ES2821649T3 - Método para preparar derivados de imidazol e intermedios de los mismos y formas cristalinas - Google Patents
Método para preparar derivados de imidazol e intermedios de los mismos y formas cristalinas Download PDFInfo
- Publication number
- ES2821649T3 ES2821649T3 ES16806806T ES16806806T ES2821649T3 ES 2821649 T3 ES2821649 T3 ES 2821649T3 ES 16806806 T ES16806806 T ES 16806806T ES 16806806 T ES16806806 T ES 16806806T ES 2821649 T3 ES2821649 T3 ES 2821649T3
- Authority
- ES
- Spain
- Prior art keywords
- crystalline form
- liters
- compound
- formula
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 21
- 239000000543 intermediate Substances 0.000 title description 4
- 150000002460 imidazoles Chemical class 0.000 title description 3
- 229940079865 intestinal antiinfectives imidazole derivative Drugs 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 62
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 90
- 238000006243 chemical reaction Methods 0.000 claims description 59
- 239000000203 mixture Substances 0.000 claims description 56
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 51
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 42
- 239000002904 solvent Substances 0.000 claims description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 18
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 17
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 14
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 14
- 238000004519 manufacturing process Methods 0.000 claims description 11
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 10
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 8
- 239000012046 mixed solvent Substances 0.000 claims description 8
- 238000001228 spectrum Methods 0.000 claims description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 7
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 7
- 239000003960 organic solvent Substances 0.000 claims description 7
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- 239000012442 inert solvent Substances 0.000 claims description 6
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 6
- 229940011051 isopropyl acetate Drugs 0.000 claims description 6
- 239000003495 polar organic solvent Substances 0.000 claims description 6
- 239000013078 crystal Substances 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 230000009385 viral infection Effects 0.000 claims description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 4
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 claims description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 4
- 125000005233 alkylalcohol group Chemical group 0.000 claims description 4
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 claims description 4
- 150000004292 cyclic ethers Chemical class 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 238000010438 heat treatment Methods 0.000 claims description 4
- 150000002576 ketones Chemical class 0.000 claims description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Chemical group C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- 239000004480 active ingredient Substances 0.000 claims description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 3
- 230000000241 respiratory effect Effects 0.000 claims description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 claims description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 2
- 239000002253 acid Substances 0.000 claims description 2
- 125000000217 alkyl group Chemical group 0.000 claims description 2
- 125000004429 atom Chemical group 0.000 claims description 2
- 238000001816 cooling Methods 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 150000002148 esters Chemical group 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 2
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 claims description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 2
- 238000010992 reflux Methods 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 claims description 2
- 239000008096 xylene Substances 0.000 claims description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims 3
- 238000004458 analytical method Methods 0.000 claims 2
- 239000007787 solid Substances 0.000 description 26
- 239000010410 layer Substances 0.000 description 24
- 239000012044 organic layer Substances 0.000 description 23
- 239000000243 solution Substances 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 238000001514 detection method Methods 0.000 description 13
- 238000004128 high performance liquid chromatography Methods 0.000 description 13
- 239000000523 sample Substances 0.000 description 12
- 241000725643 Respiratory syncytial virus Species 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 241000700605 Viruses Species 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000012065 filter cake Substances 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 238000002411 thermogravimetry Methods 0.000 description 4
- AAMCPTRELXTKPF-UHFFFAOYSA-N 1'-(2,2-diethoxyethyl)-1-(2-methylpropanoyl)spiro[azetidine-3,3'-indole]-2'-one Chemical compound C(C)OC(CN1C(C2(C3=CC=CC=C13)CN(C2)C(C(C)C)=O)=O)OCC AAMCPTRELXTKPF-UHFFFAOYSA-N 0.000 description 3
- WGXLLSUWCXNWHF-UHFFFAOYSA-N 1'-(2-methylpropanoyl)spiro[1H-indole-3,3'-azetidine]-2-one Chemical compound C(C(C)C)(=O)N1CC2(C(NC3=CC=CC=C23)=O)C1 WGXLLSUWCXNWHF-UHFFFAOYSA-N 0.000 description 3
- OVBJWUPRIYESCO-UHFFFAOYSA-N 1-(2-methylpropanoyl)-1'-(4,5,6,7-tetrahydro-1H-benzimidazol-2-ylmethyl)spiro[azetidine-3,3'-indole]-2'-one Chemical compound C(C(C)C)(=O)N1CC2(C(N(C3=CC=CC=C23)CC2=NC3=C(N2)CCCC3)=O)C1 OVBJWUPRIYESCO-UHFFFAOYSA-N 0.000 description 3
- HMNNTQIOMPFTFY-UHFFFAOYSA-N 1-(2-methylpropanoyl)-1'-[[1-(4,4,4-trifluorobutyl)-4,5,6,7-tetrahydrobenzimidazol-2-yl]methyl]spiro[azetidine-3,3'-indole]-2'-one Chemical compound C(C(C)C)(=O)N1CC2(C(N(C3=CC=CC=C23)CC2=NC3=C(N2CCCC(F)(F)F)CCCC3)=O)C1 HMNNTQIOMPFTFY-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 3
- RPYYRMZTTCUCLS-UHFFFAOYSA-N 2-[1-(2-methylpropanoyl)-2'-oxospiro[azetidine-3,3'-indole]-1'-yl]acetaldehyde Chemical compound C(C(C)C)(=O)N1CC2(C(N(C3=CC=CC=C23)CC=O)=O)C1 RPYYRMZTTCUCLS-UHFFFAOYSA-N 0.000 description 3
- -1 4,4,4-trifluorobutyl Chemical group 0.000 description 3
- 229910002483 Cu Ka Inorganic materials 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 206010061603 Respiratory syncytial virus infection Diseases 0.000 description 3
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 230000000840 anti-viral effect Effects 0.000 description 3
- 239000006143 cell culture medium Substances 0.000 description 3
- 238000000113 differential scanning calorimetry Methods 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 229960000329 ribavirin Drugs 0.000 description 3
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 3
- FQZGASUXNNVJOM-UHFFFAOYSA-N tert-butyl 1'-[(4-methoxyphenyl)methyl]-2'-oxospiro[azetidine-3,3'-indole]-1-carboxylate Chemical compound C1=CC(OC)=CC=C1CN1C2=CC=CC=C2C2(CN(C2)C(=O)OC(C)(C)C)C1=O FQZGASUXNNVJOM-UHFFFAOYSA-N 0.000 description 3
- BFTCPUNZAOONFL-UHFFFAOYSA-N tert-butyl 3-[(2-bromophenyl)carbamoyl]azetidine-1-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC1C(=O)NC1=CC=CC=C1Br BFTCPUNZAOONFL-UHFFFAOYSA-N 0.000 description 3
- ANQAHRBJLMNFAK-UHFFFAOYSA-N 1-cyclopropyl-3-[[1-(3-hydroxypropyl)benzimidazol-2-yl]methyl]imidazo[4,5-c]pyridin-2-one Chemical compound N=1C2=CC=CC=C2N(CCCO)C=1CN(C1=O)C2=CN=CC=C2N1C1CC1 ANQAHRBJLMNFAK-UHFFFAOYSA-N 0.000 description 2
- GCDCOBYBHQJKDU-UHFFFAOYSA-N 4,4,4-trifluorobutyl 4-methylbenzenesulfonate Chemical compound CC1=CC=C(S(=O)(=O)OCCCC(F)(F)F)C=C1 GCDCOBYBHQJKDU-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- 108010087230 Sincalide Proteins 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- KVVMXWRFYAGASO-UHFFFAOYSA-N azetidine-1-carboxylic acid Chemical compound OC(=O)N1CCC1 KVVMXWRFYAGASO-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 238000010609 cell counting kit-8 assay Methods 0.000 description 2
- 239000013068 control sample Substances 0.000 description 2
- 230000000120 cytopathologic effect Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000011888 foil Substances 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000011081 inoculation Methods 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 238000010183 spectrum analysis Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 229940036185 synagis Drugs 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- 0 *C(Cc1c(C23CN(*)C2)cccc1)C3=O Chemical compound *C(Cc1c(C23CN(*)C2)cccc1)C3=O 0.000 description 1
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 1
- NCADHSLPNSTDMJ-UHFFFAOYSA-N 1-[(2-methylpropan-2-yl)oxycarbonyl]azetidine-3-carboxylic acid Chemical compound CC(C)(C)OC(=O)N1CC(C(O)=O)C1 NCADHSLPNSTDMJ-UHFFFAOYSA-N 0.000 description 1
- LILXDMFJXYAKMK-UHFFFAOYSA-N 2-bromo-1,1-diethoxyethane Chemical compound CCOC(CBr)OCC LILXDMFJXYAKMK-UHFFFAOYSA-N 0.000 description 1
- AOPBDRUWRLBSDB-UHFFFAOYSA-N 2-bromoaniline Chemical compound NC1=CC=CC=C1Br AOPBDRUWRLBSDB-UHFFFAOYSA-N 0.000 description 1
- JQPFYXFVUKHERX-UHFFFAOYSA-N 2-hydroxy-2-cyclohexen-1-one Natural products OC1=CCCCC1=O JQPFYXFVUKHERX-UHFFFAOYSA-N 0.000 description 1
- DGMOBVGABMBZSB-UHFFFAOYSA-N 2-methylpropanoyl chloride Chemical compound CC(C)C(Cl)=O DGMOBVGABMBZSB-UHFFFAOYSA-N 0.000 description 1
- VKRFUGHXKNNIJO-UHFFFAOYSA-N 4,4,4-trifluorobutan-1-ol Chemical compound OCCCC(F)(F)F VKRFUGHXKNNIJO-UHFFFAOYSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 206010006448 Bronchiolitis Diseases 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- ZKLPARSLTMPFCP-UHFFFAOYSA-N Cetirizine Chemical compound C1CN(CCOCC(=O)O)CCN1C(C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZKLPARSLTMPFCP-UHFFFAOYSA-N 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010024971 Lower respiratory tract infections Diseases 0.000 description 1
- XLVALKXFDVIVJH-UHFFFAOYSA-N O.[Na].[Na].[Na].N1=C(N=C(N=C1S)S)S Chemical compound O.[Na].[Na].[Na].N1=C(N=C(N=C1S)S)S XLVALKXFDVIVJH-UHFFFAOYSA-N 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 1
- 206010070863 Toxicity to various agents Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000001387 anti-histamine Effects 0.000 description 1
- 239000012296 anti-solvent Substances 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- VJBCNMFKFZIXHC-UHFFFAOYSA-N azanium;2-(4-methyl-5-oxo-4-propan-2-yl-1h-imidazol-2-yl)quinoline-3-carboxylate Chemical compound N.N1C(=O)C(C(C)C)(C)N=C1C1=NC2=CC=CC=C2C=C1C(O)=O VJBCNMFKFZIXHC-UHFFFAOYSA-N 0.000 description 1
- 150000001556 benzimidazoles Chemical class 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 125000003739 carbamimidoyl group Chemical class C(N)(=N)* 0.000 description 1
- 229960001803 cetirizine Drugs 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000007821 culture assay Methods 0.000 description 1
- OILAIQUEIWYQPH-UHFFFAOYSA-N cyclohexane-1,2-dione Chemical compound O=C1CCCCC1=O OILAIQUEIWYQPH-UHFFFAOYSA-N 0.000 description 1
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- VUVGMQNYJPPCCC-UHFFFAOYSA-N pyrido[1,2-a]indole Chemical compound C1=CC=CC2=CC3=CC=CC=C3N21 VUVGMQNYJPPCCC-UHFFFAOYSA-N 0.000 description 1
- HOHWYQSDLRTVDK-UHFFFAOYSA-N pyrimido[1,2-a]benzimidazole Chemical compound N1=CC=CN2C3=CC=CC=C3N=C21 HOHWYQSDLRTVDK-UHFFFAOYSA-N 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- NZHWYOLXENRFJZ-UHFFFAOYSA-N spiro[1h-indole-3,3'-azetidine]-2-one Chemical compound O=C1NC2=CC=CC=C2C11CNC1 NZHWYOLXENRFJZ-UHFFFAOYSA-N 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- TTYKXUAEAUBKCT-UHFFFAOYSA-N tert-butyl 3-[(2-bromophenyl)-[(4-methoxyphenyl)methyl]carbamoyl]azetidine-1-carboxylate Chemical compound C1=CC(OC)=CC=C1CN(C=1C(=CC=CC=1)Br)C(=O)C1CN(C(=O)OC(C)(C)C)C1 TTYKXUAEAUBKCT-UHFFFAOYSA-N 0.000 description 1
- 150000003918 triazines Chemical class 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- TVBIVRGNYNBFCD-UHFFFAOYSA-N triethylazanium;trifluoromethanesulfonate Chemical compound CC[NH+](CC)CC.[O-]S(=O)(=O)C(F)(F)F TVBIVRGNYNBFCD-UHFFFAOYSA-N 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/0004—Crystallisation cooling by heat exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D9/00—Crystallisation
- B01D9/005—Selection of auxiliary, e.g. for control of crystallisation nuclei, of crystal growth, of adherence to walls; Arrangements for introduction thereof
- B01D9/0054—Use of anti-solvent
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B41/00—Formation or introduction of functional groups containing oxygen
- C07B41/06—Formation or introduction of functional groups containing oxygen of carbonyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B43/00—Formation or introduction of functional groups containing nitrogen
- C07B43/04—Formation or introduction of functional groups containing nitrogen of amino groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/96—Spiro-condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un proceso para preparar un compuesto de fórmula (I), **(Ver fórmula)** que comprende la siguiente etapa: **(Ver fórmula)**
Description
DESCRIPCIÓN
Método para preparar derivados de imidazol e intermedios de los mismos y formas cristalinas
Campo de la invención
La presente invención se refiere a un método para preparar un derivado de imidazol, forma cristalina A, forma cristalina B, y el correspondiente proceso de preparación de los mismos, la presente invención también se refiere a un método para preparar un intermedio del compuesto de fórmula (I).
Técnicas anteriores
El virus sincitial respiratorio (VSR) es la principal causa de infección grave del tracto respiratorio inferior en bebés, niños, ancianos y personas con inmunidad deteriorada. Las infecciones virales graves pueden causar bronquiolitis o neumonía que requieren hospitalización o causar la muerte (JAMA, 1997, 277, 12). Recientemente, se ha aprobado la ribavirina para el tratamiento de la infección por virus. La ribavirina es un análogo de nucleósido que se administra por vía intranasal en forma de aerosol. Y la toxicidad del fármaco es considerable y su eficacia es controvertida. Además de la ribavirina, RespiGam y Synagis son inmunoglobulinas y anticuerpos monoclonales que neutralizan el VSR, respectivamente. Son los dos agentes biológicos aprobados para la prevención de la infección por VSR en niños de alto riesgo. RespiGam y Synagis son muy costosos y requieren administración parenteral.
Se sabe que muchos fármacos son útiles para inhibir el virus sincitial respiratorio (De Clercq, int. J. Antiviral Agent, 1996, 7, 193). Y. Tao et al. (documento EP0058146A1, 1998) reveló que el antihistamínico conocido, cetirizina, mostró actividad anti-VSR. Una serie de compuestos amidino como inhibidores de VSR fueron reportados por Tidwell et al. en J. Med. Chem. 1983, 26,294 (US 4,324,794,1982) y Dubovi et al., en Antimicrobial Agents and Chemotherapy, 1981, 19, 649. Una serie de 6-aminopirimidonas que tienen actividad antiviral anti-VSR también fueron descritas por Hsu et al. en la patente de EE. UU. N.° 5,256,668 (1993). Una serie de compuestos que contienen triazina para el tratamiento y/o prevención de la infección por VSR fueron descritos por Y. Gluzman et al. (patente de AU, Au-A-14, 704, 1997) y P.R. Wey et al. (Antiviral Res. 1998, 38, 31). El pirido [1,2-a] benzopirrol y el pirimido [1,2a] bencimidazol fueron descritos por S. Shigeta et al. en Antiviral Chem. & Amp; Chemother. 1992, 3,171. Se ha demostrado que estos compuestos inhiben la replicación del ortomixovirus y el paramixovirus en las células de HeLa. Se informó que los bisbenzimidazoles con enlaces de etilenglicol también eran potentes inhibidores de rinovirus (Roderick et al., J. Med. Chem. 1972, 15, 655). Otros compuestos estructuralmente relacionados fueron bis-benzimidazoles con actividad antifúngica (B. Cakir et al., Eczacilik Fak Derg. 1988, 5, 71). Recientemente, una serie de benzimidazoles (documento WO 00/04900) para el tratamiento y prevención de la infección por VSR fueron descubiertos por Yu et al. Además, Theodore Nitz también encontró una serie de compuestos de fórmula III (documento WO 99/38508) que inhiben el VSR en ensayos de cultivo de tejidos de células Hep-2.
En la actualidad, BMS433771 se describe por BMS que tiene una estructura de fórmula general (B-1):

El documento WO2013068769A1 de Viral describe una clase de compuestos que tienen la estructura de fórmula general (B-II):

El documento WO2014060411A1 de Janssen R & D Ireland describe una clase de compuestos que tienen una estructura de fórmula general(R1) (B-III):

en donde Het representa un anillo heterocíclico tal como la estructura (a), (b), (c) o (d), mostrado por la fórmula (B-IV) en la fórmula X representa un anillo aromático que contiene al menos dos átomos de C, o átomos de N.

El documento WO2010103306A1 de AstraZeneca describe una clase de compuestos que tienen una estructura de fórmula general (B-V):

Aunque los compuestos anteriores presentes en la técnica anterior pueden usarse para inhibir el virus sincitial respiratorio, todavía se requiere una mejora en la actividad y solubilidad, etc.
Contenido de la presente invención
La presente invención proporciona un proceso para preparar un compuesto de fórmula (I):

que comprende el siguiente etapa:

En algunas realizaciones de la presente invención, el proceso anterior comprende el siguiente etapa:

en donde, R se selecciona de

cada uno de R1 y R2 se selecciona independientemente de un alquilo C1-3;
opcionalmente, R1 y R2 están conectados al mismo átomo para formar un anillo de 5 a 6 miembros opcionalmente sustituido con 1,2, 3 o 4 de R'(s),
R' se selecciona entre metilo, etilo, isopropilo o n-propilo.
En algunas realizaciones de la presente invención, el R anterior se selecciona de

o el ácido se selecciona del grupo que consiste en ácido trifluoroacético, ácido acético, ácido clorhídrico, ácido sulfúrico diluido y ácido p-toluensulfónico.
El proceso para preparar el compuesto de fórmula (I) de la presente invención también comprende la siguiente ruta de reacción:

en donde,
HA se selecciona de ácido trifluorometanosulfónico;
el disolvente de inserción es un disolvente único o un disolvente mixto para varios disolventes seleccionados del grupo que consiste en diclorometano, acetato de etilo, acetato de isopropilo, tetrahidrofurano, dioxano, 2-metiltetrahidrofurano, éter dietílico, metil ter-butil éter, pentano, n-hexano, ciclohexano, n-heptano y octano;
la relación en peso del compuesto de fórmula (VII) al disolvente inerte es de 1:1 a 1:10.
En algunas realizaciones de la presente invención, el disolvente de inserción es un disolvente único o un disolvente mixto para varios disolventes seleccionados del grupo que consiste en acetato de etilo, acetato de isopropilo, metil terbutil éter, ciclohexano y n-heptano; o el disolvente de inserción es un disolvente mixto de acetato de etilo y n-heptano. En algunas realizaciones de la presente invención, el proceso para preparar el compuesto de fórmula (I) comprende la siguiente ruta de reacción:

y/o

La presente invención también proporciona un compuesto de la siguiente fórmula como intermedio para preparar el compuesto de fórmula (I):

La presente invención proporciona la forma cristalina A del compuesto de fórmula (I), el espectrograma de XRPD se muestra en la Figura 1 y el análisis de espectros de XRPD se muestra en la Tabla 1.
Tabla 1. Forma cristalina A del compuesto de fórmula (I)

La presente invención proporciona la forma cristalina B del compuesto de fórmula (I), el espectrograma de XRPD se muestra en la Figura 2 y el análisis de espectros de XRPD se muestra en la Tabla 2.
Tabla 2. Forma cristalina B del compuesto de fórmula (I)

La presente invención proporciona un proceso para preparar la forma cristalina A, que comprende añadir el compuesto de fórmula (I) a un disolvente orgánico, calentar a 30 °C hasta la temperatura de reflujo para disolverlo y después enfriarlo a 0 a 20 °C dentro de 0,5 a 10 horas para precipitar el cristal.
En algunas realizaciones de la presente invención, el disolvente orgánico anterior se selecciona del grupo que consiste en dicloroetano, un alcohol alquílico C1-6, un éter C4-10 o éter cíclico, una cetona C2-6, un éster C2-6, benceno opcionalmente sustituido con un(os) átomo(s) de metilo o etilo o halógeno, en donde el número de sustituyente(s) se selecciona(n) entre 1, 2 y/o 3.
En algunas realizaciones de la presente invención, el disolvente orgánico se selecciona del grupo que consiste en diclorometano, metanol, etanol, isopropanol, tetrahidrofurano, dioxano, 2-metiltetrahidrofurano, acetona, acetato de etilo, acetato de isopropilo, tolueno, xileno y/o clorobenceno.
La presente invención proporciona un proceso para preparar la forma cristalina B, que comprende añadir el compuesto de fórmula (I) a un disolvente orgánico polar y después dejar caer lentamente agua para precipitar el cristal.
En algunas realizaciones de la presente invención, el disolvente orgánico polar anterior se selecciona del grupo que consiste en un alcohol alquílico C1-6, un éter C4-10 o éter cíclico y una cetona C2-6; opcionalmente el disolvente orgánico polar se selecciona del grupo que consiste en metanol, etanol, isopropanol, tetrahidrofurano, 2-metiltetrahidrofurano, dioxano y acetona.
La presente invención también proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto anterior o la forma cristalina anterior como ingrediente activo y un vehículo farmacéuticamente aceptable.
La presente invención también proporciona la forma cristalina anterior para su uso en el tratamiento de la infección por virus respiratorios.
La presente invención también proporciona la composición farmacéutica anterior para uso en el tratamiento de la infección por virus respiratorios.
Efectos técnicos
El compuesto de fórmula (V) es un fragmento importante del compuesto de fórmula (I), la operación de reacción y el postratamiento de preparar el compuesto de fórmula (V) a partir del compuesto de fórmula (IV) son simples y la pureza del producto es alta, el compuesto de fórmula (II) precipita un sólido en un disolvente inerte y el postratamiento es simple.
La forma cristalina A y la forma cristalina B del compuesto de fórmula (I) tienen buena estabilidad y buenas perspectivas de aplicación.
Definiciones y descripciones
A menos que se especifique lo contrario, los siguientes términos y frases utilizados en la presente memoria pretenden tener los siguientes significados. Un término o frase en particular no debe considerarse incierto o poco claro en ausencia de una definición específica, mientras que debe entenderse según el significado corriente. Cuando en la presente memoria aparece un nombre comercial, se refiere al producto correspondiente o su ingrediente activo.
El compuesto de la presente invención se puede preparar a través de muchos métodos sintéticos que son bien conocidos por el experto en la técnica, incluyendo las realizaciones específicas enumeradas a continuación y su combinación con otros métodos de síntesis química y los métodos alternativos equivalentes que son conocidos por los expertos en la técnica, las realizaciones preferidas incluyen, pero no se limitan a, las realizaciones de la presente invención.
Los disolventes usados en la presente invención están disponibles comercialmente. Los compuestos disponibles comercialmente se nombran según el catálogo de proveedores. Cuando se añade el disolvente mixto a la disolución de reacción, cada uno de los disolventes puede mezclarse primero y después añadirse a la disolución de reacción, o cada uno de los disolventes se añade secuencialmente a la disolución de reacción y se mezcla en el sistema de reacción.
El método de difracción de rayos X en polvo es el siguiente:
Instrumento: Difractómetro de rayos X Bruker D8 ADVANCE; Objetivo: Cu: K-Alpha; Longitud de onda A = 1,54179 Á; Voltaje del tubo: 40 kV; Corriente del tubo: 40 mA; Alcance de escaneo: 4-40°; Velocidad de rotación de la muestra: 15 rpm; Velocidad de escaneo: 10°/min.
El método de análisis termogravimétrico (TGA) de la presente invención:
Instrumento: Analizador termogravimétrico TA Q5000;
Métodos: Se pesan aproximadamente 2-5 mg de la muestra y se colocan en un recipiente TGA Platinum para su prueba. El método es: temperatura ambiente-300 °C, la velocidad de calentamiento es de 10 °C/min.
El método de calorimetría diferencial de barrido (DSC) de la presente invención:
Instrumento: Calorímetro de barrido diferencial TA Q2000;
Métodos: Se pesa con precisión aproximadamente 1 mg de muestra y se coloca en una olla de aluminio DSC para realizar la prueba, el método es: la temperatura es de 25 °C a 300 °C y la velocidad de calentamiento es de 10 °C/min. Breve descripción de los dibujos
La figura 1 es un espectro XRPD de radiación Cu-Ka de la forma cristalina A del compuesto de fórmula (I). La figura 2 es un espectro XRPD de radiación Cu-Ka de la forma cristalina B del compuesto de fórmula (I). La figura 3 es un espectro XRPD de radiación Cu-Ka de la forma cristalina A del compuesto de fórmula (I) a diferentes temperaturas y condiciones de humedad.
La figura 4 es un espectro de DSC de la forma cristalina A del compuesto de fórmula (I).
La figura 5 es un espectro de TGA de la forma cristalina A del compuesto de fórmula (I).
Descripción detallada de la realización preferida
Preparación de compuestos de fórmula (I):

1 -Isobutiril-1 ’-((1 -(4,4,4-trifluorobutil)-4,5,6,7-tetrahidro-1 H-benzo[d]imida azol-2-il)metil)espiro[azetidina-3,3'-indolin]-2'-ona

Etapa 1
Ter-butil 3-((2-bromofenil)carbamoil)azetidina-1-carboxilato
Se añadieron ácido 1 -(ter-butoxicarbonil)azetidina-3-carboxílico 1 (3,67 kg, 21,33 mol) y 2-bromoanilina (4,72 kg, 23,25 mol) a una caldera de reacción de 30 L y se añadieron 7,4 L de acetato de etilo, trietilamina (4,46 L, 32,0 mol). La temperatura se enfrió a 0-10°C, anhídrido de ácido propilfosfónico (T3P, disolución al 50% en peso en acetato de etilo, 15,23 l, 25,6 mol), la temperatura se controló por debajo de 35°C. Una vez completada la adición, la mezcla se agitó a temperatura ambiente 28-32 °C durante 16 horas. La relación de 1/2 fue de 0,3% mediante detección por HPLC, se terminó la reacción. La temperatura se controló por debajo de 35 °C, se añadieron lentamente gota a gota 5 litros de disolución acuosa de hidróxido de sodio 5 mol/L y se inactivó la reacción. La disolución de reacción se transfirió a una caldera de 50 L y se añadieron 7 litros de disolución acuosa de hidróxido de sodio 5 mol/L, el pH de la fase acuosa fue 7-8 por detección, se dejó reposar la mezcla y se separó en capas. Se añadieron 5 litros de disolución acuosa de hidróxido de sodio 5 mol/L a la capa orgánica, y el pH de la fase acuosa fue de 8-9 por detección, la mezcla se dejó reposar y se separó en capas. Se añadieron 500 g de sólidos de hidróxido de sodio a la capa acuosa y el pH de la fase acuosa fue de 9 a 10 por detección. La mezcla se extrajo con 5 litros de acetato de etilo. Las capas orgánicas se combinaron y se añadieron lentamente con agitación 8 litros de ácido clorhídrico 8 mol/L. El pH de la fase acuosa fue de 6-7 por detección, la mezcla se dejó reposar y se separó en capas. Se añadieron 8 litros de ácido clorhídrico 0,3 mol/l a la capa orgánica con agitación y la mezcla se dejó reposar y se separó en capas. Se añadieron 10 litros de cloruro de sodio saturado a la capa orgánica y se agitó para separar las capas. Se añadieron 10 litros de cloruro de sodio saturado a la capa orgánica y se agitó para separar las capas. La capa orgánica se concentró a 2 litros a presión reducida y se concentró a sequedad mediante la adición de 5 litros de n-heptano para dar ter-butil 3-((2-bromofenil)carbamoil)azetidina-1-carboxilato 2 (7,35 kg sólido amarillo pálido, bruto), que se utilizó directamente en la siguiente etapa sin purificación.
MS m/z (ESI): 355,1 [M 1].
1H RMN (400 MHz, CDCh), 58,35 (d, J = 7,9 Hz, 1 H), 7,66 (br. s., 1 H), 7,55 (d, J = 7,9 Hz, 1 H), 7,34 (t, J = 7,7 Hz, 1 H), 7,02 (t, J = 7,5 Hz, 1 H), 4,29-4,14 (m, 4 H), 3,46-3,34 (m, 1 H), 1,46 (s, 9 H).
Etapa 2
T er-butil 3-((2-bromofenil)(4-metoxibencil)carbamoil)azetidina-1 -carboxilato
Se añadió ter-butil 3-((2-bromofenil)carbamoil)azetidina-1-carboxilato 2 (7,0 kg, 19,71 mol) a una caldera de reacción de 30 L, se añadieron 14,0 litros de DMF, carbonato de cesio (7,7 kg, 23,65 mol). La temperatura se controló por debajo de 60 °C y se añadió gota a gota cloruro de p-metoxibencilo (3,24 kg, 20,69 mol). Una vez completada la adición, la temperatura de la reacción se controló a 98-102 °C y la reacción se llevó a cabo durante 17 horas. Las materias primas se hicieron reaccionar completamente mediante detección por TLC. La disolución de reacción se transfirió a una caldera de 50 L, se añadieron 15 litros de acetato de etilo y 10 litros de agua y la mezcla se agitó durante 10 minutos y se dejó reposar y se separó en capas. La capa acuosa se extrajo con 15 litros de acetato de etilo. Las capas orgánicas se combinaron y lavaron con 10 litros de agua y 10 litros de salmuera al 15%, respectivamente. La capa orgánica se concentró a sequedad a presión reducida, y se añadieron 20 litros de n- heptano y la mezcla se trituró durante 3 horas, y la torta del filtro se secó a vacío para producir ter-butil 3-((2-bromofenil)(4-metoxibencil)carbamoil)azetidina-1-carboxilato 3 (8,78 kg, sólido blanco), que se utilizó directamente en la siguiente etapa sin purificación.
MS m/z (ESI): 475,2 [M 1].
1H RMN (400 MHz, CDCI3) 57,69 (dd, J = 1,9, 7,7 Hz, 1 H), 7,25-7,16 (m, 2 H), 7,10 (d, J = 8,5 Hz, 2 H), 6,79 (d, J = 8,8 Hz, 2 H), 6,65 (dd, J = 2,0, 7,3 Hz, 1 H), 5,56 (d, J = 14,1 Hz, 1 H), 4,29 (br. s., 1 H), 4,00 (d, J = 14,3 Hz, 2 H), 3,79 (s, 3 H), 3,72 (br. s., 1 H), 3,56 (br. s., 1 H), 3,10-3,01 (m, 1 H), 1,41 (s, 9 H).
Etapa 3
Ter-butil 1 ’-(4-metoxibencil)-2'-oxospiro[azetidina-3,3'-indolina]-1 -carboxilato
Ter-ButiIo
Se añadió 3-((2-bromofenil)(4-metoxibencil)carbamoil)azetidina-1-carboxilato 3 (3,0 kg, 6,31 mol) a una caldera de reacción de 30 L, se añadieron 12 litros de 1,4-dioxano y se añadió ter-butóxido de sodio (0,91 kg, 9,47 mol). La caldera de reacción se selló, se puso a vacío durante 5 minutos, se llenó con nitrógeno, que se repitió 3 veces. El puerto de escape de la caldera de reacción se selló con líquido y se añadieron acetato de paladio (70,84 g, 0,32 mol), triciclohexilfosfina (176,97 g, 0,63 mol) bajo flujo continuo de nitrógeno. El nitrógeno se mantuvo lleno y la temperatura exterior de la caldera de reacción se ajustó a 75 °C. Cuando la temperatura interna se elevó a 60 °C, la pared interna del recipiente de reacción comenzó a condensarse, lo que duró 10 minutos. En este momento la temperatura interna es de 65 °C. Cuando la temperatura exterior de la caldera de reacción se fijó en 90 °C, la temperatura interna aumentó lentamente a 78 °C, el color de la disolución de reacción cambió gradualmente de marrón oscuro a gris oscuro, mientras que la condensación de la pared interna de la caldera de reacción se agravó y se aceleró el aumento de temperatura. Al mismo tiempo, se aumentó la velocidad de entrada de nitrógeno y se abrió el puerto de escape de la caldera de reacción en el sello de líquido. La temperatura interna continuó subiendo a 103 °C y comenzó a enfriarse después de 103 °C durante 10 minutos. La velocidad de entrada de nitrógeno se redujo y el sistema de reacción se selló con líquido, la temperatura interna se redujo gradualmente a 90 °C. La reacción continuó a 90 °C durante 0,5 horas. Se tomó y controló la muestra, la HPLC mostró que 3 desapareció por completo, apareció un subproducto de aproximadamente el 10%. La caldera de reacción se enfrió a temperatura ambiente, se añadieron 10 litros de acetato de etilo, se añadieron 15 litros de agua y se agitó durante 5 minutos, la mezcla se dejó reposar y se separó en capas. La capa acuosa se extrajo con 10 litros de acetato de etilo y las capas orgánicas se combinaron. La capa orgánica se lavó una vez con 10 litros de salmuera al 15%, después de la separación, la mezcla se concentró a presión reducida a 10 litros de disolvente y se añadieron 5 litros de n-heptano y la mezcla se concentró a sequedad para dar 3 kg de peso húmedo en bruto. El peso húmedo en bruto se transfirió a una caldera de reacción, se añadieron 9 litros de acetato de etilo, 0,3 kg de sal trisódica monohidrato de 1,3,5-triazina-2,4,6-tritiol y la mezcla se calentó a 80 °C y se agitó durante 2 horas y después se filtró la mezcla caliente. El filtrado se concentró hasta sequedad a presión reducida para dar 2,8 kg de producto bruto. El producto bruto se transfirió a una caldera de reacción, se añadieron 1,125 litros de acetato de etilo y la mezcla se calentó a 80 °C y se agitó durante 1 hora. Se añadieron 4,5 litros de n-heptano lentamente. Una vez completada la adición, la mezcla se agitó a 80 °C durante 1 hora. La mezcla se enfrió a 60 °C y se mantuvo a 60 °C durante 1 hora. La caldera de reacción se enfrió a 20 °C en 7 horas y la mezcla se agitó a 20 °C durante 8 horas. La mezcla se filtró para producir una torta de filtración de ter-butil 1'-(4-metoxibencil)-2'-oxospiro[azetidina-3,3'-indolina]-1-carboxilato 4 (1,9 kg, sólido de color marrón claro), que se utilizó directamente en el siguiente etapa sin purificación.
MS m/z (ESI): 395,2 [M 1].
1H RMN (400 MHz, CDCla) 57,59 (d, J = 7,3 Hz, 1 H), 7,32-7,26 (m, 3 H), 7,15 (t, J = 8,0 Hz, 1 H), 6,89 (día, J = 8,5 Hz, 2 H), 6,82 (d, J = 7,8 Hz, 1 H), 4,89 (s, 2 H), 4,47 (d, J = 8,3 Hz, 2 H), 4,13 (d, J = 8,0 Hz, 2 H), 3,82 (s, 3 H), 1,55 (s, 9 H).
Etapa 4
1-Isobutirilspiro[azetidina-3,3'-indolin]-2'-ona
Se añadieron 3,3 litros de anisol a una caldera de reacción de 30 L, se añadió ter-butil 1'-(4-metoxibencil)-2'-oxospiro[azetidina-3,3'-indolina]-1-carboxilato 4 (3,3 kg, 8,37 mol). La temperatura se controló a no más de 15 °C y se añadieron 6,6 litros de ácido trifluoroacético. Una vez completada la adición, la reacción se mantuvo a 15 °C y se hizo reaccionar durante 0,5 h. La temperatura se controló a no más de 15 °C y se añadieron gota a gota 2,22 litros de ácido trifluorometanosulfónico. Una vez completada la adición, la mezcla se mantuvo a 15 °C y se agitó durante 14 horas. La temperatura se controló a no más de 15 °C, se añadieron 6,6 litros de acetato de etilo al recipiente de reacción y la mezcla se agitó durante 0,25 horas. La disolución de reacción se transfirió a una caldera de 50 L y se añadieron gota a gota 12 litros de n-heptano a la caldera de 50 L para precipitar un sólido, se continuó agitando la mezcla durante 0,5 horas. La mezcla se filtró y la torta de filtración pesó 4,0 kg. La torta de filtración se transfirió a una caldera de reacción de 30 L, se añadieron 13,2 litros de diclorometano, la temperatura se controló a no más de 15 °C y se añadió trietilamina (4,08 L, 29,28 mol), la temperatura se controló a no más de 5 °C, y se añadió gota a gota cloruro de isobutirilo (0,865 L, 8,37 mol) diluido con 0,8 litros de diclorometano en aproximadamente 2 horas. Una vez completada la adición, la temperatura se mantuvo a 5 °C durante 0,25 horas. La pureza del producto principal fue del 91,5% mediante detección por HPLC. La temperatura no superó los 15 °C, se añadieron 0,13 litros de metanol y la mezcla se agitó durante 0,25 horas. La temperatura se controló a no más de 15 °C, se añadieron 5 litros de ácido clorhídrico 2 mol/L. El pH de la fase acuosa fue de aproximadamente 3 por detección. La mezcla se dejó reposar y se separó en capas, la capa
acuosa se extrajo cuatro veces con 3 litros de diclorometano/metanol (v/v = 10/1). Las capas orgánicas se combinaron y lavaron con 5 litros de agua. La capa orgánica se concentró a 1,5 litros a presión reducida, se añadieron 10 litros de acetato de etilo y la mezcla se concentró a 4 litros, y después el concentrado se transfirió a una caldera de reacción de 30 litros, se calentó a 70 °C y se añadieron 4 litros de n-heptano. La temperatura del recipiente de reacción se controló a 70 °C y se agitó durante 6 horas. Se enfrió la temperatura a 5 °C en 10 horas y se continuó agitando la mezcla a 5 °C durante 50 horas. La mezcla se filtró para dar 2,6 kg de sólido. El sólido contenía trifluorometanosulfonato de trietilamonio por detección de 1H RMN. El sólido se transfirió a una caldera de reacción de 30 L, se añadieron 7,5 litros de agua destilada, la mezcla se agitó durante 16 horas y se filtró para dar 2,95 kg de polvo de peso húmedo, que se secó a 50 °C durante 48 horas a vacío para dar 1 -isobutirilspiro[azetidina-3,3'-indolin]-2'-ona 5 (1,54 kg, sólido amarillo pálido, 99% de pureza por HPLC).
MS m/z (ESI): 245,2 [M 1].
1RMN H (400 MHz, CDCla) 59,03 (br. s., 1H), 7,48 (d, J = 7,5 Hz, 1 H), 732-7,23 (m, 1 H), 7,16-7,05 (m, 1 H), 6,96 (d, J = 8,0 Hz, 1 H), 4,59 (d, J = 8,0 Hz, 1 H), 4,44 (d, J = 9,5 Hz, 1 H), 4,30 (d, J = 8,0 Hz, 1 H), 4,19 (d, J = 9,5 Hz, 1 H), 2,53 (td, J = 6,8, 13,6 Hz, 1 H), 1,19 (dd, J = 2,5, 6,5 Hz, 6 H).
Etapa 5
1 '-(2,2-Dietoxietil)-1 -isobutirilspiro [azetidina-3,3'-indolin]-2'-ona
Se añadieron 15 litros de DMF a una caldera de reacción de 30 L, se añadieron 1 -isobutirilspiro[azetidina-3,3'-indolin]-2'-ona 5 (2,94 kg, 11,35 mol), carbonato de cesio (5,55 kg, 17,02 mol), 2-bromo-1,1-dietoxietano (2,50 kg, 12,48 mol). La temperatura se controló a 88-92 °C y se hizo reaccionar durante 18 horas. La muestra fue tomada y detectada por HPLC, la relación de 5/6 fue del 0,7%, la reacción se completó. La temperatura se enfrió a 20 °C, el volumen total de la mezcla de reacción fue de aproximadamente 20 litros. La disolución de reacción se transfirió a dos calderas de 50 litros, 10 litros por caldera. Se añadieron 12 litros de acetato de etilo, 28 litros de agua, respectivamente, a la caldera de reacción de 50 litros, y la mezcla se agitó durante 5 mezclas, se dejó reposar y se separó en capas. La capa acuosa se extrajo dos veces con acetato de etilo y 7 litros cada vez. Las capas orgánicas se combinaron, se lavaron dos veces con salmuera al 10%, 5 litros cada vez, y se lavaron de nuevo con 5 litros de agua. Todas las capas orgánicas se combinaron y después se concentraron a 7 litros a 45 °C bajo presión reducida, se añadieron 7 litros de diclorometano, se concentró a 4 litros a 45 °C, se añadieron 4 litros de diclorometano y se concentró a sequedad bajo presión reducida a 45 °C para dar 1’-(2,2-dietoxietil)-1-isobutirilspiro[azetidina-3,3'-indolin]-2'-ona 6 (4,17 kg, aceite amarillo brillante, 93,9% de pureza por HPLC, contenido de peso magnético nuclear 91,0%).
MS m/z (ESI): 361,2 [M 1]
1RMN H (400 MHz, CDCla) 57,46 (d, J = 7,3 Hz, 1 H), 7,30 (dt, J = 1,0, 7,8 Hz, 1H), 7,16-7,08 (m, 1H), 7,05 (d, J = 8,0 Hz, 1 H), 4,69 (t, J = 5,3 Hz, 1 H), 4,56 (d, J = 8,0 Hz, 1 H), 4,40 (d, J = 9,3 Hz, 1 H), 4,26 (d, J = 8,0 Hz, 1 H), 4,15 (d, J = 9,3 Hz, 1 H), 3,88-3,78 (m, 2H), 3,78-3,69 (m, 2H), 3,55-3,45 (m, 2H), 2,51 (td, J = 6,8, 13,7 Hz, 1 H), 1,19-1,10 (m, 12 H).
Etapa 6
2-(1-isobutiril-2'-oxospiro[azetidina-3,3'-indolin]-1'-il)acetaldehído
Se disolvió 1'-(2,2-dietoxietil)-1-isobutirilspiro[azetidina-3,3'-indolin]-2'-ona 6 (4,16 kg, 10,5 mol) en 6,24 litros de diclorometano y se añadió a una caldera de reacción de 30 litros y se añadieron 2,08 litros de agua. La temperatura se controló a 0-5 °C, se añadieron 6,24 litros de ácido trifluoroacético. Una vez completada la adición, se llevó a cabo la reacción a 28-32 °C durante 3 horas. La muestra fue tomada y detectada por HPLC, la relación de 6/7 fue del 0,95% y la reacción se completó. La mezcla de reacción se enfrió a 15 °C y el volumen total de la disolución de reacción fue de 18 litros. Se añadieron 12 litros de diclorometano y el volumen total fue de 30 litros, que se transfirió a dos calderas de 50 litros, 15 litros por caldera. Se añadieron 30 litros de agua al caldera de 50 litros, respectivamente, y la mezcla se agitó durante 10 minutos y se dejó reposar y se separó en capas. La capa acuosa se extrajo una vez con 10 litros de diclorometano, las capas orgánicas se combinaron y se lavaron con salmuera al 10% tres veces, 8 litros cada vez, la capa orgánica se lavó una vez con 8 litros de bicarbonato de sodio saturado y 8 litros de agua. La salmuera al 10%, el bicarbonato de sodio saturado y la capa acuosa después del lavado se combinaron y extrajeron con 10 litros de diclorometano. Todas las capas orgánicas extraídas se combinaron, se concentraron a 5 litros a presión reducida a 35 °C y se concentraron hasta sequedad mediante la adición de 5 litros de acetato de etilo para producir 2-(1 -isobutiril-2'-oxospiro [azetidina-3,3'-indolin]-1'-il) acetaldehído 7 (3,41 kg, aceite amarillo bruto), que se utilizó directamente en la siguiente etapa sin purificación.
MS m/z (ESI): 287,1 [M 1].
1RMN H (400 MHz, CDCh) 59,70 (s, 1 H), 7,54 (d, J = 7,0 Hz, 1 H), 7,32 (dt, J = 1,0, 7,8 Hz, 1 H), 7,22-7,16 (m, 1 H), 6,67 (d, J = 7,8 Hz, 1 H), 4,66-4,49 (m, 3 H), 4,44 (d, J = 9,5 Hz, 1 H), 4,33 (d, J = 8,0 Hz, 1 H), 4,21 (d, J = 9,5 Hz, 1 H), 2,53 (quin, J = 6,8 Hz, 1 H), 1,18 (dd, J = 3,5, 6,8 Hz, 6 H).
Etapa 7
1 -Isobutiril-1 ’-((4,5,6,7-tetrahidro-1 H-benzo[d] imidazol-2-il) metil)espiro[azetidina-3,3'-indolin]-2'-ona
Se disolvió 2-(1 -isobutiril-2'-oxospiro[azetidina-3,3'-indolin]-1 '-il)acetaldehído 7 (3,4 kg, bruto) en 17 litros de DMF y se añadió a una caldera de reacción de 30 litros, se añadieron 1,2-ciclohexanodiona (1,34 kg, 11,96 mol), acetato de amonio (3,84 kg, 49,81 mol) y la mezcla se hizo reaccionar. a 68-72 °C durante 2 horas. La muestra se tomó y se detectó mediante HPLC y la reacción se completó. La mezcla de reacción se enfrió a 15 °C y el volumen total de la disolución de reacción fue de 20 litros, que se transfirió a dos calderas de 50 litros, 10 litros por caldera. Se añadieron 9 litros de diclorometano, 4,5 litros de acetato de etilo, 17 litros de hidróxido de sodio 1,5 mol/L, 17 litros de salmuera al 10%, respectivamente, a la caldera de 50 litros y la mezcla se agitó durante 10 minutos y se dejó reposar y se separó en capas. La capa orgánica se lavó tres veces con salmuera al 10%, 9 litros cada vez y se lavó de nuevo con 9 litros de agua. Todas las capas orgánicas extraídas se combinaron, se concentraron a 10 litros a 40 °C bajo presión reducida y se añadieron 17 litros de acetato de etilo y la mezcla se concentró hasta sequedad a 40 °C bajo presión reducida para dar un producto bruto. El producto bruto se transfirió a una caldera de reacción de 30 L y se añadieron 13 litros de acetato de etilo y la mezcla se agitó a 70 °C durante 1 hora. Se añadieron 13 litros de n-heptano y la mezcla se agitó a 70 °C durante 1 hora. Se controló la temperatura para enfriar de 70 °C a 0 °C en 2 horas y se agitó la mezcla a 0 °C durante 1 hora. La mezcla se filtró y la torta del filtro se secó a vacío a 60 °C durante 18 horas para producir 1 -isobutiril-1 ’-((4,5,6,7-tetrahidro-1 H-benzo[d] imidazol-2-il)metil)espiro[azetidina-3,3'-indolin]-2'-ona 8 (3,3 kg, sólido amarillo pálido, 95,9% de pureza por HPLC, contenido magnético nuclear 93,6%).
MS m/z (ESI): 379,2 [M 1]
1RMN H (400 MHz, CDCls) 59,62 (br. s., 1H), 7,46 (d, J = 7,3 Hz, 1 H), 7,36-7,28 (m, 1 H), 7,20 (d, J = 7,8 Hz, 1 H), 7,17-7,10 (m, 1 H), 4,94-4,79 (m, 2 H), 4,54 (d, J = 8,0 Hz, 1 H), 4,38 (d, J = 9,3 Hz, 1 H), 4,26 (d, J = 8,0 Hz, 1 H), 4,13 (d, J = 9,5 Hz, 1 H), 2,50 (d, J = 6,8 Hz, 4 H), 1,75 (br. s., 4 H), 1,17 (d, J = 6,8 Hz, 6 H).
Etapa 8
4-Metilbencenosulfonato de 4,4,4-trifluorobutilo
Se disolvió 4,4,4-trifluorobutan-1 -ol 9 (1,5 kg, 11,71 mol) en 7,5 litros de tetrahidrofurano y se añadió a una caldera de reacción de 30 litros. Se añadió lentamente gota a gota una disolución de hidróxido de potasio al 20% (1,97 kg, 7,5 L) a 0-5 °C. Una vez completada la adición, se añadió cloruro de p-toluenosulfonilo (3,35 kg, 17,57 mol) en lotes a 0-5 °C. Una vez completada la adición y la mezcla se hizo reaccionar a 10 °C durante 2 horas. La reacción se completó mediante detección por TLC. Se añadieron 4,5 litros de acetato de etilo, 4,5 litros de agua y la mezcla se agitó durante 5 minutos y se dejó reposar y se separó en capas. La capa acuosa se extrajo dos veces con acetato de etilo y 4,5 litros cada vez. La capa orgánica se concentró a presión reducida para proporcionar 4,4,4-trifluorobutil 4-metilbencenosulfonato 10 (2,95 kg, aceite incoloro).
1RMN H (400 MHz, CDCla) 57,77 (d, J = 8,0 Hz, 2 H), 7,35 (d, J = 8,0 Hz, 2 H), 4,07 (t, J = 6,0 Hz, 2 H), 2,45 (s, 3 H), 2,21-2,12 (m, 2 H), 1,93-1,89 (m, 2 H).
Etapa 9
1 -Isobutiril-1 ’-((1-(4,4,4-trifluorobutil)-4,5,6,7-tetrahidro-1 H-benzo[d]imidazol-2-il)metil)espiro[azetidina-3,3'-indolin]-2'-ona (I)
Se añadieron 12 litros de DMF a un caldera de reacción de 30 L, se añadieron 1 -isobutiril-1 ’-((4,5,6,7-tetrahidro-1 H-benzo[d] imidazol-2-il) metil)espiro[azetidina-3,3'-indolin]-2'-ona 8 (3,2 kg, 8,81 mol), carbonato de cesio (3,87 kg, 11,87 mol), 4,4,4-trifluorobutil-4-metilbencenosulfonato 10 (2,51 kg, 8,86 mol) y la mezcla se hizo reaccionar a 88-92 °C durante 3,5 horas. La muestra fue tomada y detectada por HPLC, la relación de 10/8 fue del 0% y la reacción se completó. La mezcla de reacción se enfrió a 15 °C y el volumen total de la disolución de reacción fue de 18 litros, que se transfirió a dos calderas de 50 litros, 9 litros por caldera. Se añadieron 12 litros de acetato de etilo, 24 litros de agua destilada a la caldera de 50 litros, respectivamente, y la mezcla se agitó durante 10 minutos y se dejó reposar y se separó en capas. La capa acuosa se extrajo de nuevo con 12 litros de acetato de etilo y las capas orgánicas se combinaron. La capa orgánica se lavó tres veces con agua destilada, 8 litros cada vez. Todas las capas orgánicas extraídas se combinaron y concentraron hasta sequedad a 45 °C bajo presión reducida y se secaron a vacío para dar 3,8 kg de producto bruto, 93,5% de pureza por HPLC. Se tomaron 2,8 kg del producto bruto y se añadieron a una caldera de reacción de 30 L, se añadieron 1,68 litros de etanol y la mezcla se calentó a 65 °C para disolver completamente. Se añadieron 3,36 litros de n-heptano y se continuó agitando la mezcla a 65 °C durante 10 minutos. La mezcla caliente se filtró, la bolsa de tela filtrante se lavó con 1,26 litros de etanol/n-heptano (v/v = 1/2), que se combinó en las aguas madres. Las aguas madres se transfirieron a una caldera de reacción de 30 L, se calentó a 75 °C, se añadieron 8,4 litros de n-heptano y se continuó agitando la mezcla durante 10 minutos. La mezcla se enfrió a 55-57 °C, se añadieron 3 g de cristales de siembra y la mezcla se continuó agitando durante 1 hora a 55-57 °C. La temperatura de la mezcla se enfrió a 2-4 °C en 4 horas y se mantuvo a 2-4 °C y se agitó durante 16 horas. La mezcla se filtró y la torta del filtro se enjuagó con 2,8 litros de etanol/n-heptano (v/v = 1/6) y se secó a vacío a 45 °C durante 16-24 horas para producir un polvo amarillo pálido de 2,1 kg y la pureza por HPLC fue del 98,9%. El polvo se pulverizó
mediante un molino de chorro para dar 1 -isobutiril-1 ’-((1 -(4,4,4-trifluorobutil)-4,5,6,7-tetrahidro-1 H-benzo[d]imidazol-2-il)metil)espiro[azetidina-3,3'-indolin]-2'-ona (I) (2,05 kg, sólido blanco amarillo claro, 98,7% de pureza por HPLC, contenido de peso magnético nuclear 98,2%).
MS m/z (ESI): 489,3 [M 1].
1RMN H (400 MHz, CD3OD) 5 7,61 (d, J = 8,03 Hz, 1 H), 7,49 (d, J = 7,28 Hz, 1 H), 7,37 (t, J = 7,78 Hz, 1 H), 7,21 7,15 (m, 1 H), 5,05-4,92 (m, 2 H), 4,56 (d, J = 8,03 Hz, 1 H), 4,41 (d, J = 9,29 Hz, 1 H), 4,28 (d, J = 7,78 Hz, 1 H), 4,18 (d, J = 9,54 Hz, 1 H), 4,10-3,91 (m, 2 H), 2,63-2,44 (m, 5 H), 2,31-2,05 (m, 2 H), 1,81 (br. s., 6 H), 1,20 (d , J = 6,78 Hz, 6 H).
Preparación de la forma cristalina A
Se pesaron aproximadamente 0,6 g del compuesto de fórmula (I) y se añadieron a 5 ml de acetato de etilo, la mezcla se calentó a 60 °C y se agitó para disolverla y convertirla en una disolución saturada. La disolución caliente se filtró y el filtrado se enfrió de forma natural a 10 a 15 °C y se tardó de 1,5 a 2 horas. El sólido precipitado se recogió por filtración y se secó a vacío a 40 °C durante 20 horas para proporcionar 0,39 g de polvo sólido. El resultado de la detección de XRPD fue la forma cristalina A.
Preparación de la forma cristalina B
Se pesaron aproximadamente 0,25 g del compuesto de fórmula (I) y se añadieron 2,5 ml de tetrahidrofurano a temperatura ambiente 10-15 °C para formar una disolución saturada. La disolución se filtró y el agua desionizada antidisolvente se añadió gradualmente gota a gota a 6 ml para precipitar gradualmente el sólido blanco lechoso. Se continuó agitando la mezcla durante 16 a 20 horas. El sólido precipitado se recogió por filtración y se secó a vacío a 40 °C durante 20 horas para proporcionar 0,13 g de polvo sólido. El resultado de la detección de XRPD fue la forma cristalina B.
El experimento de estabilidad de la forma cristalina A del compuesto de fórmula (I) en diferentes disolventes.
Se pesaron en paralelo una pluralidad de 50 mg de la forma cristalina A del compuesto de fórmula (I), se añadieron de 0,2 a 0,3 ml del disolvente simple o mixto de la tabla siguiente, respectivamente. La mezcla se agitó a 40 °C. Si el compuesto estaba completamente disuelto, la muestra se dejaba enfriar a temperatura ambiente, y si el sólido precipitaba, la mezcla continuaba agitando; y si la muestra permaneció en disolución, el disolvente se evaporó naturalmente para eliminarlo. Después de agitar todas las suspensiones durante 2 días, se recogieron los sólidos de todas las muestras y se utilizó XRPD para detectar el estado de la forma cristalina. Los resultados se muestran en la Tabla 3.
Tabla 3. El experimento de estabilidad de la forma cristalina A en diferentes disolventes

El experimento de estabilidad física sólida de la forma cristalina A del compuesto de fórmula (I) en diferentes condiciones de temperatura y humedad.
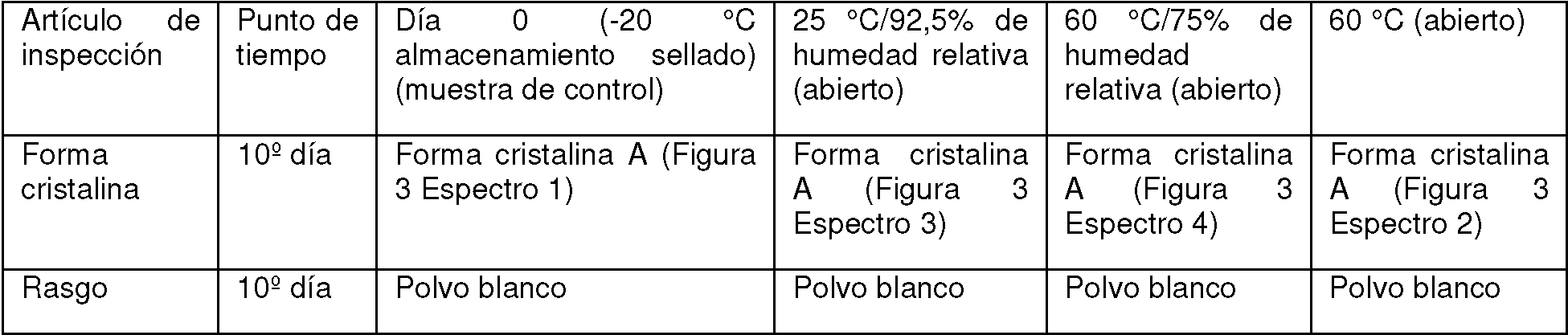
Las partes 3 de la forma cristalina A del compuesto de fórmula (I) sólido se pesaron en paralelo, cada una de aproximadamente 50 mg, y después se colocaron en el fondo de los viales de vidrio, se extendieron para formar una capa delgada, los cuellos de botella se sellaron con papel de aluminio, y se hicieron algunos pequeños orificios en el papel de aluminio para asegurar que la muestra pueda estar completamente en contacto con el aire ambiente. Las tres muestras preparadas se colocaron a 25 °C/92,5% de humedad relativa, 60 °C y 60 °C/75% de humedad relativa, respectivamente, y se observó la estabilidad física de las muestras durante 10 días. Al mismo tiempo, se pesó por separado aproximadamente 50 mg de la forma cristalina A del compuesto de fórmula (I) sólido, se colocó en el fondo de un vial de vidrio, se selló con una tapa roscada y se almacenó a-20 °C como muestra de control. Al décimo día, se retiraron todas las muestras y se volvieron a poner a temperatura ambiente. Se observaron los cambios de apariencia de las muestras y la forma cristalina de las muestras se detectó mediante XRPD. La estabilidad física sólida de la forma cristalina A del compuesto de fórmula (I) se determinó comparando la muestra acelerada con la muestra de control. Los resultados experimentales para la estabilidad física sólida de la forma cristalina A se muestran en la tabla 4 a continuación.
Tabla 4. El experimento de estabilidad física sólida de la forma cristalina A en diferentes condiciones de temperatura y humedad
La prueba de solubilidad aproximada de la forma cristalina A del compuesto de fórmula (I).
A temperatura ambiente (20 a 30 °C), se añadieron de 1 a 1,5 mg de la forma cristalina A del compuesto de fórmula (I) a un matraz aforado, y se añadió varias veces un único disolvente orgánico o un disolvente mixto en una pequeña cantidad hasta que la disolución visual fue clara o sin partículas sólidas presentes. Se determinó la solubilidad aproximada de la forma cristalina A en diferentes disolventes y los resultados se muestran en la Tabla 5.
Tabla 5. La solubilidad de la forma cristalina A del compuesto de fórmula (I) en diferentes disolventes

Evaluación de la Realización 1 in vitro.
Ensayo de CPE largo de VSR
Propósito experimental:
Los valores de EC50 y CC50 de los compuestos del virus sincitial respiratorio anti-VSR se detectaron por citopatología. Materiales experimentales:
Línea celular: Hep2
Cepa de virus: virus sincitial de aspiración VSR (una cepa larga)
Medio de cultivo celular (se añadieron DMEM/F12, Gibco # 11330, 10% de suero Gibco # 16140 y 1% de penicilinaestreptomicina (penicilina 5000 UI/ml, estreptomicina 10 mg/ml), Gibco # 15140)
T ripsina (Gibco # 12605010)
PBS (Thermo # SH30264,01)
Azul tripán (Cat. Invitrogen 15250061)
CCK-8 (Dojindo # CK04-20)
CO2 Incubadora, Thermo 240 I
Dispensador automático multipunto, Thermo
Sistema de pretratamiento de microplacas totalmente automático ensamblador de placas POD 810, Labcyte Contador de células automatizado de mano Sceptre, Millipore
Espectrofotómetro de microplacas, dispositivo molecular.
Procedimientos y métodos experimentales:
a) Inoculación celular (célula Hep2)
1) El medio de cultivo celular se retiró y se lavó con 10 mL de PBS;
2) La tripsina precalentada se añadió al matraz de cultivo limpio y se hizo girar el matraz para permitir que la tripsina cubriera uniformemente el matraz de cultivo. Y después lo succionó en una incubadora a 37 °C, 5% CO2 para digerir.
3) Cada T150 se suspendió en 10 a 15 ml de medio de cultivo, se tomaron 0,1 ml y después se diluyó dos veces con una disolución de azul tripán.
4) Las células se diluyeron a 5 x 102 *4/mL con el medio de cultivo y las células diluidas se añadieron a las placas Corning 384 (Cat 3701) (30 |uL/pocillo, 1500 células/pocillo) con un dispensador automático (Thermo Scientific). Las células se centrifugaron (300 rpm) y las células se adhirieron a la pared y se colocaron en una incubadora a 37 °C, 5% CO2 durante la noche.
b) Adición de compuestos:
1) El compuesto disuelto en DMSO al 100% se diluyó en medio logaritmo y se añadió a la placa celular usando un manipulador de líquidos Echo. Para asegurar que la concentración final de DMSO fuera del 1 %;

2) Pocillo de control celular: sin compuesto ni virus; pocillo de control de virus: sin compuesto.
c) Inoculación del virus:
El virus VSR se diluyó a 100 TCID50/30 |uL con un medio de cultivo celular cultivado a 4 °C, y el virus diluido se añadió a las placas de células (30 |uL/pocillo) con un dispensador automático Multidrop® y se colocó en una incubadora a 37 °C, 5% CO2 durante 5 días.
d) Detección de efectos citopáticos:
1) Después de 5 días, se observó la citopática de cada pocilio. En circunstancias normales, no hubo citopatía en los pocilios de control celular, las células en los pocillos de control de virus eran completamente patologías.
2) Se añadió CCK-8 (Dojindo-CK04-20, 6 pL/pocillo) a placas de 384 pocillos con dispensador automático Multidrop®.
3) La mezcla se colocó en una incubadora a 37 °C, 5% CO2 durante 3 a 4 horas, los valores de absorbancia se leyeron a la longitud de onda de 450 nm y 630 nm utilizando un lector de microplacas (SPECTRA max 340PC_Molecular device).
4) Se analizaron los datos.
Los resultados experimentales se muestran en la Tabla 6:
Tabla 6. Resultados del ensayo CPE de prueba EC50/CC50

Conclusiones: Comparado con BMS433771, el compuesto de fórmula (I) tuvo mejor actividad in vitro y ninguno de los dos tiene citotoxicidad.
Claims (15)
1. Un proceso para preparar un compuesto de fórmula (I),

que comprende la siguiente etapa:

2. El proceso según la reivindicación 1, que comprende la siguiente etapa:

en donde, R se selecciona de

cada uno de R1 y R2 se selecciona independientemente de un alquilo C1-3;
opcionalmente, R1 y R2 están conectados al mismo átomo para formar un anillo de 5 a 6 miembros opcionalmente sustituido con 1,2, 3 o 4 de R’(s),
R’ se selecciona entre metilo, etilo, isopropilo o n-propilo.
3. El proceso según la reivindicación 2, en donde R se selecciona de


o el ácido se selecciona del grupo que consiste en ácido trifluoroacético, ácido acético, ácido clorhídrico, ácido sulfúrico diluido y ácido p-toluenosulfónico.
4. El proceso según la reivindicación 1, que comprende la siguiente ruta de reacción:

en donde,
HA se selecciona de ácido trifluorometanosulfónico;
el disolvente inerte es un disolvente único o una mezcla de disolventes para varios disolventes seleccionados del grupo que consiste en diclorometano, acetato de etilo, acetato de isopropilo, tetrahidrofurano, dioxano, 2 metiltetrahidrofurano, éter dietílico, metil ter-butil éter, pentano, n-hexano, ciclohexano, n-heptano y octano; la relación en peso del compuesto de fórmula (VII) al disolvente inerte es de 1:1 a 1:10.
5. El proceso según la reivindicación 4, en donde el disolvente inerte es un disolvente único o un disolvente mixto para varios disolventes seleccionados del grupo que consiste en acetato de etilo, acetato de isopropilo, metil ter-butil éter, ciclohexano y n-heptano, o el disolvente inerte es un disolvente mixto de acetato de etilo y n-heptano.
6. El proceso según la reivindicación 1, que comprende la siguiente ruta de reacción:
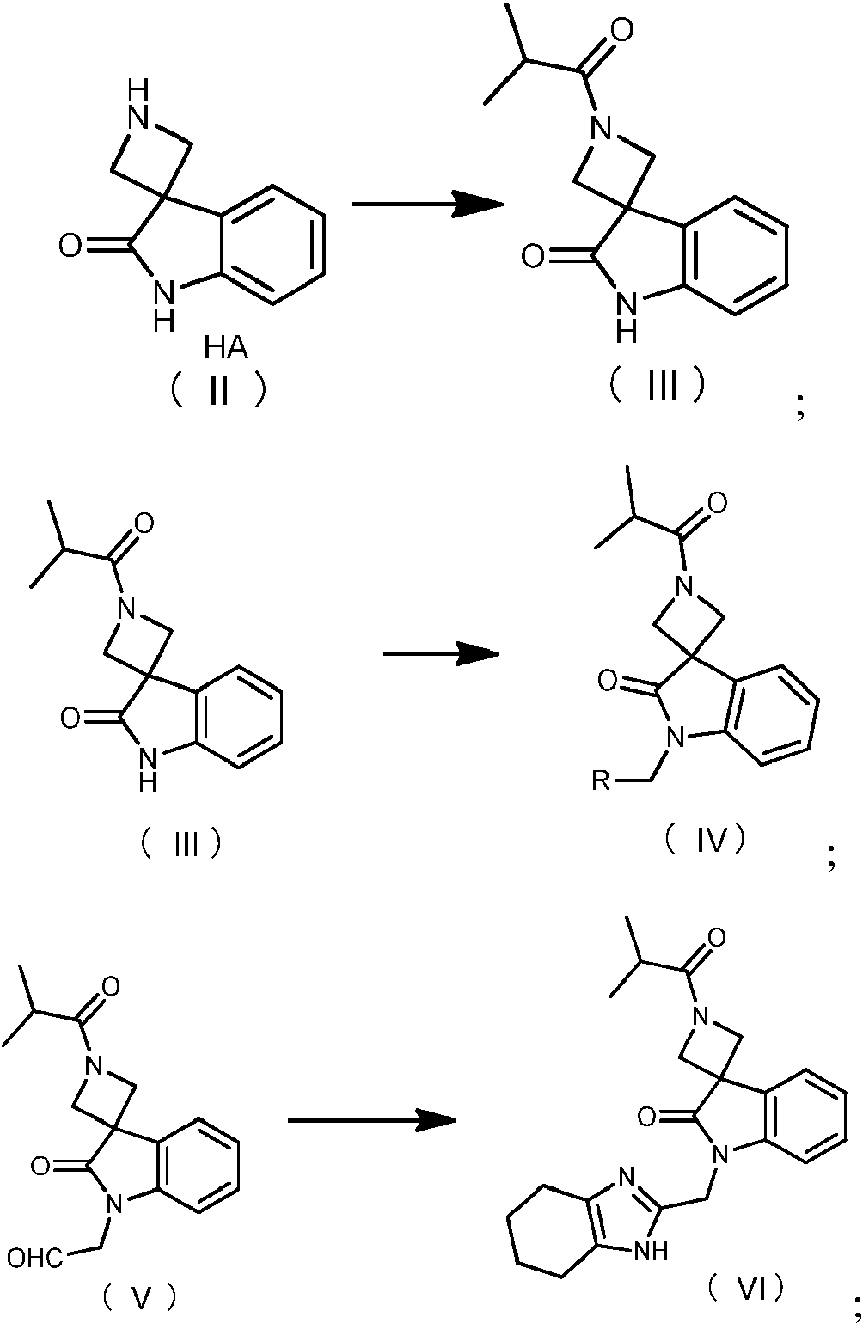
y/o

7. Un compuesto de la siguiente fórmula como intermedio para preparar el compuesto de fórmula (I):

8. Forma cristalina A o forma cristalina B del compuesto de fórmula (I),

el análisis de espectros XRPD de la forma cristalina A se muestra en la Tabla 1;
Tabla 1


el análisis de espectros XRPD de la forma cristalina B se muestra en la Tabla 2;
Tabla 2

9. Un proceso para preparar la forma cristalina A según la reivindicación 8, que comprende añadir el compuesto de fórmula (I) a un disolvente orgánico, calentar a 30 °C hasta la temperatura de reflujo para disolver y después enfriar a 0 a 20 °C dentro de 0,5 a 10 horas para precipitar el cristal.
10. El proceso para preparar la forma cristalina A según la reivindicación 9, en donde el disolvente orgánico se selecciona del grupo que consiste en dicloroetano, un alcohol alquílico C1-6, un éter C4-10 o éter cíclico, una cetona C2-, un éster C2-6, benceno opcionalmente sustituido con un(os) átomo(s) de metilo o etilo o halógeno, en donde el número de sustituyentes se selecciona entre 1,2 y/o 3.
11. El proceso para preparar la forma cristalina A según la reivindicación 10, el disolvente orgánico se selecciona del grupo que consiste en diclorometano, metanol, etanol, isopropanol, tetrahidrofurano, dioxano, 2-metiltetrahidrofurano, acetona, acetato de etilo, acetato de isopropilo, tolueno, xileno y/o clorobenceno.
12. Un proceso para preparar la forma cristalina B según la reivindicación 8, que comprende añadir el compuesto de fórmula (I) a un disolvente orgánico polar y después dejar caer lentamente agua para precipitar el cristal.
13. El proceso para preparar la forma cristalina B según la reivindicación 12, en donde el disolvente orgánico polar se selecciona del grupo que consiste en un alcohol alquílico C1-6, un éter C4-10 o éter cíclico, una cetona C2-6, opcionalmente, el disolvente orgánico polar se selecciona del grupo que consiste en metanol, etanol, isopropanol, tetrahidrofurano, 2-metiltetrahidrofurano, dioxano, acetona.
14. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de la forma cristalina según la reivindicación 8 como ingrediente activo y un vehículo farmacéuticamente aceptable.
15. La forma cristalina según la reivindicación 8 o la composición farmacéutica según la reivindicación 14 para uso en el tratamiento de la infección por virus respiratorios.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510309106 | 2015-06-08 | ||
| PCT/CN2016/085104 WO2016197908A1 (zh) | 2015-06-08 | 2016-06-07 | 咪唑衍生物及其中间体的制备方法和晶型 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2821649T3 true ES2821649T3 (es) | 2021-04-27 |
Family
ID=57502897
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16806806T Active ES2821649T3 (es) | 2015-06-08 | 2016-06-07 | Método para preparar derivados de imidazol e intermedios de los mismos y formas cristalinas |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US10227309B2 (es) |
| EP (1) | EP3305790B1 (es) |
| JP (1) | JP6548751B2 (es) |
| CN (1) | CN107709328B (es) |
| ES (1) | ES2821649T3 (es) |
| PT (1) | PT3305790T (es) |
| TW (1) | TWI695005B (es) |
| WO (1) | WO2016197908A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107709328B (zh) | 2015-06-08 | 2021-03-05 | 山东丹红制药有限公司 | 咪唑衍生物及其中间体的制备方法和晶型 |
| CN112057453B (zh) * | 2019-08-26 | 2022-06-28 | 中检科医药科技(北京)集团有限公司 | Enc002及其类似物在治疗或预防肠道病毒感染中的应用 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4175289A (en) | 1978-06-05 | 1979-11-20 | International Business Machines Corporation | Serrated Y-bar magnetic bubble switch |
| US4324794A (en) | 1980-08-26 | 1982-04-13 | Research Triangle Institute | Inhibition of respiratory syncytial virus-induced cell fusion by amidino compounds |
| DK154078C (da) | 1981-02-06 | 1989-05-22 | Ucb Sa | Analogifremgangsmaade til fremstilling af 2-(2-(4-(diphenyl-methyl)-1-piperazinyl)ethoxy)-acetamider eller syreadditionssalte heraf |
| US5256668A (en) | 1993-03-17 | 1993-10-26 | American Home Products Corporation | Aminopyrimidine derivatives as antiviral agents for respiratory syncytial virus |
| BR9700939A (pt) | 1996-02-13 | 1998-09-01 | American Cyanamid Co | Compostos aniônicos contendo traiazina úteis como agentes antivirais |
| AU759772B2 (en) | 1998-01-29 | 2003-05-01 | Viropharma Incorporated | Compounds, compositions and methods for treating or preventing pneumovirus infection and associated diseases |
| AU741946B2 (en) | 1998-07-20 | 2001-12-13 | Bristol-Myers Squibb Company | Substituted benzimidazole antiviral agents |
| WO2010103306A1 (en) | 2009-03-10 | 2010-09-16 | Astrazeneca Uk Limited | Benzimidazole derivatives and their use as antivaral agents |
| CN103269712B (zh) | 2010-11-03 | 2016-09-21 | 伊缪诺金公司 | 包含新型安丝菌素衍生物的细胞毒性剂 |
| TWI527814B (zh) * | 2010-12-16 | 2016-04-01 | 健生科學愛爾蘭無限公司 | 作為呼吸道融合病毒抗病毒劑之氮雜苯并咪唑類 |
| GB201119538D0 (en) | 2011-11-10 | 2011-12-21 | Viral Ltd | Pharmaceutical compounds |
| BR112015008325B1 (pt) * | 2012-10-16 | 2022-01-25 | Janssen Sciences Ireland Uc | Compostos antivirais contra o rsv, seu uso e composição farmacêutica que os compreende |
| CN103319599B (zh) | 2013-05-27 | 2015-02-18 | 上海交联药物研发有限公司 | 一种抗人ErbB2抗体—美登木素偶联物及其应用 |
| WO2015085844A1 (zh) * | 2013-12-10 | 2015-06-18 | 南京明德新药研发股份有限公司 | 作为抗病毒剂的咪唑衍生物及其制药用途 |
| TWI671299B (zh) * | 2014-04-14 | 2019-09-11 | 愛爾蘭商健生科學愛爾蘭無限公司 | 作為rsv抗病毒化合物之螺脲化合物 |
| GB201417707D0 (en) * | 2014-10-07 | 2014-11-19 | Viral Ltd | Pharmaceutical compounds |
| CN107709328B (zh) * | 2015-06-08 | 2021-03-05 | 山东丹红制药有限公司 | 咪唑衍生物及其中间体的制备方法和晶型 |
-
2016
- 2016-06-07 CN CN201680031461.2A patent/CN107709328B/zh active Active
- 2016-06-07 EP EP16806806.2A patent/EP3305790B1/en active Active
- 2016-06-07 US US15/580,167 patent/US10227309B2/en active Active
- 2016-06-07 ES ES16806806T patent/ES2821649T3/es active Active
- 2016-06-07 WO PCT/CN2016/085104 patent/WO2016197908A1/zh not_active Ceased
- 2016-06-07 JP JP2017564539A patent/JP6548751B2/ja active Active
- 2016-06-07 PT PT168068062T patent/PT3305790T/pt unknown
- 2016-06-08 TW TW105118076A patent/TWI695005B/zh active
-
2019
- 2019-01-08 US US16/242,753 patent/US10633347B2/en active Active
-
2020
- 2020-03-20 US US16/824,877 patent/US10858322B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP3305790A1 (en) | 2018-04-11 |
| TWI695005B (zh) | 2020-06-01 |
| US20200223802A1 (en) | 2020-07-16 |
| EP3305790B1 (en) | 2020-08-05 |
| JP2018517719A (ja) | 2018-07-05 |
| US10227309B2 (en) | 2019-03-12 |
| EP3305790A4 (en) | 2018-10-03 |
| PT3305790T (pt) | 2020-10-19 |
| TW201704238A (zh) | 2017-02-01 |
| JP6548751B2 (ja) | 2019-07-24 |
| HK1244807A1 (zh) | 2018-08-17 |
| US20190218189A1 (en) | 2019-07-18 |
| US20180141915A1 (en) | 2018-05-24 |
| WO2016197908A1 (zh) | 2016-12-15 |
| CN107709328B (zh) | 2021-03-05 |
| US10633347B2 (en) | 2020-04-28 |
| US10858322B2 (en) | 2020-12-08 |
| CN107709328A (zh) | 2018-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2887303T3 (es) | Derivados de pirimidina para el tratamiento de infecciones víricas | |
| CA2879789C (en) | Fused pyrimidines as inhibitors of p97 complex | |
| CA3221608A1 (en) | Fused heteroaryl compounds useful as anticancer agents | |
| CA2936559C (en) | Fused pyrimidines as inhibitors of p97 complex | |
| CA3023922C (en) | Heterocyclic derivatives for the treatment of rsv | |
| US20090227619A1 (en) | Harmine derivatives, intermediates used in their preparations, preparation processes and use therefo | |
| PH12015500983B1 (en) | Heterocyclic substituted 2-amino-quinazoline derivatives for the treatment of viral infections | |
| AU2010214440A1 (en) | Hetero ring derivative | |
| CN118206550A (zh) | Shp2抑制剂及其应用 | |
| ES2821649T3 (es) | Método para preparar derivados de imidazol e intermedios de los mismos y formas cristalinas | |
| ES2511917T3 (es) | Derivados de 2-piridin-2-il-pirazol-3(2H)-ona, su preparación y su aplicación en terapéutica | |
| AU2022289202A1 (en) | Compound as cdk kinase inhibitor and use thereof | |
| ES2971892T3 (es) | Formas cristalinas del inhibidor de Syk sal hidrocloruro de 5-fluoro-1-metil-3-[[5-[4-(3-oxetanil)-1-piperazinil]-2-piridinil]amino]-6-(1H-pirazol-3-il)-2(1H)-quinolinona (1:1) | |
| WO2016133888A1 (en) | Compounds for treating respiratory syncytial virus infections | |
| BR112015028890B1 (pt) | Derivados de piridona, composição farmacêutica que os compreende e uso dos mesmos | |
| AU2019308941A1 (en) | BRD4 inhibitor, preparation method and use thereof | |
| CA2963054C (en) | Piperidine substituted tricyclic pyrazolo[1,5-a]pyrimidine derivatives with inhibitory activity on the replication of the respiratory syncytial virus (rsv) | |
| WO2017196982A1 (en) | Bicyclic fused pyrazole derivatives for the treatment of rsv | |
| ES2459301T3 (es) | Nuevos polimorfos de lopinavir | |
| CN115919872A (zh) | 三环类化合物的用途及包含其的药物组合物 | |
| HK1244807B (zh) | 咪唑衍生物及其中间体的制备方法和晶型 | |
| ES2835061T3 (es) | Forma de sal y forma cristalina de 1,2,5-tiadiazolidina 1,1-dióxido, método de preparación de las mismas e intermedios | |
| UA51666C2 (uk) | Аналоги 20(s)-камптотецину, спосіб їх одержання, фармацевтична композиція та спосіб лікування раку, лейкозу або станів пов'язаних з віл | |
| WO2024174953A1 (zh) | 苯并咪唑类衍生物及其医药用途 | |
| CN104861026A (zh) | 含呋喃骨架的二氢吡唑氧肟酸c21甾体皂苷苷元衍生物、其制备方法及应用 |