ES2877563T3 - Medios de cromotografía que comprenden conjuntos porosos discretos de nanofibrillas - Google Patents
Medios de cromotografía que comprenden conjuntos porosos discretos de nanofibrillas Download PDFInfo
- Publication number
- ES2877563T3 ES2877563T3 ES15838269T ES15838269T ES2877563T3 ES 2877563 T3 ES2877563 T3 ES 2877563T3 ES 15838269 T ES15838269 T ES 15838269T ES 15838269 T ES15838269 T ES 15838269T ES 2877563 T3 ES2877563 T3 ES 2877563T3
- Authority
- ES
- Spain
- Prior art keywords
- fiber
- fibers
- chromatography
- core
- pla
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000003491 array Methods 0.000 title description 3
- 239000000835 fiber Substances 0.000 claims abstract description 336
- 239000000203 mixture Substances 0.000 claims abstract description 98
- 229920000747 poly(lactic acid) Polymers 0.000 claims abstract description 98
- 229920001778 nylon Polymers 0.000 claims abstract description 76
- 239000004677 Nylon Substances 0.000 claims abstract description 59
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims abstract description 54
- 238000000605 extraction Methods 0.000 claims abstract description 44
- 238000000034 method Methods 0.000 claims abstract description 40
- 229920000642 polymer Polymers 0.000 claims abstract description 19
- 238000004587 chromatography analysis Methods 0.000 claims abstract description 15
- 238000001125 extrusion Methods 0.000 claims abstract description 14
- 239000012501 chromatography medium Substances 0.000 claims abstract description 13
- 238000004191 hydrophobic interaction chromatography Methods 0.000 claims abstract description 6
- 238000001042 affinity chromatography Methods 0.000 claims abstract description 4
- 238000004255 ion exchange chromatography Methods 0.000 claims abstract description 4
- 229920002959 polymer blend Polymers 0.000 claims abstract 2
- 238000010828 elution Methods 0.000 claims description 19
- 238000000746 purification Methods 0.000 claims description 18
- 238000005341 cation exchange Methods 0.000 claims description 12
- 239000000155 melt Substances 0.000 claims description 7
- -1 sulfopropyl groups Chemical group 0.000 claims description 6
- 238000005349 anion exchange Methods 0.000 claims description 4
- 241000700605 Viruses Species 0.000 claims description 2
- 230000001747 exhibiting effect Effects 0.000 abstract description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 abstract 2
- 239000002253 acid Substances 0.000 abstract 1
- 239000004310 lactic acid Substances 0.000 abstract 1
- 235000014655 lactic acid Nutrition 0.000 abstract 1
- 239000003361 porogen Substances 0.000 description 83
- 230000000052 comparative effect Effects 0.000 description 58
- 239000011162 core material Substances 0.000 description 54
- 239000000243 solution Substances 0.000 description 46
- 239000000463 material Substances 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 27
- 239000007787 solid Substances 0.000 description 24
- 229920002292 Nylon 6 Polymers 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- 238000002474 experimental method Methods 0.000 description 22
- 238000001878 scanning electron micrograph Methods 0.000 description 22
- 102000004169 proteins and genes Human genes 0.000 description 21
- 108090000623 proteins and genes Proteins 0.000 description 21
- 238000012986 modification Methods 0.000 description 20
- 230000004048 modification Effects 0.000 description 20
- 239000002131 composite material Substances 0.000 description 19
- 229910052500 inorganic mineral Inorganic materials 0.000 description 19
- 235000010755 mineral Nutrition 0.000 description 19
- 239000011707 mineral Substances 0.000 description 19
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 18
- 239000003446 ligand Substances 0.000 description 18
- 239000002609 medium Substances 0.000 description 18
- 230000003068 static effect Effects 0.000 description 17
- 238000012856 packing Methods 0.000 description 16
- 230000008569 process Effects 0.000 description 16
- 239000008187 granular material Substances 0.000 description 15
- 239000008367 deionised water Substances 0.000 description 14
- 229910021641 deionized water Inorganic materials 0.000 description 14
- 238000009987 spinning Methods 0.000 description 14
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 239000011324 bead Substances 0.000 description 12
- 238000005259 measurement Methods 0.000 description 12
- 238000005342 ion exchange Methods 0.000 description 11
- 238000002074 melt spinning Methods 0.000 description 11
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- 238000011084 recovery Methods 0.000 description 9
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 9
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 238000005277 cation exchange chromatography Methods 0.000 description 8
- 239000011248 coating agent Substances 0.000 description 8
- 238000000576 coating method Methods 0.000 description 8
- 229910017604 nitric acid Inorganic materials 0.000 description 8
- 230000035699 permeability Effects 0.000 description 8
- 238000007906 compression Methods 0.000 description 7
- 230000006835 compression Effects 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 238000010559 graft polymerization reaction Methods 0.000 description 7
- 239000008188 pellet Substances 0.000 description 7
- 239000011148 porous material Substances 0.000 description 7
- 238000004626 scanning electron microscopy Methods 0.000 description 7
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 239000007983 Tris buffer Substances 0.000 description 6
- 239000012539 chromatography resin Substances 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 238000003828 vacuum filtration Methods 0.000 description 6
- 238000004438 BET method Methods 0.000 description 5
- 239000004593 Epoxy Substances 0.000 description 5
- 238000005571 anion exchange chromatography Methods 0.000 description 5
- 239000011521 glass Substances 0.000 description 5
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- KFNGWPXYNSJXOP-UHFFFAOYSA-N 3-(2-methylprop-2-enoyloxy)propane-1-sulfonic acid Chemical compound CC(=C)C(=O)OCCCS(O)(=O)=O KFNGWPXYNSJXOP-UHFFFAOYSA-N 0.000 description 4
- 229920001410 Microfiber Polymers 0.000 description 4
- 208000034530 PLAA-associated neurodevelopmental disease Diseases 0.000 description 4
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 4
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 4
- 235000010323 ascorbic acid Nutrition 0.000 description 4
- 229960005070 ascorbic acid Drugs 0.000 description 4
- 239000011668 ascorbic acid Substances 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 239000003658 microfiber Substances 0.000 description 4
- 239000002121 nanofiber Substances 0.000 description 4
- 239000011257 shell material Substances 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 229910052684 Cerium Inorganic materials 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 239000004952 Polyamide Substances 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 239000008351 acetate buffer Substances 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 229910000019 calcium carbonate Inorganic materials 0.000 description 3
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 description 3
- 239000013068 control sample Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 238000007689 inspection Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 238000004806 packaging method and process Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 229920000083 poly(allylamine) Polymers 0.000 description 3
- 229920002647 polyamide Polymers 0.000 description 3
- 238000007717 redox polymerization reaction Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 238000001179 sorption measurement Methods 0.000 description 3
- 125000000542 sulfonic acid group Chemical group 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229920001169 thermoplastic Polymers 0.000 description 3
- XHZPRMZZQOIPDS-UHFFFAOYSA-N 2-Methyl-2-[(1-oxo-2-propenyl)amino]-1-propanesulfonic acid Chemical compound OS(=O)(=O)CC(C)(C)NC(=O)C=C XHZPRMZZQOIPDS-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 2
- 241000533950 Leucojum Species 0.000 description 2
- 102000016943 Muramidase Human genes 0.000 description 2
- 108010014251 Muramidase Proteins 0.000 description 2
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229940049595 antibody-drug conjugate Drugs 0.000 description 2
- 238000000429 assembly Methods 0.000 description 2
- 230000000712 assembly Effects 0.000 description 2
- PCCNIENXBRUYFK-UHFFFAOYSA-O azanium;cerium(4+);pentanitrate Chemical compound [NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O PCCNIENXBRUYFK-UHFFFAOYSA-O 0.000 description 2
- 229920005601 base polymer Polymers 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 238000013375 chromatographic separation Methods 0.000 description 2
- 238000011097 chromatography purification Methods 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000012149 elution buffer Substances 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 229920001002 functional polymer Polymers 0.000 description 2
- 229920000578 graft copolymer Polymers 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 210000004408 hybridoma Anatomy 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 239000004325 lysozyme Substances 0.000 description 2
- 235000010335 lysozyme Nutrition 0.000 description 2
- 229960000274 lysozyme Drugs 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 2
- 229960000907 methylthioninium chloride Drugs 0.000 description 2
- 238000001000 micrograph Methods 0.000 description 2
- 239000012764 mineral filler Substances 0.000 description 2
- 230000007505 plaque formation Effects 0.000 description 2
- 229920002454 poly(glycidyl methacrylate) polymer Polymers 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- 238000007142 ring opening reaction Methods 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 239000004416 thermosoftening plastic Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000004804 winding Methods 0.000 description 2
- 229920002818 (Hydroxyethyl)methacrylate Polymers 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- QZPSOSOOLFHYRR-UHFFFAOYSA-N 3-hydroxypropyl prop-2-enoate Chemical compound OCCCOC(=O)C=C QZPSOSOOLFHYRR-UHFFFAOYSA-N 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 101710141544 Allatotropin-related peptide Proteins 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102100026735 Coagulation factor VIII Human genes 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 238000012855 HCP-ELISA Methods 0.000 description 1
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 1
- 229920000299 Nylon 12 Polymers 0.000 description 1
- 229920002302 Nylon 6,6 Polymers 0.000 description 1
- 229920000572 Nylon 6/12 Polymers 0.000 description 1
- 229910002054 SYLOID® 244 FP SILICA Inorganic materials 0.000 description 1
- 241001613925 Staphylococcus virus 96 Species 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- AOBHTXFZZPUOGU-UHFFFAOYSA-N [2-(trifluoromethyl)-1,3-dioxolan-2-yl]methanol Chemical compound OCC1(C(F)(F)F)OCCO1 AOBHTXFZZPUOGU-UHFFFAOYSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 239000000611 antibody drug conjugate Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 238000010560 atom transfer radical polymerization reaction Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 239000006177 biological buffer Substances 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000005253 cladding Methods 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- 230000005251 gamma ray Effects 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000000879 optical micrograph Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- PNOXUQIZPBURMT-UHFFFAOYSA-M potassium;3-(2-methylprop-2-enoyloxy)propane-1-sulfonate Chemical compound [K+].CC(=C)C(=O)OCCCS([O-])(=O)=O PNOXUQIZPBURMT-UHFFFAOYSA-M 0.000 description 1
- 229940088417 precipitated calcium carbonate Drugs 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 238000005086 pumping Methods 0.000 description 1
- 239000005297 pyrex Substances 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000012712 reversible addition−fragmentation chain-transfer polymerization Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000012799 strong cation exchange Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000004441 surface measurement Methods 0.000 description 1
- 238000006557 surface reaction Methods 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940035024 thioglycerol Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/282—Porous sorbents
- B01J20/285—Porous sorbents based on polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/32—Bonded phase chromatography
- B01D15/325—Reversed phase
- B01D15/327—Reversed phase with hydrophobic interaction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/36—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving ionic interaction, e.g. ion-exchange, ion-pair, ion-suppression or ion-exclusion
- B01D15/361—Ion-exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/36—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving ionic interaction, e.g. ion-exchange, ion-pair, ion-suppression or ion-exclusion
- B01D15/361—Ion-exchange
- B01D15/362—Cation-exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/36—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving ionic interaction, e.g. ion-exchange, ion-pair, ion-suppression or ion-exclusion
- B01D15/361—Ion-exchange
- B01D15/363—Anion-exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
- B01D15/3804—Affinity chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
- B01D15/3804—Affinity chromatography
- B01D15/3809—Affinity chromatography of the antigen-antibody type, e.g. protein A, G or L chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28002—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their physical properties
- B01J20/28004—Sorbent size or size distribution, e.g. particle size
- B01J20/28007—Sorbent size or size distribution, e.g. particle size with size in the range 1-100 nanometers, e.g. nanosized particles, nanofibers, nanotubes, nanowires or the like
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
- B01J20/28023—Fibres or filaments
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
- B01J20/28028—Particles immobilised within fibres or filaments
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3202—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the carrier, support or substrate used for impregnation or coating
- B01J20/3206—Organic carriers, supports or substrates
- B01J20/3208—Polymeric carriers, supports or substrates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3231—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the coating or impregnating layer
- B01J20/3242—Layers with a functional group, e.g. an affinity material, a ligand, a reactant or a complexing group
- B01J20/3268—Macromolecular compounds
- B01J20/3278—Polymers being grafted on the carrier
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3291—Characterised by the shape of the carrier, the coating or the obtained coated product
- B01J20/3293—Coatings on a core, the core being particle or fiber shaped, e.g. encapsulated particles, coated fibers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J39/00—Cation exchange; Use of material as cation exchangers; Treatment of material for improving the cation exchange properties
- B01J39/08—Use of material as cation exchangers; Treatment of material for improving the cation exchange properties
- B01J39/16—Organic material
- B01J39/18—Macromolecular compounds
- B01J39/19—Macromolecular compounds obtained otherwise than by reactions only involving unsaturated carbon-to-carbon bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J39/00—Cation exchange; Use of material as cation exchangers; Treatment of material for improving the cation exchange properties
- B01J39/26—Cation exchangers for chromatographic processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J41/00—Anion exchange; Use of material as anion exchangers; Treatment of material for improving the anion exchange properties
- B01J41/08—Use of material as anion exchangers; Treatment of material for improving the anion exchange properties
- B01J41/12—Macromolecular compounds
- B01J41/13—Macromolecular compounds obtained otherwise than by reactions only involving unsaturated carbon-to-carbon bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J41/00—Anion exchange; Use of material as anion exchangers; Treatment of material for improving the anion exchange properties
- B01J41/20—Anion exchangers for chromatographic processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J47/00—Ion-exchange processes in general; Apparatus therefor
- B01J47/12—Ion-exchange processes in general; Apparatus therefor characterised by the use of ion-exchange material in the form of ribbons, filaments, fibres or sheets, e.g. membranes
- B01J47/127—Ion-exchange processes in general; Apparatus therefor characterised by the use of ion-exchange material in the form of ribbons, filaments, fibres or sheets, e.g. membranes in the form of filaments or fibres
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/18—Ion-exchange chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/20—Partition-, reverse-phase or hydrophobic interaction chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
Landscapes
- Chemical & Material Sciences (AREA)
- Analytical Chemistry (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Nanotechnology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Virology (AREA)
- General Engineering & Computer Science (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Treatment Of Liquids With Adsorbents In General (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Artificial Filaments (AREA)
- Chemical Or Physical Treatment Of Fibers (AREA)
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
Abstract
Medios de cromatografía que comprenden fibras que presentan una estructura enrevesada compuesta por conjuntos discretos de nanofibrillas porosas muy entrelazadas y en donde las fibras presentan un área superficial en el intervalo de 1 a 12 metros cuadrados por gramo, dichas nanofibrillas dentro de dichos conjuntos porosos discretos tienen diámetros menores que 1 micra y a dichas nanofibrillas se les ha impartido una funcionalidad que permite una cromatografía seleccionada del grupo que consiste en cromatografía de intercambio iónico, cromatografía de interacciones hidrófobas y cromatografía de afinidad, en donde las fibras se preparan mediante un método que incluye la extrusión por fusión de fibras de dos componentes en configuración núcleo/envoltura con una relación núcleo/envoltura de 50:50, en donde la envoltura está formada por poli(ácido láctico) y el núcleo está formado por una mezcla de nailon y poli(ácido láctico), en donde los polímeros son inmiscibles, el estiramiento de las fibras y subsecuentemente la extracción del poli(ácido láctico) de dicha envoltura y dicha mezcla con una solución de hidróxido de sodio, en donde la carga de poli(ácido láctico) en la mezcla es mayor del 35 % en peso y menor del 50 % en peso de la mezcla polimérica.
Description
DESCRIPCIÓN
Medios de cromatografía que comprenden conjuntos porosos discretos de nanofibrillas
Campo
Las modalidades descritas en la presente descripción se refieren a fibras porosas de gran área superficial que comprenden conjuntos porosos discretos de nanofibrillas altamente entrelazadas que son adecuadas para el servicio como fase estacionaria cromatográfica para la purificación de unión/elución de proteínas en un modo de cromatografía de intercambio catiónico, por ejemplo.
Antecedentes
La purificación a escala comercial de diversas biomoléculas terapéuticas, tales como los anticuerpos monoclonales, se realiza actualmente mediante el uso de resinas de cromatografía a base de perlas. Los anticuerpos monoclonales continúan ganando importancia como agentes terapéuticos y de diagnóstico. El proceso de cribado de bibliotecas de hibridomas en busca de mAb candidatos requiere mucho tiempo y trabajo. Una vez que se establece una línea celular de hibridoma que expresa un mAb adecuado, debe desarrollarse una metodología de purificación para producir suficiente mAb para una caracterización adicional. Un método tradicional de purificación implica el uso de cromatografía de afinidad de proteína A o proteína G, así como también cromatografía de intercambio iónico. El anticuerpo purificado se desala y se intercambia en un tampón biológico mediante el uso de diálisis. Típicamente, todo el proceso requiere varios días para completarse y puede ser particularmente oneroso si se van a evaluar múltiples mAb en paralelo.
Las resinas de cromatografía se preparan actualmente con diversas estructuras de ligandos que permiten que las perlas funcionen en modos de afinidad, intercambio catiónico o intercambio aniónico. Estas resinas demuestran una gran porosidad y grandes áreas superficiales que proporcionan materiales con capacidades de adsorción suficientes para el procesamiento por lote de biomoléculas a escalas de producción (por ejemplo, 10000 litros). Las resinas de cromatografía presentan típicamente una estructura esférica que permite un empaque de la columna eficiente con irregularidades de flujo mínimas. Los espacios intersticiales entre las perlas proporcionan canales de flujo para el transporte convectivo a través de la columna de cromatografía. Esto permite que las columnas de cromatografía se ejecuten con grandes profundidades de lecho a una alta velocidad lineal con una caída de presión mínima. La combinación de estos factores permite que las resinas de cromatografía presenten la eficiencia requerida, alta permeabilidad y la capacidad de unión suficiente que se requieren para la purificación a gran escala de biomoléculas.
En la cromatografía basada en perlas, la mayor parte del área superficial disponible para la adsorción es interna a la perla. En consecuencia, el proceso de separación es intrínsecamente lento, ya que la velocidad de transporte de masa se controla típicamente mediante la difusión de los poros. Para minimizar esta resistencia a la difusión y maximizar concomitantemente la capacidad de unión dinámica, pueden emplearse perlas de diámetro pequeño. Sin embargo, el uso de perlas de diámetro pequeño tiene el costo de una mayor caída de presión de la columna. En consecuencia, la optimización de las separaciones cromatográficas preparativas a menudo implica un compromiso entre la eficiencia/capacidad dinámica (se prefieren las perlas pequeñas) y la caída de presión de la columna (se prefieren las perlas grandes).
Los medios de cromatografía típicamente tienen un costo muy alto (> $ 1000/L) y se requieren cantidades significativas para las columnas de producción a gran escala. Como resultado, los fabricantes biofarmacéuticos reciclan las resinas de cromatografía cientos de veces. Cada uno de estos ciclos de regeneración consume cantidades sustanciales de medio tampón, y cada etapa incurre en costos adicionales asociados con la validación de cada operación de limpieza, esterilización y empaque de la columna.
Varias tecnologías se describen en la bibliografía de patentes y se comercializan comercialmente para separaciones biofarmacéuticas basadas en medios fibrosos funcionalizados y/o compuestos. La mayoría se basa en la incorporación de un gel poroso en la matriz de la fibra, proporcionando el gel el área superficial necesaria para obtener capacidades de unión razonables. Sin embargo, en tales construcciones, la uniformidad deficiente en la ubicación y la masa del gel generalmente conduce a pobres eficiencias (frentes de elución y avance superficial). Además, la resistencia al flujo puede ser alta incluso para lechos de poca profundidad, un problema que a menudo se ve agravado por la compresión del gel bajo cargas de presión moderadas. Otro enfoque adoptado ha sido la incorporación de partículas dentro de la matriz de la fibra, las partículas a menudo porosas y que poseen una funcionalidad adsorbente nativa, por ejemplo, carbón activado y gel de sílice. Un portador de adsorción adecuado para columnas de procesamiento de sangre se describe en Estados Unidos 2010/176051)
Recientemente, EMD Millipore ha desarrollado un medio de cromatografía basado en fibras para aplicaciones de purificación de biomoléculas que utiliza una fibra alada funcionalizada en la superficie como medio adsorbente. Las proyecciones aladas en la superficie de la fibra proporcionan un área superficial mucho mayor que las fibras redondas ordinarias de dimensiones similares. El medio de fibra funcionalizado en su superficie resultante también tiene una
capacidad de unión a proteínas mucho mayor que las fibras funcionalizadas de manera similar que carecen de tales proyecciones aladas. Las fibras se describen en el documento WO 2014/120387 A1.
Actualmente se están desarrollando otras tecnologías emergentes para aplicaciones de purificación de proteínas, entre las que se incluyen adsorbedores de membrana, monolitos y métodos de purificación de adsorbedores de flujo continuo mediante el uso de sistemas de resinas comerciales. Mientras los adsorbedores de membrana y monolitos pueden proporcionar capacidades de unión aceptables para estas aplicaciones, estas tecnologías típicamente tienen sus propias limitaciones de escala y el costo extremadamente alto de tales medios de purificación puede limitar aún más su adopción en una industria sensible al precio con una plantilla de proceso de purificación existente.
Resumen
Para abordar muchas de las limitaciones de las tecnologías de purificación actualmente conocidas en la técnica, la presente invención proporciona un medio de cromatografía como se define en la reivindicación 1. Las modalidades descritas en la presente descripción se refieren a medios de cromatografía que comprenden una fibra termoplástica de gran área superficial y bajo costo y una funcionalidad de ligando de intercambio iónico en la superficie de esa fibra. En determinadas modalidades, el ligando de intercambio iónico es capaz de unirse selectivamente a proteínas de una corriente de alimentación biológica. La proteína unida puede liberarse subsecuentemente del medio cromatográfico tras un cambio en las condiciones de la solución, por ejemplo, mediante el uso de un tampón de elución con una fuerza iónica superior. En ciertas modalidades, los grupos funcionales colgantes de la superficie se añaden al medio que proporciona la funcionalidad de intercambio catiónico o de intercambio aniónico a las fibras de gran área superficial. Esta funcionalidad colgante es útil para la purificación cromatográfica de intercambio iónico de biomoléculas, tales como proteínas de fusión recombinantes, proteínas que contienen Fc, ADC (conjugados de anticuerpos y fármacos, vacunas, proteínas plasmáticas (IgM, factores de coagulación sanguínea, etc.) y anticuerpos monoclonales (mAb).
La estructura de los medios de cromatografía se describe en la reivindicación 1. Los medios de cromatografía comprenden fibras que muestran una estructura enrevesada compuesta por conjuntos discretos de nanofibrillas muy entrelazadas. Dichas nanofibrillas dentro de dichos conjuntos porosos discretos tienen diámetros menores de 1 micra. Las nanofibrillas les han impartido una funcionalidad que permite una cromatografía seleccionada del grupo que consiste en cromatografía de intercambio iónico, cromatografía de interacciones hidrófobas y cromatografía infinita.
Estas fibras presentan un área superficial en el intervalo de 1 a 12 metros cuadrados por gramo. Las fibras pueden prepararse mediante un método como se define en la reivindicación 1, que incluye la extrusión por fusión de una mezcla de dos polímeros inmiscibles que incluyen nailon, tales como poliamida 6, poliamida 6,6, poliamida 4,6, poliamida, poliamida 12, poliamida 6,12, y copolímeros o mezclas de diversas poliamidas y poli(ácido láctico), PLA. Después de la extrusión de la masa fundida, se estiran las fibras. El componente porógeno de polímero PLA se extrae subsecuentemente mediante tratamiento con solución de hidróxido de sodio, dejando cavidades o canales alargados a lo largo de la microfibra de nailon. El medio de fibra resultante tiene la apariencia de un conjunto de nanofibras de nailon altamente enredadas que están alineadas de manera suelta en un arreglo colineal. Estos conjuntos poseen las propiedades de flujo de las microfibras ordinarias y también demuestran una alta permeabilidad en un formato de lecho empacado. Por el contrario, las nanofibras individuales que no están agrupadas demuestran una permeabilidad muy baja en un formato de lecho empacado. Este arreglo único de nanofibras proporciona un sustrato de alta permeabilidad y gran área superficial que permite una alta capacidad de unión a proteínas después de la modificación de la superficie con el ligando de intercambio iónico apropiado. Las fibras modificadas con una funcionalidad de intercambio iónico colgante son útiles para la purificación cromatográfica de proteínas, tales como los anticuerpos monoclonales.
En determinadas modalidades, una fibra mejorada en su área superficial (SAE) se modifica con ligandos sulfopropilo (SP) de superficie funcional y se usa en una aplicación de cromatografía de intercambio catiónico de unión/elución para la purificación de IgG. El medio de fibra SAE puede modificarse en la superficie para instalar ligandos de intercambio iónico colgantes, tales como los grupos sulfopropilo (SP). Los medios funcionalizados pueden empaquetarse en un dispositivo adecuado, tal como una columna de cromatografía, y comprimirse a una densidad de empaquetamiento objetivo. Subsecuentemente, puede pasarse una solución de proteína a purificar a través de este empaquetamiento de fibra, con lo cual la proteína de interés puede unirse a ligandos en la superficie de las fibras SAE a través de un proceso de intercambio iónico. Por ejemplo, a pH 5, los grupos sulfopropilo están fuertemente cargados negativamente y se unirán a proteínas, tales como IgG, cuyo pI es mayor que aproximadamente 7. Después de la unión de la proteína diana (por ejemplo, IgG), la columna se lava típicamente con un tampón adecuado, tal como tampón acetato 50 mM (pH 5) para eliminar las impurezas no unidas. Posteriormente, se aumenta la fuerza iónica del tampón, por ejemplo, con una solución de cloruro de sodio 0,5 M/acetato 50 mM (pH 5) para eluir la IgG unida de la columna de fibra SAE. A continuación, la columna de fibras puede regenerarse mediante lavado, por ejemplo con 5 10 volúmenes de columna de hidróxido de sodio 0,5 M y 5-10 volúmenes de columna de tampón acetato 50 mM (pH 5). El medio de fibra SAE ahora está listo para otro ciclo de unión/elución CEX. En consecuencia, las modalidades descritas en la presente descripción se refieren a métodos para el aislamiento, purificación o separación de biomoléculas con medios que comprenden una fibra porosa funcionalizada de gran área superficial.
Breve descripción de las figuras
La Figura 1(a) (comparativa) es una imagen SEM de un monofilamento de nailon no poroso ordinario, fibra de 20 mieras de diámetro, con un aumento de 4000x ;
La Figura 1(b) (comparativa) es una imagen SEM de una fibra de nailon de 15 alas después de la eliminación completa de los materiales de la envoltura, fibra de 15 micras de diámetro, con un aumento de 5000x ;
La Figura 1(c) (comparativa) es una imagen SEM de un monofilamento extruido con la composición de mezcla PA6/Albafil® CaCO375/25 después de la eliminación completa de los porógenos incrustados y los materiales de la envoltura, relación de estiramiento 2:1, con un aumento de 4000x ;
La Figura 1(d) (comparativa)es una imagen de sección transversal crio-SEM después de la eliminación completa de los porógenos incrustados y los materiales de la envoltura, de una fibra de 15 alas con una composición de mezcla de núcleo nailon/PLA 70/30, con un aumento de 10000x ;
La Figura 1(e) es una imagen de sección transversal crio-SEM después de la eliminación completa de los porógenos incrustados y los materiales de la envoltura, de un núcleo/envoltura de fibra de área superficial mejorada (SAE) con una composición de mezcla de núcleo de nailon/PLA 60/40, con un aumento 5000x
La Figura 1(f) (comparativa) es una imagen de sección transversal crio-SEM después de la eliminación completa de los porógenos incrustados y los materiales de la envoltura, de una fibra "islas conectadas en el mar" (CIST) con una composición de "islas" PA6 nylon 100/0, composición de "mar" nailon PA6/PLA 55/45, relación i/S, 1/1, con un aumento de 5000x
La Figura 2(a) (comparativa) es una imagen SEM de monofilamentos extruidos de nailon PA6, relación de estiramiento 2 :1, aumento de 1000x
La Figura 2(b) (comparativa) es una imagen SEM de monofilamentos extruidos de nailon PA6, relación de estiramiento 2:1, aumento de 4000x
La Figura 2(c) (comparativa) es una imagen SEM después de la extracción de porógenos minerales de monofilamentos extruidos con una composición de mezcla de PA6/Multifex-MM ™ CaCO375/25, relación de estiramiento de 1,3:1, aumento de 1000x ;
La Figura 2(d) (comparativa) es una imagen SEM después de la extracción de porógenos minerales de monofilamentos extruidos con una composición de mezcla de PA6/Multifex-MM ™ CaCO375/25, relación de estiramiento de 1,3:1, aumento de 4000x ;
La Figura 2(e) (comparativa) es una imagen SEM después de la extracción de porógenos minerales de monofilamentos extruidos con una composición de mezcla de PA6/Albafil® CaCO375/25, relación de estiramiento 2 :1, aumento de 1000x
La Figura 2(f) (comparativa) es una imagen SEM después de la extracción de porógenos minerales de monofilamentos extruidos con una composición de mezcla de PA6/Albafil® CaCO375/25, relación de estiramiento 2:1, aumento de 4000x
La Figura 3(a) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos de filamentos compuestos extruidos (extrusora de compuestos) con una composición de mezcla de nailon/PLA 65/35, aumento de 2000x
La Figura 3(b) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos de filamentos compuestos extruidos (extrusora de compuestos) con una composición de mezcla de nailon/PLA 60/40, aumento de 80x de acuerdo con la invención ;
La Figura 3(c) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos de filamentos compuestos extruidos (extrusora de compuestos) con una composición de mezcla de nailon/PLA 55/45, aumento de 75x de acuerdo con la invención ;
La Figura 3(d) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos de filamentos compuestos extruidos (extrusora de compuestos) con una composición de mezcla de nailon/PLA 50/50, aumento de 65x
La Figura 4(a) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos y la envoltura de PLA de una fibra de núcleo/envoltura con una composición de mezcla de núcleo nailon/PLA 65/35, aumento de 2000x
La Figura 4(b) es una imagen SEM después de la extracción del porógenos poliméricos y la envoltura de PLA de una fibra de núcleo/envoltura con una composición de mezcla de núcleo nailon/PLA 60/40, aumento de 400x. de acuerdo con la invención;
La Figura 4(c) es una imagen SEM después de la extracción del porógenos poliméricos y la envoltura de PLA de una fibra de núcleo/envoltura con una composición de mezcla de núcleo nailon/PLA 55/45, aumento de 2000x de acuerdo con la invención;
La Figura 4(d) (comparativa) es una imagen SEM después de la extracción de porógenos poliméricos y la envoltura de PLA de una fibra de núcleo/envoltura con una composición de mezcla de núcleo nailon/PLA 50/50, aumento de 400x
La Figura 5(a) es una imagen de sección transversal crio-SEM de una fibra de área superficial mejorada (SAE) de acuerdo con la invención;
La Figura 5(b) es una imagen de sección transversal crio-SEM de una fibra de nailon/PLA 60/40 de área superficial mejorada (SAE) de acuerdo con la invención;
La Figura 6 es un gráfico de las mediciones del área superficial de Kr BET para fibras seleccionadas como comparación y de acuerdo con la invención;
La Figura 7(a) (comparativa) es una imagen de sección transversal crio-SEM después de la extracción de porógenos poliméricos y la envoltura de PLA de una fibra de 15 alas con composición de mezcla de núcleo PA6/PLA 70/30, relación de bombeo de fusión S:C 2:1, aumento de 10000x;
La Figura 7(b) (comparativa) es una imagen de sección transversal crio-SEM después de la extracción de porógeno polimérico y la envoltura de PLA de una fibra de 15 alas con composición de mezcla de núcleo PA6/PLA 70/30, relación de bombeo de fusión S:C 2:1, aumento de 20000x;
La Figura 8(a) es una imagen en sección transversal crio-SEM después de la extracción de porógeno polimérico y la envoltura de PLA de una fibra de núcleo/vaina con la composición de mezcla de núcleos PA6/PLA 60/40, aumento 5000x, de acuerdo con la invención;
La Figura 8(b) (comparativa) es una imagen de sección transversal crio-SEM después de la extracción de porógeno polimérico y la envoltura de PLA de una fibra de 15 alas con composición de mezcla de núcleo PA6/PLA 60/40, relación de bombeo de fusión S:C 2:1, aumento de 5000x,
La Figura 8(c) es una imagen de sección transversal crio-SEM después de la extracción de porógeno polimérico y la envoltura de PLA de una fibra de 15 alas con composición de mezcla de núcleo PA6/PLA 60/40, relación de bombeo de fusión S:C 1:1, aumento de 5000x, de acuerdo con la invención;
La Figura 9 es un gráfico de IgG SBC para fibras SAE modificadas en la superficie con SP (izquierda) de acuerdo con la invención frente a fibras de 15 alas modificadas con SP de manera similar (derecha) (comparativo);
La Figura 10 es un gráfico de lisozima SBC para fibras SAE modificadas en la superficie con Sp (izquierda) de acuerdo con la invención frente a fibras de 15 alas modificadas con SP de manera similar (derecha) (comparativo); La Figura 11 es un gráfico de SBC de azul de metileno para fibras SAE modificadas en la superficie con SP (izquierda) de acuerdo con la invención frente a las fibras de 15 alas modificadas con SP de manera similar (derecha) (comparativo);
La Figura 12(a) es una imagen SEM de una fibra SAE con una composición de mezcla de núcleo nailon/PLA 60/40, aumento de 400x, de acuerdo con la invención;
La Figura 12(b) es una imagen en sección transversal crio-SEM de una fibra SAE con una composición de mezcla de núcleo nailon/PLA 60/40, aumento de 5000x, de acuerdo con determinadas modalidades;
La Figura 12(c) (comparativa) es una imagen SEM de una fibra CIST con una composición de "isla" de nailon PA6 100/0, una composición de "mar" nailon/PLA 55/45, relación I/S 1/1, aumento de 1000x;
La Figura 12(d) (comparativo) es una imagen en sección transversal crio-SEM de una fibra CIST con una composición de "isla" de nailon PA6 100/0, una composición de "mar" nailon/PLA 55/45, relación I/S 1/1, 5000x aumento,
La Figura 13(a) (comparativa) es una micrografía óptica de fibra fractal 1;
La Figura 13(b) (comparativa) es una micrografía óptica de fibra fractal 2;
La Figura 13(c) (comparativa) es una micrografía óptica de una fibra de copo de nieve;
La Figura 13(d) (comparativa) es una micrografía SEM de fibra fractal 2; y
La Figura 13(e) (comparativa) es una micrografía SEM de una fibra de copo de nieve.
Descripción detallada
Las modalidades descritas en la presente descripción incluyen fibras de gran área superficial que son adecuadas para la purificación de unión/elución de proteínas. Las fibras son porosas y pueden prepararse mediante la extracción en la fibra durante su fabricación en un proceso de extrusión por fusión.
El porógeno polimérico soluble es poli(ácido láctico), PLA. Este polímero se disolverá en una solución acuosa de hidróxido de sodio. Los porógenos poliméricos solubles se incorporan a la fibra en cargas que oscilan entre el 35 y el 50 % en peso • Las cargas de porógenos poliméricos solubles inferiores al 25 % en peso proporcionan solo una mejora mínima del área superficial de la fibra y estas fibras también carecen de las características superficiales porosas o fibriladas deseadas después de la extracción del porógeno. Las cargas de porógeno polimérico soluble superiores a aproximadamente el 65 % en peso pueden comprometer la integridad estructural de la fibra después de la extracción del porógeno.
La carga puede medirse mediante el peso en seco preprocesado de los diferentes materiales que se introducirán en la extrusora, o comparando el peso de la fibra antes y después de la eliminación de porógenos.
Los materiales adecuados para las fibras incluyen nailon PA6.
Estos náilones pueden obtenerse en forma de gránulos o polvos y estos materiales pueden procesarse subsecuentemente en las fibras del producto por medio de un equipo comercial de procesamiento de extrusión de fibras fundidas y de composición en estado fundido. Estas fibras presentan un área superficial mucho mayor que las microfibras redondas ordinarias o las fibras aladas como se ilustra en la Figura. 1a y 1b, respectivamente. En determinadas modalidades, las fibras pueden modificarse en la superficie para instalar la funcionalidad de ligando de intercambio catiónico colgante apropiada para la unión/elución o purificación de flujo continuo de proteínas,
anticuerpos monoclonales u otras biomoléculas de interés. Los ligandos adecuados que pueden desplegarse en las superficies de las fibras incluyen grupos sulfopropilo para aplicaciones de cromatografía de intercambio catiónico, haluros de tetraalquilamonio, aminas primarias y aminas secundarias para aplicaciones de cromatografía de intercambio aniónico, y cadenas n-alquilo, fenilo, bencilo u otros grupos aromáticos para aplicaciones de cromatografía de fase reversa y cromatografía de interacciones hidrófobas. Los ligandos pueden instalarse sobre las superficies de las fibras mediante polimerizaciones de injerto redox cérico, ATRP, RAFt o polimerizaciones de radicales libres iniciadas por fuentes de radiación de rayos e, UV o gamma.
En determinadas modalidades, un polímero termoplástico adecuado se mezcla con uno o más aditivos porógenos adecuados, tal como con una extrusora de compuestos. El polímero y/o el porógeno pueden secarse previamente y mezclarse en seco. A continuación, la mezcla puede introducirse en una extrusora, desde la que puede extrudirse de un troquel de una sola hebra en un baño de agua y luego se granula. Alternativamente, el polímero base y los gránulos o polvos porógenos pueden mezclarse en seco y alimentar directamente a una máquina de hilar fibras o filamentos sin precompuestos. A continuación, los gránulos pueden hilarse en estado fundido en filamentos de dos componentes mediante el uso una máquina de hilar fibra debidamente equipada. El polímero base mezclado/material porógeno forma el núcleo y el polímero porógeno forma la envoltura exterior. Después de hilar, estirar y enrollar la fibra, el porógeno puede extraerse de los filamentos de dos componentes con un agente de extracción adecuado, tal como una solución de ácido clorhídrico 1 M o una solución de hidróxido de sodio 1,5 N, dependiendo de la naturaleza del porógeno usado.
En la Figura 1c, 1d y 1f, se muestran varios ejemplos comparativos de fibras de gran área superficial, siendo la Figura 1(e) ilustrativa de la invención. La Figura 1c es una imagen SEM de superficie de una microfibra porosa preparada usando un método de porógeno mineral soluble. Este monofilamento extruido se preparó a partir de una mezcla compuesta fundida de nailon y carbonato cálcico precipitado al 25 % en peso (Albafil® PCC). Después de hilar, estirar la fibra y extraer el porógeno de carbonato de calcio con ácido clorhídrico de la fibra, se observan numerosos poros en la superficie de la fibra. La medición del área superficial Kr BET indica una ganancia de aproximadamente 300 % en el área superficial BET para este material sobre una fibra de nailon no porosa ordinaria de aproximadamente el mismo diámetro de fibra.
La Figura 1d muestra una imagen en sección transversal crio-SEM de una fibra porosa de 15 alas preparada mediante el uso un método de porógeno polimérico soluble. Esta fibra de dos componente extruida se preparó a partir de una mezcla compuesta en fusión de nailon y 30 % en peso de PLA que constituye el núcleo de la fibra alada y una envoltura de PLA soluble que rodea el núcleo de la fibra y estabiliza las proyecciones aladas durante el hilado en fusión (no mostrado).Después de hilar, estirar la fibra y extraer el porógeno polimérico PLA de la vaina de la fibra y dentro del interior del núcleo de la fibra con solución de hidróxido de sodio, se observan numerosos poros en toda la sección transversal de la fibra alada.
La Figura 1e muestra una imagen en sección transversal crio-SEM de una fibra de núcleo/envoltura porosa preparada mediante el uso de un método de porógeno polimérico soluble. Esta arquitectura de fibra se denomina fibra de tipo de área superficial mejorada (SAE). Esta fibra de dos componente extruida se preparó a partir de una mezcla compuesta en fusión de nailon y 40 % en peso de PLA que constituye el núcleo de la fibra y una envoltura de PLA soluble que rodea el núcleo de la fibra y estabiliza el material durante el hilado en fusión (no mostrado). Después de hilar la fibra, estirar y extraer el porógeno polimérico PLA de la envoltura de la fibra y dentro del interior del núcleo de la fibra con solución de hidróxido de sodio, se observa un arreglo agrupada de nanofibrillas de nailon ligeramente alineadas en toda la sección transversal de la fibra SAE. Esta arquitectura mejorada del área superficial aumenta en gran medida el área de superficie de la fibra y las mediciones del área de superficie de Kr BET indican que valores tan altos como 10,6 m2/ g son alcanzables con este enfoque. Por el contrario, una fibra de 15 alas no porosa tiene un área superficial modesta de solo 1,4 m2/g.
La Figura 1f muestra una imagen en sección transversal crio-SEM de otro tipo de fibra porosa de dos componentes comparativa preparada mediante el uso de un método de porógeno polimérico soluble. Esta arquitectura de fibra se conoce como fibra de tipo "islas conectadas en el mar" (CIST). Esta fibra de dos componentes extruida se preparó a partir de una mezcla compuesta fundida de nailon y 45 % en peso de PLA que constituye el dominio "marino" de la fibra y una serie de 36 "islas" continuas de nailon. Después de hilar, estirar la fibra y extraer el porógeno polimérico PLA del dominio 'marino' de la fibra con una solución de hidróxido de sodio, se observa un arreglo de nanofibrillas de nailon alineadas de forma suelta e islas de nailon de tamaño micrométrico más grandes en toda la sección transversal del CIST de la fibra. Esta arquitectura de 'islas conectadas en el mar' aumenta en gran medida el área superficial de la fibra y las mediciones de superficie N2BET indican que valores de hasta 7 m2/ g son alcanzables con este enfoque.
En determinadas modalidades, las fibras de gran área superficial que son adecuadas para la purificación de unión/elución de proteínas son fibras sólidas con secciones transversales de diferentes formas. Estas fibras conformadas con ligandos de intercambio iónico exhiben un área superficial suficiente y propiedades de flujo aceptables para usarse en separaciones cromatográficas.
La funcionalización de la superficie de las fibras porosas de gran área superficial puede llevarse a cabo mediante la deposición de un recubrimiento de polímero con función epoxi sobre la superficie de la fibra, seguido de calentamiento
para unir covalentemente el recubrimiento de polímero a la superficie de la fibra, y un proceso subsecuente de abertura del anillo epoxi para instalar la funcionalidad de ácido sulfónico en la superficie de la fibra, por ejemplo.
En otras modalidades, se puede llevarse a cabo la modificación de fibras de tipo SAE con un ligando de intercambio iónico injertado en la superficie para aplicaciones de cromatografía de intercambio catiónico de unión/elución. Puede llevarse a cabo la activación de la superficie de la fibra SAE con un recubrimiento de polímero HPA/MBAm 95/5 reticulado para proporcionar un recubrimiento con funcionalidad hidroxi altamente reactivo en la superficie de la fibra, seguido de una polimerización redox de ion cérico, tal como con 2-acrilamido- sal sódica del ácido 2-metil-1-propanosulfónico, para proporcionar un sustrato de fibra injertada con polímero.
Una densidad de relleno de columna adecuada de entre aproximadamente 0,1 y 0,4 g/mL, preferiblemente aproximadamente 0,35 g/mL, proporcionará suficiente uniformidad de flujo para un rendimiento aceptable en una evaluación cromatográfica.
En ciertas modalidades, los medios (fibras empaquetadas funcionalizadas) pueden entregarse al usuario en un formato preempacado, a diferencia de los medios basados en perlas. Las fibras pueden fusionarse por medios térmicos o químicos para formar una estructura semirrígida que puede alojarse en un recipiente a presión. Mediante tal construcción, el medio y el dispositivo que lo acompaña pueden estar listos para usar. Los medios cromatográficos basados en perlas se suministran generalmente como material suelto (húmedo) en donde se requiere que el usuario cargue un recipiente a presión (columna) y, por diversos medios, cree un lecho bien empaquetado sin huecos ni canales. Generalmente se requieren pruebas de seguimiento para asegurar la uniformidad del empaque. Por el contrario, de acuerdo con ciertas modalidades, el usuario no requiere embalaje ya que el producto llega listo para el servicio.
El medio de fibra porosa funcionalizada en la superficie de las modalidades descritas en la presente descripción demuestra una alta permeabilidad en un formato de lecho empacado. En dependencia de la densidad de empaquetamiento, la permeabilidad del lecho puede variar de 2500 mDarcy a menos de 100 mDarcy. El lecho de fibras empaquetadas no se comprime a alta velocidad lineal.
El medio de fibra de área superficial mejorada de las modalidades descritas en la presente descripción puede configurarse en un formato de lecho empacado dentro de una carcasa adecuada, tal como una columna de cromatografía u otro dispositivo. Pueden prepararse lechos de fibras empaquetadas de fibras cortadas con área superficial mejoradas cargando una suspensión acuosa diluida de las fibras cortadas en una columna de cromatografía y una compresión axial subsecuente del cabezal de distribución de solvente superior de la columna de cromatografía hasta una profundidad del lecho objetivo de entre 1 y 10 cm. La compresión axial se define como la reducción de la profundidad del lecho de una empaquetadura de fibras cortadas ubicada dentro de una columna cromatográfica u otra carcasa adecuada para aumentar la densidad de empaquetadura del empaque de fibra cortadas a un valor objetivo de entre 0,1 y 0,4 g/mL. Esta compresión se logra mediante el desplazamiento mecánico de los cabezales de distribución de flujo para proporcionar un volumen de columna o dispositivo más pequeño y un aumento correspondiente en la densidad de empaquetamiento del medio cromatográfico. En este contexto, el eje que se comprime es el eje vertical de la columna en la que se empaquetan las fibras cortadas. Dado que las fibras cortadas son comprimibles, la densidad de empaquetamiento de las fibras cortadas aumenta correspondientemente cuando se realiza tal compresión axial. Por el contrario, la compresión radial se define como la reducción del diámetro interno de una empaquetadura de fibra cortada dentro de una columna cromatográfica u otra carcasa adecuada para aumentar la densidad de empaquetadura del empaque de la fibra cortada a un valor objetivo de entre 0,1 y 0,4 g/mL. Las operaciones de compresión radial no cambian la profundidad del lecho del empaque del medio de fibra.
Ejemplos
Ejemplo comparativo 1. Fundir la mezcla con porógenos minerales o poliméricos. En este experimento, el nailon se mezcló con una variedad de aditivos porógenos minerales o poliméricos tales como carbonato de calcio, sílice o polímero de poli(ácido láctico) (PLA). También se prepararon mezclas ternarias de porógenos poliméricos de nailon, minerales y PLA. Estas mezclas se usaron subsecuentemente para experimentos de extrusión de fibras.
Se prepararon varias mezclas de nailon 6 y cargas minerales mediante el uso de una extrusora de compuestos. También se prepararon mezclas adicionales que contienen una mezcla ternaria de nailon, PLA y porógenos minerales. Se examinaron cuatro tipos diferentes de cargas minerales: Albafil A-0-255-12 de SMI, Vicality Heavy de SMI, Multifex-MM ™ de SMI y Syloid 244FP de WR Grace. Los materiales presecados se pesaron y se mezclaron en seco. La mezcla seca se colocó sobre el transportador de alimentación que se ajustó para alimentar suficientemente el microtorreador. El material se extruyó de un troquel de una sola hebra en un baño de agua y luego se granuló. Ciertas formulaciones de nailon/porógeno empleadas en este trabajo se resumen en la Tabla 1 más abajo.
Tabla 1. Resumen de formulaciones de nailon compuestas con varios porógenos minerales y poliméricos.

Ejemplo comparativo 2.
La extrusión por fusión de monofilamentos cargados de porógenos minerales. En este experimento, se proporciona una descripción general del proceso para hilar en fusión los gránulos mezclados de nailon/porógeno mineral en fibras de monofilamento de aproximadamente 20 micrómetros de diámetro.
Los gránulos mezclados de nailon/porógeno mineral se hilaron por fusión en monofilamentos mediante el uso de una máquina de hilar fibras. La máquina de hilar fibras es un sistema LBS de Hills Inc. (Melbourne, FL). Las muestras de fibras de monofilamento extruido se estiraron hasta un diámetro de aproximadamente 20 micras. Después de hilar, estirar y enrollar la fibra, el porógeno mineral se extrajo subsecuentemente de los monofilamentos de acuerdo con el procedimiento que se describe más abajo.
Ejemplo comparativo 3.Extracción de porógenos minerales a partir de monofilamentos extruidos.
En este experimento, se describe el proceso para la extracción de porógenos minerales de fibras de monofilamento extruidas mediante el uso una solución de ácido clorhídrico 1 M. Subsecuentemente, las fibras se neutralizan, lavan y las superficies de las fibras se inspeccionan mediante microscopía electrónica de barrido (SEM). También se realizan mediciones del área superficial de Kr BET.
En un frasco de vidrio de 100 mL con tapa se añadieron 1,0 g de monofilamento extruido (aproximadamente 20 pm de diámetro) y 50 mL de HCl 1,0 M (50 mmol). La suspensión se agitó a 30 °C toda la noche. Los sólidos de fibra se aislaron mediante filtración al vacío y se lavaron con Tris-HCl 0,5 M (1 x 100 mL), agua desionizada (1 x 100 mL) y etanol (1 x 100 mL). El material se colocó en un horno para que se secara a 40 °C durante 18 horas. Los resultados de los experimentos de extracción de porógenos minerales se muestran en la Tabla 2 más abajo. La inspección por SEM de la morfología de la superficie de la fibra se realizó después de la extracción de los porógenos minerales y estas imágenes se muestran en la Figura 2. El monofilamento que contiene Albafil® produce poros o cavidades de tamaño micrométrico grandes en las superficies de las fibras después de la extracción del porógeno. Debido al tamaño de partícula más pequeño del porógeno Multifex-MM ™ (< 0,2 micras), se observan poros mucho más pequeños en la superficie de la fibra después de la extracción del porógeno. Estos poros no son evidentes en una muestra de control de fibra de nailon tratada de forma similar. Las mediciones del área superficial de Kr BET revelan una ganancia significativa (~300 %) en el área superficial BET para el material preparado mediante el uso del porógeno Albafil® sobre la muestra de control de fibra de nailon no porosa.
Tabla 2. Extracción de porógenos minerales de carbonato de calcio.

Ejemplo comparativo 4. Fundir la mezcla con porógenos poliméricos. En este experimento, se mezcló nailon con varias cantidades de un porógeno polimérico, polímero de poli(ácido láctico). Estas mezclas se usaron subsecuentemente para experimentos de extrusión de fibras.
Se generó una variedad de muestras de mezcla de nailon/PLA mediante el uso una extrusora de compuestos. Se preparó una gama de mezclas de PLA y Nylon 6 que se muestran en la Tabla 3 más abajo. Se pesó la cantidad apropiada de gránulos secos y se mezcló en seco. La mezcla combinada seca se añadió luego a la cinta transportadora de alimentación en la extrusora de compuestos. La cinta de alimentación se ajustó para alimentar suficientemente la extrusora de compuestos. El material se extruyó de un troquel de una sola hebra en un baño de agua y luego se granuló.
Tabla 3. Resumen de formulaciones de nailon compuestas con PLA como porógeno polimérico.

Ejemplo comparativo 5. Extracción de porógenos PLA a partir de filamentos extruidos. En este experimento, se describe el proceso para la extracción del porógeno PLA de los filamentos extruidos del mezclador de masa fundida mediante el uso de una solución de hidróxido de sodio 1,5 N. Subsecuentemente, las fibras se neutralizan, lavan, se realiza un ensayo gravimétrico y las superficies de las fibras se inspeccionan mediante microscopía electrónica de barrido (SEM).
En un frasco de vidrio de 250 mL con tapa se añadieron 2,0 g de filamento extruido (aproximadamente 2,0 mm de diámetro) y 0,2 L de NaOH 1,5 N (0,75 mol). La suspensión se agitó a temperatura ambiente toda la noche. Los sólidos de las fibras se aislaron mediante filtración al vacío y se lavaron con agua desionizada (3 x 250 mL) y etanol (1 x 250 mL). El material se colocó en un horno para que se secara a 60 °C durante 3 horas. Los resultados de los experimentos de extracción del porógeno polimérico PLA de los filamentos extruidos de 2 mm se muestran en la Tabla 4 más abajo. A partir de estos datos, puede verse que el porógeno de PLA se extrae más fácilmente para la muestra de alta carga de PLA (50 % en peso de PLA) que para la muestra de carga de PLA baja (35 % en peso de PLA). La gran diferencia entre los rendimientos reales y esperados se debe al acceso restringido al interior de estos grandes filamentos de 2 mm de diámetro. La inspección por SEM de la morfología de la superficie del filamento se realizó después de la extracción del porógeno polimérico PLA y estas imágenes se muestran en la Figura 3. Estos datos muestran la aparición de una morfología superficial fibrilada después de la extracción del porógeno PLA de los filamentos. Estas características de la superficie son bastante pronunciadas para cargas de PLA iguales o superiores al 40 % en peso. Se espera que las morfologías de la superficie fibrilada tales como estas aumenten en gran medida el área superficial de un sustrato de fibra.
Tabla 4. Extracción de porógenos PLA a partir de filamentos extruidos.

Ejemplo 6. Extrusión por fusión de fibras cargadas de porógenos poliméricos. En este experimento, se proporciona una descripción general del proceso para hilar en fusión los gránulos de porógeno de polímero de nailon/PLA compuestos en núcleo/envoltura o fibras de dos componentes de 15 alas de aproximadamente 20 micrómetros de diámetro.
Los gránulos de nailon/PLA mezclados se hilaron por fusión en fibras mediante el uso de una máquina de hilar fibras de dos componentes. La máquina de hilar fibras de dos componentes es un sistema LBS de Hills Inc. (Melbourne, FL). Las muestras de fibra extruida eran núcleo/envoltura y fibras de 15 alas con una envoltura de PLA y los gránulos mezclados en el núcleo (o viceversa). Las muestras se resumen en la Tabla 5 más abajo.
Tabla 5. Tabla resumen de fibras extruidas que contienen porógenos poliméricos ubicados dentro del núcleo de la fibra o como revestimiento externo cargado de porógenos Las fibras de núcleo/envoltura que comprenden del 35 al 50 % en peso de PLA en el núcleo son de acuerdo con la invención, las otras fibras son comparativas.

Ejemplo 7. Extrusión por fusión mediante el uso de muestras mezcladas a mano. En este experimento, se proporciona una descripción general del proceso para hilar en fusión una mezcla mezclada a mano de nilón y gránulos porógenos de polímero PLA en fibras de dos componentes de núcleo/envoltura de aproximadamente 20 micras de diámetro.
Se generó una variedad de muestras de fibra extruidas en fusión mediante hilado en fusión mediante el uso de una extrusora de dos componentes a escala de laboratorio de Hills Inc. (Melbourne, FL) montada con un paquete de hilado de núcleo/envoltura. El lado de la envoltura del troquel se alimentó con gránulos de ácido poliláctico (PLA). Para el lado del núcleo del troquel, se mezclaron gránulos de polímero en diversas proporciones mediante simple agitación en una bolsa antes de introducirlos en la extrusora. Las fibras se estiraron y enrollaron en un núcleo para su posterior procesamiento y análisis. Las muestras se resumen en la Tabla 6 más abajo.
Tabla 6. Tabla resumen de fibras extruidas que contienen porógeno polimérico mezclado a mano/mezclas de nailon ubicadas dentro del núcleo de la fibra Las fibras de núcleo/envoltura que comprenden del 35 al 50 % en peso de PLA en el núcleo son de acuerdo con la invención, las otras fibras son comparativas.

(continuación)

Ejemplo 8. Procedimiento general para la extracción de PLA. En este experimento, se describe el proceso para la extracción de porógenos de PLA a partir de fibras de núcleo/envoltura de dos componentes extruidas mediante el uso de una solución de hidróxido de sodio 1,5 N. Subsecuentemente, las fibras se neutralizan, lavan, se realiza un ensayo gravimétrico y las superficies de las fibras se inspeccionan mediante microscopía electrónica de barrido (SEM). También se realizan mediciones del área superficial de Kr BET.
En una botella Pyrex de 1 litro con tapa se añadieron 5,0 g de fibra cortada (1,0 mm de longitud) y 0,5 L de NaOH 1,5 N (0,75 mol). La suspensión se agitó a temperatura ambiente toda la noche. Los sólidos de las fibras se aislaron mediante filtración al vacío y se lavaron con agua desionizada (3 x 250 mL) y etanol (1 x 250 mL). El material se colocó en un horno para que se secara a 60 °C durante 18 horas. Los resultados de los experimentos de extracción del porógeno polimérico PLA a partir de fibras extruidas de dos componentes (20 micras de diámetro) se muestran en la Tabla 7 más abajo. A partir de estos datos, encontramos que el porógeno PLA se extrajo tanto de la envoltura de la fibra como dentro del núcleo de la fibra para todas las muestras de fibras. La pequeña diferencia entre los rendimientos reales y esperados es evidencia de una extracción completa del porógeno PLA desde dentro de la fibra de dos componentes. La inspección por SEM de la morfología de la superficie de la fibra se realizó después de la extracción del porógeno polimérico PLA y estas imágenes se muestran en la Figura 4. A partir de estos datos, encontramos la aparición de una morfología superficial fibrilada después de la extracción del porógeno PLA de las fibras con cargas de PLA superiores al 35 % en peso. Estas fibras parecen estar construidas a partir de conjuntos de nanofibrillas de nailon muy entrelazadas. Para cargas de porógeno de PLA superiores a aproximadamente el 50 % en peso, hay un aparente deshilachamiento de la estructura de la fibra para producir nanofibras de nailon individuales. Las imágenes de la sección transversal de Cryo-SEM para la muestra de PLA al 40% en peso se muestran en la Figura 5. A partir de estas imágenes de sección transversal, encontramos que esta fibra parece estar construida con cientos de nanofibrillas de nailon alineadas axialmente sin apretar y hay una porosidad significativa dentro de la sección transversal de la fibra. Se espera que las morfologías de la superficie fibrilada tales como estas aumenten en gran medida el área superficial de un sustrato de fibra. En la Figura 6, se muestra que se miden áreas superficiales de Kr BET altas para las muestras de fibra de nailon extraídas que se construyeron con cargas de porógeno polimérico PLA superiores al 25 % en peso.
Tabla 7. Extracción de porógenos PLA y eliminación de envolturas Las fibras de núcleo/envoltura que comprenden del 35 al 50 % en peso de PLA en el núcleo son de acuerdo con la invención, las otras fibras son comparativas.

Ejemplo comparativo 9. Extrusión por fusión de fibras aladas que tienen un núcleo poroso. En este experimento, se proporciona una descripción general del proceso para hilar en fusión los gránulos de porógeno de polímero de nailon/PLA compuestos en fibras 15 alas de dos componentes de aproximadamente 15 micras diámetro. Subsecuentemente, el PLA se extrae de la envoltura de la fibra así como también de dentro del núcleo de la fibra. Como resultado, estas fibras de 15 alas presentan una estructura de núcleo porosa.
Los gránulos de nailon/PLA mezclados se hilaron por fusión en fibras mediante el uso de una máquina de hilar fibras de dos componentes. La máquina de hilar fibras de dos componentes es un sistema LBS de Hills Inc. (Melbourne, FL). Las muestras de fibra extruida eran fibras de 15 alas y se prepararon con una envoltura de PLA y los gránulos
mezclados como núcleo de fibra. Después de la extracción del PLA de la envoltura de la fibra y dentro del núcleo de la fibra, se proporciona una fibra de 15 a las que tiene una estructura porosa. El procedimiento general para la extracción del porógeno polimérico PLA se describe en el ejemplo anterior. Varias muestras de fibra de 15 alas preparadas mediante el uso de mezclas de nailon/PLA 70/30 y 60/40 como composición del núcleo de la fibra se ilustran en la Figura 7 y Figura 8. Estas imágenes muestran la apariencia de poros o cavidades cilíndricas que se extienden dentro de las secciones transversales de las fibras aladas, pero que no son estructuras fibriladas. Sin embargo, se espera que tales características aumenten aún más el área superficial de las fibras aladas.
Ejemplo comparativo 10. Procedimiento general para la modificación de la superficie de fibras SAE. En este experimento, se describe un procedimiento general para la modificación de la superficie de fibras SAE con grupos funcionales colgantes de intercambio catiónico fuerte. Este procedimiento implica la deposición de un recubrimiento de polímero con función epoxi sobre la superficie de la fibra, una etapa de calentamiento para unir covalentemente el recubrimiento de polímero a la superficie de la fibra y un proceso posterior de abertura del anillo epoxi para instalar una funcionalidad de ácido sulfónico en la superficie de la fibra.
Se prepararon veinticinco gramos de una solución al 1 % en peso de poli (metacrilato de glicidilo) en metiletilcetona (MEK) en un vial de vidrio de 30 mL. En un vial de vidrio de 30 mL separado, se añadieron 0,2 gramos de fibra y 12,5 g de la solución de polímero de poli (metacrilato de glicidilo) al 1 %. La suspensión se agitó a temperatura ambiente toda la noche. Los sólidos de la fibra se aislaron subsecuentemente mediante filtración al vacío y se colocaron en un horno a 100 °C durante 30 minutos. Los sólidos de la fibra se retiraron del horno y se resuspendieron en 40 mL de MEK durante 1 hora a temperatura ambiente. Los sólidos de la fibra se aislaron mediante filtración al vacío y luego se suspendieron en 15 mL de una solución de sulfito de sodio 1 M/bisulfato de tetra-n-butilamonio 0,4 M. La suspensión se roció con N2durante 5 minutos, el vial se selló y se calentó a 80 °C toda la noche. La suspensión se enfrió a temperatura ambiente. Los sólidos de la fibra se aislaron mediante filtración al vacío y se lavaron con agua desionizada (5 x 30 mL) y etanol (1 x 30 mL). Las fibras se secaron a 60 ° C durante 2 horas. Los resultados para la modificación de la superficie de una fibra de núcleo/envoltura mejorada en su área superficial (SAE) así como también para una de 15 alas no porosa de muestra control se presentan en la Tabla 8 más abajo. También se realizaron mediciones de la capacidad de unión estática para estas dos fibras funcionalizadas con ácido sulfónico mediante el uso de IgG, lisozima y azul de metileno como sondas de proteína grande, proteína pequeña y molécula pequeña, respectivamente. Se emplearon condiciones de unión de intercambio catiónico estándar para todas estas pruebas de capacidad de unión estática y los resultados se resumen en las Figuras. 9, 10, 11 y en la Tabla 9 más abajo. A partir de estos datos, se muestra una mayor capacidad de unión estática para la fibra SAE sobre la fibra de 15 alas, así como también que la ventaja de la capacidad de unión para la fibra SAE aumenta con la disminución del tamaño molecular.
Tabla 8. Modificación de la superficie de fibras de nailon SAE y de 15 alas, área superficial BET y datos de recuperación.

Tabla 9. Resumen de los datos de capacidad de unión estática para fibras SAE modificado con SP y de 15 alas.

Ejemplo comparativo 11. Modificación de superficie de fibras SAE (7895-136).En este experimento, se describe un procedimiento para la modificación de fibras de tipo SAE con un ligando de intercambio iónico injertado en la superficie para aplicaciones de cromatografía de intercambio catiónico de unión/elución. La primera etapa de este proceso implica la activación de la superficie de la fibra SAE con un recubrimiento de polímero HPA/MBAm 95/5 reticulado. Esta etapa proporciona un recubrimiento con función hidroxilo altamente reactivo en la superficie de la fibra que es
muy adecuado para un proceso de injerto de polímero posterior. En una segunda etapa, la fibra modificada con HPA/MBAm se somete a una polimerización redox de ion cérico con sal sódica del ácido 2-acrilamido-2-metil-1-propanosulfónico para proporcionar un sustrato de fibra injertada con polímero. El polímero injertado proporciona grupos funcionales de ácido sulfónico colgantes para aplicaciones de cromatografía de intercambio catiónico. Modificación de la superficie de la fibra de nailon SAE con HPA/MBAm 95/5.En una botella Pyrex de 500 mL se añadieron acrilato de hidroxipropilo (HPA, 4,9 g, 38 mmol), N, N'-metilenbis (acrilamida) (MBAm, 0,28 g, 2 mmol) y agua (253 mL). Se añadieron a la mezcla 6,0 g de fibras de nailon con área superficial mejorada (SAE). Se añadió persulfato de amonio (0,63 g, 3 mmol). Los sólidos húmedos se calentaron a 80 °C durante 4 horas.
Después de enfriar a temperatura ambiente, los sólidos se transfirieron a un embudo Buchner y se lavaron con agua caliente (4 x 200 mL) y etanol (1 x 200 mL). Se dejó secar el material al vacío durante 20 minutos. El material se transfirió a un horno y se secó a 60 °C durante 18 horas.
Se obtuvieron 6,46 g en forma de fibras blancas.
Polimerización por injerto de fibras de nailon modificadas con HPA/HBAm. En 3 frascos de vidrio de 125 mL se agregaron sal sódica del ácido 2-acrilamido-2-metil-1-propanosulfónico (AMPS-Na), agua, fibras de nailon SAE modificadas con HPA/MBAm (ver arriba) y solución de HNO3 1 M (en el cantidades descritas en la tabla más abajo). Se añadió a cada botella una solución 0,4 M de nitrato de amonio cerio (IV) (CAN) en HNO3 1 M. Las botellas de reacción se taparon, se rociaron con nitrógeno y las mezclas se calentaron a 35 °C durante 18 horas.
Después de enfriar a temperatura ambiente, los sólidos se lavaron con una solución de ácido ascórbico 0,2 M en ácido sulfúrico 0,5 M (3 x 80 mL), agua desionizada (3 x 80 mL), solución de hidróxido de sodio 0,5 M (3 x 80 mL), agua desionizada (3 x 80 mL) y etanol (1 x 80 mL). El material se colocó en un horno para que se secara a 60 °C durante 18 horas. Se obtuvieron muestras de un sólido fibroso blanco (consulte la tabla para conocer los datos adicionales de recuperación y peso).
Tabla 10. Composiciones de polimerización por injerto redox de cerio y datos de recuperación.

Ejemplo comparativo 12. Medición de la capacidad de unión estática. En este experimento, se presenta la capacidad de unión estática de IgG de las fibras SAE modificadas en la superficie con SP en un modo de intercambio catiónico.
Los resultados de las mediciones de la capacidad de unión estática de IgG para las fibras de nailon SAE modificadas con SP se proporcionan en la Tabla 11 más abajo. A partir de estos datos, se muestran capacidades sustanciales de unión estática de IgG para las fibras SAE modificadas en la superficie con SP y estos valores de SBC de IgG son comparables a las resinas comerciales de cromatografía de intercambio catiónico basadas en perlas.
Tabla 11. Datos de la capacidad de unión estática (SBC) de IgG para fibras de nailon SAE modificadas con SP. Reto: 2 g/L de IgG humana policlonal (SeraCare Life Sciences, Milford, MA) en acetato de sodio 50 mM (pH 5).

Ejemplo comparativo 13. Medición de la capacidad de unión dinámica. En este experimento, se describe el empaque de las fibras SAE modificadas en la superficie con SP en una columna de cromatografía y la permeabilidad del lecho de fibras empaquetadas. También se presenta la capacidad de unión dinámica de IgG de las fibras SAE de superficie modificada con SP en un modo de intercambio catiónico.
Los resultados de las mediciones de la capacidad de unión dinámica de IgG para los medios de fibra SAE funcionalizados con SP del ejemplo 7895-136B se proporcionan en la Tabla 12 más abajo. Se empaquetó 1,0 g del medio en una columna Vantage de 11 mm de diámetro interno y se comprimió hasta una profundidad de lecho de 3,0 cm (volumen de columna de 2,85 mL, densidad de relleno de fibra de 0,35 g/mL). Se determinó que la permeabilidad de la fibra compactada a 0,35 g/mL era 200 mDa mediante el uso de tampón acetato 50 mM (pH 5). Las mediciones de la capacidad de unión dinámica se realizaron en un intervalo de velocidades lineales de 200 cm/hora a 60 cm/hora. Estas velocidades corresponden a tiempos de residencia de 54 segundos a 3 minutos. El medio de fibra del ejemplo 7895-136B demuestra capacidades de unión dinámica de IgG en el intervalo de 50 mg/mL.
Tabla 12. Valores de DBC de IgG para los medios de intercambio catiónico de fibra SAE funcionalizados con SP al 1, 5, 10 y 50 % de avance a velocidades lineales variables (RT = tiempo de residencia). Reto: 2,0 g/L de IgG humana policlonal (SeraCare Life Sciences, Milford, MA) en acetato 50 mM, pH 5.

Ejemplo comparativo 14. Extrusión por fusión de fibras de "islas conectadas en el mar" (CIST).En este experimento, se describe una descripción general del proceso para hilar en fundido mezclas mezcladas a mano de nilón y gránulos porógenos de polímero PLA en fibras de tipo "islas conectadas en el mar" (CIST) de aproximadamente 20 micrones de diámetro. Subsecuentemente, las fibras se neutralizan, lavan, se realiza un ensayo gravimétrico y las superficies de las fibras se inspeccionan mediante microscopía electrónica de barrido (SEM). También se realizan mediciones de área superficial N2BET.
Se generó una variedad de muestras de fibras extruidas en fusión mediante hilado en fusión mediante el uso de una extrusora de dos componentes a escala de laboratorio de Hills Inc. (Melbourne, FL) montada con un paquete de hilado de "islas en el mar" (configuración de 36 islas). El lado de la "isla" del troquel se alimentó con gránulos de nailon 6. Para el lado del "mar" del troquel, los gránulos de polímero se mezclaron en diversas relaciones mediante simple agitación en una bolsa antes de introducirlos en la extrusora. Para este ejemplo, los componentes de la mezcla fueron PLA y nailon 6. Las fibras se estiraron y enrollaron en un núcleo para su posterior procesamiento y análisis. Las muestras se resumen en la Tabla más abajo. Después de la extracción del porógeno polimérico PLA del interior del dominio "marino" de la fibra de dos componentes extruida, se proporciona una fibra CIST que tiene una estructura porosa. En la Tabla 14,más abajo, se resumen varios ejemplos de fibras de tipo CIST. El procedimiento general para la extracción del porógeno polimérico PLA se describe en el ejemplo anterior. Se prepararon varias muestras de fibra CIST preparadas mediante el uso de un intervalo de mezclas de porógenos de nailon/PLA como la composición del dominio "marino" de la fibra. Las imágenes de la superficie y de la sección transversal crio-SEM de una fibra CIST y una fibra de tipo SAE se proporcionan en la Figura 12. A partir de las imágenes de la sección transversal, puede verse que grandes canales o grietas que se extienden por todo el interior de la fibra CIST y estas características pueden hacer que la superficie interna sea considerablemente más accesible que para la fibra de tipo SAE. Se espera que tales características mejoren el acceso de grandes proteínas y biomoléculas al área de la superficie interior de tales soportes de fibra nanofibrilada.
Tabla 13. Cuadro resumen de fibras eximidas de 'islas conectadas en el mar' (CIST) que contienen 36 'islas' de nailon PA6 y mezclas poliméricas de porógeno/nailon mezcladas a mano como el componente 'mar' de la fibra.

Tabla 14. Extracción de porógenos PLA y N2BET datos para fibras de tipo "islas conectadas en el mar"
(CIST).

Ejemplo comparativo 15. Modificación de la superficie de las fibras SAE, CIST, de 15 ala y de nailon redondas. En este experimento, se describe un procedimiento para la modificación de fibras de tipo SAE, CIST y de 15 alas con un ligando de intercambio iónico injertado en la superficie para aplicaciones de cromatografía de intercambio catiónico de unión/elución. En este proceso, las superficies de las fibras se modifican en una sola etapa mediante el uso de una polimerización redox de ion cérico con sal de potasio de metacrilato de 3-sulfopropilo para proporcionar un sustrato de fibra injertada con polímero. El polímero injertado proporciona grupos funcionales colgantes de ácido sulfónico para aplicaciones de cromatografía de intercambio catiónico.
En una botella de 125 mL se añadió sal de potasio de metacrilato de 3-sulfopropilo (3-SPMA), agua, fibras de nailon CIST y solución de HNO3 1 M (en las cantidades descritas en la tabla más abajo). Se añadió a la botella una solución 0,4 M de nitrato de amonio cerio (IV) (CAN) en HNO3 1 M. La botella de reacción se tapó y la mezcla se calentó a 35 °C durante 5 horas.
Después de enfriar a temperatura ambiente, los sólidos de fibra de la botella se lavaron con una solución de ácido ascórbico 0,2 M en ácido sulfúrico 0,5 M (3 x 50 mL), agua desionizada (3 x 50 mL), solución de hidróxido de sodio 0,5 M (3 x 50 mL), agua desionizada (3 x 50 mL) y etanol (1 x 50 mL). El material se colocó en un horno para que se secara a 60 °C durante 18 horas.
Se obtuvieron muestras de un sólido fibroso blanco (consulte la tabla para conocer los datos adicionales de recuperación y peso).
Tabla 15. Composiciones de polimerización de injerto redox de cerio y datos de recuperación para las fibras con área superficial mejorada (SAE), de 'islas conectadas en el mar' (CIST), las de 15 alas no porosas y de control redondas (15 pm de diámetro).

Ejemplo comparativo 16. Medición de la capacidad de unión estática. En este experimento, se presenta la capacidad de unión estática de IgG de las fibras SAE modificadas con SP, CIST, de 15 alas y redondas simples modificadas en la superficie en un modo de intercambio catiónico. Para las fibras SAE (muestras # 7895-142A y 7895-142B), encontramos que un aumento en el monómero 3-SPMA de 7,5 mmol a 19 mmol en la etapa de injerto produce un aumento sustancial en la capacidad de unión estática de IgG de 47 mg/g hasta 212 mg/g. Las fibras CISt (entrada 7993-2A-1, 7993-2A-2) dan valores SBC de IgG comparables a los de las fibras de tipo SAE en la condición de carga de monómero 3-SPMA baja (7,5 mmol). La fibra de 15 alas (entrada 7895-62D) se modificó mediante el uso de 38 mmol de monómero 3-SPMa y esta muestra produce un valor de IgG SBC de 130 mg/g. En comparación, las fibras redondas simples de 15 micrones proporcionan valores de SBC de IgG muy bajos en todas las condiciones de injerto evaluadas. Esto puede atribuirse a la muy baja área superficial de las fibras redondas que carecen de proyecciones o estructuras porosas internas.
Los resultados de las mediciones de la capacidad de unión estática de IgG para las fibras con el área superficial mejorada (SAE) modificada con SP, las de 'islas conectadas en el mar' (CIST), las de 15 alas no porosas y las de control redondas (15 pm de diámetro) se proporcionan en la Tabla 16 más abajo.
Tabla 16. Datos de capacidad de unión estática de IgG para las fibras con el área superficial mejorada (SAE) modificada con SP, las de "islas conectadas en el mar" (CIST), las de 15 alas no porosas y las de control redondas (15 pm de diámetro). Reto: 2 g/L de IgG humana policlonal (SeraCare Life Sciences, Milford, MA) en acetato de sodio 50 mM (pH 5).

(continuación)
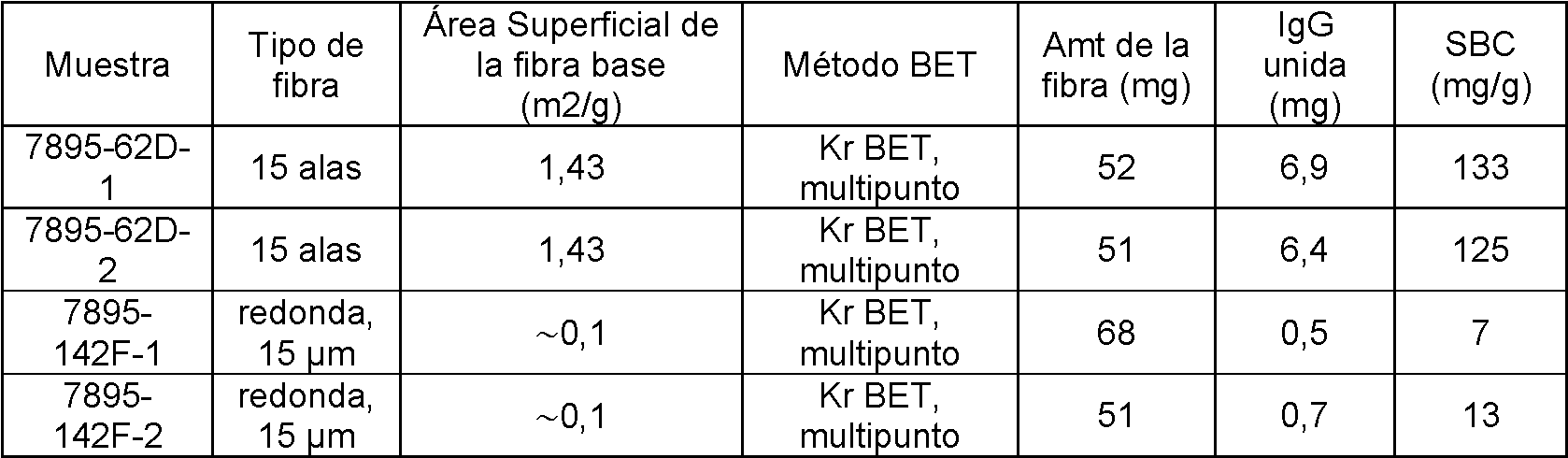
Ejemplo comparativo 17.Extrusión por fusión de fibras conformadas Las fibras conformadas se preparan mediante el uso de un proceso de hilado en fusión de dos componentes. La fibra de dos componentes tiene un núcleo de un material y una envoltura de un segundo polímero. Estos materiales de núcleo y envoltura podrían ser cualquier tipo de termoplástico procesable por fusión conocido por los estudiados en la técnica. Se usan una serie de placas de troquel para dividir y redirigir las dos corrientes de alimentación de polímero en un número determinado de fibras y la forma de sección transversal deseada. Las fibras se estiran al tamaño apropiado después del hilado por fusión. Las características de las fibras se resumen en la Tabla 17.
Tabla 17.-Resumen de formas de las fibras conformadas, áreas superficiales BET y diámetro de fibra.

Ejemplo comparativo 18. Procedimiento general para la modificación superficial de fibras conformadas. Las fibras conformadas se prepararon de acuerdo con el Ejemplo 10. Consulte las tablas para obtener datos sobre la capacidad de unión estática de IgG.
Tabla 1817. Resumen de los datos de capacidad de unión estática para formas modificadas con SP y fibras de 15 alas.

Ejemplo comparativo 19. Modificación de la superficie de las fibras de nailon conformadas. Las fibras conformadas se prepararon de acuerdo con el Ejemplo 11. Consulte la tabla para conocer los datos adicionales de recuperación y peso.
Tabla 19. Composiciones de polimerización por injerto redox de cerio y datos de recuperación para fibra fractal.
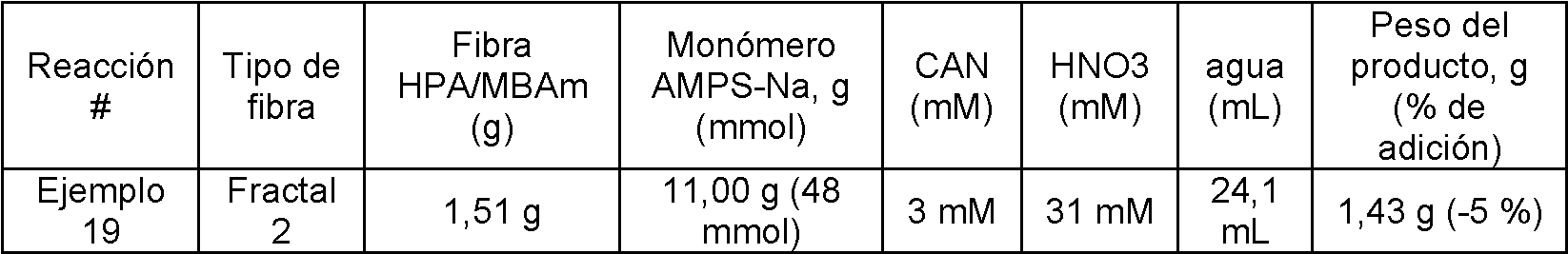
Ejemplo comparativo 20. Medición de la capacidad de unión dinámica. Las fibras fractales de superficie modificada del Ejemplo 19 anterior se empaquetaron de acuerdo con el método descrito en el Ejemplo 13.
Las mediciones de la capacidad de unión dinámica se realizaron en un intervalo de velocidades lineales de 200 cm/hora a 600 cm/hora. Estas velocidades corresponden a tiempos de residencia de 54 segundos a 18 segundos. El medio de fibra del Ejemplo 19 demuestra capacidades de unión dinámica de IgG en el intervalo de 72 mg/mL.
Tabla 20. Valores de DBC de IgG para los medios de intercambio catiónico de fibras conformadas funcionalizadas con SP al 1, 5, 10 y 50 % de avance a velocidades lineales variables (RT=tiempo de residencia). Reto: 2,0 g/L de IgG humana policlonal (SeraCare Life Sciences, Milford, MA) en acetato 50 mM, pH 5.

Ejemplo comparativo 21.Polimerización por injerto de fibras SAE no modificadas. Modificación de la superficie de las fibras SAE con una funcionalidad de ligando polimérico de tetraalquilamonio (tipo Q) para aplicaciones de cromatografía de intercambio aniónico (AEX). En una botella de 500 mL se añaden metacrilato de glicidilo (GMA, 1,70 g, 12 mmol) y agua (232,8 mL). Se añaden 5 g de fibras SAE a la solución. Se añade una solución de HNO3 (7,22 mL, 7,2 mmol) 1 M a la mezcla de reacción, seguido de la adición de una solución de nitrato de amonio y cerio (IV) 0,4 M en HNO3(0,602 mL, 0,240 mmol) 1 M.
La mezcla de reacción se calienta a 35 °C durante 1 hora.
Después de enfriar a temperatura ambiente, los sólidos se lavan con agua desionizada (3 x 100 mL) y el material húmedo (12,21 g) se usa inmediatamente en la siguiente etapa.
Funcionalización Q de fibras SAE funcionalizadas con epoxi. En una botella de 250 mL se añaden las fibras SAE funcionalizadas con GMA húmedas del ejemplo anterior y una solución de trimetilamina (acuosa) al 50 % en peso en metanol (en las cantidades descritas en la Tabla 21 más abajo). La mezcla se agita a temperatura ambiente durante 18 horas.
Los sólidos de fibra se lavan subsecuentemente con una solución de ácido ascórbico 0,2 M en ácido sulfúrico 0,5 M (3 x 50 mL), agua desionizada (3 x 50 mL), solución de hidróxido de sodio 1 M (3 x 50 mL), agua desionizada (3 x 50 mL) y etanol (1 x 50 mL). El material se coloca en un horno para que se seque a 40 °C durante 12 horas.
Se obtienen muestras de un sólido fibroso blanco.
Tabla 21. Composición para la modificación de fibras SAE funcionalizadas con epoxi con trimetilamina.

Ejemplo comparativo 22.Polimerización por injerto de fibras SAE no modificadas. Modificación de la superficie de fibras SAE con una funcionalidad de polímero de poli (hidroxietilmetacrilato) para aplicaciones de cromatografía de interacciones hidrófobas (HIC). En una botella de 500 mL se añaden metacrilato de hidroxietilo (HEMA, 1,69 g, 13 mmol) y agua (232,5 mL). Se añaden 5,00 g de fibras SAE a la solución. Se añade una solución de HNO3 (7,21 mL, 7,2 mmol) 1 M a la mezcla de reacción, seguido de la adición de una solución de nitrato de amonio y cerio (IV) 0,4 M en HNO3(0,601 mL, 0,240 mmol) 1 M.
La mezcla de reacción se calienta a 35 °C durante 1 hora.
Después de enfriar a temperatura ambiente, los sólidos se lavan con una solución de ácido ascórbico 0,2 M en ácido sulfúrico 0,5 M (3 x 100 mL), agua desionizada (3 x 100 mL), solución de hidróxido de sodio 1M (3 x 100 mL), agua
desionizada (3 x 100 mL) y etanol (1 x 100 mL). El material se coloca en un horno para que se seque a 40 °C durante 12 horas.
Ejemplo comparativo 23. Modificación de la superficie de la fibra SAE con ligando de afinidad de proteína A recombinante, rSPA. Modificación de la superficie de las fibras SAE con un ligando de afinidad de proteína A recombinante para aplicaciones de cromatografía de afinidad. En una botella de 250 ml se añaden bicarbonato de sodio 1 M (100 mL), proteína A recombinante (rSPA # RN091139, 150 mg, como una solución de 47,5 mg/mL en agua) y agua (90 mL). Se añaden fibras SAE injertadas con GMA (350 mg) del Ejemplo 21 anterior a la mezcla de reacción. La mezcla se calienta a 37 °C durante 2,5 horas.
Después de enfriar a temperatura ambiente, los sólidos se transfieren a un embudo Buchner y se lavan con bicarbonato de sodio 0,1 M (3 x 100 mL). Los sólidos de fibra húmeda se suspenden en 100 mL de una solución de tioglicerol al 10 % en peso en bicarbonato de sodio 0,2 M/solución de cloruro de sodio 0,5 M. La mezcla se agita a temperatura ambiente toda la noche.
Los sólidos se transfieren a un embudo Buchner y se lavan con una solución de base TRIZMA 0,1 M con cloruro de sodio 0,15 M (1 x 75 mL), solución de ácido acético 0,05 M (1 x 75 mL). Los ciclos de lavado con base TRIZMA y ácido acético se repiten dos veces adicionales. Los sólidos de fibra SAE se lavan finalmente con agua desionizada (1 x 75 mL) y etanol al 20 % en peso (1 x 75 mL). Los sólidos de fibra SAE se almacenan en una solución de etanol al 20 % en peso.
Ejemplo comparativo 24. Modificación con poli (alilamina) de fibras funcionalizadas con epoxi. Modificación de la superficie de las fibras SAE con una funcionalidad de ligando polimérico de poli (alilamina) para aplicaciones de cromatografía de intercambio aniónico (AEX). En una botella de 30 mL se añaden fibras SAE injertadas con GMA del Ejemplo 21 anterior (0,5 g), agua (10 mL), solución de hidrocloruro de poli (alilamina) al 40 % en peso (1,25 g de solución al 40 % en peso) e hidróxido de sodio 1,0 M (10 mL). La mezcla de reacción se calienta a 35 °C durante 18 horas.
Después de enfriar a temperatura ambiente, los sólidos se lavan con agua desionizada (3 x 50 mL) y acetona (1 x 50 mL).
El material húmedo se coloca en un horno para que se seque a 40 °C durante 12 horas.
Ejemplo comparativo 25. Aclaramiento de proteínas de la célula huésped de flujo continuo. Los medios de fibra SAE funcionalizados con Q preparados de acuerdo con el Ejemplo 21 se evalúan para determinar la actividad de eliminación de HCP en un modo de pulido de flujo continuo. Se empaquetan 0,34 g del medio de fibra funcionalizado con Q en una columna de 14,5 mm de diámetro interno y se comprime hasta una profundidad del lecho de 0,6 cm (volumen de columna de 1,00 mL, densidad de relleno de fibra de 0,34 g/ml).
Se clarifica un medio de cultivo celular que contiene anticuerpo monoclonal y luego se aísla mediante el uso de cromatografía en columna de Proteína A y el pH de la solución se ajusta a pH 5. El pH de la elución de Proteína A se ajusta subsecuentemente a pH 8 con base TRIZMA y luego se filtra a través de una membrana de 0,2 micras.
La columna de medios de fibra SAE funcionalizada con Q se equilibra con una solución tampón (Tris 25 mM a pH 8). Cien mililitros del anticuerpo monoclonal de 8,2 g/L (pH 8) de elución de la proteína A se pasan a través de la columna a un régimen de flujo de 1,0 mL/min. Se recogen diez facciones de 10 mL. E1HCP unido se eluye mediante el uso de una solución de cloruro de sodio 1 M en Tris 25 mM pH 8 como tampón de elución. También se recogen dos fracciones de elución de 10 mL. Las diez fracciones de flujo continuo y las dos fracciones de elución se analizan mediante HCP-ELISA y HPLC de proteína A para determinar el nivel de aclaramiento de HCP y la recuperación de anticuerpos monoclonales, respectivamente.
Ejemplo comparativo 26. Capacidad de medios de fibra SAE para el unión/eluir purificación de virus. Los experimentos de capacidad de unión estática y recuperación de elución para el bacteriófago 96 se llevan a cabo como se muestra más abajo. Las operaciones de unión/elución en modo de intercambio aniónico también pueden realizarse en un formato de columna empaquetada de acuerdo con procedimientos similares a los descritos en el Ejemplo 13. En 5 tubos de centrífuga de plástico se añaden los medios de fibra SAE funcionalizados con Q del Ejemplo 21. Cada una de las muestras de fibra SAE se equilibra con 5 mL de tampón Tris 25 mM (pH 8, con 0,18 mg/mL de HSA) con agitación durante 10 minutos. Los tubos se centrifugan a temperatura ambiente en una centrífuga de mesa a 4000 rpm durante 10 minutos para sedimentar el medio de fibra SAE. Se eliminan 2,5 mL del sobrenadante y se añaden a cada tubo 2,5 mL de una solución de 1,7x10796 de ufp/mL en tampón Tris 25 mM (pH 8, con 0,18 mg/mL de HSA). Las muestras se agitan a temperatura ambiente durante 1 hora. Posteriormente, los tubos se centrifugan a temperatura ambiente en una centrífuga de mesa a 4000 rpm durante 15 minutos para sedimentar el medio de fibra SAE. Se eliminan 2,5 mL del sobrenadante y estas muestras se analizan para determinar el 96 no unido mediante un ensayo de formación de placa. Los tubos se lavan 3 veces con lavados de 2,5 mL de tampón Tris 25 mM (pH 8, con 0,18 mg/mL de HSA) con centrifugación para sedimentar el medio de fibra SAE entre cada lavado y eliminación de 2,5 mL del sobrenadante. Después del lavado, se añaden a cada tubo 2,5 mL de una solución de NaCl 1,0 M en tampón Tris
25 mM (pH 8, con 0,18 mg/mL de HSA) (el volumen total es de 5 mL, la concentración final de NaCI es 0,5 M). Las muestras se agitan a temperatura ambiente durante 10 minutos. Posteriormente, los tubos se centrifugan a temperatura ambiente en una centrífuga de mesa a 4000 rpm durante 10 minutos para sedimentar el medio de fibra SAE. Se eliminan 2,5 mL del sobrenadante y estas muestras de elución se analizan para determinar el 96 eluido mediante un ensayo de formación de placa. El medio de fibra SAE con funcionalidad Q puede integrarse en un formato de dispositivo preempacado o en una columna de cromatografía para aplicaciones de purificación viral de flujo continuo o de unión/elución.
Claims (8)
1. Medios de cromatografía que comprenden fibras que presentan una estructura enrevesada compuesta por conjuntos discretos de nanofibrillas porosas muy entrelazadas y en donde las fibras presentan un área superficial en el intervalo de 1 a 12 metros cuadrados por gramo, dichas nanofibrillas dentro de dichos conjuntos porosos discretos tienen diámetros menores que 1 micra y a dichas nanofibrillas se les ha impartido una funcionalidad que permite una cromatografía seleccionada del grupo que consiste en cromatografía de intercambio iónico, cromatografía de interacciones hidrófobas y cromatografía de afinidad, en donde las fibras se preparan mediante un método que incluye la extrusión por fusión de fibras de dos componentes en configuración núcleo/envoltura con una relación núcleo/envoltura de 50:50, en donde la envoltura está formada por poli(ácido láctico) y el núcleo está formado por una mezcla de nailon y poli(ácido láctico), en donde los polímeros son inmiscibles, el estiramiento de las fibras y subsecuentemente la extracción del poli(ácido láctico) de dicha envoltura y dicha mezcla con una solución de hidróxido de sodio, en donde la carga de poli(ácido láctico) en la mezcla es mayor del 35 % en peso y menor del 50 % en peso de la mezcla polimérica.
2. Los medios de acuerdo con la reivindicación 1, en donde dicha funcionalidad es la funcionalidad de intercambio catiónico.
3. Los medios de acuerdo con la reivindicación 1, en donde dicha funcionalidad es una funcionalidad de intercambio aniónico.
4. Los medios de acuerdo con la reivindicación 1, en donde las superficies de dichas nanofibrillas están modificadas con grupos sulfopropilo colgantes.
5. Un método para purificar una biomolécula en una muestra, que comprende poner en contacto dicha muestra con los medios de cromatografía de cualquiera de las reivindicaciones 1 a 4.
6. El método de la reivindicación 5, en donde dicha biomolécula comprende un virus.
7. El método de la reivindicación 5, en donde dicha funcionalidad se injerta en dichas nanofibrillas.
8. El método de la reivindicación 5, en donde dicha funcionalidad permite la purificación en un modo de unión/elución.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462044630P | 2014-09-02 | 2014-09-02 | |
| PCT/US2015/045873 WO2016036508A1 (en) | 2014-09-02 | 2015-08-19 | High surface area fiber media with nano-fibrillated surface features |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2877563T3 true ES2877563T3 (es) | 2021-11-17 |
Family
ID=55440257
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15838269T Active ES2877563T3 (es) | 2014-09-02 | 2015-08-19 | Medios de cromotografía que comprenden conjuntos porosos discretos de nanofibrillas |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US10449517B2 (es) |
| EP (1) | EP3188816B1 (es) |
| JP (2) | JP6652554B2 (es) |
| KR (1) | KR102072529B1 (es) |
| CN (1) | CN106794389A (es) |
| CA (1) | CA2954425C (es) |
| ES (1) | ES2877563T3 (es) |
| SG (1) | SG11201700030UA (es) |
| WO (1) | WO2016036508A1 (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101997543B1 (ko) | 2010-07-30 | 2019-07-09 | 이엠디 밀리포어 코포레이션 | 크로마토그래피 매질 및 방법 |
| ES2684527T3 (es) | 2013-02-14 | 2018-10-03 | Nanopareil, Llc | Fieltro híbrido de nanofibras electrohiladas, método de preparación del mismo y método de purificación de biomoléculas |
| US20170298091A1 (en) | 2014-12-08 | 2017-10-19 | Emd Millipore Corporation | Mixed Bed Ion Exchange Adsorber |
| US11214789B2 (en) | 2016-05-03 | 2022-01-04 | Flodesign Sonics, Inc. | Concentration and washing of particles with acoustics |
| US20190284321A1 (en) * | 2016-07-18 | 2019-09-19 | North Carolina State University | Heat-Induced Grafting Of Nonwovens For High Capacity Ion Exchange Separation |
| GB201616758D0 (en) * | 2016-10-03 | 2016-11-16 | Ge Healthcare Bio Sciences Ab | Novel chromatography media |
| KR101863955B1 (ko) * | 2016-12-01 | 2018-06-01 | 강원대학교 산학협력단 | 역미셀을 이용하여 다공성 나노섬유를 제조하는 방법 및 상기 방법에 의해 제조된 다공성 나노섬유 |
| US20210229004A1 (en) * | 2020-01-23 | 2021-07-29 | Jonell filtration Products, Inc. | Tubular filter with nonwoven media and method |
| CN114717749A (zh) * | 2022-04-19 | 2022-07-08 | 东华大学 | 含有毛刷结构微/纳米纤维的水刺非织造材料及其制备方法 |
| DE102023107371A1 (de) | 2023-03-23 | 2024-09-26 | Constructor University Bremen Ggmbh | Funktionalisierte, polymer-konjugierte Chromatographiemedien auf Polyamidbasis |
| WO2025072286A1 (en) * | 2023-09-25 | 2025-04-03 | The Regents Of The University Of Michigan | Chromatographic separation of chiral biological and man-made nanostructures on a chiral stationary phase |
Family Cites Families (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3382305A (en) | 1954-10-29 | 1968-05-07 | Du Pont | Process for preparing oriented microfibers |
| US3936394A (en) | 1972-04-13 | 1976-02-03 | Asahi Kasei Kogyo Kabushiki Kaisha | Polymer adsorbents and method for manufacture thereof |
| US4169790A (en) | 1975-05-02 | 1979-10-02 | South African Inventions Development Corporation | Solid surface texture suitable for a stationary phase for chromatography |
| US4384957A (en) | 1980-09-08 | 1983-05-24 | Amf Incorporated | Molecular separation column and use thereof |
| US4512897A (en) | 1981-07-28 | 1985-04-23 | Amf Incorporated | Molecular separation column and use thereof |
| US4496461A (en) | 1983-06-17 | 1985-01-29 | Amf Incorporated | Chromatography column |
| US4657742A (en) | 1985-07-01 | 1987-04-14 | Ppg Industries, Inc. | Packed fiber glass reaction vessel |
| US4721572A (en) | 1985-07-12 | 1988-01-26 | Miles Laboratories, Inc. | Purfication of blood clotting factors and other blood proteins on non-carbohydrate sulfated matrices |
| AU598606B2 (en) | 1986-11-27 | 1990-06-28 | Unitika Ltd. | Adsorptive fiber sheet |
| US5162074A (en) | 1987-10-02 | 1992-11-10 | Basf Corporation | Method of making plural component fibers |
| US5360540A (en) | 1990-04-23 | 1994-11-01 | Andelman Marc D | Chromatography system |
| US5354472A (en) | 1991-12-04 | 1994-10-11 | Cobe Cardiovascular, Inc. | Anion exchange materials comprised of derivatized cellulose-polyester composites |
| JPH05239709A (ja) | 1992-02-24 | 1993-09-17 | Toyobo Co Ltd | 極細超異形繊維 |
| US5468847A (en) | 1994-03-10 | 1995-11-21 | Minnesota Mining And Manufacturing Company | Method of isolating and purifying a biomacromolecule |
| US5502022A (en) | 1994-05-16 | 1996-03-26 | Biosepra, Inc. | Chromatography adsorbents utilizing mercapto heterocyclic ligands |
| JPH08108069A (ja) | 1994-10-07 | 1996-04-30 | Toyobo Co Ltd | 幹細胞分離方法 |
| JPH08170958A (ja) | 1994-12-19 | 1996-07-02 | Makoto Kawabata | 分離剤 |
| FR2737730B1 (fr) | 1995-08-10 | 1997-09-05 | Pasteur Merieux Serums Vacc | Procede de purification de virus par chromatographie |
| AUPN520395A0 (en) | 1995-09-04 | 1995-09-28 | Steripak Pty Ltd | Manufacturing process for polymers with fractal structure |
| US5906747A (en) | 1995-11-13 | 1999-05-25 | Biosepra Inc. | Separation of molecules from dilute solutions using composite chromatography media having high dynamic sorptive capacity at high flow rates |
| DE69733268T2 (de) | 1996-01-30 | 2006-01-19 | Novagent Oy | Zusammensetzung für die kontrollierte Freisetzung eines Arzneimittels bei der transdermalen Verabreichung |
| US5800706A (en) | 1996-03-06 | 1998-09-01 | Hyperion Catalysis International, Inc. | Nanofiber packed beds having enhanced fluid flow characteristics |
| AU714882B2 (en) | 1996-04-09 | 2000-01-13 | Therakos, Inc. | Method for removal of psoralens from biological fluids |
| CN1211199C (zh) * | 1996-05-15 | 2005-07-20 | 海珀里昂催化国际有限公司 | 刚性多孔碳结构材料、其制法、用法及含该结构材料的产品 |
| KR100522108B1 (ko) | 1996-05-15 | 2006-01-27 | 하이페리온 커탤리시스 인터내셔널 인코포레이티드 | 고표면적나노섬유 |
| ATE336610T1 (de) * | 1996-05-15 | 2006-09-15 | Hyperion Catalysis Int | Nanofasern mit grossen oberflächen |
| US5948528A (en) | 1996-10-30 | 1999-09-07 | Basf Corporation | Process for modifying synthetic bicomponent fiber cross-sections and bicomponent fibers thereby produced |
| AT406373B (de) | 1997-02-27 | 2000-04-25 | Immuno Ag | Verfahren zur reinigung von faktor viii/vwf-komplex mittels kationenaustauscherchromatographie |
| US5886154A (en) | 1997-06-20 | 1999-03-23 | Lebing; Wytold R. | Chromatographic method for high yield purification and viral inactivation of antibodies |
| JP2002500088A (ja) | 1998-01-12 | 2002-01-08 | ハー マジェスティ イン ライト オブ カナダ アズ リプレゼンティッド バイ ザ ミニスター オブ アグリカルチャー アンド アグリ−フード カナダ | 非極性抽出物の単離、回収及び精製方法 |
| ES2247791T3 (es) | 1998-02-27 | 2006-03-01 | Bia Separations D.O.O. | Cromatografo. |
| JPH11279945A (ja) | 1998-03-23 | 1999-10-12 | Japan Atom Energy Res Inst | 放射線グラフト重合されたポリエチレン材料 |
| FR2787465A1 (fr) | 1998-12-21 | 2000-06-23 | Transgene Sa | Procede d'inactivation des virus enveloppes dans une preparation virale de virus non enveloppes |
| CA2356373A1 (fr) | 1998-12-31 | 2000-07-13 | Aventis Pharma S.A. | Methode de separation de particules virales |
| TW522042B (en) | 1999-10-20 | 2003-03-01 | Honeywell Int Inc | Devices and methods for chemical reactive filtration |
| DE19962577A1 (de) | 1999-12-23 | 2001-07-12 | Tittgen Biotechnologie Dr | Chromatographiematerial und Verfahren unter Verwendung desselben |
| DK1285080T3 (da) | 2000-05-31 | 2008-12-01 | Novartis Vaccines & Diagnostic | Fremgangsmåde til oprensning af alphavirusreplikonpartikler |
| JP4520621B2 (ja) | 2000-11-01 | 2010-08-11 | 信和化工株式会社 | クロマトグラフィー用分離カラム、固相抽出用媒体、及びクロマトグラフィーの試料注入システム |
| GB2377447B (en) | 2001-04-18 | 2003-06-25 | Amersham Biosciences Ab | Isolation of dna molecules |
| CN100347355C (zh) | 2001-06-15 | 2007-11-07 | 可乐丽股份有限公司 | 复合纤维 |
| US6838005B2 (en) | 2001-06-22 | 2005-01-04 | Frederick Tepper | Nanosize electropositive fibrous adsorbent |
| US7374673B2 (en) | 2001-09-10 | 2008-05-20 | Clemson University | Channeled polymer fibers as stationary/support phases for chemical separation by liquid chromatography and for waste stream clean-up |
| CN1285779C (zh) | 2001-09-28 | 2006-11-22 | 纳幕尔杜邦公司 | 可拉伸非织造纤网及其制造方法 |
| WO2003066669A2 (en) | 2002-02-04 | 2003-08-14 | Millipore Corporation | Process for removing protein aggregates and virus from a protein solution |
| SE0200543D0 (sv) | 2002-02-21 | 2002-02-21 | Amersham Biosciences Ab | Method of separation using aromatic thioether ligands |
| US20030180936A1 (en) * | 2002-03-15 | 2003-09-25 | Memarzadeh Bahram Eric | Method for the purification, production and formulation of oncolytic adenoviruses |
| CN1312335C (zh) | 2002-08-05 | 2007-04-25 | 东丽株式会社 | 多孔纤维 |
| US7510848B2 (en) | 2003-04-04 | 2009-03-31 | North Carolina State University | Prion protein binding materials and methods of use |
| EP1651331B1 (en) | 2003-08-05 | 2007-05-30 | Millipore Corporation | Electrodeionization module and apparatus comprising it |
| WO2005021844A2 (en) | 2003-08-21 | 2005-03-10 | Filtrona Richmond, Inc. | Polymeric fiber rods for separation applications |
| US7622306B2 (en) | 2004-04-20 | 2009-11-24 | Board Of Regents, The University Of Texas System | Using plasma proteomic pattern for diagnosis, classification, prediction of response to therapy and clinical behavior, stratification of therapy, and monitoring disease in hematologic malignancies |
| DE602005021485D1 (de) | 2004-05-07 | 2010-07-08 | Wisconsin Alumni Res Found | Verfahren und zusammensetzungen mit molkeproteinisolaten |
| US7261813B2 (en) | 2004-08-10 | 2007-08-28 | Clemson University | Monolithic structures comprising polymeric fibers for chemical separation by liquid chromatography |
| CA2576221A1 (en) | 2004-08-13 | 2006-02-16 | Mcmaster University | Composite material comprising a non-crosslinked gel polymer |
| US20060070950A1 (en) | 2004-10-01 | 2006-04-06 | 3M Innovative Properties Company | Composite filtration article |
| EP1815029A4 (en) | 2004-11-03 | 2009-06-10 | Introgen Therapeutics Inc | NEW PROCESS FOR THE PRODUCTION AND PURIFICATION OF ADENOVIRAL VECTORS |
| US7311825B2 (en) | 2005-05-02 | 2007-12-25 | Varian, Inc. | Polymer modified porous substrate for solid phase extraction |
| WO2007053235A1 (en) | 2005-11-04 | 2007-05-10 | Sachem, Inc. | Cation-exchange displacement chromatography process and cationic organic compounds for use as displacer compounds in cation-exchange displacement chromatography process |
| CN101390415B (zh) | 2005-12-28 | 2012-07-18 | 梵提克斯公司 | 用于多媒体会话的多用户实时代码转换系统与方法 |
| US7490272B2 (en) * | 2006-06-07 | 2009-02-10 | Oxlo Systems | Method for validating the proper operation of a transactional management system |
| JP2007325543A (ja) * | 2006-06-08 | 2007-12-20 | Toray Ind Inc | 細胞足場材料およびその製造方法 |
| US20100069815A1 (en) | 2006-06-27 | 2010-03-18 | Asahi Kasei Kabushiki Kaisha | Substrate for biological fluid treatment |
| EP1878791A1 (en) | 2006-07-11 | 2008-01-16 | Bia Separations D.O.O. | Method for influenza virus purification |
| KR101340932B1 (ko) | 2006-08-31 | 2013-12-13 | 도레이 카부시키가이샤 | 복합 섬유를 포함하는 흡착 담체 |
| EP2066418A4 (en) | 2006-09-29 | 2011-12-28 | Ge Healthcare Bio Sciences Ab | SEPARATION MATRIX FOR VIRAL PURIFICATION |
| US8129019B2 (en) | 2006-11-03 | 2012-03-06 | Behnam Pourdeyhimi | High surface area fiber and textiles made from the same |
| WO2008058349A1 (en) | 2006-11-17 | 2008-05-22 | Gareth Michael Forde | Materials, methods and systems for purification and/or seperation |
| JP4805132B2 (ja) * | 2006-12-27 | 2011-11-02 | 日本水産株式会社 | キチン・キトサン系材料を含む積層体 |
| CN101036876B (zh) * | 2007-01-25 | 2010-09-01 | 天津大学 | 超大孔纤维素微球蛋白质吸附介质的制备方法 |
| FR2911883B1 (fr) | 2007-01-29 | 2009-05-01 | Intissel Soc Par Actions Simpl | Procede de greffage par rayonnement ionisant au moyen d'une molecule tensioactive reactive, substrat textile et separateur de batterie obtenus par greffage |
| US9433922B2 (en) | 2007-08-14 | 2016-09-06 | Emd Millipore Corporation | Media for membrane ion exchange chromatography based on polymeric primary amines, sorption device containing that media, and chromatography scheme and purification method using the same |
| US8710111B2 (en) | 2007-11-09 | 2014-04-29 | 3M Innovative Properties Company | Porous polymeric resins |
| US20090130738A1 (en) | 2007-11-19 | 2009-05-21 | Mikhail Kozlov | Media for membrane ion exchange chromatography |
| JP2011016036A (ja) | 2007-11-26 | 2011-01-27 | Asahi Kasei Chemicals Corp | タンパク吸着材料およびその製造方法 |
| CN102257134B (zh) | 2008-02-12 | 2014-03-05 | 赛诺菲巴斯德有限公司 | 使用离子交换和凝胶过滤色谱进行痘病毒纯化的方法 |
| CN102083897B (zh) * | 2008-05-30 | 2013-06-12 | 3M创新有限公司 | 制备配体官能化基底的方法 |
| CN102105213B (zh) | 2008-05-30 | 2013-12-18 | 3M创新有限公司 | 配体官能化基底 |
| JP2011523986A (ja) | 2008-06-10 | 2011-08-25 | パソジェン リムーバル アンド ダイアグノスティック テクノロジーズ インコーポレーテッド | ポリマー不織布の改質方法、及び改質ポリマー不織布 |
| CN102215865B (zh) | 2008-09-24 | 2013-10-30 | 米迪缪尼有限公司 | 病毒纯化方法 |
| MX2011004201A (es) | 2008-10-20 | 2011-05-24 | Abbott Lab | Desactivacion viral durante purificacion de anticuerpos. |
| ES2488542T3 (es) | 2008-12-22 | 2014-08-27 | F. Hoffmann-La Roche Ag | Purificación de inmunoglobulinas |
| CN101768206B (zh) | 2008-12-31 | 2013-05-15 | 华北制药集团新药研究开发有限责任公司 | 一种重组人血清白蛋白的纯化方法及其应用 |
| WO2010096704A2 (en) | 2009-02-19 | 2010-08-26 | Alex Garfield Bonner | Porous interpenetrating polymer network |
| JP5716297B2 (ja) | 2009-06-25 | 2015-05-13 | Jnc株式会社 | クロマトグラフィー用充填剤、その製造方法、およびそれを用いたウイルス用ワクチンの製造方法 |
| WO2011038053A1 (en) * | 2009-09-25 | 2011-03-31 | Armark Authentication Technologies, Llc | Tissue fiber scaffold and method for making |
| US20110142863A1 (en) | 2009-12-16 | 2011-06-16 | Millipore Corporation | Flow through purification processes for large biomolecules |
| US20120077249A1 (en) * | 2010-04-20 | 2012-03-29 | Millipore Corporation | Separation Of Virus And/Or Protein From Nucleic Acids By Primary Amines |
| US8300995B2 (en) * | 2010-06-30 | 2012-10-30 | Jds Uniphase Corporation | M X N WSS with reduced optics size |
| KR101997543B1 (ko) * | 2010-07-30 | 2019-07-09 | 이엠디 밀리포어 코포레이션 | 크로마토그래피 매질 및 방법 |
| US8889572B2 (en) | 2010-09-29 | 2014-11-18 | Milliken & Company | Gradient nanofiber non-woven |
| US8795561B2 (en) | 2010-09-29 | 2014-08-05 | Milliken & Company | Process of forming a nanofiber non-woven containing particles |
| US20120074611A1 (en) | 2010-09-29 | 2012-03-29 | Hao Zhou | Process of Forming Nano-Composites and Nano-Porous Non-Wovens |
| US20120077406A1 (en) | 2010-09-29 | 2012-03-29 | Scrivens Walter A | Nanofiber Non-Wovens Containing Particles |
| US20120077405A1 (en) | 2010-09-29 | 2012-03-29 | Hao Zhou | Core/Shell Nanofiber Non-Woven |
| WO2012044382A1 (en) | 2010-09-29 | 2012-04-05 | Milliken & Company | Process of forming nano-composites and nano-porous non-wovens |
| WO2012051147A1 (en) | 2010-10-11 | 2012-04-19 | Abbott Laboratories | Processes for purification of proteins |
| GB2485384A (en) * | 2010-11-12 | 2012-05-16 | Ngee Ann Polytechnic | Porous fibre encapsulating biological material |
| WO2012068442A1 (en) | 2010-11-19 | 2012-05-24 | Pathogen Removal And Diagnostic Technologies Inc. | High-surface area fibers and nonwoven membranes for use in bioseparations |
| WO2012141791A1 (en) | 2011-02-22 | 2012-10-18 | Avaxia Biologics, Inc. | Polyclonal antibody compositions |
| US9487566B2 (en) | 2011-06-29 | 2016-11-08 | Kyowa Hakko Kirin Co., Ltd | Method for purifying protein |
| US8722757B2 (en) | 2011-07-22 | 2014-05-13 | Ut-Battelle, Llc | Fiber-based adsorbents having high adsorption capacities for recovering dissolved metals and methods thereof |
| JP2013253949A (ja) * | 2011-10-21 | 2013-12-19 | Kuraray Co Ltd | セシウムを吸着する濾材及びその濾材から形成された浄水フィルタ |
| GB201119192D0 (en) * | 2011-11-07 | 2011-12-21 | Ucl Business Plc | Chromatography medium |
| US20130115837A1 (en) | 2011-11-09 | 2013-05-09 | Dale S. Kitchen | Nanofiber nonwovens and nanofiber nonwoven composites containing roped fiber bundles |
| US20130112625A1 (en) * | 2011-11-09 | 2013-05-09 | Pradipkumar Bahukudumbi | Consolidated nanofiber nonwovens and consolidated nanofiber nonwoven composites containing roped fiber bundles |
| WO2013123137A1 (en) * | 2012-02-16 | 2013-08-22 | Cornell University | Ordered porous nanofibers, methods, and applications |
| SG10201701224UA (en) * | 2012-03-12 | 2017-04-27 | Merck Patent Gmbh | Removal of protein aggregates from biopharmaceutical preparations in a flowthrough mode |
| HUP1200172A2 (en) | 2012-03-19 | 2013-10-28 | Richter Gedeon Nyrt | Methods for refolding g-csf from inclusion bodies |
| US9650411B2 (en) | 2012-08-07 | 2017-05-16 | Kyowa Hakko Kirin Co., Ltd. | Method of purifying protein |
| US9284663B2 (en) * | 2013-01-22 | 2016-03-15 | Allasso Industries, Inc. | Articles containing woven or non-woven ultra-high surface area macro polymeric fibers |
| SG11201505464QA (en) | 2013-01-31 | 2015-08-28 | Emd Millipore Corp | Chromatography media for purifying vaccines and viruses |
| ES2684527T3 (es) * | 2013-02-14 | 2018-10-03 | Nanopareil, Llc | Fieltro híbrido de nanofibras electrohiladas, método de preparación del mismo y método de purificación de biomoléculas |
| JP6320429B2 (ja) * | 2013-02-22 | 2018-05-09 | エイジェンシー・フォー・サイエンス,テクノロジー・アンド・リサーチ | 負帯電粒子を用いるウイルス調製物のクロマトグラフィー精製 |
| US20170298091A1 (en) | 2014-12-08 | 2017-10-19 | Emd Millipore Corporation | Mixed Bed Ion Exchange Adsorber |
-
2015
- 2015-08-19 ES ES15838269T patent/ES2877563T3/es active Active
- 2015-08-19 SG SG11201700030UA patent/SG11201700030UA/en unknown
- 2015-08-19 CN CN201580047255.6A patent/CN106794389A/zh active Pending
- 2015-08-19 EP EP15838269.7A patent/EP3188816B1/en active Active
- 2015-08-19 WO PCT/US2015/045873 patent/WO2016036508A1/en not_active Ceased
- 2015-08-19 CA CA2954425A patent/CA2954425C/en active Active
- 2015-08-19 US US15/323,471 patent/US10449517B2/en not_active Expired - Fee Related
- 2015-08-19 KR KR1020177000772A patent/KR102072529B1/ko not_active Expired - Fee Related
- 2015-08-19 JP JP2017511980A patent/JP6652554B2/ja not_active Expired - Fee Related
-
2020
- 2020-01-21 JP JP2020007673A patent/JP6922006B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CA2954425A1 (en) | 2016-03-10 |
| KR20170015997A (ko) | 2017-02-10 |
| EP3188816A4 (en) | 2018-05-16 |
| CN106794389A (zh) | 2017-05-31 |
| JP6652554B2 (ja) | 2020-02-26 |
| EP3188816B1 (en) | 2021-06-09 |
| JP6922006B2 (ja) | 2021-08-18 |
| US10449517B2 (en) | 2019-10-22 |
| SG11201700030UA (en) | 2017-02-27 |
| EP3188816A1 (en) | 2017-07-12 |
| WO2016036508A1 (en) | 2016-03-10 |
| JP2020089882A (ja) | 2020-06-11 |
| US20170165638A1 (en) | 2017-06-15 |
| JP2017534839A (ja) | 2017-11-24 |
| KR102072529B1 (ko) | 2020-02-25 |
| CA2954425C (en) | 2019-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2877563T3 (es) | Medios de cromotografía que comprenden conjuntos porosos discretos de nanofibrillas | |
| CN108404453B (zh) | 层析介质和方法 | |
| CN113195519B (zh) | 亲和膜及其制备方法 | |
| ES2621945T3 (es) | Medio de cromatografía | |
| ES2596583T3 (es) | Método de separación usando perlas adsorbentes porosas asimétricas | |
| EP3897696B1 (en) | Extracorporeal devices for methods for treating diseases associated with anti-neutrophil cytoplasmic antibodies | |
| KR20190051946A (ko) | 혈액정화용 재료 | |
| WO2010005364A1 (en) | Separation medium for chromatography of various biomolecules | |
| EP3367093B1 (en) | Affinity chromatography carrier and its use in a biological substance purification method | |
| WO2011046494A1 (en) | Separation matrices | |
| KR20150099846A (ko) | 백신 및 바이러스를 정제하기 위한 크로마토그래피 매질 | |
| Zhu et al. | Bio-functionalized nanofibrous membranes as a hybrid platform for selective antibody recognition and capturing | |
| Sakata et al. | γ-Cyclodextrin–polyurethane copolymer adsorbent for selective removal of endotoxin from DNA solution | |
| KR20120123578A (ko) | 리간드 고정화용 기재 및 그 제조 방법 | |
| JP6064402B2 (ja) | 抗体単量体の分離用分離剤及び分離方法 | |
| CN121873169A (zh) | 一种腺病毒载体纯化介质及其制备方法和应用 |