ES2877659T3 - Agonista del receptor de glucocorticoides y sus inmunoconjugados - Google Patents
Agonista del receptor de glucocorticoides y sus inmunoconjugados Download PDFInfo
- Publication number
- ES2877659T3 ES2877659T3 ES18833113T ES18833113T ES2877659T3 ES 2877659 T3 ES2877659 T3 ES 2877659T3 ES 18833113 T ES18833113 T ES 18833113T ES 18833113 T ES18833113 T ES 18833113T ES 2877659 T3 ES2877659 T3 ES 2877659T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- tnfa
- formula
- mmol
- drug
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCCC(C)C(*)(CC)CC(NCC(N[C@@](CCCCN)C(Nc1cccc(Cc2ccc([C@](O[C@@]3C[C@@]([C@](CC4)[C@@]5[C@@](C)(C=C6)C4=CC6=O)[C@]4(C)C[C@@]5O)O[C@]34C(CO*(O)O)=O)cc2)c1)=O)=O)=O Chemical compound CCCC(C)C(*)(CC)CC(NCC(N[C@@](CCCCN)C(Nc1cccc(Cc2ccc([C@](O[C@@]3C[C@@]([C@](CC4)[C@@]5[C@@](C)(C=C6)C4=CC6=O)[C@]4(C)C[C@@]5O)O[C@]34C(CO*(O)O)=O)cc2)c1)=O)=O)=O 0.000 description 4
- KGDYKUMSFJMVQR-UHFFFAOYSA-N CCC(NCC(NC(CCC(O)=O)C(C)=O)=O)=O Chemical compound CCC(NCC(NC(CCC(O)=O)C(C)=O)=O)=O KGDYKUMSFJMVQR-UHFFFAOYSA-N 0.000 description 1
- HONOLJNPEQFUGW-WWZAFPNPSA-N C[C@@]1(C[C@@H]2O)[C@]3(C(COP(O)(O)=O)=O)O[C@H](C4=CCC(Cc5ccccc5)C=C4)O[C@@H]3C[C@H]1[C@H](C[C@@H]1F)C2[C@@](C)(C=C2)C1=CC2O Chemical compound C[C@@]1(C[C@@H]2O)[C@]3(C(COP(O)(O)=O)=O)O[C@H](C4=CCC(Cc5ccccc5)C=C4)O[C@@H]3C[C@H]1[C@H](C[C@@H]1F)C2[C@@](C)(C=C2)C1=CC2O HONOLJNPEQFUGW-WWZAFPNPSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/191—Tumor necrosis factors [TNF], e.g. lymphotoxin [LT], i.e. TNF-beta
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6845—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a cytokine, e.g. growth factors, VEGF, TNF, a lymphokine or an interferon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/241—Tumor Necrosis Factors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/249—Interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Rheumatology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Steroid Compounds (AREA)
Abstract
Un conjugado de anticuerpo-fármaco que comprende: (a) un anticuerpo anti-TNFα que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4; y (b) un agonista del receptor de glucocorticoides que comprende un radical representado por la fórmula: **(Ver fórmula)** en donde el anticuerpo se conjuga con el agonista del receptor de glucocorticoides mediante un enlazador representado por la fórmula: **(Ver fórmula)**
Description
DESCRIPCIÓN
Agonista del receptor de glucocorticoides y sus inmunoconjugados
Solicitudes relacionadas
Esta solicitud reclama prioridad a la Solicitud Provisional de EE. UU. núm. 62/593,776, presentada el 1 de diciembre de 2017 y la Solicitud Provisional de EE. UU. núm. 62/595,054, presentada el 5 de diciembre de 2017.
Listado de secuencias
La presente solicitud contiene un Listado de Secuencias que se ha enviado electrónicamente en formato ASCII y se incorpora aquí como referencia en su totalidad. Dicha copia ASCII, creada el 28 de noviembre de 2018, se denomina A103017_1490WO_SL.txt y tiene un tamaño de 14758 bytes.
Campo técnico
El Factor de Necrosis Tumoral a (TNFa) juega un papel central en la fisiopatología de varios trastornos humanos, y los agentes anti-TNFa tienen una utilidad terapéutica clínicamente validada en el tratamiento de trastornos autoinmunitarios e inflamatorios, tales como la artritis reumatoide, la psoriasis y la enfermedad inflamatoria intestinal. A pesar de su éxito en la clínica, los biológicos anti-TNFa todavía tienen la eficacia máxima que pueden lograr en los pacientes limitada, lo que requiere la identificación y el desarrollo de terapias más potentes y efectivas. Los pacientes tratados con biológicos anti-TNFa también pueden desarrollar una respuesta inmunogénica al terapéutico, lo que limita su eficacia. Por lo tanto, las terapias anti-TNFa con menor inmunogenicidad y alta eficacia serían útiles para controlar aún más la enfermedad.
Los agonistas del receptor de glucocorticoides sintéticos son una clase potente de moléculas pequeñas que se usan en el tratamiento de trastornos inflamatorios, pero su utilidad en el tratamiento crónico de enfermedades está limitada debido a los efectos secundarios graves. Existe la necesidad de desarrollar agentes terapéuticos con una eficacia mejorada y una acción de mayor duración en comparación con los anticuerpos anti-TNF y con mínimos efectos no deseados.
Se conoce de Patel, Dermatologic Therapy, Vol. 17, 2004, 427-431, que el anticuerpo anti-TNFa adalimumab se emplea en el tratamiento de la artritis reumatoide y la psoriasis.
Resumen
La presente descripción proporciona inmunoconjugados agonistas del receptor de glucocorticoides útiles para el tratamiento de enfermedades autoinmunitarias 11.
La invención para la cual se solicita protección se define mediante las reivindicaciones. Cualquier tema que quede fuera del alcance de las reivindicaciones se proporciona solo con fines informativos. Cualquier referencia en la descripción a métodos de tratamiento se refiere a los conjugados de la presente invención para usar en un método para el tratamiento del cuerpo humano o animal mediante terapia.
En un aspecto, la presente descripción proporciona un conjugado de anticuerpo-fármaco que comprende: (a) un anticuerpo anti-TNFa que comprende una cadena pesada indicada como s Eq ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4; y (b) un agonista del receptor de glucocorticoides que comprende un radical representado por la fórmula:

y en donde el anticuerpo se conjuga con el agonista del receptor de glucocorticoides mediante un enlazador representado por la fórmula:

En un aspecto, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es el anticuerpo y n es un número entero de 1-10.
En un aspecto, la presente descripción proporciona un conjugado de anticuerpo-fármaco que comprende:
(a) un anticuerpo anti-TNFa que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4; y (b) un agonista del receptor de glucocorticoides que comprende un radical representado por la fórmula:

y en donde el anticuerpo se conjuga con el agonista del receptor de glucocorticoides mediante un enlazador representado por la fórmula:

En un aspecto, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es el anticuerpo y n es un número entero de 1-10.
En un aspecto, la presente descripción proporciona el conjugado de anticuerpo-fármaco de cualquier aspecto anterior, en donde la carga de fármaco es 1, 2, 3, 4, 5, 6, 7, 8, 9 o 10. En un aspecto, la presente descripción proporciona el conjugado de anticuerpo-fármaco de cualquier aspecto anterior, en donde la carga de fármaco es 4, por ejemplo, n en la fórmula anterior del conjugado anticuerpo-fármaco es igual a 4. En un aspecto, la presente descripción proporciona el conjugado de anticuerpo-fármaco de cualquier aspecto anterior en donde la carga de fármaco es 2, por ejemplo, n en la fórmula anterior de conjugado de anticuerpo-fármaco es igual a 2.
En un aspecto, la presente descripción proporciona un método para preparar el conjugado de anticuerpo-fármaco de cualquier aspecto anterior, que comprende la etapa de conjugar el anticuerpo con el agonista del receptor de glucocorticoides. En un aspecto, la presente descripción proporciona el método del aspecto anterior, que comprende además la etapa de introducir un resto PO4 en el agonista del receptor de glucocorticoides antes de conjugar el anticuerpo con el agonista del receptor de glucocorticoides. En un aspecto, la presente descripción proporciona el método de cualquier aspecto anterior, en donde la conjugación comprende reducir parcialmente el anticuerpo y alquilar el anticuerpo parcialmente reducido con un compuesto de acuerdo con la fórmula:

En un aspecto, la presente descripción proporciona una composición farmacéutica que comprende el conjugado de anticuerpo-fármaco de cualquier aspecto anterior y un portador farmacéuticamente aceptable. En un aspecto, la presente descripción proporciona la composición farmacéutica de cualquiera de los aspectos anteriores, que comprende una relación de fármaco a anticuerpo (DAR) de 1-10.
En una modalidad preferida, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es adalimumab y n es 4.
En una modalidad preferida, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es un anticuerpo anti-TNFa que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4, y n es 4.
En otra modalidad preferida, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es adalimumab y n es 2. En otra modalidad preferida, la presente descripción proporciona un conjugado de anticuerpo-fármaco de acuerdo con la fórmula:

en donde A es un anticuerpo anti-TNFa que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4, y n es 2.
En un aspecto preferido, la presente descripción proporciona la composición farmacéutica de cualquier aspecto anterior, que comprende una relación de fármaco a anticuerpo (DAR) de 2,0.
En un aspecto preferido, la presente descripción proporciona la composición farmacéutica de cualquier aspecto anterior, que comprende una relación de fármaco a anticuerpo (DAR) de 4,0.
En un aspecto, la presente descripción proporciona el conjugado de anticuerpo-fármaco de cualquier aspecto anterior o la composición farmacéutica de cualquier aspecto anterior para usar en el tratamiento de una afección seleccionada entre artritis reumatoide, espondilitis anquilosante, artritis psoriásica, psoriasis en placas, colitis ulcerativa, enfermedad de Crohn, enfermedad de Crohn pediátrica, uveítis, hidradenitis supurativa y artritis idiopática juvenil.
En un aspecto, la presente descripción proporciona el uso del conjugado de anticuerpo-fármaco de cualquier aspecto anterior o la composición farmacéutica de cualquier aspecto anterior para la preparación de un medicamento para tratar una afección seleccionada entre artritis reumatoide, espondilitis anquilosante, artritis psoriásica, psoriasis en placas, colitis ulcerativa, enfermedad de Crohn del adulto, enfermedad de Crohn pediátrica, uveítis, hidradenitis supurativa y artritis idiopática juvenil.
En un aspecto, la presente descripción proporciona un kit que comprende: (a) un recipiente que comprende el conjugado de anticuerpo-fármaco de cualquier aspecto anterior o la composición farmacéutica de cualquier aspecto anterior; y (b) una etiqueta o prospecto en o asociado con el uno o más envases, en donde la etiqueta o prospecto indica que el conjugado de anticuerpo-fármaco o composición farmacéutica se usa para tratar una afección seleccionada entre artritis reumatoide, espondilitis anquilosante, artritis psoriásica, psoriasis en placas, colitis ulcerativa, enfermedad de Crohn del adulto, enfermedad de Crohn pediátrica, uveítis, hidradenitis supurativa y artritis idiopática juvenil.
En un aspecto, la presente descripción proporciona un método para administrar un agonista del receptor de glucocorticoides a una célula que expresa TNFa, que comprende la etapa de poner en contacto la célula con el conjugado de anticuerpo-fármaco de cualquier aspecto anterior. En un aspecto, la presente descripción proporciona un método para determinar la actividad antiinflamatoria de un conjugado de anticuerpo-fármaco que comprende: (a) poner en contacto una célula que expresa TNFa con el conjugado de anticuerpo-fármaco de cualquier aspecto anterior; y (b) determinar la liberación de citocinas proinflamatorias de la célula en comparación con una célula de control.
Breve descripción de los dibujos
La Figura 1 proporciona una resolución cromatográfica del modulador del receptor de BrAc-Gly-Glu-glucocorticoide (GRM)-PO4, como se realizó y describió en el Ejemplo 7. Como se muestra, el ADC es una mezcla de ADC heterogénea que contiene ADC con dos moléculas enlazadoras de fármacos unidas y ADC con cuatro moléculas enlazadoras de fármacos unidas.
La Figura 2 expone datos MS deconvolucionados de adalimumab conjugado con BrAc-Gly-Glu-glucocorticosteroide-PO4. Como se muestra, se logró la conjugación.
La Figura 3 proporciona un gráfico que demuestra la eficacia de una dosis alta y baja de ADC1 en comparación con el mAb anti-TNFa (dosis alta) o vehículo en un modelo de artritis inducida por colágeno (CIA) en ratón, como se realizó y describió en el Ejemplo 7. Como se muestra, una dosis única de glucocorticosteroide ADC1 anti-TNFa exhibió una duración de acción prolongada a través de la mejora de la hinchazón de la pata durante ~28 días en comparación con mAb anti-TNFa o vehículos solos.
La Figura 4 es un gráfico de concentración (ug/mL) para ADC de anillo abierto y cerrado en monos cynomolgus a lo largo del tiempo, como se realizó y describió en el Ejemplo 7. Como se muestra, la forma de anillo cerrado es susceptible a la reacción de Michael inversa y la subsiguiente pérdida de fármaco-enlazador in vivo.
Descripción detallada
En la presente descripción se proporcionan inmunoconjugados agonistas del receptor de glucocorticoides, agonistas del receptor de glucocorticoides y métodos para preparar y usar los mismos.
La invención para la cual se solicita protección se define mediante las reivindicaciones. Cualquier tema que quede fuera del alcance de las reivindicaciones se proporciona solo con fines informativos.
En la presente descripción se proporciona un conjugado de anticuerpo fármaco de acuerdo con la fórmula:

en donde A es adalumimab y n es 4. Como se demuestra en el Ejemplo 7 más abajo, este ADC (es decir, ADC4 más abajo) demuestra actividad in vitro, estabilidad en plasma y agregación mínima.
También se proporcionan métodos de fabricación y métodos de uso de ADC4.
I. Definiciones
Para facilitar la comprensión de la presente descripción, más abajo se definen varios términos y frases.
El término "proteína anti-TNFa" se refiere a proteínas que son capaces de (i) unirse a TNFa e (ii) inhibir la unión de TNFa soluble a receptores de TNF de superficie celular (p55 y/o p75) y/o lisar las células que expresan TNFa de superficie o receptor de TNFa in vitro en presencia de complemento. En algunos aspectos, el anticuerpo anti-TNF, puede unirse al TNF alfa en la superficie de una célula e internalizarse. Por ejemplo, el documento US 2014/0294813 describe anticuerpos anti-TNF que exhiben internalización celular al unirse al TNF humano de la superficie celular. Las proteínas anti-TNFa incluyen, por ejemplo, anticuerpos anti-TNFa (por ejemplo, adalimumab, infliximab y golimumab). Los anticuerpos anti-TNFa se internalizan activamente al unirse al TNF transmembrana en las DC derivadas de monocitos y entran rápidamente en los lisosomas donde se degradan. (Deora y otros, MABS, 2017, Vol. 9, No. 4, 680-694).
El término "anticuerpo", como se usa en la presente descripción, pretende hacer referencia a moléculas de inmunoglobulina compuestas por cuatro cadenas polipeptídicas, dos cadenas pesadas (H) y dos cadenas ligeras (L) interconectadas por enlaces disulfuro. Cada cadena pesada está compuesta por una región variable de cadena pesada (abreviada aquí como HCVR o VH) y una región constante de cadena pesada. La región constante de la cadena pesada se compone de tres dominios, CHI, CH2 y CH3. Cada cadena ligera está compuesta por una región variable de cadena ligera (abreviada aquí como LCVR o VL) y una región constante de cadena ligera. La región constante de la cadena ligera está compuesta por un dominio, CL. Las regiones VH y VL pueden subdividirse, además, en regiones de hipervariabilidad, denominadas regiones determinantes de complementariedad (CDR), intercaladas con regiones que están más conservadas, denominadas regiones marco (FR). Cada VH y VL está compuesto por tres CDR y cuatro FR, dispuestos desde el extremo amino al extremo carboxi en el siguiente orden: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
El término "anticuerpo anti-TNFa" o "un anticuerpo que se une a TNFa" se refiere a un anticuerpo que es capaz de unirse a TNFa, por ejemplo, con suficiente afinidad como para que el anticuerpo sea útil como agente terapéutico para dirigirse al TNFa. El grado de unión de un anticuerpo anti-TNFa a una proteína no relacionada, no TNFa puede ser menor que aproximadamente el 10 % de la unión del anticuerpo a TNFa medida, por ejemplo, mediante un radioinmunoensayo (RIA). En cierto aspecto, un anticuerpo que se une a TNFa tiene una constante de disociación (Kd) de <1 pM, <100 nM, <10 nM, <1 nM, or <0,1 nM.
El término "inmunoconjugado," "conjugado," "conjugado anticuerpo-fármaco" o "ADC" como se usa en la presente descripción, se refiere a un compuesto o derivado del mismo que está conjugado con una proteína, como un agente de unión celular (por ejemplo, un anti-anticuerpo TNFa). Dichos inmunoconjugados pueden definirse mediante una fórmula genérica: (SM-LQ)n-A, en donde SM = radical derivado de un agonista del receptor de glucocorticoides de molécula pequeña, por ejemplo, un glucocorticosteroide, L = enlazador, Q = grupo heterobifuncional o está ausente y A = una proteína (por ejemplo, un anticuerpo), y n = 1-10. Los inmunoconjugados también pueden definirse mediante la fórmula genérica en orden inverso: A-(Q-L-SM)n.
En la presente descripción, el término "enlazador" se refiere a un resto químico capaz de enlazar la proteína anti-TNFa (por ejemplo, un anticuerpo) a un glucocorticosteroide. Los enlazadores pueden ser susceptibles de escisión (un "enlazador escindible") lo que facilita así la liberación del glucocorticosteroide. Por ejemplo, tales enlazadores escindibles pueden ser susceptibles de escisión inducida por peptidasa, en condiciones en las que el glucocorticosteroide y/o el anticuerpo permanecen activos.
En particular, el componente enlazador escindible descrito en la presente descripción comprende un péptido que comprende de dos a tres residuos de aminoácidos (un dipéptido o tripéptido) y específicamente a dipéptidos y tripéptidos seleccionados del grupo que consiste en alanina-alanina (Ala-Ala), glicina-ácido glutámico (Gly-Glu), ácido glutámico-alanina-alanina (Glu-Ala-Ala) y glicina-lisina (Gly-Lys). El péptido permite la escisión del enlazador por una proteasa, lo que facilita así la liberación del glucocorticosteroide tras la exposición a proteasas intracelulares, tales como enzimas lisosomales (Doronina y otros, (2003) Nat. Biotechnol. 21:778-784).
En la presente descripción, el término "glucocorticosteroide" se refiere a una hormona esteroidea de origen natural o sintético que interactúa con los receptores de glucocorticoides, y los glucocorticosteroides específicos se describen en detalle en la presente descripción. Un "radical de un glucocorticosteroide" se obtiene mediante la eliminación de uno o más átomos de hidrógeno de un glucocorticosteroide original. La eliminación de los átomos de hidrógeno facilita la unión del glucocorticosteroide original a un enlazador. En la presente descripción, el átomo de hidrógeno se elimina de cualquier grupo -NH2 adecuado del glucocorticosteroide original. En particular, el "radical de un glucocorticosteroide" es un radical monovalente derivado de la eliminación de un átomo de hidrógeno de un glucocorticosteroide original.
En la presente descripción, el término "grupo heterobifuncional" se refiere a un resto químico que conecta el enlazador y la proteína anti-TNFa (por ejemplo, anticuerpo). Los grupos heterobifuncionales se caracterizan por tener diferentes grupos reactivos en cada extremo del resto químico.
El término "relación fármaco-anticuerpo" o "DAR" se refiere al número de SM (por ejemplo, radicales derivados de un
agonista del receptor de glucocorticoides de molécula pequeña, por ejemplo, un glucocorticoide) unidos a A (por ejemplo, un anticuerpo). Así, en el inmunoconjugado que tiene la fórmula genérica (SM-LQ)n-A, el DAR se define por la carga de fármaco por conjugado de anticuerpo-fármaco, por ejemplo, "n".
Cuando se hace referencia a un compuesto que tiene la fórmula (SM-LQ)n-A que representa un inmunoconjugado individual, el término "compuesto DAR" se refiere al número de SM vinculados al A individual (por ejemplo, carga de fármaco o n como un número entero de 1 a 10).
Cuando se hace referencia a un compuesto que tiene la fórmula (SM-LQ)n-A que representa una población de inmunoconjugados, el término "población DAR" se refiere al número promedio de SM enlazados a los A (por ejemplo, carga de fármaco o n como un número entero o fracción de 1 a 10 ± 0,5, ± 0,4, ± 0,3, ± 0,2, ± 0,1).
El término "sujeto" se refiere a humanos, primates no humanos y similares, que debe ser el receptor de un tratamiento particular. Típicamente, los términos "sujeto" y "paciente" se usan indistintamente en la presente descripción en referencia a un sujeto humano.
El término "formulación farmacéutica" se refiere a una preparación que está en una forma tal que permite que la actividad biológica del ingrediente activo sea eficaz y que no contiene componentes adicionales que sean inaceptablemente tóxicos para un sujeto al que se administraría la formulación. La formulación puede ser estéril. Una "cantidad eficaz" de un inmunoconjugado como se describe en la presente descripción es una cantidad suficiente para llevar a cabo un propósito específicamente establecido. Puede determinarse una "cantidad efectiva" en relación con el propósito establecido.
El término "cantidad terapéuticamente eficaz" se refiere a una cantidad de un inmunoconjugado eficaz para "tratar" una enfermedad o trastorno en un sujeto o mamífero. Una "cantidad profilácticamente eficaz" se refiere a una cantidad eficaz para lograr el resultado profiláctico deseado.
Términos como "tratar" o "tratamiento" o "tratar" o "aliviar" se refieren a medidas terapéuticas que curan, ralentizan, disminuyen uno o más síntomas de, y/o ralentizan o detienen la progresión de una afección patológica diagnosticada o trastorno ("tratamiento terapéutico"). Por tanto, los que necesitan tratamiento terapéutico incluyen a aquellos ya diagnosticados o que se sospecha que tienen el trastorno. Las medidas profilácticas o preventivas se refieren a medidas que previenen el desarrollo de una afección o trastorno patológico específico ("tratamiento profiláctico"). Por tanto, aquellos que necesitan un tratamiento profiláctico incluyen aquellos propensos a padecer el trastorno y aquellos en los que se debe prevenir el trastorno.
II. Proteínas para la unión a agonistas del receptor de glucocorticoides
La presente descripción proporciona inmunoconjugados que contienen agonistas del receptor de glucocorticoides unidos a proteínas, por ejemplo, anticuerpos. En algunos aspectos, el anticuerpo es humano, humanizado, quimérico o murino. En algunos aspectos, la proteína, por ejemplo, el anticuerpo, puede unirse a una diana en la superficie de una célula e internalizarse.
La presente descripción también proporciona inmunoconjugados que contienen agonistas del receptor de glucocorticoides unidos a proteínas anti-TNFa. En ciertos aspectos, las proteínas anti-TNFa son anticuerpos. En ciertos aspectos, las proteínas anti-TNFa son anticuerpos que se unen a TNFa (por ejemplo, TNFa soluble y/o TNFa unido a membrana). En ciertos aspectos, las proteínas anti-TNFa son proteínas del receptor de TNF soluble, por ejemplo, proteínas del receptor de TNF soluble fusionadas a una constante de cadena pesada. En algunos aspectos, la proteína anti-TNFa, por ejemplo, el anticuerpo anti-TNFa, se une al TNFa en la superficie de una célula y se internaliza. Por ejemplo, la publicación de solicitud de patente de EE. UU. núm. 2014/0294813 describe proteínas anti-TNFa que exhiben internalización celular al unirse al TNFa humano de la superficie celular.
En ciertos aspectos, los anticuerpos se unen al TNFa humano y/o de ratón.
La secuencia de aminoácidos de longitud completa para el TNFa humano unido a la membrana es:
MSTESMIRDVELAEEALPKKTGGPQGSRRCLFLSLFSFLIVAGATTLFCLLHFGVIGPQRE EFPRDLSLISPLAQAVRSSSRTPSDKPVAHVVANPQAEGQLQWLNRRANALLANGVELRDNQLVVPSEGLYLIYSQVL FKGQGCPSTHVLLTHTISRIAVSYQTKVNLLSAIKSPCQRETPEGAEAKPWYEPIY LGGVFQLE-KGDRLSAEINRPDYLDFAESGQVYFGIIAL (SEQ ID NO:1). El TNFa soluble humano contiene los aminoácidos 77 233 de SEQ ID NO: 1. La secuencia de aminoácidos de longitud completa para el TNFa murino unido a la membrana es:
MSTESMIRDVELAEEALPQKMGGFQNSRRCLCLSLFSFLLVAGATTLFCLLNFGVIGPQRDEKFPNGLPLISSMAQTLT LRSSSQNSSDKPVAHVVANHQVEEQLEWLSQRANALLANGMDLKDNQLVVPADGLYLVYSQVLFKGQGCPDYVLLT HTVSRFAISYQEKVNLLSAVKSPCPKDTPEGAELKPWYEPIYLGGVFQLEKGDQLSAEVNLPKYLDFAESGQVYFGVI
AL (SEQ ID NO:2). El TNFa murino soluble contiene los aminoácidos 80-235 de SEQ ID NO: 2.
En algunos aspectos, el anticuerpo anti-TNFa se une al TNFa humano.
En algunos aspectos, el anticuerpo anti-TNFa se une al TNFa murino.
En ciertos aspectos, el anticuerpo anti-TNFa tiene uno o más de los siguientes efectos: neutraliza la citotoxicidad del TNFa humano en un ensayo de L929 in vitro con una IC50 de 1X10-7 M o menos; bloquea la interacción del TNFa con los receptores de la superficie celular p55 y p75; y/o lisa células que expresan TNF de superficie in vitro en presencia de complemento.
En ciertos aspectos, el anticuerpo anti-TNFa no se une a TNF-beta.
Los anticuerpos anti-TNFa incluyen, por ejemplo, adalimumab, que es un anticuerpo humano recombinante. Las secuencias de aminoácidos correspondientes a la CDR y las regiones variables de adalimumab se describen en la patente de EE.UU. núm. 6,258,562 con referencia al anticuerpo D2E7, es decir, SEQ ID Nos: 1 a 8.
La denominación común internacional (INN) adalimumab se proporciona en el sitio de listado de INN de la OMS: Año 2000, Lista 44 (Información sobre medicamentos de la OMS (2000) Vol. 14 (3)).
En ciertos aspectos, un anticuerpo anti-TNFa comprende secuencias de adalimumab, por ejemplo, las regiones determinantes de complementariedad (CDR), el dominio variable pesado (VH) y/o el dominio variable ligero (VL). Las secuencias ilustrativas se proporcionan en la Tabla 1.
Tabla 1: Secuencias ilustrativas de la región del anticuerpo adalimumab

En ciertos aspectos, el anticuerpo anti-TNFa comprende las CDR de las SEQ ID NO: 3 y 4. En algunos aspectos, las CDR comprenden las SEQ ID NO: 7 u 8, 9, 10 u 11, 12, 13 y 14. En ciertos aspectos, el anticuerpo anti-TNFa comprende la cadena pesada de SEQ ID NO: 3 y/o la cadena ligera de SEQ ID NO: 4.
La presente descripción abarca, además, variantes y equivalentes que son sustancialmente homólogos a los anticuerpos anti-TNFa expuestos en la presente descripción. Estos pueden contener, por ejemplo, mutaciones de sustitución conservadora, es decir, la sustitución de uno o más aminoácidos por aminoácidos similares. Por ejemplo,
la sustitución conservadora se refiere a la sustitución de un aminoácido por otro dentro de la misma clase general como, por ejemplo, un aminoácido ácido por otro aminoácido ácido, un aminoácido básico por otro aminoácido básico o un aminoácido neutro por otro aminoácido neutro. Lo que se pretende con una sustitución conservadora de aminoácidos se conoce bien en la técnica.
Los anticuerpos anti-TNFa aislados descritos en la presente descripción pueden producirse mediante cualquier método adecuado conocido en la técnica. Dichos métodos van desde métodos directos de síntesis de proteínas hasta la construcción de una secuencia de ADN que codifica secuencias polipeptídicas aisladas y que expresan esas secuencias en un hospedero transformado adecuado. En algunos aspectos, una secuencia de ADN se construye mediante el uso de tecnología recombinante al aislar o sintetizar una secuencia de ADN que codifica una proteína de interés de tipo silvestre. Opcionalmente, la secuencia puede mutagenizarse mediante mutagénesis específica de sitio para proporcionar sus análogos funcionales. Ver, por ejemplo, Zoeller y otros, Proc. Nat'l. Acad. Sci. USA 81:5662-5066 (1984) y la patente de EE.UU. núm. 4,588,585.
En algunos aspectos, se construiría una secuencia de ADN que codifica un anticuerpo de interés mediante síntesis química mediante el uso de un sintetizador de oligonucleótidos. Dichos oligonucleótidos pueden diseñarse con base en la secuencia de aminoácidos del polipéptido deseado y al seleccionar aquellos codones que están favorecidos en la célula hospedera en la que se producirá el polipéptido recombinante de interés. Pueden aplicarse métodos estándar para sintetizar una secuencia polinucleotídica aislada que codifica un polipéptido aislado de interés.
En ciertos aspectos, los vectores de expresión recombinantes se usan para amplificar y expresar ADN que codifica anticuerpos anti-TNFa. Puede emplearse una amplia variedad de combinaciones de expresión vector/hospedador. Los vectores de expresión útiles para hospedadores eucariotas incluyen, por ejemplo, vectores que comprenden secuencias de control de la expresión de SV40, virus del papiloma bovino, adenovirus y citomegalovirus. Los vectores de expresión útiles para hospedadores bacterianos incluyen plásmidos bacterianos conocidos, tales como plásmidos de Escherichia coli, que incluyen pCR 1, pBR322, pMB9 y sus derivados, plásmidos de más amplia gama de hospedadores, tales como M13 y fagos de ADN monocatenario filamentoso.
Las células hospedadoras adecuadas para la expresión de anticuerpos anti-TNFa incluyen células procariotas, levaduras, células de insectos o eucariotas superiores bajo el control de promotores apropiados. Los procariotas incluyen organismos gramnegativos o grampositivos, por ejemplo E. coli o bacilos. Las células eucariotas superiores incluyen líneas celulares establecidas de origen mamífero. También se podrían emplear sistemas de traducción sin células. Pouwels y otros describen vectores de clonación y expresión apropiados para usar con hospedadores celulares bacterianos, fúngicos, de levadura y de mamíferos. (Cloning Vectors: A Laboratory Manual, Elsevier, N.Y., 1985). Puede encontrarse información adicional con respecto a métodos de producción de proteínas, incluida la producción de anticuerpos, por ejemplo, en la Publicación de Patente de EE.UU. núm. 2008/0187954, las Patentes de EE.UU. núms. 6,413,746 y 6,660,501 y la Publicación de Patente Internacional núm. WO 04009823.
III. Inmunoconjugados que contienen agonistas del receptor de glucocorticoides
También se proporcionan inmunoconjugados que contienen agonistas del receptor de glucocorticoides. En algunos aspectos, un inmunoconjugado se une al receptor Fc gamma. En algunos aspectos, un inmunoconjugado es activo en el ensayo indicador de GRE y TNFa transmembrana (como se usa en la presente descripción el "ensayo indicador de GRE y TNFa transmembrana" se refiere al ensayo usado en el Ejemplo 7 más abajo). En algunos aspectos, un inmunoconjugado muestra una inmunogenicidad reducida (respuesta inmunitaria anti-fármaco reducida (ADA)) en comparación con la proteína en el inmunoconjugado (por ejemplo, el anticuerpo) solo.
En un aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a:
(SM-L-Q)n-A I-a,
en donde:
A es un anticuerpo a anti-factor de necrosis tumoral (TNF), un anticuerpo monoclonal anti-TNFa o adalimumab; L es un enlazador;
Q es un grupo heterobifuncional; o
Q está ausente;
n es 1-10; y
SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de:
(1) Fórmula Il-a:

(2) Fórmula ll-b:

(3) Fórmula II-c:

o
(4) Fórmula ll-d:

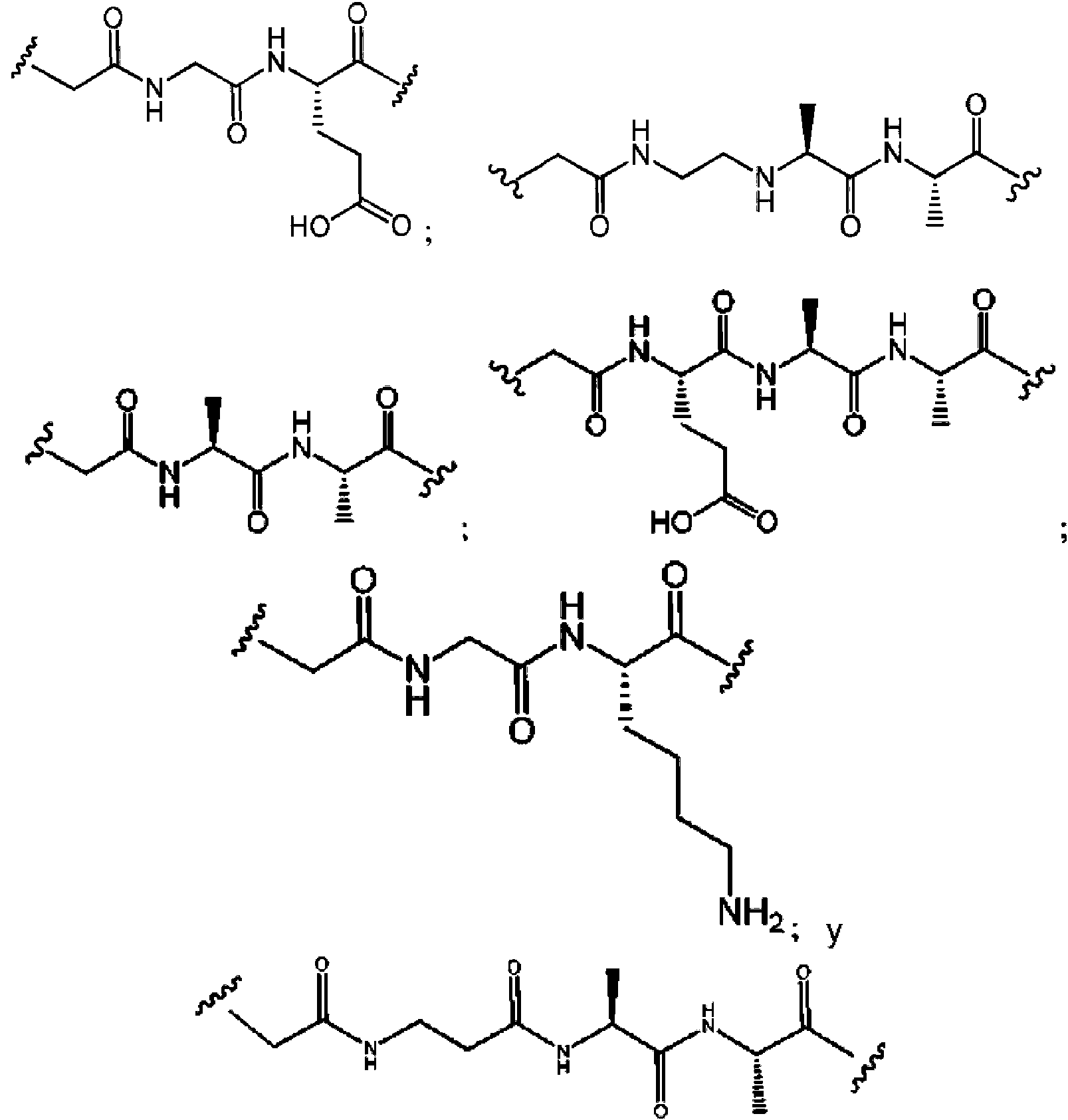
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas ll-a, ll-b, ll-c o ll-d, en donde L es un enlazador escindible que comprende un dipéptido o tripéptido, Q es un grupo heterobifuncional o Q está ausente y n es 1-10. En particular, L comprende un dipéptido o tripéptido seleccionado del grupo que consiste en alanina-alanina (Ala-Ala), glicina-ácido glutámico (Gly-Glu), ácido glutámico-alanina-alanina (Glu-Ala-Ala) y glicina -lisina (Gly-Lys).
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula l-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas ll-a, ll-b, ll-c y ll-d, en donde Q es un grupo heterobifuncional. representado por:

en donde m es 0 o 1.
En otro aspecto, m es 0 y Q está representado por:

En otro aspecto, m es 1 y Q está representado por:

En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas Il-a, Il-b, II-c y Il-d, en donde -LQ- es cualquiera de las estructuras químicas de la Tabla 2:
Tabla 2
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un
compuesto que tiene la Fórmula I-a en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas Il-a, Il-b, II-c y Il-d, en donde n es 2-8. En otro aspecto, n es 1-5. En otro aspecto, n es 2 5. En otro aspecto, n es 1. En otro aspecto, n es 2. En otro aspecto, n es 3. En otro aspecto, n es 4. En otro aspecto, n es 5. En otro aspecto, n es 6. En otro aspecto, n es 7. En otro aspecto, n es 8.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A es un anticuerpo.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde el anticuerpo es murino, quimérico, humanizado o humano. En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A inhibe competitivamente la unión de un anticuerpo seleccionado del grupo que consiste en adalimumab, infliximab, certolizumab pegol y golimumab a TNFa.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A se une al mismo epítopo de TNFa que un anticuerpo seleccionado del grupo que consiste en adalimumab, infliximab, certolizumab pegol, afelimomab, nerelimomab, ozoralizumab, placulumab y golimumab.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A comprende las secuencias de CDR1, CDR2 y CDR3 de cadena pesada variable SEQ ID NO: 7 u 8, SEQ ID NO: 9 y SEQ ID NO: 10 u 11, respectivamente, y las secuencias de CDR1, CDR2 y CDR3 de cadena ligera variable SEQ ID NO: 12, SEQ ID NO: 13 y SEQ ID NO: 14, respectivamente.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A es un anticuerpo anti- TNFa que comprende una región variable de cadena pesada indicada como SEQ ID NO: 5 y una región variable de cadena ligera indicada como SEQ ID NO: 6.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A es un anticuerpo anti-TNFa que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A bloquea la interacción de TNFa con los receptores de la superficie celular p55 y p75.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A lisa las células que expresan TNF de superficie in vitro en presencia de complemento.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, por ejemplo, un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A es etanercept.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, c.g., un compuesto que tiene la Fórmula I-a, en donde SM es un radical monovalente de un glucocorticosteroide que tiene cualquiera de las Fórmulas II-a, II-b, II-c y II- d, en donde A es adalimumab.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula I-a, que es cualquiera de las estructuras químicas de la Tabla 3:
Ċ
Tabla 3



en la presente descripción, n es 1-5 y A es adalimumab.
En otro aspecto, se describe en la presente descripción un conjugado de anticuerpo-fármaco de acuerdo con la
fórmula:

en donde A es adalumimab y n es 4. Como se demuestra en el Ejemplo 7 más abajo, este ADC (es decir, ADC4 más abajo) demuestra actividad in vitro, estabilidad en plasma y agregación mínima.
IV. Métodos para fabricar inmunoconjugados e intermediarios sintéticos
La síntesis general de varios inmunoconjugados de la descripción implica hacer reaccionar una pequeña molécula (SM) de NH2-funtionalizada de cualquiera de las Fórmulas III-a, III-b, III-c, o III-d más debajo con una porción de enlazador y funcionalizar el compuesto resultante para dar un intermediario funcionalizado de bromoacetamida. El intermediario funcionalizado de bromoacetamida se hace reaccionar luego con HS-A, en donde HS-A es un anticuerpo, por ejemplo adilumimab, que tiene un número limitado de enlaces disulfuro intercatenarios reducidos. (1) Fórmula III-a:

(2) Fórmula III-b:

(3) Fórmula III-c:

o
(4) Fórmula IlI-d:

En otro aspecto, se describe en la presente descripción un método para preparar un compuesto que tiene la Fórmula IV-a:

en donde:
A es adalimumab;
L es un enlazador;
n es 1-10; y
SM es un radical de un glucocorticosteroide que tiene cualquiera de las Fórmulas lll-a-lll-d;
el método comprende:
a) conjugar un compuesto que tiene la Fórmula V:
SM-L
Y ' Br v
con una proteína a anti-factor de necrosis tumoral (TNF) o una proteína; y
b) aislar el compuesto que tiene la Fórmula lV-a.
En algunos aspectos, el método descrito comprende además purificar el compuesto de Fórmula lV-a. En ciertos aspectos, se usa la cromatografía de intercambio aniónico (AEC), que (debido a restos cargados en la porción
peptídica del enlazador en algunos aspectos y/o el grupo fosfato en el SM en algunos aspectos) puede proporcionar una separación eficiente de diferentes especies de DAR.
En algunos aspectos, el método descrito requiere menos etapas sintéticas que los métodos que se basan en la química de enlace estándar basada en maleimida. En particular, los métodos que se basan en la química de enlace estándar basada en maleimida pueden requerir una etapa posterior de hidrólisis de apertura de anillo de succinimida que se realiza después de la purificación de las especies DAR apropiadas. Como tal, en ciertos aspectos, el método descrito acorta significativamente el protocolo de conjugación con respecto a la química estándar de enlace basada en maleimida.
En otro aspecto, se describe en la presente descripción un método para preparar un compuesto que tiene la Fórmula Vl-a:

en donde:
A es adalimumab; y
n es 1-10,
el método comprende:
a) conjugar un compuesto de Fórmula Vll-a:

con adalimumab parcialmente reducido; y
b) aislar, por ejemplo, mediante cromatografía, el compuesto que tiene la Fórmula Vl-a.
En otro aspecto, se describe en la presente descripción un método para preparar un compuesto que tiene la Fórmula IV-a o la Fórmula Vl-a, en donde n es 1-7. En otro aspecto, n es 1-5. En otro aspecto, n es 2-4. En otro aspecto, n es 1. En otro aspecto, n es 2. En otro aspecto, n es 3. En otro aspecto, n es 4. En otro aspecto, n es 5. En otro aspecto, n es 6. En otro aspecto, n es 7. En otro aspecto, n es 8.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula IV-a o Vl-a, en donde: A es adalimumab; y
n es 1-10.
En otro aspecto, se describe en la presente descripción un compuesto que tiene la Fórmula IV-a o Vl-a, en donde n es 1-7. En otro aspecto, n es 1-5. En otro aspecto, n es 2-4. En otro aspecto, n es 1. En otro aspecto, n es 2. En otro aspecto, n es 3. En otro aspecto, n es 4. En otro aspecto, n es 5. En otro aspecto, n es 6. En otro aspecto, n es 7. En otro aspecto, n es 8.
También se proporcionan en la presente descripción intermediarios sintéticos que son útiles para la preparación de inmunoconjugados.
En un aspecto, el intermediario sintético descrito en la presente descripción es un compuesto que tiene cualquiera de las fórmulas V o VlI-a.
VI. Métodos de uso y composiciones farmacéuticas
En la presente descripción se proporcionan conjugados que tienen la Fórmula I-a (por ejemplo, que tienen las fórmulas que se muestran en la Tabla 3) que pueden usarse in vitro o in vivo. En consecuencia, también se proporcionan composiciones, por ejemplo, composiciones farmacéuticas para ciertos usos in vivo, que comprenden un conjugado o un agonista del receptor de glucocorticoides que tiene el grado de pureza deseado en un portador, excipiente o estabilizador fisiológicamente aceptable (Remington's Pharmaceutical Sciences (1990) Mack Publishing Co., Easton, PA). Los portadores, excipientes o estabilizadores aceptables no son tóxicos para los receptores en las dosis y concentraciones empleadas.
Las composiciones (por ejemplo, composiciones farmacéuticas) que se usarán para la administración in vivo pueden ser estériles, lo que puede lograrse mediante filtración a través de, por ejemplo, membranas de filtración estériles. Las composiciones (por ejemplo, composiciones farmacéuticas) que se usarán para la administración in vivo pueden comprender un conservante.
Los conjugados de anticuerpo-fármaco y/o las composiciones farmacéuticas que comprenden conjugados de anticuerpo-fármaco descritos en la presente descripción pueden ser útiles para lisar una célula que expresa TNFa de superficie (in vitro o in vivo) y/o para el tratamiento de enfermedades o trastornos caracterizados por un aumento de TNFa (por ejemplo, TNFa aumentado en líquido sinovial). En algunos aspectos, los conjugados de anticuerpofármaco y/o composiciones son útiles para inhibir la liberación de citocinas (in vitro o in vivo) y/o para el tratamiento de enfermedades autoinmunes o inflamatorias. En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la enfermedad de Crohn (por ejemplo, enfermedad de Crohn activa de moderada a grave que afecta al íleon y/o el colon ascendente y/o el mantenimiento de la remisión clínica de la enfermedad de Crohn activa moderada a grave que afecta el íleon y/o el colon ascendente hasta por 3 meses). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la colitis ulcerativa (por ejemplo, para la inducción de la remisión en pacientes con colitis ulcerativa activa, de moderada a grave). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la artritis reumatoide (RA). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la artritis idiopática juvenil (JA). En algunos aspectos, los conjugados de anticuerpofármaco y/o composiciones se usan para el tratamiento de la artritis psoriásica (PsA). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de una espondiloartropatía como la espondilitis anquilosante (AS) o la espondiloartritis axial (axSpA). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la enfermedad de Crohn (CD) del adulto. En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la enfermedad de Crohn pediátrica. En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la colitis ulcerativa (UC). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la psoriasis en placas (Ps). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la hidradenitis supurativa (HS). En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la uveítis. En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la enfermedad de Behcet. En algunos aspectos, los conjugados de anticuerpo-fármaco y/o composiciones se usan para el tratamiento de la psoriasis, incluida la psoriasis en placas. Algunos aspectos comprenden el uso de conjugados de fármacos y/o composiciones farmacéuticas para la preparación de un medicamento para tratar las enfermedades o trastornos descritos en la presente descripción.
Algunos aspectos comprenden métodos para administrar un agonista del receptor de glucocorticoides a una célula que expresa TNFa. Dichos métodos pueden incluir una etapa de poner en contacto una célula que expresa TNFa con un conjugado de anticuerpo-fármaco como se describe en la presente descripción. Algunos aspectos comprenden un método in vitro para administrar un agonista del receptor de glucocorticoides a una célula que expresa TNFa.
También se proporcionan métodos para determinar la actividad antiinflamatoria de un conjugado de anticuerpofármaco. Dichos métodos pueden incluir una etapa de poner en contacto una célula que expresa TNFa con un conjugado de anticuerpo-fármaco como se describe en la presente descripción. Algunos aspectos comprenden poner en contacto una célula que expresa TNFa con un conjugado de anticuerpo-fármaco como se describe en la presente descripción y determinar la liberación reducida de citocinas proinflamatorias de la célula en comparación con una célula de control. Algunos aspectos comprenden un método in vitro para determinar la actividad antiinflamatoria de un conjugado de anticuerpo-fármaco.
Algunos aspectos comprenden métodos de cribado (por ejemplo, Métodos in vitro) que incluyen poner en contacto;
directa o indirectamente, células (por ejemplo, células que expresan TNFa) con un conjugado de anticuerpo-fármaco y determinar si el conjugado de anticuerpo-fármaco modula una actividad o función de las células, como se refleja, por ejemplo, por cambios en la morfología o viabilidad celular, expresión de un marcador, diferenciación o desdiferenciación, respiración celular, actividad mitocondrial, integridad de la membrana, maduración, proliferación, viabilidad, apoptosis o muerte celular. Un ejemplo de interacción directa es la interacción física, mientras que una interacción indirecta incluye, por ejemplo, la acción de una composición sobre una molécula intermediaria que, a su vez, actúa sobre la entidad referenciada (por ejemplo, célula o cultivo celular).
VII. Artículos de manufactura
La descripción también incluye paquetes y kits farmacéuticos que comprenden uno o más recipientes, en donde un recipiente puede comprender una o más dosis de un conjugado de anticuerpo-fármaco o composición como se describe en la presente descripción. En ciertos aspectos, el paquete o kit contiene una dosis unitaria, lo que significa una cantidad predeterminada de una composición o conjugado de anticuerpo-fármaco, con o sin uno o más agentes adicionales.
El kit puede comprender uno o varios recipientes y una etiqueta o prospecto en, sobre o asociado con el recipiente o recipientes, lo que indica que la composición adjunta se usa para tratar la enfermedad de elección. Los recipientes adecuados incluyen, por ejemplo, botellas, viales, jeringas, etc. Los recipientes pueden estar formados por una variedad de materiales tales como vidrio o plástico. Los recipientes pueden comprender un puerto de acceso estéril, por ejemplo, el recipiente puede ser una bolsa de solución intravenosa o un vial que tiene un tapón que puede perforarse con una aguja de inyección hipodérmica.
En algunos aspectos, el kit puede contener un medio por el cual administrar el anticuerpo y cualquier componente opcional a un paciente, por ejemplo, una o más agujas o jeringas (precargadas o vacías), un gotero, una pipeta u otro aparato similar, a partir del cual la formulación puede inyectarse o introducirse en el sujeto o aplicarse a un área enferma del cuerpo. Los kits de la descripción también incluirán típicamente un medio para contener los viales, o similares, y otros componentes en un confinamiento estrecho para la venta comercial, como, por ejemplo, envases de plástico moldeados por soplado en los que se colocan y se retienen los viales deseados y otros aparatos.
Ejemplos
Se entiende que los ejemplos y modalidades descritos en la presente descripción son solo para fines ilustrativos y que se sugerirán a los expertos en la técnica diversas modificaciones o cambios a la luz de los mismos y se incluirán dentro del espíritu y alcance de esta descripción.
Los materiales de partida están disponibles comercialmente, pueden prepararse mediante los procedimientos descritos en la presente descripción, mediante procedimientos en la bibliografía o mediante procedimientos que serían bien conocidos por un experto en la técnica de la química orgánica. Los nombres de los reactivos/reactantes indicados son los que aparecen en el frasco comercial o los generados por las convenciones de la IUPAC, CambridgeSoft® ChemDraw Ultra 12.0, CambridgeSoft® Chemistry E-Notebook 11 o AutoNom 2000. Se entiende que los ejemplos y modalidades descritos en la presente descripción son solo para fines ilustrativos y que se sugerirán a los expertos en la técnica diversas modificaciones o cambios a la luz de los mismos y se incluirán dentro del espíritu y alcance de esta descripción.
Métodos Analíticos para la Síntesis y Caracterización de Compuestos
Los datos analíticos se incluyen dentro de los procedimientos más abajo, en las ilustraciones de los procedimientos generales o en las tablas de ejemplos. A menos que se indique de cualquier otra manera, todos los datos de RMN de 1H y 13C se recopilaron en un instrumento Varian Mercury Plus 400 MHz o Bruker AVIII 300 MHz; los cambios químicos se señalan en partes por millón (ppm). Los datos analíticos de HPLC se detallan dentro del experimento o se hace referencia a la tabla de condiciones de LC/MS y HPLC, mediante el uso del método proporcionado en la Tabla 4.
Tabla 4

Las abreviaturas usadas en los ejemplos siguientes son:

Ejemplo 1 (Ejemplo de referencia): Síntesis de (2S,6aS,6bR,7S,8aS,8bS,10S,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenyl)-2,6b-difluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12bdodecahidro-4H-nafto[2',1':4,5]mdeno[1,2-d][1,3]dioxol-4-ona
Etapa 1: Síntesis de 4-(bromometil)benzaldehído

Se añadió gota a gota hidruro de diisobutilaluminio (153 mL, 153 mmol, 1 M en tolueno) a una solución a 0 °C de 4-(bromometil) benzonitrilo (20 g, 102 mmol) en tolueno (400 mL) durante 1 hora. Se prepararon dos viales adicionales como se describió anteriormente. Se combinaron las tres mezclas de reacción. A la mezcla se le añadió HCl acuoso al 10 % (1,5 L). La mezcla se extrajo con DCM (3 X 500 mL). La capa orgánica se secó sobre Na2SÜ4, se filtró y se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (eluido con PE: EtoAc = 10: 1) para obtener el compuesto del título (50 g, rendimiento del 82 %) como un sólido blanco. 1H NMR (400MHz, CDCla) ó 10,02 (s, 1H), 7,91 - 7,82 (m, 2H), 7,56 (d, J=7,9 Hz, 2H), 4,55 - 4,45 (m, 2H).
Etapa 2: Síntesis de 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) anilina

A una solución de 3-bromoanilina (40 g, 233 mmol) en 1,4-dioxano (480 mL) se le añadió 4,4,4',4',5,5,5',5'-tetrametil-2,2'-bi (1,3,2-dioxaborolano) (94 g, 372 mmol), acetato de potasio (45,6 g, 465 mmol), 2-diciclohexilfosfino-2',4',6'-trii-propil-1, 1 '-bifenilo (8,07 g, 13,95 mmol) y tris(dibencilidenacetona) dipaladio (0) (8,52 g, 9,30 mmol). La mezcla resultante se calentó a 80 °C durante 4 horas bajo nitrógeno. Se preparó un vial adicional como se describió anteriormente. Las dos mezclas de reacción se combinaron y concentraron y el residuo se purificó por cromatografía en columna sobre gel de sílice (eluído con PE: EtOAc = 10:1) para obtener el compuesto del título (60 g, rendimiento 55,4 %) como un sólido amarillo claro. 1H NMR (400MHz, CDCla) ó 7,23 - 7,13 (m, 3H), 6,80 (d, J=7,5 Hz, 1H), 3,82 -3,38 (m, 2H), 1,34 (s, 12H).
Etapa 3: Síntesis de carbamato de (3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil) terc-butilo

oc
Se mezclaron 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) anilme (Ejemplo 1, Etapa 2) (30 g, 137 mmol) y dicarbonato de di-terc-butilo (38,9 g, 178 mmol) en tolueno (600 mL) a 100 °C durante 24 horas. Se preparó otro vial como se describió anteriormente. Las dos mezclas de reacción se combinaron y la mezcla se evaporó, se disolvió en EtOAc (1,5 L), se lavó con HCl 0,1 N (3 X 2 L) y salmuera (3 L), se secó sobre Na2SO4, se filtró y se concentró a una presión reducida para dar el compuesto del título (50 g, rendimiento del 57 %) como un sólido rojo. 1H NMR (400MHz, CDCla) d7,63 (br m, 2H), 7,48 (d, J=7,1 Hz, 1H), 7,37 - 7,28 (m, 1H), 1,52 (s, 9H), 1,34 (s, 12H).
Etapa 4: Síntesis de carbamato de (3-(4-formilbencil)fenil) terc-butilo

Una mezcla de 4-(bromometil)benzaldehído (Ejemplo 1, Etapa 1) (24,94 g, 125 mmol), complejo de 1,1 '-bis (difenilfosfino) ferrocenodicloro paladio (II) Dc M (13,75 g, 18,80 mmol), carbamato de (3- (4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) fenil)terc-butilo (del Ejemplo 1, Etapa 3) (20 g, 62,7 mmol) y carbonato de potasio (43,3 g, 313 mmol) en tetrahidrofurano (400 mL) se calentó a 80 °C durante 12 horas. Se preparó otro vial como se describió anteriormente. Las dos mezclas de reacción se combinaron y se diluyeron con agua (500 mL). La mezcla acuosa se extrajo con EtOAc (3 X 500 mL). Las capas orgánicas se combinaron y se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en columna sobre gel de sílice (eluído con PE: EtOAc = 10:1) para obtener el compuesto del título (15 g, rendimiento 38,4 %) como un sólido blanco. 1H NMR (400MHz, CDCla) d9,95 (s, 1H), 7,78 (d, J=7,9 Hz, 2H), 7,33 (d, J=7,9 Hz, 2H), 7,27 - 7,13 (m, 3H), 6,82 (d, J=7,1 Hz, 1H), 6,47 (br. s., 1H), 4,00 (s, 2H), 1,48 (s, 9H).
Etapa 5: Síntesis de (6S,8S,9R,10S,11S,13S,14S,16R,17S)-6,9-difluoro-11,16,17-trihidroxi-17-(2-hidroxiacetil)-10,13-dimetil-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-cyclopenta[a]fenantren-3-ona

Se suspendió (2S,6aS,6bR,7S,8aS,8bS,11aR,12aS,12bS)-2,6b-Difluoro-7-hidroxi-8b-(2-hidroxyacetil)-6a,8a,10,10-tetrametil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-4H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-4-ona (20 g, 44,2 mmol) en HBF4 acuoso al 40 % (440 mL) y la mezcla se agitó a 25 °C durante 48 horas. Una vez completada la reacción, se añadieron 2 L de agua y el sólido se recogió por filtración. Este sólido se lavó con agua (1 L) y luego con MeOH (200 mL) para dar el compuesto del título (11 g, rendimiento del 60,3 %) como un sólido blanco. 1H NMR (400MHz, DMSO-d6) d7,25 (d, J=10,1 Hz, 1H), 6,28 (d, J=10,1 Hz, 1H), 6,10 (s, 1H), 5,73 - 5,50 (m, 1H), 5,39 (br. s., 1H), 4,85 - 4,60 (m, 2H), 4,50 (d, J=19,4 Hz, 1H), 4,20 - 4,04 (m, 2H), 2,46 - 2,06 (m, 6H), 1,87 - 1,75 (m, 1H), 1,56 -1,30 (m, 6H), 0,83 (s, 3H).
Etapa 6: Síntesis de (2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenil)-2,6b-difluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-4H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-4-ona.

Una suspensión de (6S,8S,9R,10S,11S,13S,14S,16R,17S)-6,9-difluoro-11,16,17-trihidroxi-17-(2-hidroxi-acetil)-10,13-dimetil-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-3-ona (Ejemplo 1, Etapa 5) (4,4 g, 10,67 mmol) y MgSO4 (6,42 g, 53,3 mmol) en MeCN (100 mL) se agitó a 20 °C durante 1 hora. Se añadió una
solución de carbamato de (3-(4-formilbenzil)fenil) terc-butilo (Ejemplo 1, Etapa 4) (3,65 g, 11,74 mmol) en MeCN (100 mL) en una porción. Se añadió gota a gota ácido trifluorometanosulfónico (9,01 ml, 53,3 mmol) mientras se mantenía una temperatura interna por debajo de 25 °C mediante el uso de un baño de hielo. Después de la adición, la mezcla se agitó a 20 °C durante 2 horas. Se prepararon tres viales adicionales como se describió anteriormente. Las cuatro mezclas de reacción se combinaron y se concentraron y el residuo se purificó mediante HPLC preparativa para dar el compuesto del título (4,5 g, rendimiento del 14,2 %) como un sólido amarillo. LCMS (Método a, Tabla 4) Rt = 2,65 min; MS m/z = 606,2 (M+H)+; 1H NMR (400MHz, DMSO-d6) d7,44 - 7,17 (m, 5H), 6,89 (t, J=7,7 Hz, 1H), 6,44 - 6,25 (m, 4H), 6,13 (br. s., 1H), 5,79 - 5,52 (m, 2H), 5,44 (s, 1H), 5,17 - 4,89 (m, 3H), 4,51 (d, J=19,4 Hz, 1H), 4,25 - 4,05 (m, 2H), 3,73 (s, 2H), 3,17 (br. s., 1H), 2,75 - 2,55 (m, 1H), 2,36 - 1,97 (m, 3H), 1,76 - 1,64 (m, 3H), 1,59 -1,39 (m, 4H), 0,94 - 0,78 (m, 3H). Método de HPLC preparativa: Instrumento: sistema de HPLC semipreparativa Gilson 281; Fase móvil: A: Acido fórmico/H2O=0,01 % v/v; B: MeCN; Columna: Luna C18 150*25 5 micrones; Velocidad de flujo: 25 mL/min; Monitor de longitud de onda: 220 y 254 nm.

Ejemplo 2: Síntesis de (6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenil)-7-hidroxi-8b-(2-hidroxiaceti|)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-4H-nafto[2',1':4,5]mdeno[1,2-d][1,3]dioxol-4-ona.

El Ejemplo 2 se sintetizó en un procedimiento similar al Ejemplo 1 mediante el uso de (8S,9S,10R,11S,13S,14S,16R,17S)-11,16,17-trihidroxi-17-(2-hidroxiacetil)-10,13-dimetil-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-3-ona.
1H NMR (400MHz, DMSO-d6) d7,36 (d, J=7,9 Hz, 2H), 7,31 (d, J=10,1 Hz, 1H), 7,20 (d, J=7,9 Hz, 2H), 6,89 (t, J=7,9 Hz, 1H), 6,39 - 6,28 (m, 3H), 6,16 (dd, J=1,5, 9,9 Hz, 1H), 5,93 (s, 1H), 5,39 (s, 1H), 5,08 (t, J=5,7 Hz, 1H), 4,98 -4,87 (m, 3H), 4,78 (d, J=3,1 Hz, 1H), 4,49 (dd, J=6,2, 19,4 Hz, 1H), 4,29 (br. s., 1H), 4,17 (dd, J=5,5, 19,6 Hz, 1H), 3,74 (s, 2H), 2,61 - 2,53 (m, 1H), 2,36 - 2,26 (m, 1H), 2,11 (d, J=11,0 Hz, 1H), 2,07 (s, 1H), 2,02 (d, J=12,8 Hz, 1H), 1,83 - 1,54 (m, 5H), 1,39 (s, 3H), 1,16 - 0,96 (m, 2H), 0,85 (s, 3H). LCMS (Método a, Tabla 4) Rt = 2,365 min; m/z = 570,2 (M+H)+.
Ejemplo 3 (Ejemplo de Referencia): Síntesis de (6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenil)-6b-fluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12bdodecahidro-4H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-4-ona.

El Ejemplo 3 se sintetizó en un procedimiento similar al Ejemplo 1 mediante el uso de (8S,9R,10S,11S,13S,14S,16R,17S)-9-fluoro-11,16,17-trihidroxi-17-(2-hidroxiacetil)-10,13-dimetil-6,7,8,9,10,11,12,13,14,15,16,17-dodecahidro-3H-ciclopenta[a]fenantren-3-ona.
1H NMR (400MHz, DMSO-d6) d7,37 - 7,26 (m, 3H), 7,21 (d, J=7,9 Hz, 2H), 6,89 (t, J=7,7 Hz, 1H), 6,43 - 6,30 (m, 3H), 6,23 (d, J=10,1 Hz, 1H), 6,04 (s, 1H), 5,75 (s, 1H), 5,44 (s, 2H), 5,09 (t, J=5,7 Hz, 1H), 4,93 (br. s., 3H), 4,50 (dd, J=6,2, 19,4 Hz, 1H), 4,28 - 4,09 (m, 2H), 3,74 (s, 2H), 2,73 - 2,54 (m, 2H), 2,35 (d, J=13,2 Hz, 1H), 2,25 - 2,12 (m, 1H), 2,05 (d, J=15,0 Hz, 1H), 1,92 - 1,77 (m, 1H), 1,74 - 1,58 (m, 3H), 1,50 (s, 3H), 1,45 - 1,30 (m, 1H), 0,87 (s, 3H). LCMS (Método a, Tabla 4) Rt = 2,68 min; m/z = 588,1 (M+H)+
Ejemplo 4: Síntesis de ácido (S)-4-(2-(2-bromoacetamido)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-7-hidroxi-6a,8a-dimetil-4-oxo-8b-(2-(fosphonooxi)acetil)-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]mdeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-5-oxopentanoico
Etapa 1: Síntesis del ácido (S)-2-(2-((((9H-fluoren-9-yl)metoxi)carbonil)amino)acetamido)-5-(terc-butoxi)-5-oxopentanoico.

Una mezcla de resina de cloruro de 2-clorotritilo (30 g, 92 mmol), trietilamina (46,4 g, 458 mmol) y ácido (S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-5-(terc-butoxi)-5-oxopentanoico (25,5 g, 60 mmol) en DCM seco (200 mL) se burbujeó con N2 a 20 °C durante 8 horas. La mezcla se filtró y la resina se lavó con DCM (2 X 200 mL), MeOH ( 2 X 200 mL) y DMF (2 X 200 mL). A la resina se le añadió una solución de piperidina: DMF (1:4, 400 mL) y la mezcla se burbujeó con N2 durante 8 minutos y luego se filtró. Esta operación se repitió cinco veces para dar la eliminación completa del grupo protector Fmoc. La resina se lavó con DMF (5 X 500 mL) para producir ácido (S)-2-amino-5-(tercbutoxi)-5-oxopentanoico unido a resina. Una mezcla de ácido 2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acético (13,38 g, 45,0 mmol), W,W-diisopropiletilamina (7,86 ml, 45 mmol), hidroxibenzotriazol (6,89 g, 45 mmol), 2-(6-cloro-1H-benzo[d][1,2,3]triazol-1-il)-1,1,3,3-tetrametilisouronio hexafluorofosfato (V) (18,62 g, 45,0 mmol) en d Mf (200 mL) se agitó a 20 °C durante 30 min. A la mezcla se añadió el ácido (S)-2-amino-5-(terc-butoxi)-5-oxopentanoico unido a la resina y la mezcla resultante se burbujeó con N2 a 25 °C durante 1,5 horas. La mezcla se filtró y la resina se lavó con DMF (4 X 500 mL) y DCM (2 X 500 mL). A la mezcla se le añadió TFA al 1 % / DCM (5 x 500 mL) y se burbujeó con N2 durante 5 minutos. La mezcla se filtró y el filtrado se añadió directamente a una solución saturada de NaHCO3 (200 mL). La mezcla combinada se separó y la fase orgánica se lavó con una solución acuosa saturada de ácido cítrico (4 x 400 mL) y salmuera (2 x 300 mL). La solución orgánica final se secó sobre Na2SO4 (20 g), se filtró y se concentró a presión reducida para producir el compuesto del título (10 g, rendimiento del 20 %) en forma de un sólido amarillo claro.
1H NMR: (CDCla, 400 MHz) 6 = 7,75 (d, J = 7,5 Hz, 2H), 7,59 (br d, J = 7,5 Hz, 2H), 7,41 - 7,36 (m, 2H), 7,30 (t, J = 7,0 Hz, 2H), 5,82 (br s, 1H), 4,57 (br d, J = 4,8 Hz, 1H), 4,38 (br d, J = 7,5 Hz, 2H), 4,27 - 4,15 (m, 1H), 4,06 - 3,83 (m, 2H), 2,50 - 2,29 (m, 2H), 2,26 - 2,13 (m, 1H), 2,06 - 2,02 (m, 1H), 1,43 (s, 9H).
Etapa 2: Síntesis de terc-butyl (S)-4-(2-((((9H-fluoren-9-yl)metoxi)carbonil)amino)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-5-oxopentanoato
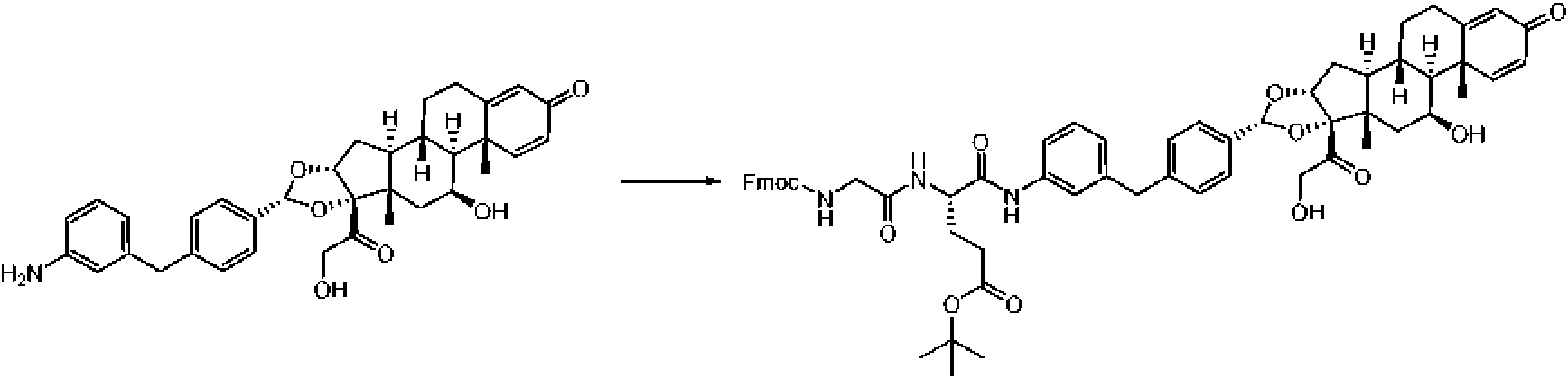
A una solución de ácido (S)-2-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-5-(terc-butoxi)-5-oxopentanoico (Ejemplo 4, Etapa 1) (424 mg, 0,878 mmol) en DMF (3,5 ml) se añadió (6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenil)-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-4H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-4-ona (Ejemplo 2) (500 mg, 0,878 mmol) y trietilamina (0,3 mL, 2,63 mmol) a 25 °C. La solución se enfrió a 0 °C y luego se añadió 2,4,6-tripropil-1,3,5,2,4,6-trioxatrifosfinano 2,4,6-trióxido (1,12 g, 1,755 mmol). La mezcla de reacción se agitó durante 12 horas a 25 °C. LCMS mostró que la reacción fue completa. Se prepararon catorce viales adicionales como se describió anteriormente. Se combinaron las quince mezclas de reacción. La mezcla se purificó mediante una columna de fase inversa para proporcionar el compuesto del título (5 g, rendimiento del 38,4 %) como un sólido amarillo. Método de columna de fase inversa: Instrumento: HPLC preparativa Shimadzu LC-8A; Columna: Phenomenex Luna C18200*40 mm*10 mm; Fase móvil: A para H2O (TfA al 0,05 %) y B para MeCN; Gradiente: B de 30 % a 100 % en 30 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm.
LCMS (Método a, Tabla 4) Rt = 1,34 min; m/z 1016,6 (M+H-18)+.
Etapa 3: Síntesis de terc-butil (S)-4-(2-((((9H-fluoren-9-yl)metoxi)carbonil)amino)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-5-oxopentanoato.

A una solución de terc-butil (S)-4-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-5-oxopentanoato (Ejemplo 4, etapa 2) (400 mg, 0,387 mmol) en DMF (2,5 mL) se añadió 1 H-tetrazol (271 mg 3,87 mmol) y di-terc-butil dietilfosforamidita (1,16 g, 4,64 mmol). La reacción se agitó a temperatura ambiente durante 2,5 horas y luego se enfrió a 0 °C. Se añadió peróxido de hidrógeno (241 mg, 2,127 mmol) a la mezcla resultante, se dejó calentar hasta temperatura ambiente y se agitó durante 1 hora, después de lo cual la LCMS mostró que la reacción fue completa. Se prepararon once viales adicionales como se describió anteriormente. Se combinaron las doce mezclas de reacción. La mezcla se purificó mediante una columna de fase inversa para proporcionar el compuesto del título (4,4 g, rendimiento del 64,2 %) como un sólido amarillo. Método de columna de fase inversa: Instrumento: HPLC preparativa Shimadzu LC-8A; Columna: Phenomenex Luna C18200*40mm*10 mm; Fase móvil: A para H2O y B para MeCN; Gradiente: B de 50 % a 100 % en 30 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm. LCMS (Método a, Tabla 4) Rt = 1,41 min; m/z 1226,7 (M+H)+.
Etapa 4: Síntesis de terc-butil (S)-4-(2-aminoacetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-do-decahidro-1H-nafto[2',1 ':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-5-oxopentanoato.

A una solución de terc-butil (S)-4-(2-((((9H-fluoren-9-yl)metoxi)carbonil)amino)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-5-oxopentanoato (Ejemplo 4, Etapa 3) (1,1 g, 0,897 mmol) en MeCN (6 ml) se añadió piperidina (0,75 mL, 7,58 mmol) a 25 °C. La reacción se agitó a temperatura ambiente durante 20 minutos, después de lo cual la LCMS mostró que la reacción fue completa. Se prepararon tres viales adicionales como se describió anteriormente. Se combinaron las cuatro mezclas de reacción. La mezcla se concentró para producir un residuo, que se trató con PE (10 ml) con agitación durante 2 horas. El sólido resultante se recogió por filtración y se secó a presión reducida para producir el compuesto del título (3,8 g, rendimiento del 90 %) como un sólido amarillo.
LCMS (Método a, Tabla 4) Rt = 1,16 min; m/z 1004,6 (M+H)+.
Etapa 5: Síntesis de terc-butil (S)-4-(2 (2-bromoacetamido) acetamido)-5-((3-(4-((6aR, 6bS, 7S, 8aS, 8bS, 10R, 11aR, 12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)bencil)fenil)amino)-5-oxopentanoato.

A una solución de ácido 2-bromoacético (97 mg, 0,697 mmol) en DMF (2,5 mL) se le añadió EEDQ (172 mg, 0,697 mmol) a temperatura ambiente. La mezcla se agitó a temperatura ambiente durante 1 hora. Se añadió terc-butil (S)-4- (2-aminoacetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',I':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)phenil)amino)-5-oxopentanoato (Ejemplo 4, Etapa 4) (350 mg, 0,349 mmol) y la solución resultante se agitó durante 2,5 horas, después de lo cual la LCMS mostró que la reacción fue completa. Se prepararon siete viales adicionales como se describió anteriormente. Se combinaron las ocho mezclas de reacción. La reacción se diluyó con DCM (100 mL), se lavó con HBr acuoso (1 M, 2 X 80 mL), NaHCO3 acuoso (60 mL), y salmuera (60 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir el compuesto del título (2 g, rendimiento 63,7 %) como un aceite amarillo.
LCMS (Método a, Tabla 4) Rt = 1,30 min; m/z 1124,2, 1125,9 (M+H)+.
Etapa 6: Síntesis de ácido (S)-4-(2-(2-bromoacetamido)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-7-hidroxi-6a,8a-dimetil-4-oxo-8b-(2-(fosfonooxi)acetil)-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d)[1,3]dioxol-10-yl)benzil)fenil)amino)-5- oxopentanoico

A una solución de terc-butil (S)-4-(2-(2-bromoacetamido)acetamido)-5-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)phenil)amino)-5-oxopentanoato (Ejemplo 4, Etapa 5) (2 g, 1,778 mmol) en DCM (16 mL) se añadió TFA (8 mL, 104 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 40 minutos, después de lo cual LCMS mostró que la reacción fue completa. El disolvente se eliminó a presión reducida. El residuo resultante se purificó mediante HPLC preparativa. La fase móvil se liofilizó directamente para producir el compuesto del título (640 mg, rendimiento del 35,3 %) como un sólido amarillo. Método de HPLC preparativa: Instrumento: HPLC preparativa Shimadzu LC-8A; Columna: Phenomenex Luna C18200*40 mm*10 pm; Fase móvil: A para H2O (TFA al 0,09 %) y B para MeCN; Gradiente: B de 30 % a 40 % en 20 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm.
1H NMR: (DMSO-d6, 400 MHz) ó = 9,88 (s, 1H), 8,52 (s, 1H), 8,24 (br d, J = 8,4 Hz, 1H), 7,46 (br d, J = 7,9 Hz, 1H), 7,42 (s, 1H), 7,36 (br d, J =7,9 Hz, 2H), 7,30 (br d, J = 9,7 Hz, 1H), 7,23 - 7,17 (m, 3H), 6,90 (br d, J = 6,8 Hz, 1H), 6,16 (br d, J = 10,4 Hz, 1H), 5,93 (s, 1H), 5,47 (s, 1H), 4,96 - 4,85 (m, 3H), 4,58 (br dd, J = 7,9, 18,7 Hz, 1H), 4,38 (br d, J = 5,3 Hz, 1H), 4,29 (br s, 1H), 3,93 (s, 2H), 3,89 (s, 2H), 3,80 (br s, 2H), 2,30 - 2,22 (m, 2H), 2,16 - 1,91 (m, 4H), 1,85 - 1,62 (m, 6H), 1,39 (s, 3H), 1,00 (br s, 2H), 0,87 (s, 3H). LCMS (Método a, Tabla 4) Rt = 2,86 min; m/z 956,0, 958,0 (M+H)+.
Ejemplo 5 (Ejemplo de referencia): Síntesis de 2-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-((S)-6-amino-2-(2-(2-bromoacetamido)acetamido)hexanamido)benzil)fenil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-1,2,4,6a,6b,7,8,8a,11a,12,12a,12b-dodecahidro-8bH-nafto[2',1':4,5]indeno[1,2-d][l,3]dioxol-8b-yl)-2-oxoetil dihidrógeno fosfato.
Etapa 1: Síntesis de carbamato de ((S)-5-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-2,6b-difluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1”:4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-6-oxohexil)terc-butilo.
A una solución de N2-((((9H-fluoren-9-il)methoxi)carbonil)glycyl)-N6-(terc-butoxycarbonil)-L-lisina (5,58 g, 8,26 mmol) en DMF (60 mL) a 0 °C se añadió 2,4,6-tripropyl-1,3,5,2,4,6-trioxatrifosfinan 2,4,6-trióxido (10,51 g, 16,51 mmol) y trietilamina (3,45 mL, 24,77 mmol). La mezcla resultante se agitó a temperatura ambiente durante 1 hora y se añadió (2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-aminobenzil)fenil)-2,6b-difluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-1,2,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-4H-nafto[2',1 ':4,5]mdeno[1,2-d][1,3]dioxol-4-ona (Ejemplo 1, Etapa 6) (5 g, 8,26 mmol). La mezcla resultante se agitó durante 5 horas a temperatura ambiente, después de lo cual la LCMS mostró que la reacción fue completa. Se prepararon seis viales adicionales como se describió anteriormente. Se combinaron las siete mezclas de reacción. La reacción se purificó mediante una columna de fase inversa para proporcionar el compuesto del título (24 g, rendimiento del 24,62 %) como un sólido blanco. Método de columna de fase inversa: Instrumento: HPLC preparativa Shimadzu LC-8A; Columna: Phenomenex Luna C18 200*40 mm*10 mm; Fase móvil: A para H2O (TfA al 0,05 %) y B para MeCN; Gradiente: B de 30 % a 100 % en 30 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm.
LCMS (Método a, Tabla 4) Rt = 1,29 min; m/z 1095,6 (M+H-18)+.
Etapa 2: Síntesis de carbamato de ((S)-5-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-yl)benzil)fenil)amino)-6-oxohexil)terc-butilo.

A una solución de carbamato de ((S)-5-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-2,6b-difluoro-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahydro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-6-oxohexil) terc-butilo (Ejemplo 5, Etapa 1) (3 g, 2,69 mmol) en DMF (30 mL) se añadió 1H-tetrazol (1,888 g, 26,9 mmol) y di-terc-butil dietilfosforamidito (8,06 g, 32,3 mmol) y la reacción se agitó a temperatura ambiente durante 3,5 horas. Se añadió peróxido de hidrógeno (224 mg, 1,97 mmol) a la reacción y se agitó durante 0,5 horas, después de lo cual la LCMS mostró que la reacción fue completa. Se prepararon seis viales adicionales como se describió anteriormente. Se combinaron las siete mezclas de reacción. La reacción se purificó mediante una columna de fase inversa para producir el compuesto del título (10 g, pureza: 78 %, rendimiento 37,1 %) como un sólido blanco. Método de columna de fase inversa: Instrumento: HPLC preparativa Shimadzu LC-8A; Columna: Phenomenex Luna C18 200*40mm*10 mm; Fase móvil: A para H2O y B para MeCN; Gradiente: B de 50 % a 100 % en 30 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm.
LCMS (Método a, Tabla 4) Rt = 1,42 min; m/z 1305,7 (M+H)+.
Etapa 3: Síntesis de carbamato de ((S)-5-(2-aminoacetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-6-oxohexil)terc-butilo.

A una solución de carbamato de ((S)-5-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)acetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)ammo)-6-oxohexil)terc-butilo (Ejemplo 5, Etapa 2) (2,5 g, 1,969 mmol) en MeCN (10 mL) se le añadió piperidina (2 mL, 1,969 mmol) y la reacción se agitó a temperatura ambiente durante 1 hora, después de lo cual la LCMS mostró que la reacción se fue completa. Se prepararon tres viales adicionales como se describió anteriormente. Se combinaron las cuatro mezclas de reacción. La reacción se concentró para proporcionar un producto bruto, que se agitó en PE (30 mL) durante 2 horas. El sólido resultante se recogió por filtración y se secó a presión reducida para producir el compuesto del título (7 g, pureza: 83 %, rendimiento 70,4 %) como un sólido amarillo.
LCMS (Método a, Tabla 4) Rt = 1,17 min; m/z 1083,5 (M+H)+.
Etapa 4: Síntesis de carbamato de ((S)-5-(2-(2-bromoacetamido)acetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxi)acetil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-6-oxohexil)terc-butilo.

A una solución de ácido 2-bromoacético (0,929 g, 6,68 mmol) en DMF (35 mL) se le añadió 2-etoxi-1-etoxicarbonil-1,2-dihidroquinolina (1,653 g, 6,68 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 1 hora. Se añadió el producto del Ejemplo 5, Etapa 3 (3,5 g, 3,34 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 2 horas. La LCMs mostró que la reacción fue completa. La reacción se diluyó con DCM (100 mL), se lavó con HBr acuoso (1 M, 2 X 80 mL), NaHCO3 acuoso (60 mL) y salmuera (60 mL). La capa orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir el compuesto del título (2 g, rendimiento del 51,2 %) como un aceite amarillo.
LCMS (Método a, Tabla 4) Rt = 1,32 min; m/z 1205,5 (M+H)+.
Etapa 5: Síntesis de 2-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-10-(4-(3-((S)-6-amino-2-(2-(2-bromoacetamido)acetamido)hexanamido)benzil)fenil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-1,2,4,6a,6b,7,8,8a,11a,12,12a,12b-dodecahidro-8bH-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-8b-il)-2-oxoetil dihidrógeno fosfato.

A una solución de carbamato de ((S)-5-(2-amiaoacetamido)-6-((3-(4-((2S,6aS,6bR,7S,8aS,8bS,10R,11aR,12aS,12bS)-8b-(2-((di-terc-butoxifosforil)oxy)acetil)-2,6b-difluoro-7-hidroxi-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-6-oxohexil)terc-butilo (Ejemplo 5, Etapa 3) (2 g, 1,661 mmol) en DCM (10 mL) se añadió TFA (5 mL, 64,9 mmol) y la reacción se agitó a temperatura ambiente durante 40 minutos, después del cual LCMS mostró que la reacción fue completa. El disolvente se eliminó a presión reducida y el producto crudo se purificó mediante HPLC preparativa. La fase móvil se liofilizó directamente para producir el compuesto del título (550 mg, pureza: 96,9 %, rendimiento 32,3 %) como un sólido blanquecino. Método de HPLC preparativa: Instrumento: Shimadzu LC-8a de HPLC preparativa; Columna: Phenomenex Luna C18200*40 mm*10 mm; Fase móvil: A para H2O (TFA al 0,09 %) y B para MeCN; Gradiente: B de 30 % a 40 % en 20 min; Velocidad de flujo: 60 mL/min; Longitud de onda: 220 & 254 nm.1
1H NMR: (DMSO-d6, 400 MHz) ó ppm 0,90 (s, 3 H) 1,19 - 1,41 (m, 2 H) 1,43 - 1,62 (m, 7 H) 1,64 - 1,77 (m, 3 H) 1,84 (br d, J=14,55 Hz, 1 H) 1,95 - 2,07 (m, 1H) 2,18 - 2,36 (m, 3 H) 2,65 - 2,78 (m, 3 H) 3,71 - 3,86 (m, 3 H) 3,89 (s, 2 H) 3,93 (s, 2 H) 4,20 (br d, J=9,48 Hz, 1 H) 4,33 - 4,41 (m, 1H) 4,59 (br dd, J=18,41, 8,05 Hz, 1 H) 4,81 (br dd, J=18,52, 8,60 Hz, 1 H) 4,94 (d, J=4,63 Hz, 1 H) 5,50 (s, 1 H) 5,54 - 5,76 (m, 1 H) 6,13 (s, 1 H) 6,29 (dd, J=10,14, 1,32 Hz, 1 H)
6,95 (d, J=7,72 Hz, 1 H) 7,15 - 7,28 (m, 4 H) 7,30 - 7,41 (m, 3 H) 7,51 (br d, J=7,94 Hz, 1 H) 7,72 (br s, 3 H) 8,21 (br d, J=7,72 Hz, 1H) 8,54 (t, J=5,62 Hz, 1H) 9,93 (br d, J=2,65 Hz, 1H) LCMS (Método a, Tabla 4) Rt = 2,31 min.
Ejemplo 6 (Ejemplo de referencia): Síntesis de (S)-2-((2-(2-bromoacetamido)etil)amino)-N-((S)-1-((3-(4-((6aR,6bS,7S,8aS,8bS,10R,11aR,12aS,12bS)-7-hidroxi-8b-(2-hidroxiacetil)-6a,8a-dimetil-4-oxo-2,4,6a,6b,7,8,8a,8b,11a,12,12a,12b-dodecahidro-1H-nafto[2',1':4,5]indeno[1,2-d][1,3]dioxol-10-il)benzil)fenil)amino)-1-oxopropan-2-yl)propanamida.
El producto del Ejemplo 6 puede sintetizarse a partir del acoplamiento de N-(2-((((9H-fluoren-9-il)metoxi)carbonil)amino)etil)-N-(terc-butoxicarbonil)-L-alanil-L-alanina (el producto de las Etapas S1 y S2 más abajo) al producto amino del Ejemplo 2, seguido de las Etapas S4-S6: (1) desprotección de Fmoc, (2) acoplamiento con ácido 2-bromoacético y (3) desprotección de Boc. Fmoc = fluorenilmetiloxicarbonilo; Boc = tercbutoxicarbonilo.

Protocolo general de conjugación de cisteína
Se preparó una solución de aproximadamente 5-20 mg/mL del anticuerpo deseado en tampón PBS, pH 6 - 7,4. Un agente reductor de elección, como TCEP, se diluyó o disolvió en disolventes como H2O, DMSO, DMA o DMF para dar una solución con un intervalo de concentración entre 1 y 25 mM. Los anticuerpos (anti-hTNF hIgG1 (D2E7) o anti-mTNF mIgG2a (8C11; McRae BL y otros, J Crohns Colitis 10 (1): 69-76 (2016)) se redujeron parcialmente mediante la adición de aproximadamente 2-3,5 equivalentes de agente de reducción, al mezclar brevemente e incubar durante toda la noche a 0 - 4 °C. Luego se añadió tampón Tris, pH 8-8,5 (20-50 mM), seguido del fármaco enlazador en DMSO o DMA (menos del 15 % en total) y la mezcla se incubó durante 2-3 horas a temperatura ambiente. Luego se eliminó el exceso de fármaco enlazador y disolvente orgánico mediante purificación. Luego se analizaron las muestras de ADC purificadas mediante SEC, HIC y espectrometría de masa reducida.
Procedimientos analíticos de ADC
Se obtuvieron los perfiles de ADC mediante cromatografía de intercambio aniónico (AEC) o cromatografía de interacción hidrófoba (HIC) para determinar el grado de conjugación y pureza de ADC.
AEC. Se cargaron aproximadamente 20 pg de ADC en un sistema Ultimate 3000 Dual LC (Thermo Scientific) equipado con una columna Propac™ WAX-10 de 4 x 250 mm (Tosoh Bioscience, cat. 054999). La columna se equilibró con tampón A al 100 % y se eluyó mediante el uso de un gradiente lineal a partir de tampón A al 100 % hasta tampón B al 100 % durante 18 min a 1,0 ml/min, donde el tampón A es MES 20 mM, pH 6,7 y el tampón B es MES 20 mM, 500 cloruro de sodio, pH 6,7.
HIC. Se cargaron aproximadamente 20 mg del ADC en un sistema Ultimate 3000 Dual LC (Thermo Scientific) equipado con una columna de butil-NPR de 4,6 x 35 mm (Tosoh Bioscience, cat. 14947). La columna se equilibró en tampón A al 100 % y se eluyó mediante el uso de un gradiente lineal a partir de tampón A al 100 % hasta tampón B al 100 % durante 12 min a 0,8 ml/min, donde el tampón A es fosfato de sodio 25 mM, sulfato de amonio 1,5 M, pH 7,0 y el tampón B es fosfato de sodio 25 mM, isopropanol al 25 %, pH 7,0.
SEC. Las distribuciones de tamaño de los ADC se perfilaron mediante SEC de exclusión por tamaño mediante el uso de un sistema Ultimate 3000 Dual LC (Thermo Scientific) equipado con una columna TSK-gel 3000SWxl de 7,8 x 300 mm (Tosoh Bioscience, cat. 08541). Se cargaron aproximadamente 20 pg de ADC en la columna y se eluyeron durante 17 min mediante el uso de un gradiente isocrático de sulfato de sodio 100 mM, fosfato de sodio 100 mM, pH 6,8 a una velocidad de flujo de 1,0 mL/min.
MS. Se inyectaron muestras reducidas (10 pL) en un sistema LC/MS Agilent 6550 QTof a través de un
automuestreador CTC a temperatura controlada (5 °C). La elución de la muestra se logró en un Waters C-4, 3,5 pm, 300 A, 2,1 x 50 mm i.d. Columna de HPLC. Las fases móviles fueron: A: ácido fórmico al 0,1 % en agua y B: ácido fórmico al 0,1 % en MeCN; la velocidad de flujo fue de 0,45 mL/min, y el compartimento de la columna se mantuvo a 40 °C.
El gradiente de HPLC es como se establece en la Tabla 5:
Tabla 5
Tiempo (min) %A %B
0 95 5
0,6 95 5
1,1 10 90
2,2 10 90
2,4 95 5
3,5 95 5
Ejemplo 7: Preparación de adalimumab conjugado con un glucocorticosteroide para proporcionar un conjugado de anticuerpo-fármaco
Adalimumab BrAc-Gly-Glu-Steroid-PÜ4 ADC que tiene una población DAR 4,0 se preparó mediante un proceso químico de dos etapas: reducción con disulfuro de adalimumab seguida de alquilación (conjugación) con bromoacetamido glicina-esteroide de ácido glutámico Ejemplo 4.

Se redujeron 100 mg de adalimumab a una concentración de 20 mg/mL con ácido difenilfosfinoacético (2,9 - 3,0 eq) a 0 °C durante toda la noche. A continuación, se conjugó adalimumab parcialmente reducido con el Ejemplo 4 (10 eq) en DMSO durante 3 horas a temperatura ambiente. La mezcla de conjugación se cambió primero con tampón en tampón Tris 20 mM, NaCl 50 mM, pH 7,8 mediante el uso de múltiples columnas de desalación NAP 25. La solución de ADC desalada fue purificada por AEC para producir los componentes DAR4 del ADC. Método de cromatografía AEC: Instrumento: Akta puro; Columna: 2X Hitrap Q HP 5 mL; Fase móvil: A para tampón Tris 20 mM, pH 7,8; B para tampón Tris 20 mM, NaCl 1 M, pH 7,8; Gradiente: B de 0 % a 25 % en 60 min; Velocidad de flujo: 5 mL/min; Longitud de onda: 280 & 214 nm.
Con referencia a la Figura 1, que muestra una resolución cromatográfica de la preparación de ADC resultante, el ADC es una mezcla heterogénea que contiene anticuerpos que tienen dos moléculas enlazadoras de fármacos unidas (pico "DAR2"), cuatro moléculas enlazadoras de fármacos unidas (pico "DAR4"), en dependencia del número de enlaces disulfuro entre cadenas reducido. Las condiciones de AEC que se usan en la Figura 1 fueron las siguientes: La columna fue Propac™ WAX-10, 4 x 250 mm (Thermo Fisher Scientific, cat. 054999) y la temperatura de la columna fue de 37 °C. La longitud de onda fue de 280 nm, el tiempo de corrida fue de 18 minutos, la cantidad de inyección fue de 20 pg y la velocidad de flujo fue de 1,0 mL/minuto. Fase móvil A: MES 20 mM, pH 6,7, Fase móvil B: MES 20 mM, NaCl 500 mM, pH 6,7. Perfil de gradiente (Tabla 6):
Tabla 6

La Figura 2 muestra un espectro de masas deconvolucionado del ADC purificado. Este ADC tiene 4 moléculas
enlazadoras de fármacos conjugadas con cada anticuerpo. El pico de la izquierda tiene un peso molecular de 24284,74 Da, que es el resultado de un enlazador de fármaco unido a una sola cadena ligera. El pico de la derecha tiene un peso molecular de 51513,96 Da, que es el resultado de un enlazador de fármaco unido a una sola cadena pesada. Los ADC de la Tabla 7 y 8 se prepararon de acuerdo con el procedimiento descrito anteriormente. Los ADC de las Tablas 7 y 8 que no están cubiertos por la reivindicación 1 se proporcionan solo como referencia.
Tabla 7: Conjugados de fármaco-anticuerpo anti-TNFa de ratón. X se refiere al anticuerpo anti-TNFa murino 8C11

Tabla 8: Conjugados de fármaco-anticuerpo anti-TNFa humano. X se refiere al anticuerpo anti-TNFa humano adalimumab

Generación de líneas celulares indicadoras de GRE y TNF-alfa transmembrana humanas y de ratón
Con el fin de crear una línea celular parental, se sembraron células K562 en una placa de 6 pocilios (Costar: 3516) con 2 mL de medio de crecimiento completo (RPMI, 10 % de FBS, 1 % de L-glutamina, 1 % de piruvato de Na y 1 % de MEM NEAA) a 500,000 células por pocillo durante 24 horas a 37 °C, 5 % de CO2. Al día siguiente, 1,5 mg de pGL4.36 [Luc2P/MMTV/Hygro] (Promega: E316), 1,5 ug pG14.75 [hRLuc/CMV] (Promega: E639A) y 3 uL de reactivo PLUS (Invitrogen: 10964-021) se diluyeron en 244 mL de Opti-MEM (Gibco: 31985-070) y se incubaron a temperatura ambiente durante 15 minutos. El vector pGL4.36[/uc2P/MMTV/Hygro] contiene MMTV LTR (Repetición terminal larga del virus del tumor mamario murino) que impulsa la transcripción del gen indicador de luciferasa /uc2P en respuesta a la activación de varios receptores nucleares como el receptor de glucocorticoides y el receptor de andrógeno. El vector pGL4.75 [hR/uc/CMV] codifica el gen indicador de luciferasa hR/uc (Reni//a reniformis) y está diseñado para una alta expresión y una transcripción anómala reducida.
Después de la incubación, la solución de ADN diluida se preincubó con una solución de Lipofectamina LTX 1:1 (Invitrogen: 94756) (13,2 mL 256,8 mL de Opti-MEM) y se incubó a temperatura ambiente durante 25 minutos para formar complejos de ADN-Lipofectamina LTX. Después de la incubación, se añadieron directamente al pocillo que contenía las células 500 mL de complejos de ADN-Lipofectamina. Se transfectaron células K562 durante 24 horas a 37 °C, 5% de CO2. Después de la incubación, las células se lavaron con 3 mL de PBS y se seleccionaron con medio de crecimiento completo que contenía 125 mg/mL de higromicina B (Invitrogen: 10687-010) durante dos semanas. Se produjeron células "K562 pGL4.36[Luc2P/MMTV/Hygro]_pGL4.75[hRLuc/CMV]".
Para crear una línea celular indicadora de GRE y TNF-alfa transmembrana murina, las células parentales, K562 pGL4.36[Luc2P/MMTV/Hygro]_pGL4.75[hRLuc/CMV], se sembraron en placas de 6 pocillos (Costar: 3516) con 2 mL de medio de crecimiento completo (RPMI, 10 % FBS, 1 % L-glutamina, 1 % Na piruvato y 1 % MEM NEAA) a 500,000 células por pocillo durante 24 h a 37 °C, 5 % CO2. Al día siguiente, se diluyeron 3 mg de ADN de mFL_TNFa (Origene: MC208048), que codifica TNF de ratón no etiquetado, y 3 mL de reactivo PLUS (Invitrogen: 10964-021) en 244 mL de Opti-MEM (Gibco: 31985-070) y se incubó a temperatura ambiente durante 15 minutos. Después de la incubación, la solución de ADN diluida se preincubó con una solución de Lipofectamina LTX 1:1 (Invitrogen: 94756) (13,2 ml 256,8 ml de Opti-MEM) y se incubó a temperatura ambiente durante 25 minutos para formar complejos de ADN-Lipofectamina LTX. Después de la incubación, se añadieron directamente al pocillo que contenía las células 500 mL de complejos de ADN-Lipofectamina. Las células parentales K562 pGL4.36[Luc2P/MMTV/Hygro]_pGL4.75[hRLuc/CMV] se transfectaron durante 24 h a 37 °C, 5 % de CO2. Después de la incubación, las células se lavaron con 3 mL de PBS y se seleccionaron con medio de crecimiento completo que contenía 125 mg/mL de higromicina B (Invitrogen: 10687-010) y 250 mg/mL de G418 (Gibco: 10131-027) durante dos semanas. Se produjeron células "K562 FL-TNFa GRE (pGL4.36[luc2P/MMTV/Hygro])" de ratón.
Para crear una línea celular indicadora de GRE y TNF-alfa transmembrana humana, las células parentales, K562 pGL4.36[Luc2P/MMTV/Hygro]_pGL4.75[hRLuc/CMV], se transfectaron con el plásmido hTNF delta 1-12 C-Myc pcDNA3,1 (-) constructo de plásmido. Este plásmido es pcDNA 3.1 (Thermofisher cat # V79020) que codifica t Nf transmembrana resistente a tace (es decir, SEQ ID NO: 1 que carece de los aminoácidos 77-88). (Ver Perez C y otros, Cell 63 (2): 251-8 (1990) que discuten el TNF transmembrana resistente a tace). Estas líneas celulares se usaron luego en los ensayos de indicador de TNF-alfa descritos en los ejemplos siguientes.
Actividad de inmunoconjugados anti-TNF-alfa en ensayos indicadores de GRE y TNF-alfa transmembrana
Se sembraron células K562 parentales GRE (pGL4.36[luc2P/MMTV/Hygro]) cells and K562 mFL-TNF-a or hTNF delta 1-12 GRE (pGL4.36[luc2P/MMTV/Hygro]) en placas blancas de 96 pocillos tratadas para cultivo de tejido (Costar: 3917) a 50,000 células por pocillo en 50 pl de medio de ensayo (RPMI, CSFBS al 1 %, L-glutamina al 1 %, Piruvato de sodio al 1 % y MEAA al 1 %). Las células se trataron con 25 pL de conjugados de anticuerpo-fármaco anti-TNF murino o humano diluido en serie 3X en medio de ensayo, compuesto esteroide, o medio solo y se incubaron durante 48 horas a 37 °C, 5 % de CO2. Después de 48 horas de incubación, las células se trataron con 75 pl de Dual-Glo Luciferase Assay System (Promega-E2920) durante 10 min y se analizaron para determinar la luminiscencia mediante el uso de Microbeta (PerkinElmer). Los datos se analizaron mediante el uso de un ajuste de curva de cuatro parámetros para generar los valores de EC50. El % de activación máxima se normalizó a dexametasona 100 nM. Los resultados obtenidos mediante el uso de la línea celular de TNF-alfa murino se muestran en la Tabla 9 más abajo, y los resultados obtenidos mediante el uso de la línea celular de TNF-alfa humano se muestran en la Tabla 10 más abajo. En la Tabla 9 más abajo, el anticuerpo en el ADC es el anticuerpo anti-TNFa murino 8C11. En la Tabla 10 más abajo, el anticuerpo en el ADC es el anticuerpo anti-TNFa humano adalimumab. La SEC determinó el porcentaje (%) de monómero como se describió anteriormente (consulte los procedimientos analíticos de ADC).
Tabla 9: Actividad in vitro del conjugado de anticuerpo-fármaco anti-TNFa murino en el ensayo indicador de GRE y TNFa transmembrana de ratón

Tabla 10: Actividad in vitro del conjugado de anticuerpo-fármaco anti-TNFa humano en el ensayo indicador de GRE y TNFa transmembrana humano

Actividad de los inmunoconjugados anti-hTNF alfa en el ensayo de liberación de citocinas PBMC humanas estimuladas por lipopolisacáridos
Se adquirieron células mononucleares de sangre periférica humana primaria (PBMC) de Biological Specialty Corporation (núm. de cat. 214-00-10), se lavaron en 50 mL de PBS, se resuspendieron en FBS con DMSO al 5 %, se dividieron en alícuotas y se criopreservaron en nitrógeno líquido hasta su uso. Las PBMC se descongelaron, se resuspendieron en medio RPMI suplementado con FBS al 2 % y Penicilina-Estreptomicina al 1 %, y se colocaron en placas en una placa de ensayo celular (Costar núm. 3799). Luego, las células se incubaron a concentraciones variables de anti-TNF ADC a 37 °C y 5 % de CO2 durante 4 horas. Luego las células se estimularon con 100 ng/mL de LPS durante toda la noche. Al día siguiente, la placa se centrifugó durante cinco minutos a 1000 rpm y se transfirieron directamente 100 pL de medio sobrenadante a una placa adicional de 96 pocillos y se analizaron para detectar las concentraciones de IL-6 (MSD, # K151AKB) e IL-1 beta (MSD, # K151AGB). Los datos de dosis respuesta se ajustaron a una curva sigmoidal mediante el uso de regresión no lineal, y los valores de IC50 se calcularon con la ayuda de GraphPad 5,0 (GraphPad Software, Inc.). Los resultados que se muestran en la Tabla 11 demuestran que el ADC anti-TNF tiene una actividad potente para inhibir la liberación de citocinas proinflamatorias IL-6 y 11-1 beta a partir de células inmunitarias primarias activadas.
Tabla 11. Actividad in vitro de ADC esteroide anti-TNF en el ensayo de liberación de citocinas estimuladas por
PBMC
Actividad del inmunoconjugado anti-mTNF-alfa en modelo de hipersensibilidad de contacto
El ADC de esteroides anti-TNFa se evaluó en un modelo agudo de hipersensibilidad de contacto, una provocación de inflamación aguda de la piel mediante el uso de una respuesta de hipersensibilidad de tipo retardado (DTH) (impulsada por células T) mediante la aplicación de un agente sensibilizante (isotiocianato de fluoresceína (FTTC)). La eficacia de los ADC de esteroides anti-TNFa se midió por la capacidad de reducir la hinchazón de la oreja. Los biomarcadores de esteroides corticosterona y propéptido N-terminal de procolágeno tipo 1 (P1NP) se incluyeron como lecturas para evaluar el impacto putativo del tratamiento con ADC de esteroides anti-TNFa sobre el eje Hipotálamo-Pituitario-Adrenal (HPA) y el recambio óseo, respectivamente.
Hinchazón de la oreja
El día 0, a los ratones se les aplicó anestesia general y se les afeitó el abdomen. Mediante el uso de una micropipeta, los ratones se sensibilizaron mediante la aplicación epicutánea de 400 pl de solución de FITC (solución al 1,5 % en acetona:DBP 1:1) en el abdomen. Seis días después, a los ratones se les administró el vehículo o el agente terapéutico 1 hora antes del reto a la oreja con FITC. Para el reto de la oreja, los ratones se sometieron a anestesia general y se retaron con 20 pL de FITC aplicados en la oreja derecha. Veinticuatro horas después del reto, los ratones se sometieron a anestesia general y se midió el grosor de sus orejas con un pie de rey. Se calculó la diferencia entre orejas retadas y no retadas. Setenta y dos horas después del reto a las orejas, se inyectó a los ratones con ACTH a 1 mpk IP y se les desangró totalmente a los 30 minutos después de la ACTH. Se recogió plasma y se analizaron los niveles de P1NP, corticosterona, esteroides libres y moléculas grandes.
Cuantificación de esferoides libres liberados y corticosterona endógena
La curva de calibración del esteroide se preparó en plasma de ratón con concentraciones finales de 0,03 nM a 0,1 pM a 8 niveles de concentración diferentes. Se preparó una curva de calibración de corticosterona que variaba de concentraciones finales de corticosterona de 0,3 nM a 1 pM en 70 mg/mL de solución de albúmina de suero bovino en tampón PBS. Se añadió una solución de 160 pL de MeCN con ácido fórmico al 0,1 % a 40 pL de muestras de plasma de estudio o patrones de calibración. Los sobrenadantes se diluyeron con agua destilada y se inyectaron 30 pl de solución de muestra final para análisis LC/MS.
La cuantificación del esteroide libre liberado y la corticosterona se llevó a cabo en un espectrómetro de masas triple cuádruple AB Sciex 5500 conectado a un sistema de HPLC Shimadzu AC20 interconectado con una fuente de ionización por electropulverización que funciona en modo positivo. Se utilizó una columna de 3,5 mm Waters XBridge BEH C18, 2,1x30 mm, para la separación por cromatografía. La fase móvil A era ácido fórmico al 0,1 % en agua de HPLC Milli Q, y la fase móvil B era ácido fórmico al 0,1 % en MeCN. Se aplicó un gradiente lineal del 2 % de la fase móvil B al 98 % de la fase móvil B de 0,6 a 1,2 min. El tiempo de total de corrida fue de 2,6 min a una velocidad de flujo de 0,8 mL/min. El espectrómetro de masas se hizo funcionar en modo MRM positivo a una temperatura de la fuente de 700 °C.
Cuantificación de P1NP en plasma
La cuantificación de P1NP en plasma se realizó en una plataforma LCMS basada en la digestión de proteínas con tripsina. Las muestras de plasma se precipitaron parcialmente y se redujeron completamente mediante la adición de una mezcla de MeCN/bicarbonato de amonio 0,1 M/DTT. El sobrenadante se recogió y se alquiló al añadir ácido yodoacético. Las proteínas alquiladas se digirieron con tripsina y los péptidos trípticos resultantes se analizaron mediante LCMS. La curva de calibración se generó mediante el uso de péptido tríptico sintético añadido en suero de caballo (matriz sustituta que no interfiere). Se usó péptido flanqueante marcado con isótopos estables (extensión de 3-6 aminoácidos en ambos extremos del péptido tríptico) como patrón interno añadido en la mezcla de precipitación de proteínas MeCN TT para normalizar tanto la eficacia de la digestión como la inyección de LCMS.
Se usó una columna de 5 pm Columnex Chromenta BB-C18, 2,1x150 mm, para la separación por cromatografía. La fase móvil A era ácido fórmico al 0,1 % en agua de HPLC Milli Q y la fase móvil B era ácido fórmico al 0,1 % en MeCN. Se aplicó un gradiente lineal del 2 % de la fase móvil B al 65 % de la fase móvil B de 0,6 a 3 min. El tiempo total de corrida fue de 8 min a una velocidad de flujo de 0,45 mL/min. Se usó un espectrómetro de masas de trampa AB Sciex 4000Q en modo MRM positivo para cuantificar péptidos P1NP, a una temperatura original de 700 °C. Resultados
Los resultados se muestran en la Tabla 12 a continuación:
Tabla 12: Comparación de la actividad ADC de esteroides alfa anti-mTNF sobre la hinchazón de la oreja y los biomarcadores de esteroides en el modelo de inflamación de CHS

Actividad de inmunoconjugados anti-mTNF-alfa en artritis inducida por colágeno
La capacidad del ADC de esteroides anti-mTNFa (ADC1) para afectar la enfermedad se evaluó en el modelo de artritis inducida por colágeno (CIA).
En estos experimentos, los ratones macho DBA/1J se obtuvieron de Jackson Labs (Bar Harbor, ME). Los ratones se usaron entre las 6 y las 8 semanas de edad. Todos los animales se mantuvieron a temperatura y humedad constantes bajo un ciclo de luz/oscuridad de 12 horas y se alimentaron con comida para roedores (Lab Diet 5010 PharmaServ, Framingham, MA) y agua ad libitum. AbbVie está acreditada por la AAALAC (Asociación para la Evaluación y Acreditación del Cuidado de Animales de Laboratorio), y todos los procedimientos fueron aprobados por el Comité Institucional de Cuidado y Uso de Animales (IACUC) y monitoreados por un veterinario a cargo de la atención. Se controlaron el peso corporal y la afección, y los animales se sacrificaron si presentaban una pérdida de peso> 20%.
Los ratones macho DBA/J se inmunizaron por vía intradérmica (i.d.) en la base de la cola con 100 pL de emulsión que contenía 100 pg de colágeno bovino tipo II (MD Biosciences) disuelto en ácido acético 0,1 N y 200 pg de Mycobacterium tuberculosis H37Ra inactivado por calor (Adyuvante Completo de Freund, Difco, Laurence, KS). Veintiún días después de la inmunización con colágeno, los ratones se reforzaron IP con 1 mg de Zymosan A (Sigma, St. Louis, MO) en PBS. Después del refuerzo, los ratones se monitorearon de 3 a 5 veces por semana para
detectar artritis. Se evaluó la hinchazón de las patas traseras mediante el uso de pie de rey de resorte Dyer (Dyer 310-115)
Los ratones se inscribieron entre los días 24 y 28 ante los primeros signos clínicos de la enfermedad y se distribuyeron en grupos de gravedad artrítica equivalente. El tratamiento terapéutico temprano comenzó en el momento de la inscripción.
Los animales se dosificaron una vez por vía intraperitoneal (i.p.) con mAb anti-mTNF (dosis alta) o ADC de esteroide anti-mTNF (dosis alta y baja - mpk) en solución salina al 0,9 %. Se recogió sangre para la exposición al anticuerpo mediante un corte en la cola a las 24 y 72 horas después de la dosificación. Las patas se recogieron en el punto de tiempo terminal para histopatología. La sangre se recogió en el punto de tiempo terminal mediante punción cardíaca para realizar recuentos sanguíneos completos (Sysmex XT-2000iV). La significación estadística se determinó mediante ANOVA.
Los resultados se muestran en la Figura 3 y demuestran que una dosis única de ADC de esteroide anti-TNFa puede exhibir una duración de acción prolongada mediante la mejora de la hinchazón de la pata durante ~28 días en comparación con mAb anti-TNFa o vehículo solo.
Estabilidad de ADC en plasma
Aunque se ha empleado la hidrólisis para aumentar la estabilidad in vivo de los enlazadores basados en maleimida, generalmente requiere exposición a pH básico, lo que puede conducir a modificaciones en el anticuerpo (por ejemplo, desamidación), aumento de heterogeneidad, menor rendimiento y similares. (Shen y otros, Nature Biotechnology 30:184-189 (2012) (la hidrolización de maleimidas evita la liberación prematura del fármaco y la exposición sistémica al fármaco); Strop y otros, Chemistry & Biology 20(2):161-167 (2013) (un estudio similar); Tumey y otros, Bioconjugate Chem 25(10):1871-1880 (2014) (uso de una cadena de PEG proximal para permitir la hidrólisis del anillo en condiciones básicas); Lyon y otros, Nature Biotechnology 32:1059-1062 (2014) (procesos para facilitar la creación de succinimida hidrolizada); Christie y otros, J Control Release 220(PtB):660-70 (28 Dec 2015) (uso de N-aril maleimidas para facilitar la hidrólisis del anillo de succinimida en condiciones básicas); Dovgan y otros, Scientific Reports 6:1 (2016)(uso de 2-(maleimidometil)-1,3-dioxano para facilitar la hidrólisis del anillo en condiciones moderadamente básicas y durante un período de tiempo prolongado); y J Pharm Sci 2013: 102 (6) 1712-1723 (dependencia de la desamidación de asparagina del tipo de tampón, pH y temperatura). Por el contrario, las condiciones típicas para la conjugación mediante el uso de bromo acetamida se completan en unas pocas horas, en comparación con los múltiples días para la maleimida (conjugación y posterior hidrólisis del anillo a pH básico). Además, la 2-mercaptoacetamida formada durante la reacción de la cisteína con la bromo acetamida no es susceptible a la reacción de Michael inversa y proporciona una unión estable del enlazador al anticuerpo como se demuestra en la Tabla 13 más abajo para ADC4.
Tabla 13

Estabilidad de ADC en solución
Para evaluar la estabilidad a largo plazo, un biológico se somete a una prueba de esfuerzo acelerada. El protocolo para esta prueba es 100 mg/mL del biológico en 15 mM de histidina a 40 °C durante 21 días. Cuando el ADC4 se sometió a esta prueba, el porcentaje de aumento en el agregado fue <5 % en comparación con el ADC203 de la Publicación de Solicitud de Patente de EE. UU. Núm. 2018/012600, publicada el 10 de mayo de 2018, que registró un aumento en la agregación del 18 % (Tabla 14). Esto demuestra las propiedades mejoradas impartidas a ADC4 por el enlazador Gly-Glu con el profármaco fosfato de la carga útil. ADC203 de US2018/0126000 es:
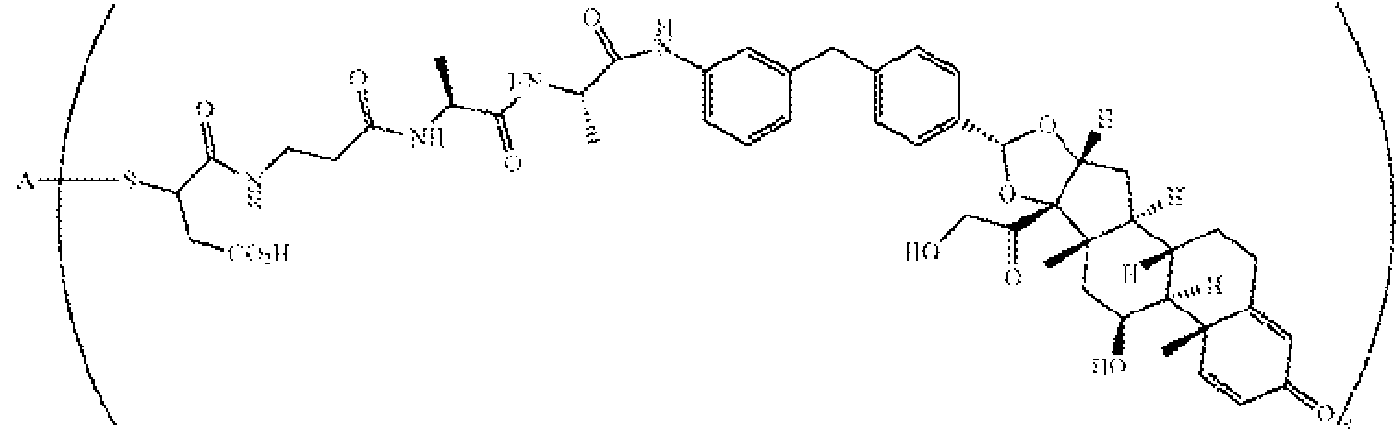
donde n=4 y A se refiere al anticuerpo anti-TNFa humano adalimumab.
Tabla 14

Un beneficio adicional del profármaco de fosfato es que permite el uso de cromatografía de intercambio aniónico para la purificación de DAR. Esto da como resultado una resolución de picos mejorada en comparación con la cromatografía de interacción hidrófoba que da como resultado mayores rendimientos de ADC purificado con DAR. En el tampón de formulación, el anillo de succinimida hidrolizado de ADC203 está en equilibrio con la forma de anillo cerrado. La forma de anillo cerrado es susceptible a la reacción de Michael inversa y la subsiguiente pérdida de fármaco-enlazador in vivo en monos cynomolgus (Figura 4).

En condiciones nominales de almacenamiento de líquido, la unión de succinimida en la conformación abierta reformará la conformación cerrada en más del 5 % a 5 °C y en más del 15 % a 25 °C después de 6 meses (Tabla 15).
Tabla 15

Debe apreciarse que la sección Descripción detallada, y no las secciones de Resumen, está destinada a usarse para interpretar las reivindicaciones. Las secciones de Resumen establecen una o más, pero no todas, las modalidades ilustrativas de la presente descripción como las contemplan los inventores y así, no pretenden limitar la presente descripción y las reivindicaciones adjuntas de ninguna manera.
La presente descripción se ha descrito anteriormente con la ayuda de bloques de construcción funcionales que ilustran la implementación de funciones específicas y sus relaciones. Los límites de estos bloques de construcción funcionales se han definido arbitrariamente en la presente descripción por conveniencia de la descripción. Pueden definirse límites alternativos siempre que las funciones especificadas y las relaciones de las mismas se realicen de manera apropiada.
Claims (6)
1. Un conjugado de anticuerpo-fármaco que comprende:
(a) un anticuerpo anti-TNFa que comprende una cadena pesada indicada como SEQ ID NO: 3 y una cadena ligera indicada como SEQ ID NO: 4; y
(b) un agonista del receptor de glucocorticoides que comprende un radical representado por la fórmula:

en donde el anticuerpo se conjuga con el agonista del receptor de glucocorticoides mediante un enlazador representado por la fórmula:

2. Un conjugado de anticuerpo-fármaco de acuerdo con la reivindicación 1, de acuerdo con la fórmula:

en donde A es el anticuerpo y n es un número entero de 1-10.
3. Un conjugado de anticuerpo-fármaco de acuerdo con la reivindicación 2, en donde n es 4.
4. Un conjugado de anticuerpo-fármaco de acuerdo con la reivindicación 2, en donde n es 2.
5. Un conjugado de anticuerpo-fármaco de acuerdo con la reivindicación 1 de acuerdo con la fórmula:

en donde A es adalimumab y n es 4.
6. Un conjugado de anticuerpo-fármaco de acuerdo con la reivindicación 1 de acuerdo con la fórmula:

en donde A es adalimumab y n es 2.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762593776P | 2017-12-01 | 2017-12-01 | |
| US201762595054P | 2017-12-05 | 2017-12-05 | |
| PCT/IB2018/059482 WO2019106609A1 (en) | 2017-12-01 | 2018-11-29 | Glucocorticoid receptor agonist and immunoconjugates thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2877659T3 true ES2877659T3 (es) | 2021-11-17 |
Family
ID=65010803
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18833113T Active ES2877659T3 (es) | 2017-12-01 | 2018-11-29 | Agonista del receptor de glucocorticoides y sus inmunoconjugados |
Country Status (35)
| Country | Link |
|---|---|
| US (2) | US10772970B2 (es) |
| EP (2) | EP3884962A1 (es) |
| JP (2) | JP6813712B2 (es) |
| KR (1) | KR20200095477A (es) |
| CN (1) | CN111417410B (es) |
| AU (1) | AU2018374634A1 (es) |
| BR (1) | BR112020010694A2 (es) |
| CA (1) | CA3082356A1 (es) |
| CL (2) | CL2020001452A1 (es) |
| CO (1) | CO2020006446A2 (es) |
| CR (1) | CR20200239A (es) |
| CY (1) | CY1124230T1 (es) |
| DK (1) | DK3658192T3 (es) |
| DO (1) | DOP2020000116A (es) |
| EC (1) | ECSP20029001A (es) |
| ES (1) | ES2877659T3 (es) |
| HR (1) | HRP20210917T1 (es) |
| HU (1) | HUE054428T2 (es) |
| IL (1) | IL274651B (es) |
| LT (1) | LT3658192T (es) |
| MA (1) | MA49796A (es) |
| MX (1) | MX2020005583A (es) |
| PE (1) | PE20201286A1 (es) |
| PH (1) | PH12020550779A1 (es) |
| PL (1) | PL3658192T3 (es) |
| PT (1) | PT3658192T (es) |
| RS (1) | RS61994B1 (es) |
| RU (1) | RU2020117698A (es) |
| SG (1) | SG11202004867WA (es) |
| SI (1) | SI3658192T1 (es) |
| TW (1) | TWI803542B (es) |
| UA (1) | UA126595C2 (es) |
| UY (1) | UY37991A (es) |
| WO (1) | WO2019106609A1 (es) |
| ZA (1) | ZA202003325B (es) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES3046537T3 (en) | 2016-11-08 | 2025-12-02 | Regeneron Pharma | Steroids and protein-conjugates thereof |
| KR20200007905A (ko) | 2017-05-18 | 2020-01-22 | 리제너론 파마슈티칼스 인코포레이티드 | 사이클로덱스트린 단백질 약물 접합체 |
| BR112020010694A2 (pt) * | 2017-12-01 | 2020-11-10 | Abbvie Inc. | agonista de receptor de glicocorticoides e imunoconjugados do mesmo |
| EP4136118A4 (en) * | 2020-04-22 | 2024-10-09 | Immunext, Inc. | ANTI-HUMAN VISTA ANTIBODIES AND USE THEREOF |
| AU2022205664A1 (en) * | 2021-01-07 | 2023-08-17 | Immunext, Inc. | NOVEL STEROID PAYLOADS, STEROID LINKERS, ADCs CONTAINING AND USE THEREOF |
| CA3207416A1 (en) * | 2021-02-04 | 2022-08-11 | Lingjian ZHU | Drug conjugate of glucocorticoid receptor agonist, and application thereof in medicine |
| AR125084A1 (es) | 2021-03-23 | 2023-06-07 | Lilly Co Eli | Agonistas del receptor de glucocorticoides novedosos |
| CN117043172A (zh) * | 2021-03-23 | 2023-11-10 | 伊莱利利公司 | 糖皮质激素受体激动剂 |
| TWI899455B (zh) | 2021-03-23 | 2025-10-01 | 美商美國禮來大藥廠 | 糖皮質素受體激動劑 |
| AR125079A1 (es) | 2021-03-23 | 2023-06-07 | Lilly Co Eli | Agonistas de receptores de glucocorticoides sustituidos con carboxi |
| CN117580849A (zh) * | 2021-06-24 | 2024-02-20 | 江苏先声药业有限公司 | 甾体类化合物、其药物组合物及其应用 |
| CN120463762A (zh) * | 2021-08-26 | 2025-08-12 | 映恩生物科技(上海)有限公司 | 一种甾体化合物及其缀合物 |
| US20250179114A1 (en) * | 2021-09-14 | 2025-06-05 | Duality Biologics (Suzhou) Co., Ltd. | Anti-Inflammatory Compound and Use Thereof |
| MX2024009080A (es) * | 2022-01-29 | 2024-07-30 | Shanghai Shengdi Pharmaceutical Co Ltd | Conjugado farmacologico de glucocorticoide. |
| CN120239705A (zh) | 2022-07-21 | 2025-07-01 | 萤火虫生物股份有限公司 | 糖皮质激素受体激动剂和其缀合物 |
| JP2025532110A (ja) * | 2022-09-22 | 2025-09-29 | イーライ リリー アンド カンパニー | グルココルチコイド受容体アゴニスト |
| CN118236514B (zh) * | 2022-12-24 | 2025-10-31 | 津药生物科技(天津)有限公司 | 抗体-糖皮质激素偶联物及其用途 |
| TW202430226A (zh) * | 2022-12-28 | 2024-08-01 | 大陸商上海盛迪醫藥有限公司 | 抗cd40抗體藥物偶聯物、其製備方法及其醫藥用途 |
| KR20250134584A (ko) * | 2023-01-10 | 2025-09-11 | 쓰촨 케룬-바이오테크 바이오파마수티컬 컴퍼니 리미티드 | 글루코코르티코이드 수용체 작용제 및 이의 공액체 |
| EP4652183A1 (en) * | 2023-01-18 | 2025-11-26 | Shanghai MicuRx Pharmaceutical Co., Ltd. | Peptide-drug conjugates for targeted therapy of renal diseases |
| CN121568722A (zh) * | 2023-07-28 | 2026-02-24 | 江苏恒瑞医药股份有限公司 | 一种含有糖皮质激素的抗体药物偶联物的药物组合物 |
Family Cites Families (134)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3126375A (en) | 1964-03-24 | Chioacyl | ||
| BE585432A (fr) | 1958-12-08 | 1960-06-08 | American Cyanamid Co | Stéroïdes fluorés. |
| IT1057859B (it) | 1972-04-28 | 1982-03-30 | Sigma Tao Ind Farmaceutiche Sp | Perivato del triamoinolone |
| US4588585A (en) | 1982-10-19 | 1986-05-13 | Cetus Corporation | Human recombinant cysteine depleted interferon-β muteins |
| US5681718A (en) | 1986-03-14 | 1997-10-28 | Celltech Limited | Methods for enhanced production of tissue plasminogen activator in cell culture using alkanoic acids or salts thereof |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| ATE106249T1 (de) | 1987-11-05 | 1994-06-15 | Hybritech Inc | Polysaccharidmodifizierte immunglobuline mit reduziertem immunogenem potential oder verbesserter pharmakokinetik. |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5010176A (en) | 1988-11-10 | 1991-04-23 | Eli Lilly And Company | Antibody-drug conjugates |
| GB9109645D0 (en) | 1991-05-03 | 1991-06-26 | Celltech Ltd | Recombinant antibodies |
| US6284471B1 (en) | 1991-03-18 | 2001-09-04 | New York University Medical Center | Anti-TNFa antibodies and assays employing anti-TNFa antibodies |
| US7192584B2 (en) | 1991-03-18 | 2007-03-20 | Centocor, Inc. | Methods of treating psoriasis with anti-TNF antibodies |
| DK0701565T3 (da) | 1993-04-02 | 1999-07-12 | Byk Gulden Lomberg Chem Fab | Nye prednisolonderivater |
| WO1994025478A1 (en) | 1993-04-29 | 1994-11-10 | Instytut Farmaceutyczny | 16α,17α-ACETAL GLUCOCORTICOSTEROIDAL DERIVATIVES |
| ATE198074T1 (de) | 1994-03-09 | 2000-12-15 | Byk Gulden Lomberg Chem Fab | Neue silylverbindungen und ihre verwendung |
| JP2749778B2 (ja) | 1994-09-05 | 1998-05-13 | 日清食品株式会社 | トリアムシノロン誘導体 |
| US6090382A (en) | 1996-02-09 | 2000-07-18 | Basf Aktiengesellschaft | Human antibodies that bind human TNFα |
| US5792758A (en) | 1995-12-08 | 1998-08-11 | G. D. Searle & Co. | Steroid nitrite ester derivatives useful as anti-inflammatory drugs |
| MX336813B (es) | 1996-02-09 | 2016-02-02 | Abbvie Biotechnology Ltd | Anticuerpos humanos que ligan el tnfa humano. |
| US5824669A (en) | 1996-03-22 | 1998-10-20 | Nitromed, Inc. | Nitrosated and nitrosylated compounds and compositions and their use for treating respiratory disorders |
| DE19635498A1 (de) | 1996-09-03 | 1998-03-26 | Byk Gulden Lomberg Chem Fab | Verfahren zur Epimerenanreicherung |
| WO2000049993A2 (en) | 1999-02-24 | 2000-08-31 | Nitromed, Inc. | Nitrosated and nitrosylated steroids for the treatment of cardiovascular diseases and disorders |
| US8383081B2 (en) | 1999-05-10 | 2013-02-26 | Immunomedics, Inc. | Anti-CD74 immunoconjugates and methods of use |
| GB0013810D0 (en) | 2000-06-06 | 2000-07-26 | Celltech Chiroscience Ltd | Biological products |
| ES2351184T3 (es) | 2000-11-10 | 2011-02-01 | Nycomed Gmbh | Procedimiento para la producción de 16,17-[(ciclohexilmetilen)bis(oxil)]-11,21-dihidroxi-pregna-1,4-dien-3,20-diona o su 21-isobutirato mediante transcetalización. |
| DE10055820C1 (de) | 2000-11-10 | 2002-07-25 | Byk Gulden Lomberg Chem Fab | Verfahren zur Herstellung eines Glucocorticoids |
| CA2817619A1 (en) | 2001-06-08 | 2002-12-08 | Abbott Laboratories (Bermuda) Ltd. | Methods of administering anti-tnf.alpha. antibodies |
| US7595378B2 (en) | 2001-06-13 | 2009-09-29 | Genmab A/S | Human monoclonal antibodies to epidermal growth factor receptor (EGFR) |
| US20060073141A1 (en) | 2001-06-28 | 2006-04-06 | Domantis Limited | Compositions and methods for treating inflammatory disorders |
| MXPA05000511A (es) | 2001-07-12 | 2005-09-30 | Jefferson Foote | Anticuepros super humanizados. |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| ES2411007T3 (es) | 2001-10-10 | 2013-07-04 | Novo Nordisk A/S | Remodelación y glicoconjugación de péptidos |
| ATE469176T1 (de) | 2001-11-12 | 2010-06-15 | Merck Patent Gmbh | Modifizierter anti-tnf antikörper |
| GB0216648D0 (en) | 2002-07-18 | 2002-08-28 | Lonza Biologics Plc | Method of expressing recombinant protein in CHO cells |
| EP1562627A4 (en) | 2002-08-23 | 2006-11-02 | Mclean Hospital Corp | CORTICOSTEROID CONJUGATES AND ITS USES |
| AU2003301516A1 (en) | 2002-10-24 | 2004-05-13 | Goldstein, Jay A | Antifungal formulations |
| MY150740A (en) | 2002-10-24 | 2014-02-28 | Abbvie Biotechnology Ltd | Low dose methods for treating disorders in which tnf? activity is detrimental |
| EP1569925A1 (en) | 2002-12-13 | 2005-09-07 | Neurogen Corporation | 2-substituted quinazolin-4-ylamine analogues as capsaicin receptor modulators |
| AU2004207003A1 (en) | 2003-01-30 | 2004-08-12 | Medimmune, Llc | Anti-integrin alphanubeta3 antibody formulations and uses thereof |
| MY143936A (en) | 2003-03-27 | 2011-07-29 | Nycomed Gmbh | Process for preparing crystalline ciclesonide with defined particle size |
| WO2005044759A2 (en) | 2003-08-14 | 2005-05-19 | Sun Pharmaceutical Industries Limited | Acetalization process for preparation of steroid compounds |
| PT1670482E (pt) | 2003-09-16 | 2014-03-12 | Takeda Gmbh | Utilização de ciclesonida para o tratamento de doenças respiratórias |
| WO2005028495A1 (en) | 2003-09-24 | 2005-03-31 | Glaxo Group Limited | Anti-inflammatory glucocorticoids |
| CA2542834C (en) | 2003-10-21 | 2012-04-24 | Igf Oncology, Llc | Conjugates or co-administration of igf-1 receptor ligands with anti-cancer chemotherapeutic agents |
| RU2348632C2 (ru) | 2003-11-13 | 2009-03-10 | Ф.Хоффманн-Ля Рош Аг | Гидроксиалкилзамещенные пиридо-7-пиримидин-7-оны |
| WO2005063761A1 (en) | 2003-12-19 | 2005-07-14 | Bristol-Myers Squibb Company | Azabicyclic heterocycles as cannabinoid receptor modulators |
| WO2005063777A1 (en) | 2003-12-23 | 2005-07-14 | Corus Pharma | Benzylphosphate and substituted benzylphosphate prodrugs for the treatment of pulmonary inflammation |
| AU2004311478A1 (en) | 2003-12-31 | 2005-07-21 | Cydex Pharmaceuticals, Inc. | Inhalant formulation containing sulfoalkyl ether cyclodextrin and corticosteroid |
| GB0402797D0 (en) | 2004-02-09 | 2004-03-10 | Novartis Ag | Organic compounds |
| JP4942643B2 (ja) | 2004-03-02 | 2012-05-30 | シアトル ジェネティックス, インコーポレイテッド | 部分的に付加された抗体およびそれらの結合体化方法 |
| US20080187954A1 (en) | 2004-03-10 | 2008-08-07 | Lonza Ltd. | Method For Producing Antibodies |
| EP2471813B1 (en) | 2004-07-15 | 2014-12-31 | Xencor, Inc. | Optimized Fc variants |
| CA2581208A1 (en) | 2004-08-30 | 2006-03-09 | Lonza Biologics Plc. | Affinity- plus ion exchange- chromatography for purifying antibodies |
| WO2006026759A2 (en) | 2004-09-03 | 2006-03-09 | Genentech, Inc. | Humanized anti-beta7 antagonists and uses therefor |
| CA2579007A1 (en) | 2004-09-10 | 2006-03-16 | Altana Pharma Ag | Ciclesonide and syk inhibitor combination and methods of use thereof |
| JO3000B1 (ar) | 2004-10-20 | 2016-09-05 | Genentech Inc | مركبات أجسام مضادة . |
| JP2008521828A (ja) | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| WO2006097458A1 (en) | 2005-03-15 | 2006-09-21 | Nycomed Gmbh | Novel combination |
| WO2006110593A2 (en) | 2005-04-07 | 2006-10-19 | Macrogenics, Inc. | Biological targets for the diagnosis, treatment and prevention of cancer |
| EP1712220A1 (en) | 2005-04-15 | 2006-10-18 | PARI GmbH Spezialisten für effektive Inhalation | Pharmaceutical aerosol composition |
| WO2006135479A2 (en) | 2005-05-10 | 2006-12-21 | Angiotech International Ag | Anti-scarring agents, therapeutic compositions, and use thereof |
| WO2006138212A1 (en) | 2005-06-14 | 2006-12-28 | Gilead Sciences, Inc. | SUBSTITUTED PHENYLPHOSPHATES AS MUTUAL PRODRUGS OF STEROIDS AND β -AGONISTS FOR THE TREATMENT OF PULMONARY INFLAMMATION AND BRONCHOCONSTRICTION |
| DK1912677T3 (da) | 2005-06-20 | 2014-01-13 | Psma Dev Company L L C | PSMA-antistof-lægemiddel-konjugater |
| WO2007054974A2 (en) | 2005-09-28 | 2007-05-18 | Arch Pharmalab Limited | A green chemistry process for the preparation of pregnadiene esters |
| WO2007117685A2 (en) | 2006-04-07 | 2007-10-18 | Nektar Therapeutics Al, Corporation | Conjugates of an anti-tnf-alpha antibody |
| WO2008015696A2 (en) | 2006-05-23 | 2008-02-07 | Cadila Healthcare Limited | Process for preparing ciclesonide |
| EP2032606B1 (en) | 2006-05-30 | 2013-11-27 | Genentech, Inc. | Antibodies and immunoconjugates and uses therefor |
| CA2663737A1 (en) | 2006-09-19 | 2008-03-27 | Cipla Limited | Processes for the preparation of ciclesonide and its crystal form |
| WO2008052350A1 (en) | 2006-11-03 | 2008-05-08 | Qlt Inc. | Photodynamic therapy for the treatment of hidradenitis suppurativa |
| NO331891B1 (no) | 2007-03-20 | 2012-04-30 | Clavis Pharma Asa | Kjemiske forbindelser, et farmasoytisk preparat inneholdende slike forbindelser, samt anvendelse derav for behandling av kreft, inflammasjon og KOLS |
| UA100120C2 (en) | 2007-04-03 | 2012-11-26 | Анадис Фармасьютикалз, Инк. | 5,6-dihydro-1h-pyridin-2-one compounds |
| CN106038481A (zh) | 2007-06-28 | 2016-10-26 | 锡德克斯药物公司 | 皮质类固醇水溶液的鼻部和眼部给药 |
| JP4686650B2 (ja) | 2007-11-30 | 2011-05-25 | ファイザー・リミテッド | 新規なグルココルチコイド受容体アゴニスト |
| WO2009085879A2 (en) | 2007-12-21 | 2009-07-09 | Schering Corporation | C20-c21 substituted glucocorticoid receptor agonists |
| EP2235035A2 (en) | 2007-12-21 | 2010-10-06 | Schering Corporation | C-21 thioethers as glucocorticoid receptor agonists |
| EP2238170B1 (en) | 2008-01-31 | 2016-11-23 | INSERM - Institut National de la Santé et de la Recherche Médicale | Antibodies against human cd39 and use thereof for inhibiting t regulatory cells activity |
| WO2009108118A1 (en) | 2008-02-27 | 2009-09-03 | Astrazeneca Ab | 16 alpha, 17 alpa-acetal glucocorticosteroidal derivatives and their use |
| JP2011513469A (ja) | 2008-03-13 | 2011-04-28 | ファルマビオス ソシエタ ペル アチオニ | プレグナン誘導体の製造法 |
| TW201010707A (en) | 2008-06-10 | 2010-03-16 | Gilead Sciences Inc | Corticosteroid linked beta-agonist compounds for use in therapy |
| CA2727278A1 (en) | 2008-06-16 | 2010-01-21 | Immunogen, Inc. | Novel synergistic effects |
| CN102076670B (zh) | 2008-06-24 | 2013-05-22 | Irm责任有限公司 | 调节g蛋白偶联受体的化合物和方法 |
| WO2010054158A2 (en) | 2008-11-07 | 2010-05-14 | Auspex Pharmaceuticals, Inc. | Steroid modulators of glucocorticoid receptor |
| RU2011127198A (ru) | 2008-12-04 | 2013-01-10 | Эбботт Лэборетриз | Иммуноглобулины с двойными вариабельными доменами и их применение |
| DK2786762T3 (da) | 2008-12-19 | 2019-05-06 | Macrogenics Inc | Kovalente diabodies og anvendelser deraf |
| WO2010075249A2 (en) | 2008-12-22 | 2010-07-01 | Genentech, Inc. | A method for treating rheumatoid arthritis with b-cell antagonists |
| WO2010104187A1 (ja) | 2009-03-09 | 2010-09-16 | 三笠製薬株式会社 | ステロイド化合物 |
| WO2010126953A1 (en) | 2009-04-29 | 2010-11-04 | Gilead Sciences, Inc. | Corticosteroid linked beta-agonist compounds for use in therapy |
| WO2010132743A1 (en) | 2009-05-15 | 2010-11-18 | Gilead Sciences, Inc. | Corticosteroid beta-agonist compounds for use in therapy |
| CA2760284A1 (en) | 2009-05-29 | 2010-12-02 | Pfizer Limited | Novel glucocorticoid receptor agonists |
| GB0917044D0 (en) | 2009-09-29 | 2009-11-18 | Cytoguide As | Agents, uses and methods |
| CN102770456B (zh) | 2009-12-04 | 2018-04-06 | 弗·哈夫曼-拉罗切有限公司 | 多特异性抗体、抗体类似物、组合物和方法 |
| CN102665757A (zh) | 2009-12-09 | 2012-09-12 | 免疫医疗公司 | 通过双特异性抗体预靶向的用于细胞毒性药物的递送系统 |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| CN102167741B (zh) | 2010-02-25 | 2014-05-14 | 上海百迈博制药有限公司 | 一种全人源抗TNF-α单克隆抗体、其制备方法及用途 |
| KR20130010123A (ko) | 2010-04-07 | 2013-01-25 | 아비에 인코포레이티드 | TNF-α 결합 단백질 |
| KR101860963B1 (ko) | 2010-04-23 | 2018-05-24 | 제넨테크, 인크. | 이종다량체 단백질의 생산 |
| US20110300150A1 (en) | 2010-05-18 | 2011-12-08 | Scott Eliasof | Compositions and methods for treatment of autoimmune and other disease |
| EP2575884B1 (en) | 2010-06-03 | 2018-07-18 | AbbVie Biotechnology Ltd | Uses and compositions for treatment of hidradenitis suppurativa (hs) |
| CA2803392A1 (en) | 2010-06-24 | 2011-12-29 | Abbvie Inc. | Dual variable domain immunoglobulins and uses thereof |
| EP2618818A4 (en) | 2010-09-22 | 2014-10-29 | Map Pharmaceuticals Inc | CORTICOSTEROID PARTICLES AND METHOD FOR PRODUCING THE SAME |
| AR084210A1 (es) | 2010-12-08 | 2013-05-02 | Abbott Lab | PROTEINAS DE UNION AL TNF-a |
| WO2012082947A1 (en) | 2010-12-16 | 2012-06-21 | Irm Llc | Compounds and compositions as tgr5 agonists |
| WO2012089247A1 (en) | 2010-12-29 | 2012-07-05 | Nokia Siemens Networks Oy | A method and apparatus for transmitting an identity |
| MX361242B (es) | 2011-03-30 | 2018-11-30 | Ablynx Nv | Anticuerpos de dominio sencillo contra tnf-alfa y usos de los mismos. |
| PE20141545A1 (es) | 2011-10-24 | 2014-11-26 | Abbvie Inc | Inmunoenlazadores dirigidos contra el tnf |
| RU2014121043A (ru) | 2011-10-24 | 2015-12-10 | Эббви Инк. | Биспецифические иммуносвязывающие средства, направленные против tnf и il-17 |
| US20130115269A1 (en) | 2011-11-04 | 2013-05-09 | Henry John Smith | Anti-tumor necrosis factor alpha (TNF-a) antibody used as a targeting agent to treat arthritis and other diseases |
| EP2791170A1 (en) | 2011-12-16 | 2014-10-22 | Synthon Biopharmaceuticals B.V. | Compounds and methods for treating inflammatory diseases |
| US9109005B2 (en) | 2012-02-23 | 2015-08-18 | Boehringer Ingelheim International Gmbh | Method for manufacturing of ciclesonide |
| EP2641900A1 (en) | 2012-03-20 | 2013-09-25 | Almirall, S.A. | Novel polymorphic Crystal forms of 5-(2-{[6-(2,2-difluoro-2-phenylethoxy) hexyl]amino}-1-(R)-hydroxyethyl)-8-hydroxyquinolin-2(1h)-one, heminapadisylate as agonist of the ß2 adrenergic receptor. |
| EP2647627A1 (en) | 2012-04-02 | 2013-10-09 | Almirall, S.A. | Salts of 5-[(1r)-2-({2-[4-(2,2-difluoro-2-phenylethoxy)phenyl] ethyl}amino)-1-hydroxyethyl]-8-hydroxyquinolin-2(1h)-one. |
| CN103421075B (zh) | 2012-05-16 | 2017-07-28 | 上海特化医药科技有限公司 | 孕烷衍生物16,17‑缩醛(酮)的制备方法 |
| EP2855745A4 (en) | 2012-06-01 | 2016-01-20 | Momenta Pharmaceuticals Inc | METHODS RELATING TO ADALIMUM AB |
| CN104854133B (zh) | 2012-10-12 | 2018-10-30 | 新加坡科技研究局 | 用于制备重组抗体治疗剂的最佳重链和轻链信号肽 |
| CA2891280C (en) | 2012-11-24 | 2018-03-20 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
| WO2015012904A2 (en) | 2012-12-13 | 2015-01-29 | Immunomedics, Inc. | Antibody-sn-38 immunoconjugates with a cl2a linker |
| LT3473255T (lt) | 2012-12-21 | 2022-03-10 | Boehringer Ingelheim Vetmedica Gmbh | Farmacinė vaisto forma, apimanti ciklezonidą |
| AU2014239972A1 (en) | 2013-03-15 | 2015-10-08 | Abbvie, Inc. | Improved TNF binding proteins |
| WO2015047510A1 (en) | 2013-09-27 | 2015-04-02 | Immunomedics, Inc. | Anti-trop-2 antibody-drug conjugates and uses thereof |
| US20150139988A1 (en) | 2013-11-15 | 2015-05-21 | Abbvie, Inc. | Glycoengineered binding protein compositions |
| US10464955B2 (en) | 2014-02-28 | 2019-11-05 | Hangzhou Dac Biotech Co., Ltd. | Charged linkers and their uses for conjugation |
| US10550190B2 (en) * | 2014-04-04 | 2020-02-04 | Merck Sharp & Dohme Corp. | Phosphate based linkers for intracellular delivery of drug conjugates |
| CA2948013A1 (en) | 2014-06-30 | 2016-01-07 | Immunomedics, Inc. | Antibodies reactive with an epitope located in the n-terminal region of muc5ac comprising cysteine-rich subdomain 2 (cys2) |
| WO2016042163A2 (en) | 2014-09-19 | 2016-03-24 | Medterials, Inc. | Ophthalmic drug compositions |
| EP3250211B1 (en) | 2015-01-30 | 2021-12-22 | Coral Drugs Pvt. Ltd. | Novel process for preparation of glucocorticoid steroids |
| JP2016190798A (ja) | 2015-03-31 | 2016-11-10 | 三笠製薬株式会社 | アレルギー性鼻炎治療用局所点鼻薬 |
| CA2989269C (en) | 2015-06-15 | 2020-09-22 | Robert Yongxin Zhao | Hydrophilic linkers for conjugation of a cytotoxic agent or chromophore molecule to a cell-binding molecule |
| WO2015151079A2 (en) | 2015-06-20 | 2015-10-08 | Hangzhou Dac Biotech Co, Ltd | Auristatin analogues and their conjugates with cell-binding molecules |
| JP6817288B2 (ja) | 2015-08-10 | 2021-01-20 | ハンジョウ ディーエーシー バイオテック シーオー.,エルティディ.Hangzhou Dac Biotech Co.,Ltd. | 新規な連結体及び生体分子と薬物との特異的共役におけるその使用 |
| JP2017066075A (ja) | 2015-09-30 | 2017-04-06 | 三笠製薬株式会社 | リウマチ治療薬 |
| US20180126000A1 (en) | 2016-06-02 | 2018-05-10 | Abbvie Inc. | Glucocorticoid receptor agonist and immunoconjugates thereof |
| JP6767796B2 (ja) | 2016-07-08 | 2020-10-14 | 株式会社日立情報通信エンジニアリング | 通話管理システム及びその音声認識制御方法 |
| ES3046537T3 (en) | 2016-11-08 | 2025-12-02 | Regeneron Pharma | Steroids and protein-conjugates thereof |
| BR112020010694A2 (pt) * | 2017-12-01 | 2020-11-10 | Abbvie Inc. | agonista de receptor de glicocorticoides e imunoconjugados do mesmo |
-
2018
- 2018-11-29 BR BR112020010694-1A patent/BR112020010694A2/pt unknown
- 2018-11-29 SI SI201830305T patent/SI3658192T1/sl unknown
- 2018-11-29 RS RS20210761A patent/RS61994B1/sr unknown
- 2018-11-29 DK DK18833113.6T patent/DK3658192T3/da active
- 2018-11-29 ES ES18833113T patent/ES2877659T3/es active Active
- 2018-11-29 TW TW107142830A patent/TWI803542B/zh not_active IP Right Cessation
- 2018-11-29 MX MX2020005583A patent/MX2020005583A/es unknown
- 2018-11-29 CR CR20200239A patent/CR20200239A/es unknown
- 2018-11-29 RU RU2020117698A patent/RU2020117698A/ru unknown
- 2018-11-29 PH PH1/2020/550779A patent/PH12020550779A1/en unknown
- 2018-11-29 EP EP21162680.9A patent/EP3884962A1/en active Pending
- 2018-11-29 WO PCT/IB2018/059482 patent/WO2019106609A1/en not_active Ceased
- 2018-11-29 JP JP2020512845A patent/JP6813712B2/ja active Active
- 2018-11-29 LT LTEP18833113.6T patent/LT3658192T/lt unknown
- 2018-11-29 MA MA049796A patent/MA49796A/fr unknown
- 2018-11-29 HU HUE18833113A patent/HUE054428T2/hu unknown
- 2018-11-29 SG SG11202004867WA patent/SG11202004867WA/en unknown
- 2018-11-29 US US16/204,825 patent/US10772970B2/en active Active
- 2018-11-29 HR HRP20210917TT patent/HRP20210917T1/hr unknown
- 2018-11-29 CN CN201880077432.9A patent/CN111417410B/zh active Active
- 2018-11-29 CA CA3082356A patent/CA3082356A1/en active Pending
- 2018-11-29 KR KR1020207015633A patent/KR20200095477A/ko active Pending
- 2018-11-29 PL PL18833113T patent/PL3658192T3/pl unknown
- 2018-11-29 AU AU2018374634A patent/AU2018374634A1/en not_active Abandoned
- 2018-11-29 EP EP18833113.6A patent/EP3658192B1/en active Active
- 2018-11-29 PE PE2020000608A patent/PE20201286A1/es unknown
- 2018-11-29 PT PT188331136T patent/PT3658192T/pt unknown
- 2018-11-29 UA UAA202003245A patent/UA126595C2/uk unknown
- 2018-12-03 UY UY0001037991A patent/UY37991A/es not_active Application Discontinuation
-
2020
- 2020-05-13 IL IL274651A patent/IL274651B/en active IP Right Grant
- 2020-05-27 CO CONC2020/0006446A patent/CO2020006446A2/es unknown
- 2020-05-29 EC ECSENADI202029001A patent/ECSP20029001A/es unknown
- 2020-05-29 CL CL2020001452A patent/CL2020001452A1/es unknown
- 2020-06-03 ZA ZA2020/03325A patent/ZA202003325B/en unknown
- 2020-06-15 DO DO2020000116A patent/DOP2020000116A/es unknown
- 2020-07-31 US US16/945,069 patent/US20210015938A1/en not_active Abandoned
- 2020-12-17 JP JP2020209056A patent/JP2021054845A/ja active Pending
-
2021
- 2021-04-26 CL CL2021001075A patent/CL2021001075A1/es unknown
- 2021-06-18 CY CY20211100547T patent/CY1124230T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2877659T3 (es) | Agonista del receptor de glucocorticoides y sus inmunoconjugados | |
| KR20200095493A (ko) | 항-cd40 항체 약물 공액체 | |
| ES2877548T3 (es) | Agonista del receptor de glucocorticoides e inmunoconjugados del mismo | |
| ES2737998T3 (es) | Moléculas de anticuerpo que tienen especificidad por OX40 humano | |
| ES2750649T3 (es) | Construcciones de anticuerpos multiespecíficos | |
| KR20220156533A (ko) | IL-7Rα 결합 화합물 | |
| ES2729278T3 (es) | Anticuerpos neutralizantes de las exotoxinas principales tcda y tcdb de Clostridium difficile | |
| TW202015739A (zh) | 剪接調節抗體-藥物結合物及其使用方法 | |
| KR20200007905A (ko) | 사이클로덱스트린 단백질 약물 접합체 | |
| US20240252670A1 (en) | Antibody drug conjugate, preparation method therefor and application thereof | |
| ES2750563T3 (es) | Moléculas con una estructura basada en fibronectina estabilizada | |
| CA3172359A1 (en) | Il-2 receptor binding compounds | |
| KR20220139918A (ko) | IL-7Rαγc 결합 화합물 | |
| CA3222358A1 (en) | Checkpoint inhibitors conjugated to il-2, and uses thereof | |
| WO2021161263A1 (en) | Glucocorticoid receptor agonist and immunoconjugates thereof | |
| US20240417436A1 (en) | Conditionally activated immunocytokines and methods of use | |
| HK40031135B (en) | Glucocorticoid receptor agonist and immunoconjugates thereof | |
| HK40031135A (en) | Glucocorticoid receptor agonist and immunoconjugates thereof | |
| HK40032698A (en) | Anti-cd40 antibody drug conjugates |