredientes de manera convencional:
Ingredientes Por comprimido
Núcleo:
Compuesto de fórmula I 10,0 mg 200,0 mg
Celulosa microcristalina 23,5 mg 43,5 mg
Lactosa hidratada 60,0 mg 70,0 mg
Povidona K30 12,5 mg 15,0 mg
Glicolato sódico de almidón 12,5 mg 17,0 mg
Estearato de magnesio 1,5 mg 4,5 mg
(Peso del núcleo) 120,0 mg 350,0 mg
Recubrimiento con película:
Hidroxipropilmetilcelulosa 3,5 mg 7,0 mg
Polietilenglicol 6000 0,8 mg 1,6 mg
Talco 1,3 mg 2,6 mg
Óxido de hierro (amarillo) 0,8 mg 1,6 mg
Dióxido de titanio 0,8 mg 1,6 mg
Se tamiza el ingrediente activo y se mezcla con celulosa microcristalina y se granula la mezcla con una solución de polivinilpirrolidona en agua. Se mezcla el granulado con glicolato sódico de almidón y estearato de magnesio y se comprime para proporcionar núcleos de 120 o 350 mg respectivamente. Se lacan los núcleos con una solución/suspensión acuosa del recubrimiento de película mencionado anteriormente.
Ejemplo C2
Se pueden fabricar cápsulas que contienen los siguientes ingredientes de manera convencional:
Ingredientes Por cápsula
Compuesto de fórmula I 25,0 mg
Lactosa 150,0 mg
Almidón de maíz 20,0 mg
Talco 5,0 mg
Se tamizan los componentes y se mezclan y rellenan en cápsulas de tamaño 2.
Ejemplo C3
Las soluciones inyectables pueden tener la siguiente composición:
Compuesto de fórmula I 3,0 mg
Polietilenglicol 400 150,0 mg
Ácido acético c.s. hasta pH 5,0
Agua para soluciones inyectables hasta 1,0 ml
Se disuelve el ingrediente activo en una mezcla de polietilenglicol 400 y agua para inyectables (parte). Se ajusta el pH a 5,0 por ácido acético. Se ajusta el volumen a 1,0 ml por adición de la cantidad residual de agua. Se filtra la solución, se rellena en viales usando un excedente apropiado y se esteriliza.
Los siguientes ejemplos sirven para ilustrar la presente invención con más detalle.
Ejemplos
Ejemplo 1
1-(2-(2-am¡noetox¡)-2-met¡lprop¡l)-2-(etox¡metil)-1H-¡m¡dazo[4.5-c1qu¡nol¡n-4-am¡na
a) 1-(2-(etox¡met¡l)-4-(tr¡t¡lam¡no)-1H-¡m¡dazo[4,5-c]quinol¡n-1-¡l)-2-met¡lpropan-2-ol

Se combinó 4-amino-2-(etoximetil)-a,a-dimetil-1H-imidazo[4,5-c]quinolina-1-etanol (CAN 144875-48-9, 1,6 g, 5,09 mmol) con acetonitrilo (60 ml) para dar una suspensión blanca. A continuación, se añadieron trietilamina (1,77 ml, 12,7 mmol) y cloruro de tritilo (1,7 g, 6,11 mmol) en argón con agitación. Se irradió la mezcla de reacción en un reactor de microondas a 100 °C durante 30 minutos. Tras agitar la mezcla, el producto precipitó y se aisló por filtración a 0 °C, se lavó con acetonitrilo frío y se secó para proporcionar el compuesto del título (2,37 g, 83 %) como un sólido blanco; EM (ESI): 557,5 (MH+).
b) 2-(1-(2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-iloxi)etilcarbamato de tere-butilo

Se combinó la dispersión de hidruro de sodio en aceite al 60 % (173 mg, 4,32 mmol) con DMF (15 ml) para dar una suspensión incolora. Se enfrió la mezcla a 0 °C con agitación, y a esta temperatura se añadió gota a gota una solución de 1-(2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ol (1,85 g, 3,32 mmol) en DMF (15 ml) durante un período de 10 min. Después, se agitó la mezcla de reacción durante 1 hora a 0 °C y durante 30 minutos a temperatura ambiente para dar una solución amarilla. A esta solución se le añadió a 0 °C éster 1,1-dimetiletílico del ácido 2,2-dióxido-1,2,3-oxatiazolidina-3-carboxílico (CAN 459817-82-4, 964 mg, 4,32 mmol). Se dejó elevar la temperatura hasta temperatura ambiente y se agitó la mezcla durante la noche. Se vertió la mezcla en agua con hielo y se extrajo con acetato de etilo. Se lavaron las capas orgánicas con agua/salmuera (2:1), se
combinaron, se secó sobre Na2SÜ4, se filtró y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 a 100 % de acetato de etilo en heptano) para dar el compuesto del título (1,47 g, 63 %) como una espuma blanca; CL-EM (área de pico de UV, ESI) 98,7 %, 700,3850 (MH+).
c) 1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina

Se combinó 2-(1-(2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-iloxi)etilcarbamato de ferc-butilo (1,45 g, 2,07 mmol) con diclorometano (DCM, 12 ml) para dar una solución incolora. Se añadió TFA (6,0 ml, 77,9 mmol) y se agitó la mezcla durante 3 horas a temperatura ambiente. Se enfrió la mezcla de reacción a 0 °C, se añadió solución de hidróxido de sodio 2 N (40 ml), y se extrajo la solución básica con diclorometano. Se combinaron las capas orgánicas, se secó sobre Na2SÜ4, se filtró y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 a 10 % de metanol en DCM) para dar el compuesto del título (0,62 g, 83 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 97,5 %, 358,2238 (MH+).
Ejemplo 2
2-aminoacetato de 1-(4-am¡no-2-(etox¡met¡l)-1H-¡m¡dazo[4.5-c1quinol¡n-1-¡l)-2-met¡lpropan-2-¡lo
a) 2-(2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5-c1quinolin-4-il)isoindolina-1,3-diona

Se combinó 4-am¡no-2-(etox¡met¡l)-a,a-dimet¡l-1H-¡m¡dazo[4,5-c]qu¡nol¡na-1-etanol (CAN 144875-48-9, 3,0 g, 9,54 mmol) con tolueno (21,0 ml) para dar una suspensión blanca. Se añadieron 1,4-diazabiciclo[2.2.2]octano (3,21 g, 28,6 mmol) y cloruro de ftaloílo (1,65 ml, 11,5 mmol) con agitación y se agitó la mezcla de reacción a 110 °C durante 4 horas. Después de enfriar, se diluyó la mezcla con acetato de etilo (300 ml) y se lavó con ácido clorhídrico 1 N. Se separaron las fases y se extrajo la capa acuosa con acetato de etilo. Se combinaron las capas orgánicas, se secó sobre MgSÜ4, se filtró y se concentró a vacío. Tras agitar el residuo con acetato de etilo (50 ml), el producto precipitó, se filtró y se secó a vacío (1,9 g). Se concentraron las aguas madres y se proporcionó, después de cromatografía ultrarrápida (gel de sílice, 50 g, de 0 % a 100 % de EtOAc en heptano), otro lote de producto (0,59 g). En total, se aislaron 2,49 g (59 %) del compuesto del título como un sólido blanco; CL-EM (área de pico de UV, ESI) 96 %, 445,2 (MH+).
b) 2-(1,3-dioxoisoindolin-2-il)acetato de 1-(4-(1,3-dioxoisoindolin-2-il)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo

Se combinaron 2-(2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-'/H-imidazo[4,5-c]quinolin-4-il)isoindolina-1,3-diona (1600 mg, 3,6 mmol), ácido 1,3-dihidro-1,3-dioxo-2H-isoindol-2-acético (CAN 4702-13-0, 2,22 g, 10,8 mmol) y 4-(1-pirrolidinil)-piridina (800 mg, 5,4 mmol) con DCM (36 ml) para dar una suspensión blanca. Se añadieron N,N'-diisopropilcarbodiimida (1,68 ml, 10,8 mmol) y tamices moleculares. Se calentó la mezcla de reacción a 50 °C y se agitó durante 2 horas y, después de enfriar, se filtró. Se diluyó el filtrado con DCM (150 ml) y se lavó con ácido clorhídrico 1 N y agua. Se extrajeron las fases acuosas con DCM, se combinaron las fases orgánicas, se secó sobre MgSO4, se filtró y se concentró a vacío.
Se retiró diisopropilurea después de la trituración con DCM/metanol por filtración, se concentró el filtrado y se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 % a 100 % de acetato de etilo en heptano) para dar un sólido amarillo claro. La cristalización con DMSO proporcionó una primera tanda de producto (1,95 g), y las aguas madres después de la concentración y HPLC preparativa otros 0,13 g de producto. En total, se aislaron 2,08 g (91 %) del compuesto del título como un sólido blanco; Cl-EM (área de pico de Uv , ESI) 95,9 %, 632,2157 (MH+). c) 2-aminoacetato de 1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo

Se combinó 2-(1,3-dioxoisoindolin-2-il)acetato de 1-(4-(1,3-dioxoisoindolin-2-il)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo (300 mg, 475 pmol) con THF (5 ml) para dar una suspensión blanca. A esta suspensión se le añadió hidracina en agua (1000 pl, 11,2 mmol) y se agitó la solución durante 0,75 horas a temperatura ambiente. Se enfrió la mezcla hasta 0 °C, se añadieron ácido clorhídrico 1 N (30 ml) y DCM (50 ml). Se retiró el precipitado blanco a (ftalilhidracida) por filtración y se desechó la fase orgánica. Se liofilizó la fase acuosa y se unió el residuo con acetonitrilo (20 ml) y DIEA (pH 10). Después de la evaporación del disolvente, se purificó el residuo por HPLC preparativa (Gemini NX 3u 50x4,6 mm; acetonitrilo/agua/EtsN) para dar el compuesto del título (111 mg, 63 %) como un sólido amorfo blanco; CL-EM (área de pico de UV, ESI) 99 %, 372,4 (MH+).
Ejemplo 3
N-(2-(1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-iloxiletil)nicotinamida

Se combinó ácido nicotínico (11,4 mg, 92,3 pmol) con DMF (1,0 ml) para dar una solución incolora. A esta solución se le añadió TBTU (29,6 mg, 92,3 pmol) y DIeA (54,2 mg, 71,8 pl). Finalmente, se añadió 1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina (30 mg, 83,9 pmol) y se agitó la mezcla de reacción durante 1 hora a temperatura ambiente. Se retiraron los volátiles a alto vacío a 40 °C. Se disolvió el residuo en una mezcla de acetato de etilo (5 ml) y metanol (0,5 ml). Se añadió solución de hidróxido de sodio 1 N (1,5 ml) y después de unos minutos de agitación se separaron las capas. Se extrajo la capa acuosa con acetato de etilo. Se combinaron las fases orgánicas, se secó sobre Na2SÜ4, se filtró y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice-NH2, de 0 % a 10 % de metanol en DCM) para dar el producto del título (33 mg, 85 %) como una espuma blanca; CL-EM (área de pico de UV, ESI) 88 %, 463,2459 (MH+).
Ejemplo 4
N-(2-(1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-iloxi)etil)acetamida

Se combinó 1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina (32 mg, 89,5 pmol) con DCM (1,0 ml) para dar una solución incolora. A esta solución se le añadió trietilamina (25,0 pl, 179 pmol) y cloruro de acetilo (7,00 pl, 98,5 pmol) y se agitó la mezcla de reacción 18 horas a temperatura ambiente. Después de concentrar la mezcla a vacío, se purificó el residuo por cromatografía ultrarrápida (gel de sílice-NH2 , de 0 % a 10 % de metanol en DCM) para dar el compuesto del título (19 mg, 53 %) como una espuma blanca; CL-EM (área de pico de UV, ESI) 97 %, 400,2348 (MH+).
Ejemplo 5
3-(2-(1-(4-am¡no-2-(etox¡met¡l)-1H-¡m¡dazo[4.5-c1qu¡nol¡n-1-¡l)-2-met¡lpropan-2-¡lox¡)et¡lam¡no)propan-1-ol a) 2-(etoximetil)-1-[2-metil-2-[2-(3-tritiloxipropilamino)etoxi]propil]-imidazo[4,5-c]quinolin-4-amina

Se combinaron 1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina (50 mg, 140 pmol) y 3-(trifenilmetoxi)-propanal (CAN 67057-68-5; 44,3 mg, 140 pmol) con etanol (500 pl) para dar una suspensión amarillo claro. Se agitó la mezcla durante 2 horas a temperatura ambiente. Después, se añadió borohidruro de sodio (5,82 mg, 154 pmol) y continuó la agitación a temperatura ambiente durante la noche. Se agitó la mezcla con agua (2 ml) y acetato de etilo (5 ml), se secó por paso a través de un cartucho ChemElut® y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice-NH2, de 0 % a 100 % de acetato de etilo en
heptano) seguido de otra cromatografía ultrarrápida (gel de sílice, de 0 a 10 % de metanol en acetato de etilo) para dar el compuesto del título (10 mg, 11 %) como una goma amarillenta; CL-EM (área de pico de UV, ESI) 89 %, 658,5 (MH+).
b) 3-(2-(1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-iloxi)etilamino)propan-1-ol

Se combinó 2-(etoximetil)-1-[2-metil-2-[2-(3-tritiloxipropilamino)etoxi]propil]-imidazo[4,5-c]quinolin-4-amina (10 mg, 15,2 pmol) con DCM (1 ml) para dar una solución incolora. Se añadió TFA (100 pl, 1,3 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 5 horas. Se concentró la mezcla a vacío y se purificó el residuo por cromatografía ultrarrápida (gel de sílice-NH2, de 0 % a 30 % de metanol en acetato de etilo) para dar el compuesto del título (2,3 mg, 36 %) como una goma incolora; CL-EM (área de pico de UV, ESI) 95,6 %, 416,2661 (MH+).
Ejemplo 6
6-(2-(1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c1quinolin-1-il)-2-metilpropan-2-iloxi)etilamino)-6-oxohexilcarbamato de tere-butilo

Se combinó ácido 6-(fere-butoxicarbonilamino)hexanoico (197 mg, 850 pmol) con DMF (10,9 ml) para dar una solución incolora. Se añadieron tetrafluoroborato de 0-(benzotriazol-1-il)-W,W,W,W-tetrametiluronio (TBTU, 300 mg, 936 pmol) y W,W-diisopropiletilamina (DIEA, 728 pl, 4,25 mmol) con agitación en una atmósfera inerte. A continuación, se añadió 1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina (ejemplo 1c, 304 mg, 850 pmol) y se agitó la mezcla durante 2 horas a temperatura ambiente. Se concentró la mezcla a vacío, se disolvió en acetato de etilo (5 ml), se agitó durante 1 min con solución fría de hidróxido de sodio (1 N) y se secó pasando a través de ChemElut® (10 g). Se concentró la fase orgánica a vacío nuevamente y se purificó el residuo por cromatografía ultrarrápida (gel de sílice, metanol al 10 % en diclorometano) para dar el compuesto del título (0,135 g, 23 %) como un aceite marrón claro; CL-EM (área de pico de UV, ESI) 83 %, 571,5 (MH+).
Ejemplo 7
3-(4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c1quinolin-7-il)acrilato de (E)-etilo a) 5-amino-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazol-4-carbonitrilo

Se añadió trietilamina (5,28 ml, 37,9 mmol) con agitación a una suspensión de 4-metilbencenosulfonato de 2aminomalononitrilo (8 g, 31,6 mmol) en THF (120 ml) para dar una solución marrón claro. Se añadió 1,1,1,2-tetraetoxietano (7,82 g, 37,9 mmol) y se agitó la mezcla de reacción en argón a temperatura de reflujo. Después de 4 h, se añadió una cantidad adicional de 2,3 g de 1,1,1,2-tetraetoxietano y se calentó la mezcla durante otras 2 h. Se dejó enfriar la mezcla hasta temperatura ambiente, se añadieron trietilamina (5,28 ml, 37,9 mmol) y 1-amino-2-metilpropan-2-ol (3,63 ml, 37,9 mmol) y se agitó la reacción durante la noche. Se concentró la mezcla a vacío, y se repartió el residuo entre acetato de etilo (200 ml, 2x150 ml) y solución de bicarbonato de sodio (2 M, 100 ml). Se combinaron las fases orgánicas, se secó sobre MgSO4, se concentró a vacío y se purificó por cromatografía ultrarrápida (gel de sílice, de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título después de una etapa de cristalización con heptano (4,47 g, 59 %) como un sólido cristalino blanco; CL-EM (área de pico de UV, ESI) 100 %, 239,1514 (MH+).
b) 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-5-yodo-1H-imidazol-4-carbonitrilo

Se añadió diyodometano (14,5 ml, 180 mmol) en argón con agitación a una suspensión de 5-amino-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazol-4-carbonitrilo (4,36 g, 18,3 mmol) en cloroformo (170 ml). Se calentó la mezcla de reacción hasta 80 °C, se añadió una solución de nitrito de isoamilo (9,86 ml, 73,2 mmol) en cloroformo (30 ml) con una bomba de jeringa durante un período de 40 min y se agitó la mezcla durante otros 30 min a 80 °C. Después de enfriar a temperatura ambiente, se concentró la mezcla a vacío y se purificó por cromatografía ultrarrápida (gel de sílice, de 20 % a 50 % de acetato de etilo en heptano) para dar el compuesto del título (3,66 g, 57 %) como un aceite marrón; CL-EM (área de pico de UV, ESI) 90 %, 350,0374 (MH+).
c) 3-amino-4-[5-ciano-2-(etoximetil)-3-(2-hidroxi-2-metilpropil)-1H-imidazol-4-il]benzoato de metilo

En una atmósfera inerte se combinó 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-5-yodo-1H-imidazol-4-carbonitrilo (3,66 g, 10,5 mmol) con dimetoxietano (43 ml) para dar una solución marrón claro. A esta solución se le añadieron Pd(OAc)2 (118 mg, 524 pmol), trifenilfosfina (275 mg, 1,05 mmol) y 3-amino-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzoato de metilo (4,36 g, 15,7 mmol) con agitación. Finalmente se añadió Na2CO3 (2 M, 15,7 ml, 31,4 mmol). Se calentó la mezcla de reacción hasta 100 °C y se agitó durante 1 hora. Después de enfriar hasta temperatura ambiente, se repartió la mezcla entre acetato de etilo (70 ml, 2x50 ml) y agua (70 ml). Se combinaron las fases orgánicas, se secó sobre MgSO4, se concentró a vacío y se purificaron por cromatografía ultrarrápida (gel de sílice, de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (2,79 g, 71 %) como espuma marrón claro que se usó en la siguiente etapa sin purificación adicional.
d) 4-amino-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5-c]quinolina-7-carboxilato de metilo

Se combinó 3-amino-4-(4-ciano-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazol-5-il)benzoato de metilo (2,79 g, 7,49 mmol) con una solución de HCl en dioxano (4 M, 56,2 ml, 225 mmol) para dar una solución naranja. Se calentó la mezcla de reacción hasta 90 °C en argón con agitación. Después de 1 h a 90 °C, se enfrió la mezcla hasta temperatura ambiente y se concentró a vacío para obtener un sólido beige. Se disolvió el sólido en acetato de etilo (500 ml), se lavó con una mezcla de agua (100 ml) y una solución saturada de bicarbonato de sodio (250 ml). Se extrajo la fase acuosa con acetato de etilo (2x250 ml), se combinaron las fases orgánicas, se secó con MgSO4 y se concentró a vacío para obtener un sólido amarillo (3,05 g). Se recristalizó el material sólido en acetato de etilo/heptano para dar el compuesto del título (2,4 g, 86 %) como un sólido amarillo claro; CL-EM (área de pico de UV, ESI) 99 %, 373,1884 (MH+).
e) 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxilato de metilo

Se sintetizó el compuesto del título de forma análoga al ejemplo 1a, usando 4-amino-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5-c]quinolina-7-carboxilato de metilo como material de partida y se aisló (0,77 g, 96 %) como un sólido marrón claro; Cl-EM (área de pico de UV, ESI) 96 %, 615,4 (MH+).
f) ácido 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxílico

Se añadió solución de hidróxido de sodio (1 N, 3,51 ml) a 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxilato de metilo (540 mg, 878 pmol) disuelto en THF (6,21 ml) y metanol (621 pl). Se agitó la mezcla de reacción durante 120 horas a temperatura ambiente y se concentró a vacío. Se repartió el residuo entre acetato de etilo (30 ml) y solución fría de HCl 1 M (5 ml). Se extrajo la capa acuosa con acetato de etilo (2x 50 ml). Se lavaron las capas orgánicas con agua (20 ml) y salmuera (20 ml), se combinaron, se secó con MgSÜ4, y se concentró a vacío para obtener el compuesto del título (0,499 g, 94 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 99 %, 601,3 (MH+).
g) 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-W-metoxi-W-metil-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxamida

A una solución de ácido 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxílico (484 mg, 741 pmol) en DMF (5,19 ml) en una atmósfera inerte se le añadió TBTU (357 mg, 1,11 mmol), DIEA (388 pl, 2,22 mmol) y clorhidrato de W,0-dimetilhidroxilamina (108 mg, 1,11 mmol) con agitación. Se agitó la mezcla de reacción durante 1 h a temperatura ambiente y después, se concentró la mezcla a vacío. Se repartió el residuo entre acetato de etilo (40 ml, 2x20 ml) y agua (40 ml). Se lavaron las fases orgánicas con agua (2x20 ml), se combinaron, se secó sobre MgSÜ4, se concentró a vacío y se purificó por cromatografía ultrarrápida (gel de sílice, de 50 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (0,53 g, cuant.) como una espuma blanca; CL-EM (área de pico de UV, ESI) 92 %, 644,4 (MH+).
h) 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carbaldehído

A una solución de 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-W-metoxi-W-metil-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carboxamida (520 mg, 808 pmol) en THF (7 ml) en una atmósfera inerte se le añadió con agitación a 0 °C una solución de hidruro de litio y aluminio en THF (1 M, 404 pl, 404 pmol). Se agitó la mezcla 1 h a 0 °C, se añadió lentamente solución saturada de cloruro de amonio (10 ml) y finalmente se añadieron agua (20 ml) y acetato de etilo (30 ml). Se separaron las fases, se extrajo la capa acuosa con acetato de etilo (30 ml) y se lavaron las fases orgánicas con agua (20 ml), se combinaron, se secó con MgSÜ4, y se concentró a vacío para dar el compuesto del título (0,49 g, cuant.) como una espuma blanca (CL-EM (área de pico de UV, ESI) 83 %, 585,4 (MH+)) que se usó en la siguiente etapa sin purificación adicional.
i) (E)-3-[2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il]prop-2-enoato de etilo

Se añadió (carbetoximetilen)trifenilfosforano (422 mg, 1,21 mmol) a una solución de 2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolina-7-carbaldehído (472 mg, 808 pmol) en diclorometano (8 ml) en argón. Se agitó la mezcla de reacción durante 20 h a temperatura ambiente y se concentró a vacío. La cristalización en diclorometano y éter dietílico produjo el compuesto del título (0,399 g, 75 %) como un sólido blanco, CL-EM (área de pico de UV, ESI) 89 %, 655,4 (MH+).
j) (E)-3-[1-[2-[2-(terc-butoxicarbonilamino)etoxi]-2-metilpropil]-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin7-il]prop-2-enoato de etilo

Se sintetizó el compuesto del título de forma análoga al ejemplo 1b, usando (E)-3-[2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il]prop-2-enoato de etilo como material de partida y se aisló (0,28 g, 42 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 76 %, 798,6 (MH+).
k) (E)-3[4-am¡no-1-[2-(2-am¡noetox¡)-2-met¡lprop¡l1-2-(etox¡met¡l)¡m¡dazo[4.5-c1qu¡nol¡n-7-¡l]prop-2-enoato de etilo

Se sintetizó el compuesto del titulo de forma análoga al ejemplo 1c, usando (E)-3-[1-[2-[2-(fercbutoxicarbonilamino)etoxi]-2-metilpropil]-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il]prop-2-enoato de etilo como material de partida y se aisló (21 mg, 34 %) como un sólido blanco; CL-Em (área de pico de UV, ESI) 93 %, 456,4 (MH+).
Ejemplo 8
Clorhidrato de 3-(4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo
a) 3-[1-[2-[2-(ferc-butoxicarbonilamino)etoxi]-2-metilpropil]-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il]propanoato de etilo

Se añadió paladio sobre carbono al 10 % (100 mg, 940 pmol) en etanol (10 ml) a una suspensión de 3-(1-(2-(2-(tercbutoxicarbonilamino)etoxi)-2-metilpropil)-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il)acrilato de (E)-etilo (280 mg, 351 pmol) en acetato de etilo (30 ml). Se agitó la mezcla durante 6 h en una atmósfera de hidrógeno a temperatura ambiente, se filtró y se retiró el disolvente a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice-NH2 , de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (0,163 g, 58 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 74 %, 800,5 (MH+).
b) Clorhidrato____ de____ 3-(4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo

Se sintetizo el compuesto del titulo de forma analoga al ejemplo 1c, usando 3-[1-[2-[2-(terc-butoxicarbonilamino)etoxi]-2-metilpropil]-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-7-il]propanoato de etilo como mater¡al de part¡da y se a¡sló (15 mg, 56 %) como un sól¡do blanco; CL-EM (area de p¡co de UV, ESI) 93 %, 458,4 (MH+).
Ejemplo 9
3-(4-amino-1-(2-(2-aminoacetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo a) 3-[4-[bis(benciloxicarbonil)amino]-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5-c]quinolin-7-il]propanoato de etilo

A una suspensión de clorhidrato de 3-(4-amino-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo (300 mg, 665 pmol), DMAP (15 mg, 123 pmol) y DIEA (1,86 ml, 10,6 mmol) en diclorometano (6 ml), se le añadió cloroformiato de bencilo (380 pl, 2,66 mmol). Se agitó la mezcla de reacción a temperatura ambiente durante 20 h, se añadió mas cloroformiato de bencilo (908 60 pl, 5,32 mmol) y se continuó la agitación durante otras 6 h. Después, se vertió la mezcla en diclorometano (50 ml), se extrajo con HCl (1 M, 20 ml) y agua (20 ml). Se extrajo la capa acuosa con diclorometano (50 ml), se combinaron las fases orgánicas, se secó con MgSÜ4 y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (0,459 g, cuant.) como un aceite incoloro; CL-EM (área de pico de UV, ESI) 86 %, 683,5 (MH+).
b) 3-[4-(benciloxicarbonilamino)-1-[2-[2-(dibencilamino)acetil]oxi-2-metilpropil]-2-(etoximetil)-1H-imidazo[4,5-c1qu¡nol¡n-7-¡l1propanoato de etilo

A una suspensión de 3-(4-(bis(benciloxicarbonil)amino)-2-(etoximetil)-1-(2-hidroxi-2-metilpropil)-1H-imidazo[4,5
c]quinolin-7-il)propanoato de etilo (300 mg, 439 pmol), ácido 2-(dibencilamino)acético (337 mg, 1,32 mmol) y DMAP (107 mg, 879 pmol) en diclorometano (4 ml) se le añadió 1-etil-3-(3-dimetilaminopropil)carbodiimida (253 mg, 1,32 mmol) y tamices moleculares (300 mg). Después de 1 h de agitación a temperatura ambiente, se disolvieron los materiales de partida, se continuó la agitación a temperatura ambiente durante 6 h y se dejó la mezcla en reposo durante el fin de semana. Se diluyó el residuo con diclorometano (50 ml) y se lavó con HCl (1 M, 20 ml) y agua (20 ml). Se extrajo la fase acuosa con diclorometano (50 ml), se combinaron las fases orgánicas, se secó con MgSÜ4 y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (182 mg, 52 %) como un aceite amarillo claro; CL-EM (área de pico de UV, ESI) 98 %, 784,6 (MH+).
c) 3-(4-amino-1-(2-(2-aminoacetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo

Se combinó 3-(4-(benciloxicarbonilamino)-1-(2-(2-(dibencilamino)acetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo (182 mg, 232 pmol) con acetato de etilo (20 ml) para dar una solución amarillo claro. Se añadió paladio sobre carbono al 10 % (600 mg, 232 pmol) y se agitó la mezcla durante 20 h a temperatura ambiente en H2. Después, se filtró la mezcla a través de Celite® y se concentró a vacío. Se purificó el material bruto por cromatografía ultrarrápida (gel de sílice, 20 g, de 0 % a 30 % de metanol en THF) hasta 27 mg de una muestra impura que se purificó adicionalmente por HPLC preparativa para dar el compuesto del título (6 mg, 5 %) como un aceite amarillo claro; CL-EM (área de pico de UV, ESI) 79 %, 516,5 (MHCOO').
Ejemplo 10
1-(2-(2-aminoetoxi)-2-metilpropil)-2-pentil-1H-imidazo[4,5-c]quinolin-4-amina
a) 2-metil-1-(2-pentil-1H-imidazo[4,5-c]quinolin-1-il)propan-2-ol

A una solución de 1-(3-aminoquinolin-4-ilamino)-2-metilpropan-2-ol (CAN 129655-59-0, 0,94 g, 4,06 mmol) en diclorometano (9,5 ml) en una atmósfera inerte se le añadió gota a gota con agitación a temperatura ambiente durante un período de 6 min, una solución de cloruro de hexanoílo (422 pl, 4,47 mmol) en diclorometano (6,4 ml). Después de 3 h, se añadió cloruro de hexanoílo adicional (192 pl, 2,03 mmol) y se continuó la agitación durante otra hora y finalmente, se concentró la mezcla a vacío. Se volvió a disolver el residuo en etanol (13,5 ml) y, después de la adición de solución de hidróxido de sodio (1 M, 10,8 ml), se calentó la mezcla con agitación en argón hasta 90 °C. Después de 75 min, se dejó enfriar la mezcla hasta temperatura ambiente y se concentró a vacío. Se agitó el residuo con acetato de etilo (30 ml) y agua (5 ml), se secó por filtración sobre ChemElut® y se retiró el acetato de etilo a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice-NH2 , de 0 % a 100 % de acetato de etilo en heptano) para dar el compuesto del título (747 mg, 89 %) como un aceite amarillo; CL-EM (área de pico de UV, ESI) 95 %, 312,2 (MH+).
b) 2-metil-1-(5-óxido-2-pentil-1H-imidazo[4,5-c]quinolin-5-io-1-il)propan-2-ol

A una solución de 2-metil-1-(2-pentil-1H-imidazo[4,5-c]quinolin-1-il)propan-2-ol (457 mg, 1,36 mmol) en diclorometano (21,2 ml) se le añadió con agitación ácido 3-cloroperoxibenzoico (283 mg, 1,64 mmol) en una porción. Se agitó la mezcla de reacción durante 16 h a temperatura ambiente. Después, se repartió la mezcla entre diclorometano frío (50 ml) y solución de hidróxido de sodio (1 M, 20 ml). Se lavó la fase orgánica con agua fría (20 ml) y salmuera (20 ml) y se extrajeron las fases acuosas con diclorometano (2x50 ml). Se combinaron todas las fases orgánicas, se secó con MgSO4, se filtró y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 % a 20 % de metanol en diclorometano) para dar el compuesto del título (360 mg, 80 %) como un sólido amarillo claro; CL-EM (área de pico de UV, ESI) 99 %, 328,2 (MH+).
c) 1-(4-amino-2-pentil-1H-imidazo[4,5-c]quinolin-1-il)-2-metil-propan-2-ol

A una solución de 2-metil-1-(5-óxido-2-pentil-1H-imidazo[4,5-c]quinolin-5-io-1-il)propan-2-ol (353 mg, 1,08 mmol) en diclorometano anhidro (47,6 ml) se le añadió isocianato de benzoílo (90 %, 286 pl, 2,05 mmol) con agitación en atmósfera de argón a temperatura ambiente. Se agitó la mezcla de reacción a temperatura de reflujo durante 35 min. A continuación, se concentró la mezcla a vacío. A continuación, se combinó el residuo con metanol anhidro (17,0 ml) para dar una suspensión blanca. Se añadió solución de metóxido de sodio en metanol (1,7 ml, 9,16 mmol) y se agitó la mezcla durante 1 h a 70 °C. Después, se enfrió la mezcla hasta temperatura ambiente y se retiraron los disolventes a vacío. Se repartió el residuo entre acetato de etilo (30 ml) y agua (15 ml), se secó por paso a través de un cartucho ChemElut® y se concentró a vacío. Se purificó el residuo por cromatografía ultrarrápida (gel de sílice, de 0 % a 10 % de metanol en diclorometano) para dar el compuesto del título (221 mg, 61 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 98 %, 327,2185 (MH+).
d) 2-metil-1-[2-pentil-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]propan-2-ol

Se sintetizó el compuesto del título de forma análoga al ejemplo 1a, usando 1-(4-amino-2-pentil-1H-imidazo[4,5-c]quinolin-1-il)-2-metil-propan-2-ol como material de partida y se aisló (230 mg, 71 %) como un sólido cristalino blanco; CL-EM (área de pico de UV, ESI) 99 %, 569,3 (MH+).
f) N-[2-[1,1-dimetil-2-[2-pentil-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]etoxi]etil]carbamato de tere-butilo

Se sintetizó el compuesto del título de forma análoga al ejemplo 1b, usando 2-metil-1-[2-pentil-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]propan-2-ol como material de partida y se aisló (306 mg, cuant.) como un aceite amarillo claro; CL-EM (área de pico de UV, ESI) 53 %, 712,2 (MH+).
g) 1-(2-í2-aminoetoxi)-2-metilpropil)-2-pentil-1H-imidazo[4,5-c]quinolin-4-amina
Se sintetizó el compuesto del título de forma análoga al ejemplo 1c, usando W-[2-[1,1-dimetil-2-[2-pentil-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]etoxi]etil]carbamato de tere-butilo como material de partida y se aisló (30 mg, 36 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 98 %, 370,2622 (MH+).
Ejemplo 11
Clorhidrato de 1-(2-(2-aminoetoxi)-2-metilpropil)-7-bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina a) 1-[7-bromo-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]-2-metilpropan-2-ol

Se sintetizó el compuesto del título de forma análoga al ejemplo 1a, usando 1-(4-amino-7 bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ol como material de partida y se aisló (100 mg, 88 %) como un sólido cristalino marrón claro; CL-EM (área de pico de UV, ESI) 95 %, 637,3 (MH+).
b) N-[2-[2-[7-bromo-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]-1,1-dimetil-etoxil]etil]carbamato de tere-butilo

Se sintetizó el compuesto del título de forma análoga al ejemplo 1b, usando 1-[7-bromo-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]-2-metilpropan-2-ol como material de partida y se aisló (20 mg, 44 %) como un sólido blanco que se usó sin purificación adicional en la siguiente etapa.
c) Clorhidrato de 1-(2-(2-aminoetoxi)-2-metilpropil)-7-bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina

Se sintetizó el compuesto del título de forma análoga al ejemplo 1c, usando W-[2-[2-[7-bromo-2-(etoximetil)-4-(tritilamino)-1H-imidazo[4,5-c]quinolin-1-il]-1,1-dimetil-etoxi]etil]carbamato de tere-butilo como material de partida y se aisló (7 mg, 58 %) como un sólido blanco; CL-EM (área de pico de UV, ESI) 99 %, 438,2 (MH+).
Claims (11)
1. Un compuesto de fórmula

en la que
R1 es alquilo C1-7 o alcoxi Cwalquilo C1-7;
R2 se selecciona del grupo que consiste en hidrógeno, halógeno, alcoxicarbonil Cwalquilo C1-7 y alcoxicarbonil C1-7-alquenilo C2-7;
R3 es hidrógeno;
R4 se selecciona del grupo que consiste en
-O-(CH2)m-NHR5 y
-O-(CO)-(CH2)n-NHR6,
en las que
m es 2,
n es 1,
R5 es hidrógeno y
R6 es hidrógeno;
o sales farmacéuticamente aceptables del mismo.
2. Un compuesto de fórmula I de acuerdo con la reivindicación 1, en la que R1 es alcoxi C1-7-alquilo C1-7.
3. Un compuesto de fórmula I de acuerdo con las reivindicaciones 1 o 2, en la que R1 es etoxietilo.
4. Un compuesto de fórmula I de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en la que R2 es hidrógeno.
5. Un compuesto de fórmula I de acuerdo con la reivindicación 1, seleccionado del grupo que consiste en
1- (2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
2- aminoacetato de 1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo,
(E)-3-[4-amino-1-[2-(2-aminoetoxi)-2-metilpropil]-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il]prop-2-enoato de etilo, 3- (4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, 3-(4-amino-1-(2-(2-aminoacetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, 1-(2-(2-aminoetoxi)-2-metilpropil)-2-pentil-1H-imidazo[4,5-c]quinolin-4-amina,
1-(2-(2-aminoetoxi)-2-metilpropil)-7-bromo-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
y sales farmacéuticamente aceptables del mismo.
6. Un compuesto de fórmula I de acuerdo con la reivindicación 1, que es
1- (2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-4-amina,
y sales farmacéuticamente aceptables del mismo.
7. Un compuesto de fórmula I de acuerdo con la reivindicación 1, seleccionado del grupo que consiste en
2- aminoacetato de 1-(4-amino-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-1-il)-2-metilpropan-2-ilo,
3- (4-amino-1-(2-(2-aminoetoxi)-2-metilpropil)-2-(etoximetil)-1H-imidazo[4,5-c]quinolin-7-il)propanoato de etilo, y sales farmacéuticamente aceptables del mismo.
8. Un compuesto de fórmula I de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso como medicamento.
9. Un compuesto de fórmula I de acuerdo con una cualquiera de las reivindicaciones 1 a 7 para su uso como medicamento para el tratamiento de cánceres o enfermedades autoinmunitarias o enfermedades infecciosas seleccionadas del grupo que consiste en enfermedades víricas, enfermedades bacterianas, enfermedades fúngicas y enfermedades parasitarias.
10. Una composición farmacéutica que comprende un compuesto de fórmula I de acuerdo con una cualquiera de las reivindicaciones 1 a 7 y un vehículo y/o coadyuvante farmacéuticamente aceptable.
11. Un procedimiento para la fabricación de un compuesto de fórmula I como se define en la reivindicación 1, procedimiento que comprende
a) hacer reaccionar un compuesto de fórmula II

en la que R1, R2 y R3 son como se define en la reivindicación 1 y GP es un grupo protector, con un compuesto de fórmula III

en la que m es como se define en la reivindicación 1, en condiciones básicas, y retirar los grupos protectores GP y Boc en condiciones ácidas para obtener un compuesto de fórmula I-a

en la que de R1 a R3 y m son como se define en la reivindicación 1, y opcionalmente acoplar además el compuesto de fórmula I-a con un alcohol o ácido de fórmula R5-OH o un aldehído de fórmula R5=O para obtener un compuesto de fórmula I-c

en la que de R1 a R3, m y R5 son como se define en la reivindicación 1 y, si se desea, convertir el compuesto obtenido en una sal farmacéuticamente aceptable, o
b) hacer reaccionar un compuesto de fórmula Il-a

en la que R1, R2 y R3 son como se define en la reivindicación 1 y GP' es un grupo protector, con un ácido carboxílico de fórmula IV
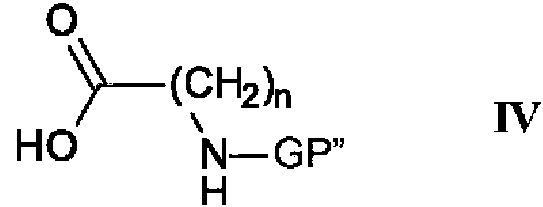
en la que n es como se define en la reivindicación 1 y GP" es un grupo protector, en presencia de un agente de esterificación, y retirar los grupos protectores GP' y GP" con un agente reductor suave para obtener un compuesto de fórmula I-b

en la que de R1 a R3 y n son como se define en la reivindicación 1, y opcionalmente acoplar además el compuesto
de fórmula I-a con un alcohol o ácido de fórmula R6-OH o un aldehido de fórmula R6=O para obtener un compuesto de fórmula I-d

en la que de R1 a R3, m y R6 son como se define en la reivindicación 1 y, si se desea, convertir el compuesto obtenido en una sal farmacéuticamente aceptable.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14165349 | 2014-04-22 | ||
| PCT/EP2015/058465 WO2015162075A1 (en) | 2014-04-22 | 2015-04-20 | 4-amino-imidazoquinoline compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2804101T3 true ES2804101T3 (es) | 2021-02-03 |
Family
ID=50489008
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15716824T Active ES2804101T3 (es) | 2014-04-22 | 2015-04-20 | Compuestos de 4-amino-imidazoquinolina |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US9334268B2 (es) |
| EP (1) | EP3134402B1 (es) |
| JP (1) | JP6367366B2 (es) |
| KR (1) | KR101905292B1 (es) |
| CN (1) | CN106232599B (es) |
| AR (1) | AR100137A1 (es) |
| BR (1) | BR112016017261B1 (es) |
| CA (1) | CA2938280C (es) |
| DK (1) | DK3134402T3 (es) |
| ES (1) | ES2804101T3 (es) |
| HR (1) | HRP20201043T1 (es) |
| HU (1) | HUE049656T2 (es) |
| LT (1) | LT3134402T (es) |
| MA (1) | MA39898B1 (es) |
| MX (1) | MX377916B (es) |
| PL (1) | PL3134402T3 (es) |
| PT (1) | PT3134402T (es) |
| RS (1) | RS60430B1 (es) |
| RU (1) | RU2698902C2 (es) |
| SI (1) | SI3134402T1 (es) |
| TW (1) | TWI607007B (es) |
| WO (1) | WO2015162075A1 (es) |
Families Citing this family (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2018007319A (es) * | 2015-12-14 | 2018-09-06 | Glaxosmithkline Biologicals Sa | Imidazoquinolinas pegiladas como agonistas tlr7 y tlr8. |
| PT3889145T (pt) | 2015-12-17 | 2024-04-02 | Merck Patent Gmbh | 8-ciano-5-piperidino-quinolinas como antagonistas de tlr7/8 e suas utilizações para tratamento de distúrbios imunitários |
| JP6918838B2 (ja) | 2016-05-23 | 2021-08-11 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | 第三級アミド基を有するベンズアゼピンジカルボキサミド化合物 |
| CN109311854B (zh) | 2016-05-23 | 2021-08-10 | 豪夫迈·罗氏有限公司 | 具有仲酰胺官能团的苯并氮杂卓二甲酰胺化合物 |
| JP7012668B2 (ja) | 2016-06-12 | 2022-02-14 | エフ・ホフマン-ラ・ロシュ・アクチェンゲゼルシャフト | ジヒドロピリミジニルベンズアゼピンジカルボキサミド化合物 |
| CN109843327B (zh) | 2016-07-07 | 2022-05-13 | 小利兰·斯坦福大学托管委员会 | 抗体佐剂缀合物 |
| EP4198031A1 (en) | 2016-08-08 | 2023-06-21 | Merck Patent GmbH | Tlr7/8 antagonists and uses thereof |
| KR20230010826A (ko) | 2016-10-14 | 2023-01-19 | 프리시젼 바이오사이언시스 인코포레이티드 | B형 간염 바이러스 게놈 내의 인식 서열에 대해 특이적인 조작된 메가뉴클레아제 |
| WO2018095426A1 (zh) * | 2016-11-28 | 2018-05-31 | 江苏恒瑞医药股份有限公司 | 吡唑并杂芳基类衍生物、其制备方法及其在医药上的应用 |
| US10757485B2 (en) * | 2017-08-25 | 2020-08-25 | Honda Motor Co., Ltd. | System and method for synchronized vehicle sensor data acquisition processing using vehicular communication |
| WO2019084060A1 (en) | 2017-10-24 | 2019-05-02 | Silverback Therapeutics, Inc. | CONJUGATES AND METHODS OF USE FOR THE SELECTIVE DELIVERY OF IMMUNOMODULATORY AGENTS |
| JP2021506827A (ja) | 2017-12-15 | 2021-02-22 | シルバーバック セラピューティックス インコーポレイテッド | 肝炎の治療用の抗体コンストラクト−薬物コンジュゲート |
| WO2019123339A1 (en) | 2017-12-20 | 2019-06-27 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2019123178A1 (en) | 2017-12-20 | 2019-06-27 | 3M Innovative Properties Company | Amide substitued imidazo[4,5-c]quinoline compounds with a branched chain linking group for use as an immune response modifier |
| EP3728283B1 (en) | 2017-12-20 | 2023-11-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| CN108299421B (zh) * | 2018-01-30 | 2020-09-29 | 中国医学科学院药用植物研究所 | 一种瑞喹莫德的酰化衍生物及制备方法与应用 |
| JP7050165B2 (ja) | 2018-02-26 | 2022-04-07 | ギリアード サイエンシーズ, インコーポレイテッド | Hbv複製阻害剤としての置換ピロリジン化合物 |
| ES3003782T3 (en) | 2018-02-28 | 2025-03-11 | Solventum Intellectual Properties Company | Substituted imidazo[4,5-c]quinoline compounds with an n-1 branched group |
| US10870691B2 (en) | 2018-04-05 | 2020-12-22 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis B virus protein X |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| US11142750B2 (en) | 2018-04-12 | 2021-10-12 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| US20190359645A1 (en) | 2018-05-03 | 2019-11-28 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides comprising carbocyclic nucleotide |
| JP7394790B2 (ja) | 2018-05-24 | 2023-12-08 | スリーエム イノベイティブ プロパティズ カンパニー | N-1分枝状シクロアルキル置換イミダゾ[4,5-c]キノリン化合物、組成物、及び方法 |
| MX2020012651A (es) * | 2018-05-25 | 2021-02-02 | Jiangsu Hengrui Medicine Co | Forma de cristal de clorhidrato de derivado de pirazoloheteroarilo, y metodo de preparacion. |
| CN110526918B (zh) * | 2018-05-25 | 2021-09-03 | 江苏恒瑞医药股份有限公司 | 一种吡唑并杂芳基类衍生物的晶型及制备方法 |
| CN110526917B (zh) * | 2018-05-25 | 2021-09-03 | 江苏恒瑞医药股份有限公司 | 一种吡唑并杂芳基类衍生物的可药用盐、晶型及其制备方法 |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020037094A1 (en) | 2018-08-16 | 2020-02-20 | Innate Tumor Immunity, Inc. | Substitued 4-amino-1h-imidazo[4,5-c]quinoline compounds and improved methods for their preparation |
| AU2019335366A1 (en) * | 2018-09-07 | 2021-03-25 | Birdie Biopharmaceuticals, Inc. | Imidazoquinoline compounds and uses thereof |
| MX2021002764A (es) | 2018-09-12 | 2021-05-12 | Silverback Therapeutics Inc | Composiciones para el tratamiento de enfermedades con conjugados inmunoestimuladores. |
| WO2020056192A1 (en) * | 2018-09-12 | 2020-03-19 | Silverback Therapeutics, Inc. | Antibody conjugates of toll-like receptor agonists |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US11203591B2 (en) | 2018-10-31 | 2021-12-21 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds |
| EP3917926B1 (en) * | 2019-02-01 | 2025-10-01 | Canwell Biotech Limited | Imidazoquinoline amine derivatives, pharmaceutical composition, use thereof |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| EP3934757B1 (en) | 2019-03-07 | 2023-02-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020190725A1 (en) | 2019-03-15 | 2020-09-24 | Bolt Biotherapeutics, Inc. | Immunoconjugates targeting her2 |
| TW202212339A (zh) | 2019-04-17 | 2022-04-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TWI751516B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| EP3972695A1 (en) | 2019-05-23 | 2022-03-30 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| KR20220035908A (ko) | 2019-06-19 | 2022-03-22 | 실버백 테라퓨틱스, 인크. | 항-메소텔린 항체 및 이의 면역접합체 |
| CN112174903B (zh) * | 2019-07-01 | 2022-07-12 | 清华大学 | Tlr8的小分子调节剂 |
| US11339159B2 (en) * | 2019-07-17 | 2022-05-24 | Pfizer Inc. | Toll-like receptor agonists |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| DK4037708T3 (da) | 2019-09-30 | 2024-09-30 | Gilead Sciences Inc | HBV-vacciner og fremgangsmåder til at behandle HBV |
| CA3151322A1 (en) | 2019-10-01 | 2021-04-08 | Silverback Therapeutics, Inc. | Combination therapy with immune stimulatory conjugates |
| BR112022008039A2 (pt) * | 2019-10-29 | 2022-07-12 | Prime Reach Trading Ltd | Compostos de 4-amino-imidazoquinolina e usos relacionados |
| DK4069729T3 (da) | 2019-12-06 | 2025-04-07 | Prec Biosciences Inc | Optimerede, modificerede meganukleaser med specificitet for en genkendelsessekvens i hepatitis b-virusgenomet |
| JP7735291B2 (ja) | 2020-02-21 | 2025-09-08 | エーアールエス ファーマシューティカルズ、インコーポレイテッド | ネクチン-4抗体コンジュゲートおよびその使用 |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| WO2021213946A1 (en) | 2020-04-19 | 2021-10-28 | Englmeier Ludwig | Prophylaxis and treatment of coronavirus infection |
| TW202200139A (zh) * | 2020-05-14 | 2022-01-01 | 德商馬克專利公司 | 用於治療冠狀病毒感染之tlr7/8拮抗劑 |
| WO2021237055A1 (en) * | 2020-05-21 | 2021-11-25 | Ohio State Innovation Foundation | Functional lipid derivatives and uses thereof |
| JP2023532304A (ja) | 2020-07-01 | 2023-07-27 | エーアールエス ファーマシューティカルズ オペレーションズ,インク. | 抗asgr1抗体コンジュゲートおよびその使用 |
| WO2022153157A1 (en) * | 2021-01-13 | 2022-07-21 | Pfizer Inc. | A crystalline form of 2-((4-amino-2-(ethoxymethyl)-6,7-dimethyl-1 h-imidazo[4,5-c]pyridin-1-yl)methyl)-2-methylpropane-1,3-diol free base |
| BR112023015561A2 (pt) * | 2021-02-03 | 2023-11-14 | Univ Minnesota | Compostos imunoestimuiladores e conjugados |
| US20240209080A1 (en) | 2021-04-10 | 2024-06-27 | Profoundbio Us Co. | Folr1 binding agents, conjugates thereof and methods of using the same |
| CA3216459A1 (en) | 2021-04-23 | 2022-10-27 | Profoundbio Us Co. | Anti-cd70 antibodies, conjugates thereof and methods of using the same |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| MX2023014762A (es) | 2021-06-23 | 2024-01-15 | Gilead Sciences Inc | Compuestos moduladores de diacilglicerol quinasa. |
| JP7686091B2 (ja) | 2021-06-23 | 2025-05-30 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| AU2022299051B2 (en) | 2021-06-23 | 2025-03-13 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| JP7651018B2 (ja) | 2021-06-23 | 2025-03-25 | ギリアード サイエンシーズ, インコーポレイテッド | ジアシルグリセロールキナーゼ調節化合物 |
| TW202320857A (zh) | 2021-07-06 | 2023-06-01 | 美商普方生物製藥美國公司 | 連接子、藥物連接子及其結合物及其使用方法 |
| WO2023081237A1 (en) * | 2021-11-03 | 2023-05-11 | Regents Of The University Of Minnesota | Toll-like receptor agonists and antagonists and uses thereof |
| JP2025525904A (ja) * | 2022-08-03 | 2025-08-07 | シージェン インコーポレイテッド | 免疫刺激性抗pd-l1-薬物コンジュゲート |
| AU2023376684A1 (en) * | 2022-11-09 | 2025-06-19 | Merck Patent Gmbh | Toll-like receptor 7 agonists as immune-stimulators to elicit the innate antitumor immunity |
| WO2024112932A1 (en) * | 2022-11-23 | 2024-05-30 | Regents Of The University Of Minnesota | Immunomodulators and immunomodulator conjugates |
| AR133955A1 (es) | 2023-09-26 | 2025-11-19 | Profoundbio Us Co | Agentes de unión a ptk7, conjugados de éstos y métodos de uso de los mismos |
| WO2025149661A1 (en) | 2024-01-10 | 2025-07-17 | Genmab A/S | Slitrk6 binding agents, conjugates thereof and methods of using the same |
| US20250381289A1 (en) | 2024-02-29 | 2025-12-18 | Genmab A/S | Egfr and c-met bispecific binding agents, conjugates thereof and methods of using the same |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69229114T2 (de) | 1991-03-01 | 1999-11-04 | Minnesota Mining And Mfg. Co., Saint Paul | 1,2-substituierte 1h-imidazo[4,5-c]chinolin-4-amine |
| US6451810B1 (en) * | 1999-06-10 | 2002-09-17 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6664265B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6861440B2 (en) * | 2001-10-26 | 2005-03-01 | Hoffmann-La Roche Inc. | DPP IV inhibitors |
| EP1455700A4 (en) * | 2001-11-16 | 2007-02-14 | 3M Innovative Properties Co | METHODS AND COMPOSITIONS RELATED TO MRI COMPOUNDS AND TO TOLL-TYPE RECEPTOR (TLR) PATHWAYS |
| JP2007504232A (ja) * | 2003-09-05 | 2007-03-01 | アナディス ファーマシューティカルズ インク | C型肝炎ウイルス感染治療用のtlr7リガンド及びそのプロドラッグの投与 |
| JP2007513170A (ja) * | 2003-12-04 | 2007-05-24 | スリーエム イノベイティブ プロパティズ カンパニー | スルホン置換イミダゾ環エーテル |
| TW201402124A (zh) | 2005-08-19 | 2014-01-16 | Array Biopharma Inc | 作為類鐸受體(toll-like receptor)調節劑之8-經取代苯并氮雜呯 |
| CN102015651B (zh) | 2008-03-03 | 2014-12-31 | Irm责任有限公司 | 作为tlr活性调节剂的化合物和组合物 |
| ES2545275T3 (es) | 2008-11-06 | 2015-09-09 | Ventirx Pharmaceuticals, Inc. | Métodos de síntesis de derivados de benzazepinas |
| KR20110117705A (ko) | 2009-02-11 | 2011-10-27 | 더 리전트 오브 더 유니버시티 오브 캘리포니아 | 톨-유사 수용체 조정제 및 질병의 치료 |
| MX2012001524A (es) | 2009-08-07 | 2012-02-29 | Glaxosmithkline Biolog Sa | Derivados de oxoadenina lipidada. |
| JP2013512859A (ja) | 2009-12-03 | 2013-04-18 | 大日本住友製薬株式会社 | トール様受容体(tlr)を介して作用するイミダゾキノリン |
| JP2013525431A (ja) | 2010-04-30 | 2013-06-20 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 合成tlr7アゴニストのリン脂質結合体の使用 |
| EP3195868A3 (en) | 2010-10-01 | 2017-08-02 | VentiRx Pharmaceuticals, Inc. | Therapeutic use of a tlr agonist and combination therapy |
| WO2012066336A1 (en) | 2010-11-19 | 2012-05-24 | Astrazeneca Ab | Benzylamine compounds as toll -like receptor 7 agonists |
| BR112013031039B1 (pt) | 2011-06-03 | 2020-04-28 | 3M Innovative Properties Co | compostos de hidrazino 1h-imidazoquinolina-4-aminas, conjugados feitos destes compostos, composição e composição farmacêutica compreendendo ditos compostos e conjugados, usos dos mesmos e método de fabricação do conjugado |
| WO2012167801A1 (ru) | 2011-06-07 | 2012-12-13 | Uglovsky Sergey Evgenievich | Импульсная установка для преобразования низкопотенциальной тепловой энергии в электрическую |
| US9034336B2 (en) | 2011-08-30 | 2015-05-19 | Regents Of The University Of Minnesota | Immunomodulators and immunomodulator conjugates |
| AU2012334825B2 (en) * | 2011-11-09 | 2017-08-24 | Ascend Biopharmaceuticals Ltd | Immunomodulatory conjugates |
| WO2013166110A1 (en) | 2012-05-02 | 2013-11-07 | Yale University | Tlr-agonist-conjugated antibody recruiting molecules (tlr_arms) |
-
2015
- 2015-04-20 PT PT157168246T patent/PT3134402T/pt unknown
- 2015-04-20 EP EP15716824.6A patent/EP3134402B1/en active Active
- 2015-04-20 MX MX2016013689A patent/MX377916B/es unknown
- 2015-04-20 SI SI201531273T patent/SI3134402T1/sl unknown
- 2015-04-20 RU RU2016144386A patent/RU2698902C2/ru active
- 2015-04-20 JP JP2016564006A patent/JP6367366B2/ja active Active
- 2015-04-20 CA CA2938280A patent/CA2938280C/en active Active
- 2015-04-20 HU HUE15716824A patent/HUE049656T2/hu unknown
- 2015-04-20 RS RS20200734A patent/RS60430B1/sr unknown
- 2015-04-20 BR BR112016017261-2A patent/BR112016017261B1/pt not_active IP Right Cessation
- 2015-04-20 HR HRP20201043TT patent/HRP20201043T1/hr unknown
- 2015-04-20 KR KR1020167032523A patent/KR101905292B1/ko active Active
- 2015-04-20 ES ES15716824T patent/ES2804101T3/es active Active
- 2015-04-20 PL PL15716824T patent/PL3134402T3/pl unknown
- 2015-04-20 MA MA39898A patent/MA39898B1/fr unknown
- 2015-04-20 LT LTEP15716824.6T patent/LT3134402T/lt unknown
- 2015-04-20 CN CN201580020712.2A patent/CN106232599B/zh active Active
- 2015-04-20 WO PCT/EP2015/058465 patent/WO2015162075A1/en not_active Ceased
- 2015-04-20 DK DK15716824.6T patent/DK3134402T3/da active
- 2015-04-20 AR ARP150101179A patent/AR100137A1/es active IP Right Grant
- 2015-04-21 TW TW104112767A patent/TWI607007B/zh active
- 2015-04-22 US US14/692,968 patent/US9334268B2/en active Active
-
2016
- 2016-04-07 US US15/093,481 patent/US9447097B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2804101T3 (es) | Compuestos de 4-amino-imidazoquinolina | |
| JP6893501B2 (ja) | スルフィニルフェニル又はスルホンイミドイルフェニルベンザゼピン | |
| JP7372255B2 (ja) | 免疫調節剤としての複素環式化合物 | |
| CN109153648B (zh) | 具有叔酰胺官能团的苯并氮杂卓二甲酰胺化合物 | |
| CN109311851B (zh) | 二氢嘧啶基苯并氮杂䓬甲酰胺化合物 | |
| TWI758300B (zh) | 免疫調節劑化合物 | |
| CN111225665B (zh) | 大环免疫调节剂 | |
| ES2929415T3 (es) | Compuestos heterocíclicos tricíclicos como activadores de STING | |
| ES2712488T3 (es) | Compuestos de benzacepina-dicarboxamida | |
| TWI856340B (zh) | 作為pd-l1相互作用的免疫調節劑的雜環化合物 | |
| HK40003985A (en) | Dihydropyrimidinyl benzazepine carboxamide compounds | |
| HK40003985B (zh) | 二氢嘧啶基苯并氮杂䓬甲酰胺化合物 | |
| HK1227845A1 (en) | 4-amino-imidazoquinoline compounds | |
| HK1227845B (zh) | 4-氨基-咪唑并喹啉化合物 | |
| HK40002463B (en) | Benzazepine dicarboxamide compounds with tertiary amide function | |
| HK40002463A (en) | Benzazepine dicarboxamide compounds with tertiary amide function |