ES2832504T3 - Formulaciones para administración de antígenos de norovirus y rsv en el intestino delgado - Google Patents
Formulaciones para administración de antígenos de norovirus y rsv en el intestino delgado Download PDFInfo
- Publication number
- ES2832504T3 ES2832504T3 ES16808196T ES16808196T ES2832504T3 ES 2832504 T3 ES2832504 T3 ES 2832504T3 ES 16808196 T ES16808196 T ES 16808196T ES 16808196 T ES16808196 T ES 16808196T ES 2832504 T3 ES2832504 T3 ES 2832504T3
- Authority
- ES
- Spain
- Prior art keywords
- vaccine
- rsv
- subjects
- poly
- methacrylic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A61K48/0058—Nucleic acids adapted for tissue specific expression, e.g. having tissue specific promoters as part of a contruct
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
- A61K39/155—Paramyxoviridae, e.g. parainfluenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2059—Starch, including chemically or physically modified derivatives; Amylose; Amylopectin; Dextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2813—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/282—Organic compounds, e.g. fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/525—Virus
- A61K2039/5256—Virus expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
- A61K2039/541—Mucosal route
- A61K2039/542—Mucosal route oral/gastrointestinal
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/575—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 humoral response
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10041—Use of virus, viral particle or viral elements as a vector
- C12N2710/10043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10341—Use of virus, viral particle or viral elements as a vector
- C12N2710/10343—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/18011—Paramyxoviridae
- C12N2760/18511—Pneumovirus, e.g. human respiratory syncytial virus
- C12N2760/18534—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/16011—Caliciviridae
- C12N2770/16034—Use of virus or viral component as vaccine, e.g. live-attenuated or inactivated virus, VLP, viral protein
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Inorganic Chemistry (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Pulmonology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una composición inmunogénica para provocar una respuesta inmunitaria en un ser humano que comprende un agente biológico inmunogénico abarcado por un recubrimiento entérico que dirige la administración del agente biológico inmunogénico al íleon del ser humano, en donde el agente biológico inmunogénico es un vector adenovírico que codifica la proteína vírica 1 de norovirus o la proteína de fusión (F) del virus respiratorio sincitial (RSV), en donde el recubrimiento entérico tiene un pH umbral de 5,8-6,8.
Description
DESCRIPCIÓN
Formulaciones para administración de antígenos de norovirus y RSV en el intestino delgado
Referencias cruzadas a solicitudes relacionadas
La presente solicitud reivindica la prioridad de la Solicitud Provisional de Estados Unidos N.° 62/175.081, presentada el viernes, 12 de junio de 2015.
REFERENCIA AL ENVÍO DE UN LISTADO DE SECUENCIAS COMO ARCHIVO DE TEXTO
El listado de secuencias escrito en el archivo 1011223_ST25.txt, creado el 13 de mayo de 2016, de 62.317 bytes, formato de máquina IBM-PC, sistema operativo MS-Windows, se incorpora en el presente documento como referencia.
Antecedentes de la invención
Las vacunas son un medio importante para prevenir y/o tratar una serie de enfermedades y trastornos (por ejemplo, infección vírica, infección bacteriana y cáncer). La vacunación se realiza normalmente mediante inyección, lo que reduce la participación debido a la inconveniencia de viajar a un sitio de vacunación y la aversión a las inyecciones. Además, la inyección de vacunas requiere el uso de un kit estéril, tal como jeringas y agujas, y un médico capacitado para la administración.
Para la vacuna contra la gripe, se realizan campañas anuales a gran escala para recolectar suficientes huevos fecundados para cosechar y procesar suficientes virus para satisfacer las necesidades del mercado. La hemaglutinina (HA) procedente de cultivo celular o plantas pueden reducir la carga de la adquisición y el procesamiento de huevos, pero estos enfoques aún requieren un llenado y un acabado estériles costosos para producir agujas de jeringa individuales, que deben eliminarse como peligro biológico. Durante una pandemia, las escuelas pueden cerrarse y el distanciamiento social es obligatorio, sin embargo, la inmunización masiva contra la gripe generalmente requiere alinear a los sujetos en las clínicas para recibir inyecciones. Vacunas orales, para la gripe u otros patógenos, podrían enviarse por correo, evitando así la mayoría de los contactos humanos. Adicionalmente, la formación de comprimidos es un proceso sanitario rápido que no requiere el costoso proceso de llenado y acabado estéril que requieren las vacunas inyectadas.
Las vacunas que se pueden administrar de manera no parenteral, por ejemplo, por vía oral o mucosa, se describen en la patente de Estados Unidos n.° 8.222.224
El documento US 2010/0111989 divulga comprimidos de una proteína vírica inmunogénica de RSV.
Breve sumario de la invención
En el presente documento se proporcionan composiciones y su uso en métodos para una vacunación más eficaz de un sujeto (ser humano o no humano) que implica la administración de un agente biológico inmunogénico específicamente al íleon del sujeto. Por tanto, la presente divulgación proporciona vacunas más eficientes y eficaces y demuestra su eficacia en seres humanos.
En el presente documento se proporcionan composiciones inmunogénicas para provocar una respuesta inmunitaria en un sujeto que comprenden: un agente biológico inmunogénico abarcado por un agente de administración que dirige la administración del agente biológico inmunogénico al íleon del sujeto. En algunas realizaciones, el sujeto es un ser humano. En algunas realizaciones, el sujeto es un animal no humano, por ejemplo, primate, ratón, rata, conejo, caballo, perro, gato o ave de corral. El agente biológico inmunogénico es un vector adenovírico que codifica la proteína vírica 1 de norovirus o la proteína de fusión (F) del virus respiratorio sincitial (RSV, por sus siglas en inglés). En algunas realizaciones, la proteína vírica 1 de norovirus es la SEQ ID NO: 2 o SEQ ID NO: 4. En algunas realizaciones, la proteína de fusión (F) del RSV es la SEQ ID NO: 6. En algunas realizaciones, el vector adenovírico comprende una secuencia de nucleótidos de SEQ ID NO: 7.
En algunas realizaciones, el agente biológico inmunogénico es un vector de expresión que codifica un polipéptido inmunogénico. En algunas realizaciones, el vector de expresión es un vector vírico (por ejemplo, adenovírico, VAA, retrovírico o lentivírico). En algunas realizaciones, el vector vírico está atenuado o la replicación es incompetente. En algunas realizaciones, el vector de expresión comprende un promotor (por ejemplo, CMV, SV40 temprano o tardío, pactina, etc.) unido operativamente a la secuencia que codifica el polipéptido inmunogénico. En algunas realizaciones, el vector de expresión además codifica bicatenario (ARNbc). En algunas realizaciones, la secuencia que codifica ARNbc está unida operativamente a un promotor, por ejemplo, ya sea el mismo promotor (usando un sitio de entrada ribosómico interno (IRES, por sus siglas en inglés)) o un promotor diferente como el promotor unido operativamente a la secuencia codificante del polipéptido inmunogénico.
En algunas realizaciones, la composición inmunogénica comprende además al menos un adyuvante, por ejemplo, un
agonista de TLR3. En algunas realizaciones, el agonista de TLR3 es ARNbc o un mimético de ARNbc.
En algunas realizaciones, al menos un 50 % del agente biológico inmunogénico se administra (libera) en el íleon, por ejemplo, al menos un 60 %, 70 %, 75 %, 80 %, 90 %, 95 % o más del agente biológico inmunogénico presente en la composición administrada. En algunas realizaciones, el recubrimiento o matriz entérica comienza a disolverse antes de que la composición inmunogénica alcance el íleon, pero conserva al menos un 50 % del agente biológico inmunogénico hasta que la composición inmunogénica alcanza el íleon. En algunas realizaciones, el recubrimiento entérico conserva el agente biológico inmunogénico a través del estómago, duodeno y yeyuno, pero libera el agente biológico inmunogénico en el íleon.
El agente biológico inmunogénico está cubierto por un recubrimiento entérico. Dicho recubrimiento entérico tiene un pH umbral de 5,8 a 6,8. En algunas realizaciones, el recubrimiento entérico se selecciona del grupo que consiste en copolímero de ácido metacrílico-acrilato de etilo (por ejemplo, 1:1), tipo A; copolímero de ácido metacrílico, tipo C; una mezcla de copolímeros metacrílicos de tipos A y C; y Time Clock®. En algunas realizaciones, el recubrimiento entérico no incluye ftalato acetato de celulosa (fAc ). En algunas realizaciones, el recubrimiento entérico es de un grosor que da como resultado la liberación del agente biológico inmunogénico en el íleon. En algunas realizaciones, el recubrimiento entérico está basado en un copolímero de ácido metacrílico con una cobertura de 5,5 a 10 miligramos por centímetro cuadrado. En algunas realizaciones, el agente de administración es una cápsula controlada por radio.
En algunas realizaciones, el recubrimiento entérico comprende poli(ácido metacrílico-co-metacrilato de metilo) 1:1. En algunas realizaciones, el recubrimiento entérico comprende Eudragit® L-100. En algunas realizaciones, el recubrimiento entérico comprende Eudragit® L-100, citrato de trietilo y talco, por ejemplo, 1, 2, 3, 4 o 1-4 partes de Eudragit® L-100, 1-2 partes de citrato de trietilo y 1-2 partes de talco. En algunas realizaciones, el recubrimiento entérico comprende una mezcla de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 y poli(ácido metacrílico-coacrilato de etilo) 1:1. En algunas realizaciones, la relación de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 a poli(ácido metacrílico-co-acrilato de etilo) 1:1 es de 1:4 a 4:1, por ejemplo, 1:3, 1:2, 1:1, 2:1, 3:1. En algunas realizaciones, el recubrimiento entérico comprende una mezcla de Eudragit® L-100 y Eudragit®L100-55. En algunas realizaciones, el recubrimiento entérico comprende Eudragit® L-100 y Eudragit®L100-55, citrato de trietilo y talco, por ejemplo, 1-4 partes de Eudragit® L-100 y Eudragit®L100-55, 1-2 partes de citrato de trietilo y 1-2 partes de talco. En algunas realizaciones, el recubrimiento entérico comprende poli(ácido metacrílico-co-metacrilato de metilo) 1:1 y poli(ácido metacrílico-co-metacrilato de metilo) 1:2. En algunas realizaciones, la relación de poli(ácido metacrílico-cometacrilato de metilo) 1:1 a poli(ácido metacrílico-co-acrilato de metilo) 1:2 es de 1:2 a 2:1. En algunas realizaciones, el recubrimiento entérico comprende una mezcla de Eudragit® L-100 y Eudragit®S100. En algunas realizaciones, el recubrimiento entérico comprende Eudragit® L-100 y Eudragit®S100, citrato de trietilo y talco, por ejemplo, 1-4 partes de Eudragit® L-100 y Eudragit®S100, 1-2 partes de citrato de trietilo y 1-2 partes de talco. En algunas realizaciones, el recubrimiento entérico comprende una mezcla de poli(ácido metacrílico-co-metacrilato de metilo) 1:2 y poli(ácido metacrílico-co-acrilato de etilo) 1:1. En algunas realizaciones, la proporción de poli(ácido metacrílico-co-metacrilato de metilo) 1:2 y poli(ácido metacrílico-co-acrilato de etilo) 1:1 es de 1:4 a 4:1, por ejemplo, 1:3, 1:2, 1:1, 2:1 o 3:1. En algunas realizaciones, el recubrimiento entérico comprende una mezcla de Eudragit® L-100-55 y Eudragit®S100. En algunas realizaciones, el recubrimiento entérico comprende Eudragit® L-100-55 y Eudragit®S100, citrato de trietilo y talco, por ejemplo, 1-4 partes de Eudragit® L-100-55 y Eudragit®S100, 1-2 partes de citrato de trietilo y 1-2 partes de talco.
En algunas realizaciones, la composición inmunogénica está en forma de un comprimido o cápsula, por ejemplo, en forma de comprimido cubierto por un recubrimiento entérico. En algunas realizaciones, la composición inmunogénica está encapsulada en una cápsula polimérica que comprende gelatina, hidroxipropilmetilcelulosa, almidón o pululano. En algunas realizaciones, la composición inmunogénica está en forma de micropartículas de menos de 2 mm de diámetro, por ejemplo, cada micropartícula cubierta con recubrimiento entérico como se describe en el presente documento.
Se proporciona además la composición inmunogénica reivindicada para usar en un método de administración al íleon de un sujeto que comprende la administración oral de la composición inmunogénica como se describe anteriormente (es decir, un agente biológico inmunogénico abarcado por un agente de administración que dirige la administración del agente biológico inmunogénico al íleon, incluyendo opcionalmente un adyuvante) al sujeto. En algunas realizaciones, el sujeto es un ser humano. En algunas realizaciones, el sujeto es un animal no humano. En algunas realizaciones, el método da como resultado una respuesta inmunitaria en el sujeto que es al menos un 10 % mayor, por ejemplo, al menos un 20 %, 30 %, 40 %, 50 %, 60 %, 75 %, 80 %, 100 % o más, que la respuesta inmunitaria en un sujeto (ya sea el mismo sujeto en un momento diferente, o un sujeto diferente) que recibe la misma composición inmunogénica no dirigida al íleon. En algunas realizaciones, la respuesta inmunitaria en el sujeto es al menos 1,5 veces mayor (por ejemplo, 2 veces, 2,5 veces, 5 veces o más) que la respuesta inmunitaria en un sujeto (ya sea el mismo sujeto en un momento diferente, o un sujeto diferente) que recibe la misma composición inmunogénica no dirigida al íleon. En algunas realizaciones, la respuesta inmunitaria es un aumento de anticuerpos específicos para el agente biológico inmunogénico. En algunas realizaciones, la respuesta inmunitaria es una respuesta inmunitaria celular, por ejemplo, un aumento de citocinas tales como IFN-y. En algunas realizaciones, la respuesta inmunitaria es inmunización (por ejemplo, el sujeto es resistente a una infección por el virus, bacterias, etc. de los que procedía el agente biológico inmunogénico).
Se proporciona además la composición inmunogénica descrita anteriormente (es decir, un agente biológico inmunogénico abarcado por un agente de administración que dirige la administración del agente biológico inmunogénico al íleon, incluyendo opcionalmente un adyuvante) para usar en un método para provocar una respuesta inmunitaria aumentada en el sujeto, por ejemplo, un sujeto humano. En algunas realizaciones, la respuesta inmunitaria aumenta en al menos un 10 %, por ejemplo, al menos un 20 %, 30 %, 40 %, 50 %, 60 %, 75 %, 80 %, 100 % o más, en comparación con la respuesta inmunitaria en un sujeto (ya sea el mismo sujeto en un momento diferente, o un sujeto diferente) que recibe la misma composición inmunogénica no dirigida al íleon. En algunas realizaciones, la respuesta inmunitaria en el sujeto aumenta al menos 1,5 veces (por ejemplo, 2 veces, 2,5 veces, 5 veces o más) en comparación con la respuesta inmunitaria en un sujeto (ya sea el mismo sujeto en un momento diferente, o un sujeto diferente) que recibe la misma composición inmunogénica no dirigida al íleon. En algunas realizaciones, la respuesta inmunitaria es un aumento de anticuerpos específicos para el agente biológico inmunogénico. En algunas realizaciones, la respuesta inmunitaria es una respuesta inmunitaria celular, por ejemplo, un aumento de citocinas tales como IFN-y. En algunas realizaciones, la respuesta inmunitaria es inmunización (por ejemplo, el sujeto es resistente a la infección por el virus, bacterias, etc. de los que procedía el agente biológico inmunogénico).
Breve descripción de los dibujos
Figura 1. Se midieron células secretoras de anticuerpos (ASC, por sus siglas en inglés) específicas para HA en la sangre periférica 7 días después de que los sujetos recibieron una cápsula radiocontrolada que contenía rAd-HA-ARNbc. Los sujetos se asignaron al azar para que se les liberara la vacuna en el íleon o en el yeyuno. (N = 12 por grupo). Los resultados muestran que 12 de los 12 sujetos a los que se les administró la vacuna en el íleon fueron capaces de generar linfocitos B secretores de anticuerpos que reconocen HA, mientras que solo 9 de los 12 sujetos que recibieron la vacuna en el yeyuno fueron capaces de generar linfocitos B específicos de antígeno. El número medio de ASC IgA e IgG fue significativamente mayor para el íleon que para el yeyuno.
Figura 2. La respuesta de los linfocitos T a rAd-HA-ARNbc se determinó mediante la detección de los niveles de IFN-y 7 días después de la administración. Todos los individuos del grupo de administración en el íleon mostraron niveles más altos de IFN-y, en comparación con un 75 % del grupo de administración en el yeyuno. El nivel medio de IFN-y también fue significativamente más alto en el grupo de administración en el íleon.
Figura 3. Se midieron las respuestas de anticuerpos microneutralizantes (MN) a la gripe A/CA/07/2009 el día 0 y el día 28 después de la inmunización. El aumento de veces en los valores de MN se representó para sujetos individuales que tenían un valor de MN inicial menor o igual a 40. Los resultados mostraron que la administración en el íleon dio como resultado una alta proporción de sujetos (9 de 10) con valores aumentados de MN después de la inmunización en comparación con la administración en el yeyuno (6 de 10).
Figura 4. Se elaboraron comprimidos con celulosa microcristalina y almidón, con sulfato de bario al 10 % como material radiopaco. Estos comprimidos se recubrieron entéricamente con Eudragit L100® y se administraron a macacos cangrejeros hembra mediante sonda gástrica oral. Se tomaron radiografías a lo largo del tiempo posterior a la administración. A. Comprimido en el estómago con la flecha apuntando hacia el comprimido. B. Una hora después, el comprimido se puede ver en el intestino, la mancha blanca a la izquierda de la columna vertebral con una flecha apuntando hacia ella. Se disolvió en el intestino en las siguientes dos horas y no se puede ver.
Figura 5. Los números de ASC se informan los días 7 y 35, 7 días después de cada inmunización. Las ASC de base en los días 0 y 28 eran minúsculas y no estaban representadas. Las respuestas promedio para el día 7 se muestran para cada grupo tratado con una línea horizontal.
Figura 6. Aumento de veces en los valores de MN para sujetos individuales. Las columnas sombreadas oscuras indican dónde aumentaron los valores entre los días 28 y 56, mientras que las columnas sombreadas claras muestran la respuesta después de la inmunización inicial. Se trazó una línea con aumentos de dos veces en MN para mostrar qué sujetos tenían una respuesta de anticuerpos neutralizantes detectable. Ningún sujeto del grupo placebo respondió, mientras que 3 sujetos en el grupo de dosis baja y 7 sujetos en el grupo de dosis alta tuvieron una respuesta de anticuerpos neutralizantes de 2 veces o más a la gripe después de la inmunización. Placebo N = 10, dosis baja y dosis alta N = 11.
Figura 7. Respuestas de anticuerpos después de una única inmunización oral. A. Los valores de anticuerpos de inhibición de la hemaglutinación ("HAI", por sus siglas en inglés) antes y después de la inmunización (días 0 y 28 respectivamente) se muestran para sujetos individuales. B. Valores de la media geométrica (GMT, por sus siglas en inglés) de HAI frente al tiempo. Los valores de HAI se midieron a los 0, 1 y 6 meses después de la inmunización para evaluar la durabilidad de la respuesta de anticuerpos. C. Se muestran valores de m N antes y después de la inmunización para sujetos individuales. D. Respuestas de ASC después de la inmunización. Se informan las cantidades de ASC IgG e IgA (por 106 PMBC) 7 días después de la inmunización.
Figura 8. Valores de IgG de ELISA en suero frente a dosis de VXA-G1.1-NN en ratones. Se inmunizaron ratones con VXA-G1.1-NN de 1x108, 5x108 y 1x109. VXA-G1.1-NN se administró por vía oral mediante sonda gástrica los
días 0 y 28. Las respuestas de IgG en suero contra Norwalk VP1 se midieron mediante ELISA en la semana 8. N = 6. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el GMT. Como la dosis se incrementó de 1x108 hasta 5x108 hasta 1x109, el valor de IgG de VP1 en suero mostró un aumento dependiente de la dosis de 2x103 a 1x104 a 5x105.
Figura 9. Valores de ELISA de SIgA fecal frente a dosis de VXA-G1.1-NN en ratones. Se inmunizaron ratones con VXA-G1.1-NN de 1x108, 5x108 y 1x109. VXA-G1.1-NN se administró por vía oral mediante sonda gástrica los días 0 y 28. Las respuestas de SIgA fecal contra Norwalk VP1 se midieron mediante ELISA en la semana 8. Cada estudio tiene un total de 6 ratones. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el GMT. Como la dosis se incrementó de 1x108 hasta 5x108 hasta 1x109, el valor de sIgA de VP1 fecal mostró un aumento dependiente de la dosis de 1x103 a 2x103 a 3x104.
Figura 10. Valores de IgG de ELISA en suero de VXA-G1.1-NN frente a la proteína VP1 en ratones. Se inmunizaron ratones con VXA-G1.1-NN. de 1x108 por vía oral o proteína VP1 del virus de Norwalk (1 jg) por vía intramuscular los días 0 y 28. Las respuestas de IgG en suero contra VP1 de Norwalk se midieron mediante ELISA en las semanas 4 y 8. Cada estudio tiene un total de 6 ratones. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el valor medio geométrico. La vacuna oral generó valores de título de IgG en suero ligeramente más altos que la vacuna de proteína im.
Figura 11. Valores de ELISA de SIgA fecal frente a dosis de VXA-G1.1-NN en ratones. Se inmunizaron ratones con VXA-G1.1-NN de 1x108 por vía oral o con proteína VP1 del virus de Norwalk (1 |jg) por vía intramuscular los días 0 y 28. Las respuestas de IgA fecal contra VP1 de Norwalk se midieron mediante ELISA en las semanas 4 y 8. Cada estudio tiene un total de 6 ratones. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el valor medio geométrico. La vacuna oral generó una respuesta inmunitaria de IgA fecal notablemente más alta que la vacuna de proteína im.
Figura 12. Inmunización oral de VXA-G1.1-NN en comparación con la inmunización con proteína VP1 para valores de ELISA en suero. Se inmunizaron ratones con VXA-G1.1-NN de 1x108 por vía oral o con la proteína VP1 del virus Norwalk (1 jg ) en presencia de adyuvante, hidróxido de aluminio por vía intramuscular los días 0 y 28. Las respuestas de IgG en suero contra VP1 de Norwalk se midieron mediante ELISA en las semanas 4 y 8. Cada estudio tiene un total de 6 ratones. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el valor medio geométrico. La inyección intramuscular con la proteína VP1 junto con el alumbre generó un valor en suero mucho más alto.
Figura 13. Inmunización oral de VXA-G1.1-NN en comparación con la inmunización con proteína VP1 para valores de ELISA de SIgA fecal. Se inmunizaron ratones con VXA-G1.1-NN de 1x108 por vía oral o proteína VP1 del virus Norwalk (1 jg ) en presencia de adyuvante, hidróxido de aluminio por vía intramuscular los días 0 y 28. Las respuestas de IgA fecal contra VP1 de Norwalk se midieron mediante ELISA en las semanas 4 y 8. Cada estudio tiene un total de 6 ratones. Cada icono representa un ratón individual. La parte superior de la barra de cada estudio indica el valor medio geométrico. Incluso en presencia de adyuvante, hidróxido de aluminio, la vacuna oral generó una respuesta inmunitaria SIgA fecal más alta que la vacuna de proteína im.
Figura 14. Valores de IgA sérica y fecal después de la inmunización oral de VXA-G2.4-NS en ratones. Se inmunizaron ratones con VXA-G2.4-NS de 1x108 por vía oral los días 0 y 28. Para comparación, se presentaron nuevamente los grupos de administración oral del estudio n.° 3. Las respuestas de IgG en suero contra VP1 de Sydney se midieron mediante ELISA en las semanas 4 y 8 (A). La respuesta de SIgA fecal contra la VP1 de Sydney se midió mediante ELISA en la semana 8 (B). A las 4 semanas, la vacuna de la cepa Sydney generó mejores valores de título de IgG que la vacuna Norwalk. Asimismo, incluso a las 4 semanas, los valores de título de la cepa Sydney fueron ligeramente más altos que los valores de Norwalk a las 8 semanas (A). La vacuna de Sydney generó valores de título de IgA de VP1 fecal ligeramente más altos que la vacuna de Norwalk.
Figura 15. Valores de IgA sérica y fecal después de la inmunización oral de VXA-G2.4-NS en hurones. A los hurones se les administró VXA-G2.4-NS por vía endoscópica los días 0 y 2 (grupo 1) o los días 0 y 28 (grupo 2). A los hurones del grupo 3 se les administró por vía intramuscular la proteína VP1 recombinante de la cepa Sydney de norovirus los días 0 y 28. Mientras que el grupo 1 generó valores de título de IgG más altos que el grupo 2, el grupo 2 generó valores de título de SIgA más altos que el grupo 1. El grupo 3 no pudo generar una respuesta de SIgA fecal, aunque se generaron respuestas de IgG en suero.
Figura 16. IgG sérica después de la inmunización oral de VXA-G2.4-NS en primates no humanos (PNH, macaco cangrejero). A los PNH se administraron por vía endoscópica VXA-G2.4-NS los días 0 y 56 (el grupo "Noro"). Los valores de IgG en suero se midieron mediante ELISA los días 0, 7, 28, 56 y 72.
Figura 17. Valores de anticuerpos anti-RSVF en ratas algodoneras 4 semanas después de la vacunación con vectores de vacuna Ad-RSVF o RSV inactivados con formalina (FIRSV, por sus siglas en inglés) de control o RSVA2 vivo de tipo silvestre. Los valores se determinaron usando un ELISA de IgG anti-RSVF.
Figura 18. Competencia de Palivizumab utilizando sueros de ratas algodoneras vacunadas con la vacuna Ad-RSVF. Se recubrieron placas de ELISA con un lisado de RSVA2 a 1 |jg/ml. Se mezcló Palivizumab biotinilado (10 ng/ml) con diluciones en serie al doble de los sueros de control y de prueba. Se utilizaron sueros sin tratamiento previo para determinar la unión al 100% de Palivizumab. Se utilizó Palivizumab sin marcar como control positivo. Se utilizó HRP-estrepavidina con sustrato TMB para detectar Palivizumab biotinilado. La inhibición se puntuó en relación con el 100 % de unión a Palivizumab biotinilado. La dilución máxima que dio un 50 % o más de actividad de neutralización se asignó como valor de competencia.
Figura 19. Criterios de inscripción para el estudio de "Anticuerpos neutralizantes de alto título contra la gripe después de la inmunización con comprimidos orales: Un ensayo aleatorizado controlado con placebo". Los principales criterios de exclusión son:
• positivo para gripe H1 por HAI;
• ha recibido una vacuna contra la gripe en los últimos 2 años;
• historial actual de consumo crónico de alcohol y/o uso de drogas ilícitas y/o recreativas;
• antecedentes de cualquier afección inmunodeficiente o inmunodepresora confirmada o sospechada;
• serología positiva para VIH, VHC o VHB;
• reacciones graves anteriores a la vacunación, tales como anafilaxia, problemas respiratorios, urticaria o dolor abdominal;
• antecedentes de enfermedad del intestino irritable u otras afecciones inflamatorias digestivas o gastrointestinales que podrían afectar la distribución prevista de la vacuna dirigida a la mucosa del intestino delgado;
• uso de inhibidores de la bomba de protones (Nexium, Prilosec); y
• muestra de heces con sangre oculta en el examen inicial.
Figuras 20A-20D. Respuestas de anticuerpos después de una única inmunización oral de una vacuna oral basada en Ad serotipo 5 recombinante (rAd5) contra la gripe estacional HI. La Figura 20A muestra los valores de anticuerpos de HAI antes y después de la inmunización (días 0 y 28 respectivamente) para sujetos individuales. La Figura 20B muestra GMT de HAI frente al tiempo. Los valores de HAI se midieron a los 0, 1 y 6 meses después de la inmunización para evaluar la durabilidad de la respuesta del anticuerpo. La Figura 20C muestra valores de MN antes y después de la inmunización para sujetos individuales. La Figura 20D muestra la respuesta de ASC después de la inmunización. El número de ASC IgG e IgA se informa (por 1e6 PMBC) 7 días después de la inmunización.
Figuras 21A-21B. Inmunidad anti-Ad5 y efectos sobre respuestas de anticuerpos neutralizantes. La Figura 21A muestra el cambio de veces en MN frente al valor anti-Ad5 inicial para sujetos tratados con vacuna. Los sujetos no fueron evaluados previamente para los valores anti-Ad5, pero se midieron retrospectivamente. La Figura 21B muestra el cambio de veces en las respuestas de HAI que se trazaron para el valor de Ad5 tanto positivo como negativo, análogo a lo realizado en la Figura 2. No se observó ninguna tendencia para los valores de partida de Ad5 sobre la capacidad de provocar una respuesta de anticuerpos neutralizantes (por MN o HAI) al virus de la gripe.
La Figura 22 muestra que los ratones obtienen valores de anticuerpos robustos contra el RSV cuando se les administra VXA-RSV-f. Estudio N.° WCB254: Se inmunizaron ratones Balb/c con VXA-RSV-f a las 0 y 3 semanas utilizando tres vías de administración diferentes. Los valores de anticuerpos IgG de ELISA se midieron en la semana 7. Todas las vías de administración generaron respuestas inmunitarias significativas contra el RSV; sin embargo, fueron más eficaces in e im para producir valores más altos que la oral (p = 0,04, o 0,02 por Mann-Whitney). N = 6 por grupo. Como controles negativos, se utilizaron sueros de ratón de animales que recibieron una vacuna contra norovirus.
Las Figuras 23A-23C muestran que la inmunización de ratas algodoneras con la vacuna VXA-RSV-f induce respuestas de anticuerpos al RSV. (Experimento XV-95). La Figura 23A muestra que se inmunizaron ratas algodoneras hembra en la semana 0 y 4, y se midieron los valores de anticuerpos contra RSV-f en la semana 8. La inmunización con VXA-RSV-f fue significativamente mejor que usar virus RSV2 o FI-RSV para inducir valores ELISA de IgG para RSV. (p < 0,0022 por Mann-Whitney) La Figura 23B muestra que los valores de ELISA competitivos de palivizumab se evaluaron utilizando muestras de suero combinadas de cada uno de los grupos descritos anteriormente. La vacuna FI-RSV no fue capaz de inducir anticuerpos específicos de epítopo que compiten por la unión de palivizumab, pero la vacuna VXA-RSV-f y los grupos tratados con RSV2 fueron capaces de inducir estos valores. La Figura 23c muestra las respuestas de anticuerpos neutralizantes inducidas por la vacuna VXA-RSV-f contra RSV después de la inmunización de ratas algodoneras. Los grupos VXA-RSV-f y RSV2 fueron superiores al FI-RSV y los controles sin vacuna (sin infección y grupos tampón) en la inducción de valores neutralizantes contra el RSV. N = 6 por grupo, excepto el control "sin infección" (N = 3).
Las Figuras 24A-24B muestran que la inmunización oral induce potentes anticuerpos contra RSV en ratas algodoneras. (Experimento XV-112). En la Figura 24A, se inmunizaron ratas algodoneras hembra con VXA-RSV-f en las semanas 0 y 4, y se midieron los valores ELISA de anticuerpos IgG totales en las semanas 4 y 8. Se
observaron respuestas de anticuerpos dependientes de la dosis de 1x109 y 1x1010 con una tendencia mejor que el grupo de dosis de 1x108 UI. En la Figura 24B, se evaluaron valores ELISA competitivos de Palivizumab para los mismos grupos descritos anteriormente, pero en la semana 8. Las dosis de vacuna más altas tendieron a tener valores de anticuerpos competitivos de Palivizumab más altos, pero los resultados no fueron significativamente diferentes. La Figura 24C muestra que la inmunización oral de VXA-RSV-f también indujo valores de anticuerpos neutralizantes dependientes de la dosis contra el RSV, con el grupo de 1x1010 comportándose significativamente mejor que el grupo de 1x108 (p = 0,018 por Mann-Whitney). N = 8 por grupo oral, N = 6 para el grupo de control con tampón.
Las Figuras 25A-25C muestran que VXA-RSV-f protege contra la replicación del RSV y las mejoras inmunitarias adaptativas de la enfermedad del RSV. (Experimento XV-95) LA Figura 25A muestra que los animales inmunizados con VXA-RSV-f antes de la exposición al RSV estaban completamente protegidos contra la replicación del RSV en muestras de pulmón y nasales. Los animales inmunizados con tampón, adyuvante solo o FI-RSV no estaban protegidos. Se administraron dos dosis diferentes de vacuna para los animales tratados in (1x108 = bajo). N = 6, excepto los animales de control sin infección (N = 3). En la Figura 25B, la inflamación en los pulmones se puntuó mediante inmunohistología. Los grupos de vacuna (VXA-RSV-f y RSV2) no causaron inflamación mejorada inmunitaria adaptativa (PB, PA, A, iP) al igual que el grupo FI-RSV. En la Figura 25C, se midieron niveles de abundancia de citocinas mediante qRT-PCR. Solo el grupo inmunizado con FI-RSV tuvo aumentos sustanciales en la abundancia relativa de IL-4 e IL-13, las citocinas Th2 medidas.
Las Figuras 26A-26C muestran que la inmunización oral induce protección contra la replicación y la enfermedad del RSV. (Experimento XV-112). En la Figura 26A, los animales inmunizados por vía oral muestran respuestas inmunitarias dependientes de la dosis a la infección por RSV, con la dosis más alta que conduce a una protección completa en los pulmones y una protección casi completa en la nariz. En cambio, no hubo protección en el grupo de control con tampón. En la Figura 26B, se comparó la inflamación entre dosis de vacuna y el grupo de control con tampón. En la Figura 26C, se midieron niveles de abundancia de citocinas mediante qRT-PCR. Ningún grupo tuvo aumentos sustanciales en la abundancia relativa de IL-4 o IL-13, las citocinas Th2 medidas. (N = 8 con la excepción del control con tampón N = 6).
Descripción detallada de la invención
Los inventores han descubierto que la administración de un agente biológico inmunogénico a una parte particular del intestino delgado, es decir, el íleon, da como resultado una respuesta terapéutica mucho mayor que cuando el agente no se dirige o se dirige a un sitio diferente. Esto permite diseñar vacunas más eficaces, costes reducidos de materiales y efectos secundarios reducidos para el receptor.
I. Definiciones
El término "inmunogénico" se refiere a la capacidad de un agente para dar lugar a una respuesta inmunitaria en un hospedador, ya sea humoral o mediada por células. Los agentes inmunogénicos son normalmente "extraños" para el hospedador, por ejemplo, de una especie diferente, o de una bacteria, virus u hongos. Un agente no extraño puede ser inmunogénico, por ejemplo, en el caso de una respuesta autoinmunitaria. Determinados agentes específicos de células cancerosas se pueden explotar como agentes inmunogénicos, permitiendo que el sistema inmunitario del hospedador ataque el cáncer.
La expresión "agente biológico" se refiere a un ácido nucleico, polipéptido, glucoproteína, hidrato de carbono, lípido, o una forma modificada de los mismos (por ejemplo, metilado, glucosilado, marcado de forma detectable). Los agentes biológicos se distinguen de los fármacos de moléculas pequeñas en que se pueden crear mediante procesos biológicos (incluyendo técnicas recombinantes) en lugar de síntesis química. Los agentes biológicos se pueden, sin embargo, modificar químicamente o incluir nucleótidos o aminoácidos no naturales. Los agentes biológicos también pueden ser de origen no natural, por ejemplo, entidades recombinantes o quiméricas.
Como se utiliza en el presente documento, un "agente biológico inmunogénico" se refiere a un agente que actúa directamente como un antígeno (por ejemplo, se reconoce mediante un anticuerpo o receptor de linfocitos T), o un agente que, una vez expresado en una célula, actúa como un antígeno. Por ejemplo, un agente biológico inmunogénico puede incluir un vector de expresión que codifica un polipéptido inmunogénico.
El término "antígeno" se refiere a un polipéptido, glucoproteína, lipoproteína, lípido, hidrato de carbono u otro agente que está unido (por ejemplo, reconocido como "extraño") por un receptor de linfocitos T y/o un anticuerpo. Los antígenos proceden comúnmente de fuentes bacterianas, víricas o fúngicas. La expresión "procede de" indica que el antígeno es esencialmente como existe en su contexto antigénico natural, o que ha sido modificado para expresarse en determinadas condiciones, para incluir solo la parte más inmunogénica, o para eliminar otros componentes asociados posiblemente dañinos, etc.
Una "dosis o cantidad inmunogénicamente eficaz" de una composición como se describe en el presente documento es una cantidad que provoca o modula una respuesta inmunitaria específica para un antígeno seleccionado para
vacunación. Respuestas inmunitarias incluyen respuestas inmunitarias humorales y respuestas inmunitarias mediadas por células. Una composición inmunogénica se puede usar terapéutica o profilácticamente para tratar o evitar una enfermedad en cualquier etapa.
Las "respuestas inmunitarias humorales" están mediadas por componentes libres de células de la sangre, por ejemplo, plasma o suero; la transferencia del suero o plasma de un individuo a otro transfiere inmunidad humoral. Las respuestas inmunitarias humorales normalmente están mediadas por linfocitos B, por ejemplo, producción de anticuerpos.
Las "respuestas inmunitarias mediadas por células" están mediadas por linfocitos específicos de antígeno; la transferencia de linfocitos específicos de antígeno de un individuo a otro transfiere inmunidad. Las respuestas inmunitarias mediadas por células están mediadas al menos en parte por linfocitos T, y se pueden detectar, por ejemplo, mediante la detección de citocinas específicas de linfocitos T o el aumento de la proliferación de linfocitos T.
El "íleon" es el más largo de los tres segmentos que forman el intestino delgado, junto con el duodeno y el yeyuno. Constituye la parte terminal, entre el yeyuno y el ciego.
Un recubrimiento entérico es una barrera aplicada a los medicamentos orales que evita que el agente terapéutico en el interior sea digerido en el ambiente de pH bajo del estómago y el duodeno (~ pH 3).
Un agente de administración, tal como un recubrimiento entérico, matriz, o cápsula, se dice que conserva un agente terapéutico incluido o integrado cuando al menos un 60 %, por ejemplo, al menos aproximadamente un 70 %, 75 %, 80 %, 85 %, 90 %, 95 % o 100 % de la cantidad original administrada de agente terapéutico permanece incluida o integrada dentro del agente. El agente de administración, por ejemplo, matriz o recubrimiento entérico, está diseñado normalmente para desintegrarse bajo determinadas condiciones y liberar el agente terapéutico. La desintegración puede ser gradual, por ejemplo, en el caso de un recubrimiento más grueso o químicamente más complejo. Se dice que el recubrimiento entérico se "desintegra" una vez que el espesor del recubrimiento se reduce al menos en un 10 %, por ejemplo, al menos un 25 %, 50 % o 75 % en comparación con el espesor original administrado. La desintegración no es un término absoluto, ya que puede ocurrir en un transcurso de tiempo diferente dependiendo de las condiciones. Por ejemplo, un recubrimiento diseñado para desintegrarse en 5 minutos a pH 6,5 puede desintegrarse, aunque lentamente, a pH 6 (por ejemplo, en 1 hora). La desintegración no indica necesariamente que se libere el agente terapéutico incluido o integrado. El agente terapéutico puede, sin embargo, comenzar a liberarse antes de que la matriz o recubrimiento entérico se desintegre por completo.
El término "quimérico" o "recombinante" como se usa en el presente documento con referencia, por ejemplo, a un ácido nucleico, proteína, o vector, indica que el ácido nucleico, proteína o vector, se ha modificado mediante la introducción de un ácido nucleico o proteína heteróloga o la alteración de un ácido nucleico o proteína nativa. Por tanto, por ejemplo, vectores quiméricos y recombinantes incluyen secuencias de ácido nucleico que no se encuentran dentro de la forma nativa (no quimérica o no recombinante) del vector. Un vector de expresión vírico quimérico se refiere a un vector de expresión vírico que comprende una secuencia de ácido nucleico que codifica un polipéptido heterólogo (por ejemplo, inmunogénico).
Un "vector de expresión" es una construcción de ácido nucleico, generada de forma recombinante o sintética, con una serie de elementos de ácidos nucleicos especificados que permiten la transcripción de un ácido nucleico particular en una célula hospedadora. El vector de expresión puede ser parte de un plásmido, virus o fragmento de ácido nucleico. Normalmente, el vector de expresión incluye un ácido nucleico a transcribir unido operativamente a un promotor. Los vectores de expresión víricos normalmente se vuelven incompetentes o atenuados para la replicación. Un vector procedente de virus puede incluir los componentes del vector de expresión necesarios para la expresión de una secuencia deseada, pero omite a los implicados en, por ejemplo, replicación u otros efectos patogénicos.
Las expresiones "promotor" y "secuencia de control de la expresión" se utilizan en el presente documento para hacer referencia a una secuencia de control de ácido nucleico que dirige la transcripción de un ácido nucleico. Las secuencias promotoras normalmente están cerca del sitio de inicio de la transcripción, tal como un elemento TATA en el caso de un promotor de tipo polimerasa II. Un promotor también puede incluir elementos potenciadores o represores distales, que pueden ubicarse hasta a varios miles de pares de bases del sitio de inicio de la transcripción. Los promotores incluyen promotores constitutivos e inducibles. Un promotor "constitutivo" es un promotor que está activo en la mayoría de las condiciones ambientales y de desarrollo. Un promotor "inducible" es un promotor que está activo bajo regulación ambiental o de desarrollo. La expresión "unido operativamente" se refiere a un enlace funcional entre una secuencia de control de la expresión de ácido nucleico (tal como un promotor o una matriz de sitios de unión a factores de transcripción) y una segunda secuencia de ácido nucleico, en donde la secuencia de control de la expresión dirige la transcripción del ácido nucleico correspondiente a la segunda secuencia.
El término "heterólogo" cuando se utiliza con referencia a las partes de un ácido nucleico indica que el ácido nucleico comprende dos o más subsecuencias que no se encuentran en la misma relación entre sí en la naturaleza. Por ejemplo, el ácido nucleico se ha producido, de forma típica, de manera recombinante, teniendo dos o más secuencias de genes no relacionados dispuestos para fabricar un ácido nucleico funcional nuevo, por ejemplo, un promotor
procedente de una fuente y una región de codificación procedente de otra fuente. De forma similar, las porciones heterólogas de una proteína indican que la proteína comprende dos o más subsecuencias que no se encuentran en la misma relación entre sí en la naturaleza (por ejemplo, una proteína de fusión). Un ácido nucleico o proteína heterólogo es uno que no se encuentra en un ambiente particular de la naturaleza, por ejemplo, una proteína de ratón heteróloga en una célula humana.
Los términos "ácido nucleico" y "polinucleótido" se usan indistintamente en el presente documento para hacer referencia a polímeros de desoxirribonucleótidos o ribonucleótidos en forma monocatenaria o bicatenaria. Los términos abarcan genes, ADNc, ARN y oligonucleótidos (polinucleótidos cortos). Los términos abarcan ácidos nucleicos que contienen análogos de nucleótidos conocidos o restos o enlaces modificados de la cadena principal, que son sintéticos, de origen natural y de origen no natural, que tienen propiedades de unión similares a las del ácido nucleico de referencia y que se metabolizan de forma similar a los nucleótidos de referencia. Ejemplos de dichos análogos incluyen, sin limitación, fosforotioatos, fosforoamidatos, metil fosfonatos, metil fosfonatos quirales, 2-O-metil ribonucleótidos, ácidos nucleicos peptídicos (ANP). El término "nucleótido" se refiere normalmente a un monómero de ácido nucleico.
A menos que se indique otra cosa, una secuencia de ácido nucleico particular también abarca variantes modificadas de forma conservadora de la misma (por ejemplo, sustituciones de codones degenerados) y secuencias complementarias, así como la secuencia indicada explícitamente. Específicamente, las sustituciones de codones degenerados se pueden lograr mediante la generación de secuencias en las que la tercera posición de uno o más codones seleccionados (o todos) se sustituye con restos de base mixta y/o de desoxiinosina (Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); Rossolini et al., Mol. Cell. Probes 8:91-98 (1994)).
Una "dosis terapéutica" o "cantidad terapéuticamente eficaz" o "cantidad eficaz" de una composición como se describe en el presente documento es una cantidad que evita, alivia, disminuye o reduce la intensidad de los síntomas de enfermedades y trastornos asociados con la fuente del antígeno seleccionado para la vacunación (por ejemplo, un virus, bacterias, un parásito o un cáncer).
El término "anticuerpo" se refiere a un polipéptido codificado por un gen de inmunoglobulina o fragmentos del mismo que se unen específicamente y reconocen un antígeno. Las secuencias de inmunoglobulinas incluyen secuencias de región constante kappa, lambda, alfa, gamma, delta, épsilon y mu, así como innumerables secuencias de región variable de inmunoglobulinas. Las cadenas ligeras se clasifican en kappa o lambda. Las cadenas pesadas se clasifican en gamma, mu, alfa, delta o épsilon, que a su vez definen las clases de inmunoglobulina, IgG, IgM, IgA, IgD e IgE, respectivamente.
Los linfocitos T se refieren a una clase particular de linfocitos que expresan un receptor específico (receptor de linfocitos T) codificado por una familia de genes. Los genes receptores de linfocitos T reconocidos incluyen loci alfa, beta, delta y gamma, y los receptores de linfocitos T reconocen normalmente (pero no universalmente) una combinación de MHC más un péptido corto. Los linfocitos T normalmente se clasifican en términos generales como linfocitos T auxiliares (CD4+) y linfocitos T citotóxicos (CD8+). Los anticuerpos se producen de manera natural mediante linfocitos B, por ejemplo, células secretoras de anticuerpos (ASC). Los linfocitos B maduros pueden ser indiferenciados, linfocitos B plasmáticos (activados y productores de anticuerpos), de memoria, B-1, linfocitos B de la zona marginal, linfocitos B foliculares y linfocitos B reguladores.
Una respuesta inmunitaria adaptativa se refiere al reconocimiento de antígeno por linfocitos T y/o linfocitos B y/o anticuerpos.
Las células presentadoras de antígenos (APC, por sus siglas en inglés) son células capaces de presentar péptidos inmunogénicos o fragmentos de los mismos a los linfocitos T para activar o mejorar una respuesta inmunitaria. Las APC incluyen células dendríticas, macrófagos, linfocitos B, monocitos y otras células que pueden genomanipularse para ser APC eficaces. Dichas células pueden, pero no necesariamente, modificarse genéticamente para aumentar la capacidad de presentar el antígeno, para mejorar la activación y/o el mantenimiento de la respuesta de linfocitos T, para tener efectos antitumorales por sí mismas y/o ser inmunológicamente compatibles con el receptor (es decir,, haplotipo HLA emparejado). Las APC se pueden aislar de cualquiera de una variedad de órganos y líquidos biológicos, incluyendo la médula ósea, sangre periférica, tejidos tumorales y peritumorales, y pueden ser células autólogas, alogénicas, singénicas o xenogénicas. Las APC normalmente utilizan un receptor del locus de histocompatibilidad principal (MHC, por sus siglas en inglés) para presentar polipéptidos cortos a linfocitos T.
Un adyuvante es un potenciador de la respuesta inmunitaria no específico. Los adyuvantes adecuados incluyen, por ejemplo, toxina colérica, monofosforil lípido A (MPL, por sus siglas en inglés), adyuvante completo de Freund, adyuvante incompleto de Freund, Quil A y Al(OH). Los adyuvantes también pueden ser aquellas sustancias que causan la activación de APC y una presentación mejorada de linfocitos T a través de moléculas de señalización secundarias como los receptores tipo Toll, por ejemplo, a Rn bicatenario (ARNbc), miméticos de ARNbc, flagelos bacterianos, LPS, ADN de C'pG y lipopéptido bacteriano (revisado recientemente en [Abreu et al., J Immunol, 174(8), 4453-4460 (2005)]).
Los términos "polipéptido", "péptido" y "proteína" se utilizan indistintamente en el presente documento para referirse a un polímero de aminoácidos. Los términos se aplican a polímeros de aminoácidos en donde uno o más restos de aminoácidos son un mimético químico artificial de un aminoácido correspondiente de origen natural, así como a polímeros de aminoácidos de origen natural y a polímeros de aminoácidos de origen no natural.
El término "aminoácido" se refiere a aminoácidos de origen natural o sintético, así como a análogos y miméticos de aminoácidos que funcionan de una manera similar a los aminoácidos de origen natural. Los aminoácidos de origen natural son los codificados mediante el código genético, así como aquellos aminoácidos que se modifican posteriormente, por ejemplo, hidroxiprolina, Y-carboxiglutamato, y O-fosfoserina. Análogos de aminoácidos se refiere a compuestos que tienen la misma estructura química básica que la de un aminoácido de origen natural, es decir, un carbono que está unido a un hidrógeno, un grupo carboxilo, un grupo amino, y un grupo R, por ejemplo, homoserina, norleucina, metionina sulfóxido, metionina metil sulfonio. Dichos análogos tienen grupos R modificados (por ejemplo, norleucina) o cadenas peptídicas modificadas, pero conservan la misma estructura química básica que un aminoácido de origen natural. Miméticos de aminoácidos se refiere a compuestos químicos que tienen una estructura que es diferente de la estructura química general de un aminoácido, pero que funcionan de manera similar a un aminoácido de origen natural.
Los aminoácidos se pueden citar en el presente documento bien por sus símbolos habitualmente conocidos de tres letras, o mediante los símbolos de una letra recomendados por la Comisión de Nomenclatura bioquímica de la IUPAC-IUB. Los nucleótidos, asimismo, se pueden citar por sus códigos de una letra habitualmente aceptados.
"Variantes modificadas de forma conservadora" se aplica a secuencias tanto de aminoácidos como de ácido nucleico. Con respecto a secuencias de ácidos nucleicos concretas, las variantes modificadas de forma conservadora se refieren a aquellos ácidos nucleicos que codifican secuencias de aminoácidos idénticas o esencialmente idénticas, o si el ácido nucleico no codifica una secuencia de aminoácidos, a secuencias prácticamente idénticas. A causa de la degeneración del código genético, un gran número de ácidos nucleicos funcionalmente idénticos codifican cualquier proteína dada. Por ejemplo, los codones GCA, GCC, GCG y GCU codifican, todos ellos, el aminoácido alanina. Por tanto, en cada posición donde una alanina se especifica mediante un codón, el codón se puede alterar a cualquiera de los correspondientes codones descritos sin alterar el polipéptido codificado. Dichas variaciones de ácido nucleico se denominan "variaciones silenciosas", que son un tipo de variaciones modificadas de forma conservadora. Cada secuencia de ácido nucleico en el presente documento que codifica un polipéptido también describe todas y cada una de las posibles variaciones silenciosas del ácido nucleico. Un experto reconocerá que cada codón en un ácido nucleico (excepto AUG, que normalmente es el único codón para metionina, y TGG, que es normalmente el único codón del triptófano) se puede modificar con el fin de conseguir una molécula funcionalmente idéntica. En consecuencia, cada variación silenciosa de un ácido nucleico que codifica un polipéptido está implícita en cada secuencia descrita.
Como secuencias de aminoácidos, un experto reconocerá que sustituciones, eliminaciones o adiciones individuales a una secuencia de ácido nucleico, péptido, polipéptido o secuencia de proteínas que altera, añade o elimina un único aminoácido o un pequeño porcentaje de aminoácidos de la secuencia codificada es una "variante modificada de forma conservadora" donde la alteración da como resultado la sustitución de un aminoácido por un aminoácido químicamente similar. Las tablas de sustituciones conservadoras que proporcionan aminoácidos funcionalmente similares son bien conocidas en la materia. Dichas variantes modificadas de forma conservadora son adicionales, y no excluyen, las variantes polimórficas, homólogos interespecíficos, y alelos de la invención.
Los ocho grupos siguientes contienen, cada uno de ellos, aminoácidos que son sustituciones conservadoras entre sí: 1) Alanina (A), Glicina (G); 2) Ácido aspártico (D), Ácido glutámico (E); 3) Asparagina (N), Glutamina (Q); 4) Arginina I, Lisina (K); 5) Isoleucina (I), Leucina (L), Metionina (M), Valina (V); 6) Fenilalanina (F), Tirosina (Y), Triptófano (W); 7) Serina (S), Treonina (T); y 8) Cisteína (C), Metionina (M) (véase, por ejemplo, Creighton, Proteins (1984)).
La expresión "hibrida selectivamente (o específicamente) con" se refiere a la unión, duplicación o hibridación de nucleótidos complementarios (o en gran parte complementarios) en una mezcla compleja (por ejemplo, ADN o ARN celular total o de biblioteca).
Los polinucleótidos pueden comprender una secuencia nativa (es decir, una secuencia endógena que codifica un polipéptido individual o ARNbc o una parte del mismo) o pueden comprender una variante de dicha secuencia. Las variantes polinucleotídicas pueden contener una o más sustituciones, adiciones, eliminaciones y/o inserciones de modo que no disminuya al menos una actividad biológica del polipéptido codificado (por ejemplo, inmunogenicidad), con respecto a un polipéptido que comprende antígenos nativos. Las variantes polinucleotídicas pueden contener una o más sustituciones, adiciones, eliminaciones y/o inserciones de manera que no disminuya la actividad adyuvante de un ARNbc codificado, relativo a un ARNbc que no contiene las sustituciones, adiciones, eliminaciones y/o inserciones. Las variantes presentan preferentemente al menos aproximadamente un 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 % o 99 % de identidad de secuencia con una secuencia polinucleotídica que codifica un polipéptido nativo o una porción del mismo o un ARNbc.
Los términos "idéntico" o porcentaje de "identidad", en el contexto de dos o más secuencias polinucleotídicas o polipeptídicas, se refiere a dos o más secuencias o subsecuencias que son iguales o tienen un porcentaje especificado
de restos de aminoácidos o nucleótidos que es igual (es decir, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 %, o más de identidad en una región especificada), cuando se compara y alinea para correspondencia máxima en una ventana de comparación, o región designada como medida utilizando uno de los siguientes algoritmos de comparación de secuencias o mediante alineación manual e inspección visual. Dichas secuencias se denominan, por tanto, "prácticamente idénticas". Esta definición también se refiere al complemento de una secuencia polinucleotídica de prueba. Opcionalmente, la identidad existe en una región que es de al menos aproximadamente 10 a aproximadamente 100, de aproximadamente 20 a aproximadamente 75, de aproximadamente 30 a aproximadamente 50 aminoácidos o nucleótidos de longitud.
Una muestra o valor "de control" se refiere a una muestra que sirve como referencia, normalmente una referencia conocida, para comparación con una muestra de ensayo. Por ejemplo, una muestra de ensayo se puede tomar a partir de una condición de ensayo, por ejemplo, en presencia de un compuesto o tratamiento de ensayo, y en comparación con muestras procedentes de condiciones conocidas, por ejemplo, en ausencia del compuesto de ensayo (control negativo), o en presencia de un compuesto conocido (control positivo). En el contexto de la presente divulgación, un ejemplo de un control negativo sería una muestra biológica de un individuo sano (no infectado) conocido, y un ejemplo de un control positivo sería una muestra biológica de un paciente infectado conocido. Un control también puede representar un valor promedio o un intervalo recogido entre un número de ensayos o resultados. Un experto en la materia reconocerá que los controles se pueden diseñar para evaluar cualquier número de parámetros. Por ejemplo, se puede concebir un control para comparar el beneficio terapéutico basándose en datos farmacológicos (por ejemplo, semivida) o medidas terapéuticas (por ejemplo, comparación de beneficios y/o efectos secundarios). Los controles se pueden diseñar para aplicaciones in vitro. Un experto en la materia sabrá qué controles son valiosos en una situación dada, y es capaz de analizar los datos basándose en comparaciones con valores de control. Los controles también son valiosos para determinar la significación de los datos. Por ejemplo, si los valores de un parámetro dado son ampliamente variables en los controles, la variación en las muestras de ensayo no se considerará significativas.
El término "diagnóstico" se refiere a una probabilidad relativa de que un sujeto tenga un trastorno tal como una infección o cáncer. De forma similar, el término "pronóstico" se refiere a una probabilidad relativa de que un determinado resultado futuro pueda ocurrir en el sujeto. No se pretende que los términos sean absolutos, como apreciará cualquier experto en el campo del diagnóstico médico.
Los términos "terapia", "tratamiento", y "mejora" se refieren a cualquier reducción en la intensidad de los síntomas. En el contexto de una infección, el tratamiento puede referirse a una reducción del agente infeccioso, síntomas reducidos, etc. En el caso de tratar un cáncer, el tratamiento puede referirse a, por ejemplo, reducción del tamaño del tumor, número de células cancerosas, tasa de crecimiento, actividad metastásica, reducción de la muerte celular de las células no cancerosas, etc. Los términos "tratar" y "prevenir" no pretenden ser términos absolutos. El tratamiento y la prevención pueden referirse a cualquier reducción comparativa o ausencia aparente del agente infeccioso, retraso en el inicio, mejora de los síntomas, mejora en la supervivencia del paciente, aumento del tiempo o tasa de supervivencia, etc. El tratamiento y la prevención pueden ser completos (niveles indetectables de agente infeccioso o células neoplásicas) o parciales, de manera que se encuentren en un paciente menos agentes infecciosos o células neoplásicas de las que se habrían producido sin los agentes biológicos inmunogénicos descritos actualmente. El efecto del tratamiento se puede comparar con un individuo o grupo de individuos que no reciben el tratamiento, o con el mismo paciente antes del tratamiento o en un momento diferente durante el tratamiento. En algunos aspectos, la intensidad de la infección o enfermedad se reduce en al menos un 10 %, en comparación, por ejemplo, con el individuo antes de la administración o con un individuo de control que no se somete al tratamiento. En algunos aspectos, la intensidad de la infección o enfermedad se reduce al menos en un 25 %, 50 %, 75 %, 80 %, o 90 %, o en algunos casos, ya no es detectable utilizando técnicas de diagnóstico estándar.
"Sujeto", "paciente", "individuo" y términos similares se utilizan indistintamente y se refieren a, excepto cuando se indique, mamíferos tales como seres humanos y primates no humanos, así como conejos, ratas, ratones, cabras, cerdos y otras especies de mamíferos. El término no indica necesariamente que el sujeto haya sido diagnosticado con una enfermedad en particular, pero normalmente se refiere a un individuo bajo supervisión médica. Un paciente puede ser un individuo que busca tratamiento, control, ajuste o modificación de un régimen terapéutico existente, etc.
II. Agentes biológicos inmunogénicos
Un agente biológico inmunogénico es cualquier agente biológico que provoca una respuesta inmunitaria en el hospedador, por ejemplo, un hospedador humano. Por tanto, el agente biológico inmunogénico puede ser un polipéptido (por ejemplo, glucoproteína, fosfoproteína u otra forma modificada), hidrato de carbono, lípido, polinucleótido (por ejemplo, cromatina, polinucleótido metilado u otra forma modificada). En algunas realizaciones, el agente biológico inmunogénico provoca directamente una respuesta inmunitaria, por ejemplo, es en sí mismo un inmunógeno (antígeno) diana. En algunas realizaciones, el agente biológico inmunogénico es un polinucleótido que codifica el inmunógeno diana. Por ejemplo, cuando un polinucleótido que codifica un antígeno diana se expresa en una célula presentadora de antígeno (APC), se monta una respuesta inmunitaria contra el antígeno expresado. El agente biológico inmunogénico se puede administrar solo, en combinación con un segundo, tercer y/o cuarto agente biológico inmunogénico (por ejemplo, en el caso de una vacuna preventiva de múltiples dianas) y/o en combinación con un adyuvante para aumentar la respuesta inmunitaria.
A. Vectores de expresión
Los vectores de expresión para usar como se describe en el presente documento pueden incluir vectores procedentes de virus, por ejemplo, vectores de virus adenoasociados (VAA) recombinantes, vectores retrovíricos, vectores adenovíricos, vectores modificados de vacuna Ankara (MVA) y vectores lentivíricos (porejemplo, procedentes de HSV-1) (véase, por ejemplo, Brouard et al. (2009) British J. Pharm. 157: 153). Los vectores procedentes de virus para uso terapéutico normalmente se vuelven incompetentes o atenuados para la replicación. Por ejemplo, en el caso de un vector adenovírico, el genoma adenovírico se puede modificar para eliminar los genes E1 y E3. Para la producción, el vector de replicación deficiente se puede administrar a una célula que expresa el gen E1 de manera que la célula produzca adenovirus recombinante (rAd, por sus siglas en inglés). Este rAd se puede recolectar y usar para una única ronda de infección para administrar la composición transgénica a otra célula dentro de un mamífero con el fin de provocar respuestas inmunitarias a un antígeno polipeptídico codificado.
Ejemplos de vectores víricos adecuados incluyen adenovirus 5, incluyendo, por ejemplo, Ad5 con eliminaciones de las regiones E1/E3 y Ad5 con una eliminación de la región E4 como se describe en la Patente de Estados Unidos N.° 8.222.224 y Scallan et al. Clinical and Vaccine Immunology 2013; 20(1): 85-94. En la SEQ ID NO: 7 se proporciona una cadena principal de vector vírico Ad5 de ejemplo. Otros vectores adenovíricos adecuados incluyen las cepas 2, las cepas 4 y 7 probadas por vía oral, adenovirus entéricos 40 y 41, y otras cepas (por ejemplo, Ad34, Ad26 o Ad35) que son suficientes para administrar un antígeno y provocar una respuesta inmunitaria adaptativa al antígeno transgénico [Lubeck et al., Proc Natl Acad Sci USA, 86(17), 6763-6767 (1989); Shen et al., J Virol, 75(9), 4297-4307 (2001); Bailey et al., Virology, 202(2), 695-706 (1994)]. El vector vírico no necesita haber sido aislado de seres humanos, pero puede provenir de un no humano tal como el adenovirus 3 del chimpancé (ChAd3, por sus siglas en inglés) (véase, por ejemplo, Colloca et al. (2012) Sci. Transl. Med. 4:115; Stanley et al. (2014) Nat. Med. doi:10.1038/nm.3702). En algunas realizaciones, el vector adenovírico es un vector adenovírico vivo incompetente para la replicación (tal como Ad5r con E1 y E3 eliminados), un vector adenovírico vivo y atenuado (tal como el virus con eliminación de E1B55K) o un vector adenovírico vivo con replicación de tipo silvestre.
Las secuencias de control de la transcripción y la traducción en vectores de expresión para usar como se describe en el presente documento se pueden proporcionar mediante fuentes víricas. Por ejemplo, promotores y potenciadores de uso común proceden, por ejemplo, de beta actina, adenovirus, virus de simio (SV40) y citomegalovirus humano (CMV). Por ejemplo, son adecuados vectores que permiten la expresión de proteínas bajo la dirección del promotor de CMV, promotor temprano de SV40, promotor tardío de SV40, promotor de metalotioneína, promotor del virus de tumor mamario murino, promotor del virus del sarcoma de Rous, promotor transductor u otros promotores que se han mostrado eficaces para la expresión en células de mamífero. Se pueden usar secuencias de señal, control y/o promotoras víricas o no víricas adicionales, siempre que dichas secuencias de control sean compatibles con las células hospedadoras que se van a transfectar.
B. Inmunógenos
Los inmunógenos para usar como se describe en el presente documento pueden proceder de antígenos, tales como, por ejemplo, antígenos víricos, antígenos bacterianos, antígenos de cáncer, antígenos de hongos o antígenos de parásitos (véase, por ejemplo, la Patente de Estados Unidos N.° 8.222.224 para una lista de antígenos que pueden usarse como se describe en el presente documento). Los agentes biológicos inmunogénicos que forman parte de la invención son vectores adenovíricos que codifican la proteína vírica 1 de norovirus de la proteína de fusión (F) del RSV.
Los antígenos que se utilizan como se describen en el presente documento son aquellos que proceden de norovirus (por ejemplo, VP1) y virus respiratorio sincitial (RSV) (porejemplo,). Otros antígenos adecuados, que no se reivindican, incluyen los del virus de la gripe (por ejemplo, HA, NA, M1, NP), virus de la inmunodeficiencia humana (VIH, por ejemplo, gag, pol, env, etc.), virus del papiloma humano (VPH, por ejemplo, proteínas de la cápside tales como L1), virus de la encefalomielitis equina venezolana (EEV), virus de Epstein Barr, virus del herpes simple (VHS), virus del herpes humano, rinovirus, virus de Coxsackie, enterovirus, hepatitis A, B, C, E y G (VHA, VHB, VHC, VHE, VHG por ejemplo, antígeno de superficie), virus de las paperas, virus de la rubeola, virus del sarampión, poliovirus, virus de la viruela, virus de la rabia y virus de la varicela-zóster.
Antígenos víricos adecuados, que no se reivindican, también incluyen proteínas víricas no estructurales, por ejemplo, proteínas codificadas por ácido nucleico vírico que no codifican polipéptidos estructurales, en contraste con los que producen la cápside o la proteína que rodea a un virus. Proteínas no estructurales incluyen aquellas proteínas que promueven la replicación del ácido nucleico vírico, expresión genética vírica, o procesamiento postraduccional, tales como, por ejemplo, proteínas no estructurales 1, 2, 3 y 4 (NS1, NS2, NS3 y NS4, respectivamente) de encefalitis equina venezolana (EEV), de encefalitis equina del este (EEE), o del bosque Semliki.
Los antígenos bacterianos pueden proceder de, por ejemplo, Staphylococcus aureus, Staphylococcus epidermis, Helicobacter pylori, Streptococcus bovis, Streptococcus pyogenes, Streptococcus pneumoniae, Listeria monocytogenes, Mycobacterium tuberculosis, Mycobacterium leprae, Corynebacterium diphtheriae, Borrelia
burgdorferi, Bacillus anthracis, Bacillus cereus, Clostridium botulinum, Clostridium difficile, Salmonella typhi, Vibrio chloerae, Haemophilus influenzae, Bordetella pertussis, Yersinia pestis, Neisseria gonorrhoeae, Treponema pallidum, Mycoplasm sp., Legionella pneumophila, Rickettsia typhi, Chlamydia trachomatis y Shigella dysenteriae, Vibrio cholera (porejemplo, subunidad B de la toxina del cólera, pilus corregulado por la toxina del cólera (PCT)); Helicobacterpylorii (por ejemplo, VacA, CagA, NAP, Hsp, catalasa, ureasa); E. coli (por ejemplo, enterotoxina termolábil, antígenos fimbriales).
Los antígenos parásitos pueden proceder de, por ejemplo, Giardia lamblia, Leishmania sp., Trypanosoma sp., Trichomonas sp., Plasmodium sp. (porejemplo, antígenos de proteína de superficie de P. falciparum tales como pfs25, pfs28, pfs45, pfs84, pfs 48/45, pfs 230, Pvs25 y Pvs28); Schistosoma sp.; tuberculosis micobacteriana (por ejemplo, Ag85, MPT64, ESAT-6, CFP10, R8307, MTB-32 MTB-39, CSP, LSA-1, LSA-3, EXP1, SSP-2, SALSA, STARP, GLURP, MSP-1, MSP-2, MSP-3, MSP-4, MSP-5, MSP-8, MSP-9, AMA-1, proteína de membrana integral tipo 1, RESA, EBA-175 y DBA).
Los antígenos fúngicos pueden proceder de, por ejemplo, Tinea pedis, Tinea corporus, Tinea cruris. Tinea unguium, Cladosporium carionii, Coccidioides immitis, Candida sp., Aspergillus fumigatus y Pneumocystis carinii.
Antígenos del cáncer incluyen, por ejemplo, antígenos expresados o sobreexpresados en el cáncer de colon, cáncer de estómago, cáncer de páncreas, cáncer de pulmón, cáncer de ovario, cáncer de próstata, cáncer de mama, cáncer de piel (por ejemplo, melanoma), leucemia o linfoma. Antígenos de cáncer de ejemplo incluyen, por ejemplo, VPH L1, VPH L2, VPH E1, VPH E2, fosfatasa alcalina placentaria, AFP, BRCA1, Her2/neu, CA 15-3, CA 19-9, CA-125, CEA, Hcg, activador de plasminógeno de tipo urocinasa (Upa), inhibidor activador del plasminógeno, CD53, CD30, CD25, C5, CD11a, CD33, CD20, ErbB2, CTLA-4. Véase Sliwkowski y Mellman (2013) Science 341: 6151 para conocer dianas adicionales del cáncer.
C. Adyuvantes
En algunas realizaciones, las composiciones comprenden además al menos un adyuvante. Los adyuvantes adecuados incluyen, por ejemplo, los compuestos lipídicos y no lipídicos, toxina del cólera (TC), TC subunidad B, derivado CTK63 de la TC, enterotoxina termolábil (TL) de E. coli, derivado LTK63 de TL, Al(OH)3 y ácidos orgánicos poliiónicos como se describe en, por ejemplo, el documento WO2004/020592, Anderson y Crowle, Infect. Immun. 31(1):413-418 (1981), Roterman y col., J. Physiol. Pharmacol., 44(3):213-32 (1993), Arora y Crowle, J. Reticuloendothel. 24(3):271-86 (1978), y Crowle y May, Infect. Immun. 38(3):932-7 (1982)). Los ácidos orgánicos poliiónicos adecuados incluyen, por ejemplo, 6,6'-[3,3'-demitil[1,1'-bifenil]-4,4'-diil]bis(azo)bis[4-amino-5-hidrox-y-1,3-naftaleno-ácido disulfónico] (Azul de Evans) y 3,3'-[1,1'bifenil]-4,4'-diilbis(azo)bis[4-aminonaftalen-1-ácido sulfónico] (Rojo Congo). Los expertos en la materia apreciarán que los ácidos orgánicos poliiónicos pueden usarse para cualquier método de vacunación basado en ácidos nucleicos junto con cualquier tipo de administración.
También se pueden usar agonistas de TLR-3 (por ejemplo, ARNbc, y miméticos del mismo tales como poliI:C, poli A:U y poliiipoliC). Los agonistas de TLR-3 incluyen, por ejemplo, ARN de horquilla corto, ARN de origen vírico, segmentos cortos de ARN que pueden formar ARN bicatenario o de horquilla corta y ARN de interferencia pequeño (ARNip). En algunas realizaciones, el agonista de TLR-3 es ARNbc procedente de virus, por ejemplo, ARNbc procedente de un virus Sindbis o intermedios víricos de ARNbc (Alexopoulou et al. (2001) Nature 413:732). En algunas realizaciones, el agonista de TLR-3 es un ARN de horquilla corto. Las secuencias de ARN de horquilla corto normalmente comprenden dos secuencias complementarias unidas mediante una secuencia enlazadora. La secuencia enlazadora particular no es un aspecto crítico de la invención. Se puede usar cualquier secuencia enlazadora apropiada siempre que no interfiera con la unión de las dos secuencias complementarias para formar un ARNbc. Los agonistas de TLR-3 pueden dar como resultado la liberación de citocinas proinflamatorias (por ejemplo, IL-6, IL-8, TNF-alfa, IFN-alfa, IFN-beta) cuando se ponen en contacto con una célula de respuesta (por ejemplo, una célula dendrítica, una célula mononuclear de sangre periférica o un macrófago) in vitro o in vivo.
Otros adyuvantes adecuados incluyen inmunomoduladores tópicos tales como, miembros de la familia de las imidazoquinolinas tales como, por ejemplo, imiquimod y resiquimod (véase, por ejemplo, Hengge et al., Lancet Infect. Dis. 1(3):189-98 (2001).
Adyuvantes adecuados adicionales están disponibles comercialmente como, por ejemplo, adyuvantes adicionales a base de alumbre (por ejemplo, Alhydrogel, Rehydragel, fosfato de aluminio, Algamulina); adyuvantes a base de aceite (adyuvante incompleto y adyuvante completo de Freund (Difco Laboratories, Detroit, Mich.), Specol, RIBI, TiterMax, Montanide ISA50 o Seppic MONTANIDE ISA 720); adyuvantes a base de copolímeros de bloqueo no iónicos, citocinas (porejemplo, GM-CSF o ligando Flat3); Merck Adjuvant 65 (Merck and Company, Inc., Rahway, N.J.); AS-2 (SmithKline Beecham, Filadelfia, Pa.); sales de calcio, hierro o zinc; una suspensión insoluble de tirosina acilada; azúcares acilados; polisacáridos derivatizados catiónica o aniónicamente; polifosfacenos; microesferas biodegradables; monofosforil lípido A y Quil A. Citocinas, tales como GM-CSF o interleucina-2, -7 o -12, también son adyuvantes adecuados. También se pueden utilizar como adyuvantes hemocianinas (por ejemplo, hemocianina de lapa californiana) y hemoeritrinas. Adyuvantes de polisacáridos tales como, por ejemplo, quitina, quitosano y quitina desacetilada también son adecuados como adyuvantes. Otros adyuvantes adecuados incluyen peptidoglucanos
bacterianos de muramil dipéptido (MDP, N acetilmuramil L alanil D isoglutamina) y sus derivados (por ejemplo, treonil-MDP y MTPPE). El esqueleto de la pared celular (EPC) de BCG y BCG se pueden usar como adyuvantes, con o sin dimicolato de trehalosa. El dimicolato de trehalosa se puede utilizar solo (véase, por ejemplo, la Patente de Estados Unidos N.° 4.579.945). Las endotoxinas destoxificadas también son útiles como adyuvantes solos o en combinación con otros adyuvantes (véase, por ejemplo, las Patentes de Estados Unidos N.° 4.866.034; 4.435.386; 4.505.899; 4.436.727; 4.436.728; 4.505.900; y 4.520.019). Las saponinas QS21, QS17, QS7 también son útiles como adyuvantes (véase, por ejemplo, la patente de Estados Unidos n.° 5.057.540; los documentos EP 0362279; WO 96/33739; y WO 96/11711). Otros adyuvantes adecuados incluyen Montanide ISA 720 (Seppic, Francia), SAF (Chiron, Calif, Estados Unidos), ISCOMS (Cs L), MF-59 (Chiron), la serie SBAS de adyuvantes (por ejemplo, SbAS-2, SBAS-4 o SBAS-6 o variantes de los mismos, disponibles en SmithKline Beecham, Rixensart, Bélgica), Detox (Corixa, Hamilton, Mont.) y RC-529 (Corixa, Hamilton, Mont.).
También se contempla el uso de superantígenos como adyuvantes en la presente invención. Los superantígenos incluyen exoproteínas de Staphylococcus, tales como las enterotoxinas alfa, beta, gamma y delta de S. aureus y S. epidermidis, y las exotoxinas alfa, beta, gamma y delta de E. coli. Las enterotoxinas de Staphylococcus comunes se conocen como enterotoxina A estafilocócica (EAE) y enterotoxina B estafilocócica (EBE), y se describen enterotoxinas hasta E (EEE) (Rott et al., 1992). También se pueden utilizar Streptococcus pyogenes B (SEB), enterotoxina de Clostridium perfringens (Bowness et al., 1992), proteína asociada a la membrana citoplasmática (CAP, por sus siglas en inglés) de S. pyogenes (Sato et al., 1994) y toxina 1 del síndrome de choque tóxico (TSST 1) de S. aureus (Schwab et al., 1993).
Para las composiciones farmacéuticas proporcionadas en el presente documento, los adyuvantes pueden diseñarse para inducir, por ejemplo, una respuesta inmunitaria predominantemente del tipo Th1 o Th2. Niveles altos de citocinas de tipo Th1 (por ejemplo, IFN-gamma, TNF-alfa, IL-2 e IL-12) tienden a favorecer la inducción de respuestas inmunitarias mediadas por células a un antígeno administrado. En cambio, altos niveles de citocinas de tipo Th2 (por ejemplo, IL-4, IL-5, IL-6 e IL-10) tienden a favorecer la inducción de respuestas inmunitarias humorales. Después de la administración oral de una composición que comprende un polipéptido inmunogénico como se proporciona en el presente documento, normalmente se provocará una respuesta inmunitaria que incluye respuestas de tipo Th1 y Th2.
III. Sistemas de administración dirigidos
Las composiciones y métodos descritos actualmente para la administración ileal pueden depender de recubrimientos, matrices y dispositivos apropiados tales como los que se describen a continuación. De acuerdo con la invención, el agente biológico inmunogénico está abarcado por un recubrimiento entérico que tiene un pH umbral de 5,8 a 6,8.
A. Recubrimientos entéricos, matrices y dispositivos
Los recubrimientos entéricos se utilizan para proteger sustancias del entorno de pH bajo del estómago y retrasar la liberación de la sustancia encerrada hasta que alcanza una diana deseada más adelante en el tracto digestivo. Los recubrimientos entéricos son conocidos y están disponibles comercialmente. Los ejemplos incluyen polímeros sensibles al pH, polímeros biodegradables, hidrogeles, sistemas de liberación prolongada y sistemas de administración osmótica (véase, por ejemplo, Chourasia y Jain (2003) J. Pharm. Pharmaceutical Sci. 6:33).
El pH del tracto gastrointestinal (TGI) progresa desde muy ácido en el estómago (pH ~ 2) a más neutro en el íleon (pH ~ 5,8-7,0). Se pueden usar recubrimientos sensibles al pH que se disuelven en el íleon o justo antes del íleon. Los ejemplos incluyen los polímeros L y S Eudragit® (valores de pH de umbral que varían entre 5,5 y 7,0); ftalato de acetato de polivinilo (pH 5,0), ftalato de hidroxipropil metilcelulosa 50 y 55 (pH 5,2 y 5,4, respectivamente) y ftalato de acetato de celulosa (pH 5,0). Thakral et al. (2013) Expert Opin. Drug Deliv. 10:131 revisan las formulaciones de Euragit® para la administración ileal, en particular, combinaciones de L y S que aseguran la administración a pH < 7,0. Crotts et al. (2001) Eur. J Pharm. Biol. 51:71 describen formulaciones de Eudragit® con propiedades de desintegración apropiadas. Vijay et al. (2010) J. Mater. Sci. Mater. Med. 21:2583 revisan los copolímeros a base de ácido acrílico (AA)-metil metacrilato (MMA) para la administración ileal a pH 6,8.
Para la administración ileal, el recubrimiento de polímero normalmente se disuelve a aproximadamente pH 6,8 y permite la liberación completa en aproximadamente 40 minutos (véase, por ejemplo, Huyghebaert et al. (2005) Int. J. Pharm. 298:26). Para conseguir esto, una sustancia terapéutica se puede cubrir con capas de diferentes recubrimientos, por ejemplo, de modo que la capa más externa protege la sustancia a través de condiciones de pH bajo y se disuelve cuando el comprimido sale del estómago, y al menos una capa interna que se disuelve cuando el comprimido pasa a un pH creciente. Se describen ejemplos de recubrimientos en capas para la administración al íleon distal, por ejemplo, en el documento WO2013148258.
Polímeros biodegradables (por ejemplo, pectina, polímeros azo) se basan normalmente en la actividad enzimática de la microflora que vive en el TGI. El íleon alberga una mayor cantidad de bacterias que las etapas anteriores, incluyendo lactobacilos y enterobacterias.
Sistemas de administración oral de liberación controlada osmótica (OROS®; Alza) es un ejemplo de un sistema
osmótico que se degrada con el tiempo en condiciones acuosas. Dichos materiales pueden manipularse con otros recubrimientos o en diferentes espesores, para administrar específicamente al íleon (véase, por ejemplo, Conley et al. (2006) Curr. Med. Res. Opin. 22:1 879).
Polímeros combinados para la administración al íleon se describen en el documento WO2000062820. Los ejemplos incluyen Eudragit® L100-55 (25 mg/cápsula) con citrato de trietilo (2,4 mg/cápsula) y Povidona K-25 (20 mg/comprimido) seguido de Eudragit® FS30D (30 mg/comprimido). Se pueden aplicar polímeros sensibles al pH para efectuar la administración al íleon, como se describe arriba y, por ejemplo, copolímeros de ácido metacrílico (por ejemplo, poli(ácido metacrílico-co-metacrilato de metilo) 1:1), ftalato de acetato de celulosa, ftalato de hidroxipropil metil celulosa, acetato succinato de hidroxipropil metilcelulosa, ftalato de acetato de polivinilo, trimelitato de acetato de celulosa, carboximetil etilcelulosa, goma laca u otros polímeros adecuados. La capa de recubrimiento también puede estar compuesta por polímeros formadores de película que son sensibles a otros componentes luminales además del pH, tales como la degradación bacteriana o un componente que tiene dicha sensibilidad cuando se mezcla con otro polímero formador de película. Ejemplos de dichos componentes que proporcionan liberación retardada al íleon son polímeros que comprenden enlace(s) azo, polisacáridos tales como la pectina y sus sales, galactomananos, amilosa y condroitina, polímeros disulfuro y glucósidos.
Componentes con pH variable, agua y sensibilidades enzimáticas se pueden usar en combinación para dirigir una composición terapéutica al íleon. El espesor del recubrimiento también se puede utilizar para controlar la liberación. Los componentes también se pueden utilizar para formar una matriz, en el que está integrada la composición terapéutica. Véase, en general, Frontiers in Drug Design & Discovery (Bentham Science Pub. (2009), vol. 4.
B. Cápsulas de frecuencia o radiocontroladas
Como alternativa a la disolución de recubrimientos y matrices, la administración en un sitio específico puede ser a través de cápsulas que se liberan con una señal generada externamente. Los primeros modelos se liberaban para una señal de alta frecuencia (AF), como se divulga en Digenis et al. (1998) Pharm. Sci. Tech. Today 1:160. Desde entonces, el concepto de cápsula de AF original se ha actualizado y el resultado se comercializa como InteliSite®. La cápsula actualizada es un sistema de administración que no se desintegra y se activa por radiofrecuencia. El radiomarcaje de la cápsula permite determinar la ubicación de la cápsula dentro de una región específica del tracto GI mediante gammagrafía gamma. Cuando la cápsula llega a la ubicación deseada en el tracto GI, la activación externa abre una serie de ventanas del depósito de la cápsula del fármaco.
El agente biológico inmunogénico se puede encerrar en una cápsula radiocontrolada, de modo que la cápsula sea rastreada y señalada una vez llega al íleon. En algunas realizaciones, la cápsula se señala en un momento dado después de la administración que corresponde al momento en que se espera que llegue al íleon, con o sin detección.
C. Formulaciones
Las composiciones farmacéuticas se pueden usar con fines profilácticos y terapéuticos como se describe en el presente documento. Tal como se ha explicado anteriormente, se pueden preparar composiciones farmacéuticas para proteger contra la degradación del estómago de manera que el agente biológico inmunogénico administrado alcance la ubicación deseada. Se describen métodos de microencapsulación de ADN y fármacos para administración oral, por ejemplo, en el documento US2004043952.
Una composición farmacéutica inmunogénica puede contener sales farmacéuticamente aceptables del agente biológico inmunogénico (por ejemplo, polipéptido inmunogénico o polinucleótido que codifica un polipéptido inmunogénico). Se pueden preparar dichas sales a partir de bases no tóxicas farmacéuticamente aceptables, incluyendo bases orgánicas (por ejemplo, sales de aminas primarias, secundarias y terciarias y aminoácidos básicos) y bases inorgánicas (por ejemplo, sales de sodio, potasio, litio, amonio, calcio y magnesio). Algunos ejemplos particulares de sales incluyen solución salina tamponada con fosfato y solución salina (por ejemplo, por ingestión, administración nasal o inyección).
Un recubrimiento de liberación retardada o un recubrimiento adicional de la formulación puede contener otros polímeros formadores de película que no son sensibles a las condiciones luminales por razones técnicas o control cronográfico de la liberación del fármaco. Los materiales que se utilizarán para dicho fin incluyen, aunque no de forma limitativa; azúcar, polietilenglicol, polivinilpirrolidona, alcohol polivinílico, acetato de polivinilo, hidroxipropilcelulosa, metilcelulosa, etilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa sódica y otros, utilizados solo o en mezclas.
Aditivos tales como dispersantes, colorantes, pigmentos, polímeros adicionales, por ejemplo, poli(etilacrilato, metilmetacrilato), agentes antiadherentes y antiespumantes se pueden incluir en una capa de recubrimiento. Se pueden añadir otros compuestos para aumentar el espesor de la película y disminuir la difusión de jugos gástricos ácidos en el material del núcleo. Las capas de recubrimiento también pueden contener plastificantes farmacéuticamente aceptables para obtener propiedades mecánicas deseadas. Dichos plastificantes son, por ejemplo, aunque sin limitación, triacetina, ésteres de ácido cítrico, ésteres de ácido ftálico, sebacato de dibutilo, alcohol cetílico, polietilenglicoles, monoésteres de glicerol, polisorbatos u otros plastificantes y mezclas de los mismos. La cantidad de
plastificante se puede optimizar para cada fórmula, y en relación con el polímero(s) seleccionado(s), plastificante(s) seleccionado(s) y la cantidad aplicada de dicho(s) polímero(s).
En las composiciones farmacéuticas de esta invención se pueden emplear otros ingredientes farmacéuticos adecuados conocidos en la materia. Los vehículos adecuados incluyen, por ejemplo, agua, solución salina, alcohol, una grasa, una cera, un tampón, un vehículo sólido, tal como manitol, lactosa, almidón, estearato de magnesio, sacarina sódica, talco, celulosa, glucosa, sacarosa y carbonato de magnesio o microesferas biodegradables (por ejemplo, poliglicolato de polilactato). Microesferas biodegradables adecuadas se divulgan, por ejemplo, en las Patentes de los Estados Unidos N.° 4.897.268; 5.075.109; 5.928.647; 5.811.128; 5.820.883. El polipéptido inmunogénico y/o vector de expresión transportador pueden encapsularse dentro de la microesfera biodegradable o asociarse con la superficie de la microesfera.
Dichas composiciones también pueden comprender tampones no inmunogénicos (por ejemplo, solución salina tamponada neutra o solución salina tamponada con fosfato), hidratos de carbono (por ejemplo, glucosa, manosa, sacarosa o dextranos), manitol, proteínas, polipéptidos o aminoácidos tales como glicina, antioxidantes, bacteriostáticos, agentes quelantes tales como Ed Ta o glutatión, adyuvantes (por ejemplo, hidróxido de aluminio), agentes de suspensión, agentes espesantes y/o conservantes. Como alternativa, las composiciones de la presente invención se pueden formular como liofilato. Los compuestos también pueden encapsularse dentro de liposomas usando tecnología bien conocida.
IV. Respuestas inmunitarias y vacunas
Las composiciones farmacéuticas para administración al íleon como se describen en el presente documento están diseñadas para provocar una respuesta inmunitaria de un individuo que es específica para un agente biológico inmunogénico incluido en la composición farmacéutica. La composición farmacéutica se puede utilizar de forma profiláctica o terapéutica como vacuna para evitar o reducir una infección vírica, infección bacteriana, infección parasitaria, infección por hongos o cáncer. Las composiciones farmacéuticas se pueden utilizar para tratar en cualquier etapa, por ejemplo, en las etapas precancerosa, cancerosa o metastásica, o para prevenir una enfermedad o infección.
Por ejemplo, las composiciones descritas en el presente documento pueden usarse para prevenir o tratar infecciones, tales como la gripe, hepatitis o VIH, o para la prevención o el tratamiento del cáncer. Dentro de dichos métodos, las composiciones farmacéuticas se administran normalmente a un individuo que puede o no estar afectado por la enfermedad, trastorno o infección. En algunas realizaciones, una enfermedad, trastorno o infección se diagnostica antes de la administración, por ejemplo, utilizando criterios generalmente aceptados en la materia. Por ejemplo, una infección vírica puede diagnosticarse mediante la medición del valor vírico en una muestra del paciente, una infección bacteriana puede diagnosticarse mediante la detección de las bacterias en una muestra del paciente, y el cáncer puede diagnosticarse mediante la detección de la presencia de un tumor maligno. Las composiciones farmacéuticas se pueden administrar antes o después de la extirpación quirúrgica de tumores primarios y/o tratamientos tales como la administración de radioterapia o fármacos quimioterapéuticos convencionales.
La inmunoterapia es normalmente inmunoterapia activa, en la que el tratamiento se basa en la estimulación in vivo del sistema inmunitario endógeno del hospedador para reaccionar contra, por ejemplo, tumores o células infectadas por bacterias o virus, con la administración de agentes modificadores de la respuesta inmunitaria (por ejemplo, agentes biológicos inmunogénicos).
La frecuencia de administración de las composiciones profilácticas o terapéuticas descritas en el presente documento, así como la dosificación, variará de un individuo a otro y se puede establecer fácilmente utilizando técnicas estándar. Normalmente, se pueden administrar entre 1 y 52 dosis durante un periodo de 52 semanas. En algunas realizaciones, se administran 3 dosis, a intervalos de 1 mes, o se administran 2-3 dosis cada 2-3 meses. En algunas realizaciones, se puede administrar una combinación de más de un antígeno simultánea o secuencialmente, por ejemplo, una vacuna anual contra la gripe que contiene componentes individuales dirigidos a cada subtipo de gripe o múltiples clados dentro de un subtipo. En algunas realizaciones, los intervalos son más como una vez al año, por ejemplo, una vacuna anual contra la gripe basada en la cepa actual en particular. Las vacunas de refuerzo se pueden administrar periódicamente a partir de entonces. Los protocolos alternativos pueden ser apropiados para pacientes individuales y enfermedades y trastornos particulares.
Una dosis adecuada es una cantidad de un agente biológico inmunogénico que, cuando se administra como se ha descrito anteriormente, es capaz de promover, por ejemplo, una respuesta inmunitaria antitumoral, antivírica o antibacteriana, y está al menos un 15-50 % por encima del nivel basal (sin tratar), o al menos un 5-50 % (por ejemplo, 5 %, 10 %, 20 %, 30 %, 50 %, 1,5 veces, 2 veces o más) por encima del nivel del tratamiento no dirigido al íleon. Dicha respuesta se puede controlar mediante la medición de los anticuerpos antitumorales en un paciente o mediante la generación dependiente de la vacuna de linfocitos T citolíticos capaces de destruir, por ejemplo, las células tumorales del paciente, las células infectadas por virus del paciente o las células infectadas por bacterias del paciente in vitro. Estas vacunas también pueden generar una respuesta inmunitaria que conduce a un mejor resultado clínico (por ejemplo, supervivencia libre de enfermedad completa, parcial o más prolongada, valores víricos reducidos) en pacientes vacunados en comparación con pacientes no vacunados, o pacientes que reciben tratamiento no dirigido al
íleon.
En general, un régimen de dosificación y tratamiento adecuado proporciona el compuesto(s) activo(s) en una cantidad suficiente para proporcionar beneficio terapéutico y/o profiláctico. Dicha respuesta se puede controlar mediante el establecimiento de un resultado clínico mejorado (por ejemplo, valor vírico reducido o negativo, remisiones más frecuentes, supervivencia libre de enfermedad completa o parcial o más prolongada) en pacientes tratados en comparación con pacientes tratados con tratamiento no dirigido al íleon o pacientes no tratados. Dichas respuestas inmunitarias generalmente se pueden evaluar usando ensayos estándar de proliferación, citotoxicidad o citocinas descritos anteriormente, que se pueden realizar usando muestras obtenidas de un paciente antes y después del tratamiento.
Por ejemplo, la detección de inmunocomplejos formados entre un polipéptido inmunogénico y anticuerpos en el líquido corporal, que son específicos para el polipéptido inmunogénico, puede usarse para controlar la eficacia de la terapia, por ejemplo, para una enfermedad o trastorno en el que está asociado el polipéptido inmunogénico. Muestras de líquido corporal tomadas de un individuo antes y después del inicio de la terapia (por ejemplo, terapia dirigida al íleon) pueden analizarse para los inmunocomplejos usando métodos conocidos. En resumen, se compara el número de inmunocomplejos detectados en ambas muestras. Un cambio significativo en el número de inmunocomplejos en la segunda muestra (terapia post-dirigida) en relación con la primera muestra (terapia pre-dirigida) refleja una terapia exitosa.
V. Ejemplos
Ejemplos que comprenden un vector adenovírico que codifica la proteína vírica 1 de norovirus o la proteína de fusión (F) del RSV, abarcado por un recubrimiento entérico que tiene un pH umbral de 5,8-6,8, son de acuerdo con la presente invención.
Se conocen métodos farmacéuticos para administrar moléculas pequeñas al intestino, pero la capacidad de administrar un producto biológico grande al intestino para reconocimiento inmunológico adecuado es poco conocida. Los ratones no son capaces de tragar pastillas, por lo que es difícil realizar estudios con comprimidos en modelos animales. Adicionalmente, la ubicación del mejor lugar para administrar el vector de vacuna a fin de provocar una respuesta al antígeno transgénico no se ha caracterizado en seres humanos. En ovejas, se demostró que el yeyuno es la diana más eficaz para provocar una respuesta inmunitaria a un antígeno transgénico codificado por adenovirus (Mutwari et al. (1999) Immunology 97:455). Aquí se muestra el resultado de varios estudios en primates humanos o no humanos con formas farmacéuticas orales humanas mejoradas para la administración de agentes biológicos.
Ejemplo 1
Para determinar qué región del intestino delgado es más activa para inducir una respuesta inmunitaria al antígeno, se realizaron pruebas en seres humanos. Se administraron cápsulas radiocontroladas a voluntarios sanos normales, con la vacuna liberada de manera temprana en el intestino delgado (yeyuno) o más tarde en el intestino delgado (íleon). Se ha descrito el uso de cápsulas radiocontroladas para la administración de fármacos de molécula pequeña, pero no para la administración de vacunas (Digenis et al. (1991) Crit. Rev. Ther. Drug Carrier Syst. 7:309).
La vacuna estaba compuesta por adenovirus recombinantes que expresaban el antígeno HA de la gripe de A/CA/04/2009 (rAd-HA-ARNbc) (véase, por ejemplo, el documento US2012/0244185). Se administraron un total de 1011 unidades infecciosas (UI) a cada sujeto el día 0. El número de linfocitos B preplasmáticos circulantes en sangre periférica se midió mediante el ensayo de células secretoras de anticuerpos (ASC) los días 0 y 7 después de la administración de la vacuna. Los resultados solo miden el número de ASC que reconocen el antígeno HA.
Los resultados muestran que las ASC se pudieron medir 7 días después de la inmunización en cada uno de los grupos tratados (Figura 1). Las respuestas promedio fueron más altas en el grupo dosificado al íleon que en el grupo dosificado al yeyuno. Las ASC de fondo en el día 0 fueron insignificantes. Para el íleon, se observó un promedio de 340 /- 111 (error estándar) ASC IgG y 74 /- 18 IgA en el día 7. Para el yeyuno, las respuestas promedio y de error estándar fueron 118 /- 30 ASC IgG y 28 /- 8 IgA. El grupo de íleon fue significativamente diferente del placebo (P = 0,03 el día 7 para ASC IgA, y tuvo una tendencia más alta para ASC IgG p = 0,07). Al contrario de los resultados en ovejas, los resultados en seres humanos indican que la administración en el íleon es más potente para provocar una respuesta de anticuerpos IgG o IgA que la administración en el yeyuno.
Las respuestas de linfocitos T también se determinaron mediante la detección de la liberación de interferón-Y (IFN-y) usando el ensayo ELISPOT®. La Figura 2 muestra que 12/12 del grupo con dosis en el íleon tenían niveles aumentados de IFN-y, en comparación con 8/12 del grupo con dosis en el yeyuno 7 días después de la administración. Asimismo, los niveles de IFN-y fueron significativamente más altos en el grupo con dosis en el íleon que en el grupo con dosis en el yeyuno.
Se midieron los valores de anticuerpos microneutralizantes (MN) de la gripe A/CA/07/2009. Los niveles aumentados de anticuerpos MN son indicativos de una respuesta de anticuerpos neutralizantes. Después de excluir a los sujetos
que tenían una respuesta inicial de anticuerpos neutralizantes superior a 40 (Faix et al. (2012) PloS One 7:e34581), se trazaron los aumentos de veces en los valores de MN para sujetos individuales. El número de sujetos con un aumento positivo fue de 9 de 10 para la vacuna administrada en el íleon frente a 6 de 10 para la vacuna administrada en el yeyuno (Figura 3). Los valores de la media geométrica (GMT) fueron similares entre los dos grupos, con GMT en el íleon que aumentaron de 22 a 92 frente a GMT en el yeyuno que aumentaron de 18 a 90. Los resultados indican que la liberación en el íleon es más fiable para inducir respuestas de anticuerpos neutralizantes a la gripe, lo que conduce posiblemente a un mayor porcentaje de sujetos protegidos contra la gripe.
Ejemplo 2
Se fabricaron a mano comprimidos utilizando celulosa microcristalina (PH-101, FMC) y almidón (Starch 1500, Colorcon) que incorporan sulfato de bario al 10 % como material radiopaco que contenía sílice ahumada como auxiliar de flujo y estearato de magnesio como lubricante de comprimidos. Los comprimidos de 7,14 mm de diámetro y 150 mg de peso se recubrieron con Eudragit® L-100 en una bandeja de recubrimiento usando un aumento de peso sólido de un 10 % de recubrimiento como guía para saber si se añadió el recubrimiento entérico; los sólidos de recubrimiento contenían 4 partes de polímero Eudragit® por una parte de citrato de trietilo y 1 parte de talco. Como prueba inicial de rendimiento del recubrimiento entérico, a cuatro macacos cangrejeros se les administraron comprimidos mediante una sonda gástrica oral. La sonda gástrica oral es sólida y rígida, pero ahuecada en el medio para instilar líquidos. Tiene un tubo de silicona flexible en el extremo delantero de la sonda rígida que puede sostener un comprimido pequeño en su lugar. El tubo y el aparato de la pastilla se deslizaron por el esófago de los monos inmovilizados hasta que el extremo anterior pasó a través del esfínter cardíaco y llegó al estómago. Se utilizó un chorro de zumo de naranja para desalojar la pastilla en el estómago. Se tomaron radiografías en puntos temporales establecidos y se examinaron para determinar la ubicación y disolución del comprimido. La Tabla 1 resume los resultados.
Tabla 1: Rendimiento del recubrimiento L-100

La Figura 4 muestra que los comprimidos estaban completamente inalterados en el entorno de pH bajo del estómago; no hubo evidencia de disolución prematura de los comprimidos. Aunque grande para un mono, los comprimidos fueron capaces de pasar inalterados a través del estómago al intestino. En el intestino, se disolvieron a una velocidad razonable y se disolvieron completamente en 3 de cada 4 monos. En el 4o mono, la pastilla salió del estómago después de 3 horas y la pastilla no se había disuelto en el momento de la última radiografía. En general, los comprimidos funcionaron de manera aceptable y se seleccionó el recubrimiento Eudragit® L-100 para futuros estudios en seres humanos.
Ejemplo 3
Se completó un estudio clínico de fase 1, inscrito secuencialmente, con una cohorte aleatorizada y controlada con placebo para evaluar la seguridad y la inmunogenicidad de una vacuna oral basada en Ad serotipo 5 recombinante (rAd5) contra la gripe estacional HI. El vector rAd5 (rAd-HA-ARNbc con HA de A/CA/04/2(09) se describió en el Ejemplo 1. El estudio tuvo una fase activa de aproximadamente 3 meses y se llevó a cabo de acuerdo con las directrices de Good Clinical Practice aplicables, el United States Code of Federal Regulations, y las directrices del International Conference on Harmonization. Se obtuvo el consentimiento informado de todos los sujetos después de discutir los riesgos. Se otorgó la aprobación del IRB antes de la dosificación de los sujetos.
Se produjo rAd-HA-ARNbc de grado de buenas prácticas de fabricación (GMP, por sus siglas en inglés) en bolsas Wave® (GE Healthcare, Waukesha, WI) en Lonza Biologicals (Houston, TX). La purificación se realizó mediante cromatografía de intercambio iónico, seguido de intercambio de tampón. El vector purificado se mezcló con excipientes, liofilizados y luego se formaron comprimidos en Lonza utilizando celulosa microcristalina y almidón como volumen de comprimidos. Los comprimidos se recubrieron entéricamente con Eudragit® L 100 (Evonik Industries, Darmstadt, Alemania) utilizando un revestidor Vector HiCoater® LDCS-5 (Vector Freund, Cedar Rapids, IA). El producto final se liberó en un lote y se valoró mediante un ensayo de UI estándar. El placebo se preparó como comprimidos de tamaño y forma similares que contenían 150 mg de celulosa microcristalina, sin recubrimiento entérico. El estudio comparó sujetos tratados con 109 UI, 1010 UI y placebo para la capacidad de provocar una respuesta inmunitaria al transgén. Los sujetos recibieron comprimidos los días 0 y 28.
El número de linfocitos B preplasmáticos circulantes en sangre periférica se midió mediante ensayos de ASC los días 0 y 7 después de la dosis inicial, y los días 28 y 35 después de la segunda dosis (la segunda dosis se administró el día 28). Los resultados muestran que los recuentos de ASC se pudieron medir 7 días después de cada inmunización en los grupos tratados, pero no en el grupo placebo (Figura 5). Las respuestas promedio fueron más altas en el día 7
y más altas en el grupo de dosis alta que en el grupo de dosis baja. Las ASC de fondo en los días 0 y 28 fueron insignificantes e insignificantes para el grupo de placebo en todos los puntos temporales. Para el grupo de dosis alta, se encontró un promedio de 105 /- 33 y 27 /- 12 ASC para los días 7 y 35 respectivamente. Para el grupo de dosis baja, las ASC promedio fueron 41 /- 32 y 14 /- 8 para los días 7 y 35 respectivamente. El grupo placebo tuvo un promedio de 0,3 /- 0,3 y 0, para los días 7 y 35 respectivamente. El grupo de dosis alta fue significativamente más alto que el placebo (P = 0,01 y 0,05 para los días 7 y 35 respectivamente).
Las respuestas de anticuerpos neutralizantes a la gripe se midieron mediante ensayo de MN. Los resultados muestran un aumento dependiente de la dosis en los valores de MN en los grupos tratados frente al control con placebo (Figura 6). La frecuencia de pacientes que responden al tratamiento de MN con al menos un aumento de 2 veces en el grupo de dosis alta fue significativamente diferente a la del grupo de placebo (P = 0,003 según la prueba exacta de Fisher), mientras que la dosis baja tendió a aumentar, pero no fue significativamente mayor que el placebo (P = 0,2). Después de eliminar a los sujetos que tenían valores de MN superiores a 40, los valores de la media geométrica (GMT) se calcularon en los sujetos restantes (Tabla 2). También se calculó la respuesta del valor de veces de la media geométrica (GMFR, por sus siglas en inglés) del día 56 (Tabla 2). Estos resultados muestran que la inmunización oral genera valores de anticuerpos neutralizantes contra la gripe, con un aumento de más de 3 veces en el GMT después de la inmunización en el grupo de dosis alta. Estos resultados muestran que los comprimidos recubiertos con L 100 se pueden utilizar para la administración de vacunas al intestino.
T l 2: m i l MT n l v l r MN r n MN < 40

Ejemplo 4
Se probaron parámetros para recubrimientos entéricos in vitro para determinar los tiempos de disolución con pH variable y porcentaje de recubrimiento. Los datos proporcionan pautas para la administración ileal después de la exposición gástrica a pH bajo (como en el estómago) y el tránsito posterior a través de un gradiente de pH creciente (como se encuentra en el duodeno y yeyuno) antes de llegar al íleon.
La desintegración del comprimido se probó con comprimidos de 150 mg preparados como se describe anteriormente, y recubiertos con un aumento de peso sólido total de un 8, 10 o 12 %, utilizando Eudragit® L100, Eudragit® L100-55, o mezcla 1:1 (p/p) de polímeros L100 y L100-55, aplicado como una suspensión de disolvente orgánico. Por duplicado, los comprimidos preparados con cada polímero de recubrimiento, y en cada nivel de aplicación de recubrimiento, se expusieron previamente a líquido gástrico simulado USP (SGF, pH 1,6, sin pepsina) durante 120 minutos en un aparato de prueba de disolución de cilindro alternativo VanKel Bio-Dis III a 37 °C a una velocidad de movimiento alternativo de 10 inmersiones por minuto (IPM). Luego, los comprimidos se transfirieron a líquido intestinal simulado USP (SIF, pH 6,8, sin pancreatina). Se observó la desintegración de los comprimidos y se registró el tiempo hasta la desintegración completa de ambos comprimidos con una precisión de 5 minutos. Los datos indican que el tiempo de desintegración está influenciado tanto por la composición del polímero como por el espesor y proporcionan una guía con respecto a la selección adecuada de la composición de recubrimiento para influir en el comportamiento de los recubrimientos después de que los comprimidos salen del estómago.

El efecto del pH sobre el tiempo de desintegración se probó con comprimidos de 150 mg, recubiertos hasta un aumento de peso sólido total de un 10 % con Eudragit® L100 o Eudragit® L100-55. Se preparó una serie de tampones ajustando el pH de USP SIF (sin pancreatina) a valores que abarcan la especificación USP de 6,8. Los comprimidos se expusieron previamente a USP SGF (sin pepsina) durante 120 minutos a 37 °C y 10 IPM, luego se transfirieron a las soluciones USP SIF de pH modificado. Se observó la desintegración de los comprimidos y se registró el tiempo hasta la desintegración completa con una precisión de 5 minutos. Los datos indican que la velocidad de desintegración está influenciada por el pH ambiental y difiere entre los dos polímeros. De nuevo, los resultados se pueden usar para la selección adecuada de una composición de recubrimiento para lograr la retención del fármaco a través del estómago y la parte superior del intestino delgado.

continuación

Ejemplo 5
Se llevó a cabo un estudio de fase 1, inscrito secuencialmente, con una cohorte aleatorizada y controlada con placebo para evaluar la seguridad y la inmunogenicidad de una vacuna oral basada en Ad serotipo 5 recombinante (rAd5) contra la gripe estacional HI. Los comprimidos que contienen la vacuna se recubrieron como se describe en el presente documento para disolverse en el íleon. Los datos muestran que una vacuna oral en comprimidos sería competitiva con las vacunas existentes en términos de provocar respuestas de anticuerpos neutralizantes a la gripe.
Las respuestas de inhibición de la hemaglutinación (HAI) se midieron los días 0 y 28 (Figura 7A). Ningún sujeto tratado con placebo seroconvirtió, pero un sujeto con placebo pasó por alto la selección y tuvo un valor alto en el día 0. Ninguno de los sujetos vacunados tenía un valor de HAI inicial > 20. Después de la inmunización, nueve sujetos en el grupo de vacuna alcanzaron niveles seroprotectores (HAI > 40) (Figura 7A). El valor de la media geométrica (GMT) para el grupo fue 611 (IC del 95 %: 30 - 124), un aumento de 7,7 veces la media geométrica (GMFR) sobre el GMT inicial de 79 (IC del 95 %: 6 - 11). De los once que aumentaron 4 veces (92 %), nueve seroconvertidos (SC) y los otros 2 sujetos mostraron un aumento de 4 veces en el valor de HAI de 5 a 20. El grupo de vacuna tuvo un aumento estadísticamente significativo en el número de los que respondieron 4 veces frente al placebo (11 frente a 0, con P < 0 0000 según la prueba exacta de Fisher). Los sujetos que recibieron placebo tuvieron un GMT de 11,9 (IC del 95 %: 6 - 25) el día 28 frente a un GMT de 11,0 el día 0 (IC del 95 %: 5 - 23).
La durabilidad de la respuesta de anticuerpos se midió mediante el examen de las respuestas de HAI 180 días después de la inmunización. En el grupo inmunizado con vacuna, un 75 % (9 de 12) de los sujetos estaban seroprotegidos el día 28 y un 75 % (9 de 12) todavía estaban seroprotegidos el día 180. Se representaron gráficamente los GMT de la HAI (Figura 7B), y se encontró que la disminución del GMT era de un 28 % entre los días 28 y 180 después de la inmunización.
Las respuestas de anticuerpos neutralizantes a la gripe se midieron mediante ensayo de MN. Se observaron aumentos significativos en los valores de MN en el grupo tratado frente al control con placebo (Figura 7C). La frecuencia de los que responden 4 veces de MN en el grupo tratado con la vacuna fue significativamente diferente a la del grupo placebo, con 11 sujetos que respondieron en el grupo tratado con la vacuna frente a 0 en el grupo de placebo (P < 00000 según la prueba exacta de Fisher).
Después de eliminar a los sujetos que tenían valores de MN iniciales (y valores de HAI) superiores a 40, los valores de la media geométrica (GMT) se calcularon en los sujetos restantes los días 0 y 28 como se muestra en la siguiente tabla. El GMT para el grupo de vacuna aumentó a 247 (IC 95: 89-685) frente a ningún aumento en el placebo durante un GMT el día 28 de 9-6 (IC 95: 5-18). Estos cálculos no tuvieron ningún impacto en el grupo de vacuna, ya que ninguno de los sujetos tenía valores iniciales altos de MN o HAI. Estos resultados muestran que los valores de anticuerpos neutralizantes de la gripe se generan mediante inmunización oral, con un aumento de más de 20 veces en el g Mt después de la inmunización en el grupo tratado con la vacuna.
<

Para medir las respuestas totales de anticuerpos a HA, el número de linfocitos B preplasmáticos circulantes en sangre periférica se midió mediante ensayo de ASC los días 0 y 7 después de la inmunización. Los resultados muestran que las ASC se pudieron medir de forma fiable el día 7 en el grupo tratado con la vacuna (Figura 7D). Las ASC de fondo fueron generalmente insignificantes el día 0. Para el grupo tratado con la vacuna, un promedio de 992 (+/- error est.
209, IC del 95 %: 532-1452) ASC IgG y 337 ASC IgA (+/- error est. 104, IC del 95 %: 117-580) cada uno por 1 x 106 PBMC se encontraron para el día 7, con solo un sujeto de 12 que no tiene respuesta de ASC detectable. El grupo de placebo no tenía manchas de IgA el día 7, pero un sujeto tenía un frotis de fondo alto y una respuesta de ASC IgG medible con manchas más pequeñas que las observadas normalmente. El grupo tratado fue significativamente diferente del placebo en términos de la capacidad de provocar una respuesta de ASC IgG o IgA en el día 7 (P = 0-0007 y P = 0,008 respectivamente mediante la prueba T).
Se midió retrospectivamente a los sujetos sus valores de antivector antes y después de la inmunización. Después de
la inmunización oral, algunos sujetos tratados con la vacuna tuvieron un aumento en las respuestas de anticuerpos neutralizantes a Ad5, lo que llevó a un aumento de 26 veces en los valores de la GM de anticuerpos neutralizantes, en comparación con el aumento de veces de la GM de 1-0 veces en los sujetos tratados con placebo. En el grupo de vacuna, las respuestas de HAI y MN mostraron una tendencia similar para los sujetos individuales. Ocho sujetos fueron negativos para Ad5 antes de la inmunización y cuatro fueron positivos para Ad5 antes de la inmunización. Un sujeto que fue positivo para Ad5 no seroconvirtió HAI, sin embargo, un sujeto que fue positivo para Ad5 tuvo el mayor aumento en los valores de HAI (64 veces) de cualquiera de los sujetos del estudio. Este mismo sujeto tuvo una ganancia en los valores de MN de 362 veces sin ningún aumento en los valores de anticuerpos neutralizantes de Ad5 antes y después de la inmunización. No se observó una correlación entre los valores de Ad5 iniciales frente al número de veces de respuesta MN (o respuesta HAI) para los sujetos inmunizados con la vacuna en comprimidos.
Además, la vacuna en comprimidos descrita actualmente es estable a temperatura ambiente durante más de 270 días y puede tolerar excursiones a corto plazo a temperaturas más altas, lo que hace que este enfoque sea técnicamente viable.
Ejemplo 5 Discusión
El ejército de Estados Unidos realizó un estudio independiente para medir los efectos de sus campañas de vacunación estacional sobre las respuestas de anticuerpos neutralizantes en el personal militar, e informó un GMFR de valores de MN de 5,6 después de la inyección de vacuna trivalente inactivada (TIV, por sus siglas en inglés) y un GMFR de 2,2 después de la administración intranasal de vacuna gripe viva y atenuada (LAIV, por sus siglas en inglés), después de considerar los sujetos que tenían valores de MN superiores a 40 para comenzar (Faix et al. (2012) PloS one 7: e34581). En otro estudio, se encontró que la tasa de SC para H1N1 era de un 45 % para una inyección de 45 |jg de proteína HA (sin adyuvante) (Gordon et al. (2012) Vaccine 30:5407), mientras que en otro, la vacuna H1N1 fue altamente inmunogénica con una tasa de SC de un 78 % observada después de 1 dosis de una vacuna dividida (Greenberg et al. (2009) 361:2405).
En contraste con los resultados variables observados con vacunas inyectadas, en el presente estudio, el GMFR de MN se calculó en 29 para los 12 sujetos tratados con la vacuna y un 92 % de los sujetos mostró un aumento de más de 4 veces en los valores de MN. En el presente estudio de comprimidos, la tasa de SC por HAI entre los sujetos tratados con vacuna fue de un 75 % y más de un 92 % de los sujetos tuvieron un aumento de 4 veces en los valores de HAI (Figura 7A). Los valores de MN fueron más altos que los valores de HAI. Es posible que el ensayo de MN sea más sensible o que la vacuna oral basada en rAd provoque respuestas neutralizantes más fuertes fuera de la región de la cabeza que las vacunas inyectadas con proteínas.
Las respuestas de HAI se provocan con vacunas comerciales inyectadas, pero se sabe que los valores de HAI disminuyen. Por ejemplo, los voluntarios no infectados por VIH tuvieron una caída de un 67 % en los valores GMT de HAI entre 1 y 6 meses después de la inmunización (Crum-Cianflone et al. (2011) Vaccine 29:3183). De forma similar, el porcentaje de sujetos seroprotegidos se redujo de un 75 % a un 56 % para los sujetos VIH negativos que se inscribieron con valores de HAI seronegativos (< 1:10). Los estudios con vacunas contra la gripe pandémica también han demostrado una disminución en la durabilidad. En el estudio de la vacuna contra la gripe aviar AS03, el GMT alcanzó 563 después de 2 dosis de vacuna, pero a los 6 meses después de la inmunización, el GMT había bajado a 18, una disminución de un 96 % (Leroux-Roels et al. (2010) Vaccine 28:849). En el presente estudio de vacuna en comprimidos, el porcentaje de sujetos seroprotegidos permaneció constante a un 75 % 1 y 6 meses después de la inmunización, y la caída del valor GMT de HAI fue menos dramática mostrando solo una disminución de un 28 % (Figura 7B). Una posibilidad es que la durabilidad sea mejor para las vacunas basadas en vectores debido a las respuestas mejoradas de linfocitos T.
Ejemplo 5 Materiales y métodos
Protocolo clínico e inscripción. Los sujetos se evaluaron previamente para valores de inhibición de hemaglutinación (HAI) dentro de los 45 días posteriores a la inscripción. Para poder participar en el estudio, los sujetos tenían que tener un título de HAI inicial de < 1:20, tener entre 18 y 49 años y gozar de buena salud. La fase activa del ensayo fue hasta el día 28, con la fase de seguimiento para controlar la seguridad para continuar durante 1 año.
Se inscribieron 24 sujetos. Todos los sujetos que se inscribieron completaron evaluaciones de seguridad e inmunogenicidad durante la fase activa y hasta el día 180 de la fase de control.
Aleatorización y enmascaramiento. El estudio fue diseñado para evaluar la vacuna (VXA-A11) en 12 sujetos a una dosis única de 1 x 1011 unidades infecciosas (UI) con 12 sujetos que recibieron un control de placebo. Hubo 3 sujetos tratados con vacuna centinela inscritos secuencialmente, con cada sujeto dosificado con una frecuencia no mayor a una cada 24 h. Después de una semana de control de las toxicidades relacionadas con la vacuna, los sujetos restantes en la cohorte tratada (9) fueron asignados al azar junto con 12 controles de placebo. La aleatorización se realizó mediante asignación generada por computadora, y el farmacéutico no ciego distribuyó el fármaco del estudio con identidad oculta al personal ciego. Todo el personal del centro de investigación, así como las personas directamente implicadas con los ensayos inmunológicos o la evaluación de la seguridad clínica, permanecieron ciegos a las
asignaciones de tratamiento. Todos los sujetos estaban ciegos en el estudio.
Vacuna. El vector rAd (Ad5 no replicante) transporta ADN que codifica el transgén HA (A/CA/04/2009) cuya expresión está dirigida por un promotor de CMV y una horquilla de ARNbc molecular dirigida por un promotor separado. La sustancia farmacéutica de GMP se produjo en bolsas Wave (GE Healthcare, Waukesha, WI) en Lonza Biologicals (Houston, TX). La purificación se realizó mediante cromatografía de intercambio iónico, seguido de intercambio de tampón. El vector purificado se mezcló con excipientes, liofilizados y luego se formaron comprimidos en Lonza utilizando celulosa microcristalina y almidón como volumen de comprimidos. Los comprimidos se recubrieron entéricamente con Eudragit L100® (Evonik Industries, Darmstadt, Alemania) utilizando un sistema Vector Hi-Coater (Vector Freund, Cedar Rapids, IA). El producto final se liberó en un lote y se valoró mediante un ensayo estándar de UI en Lonza. El placebo se preparó como comprimidos de tamaño y forma similares que contenían 150 mg de celulosa microcristalina, sin recubrimiento entérico.
Criterios de valoración. El criterio de valoración principal de este estudio es la seguridad y el criterio de valoración secundario es la inmunogenicidad a través de la fase activa, principalmente mediante valores de HAI y seroconversiones de HAI. Los criterios de valoración inmunológicos adicionales incluyen valores de MN y ASC. Hubo 5 acontecimientos adversos en el grupo de placebo y 4 en el grupo de vacuna, todos los cuales fueron de grado 1 en intensidad. No se informaron acontecimientos adversos graves en el estudio.
Aislamiento y crioconservación de PBMC. Se recogió sangre en tubos K3 EDTA Vacutainer® (BD, Franklin Lakes, NJ) y se aislaron PBMC el mismo día usando tubos Lymphoprep™ (Axis-Shield, Noruega). Las PBMC se congelaron y descongelaron utilizando reactivos sin suero de acuerdo con las instrucciones del fabricante (Cellular Technology Ltd [CTL], Shaker Heights, OH).
Células secretoras de anticuerpos (ASC). Los kits de inmunoabsorción ligado a enzimas (ELISpot) para linfocitos B secretores de IgG e IgA se realizaron de acuerdo con las instrucciones del fabricante (Mabtech, Mariemont, OH). Se cultivaron células (entre 15 X 104 hasta 5 X 105 células por pocillo) en pocillos triplicados, en medio CTL-Test para optimizar las manchas. La proteína HA (Protein Sciences Corp, Meriden, CT) se biotiniló y cuantificó usando un kit de biotinilación (Pierce, Rockford, IL).
Ensayos de anticuerpos. Se realizaron valores de HAI y microneutralización (MN) y se midieron frente a A/CA/07/2009 procedente de MDCK y A/CA/07/2009 procedente de huevo, respectivamente. Los valores de HAI y MN inferiores a 10 se marcaron como 5 según lo sugerido por el asesoramiento reglamentario.
Análisis estadístico. Se realizaron pruebas "t" de Students no apareado para comprobar las diferencias significativas entre grupos. Se utilizó una prueba exacta de Fisher de dos colas para determinar si las frecuencias observadas eran diferentes para algunos análisis, como se indica en el texto. Para ambas pruebas, se consideraron significativos valores de p < 0,05. Se proporcionaron intervalos de confianza del 95 por ciento (IC 95) para valores medidos.
Ejemplo 6
Estudios no clínicos de una vacuna contra norovirus.
Introducción
Las vacunas de norovirus VP1 (VXA-G2.4-NS y VXA-G1.1-NN) de la invención tienen la misma cadena principal del vector vírico de replicación defectuosa y la misma secuencia de ARN adyuvante que se describe en la Patente de Estados Unidos N.° 8.222.224 y Scallan et al. Clinical and Vaccine Immunology 2013; 20(1): 85-94. La secuencia de la cadena principal del vector se proporciona en la SEQ ID NO: 7.
VXA-G2.4-NS es un vector de adenovirus de serotipo 5 incompetente para la replicación con eliminación de E1/E3 diseñado para usar como una vacuna para la prevención de la enfermedad por norovirus (NVD, por sus siglas en inglés). El vector de adenovirus recombinante (rAd) codifica un gen de 1,6 kb de la proteína vírica 1 (VP1, por sus siglas en inglés) de norovirus (cepa G2.4 Sydney) y una secuencia de ARNbc adyuvante que mejora la inmunogenicidad del antígeno expresado en la mucosa intestinal a través de su actividad agonista de TLR3. Se ha optimizado por codones el gen VP1 para la expresión en células de mamífero y se expresa utilizando un potenciador/promotor de la región temprana intermedia del citomegalovirus humano (hCMVIE, por sus siglas en inglés) y una señal de poliadenilación (pA) de la hormona del crecimiento bovino. Este casete de expresión también incluye el primer intrón de la p-globina humana para potenciar la expresión del transgén. Se usa un segundo promotor de hCMVIE para expresar la secuencia de ARN adyuvante. La secuencia adyuvante procede de una secuencia de luciferasa y se ha informado que estimula la inducción de interferones de tipo I in vitro (2). El adyuvante se expresa como un ARN de horquilla corta, que comprende una secuencia de 21 nucleótidos (GAAACGATATGGGCTGAATAC) como una secuencia en tándem en orientaciones directa e inversa separadas por seis nucleótidos que comprenden el bucle del ARN. Las secuencias de ARN directo e inverso de 21 nucleótidos se aparean para formar el tallo del bucle. Este casete de adyuvante utiliza poli A sintético (SPA, por sus siglas en inglés).
Como se describe a continuación, los estudios clínicos previos en ratones y hurones muestran que la vacunación con VXA-G2.4-NS (y el VXA-G1.1-NN relacionado) provocó respuestas de anticuerpos sustanciales y fiables tanto en la IgG sérica sistémica como en la respuesta de IgA intestinal (fecal) en animales de prueba. Una respuesta inmunitaria única provocada por la vacunación oral es la inducción de anticuerpos procedentes de fuentes locales de linfocitos B. Después de una vacunación oral o una infección entérica, los linfocitos B que residen en la lámina propia (debajo de las células epiteliales del intestino) producen predominantemente anticuerpos diméricos del isotipo IgA. Los anticuerpos IgA atraviesan las células epiteliales intestinales hacia la luz a través del transporte facilitado, dejando un componente secretor unido a la IgA dimérica. La molécula resultante se conoce como IgA secretora o SIgA. SIgA actúa como una barrera externa adicional para bloquear la infección entérica. Las SIgA finalmente se eliminan del sistema y se pueden detectar en muestras fecales.
Farmacología no clínica
Introducción
El objetivo principal de los estudios de inmunogenicidad fue demostrar que las construcciones adenovíricas orales que expresan VP1 de dos especies diferentes de norovirus podrían provocar SIgA intestinal específica, así como respuestas inmunitarias de IgG sérica contra el antígeno de norovirus (VP1) después de la inmunización oral de la vacuna contra norovirus.
El objetivo secundario de los estudios de inmunogenicidad fue comparar dos vías de inmunización (oral con VXA-G2.4-NS frente a im con proteína VP1). Se comparó la proteína VP1 ya que la vacuna inyectable basada en VP1 VLP es una de las pocas vacunas contra norovirus en desarrollo. En general, las SIgA se inducen mediante inmunización de las mucosas, pero en mucho menor grado mediante inmunización parenteral. Por lo tanto, se investigó la respuesta intestinal de SIgA inducida mediante administración oral de VXA-G2.4-NS en comparación con vacunas inyectables basadas en la proteína VP1 recombinante.
Tabla 4: Estudios de farmacolo ía no clínica
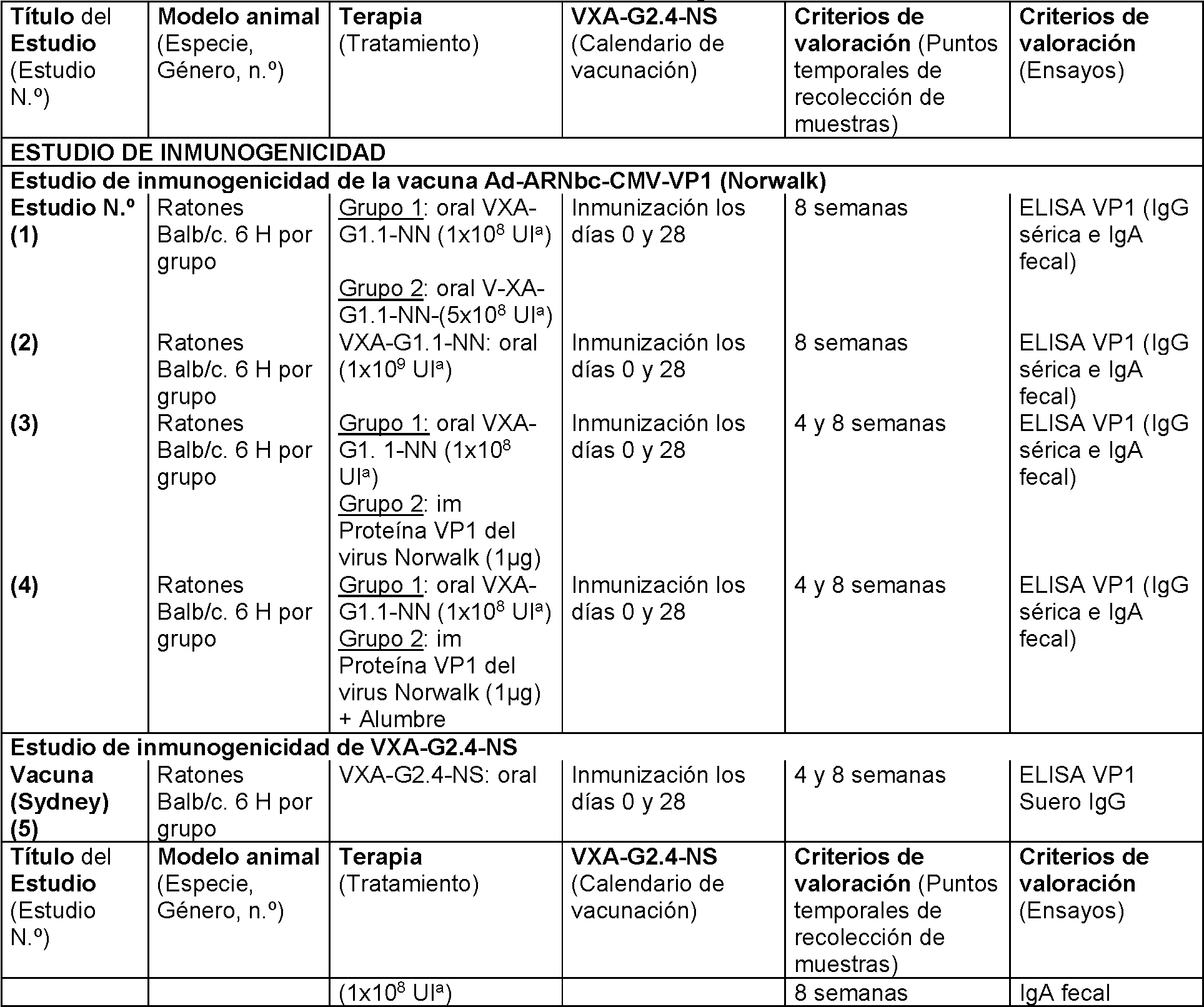
continuación

Estudios de inmunogenicidad
El objetivo principal de los estudios iniciales de inmunogenicidad en ratones fue determinar si la cadena principal del vector que expresa una VP1 de norovirus del virus Norwalk o la cepa Sydney podría inducir una respuesta de anticuerpos a VP1 del virus Norwalk o de la cepa Sydney medida por ELISA.
El rAd que expresa la cepa de Norwalk VP2 y el adyuvante de ARNbc (VXA-G1.1-NN) se administró por vía oral mediante sonda en los días 0 y 28. Se realizó una valoración de la dosis (estudio n.° 1 y estudio n.° 2). Las respuestas de IgG y sIgA específicas de VP1 del virus Norwalk se midieron a partir de muestras de suero y heces, respectivamente a las 8 semanas. Como cabía esperar, como la dosis se incrementó de 1x108 hasta 5x108 hasta 1x109, el valor de IgG de VP1 en suero mostró un aumento dependiente de la dosis del valor de la media geométrica (GMT) de 2x103 hasta 1x104 hasta 5x105 (Figura 8). También se observó un aumento dependiente de la dosis similar pero ligeramente más gradual de 1x103 hasta 2x103 hasta 3x104 en la respuesta de IgA de VP1 fecal (Figura 9). Los estudios con ratones mostraron que la vacuna VXA-G1.1-NN es inmunogénica y generó respuestas de anticuerpos VP1 fecales y séricos dependientes de la dosis.
En el siguiente estudio (estudio n.° 3), se compararon las respuestas inmunitarias a través de dos vías de administración con administración oral mediante sonda con VXA-G1.1-NN (grupo 1) e inyección intramuscular con la proteína VP1 de Norwalk (grupo 2) (Figuras 2.2.3 y 2.2.4). Las respuestas de IgG y SIgA específicas de VP1 del virus Norwalk se midieron a partir de muestras de suero y heces, respectivamente a las 4 y 8 semanas. La vacuna oral de la invención generó valores de título de IgG en suero ligeramente más altos que la vacuna de proteína intramuscular (Figura 10). En el estudio fecal, la vacuna oral generó una respuesta inmunitaria de SIgA intestinal notablemente más alta que la vacuna de proteína intramuscular (Figura 11).
Se llevó a cabo un estudio similar (estudio n.° 4) con la vacuna de proteína VP1 del virus Norwalk con un adyuvante de vacuna, hidróxido de aluminio (Figuras 12 y 13). La inyección intramuscular con la proteína VP1 junto con alumbre generó un valor en suero mucho mayor (Figura 12). Sin embargo, la vacuna oral fue claramente superior en la generación de SIgA, como lo indica el valor de título de IgA específico de VP1 fecal más alto (Figura 13). Con la presencia de alumbre, que es un adyuvante fuerte, unos pocos animales produjeron IgA de VP1 fecal después de la inmunización intramuscular, mientras que la IgA de VP1 fecal de muestras sin alumbre fue casi no detectable. Los datos sugieren que la vacuna oral de la invención tiene una ventaja inmunológica sobre la vacuna inyectable para producir una respuesta inmunitaria intestinal de SIgA.
Se probó la variante de norovirus que circula actualmente, la vacuna de la cepa Sydney (VXA-G2.4-NS) (estudio n.° 5 Figura 2.2.7). A las 4 semanas, la vacuna de la cepa Sydney generó valores de título mucho mejores que la vacuna contra el virus Norwalk. Asimismo, incluso a las 4 semanas, el valor del título de la vacuna de la cepa Sydney fue ligeramente superior al valor de Norwalk a las 8 semanas (Figura 14A). La vacuna de la cepa Sydney generó valores de título de SIgA de VP1 fecal ligeramente más altos que la vacuna Norwalk (Figura 14B). La vacuna de la cepa Sydney parece ser más inmunogénica. En consecuencia, se eligió la cepa Sydney para el siguiente estudio en hurones.
El objetivo del estudio en hurones (estudio n.° 6) fue (1) determinar si la vacuna oral de la invención podría inducir una respuesta de IgG sérica sistémica y de SIgA intestinal en comparación con la proteína VP1 recombinante de Sydney y (2) comparar la respuesta de SIgA e IgG con dos calendarios de vacunación diferentes. Se aleatorizaron veinte hurones en 3 grupos (4 machos y 4 hembras en cada uno de los grupos 1 y 2 y 2 machos y 2 hembras en el Grupo
3). En los días 0 y 2, a los animales del grupo 1 se les administró VXA-G2.4-NS por vía endoscópica. En los días 0 y 28, a los animales del grupo 2 se les administró por vía endoscópica VXA-G2.4-NS y a los animales del Grupo 3 se les administró por vía intramuscular la proteína VP1 recombinante del norovirus de Sydney. Los animales del grupo 1 produjeron un valor de título de IgG más alto que el grupo 2 (3x104 frente a 5x103). Mientras que los animales del grupo 2 generaron valores de título de SIgA más altos que el grupo 1 (247 frente a 92). De manera similar al estudio con ratones (Figura 4), la vacuna de proteína VP1 intramuscular no generó una respuesta de SIgA intestinal aunque la respuesta de IgG de VP1 en suero se generó bien (8x104) (Figura 15). En general, la vacuna oral de la invención genera niveles comparables de IgG en suero y niveles superiores de IgA fecal a la vacuna inyectable de proteína VP1.
Sumario de estudios de farmacología
Los estudios resumidos en esta sección demostraron que la administración oral de VXA-G2.4-NS podría provocar respuestas sustanciales de anticuerpos a la VP1 de norovirus mediante ELISA en ratones y hurones. La inmunidad local podría desempeñar un papel muy importante en la prevención de infecciones y enfermedades, y SIgA probablemente sería la respuesta inmunitaria intestinal más eficaz para combatir un patógeno intestinal como el norovirus. Estos estudios demuestran que la administración oral de VXA-G2.4-NS generó una respuesta de SIgA intestinal y niveles superiores de SIgA a la vacuna inyectable de proteína VP1.
La duración de la infección por norovirus es relativamente corta. El norovirus tiene alrededor de 2 días de tiempo de incubación, seguido de enfermedad con vómitos y diarrea, que dura de 2 a 4 días. Las respuestas de linfocitos T y B comienzan a producirse alrededor de 4 días después de la infección. Curiosamente, la eliminación de norovirus ocurre justo después del desarrollo de la etapa inicial de activación de linfocitos T y B. Sin embargo, la vacuna oral de la invención podría manipular la respuesta inmunitaria para beneficiar al individuo. Antes de la infección por norovirus, la vacuna oral inducirá linfocitos B que se dirigen al intestino para que secreten anticuerpo SIgA específico de VP1. En estudios anteriores se detectaron linfocitos B dirigidos al intestino enriquecidos con IgA en PBMC. SIgA pasará a través de las células epiteliales intestinales hasta el tracto digestivo (luz intestinal). Después de la infección por norovirus, SIgA actuará como un anticuerpo de bloqueo (neutralizador) contra la infección por norovirus. En conclusión, un patógeno intestinal es una combinación excelente para la plataforma oral porque el método de administración oral activa las células inmunitarias con la propiedad de localización intestinal deseada y la SIgA intestinal es la respuesta inmunitaria intestinal más eficaz para combatir un patógeno intestinal.
Ejemplo 7
Sinopsis del estudio VXA-G24-NS de fase 1
Este estudio es un ensayo de fase 1, aleatorizado, doble ciego, controlado con placebo, de intervalo de dosis para determinar la seguridad e inmunogenicidad de una vacuna de norovirus basada en vectores adenovíricos (vxa-g2.4-ns) que expresa gii.4 vp1 y adyuvante de ARNbc administrado por vía oral a voluntarios sanos. El estudio se llevará a cabo en 1 o 2 sitios de Estados Unidos en voluntarios adultos sanos de entre 18 y 49 años. La participación de los sujetos en el estudio durará ~ 1 año después de la selección e inscripción exitosas. Después de la vacunación, se seguirá a los sujetos durante un mes para determinar la eficacia (inmunogenicidad) y durante 12 meses para determinar la seguridad.
Producto de investigación
VXA-G2.4-NS es un vector de vacuna de adenovirus serotipo 5 con replicación defectuosa con deleción E1/E3 para la prevención de la gastroenteritis norovírica causada por Norovirus GII.4. El vector de vacuna codifica un gen VP1 (proteína principal de la cápside) de longitud completa de Norovirus GII.4 Sydney y se describe en detalle en el Ejemplo 6.
El gen GII.4 VP1 se expresa usando un potenciador/promotor de la región temprana intermedia del citomegalovirus (CMV) humano (hCMVie, por sus siglas en inglés). Además del casete transgénico, se usa un segundo promotor de hCMVie para expresar una secuencia de ARN bicatenario (ARNbc) que actúa como adyuvante basado en TLR3.
Producto de control
Una formulación de dosificación de comprimidos de placebo indistinguible en apariencia y número de los comprimidos de vacuna
Régimen y dosificación
Administración única de VXA-G2.4-NS a una dosis baja de 1x1010 UI, una dosis alta de 1x1011 UI o placebo. El número de comprimidos por dosis se calculará en función de los resultados del ensayo de liberación del medicamento. Dos grupos centinela inscribirán a tres sujetos cada uno de forma abierta (Cohortes 1 y 3) para recibir VXA-G2.4-NS antes de inscribir a cualquiera de los aleatorizados, cohortes controladas (cohortes 2 y 4). Dentro de los grupos doble ciego (cohortes 2 y 4), los sujetos con placebo recibirán el mismo número de comprimidos que los sujetos con vacuna. Los
sujetos se inscribirán y se dosificarán en el grupo de dosis baja antes de iniciar la dosificación en el grupo de dosis alta.
Cohorte 1: 1x1010 UI ± 0,5 log (n = 3)
Cohorte 2: 1x1010 UI ± 0,5 log (n = 20) o placebo (n = 10)
Cohorte 3: 1x1011 UI ± 0,5 log (n = 3)
Cohorte 4: 1x1011 UI ± 0,5 log (n = 20) o placebo (n = 10)
Los sujetos de las cohortes 2 y 4 se aleatorizarán en una proporción de 2:1 a VXA-G2.4-NS en 1x1010 UI (dosis baja) o 1x1011 UI (dosis alta), respectivamente, o placebo.
Objetivos
El objetivo principal es determinar la seguridad de un candidato a vacuna de norovirus VXA-G2.4-NS. El objetivo secundario es determinar la inmunogenicidad de un candidato a vacuna de norovirus VXA-G2.4-NS en dos niveles de dosis
Diseño del estudio
Este es un estudio de fase 1, aleatorizado, controlado con placebo, doble ciego (después de la fase inicial abierta), de intervalo de dosis para evaluar la seguridad, reactogenicidad e inmunogenicidad de una vacuna oral de Norovirus GII.4 basada en un vector adenovírico y adyuvante de ARNbc. Todos los sujetos recibirán una única administración de vacuna.
El estudio se inscribirá en cuatro cohortes:
Cohorte 1: VXA-G2.4-NS a 1x1010 UI (dosis baja) centinela
Cohorte 2: VXA-G2.4-NS a 1x1010 UI (dosis baja) o placebo
Cohorte 3: VXA-G2.4-NS a 1x1011 UI (dosis alta) centinela
Cohorte 4: VXA-G2.4-NS a 1x1011 UI (dosis alta) o placebo
La cohorte 1 (grupo centinela de dosis baja) inscribirá a 3 sujetos para recibir una dosis única de VXA-G2.4-NS a 1x1010 UI el día 0. Los 3 sujetos se inscribirán secuencialmente (uno por día), en forma de estudio abierto. Al completar la visita del día 7 en los 3 sujetos, si no se observan toxicidades limitadas por la dosis en estos sujetos centinela (véase Reglas de detención a continuación), comenzará la inscripción de la cohorte 2.
La cohorte 2 (grupo aleatorizado de dosis baja) aleatorizará a 30 sujetos en una proporción de 2:1 para recibir VXA-G2.4-NS a 1x1010 UI (dosis baja) (n = 20) o placebo (n = 10) de una manera doble ciego.
El estudio se inscribirá continuamente durante esta fase a menos que se cumplan los criterios para las reglas de interrupción preestablecidas (véase más abajo). Si esto sucediera, la inscripción de sujetos posteriores no se iniciará hasta que el Comité de Control de Seguridad (SMC, por sus siglas en inglés) del estudio haya completado la revisión de los datos de seguridad y haya ofrecido la recomendación para continuar. La evaluación de seguridad se realizará con las asignaciones de tratamiento codificadas en la Cohorte 2. Si el SMC necesita información sobre el tratamiento para evaluar un EA/SAE, se revelará el código para ese sujeto.
Al completar la visita del día 7 en los 30 sujetos, si no se observan toxicidades limitadas por la dosis en estos sujetos (véase Reglas de detención a continuación), comenzará la inscripción de la cohorte 3. La cohorte 3 (grupo centinela de dosis alta) inscribirá a 3 sujetos para recibir una dosis única de VXA-G2.4-NS a 1x1011 UI el día 0. Los 3 sujetos se inscribirán secuencialmente (uno por día), en forma de estudio abierto. Al completar la visita del día 7 en los 3 sujetos, si no se observan toxicidades limitadas por la dosis en estos sujetos centinela (véase Reglas de detención a continuación), la inscripción se hará en la Cohorte 4.
La cohorte 4 (grupo aleatorizado de dosis alta) aleatorizará a 30 sujetos en una proporción de 2:1 para recibir una dosis única de VXA-G2.4-NS a 1x1011 UI (dosis alta) (n = 20) o placebo (n = 10 ) de manera doble ciego.
El estudio se inscribirá continuamente durante esta fase a menos que se cumplan los criterios para las reglas de interrupción preestablecidas (véase Reglas de detención a continuación). Si esto sucediera, la inscripción de sujetos posteriores no se iniciará hasta que el SMC haya completado la revisión de los datos de seguridad y haya ofrecido la recomendación para continuar. La evaluación de seguridad se realizará con las asignaciones de tratamiento codificadas en la Cohorte 4. Si el SMC necesita información sobre el tratamiento para evaluar un EA/SAE, se revelará el código para ese sujeto.
Todos los sujetos que reciben el fármaco del estudio (vacuna o placebo) tendrán evaluaciones de seguridad e inmunogenicidad completadas durante un mes después de la vacunación. Las evaluaciones de inmunogenicidad se obtendrán al inicio del estudio antes de la vacunación y en los días 7 y 28 después de la vacunación. También se
evaluará a los sujetos para determinar la inmunogenicidad persistente el día 180 y se les hará un seguimiento de seguridad durante los 12 meses después de la vacunación.
Tamaño de la muestra
La inscripción prevista en este estudio es de 66 sujetos de la siguiente manera:
Cohorte 1: VXA-G2.4-NS (1x1010 UI ± 0,5 log): n = 3, centinela
Cohorte 2: VXA-G2.4-NS (1x1010 UI ± 0,5 log): n = 20, o placebo: n = 10; total 30 sujetos, relación 2:1 Cohorte 3: VXA-G2.4-NS (1x1011 UI ± 0,5 log): n = 3, centinela
Cohorte 4: VXA-G2.4-NS (1x1011 UI ± 0,5 log): n = 20, o placebo: n = 10; total 30 sujetos, relación 2:1
Se pueden inscribir sujetos adicionales para reemplazar a los desertores.
Población del estudio
Voluntarios sanos masculinos o femeninos, 18 a 49 años de edad.
Criterios de inclusión/exclusión
Los criterios de inclusión incluyen:
1. Voluntarios masculinos o femeninos de entre 18 y 49 años de edad, ambos inclusive
2. Capaces de dar consentimiento informado por escrito
3. Saludable (sin problemas de salud clínicamente significativos), según lo determine el historial médico, examen físico, ECG de 12 derivaciones y signos vitales en la selección
4. Valores de laboratorio de seguridad dentro de los siguientes criterios de intervalo al inicio:
a. Intervalo normal de fosfatasa alcalina (ALP), alanina aminotransferasa (ALT), aspartato aminotransferasa (AST), bilirrubina, fósforo (hipofosfatemia), neutrófilos, sangre oculta, glóbulos blancos (WBC, por sus siglas en inglés) y proteína de la orina;
b. Anomalía normal o de grado 1 sin importancia clínica (NCS, por sus siglas en inglés) para la albúmina, amilasa, nitrógeno de urea en sangre (BUN, por sus siglas en inglés), calcio, creatina fosfocinasa (CPK), creatinina, glucosa, magnesio, potasio, sodio, proteína total, eosinófilos (aumento), hemoglobina, linfocitos (disminución) y plaquetas;
c. Negativo o positivo NCS para sangre y orina
5. Índice de masa corporal entre 17 y 35 en la selección
6. Comprensión de los requisitos del estudio con capacidad y disposición para completar todas las evaluaciones y cumplir con las visitas y contactos programados
7. Las mujeres participantes deben tener una prueba de embarazo negativa al inicio del estudio y cumplir uno de los siguientes criterios:
a. Al menos un año después de la menopausia;
b. Estéril quirúrgicamente;
c. Estar dispuesta a usar anticonceptivos orales, implantables, transdérmicos o inyectables durante 30 días antes y hasta 60 días después de la vacunación;
i. El investigador debe aprobar un método anticonceptivo fiable (por ejemplo, método de doble barrera, Depo-Provera, dispositivo intrauterino, Norplant, anticonceptivos orales, parches anticonceptivos, abstinencia)
Los criterios de exclusión incluyen:
1. Recepción de cualquier vacuna contra el norovirus en los dos años anteriores a la vacunación de estudio 2. Administración de cualquier vacuna, medicamento o dispositivo en investigación dentro de las 8 semanas anteriores a la vacunación, o el uso planificado de lo mencionado anteriormente durante el estudio hasta el seguimiento de seguridad de 12 meses
3. Administración de cualquier vacuna autorizada dentro de los 30 días anteriores a la vacunación
4. Presencia de una enfermedad médica o psiquiátrica significativa no controlada (aguda o crónica), incluyendo la institución de un nuevo tratamiento médico/quirúrgico o una alteración significativa de la dosis por síntomas no controlados o toxicidad por fármacos dentro de los 3 meses posteriores a la detección y reconfirmada al inicio 5. Cualquiera de los siguientes hallazgos de ECG dentro de los 30 días anteriores a la vacunación:
a. Duración del intervalo QTc (Bazett) > 450 ms (hombre) o > 470 ms (mujer),
b. Intervalo QRS superior a 120 ms,
c. Intervalo PR superior a 220 ms,
d. Cambios clínicamente significativos en la onda ST-T u ondas Q patológicas
6. Serología positiva para VIH-1 o VIH-2, o anticuerpos HBsAg o VHC
7. Cáncer, o tratamiento para el tratamiento del cáncer, en los últimos 3 años (excluyendo el carcinoma de células basales o el carcinoma de células escamosas)
8. Presencia de inmunodepresión o afección médica posiblemente asociada con una respuesta inmunitaria alterada, incluyendo diabetes mellitus
9. Administración de cualquier medicamento o tratamiento que pueda afectar negativamente al sistema inmunitario, tal como inyecciones para alergias, inmunoglobulina, interferón, inmunomoduladores, medicamentos citotóxicos u otros medicamentos que se sabe que están asociados con una toxicidad significativa en órganos importantes, o corticosteroides sistémicos (orales o inyectables) durante los 3 meses anteriores a la vacunación. Se permiten corticosteroides inhalados y tópicos
10. Presencia de miembros del hogar que hayan recibido las vacunas Ad4 o Ad7 en los 2 meses anteriores a la vacunación
11. Presencia de miembros del hogar que son recién nacidos, mujeres embarazadas o receptores de trasplantes de células madre hematopoyéticas o de trasplantes de órganos sólidos
12. Historial de abuso de drogas, alcohol o sustancias químicas durante 1 año anterior a la vacunación
13. Recepción de sangre o hemoderivados 6 meses previos a la vacunación o administración planificada durante el período de seguimiento del estudio
14. Donación de sangre o hemoderivados dentro de las 4 semanas previas a la vacunación o donación planificada durante el período del estudio
15. Enfermedad aguda dentro de las 72 horas previas a la vacunación definida como la presencia de una enfermedad moderada o intensa con o sin fiebre (según lo determine el investigador a través de la historia clínica y el examen físico)
16. Presencia de fiebre > 38 °C medida por vía oral al inicio del estudio
17. Muestra de heces con sangre oculta en la selección
18. Prueba de detección de drogas en orina positiva para detectar abuso de drogas
19. Tabaquismo constante/habitual en los 2 meses previos a la vacunación
20. Antecedentes de reacciones graves a la vacunación, tales como anafilaxia, problemas respiratorios, urticaria o dolor abdominal
21. Trastorno hemorrágico diagnosticado o hematomas importantes o dificultades hemorrágicas que podrían hacer que la extracción de sangre sea problemática
22. Antecedentes de enfermedad del intestino irritable u otra afección inflamatoria digestiva o gastrointestinal que podría afectar la distribución/evaluación de seguridad de una vacuna administrada por vía oral dirigida a la mucosa del intestino delgado.
Dichas afecciones pueden incluir pero no se limitan a:
26. Cualquier afección que, en opinión del investigador, podría interferir con la capacidad para evaluar los objetivos primarios del estudio CALENDARIO DEL ESTUDIO Las siguientes visitas/llamadas de estudio se realizarán durante el estudio:
Período de selección (dentro de los 30 días anteriores a la vacunación)
Día -2 (Recogida de muestras de laboratorio de seguridad de referencia)
Día 0 Visita (Valores iniciales; día de la vacunación)
Día 2 Visita
Día 7 Visita
Día 28 Visita
Día 180 Visita
Día 365 Contacto de fin de estudio
Se seguirán a los sujetos a través de una llamada telefónica todos los días los días 1, 3 al 6 y 14. También serán contactados mensualmente entre las visitas del día 28 y el día 180 y también después de la visita del día 180 hasta el día 365 (fin del estudio). Véase la Tabla 1 para conocer el calendario detallado de los procedimientos del estudio.
Evaluaciones de seguridad e inmunogenicidad
Seguridad:
La seguridad y la tolerabilidad se evaluarán mediante: reactogenicidad local (oral, esofágica y gastrointestinal) y sistémica (síntomas solicitados), y evaluaciones clínicas y de laboratorio. Los exámenes físicos, análisis de orina de rutina, recuentos sanguíneos completos y las químicas séricas se recolectarán antes de la dosis en la selección y el inicio (laboratorios de seguridad en el día 2) y en los días de estudio 2, 7 y 28. Los signos vitales se registrarán antes de la dosis en la selección y el inicio y en los días de estudio 2, 7, 28 y 180.
La seguridad se evaluará utilizando la química sanguínea, hematología y análisis de orina estándar y análisis realizados mediante métodos estadísticos a continuación. Cualquier sujeto que experimente síntomas agudos de
conjuntivitis, infección de las vías respiratorias superiores, heces blandas y/o diarrea dentro de los 14 días posteriores a la vacunación inicial, se le pedirá que regrese para una evaluación médica y una evaluación de la infección por adenovirus 5. En estos sujetos, se recolectarán cultivos de adenovirus de frotis de garganta y rectales.
Inmunogenicidad:
La inmunogenicidad se evaluará utilizando ensayos de función inmunitaria humoral y celular de muestras de sangre obtenidas al inicio (antes de la dosis) y en los días de estudio 7 y/o 28, según el ensayo. Asimismo, se realizará una evaluación final de inmunogenicidad persistente el día 180. Se realizarán las siguientes evaluaciones: IgG sérica de VP1; Anticuerpos de bloqueo de antígenos de histo-grupo sanguíneo (BT50); ASC IgG de VP1; ASC IgA de VP1; Inmunofenotipado de linfocitos B mediante citometría de flujo; Cultivo de linfocitos B pre-plasma para IgG de VP1 e IgA de VP1; IgA fecal de VP1; HAI; y anti-Ad5.
Reglas de detención
El estudio se detendrá (no se realizarán nuevas inscripciones ni se administrarán más productos de investigación en espera de una revisión de seguridad completa por el SMC) si ocurre cualquiera de las siguientes situaciones: Para las Cohortes 1 y 3 (3 sujetos centinela de estudio abierto lideran en grupos):
1. Uno o más sujetos experimentan un SAE relacionado con la vacuna de cualquier grado;
2. Uno o más sujetos experimenta una anomalía de laboratorio o un EA clínico de Grado 3 o superior;
3. Dos o más sujetos experimentan cualquier anomalía de laboratorio o EA clínico de Grado 2 relacionado con la vacuna.
Para las cohortes 2 y 4 (grupos aleatorios controlados con placebo):
1. Uno o más sujetos experimentan un SAE relacionado con la vacuna de cualquier grado;
2. Dos o más sujetos experimentan EA clínicos o de laboratorio de Grado 3,
3. Uno o más sujetos experimentan un EA clínico de Grado 4 o una anomalía de laboratorio de Grado 4;
4. Tres o más sujetos presentan síntomas de infección por adenovirus y virus de vacuna adenovirus 5 con capacidad de replicación detectable. Si tres o más sujetos presentan síntomas de infección por adenovirus, la inscripción se detendrá en espera de los resultados de la detección del virus de la vacuna del adenovirus 5.
Parámetros de criterios de valoración
Los análisis de seguridad incluyen: 1) Demografía descriptiva estándar; 2) Se tabulará la proporción de sujetos en cada grupo de tratamiento para cada acontecimiento de reactogenicidad solicitado local y sistémico y cualquier EA no solicitado. La intensidad del EA se clasificará utilizando criterios estandarizados adaptados de la Guía de la FDA de septiembre de 2007 titulada "Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical T rials"; 3) Los sujetos con EA (incluyendo anomalías de laboratorio clínico) se resumirán mediante (1) Sistema de órganos del cuerpo MedDRA y término preferido; (2) intensidad; (3) parentesco; y, por separado, (4) gravedad; 4) La proporción de sujetos en cada grupo de tratamiento con informes de EA dentro de cada sistema de órganos del cuerpo se comparará de la misma manera. Se probará la heterogeneidad significativa en el nivel de término preferido
Los análisis de inmunogenicidad incluyen: 1) Anticuerpos de bloqueo de antígenos de histo-grupo sanguíneo (BT50) e IgG en suero de VP1; 2) Los análisis exploratorios adicionales incluirán ASC IgG de VP1; ASC IgA de VP1; Inmunofenotipado de 13 células mediante citometría de flujo; Cultivo de linfocitos B pre-plasma para IgG de VP1 e IgA de VP1; IgA fecal de VP1; HAI; Anti-Ad5.
Métodos estadísticos
Tamaño y potencia de la muestra:
Este es el primer ensayo clínico en seres humanos con VXA-G2.4-NS que será realizado por el patrocinador. Actualmente no hay información clínica sobre el fármaco de estudio. Por lo tanto, el tamaño de la muestra se determinó en función de la experiencia de un estudio normal de vacuna de fase 1. Se predice que el número de voluntarios por grupo en las cohortes 2 y 4 producirá resultados de inmunogenicidad significativos. Un tamaño de muestra de 20 en el grupo de vacuna y 10 en el grupo de placebo (es decir, una proporción de aleatorización de 2:1) proporcionará aproximadamente un 86 % de poder para detectar una diferencia de grupo, suponiendo que la proporción de respuesta (observada en IgG sérica de VP1) en el grupo de vacuna es de un 50 % y en el placebo es 0, utilizando la prueba bilateral exacta de Fisher de dos grupos a un nivel de significancia de 0,05.
Análisis de datos:
Seguridad:
La seguridad se resumirá por grupo de tratamiento. La reactogenicidad local y sistémica, los AE, los resultados de laboratorio clínico y los signos vitales se resumirán de forma descriptiva mediante visita de estudio. El número y porcentaje de sujetos que experimentan síntomas agudos de conjuntivitis, infección de las vías respiratorias superiores, heces blandas y/o la diarrea dentro de los 14 días posteriores a la vacunación inicial se compararán por grupo de tratamiento utilizando la prueba exacta de Fisher.
Inmunogenicidad:
Los resultados de inmunogenicidad evaluados mediante ensayos de función inmunitaria humoral y celular a partir de muestras de sangre recolectadas en visitas de estudio preseleccionadas se resumirán de manera descriptiva. El análisis de covarianza (ANCOVA) se utilizará en el análisis de los valores de anticuerpos, con el valor logarítmico posinicial como variable dependiente, el tratamiento como factor y el valor logarítmico inicial como covariable. Las medias de mínimos cuadrados (LS, por sus siglas en inglés) y el IC del 95 % de las medias LS se obtendrán del modelo. Se informará el valor de la media (LS) geométrica (GMT) posterior al inicio para el grupo VXA-G2.4-NS y el aumento de veces de la media (LS) geométrica (GMFR) sobre el GMT inicial en el inicio.
Ejemplo 8
Antecedentes del RSV
El virus respiratorio sincitial (RSV) es la causa más importante de infección del tracto respiratorio inferior (LRI, por sus siglas en inglés) en bebés y niños pequeños y es una de las principales causas de LRTI en los ancianos y en pacientes inmunodeprimidos, donde puede tener efectos devastadores, causando morbilidad y mortalidad significativas. Se estima que infecta a un 5-10 % de los residentes de residencias de ancianos por año, con tasas de neumonía o muerte de un 10-20 % y 2-5 % respectivamente (Falsey et al. 2000). No existe una vacuna aprobada, aunque existe un anticuerpo monoclonal profiláctico aprobado, Palivizumab, para la prevención de enfermedades en lactantes de alto riesgo.
Dada la falta de una vacuna, Vaxart está abordando esta gran necesidad médica insatisfecha mediante el inicio de un programa preclínico para evaluar una vacuna contra el RSV administrada utilizando su plataforma de vector de adenovirus oral. Esta plataforma se ha utilizado anteriormente para administrar vacunas contra la gripe a pacientes en los que se ha demostrado su eficacia e inducción de inmunidad significativa. Una estrategia importante en este camino preclínico es demostrar la protección contra enfermedades en un modelo de exposición al RSV de rata algodonera. Con este fin, se ha comenzado dicha evaluación y pronto se tendrá un conjunto de datos completo. Se presentan datos preliminares en este informe, lo que demuestra que se genera una inmunidad significativa incluso después de una sola vacunación. Con base en esta respuesta inmunitaria, se espera que la vacuna sea protectora.
Evaluación de la vacuna RSVF en ratas algodoneras
Se generó un vector de vacuna (Ad-RSVF) que expresa la proteína de fusión (F) de RSV y un adyuvante de ARNbc como se describió anteriormente. El vector de la vacuna Ad se produjo en 293 células, se purificó y se determinó un valor. Luego, el vector se evaluó primero en ratones donde se provocó una respuesta inmunitaria significativa a la proteína de fusión (datos no mostrados) y luego en el modelo animal de RSV relevante, de ratas algodoneras. Como un método de administración oral no se ha optimizado en ratas algodoneras, se eligió la ruta intranasal como una ruta alternativa de administración a las mucosas. Se administraron dos dosis, una dosis baja (1e8 UI) y una dosis alta (1e9 UI). Los animales no infectados se utilizaron como control negativo y el virus RSV de tipo silvestre (RSVA2) se utilizó como control positivo. El RSV inactivado con formalina (FIRSV) se utilizó como control de los efectos negativos no deseados asociados con una vacuna de RSV desarrollada en la década de 1960. Después de una única vacunación, la Ad-RSVF en dosis bajas y altas indujo valores más altos en comparación con la infección por RSV de tipo silvestre (Figura 17) mientras que el FIRSV indujo una respuesta inmunitaria muy débil (200-300 veces menor). Estos datos indican que la presente vacuna es superior a la inducción de inmunidad que el virus WTRSV y más eficaz que la vacuna FIRSVF anterior.
Además de los anticuerpos anti-proteína de fusión totales (Figura 17), también se analizaron los anticuerpos generados por la presente vacuna que podrían competir con el anticuerpo Palivizumab aprobado. En un ensayo de competencia utilizando sueros combinados de cada uno de los grupos de tratamiento, solo la vacuna Ad-RSVF generó valores de anticuerpos detectables (Figura 18). Estos datos indican que la vacuna produce anticuerpos protectores contra el RSV.
Estudios no clínicos de VXA-RSV-f
8.1 Introducción
VXA-RSV-f es un vector de adenovirus de serotipo 5 incompetente para la replicación con eliminación E1/E3 diseñado para usar como vacuna para la prevención del RSV y administrado por vía oral. El vector de adenovirus recombinante
(rAd) codifica 1) el gen de fusión (F) de la cepa A2 de RSV (Genbank n.° HQ317243.1) y 2) una secuencia de ARNbc adyuvante que mejora la inmunogenicidad del antígeno F expresado en la mucosa intestinal a través de su actividad agonista de TLR3. La cadena principal de la vacuna es idéntica a la de VXA-A1.1, VXA-BYW y ND1.1 de Vaxart. 10 vacunas para la gripe pandémica y estacional A y B, respectivamente, actualmente en desarrollo clínico; el único cambio es que VXA-RSV-f tiene una proteína de superficie diferente (proteína F) que se expresa. El gen de la proteína F de RSV se ha optimizado por codones para la expresión en células de mamíferos y se expresa utilizando un potenciador/promotor de la región temprana intermedia del citomegalovirus humano (hCMVIE) y una señal de poliadenilación (pA) de la hormona del crecimiento bovino. Este casete de expresión también incluye el primer intrón de la p-globina humana para potenciar la expresión del transgén. Se usa un segundo promotor de hCMVIE para expresar la secuencia de ARN adyuvante. La secuencia adyuvante procede de una secuencia de luciferasa y se ha informado que estimula la inducción de interferones de tipo I in vitro (1). El adyuvante se expresa como un ARN de horquilla corta, que comprende una secuencia de 21 nucleótidos (GAAACGATATGGGCTGAATAC) como una secuencia en tándem en orientaciones directa e inversa separadas por seis nucleótidos que comprenden el bucle del ARN. Las secuencias de ARN directo e inverso de 21 nucleótidos se aparean para formar el tallo del bucle. Este casete de adyuvante utiliza poli A sintético (SPA, por sus siglas en inglés).
Vaxart ha realizado estudios clínicos previos para determinar el potencial inmunogénico de VXA-RSV-f en ratones y ratas algodoneras. Estos estudios mostraron que la vacunación con VXA-RSV-f provocó respuestas de IgG séricas sistémicas sustanciales en animales de prueba.
Como se ha descrito anteriormente, la vacuna de RSV de Vaxart (VXA-RSV-f) utilizará la misma cadena principal del vector vírico con replicación defectuosa y la misma secuencia de ARN adyuvante que los programas de virus de gripe pandémica y estacional de la compañía. La vacuna contra la gripe pandémica A/Indonesia/05/2005 (H5N1), ND1.1, actualmente se está estudiando bajo BB-INDs 14660 y 15122, y la vacuna contra la gripe estacional A/Califomia/04/2009 (H1N1), VXA-A1.1, se está estudiando actualmente bajo BB-IND 15198 y 15285. La vacuna B/Wisconsin/1/2010 (Yamagata) se está estudiando actualmente bajo BB-IND 16611. Debido a que la única diferencia entre las vacunas es el gen del antígeno [la proteína F en VXA-RSV-f frente a HA en VXA-A1.1, ND1.1 y VXA-BYW.10], los estudios clínicos previos de ND1.1 y VXA-A1.1 son relevantes y respaldan el desarrollo clínico de la vacuna VXA-RSV-f para la prevención de la enfermedad por RSV.
8.2 Farmacología no clínica
8.2.1 Introducción
El objetivo principal de los estudios de inmunogenicidad de la vacuna candidata VXA-RSV-f contra el VSR era demostrar que la construcción del vector podía provocar respuestas de anticuerpos en ratones y ratas algodoneras, y que las respuestas inmunitarias adaptativas generadas no conducían a un aumento de la enfermedad por VSR, tal como el que se sabe que ocurre con la vacuna contra el RSV inactivada con formalina. Adicionalmente, se realizaron estudios en animales para demostrar el valor del adyuvante para inducir la potenciación inmunitaria específica de antígeno.
Tabla 5 Estudios de farmacolo ía no clínica

_________
continuación

8.2.2 Estudios de inmunogenicidad y exposición
El objetivo principal del estudio inicial de inmunogenicidad en ratón (estudio n.° WCB254) fue para determinar si la cadena principal del vector Vaxart que expresa la proteína F del RSV podría inducir una respuesta de anticuerpos al RSV medida por ELISA. Una vez completado el estudio con ratones, se realizaron dos estudios con ratas algodoneras. Los objetivos de los estudios con ratas algodoneras eran demostrar que VXA-RSV-f podía provocar potentes anticuerpos contra el RSV y que la vacuna podía provocar respuestas inmunitarias protectoras contra el RSV. Adicionalmente, los estudios con ratas de algodón se utilizaron para determinar que las respuestas inmunitarias adaptativas inducidas por VXA-RSV-f no aumentaron la enfermedad por RSV, tal como los registrados para la antigua vacuna contra el RSV inactivada con formalina (FI-RSV).
Estudios de inmunogenicidad en ratones
Se inmunizaron ratones (6 hembras/grupo) con VXA-RSV-f usando tres vías de administración diferentes para determinar si la construcción era inmunogénica. Se inmunizaron animales con 1x108 UI en las semanas 0 y 3 utilizando tres vías de administración diferentes (intranasal, intramuscular y oral). Los valores de anticuerpos contra el VSR se midieron en la semana 7. Los resultados muestran que tanto la vía de administración in como la im fueron potentes para provocar respuestas de anticuerpos, y que la administración oral fue capaz de provocar algo de inmunidad, pero no fue tan potente en ratones como las otras dos vías de administración. Véase la Figura 22.
Estudios de inmunogenicidad y exposición en ratas algodoneras
Los objetivos del primer estudio con ratas algodoneras (Estudio N.° XV-95) fueron determinar la capacidad de VXA-RSV-f para inducir respuestas de anticuerpos y para proteger contra la enfermedad del RSV y la replicación vírica. La rata algodonera se considera un modelo importante para el desarrollo clínico previo de vacunas contra el VSR debido a la susceptibilidad del animal a la infección por el VSR y la reproducibilidad del fenotipo de inflamación pulmonar/sesgo de citocinas cuando se administra la vacuna contra el VSR inactivada con formalina (FI-RSV) seguida por la exposición al VSR (2). Se inmunizaron ratas algodoneras hembras (N = 6 por grupo) con VXA-RSV-f mediante administración intranasal (in) e intramuscular (im) en las semanas 0 y 4 a 1x109 U i. También se utilizó una dosis más baja en el grupo de administración in a 1x108 UI de VXA-RSV-f (VXA-RSV-f baja). Los animales tratados con la vacuna VXA-RSV-f se compararon para los valores ELISA de IgG en la semana 8 con un grupo de control sin infección/sin vacuna (Sin infección), un grupo de tampón de almacenamiento de adenovirus solo (Tampón) y un grupo de vacuna FI-RSV a una dilución 1:100 de una reserva (lote n.° 100) fabricada por Pfizer a partir de la cepa RSV-A2 (3). Para mostrar que los resultados posteriores a la exposición fueron específicos de antígeno, se administró por vía in un rAd que expresa el adyuvante sin el antígeno a 1x109 UI (Ad-Adj) y se utilizó como control. Como control positivo de inmunogenicidad y protección adaptativa mediada por inmunidad, se administró una dosis única de la cepa A2 de RSV de tipo silvestre (RSV2) a 1x105 UFP en la semana 0. Véase la Tabla 5 para el sumario de los grupos de tratamiento de estudio, puntos temporales y criterios de valoración.
Todos los animales inmunizados con VXA-RSV-f indujeron valores significativos de anticuerpos IgG contra RSV, con valores de anticuerpos promedio de grupo superiores a 1x105 (Figura 23A) en la semana 8. Los animales inmunizados con FI-RSV y RSV2 también indujeron valores significativos de anticuerpos IgG, con valores promedio que alcanzan 1x104 en la semana 8 (Figura 23a ). Los animales tratados con la vacuna VXA-RSV-f también fueron capaces de inducir anticuerpos que competían por el sitio de unión a palivizumab en la proteína F del virus RSV, medido por ELISA de unión competitiva, mientras que la vacuna FI-RSV no indujo anticuerpos capaces de reconocer el sitio de unión a palivizumab (Figura 23B). Hubo cierta capacidad del grupo RSV2 de tipo silvestre (control positivo) para inducir anticuerpos que compiten por el sitio de unión a palivizumab. Los anticuerpos neutralizantes del RSV se midieron mediante el ensayo PRNT. Se observó una tendencia similar al análisis de palivizumab, con todos los grupos VXA-RSV-f capaces de inducir anticuerpos neutralizantes significativos contra el RSV, mientras que la vacuna FI-RSV no fue capaz de inducir valores neutralizantes sustanciales (Figura 23C). La vacuna de control RSV2 fue capaz de provocar valores neutralizantes similares a los de los grupos VXA-RSV-f estadísticamente hablando, teniendo la VXA-RSV-f (im) el valor de la media geométrica más alto (p = 0,47 según Mann-Whitney).
Los objetivos del segundo estudio con ratas algodoneras (Estudio N.° XV-112) fueron determinar si las ratas algodoneras podrían inmunizarse por vía oral con la vacuna VXA-RSV-f e inducir respuestas inmunitarias significativas. La exposición fue opcional, posterior a una inmunización oral eficaz. La inmunización oral puede ser difícil en animales y no se disponía de experiencia previa documentada para la dosificación oral de rAd en ratas algodoneras. Por este motivo, se realizó un ajuste posológico de la vacuna VXA-RSV-f con dosis de 1x108, 1x109, y 1x1010 UI administradas por sonda oral a ratas algodoneras con el estómago neutralizado para aproximar la administración de comprimidos orales en seres humanos. Se inmunizaron animales en las semanas 0 y 4 y los valores de anticuerpos se midieron en las semanas 4 y 8. Se usó un grupo de solo tampón como control, para mostrar los efectos de fondo de la falta de inmunización.
Los resultados muestran que tanto la dosis de 1x109 como la de 1x1010 podrían inducir valores de anticuerpos significativos contra RSV-F en la semana 4, con un aumento significativo en la semana 8 (Figura 24A). El grupo de la vacuna de 1x108 UI mostró una tendencia más baja en términos de respuestas de anticuerpos IgG totales al RSV. La medición de la capacidad de VXA-RSV-f oral para inducir anticuerpos que podrían competir por el sitio de unión de palivizumab mostró un efecto dependiente de la dosis con dosis más altas que muestran valores de competencia promedio más altos que la dosis más baja de 1x108 UI (Figura 24B). La inducción de anticuerpos neutralizantes del RSV aumentó con dosis más altas de vacuna, con dosis de vacuna VXA-RSV-f de 1x109 y 1x1010 UI que provocan los valores de neutralización más altos (Figura 24C).
Después de la inmunización en el primer experimento con ratas algodoneras (XV-95, Figura 23), las ratas algodoneras recibieron la cepa A2 del RSV de tipo silvestre a 1x105 UFP en el día 56. Los pulmones y las fosas nasales se recolectaron 5 días después y se analizaron para determinar la capacidad de las vacunas para proteger contra la replicación y la enfermedad del RSV en tejido de rata algodonera. La inmunización con la vacuna VXA-RSV-f proporcionó una protección completa contra la replicación del RSV tanto en los pulmones como en la nariz, mientras que los grupos de vacuna inactivada con formalina (FI-RSV) y adyuvante solo (Ad-Adj) no proporcionaron protección contra la replicación del RSV en la nariz y solo una protección limitada en los pulmones con la vacuna FI-RSv (Figura 25A). La replicación máxima (como se ve en el grupo de control de tampón) tuvo un promedio de 4,9 logi0 UFP del RSV/g de tejido pulmonar y la inmunidad inducida por la vacuna fue capaz de reducir los valores del RSV posteriores a la exposición por debajo del nivel detectable, una disminución mayor a 3 logi0.
La inflamación pulmonar se midió mediante inmunohistología y análisis de qRT-PCR el día 5 después de la infección por RSV. Se evaluaron cuatro regiones diferentes mediante inmunohistología para determinar si la vacuna conducía a una mejora inmunitaria adaptativa de la enfermedad. La vacuna VXA-RSV-f no indujo un aumento significativo en las puntuaciones de patología pulmonar para peribroncolitis (PB), perivasculitis (PV), neumonía intersticial (IP, por sus
siglas en inglés) y alveolitis (A) en comparación con la vacuna FI-RSV, el control "positivo" para la inflamación pulmonar (Figura 25B), y no dio lugar a aumentos en la abundancia relativa de IL-4 o IL-13 (Figura 25C). Los grupos administrados por vía in tuvieron una tendencia más alta para PB y PV, incluyendo los grupos de control de tampón, RSV2 y adyuvante, pero no necesariamente para IP y A (Figura 25B). El grupo adyuvante (sin expresión de la proteína F de RSV) indujo un alto nivel de PB después de la exposición al RSV, pero indujo solo niveles bajos de IP y A. Los grupos de control con tampón y adyuvante solo no indujeron un aumento significativo en la abundancia relativa de ARNm de IL-4 o IL-13 como lo hizo el grupo de FI-RSV (Figura 25B). El grupo de vacuna FI-RSV indujo una abundancia relativa promedio superior a 1 para IL-4 y superior a 3 para IL-13, en comparación con menos de 0,1 y 0,3 respectivamente para los grupos de vacunas VXA-RSV -f y RSV2 (Figura 25C).
El estudio de inmunogenicidad oral en ratas algodoneras (Estudio N.° XV-112) se expuso con RSV de tipo silvestre debido a los potentes valores de anticuerpos neutralizantes observados. Después de la inmunización, a las ratas de algodón se les dio la cepa A2 del RSV de tipo silvestre a 1x105 UFP en el día 56. Los pulmones y las fosas nasales se recolectaron 5 días después y se analizaron para determinar la capacidad de las vacunas para proteger contra la replicación y la enfermedad del RSV en tejido de rata algodonera. La inmunización oral con la vacuna VXA-RSV-f indujo una protección dependiente de la dosis contra la replicación del RSV tanto en los pulmones como en la nariz, con la dosis más alta de la vacuna que induce una protección completa en los pulmones y una protección casi completa en la nariz; 8 de 8 animales fueron negativos para valores de RSV en los pulmones y 7 de 8 animales fueron negativos para la replicación de RSV en la nariz (Figura 26A). La replicación máxima (como se ve en el grupo de control de tampón) tuvo un promedio de 5,2log10 UFP de RSV/g de tejido pulmonar, y la inmunidad inducida por la vacuna fue capaz de reducir los valores de RSV posteriores a la exposición superiores a 3 log10 para los grupos de vacunas de 1x109 y 1x1010 UI (Figura 26A). El grupo de dosis más baja (1x108 UI) también demostró una protección sustancial, con 6 de 8 animales que tienen protección completa. La inflamación se evaluó como antes. Los grupos de vacuna mostraron una mayor tendencia a la inflamación en comparación con el grupo de control de tampón, pero no fueron estadísticamente diferentes (Figura 26B). El análisis de citocinas de los pulmones después de la exposición no vio un aumento sustancial en la abundancia relativa del ARNm de IL-4 o IL-13 medido mediante qRT-PCR. Todas las vacunas y los grupos de control de tampón tenían niveles de abundancia relativa de ARNm de IL-4 e IL-13 por debajo de 0,04 y 0,005, respectivamente (Figura 26C). Estos valores son extremadamente bajos comparados con los niveles de abundancia relativa de 1 y 3 para IL-4 e IL-13 respectivamente inducidos por el grupo FI-RSV en el Estudio N.° XV-95 (Figura 25C), lo que sugiere que no se ha producido una inducción significativa de estas citocinas después de la administración oral de VXA-RSV-f.
Ejemplo 9
Anticuerpos neutralizantes de alto valor contra la gripe después de la inmunización con comprimidos orales: Un ensayo aleatorizado controlado con placebo
Introducción
La vacunación contra la gripe estacional requiere campañas anuales sustanciales para recolectar suficientes huevos fecundados y maquinaria masiva para recolectar y procesar cada mini biorreactor de huevos. La hemaglutinina (HA) procedente de cultivo celular o plantas pueden reducir la carga de la adquisición y el procesamiento de huevos, pero estos enfoques aún requieren un llenado y un acabado estériles costosos para producir agujas de jeringa individuales, que deben eliminarse como peligro biológico. Durante una pandemia, las escuelas pueden cerrarse y el distanciamiento social es obligatorio, sin embargo, las campañas masivas de inmunización contra la gripe normalmente requieren alinear a los sujetos en las clínicas para recibir inyecciones. Para sortear este dilema, las vacunas orales contra la gripe podrían enviarse por correo, evitando así el contacto entre seres humanos. La entrega por correo ya se usa para una amplia variedad de medicamentos orales y ya se ha sugerido como un medio para entregar medicamentos críticos a los veteranos durante una pandemia. Adicionalmente, la formación de comprimidos es rápida, proceso sanitario que no requiere el costoso proceso de llenado y acabado estéril que requieren las vacunas inyectadas.
Se han intentado varios enfoques de vectores adenovíricos para permitir la inmunización oral contra la gripe. En 2011, se inició un ensayo clínico en el que se utilizó una vacuna oral contra la gripe aviar (H5) vectorizada por adenovirus producida en cultivo celular. Las respuestas de linfocitos T a HA de la gripe H5 se midieron en más de un 75 % de los sujetos, pero no se observaron respuestas de anticuerpos neutralizantes (Peters et al. Vaccine 2013; 31: 1752-8). Después de que se realizó el desarrollo adicional de la formulación y la optimización de la dosis, el ensayo clínico objeto de este informe se inició para la gripe estacional, utilizando un formato de administración en comprimidos en lugar de una cápsula. Se utilizó la misma cadena principal del vector que antes, pero con una nueva secuencia de HA de una cepa similar a la cepa H1N1 de la vacuna comercial actual contra la gripe estacional (A/California/04/2009 (H1N1)) y con una dosis 10 veces mayor. Se probó una dosis única de rAd-HA(A/CA/04/2009) -ARNbc para determinar su seguridad e inmunogenicidad, en un estudio clínico controlado, aleatorizado y doble ciego. Este informe resume los hallazgos de este ensayo.
Materiales y métodos
Protocolo clínico e inscripción
Este fue un estudio de fase 1, inscrito secuencialmente, con una cohorte aleatorizada y controlada con placebo para evaluar la seguridad y la inmunogenicidad de una vacuna oral basada en Ad serotipo 5 recombinante (rAd5) contra la gripe estacional H1. El estudio se realizó de acuerdo con las directrices aplicables de Good Clinical Practice, el United States Code of Federal Regulations, y las directrices del International Conference on Harmonization. La aprobación del IRB se obtuvo de Aspire IRB (Santee, California; AAHRPP acreditado) antes de la inscripción de los sujetos. Los participantes del estudio fueron reclutados utilizando la base de datos de voluntarios existente de la Unidad de CRO/Fase 1, así como también utilizando publicidad aprobada por el IRB (anuncios impresos, anuncios de radio y redes sociales). Se obtuvo el consentimiento informado de todos los sujetos después de discutir los procedimientos del estudio y los posibles riesgos.
Los sujetos se evaluaron previamente para valores de inhibición de hemaglutinación (HAI) dentro de los 45 días posteriores a la inscripción. Para poder participar en el estudio, los sujetos tenían que tener un valor de HAI inicial de < 1:20, tener entre 18 y 49 años y gozar de buena salud. Los criterios de inscripción adicionales se enumeran en clinicaltrials.gov en NCT01 688297. La fase activa del ensayo fue hasta el día 28, con la fase de seguimiento para el control de la seguridad para continuar durante 1 año.
Aleatorización y enmascaramiento
El estudio se diseñó para evaluar la vacuna (VXA-A1-1) en 12 sujetos en una dosis única de 1 x 1011 unidades infecciosas (UI) con 12 sujetos que recibieron un control de placebo. Hubo 3 sujetos tratados con vacuna centinela inscritos secuencialmente, con cada sujeto dosificado con una frecuencia no mayor a una cada 24 h. Después de una semana de control de las toxicidades relacionadas con la vacuna, los sujetos restantes en la cohorte tratada (9) fueron asignados al azar junto con 12 controles de placebo. La aleatorización se realizó mediante asignación generada por computadora, y el farmacéutico no ciego distribuyó el fármaco del estudio con identidad oculta al personal ciego. Todo el personal del centro de investigación, así como las personas directamente implicadas con los ensayos inmunológicos o la evaluación de la seguridad clínica, permanecieron ciegos a las asignaciones de tratamiento. Todos los sujetos estaban ciegos en el estudio.
Tamaño de la muestra
Se predijo que el número total de voluntarios por grupo de prueba (n = 12) produciría resultados significativos. Esto se definió en un estudio anterior como la observación de un 50 % de pacientes que responde al tratamiento en el grupo de vacuna y ninguno en el grupo de placebo. Con un tamaño de muestra de 12 por grupo, hay un 80 % de potencia para detectar una diferencia de grupo, asumiendo que la proporción de respuesta (definida como HAI > 40) en el grupo de vacuna es de un 50 % y en el placebo es 0, utilizando la prueba bilateral exacta de Fisher de dos grupos a un nivel de significancia de 0,05.
Vacuna
El vector rAd (Ad5 no replicante) transporta ADN que codifica el transgén HA (A/CA/04/2009) cuya expresión es impulsada por un promotor de CMV y una horquilla de ARNbc molecular impulsada por un promotor separado, como se describió anteriormente (Scallan et al. Clinical and Vaccine Immunology 2013; 20(1): 85-94). La sustancia farmacéutica GMP se produjo en biorreactores desechables Wave (GE Healthcare, Waukesha, WI) en Lonza Biologicals (Houston, TX). La purificación se realizó mediante cromatografía de intercambio iónico, seguido de intercambio de tampón. El vector purificado se mezcló con excipientes, liofilizados y luego se formaron comprimidos en Lonza utilizando celulosa microcristalina y almidón como volumen de comprimidos. Los comprimidos se recubrieron entéricamente con Eudragit L100® (Evonik Industries, Darmstadt, Alemania) utilizando un sistema Vector Hi-Coater (Vector Freund, Cedar Rapids, IA). El producto final se liberó en un lote y se valoró mediante un ensayo estándar de UI en Lonza. El placebo se preparó como comprimidos de tamaño y forma similares que contenían 150 mg de celulosa microcristalina, sin recubrimiento entérico.
Evaluaciones de seguridad
El investigador principal (IP) evaluó los acontecimientos adversos (EA, por sus siglas en inglés) solicitados y no solicitados de manera cegada. El SMC supervisó la seguridad del estudio pero no participó en la clasificación de EA. Los EA solicitados (reactogenicidad) se recopilaron con la ayuda de una tarjeta de diario de síntomas solicitados de 7 días. Los EA no solicitados (todos los demás EA clínicos) se recopilaron con la ayuda de una tarjeta de diario de no solicitados hasta el día 28. Los investigadores utilizaron la Center for Biologies Evaluation and Research (CBER) Guidance for Industry: Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Trials, Septiembre de 2007 para calificar EA.
Debido al nuevo adyuvante, se recopilaron datos sobre la aparición de EA de especial interés (AESI, por sus siglas en inglés) y de enfermedades crónicas de nueva aparición (NOCI, por sus siglas en inglés). Estas incluyen trastornos neuroinflamatorios, trastornos musculoesqueléticos, trastornos gastrointestinales, enfermedades metabólicas,
trastornos de la piel y otros trastornos autoinmunitarios. No se han notificado AESI ni NOCI hasta el día 180 después de la inmunización.
Criterios de valoración
El criterio de valoración principal de este estudio es la seguridad y el criterio de valoración secundario es la inmunogenicidad a través de la fase activa, principalmente mediante valores de HAI y seroconversiones de HAI. Los criterios de valoración inmunológicos adicionales incluyen valores de MN y ASC.
Aislamiento y crioconservación de PBMC
Se recogió sangre en tubos K3 EDTA Vacutainer® (BD, Franklin Lakes, NJ) y se aislaron PBMC el mismo día usando tubos Lymphoprep™ (Axis-Shield, Noruega).
Se congelaron y descongelaron PBMC utilizando reactivos sin suero de acuerdo con las instrucciones del fabricante (Cellular Technology Ltd [CTL], Shaker Heights, OH).
Células secretoras de anticuerpos (ASC)
Los kits de inmunoabsorción ligado a enzimas (ELISpot) para linfocitos B secretores de IgG e IgA se realizaron de acuerdo con las instrucciones del fabricante (Mabtech, Mariemont, OH). Se cultivaron células (entre 15 X 104 hasta 5 X 105 células por pocillo) en pocillos triplicados, en medio CTL-Test para optimizar las manchas. La proteína HA (Protein Sciences Corp, Meriden, CT) se biotiniló y cuantificó de Vaxart usando un kit de biotinilación (Pierce, Rockford, IL). Se contaron manchas en Zellnet Consulting Inc (Fort Lee, Nueva Jersey).
Ensayos de anticuerpos
Los valores de HAI y MN se realizaron mediante Focus Diagnostics (Cypress, CA) de manera similar a como se describió anteriormente (Greenberg et al. The New England Journal of Medicine 2009; 361(25): 2405-13). Se midieron HAI y MN contra A/CA/07/2009 procedente de MDCK y A/CA/07/2009 procedente de huevo respectivamente. Los valores de HAI y MN inferiores a 10 se marcaron como 5 según lo sugerido por el asesoramiento reglamentario. Los valores de neutralización de adenovirus se midieron como se describe anteriormente 2.
Análisis estadístico
En general, la estadística descriptiva para las variables continuas incluyó el número de sujetos con datos a resumir (n), media, error estándar (error est.) e intervalos de confianza del 95 % (IC 95). Los valores se informaron con medias geométricas e IC 95. Las variables categóricas se presentaron mediante recuentos de frecuencia y porcentajes. Las diferencias de los grupos de tratamiento se compararon mediante la prueba t de dos grupos en las variables continuas y la prueba exacta de Fisher en las variables categóricas. Todas las pruebas estadísticas fueron de dos caras a un nivel de significancia de 0,05 sin ajustar por multiplicidad. Se utilizó un modelo de análisis de covarianza (ANCOVA) para los valores de anticuerpos de HAI (transformados logarítmicamente), con el valor logarítmico del día 28 como variable dependiente, el tratamiento como factor y el valor logarítmico del día 0 como covariable. Las medias de mínimos cuadrados (LS), el IC 95 de las medias LS, la diferencia de las medias LS y el IC 95 de la diferencia de las medias LS se obtuvieron del modelo. Como análisis exploratorio para el valor de anticuerpos de HAI, también se realizó otro modelo ANCOVA que incluyó la edad, el sexo y el índice de masa corporal (IMC) como covariables.
Resultados
Demografía
Se seleccionaron 365 sujetos y se inscribieron 24 sujetos. Todos los sujetos que se inscribieron completaron evaluaciones de seguridad e inmunogenicidad durante la fase activa y hasta el día 180 de la fase de control (Figura 19). Los datos demográficos se describen en la Tabla 8 tanto para los sujetos tratados con placebo como con la vacuna.
Sumario de acontecimientos adversos
En los primeros 7 días posteriores a la administración del artículo de prueba, se informaron 8 acontecimientos adversos (AE) solicitados en total en los grupos de vacuna VXA-A11 y placebo (Tabla 9). Todos estos EA fueron de grado 1 en intensidad. También se indica la evaluación del investigador sobre si el EA estaba relacionado con el tratamiento (Tabla 9). El EA más frecuente fue dolor de cabeza (2 en el grupo placebo y 1 en el grupo de vacuna). Todos los demás EA solicitados fueron acontecimientos únicos (Tabla 9). Hubo un total de 8 EA no solicitados en los grupos de vacuna y placebo en los 28 días posteriores a la inmunización, con 3 acontecimientos ocurridos en el grupo de placebo y 5 acontecimientos ocurridos en el grupo de vacuna. No se informaron acontecimientos adversos graves en el estudio.
Las anomalías de laboratorio clínico se distribuyeron entre los grupos de vacuna y placebo. Cabe destacar que, hubo 6 acontecimientos neutropénicos en el grupo de vacuna y 4 en el grupo de placebo. Estos acontecimientos ocurrieron en un total de 8 sujetos, 4 de los cuales tenían recuentos sanguíneos neutropénicos previos al tratamiento. Cinco de estos sujetos también eran negros o japoneses, que son grupos étnicos que tienen una frecuencia relativamente alta de neutropenia étnica benigna (BEN, por sus siglas en inglés). Como es el caso con (BEN), no hubo manifestaciones clínicas que resultaran de ninguno de los acontecimientos neutropénicos notificados.
Resultados de inmunogenicidad
Las respuestas de HAI se midieron los días 0 y 28 (Figura 20A). Ningún sujeto tratado con placebo seroconvirtió, pero un placebo tuvo un valor alto en el día 0 (que habría excluido al sujeto si se hubiera medido en la selección). Ninguno de los sujetos vacunados tenía un valor de HAI inicial > 20. Después de la inmunización, nueve sujetos en el grupo de vacuna alcanzaron niveles seroprotectores (HAI > 40) (Figura 20A). De los once que aumentaron 4 veces (92 %), nueve seroconvertidos (SC) y los otros 2 sujetos mostraron un aumento de 4 veces en el valor de HAI de 5 a 20. El grupo de vacuna tuvo un aumento estadísticamente significativo en el número de los que respondieron 4 veces frente al placebo (11 frente a 0, con P < 00000 según la prueba exacta de Fisher). Utilizando un modelo ANCOVA que representa el valor logarítmico del día 0 como covariable, se calculó que el valor de la media (LS) geométrica (GMT) para el grupo de vacuna era 715 (IC 95: 45-114) el día 28, un aumento de veces de la media (LS) geométrica (GMFR) de 77 veces sobre el GMT inicial de 79 (IC 95: 46 - 136) el día 0. El GMT del día 28 para el grupo de placebo fue 101 (IC 95: 64 - 162) el día 28, un GMFR de 1,1 veces superior al GMT inicial de 110 (IC 95: 64 - 189) el día 0. En comparación con el placebo, el grupo de vacuna tuvo un aumento estadísticamente significativo en GMT el día 28 (valor p < 0,001). El efecto de la covariable al inicio también fue estadísticamente significativo (valor p < 0,001). También se llevó a cabo un análisis exploratorio utilizando otro modelo ANCOVA, donde se incluyeron covariables adicionales, edad, sexo e IMC. Los efectos de estas covariables no fueron estadísticamente significativos el día 28 [valores p: 0,993 (para edad), 0,696 (por sexo), 0,201 (para IMC)].
La durabilidad de la respuesta de anticuerpos se midió mediante el examen de las respuestas de HAI 180 días después de la inmunización. En el grupo inmunizado con vacuna, un 75 % (9 de 12) de los sujetos estaban seroprotegidos el día 28 y un 75 % (9 de 12) todavía estaban seroprotegidos el día 180. Se representaron gráficamente los GMT de la HAI (Figura 20B), y se encontró que la disminución del GMT era de un 29 % entre los días 28 y 180 después de la inmunización.
Las respuestas de anticuerpos neutralizantes a la gripe se midieron mediante ensayo de MN. Se observaron aumentos significativos en los valores de MN en el grupo tratado frente al control con placebo (Figura 20C). La frecuencia de los que responden 4 veces de MN en el grupo tratado con la vacuna fue significativamente diferente a la del grupo placebo, con 11 sujetos que respondieron en el grupo tratado con la vacuna frente a 0 en el grupo de placebo (P < 00000 según la prueba exacta de Fisher).
Después de eliminar a los sujetos que tenían valores de MN iniciales (y valores de HAI) superiores a 40, los valores de la media geométrica (GMT) se calcularon en los sujetos restantes los días 0 y 28 (Tabla 10). El GMT para el grupo de vacuna aumentó a 247 (IC 95: 89-685) frente a ningún aumento en el placebo durante un GMT el día 28 de 96 (IC 95: 5-18). Estos cálculos no tuvieron ningún impacto en el grupo de vacuna, ya que ninguno de los sujetos tenía valores iniciales altos de MN o HAI. Estos resultados muestran que los valores de anticuerpos neutralizantes de la gripe se generan mediante inmunización oral, con un aumento de más de 20 veces en el GMT después de la inmunización en el grupo tratado con la vacuna.
Para medir las respuestas totales de anticuerpos a HA, el número de linfocitos B preplasmáticos circulantes en sangre periférica se midió mediante ensayo de ASC los días 0 y 7 después de la inmunización. Los resultados muestran que las ASC se pudieron medir de forma fiable el día 7 en el grupo tratado con la vacuna (Figura 20D). Las ASC de fondo fueron generalmente insignificantes el día 0. Para el grupo tratado con la vacuna, un promedio de 992 (+/- error est.
209, IC 95: 532-1452) ASC IgG y 337 ASC IgA (+/- error est. 104, IC 95: 117-580) cada uno por 1 x 106 PBMC se encontraron para el día 7, con solo un sujeto de 12 que no tiene respuesta de ASC detectable. El grupo de placebo no tenía manchas de IgA el día 7, pero un sujeto tenía un frotis de fondo alto y una respuesta de ASC IgG medible con manchas más pequeñas que las observadas normalmente. El grupo tratado fue significativamente diferente del placebo en términos de la capacidad de provocar una respuesta de ASC IgG o IgA en el día 7 (P = 00007 y P = 0,008 respectivamente mediante la prueba T).
Se midió en los sujetos sus valores de antivector antes y después de la inmunización. Después de la inmunización oral, algunos sujetos tratados con la vacuna tuvieron un aumento en las respuestas de anticuerpos neutralizantes a Ad5, lo que llevó a un aumento de 26 veces en los valores de la GM de anticuerpos neutralizantes, en comparación con el aumento de veces de la GM de 1-0 veces en los sujetos tratados con placebo. En el grupo de vacuna, las respuestas de HAI y MN mostraron una tendencia similar para los sujetos individuales. Ocho sujetos fueron negativos para Ad5 antes de la inmunización y cuatro fueron positivos para Ad5 antes de la inmunización. Un sujeto que fue positivo para Ad5 no seroconvirtió HAI, sin embargo, un sujeto que fue positivo para Ad5 tuvo el mayor aumento en los valores de HAI (64 veces) de cualquiera de los sujetos del estudio (Figura 21B). Este mismo sujeto tuvo una ganancia en los valores de MN de 362 veces (Figura 21A) sin ningún aumento en los valores de anticuerpos
neutralizantes de Ad5 antes y después de la inmunización. No se observó una correlación entre los valores de Ad5 iniciales frente al número de veces de respuesta MN (o respuesta HAI) para los sujetos inmunizados con la vacuna en comprimidos (Figura 21A y 21B).
Análisis
Cuando el ejército de Estados Unidos realizó un estudio independiente para medir los efectos de sus campañas de vacunación estacional sobre las respuestas de anticuerpos neutralizantes en el personal militar, informaron un GMFR del valor de MN de 56 después de la inyección de vacuna trivalente inactivada (TIV) y un GMFR de 22 después de la administración intranasal de vacuna viva atenuada contra la gripe (LAIV) después de tener en cuenta los sujetos que tenían valores de MN superiores a 40 al inicio (Faix et al. PloS one 2012; 7(4): e34581). En este estudio, el GMFR de MN se calculó en 29 para los 12 sujetos tratados con la vacuna (Tabla 10), con un 92 % de los sujetos que muestran un aumento de más de 4 veces en los valores de MN. En el estudio de Gordon, et al, se encontró que la tasa de SC para H1N1 era de un 45 % para una inyección de 45 |jg de proteína HA (sin adyuvante) (Gordon et al. Vaccine 2012; 30(36): 5407-16). Esto contrasta con los resultados publicados por Greenberg, et al, (mencionado anteriormente) donde la vacuna H1N1 fue altamente inmunogénica y se observó una tasa de SC de un 78 % después de 1 dosis de una vacuna dividida. En nuestro estudio de comprimidos, la tasa de SC HAI entre los sujetos tratados con vacuna fue de un 75 % y más de un 92 % de los sujetos tuvieron un aumento de 4 veces en los valores de HAI (Figura 20A). No está claro por qué los presentes valores de MN son mucho más altos que los valores de HAI. Es posible que el ensayo de MN sea simplemente más sensible o que la vacuna oral basada en rAd provoque respuestas neutralizantes más fuertes fuera de la región de la cabeza que las vacunas inyectadas con proteínas. En cualquier caso, los presentes resultados de etapa inicial sugieren que una vacuna en comprimidos orales sería competitiva con las vacunas existentes en términos de provocar respuestas de anticuerpos neutralizantes a la gripe.
Los individuos con inmunidad preexistente a la gripe H1N1 fueron excluidos de la participación en el estudio. Esto fue útil en el análisis de las respuestas inmunitarias en este estudio de fase 1 para comprender mejor los efectos de la vacuna. En la práctica, en el "mundo real", los individuos con y sin inmunidad preexistente a la gripe están inmunizados. La inscripción en los estudios de Fase 2 y 3 incluirán individuos con y sin niveles de anticuerpos detectables contra el antígeno de la vacuna al inicio del estudio.
Las respuestas de HAI se provocan con vacunas comerciales inyectadas, pero se sabe que los valores de HAI disminuyen. En un estudio de Crum-Cianflone, et al, voluntarios no infectados por VIH tuvieron una caída de un 67 % en los valores GMT de HAI entre 1 y 6 meses después de la inmunización (Crum-Cianflone et al. Vaccine 2011; 29(17): 3183-91). De forma similar, el porcentaje de sujetos seroprotegidos se redujo de un 75 % a un 56 % para los sujetos VIH negativos que se inscribieron con valores de HAI seronegativos (< 1:10)10. Los estudios con vacunas contra la gripe pandémica también han demostrado una disminución en la durabilidad. En el estudio de la vacuna contra la gripe aviar AS03 realizado por Leroux-Roels, et al, el GMT alcanzó 563 después de 2 dosis de vacuna, pero a los 6 meses después de la inmunización, el GMT había bajado a 18, una disminución de un 96 % (Leroux-Roels et al. Vaccine 2010; 28(3): 849-57). En el presente estudio de vacuna en comprimidos que reclutó sujetos seronegativos (todos los sujetos < 1:20), el porcentaje de sujetos seroprotegidos permaneció constante en un 75 % en 1 y 6 meses después de la inmunización, y la caída del valor GMT de HAI fue menos notable mostrando solo una disminución de un 29 % (Figura 20B). Aunque no ha sido probado, una posibilidad es que la durabilidad sea mejor para las vacunas basadas en vectores debido a las respuestas mejoradas de linfocitos T. Estos datos preliminares son al menos alentadores de que la vacuna en comprimidos puede proporcionar durabilidad de anticuerpos.
El número de acontecimientos adversos notificados clínicamente fue similar a los observados antes para otras vacunas de la invención basadas en vectores adenovíricos. En este estudio, se informaron 17 acontecimientos adversos clínicos hasta el día 28 después de la inmunización y se distribuyen de manera razonablemente uniforme entre los grupos de placebo y tratados. En un estudio publicado de una vacuna de adenovirus recombinante EBOV inyectada en seres humanos, la frecuencia de cualquier acontecimiento adverso fue de un 55 % entre los receptores de la vacuna y de un 25 % entre los receptores de placebo, siendo el acontecimiento adverso más común los dolores de cabeza (55 %), mialgia (46 %) y escalofríos (27 %) en el grupo de vacuna de dosis alta (Ledgerwood et al, Vaccine 2010; 29(2): 304-13). El acontecimiento adverso informado con más frecuencia en el presente estudio de comprimidos de vacuna fue dolor de cabeza (informado en 1 sujeto con vacuna y 2 sujetos con placebo).
Aunque la inmunidad al Ad5 puede ser un problema con las vacunas inyectadas, puede que no sea el caso de la inmunización oral con un vector no replicante donde los valores de anticuerpos neutralizantes no parecen obstaculizar el rendimiento (Xiang et al. Journal of virology 2003; 77(20): 10780-9). La capacidad de provocar una respuesta inmunitaria neutralizante a la gripe no pareció verse afectada después de la inmunización oral con el comprimido de vacuna (Figura 21). Uno de los sujetos con el valor de anti-Ad5 más alto al comienzo tuvo el mayor aumento medido en respuestas de anticuerpos neutralizantes mediante el ensayo de MN y HAI (Figura 21). Si bien la inmunización im puede causar seroconversión de Ad5 de un 100 % y que los GMT aumenten más de 50 veces (estimado a partir de la Fig2b de O'Brien et al. Nat Med 2009; 15(8): 873-5), el aumento en los valores de neutralización con el comprimido de 1 x 1011 UI fue mucho más modesto. La inmunización oral parece conducir a un aumento mucho más selectivo de la respuesta inmunitaria al transgén en comparación con el vector con un aumento del valor GMT de MN de 29 en comparación con el aumento del valor de Ad5 de 26. En contraste con los resultados obtenidos con la vacuna basada
Claims (15)
1. Una composición inmunogénica para provocar una respuesta inmunitaria en un ser humano que comprende un agente biológico inmunogénico abarcado por un recubrimiento entérico que dirige la administración del agente biológico inmunogénico al íleon del ser humano,
en donde el agente biológico inmunogénico es un vector adenovírico que codifica la proteína vírica 1 de norovirus o la proteína de fusión (F) del virus respiratorio sincitial (RSV), en donde el recubrimiento entérico tiene un pH umbral de 5,8-6,8.
2. La composición inmunogénica de la reivindicación 1, en donde el recubrimiento entérico se desintegra al menos un 75 % en comparación con su espesor original en 110 minutos a pH 5,8-6,8.
3. La composición inmunogénica de la reivindicación 1 o 2, en donde el recubrimiento entérico comprende poli(ácido metacrílico-co-metacrilato de metilo) 1:1.
4. La composición inmunogénica de la reivindicación 1 o 2, en donde el recubrimiento entérico comprende una mezcla de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 y poli(ácido metacrílico-co-acrilato de etilo) 1:1.
5. La composición inmunogénica de la reivindicación 1 o 2, en donde el recubrimiento entérico comprende:
i) poli(ácido metacrílico-co-metacrilato de metilo) 1:1, citrato de trietilo y talco;
ii) poli(ácido metacrílico-co-metacrilato de metilo) 1:1 y poli(ácido metacrílico-co-metacrilato de metilo) 1:2; o iii) una mezcla de poli(ácido metacrílico-co-metacrilato de metilo) 1:2 y poli(ácido metacrílico-co-acrilato de etilo) 1:1.
6. La composición inmunogénica de la reivindicación 5(i), que comprende 1-4 partes de poli(ácido metacrílico-cometacrilato de metilo) 1:1, 1-2 partes de citrato de trietilo y 1-2 partes de talco.
7. La composición inmunogénica de la reivindicación 4, en donde la relación de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 a poli(ácido metacrílico-co-acrilato de etilo) 1:1 es de 1:4 a 4:1.
8. La composición inmunogénica de la reivindicación 5(ii), en donde la relación de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 a poli(ácido metacrílico-co-acrilato de metilo) 1:2 es de 1:2 a 2:1.
9. La composición inmunogénica de la reivindicación 5(ii) u 8, en donde el recubrimiento entérico comprende de 1 a 4 partes de poli(ácido metacrílico-co-metacrilato de metilo) 1:1 y poli(ácido metacrílico-co-acrilato de metilo) 1:2; de 1 a 2 partes de citrato de trietilo; y de 1 a 2 partes de talco.
10. La composición inmunogénica de la reivindicación 5(iii), en donde la relación de poli(ácido metacrílico-cometacrilato de metilo) 1:2 a poli(ácido metacrílico-co-acrilato de etilo) 1:1 es de 1:4 a 4:1.
11. La composición inmunogénica de la reivindicación 5(iii) o 10, en donde el recubrimiento entérico comprende de 1 a 4 partes de poli(ácido metacrílico-co-metacrilato de metilo) 1:2 y poli(ácido metacrílico-co-acrilato de etilo) 1:2; de 1 a 2 partes de citrato de trietilo; y de 1 a 2 partes de talco.
12. La composición inmunogénica de una cualquiera de las reivindicaciones 1-11, en donde el vector adenovírico codifica además ARNbc.
13. La composición inmunogénica de una cualquiera de las reivindicaciones anteriores, en donde la composición está en forma de comprimido.
14. La composición inmunogénica de una cualquiera de las reivindicaciones anteriores para usar en un método para administrar la composición inmunogénica al íleon de un ser humano que comprende la administración oral de la composición inmunogénica al ser humano.
15. La composición inmunogénica de una cualquiera de las reivindicaciones 1-13 para usar en un método para provocar una respuesta inmunitaria en un ser humano que comprende administrar la composición al ser humano, en donde la respuesta inmunitaria es específica para el agente biológico inmunogénico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562175081P | 2015-06-12 | 2015-06-12 | |
| PCT/US2016/036461 WO2016200951A1 (en) | 2015-06-12 | 2016-06-08 | Formulations for small intestinal delivery of rsv and norovirus antigens |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2832504T3 true ES2832504T3 (es) | 2021-06-10 |
Family
ID=57503876
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16808196T Active ES2832504T3 (es) | 2015-06-12 | 2016-06-08 | Formulaciones para administración de antígenos de norovirus y rsv en el intestino delgado |
Country Status (12)
| Country | Link |
|---|---|
| US (3) | US11433146B2 (es) |
| EP (2) | EP3307239B1 (es) |
| JP (2) | JP6912394B2 (es) |
| KR (2) | KR102678401B1 (es) |
| CN (2) | CN108024955A (es) |
| AU (1) | AU2016274599B2 (es) |
| CA (1) | CA2988761A1 (es) |
| DK (1) | DK3307239T3 (es) |
| EA (1) | EA201792512A1 (es) |
| ES (1) | ES2832504T3 (es) |
| WO (1) | WO2016200951A1 (es) |
| ZA (1) | ZA201708434B (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EA201792512A1 (ru) | 2015-06-12 | 2018-06-29 | Вэксарт, Инк. | Составы для доставки антигенов респираторного синтициального вируса и норовируса в тонкий кишечник |
| CA3048313A1 (en) * | 2017-01-06 | 2018-07-12 | Stabilitech Biopharma Ltd | Virus |
| WO2019104439A1 (en) | 2017-11-30 | 2019-06-06 | Medicago Inc. | Modified norovirus vp1 proteins and vlps comprising modified norovirus vp1 proteins |
| CN108904795B (zh) * | 2018-08-29 | 2022-07-05 | 温氏食品集团股份有限公司 | 一种猪流行性腹泻病毒的口服疫苗的制备方法 |
| US20210311600A1 (en) * | 2020-04-06 | 2021-10-07 | Kirk David Bacon | Seat Selection Application For Social Distancing Compliance |
| WO2024193965A1 (en) * | 2023-03-17 | 2024-09-26 | Glaxosmithkline Biologicals Sa | Rsv-f-encoding nucleic acids |
Family Cites Families (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4152413A (en) | 1978-08-18 | 1979-05-01 | Chromalloy American Corporation | Oral vaccine for swine dysentery and method of use |
| US4520019A (en) | 1982-04-29 | 1985-05-28 | Ribi Immunochem Research, Inc. | Stable composition and preparation thereof |
| US4579945A (en) | 1982-04-29 | 1986-04-01 | Ribi Immunochem Research, Inc. | Purification of trehalose dimycolates |
| US4436727A (en) | 1982-05-26 | 1984-03-13 | Ribi Immunochem Research, Inc. | Refined detoxified endotoxin product |
| US4436728A (en) | 1982-05-26 | 1984-03-13 | Ribi Immunochem Research, Inc. | Refined detoxified endotoxin product |
| US4435386A (en) | 1982-05-26 | 1984-03-06 | Ribi Immunochem Research, Inc. | Refined detoxified endotoxin product |
| US4505900A (en) | 1982-05-26 | 1985-03-19 | Ribi Immunochem Research, Inc. | Refined detoxified endotoxin product |
| US4505899A (en) | 1982-05-26 | 1985-03-19 | Ribi Immunochem Research, Inc. | Refined detoxified endotoxin product |
| US4866034A (en) | 1982-05-26 | 1989-09-12 | Ribi Immunochem Research Inc. | Refined detoxified endotoxin |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US6024983A (en) | 1986-10-24 | 2000-02-15 | Southern Research Institute | Composition for delivering bioactive agents for immune response and its preparation |
| US5057540A (en) | 1987-05-29 | 1991-10-15 | Cambridge Biotech Corporation | Saponin adjuvant |
| CA1331443C (en) | 1987-05-29 | 1994-08-16 | Charlotte A. Kensil | Saponin adjuvant |
| US4897268A (en) | 1987-08-03 | 1990-01-30 | Southern Research Institute | Drug delivery system and method of making the same |
| DK0590060T3 (da) | 1991-06-21 | 1998-05-11 | Univ Cincinnati | Oralt indgivelige terapeutiske proteiner samt fremgangsmåde til fremstilling |
| DK0678034T3 (da) | 1993-01-11 | 1999-11-08 | Dana Farber Cancer Inst Inc | Induktion af cytotoksiske T-lymfocytreaktioner |
| US6224911B1 (en) | 1993-03-16 | 2001-05-01 | Syntex (U.S.A.) Llc | Process for the preparation of enteric coated pharmaceutical dosage forms |
| WO1995003035A1 (en) | 1993-07-23 | 1995-02-02 | Massachusetts Institute Of Technology | Polymerized liposomes with enhanced stability for oral delivery |
| GB9412394D0 (en) | 1994-06-21 | 1994-08-10 | Danbiosyst Uk | Colonic drug delivery composition |
| AUPM873294A0 (en) | 1994-10-12 | 1994-11-03 | Csl Limited | Saponin preparations and use thereof in iscoms |
| NZ295998A (en) | 1994-10-24 | 1999-10-28 | Ophidian Pharm Inc | Neutralizing antitoxins against clostridium difficile and clostidium botulinum toxins |
| UA56132C2 (uk) | 1995-04-25 | 2003-05-15 | Смітклайн Бічем Байолоджікалс С.А. | Композиція вакцини (варіанти), спосіб стабілізації qs21 відносно гідролізу (варіанти), спосіб приготування композиції вакцини |
| US6676935B2 (en) | 1995-06-27 | 2004-01-13 | Cell Genesys, Inc. | Tissue specific adenoviral vectors |
| AU4133397A (en) | 1996-10-28 | 1998-05-22 | Pfizer Inc. | Oral vaccines for young animals with an enteric coating |
| FR2759293B1 (fr) | 1997-02-11 | 1999-04-30 | Ethypharm Lab Prod Ethiques | Microgranules contenant du cisplatine, procede de fabrication, preparation pharmaceutique et utilisation en polychimiotherapie ou en association avec une radiotherapie |
| AUPO856097A0 (en) | 1997-08-14 | 1997-09-04 | Commonwealth Scientific And Industrial Research Organisation | Vector |
| US6328967B1 (en) | 1998-03-12 | 2001-12-11 | Allergenics, Inc. | Delivery system to modulate immune response |
| GB9818591D0 (en) | 1998-08-27 | 1998-10-21 | Danbiosyst Uk | Pharmaceutical composition |
| US7264771B2 (en) | 1999-04-20 | 2007-09-04 | Baxter International Inc. | Method and apparatus for manipulating pre-sterilized components in an active sterile field |
| US6355270B1 (en) | 1999-01-11 | 2002-03-12 | The Regents Of The University Of California | Particles for oral delivery of peptides and proteins |
| US20010026800A1 (en) | 2000-01-07 | 2001-10-04 | Michael Jacob G. | Selective activation of a TH1 or TH2 lymphocyte regulated immune response |
| AU2003240978A1 (en) | 2002-05-31 | 2003-12-19 | Genteric, Inc. | Multi-functional polyamines for delivery of biologically-active polynucleotides |
| AU2003262922A1 (en) | 2002-08-30 | 2004-03-19 | Genteric, Inc. | Retroductal salivary gland genetic vaccination |
| GB2407500A (en) | 2003-11-03 | 2005-05-04 | Ist Superiore Sanita | Use of microparticles for antigen delivery |
| US20060128653A1 (en) | 2004-12-10 | 2006-06-15 | Chunlin Tang | Pharmaceutical formulation of decitabine |
| TW200626185A (en) | 2004-12-17 | 2006-08-01 | Bpsi Holdings Inc | Enteric film coating composition containing entericpolymer micronized with detackifier |
| CN103122360A (zh) | 2006-02-28 | 2013-05-29 | 瓦克萨特公司 | 嵌合腺病毒载体 |
| DE102006060799A1 (de) | 2006-12-21 | 2008-06-26 | RUHR-UNIVERSITäT BOCHUM | RSV F-Protein und seine Verwendung |
| JP2009298707A (ja) | 2008-06-11 | 2009-12-24 | Ohara Yakuhin Kogyo Kk | 粒子加工方法 |
| US20110305768A1 (en) | 2008-07-01 | 2011-12-15 | The Johns Hopkins University | Quick-dissolving oral thin film for targeted delivery of therapeutic agents |
| US20100221329A1 (en) | 2008-12-03 | 2010-09-02 | Synergy Pharmaceuticals, Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| US20120276167A1 (en) | 2009-07-13 | 2012-11-01 | Vaxgene Corporation | Immunoprotection by oral administration of recombinant lactococcus lactis mini-capsules |
| WO2011026111A1 (en) * | 2009-08-31 | 2011-03-03 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Oral delivery of a vaccine to the large intestine to induce mucosal immunity |
| JP2013505022A (ja) | 2009-09-16 | 2013-02-14 | バクサート インコーポレーティッド | H1n1感染を予防するための免疫戦略の方法 |
| US20130273154A1 (en) | 2011-03-02 | 2013-10-17 | Joseph M. Fayad | Oral formulations Mimetic of Roux-en-Y gastric bypass actions on the ileal brake; Compositions, Methods of Treatment, Diagnostics and Systems for treatment of metabolic syndrome manifestations including insulin resistance, fatty liver disease, hpperlipidemia, and type 2 diabetes |
| KR20130010121A (ko) * | 2010-03-23 | 2013-01-25 | 인트렉손 코포레이션 | 치료적 단백질을 조건부로 발현하는 벡터,상기 벡터를 포함하는 숙주 세포 및 이의 용도 |
| AU2011276328C1 (en) | 2010-07-06 | 2016-01-21 | Novartis Ag | Norovirus derived immunogenic compositions and methods |
| CN103153289B (zh) | 2010-08-25 | 2016-08-31 | 格兰特·鲁弗斯·斯柏林二世 | 改进型肠溶活性物质给药 |
| EP2680859B1 (en) | 2011-03-02 | 2022-04-27 | Jerome Schentag | Compositions, methods of treatment and diagnostics for treatment of hepatic steatosis alone or in combination with a hepatitis c virus infection |
| CA3008794C (en) * | 2012-03-29 | 2021-03-16 | Therabiome, Llc | Gastrointestinal site-specific oral vaccination formulations active on the ileum and appendix |
| DK2833914T3 (en) | 2012-04-04 | 2019-03-11 | Vaxform Llc | ADMINISTRATIVE SYSTEM FOR ORAL VACCINE ADMINISTRATION |
| DK3107568T3 (da) | 2014-02-20 | 2024-06-03 | Vaxart Inc | Formuleringer til indgivelse til tyndtarmen |
| EA201792512A1 (ru) | 2015-06-12 | 2018-06-29 | Вэксарт, Инк. | Составы для доставки антигенов респираторного синтициального вируса и норовируса в тонкий кишечник |
-
2016
- 2016-06-08 EA EA201792512A patent/EA201792512A1/ru unknown
- 2016-06-08 KR KR1020187000421A patent/KR102678401B1/ko active Active
- 2016-06-08 WO PCT/US2016/036461 patent/WO2016200951A1/en not_active Ceased
- 2016-06-08 ES ES16808196T patent/ES2832504T3/es active Active
- 2016-06-08 CA CA2988761A patent/CA2988761A1/en active Pending
- 2016-06-08 KR KR1020247005811A patent/KR102773575B1/ko active Active
- 2016-06-08 CN CN201680046440.8A patent/CN108024955A/zh active Pending
- 2016-06-08 DK DK16808196.6T patent/DK3307239T3/da active
- 2016-06-08 CN CN202311287963.XA patent/CN118045170A/zh active Pending
- 2016-06-08 EP EP16808196.6A patent/EP3307239B1/en active Active
- 2016-06-08 US US15/580,651 patent/US11433146B2/en active Active
- 2016-06-08 EP EP20195995.4A patent/EP3791859A1/en active Pending
- 2016-06-08 AU AU2016274599A patent/AU2016274599B2/en active Active
- 2016-06-08 JP JP2017564358A patent/JP6912394B2/ja active Active
-
2017
- 2017-12-12 ZA ZA2017/08434A patent/ZA201708434B/en unknown
-
2021
- 2021-07-08 JP JP2021113391A patent/JP7269285B2/ja active Active
-
2022
- 2022-08-02 US US17/816,788 patent/US12186407B2/en active Active
-
2024
- 2024-11-22 US US18/956,413 patent/US20250082784A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| US20220378944A1 (en) | 2022-12-01 |
| US20180161458A1 (en) | 2018-06-14 |
| JP7269285B2 (ja) | 2023-05-08 |
| CA2988761A1 (en) | 2016-12-15 |
| KR102678401B1 (ko) | 2024-06-26 |
| US12186407B2 (en) | 2025-01-07 |
| AU2016274599A1 (en) | 2018-01-18 |
| WO2016200951A1 (en) | 2016-12-15 |
| CN118045170A (zh) | 2024-05-17 |
| EP3307239A1 (en) | 2018-04-18 |
| JP2018521030A (ja) | 2018-08-02 |
| EP3307239B1 (en) | 2020-09-16 |
| AU2016274599B2 (en) | 2021-09-02 |
| US11433146B2 (en) | 2022-09-06 |
| CN108024955A (zh) | 2018-05-11 |
| JP2021169480A (ja) | 2021-10-28 |
| US20250082784A1 (en) | 2025-03-13 |
| EP3791859A1 (en) | 2021-03-17 |
| ZA201708434B (en) | 2020-02-26 |
| DK3307239T3 (da) | 2020-11-23 |
| EA201792512A1 (ru) | 2018-06-29 |
| KR20180018657A (ko) | 2018-02-21 |
| NZ738382A (en) | 2025-02-28 |
| EP3307239A4 (en) | 2019-02-20 |
| KR20240031420A (ko) | 2024-03-07 |
| KR102773575B1 (ko) | 2025-02-27 |
| JP6912394B2 (ja) | 2021-08-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7269285B2 (ja) | Rsvおよびノロウイルス抗原の小腸送達のための製剤 | |
| US20250032602A1 (en) | Formulations for small intestinal delivery | |
| US20230210980A1 (en) | Chimeric adenoviral vectors | |
| US20240093234A1 (en) | Chimeric adenoviral vectors |