ES2852098T3 - Compuesto cíclico que actúa como inhibidor de PDE4 - Google Patents
Compuesto cíclico que actúa como inhibidor de PDE4 Download PDFInfo
- Publication number
- ES2852098T3 ES2852098T3 ES17842893T ES17842893T ES2852098T3 ES 2852098 T3 ES2852098 T3 ES 2852098T3 ES 17842893 T ES17842893 T ES 17842893T ES 17842893 T ES17842893 T ES 17842893T ES 2852098 T3 ES2852098 T3 ES 2852098T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- group
- mmol
- room temperature
- pmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCc(c*1)cc*1F Chemical compound CCc(c*1)cc*1F 0.000 description 5
- RBRRRALIDXHNKP-GOSISDBHSA-N CCOC(c(cc1)cc(N2)c1N([C@H](CS(C)(=O)=O)c(cc1OCC)ccc1OC)C2=O)=O Chemical compound CCOC(c(cc1)cc(N2)c1N([C@H](CS(C)(=O)=O)c(cc1OCC)ccc1OC)C2=O)=O RBRRRALIDXHNKP-GOSISDBHSA-N 0.000 description 1
- SKXZZDHJQLRXSJ-GOSISDBHSA-N CCOc(cc([C@@H](CS(C)(=O)=O)N(c(cccc1-[n]2nccc2)c1N1)C1=O)cc1)c1OC Chemical compound CCOc(cc([C@@H](CS(C)(=O)=O)N(c(cccc1-[n]2nccc2)c1N1)C1=O)cc1)c1OC SKXZZDHJQLRXSJ-GOSISDBHSA-N 0.000 description 1
- PNETWKSJIDVOJS-OAHLLOKOSA-N CCOc1cc([C@@H](CS(C)(=O)=O)N(c(c(F)cc(C(F)(F)F)c2)c2N2)C2=O)ccc1OC Chemical compound CCOc1cc([C@@H](CS(C)(=O)=O)N(c(c(F)cc(C(F)(F)F)c2)c2N2)C2=O)ccc1OC PNETWKSJIDVOJS-OAHLLOKOSA-N 0.000 description 1
- OOFHEXZJUUUYCV-MRXNPFEDSA-N CCOc1cc([C@@H](CS(C)(=O)=O)N(c(ccc(C(O)=O)c2)c2N2)C2=O)ccc1OC Chemical compound CCOc1cc([C@@H](CS(C)(=O)=O)N(c(ccc(C(O)=O)c2)c2N2)C2=O)ccc1OC OOFHEXZJUUUYCV-MRXNPFEDSA-N 0.000 description 1
- PMZDDYGZGWORPV-MRXNPFEDSA-N CCOc1cc([C@@H](CS(C)(=O)=O)N(c(ccc(Cl)c2)c2N2)C2=O)ccc1OC Chemical compound CCOc1cc([C@@H](CS(C)(=O)=O)N(c(ccc(Cl)c2)c2N2)C2=O)ccc1OC PMZDDYGZGWORPV-MRXNPFEDSA-N 0.000 description 1
- OORBDHOQLZRIQR-UHFFFAOYSA-N Cc(cc1)cc([N+]([O-])=O)c1F Chemical compound Cc(cc1)cc([N+]([O-])=O)c1F OORBDHOQLZRIQR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/26—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/423—Oxazoles condensed with carbocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/52—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings condensed with carbocyclic rings or ring systems
- C07D263/54—Benzoxazoles; Hydrogenated benzoxazoles
- C07D263/58—Benzoxazoles; Hydrogenated benzoxazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto como se muestra en la fórmula (I) o una sal farmacéuticamente aceptable del mismo, **(Ver fórmula)** en la que, X es O o N (R2); R2 es H, F, Cl, Br, I, OH, NH2 o R3-L1-, o seleccionado del grupo formado por alquilo C1-6, heteroalquilo C1-6, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; R3 se selecciona del grupo que consiste de cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; L1 se selecciona del grupo que consiste de -CH2-, -CH2CH2-, O, S, NH y -C(=O)-; n se selecciona del grupo que consiste en 1, 2 y 3; R1 es H, F, Cl, Br, I, OH, NH2, COOH, R4-L2-, o seleccionado del grupo que consiste de alquilo C1-6, heteroalquilo C1- 6, alquenilo C2-6, cicloalquenilo C3-6, heterocicloalquenilo de 3-6 miembros, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; R4 se selecciona del grupo que consiste de cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, heterocicloalquenilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; L2 se selecciona del grupo que consiste de -CH2-, -CH2CH2-, O, S, NH, -C(=O)NH-, -C(=O)O- y -C(=O)-; R es H, halógeno, OH, NH2, CN, o seleccionado del grupo que consiste de alquilo C1-6, heteroalquilo C1-6, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; R' se selecciona del grupo que consiste en H, F, Cl, Br, I, OH, CN, NH2, Me, Et, CF3, CHF2, CH2F, NHCH3, N(CH3)2; el "hetero" en el heteroalquilo C1-6, heterocicloalquilo de 3-6 miembros, heteroarilo de 5-6 miembros y heterocicloalquenilo de 3-6 miembros se selecciona de -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)2NH-, -S(=O) NH-, -O-, -S- , =O, =S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- y NHC(=O)NH-; en cualquiera de los casos anteriores, el número del heteroátomo o del grupo heteroátomo se selecciona independientemente entre 1, 2 o 3.
Description
DESCRIPCIÓN
Compuesto cíclico que actúa como inhibidor de PDE4
Campo de la invención
La presente invención se refiere a una clase de inhibidor de la fosfodiesterasa-4 (PDE4) y su uso en la fabricación de un medicamento para tratar una enfermedad relacionada con la PDE4. Específicamente relacionado está un compuesto como se muestra en la fórmula (I) y una sal farmacéuticamente aceptable del mismo.
Técnicas anteriores
Opinión de expertos sobre patentes terapéuticas, 2013, 997-1016 proporciona una revisión de los desarrollos actuales en los inhibidores de la fosfodiesterasa-4.
El factor de necrosis tumoral alfa (TNF-a) es una citocina liberada principalmente por fagocitos mononucleares en respuesta a inmunoestimuladores. El TNF-a es capaz de potenciar la mayoría de los procesos celulares, como la diferenciación, el reclutamiento, la proliferación y la degradación proteolítica. A niveles bajos, TNF-a confiere protección contra agentes infecciosos, tumores y daño tisular. Sin embargo, la liberación excesiva de TNF-a también causa enfermedades, cuando se administra a mamíferos o humanos, el TNF-a causa o agrava inflamación, fiebre, efectos cardiovasculares, hemorragia, coagulación y respuestas agudas similares a las que se observan durante infecciones agudas y estados de conmoción. La producción aumentada o descontrolada de TNF-a en animales o humanos a menudo indica una serie de enfermedades, por ejemplo, endotoxemia y/o síndrome de choque tóxico, caquexia, síndrome de dificultad respiratoria del adulto, cánceres como tumores sólidos y tumores hematológicos, enfermedades cardíacas tal como insuficiencia cardiaca congestiva; infección viral y enfermedades genéticas, inflamatorias, alérgicas o autoinmunes.
El cáncer es una enfermedad particularmente devastadora y el aumento de los niveles de TNF-a en sangre indica cáncer o riesgo de propagación del cáncer. Normalmente, en sujetos sanos, las células cancerosas no logran sobrevivir en el sistema circulatorio, una de las razones es que la pared interna de los vasos sanguíneos actúa como una barrera para la extravasación de células tumorales. Se demostró que ELAM-1 en células endoteliales medía la adhesión incrementada de células de cáncer de colon al endotelio tratado con citocinas.
El monofosfato de adenosina cíclico (AMPc) también desempeña un papel en muchas enfermedades y afecciones. Se ha demostrado que la elevación de AMPc en leucocitos inflamatorios inhibe su activación y luego libera mediadores inflamatorios, incluidos TNF-a y factor nuclear kB (NF-kB). Los niveles elevados de AMPc también conducen a la relajación del músculo liso de las vías respiratorias.
Se cree que el mecanismo celular primario para la inactivación de AMPc es la degradación de AMPc por una familia de isoenzimas denominadas fosfodiesterasas de nucleótidos cíclicos (PDE). Hay once miembros conocidos de la familia de las PDE. Hasta ahora se ha reconocido que la inhibición de la PDE tipo IV (PDE4) es particularmente eficaz tanto en la inhibición de la liberación de mediadores inflamatorios como en la relajación del músculo liso de las vías respiratorias. Por lo tanto, la PDE4 se ha convertido en uno de los objetivos farmacológicos más populares. La familia de PDE-4 se puede dividir en cuatro subtipos (PDE-4A, B, C, D) en función de diferentes códigos genéticos, en los que PDE-4A, PDE-4B y PDE-4D se expresan más ampliamente en células inflamatorias (tal como células B, células T y neutrófilos) que PDE-4C. La inhibición de PDE4 conduce a un aumento de los niveles de AMPc, regulando así los niveles de TNF-a con fines terapéuticos.
Contenido de la presente invención
La presente invención proporciona un compuesto como se muestra en la fórmula (I) y una sal farmacéuticamente aceptable del mismo,

en el que,
X es O N (R2);
R2 es H, F, Cl, Br, I, OH, NH2 , R3-L1-, o seleccionado del grupo que consiste de alquilo C1-6, heteroalquilo C1-6, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
R3 se selecciona del grupo que consiste de cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
L1 es -CH2-, -CH2CH2-, O, S, NH o -C(=O) -;
n es 1, 2 o 3;
R1 es H, F, Cl, Br, I, OH, NH2 , o COOH, R4-L2-, o seleccionado del grupo que consiste de alquilo C1-6, heteroalquilo C1-6 , alquenilo C2-6, cicloalquenilo C3-6, heterocicloalquenilo de 3-6 miembros, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; R4 se selecciona del grupo que consiste de cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, heterocicloalquenilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
L2 es -CH2-, -CH2CH2-, O, S, NH, -C(=O)NH-, -C(=O)O-, o -C(=O)-;
R es H, halógeno, OH, NH2 , CN, o seleccionado del grupo que consiste de alquilo C1-6, heteroalquilo C1-6, cicloalquilo C3-6, heterocicloalquilo de 3-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1 , 2 o 3 R;
R' es H, F, Cl, Br, I, OH, CN, NH2 , Me, Et, CF3 , CHF2 , CH2F, NHCH3 , o N(CH3)2 ;
El "hetero" en el heteroalquilo C1-6, heterocicloalquilo de 3-6 miembros, heteroarilo de 5-6 miembros y heterocicloalquenilo de 3-6 miembros se selecciona del grupo que consiste en -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)2NH-, -S(=O)NH-, -O-, -S-, =O, =S, -ON=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)2- y -NHC(=O)NH-;
En cualquiera de los casos anteriores, el número del heteroátomo o del grupo heteroátomo es independientemente 1, 2 o 3.
En algunas realizaciones de la presente invención, R es H, F, Cl, Br, I, OH, CN o NH2 , o seleccionado del grupo que consiste en alquilo C1-3, alcoxi C1-3, alquiltio C1-3, alquilamino C1- 3 , alquilo C1-4-OC(=O)-, N,N'-di(alquilo C1-3) amino, cicloalquilo C3-6, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
En algunas realizaciones de la presente invención, R es H, F, Cl, Br, I, OH, CN, NH2 , Me, CF3 , CHF2 , CH2F, Et,

En algunas realizaciones de la presente invención, R3 se selecciona del grupo que consiste en ciclopropilo, ciclobutilo, fenilo, piridilo, pirimidinilo, pirazinilo, tienilo, imidazolilo, pirazolilo, oxazolilo, tiazolilo, isoxazolilo, isotiazolilo, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R;
En algunas realizaciones de la presente invención, R3 es

En algunas realizaciones de la presente invención, R3-L1- es

En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, NH2 o R3-L1-, o seleccionado del grupo que consiste de alquilo C1-3, alquilo C1-3- -S(=O)2-C1-3 alquilo, ciclopropilo, ciclobutilo, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R.
En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, NH2 o R3-L1-, o seleccionado del grupo que consiste en Me, Et,

cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R.
En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, CN, NH2, COOH, Me, Et,

En algunas realizaciones de la presente invención, X es
¡ H N '' ^ N '
0» ! !

En algunas realizaciones de la presente invención, R4 se selecciona del grupo que consiste en fenilo, pirrolidinilo, piridilo, pirimidinilo, pirazinilo y piridazinilo, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R. En algunas realizaciones de la presente invención, R4 se selecciona del grupo que consiste de

cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R.
En algunas realizaciones de la presente invención, R4 es

En algunas realizaciones de la presente invención, R4-L2- es

En algunas realizaciones de la presente invención, Ri es H, F, Cl, Br, I, OH, NH2 , COOH o R4-L2-, o seleccionado del grupo que consiste de alquilo C1-3, alquilo C1-3-C(=O)-, alquilo C1-3 -OC(=O)-, alquenilo C2-4, cicloalquenilo C4-6, 3,6-dihidro-2H-piranilo, cicloalquilo C3-6, tetrahidropiranilo, fenilo, piridilo, pirimidinilo, pirazinilo, piridazinilo, pirazolilo, imidazolilo, tienilo, tiazolilo, isotiazolilo, oxazolilo e isoxazolilo, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R.
En algunas realizaciones de la presente invención, R1 es H, F, Cl, Br, I, OH, NH2 , COOH o R4-L2-, o seleccionado del grupo que consiste en Me, Et,

cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R.
En algunas realizaciones de la presente invención, R1 es H, F, Cl, Br, I, OH, NH2 , COOH,

En algunas realizaciones de la presente invención, R es H, F, Cl, Br, I, OH, CN o NH2 , o seleccionado del grupo que consiste en alquilo C1-3, alcoxi C1-3, alquiltio C1-3, alquilamino C1-3, alquilo C1-4-OC(=O)-, N, N'-di(alquilo C1-3)amino, cicloalquilo C3-6, cada uno de los cuales está opcionalmente sustituido con 1 ,2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R es H, F, Cl, Br, I, OH, CN, NH2 , Me, CF3 , CHF2 , CH2F, Et,

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R3 se selecciona del grupo que consiste en ciclopropilo, ciclobutilo, fenilo, piridilo, pirimidinilo, pirazinilo, tienilo, imidazolilo, pirazolilo, oxazolilo, tiazolilo, isoxazolilo e isotiazolilo, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R, y otras variables son como se definieron anteriormente. En algunas realizaciones de la presente invención, R3 es

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R3-L1- es

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, NH2 o R3-L1-, o seleccionado del grupo que consiste en alquilo C1-3, alquilo C1-3 -S(=O)2- alquilo C1-3, ciclopropilo, ciclobutilo, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, NH2 o R3-L1-, o seleccionado del grupo que consiste en Me, Et,

cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R2 es H, F, Cl, Br, I, OH, NH2 , COOH, Me, Et,

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, X es

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R4 se selecciona del grupo que consiste en fenilo, pirrolidinilo, piridilo, pirimidinilo, pirazinilo y piridazinilo, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R4 se selecciona del grupo que consiste en

cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R4 es

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R4-L2- es

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R1 es H, F, Cl, Br, I, OH, NH2 , COOH o R4-L2-, o seleccionadas del grupo que consiste de alquilo C1-3, alquilo C1-3 -C(=O)-, alquilo C1-3 -OC(=O)-, alquenilo C2-4, cicloalquenilo C4-6, 3,6-dihidro-2 H-piranilo, cicloalquilo C3-6, tetrahidropiranilo, fenilo, piridilo, pirimidinilo, pirazinilo, piridazinilo, pirazolilo, imidazolilo, tienilo, tiazolilo, isotiazolilo, oxazolilo e isoxazolilo, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R1 es H, F, Cl, Br, I, OH, NH2 , COOH o R4-L2-, o seleccionado del grupo que consiste en Me, Et,


cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R, y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, R1 es H, F, Cl, Br, I, OH, NH2, COOH,

y otras variables son como se definieron anteriormente.
En algunas realizaciones de la presente invención, el compuesto o la sal farmacéuticamente aceptable del mismo es

en el que, Ri y R2 se definieron como anteriormente.
En algunas realizaciones de la presente invención, el compuesto o la sal farmacéuticamente aceptable del mismo es

en el que, R1 y R2 se definieron como anteriormente.
Las variables anteriores se pueden combinar arbitrariamente, luego se obtienen otras realizaciones de la presente invención.
La presente invención también proporciona un compuesto como se muestra en la fórmula siguiente o una sal farmacéuticamente aceptable del mismo, que se selecciona del grupo que consiste en




En algunas realizaciones de la presente invención, el compuesto o la sal farmacéuticamente aceptable del mismo se selecciona del grupo que consiste en







La presente invención también proporciona una composición farmacéutica, que comprende una cantidad terapéuticamente eficaz del compuesto o la sal farmacéuticamente aceptable del mismo como principio activo, y un vehículo farmacéuticamente aceptable.
La presente invención también proporciona el uso del compuesto o la sal farmacéuticamente aceptable del mismo en la fabricación de un medicamento para tratar una enfermedad relacionada con PDE4.
Efecto técnico
Comparado con Apremilast, el compuesto de la presente invención reduce la distribución en el cerebro, reduciendo potencialmente los vómitos y los efectos secundarios asociados con el cerebro. Aumenta significativamente el efecto inhibidor del compuesto de la presente invención sobre TNF-a en hPBMC y reduce la dosis terapéuticamente eficaz en experimentos con animales, reduciendo así la dosis terapéuticamente eficaz para humanos y aumentando el factor de seguridad para la misma. Ha mejorado las características de la farmacocinética y se espera que se administre a un ser humano una vez al día.
Definición y descripción
A menos que se indique lo contrario, los siguientes términos cuando se usan en las descripciones y las reivindicaciones de la presente invención tienen los siguientes significados. Un término o frase específica no debe considerarse indefinido o poco claro en ausencia de una definición particular, sino que debe entenderse en el sentido ordinario. Cuando un nombre comercial aparece en el presente documento, se pretende que se refiera a su correspondiente producto básico o principio activo del mismo. El término "farmacéuticamente aceptable" se usa en el presente documento en términos de aquellos compuestos, materiales, composiciones y o formas de dosificación, que son adecuadas para su uso en contacto con tejidos humanos y animales dentro del alcance de un juicio médico confiable, sin toxicidad excesiva, irritación, reacción alérgica u otros problemas o complicaciones, acordes con una relación beneficio/riesgo razonable.
El término "sal farmacéuticamente aceptable" se refiere a una sal del compuesto de la presente invención que se prepara haciendo reaccionar el compuesto que tiene un sustituyente específico de la presente invención con un ácido o base relativamente no tóxico. Cuando el compuesto de la presente invención contiene un grupo funcional relativamente ácido, se puede obtener una sal de adición de base poniendo en contacto la forma neutra del compuesto con una cantidad suficiente de base en una solución pura o un disolvente inerte adecuado. La sal de adición de base farmacéuticamente aceptable incluye una sal de sodio, potasio, calcio, amonio, amina orgánica o magnesio o sales similares. Cuando el compuesto de la presente invención contiene un grupo funcional relativamente básico, se puede obtener una sal de adición de ácido poniendo en contacto la forma neutra del compuesto con una cantidad suficiente de ácido en una solución pura o un disolvente inerte adecuado. Los ejemplos de la sal de adición de ácido farmacéuticamente aceptable incluyen una sal de ácido inorgánico, en la que el ácido inorgánico incluye, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido nítrico, ácido carbónico, bicarbonato, ácido fosfórico, fosfato monohidrógeno, fosfato dihidrógeno, ácido sulfúrico, hidrógensulfato, ácido yodhídrico, ácido fosforoso y similares; y una sal de ácido orgánico, en la que el ácido orgánico incluye, por ejemplo, ácido acético, ácido propiónico, ácido isobutírico, ácido maleico, ácido malónico, ácido benzoico, ácido succínico, ácido subérico, ácido fumárico, ácido láctico, ácido mandélico, ácido ftálico, ácido bencenosulfónico, ácido p-toluensulfónico, ácido cítrico, ácido tartárico y ácido metanosulfónico y similares; y una sal de aminoácido (tal como arginina y similares), y una sal de un ácido orgánico como ácido glucurónico y similares (consulte Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Ciertos compuestos específicos de la presente invención que contienen grupos funcionales tanto básicos como ácidos pueden convertirse en cualquier sal de adición de ácido o base.
Preferiblemente, poniendo la sal en contacto con una base o un ácido de una manera convencional, y luego separando el compuesto original, la forma neutra del compuesto se regenera de ese modo. La diferencia entre la forma original del compuesto y sus diversas formas de sal radica en propiedades físicas específicas, tal como la diferente solubilidad en un disolvente polar.
La "sal farmacéuticamente aceptable" utilizada en la presente pertenece a un derivado del compuesto de la presente invención, en el que el compuesto original se modifica formando una sal con un ácido o una base. Los ejemplos de sal farmacéuticamente aceptable incluyen, pero no se limitan a, un ácido inorgánico o una sal de ácido orgánico de un resto básico tal como una amina, una sal de metal alcalino o una sal orgánica de un resto ácido tal como ácido carboxílico y similares. La sal farmacéuticamente aceptable incluye sal no tóxica convencional o sal de amonio cuaternario del compuesto original, tal como una sal formada por un ácido inorgánico no tóxico o un ácido orgánico. La sal no tóxica convencional incluye, pero no se limita a, la sal derivada de un ácido inorgánico y un ácido orgánico, en el que el ácido inorgánico o ácido orgánico se selecciona del grupo que consiste en ácido 2-acetoxibenzoico, ácido 2-hidroxietanosulfónico, ácido acético, ácido ascórbico, ácido bencenosulfónico, ácido benzoico, bicarbonato, ácido carbónico, ácido cítrico, ácido edético, ácido etanodisulfónico, ácido etanosulfónico, ácido fumárico, glucoheptosa, ácido glucónico, ácido glutámico, ácido glicólico, ácido bromhídrico, ácido clorhídrico, yodohidrato, hidroxilo, hidroxinaftaleno, ácido isetiónico, ácido láctico, lactosa, ácido dodecilsulfónico, ácido maleico, ácido málico, ácido mandélico, ácido metanosulfónico, ácido nítrico, ácido oxálico, ácido pamoico, ácido pantoténico, ácido fenilacético, ácido fosfórico, ácido poligalactanal, ácido propiónico, ácido salicílico, ácido esteárico, ácido subacético, ácido succínico, ácido sulfámico, ácido sulfanílico, ácido sulfúrico, tanino, ácido tartárico y ácido p-toluenosulfónico.
La sal farmacéuticamente aceptable de la presente invención se puede preparar a partir del compuesto original que contiene un resto ácido o básico mediante un método químico convencional. Generalmente, dicha sal se puede preparar haciendo reaccionar la forma de ácido o base libre del compuesto con una cantidad estequiométrica de una base o ácido apropiado en agua o un disolvente orgánico o una mezcla de los mismos. Generalmente, se prefieren los medios no acuosos tales como éter, acetato de etilo, etanol, isopropanol o acetonitrilo.
Además de la forma de sal, también puede existir un compuesto en forma de profármaco. El profármaco de un compuesto es un compuesto que sufre fácilmente un cambio químico en condiciones fisiológicas para convertirse en el compuesto. Además, el profármaco puede convertirse en el compuesto mediante un método químico o bioquímico en un entorno in vivo.
Ciertos compuestos de la presente invención pueden existir en forma no solvatada o solvatada, incluida la forma hidratada. Generalmente, la forma solvatada es equivalente a la forma no solvatada y ambas están incluidas dentro del alcance de la presente invención.
Ciertos compuestos de la presente invención pueden tener un átomo de carbono asimétrico (centro óptico) o un doble enlace. El racemato, diastereoisómero, isómero geométrico e isómero individual están todos incluidos dentro del alcance de la presente invención.
A menos que se especifique lo contrario, la configuración absoluta de un centro estereogénico está representada por un enlace sólido acuñado ) y un enlace discontinuo acuñado y la configuración relativa de un centro estereogénico está representada por un enlace sólido recto ) y un enlace punteado recto ('**' ). Cuando el compuesto descrito en este documento contiene un doble enlace olefínico u otros centros geométricos asimétricos, los isómeros geométricos E y Z se incluyen a menos que se especifique lo contrario. Asimismo, todas las formas tautoméricas están comprendidas dentro del alcance de la presente invención.
y la configuración relativa de un centro estereogénico está representada por un enlace sólido recto ) y un enlace punteado recto ('**' ). Cuando el compuesto descrito en este documento contiene un doble enlace olefínico u otros centros geométricos asimétricos, los isómeros geométricos E y Z se incluyen a menos que se especifique lo contrario. Asimismo, todas las formas tautoméricas están comprendidas dentro del alcance de la presente invención.
El compuesto de la presente invención puede tener una forma geométrica o estereoisomérica específica. La presente invención contempla todos estos compuestos, incluyendo el isómero cis y trans, enantiómero (-)- y (+)- enantiómero (R)- y (S), diastereoisómero, isómero (D), isómero (L), y mezcla racémica y otras mezclas, por ejemplo, una mezcla enriquecida en enantiómero o diastereoisómero, todas las cuales están comprendidas dentro del alcance de la presente invención. El sustituyente tal como alquilo puede tener un átomo de carbono asimétrico adicional. Todos estos isómeros y mezclas de los mismos están comprendidos dentro del alcance de la presente invención.
Los isómeros (R)- y (S) ópticamente activos, y los isómeros D y L se pueden preparar usando síntesis quiral o reactivos quirales u otras técnicas convencionales. Si se va a obtener un tipo de enantiómero de cierto compuesto de la presente invención, el enantiómero puro deseado puede obtenerse mediante síntesis asimétrica o acción derivada del auxiliar quiral seguido de la separación de la mezcla diastereomérica resultante y la escisión del grupo auxiliar. Alternativamente, cuando la molécula contiene un grupo funcional básico (tal como amino) o un grupo funcional ácido (tal como carboxilo), el compuesto reacciona con un ácido o base ópticamente activo apropiado para formar una sal del isómero diastereomérico que luego se somete a resolución diastereomérica a través del método convencional en la técnica para dar el enantiómero puro. Además, el enantiómero y el diastereoisómero generalmente se aíslan mediante cromatografía que usa una fase estacionaria quiral y opcionalmente se combina con un método de derivado químico (tal como carbamato generado a partir de amina).
El compuesto de la presente invención puede contener una proporción no natural de isótopo atómico en uno o más de un átomo(s) que constituyen el compuesto. Por ejemplo, el compuesto puede radiomarcarse con un isótopo radiactivo, como el tritio (3H), yodo-125 (125I) o C-14 (14C). Todas las variaciones isotópicas del compuesto de la presente invención, ya sean radiactivas o no, están incluidas dentro del alcance de la presente invención.
El término "vehículo farmacéuticamente aceptable" se refiere a cualquier agente o medio vehículo que sea capaz de suministrar una cantidad eficaz de la sustancia activa de la presente invención, no interfiera con la actividad biológica de la sustancia activa y no tenga efectos secundarios tóxicos sobre el huésped o paciente. El vehículo representativo incluye agua, aceite, vegetales y minerales, base de crema, base de loción, base de ungüento y similares. La base incluye un agente de suspensión, un espesante, un potenciador de la penetración y similares. Sus formulaciones son bien conocidas por los expertos en el campo cosmético o farmacéutico tópico.
El término "excipiente" generalmente se refiere a un vehículo, un diluyente y/o un medio necesario para formular una composición farmacéutica eficaz.
Para un medicamento o un agente farmacológicamente activo, el término "cantidad eficaz" o "cantidad terapéuticamente eficaz" se refiere a una cantidad no tóxica pero suficiente para lograr un efecto deseado del medicamento o del agente. Para la forma de dosificación oral de la presente invención, una "cantidad eficaz" de la sustancia activa en la composición se refiere a una cantidad requerida para lograr un efecto deseado cuando se combina con otra sustancia activa en la composición. La cantidad efectiva varía de una persona a otra y se determina en función de la edad y el estado general del receptor, así como de la sustancia activa específica. Los expertos en la técnica pueden determinar la cantidad eficaz apropiada en un caso individual basándose en un experimento de rutina.
El término "principio activo", "agente terapéutico", "sustancia activa" o "agente activo" se refiere a una entidad química que puede tratar eficazmente el trastorno, enfermedad o afección diana.
"Opcional" u "opcionalmente" significa que el evento o condición subsiguiente puede ocurrir pero no es un requisito, que el término incluye el caso en el que ocurre el evento o condición y el caso en el que no ocurre el evento o condición.
El término "sustituido" significa que uno o más de un átomo(s) de hidrógeno en un átomo específico están sustituidos con el sustituyente, incluyendo deuterio y variantes de hidrógeno, siempre que la valencia del átomo específico sea normal y el compuesto sustituido sea estable. Cuando el sustituyente es una cetona (es decir, =O), significa que dos átomos de hidrógeno están sustituidos. Las posiciones en un anillo aromático no se pueden sustituir por una cetona. El término "opcionalmente sustituido" significa que un átomo puede estar sustituido con un sustituyente o no, a menos que se especifique lo contrario, el tipo y número del sustituyente puede ser arbitrario siempre que sea químicamente alcanzable.
Cuando cualquier variable (tal como R) aparece en la constitución o estructura del compuesto más de una vez, la definición de la variable en cada caso es independiente. Así, por ejemplo, si un grupo está sustituido con 0-2 R, el grupo puede estar opcionalmente sustituido con hasta dos R, en el que la definición de R en cada caso es independiente. Además, se permite una combinación del sustituyente y/o la variante del mismo solo cuando la combinación da como resultado un compuesto estable.
Cuando el número de un grupo de enlace es 0, como -(CRR)0-, significa que el grupo de enlace es un enlace sencillo.
Cuando una de las variables se selecciona de un enlace sencillo, significa que los dos grupos vinculados por el enlace sencillo están conectados directamente. Por ejemplo, cuando L en A-L-Z representa un enlace sencillo, la estructura de A-L-Z es en realidad A-Z.
Cuando un sustituyente está vacante, significa que el sustituyente no existe. Por ejemplo, cuando X está vacante en A-X, la estructura de A-X es en realidad A. Cuando un enlace de un sustituyente puede reticularse con más de un átomo en un anillo, dicho sustituyente puede enlazarse a cualquier átomo del anillo. Cuando un sustituyente enumerativo no indica por qué átomo está unido a un compuesto incluido en la fórmula química general pero no mencionado específicamente, tal sustituyente puede estar unido por cualquiera de sus átomos. Se permite una combinación de sustituyentes y/o variantes de los mismos solo cuando tal combinación puede dar como resultado un compuesto estable. Por ejemplo, la unidad estructural

significa que el sustituyente R puede estar ubicado en cualquier posición del ciclohexilo o ciclohexadieno.
A menos que se especifique lo contrario, el término "hetero" representa un heteroátomo o un grupo heteroátomo (por ejemplo, un grupo atómico que contiene un heteroátomo), incluido el átomo excepto el carbono (C) y el hidrógeno (H) y el grupo atómico que contiene el heteroátomo anterior, por ejemplo, incluyendo oxígeno (O), nitrógeno (N), azufre (S), silicio (Si), germanio (Ge), aluminio (Al), boro (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2-, y el grupo que consiste de -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N H)- y -S(=O)N(H)-, cada uno de los cuales está opcionalmente sustituido.
A menos que se especifique lo contrario, el término "anillo" se refiere a un cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo, cicloalquinilo, heterocicloalquinilo, arilo o heteroarilo sustituidos o no sustituidos. El llamado anillo incluye un solo anillo, un conjunto de anillo, un anillo en espiral, un anillo fusionado o un anillo con puente. El número del átomo en el anillo se define normalmente como el número de miembro del anillo, por ejemplo, un "anillo de 5-7 miembros" significa que de 5 a 7 átomos están dispuestos en un anillo. A menos que se especifique lo contrario, el anillo contiene opcionalmente de 1 a 3 heteroátomos. Por lo tanto, un "anillo de 5-7 miembros" incluye, por ejemplo, fenilo, piridinilo y piperidinilo; por otro lado, el término "anillo heterocicloalquilo de 5-7 miembros" incluye piridilo y piperidinilo, pero excluyendo fenilo. El término "anillo" también incluye un sistema de anillos que contiene al menos un anillo, en el que cada anillo cumple independientemente la definición anterior.
A menos que se especifique lo contrario, el término "heterociclo" o "heterociclo" se refiere a un anillo monocíclico, bicíclico o tricíclico estable que contiene un heteroátomo o un grupo heteroátomo, que puede ser saturado, parcialmente insaturado o insaturado (aromático) y puede contener átomos de carbono y 1,2, 3 o 4 heteroátomos de anillo seleccionados independientemente de N, O y S, en los que cualquiera de los heterociclo anteriores puede fusionarse con un anillo de benceno para formar un anillo bicíclico. Los heteroátomos de nitrógeno y azufre se pueden oxidar opcionalmente (es decir, NO y S(O)p, p es 1 o 2). El átomo de nitrógeno puede estar sustituido o no sustituido (es decir, N o NR, en el que R es H u otros sustituyentes ya definidos aquí). El heterociclo se puede unir al grupo colgante de cualquier heteroátomo o átomo de carbono para formar una estructura estable. Si el compuesto resultante es estable, el heterociclo descrito en este documento puede tener una sustitución en una posición de carbono o nitrógeno. El átomo de nitrógeno en el heterociclo está opcionalmente cuaternizado. En una realización preferida, cuando el número total de átomos de S y O del heterociclo es más de 1, el heteroátomo no es adyacente entre sí. En otra realización preferida, el número total de átomos de S y O del heterociclo no es más de 1. Como se usa en este documento, el término "grupo heterocíclico aromático" o "heteroarilo" se refiere a un anillo aromático monocíclico o bicíclico de 5, 6 o 7 miembros o heterocíclico bicíclico de 7, 8, 9 o 10 miembros estable. o que contiene átomos de carbono y 1,2, 3 o 4 heteroátomos de anillo seleccionados independientemente entre N, O y S. El átomo de nitrógeno puede estar sustituido o no sustituido (es decir, N o NR, en el que R es H u otros sustituyentes ya definidos en el presente documento). Los heteroátomos de nitrógeno y azufre se pueden oxidar opcionalmente (es decir, NO y S(O)p, p es 1 o 2). Vale la pena señalar que el número total de átomos de S y O de un heterociclo aromático no es más de uno. El anillo con puente también se incluye en la definición del heterociclo. Se forma un anillo con puente cuando uno o más de un átomo (es decir, C, O, N o S) enlazan dos átomos de carbono o nitrógeno no adyacentes. Un anillo con puente preferido incluye, pero no se limita a, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo carbono-nitrógeno. Vale la pena señalar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. En un anillo con puente, el sustituyente del anillo también puede estar presente en el puente.
Los ejemplos del compuesto heterocíclico incluyen, pero no se limitan a: acridinilo, azocinilo, bencimidazolilo, benzofuranilo, benzomercaptofuranilo, benzomercaptofenilo, benzoxazolilo, benzoxazolinilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidasolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromeno, cinolinilo, decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro[2,3-b]tetrahidrofuranilo, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1 H-indazolilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isobenzofuranilo, isoindolilo, isoindolinilo, isoquinolinilo, isotiazolilo, isoxazolilo, metilendioxifenilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1.2.5- oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, hidroxindolilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazina, fenotiazina, benzoxantinilo, fenoloxazinilo, ftalazinilo, piperazinilo, piperidinilo, piperidonilo, 4-piperidonilo, piperonilo, pteridinilo, purinilo, piralino, pirazinilo, pirazolidinilo, pirazolinilo, pirazolilo, piridazinilo, pirido-oxazolilo, pirido-imidazolilo, pirido-tiazolilo, piridinilo, pirrolidinilo, pirrolinilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, tetrazolilo, 6H-1.2.5- tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, isotiazoliltienilo, tieno-oxazolilo, tieno-tiazolilo, tieno-imidazolilo, tienilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo y xantenilo. También se incluyen compuestos de anillo condensado y compuestos espiro.
A menos que se especifique lo contrario, el término "hidrocarbilo" o sus hipónimos (por ejemplo, alquilo, alquenilo, alquinilo y arilo, etc.), por sí mismo o como parte de otro sustituyente, se refiere a un radical hidrocarburo de cadena lineal, o ramificada o cíclico o cualquier combinación del mismo. Pueden estar completamente saturados (por ejemplo, alquilo), mono o poliinsaturados (por ejemplo, alquenilo, alquinilo y arilo), pueden estar mono, di o polisustituidos, pueden ser monovalentes (por ejemplo, metilo), divalentes (por ejemplo, metileno) o multivalente (por ejemplo, metenilo), también puede incluir un grupo divalente o multivalente, tener un número específico de átomos de carbono (por ejemplo, C1-C12 indica de 1 a 12 átomos de carbono, C1-12 se selecciona de C1, C2 , C3 , C4 , C5 , C6 , C7 , C8 , C9 , C10, C11 y C12; C3-12 se selecciona de C3 , C4 , C5 , C6 , C7 , C8 , C9 , C10, C11 y C12). El término "hidrocarbilo" incluye, pero no se limita a, hidrocarbilo alifático e hidrocarbilo aromático. El hidrocarbilo alifático incluye hidrocarbilo lineal y cíclico, específicamente incluye pero no se limita a alquilo, alquenilo y alquinilo. El hidrocarbilo aromático incluye, pero no se limita a, hidrocarbilo aromático de 6-12 miembros tal como fenilo, naftilo y similares. En algunas realizaciones, el término "hidrocarbilo" se refiere a un grupo lineal o ramificado o una combinación de los mismos que puede estar completamente saturado, monoinsaturado o poliinsaturado y puede incluir un grupo divalente o multivalente. Los ejemplos del grupo hidrocarbilo saturado incluyen, pero no se limitan a, metilo, etilo, n-propilo, isopropilo, n-butilo, terc-butilo, isobutilo, sec-butilo, ciclohexilo, (ciclohexil)metilo, ciclopropilmetilo y el homólogo o isómero de n-amilo, n-hexilo, n-heptilo, n-octilo y otros grupos de átomos. El hidrocarbilo insaturado tiene uno o más de un doble o triple enlace. Los ejemplos de alquilo insaturado incluyen, pero no se limitan a, vinilo, 2-propenilo, butenilo, crotilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo, 3-(1,4-pentadienilo), etinilo, 1- y 3-propinilo, 3-butinilo y más homólogos e isómeros superiores.
A menos que se especifique lo contrario, el término "heterohidrocarbilo" o sus hipónimos (tales como heteroalquilo, heteroalquenilo, heteroalquinilo y heteroarilo, etc.), por sí mismo o como parte de otro sustituyente, se refiere a un grupo hidrocarburo estable lineal, ramificado o cíclico o cualquier combinación de los mismos, que tiene un número especificado de átomos de carbono y al menos un heteroátomo. En algunas realizaciones, el término "heteroalquilo" por sí mismo o en combinación con otro término se refiere a un radical hidrocarbonado ramificado, de cadena lineal estable o una combinación de los mismos que tiene un número especificado de átomos de carbono y al menos un heteroátomo. En una realización específica, un heteroátomo se selecciona de B, O, N y S, en el que los átomos de nitrógeno y azufre están opcionalmente oxidados y el átomo de nitrógeno opcionalmente está cuaternizado. El heteroátomo o grupo heteroátomo puede estar ubicado en cualquier posición interior de un heterohidrocarbilo, incluida la posición en la que el hidrocarbilo se une a la parte restante de la molécula. Pero los términos "alcoxi", "alquilamino" y "alquiltio" (o tioalquilo) se usan con el significado convencional y se refieren a un grupo alquilo conectado al resto de la molécula a través de un átomo de oxígeno, un amino o un átomo de azufre, respectivamente. Los ejemplos incluyen, pero no se limitan a, -CH2-CH2-O-CH3 , -CH2-CH2-NH-CH3 , -CH2-CH2-N(CH3)-CH3 , -CH2-S-CH2-CH3 , -CH2-CH2 , -S(O)-CH3 , -CH2-CH2-S(O)2,-CH3 , -CH=CH-O-CH3 , -CH2-CH=N-OCH3 y -CH=CH-N(CH3)-CH3. Pueden estar presentes hasta dos heteroátomos consecutivos, tal como, -CH2 -NH-OCH3.
A menos que se especifique lo contrario, el término "ciclohidrocarbilo", "heterociclohidrocarbilo" o sus hipónimos (tales como arilo, heteroarilo, cicloalquilo, heterocicloalquilo, cicloalquenilo, heterocicloalquenilo, cicloalquinilo, heterocicloalquinilo, etc.) por sí mismo o en combinación con otro término se refiere a “hidrocarbilo” cicladoo "heterohidrocarbilo". Además, para heterohidrocarbilo o heterociclohidrocarbilo (por ejemplo, heteroalquilo y heterocicloalquilo), un heteroátomo puede ocupar la posición en la que el heterociclo se une a la posición restante de la molécula. Los ejemplos del cicloalquilo incluyen, pero no se limitan a, ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo y similares. Los ejemplos no limitantes de heterocicloalquilo incluyen 1 -(1,2,5,6-tetrahidropiridilo), 1 -piperidinilo, 2-piperidinilo, 3-piperidinilo, 4-morfolinilo, 3-morfolinilo, tetrahidrofuran-2-ilo, tetrahidrofuran-3-ilo, tetrahidro-tiofen-2-ilo, tetrahidro-tiofen-3-ilo, 1 -piperazinilo y 2-piperazinilo.
A menos que se especifique lo contrario, el término "alquilo" se refiere a un grupo hidrocarburo saturado ramificado o de cadena lineal, que puede estar monosustituido (por ejemplo, -CH2F) o polisustituido (por ejemplo, -CF3), puede ser monovalente (por ejemplo, metilo), divalente (por ejemplo, metileno) o multivalente (por ejemplo, metenilo). Ejemplos de alquilo incluyen metilo (Me), etilo (Et), propilo (tal como n-propilo e isopropilo), butilo (como n-butilo, isobutilo, s-butilo, t-butilo), pentilo (tal como n-pentilo, isopentilo, neopentilo) y similares.
A menos que se especifique lo contrario, el término "alquenilo" se refiere a un grupo alquilo que tiene uno o más de un doble enlace carbono-carbono en cualquier posición de la cadena, puede ser monosustituido o polisustituido y puede ser monovalente, divalente o multivalente. Los ejemplos de alquenilo incluyen etenilo, propenilo, butenilo, pentenilo, hexenilo, butadienilo, pentadienilo, hexadienilo y similares.
A menos que se especifique lo contrario, el término "alquinilo" se refiere a un grupo alquilo que tiene uno o más de un triple enlace carbono-carbono en cualquier posición de la cadena, puede ser monosustituido o polisustituido y puede ser monovalente, divalente o multivalente. Los ejemplos de alquinilo incluyen etinilo, propinilo, butinilo, pentinilo, hexinilo y similares.
A menos que se especifique lo contrario, cicloalquilo incluye cualquier hidrocarbilo cíclico o policíclico estable, y cualquier átomo de carbono es saturado, puede ser monosustituido o polisustituido y puede ser monovalente, divalente o multivalente. Los ejemplos de cicloalquilo incluyen, pero no se limitan a, ciclopropilo, norbonanilo, [2.2.2]biciclooctano, [4.4.0]biciclodecanilo y similares.
A menos que se especifique lo contrario, cicloalquenilo incluye cualquier hidrocarbilo cíclico o policíclico estable que tenga uno o más de un enlace sencillo carbono-carbono insaturado en cualquier posición del anillo, puede ser monosustituido o polisustituido y puede ser monovalente, divalente o multivalente. Los ejemplos del cicloalquenilo incluyen, pero no se limitan a, ciclopentenilo, ciclohexenilo y similares.
A menos que se especifique lo contrario, cicloalquinilo incluye cualquier hidrocarbilo cíclico o policíclico estable que tenga uno o más triples enlaces carbono-carbono en cualquier posición del anillo, puede ser monosustituido o polisustituido y puede ser monovalente, divalente o multivalente.
A menos que se especifique lo contrario, el término "halo" o "halógeno" por sí mismo o como parte de otro sustituyente se refiere a un átomo de flúor, cloro, bromo o yodo. Además, el término "haloalquilo" pretende incluir monohaloalquilo y polihaloalquilo. Por ejemplo, el término "haloalquilo(C1-C4)" incluye, pero no se limita a, trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo y similares. Ejemplos de haloalquilo incluyen, pero no se limitan a, trifluorometilo, triclorometilo, pentafluoroetilo y pentacloroetilo.
El término "alcoxi" representa cualquier alquilo definido anteriormente que tiene un número específico de átomos de carbono unidos por un puente de oxígeno. A menos que se especifique lo contrario, alcoxi C1-6 incluye alcoxi C1, C2 , C3 , C4 , C5 y C6. Los ejemplos de alcoxi incluyen, pero no se limitan a, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, sec-butoxi, terc-butoxi, n-pentiloxi y S-pentoxi. A menos que se especifique lo contrario, el término "arilo" se refiere a un sustituyente aromático poliinsaturado, puede ser mono, di o polisustituido, puede ser monovalente, divalente o multivalente, puede ser un anillo único o un anillo múltiple (por ejemplo, uno a tres anillos; en el que al menos un anillo es aromático), que están fusionados entre sí o conectados covalentemente. El término "heteroarilo" se refiere a un arilo (o anillo) que contiene de uno a cuatro heteroátomos. En un ejemplo ilustrativo, el heteroátomo se selecciona de B, O, N y S, en el que los átomos de nitrógeno y azufre están opcionalmente oxidados y el átomo de nitrógeno opcionalmente está cuaternizado. Un heteroarilo puede unirse al resto de una molécula a través de un heteroátomo. Los ejemplos no limitantes de arilo o heteroarilo incluyen fenilo, naftilo, bifenilo, pirrolilo, pirazolilo, imidazolilo, pirazinilo, oxazolilo, feniloxazolilo, isoxazolilo, tiazolilo, furanilo, tienilo, piridilo, pirimidinilo benzotiazolilo, purinilo, benzimidazolilo, indolilo, isoquilonilo, quinoxalinilo, quinolilo, 1 -naftilo, 2-naftilo, 4-bifenilo, 1 -pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidilo, 4-pirimidilo, 5-benzotiazolilo, purinilo, 2-bencimidazolilo, 5-indolilo, 1 -isoquinolilo, 5-isoquinolilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolilo y 6-quinolilo. El sustituyente de cualquiera de los sistemas de anillo arilo y heteroarilo anteriores se selecciona del sustituyente aceptable descrito a continuación.
A menos que se especifique lo contrario, cuando arilo se combina con otros términos (tales como ariloxi, ariltio, arilalquilo), el arilo incluye el anillo de arilo y heteroarilo como se definió anteriormente. Por lo tanto, el término "aralquilo" incluye el grupo (por ejemplo, bencilo, fenetilo, piridilmetilo, etc.) donde un arilo está unido a un alquilo, incluyendo un alquilo donde el átomo de carbono (por ejemplo, metileno) ha sido reemplazado por un átomo tale como oxígeno, por ejemplo, fenoximetilo, 2-piridiloxi, 3-(1-naftiloxi)propilo y similares.
El término "grupo saliente" se refiere a un grupo funcional o átomo que puede ser reemplazado por otro grupo funcional o átomo mediante una reacción de sustitución (tal como una reacción de sustitución por afinidad). Por ejemplo, los grupos salientes representativos incluyen triflato; cloro, bromo y yodo; grupo sulfonato, tal como mesilato, tosilato, p-bromobencenosulfonato, p-toluenosulfonatos y similares; aciloxi, tal como acetoxi, trifluoroacetoxi y similares.
El término "grupo protector" incluye, pero no se limita a, "grupo protector de amino", "grupo protector hidroxi" o "grupo protector tio". El término "grupo protector amino" se refiere a un grupo protector adecuado para bloquear la reacción secundaria en el nitrógeno de un amino. Los grupos protectores de amino representativos incluyen, pero no se limitan a: formilo; acilo, tal como alcanoílo (por ejemplo, acetilo, tricloroacetilo o trifluoroacetilo); alcoxicarbonilo, como terc-butoxicarbonilo (Boc); arilmetoxicarbonilo tal como benciloxicarbonilo (Cbz) y 9-fluorenilmetoxicarbonilo (Fmoc); arilmetilo tal como bencilo (Bn), tritilo (Tr), 1,1-bis-(4'-metoxifenil)metilo; sililo tal como trimetilsililo (TMS) y tercbutildimetilsililo (TBS) y similares. El término "grupo protector hidroxi" se refiere a un grupo protector adecuado para
bloquear la reacción secundaria en hidroxi. Los grupos protectores hidroxi representativos incluyen, pero no se limitan a: alquilo tal como metilo, etilo y tere-butilo; acilo tal como alcanoílo (por ejemplo, acetilo); arilmetilo tal como bencilo (Bn), p-metoxibencilo (PMB), 9-fluorenilmetilo (Fm) y difenilmetilo (benzhidrilo, DPM); sililo como trimetilsililo (TMS) y terc-butildimetilsililo (TBS) y similares.
El compuesto de la presente invención se puede preparar mediante una variedad de métodos sintéticos bien conocidos por los expertos en la técnica, incluida la siguiente realización enumerativa, la realización formada por la siguiente realización enumerativa en combinación con otros métodos de síntesis química y el reemplazo equivalente bien conocido por los expertos en la técnica. La realización preferida incluye, pero no se limita a, la realización de la presente invención.
Todos los disolventes usados en la presente invención están disponibles comercialmente. Esta presente invención adopta las siguientes palabras abreviadas: aq se refiere a acuoso; "HATU" se refiere a hexafluorofosfato de 2-(7-Azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio; "EDC" se refiere a clorhidrato de n-(3-dimetilaminopropil)-W-etilcarbodiimida; "m-CPBA" se refiere a ácido 3-cloroperoxibenzoico; "eq" se refiere a igual; "CDI" se refiere a carbonildiimidazol; "DCM" se refiere a diclorometano; "PE" se refiere a éter de petróleo; "DIAD" se refiere a azodiformiato de diisopropilo; "DMF" se refiere a N,N-dimetilformamida; "DMSO" se refiere a dimetilsulfóxido; "EtOAc" se refiere a acetato de etilo; "EtOH" se refiere a etanol; "MeOH" se refiere a metanol; "CBz" se refiere a carbobenciloxi, un tipo de grupo protector para amina; "BOC" se refiere a t-butiloxicarbonilo; "HOAc" se refiere a ácido acético; "NaCNBH3" se refiere a cianoborohidruro de sodio; t.a. se refiere a temperatura ambiente; O/N se refiere a durante noche a; "THF" se refiere a tetrahidrofurano;"Boc2O"se refiere a dicarbonato de di-terc-butilo; "TFA" se refiere a ácido trifluoroacético; "DIPEA" se refiere a etildiisopropilamina; "SOCL" se refiere a cloruro de tionilo; "CS2" se refiere a disulfuro de carbono; TsOH se refiere a ácido p-toluenosulfónico; "NFSI" se refiere a W-Fluorobencenosulfonimida; "NCS" se refiere a 1 -cloropirrolidin-2,5-dicetona; "n-Bu4NF" se refiere a fluoruro de tetrabutilamonio; "iPrOH" se refiere a 2-propanol; "pf" se refiere al punto de fusión. "LDA" se refiere a diisopropilamida de litio.
Los compuestos se nombran manualmente o mediante el software ChemDraw®, los compuestos disponibles comercialmente utilizan sus nombres de directorio de proveedores.
Descripción detallada de la realización preferida
Los siguientes ejemplos ilustran adicionalmente la presente invención, pero la presente invención no se limita a ellos. La presente invención se ha descrito en detalle en el texto, y sus realizaciones específicas también se han divulgado, para un experto en la técnica, es obvio modificar y mejorar las realizaciones de la presente invención dentro del espíritu y alcance de la presente invención.
Realización 1: WX001

Ruta de síntesis:

Etapa 1: preparación del compuesto WX001-3
El compuesto WX001-2 (285.00 mg, 1.04 mmol) y el compuesto WX001-1 (250.00 mg, 1.05 mmol) se disolvieron en acetonitrilo (15 ml) a temperatura ambiente, seguido de la adición de carbonato de potasio (199.80 mg, 1.45 mmol). La mezcla de reacción se calentó a 50 °C y se agitó durante 6 horas. Después de la reacción, la mezcla se enfrió a
temperatura ambiente. Los insolubles se eliminaron por filtración y la torta del filtro se lavó con diclorometano (10 ml x 2). El filtrado se combinó y se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/1 -2/1, relación en volumen) para obtener el producto diana WX001-3. 1H RMN (400 MHz, CDCta) ó: 8.21-8.01 (m, 2H), 7.39-7.23 (m, 1H), 6.83 (d, J=12.5 Hz, 3H), 5.61-5.50 (m, 1H), 4.16-3.98 (m, 2H), 3.85 (s, 3H), 3.70-3.57 (m, 1H), 3.55-3.43 (m, 1H), 2.76 (s, 3H), 1.46 (t, J=6.9 Hz, 3H).
Etapa 2: preparación del compuesto WX001-4
Compuesto WX001-3 (270.00 mg, 549.53 pmol) se disolvió en metanol (20 ml) a temperatura ambiente, seguido de la adición de polvo de zinc (359.34 mg, 5.50 mmol) y cloruro de amonio (293.94 mg, 5.50 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1.5 horas. Después de la reacción, los insolubles se eliminaron por filtración y la torta del filtro se lavó con metanol (10 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto crudo WX001-4.
Etapa 3: preparación del compuesto WX001
Compuesto WX001-4 (250.00 mg, 541.90 pmol) y trietilamina (329.01 mg, 3.25 mmol) se disolvieron en tetrahidrofurano (50 ml) a 0 °C, seguido de la adición de trifosgeno (192.97 mg, 650.28 pmol). La mezcla de reacción se agitó a 0° C durante 2 horas. Después de la reacción, la mezcla se apagó con la solución acuosa de trietilamina (1 ml en 20 ml), se diluyó con salmuera saturada (20 ml) y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX001. MS-ESI m/z: 487.0 [M H]+, 489.0 [M H 2]+. 1H RMN (400 MHz, CDCh) 5: 10.32 (br s, 1H), 7.25-6.91 (m, 4H), 6.84 (d, J=8.3 Hz, 1H), 6.18 (br s, 1H), 4.73 (br s, 1H), 4.08 (q, J=7.0 Hz, 2H), 3.94-3.79 (m, 4H), 2.88 (s, 3H), 1.44 (t, J=6.9 Hz, 3H).
Realización 2: WX002

Ruta de síntesis:

Etapa 1: preparación del compuesto WX002-2
Compuesto WX002-1 (151.52 mg, 671.80 pmol) y la sal de N-acetil-L-leucinato del compuesto WX001-2 (183.00 mg, 671.80 pmol) y carbonato de potasio (185.70 mg, 1.34 mmol) se añadieron a N, N-dimetilformamida (3.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 70° C y se agitó durante 3 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con agua (8 ml) y se extrajo con acetato de etilo (5 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna ultrarrápida en gel de sílice (columna de gel de sílice de 4 g, eluyente: éter de petróleo acetato de etilo=9/1-1/1, relación en volumen) para obtener el producto diana WX002-2. MS-ESI m/z: 526.0 [M CH3CNNa]+.
Etapa 2: preparación del compuesto WX002-3
El compuesto WX002-2 (70.00 mg, 151.37 pmol), zinc en polvo (79.18 mg, 1.21 mmol) y cloruro de amonio (80.97 mg, 1.51 mmol) se añadieron a metanol (3.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura
ambiente durante 16 horas. Después de la reacción, se añadió acetato de etilo (8 ml) y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. Se añadió agua (10 ml) al residuo y se extrajo con acetato de etilo (8 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo. WX002-3. MS-ESI m/z: 455.0 [M+Na]+.
Etapa 3: preparación del compuesto WX002
El compuesto WX002-3 (30.00 mg, 69.37 pmol) y trietilamina (42.12 mg, 416.22 pmol) se disolvieron en tetrahidrofurano (3.00 ml) a 0 °C, seguido de la adición de trifosgeno (10.29 mg, 34.69 pmol). La mezcla de reacción se agitó a 0 °C durante 0.5 horas. Después de la reacción, la mezcla se apagó con agua (10 ml) y se extrajo con acetato de etilo (8 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante HPLC preparativa para obtener el producto diana WX002. MS-ESI m/z: 459.0 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.47 (br s, 1H), 7.44-7.31 (m, 2H), 7.26-7.00 (m, 3H), 6.85 (d, J=7.3 Hz, 1H), 5.90 (br s, 1H), 4.74 (br s, 1 H), 4.09 (m, 2H), 3.95-3.82 (m, 4H), 2.85 (br s, 3H), 1.45 (t, J=6.0 Hz, 3H).
Realización 3: WX003

Ruta de síntesis:

Etapa 1: preparación del compuesto WX003-2
El compuesto WX003-1 (5.00 g, 28.48 mmol), el compuesto WX001-2 (7.79 g, 28.48 mmol) y diisopropiletilamina (11.04 g, 85.44 mmol, 14.92 ml) se disolvieron en N,N-dimetilformamida (100.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 100 °C y se agitó durante 3 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. Se añadió metanol (150 ml) al residuo y se agitó durante 30 minutos, seguido de filtración. La torta del filtro se lavó con metanol (50 ml) y se secó a 40 °C al vacío para obtener el producto diana WX003-2. MS-ESI m/z: 451.0 [M+Na]+.
Etapa 2: preparación del compuesto WX003-3
El compuesto WX003-2 (5.00 g, 11.66 mmol), zinc en polvo (6.10 g, 93.28 mmol) y cloruro de amonio (6.24 g, 116.60 mmol) se añadieron a metanol (100.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (100 ml) al residuo obtenido y se agitó durante 15 minutos, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto diana WX003-3. MS-ESI m/z: 421.0 [M+Na]+.
Etapa 3: preparación del compuesto WX003
El compuesto WX003-3 (4.00 g, 10.03 mmol) y trietilamina (3.04 g, 30.08 mmol, 4.17 ml) se disolvieron en tetrahidrofurano (50 ml) a 0 °C, seguido de la adición de trifosgeno en forma de porciones (1.19 g, 4.01 mmol). La mezcla de reacción se agitó a 0-5 °C durante 1 hora. Después de la reacción, se agregaron agua (20 ml) y acetato de
etilo (100 ml). Las fases orgánicas se separaron, se lavaron con salmuera saturada (30 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=5/1 -1/1, relación en volumen) para obtener el producto diana WX003. MS-ESI m/z: 425.1 [M+H]+ .1H RMN (400 MHz, CD3OD) 5: 7.32-7.10 (m, 2H), 7.10 6.98 (m, 3H), 6.94 (d, J=8.3 Hz, 1H), 6.03 (dd, J=3.8, 10.3 Hz, 1H), 4.59 (dd, J=10.7, 14.4 Hz, 1H), 4.13-3.96 (m, 3H), 3.81 (s, 3H), 2.97 (s, 3H), 1.37 (t, J=6.9 Hz, 3H).
Realización 4: WX004

Ruta de síntesis:

Etapa 1: preparación del compuesto WX004-2
El compuesto WX004-1 (142.50 mg, 895,74 pmol), la sal de N-acetil-L-leucinato del compuesto WX004-1 (200.00 mg, 447.87 pmol) y carbonato de potasio (185.70 mg, 1.34 mmol) se añadieron a N,N-dimetilformamida (4.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 70 °C y se agitó durante 3 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Se añadió agua (8 ml) y se extrajo con acetato de etilo (10 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: acetato de etilo/éter de petróleo = 1/1, relación en volumen) para obtener el producto diana WX004-2. MS-e S i m/z: 413.1 [M+H]+ .1H RMN (400 MHz, CDCh) 5: 8.51 (d, J=6.5 Hz, 1H), 7.91 (dd, J=2.,8, 8.8 Hz, 1H), 7.21-7.15 (m, 1H), 6.97-6.92 (m, 1H), 6.89 6.84 (m, 2H), 6.80 (dd, J=4.3, 9.3 Hz, 1H), 5.21-5.14 (m, 1 H), 4.12-4.06 (m, 2H), 3.86 (s, 3H), 3.63 (dd, J=8.3, 14.8 Hz, 1H), 3.51 -3.44 (m, 1 H), 2.80 (s, 3H), 1.46 (t, J=6.8 Hz, 3H).
Etapa 2: preparación del compuesto WX004-3
El compuesto WX004-2 (70.00 mg, 169.73 pmol), cloruro de amonio (72.63 mg, 1.36 mmol) y polvo de hierro (47.40 mg, 848.63 pmol) se añadieron a metanol (3.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 50 °C y se agitó durante 1 hora. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Se añadió acetato de etilo (5 ml) y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. Se añadió agua (8 ml) al residuo y se extrajo con acetato de etilo (5 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto diana WX004-3. MS-ESI m/z: 423.0 [M+H2O+Na]+ .
Etapa 3: preparación del compuesto WX004
El compuesto WX004-3 (25.00 mg, 65.37 pmol) y trietilamina (39.69 mg, 392.21 pmol) se disolvieron en tetrahidrofurano (3.00 ml) a 0 °C, seguido de la adición de trifosgeno (9.70 mg, 32.68 pmol). La mezcla de reacción se agitó a 0 °C durante 0.5 horas. Después de la reacción, se añadió agua (8 ml) a la mezcla y se extrajo con acetato de etilo (5 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX004. MS-ESI m/z: 409.1 [M+H]+ .1H RMN (400 MHz, CD3OD) 5: 7.20-7.06 (m, 3H), 6.98-6.73 (m, 3H), 6.03 (d, J=7.5 Hz, 1H), 4.68-4.59 (m, 2H), 4.11-4.04 (m, 2H), 3.82 (br s, 3H), 2.97 (br s, 3H), 1.38 (br s, 3H).
Realización 5: WX005

Ruta de síntesis:

Etapa 1: preparación del compuesto WX005-2
El N-acetil-L-leuccinato (735.00 mg, 2.69 mmol) de compuesto WX001-2, compuesto WX005-1 (952.69 mg, 5.38 mmol) y carbonato de potasio (1.12 g, 8.07 mmol) se añadieron a N,N-dimetilformamida (6.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 70 °C y se agitó durante 1.5 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se le añadió acetato de etilo (5 ml), seguido de filtración. El filtrado se concentró a presión reducida. Se añadió agua (8 ml) al residuo obtenido y se extrajo con acetato de etilo (5 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=1/0-1/1, relación en volumen) para obtener el producto diana WX005-2. 1H RMN (400 MHz, CDCl3) ó: 7.85 (d, J=9.0 Hz, 1H), 7.68 (td, J=2.4, 8.5 Hz, 1H), 7.06 (ddd, J=3.0, 7.5, 12.8 Hz, 1H), 6.85-6.75 (m, 3H), 5.49 (dt, J=5.5, 8.5 Hz, 1H), 4.11-4.01 (m, 2H), 3.85 (s, 3H), 3.63 (dd, J=8.0, 14.8 Hz, 1H), 3.47 (dd, J=5.4, 14.7 Hz, 1H), 2.79 (s, 3H), 1.45 (t, J=7.0 Hz, 3H).
Etapa 2: preparación del compuesto WX005-3
El compuesto WX005-2 (800.00 mg, 1.86 mmol), cloruro de amonio (994.19 mg, 18.60 mmol) y polvo de zinc (972.30 mg, 14.88 mmol) se añadieron a metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 15 horas. Después de la reacción, se añadió acetato de etilo (10 ml), seguido de filtración y el filtrado se concentró a presión reducida. Se añadió agua (8 ml) al residuo obtenido y se extrajo con acetato de etilo (10 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto diana WX005-3. MS-ESI m/z: 422.9 [M+Na]+.
Etapa 3: preparación del compuesto WX005
El compuesto WX005-3 (580.00 mg, 1.45 mmol) y trietilamina (586.90 mg, 5,80 mmol) se disolvieron en tetrahidrofurano (6.00 ml) a 0 °C, seguido de la adición de trifosgeno (172.11 mg, 580.00 pmol). La mezcla de reacción se agitó a 0 °C durante 0.5 horas. Después de la reacción, se añadió agua (10 ml) a la mezcla y se extrajo con acetato de etilo (8 ml x 2). Las fases orgánicas se combinaron y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=1/0-1/2, relación en volumen) para obtener el producto diana WX005. MS-ESI m/z: 427.0 [M+H]+. 1H RMN (400 MHz, MeOD) 5: 7.08 (br s, 1H), 7.02-6.87 (m, 2H), 6.79-6.61 (m, 2H), 6.21 (d, J=8.0 Hz, 1 H), 4.49 (br s, 1H), 4.14-3.93 (m, 3H), 3.81 (s, 3H), 3.00 (s, 3H), 1.36 (t, J=6.9 Hz, 3H).
Realización 6: WX006

Ruta de síntesis:

Etapa 1: preparación del compuesto WX006-2
El compuesto WX006-1 (500.00 mg, 2.20 mmol) se disolvió en N,N-dimetilformamida (10.00 ml) a temperatura ambiente, seguido de la adición de diisopropiletilamina (853.67 mg, 6.60 mmol, 1.15 ml) y el compuesto WX001-2 (601.85 mg, 2.20 mmol). La mezcla de reacción se calentó a 120 °C y se agitó durante 12 horas bajo una atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. Se añadió metanol (30 ml) al residuo obtenido, se agitó durante 10 minutos y se filtró para obtener el producto crudo WX006-2.
Etapa 2: preparación del compuesto WX006-3
El Compuesto WX006-2 (1.20 g, 2.50 mmol) y cloruro de amonio (1.18 g, 22.11 mmol) se añadieron a metanol (30.00 ml) a 0 °C, seguido de la adición de polvo de zinc (828.08 mg, 12.66 mmol). La mezcla de reacción se agitó a 0 °C durante 1 hora en atmósfera de nitrógeno. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (50 ml) al residuo obtenido y se agitó durante 10 min, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX006-3. MS-ESI m/z: 472.9 [M+Na]+.
Etapa 3: preparación del compuesto WX006
El compuesto WX006-3 (200.00 mg, 444.00 pmol) y trietilamina (247.11 mg, 2.44 mmoles, 338.50 pl) se disolvieron en tetrahidrofurano (2.00 ml) a 0 °C, seguido de la adición de trifosgeno (158.11 mg, 532.80 pmol). La mezcla de reacción se agitó a 0 °C durante 1 hora en atmósfera de nitrógeno. Después de la reacción, la mezcla se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX006.
MS-ESI m/z: 499.0 [M+Na]+. 1H RMN (400 MHz, CDCh) 5: 10.15 (br, s, 1 H), 7.19-7.02 (m, 4H), 6.81 (d, J=8.3 Hz, 1 H), 6.23 (br s, 1H), 4.17-4.09 (m, 1 H), 4.06 (q, J=6.9 Hz, 2H), 3.91 -3.78 (m, 4H), 2.88 (s, 3H), 1.41 (t, J= 6.9 Hz, 3H).
Realización 7: WX007

Ruta de síntesis:

Etapa 1: preparación del compuesto WX007-2
El compuesto WX007-1 (5.03 g, 22.86 mmol) y compuesto WX001-2 (5.00 g, 18.29 mmol) se disolvieron en N,N-dimetilformamida (100.00 ml) a temperatura ambiente, seguido de la adición gota a gota de diisopropiletilamina (7.99 g, 54.87 mmol). La mezcla de reacción se calentó a 90 °C y se agitó durante 30 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se diluyó con agua (20 ml) y se extrajo con acetato de etilo (50 ml x 4). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna de gel de sílice (eluyente: acetato de etilo/éter de petróleo=0/1-1/1, relación en volumen) para obtener el producto diana WX007-2. 1H RMN (400 MHz, CDCh) 5: 7.08-7.03 (m, 1H), 7.02 6.98 (m, 1 H), 6.93-6.89 (m, 1H), 6.87-6.83 (m, 2H), 6.68 (d, J=7.5 Hz, 1 H), 6.23 (d, J=6.0 Hz, 1 H), 5.07 (td, J=5.2, 8.4 Hz, 1H), 4.10-4.03 (m, 2H), 3.85 (s, 3H), 3.57-3.49 (m, 1 H), 3.44-3.36 (m, 1 H), 2.78 (s, 3H), 1.44 (t, J=7.0 Hz, 3H).
Etapa 2: preparación del compuesto WX007-3
El compuesto WX007-2 (1.35 g, 2.85 mmol) y cloruro de amonio (1.52 g, 28.50 mmol) se añadieron a agua (3.00 ml) y etanol (30.00 ml) a temperatura ambiente, seguido de la adición de polvo de hierro (796.44 mg, 14.25 mmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 1 hora en atmósfera de nitrógeno. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (100 ml) al residuo obtenido, seguido de filtración y el filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante columna de cromatografía (eluyente: acetato de etilo/éter de petróleo=1/4-4/1, relación en volumen) para obtener el producto diana WX007-3. 1H RMN (400 MHz, CDCh) 5: 6.93 (dd, J=8.0, 14.6 Hz, 2H), 6.89-6.82 (m, 2H), 6.52-6.44 (m, 1H), 6.44-6.35 (m, 1H), 4.92 (br s,1 H), 4.47 (br s, 1 H), 4.06 (q, J=6.7 Hz, 2H), 3.85 (s, 3H), 3.60-3.49 (m, 1H), 3.47-3.37 (m, 1H), 2.81 (s, 3H), 1.43 (t, J=6.8 Hz, 3H).
Etapa 3: preparación del compuesto WX007
El compuesto WX007-3 (50.00 mg, 112.78 pmol) y trietilamina (57.06 mg, 563.89 pmol, 78.16 ul) se disolvieron en tetrahidrofurano (5.00 ml) a temperatura ambiente, luego la mezcla de reacción se enfrió a 0 °C, seguido de la adición de trifosgeno (16.73 mg, 56.39 pmol). La mezcla de reacción se agitó a 0 °C durante 30 minutos en atmósfera de nitrógeno. Después de la reacción, la mezcla se apagó con agua (20 ml), la fase orgánica se separó y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (20 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=0/1-4/1, relación en volumen) para obtener el producto diana WX007. MS-ESI m/z: 469.0 [M+H]+, 471.0 [M+H+2]+. 1H RMN (400 MHz, CDCl3) 5: 8.40 (s, 1H), 7.21 (d, J=7.5 Hz, 1H), 7.14 (d, J=2.0 Hz, 1H), 7.07-7.02 (m, 2H), 7.02-6.96 (m, 1H), 6.82 (d, J=8.5 Hz, 1 H), 5.75 (dd, J=4.5, 9.5 Hz, 1H), 4.75 (dd, J=9.5, 14.8 Hz, 1H), 4.07 (q, J=7.0 Hz, 2H), 3.88-3.82 (m, 4H), 2.79 (s, 3H), 1.44 (t, J=6.9 Hz, 3H).
Realización 8: WX008

Ruta de síntesis:

Etapa 1: preparación del compuesto WX008-2
El compuesto WX008-1 (963.27 mg, 5.49 mmol) se disolvió en N,N-dimetilformamida (15.00 ml) a temperatura ambiente, seguido de la adición de diisopropiletilamina (2.13 g, 16.47 mmol, 2.88 ml) y el compuesto WX001-2 (1.50 g, 5.49 mmol). La mezcla de reacción se calentó a 130 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. Se añadió agua (20 ml) al residuo obtenido, se extrajo con acetato de etilo (15 ml x 3) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX008-2. MS-ESI m/z: 451.1 [M+Na]+.
Etapa 2: preparación del compuesto WX008-3
El compuesto WX008-2 (2.00 g, 4.66 mmol) y cloruro de amonio (2.49 g, 46.60 mmol, 1.63 ml) se añadieron a metanol (20.00 ml) a temperatura ambiente, seguido de la adición en porciones de polvo de zinc (1.52 g, 23.30 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 10 minutos en atmósfera de nitrógeno. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadieron agua (50 ml) y acetato de etilo (50 ml) al residuo obtenido y se separó la fase orgánica. Las fases acuosas se extrajeron con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX008-3. MS-ESI m/z: 421.1 [M+Na]+.
Etapa 3: preparación del compuesto WX008
El compuesto WX008-3 (100.00 mg, 250.69 pmol) y trietilamina (139.52 mg, 1.38 mmol, 191.12 pl) se disolvieron en tetrahidrofurano (1.00 ml) a 0 °C, seguido de la adición de trifosgeno (40.92 mg, 137.88 pmol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 12 horas. Después de la reacción, la mezcla se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX008.
MS-ESI m/z: 425.0 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.30 (br s, 1H), 7.12 (d, J=2.0 Hz, 1H), 7.11-7.01 (m, 4H), 6.82 (d, J=8.3 Hz, 1 H), 5.79 (dd, J=4.5, 9.5 Hz, 1 H), 4.73 (dd, J=9.7, 14.9 Hz, 1H), 4.06 (q, J=6.9 Hz, 2H), 3.91 -3.79 (m, 4H), 2.80 (s, 3H), 1.42 (t, J=7.0 Hz, 3H).
Realización 9: WX009
Ruta de síntesis:

Etapa 1: preparación del compuesto WX009-2
La sal de N-acetil-L-leucinato (500.00 mg, 1.12 mmol) del compuesto WX001-2 y compuesto WX009-1 (248.86 mg, 1.13 mmol) se disolvieron en N,N-dimetilformamida (5.00 ml) a temperatura ambiente, seguido de la adición de carbonato de potasio (300.21 mg, 2.17 mmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 4 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con salmuera saturada (10 ml), se diluyó con agua (30 ml) y se extrajo con diclorometano (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (100 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: diclorometano/éter de petróleo=3/1 -5/1, relación en volumen) para obtener el producto diana WX009-2. 1H RMN (400 MHz, DMSO_ate) 5: 8.67 (d, J=6.0 Hz, 1H), 8.18 (br s, 1H), 7.59 (d, J=8.3 Hz, 1H), 7.16 (br s, 1H), 7.08-6.77 (m, 3H), 5.24 (br s, 1H), 4.26 4.12 (m, 1H), 4.08-3.94 (m, 2H), 3.71 (br s, 4H), 2.96 (br s, 3H), 1.31 (d, J=6.0 Hz, 3H).
Etapa 2: preparación del compuesto WX009-3
El compuesto WX009-2 (200.00 mg, 422.53 pmol) en metanol (5.00 ml) a temperatura ambiente, seguido de la adición de polvo de zinc (276.29 mg, 4.23 mmol) y cloruro de amonio (226.01 mg, 4.23 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Después de la reacción, los insolubles se eliminaron por filtración y la torta del filtro se lavó con metanol (10 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto crudo WX009-3. 1H RMN (400 MHz, DMSO_ote) 5: 7.15-6.95 (m, 1H), 6.87 (br s, 2H), 6.69 (br s, 1H), 6,46 (d, J=7.5 Hz, 1H), 6.28 (d, J=8.0 Hz, 1H), 5.24 (d, J=8.3 Hz, 1 H), 4.3 (br s, 2 H), 4.81 (br s, 1 H), 4.19-3.86 (m, 2H), 3.70 (br s, 4H), 3.09-2.88 (m, 3H), 1.31 (d, J=6.5 Hz, 3H).
Etapa 3: preparación del compuesto WX009
El compuesto WX009-3 (150.00 mg, 338.33 pmol) y trietilamina (188.30 mg, 1.86 mmol) se disolvieron en tetrahidrofurano (20.00 ml) a 0 °C, seguido de la adición de trifosgeno (120.48 mg, 406.00 pmol). La mezcla de reacción se agitó a 0 °C durante 2 horas. Después de la reacción, la mezcla se apagó con salmuera saturada (15 ml), se diluyó con agua (15 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (25 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX009. MS-ESI m/z: 469.0 [M+H]+, 471.0 [M+H+2]+. 1H RMN (400 MHz, DMSO_ate) 5: 11.16 (s, 1H), 7.24-7.06 (m, 4H), 7.02-6.86 (m, 2H), 5.91 (d, J=6.8 Hz, 1H), 4.56-4.39 (m, 1 H), 4.23-4.11 (m, 1 H), 4.05-3.90 (m, 2H), 3.71 (s, 3H), 3.01 (s, 3H), 1.30 ( t, J=6.8 Hz, 3H).
Realización 10: WX010
Ruta de síntesis:

El compuesto WX009 (2.00 g, 4.26 mmol) se disolvió en metanol (40.00 ml) a temperatura ambiente, seguido de la adición de carbón de paladio (0.50 g, 10 % de pureza) bajo atmósfera de argón. El sistema de reacción se despresurizó a vacío y se llenó con hidrógeno. El procedimiento anterior se repitió tres veces. La mezcla de reacción se agitó a temperatura ambiente bajo una atmósfera de hidrógeno (30 psi) durante 10 horas. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX010. MS-ESI m/z: 391.1 [M+H]
+
.
1
H RMN (400 MHz, CDCl
3
) 5: 9.71 (br s, 1 H), 7.22-6.95 (m, 6 H), 6.81 (d, J=7.0 Hz, 1 H), 5.86 (br s, 1H), 4.76 (br s, 1 H), 4.06 (d, J=6.5 Hz, 2H), 3.91 (d, J=13.3 Hz, 1H), 3.83 (br s, 3H), 2.76 (br s, 3H), 1.41 (t, J=5.9 Hz, 3H).
Realización 11: WX011

Ruta de síntesis:

El compuesto WX009 (280 mg, 596.57 pmol) y ácido o-fluorofenilborónico (125.21 mg, 894.86 pmol) se disolvieron en dioxano (5.00 ml) y agua (2.00 ml) a temperatura ambiente, seguido de la adición de tetrakistrifenilfosfina paladio (68.94 mg, 59.66 pmol) y carbonato de potasio (90.70 mg, 656.23 pmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con salmuera saturada (20 ml), se diluyó con agua (20 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (30 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX011. MS-ESI m/z: 485.2 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.20 (br s, 1H), 7.39 (t, J=7.7 Hz, 1H), 7.44-7.28 (m, 1H), 7.35-7.24 (m, 4H), 7.24-7.03 (m, 5H), 7.24-7.00 (m, 1H), 6.82 (d, J=8.3 Hz, 1H), 5.87 (d, J=4.8 Hz, 1H), 4.78 (dd, J=9.7, 14.2 Hz, 1H), 4.07 (q, J=6.8 Hz, 2H), 3.96-3.74 (m, 4H), 2.81 (s, 3H), 1.42 (t, J 6.9 Hz, 3H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 11.
Tabla 1


Datos de LCMS y 1H RMN de cada realización
Tabla 2

Realización 20: WX020
Ruta de síntesis:

El compuesto WX003 (50.00 mg, 117.67 pmol) y ácido ciclopropilborónico (20.22 mg, 235.34 pmol) se disolvieron en diclorometano (2.0 ml) a temperatura ambiente, seguido de la adición de 2,2'-bipiridina (18.38 mg, 117.67 pmol) y acetato de cobre (42.74 pmol) mg, 235.34 pmol). La mezcla de reacción se agitó a temperatura ambiente durante 24 horas. Después de la reacción, se añadió agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (10 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX020. MS-ESI m/z: 465.1 [M H]+. 1H RMN (400 MHz, MeOD) 5: 7.30 (d, J=1.5 Hz, 1H), 7.17-7.10 (m, 2H), 7.07 (d, J=8.5 Hz, 2H), 6.95 (d, J=8.5 Hz, 1H), 6.02 (dd, J=3.8, 10.8 Hz, 1 H), 4.56 (dd, J=10.8, 14.3 Hz, 1H), 4.16-3.97 (m, 3H), 3.82 (s, 3H), 3.04-2.87 (m, 4H), 1.39 (t, J=6.8 Hz, 3H), 1.20-1.06 (m, 2H), 1.01-0.93 (m, 2H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 20.
Tabla 3

Datos de LCMS y 1HRMN de cada realización
Tabla 4


Realización 24: WX024

Ruta de síntesis:

Etapa 1: preparación de WX024-1
El compuesto WX009 (3.00 g, 6.39 mmol) y bis (pinacolato) diboro (1.78 g, 7.03 mmol) se disolvieron en dioxano (60.00 ml) a temperatura ambiente, seguido de la adición de complejo (1,1 '-bis(difenilfosfina)ferroceno). dicloruro de paladio en diclorometano (521.83 mg, 639.00 pmol) y acetato de potasio (1.88 g, 19.17 mmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 4 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna ultrarrápida de gel de sílice (eluyente: acetato de etilo éter de petróleo=0/1 -17/1, relación en volumen) para obtener el producto diana WX024-1. 1H RMN (400 MHz, CDCh) 5: 8.67 (br s, 1 H), 7.60 (dd, J=0.9, 7.9 Hz, 1H), 7.50 (s, 1H), 7.21-7.10 (m, 2H), 7.06 (dd, J=2.0, 8.3 Hz, 1H), 6.87-6.74 (m, 1H), 5.82 (dd, J=4.8, 9.3 Hz, 1H), 4.74 (dd, J=9.4, 14.9 Hz, 1H), 4.07 (q, J=7.0 Hz, 2H), 3.90 (dd, J=4.8, 14.8 Hz, 1H), 3.84 (s, 3H), 2.73 (s, 3H), 1.34 (s, 12H), 1.31-1.23 (m, 3H).
Etapa 2: preparación de WX024
El compuesto WX024-1 (50.00 mg, 96.82 pmol) y 1-bromo-2-etoxibenceno (38.93 mg, 193.64 pmol) se disolvieron en dioxano (3.00 ml) y agua (1.00 ml) a temperatura ambiente, seguido de la adición de carbonato de potasio (26.76 mg, 193.64 pmol) y complejo (1,1'-bis(difenilfosfino)ferroceno). cloruro de paladio en diclorometano (3.54 mg, 4.84 pmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se le añadió agua (5.00 ml), se agitó durante 5 minutos y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (8 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: acetato de etilo/éter de petróleo=2/1, relación en volumen) y se purificó adicionalmente mediante HPLC preparativa para obtener el producto diana WX024. MS-ESI m/z: 511.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.50 (br s, 1H), 7.35-7.26 (m, 4H), 7.19-7.12 (m, 2H), 7.07 (d, J=8.3 Hz, 1 H), 7.04-6.93 (m, 2H), 6.81 (d, J=8.3 Hz, 1H), 5.99-5.79 (m, 1H), 4.71 (dd, J=9.5, 14.1 Hz, 1H), 4.10-4.00 (m, 4H), 3.92 (d, J=14.6 Hz, 1H), 3.82 (br s, 3 H), 2.77 (br s, 3H), 1.44-1.39 (m, 3H), 1.36-1.30 (m, 3H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 24.
Tabla 5

Datos de LCMS y de 1HRMN de cada realización
Tabla 6

Realización 31: WX031

Ruta de síntesis:
El Compuesto WX024-1 (150.00 mg, 290.47 pmol), trifenilfosfina (7.62 mg, 29.05 pmol) y benzofenona (52.93 mg, 290.47 pmol) se disolvieron en etanol (1.00 ml) a temperatura ambiente, seguido de la adición de acetato de paladio (3.26 mg, 14.52 pmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 horas bajo una atmósfera de monóxido de carbono (15 psi). Los insolubles se eliminaron por filtración y el filtrado se purificó directamente mediante HPLC preparativa para obtener el producto diana WX031. MS-ESI m/z: 463.1 [M+H]+ . 1H RMN (400 MHz, CDCl3) 5: 9.96 (br s, 1H), 7.84 (dd, J=1.4, 8.4 Hz, 1 H), 7.75 (d, J=1.0 Hz, 1 H), 7.18-7.09 (m, 2H), 7.05 (dd, J=1.6 , 8.4 Hz, 1H), 6.82 (d, J=8.3 Hz, 1H), 5.88 (dd, J=4.5, 9.3 Hz, 1H), 4.71 (dd, J=9.5, 14.6 Hz, 1H), 4.35 (q, J=7.2 Hz, 2H), 4.04 (q, J=6.9 Hz, 2H), 3.92 (dd, J=4.5, 14.6 Hz, 1H), 3.82 (s, 3H), 2.80 (s, 3H), 1.39 (td, J=6.9, 10.5 Hz, 6 H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 31.
Tabla 7

Datos de LCMS Y 1HRMN de cada realización
Tabla 8

Realización 34: WX034
Ruta de síntesis:

El compuesto WX031 (255.00 mg, 551.33 pmol) se disolvió en metanol (15.00 ml) y agua (15.00 ml) a temperatura ambiente, seguido de la adición monohidrato de hidróxido de litio (69.40 mg, 1.65 mmol). La mezcla de reacción se calentó a 60 °C y se agitó durante 0.5 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se extrajo con diclorometano (15 ml x 3). Las fases orgánicas se descartaron y las fases acuosas se acidificaron con ácido clorhídrico acuoso diluido (3 M) a pH = 4 y se extrajeron con acetato de etilo (15 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (45 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX034. MS-ESI m/z: 435.0 [M H]+. 1H RMN (400 MHz, CDCh) 5: 11.23 (s, 1H), 7.61 (dd, J=1.1,8.4 Hz, 1H), 7.50 (s, 1H), 7.35 (d, J=8.3 Hz, 1H), 7.15 (d, J=1.5 Hz, 1H), 7.06-6.97 (m, 1 H), 6.96-6.89 (m, 1 H), 5.97 (dd, J=4.0, 10.0 Hz, 1H), 4.53 (br dd, J= 10.4, 14.4 Hz, 1 H), 4.20 (br dd , J= 4.0, 14.6 Hz, 1H), 4.00 (br t, J= 6,8 Hz, 2H), 3,72 (s, 3H), 3,02 (s, 3H), 1,31 (t, J=7,0 Hz, 3H).
Realización 35: WX035

Ruta de síntesis:

Etapa 1: preparación de WX035-2
El compuesto WX035-1 (2.16 g, 12.81 mmol) se agregó a ácido sulfúrico concentrado (17 ml, 98 %) a temperatura ambiente. La mezcla de reacción se enfrió a -15 °C, seguido de la adición gota a gota de la mezcla de ácido nítrico concentrado (0.915 ml, 63 %) y ácido sulfúrico concentrado (3 ml, 98 %). La reacción se llevó a cabo durante 30 minutos con agitación. Después de la reacción, la mezcla de reacción se vertió en agua helada (50 ml), seguido de la adición de acetato de etilo (100 ml). Las fases orgánicas se lavaron con salmuera saturada (30 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/5, relación en volumen) para obtener el producto diana WX035-2. 1H RMN (400 MHz, CDCh) 5: 8.43 (d, J=1.6 Hz, 1H), 8.09 (dd, J=2.0, 8.4 Hz, 1H)), 7.67 (d, J=8.0 Hz, 1H), 3.02 (q, J=7.6 Hz, 2H), 1.26 (t, J=7.2 Hz, 3H).
Etapa 2: preparación de WX035-3
El compuesto WX035-2 (2.00 g, 9.36 mmol) y trifluoruro de dietilaminosulfuro (7.55 g, 46.81 mmol) se disolvieron en diclorometano (50.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 60 °C y se agitó durante 16 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se vertió en agua helada (30 ml), seguido de la adición de diclorometano (100 ml) con agitación durante 10 minutos. Las fases
orgánicas se lavaron con salmuera saturada (30 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/20, relación en volumen) para obtener el producto diana WX035-3. 1H RMN (400 MHz, CDCta) ó: 7.98 (s, 1H), 7.65-7.60 (m, 2H), 2.23-2.09 (m, 2H), 1.02 (t, J=7.6 Hz, 3H).
Etapa 3: preparación de WX035-4
El compuesto WX035-3 (950.00 mg, 4.03 mmol), compuesto WX001-2 (1.65 g, 6.05 mmol) y diisopropiletilamina (1.56 g, 12.10 mmol, 2.11 ml) se disolvieron en N,N-dimetilformamida (20.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 100 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. Se añadieron acetato de etilo (100 ml) y agua (30 ml) y se lavó con salmuera saturada (30 ml x 2) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: acetato de etilo/éter de petróleo=1/1, relación en volumen) para obtener el producto diana WX035-4. MS-ESI m/z: 495.2 [M+Na]+.
Etapa 4: preparación de WX035-5
El compuesto WX035-4 (280.00 mg, 592.59 pmol), polvo de zinc (310.00 mg, 4.74 mmol) y cloruro de amonio (253.58 mg, 4.74 mmol) se añadieron a metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas en atmósfera de nitrógeno. Los insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (50 ml) al residuo y se agitó durante 15 minutos. Los insolubles se eliminaron por filtración de nuevo y el filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX035-5. MS-ESI m/z: 443.1 [M+Na]+.
Etapa 5: Preparación de WX035
El compuesto WX035-5 (30.00 mg, 71.34 pmol), trifosgeno (10.59 mg, 35.67 pmol) y trietilamina (21.66 mg, 214.02 pmol, 29.67 pl) se disolvieron en tetrahidrofurano (2.00 ml) a 0-5 °C. La mezcla de reacción se agitó a 0-5 °C durante 2 horas en atmósfera de nitrógeno. Después, la mezcla de reacción se vertió en agua helada (10 ml) con agitación a temperatura ambiente durante 10 minutos y luego se extrajo con acetato de etilo (30 ml x 2). Las fases orgánicas se separaron y la fase acuosa se extrajo con acetato de etilo (30 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX035. MS-ESI m/z: 447.0 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 8,54 (s, 1H), 7.77 (d, J=8.4 Hz, 1H), 7.70 (s, 1H), 7.15-7.05 (m, 3H), 6.84 (d, J=8.4 Hz 1H), 5.85 (dd, J=4.0, 9.2 Hz, 1H), 4.73 (dd, J=9.6, 14.8 Hz, 1H), 2.98 (q, J=7.2 Hz, 2H), 3.91-3.85 (m, 4H), 2.98 (q, J=7.2 Hz, 2H), 2.81 (s, 3H), 1.44 (t, J=6.8 Hz, 3H), 1.23 (t, J=7.2 Hz, 3H).
Realización 36: WX036

Ruta de síntesis:

El compuesto WX003 (300.00 mg, 706.05 pmol), 2,2-difluoroetil-4-metilbencenosulfonato (498.25 mg, 2.12 mmol) y carbonato de potasio (292.75 mg, 2.12 mmol) se disolvieron en N,N-dimetilformamida (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 12 horas en atmósfera de nitrógeno. Después de la reacción, se añadieron a la mezcla acetato de etilo (100 ml) y agua (30 ml) y se separaron las fases orgánicas. Las fases acuosas se lavaron con salmuera saturada (30 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1 /5-1/1) para obtener el producto diana WX036. MS-ESI m/z: 511.0 [M+Na]+. 1H RMN (400 MHz, CDCh) 5: 7.09-6.82 (m, 6H), 6.03-6.02 (m, 1H), 5.80-5.76 (m, 1H), 4.71-4.66 (m, 1H), 4.22-4.03 (m, 4H), 3.85- 3.81 (m, 4H), 2.77 (s, 3H), 1.45 (t, J=6.4 Hz, 3H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 36.
Tabla 9





Datos de LCMS Y 1HRMN de cada realización Tabla 10



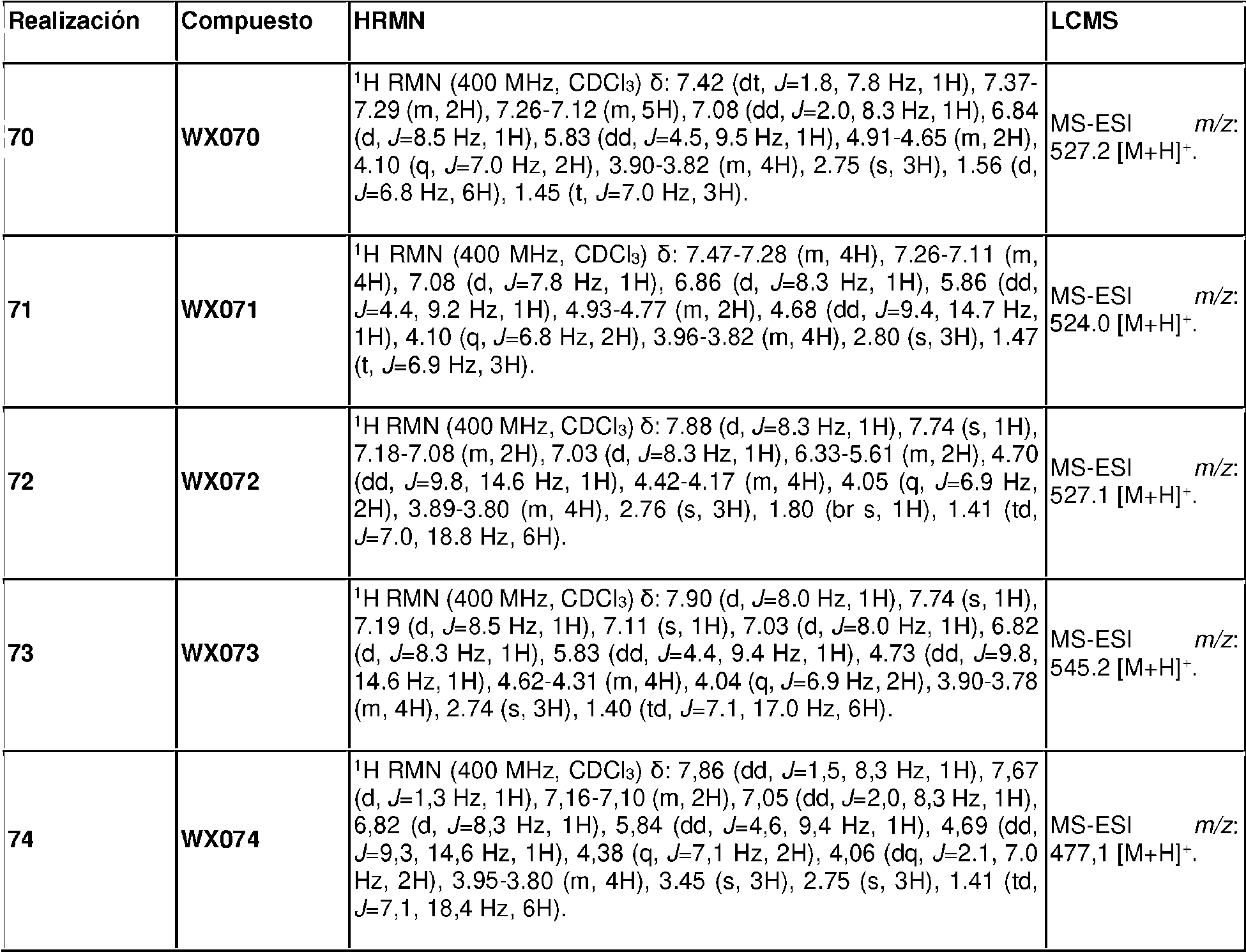
Realización 75: WX075
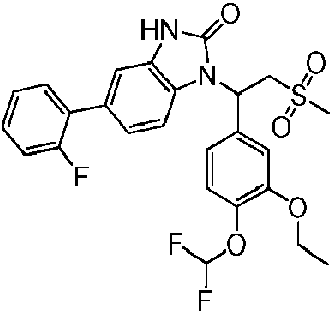
Ruta de síntesis:
Etapa 1: preparación de WX075-2
El compuesto WX075-1 (29.00 g, 177,73 mmol), difluorocloroacetato de sodio (62.32 g, 408.78 mmol) y carbonato de cesio (86.86 g, 266.60 mmol) se añadieron a N,N-dimetilformamida (600 ml) y agua (150 ml). La mezcla de reacción se calentó a 100 °C y se agitó durante 3 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se añadieron acetato de etilo (1000 ml) y agua (200 ml). Las fases orgánicas se separaron, se lavaron con salmuera saturada (200 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/10-1/3, relación en volumen) para obtener el producto diana WX075-2. 1H RMN (400 MHz, CDCla ) 5: 7.20-7.13 (m, 3H), 6.58 (t, J=74.4 Hz, 1H), 4.05 (q, J=6.8 Hz, 2H), 1.41 (t, J=6.8 Hz, 3H).
Etapa 2: preparación de WX075-3
Se disolvió dimetilsulfona (9.71 g, 103.20 mmol, 8.37 ml) en tetrahidrofurano (300 ml) a temperatura ambiente, después de enfriar a 0 °C, se añadió gota a gota n-butillitio (2.5 M, 41.28 ml). La mezcla de reacción se agitó a 0-5 °C durante 1 hora en atmósfera de nitrógeno. El compuesto WX075-2 (20.00 g, 93.82 mmol) en tetrahidrofurano (200 ml) se añadió gota a gota a la mezcla de reacción anterior. Posteriormente, la mezcla de reacción se agitó a 0-5 °C durante 30 minutos y luego se calentó a temperatura ambiente y se agitó durante 1.5 horas. Se añadió borohidruro de sodio (4.61 g, 121.97 mmol) a la mezcla de reacción anterior a temperatura ambiente. Después de agitar durante 30 minutos, la mezcla se enfrió a 0° C y se añadió ácido acético (25.92 g, 431.57 mmol, 24.69 ml). La mezcla de reacción se agitó a 0-5 °C durante 2 horas. Finalmente, se añadió una solución acuosa de hidróxido de sodio (2.5 M, 123.84 ml) al sistema de reacción. La mezcla de reacción se agitó a 0-5 °C durante 30 minutos y luego se calentó a 60 °C y se agitó durante 12 horas más. Después de que la reacción y la mezcla se enfriaran a temperatura ambiente, se añadieron acetato de etilo (1000 ml) y agua (200 ml). Las fases orgánicas se separaron, se lavaron con salmuera saturada (200 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/100-1/10, relación en volumen) para obtener el producto diana WX075-3. 1H RMN (400 MHz, CDCl3) 5: 7.08-6.84 (m, 3H), 6.50 (t, J=75.2 Hz, 1H), 4.58 (dd, J=2.8, 9.6 Hz, 1H), 4.04 (dd, J=6.8, 13.2 Hz, 2H), 3.27-3.12 (m, 2H), 2.90 (s, 3H), 1.38 (t, J=7.2 Hz, 3H).
Etapa 3: preparación de WX075-4
El compuesto WX075-3 (400.00 mg, 1.29 mmol) y el compuesto WX009-1 (426.73 mg, 1.94 mmol) se disolvieron en N,N-dimetilformamida (15.00 ml) a temperatura ambiente, seguido de la adición de diisopropiletilamina (501.37 mg, 3.88 mmol). La mezcla de reacción se calentó a 90 °C y se agitó durante 15 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla de reacción se enfrió a temperatura ambiente, se diluyó con agua (30 ml) y se extrajo con acetato de etilo (40 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (20 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/9-1/1, relación en volumen) para obtener el producto diana WX075-4. 1H Rm N (400 MHz, CDCl3) 5: 8.65 (d, J=6.3 Hz, 1H), 8.34 (d, J=2.3 Hz, 1H), 7.45 (dd, J=2.3, 9.0 Hz, 1H), 7.18 (d, J=8.5 Hz, 1H), 6.97-6.92 (m, 2H), 6.75 (s, 0.25 H), 6.66 (d, J=9.3 Hz, 1H), 6.57 (s, 0.5H), 6.38 (s, 0.25H), 5.27-5.19 (m, 1H), 4.10-4.00 (m, 2H), 3.63 (dd, J=8.8 , 14.6 Hz, 1H), 3.46 (dd, J=4.0, 14.6 Hz, 1H), 2.88 (s, 3H), 1.44 (t, J=6.9 Hz, 3H).
Etapa 4: preparación de WX075-5
El compuesto WX075-4 (200.00 mg, 392.68 pmol) y cloruro de amonio (210.04 mg, 3.93 mmol) se disolvieron en etanol (10.00 ml) y agua (1.00 ml) a temperatura ambiente, seguido de la adición de polvo de hierro (109.66 mg, 1.96 mmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 1 hora en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. Se añadió diclorometano (50 ml) al residuo y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/4-4/1, relación en volumen) para obtener el producto diana WX075-5. 1H Rm N (400 MHz, CDCl3) 5: 7.15 (d, J=7.8 Hz, 1H), 6.96-6.90 (m, 2H), 6.84 (d, J=2.0 Hz, 1H), 6.75 (s, 0.25H), 6.70 (dd, J=2.1,8.4 Hz, 1H), 6.56 (s, 0.5H), 6.37 (s, 0.25H), 6.22 (d, J=8.5 Hz, 1H), 4.92 (td, J=4.4, 9.5 Hz, 1H), 4.43 (d, J=5.0 Hz, 1H), 4.04 (dq, J=2.1,7.0 Hz, 2H), 3.65 (s, 2H), 3.54-3.45 (m, 1H), 3.43 3.37 (m, 1H), 2.90 (s, 3H), 1.41 (t, J=7.0 Hz, 3H).
Etapa 5: Preparación de WX075-6
El compuesto WX075-5 (190.00 mg, 396.38 pmol.), o-Acido fluorofenilborónico (72.10 mg, 515.29 pmol), complejo (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (16.18 mg, 19.82 pmol) y carbonato de potasio (136.96 mg, 990.95 pmol) se agregaron a dioxano (4.50 ml) y agua (1.50 ml) a temperatura ambiente. La mezcla de reacción se calentó a 70 °C y se agitó durante 15 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se diluyó con agua (20 ml) y acetato de etilo (30 ml) y se filtró para eliminar los insolubles. Las fases orgánicas se separaron y las fases acuosas se extrajeron con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron y se lavaron con salmuera saturada (20 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía
en columna (eluyente: acetato de etilo/éter de petróleo=0/1-3/2, relación en volumen) para obtener el producto diana WX075-6. 1H RMN (400 MHz, CDCh) 5: 7.34 (dt, J=1.6, 7.7 Hz, 1H), 7.25-7.19 (m, 1H), 7.19-7.12 (m, 2H), 7.12-7.06 (m, 1H), 7.03-6.95 (m, 3H), 6.85 (d, J=8.0 Hz, 1H), 6.75 (s, 0.25H), 6.57 (s, 0.5H), 6.47 (d, J=8.0 Hz, 1H), 6.38 (s, 0.25H), 5.02 (dd, J=3.6, 9.4 Hz, 1H), 4.07 (q, J=6.9 Hz, 2H), 3.58-3.51 (m, 1H), 3.46-3.41 (m, 1H), 2.91 (s, 3H), 1.42 (t, J=6.9 Hz, 3H).
Etapa 6: Preparación de WX075
El compuesto WX075-6 (100.00 mg, 202.21 pmol) y trietilamina (122.77 mg, 1.21 mmol, 168.18 pl) se disolvieron en tetrahidrofurano (10.00 ml). La mezcla de reacción se enfrió a 0° C, seguido de la adición de trifosgeno (36.00 mg, 121.33\ pmol). La mezcla de reacción se agitó a 0 °C durante 30 minutos en atmósfera de nitrógeno. Después de la reacción, la mezcla se apagó con agua (3 ml) y se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX075. MS-ESI m/z: 521.0 [M+H]+ . 1H RMN (400 MHz, CD3OD) 5: 7.45 (dt, J=1.6 , 7.8 Hz, 1H), 7.36-7.30 (m, 1H), 7.29-7.27 (m, 1H), 7.27-7.23 (m, 3H), 7.22 (d, J=1.0 Hz, 1H), 7.18 (d, J=1.0 Hz, 1H), 7.14 (s, 1H), 7.13-7.09 (m, 1H), 6.90 (s, 0.25H), 6.71 (s, 0.5H), 6.52 (s, 0.25H), 6.13 (dd, J=3.8, 10.5 Hz, 1H), 4.68 (dd, J=10.5, 14.8 Hz, 1H), 4.16-4.05 (m, 3H), 2.99 (s, 3H), 1.39 (t, J=7.0 Hz, 3H).
Realización 76: WX076

Ruta de síntesis:

Etapa 1: preparación de WX076-2
Se disolvió dimetilsulfona (848.34 mg, 9.01 mmol, 731.33 pl) en tetrahidrofurano (10.00 ml), se enfrió a 0° C, seguido de la adición gota a gota de n-butillitio (2.5 M, 3.30 ml) en atmósfera de nitrógeno. La mezcla de reacción se agitó a 0 °C durante 1 hora. Para el sistema de reacción anterior, una solución de compuesto WX076-1 (1.00 g, 7.51 mmol) en tetrahidrofurano (10.00 ml), se añadió gota a gota y la mezcla de reacción se agitó a 0 °C durante 15 minutos, luego se calentó a temperatura ambiente y se agitó durante otras 1.5 horas. La mezcla de reacción se enfrió de nuevo a 0° C y se le añadió borohidruro de sodio (369.35 mg, 9.76 mmol) con agitación durante 10 minutos. Luego, se le añadió ácido acético (2.03 g, 33.80 mmol, 1.93 ml) a 0 °C con agitación durante 2 horas. Finalmente, se le añadió una solución acuosa de hidróxido de sodio (2.5 M, 9.,91 ml) a 0 °C con agitación durante 15 minutos y luego se calentó a 60 °C con agitación durante 12 horas más. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con una solución saturada de cloruro de amonio (5 ml), se extrajo con acetato de etilo (10 ml x 3) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX076-2.
Etapa 2: preparación de WX076-3
El compuesto WX076-2 (1.00 g, 4.38 mmol) y piridina (5.20 g, 65.70 mmol, 5.31 ml) se disolvieron en etanol (10.00 ml) a temperatura ambiente, seguido de la adición de hidrocloruro de O-metilhidroxilamina (1.10 g, 13.14 mmol). La mezcla de reacción se calentó a 80 °C y se agitó durante 4 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se diluyó con salmuera saturada (40 ml) y se extrajo con diclorometano (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con ácido clorhídrico diluido al 10 % (30 ml) y agua (30 ml) respectivamente y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX076-3.
Etapa 3: preparación de WX076-4
El compuesto WX076-3 (2.00 g, 7.77 mmol) se disolvió en tetrahidrofurano (40.00 ml) a temperatura ambiente y se enfrió a 0 °C, seguido de la adición gota a gota de una solución de borano dimetil sulfuro (10 M, 3.89 ml). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 2.5 horas en atmósfera de nitrógeno. Después de la reacción, se añadió lentamente metanol a la mezcla para apagar la reacción hasta que no se produjo gas. Luego, la mezcla se concentró a presión reducida y se le añadió una solución acuosa de hidróxido de sodio (2 M, 10 ml) y se extrajo con acetato de etilo (15 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX076-4.
Etapa 4: preparación de WX076-5
El compuesto WX076-4 (1.10 g, 3.84 mmol) y 4-bromo-1-fluoro-2-nitrobenceno (1.69 g, 7.68 mmol) se disolvieron en N,N-dimetilformamida (30.00 ml) a temperatura ambiente, seguido de la adición de diisopropiletilamina (1.49 g, 11.52 mmol, 2.01 ml). La mezcla de reacción se calentó a 80 °C y se agitó durante 4 horas. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con salmuera saturada (20 ml), se diluyó con agua (40 ml) y se extrajo con acetato de etilo (20 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (40 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1 /8-1 /2 , relación en volumen) para obtener el producto diana WX076-5. 1H r Mn (400 MHz, CDCh) 5: 8.64 (d, J=6.8 Hz, 1H), 8.34 (d, J=2.3 Hz, 1H), 7.44 (dd, J=2.4, 9.2 Hz, 1H), 7.36-7.29 (m, 2H), 7.00-6.85 (m, 2H), 6.72 (d, J=9.0 Hz, 1H), 5.29 5.16 (m, 1H), 3.81 (s, 3H), 3.68-3.58 (m, 1H), 3.54-3.44 (m, 1H), 2.77 (s, 3H).
P aso 5: Preparación de WX076-6
El compuesto WX076-5 (1.35 g, 3.14 mmol) se disolvió en metanol (25.00 ml) a temperatura ambiente, seguido de la adición de polvo de zinc (1.03 g, 15.70 mmol) y cloruro de amonio (1.68 g, 31.40 mmol, 1.10 ml). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. Después de la reacción, los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. Se añadió diclorometano (20 ml) al residuo con agitación durante 10 minutos, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX076-6. 1H RMN (400 MHz, CDCla ) 5: 7.28 (d, J=7.8 Hz, 2H), 6.90 (d, J=8.5 Hz, 2H), 6.83 (d, J=2.3 Hz, 1H), 6.70 (dd, J=2.1, 8.4 Hz, 1H), 6.28 (d, J=8.5 Hz, 1H), 5.01-4.84 (m, 1H), 4.34 (br. s., 1H), 3,80 (s, 3H), 3,74-3,58 (m, 2H), 3,58-3,36 (m, 2H), 2,81 (s, 3H).
Etapa 6: Preparación de WX076-7
El compuesto WX076-6 (1.25 g, 3,13 mmol) y trietilamina (1,27 g, 12,52 mmol, 1,74 ml) se disolvieron en tetrahidrofurano (40,00 ml) a temperatura ambiente y se enfriaron a 0 °C, seguido de la adición en porciones de trifosgeno (371,59 mg, 1,25 mmol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 6 horas. Después de la reacción, la mezcla se apagó con salmuera saturada (10 ml), se diluyó con agua (50 ml) y se extrajo con acetato de etilo (15 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/3-1/1, relación en volumen) para obtener el producto diana WX076-7.
Etapa 7: Preparación de WX076
El compuesto WX076-7 (250.00 mg, 587.82 pmol) y ácido o-fluorofenilborónico (99.52 mg, 711.26 pmol) se disolvieron en dioxano (10.00 ml) a temperatura ambiente, seguido de la adición de complejo (1 ,1 '-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (48.00 mg, 58.78 pmol), carbonato de potasio (121.86 mg, 881.73 pmol) y agua (3.00 ml). La mezcla de reacción se calentó a 80 °C y se agitó durante 3 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se apagó con salmuera saturada (5 ml), se diluyó con agua (25 ml) y se extrajo con acetato de etilo (5 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (20 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX076. MS-ESI m/z: 441.1 [M+H]+ . 1H RMN (400 MHz, CDCla ) 5: 8.61 (br s, 1H), 7.49 (d, J=8.5 Hz, 2H), 7.44-7.37 (m, 1H), 7.31 (d, J=7.0 Hz,
2H), 7.26 (br s, 1 H), 7.24-7.09 (m, 3H), 6.90 (d, J=8.5 Hz, 2H), 5.90 (dd, J=4.3, 9.5 Hz, 1H), 4.79 (dd, J=9.8, 14.8 Hz, 1H), 3.87 (dd, J=3.5, 14.3 Hz, 1H), 3.79 (s, 3H), 2.80 (s, 3H).
Realización 77: WX077

Se disolvió dimetilsulfona (5.22 g, 55.50 mmol) en tetrahidrofurano (50.00 ml) en atmósfera de nitrógeno y se enfrió a 0 °C, seguido de la adición gota a gota de n-butiNitio (2.5 M, 22.20 ml) con agitación a 0 °C durante 1 hora. Luego, una solución del compuesto WX077-1 (5.00 g, 27.75 mmol) en tetrahidrofurano (20.00 ml) se añadió gota a gota al sistema anterior y la mezcla se agitó a 0 °C durante 1.5 horas. Después de la reacción, la mezcla se calentó a temperatura ambiente y se apagó con una solución saturada de cloruro de amonio (50 ml), se diluyó con agua (150 ml) y se extrajo con acetato de etilo (50 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (100 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se recristalizó con acetato de etilo (20 ml) para obtener el producto diana WX077-2. 1H RMN (400 MHz, DMSO_de) 5: 6.99 (s, 1 H), 6.91 (s, 2H), 5.83 (d, J=4.0 Hz, 1H), 4.96 (ddd, J=2.8, 4.3, 10.0 Hz, 1H), 4.02 (q, J=7.0 Hz, 2H), 3.73 (s, 3H), 3.57 (dd, J=10.0, 14.6 Hz, 1H), 3.15 (d, J=14.6 Hz, 1H), 3.01 (s, 3H), 1.33 (t, J=7.0 Hz, 3H).
Etapa 2: preparación de WX077
El compuesto WX077-2 (300.00 mg, 1.09 mmol), el compuesto WX077-3 (233.28 mg, 1.09 mmol) y trifenilfosfina (343.08 mg, 1.31 mmol) se disolvieron en tetrahidrofurano (5.00 ml) en atmósfera de nitrógeno y se enfriaron a 0 °C, seguido de la adición gota a gota de azodicarboxilato de diisopropilo (264.49 mg, 1.31 mmol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 24 horas. Después de la reacción, la mezcla se apagó con salmuera saturada (10 ml), se diluyó con agua (20 ml) y se extrajo con acetato de etilo (15 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (25 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: metanol/diclorometano=0/1-1/30, relación en volumen), seguido de HPLC preparativa para obtener el producto diana WX077. MS-ESI m/z: 492.0 [M+Na]+, 494.0 [M+Na+2]+. 1H RMN (400 MHz, DMSO_de) 5: 7.67 (s, 1H), 7.38 (s, 2H), 7.14 (d, J=1.5 Hz, 1 H), 7.07 (d, J=8.3 Hz, 1H), 6.95 (d, J=8.3 Hz, 1H), 5.89 (dd, J=4.0, 10.5 Hz, 1H), 4.43 (dd, J=10.8, 14.6 Hz, 1H), 4.24 (dd, J=4.0, 14.6 Hz, 1H), 4.06-3.96 (m, 2H), 3.73 (s, 3H), 3.06 (s, 3H), 1.31 (t, J=6.9 Hz, 3H). Realización 78: WX078
Ruta de síntesis:

Etapa 1: preparación de WX078-1
El compuesto WX007-2 (450.00 mg, 950.69 pmol), 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolano-2-il)pirazol (296.70 mg, 1.43 mmol) y carbonato de potasio (394.18 mg, 2.85 mmol) se disolvieron en dioxano (14.00 ml) y agua (4.30 ml) a temperatura ambiente, seguido de la adición de complejo (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (116.46 mg, 142.60 pmol). La mezcla de reacción se calentó a 75 °C y se agitó durante 13 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se le añadieron agua (30 ml) y acetato de etilo (40 ml) con agitación durante 5 minutos. Los materiales insolubles se eliminaron por filtración y la torta del filtro se lavó con acetato de etilo (30 ml). El filtrado se combinó y la fase orgánica se separó, se lavó con agua (50 ml x 3) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante cromatografía en columna (eluyente: acetato de etilo/éter de petróleo=1/2-2/1, relación en volumen) para obtener el producto diana WX078-1. 1H RMN (400 MHz, CDCh) 5: 7.40-7.51 (m, 2H), 7.18 (t, J=8.0 Hz, 1H), 6.92 (br d, J=1.8 Hz, 1 H), 6.83-6.89 (m, 2 H), 6.74 (d, J=7.3 Hz, 1 H), 6.65 (d, J=8.3 Hz, 1 H), 5.95 (d, J=6.0 Hz, 1 H), 5.06-5.12 (m, 1 H), 4.02-4.09 (m, 2 H), 3.90 (s, 3 H), 3.84 (s, 3 H), 3.36 3.60 (m, 2 H), 2.80 (s, 3 H), 1.42 (t, J=7.0 Hz, 3 H).
Etapa 2: preparación de WX078-2
El compuesto WX078-1 (415.00 mg, 874.55 pmol) y cloruro de amonio (467.80 mg, 8.75 mmol) se disolvieron en metanol (10.00 ml) a temperatura ambiente, seguido de la adición de polvo de zinc (285.93 mg, 4.37 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora en atmósfera de nitrógeno. Los insolubles se eliminaron por filtración y la torta del filtro se añadió a una mezcla de disolvente de acetato de etilo (50 ml) y diclorometano (5 ml) con agitación a temperatura ambiente durante 15 minutos, seguido de filtración. Los insolubles se eliminaron por filtración. El filtrado se combinó, se lavó con salmuera saturada (50 ml x 2) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX078-2. MS-ESI m/z: 467.1 [M+Na]+.
Etapa 3: preparación de WX078
El compuesto WX078-2 (450.00 mg, 1.01 mmol) y trietilamina (563.37 mg, 5.57 mmol, 771.74 pl) se disolvieron en tetrahidrofurano (15.00 ml) a temperatura ambiente, seguido de la adición de trifosgeno (240.31 mg, 809.81 pmol). La mezcla de reacción se agitó a temperatura ambiente durante 15 horas en atmósfera de nitrógeno. Después de la reacción, el disolvente se eliminó por concentración a presión reducida. Se añadió agua (40 ml) al residuo obtenido y se extrajo con acetato de etilo (30 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (60 ml) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX078. MS-ESI m/z: 471.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.95 (br s, 1 H), 7.86 (s, 1 H), 7.70 (s, 1 H), 7.02-7.17 (m, 5 H), 6.82 (d, J=8.3 Hz, 1 H), 5.85 (dd, J=9.0, 4.8 Hz, 1 H), 4.71 (dd, J=14.7, 9.4 Hz, 1 H), 4.04 (q, J 6.9 Hz, 2 H), 3.97 (s, 3 H), 3.90-3.96 (m, 1 H), 3.83 (s, 3H), 2.74 (s, 3 H), 1.41 (t, J=6.9 Hz, 3 H).
Realización 79: WX079
Ruta de síntesis:

Etapa 1: preparación de WX079-1
El compuesto WX007-2 (150.00 mg, 316.90 pmol), trifluoroborato de isopropenil potasio (70.34 mg, 475.35 pmol), complejo (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (25.88 mg, 31.69 pmol) y carbonato de potasio (87.60 mg, 633.80 pmol) se disolvieron en dioxano (1.00 ml) y agua (300.00 pl) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 10 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se añadió agua (20 ml) y se extrajo con acetato de etilo (20 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (20 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: acetato de etilo/éter de petróleo=1/1, relación en volumen) para obtener el producto diana WX079-1. MS-ESI m/z: 457.2 [M+Na]+.
Etapa 2: preparación de WX079-2
El compuesto WX079-1 (110.00 mg, 253.16 pmol) e hidróxido de paladio (40.89 mg, 58.23 pmol, 20 % de pureza) se disolvieron en etanol (1.50 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 12 horas bajo una atmósfera de hidrógeno (30 psi). Después de la reacción, los insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida para obtener el producto diana WX079-2. MS-ESI m/z: 429.1 [M+Na]+.
Etapa 3: preparación de WX079
El compuesto WX079-2 (100.00 mg, 245.98 pmol), trietilamina (124.45 mg, 1.23 mmol, 170.48 pl) y trifosgeno (29.20 mg, 98.39 pmol) se disolvieron en tetrahidrofurano (1.00 ml) a temperatura ambiente. La mezcla de reacción se enfrió a 0-5 °C y se agitó durante 10 horas bajo una atmósfera de nitrógeno. Después de la reacción, se añadió agua (10 ml) y se extrajo con acetato de etilo (10 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (10 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX079. MS-ESI m/z: 433.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 8.55 (br s, 1H), 7.16 (s, 1H), 7.07 (t, J=7.8 Hz, 2H), 7.00-6.95 (m, 2H), 6.80 (d, J=8.3 Hz, 1H), 5.78 (dd, J=4.6, 8.9 Hz, 1 H), 4.76 (dd, J=9.2, 14.9 Hz, 1H), 4.07 (q, J=6.9 Hz, 2H), 3.89 (dd, J=4.6, 14.9 Hz, 1H), 3.82 (s, 3H), 2.73 (s, 3H), 1.42 (t, J=6.9 Hz, 3H), 1.30 (t, J=6.8 Hz, 7H).
Realización 80: WX080

Ruta de síntesis:

Etapa 1: preparación de WX080-1
El compuesto WX007-2 (500.00 mg, 1.06 mmol), 2-(3,6-dihidro-2H-piran-4-il)-4,4,5,5-tetrametil-1,3,2-dioxolpentano (334.03 mg, 1.59 mmol), complejo (1,1 '-bis (difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (86.56 mg, 106.00 pmol) y carbonato de potasio (293.01 mg, 2.12 mmol) se disolvieron en 1,4-dioxano (1.00 ml) y agua (300.00 pl) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 4 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se le añadió agua (20 ml) y se extrajo con acetato de etilo (20 x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (25 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=1/0-3/1-1/1, relación en volumen) para obtener el producto diana WX080-1. MS-ESI m/z: 499.1 [M+Na]+. 1H RMN (400 MHz, CDCh) 5: 7.22 (t, J=8.0 Hz, 1 H), 6.96-6.85 (m, 4H), 6.71 (d, J=8.5 Hz, 1 H), 6.58 (d, J=7.5 Hz, 1 H), 5.61 (br s, 1H), 5.14-5.08 (m, 1 H), 4.24 (d, J=2.5 Hz, 2H), 4.13-4.05 (m, 2H), 3.89 (t, J=5.3 Hz, 2H), 3.86 (s, 3H), 3.58-3.51 (m, 1H), 3.45-3.38 (m, 1H), 2.78 (s, 3H), 2.31 (br s, 2H), 1.45 (t, J=7.0 Hz, 3H).
Etapa 2: preparación de WX080-2
Se añadieron polvo de zinc (315.60 mg, 4.83 mmol) y cloruro de amonio (258.17 mg, 4.83 mmol) a una solución de compuesto WX080-1 (230.00 mg, 482.65 pmol) en metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 12 horas. Después de la reacción, los insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida para eliminar el disolvente. El residuo obtenido se añadió a diclorometano (30 ml) con agitación durante 30 min, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto diana WX080-2. MS-ESI m/z: 469.1 [M+Na]+.
Etapa 3: preparación de WX080
Una solución de compuesto WX080-2 (180.00 mg, 403.08 pmol), trietilamina (203.94 mg, 2.02 mmol) y trifosgeno (47.85 mg, 161.23 pmol) en tetrahidrofurano (10.00 ml) se agitaron a 0-5 °C durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se vertió en agua (50 ml) y se extrajo con acetato de etilo (50 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (50 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX080. MS-ESI m/z: 473.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.57 (s, 1H), 7.17 (d, J=1.8 Hz, 1H), 7.12-7.06 (m, 2H), 7.05-7.01 (m, 1H), 6.96 (d, J=7.5 Hz, 1H), 6.82 (d, J=8.3 Hz, 1H), 5.99 (br s, 1H), 5.78 (dd, J=5.3, 8.8 Hz, 1H), 4.67 (dd, J=8.8 , 14.8 Hz, 1H), 4.34 (d, J=2.5 Hz, 2H), 4.06 (q, J=6.9 Hz, 3H), 3.95 (q, J=5.5 Hz, 2H), 3.83 (s, 3H), 2.73 (s, 3H), 2.50 (d, J=1.8 Hz, 2H), 1.41 (t, J=7.0 Hz, 3H).
Realización 81: WX081
Ruta de síntesis:

Etapa 1: preparación de WX081-1
Una solución de compuesto WX080-1 (200.00 mg, 419.69 pmol) e hidróxido de paladio (67.78 mg, 96.53 pmol, 20 % de pureza) en etanol (10.00 ml) se agitó a temperatura ambiente durante 12 horas bajo una atmósfera de hidrógeno (30 psi). Después de la reacción, los insolubles se eliminaron por filtración y luego el filtrado se concentró a presión reducida para eliminar el disolvente y obtener el producto crudo WX081-1. MS-ESI m/z: 449.2 [M+H]+.
Etapa 2: preparación de WX081
Una solución de compuesto WX081-1 (188.00 mg, 419.10 pmol), trietilamina (212.04 mg, 2.10 mmol) y trifosgeno (49.75 mg, 167.64 pmol) en tetrahidrofurano (10.00 ml) y se agitó a 0-5 °C durante 12 horas bajo una atmósfera de nitrógeno. Después de la reacción, la mezcla se vertió en agua (50 ml) y se extrajo con acetato de etilo (50 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera (50 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX081. MS-ESI m/z: 475.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 10.96 (br s, 1H), 7.22-7.13 (m, 2H), 7.12-7.02 (m, 2H), 6.97 (d, J=7.0 Hz, 1H), 6.82 (d, J=8.3 Hz, 1H), 5.80 (br s, 1H), 4.62 (dd, J=8.9, 13.7 Hz, 1H), 4.27-4.18 (m, 1H), 4.17-3.98 (m, 4H), 3.82 (s, 3H), 3.67-3.49 (m, 2H), 3.01-2.86 (m, 1H) ), 2.70 (s, 3H), 2.02 (t, J=12.2 Hz, 2H), 1.74 (d, J=12.0 Hz, 2H), 1.40 (t, J=6.7 Hz, 3H).
Realización 82: WX082

Ruta de síntesis:

Etapa 1: preparación de WX082-1
Se añadieron ácido oxibenceno borónico (77.05 mg, 507.04 pmol) y (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio (15.46 mg, 21.13 pmol) a una solución del compuesto WX007-2 (200.00 mg, 422.53 pmol) y carbonato de potasio (175.19 mg, 1.27 mmol) en 1,4-dioxano (6.00 ml) y agua (2.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 16 horas bajo una atmósfera de nitrógeno. Después de la reacción, la mezcla de reacción se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera (10 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante TLC preparativa (eluyente: éter de petróleo/acetato de etilo=2/1, relación en volumen) para obtener el producto diana WX082-1. MS-ESI m/z: 523.1 [M+Na]+. 1H RMN (400 MHz, CDCh) 5: 7.41-7.29 (m, 2 H), 7.12-6.77 (m, 7 H), 6.69 (d, J=7.5 Hz, 1 H), 5.16 (s, 1 H), 4.19-4.03 (m, 1 H), 3.87 (s, 3 H), 3.71 (s, 3 H), 3.63-3.54 (m, 1 H), 3.52-3.38 (m, 2 H), 2.90-2.59 (m, 3 H), 1.46 (s, 3 H).
Etapa 2: preparación de WX082-2
Se añadieron polvo de zinc (248.20 mg, 3.80 mmol) y cloruro de amonio (203.03 mg, 3.80 mmol) a una solución de compuesto WX082-1 (190.00 mg, 379.57 pmol) en metanol (10.00 ml) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 0.1 horas. Después de la reacción, los materiales insolubles se eliminaron por filtración y la torta del filtro se lavó con diclorometano (20 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto diana WX082-2. MS-ESI m/z: 471.1 [M+H]+.
Etapa 3: preparación de WX082
Se añadió trifosgeno (42.88 mg, 144.50 pmol) de una vez a una solución de compuesto WX082-2 (170.00 mg, 361.26 pmol) y trietilamina (182.78 mg, 1.81 mmol) en tetrahidrofurano (25.00 ml) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 2.5 horas. Después de la reacción, la mezcla se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera (10 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX082. MS-ESI m/z: 497.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 8.13 (s., 1 H), 7.40-7.46 (m, 1 H), 7.36 (dd, J=7.53, 1.8 Hz, 1 H), 7.24-7.16 (m, 2 H), 7.15-7.02 (m, 5 H), 6.85 (d, J=8.28 Hz, 1 H), 5.80 (s., 1 H), 4.76 (d, J=4.8 Hz, 1 H), 4.22-4.04 (m, 2 H), 3.96-3.67 (m, 7 H), 2.70 (s, 3 H), 1.46 (t, J=6.9 Hz, 3 H).
Realización 83: WX083

Ruta de síntesis:

Etapa 1: preparación de WX083-1
Se añadieron ácido benzoborónico (68.94 mg, 507.04 pmol) y (1,1'-bis(difenilfosfino)ferroceno).dicloruro de paladio (15.46 mg, 21.13 pmol) a una solución del compuesto WX007-2 (200.00 mg, 422.53 pmol) y carbonato de potasio (175.19 mg, 1.27 mmol) en 1,4-dioxano (6.00 ml) y agua (2.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 16 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron y concentraron a presión reducida. El residuo obtenido se aisló mediante TLC preparativa (eluyente: éter de petróleo/acetato de etilo=2/1, relación en volumen) para obtener el producto diana WX083-1. MS-ESI m/z: 507.2 [M+Na]+. 1H RMN (400 MHz, CDCh) 5: 7.34-7.28 (m, 1 H), 7.25-7.14 (m, 2 H), 7.07 (m, 1 H), 7.01-6.95 (m, 1 H), 6.94 6.87 (m, 2 H), 6.81 (dd, J=8.0, 4.3 Hz, 1 H), 6.75 (t, J=6.7 Hz, 2 H), 6.62-6.56 (m, 1 H), 5.22-5.10 (m, 1 H), 4.13-4.02 (m, 2 H), 3.88 (s, 3 H), 3.63-3.53 (m, 1 H), 3.51-3.38 (m, 1 H), 2.78 (d, J=10.6 Hz, 3 H), 2.23-2.09 (m, 3H), 1.46 (q, J=7.2 Hz, 3 H).
Etapa 2: preparación de WX083-2
Se añadieron polvo de zinc (242.90 mg, 3.71 mmol) y cloruro de amonio (198.70 mg, 3.71 mmol) a una solución de compuesto WX083-1 (180.00 mg, 371.46 pmol) en metanol (10.00 ml) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 0.1 horas. Después de la reacción, los insolubles se eliminaron por filtración y la torta del filtro se lavó con diclorometano (20 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto diana WX083-2. (20 ml, EtOAc). Ms -ESI m/z: 477.2 [M+Na]+.
Etapa 3: preparación de WX083
Se añadió trifosgeno (37.86 mg, 127.59 pmol) de una vez a una solución de compuesto WX083-2 (145.00 mg, 318.98 gmol.) y trietilamina (161.39 mg, 1.59 mmol) en tetrahidrofurano (25.00 ml) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 2.5 horas. Después de la reacción, la solución se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera (10 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida para eliminar el disolvente. El residuo obtenido se batió con metanol (10 ml), seguido de filtración y la torta del filtro se lavó con metanol (10 ml x 3). La torta de filtración se concentró a presión reducida para obtener el producto diana WX083. MS-ESI m/z: 481.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 7.37-7.27 (m, 3 H), 7.25-7.0 (m, 1 H), 7.18-7.05 (m, 4 H), 6.98 (dd, J=7.3, 1.0 Hz, 1 H), 6.84 (d, J=8.3 Hz, 1 H), 5.97-5.58 (m, 1 H), 4.65 (dd, J=14.2, 9.2 Hz, 1H), 4.06 (q, J=6.8 Hz, 2H), 3.92-3.77 (m, 4H), 2.66 (s, 3H), 2.18 (s, 3H), 1.43 (t, J=6.90 Hz, 3H).
Realización 84: WX084

Ruta de síntesis:

Etapa 1: preparación de WX084-1
Yoduro cuproso (40.24 mg, 211.26 pmol), (1S, 2S) -ciclohexano-1,2-diamina (48.25 mg, 422.53 pmol) y carbonato de potasio (218.99 mg, 1.58 mmol) se añadieron a una solución del compuesto WX007-2 (500.00 mg, 1.06 mmol) y pirazol (107.87 mg, 1.58 mmol) en 1,4-dioxano (5.00 ml) a temperatura ambiente. La reacción se calentó a 80 °C y se agitó durante 4 horas bajo una atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y se le añadieron salmuera (10 ml) y agua (30 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con agua (50 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=2/1 -1 /1, relación en volumen) para obtener el producto diana WX084-1.1H RMN (400 MHz, CDCl3) 5: 7.70 (dd, J=2.0, 5.8 Hz, 2H), 7.33 (t, J=8.2 Hz, 1H), 6.98-6.91 (m, 1H), 6.90-6.84 (m, 2H), 6.81 (d, J=8.0 Hz, 2H), 6.60 (d, J=6.5 Hz, 1 H), 6.46 (t, J=2.1 Hz, 1 H), 5.21-5.10 (m, 1 H), 4.13-4.03 (m, 2H), 3.87 (s, 3H), 3.66 3.51 (m, 1H), 3.50-3.38 (m, 1 H), 2.80 (s, 3H), 1.46 (t, J=6.9 Hz, 3H).
Etapa 2: preparación de WX084-2
Se añadieron polvo de zinc (567.99 mg, 8.69 mmol) y cloruro de amonio (464.62 mg, 8.69 mmol) a una solución del compuesto WX084-1 (400.00 mg, 868.62 pmol) en metanol (10.00 ml) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 1 hora. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (15 ml) al residuo con agitación a temperatura ambiente durante 5 minutos, seguido de filtración para eliminar los insolubles. El filtrado se concentró a presión reducida para obtener el producto diana WX084-2.
Etapa 3: preparación de WX084
Se añadió lentamente trifosgeno (110.29 mg, 371.64 pmol) a una solución de compuesto WX084-2 (400.00 mg, 929.11 pmol) y trietilamina (470.08 mg, 4.65 mmol) en tetrahidrofurano (30.00 ml) a 0 °C. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 6 horas. Después de la reacción, se filtró la mezcla y se concentró el filtrado. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX084. MS-ESI m/z: 457.1 [M+H]+ .1H RMN (400 MHz, CDCh) 5: 9.87 (s, 1H), 8.03 (d, J=2.3 Hz, 1H), 7.76 (d, J=1.8 Hz, 1H), 7.24-7.11 (m, 3H),
7.09-6.98 (m, 2H), 6.83 (d, J=8.3 Hz, 1H), 6.50 (t, J=2.1 Hz, 1H), 5.83 (dd, J=4.4, 9.7 Hz, 1H), 4.83 (dd, J=9.7, 14.9 Hz, 1H), 4.08 (q, J=6.9 Hz, 2H), 3.90-3.80 (m, 4H), 2.78 (s, 3H), 1.45 (t, J=7.0 Hz, 3 H).
Realización 85: WX085

Ruta de síntesis:

Etapa 1: preparación de WX085-1
Se añadieron trietilamina (292.00 mg, 2.89 mmol) y complejo (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (224.58 mg, 275.00 pmol) a una solución del compuesto WX007-2 (1.30 g, 2.75 mmol) y pirrolidina (254.25 mg, 3.58 mmol) en N,N-dimetilformamida (30.00 ml). La mezcla de reacción se calentó a 80 °C y se agitó durante 24 horas bajo una atmósfera de monóxido de carbono (50 psi). Después de la reacción, la mezcla se enfrió a temperatura ambiente y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida y el residuo obtenido se separó por cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=10/1 -1 /1, relación en volumen) para obtener el producto diana WX085-1. 1H RMN (400 MHz, CDCh) 5: 8.36 (d, J=6.8 Hz, 1H), 7.41 -7.30 (m, 1H), 6.99-6.76 (m, 4H), 6.64 (dd, J=1.0, 7.3 Hz, 1 H), 5.26-5.11 (m, 1H), 4.10 (q, J=7.0 Hz, 2H), 3.88 (s, 3H), 3.73-3.57 (m, 3H), 3.51-3.42 (m, 1H), 3.26-3.15 (m, 2H), 2.79 (s, 3H), 2.03-1.86 (m, 4H), 1.48 (t, J=6.9 Hz, 3H).
Etapa 2: preparación de WX085-2
Se añadió lentamente polvo de zinc (186.24 mg, 2.85 mmol) a una solución de compuesto WX085-1 (280.00 mg, 569.62 pmol) y cloruro de amonio (304.69 mg, 5.70 mmol) en metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas bajo una atmósfera de nitrógeno. Después de la reacción, los insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (15 ml) al residuo obtenido con agitación a temperatura ambiente durante 5 minutos, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto diana WX085-2.
Etapa 3: preparación de WX085
Se añadió lentamente trifosgeno (72.01 mg, 242.65 pmol) a una solución de compuesto WX085-2 (280.00 mg, 606.63 pmol) y trietilamina (306.92 mg, 3.03 mmol) en tetrahidrofurano (20.00 ml) a 0 °C. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 12 horas. Después de la reacción, los insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida para eliminar el disolvente. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX085. EM-ESI m/z: 488.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 9.50 (br s, 1H), 7.27 (t, J=3.9 Hz, 1H), 7.23-7.16 (m, 1H), 7.15-7.00 (m, 3H), 6.82 (d, J=8.3 Hz, 1H), 5.81 (dd, J=4.3, 9.5 Hz, 1H), 4.79 (dd, J=9.8, 14.8 Hz, 1H), 4.15-3.97 (m, 2H), 3.93-3.76 (m, 4H), 3.68 (t, J=6.4 Hz, 4H), 2.80 (s, 3H), 1.96 (d, J=11.8 Hz, 4H), 1.44 (t, J=7.0 Hz, 3H).
Realización 86 : WX086

Ruta de síntesis:

Una solución de compuesto WX007 (300.00 mg, 639.18 pmol), tributil(1-etoxivinil)estaño (1.32 g, 3.65 mmol) y tetraquistrifenilfosfina paladio (147.72 mg, 127.84 pmol) en tolueno (50.00 ml) se calentó a 100-110 °C y se agitó durante 12 horas bajo una atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida para eliminar el disolvente. El residuo obtenido se vertió en acetato de etilo (50 ml) y ácido clorhídrico/acetato de etilo (4 M, 1.60 ml) con agitación a temperatura ambiente durante 1.5 horas. Luego, la mezcla se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX086. MS-ESI m/z: 433.1 [M+H] + . 1 H RMN (400 MHz, CDCl 3 ) 5: 9.62 (s, 1 H), 7.57 (d, J=8.0 Hz, 1 H), 7.31 -7.29 (m, 1H), 7.18-7.16 (m, 3H), 6.84 (d, J=8.0 Hz, 1 H), 5.80 (dd, J=4.0, 9.2 Hz, 1 H), 4.82 (dd, J=10.0, 14.8 Hz, 1 H), 4.12-4.07 (m, 2H), 3.88-3.84 (m, 4H), 2.79 (s, 3H), 2.64 (s, 3H), 1.46 (t, J=6.8 Hz, 3H).
Realización 87: WX087

Ruta de síntesis:

Una solución de compuesto WX007 (50.00 mg, 106.53 pmol), ácido o-fluorofenilborónico (19.38 mg, 138.49 pmol.), carbonato de potasio (36.81 mg, 266.33 pmol) y complejo (1, 1 ’-bis (difenilfosfino)ferroceno).dicloruro de paladio en diclorometano (4.35 mg, 5.33 pmol) en 1,4-dioxano (3.00 ml) y agua (1.00 ml) se calentó a 70 °C y se agitó durante 10 horas en una atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Se le añadieron agua (5 ml) y acetato de etilo (5 ml) y los insolubles se eliminaron por filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX087. MS-ESI m/z: 485.1 [M+H]
+
.
1
H RMN (400 MHz, CD
3
OD) 5: 7.49-7.39 (m, 2 H), 7.31-7.25 (m, 1H), 7.25-7.18 (m, 2H), 7.16-7,07 (m, 3H), 7.06-7.02 (m, 1 H), 6.95 (d, J=8.5 Hz, 1H), 6.06 (dd, J=4.1, 10.2 Hz, 1H), 4.65 (dd, J=10.4, 14.7 Hz, 1H), 4.12-3.99 (m, 3H), 3.81 (s, 3H), 2.94 (s, 3H), 1.36 (t, J=7.0 Hz, 3H).
Los compuestos de cada realización en la siguiente tabla se prepararon de acuerdo con el método de realización 87.
Tabla 11

Datos de LCMS Y 1HRMN de cada realización
Tabla 12


Realización 94: WX094

Ruta de síntesis:

Etapa 1: preparación de WX094-1
Una solución de compuesto WX007-2 (2.62 g, 5.54 mmol), bis(pinacolato)diboro (2.81 g, 11.08 mmol), acetato de potasio (1.63 g, 16.62 mmol) y complejo (1,1'-bis(difenilfosfino)ferroceno).dicloruro de paladio en diclorometano (452.02 mg, 554.00 pmol) en 1,4-dioxano (30.00 ml) se calentó a 80 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Se le añadió agua (100 ml) y se extrajo con acetato de etilo (100 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (150 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante cromatografía en columna preparativa (eluyente: éter de petróleo/acetato de etilo=1/ 0-4/1 -2/1-1/1, relación en volumen) para obtener el producto diana WX094-1. MS-ESI m/z: 543.0 [M+Na] + . 1 H RMN (400 MHz, CDCl 3 ) 5: 8.63 (d, J=7.0 Hz, 1H), 7.41-7.33 (m, 1H), 6.92-6.88 (m, 1H), 6.87-6.79 (m, 3H), 6.75 (d, J=6.8 Hz, 1H), 5.26-5.19 (m, 1H), 4.09-4.04 (m, 2H), 3.86 (s, 3H), 3.66-3.57 (m, 1H), 3.50-3.42 (m, 1H), 2.77 (s, 3H), 1.46 (t, J=6.9 Hz, 3H), 1.40 (s, 12H).
Etapa 2: preparación de WX094-2
El compuesto WX094-1 (500.00 mg, 960.80 pmol), bromuro de o-fluorobencilo (363.24 mg, 1.92 mmol, 231.36 pl), fosfato de potasio (407.90 mg, 1.92 mmol) y tetratrifenilfosfina paladio (1.11 g, 960.80 pmol) se disolvieron en etilenglicol dimetiléter (4.00 ml), etanol (1.00 ml) y agua (1.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 90 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Luego se le añadió agua (30 ml) y se extrajo con acetato de etilo (40 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se aisló mediante cromatografía en columna preparativa (eluyente: éter de petróleo acetato de etilo=9/1 -4/1 -2/1, relación en volumen) para obtener el producto diana WX094-2. MS-ESI m/z: 525.1 [M+Na]+.1H RMN (400 MHz, CDCh) 5: 7.16 (t, J=8.2 Hz, 1H), 7.07-7.03 (m, 2H), 7.03-6.99 (m, 1H), 6.95-6.91 (m, 1 H), 6.88-6.84 (m, 2H), 6.70-6.64 (m, 2H) ), 6.51 (d, J=7.5 Hz, 1H), 5.13-5.07 (m, 1H), 4.16-4.12 (m, 2H), 4.07 (dquin, J=2.6, 7.1 Hz, 2H), 3.86 (s, 3H), 3.58-3.49 (m, 1 H), 3.43 (d, J=5.0 Hz, 1 H), 2.76 (s, 3H), 1.48-1.41 (m, 3H).
Etapa 3: preparación de WX094-3
Se agregaron polvo de zinc (299.26 mg, 4.58 mmol) y cloruro de amonio (244.80 mg, 4.58 mmol) a una solución de compuesto WX094-2 (230.00 mg, 457.66 pmol) en metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 12 horas. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (30 ml) al residuo obtenido con agitación durante 30 minutos, seguido de filtración para eliminar los insolubles. El filtrado se concentró a presión reducida para eliminar el disolvente y obtener el producto diana WX094-3. MS-ESI m/z: 473.1 [M+H]+.
Etapa 4: preparación de WX094
El compuesto WX094-3 (210.00 mg, 444.38 pmol), trietilamina (224.83 mg, 2.22 mmol) y trifosgeno (52.75 mg, 177.75 pmol) se disolvieron en tetrahidrofurano (10.00 ml) en atmósfera de nitrógeno. La mezcla de reacción se agitó a -5 °C durante 2.5 horas. Después de la reacción, se le añadió agua (40 ml) y se extrajo con acetato de etilo (40 ml x 2). Las fases orgánicas se lavaron con salmuera saturada (45 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX094. MS-ESI m/z: 499.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 10.44 (br s, 1 H), 7.41 -7.29 (m, 1 H), 7.24-6.97 (m, 8H), 6.90 (d, J=7.0 Hz, 1 H), 5.88 (br s, 1H), 4.78 (d, J=12.5 Hz, 1 H), 4.29-4.12 (m, 4H), 4.04 (d, J=12.3 Hz, 1H), 3.93 (br s, 3H), 2.97-2.58 (m, 3H), 1.51 (br s, 3H).
Realización 95: WX095

Ruta de síntesis:

Etapa 1: preparación de WX095-1
Se añadieron 2- bromotiazol (75.64 mg, 461.18 pmol) y dicloruro de 1,1'-bis(difenilfosfino)ferroceno paladio (14.06 mg, 19.22 pmol) a una solución mixta del compuesto WX080-1 (200.00 mg, 384.32 pmol) y carbonato de potasio (159.35 mg, 1.15 mmol) en 1,4-dioxano (3.00 ml) y agua (1.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 2 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente, se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). La fase orgánica se combinó, se lavó con salmuera saturada (10 ml x 2) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: éter de petróleo/acetato de etilo=2 /1, relación en volumen) para obtener el producto diana WX095-1. m S-ESI m/z: 500.1 [M+Na]+.
Etapa 2: preparación de WX095-2
Se añadieron polvo de zinc (205.39 mg, 3.14 mmol) y cloruro de amonio (168.01 mg, 3.14 mmol) a una solución del compuesto WX095-1 (150.00 mg, 314.10 pmol) en metanol (10.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 0.1 horas. Después de la reacción, los insolubles se eliminaron por filtración y la torta del filtro se lavó con diclorometano (20 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto crudo WX095-2. MS-ESI m/z: 448.1 [M+H]+.
Etapa 3: preparación de WX095
Se añadió trifosgeno (31.83 mg, 107.25 pmol) de una vez a una solución de compuesto WX095-2 (120.00 mg, 268.11 pmol) y trietilamina (135.65 mg, 1.34 mmol) en tetrahidrofurano (25.00 ml) a 0 °C. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 16 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se vertió en agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). La fase orgánica se combinó, se lavó con agua (10 ml x 2) y se secó sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX095. MS-ESI m/z: 474.0 [M+H]+. 1H RMN (400 MHz, CDCh) ó: 9.99 (s, 1 H), 7.88 (d, J=3.0 Hz, 1 H), 7.46 (t, J=4.4 Hz, 1 H), 7.34 (d, J=3.0 Hz, 1 H), 7.22-7.10 (m, 3 H), 7.06 (d, J=7.8 Hz, 1 H), 6.82 (d, J =8.3 Hz, 1 H), 5.81 (d, J=5.3 Hz, 1 H), 4.85 (dd, J=9.8 Hz, 1 H), 4.08 (q, J=6.9 Hz, 2 H), 3.92-3.75 (m, 4 H), 2.77 (s, 3 H), 1.44 (t, J=6.9 Hz, 3 H).
Realización 96: WX096

Ruta de síntesis:

Etapa 1: preparación de WX096-1
Se añadieron 2-cloro-5-fluoro-piridina (61.12 mg, 461.18 pmol) y complejo (1,1'-bis(difenilfosfino)ferroceno). dicloruro de paladio en diclorometano (14.06 mg, 19.22 pmol) a una solución mixta del compuesto WX080-1 (200.00 mg, 384.32 pmol) y carbonato de potasio (159.35 mg, 1.15 mmol) en agua (2.00 ml) y 1,4-dioxano (6.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 2 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla se enfrió a temperatura ambiente. Se le añadió agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (10 ml x 2) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante TLC preparativa (eluyente: éter de petróleo/acetato de etilo 2/1, relación en volumen) para obtener el producto diana WX096-1. MS-ESI m/z: 513.0 [M+Na]+.
Etapa 2: preparación de WX096-1
Se añadieron cloruro de amonio (229.01 mg, 4.28 mmol) y polvo de zinc (279.96 mg, 4.28 mmol) de una vez a una solución de compuesto WX096-1 (210.00 mg, 428.13 pmol) en metanol (10.00 ml) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 0.1 horas. Después de la reacción, los insolubles se eliminaron por filtración
y la torta del filtro se lavó con diclorometano (20 ml x 2). El filtrado se combinó y se concentró a presión reducida para obtener el producto crudo WX096-2. MS-ESI m/z: 461.0 [M+H]+.
Etapa 3: preparación de WX096
Se añadió trifosgeno (43.82 mg, 147.66 pmol) de una vez a una solución de compuesto WX096-2 (170.00 mg, 369.15 pmol) y trietilamina (186.77 mg, 1.85 mmol) en tetrahidrofurano (25.00 ml) a 0 °C. La mezcla de reacción se agitó a 0 °C durante 2.5 horas. Después de la reacción, se añadió agua (10 ml) y se extrajo con acetato de etilo (10 ml x 3). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (10 ml x 2 ) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX096. MS-ESI m/z: 487.0 [M+H]+. 1H RMN (400 MHz, CDCh) ó: 9.97 (s, 1 H), 8.68 (s, 2 H), 8.18-8.04 (m, 1 H), 7.25-7.14 (m, 3 H), 7.07 (d, J=8.3 Hz, 1 H), 6.83 (d, J=8.0 Hz, 1 H), 5.84 (d, J=5.3 Hz, 1 H), 4.85 (d, J=4.3 Hz, 1 H), 4.08 (q, J=6.9 Hz, 2 H), 3.84 (m, 4 H), 2.78 (s, 3 H), 1.45 (t, J=6.8 Hz, 3 H).
Realización 97: WX097

Ruta de síntesis:

Etapa 1: preparación de WX097-1
El compuesto WX007-2 (500.00 mg, 960.80 pmol), 2-cloro-3-3-fluoro-piridina (189.58 mg, 1.44 mmol), complejo (1,1'-bis(difenilfosfino)ferroceno). cloruro de paladio en diclorometano (78.46 mg, 96.08 pmol) y carbonato de potasio (265.58 mg, 1.92 mmol) se disolvieron en 1,4-dioxano (5.00 ml) y agua (1.50 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 12 horas en atmósfera de nitrógeno. Después de la reacción, la mezcla de reacción se enfrió a temperatura ambiente. Se le añadió agua (30 ml) y se extrajo con acetato de etilo (30 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (20 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El filtrado se concentró a presión reducida. El filtrado se concentró a presión reducida para obtener el producto crudo WX097-1. MS-ESI m/z: 512.1 [M+Na]+.
Etapa 2: preparación de WX097-2
El compuesto WX097-1 (560.00 mg, 1.14 mmol), zinc en polvo (745.45 mg, 11.40 mmol) y cloruro de amonio (609.79 mg, 11.40 mmol) se disolvieron en metanol (5.00 ml) a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas en atmósfera de nitrógeno. Después de la reacción, los materiales insolubles se eliminaron por filtración y el filtrado se concentró a presión reducida. Se añadió diclorometano (20 ml) al residuo obtenido con agitación a temperatura ambiente durante 30 minutos, seguido de filtración. El filtrado se concentró a presión reducida para obtener el producto crudo WX097-2. MS-ESI m/z: 482.2 [M+Na]+.
Etapa 3: preparación de WX097
El compuesto WX097-2 (470.00 mg, 1.02 mmol), trietilamina (516.07 mg, 5.10 mmol) y trifosgeno (121.07 mg, 408.00 pmol) se disolvieron en tetrahidrofurano (5.00 ml) a 0-5 °C. La mezcla de reacción se agitó a 0-5 °C durante 1.5 horas en atmósfera de nitrógeno. Después de la reacción, se añadió agua (30 ml) y se extrajo con acetato de etilo (30 ml x 2). Las fases orgánicas se combinaron, se lavaron con salmuera saturada (30 ml x 3) y se secaron sobre sulfato de sodio anhidro, seguido de filtración. El residuo obtenido se purificó mediante HPLC preparativa para obtener el producto diana WX097. MS-ESI m/z: 486.1 [M+H]+. 1H RMN (400 MHz, CDCh) 5: 10.62 (br s, 1H), 8.52 (br s, 1H), 7.92 (br s, 1 H), 7.59-7.50 (m, 1H), 7.33-7.28 (m, 1H), 7.20 (d, J=6.0 Hz, 3H), 7.06 (d, J=7.5 Hz, 1H), 6.82 (d, J=8.0 Hz, 1H), 5.85 (d, J=4.8 Hz, 1H), 4.84 (dd, J=9.4, 14.7 Hz, 1 H), 4.08 (q, J=6.6 Hz, 2H), 3.89-3.82 (m, 4H), 2.76 (s, 3H), 1.44 (t, J=6.8 Hz, 3H).
Realización 98: WX098

Ruta de síntesis:

Se añadió complejo (1,1'-bis(difenilfosfino)ferroceno).dicloruro de paladio en diclorometano (46.98 mg, 57.53 pmol) a una solución de WX007 (270.00 mg, 575.26 pmol) y trietilamina (174.63 mg, 1.73 mmol) en etanol (15.00 ml) a temperatura ambiente. La mezcla de reacción se calentó a 80 °C y se agitó durante 12 horas bajo una atmósfera de monóxido de carbono. Después de la reacción, la mezcla se enfrió a temperatura ambiente y los insolubles se eliminaron por filtración. El residuo obtenido se aisló mediante cromatografía en columna (eluyente: éter de petróleo/acetato de etilo=1/0-7/10, relación en volumen) para obtener el producto diana WX098. MS-ESI m/z: 463.1 [M+H] + . 1 H RMN (400 MHz, CD 3 OD) 5: 7.63 (dd, J=0.9, 8.2 Hz, 1H), 7.37 (dd, J=0.9, 7.9 Hz, 1H), 7.13-7.05 (m, 3H), 6.93 (d, J=8.5 Hz, 1H), 6.06 (dd, J=4.0, 10.5 Hz, 1H), 4.62 (d, J=3.8 Hz, 1 H), 4.42 (q, J=7.2 Hz, 2H), 4.12-3.97 (m, 3H), 3.80 (s, 3H), 2.97 (s, 3H), 1.37 (td, J=.7.0, 16.9 Hz, 6H).
Ensayos biológicos
Actividad inhibidora de los compuestos frente a PDE 4B
El ensayo se basa en la medición de la polarización de fluorescencia de la producción de AMP/GMP como resultado de la actividad enzimática mediante el desplazamiento de la unión del trazador al anticuerpo AMP/GMP.
Reactivos:
Tampón de reacción: 10 mM Tris-HCl(pH 7.5), MgCl
2
5 mM, Brij 35 al 0.01 %, DTT 1 mM y DMSO al 1 %.
Enzima: La PDE4B humana recombinante (número de acceso al gen: NM_002600; extremo 305 del aminoácido) se expresó mediante baculovirus en células de insecto Sf9 usando una etiqueta GST N-terminal. PM = 78 kDa.
Sustrato enzimático: AMPc 1 pM
Detección: Anticuerpo Transcreener®AMP
2
/GMP
2
y Trazador AMP
2
/GMP
2
AlexaFluor 633
Procedimiento de reacción:
i) Se disolvieron la PDE4B humana recombinante y el sustrato enzimático (AMPc 1 pM) en la solución tampón recién preparada, respectivamente.
ii) La solución tampón de PDE4B definida anteriormente se transfirió a los pocillos de reacción.
iii) El compuesto que se disolvió en DMSO al 100 % se añadió a los pocillos de reacción de la solución tampón PDE4B mediante tecnología acústica (Echo550; intervalo de nanolitros) y se incubó a temperatura ambiente durante 10 minutos.
iv) A continuación, se añadió la solución tampón enzimática a los pocillos de reacción definidos anteriormente para iniciar la reacción.
v) La reacción se incubó a temperatura ambiente durante 1 hora.
vi) La reacción se terminó agregando mezcla de detección (anticuerpo Transcreener® AMP
2
/ GMP
2
y Trazador AMP
2
/ GMP
2
AlexaFluor 633) y se incubó durante 90 minutos con una mezcla suave. El rango de medición de la polarización de fluorescencia es Ex/Em = 620/688.
Análisis de los datos: La señal de polarización de fluorescencia se convirtió en nM basándose en la curva estándar de AMP/GMP y el porcentaje de actividad enzimática en relación con DMSO calculado por Excel. Se utilizó GraphPad Prism para trazar las curvas (Drawing Medicine Icon).
Tabla 13 Resultados de la prueba de cribado in vitro de los compuestos de la presente invención


Conclusión: Todos los compuestos de la presente invención exhiben una excelente actividad inhibidora in vitro contra PDE4B.
Claims (15)
1. Un compuesto como se muestra en la fórmula (I) o una sal farmacéuticamente aceptable del mismo,

en la que,
X es O o N (R
2
);
R
2
es H, F, Cl, Br, I, OH, NH
2
o R
3
-L
1
-, o seleccionado del grupo formado por alquilo C
1-6
, heteroalquilo C
1-6
, cicloalquilo C
3-6
, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
R
3
se selecciona del grupo que consiste de cicloalquilo C
3-6
, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
L
1
se selecciona del grupo que consiste de -CH
2
-, -CH
2
CH
2
-, O, S, NH y -C(=O)-;
n se selecciona del grupo que consiste en 1,2 y 3;
R
1
es H, F, Cl, Br, I, OH, NH
2
, COOH, R
4
-L
2
-, o seleccionado del grupo que consiste de alquilo C
1-6
, heteroalquilo C
1-6
, alquenilo C
2-6
, cicloalquenilo C
3-6
, heterocicloalquenilo de 3-6 miembros, cicloalquilo C
3-6
, heterocicloalquilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; R
4
se selecciona del grupo que consiste de cicloalquilo C
3-6
, heterocicloalquilo de 3-6 miembros, heterocicloalquenilo de 3-6 miembros, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R;
L
2
se selecciona del grupo que consiste de -CH
2
-, -CH
2
CH
2
-, O, S, NH, -C(=O)NH-, -C(=O)O- y -C(=O)-;
R es H, halógeno, OH, NH
2
, CN, o seleccionado del grupo que consiste de alquilo C
1-6
, heteroalquilo C
1-6
, cicloalquilo C
3-6
, heterocicloalquilo de 3-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R;
R' se selecciona del grupo que consiste en H, F, Cl, Br, I, OH, CN, NH
2
, Me, Et, CF
3
, CHF
2
, CH
2
F, NHCH
3
, N(CH
3
)
2
; el "hetero" en el heteroalquilo C
1-6
, heterocicloalquilo de 3-6 miembros, heteroarilo de 5-6 miembros y heterocicloalquenilo de 3-6 miembros se selecciona de -C(=O)NH-, -NH-, -C(=NH)-, -S(=O)
2
NH-, -S(=O) NH-, -O-, -S-, =O, =S, -O-N=, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O)-, -S(=O)
2
- y NHC(=O)NH-;
en cualquiera de los casos anteriores, el número del heteroátomo o del grupo heteroátomo se selecciona independientemente entre 1, 2 o 3.
2. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1, en el que R es H, F, Cl, Br, I, OH, CN o NH 2 , o seleccionado del grupo que consiste en alquilo C 1-3 , alcoxi C 1-3 , alquiltio C 1-3 , alquilamino C 1-3 , alquilo C 1-4 - -OC(=O)-, N, N'-di(alquilo C 1-3 )amino, cicloalquilo C 3-6 , cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R se selecciona del grupo que consiste en H, F, Cl, Br, I, OH, Cn , NH 2 , Me, CF 3 , CHF 2 , CH 2 F, Et,

3. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 2, en el que R 3 se selecciona del grupo que consiste en ciclopropilo, ciclobutilo, fenilo, piridilo, pirimidinilo, pirazinilo, tienilo, imidazolilo, pirazolilo, oxazolilo, tiazolilo, isoxazolilo e isotiazolilo, cada uno de los cuales está opcionalmente sustituido con 1 , 2 o 3 R; opcionalmente, R 3 se selecciona del grupo que consiste de
A. o -
Ó "

4. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 3, en el que R
3
-L
i
- es

5. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 2, en el que R
2
es H, F, Cl, Br, I, OH, NH
2
, R
3
-L
1
-, o seleccionado del grupo que consiste de alquilo C
1-3
, alquilo C
1-3
- S(=O)
2
- alquilo C
1-3
, ciclopropilo, ciclobutilo, fenilo y heteroarilo de 5-6 miembros, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R
2
es H, F, Cl, Br, I, OH, NH
2
, R
3
-L
1
-, o seleccionado del grupo que consiste en Me, Et,

cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R
2
es H, F, Cl, Br, I, OH, CN, NH
2
, COOH, Me, Et,

6. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 2, en el que X es

7. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 2, en el que R4 se selecciona del grupo que consiste en fenilo, pirrolidinilo, piridilo, pirimidinilo, pirazinilo, piridazinilo, cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R4 se selecciona del grupo que consiste en

cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R4 es

8. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 7, en el que R 4 -L 2 - es

9. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 1 o 2, en el que R
1
es H, F, Cl, Br, I, OH, NH
2
, COOH, R
4
-L
2
-, o seleccionado del grupo que consiste de alquilo C
1-3
, alquilo C
1-3
-C(=O)-, alquilo C
1-3
-OC(=O)-, alquenilo C
2-4
, cicloalquenilo C
4-6
, 3,6-dihidro-2H-piranilo, cicloalquilo C
3-6
, tetrahidropiranilo, fenilo, piridilo, pirimidinilo, pirazinilo, piridazinilo, pirazolilo, imidazolilo, tienilo, tiazolilo, isotiazolilo, oxazolilo, isoxazolilo, cada uno de los cuales está opcionalmente sustituido con 1, 2 o 3 R; opcionalmente, R
1
es H, F, Cl, Br, I, OH, NH
2
, COOH, R
4
-L
2
-, o seleccionado del grupo que consiste en Me, Et,

cada uno de los cuales está opcionalmente sustituido con 1,2 o 3 R; opcionalmente, R
1
es H, F, Cl, Br, I, OH, NH
2
, COOH,

10. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con cualquiera de las reivindicaciones 1-9, que es

en el que, R1 y R2 se definen como cualquiera de las reivindicaciones 1-9.
11. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 10, que es


en el que, Ri y R2 se definen como cualquiera de las reivindicaciones 1-9.
12. El compuesto o la sal farmacéuticamente aceptable del mismo, que se selecciona del grupo que consiste en



13. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la reivindicación 12, que se selecciona del grupo que consiste en







14. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz del compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1-13 como principio activo y un vehículo farmacéuticamente aceptable.
15. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1-13 o la composición farmacéutica de acuerdo con la reivindicación 14 para su uso en el tratamiento de una enfermedad relacionada con la PDE4.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610706073 | 2016-08-22 | ||
| PCT/CN2017/098462 WO2018036470A1 (zh) | 2016-08-22 | 2017-08-22 | 作为pde4抑制剂的并环类化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2852098T3 true ES2852098T3 (es) | 2021-09-13 |
Family
ID=61246426
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17842893T Active ES2852098T3 (es) | 2016-08-22 | 2017-08-22 | Compuesto cíclico que actúa como inhibidor de PDE4 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10532986B2 (es) |
| EP (1) | EP3502099B1 (es) |
| JP (1) | JP7050054B2 (es) |
| CN (1) | CN109563051B (es) |
| ES (1) | ES2852098T3 (es) |
| WO (1) | WO2018036470A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3089414A1 (en) * | 2018-01-29 | 2019-08-01 | Shijiazhuang Sagacity New Drug Development Company, Ltd. | Crystal form of 1h-imidazo[4,5-b]pyridine-2(3h)-one compound and preparation process therefor |
| JP2022539257A (ja) * | 2019-07-04 | 2022-09-07 | メッドシャイン ディスカバリー インコーポレイテッド | ベンズイミダゾール-2-オン系化合物の結晶体、溶媒和物、溶媒和物の結晶体、およびそれらの調製方法 |
| JP2023531113A (ja) * | 2020-02-24 | 2023-07-21 | スーヂョウ ロングボタイ ファーマシューティカルズ カンパニー リミテッド | Pde4阻害剤化合物及びその医薬用途 |
| EP4326713A4 (en) * | 2021-04-12 | 2025-03-19 | Impact Therapeutics (Shanghai), Inc. | Substituted fused bicyclic compounds as parp inhibitors and the use thereof |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AP2002002460A0 (en) | 2001-03-09 | 2002-06-30 | Pfizer Prod Inc | Novel benzimidazole anti-inflammatory compounds. |
| US7115644B2 (en) * | 2002-09-13 | 2006-10-03 | Boehringer Ingelheim Pharmaceuticals Inc. | Heterocyclic compounds |
| US8119676B2 (en) * | 2006-08-26 | 2012-02-21 | Abbott GmbH & Co. HG | Substituted benzimidazolone derivatives, medicaments comprising them and their use |
| WO2011021678A1 (ja) * | 2009-08-21 | 2011-02-24 | 武田薬品工業株式会社 | 縮合複素環化合物 |
| EP2603079B1 (en) * | 2010-08-12 | 2018-09-19 | Merck Sharp & Dohme Corp. | Positive allosteric modulators of mglur2 |
| BR112015019276A2 (pt) | 2013-02-19 | 2017-07-18 | Pfizer | compostos de azabenzimidazol como inibidores de isoenzimas de pde4 para o tratamento de distúrbios do snc e outros distúrbios |
| SI3010503T1 (sl) | 2013-06-21 | 2020-07-31 | Zenith Epigenetics Ltd. | Novi biciklični inhibitorji bromodomene |
| JP2014114308A (ja) | 2014-01-31 | 2014-06-26 | Celgene Corp | (+)−2−[1−(3−エトキシ−4−メトキシフェニル)−2−メチルスルホニルエチル]−4−アセチルアミノイソインドリン−1,3−ジオンを含む固形物形態、その組成物およびその使用 |
| AR101196A1 (es) | 2014-07-16 | 2016-11-30 | Gruenenthal Gmbh | Compuestos de pirimidina sustituidos |
-
2017
- 2017-08-22 CN CN201780046018.7A patent/CN109563051B/zh active Active
- 2017-08-22 WO PCT/CN2017/098462 patent/WO2018036470A1/zh not_active Ceased
- 2017-08-22 ES ES17842893T patent/ES2852098T3/es active Active
- 2017-08-22 EP EP17842893.4A patent/EP3502099B1/en active Active
- 2017-08-22 US US16/325,626 patent/US10532986B2/en active Active
- 2017-08-22 JP JP2019510933A patent/JP7050054B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN109563051B (zh) | 2024-06-11 |
| JP7050054B2 (ja) | 2022-04-07 |
| EP3502099B1 (en) | 2020-12-09 |
| JP2019529364A (ja) | 2019-10-17 |
| US10532986B2 (en) | 2020-01-14 |
| WO2018036470A1 (zh) | 2018-03-01 |
| EP3502099A1 (en) | 2019-06-26 |
| EP3502099A4 (en) | 2020-03-04 |
| US20190210975A1 (en) | 2019-07-11 |
| CN109563051A (zh) | 2019-04-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2969284T3 (es) | Derivado de piridona-pirimidina que actúa como inhibidor de la muteína krasg12c | |
| JP7644517B2 (ja) | Kras突然変異型タンパク質阻害剤 | |
| ES2919474T3 (es) | Derivado de [1,2,4]triazolo[1,5-c]pirimidina como inhibidor del receptor A2A | |
| ES2870524T3 (es) | Derivado de piridina como inhibidor de ASK1, y método de preparación y uso del mismo | |
| EP3719012B1 (en) | N-[4-[(2-amino-4-pyridinyl)oxy]-3-fluorophenyl]-3-(4-fluorophenyl)-1,2,3,4-tetrahydro-2,4-dioxo-5-pyrimidinecarboxamide derivatives as c-met/axl inhibitors for the treatment of tumors | |
| AU2019342102B2 (en) | Compounds for treating certain leukemias | |
| ES2746207T3 (es) | Derivado cristalino de 6,7-insaturado-7-carbamoilmorfinano y método de producción del mismo | |
| KR20240159579A (ko) | Parg 억제제 | |
| CN112839655B (zh) | 甲状腺激素受体β促效剂化合物 | |
| RU2743126C2 (ru) | Ингибитор pde4 | |
| ES2934432T3 (es) | Compuestos de imidazopirazina, métodos de preparación y usos de los mismos | |
| ES2852098T3 (es) | Compuesto cíclico que actúa como inhibidor de PDE4 | |
| ES2927086T3 (es) | Derivado de anillo condensado como inhibidor del receptor A2A | |
| ES2984158T3 (es) | Inhibidores y moduladores de WDR5 | |
| CN116940581A (zh) | 一类新型蛋白降解剂及其应用 | |
| EP4538274A1 (en) | Hpk1 inhibitor and medical use thereof | |
| ES3061587T3 (en) | Azaheteroaryl compound, preparation method therefor, and application thereof | |
| BR112016005361B1 (pt) | Composto, composição, e, uso de um composto | |
| CN111440146B (zh) | 一种具有par4拮抗活性的苯并三嗪类化合物及其应用 | |
| JP2022537415A (ja) | Fgfr4を阻害するための化合物 | |
| TWI894448B (zh) | Ctla-4小分子降解劑及其應用 | |
| CN121311480A (zh) | Lzk抑制剂 | |
| WO2023015240A1 (en) | Tricyclic fused pyrimidine compounds for use as her2 inhibitors | |
| ES2882531T3 (es) | Compuesto anti-virus CMVH | |
| WO2018220252A1 (es) | Derivados de piridoquinazolina eficaces como inhibidores de proteína quinasa |