ES2818185T3 - Proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio - Google Patents
Proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio Download PDFInfo
- Publication number
- ES2818185T3 ES2818185T3 ES16784446T ES16784446T ES2818185T3 ES 2818185 T3 ES2818185 T3 ES 2818185T3 ES 16784446 T ES16784446 T ES 16784446T ES 16784446 T ES16784446 T ES 16784446T ES 2818185 T3 ES2818185 T3 ES 2818185T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- following formula
- solvent
- stage
- quinine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 title claims abstract description 32
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 23
- 238000002360 preparation method Methods 0.000 title claims abstract description 13
- 150000001875 compounds Chemical class 0.000 claims abstract description 136
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 claims abstract description 112
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims abstract description 72
- 235000001258 Cinchona calisaya Nutrition 0.000 claims abstract description 43
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 claims abstract description 43
- 229960000948 quinine Drugs 0.000 claims abstract description 43
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims abstract description 38
- 239000002904 solvent Substances 0.000 claims abstract description 29
- 239000002798 polar solvent Substances 0.000 claims abstract description 24
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims abstract description 22
- 238000002425 crystallisation Methods 0.000 claims abstract description 18
- 230000008025 crystallization Effects 0.000 claims abstract description 18
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 239000003960 organic solvent Substances 0.000 claims abstract description 15
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims abstract description 15
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims abstract description 11
- 235000019253 formic acid Nutrition 0.000 claims abstract description 11
- MNOALXGAYUJNKX-UHFFFAOYSA-N s-chloro chloromethanethioate Chemical compound ClSC(Cl)=O MNOALXGAYUJNKX-UHFFFAOYSA-N 0.000 claims abstract description 8
- 239000003586 protic polar solvent Substances 0.000 claims abstract description 7
- 239000013078 crystal Substances 0.000 claims abstract description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims abstract description 5
- 238000005516 engineering process Methods 0.000 claims abstract description 5
- 150000001450 anions Chemical class 0.000 claims abstract description 4
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 claims abstract description 4
- 239000000203 mixture Substances 0.000 claims description 52
- 239000000243 solution Substances 0.000 claims description 52
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 42
- 238000000034 method Methods 0.000 claims description 41
- 239000000047 product Substances 0.000 claims description 34
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 32
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 29
- 230000008569 process Effects 0.000 claims description 25
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 17
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 claims description 15
- 239000011541 reaction mixture Substances 0.000 claims description 14
- 239000000010 aprotic solvent Substances 0.000 claims description 13
- 239000008346 aqueous phase Substances 0.000 claims description 13
- -1 diisopropyl-ethylamine hexafluorophosphate Chemical compound 0.000 claims description 13
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 12
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 claims description 12
- 238000011084 recovery Methods 0.000 claims description 12
- 239000007787 solid Substances 0.000 claims description 10
- BJCNUZJDFWFVBY-VITQDTLGSA-N 1-ethoxycarbonyloxyethyl 2-[[(2s)-2-[[[(2s)-2-amino-4-methylsulfanylbutyl]disulfanyl]methyl]-3-phenylpropanoyl]amino]acetate Chemical compound CCOC(=O)OC(C)OC(=O)CNC(=O)[C@@H](CSSC[C@@H](N)CCSC)CC1=CC=CC=C1 BJCNUZJDFWFVBY-VITQDTLGSA-N 0.000 claims description 8
- 239000007864 aqueous solution Substances 0.000 claims description 8
- 239000001530 fumaric acid Substances 0.000 claims description 8
- 239000012044 organic layer Substances 0.000 claims description 8
- 238000005112 continuous flow technique Methods 0.000 claims description 7
- 238000001914 filtration Methods 0.000 claims description 7
- YVRGKFXJZCTTRB-UHFFFAOYSA-N 1-chloroethyl ethyl carbonate Chemical compound CCOC(=O)OC(C)Cl YVRGKFXJZCTTRB-UHFFFAOYSA-N 0.000 claims description 6
- 239000003208 petroleum Substances 0.000 claims description 6
- 238000005063 solubilization Methods 0.000 claims description 6
- 230000007928 solubilization Effects 0.000 claims description 6
- VRPJIFMKZZEXLR-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)NCC(O)=O VRPJIFMKZZEXLR-UHFFFAOYSA-N 0.000 claims description 5
- 238000005349 anion exchange Methods 0.000 claims description 5
- 239000000725 suspension Substances 0.000 claims description 5
- 239000012141 concentrate Substances 0.000 claims description 4
- 238000001816 cooling Methods 0.000 claims description 4
- 238000000605 extraction Methods 0.000 claims description 4
- 238000010306 acid treatment Methods 0.000 claims description 3
- 238000005904 alkaline hydrolysis reaction Methods 0.000 claims description 3
- 239000012267 brine Substances 0.000 claims description 3
- 239000003880 polar aprotic solvent Substances 0.000 claims description 3
- 239000002244 precipitate Substances 0.000 claims description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 claims description 3
- 239000007810 chemical reaction solvent Substances 0.000 claims description 2
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 abstract 3
- 230000015572 biosynthetic process Effects 0.000 description 23
- 239000012074 organic phase Substances 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 17
- 238000003786 synthesis reaction Methods 0.000 description 14
- 238000003756 stirring Methods 0.000 description 13
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N dichloromethane Natural products ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 10
- 238000005755 formation reaction Methods 0.000 description 10
- 238000004128 high performance liquid chromatography Methods 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- 238000012546 transfer Methods 0.000 description 9
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 235000011087 fumaric acid Nutrition 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 239000002253 acid Substances 0.000 description 7
- BCAAXVOKLXDSPD-UHFFFAOYSA-N 2-(acetylsulfanylmethyl)-3-phenylpropanoic acid Chemical compound CC(=O)SCC(C(O)=O)CC1=CC=CC=C1 BCAAXVOKLXDSPD-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- 238000004090 dissolution Methods 0.000 description 6
- 239000012535 impurity Substances 0.000 description 6
- 238000001953 recrystallisation Methods 0.000 description 6
- BCAAXVOKLXDSPD-LLVKDONJSA-N (2s)-2-(acetylsulfanylmethyl)-3-phenylpropanoic acid Chemical compound CC(=O)SC[C@H](C(O)=O)CC1=CC=CC=C1 BCAAXVOKLXDSPD-LLVKDONJSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 239000006227 byproduct Substances 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 230000010354 integration Effects 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 238000006386 neutralization reaction Methods 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 3
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000010924 continuous production Methods 0.000 description 3
- 238000011018 current good manufacturing practice Methods 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 150000004675 formic acid derivatives Chemical class 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- IMUSLIHRIYOHEV-ZETCQYMHSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-methylsulfanylbutanoic acid Chemical compound CSCC[C@@H](C(O)=O)NC(=O)OC(C)(C)C IMUSLIHRIYOHEV-ZETCQYMHSA-N 0.000 description 2
- UOVSNFYJYANSNI-UHFFFAOYSA-N 2-acetylsulfanyl-3-phenylpropanoic acid Chemical compound CC(=O)SC(C(O)=O)CC1=CC=CC=C1 UOVSNFYJYANSNI-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- OQFDYVOPDGPDIC-UHFFFAOYSA-N [2-(1-ethoxycarbonyloxyethoxy)-2-oxoethyl]azanium chloride Chemical compound [Cl-].C(C)OC(=O)OC(C)OC(C[NH3+])=O OQFDYVOPDGPDIC-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000000202 analgesic effect Effects 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000002051 biphasic effect Effects 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- JNGZXGGOCLZBFB-IVCQMTBJSA-N compound E Chemical compound N([C@@H](C)C(=O)N[C@@H]1C(N(C)C2=CC=CC=C2C(C=2C=CC=CC=2)=N1)=O)C(=O)CC1=CC(F)=CC(F)=C1 JNGZXGGOCLZBFB-IVCQMTBJSA-N 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 238000007127 saponification reaction Methods 0.000 description 2
- 239000012265 solid product Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- NSPBTQHSHXESOT-YFKPBYRVSA-N (2s)-2-amino-4-methylsulfanylbutane-1-thiol Chemical compound CSCC[C@H](N)CS NSPBTQHSHXESOT-YFKPBYRVSA-N 0.000 description 1
- FKGMQENMUNWFBJ-SJORKVTESA-N (2s)-2-benzyl-3-[[(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-methylsulfanylbutyl]disulfanyl]propanoic acid Chemical compound CC(C)(C)OC(=O)N[C@@H](CCSC)CSSC[C@H](C(O)=O)CC1=CC=CC=C1 FKGMQENMUNWFBJ-SJORKVTESA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- JYWKEVKEKOTYEX-UHFFFAOYSA-N 2,6-dibromo-4-chloroiminocyclohexa-2,5-dien-1-one Chemical compound ClN=C1C=C(Br)C(=O)C(Br)=C1 JYWKEVKEKOTYEX-UHFFFAOYSA-N 0.000 description 1
- UWCWUCKPEYNDNV-LBPRGKRZSA-N 2,6-dimethyl-n-[[(2s)-pyrrolidin-2-yl]methyl]aniline Chemical compound CC1=CC=CC(C)=C1NC[C@H]1NCCC1 UWCWUCKPEYNDNV-LBPRGKRZSA-N 0.000 description 1
- ZUEBVBPVXLQMQR-SECBINFHSA-N 2-thiomethyl-3-phenylpropanoic acid Chemical compound OC(=O)[C@@H](CS)CC1=CC=CC=C1 ZUEBVBPVXLQMQR-SECBINFHSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- 102000004400 Aminopeptidases Human genes 0.000 description 1
- 108090000915 Aminopeptidases Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical group C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- 206010061380 Therapeutic reaction time decreased Diseases 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 108010027597 alpha-chymotrypsin Proteins 0.000 description 1
- 150000001408 amides Chemical group 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000001944 continuous distillation Methods 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 125000000107 disulfanyl group Chemical group [*]SS[H] 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000002792 enkephalinase inhibitor Substances 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 238000011194 good manufacturing practice Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000009533 lab test Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- IYUKFAFDFHZKPI-DFWYDOINSA-N methyl (2s)-2-aminopropanoate;hydrochloride Chemical compound Cl.COC(=O)[C@H](C)N IYUKFAFDFHZKPI-DFWYDOINSA-N 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 238000005897 peptide coupling reaction Methods 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000004886 process control Methods 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 238000010966 qNMR Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000010517 secondary reaction Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DRDVJQOGFWAVLH-UHFFFAOYSA-N tert-butyl n-hydroxycarbamate Chemical compound CC(C)(C)OC(=O)NO DRDVJQOGFWAVLH-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000008646 thermal stress Effects 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- LJJKNPQAGWVLDQ-SNVBAGLBSA-N thiorphan Chemical compound OC(=O)CNC(=O)[C@@H](CS)CC1=CC=CC=C1 LJJKNPQAGWVLDQ-SNVBAGLBSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/22—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of hydropolysulfides or polysulfides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/02—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of thiols
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/18—Stationary reactors having moving elements inside
- B01J19/1862—Stationary reactors having moving elements inside placed in series
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/22—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of hydropolysulfides or polysulfides
- C07C319/24—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of hydropolysulfides or polysulfides by reactions involving the formation of sulfur-to-sulfur bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/26—Separation; Purification; Stabilisation; Use of additives
- C07C319/28—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/56—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/60—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton with the carbon atom of at least one of the carboxyl groups bound to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C327/00—Thiocarboxylic acids
- C07C327/20—Esters of monothiocarboxylic acids
- C07C327/32—Esters of monothiocarboxylic acids having sulfur atoms of esterified thiocarboxyl groups bound to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/24—Oxygen atoms attached in position 8
- C07D215/26—Alcohols; Ethers thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00027—Process aspects
- B01J2219/00029—Batch processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00027—Process aspects
- B01J2219/0004—Processes in series
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/39—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton at least one of the nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom
- C07C323/40—Y being a hydrogen or a carbon atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Proceso industrial para la preparación de sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18- trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio de la fórmula (I) siguiente: **(Ver fórmula)** que comprende las etapas sintéticas sucesivas siguientes realizadas en solventes orgánicos desgasificados polares o apolares, próticos o apróticos: 1) preparar el compuesto (E) de la fórmula siguiente con un exceso enantiomérico superior a 95% **(Ver fórmula)** 1a) haciendo reaccionar el compuesto (A) de la fórmula siguiente **(Ver fórmula)** con 0.5 - 0.6 equivalentes molares de quinina en solventes orgánicos polares y apróticos, preferentemente en acetato de etilo; 1b) cristalizar la sal de quinina resultante a una temperatura que varía de 10°C a 20°C, en el mismo solvente orgánico que el utilizado en la etapa 1a, en el que la cristalización se inicia añadiendo unos cuantos cristales de la sal de enantiómero deseada para iniciar la cristalización, a continuación; 1c) recristalizar la sal obtenida después de la etapa 1b al mismo intervalo de temperatura y el mismo solvente que el utilizado en la etapa 1b; 1d) recuperar el compuesto (E): 1d.1)recuperando el compuesto (D) de la fórmula siguiente **(Ver fórmula)** 1d.2)desproteger el tiolacetato en un solvente polar y prótico tal como MeOH; 1e) recuperar la quinina; 2) preparar el compuesto (F) de la fórmula siguiente **(Ver fórmula)** 2a) haciendo reaccionar los primeros 1.1 equivalentes molares de dicho compuesto (E) con 1 equivalente molar de cloruro de clorocarbonil sulfenilo, en un solvente polar y aprótico, a continuación; 2b) hacer reaccionar el intermedio obtenido después de la etapa 2a con 0.9 equivalentes molares del compuesto (B) de la fórmula siguiente **(Ver fórmula)** en solución con 1 equivalente molar de Et3N en el mismo solvente que el utilizado en la etapa 2a; 3) preparar el compuesto (G) de la fórmula siguiente **(Ver fórmula)** haciendo reaccionar dicho compuesto (F) con el amino-éster C de la fórmula siguiente, en el que Y- es un anión: **(Ver fórmula)* en solvente polar; 4) a continuación, recuperar la sal (I) de la fórmula siguiente **(Ver fórmula)** 4a) añadiendo 5 equivalentes molares de ácido fórmico en dicho compuesto (G); 4b) intercambiar el formiato por un fumarato utilizando una tecnología de flujo continuo.
Description
DESCRIPCIÓN
Proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metiM1,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio
Campo técnico
La presente invención se refiere a un nuevo proceso industrial para la preparación de sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio, combinando la porción de ácido 3-(acetiltio)-2-bencilpropanoico y 1-mercapto-4-(metiltio)butan-2-ilcarbamato de (S)-terc-butilo y cloruro de 2-(1-(etoxicarboniloxi)etoxi)-2-oxoetanaminio.
Antecedentes de la invención
La sal de (E)-3-carboxiacrilato de (5S, 10S)-10-benciM6-metiM1,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio, a la que se hace referencia en la presente memoria como el “Compuesto (I)” de la siguiente fórmula:

es un profármaco del disulfuro asimétrico que está compuesto por el inhibidor selectivo de aminopeptidasa neutral (APN), ((S)-1-mercapto-4-(metiltio)butan-2-aminio), y por el inhibidor selectivo de neprilisina (n Ep ), ácido ((S)-2-(2-bencil-3-mercaptopropanamido)acético).
Se ha demostrado que el Compuesto (I) es un analgésico eficiente, como describe Poras et al. en J Med Chem, 2014, 57, 5748-5763.
El Compuesto (I) y su utilización como analgésico fueron divulgados primero en la solicitud de patente WO2007/048787. El proceso ejemplificado en el documento WO 2007/048787 permite la preparación del compuesto (I), en 4 etapas desde ácido 3-(acetiltio)-2-bencilpropanoico (A), 1-mercapto-4-(metiltio)butan-2-ilcarbamato de (S)-terc-butilo (B) y cloruro de 2-(1-(etoxicarboniloxi)etoxi)-2-oxoetanaminio (C). Las especificaciones sintéticas técnicas, particularmente la resolución enzimática, números de equivalentes, solventes y/o técnicas de purificación involucradas en este proceso, no permiten que éste sea convertido de manera eficiente y fácil a una escala industrial.
Un objetivo permanente en la síntesis orgánica es crear procesos de síntesis que puedan ser traspuestos en condiciones industriales. Para cumplir con los requerimientos para procesos industriales, deberán de ser optimizados diferentes parámetros de la síntesis. En primer lugar, los solventes deben de ser lo menos volátiles posible, para que sean fácilmente recuperables.
Las temperaturas involucradas permanecen preferentemente en un intervalo fácilmente accesible, y se deberá privilegiar una purificación fácil de proceder. Por último, las mezclas de reacciones y los productos aislados preferentemente son térmicamente estables.
Se ha definido una Current Good Manufacturing Practice (c-GMP) para la preparación de productos de fármaco para su administración a seres humanos o animales. Las regulaciones de GMP requieren un enfoque de calidad para la fabricación, haciendo posible que las compañías minimicen o eliminen los casos de contaminación, confusiones y errores. Hasta donde sabe el solicitante, hasta ahora no se ha descrito ningún proceso industrialmente aplicable para sintetizar el compuesto (I). Por lo tanto, sigue existiendo la necesidad de un proceso para preparar el compuesto (I) que pueda ser adaptado fácil y eficientemente a escala industrial, en particular un proceso en el que no se utilicen solventes tóxicos, tal como solventes clorados, resolución de enzima y cromatografía de columna.
Sumario de la invención
La presente invención se refiere a un proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio de la siguiente fórmula (I):

Este procedimiento comprende las etapas siguientes sintéticas sucesivas que se realizan en solventes orgánicos polares o apolares, próticos o apróticos desgasificados:
1) Preparar el compuesto E de la siguiente fórmula con un exceso enantiomérico superior a 95%

con de 0.5 - 0.6 equivalentes molares de quinina en solventes orgánicos polares y apróticos;
1b) Cristalizando la sal de quinina resultante a una temperatura que varía de 10°C a 20°C, en el mismo solvente orgánico que se usó en la etapa 1a, en el que la cristalización se inicia al añadir unos cuantos cristales de la sal de enantiómero deseada para iniciar la cristalización, a continuación;
1c) Recristalizar la sal obtenida después de la etapa 1b a la misma escala de temperatura y el mismo solvente utilizado en la etapa 1b;
1d) Recuperar el compuesto (E):
1d.1) recuperando el compuesto (D) de la siguiente fórmula

1d.2) desprotegiendo el tiolacetato en el solvente polar y prótico como MeOH;
1e) Recuperar la quinina;
2) Preparar el compuesto (F) de la siguiente fórmula

2a) Haciendo reaccionar los primeros 1.1 equivalentes molares de dicho compuesto (E) con 1 equivalente molar de cloruro de clorocarbonil sulfenilo, en un solvente polar y aprótico, a continuación;
2b) Hacer reaccionar el intermedio obtenido después de la etapa 2a con 0.9 equivalentes molares del compuesto (B) de la siguiente fórmula

En solución con 1 equivalente molar de Et3N en el mismo solvente que se utilizó en la etapa 2a;
) Preparar el compuesto (G) de la siguiente fórmula

Haciendo reaccionar d icho compuesto (F) con el amino-éster (C) de la siguiente fórmula, en el que Y- es un anión:
©
Y H3N ^ r 0 Y 0 Y 0E t
nO o (C)
En solvente polar;
) A continuación, recuperar la sal (I) de la siguiente fórmula
c 
4a) agregando 5 equivalentes molares de ácido fórmico en dicho compuesto (G);
4b) intercambiando el formiato por un fumarato utilizando una tecnología de flujo continuo.
Descripción detallada de la invención
En la presente memoria, la siguiente abreviación significa:
- AcOEt: acetato de etilo
- Bn: bencilo
- Boc: ferc-Butoxicarbonilo
- DCM: diclorometano
- DME: dimetoxietano
- DMF: dimetil formamida
- e.e.: exceso enantiomérico
- EtOH: etanol
- HBTU: hexafluorofosfato de O-benzotriazol-1-il-N,N,N',N'-tetrametiluronio
- HPLC: cromatografía líquida de alto rendimiento
- IPC: control en proceso
- MeOH: metanol
- THF: tetrahidrofurano
Uno de los objetos de la presente invención se refiere a un método general para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio de la siguiente fórmula (I)

Que comprende las etapas siguientes:

Etapa (1): Síntesis del compuesto (E), por una resolución cinética de (A) con quinina para proporcionar ácido (S)-3-(acetiltio)-2-bencilpropanoico (D), seguida por una hidrólisis alcalina de la porción acetilo de (D) proporcionando ácido (S)-2-bencil-3-mercaptopropanoico (E), y recuperar la quinina;
Etapa (2): Síntesis del ácido (S)-2-bencil-3-(((S)-2-(terc-butoxicarbonilamino)-4-(metiltio)butil)disulfanil)propanoico (F) de disulfuro asimétrico, compuesto por (B) y (E) por la activación con clorosulfonilcloruro en un solvente aprótico polar;
Etapa (3): Síntesis de 11-bencil-2,2-dimetil-6-(2-(metiltio)etil)-4,12-dioxo-3-oxa-8,9-ditia-5,13-diazapentadecan-15-oato de (6S,11S)-1-(etoxicarboniloxi)etilo (G) por un acoplamiento de péptidos entre el compuesto (F) obtenido después de la etapa (2) y éster glicina en cascada (C);
Etapa (4): Recuperar la sal (I) por la desprotección del grupo protector Boc, seguido por una reacción de intercambio de flujo continuo de aniones.
Etapa (1)

En el documento WO2007/048787, se obtuvo ácido (S)-3-(acetiltio)-2-bencilpropanoico (D) por una resolución enzimática del compuesto racémico con alfa-quimotripsina. La resolución también se puedo realizar con tripsina. Aunque esto permitió producir el compuesto (I) con una buena pureza quiral, el proceso fue difícil de realizar en una planta a escala industrial.
En la invención, la etapa 1a implica una resolución cinética del ácido 2-acetiltio-3-fenilpropiónico (A) racémico, disponible comercialmente, utilizando un agente de resolución química, preferentemente realizada con quinina de la siguiente fórmula:
en un solvente polar y aprótico.
La quinina es un agente de resolución descrito por ejemplo para la resolución cinética de un compuesto amino racémico en la publicación de Babak Kaboudin et al. (Chirality, 2015, 27, p. 71-74) en la que la resolución se alcanza en etanol de reflujo con dos equivalentes de quinina. La quinina se describe asimismo como un agente de resolución en Campbell et al. (Journal of the science of food and agriculture, 1952, 3, p. 189-192) para la resolución cinética de un derivado de ácido carboxílico, alcanzada en etanol y con una cantidad estequiométrica de quinina. En una forma de realización preferida, la resolución cinética se realiza en AcOEt.
En la etapa 1b, la sal de quinina resultante es cristalizada a una temperatura que varía de 10°C a 20°C, en el mismo solvente orgánico que se utilizó en la etapa 1a, en la que la cristalización se inicia agregando unos cuantos cristales del enantiómero deseado con quinina para iniciar la cristalización. Se demostró que el uso de una semilla era importante para asegurar la robustez de la cristalización inicial, a una temperatura que varía de 10°C a 20°C. En la presente descripción, “semilla” significa unos pocos cristales del enantiómero deseado del compuesto (D) con quinina para iniciar la cristalización.
Ventajosamente, la cristalización en la etapa 1b comprende las etapas sucesivas siguientes:
1b.1) disolución de la sal de quinina a temperatura de solubilización, a continuación;
1b.2) enfriar la mezcla obtenida en la etapa 1b.1 hasta una temperatura que varía de 10°C a 20°C;
1b.3) aislar la sal de quinina obtenida después de la etapa 1b.2 por filtración.
En una forma de realización preferida, la cristalización en la etapa 1b se realiza calentado una mezcla de salquinina hasta completar la disolución de dicha sal de quinina en suspensión en un solvente polar y aprótico. En la etapa 1b.1, la sal de quinina es ventajosamente calentada hasta completar la disolución. En particular, en la etapa
1b.1 la temperatura de solubilización varía de 38°C a 50°C.
Después de la etapa 1b.1, la mezcla es enfriada a continuación hasta una temperatura que varía de 10°C a 20°C. Ventajosamente en la etapa 1b.2 el enfriamiento se realiza a una velocidad de 3-10°C/h.
Una vez alcanzada la temperatura, la mezcla se mantiene ventajosamente a la temperatura durante un período de 10 a 30 horas, en particular de 10 a 20 horas.
Después de una filtración se obtiene la sal enantiomérica de quinina cristalizada enriquecida, y después se pone en una etapa de recristalización 1c. La etapa 1c comprende la recristalización de la sal obtenida después de la etapa 1b al mismo intervalo de temperatura y el mismo solvente que se usó en la etapa 1b. Desde el punto de vista industrial, realmente es más eficiente y económico usar el mismo solvente.
En una forma de realización preferida, la recristalización en la etapa 1c comprende las etapas sucesivas siguientes:
1c.1) disolver la sal de quinina a la temperatura de solubilización, a continuación;
1c.2) enfriar la mezcla obtenida en la etapa 1c.1 hasta una temperatura que varía de 10°C a 20°C;
1c.3) aislar la sal de quinina obtenida después de la etapa 1c.2 por filtración;
lc . 4) las etapas 1c.1 a 1c.3 se repiten opcionalmente.
En particular, en la etapa 1c.1, la mezcla es calentada hasta completar la disolución de dicha sal de quinina. Ventajosamente, en la etapa 1c.1 la temperatura de solubilización varía de 45°C a 65°C.
En una forma de realización ilustrativa, la resolución cinética con quinina proporciona 90% de exceso enantiomérico después de una sola cristalización, y se podría obtener 95.5% de exceso enantiomérico después de la recristalización de la sal de quinina. La relación molar del agente de resolución química y el compuesto racémico (A) debe ser de 0.5-0.6/1, en particular de 0.55/1.
Todos estos parámetros clave permiten obtener el compuesto (D) en una forma consistente con una pureza quiral superior a 95%, pureza superior a 98% y un rendimiento molar promedio de 38% partiendo desde el compuesto racémico (A), en comparación con un rendimiento de 27% partiendo desde el compuesto racémico (A) en caso de resolución enzimática (WO2007/048787).
En particular, las etapas (1a) a (1c) comprenden típicamente las etapas sucesivas siguientes:
- La adición de quinina a una solución de ácido rac-3-(acetiltio)-2-bencilpropanoico en AcOEt;
- La disolución completa por el calentamiento a una temperatura que varía de 38°C a 50°C;
- Enfriar a una temperatura de 5°C y a continuación sembrar, a continuación;
- Enfriar en 5 horas (tasa de 3-10°C) a una temperatura que varía de 10°C a 20°C, manteniendo a continuación la mezcla de reacción al mismo intervalo de temperatura durante 16 horas;
- Aislamiento de la sal de quinina de ácido (S)-3-(acetiltio)-2-bencilpropanoico por filtración;
- Suspensión de dicha sal en AcOEt;
- La disolución completa por calentamiento hasta alcanzar una temperatura de solubilización (que varía de 45°C a 60°C) de dicha sal;
- Enfriar en 9 horas a una temperatura que varía de 10°C a 20°C;
- Aislamiento de la sal de quinina de ácido (S)-3-(acetiltio)-2-bencilpropanoico por filtración.
La recuperación del compuesto E comprende las etapas sucesivas siguientes:
ld . 1) recuperar el compuesto (D) de la siguiente fórmula

1d.2) desproteger el tiolacetato en un solvente polar y prótico como MeOH
En una forma de realización preferida, la etapa 1d.1 comprende además las etapas sucesivas siguientes:
1d.1.1) suspensión de la sal de quinina obtenida en la etapa 1c.3 o 1c.4 en agua acidificada hasta un pH que varía de 1 a 3, en particular con una mezcla de HCl/agua, a continuación;
1d.1.2) extracción del compuesto (D) de la siguiente fórmula

un solvente aprótico y apolar, en particular con AcOEt, a continuación;
1d.1.3) concentración al vacío para obtener un aceite.
Ventajosamente, la etapa 1d.2 comprende las etapas sucesivas siguientes:
1d.2.1) Hidrólisis alcalina en un solvente polar y prótico, a continuación;
1d.2.2) tratamiento ácido, a continuación;
ld . 2.3) Extracción del compuesto (E) con un solvente orgánico, en particular con AcOEt.
Preferentemente, la hidrólisis en la etapa 1d.2.1 se lleva a cabo con 0.5M de una solución acuosa de NaOH en un solvente polar y prótico, en particular metanol. En una forma de realización preferida, la reacción se realiza a una temperatura que varía de 15°C a 28°C durante 20-40 min., en particular 20 min. Ventajosamente, el tratamiento ácido en la etapa 1d.2.2 se realiza de la siguiente manera:
- la mezcla de reacción es acidificada hasta que el pH sea inferior a 7, en particular con una mezcla de 6N HCI en agua, a continuación;
- se agrega polvo de cinc (0.15 - 0.20 g, en particular 0.18 g para 1g del Compuesto (D)), y a continuación; - agitar la mezcla a una temperatura que varía de 2°C a 8°C, en particular a 5°C, hasta que todo el producto secundario de disulfuro se haya convertido en el producto tiol.
Dado que ni el acetato de etilo ni el agua interfieren en la etapa subsiguiente, la solución final no se seca y el producto (E) se mantiene disuelto a una concentración de 40% de su volumen inicial, para obtener un producto fácil de manipular. Como se esperaba, dicho producto (E) es sensible al aire, debido a la formación del producto secundario de disulfuro. Así, todos los líquidos utilizados fueron desgasificados bajo una atmósfera de gas inerte. El compuesto (E), si se almacena en forma adecuada, es estable durante por lo menos 3 días a temperatura ambiente. Debido a la dificultad para obtener y almacenar un estándar analítico del producto, el análisis cuantitativo de la solución concentrada se realiza por medio de RMN, mientras que la pureza es revisada por HPLC.
Por último, la recuperación de quinina se realiza en la etapa 1e.
En una forma de realización preferida, la etapa 1e comprende las etapas sucesivas siguientes:
le . 1) Combinar las fases acuosas obtenidas en la etapa 1d.2.1 y en la etapa 1d.2.2, a continuación;
1e.2) Añadir 20% en peso de una solución acuosa de NaOH en agua para ajustar el pH a 12, a continuación; 1e.3) Extraer la mezcla resultante obtenida en la etapa 1e.2 con AcOEt, a continuación;
1e.4) Concentrar al vacío la capa orgánica resultante obtenida en la etapa 1e.3, a continuación;
1e.5) Añadir éter de petróleo a una temperatura que varía de 10°C a 20°C, a continuación;
1e.6) Filtrar el sólido resultante obtenido al final de la etapa 1e.5 y recuperar la quinina.
La posibilidad de reutilizar la quinina es una ventaja adicional del proceso de la invención, que conduce a un proceso más económico.
En una forma de realización ilustrativa, la etapa 1 se lleva a cabo partiendo de 195 kg de ácido 2-acetiltio-3-fenilpropiónico (A), el compuesto (D) se pudo obtener a un rendimiento de 68 % usando quinina como agente de resolución. Se obtuvieron 154.6 kg sal de quinina de ácido S-2-acetiltio-3-fenilpropiónico (pureza quiral de 97.5%). El reciclado potencial de la base de quinina fue demostrado a una escala de laboratorio (rendimiento de 80%). Etapa (2)

En la solicitud WO2007/048787, (E) reaccionó con (B) que fue activado con clorosulfonilcloruro, en MeOH/THF, para proporcionar ácido (S)-2-bencil-3-(((S)-2-(terc-butoxicarbonilamino)-4-(metiltio)butil)disulfanil)propanoico (F). En la presente invención, la preparación del compuesto (F) de la siguiente fórmula, comprende las etapas sucesivas siguientes

2a)Hacer reaccionar los primeros 1.1 equivalentes molares de dicho compuesto (E) con 1 equivalente molar de cloruro de clorocarbonil sulfenilo, en un solvente polar y aprótico desgasificado, a continuación;
2b)Hacer reaccionar el intermedio obtenido después de la etapa 2a con 0.9 equivalentes molares del compuesto (B) de la siguiente fórmula

En solución con 1 equivalente molar de Et3N en el mismo solvente desgasificado que se usó en la etapa 2a; En particular, en la etapa 2a, el solvente polar y aprótico es AcOEt.
En una forma de realización preferida, el cloruro de clorocarbonil sulfenilo útil en la etapa 2a, es activado en THF, particularmente a una concentración de 0.2M-0.4M, preferentemente a una concentración de 0.33M, antes de su
adición, en la etapa 2a, a una solución del compuesto (E) a 0.1M-0.2M, preferentemente a 0.15M en solvente polar y aprótico, preferentemente dicho solvente es AcOEt.
Antes de dicha adición, se enfría la mezcla de reacción de cloruro de clorocarbonil sulfenilo a una temperatura que varía de -2°C a 5°C, preferentemente a 0°C bajo una atmósfera inerte durante 10-20 min, en particular 15 min. Ventajosamente, la mezcla de cloruro de clorocarbonil sulfenilo activado se añade de una sola vez, y a continuación la solución resultante se devuelve a una temperatura que varía de 15°C a 25°C, preferentemente de 22°C, y se agita adicionalmente durante 20-40 min., preferentemente 30 min.
En una forma de realización preferida, en la etapa 2b, la mezcla resultante obtenida después de la etapa 2a se agrega gota a gota a una solución del compuesto (B) a una concentración de 0.6M-1.0M, preferentemente a 0.9M, con 1 equivalente molar de Et3N a cloruro de clorocarbonil sulfenilo, en un solvente polar y aprótico, en particular en THF. Después la solución resultante es agitada durante 1 a 2 horas, preferentemente 1 hora, a una temperatura que varía de 15°C a 25°C, preferentemente a 22°C.
La cantidad específica de equivalentes molares del compuesto (B) en la etapa 2b permite evitar la impureza del disulfuro simétrico. Esto es importante dado que dicha impureza es muy difícil de eliminar del producto deseado. Al final de la etapa 2b, el solvente se evapora ventajosamente bajo una presión reducida y el producto crudo resultante es recogido en AcOEt.
Ventajosamente, después de la etapa 2b se llevan a cabo las etapas sucesivas siguientes:
2b.1) añadir agua que comprende 10% en peso de ácido cítrico a la mezcla de reacción obtenida después de la etapa 2b, hasta un pH<7, a continuación;
2b.2) extraer el compuesto (F) con AcOEt.
En una forma de realización preferida, después de la etapa 2b.2, el compuesto (F) es precipitado en hexano y AcOEt y dicho compuesto (F) puede ser recristalizado en una mezcla de hexano/AcOEt, en 5.5/1 - 7.5/1 en una proporción en volumen, preferentemente 6.5/1 en una proporción en volumen.
Todos los solventes fueron desgasificados bajo una atmósfera inerte, para evitar la oxidación de sulfuro.
En la presente divulgación, la síntesis del compuesto (B), útil para la síntesis del compuesto (F), cambia de la de la solicitud WO, y el proceso comprende las etapas siguientes sintéticas:
u Formación de ester activado
después, Agente reductivo
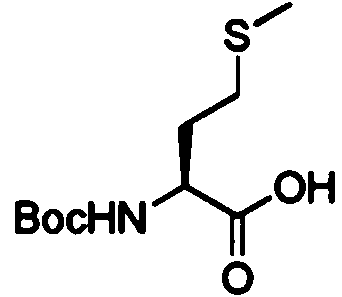

X■ c h 3c o s k
6. Recuperación de B
El compuesto (B) fue obtenido con un rendimiento de 60% molar mediante un proceso de 5 etapas, partiendo desde la Boc-L-Metionina (de origen sintético) disponible comercialmente como se describe en la solicitud de patente WO2007/048787.
En la primera etapa a, la porción de ácido carboxílico de Boc-L-metionina reacciona con cloroformiato de /-butilo en la presencia de N-metilmorfolina en DME de acuerdo con el procedimiento de J. Med. Chem., 35, 1992, 2473. El éster activado resultante no está aislado y en consecuencia reacciona con un agente selectivo reductivo de ésteres. En particular, el reactivo reductivo es, por ejemplo NaBH4.
En la etapa p, el Boc-amino alcohol obtenido después de la etapa a, es protegido a continuación por un grupo protector hidroxilo, en particular un grupo mesilo usando el cloruro de mesilo correspondiente.
En la etapa x, se realiza después una sustitución nucleófila del grupo protector hidroxilo, en presencia de tioacetato de potasio.
Por último, en la etapa 5, el compuesto (B) es recuperado después de una reacción de tipo saponificación. Ventajosamente la saponificación se realiza usando una solución de 1M NaOH en MeOH.
En una forma de realización preferida en la etapa 5, el volumen de metanol usado para la hidrólisis se ha reducido en 30%, en comparación con la cantidad utilizada en el procedimiento del documento WO2007/048787.
Los equivalentes molares de NaOH, en la etapa 5, se han reducido ventajosamente de 3 a 2; y este cambio permite reducir la cantidad de ácido que se necesita para la neutralización con una ganancia general en la productividad del proceso.
En una forma de realización preferida, se observó en la etapa 5, que una adición rápida de la base reduce la cantidad de disulfuro formado, siempre y cuando la temperatura permanezca por debajo de 10°C ventajosamente en un intervalo de 0°C-10°C.
En particular, siguiendo a la adición de la base y después de agitar durante 1-2 h la mezcla básica resultante, la temperatura de la reacción, en la etapa 5, también había aumentado de 18°C-25°C a 28°C-35°C, de esta forma el tiempo de reacción se redujo a 0.5 h, con respecto a 2 h del procedimiento inicial descrito en el documento WO2007/048787.
En este caso, la elaboración ha cambiado ventajosamente: la concentración de metanol se realiza a un pH de aproximadamente 7, ya que se ha observado que el grupo protector Boc es lábil tanto en condiciones básicas como acídicas. Además, para reducir el tiempo general del proceso, ventajosamente únicamente se realiza una extracción en solvente orgánico, y no se realizan lavados acuosos de la fase orgánica, en comparación con el procedimiento inicial en el documento WO2007/048787.
En particular, la solución final no se concentra al residuo oleoso, sino a una concentración molar de aproximadamente 35% (según lo determinado por la RMN cuantitativa), ya que el solvente de la etapa subsiguiente ha sido cambiado.
Este proceso fue escalado a una escala de 50 kg. Como se esperaba, el tiol libre es altamente sensible al oxígeno, dando lugar al correspondiente disulfuro, para evitar la formación de dicho producto secundario de disulfuro, todos los solventes y reactivos fueron desgasificados con varios ciclos de vacío/gas inerte.
Etapa (3):

En la presente invención, el compuesto G de la siguiente fórmula

es sintetizado, en un solvente polar, haciendo reaccionar dicho compuesto F con el amino-éster C de la siguiente fórmula, en donde Y' es un anión:

En una forma de realización preferida, la etapa 3 comprende las etapas sucesivas siguientes:
3a) solubilizar el compuesto F en un solvente polar y aprótico, a continuación;
3b) a la mezcla de reacción obtenida después de la etapa 3a, añadir 1.2 equivalentes molares de hexafluorofosfato de O-benzotriazol-1-il-N,N,N',N'-tetrametiluronio y diisopropil-etilamina, a continuación; 3c) a la mezcla de reacción obtenida después de la etapa 3b, añadir 1.3 equivalentes molares de aminoéster (C).
En la etapa 3a, el compuesto (F) se pone ventajosamente en un solvente orgánico a una concentración de 0.05M-0.3M, preferentemente a 0.1M. En una forma de realización preferida, el compuesto (F) es solubilizado en DMF. En una forma de realización preferida, la mezcla se agita durante 5-10 min. a una temperatura que varía de 2 a 10°C, para evitar la formación de producto secundario.
Después de la etapa 3c y antes de la etapa 4 preferentemente se obtiene el compuesto (G) por medio de las etapas sucesivas siguientes:
3c.1) recuperar la capa orgánica que contiene el compuesto (G);
3c.2) precipitar el compuesto (G) presente en la capa orgánica de la etapa 3c.1 añadiendo una mezcla de éter de petróleo (n-hexano)/AcOEt en una proporción en volumen de 8/1-6/1, preferentemente una proporción en volumen de 7/1.
En una forma de realización preferida, la etapa 3c.1 comprende las etapas sucesivas siguientes:
3c.1.1)añadir agua a la mezcla resultante obtenida en la etapa 3c, a continuación;
3c.1.2)sin concentrar el solvente de reacción, extraer el producto obtenido después de la etapa 3c.1.1 con un solvente aprótico polar, preferentemente AcOEt.
En particular en la etapa 3c.1.2, el producto es purificado por precipitación con una mezcla de éter de petróleo (nhexano) y AcOEt a una temperatura que varía de 20°C a 25°C sin cromatografía de columna. El compuesto (G) es estable a una temperatura que varía de 18°C a 25°C, y fue obtenido con un rendimiento de 87% molar de hasta una escala de 43.7 kg.
En la presente invención, la síntesis del compuesto (C), útil en la etapa 3c, cambia a partir de la solicitud WO, y el proceso comprende las etapas siguientes:

En una forma de realización preferida, el compuesto (C) se prepara por medio de un proceso que comprende las siguientes etapas sintéticas sucesivas:
£) hacer reaccionar 1.1 equivalentes molares de Boc-glicina con 1.2 equivalentes molares de Et3N en un solvente polar y aprótico, a continuación;
O) hacer reaccionar el producto obtenido en la etapa £ con 1 equivalente molar de etil-1-cloroetilcarbonato, y 0.2 equivalentes molares de yoduro de potasio en el mismo solvente de la etapa £, a continuación;
Y) hacer reaccionar el producto obtenido en la etapa O con 2 equivalentes molares de gas de HCl en AcOEt a una temperatura que varía de 5°C a 10°C, y recuperar C.
En una forma de realización preferida en la etapa £, la estoquiometría ha cambiado y la cantidad de Boc-glicina aumentó a 1.1 equivalentes molares, con respecto a 0.95 equivalentes molares en el documento WO2007/048787, y la de Et3N a 1.2 equivalentes molares desde 1 equivalente molar. El solvente polar y aprótico usado en la etapa £, preferentemente es una mezcla de AcOEt/DMF en una proporción de 10/1 en peso/peso. La introducción de
DMF como cosolvente se realiza para aumentar la polaridad y el punto de ebullición de la mezcla de reacción. La temperatura de las etapas £ y O es ventajosamente la temperatura de reflujo. Al final de la etapa O, únicamente se un lavado ácido con l0% en peso de ácido cítrico en agua se realiza preferentemente para eliminar el exceso de base, en comparación con el procedimiento del documento WO2007/048787, mientras que el segundo lavado básico con 10% en peso de NaHCO3 en agua, permite la eliminación completa del exceso de Boc-Glicina.
Debido a la inestabilidad del producto obtenido después de la etapa O, en gel de sílice, se realiza un control de proceso por medio de RMN en lugar de TLC. El producto es un aceite amarillo y es estable a una temperatura que varía de 18°C a 25°C.
En particular, en la etapa O, se utilizó yoduro de potasio en lugar de la correspondiente sal sodio, debido a que presenta una solubilidad superior en AcOEt y está más fácilmente disponible en el mercado. Debido a esta propiedad, la cantidad molar de cristales de yoduro de potasio ha sido ventajosamente disminuida a 0.2 equivalentes molares (en lugar de los 0.3 equivalentes molares usados en el documento WO2007/048787). Una cantidad de yoduro de potasio inferior a 0.2 equivalentes molares disminuye la velocidad de reacción.
En la etapa y, para la preparación de la sal de éster etílico de «cascada» de glicina (C), la presencia de agua en la solución de partida produce una pérdida de rendimiento, debido a la solubilidad del producto. Por esta razón, preferentemente se introduce un IPC para medir la cantidad de agua residual en la solución orgánica, antes de la desprotección del grupo Boc-amino en la etapa y (el contenido de agua en AcOEt deberá ser no superior a 0.1% en peso).
Preferentemente para la etapa y, la introducción de AcOEt como solvente en lugar de DCM con respecto al procedimiento del documento WO2007/048787, hace posible la disminución del efecto del agua residual en la solución de partida.
En particular, el ácido utilizado en la etapa y, es un gas HCl seco, y de esta forma, es posible usar únicamente 2 equivalentes molares de dicho ácido. Probablemente debido a la mayor solubilidad del gas HCl seco en AcOEt, se encontró en particular en la etapa y que, con 2 equivalentes molares de ácido, la conversión es superior a una temperatura que varía de 5°C a 10 °C que a una temperatura que varía de 20°C a 25 °C.
El orden de adición fue invertido ventajosamente, es decir, una solución del aminoácido protegido, obtenido después de la etapa y, fue añadida a una solución fría de dicho ácido. Otra ventaja de la adición inversa es que el producto empieza a precipitar realmente rápido, conservando así la calidad del producto aislado (C).
En una forma de realización preferida, se observó que la adición del compuesto amino protegido obtenido después de la etapa p, en solución ácida en 2-4 h, preferentemente en 3 h, permitió una reacción más completa.
En este caso, debido a la naturaleza del producto, se eligió la RMN como técnica analítica para la evaluación de la conversión. Si la cantidad de impureza determinada por RMN es de por lo menos 0.5%, se debe hacer la purificación del producto por lavado con isopropanol. El producto es un sólido de blanco a amarillo claro. Es estable a temperatura ambiente pero es sensible a la humedad.
En una forma de realización ilustrativa la síntesis del compuesto (C) fue escalada a una escala de 30 kg sin ningún problema en un rendimiento de 69% partiendo de Boc-Glicina disponible comercialmente.
Preferentemente, después del procedimiento de Barcelo et al. (Synthesis, 1986, 627), en el caso de la síntesis de 1-cloroetilcarbonato de etilo útil en la etapa O, se usó AcOEt como solvente en lugar de DCM.
En una forma de realización preferida, es preferida la utilización de piridina en lugar de Et3N para disminuir la cantidad de impurezas durante la síntesis de 1-cloroetilcarbonato de etilo.
En particular, ha cambiado la estequiometría de la reacción: la cantidad de etanol y piridina aumentó a 1.05 y 1.1 equivalentes molares, respectivamente. Esta modificación permitió una mayor conversión del material de partida, sin afectar la calidad del 1-cloroetilcarbonato de etilo deseado al final de la elaboración.
La temperatura de la mezcla de reacción durante la adición de piridina ha aumentado ventajosamente, manteniéndola por debajo de los 10°C. Esto permite un control de temperatura más fácil en una producción a gran escala: por lo tanto, la temperatura de reacción se mantiene en un intervalo de 2°C a 8°C durante el tiempo de reacción. Debido al aumento en la cantidad molar de etanol y piridina añadidos, el tiempo de reacción disminuyó a aproximadamente 1 hora de tiempo de reacción.
Preferentemente, la elaboración cambia, en particular se ha evitado la filtración de las sales y únicamente se realizan dos lavados (uno ácido y uno básico) de la fase orgánica.
Debido a la inestabilidad del 1-cloroetilcarbonato de etilo y a la ausencia de grupos cromóforos, se selecciona RMN
como técnica analítica. El producto es un aceite amarillo claro y es estable a una temperatura que varía de 18°C a 28°C, preferentemente a 22°C.
Etapa (4)

En la presente invención, la recuperación de la sal (I) de la siguiente fórmula

comprende las etapas sucesivas siguientes:
4a) añadir 5 equivalentes molares de ácido fórmico en dicho compuesto G
4b) intercambiar el formiato por un fumarato usando una tecnología de flujo continuo.
La desprotección del grupo Boc-amino de (G) se realiza en condición acídica, usando ácido fórmico, para proporcionar la sal formiato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio (H).
En una forma de realización preferida, en la etapa 4a, después de añadir ácido fórmico, el proceso comprende además las etapas sucesivas siguientes:
4a.1) coevaporar el producto obtenido después de añadir ácido fórmico con tolueno, proporcionando el compuesto (H) y la capa orgánica con tolueno, a continuación;
4a.2) añadir AcOEt al compuesto (H) obtenido después de la etapa 4a.1, y lavar la mezcla resultante con salmuera a una temperatura que varía de 0°C a 10°C.
En la etapa 4a.1, el compuesto (H) obtenido es coevaporado en particular varias veces con tolueno, hasta haber eliminado el exceso de ácido fórmico.
En una forma de realización preferida en la etapa 4b, se lleva a cabo un intercambio de aniones por medio de un proceso de flujo continuo que comprende las etapas sucesivas siguientes:
4b.1) a una temperatura que varía de 0°C a 10°C, mantener una solución de 3.3% en peso del compuesto (H) en AcOEt a un pH que varía de 8 a 9, preferentemente agregando una solución de 2% NaOH en peso en agua a dicha solución del compuesto (H), a continuación;
4b.2) añadir una solución de ácido fumárico al 5% en peso en EtOH a la mezcla obtenida después de la etapa 4b.1 para aislar el compuesto (I) cristalizado usando un proceso de flujo continuo.
El proceso de flujo continuo es un procedimiento automático en el que se añaden los reactivos en continuo en un reactor, en lugar de ser cargados únicamente una vez. Este método presenta varias desventajas, como el control de la estequiometría por flujo relativo, la mezcla es rápida y completa y la temperatura es uniforme. En el presente proceso, esto permite que exista un intermedio inestable en una cantidad baja y en una manera de transición. Al final de la etapa 4a.2, se preparó una solución de 3.3% en peso del compuesto (H) en AcOEt, y se usó a continuación en una reacción de intercambio aniónico en la etapa 4b con ácido fumárico.
La reacción de intercambio aniónico en la etapa 4b representa la fase clave del proceso, debido a que la base
amino libre, obtenida ventajosamente después de la etapa 4b.1, una vez generada a partir de la correspondiente sal formiato (H), preferentemente debe ser convertida lo más pronto posible en la sal fumarato (I) que a su vez debe ser rápidamente cristalizada, para evitar reacciones secundarias.
Se observó, durante la síntesis en lotes de c-GMP preliminar, que al realizar estas operaciones en lotes, aumentó la formación de productos secundarios con la escala de la preparación. En consecuencia, la configuración de estas operaciones en un modo continuo, permite resolver el problema de escalar el proceso.

Esquema 4: Vía de acceso posible para la descomposición de la base amino libre del compuesto (I). La fase crítica en la formación del compuesto (I) es la etapa 4b en la que el compuesto (H) es tratado en condiciones básicas. Dicha etapa 4c proporciona la base amino libre del compuesto (I), que es inestable. La base amino libre puede evolucionar a éster etílico, dímero y compuesto sulfuro oxidado. El proceso de flujo continuo permite evitar estas formaciones de impurezas.
Descripción de las figuras
La figura 1 describe el diagrama de bloques de la etapa 4b exponiendo el proceso de flujo continuo.
Para la realización de la etapa 4b, la solución de 3.3% en peso del compuesto (H) en AcOEt fue introducida a través de 1. Consecutivamente se agregó la solución de 2% NaOH en peso en agua a través de 2.
Etapa 4b.1: la mezcla reaccionó continuamente a 5°C y a un pH que varía de 8-9, permitiendo una conversión de la sal formiato (H) en el compuesto de base libre (I).
En la sección 3, se obtuvo una mezcla bifásica, que después fue extraída continuamente, a 5°C en 4. La fase acuosa fue almacenada en un contenedor en 5, y la fase orgánica obtenida en 6, fue agregada en la etapa 4b.2. Una solución de 5% en peso de ácido fumárico en EtOH fue introducida a través de 7, a la etapa 4b.2. La mezcla en la etapa 4b.2 reaccionó continuamente a 5°C. Se obtuvo una solución del compuesto (I) en 8. Después se realizó una destilación continua del solvente en 9. El solvente destilado fue almacenado en un contenedor en 10, y se obtuvo el compuesto (I) en 11.
La figura 2 describe el aparato continuo para la preparación del compuesto (I).
Para la realización de la etapa 4b a una escala industrial, se ensambló el aparato continuo que consiste en un mezclador decantador y un reactor de tanque agitado continuo (CSTR2). El mezclador decantador estaba formado por un reactor de tanque agitado continuo (CSTR1) y un decantador 4. El CSTR1 fue el reactor en el que se realizó la etapa 4b.1. El CSTR2 fue el reactor en el que se realizó la etapa 4b.2.
El tanque 1 fue cargado con la solución de 3.3% en peso del compuesto (H) en AcOEt, y dicha solución fue agregada a un reactor CSTR1 con una alimentación F1 a 1095 g/h. En consecuencia, el tanque 2 fue cargado con la solución de 2% NaOH en peso en agua y después dicha solución fue agregada al reactor CRST1 con una alimentación F2 a 348 g/h. El pH fue controlado a través de 1 durante toda la agitación para mantenerla a un intervalo de 8 a 8.5. La mezcla de reacción bifásica se desbordó desde CSTR1 hacia el decantador en el que se separaron las fases. La fase acuosa fue almacenada en el tanque 4 con una salida 3 de 375 g/h. La fase orgánica que contiene la base libre del compuesto (I) fue agregada al reactor CSTR2 con una alimentación F3 a 1068 g/h.
En consecuencia, el tanque 3 fue cargado con la solución de ácido fumárico al 5% en peso en EtOH y después dicha solución fue agregada al reactor CRST2 con una alimentación F4 a 142 g/h.
F4 fue regulada para presentar una relación molar de 0.95 entre una cantidad teórica del compuesto (I) contenido en F3 y ácido fumárico. La mezcla de reacción que contenía la solución del compuesto (I) desbordó desde CSTR2 al tanque 5 con una salida 5 de 1201 g/h, en el que fue recogida dicha solución que contenía el compuesto (I) y almacenada durante la duración del experimento.
Todo el equipo fue termostatizado a una temperatura de 5°C.
La concentración de la solución de fumarato del compuesto (I) en bruto resultante del proceso continuo, fue completada por medio de destilación al vacío en lotes, y después se hizo una cristalización en diisopropil éter. El compuesto (I) fue obtenido como un sólido blanco, con un rendimiento molar general, a partir del compuesto (F), de 84%.
El compuesto aislado (I), obtenido de acuerdo con el método desarrollado, fue analizado por HPLC, y se comparó con la muestra del compuesto (I), obtenido de acuerdo con el procedimiento en lotes previo (WO2007/048787). El contenido de impurezas es significativamente menor en la muestra del compuesto (I) del proceso continuo. En una forma de realización ilustrativa, se realizó un proceso de flujo continuo con 0.15kg del compuesto (G) tamaño a escala.
Ejemplos
En la presente invención, “temperatura ambiente” significa una temperatura que varía de 18°C a 28°C, preferentemente que varía de 20°C a 25°C.
1- Estudios en una cantidad de quinina útil para la etapa 1.
Fueron probados diferentes proveedores de quinina como agente de resolución, en las condiciones descritas anteriormente para la resolución cinética en la etapa 1 (tabla 1). Los resultados obtenidos presentaron una ligera diferencia en la pureza quiral con la muestra Vital Health Care.
Tabla 1: diferentes proveedores de quinina probados para la resolución cinética en la etapa 1

Se asumió que las dos muestras de quinina tenían un ensayo diferente y por lo tanto se utilizó un equivalente diferente de “quinina pura” en las dos pruebas.
Utilizando la muestra Buchler, la resolución fue probada con tres equivalentes molares diferentes de quinina (0.5, 0.55 y 0.6) partiendo de una mezcla racémica del compuesto (A), y proporcionando el compuesto (D) aislado como aceite (tabla 2).
Tabla 2: Resolución cinética con diferentes equivalentes molares de quinina del proveedor Buchler

Estas pruebas demostraron que un exceso de quinina en comparación con los 0.5 equivalentes molares del S-enantiómero, produjo una pérdida de pureza quiral.
En una tendencia opuesta, la disminución equivalente de quinina a 0.5 equivalentes molares y usando 1 equivalente molar del compuesto (A) condujo a una disminución en la recuperación de S enantiómero (63.4% vs 66.6%) sin ninguna ganancia significativa en la pureza quiral. Estos resultados podrían explicar por qué se podría obtener una mejor pureza quiral usando 0.6 equivalente molar de quinina de un ensayo inferior (tabla 2). Dado que la muestra de quinina de Buchler fue tomada de un lote industrial en existencia y dado su perfil de calidad superior, fue seleccionado Buchler como el proveedor.
En una forma de realización ilustrativa, se retuvieron preferentemente 0.55 equivalentes molares de quinina de los ensayos de laboratorio como compromiso para la pureza quiral y la recuperación de enantiómero.
La sal de quinina sólida fue probada por tensión térmica a 100°C durante 36 horas y se asume que es estable en esta etapa. A pesar de un cambio de aspecto de cristalino blanco a sólido beis semifundido, la muestra no presentó ninguna degradación visible por HPLC.
Desde un punto de vista industrial, el almacenamiento del compuesto (D) se hizo bajo su correspondiente sal de quinina, y dicha sal de quinina después se añade en la siguiente etapa 1d.2 en la que el solvente es cambiado de acetato de etilo a metanol.
2- Etapa a: resolución/recristalización
En la tabla 3 se describe una resolución en una escala de 5kg.
Tabla 3: materiales utilizados para la resolución de 5 kg del compuesto (A) con quinina

Volumen máximo: 103 l
Procedimiento general para la resolución cinética industrial
Fueron evaluados diferentes parámetros y a continuación se describe un procedimiento típico.
Procedimiento industrial preferido para cristalización:
1) Cargar 90 l de AcOEt, 5.0 kg (21 mol) del compuesto (A) y 4.08 kg (12.6 mol) de quinina en un reactor. 2) Enjuagar el embudo de adición con 10 l de AcOEt para retirar el sólido detenido en la pared del embudo hacia el reactor, y agitar la mezcla a una temperatura que varía de 10°C a 15oC durante 20 min;
3) Calentar la mezcla a 45oC y agitar la mezcla a 45oC hasta formar una solución clara;
4) Enfriar la solución a una velocidad de 5oC/h a 40oC;
5) Añadir 1 g de semilla que presenta 86% de pureza quiral;
6) Enfriar la mezcla a una velocidad de 5oC/h hasta una temperatura que varía de 10°C a 20oC;
7) Agitar la mezcla a una temperatura que varía de 10°C a 15oC durante 16 horas adicionales;
8) Filtrar el producto de sal de quinina después de la cristalización y mantener el filtrado en un contenedor (la temperatura ambiente es de 11°C cuando la mezcla es realizada y filtrada);
9) Analizar la torta de filtrado con HPLC para verificar la pureza quiral, preferentemente se obtiene 84% de pureza quiral;
Procedimiento industrial preferido para recristalización:
10) Cargar la torta húmeda obtenida en la operación 9 en otro reactor;
11) Añadir 70 l de AcOEt;
12) Calentar la mezcla a 60oC y bajo agitación hasta que se disuelva todo el sólido;
13) Enfriar la solución a una velocidad de 5oC/h a temperatura ambiente (cuando la temperatura alcanza los
40oC, empieza a formarse el precipitado);
14) Agitar la mezcla a una temperatura que varía de 10°C a 15oC durante una noche (16 h);
15) Filtrar la sal de quinina recristalizada (la temperatura ambiente es de 11 oC cuando la mezcla es llevada a cabo y filtrada) y mantener el filtrado en un contenedor;
16) Analizar la torta de filtrado con HPLC para verificar la pureza quiral, preferentemente se obtiene 95.5% de pureza quiral;
Recuperación del solvente de AcOEt utilizado en en el proceso anterior (operaciones 1 a 16):
17) Cargar 30 l del filtrado obtenido en la operación 15 en un reactor;
18) Añadir 10 l de una solución acuosa de 0.5 M de NaOH en agua, hasta pH>10, y agitar la mezcla a temperatura ambiente durante 20 min;
19) Separar la fase orgánica y almacenar la fase acuosa en un contenedor;
20) Analizar la fase orgánica con HPLC.
Si no se puede detectar ácido 2-acetiltiometil-3-fenilpropiónico (A), ir a la siguiente operación. Si se puede detectar, lavar la fase orgánica con agua hasta que ya no se pueda detectar ácido 2-acetiltiometil-3-fenilpropiónico (A) en la fase orgánica;
Recuperación del compuesto libre (D):
21) Transferir la fase orgánica obtenida en la operación 19 a un reactor;
22) Añadir la torta húmeda obtenida en la operación 15;
23) Añadir 10 l de agua y 2 l de una solución acuosa de HCl 12 N y agitar la mezcla a temperatura ambiente, durante 30 minutos adicionales (pH de la fase acuosa es ~1);
24) Separar la fase orgánica del producto y mantener la fase acuosa en un contenedor;
25) Lavar la fase orgánica con aproximadamente 5 l de agua, y monitorizar el lavado por medio de HPLC para detectar quinina en la fase orgánica;
26) Concentrar la fase orgánica a una temperatura que varía de 40°C a 45oC y bajo vacío para remover el solvente de la manera más completa posible;
27) Añadir 2.5 l de metanol en el residuo y volver a concentrar la mezcla a una temperatura que varía de 40°C a 45oC y al vacío para eliminar el solvente restante de AcOEt;
28) Repetir una vez la operación 27 para proporcionar 2.1 kg de producto de aceite del compuesto (D); 29) El análisis por RMN muestra una conversión de sal de quinina de (D) en el compuesto libre (D) de 89.8%.
De esta forma se obtienen 1884 g del compuesto (D) con un rendimiento de 75.4% con relación al (S) enantiómero, y 98.9% de pureza química.
2.2 Etapa b: Recuperación de quinina al final de la etapa 1.
Otro punto atractivo de la resolución química es la recuperación del agente quiral, como se describe a continuación:
1) Cargar el filtrado restante (aproximadamente 30 l) obtenido en la operación de 15 en la etapa a en un reactor;
2) Añadir 10 l de una solución acuosa de 0.5 M de NaOH en agua y agitar la mezcla a temperatura ambiente, durante 20 minutos adicionales (el pH de la fase acuosa es de aproximadamente 12);
3) Separar la fase orgánica y almacenar la fase acuosa en un contenedor;
4) Lavar la fase orgánica con agua y monitorizar el lavado por medio de HPLC para detectar ácido 2-acetiltiometil-3-fenilpropiónico en la fase orgánica;
5) Mantener la fase orgánica en un contenedor;
6) Cargar el filtrado (aproximadamente 90 l) obtenido en la operación 8 en la etapa a a otro reactor 7) Añadir 6 l de una solución acuosa concentrada de HCl para ajustar el pH a 1~2
8) Separar la fase acuosa y almacenar la fase orgánica en un contenedor para recuperar el AcOEt por destilación
9) Combinar la fase acuosa obtenida en las operaciones 19 y 24 en la etapa 1 y la fase acuosa obtenida en las operaciones 3 y 8 en la etapa b;
10) Añadir 20% en peso de una solución acuosa de NaOH en agua para ajustar el pH a 12;
11) Añadir el AcOEt obtenido en la operación 5 y agitar la mezcla a temperatura ambiente durante 20 minutos adicionales;
12) Separar la fase orgánica y extraer la fase acuosa con 10 l de AcOEt;
13) Combinar las soluciones orgánicas y concentrarlas a una temperatura que varía de 40°C a 45oC y al vacío a un volumen de aproximadamente 3.0 l;
14) Bajo una agitación vigorosa, añadir 10 l de éter de petróleo a una temperatura que varía de 10°C a 20°C; 15) Agitar la mezcla a una temperatura que varía de 10°C a 20°C durante 1 hora adicional;
16) Filtrar la mezcla para aislar el producto de quinina sólida;
17) Secar la torta de filtrado a una temperatura que varía de 50°C a 55oC y al vacío para proporcionar 1832 g de producto sólido blanco de quinina (80% de recuperación).
Usando un procedimiento similar, se fabricó un lote tal como 150 kg de sal de quinina.
2.3 Determinación del valor de e.e. del compuesto (D)
Procedimiento:
1) Mezclar L-Ala-OMe.HCl con 1.0 equivalente molar del compuesto (D) en DCM, a continuación;
2) Agitar a una temperatura que varía de 10°C a 20oC hasta formar una solución, a continuación;
3) Añadir 1.5 equivalentes molares de EDCI y 2 equivalentes molares de Et3N a temperatura ambiente, y agitar durante 1-2 minutos adicionales, a continuación;
4) Remover el solvente al vacío, a continuación;
5) Añadir AcOEt para disolver el residuo, a continuación;
6) Lavar consecutivamente la solución con 10% en peso de ácido cítrico en agua, a continuación con una solución acuosa de bicarbonato de sodio, a continuación con agua y a continuación con salmuera, a continuación;
7) Remover el AcOEt y recuperar un producto sólido.
8) Disolver dicho producto sólido obtenido en la operación 7 en CDCh para un análisis de RMN de 1H Métodos para calcular el exceso enantiomérico:
1) Integración del pico a 6.0 ppm - Integración del pico a 5.85 ppm (sobre la base del protón de amida), o; 2) Integración del pico a 1.34 ppm - Integración del pico a 1.04 ppm (sobre la base del protón de metilo en la parte de alanina).
Usando el protocolo que se describe en la figura 2, en la tabla 4 se describe una escala de tecnología continua.
Tabla 4: Materiales utilizados para el proceso continuo


1) Cargar un matraz de fondo redondo de 1 l con 0.565 kg de ácido fórmico;
2) Mantener la temperatura a 25°C;
3) Purgar un matraz de fondo redondo de 1 l con N2;
4) Cargar el matraz de fondo redondo de 1 l con 0.15 kg del compuesto (G), y mantener la temperatura a 25°C; 5) Calentar el matraz de fondo redondo de 1 l unitario a 30°C;
6) Hacer reaccionar el matraz de fondo redondo de 1 l unitario por medio de la desprotección, durante 5 horas adicionales;
7) La temperatura final del lote es de 30°C;
8) Se hace la prueba de QC al material en el matraz de fondo redondo de 1 l unitario en 30 min., y la especificación obtenida muestra: (G) <2%.
Si no se cumple la especificación, la prueba sigue durante 1 hora más y se repite.
9) Destilar el lote en el matraz de fondo redondo de 1 l unitario.
10) La presión de fondo es de 80 mm-Hg y la temperatura máxima es de 40°C.
11) Destilar aproximadamente 65% de la masa inicial;
12) - Cargar un matraz de fondo redondo de 1 l con 0.275 kg de tolueno.
13) Destilar el lote en el matraz de fondo redondo de 1 l unitario;
14) La presión de fondo es de 65 mm-Hg, y la temperatura máxima es de 40°C.
15) Destilar aproximadamente 63% de la masa inicial;
16) Cargar un matraz de fondo redondo de 1 l con 0.275 kg de tolueno;
17) Destilar el lote en el matraz de fondo redondo de 1 l unitario;
18) La presión de fondo es de 50 mm-Hg, y la temperatura máxima es de 40°C.
19) Destilar aproximadamente 60% de la masa inicial;
20) Transferir el contenido del matraz de fondo redondo de 1 l unitario a un matraz de fondo redondo de 5 l; 21) Transferir 100% del contenido del recipiente;
22) Enfriar un matraz de fondo redondo de 5 l unitario a una temperatura que varía de 5°C;
23) Cargar el matraz de fondo redondo de 5 l con 1.99 kg de AcOEt;
24) Mantener la temperatura a un intervalo de 0°C a 10°C;
25) Cargar el matraz de fondo redondo de 5 l con 1.09 kg de una solución de NaCI al 7.2% en peso.
26) Mantener la temperatura a un intervalo de 5°C;
27) Extraer en el matraz de fondo redondo de 5 l unitario durante 10 minutos;
28) La corriente de la capa inferior, denominada fase de agua del lavado, se envía al drenaje.
29) Cargar un matraz de fondo redondo de 5 l con 1.974 kg de AcOEt;
30) Mantener la temperatura a un intervalo de 5°C;
31) Transferir 100% del contenido del recipiente del matraz de fondo redondo de 5 l unitario a un tanque 1; 32) Cargar un matraz de 1 l con 0.56 kg de etanol;
33) Mantener la temperatura a 25°C;
34) Cargar un matraz de 1 l con 0.0295 kg de ácido (2E)-but-2-enedioico;
35) Mantener la temperatura a 25°C;
36) Transferir 100% del contenido de recipiente del matraz de 1 l unitario a un tanque 3;
37) Hacer reaccionar continuamente la mezcla del tanque 1 unitario en CSTR 1 unitario a través de neutralización de reacción.
38) La velocidad de alimentación de la mezcla es de 1.097 kg/h.
39) La corriente se denomina formiato PL37 en AcOEt a sección continua.
40) La temperatura final es de 5°C.
41) La corriente de producto, denominada mezcla bifásica de neutralización, es enviada a un decantador.
42) Añadir continuamente una solución de NaOH al 2% en peso en agua desde el tanque 2 a una velocidad de 0.348 kg/h y la alimentación se denomina 2% NaOH a sección continua;
43) Extraer continuamente la mezcla de CSTR 1 en el decantador unitario;
44) La capa superior, denominada fase orgánica PL37 de neutralización, es enviada a CSTR 2.
45) La capa inferior es enviada al desagüe.
46) Hacer reaccionar continuamente la mezcla del decantador en CSTR 2 unitario a través de una reacción de salificación, con una solución de ácido fumárico al 5% en peso en EtOH.
47) La temperatura final varía de 5°C.
48) La corriente de producto es enviada al tanque 5.
49) Añadir continuamente el material del tanque 3 a una velocidad de 0.142 kg/h, y la alimentación es denominada solución de ácido fumárico a salificación continua;
50) Para alcanzar el estado estable, el aparato continuo se hace funcionar durante 20 min, antes de empezar a recoger la solución del compuesto (I) en el tanque 5. En la siguiente tabla se encuentran las cantidades de cada solución utilizadas para alcanzar el estado estable.
Tabla 5: Cantidad de solución usada para realizar la formación continua del compuesto (I) en los tanques 1 a 3

A continuación en aparato continuo se hace funcionar durante 3.45 h. En la siguiente tabla están señaladas las
corrientes de entrada y de salida.
Tabla 6: Corriente de entrada/salida en la formación continua del compuesto (I) en los tanques 1 a 5

51) Destilar continuamente la mezcla del tanque 5 unitario en un evaporador de película delgada unitario. La temperatura de camisa es de 40°C. La presión residual es de 50 mm-Hg. La temperatura general del condensador es de -5°C.
52) La corriente de destilado es enviada al desagüe, y la corriente inferior es enviada a un matraz de fondo redondo de 3 l.
Tabla 7: Corriente de entrada/salida en la formación continua del compuesto (I) en los tanques 5 a 7

53) Destilar el lote en el matraz de fondo redondo de 3 l unitario. La parte superior es enviada al desagüe. La presión de fondo es de 100 mm-Hg, y la temperatura máxima es de 30°C.
54) Enfriar el matraz de fondo redondo de 3 l unitario a una temperatura que varía de 20°C.
55) Cargar el matraz de fondo redondo de 3 l con 1.56 kg de diisopropil éter,
56) El tiempo de carga es de 70 min.
57) La semilla es cargada para cristalización.
58) Mantener la temperatura que varía desde 20°C.
59) Enfriar un matraz de fondo redondo de 3 l unitario a un intervalo desde 5°C.
60) Cristalizar el lote en el matraz de fondo redondo de 3 l unitario, durante 3h.
61) Filtrar el lote del matraz de fondo redondo de 3 l unitario en un filtro.
62) El tiempo de transferencia de la suspensión es de 1 hora, y el licor madre es enviado al desagüe.
63) Lavar 2 veces la torta en un filtro unitario, en particular para cada lavado, usando 0.10 kg de diisopropil éter.
64) Transferir 100% del contenido de recipiente del filtro unitario a un secador.
65) Secar el lote en un secador unitario durante un tiempo adicional de 16 horas, a una temperatura que varía desde 25°C, y la presión de secado es de 50 mm-Hg.
66) Transferir el contenido del secador unitario al almacenamiento.
67) La corriente de transferencia es denominada PL37 fumarato seco, que se obtiene en 118 g, y con un rendimiento general de 84% partiendo del compuesto (G).
Claims (15)
1. Proceso industrial para la preparación de sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio de la fórmula (I) siguiente:

que comprende las etapas sintéticas sucesivas siguientes realizadas en solventes orgánicos desgasificados polares o apolares, próticos o apróticos:
1) preparar el compuesto (E) de la fórmula siguiente con un exceso enantiomérico superior a 95%

con 0.5 - 0.6 equivalentes molares de quinina en solventes orgánicos polares y apróticos, preferentemente en acetato de etilo;
lb ) cristalizar la sal de quinina resultante a una temperatura que varía de 10°C a 20°C, en el mismo solvente orgánico que el utilizado en la etapa 1a, en el que la cristalización se inicia añadiendo unos cuantos cristales de la sal de enantiómero deseada para iniciar la cristalización, a continuación;
lc ) recristalizar la sal obtenida después de la etapa 1b al mismo intervalo de temperatura y el mismo solvente que el utilizado en la etapa 1b;
ld ) recuperar el compuesto (E):
1d.1)recuperando el compuesto (D) de la fórmula siguiente

1d.2)desproteger el tiolacetato en un solvente polar y prótico tal como MeOH;
1e) recuperar la quinina;
2) preparar el compuesto (F) de la fórmula siguiente

2a) haciendo reaccionar los primeros 1.1 equivalentes molares de dicho compuesto (E) con 1 equivalente molar de cloruro de clorocarbonil sulfenilo, en un solvente polar y aprótico, a continuación;
2b) hacer reaccionar el intermedio obtenido después de la etapa 2a con 0.9 equivalentes molares del compuesto (B) de la fórmula siguiente
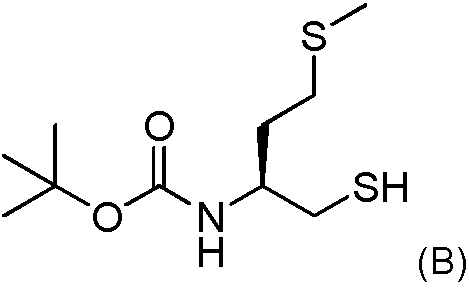
en solución con 1 equivalente molar de Et3N en el mismo solvente que el utilizado en la etapa 2a;
3) preparar el compuesto (G) de la fórmula siguiente

haciendo reaccionar dicho compuesto (F) con el amino-éster C de la fórmula siguiente, en el que Y- es un anión:
Y H3N ^ r 0Y 0Y 0Et
O 1 O (C)
en solvente polar;
4) a continuación, recuperar la sal (I) de la fórmula siguiente

4a) añadiendo 5 equivalentes molares de ácido fórmico en dicho compuesto (G);
4b) intercambiar el formiato por un fumarato utilizando una tecnología de flujo continuo.
2. Proceso industrial según la reivindicación 1, en el que la cristalización en la etapa 1b comprende las etapas sucesivas siguientes:
1b.1)disolver la sal de quinina a temperatura de solubilización, a continuación;
1b.2)enfriar la mezcla obtenida en la etapa 1b.1, preferentemente a una velocidad de 3-10°C/h, hasta una temperatura que varía de 10°C a 20°C;
1b.3)aislar la sal de quinina obtenida después de la etapa 1b.2 por filtración.
3. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que la etapa 1d.1 comprende además las etapas sucesivas siguientes:
1d.1.1) la suspensión de la sal de quinina obtenida en la etapa 1c.3 o 1c.4 en HCl en solución en agua, a continuación;
1d.1.2) la extracción del compuesto (D) de la fórmula siguiente

con un solvente aprótico y polar, en particular con acetato de etilo, a continuación;
1d.1.3) la concentración al vacío para obtener un aceite.
4. Proceso industrial según la reivindicación 3, que comprende además después de la etapa 1d.1, las etapas sucesivas siguientes:
1d.2.1) la hidrólisis alcalina en un solvente polar y prótico, a continuación
1d.2.2) el tratamiento ácido, a continuación;
ld . 2.3) la extracción del compuesto (E) con un solvente orgánico.
5. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que la recuperación de la quinina en la etapa 1e comprende las etapas sucesivas siguientes:
le . 1) combinar las fases acuosas obtenidas en la etapa 1d.2.1 y en la etapa 1d.2.2, a continuación;
1e.2) añadir 20% en peso de una solución acuosa de NaOH en agua para ajustar el pH a 12, a continuación; 1e.3) extraer la mezcla resultante obtenida en la etapa 1e.2 con AcOEt, a continuación;
1e.4) concentrar al vacío la capa orgánica resultante obtenida en la etapa 1e.3, a continuación;
1e.5) añadir éter de petróleo a una temperatura que varía de 10°C a 20°C, a continuación;
1e.6) filtrar el sólido resultante obtenido al final de la etapa 1e.5 y recuperar la quinina.
6. Proceso industrial según cualquiera de las reivindicaciones anteriores, que comprende además después de la etapa 2b y antes de la etapa 3 las etapas sucesivas siguientes:
2b.1) añadir agua que comprende 10% en peso de ácido cítrico a la mezcla de reacción obtenida después de la etapa 2b, hasta un pH<7, a continuación;
2b.2) extraer el compuesto (F) con AcOEt.
7. Proceso industrial según la reivindicación 6, en el que después de la etapa 2b.2 el compuesto (F) es precipitado en hexano y puede ser recristalizado a partir de hexano/AcOEt, en 5.5/1 - 7.5/1 en una proporción en volumen, preferentemente 6.5/1 en una proporción en volumen.
8. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que la etapa 3 comprende las etapas siguientes:
3a) solubilizar el compuesto (F) en un solvente polar y aprótico, a continuación
3b) a la mezcla de reacción obtenida después de la etapa 3a, añadirle 1.2 equivalentes molares de hexafluorofosfato de O-benzotriazol-1-il-N,N,N',N-tetrametiluronio y diisopropil-etilamina,
3c) a la mezcla de reacción obtenida después de la etapa 3b, añadirle 1.3 equivalentes molares del aminoéster (C).
9. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que la etapa 3 se realiza a una temperatura comprendida entre 2°C y 10°C.
10. Proceso industrial según la reivindicación 8, en el que en la etapa 3a, el compuesto (F) se coloca en un solvente orgánico a una concentración de 0.05M-0.3M, preferentemente a 0.1M.
11. Proceso industrial según la reivindicación 8, en el que después de la etapa 3c se obtiene el compuesto (G) mediante las etapas sucesivas siguientes:
3c.1) recuperar la capa orgánica que contiene el compuesto (G);
3c.2) precipitar el compuesto (G) presente en la capa orgánica de la etapa 3c.1 añadiendo una mezcla de éter de petróleo (hexano)/AcOEt en una proporción en volumen de 8/1-6/1, preferentemente una proporción en volumen de 7/1.
12. Proceso industrial según la reivindicación 11, en el que la etapa 3c.1 comprende las etapas sucesivas siguientes:
3c.1.1) añadir agua a la mezcla resultante obtenida en la etapa 3c, a continuación;
3c.1.2) sin concentrar el solvente de reacción, extraer el producto obtenido después de la etapa 3c.1.1 con un solvente aprótico polar, preferentemente AcOEt.
13. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que el compuesto (C) se prepara mediante un proceso que comprende las etapas sintéticas sucesivas siguientes:
£) hacer reaccionar 1.1 equivalentes molares de Boc-glicina con 1.2 equivalentes molares de Et3N en acetato de etilo, a continuación;
O) hacer reaccionar el producto obtenido en la etapa £) con 1 equivalente molar de etil-1-cloroetilcarbonato, y 0.2 equivalentes molares de yoduro de potasio;
Y) hacer reaccionar el producto obtenido en la etapa O) con 2 equivalentes molares de gas de HCl en acetato de etilo a una temperatura que varía de 5°C a 10°C, y recuperar (C).
14. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que después de añadir ácido fórmico en la etapa 4a, el proceso comprende además las etapas sucesivas siguientes:
4a.1) coevaporar el producto obtenido después de añadir ácido fórmico con tolueno, proporcionando el compuesto (H) y la capa orgánica con tolueno, a continuación;
4a.2) al compuesto (H) obtenido después de la etapa 4a, añadir acetato de etilo, y a continuación lavar la mezcla resultante con salmuera a una temperatura que varía de 0°C a 10°C.
15. Proceso industrial según cualquiera de las reivindicaciones anteriores, en el que se realiza un intercambio de aniones por medio de las etapas siguientes:
4b.1) añadir una solución de NaOH al 2% en peso en agua a un producto obtenido después de la etapa 4a.2 a una temperatura que varía de 0°C a 10°C, a continuación;
4d) añadir una solución de ácido fumárico al 5% en peso en EtOH a la mezcla obtenida después de la etapa 4b.1 para aislar el compuesto (I) cristalizado utilizando un proceso de flujo continuo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15306626 | 2015-10-14 | ||
| PCT/EP2016/074709 WO2017064250A1 (en) | 2015-10-14 | 2016-10-14 | Industrial process for the preparation of (5s,10s)-10-benzyl-16-methyl-11,14,18-trioxo-15,17,19-trioxa-2,7,8-trithia-12-azahenicosan-5-aminium (e)-3-carboxyacrylate salt |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2818185T3 true ES2818185T3 (es) | 2021-04-09 |
Family
ID=54330703
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16784446T Active ES2818185T3 (es) | 2015-10-14 | 2016-10-14 | Proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US10399936B2 (es) |
| EP (1) | EP3362433B1 (es) |
| JP (2) | JP6998864B2 (es) |
| KR (1) | KR20180095507A (es) |
| CN (1) | CN108430968B (es) |
| AU (1) | AU2016336891B2 (es) |
| BR (1) | BR112018007505B1 (es) |
| CA (1) | CA3016962A1 (es) |
| DK (1) | DK3362433T3 (es) |
| ES (1) | ES2818185T3 (es) |
| IL (1) | IL258663B (es) |
| MX (1) | MX381308B (es) |
| TW (1) | TWI712582B (es) |
| WO (1) | WO2017064250A1 (es) |
| ZA (1) | ZA201803105B (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107814708A (zh) * | 2017-11-27 | 2018-03-20 | 杭州新博思生物医药有限公司 | 一种拆分剂奎宁的回收套用方法 |
| FR3077201B1 (fr) | 2018-01-26 | 2020-01-17 | Pharmaleads | Derives aminoacides contenant un groupement disulfanyle sous forme d'un inhibiteur de nep et d'apn pour la prevention et le traitement des douleurs relatives au nerf trijumeau |
| CN113651745B (zh) * | 2021-09-09 | 2023-06-02 | 上海医药工业研究院 | 布瓦西坦中间体及其制备方法和纯化方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2651229B1 (fr) * | 1989-08-24 | 1991-12-13 | Inst Nat Sante Rech Med | Nouveaux derives d'amino-acides, leur procede de preparation et leur application therapeutique. |
| US5552397A (en) * | 1992-05-18 | 1996-09-03 | E. R. Squibb & Sons, Inc. | Substituted azepinone dual inhibitors of angiotensin converting enzyme and neutral exdopeptidase |
| JPH11228532A (ja) * | 1998-02-16 | 1999-08-24 | Ajinomoto Co Inc | 光学活性2−アセチルチオメチル−3−フェニルプロピオン酸の製造方法 |
| ES2369256T3 (es) * | 2000-06-26 | 2011-11-28 | Helen Of Troy Limited | Sistema de lámpara recargable. |
| AR036237A1 (es) * | 2001-07-27 | 2004-08-25 | Bayer Corp | Derivados del acido indan acetico, intermediarios, y metodo para su preparacion, composicion farmaceutica y el uso de dichos derivados para la manufactura de un medicamento |
| WO2007048787A1 (fr) * | 2005-10-25 | 2007-05-03 | Pharmaleads | Derives d'amino-acides contenant un groupement disulfanyle comme inhibiteurs mixtes de la neprilysine et de l 'aminopeptidase n |
| CN100497296C (zh) * | 2006-07-07 | 2009-06-10 | 沈阳药科大学 | 旋光活性的苯乙醇胺类化合物及其制法 |
| FR2931151A1 (fr) * | 2008-05-13 | 2009-11-20 | Pharmaleads Soc Par Actions Si | Nouveaux derives d'amino-acides, leur procede de preparation et leur utilisation therapeutique |
| FR2997080B1 (fr) * | 2012-10-23 | 2015-11-27 | Pharmaleads | Inhibiteurs de neprilysine |
-
2016
- 2016-10-13 TW TW105133061A patent/TWI712582B/zh active
- 2016-10-14 MX MX2018004631A patent/MX381308B/es unknown
- 2016-10-14 US US15/767,966 patent/US10399936B2/en active Active
- 2016-10-14 KR KR1020187013628A patent/KR20180095507A/ko not_active Ceased
- 2016-10-14 CA CA3016962A patent/CA3016962A1/en active Pending
- 2016-10-14 JP JP2018519343A patent/JP6998864B2/ja active Active
- 2016-10-14 CN CN201680068001.7A patent/CN108430968B/zh active Active
- 2016-10-14 AU AU2016336891A patent/AU2016336891B2/en active Active
- 2016-10-14 DK DK16784446.3T patent/DK3362433T3/da active
- 2016-10-14 EP EP16784446.3A patent/EP3362433B1/en active Active
- 2016-10-14 BR BR112018007505-1A patent/BR112018007505B1/pt active IP Right Grant
- 2016-10-14 WO PCT/EP2016/074709 patent/WO2017064250A1/en not_active Ceased
- 2016-10-14 ES ES16784446T patent/ES2818185T3/es active Active
-
2018
- 2018-04-12 IL IL258663A patent/IL258663B/en active IP Right Grant
- 2018-05-11 ZA ZA2018/03105A patent/ZA201803105B/en unknown
-
2021
- 2021-09-15 JP JP2021149882A patent/JP2021193127A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| ZA201803105B (en) | 2019-02-27 |
| US10399936B2 (en) | 2019-09-03 |
| MX2018004631A (es) | 2018-11-09 |
| BR112018007505B1 (pt) | 2020-11-17 |
| JP6998864B2 (ja) | 2022-02-04 |
| DK3362433T3 (da) | 2020-09-21 |
| CN108430968B (zh) | 2021-01-05 |
| IL258663B (en) | 2020-08-31 |
| EP3362433B1 (en) | 2020-07-08 |
| US20180305308A1 (en) | 2018-10-25 |
| JP2018531946A (ja) | 2018-11-01 |
| AU2016336891B2 (en) | 2020-07-09 |
| CN108430968A (zh) | 2018-08-21 |
| TWI712582B (zh) | 2020-12-11 |
| TW201718479A (zh) | 2017-06-01 |
| IL258663A (en) | 2018-06-28 |
| MX381308B (es) | 2025-03-12 |
| KR20180095507A (ko) | 2018-08-27 |
| JP2021193127A (ja) | 2021-12-23 |
| WO2017064250A1 (en) | 2017-04-20 |
| EP3362433A1 (en) | 2018-08-22 |
| AU2016336891A1 (en) | 2018-05-31 |
| CA3016962A1 (en) | 2017-04-20 |
| BR112018007505A2 (pt) | 2018-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2818185T3 (es) | Proceso industrial para la preparación de la sal de (E)-3-carboxiacrilato de (5S,10S)-10-bencil-16-metil-11,14,18-trioxo-15,17,19-trioxa-2,7,8-tritia-12-azahenicosan-5-aminio | |
| ES2847239T3 (es) | Procedimientos para la preparación de 4-alcoxi-3-(acil o alquil)oxipicolinamidas | |
| ES2879294T3 (es) | Formas polimórficas de Belinostat y procesos para la preparación de las mismas | |
| ES2903434T3 (es) | Forma cristalina de trihidrato de (3S,3S')-4,4'-disulfanodiilbis(ácido 3-aminobutano-1-sulfónico) | |
| WO2018125820A1 (en) | Processes for the preparation of pesticidal compounds | |
| ES2198389T3 (es) | Procedimiento para la sintesis de derivados n-(mercaptoacil)-aminoacidos a partir de acidos alfa-substituidos. | |
| TWI432416B (zh) | 截短側耳素類(pleuromutilins)的製備方法 | |
| JPWO2010122682A1 (ja) | N−アルコキシカルボニル−tert−ロイシンの製造法 | |
| CN111410622A (zh) | L-胱硫醚盐酸盐的合成方法 | |
| ES2314464T3 (es) | Procedimiento mejorado para preparar bencihidriltioacetamida. | |
| US20100228037A1 (en) | Process for Preparing Allylmercaptocaptopril (Cpssa) and Related Asymmetrical Disulfides | |
| ES2562234T3 (es) | Proceso en un recipiente para producir 1,2-bencisoxazol-3-metanosulfonamida | |
| ES2293587T3 (es) | Procedimiento de sintesis e intermediarios de benzoxatiepinas. | |
| HK1259577B (en) | Industrial process for the preparation of (5s,10s)-10-benzyl-16-methyl-11,14,18-trioxo-15,17,19-trioxa-2,7,8-trithia-12-azahenicosan-5-aminium (e)-3-carboxyacrylate salt | |
| HK1259577A1 (en) | Industrial process for the preparation of (5s,10s)-10-benzyl-16-methyl-11,14,18-trioxo-15,17,19-trioxa-2,7,8-trithia-12-azahenicosan-5-aminium (e)-3-carboxyacrylate salt | |
| JPH06192207A (ja) | ウレタン化合物の製造方法 | |
| JPS5846065A (ja) | ラセミ化合物s−(カルボキシメチル)−(rs)−システインの分割法 | |
| JP2005232086A (ja) | N−スルフェニルアミノ酸エステル化合物の製造方法 | |
| JPH07101928A (ja) | N−アルコキシカルボニルアミノ酸エステルの製造方法 |