ES2857629T3 - Forma cristalina de un inhibidor de PDE4 - Google Patents
Forma cristalina de un inhibidor de PDE4 Download PDFInfo
- Publication number
- ES2857629T3 ES2857629T3 ES17192278T ES17192278T ES2857629T3 ES 2857629 T3 ES2857629 T3 ES 2857629T3 ES 17192278 T ES17192278 T ES 17192278T ES 17192278 T ES17192278 T ES 17192278T ES 2857629 T3 ES2857629 T3 ES 2857629T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- reaction
- solution
- crystalline form
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(*c1c(CCCCC2CC2)cc([C@@](C)CC(C(*)=CCC(C)=C2)=C2I)cc1)I=C Chemical compound CCC(*c1c(CCCCC2CC2)cc([C@@](C)CC(C(*)=CCC(C)=C2)=C2I)cc1)I=C 0.000 description 4
- IGFDIFLMMLWKKY-UHFFFAOYSA-N OC(c(cc1)cc(OCC2CC2)c1OC(F)F)=O Chemical compound OC(c(cc1)cc(OCC2CC2)c1OC(F)F)=O IGFDIFLMMLWKKY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/49—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups
- C07C205/57—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C205/59—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton the carbon skeleton being further substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/08—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Dermatology (AREA)
- Diabetes (AREA)
- Otolaryngology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Endocrinology (AREA)
- Transplantation (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Emergency Medicine (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Oncology (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Una forma cristalina del compuesto de fórmula (I) **(Ver fórmula)** en la que n es 1, caracterizada por los siguientes picos de XRPD característicos: 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19,1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ± 0,2 grados/2 theta (CuKα2).
Description
DESCRIPCIÓN
Forma cristalina de un inhibidor de PDE4
Campo de la invención
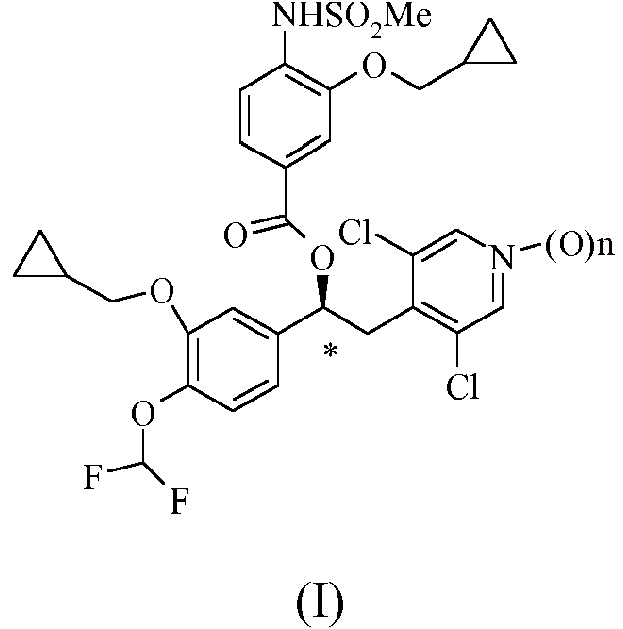
La presente invención se refiere a un forma cristalina de un compuesto de fórmula (I) cuando n es 1. El producto sintetizado es apropiado para su uso en aplicaciones farmacéuticas por ejemplo en el tratamiento de enfermedades respiratorias.
Antecedentes de la invención
Se pueden usar compuestos de fórmula (I) en la que n es 0 o 1

con nombres químicos éster 1-(3-ciclopropilmetoxi-4-difluorometoxi-fenil)-2-(3,5-dicloro-1-oxi-piridin-4-il)-etílico de ácido (S)-3-ciclopropilmetoxi-4-metanosulfonilaminobenzoico y éster 1-(3-ciclopropilmetoxi-4-difluorometoxifenil)-2-(3,5-dicloro-piridin-4-il)-etílico de ácido (S)-3-ciclopropilmetoxi-4-metanosulfónilamino-benzoico, obtenidos de acuerdo con la invención, con fines profilácticos o para la liberación sintomática en una amplia gama de afecciones que incluyen trastornos respiratorios tales como bronquitis crónica, enfermedad pulmonar obstructiva crónica (COPD), asma de todos los tipos y estados de enfermedad alérgicos tales como dermatitis atópica y rinitis alérgica. Dichos compuestos se divulgaron en el documento WO 2010/089107 como potentes inhibidores de PDE4 que presentan una excelente selectividad de LDPE-4.
Los procedimientos para la preparación de compuestos de fórmula (I) en la que n es 0 o 1 y análogos de los mismos, se divulgaron en el documento WO 2010/089107.
Sumario de la invención
El objeto de la presente invención es una forma cristalina del compuesto de fórmula (I):

en la que n es 1, caracterizado por los picos de DRXP característicos: 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19,1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ± 0,2 grados /2 theta (CuKa2).
Se desvela un procedimiento para la preparación de los compuestos de fórmula (I), en la que n es 0 o 1 y el átomo de carbono quiral marcado con un asterisco en la fórmula siguiente muestra una configuración (S).
Dichos compuestos son terapéuticamente útiles debido a su acción como inhibidores de PDE4, de manera que las composiciones farmacéuticas relacionadas que los comprenden se pueden usar en la prevención y tratamiento de enfermedades respiratorias tales como COPd (bronquitis crónica y enfisema), asma, rinitis alérgica y dermatitis atópica; estados de enfermedad alérgicos, artritis inflamatoria; enfermedad de Crohn; lesiones de reperfusión de miocardio y cerebro; fibrosis quística, restenosis arterial, ateroesclerosis, queratosis, espondilitis reumática, osteoartritis, piresis, diabetes melitus, neumoconiosis, eczema de contacto alérgico y tóxico; lupus sistémico eritematoso, piodermias foliculares y de gran alcance, acné endógeno y exógeno, acné rosácea, enfermedad de Beghet, nefritis púrpura anafilactoide, enfermedad intestinal inflamatoria, leucemia, esclerosis múltiple, enfermedades gastrointestinales, enfermedad autoinmunitaria; trastornos siquiátricos y neurológicos; accidente cerebrovascular y lesiones de médula espinal.
Se desvela un procedimiento particularmente eficaz para la preparación de los compuestos de fórmula (I) alternativo al divulgado en el documento previamente citado de la técnica anterior.
Este procedimiento es particularmente ventajoso en comparación con uno conocido ya que proporciona un procedimiento más simple y seguro, con un control mejorado de los parámetros de procedimiento y la reproducibilidad, número reducido de etapas de síntesis y aislamiento intermedio, mayor eficiencia atómica, menores cantidades de disolvente, mayores rendimientos de la formación de productos y menores impurezas.
Este procedimiento también es particularmente apropiado para la fabricación a escala industrial.
Se puede obtener una forma cristalina termodinámicamente estable del compuesto de fórmula (I), en la que n es 1, que posteriormente se denomina Forma A, caracterizada por un nivel elevado de pureza química y cristalinidad, así como buenas calidades de manipulación para uso farmacéutico, de acuerdo con el procedimiento desvelado.
Se puede producir selectivamente una Forma cristalina A de la invención, para la cual se proporcionan sus picos característicos en el patrón de difracción de rayos-X en forma de polvo (XRPD) y el intervalo de fusión, a través de cristalización mediante el uso de disolventes apropiados y condiciones operativas, como se muestran en la siguiente sección detallada.
Por consiguiente, la divulgación proporciona procedimientos para la preparación de dicha Forma A, que comprenden cristalización o recristalización en condiciones seleccionadas.
Dado que dicha forma cristalina A se puede usar con fines profilácticos o terapéuticos, se desvela además el uso de la forma cristalina A del compuesto de fórmula (I), en la que n es 1, en la fabricación de un medicamento para la prevención y/o tratamiento de una enfermedad respiratoria inflamatoria u obstructiva tal como asma o enfermedad pulmonar obstructiva crónica (EPOC).
En aún un aspecto más, se desvela un procedimiento para prevenir y/o tratar una enfermedad respiratoria inflamatoria u obstructiva tal como asma o enfermedad pulmonar obstructiva crónica (EPOC), que comprende la administración inhalatoria de una cantidad eficaz de la forma cristalina A.
Los solvatos del compuesto de fórmula (I) en la que n es 1 se obtienen también operando con disolventes apropiados.
En particular se obtiene un solvato de un compuesto de fórmula (I) a partir de etanol y se puede distinguir basándose en sus picos característicos en el patrón de difracción de rayos X de polvo (DRXP) y su intervalo de fusión característico.
Definiciones
A menos que se defina lo contrario, todos los términos científicos y técnicos usados en la presente memoria tienen el mismo significado que comúnmente se comprende por parte del experto en la técnica a la cual pertenece la materia objetivo.
La expresión "alto nivel de pureza química" se refiere a una forma cristalina en la que la cantidad total de impurezas fácilmente detectables, tal y como viene determinado por medio de procedimientos de análisis convencionales, tales como cromatografía en capa fina (TLC) o cromatografía de líquidos de alto rendimiento (HPLC), es menor de un 5 %, de manera ventajosa menor de un 2,5 %, incluso menor de un 1,0, o más preferentemente incluso menor de un 0,5 % en peso/peso.
La expresión "alto nivel de cristalinidad" se refiere a una forma cristalina en la que el porcentaje de cristalinidad es igual o mayor de un 90 %, preferentemente mayor de un 95 % en peso/peso, tal y como viene determinado por medio de procedimientos de análisis convencionales, tales como microcalorimetría o difracción de rayos-X en forma de polvo.
Breve descripción de los dibujos
La Figura 1 es la traza térmica de calorimetría de barrido diferencial (DSC) del solvato a partir de etanol del compuesto de fórmula (I) en la que n es 1.
La Figura 2 es el espectro de Raman del solvato a partir de etanol del compuesto de fórmula (I) en la que n es 1. La Figura 3 es el patrón de XRPD del solvato a partir de etanol del compuesto de fórmula (I) en la que n es 1. La Figura 4 es la traza térmica de calorimetría de barrido diferencial (DSC) de la Forma cristalina A a partir de acetato de etilo/n-heptano.
La Figura 5 es el espectro de Raman de la Forma cristalina A a partir de acetato de etilo/n-heptano.
La Figura 6 es el patrón de XRPD de la Forma cristalina A a partir de acetato de etilo/n-heptano, registrada en un Bruker D8 Advance con Tubo de Difracción de rayos X de tipo Cu 2k.
La Figura 7 es patrón de XRPD de la Forma cristalina A a partir de acetato de isopropilo.
Descripción detallada de la invención
Es objeto de la presente invención una forma cristalina del compuesto de fórmula (I):
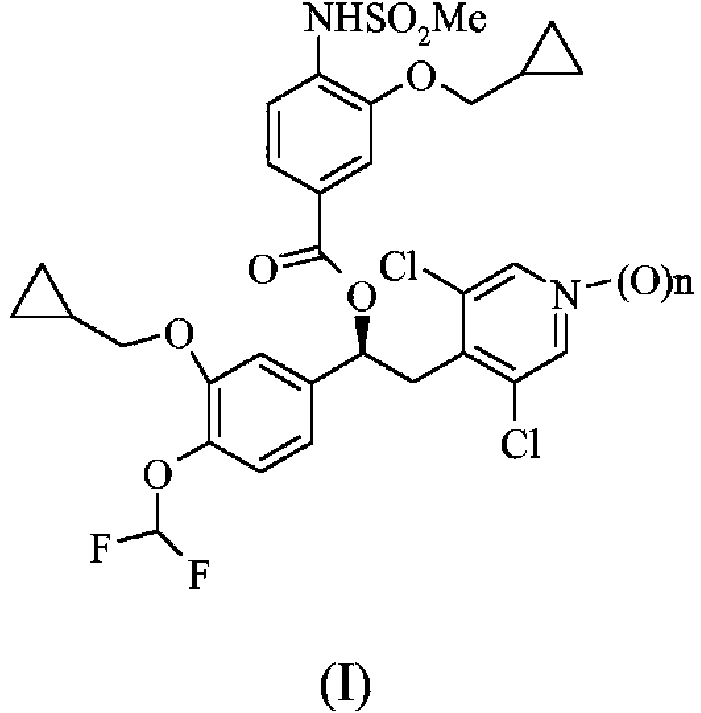
en la que n es 1, caracterizado por los siguientes picos de DRXP característicos: 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19,1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ± 0,2 grados/2 theta (CuKa2).
Se desvela un procedimiento para la preparación de un compuesto de fórmula (I)

en la que n es 0 o 1, comprendiendo el procedimiento:
a) hacer reaccionar un compuesto de fórmula (II)

en la que n es 0 o 1, con un compuesto de fórmula (III)
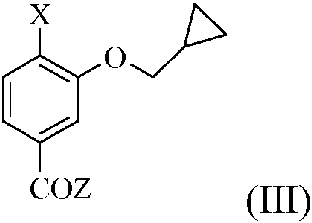
en la que X está seleccionado entre -NHSO2Me y -NO2 y Z está seleccionado entre -OH, cloro, bromo, alcoxi (Ci-C6) lineal o ramificado, ariloxi, arilalcoxi, alquil (Ci-C6) carboniloxi, arilcarboniloxi y arilalquil (Ci-C6) carboniloxi, para obtener un compuesto de fórmula (I) en la que n es 0 o 1 o un compuesto de fórmula (IV)

en la que n tiene el significado comentado anteriormente; y, cuando se obtiene un compuesto de fórmula (IV) en la etapa (a):
b) reducción del mismo al correspondiente compuesto de fórmula (V)

en la que n es 0 o 1, y reacción del mismo con haluro de metanosulfonilo para obtener un compuesto de fórmula (I) en la que n tiene el significado comentado anteriormente;
y en la que el compuesto de fórmula (II) de la etapa (a) se obtiene de acuerdo con una cualquiera de las etapas alternativas (c1) o (c2) o (c3) por medio de:
c1) oxidación de un compuesto de fórmula (VI)

en la que n es 0 o 1, para obtener un compuesto de fórmula (VII)

en la que n es 0 o 1, y posterior reducción enantioselectiva del mismo para obtener un compuesto de fórmula (II) en la que n tiene el significado anteriormente comentado; o
c2) separación cromatográfica de un compuesto de fórmula (VI) en la que n es 0 o 1, para obtener tanto un compuesto de fórmula (II) como un compuesto de fórmula (VIII)

en la que n tiene el significado anteriormente comentado;
y oxidación opcional del compuesto de fórmula (VIII) obtenido en la etapa (c2) hasta un compuesto correspondiente de fórmula (VII) para reducción posterior hasta un compuesto de fórmula (VI) en el que n es 0 o 1 y reprocesado en el siguiente procedimiento de separación cromatográfica; o
c3) reacción de un intermedio de fórmula B"

con un intermedio de fórmula D

en la que R es un grupo alquilo (C1-C6) lineal o ramificado o un grupo arilalquilo y n tiene el significado anteriormente comentado, para obtener directamente un compuesto de fórmula (VII) y posterior reducción enantioselectiva del mismo para obtener un compuesto de fórmula (II) en la que n tiene el significado anteriormente mencionado;
y en el que todos los compuestos de formula (I), (II), (IV), (V), (VI), (VII) o (VIII) en las que n es 1 se pueden obtener por medio de oxidación de los correspondientes compuestos en los que n es 0.
En la presente descripción, y a menos que se indique lo contrario, el enlace con el símbolo

en la fórmula (VI) indica una mezcla racémica de los dos enantiómeros (R) y (S).
El enlace con el símbolo

en las fórmulas (I) y (II) indica el enantiómero (S), mientras que el enlace con el símbolo
en la fórmula (VIII), indica el enantiómero (R).
La expresión grupo alquilo (C1-C6) lineal o ramificado representa un grupo alquilo lineal o ramificado con 1 a 6 átomos de carbono, por ejemplo, metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, terc-butilo, n-pentilo, n-hexilo y similares.
La expresión arilalquilo (C1-C6) se refiere a grupos alquilo (C1-C6) que además están sustituidos por arilo.
La expresión grupo alcoxi (C1-C6) lineal o ramificado significa una cadena alquiloxi en la que alquilo representa un grupo alquilo lineal o ramificado con de 1 a 6 átomos de carbono, por ejemplo, metoxi, etoxi, n-propiloxi, isopropiloxi, n-butoxi, isobutoxi, sec-butoxi, terc-butoxi, n-pentiloxi, n-hexiloxi y similares, preferentemente metoxi.
La expresión grupo ariloxi significa un grupo arilo ligado al resto de la molécula a través de un átomo de oxígeno, es decir, un grupo aril-O-. En este sentido, y a menos que se indique lo contrario, arilo representa un anillo carbocíclico aromático o anillo heterocíclico aromático, que comprende por ejemplo anillos de 5 o 6 miembros con 1 a 3 heteroátomos o grupos heteroatómicos seleccionados entre N, NH, O S. Se prefiere el grupo fenoxi.
El término arilalcoxi significa un alcoxi (C1-C6) sustituido por uno o más grupos arilo, como se ha definido con anterioridad. Se prefiere benciloxi.
El término arilalquilcarboniloxi significa alquil (C1-C6) carboniloxi sustituido por uno o más grupos arilo, como se ha definido con anterioridad, preferentemente bencilcarboniloxi.
El término haluro, cuando se refiere a haluro de metanosulfonilo en la etapa (b) del procedimiento de la invención,
significa cloruro y bromuro.
En una realización preferida, se desvela un procedimiento para la preparación de un compuesto de fórmula (I) en la que n es 0 o 1, comprendiendo el procedimiento la reacción, en la etapa (a), de un compuesto de fórmula (II) en la que n tiene el significado previamente comentado, con un compuesto de fórmula (III) en la que X es NHSO2Me y Z tiene el significado anteriormente mencionado.
De acuerdo con una realización preferida alternativa, se desvela un procedimiento para la preparación de un compuesto de fórmula (I) en la que n es 0 o 1, comprendiendo el procedimiento la reacción, en la etapa (a), de un compuesto de fórmula (II) en la que n tiene el significado previamente comentado, con un compuesto de fórmula (III) en la que X es -NO2 y Z tiene el significado anteriormente mencionado.
De acuerdo con una realización más preferida, se desvela un procedimiento para la preparación de un compuesto de fórmula (I) en la que n es 0 o 1, comprendiendo el procedimiento la reacción del compuesto de fórmula (II) que se obtiene en la etapa (c1), mediante oxidación de un compuesto de fórmula (VI) hasta un compuesto de fórmula (VII), y mediante reducción enantioselectiva de este último hasta un compuesto de fórmula (II), en la que n tiene el significado anteriormente comentado.
De acuerdo con una realización más preferida, se desvela un procedimiento para la preparación de un compuesto de fórmula (I) en la que n es 0 o 1, comprendiendo el procedimiento la reacción del compuesto de fórmula (II) que se obtiene en la etapa (c2), por medio de separación cromatográfica de un compuesto de fórmula (VI) para obtener tanto un compuesto de fórmula (II) como de fórmula (VIII), en el que n tiene el significado anteriormente comentado. Incluso más preferentemente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I) en la que n es 0 o 1, comprendiendo el procedimiento la reacción del compuesto de fórmula (II) que se obtiene en la etapa (c2), por medio de separación cromatográfica de un compuesto de fórmula (VI) para obtener tanto un compuesto de fórmula (II) como de fórmula (VIII), en el que n tiene el significado anteriormente comentado, y posterior oxidación del compuesto de fórmula (VIII) hasta un compuesto correspondiente de fórmula (VII) para reducirlo posteriormente hasta un compuesto de fórmula (VI) que se puede reciclar en una separación cromatográfica adicional.
De acuerdo con una realización más preferida, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), en la que n es 0 o 1, comprendiendo el procedimiento la reacción del compuesto de fórmula (II) que se obtiene en la etapa (c3), haciendo reaccionar un intermedio de fórmula B"

con un intermedio de fórmula D

para obtener directamente un compuesto de fórmula (VII) y posterior reducción enantioselectiva del mismo para obtener un compuesto de fórmula (II) en la que n tiene el significado anteriormente comentado.
De acuerdo con una realización adicional, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), en la que n es 1, comprendiendo el procedimiento la oxidación de un compuesto de fórmula (I) en la que n es 0.
Alternativamente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), en la que n es 1 partiendo de un compuesto de fórmula (II) en la que n es 1, obteniéndose este último por medio de oxidación del correspondiente compuesto de fórmula (II) en la que n es 0.
Alternativamente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), en la que n es 1 partiendo de un compuesto de fórmula (IV) en la que n es 1, obteniéndose este último por medio de oxidación del correspondiente compuesto de fórmula (IV) en la que n es 0.
Alternativamente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), en la que n es 1
partiendo de un compuesto de fórmula (V) en la que n es 1, obteniéndose este último por medio de oxidación del correspondiente compuesto de fórmula (V) en la que n es 0.
Alternativamente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), , en la que n es 1 partiendo de un compuesto de fórmula (VI) en la que n es 1, obteniéndose este último por medio de oxidación del correspondiente compuesto de fórmula (VI) en la que n es 0.
Alternativamente, se desvela un procedimiento para la preparación de un compuesto de fórmula (I), , en la que n es 1 partiendo de un compuesto de fórmula (VII) en la que n es 1, obteniéndose este último por medio de oxidación del correspondiente compuesto de fórmula (VII) en la que n es 0.
De acuerdo con la etapa (a) del procedimiento, el procedimiento proporciona la preparación de un compuesto de fórmula (I) o de fórmula (IV) mediante reacción de un compuesto de fórmula (II) con un compuesto de fórmula (III) en la que n, X y Z tienen los significados anteriormente mencionados.
Más en particular, cuando se usa el compuesto de fórmula (III) en el que Z es -OH, la reacción se lleva a cabo en presencia de un reactivo de acoplamiento seleccionado entre DCC, Cd I, HATU, HBTU, TBTU, DMTMM, COMU, EDCI, con o sin HOBt, con o sin una base orgánica como TEA, DIPEA, NMM, DBU, BDO, piridina y DMAP, en un disolvente seleccionado entre sulfóxido de dimetilo, sulfolano, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter diisopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, cloroformo, clorobenceno y mezclas de los mismos.
Cuando el compuesto de fórmula (III) es un cloruro o bromuro de acilo, o un éster activado y un anhídrido mixto, la reacción se lleva a cabo como se ha descrito anteriormente sin la presencia de un reactivo de acoplamiento.
Preferentemente, la reacción anterior con un compuesto de fórmula (III) en la que X es -NHSO2Me se lleva a cabo con CDI y DBU en acetato de etilo.
En una realización preferida alternativa, cuando la reacción se lleva a cabo con un compuesto de fórmula (III) en la que X es -NO2, para proporcionar un compuesto de fórmula (IV), la reacción anterior se lleva a cabo con EDCI y DMAP en DMF.
De acuerdo con la etapa (b) del procedimiento, para llevarlo a cabo opcionalmente cuando se parte de un compuesto de fórmula (III) en la que X es -NO2 en la etapa (a), el compuesto de fórmula (IV) en la que n tiene el significado anterior se reduce en primer lugar hasta el correspondiente derivado amino de fórmula (V) y posteriormente se hace reaccionar de manera apropiada con un haluro de metanosulfonilo para obtener el compuesto de fórmula (I).
Preferentemente, la etapa de reducción se lleva a cabo con un agente reductor seleccionado entre hidrógeno, ciclohexadieno, formiato de amonio, ácido fórmico, hierro, dicloruro de estaño, estaño, cloruro de níquel, níquel, hidruro de litio y aluminio, hidruro de sodio y aluminio, borohidruro de litio, borohidruro de sodio, borohidruro de potasio e hidrosulfito de sodio.
En una realización incluso preferida, cuando se lleva a cabo la reacción con hidrógeno, ciclohexadieno, formiato de amonio y ácido fórmico, entonces la reacción tiene lugar en presencia de un catalizador seleccionado entre un catalizador basado en paladio, paladio o níquel, o seleccionado entre el grupo que consiste en paladio sobre carbono, paladio sobre sulfato de bario y paladio sobre carbonato de calcio.
En una realización incluso más preferida, cuando se usa ácido fórmico, la reacción se lleva a cabo en presencia de amoníaco o de una amina, preferentemente trietilamina.
El disolvente apropiado para la etapa de reducción anterior está seleccionado entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo y mezclas de los mismos.
Más preferentemente, la reacción se lleva a cabo con hidrógeno con paladio sobre carbón vegetal en acetato de etilo.
La reacción posterior del compuesto de fórmula (V) con haluro de metanosulfonilo se lleva a cabo en presencia de disolventes apropiados tales como tolueno, benceno, xileno, tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butilmetílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, cloroformo, clorobenceno y mezclas de los mismos y una base seleccionada preferentemente entre hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidruro de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina), DIPEA (base de Hünig, diisopropiletil-amina), NMM (N-metilmorfolina), DBU (1,8-diazabiciclo[5.4.0]unde-7-eno), DBO (1,4-diazabiciclo[2.2.2]octano), piridina y DMAP (4-dimetilaminopiridina); en
caso de usar piridina en exceso se pueden evitar otros disolventes.
Preferentemente, la reacción se lleva a cabo con trietilamina en diclorometano.
De acuerdo con la etapa (c1) para la preparación del compuesto de fórmula (II), en primer lugar, se oxida el compuesto (VI) hasta el correspondiente derivado ceto de fórmula (VII) que posteriormente se reduce de forma enantioselectiva hasta el compuesto de fórmula (II).
Preferentemente, la oxidación se lleva a cabo en presencia de un agente oxidante seleccionado entre un óxido metálico tal como MnO2, un yodo hipervalente, como ácido 2-yodoxibenzoico (IBX) o peryodinano de Dess-Martin, oxidantes basados en sulfóxido de dimetilo (Swern) como el complejo de piridina y trióxido de azufre, en un disolvente seleccionado entre agua, dimetil formamida, dimetil acetamida, N-metil pirrolidona, sulfóxido de dimetilo, sulfolano, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, THF, dioxano y mezclas de los mismos.
Incluso más preferentemente, la reacción se lleva a cabo con MnO2 en tolueno o con un oxidante Swern en DMSO. El compuesto de fórmula (VI) se puede preparar a partir de un intermedio de fórmula B

y un intermedio de fórmula D en la que n = 0.

tal y como se describen en el documento WO 2010/089107.
De acuerdo con la etapa (c3) para la preparación del compuesto de fórmula (II), el intermedio de fórmula B'

se convierte en el intermedio de fórmula B"

por medio de reacción con cloruro de tionilo, ácido clorhídrico, ácido sulfúrico en metanol, etanol, isopropanol, nbutanol, t-butanol, alcohol bencílico con o sin otros disolventes, o mediante reacción con un haluro de alquilo relativo en presencia de disolventes apropiados tales como metanol, etanol, isopropanol, n-butanol, t-butanol, dimetilformamida, dimetilacetamida, N-metil pirrolidona, tetrahidrofurano, dioxano, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano y mezclas de los mismos, y una base seleccionada preferentemente entre hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina), DIPEA (base de Hünig, diisopropiletilamina), NMM (N-metilmorfolina), piridina.
Más preferentemente, la reacción anterior se lleva a cabo con carbonato de potasio en dimetil formamida o dimetil acetamida.
El intermedio B' se puede obtener por medio de oxidación del intermedio B con un agente oxidante seleccionado entre agua oxigenada, un perácido orgánico, tal como ácido peracético, o ácido m-cloroperbenzoico, o un perácido mineral como ácido persulfúrico u Oxone® (KHSO5*1/2KHSO4*1/2K2SO4), en presencia de disolventes apropiados tales como agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tetrahidrofurano, dioxano, éter 2-metoxietílico, acetato de isopropilo, acetonitrilo y mezclas de los mismos. Más preferentemente, la reacción anterior se lleva a cabo con Oxone® en metanol.
Alternativamente, el intermedio B" se puede preparar directamente a partir del intermedio B por medio de oxidación con Oxone® en el correspondiente alcohol alquílico como disolvente.
Alternativamente, el intermedio B" se puede preparar a partir de la conversión del intermedio C' en el intermedio C".

por medio de reacción de Pinner con ácido sulfúrico en el correspondiente alcohol alquílico como disolvente, seguido de alquilación con bromuro de ciclopropillo en presencia de disolventes apropiados tales como tolueno, benceno, xileno, tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butilmetílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, cloroformo, clorobenceno y mezclas de los mismos y una base seleccionada preferentemente entre hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidruro de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina), DIPEA (base de Hünig, diisopropiletil-amina), NMM (N-metilmorfolina), DBU (1,8-diazabiciclo[5.4.0]unde-7-eno), DBO (1,4-diazabiciclo[2.2.2]octano), piridina y DMAP (4-dimetilaminopiridina).
Posteriormente, se convierte el intermedio B" en el correspondiente derivado ceto de fórmula (VII) mediante reacción con el intermedio D en presencia de una base preferentemente seleccionada entre diispropilamida de litio (LDA), butil litio, hexil litio, pentil litio, bis(trimetilsilil)amida de litio (LHMDS), bis(trimetilsilil)amida de sodio, t-butilato de potasio, en presencia de disolventes apropiados tales como tolueno, benceno, xileno, tetrahidrofurano, metiltetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butilmetílico y mezclas de los mismos.
Más preferentemente, la reacción anterior se lleva a cabo con LHMDS en THF. Preferentemente, la etapa de reducción enantioselectiva posterior se lleva a cabo con un agente reductor seleccionado entre hidrógeno en presencia de un complejo quiral de metal pesado pre-conformado o formado in situ. La formación in situ puede tener lugar mediante reacción de un complejo de Ru, Rh o Ir, tal como RuCl2(PPh3)3, [Ru (p-cimeno)Cl2]2, [RhCI2(Cp*)]2 o [IrCl2(Cp*)]2 con un ligando quiral tal como SL-N004-1 ((R)-4-terc-butil-2-[(R)-2-(bis(1-fenil)fosfino)ferrocen-1-il]oxazolina,), SL-N003-1 ((R)-4-isopropil-2-[(R)-2-(difenilfosfino)-ferrocen-1-il]oxazolina), (S,S)-Ts-DPEN ((1S,2S)-(-)-N-p-tosil-1,2-difeniletilendiamina), (S,S)-Ms-DPEN ((1S,2S)-(-)-N-Mesil-1,2-difeniletilendiamina), (R)-DAIPEN ((2R)-(-)-1,1 -Bis(4-metoxifenil)-3-metil-1,2-butanodiamina), (1R, 2S)-1 -amino-2-indanol.
Preferentemente, la reacción de reducción anterior se lleva a cabo en presencia de una base, preferentemente seleccionada entre hidróxido de sodio, carbonato de sodio, alcoholatos Ci-C4 de sodio, bicarbonato de sodio, hidruro de sodio, hidróxido de potasio, carbonato de potasio, alcoholatos C1-C4 de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, alcoholatos C1-C4 de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, trietil amina, piridina y 4-dimetilaminopiridina.
En una realización incluso más preferida, la reacción se lleva a cabo con el complejo formado in situ por medio de reacción de RuCh(PPh3)3 y el ligando quiral SL-N004-1 en tolueno y en presencia de hidróxido de sodio acuoso.
Alternativamente, los compuestos de fórmula (II) y (VIII) se pueden separar por medio de cromatografía quiral de preparación; se puede adoptar un procedimiento por lotes que rellena la columna quiral con una solución de (VI) racémico en varias veces y recoge las fracciones eluidas de enantiómeros separados. Se debería considerar un procedimiento de lecho móvil simulado (SMB) para separar una gran cantidad de material.
Ventajosamente, de acuerdo con una realización alternativa del procedimiento desvelado, una vez que los compuestos de fórmula (II) y (VIII) se han separado a través de técnicas de HPLC quiral de preparación, el compuesto de fórmula (VIII) se puede reconvertir de manera apropiada en el compuesto de fórmula (VI) a través de oxidación hasta el correspondiente derivado de fórmula (VII) y posterior reducción y reprocesado en el siguiente procedimiento de separación cromatográfica, como se ha presentado con anterioridad.
La reducción se puede llevar a cabo sobre hidruro de litio y aluminio, hidruro de sodio y aluminio, borohidruro de litio, borohidruro de sodio, borohidruro de potasio en un disolvente como agua, metanol, etanol, isopropanol, n-butanol, tbutanol, tolueno, benceno, xileno, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil tbutílico y mezclas de los mismos.
Se debería comprender que todos los compuestos en los que n es 0 se pueden transformar en los correspondientes compuestos en los que n es 0 por medio de oxidación con un agente oxidante seleccionado entre agua oxigenada, un perácido orgánico, como ácido peracético, o ácido m-cloroperbenzoico, o un perácido mineral como ácido persulfúrico u Oxone® (KHSO5*1/2KHSO4*1/2K2SO4), en un disolvente seleccionado entre el grupo que consiste en agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, acetato de etilo, acetato de isopropilo, acetonitrilo, ácido acético y mezclas de los mismos.
Más preferentemente, la reacción anterior se lleva a cabo sobre (I) o sobre (II), en la que n es 0 con Oxone® en agua y metanol.
A partir de todo lo anterior, resulta evidente que cuando se preparan los compuestos de fórmula (I) de acuerdo con unas de las variantes de procedimiento anteriormente mencionadas, los grupos funcionales opcionales con los materiales de partida o los intermedios de los mismos y que podrían dar lugar a reacciones secundarias no deseadas, precisan protección apropiada de acuerdo con técnicas convencionales. De igual forma, la conversión de estos últimos en los compuestos desprotegidos libres se puede llevar a cabo de acuerdo con procedimientos conocidos.
Los compuestos de fórmula (VI), como materiales de partida del presente procedimiento, se conocen o se pueden preparar de acuerdo con procedimientos conocidos.
A modo de ejemplo, los compuestos de fórmula (VI) y su preparación se desvelan en el documento WO 2010/089107.
El compuesto de fórmula (III) en la que X es -NHSO2Me y Z es -OH representa un objetivo adicional de la invención. Los otros materiales de partida de fórmula (III) se conocen o se pueden preparar fácilmente de acuerdo con procedimientos conocidos.
A modo de ejemplo adicional, los compuestos de fórmula (III) en la que X es -NHSO2Me se pueden preparar a partir de los correspondientes derivados en los que X es -NO2 por medio de reducción de estos últimos a los derivados amino y su reacción posterior con un haluro de metanosulfonilo, esencialmente como se ha presentado con anterioridad.
De igual forma, la preparación de los compuestos de fórmula (III) en la que X es -OH se puede obtener a través de hidrólisis convencional de los correspondientes derivados de éster.
En este sentido, la reacción de hidrólisis por ejemplo que tiene lugar sobre un compuesto de fórmula (III) en la que Z es metoxi se puede conseguir de forma sencilla en presencia de una base apropiada seleccionada a partir de hidróxido de sodio, carbonato de sodio, hidróxido de potasio, carbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio; estando el disolvente seleccionado entre agua sola o en forma de mezcla con metanol, etanol, isopropanol, n-butanol, t-butanol, sulfóxido de dimetilo, sulfolano, tolueno, benceno, xileno, THF, dioxano y mezclas de los mismos.
Más preferentemente, la reacción de hidrólisis de los ésteres para dar lugar al ácido libre en la que Z es -OH se lleva a cabo con NaOH en THF y agua.
De igual forma, la preparación de los compuestos de fórmula (III) en la que Z es diferente de -OH se puede lograr de acuerdo con técnicas bien conocidas de esterificación o transesterificación o partiendo del éster relativo del ácido 3-hidroxi-4-nitrobenzoico.
Se desvela también un procedimiento para la preparación de compuestos adicionales de fórmula (VI) que, con respecto a los compuestos anteriores de fórmula (I), portan grupos R1 y R2 adicionales en lugar de los grupos ciclopropilmetilo y difluorometilo de fórmula (I).
Dichos compuestos de fórmula (XI) se pueden usar con fines profilácticos o para el alivio sintomático de una amplia gama de afecciones que incluyen trastornos respiratorios tales como bronquitis crónica, enfermedad pulmonar obstructiva crónica (COPD), asma de todos los tipos y estados de enfermedad alérgicos tales como dermatitis atópica y rinitis alérgica.
Por consiguiente, se desvela también un procedimiento para la preparación de compuestos de fórmula (XI)

en el que n es 0 o 1;
y Ri y R2, están seleccionados independientemente en un grupo que consiste en H, alquilo (Ci-C6) lineal o ramificado, opcionalmente sustituido por uno o más sustituyentes seleccionados entre átomos de halógeno, cicloalquilo (C3-C7); cicloalquenilo (C5-C7); alquenilo (C2-C6) lineal o ramificado; arilalquenilo (C2-C6) y alquinilo (C2-C6) lineal o ramificado, comprendiendo el procedimiento:
a) hacer reaccionar un compuesto de fórmula (X)

en la que n es 0 o 1, con un compuesto de fórmula (III)
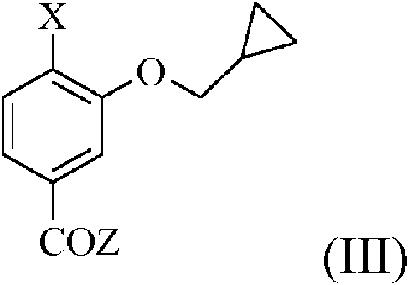
en la que X está seleccionado entre -NHSO2Me y -NO2 y Z está seleccionado entre -OH, cloro, bromo, alcoxi (C1-C6) lineal o ramificado, ariloxi, arilalcoxi, alquil (C1-C6)carboniloxi, arilcarboniloxi y arilalquil (C1-C6)carboniloxi, para obtener un compuesto de fórmula (XI) en la que n es 0 o 1 o un compuesto de fórmula (XII)

en la que R1, R2 y n tienen los significados anteriormente mencionados; y, cuando se obtiene un compuesto de fórmula (XII) en la etapa (a):
b) reducción del mismo hasta un compuesto correspondiente de fórmula (XIII)

en la que R1 y R2 y n tienen los significados anteriormente mencionados, y reacción del mismo con haluro de metanosulfonilo para obtener un compuesto de fórmula (XI) en la que n tiene los significados anteriormente mencionados;
y en la que el compuesto de fórmula (X) de la etapa (a) se obtiene de acuerdo con una cualquiera de las etapas alternativas (c1) o (c2) por medio de:
c1) oxidación de un compuesto de fórmula (XIV)
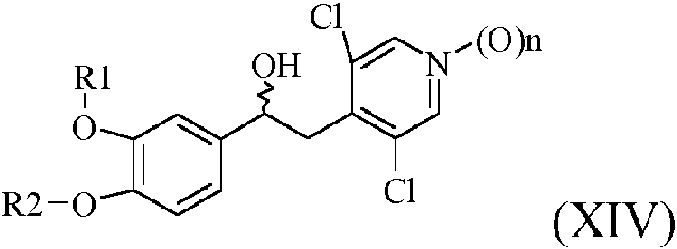
en la que n es 0 o 1, para obtener un compuesto de fórmula (XV)

en la que n es 0 o 1, y posterior reducción enantioselectiva del mismo para obtener un compuesto de fórmula (X) en la que n tiene el significado anteriormente comentado; o
c2) separación cromatográfica de un compuesto de fórmula (XIV) en la que n es 0 o 1, para obtener tanto un compuesto de fórmula (X) como un compuesto de fórmula (XVI)

en la que n tiene el significado anteriormente comentado;
y oxidación opcional del compuesto de fórmula (XVI) obtenido en la etapa (c2) hasta un compuesto correspondiente de fórmula (XV) para reducción posterior hasta un compuesto de fórmula (XIV) en la que n es 0 o 1 y reprocesado en el siguiente procedimiento de separación cromatográfica;
y en el que todos los compuestos de formula (XI), (X), (XII), (XIII), (XIV), (XV) o (XVI) en las que n es 1 se pueden obtener por medio de oxidación de los correspondientes compuestos en los que n es 0.
A partir de todo lo anterior, resulta evidente que las condiciones de operación aplicables a las etapas anteriormente mencionadas del procedimiento para la preparación de los compuestos de fórmula (I) se pueden aplicar también a la preparación de los compuestos de fórmula (XI).
Los materiales de partida de fórmula (X) se conocen o se pueden preparar fácilmente de acuerdo con procedimientos conocidos.
En una realización incluso más preferida adicional, cuando se obtiene el compuesto (I) en el que n es 0 o 1, se puede purificar por medio de cristalización o trituración a partir de uno o más disolventes preferentemente seleccionados entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter
isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, diclorometano, un hidrocarburo alifático o aromático preferentemente escogido entre el grupo que consiste en pentano, hexano, heptano, ciclohexano y metilciclohexano o mezclas de los mismos.
Preferentemente, la reacción se lleva a cabo en acetato de etilo con n-heptano.
En otra realización preferida se desvela el procedimiento de aislamiento por medio de cristalización del compuesto (I) y para su uso para la preparación de composiciones farmacéuticas para inhalación en combinación con portadores o vehículos adecuados.
En otra realización preferida se desvela un procedimiento para la preparación de la Forma cristalina A partir de acetato de etilo y n-heptano caracterizado por los siguientes picos XRPD característicos: 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19, 1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ± 0,2 grados/2 theta.
En otra realización preferida la invención se refiere al uso de una forma cristalina A para la prevención y/o tratamiento de una enfermedad respiratoria inflamatoria u obstructiva tal como asma o enfermedad pulmonar obstructiva crónica (EPOC).
En aún otro aspecto, se desvela un procedimiento para prevenir y/o tratar una enfermedad respiratoria inflamatoria u obstructiva tal como asma o enfermedad pulmonar obstructiva crónica (EPOC), que comprende la administración por inhalación de una cantidad eficaz de la forma cristalina A.
En otra realización preferida la invención se refiere al procedimiento para la preparación de solvatos de un compuesto de fórmula (I).
En otra realización preferida se desvela un procedimiento para la preparación de un solvato de un compuesto de fórmula (I) a partir de etanol, caracterizado por los siguientes picos de DRXP característicos: 7,45; 7,87; 8,51; 10,12; 10,28; 12,66; 13,29; 13,45; 14,95; 16,14; 16,34; 17,05; 17,74; 18,05; 18,48; 18,88; 19,05; 19,33; 19,85; 20,18; 20,65; 21,3; 22,96; 23,55; 23,87; 24,41; 24,66; 24,88; 25,62; 25,82; 26,45; 28,12 y 28,53 ± 0,2 grados/2 theta.
Las composiciones farmacéuticas se pueden preparar mezclado los compuestos de fórmula (I) en la que n es 0 o 1, preparados de acuerdo con la invención, y uno o más excipientes farmacéuticamente aceptables. Dependiendo de la naturaleza de la enfermedad o afección médica a tratar y del tipo de paciente, las composiciones farmacéuticas se pueden formular para ser administrado por cualquier vía adecuada, incluyendo oral, intravenosa, parenteral, inhalación, intranasal, tópica, subcutánea, intramuscular, rectal, vaginal. Las formas de dosificación adecuadas incluyen formulaciones conocidas tales como comprimidos, cápsulas, polvos, formulaciones de liberación sostenida, pomadas, geles, cremas, supositorios, gotas oculares, parches transdérmicos, jarabes, soluciones, suspensiones, aerosoles, soluciones para nebulizadores, aerosoles nasales, etc. En una realización preferida la composición se formula para suministró por inhalación o vías intranasales, por ejemplo en una solución o suspensión en aerosol, como un polvo seco para inhalación o en un aerosol nasal.
Los excipientes adecuados incluyen vehículos, diluyentes, agentes humectantes, agentes emulsionantes, aglutinantes, recubrimientos, cargas, fluidificantes, lubricantes, disgregantes, conservantes, tensioactivos, sustancias tamponadoras del pH y similares. Se proporcionan ejemplos de los excipientes y su uso en e1Handbook of Pharmaceutical Excipients, 5a ed. (2006), Ed. Rowe y col., Pharmaceutical Press.
Las dosis de los compuestos de la invención pueden depender de una diversidad de factores que incluyen la enfermedad particular a tratar, la gravedad de los síntomas, la vía de administración, la frecuencia del intervalo de dosificación, el compuesto particular utilizado, la eficacia, el perfil toxicológico y el perfil farmacocinético del compuesto.
Ventajosamente, los compuestos de fórmula (I) en la que n es 0 o 1 se puede administrar, por ejemplo, a una dosis comprendida entre 0,001 y 1000 mg/día, preferentemente entre 0,1 y 500 mg/día, aún más preferentemente entre 0,2 y 2000 mg/día y aún más preferentemente entre 0,1 y 4000 mg/día.
Los compuestos de fórmula (I) en la que n es 0 o 1, obtenidos de acuerdo con la invención, se pueden usar con fines profilácticos o para alivio sintomático para un amplio espectro de afecciones que incluye: trastornos respiratorios tales como bronquitis crónica, enfermedad pulmonar obstructiva crónica (EPOC) y asma de todos los tipos. De todos modos, los compuestos de fórmula (I) en la que n es 0 o 1 se pueden administrar para la prevención y/o tratamiento de cualquier enfermedad en la que la actividad de los receptores PDE4 está implicada y se desea la inhibición de la actividad del receptor PDE4 o un estado patológico que está mediado por la actividad de PDE4 (por ejemplo, un estado patológico en el que PDE4 está sobreexpresado o sobreactivado). Los ejemplos de dichas enfermedades incluyen: estados patológicos alérgicos tales como dermatitis atópica, urticaria, rinitis alérgica, conjuntivitis alérgica, conjuntivitis primaveral, granuloma eosinofílico, psoriasis, artritis inflamatoria, artritis reumatoide, choque séptico, colitis ulcerosa, enfermedad de Crohn, lesión por reperfusión del miocardio y el cerebro, glomerulonefritis crónica, choque endotóxico, fibrosis quística, reestenosis arterial, aterosclerosis, queratosis, espondilitis reumatoide, osteroartritis, piresis, diabetes mellitus, pneumoconiosis, eczema por contacto tóxico y alérgico, eczema atópico,
eczema seborreico, liquen simple, quemadura solar, picazón en el área anogenital, alopecia areata, cicatrices hipertróficas, lupus eritematoso discoide, lupus eritematoso sistémico, piodermias foliculares y extensivas, acné endógeno y exógeno, acné rosácea, enfermedad de Behcet, nefritis púrpura anafilactoide, enfermedad del intestino irritado, leucemia, esclerosis múltiple, enfermedades gastrointestinales, enfermedades autoinmunes y similares. Estas incluyen también trastornos neurológicos y siquiátricos tales como enfermedad de Alzheimer, esclerosis múltiple, amiloaterosclerosis (ALS), atrofia sistémica múltiple (ASM), esquizofrenia, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Pick, depresión, ictus y lesión en la médula espinal.
En una realización, se desvela el uso de compuestos de fórmula (I) en la que n es 0 o 1, preparado de acuerdo con cualquiera de los procedimientos de la invención, en la fabricación de un medicamente para la prevención o tratamiento de cualquiera de bronquitis crónica, enfermedad pulmonar obstructiva crónica (EPOC), asma de todos los tipos, dermatitis atópica y rinitis alérgica.
En un aspecto más, se desvela un procedimiento para la prevención o tratamiento de cualquiera de bronquitis crónica, enfermedad pulmonar obstructiva crónica (EPOC), asma de todos los tipos, dermatitis atópica y rinitis alérgica en un paciente que comprende la administración al paciente de una cantidad terapéuticamente eficaz de compuestos de fórmula (I) en la que n es 0 o 1, preparado de acuerdo con cualquiera de los procedimientos de la invención.
Una "cantidad terapéuticamente eficaz" de una sustancia se define en el presente documento como una cantidad que conduce a una mejora detectable en uno o más de los síntomas clínicos de una afección tratada o reduce hasta cierto grado la probabilidad de desarrollo de una afección patológica o sus síntomas.





Descripción detallada de la invención
La presente divulgación proporciona un procedimiento para la preparación de un compuesto de fórmula general (I) en la que n es 0 o 1 de acuerdo con las siguientes etapas.
Ruta A - se oxida el intermedio (VI) en la que n es 0 o 1, obtenido de acuerdo con el procedimiento descrito en el documento WO 2010/089107 Ejemplo 1, hasta (VII) en la que n es 0 o 1, en presencia de un agente oxidante seleccionado entre un óxido metálico tal como MnO2, un yodo hipervalente, como ácido 2-yodoxibenzoico (IBX) o peryodinato de Dess-Martin, oxidantes basados en sulfóxido de dimetilo (Swern) como el complejo de piridina y trióxido de azufre. Preferentemente, la síntesis se lleva a cabo en un disolvente seleccionado entre agua, dimetil formamida, dimetil acetamida, N-metil pirrolidona, sulfóxido de dimetilo, sulfolano, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, tetrahidrofurano (THF) dioxano y mezclas de los mismos. Preferentemente, la reacción se lleva a cabo con MnO2 en tolueno o con un oxidante Swern en DMSO.
Alternativamente, el compuesto de fórmula (VII) se puede obtener por reacción de un intermedio de fórmula B"

en la que R es un grupo alquilo (C1-C6) lineal o ramificado o un grupo arilalquilo, con un intermedio de fórmula D

en la que n tiene el significado anteriormente presentado, en presencia de una base preferentemente seleccionada entre diisopropilamida de litio (LDA), butil litio, hexil litio, pentil litio, bis(trimetilsilil)amida de litio (LHMDS), bis(trimetilsilil)amida de sodio, t-butilato de potasio, en presencia de disolventes apropiados tales como tolueno, benceno, xileno, tetrahidrofurano, metil-tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butilmetílico y mezclas de los mismos.
Más preferentemente, R es metilo y la reacción anterior se lleva a cabo con LHMDS y THF.
El compuesto B" se puede obtener a partir del compuesto B' por medio de reacción con cloruro de tionilo, ácido clorhídrico, ácido sulfúrico en metanol, etanol, isopropanol, n-butanol, t-butanol, alcohol bencílico con o sin otros disolventes, o mediante reacción con un haluro de alquilo relativo en presencia de disolventes apropiados tales como metanol, etanol, isopropanol, n-butanol, t-butanol, dimetilformamida, dimetilacetamida, N-metil pirrolidona, tetrahidrofurano, dioxano, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano y mezclas de los mismos, y una base seleccionada preferentemente entre hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina), DIPEA (base de Hünig, diisopropiletilamina), NMM (N-metilmorfolina), piridina.
Más preferentemente, la reacción anterior se lleva a cabo con carbonato de potasio en dimetil formamida o dimetil acetamida.
El compuesto B' se puede obtener a partir del compuesto B con un agente oxidante seleccionado entre agua oxigenada, un perácido orgánico, tal como ácido peracético, o ácido m-cloroperbenzoico, o un perácido mineral como ácido persulfúrico u Oxone® (KHSO5*1/2KHSO4*1/2K2SO4), en presencia de disolventes apropiados tales como agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tetrahidrofurano, dioxano, éter 2-metoxietílico, acetato de isopropilo, acetonitrilo y mezclas de los mismos. Más preferentemente, la reacción anterior se lleva a cabo con Oxone® en metanol.
Alternativamente, el intermedio de fórmula B" se puede obtener a partir del intermedio de fórmula C" por medio de alquilación con bromo-metilciclopropano en presencia de una base preferentemente seleccionada entre hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina),
DIPEA (base de Hünig, diisopropiletil-amina), NMM (N-metilmorfolina), piridina, DBU, DBO, DMAP y en disolventes apropiados tales como metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano y mezclas de los mismos. Más preferentemente, la reacción anterior se lleva a cabo con carbonato de potasio en dimetil formamida.
El intermedio C" se puede obtener a partir del intermedio C' por medio de reacción de Pinner en presencia de un alcohol y un ácido de Lewis seleccionado entre ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácidos alcano sulfónicos tales como ácido metano sulfónico, ácidos aril sulfónicos tales como ácido benceno sulfónico, tribromuro de aluminio, tricloruro de aluminio, tetracloruro de titanio (IV), isopropóxido de titanio (IV), cloruro de estaño (IV), trifluoruro de boro, tricloruro de boro, cloruro de hierro (III), bromuro de hierro (III), isopropóxido de aluminio, cloruro de tionilo, cloruro de oxalilo, cloruro de trimetilsililo (TMSCI), triflato de trimetilsililo (Me3SiOTf), con o sin un disolvente apropiado tal como dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butil metílico y mezclas de los mismos. Más preferentemente, la reacción anterior se lleva a cabo con ácido sulfúrico en metanol.
La posterior reducción enantioselectiva de (VII) en la que n es 0 o 1 proporciona el enantiómero individual (II) en el que n es 0 o 1.
El agente reductor está seleccionado entre hidrógeno en presencia de un complejo quiral de metal pesado pre conformado o formado in situ haciendo reaccionar un complejo de Ru, Rh o Ir, tal como RuCh(PPh3)3, [Ru (pcimeno)Cl2]2, [RhCI2(Cp*)]2 o [IrCh(Cp*)]2 con un ligando quiral tal como SL-N004-1 ((R)-4-terc-butil-2-[(R)-2-(bis(1-fenil)fosfino)ferrocen-1-il]oxazolina,), SL-N003-1 ((R)-4-isopropil-2-[(R)-2-(difenilfosfino)-ferrocen-1-il]oxazolina), (S,S)-Ts-DPEN ((1S,2S)-(-)-N-p-tosil-1,2-difeniletilendiamina), (S,S)-Ms-DPEN ((1S,2S)-(-)-N-Mesil-1,2-difeniletilendiamina), (R)-DAiPEn ((2R)-(-)-1,1-Bis(4-metoxifenil)-3-metil-1,2-bu- tandiamina), (1R, 2S)-1-amino-2-indanol. La reacción se lleva a cabo en presencia de una base, preferentemente seleccionada entre hidróxido de sodio, carbonato de sodio, alcoholatos C1-C4 de sodio, bicarbonato de sodio, hidruro de sodio, hidróxido de potasio, carbonato de potasio, alcoholatos C1-C4 de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, alcoholatos C1-C4 de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, trietil amina, piridina y 4-dimetilaminopiridina.
Preferentemente, la síntesis se lleva a cabo en un disolvente seleccionado entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo y mezclas de los mismos.
Preferentemente, la reacción se lleva a cabo con el complejo formado in situ por medio de reacción de RuCh(PPh3)3 y el ligando quiral SL-N004-1 en tolueno y en presencia de hidróxido de sodio acuoso.
Alternativamente, (II) en la que n es 1 se obtiene por medio de oxidación de (II) en la que n es 0 con un agente oxidante seleccionado entre agua oxigenada, un perácido orgánico, como ácido peracético, o ácido mcloroperbenzoico, o un perácido mineral como ácido persulfúrico u Oxone® (KHSO5M/2 KHSO4M/2 K2SO4). El disolvente de reacción está seleccionado entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, acetato de etilo, acetato de isopropilo, acetonitrilo, ácido acético y mezclas de los mismos. Preferentemente, la reacción se lleva a cabo con Oxone® en agua y metanol.
Ruta B - Como alternativa a la Ruta A, se obtienen los intermedios (II) y (VIII) en los que n es 0 o 1 a partir de (VI), en la que n es 0 o 1, por medio de separación de HPLC de preparación de los enantiómeros.
Se puede adoptar un procedimiento por lotes rellenando la columna quiral con una solución de (VI) racémico en varias veces y recogiendo las fracciones eluidas de los enantiómeros separados. Se debería considerar un procedimiento de lecho móvil simulado (SMB) para separar una gran cantidad de material.
Una vez que los compuestos de fórmula (II) y (VIII) se han separado a través de técnicas de HPLC quiral de preparación, el compuesto de fórmula (VIII) se puede reconvertir de manera apropiada en el compuesto de fórmula (VI) a través de oxidación hasta el correspondiente derivado de fórmula (VII) y posterior reducción y reprocesado en el procedimiento de separación cromatográfica, como se ha presentado con anterioridad.
De esta forma, por medio de reciclaje de (VIII), los rendimientos finales del compuesto de fórmula (I) se pueden mejorar de forma adicional.
En el intermedio (III), en el que X es -NHSO2Me y Z está seleccionado entre -OH, cloro, bromo, un alcoxi (C1-C6) lineal o ramificado, ariloxi, arilalcoxi, alquil (C1-C6)carboniloxi, arilcarboniloxi y arilalquil (C1-C6)carboniloxi, Z es un grupo protector que se puede introducir y retirar usando procedimientos convencionales de acuerdo con "Protective Groups in Organic Synthesis" de Theodora W. Greene (Wiley-Interscience, Nueva York, 1981) y "Protective Groups in Organic Chemistry" de J. F. W. McOmie (Plenum Press, Londres, 1973).
El intermedio (III), en el que -NHSO2Me y Z es como se ha definido anteriormente, se puede obtener en condiciones bien conocidas partiendo de éster metílico de ácido 3-cidopropilmetoxi-4-metanosulfonilaminobenzoico, obtenido como se describe en el documento WO 2007/089107, Ejemplo 18 o de acuerdo con la misma ruta de síntesis partiendo del éster relativo de ácido 3-hidroxi-4-nitrobenzoico.
El intermedio (III), en el que X es -NHSO2Me y Z es como se ha definido anteriormente, se convierte en (III) en la que Z es -OH por medio de hidrólisis en una base, preferentemente seleccionada entre el grupo que consiste en hidróxido de sodio, carbonato de sodio, hidróxido de potasio, carbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio; estando el disolvente seleccionado entre agua sola o en forma de mezcla con metanol, etanol, isopropanol, n-butanol, t-butanol, sulfóxido de dimetilo, sulfolano, tolueno, benceno, xileno, THF, dioxano y mezclas de los mismos. En una realización preferida, la reacción se lleva a cabo con NaOH en THF y agua.
Ruta C - el compuesto (I) en el que n es 0 o 1 se obtiene por medio de condensación del intermedio (III) en el que X es -NHSO2Me y Z es -OH, con (II) en el que n es 0 o 1 en presencia de un agente de acoplamiento seleccionado entre CDI (1,1'-carbonildiimidazol), HATU hexafluorofosfato de (1-[bis(dimetilamino)metilen]-1H-1,2,3-triazolo[4,5-b]piridinio 3-oxid), HBTU hexafluorofosfato de (O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio), TBTU tetrafluoroborato de (O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio), DMTm M cloruro de (4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio), COMU hexafluorofosfato de ((1-ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilaminomorfolino-carbenio), EDCI clorhidrato de (N-(3-dimetilaminopropil)-N'-etilcarbodiimida) y DCC (N,N'-diciclohexilcarbodiimida) o un reactivo que puede convertir el ácido carboxílico en un cloruro de acilo, un bromuro de acilo, un éster activado o un anhídrido mixto, con o sin HOBt (1-hidroxibenzotriazol), con o sin una base orgánica como TEA, DIPEA, NMM, DBU, DBO, piridina y DMAP en un disolvente seleccionado entre el grupo que consiste en sulfóxido de dimetilo, sulfolano, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, cloroformo, clorobenceno y mezclas de los mismos.
Cuando el compuesto de fórmula (III) es un cloruro o bromuro de acilo, o un éster activado y un anhídrido mixto, la reacción se lleva a cabo como se ha descrito anteriormente sin la presencia de un reactivo de acoplamiento.
En una realización preferida, la reacción se lleva a cabo con CDI y DBU en acetato de etilo.
El intermedio (IV), en el que n es 0 o 1, se obtiene por medio de condensación de (III), en la que X es -NO2, con (II) en la que n es 0 o 1, en las mismas condiciones descritas anteriormente para la condensación de (III) en la que X es -NHSO2Me con (II). En una realización preferida, la reacción se lleva a cabo con EDCI y DMAP en DMF.
El intermedio (V), en el que n es 0 o 1, se obtiene por medio de reducción de (IV), en la que n es 0 o 1, con un agente reductor seleccionado entre el grupo que consiste en hidrógeno, ciclohexadieno, formiato de amonio, ácido fórmico, hierro, dicloruro de estaño, estaño, cloruro de níquel, níquel, hidruro de aluminio y litio, hidruro de aluminio y sodio, borohidruro de litio, borohidruro de sodio y borohidruro de potasio e hidrosulfito de sodio. En caso de emplear hidrógeno, ciclohexadieno, formiato de amonio y ácido fórmico, la reacción se lleva a cabo en presencia de un catalizador, preferentemente basado en paladio, platino o níquel, más preferentemente seleccionado entre paladio sobre carbono, paladio sobre sulfato de bario y paladio sobre carbonato de calcio. Cuando se usa ácido fórmico, la reacción se lleva a cabo en presencia de amoníaco o una amina, preferentemente trietilamina.
Los disolventes apropiados para las etapas de reducción anteriores están seleccionados entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, dimetil formamida, dimetil acetamida, N-metil pirrolidona, tolueno, benceno, xileno, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, acetonitrilo y mezclas de los mismos. En una realización preferida, la reacción se lleva a cabo con hidrógeno con paladio al 5 % sobre polvo de carbono activado, tipo A103038 sometido a sulfuración en acetato de etilo.
En otra realización preferida, la reacción se lleva a cabo con hidrógeno con paladio sobre carbón vegetal en acetato de etilo.
El compuesto (I), en el que n es 0 o 1, se obtiene por medio de reacción de (V), en la que n es 0 o 1, con cloruro de metanosulfonilo en presencia de disolventes apropiados seleccionados entre tolueno, benceno, xileno, tetrahidrofurano, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter t-butilmetílico, acetato de etilo, acetato de isopropilo, acetonitrilo, diclorometano, cloroformo, clorobenceno y mezclas de los mismos y una base preferentemente seleccionada entre el grupo que consiste en hidróxido de sodio, carbonato de sodio, bicarbonato de sodio, hidruro de sodio, hidróxido de potasio, carbonato de potasio, bicarbonato de potasio, hidróxido de litio, carbonato de litio, hidróxido de cesio, carbonato de cesio, bicarbonato de cesio, TEA (trietilamina), DIPEA (base Hünig, diisopropiletil-amina), NMM (N-metilmorfolina), DBU (1,4-diazabiciclo'5.4.0]undec-7-eno), DBO (1,4-diazabiciclo[2.2.2]octano), piridina y DMAP (4-dimetilaminopiridina), piridina; en caso de usar piridina en exceso se pueden evitar otros disolventes.
Preferentemente, la reacción se lleva a cabo con trietilamina en diclorometano.
Todos los compuestos de formula (I), (II), (IV), (V), (VI), (VII) o (VIII) en las que n es 1 se pueden obtener por medio de oxidación de los correspondientes compuestos en los que n es 0, como se describe anteriormente para la oxidación del compuesto (II) en el que n es 0 hasta obtener un compuesto (II) en el que n es 1.
Cuando se obtiene el compuesto (I) en el que n es 0 o 1, se puede purificar por medio de cristalización o trituración a partir de uno o más disolventes preferentemente seleccionados entre agua, metanol, etanol, isopropanol, n-butanol, t-butanol, tolueno, benceno, xileno, acetona, isopropil cetona, etil metil cetona, isobutil metil cetona, THF, dioxano, éter 2-metoxietílico, éter dietílico, éter isopropílico, éter metil t-butílico, acetato de etilo, acetato de isopropilo, diclorometano, un hidrocarburo alifático o aromático preferentemente escogido entre el grupo que consiste en pentano, hexano, heptano, ciclohexano y metilciclohexano o mezclas de los mismos. Preferentemente, la reacción se lleva a cabo en acetato de etilo con n-heptano.
De este modo, por ejemplo, se puede preparar la forma A cristalina en presencia de acetato de etilo/heptano o acetato de isopropilo.
La reacción se puede llevar a cabo en un reactor, en el que el compuesto de fórmula (I) se introduce junto con uno o más disolventes seleccionados entre el listado anterior, y la suspensión se puede agitar al tiempo que se calienta hasta una temperatura de 50-90 °C, hasta disolver el sólido por completo. La suspensión se puede enfriar entre 0 5 °C durante 1-5 horas, se filtra y se seca.
Cuando se lleva a cabo la cristalización en presencia de etanol, se puede obtener un solvato de un compuesto de fórmula (I).
La reacción se puede llevar a cabo partiendo de un compuesto de fórmula (I), en uno o más disolventes seleccionados entre el grupo que consiste en pentano, hexano, heptano, ciclohexano, metilciclohexano y diclorometano, obteniéndose una solución que se puede concentrar y posteriormente se añade etanol. La solución se puede concentrar y la suspensión obtenida se puede enfriar a una temperatura entre 0-10 °C y agitar durante 1-5 horas. El sólido se filtra, se lava con etanol y se seca a una temperatura entre 25-55 °C durante 10-30 horas.
A continuación, se ilustra la invención con más detalle en los ejemplos siguientes.
Ejemplo 1
Preparación de ácido 3-(ciclopropilmetoxi)-4-(metilsulfonamido)benzoico (intermedio (III), X = -NHSO
2
Me, Z = -OH)

(III), X = -NHSO2Me, Z =-OMe (III), X = - NHSO2Me, Z = -OH
Se obtuvo (III) en la que X es -NHSO2Me y Z es -OMe como se describe en el documento WO 2010/08910, Ejemplo 18. Se introdujeron (6,0 Kg) y 18 l de THF en un reactor. Por separado, se mezclaron 6,6 kg de hidróxido de sodio al 35 % en peso/peso y 21 l de agua purificada y se transfirieron al reactor y se calentó la mezcla a 65 °C al tiempo que se destiló todo el THF. Tras completar la reacción hidrolítica, se transfirió lentamente la solución básica a otro reactor que contenía una solución de 24 l de agua purificada y 7,2 kg de ácido clorhídrico al 37 % en peso/peso, manteniendo la temperatura por debajo de 40 °C, y agitando durante 15 minutos. El sólido obtenido se filtró y lavó con 24 l de agua. El sólido húmedo (III) (16,6 kg humedad) se reintrodujo en el reactor junto con 60 l de acetato de etilo, posteriormente se calentó a reflujo para destilar 30 l de disolvente. Se introdujeron 12,6 l de heptano en el reactor y se mantuvo la mezcla en agitación durante 15-30 minutos. Se enfrió posteriormente hasta 5 °C y se mantuvo en agitación durante 2 horas. Se filtró el sólido obtenido y se lavó el reactor y la torta con 12 l de heptano. Se secó el sólido húmedo a vacío en un dispositivo de secado estático de bandejas. Se obtuvieron 6235 g de sólido blanco (93,9 % de rendimiento).
RMN 1H (400 MHz, DMSO-cfe) 8 ppm 12.85 (s a, 1 H), 9,03 (s, 1 H), 7,40 - 7,71 (m, 2 H), 7,35 (d, J=8,16 Hz, 1 H), 3,91 (d, J=6,84 Hz, 2 H), 3,07 (s, 3 H), 1,11 -1,42 (m, 1 H), 0,50 -0,67 (m, 2 H), 0,18 -0,41 (m, 2 H).
Ejemplo 2
Preparación de 1-(3-(cidopropMmetoxi)-4-(difluorometoxi)fenM)-2-(3,5-didoro-1-pirídm-4-N)etanona (intermedio (VII), n = 0)
Se obtuvo el intermedio (VI) en el que n es 0, de acuerdo con el procedimiento de fabricación descrito en el documento WO 2010/089107, Ejemplo 1.

Procedimientos alternativos para obtener el intermedio (VII), n = 0
Procedimiento con MnO
2
Se disolvieron 5 kg de (VI) en la que n es 0, en 30 l de tolueno en un reactor; se añadieron 3,15 kg de MnO2 activado en la mezcla orgánica y se calentó la suspensión hasta reflujo durante 3 horas. Se enfrió la mezcla hasta 50 °C y se filtró MnO2 sobre un lecho de celite. Se introdujo la solución orgánica en el reactor y se destiló tolueno hasta 3 volúmenes residuales. Se añadieron 20 l de 2-propanol en el reactor y se concentró de nuevo hasta 2 volúmenes residuales, con el fin de retirar toda la cantidad de tolueno. Además, se introdujeron 20 l de 2-propanol y se destiló parcialmente el disolvente para tener 4 volúmenes residuales en el reactor. Se enfrió la suspensión y se mantuvo en agitación a 10 °C durante la noche. Se filtró el sólido y el sólido húmedo se secó en un horno a T= 50 °C durante 12 horas obteniéndose un sólido blanco (4,12 kg, 82,8 % de rendimiento).
La caracterización del producto se describe en el documento WO 2009018909, Ejemplo 2 (intermedio 1b).
Procedimiento Swern
Se añadió trietilamina (4,5 ml, 32 mmol) gota a gota a una solución de alcohol (VI) en la que n es 0, (5,0 g, 12,4 mmoles) en DMSO (15 mi), agitando a 25 °C. Se añade complejo de SO3 de piridina (5,0 g, 31 mmol) por partes en aproximadamente 1 horas, de manera que la temperatura interna del lote no aumente por encima de 35 °C. Se agita la mezcla de reacción a 25 °C durante 4 horas y posteriormente se inactiva con agua (60 ml) y H2SO4 acuoso al 10 % (10 ml). Se agita la mezcla de reacción a 25 °C y se filtra y seca el sólido a 50 °C a presión reducida, para permitir la obtención de 4,6 g (rendimiento de un 92 %) de cetona pura (VII) en forma de sólido incoloro.
Procedimiento con IBX
Se añadió (VI) en la que n es 0, (1,0 g, 2,5 mmol) de una vez a una suspensión de ácido 2-yodoxibenzoico (IBX) (0,9 g, 3,2 mmol), preparado de acuerdo con la bibliografía (documento JOC 1999 pág. 4537), en DMSO (5 ml), y se agitó la mezcla resultante a 25 °C durante 1 hora y posteriormente a 50 °C durante 2 horas. Se inactivó la reacción con una solución de carbonato de potasio acuosa al 10 % (40 ml) tras enfriar hasta 25 °C y se filtró el sólido para permitir la obtención de acetona (VII) con rendimiento cuantitativo.
Procedimiento con sIBX®
Se añadió sIBX® comercialmente disponible ("IBX estabilizado", una formulación de polvo blanco de IBX formada por una mezcla de ácido benzoico (22 %), ácido isoftálico (29 %) y ácido o-yodoxibenzoico (49 %) a partir de SIMAFEX) (2,0 g, 3,2 mmol) de una vez a una solución de (VI) en la que n es 0 (1,0 g, 2,5 mmol) en acetona (15 ml) a 25 °C, se sometió a reflujo la mezcla resultante durante 2,5 horas, se enfrió a 25 °C y posteriormente se inactivó con una solución acuosa al 10 % de sulfito de sodio (10 ml) y una solución acuosa al 10 % de carbonato de potasio (40 ml). Se agitó la mezcla a 25 °C durante 0,5 horas y se filtró el sólido, para permitir la obtención de una cetona (VI) en la que n es 0 con rendimiento cuantitativo.
Procedimiento con DMP
Se añadió peryodinano de Dess-Martin (DMP) (1,3 g, 0,31 mmol) de una vez a una solución de alcohol (VI) en la que n es 0 (1,0 g, 2,5 mmol) en acetona (5 ml). Se agita la mezcla de reacción a 25-30 °C durante 1 horas y se inactiva con una solución acuosa al 10 % de metabisulfito de sodio (10 ml) y una solución acuosa al 15 % de carbonato de potasio (30 ml). Se agitó la mezcla a 25 °C durante 0,5 horas y se filtró el sólido, para permitir la obtención de (VII) en la que n es 0 con rendimiento cuantitativo.
Ejemplo 2A
Preparación de 3,5-dicloro-4-metil-1-oxi-piridina (Intermedio A)

Se suspendieron 3,5-didoro-4-metil-piridina (0,5 g, 3,08 mmol) y Oxone ® (1,5 g, 4,62 mmol) en una mezcla 8:3 de metanol y agua (5,5 ml) en un matraz de 25 ml. Se agitó la suspensión y se calentó a 55 °C durante 10-15 horas. Se retiró el disolvente a presión reducida y se suspensión el sólido bruto obtenido con agitación en tolueno caliente (80 °C) durante 20 minutos. La solución caliente heterogénea se filtró posteriormente y se enfriaron los licores-madre a temperatura ambiente obteniéndose la precipitación de un sólido. Se obtuvo el producto puro en forma de sólido blanco tras agitación a 0-5 °C durante 30 minutos y se filtró (0,43 g, 78 % de rendimiento).
Ejemplo 2B
Preparación de (R/S)-1-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-dicloro-1-oxi-piridin-4- il)etanol (intermedio (VI), n = 1)

En un matraz de 50 ml de tres bocas bajo nitrógeno, se añadieron el intermedio A (0,4 g, 2,25 mmol) y el intermedio B (0,78 g, 3,22 mmol) y se disolvieron en THF seco (5 ml). Se enfrió la solución agitada hasta -35 °C. Se añadió por partes terc-butilado de potasio (0,3 g, 2,67 mmol) a la solución en 10 minutos. Trascurridos 60 minutos de reacción a -35 °C, se inactivó la solución con una solución acuosa al 25 % de NH4Cl (10 ml). Se añadieron EtOAC (8 ml) y agua (8 ml) a la suspensión y se agitó, se separaron las fases y se extrajo la fase orgánica y se lavó con solución acuosa al 5 % de NaCl (10 ml). A continuación, se sometió el disolvente orgánico a tratamiento de anhidrificación sobre Na2SO4 y se retiró a presión reducida obteniéndose un sólido bruto blanco. Esto se cristalizó a partir de tolueno caliente obteniéndose un sólido blanco (0,40 g, 42 % de rendimiento).
Ejemplo 3
Preparación de (R)-1 -(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-dicloropiridin-4-il)etanol (intermedio (II), n = 0)

Se disolvieron 2,40 kg de (VII) en la que n es 0 en 23 l de tolueno en un reactor. El reactor se desgasificó con nitrógeno. Se colocaron ligando propiedad de Solvias SL-N004-1 y RuCl2(PPh3)3 en un matraz de Schlenk de 2 l, se secó y se introdujo tolueno desgasificado (1,2, l). Se calentó la mezcla a 80 °C durante 1 hora y posteriormente se permitió que alcanzar temperatura ambiente (RT). Se añadieron posteriormente al reactor la solución de catalizador y 298 ml de una solución de NaOH 0,5 M desgasificada. Se cerró el reactor y se desgasificó con nitrógeno y se ajustó bajo 10 bar de hidrógeno. Se calentó la mezcla a 35 °C a presión constante de 10 bar. Trascurrido un tiempo total de 19 horas, se desconectó el dispositivo de calentamiento. Se enfrió el reactor a RT y se retiró la fase acuosa. Se lavó dos veces la fase orgánica con 0,5 l de agua; se volvió a extraer la fase acuosa con 1l de tolueno que se añadió a la fase orgánica. Se añadieron 240 g de carbono decolorante (Norit CAP Super) a la solución de tolueno y se agitó la mezcla durante la noche a RT. Se filtró el carbono y se lavó la torta filtrante con 1,5 l de acetato de etilo. Se concentró la solución amarillenta a sequedad a presión reducida para producir 2,38 kg de material húmedo bruto.
Se disolvió en 1,5 l de acetato de isopropilo a 60 °C bajo agitación, se añadieron 9 l de heptano precalentado (50 °C) y se agitó la mezcla a 60 °C. Se sembró la solución y se enfrió lentamente hasta RT bajo agitación. Se continuó la agitación a temperatura ambiente durante la noche, posteriormente se enfrió la mezcla a 0 °C durante 1 hora. Se filtró el sólido y se secó. El rendimiento fue de 2,1 kg (rendimiento de un 87 %, 95,0 % ee).
Ejemplo 4
Separación de (R)-1-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-didoro-1-piridin-4-il)etanol y (S)-1-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-didoro-1-piridin-4-l)etanol(intermedios (VIII) y (II), n = 0)

Se llevó a cabo la separación cromatográfica sobre 3000 g de (VI) en la que n es 0, en lote usando un columna Chiralpak IC 20mm-250*76 mm y diclorometano/etanol 95/5 v/v como fase móvil. Se introdujo una solución de (VI) racémico en varias veces en la parte superior de la columna quiral y se recogieron las fracciones eluidas de los enantiómeros separados en la parte inferior de la columna. Se cristalizó (II) a partir de la mezcla de elución DCM/EtOH concentrado enriquecida con etanol. Se obtuvieron 1440 g (48 % de rendimiento) del enantiómero deseado (II) en el que n es 0 con pureza de HPLC > 95 % y pureza de HPLC quiral > 99,5 %. También se obtuvieron 1470 g (49% de rendimiento) del otro enantiómero (VIII) en el que n es 0 con pureza de HPLC > 99 % y pureza de HPLC quiral > 99%.
Ejemplo 4A
Separación de (R)-1-(3-(ddopropNmetoxi)-4-(difluorometoxi)fenN)-2-(3,5-didoro-1-oxi-piridm-4-N)etanol y (S)-1-(3-(cidopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-didoro-1-oxipiridin-4-l)etanol(intermedios (VIII) y (II), n = 1)

Por analogía con el Ejemplo 4 se puede llevar a cabo una separación cromatográfica usando una columna Chiralpak IC 20 mm-250*76 mm y metanol como fase móvil sobre una solución de (VI) racémico en la que n es 1, para obtener el enantiómero deseado (II) en el que n es 1 con elevada pureza de HPLC y pureza de HpLC quiral. También se puede obtener el otro enantiómero (VIII) en el que n es 1 con elevada pureza de HPLC y pureza de HPLC quiral.
Preparación de éster 1-(3-ddopropMmetoxi-4-di-fluorometoxi-fenM)-2-(3,5-didoro-1-piridm-4-M)etíMco de ácido (S)-3-ddopropNmetoxi-4-metanosulfonNammobenzoico (compuesto (I), n = 0)

Se suspendieron 75 g de (111) en la que X es -NHSO2Me y Z es -OH en 750 ml de DCM; se añadieron 42,5 g de N,N-carbonil-diimidazol por partes y se agitó la solución obtenida a RT durante 30 minutos. Se añadieron 375 ml de tolueno, seguido de 85 g de (II) en la que n es 0 y se calentó la mezcla de reacción a reflujo. Se retiró DCM por medio de destilación, posteriormente se agitó la suspensión a 100 °C durante la noche. Se enfrió la solución obtenida a 40 °C, se añadieron 500 ml de acetato de etilo y se lavó con una solución de NaHCO3 y salmuera. Se aisló el producto por medio de cristalización a partir de acetato de etilo/heptano y se recristalizó con la misma mezcla de disolvente para obtener un sólido blanco (recuperación de 129 g, 73 % de rendimiento).
La caracterización del producto se describe en el documento WO 2010089107, Ejemplo 15.
Ejemplo 6
Preparación de éster 1-(3-ddopropMmetoxi-4-di-fluorometoxi-fenM)-2-(3,5-didoro-1-oxi-piridm-4-M)etíMco de ácido (S)-3-cidopropilmetoxi-4-metanosulfonilaminobenzoico (compuesto (I), n = 1)

Procedimiento

Se introdujeron 73 g de (I) en la que n es 0, en un matraz, seguido de 150 ml de tolueno y 290 ml de ácido acético y 75 ml de H2O2 al 35 % y se calentó la mezcla hasta 80 °C durante 8 horas. Se enfrió la mezcla a 50 °C, se añadieron 750 ml de acetato de etilo y se retiró la fase acuosa; se lavó la fase orgánica con agua y una solución acuosa de NaHCO3 al 10 % hasta un pH alcalino y se retiró el disolvente por medio de destilación. Se purificó el material bruto por medio de cristalización a partir de 375 ml de acetato de etilo y 225 ml de n-heptano y se secó en un dispositivo de secado estático de bandejas para obtener un sólido blanco (recuperación 65,1 g, rendimiento de 87,1 %).
La caracterización del producto se describe en el documento WO 2010089107, Ejemplo 17.
Preparación de (SH3-cidopropNmetoxi-4-difluorometoxifenN)-2-(3,5-didoro-1-oxi-piridm-4-M)-etanol

Procedimiento con H
2
O
2
/ácido acético
Se introdujeron 490 g de (II) en la que n es 0, en un reactor junto con 1960 ml de ácido acético glacial. Se calentó la mezcla a 50 °C, posteriormente se añadieron gradualmente 980 ml de agua oxigenada 30-35 % en agua y se mantuvo la mezcla en agitación a la misma temperatura durante 16 horas. Se añadieron lentamente 2000 ml de agua purificada y (II) en la que n = 1 precipitó en forma de sólido. Se enfrió la suspensión hasta 10 °C y se mantuvo en agitación durante 3 horas. A continuación, se filtró el sólido y se lavó el sólido obtenido con 1000 ml de agua. Se resuspendió el sólido húmedo (II) en la que n es 1 en 2000 ml de agua durante 2 horas y en 2000 ml de éter diisopropílico durante 3 horas. Se secó el sólido húmedo a vacío. Se obtuvieron 433 g de sólido blanco (85% de rendimiento).
La caracterización del producto se describe en el documento WO 2010089107, Ejemplo 7.
Procedimiento con Oxone®
Se añadieron 456 g de Oxone® (KHSO5*1/2KHSO4*1/2K2SO4) y 1,2 l de agua en un reactor y se agitó la mezcla a RT. Se añadieron 400 g de (II) en la que n es 0, y 3,2 l de metanol y se calentó la mezcla a 70 °C durante 3 horas. Se añadieron otros 50 g de Oxone® y después de 1,5 horas se completó la reacción. Se destiló el alcohol y se añadieron 4 l de agua y 2 l de acetato de etilo a 50 °C. Se descargó la fase acuosa y se lavó la fase orgánica con 800 ml de agua y se concentró a vacío hasta 1,5 l. Se añadieron 4 l de tolueno y se concentró la mezcla a vacío hasta 2,5 l al tiempo que comenzó la precipitación del producto. Se enfrió la suspensión hasta 10 °C y se mantuvo en agitación durante 1,5 horas. El sólido obtenido se filtró y se lavó con 800 ml de tolueno. Se secó el sólido húmedo a vacío en un dispositivo de secado estático de bandejas. Se obtuvieron 288 g de sólido blanco (72% de rendimiento).
La caracterización del producto se describe en el documento WO 2010089107, Ejemplo 7.
Ejemplo 8
Preparación de éster 1-(3-ciclopropilmetoxi-4-di-fluorometoxi-fenil)-2-(3,5-didoro-1-oxi-piridin-4-il)etílico de ácido (S)-3-ciclopropilmetoxi-4-metanosulfonilaminobenzoico (compuesto (I), n = 1)

Se introdujeron 100 g de (III) en la que X es NHSO2Me y Z es -OH y 1 l de acetato de etilo en un reactor. Se añadieron 57 g de carbonildiimidazol por partes bajo agitación a 40 °C, posteriormente se agitó la mezcla durante 60 minutos. Se añadieron 123 g de (II) en la que n =1 y 3,7 ml de 1,8-diazabiciclo[5.4.0]undec-7-eno y se calentó la mezcla a 75 °C durante aproximadamente 4 horas. Se lavó la solución orgánica con 500 ml de HCl 1 M en agua, con 500 ml de solución acuosa de NaHCO3 al 5 % y con 500 ml de solución acuosa de NaCl al 10 %. Se calentó la mezcla hasta 70 °C a vacío y se concentró hasta 600 ml. Se enfrió la mezcla a 50 °C y se añadieron 300 ml de nheptano. Se sembró la solución, se enfrió a 5 °C y se mantuvo la agitación durante 1,5 horas. Se filtró el sólido obtenido y se secó a vacío. Se obtuvieron 168 g de sólido bruto (82% de rendimiento).
La caracterización del producto se describe en el documento WO 2010089107, Ejemplo 17.
Ejemplo 9
Preparación de éster 1-(3-ciclopropNmetoxi-4-di-fluorometoxi-fenM)-2-(3,5-dicloro-1-oxi-piridin-4-N)etflico de ácido (S)-3-ciclopropilmetoxi-4-metanosulfonilaminobenzoico (compuesto (I), n = 1)- solvatado a partir de etanol
Se introdujo una solución de compuesto bruto (I) en la que n es 1 en un rector de 1 l. Se añadieron DCM (90 ml) y EtOH (300 ml), se agitó la suspensión blanca y se calentó a reflujo hasta disolver por completo. Se destiló DCM y comenzó a precipitar un sólido blanco. Se concentró de forma adicional la solución etanólica hasta 6-7 volúmenes destilando parte de EtOH y posteriormente se enfrió a 0-5 °C y se agitó durante 120 minutos. El sólido obtenido se filtró y se lavó con 30 ml de EtOH. Se secó el sólido húmedo a vacío en un dispositivo de secado estático de bandejas. Se obtuvieron 28,55 g de sólido blanco (95% de rendimiento).
Se investigó el compuesto de fórmula (I) en el que n es 1 obtenido como solvato como en el Ejemplo 9, para determinar el punto de fusión por medio de Calorimetría de Barrido Diferencial (DSC), espectroscopia de Raman para observar los modos vibracional, rotacional y de baja frecuencia y el patrón de difracción de rayos-X en forma de polvo (XRPD).
Se caracteriza por:
un intervalo de fusión de 87°-101 °C determinado por medio de DSC a una tasa de barrido de 10 °C/min.; un patrón de difracción de rayos-X caracterizado por los siguientes picos XRPD (Bruker D8 Advance con tubo de difracción de rayos-X tipo KFL CuKa2) 7,45; 7,87; 8,51; 10,12; 10,28; 12,66; 13,29; 13,45; 14,95; 16,14; 16,34; 17,05; 17,74; 18,05; 18,48; 18,88; 19,05; 19,33; 19,85; 20,18; 20,65; 21,3; 22,96; 23,55; 23,87; 24,41; 24,66; 24,88; 25,62; 25,82; 26,45; 28,12 y 28,5360,2 grados/2 theta.
Ejemplo 10
Cristalización del compuesto (I) en el que n es 1 - Forma A
Procedimiento a partir de acetato de etilo/heptano
Se introdujeron 5 g de (I) bruto en la que n es 1 en un reactor junto con 30 ml de acetato de etilo y se agitó la suspensión al tiempo que se calentó hasta 75 °C hasta que se disolvió el sólido por completo. Se añadieron 15 ml de n-heptano y se permitió que la solución alcanzara RT. Se enfrió la suspensión a 5 °C durante 2 horas, se filtró y se secó a vacío. Se obtuvo un sólido blanco, la denominada Forma A, (3,6 g, 72 % de rendimiento).
Se investigó el compuesto de fórmula (I) en el que n es 1 obtenido como Forma A como en el Ejemplo 10, para determinar el punto de fusión por medio de Calorimetría de Barrido Diferencial (DSC), espectroscopia de Raman para observar los modos vibracional, rotacional y de baja frecuencia y el patrón de difracción de rayos-X en forma de polvo (XRPD).
Se caracteriza por: un intervalo de fusión de 144°-147 °C determinado por medio de DSC a una tasa de barrido de 10 °C/min.; un patrón de difracción de rayos-X caracterizado por los siguiente picos XRPD (Bruker D8 Advance con tubo de difracción de rayos X tipo KFL CuKa2): 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19, 1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ±0,2 grados/2 theta.
Procedimiento a partir de acetato de isopropilo
Se introdujeron 5 g de (I) bruto en la que n es 1 en un matraz junto con 20 ml de acetato de isopropilo y se calentó la suspensión a reflujo hasta disolver por completo. Se enfrió la mezcla a 0 °C y se agitó durante 2 horas. Se filtró el sólido obtenido y se lavó con 10 ml de acetato de isopropilo. Se secó el sólido húmedo a vacío. Se obtuvieron 4,05 g de sólido blanco, la Forma A cristalina, (81 % de rendimiento).
La caracterización del producto se describe en el documento WO 2010089107 Ejemplo
Oxidación del intermedio (VII), n=0 hasta 1-(3-(cidopropMmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-didoro-1- oxipiridin-4-il)etanona (intermedio (VII), n=1)
Procedimiento con H
2
O
2
/ácido acético
Se introdujeron 0,5 g de (VII) en la que n es 0 en un matraz de 50 ml junto con 3 ml de ácido acético glacial. Se calentó la solución homogénea hasta 50 °C, posteriormente se añadieron gradualmente 1 ml de agua oxigenada 30 35 % en agua y se mantuvo la mezcla en agitación a la misma temperatura durante 21 horas. Se retiró posteriormente el disolvente a presión reducida y se purificó el sólido bruto en una columna cromatográfica con una elución por gradiente (hexano/EtOAc 85/15 hasta EtOAc 100 %), dando un producto puro en forma de sólido blanco. (Rendimiento 50 %).
Procedimiento con Oxone®
Se introdujeron 10g de (VII) en la que n es 0, en un matraz junto con 11,44 g de Oxone®, 80 ml de metanol y 30 ml de agua. Se calentó la mezcla hasta 65 °C durante 5 horas y a RT durante 48 horas. Se destiló el alcohol y se añadieron 50 ml de agua y 100 ml de tolueno. Se calentó la mezcla hasta disolver por completo el sólido, se concentró la fase acuosa descargada y la fase orgánica a vacío hasta 70 ml. Se enfrió la suspensión hasta 0 °C y se mantuvo en agitación durante 1,5 horas. Se filtró el sólido obtenido y se secó a vacío en un dispositivo de secado estático de bandejas. Se obtuvieron 6,7 g de sólido blanco (60% de rendimiento).
Procedimiento con MCPBA
Se disolvieron 0,5 de (VII) en la que n es 0 en 10 ml de THF, se añadieron 0,34 g de MCPBA (ácido 3-cloroperoxibenzoico, ensayo al 77 %) y se agitó la mezcla a RT durante la noche. El control por HPLC confirmó la conversión casi completa. La solución se separó entre 100 ml de acetato de etilo y 50 ml de una solución acuosa al 5 % de hidrogenocarbonato de potasio. Se lavó la fase orgánica con 50 ml adicionales de solución básica y se secó a vacío. Se purificó la sustancia bruta sobre un lecho de sílice con una mezcla de acetato de etilo y diclorometano como eluyente. Se obtuvieron 0,22 g de (VII) en la que n = 1 (42 % de rendimiento)
Ejemplo 12
Oxidación del intermedio (VI), n=1 hasta 1-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-dicloro-1- oxipiridin-4-il)etanona (intermedio (VII), n=1)

Procedimiento con DMP
Se suspendió alcohol (VI) en la que n es 1 (1,0 g, 2,38 mmol) en acetona (15 ml). Se enfrió la suspensión bajo agitación a 0-5 °C con un baño de hielo. Posteriormente se añadió peryodinano de Dess-Martin (1,4 g, 3,3 mmol) en una parte individual. Inicialmente la reacción fue exotérmica y trascurrida 1 h se dejó que alcanzar RT. Después de 20 horas la reacción se completó y se inactivó con la adición de 10 ml de una solución acuosa al 10 % de metabisulfito de sodio y 30 ml de una solución acuosa al 15 % de carbonato de potasio. Se agitó la mezcla a 25 °C durante 0,5 horas y se filtró el sólido, para permitir la obtención de una cetona (VII) en la que n es 1 con rendimiento cuantitativo.
Preparación de ácido 3-(ddopropNmetoxi)-4-nitrobenzoico (intermedio (III), X = -NO
2
, Z = -OH)

Se preparó (III) en la que X es -NO2 y Z es -OMe de acuerdo con el procedimiento descrito en el documento WO 2010/089107, Ejemplo 18. Se introdujeron 550 g de (III) en la que X es -NO2 y Z es -OMe en un reactor, seguido de 1,65 l de THF y 2,85 l de una solución acuosa 1 M de hidróxido de litio. Se calentó la mezcla hasta 40 °C durante 1,5 horas, posteriormente se enfrió a RT. Se añadieron 4,4 l de acetato de etilo, seguido de 240 ml de una solución acuosa al 37 % de HCl. Se descargó la fase acuosa y se lavó la fase orgánica dos veces con 2,75 l de agua y posteriormente se concentró a vacío a 50 °C, se añadieron 1,65 l de n-heptano a la misma temperatura y se enfrió la suspensión hasta RT. Se filtró el sólido y se secó a vacío en un dispositivo de secado de bandejas a vacío obteniéndose 337 g de (III) en la que x es -NO2 y Z es -OH (rendimiento de 73 %).
Ejemplo 14
Preparación de éster 1-(3-ddopropMmetoxi-4-di-fluorometoxi-fenM)-2-(3,5-didoro-1-piridin-4-N)etflico de ácido (S)-3-ddopropNmetoxi-4-nitrobenzoico (intermedio (IV), n = 0)

Se mezclaron el intermedio (III) en la que X es -NO2, Z es -OH (80 g, 0,34 mol, ref.) y (II) en la que n es 0 (109,1 g, 0,27 mol, 0,9 eq), EDC.HCl (193,9 g, 1,01 mmol, 3 eq), DMAP (20,6 g, 0,17 mol, 0,5 eq) y DMF (400 ml, 5 vol) de manera conjunta y se calentó a 75 °C durante la noche. Se separó la solución entre agua y acetato de etilo, se lavó la fase orgánica con una solución acuosa ácida y básica y se concentró a vacío. Se cristalizó el material bruto con EtOH (1200 ml), acetona (100 ml). Se obtuvo un sólido blanco (101 g, 60 % de rendimiento con respecto a (VIII)). RMN 1H (400 MHz, DMSO-CÍ6) 8 ppm 8,60 (s, 2 H), 7,97 (d, J=8,38 Hz, 1 H), 7,61 - 7,80 (m, 2 H), 7,18 - 7,32 (m, 2 H), 7,02 - 7,14 (m, 2 H), 6,27 (dd, J=9,70, 3,97 Hz, 1 H), 4,04 - 4,21 (m, 2 H), 3,89 - 4,02 (m, 2 H), 3,74 (dd, J=14,11, 9,70 Hz, 1 H), 3,45 (dd, J=13,89, 4,19 Hz, 1 H), 1,10 - 1,30 (m, 2 H), 0,49 - 0,65 (m, 4 H), 0,36 (qd, J=5,44, 5,29 Hz, 4 H).
Ejemplo 15
Preparación de éster 1-(3-ciclopropilmetoxi-4-di-fluorometoxi-fenil)-2-(3,5-dicloro-1-piridin-4-il)etílico de ácido (S)-3-ciclopropilmetoxi-4-nitrobenzoico (intermedio (IV), n = 1)

Se mezclaron (III) en la que X es -NO2, Z es -OH (80 g, 0,34 mol, ref.), (II) en la que n es 1 (113,9 g, 0,27 mol, 0,9 eq), EDC.HCl (193,9 g, 1,01 mmol, 3 eq), DMAP (20,6 g, 0,17 mol, 0,5 eq) y DMF (400 ml, 5 vol) de manera conjunta y se calentó a 100 °C durante la noche. Se separó la solución entre agua y acetato de etilo, se lavó la fase orgánica
con una solución acuosa ácida y básica y se concentró a vacío. Se cristalizó el material bruto con EtOH (600 ml), acetona (200 ml) y heptano (200 ml). Se obtuvo un sólido blanco (71 g, 41% de rendimiento con respecto a (II) en la que n es 1).
RMN 1H (400 MHz, DMSO-Ce) 8 ppm 8,56 (s, 2 H), 7,97 (d, J=8,38 Hz, 1 H), 7,62 - 7,83 (m, 2 H), 7,16 - 7,32 (m, 2 H), 7,04 - 7,14 (m, 2 H), 6,20 (dd, J=9,26, 4,41 Hz, 1 H), 4,11 (dd, J=7,06, 3,53 Hz, 2 H), 3,93 (d, J=6,62 Hz, 2 H), 3,62 (d, J=9,26 Hz, 1 H), 3,32 (d, J=9,26 Hz, 1 H), 1,17 -1,26 (m, 2 H), 0,49 - 0,67 (m, 4 H), 0,24 - 0,43 (m, 4 H).
Ejemplo 16
Preparación de éster 1-(3-ddopropNmetoxi-4-di-fluorometoxi-fenN)-2-(3,5-didoro-1-piridin-4-N)etíNco de ácido (S)-3-cidopropMmetoxi-4-aminobenzoico (intermedio (V), X =NH
2
y n = 0)

Procedimiento de hidrogenación
Se introdujeron 2,5 g de (IV) en la que n es 0, 119 mg de catalizador Pd/C y 25 ml de acetato de etilo en el reactor. Posteriormente se selló el reactor y se calentó con agitación suave hasta la temperatura interna de 40 °C. Se introdujo hidrógeno en el reactor a 4 bar. Tras 4 horas, la conversión fue completa. Se retiró el catalizador por medio de filtración y el disolvente se destiló a presión reducida. Se recuperaron 2,20 g de producto (90 % de rendimiento). RMN 1H (400 MHz, CDCl3) 8 ppm 8,50 (s, 2 H), 7,49-7,56 (m, 1 H), 7,30 - 7,36 (m, 2 H), 7,10 - 7,19 (m, 1 H), 7,00 -7,08 (m, 2 H), 6,58-6,68 (m, 1 H), 6,20-6,28 (m, 1 H), 4,11 (sa, 2 H), 3,78-3,92 (m, 4 H), 3,69-3,79 (m, 1 H), 3,30-3,37 (m, 1 H), 1,178 -1,32 (m, 2 H), 0,58 -0,71 (m, 4 H), 0,28 -0 ,35 (m, 4 H).
Procedimiento con SnCh
Se disolvieron 2 g de (IV) en la que n es 0 en 20 ml de THF y se añadieron 4,34 g de cloruro de estaño (II) dihidratado. Se agitó la solución a 80 °C durante la noche. La solución se separó entre 100 ml de acetato de etilo y 100 ml de una solución acuosa de KHCO3 al 5 %. Se filtró la mezcla para retirar las sales precipitadas y se descargó la fase acuosa. Se lavó la fase orgánica con KHCO3 adicional y salmuera. Se retiró el disolvente orgánico a vacío aislando (V) en la que n es 0 en forma de aceite amarillo (1,84 g, 97 % de rendimiento).
Ejemplo 17
Preparación de éster 1-(3-ddopropNmetoxi-4-di-fluorometoxi-fenN)-2-(3,5-didoro-1-piridin-4-N)etíNco de ácido (S)-3-ciclopropilmetoxi-4-aminobenzoico (intermedio (V), n = 1)

Procedimiento de hidrogenación
Se introdujeron 400 mg de (IV) en la que n =1, 8 mg de catalizador de Pd/C al 1 % y 4 ml de acetato de etilo en el reactor, se selló y se calentó con agitación suave a 60 °C, se introdujo hidrógeno a 4 bar y se continuó la agitación durante 4 horas. Se filtró la mezcla para retirar el catalizador y se secó a vacío.
Procedimiento con SnCl
2
Se disolvió 1g de (IV) en la que n =1 en 10 ml de THF y se añadieron 1,06 g de cloruro de estaño (II) dihidratado. Se agitó la solución a RT durante la noche. Se evaporó el disolvente a vacío y se añadieron 10 ml de acetato de etilo y 10 ml de una solución acuosa 1 M de NaOH a la sustancia bruta. Se descargó la fase acuosa y se lavó la fase orgánica con 10 ml de una solución acuosa de NaCl al 10 %. Se retiró el disolvente orgánico y se suspendió la sustancia bruta en éter dietílico y se agitó hasta obtener un sólido; se filtró, se lavó con 4 ml de éter dietílico y se secó en un dispositivo de secado estático de bandejas. Se obtuvo un sólido blanco (0,68 g, 71,3%).
RMN 1H (400 MHz, DMSO-CÍ6) 8 ppm 8,55 (s, 2 H), 7,40 (dd, J=8,38, 1,76 Hz, 1 H), 7,28 (d, J=1,76 Hz, 1 H), 7,15 -7,21 (m, 2 H), 6,99 - 7,08 (m, 2 H), 6,64 (d, J=8,38 Hz, 1 H), 6,14 (dd, J=9,59, 4,30 Hz, 1 H), 5,63 (s, 2 H), 3,88 - 3,96 (m, 2 H), 3,70 - 3,88 (m, 2 H), 3,55 (dd, J=14,11, 9,92 Hz, 1 H), 3,24 -3,31 (m, 1 H), 1,11 - 1,34 (m, 2 H), 0,47 -0,65 (m, 4 H), 0,19 -0,41 (m, 4 H).
Ejemplo 18
Preparación de éster 1-(3-ddopropMmetoxi-4-di-fluorometoxi-fenM)-2-(3,5-didoro1-piridin-4-M)etflico de ácido (S)-3-cidopropNmetoxi-4-metanosulfonMaminobenzoico (compuesto (I), n = 0)

Se disolvieron 0,5 g de (V) en la que n es 0 (0,84 mmol) en DCM (7 ml) y TEA (0,17 ml, 1,26 mmol), posteriormente se añadió lentamente cloruro de metanosulfonilo (0,11 g, 0,93 ml), y se agitó la solución a RT durante 20 horas. A continuación, se inactivó la reacción con agua (20 ml) y se extrajo el disolvente orgánico y se lavó con una solución acuosa de NaCl al 5 % (10 ml). Se retiró el disolvente y se purificó la sustancia bruta en cromatografía en columna con una elución por gradiente (Hexano al 100 % hasta Hexano/EtOAc 60/40), dando lugar al producto puro en forma de aceite incoloro (Rendimiento de 30 %).
Ejemplo 19
Oxidación de intermedio (IV) en la que n es 0 para obtener éster 1-(3-cidopropilmetoxi-4-difluorometoxifenil)-2-(3,5-didoro-1-oxipiridin-4-il)-etílico de ácido (S)-3-ciclopropilmetoxi-4-nitrobenzoico (intermedio (IV), n = 1)

Procedimiento con H2O2/ácido acético
Se introdujeron 0,5 g de (IV) en la que n es 0 en un matraz de 50 ml junto con 3 ml de ácido acético glacial. Se calentó la solución hasta 55 °C, posteriormente se añadieron gradualmente 1 ml de agua oxigenada (35 %) y se mantuvo la mezcla en agitación a la misma temperatura durante 48 horas. Se añadieron 5 ml de agua, se extrajo el producto con acetato de etilo y se retiró el disolvente orgánico a presión reducida dando lugar al producto en forma de aceite amarillo (Rendimiento 74 %).
Procedimiento con Oxone®
Se introdujeron 0,3 g de (IV) en la que n es 0 en un matraz de 50 ml, seguido de 2,4 ml de metanol, 1 ml de agua y 215 mg de Oxone®. Se agitó la suspensión a 55 °C durante 48 h y a 40 °C durante 72 horas. Se retiró el metanol a presión reducida y se añadieron 5 ml de acetato de etilo. Se extrajo la fase acuosa con acetato de etilo (3 x 5 ml), se secó la fase orgánica con NaSO4 y se retiró el disolvente a presión reducida dando lugar al producto en forma de aceite amarillento (rendimiento 96 %).
Procedimiento con MCPBA
Se disolvieron 0,5 de (IV) en la que n es 0 en 10 ml de THF, se añadieron 0,22 g de MCPBA (ácido 3-cloroperoxibenzoico, ensayo al 77 %) y se agitó la mezcla a RT durante la noche. El control por HPLC confirmó la conversión casi completa. La solución se separó entre 100 ml de acetato de etilo y 50 ml de una solución acuosa al 5 % de hidrogenocarbonato de potasio. Se lavó la fase orgánica con 50 ml adicionales de solución básica y se secó a vacío. Se purificó la sustancia bruta sobre un lecho de sílice con una mezcla de acetato de etilo y diclorometano como eluyente. Se obtuvieron 0,19 g de (VII) (37% de rendimiento).
Ejemplo 20
Metanosulfonilación de (V) en la que n es 1 para obtener éster 1-(3-ciclopropilmetoxi-4-difluorometoxi-fenil)-2-(3,5-dicloro-1-oxipiridin-4-il)-etílico de ácido (S)-3-ciclopropilmetoxi-4-metanosulfonilaminobenzoico (compuesto (I) en la que n es 1)

Se disolvieron 0,2 g de (V) en la que n es 1 (0,33 mmol) en DCM (3 ml) y TEA (0,05 ml, 0,39 mmol), posteriormente se añadió lentamente cloruro de metanosulfonilo (0,045 g, 0,07 ml), y se agitó la solución a RT durante 20 horas. A continuación, se inactivó la reacción con HCl 1 N (10 ml) y se extrajo el disolvente orgánico y se lavó con una solución acuosa de NaCl al 5 % (10 ml). Se retiró el disolvente y se purificó la sustancia bruta en cromatografía en columna con una elución por gradiente ( Hexano/EtOAc 85/15 hasta EtOAc al 100 %), dando lugar al producto puro en forma de aceite incoloro (Rendimiento de 30 %).
Ejemplo 21
Preparación de éster metílico de ácido 3-hidroxi-4-(difluorometoxi)benzoico

Se disolvieron 100 g de 3-hidroxi-4-(difluorometoxi)-benzaldehído (0,53 mol) en MeOH (600 ml), se añadió por partes Oxone® sólido (325 g, 1,06 mol) en 1 hora y se agitó la solución y se calentó a 50-55 °C durante 2 horas. Se concentró el disolvente a vacío hasta 200 ml y se añadió agua (1 l). Se agitó la solución heterogénea resultante a 50-55 °C, posteriormente se añadió tolueno (500 ml), y se agitó vigorosamente la mezcla bifásica. Se descargó la fase acuosa y se lavó la fase orgánica con agua (500 ml). Se añadió carbón vegetal activo (10 g) y se agitó la solución orgánica durante 20 minutos. Se filtró sobre un lecho de celite, se concentró el disolvente a vacío hasta 2-3 volúmenes y se calentó la solución obtenida hasta 80-90 °C. Se añadió n-heptano (400 ml) de forma lenta. Se enfrió la mezcla hasta 0 °C y se agitó la suspensión a 0 °C durante la noche. Se filtró el sólido sobre un embudo de Buchner y se lavó con n-heptano (100 ml). Se secó el sólido blanco obtenido a vacío a temperatura ambiente. (Rendimiento 70%).
Ejemplo 22
Preparación de éster metílico de ácido 3-(cidopropilmetoxi)-4-(difluorometoxi)benzoico

Se disolvieron 873 g de 3-(ciclopropilmetoxi)-4-(difluorometoxi)-benzaldehído (3,61 mol) en MeOH (4,4 l), posteriormente se añadió por partes Oxone® sólido (1,86 Kg, 6,06 mol) en 1 hora y se agitó la solución y se calentó a 55-60 °C durante 2 horas. Se concentró el disolvente a vacío hasta 1,6 l y se añadió agua (7 l). Se agitó la solución heterogénea resultante a 50-55 °C, posteriormente se añadió tolueno (3 l), y se agitó vigorosamente la mezcla bifásica. Se descargó la fase acuosa y se lavó la fase orgánica con agua (3 l). Se concentró el disolvente a vacío hasta 2-3 volúmenes y se calentó la solución obtenida hasta 80-90 °C. Se añadió n-heptano (5,5 l) de forma lenta. Se enfrió la mezcla hasta -10 °C y se agitó la suspensión a -10 °C durante la noche. Se filtró el sólido sobre un embudo de Buchner y se lavó con n-heptano (1 l). Se secó el sólido blanco obtenido a vacío a temperatura ambiente. (Rendimiento 52%).
Ejemplo 23
Preparación de 1-(3-(ciclopropilmetoxi)-4-(difluorometoxi)fenil)-2-(3,5-dicloro-1-piridin-4-il)etanona (intermedio (VII), n = 0)

Se introdujo éster metílico de ácido 3-(ciclopropilmetoxi)-4-(difluorometoxi)-benzoico (30 g, 0,11 mol) y 3,5-dicloro-4-metil-piridina (21,4 g, 0,13 mol) en un reactor de 1 l y se disolvió en THF (120 ml). Se enfrió la solución homogénea a -10 °C bajo agitación. Se añadió lentamente una solución 1 M de bis(trimetilsilil)amida de litio en THF (0,22 mol, 220 ml) en 30 minutos. Se agitó la mezcla a baja temperatura durante 15-30 minutos, posteriormente se inactivó con una solución acuosa de HCl al 10 % (250 ml) y se calentó a temperatura ambiente. Se añadió acetato de etilo (300 ml) y se agitó vigorosamente la mezcla bifásica durante 15-20 minutos. Se reextrajo la fase acuosa con acetato de etilo (150 ml). Se concentraron las fases orgánicas reunidas hasta 2 volúmenes. Se añadió isopropanol (240 ml) y se concentró la solución de nuevo hasta 2-3 volúmenes. Se añadió isopropanol (150 ml) y se enfrió la solución hasta 0 °C obteniéndose la precipitación de un sólido amarillo pálido. Trascurridas 3 horas, se filtró el sólido en un embudo de Buchner y se lavó con un volumen de isopropanol frío. Se secó el sólido obtenido a vacío a RT. (Rendimiento 90,5%).
Preparación de éster metílico de ácido 3-(cidopropilmetoxi)-4-(difluorometoxi)benzoico

Procedimiento con DMF y Carbonato de potasio
Se disolvieron éster metílico de ácido 3-hidroxi-4-(difluorometoxi)-benzoico (5 g, 22,9 mmol), K2CO3 (4,75 g, 34,4 mmol), NaI (0,34 g, 2,3 mmol) y bromo-metilciclopropano (3,7 g, 27,5 mmol) en DMF (25 ml) y se agitó la mezcla heterogénea y se lavó a 80 °C durante dos horas. Se enfrió la suspensión a temperatura ambiente y se añadió agua (50 ml) bajo agitación. Se enfrió la mezcla heterogénea a 0-5 °C durante 60-90 minutos y se filtró el sólido sobre un embudo de gooch y se lavó con agua (50 ml). Se obtuvo un sólido naranja. Se secó a vacío a temperatura ambiente. (Rendimiento 95,8%).
Ejemplo 25
Preparación de éster metílico de ácido 3-(ciclopropilmetoxi)-4-(difluorometoxi)benzoico

Procedimiento con DMA, carbonato de potasio y MeI
Se suspendieron ácido 3-(ciclopropilmetoxi)-4-(difluorometoxi)benzoico (50 g, 193,6 mmol) y K2CO3 (28,1 g, 203,3 mmol) en DMA (400 ml) y se calentó la suspensión a 75-85 °C. Se añadió una solución de MeI (32,97 g, 232,0 mmol) en DMA (100 ml) a través de un embudo de adición en 1 hora. A final de la adición se enfrió la suspensión a 0-5 °C y se añadió agua (500 ml) bajo agitación. Tuvo lugar la precipitación de un sólido blanco. Se agitó la mezcla fría heterogénea durante 60-90 minutos y se filtró el sólido en un embudo de gooch y se lavó con agua (50 ml). Se obtuvo un producto como sólido blanco. Se secó a vacío a temperatura ambiente. (Rendimiento 98,1%).
Ejemplo 26
Preparación de éster metílico de ácido 3-hidroxi-4-(difluorometoxi)benzoico

En un matraz de 50 ml se disolvió 3-(ciclopropilmetoxi)-4-(difluorometoxi)-benzonitrilo (0,5 g, 2,7 mmol), en MeOH (3 ml) y se agitó la solución homogénea a temperatura ambiente. Se añadió lentamente por partes H2SO4 al 91 % (1 ml) y se calentó la solución a 50 °C durante una semana. Se enfrió la solución a 0-5 °C y se añadió agua (10 ml) y se agitó la suspensión obtenida a baja temperatura durante 1 hora. Se filtró la suspensión en un embudo de gooch. Se obtuvo el producto en forma de sólido blanco (78 % de rendimiento).
Ejemplo 27
Preparación de (R/S)-1-(3-(ciclopropNmetoxi)-4-(difluorometoxi)fenN)-2-(3,5-dicloro-I-oxi-pindm-4- il)etanol (intermedio (VI), n = 1)

En un matraz de fondo redondo de 100 ml se añadió (VII) en la que n es 1 (0,2 g, 0,48 mmol) bajo atmósfera de nitrógeno, se suspendió en MeOH (10 ml) y se enfrió a 0-5 °C. Se añadió NaBH4 (18,0 mg, 0,48 mmol) y se agitó la suspensión durante 1,5 horas. Se inactivó la reacción con H2O (25 ml) y se calentó hasta temperatura ambiente. Se extrajo la solución acuosa dos veces con acetato de etilo (2 x 15 ml) y se secaron las fases orgánicas reunidas sobre Na2SO4. Se evaporó el disolvente a presión reducida obteniéndose un sólido bruto. Se disolvió en tolueno (10 ml, 85-90 °C9 y se cristalizó enfriando la solución a 0-5 °C durante 2 horas. Se filtró el sólido obtenido, se lavó con 10 ml de tolueno y se secó a vacío en un dispositivo de secado estático de bandejas. Se obtuvieron 164,5 mg de sólido blanco. (Rendimiento de 81,6 %).
Claims (10)
1. Una forma cristalina del compuesto de fórmula (I)

en la que n es 1, caracterizada por los siguientes picos de XRPD característicos: 7,48; 7,93; 10,15; 10,32; 12,72; 13,51; 16,18; 16,46; 18,08; 18,53; 18,94; 8,55; 17,79; 19,89; 19,1; 20,2; 21,37; 22,96; 23,63; 24,87; 26,51; 28,09; 28,61 y 25,82 ± 0,2 grados/2 theta (CuKa2).
2. La forma cristalina de acuerdo con la reivindicación 1, en la que el porcentaje de cristalización es igual o superior al 90 %.
3. La forma cristalina de acuerdo con la reivindicación 1, en la que el porcentaje de cristalización es igual o superior al 95 %.
4. La forma cristalina de acuerdo con la reivindicación 1, en la que la cantidad de impurezas detectables con facilidad es inferior al 1.0% p/p.
5. La forma cristalina de acuerdo con la reivindicación 1, en la que la cantidad de impurezas detectables con facilidad es inferior al 0.5% p/p.
6. La forma cristalina de acuerdo con las reivindicaciones 1 a 5 para su uso en la prevención y/o tratamiento de una enfermedad respiratoria inflamatoria u obstructiva.
7. La forma cristalina para su uso de acuerdo con la reivindicación 6 en la que la enfermedad respiratoria inflamatoria u obstructiva es asma o enfermedad pulmonar obstructiva crónica (EPOC).
8. Una composición farmacéutica para inhalación que contiene la forma cristalina de acuerdo con las reivindicaciones 1 a 5 en combinación con portadores o vehículos adecuados.
9. Las composiciones farmacéuticas para inhalación de acuerdo con la reivindicación 8 para su uso para prevenir y/o tratar una enfermedad respiratoria inflamatoria u obstructiva.
10. Las composiciones farmacéuticas para inhalación para su uso de acuerdo con la reivindicación 9, en las que la enfermedad respiratoria inflamatoria u obstructiva es asma o enfermedad pulmonar obstructiva crónica (EPOC).
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13189784 | 2013-10-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2857629T3 true ES2857629T3 (es) | 2021-09-29 |
Family
ID=49385193
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19183016T Active ES2980505T3 (es) | 2013-10-22 | 2014-10-17 | Procedimiento para la preparación de un inhibidor de PDE4 |
| ES14795565T Active ES2759514T3 (es) | 2013-10-22 | 2014-10-17 | Procedimiento de preparación de un inhibidor de PDE4 |
| ES17192278T Active ES2857629T3 (es) | 2013-10-22 | 2014-10-17 | Forma cristalina de un inhibidor de PDE4 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19183016T Active ES2980505T3 (es) | 2013-10-22 | 2014-10-17 | Procedimiento para la preparación de un inhibidor de PDE4 |
| ES14795565T Active ES2759514T3 (es) | 2013-10-22 | 2014-10-17 | Procedimiento de preparación de un inhibidor de PDE4 |
Country Status (29)
| Country | Link |
|---|---|
| US (7) | US9434691B2 (es) |
| EP (5) | EP4059921A1 (es) |
| JP (5) | JP6458957B2 (es) |
| KR (3) | KR102287598B1 (es) |
| CN (5) | CN120136784A (es) |
| AR (4) | AR098128A1 (es) |
| AU (1) | AU2014339136C1 (es) |
| BR (1) | BR112016008161B1 (es) |
| CA (3) | CA3115587C (es) |
| CY (1) | CY1123811T1 (es) |
| DK (3) | DK3587400T3 (es) |
| ES (3) | ES2980505T3 (es) |
| FI (1) | FI3587400T3 (es) |
| HR (3) | HRP20240783T1 (es) |
| HU (3) | HUE067681T2 (es) |
| IL (4) | IL285528B2 (es) |
| LT (2) | LT3293176T (es) |
| MX (4) | MX389002B (es) |
| MY (1) | MY182559A (es) |
| PH (3) | PH12020500162B1 (es) |
| PL (3) | PL3587400T3 (es) |
| PT (3) | PT3060551T (es) |
| RS (2) | RS61558B1 (es) |
| RU (1) | RU2682660C9 (es) |
| SG (4) | SG10202112587RA (es) |
| SI (3) | SI3060551T1 (es) |
| TW (3) | TWI685486B (es) |
| UA (2) | UA127453C2 (es) |
| WO (1) | WO2015059050A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112013002506A2 (pt) | 2010-08-03 | 2016-05-31 | Chiesi Farma Spa | formulação em pó seco inalável, uso de uma formulação em pó seco inalável, inalador de pó seco inalável e embalagem |
| HUE067681T2 (hu) * | 2013-10-22 | 2024-11-28 | Chiesi Farm Spa | Eljárás PDE4-inhibitor elõállítására |
| WO2018017713A1 (en) * | 2016-07-20 | 2018-01-25 | Allergan, Inc. | Methods, compositions, and compounds for treatment of dermatological and ocular conditions |
| JP7361584B2 (ja) * | 2018-12-19 | 2023-10-16 | 高砂香料工業株式会社 | アミドの還元によるアミンの製造方法 |
| JP6780763B2 (ja) | 2018-12-25 | 2020-11-04 | トヨタ自動車株式会社 | 内燃機関の制御装置 |
| US11149615B2 (en) | 2018-12-25 | 2021-10-19 | Toyota Jidosha Kabushiki Kaisha | Control device for internal combustion engine |
| CN111808029A (zh) * | 2020-08-31 | 2020-10-23 | 兰晟生物医药(苏州)有限公司 | 一种pde4抑制剂氯比普兰的制备方法 |
| EP4452223A1 (en) | 2021-12-21 | 2024-10-30 | Chiesi Farmaceutici S.p.A. | Dry powder formulations filled in an inhaler with improved resistance to humidity |
| US20250288568A1 (en) | 2022-04-27 | 2025-09-18 | Chiesi Farmaceutici S.P.A. | New crystal form of a pde4 inhibitor |
| CN115304542B (zh) * | 2022-07-18 | 2024-02-02 | 湖南华纳大药厂手性药物有限公司 | 一种3-羟基吡啶的合成工艺 |
| WO2024027901A1 (en) | 2022-08-02 | 2024-02-08 | Chiesi Farmaceutici S.P.A. | Predictive biomarker of clinical response to a pde4 inhibitor |
| EP4590271A1 (en) | 2022-09-22 | 2025-07-30 | Chiesi Farmaceutici S.p.A. | Capsule inhaler for the administration of a phosphodiesterase-4 inhibitor |
| GEAP202516739A (en) | 2022-09-22 | 2025-05-27 | Chiesi Farm Spa | Capsule inhaler for the administration of a phosphodiesterase-4 inhibitor |
| CN119947705A (zh) | 2022-09-22 | 2025-05-06 | 奇斯药制品公司 | 用于施用磷酸二酯酶-4抑制剂的胶囊吸入器 |
| KR102925863B1 (ko) | 2023-11-27 | 2026-02-09 | 국립한국해양대학교산학협력단 | 복수의 학습모델을 이용한 이미지 기반의 강도 추정 장치 및 방법 |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5935978A (en) * | 1991-01-28 | 1999-08-10 | Rhone-Poulenc Rorer Limited | Compounds containing phenyl linked to aryl or heteroaryl by an aliphatic- or heteroatom-containing linking group |
| WO2001074829A1 (en) * | 2000-03-30 | 2001-10-11 | Chirotech Technology Limited | Ruthenium-diphosphine complexes and their use as catalysts |
| WO2005026095A1 (en) | 2003-09-12 | 2005-03-24 | Ranbaxy Laboratories Limited | Process for the preparation of roflumilast |
| EA010198B1 (ru) | 2004-04-19 | 2008-06-30 | Крка, Товарна Здравил, Д.Д., Ново Место | Способы получения полиморфной формы i гидросульфата клопидогрела |
| JP4685861B2 (ja) * | 2004-04-29 | 2011-05-18 | アベンティス・ファーマスーティカルズ・インコーポレイテツド | ドーパミンアゴニストとしての3−ピペリジニルイソクロマン−5−オール |
| KR20070079278A (ko) | 2006-02-01 | 2007-08-06 | 삼성전자주식회사 | 수신자 제한 시스템을 이용하는 디지털 멀티미디어시스템에서 수신기의 채널 전환 시간 단축 장치 및 방법 |
| EP2044023B1 (en) * | 2006-07-14 | 2011-01-19 | CHIESI FARMACEUTICI S.p.A. | Derivatives of 1-phenyl-2-pyridynyl alkylene alcohols as phosphodiesterase inhibitors |
| BRPI0721113A2 (pt) * | 2006-12-22 | 2014-10-07 | Leo Pharma As | Composto, composição farmacêutica, uso de um composto, e, método para prevenir, tratar ou melhorar doenças ou condições dérmicas ou distúrbios |
| EP2022783A1 (en) * | 2007-08-08 | 2009-02-11 | CHIESI FARMACEUTICI S.p.A. | "Derivatives of 1-phenyl-2-pyridinyl alkyl alcohols as phosphodiesterase inhibitors" |
| US20100015430A1 (en) | 2008-07-16 | 2010-01-21 | Outlast Technologies, Inc. | Heat Regulating Article With Moisture Enhanced Temperature Control |
| EP2216327A1 (en) * | 2009-02-06 | 2010-08-11 | CHIESI FARMACEUTICI S.p.A. | Benzoic acid (1-phenyl-2-pyridin-4-yl)ethyl esters as phosphodiesterase inhibitors |
| KR101736529B1 (ko) | 2010-06-14 | 2017-05-17 | 키에시 파르마슈티시 엣스. 피. 에이. | 글리코피로늄 클로라이드의 결정형 |
| BR112013002506A2 (pt) * | 2010-08-03 | 2016-05-31 | Chiesi Farma Spa | formulação em pó seco inalável, uso de uma formulação em pó seco inalável, inalador de pó seco inalável e embalagem |
| KR101803121B1 (ko) * | 2010-08-03 | 2017-11-29 | 키에시 파르마슈티시 엣스. 피. 에이. | 포스포디에스테라제 억제제를 포함하는 약학적 제제 |
| RU2628560C2 (ru) | 2010-11-23 | 2017-08-18 | Эббви Инк. | Соли и кристаллические формы индуцирующего апоптоз агента |
| WO2012097479A1 (en) * | 2011-01-21 | 2012-07-26 | Abbott Laboratories | Bicyclic inhibitors of anaphastic lymphoma kinase |
| CN103827088B (zh) * | 2011-09-26 | 2017-10-13 | 奇斯药制品公司 | 作为磷酸二酯酶抑制剂的1‑苯基‑2‑吡啶基烷基醇的衍生物 |
| AR089232A1 (es) * | 2011-12-16 | 2014-08-06 | Chiesi Farma Spa | Potenciacion inducida por inhibidores de pde4 en el tratamiento de la leucemia |
| BR112015012724A2 (pt) * | 2012-12-05 | 2017-07-11 | Chiesi Farm Spa | derivados de álcoois 1-fenil-2-piridinil alquílicos como inibidores da fosfodiesterase |
| CN103304408B (zh) * | 2013-06-05 | 2016-10-05 | 威海迪素制药有限公司 | 罗氟司特中间体3-环丙甲氧基-4-二氟甲氧基苯甲酸的制备 |
| US9427376B2 (en) * | 2013-10-10 | 2016-08-30 | Chiesi Farmaceutici S.P.A. | Process for preparing pharmaceutical formulations for inhalation comprising a high-dosage strength active ingredient |
| HUE067681T2 (hu) * | 2013-10-22 | 2024-11-28 | Chiesi Farm Spa | Eljárás PDE4-inhibitor elõállítására |
| KR101591785B1 (ko) | 2013-12-24 | 2016-02-04 | 주식회사 투게더 | 데이터 입력 장치, 이를 이용한 데이터 전송 시스템 및 방법 |
| MA51413A (fr) * | 2017-12-28 | 2021-04-28 | Chiesi Farm Spa | Utilisation de dérivés d'alcool alkylique de 1-phényl-2-pyridinyl pour le traitement de la fibrose kystique |
| EP4452223A1 (en) * | 2021-12-21 | 2024-10-30 | Chiesi Farmaceutici S.p.A. | Dry powder formulations filled in an inhaler with improved resistance to humidity |
-
2014
- 2014-10-17 HU HUE19183016A patent/HUE067681T2/hu unknown
- 2014-10-17 EP EP22169035.7A patent/EP4059921A1/en active Pending
- 2014-10-17 AU AU2014339136A patent/AU2014339136C1/en active Active
- 2014-10-17 EP EP14795565.2A patent/EP3060551B1/en active Active
- 2014-10-17 PT PT147955652T patent/PT3060551T/pt unknown
- 2014-10-17 EP EP19183016.5A patent/EP3587400B1/en active Active
- 2014-10-17 IL IL285528A patent/IL285528B2/en unknown
- 2014-10-17 FI FIEP19183016.5T patent/FI3587400T3/fi active
- 2014-10-17 ES ES19183016T patent/ES2980505T3/es active Active
- 2014-10-17 PL PL19183016.5T patent/PL3587400T3/pl unknown
- 2014-10-17 UA UAA201909177A patent/UA127453C2/uk unknown
- 2014-10-17 DK DK19183016.5T patent/DK3587400T3/da active
- 2014-10-17 PL PL14795565T patent/PL3060551T3/pl unknown
- 2014-10-17 CA CA3115587A patent/CA3115587C/en active Active
- 2014-10-17 PH PH1/2020/500162A patent/PH12020500162B1/en unknown
- 2014-10-17 RS RS20210247A patent/RS61558B1/sr unknown
- 2014-10-17 IL IL285532A patent/IL285532B2/en unknown
- 2014-10-17 LT LTEP17192278.4T patent/LT3293176T/lt unknown
- 2014-10-17 HR HRP20240783TT patent/HRP20240783T1/hr unknown
- 2014-10-17 US US14/516,976 patent/US9434691B2/en active Active
- 2014-10-17 ES ES14795565T patent/ES2759514T3/es active Active
- 2014-10-17 CN CN202510293709.3A patent/CN120136784A/zh active Pending
- 2014-10-17 PH PH1/2021/550257A patent/PH12021550257A1/en unknown
- 2014-10-17 BR BR112016008161-7A patent/BR112016008161B1/pt active IP Right Grant
- 2014-10-17 CA CA2928242A patent/CA2928242C/en active Active
- 2014-10-17 EP EP17192278.4A patent/EP3293176B1/en active Active
- 2014-10-17 PT PT171922784T patent/PT3293176T/pt unknown
- 2014-10-17 CN CN202210366235.7A patent/CN114621139B/zh active Active
- 2014-10-17 LT LTEP19183016.5T patent/LT3587400T/lt unknown
- 2014-10-17 CN CN202510734070.8A patent/CN120607463A/zh active Pending
- 2014-10-17 MX MX2019014408A patent/MX389002B/es unknown
- 2014-10-17 KR KR1020217004799A patent/KR102287598B1/ko active Active
- 2014-10-17 DK DK17192278.4T patent/DK3293176T3/da active
- 2014-10-17 SG SG10202112587RA patent/SG10202112587RA/en unknown
- 2014-10-17 SI SI201431364T patent/SI3060551T1/sl unknown
- 2014-10-17 MX MX2016004834A patent/MX370104B/es active IP Right Grant
- 2014-10-17 DK DK14795565T patent/DK3060551T3/da active
- 2014-10-17 CN CN201910622800.XA patent/CN110256338A/zh active Pending
- 2014-10-17 ES ES17192278T patent/ES2857629T3/es active Active
- 2014-10-17 SG SG10201912852YA patent/SG10201912852YA/en unknown
- 2014-10-17 PT PT191830165T patent/PT3587400T/pt unknown
- 2014-10-17 CN CN201480058065.XA patent/CN105658629B/zh active Active
- 2014-10-17 SI SI201432068T patent/SI3587400T1/sl unknown
- 2014-10-17 RS RS20240463A patent/RS65452B1/sr unknown
- 2014-10-17 HU HUE14795565A patent/HUE046637T2/hu unknown
- 2014-10-17 KR KR1020217004798A patent/KR102287596B1/ko active Active
- 2014-10-17 MY MYPI2016000681A patent/MY182559A/en unknown
- 2014-10-17 SG SG11201603130TA patent/SG11201603130TA/en unknown
- 2014-10-17 KR KR1020167010773A patent/KR102240865B1/ko active Active
- 2014-10-17 UA UAA201604362A patent/UA121853C2/uk unknown
- 2014-10-17 SG SG10201903575UA patent/SG10201903575UA/en unknown
- 2014-10-17 CA CA3115570A patent/CA3115570C/en active Active
- 2014-10-17 PL PL17192278T patent/PL3293176T3/pl unknown
- 2014-10-17 RU RU2016115246A patent/RU2682660C9/ru active
- 2014-10-17 JP JP2016519348A patent/JP6458957B2/ja active Active
- 2014-10-17 EP EP20207308.6A patent/EP3798209A1/en active Pending
- 2014-10-17 SI SI201431775T patent/SI3293176T1/sl unknown
- 2014-10-17 HU HUE17192278A patent/HUE053112T2/hu unknown
- 2014-10-17 HR HRP20192152TT patent/HRP20192152T1/hr unknown
- 2014-10-17 WO PCT/EP2014/072334 patent/WO2015059050A1/en not_active Ceased
- 2014-10-20 TW TW107133858A patent/TWI685486B/zh active
- 2014-10-20 AR ARP140103938A patent/AR098128A1/es active IP Right Grant
- 2014-10-20 TW TW103136112A patent/TWI651305B/zh active
- 2014-10-20 TW TW109102549A patent/TWI720803B/zh active
-
2016
- 2016-04-14 MX MX2022000036A patent/MX2022000036A/es unknown
- 2016-04-14 MX MX2022000030A patent/MX2022000030A/es unknown
- 2016-04-20 IL IL245220A patent/IL245220A/en active IP Right Grant
- 2016-04-20 PH PH12016500737A patent/PH12016500737B1/en unknown
- 2016-07-22 US US15/217,468 patent/US9890122B2/en active Active
-
2017
- 2017-06-11 IL IL252834A patent/IL252834B/en unknown
- 2017-11-03 US US15/802,737 patent/US10323003B2/en active Active
-
2018
- 2018-10-05 JP JP2018189627A patent/JP2019038813A/ja active Pending
-
2019
- 2019-04-23 US US16/391,952 patent/US10759761B2/en active Active
-
2020
- 2020-02-17 AR ARP200100438A patent/AR118124A2/es active IP Right Grant
- 2020-07-21 US US16/934,527 patent/US11352327B2/en active Active
- 2020-10-14 JP JP2020173216A patent/JP7051970B2/ja active Active
-
2021
- 2021-02-15 CY CY20211100122T patent/CY1123811T1/el unknown
- 2021-03-01 HR HRP20210336TT patent/HRP20210336T1/hr unknown
- 2021-05-27 AR ARP210101431A patent/AR122183A2/es unknown
- 2021-05-27 AR ARP210101430A patent/AR122182A2/es unknown
-
2022
- 2022-02-22 JP JP2022025240A patent/JP7345581B2/ja active Active
- 2022-02-22 JP JP2022025300A patent/JP7534346B2/ja active Active
- 2022-05-06 US US17/738,630 patent/US11981639B2/en active Active
-
2024
- 2024-01-02 US US18/401,832 patent/US20240166605A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2857629T3 (es) | Forma cristalina de un inhibidor de PDE4 | |
| AU2014339136A1 (en) | Process for the preparation of a PDE4 inhibitor | |
| HK1223926B (zh) | 用於制备pde4抑制剂的方法 |