ES2883274T3 - Moduladores heterocíclicos de la síntesis de lípidos - Google Patents
Moduladores heterocíclicos de la síntesis de lípidos Download PDFInfo
- Publication number
- ES2883274T3 ES2883274T3 ES17153778T ES17153778T ES2883274T3 ES 2883274 T3 ES2883274 T3 ES 2883274T3 ES 17153778 T ES17153778 T ES 17153778T ES 17153778 T ES17153778 T ES 17153778T ES 2883274 T3 ES2883274 T3 ES 2883274T3
- Authority
- ES
- Spain
- Prior art keywords
- virus
- alkyl
- compound
- cycloalkyl
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C*c1cnc(C(*)(CC2)CCN2C(c2cc(I(C(c3ccc(N4CCC4)nc3)=[U])I)c(C)cc2)=I)cc1 Chemical compound C*c1cnc(C(*)(CC2)CCN2C(c2cc(I(C(c3ccc(N4CCC4)nc3)=[U])I)c(C)cc2)=I)cc1 0.000 description 28
- BONZEVXQXNICCZ-UHFFFAOYSA-N CCc1nnc(-c2c(C3CCC3)cc(C)c(C(OC)=O)c2)[nH]1 Chemical compound CCc1nnc(-c2c(C3CCC3)cc(C)c(C(OC)=O)c2)[nH]1 BONZEVXQXNICCZ-UHFFFAOYSA-N 0.000 description 2
- ZFJOMUKPDWNRFI-UHFFFAOYSA-N Cc(ccc(C(O)=O)c1)c1Br Chemical compound Cc(ccc(C(O)=O)c1)c1Br ZFJOMUKPDWNRFI-UHFFFAOYSA-N 0.000 description 2
- DLRCTZUOPMFIDN-IJQSQKGZSA-N C#C/N=C(\C=C(/CN)\N)/N1CCCC1 Chemical compound C#C/N=C(\C=C(/CN)\N)/N1CCCC1 DLRCTZUOPMFIDN-IJQSQKGZSA-N 0.000 description 1
- VYRKLCSYFBVVCD-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O VYRKLCSYFBVVCD-UHFFFAOYSA-N 0.000 description 1
- SXVRARNYAVGEEZ-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(c(cc1)cnc1Br)O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(c(cc1)cnc1Br)O)=O SXVRARNYAVGEEZ-UHFFFAOYSA-N 0.000 description 1
- XROLGGDBCWEWOK-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1(c(cc1)cnc1C#N)O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1(c(cc1)cnc1C#N)O)=O XROLGGDBCWEWOK-UHFFFAOYSA-N 0.000 description 1
- HGOWOVVULOPYQT-UHFFFAOYSA-N CC(C)Nc(nc1)ccc1C(Nc1c(C)ccc(C(N(CC2)CCC2(C(CC2C)=CC=C2C#N)F)=O)c1)=O Chemical compound CC(C)Nc(nc1)ccc1C(Nc1c(C)ccc(C(N(CC2)CCC2(C(CC2C)=CC=C2C#N)F)=O)c1)=O HGOWOVVULOPYQT-UHFFFAOYSA-N 0.000 description 1
- ZMRCOHMOBLASFC-ZCFIWIBFSA-N CC(C)[C@H](C)C#N Chemical compound CC(C)[C@H](C)C#N ZMRCOHMOBLASFC-ZCFIWIBFSA-N 0.000 description 1
- VFVRJHBRAWHZHL-UHFFFAOYSA-N CC(CC(C=C1)=O)N1C(OC(C)(C)C)=O Chemical compound CC(CC(C=C1)=O)N1C(OC(C)(C)C)=O VFVRJHBRAWHZHL-UHFFFAOYSA-N 0.000 description 1
- ZUTZHWKSOIKHER-UHFFFAOYSA-N CC1=CCCC=C1C(OC)=O Chemical compound CC1=CCCC=C1C(OC)=O ZUTZHWKSOIKHER-UHFFFAOYSA-N 0.000 description 1
- FVEQEKNHMGLETE-UHFFFAOYSA-N CCc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CC2)c1C(OC)=O Chemical compound CCc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CC2)c1C(OC)=O FVEQEKNHMGLETE-UHFFFAOYSA-N 0.000 description 1
- MMIFDLWBNUTIJJ-UHFFFAOYSA-N CCc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(CC)c1-c1nnc(N)[o]1 Chemical compound CCc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(CC)c1-c1nnc(N)[o]1 MMIFDLWBNUTIJJ-UHFFFAOYSA-N 0.000 description 1
- ZPWVAIOAAWNUIB-UHFFFAOYSA-N CCc(c(C(N(CCC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1C(NN)=O Chemical compound CCc(c(C(N(CCC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1C(NN)=O ZPWVAIOAAWNUIB-UHFFFAOYSA-N 0.000 description 1
- RWLANPNMVYYZHB-UHFFFAOYSA-N CCc(c(C(O)=O)c1)cc(C2CCC2)c1C(OC)=O Chemical compound CCc(c(C(O)=O)c1)cc(C2CCC2)c1C(OC)=O RWLANPNMVYYZHB-UHFFFAOYSA-N 0.000 description 1
- WCPUOOZFROIMKZ-UHFFFAOYSA-N CCc(cc(C)c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1-c1nc(CCOC2)c2[nH]1 Chemical compound CCc(cc(C)c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1-c1nc(CCOC2)c2[nH]1 WCPUOOZFROIMKZ-UHFFFAOYSA-N 0.000 description 1
- NMMBGQQTZGTBBP-UHFFFAOYSA-N CCc(cc(C)c(C=O)c1)c1C(OC)=O Chemical compound CCc(cc(C)c(C=O)c1)c1C(OC)=O NMMBGQQTZGTBBP-UHFFFAOYSA-N 0.000 description 1
- SQLZYUPLNUVBGX-UHFFFAOYSA-N CCc(cc(C1CC1)c(C(NN)=O)c1)c1C(N(CC1)CCC1c(cc1)ccc1C#N)=O Chemical compound CCc(cc(C1CC1)c(C(NN)=O)c1)c1C(N(CC1)CCC1c(cc1)ccc1C#N)=O SQLZYUPLNUVBGX-UHFFFAOYSA-N 0.000 description 1
- RIPIRVSADCDFQA-UHFFFAOYSA-N CCc(cc(C1CC1)c(C(OC)=O)c1)c1C(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O Chemical compound CCc(cc(C1CC1)c(C(OC)=O)c1)c1C(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O RIPIRVSADCDFQA-UHFFFAOYSA-N 0.000 description 1
- LGQLMRVUXNTEHZ-UHFFFAOYSA-N CCc(cc(C1CCC1)c(C(OC)=O)c1)c1C(N(CC1)CCC1c(cc1)ccc1C#N)=O Chemical compound CCc(cc(C1CCC1)c(C(OC)=O)c1)c1C(N(CC1)CCC1c(cc1)ccc1C#N)=O LGQLMRVUXNTEHZ-UHFFFAOYSA-N 0.000 description 1
- PZJGKKUJCUHJON-UHFFFAOYSA-N CCc(cc(CC)c(-c([nH]1)nnc1OCC)c1)c1C(N(CC1)CCC1c(cc1)ccc1C(N)=O)=O Chemical compound CCc(cc(CC)c(-c([nH]1)nnc1OCC)c1)c1C(N(CC1)CCC1c(cc1)ccc1C(N)=O)=O PZJGKKUJCUHJON-UHFFFAOYSA-N 0.000 description 1
- AMCNKRFLMQTXAC-UHFFFAOYSA-N CCc1cc(F)ccc1C(OC)=O Chemical compound CCc1cc(F)ccc1C(OC)=O AMCNKRFLMQTXAC-UHFFFAOYSA-N 0.000 description 1
- DGWFAVPDFKSGQI-UHFFFAOYSA-N CCc1nnc(-c2c(C3CCC3)cc(C)c(C(O)=O)c2)[nH]1 Chemical compound CCc1nnc(-c2c(C3CCC3)cc(C)c(C(O)=O)c2)[nH]1 DGWFAVPDFKSGQI-UHFFFAOYSA-N 0.000 description 1
- YLMVLDBSYHNJBC-UHFFFAOYSA-N CN(CC1)CCc2c1[nH]c(-c1c(C3CCC3)ccc(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c1)n2 Chemical compound CN(CC1)CCc2c1[nH]c(-c1c(C3CCC3)ccc(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c1)n2 YLMVLDBSYHNJBC-UHFFFAOYSA-N 0.000 description 1
- ATKQSBPFRCWHFD-UHFFFAOYSA-N COC(C1=CCC(C2CCC2)C=C1)=O Chemical compound COC(C1=CCC(C2CCC2)C=C1)=O ATKQSBPFRCWHFD-UHFFFAOYSA-N 0.000 description 1
- JENBPOJAZCPSEW-UHFFFAOYSA-N COC(c(ccc(F)c1)c1Br)=O Chemical compound COC(c(ccc(F)c1)c1Br)=O JENBPOJAZCPSEW-UHFFFAOYSA-N 0.000 description 1
- KTHYNBDPCSXGSI-UHFFFAOYSA-N COC1CC=CC1 Chemical compound COC1CC=CC1 KTHYNBDPCSXGSI-UHFFFAOYSA-N 0.000 description 1
- HTNUUDFQRYBJPH-UHFFFAOYSA-N COCCC(NN)=O Chemical compound COCCC(NN)=O HTNUUDFQRYBJPH-UHFFFAOYSA-N 0.000 description 1
- QQFDHIRJXYDYAE-MXWKQRLJSA-N COc1ccc([C@@H](C[C@H]2C(O)=O)[C@@H]2C(O)=O)cc1 Chemical compound COc1ccc([C@@H](C[C@H]2C(O)=O)[C@@H]2C(O)=O)cc1 QQFDHIRJXYDYAE-MXWKQRLJSA-N 0.000 description 1
- IWMHCJCJBIGUMG-JLTDHHPGSA-N Cc(c(C(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O)c1)cc(C2CCC2)c1/C(/C=N)=N/N Chemical compound Cc(c(C(N(CC1)CCC1(c(cc1)ccc1C#N)F)=O)c1)cc(C2CCC2)c1/C(/C=N)=N/N IWMHCJCJBIGUMG-JLTDHHPGSA-N 0.000 description 1
- WCAHZOCWDQEZQP-UHFFFAOYSA-N Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1nc(CCOC2)c2[s]1 Chemical compound Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1nc(CCOC2)c2[s]1 WCAHZOCWDQEZQP-UHFFFAOYSA-N 0.000 description 1
- XGFNYSTVGCMISH-SANMLTNESA-N Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1ncc([C@H]2OCCC2)[nH]1 Chemical compound Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1ncc([C@H]2OCCC2)[nH]1 XGFNYSTVGCMISH-SANMLTNESA-N 0.000 description 1
- PUHZSUMDEXWFSE-DEOSSOPVSA-N Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1nnc([C@H]2OCCC2)[nH]1 Chemical compound Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C)c1-c1nnc([C@H]2OCCC2)[nH]1 PUHZSUMDEXWFSE-DEOSSOPVSA-N 0.000 description 1
- GXSTYTDMNRSYHN-UHFFFAOYSA-N Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1-c1ncc(C(F)(F)F)[nH]1 Chemical compound Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1-c1ncc(C(F)(F)F)[nH]1 GXSTYTDMNRSYHN-UHFFFAOYSA-N 0.000 description 1
- RMRMEYVSNQVRHU-MHZLTWQESA-N Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1-c1nnc([C@H]2OCCC2)[nH]1 Chemical compound Cc(c(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)cc(C2CCC2)c1-c1nnc([C@H]2OCCC2)[nH]1 RMRMEYVSNQVRHU-MHZLTWQESA-N 0.000 description 1
- GPKJCNPCJJJOOO-UHFFFAOYSA-N Cc(c(C(N(CC1)CCC1c1ccc(C#N)cc1)=O)c1)cc(C)c1-c([nH]1)nnc1N1CCOCC1 Chemical compound Cc(c(C(N(CC1)CCC1c1ccc(C#N)cc1)=O)c1)cc(C)c1-c([nH]1)nnc1N1CCOCC1 GPKJCNPCJJJOOO-UHFFFAOYSA-N 0.000 description 1
- NSFUDNCNTRWWJE-UHFFFAOYSA-N Cc(c(C(O)=O)c1)cc(C)c1-c1ncc(C(N(C)C)=O)[nH]1 Chemical compound Cc(c(C(O)=O)c1)cc(C)c1-c1ncc(C(N(C)C)=O)[nH]1 NSFUDNCNTRWWJE-UHFFFAOYSA-N 0.000 description 1
- NPVQGEIJWDFHJA-AWEZNQCLSA-N Cc(c(C(O)=O)c1)cc(C)c1-c1ncc([C@H]2OCCC2)[nH]1 Chemical compound Cc(c(C(O)=O)c1)cc(C)c1-c1ncc([C@H]2OCCC2)[nH]1 NPVQGEIJWDFHJA-AWEZNQCLSA-N 0.000 description 1
- JFISQRPYNOQTAD-UHFFFAOYSA-N Cc(c(C(O)=O)c1)cc(Cl)c1I Chemical compound Cc(c(C(O)=O)c1)cc(Cl)c1I JFISQRPYNOQTAD-UHFFFAOYSA-N 0.000 description 1
- JSGDDHAOPILSEU-UHFFFAOYSA-N Cc(c(C(OC)=O)c1)cc(C)c1-c1ncc(C(N(C)C)=O)[nH]1 Chemical compound Cc(c(C(OC)=O)c1)cc(C)c1-c1ncc(C(N(C)C)=O)[nH]1 JSGDDHAOPILSEU-UHFFFAOYSA-N 0.000 description 1
- ABUFGODMARUBDI-UHFFFAOYSA-N Cc(c(C(OC)=O)c1)cc(C)c1C#N Chemical compound Cc(c(C(OC)=O)c1)cc(C)c1C#N ABUFGODMARUBDI-UHFFFAOYSA-N 0.000 description 1
- MWAKQPHEWXHLIJ-UHFFFAOYSA-N Cc(c(C(OC)=O)c1)cc(C2CCC2)c1I Chemical compound Cc(c(C(OC)=O)c1)cc(C2CCC2)c1I MWAKQPHEWXHLIJ-UHFFFAOYSA-N 0.000 description 1
- REPNRLBITGLALU-UHFFFAOYSA-N Cc(c(C=O)c1)ccc1C(O)=O Chemical compound Cc(c(C=O)c1)ccc1C(O)=O REPNRLBITGLALU-UHFFFAOYSA-N 0.000 description 1
- YLXYCSUVACXFLH-UHFFFAOYSA-N Cc(cc(C)c(-c1nc(CCOC2)c2[s]1)c1)c1C(O)=O Chemical compound Cc(cc(C)c(-c1nc(CCOC2)c2[s]1)c1)c1C(O)=O YLXYCSUVACXFLH-UHFFFAOYSA-N 0.000 description 1
- ZETITKGSTYFZFM-UHFFFAOYSA-N Cc(cc(C)c(I)c1)c1C(OC)=O Chemical compound Cc(cc(C)c(I)c1)c1C(OC)=O ZETITKGSTYFZFM-UHFFFAOYSA-N 0.000 description 1
- XPLHGPYPIQETMB-UHFFFAOYSA-N Cc(cc(C)c(N)c1)c1C(O)=O Chemical compound Cc(cc(C)c(N)c1)c1C(O)=O XPLHGPYPIQETMB-UHFFFAOYSA-N 0.000 description 1
- NTCJLNNFMMTEIM-UHFFFAOYSA-N Cc(cc(C)c(NC)c1)c1C(OC)=O Chemical compound Cc(cc(C)c(NC)c1)c1C(OC)=O NTCJLNNFMMTEIM-UHFFFAOYSA-N 0.000 description 1
- RVBZNAWVJWDXHN-UHFFFAOYSA-N Cc(cc(C1CCC1)c(-c1nnc(CCOC)[nH]1)c1)c1C(N(CC1)CCC1c1ccc(C(F)(F)F)cc1)=O Chemical compound Cc(cc(C1CCC1)c(-c1nnc(CCOC)[nH]1)c1)c1C(N(CC1)CCC1c1ccc(C(F)(F)F)cc1)=O RVBZNAWVJWDXHN-UHFFFAOYSA-N 0.000 description 1
- YHHILKGSJUXDPL-UHFFFAOYSA-N Cc(ccc(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1-c1nc(CCN(C)C2)c2[nH]1 Chemical compound Cc(ccc(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1-c1nc(CCN(C)C2)c2[nH]1 YHHILKGSJUXDPL-UHFFFAOYSA-N 0.000 description 1
- ADWMYAUQOMTCNJ-DZFIZOCASA-N Cc(ccc(C(N(C[C@H]12)C[C@@H]1[C@H]2c(cc1)ccc1OC)=O)c1)c1N Chemical compound Cc(ccc(C(N(C[C@H]12)C[C@@H]1[C@H]2c(cc1)ccc1OC)=O)c1)c1N ADWMYAUQOMTCNJ-DZFIZOCASA-N 0.000 description 1
- XKFIFYROMAAUDL-UHFFFAOYSA-N Cc(ccc(C(O)=O)c1)c1N Chemical compound Cc(ccc(C(O)=O)c1)c1N XKFIFYROMAAUDL-UHFFFAOYSA-N 0.000 description 1
- MASRAGFWFYHMFI-UHFFFAOYSA-N Cc(ccc(C(OC)=O)c1)c1Br Chemical compound Cc(ccc(C(OC)=O)c1)c1Br MASRAGFWFYHMFI-UHFFFAOYSA-N 0.000 description 1
- CTOLEJCBAPVMBY-UHFFFAOYSA-N Cc(ccc(C(OC)=O)c1)c1C(SC)=N Chemical compound Cc(ccc(C(OC)=O)c1)c1C(SC)=N CTOLEJCBAPVMBY-UHFFFAOYSA-N 0.000 description 1
- JLUQRVNDIUXTLW-UHFFFAOYSA-N Cc(ncc(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1NC(c1ccc(N2CCCC2)nc1)=O Chemical compound Cc(ncc(C(N(CC1)CCC1c(cc1)ccc1C#N)=O)c1)c1NC(c1ccc(N2CCCC2)nc1)=O JLUQRVNDIUXTLW-UHFFFAOYSA-N 0.000 description 1
- AGOHSSNXGVWQKJ-UHFFFAOYSA-N Cc1c(C2CCC2)cc(C)c(C(OC)=O)c1 Chemical compound Cc1c(C2CCC2)cc(C)c(C(OC)=O)c1 AGOHSSNXGVWQKJ-UHFFFAOYSA-N 0.000 description 1
- CECHZZCOMLPSPY-UHFFFAOYSA-N Cc1c(CCI)[nH]c(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 Chemical compound Cc1c(CCI)[nH]c(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 CECHZZCOMLPSPY-UHFFFAOYSA-N 0.000 description 1
- MYNGEWWHNIMIII-UHFFFAOYSA-N Cc1c(CN(C)C)[nH]c(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 Chemical compound Cc1c(CN(C)C)[nH]c(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 MYNGEWWHNIMIII-UHFFFAOYSA-N 0.000 description 1
- DMIYCAFESLIDFW-UHFFFAOYSA-N Cc1c(CNC)[nH]c(-c2c(C)ccc(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 Chemical compound Cc1c(CNC)[nH]c(-c2c(C)ccc(C(N(CC3)CCC3c(cc3)ccc3C#N)=O)c2)n1 DMIYCAFESLIDFW-UHFFFAOYSA-N 0.000 description 1
- JTCXJKGNMVMVBS-UHFFFAOYSA-N Cc1cc(C(OC)=O)ccc1C1CCC1 Chemical compound Cc1cc(C(OC)=O)ccc1C1CCC1 JTCXJKGNMVMVBS-UHFFFAOYSA-N 0.000 description 1
- DJTUYAFJAGLQCK-UHFFFAOYSA-N Cc1ccc(C(OC)=O)c(Br)c1 Chemical compound Cc1ccc(C(OC)=O)c(Br)c1 DJTUYAFJAGLQCK-UHFFFAOYSA-N 0.000 description 1
- UCMAFTYOIOQEML-UHFFFAOYSA-N Cc1cnc(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3NC)=O)c2)[nH]1 Chemical compound Cc1cnc(-c2c(C3CCC3)cc(C)c(C(N(CC3)CCC3c(cc3)ccc3NC)=O)c2)[nH]1 UCMAFTYOIOQEML-UHFFFAOYSA-N 0.000 description 1
- GCBYFMLJKXJBDZ-QHCPKHFHSA-N N#Cc1ccc(C(CC2)(CCN2C(c2cc(NC(N[C@@H]3COCC3)=O)c(C3CCC3)cc2)=O)F)cc1 Chemical compound N#Cc1ccc(C(CC2)(CCN2C(c2cc(NC(N[C@@H]3COCC3)=O)c(C3CCC3)cc2)=O)F)cc1 GCBYFMLJKXJBDZ-QHCPKHFHSA-N 0.000 description 1
- GCBYFMLJKXJBDZ-HSZRJFAPSA-N N#Cc1ccc(C(CC2)(CCN2C(c2cc(NC(N[C@H]3COCC3)=O)c(C3CCC3)cc2)=O)F)cc1 Chemical compound N#Cc1ccc(C(CC2)(CCN2C(c2cc(NC(N[C@H]3COCC3)=O)c(C3CCC3)cc2)=O)F)cc1 GCBYFMLJKXJBDZ-HSZRJFAPSA-N 0.000 description 1
- BXFNUGNJERSDQT-UHFFFAOYSA-N N#Cc1ccc(C(CC2)(CCN2C(c2ccc(C(F)(F)F)c(NC(NC3COCC3)=O)c2)=O)F)cc1 Chemical compound N#Cc1ccc(C(CC2)(CCN2C(c2ccc(C(F)(F)F)c(NC(NC3COCC3)=O)c2)=O)F)cc1 BXFNUGNJERSDQT-UHFFFAOYSA-N 0.000 description 1
- BPKOJZITNVDWCR-UHFFFAOYSA-N N#Cc1ccc(C2(CCNCC2)F)cc1 Chemical compound N#Cc1ccc(C2(CCNCC2)F)cc1 BPKOJZITNVDWCR-UHFFFAOYSA-N 0.000 description 1
- WIAZPDPJRACUDP-UHFFFAOYSA-N N#Cc1ccc(C2CCNCC2)cc1 Chemical compound N#Cc1ccc(C2CCNCC2)cc1 WIAZPDPJRACUDP-UHFFFAOYSA-N 0.000 description 1
- WHZBRFVNSCZSNS-UHFFFAOYSA-N NNN(CC1)CCC1c(cc1)ccc1C#N Chemical compound NNN(CC1)CCC1c(cc1)ccc1C#N WHZBRFVNSCZSNS-UHFFFAOYSA-N 0.000 description 1
- YKWBEPUOVBMENG-UHFFFAOYSA-N Nc1cc(Cl)ncc1[N+]([O-])=O Chemical compound Nc1cc(Cl)ncc1[N+]([O-])=O YKWBEPUOVBMENG-UHFFFAOYSA-N 0.000 description 1
- KXRLXNKGRHCBDI-UHFFFAOYSA-N Nc1cc(N2CCCC2)ncc1[N+]([O-])=O Chemical compound Nc1cc(N2CCCC2)ncc1[N+]([O-])=O KXRLXNKGRHCBDI-UHFFFAOYSA-N 0.000 description 1
- GSVGTEORFZQAHO-UHFFFAOYSA-N O=C(CCOCC1)C1Br Chemical compound O=C(CCOCC1)C1Br GSVGTEORFZQAHO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4525—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/212—IFN-alpha
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Gastroenterology & Hepatology (AREA)
- Biotechnology (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Zoology (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Emergency Medicine (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Un compuesto de Fórmula (X): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, donde: R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) donde: el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; y cuando R1 no es H, -CN o halógeno, está sustituido opcionalmente por uno o más halógenos; cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 lineal o ramificado; R3 es H, -OH o halógeno; L3 es C(R60)2, O o NR50; cada R60 es independientemente H, -OH, -CN, -Ot-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado), o -C(O)- N(R601)2 en donde: t es 0 o 1, y el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; cada R50 es independientemente H, -C(O)-Ot-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C3-C5 cíclico), - alquilo C3-C5 cíclico conteniendo opcionalmente un heteroátomo de oxígeno o nitrógeno, -C(O)-N(R501)2, o alquilo C1- C4 lineal o ramificado, en donde: t es 0 o 1, y el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; n es 1, 2 o 3; m es 1 o 2; R21 es H, halógeno, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 donde el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; R22 es H, halógeno, alquilo C1-C2; cada R26 es independientemente -OH, -CN, halógeno, alquilo C1-C4 lineal o ramificado, alquilo C1-C4)t-Ot-cicloalquilo C3-C5, -(alquilo C1-C4)t-O-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C1-C4), o -C(O)-N(R501)2 en donde t es 0 o 1, y el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; s es 0, 1 o 2; cada R601 y R501 es independientemente H o alquilo C1-C4 lineal o ramificado; y en donde dos de R26, R60, R50, R501 y R601 se unen opcionalmente para formar un anillo en donde los dos de R26, R60, R50, R501 y R601 pueden ser dos R26, dos R60, dos R50, dos R501 o dos R601.
Description
DESCRIPCIÓN
Moduladores heterocíclicos de la síntesis de lípidos
CAMPO
La presente descripción se refiere, en general, a moduladores heterocíclicos de la síntesis de lípidos y procedimientos de uso de los mismos. Los presentes moduladores heterocíclicos de la síntesis de lípidos pueden usarse para el tratamiento de trastornos caracterizados por la desregulación en la función de ácido graso sintasa en un sujeto modulando la ruta de ácido graso sintasa y/o la función de ácido graso sintasa.
ANTECEDENTES
La enfermedad vírica es un problema sanitario significativo que amenaza a grandes segmentos de poblaciones humanas. Algunas de las características relacionadas con una infección vírica que son preocupantes para los profesionales sanitarios incluyen su naturaleza altamente contagiosa (por ejemplo, VIH, SRAG, etc.) y alta mutabilidad. Algunos virus también son oncógenos (tales como VPH, VEB y VHB). Aunque los virus están estructuralmente entre los organismos más sencillos, se considera que están entre los más difíciles de controlar y presentan un desafío formidable para la I+D en fármacos antivirales.
Hasta la fecha, ha habido unos pocos fármacos antivirales usados ampliamente en pacientes, tales como Amantadina y Oseltamivir para la gripe, Aciclovir para infecciones relacionadas con VHS, Ganciclovir para infección por CMV, y múltiples agentes que incluyen fármacos co-formulados (Efavirenz, emtricitabina y tenofovir disoproxil fumarato) para tratamientos del SIDA. Estos fármacos poseen diversos efectos secundarios neurológicos, metabólicos e inmunológicos indeseables. Por lo tanto, el desarrollo de una nueva terapia antiviral se ha convertido en un foco principal de investigación y desarrollo médico y farmacéutico.
La infección por el virus de la hepatitis C (VHC) es un grave problema de salud. Se estima que 170 millones de personas en todo el mundo están infectados de forma crónica con VHC. La infección por VHC puede causar hepatitis crónica, cirrosis, insuficiencia hepática y carcinoma hepatocelular. La infección crónica por VHC es, por lo tanto, una causa principal en el mundo de mortalidad prematura relacionada con el hígado.
El presente estándar de régimen de tratamiento sanitario para infección por VHC implica terapia de combinación con interferón-alfa y ribavirina, a menudo con la adición de un inhibidor de proteasa de acción directa (Telaprevir o Boceprevir). El tratamiento es engorroso y algunas veces tiene efectos secundarios debilitantes y graves. Por esta razón, muchos pacientes no son tratados en las fases tempranas de la enfermedad. Adicionalmente, algunas poblaciones de pacientes no responden de forma duradera al tratamiento. Se necesitan urgentemente procedimientos eficaces de tratamiento de infección por VHC.
Los enfoques terapéuticos dominantes que se emplean actualmente para tratar el cáncer incluyen la extirpación quirúrgica de tumores primarios, irradiación de tumores y aplicación parenteral de agentes citotóxicos anti-mitóticos. Desafortunadamente, solamente una muestra representativa relativamente pequeña de pacientes de cáncer presenta tumores que son "dependientes" de una ruta específica, y pueden ser tratados, por lo tanto, con agentes dirigidos más nuevos. La preponderancia continuada de estas terapias establecidas largas es reflejada por la falta de mejora en las tasas de supervivencia para la mayoría de los cánceres. Además del limitado éxito clínico, efectos secundarios devastadores acompañan a las terapias clásicas. Las terapias basadas tanto en radiación como citotóxicas dan como resultado la destrucción de células epiteliales hematopoyéticas e intestinales que se dividen rápidamente, lo que causa función inmunitaria comprometida, anemia y absorción de nutrientes alterada. La intervención quirúrgica a menudo da como resultado una liberación de células tumores a los sistemas circulatorio o linfático, a partir de las cuales se pueden establecer posteriormente tumores metastásicos. Se necesitan procedimientos mejorados para el tratamiento del cáncer.
El documento US 2009/105305 A1 divulga inhibidores de sintetasa de ácido graso para uso en el tratamiento de cáncer, infección, y enfermedades metabólicas.
RESUMEN
En diversos aspectos, la presente descripción proporciona compuestos de estructura (VI-J)aborda las deficiencias para tratamientos antivirales y anticáncer proporcionando novedosos moduladores heterocíclicos de la síntesis de lípidos que tienen actividades antivirales y anticáncer mejoradas.
En diversos aspectos, se proporcionan compuestos de estructura IX:
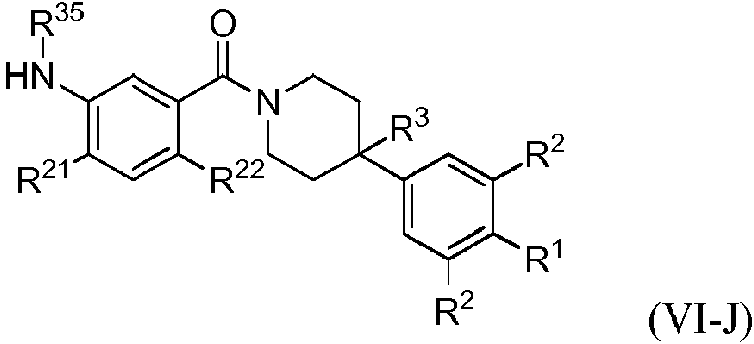
o una sal farmacéuticamente aceptable de los mismos, donde:
R1 es H, -CN, halógeno, alquilo Ci-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo Ci-C4 lineal o ramificado)
donde:
el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -Cn o halógeno, está sustituido opcionalmente por uno o más halógenos;
cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;
R3 es H, -OH o halógeno;
R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;
R22 es H, halógeno o alquilo C1-C2;
R35 es -C(O)-NHR351; y
R351 es alquilo C1-C6 lineal o ramificado, cicloalquilo, heterociclilo, arilo o heteroarilo.
En algunos aspectos de Estructura (VI-J), R3 es H o halógeno.
En algunos aspectos de Estructura En algunos aspectos de Estructura(VI-J), R1 es halógeno, -CN o haloalquilo C1-C2.
En algunos aspectos de Estructura (VI-J), R22 es un alquilo C1-C2.
En algunos aspectos de Estructura (VI-J), R21 es un ciclobutilo y R22 es un alquilo C1-C2.
En algunos aspectos de Estructura (VI-J), R21 es ciclobutilo.
En algunos aspectos de Estructura (VI-J), R3 es H o F.
En algunos aspectos de Estructura (VI-J), R1 es -CN.
En algunos aspectos de Estructura (VI-J), R1 es -CF3.
En algunos aspectos de Estructura (VI-J), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (VI-J), R22 es H.
En algunos aspectos de Estructura (VI-J), R22 es metilo.
En algunos aspectos de Estructura (VI-J), R35 es -C(O)-NHR351.
En algunos aspectos de Estructura (VI-J), R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-il)metilo, (S)-(tetrahidrofuran-2-il)metilo, (R)-tetrahidro-2H-piran-3-ilo o (S)-tetrahidro-2H-piran-3-ilo.
En algunos aspectos de Estructura (VI-J), R351 es (R)-(tetrahidrofuran-2-il)metilo o (S)-(tetrahidrofuran-2-il)metilo. En algunos aspectos de Estructura (VI-J), R1 es -CN, cada R2 es hidrógeno, R3 es H o F, R21 es cicloalquilo C3-C4 , R22 es H, R35 es -C(O)-NHR351 en donde R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-il)metilo, (S)-(tetrahidrofuran-2-il)metilo, (R)-tetrahidro-2H-piran-3-ilo, o (S)-tetrahidro-2H-piran-3-ilo. En algunos aspectos de Estructura (VI-J), R35 es -C(O)-O-R351.
En algunos aspectos de Estructura (VI-J), R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-ilo)metilo, (S)-(tetrahidrofuran-2-ilo-ilo)metilo, (R)-tetrahidro-2H-piran-3-ilo-ilo, o (S)-tetrahidro-2H-piran-3-ilo-ilo.
En algunos aspectos de Estructura (VI-J), R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4 , R22 es H, R35 es -C(O)-O-R351 donde R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-ilo-ilo)metilo, (S)-(tetrahidrofuran-2-ilo-ilo)metilo, (R)- tetrahidro-2H-piran-3-ilo-ilo, o (S)-tetrahidro-2H-piran-3-ilo-ilo.
En algunos aspectos de Estructura (VI-J), R351 es (R)-3-tetrahidrofuranilo o (S)-3-tetrahidrofuranilo.
En algunos aspectos de Estructura (VI-J), los compuestos tienen una estructura seleccionada del grupo que consiste en:

En diversos aspectos, se proporcionan compuestos de Estructura (X):

o una sal farmacéuticamente aceptable de los mismos, en donde:
R1 es H, -CN, halógeno, alquilo C1-C4 lineal o o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) en donde:
El cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno, y opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;
cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 lineal o ramificado;
R3 es H, -OH o halógeno;
L3 es C(R60)2, O o NR50;
cada R60 es independientemente H, -OH, -CN, -Ot-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado), o -C(O)-N(R601)2 en donde:
t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;
cada R50 es independientemente H, -C(O)-Ot-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(cicloalquilo C3-C5), alquilo-C3-C5 cíclico que contiene opcionalmente un heteroátomo de oxígeno o nitrógeno, -C(O)-N(R501)2, alquilo C1-C4 lineal o ramificado en donde:
t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;
n es 1, 2 o 3;
m es 1 o 2 ;
R21 es H, halógeno, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 alquilo en donde el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno
R22 es H, halógeno, alquilo C1-C2;
cada R26 es independientemente -OH, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -(alquilo C1-C4)t-Ot-(cicloalquilo C3-C5), -(alquilo C1-C4)t-O-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C1-C4), o -C(O)-N(R501)2 en donde: t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; s es 0 , 1 o 2 ;
cada R601 y R501 es independientemente H o alquilo C1-C4 lineal o ramificado; y
en donde dos de R26, R60, R5 0 , R501 y R601 opcionalmente se une para formar un anillo en donde los dos de R26, R60, r 50, r501 y r 601 pueden ser dos R26, dos R60, dos R50, dos R501 o dos R601.
En algunos aspectos de Estructura (X), R21 es halógeno, alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C5.
En algunos aspectos de Estructura (X), R3 es H o halógeno.
En algunos aspectos de Estructura (X), R1 es -CN o haloalquilo C1-C2.
En algunos aspectos de Estructura (X), R3 es H o F.
En algunos aspectos de Estructura (X), R1 es -CN.
En algunos aspectos de Estructura (X), R1 es-CF3.
En algunos aspectos de Estructura (X), n es 1.
En algunos aspectos de Estructura (X), n es 2.
En algunos aspectos de Estructura (X), m es 1
En algunos aspectos de Estructura (X), m es 2.
En algunos aspectos de Estructura (X), R21 es alquilo Ci-C2 o cicloalquilo C3-C5 y R22 es alquilo Ci-C2.
En algunos aspectos de Estructura (X), R21 es cicloalquilo C3-C5 y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (X), n es 2, m es 1, L3 es -N-C(O)-O-(alquilo C1-C2).
En algunos aspectos de Estructura (X), L3 es NR50; R50 es alquilo C1-C2; R21 es ciclobutilo; R22 es H o metilo; R3 es H; R1 es -CN; m es 2 y n es 1 o 2.
En algunos aspectos de Estructura (X), n es 2, m es 1, L3 es O y s es 0.
En algunos aspectos de Estructura (X), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (X), R22 es metilo.
En algunos aspectos de Estructura (X), R22 es H.
En algunos aspectos de Estructura (X), R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4, R22 es metilo, n es 2 y L3 es NR50 donde R50 es metilo o etilo.
En algunos aspectos de Estructura (X), R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4 , R22 es metilo, n es 2 y L3 es O.
En algunos aspectos de Estructura (X), el compuesto tiene una estructura seleccionada del grupo que consiste en:

En diversos aspectos, se proporcionan compuestos de Estructura (XI):

o una sal farmacéuticamente aceptable de los mismos, en donde:
R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) en donde:
el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, opcionalmente sustituido con un o más halógenos;
cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;
R3 es H, -OH, o halógeno;
R21 es ciclobutilo, azetidin-1-ilo-ilo, o ciclopropilo;
R22 es H, halógeno, alquilo C1-C2; y
R351 es alquilo C1-C2 o C2-O-(alquilo C1 o C2).
En algunos aspectos de Estructura (XI), R3 es H o halógeno.
En algunos aspectos de Estructura (XI), R1 es halógeno, -CN o haloalquilo C1-C2.
En algunos aspectos de Estructura (XI), R21 es cicloalquilo C3-C4 y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (XI), R21 es ciclobutilo y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (XI), R21 es ciclobutilo.
En algunos aspectos de Estructura (XI), R3 es H o F.
En algunos aspectos de Estructura (XI), R1 es -CN.
En algunos aspectos de Estructura (XI), R1 es -CF3.
En algunos aspectos de Estructura (XI), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (XI), R22 es H.
En algunos aspectos de Estructura (XI), R22 es metilo.
En algunos aspectos de Estructura (XI), R1 es -CN, cada R2 es H, R3 es H o F, R21 es ciclobutilo, R22 es metilo y R351 es metilo o etilo.
En algunos aspectos de Estructura (XI), el compuesto tiene una estructura seleccionada del grupo que consiste en:

En diversos aspectos, la presente descripción proporciona composiciones farmacéuticas que comprenden uno cualquiera de los compuestos de las Estructuras (VI-J), (X) o (XI) y un vehículo, excipiente o diluyente farmacéuticamente aceptable.
En diversos aspectos, la presente descripción proporciona una cantidad terapéuticamente efectiva de un compuesto de cualquiera de las Estructuras (VI-J), (X) o (XI) para uso en el tratamiento de una infección viral en un sujeto, una afección caracterizada por desregulación de una función sintasa de ácido graso en un sujeto, cáncer en un sujeto. En diversos aspectos, la infección viral es hepatitis C.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La figura 1 ilustra una correlación entre la inhibición de FASN y la inhibición de VHC.
DESCRIPCIÓN DETALLADA
La invención se define por las reivindicaciones anejas.
La presente descripción aborda las deficiencias en el tratamiento de afecciones caracterizadas por desregulación de la función de FASN en un sujeto, tal como infección vírica, cáncer y trastornos metabólicos, proporcionando novedosos moduladores heterocíclicos de la síntesis de lípidos.
En ciertos aspectos, la presente descripción proporciona composiciones para el tratamiento de infecciones víricas. En general, las composiciones para el tratamiento de infecciones víricas están dirigidos a la modulación de la ruta de síntesis de ácidos grasos. La ruta de síntesis de ácidos grasos está implicada en la replicación de virus en las células huésped. La presente invención materializa composiciones para el tratamiento de infección vírica, tal como infecciones por hepatitis C, infecciones por fiebre amarilla e infecciones por rinovirus humano, o cualquier virus que se dirige a la ruta de síntesis de ácidos grasos.
En ciertos aspectos, la presente descripción proporciona composiciones para el tratamiento de cáncer. La ácido graso sintasa es responsable de la conversión de malonil-CoA en ácidos grasos de cadena larga, que es una reacción temprana en la biosíntesis de ácidos grasos. La ácido graso sintasa se sobreexpresa en muchas células cancerosas. Sin desear quedar ligados a ninguna teoría particular, se formula la hipótesis de que la inhibición de la expresión de ácido graso sintasa o la selectividad de actividad de ácido graso sintasa suprime la proliferación e induce la muerte celular de células cancerosas, con poca toxicidad hacia células normales.
Además, la presente descripción proporciona compuestos para modular dianas de células huésped a las que se dirigen los virus. Dicha modulación de dianas de células huésped puede incluir activación o inhibición de las dianas de células huésped. Por consiguiente, compuestos que modulan, por ejemplo, inhiben, la actividad de una proteína no viral, por ejemplo, una proteína de célula huésped, por ejemplo, componentes de la ruta de síntesis de ácidos grasos, pueden usarse como agentes farmacéuticos antivirales.
Definiciones
Fracciones químicas denominadas fracciones químicas univalentes (por ejemplo, alquilo, arilo, etc.) también abarcan fracciones multivalentes estructuralmente permisibles, tal como lo entienden los expertos en la materia. Por ejemplo, aunque una fracción "alquilo" se refiere, en general, a un radical monovalente (por ejemplo, CH3CH2-), en circunstancias apropiadas una fracción "alquilo" también puede referirse a un radical divalente (por ejemplo, -CH2CH2-, que es equivalente a un grupo "alquileno"). Análogamente, en circunstancias donde se requiere una fracción divalente, los expertos en la técnica entenderán que el término "arilo" se refiere al grupo arileno divalente correspondiente.
Se entiende que todos los átomos tienen su número normal de valencias para la formación de enlaces (por ejemplo, 4 para carbono, 3 para N, 2 para O, y 2 , 4 o 6 para S, dependiendo del estado de oxidación del átomo). En ocasiones una fracción puede definirse, por ejemplo, con (A)aB, donde a es 0 o 1. En dichos aspectos, cuando a es 0 la fracción es B y cuando a es 1 la fracción es AB.
Donde un sustituyente puede variar en el número de átomos o grupos del mismo tipo (por ejemplo, los grupos alquilo pueden ser C1, C2 , C3, etc.), el número de átomos o grupos repetidos puede estar representado por un intervalo (por ejemplo, alquilo C1-C6) que incluye todos y cada uno de los números en el intervalo y todos y cada uno de los subintervalos. Por ejemplo, alquilo C1-C3 incluye alquilo C1, C2 , C3, C1-2, C1 -3 y C2-3.
"Alcanoílo" se refiere a un grupo carbonilo con un grupo alquilo inferior como sustituyente.
"Alquilamino" se refiere a un grupo amino sustituido por un grupo alquilo.
"Alcoxi" se refiere a un átomo de O sustituido por un grupo alquilo tal como se define en el presente documento, por ejemplo, metoxi [-OCH3, un alcoxi C1]. La expresión "alcoxi C1-6" abarca alcoxi C1, alcoxi C2 , alcoxi C3, alcoxi C4 , alcoxi C5 , alcoxi C6 y cualquier subintervalo de los mismos.
"Alcoxicarbonilo" se refiere a un grupo carbonilo con un grupo alcoxi como sustituyente.
"Alquilo," "alquenilo" y "alquinilo," se refieren a grupos alifáticos de cadena lineal o ramificada, opcionalmente sustituidos que tienen de 1 a 30 átomos de carbono, o preferentemente de 1 a 15 átomos de carbono, o más preferentemente de 1 a 6 átomos de carbono. Los ejemplos de grupos alquilo incluyen metilo, etilo, propilo, isopropilo, butilo, tere-butilo, isobutilo, pentilo, hexilo, vinilo, alilo, isobutenilo, etinilo y propinilo. El término "heteroalquilo", tal como
se usa en el presente documento, contempla un alquilo con uno o más heteroátomos.
"Alquileno" se refiere a un radical divalente opcionalmente sustituido que es un fragmento de hidrocarburo ramificado o sin ramificar que contiene el número especificado de átomos de carbono, y que tiene dos puntos de fijación. Un ejemplo es propileno [-CH2CH2CH2-, un alquileno C3].
"Amino" se refiere al grupo -NH2.
"Arilo" se refiere a grupos aromáticos opcionalmente sustituidos que tienen al menos un anillo que tiene un sistema de electrones pi conjugados e incluye grupos arilo carbocíclico y biarilo, todos los cuales pueden estar opcionalmente sustituidos. Los grupos fenilo y naftilo son grupos arilo carbocíclico preferidos.
"Aralquilo" o "arilalquilo" se refieren al grupos arilo sustituidos con alquilo. Los ejemplos de grupos aralquilo incluyen butilfenilo, propilfenilo, etilfenilo, metilfenilo, 3,5-dimetilfenilo, terc-butilfenilo.
"Carbamoílo", tal como se usa en el presente documento, contempla un grupo de la estructura
O
------- C------ NRn2
donde RN se selecciona de entre el grupo que consiste en hidrógeno, -OH, alquilo C1 a C12, heteroalquilo C1 a C12, alquenilo, alquinilo, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoílo, carbamoílo, sulfonilo, sulfonato y sulfonamida.
"Carbonilo" se refiere a un grupo de la estructura
O
--------C--------
"Cicloalquilo" se refiere a un anillo opcionalmente sustituido, que puede estar saturado o sin saturar y ser monocíclico, bicíclico o tricíclico formado enteramente de átomos de carbono. Un ejemplo de un grupo cicloalquilo es el grupo ciclo pentenilo (C5H7-), que es un grupo cicloalquilo insaturado de cinco carbonos (C5).
"Heterociclo" se refiere a un sistema de anillo cicloalquilo de 5 a 7 miembros opcionalmente sustituido que contiene 1, 2 o 3 heteroátomos, que pueden ser iguales o diferentes, seleccionados de entre N, O o S, y que opcionalmente contiene un doble enlace.
"Halógeno" se refiere a un radical átomo de cloro, bromo, flúor o yodo. El término "halógeno" también contempla términos "halo" o "haluro".
"Heteroátomo" se refiere aun átomo no de carbono, donde boro, nitrógeno, oxígeno, azufre y fósforo son heteroátomos preferidos, con nitrógeno, oxígeno y azufre siendo heteroátomos particularmente preferidos en los compuestos de la presente descripción.
"Heteroarilo" se refiere a grupos arilo opcionalmente sustituidos que tienen de 1 a 9 átomos de carbono y el resto de los átomos son heteroátomos, e incluye aquellos sistemas heterocíclicos descritos en "Handbook of Chemistry and Physics," 49a Edición, 1968, R. C. Weast, editor; The Chemical Rubber Co., Cleveland, Ohio. Véase particularmente la Sección C, Normas para nombrar compuestos orgánicos, B. Sistemas heterocíclicos fundamentales. Los heteroarilos adecuados incluyen tienilo, pirrilo, furilo, piridilo, pirimidilo, pirazinilo, pirazolilo, oxazolilo, isoxazolilo, imidazolilo, tiazolilo, piranilo, tetrazolilo, pirrolilo, pirrolinilo, piridazinilo, triazolilo, indolilo, isoindolilo, indolizinilo, benzimidazolilo, quinolilo, isoquinolilo, indazolilo, benzotriazolilo, tetrazolopiridazinilo, oxadiazolilo, benzoxazolilo, benzoxadiazolilo, tiadiazolilo, benzotiazolilo, benzotiadiazolilo y similares.
Una fracción "opcionalmente sustituida" puede estar sustituida por de uno a cuatro, o preferentemente de uno a tres, o más preferentemente uno o dos sustituyentes no hidrógeno. A menos que se especifique otra cosa, cuando el sustituyente está en un carbono, este se selecciona de entre el grupo que consiste en -OH, -CN, -NO2, halógeno, alquilo C1 a C12, heteroalquilo C1 a C12, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoílo, carbamoílo, sulfonilo sustituido, sulfonato, sulfonamida y amino, ninguno de los cuales están sustituidos adicionalmente. A menos que se especifique otra cosa, cuando el sustituyente está en un nitrógeno, este se selecciona de entre el grupo que consiste en alquilo C1 a C12, heteroalquilo C1 a C12, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoílo, carbamoílo, sulfonilo, sulfonato y sulfonamida, ninguno de los cuales están sustituidos adicionalmente.
El término "sulfonamida", tal como se usa en el presente documento, contempla un grupo que tiene la estructura
O
------- S-------NRn2
O
donde RN se selecciona de entre el grupo que consiste en hidrógeno, -OH, alquilo C1 a C12, heteroalquilo C1 a C12, alquenilo, alquinilo, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoílo, carbamoílo, sulfonilo sustituido, sulfonato y sulfonamida.
El término "sulfonato", tal como se usa en el presente documento, contempla un grupo que tiene la estructura
o
--------S-------O Rs
O
donde Rs se selecciona de entre el grupo que consiste en hidrógeno, alquilo C1-C10, alquenilo C2-C10, alquinilo C2-C10, alcanoílo C1-C10 o alcoxicarbonilo C1-C10.
"Sulfonilo", tal como se usa en el presente documento en solitario o como parte de otro grupo, se refiere a un grupo SO2. La fracción SO2 está opcionalmente sustituida.
Los compuestos de la presente descripción pueden existir como estereoisómeros, donde están presentes centros asimétricos o quirales. Los estereoisómeros se designan (R) o (S) dependiendo de la configuración de sustituyentes alrededor del átomo de carbono quiral. Los términos (R) y (S) usados en el presente documento son configuraciones tal como se definen en IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, Pure Appl. Chem., (1976), 45: 13-30. La presente descripción contempla diversos estereoisómeros y mezclas de los mismos y están específicamente incluidos dentro del alcance de la presente descripción. Los estereoisómeros incluyen enantiómeros, diastereómeros y mezclas de enantiómeros o diastereómeros. Los estereoisómeros individuales de compuestos de la presente descripción pueden prepararse de forma sintética a partir de materiales de partida disponibles en el mercado que contienen centros asimétricos o quirales o mediante la preparación de mezclas racémicas seguida por resolución, bien conocida por los expertos en la materia. Estos procedimientos de resolución se ejemplifican mediante (1) fijación de una mezcla de enantiómeros a un auxiliar quiral, separación de la mezcla de diastereómeros resultante mediante recristalización o cromatografía y liberación del producto ópticamente puro del auxiliar o (2 ) separación directa de la mezcla de enantiómeros ópticos en columnas cromatográficas quirales.
Además, las fracciones descritas en el presente documento que existen en múltiples formas tautoméricas incluyen todas dichas formas abarcadas por una estructura tautomérica dada.
Los átomos individuales en los compuestos descritos pueden ser cualquier isótopo de ese elemento. Por ejemplo, el hidrógeno puede estar en forma de deuterio.
"Farmacéuticamente aceptable" significa aprobado o aprobable por una agencia reguladora del gobierno federal o estatal o enumerada en la Farmacopea estadounidense u otra farmacopea reconocida en general para uso en animales, y más particularmente en seres humanos. Puede ser material que no es biológicamente o de otra forma indeseable, es decir, el material puede ser administrado a un individuo sin causar ningún efecto biológico indeseable o interactuar de manera perjudicial con ninguno de los componentes de la composición en la que está contenido. La expresión "sal farmacéuticamente aceptable" de un compuesto significa una sal que es farmacéuticamente aceptable y que posee la actividad farmacológica deseada del compuesto parental. Dichas sales incluyen, por ejemplo, sales de adición de ácidos y sales de adición de bases.
"Sales de adición de ácidos" de acuerdo con la presente descripción, están formadas con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, y similares; o formadas con ácidos orgánicos tales como ácido acético, ácido propiónico, ácido hexanoico, ácido ciclopentanopropiónico, ácido glicólico, ácido pirúvico, ácido láctico, ácido malónico, ácido succínico, ácido málico, ácido maleico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido 3-(4-hidroxibenzoil)benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2-etanodisulfónico, ácido 2-hidroxietanosulfónico, ácido bencenosulfónico, ácido 2-naftalenosulfónico, ácido 4-metilbiciclo-[2.2.2]oct-2-eno-1-carboxílico, ácido glucoheptónico, ácido 4,4'-metilenbis-(3-hidroxi-2-eno-1-carboxílico), ácido 3-fenilpropiónico, ácido trimetilacético, ácido butilacético terciario, ácido lauril sulfúrico, ácido glucónico, ácido glutámico, ácido hidroxinaftoico, ácido salicílico, ácido esteárico, ácido mucónico y similares.
"Sales de adición de bases" de acuerdo con la presente descripción se forman cuando un protón ácido presente en el compuesto parental se sustituye por un ion metálico, por ejemplo, un ion de metal alcalino, un ion alcalinotérreo, o un ion de aluminio; o se coordina con una base orgánica. Las bases orgánicas aceptables incluyen etanolamina, dietanolamina, trietanolamina, trometamina, N-metilglucamina y similares. Las bases inorgánicas aceptables incluyen hidróxido de aluminio, hidróxido de calcio, hidróxido de potasio, carbonato de sodio, hidróxido de sodio y similares. Debe entenderse que una referencia a una sal farmacéuticamente aceptable incluye las formas de adición de disolvente o formas cristalinas de la misma, particularmente solvatos o polimorfos. Los solvatos contienen cantidades estequiométricas o no estequiométricas de un disolvente y a menudo se forman durante el proceso de cristalización. Los hidratos se forman cuando el disolvente es agua, o los alcoholatos se forman cuando el disolvente es alcohol. Los polimorfos incluyen las diferentes disposiciones de empaquetamiento cristalino de la misma composición elemental de un compuesto. Los polimorfos habitualmente tienen diferentes patrones de difracción de rayos X, espectros infrarrojos, puntos de fusión, densidad, dureza, forma cristalina, propiedades ópticas y eléctricas, estabilidad y solubilidad. Diversos factores tales como el disolvente de recristalización, la velocidad de cristalización y la temperatura de almacenamiento pueden hacer que una forma de cristal único domine.
El término "tratamiento" incluye la administración de los compuestos o agentes de la presente invención a un sujeto para prevenir o retardar, para aliviar, o para detener o inhibir el desarrollo de los síntomas o afecciones asociadas con trastornos asociados con la ácido graso sintasa, por ejemplo, crecimiento tumoral asociado con cáncer. Un facultativo médico experto sabrá cómo usar procedimientos estándar para determinar si un paciente está padeciendo una enfermedad asociada con la actividad de ácido graso sintasa, por ejemplo, examinando al paciente y determinando si
el paciente está padeciendo una enfermedad que se sabe que está asociada con la actividad de ácido graso sintasa o determinando cuantitativamente los niveles de ácido graso sintasa en plasma sanguíneo o tejido del individuo que se sospecha que padece una enfermedad asociada con la ácido graso sintasa y comparando los niveles de ácido graso sintasa en el plasma sanguíneo o el tejido del individuo que se sospecha que padece una enfermedad asociada con ácido graso sintasa con los niveles de ácido graso sintasa en el plasma sanguíneo o el tejido de un individuo sano. Los niveles de securina incrementados son indicativos de enfermedad. Por consiguiente, la presente invención proporciona, entre otros, procedimientos de administración de un compuesto de la presente invención a un sujeto y determinación de la actividad de ácido graso sintasa en el sujeto. La actividad de ácido graso sintasa en el sujeto puede determinarse antes y/o después de la administración del compuesto.
Una "cantidad terapéuticamente efectiva" o "cantidad farmacéuticamente efectiva" significa la cantidad que, cuando se administra a un sujeto, produce efectos para los cuales se administra. Por ejemplo, una "cantidad terapéuticamente efectiva", cuando se administra a un sujeto para inhibir la actividad de ácido graso sintasa, es suficiente para inhibir la actividad de ácido graso sintasa. Una "cantidad terapéuticamente efectiva", cuando se administra a un sujeto para tratar una enfermedad, es suficiente para efectuar el tratamiento para esa enfermedad.
Excepto cuando se indica, los términos "sujeto" o "paciente" se usan de forma intercambiable y se refieren a mamíferos tales como pacientes humanos y primates no humanos, así como animales experimentales tales como conejos, ratas y ratones, y otros animales. Por consiguiente, el término "sujeto" o "paciente", tal como se usa en el presente documento, significa cualquier paciente o sujeto mamífero al que se le pueden administrar los compuestos de la invención. En un aspecto ejemplar de la presente invención, para identificar pacientes sujeto para tratamiento de acuerdo con los procedimientos de la invención, se emplean procedimientos de cribado aceptados para determinar factores de riesgo asociados con una enfermedad o afección diana o que se sospecha o para determinar el estado de una enfermedad o afección existente en un sujeto. Estos procedimientos de cribado incluyen, por ejemplo, metodologías diagnósticas convencionales para determinar factores de riesgo que están asociados con la enfermedad o afección diana o que se sospecha. Estos y otros procedimientos rutinarios permiten al facultativo seleccionar pacientes que necesitan terapia usando los procedimientos y las formulaciones de la presente invención.
Moduladores de la ruta de FASN
Un aspecto de la presente descripción incluye compuestos para uso en un procedimiento de inhibición de una infección vírica o tratamiento de cáncer poniendo en contacto una célula con un agente que modula la ruta de síntesis de ácidos grasos. Este procedimiento de inhibición de una infección vírica o tratamiento de cáncer puede realizarse in vitro poniendo en contacto células infectadas viralmente/cancerosas con un agente que modula la ruta de síntesis de ácidos grasos, o in vivo administrando un agente que modula la ruta de síntesis de ácidos grasos a un sujeto infectado con un virus/que tiene cáncer. En un aspecto, un agente puede ser un inhibidor de la ruta de síntesis de ácidos grasos. Los ejemplos de inhibidores de la ruta de síntesis de ácidos grasos que pueden usarse en los procedimientos y composiciones de la presente descripción se describen a continuación.
Compuestos de estructura (VI)
En diversos aspectos, la presente descripción proporciona compuestos de Estructura (Vl-J):

o una sal farmacéuticamente aceptable de los mismos, donde:
R1 es H, -CN, halógeno, alquilo Ci-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo Ci-C4 lineal o ramificado) en donde:
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;
cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;
R3 es H, -OH, o halógeno;
R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;
R22 es H, halógeno, o alquilo C1-C2;
R35 es -C(O)-R351, -C(O)-NHR351, -C(O)-O-R351 o S(O)2R351; y
R351 es alquilo C1-C6 lineal o ramificado, cicloalquilo, heterociclilo, arilo o heteroarilo, cada uno de los cuales está opcionalmente sustituido.
En algunos aspectos de Estructura (VI-J), R3 es H o halógeno.
En algunos aspectos de Estructura (VI-J), R1 es halógeno, -CN o haloalquilo C1-C2
En algunos aspectos de Estructura (VI-J), R22 es alquilo C1-C2.
En algunos aspectos de Estructura (VI-J), R21 es ciclobutilo y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (VI-J), R21 es ciclobutilo.
En algunos aspectos de Estructura (VI-J), R3 es H o F.
En algunos aspectos de Estructura (VI-J), R1 es -CN.
En algunos aspectos de Estructura (VI-J), R1 es -CF3.
En algunos aspectos de Estructura (VI-J), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (VI-J), R22 es H.
En algunos aspectos de Estructura (VI-J), R22 es metilo.
En algunos aspectos de Estructura (VI-J), R35 es -C(O)-NHR351.
En algunos aspectos de Estructura (VI-J), R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo,
(R)-(tetrahidrofuran-2-ilo)metilo, (S)-(tetrahidrofuran-2-ilo)metilo, (R)- tetrahidro-2H-piran-3-ilo o (S)-tetrahidro-2H-piran-3-ilo.
En algunos aspectos de Estructura (VI-J), R351 es (R)-(tetrahidrofuran-2-ilo)metilo o (S)-(tetrahidrofuran-2-ilo)metilo.
En algunos aspectos de Estructura (VI-J), R1 es -Cn , cada R2 es H, R3 es H o F, R21 es cicloalquilo R35 es -C(O)-NHR351 donde R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-ilo)metilo, (S)-(tetrahidrofuran-2-ilo)metilo, (R)-tetrahidro-2H-piran-3-ilo, o (S)-tetrahidro-2H-piran-3-ilo.
En algunos aspectos de Estructura (VI-J), R35 es -C(O)-O-R351.
En algunos aspectos de Estructura (VI-J), R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo,
(R)-(tetrahidrofuran-2-ilo)metilo, (S)-(tetrahidrofuran-2-ilo)metilo, (R)- tetrahidro-2H-piran-3-ilo, o (S)-tetrahidro-2H-piran-3-ilo.
En algunos aspectos de Estructura (VI-J), R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4 , R22 es H,
R35 es -C(O)-O-R51 donde R351 es isopropilo, isobutilo, (R)-3-tetrahidrofuranilo, (S)-3-tetrahidrofuranilo, (R)-(tetrahidrofuran-2-ilo)metilo, (S)-(tetrahidrofuran-2-ilo)metilo, (R)- tetrahidro-2H-piran-3-ilo, o (S)-tetrahidro-2H-piran-3-ilo.
En algunos aspectos de Estructura (VI-J), R351 es (R)-3-tetrahidrofuranilo o (S)-3-tetrahidrofuranilo.
En algunos aspectos de Estructura (VI-J), los compuestos tienen una estructura seleccionada del grupo que consiste en:

En diversos aspectos, se proporcionan compuestos de Estructura (X):

o una sal farmacéuticamente aceptable de los mismos, en donde:
R1 es H, -CN, halógeno, alquilo C i-C4 lineal o ramificado, -O-(cicloalquilo C3-C5 ), -O-(alquilo Ci-C4 lineal o ramificado) en donde:
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;
cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 lineal o ramificado;
R3 es H, -OH o halógeno;
L3 es C(R60)2, O o NR50;
cada R60 es independientemente H, -OH, -CN, -Ot-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado), o -C(O)-N(R601)2 en donde:
t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno
cada R50 es independientemente H, -C(O)-Ot-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C3-C5 cíclico), -alquilo C3-C5 cíclico que contiene opcionalmente un heteroátomo de oxígeno o nitrógeno, -C(O)-N(R501)2 , alquilo C1-C4 lineal o ramificado en donde:
t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;
n es 1, 2 o 3;
m es 1 o 2 ;
R21 es H, halógeno, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 en donde el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno
R22 es H, halógeno, alquilo C1-C2;
cada R26 es independientemente -OH, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -(alquilo C1-C4)t-Ot-(cicloalquilo C3-C5), -(alquilo C1-C4)t-O-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot(alquilo C1-C4), o -C(O)-N(R501)2 en donde: t es 0 o 1, y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno un heteroátomo de oxígeno o nitrógeno; s es 0 , 1 o 2 ;
cada R601 and R501 es independientemente H o alquilo C1-C4 lineal o ramificado; y
en donde dos de R26, R60, R50, R501 y R601 se unen opcionalmente para formar un anillo en donde los dos de R26, R60, R50, R501 y R601 pueden ser dos R26, dos R60, dos R50, dos R501 o dos R601.
En algunos aspectos de Estructura (X), R21 es halógeno, alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C5.
En algunos aspectos de Estructura (X), R3 es H o halógeno.
En algunos aspectos de Estructura (X), R1 es -CN o haloalquilo C1-C2.
En algunos aspectos de Estructura (X), R3 es H o F.
En algunos aspectos de Estructura (X), R1 es -CN.
En algunos aspectos de Estructura (X), R1 es-CF3.
En algunos aspectos de Estructura (X), n es 1.
En algunos aspectos de Estructura (X), n es 2.
En algunos aspectos de Estructura (X), m es 1
En algunos aspectos de Estructura (X), m es 2.
En algunos aspectos de Estructura (X), R21 es alquilo C1-C2 o cicloalquilo C3-C5 y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (X), R21 es cicloalquilo C3-C5 y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (X), n es 2, m es 1, L3 es -N-C(O)-O-(alquilo C1-C2 ).
En algunos aspectos de Estructura (X), L3 es NR51 ; R50 es alquilo C1-C2 ; R21 es ciclobutilo; R22 es H o metilo; R3 es H; R1 es -CN; m es 2 y n es 1 o 2.
En algunos aspectos de Estructura (X), n es 2, m es 1, L3 es O y s es 0.
En algunos aspectos de Estructura (X), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (X), R22 es metilo.
En algunos aspectos de Estructura (X), R22 es H.
En algunos aspectos de Estructura (X), R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4 , R22 es metilo,
n es 2 y L3 es NR50 donde R50 es metilo o etilo.
En algunos aspectos de Estructura (X), R1 es -CN, cada  cicloalquilo C3-C4 , R22 n es 2 y L3 es O.
cicloalquilo C3-C4 , R22 n es 2 y L3 es O.
En algunos aspectos de Estructura (X), el compuesto tiene una estructura seleccionada del grupo que consiste en:

En diversos aspectos, se proporcionan compuestos de Estructura (XI):

o a pharmaceutically acceptable salt thereof, wherein:
R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado , -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado
) en donde:
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;
cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado alquilo;
R3 es H, -OH, o halógeno;
R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;
R22 es H, halógeno, alquilo C1-C2 ; y
R351 es alquilo C1-C2 o C2-O-(alquilo C1 o C2 ).
En algunos aspectos de Estructura (XI), R3 es H o halógeno.
En algunos aspectos de Estructura (XI), R1 es halógeno, -CN o haloalquilo C1-C2 .
En algunos aspectos de Estructura (XI), R21 es cicloalquilo C3-C4 y R22 es alquilo C1-C2.
En algunos aspectos de Estructura (XI), R21 es ciclobutilo and R22 es alquilo C1-C2 .
En algunos aspectos de Estructura (XI), R21 es ciclobutilo.
En algunos aspectos de Estructura (XI), R3 es H o F.
En algunos aspectos de Estructura (XI), R1 es -CN.
En algunos aspectos de Estructura (XI), R1 es -CF3.
En algunos aspectos de Estructura (XI), R22 es H, metilo o etilo.
En algunos aspectos de Estructura (XI), R22 es H.
En algunos aspectos de Estructura (XI), R22 es metilo.
En algunos aspectos de Estructura (XI), R1 es -CN, cada R2 es H, R3 es H o F, R21 es ciclobutilo, R22 es metilo and
R351 es metilo o etilo.
En al unos aspectos de Estructura XI , el compuesto tiene una estructura seleccionada del grupo que consiste en:

En el presente documento también se describen procedimientos de síntesis de los compuestos de la presente descripción. Los compuestos de la presente descripción pueden sintetizarse tal como se indica en los ESQUEMAS SINTÉTICOS 1-13 a continuación.

R" es hidrógeno o alquilo;
Ri es hidrógeno, ciano, halo, alquilo Ci -6, alcoxi Ci -6, -C(=O)N(Ri3)(Ri4), - (CH2)qC(=O)N(Ri3)(Ri4), CF3, -OCF3 o -S(=O)2R20;
q es 0, 1, 2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6 o N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6 o alquilo C1.6;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son, cada uno independientemente, hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3, -OCF3 o -S(=O)2R20; R13 y R14 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20;
R15 y R16 son, cada uno independientemente, hidrógeno, alquilo C1.6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino; y
R17 es hidrógeno o alquilo.

Ri es hidrógeno, ciano, halo, alquilo Ci -6, alcoxi Ci -6, -C(=O)N(Ri3)(Ri4), - (CH2)qC(=O)N(Ri3)(Ri4), CF3, -OCF3 o -S(=O)2R2o;
q es 0, 1, 2, 3 o 4;
R20 es hidrógeno o alquilo C1.6, alcoxi C1-6 o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1.6 o alquilo C1-6;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son, cada uno independientemente, hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3, -OCF3 o -S(=O)2R20; R13 y R14 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20;
R15 y R16 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino;
R23 es hidrógeno, N(R13)(Rm ), alquilo C1.6, alcoxi C1.6, está ausente si L1 es N, o R23 y R24 tomados juntamente con los átomos a los que están fijados, se unen entre sí para formar un heterociclilo, heteroarilo o cicloalquilo; y R24 es hidrógeno, -N(R13)(R14), alquilo C1-6, alcoxi C1-6, -(alcoxi C1-6)(heterociclilo), heterociclilo, o R23 y R24 tomados juntamente con los átomos a los que están fijados, se unen entre sí para formar un heterociclilo, heteroarilo o cicloalquilo

LG es un grupo saliente;
Nu es un nucleófilo;
L2, L3, L4 y L4' son, cada uno independientemente, CH o N;
Ri es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(R13)(R14), - (CH2)qC(=O)N(R13)(R14), CF3, -OCF3 o -S(=O)2R20;
q es 0, 1, 2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6 o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6 o alquilo C1.6;
R3 es hidrógeno, hidroxilo, halo, alquilo C1-6 o alcoxi C1.6;
R21 y R22 son, cada uno independientemente, hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3, -OCF3 o -S(=O)2R20; R13 y R14 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20;
R15 y R16 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino; y
R17 es hidrógeno o alquilo.

Ri es hidrógeno, ciano, halo, alquilo Ci -6, alcoxi Ci -6, -C(=O)N(Ri3)(Ri4), - (CH2)qC(=O)N(Ri3)(Ri4), CF3, -OCF3 o -S(=O)2R2o;
q es 0, 1, 2, 3 o 4;
R20 es hidrógeno o alquilo C1.6, alcoxi C1-6 o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1.6 o alquilo C1-6;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son, cada uno independientemente, hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3, -OCF3 o -S(=O)2R20; R13 y R14 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20;
R15 y R16 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino;
R17 es hidrógeno o alquilo; y
R24 es hidrógeno, -N(R13)(R14), alquilo C1.6, alcoxi C1.6, -(alcoxi C1-6)(heterociclilo) o heterociclilo.

donde:
Ri es hidrógeno, ciano, halo, alquilo Ci -6, alcoxi Ci -6, -C(=O)N(Ri3)(Ri4), - (CH2)qC(=O)N(Ri3)(Ri4), CF3, -OCF3 o -S(=O)2R2o;
q es 0, 1, 2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6 o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6 o alquilo C1.6;
R3 es hidrógeno, hidroxilo, halo, alquilo C1-6 o alcoxi C1.6;
R21 y R22 son, cada uno independientemente, hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6,
CF3,-OCF3 o-S(=O)2R20;
R13 y R14 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20;
R15 y R16 son, cada uno independientemente, hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo,
hidroxialquMo o alquilamino;
R17 es hidrógeno o alquilo;
R24 es hidrógeno, -N(Ri3)(Ri4), alquilo Ci -6, alcoxi Ci -6, -(alcoxi Ci -6)(heterociclilo) o heterociclilo;
R29 es hidrógeno, alquilo C1-6, alcoxi Ci -6, hidroxialquilo, heteroarilo, heterociclilo, -N(Ri5Ri6), -C(=O)R46 o -R48C(=O)R47;
R34 es hidrógeno, alquilo Ci -6, alcoxi Ci -6, ciclo alquilo, hidroxilo, hidroxialquilo, arilo, heterociclilo, heteroarilo, alquilamino, CF3, -OCF3, -S(=O)2R20 o -N(Ri5Ri6); y

Esquema 8
Esquema 11

Esquema 12
BuLi: a continuación POCI
A r-B r/l E3ocN B ocN *^íi 1) H2, Pd/C 



Procedimientos adicionales para producir compuestos particulares de acuerdo con la presente descripción están provistos en los EJEMPLOS. Un experto en la técnica reconocerá que otros compuestos de estructuras pueden prepararse mediante modificaciones a los esquemas descritos específicamente que emplean procedimientos conocidos por los expertos en la materia. Pueden encontrarse ejemplos adicionales en la tabla 1. Los ejemplos que no caen dentro del alcance de las reivindicaciones solo se proporcionan con propósitos ilustrativos.
Muchas de dichas técnicas son bien conocidas en la técnica. Sin embargo, muchas de las técnicas conocidas se elaboran en Compendium of Organic Synthetic Methods (Vol. 1, 1971; Vol. 2, 1974; Vol. 3, 1977; Vol. 4, 1980; Vol. 5, 1984; y Vol. 6 así como March in Advanced Organic Chemistry (1985); Comprehensive Organic Synthesis. Selectivity, Strategy & Efficiency in Modern Organic Chemistry. En 9 volúmenes (1993); Advanced Organic Chemistry Part B: Reactions and Synthesis, Second Edition (1983); Advanced Organic Chemistry, Reactions, Mechanisms, and Structure, Second Edition (1977); Protecting Groups in Organic Synthesis, Second Edition; y Comprehensive Organic Transformations (1999).
Rutas de infección vírica
Las dianas de célula huésped inhibidas por los presentes compuestos y procedimientos desempeñan un papel en las rutas de replicación y/o infección vírica. El direccionamiento a dichas dianas de célula huésped modula las rutas de replicación y/o infección de los virus. En aspectos preferidos, las dianas de célula huésped identificadas se modulan directa o indirectamente usando las composiciones de la presente descripción. La modulación de dichas dianas de célula huésped también puede realizarse dirigiéndose a entidades en las rutas de señalización cadena arriba o cadena abajo de las dianas de célula huésped.
De acuerdo con la presente descripción, la infección vírica puede tratarse dirigiéndose a la ruta de síntesis de ácidos grasos, y en particular ácido graso sintasa. El RVH es representativo de virus que pueden tratarse de acuerdo con la presente descripción. Como otros virus, la replicación de RVH implica seis fases; transmisión, entrada, replicación, biosíntesis, ensamblaje y salida. La entrada se produce mediante endocitosis, la replicación y el ensamblaje de la vRNP tienen lugar en el núcleo, y el virus surge de la membrana plasmática. En el paciente infectado, el virus se dirige a células epiteliales de las vías respiratorias. Los presentes compuestos y procedimientos se dirigen a y modulan al menos una diana de célula huésped implicada en dichas rutas.
Para algunos virus, se ha realizado un buen progreso en la elucidación de las etapas implicadas durante la infección de células huésped. Por ejemplo, experimentos iniciados a principios de los años 1980 mostraron que los virus de la gripe siguen una programa de entrada endocítica por etapas con elementos compartidos con otros virus tales como alfa- y rabdovirus (Marsh y Helenius 1989; Whittaker 2006). Las etapas incluyen: 1) Fijación inicial a receptores glucoconjugados que contienen ácido siálico sobre la superficie celular; 2) señalización inducida por la partícula de virus; 3) endocitosis mediante mecanismo celular dependiente de clatrina e independiente de clatrina; 4) penetración inducida por ácido, mediada por hemaglutinina (HA) a partir de endosomas tardíos; 5) desencapsulación de la cápsida dependiente de M2 y la proteína de la matriz (M1), activada por ácido; y, 6) transporte intra-citosólico e importación nuclear de la vRNP. Estas etapas dependen de la asistencia por parte de la célula huésped en forma de receptores de clasificación, maquinaria de formación de vesículas, regulación mediada por quinasa, acidificación de orgánulos y, de la forma más probable, actividades del citoesqueleto.
La fijación de la gripe a la superficie de las células se produce mediante unión de la subunidad HA1 a glucoproteínas y glucolípidos de la superficie celular que portan fracciones de oligosacáridos con residuos de ácido siálico terminales (Skehel and Wiley 2000). El enlace mediante el cual el ácido siálico está conectado al siguiente sacárido contribuye a la especificidad por especies. Las cepas aviares, incluyendo H5N1, prefieren un enlace a-(2,3) y las cepas humanas un enlace a-(2,6) (Matrosovich 2006). En las células epiteliales, la unión se produce preferentemente a microvellosidades en la superficie apical, y la endocitosis se produce en la base de estas extensiones (Matlin 1982). Aún no se conoce si la unión al receptor induce señales que preparan a la célula para la invasión, pero es probable
dado que se requieren activación de proteína quinasa C y síntesis de fosfatidilinositol-3-fosfato (PI3P) para una entrada eficiente (Sieczkarski et al. 2003; Whittaker 2O06).
La internalización endocítica se produce en el plazo de pocos minutos después de la unión (Matlin 1982; Yoshimura y Ohnishi 1984). En células de cultivo tisular, el virus de la gripe utiliza tres tipos diferentes de procesos celulares; 1) hoyos revestidos de clatrina preexistentes, 2) hoyos revestidos de clatrina inducidos por virus y 3) endocitosis en vesículas sin revestimiento visible (Matlin 1982; Sieczkarski y Whittaker 2002; Rust et al. 2004). La videomicroscopía usando virus fluorescentes mostró que las partículas de virus que experimentan movimiento rápido mediado por actina en la periferia celular, seguido por transporte mediado por microtúbulos, dirigido al extremo menos, hasta la zona perinuclear de la célula. La imaginología de células vivas indicaba que las partículas de virus entraban en primer lugar en una subpoblación de endosomas tempranos periféricos, móviles que los transportan a mayor profundidad en el citoplasma antes de que tenga lugar la penetración (Lakadamyali et al. 2003; Rust et al. 2004). El proceso endocítico está regulado por proteína y lípido quinasas, el proteasoma, así como por Rabs y factores de clasificación dependientes de ubiquitina (Khor et al. 2003; Whittaker 2006).
La etapa de penetración en la membrana está mediada por activación mediada por pH bajo de la HA metaestable trimérica, y la conversión de esta proteína de fusión viral de tipo I en una conformación competente para fusión a membranas (Maeda et al. 1981; White et al. 1982). Esto se produce aproximadamente 16 minutos después de la internalización, y el umbral de pH varía entre cepas en el intervalo 5,0-5,6. La membrana diana es la membrana limitante de endosomas intermedios o tardíos. El mecanismo de fusión ha sido estudiado exhaustivamente (Kielian y Rey 2006). Además, se observó que la propia fusión no parece requerir ningún componente de la célula huésped excepto una membrana de bicapa lipídica y un sistema de acidificación funcional (Maeda y col, 1981; White et al.
1982). La etapa de penetración es inhibida por agentes tales como bases débiles lisosomotrópicas, ionóforos carboxílicos e inhibidores de bomba de protones (Matlin 1982; Whittaker 2006).
Para permitir la importación nuclear de las vRNP entrantes, la cápsida debe ser desmontada. Esta etapa implica la acidificación del interior viral a través de canales M2 sensibles a amantadina, causa la disociación de M1 a partir de las vRNP (Bukrinskaya et al. 1982; Martin y Helenius 1991; Pinto et al. 1992). El transporte de las vRNP individuales a los complejos de poro nuclear y transferencia al interior del núcleo depende de los receptores del transporte nuclear celular (O'Neill et al. 1995; Cros et al. 2005). La replicación de los ARN virales (síntesis de cadenas positivas y negativas), y la transcripción se produce en complejos estrechamente asociados con la cromatina en el núcleo. Es evidente que, aunque muchas de las etapas están catalizadas por la polimerasa viral, están implicados factores celulares que incluyen factores de activación de ARN polimerasa, una chaperona HSP90, hCLE y un factor de splicing humano UAP56. La expresión de genes virales está sujeta a complejo control celular a nivel transcripcional, un sistema de control dependiente de quinasas celulares (Whittaker 2006).
El ensamblaje final de una partícula de la gripe se produce durante un proceso de gemación en la membrana plasmática. En células epiteliales, la gemación se produce en el dominio de membrana apical solamente (Rodriguez-Boulan 1983). En primer lugar, las vRNP de la progenia son transportadas dentro del nucleoplasma a la envuelta nuclear, a continuación desde el núcleo al citoplasma, y finalmente se acumulan en la periferia celular. La salida del núcleo depende de la proteína viral NEP y M1, y diversas proteínas celulares incluyendo CRM1 (un receptor de exportación nuclear), caspasas y posiblemente algunas chaperonas de proteína nuclear. La fosforilación desempeña un papel en la exportación nuclear regulando la síntesis de M1 y NEP, y también a través del sistema MAPK/ERK (Bui et al. 1996; Ludwig 2006). La señalización de proteína G y proteína quinasa está implicada en la gemación del virus de la gripe a partir de células infectadas (Hui E. y Nayak D, 2002).
Las tres proteínas de membrana del virus se sintetizan, se pliegan y se ensamblan en oligómeros en el ER (Doms et al. 1993). Pasan a través del complejo de Golgi; experimentan maduración a través de modificación de sus fracciones de carbohidratos y escisión proteolítica. Después de alcanzar la membrana plasmática, se asocian con M1 y las vRNP en un proceso de gemación que da como resultado la inclusión de las ocho vRNP y la exclusión de la mayoría de los componentes de célula huésped excepto lípidos.
La infección por gripe está asociada con la activación de varias cascadas de señalización que incluyen la ruta MAPK (ERK, JNK, p38 y BMK-1/ERK5), el módulo de señalización IkB/NF-kB, la cascada Raf/MEK/ERK, y muerte celular programada (Ludwig 2006). Estas dan como resultado diversos efectos que limitan el progreso de la infección tales como la activación transcripcional de IFNb, muerte celular apoptótica, y un bloqueo en el escape en virus a partir de endosomas tardíos (Ludwig 2006).
La mayoría de los estudios previos sobre interacciones virus-célula se realizaron en cultivo tisular usando cepas de virus adaptadas al crecimiento en cultivo tisular o en huevos. Los virus en estos ejemplos se adaptaron de tal manera que se indujeron cambios que afectaron a la unión al receptor y al tropismo (Matrosovich 2006). La infección con cepas patógenas de tipo silvestre proporciona un panorama más natural de la infección vírica con proteínas del huésped. Se sabe que en las vías respiratorias humanas la gripe A y B infectan principalmente células epiteliales no ciliadas en las vías respiratorias superiores que portan NeuSAc a-(2,6)-Gal, mientras que las cepas aviares infectan células epiteliales ciliadas con ácidos siálicos enlazados en a-(2,3) a mayor profundidad en las vías respiratorias (Matrosovich et al. 2004a).
Adicionalmente, se han realizado progresos en la elucidación de las etapas implicadas durante la infección por RVH de células huésped. Puede considerarse que acontecimientos seleccionados en infección por rinovirus de las vías respiratorias humanas normales se producen secuencialmente. Se cree que las etapas iniciales en la patogenia de rinovirus incluyen entrada viral a través de la nariz, transporte mucociliar de virus a la faringe posterior, e inicio de la infección en células epiteliales ciliadas y no ciliadas de las vías respiratorias superiores. La replicación viral alcanza
un máximo de promedio en el plazo de 48 h desde el inicio de la infección y persiste durante hasta 3 semanas. La infección viene seguida por activación de varios mecanismos inflamatorios, que pueden incluir liberación o generación de interleucinas, bradiquininas, prostaglandinas, y posiblemente histamina y estimulación de reflejos parasimpáticos. Se inician procesos patofisiológicos, que incluyen vasodilatación de vasos sanguíneos nasales, trasudación de plasma, secreción glandular y estimulación de fibras nerviosas, causando dolor y desencadenando reflejos de estornudo y tos. La dolencia clínica resultante es una rinosinusitis, faringitis y bronquitis, que, de promedio, dura una semana.
Se han identificado cambios en los perfiles de expresión génica durante infecciones por rinovirus in vivo (Proud D. et al. Am J Respir Crit Care Med Vol 178. págs. 962-968, 2008). Se obtuvieron raspados epiteliales nasales antes de y durante infección por rinovirus experimental, y la expresión génica se evaluó mediante micromatriz. La viperina está identificada como una proteína antiviral inducida por interferón (IFN), infecciones víricas y moléculas asociadas a patógenos. Se usaron infecciones por rinovirus adquiridas de forma natural, células epiteliales humanas cultivadas e inactivación de ARN interferente pequeño evaluar adicionalmente el papel de la viperina en las infecciones por rinovirus. Se midieron puntuaciones de síntomas y valores cuantitativos virales en sujetos inoculados con rinovirus o un control simulado, y los cambios en la expresión génica se valoraron 8 y 48 horas después de la inoculación. No se observaron cambios inducidos por rinovirus en la expresión génica 8 horas después de la infección vírica, pero 11887 transcritos génicos fueron alterados significativamente en raspados obtenidos 2 días post-inoculación. Los grupos principales de genes regulados positivamente incluyen quimiocinas, moléculas de señalización, genes sensibles a interferón y antivirales. La infección por rinovirus altera significativamente la expresión de muchos genes asociados con la respuesta inmunitaria, incluyendo quimiocinas y antivirales. Algunos de los genes inducidos marcadamente por infección por RVH-16 incluyen CCL2, CCL8 , CXCL11, CXCL10, CXCL13, CXCL9, CCL20, IFIT2, GBP1, IFIT1, GIP2, IFIT4, IL28B, IRF7, CIG5, NOS2A, OAS3, OASL, OAS2, OAS1, MX2, MX1, PLSCR1, SOCS1, SOCS2, MDA5, RIGI, SOCS3, ICAM-1, HAPLN3, MMP12, EPSTI1 y TNC.
Ruta de síntesis de ácidos grasos
Diversos aspectos de la presente descripción se refieren a composiciones y procedimientos que modulan la actividad de la ruta de síntesis de ácidos grasos para tratar una infección vírica o tratar el cáncer. La ruta de síntesis de ácidos grasos en seres humanos puede usar cuatro enzimas: 1) acetil-CoA carboxilasa (ACC), que puede sintetizar malonil-CoA; 2) enzima málica, que puede producir NADPH; 3) citrato liasa, que puede sintetizar acetil-CoA; y 4) ácido graso sintasa, que puede catalizar la síntesis de ácidos grasos dependiente de NADPH a partir de acetil-CoA y malonil-CoA. En diversos aspectos, la presente descripción se refiere al tratamiento de infecciones víricas y cáncer modulando la actividad de la proteína ácido graso sintasa.
Los productos finales de ácido graso sintasa son ácidos grasos libres que pueden usar derivatización enzimática separada con coenzima-A para incorporación en otros productos. En seres humanos, la síntesis de ácidos grasos puede producirse en dos lugares: el hígado, donde se puede preparar ácido palmítico (Roncari, (1974) Can. J. Biochem., 52:221-230) y glándula mamaria durante la lactancia, donde pueden prepararse ácidos grasos C10-C14 (Thompson, et al., (1985) Pediatr. Res., 19: 139-143).
Los ácidos grasos pueden sintetizarse en el citoplasma a partir de acetil-CoA. La acetil-CoA puede generarse a partir de piruvato por piruvato deshidrogenasa (PDH) y por p-oxidación de ácidos grasos en la mitocondria. Una "lanzadera de citrato" puede transportar acetil-CoA desde la mitocondria al citoplasma. La acetil-CoA puede reaccionar con oxaloacetato para dar citrato, y una tricarboxilato translocasa puede transportar citrato desde la mitocondria al citosol. En el citoplasma, el citrato puede escindirse de vuelta a oxaloacetato y acetil-CoA, una reacción que puede estar catalizada por ATP-citrato liasa. El oxaloacetato puede volver a convertirse en piruvato para reentrada en la mitocondria.
La acetil-CoA puede convertirse en malonil-CoA. La acetil-CoA carboxilasa (ACC) es un complejo sistema enzimático multifuncional, que contiene biotina que puede catalizar la carboxilación de acetil-CoA a malonil-CoA. Esta conversión es una etapa limitante de la velocidad e irreversible en la síntesis de ácidos grasos. La ACC puede llevar a cabo tres funciones: proteína vehículo de biotina carboxilo, biotina carboxilasa y carboxiltransferasa. La carboxilación dependiente de ATP de biotina, un grupo prostético (cofactor) puede venir seguida por la transferencia del grupo carboxilo a acetil-CoA.
HCO3' ATP acetil-CoA -> ADP Pi malonil-CoA
Existen dos formas de ACC, alfa y beta, codificadas por dos genes diferentes ACC-alfa (también conocido como ACC, ACAC, ACC1, ACCA y ACACA) puede codificar proteína altamente enriquecida en tejidos lipógenos. Para este gen se han descubierto múltiples variantes de transcrito sometidas a splicing de forma alternativa divergentes en la secuencia y que codifican distintas isoformas. ACC-beta (también conocido como ACC2, ACCB, HACC275 y ACACB) puede codificar proteína que se piensa que controla la oxidación de ácidos grasos por medio de la capacidad de malonil-CoA de inhibir la carnitina-palmitoil-CoA transferasa I, la etapa limitante de la velocidad en una captación y oxidación de ácidos grasos por la mitocondria. ACC-beta puede estar implicada en la regulación de la oxidación de ácidos grasos, en lugar de la biosíntesis de ácidos grasos. Existen indicios de la presencia de dos isoformas de ACC-beta.
La ACC puede estar regulada por la fosforilación/desfosforilación de residuos de serina diana. Por ejemplo, la quinasa activada por AMP (AMPK) puede fosforilar ACC, y esta fosforilación puede inhibir la capacidad de ACC para producir malonil-CoA. En a Ca CA, la AMPK puede fosforilar Ser79, Ser1200 y Ser1215 (Park S.H. et al. (2002) J. Appl. Physiol.
92:2475-82). La AMPK puede fosforilar Ser218 en ACACB (Hardie D.G. (1992) Biochim. Biophys. Acta 1123:231-8). Además, la proteína quinasa dependiente de cAMP (proteína quinasa A, o PKA) puede fosforilar ACC.
La ACC puede regularse mediante transformación alostérica mediante citrato o palmitoil-CoA. Por ejemplo, el citrato puede ser un efector positivo (es decir, el citrato puede activar alostéricamente la ACC). La concentración de citrato puede ser alta cuando entra el acetil-CoA adecuado en el ciclo de Krebs. El exceso de acetil-CoA puede convertirse a continuación mediante malonil-CoA en ácidos grasos. El palmitoil-CoA puede ser un efector negativo. El palmitoil-CoA, que es el producto de la ácido graso sintasa (FASN), puede promover la conformación inactiva de ACC, que puede reducir la producción de malonil-CoA (un proceso de inhibición de la retroalimentación). El AMP puede regular la síntesis de ácidos grasos regulando la disponibilidad de malonil-CoA. La unión de insulina a un receptor puede activar una fosfatasa para desfosforilar ACC, lo que puede eliminar el efecto inhibidor.
El gen de la ácido graso sintasa (también conocido como FAS, OA-519, SDR27X1; MGC14367; MGC15706; FASN) está implicado en la síntesis de ácidos grasos. La enzima codificada por este gen es una proteína multifuncional de aproximadamente 272 kDa con múltiples dominios, cada uno con distintas actividades enzimáticas que pueden desempeñar un papel en la biosíntesis de ácidos grasos. La FASN puede catalizar la síntesis de palmitato a partir de acetil-CoA y malonil-CoA, en presencia de NADPH, en ácidos grasos saturados de cadena larga. En algunas líneas celulares de cáncer, se ha descubierto que la proteína FASN está fusionada con el receptor de estrógenos alfa (ER-alfa), en el que el extremo N de FASN se fusiona en marco con el extremo C de ER-alfa.
La proteína FASN puede existir en el citosol como un dímero de subunidades idénticas. FASN consiste en tres dominios catalíticos en la sección N-terminal (-cetoacil sintasa (KS), malonil/acetiltransferasa (MAT) y deshidrasa (DH)). La sección N-terminal está separada por una región central de aproximadamente 600 aminoácidos de cuatro dominios C-terminales (enoil reductasa (ER), -cetoacil reductasa (KR), proteína vehículo de acilo (ACP) y tioesterasa (TE)). La estructura cristalina de una ácido graso sintasa de mamífero se ha descrito (Maier T. et al. (2008) Science 321: 1315-1322). Cada uno de los dominios catalíticos de FASN puede ser seleccionado como diana en los procedimientos de direccionamiento a una infección vírica de la invención proporcionada.
Las etapas enzimáticas de la síntesis de ácidos grasos pueden implicar condensación descarboxilativa, reducción, deshidratación, y otra reducción y pueden dar como resultado una fracción acilo saturada. El NADPH puede ser un donador de electrones en reacciones reductoras.
Actividad antiviral
En diversos aspectos, la presente descripción proporciona compuestos para uso en procedimientos para tratar la infección vírica en un sujeto, comprendiendo el procedimiento administrar a un sujeto que necesita dicho tratamiento una cantidad efectiva de un compuesto de Estructuras (VI-J), (X), o (XI).En diversos aspectos, la descripción proporciona compuestos para uso en procedimientos para tratar una infección vírica, comprendiendo el procedimiento administrar los compuestos de la presente descripción a un sujeto que necesita el agente.
La presente descripción contempla el tratamiento de cualquier infección vírica que se dirige a la ruta de síntesis de ácidos grasos en un huésped y, en particular, modulando la actividad de la ácido graso sintasa. Por ejemplo, los presentes procedimientos pueden usarse para tratar infección por gripe, infección por adenovirus, infección por virus respiratorio sincitial, infección por poxvirus, infección por poliomielitis, infección por hepatitis C, infección por fiebre amarilla, infección por fiebre del dengue, infección por rinovirus, y similares.
En diversos aspectos, la presente descripción proporciona procedimientos para tratar infección por hepatitis C administrando al sujeto uno o más compuestos descritos en el presente documento. En la modulación de la ruta de FASN en el sujeto, la infección por hepatitis C es tratada. Se ha demostrado que la expresión de FASN está regulada positivamente en la línea celular de hepatoma humano Huh7 cuando estás células son infectadas con VHC. La inhibición de la producción de FASN con un inhibidor de FASN redujo la producción de VHC. Por lo tanto, la administración a un sujeto de los compuestos de la presente descripción. (Yang, W. et al.(2008) Hepatology 48(5):1396-1403). Se ha demostrado en los EJEMPLOS que la inhibición de FASN se correlaciona con la inhibición del VHC.
En ciertos aspectos, los procedimientos de inhibición de una infección vírica pueden realizarse in vitro. En aspectos adicionales, los procedimientos de inhibición de una infección vírica pueden realizarse in vivo.
En ciertos aspectos, los compuestos de la presente descripción pueden usarse en combinación con otros tratamientos antivirales en el tratamiento de una infección vírica.
En diversos aspectos, la infección vírica es una infección por fiebre amarilla humana. En aspectos adicionales, la infección vírica es una infección por hepatitis C humana. En otros aspectos adicionales, la infección vírica es una infección rinovírica humana.
En diversos aspectos, los compuestos de la presente descripción pueden usarse para el tratamiento de infección de un sujeto animal, tal como un ser humano, por cualquiera de una plétora de virus.
En ciertos aspectos, los compuestos de la presente descripción pueden usarse para la inhibición de un huésped por un virus respiratorio. Los virus respiratorios son transmitidos de la forma más común por gotas o secreciones nasales transportadas por el aire y pueden causar un amplio espectro de dolencias. Los virus respiratorios incluyen el virus respiratorio sincitial (VRS), virus de la gripe, coronavirus tales como SRAG, adenovirus, virus paragripales y rinovirus (RVH).
De acuerdo con un aspecto, la presente descripción puede usarse para tratar infección por RVH. El género de los rinovirus es un miembro de la familia de virus Picornaviridae. Los géneros dentro de la familia incluyen el género Enterovirus, Rhinovirus, Cardiovirus, Aphthovirus, Hepatovirus, Parechovirus, Erbovirus, Kobuvirus, Teschovirus. Los rinovirus humanos (RVH) incluyen los virus más comunes que infectan a seres humanos y pueden causar el resfriado común. Los RVH son de naturaleza lítica. Los rinovirus tienen genomas de ARN positivo monocatenario de entre 7,2 y 8,5 kb de longitud. En el extremo 5' de estos genomas hay una proteína codificada por el virus, y al igual que el
ARNm de mamíferos, también hay una cola poli-A en 3'. El UMP 5'-terminal del ARN viral está enlazado covalentemente a la proteína viral pequeña VPg (Paul AV, et al. Nature 1998, 393(6682):280-284). El 5'UTR contiene dos elementos estructurales. Uno es la estructura de hoja de trébol en 5' implicada en la síntesis de ARN de cadena y en el proceso de cambio de traducción a replicación (Huang H, et al Biochemistry 2001,40(27):8055-8064). El otro es el sitio interno de entrada al ribosoma (IRES) que promueve la traducción de la poliproteína. Además, se han identificado elementos de replicación que actúan en cis internos específicos de especie (cre) en enterovirus humanos (EVH), RVH-A y RVH-B (Gerber K, Wimmer E, Paul AV, J Virol 2001, 75(22):10979-10990). Las propias partículas virales no están envueltas y son de estructura icosaédrica. Los rinovirus también crecen de la mejor manera a temperaturas entre 33-35 °C. También son sensibles a un entorno ácido.
Las proteínas virales de RVH se transcriben como un único polipéptido largo, que se escinde en las proteínas estructurales y no estructurales virales. Los rinovirus están compuestos por una cápsida que contiene cuatro proteínas virales VP1, VP2, VP3 y VP4 (Rossmann M, et al. 1985 Nature 317 (6033): 145-53; Smith T, et al. 1986, Science 233 (4770): 1286-93). La nucleocápsidas isométricas tienen 22-40 nm de diámetro. VP1, VP2 y VP3 forman la mayor parte de la cápsida de proteínas. La proteína VP4 mucho más pequeña tiene una estructura más extendida y está en la interfaz entre la cápsida y el genoma de ARN. Hay 60 copias de cada una de estas proteínas ensambladas como un icosaedro. Los anticuerpos humanos que se dirigen a epítopos que se encuentran en las regiones exteriores de VP1-VP3 desempeñan un papel en la respuesta inmunitaria a los RVH.
Los RVH tienen dos modos de transmisión generales: 1) mediante aerosoles de gotas respiratorias y 2) a partir de superficies contaminadas, incluyendo contacto directo persona a persona. La vía de entrada primaria para los rinovirus son las vías respiratorias superiores. Seguidamente, un RVH se une a receptores de ICAM-1 (Molécula de adhesión intercelular 1) también conocida como CD54 (Grupo de diferenciación 54) en células epiteliales respiratorias. A medida que el virus se replica y se propaga, las células infectadas liberan quimiocinas y citocinas, que, a su vez, activan mediadores inflamatorios. La infección se produce rápidamente, con el rinovirus adhiriéndose a receptores de la superficie en el plazo de 15 minutos desde la entrada en las vías respiratorias. El periodo de incubación es generalmente de 8-10 horas antes de que comiencen a producirse los síntomas. Los RVH son la causa más frecuente de infección entre todos los grupos de edad de la población humana. La replicación a menudo está restringida a las vías respiratorias superiores, lo que causa dolencias auto-limitadas tales como el resfriado común. Sin embargo, las infecciones por RVH también pueden exacerbar trastornos de las vías respiratorias preexistentes, invaden las vías respiratorias inferiores y causan graves complicaciones.
En otro aspecto, los compuestos de la presente descripción pueden usarse para el tratamiento de infección por el virus de la gripe dirigiéndose a las rutas de las que depende el virus para infección o replicación. Los virus de la gripe pertenecen a la familia de virus Orthomyxoviridae. Esta familia también incluye virus Thogoto y Dhorivirus. Existen varios tipos y subtipos de virus de la gripe conocidos que infectan seres humanos y otras especies. Los virus de la gripe de tipo A infectan personas, aves, cerdos, caballos, focas y otros animales, pero las aves salvajes son los huéspedes naturales para estos virus. Los virus de la gripe de tipo A se dividen en subtipos y se nombran tomando como base dos proteínas sobre la superficie de los virus: hemaglutinina (HA) y neuraminidasa (NA). Por ejemplo, un "virus H7N2" designa un subtipo de gripe A que tiene una proteína HA 7 y una proteína NA 2. Análogamente, un virus "H5N1" tiene una proteína HA 5 y una proteína NA 1. Existen 16 subtipos de HA conocidos y 9 subtipos de NA conocidos. Son posibles muchas combinaciones diferentes de proteínas HA y NA. Solamente algunos subtipos de gripe A (es decir, H1N1, H1N2 y H3N2) están actualmente en circulación general entre la gente. Otros subtipos se encuentran de la forma más común en otras especies animales. Por ejemplo, los virus H7N7 y H3N8 causan dolencia en caballos, y el H3N8 también ha demostrado recientemente causar dolencia en perros (véase www.cdc.gov/flu/avian/gen-info/flu-viruses.htm).
Los agentes antivirales que se dirigen a proteínas de célula huésped implicadas en infección por gripe pueden usarse para proteger a grupos de alto riesgo (unidades hospitalarias, centros para el cuidado de ancianos, individuos inmunosuprimidos), y caso por caso. Un uso potencial para agentes antivirales es limitar la propagación y la gravedad de las futuras pandemias ya sean causadas por un H5N1 aviar u otras cepas de virus de la gripe. Los virus de la gripe A aviar de los subtipos H5 y H7, incluyendo virus H5N1, H7N7 y H7N3, se han usado con elevada patogenicidad, y la infección humana con estos virus ha variado desde enfermedad leve (H7N3, H7N7) a grave y mortal (H7N7, H5N1). La dolencia humana debida a infección con virus de baja patogenicidad se ha documentado, incluyendo síntomas muy leves (por ejemplo, conjuntivitis) para dolencia similar a la gripe. Los ejemplos de virus de baja patogenicidad que han infectado a seres humanos incluyen H7N7, H9N2 y H7N2 (véase www.cdc.gov/flu/avian/gen-info/flu-viruses.htm). Los virus de la gripe B se encuentran habitualmente en seres humanos pero también pueden infectar focas. A diferencia de los virus de la gripe A, estos virus no están clasificados de acuerdo con el subtipo. Los virus de la gripe B pueden causar morbilidad y mortalidad entre seres humanos, pero en general están asociados con epidemias menos graves que los virus de la gripe A. Aunque los virus de la gripe de tipo B pueden causar epidemias humanas, no han causado pandemias. (Véase www.cdc.gov/flu/avian/gen-info/fluviruses.htm).
Los virus de la gripe de tipo C causan dolencia leve en seres humanos y no causan epidemias o pandemias. Estos virus también pueden infectar perros y cerdos. Estos virus no están clasificados de acuerdo con el subtipo. (véase www.cdc.gov/flu/avian/gen-info/flu-viruses.htm).
Los virus de la gripe difieren entre sí con respecto a especificidad por receptor de la superficie celular y tropismo celular, sin embargo usan rutas de entrada comunes. Los compuestos de la presente descripción se dirigen ventajosamente a rutas que son comunes a múltiples virus, dando origen a una actividad antiviral más amplia. Por lo tanto, los presentes compuestos también pueden mostrarse útiles contra virus no relacionados que usan rutas
similares. Por ejemplo, los agentes pueden proteger las células epiteliales de las vías respiratorias contra una serie de virus diferentes además de los virus de la gripe.
En ciertos aspectos, los compuestos de la presente descripción pueden usarse para el tratamiento de infección por adenovirus. La mayoría de los adenovirus causan comúnmente una dolencia respiratoria; los síntomas de una dolencia respiratoria causada por infección por adenovirus varían entre el resfriado común y neumonía, anginas y bronquitis. Los pacientes con sistemas inmunitarios comprometidos son especialmente susceptibles a complicaciones graves de infección por adenovirus. La enfermedad respiratoria aguda (ERA), reconocida en primer lugar entre reclutas militares durante la Segunda Guerra Mundial, puede ser causada por infecciones por adenovirus durante condiciones de hacinamiento y estrés. Los adenovirus son virus icosaédricos no envueltos, de tamaño medio (90-100 nm), que contienen ADN bicatenario. Existen 49 tipos inmunológicamente distintos (6 subgéneros: A a F) que pueden causar infecciones en ser humano. Los adenovirus son habitualmente estables a agentes químicos o físicos y condiciones de pH adversas, permitiendo una prolongada supervivencia fuera del cuerpo. Algunos adenovirus, tales como AD2 y Ad5 (especie C) usan endocitosis y macropinocitosis mediadas por clatrina para entrada infecciosa. Otros adenovirus, tales como Ad3 (especie B) usan endocitosis y macropinocitosis dependientes de dinamina para entrada infecciosa. En ciertos aspectos, los compuestos de la presente descripción pueden usarse para el tratamiento de infección por virus respiratorio sincitial (VRS). El VRS es la causa más común de bronquiolitis y neumonía entre lactantes y niños de menos de 1 año. La dolencia comienza de la forma más frecuente con fiebre, secreción nasal, tos y, algunas veces, sibilancia. Durante su primera infección por VRS, entre el 25 % y el 40 % de los lactantes y niños pequeños presentan signos o síntomas de bronquiolitis o neumonía, y del 0,5% al 2% requieren hospitalización. La mayoría de los niños se recuperan de la dolencia en de 8 a 15 días. La mayoría de los niños hospitalizados por infección por VRS tienen menos de 6 meses de edad. El VRS también causa infecciones repetidas durante toda la vida, habitualmente asociadas con síntomas similares a un resfriado de moderados a graves; sin embargo, una enfermedad grave de las vías respiratorias inferiores puede producirse a cualquier edad, especialmente entre los ancianos o entre aquellos con sistemas cardiacos, pulmonares o inmunitarios comprometidos. El VRS es un virus de ARN con envuelta y negativo. El virión es de forma y tamaño variables (diámetro promedio de entre 120 y 300 nm), es inestable en el entorno (sobreviviendo solamente unas pocas horas en superficies del entorno), y es fácilmente inactivado con agua y jabón y desinfectantes.
En ciertos aspectos, los compuestos de la presente descripción pueden usarse para el tratamiento de infección por virus paragripal humano (VPGh ). Los VPGH van en segundo lugar tras el virus respiratorio sincitial (VRS) como una causa común de enfermedad de las vías respiratorias inferiores en niños pequeños. Similar al VRS, los VPGH pueden causar infecciones repetidas durante toda la vida, habitualmente manifestadas por una dolencia de las vías respiratorias superiores (por ejemplo, un refriado y/o dolor de garganta). Los VPGH también pueden causar enfermedad grave de las vías respiratorias inferiores con infección repetida (por ejemplo, neumonía, bronquitis y bronquiolitis), especialmente entre los ancianos, y entre pacientes con sistemas inmunitarios comprometidos. Cada uno de los cuatro VPGH presenta diferentes características clínicas y epidemiológicas. La característica clínica más distintiva de VPGH-1 y VPGH-2 son anginas (es decir, laringotraqueobronquitis); el VPGH-1 es la causa principal de anginas en niños, mientras que el VPGH-2 se detecta de forma menos frecuente. Tanto VPGH-1 como -2 pueden causar otras dolencias de las vías respiratorias superior e inferior. VPGH-3 está asociado más a menudo con bronquiolitis y neumonía. VPGH-4 se detecta de forma infrecuente, posiblemente debido a que es menos probable que cause una enfermedad grave. El periodo de incubación para los VPGH es generalmente de 1 a 7 días. Los VPGH son virus de ARN monocatenario negativo que poseen "púas" de glucoproteína de fusión y hemaglutininaneuraminidasa en su superficie. Existen cuatro tipos de serotipo de VPGH (1 a 4) y dos subtipo (4a y 4b). El virión varía en tamaño (diámetro promedio de entre 150 y 300 nm) y forma, es inestable en el entorno (sobreviviendo solamente unas pocas horas en superficies del entorno), y es fácilmente inactivado con agua y jabón.
En diversos aspectos, los compuestos de la presente descripción pueden usarse para el tratamiento de infección por coronavirus. Coronavirus es un género de virus animal que pertenece a la familia Coronaviridae. Los coronavirus son virus con envuelta con un genoma de ARN monocatenario positivo y simetría helicoidal. El tamaño genómico de los coronavirus varía entre aproximadamente 16 y 31 kilobases, extraordinariamente grande para un virus de ARN. El nombre "coronavirus" se deriva del latín "corona", que significa corona, dado que la envuelta del virus parece, en microscopía electrónica, estar coronado por un anillo característico de pequeñas estructuras bulbosas. Esta morfología está formada en realidad por los peplómeros de púas virales, que son proteínas que pueblan la superficie del virus y determinan el tropismo en el huésped. Los coronavirus están agrupados en el orden Nidovirales, que reciben su nombre del latín "nidus", que significa nido, dado que todos los virus en este orden producen un conjunto anidado coterminal en 3' de ARNm subgenómicos durante la infección. Las proteínas que contribuyen a la estructura global de todos los coronavirus son la púa, envuelta, membrana y nucleocápsida. En el caso específico de SRAG, un dominio de unión al receptor definido en S media en la fijación del virus a su receptor celular, la enzima convertidora de angiotensina 2.
La presente descripción contempla el tratamiento de cualquier infección vírica que se dirige a la ruta de síntesis de ácidos grasos en un huésped y, en particular, modulando la actividad de la ácido graso sintasa. Por ejemplo, los presentes procedimientos pueden usarse para tratar infecciones causadas por el virus de la leucemia de Abelson, virus de la leucemia murina de Abelson, virus de Abelson, virus de laringotraqueobronquitis aguda, virus Adelaide River, grupo de virus adenoasociados, adenovirus, virus de la peste equina africana, virus de la fiebre porcina africana, virus del SlDA, parvovirus de enfermedad Aleutiana del visón, alfarretrovirus, alfvirus, virus relacionado con ALV, virus Amapari, Aftovirus, Aquareovirus, Arbovirus, Arbovirus C, grupo A de arbovirus, grupo B de arbovirus, grupo de
Arenavirus, virus de la fiebre hemorrágica argentina, virus de la fiebre hemorrágica argentina, Arterivirus, Astrovirus, grupo del herpesvirus ateles, virus de la enfermedad de Aujezky, virus Aura, virus de la enfermedad de Ausduk, lyssavirus de murciélagos australianos, virus Aviadenovirus, virus de la eritroblastosis aviar, virus de la bronquitis infecciosa aviar, virus de la leucemia aviar, virus de la leucosis aviar, virus de la linfomatosis aviar, virus de la mieloblastosis aviar, paramixovirus aviar, virus de la neumoencefalitis aviar, virus de la reticuloendoteliosis aviar, virus del sarcoma aviar, grupo de retrovirus aviar de tipo C, Avihepadnavirus, Avipoxvirus, virus B, virus B19, virus Babanki, herpesvirus de babuino, baculovirus, virus del bosque de Barmah, virus Bebaru, virus Berrimah, Betarretrovirus, Birnavirus, virus Bittner, virus BK, virus del canal Black Creek, virus de la lengua azul, virus de la fiebre hemorrágica boliviana, virus de la enfermedad de Boma, virus de la enfermedad fronteriza de las ovejas, virus borna, alfaherpesvirus bovino 1, alfaherpesvirus bovino 2 , coronavirus bovino, virus de la fiebre efímera bovina, virus de inmunodeficiencia bovina, virus de leucemia bovina, virus de leucosis bovina, virus de mamilitis bovina virus del papiloma bovino, virus de la estomatitis papular bovina, parvovirus bovino, virus bovino sincitial, oncovirus bobino de tipo C, virus de la diarrea viral bovina, virus de Buggy Creek, grupo de virus en forma de bala, supergrupo de virus Bunyamwera, Bunyavirus, virus de la linfoma de Burkitt, fiebre Bwamba, virus CA, calicivirus, virus de encefalitis de California, virus de la viruela del camello, virus de la viruela del canario, herpesvirus de cánidos, Coronavirus canino, virus del moquillo canino, herpesvirus canino, virus diminuto canino, parvovirus canino, virus Cano Delgadito, virus de la artritis caprina, virus de la encefalitis caprina, herpesvirus caprino, virus de la viruela caprina, Cardiovirus, herpesvirus caviido 1, herpesvirus cercopitécido 1, herpesvirus cercopitecino 1, herpesvirus cercopitecino 2, virus Chandipura, virus Changuinola, virus del siluro del canal, virus de Charleville, virus de la varicela, virus Chikungunya, herpesvirus de chimpancé, reovirus del barbo, virus del salmón keta, virus cocal, reovirus del salmón coho, virus del exantema coital, virus de la fiebre de la garrapata de Colorado, coltivirus, virus de la fiebre de Columbia, virus del resfriado común, virus de ectima contagiosa, virus de la dermatitis pustular contagiosa, Coronavirus, virus Comparta, virus de coriza, virus de la viruela vacuna, virus Coxsackie, VCP (virus de la poliedrosis citoplásmica), virus de la parálisis del grillo, virus de la fiebre hemorrágica de Crimea-Congo, virus asociado al crup, Criptovirus, Cipovirus, Citomegalovirus, Grupo de citomegalovirus, virus de poliedrosis citoplásmica, virus del papiloma de venado, Deltarretrovirus, virus del dengue, Densovirus, Dependovirus, virus Dhori, diplornavirus, virus Drosophila C, Virus de la hepatitis B de pato, virus de hepatitis B de pato 1, virus de hepatitis de pato 2, duovirus, virus Duvenhage, virus de ala deformada v Ad , virus de la encefalitis equina oriental, virus EB, virus del Ébola, virus similar al Ébola, virus echo, echovirus, echovirus 10, echovirus 28, echovirus 9, virus de ectromelia, virus EEE, virus EIA, virus EIA, virus de encefalitis, virus del grupo de la encefalomiocarditis, virus de encefalomiocarditis, enterovirus, virus elevador de enzimas, virus elevador de enzimas (LDH), virus de la fiebre hemorrágica epidémica, virus epizoótico de la enfermedad hemorrágica, virus de Epstein-Barr, alfaherpesvirus equino 1, alfaherpesvirus equino 4, herpesvirus equino 2, virus del aborto equino, virus de la arteritis equina, virus de la encefalosis equina, virus de la anemia infecciosa equina, morbilivirus equino, virus de la rinoneumonitis equina, rinovirus equino, virus Eubenangu, papilomavirus del alce europeo, virus de la peste porcina europea, virus Everglades, virus Eyach, herpesvirus felino 1, calicivirus felino, virus del fibrosarcoma felino, herpesvirus felino, virus de la inmunodeficiencia felina, Virus de la peritonitis infecciosa felina, virus de la leucemia felina/sarcoma, virus de la leucemia felina, virus de la panleucopenia felina, parvovirus felino, virus del sarcoma felino, virus sincitial felino, Filovirus, virus de Flanders, Flavivirus, virus de la fiebre aftosa, virus Fort Morgan, hantavirus Four Corners, adenovirus 1 de aves de corral, virus de la viruela aviar, virus de Friend, Gammaretrovirus, virus de hepatitis GB, virus GB, virus de sarampión alemán, virus Getah, virus de leucemia de mono gibón, virus de fiebre glandular, virus de la viruela caprina, Virus Gonometa, parvovirus de ganso, virus de granulosis, virus Gross, virus de la hepatitis B de ardilla, arbovirus del grupo A, virus Guanarito, citomegalovirus de cobaya, virus de cobaya de tipo C, virus de Hantaan, Hantavirus, reovirus de almeja dura, virus de fibroma de liebre, el CMVH (citomegalovirus humano), virus de hemadsorción 2, el virus hemaglutinante de Japón, virus de la fiebre hemorrágica, virus hendra, Henipavirus, Hepadnavirus, virus de la hepatitis A, grupo de virus de la hepatitis B, virus de la hepatitis C, virus de la hepatitis D, virus de la hepatitis delta, virus de la hepatitis E, virus de la hepatitis F, virus de la hepatitis G, virus de la hepatitis no A no B, virus de la hepatitis, virus de la hepatitis (no humano), reovirus de hepatoencefalomielitis 3, Hepatovirus, virus de la hepatitis B de la garza, herpesvirus B, virus del herpes simple, virus del herpes simple 1, virus del herpes simple 2, herpesvirus, herpesvirus 7, Herpesvirus ateles, Herpesvirus hominis, Herpesvirus, Herpesvirus saimiri, Herpesvirus suis, Herpesvirus varicellae, virus J de las tierras altas, Hirame rhabdovirus, virus del cólera porcino, adenovirus humano 2, alfaherpesvirus humano 1, alfaherpesvirus humano 2, alfaherpesvirus humano 3, virus linfotrópico B humano, betaherpesvirus humano 5, coronavirus humano, grupo de citomegalovirus humano, virus espumoso humano, gammaherpesvirus humano 4, gammaherpesvirus humano 6 , virus de hepatitis A humano, grupo de herpesvirus humano 1, Grupo de herpesvirus humano 2, grupo de herpesvirus humano 3, grupo de herpesvirus humano 4, herpesvirus humano 6 , herpesvirus humano 8 , virus de inmunodeficiencia humana, virus de inmunodeficiencia humana 1, virus de inmunodeficiencia humana 2 , virus del papiloma humano, virus de leucemia de células T humanas, virus de leucemia de células T humanas I, virus de leucemia de células T humanas II, Virus de leucemia de células T humanas III, virus de linfoma de células T humanas I, virus de linfomas de células T humanas II, virus linfotrópico de células T humanas de tipo 1, virus linfotrópico de células T humanas de tipo 2, virus linfotrópico T humano I, virus linfotrópico T humano II, virus linfotrópico T humano III, icnovirus, virus de la gastroenteritis infantil, virus de la rinotraqueítis infecciosa bovina, virus de la necrosis hematopoyética infecciosa, virus de la necrosis pancreática infecciosa, virus de la gripe A, virus de la gripe B, virus de la gripe C, virus de la gripe D, virus de la gripe pr8 , virus iridiscente de los insectos, virus de insecto, iridovirus, virus B japonés, virus de la encefalitis japonesa, virus JC, virus Junin, herpesvirus asociado al sarcoma de Kaposi, virus Kemerovo, virus de la rata Kilham, virus Klamath,
virus Kolongo, virus de la fiebre hemorrágica coreana, virus de Kumba, virus de la enfermedad del bosque de Kysanur, virus de Kyzylagach, virus La Crosse, virus elevador de la deshidrogenasa láctica, virus de la deshidrogenasa láctica, virus del murciélago de Lagos, virus Langur, parvovirus de conejo, virus de la fiebre de Lassa, virus Lassa, virus de CML, virus de la rata latente, virus Leaky, Lentivirus, Leporipoxvirus, virus de la leucemia, leucovirus, virus de la dermatosis nodular, virus asociado a linfadenopatía, linfocriptovirus, virus de la coriomeningitis linfocítica, grupo de virus linfoproliferativo, virus Machupo, virus de pseudorrabia, grupo de oncovirus de tipo B de mamífero, retrovirus de tipo B de mamífero, grupo de retrovirus de tipo C de mamífero, retrovirus de tipo D de mamífero, virus de tumor mamario, virus Mapuera, virus de Marburg, virus de tipo Marburg, virus de mono Mason Pfizer, Mastadenovirus, Virus Mayaro, virus ME, virus del sarampión, virus Menangle, virus Mengo, Mengovirus, virus Middelburg, virus del nódulo de los ordeñadores, virus de la enteritis de los mineros, virus diminutos de ratones, virus relacionados con MLV, virus MM, virus Mokola, Molluscipoxvirus, virus del molusco contagioso, virus B de mono, virus de la viruela de mono, Mononegavirales, Morbilivirus, virus de murciélago del Monte Elgon, citomegalovirus de ratón, virus de encefalomielitis de ratón, virus de hepatitis de ratón, virus K de ratón, virus de leucemia de ratón, virus de ratón tumoral mamario, virus diminuto de ratón, virus de la neumonía del ratón, virus de la poliomielitis del ratón, poliomavirus del ratón, virus del sarcoma del ratón, virus de la viruela del ratón, virus de Mozambique, virus Mucambo, virus de enfermedad de la mucosa, virus de la parotiditis, herpesvirus murino 1, citomegalovirus murino 2 , grupo de citomegalovirus murino, virus de la encefalomielitis murina, virus de la hepatitis murina, virus de la leucemia murina, virus inductor de nódulos murinos, poliomavirus murino, virus del sarcoma murino, Muromelgalovirus, virus de la encefalitis del valle de Murray, virus del mixoma, Mixovirus, Mixovirus multiforme, mixovirus parotitidis, virus de la enfermedad de las ovejas de Nairobi, Nairovirus, Nanirnavirus, virus Nariva, virus Ndumo, virus Neethling, virus Nelson Bay, virus neurotrópico, arenavirus del Nuevo Mundo, virus de la neumonitis del recién nacido, virus de la enfermedad de Newcastle, virus Nipah, virus no citopatógeno, virus de Norwalk, virus de la poliedrosis nuclear (VPN), virus del cuello del pezón, virus O'nyong'nyong, virus Ockelbo, virus oncogénico, partícula similares a virus oncogénicos, oncornavirus, Orbivirus, virus Orf, virus Oropouche, Ortohepadnavirus, Ortomixovirus, Ortopoxvirus, Ortorreovirus, Orungo, virus del papiloma ovino, virus de la fiebre catarral ovina, herpesvirus del mono con cara de búho, virus Palyam, papilomavirus, Papilomavirus sylvilagi, papovavirus, virus paragripal, virus paragripal tipo 1, virus paragripal tipo 2, virus paragripal tipo 3, virus paragripal tipo 4, paramixovirus, parapoxvirus, virus paravaccinia, parvovirus, parvovirus B19, grupo de parvovirus, pestivirus, flebovirus, virus del moquillo porcino, picodnavirus, picornavirus, ciromegalovirus porcino - virus de viruela de palomas, virus Piry, virus Pixuna virus, virus de neumonía de ratones, neumovirus, virus de la poliomielitis, poliovirus, Polidnavirus, virus polihédirco, polyomavirus, Polyomavirus, Polyomavirus bovis, Polyomavirus cercopitheci, Polyomavirus hominis 2, Polyomavirus maccacae 1, Polyomavirus muris 1, Polyomavirus muris 2, Polyomavirus papionis 1, Polyomavirus papionis 2, Polyomavirus sylvilagi, Pongine herpesvirus 1, virus de la diarrea epidémica porcina, virus de la encefalomielitis hemaglutinante porcina, parvovirus porcino, virus de la gastroenteritis porcina transmisible, virus de tipo C porcino, virus de la viruela, poxvirus, poxvirus variolae, virus de Prospect Hill, Provirus, virus pseudoviruela vacuna, virus pseudorabies, virus psittacinepox, virus quailpox, virus de fibroma de conejo, virus vacuolizante de riñón de conejo, papilomavirus de conejo, virus de la rabia, parvovirus de mapache, virus de la viruela de mapache, virus de Ranikhet, citomegalovirus de rata, virus de rata, virus de Rauscher, virus vaccinia recombinante , virus recombinante, reovirus, reovirus 1, reovirus 2, reovirus 3, virus reptiliano de tipo C, virus de la infección respiratoria, virus respiratorio sincitial, virus respiratorio, virus de reticuloendoteliosis, Rabdovirus, Rabdovirus carpia, Radinovirus, Rinovirus, Rizidovirus, virus de la fiebre del valle del Rift, Virus de Riley, virus de la peste bovina, virus de tumor de ARN, virus del río Ross, virus Rotavirus, virus del sarampión, virus del sarcoma de Rous, virus de la rubéola, virus de la rubeola, rubivirus, virus de la encefalitis otoñal rusa, virus del simio SA 11, virus SA2, virus Sabia, virus Sagiyama, herpesvirus 1 de Saimirine, virus de la glándula salivar, grupo de virus de la fiebre estival, virus Sandjimba, virus SRAG, VSDA (virus de sialodacrioadenitis), virus de la viruela de focas, virus del bosque de Semliki, virus de Seúl, virus de viruela de oveja, virus de fibroma de Shope, virus de papiloma de Shope, virus espumoso de simio, virus de hepatitis A de simio, virus de inmunodeficiencia humana de simio, virus de inmunodeficiencia de simio, virus paragripal de simio, virus linfotrófico de células T de simio, virus de simio, virus de simio 40, Simplexvirus, virus Sin Nombre, virus Sindbis virus, virus de la viruela, virus de la fiebre hemorrágica de América del Sur, virus de la viruela del gorrión, espumavirus, virus del fibroma de la ardilla, retrovirus del mono ardilla, grupo de virus VSS 1,, VTLS (virus T linfotrópico de simio) tipo I, VTLS (virus T linfotrópico de simio) tipo II, VTLS (virus T linfotrópico de simio) tipo III, virus de la estomatitis papulosa, virus submaxilar, alfaherpesvirus porcino 1, herpesvirus porcino 2, Suipoxvirus, virus de la fiebre del pantano, virus de la viruela porcina, virus suizo de la leucemia del ratón, virus TAC, virus del complejo Tacaribe, virus de Tacaribe, virus de Tanapox, virus de Taterapox, reovirus de Tench, virus de encefalomielitis de Theiler, virus de Theiler, virus Thogoto, virus Thottapalayam, virus de la encefalitis transmitida por garrapatas, virus Tioman, Togavirus, Torovirus, virus tumoral, virus Tupaia, virus de la rinotraqueítis del pavo, virus de la viruela del pavo, retrovirus tipo C, oncovirus tipo D, grupo del retrovirus tipo D, rabdovirus de enfermedad ulcerativa, virus Una, grupo virus Uukuniemi, virus Vaccinia, virus vacuolizante, virus de varicela zoster, Varicelovirus, virus Varicola, virus de la viruela mayor, virus de la viruela, virus Vasin Gishu, virus VEE, virus de la encefalitis equina venezolana, virus de la encefalomielitis equina venezolana, virus de la fiebre hemorrágica venezolana, virus de la estomatitis vesicular, vesiculovirus, virus Vilyuisk, retrovirus de víbora, virus de la septicemia hemorrágica vírica, virus Visna Maedi, virus Visna, virus volepox, VEV (virus de la estomatitis vesicular), virus Wallar, virus Warrego, virus de las verrugas, el virus WEE, el virus del Nilo Occidental, virus de la encefalitis equina occidental, virus de la encefalomielitis equina occidental, virus Whataroa, virus del vómito invernal, virus de la hepatitis B de la marmota, virus del sarcoma del mono lanudo, Virus de tumor de herida, virus de WRSV, virus de tumor de mono de
Yaba, virus de Yaba, Yatapoxvirus, virus de la fiebre amarilla y virus Yug Bogdanovic.
Utilidad en trastornos metabólicos
En diversos aspectos, los compuestos de la presente descripción presentan utilidad en el tratamiento de enfermedades metabólicas. La FASN ha demostrado estar implicada en la regulación del metabolismo de glucosa, lípidos y colesterol. Los ratones con una inactivación específica del hígado de FASN tienen fisiología normal a menos que sean alimentados con una dieta con cero grasas, en cuyo caso desarrollan hipoglucemia e hígado graso, ambos de los cuales se invierten con grasa en la dieta. (Chakravarthy, M. V., etal.(2005) Cell Metabolism 1:309-322). Ratones Db/+ alimentados con una dieta rica en fructosa muestran niveles de triglicéridos en el hígado reducidos y sensibilidad a insulina mejorada cuando son tratados durante 28 días con platensimicina, un inhibidor covalente de FASN. (Wu, M. et al.(2011) PNAS 108(13):5378-5383). Los niveles de glucosa ambiente también se reducen en ratones db/db después del tratamiento con platensimicina. Estos resultados proporcionan indicios de que la inhibición de FASN puede producir beneficios terapéuticamente relevantes en modelos en animales de diabetes y trastornos metabólicos relacionados. De este modo, los inhibidores de FASN descritos son útiles en el tratamiento de trastornos caracterizados por desregulación en estos sistemas. Los ejemplos incluyen esteatosis y diabetes.
Actividad anticáncer
En diversos aspectos, la presente descripción proporciona compuestos para uso en procedimientos para tratar cáncer en un sujeto, comprendiendo el procedimiento administrar a un sujeto que necesita dicho tratamiento una cantidad efectiva de un compuesto de estructuras ((VI-J), (X), (XI). En aspectos adicionales, pueden usarse compuestos que tienen la estructura (VI-J), (X), o (XI) para la fabricación de un medicamento para tratar cáncer.
En ciertos aspectos, la presente descripción proporciona compuestos para uso en un procedimiento para inhibir el crecimiento de células tumorales en un sujeto, comprendiendo el procedimiento administrar a un sujeto que necesita dicho tratamiento una cantidad efectiva de un compuesto de estructura ( VI-J), (X), o (XI). En aspectos adicionales, el tumor puede derivarse de ovario, mama, pulmón, tiroides, ganglio linfático, riñón, uréter, vejiga, ovario, testículo, próstata, hueso, músculo esquelético, médula ósea, estómago, esófago, intestino delgado, colon, recto, páncreas, hígado, músculo liso, cerebro, médula espinal, nervios, oído, ojo, nasofaringe, orofaringe, glándulas salivales o tejido cardíaco. En ciertos aspectos, los presentes compuestos pueden administrarse de forma concurrente con uno o más tratamientos anticáncer adicionales.
Células cancerosas que proliferan rápidamente activan la ruta de síntesis de ácidos grasos para suministrar los altos niveles de lípidos necesarios para el ensamblaje de membranas y el metabolismo oxidativo. (Flavin, R. et al. (2010) Future Oncology. 6(4):551-562) Los inhibidores de la síntesis de ácidos grasos han demostrado actividad in vivo en modelos de cáncer preclínicos. (Orita, H. et al. (2007) Clinical Cancer Research. 13(23):7139-7145 y Puig, T. et al.(2011) Breast Cancer Research, 13(6):R131) Adicionalmente, la síntesis de ácidos grasos apoya la formación de nuevos vasos sanguíneos y los inhibidores de esta ruta tienen actividad en modelos in vitro de angiogénesis. (Browne, C.D., et al.(2006) The FASEB Journal, 20(12):2027-2035). Los compuestos descritos actualmente demostraron la capacidad para inducir selectivamente parada del ciclo celular en células HUVEC sin causar muerte celular general por apoptosis. Véase EJEMPLOS.
El tratamiento de cáncer de la presente descripción incluye un efecto anti-tumoral que puede ser valorado mediante medios convencionales tales como la tasa de respuesta, el tiempo hasta la progresión de la enfermedad y/o la tasa de supervivencia. Los efectos anti-tumorales de la presente invención incluyen la inhibición del crecimiento tumoral, retraso del crecimiento tumoral, regresión de tumores, retracción de tumores, tiempo incrementado hasta el recrecimiento del tumor tras el cese del tratamiento y ralentización de la progresión de la enfermedad. Por ejemplo, se espera que, cuando la combinación de la presente invención se administra a un animal de sangre caliente tal como un ser humano, que necesita tratamiento para cáncer que implica un tumor sólido, dicho procedimiento de tratamiento producirá un efecto, según lo medido mediante, por ejemplo, uno o más de: el alcance del efecto anti-tumoral, la tasa de respuesta, el tiempo hasta la progresión de la enfermedad y la tasa de supervivencia.
En el presente documento también se proporcionan composiciones farmacéuticas que comprende los compuestos de la presente descripción. Las presentes composiciones tienen actividad antiviral y/o anticáncer.
En diversos aspectos, la presente descripción proporciona composiciones farmacéuticas que comprenden cualquiera de los compuestos de Estructuras (VI-J), (X) o (XI) y un vehículo, excipiente o diluyente farmacéuticamente aceptable. Ciertos aspectos de la presente descripción se refieren a procedimientos de uso de composiciones farmacéuticas y kits que comprenden uno o más agentes que inhiben la ruta de síntesis de ácidos grasos para inhibir o reducir una infección vírica o para el tratamiento de cáncer. Ciertos aspectos de la presente descripción se refieren a procedimientos de uso de composiciones farmacéuticas y kits que comprenden uno o más agentes que inhiben la ácido graso sintasa para inhibir o reducir una infección vírica o para el tratamiento de cáncer. Otro aspecto de la presente invención proporciona procedimientos, composiciones farmacéuticas y kits para el tratamiento de sujetos animales que tienen una infección vírica o cáncer o en riesgo de desarrollar una infección vírica o cáncer. El término "sujeto", tal como se usa en el presente documento, incluye seres humanos, así como otros mamíferos. El término "tratamiento", tal como se usa en el presente documento, incluye conseguir un beneficio terapéutico y/o un beneficio profiláctico. Por beneficio terapéutico se entiende erradicación o mejora de la infección vírica subyacente. Además, se consigue un beneficio terapéutico con la erradicación o mejora de uno o más de los síntomas fisiológicos asociados con la infección vírica subyacente, de modo que se observa una mejora en el sujeto animal, a pesar del hecho de que el sujeto aún pueda estar afectado por el virus subyacente.
Para aspectos donde se desea un beneficio profiláctico, una composición farmacéutica de la invención puede
administrarse a un paciente en riesgo de desarrollar infección vírica tal como RVH, o VIH, o a un paciente que notifica uno o más de los síntomas fisiológicos de una infección vírica, incluso aunque puede no haberse realizado un diagnóstico de la afección. La administración puede impedir que la infección vírica se desarrolle, o puede reducir, rebajar, acortar y/o mejorar de otro modo la infección vírica que se desarrolla. La composición farmacéutica puede modular la ruta de síntesis de ácidos grasos, por ejemplo, la expresión del gen FASN o la actividad de la proteína FASN. Donde, el término modular incluye inhibición de la ruta de síntesis de ácidos grasos, por ejemplo, la expresión del gen FASN o la actividad de la proteína FASN o, como alternativa, activación de la ruta de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN.
Reducir la actividad de la ruta de síntesis de ácidos grasos, por ejemplo, la expresión del gen FASN o la actividad de la proteína FASN, también se denomina "inhibir" la ruta de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN. El término "inhibe" y sus conjugaciones gramaticales, tales como "inhibidor," no requieren inhibición completa, sino que se refieren a una reducción de la actividad de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN. En otro aspecto, dicha reducción es de al menos un 50 %, al menos un 75 %, al menos un 90 %, y puede ser de al menos un 95 % de la actividad de la enzima en ausencia del efecto inhibidor, por ejemplo, en ausencia de un inhibidor. A la inversa, la frase "no inhibe" y sus conjugaciones gramaticales se refieren a situaciones donde hay menos del 20 %, menos del 10 %, y puede ser menos del 5 %, de reducción de la actividad enzimática en presencia del agente. Además, la frase "no inhibe sustancialmente" y sus conjugaciones gramaticales se refieren a situaciones donde hay menos del 30 %, menos del 20 %, y, en algunos aspectos, menos del 10 %, de reducción de la actividad enzimática en presencia del agente.
Incrementar la actividad de la ruta de síntesis de ácidos grasos, por ejemplo, la expresión del gen FASN o la actividad de la proteína FASN, también se denomina "activar" la ruta de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN. El término "activado" y sus conjugaciones gramaticales, tales como "activar," no requieren activación completa, sino que se refieren a un incremento de la actividad de la ruta de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN. En otro aspecto, dicho incremento es de al menos un 50 %, al menos un 75 %, al menos un 90 %, y puede ser de al menos un 95 % de la actividad de la enzima en ausencia del efecto de activación, por ejemplo, en ausencia de un activador. A la inversa, la frase "no activa" y sus conjugaciones gramaticales se refieren a situaciones donde hay menos del 20 %, menos del 10 %, y puede ser menos del 5 %, de un incremento de la actividad enzimática en presencia del agente. Además, la frase "no activa sustancialmente" y sus conjugaciones gramaticales se refieren a situaciones donde hay menos del 30 %, menos del 20 %, y, en otro aspecto, menos del 10 %, de un incremento de la actividad enzimática en presencia del agente.
La capacidad de reducir la actividad enzimática es una medida de la potencia o la actividad de un agente, o combinación de agentes, hacia o contra la enzima. La potencia puede medirse mediante ensayos sin células, con células completas y/o in vivo en términos de valores de CI50, Ki y/o DE50. Un valor de CI50 representa la concentración de un agente requerida para inhibir la actividad enzimática a la mitad (50 %) en un conjunto de condiciones dadas. Un valor de Ki representa la constante de afinidad en equilibrio para la unión de un agente de inhibición a la enzima. Un valor de DE50 representa la dosis de un agente requerida para efectuar la mitad de una respuesta máxima en un ensayo biológico. Detalles adicionales de estas medidas serán apreciados por los expertos en la técnica y pueden encontrarse en textos estándar sobre bioquímica, enzimología y similares.
La presente invención también incluye kits que pueden usarse para tratar infecciones víricas o tratar un cáncer. Estos kits comprenden un agente o combinación de agentes, como se describen en el presente documento, que inhiben la ruta de síntesis de ácidos grasos, por ejemplo, expresión del gen FASN o actividad de la proteína FASN, y opcionalmente instrucciones que enseñan el uso del kit de acuerdo con los diversos procedimientos y enfoques descritos en el presente documento. Dichos kits también pueden incluir información, tal como referencias de bibliografía científica, materiales de inserto en el envase, resultados de ensayos clínicos y/o resúmenes de estos y similares que indican o establecen las actividades y/o ventajas del agente. Dicha información puede basarse en los resultados de diversos estudios, por ejemplo, estudios que usan animales experimentales que implican modelos in vivo y estudios basados en ensayos clínicos en ser humano. Los kits descritos en el presente documento pueden proporcionarse, comercializarse y/o promoverse a profesionales sanitarios, incluyendo facultativos, enfermeros, farmacéuticos, responsables de vademecum y similares.
Formulaciones, vías de administración y dosis eficaz
Otro aspecto más de la presente descripción se refiere a formulaciones, vías de administración y dosis eficaces para composiciones farmacéuticas que comprenden un agente o combinación de agentes de la presente invención. Dichas composiciones farmacéuticas pueden usarse para tratar infecciones víricas tal como se ha descrito anteriormente. Los compuestos de la invención pueden administrarse como formulaciones farmacéuticas que incluyen aquellas adecuadas para administración oral (incluyendo bucal y sublingual), rectal, nasal, tópica, mediante parche transdérmico, pulmonar, vaginal, mediante supositorio o parenteral (incluyendo intramuscular, intraarterial, intratecal, intradérmica, intraperitoneal, subcutánea e intravenosa) o en una forma adecuada para administración mediante aerosolización, inhalación o insuflación. Información general sobre sistemas de administración de fármacos puede encontrarse en Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems (Lippencott Williams & Wilkins, Baltimore Md. (1999).
En diversos aspectos, la composición farmacéutica incluye vehículos y excipientes (incluyendo tampones, hidratos de carbono, manitol, proteínas, polipéptidos o aminoácidos tales como glicina, antioxidantes, bacteriostáticos, agentes quelantes, agentes de suspensión, agentes espesantes y/o conservantes), agua, aceites, incluyendo los de origen del
petróleo, animal, vegetal o sintético, tales como aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares, soluciones salinas, soluciones acuosas de dextrosa y glicerol, agentes aromatizantes, agentes colorantes, eliminadores de pegajosidad y otros aditivos aceptables, adyuvantes, o aglutinantes, otras sustancias auxiliares farmacéuticamente aceptables según se requiera para aproximarse a las condiciones fisiológicas, tales como agentes tamponantes del pH, agentes de ajuste de tonicidad, agentes emulsionantes, agentes humectantes y similares. Los ejemplos de excipientes incluyen almidón, glucosa, lactosa, sacarosa, gelatina, malta, arroz, harina, tiza, gel de sílice, estearato de sodio, monoestearato de glicerol, talco, cloruro de sodio, leche desnatada en polvo, glicerol, propileno, glicol, agua, etanol y similares. En otro aspecto, la preparación farmacéutica está sustancialmente libre de conservantes. En otro aspecto, la preparación farmacéutica puede contener al menos un conservante. Metodología general sobre formas de dosificación farmacéuticas puede encontrarse en Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems (Lippencott Williams & Wilkins, Baltimore Md. (1999)). Se reconocerá que, aunque puede emplearse cualquier vehículo adecuado conocido por los expertos en la técnica para administrar las composiciones de esta invención, el tipo de transportador variará dependiendo de la vía de administración.
Los compuestos también pueden encapsularse dentro de liposomas usando tecnología bien conocida. También pueden emplearse microesferas biodegradables como vehículos para las composiciones farmacéuticas de esta invención. Microesferas biodegradables adecuadas se describen, por ejemplo, en las patentes de Estados Unidos núm. 4,897,268; 5,075,109; 5,928,647; 5,811,128; 5,820,883; 5,853,763; 5,814,344 y 5,942,252.
El compuesto puede administrarse en liposomas o microesferas (o micropartículas). Procedimientos para preparar liposomas y microesferas para administración a un paciente son bien conocidos por los expertos en la materia. La patente de Estados Unidos núm. 4,789,734, describe procedimientos para encapsular materiales biológicos en liposomas. Esencialmente, el material se disuelve en una solución acuosa, los fosfolípidos y lípidos apropiados se añaden, junto con tensioactivos si se requiere, y el material se dializa o se sonica, según sea necesario. Una revisión de procedimientos conocidos es proporcionada porG. Gregoriadis, Capítulo 14, "Liposomes," Drug Carriers in Biology and Medicine, págs. 2. sup. 87-341 (Academic Press, 1979).
Las microesferas formadas por polímeros o proteínas son bien conocidas por los expertos en la materia, y pueden diseñarse a medida para el paso a través del tracto gastrointestinal directamente al torrente sanguíneo. Como alternativa, el compuesto puede estar incorporado y las microesferas, o material compuesto de microesferas, implantarse para liberación lenta durante un periodo de tiempo que varía entre días y meses. Véase, por ejemplo, las patentes de Estados Unidos núm. 4,906,474, 4,925,673 y 3,625,214, y Jein, TIPS 19: 155-157 (1998).
La concentración de fármaco puede ajustarse, el pH de la solución tamponarse y la isotonicidad ajustarse para ser compatible con inyección intravenosa, tal como se conoce bien en la técnica.
Los compuestos de la invención pueden formularse como una solución o suspensión estéril, en vehículos adecuados, bien conocidos en la técnica. Las composiciones farmacéuticas pueden esterilizarse mediante técnicas de esterilización bien conocidas, convencionales, o pueden ser filtradas estériles. Las soluciones acuosas resultantes pueden envasarse para uso tal cual, o liofilizadas, combinándose la preparación liofilizada con una solución estéril antes de la administración. Formulaciones adecuadas y vehículos adicionales se describen en Remington "The Science and Practice of Pharmacy" (20th Ed., Lippincott Williams & Wilkins, Baltimore MD).
Los agentes o sus sales farmacéuticamente aceptables pueden proporcionarse en solitario o en combinación con uno o más agentes diferentes o con una o más formas diferentes. Por ejemplo, una formulación puede comprender uno o más agentes en proporciones particulares, dependiendo de las potencias relativas de cada agente y la indicación pretendida. Por ejemplo, en composiciones para dirigirse a dos dianas de huésped diferentes, y donde las potencias son similares, puede usarse una proporción de agentes de aproximadamente 1:1. Las dos formas pueden formularse conjuntamente, en la misma unidad de dosificación por ejemplo, en una crema, supositorio, comprimido, cápsula, pulverizador en aerosol, o sobrecito de polvo a disolver en una bebida; o cada forma puede formularse en una unidad independiente, por ejemplo, dos cremas, dos supositorios, dos comprimidos, dos cápsulas, un comprimido y un líquido para disolver el comprimido, dos pulverizadores en aerosol, o un paquete de polvo y un líquido para disolver el polvo, etc.
La expresión "sal farmacéuticamente aceptable" significa aquellas sales que conservan la eficacia y propiedades biológicas de los agentes usados en la presente invención y que no son biológicamente, o por otro motivo, indeseables. Por ejemplo, una sal farmacéuticamente aceptable no interfiere en el efecto beneficioso de un agente de la invención en la inhibición de la ruta de síntesis de ácidos grasos, por ejemplo, inhibición de la expresión del gen FASN o la actividad de la proteína FASN.
Las sales típicas son aquellas de los iones inorgánicos, tales como, por ejemplo, iones de sodio, potasio, calcio, magnesio, y similares. Dichas sales incluyen sales con ácidos inorgánicos u orgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido nítrico, ácido sulfúrico, ácido metanosulfónico, ácido p-toluenosulfónico, ácido acético, ácido fumárico, ácido succínico, ácido láctico, ácido mandélico, ácido málico, ácido cítrico, ácido tartárico o ácido maleico. Además, si el uno o varios agentes contienen un grupo carboxi u otro grupo ácido, se puede convertir en una sal de adición farmacéuticamente aceptable con bases inorgánicas u orgánicas. Los ejemplos de bases adecuadas incluyen hidróxido de sodio, hidróxido de potasio, amoniaco, ciclohexilamina, diciclohexilamina, etanolamina, dietanolamina, trietanolamina, y similares.
Un éster o amida farmacéuticamente aceptable se refiere a aquellas que conservan la eficacia y propiedades biológicas de los agentes usados en la presente invención y que no son biológicamente, o por otro motivo, indeseables. Por ejemplo, el éster o amida no interfiere en el efecto beneficioso de un agente de la invención en la inhibición de la ruta de síntesis de ácidos grasos, por ejemplo, inhibición de la expresión del gen FASN o la actividad de la proteína
FASN. Los ésteres típicos incluyen etilo, metilo, isobutilo, etilenglicol, y similares. Las amidas típicas incluyen amidas no sustituidas, alquilamidas , dialquilamidas, y similares.
En otro aspecto, puede administrarse un agente en combinación con uno o más compuestos, formas y/o agentes diferentes, por ejemplo, tal como se ha descrito anteriormente. Las composiciones farmacéuticas que comprenden combinaciones de un inhibidor de la ruta de síntesis de ácidos grasos por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN con uno o más agentes activos diferentes, puede formularse para comprender ciertas proporciones molares. Por ejemplo, pueden usarse proporciones molar de aproximadamente 99:1 a aproximadamente 1:99 de inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, con respecto al otro agente activo. En algún subconjunto de los aspectos, el intervalo de proporciones molares de inhibidor de la ruta de síntesis de ácidos grasos por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN: otro agente activo se selecciona de entre aproximadamente 80:20 y aproximadamente 20:80; aproximadamente 75:25 y aproximadamente 25:75, aproximadamente 70:30 y aproximadamente 30:70, aproximadamente 66:33 y aproximadamente 33:66, aproximadamente 60:40 y aproximadamente 40:60; aproximadamente 50:50; y aproximadamente 90:10 y aproximadamente 10:90. La proporción molar de inhibidor de la ruta de síntesis de ácidos grasos por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN: otro agente activo puede ser de aproximadamente 1:9, y en otro aspecto puede ser de aproximadamente 1:1. Los dos agentes, formas y/o compuestos pueden formularse conjuntamente, en la misma unidad de dosificación por ejemplo, en una crema, supositorio, comprimido, cápsula, o paquete de polvo a disolver en una bebida; o cada agente, forma y/o compuesto puede formularse en un unidades independientes, por ejemplo, dos cremas, dos supositorios, dos comprimidos, dos cápsulas, un comprimido y un líquido para disolver el comprimido, un pulverizador en aerosol, un paquete de polvo y un líquido para disolver el polvo, etc.
Si fuera necesario o deseable, los agentes y/o combinaciones de agentes pueden administrarse con otros agentes más. La selección de agentes que pueden coadministrarse con los agentes y/o combinaciones de agentes de la presente invención puede depender, al menos en parte, de la afección que está siendo tratada. Los agentes de uso particular en las formulaciones de la presente invención incluyen, por ejemplo, cualquier agente que tenga un efecto terapéutico para una infección vírica, incluyendo, por ejemplo, fármacos usados para tratar afecciones inflamatorias. Por ejemplo, en tratamientos para RVH, en algunos aspectos, formulaciones de la presente invención pueden contener adicionalmente uno o más fármacos antiinflamatorios convencionales, tales como un AINE, por ejemplo, ibuprofeno, naproxeno, acetaminofeno, ketoprofeno o aspirina. En algunos aspectos alternativos para el tratamiento de la gripe, formulaciones de la presente invención pueden contener adicionalmente uno o más agentes antivirales para gripe convencionales, tales como amantadina, rimantadina, zanamivir y oseltamivir. En tratamientos para infecciones retrovíricas , tales como VIH, las formulaciones de la presente invención pueden contener adicionalmente uno o más fármacos antivirales convencionales, tales como inhibidores de la proteasa (lopinavir/ritonavir (Kaletra), indinavir (Crixivan), ritonavir (Norvir), nelfinavir (Viracept), cápsulas de gelatina dura de saquinavir (Invirase), atazanavir (Reyataz), amprenavir (Agenerase), fosamprenavir (Telzir), tipranavir (Aptivus)), inhibidores de transcriptasa inversa, incluyendo inhibidores no nucleósidos y nucleósidos/nucleótidos (AZT (zidovudina, Retrovir), ddI (didanosina, Videx), 3TC (lamivudina, Epivir), d4T (estavudina, Zerit), abacavir (Ziagen), emtricitabina (FTC, Emtriva), tenofovir (Viread), efavirenz (Sustiva) y nevirapina (Viramune)), inhibidores de fusión T20 (enfuvirtida, Fuzeon), inhibidores de integrasa (MK-0518 y GS-9137), e inhibidores de maduración (PA-457 (bevirimat)). Como otro ejemplo, las formulaciones pueden contener adicionalmente uno o más suplementos, tales como vitamina C, E u otros antioxidantes.
En ciertos aspectos, los compuestos de la presente descripción pueden administrarse en combinación con un terapéutico para cáncer conocido. Por ejemplo, los compuestos pueden administrarse en combinación con paclitaxel (disponible en el mercado como Taxol, Bristol-Myers Squibb), doxorrubicina (también conocida con el nombre comercial Adriamicina), vincristina (conocida con los nombres comerciales Oncovin, Vincasar PES y Vincrex) , actinomicina D, altretamina, asparaginasa, bleomicina, busulfán, capecitabina, carboplatino, carmustina, clorambucilo, cisplatino, ciclofosfamida, citarabina, dacarbazina, daunorrubicina, epirrubicina, etopósido, fludarabina, fluorouracilo, gemcitabina, hidroxiurea, idarrubicina, ifosfamida, irinotecán, lomustina, melfalán, mercaptopurina, metotrexato, mitomicina, mitoxantrona, oxaliplatino, procarbazina, esteroides, estreptozocina, taxotere, tamozolomida, tioguanina, tiotepa, tomudex, topotecán, treosulfán, UFT (uracilo-tegafur), vinblastina y vindesina, o similares.
El agente(s) (o sales, ésteres o amidas farmacéuticamente aceptables de los mismos) pueden administrarse per se o en forma de una composición farmacéutica donde el uno o varios agentes activos está mezclado o en mezcla con uno o más vehículos farmacéuticamente aceptables. Una composición farmacéutica, tal como se usa en el presente documento, puede ser cualquier composición preparada para administración a un sujeto. Las composiciones farmacéuticas para uso de acuerdo con la presente invención pueden formularse de manera convencional usando uno o más vehículos fisiológicamente aceptables, que comprenden excipientes, diluyentes y/o auxiliares, por ejemplo, que facilitan el procesamiento de los agentes activos en preparaciones que pueden administrarse. La formulación apropiada puede depender al menos en parte de la vía de administración elegida. El uno o varios agentes útiles en la presente invención, o sales, ésteres o amidas farmacéuticamente aceptables de los mismos, pueden administrarse a un paciente usando una serie de rutas o vías de administración, incluyendo administración aplicaciones oral, bucal, tópica, rectal, transdérmica, transmucosal, subcutánea, intravenosa e intramuscular, así como por inhalación Para la administración oral, los agentes se pueden formular fácilmente combinando el uno o varios agentes activos con vehículos farmacéuticamente aceptables bien conocidos en la técnica. Dichos vehículos permiten que los agentes de la invención se formulen como comprimidos, incluyendo comprimidos masticables, píldoras, grageas, cápsulas,
pastillas, caramelos duros, líquidos, geles, jarabes, pastas semilíquidas, polvos, suspensiones, elixires, obleas, y similares, para ingestión oral por un paciente a tratar. Dichas formulaciones pueden comprender vehículos farmacéuticamente aceptables incluyendo diluyentes o cargas sólidas, medios acuosos estériles y diversos disolventes orgánicos no tóxicos. Un vehículo sólido puede ser una o más sustancias que también pueden actuar como diluyentes, agentes aromatizantes, solubilizantes, lubricantes, agentes de suspensión, aglutinantes, conservantes, agentes disgregantes de comprimidos o un material encapsulante. En polvos, el vehículo generalmente es un sólido finamente dividido que es una mezcla con el componente activo finamente dividido. En los comprimidos, el componente activo generalmente se mezcla con el vehículo que tiene la capacidad de unión necesaria en proporciones adecuadas y se compacta en la forma y tamaño deseados. Los polvos y comprimidos contienen preferentemente de aproximadamente el uno (1) a aproximadamente el setenta (70) por ciento del compuesto activo. Los vehículos adecuados incluyen carbonato de magnesio, estearato de magnesio, talco, azúcar, lactosa, pectina, dextrina, almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa sódica, una cera de bajo punto de fusión, manteca de cacao y similares. En general, los agentes de la invención se incluirán a niveles de concentración que varían de aproximadamente el 0,5 %, aproximadamente el 5 %, aproximadamente el 10 %, aproximadamente el 20 %, o aproximadamente el 30 % a aproximadamente el 50 %, aproximadamente el 60 %, aproximadamente el 70 %, aproximadamente el 80 % o aproximadamente el 90 % en peso de la composición total de formas de dosificación oral, en una cantidad suficiente para proporcionar una unidad deseada de dosificación.
Las suspensiones acuosas para uso oral pueden contener uno o varios agentes de esta invención con excipientes farmacéuticamente aceptables, tales como un agente de suspensión (por ejemplo, metilcelulosa), un agente humectante (por ejemplo, lecitina, lisolecitina y/o un alcohol graso de cadena larga), así como agentes colorantes, conservantes, agentes aromatizantes y similares.
En otro aspecto, pueden requerirse aceites o disolventes no acuosos para poner los agentes en solución, debido a, por ejemplo, la presencia de grandes fracciones lipófilas. Como alternativa, pueden usarse emulsiones, suspensiones u otras preparaciones, por ejemplo, preparaciones liposómicas. Con respecto a las preparaciones liposómicas, puede usarse cualquiera de los procedimientos conocidos para la preparación de liposomas para el tratamiento de una afección. Véase, por ejemplo, Bangham et al., J. Mol. Biol. 23: 238-252 (1965) y Szoka et al., Proc. Natl Acad. Sci. EE. UU. 75: 4194-4198 (1978). También pueden fijarse ligandos a los liposomas para dirigir estas composiciones a sitios de acción particulares. Los agentes de esta invención también se pueden integrar en productos alimenticios, por ejemplo, queso crema, mantequilla, aliños para ensaladas, helado para facilitar la solubilización, la administración y/o el cumplimiento terapéutico de ciertas poblaciones de pacientes.
Las preparaciones farmacéuticas para uso oral se pueden obtener como un excipiente sólido, moliendo opcionalmente una mezcla resultante y procesando la mezcla de gránulos, después de añadir auxiliares adecuados, si se desea, para obtener núcleos de comprimidos o de grageas. Los excipientes adecuados son, en particular, cargas tales como azúcares, incluyendo lactosa, sacarosa, manitol o sorbitol; elementos aromatizantes, preparaciones de celulosa tales como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma de tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa sódica y/o polivinilpirrolidona (PVP). Si se desea, se pueden añadir agentes disgregantes, tales como la polivinilpirrolidona reticulada, agar, o ácido algínico o una sal del mismo, tal como alginato de sodio. Los agentes también se pueden formular como una preparación de liberación sostenida.
Los núcleos de gragea pueden estar dotados de revestimientos adecuados. Para este fin, pueden usarse soluciones de azúcar concentradas, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y disolventes orgánicos o mezclas de disolventes adecuados. Pueden añadirse colorantes o pigmentos a los revestimientos de comprimidos o de grageas para identificación o para caracterizar diferentes combinaciones de agentes activos.
Las preparaciones farmacéuticas que pueden usarse por vía oral incluyen cápsulas duras hechas de gelatina, así como cápsulas blandas, selladas hechas de gelatina y un plastificante, tal como glicerol o sorbitol. Las cápsulas duras pueden contener los ingredientes activos mezclados con una carga tal como lactosa, aglutinantes tales como almidones y/o lubricantes tales como talco o estearato de magnesio y, opcionalmente, estabilizantes. En las cápsulas blandas, los agentes activos pueden disolverse o suspenderse en líquidos adecuados, tales como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, pueden añadirse estabilizantes. Todas las formulaciones para administración oral deben estar en dosis adecuadas para la administración.
Otras formas adecuadas para la administración oral incluyen preparaciones en forma líquida incluyendo emulsiones, jarabes, elixires, soluciones acuosas, suspensiones acuosas, o preparaciones en forma sólida que están concebidas para convertirlas poco antes del uso en preparaciones en forma líquida. Las emulsiones pueden prepararse en soluciones, por ejemplo, en soluciones acuosas de propilenglicol o pueden contener agentes emulsionantes, por ejemplo, tales como lecitina, monooleato de sorbitán o goma arábiga. Las soluciones acuosas pueden prepararse disolviendo el componente activo en agua y añadiendo colorantes, sabores, estabilizantes y agentes espesantes adecuados. Las suspensiones acuosas pueden prepararse dispersando el componente activo finamente dividido en agua con material viscoso, tal como gomas naturales o sintéticas, resinas, metilcelulosa, carboximetilcelulosa sódica, y otros agentes de suspensión bien conocidos. Cargas o vehículos adecuados con los que se pueden administrar las composiciones incluyen agar, alcohol, grasas, lactosa, almidón, derivados de celulosa, polisacáridos, polivinilpirrolidona, sílice, solución salina estéril y similares, o mezclas de los mismos usadas en cantidades adecuadas. Las preparaciones en forma sólida incluyen soluciones, suspensiones y emulsiones, y pueden contener, además del componente activo, colorantes, sabores, estabilizantes, tampones, edulcorantes artificiales y naturales,
dispersantes, espesantes, agentes solubilizantes, y similares.
Un jarabe o suspensión pueden prepararse añadiendo el compuesto activo a una solución acuosa concentrada de un azúcar, por ejemplo, sacarosa, a la que también se puede añadir cualquier ingrediente accesorio. Dichos ingredientes accesorios pueden incluir aromatizante, un agente para retardar la cristalización del azúcar o un agente para incrementar la solubilidad de cualquier otro ingrediente, por ejemplo, tal como un alcohol polihídrico, por ejemplo, glicerol o sorbitol.
Cuando la formulación de compuestos de la invención para la administración oral, puede ser deseable utilizar formulaciones de retención gástrica para mejorar la absorción desde el tracto gastrointestinal (GI). Una formulación que es retenida en el estómago durante varias horas puede liberar compuestos de la invención lentamente y proporcionar una liberación sostenida que puede usarse en los procedimientos de la invención. La descripción de dichas formulaciones de retención gástrica se encuentra en Klausner, E.A.; Lavy, E.; Barta, M.; Cserepes, E.; Friedman, M.; Hoffman, A. 2003 "Novel gastroretentive dosage forms: evaluation of gastroretentivity and its effect on levodopa in humans." Pharm. Res. 20, 1466-73, Hoffman, A.; Stepensky, D.; Lavy, E.; Eyal, S. Klausner, E.; Friedman, M. 2004 "Pharmacokinetic and pharmacodynamic aspects of gastroretentive dosage forms" Int. J. Pharm. 11, 141-53, Streubel, A.; Siepmann, J.; Bodmeier, R.; 2006 "Gastroretentive drug delivery systems" Expert Opin. Drug Deliver.
3,217-3, y Chavanpatil, M.D.; Jain, P.; Chaudhari, S.; Shear, R.; Vavia, P.R. "Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for olfoxacin" Int. J. Pharm. 2006 publicación en línea 24 de marzo. Pueden utilizarse técnicas expansibles, flotantes y bioadhesivas para maximizar la absorción de los compuestos de la invención.
Los compuestos de la invención pueden formularse para administración parenteral (por ejemplo, por inyección, por ejemplo inyección en embolada o infusión continua) y pueden presentarse en forma de dosis unitaria en ampollas, jeringas precargadas, infusión de pequeño volumen o en recipientes de dosis múltiples con un conservante añadido. Las composiciones pueden asumir formas tales como suspensiones, soluciones, o emulsiones en vehículos oleosos o acuosos, por ejemplo soluciones en polietilenglicol acuoso.
Para formulaciones inyectables, el vehículo puede seleccionarse de entre los que se sabe en la técnica que son adecuados, incluyendo soluciones acuosas o suspensiones o emulsiones oleosas, con aceite de sésamo, aceite de maíz, aceite de semilla de algodón, o aceite de cacahuete, así como elixires, manitol, dextrosa, o una solución acuosa estéril, y vehículos farmacéuticos similares. La formulación también puede comprender composiciones poliméricas que son biocompatibles, biodegradables, tales como ácido poli(láctico-co-glicólico). Estos materiales se pueden preparar en micro o nanoesferas, cargadas con el fármaco y además revestidas o derivatizadas para proporcionar un rendimiento de liberación sostenida superior. Los vehículos adecuados para inyección periocular o intraocular incluyen, por ejemplo, suspensiones de agente terapéutico en agua de calidad para inyección, liposomas y vehículos adecuados para las sustancias lipófilas. Otros vehículos para inyección periocular o intraocular son bien conocidos en la técnica.
En un aspecto preferido, la composición se formula de acuerdo con procedimientos rutinarios como una composición farmacéutica adaptada para administración intravenosa a seres humanos. Normalmente, las composiciones para administración intravenosa son soluciones en tampón acuoso isotónico estéril. Cuando sea necesario, la composición también puede incluir un agente solubilizante y un anestésico local tal como lidocaína para aliviar el dolor en el sitio de la inyección. Generalmente, los ingredientes se suministran por separado o mezclados juntos en forma de dosificación unitaria, por ejemplo, como un polvo liofilizado seco o concentrado libre de agua en un recipiente sellado herméticamente, tal como una ampolla o bolsita que indica la cantidad de agente activo. Cuando la composición debe administrarse por infusión, se puede dispensar con una botella de infusión que contiene agua o solución salina de calidad farmacéutica estéril. Cuando la composición se administra por inyección, se puede proporcionar una ampolla de agua o solución salina estéril para inyección, de modo que los ingredientes se puedan mezclar antes de la administración.
Cuando la administración es por inyección, el compuesto activo se puede formular en soluciones acuosas, específicamente en tampones fisiológicamente compatibles tales como solución de Hank, solución de Ringer, o tampón de solución salina fisiológica. La solución puede contener agentes de formulación tales como agentes de suspensión, estabilizantes y/o dispersantes. Como alternativa, el compuesto activo puede estar en forma de polvo para constitución con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso. En otro aspecto, la composición farmacéutica no comprende un adyuvante o cualquier otra sustancia que se añade para intensificar la respuesta inmunitaria estimulada por el péptido. En otro aspecto, la composición farmacéutica comprende una sustancia que inhibe una respuesta inmunitaria al péptido. Los procedimientos de formulación son conocidos en la técnica, por ejemplo, tal como se describe en Remington's Pharmaceutical Sciences, última edición, Mack Publishing Co., Easton P.
Además de las formulaciones descritas anteriormente, los agentes también pueden formularse como una preparación de absorción lenta. Dichas formulaciones de acción prolongada pueden administrarse mediante implantación o administración transcutánea (por ejemplo por vía subcutánea o por vía intramuscular), inyección intramuscular o uso de un parche transdérmico. De este modo, por ejemplo, los agentes pueden formularse con materiales poliméricos o hidrófobos adecuados (por ejemplo como una emulsión en un aceite aceptable) o resinas de intercambio iónico, o como derivados moderadamente solubles, por ejemplo, como una sal moderadamente soluble
En otro aspecto, composiciones farmacéuticas que comprenden uno o más agentes de la presente invención ejercen efectos locales y regionales cuando se administran por vía tópica o se inyectan en o cerca de sitios de infección particulares. La aplicación tópica directa, por ejemplo, de un líquido viscoso, solución, suspensión, soluciones basadas
en dimetilsulfóxido (DMSO), formulaciones liposómicas, gel, gelatina, crema, loción, pomada, supositorio, espuma o pulverizador de aerosol, puede usarse para la administración local, para producir, por ejemplo, efectos locales y/o regionales. Los vehículos farmacéuticamente apropiados para dicha formulación incluyen, por ejemplo, alcoholes alifáticos inferiores, poliglicoles (por ejemplo, glicerol o polietilenglicol), ésteres de ácidos grasos, aceites, grasas, siliconas, y similares. Dichas preparaciones también pueden incluir conservantes (por ejemplo, ésteres del ácido phidroxibenzoico) y/o antioxidantes (por ejemplo, ácido ascórbico y tocoferol). Véase también Dermatological Formulations: Percutaneous absorption, Barry (Ed.), Marcel Dekker Incl, 1983. En otro aspecto, se usan formulaciones locales/tópicas que comprenden un inhibidor de la ruta de síntesis de ácidos grasos por ejemplo, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, para tratar infecciones víricas epidérmicas o mucosales. Las composiciones farmacéuticas de la presente invención pueden contener un vehículo cosmética o dermatológicamente aceptable. Dichos vehículos son compatibles con la piel, las uñas, las mucosas, los tejidos y/o el pelo, y pueden incluir cualquier vehículo cosmético o dermatológico portador usado convencionalmente que cumpla estos requisitos. Dichos vehículos pueden ser seleccionados fácilmente por un experto en la técnica. En la formulación de pomadas para la piel, un agente o combinación de agentes de la presente invención puede formularse en una base de hidrocarburos oleaginosos, una base de absorción anhidra, una base de absorción de agua en aceite, una base que se retira con agua de aceite en agua y/o una base soluble en agua. Los ejemplos de dichos transportadores y excipientes incluyen humectantes (por ejemplo, urea), glicoles (por ejemplo, propilenglicol), alcoholes (por ejemplo, etanol), ácidos grasos (por ejemplo, ácido oleico), tensioactivos (por ejemplo, miristato de isopropilo y lauril sulfato sódico), pirrolidonas, monolaurato de glicerol, sulfóxidos, terpenos (por ejemplo, mentol), aminas, amidas, alcanos, alcanoles, agua, carbonato cálcico, fosfato de calcio, diversos azúcares, almidones, derivados de celulosa, gelatina y polímeros tales como polietilenglicoles.
Las pomadas y cremas pueden, por ejemplo, formularse con una base acuosa u oleosa con la adición de agentes espesantes y/o gelificantes adecuados. Las lociones pueden formularse con una base acuosa u oleosa y, en general, también contendrán uno o más agentes emulsionantes, agentes estabilizantes, agentes dispersantes, agentes de suspensión, agentes espesantes, o agentes colorantes. La construcción y el uso de parches transdérmicos para la administración de agentes farmacéuticos es bien conocida en la técnica. Véase, por ejemplo, las patentes de Estados Unidos núm. 5,023,252, 4,992,445 y 5,001,139. Dichos parches pueden construirse para administración continua, pulsátil o bajo demanda de agentes farmacéuticos.
Los lubricantes que pueden usarse para formar composiciones farmacéuticas y formas de dosificación de la invención incluyen estearato de calcio, estearato de magnesio, aceite mineral, aceite mineral ligero, glicerina, sorbitol, manitol, polietilenglicol, otros glicoles, ácido esteárico, laurilsulfato sódico, talco, aceite vegetal hidrogenado (por ejemplo, aceite de cacahuete, aceite de semilla de algodón, aceite de girasol, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja), estearato de zinc, oleato de etilo, laurato de etilo, agar o mezclas de los mismos. Lubricantes adicionales incluyen, por ejemplo, un gel de sílice Syloid, un aerosol coagulado de sílice sintética o mezclas de los mismos. Un lubricante puede añadirse opcionalmente, en una cantidad de menos de aproximadamente el 1 por ciento en peso de la composición farmacéutica.
Las composiciones de acuerdo con la presente invención pueden estar en cualquier forma adecuada para aplicación tópica, incluyendo soluciones acuosas, acuosas-alcohólicas u oleosas, dispersiones de loción o suero, geles acuosos, anhidros u oleosos, emulsiones obtenidas por dispersión de una fase grasa en una fase acuosa (Ac/Ag o aceite en agua) o, a la inversa, (Ag/Ac o agua en aceite), microemulsiones o, como alternativa, microcápsulas, micropartículas o dispersiones de vesículas lipídicas de tipo iónico y/o no iónico. Estas composiciones pueden prepararse de acuerdo con procedimientos convencionales. Aparte de los agentes de la invención, las cantidades de los diversos constituyentes de las composiciones de acuerdo con la invención son las usadas convencionalmente en la técnica. Estas composiciones, en particular, constituyen cremas, leches, lociones, geles o espumas de protección, de tratamiento o de cuidado para la cara, para las manos, para el cuerpo y/o para las mucosas, o para la limpieza de la piel. Las composiciones también pueden consistir en preparaciones sólidas que constituyen jabones o pastillas de jabón para limpieza.
Las composiciones de la presente invención también pueden contener adyuvantes comunes a los campos cosmético y dermatológico, tales como agentes gelificantes hidrófilos o lipófilos, agentes activos hidrófilos o lipófilos, agentes conservantes, antioxidantes, disolventes, perfumes, cargas, filtros solares, absorbentes del olor y colorantes. Las cantidades de estos diferentes adyuvantes son las usadas convencionalmente en los campos considerados y, por ejemplo, son de aproximadamente el 0 ,01 % a aproximadamente el 20 % del peso total de la composición. Dependiendo de su naturaleza, estos adyuvantes pueden ser introducidos en la fase grasa, en la fase acuosa y/o en las vesículas lipídicas.
En otro aspecto, las infecciones víricas oculares pueden ser tratadas eficazmente con soluciones, suspensiones, pomadas o insertos oftálmicos que comprenden un agente o combinación de agentes de la presente invención. Pueden prepararse colirios disolviendo el ingrediente activo en una solución acuosa estéril tal como solución salina fisiológica, solución tampón, etc. o combinando composiciones de polvo que se disolverán antes de usar. Pueden seleccionarse otros vehículos, tal como se conoce en la técnica, incluyendo: solución de sales en equilibrio, solución salina, poliéteres solubles en agua tales como polietilenglicol, polivinilos, como alcohol polivinílico y povidona, derivados de celulosa tales como metilcelulosa e hidroxipropilmetilcelulosa, derivados del petróleo tales como aceite mineral y vaselina blanca, grasas animales tales como lanolina, polímeros de ácido acrílico tales como gel de carboxipolimetileno, grasas vegetales tales como aceite de cacahuete y polisacáridos tales como dextranos, y glucosaminoglucanos tales como hialuronato sódico. Si se desea, se pueden añadir aditivos usados normalmente en
los colirios. Dichos aditivos incluyen agentes isotonizantes (por ejemplo, cloruro sódico, etc.), un agente tampón (por ejemplo, ácido bórico, monohidrógeno fosfato sódico, dihidrógeno fosfato sódico, etc.), conservantes (por ejemplo, cloruro de benzalconio, cloruro de bencetonio, clorobutanol, etc.) , espesantes (por ejemplo, sacáridos tales como lactosa, manitol, maltosa, etc.; por ejemplo, ácido hialurónico o su sal, tal como hialuronato sódico, hialuronato potásico, etc.; por ejemplo, mucopolisacárido tal como condroitín sulfato, etc.; por ejemplo, poliacrilato sódico, polímero de carboxivinilo, poliacrilato reticulado, alcohol polivinílico, polivinilpirrolidona, metilcelulosa, hidroxipropilmetilcelulosa, hidroxietilcelulosa, carboximetilcelulosa, hidroxipropilcelulosa u otros agentes conocidos por los expertos en la materia).
La solubilidad de los componentes de las presentes composiciones se puede mejorar mediante un tensioactivo u otro codisolvente apropiado en la composición. Dichos codisolventes incluyen polisorbato 20, 60 y 80, Pluronic F68, F-84 y P-103, ciclodextrina, u otros agentes conocidos por los expertos en la materia. Dichos codisolventes pueden emplearse a un nivel de aproximadamente el 0,01 % al 2 % en peso.
Las composiciones de la invención pueden envasarse en forma de dosis múltiple. Pueden preferirse conservantes para prevenir la contaminación microbiana durante el uso. Los conservantes adecuados incluyen: cloruro de benzalconio, timerosal, clorobutanol, metilparabeno, propilparabeno, alcohol feniletílico, edetato disódico, ácido sórbico, Onamer M u otros agentes conocidos por los expertos en la materia. En los productos oftálmicos de la técnica anterior, dichos conservantes pueden emplearse a un nivel del 0,004 % al 0,02 %. En las composiciones de la presente solicitud, el conservante, preferentemente cloruro de benzalconio, puede emplearse a un nivel del 0,001 % a menos del 0,01 %, por ejemplo del 0,001 % al 0,008 %, preferentemente de aproximadamente el 0,005 % en peso. Se ha descubierto que una concentración de cloruro de benzalconio del 0,005 % puede ser suficiente para preservar las composiciones de la presente invención del ataque microbiano.
En otro aspecto, las infecciones víricas del oído pueden ser tratadas eficazmente con soluciones, suspensiones, pomadas o insertos óticos que comprenden un agente o combinación de agentes de la presente invención.
En otro aspecto, los agentes de la presente invención se administran en forma soluble en lugar de en suspensión, lo que permite una absorción más rápida y cuantitativa a los sitios de acción. En general, las formulaciones tales como gelatinas, cremas, lociones, supositorios y pomadas pueden proporcionar una zona con una exposición más prolongada a los agentes de la presente invención, mientras que las formulaciones en solución, por ejemplo, pulverizadores, proporcionan, una exposición a corto plazo, más inmediata.
En otro aspecto relacionado con la aplicación tópica/local, las composiciones farmacéuticas pueden incluir uno o más potenciadores de la penetración. Por ejemplo, las formulaciones pueden comprender transportadores o excipientes en fase sólida o de gel adecuados que incrementan la penetración o ayudan a la administración de agentes o combinaciones de agentes de la invención a través de una barrera de permeabilidad, por ejemplo, la piel. Muchos de estos compuestos potenciadores de la penetración son conocidos en la técnica de la formulación tópica, e incluyen, por ejemplo, agua, alcoholes (por ejemplo, terpenos como metanol, etanol, 2-propanol), sulfóxidos (por ejemplo, dimetilsulfóxido, decilmetilsulfóxido, tetradecilmetilsulfóxido), pirrolidonas (por ejemplo, 2-pirrolidona, N-metil-2-pirrolidona, N-(2-hidroxietil)pirrolidona), laurocapram, acetona, dimetilacetamida, dimetilformamida, alcohol tetrahidrofurfurílico, ácidos L-a-amino, tensioactivos aniónicos, catiónicos, anfóteros o no iónicos (por ejemplo, miristato de isopropilo y laurilsulfato sódico), ácidos grasos, alcoholes grasos (por ejemplo, ácido oleico), aminas, amidas, amidas de ácido clofíbrico, hexametilenlauramida, enzimas proteolíticas, a-bisabolol, d-limoneno, urea y N,N-dietil-m-toluamida, y similares. Ejemplos adicionales incluyen humectantes (por ejemplo, urea), glicoles (por ejemplo, propilenglicol y polietilenglicol), monolaurato de glicerol, alcanos, alcanoles, ORGELASE, carbonato cálcico, fosfato cálcico, diversos azúcares, almidones, derivados de celulosa, gelatina, y/u otros polímeros. En otro aspecto, las composiciones farmacéuticas incluirán uno o más de dichos potenciadores de la penetración.
En otro aspecto, las composiciones farmacéuticas para aplicación local/tópica pueden incluir uno o más conservantes antimicrobianos tales como compuestos de amonio cuaternario, mercuriales orgánicos, p-hidroxibenzoatos, alcoholes aromáticos, clorobutanol, y similares.
Las infecciones víricas gastrointestinales pueden ser tratadas eficazmente con soluciones, suspensiones, pomadas, enemas y/o supositorios administrados por vía oral o por vía rectal, que comprenden un agente o combinación de agentes de la presente invención.
Las infecciones víricas respiratorias pueden ser tratadas eficazmente con soluciones en aerosol, suspensiones o polvos secos que comprenden un agente o combinación de agentes de la presente invención. La administración por inhalación es particularmente útil en el tratamiento de infecciones víricas del pulmón, tales como una infección por RVH. El aerosol se puede administrar a través del sistema respiratorio o las fosas nasales. Por ejemplo, un experto en la técnica reconocerá que una composición de la presente invención se puede suspender o disolver en un transportador apropiado, por ejemplo, un propulsor farmacéuticamente aceptable, y administrarse directamente a los pulmones usando un pulverizador o inhalador nasal. Por ejemplo, una formulación de aerosol que comprende un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, puede disolverse, suspenderse o emulsionarse en un propulsor o una mezcla de disolvente y propulsor, por ejemplo, para administración como un pulverizador o inhalador nasal. Las formulaciones en aerosol pueden contener cualquier propulsor aceptable a presión, tal como un propulsor cosmética o dermatológica o farmacéuticamente aceptable, tal como se usa convencionalmente en la técnica.
Una formulación en aerosol para administración nasal es, generalmente, una solución acuosa diseñada para ser administrada a las fosas nasales en gotas o pulverizadores. Las soluciones nasales pueden ser similares a las secreciones nasales ya que son generalmente isotónicas y están ligeramente tamponadas para mantener un pH de
aproximadamente 5,5 a aproximadamente 6,5, aunque, adicionalmente, se pueden usar valores de pH fuera de este intervalo. Los agentes antimicrobianos o conservantes también pueden incluirse en la formulación.
Una formulación en aerosol para inhalaciones e inhaladores puede diseñarse de modo que el agente o combinación de agentes de la presente invención sea transportado al árbol respiratorio del sujeto cuando se administra por la vía nasal o respiratoria oral. Las soluciones para inhalación pueden administrarse, por ejemplo, mediante un nebulizador. Las inhalaciones o insuflaciones, que comprenden fármacos en polvo fino o líquidos, pueden administrarse al aparato respiratorio como un aerosol farmacéutico de una solución o suspensión del agente o combinación de agentes en un propulsor, por ejemplo, para ayudar a la distribución de la carga. Los propulsores puede ser gases licuados, incluyendo halocarburos, por ejemplo, fluorocarbonos tales como hidrocarburos clorados y fluorados, hidroclorofluorocarbonos e hidroclorocarbonos, así como hidrocarburos y éteres de hidrocarburos.
Los propulsores de halocarbono útiles en la presente invención incluyen propulsores de fluorocarbono en los que todos los hidrógenos se sustituyen por flúor, propulsores de clorofluorocarbono en los que todos los hidrógenos se sustituyen por cloro y al menos un flúor, propulsores de fluorocarbono que contienen hidrógeno, y propulsores de clorofluorocarbono que contienen hidrógeno. Los propulsores de halocarbono se describen en Johnson, patente de Estados Unidos núm. 5,376,359, expedida el 27 de diciembre de 1994; Byron et al., patente de Estados Unidos núm.
5,190,029, expedida el 2 de marzo de 1993; y Purewal et al., patente de Estados Unidos núm. 5,776,434, expedida el 7 de julio de 1998. Los propulsores de hidrocarburo útiles en la invención incluyen, por ejemplo, propano, isobutano, n-butano, pentano, isopentano y neopentano. También puede usarse una mezcla de hidrocarburos como propulsor. Los propulsores de éter incluyen, por ejemplo, éter dimetílico, así como los éteres. Una formulación en aerosol de la invención también puede comprender más de un propulsor. Por ejemplo, la formulación de aerosol puede comprender más de un propulsor de la misma clase, tal como dos o más fluorocarburos; o más de uno, más de dos, más de tres propulsores de diferentes clases, tales como un fluorohidrocarburo y un hidrocarburo. Las composiciones farmacéuticas de la presente invención también pueden dispensarse con un gas comprimido, por ejemplo, un gas inerte tal como dióxido de carbono, óxido nitroso o nitrógeno.
Las formulaciones en aerosol también pueden incluir otros componentes, por ejemplo, etanol, isopropanol, propilenglicol, así como tensioactivos u otros componentes, tales como aceites y detergentes. Estos componentes pueden servir para estabilizar la formulación y/o lubricar componentes de válvula.
La formulación en aerosol puede envasarse a presión y puede formularse como un aerosol usando soluciones, suspensiones, emulsiones, polvos y preparaciones semisólidas. Por ejemplo, una formulación en aerosol en solución puede comprender una solución de un agente de la invención, tal como un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, en propulsor (sustancialmente) puro o como una mezcla de propulsor y disolvente. El disolvente puede usarse para disolver el agente y/o retardar la evaporación del propulsor. Los disolventes útiles en la invención incluyen, por ejemplo, agua, etanol y glicoles. Se puede usar cualquier combinación de disolventes adecuados, opcionalmente en combinación con conservantes, antioxidantes y/u otros componentes de aerosol.
Una formulación en aerosol también puede ser una dispersión o suspensión. Una formulación en aerosol en suspensión puede comprender una suspensión de un agente o combinación de agentes de la presente invención, por ejemplo, un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, y un agente dispersante. Los agentes dispersantes útiles en la invención incluyen, por ejemplo, trioleato de sorbitán, alcohol oleílico, ácido oleico, lecitina y aceite de maíz. Una formulación en aerosol en suspensión también puede incluir lubricantes, conservantes, antioxidantes y/u otros componentes de aerosol. Una formulación en aerosol puede formularse, análogamente, como una emulsión. Una formulación en aerosol en emulsión puede incluir, por ejemplo, un alcohol tal como etanol, un tensioactivo, agua y un propulsor, así como un agente o combinación de agentes de la invención, por ejemplo, un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN. El tensioactivo usado puede ser no iónico, aniónico o catiónico. Un ejemplo de una formulación en aerosol en emulsión comprende, por ejemplo, etanol, tensioactivo, agua y propulsor. Otro ejemplo de una formulación en aerosol en emulsión comprende, por ejemplo, aceite vegetal, monoestearato de glicerilo y propano.
Los compuestos de la invención pueden formularse para administración como supositorios. Una cera de bajo punto de fusión, tal como una mezcla de triglicéridos, glicéridos de ácidos grasos, Witepsol S55 (marca registrada de Dynamite Nobel Chemical, Alemania), o manteca de cacao se funde en primer lugar y el componente activo se dispersa de forma homogénea, por ejemplo, mediante agitación. La mezcla homogénea fundida se vierte a continuación en moldes de tamaño conveniente, se deja enfriar y solidificar.
Los compuestos de la invención pueden formularse para administración vaginal. Pesarios, tampones, cremas, geles, pastas, espumas o pulverizadores que contienen, además del ingrediente activo, transportadores que se sabe en la técnica que son apropiados.
Se prevé adicionalmente que los compuestos de la invención se puedan fijar de forma liberable a polímeros biocompatibles para uso en formulaciones de liberación sostenida en, o fijados a insertos para administración tópica, intraocular, periocular o sistémica. La liberación controlada a partir de un polímero biocompatible se puede utilizar con un polímero soluble en agua para formar una formulación instilable, también. La liberación controlada de un polímero biocompatible, tal como, por ejemplo, microesferas o nanoesferas de PLGA, se puede utilizar en una formulación adecuada para implantación o inyección intraocular para administración de liberación sostenida, también. Puede usarse cualquier polímero biodegradable y biocompatible adecuado.
Las composiciones farmacéuticas adecuadas para uso en la presente invención incluyen composiciones donde los
ingredientes activos están presentes en una cantidad efectiva, es decir, en una cantidad efectiva para conseguir un beneficio terapéutico y/o profiláctico en un huésped con al menos una infección vírica o en un sujeto que tiene cáncer. La cantidad real efectiva para una aplicación particular dependerá de la afección o afecciones a tratar, el estado del sujeto, la formulación y la vía de administración, así como de otros factores conocidos por los expertos en la materia. La determinación de una cantidad efectiva de un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, está bien dentro de las capacidades de los expertos en la materia, a la luz de la descripción en el presente documento, y se determinará usando técnicas de optimización rutinarias.
La cantidad efectiva para uso en seres humanos puede determinarse a partir de modelos en animales. Por ejemplo, una dosis para seres humanos puede formularse para conseguir concentraciones circulantes, en el hígado, tópicas y/o gastrointestinales que se ha descubierto que son efectivas en animales. Un experto en la técnica puede determinar la cantidad efectiva para uso humano, especialmente a la luz de los datos experimentales de modelos en animales descritos en el presente documento. Basándose en los datos en animales y otros tipos de datos similares, los expertos en la técnica pueden determinar las cantidades efectivas de las composiciones de la presente invención apropiadas para seres humanos.
La cantidad efectiva cuando se hace referencia a un agente o combinación de agentes de la invención generalmente significará los intervalos de dosis, vías de administración, formulaciones, etc., que han sido recomendados o aprobados por cualquiera de los diversos organismos reguladores o de asesoramiento en la técnica médica o farmacéutica (por ejemplo, FDA, AMA) o por el fabricante o proveedor.
Además, las dosis apropiadas para un inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, pueden determinarse basándose en resultados experimentales in vitro. Por ejemplo, la potencia in vitro de un agente en la inhibición de un componente inhibidor de la ruta de síntesis de ácidos grasos, por ejemplo, un inhibidor de la expresión del gen FASN o la actividad de la proteína FASN, proporciona información útil en el desarrollo de dosificación in vivo efectivas para conseguir efectos biológicos similares.
En otro aspecto, la administración de agentes de la presente invención puede ser intermitente, por ejemplo, administración una vez cada dos días, cada tres días, cada cinco días, una vez a la semana, una vez o dos veces al mes, y similares. En otro aspecto, la cantidad, formas y/o cantidades de las diferentes formas pueden variar en diferentes momentos de administración.
Un experto en la técnica sería capaz de monitorizar en un paciente el efecto de la administración de un agente particular. Por ejemplo, los niveles de carga viral de VIH pueden determinarse mediante técnicas estándar en la técnica, tales como la medición de los recuentos de células CD4, y/o los niveles virales, tal como se detectan por PCR. Otras técnicas serían evidentes para un experto en la técnica.
Habiendo descrito ahora, en general, diversos aspectos y aspectos de la invención, la misma se entenderá más fácilmente mediante la referencia a los siguientes ejemplos que se proporcionan a modo de ilustración.
EJEMPLOS
Ejemplo - Síntesis de Compuestos de la presente descripción
Los ejemplos que no están dentro del alcance de las reivindicaciones son para fines ilustrativos solamente General: Todas las reacciones y manipulaciones descritas se llevaron a cabo en campanas extractoras bien ventiladas. Las operaciones y reacciones llevadas a cabo a presión elevada o reducida se llevaron a cabo detrás de pantallas protectoras contra explosiones. Abreviaturas: ACN, acetonitrilo; AcOH, ácido acético; AIBN, azobisisobutironitrilo; BuLi, butillitio; CDI, 1,1' carbonildiimidazol; DBU, 1,8-diazabiciclo[5.4.0]undec-7-eno; DCE, 1,2-dicloroetano; DCM, diclorometano o cloruro de metileno; DIEA, N,N-diisopropiletilamina; DMAP, 4-dimetilaminopiridina; DMF, N,N-dimetilformamida; DMSO, dimetilsulfóxido; Ed C, 1-etil-3-(3-dimetilaminopropil)carbodiimida; EDCI, clorhidrato de 1 -etil-3- (3-dimetilaminopropil)carbodiimida; EtOAc, acetato de etilo; EtOH, etanol; HATU, 2-(1H-7-azabenzotriazol-1-il) -1,1,3,3-tetrametil-uronio; HBTU, O-benzotriazol-N,N,N',N'-tetrametil-uronio-hexafluorofosfato o 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametillaminio hexafluorofosfato; HMPA, hexametilfosforamida; HOAc, ácido acético; HOBT, 1-hidroxibenzotriazol; LDA, diisopropilamina de litio; MeOH, metanol; MsCl, cloruro de metanosulfonilo; MsOH, ácido metanosulfónico; NBS, N-bromosuccinimida; NIS, N-yodosuccinimida; PE, éter de petróleo; PTAT, tribromuro de feniltrimetilamonio; PTSA, ácido p-toluenosulfónico; Py, piridina; Pyr, piridina; TEA, trietilamina; TFA, ácido trifluoroacético; THF, tetrahidrofurano; TMSC1, clorotrimetilsilano; TsOH, ácido p-toluenosulfónico.

Compuesto 1.1. 4-(4-bromofenil)-4-hidroxipiperidin-1-carboxilato de terc-butilo. A una solución agitada de 1-bromo-4-yodobenceno (93,7 g, 331,21 mmol, 1,10 equiv) en tetrahidrofurano (800 ml) en nitrógeno a -78 <o>C se le añadió gota a gota una solución de butillitio (150 ml, 2,43 M en THF, 1,05 equiv) durante 30 minutos. La solución resultante se agitó durante 2 h a -78<o>C. A esto se le añadió a continuación una solución de 4-oxopiperidin-1-carboxilato de terc-butilo (60 g, 301,13 mmol, 1,00 equiv) en tetrahidrofurano (800 ml) gota a gota con agitación a -78<o>C durante 30 minutos. Después de agitar durante 1 h a -78<o>C, la reacción se inactivó cuidadosamente con 350 ml de H2O. La mezcla resultante se extrajo con 2x400 ml de acetato de etilo y las capas orgánicas combinadas
se secaron (Na2SO4) y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:200-1:10) como eluyente para dar 91 g (85%) del compuesto del título como un aceite de color amarillo

Compuesto 1.2.4-(4-cianofenil)-4-hidroxipiperidin-1-carboxilato de tere-butilo. Una solución de 4-(4-bromofenil)-4-hidroxipiperidin-1-carboxilato de terc-butilo (compuesto 1.1, 36 g, 101,05 mmol, 1,00 equiv), Pd(PPh3)4 (11,7 g, 10,12 mmol, 0,05 equiv ) y Zn(CN)2 (17,9 g, 152,44 mmol, 1,51 equiv) en DMF (400 ml) en nitrógeno se agitó durante una noche a 80 °C. Después de alcanzar la temperatura ambiente, la reacción se inactivó mediante la adición de 600 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 300 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x200 ml de carbonato potásico (ac., sat.) seguido por 1x200 ml de solución saturada de cloruro sódico, se secaron (Na2SO4) y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:200-1:5) como eluyente para dar 23 g (75 %) del compuesto del título como un sólido de color blanco

Compuesto 1.3. 4-(4-cianofenil)-5,6-dihidropiridin-1(2H)-carboxilato de terc-butilo. En un matraz de fondo redondo, se colocó una solución de 4-(4-cianofenil)-4-hidroxipiperidin-1-carboxilato de terc-butilo (compuesto 1.2, 2 g, 6,61 mmol, 1,00 equiv) en piridina (40 ml). Se añadió POCl3 cuidadosamente (10,16 g, 66,26 mmol, 10,02 equiv). La mezcla resultante se agitó en nitrógeno durante una noche a temperatura ambiente y a continuación se concentró al vacío. El residuo se recogió en 20 ml de DCM, se lavó con 2x20 ml de bicarbonato sódico (ac), se secó (Na2SO4) y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con PE/EtOAc (100:1-30:1) como eluyente para dar 1,4 g (74%) del compuesto del título como un sólido de color blanco

Compuesto 1.4. 4-(4-cianofenil)piperidin-1-carboxilato de terc-butilo. Un matraz de fondo redondo, que contenía una solución de 4-(4-cianofenil)-5,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (compuesto 1.3, 500 mg, 1,76 mmol, 1,00 equiv) en acetato de etilo (20 ml) se purgó con nitrógeno gaseoso. A la solución se añadió paladio sobre carbono (0,1 g, 10 %, 60 % de agua) y el matraz se purgó a continuación adicionalmente con nitrógeno. La atmósfera se cambió a continuación a hidrógeno y la mezcla se agitó durante una noche a temperatura ambiente. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida para dar 0,2 g (40 %) del compuesto del título como un aceite de color amarillo.

Compuesto 1.5. Clorhidrato de 4-(piperidin-4-il)benzonitrilo. En un matraz de fondo redondo de 3 bocas de 100 ml, se colocó una solución de 4-(4-cianofenil)piperidin-1-carboxilato de terc-butilo (compuesto 1.4, 4 g, 13,99 mmol, 1,00 equiv) en acetato de etilo (60 ml). Se burbujeó cloruro de hidrógeno (gaseoso) a través de la solución y la mezcla resultante se agitó durante 1 h a temperatura ambiente. El precipitado formado se recogió por filtración y se secó para dar 2,2 g (71 %) del compuesto del título como un sólido de color blanco m/z (ES+) 187 (M+H)+. 1H RMN (300 Mh z , CD3OD): ó 7,72 (d, J = 8,4 Hz, 2H), 7,50 (d, J = 8,4 Hz, 2H), 3,54 (d con estructura fina, J = 12,6 Hz, 2H), 3,18 (t con estructura fina, J = 12,2 Hz, 2H), 3,12-1,97 (m, 1H), 2,11 (d con estructura fina, J = 14,1 Hz, 2H), 2,04-1,84 (m, 2H).

Compuesto 1.6. 3-Bromo-4-metilbenzoato de metilo. Una solución de ácido 3-bromo-4-metilbenzoico (20 g, 93,00 mmol, 1,00 equiv) y ácido sulfúrico (20 ml) en metanol (100 ml) se agitó durante una noche a 80 °C. A continuación, la mezcla se concentró a presión reducida y el residuo se diluyó con 500 ml de acetato de etilo. La mezcla resultante se lavó con 3x200 ml de agua, 1x200 ml de bicarbonato sódico (acuoso), seguido por 1x200 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro, se concentró a presión reducida, y se secó
para dar 20 g (94%) del compuesto del título como un aceite de color rojo oscuro.

Compuesto 1.7.3-Ciano-4-metilbenzoatode metilo. Una mezcla de 3-bromo-4-metilbenzoato de metilo (compuesto 1.6, 18 g, 78,58 mmol, 1,00 equiv), Zn(CN)2 (11,1 g, 94,87 mmol, 1,20 equiv), y Pd(PPh3)4 (7,3 g, 6,32 mmol, 0,08 equiv) en N,N-dimetilformamida (250 ml) se agitó en una atmósfera de nitrógeno a 100 °C durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición cuidadosa de 200 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 500 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3x200 ml de solución saturada de cloruro sódico, se secaron sobre (Na2SO4), y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) como eluyente para dar 11 g (76 %) del compuesto del título como un sólido de color blanquecino

Compuesto 1.8. 3-(-N'-hidroxicarbamimidoil)4-metilbenzoato de metilo. Una mezcla de 3-ciano-4-metilbenzoato de metilo (compuesto 1.7, 8 g, 43,38 mmol, 1,00 equiv, 95%), NH2 OH.HCl (3,785 g, 54,86 mmol, 1,20 equiv), y N,N-diisopropiletilamina (DIEA, 17,7 g, 136,95 mmol, 3,00 equiv) en tetrahidrofurano (100 ml) se agitó durante una noche a 70 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se recogió en agua y el pH se ajustó a 2-3 con cloruro de hidrógeno (acuoso, 1 M). Después de lavar la mezcla con 3x40 ml de acetato de etilo, el pH de la capa acuosa se ajustó a 8-9 con NaOH (acuoso, 2 M) seguido de extracción con 3x30 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre (Na2SO4) y se concentraron a presión reducida para dar 2,76 g (29 %) del compuesto del título como un sólido de color blanco.

Compuesto 1.9. 3-Carbamimidoil-4-metilbenzoato de metilo. Un matraz de fondo redondo, que contenía una solución de 3-(N'-hidroxicarbamimidoil)-4-metilbenzoato de metilo (compuesto 1.8, 8 g, 36,50 mmol, 1,00 equiv, 95%) en metanol (150 ml) se purgó con nitrógeno gaseoso. A la solución se añadió paladio sobre carbono (9 g, 10 %, 60 % de agua) y el matraz se purgó a continuación adicionalmente con nitrógeno gaseoso. La atmósfera se cambió a continuación a hidrógeno y la mezcla se agitó durante una noche a 25 °C bajo un globo. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida para dar 4 g (54 %) del compuesto del título como un sólido de color marrón.

Compuesto 1.10.13-Bromodihidro-2H-piran-4(3H)-ona. A una mezcla de dihidro-2H-piran-4 (3H)-ona (10 g, 99,88 mmol, 1,00 equiv) y NH4OAc (3,56 g, 46,23 mmol) en tetrahidrofurano (100 ml) a 0 °C en nitrógeno se le añadió en varios lotes N-bromosuccinimida (17,8 g, 100,00 mmol, 1,00 equiv). La mezcla resultante se agitó durante una noche a 30 °C en nitrógeno. La mezcla de reacción se filtró a continuación y el filtrado se concentró. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1/10-1/5) como eluyente para dar 15 g (50 %) del compuesto del título como un aceite de color marrón.

Compuesto 1.10. 4-Metil-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoato de metilo. Una mezcla de 3-carbamimidoil-4-metilbenzoato de metilo (compuesto 1.9, 400 mg, 1,98 mmol, 1,00 equiv, 95 %), 3-bromodihidro-2H-piran-4(3H)-ona (compuesto 1.10.1, 750 mg , 2,09 mmol, 1,00 equiv), y carbonato potásico (580 mg, 4,20 mmol, 2,00 equiv) en CH3CN (15 ml) se agitó durante una noche a 80 °C en nitrógeno. Después de enfriar a temperatura ambiente, la mezcla de reacción se concentró a presión reducida. El residuo se disolvió en 50 ml de acetato de etilo y se lavó con 2x10 ml de H2O. La fase orgánica se secó sobre (Na2SO4) y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice y se purificó usando acetato de etilo/éter de petróleo (1/3) para dar 0,21 g (37 %) del compuesto del título como un sólido de color blanco.

Compuesto 1.11. Acido 4-metil-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoico. Una mezcla de 4-metil-3-(3,4,6,7-tetrahidropirano [3,4-d] imidazol-2-il)benzoato de metilo (compuesto 1.10, 210 mg, 0,73 mmol, 1,00 equiv, 95%) e hidróxido sódico (92,65 mg, 2,32 mmol, 3,00 equiv) en 12 ml de metanol/H2O (2:1) se agitó durante a 60 °C en un baño de aceite. Después de 2 h, la mezcla de reacción se concentró a presión reducida y el residuo se recogió en 5 ml de H2O. La mezcla resultante se lavó con 3x10 ml de acetato de etilo y el pH de la capa acuosa se ajustó a continuación a 2-3 usando cloruro de hidrógeno (ac., 2 M). La mezcla resultante se extrajo con 3x30 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron a presión reducida para dar 0,21 g (89 %) del compuesto del título como un sólido de color blanco.

Compuesto 1. Metil 4-(1-(4-metM-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-M)benzoM)piperidm-4-il)benzonitrilo. Una mezcla de ácido 4-metil-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoico (compuesto 1.11, 60 mg, 0,22 mmol, 1,00 equiv, 95%) , EDCI (88,4 mg, 0,46 mmol, 2,00 equiv), DMAP (85,12 mg, 0,70 mmol, 3,00 equiv), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 52 mg, 0,23 mmol, 1,00 equiv) en DMF se agitó a temperatura ambiente. Después de 3 h, la mezcla de reacción se diluyó con 40 ml de DCM y se lavó con 2x10 ml de NH4Cl (ac. sat.) seguido de 2x10 ml de solución saturada de cloruro sódico. La capa orgánica se secó (Na2SO4) y se concentró a presión reducida. El producto impuro (~100 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con TFA al 0,05 % y CH3CN (CH3CN al 15,0 % asciende al 42,0 % en 12 minutos, asciende al 100,0 % en 1 minuto); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 32,9 mg (34 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 427 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,73-7,65 (m, 4H), 7,61 (d, J = 5,7 Hz, 1H), 7,50 (d, J = 6,0 Hz, 2H), 4,89-4,78 (1H parcialmente oscurecido por pico de agua), 4,81 (s, 2H), 4,10 (t, J = 4,1 Hz, 2H), 3,94-3,81 (m, 1H), ~3,35 (1H parcialmente oscurecido por pico de disolvente metanol), 3,08-1,95 (m, 2H), 2,92 (t, J = 4,1 Hz, 2H), 2,52 (s, 3H), 2,08-1,92 (m, 1H), 1,92-1,65 (m, 3H). 1H RMN (400 MHz, CDCla): ó 7,63 (d, J =8,3 Hz, 2H), 7,34 (d, J =8,3 Hz, 2H), 7,30 (s, 1H)7,50(m, 2H), 4,89 (d, J =13,0
), 2
Compuesto 2.1. cido 5-yodo-2,4-dimetilbenzoico. Una solución de ácido 2,4-dimetilbenzoico (20 g, 133,18 mmol, 1,00 equiv), peryodato sódico (14,27 g, 66,72 mmol, 0,50 equiv), yodo (37,25 g, 146,76 mmol, 1,10 equiv) y ácido sulfúrico (1,96 g, 19,98 mmol, 0,15 equiv) en ácido acético (150 ml) se agitó a 110 °C en un baño de aceite. Después de 6 h, la mezcla de reacción se dejó alcanzar la temperatura ambiente y a continuación se diluyó con 1,2 l de agua. A esto se le añadieron cuidadosamente 800 ml de Na2S2O3 acuoso (ac., sat.). Los sólidos resultantes se recogieron por filtración, se disolvieron en 1,2 l de acetato de etilo, y se lavaron con 1x300 ml de Na2S2O3 (ac., sat.) seguido por 1x400 ml de solución saturada de cloruro sódico. La capa orgánica se secó (Na2SO4) y se concentró a presión reducida. El residuo impuro se recristalizó a partir de etanol:H2O (2:1) para dar 30 g (82 %) del compuesto del título como un sólido blanco
Compuesto 2.2. 5-Yodo-2,4-dimetilbenzoato de metilo. Una solución de ácido 5-yodo-2,4-dimetilbenzoico (compuesto 2.1, 10 g, 32,60 mmol, 1,00 equiv, 90 %) y ácido sulfúrico (10 ml) en metanol (100 ml) se agitó durante una noche a 80 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida y el residuo se diluyó con 200 ml de acetato de etilo. La mezcla resultante se lavó con 3x50 ml de agua, 2x50 ml de bicarbonato sódico (ac., sat.), seguido por 2x50 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 9,2 g (88 %) del compuesto del título como un aceite de color amarillo.
Compuesto 2.3. 5-Ciano-2,4-dimetilbenzoato de metilo. Una solución de 5-yodo-2,4-dimetilbenzoato de metilo (compuesto 2.2, 9,2 g, 31,71 mmol, 1,00 equiv), Zn(CN)2 (4,46 g, 38,12 mmol, 1,2o equiv), y Pd(PPh3)4 (2,93 g, 2,54 mmol, 0,08 equiv) en N,N-dimetilformamida (250 ml) se agitó en una atmósfera de nitrógeno a 100 °C durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición cuidadosa de 100 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2x20 ml de solución saturada de cloruro sódico, se secaron sobre (Na2SO4), y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para dar 6,2 g (93 %) del compuesto del título como un sólido de color blanco

Compuesto 2.4. 5-(-N'-hidroxicarbamimidoil)-2,4-dimetilbenzoato de metilo. Una solución de 5-ciano-2,4-dimetilbenzoato de metilo (compuesto 2.3, 6 g, 28,54 mmol, 1,00 equiv, 90%) y NH2OH (10 ml, 5,00 equiv, 50% en agua) en EtOH (20 ml ) se agitó a 100 °C en un baño de aceite. Después de 2 h, la mezcla se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se diluyó con 100 ml de acetato de etilo, se lavó con 2x20 ml de solución saturada de cloruro sódico, se secó (Na2SO4), y se concentró a presión reducida para dar 4,66 g (66 %) del compuesto del título como un sólido de color blanco.

Compuesto 2.5. Clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo. Una solución de 5-(N'-hidroxicarbamimidoil)-2,4-dimetilbenzoato de metilo (compuesto 2.4, 4,66 g, 18,87 mmol, 1,00 equiv, 90 %) y Ac2O (2,36 g, 23,09 mmol, 1,10 equiv) en AcOH (21 ml) se agitó a temperatura ambiente. Después de 5 minutos, el matraz se purgó con nitrógeno y se añadieron HCOOK (8,8 g, 104,76 mmol, 5,00 equiv) y paladio sobre carbono (10 %, 2,33 g). El matraz se purgó con nitrógeno seguido de hidrógeno. La mezcla se agitó en una atmósfera de hidrógeno (globo) a temperatura ambiente durante 4 h. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración, y el filtrado se concentró a presión reducida. El residuo se disolvió en 50 ml de etanol y el pH se ajustó a 5-6 con cloruro de hidrógeno (ac., 5 M). Los sólidos resultantes se retiraron por filtración y el filtrado se concentró a presión reducida para dar 4 del compuesto del título como un sólido de color blanco.

Compuesto 2.6. 2-(5-(metoxicarbonil)-2,4-dimetilfenil)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de terc-butilo. Una mezcla de clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5, 500 mg, 90 %), 3-bromo-4-oxopiperidin-1-carboxilato de terc-butilo (860 mg, 2,78 mmol), y carbonato potásico (570 mg, 4,12 mmol) en CH3CN (15 ml) se agitó durante una noche en nitrógeno a 80 °C. Después de enfriar a temperatura ambiente, la mezcla de reacción se concentró a presión reducida. El residuo se disolvió en 25 ml de acetato de etilo y se lavó con 2x10 ml de H2O. La fase orgánica se secó (Na2SO4) y se concentró a presión reducida. El producto impuro obtenido de este modo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:3) como eluyente para dar 0,3 g del compuesto del título como un sólido de color blanco

Compuesto 2.7. Ácido 5-(5-(terc-butoxicarbonil)4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-2,4-dimetilbenzoico. Una mezcla de 2-(5-(metoxicarbonil)-2,4-dimetil-fenil)-6,7-dihidro-3H-imidazo [4,5-c]piridin-5(4H)-carboxilato de terc-butilo (compuesto 2.6, 300 mg, 0,70 mmol, 1,00 equiv, 90 %) e hidróxido sódico (62 mg, 1,55 mmol, 2,00 equiv) en 15 ml de metanol/H2O (2:1) se agitó durante a 40 °C en un baño de aceite. Después de 2 h, la mezcla de reacción se concentró a aproximadamente 1/3 del volumen, a presión reducida. El valor del pH de la mezcla restante se ajustó a 3-4 con cloruro de hidrógeno (ac., 1 M). Los sólidos resultantes se recogieron por filtración y se
secaron en un horno a presión reducida para dar 0,2 g (69 %) del compuesto del título como un sólido de color amarillo

Compuesto 2.8. 2-(5-(4-(4-cianofenM)piperidm-1-carbonM)-2,4-dimetMfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de terc-butilo. Una solución del compuesto 2.7, (125 mg, 0,30 mmol, 1,00 equiv, 90%) , DIEA (130,5 mg, 1,01 mmol, 3,00 equiv), HBTU (256,2 mg, 0,68 mmol, 2,00 equiv), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 75 mg, 0,30 mmol, 1,00 equiv) en DMF (5 ml) se agitó durante una noche a temperatura ambiente. La mezcla de reacción se diluyó a continuación con 50 ml de acetato de etilo, se lavó con 2x20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice y se purificó usando acetato de etilo/éter de petróleo (1:1) para dar 0,1 g (55%) del compuesto del título como un sólido de color amarillo.

Compuesto 2.9. 4-(1-(2,4-DimetM-5-(4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-M)benzoM)piperidm-4-il)benzonitrilo. A una solución de 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dimetilfenil)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5 (4H)-carboxilato de terc-butilo (compuesto 2.8, 100 mg, 0,17 mmol, 1,00 equiv, 90%) en diclorometano (3 ml) se le añadió ácido trifluoroacético (1 ml). La mezcla resultante se agitó a temperatura ambiente. Después de 2 h, la mezcla de reacción se concentró a presión reducida. El residuo se recogió en diclorometano y se lavó con bicarbonato de sodio (ac., sat.). La fase orgánica se secó (Na2SO4) y se concentró a presión reducida para dar 0,1 g (68 %) del compuesto del título como un sólido de color amarillo.

Compuesto 2. 4-(1-(5-(5-Acetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. A una mezcla agitada de 4-(1-(2,4-dimetil-5-(4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 2.9, 20 mg, 0,02 mmol, 1,00 equiv, 50%) en DMF (2 ml) en nitrógeno se le añadió una solución de AC2O (2,4 mg, 0,02 mmol, 1,00 equiv) en DMF ( 0,2 ml) gota a gota a 0 °C. La solución resultante se agitó durante 1 h a 0~3 °C en un baño de agua/hielo. A continuación se diluyó la mezcla de reacción con 50 ml de acetato de etilo y se lavó con 2x20 ml de solución saturada de cloruro sódico. La fase orgánica se secó (Na2SO4) y se concentró a presión reducida. El residuo impuro (50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): Columna, Xbridge C18; fase móvil, agua con el 0,05 % de NH3.H2O y CH3CN (se mantiene el 5 % de CH3CN en 2 minutos, desciende al 20 %-46 % en 10 minutos, asciende al 100 % en 12 minutos, desciende al 20 % en 14 minutos); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 5,5 mg del compuesto del título como un sólido de color blanco. m/z (ES+) 482 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,69 (d, J = 8,1 Hz, 2H), 7,48 (d, J = 8,1 Hz, 2H), 7,39 - 7,28 (m, 2H), 4,90 (m, 1H, parcialmente oscurecido por el pico de disolvente), 4,67 y 4,62 (2 singletes, rotámeros de acetamida, MeCONCH2-imidazol, 2H), 3,92 (m, 2H), 3,66 (m, 1H), 3,23 (m, 1H), 3,00 (m, 2H), 2,85-2,76 (m, 2H), 2,48 (s, 3H), 2,41 y 2,31 (2 singletes, rotámeros de aril amida, ArCH3, 3H), 2,25 y 2,22 (2 singletes, rotámeros de acetamida,

Compuesto 3. 4-(1-(3-(5-Acetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 468 (M+H)+.

Compuesto 4.4-(1-(4-Metil-3-(5-(metilsulfonil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. miz (ES+) 504 (M+H)+.

Compuesto 5. 4-(1-(5-(5-(IsopropNsulfonM)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 546 (M+H)+.
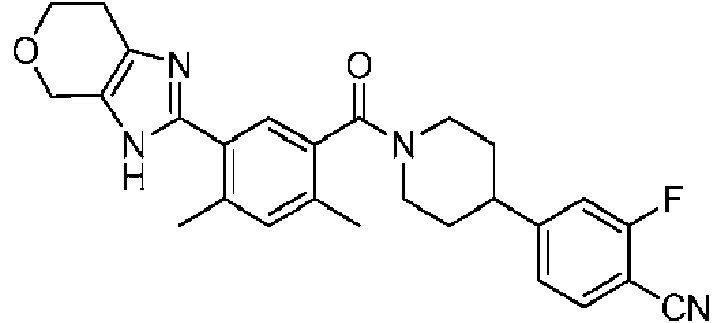
Compuesto 6. 4-(1-(2,4-DimetM-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-M)benzoM)piperidm-4-M)-2-fluorobenzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 459 (M+H)+.

Compuesto 7. 2-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2,4-dimetNfeml)-N,N-dimetN-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-sulfonamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 547 (M+H)+.

Compuesto 8. (2,4-DimetN-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)fenM)(4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 484 (M+H)+.
Compuesto 9. 2-(5-(4-(4-CianofenM)piperidm-1-carboml)-2,4-dimetNfenM)-N-metM-4,5,6,7-tetrahidro-1H-benzo[d]imidazol-6-carboxamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m iz (ES+) 496 (M+H)+.

Compuesto 11.1. 4-(4-cianofenil)-4-fluoropiperidin-1-carboxilato de terc-butilo. A una solución agitada del compuesto 1.2 (5 g, 16,54 mmol, 1,00 equiv) en diclorometano (250 ml) en nitrógeno a -78 °C se le añadió gota a gota Deoxo-Fluor® (4,4 g, 19,89 mmol, 1,20 equiv). La mezcla resultante se agitó durante 1 h a -78 °C. La mezcla de reacción se inactivó cuidadosamente a continuación mediante la adición de 50 ml de bicarbonato sódico (ac. sat.) y se extrajo con 3x100 ml de diclorometano. Las capas orgánicas combinadas se lavaron con 150 ml de solución saturada de cloruro sódico, se secaron sobre sulfato de magnesio anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:30) como
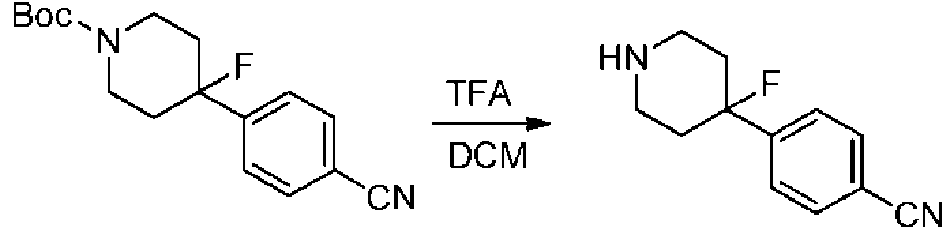
Compuesto 11.2. 4-(4-Fluoropiperidin-4-il)benzonitrilo. A una solución agitada del compuesto 11.1 (620 mg, 1,02 mmol, 1,00 equiv, 50 %) en DCM (40 ml) se le añadió gota a gota ácido trifluoroacético (6 g, 52,62 mmol, 51,66 equiv). Después de agitar a temperatura ambiente durante 2 h, la mezcla se concentró a presión reducida. El residuo se recogió en DCM y se trató con bicarbonato sódico acuoso. Las fases se separaron y la capa acuosa se extrajo con 2x50 ml de DCM. Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron a presión reducida para dar 0,4 g del compuesto del título como un sólido de color amarillo claro. m /z (ES+) 205 (M+H)+.
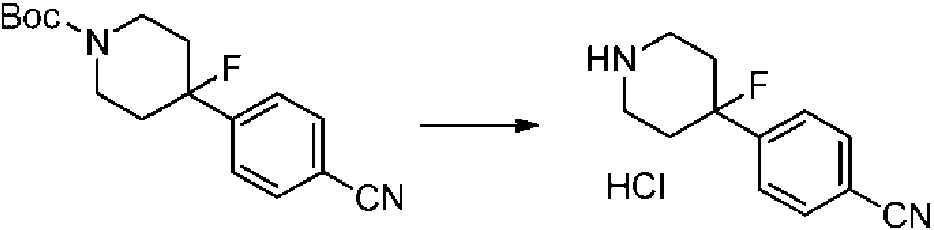
Compuesto 11.2, sal HCl. Clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y un procedimiento similar al usado para la preparación del compuesto 1.5 y usando el compuesto 11.1 en lugar del compuesto 1.4. m /z (ES+) 205 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,83

Compuesto 11. 4-(1-(2,4-DimetN-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 y usando el compuesto 11.2 en lugar del compuesto 1.5. m /z (ES+) 459 (M+H)+.

Compuesto 12. (4-(4-Clorofenil)piperidin-1-il)(2,4-dimetil-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)fenil)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m /z (ES+) 450 (M+H)+.

Compuesto 13. 2-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2,4-dimetNfeml)-N-metM-1,4,5,6-tetrahidrociclopenta[d]imidazol-5-carboxamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 482 (M+H)+.

Compuesto 14. 4-(1-(2,4-DimetM-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-M)benzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos
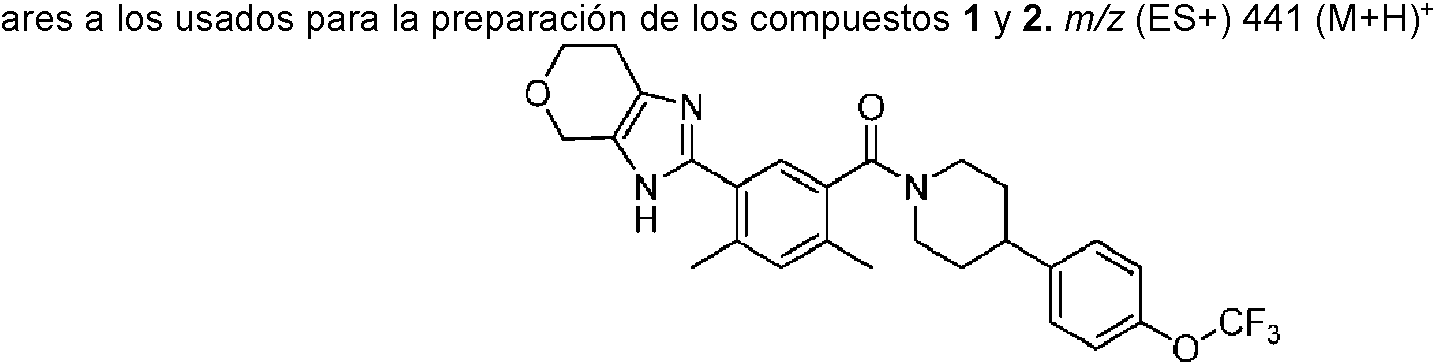
Compuesto 15. (2,4-DimetN-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)fenM)(4-(4-(trifluorometoxi)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2. m/z (ES+) 500 (M+H)+.

Compuesto 16.1. N2-(2-Metoxietil)-N2-metil-5-nitropiridin-2,4-diamina. Una solución de 2-cloro-5-nitropiridin-4-amina (500 mg, 2,88 mmol, 1,00 equiv), trietilamina (1,167 g, 11,53 mmol, 4,00 equiv) y (2-metoxietil)(metil)amina (514 mg , 5,77 mmol, 2,00 equiv) en DMF (20 ml) se agitó en un tubo sellado a 55 °C detrás de una pantalla protectora contra explosiones durante una noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 100 ml de agua y se extrajo con 3x150 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron a presión reducida para dar 723 mg (impuros) del compuesto del título como un aceite de color rojo oscuro.

Compuesto 16.2. N2-(2-Metoxietil)-N2-metilpiridin-2,4,5-triamina. Un matraz de fondo redondo, que contenía una solución del compuesto 16.1 (400 mg, 1,77 mmol, 1,00 equiv)) en metanol (30 ml) se purgó con nitrógeno gaseoso. A la solución se le añadió a continuación paladio sobre carbono (40 mg, 10%, 60% de agua). El matraz se purgó adicionalmente con nitrógeno y la atmósfera se cambió a continuación a hidrógeno. La mezcla se agitó a temperatura ambiente en un globo con hidrógeno durante 4 h. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida para dar 300 mg (86 %) del compuesto del título como un sólido de color marrón claro. Se descubrió que el producto impuro era inestable y se usó inmediatamente.
Compuesto 16.3. Ácido 5-formil-2,4-dimetilbenzoico. A una solución agitada de ácido 5-yodo-2,4-dimetilbenzoico (compuesto 2.1, 5 g, 18,11 mmol, 1,00 equiv) en tetrahidrofurano (150 ml) en nitrógeno a -78 °C se le añadió n-BuLi (2,5 M en THF, 18 ml, 2,50 equiv) gota a gota. Después de agitar a -78 °C durante 1 h, se añadió DMF (5 g, 68,4 mmol, 3,80 equiv) gota a gota. La mezcla resultante se agitó a -78 °C durante 0,5 h adicional y a continuación se inactivó cuidadosamente mediante adición lenta de 50 ml de agua. El pH se ajustó a continuación a ~3-4 usando HCl acuoso (ac., 6 M). La mezcla se extrajo con 3x200 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron a presión reducida. El residuo obtenido de este modo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10-1:5) como eluyente para dar 2,4 g (74 %) del

Compuesto 16.4. 4-(1-(5-Formil-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. A una solución agitada de ácido 5-formil-2,4-dimetilbenzoico (compuesto 16.3, 950 mg, 5,33 mmol, 1,10 equiv) en DMF (15 ml) se le añadió DIEA (2,48 g, 19,19 mmol, 4,00 equiv) seguido de HBTU (3,67 g, 9,68 mmol, 2,00 equiv), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 1,07 g, 4,80 mmol, 1,00 equiv). Después de agitar durante una noche a temperatura ambiente, la reacción se inactivó mediante la adición de 60 ml de agua. La mezcla resultante se extrajo con 3x150 ml de acetato de etilo y las capas orgánicas se combinaron, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter

Compuesto 16. 4-(1-(5-(6-((2-MetoxietN)(metM)ammo)-3H-imidazo[4,5-c]piridm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. Una solución del compuesto 16.2 (300 mg, 1,53 mmol, 1,30 equiv), el compuesto 16.4 (407 mg, 1,17 mmol, 1,00 equiv), y acetato de amonio (362 mg, 4,70 mmol, 4,00 equiv) en etanol (20 ml) se agitó durante una noche a 70 °C en un baño de aceite. A continuación, la mezcla resultante se concentró al vacío y el residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10-2:1) como eluyente para dar~200 mg de producto que se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (se mantiene el 5,0 % de CH3CN en 2 minutos, asciende al 25,0 % en 1 minuto, asciende al 55,0 % en 12 minutos, asciende al 100,0 % en 1 minuto); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 150 mg (24 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 523 (M+H)+. 1H RMN (300 MHz, CDCh): 88,57-8,38 (m, 1H), 7,78-7,53 (m, 4H), 7,34 (d, J = 7,5 Hz, 2H), 7,28-7,12 (m, 2H), 5,06-4,88 (m, 1H), 3,71 (s aparente, 5H), 3,50 (s, 3H), 3,41-3,05 (m, 4H), 3,01-2,75 (m, 2H), 2,68 (s, 3H), 2,42 y 2,31 (2 singletes, rotámeros de amida, A C H 3, 3H), 2,12-1,93 (m, 1H), 1,93-1,39
Compuesto 17. 4-(1-(2,4-Dimetil-5-(6-morfolino-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo.
El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados
para la preparación del compuesto 16. m/z ES+ 521 M+H .

Compuesto 18. 4-(1-(2,4-Dimetil-5-(6-(4-metilpiperazin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 534 (M+H)+. 1H RMN (300 MHz, DMSO-d6): 8,73 (s, 1H), 7,76 (d, J = 7,5 Hz, 2H), 7,66 y 7,56 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,49 (d, J = 8,1 Hz, 2H), 7,37 (s, 1H), 7,12 (s, 1H), 4,81-4,67 (m, 1H), 4,30 (s ancho, 2H), 3,70-3,37 (m, 3H), 3,37-3,05 (m, 5H), 3,05-1,80 (m, 5H), 2,60 (s, 3H), 2,37 y 2,27 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,02-1,87 (m, 1H), 1,87-2,40 (m, 3H).

Compuesto 19. 4-(1-(2,4-DimetN-5-(6-(piperazm-1-M)-3H-imidazo[4,5-c]pmdm-2-N)benzoN)piperídm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z ES+ 519 M+H .

Compuesto 20.1.5-Nitro-2-(pirrolidin-1-il)piridin-4-amina. Un matraz de fondo redondo de 500 ml equipado con un condensador de reflujo se cargó con 2-cloro-5-nitropiridin-4-amina (2,00 g, 11,5 mmol, 1,0 equiv), pirrolidina (2,86 ml, 34,6 mmol, 3,0 equiv), carbonato potásico (4,78 g, 34,6 mmol, 3,0 equiv) y acetonitrilo (50 ml). La mezcla se calentó a 70 °C durante una noche en nitrógeno y a continuación se enfrió a temperatura ambiente. Los sólidos de color amarillo se recogieron por filtración y se lavaron con acetonitrilo, agua y hexanos. Los sólidos se secaron para obtener el compuesto del título como un sólido de color amarillo brillante (1,43 g, 1a cosecha). Los disolventes del filtrado se retiraron y la fase acuosa se extrajo con acetato de etilo. Los extractos orgánicos se secaron (Na2SO4), se filtraron y se retiraron al vacío para obtener producto adicional como un sólido de color naranja/amarillo (820 mg, 2a cosecha). La 2a cosecha de producto se trituró con acetato de etilo (3 ml) y a continuación se filtró y se lavó con acetato de etilo (2 x 1 ml) y éter dietílico (3 ml) para obtener un sólido de color amarillo brillante (751 mg). El rendimiento total de 5-nitro-2- (pirrolidin-1-il)piridin-4-amina obtenida fue de 2,18 g (91 %). m/z (ES+) 209,1 (M+H)+. 1H RMN (400 MHz, DMSO-de): ó 8,79 (s, 1H), 7,39 s ancho, 2H , 5,63 s, 1H, 3,40 s ancho, 4H , 1,93 s ancho, 4H).

Compuesto 20.2. 6-(Pirrolidin-1-il)piridin-3,4-diamina. A un matraz de fondo redondo de 100 ml se le añadió 5-nitro-2-(pirrolidin-1-il)piridin-4-amina (compuesto 20.1, 800 mg, 3,84 mmol, 1,0 equiv) y Pd sobre carbono (10% en peso de Pd, 400 mg, 0,38 mmol, 0,1 equiv). El sistema se purgó con nitrógeno y se cargó con metanol (19 ml) seguido de hidrógeno. La mezcla se agitó a temperatura ambiente en hidrógeno durante 4 horas y a continuación se purgó con nitrógeno. La mezcla se filtró a través de celite y se lavó exhaustivamente con MeOH. Los disolventes se eliminaron al vacío para obtener el compuesto del título como un sólido de color púrpura (641 mg, 94%). m/z (ES+) 179,3 (M+H)+. 1H RMN (400 MHz, DMSO-d6): ó 7,27 (s, 1H), 5,62 (s, 1H), 5,14 (sancho, 2H), 3,67 (sancho, 2H), 3,23 3,13 (m, 4H), 1,91-1,79 (m, 4H).

Compuesto 20. 4-(1-(2,4-Dimetil-5-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo de 100 ml se le añadieron 6-(pirrolidin-1-il)piridin-3,4-diamina (compuesto 20.2, 641 mg, 3,60 mmol, 1,0 equiv), 4-(1-(5-formil-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo (compuesto 16.4, 1,25 g, 3,60 mmol, 1,0 equiv), metabisulfito sódico (890 mg, 4,68 mmol, 1,3 equiv) y DMF (36 ml). La mezcla se calentó a 100 °C en aire durante 22 horas y a continuación se dejó enfriar a temperatura ambiente. Se añadió agua (80 ml) lentamente a la solución agitada hasta que ya no precipitó más producto. Los sólidos se filtraron y se lavaron con agua (2 x 15 ml) y se secaron a un sólido marrón (1,42 g). Producto adicional precipitó desde el filtrado al reposar, que se filtró y se lavó con agua (2 x 10 ml) para obtener un sólido de color blanquecino (241 mg). La reacción anterior se repitió una segunda vez de la manera y las cantidades exactas como se han descrito para dar el producto adicional (1,26 g de sólido de color marrón tras la precipitación inicial, más 288 mg de sólido de color blanquecino a partir de la precipitación adicional). Lo filtrados acuosos de las dos reacciones se combinaron y se añadió NaHCO3 acuoso para ajustar el pH a 7. La fase acuosa se extrajo con DCM/MeOH al 2% (300 ml), se secó (Na2SO4), se filtró y se evaporó para obtener un sólido de color marrón (547 mg). Todos los productos obtenidos se combinaron y se purificaron mediante cromatografía en gel de sílice (DCM/MeOH) para obtener el compuesto del título como un sólido de color amarillo (2,37 g, 65% basándose en el rendimiento teórico para las dos reacciones). m/z (ES+) 505,1 (M+H)+. 1H RMN (400 MHz, DMSO-d6): 812,41 (s ancho, 1H), 8,52 (s, 1H), 7,77 (d, J = 8,2 Hz, 2H), 7,66 y 7,56 (2 singletes anchos, rotámeros de amida, ArH, 1H), 7,49 (s, J = 8,2 Hz, 2H), 7,29 (s, 1H), 6,31 (s ancho, 1H), 4,79-4,67 (m, 1H), 3,54-3,34 (m, 5H), 3,17 (t aparente, J = 11,8, 1H), 2,99-2,82 (m, 2H), 2,63 (s, 3H), 2,33 y 2,24 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,03-1,87 (m, 5H), 1,82-1,38 (m, 3H). Análisis elemental (C3-iH32N6O^1,3

Compuesto 20, sal HCl. Clorhidrato de 4-(1-(2,4-dimetN-5-(6-(pirroNdm-1-M)-3H-imidazo[4,5-c]piridm-2-il)benzoil)piperidin-4-il)benzonitrilo. Se añadió diclorometano (~50 ml) a 4-(1-(2,4-dimetil-5- (6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il) benzoil)piperidin-4-il)benzonitrilo (compuesto 20, base libre, 2,31 g, 4,58 mmol) hasta que se disolvió completamente y a continuación HCl 4 M en dioxano (5 ml, 20 mmol, 4,3 equiv). Los disolventes se retiraron y el residuo se disolvió en DCM y se retiró al vacío. La sal de clorhidrato resultante se recristalizó a partir de etanol en ebullición (180 ml) y se dejó enfriar a temperatura ambiente lentamente. La mezcla se dejó reposar a temperatura ambiente durante una noche y a continuación se introdujo en el congelador durante 4 horas. Los sólidos resultantes se filtraron y se lavaron con etanol frío (25 ml) seguido de éter dietílico (25 ml). Los sólidos se secaron para obtener un sólido de color blanquecino a gris claro (1,82 g, 73%). 1H RMN (400 MHz, DMSO-d6): 813,59 y 13,46 (2 singletes anchos, tautómeros de imidazol NH, 1H), 13,29 (s ancho, 1H), 8,62 (s ancho, 1H), 7,83 & 7,67 (2 singletes, rotámeros de amida, ArH, 1H,), 7,78 (d, J = 7,8 Hz, 2H), 7,58-7,45 (m, 2H), 7,37 (s, 1H), 6,71 (s anchos, 1H), 4,80-4,68 (m, 1H), 3,65-3,38 (m, 5H), 3,25-3,12 (m, 1H), 3,00-2,82 (m, 2H), 2,66 (s, 3H), 2,37 y 2,27 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,13-2,01 (m, 4H), 1,98-1,87 (m, 1H), 1,82-1,40 (m, 3H). Análisis elemental (C3-iH32N6O^1,0 HCh2,4 H2O, 584,32), descubierto (calculado), C: 63,80 (63,72); H: 6,32 (6,52); N: 14,25 (14,38).
Compuesto 20 sal MsOH. Metanosulfonato de 4-(1-(2,4-dimetN-5-(6-(pirroNdm-1-M)-3H-imidazo[4,5-c]pmdm-2-il)benzoil)piperidin-4-il)benzonitrilo. Se añadió clorhidrato de 4-(1-(2,4-dimetil-5-(6-(pirrolidin-1-il)-3H-imidazo [4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 20, sal HCl, 1,82 g, 3,36 mmol) a agua (100 ml) más NaHCO3 saturado (50 ml) y se extrajo con 1:1 DCM/EtOAc (500 ml) más MeOH (25 ml). Una vez que todo el producto se disolvió completamente, las capas se separaron y la acuosa se extrajo con DCM/EtOAc 1:1 adicional (100 ml). Las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se evaporaron a un polvo de color amarillo claro (1,68 g, 3,33 mmol). El material de base libre se disolvió en acetonitrilo caliente (10 ml), a continuación se añadió una solución de ácido metanosulfónico 1,0 M acuoso (3,33 ml, 3,33 mmol, 1,0 equiv) y se mezcló bien. Se añadió agua adicional (6 ml) y la mezcla se congeló y se secó en el liofilizador para obtener un polvo de color amarillo (1,99 g, 99%). m/z (ES+) 505,1 (M+H)+. 1H RMN (400 MHz, DMSO-da): 6 13,29 (s ancho, 1H), 13,10 (s ancho, 1H), 8,65 (s ancho, 1H), 7,78 (d, J = 8,2 Hz, 2H), 7,74 y 7,64 (2 singletes, rotámeros de amida, ArH, 1H,), 7,50 (d, J = 8,3 Hz, 2H), 7,37 (s, 1H), 6,74 (s ancho, 1H), 4,80-4,68 (m, 1H), 3,65-3,38 (m, 5H), 3,26-3,12 (m, 1H), 3,01-2,83 (m, 2H), 2,64 (s, 3H), 2,37 y 2,27 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,32 (s, 3H) 2,12-2,00 (m, 4H), 1,98-1,88 (m, 1H), 1,82 1,39 (m, 3H). Análisis elemental ( C s ^ ^ O ^ ^ MsOH^1,7 H2O, 631,36), descubierto (calcado), C: 60,96 (60,88); H:

Compuesto 21. 4-(1-(5-(6-(Azetidm-1-M)-3H-imidazo[4,5-c]pmdm-2-M)-2,4-dimetNbenzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 491 (M+H)+.
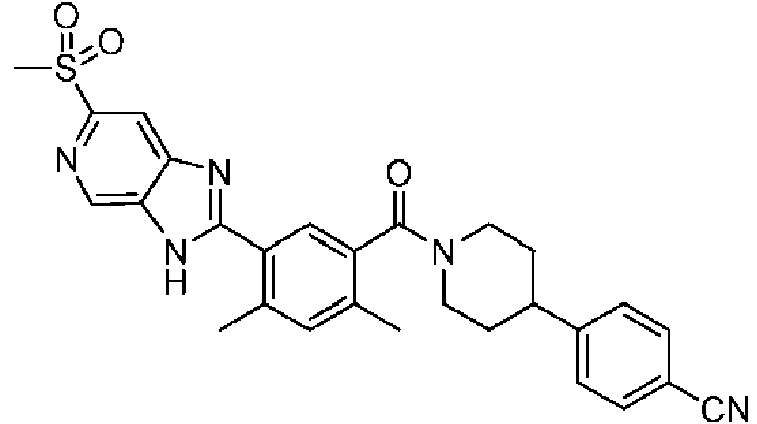
Compuesto 22. 4-(1-(2,4-DimetN-5-(6-(metNsulfonM)-3H-imidazo[4,5-c]pmdm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 514 (M+H)+.

Compuesto 23. 4-(1-(2,4-DimetN-5-(6-(metMsulfonM)-3H-imidazo[4,5-b]piridm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 514 (M+H)+. 1H RMN (300 MHz, DMSO-d6 D2O): 68,87 (d, J= 1,8 Hz, 1H), 8,52 (d, J = 2,1 Hz, 1H), 7,74 (d, J = 7,5 Hz, 2H), 7,66 y 7,56 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,48 (d, J = 8,4 Hz, 2H), 7,38 (s ancho, 1H), 4,76-4,63 (m, 1H), 3,57-3,38 (m, 1H), 3,31 (s, 3H), 3,28-3,13 (m, 1H), 3,03-2,85 (m, 2H), 2,60 (s, 3H), 2,35 y 2,27 (2 singletes, rotámeros de amida, Ar-CHs, 3H), 2,00 1,85 (m, 1H), 1,85-1,32 (m, 3H).

Compuesto 24. 4-(1-(5-(5-(Azetidm-1-M)-1H-imidazo[4,5-b]piridm-2-M)-2,4-dimetMbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16, pero usando 6-cloro-3-nitropiridin-2-amina y clorhidrato de azetidina en lugar de 2-cloro-5-nitropiridin-4-amina y (2-metoxietil)(metil)amina, respectivamente. m/z (ES+) 491 (M+H)+. 1H RMN (300 MHz, CD3OD): 88,06 (d, J = 9,0 Hz, 1H), 7,70-7,42 (m, 6H), 6,60 (d, J = 9,0 Hz, 1H), 4,90 (m, 1h , parcialmente oscurecido por el pico de disolvente), 4,32 (m, 4H), 3,65 (m, 1H), 3,03 (m, 1H), 2,62 (s, 3H), 2,57 (m, 3H), 2,48 y 2,38 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,04 (m, 1H), 1,96-1,68 (m, 4H).
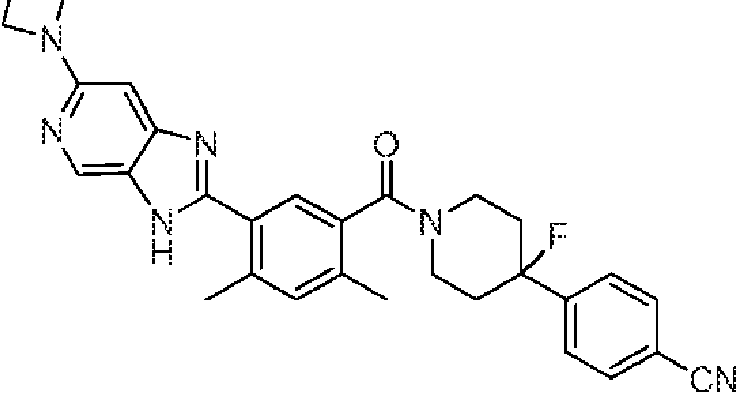
Compuesto 25. 4-(1-(5-(6-(Azetidm-1-M)-3H-imidazo[4,5-c]piridm-2-M)-2,4-dimetNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16 y usando el compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 509 (M+H)+.

Compuesto 26.1. 4-(5-bromopiridin-2- il)-4-hidroxipiperidin-1-carboxilato de terc-butilo. A una solución agitada de 2,5-dibromopiridina (10 g, 42,19 mmol, 1,00 equiv) en tolueno (1000 ml) en nitrógeno a -78°C se le añadió gota a gota n-BuLi (18 ml, 2,5 M en tolueno). Después de 1 h a -78 °C, se añadió gota a gota una solución de 4-oxopiperidin-1-carboxilato de terc-butilo (10 g, 50,25 mmol, 1,19 equiv) en tolueno (200 ml) mientras se agitaba. Después de 2 h adicionales de agitación a -78 °C, la reacción se inactivó a continuación cuidadosamente mediante la adición de 300 ml de agua. La mezcla resultante se extrajo con 3x500 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x300 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:5) para dar 6,5 g (42 %) del compuesto del título como un aceite de color amarillo.

Compuesto 26.2. 4-(5-cianopiridin-2-il)-4-hidroxipiperidin-1-carboxilato de terc-butilo. Una mezcla de 4-(5-bromopiridin-2-il)-4-hidroxipiperidin-1-carboxilato de terc-butilo (compuesto 26.1, 1 g, 2,80 mmol, 1,00 equiv), cianuro de zinc (400 mg, 3,42 mmol, 1,22 equiv), Pd(PPh3)4 (200 mg, 0,17 mmol, 0,06 equiv) en DMF (50 ml) se agitó en nitrógeno durante 2 h a 100 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición cuidadosa de 300 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x 100 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:4) para dar 0,5 g (58 %) del compuesto del título como un aceite de color amarillo.

Compuesto 26.3. 4-(5-cianopiridin-2-il)-4-fluoropiperidin-1-carboxilato de terc-butilo. A una solución de 4-(5-cianopiridin-2-il)-4-hidroxipiperidin-1-carboxilato de terc-butilo (compuesto 26.2, 1 g, 3,13 mmol, 1,00 equiv, 95%) en diclorometano (50 ml) en nitrógeno a -78 °C se le añadió gota a gota una solución de trifluoruro de bis(2-metoxietil)aminoazufre (830 mg, 3,75 mmol, 1,20 equiv) en diclorometano (10 ml) durante el transcurso de 1 minuto. La mezcla resultante se agitó durante 1 h a -78 °C. La reacción se inactivó a continuación cuidadosamente mediante adición gota a gota de agua y se lavó con 2x20 ml de bicarbonato sódico (ac) seguido de 3x20 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo impuro se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10) para

Compuesto 26.4. Trifuoroacetato de 6-(4-fluoropiperidin-4-il)nicotinonitrilo. A una solución agitada de 4-(5-cianopiridin-2-il)-4-fluoropiperidin-1-carboxilato de terc-butilo (compuesto 26.3, 1 g, 3,11 mmol, 1,00 equiv, 95%) en diclorometano (20 ml) se le añadió gota a gota TFA (3,75 g, 32,89 mmol, 10,57 equiv). Después de agitar durante 1 h a 25 °C, la mezcla se concentró a presión reducida para dar 0,5 g del compuesto del título como un aceite de color marrón.

Compuesto 26. 6-(1-(5-(6-(Azetidm-1-M)-3H-imidazo[4,5-c]piridm-2-M)-2,4-dimetNbenzoM)-4-fluoropiperidm-4-il)nicotinonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16 pero usando el compuesto 26.4 en lugar del compuesto 1.5. miz (ES+) 509 (M+H)+.

Compuesto 27. 4-(1-(4-Metil-3-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16 pero usando ácido 3-bromo-4-metilbenzoico en lugar de ácido 5-yodo-2,4-dimetilbenzoico. miz ES+ 491 M+H .

Compuesto 28. 4-(1-(3-(1H-imidazo[4,5-c]piridin-2-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la
preparación de los compuestos 16 y 27. m/z (ES+) 422 (M+H)+. 1H RMN (300 MHz, CD3OD): 59,35 (s, 1H), 8,60 (d, J = 6,6 Hz, 1H), 8,19 (d, J = 6,6 Hz, 1H), 7,92 (s, 1H), 7,73-7,58 (m, 4H), 7,50 (d, J=8,1 Hz, 1H), ~4,9(1H, parcialmente oscurecido por el pico de agua), 4,06-3,87 (m, 1H), 3,13-2,95 (m, 3H), 2,73 (s, 3H), 2,11-1,63 (m, 4H).
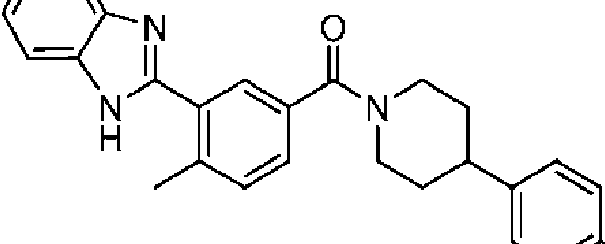
Compuesto 29. 4-(1-(3-(1H-Benzo[d]imidazol-2-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 16 y 27. m/z (ES+) 421 (M+H)+. 1H RMN (300 MHz, CD3OD): 57,87-7,77 (m, 3H), 7,75-7,60 (m, 4H), 7,60-7,47 (m, 4H), ~4,85 (1H, parcialmente oscurecido por el pico de agua), 4,04-3,89 (m, 1h ),
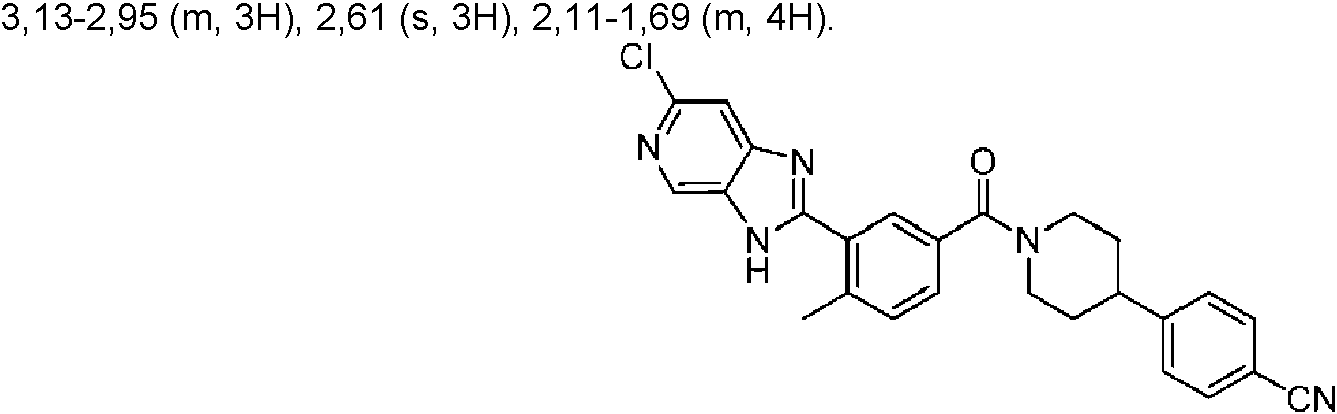
Compuesto 30. 4-(1-(3-(6-Cloro-3H-imidazo[4,5-c]piridin-2-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 16 y 27. m/z (ES+) 456 (M+H)+.
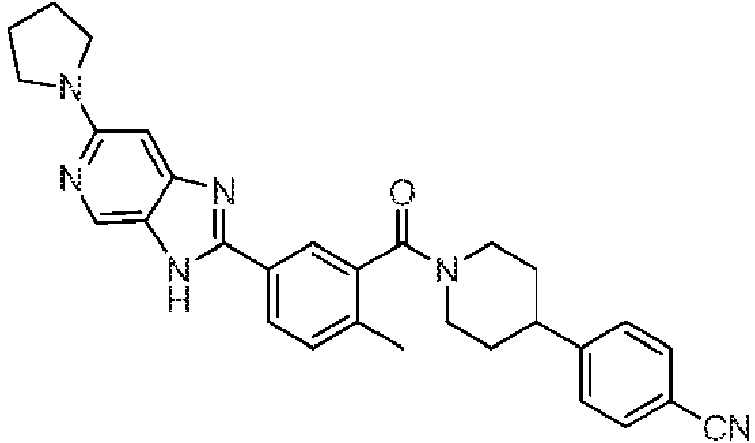
Compuesto 31. 4-(1-(2-Metil-5-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16 pero usando ácido 5-bromo-2-metilbenzoico en lugar de ácido 5-yodo-2,4-dimetilbenzoico (compuesto 2.1). m/z (ES+) 491 (M+H)+. 1H RMN (300 MHz, DMSO-d6): 512,63 (s ancho, 1H), 8,06 (d, J = 7,8 Hz, 1H), 8,01 y 7,91 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,81 (d, J = 6,6 Hz, 2H), 7,58-7,42 (m, 3H), 6,31 (s, 1H), 4,85 (m, 1H), 3,57-3,35 (m, 5H), 3,30-3,13 (m, 1H), 3,07-1,85 (m, 2H), 2,39 y 2,29 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,10-1,89 (m, 5H), 1,87-1,40 (m, 3H).
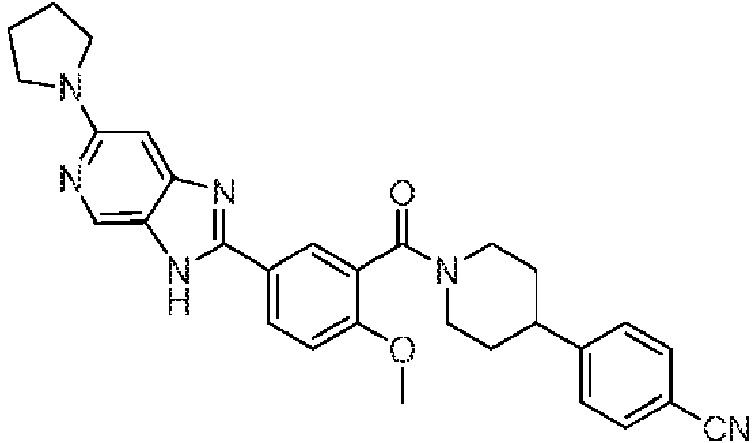
Compuesto 32. 4-(1-(2-Metoxi-5-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 507 (M+H)+.

Compuesto 33. 2-(4-(4-Cianofeml)piperidm-I-carbonM)-4-(6-(pirroMdm-1-M)-3H-imidazo[4,5-c]piridm-2-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. miz (ES+) 502 (M+H)+.

Compuesto 34. 4-(1-(2-Cloro-5-(6-(pirroMdm-1-M)-3H-imidazo[4,5-c]piridm-2-M)benzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. miz (ES+) 511 (M+H)+.
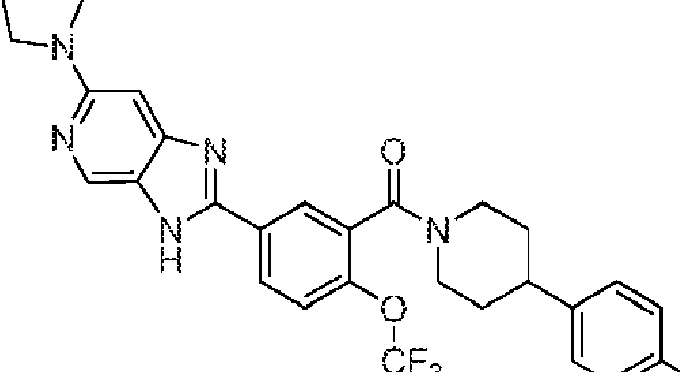
Compuesto 35. 4-(1-(5-(6-(Pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)-2-(trifluorometoxi)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. miz (ES+) 561 (M+H)+.

Compuesto 36. 4-(1-(3-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z (ES+) 477 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 8,44 (s, 1H), 8,25-8,17 (m, 2H), 7,77-7,63 (m, 4H), 7,51 (d, J = 8,1 Hz, 2H), 6,59 (s, 1H), -4 ,85 (1H, parcialmente oscurecido por el pico de agua), 3,98-3,83 (m, 1H), 3,61-3,48 (m, 4H), -3 ,4 (1H, parcialmente oscurecido por el pico de disolvente), 3,12-2,96 (m, 2H), 2,22-2,09 (m, 4H), 2,09-1,95 (m, 1H), 1,95 (m, 3H).

Compuesto 37. 4-(1-(3-(6-(isopropilamino)-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 16. m/z ES+ 465 M+H .

Compuesto 38.1. Tetrahidrofuran-3-carboxilato de metilo. Una solución de ácido tetrahidrofuran-3-carboxílico (540 mg, 4,65 mmol, 1,00 equiv) y TsOH (10 mg, 0,06 mmol, 0,01 equiv) en metanol (40 ml) se agitó a 66 °C en un baño de aceite. Después de 16 h, la mezcla resultante se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se disolvió en 10 ml de éter, se lavó con 1x20 ml de NaHCO3 (ac., sat.) seguido de 3x20 ml de solución saturada de cloruro sódico, y se concentró a presión reducida para dar 0,40 g (66 %) del compuesto del título como un aceite incoloro.

Compuesto 38.2. Tetrahidrofuran-3-carbohidrazida. En un matraz de fondo redondo, se colocó hidrato de hidrazina (20 ml). A esto se le añadió tetrahidrofuran-3-carboxilato de metilo (compuesto 38.1, 390 mg, 3,00 mmol, 1,00 equiv) gota a gota con agitación. La mezcla resultante se agitó a 50 °C en un baño de aceite. Después de 3 h, la mezcla de reacción se concentró y se secó a presión reducida para dar 0,29 g (74%) del compuesto del título como un aceite incoloro.

Compuesto 38.3. 3-Carbamotioil-4-metilbenzoato de metilo. Una solución de 3-ciano-4-metilbenzoato de metilo (compuesto 1.7, 880 mg, 5,02 mmol, 1,00 equiv) y ditiofosfato de O,O'-dietilo (1,41 g, 8,29 mmol, 1,50 equiv) en THF/H2O (40 ml) se agitó a 80 °C(ADVERTENCIA: se produce un desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en las campanas extractoras bien ventiladas). Después de 3 h, el disolvente orgánico se retiró a presión reducida y la fase acuosa residual se extrajo con 3x20 ml de acetato de etilo. Las capas orgánicas combinadas se concentraron a presión reducida para dar 0,85 g (79 %) del compuesto del título como un sólido de color amarillo claro.

Compuesto 38.4. 3-(Imino(metiltio)metil)-4-metilbenzoato de metilo. A una solución agitada de 3-carbamotioil-4-metilbenzoato de metilo (compuesto 38.3, 2,10 g, 9,85 mmol, 1,00 equiv) en tetrahidrofurano (30 ml) se le añadió yodometano (2,8 g, 19,73 mmol, 2,00 equiv) gota a gota. La mezcla resultante se agitó a temperatura ambiente. Después de 3 h, la mezcla de reacción se concentró a presión reducida y el residuo se secó al vacío para dar 1,6 g (73 %) del compuesto del título como un sólido de color amarillo claro.

Compuesto 38.5. 4-Metil-3-(5-(tetrahidrofuran-3-il)-4H-1,2,4-triazol-3-il)benzoato de metilo. Una solución de tetrahidrofuran-3-carbohidrazida (compuesto 38.2, 195 mg, 1,50 mmol, 1,50 equiv) y el compuesto 38.4 (223 mg, 1,00 mmol, 1,00 equiv) en AcOH (30 ml) se agitó a 80 °C. Después de 4 h, la mezcla de reacción se concentró a presión reducida y el residuo se secó a alto vacío para dar 153 mg (53 %) del compuesto del título como un aceite de color amarillo.

Compuesto 38.6. Ácido 4-metil-3-(5-(tetrahidrofuran-3-il)-4H-1,2,4-triazol-3-il)benzoico. A una solución agitada del compuesto 38.5 (57 mg, 0,20 mmol, 1,00 equiv) en metanol (20 ml) se le añadió gota a gota hidróxido sódico (acuoso, 1 M, 0,2 ml). La mezcla resultante se agitó a temperatura ambiente. Después de 4 h, el disolvente orgánico se retiró a presión reducida. La capa acuosa residual se lavó con 2x20 ml de acetato de etilo. El pH de la fase acuosa se ajustó a continuación a 4 con cloruro de hidrógeno (ac., 1 M) y los sólidos resultantes se recogieron por filtración y

Compuesto 38. 4-(1-(4-Metil-3-(5-(tetrahidrofuran-3-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo.
Una mezcla del compuesto 38.6, (137 mg, 0,50 mmol, 1,00 equiv, 90%), HBTU (228 mg, 0,60 mmol, 1,20 equiv), DIEA (162 mg, 1,25 mmol, 2,50 equiv), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (1.5, 111 mg, 0,50 mmol, 1,00 equiv) en DMF (20 ml) se agitó a temperatura ambiente. Después de 1 h, la reacción se inactivó mediante la adición de 20 ml de agua y la mezcla resultante se extrajo con 3x20 ml de acetato de etilo. Las fases orgánicas combinadas se secaron (Na2SÜ4) y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:3) para dar 35 mg (16 %) del compuesto del título como un sólido de color marrón, miz (ES+ 442 M+H .

Compuesto 39.1. Ácido 3-formil-4-metilbenzoico. A una solución agitada de ácido 3-bromo-4-metilbenzo¡co (2,14 g, 10,00 mmol, 1,00 equiv) en tetrahidrofurano (30 ml) en nitrógeno a -78 °C se le añadió n-BuLi (10 ml, 2,5 M en THF, 2,50 equiv) gota a gota. Después de agitar durante 1 h por debajo de -70 ° C, se añadió lentamente DMF (5 ml). La solución resultante se calentó lentamente a temperatura ambiente y se agitó durante 1 h. Después de inactivar cuidadosamente la reacción añadiendo lentamente 50 ml de agua, el pH se ajustó a ~3-4 usando HCl acuoso (6 M). La mezcla resultante se extrajo con 2x50 ml de acetato de etilo y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 1,6 g (98%) del compuesto del título como un sólido de color amarillo.

Compuesto 39.2. 4-(1-(3-Formil-4-metilbenzoil)piperidin-4-il)benzonitrilo. Una mezcla de ácido 3-formil-4-metilbenzoico (compuesto 39.1, 660 mg, 4,02 mmol, 1,00 equiv), HBTU (2 g, 5,28 mmol, 1,30 equiv) en N,N-dimetilformamida (20 ml) se agitó a temperatura ambiente. Después de 1 h, se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (1.5, 890 mg, 4,01 mmol, 1,00 equiv) y DIEA (1,03 g, 7,98 mmol, 2,00 equiv). La mezcla resultante se agitó durante 5 h a temperatura ambiente y a continuación durante una noche a 60 °C. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con 100 ml de EtOAc y se lavó con 2x50 ml de NH4Cl (ac., sat.) seguido de 2x50 ml de bicarbonato sódico (ac., sat.). La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 1 75 % del compuesto del título como un aceite de color marrón.

Compuesto 39.3. Ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilbenzoico. A una mezcla agitada de 4-(1-(3-formil-4-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 39.2, 600 mg, 1,81 mmol, 1,00 equiv) en THF (5 ml) se
le añadió gota a gota una solución de KMnO4 (1 g) en agua (10 ml). La mezcla resultante se agitó durante una noche a 60 °C en un baño de aceite. Después de enfriar a temperatura ambiente, los sólidos se retiraron por filtración y el pH del filtrado se ajustó a > 10 con hidróxido sódico (acuoso, 1 M). La mezcla resultante se lavó con 20 ml de acetato de etilo. A continuación se empleó HCl 1 M para ajustar el pH de la capa acuosa a ~4. La fase acuosa resultante se extrajo con 2x100 ml de acetato de etilo y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 500 mg (80 %) del compuesto del título como un aceite de color amarillo claro.
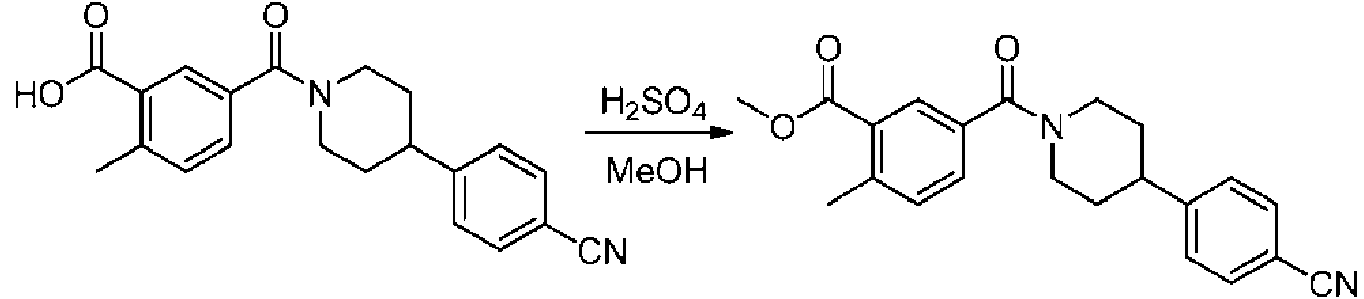
Compuesto 39.4. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilbenzoato de metilo. A una mezcla agitada de ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilbenzoico (compuesto 39.3, 1,392 g, 4,00 mmol, 1,00 equiv) en metanol (50 ml) se le añadió ácido sulfúrico (784 mg, 7,99 mmol, 2,00 equiv), gota a gota. La mezcla resultante se calentó a reflujo durante una noche en un baño de aceite. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El residuo se diluyó con 20 ml de EtOAc y se lavó con 1x100 ml de bicarbonato sódico saturado (ac) seguido de 1x100 ml de solución saturada de cloruro sódico. La capa orgánica se secó (Na2SO4) y se concentró a presión reducida para dar 1,303 g (90 %) del compuesto del título como un sólido de color blanco.

Compuesto 39.5. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilbenzohidrazida. Una solución del compuesto 39.4 (1,503 g, 4,15 mmol, 1,00 equiv) e hidrato de hidrazina (10 ml) en etanol (50 ml) se calentó a reflujo en un baño de aceite. Después de 2 h, la mezcla se concentró a presión reducida y el residuo se disolvió en 20 ml de EtOAc. La mezcla resultante se lavó con 1x50 ml de H2O y 1x50 ml de solución saturada de cloruro sódico. La capa orgánica se secó (Na2SO4) y se concentró a presión reducida para dar 1,353 g (90 %) del compuesto del título como un sólido de color blanco

Compuesto 39.6. 4-(1-(3-(5-Amino-1,3,4-oxadiazol-2-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. Una mezcla del compuesto 39.5 (380 mg, 1,05 mmol, 1,00 equiv) y bicarbonato sódico (105,8 mg, 1,26 mmol, 1,20 equiv) en dioxano/H2O (1:1) (50 ml) se agitó a temperatura ambiente. Después de 5 minutos, se añadió bromuro de cianógeno (212 mg, 2,00 mmol, 2,00 equiv). La mezcla resultante se agitó durante 3 h a temperatura ambiente. La solución resultante se agitó durante 3 h a temperatura ambiente, a continuación se inactivó con 30 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 397 mg (98 %) de un sólido de color blanquecino.

Compuesto 39. 4-(1-(3-(5-Etoxi-4H-1,2,4-triazol-3-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. Una mezcla del compuesto 39.6 (397 mg, 1,02 mmol, 1,00 equiv) e hidróxido potásico (287 mg, 5,12 mmol, 5,00 equiv) en etanol (25 ml) se calentó a reflujo durante una noche. Después de enfriar la mezcla de reacción a temperatura ambiente con un baño de agua, el pH se ajustó a ~7 con ácido acético. La mezcla resultante se concentró a presión reducida. El residuo se recogió en acetato de etilo y se lavó con agua. La capa orgánica se secó (Na2SO4) y se concentró. El residuo impuro se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5-1:0). El
producto se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): Columna, SunFire Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,05% de TFAy CH3CN (se mantiene un 5,0% de CH3CN en 2 minutos, asciende al 30,0% en 1 minuto, asciende al 59,0% en 12 minutos, asciende al 100,0% en 2 minutos); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 51,9 mg (12%) del compuesto del título como un sólido de color blanco. m/z (ES+) 416 (M+H)+.

Compuesto 40.1. 4-(4-cianofenil)-4-hidroxiazepan-1-carboxilato de terc-butilo. Se añadió una solución de 4-yodobenzonitrilo (510 mg, 2,22 mmol, 1,1 equiv) en THF (3 ml) a un matraz de fondo redondo de 10 ml y el sistema se purgó con nitrógeno. La mezcla se enfrió a -40 °C y a continuación se añadió cloruro de isopropilmagnesio (1,16 ml de una solución 2,0 M en THF, 2,32 mmol, 1,15 equiv) gota a gota durante 20 minutos. La mezcla resultante se agitó a -40 °C durante 2 horas y a continuación se añadió 4-oxoazepan-1-carboxilato de terc-butilo (431 mg, 2,02 mmol, 1,0 equiv) en THF (0,5 ml) gota a gota durante 15 minutos. La mezcla resultante se agitó a -40 °C durante 1 hora y a continuación se diluyó en acetato de etilo (15 ml) y se lavó con HCl 0,2 M (10 ml), HCl 0,1 M (5 ml), bicarbonato sódico saturado (5 ml) y a continuación solución saturada de cloruro sódico (5 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se evaporaron. Para retirar el subproducto de cetona y aldol restante, el residuo se disolvió en acetonitrilo (3 ml), a continuación se añadió hidrazina (0,2 ml) y se calentó a 60 °C durante 4 horas. La mezcla se diluyó con EtOAc (20 ml) y se lavó con agua (20 ml), seguida de ácido fosfórico 0,5 M (3 x 20 ml), bicarbonato sódico saturado (10 ml) y solución saturada de cloruro sódico (10 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se evaporaron. El residuo se purificó por cromatografía en columna de sílice (hexanos/acetato de etilo) para obtener el compuesto del título como un aceite de color rosa que se solidificó tras reposar (303 mg, 47 %). m/z (ES+) 317

Compuesto 40.2. 4-(4-Cianofenil)-2,3,6,7-tetrahidro-1H-azepin-1-carboxilato de terc-butilo y 5-(4-cianofenil)-2,3,4,7-tetrahidro-1H-azepin-1-carboxilato de terc-butilo. En un vial que contenía 4-(4-cianofenil) -4-hidroxiazepan-1-carboxilato de terc-butilo (compuesto 40.1, 200 mg, 0,63 mmol, 1,0 equiv) se añadió piridina (3 ml) y oxicloruro de fósforo (438 pl, 4,7 mmol, 7,5 equiv) y la mezcla se agitó a temperatura ambiente durante 45 horas. Los disolventes se retiraron al vacío y el residuo se disolvió en DCM (5 ml) y se lavó con agua (10 ml). La capa acuosa se extrajo con DCM (5 ml) y las fases orgánicas combinadas se lavaron con bicarbonato de sodio saturado (2 x 10 ml), se secaron (Na2SO4), se filtraron y se concentraron para obtener una mezcla de los compuestos del título como un aceite de color amarillo pálido (167 mg, 89 %). m/z (ES+) 299 (M+H)+.

Compuesto 40.3. 4-(4-cianofenil)azepan-1-carboxilato de terc-butilo. A un matraz de fondo redondo de 25 ml que contenía una mezcla de 4-(4-cianofenil)-2,3,6,7-tetrahidro-1H-azepin-1-carboxilato de terc-butilo y 5-(4-cianofenil)-2,3,4,7-tetrahidro-1H-azepin-1-carboxilato de terc-butilo (compuesto 40.2, 167 mg, 0,56 mmol) se le añadió paladio sobre carbono al 10 % (40 mg) y acetato de etilo (6 ml). El sistema se purgó con nitrógeno y a continuación se cargó con hidrógeno y se agitó a temperatura ambiente. Después de purgar el sistema con nitrógeno, la mezcla se filtró a través de celite y el filtrado se concentró. El residuo se purificó mediante TLC preparativa para obtener una cera incolora (77 mg, 45 %). m/z ES+ 245 M menos t-butilo+H .

Compuesto 40.4. 4-(Azepan-4-il)benzonitrilo. A un vial que contenía 4-(4-cianofenil)azepan-1-carboxilato de tercbutilo (compuesto 40.3, 77 mg, 0,257 mmol, 1,0 equiv) se le añadió DCM (500 pl) y ácido trifluoroacético (198 pl, 2,57 mmol, 10 equiv) y la mezcla se agitó con ventilación durante 90 minutos. La mezcla se diluyó en acetato de etilo (5 ml) y se lavó con bicarbonato sódico saturado (5 ml). La fase acuosa se ajustó a pH 10-11 y se extrajo con adición de acetato de etilo hasta que no quedó producto. Las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron para obtener el compuesto del título como un aceite de color amarillo (51 mg, teórico). m/z (ES+) 201 (M+H)+.

Compuesto 40.5. 4-(1-(3-Amino-4-metilbenzoil)azepan-4-il)benzonitrilo. A un vial de 4 ml se le añadió ácido 3-amino-4-metilbenzoico (27 mg, 0,18 mmol, 1,0equiv), 4 -(azepan-4-il)benzonitrilo (compuesto 40.4, 40 mg, 0,2 mmol, 1,1 equiv) , hidroxibenzotriazol (39 mg de h 2o al 20% en peso, 0,23 mmol, 1,3 equiv), 1-etil-3-(3-dimetilaminopropil)carbodiimida (44 mg, 0,23 mmol, 1,3 equiv), DMF (1 ml) y N,N-diisopropiletilamina (93 pl, 0,54 mmol, 3,0 equiv). La mezcla se agitó a temperatura ambiente durante 4 horas y a continuación se diluyó en acetato de etilo (10 ml) y se lavó con solución saturada de cloruro sódico (10 ml). La fase acuosa se extrajo con acetato de etilo (3 ml) y las fases orgánicas combinadas se lavaron con solución saturada de cloruro sódico (10 ml), NaH2PO41 M (5 ml) y solución saturada de cloruro sódico (10 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se concentraron para obtener el compuesto del título como un sólido de color marrón (45 mg, 74%). m/z (ES+) 334 (M+H)+.

Compuesto 40.6. N-(5-(4-(4-Cianofenil)azepan-1-carbonil)-2-metilfenil)-6-fluoronicotinamida. A un vial de 4 ml que contenía 4-(1-(3-amino-4-metilbenzoil)azepan-4-il)benzonitrilo (compuesto 40.5, 44 mg, 0,13 mmol, 1,0 equiv) se le añadió diclorometano (1 ml) y N,N-diisopropiletilamina (35 pl, 0,20 mmol, 1,5 equiv). Se añadió una solución de cloruro de 6-fluoronicotinoílo (22 mg, 0,14 mmol, 1,05 equiv) en diclorometano (1 ml) gota a gota durante aproximadamente 2 minutos. La mezcla resultante se agitó a temperatura ambiente durante 19 horas y a continuación se diluyó con diclorometano (5 ml) y se lavó con NaH2PO41 M (3 ml), bicarbonato sódico saturado (3 ml) y solución saturada de cloruro sódico (3 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se concentraron para obtener el compuesto del título como una cera de color marrón (64 mg, teórico), m/z (ES+) 457 (M+H)+.

Compuesto 40. N-(5-(4-(4-Cianofenil)azepan-1-carbonil)-2-metilfenil)-6-(isopropilamino)nicotinamida. A un vial que contenía N-(5-(4-(4-cianofenil)azepan-1-carbonil)-2-metilfenil)-6-fluoronicotinamida (compuesto 40.6, 61 mg, 0,13 mmol) se le añadió dimetilsulfóxido (1 ml) e isopropilamina (1 ml). La mezcla se agitó a 35 °C durante 2 horas y a continuación a temperatura ambiente durante 16 horas seguido de 35 °C durante 4 horas. El exceso de isopropilamina se retiró al vacío y la solución restante se purificó mediante HPLC preparativa para dar el producto como un polvo de

Compuesto 41.1. 5-Nitro-2-(pirrolidin-1-il)piridina. Una mezcla de 2-cloro-5-nitropiridina (1,58 g, 10,00 mmol, 1,00 equiv), pirrolidina (710 mg, 10,00 mmol, 1,00 equiv) y carbonato potásico (2,76 g, 20,00 mmol, 2,00 equiv) en CH3CN ( 20 ml) se agitó a 60 °C en un baño de aceite. Después de 2 h, se dejó que la mezcla de reacción alcanzara la temperatura ambiente. Los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida. El residuo se trituró con 1x20 ml de éter de petróleo para dar 1,8 93 % del compuesto del título como un sólido de color amarillo.
Compuesto 41.2. 6-(Pirrolidin-1-il)piridin-3-amina. Un matraz de fondo redondo, que contenía una solución de 5-nitro-2-(pirrolidin-1-il)piridina (compuesto 40.1, 1,93 g, 10,00 mmol, 1,00 equiv)) en etanol (30 ml) se purgó con nitrógeno. Se añadió paladio sobre carbono (0,4 g, 10 %, 60 % de agua). Después de purgar adicionalmente el sistema con nitrógeno, la atmósfera se cambió a hidrógeno y la mezcla resultante se agitó durante una noche a temperatura ambiente. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró

Compuesto 41. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metil-N-(6-(pirrolidin-1-il)piridin-3-il)benzamida. Una mezcla del compuesto 39.3 (528 mg, 1,52 mmol, 1,00 equiv) y HBTU (635 mg, 2,28 mmol, 1,50 equiv) en DMF (30 ml) se agitó durante 30 minutos a temperatura ambiente. A esto se le añadió 6-(pirrolidin-1-il)piridin-3-amina (compuesto 41.2, 299 mg, 1,82 mmol, 1,20 equiv) y DIEA (50 mg). La mezcla resultante se agitó durante una noche a 60 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con 50 ml de EtOAc y se lavó con 2x30 ml de NH4Cl (ac., sat.) seguido de 2x30 ml de bicarbonato sódico (ac., sat.). La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El producto impuro se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-016(Waters)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con 50 mmol de NH4HCO3 y CH3CN (10% de CH3CN asciende al 32% en 3 minutos, asciende al 51 % en 20 minutos, asciende al 100 % en 1 minuto, desciende al 10 % en 1 minuto); Detector UV, 220 NMnm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 50 mg (7 %) del compuesto del título como un sólido, miz ES+ 494 M+H .

Compuesto 42.1. Ácido 5-amino-2,4-dimetilbenzoico. Una mezcla de 5-amino-2,4-d¡metilbenzoato de metilo (1,2 g, 6,70 mmol, 1,00 equiv) e hidróxido sódico (1,5 g, 37,50 mmol, 5,60 equiv) en metanol / H2O (20/20 ml) se agitó durante una noche a 50 °C. Después de enfriar a temperatura ambiente, la fase orgánica se retiró a presión reducida. El pH de la capa acuosa restante se ajustó a ~4-5 con cloruro de hidrógeno (ac., 2 M). Los sólidos resultantes se

Compuesto 42.2. 4-(1-(5-Amino-2,4-dimetilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. Una mezcla de ácido 5-amino-2,4-dimetilbenzoico (compuesto 42.1, 2,264 g, 13,71 mmol, 1,00 equiv), HBTU (7,803 g, 20,59 mmol, 1,50 equiv), 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2, 2,8 g, 13,71 mmol, 1,00 equiv), y DIEA (3,541 g, 27,45 mmol, 2,00 equiv) en DMF (50 ml) se agitó a temperatura ambiente. Después de 1 h, la mezcla se diluyó con 100 ml de EtOAc y se lavó con 1x100 ml de agua, seguida de 1x100 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 3,51 g (73 %) del compuesto del título como un sólido de color blanco.

Compuesto 42.3. 6-Cloro-N-(5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)nicotinamida. A una mezcla del compuesto 42.2 (3,51 g, 9,99 mmol, 1,00 equiv) y trietilamina (2,02 g, 19,96 mmol, 2,00 equiv) en DCM (50 ml) en nitrógeno a 0 °C se le añadió gota a gota una solución de cloruro de 6-cloropiridin-3-carbonilo (1,936 g, 11,00 mmol, 1,10 equiv) en DCM (50 ml). La mezcla resultante se agitó durante 1 h a 0 °C. La mezcla de reacción se
inactivó mediante la adición cuidadosa de 50 ml de agua y se extrajo con 3x50 ml de diclorometano. Las capas orgánicas combinadas se lavaron con 2x50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 4,41 g (90 %) del compuesto del título como un sólido de color amarillo.

Compuesto 42. N-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2,4-dimetMfenM)-6-(isopropilamino)nicotinamida. Una mezcla del compuesto 42.3 (392 mg, 0,80 mmol, 1,00 equiv), propan-2-amina (472 mg, 7,99 mmol, 10,00 equiv), carbonato potásico (221 mg, 1,60 mmol, 2,01 equiv), KI (66,4 mg, 0,40 mmol, 0,50 equiv) en DMSO (10 ml) se calentó a 100 °C en un tubo sellado detrás de una pantalla protectora contra explosiones. Después de 48, la mezcla de reacción se dejó alcanzar la temperatura ambiente y a continuación se diluyó con 20 ml de EtOAc. La mezcla se lavó con 1x20 ml de agua, seguida de 1x20 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El producto impuro (~300 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-002(Agilent)): columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (15,0 % de CH3CN, asciende al 40,0 % en 6 minutos, asciende al 100,0 % en 2 minutos, desciende al 15,0 % en 2 minutos); Detector, UV 220 y 254 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 158 mg (39 %) del compuesto del título como un sólido de color blanquecino. m/z (ES+) 514 (M+H)+. 1H RMN (300 MHz, CD3OD, ppm): 88,49 (s, 1H), 8,32 (d, J = 9,9Hz, 1H), 7,79 (d, J = 8,1Hz, 2H), 7,67 (d, J = 7,5Hz, 2H), 7,40-7,22 (m, 2H), 7,07 (d, J = 9,6Hz, 1H), 4,85 (m, 1H), 4,03 (m, 1H), 3,80-3,42 (m, 2H), 3,29 (m, 1H),

Compuesto 43. N-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)-6-morfolinonicotinamida.
Una mezcla del compuesto 42.3 (392 mg, 0,80 mmol, 1,00 equiv), morfolino (348 mg, 3,99 mmol, 5,00 equiv) y carbonato potásico (221 mg, 1,60 mmol, 2,01 equiv), en DMSO (10 ml) se calentó a 100 °C en un tubo sellado detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con 20 ml de EtOAc y se lavó con 1x20 ml de agua, seguida de 1x20 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El producto impuro (300 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-002(Agilent)): Columna, 1 # -PrepC-005 (XBridge C18 19 * 15018600297917013004711303), n; fase móvil, agua con el 0,05 % de TFA y CH3CN (15,0 % de CH3CN asciende al 50,0 % en 10 minutos, asciende al 100,0 % en 1 minuto, se mantiene el 100,0 % 1 minuto, desciende al 15,0 % en 2 minutos); Detector, UV 220 y 254 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 106 mg (25 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 542 M+H .
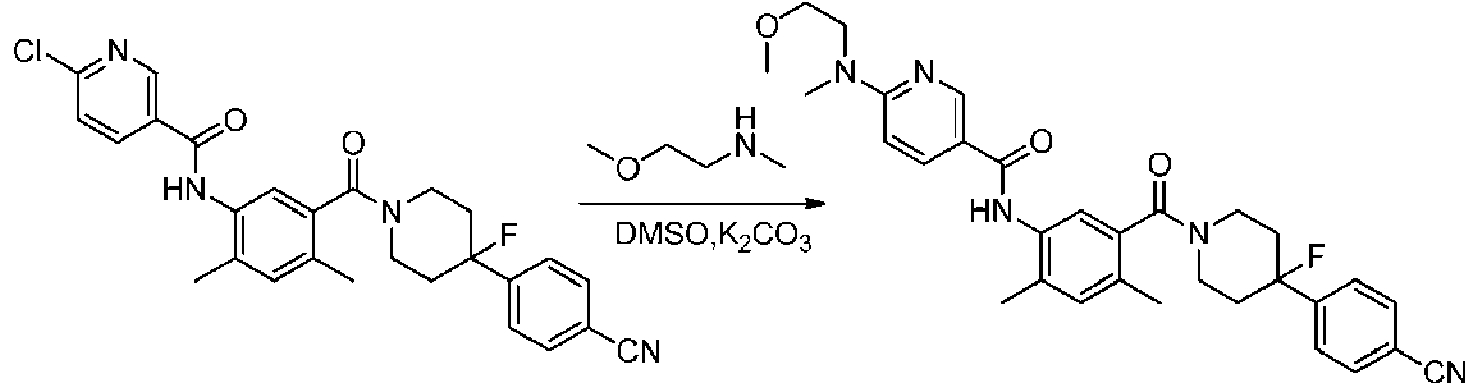
Compuesto 44. N-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2,4-dimetMfenM)-6-((2-metoxietil)(metil)amino)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 43 para dar un sólido de color blanco (219 mg, 50 %). m/z (ES+) 544 (M+H)+. 1H RMN (300 MHz, CD3OD): 88,56 (s con estructura fina, 1H), 8,41 (d con estructura fina, J = 9,9 Hz, 1H), 7,79 (d ancho, J = 8,1 Hz, 2H), 7,67 (d ancho, J = 6,9 Hz, 2H), 7,44-7,19 (m, 3H), 4,88-4,73 (m, 1H), 3,94 (t, J =5,0 Hz, 2H), 3,73 (t, J =5,0 Hz,
2H), 3,70-3,47 (m, 2H), 3,39 (s, 3H), 3,36 (s, 3H), 3,36-3,21 (m, 1H), 2,46-2,22 (m, 6H), 2,22-1,85 (m, 4H).

Compuesto 45. N-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)-6-(4-metilpiperazin-1-il)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 43 para dar un

Compuesto 46. 6-(Azetidm-1-M)-N-(5-(4-(4-cianofenM)-4-fluoropipendm-1-carbonM)-2,4-dimetilfenil)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 43

Compuesto 47.1. 6-(1-(5-Amino-4-etil-2-metilbenzoil)-4-fluoropiperidin-4-il)nicotinonitrilo. El compuesto del título se sintetizó de manera similar a la descrita para 42.2 y usando el compuesto 26.4 en lugar del compuesto 1.5

Compuesto 47.2. 6-Cloro-N-(5-(4-(5-cianopmdm-2-M)-4-fluoropiperidm-1-carbonM)-2-etN-4-metilfenil)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 42.3

Compuesto 47. 6-(Azetidm-1-M)-N-(5-(4-(5-cianopmdm-2-M)-4-fluoropiperidm-1-carbonM)-2-etN-4-metilfenil)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 43 para dar un sólido de color blanco (187 mg, 54%). m/z (ES+) 527 (M+H)+. 1H RMN (300 MHz, DMSO-da): 69,88 (s, 1H), 9,06 (s, 1H), 8,59 (d, J =1,8 Hz, 1H), 8,43 (dd, J = 8,4 Hz, J = 2,1 Hz, 1H), 8,23 (d, J = 9,3 Hz, 1H), 7,81 (d, J = 8,1 Hz, 1H), 7,22 (s, 1H), 7,17 (sancho, 1H), 6,74 (d, J = 9,0 Hz, 1H), 4,68-4,53 (m, 1H), 4,23 (t, J = 7,7 Hz, 4H), 3,54-3,32 (m, 2H), 3,22-3,05 (m, 1H, 2,59 c, J = 7,5 Hz, 2H , 2,50-2,38 m, 2H , 2,35-1,80 (m, 7H), 1,14 (t, 3H).

Compuesto 48.1. 2-Etil-4-metilbenzoato de metilo. A una mezcla agitada de ZnBr2 (13 g, 57,72 mmol, 2,00 equiv) en THF (230 ml) en nitrógeno a 0 ° C s e le añadió gota a gota EtMgBr (19,5 ml, 3 M en THF). Después de 30 minutos a 0 °C, la temperatura se hizo descender a -78 °C y se añadió Pd(dppf)Ch (2 g, 2,73 mmol, 0,09 equiv), seguido de la adición gota a gota de una solución de 2-bromo-4 -metilbenzoato de metilo (6,6 g, 28,81 mmol, 1,00 equiv) en tetrahidrofurano (200 ml). Se dejó que la mezcla resultante alcanzara lentamente la temperatura ambiente y se agitó en nitrógeno durante una noche. La mezcla de reacción se inactivó mediante la adición cuidadosa de 20 ml de NH4Cl (ac., sat.) y se extrajo con 3x100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x200 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:30) como eluyente para dar 3,7 g (72 %) del compuesto del título como un aceite incoloro.
O O
i f V V N3° H , f V 'O H
Compuesto 48.2 Á.cido 2-etil-4-m Aetilbe Anzoic /o. Una M me0eHzc,Hla2° del co Ampues Ato 48.1 (3,7 g, 20,76 mmol, 1,00 equiv) e hidróxido sódico (4 g, 100,01 mmol, 4,82 equiv) en metanol/H2O (30/20 ml) se agitó durante una noche a 50 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El pH de la capa acuosa residual se ajustó a 3-4 con cloruro de hidrógeno (ac., 1 M). El precipitado resultante se recogió por filtración y se secó para dar 3,0 g (83 %) del compuesto del título como un sólido de color blanco

Compuesto 48.3. Acido 2-etil-4-metil-5-nitrobenzoico. A una mezcla agitada de ácido 2-etil-4-metilbenzoico (compuesto 48.2, 2 g, 12,18 mmol, 1,00 equiv) en ácido sulfúrico (30 ml) a -10 °C se le añadió gota a gota una solución de ácido nítrico (1,6 g, 16,50 mmol, 2,08 equiv) en ácido sulfúrico (10 ml). Después de agitar durante 30 minutos a -10 °C, se añadieron cuidadosamente 200 ml de H2O/hielo y la mezcla resultante se extrajo con 50 ml de acetato de etilo. La fase orgánica se lavó con 2x50 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico, y se concentró a presión reducida. El producto impuro se purificó mediante recristalización a partir de acetato de etilo/éter de petróleo en la proporción de 1:10 para dar 1,0 g (37 %) de ácido 2-etil-4-metil-5-nitrobenzoico como un sólido de color amarillo claro.

Compuesto 48.4. Ácido 5-amino-2-etil-4-metilbenzoico. El compuesto del título se sintetizó a partir de 48.3 (1 g, 4,78 mmol) usando un procedimiento similar al descrito para la preparación de 1.9 para dar el compuesto del título

Compuesto 48.5. 4-(1-(5-Amino-2-etil-4-metilbenzoil)4-fluoropiperidin-4-il)benzonitrilo. Una mezcla de ácido 5-amino-2-etil-4-metilbenzoico (compuesto 48.4, 100 mg, 0,56 mmol, 1,00 equiv), 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2,140 mg, 0,69 mmol, 1,23 equiv), HBTU (320 mg, 0,84 mmol, 1,51 equiv)y DIEA(150 mg, 1,16 mmol, 2,08 equiv) en DMF (20 ml) se agitó durante 48 h a temperatura ambiente. Se añadió agua (30 ml) y la mezcla resultante se extrajo con 3x20 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x50 ml de solución saturada de cloruro sódico, se secaron sobre Na2SO4, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con PE:EtOAc (1:1) para dar 130 mg (38 %) del compuesto del título como un sólido de color amarillo claro
Compuesto 48.6. 6-Cloro-N-(5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)4-etil-2-metilfenil)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 42.3 y usando el compuesto 48.5 en lugar del compuesto 42.2 para dar un sólido de color amarillo claro (200 mg, 87 %).

Compuesto 48. N-(5-(4-(4-Cianofeml)-4-fluoropiperidm-1-carbonM)-4-etN-2-metNfenM)-6-(isopropilamino)nicotinamida. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 43 y usando el compuesto 48.6 en lugar del compuesto 42.3 para dar un sólido de color blanquecino (32 mg, 30%). m lz {ES+) 528 (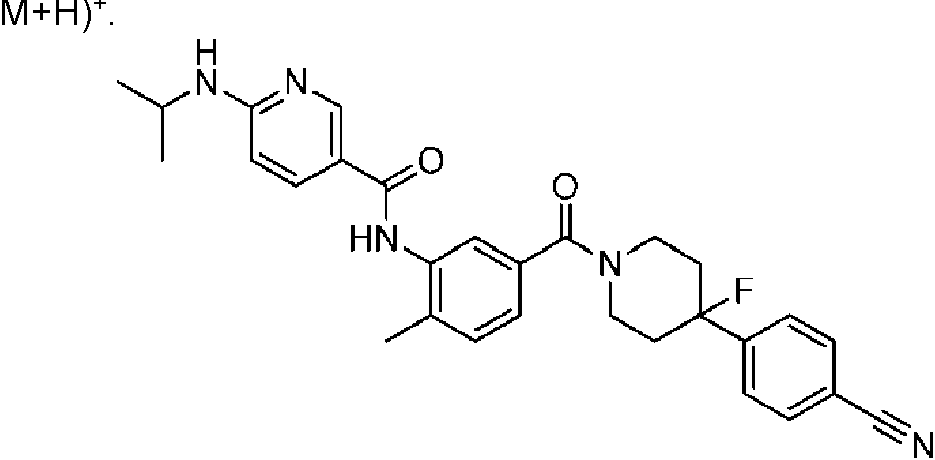
Compuesto 49. N-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2-metNfenM)-6-(isopropilamino)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y

Compuesto 50.1. 5-Ciano-5',6'-dihidro-[2,4'-bipiridin]-1'(2'H)-carboxilato de terc-butilo. A una mezcla agitada de 4-(5-cianopiridin-2-il)-4-hidroxipiperidin-1 -carboxilato de terc-butilo (compuesto 26.2, 500 mg, 1,65 mmol, 1,00 equiv) en piridina (20 ml) se le añadió gota a gota tricloruro de fosforilo (2,5 g, 16,34 mmol, 9,90 equiv) a 10-15 °C. La solución resultante se agitó durante 18 h a 15-20 °C. La reacción se inactivo a continuación cuidadosamente mediante la adición de 20 ml de agua y se extrajo con 3x100 ml de acetato de etilo. Las fases orgánicas combinadas se lavaron con 2x50 ml de HCl acuoso (1 M), seguido de 1x100 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100 a 1:7) como eluyente para dar 0,3 g (64%) del

Compuesto 50.2. 4-(5-cianopiridin-2-il)piperidin-1-carboxilato de terc-butilo. Un matraz de fondo redondo, que contenía una solución del compuesto 50.1 (300 mg, 1,05 mmol, 1,00 equiv) en acetato de etilo (20 ml) se purgó con nitrógeno gaseoso. A la solución se añadió paladio sobre carbono (40 mg, 10 %) y el matraz se purgó adicionalmente con nitrógeno gaseoso. La atmósfera se cambió a continuación a hidrógeno y la mezcla se agitó durante 16 h a 15-20 °C. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida para dar 0,2 66 % del compuesto del título como un aceite de color amarillo.

Compuesto 50.3. Clorhidrato de 6-(piperidin-4-il)nicotinonitrilo. A través de una solución de 4-(5-cianopiridin-2-il)piperidin-1-carboxilato de terc-butilo (compuesto 50.2, 200 mg, 0,70 mmol, 1,00 equiv) en acetato de etilo (20 ml) se burbujeó HCl gaseoso. La mezcla resultante se agitó durante 40 minutos a 5-10 °C. El precipitado resultante se recogió por filtración y se secó para dar 150 mg (97 %) del compuesto del título como un sólido de color blanco.

Compuesto 50. N-(5-(4-(5-Cianopiridin-2-il)piperidin-1-carbonil)-2-metilfenil)-6-(isopropilamino)nicotinamida.
El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para el compuesto 43 pero usando el compuesto 50.3 en lu ar del compuesto 11.2. m/z (ES+) 483 (M+H)+.
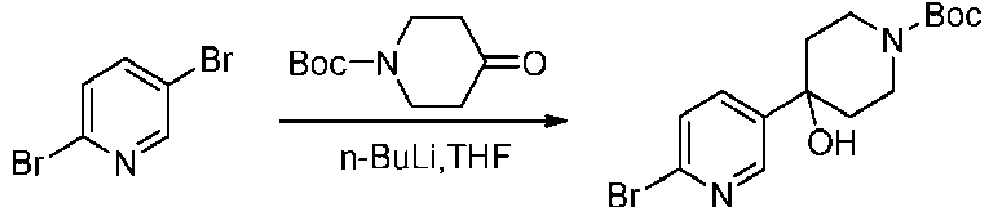
Compuesto 51.1. 4-(6-bromopiridin-3- il)-4-hidroxipiperidin-1-carboxilato de terc-butilo. A una solución de 2,5-dibromopiridina (10 g, 42,55 mmol, 1,00 equiv) en tetrahidrofurano (400 ml) en nitrógeno a -78 °C se le añadió gota a gota n-BuLi (19 ml, 2,4 M en THF). Después de 1 h a -78 °C, se añadió gota a gota una solución de 4-oxopiperidin-1-carboxilato de terc-butilo (9,5 g, 47,74 mmol, 1,12 equiv) en tetrahidrofurano (100 ml). La mezcla resultante se agitó durante una hora adicional a -78 °C. La reacción se calentó a continuación a -30 °C y se inactivó cuidadosamente mediante la adición de 300 ml de agua. La mezcla resultante se extrajo con 3x200 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 1x200 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:3) como eluyente para dar 5 g (33 %) del compuesto del título como un sólido de color amarillo

Compuesto 51.2. 4-(6-cianopiridin-3-il)-4-hidroxipiperidin-1-carboxilato de terc-butilo. A una mezcla de 4-(6-bromopiridin-3-il)-4-hidroxipiperidin-1 -carboxilato de terc-butilo (compuesto 51.1, 1 g, 2,81 mmol, 1,00 equiv) en DMF (50 ml) en nitrógeno a 50 °C se le añadió Zn(CN)2 (400 mg, 3,42 mmol, 1,22 equiv) a 50 °C seguido de Pd(PPh3)4 (200 mg, 0,17 mmol, 0,06 equiv) a 80 °C. A continuación, la mezcla resultante se agitó durante 1 h a 120 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó mediante la adición de 200 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x200 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5-1:3) como eluyente para dar 0,6 g (70 %) del compuesto del título como un aceite de color amarillo claro.
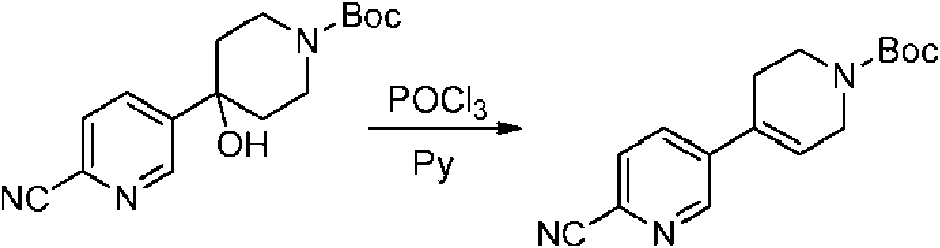
Compuesto 51.3. 6-Ciano-5',6'-dihidro-[3,4'-bipiridin]-1'(2'H)-carboxilato de terc-butilo. A una solución de 4-(6-cianopiridin-3-il)-4-hidroxipiperidin-1 -carboxilato de terc-butilo (compuesto 51.2, 600 mg, 1,98 mmol, 1,00 equiv) en piridina (15 ml) en nitrógeno a 10-15 °C se le añadió cuidadosamente POCh (3 g, 19,74 mmol, 9,97 equiv). Después de agitar durante una noche en nitrógeno en un baño de agua/hielo, la mezcla se concentró y el residuo se disolvió en 50 ml de acetato de etilo. La fase orgánica se lavó con 1x50 ml de cloruro de hidrógeno (ac. 1 M) seguido de 1x50 ml de bicarbonato sódico (ac. sat.). La capa orgánica se secó a continuación sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10) como eluyente para dar 0,26 g (46 %) del compuesto del título como un sólido de color blanco.
Compuesto 51.4. 4-(6-cianopiridin-3-il)piperidin-1-carboxilato de terc-butilo. Un matraz de fondo redondo, que contenía una solución del compuesto 51.3 (260 mg, 0,91 mmol, 1,00 equiv) en acetato de etilo (20 ml) se purgó con nitrógeno gaseoso. A la solución se le añadió a continuación paladio sobre carbono (0,1 g, 10%, 60% de agua) y el matraz se purgó adicionalmente con nitrógeno gaseoso. La atmósfera se cambió a continuación a hidrógeno y la mezcla se agitó durante 16 h a 15-20 °C. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida. El residuo impuro se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5). Esto dio como resultado 0,18 g (69%) del compuesto del título como un aceite incoloro.
Compuesto 51.5. Clorhidrato de 5-(piperidin-4-il)picolinonitrilo. A una solución enfriada (5-10 °C) de 4-(6-cianopiridin-3-il)piperidin-1 -carboxilato de terc-butilo (51.4, 180 mg, 0,63 mmol, 1,00 equiv) en acetato de etilo (30 ml) se burbujeó cloruro de hidrógeno gaseoso. La mezcla se agitó durante 30 minutos a 5-10 °C y los sólidos resultantes se recogieron por filtración y se secaron para dar 0,11 g (78 %) de clorhidrato de 5-(piperidin-4-il)picolinonitrilo como un sólido de color blanco.

Compuesto 51. N-(5-(4-(6-Cianopiridin-3-il)piperidin-1-carbonil-2-metilfenil)-6-(isopropilamino)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para

Compuesto 52. N-(5-(4-(4-Cianofenil)piperidin-1-carbonil)piridazin-3-il)-6-(isopropilamino)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 43. m/z (ES+) 470 (M+H)+.

Compuesto 53. 2-(Azetidin-1-il)-N-(5-(4-(5-cianopiridin-2-il)piperidin-1-carbonil)-2-metilfenil)pirimidin-5-carboxamida. El co m p u e s to del títu lo se s in te tizó usando re a c tivo s d isp o n ib le s fá c ilm e n te y p ro ce d im ie n to s s im ila re s a los usa do s para la p re pa rac ió n de l co m p u e s to 50. m /z (E S ) 482 (M+H)+.

Compuesto 54. 2-(Azetidin-1-il)-N-(5-(4-(5-cianopiridin-2-il)-4-fluoropiperidin-1-carbonil)-2-metilfenil)pirimidin-5-carboxamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos

Compuesto 55. 6-(Azetidin-1-il)-N-(5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-etilfenil)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 43. m/z ES+ 512 M+H .

Compuesto 56. 6-(Azetidm-1-M)-N-(5-(4-fluoro-4-(5-metoxipmdm-2-N)piperidm-1-carboml)-2-metilfenil)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y
procedimientos similares a los usados para la preparación del compuesto 43. m/z (ES+) 504 (M+H)+.

Compuesto 57. 6-(Azetidm-1-M)-N-(2-cloro-5-(4-(4-cianofeml)-4-fluoropiperidm-1-carbonM)feml)mcotmamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 43. m/z (ES+) 518 (M+H)+.

Compuesto 58. N-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-etil-4-metilfenil)-6-morfolinonicotinamida.
El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados
para la preparación del compuesto 43. m/z ES+ 556 M+H .

Compuesto 60. N-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2-etN-4-metNfenM)-6-((2-metoxietil)(metil)amino)nicotinamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente

Compuesto 61. N-(5-(4-(5-Cianopiridm-2-M)-4-fluoropiperidm-1-carboml)-2,4-dimetMfeml)-2-morfolmopirimidm-5-carboxamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 47. m/z (ES+) 544 (M+H)+.

Compuesto 62. N-(5-(4-(5-Cianopmdm-2-M)-4-fluoropiperidm-1-carboml)-2-etN-4-metNfeml)-2-morfolinopirimidin-5-carboxamida. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 47. m/z (ES+) 558 (M+H)+.

Compuesto 63. N-(5-(4-(5-Cianopmdm-2-M)-4-fluoropiperidm-1-carboml)-2-etN-4-metNfenM)-6-morfolinonicotinamida.
El co m p u e s to de l títu lo se s in te tizó u sa nd o rea c tivos d isp o n ib le s fá c ilm e n te y
p ro ce d im ie n to s s im ila re s a los u sa do s para la p re pa rac ió n del c o m p u e s to
47
. m /z (E S ) 557 (M+H)+.

Compuesto 64. (R)-1-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carboml)-2,4-dimetMfenM)-3-((tetrahidrofuran-2-il)metil)urea. Una mezcla de 4-(1-(5-amino-2,4-dimetilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo (compuesto 42.2, 150 mg, 0,43 mmol, 1,00 equiv), DIEA (560 mg, 4,33 m m ol, 10,15 equiv) y CO(OCCl3)2 (160 mg, 0,54 mmol, 1,26 equiv) en DCM (50 ml) se agitó en nitrógeno a temperatura ambiente. Después de 0,5 h, se añadió (R)-(tetrahidrofuran-2-il)metanamina (52 mg, 0,51 mmol, 1,20 equiv). Después de agitar durante 2 h a temperatura ambiente, la mezcla se lavó con 2x50 ml de agua y 1x50 ml de solución saturada de cloruro sódico. La fase orgánica se concentró a presión reducida y el residuo (-200 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05% de TFAy CH3CN (se mantiene el 5,0% de CH3CN en 2 minutos, asciende al 35,0% en 1 minuto, asciende al 65,0% en 12 minutos, asciende al 100,0% en 1 minuto); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 110 mg (54%) del compuesto del título como un sólido de color amarillo claro. m/z (ES+) 479 (M+H)+. 1H RMN (300 MHz, DMSO, ppm): 87,91 (d, J = 8,1 Hz, 2H), 7,77-7,69 (m, 4H), 7,04 (s, 1H), 6,68 (s, 1H), 4,64 (m, 1H), 3,89-3,82 (m, 2H), 3,71 (m, 1H), 3,33-3,23 (m, 3H), 3,11-3,09 (m, 2H), 2,18-2,03 (m, 8H),
Compuesto 65. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)-3-(tetrahidro-2H-piran-3-il)urea. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 64 para dar un sólido de color amarillo (111 mg, 53 %). m/z (ES+) 479 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,80 (d, J = 8,1 Hz, 2H), 7,78 7,50 (m, 3H), 7,13 (s, 1H), 4,88-4,72 (m, 1H), 3,94-8,82 (m, 1H), 3,82-3,68 (m, 2H), 3,68-3,33 (m, 3H), 3,33-3,19 (m, 2H), 2,42-2,08 m, 8H, 2,08-1,74 m, 4H, 1,74-1,54 m, 2H .

Compuesto 66. (R)-1-(5-(4-(4-Cianofeml)-4-fluoropiperidm-1-carbonM)-2,4-dimetNfeml)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 64 para dar un sólido de color amarillo claro (105 mg, 53%). m/z ES+ 465 M+H .

Compuesto 67. (R)-1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-4-etil-2-metilfenil)-3-((tetrahidrofuran-2-il)metil)urea. El compuesto del título se sintetizó de una manera similar a la descrita para el compuesto 64 pero usando el compuesto 48.5 en lugar del compuesto 42.2. m/z (ES+) 493 (M+H)+.

Compuesto 68. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-4-etil-2-metilfenil)-3-(tetrahidro-2H-piran-3-il)urea. El compuesto del título se sintetizó de una manera similar a la descrita para el compuesto 64 pero usando el compuesto 48.5 en lugar del compuesto 42.2. m/z ES+ 493 M+H .
Compuesto 69.1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-(tetrahidro-2H-piran-4-il)urea.
El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z ES+ 465 M+H .

Compuesto 70.1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-(tetrahidro-2H-piran-3-il)urea.
El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados

Compuesto 71. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-ciclopentilurea. El co m p u e s to de l títu lo se s in te tizó usando re a c tivo s d isp o n ib le s fá c ilm e n te y p ro ce d im ie n to s s im ila re s a los usa do s pa ra la p re p a ra c ió n de l co m p u e s to 64 . m /z (E S ) 449 (M+H)+.

Compuesto 72. (R)-1-(5-(4-(5-Cianopiridin-2-il)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z ES+ 452 M+H .
Compuesto 73. (R)-1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-(tetrahidrofuran-3-il)urea.
El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z ES+ 451 M+H .

Compuesto 74.1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metoxifenil)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z (ES+ 467 M+H .

Compuesto 75. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-fluorofenil)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para
Compuesto 76. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-etilfenil)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z (ES+ 465 M+H .

Compuesto 77. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-(trifluorometil)fenil)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z (ES+) 505 (M+H)+.
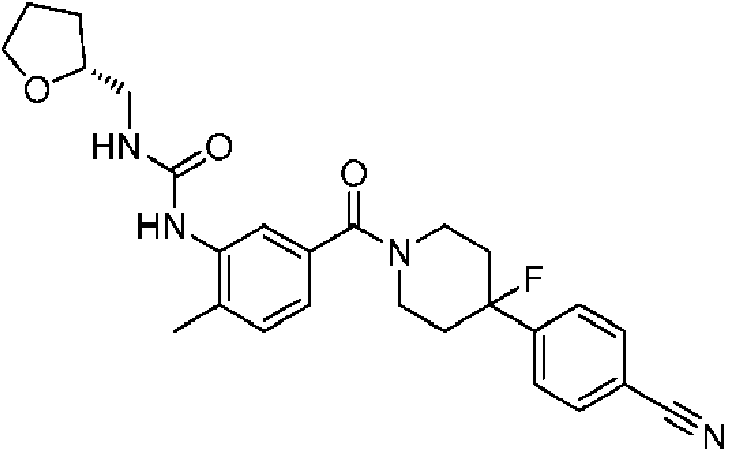
Compuesto 78. (R)-1-(5-(4-(4-Cianofeml)-4-fluoropiperidm-1-carboml)-2-metNfenM)-3-((tetrahidrofuran-2-il)metil)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares

Compuesto 79. 1-(5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)-3-(2-metoxietil)urea. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 64. m/z (ES+ 439 M+H .

Compuesto 80.1. 1H-Imidazol-1-carboxilato de (R)-tetrahidrofuran-3-ilo. Una solución de (R)-tetrahidrofuran-3-ol (500 mg, 5,68 mmol, 1,00 equiv) y CDI (2 g, 12,33 mmol, 2,17 equiv) en tetrahidrofurano (50 ml) se agitó durante una noche en nitrógeno a 60 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se disolvió en 30 ml de DCM y se lavó con 1x50 ml de H2O. La capa orgánica se secó (Na2SO4) y se concentró a presión reducida. El residuo impuro se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:1) para dar 0,95 g (92 %) del compuesto del título como un sólido de color blanco.

Compuesto 80. (5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)carbamato de (R)-tetrahidrofuran-3-ilo. Una mezcla del compuesto 42.2 (200 mg, 0,57 mmol, 1,00 equiv), 1H-imidazol-1-carboxilato de (R)-tetrahidrofuran-3-ilo (80.1, 124 mg, 0,68 mmol, 1,20 equiv) y DBU (2,6 mg , 0,02 mmol, 0,03 equiv) en DMF (50 ml) se agitó durante 16 h en nitrógeno, a 120 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó mediante la adición de 200 ml de agua. La mezcla resultante se extrajo con 3x100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1x 100 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo (200 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-002(Agilent)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (40,0 % de CH3CN asciende al 45,0 % en 8 minutos, se mantiene el 45,0 % en 2 minutos, asciende al 100,0 % en 1 minuto, desciende al 40,0 % en 2 minutos ); Detector UV, 220254 nm. Se obtuvieron 47,4 mg del producto. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 47,4

Compuesto 81. (5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2,4-dimetilfenil)carbamato de 1-acetilpirrolidin-3-ilo. El compuesto del título se sintetizó de manera similar a la descrita para el compuesto 80 para dar un sólido de color blanco (113 mg, 39 %). m/z (ES+) 507 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,80 (d, J = 8,1 Hz, 2H), 7,77-7,61 (m, 2H), 7,52 y 7,33 (2 singletes anchos, rotámeros de amida, Ar-H, 1H), 7,17 (s, 1H), 5,41-5,28 (m, 1H), 4,87-4,72 (m, 1H), 3,88-3,42 (m, 6H, 3,33-3,19 m, 1H, 2,42-1,82 m, 15H).

Compuesto 82. (5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-metilfenil)carbamato de (R)-tetrahidrofuran-3-ilo. El compuesto del título se sintetizó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 80. m/z (ES+) 452 (M+H)+.
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación de los compuestos 1 y 2:

(continuación)

(continuación)


Compuesto 105. 4-(1-(2,4-Dimetil-5-(6-(pirrolidin-1-il)-3H-imidazo[4,5-c]piridin-2-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 20 excepto que se usó sa1HCl del compuesto 11.2 en lugar

Compuesto 106. 4-(1-(5-(6-(Azetidm-1-il)-3H-imidazo[4,5-c]piridm-2-il)-4-fluoro-2-metilbenzoil)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 20 excepto que se usaron ácido 4-fluoro-2-metilbenzoico y azetidina en lugar de ácido 2,4-dimetilbenzoico y pirrolidina, respectivamente. m/z (ES+) 495 (M+H)+.
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida i ni l f ilm n r imi n imil r l r l r r i n l m 2 :

(continuación)

(continuación)

Compuesto 118.1. 2-(4-Metoxifenil)propano-1,1,3,3-tetracarboxilato de tetrametilo. Una mezcla de (E)-N-(4-metoxibenciliden)-4-metilbencenosulfonamida (2,89 g, 10 mmol), malonato de dimetilo (3,43 ml, 30 mmol), t-BuOK (2,24 g, 20 mmol) en t-BuOH anhidro (20 ml) se calentó a 50 °C durante 4 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se vertió en NH4Cl acuoso saturado y se extrajo con CH2Ch (3x). La capa orgánica combinada se secó sobre MgSO4, se filtró y a continuación se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (10 %, 20 %, 30 %, a continuación, 40 %) para dar el compuesto del título como un aceite transparente (3,255 g, 85 % de rendimiento).

Compuesto 118.2. Ácido 3-(4-metoxifenil)pentanodioico y ácido 3-(4-hidroxifenil)pentanodioico. Una mezcla de 2-(4-metoxifenil)propano-1,1,3,3-tetracarboxilato de tetrametilo (compuesto 118.1, 6,0 g, 15,7 mmol) en HCl concentrado (37 %, 80 ml) se calentó a reflujo durante una noche. Después de enfriar a temperatura ambiente, la suspensión se filtró. El filtrado se lavó con agua y se secó al vacío para dar 2,66 g de producto como una mezcla de ácido 3-(4-metoxifenil)pentanodioico (mayor, LCMS observado [M-H]- 237) y 3-(4-hidroxifenil)pentanodioico (menor, LCMS observado [M-H]- 223).

Compuesto 118.3. 3-(4-Metoxifenil)pentanodioato de dimetilo. Una mezcla del producto (compuesto 118.2) obtenido de la etapa anterior (2,66 g), K2CO3 (6,56 g, 47,5 mmol) y CH3I (3 ml, 47,5 mmol) en DMF (10 ml) se calentó a 50 °C durante una noche en un tubo a presión. Después de enfriar a temperatura ambiente, la mezcla de reacción se vertió en NaHCO3 acuoso saturado y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (20 %, 30 %, 40 %, a continuación, 50 %) para dar el compuesto del título como un aceite transparente (2,0 g, 48 % de rendimiento en dos etapas).

Compuesto 118.4. 3-(4-Metoxifenil)ciclopropano-1,2-dicarboxilato de dimetilo. A una solución de LDA (18,8 mmol) en THF (60 ml) a -78 °C se le añadió una solución de 3-(4-metoxifenil) pentanodioato de dimetilo (2,0 g, 7,52
mmol) en THF (10 ml) gota a gota. Después de agitar a -78 °C durante 1 h, se retiró el baño de hielo seco-acetona. La mezcla de reacción se agitó durante 30 minutos antes de enfriarla a -78 °C. Se añadió AgCl sólido (2,2 g, 15,4 mmol) todo de una vez. La mezcla de reacción se agitó a -78 °C durante 1 h y a continuación a temperatura ambiente durante una noche. Se añadió NH4Cl acuoso saturado. La mezcla se agitó vigorosamente durante 10 minutos. La suspensión se filtró a través de celite. El filtrado se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (10 %, 15 %, 20 %, a continuación, 30 %) para dar el compuesto del título como un sólido de color amarillo pálido (0,87 g, 44 % de rendimiento). 1HRMN (CDCla, 400 Hz) 87,09 (d, J = 8,8 Hz, 2H), 6,86 (d, J = 8,8 Hz, 2H), 3,82 (s, 3H), 3,77 (s, 6H), 3,17 (t, J = 7,6 Hz, 1H), 2,36 (d, J = 7,6 Hz, 2H).

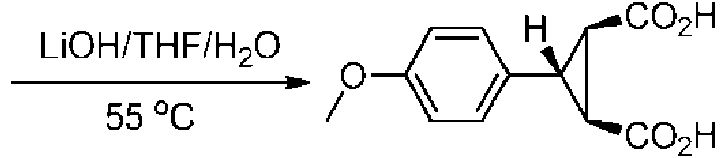
Compuesto 118.5. Ácido 3-(4-metoxifenil)ciclopropano-1,2-dicarboxílico. Una mezcla de 3-(4-metoxifenil)ciclopropano-1,2-dicarboxilato de dimetilo (0,88 g, 3,33 mmol) y LiOH (2 M en H2O, 10 ml) en THF (30 ml) se agitó a 55 °C durante una noche . Después de enfriar a temperatura ambiente, la mezcla de reacción se vertió en HCl IN y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró para dar el compuesto del título como un sólido de color amarillo claro.
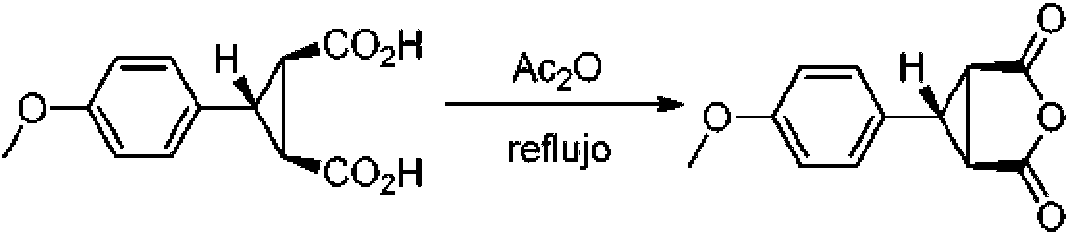
Compuesto 118.6. 6-(4-Metoxifenil)-3-oxabiciclo[3.1.0]hexano-2,4-diona. Una mezcla de ácido 3-(4-metoxifenil)ciclopropano-1,2-dicarboxílico (producto impuro procedente de la etapa anterior) en Ac2O (20 ml) se calentó a reflujo durante 1 h. El exceso de Ac2O se retiró a presión reducida. El producto impuro se usó en la siguiente etapa sin purificación adicional.

Compuesto 118.7.3-(4-Metoxibencil)-6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano-2,4-diona. Una mezcla de 6-(4-metoxifenil)-3-oxabiciclo[3.1.0]hexano-2,4-diona (producto impuro procedente de la etapa anterior) y (4-metoxifenil)metanamina se calentó a 180 °C durante 1,5 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se disolvió en CH2Ch y se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (20 %, 30 %, a continuación, 40 %) para dar el compuesto del título como un sólido de color amarillo (0,71 g, 63 % de rendimiento en tres etapas). MS [M+H]+: 338.

Compuesto 118.8. 3-(4-Metoxibencil)-6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano. Una mezcla de 3-(4-metoxibencil)-6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano-2,4-diona (0,86 g, 2,6 mmol), NaBH4 (0,296 g, 7,8 m m ol) y eterato de BF3 (1,0 ml, 7,8 mmol) en THF se calentó a reflujo durante una noche. Después de enfriar a 0 °C, se añadió una solución de piperazina (2 g) en H2O (20 ml). La mezcla se agitó a temperatura ambiente durante 2 h, se vertió en H2O y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y a continuación se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (10 %, 20 %, a continuación, 30 %) para dar un sólido blanco. A una suspensión del sólido en THF (20 ml) y H2O (10 ml) se le añadió piperazina (3 g). Después de calentar la mezcla a reflujo durante una noche, se vertió en solución saturada de cloruro sódico y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró para dar el compuesto del título como un sólido de color blanco (0,51 g, rendimiento 65 %). MS [M+H]+: 310.

Compuesto 118.9. 6-(4-Metoxifenil)-3-azabiciclo[3.1.0]hexano. A una solución de (1R,5S,6S)-3-(4-metoxibencil)-6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano (0,5 g, 1,6 mmol) en DCE (30 ml) a 0°C se le añadió cloroformiato de 1-cloroetilo (0,21 ml, 1,9 mmol, 1,2 equiv). La mezcla de reacción se agitó a 0 °C durante 30 minutos, se calentó a reflujo durante 1 h y a continuación se concentró a presión reducida. Se añadió MeOH (20 ml). La mezcla resultante se calentó a reflujo durante 40 minutos y a continuación se concentró. El residuo se purificó mediante TLC preparativa para dar el compuesto del título como un sólido cristalino de color blanco (155 mg, 51 % de rendimiento). Ms [M+H]+: 190.

Compuesto 118.10. (3-Ammo-4-metMfenM)(6-(4-metoxifenM)-3-azabicid o[3.1.0]hexan-3-M)metanona. Una mezcla de 6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano (0,124 g, 0,82 mmol), ácido 3-amino-4-metilbenzoico (0,155 g, 0,82 mmol), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDCI, 0,172 g, 0,90 mmol), 1-hidroxibenzotriazol (~20 % de H2O, 0,122 g, 0,90 mmol) y diisopropiletilamina (0,71 ml, 4,1 mmol) en DMF (3 ml) se agitó a temperatura ambiente durante una noche. La mezcla de reacción se vertió en NaHCO3 acuoso saturado y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (60 %, a continuación, 100 %) para dar el compuesto del

Compuesto 118.11. 6-Cloro-N-(5-(6-(4-metoxifenM)-3-azabid d o[3.1.0]hexano-3-carboml)-2-metilfenil)nicotinamida. A una solución de (3-amino-4-metilfenil)(6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano-3-il)metanona (150 mg, 0,47 mmol) y Et3N (0,25 ml, 1,8 mmol) en C ^ C h (4 ml) a 0 °C se le añadió cloruro de 6-cloronicotinoílo (106 mg, 0,6 mmol). El baño de hielo se retiró después de la adición. La mezcla de reacción se agitó a temperatura ambiente durante 1,5 h y a continuación se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (60 %, a continuación, 100 %) para dar el compuesto del título como una espuma de color blanco

Compuesto 118. 6-(IsopropMammo)-N-(5-(6-(4-metoxifenM)-3-azabid d o[3.1.0]hexano-3-carboml)-2-metilfenil)nicotinamida. Una mezcla de 6-cloro-N-(5-(6-(4-metoxifenil)-3-azabiciclo[3.1.0]hexano-3-carbonil)-2-metilfenil)nicotinamida (compuesto 118.11, 0,107 g, 0,23 mmol) e isopropilamina (1,5 ml) en DMSO (1,5 ml) se calentó a 120 °C en un tubo a presión sellado, durante una noche. Después de enfriar a temperatura ambiente, la mezcla de reacción se vertió en NaHCO3 acuoso saturado y se extrajo con EtOAc (3x). El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró. El residuo se trituró con hexano para dar el compuesto del título como un polvo de color blanco (40 mg, 36 % de rendimiento). MS [M+H]+: 485. 1H-RMN (DMSO-d6, 400 Hz) 89,63 (s, 1H), 8,69 (d, J = 3 Hz, 1H), 7,95-7,90 (m, 1H), 7,55-7,51 (m, 1H), 7,39-7,30 (m, 2H), 7,10-7,03 (m, 3H), 6,84 (d, J = 9,2 Hz, 2H), 6,52 (d, J = 9,2 Hz, 1H), 4,17-4,07 (m, 2H), 3,85-3,77 (m, 1H), 3,74 (s, 3H), 3,65-3,51 (m, 2H), 2,30 (s, 3H), 1,87 (s, 2H), 1,69 (t, J = 3,7 Hz, 1H), 1,20 (d, J = 7,2 Hz, 6H).

Compuesto 119. 6-(EtNammo)-N-(5-(6-(4-metoxifenM)-3-azabiddo[3.1.0]hexano-3-carboml)-2-metilfenil)nicotinamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 118. MS [M+H]+: 471. 1HRMN (DMSO-d6, 400 Hz) 69,62 (s, 1H), 8,66 (d, J = 2,4 Hz, 1H), 7,91 (dd, J =2,4, 9,0 Hz, 1H), 7,50 (s, 1H), 7,34-7,24 (m, 2H), 7,18 (t, J = 6,0Hz, 1H), 7,02 (d, J = 8,8 Hz, 2H), 6,81 (d, J = 8,8 Hz, 2H), 6,50 (d, J = 9,2 Hz, 1H), 4,08 (d, J = 12,4 Hz, 1H), 3,81-3,75 (m, 1H), 3,70 (s, 3H), 3,59 (d, J = 11,2 Hz, 1H), 3,52 (d, J = 13,2 Hz, 1H), 3,37-3,30 (m, 2H), 2,26 (s, 3H), 1,84 (s, 2H), 1,66 (t, J = 3,2 Hz, 1H), 1,16 (t, J = 8,4 Hz, 3H).

Compuesto 120. 1-(5-(6-(4-MetoxifenM)-3-azabiddo[3.1.0]hexano-3-carboml)-2-metNfenM)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se preparó usando reactivos disponibles fácilmente procedimientos similares a los

Compuesto 121.1. 4-(1-(3-Amino-4-metilbenzoil)piperidin-4-il)benzonitrilo Una solución de ácido 3-amino-4-metilbenzoico (1,36 g, 9,0 mmol), sal HCl de (4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 2,0 g, 9,0 mmol), EDCI (1,89 g, 9,9 mmol), HOBT (1,66 g, 9,9 mmol, 20 % con H2O) y DIEA (3,13 ml, 18,0 mmol) en DMF (50 ml) se agitó a temperatura ambiente durante una noche. La mezcla de reacción se vertió en agua fría (300 ml) y precipitaron sólidos de color blanquecino. El precipitado se filtró, se lavó con agua y se seca a presión reducida en un horno para producir

Compuesto 121. 6-((5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilfenil)amino)-N-isopropilnicotinamida. Una mezcla de 6-cloro-N-isopropilnicotinamida (0,055 g, 0,26 mmol), 4-(1-(3-amino-4-metil-benzoil)piperidin-4-il)benzonitrilo (121.1, 0,1 g, 0,31 mmol), K2CO3 (0,171 g, 1,24 mmol), Pd(OAc)2 (6,7 mg, 0,03 mmol) y el ligando (10,2 mg, 0,03 mmol) en tolueno (5 ml) se calentó a 90 °C en argón durante 3 h . Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con H2O y se extrajo con EtOAc. El extracto orgánico combinado se secó sobre MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía en columna ultrarrápida con EtOAc en hexano (60 %, 80 %, a continuación, 100 %) para dar el compuesto del título como un polvo de color blanquecino después de la liofilización a partir de CH3CN/H2O (11,2 mg, 9 % de rendimiento). m/z (ES+) 482 (M+H)+.

Compuesto 122. 4-(1-(4-MetN-3-((5-(4-metNpiperazm-1-carboml)pmdm-2-N)ammo)benzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 121 sustituyendo K2CO3 port-BuONa m/z (ES+) 524 (M+H)+.

Compuesto 123. N-(4-(4-(4-Cianofenil)piperidin-1-carbonil)piridin-2-il)-6-(pirrolidin-1-il)nicotinamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 43 y usando ácido 2-aminoisonicotínico en lugar de ácido 3-amino-4-metilbenzoico. m/z (ES+) 481 (M+H)+.
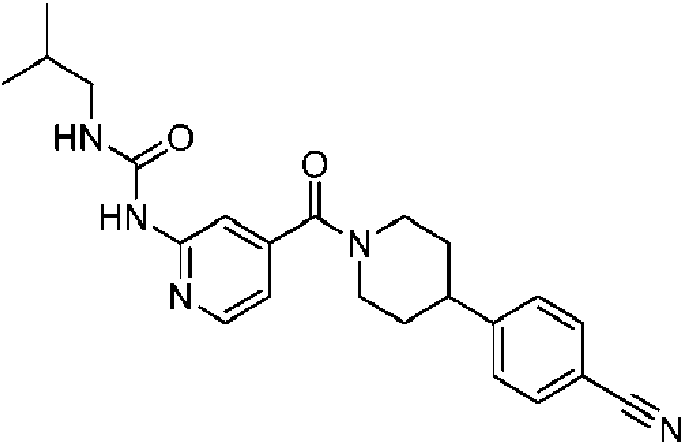
Compuesto 124. 1-(4-(4-(4-Cianofenil)piperidin-1-carbonil)piridin-2-il)-3-isobutilurea. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 64 y usando ácido 2-aminoisonicotínico en lu ar de ácido 3-amino-4-metilbenzo¡co. m/z (ES+) 406 (M+H)+.

Compuesto 125. N-(4-(4-(4-Cianofenil)piperidin-1-carbonil)piridin-2-il)pirrolidin-1-carboxamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 64 y usando ácido 2-aminoisonicotínico en lugar de ácido 3-amino-4-metilbenzoico. m/z (ES+) 404 (M+H)+.

Compuesto 126. N-(3-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2,4-dimetNfeml)-6-(isopropilamino)nicotinamida. El compuesto del título se preparó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 43 y usando ácido 3-amino-2,6-

Compuesto 127. 4-(1-(5-(5-Etoxi-4H-1,2,4-triazol-3-il)-2-etil-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 39. m/z (ES+) 445 (M+H)+.

Compuesto 128. 4-(1-(3-(5-(Metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-metil-3-(5-(tetrahidrofuran-3-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo

Compuesto 129. 4-(1-(3-(5-(Metoximetil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar procedimientos similares a los usados para la

Compuesto 130.1. 5-Carbamotioil-2,4-dimetilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-ciano-2,4-dimetilbenzoato de metilo (compuesto 2.3, 1,78 g, 9,41 mmol, 1,00 equiv) en tetrahidrofurano/H2O (30/3 ml). Se añadió ditiofosfato de O,O'-dietilo (3,30 g, 17,7 mmol, 2,00 equiv) y la mezcla resultante se agitó durante 2 días a 85 °C(ADVERTENCIA: se produce un desprendimiento de gas significativo - esta y todas las otras reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la mezcla se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico y se concentraron al vacío. El residuo impuro se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10) como eluyente para proporcionar 1,20 g (57 %) del compuesto del título como un sólido de color amarillo.
Compuesto 130.2. 5-(Imino(metiltio)metil)-2,4-dimetilbenzoato de metilo. A una solución de 5-carbamotioil-2,4-dimetilbenzoato de metilo (compuesto 130.1, 3,10 g, 12,5 mmol, 1,00 equiv, 90%) en tetrahidrofurano (30 ml) se le añadió CH3I (3,95 g, 27,8 mmol, 2,00 equiv) y la mezcla resultante se agitó durante una noche a 25 °C. La capa orgánica se lavó con 2 x 30 ml de Na2S2Ü4 (ac.) y 1 x 30 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró al vacío. Esto dio como resultado 2,10 g (64%) de 5-(imino(metiltio)metil)-2,4-dimetilbenzoato de metilo como un aceite de color amarillo.

Compuesto 130.3. 2-(Tetrahidrofuran-3-il)acetato de metilo. Una mezcla de ácido 2-(tetrahidrofuran-3-il)acético (2,00 g, 15,4 mmol, 1,00 equiv) y ácido sulfúrico (2 ml) en metanol (20 ml) se agitó durante 3 h a 80 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 50 ml de éter y se lavó con 2 x 20 ml de agua, 2 x 20 ml de bicarbonato sódico (ac., sat. Nota: desprendimiento de gas), y 2 x 20 ml de solución saturada de cloruro sódico. La fase orgánica se secó a continuación sobre sulfato sódico anhidro y se concentró al vacío para dar 1,50 g (68 %) del compuesto del título como un aceite de color amarillo.

Compuesto 130.4. 2-(Tetrahidrofuran-3-il)acetohidrazida. A un matraz de fondo redondo, se le añadió una solución de 2-(tetrahidrofuran-3-il)acetato de metilo (compuesto 130.3, 1,50 g, 10,4 mmol, 1,00 equiv) y NH2NH2 H2O (1,04 g, 20,8 m m ol, 2,00 equiv) en metanol (15 ml). La mezcla resultante se agitó durante una noche a 80 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío para dar 1,20 g (80 %) del

matraz de fondo redondo, se le añadió una solución de 2-(tetrahidrofuran-3-il)acetohidrazida (compuesto 130.4, 1,20 g, 8,32 mmol, 1,50 equiv) en ácido acético (4 ml). Se añadió 2,4-dimetil-5-(metilsulfanil)carboximidoilbenzoato de metilo (compuesto 130.2, 1,30 g, 5,48 mmol, 1,00 equiv) y la mezcla resultante se agitó durante una noche a 100 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se diluyó con 50 ml de acetato de etilo, a continuación se lavó con 2 x 20 ml de agua y 2 x 20 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (0 :1-1:10-1:1) como eluyente

Compuesto 130.6. Ácido 2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoico. A un matraz de fondo redondo, se le añadió una solución de 2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoato de metilo (compuesto 130.5, 600 mg, 1,90 mmol, 1,00 equiv) en metanol (10 ml). Se añadió una solución de hidróxido sódico (381 mg, 9,53 mmol, 5,00 equiv) en agua (5 ml) y la mezcla resultante se agitó durante 3 horas a 70 °C en un baño de aceite. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a continuación a presión reducida y el pH de la fase acuosa restante se ajustó a 3-4 con cloruro de hidrógeno (ac., 1 M). Los sólidos resultantes se recogieron mediante filtración y se secaron en un horno a presión reducida para dar 0,500 g (87 %) del

Compuesto 130. 4-(1-(2,4-DimetN-5-(5-((tetrahidrofuran-3-N)metM)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. Una mezcla del compuesto 130.6 (200 mg, 0,660 mmol, 1,00 equiv), EDCI (253 mg, 1,32 mmol, 2,00 equiv), DMAP (243 mg, 1,99 mmol, 3,00 equiv) y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 148 mg,
0,660 mmol, 1,00 equiv) en DMF (5 ml) se agitó durante 3 h a 25 °C, a continuación se diluyó con 50 ml de acetato de etilo. La capa orgánica se lavó con 2 x 10 ml de agua, 2 x 10 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El producto impuro (~300 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (28 % CH3CN asciende al 52 % en 8 minutos, asciende al 100 % en 1 minuto, desciende al 28 % en 1 minuto); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 168 mg (52 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 470 (M+H)+. 1H-RMN (400Hz, CD3OD): 87,68 (d, J = 8,0Hz, 2H), 7,61 (s, 1H), 7,50-7,49 (m, 2H), 7,33-7,31 (m, 1H), 4,90-4,88 (m, 1H), 3,95-3,92 (m, 2H), 3,90-3,81 (m, 1H), 3,79-3,77 (m, 1H), 3,69-3,55 (m, 1H), 3,28-3,25 (m, 1H), 3,03-2,94 (m, 4H), 2,81-2,73 (m, 1H), 2,53 (s, 3H), 2,43 y 2,33 (2s, rotámeros de amida, ArCH3,

Compuesto 131. 4-(1-(5-(5-(Metoxietil)-4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 3-metoxipropanohidrazida (compuesto 143.1) en lugar de 2-(tetrahidrofuran-

Compuesto 132. 4-(1-(5-(5-(Metoximetil)-4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 2-metoxiacetohidrazida (compuesto 190.6) en lugar de 2-(tetrahidrofuran-3-¡IJacetohidrazida (compuesto ) 430 M+H .
) 430 M+H .
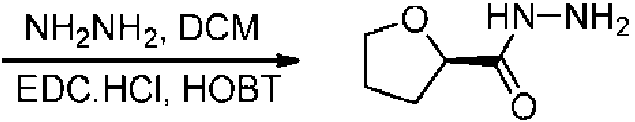
Compuesto 133.1. (R)-Tetrahidrofuran-2-carbohidrazida. Una mezcla de ácido (R)-tetrahidrofuran-2-carboxílico (5,00 g, 43,1 mmol, 1,00 equiv), EDCI (12,4 g, 64,6 mmol, 1,50 equiv) y HOBt (8,70 g, 64,4 mmol, 1,50 equiv) en diclorometano (100 ml) se agitó durante 30 minutos a 25 °C. A la mezcla se le añadió a continuación hidrazina (2,00 g, 62,4 mmol, 1,50 equiv) gota a gota. La mezcla resultante se agitó durante una noche a 25 °C. Los sólidos se retiraron por filtración, y el filtrado se concentró al vacío para proporcionar 25,0 g (impuros) de (R)-tetrahidrofuran-2-carbohidrazida como un aceite de color amarillo.

Compuesto 133. (R)-4-(1-(2,4-DimetN-5-(5-(tetrahidrofuran-2-M)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando (R)-tetrahidrofuran-2-carbohidrazida (compuesto 133.1) en lugar de 2-(tetrahidrofuran-3-il)acetoh¡draz¡da (compuesto 130.4). m/z ES+ 446 M+H)+.


Compuesto 134.1. 2-Hidrazinil-2-oxoacetato de etilo. A un matraz de fondo redondo, se le añadió una solución de oxalato de dietilo (10,0 g, 68,4 mmol, 1,00 equiv) en etanol (100 ml). Se añadió hidrato de hidrazina (2,75 g, 85,8 mmol, 1,00 equiv) y la mezcla resultante se agitó durante 3 h a 80 °C. Después de enfriar a temperatura ambiente, los sólidos se retiraron mediante filtración y el filtrado se concentró al vacío para dar 8,00 g (80 %) del compuesto del título
como un aceite incoloro.

Compuesto 134.2. 2-Hidrazinil-N-metil-2-oxoacetamida. A un matraz de fondo redondo, se le añadió una solución de 2-hidrazinil-2-oxoacetato de etilo (compuesto 134.1, 300 mg, 2,04 mmol, 1,00 equiv, 90%) en metanol (10 ml). Se añadió metilamina (10 ml, 40% en agua) y la mezcla resultante se agitó durante una noche a 70 °C. Después de enfriar a temperatura ambiente, los sólidos se recogieron por filtración y se secaron para dar 250 mg (94%) del

Compuesto 134. 5-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2,4-dimetMfenM)-N-metM-4H-1,2,4-triazol-3-carboxamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 130, usando el compuesto 134.2 en lugar del compuesto 130.4. m/z (ES+) 443 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,69 (d, J = 8,1 Hz, 2H), 7,62-7,43 (m, 3H), 7,32 (s, 1H), ~4,9 (1H parcialmente oscurecido por pico de agua), 3,73-3,58 (m, 1H), 3,32-3,18 (m, 1H parcialmente oscurecido por pico de disolvente metanol), 3,07-1,91 (m, 5H), 2,58 (s, 3H), 2,43 & 2,33 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,10-1,93 (
Compuesto 135.1. 2-Hidrazinil-A/,A/-dimetil-2-oxoacetamida. A un matraz de fondo redondo, se le añadió 2-hidrazinil-2-oxoacetato de etilo (compuesto 134.1, 2,00 g, 13,6 mmol, 1,00 equiv, 90%). Se añadió dimetilamina (10 ml) a la reacción, y a continuación la reacción se agitó durante una noche a 70 °C en un baño de aceite. La mezcla se enfrió a temperatura ambiente y se concentró al vacío para dar 1,50 g (76 %) del compuesto del título como un aceite incoloro.

Compuesto 135. 5-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2,4-dimetMfenM)-W,W-dimetN-4H-1,2,4-triazol-3-carboxamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 2-hidrazinil-W,A/-dimetil-2-oxoacetamida (compuesto 135. 1) en lugar de 2-(tetrahidrofuran-3-il)acetoh¡draz¡da (compuesto 130.4. m/z ES+) 457 (M+H)+.
-O. p n h 2n h 2, d c m
O "<0H EDC.HCI, HOBT 
Compuesto 136.1. (S)-Tetrahidrofuran-2-carbohidrazida. A un matraz de fondo redondo, se le añadió una solución de ácido (S)-tetrahidrofuran-2-carboxílico (3,00 g, 23,3 mmol, 1,00 equiv, 90 %) en diclorometano (40 ml). Se añadieron NH2NH2 (2 ml, 2,00 equiv), HOBt (5,20 g, 38,5 mmol, 1,50 equiv) y EDCI (7,50 g, 39,1 mmol, 1,50 equiv) y la mezcla resultante se agitó durante una noche a 25 °C. Los sólidos se retiraron por filtración, y el filtrado se concentró al vacío para proporcionar 2,50 g (74 %) de (S)-tetrahidrofuran-2-carbohidrazida como un aceite de color amarillo.

Compuesto 136. (S)-4-(1-(2,4-Dimetil-5-(5-(tetrahidrofuran-2-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando (S)-tetrahidrofuran-2-carbohidrazida (compuesto 136.1)
en lugar de 2-(tetrahidrofuran-3-il)a (compuesto 130.4. m/z ES+ 456 (M+H)+.
PISA


Compuesto 137.1. Tetrahidro-2H-piran-4-carboxilato de metilo. A una solución de ácido tetrahidro-2H-piran-4-carboxílico (520 mg, 4,00 mmol, 1,00 equiv) en metanol (50 ml) se le añadió PTSA(35,0 mg, 0,200 mmol). La mezcla resultante se agitó durante una noche a 80 °C en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró al vacío. El residuo se diluyó con 30 ml de agua y se extrajo con 3 x 30 ml de DCM. Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron al vacío. Esto dio como resultado 500 mg (87%) de tetrahidro-2H-piran-4-carbox¡lato de metilo como un aceite incoloro.

Compuesto 137.2. Tetrahidro-2H-piran-4-carbohidrazida. A un matraz de fondo redondo, se le añadió una solución de tetrahidro-2H-piran-4-carboxilato de metilo (compuesto 137.1, 5,00 g, 31,2 mmol, 1,00 equiv, 90%) en metanol (50 ml). Se añadió hidrato de hidrazina (5,20 g, 83,2 mmol, 3,00 equiv) y la mezcla resultante se agitó durante una noche a 40 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío para dar 4,00 g (80%) de tetrahidro-2H-piran-4-carbohidrazida como un sólido de color blanco.
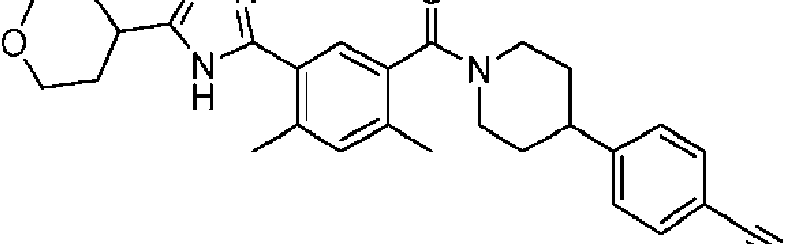
Compuesto 137. 4-(1-(2,4-DimetN-5-(5-(tetrahidro-2H-piran-4-M)-4H-1,2,4-tnazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando tetrahidro-2H-piran-4-carbohidrazida (compuesto 137.2) en lugar de 2-(tetrahidrofuran-3-il)acetoh¡draz¡da (compuesto 130.4). m/z (ES+) 470 (M+H)+.
|_|

Compuesto 138.1.2-(Tetrahi < drofur T an-2-il f )ac ' e ' tohid o redondo, se le añadió una solución de 2-(tetrahidrofuran-2-il)acetato de etilo (2,00 g, 12 tanol (20 ml). Se añadió NH2NH2 H2O (1,27 g, 25,4 mmol, 2.00 equiv) a la reacción. La sol ”razida. A un matraz de fond
,6 mmol, 1,00 equiv) en e
ución resultante se agitó durante una noche a 80 °C en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró al vacío. Esto dio como resultado 2,10 g

Compuesto 138. 4-(1-(2,4-DimetN-5-(5-((tetrahidrofuran-2-N)metM)-4H-1,2,4-tnazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 2-(tetrahidrofuran-2-il)acetohidrazida (compuesto 138.1) en lugar de 2-(tetrahidrofuran-3-il)acetoh¡draz¡da (compuesto 130.4). m/z (ES+) 470 (M+H)+.
O NH2NH2H20 °
NC- A ) ^ BOH,Et20 * NC^ V NH!
H
Compuesto 139.1. 2-Cianoacetohidrazida. A una solución de NH2NH2 H2O (3,50 g, 70,0 mmol, 1,00 equiv) en una mezcla disolvente de etanol y Et2O (35/35 ml) a 0 °Cse le añadió gota a gota una solución de 2-cianoacetato de metilo (7,00 g, 70,6 mmol, 1,00 equiv) en etanol (5 ml). La mezcla resultante se agitó durante 3 h a temperatura ambiente, a continuación se lavó con 2 x 30 ml de éter. Los sólidos se recogieron por filtración para dar 5,00 g (68 %) de 2-cianoacetohidrazida como un sólido de color blanco.

Compuesto 139. 4-(1-(5-(5-(cianometil)4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 2-cianoacetohidrazida (compuesto 139.1) en lugar de 2-(tetrahidrofuran-3-il)acetohidrazida (compuesto 130.4). m/z (ES+) 425 (M+H)+. 1H-RMN (300Hz, CD3OD): 87,70 (d, J = 8,4Hz, 2H), 7,54 7,42 (m, 3H), 7,35 (s, 1H), 4,87-4,80 (m, 1H), 4,12 (s, 2H), 3,77-3,65 (m, 1H), 3,27-3,23 (m, 1H), 3,09-2,99 (m, 2H), 2,55 (s, 3H), 2,43 y 2,33 (2 singletes, rotámeros de amida, ArCHa, 3H), 2,05-2,00 (m, 1H), 1,83-1,76 (m, 3H).


Compuesto 140.1. 3-cianopropanohidrazida. A un matraz de fondo redondo, se le añadió una solución de NH2NH2 H2O (1,25 g, 25,1 mmol, 1,00 equiv) en éter/EtOH (8/8 ml). A esto se le añadió 3-cianopropanoato de metilo (2,84 g, 25,1 mmol, 1,00 equiv) gota a gota. La solución resultante se agitó durante 2 h a temperatura ambiente, a continuación se concentró al vacío.
Esto dio como resultado 1,40 g (49%) de 3-cianopropanohidrazida como un aceite incoloro.

Compuesto 140. 4-(1-(5-(5-(2-Cianoetil)-4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-5-(5-((tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 130), usando 3-cianopropanohidrazida (compuesto 140.1) en lugar de 2-(tetrahidrofuran-3-il)acetohidrazida (compuesto 130.4). m/z (ES+) 439 (M+H)+. 1H-RMN (300Hz, CD3OD): 87,68 (d, 2H), 7,58-4,47 (m, 3H), 7,30 (s, 1H), 4,89-4,80 (m, 1H), 3,65-3,62 (m, 1H), 3,32-3,30 (m, 1H), 3,15 (t, 2H), 3,03-2,95 (m, 4H), 2,50 (s, 3H), 2,42 y 2,32 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,03-2,00 (m, 1H), 1,83-1,78 (m, 3H).

Compuesto 141. 4-(1-(2,4-Dimetil-5-(5-(oxetan-3-ilmetil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo.
El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dimetil-3-(5-(tetrahidrofuran-3-il)metil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-¡IJbenzonitrilo (compuesto 130). m/z (ES+) 456 (M+H)+.

Compuesto 142.1. 4-Ciclopropil-2-metilbenzoato de metilo. A una solución de 4-bromo-2-metilbenzoato de metilo (5,00 g, 20,7 mmol, 1,00 equiv, 95 %) en una mezcla de tolueno y H2O (20 ml /1 ml) se le añadieron carbonato potásico (6,10 g, 44,1 mmol, 2,00 equiv), ácido ciclopropilborónico (2,30 g, 26,8 mmol, 1,20 equiv), Pd(dppf)Ch (900 mg, 1,23 mmol, 0,05 equiv) y Pd(OAc)2 (250 mg, 1,12 mmol, 0,05 equiv). La mezcla de reacción se purgó con nitrógeno y se agitó a 80 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo resultante se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) como eluyente para dar 2,68 g (61 %) de 4-ciclopropil-2-metilbenzoato de metilo como un aceite incoloro.

Compuesto 142.2. 4-Ciclopropil-5-yodo-2-metilbenzoato de metilo. A una solución de 4-ciclopropil-2-metilbenzoato de metilo (compuesto 142.1, 2,68 g, 13,4 mmol, 1,00 equiv, 95%) en AcOH (50 ml) se añadieron NaIO4 (1,51 g, 7,08 mmol, 0,50 equiv), I2 (3,58 g, 14,1 mmol, 1,00 equiv) y ácido sulfúrico (201 mg, 2,01 mmol, 0,15 equiv, 98%). La mezcla de reacción se agitó a 110 °C durante una noche. Después de enfriar a temperatura ambiente, se añadieron 100 ml de agua. La mezcla resultante se diluyó con 100 ml de acetato de etilo, a continuación se lavó con 3 x 30 ml de Na2S2O3 (ac., sat.) y 1 x 30 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1/50) como eluyente para dar 2,00 g (45%) de 4-ciclopropil-5-yodo-2-metilbenzoato de metilo como un aceite incoloro.
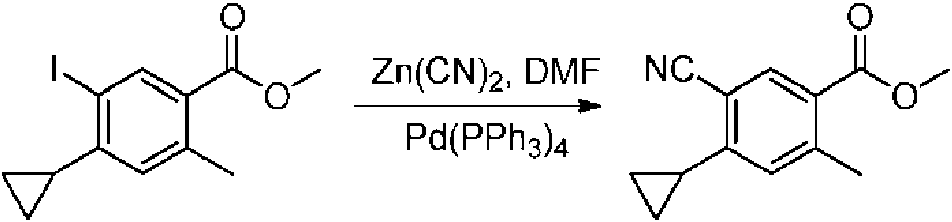
Compuesto 142.3. 5-Ciano-4-ciclopropil-2-metilbenzoato de metilo. A una solución de 4-ciclopropil-5-yodo-2-metilbenzoato de metilo (compuesto 142.2 ,2,00 g, 6,01 mmol, 1,00 equiv, 95 %) en DMF (16 ml) se le añadió Zn(CN)2 (890 mg, 7,58 mmol, 1,27 equiv) y Pd(PPh3)4 (731 mg, 0,630 mmol, 0,11 equiv). La solución resultante se agitó a 100 °C en nitrógeno durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición de 100 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 100 ml de acetato de etilo. Las fases orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1/50) como eluyente para dar 1,10 g (81 %) de 5-ciano-4-ciclopropil-2-met¡lbenzoato de metilo como un aceite de color amarillo claro.

Compuesto 142.4. 5-Carbamotioil-4-ciclopropil-2-metilbenzoato de metilo. A una solución de 5-ciano-4-ciclopropil-2-metilbenzoato de metilo (compuesto 142.3, 1,65 g, 7,28 mmol, 1,00 equiv, 95 %) en una mezcla de tetrahidrofurano y H2O (20 ml/5 ml) se le añadió 0,0-dietilfosforoditioato (3,79 g, 22,3 mmol, 2,00 equiv). La mezcla resultante se agitó a 80 °C durante una noche (ADVERTENCIA: se produce desprendimiento de gas significativo -esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la reacción se inactivó con 100 ml de agua. La solución resultante se extrajo con 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 30 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1/5) como eluyente para proporcionar 0,880 g (46 %) de 5-carbamotioil-4-ciclopropil-2-metilbenzoato de metilo como un sólido de color blanco.
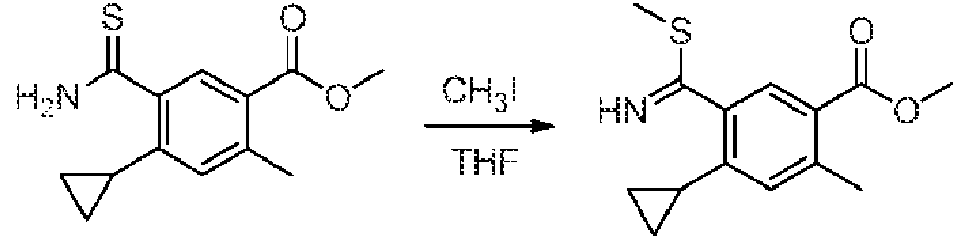
Compuesto 142.5. 4-Ciclopropil-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-carbamotioil-4-ciclopropil-2-metilbenzoato de metilo (compuesto 142.4, 880 mg, 3,35 mmol, 1,00 equiv, 95 %) en tetrahidrofurano (10 ml). Se añadió yodometano (1,00 g, 7,05 mmol, 2,00 equiv) y la mezcla resultante se agitó a temperatura ambiente durante una noche. A continuación, la mezcla se concentró al vacío para dar 0,800 g (86 %) de 4-ciclopropil-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo como un líquido incoloro.
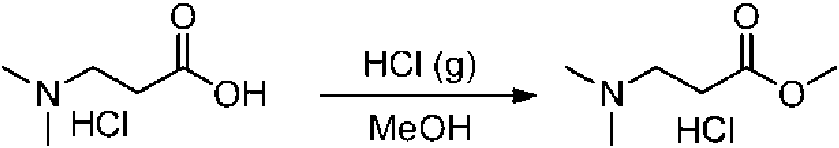
Compuesto 142.6. Clorhidrato de 3-(dimetilamino)propanoato de metilo. A un matraz de fondo redondo, se le añadió una solución de ácido 3-(dimetilamino)propanoico (2,00 g, 17,1 mmol, 1,00 equiv) en metanol (60 ml). Se burbujeó cloruro de hidrógeno (g) en la mezcla de reacción y la solución resultante se agitó durante 4 h a 25 °C. La concentración de la mezcla de reacción al vacío produjo 2,00 g del compuesto del título como un aceite incoloro.

Compuesto 142.7.3-(Dimetilamino)propanohidrazida. A una solución de clorhidrato de 3-(dimetilam¡no)propanoato de metilo (compuesto 142.6, 2,00 g, 15,3 mmol, 1,00 equiv) en metanol (40 ml) se le añadió hidrato de hidrazina (6 ml, 6,00 equiv). La mezcla de reacción se agitó a 70 °C durante 3 h. La mezcla se concentró al vacío y a continuación se disolvió en 50 ml de H2O y se lavó con 2 x 10 ml de acetato de etilo. Las capas acuosas se combinaron y se concentraron al vacío .
.
Compuesto 142.8. 4-CidopropM-5-(5-(2-(dimetMammo)etM)-4H-1,2,4-triazol-3-M)-2-metMbenzoato de metilo. Una solución de 3-(dimetilamino)propanohidrazida (compuesto 142.7, 1,20 g, 9,15 mmol, 5,00 equiv) y 4-ciclopropil-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo (compuesto 142.5 ,600 mg, 2,28 mmol, 1,00 equiv) en AcOH (30 ml) se agitó a 80 °C durante una noche. Después de enfriar a temperatura ambiente, el pH se ajustó a 8-9 con hidróxido sódico (ac., 1 M). La mezcla resultante se extrajo con 2 x 100 ml de acetato de etilo y las capas orgánicas combinadas se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de sílice con diclorometano/metanol (10/1) como eluyente para dar 504 mg (67 %) del compuesto del título como un sólido de color blanco.

Compuesto 142.9. Ácido 4-ciclopropil-5-(5-(2-(dimetilamino)etil)-4H-1,2,4-triazol-3-il)-2-metilbenzoico. A una solución del compuesto 142.8, (200 mg, 0,610 mmol, 1,00 equiv, 95%) en una mezcla de metanol y H2O (6 ml/3 ml) se le añadió hidróxido sódico (97,6 mg, 2,44 mmol, 4,00 equiv) en agua (1 ml). Después de agitar a 60 °C durante una noche, el disolvente orgánico se retiró a presión reducida. La capa acuosa residual se lavó con 20 ml de acetato de etilo. El pH se ajustó a 4-5 con HCl (ac., 3 M), y la mezcla resultante se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío para producir 280 mg (73 %) del compuesto del título como un sólido de color marrón.

Compuesto 142. 4-(1-(4-CidopropN-5-(5-(2-(dimetNammo)etM)-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo A una solución del compuesto 2661.9 (250 mg, 0,800 mmol, 1,00 equiv) en W,A/-dimetilformamida (3 ml) se le añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 175 mg, 0,790 mmol, 1,00 equiv), EDCI (302 mg, 1,58 mmol, 2,00 equiv) y DMAP (194 mg, 1,59 mmol, 2,00 equiv). La mezcla resultante se agitó a 25 °C durante una noche y a continuación se diluyó con agua. La mezcla se extrajo con 3 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 20 ml de NH4Cl (ac) y 2 x 20 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con diclorometano/metanol (10/1) como eluyente. El producto (~150 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones [(1 # Pre-HPLC-001 (Shimadzu)): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (32,0 % CH3CN asciende al 47,0 % en 7 minutos, asciende al 100,0 % en 1 minuto, desciende al 32,0 % en 1 minuto); detector, Waters 2489254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 70,3 mg (18 %) del compuesto del título como un sólido de color blanco, m/z (ES+) 483 (M+H)+.

Compuesto 143.1. 3-Metoxipropanohidrazida. Una mezcla de 3-metoxipropanoato de metilo (5,0 g, 42,33 mmol) e hidrazina (1,36 g, 42,33 mmol) se calentó a 50 °C durante dos horas. La mezcla se concentró y se secó a presión reducida para dar el producto como un aceite transparente. Rendimiento: 5,0 g, 100 %. m/z (ES+) 119 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) ó 7,86 (ancho, 1H), 4,05 - 3,71 (m, 2H), 3,63 (t, 2H), 3,34 (s, 3H), 2,42 (t, 2H).
Compuesto 143.

il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 142, usando el compuesto 143.1 en lugar de 142.7. m/z (ES+) 471 (M+H)+.
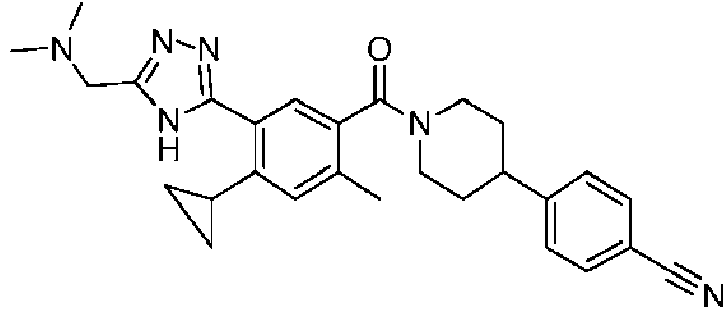
Compuesto 144. 4-(1-(4-Ciclopropil-5-(5-((dimetilamino)metil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 142. m/z (ES+) 469 (M+H)+. 1H RMN (300 MHz, CD3OD): 8 7,69 (d, J = 6,3 Hz, 2H), 7,49 (d, J = 6,0 Hz, 2H), 7,47 y 7,39 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,03 (s, 1H), ~4,9 (1H parcialmente oscurecido por pico de agua), 3,74 (s, 2H), 3,72-3,57 (m, 1H), 3,32-3,22 (m, 1H parcialmente oscurecido por pico de disolvente metanol), 3,00 (t con estructura fina, J = 8,9 Hz, 2H), 2,49-2,27 (m, 2H), 2,10-1,98

Compuesto 145. 4-(1-(5-(5-(Azet¡dm-1-NmetM)-4H-1,2,4-tr¡azol-3-M)-4-c¡clopropN-2-met¡lbenzoN)p¡per¡dm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos l compuesto 142. m/z (ES+) 481 (M+H)+.
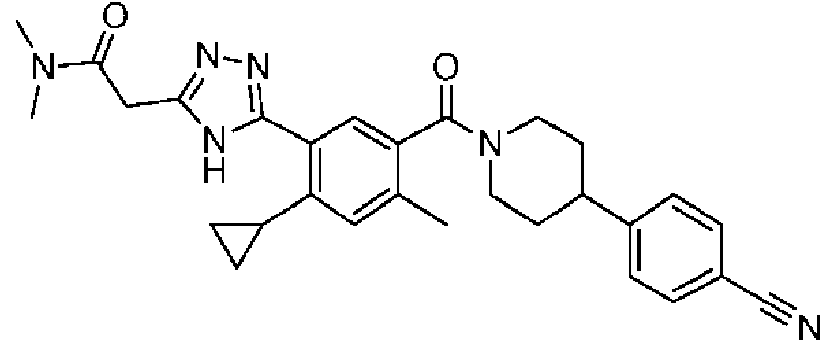
Compuesto 146.2-(5-(5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclopropil-4-metilfenil)-4H-1,2,4-triazol-3-il)-W,W-dimetilacetam¡da. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 142. m/z ES+) 497 (M+H)+.

Compuesto 147.1.4-Etil-2-metilbenzoato de metilo. A una mezcla agitada de ZnBr2 (4,50 g, 20,0 mmol, 2,00 equiv) en tetrahidrofurano (50 ml) en nitrógeno a 0°C se le añadió gota a gota EtMgBr (6,6 ml, 2,00 equiv, 3 M en THF). Después de agitar durante 30 minutos a 0 °C, la temperatura se hizo descender a -78 °C y se añadió Pd(dppf)Ch (1,08 g, 1,48 mmol, 0,30 equiv), seguido de una solución de 4-bromo-2-metilbenzoato de metilo (compuesto 152.1, 2,30 g, 10,0 mmol, 1,00 equiv) en tetrahidrofurano (20 ml). La mezcla resultante se agitó durante 30 minutos a -78 °C, se calentó a temperatura ambiente, y se agitó durante una noche. La mezcla de reacción se inactivó cuidadosamente con 60 ml de NH4Cl (ac.) y se extrajo con 3 x 50 ml de acetato de etilo. Las fases orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:5) como eluyente para proporcionar 1,50 g (84 %) de 4-etil-2-metilbenzoato de metilo como un aceite de color marrón.

Compuesto 147.2. Ácido 4-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del

Compuesto 147. 4-(1-(4-Etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. A una solución del compuesto (147.2, 100 mg, 0,350 mmol, 1,00 equiv) en DMF (10 ml) en nitrógeno, se le añadieron EDCI (132 mg, 0,690 mmol, 2,00 equiv) y DMAP (85,0 mg, 0,700 mmol, 2,00 equiv). La mezcla resultante se agitó 30 minutos a 25 °C seguido de la adición de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 129 mg, 2,77 mmol, 2,00 equiv). La mezcla de reacción se agitó durante 25 h a 25 °C, a continuación se inactivó con 40 ml de agua con hielo, y se extrajo con 3 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éterde petróleo (1:100-1:1) como eluyente. El producto impuro (50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMa Dz U)): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03% de NH3H2O y CH3CN (33 % CH3CN asciende al 52 % en 10 minutos, asciende al 100 % en 1 minuto, desciende al 33 % en 1 minuto); detector, Waters 2489254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 20,2 mg (12 %) del compuesto del título como un sólido de color blanco, miz (ES+) 458 (M+H)+.

Compuesto 148.4-(1-(4-etN-5-(5-(2-metoxietM)-4H-1,2,4-triazol-3-M)-2-metNbenzoM)-4-fluoropipeN dm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 147 y usando sal HCl del compuesto 11.2 en lugar del compuesto 1.5. miz (ES+) 476 (
Compuesto 149.4-(1-(5-(5-(EtoximetM)-4H-1,2,4-triazol-3-M)-4-etN-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 147 y 148. m/z (ES+) 476 (M+H)+.

Compuesto 150. 4-(1-(4-EtN-2-metN-5-(5-(tetrahidrofuran-3-M)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 147). m/z (ES+) 470 (M+H)+.

Compuesto 151. 4-(1-(4-EtN-5-(5-(isopropoximetM)-4H-1,2,4-triazol-3-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 147 y 148. m/z (ES+) 490 (M+H)+. 1H-RMN (300 MHz, CD3OD): 87,78 (d, 2H), 7,69-7,34 (m, 4H), 4,82-4,78 (m, 1H), 4,72 (s, 2H), 3,80 (quintuplete, 1H), 3,57-3,55 (m, 2H), 3,30-3,25 (m, 1H), 2,94 (c, 2H), 2,46 y 2,35 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,35-2,1 (m, 3H), 2,0-1,95 (m, 1H), 1,30 (d, 6H), 1,14 (t, 3H.

Compuesto 152.1. 4-Bromo-2-metilbenzoato de metilo. A una solución de ácido 4-bromo-2-metilbenzo¡co (5,11 g, 23,8 mmol, 1,0 equiv) en metanol (25 ml) se le añadió gota a gota ácido sulfúrico (2,0 ml) durante aproximadamente 3 minutos (ligeramente exotérmico). La mezcla resultante se calentó a reflujo durante 4 horas. Después de enfriar a temperatura ambiente, la mezcla de reacción se inactivó cuidadosamente en solución acuosa saturada de NaHCO3 (100 ml) (nota - desprendimiento de gas significativo) y se extrajo con diclorometano (200 ml x 1, a continuación 50 ml x 1). Las fases orgánicas combinadas se lavaron con una mezcla de solución saturada de cloruro sódico/NaHCO3 saturado de (9:1) (50 ml), se secaron (Na2SO4), y se concentraron a presión reducida para obtener el compuesto del título como un aceite incoloro (5,28 g , 97%). 1H RMN (400 MHz, CDCh): 87,78 (d, J = 8,0 Hz, 1 H), 7,42 (d, J =1,6 Hz, 1H), 7,38 (dd, J = 1,6 Hz, 1H), 3,89 (s, 3H), 2,58 s, 3H.

Compuesto 152.2. 4-Ciclobutil-2-metilbenzoato de metilo. Se añadió bromuro de ciclobutilzinc (II) (50 ml, 0,5 M en THF, 25,0 mmol) a una mezcla de 4-bromo-2-metilbenzoato de metilo (compuesto 152.1, 5,2 g, 22,7 mmol) y PdCh(dppf)CH2Cl2 (1,85 g, 2,27 mmol). La mezcla se desgasificó y el matraz se llenó con argón a través de un globo. La mezcla se calentó a 65 °C en argón durante 24 horas. La mezcla se enfrió a 0 °C y se inactivó con agua (10 ml). La mezcla se diluyó con EtOAc (200 ml), se lavó con agua y a continuación con solución saturada de cloruro sódico. La capa de EtOAc se secó (Na2SO4), se concentró a presión reducida, y se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30:1 a 20:1). Rendimiento: 4,1 g, aceite transparente, 89,1 %. 1H RMN (400 MHz, Cloroformo-d) 87,86 (d, 1H), 7,12 - 7,02 (m, 2H), 3,88 (s, 3H), 3,59 - 3,48 (m, 1H), 2,59 (s, 3H), 2,35 (m, 2H), 2,22 -1,96 (m, 3H), 1,86-1,84 (m, 1H).

Compuesto 152.3. 4-Ciclobutil-5-yodo-2-metilbenzoato de metilo. Se añadió N-yodosuccinimida (3,52 g, 15,6 mmol) por partes a una solución de 4-ciclobutil-2-metilbenzoato de metilo (compuesto 152.2 , 3,2 g, 15,6 mmol) en ácido sulfúrico concentrado (25 ml) a 0 °C. La mezcla se agitó a 0 °C durante 30 minutos y a TA durante 2 horas. La mezcla se volvió muy espesa. La mezcla se enfrió a 0 °C de nuevo y se añadió MeOH (30 ml). La mezcla se calentó a 60 °C durante 2 horas. El metanol se retiró a presión reducida y el residuo se vertió en agua con hielo (100 ml). La mezcla se extrajo con EtOAc (2x). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico, a continuación, NaHCO3 IN (nota-desprendimiento de gas significativo), se secaron (Na2SO4) y se concentraron. El residuo se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30:1 a 20:1). Rendimiento: 4, 17 g aceite de color amarillo claro, 81 %. 1H RMN (400 MHz, Cloroformo-d) 88,33 (s, 1H), 7,14 (s,
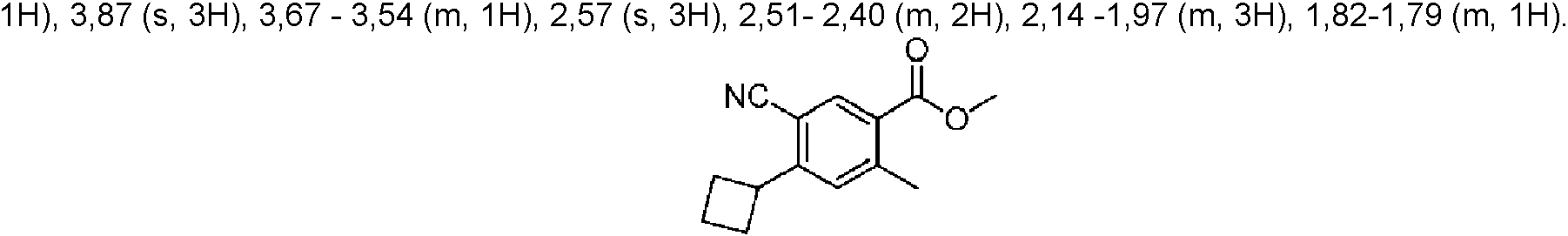
Compuesto 152.4. 5-Ciano-4-ciclobutil-2-metilbenzoato de metilo. Una mezcla de 4-ciclobutil-5-yodo-2-metilbenzoato de metilo (compuesto 152.3 ,4,17 g, 12,64 mmol), Zn(CN)2 (2,96 g, 25,21 mmol) y Pd(PPh3)4 (0,73 g,
0,63 mmol) en DMF (30 ml) se desgasificó y el matraz se llenó con argón a través de un globo. La mezcla se calentó a 100 °C en argón durante una noche. Después de enfriar a temperatura ambiente, la mezcla se inactivó con. FeSO4 ac. saturado (20 ml) y se diluyó con EtOAc (200 ml). El sólido verdoso se retiró por filtración a través de celite. El filtrado se repartió entre agua y EtOAc. La capa de EtOAc se lavó con solución saturada de cloruro sódico, se secó (Na2SO4) y se concentró. El residuo se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30: 1 a 20: 1). Rendimiento: 2,55 g, sólido de color blanco, 88%. 1H RMN (400 MHz, Cloroformo-d) 88,16 (s, 1H), 7,28 (s, 1H), 3,90 (s, 3H), 3,86 - 3,82 m, 1H, 2,68 s, 3H , 2,55- 2,45 m, 2H , 2,27 - 2,04 m, 3H), 1,89-1,87 (m, 1H).

Compuesto 152.5. 5-Carbamotioil-4-ciclobutil-2-metilbenzoato de metilo. A un matraz de fondo redondo se le añadieron 5-ciano-4-ciclobutil-2-metilbenzoato de metilo (compuesto 152.4, 3,63 g, 0,015 mol), ditiofosfato de O,O'-dietilo (10 ml) y agua (1 ml). La mezcla de reacción se calentó a 80 °C durante 3 horas (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la mezcla de reacción se repartió entre acetato de etilo (50 ml) y agua (50 ml). Las capas orgánicas combinadas se lavaron sucesivamente con NaHCO3 saturado acuoso (50 ml) y solución saturada de cloruro sódico (50 ml), se secaron sobre Na2SO4, y se concentraron al vacío. La purificación por cromatografía ultrarrápida con SiO2 (hexanos/acetato de etilo = 80/20 a 50/50) produjo 5-carbamotioil-4-ciclobutil-2-metilbenzoato de metilo como un sólido de color amarillo (3,06 g, 78% de rendimiento). m/z (ES+) 264 (M+H)+. 1H RMN (400 MHz, CDCh): 87,93 (s, 1H), 7,82 (s, 1H), 7,26 (s, 1H), 6,92 (s, 1H), 4,19 (m, 1H), 3,89 (s, 3H), 2,64 (s, 3H), 2,40 (m, 2H), 2,29-2,15 (m, 2H), 2,12-2,00 (m, 1H), 1,95-1,84 (m, 1H).

Compuesto 152.6. 4-CidobutM-5-(immo(metMtio)metM)-2-metMbenzoato de metilo. A un matraz de fondo redondo, se le añadió 5-carbamotioil-4-ciclobutil-2-metilbenzoato de metilo (compuesto 152.5 ,861 mg, 3,27 mmol) en THF (10 ml). Se añadió yodometano (912 mg, 6,42 mmol) gota a gota y la mezcla de reacción se agitó a temperatura ambiente durante 7 horas. La mezcla de reacción se concentró al vacío y se purificó por cromatografía ultrarrápida con SiO2 (acetato de etilo a acetato de etilo/metanol = 95/5) para dar 4-ciclobutil-5-(imino(metiltio)metil) -2-metilbenzoato de metilo como un aceite de color amarillento (807 mg, 89 % de rendimiento). m/z (ES+) 278 (M+H)+. 1H RMN (400 MHz, DMSO-d6): 87,67 (s, 1H), 7,40 (s, 1H), 3,88-3,71 (m, 4H), 2,57 (s, 3H), 2,44 (s, 3H), 2,22-2,19 (m, 2H), 2,12 (m, 2H), 1,98 - 1,86 (m, 1H), 1,82 - 1,70 (m, 1H).

Compuesto 152.7. 4-Ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoato de metilo. A un matraz de fondo redondo se le añadieron 4-ciclobutil-5-(imino(metiltio)metil) -2-metilbenzoato de metilo (compuesto 152.6, 556 mg, 0,002 mol) y acetohidrazida (223 mg, 0,003 mol) en 6 ml de ácido acético. La mezcla de reacción se calentó a 90 °C durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla de reacción se repartió entre agua (50 ml) y acetato de etilo (50 ml). La capa orgánica se lavó con solución saturada de cloruro sódico (2 x 50 ml), se secó sobre Na2SO4, y se concentró al vacío. La purificación por cromatografía ultrarrápida con SiO2 (hexanos/acetato de etilo = 50/50 a 30/70) produjo el compuesto del título como un sólido de color blanco (243 mg, 43 % de rendimiento). m/z (ES+) 286 (M+H)+. 1H RMN (400 MHz, CDCla): 88,23 (s, 1H), 7,32 (s, 1H), 4,24 - 4,05 (m, 1H), 3,89 (s, 3H), 2,69 (s, 3H), 2,54 (s, 3H), 2,23-2,20 (m, 2H), 2,16-2,05 (m, 2H), 2,05-1,88 (m, 1H), 1,88-1,71 (m, 1H).

Compuesto 152.8. Ácido 4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoico. A una solución de 4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoato de metilo (compuesto 152.7 , 240 mg, 0,842 mmol) en metanol (5 ml ) se le añadió NaOH acuoso (6 ml, 1 M). La mezcla resultante se calentó a 50 °C durante 6 horas. Después de enfriar a temperatura ambiente, la mezcla de reacción se acidificó con HCl IN a pH 2 y se extrajo con acetato de etilo (3 x 50 ml). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (50
ml), se secaron sobre Na2SO4, y se concentraron al vacío para proporcionar ácido 4-ciclobutil-2-metil-5- (5-metil-4H-

Compuesto 152. 4-(1-(4-Ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. A una solución de ácido 4-cidobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoico (compuesto 152.8, 260 mg, 0,95 mmol) en DMF (4 ml) se le añadieron sal clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5 , 232 mg, 1,045 mmol), EDC (272 mg, 1,425 mmol), HOBt (39 mg, 0,285 mmol), y DIEA (367,7 mg, 2,85 mmol). La mezcla resultante se agitó a temperatura ambiente durante 16 horas. La mezcla se inactivó con NaHCO3 acuoso saturado (20 ml) y se extrajo con acetato de etilo (2 x 50 ml). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (50 ml), se secaron sobre Na2SO4, se filtraron, y se concentraron al vacío. La purificación mediante cromatografía en columna con SO 2 (diclorometano/metanol = 95/5) produjo 4- (1-(4-ciclobutN-2-metN-5-(5-metN-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo como un sólido de color blanco (193 mg, 44% de rendimiento). m/z (ES+) 440 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,69 (d, J = 5,4 Hz. 2H), 7,56-7,30 (m, 4H), 1 protón oscurecido por pico de disolvente metanol, 4,10-3,98 (m, 1H), 3,64 (t, J = 10,7 Hz, 1H), 3,33-3,21 (m, 1H), 3,00 (t, J = 8,9 Hz, 2H), 2,58 (s, 3H), 2,48 y 2,38 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,28-1,92 (m, 6H), 1,92-1,55 (m, 4H). 1H RMN (400 MHz, DMSO-d6): 813,66 (s, 1H), 7,77 (d, J = 8,0 Hz, 2H), 7,62-7,34 (m, 4H), 4,78-4,63 (m, 1H), 4,31 (s ancho, 1H), 3,45 (s ancho, 1H), 3,15 (t aparente, J = 12,3 Hz, 1H), 2,99-2,78 (m, 2H), 2,44-1,80 (m, 12H), 1,80-1,37 (m, 4H).

Compuesto 153. 4-(1-(4-CidobutN-5-(5-(2-metoxietM)-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol 3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152), pero usando 3-metoxipropanohidrazida (compuesto 143.1) en lugar de acetohidrazida. m/z (ES+) 484 (M+H)+, 967 (2M+H)+. 1H RMN (400 MHz, Cloroformo-d) 811,50- 11,33 (s ancho, 1H), 7,66 - 7,44 (m, 3H), 7,33 - 7,27 (m, 3H), 4,98 (d, 1H), 4,24-4,12 (m, 1H), 3,78 (t, 2H), 3,70 (d, 1H), 3,44 (s, 3H), 3,14 - 3,03 (m, 3H), 2,90 - 2,75 (m, 2H), 2,42 y 2,34 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,17 (d, 2H), 2,08 - 1,88 (m, 3H), 1,84 -1,51 (m, 5H).

Compuesto 154.4-(1-(4-CidobutN-5-(5-(metoximetN)-4H-1,2,4-tnazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol 2-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152), pero usando 2-metoxiacetohidrazida (compuesto 190.6) en lugar de acetohidrazida. m/z (ES+) 470 (M+H)+. 1H RMN (400 MHz, CDCh): 812,15 (s ancho, 1H), 7,64-7,57 (m, 2H), 7,43 y 7,33 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,30 (d, J = 8,4 Hz, 2H), 7,20 (s, 1H), 5,04-4,92 (m, 1H), 4,65 (s, 2H), 4,18-4,03 (m, 1H), 3,63 (d ancho, J =13,2 Hz, 1H), 3,51 (s, 3H), 3,08 (t con estructura fina, J = 12,8 Hz, 1H), 2,93-2,77 (m, 2H), 2,38 y 2,30 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,25-1,84 (m, 6H), 1,84 1,43 (m, 4H).
Compuesto 155. 4-(1-(4-CidobutN-5-(5-(hidroximetM)-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152 pero usando 2-hidroxiacetohidrazida en lugar de acetohidrazida. miz (ES+) 456 (
Compuesto 156. 4-(1-(4-CidobutN-5-(5-(2-metoxietM)-4H-1,2,4-tnazol-3-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(2-metoxietil)-4H-1,2,4-triazol 3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 153), usando sal clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2, sal HCl) en lugar de sal clorhidrato de 4-(piperidin-4il)benzonitrilo (compuesto 1.5). m/z (ES+) 501,8 (M+H)+. 1H RMN (400 MHz, CDCla): 611,46 (s ancho, 1H), 7,75-7,45 (m, 3H), 7,49 (d, J =8,4 Hz, 2H), 7,32 (s, 1H), 4,89 (d ancho, J =13,2 Hz, 1H), 4,28-4,15 (m, 1H), 3,79 (t, J = 6,0 Hz, 2H), 3,70-3,45 (m, 2H), 3,46 (s, 3H), 3,31-3,17 (m, 1H), 3,13 (t, J = 6,0 Hz, 2H), 2,45 & 2,38 (2 singletes anchos, rotámeros de amida, Ar-CH3, 3H), 2,30-1,68 (m, 10H).
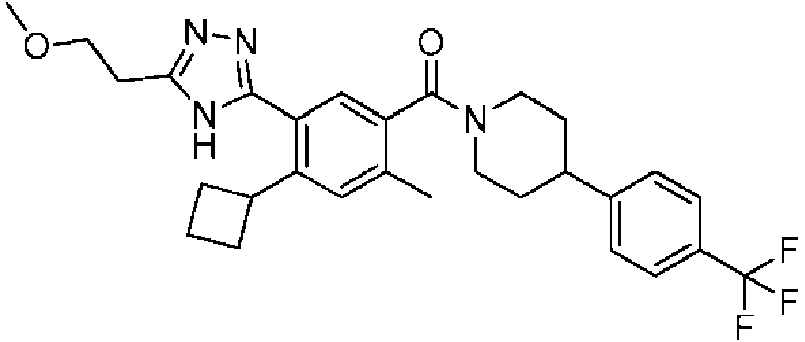
Compuesto 157. (4-CidobutN-5-(5-(2-metoxietM)-4H-1,2,4-tnazol-3-M)-2-metNfenM)(4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(2-metoxietil)-4H-1,2,4-triazol 3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 153), usando sal clorhidrato de 4-(4-(trifluorometil)fenil)piperidina en lugar de sal clorhidrato de 4-(piperidin-4il)benzonitrilo (compuesto 1.5). m/z (ES+) 527 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 611,30-11,10 (ancho, 1H), 7,60-7,47 (m, 3H), 7,35-7,28 (m, 3H), 5,10-4,93 (m, 1H), 4,24-4,13 (m, 1H), 3,78 (t, 2H), 3,70 (d, 1H), 3,45 (s, 3H), 3,12 t, 2H), 3,15-3,05 (m, 1H), 2,90-2,75 (m, 2H), 2,44 y 2,35 (2 singletes, rotámeros de amida, ArCHs, 3H), 2,25-2,13 (m, 2H), 2,13-1,85 (m, 4H), 1,87-1,66 (m, 4H).

Compuesto 158. 4-(1-(4-CidobutN-2-metN-5-(5-metN-4H-1,2,4-tnazol-3-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152), usando sal HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 458 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 613,66 (s, 1H), 7,66 (d, 2H), 7,51 (d, 3H), 7,38 (s, 1H), 4,72 (d, 1H), 4,30 (s ancho, 1H), 3,46 (s ancho, 1H), 3,16 (dd, 1H), 3,04-2,78 (m, 2H), 2,38 y 2,36 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,35-2,27 (m, 3H), 2,22 - 1,82 (m, 6H), 1,81-1,56 (m, 3H).

Compuesto 159. (4-Ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)fenil)(4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol 3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152). m/z (ES+) 483 (M+H)+.

Compuesto 160. 4-(1-(4-CidobutN-5-(5-etN-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)piperidm-4-N)benzomtrNo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152 y usando propionohidrazida en lugar de acetohidrazida. m/z (ES+) 454 (M+H)+.
1H RMN (400 MHz, Cloroformo-d) 87,75 - 7,43 (m, 3H), 7,40 - 7,17 (m, 3H), 5,18 - 4,81 (m, 1H), 4,30 - 3,91 (m, 1H), 3,84 - 3,55 (m, 1H), 3,21 - 2,99 (m, 1H), 2,92 - 2,69 (m, 4H), 2,40 y 2,32 (2 singletes, rotámeros de amida, ArCH3, 3H),
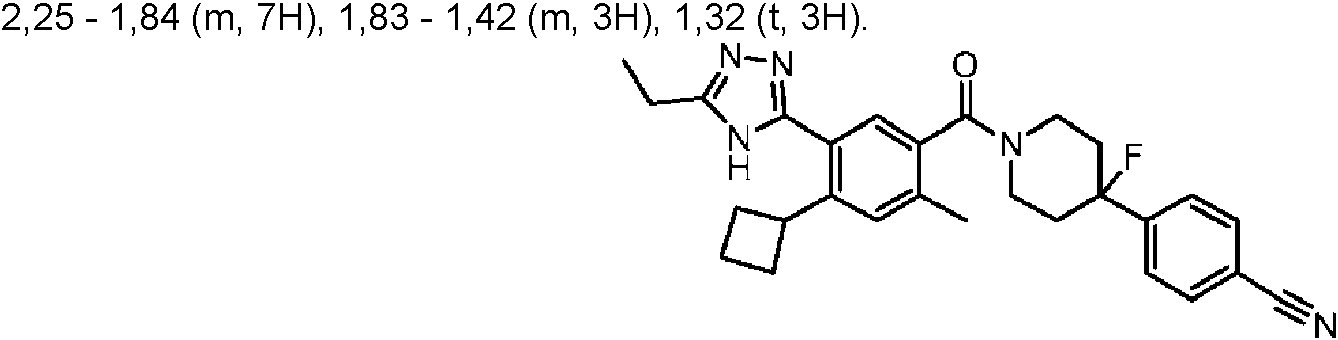
Compuesto 161. 4-(1-(4-CidobutN-5-(5-etN-4H-1,2,4-tnazol-3-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152). m/z (ES+) 472 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 8 7,79 - 7,57 (m, 2H), 7,57 - 7,18 (m, 4H), 4,86 (dd, 1H), 4,14 (s, 1H), 3,62-3,40 (m, 4H), 3,22 (t, 1H), 2,77 (c, 2H), 2,31 y 2,41 (2 singletes, rotámeros de amida, 3H), 2,29 - 1,47 (m, 8H), 1,30 (t, 3H).

Compuesto 162. 4-(1-(4-CidobutN-5-(5-isopropN-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)piperidm-4-N)benzomtrMo.
El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol 3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152). m/z (ES+) 468 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 8 13,67 (s, 1H), 7,77 (d, 2H), 7,49-7,47 (m, 3H), 7,36 (s, 1H), 4,72 (d, 1H), 4,29 (s, 1H), 3,46 (d, 1H), 3,12 (m, 2H), 2,98 - 2,75 (m, 2H), 2,31 (d, 3H), 2,23 -1,81 (m, 6H), 1,83 -1,36 (m, 4H), 1,31 (
Compuesto 163.4-(1-(4-CidobutN-5-(5-isopropN-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol 3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152). m/z (ES+) 486 M+H .

Compuesto 164.1. 4-(1-(4-Cidobutil-5-(5-isopropil-4H-1,2,4-triazol-3-il)-2-metilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.7 y usando propionohidrazida en lugar de acetohidrazida.
Compuesto 164.2. 4-ciclobutil-5-(5-etil-N-metil-4H-1,2,4-triazol-3-il)-2-metilbenzoato de metilo. Se disolvió 4-ciclobutil-5-(5-etil-4H-1,2,4-triazol-3-il)-2-metilbenzoato de metilo (compuesto 164.1, 87 mg, 0,29 mmol) en metanol y diclorometano (1:1 v/v) (6 ml). Se añadió (trimetilsilil)metil)diazometano (2,0 M en éter) (220 ul, 0,44 mmol). La mezcla se agitó durante 16 horas y se inactivó con HOAc (300 ul). Los volátiles se retiraron al vacío para producir un aceite (85 mg). El residuo se llevó impuro a la siguiente etapa sin purificación adicional, m/z (ES+) 314 (M+H)+.
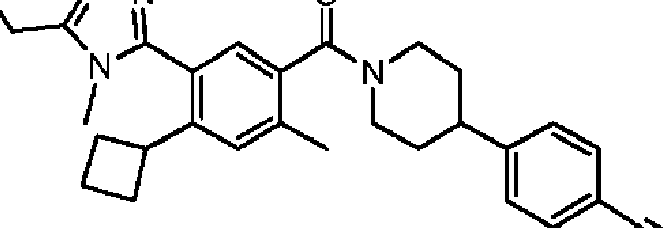
Compuesto 164. 4-(1-(4-CidobutN-5-(5-etN-N-metN-4H-1,2,4-tnazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título (mezcla de isómeros de N-metilo) se preparó usando manipulaciones químicas estándar procedimientos similares a los usados para la preparación del compuesto 152, usando el

Compuesto 165. 4-(1-(4-CidobutN-2-metN-5-(5-(trífluorometM)-4H-1,2,4-tríazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-

Compuesto 166. 4-(1-(4-Ciclobutil-5-(5-(difluorometil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil) piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152). m/z ES+ 476 M+H .

Compuesto 167. 4-(1-(4-Ciclobutil-5-(5-(difluorometil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-[1-[4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil]-4-piperidinil]benzonitrilo (compuesto 152. m/z ES+ 494 M+H .

Compuesto 168.1.4-Ciclobutilbenzoato de metilo. A una mezcla agitada de ZnBr2 (83,0 g, 368,53 mmol, 4,00 equiv) en t Hf (500 ml) en nitrógeno a 0°C, se le añadió una solución de bromo(ciclobutil)magnesio (242 ml, 364 mmol, 1,5 M en THF) gota a gota durante 20 minutos. A la mezcla resultante se le añadieron Pd(dppf)Ch (2,00 g, 0,10 equiv) y 4-bromobenzoato de metilo (20 g, 93,00 mmol, 1,00 equiv) a -40 °C. La mezcla resultante se agitó a -40 °C durante 1 h en nitrógeno, y a continuación se inactivó cuidadosamente con 500 ml de NH4Cl (ac., sat.). La mezcla se extrajo con 3 x 500 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 500 ml de solución saturada de cloruro sódico, a continuación se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 18,0 g del compuesto del título (impuro) como un aceite de color amarillo claro.

Compuesto 168.2. 4-CIclobutil-3-yodobenzoato de metilo. A una solución de 4-ciclobutilbenzoato de metilo (168.1, 2,00 g, 10,5 mmol, 1,00 equiv) en ácido acético (30 ml) se le añadió cuidadosamente peryodato sódico (1,00 g, 4,68 mmol, 0,50 equiv), yodo (3,00 g, 11,8 mmol , 1,10 equiv) y ácido sulfúrico (0,15 g, 0,15 equiv). La mezcla resultante se agitó durante una noche a 100 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición cuidadosa de 30 ml de Na2S2O3 (ac., sat.) y la mezcla resultante se extrajo con 3 x 20 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 20 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 1,50 g (45%) del compuesto del título como un aceite de color amarillo.

Compuesto 168.3. 3-Ciano-4-ciclobutilbenzoato de metilo. El compuesto del título (2,60 g (95%), sólido de color blanco) se preparó usando manipulaciones químicas estándar y un procedimiento similar al usado para la preparación del compuesto 152.4 y usando el compuesto 168.2 (4,00 g, 12,7 mmol) en lugar del compuesto 152.3.

Compuesto 168. 4-(1-(4-Ciclobutil-3-(5-etil-4H-1,2,4-triazol-3-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 152,

Compuesto 169. 4-(1-(4-Ciclobutil-3-(5-etil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 152, usando el compuesto 168.3 en lugar de 152.4. m/z (ES+) 440 (M+H)+, 1H RMN (300 MHz, CD3OD) 67,88 (d, 2H), 7,67-7,50 (m, 3H), 7,47 (d, 2H), 4,89-4,75 (m, 1H), 4,25-3,73 (m, 2H), 3,35-3,25 (m, 1H),3,16-2,75 (m, 4H), 2,30-1,94 (m, 6H), 1,93 1,56 (m, 4H), 1,51-1,32 (m, 3H).

Compuesto 170. 4-(1-(4-Ciclobutil-3-(5-metil-4H-1,2,4-triazol-3-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 152 y usando los compuestos 168.3 y 11.2, sal HCl en lugar de los compuestos 152.4 y 1.5 respectivamente. m/z (ES+) 444 (M+H)+.

Compuesto 171. 4-(1-(4-Ciclobutil-3-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación de los compuestos 152 y 156, y usando 168.3 en lugar de 152.4. m/z (ES+) 488 (M+H)+.

Compuesto 172.1. (S)-Tetrahidrofuran-2-carbohidrazida. A un matraz de fondo redondo, se le añadió una solución de ácido (S)-tetrahidrofuran-2-carboxílico (3,00 g, 25,8 mmol, 1,00 equiv) en diclorometano (40 ml). A la reacción se le añadieron EDCHCl (7,50 g, 39,1 mmol, 1,50 equiv), HOBT (5,20 g, 38,5 mmol, 1,50 equiv), e hidrato de hidrazina (2 ml, 2,00 equiv, 99%). La solución resultante se agitó durante una noche a temperatura ambiente. Los sólidos se retiraron por filtración y el filtrado se concentró al vacío para proporcionar 5,38 g (80 %) del compuesto del título como un aceite de color amarillo.

Compuesto 172. (S)-4-(1-(4-Ciclobutil-2-metil-5-(5-(tetrahidrofuran-2-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152), usando (S)-tetrahidrofuran-2-carbohidrazida (compuesto 172.1) en lugar de acetohidrazida. m/z (ES+) 496 (
Compuesto 173. (S)-4-(1-(4-CidobutN-2-metM-5-(5-(tetrahidrofuran-2-M)-4H-1,2,4-triazol-3-N)benzoM)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 172, usando sal HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 514 (M+H)+.

Compuesto 174.1. (R)-Tetrahidrofuran-2-carbohidrazida. A un matraz de fondo redondo, se le añadió una solución de ácido (R)-tetrahidrofuran-2-carboxílico (3,00 g, 25,8 mmol, 1,00 equiv) en diclorometano (60 ml). A la reacción se le añadieron EDC HCl (7,37 g, 38,5 mmol, 1,50 equiv), HOBt (5,24 g, 38,8 mmol, 1,50 equiv), e hidrato de hidrazina (2,60 g, 51,9 mmol, 2,00 equiv). La solución resultante se agitó durante una noche a 25 °C. Los sólidos se retiraron con filtración. El filtrado se concentró al vacío para dar 2,00 g (59 %) de (R)-tetrahidrofuran-2-carbohidrazida como un aceite de color amarillo.

Compuesto 174. (R)-4-(1-(4-Ciclobutil-2-metil-5-(5-(tetrahidrofuran-2-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 152), usando (R)-tetrahidrofuran-2-carbohidrazida (compuesto 174.1) en lugar de acetohidrazida. m/z (ES+) 496 (M+H)+. 1H RMN (300 MHz, CD3OD) 67,70 (d, 2H), 7,49-7,41 (m, 4H), 5,15 (t, 1H), 4,89-4,80 (m, 1H), 4,14-3,92 (m, 3H), 3,65-3,51 (m, 1H), 3,33-3,27 (m, 1H), 3,03-2,95 (m, 2H), 2,47-2,37 (m, 4H), 2,24 1,91 (m, 9H), 1,83-1,71 (m, 4H).

Compuesto 175.1. Acido 4-ciclopentil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoico. El compuesto del título se sintetizó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.8 y usando bromo(ciclopentil)magnesio en lugar de bromo(ciclobutil)magnesio.

Compuesto 175. 4-(1-(4-Ciclopentil-2-metil-5-(5-metil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se sintetizó usando un procedimiento similar al usado para la preparación del compuesto 152 y usando el compuesto 175.1 en lu ar del compuesto 152.8. miz ES+ 495 M+H .

Compuesto 176.1. 4-(4-Bromofenil)-4-hidroxi-2-metilpiperidin-1-carboxilato de íerc-butilo. El compuesto del título se sintetizó un usando procedimiento similar al usado para la preparación del compuesto 1.1 y usando 2-metil-4-oxop¡per¡d¡n-1-carbox¡lato de tere-butilo en lugar de 4-oxopiper¡d¡n-1-carboxilato de tere-butilo.
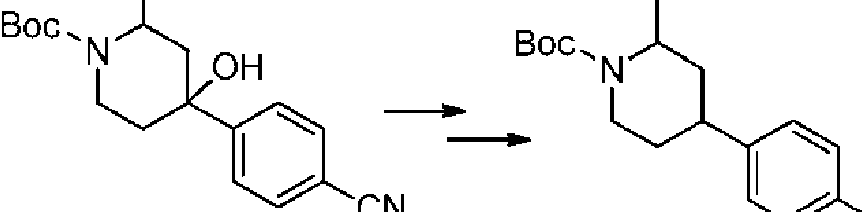
Compuesto 176.2. 4-(4-Cianofenil)-2-metilpiperidin-1-carboxilato de íerc-butilo. El compuesto del título se sintetizó usando procedimientos similares a los usados para la preparación del compuesto 1.4 y usando el compuesto 176.1 en lugar del compuesto 1.1.

Compuesto 176.3. 4-(2-Metilpiperidin-4-il)benzonitrilo. A una solución de 4-(4-cianofenil)-2-metil-piperidin-1-carboxilato de terc-butilo (compuesto 176.2, 500 mg, 1,50 mmol, 1,00 equiv, 90 %) en diclorometano (3 ml) se le añadió TFA gota a gota (1 ml). La mezcla resultante se agitó durante 1,5 h a temperatura ambiente, a continuación se diluyó con 30 ml de diclorometano. La mezcla resultante se lavó con bicarbonato sódico (ac., 1 M. Nota: desprendimiento de gas significativo). La fase acuosa se extrajo con 2 x 50 ml de diclorometano y las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con metanol/diclorometano (1:50-1:20) como eluyente para dar 280 mg (93%) de 4-(2-metilpiperidin-4-il)benzonitrilo como un aceite incoloro.

Compuesto 176.4. 4-(4-Cianofenil)-2-metilpiperidin-1-carboxilato de íerc-butilo. El compuesto del título se sintetizó usando un procedimiento similar al usado para la preparación del compuesto 152,8 y usando el compuesto

Compuesto 176. 4-(1-(4-CidobutN-5-(5-etN-4H-1,2,4-tríazol-3-M)-2-metNbenzoM)-2-metMpiperidm-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 4-(2-metilpiperidin-4-il)benzonitrilo (compuesto 176.3, 210 mg, 0,940 mmol, 1,00 equiv, 90%) en W,A/-dimetilformamida (2 ml). Se añadieron EDC ■ HCl (404 mg, 2,11 mmol, 2,00 equiv), 4-dimetilaminopiridina (257 mg, 2,10 mmol, 2,00 equiv), y ácido 4-ciclobutil-5-(5-etil-4H-1,2,4-triazol 3-il)-2-metilbenzoico (compuesto 176.4 ,300 mg, 1,05 mmol, 1,00 equiv) a la mezcla de
reacción. La solución resultante se agitó durante 4 h a 25 °C, a continuación se diluyó con 50 ml de acetato de etilo. La mezcla resultante se lavó con 2 x 30 ml de NH4Cl (ac., sat.) y 2 x 30 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (20/1) como eluyente. El producto impuro (~20 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)):
Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (48 % CH3CN asciende al 49 % en 8 minutos, asciende al 100 % en 3 minutos, desciende al 48 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 3,8 mg (1 %) del compuesto del título como un sólido de color blanco, m/z (ES+) 468 (M+H)+.
O
[ r V 'O H h . Naio, ^
acoh, h 2 so 4 
Compuesto 177.1. Ácido 4-cloro-5-yodo-2-metilbenzoico. A un matraz de fondo redondo, se le añadió una solución de ácido 4-cloro-2-metilbenzoico (30,0 g, 176 mmol, 1,00 equiv) en ácido acético (300 ml). A la reacción se le añadieron NaIO4 (19,0 g, 88,8 mmol, 0,50 equiv), I2 (49,0 g, 193 mmol, 1,10 equiv), y ácido sulfúrico (3 ml). La mezcla resultante se agitó durante una noche a 110 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente con 500 ml de Na2S2O3 (ac., sat.). Los sólidos resultantes se recogieron por filtración y a continuación se disolvieron en 500 ml de acetato de etilo. La fase orgánica se lavó con 2 x 200 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. Esto dio como resultado 20,0 g (38 %) de ácido 4-cloro-5-yodo-2-metilbenzoico como un sólido de color blanco.

Compuesto 177.2. 4-Cloro-5-yodo-2-metilbenzoato de metilo. A una solución de ácido 4-cloro-5-yodo-2-metilbenzoico (compuesto 177,1, 20,0 g, 67,5 mmol, 1,00 equiv) en metanol (100 ml) se le añadió ácido sulfúrico (5 ml) gota a gota. La mezcla resultante se agitó durante una noche a 75 °C. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El valor de pH de la capa acuosa restante se ajustó cuidadosamente a 7 con bicarbonato sódico (ac., 1 M. Nota: desprendimiento de gas significativo). La fase acuosa se extrajo con 2 x 200 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2 x 100 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) como eluyente para proporcionar 20,0 g (95 %) de 4-cloro-5-yodo-2-metilbenzoato de metilo como un líquido de color amarillo claro.

Compuesto 177.3. Ácido 4-cloro-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.8 pero usando los compuestos 177.2 y 143.1 en lugar del compuesto 152.3 y acetohidrazida, respectivamente.

Compuesto 177. 4-(1-(4-Cloro-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2 metilbenzoil)piperidin-4-il)benzonitrilo.
El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152, usando el compuesto 177.3 en lugar del compuesto 152.8. m/z (ES+) 464 (M+H)+.

Compuesto 178.1. 2-Bromo-4-clorobenzoato. El compuesto del título (17,0 g, sólido de color amarillo claro, 80%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 177.2 pero usando ácido 2-bromo-4-clorobenzoico (20,0 g) en lugar del compuesto 177.1.

Compuesto 178.2. 4-Cloro-2-etilbenzoato de metilo. El compuesto del título (2,20 g, líquido incoloro claro, 55 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 48.1 pero usando el compuesto

Compuesto 178.3. Ácido 4-cloro-2-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.8 usando el compuesto 178.2 y 3-metoxipropanihidrazida (compuesto 143.1) en lugar del compuesto 152.2 y acetohidrazida, respectivamente.

Compuesto 178.4-(1-(4-Cloro-2-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando un procedimiento similar al usado para la preparación del compuesto 152,

Compuesto 179. 4-(1-(4-Cloro-2-etN-5-(5-(tetrahidrofuran-3-M)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 38 178. m/z ES+ 490 (M+H)+.

Compuesto 180.1. 4-Bromo-2-fluorobenzoato de metilo. A un matraz de fondo redondo de tres bocas de 500 mi, se le añadió una solución de ácido 4-bromo-2-fluorobenzoico (30,0 g, 137 mmol, 1,00 equiv) en metanol (200 ml). Se añadió SOCh (24,0 g, 202 mmol, 1,50 equiv) gota a gota a 0°C. La solución resultante se agitó durante 10 minutos a 0°C, durante 30 minutos a 25 °C, y a continuación durante 3 horas a 50 °C. Después de enfriar a temperatura ambiente, la mezcla resultante se concentró a presión reducida. El residuo se repartió entre agua (100 ml) y acetato de etilo (100 ml). La fase acuosa se extrajo con 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1 x 100 ml de agua, 1 x 100 ml de bicarbonato sódico (ac. sat.) y 1 x 100 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 30,0 g (94 %) del compuesto del título como un sólido de color amarillo claro.
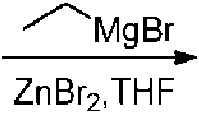


Compuesto 180.2. 4-Etil-2-fluorobenzoato de metilo. El compuesto del título (3,80 g, aceite incoloro claro, 97 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 48.1 pero usando el compuesto 180.1 (5,00 g) en lugar de 2-bromo-4-metilbenzoato de metilo.

Compuesto 180.3. Ácido 4-etil-2-fluoro-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.8 usando los compuestos 180.2 y 143.1 en lugar del compuesto 152.2 y acetohidrazida, respectivamente.

Compuesto 180. 4-(1-(4-Etil-2-fluoro-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo.
El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152, pero usando el compuesto 180.3 en lugar del compuesto 152.8. m/z (ES+) 462 (M+H)+,

Compuesto 181.1. Ácido 4-bromo-2-etilbenzoico. A un matraz de fondo redondo de tres bocas de 1 I, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió ácido 4-bromo-2-fluorobenzoico (50,0 g, 228 mmol, 1,00 equiv) en tetrahidrofurano (500 ml). Se añadió una solución de bromuro de etilmagnesio (250 ml, 3 M en THF) gota a gota a 0 °C. La solución resultante se agitó durante 3-4 h a 0 °C. La mezcla se inactivó a continuación cuidadosamente por adición gota a gota de agua a 0 °C. Después de la inactivación completa de la reacción, se añadió agua adicional y el pH se ajustó a 2-3 con cloruro de hidrógeno (acuoso, 2 M). La mezcla se extrajo con 2 x 200 ml de acetato de etilo y las capas orgánicas se combinaron. Se empleó hidróxido sódico (2 N, ac.) Para ajustar el pH a 7-8. La mezcla resultante se lavó con 2 x 200 ml de acetato de etilo. El valor de pH de la solución acuosa se ajustó a 2-3 con cloruro de hidrógeno 2 N, a continuación se extrajo con 2 x 200 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 30,0 g (57%) de ácido 4-bromo-2-etilbenzoico como un sólido de color amarillo.

Compuesto 181.2. 4-Bromo-2-etilbenzoato de metilo. El compuesto del título (25,0 g, líquido amarillo claro, 79%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 177.2 pero usando el

Compuesto 181.3. 4-Ciclobutil-2-etilbenzoato de metilo. A una mezcla agitada de ZnBr2 (33,5 g, 149 mmol, 4,00 equiv) en THF (350 ml) en nitrógeno a 0 °C se le añadió gota a gota una solución de bromuro de ciclobutilmagnesio (148 mmol en 50 ml de THF). Después de agitar a 0 °C durante 0,5 h, la temperatura se redujo a -78 °C y Pd(dppf)Ch (2,00 g, 2,73 mmol, 0,07 equiv) seguido de la adición de una solución de 4-bromo-2-benzoato de metilo (compuesto 181.2, 9,00 g, 37,0 mmol, 1,00 equiv) en tetrahidrofurano (10 ml) gota a gota a la misma temperatura. A la mezcla de reacción se le dejó alcanzar lentamente temperatura ambiente y a continuación se agitó durante una noche. La reacción se inactivó cuidadosamente con una solución acuosa de NH4Cl saturado (100 ml). La mezcla resultante se extrajo con 500 ml de acetato de etilo. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) para proporcionar 8,00 g (99 %) del producto deseado como un aceite de color amarillo claro.
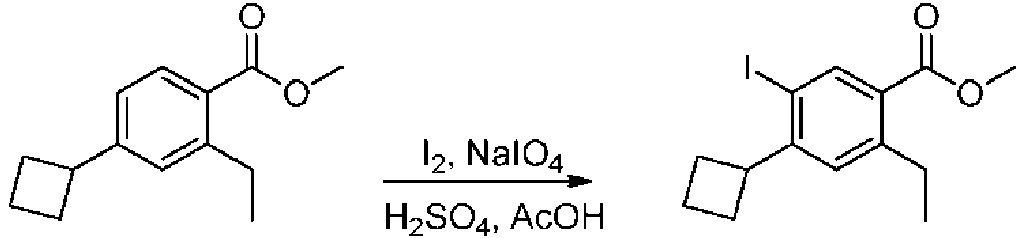
Compuesto 181.4. 4-Ciclobutil-2-etil-5-yodobenzoato de metilo. A una solución de 4-ciclobutil-2-etilbenzoato de metilo (compuesto 181.3, 7,70 g, 35,3 mmol, 1,00 equiv,) en ácido acético (80 ml) se le añadieron yodo (8,98 g, 35,4 mmol, 1,00 equiv), NaIO4 (3,78 g, 17,7 mmol, 0,50 equiv), y ácido sulfúrico (0,870 g, 8,87 mmol, 0,25 equiv). La mezcla de reacción se agitó a 60 °C durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó lentamente con Na2S2O3 (ac., sat.). La mezcla se extrajo con 200 ml de acetato de etilo y la capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) para proporcionar 10,5 g (86%) del producto deseado como un sólido incoloro.

Compuesto 181.5. 5-Ciano-4-ciclobutil-2-etilbenzoato de metilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió una solución de 4-ciclobutil-2-etil-5-yodobenzoato de metilo (compuesto 181.4, 5,50 g, 16,0 mmol, 1,00 equiv) en W,A/-dimetilformamida (150 ml). Se añadieron cianuro de zinc (2,78 g, 23,7 mmol, 1,50 equiv) y Pd(PPh3)4 (1,83 g, 1,59 mmol, 0,10 equiv) a la mezcla de reacción. La solución resultante se agitó a 100 °C durante 15 h en nitrógeno. Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente con 300 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 300 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 300 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:50) como eluyente para proporcionar 3,20 g (82%) de 5-ciano-4-ciclobutil-2-etilbenzoato de metilo como un aceite de color amarillo claro.

Compuesto 181.6. 5-Carbamotioil-4-ciclobutil-2-etilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-ciano-4-ciclobutil-2-etilbenzoato de metilo (compuesto 181.5, 3,00 g, 12,3 mmol, 1,00 equiv) en una mezcla disolvente de tetrahidrofurano y H2O (80ml/40 ml). A esto se le añadió ditiofosfato de O,O'-dietilo (6,69 g, 35,9 mmol, 3,00 equiv) gota a gota con agitación. La solución resultante se agitó a 85 °C durante 48 h (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El producto impuro se purificó por recristalización a partir de éter de petróleo para proporcionar 1,30 g (38 %) de 5-carbamotioil-4-ciclobutil-2-etilbenzoato de metilo como un sólido de color amarillo claro.

Compuesto 181.7. 4-Ciclobutil-2-etil-5-(imino(metiltio)metil)benzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-carbamotioil-4-ciclobutil-2-etilbenzoato de metilo (compuesto 181.6, 1,50 g, 5,41 mmol, 1,00 equiv) en tetrahidrofurano (30 ml). A esto le siguió la adición de yodometano (3,80 g, 26,8 mmol, 5,00 equiv) gota a gota con agitación. La solución resultante se agitó a 25 °C durante 15 h, a continuación se concentró al vacío. Esto dio como resultado 1,80 g (97 %) del compuesto del título como un aceite de color amarillo.

Compuesto 181.8. 4-Ciclobutil-2-etil-5-(5-etil-4H-1,2,4-triazol-3-il)benzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 4-ciclobutil-2-etil-5-(metilsulfamil)carboximidoilbenzoato de metilo (compuesto
181.7, 900 mg, 3,09 mmol, 1,00 equiv) en AcOH (20 ml). Se añadió propionohidrazida (880 mg, 9,99 mmol, 3,00 equiv) y la mezcla resultante se agitó a 90 °C durante 2 h. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50-1:3) como eluyente para dar 0,360 g (37 %) del compuesto como un aceite transparente.

Compuesto 181.9. Acido 4-ciclobutil-2-etil-5-(5-etil-4H-1,2,4-triazol-3-il)benzoico. A un matraz de fondo redondo, se le añadió una solución de 4-ciclobutil-2-etil-5-(5-etil-4H-1,2,4-triazol-3-il)benzoato de metilo (compuesto 181.8, 360 mg, 1,15 mmol, 1,00 equiv) en metanol (20 ml). A la mezcla de reacción se le añadió una solución de hidróxido sódico (460 mg, 11,5 mmol, 10,0 equiv) en agua (10 ml). La solución resultante se agitó a 25 °C durante 15 h. A continuación, el disolvente orgánico se retiró a presión reducida. El valor del pH de la fase acuosa restante se ajustó a 2-3 con cloruro de hidrógeno (ac., 2 M). El precipitado resultante se recogió por filtración y se secó a alto vacío para dar 320

Compuesto 181. 4-(1-(4-Ciclobutil-2-etil-5-(5-etil-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. A una solución de ácido 4-ciclobutil-2-etil-5-(5-etil-4H-1,2,4-triazol-3-il)benzoico (compuesto 181.9, 160 mg, 0,530 mmol, 1,00 equiv) en W,W-dimetilformamida (20 ml), se le añadieron EDCI (113 mg, 0,590 mmol, 1,11 equiv), DMAP (197 mg, 1,62 mmol, 3,03 equiv), y HOBT (87,5 mg, 0,650 mmol, 1,21 equiv). Después de 5 minutos, se añadió clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 110 mg, 0,590 mmol, 1,10 equiv). La mezcla resultante se agitó a 25 °C durante 15 h, a continuación se inactivó con 100 ml de agua con hielo. La mezcla se extrajo con 2x150 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3x150 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron. El producto impuro (~150 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (47 % CH3CN asciende al 61 % en 6 minutos, asciende al 100 % en 1,5 minutos, desciende al 47 % en 1,5 minutos); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 99,5 mg (40 %) del compuesto como un sólido de color blanco. m/z (ES+) 468 (M+H)+. 1H-RMN (300 MHz, CD3OD): 67,71-7,69 (m, 2H), 7,50-7,32 (m, 4H), 4,03-3,99 (m, 1H), 3,65-3,5 (m, 1H), 3,32-3,20 (m, 1H), 2,90-2,95 (m, 4H), 2,70-2,74 (m, 2H), 2,20-1,98 (m, 6H), 1,98-1,79 (m, 4H), 1,41 (t, 3H), 1,39-1,28 (m, 3H).

Compuesto 182. 4-(1-(4-Ciclopropil-2-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 143 y 181. m/z (ES+) 484 (M+H)+. 1H-RMN (300 MHz, CD3OD): 67,67 (d, 2H), 7,49-7,37 (m, 3H), 7,11-7,05 (m, 1H), 4,89-4,80 (m, 1H), 3,81 (t, 2H), 3,77-3,60 (m, 1H), 3,4 (s, 3H), 3,37-3,32 (m, 1H), 3,14 (t, 2H), 3,15-2,9 (m, 2H), 2,92-2,5 (m, 2H), 2,39-2,36 (m, 1H), 2,02-1,85 (m, 1H), 1,85-1,69 (m, 3H), 1,32-1,21 (m, 3H), 1,0-0,95 (m, 2H), 0,77-0,69 (m, 2H).

Compuesto 183. 4-(1-(4-CiclopropN-2-etN-5-(5-(metoximetM)-4H-1,2,4-tríazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 143 y 181. m/z (ES+) 470 (M+H)+.

Compuesto 184. 4-(1-(4-CidobutN-2-etN-5-(5-(metoximetM)-4H-1,2,4-triazol-3-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 143 y 181 y usando sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+ 502 M+H .
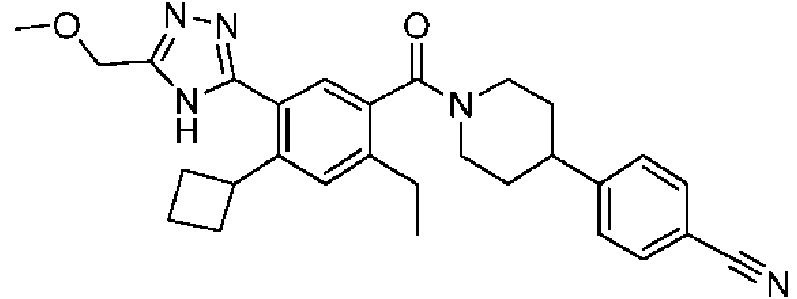
Compuesto 185. 4-(1-(4-CidobutN-2-etN-5-(5-(metoximetM)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 143 y 181. m/z (ES+) 484 (M+H)+.

Compuesto 186. 4-(1-(4-CidobutN-2-etN-5-(5-etN-4H-1,2,4-triazol-3-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 181 y usando sal HCl del compuesto 11.2 en lugar del
Compuesto 189.1. 4-(Ciclobutilmetil)-2-metilbenzoato de metilo. El compuesto del título (4,00 g, aceite de color amarillo, 84%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.3 pero usando el compuesto 152.1 (5,00 g) y bromuro de ciclobutilmetilmagnesio en lugar del compuesto 181.2 y bromuro de ciclobutilmagnesio, respectivamente.

Compuesto 189.2. 4-(Ciclobutilmetil)-5-yodo-2-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 4-(ciclobutilmetil)-2-metilbenzoato de metilo (compuesto 189.1, 8,40 g, 38,5 mmol, 1,00 equiv) en AcOH (150 ml). A la mezcla de reacción se le añadieron NaIO4 (5,00 g, 23,4 mmol, 0,50 equiv), yodo (10,0 g, 39,4 mmol, 1,00 equiv), y ácido sulfúrico (0,3 ml). La solución resultante se agitó a 60 °C durante 12 h. Después de enfriar a temperatura ambiente, la reacción se inactivó con 200 ml de NaHSO3 (ac). La mezcla se extrajo con 2 x 200 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (20/1) como eluyente para proporcionar 8,00 g (60%) de de 4-(ciclobutilmetil)-5-yodo-2-metilbenzoato de metilo como un sólido de color blanco.

Compuesto 189.3. 5-ciano-4-(ciclobutilmetil)-2-metilbenzoato de metilo. El compuesto del título (5,0 g, aceite de color amarillo claro, 88%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.5 usando el compuesto 189.2 (8,00 g) en lugar del compuesto 181.4.

Compuesto 189.4. 5-carbamotioil-4-(ciclobutilmetil)-2-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-ciano-4-(ciclobutilmetil)-2-metilbenzoato de metilo (compuesto 189.3, 5,00 g, 20,6 mmol, 1,00 equiv) en una mezcla disolvente de tetrahidrofurano y H2O (50 ml/25 ml). Al matraz de reacción se le añadió ditiofosfato de O,O'-dietilo (15,0 g, 80,5 mmol, 4,00 equiv). La solución resultante se agitó a 80 °C durante 48 h (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente con 50 ml de solución saturada de cloruro sódico. La mezcla se extrajo con 3 x 50 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:4) como eluyente para proporcionar 1,20 g (21 %) del compuesto del título como un aceite de color amarillo
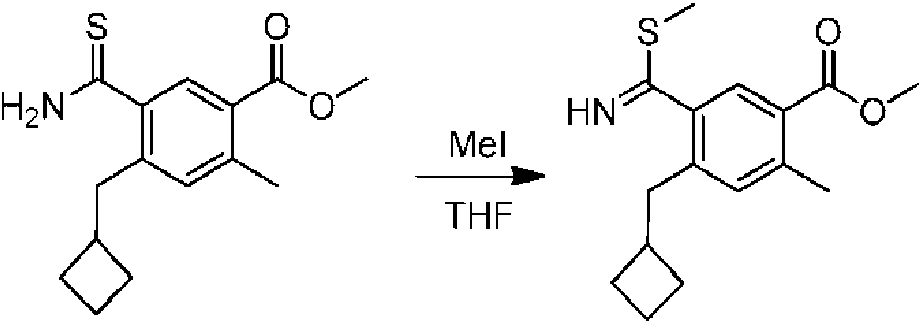
Compuesto 189.5. 4-(Ciclobutilmetil)-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-carbamotioil-4-(ciclobutilmetil)-2-metilbenzoato de metilo (compuesto 189.4, 1,20 g, 4,33 mmol, 1,00 equiv) en tetrahidrofurano (25 ml). A la mezcla de reacción se le añadió yodometano (5,00 g, 35,2 mmol, 8,00 equiv). La solución resultante se agitó a 20 °C durante 12 h y a continuación se concentró al vacío. Esto dio como resultado 1,10 g (87 %) de 4-(ciclobutilmetil)-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo como un aceite de color amarillo.

Compuesto 189.6. 4-(Ciclobutilmetil)-5-(5-etil-4H-1,2,4-triazol-3-il)-2-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 4-(cidobutilmetil)-2-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo (compuesto 189.5, 1,00 g, 3,43 mmol, 1,00 equiv) en AcOH (25 ml). Se añadió propionohidrazida (1,20 g, 13,6 mmol, 4,00 equiv) y la mezcla resultante se agitó a 100 °C durante 1 h. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para proporcionar 300 mg (28%) del compuesto del título como un sólido de color blanco

Compuesto 189.7. Ácido 4-(ciclobutNmetM)-5-(5-etN-4H-1,2,4-triazol-3-M)-2-metNbenzoico. El compuesto del título (260 mg, sólido de color blanco, 91 %) se preparó usando un procedimiento similar al usado para la preparación del

Compuesto 189. 4-(1-(4-(CiclobutNmetM)-5-(5-etN-4H-1,2,4-triazol-3-M)-2-metNbenzoM)piperidm-4-N)benzomtrMo.
El compuesto del título (28 mg, sólido de color blanco, 18%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181 usando el compuesto 189.7 (100 mg) en lugar del compuesto 181.9. m/z (ES+) 468 (M+H)+. 1H-RMN (300 MHz, CD3OD): 67,67 (d, 2H), 7,52-7,47 (m, 3H), 7,26 (s, 1H), 3,66-3,22 (m, 1H), 3,33-3,22 (m, 1H), 3,05-2,99 (m, 4H), 2,97 (c, 2H), 2,53-2,47 (m, 1H), 2,47 y 2,45 (2 singletes, rotámeros de amida, ArCH3, 3H) 2,00-1,50 (m, 10H), 1,41 (t, 3H).

Compuesto 190.1. 2-Bromo-5-yodo-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una mezcla de 2-bromo-4-metilbenzoato de metilo (2,00 g, 7,86 mmol, 1,00 equiv) en AcOH (20 ml). Se añadieron I2 (2,45 g, 9,65 mmol, 1,10 equiv), NaIO4 (950 mg, 4,42 mmol, 0,50 equiv), y ácido sulfúrico (0,1 ml, 0,15 equiv) y la mezcla resultante se agitó a 100 °C durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente con Na2S2O3 (ac., sat.). La mezcla resultante se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío para dar 2,50 g (81 %) del compuesto del título como un sólido de color marrón.

Compuesto 190.2. 2-Bromo-5-ciano-4-metilbenzoato de metilo. El compuesto del título (1,10 g, sólido de color blanco, 61 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181,5 usando el compuesto 190.1 (2,50 g) en lu ar del compuesto 181.4.
Compuesto 190.3. 5-Ciano-2-ciclopropil-4-metilbenzoato de metilo. A una solución de 2-bromo-5-ciano-4-metilbenzoato de metilo (compuesto 190.2, 600 mg, 2,13 mmol, 1,00 equiv, 90%) en tolueno (20 ml) en atmósfera de nitrógeno se le añadieron ácido ciclopropilborónico (552 mg, 6,43 mmol , 2,00 equiv), una solución de carbonato potásico (876 mg, 6,34 mmol, 2,00 equiv) en agua (1 ml), Pd(dppf)Ch (252 mg, 0,10 equiv), y Pd(OAc)2 (70 mg , 0,10 equiv). La mezcla resultante se agitó en nitrógeno a 100 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 20 ml de H2O, a continuación se extrajo con 3 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío para proporcionar 450 mg (89 %) de 5-ciano-2-ciclopropil-4-met¡lbenzoato de metilo como un sólido de color blanco.

Compuesto 190.4. 5-Carbamotioil-2-ciclopropil-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-ciano-2-ciclopropil-4-metilbenzoato de metilo (compuesto 190.3, 220 mg, 0,920 mmol, 1,00 equiv, 90%) en tetrahidrofurano (6 ml). A la solución se le añadió una solución de ditiofosfato de O,O'-dietilo (300 mg, 1,61 mmol, 2,00 equiv) en agua (1,5 ml) y la mezcla resultante se agitó durante una noche a 85 °C en un baño de aceite (ADVERTENCIA: se produce un desprendimiento de gas significativo - esta y todas las otras reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (5:1) como eluyente. Las fracciones recogidas se combinaron y se concentraron al vacío para proporcionar 100 mg (39 %) de 5-carbamotioil-2-ciclopropil-4-metilbenzoato de metilo como un sólido de color amarillo.

Compuesto 190.5. 2-Ciclopropil-4-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo. A una solución de 5-carbamotioil-2-ciclopropil-4-metilbenzoato de metilo (compuesto 190.4, 600 mg, 2,17 mmol, 1,00 equiv, 90 %) en THF (55 ml) se le añadió yodometano gota a gota (1 ml). Las mezcla resultante se agitó durante una noche a 25 °C, a continuación se concentró y se secó a presión reducida para dar 400 mg (56 %) del compuesto del título como un sólido de color amarillo.

Compuesto 190.6. 2-Metoxiacetohidrazida. A un matraz de fondo redondo, se le añadió una solución de 2-metoxiacetato de etilo (10,0 g, 76,2 mmol, 1,00 equiv, 90 %) y NH2NH2 H2O (12 ml, 3,00 equiv) en etanol (100 ml). La solución resultante se agitó durante 3 h a 80 °C en un baño de aceite, a continuación se concentró y se secó a presión reducida para dar 6 g (68 %) de 2-metoxiacetohidrazida como un sólido de color blanco.

Compuesto 190.7.2-Ciclopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoato de metilo. Una solución de 2-ciclopropil-4-metil-5-(metilsulfanil)carboximidoilbenzoato de metilo (compuesto 190.5, 400 mg, 1,37 mmol, 1,00 equiv, 90 %) y 2-metoxiacetohidrazida (compuesto 190.6, 889 mg, 7,69 mmol, 5,00 equiv) en AcOH (25 ml) se agitó durante una noche a 90 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (2:1) como eluyente. Las fracciones recogidas se combinaron y se concentraron al vacío para proporcionar 200 mg (44 %) de 2-ciclopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il -4-metilbenzoato de metilo como un sólido de color blanco.

Compuesto 190.8. Ácido 2-ciclopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoico. A un matraz de fondo redondo, se le añadió una solución de 2-ciclopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoato de metilo (compuesto 190.7, 200 mg, 0,600 mmol, 1,00 equiv, 90 %) en metanol (4 ml). A la mezcla de reacción se le añadió una solución de hidróxido sódico (106 mg, 2,65 mmol, 4,00 equiv) en agua (2 ml). La solución resultante se agitó durante 2 h a 60 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión
reducida y el pH de la capa acuosa restante se ajustó a 2-3 con cloruro de hidrógeno (ac., 6 M). Los sólidos resultantes se recogieron por filtración y se secaron para dar 170 mg (89%) del compuesto del título como un sólido de color blanco

Compuesto 190. 4-(1-(2-CidopropN-5-(5-(metoximetM)-4H-1,2,4-triazol-3-M)-4-metNbenzoN)piperidm-4-il)benzonitrilo. El compuesto del título (115,5 mg, sólido de color blanco, 51 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181 usando el compuesto 190.8 (15o mg) en lugar del compuesto 181.9. m lz {ES+) 456 (M+H)+.

Compuesto 191.1. Ácido 2-etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-4-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del

Compuesto 191. 4-(1-(2-Etil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título (74,8 mg, sólido de color blanco, 33 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181, pero usando el compuesto 191.1 (145 mg) en lugar del compuesto 181.9. m/z (ES+) 458 (M+H)+.

Compuesto 192. 4-(1-(2-Etil-4-metil-5-(5-(tetrahidrofuran-3-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 191. m/z (ES+) 470 (M+H)+.

Compuesto 193. 4-(1-(2-EtN-5-(5-(l-metoxipropan-2-M)-4H-1,2,4-triazol-3-M)-4-metNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 191. m/z (ES+) 472 (M+H)+.

Compuesto 194. 4-(I-(2-Etil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 191. m/z (Es ) 444 (M+H)+.

Compuesto 195. 4-(1-(2-CidobutM-5-(5-(metoximetM)-4H-1,2,4-triazol-3-M)-4-metMbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 190. m/z ES+ 470 M+H .


Compuesto 196.1.2-Isopropil-4-metilbenzoato de metilo. A una mezcla agitada de dibromozinc (39,0 g, 173 mmol, 4,00 equiv) en THF (500 ml) en nitrógeno a -48 °C, se le añadió gota a gota bromuro de isopropilmagnesio (3 M en THF, 170 mmol) durante un período de 30 minutos. A la mezcla de reacción anterior se le añadieron Pd(dppf)Ch (0,5 g, 0,05 equiv) y 2-bromo-4-metilbenzoato de metilo (10,0 g, 43,7 mmol, 1,00 equiv). Después de agitar durante 1 hora a -48 °C, la mezcla de reacción se inactivó mediante la adición cuidadosa de 500 ml de NH4Cl (ac.). La mezcla resultante se extrajo con 3 x 500 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 500 ml de solución saturada de cloruro sódico, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con éter de petróleo (1:1) como eluyente. Esto dio como resultado 6,50 g (77%) de 2-isopropil-4-metilbenzoato de metilo como un aceite de color amarillo.

Compuesto 196.2. 5-Yodo-2-isopropil-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 2-isopropil-4-metilbenzoato de metilo (compuesto 196.1, 6,50 g, 33,8 mmol, 1,00 equiv) en ácido acético (60 ml). A la mezcla de reacción se le añadieron yoduro (9,50 g, 37,4 mmol, 1,10 equiv), peryodato sódico (3,60 g, 16,8 mmol, 0,50 equiv), y ácido sulfúrico (0,500 g, 0,15 equiv). La solución resultante se agitó durante una noche a 100 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó con Na2S2O3 (ac., sat.). La mezcla se extrajo con 3 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 100 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 6,00 g (56%) de 5-yodo-2-isopropil-4-metilbenzoato de metilo como un aceite de color amarillo.


Compuesto 196.3. 5-Ciano-2-isopropil-4-metilbenzoato de metilo. El compuesto del título (0,8 g, sólido de color blanco, 37 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.5 usando el compuesto 196.2 (3,20 g en lu ar del compuesto 181.4.

Compuesto 196.4. 5-Carbamotioil-2-isopropil-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 5-ciano-2-isopropil-4-metilbenzoato de metilo (compuesto 196.3, 800 mg, 3,68 mmol, 1,00 equiv) en tetrahidrofurano/H2O (10/5 ml). A la mezcla de reacción se le añadió ditiofosfato de O,O'-dietilo (2,05 g, 11,0 mmol, 3,00 equiv). La solución resultante se agitó durante 15 h a 85 °C en un baño de aceite (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Después de enfriar a temperatura ambiente, la mezcla se extrajo con 2 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:50-1:10) como eluyente para proporcionar 0,800 g (86 %) de 5-carbamotioil-2-isopropil-4-metilbenzoato de metilo como un aceite de color amarillo.

Compuesto 196.5. 5-(Imino(metiltio)metil)-2-isopropil-4-metilbenzoato de metilo. El compuesto del título (0,800 g, aceite de color amarillo, 92%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.7 usando el compuesto 196.4 (820 mg) en lugar del compuesto 181.6.

Compuesto 196.6. 2-Isopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoato de metilo. Una solución de 5-(imino(metiltio)metil)-2-isopropil-4-metilbenzoato de metilo (compuesto 196.5, 800 mg, 3,01 mmol, 1,00 equiv) y 2-metoxiacetohidrazida (compuesto 190.6, 1,90 g, 18,3 mmol, 5,00 equiv) en ácido acético (30 ml) se agitó durante 2 horas a 80 °C en un baño de aceite, a continuación se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50-1:2) como eluyente para dar 250 mg (27 %) de 2-¡sopropil-5-(5-(metox¡met¡l 4H 1,2,4-triazol-3-¡l -4-metilbenzoato de metilo como un aceite transparente.

Compuesto 196.7. Ácido 2-isopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoico. El compuesto del título (0,20 g, sólido de color blanco, 84%) se preparó usando un procedimiento similar al usado para la preparación

Compuesto 196. 4-(1-(2-IsopropN-5-(5-(metoximetM)-4H-1,2,4-triazol-3-M)-4-metNbenzoN)piperidm-4-il)benzonitrilo y Compuesto 197. 4-(1-(5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metil-2-propilbenzoil)piperidin-4-il)benzonitrilo. A una solución de ácido 2-isopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-4-metilbenzoico (compuesto 196.7,200 mg, 0,690 mmol, 1,00 equiv) en DMF (20 ml), se le añadieron EDCI (200 mg, 1,04 mmol, 1,51 equiv), DMAP (250 mg, 2,05 mmol, 2,96 equiv), y HOBT (110 mg, 0,810 mmol, 1,18 equiv). Después de agitar durante 30 minutos a temperatura ambiente, se añadió el compuesto 1.5, 142 mg, 0,640 mmol, 0,92 equiv y la solución resultante se agitó durante 15 h a 25 °C, a continuación se inactivó con 100 ml de agua con hielo. La mezcla resultante se extrajo con 2 x 200 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 100 ml de solución saturada de cloruro sódico, a continuación se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50-1:1 -1:0) como eluyente. El producto impuro (~120 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (40,0 % CH3CN asciende al 55,0 % en 8 minutos, asciende al 100,0 % en 1 minuto, desciende al 40,0 % en 1 minuto); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 57,4 mg (18 %) del compuesto 196 y 11,7 mg (4 %) del compuesto 197 como sólidos de color blanco. Compuesto 196. m/z (ES+) 458 (M+H)+. 1H-RMN (300 MHz, CD3OD): 6 7,65-7,62 (m, 2H,), 7,55-7,28 (m, 4H), 4,61 (s, 2H), 3,67-3,62 (m, 1H), 3,44 (s, 3H), 3,27-3,20 (m, 1H), 3,04-2,86 (m, 3H), 2,51 (s, 3H), 1,99-1,68 (m, 5H), 1,31-1,26 (m, 6H). Compuesto 197. m/z (ES+) 458 (M+H)+. 1H-RMN (300 MHz, CD3OD): 67,66-7,63 (m, 2H), 7,55 (s, 1H), 7,46-7,37 (m, 2H), 7,28 (m, 1H), 4,61 (s, 2H), 3,62-3,58 (m, 1H), 3,44 (s, 3H), 3,19-3,05 (m, 2H), 3,04-2,91 (m, 2H), 2,73-2,45 (m, 5H), 1,98-1,54 (m, 6H), 1,05-0,85 (m, 3H).

Compuesto 198.1.4-Isopropil-2-metilbenzoato de metilo. Un matraz de fondo redondo secado, con barra agitadora se purgó con nitrógeno y se cargó con diisopropilzinc (25 ml de una solución 1 M en THF, 25 mmol, 2,0 equiv). Se
añadió 4-bromo-2-metilbenzoato de metilo (compuesto 152.1, 2,86 g, 12,5 mmol, 1,0 equiv) en 1,4-dioxano (25 ml) seguido de la adición de Pd(dppf)2ChDCM (1,02 g, 1,25 mmol, 0,1 equiv) (exotérmica después de la adición de catalizador). El sistema se purgó con nitrógeno adicional, a continuación después argón y se calentó a 100 °C durante 4 horas y a continuación se agitó a temperatura ambiente durante 12 horas. La mezcla se inactivó cuidadosamente con HCl 1 M (ac., 12 ml) (cierto burbujeo) y a continuación se diluyó con agua (100 ml) y acetato de etilo (150 ml) y se mezcló. La mezcla se filtró a través de celite y se lavó con agua y acetato de etilo (2 x 25 ml cada uno). Las capas se separaron y la acuosa se extrajo con la adición de acetato de etilo (50 ml). Los capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (50 ml), se secaron (Na2SO4), y se concentraron a presión reducida. El producto impuro se purificó por cromatografía en columna de gel de sílice (hexanos respecto a acetato de etilo 6%) para obtener el compuesto del título como un aceite incoloro (2,31 g, 96%). 1H RMN (400 MHz, CDCh): 887,86 (d con estructura fina, J = 8,8 Hz, 1 H), 7,12-7,07 (m, 2H), 3,87 (s, 3H), 2,90 (septuplete, J = 6,8 Hz, 1H), 2,59 (s, 3H), 1,25 (d, J = 6,8 Hz, 6H).

Compuesto 198.2. 5-Yodo-4-isopropil-2-metilbenzoato de metilo. A 4-isopropil-2-metilbenzoato de metilo (compuesto 198.1 , 2,31 g, 12,0 mmol, 1,0 equiv) en un matraz de fondo redondo de 100 ml, se le añadió cuidadosamente TFA(24 ml) y la mezcla se enfrió a 0 °C. Se añadió W-yodosuccinimida (2,70 g, 12,0 mmol, 1,0 equiv) por partes durante 2 minutos y se añadió la mezcla resultante en nitrógeno y se agitó a 0 °C durante 20 minutos y a continuación a temperatura ambiente durante 15 horas. La mezcla se diluyó cuidadosamente a una mezcla de diclorometano (50 ml) y se añadió a fosfato disódico acuoso saturado para mantener un pH por encima de 3 (fosfato disódico total de aproximadamente 500 ml). La mezcla se agitó bien, se separó y la fase acuosa se extrajo con DCM adicional (5 x 25 ml). Las capas orgánicas combinadas se lavaron con una mezcla de sulfito sódico saturado (10 ml) más agua (40 ml) seguida de solución saturada de cloruro sódico (50 ml), se secó (Na2SO4), se filtró y se retiró al vacío. El producto impuro se purificó por cromatografía en columna de gel de sílice (hexanos respecto a acetato de etilo 3 %) para obtener el compuesto del título como un aceite de color canela claro (3,59 g, 94 %). 1H RMN (400 MHz, CDCla): 88,35 (s, 1H), 7,08 (s, 1H), 3,87 (s, 3H), 3,17 (septuplete, J = 6,8 Hz, 1H), 2,59 (s, 3H), 1,23 (d, J = 6,8 Hz, 6H).

Compuesto 198.3. 5-Ciano-4-isopropil-2-metilbenzoato de metilo. A un matraz de fondo redondo secado, se le añadió 5-yodo-4-isopropil-2-metilbenzoato de metilo (compuesto 198.2, 2,00 g, 6,29 mmol, 1,0 equiv), cianuro de zinc (1,48 g, 12,6 mmol, 2,0 equiv), DMF (20 ml) y Pd(PPh3)4 (364 mg, 0,315 mmol, 0,05 equiv). El sistema se purgó con nitrógeno seguido de argón y la mezcla se calentó a 100 °C durante 15 horas. La reacción se dejó enfriar a temperatura ambiente, a continuación se diluyó con acetato de etilo (50 ml) y se inactivó con FeSO41 M (25 ml). La mezcla se agitó vigorosamente durante 40 minutos y a continuación se filtró a través de celite y se lavó con FeSO41 M (15 ml), agua (50 ml) y acetato de etilo (150 ml). Las capas se separaron y la fase acuosa se extrajo con acetato de etilo (50 ml). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (4 x 100 ml), se secaron (Na2SO4), se filtraron, y se concentraron al vacío. El producto se purificó por cromatografía en columna de sílice (hexanos respecto a acetato de etilo 8 %) para obtener el compuesto del título (1,14 g, 83 %). 1H RMN (400 MHz, CDCla): 88,19 (s, 1H), 7,25 (s, 1H), 3,91 (s, 3H), 3,37 (septuplete, J = 6,8 Hz, 1H), 2,67 (s, 3H), 1,32 (d, J = 6,8 Hz, 6H).
Compue  -ciano-4-isopropil-2-metilbenzoato de metilo (compuesto 198.3, 1,12 g, 5,16 mmol, 1,0 equiv), ditiofosfato de 0 ,0-dietilo (90 %) (2,0 ml, 10,7 mmol, 2,1 equiv) y agua (200 pl) a un vial de 16 ml y la mezcla se calentó con la tapa suelta a 80 °C durante 85 horas (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Nótese que ditiofosfato de 0,0'-dietilo adicional (0,5 ml, 2,7 mmol, 0,5 equiv) y agua (50 pl) se añadieron a las 6 horas y a las 14 horas. El intermedio tioamida también se observa mediante LC/m S durante la reacción y se calienta hasta que la conversión al producto deseado es completa. La mezcla de reacción se diluyó con acetato de etilo (75 ml) y se lavó con NaHCO3 acuoso saturado (20 ml) seguido de NaH2PO41 M (10 ml) y solución saturada de cloruro sódico (10
ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío. El producto impuro se purificó por cromatografía en columna de sílice (hexanos respecto a acetato de etilo 25%) para obtener un aceite incoloro (1,12 g, 78 %). miz (ES+) 280 M+H .
-ciano-4-isopropil-2-metilbenzoato de metilo (compuesto 198.3, 1,12 g, 5,16 mmol, 1,0 equiv), ditiofosfato de 0 ,0-dietilo (90 %) (2,0 ml, 10,7 mmol, 2,1 equiv) y agua (200 pl) a un vial de 16 ml y la mezcla se calentó con la tapa suelta a 80 °C durante 85 horas (ADVERTENCIA: se produce desprendimiento de gas significativo - esta y todas las demás reacciones descritas en el presente documento deben llevarse a cabo en campanas extractoras bien ventiladas). Nótese que ditiofosfato de 0,0'-dietilo adicional (0,5 ml, 2,7 mmol, 0,5 equiv) y agua (50 pl) se añadieron a las 6 horas y a las 14 horas. El intermedio tioamida también se observa mediante LC/m S durante la reacción y se calienta hasta que la conversión al producto deseado es completa. La mezcla de reacción se diluyó con acetato de etilo (75 ml) y se lavó con NaHCO3 acuoso saturado (20 ml) seguido de NaH2PO41 M (10 ml) y solución saturada de cloruro sódico (10
ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío. El producto impuro se purificó por cromatografía en columna de sílice (hexanos respecto a acetato de etilo 25%) para obtener un aceite incoloro (1,12 g, 78 %). miz (ES+) 280 M+H .
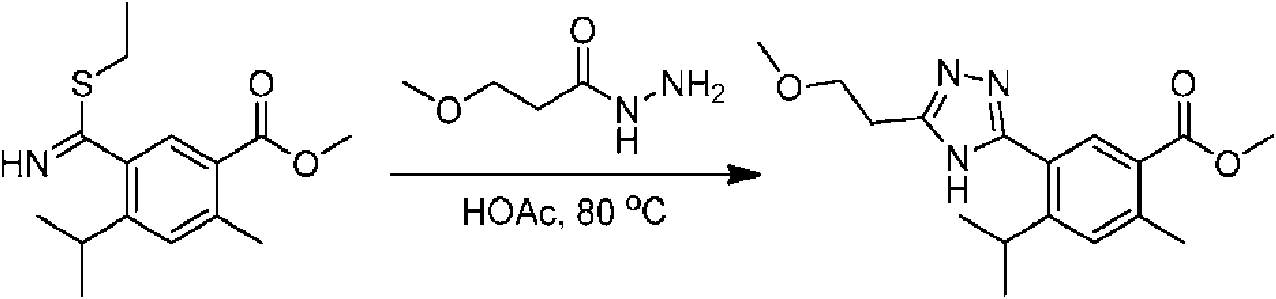
Compuesto 198.5. 4-Isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoato de metilo. Se añadieron 5-((etiltio)(imino)metil)-4-isopropil-2-metilbenzoato de metilo (compuesto 198.4, 33 mg, 0,12 mmol, 1,0 equiv), 3-metoxipropanihidrazida (compuesto 143.1, 21 mg, 0,18 mmol, 1,5 equiv) y ácido acético (1,2 ml) a un vial de 4 ml y se calentaron a 80 °C con una tapa suelta durante 4 horas. El disolvente se retiró al vacío y el residuo se disolvió en DCM (10 ml) y se lavó con NaHCO3 saturado (5 ml) y a continuación con solución saturada de cloruro sódico (5 ml), se secó

Compuesto 198.6. Ácido 4-isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoico. A 4-isopropil-5-(5-(2-metoxietil)4H-1,2,4-triazol-3-il) -2-metilbenzoato de metilo impuro (compuesto 198.5, 0,12 mmol, 1,0 equiv) de la etapa anterior en un vial de 4 ml, se le añadió hidróxido de litio monohidrato (10,1 mg, 0,24 mmol, 2,0 equiv), metanol (0,9 ml) y agua (0,3 ml). La mezcla resultante se agitó a temperatura ambiente durante 3 horas, seguido de 50 °C durante 3 horas y 40 °C durante 17 horas. Los disolventes se retiraron al vacío y el residuo se diluyó con agua (7 ml) más NaHCO3 saturado (1 ml) y la capa acuosa se lavó con éter dietílico (2 ml). La capa orgánica se extrajo de nuevo con una mezcla de agua (2 ml) más NaHCO3 saturado (0,5 ml). La capa acuosa combinada se acidificó a pH = 3 con H3PO41 M y se extrajo con diclorometano ( 3 x 5 ml). Las capas orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío para obtener el compuesto del título como un sólido ceroso de color blanco (31,5 mg, 88 % en dos etapas), miz ES+ 304 M+H .

Compuesto 198. 4-(1-(4-IsopropN-5-(5-(2-metoxietM)-4H-1,2,4-triazol-3-M)-2-metNbenzoN)piperidm-4-il)benzonitrilo. A un vial de 4 ml, se le añadió ácido 4-isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoico (compuesto 198.6, 31 mg, 0,10 mmol, 1,0 equiv), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 23 mg, 0,10 mmol, 1,0 equiv), 1-hidroxibenzotriazol hidrato (20 % en peso de agua) (22 mg, 0,13 mmol, 1,25 equiv), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (21,5 mg, 0,11 mmol, 1,1 equiv), DMF (1 ml) y DIEA (71 pl, 0,41 mmol, 4 equiv). La mezcla se agitó a temperatura ambiente durante 24 horas y a continuación se diluyó con acetato de etilo (10 ml) y se lavó con solución saturada de cloruro sódico (10 ml). La fase acuosa se extrajo de nuevo con acetato de etilo (3 ml) y las fases orgánicas combinadas se lavaron con NaHCO3 saturado (5 ml), NaH2PO4 1 M (5 ml), y solución saturada de cloruro sódico (5 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío. El residuo impuro se purificó por TLC preparativa (DCM/MeOH al 8 %) para obtener el compuesto del título como un sólido de color blanquecino (23 mg, 48 %). miz (ES+) 472 (M+H)+.

Compuesto 199. 4-(1-(4-Isopropil-5-(5-(metoximetil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 198). m/z (Es ) 458 (M+H)+.

Compuesto 200. 4-(1-(4-Isopropil-3-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1 -(4-isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 198). m/z ES+ 458 M+H .

Compuesto 201. 4-(M-(4-Isopropil-3-(5-(metoximetil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-isopropil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 198. m/z ES+ 444 M+H .

Compuesto 202.1. 5-Carbamoil-4-ciclobutil-2-metilbenzoato de metilo. A un vial de 8 ml, se le añadió 5-ciano-4-ciclobutil-2-metilbenzoato de metilo (152.4, 100 mg, 0,436 mmol, 1,0 equiv), carbonato potásico (181 mg, 1,31 mmol, 3.0 equiv) y DMSO (2,2 ml) y se inició la agitación. Se añadió peróxido de hidrógeno (50% en peso) (176 pl, 3,05 mmol, 7,0 equiv) durante aproximadamente 1 minuto. La mezcla se agitó a temperatura ambiente durante 29 horas y a continuación se diluyó en solución saturada de cloruro sódico (10 ml) más H3PO41 M (1,5 ml). La mezcla se extrajo con acetato de etilo (10 ml x 1,3 ml x 1). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (10 ml), más unas cuantas gotas de H3PO41 M seguido de una mezcla de agua (10 ml) más NaHCO3 saturado (1 ml) y finalmente con solución saturada de cloruro sódico (5 ml). Las capas orgánicas se secaron (Na2SO4), se filtraron y se evaporaron para obtener el compuesto del título como un sólido de color blanco (59 mg, 55 %). m/z (ES+) 248.0 (M+H)+.

Compuesto 202.2. 4-Ciclobutil-2-metil-5-(4H-1,2,4-triazol-3-il)benzoato de metilo. A un vial se le añadió 5-carbamoil-4-ciclobutil-2-metilbenzoato de metilo (compuesto 202.1, 59 mg, 0,24 mmol, 1,0 equiv) y se añadió N,N-dimetilformamida dimetil acetal (1 ml). La mezcla se calentó a 80 °C durante 5 horas y a continuación se retiraron los disolventes al vacío. Al residuo se le añadió ácido acético (400 pl) y una solución de hidrazina anhidra (8,2 pl, 0,26 mmol, 1,1 equiv) en ácido acético (100 pl). A la suspensión espesa resultante se le añadió ácido acético adicional (500 pl) y la mezcla se agitó a 80 °C durante 1,5 horas y a continuación los disolventes se retiraron al vacío. Se añadió acetato de etilo (5 ml) y DCM (5 ml) (algunos sólidos no disueltos). Las capas orgánicas se lavaron con NaHCO3 saturado (10 ml) y solución saturada de cloruro sódico (5 ml) y se retiraron al vacío para obtener el compuesto del título como un sólido de color blanco (rendimiento teórico), m/z (ES+) 272 (M+H)+.

Compuesto 202.3. Ácido 4-ciclobutil-2-metil-5-(4H-1,2,4-triazol-3-il)benzoico. A 4-ciclobutil-2-metil-5-(4H-1,2,4-triazol-3-il)benzoato de metilo impuro (compuesto 202.2, 0,24 mmol, 1,0 equiv) de la etapa anterior en un vial de 4 ml,
se le añadió metanol (1,5 ml) y agua (0,5 ml) e hidróxido de litio monohidrato (20 mg, 0,48 mmol, 2,0 equiv), la mezcla resultante se agitó a 40 °C durante 42 horas. La mezcla de reacción se diluyó en agua (5 ml), se acidificó a pH 3 con H3PO41 M y se extrajo con DCM ( 3 x 5 ml). Las capas orgánicas se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener el compuesto del título como un sólido de color blanco (61 mg, 98 % en dos etapas). m/z (ES+) 258 (M+H)+.

Compuesto 202. 4-(1-(4-CidobutN-2-metM-5-(4H-1,2,4-triazol-3-N)benzoN)piperidm-4-N)benzomtrNo. A un vial de 4 ml, se le añadió ácido 4-ciclobutil-2-metil-5-(4H-1,2,4-triazol-3-il)benzoico (compuesto 202.3, 31 mg, 0,12 mmol, 1,0 equiv), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 27 mg, 0,12 mmol, 1,0 equiv), 1-hidroxibenzotriazol hidrato (20% en peso de agua) (25 mg, 0,15 mmol, 1,25 equiv), clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida (25 mg, 0,13 mmol, 1,1 equiv), DMF (0,6 ml) y DieA (83 pl, 0,48 mmol, 4 equiv). La mezcla se agitó a temperatura ambiente durante 4 horas y a continuación se diluyó con acetato de etilo (10 ml) y se lavó con NaH2PO41 M (5 ml). La fase acuosa se extrajo de nuevo con acetato de etilo (3 ml) y las fases orgánicas combinadas se lavaron con solución saturada de cloruro sódico (5 ml), NaHCO3 saturado (5 ml) y solución saturada de cloruro sódico (5 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío. El residuo impuro se purificó por TLC preparativa (DCM/MeOH al 8%) para obtener el compuesto del título como un sólido de color blanco (23 mg, 46 %). m/z (ES+) 426 (M+H)+.

Compuesto 203. 4-(1-(4-CidobutN-2-metN-5-(4H-1,2,4-triazol-3-N)benzoM)-4-fluoropiperidm-4-N)benzomtrMo. A un vial de 4 ml, se le añadió ácido 4-ciclobutil-2-metil-5-(4H-1,2,4-triazol-3-il)benzoico (compuesto 202.3, 31 mg, 0,12 mmol, 1,0 equiv), 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2, 31 mg, 0,13 mmol, 1,1 equiv), 1-hidroxibenzotriazol hidrato (20% en peso de agua) (25 mg, 0,15 mmol, 1,25 equiv), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (25 mg, 0,13 mmol, 1,1 equiv), DMF (0,6 ml) y DIEA (83 pl, 0,48 mmol, 4 equiv). La mezcla se agitó a temperatura ambiente durante 4 horas y a continuación se diluyó con acetato de etilo (10 ml) y se lavó con NaH2PO41 M (5 ml). La fase acuosa se extrajo de nuevo con acetato de etilo (3 ml) y las fases orgánicas combinadas se lavaron con solución saturada de cloruro sódico (5 ml), NaHCO3 saturado (5 ml) y solución saturada de cloruro sódico (5 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se retiraron al vacío. El residuo impuro se purificó por TLC preparativa (DCM/MeOH al 8%) para obtener el compuesto del título como un sólido de color blanco (30 mg, 56 %). m/z (ES+) 444 M+H .

Compuesto 204.1.2,4-Dibromobenzoato de metilo. Una solución de ácido 2,4-dibromobenzoico (52,0 g, 176 mmol, 1,00 equiv, 95 %) y ácido sulfúrico (20 ml) en metanol (400 ml) se agitó a 90 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se diluyó con 500 ml de acetato de etilo y se lavó con 3 x 100 ml de H2O seguido de 1 x 100 ml de NaHCO3 (ac., sat. Nota: se produce desprendimiento de gas). La capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. Esto dio como resultado 45,0
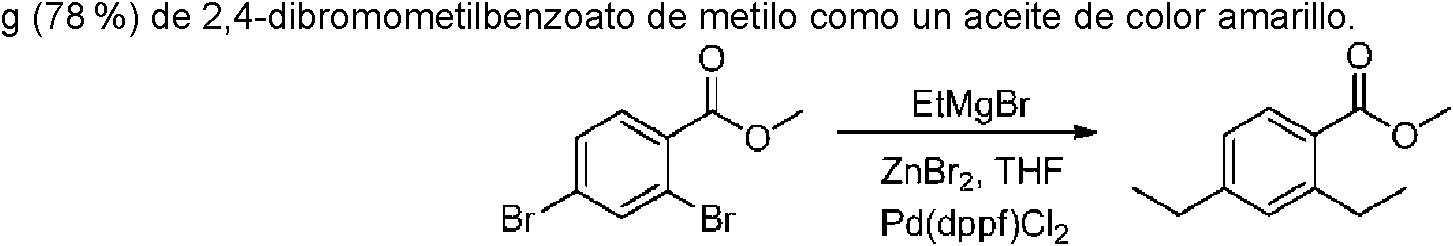
Compuesto 204.2. 2,4-Dietilbenzoato de metilo. A una mezcla agitada de ZnBr2 (40,0 g, 176 mmol, 5,44 equiv, 99 %) en THF (100 ml) en nitrógeno a 0 °C se le añadió gota a gota EtMgBr (60 ml, 3 M en THF). Después de 0,5 minutos a 0 °C, la temperatura se hizo descender a -78 °C y se añadió PdCh(dppf) (3,72 g, 5,03 mmol, 0,16 equiv, 99 %), seguido de la adición gota a gota de una solución de 2,4-dibromobenzoato de metilo (compuesto 204.1, 10,0 g, 32,3 mmol, 1,00 equiv, 95 %) en tetrahidrofurano (200 ml). La reacción se agitó durante una noche a temperatura ambiente, a continuación se inactivó cuidadosamente con agua seguida de HCl (ac., 1 M). La mezcla resultante se
extrajo con 3 x 500 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 3 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:60) como eluyente para proporcionar 6,30 g (96%) de 2,4-dietilbenzoato de metilo como un aceite incoloro

Compuesto 204.3.2,4-Dietil-5-yodobenzoato de metilo. El compuesto del título (4,0 g, aceite de color amarillo claro, 69%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.4 usando el compuesto 204.2 (3,50 g) en lugar del compuesto 181.3.

Compuesto 204.4. Ácido 2,4-dietil-5-yodobenzoico. El compuesto del título (3,24 g, sólido de color blanco, 85%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.9 usando el compuesto 204.3 (4,00 g) en lugar del compuesto 181.8.


Compuesto 204.5. Ácido 2,4-dietil-5-(metoxicarbonil)benzoico. A una solución agitada de ácido 2,4-dietil-5-yodobenzoico (compuesto 204.4., 500 mg, 1,64 mmol, 1,00 equiv, 90%) en tetrahidrofurano (20 ml) a -78 °C en nitrógeno, se le añadió gota a gota una solución de n-BuLi (1,73 ml, 4,10 mmol, 2,36 M en THF). Después de 5 minutos, se añadió una solución de cloroformiato de metilo (0,315 ml, 4,10 mmol, 2,50 equiv) en THF (5 ml) gota a gota a la reacción a -78 °C durante 5 minutos. La reacción se agitó durante otros 5 minutos a -78 °C, y a continuación se inactivó cuidadosamente con 10 ml de agua. El pH se ajustó a 1-2 con ácido clorhídrico (6 M )y la mezcla resultante se extrajo con acetato de etilo (2 x 30 ml). Las capas orgánicas combinadas se lavaron con 20 ml de solución saturada de cloruro sódico, a continuación se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo-éter de petróleo (1:20) como eluyente para dar 96 mg (25 %) del compuesto del título como un sólido de color blanquecino.

Compuesto 204.6. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2,4-dietilbenzoato de metilo. A un matraz de fondo redondo, se le añadieron una solución de ácido 2,4-dietil-5-(metoxicarbonil)benzoico (compuesto 204.5, 500 mg, 2,12 mmol, 1,00 equiv), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 470 mg, 2,12 mmol, 1 equiv), EDC-HCl (810 mg, 4,24 mmol, 2,00 equiv), y 4-dimetilaminopiridina (520 mg, 4,24 mmol, 2 equiv) en W,W-dimetilformamida (15 ml). La reacción se agitó durante la noche a 25 °C. Tras la finalización de la reacción, la mezcla de reacción se diluyó con 30 ml de acetato de etilo, a continuación se lavó con 1 x 20 ml de NH4Cl (ac.) y 1 x 20 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con una mezcla disolvente de acetato de etilo y éter de petróleo (1:1) para dar 500

Compuesto 204.7. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2,4-dietilbenzohidrazida. A una solución del compuesto 204.6, (500 mg, 1,05 mmol, 1,00 equiv, 85 %) en etanol (6 ml) se le añadió hidrato de hidrazina (3 ml). La solución resultante se agitó durante una noche a 90 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo se diluyó con 20ml de acetato de etilo, a continuación se lavó con 1 x 5 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con diclorometano-metanol (20:1) como eluyente para dar 260
mg (55 %) del compuesto deseado como un sólido de color amarillo

Compuesto 204.8.4-(1-(5-(5-Amino-1,3,4-oxadiazol-2-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dietilbenzohidrazida (compuesto 204.7, 100 mg, 0,220 mmol, 1,00 equiv, 90%) en una mezcla disolvente de agua (2 ml) y dioxano (3 ml). Se añadió bicarbonato sódico (62 mg, 0,740 mmol, 3,00 equiv) a la mezcla de reacción a temperatura ambiente, y se agitó durante 5 minutos. A la mezcla de reacción se le añadió a continuación BeCN (75 mg, 0,740 mmol, 3,00 equiv) a temperatura ambiente. La solución resultante se agitó durante 2 h a temperatura ambiente, a continuación se inactivó con 30 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 100 mg (89%) del compuesto del título como un sólido de color amarillo. El compuesto se usó en la siguiente etapa de reacción sin purificación adicional.

Compuesto 204.9. 4-(1-(5-(5-Etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzamida. Una mezcla del compuesto 204.8 (100 mg, 0,200 mmol, 1,00 equiv, 85%) e hidróxido potásico (132 mg, 2,35 mmol, 10,0 equiv) en etanol (10 ml) se calentó a 85 °C durante una noche. Después de enfriar a temperatura ambiente, el pH se ajustó a 7 con ácido acético y la solución resultante se concentró al vacío. El residuo se diluyó con 50 ml de acetato de etilo, se lavó con 2 x 20 ml de solución saturada de cloruro sódico, a continuación se concentró al vacío. El producto impuro se secó en un horno de vacío, antes cargarlo en una columna de gel de sílice, y se purificó con diclorometano-metanol

Compuesto 204. 4-(1-(5-(5-Etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 4-(1-(5-(5-etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzamida (compuesto 204.9, 100 mg, 0,170 mmol, 1,00 equiv) en diclorometano (10 ml). Se añadió trietilamina (170 mg, 1,68 mmol, 8,00 equiv) a la reacción a temperatura ambiente, seguida de la adición gota a gota de una solución de (CF3CO)2O (160 mg, 0,760 mmol, 4,50 equiv) en diclorometano (0,5 ml) de 0 a 5 °C. La solución resultante se agitó a temperatura ambiente durante otras 2 horas. La mezcla se diluyó con 60 ml de acetato de etilo, a continuación se lavó con 3 x 20 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó mediante HPLC preparativa (SunFire Prep C18, 19 * 150 mm 5 um; agua con el 0,05 % de TFA y CH3CN (53,0 % CH3CN asciende al 65,0 % en 8 minutos, asciende al 100,0 % en 2 minutos, desciende al 53,0 % en 1 minuto). Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 50 mg (50 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 458 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,63 (d, 2H), 7,45-7,30 (m, 4H), 4,89-4,80 (m, 1H, solapado), 4,36 (c, 2H), 3,60-3,57 (m, 1H), 3,29-3,23 (m, 1H), 3,19-2,85 (m, 4H), 2,74-2,56 (m, 2H), 2,00-1,82 (m, 1H), 1,77-1,66 (m, 3H), 1,40 (t, 3H), 1,30 1,21 (m, 3H), 1,11 (t, 3H).

Compuesto 205. 4-(1-(5-(5-Etoxi-4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados
para la preparación de 4-(1-(5-(5-etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo (compuesto 204). m/z (ES+) 430 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,69 (d, 2H), 7,52-7,47 (m, 3H), 7,29 (s, 1H), 4,89-4,80 (m, 1H, solapado), 4,42-4,40 (m, 2H), 3,64-3,61 (m, 1H), 3,02-2,96 (m, 2H), 2,51 (s, 3H), 2,41 y 2,31 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,05-2,02 m, 1H, 1,77-1,46 m, 3H , 1,45 t, 3H.

Compuesto 206. 4-(1-(5-(5-(2-Metoxietoxi)-4H-1,2,4-triazol-3-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(5-(5-etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo (compuesto 204). m/z (ES+) 460 (M+H)+.

Compuesto 207. 4-(1-(2,4-Dimetil-5-(5-((tetrahidrofuran-3-il)oxi)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(5-(5-etoxi-4H-1,2,4-triazol-3-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo (compuesto 204). m/z (ES+) 472 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,69 (d, J = 6,0 Hz, 2H), 7,58 7,37 (m, 3H), 7,30 (s ancho, 1H), 5,40 (s ancho, 1H), ~4,9 (1H parcialmente oscurecido por pico de agua), 4,08-3,96 (m, 3H), 3,96-3,88 (m, 1H), 3,71-3,58 (m, 1H), 3,33-3,22 (m, 1H), 3,00 (t con estructura fina, J = 9,0 Hz, 2H), 2,52 (s, 3H), 2,42 y 2,32 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,38-2,20 (m, 2H), 2,08-1,97 (m, 1H), 1,93-1,55 (m, 3H).
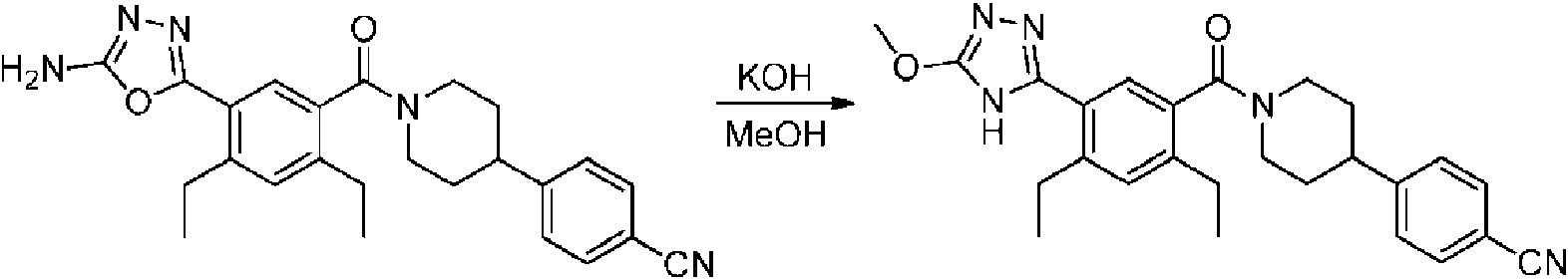
Compuesto 208. 4-(1-(2,4-Dietil-5-(5-metoxi-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 4-(1-(5-(5-amino-1,3,4-oxadiazol-2-il)-2,4-dietilbenzoil)piperidin-4-il)benzonitrilo (compuesto 204.8, 80,0 mg, 0,150 mmol, 1,00 equiv, 80%) e hidróxido potásico (104 mg, 1,85 mmol, 10,00 equiv) en metanol (10 ml). La mezcla resultante se agitó a temperatura ambiente durante una noche, a continuación se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (20/1) como eluyente. El producto impuro (50 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (40 % CH3CN asciende al 64 % en 8 minutos, asciende al 100 % en 1 minuto, desciende al 40 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 1,7 mg (3 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 444 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,63 (d, J=5,7Hz, 2H), 7,45-7,30 (m, 4H), 4,85 4,80 (m, 1H), 4,02 (s, 3H), 3,65-3,62 (m, 1H), 3,15-3,10 (m, 1H), 2,90-2,84 (m, 4H), 2,72-2,60 (m, 2H), 2,02-2,00 (m,
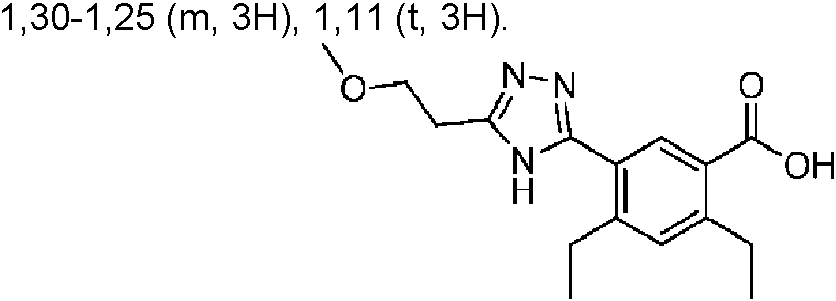
Compuesto 209.1. Ácido 2,4-dietil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 198.6), pero usando 2,4-dietil-5-yodobenzoato de metilo (compuesto 204.3) en lugar del compuesto 198.2 como material de partida.

Compuesto 209. 4-(I-(2,4-Dietil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 2,4-dietil-5-(5-(2-metoxietil)-4H-1,2,4-triazol-3-il)benzoico (compuesto 209. 1,300 mg, 0,940 mmol, 1,00 equiv, 95%) en N,N-dimetilformamida (20 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 243 mg, 1,09 mmol, 1,10 equiv), EDCHCl (380 mg, 1,98 mmol, 2,00 equiv), y 4-dimetilaminopiridina (240 mg, 1,96 mmol, 2,09 equiv) a la mezcla de reacción. La solución resultante se agitó durante una noche a temperatura ambiente, a continuación se diluyó con 100 ml de acetato de etilo. La capa orgánica se lavó con 3 x 15 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo como eluyente. El producto impuro (300 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (35 % CH3CN asciende al 60 % en 8 minutos, asciende al 100 % en 1 minuto, desciende al 35 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron Dara dar 188 ma {42%) del comDuesto del título como un sólido de color blanco, miz ÍES+) 472 (M+H)+.

Compuesto 210. 4-(1-(2,4-DietN-5-(5-(tetrahidrofuran-3-M)-4H-1,2,4-triazol-3-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2,4-dietil-5-(5-(2-metioxietil)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo (compuesto 209) pero usando tetrahidrofuran-3-carbohidrazida (compuesto 38.2) en lugar de 3-metoxipropanohidrazida (compuesto 143.1). miz (ES+) 484,05 (M+

Compuesto 211.2. 2-Etil-5-yodo-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 2-etil-4-metilbenzoato de metilo (compuesto 48.1, 12,5 g, 70,1 mmol, 1,00 equiv) en AcOH (100 ml). Se añadieron NaIO4 (7,51 g, 35,1 mmol, 0,50 equiv) e I2 (21,4 g, 84,3 mmol, 1,20 equiv) por partes a 25 °C. A continuación, se añadió ácido sulfúrico (1,60 g, 16,3 mmol, 0,20 equiv) gota a gota a la mezcla de reacción a 25 °C. La solución resultante se agitó durante 12 h a 110 °C en un baño de aceite, y a continuación se enfrió a temperatura ambiente. La reacción se inactivó con 150 ml de Na2S2O3 acuoso (ac., sat.). La fase acuosa se extrajo con 2 x 200 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3 x 300 ml de bicarbonato sódico (ac. sat.) y 1 x 150 ml de solución saturada de cloruro sódico. La mezcla se secó sobre sulfato sódico anhidro y se concentró al vacío. Esto dio como resultado 20,0 g (94%) de 2-etil-5-yodo-4-met¡lbenzoato de metilo como un aceite de color amarillo.

Compuesto 211.3. Ácido 2-etil-5-yodo-4-metilbenzoico. A un matraz de fondo redondo, se le añadió una solución de 2-etil-5-yodo-4-metilbenzoato de metilo (compuesto 211.2, 10,0 g, 32,9 mmol, 1,00 equiv) en metanol (40 ml). Se añadió gota a gota una solución de NaOH (5,26 g, 132 mmol, 4,00 equiv) en agua (20 ml). Después agitar durante 12 h a 40 °C, el disolvente orgánico se retiró a presión reducida. El pH de la fase acuosa restante se ajustó a 4 con cloruro de hidrógeno (ac., 6 M), a continuación se extrajo con 2 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 9,30 g (97 %) de ácido 2-etil-5-yodo-4-met¡lbenzo¡co como un sólido de color blanco.

Compuesto 211.4. Ácido 2-etil-5-formil-4-metilbenzoico. A una solución agitada de ácido 2-etil-5-yodo-4-metilbenzoico (compuesto 211.3., 5,00 g, 17,2 mmol, 1,00 equiv) en tetrahidrofurano (100 ml) a -78 °C en nitrógeno, se le añadió gota a gota n-BuLi (17 ml, 2 M en THF, 2,00 equiv). Después de 1 h a -78 °C, se añadió DMF (7,00 g, 95,8 mmol, 3,00 equiv) gota a gota a -78 °C. La solución resultante se calentó lentamente hasta 25 °C, a continuación
se inactivó cuidadosamente con agua, seguida de la adición de 10 ml de HCl (ac., 6 M). La fase acuosa se extrajo con 3 x 100 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (2:1) como eluyente para proporcionar 450 mg (14%) de ácido 2-etil-5-formil-4-metilbenzoico como un sólido de color amarillo.

Compuesto 211.5. 4-(1-(2-Etil-5-formil-4-metilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 2-etil-5-formil-4-metilbenzoico (compuesto 211.4, 450 mg, 2,34 mmol, 1,00 equiv) en W,W-dimetilformamida (5 ml). Se añadieron DIEA (907 mg, 7,02 mmol, 3,00 equiv) y HBTU (1,30 g, 3,43 mmol, 1,50 equiv) a la mezcla de reacción. La solución resultante se agitó durante 30 minutos a 25 °C. Una solución de clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 624 mg, 2,80 mmol, 1,20 equiv) en DIEA (2 ml) se añadió gota a gota. La solución resultante se agitó durante 30 minutos a 25 °C, a continuación se inactivo con 20 ml de agua. La fase acuosa se extrajo con 3 x 30 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (5:1) como eluyente para proporcionar 720 mg (85 %) del compuesto del título como un sólido de color amarillo.
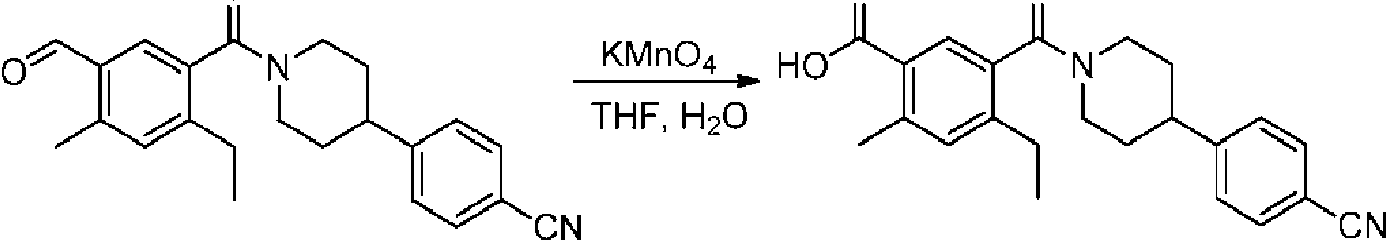
Compuesto 211.6. Ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoico. A un matraz de fondo redondo, se le añadió una solución de 4-(1-(2-etil-5-formil-4-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 211.5, 720 mg, 2,00 mmol, 1,00 equiv) en tetrahidrofurano (20 ml). Se añadió gota a gota una solución de KMnO4 (640 mg, 4,05 mmol, 2,00 equiv) en agua (20 ml). La solución resultante se agitó durante 15 horas a 60 °C y a continuación se enfrió a temperatura ambiente con un baño de agua. Los sólidos se retiraron con filtración. El filtrado se extrajo con 3 x 30 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 600 mg (80 %) de ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoico como un sólido de color amarillo claro.

Compuesto 211.7.5-(4-(4-Cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoato de metilo. Aun matraz de fondo redondo, se le añadió una solución de ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoico (compuesto 211.6, 600 mg, 1,59 mmol, 1,00 equiv) en tetrahidrofurano (30 ml). A esto se le añadió ácido sulfúrico (500 mg, 5,10 mmol, 3,20 equiv) gota a gota. La solución resultante se agitó durante 15 h a 60 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío. El residuo de diluyó con 20 ml de H2O. La fase acuosa se extrajo con 3 x 20 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 500 mg (80 %) de 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoato de metilo como un aceite de color amarillo.

Compuesto 211.8. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzohidrazida. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzoato de metilo (compuesto 211.7, 500 mg, 1,28 mmol, 1,00 equiv) en etanol (20 ml). A la mezcla de reacción se le añadió hidrazina (4 ml). La solución resultante se agitó durante 15 h a 80 °C, a continuación se concentró al vacío. El residuo se repartió entre agua y acetato de etilo. La capa acuosa se extrajo con acetato de etilo (2 x). Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío para dar 400 mg (80 %) de 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzohidrazida como un sólido de color amarillo claro.

Compuesto 211.9. W-(MorfoMn-4-carbonotioM)benzamida. A un matraz de fondo redondo de tres bocas de 50 ml, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de morfolina (1,00 g, 11,5 mmol, 1,00 equiv) en acetona (10 ml). Se añadió TEA (1,74 g, 17,2 mmol, 1,50 equiv) a la reacción, y la solución resultante se agitó durante 30 minutos a 25 °C. Se añadió isotiocianato de benzoílo (1,87 g, 11,5 mmol, 2,00 equiv) gota a gota a la reacción a 0 °C. La solución resultante se agitó durante 30 minutos a 0 °C, a continuación se inactivó con 20 ml de agua. La mezcla se extrajo con 2 x 30 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10-1:3) como eluyente para proporcionar 2,30 g (80 %) de A/-(morfolin-4-carbonotioiljbenzamida como un sólido de color amarillo.

Compuesto 211.10. Morfolin-4-carbotioamida. A un matraz de fondo redondo, se le añadió una solución de N-(morfolin-4-carbonotioil)benzamida (compuesto 211.9, 3,00 g, 12,0 mmol, 1,00 equiv) en metanol (20 ml). A la reacción se le añadió una solución de hidróxido sódico (1,44 g, 36,0 mmol, 3,00 equiv) en agua (20 ml). La solución resultante se agitó durante una noche a 60 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El residuo se extrajo con 2 x 20 ml de acetato de etilo/éter de petróleo (1:1). Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 430 mg (25 %) de morfolin-4-carbotioamida como un sólido de color amarillo.

Compuesto 211.11.4-(Metilsulfanil)carboximidoilmorfolina. Una solución de morfolin-4-carbotioamida (compuesto 211.10, 430 mg, 2,94 mmol, 1,00 equiv) y yodometano (1,25 g, 8,81 mmol, 3,00 equiv) en tetrahidrofurano (10 ml) se agitó durante 4 horas a 25 °C, a continuación, se concentró al vacío. Esto dio como resultado 471 mg (100 %) de 4-

Compuesto 211. 4-(1-(2-Etil-4-metil-5-(5-morfolino-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. A un tubo sellado de 20 ml se le añadió una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-4-etil-2-metilbenzohidrazida (compuesto 211.8, 225 mg, 0,580 mmol, 1,00 equiv) en piridina (10 ml). A la mezcla de reacción se le añadió 4-(metilsulfanil)carboximidoilmorfolina (compuesto 211.11, 200 mg, 1,25 mmol, 1,00 equiv). La solución resultante se agitó durante 3 días a 100 °C detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la mezcla se a continuación concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con cloroformo/metanol (30:1) como eluyente. El producto impuro (~100 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (32 % CH3CN asciende al 55 % en 8 minutos, asciende al 100 % en 2 minutos, desciende al 32 % en 1 minuto); detector, Waters 2489 254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 67,7 mg (24 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 485 (M+H)+. 1H-RMN (300 MHz, CD3OD): ó 7,69 (d, 2H), 7,49-7,31 (m, 4H), 4,90 4,80 (m, 1H), 3,83-3,81 (m, 4H), 3,79-3,65 (m, 1H), 3,60-3,41 (m, 4H), 3,40-3,31 (m, 1H), 3,08-2,97 (m, 2H), 2,76-2,71 (m, 2H), 2,51 (s, 3H), 2,02-1,98 (m, 1H), 1,85-1,80 (m, 3H), 1,33-1,30 (m, 3H).
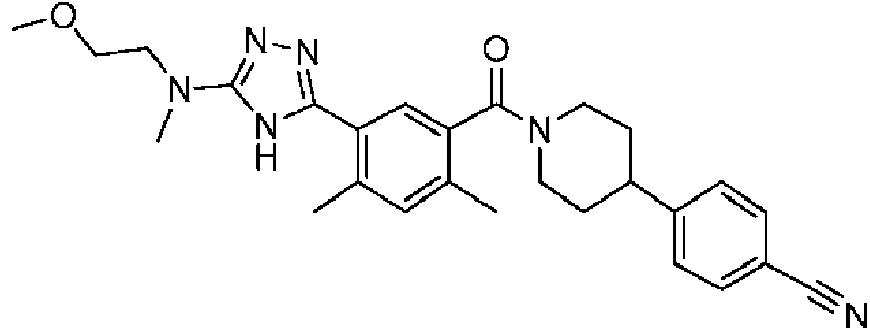
Compuesto 212. 4-(1-(5-(5-((2-MetoxietN)(metM)ammo)-4H-1,2,4-triazol-3-M)-2,4-dimetMbenzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1 -(2-etil-4-metil-5-(5-morfolino-4H-1,2,4-triazol-3-il)benzoM)piperidin-4-il)benzonitrilo (compuesto 211). m/z (ES+) 473 (M+H)+.

Compuesto 213. 4-(1-(2,4-Dimetil-5-(5-morfolino-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(2-etil-4-metil-5-(5-morfolino-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo

Compuesto 214. 4-(1-(2,4-Dimetil-5-(5-(pirrolidin-1-il)-4H-1,2,4-triazol-3-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4- 1- 2-etil-4-metil-5- 5-morfolino-4H-1,2,4-triazol-3-il benzoil)piperidin-4-il)benzonitrilo

Compuesto 215.3. Acido 4-ciclobutil-2-etil-5-yodobenzoico. A un matraz de fondo redondo, se le añadió una solución de 4-ciclobutil-2-etil-5-yodobenzoato de metilo (compuesto 181.4, 10,3 g, 30,0 mmol, 1,00 equiv) en metanol (100 ml). A la mezcla de reacción agitada se le añadió gota a gota una solución de hidróxido sódico (3,60 g, 3,00 equiv) en agua (10 ml). La mezcla resultante se agitó durante 2 h a 60 °C. Después de enfriar a temperatura ambiente, los volátiles se retiraron a presión reducida. El pH de la mezcla acuosa restante se ajustó a ~4 usando HCl (ac., 1 M). El precipitado resultante se recogió por filtración y se secó para dar 9,00 g (91 %) de ácido 4-ciclobutil-2-etil-5-yodobenzoico como un sólido de color blanco.

Compuesto 215.4. Ácido 4-ciclobutil-2-etil-5-(metoxicarbonil)benzoico. A una solución de 4-ciclobutil-2-etil-5-yodobenzoico (compuesto 215.3, 3,30 g, 10,0 mmol, 1,00 equiv) en THF (40 ml) se le añadió n -BuLi (2,5 M, 9,50 ml, 2,38 equiv) gota a gota a -78 °C en atmósfera de nitrógeno. La solución resultante se agitó durante otros 20 minutos a la misma temperatura, seguido de la adición de una solución de carbonato de dimetilo (2,70 g, 30,0 mmol, 3,00 equiv) en tetrahidrofurano (2 ml) gota a gota. La temperatura de reacción se elevó lentamente hasta 25 °C y se agitó durante 1 h más, a continuación se inactivó lentamente con 50 ml de agua. El pH se ajustó a ~4 con HCl (ac., 1 M) y la mezcla resultante se extrajo con 100 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó por cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:3) como eluyente para proporcionar 1,80 g (69 %) del producto deseado como un sólido de color blanco

Compuesto 215.5. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-etilbenzoato de metilo. A un matraz de fondo redondo, se le añadieron una solución de ácido 4-cidobutil-2-etil-5-(metoxicarbonil)benzoico (compuesto 215.4, 1,15 g, 4,38 mmol, 1,00 equiv), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 1,07 g, 4,80 mmol, 1,10 equiv), HBTU (2,50 g, 6,59 mmol, 1,50 equiv), y DIEA (1,07 g, 8,28 mmol, 3,00 equiv) en W,A/-dimetilformamida (10 ml). La solución resultante se agitó durante 1 h a 25 °C. La reacción se inactivó a continuación mediante la adición de 20 ml de agua. La mezcla resultante se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:3) para dar 1,80 g (95%) de 5-(4-(4-
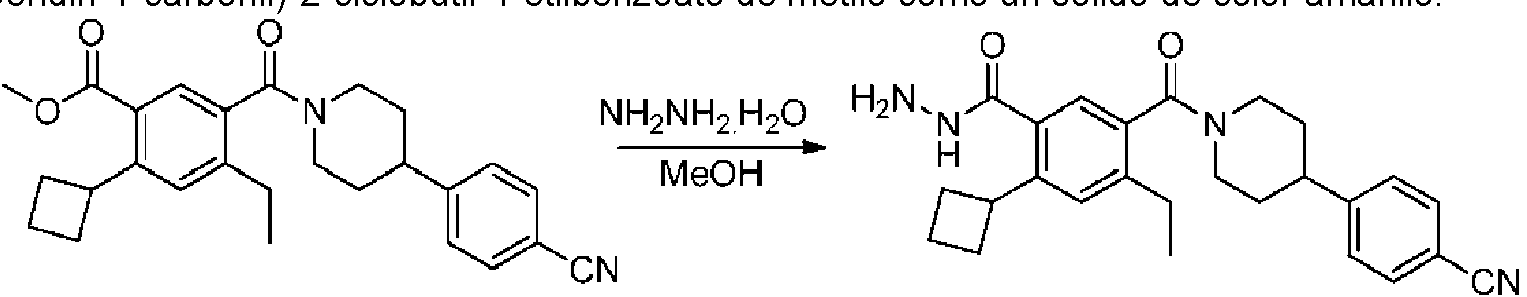
Compuesto 215.6. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-etilbenzohidrazida. A una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-etilbenzoato de metilo (compuesto 215.5, 2,15 g, 4,99 mmol, 1,00 equiv) en metanol (20 ml), se le añadió hidrato de hidrazina (80%, 15 ml) de forma discontinua. La reacción se agitó a 80 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se extrajo con diclorometano (2 x 100 ml). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con diclorometano/metanol (100:1) para proporcionar 1,13 g (53 %) del producto deseado como un sólido rosáceo.

Compuesto 215.7.4-(1-(5-(5-Ammo-1,3,4-oxadiazol-2-N)-4-ciclobutN-2-etNbenzoN)pipendm-4-M)benzomtrMo. A una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-etilbenzohidrazida (compuesto 215.6, 600 mg, 1,39 mmol, 1,00 equiv) en 1,4-dioxano (10 ml ) se le añadieron 10 ml de una solución acuosa de bicarbonato sódico (350 mg, 4,17 mmol, 1,00 equiv). A la mezcla de reacción se le añadió gota a gota bromuro de cianógeno (220 mg, 2,08 mmol, 1,50 equiv). La mezcla de reacción se agitó a 25 °C durante 2 h, a continuación se inactivó con 20 ml de FeSO4 (ac., sat.) y se diluyó con DCM. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 50 ml de DCM. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 600 mg (95 %) de 4-(1-(5-(5-amino-1,3,4-oxadiazol-2-il)-4-ciclobutil-2-etilbenzoil)piperidin-4-

Compuesto 215.8. 4-(1-(4-Ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzamida. A un tubo sellado de 10 ml, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de 4-(1-(5-(5-amino-1,3,4-oxadiazol-2-il)-4-ciclobutil-2-etilbenzoil)piperidin-4-il)benzonitrilo (compuesto 215.7, 228 mg, 0,500 mmol, 1,00 equiv) en etanol (5 ml). A esto le siguió la adición de hidróxido potásico (280 mg, 4,99 mmol, 10,0 equiv) por partes. La solución resultante se agitó durante 5 h a 80 °C detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la reacción se inactivó a continuación mediante la adición de 15 ml de agua y se extrajo con 2 x 20 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 200 mg (80 %) de 4-(1-(4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzamida como un sólido de color amarillo.

Compuesto 215. 4-(1-(4-Ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución del compuesto 215.8 (251 mg, 0,500 mmol, 1,00 equiv) en diclorometano (5 ml). A esto le siguió la adición de (CF3CO)2O (158 mg, 1,50 equiv) gota a gota con agitación. Se añadió trietilamina (101 mg, 1,00 mmol, 2,00 equiv) gota a gota con agitación. La mezcla resultante se agitó a 25 °C durante 1 h, a continuación se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (20/1) como eluyente. El producto impuro (120 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (54,0 % CH3CN asciende al 64,0 % en 6 minutos, asciende al 100,0 % en 1 minuto, desciende al 54,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 60 mg (25 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 484 (M+H)+. 1H-RMN (300 MHz, CD3OD): 87,64 (d, 2H), 7,45-7,21 (m, 4H), 4,37 (c, 2H), 4,02-3,96 (m, 1H), 3,60-3,53 (m, 1H), 3,29-3,19 (m, 2H), 2,98-2,90 (m, 2H), 2,76-2,61 (m, 2H), 2,15-2,12 (m, 2H),

Compuesto 217.1. 5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclobutil-4-etilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de ácido 4-ciclobutil-2-etil-5-(metoxicarbonil)benzoico (compuesto 215.4, 70,0 mg, 0,270 mmol, 1,00 equiv, 90 %) en W,A/-dimetilformamida (5 ml). Se añadieron HBTU (210 mg, 0,550 mmol, 2,07 equiv), DIEA (150 mg, 1,16 mmol, 4,35 equiv) a la mezcla de reacción a 0 °C, y se agitaron durante 30 minutos. A esto le siguió la adición de clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2, sa1HCl, 80,0 mg, 0,330 mmol, 1.20 equiv). La solución resultante se agitó durante 15 h a 25 °C y a continuación se inactivo con 20 ml de agua con hielo. La mezcla se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 1 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:100-1:5) como eluyente para dar 100 mg (84 %) de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclobutil-4-etilbenzoato de metilo como un sólido de color blanco.

Compuesto 217.2. 5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclobutil-4-etilbenzohidrazida. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclobutil-4-etilbenzoato de metilo (compuesto 217.1, 1,10 g, 2,45 mmol, 1,00 equiv) en etanol (40 ml). Se añadió hidrato de hidrazina (20 ml) a la mezcla de reacción, y la reacción se agitó a 80 °C durante 15 h. Después de enfriar a temperatura ambiente, la mezcla se concentró al vacío, a continuación se diluyó con 50 ml de H2O. La mezcla resultante se extrajo con 3 x 50 ml de diclorometano. Las capas orgánicas combinadas se lavaron con 2 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:1-1:0) como eluyente para dar 900
mg (82%) de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclobutil-4-etilbenzohidrazida como un sólido de color blanco.

Compuesto 217.3. 4-(1-(5-(5-Ammo-1,3,4-oxadiazol-2-M)-4-cidobutM-2-etMbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-cidobutil-4-etilbenzohidrazida (compuesto 217.2, 450 mg, 1,00 mmol, 1,00 equiv) en 1,4-dioxano (10 ml). A la mezcla de reacción se le añadió una solución de bicarbonato sódico (252 mg, 3,00 mmol, 2,99 equiv) en agua (10 ml) y BrCN (128 mg, 1,21 mmol, 1,20 equiv). La solución resultante se agitó durante 3 h a temperatura ambiente, a continuación se inactivó con 30 ml de FeSÜ4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSÜ41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 400 mg (84%) de 4-(1-(5-(5-amino-1,3,4-oxadiazol-2-il)-4-ciclobutil-2-etilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo como un sólido de color marrón claro.

Compuesto 217. 4-(1-(4-Ciclobutil-2-etil-5-(5-metoxi-4H-1,2,4-triazol-3-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió una solución de 4-(1-(5-(5-amino-1,3,4-oxadiazol-2-il)-4-ciclobutil-2-etilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo (compuesto 217.3, 200 mg, 0,420 mmol, 1,00 equiv) en metanol (20 ml). A la mezcla de reacción se le añadió hidróxido potásico (237 mg, 4,22 mmol, 10,0 equiv). La solución resultante se agitó durante 15 h a 70 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El residuo se diluyó con 30 ml de H2O, a continuación se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2 x 50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El producto impuro (50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (49,0 % CH3CN asciende al 63,0 % en 8 minutos, asciende al 100,0 % en 1 minuto, desciende al 49,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 20 mg (10 %) del compuesto del título como un de color blanco. m/z (ES+) 488 (M+H)+. 1H-RMN (300 MHz, CD3OD): ó 7,72 (d, 2H), 7,64-7,61 (m, 2H), 7,39-7,25 (m, 2H), 4,85-4,78 (m, 1H), 4,00-3,93 (m, 4H), 3,54-3,49 (m, 2H), 3,28-3,22 (m, 1H), 2,80-2,65 (m, 2H), 2,26-1,84 (m, 10H), 1,45-1,27 (m, 3H).

Compuesto 218. 4-(1-(4-Ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzonitrilo (compuesto 215) y usando sal HCl, del compuesto 11.2 en lugar del compuesto 1.5. m/z (Es ) 502 [M+H]+, 543 [M+CH3CN+H]+. 1H-RMN (300 MHz, CD3OD): ó 7,73 (d, 2H), 7,64-7,58 (m, 2H), 7,39-7,26 (m, 2H), 4,89-4,82 (m, 1H), 4,35 (c, 2H), 4,10-3,95 (m, 1H), 3,50-3,38 (m, 2H), 3,30-3,27 (m, 1H), 2,75-2,65 (m, 2H), 2,22-1,86 (m, 10H), 1,41 (t, 3H), 1,35-1,20 (m, 3H).

Compuesto 219.1 2-Bromo-4-metilbenzoato de metilo. A una solución de ácido 2-bromo-4-metilbenzoico (10 g) en MeOH (50 ml) se le añadió gota a gota ácido sulfúrico concentrado (10 ml) a 0 °C. La mezcla se calentó a 70 °C durante 2 horas. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El residuo se
vertió en agua con hielo (100 ml). La mezcla se extrajo con EtOAc (x2). Las capas orgánicas combinadas se lavaron con NaHCO3 (ac. sat. Nota: desprendimiento de gas), solución saturada de cloruro sódico, se secaron sobre MgSO4, se filtraron, y se concentraron para dar el producto como un aceite transparente. Rendimiento: 10,5 g, 99%. 1H RMN (400 MHz, Cloroformo-d) 67,73 (d, 1H), 7,50 (d, 1H, 7,19-7,11 m, 1H), 3,92 (s, 3H), 2,36 (s, 3H).

Compuesto 219.22-Ciclobutil-4-metilbenzoato de metilo. Se añadió bromuro de ciclobutilzinc (II) (50 ml, 0,5 M en THF, 25,0 mmol) a una mezcla de 2-bromo-4-metilbenzoato de metilo (compuesto 219.1, 5,0 g, 21,8 mmol) y PdCh(dppf)CH2Cl2 (1,78 g, 2,20 mmol). La mezcla se desgasificó y el matraz se llenó con argón a través de un globo. Después de que la mezcla se calentó a 65 °C en argón durante 24 horas, se enfrió a 0 °C y se inactivó con agua (10 ml). La mezcla se diluyó con EtOAc (200 ml), se lavó con agua y a continuación con solución saturada de cloruro sódico. La capa de EtOAc se secó (Na2SO4), se concentró, y se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30:1 a 20:1). Rendimiento: 3,6 g, 81 %. 1H RMN (400 MHz, Cloroformo-d) 67,68 (d, 1H), 7,23 - 7,17 (s, 1H), 7,03 (d, 1H), 4,16 (m, 1H), 3,86 (s, 3H , 2,39 s, 3H , 2,34 (m, 2H), 2,16 -1,96 (m, 3H), 1,80 (m, 1H).

Compuesto 219.3 2-Ciclobutil-5-yodo-4-metilbenzoato de metilo. Se añadió N-yodosuccinimida (5,25 g, 23,3 mmol) por partes a una solución de 2-ciclobutil-4-metilbenzoato de metilo (compuesto 219.2,4,77 g, 23,3 mmol) en ácido sulfúrico concentrado (100 ml) a 0 °C. La mezcla se agitó a 0 °C durante 30 minutos y a TA durante 2 horas. La mezcla se enfrió a 0 °C de nuevo y se añadió MeOH (100 ml). La mezcla se calentó a 60 °C durante 2 horas. El metanol se retiró a presión reducida y el residuo se vertió en agua con hielo (200 ml). La mezcla se extrajo con EtOAc (2x). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico, a continuación, NaHCO3 IN, se secaron (Na2SO4) y se concentraron. El residuo se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30:1 a 20:1). Rendimiento: 5,0 g, aceite transparente, 65 %. 1H RMN (400 MHz, Cloroformo-d) 68,19 (s, 1H), 7,24 (s, 1H), 4,17-4,04 (m, 1H), 3,86 (s, 3H), 2,48-2,44 (s, 3H), 2,40 - 2,28 (m, 2H), 2,13-1,92 (m, 3H), 1,85 - 1,75 (m, 1H).

Compuesto 219.4 5-Ciano-2-ciclobutil-4-metilbenzoato de metilo. Una mezcla de 2-ciclobutil-5-yodo-4-metilbenzoato de metilo (compuesto 219.3 , 3,0 g, 9,1 mmol), Zn(CN)2 (2,3 g, 19,6 mmol) y Pd(PPh3)4 (0,55 g, 0,47 mmol) en DMF (50 ml) se desgasificó y el matraz se llenó con argón a través de un globo. La mezcla se calentó a 100 °C en argón durante una noche. Después de enfriar a temperatura ambiente, la reacción se inactivó con. FeSO4 ac. saturado (20 ml) y se diluyó con EtOAc (200 ml). La mezcla se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico, se secaron (Na2SO4) y se concentraron. El residuo se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 30: 1 a 20: 1). Rendimiento: 2,0 g, 96 %. 1H RMN (400 MHz, Cloroformo-d) 68,03 (s, 1H), 7,34 (s, 1H), 4,26-4,13 (m, 1H), 3,89 (s, 3H), 2,59 (s, 3H), 2 ,46-2,32 (m, 2H), 2,16-1,98 m, 3H , 1,90- 1,78 (m, 1H).
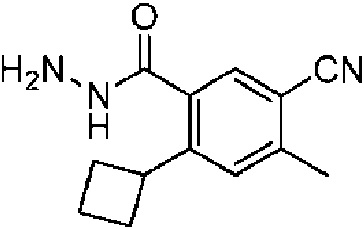
Compuesto 219.5. 5-Ciano-2-ciclobutil-4-metilbenzohidrazida. A una solución de 5-ciano-2-ciclobutil-4-metilbenzoato de metilo ( 219.4, 2,0 g, 8,73 mmol) en EtOH (10 ml) se le añadió hidrazina anhidra (2 ml, exceso) a temperatura ambiente. La mezcla se calentó a 90 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se repartió entre agua (60 ml) y EtOAc (200 ml). La capa de EtOAc se lavó con agua (x2), solución saturada de cloruro sódico, se secó con Na2SO4, y se concentró para dar el producto como un sólido de color blanco. Rendimiento: 1,9 g, 95 %. m/z (ES+) 230 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 67,52 (s, 1H), 7,32 (s, 1H), 6,91 (ancho, 1H), 4,08 (ancho, 2H), 3,89 (m, 1H), 2,61 - 2,52 (m, 3H), 2,42 - 2,28 (m, 2H), 2,18 -1,98 (m, 3H), 1,91 - 1,78 (m, 1H).

Compuesto 219.6. 5-(5-Ammo-1,3,4-oxadiazol-2-M)-4-cidobutN-2-metNbenzomtrMo. El compuesto del título (0,55 g, sólido de color blanco, 100%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 217.3 y usando el compuesto 219.5 (0,50 g) en lugar del compuesto 217.2. m/z (ES+) 255 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 87,93 (s, 1H), 7,45 (s, 1H), 5,10 (ancho, 2H), 4,38 (m, 1H), 2,61 (s, 3H), 2,48 - 2,34 (m, 2H), 2,17-1,98 (m, 3H), 1,91 -1,79 (m, 1H.

Compuesto 219.7. 4-Ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzonitrilo. A una solución de 5-(5-amino-1,3,4-oxadiazol-2-il)-4-ciclobutil-2-metilbenzonitrilo (219.6, 0,5 g, 2,0 mmol) en EtOH (40 ml), se le añadió KOH (1,11 g, 20,0 mmol). La mezcla se calentó a 85 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se neutralizó a pH 7 con HCl IN a 0 °C y a continuación se extrajo con EtOAc (x2). Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron. El residuo se purificó usando cromatografía en columna (gel de sílice) (hexanos: EtOAc 1:1 a EtOAc). Rendimiento: 0,2 g, sólido de color blanco, 34%. m/z (ES+) 283 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 87,84 (s, 1H), 7,35 (s, 1H), 4,48 (t, 2H), 4,19-4,10(m , 1H), 2,58 (s, 3H), 2,30-2,19(m , 2H), 2,11 1,95 (m, 3H), 1,85 - 1,76 (m, 1H), 1,45 (
Compuesto 219.8. 4-Ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzamida. A una solución de 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzonitrilo (219.7, 0,15 g, 0,53 mmol) en EtOH (10 ml), se le añadió NH4OH (0,18 ml, 2,66 mmol, 14,8 N en H2O), seguido de H2O2(1,8 ml, 26,6 mmol, 50% en H2O). La mezcla se agitó a temperatura ambiente durante una noche. La mezcla se enfrió a 0 °C yse inactivó cuidadosamente con una solución de Na2S2O3 IN (26 ml). La mezcla se extrajo con EtOAc (x2) y las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron, y se concentraron. El residuo se purificó con TLC preparativa (MeOH al 5 % en CH2Ch). Rendimiento: 0,1 g,

Compuesto 219.9. Acido 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoico. A una solución de 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzamida (219.8, 0,1 g, 0,33 mmol) en TFA (5 ml), se le añadió NaNO2 (0,046 g, 0,66 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 1 hora y a continuación a temperatura ambiente durante 2 horas. La mezcla se concentró a presión reducida. El residuo se repartió entre EtOAc y solución saturada de cloruro sódico. La capa acuosa se extrajo con EtOAc. Las capas orgánicas combinadas se secaron (Na2SO4) y se

Compuesto 219. 4-(1-(4-CidobutN-5-(5-etoxi-4H-1,2,4-triazol-3-N)-2-metNbenzoM)piperidm-4-N)benzomtrMo. Una solución de ácido 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoico (compuesto 219.9, 40 mg, 0,13 mmol), DIEA (0,07 ml, 0,39 mmol), HOBT (34 mg, 0,19 mmol, con el 20 % de agua), EDc I (38 mg, 0,19 mmol), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 36 mg, 0,16 mmol) en N,N- dimetilformamida (3 ml) se agitó durante una noche a temperatura ambiente. A continuación se diluyó la mezcla de reacción con 50 ml de acetato de etilo y se lavó con 2x20 ml de solución saturada de cloruro sódico. La mezcla se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo se aplicó sobre una placa de TLC preparativa de gel de sílice y se reveló usando acetato de etilo/hexanos (1:1) para dar 0,1 g (68 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 470 (M+H)+.

Compuesto 220. 4-(1-(4-CidobutN-5-(5-etoxi-4H-1,2,4-triazol-3-N)-2-metNbenzoN)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 219), usando sal HCl del compuesto 11.2, en lugar del compuesto 1.5. m/z (ES+) 488 (M+H)+. 1H RMN (400 MHz, CDCla): 811,10 (s ancho, 1H), 7,72-7,63 (m, 2H), 7,47 (d, J =8,4 Hz, 2H), 7,42 y 7,33 (2 singletes, rotámeros de amida, Ar-H, 1H), 7,25 (s, 1H), 4,86 (d ancho, J = 11,2 Hz, 1H), 4,41 (c, J

Compuesto 221. (4-CidobutN-5-(5-etoxi-4H-1,2,4-triazol-3-M)-2-metMfeml)(4-fluoro-4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 219). m/z (ES+) 531 (M+H)+.

[0635]Compuesto 222.1.4-Ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzonitrilo (compuesto 219.7), excepto que se usó MeOH como disolvente en lugar de EtOH. m/z (ES+) 269(M+H)+. 1H RMN (400 MHz, Cloroformo-d) 810.96 (ancho, 1H), 7,82 (s, 1H), 7,35 (s, 1H), 4,11 (s, 3H), 4,15-4,05 (m, 1H), 2,59 (s, 3H), 2,31 - 2,16 (m, 2H), 2,14 -1,89 (m, 3H), 1,87-1,71 (m, 1H).

Compuesto 222.2. 4-Ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzamida (compuesto 219,8) pero usando el compuesto 222.1 en lugar del compuesto 219.7. m/z (ES+) 287(M+H)+. 1H RMN (400 MHz, M etano l^) 87,50 (s, 1H), 7,33 (s, 1H), 4,03 (s, 3H), 3 ,95-4,05 (m, 1H), 2,51 (s, 3H), 2,23-2,11 m, 2H , 2,11-1,88 (m, 3H), 1,83-1,71 (m, 1H).

Compuesto 222.3. Acido 4-ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de ácido 4-ciclobutil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoico (compuesto 219,9) pero usando el compuesto 222.2
Compuesto 222. 4-(1-(4-CidobutM-5-(5-metoxi-4H-1,2,4-triazol-3-M)-2-metMbenzoM)piperidm-4-M)benzomtrMo. Una solución de ácido 4-cidobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoico (compuesto 222.3, 60 mg, 0,21 mmol), DIEA (0,11 ml, 0,63 mmol), Ho BT (53 mg, 0,32 mmol, con el 20% de agua), EDCI (60 mg, 0,32 mmol), y clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 47 mg, 0,21 mmol) en N,N- dimetilformamida (3 ml) se agitó durante una noche a temperatura ambiente. A continuación se diluyó la mezcla de reacción con 50 ml de acetato de etilo y se lavó con 2x20 ml de solución saturada de cloruro sódico. La mezcla se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo se aplicó sobre una placa de TLC preparativa de gel de sílice y se reveló usando acetato de etilo/hexanos (1:1) para dar 21,0 mg (22 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 456 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 811,54-10,86 (s ancho, 1H), 7,60 (d, 2H), 7,38-7,20 (m, 4H), 5,04-4,92 (m, 1H), 4,06 (s, 3H), 4,14-4,00 (m, 1H), 3,62 (d, 1H), 3,15-3,03 (m, 1H), 2,93-2,76 (m, 2H), 2,38 y 2,30 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,34-2,22 (m, 1H), 2,20-2,06 (m, 2H), 2,05-1,84 (m, 3H), 1,61-1,45 (m, 1 H ) .

Compuesto 223. 4-(1-(4-CidobutN-5-(5-metoxi-4H-1,2,4-triazol-3-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 222) pero usando sal HCl del compuesto 11.2, en lugar del

Compuesto 224. (4-Ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilfenil)(4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(I-(4-ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-

Compuesto 225. (4-CidobutN-5-(5-metoxi-4H-1,2,4-triazol-3-M)-2-metNfenM)(4-fluoro-4-(4-(trifluorometil)fenil)piperidin-1-il)metanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-metoxi-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (Compuesto 222). m/z(ES+) 517 (M+H)+.
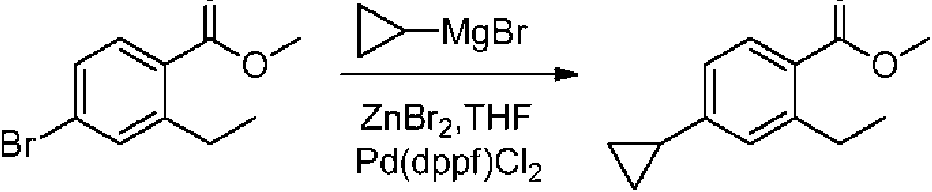
Compuesto 226.3. 4-Ciclopropil-2-etilbenzoato de metilo. A una mezcla agitada de ZnBr2 (37,0 g, 164 mmol, 3,99 equiv) en tetrahidrofurano (150 ml) en nitrógeno, se le añadió bromo(ciclopropil)magnesio (3,28 M en THF, 50 ml, 4,00 equiv) gota a gota a 0 °C. Después de agitar 15 minutos a 0 °C, se redujo la temperatura a -30 °C seguido de la adición gota a gota de una solución de Pd(dppf)Ch (2,00 g, 2,73 mmol, 0,07 equiv) en tetrahidrofurano (50 ml) y una solución de 4-bromo-2-benzoato de metilo (compuesto 181.2, 10,0 g, 41,1 mmol, 1,00 equiv) en tetrahidrofurano (50 ml). La solución resultante se calentó lentamente a 25 °C y se agitó durante 15 h, a continuación se inactivó cuidadosamente mediante la adición lenta de 100 ml de NH4Cl (ac. sat.). La mezcla se extrajo con 2 x 100 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:30) como eluyente para dar 7,50 g (89 %) de 4-ciclopropil-2-etilbenzoato de metilo como un aceite de color amarillo claro.

Compuesto 226.4. 4-Ciclopropil-2-etil-5-yodobenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 4-cidopropN-2-etilbenzoato de metilo (compuesto 226.3, 5,00 g, 24,5 mmol, 1,00 equiv) en AcOH (40 ml). A la reacción se le añadieron I2 (6,80 g, 26,8 mmol, 1,10 equiv), NaIO4 (2,60 g, 12,2 mmol, 0,50 equiv), y ácido sulfúrico (400 mg, 4,08 mmol, 0,15 equiv). La solución resultante se agitó durante 2 h a 50 °C. Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente con Na2S2O3 (ac., sat.). La mezcla se extrajo con 2 x 100 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (20:1) como eluyente para proporcionar 4,00 g (49%) de 4-ciclopropil-2-etil-5-yodobenzoato de metilo como un aceite de color amarillo

Compuesto 226.5. Ácido 4-ciclopropil-2-etil-5-yodobenzoico. A una solución de 4-ciclopropil-2-etil-5-yodobenzoato de metilo (compuesto 226.4, 2,50 g, 7,57 mmol, 1,00 equiv) en metanol (40 ml) se le añadió hidróxido sódico acuoso (3,10 g, 77,5 mmol, 10,0 equiv, en 10 ml de agua). La mezcla resultante se agitó durante 15 h a 60 °C en un baño de aceite. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida y el pH de la capa acuosa restante se ajustó a 4 con cloruro de hidrógeno (ac., 1 M). Los sólidos resultantes se recogieron por filtración y se secaron en un horno a presión reducida para dar 2,20 g (92 %) de ácido 4-ciclopropil-2-etil-5-yodobenzoico como un sólido de color blanco

Compuesto 226.6. Ácido 4-ciclopropil-2-etil-5-(metoxicarbonil)benzoico. A una solución de ácido 4-ciclopropil-2-etil-5-yodobenzoico (compuesto 226.5, 500 mg, 1,58 mmol, 1,00 equiv) en THF/Et2O (10/10 ml) en nitrógeno a -78 °C se le añadió gota a gota n-butillitio (2,5 M, 1,88 ml, 3,00 equiv) durante 3 minutos. Después de agitar durante 10 minutos a -78 °C, se añadió cloroformiato de metilo (230 mg, 2,43 mmol, 1,50 equiv) gota a gota a -78 °C. La solución resultante se agitó durante 40 minutos a -78 °C, a continuación se calentó lentamente a -35 °C. La reacción se inactivó cuidadosamente con 2 ml de agua. El pH se ajustó a ~4-5 con cloruro de hidrógeno (ac., 1 M). La mezcla resultante se extrajo con 3 x 20 ml de acetato de etilo y las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron

Compuesto 226.7.5-(4-(4-Cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclopropil-4-etilbenzoato de metilo. A una solución de ácido 4-ciclopropil-2-etil-5-(metoxicarbonil)benzoico (compuesto 226.6, 150 mg, 0,600 mmol, 1,00 equiv) en W,A/-dimetilformamida (15 ml), se le añadieron clorhidrato de 4-(4-fluoropiperidin -4-il)benzonitrilo (compuesto 11.2, 150 mg, 0,620 mmol, 1,00 equiv), DIPEA(450 mg, 3,49 mmol, 6,00 equiv) y HBTU (300 mg, 0,790 mmol, 1,30 equiv). La solución resultante se agitó durante 4 h a 20 °C, a continuación se inactivó mediante la adición de 20 ml de solución saturada de cloruro sódico. La mezcla resultante se extrajo con 4 x 30 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó mediante columna de gel de sílice con acetato de etilo/éter de petróleo (1:3) como eluyente. Esto dio como resultado 0,200 g (76 %) de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclopropil-4-etilbenzoato de metilo como un sólido de color amarillo claro.

Compuesto 226.8. 5-(4-(4-Cianofeml)-4-fluoropiperidm-1-carbonM)-2-cidopropil-4-etMbenzohidrazida. A una solución de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-2-ciclopropil-4-etilbenzoato de metilo (compuesto 226.7, 200 mg, 0,460 mmol, 1,00 equiv) en etanol (15 ml), se le añadió hidrato de hidrazina (80 %, 5 ml). La solución resultante se agitó a 80 °C durante 15 h. Después de enfriar a temperatura ambiente, la reacción se inactivó con 50 ml de agua, a continuación se extrajo con 3 x 50 ml de diclorometano. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío para dar 150 mg (75%) de 5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-

il)benzonitrilo. El compuesto del título (50 mg, sólido de color blanco, 95%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 217.3 y usando el compuesto 226.8 (50 mg) en lugar del compuesto 217.2.

Compuesto 226. 4-(1-(4-CidopropN-2-etN-5-(5-metoxi-4H-1,2,4-triazol-3-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título (50 mg, sólido de color blanco, 95 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 219.7 y usando el compuesto 226.9 (300 mg) en lugar del compuesto 219.6. El producto impuro (~30 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (46,0 % CH3CN asciende al 57,0 % en 8 minutos, asciende al 100,0 % en 1 minuto, desciende al 46,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 15 mg del compuesto del título como un sólido de color blanco. m/z (ES+) 474 (M+H)+. 1H-RMN (300MHz, CD3OD, ppm): 87,73 (d, 2H), 7,65 - 7,57 (m, 2H), 7,46-7,32 (m, 1H), 7,03 (s, 1H), 4,85-4,73 (m, 1H), 4,00 (s, 3H), 3,48-3,32 (m, 2H), 3,28-3,21 (m, 1H), 2,71-2,62 (m, 2H), 2,28-2,14 (m, 2H), 2,10-1,80 (m, 3H), 1,28-1,20

Compuesto 227. 4-(1-(4-CidopropN-5-(5-etoxi-4H-1,2,4-tnazol-3-M)-2-etMbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclopropil-2-metil-5-(5-metoxi-4H-1,2,4-triazol 3-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo (compuesto 226). m/z (ES+) 488 (M+H)+. 1H-RMN (300 MHz, CD3OD): 87,80-7,78 (m, 2H), 7,70-7,63 (m, 2H), 7,51 (m, 1H), 7,07 (s, 1H), 4,89-4,82 (m, 1H), 4,40 (t aparente, 2H), 3,57-3,52 (m, 2H), 3,28 3,25 (m, 1H), 2,80-2,50 (m, 2H), 2,40-2,10 (m, 4H), 1,95-1,80 (m, 1H), 1,45 (t, 3H), 1,32-1,22 (m, 3H), 1,01-0,99 (m, 2H), 0,73-0,73
Compuesto 228.1.5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclopropil-4-etilbenzoato de metilo. A un matraz de
fondo redondo, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió una solución de ácido 4-cidopropil-2-etil-5-(metoxicarbonil)benzoico (compuesto 226.6., 500 mg, 2,01 mmol, 1,00 equiv ) en N,N-dimetilformamida (20 ml). Se añadieron HBTU (1,53 g, 4,03 mmol, 2,00 equiv), DIEA(780 mg, 6,04 mmol, 3,00 equiv) a la solución de reacción, y esta se agitó durante 5 minutos a 25 °C. Se añadió 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, se añadió 410 mg, 2,20 mmol, 1,10 equiv) a la mezcla anterior. Después de agitar durante 15 h a 25 °C, la reacción se interrumpió mediante la adición de agua. La mezcla resultante se extrajo con 100 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 4 x 40 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (4:1) como eluyente para proporcionar 0,830 g (99%) de 5-(4-(4-cianofenil)piperidin-1 -carbonil)-2-ciclopropil-4-etilbenzoato de metilo como un aceite incoloro.

Compuesto 228.2. 5-(4-(4-Cianofeml)piperidm-1-carboml)-2-cidopropM-4-etMbenzohidrazida. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclopropil-4-etilbenzoato de metilo (compuesto 228.1, 830 mg, 1,99 mmol, 1,00 equiv) en etanol (15 ml). A la mezcla de reacción se le añadió hidrazina (5 ml, 100 equiv). La solución resultante se agitó durante 15 h a 100 °C, a continuación se concentró al vacío. La mezcla resultante se extrajo con 100 ml de diclorometano y las capas orgánicas combinadas se lavaron con 1 x 30 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 0,800 g (96 %) de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclopropil-4-etilbenzohidrazida como un sólido de color blanco.

Compuesto 228.3. 4-(1-(5-(5-Amino-1,3,4-oxadiazol-2-il)-4-ciclopropil-2-etilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título (750 mg, sólido de color marrón claro, 94 %) se preparó usando un procedimiento similar al

Compuesto 228.4. 4-(1-(4-Ciclopropil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzamida. El compuesto del título (80 mg, sólido de color amarillo, 36 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 219.7 y usando el compuesto 228.3 (200 mg) en lugar del compuesto 219.6.

Compuesto 228. 4-(1-(4-Ciclopropil-5-(5-etoxi-4H-1,2,4-triazol-3-il)-2-etilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió una solución de 4-(1-(4-ciclopropil-5-(5-etoxi-4H-1,2,4-triazol-3-il) -2-etilbenzoil)piperidin-4-il)benzamida (compuesto 228.4, 80,0 mg, 0,160 mmol, 1,00 equiv) en diclorometano (10 ml). Se añadieron anhídrido trifluoroacético (34,0 mg, 0,160 mmol, 1,00 equiv) y trietilamina (33,0 mg, 0,330 mmol, 2,00 equiv) gota a gota a la mezcla agitada. La solución resultante se agitó durante 2 h a 25 °C, a continuación se lavó con 1 x 10 ml de solución saturada de cloruro sódico. La capa acuosa se extrajo con 2 x 20 ml de diclorometano, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El producto impuro (30 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (42 % CH3CN asciende al 57 % en 8 minutos, asciende al 100 % en 1,5 minutos, desciende al 42 % en 1 minuto); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 15 mg (20 %) del compuesto del título como un sólido de color blanquecino. m/z
(ES+) 470 (M+H)+. 1H-RMN (300MHz, CD3OD, ppm): 57,64 (d, 2H), 7,46-7,28 (m, 3H), 7,02-7,01 (m, 1H), 4,89-4,80 (m, 1H), 4,40-4,33 (m, 2H), 3,58-3,56 (m, 1H), 3,27-3,25 (m, 1H), 3,04-2,90 (m, 2H), 2,73-2,58 (m, 2H), 2,31-2,29 (m, 1H), 1,94-1,96 (m, 1H), 1,82-1,78 (m, 3H), 1,45 (t, 3H), 1,32-1,12 (m, 3H), 0,98-0,88 (m, 2H), 0,71-0,68 (m, 2H).

Compuesto 229. 4-(1-(4-CidopropM-2-etM-5-(5-metoxi-4H-1,2,4-triazol-3-M)benzoM)piperidm-4-M)benzomtrMo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 228. m/z (ES+) 456 (M+H)+. 1H-RMN (300MHz, CD3OD, ppm): 57,64 (d, 2H), 7,43 7,27 (m, 3H), 7,03-7,02 (m, 1H), 4,0 (s, 3H), 3,59-3,55 (m, 1H), 3,28-3,27 (m, 1H), 3,04-2,98 (m, 2H), 2,97-2,56 (m, 2H), 2,53-2,29 (m, 1H), 2,0-1,98 m, 1H, 1,76-1,42 m, 3H , 1,30-1,16 m, 3H , 0,99-0,94 (m, 2H), 0,70-0,64 (m, 2H).

Compuesto 230.1. Ácido 4-ciclobutil-5-yodo-2-metilbenzoico. A una solución de 4-ciclobutil-5-yodo-2-metilbenzoato de metilo (compuesto 152.3, 35,0 g, 106 mmol, 1,00 equiv) en metanol (200 ml) a 0-5 °C, se le añadió gota a gota hidróxido sódico acuoso (12,7 g , 318 mmol, 3,00 equiv en 100 ml de agua). La mezcla resultante se agitó durante 3 h a 60 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El pH de la fase acuosa restante se ajustó a ~4 con cloruro de hidrógeno (acuoso, 2 M). Los sólidos resultantes se recogieron por filtración y se secaron en un horno a presión reducida para dar 31,0 g (93 %) del compuesto del título como un sólido de color blanco

Compuesto 230.2. Ácido 4-ciclobutil-5-(metoxicarbonil)-2-metilbenzoico. A una solución de ácido 4-ciclobutil-5-yodo-2-metilbenzoico (compuesto 230.1, 3,00 g, 9,49 mmol, 1,00 equiv) en THF (40 ml) en nitrógeno a -78 °C, se le añadió gota a gota n-BuLi (9,5 ml, 2,50 equiv, 2,5 M en THF). Después de 10 minutos, se añadió gota a gota una solución de carbonato de dimetilo (2,56 g, 28,4 mmol, 3,00 equiv) en THF (10 ml) a -78 °C. La solución resultante se agitó durante 1 h a -78 °C, a continuación se inactivó cuidadosamente mediante la adición lenta de 50 ml de agua. El pH se ajustó a ~4 con cloruro de hidrógeno (ac., 1 M). La mezcla se extrajo con 2 x 80 ml de acetato de etilo, las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éterde petróleo (1:10-1:2) como eluyente para proporcionar 1,30 g (55%) de ácido 4-ciclobutil-5-(metoxicarbonil)-2-metilbenzoico como un sólido de color blanco.
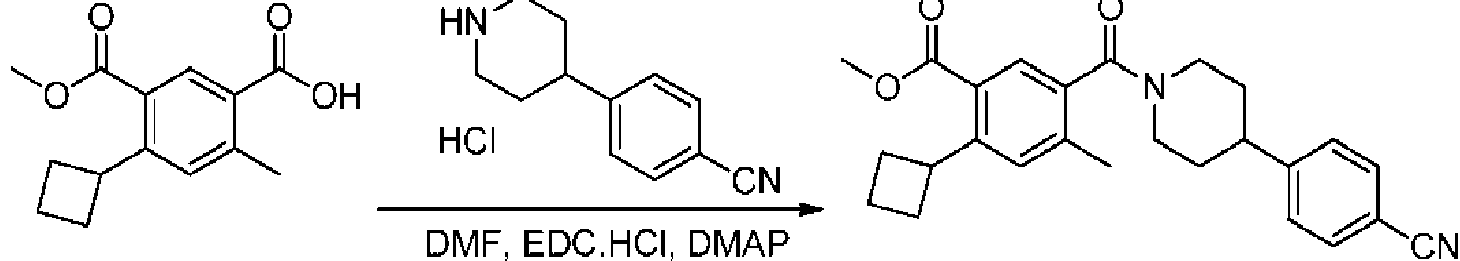
Compuesto 230.3. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de ácido 4-ciclobutil-5-(metoxicarbonil)-2-metilbenzoico (compuesto 230.2, 2,10 g, 8,46 mmol, 1,00 equiv) en W,A/-dimetilformamida (10 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 1,88 g, 8,44 mmol, 1,00 equiv), EDC HCl (3,22 g, 16,8 mmol, 2,00 equiv), y 4-dimetilaminopiridina (3,10 g, 25,4 mmol, 3,00 equiv) a la reacción. La solución resultante se agitó durante una noche a 25 °C, a continuación se diluyó con 100 ml de acetato de etilo. La mezcla se lavó con 2 x 30 ml de NH4Cl (ac., sat.) y 2 x 30 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (0:1 -1:3) como eluyente para dar 3,20 g (91 %) de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoato de metilo como un sólido de color blanco.
Compuesto 230.4. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzohidrazida. El compuesto del título (660 mg, sólido de color blanco, 66 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 219.5 y usando el compuesto 230.3 (1,00 g) en lugar del compuesto 219.4.

Compuesto 230. 4-(1-(4-Ciclobutil-5-(5-etil-1,3,4-oxadiazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzohidrazida (compuesto 230.4, 150 mg, 0,360 mmol, 1,00 equiv) en dioxano (5 ml). Se añadieron MeSOaH (7 mg, 0,07 mmol, 0,20 equiv) y 1,1,1-trietoxipropano (190 mg, 1,08 mmol, 3,00 equiv) a la reacción. La solución resultante se agitó durante 20 minutos a 110 °C, a continuación se enfrió a temperatura ambiente y se diluyó con 50 ml de acetato de etilo. La capa orgánica se lavó con 2 x 20 ml de agua, se secó sobre sulfato sódico anhidro, y se concentró al vacío. El producto impuro (200 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (64,0 % CH3CN asciende al 76,0 % en 6 minutos, asciende al 100,0 % en 4 minutos, desciende al 64,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 129 mg (79 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 455 (M+H)+.
1H-RMN(300MHz, CD3OD, ppm): 67,73-7,67 (m, 3H), 7,52-7,48 (m, 3H), 5-4,94 (m, 1H), 4,25-4,20 (m, 1H), 3,60-3,58 (m, 1H), 3,33-3,24 (m, 1H), 3,05-2,97 (m, 4H), 2,49 y 2,30 (2 singletes, rotámeros de amida, CH3 , 3H), 2,39-2,34 (m, 2H), 2,19-1,91 (m, 4H), 1,88-1,66 m, 4H , 1,49-1,42 t, 3H.

Compuesto 231.1. 4-Etenil-2-metilbenzoato de metilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera de nitrógeno, se le añadió una solución de 4-bromo-2-metilbenzoato de metilo (compuesto 152.1, 14,0 g, 61,1 mmol, 1,00 equiv) en W,W-dimetilformamida (150 ml). Se añadieron tributil(etenil)estannano (29,3 g, 92,4 mmol, 2,00 equiv) y Pd(PPh3)4 (7,10 g, 6,14 mmol, 0,10 equiv) a la mezcla de reacción. La mezcla resultante se agitó durante una noche a 100 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 400 ml de acetato de etilo. La capa orgánica se lavó con 2 x 400 ml de NH4Cl (ac.) y 2 x 400 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro y a continuación se concentraron al vacío. El producto impuro se purificó mediante CombiFlash con las siguientes condiciones (IntelFlash-1): fase móvil, éter de petróleo/acetato de etilo = 1 :0 incrementándose hasta éter de petróleo/acetato de etilo = 100:1 en el plazo de 20 minutos; Detector, UV 254 nm. Esto dio como resultado 6,81 g (63 %) de 4-etenil-2-metilbenzoato de metilo como un aceite incoloro.

Compuesto 231.2. 2-Metil-4-(3-oxociclobutil)benzoato de metilo. A una solución de W,W-dimetilacetamida (5,5 ml) en DCE (20 ml) en nitrógeno a -15 °C, se le añadió una solución de anhídrido trifluorometanosulfónico (10 ml) en DCE (50 ml) gota a gota. A continuación, la mezcla se agitó durante 10 minutos a -15 °C para preparar la solución A. A otro matraz se le añadió 4-etenil-2-metilbenzoato de metilo (compuesto 231.1, 5,30 g, 0,03 mol, 1,00 equiv). Se añadió una solución de 2,4,6-trimetilpiridina (5,5 ml) en DCE (80 ml) gota a gota a -15 °C. Se añadió la mezcla resultante a la solución A, gota a gota en una atmósfera inerte de nitrógeno. La solución resultante se agitó durante una noche a 80 °C. Después de enfriar a temperatura ambiente, la mezcla se inactivó cuidadosamente con agua. La mezcla resultante se extrajo con 2 x 200 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 3 x 400 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50-1:5) como eluyente para proporcionar 2,95 g (45 %) de 2-metil-4-(3-oxociclobutil)benzoato de metilo como un aceite de color marrón.

Compuesto 231.3. 4-(3,3-Difluorociclobutil)-2-metilbenzoato de metilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de 2-metil-4-(3-oxociclobutil)benzoato de metilo (compuesto 231.2, 3,00 g, 13,8 mmol, 1,00 equiv) en diclorometano (100 ml). A la mezcla de reacción se le añadió DAST (22,2 g, 137 mmol, 10,00 equiv). La solución resultante se agitó durante una noche a 25 °C, a continuación se inactivó cuidadosamente añadiendo lentamente (gota a gota en primer lugar) 500 ml de bicarbonato sódico (ac.) y hielo. La mezcla se extrajo con 300 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2 x 300 ml de bicarbonato sódico (ac.) y 2 x 300 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:20) como eluyente para proporcionar 3,00 g (91 %) de 4-(3,3-difluoroc¡clobut¡l -2-metilbenzoato de metilo como un aceite de color marrón.

Compuesto 231.4. 4-(3,3-Difluorociclobutil)-5-yodo-2-metilbenzoato de metilo. El compuesto del título (3,02 g, sólido de color amarillo, 66 %) se preparó usando un procedimiento similar al usado para la preparación del compuesto 181.4 y usando el compuesto 231.3 (3,00 g) en lugar del compuesto 181.3.

Compuesto 231.5. Ácido 4-(3,3-difluorociclobutil)-5-(5-etil-4H-1,2,4-triazol-3-il)-2-metilbenzoico. El compuesto del título se sintetizó usando manipulaciones químicas estándar y procedimientos similares a los usados para la

Compuesto 231. 4-(1-(4-(3,3-DifluorocidobutM)-5-(5-etN-4H-1,2,4-triazol-3-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 4-(3,3-difluorociclobutil)-5-(5-etil-4H-1,2,4-triazol-3-il)-2-metilbenzoico (compuesto 231.5, 211 mg, 0,660 mmol, 1,00 equiv) en W,A/-dimetilformamida (5 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 146 mg, 0,660 mmol, 1,00 equiv), EDCHCl (252 mg, 1,31 mmol, 2,00 equiv), y 4-dimetilaminopiridina (160 mg, 1,31 mmol, 2,00 equiv) a la reacción. La solución resultante se agitó durante 3 h a 30 °C en un baño de aceite, a continuación se diluyó con 30 ml de acetato de etilo. La mezcla se lavó con 3 x 40 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró al vacío. El producto impuro (448 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (48,0 % CH3CN asciende al 62,0 % en 6 minutos, asciende al 100,0 % en 4 minutos, desciende al 48,0 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 186 mg (58 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 490 (M+H)+. 1H-RMN (300MHz, CD3OD, ppm): ó 7,68 (d, J=8Hz, 2H), 7,59-7,44 (m, 4H), 4,89-4,80 (m, 1H), 4,03-3,98 (m, 1H), 3,67-3,61 (m, 1H), 3,28-3,24 (m, 1H), 3,04-3,00 (m, 2H), 2,97-2,89 (m, 2H), 2,87-2,83 (m, 2H), 2,62 2,60 (m, 2H), 2,48 y 2,38 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,05-2,00 (m, 1H), 1,88-1,58 (m, 3H), 1,41 (t, 3H).

Compuesto 232.1. 5-(5-(Dimetilcarbamoil)-1H-imidazol-2-il)-2,4-dimetilbenzoato de metilo. Una mezcla de 3-bromo-W,W-dimetil-2-oxopropanamida (246 mg), clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5, 237 mg), y carbonato potásico (311 mg) en acetonitrilo (12 ml) se calentó a reflujo durante 48 horas.
Después de enfriar a temperatura ambiente, la mezcla se concentró. El residuo se disolvió en EtOAc y se lavó con solución saturada de cloruro sódico, se secó sobre MgSO4, se concentró y se purificó por cromatografía ultrarrápida (SÍO2; EtOAc) para dar 88 m del compuesto del título, miz ES+ 302 M+H .

Compuesto 232.2. cido 5-(5-(dimetilcarbamoil)-1H-imidazol-2-il)-2,4-dimetilbenzoico. Se disolvió 5-(5-(dimetilcarbamoil)-1H-imidazol-2-il)-2,4-dimetilbenzoato de metilo (compuesto 232.1, 113 mg, 0,37 mmol) en LiOH acuoso 2 N (1 ml) y tetrahidrofurano (THF) ( 5 ml) y se calentó a 50 °C durante 16 horas. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró al vacío. El pH de la capa acuosa restante se ajustó con HCl 2 N a pH 3-4. El precipitado resultante se recogió por filtración y se secó para dar 56 mg de ácido 5-(5-(dimetilcarbamoil)-

Compuesto 232. 2-(5-(4-(4-C¡anofeml)p¡per¡dm-1-carboml)-2,4-d¡met¡lfeml)-W,W-d¡met¡l-1H-¡m¡dazol-5-carboxamida. Una mezcla del ácido anterior (compuesto 232.2, 56 mg, 0,2 mmol), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 45 mg, 0,2 mmol), HbTu (152 mg, 0,4 mmol) y DIEA ( 105 ul, 0,6 mmol) en DMF (2 ml) se agitó a temperatura ambiente durante 16 horas. La reacción se diluyó con solución saturada de cloruro sódico y se extrajo con EtOAc. La capa orgánica se lavó con solución saturada de cloruro sódico, se secó sobre MgSO4 y se concentró. El residuo se purificó por cromatografía ultrarrápida (SO 2; metanol al 4% en EtOAc) para dar 44 mg de
Compuesto

compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dimetilfenil)-W,W-dimetil-1H-imidazol-5-

Compuesto 234. Ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoico. A una solución de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoato de metilo (compuesto 230.3, 1,50 g, 3,60 mmol, 1,00 equiv) en metanol (10 ml), se le añadió hidróxido sódico acuoso (577 mg, 14,4 mmol, en 5 ml de agua) gota a gota a 0°C. La solución resultante se agitó durante 2 h a 60 °C en un baño de aceite. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El pH de la capa acuosa restante se ajustó a 3-4 con cloruro de hidrógeno (ac., 2 M). Los sólidos resultantes se recogieron por filtración y se secaron en un horno a presión reducida para dar 1,20 g (83 %) del compuesto del título como un sólido de color blanco miz (ES+) 403 (M+H)+.
Compuesto 2

de fondo redondo, se le añadió una solución de ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4metilbenzoico (compuesto 234, 500 mg, 1,24 mmol, 1,00 equiv) en diclorometano (5 ml). Se añadieron cloruro de oxalino (317 mg, 2,50 mmol, 2,00 equiv) y W,A/-etilformamida (~5 mg) gota a gota a la mezcla. La solución resultante se agitó durante 1 h a 40 °C, a continuación se concentró y se secó a presión reducida para dar 480 mg (92%) del compuesto del título como un aceite de color amarillo claro.

Compuesto 235.2. 4-(1-(5-(2-BromoacetM)-4-ddobutM-2-metMbenzoM)piperidm-4-M)benzomtrMo. A una solución de TMSCHN2 (2 M en hexano) (0,476 ml, 2,00 equiv) en diclorometano (10 ml) en nitrógeno a 0 °C, se le añadió gota a gota una solución de cloruro de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoílo (compuesto 235.1, 200 mg, 0,480 mmol, 1,00 equiv) en diclorometano (3 ml). La mezcla resultante se agitó durante una noche a 25 °C. A continuación se añadió HBr (40 %) (0,154 ml, 1,50 equiv) gota a gota a 0 °C y la mezcla se agitó durante otras 2 h a 0 °C, a continuación se diluyó con 50 ml de diclorometano. La capa orgánica se lavó con 2 x 20 ml de agua y 1 x 20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (0:1-1:50-1:5) como eluyente para proporcionar 200 mg (88 %) de 4-(1-(5-(2-bromoacetil)-4-ciclobutil-2-metilbenzoil)piperidin-4-il)benzon¡trilo como un aceite de color amarillo.

Compuesto 235. 4-(1-(4-CidobutM-5-(2-etiMH-imidazol-5-M)-2-metMbenzoM)piperidm-4-M)benzomtrMo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de 4-(1-(5-(2-bromoacetil)-4-ciclobutil-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 235.2, 100 mg, 0,210 mmol, 1,00 equiv) en CH3CN (5 ml). Se añadieron clorhidrato de propionimidamida (22,8 mg, 1,00 equiv) y carbonato potásico (86,6 mg, 0,63 mmol, 3,00 equiv) a la reacción. La solución resultante se agitó durante 3 h a 80 °C, a continuación se enfrió a temperatura ambiente y se concentró al vacío. El residuo se disolvió en 20 ml de agua. La mezcla se extrajo con 2 x 20 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El producto impuro (~100 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (29,0 % CH3CN asciende al 43,0 % en 7 minutos, asciende al 100,0 % en 3 minutos, desciende al 29,0 % en 2 minutos); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 43 mg (44 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 453 (M+H)+. 1H RMN (300 MHz, CD3OD): 67,68 (d con estructura fina, J = 7,8 Hz, 2H), 7,56-7,53 (m, 4H), 7,30 y 7,20 (2 singletes, rotámeros de amida, Ar-H, 1H), ~4,9 (1H parcialmente oscurecido por pico de agua), 3,84-3,70 (m, 1H), 3,70-3,53 (m, 1H), 3,33-3,19 (m, 1H parcialmente oscurecido por pico de disolvente metanol), 3,13 2,92 (m, 4H), 2,48 & 2,38 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,29-2,11 (m, 4H), 2,11-1,93 (m, 2H), 1,93 1,72 (m, 3H), 1,72-1,53 (m, 1H), 1,45
Compuesto 236.1. 4-(1-(5-(2-Bromoacetil)-4-cidobutil-2-metilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(5-(2-bromoacetil)-4-ciclobutil-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 235.2) pero usando sal HCI del compuesto 11.2, en lugar del compuesto 1.5.

Compuesto 236.4-(1-(4-CidobutN-2-metN-5-(2-metiMH-imidazol-5-N)benzoM)-4-fluoropiperídm-4-il)benzonitrilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de 4-(1-(5-(2-bromoacetil)-4-ciclobutil-2-metilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo
(compuesto 236.1, 40 mg, 0,08 mmol, 1,00 equiv) en CH3CN (10 ml). Se añadieron clorhidrato de acetimidamida (7,6 mg, 0,08 mmol, 1,00 equiv) y carbonato potásico (33,4 mg, 0,24 mmol, 3,00 equiv) a la reacción. La solución resultante se agitó durante 2 h a 80 °C en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se disolvió en 50 ml de acetato de etilo. La capa orgánica se lavó con 2 x 20 ml de agua, 2 x 20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El producto impuro (~50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMa Dz U)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (29,0 % CH3CN asciende al 43,0 % en 8 minutos, asciende al 100,0 % en 2 minutos, desciende al 29,0 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 13 mg (35 %) del compuesto del título como un sólido de color blanco. miz (ES+) 457 (M+H)+. 1H RMN (400 MHz, CD3OD): 87,78 (d, 2H), 7,65 (d, 2H), 7,50-7,46 (m, 2H), 7,35 y 7.23 (2 s, rotámeros de amida, 1H), 4,91-4,83 (m, 1H), 3,76-3,63 (m, 1H), 3,54-3,49 (m, 2H), 3,32-3,30 (m, 1H), 2,70 (s, 3H), 2,49 y 2,39 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,34-2,28 (m, 7H), 1,98-1,80

Compuesto 237.1. N'-Hidroxi-3-metoxipropanimidamida. En un matraz de fondo redondo, se colocó una solución de 3-metoxipropanonitrilo (10,0 g, 118 mmol, 1,00 equiv) en etanol (20 ml). A la reacción se le añadió NH2OH (50 % en H2O) (10 ml). La solución resultante se agitó durante una noche a 90 °C en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se diluyó con 30 ml de H2O, y se lavó con 2 x 20 ml de acetato de etilo. Las capas acuosas se combinaron y se concentraron a presión reducida para dar 10,0 g (65 %) del compuesto del título como un aceite incoloro.
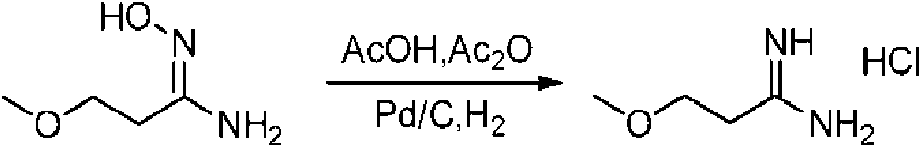
Compuesto 237.2. Clorhidrato de 3-metoxipropanimidamida. En un matraz de fondo redondo, se colocó una solución de N'-hidroxi-3-metoxipropanimidina (compuesto 237.1, 10,0 g, 76,2 mmol, 1,00 equiv, 90 %) en AcOH (50 ml). Se añadió anhídrido acético (9,50 g, 93,1 mmol, 1,10 equiv) gota a gota y la mezcla resultante se agitó durante 30 minutos a temperatura ambiente. Después de purgar el matraz con nitrógeno, se añadió paladio sobre carbono (10 %, 60 % de agua, 5 g). El matraz se purgó cuidadosamente con nitrógeno de nuevo y la atmósfera se cambió a continuación a hidrógeno. La mezcla se agitó durante una noche a 20 °C a presión atmosférica de hidrógeno. Después de purgar el sistema con nitrógeno, los sólidos se retiraron a continuación por filtración y el filtrado se concentró a presión reducida. El residuo de diluyó con 50 ml de H2O. El valor de pH de la solución se ajustó a 2-3 con cloruro de hidrógeno (12 molil), a continuación se lavó con 2x30 ml de acetato de etilo. Las capas acuosas se combinaron y se concentraron a presión reducida para proporcionar 5,00 g (40 %) del compuesto del título como un sólido de color blanquecino.

Compuesto 237.4-(1-(4-CidobutN-5-(2-(2-metoxietM)-1H-imidazol-5-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-2-metil-5-(2-metil-1H-imidazol-5-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo (compuesto 236) y usando 237.2 en lugar de clorhidrato de acetimidamida. miz (ES+) 501 (M+H)+.

Compuesto 238.1. 5-Acetil-4-ciclobutil-2-metilbenzoato de metilo. A una solución de 4-ciclobutil-5-yodo-2-metilbenzoato de metilo (152.3, 5,00 g, 15,1 mmol, 1,00 equiv) en DMSO (50 ml) en nitrógeno, se le añadieron 1-(eteniloxi)butano (3,03 g, 30,3 mmol , 2,00 equiv), DPPP (624 mg, 1,51 mmol, 0,10 equiv), Pd(OAc)2 (324 mg, 1,51 mmol, 0,10 equiv) y TEA (3,06 g, 30,2 mmol, 2,00 equiv). La mezcla resultante se agitó durante una noche a 120 °C en nitrógeno, a continuación se enfrió a temperatura ambiente y se diluyó con agua. El pH se ajustó a 1 con cloruro de hidrógeno (ac., 6 M). La mezcla resultante se diluyó con 200 ml de acetato de etilo, se lavó con solución saturada de cloruro sódico (3x200 ml), se secó (Na2SO4), y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etiloiéter de petróleo (1:30) como eluyente para proporcionar 2,28 g (61 %) del compuesto del título como un aceite de color amarillo

Compuestos 238.2 y 238.3. 5-(2-Bromoacetil)-4-ciclobutil-2-metilbenzoato de metilo y 4-ciclobutil-5-(2,2-dibromoacetil)-2-metilbenzoato de metilo. A una solución de 5-acetil-4-cidobutil-2-metilbenzoato de metilo (238.1, 500 mg, 1,83 mmol, 1,00 equiv, 90%) en cloroformo (5 ml) se le añadió gota a gota Br2 (325 mg, 2,03 mmol, 1,00 equiv) (ADVERTENCIA: reacción exotérmica con desprendimiento significativo de HBr). La mezcla resultante se agitó durante 3 h a 25°C, y a continuación se inactivó mediante la adición de 5 ml de H2O. La mezcla se extrajo a continuación con 50 ml de acetato de etilo. La fase orgánica se lavó con 2x10 ml de Na2S2O3 (ac., sat.) seguido de 1x20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida para dar 500 m de una mezcla de los compuestos del título como un aceite de color amarillo.

Compuesto 238.4. 4-Ciclobutil-5-(2-(2-metoxietil)-1H-imidazol-5-il)-2-metilbenzoato de metilo. En un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución de una mezcla de los compuestos 238.2 y 238.3 (500 mg, ~0,6 mmol, 80%), carbonato potásico (640 mg, 3,00 equiv ), clorhidrato de 3-metioxipropanimidamida (compuesto 237.2, 213 mg, 1,54 mmol) en acetonitrilo. La solución resultante se agitó durante una noche a 80 °C en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se diluyó con 60 ml de acetato de etilo. La fase orgánica se lavó con 2x25 ml de agua, 2x25 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (2:1) como eluyente para proporcionar 90 mg (43 %) del compuesto del título como un sólido de color amarillo.

Compuesto 238.5. Ácido 4-ciclobutil-5-(2-(2-metoxietil)-1H-imidazol-5-il)-2-metilbenzoico. Una mezcla del compuesto 238.4 (90 mg, 0,25 mmol, 1,00 equiv, 90 %) e hidróxido sódico acuoso (44 mg, 1,10 mmol, 4,00 equiv en 1 ml de agua) en metanol (3 ml) se agitó durante 2 h a 60 °C. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El pH de la capa acuosa residual se ajustó a 3-4 con cloruro de hidrógeno (acuoso, 2 M). La mezcla resultante se concentró a presión reducida para dar 80 mg (impuros) del compuesto del título como un sólido de color amarillo.

Compuesto 238. 4-(1-(4-Ciclobutil-5-(2-(2-metoxietil)-1H-imidazol-5-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. En un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución del compuesto 238.5 (80 mg, 0,23 mmol, 1,00 equiv, 90%) en N,N-dimetilformamida (2 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 54 mg, 0,24 mmol, 1,00 equiv), EDCHCl (93 mg, 0,49 mmol, 2,00 equiv), y 4-dimetilaminopiridina (59 mg, 0,48 mmol, 2,00 equiv) a la solución. La mezcla resultante se agitó durante 3 h a 30 °C, a continuación se diluyó con 50 ml de acetato de etilo. La fase orgánica se lavó con 2x20 ml de agua, 2x20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con cloroformo/metanol (20:1) como eluyente. El producto (~50 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones [(1#-Pre-HPLC-001 (Shimadzu): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (29 % CH3CN asciende al 43 % en 8 minutos, asciende al 100 % en 6 minutos, desciende al 29 % en 1 minuto); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 20,6 mg (19 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 483 (M+H)+.
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación de los compuestos 236, 237 y 238:


Compuesto 245.1. Clorhidrato de 5-carbamimidoil-4-ciclobutil-2-metilbenzoato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 5-(N'-hidroxicarbamimidoil)-2,4-dimetilbenzoato de metilo (compuesto 2.4) y clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5) usando 5-ciano-4-ciclobutil-2-metilbenzoato de metilo (compuesto 152.4) en lugar de 5-ciano-2,4-dimetilbenzoato de metilo (compuesto 2.3).

Compuesto 245.2. 4-Ciclobutil-5-(5-etil-1H-imidazol-2-il)-2-metilbenzoato de metilo. A un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de clorhidrato de 5-carbamimidoil-4-cidobutil-2-metilbenzoato de metilo (compuesto 245.1, 350 mg, 1,11 mmol, 1,00 equiv, 90%) en ACN (50 ml). Se añadieron 1-bromobutan-2-ona (186 mg, 1,23 mmol, 1,00 equiv) y carbonato potásico (513 mg, 3,53 mmol, 3,00 equiv, 95%) a la reacción. La solución resultante se agitó durante una noche a 80 °C. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo y se lavó con 3 x 30 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para proporcionar 290 mg (83 %) de de 4-ciclobutil-5- 5-etil-1 H-imidazol-2-¡l -2-met¡lbenzoato de metilo como un sólido de color amarillo.

Compuesto 245.3. Acido 4-ciclobutil-5-(5-etil-1H-imidazol-2-il)-2-metilbenzoico. Una mezcla de 4-ciclobutil-5-(5-etil-1H-imidazol-2-il)-2-metilbenzoato de metilo (compuesto 245.2, 200 mg, 0,600 mmol, 1,00 equiv, 90 %) e hidróxido sódico (107 mg, 2,68 mmol, 4,00 equiv) en una mezcla disolvente de metanol y agua (4/2 ml) se agitó durante 3 h a 60 °C. Después de enfriar a temperatura ambiente, el pH se ajustó a 3-4 con cloruro de hidrógeno (ac., 6 M). La mezcla resultante se concentró al vacío para dar 150 mg de un sólido de color blanco.

Compuesto 245. 4-(1-(4-Ciclobutil-5-(5-etil-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 4-ciclobutil-5-(5-etil-1H-imidazol-2-il)-2-metilbenzoico (compuesto 245.3, 153 mg, 0,480 mmol, 1,00 equiv, 90 %) en W,A/-dimetilformamida (5 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 132 mg, 0,590 mmol, 1,10 equiv), Ed C h c Í (204 mg, 1,01 mmol, 2,00 equiv, 95 %), y 4-dimetilaminopiridina (131 mg, 1,02 mmol, 2,00 equiv, 95 %) a la reacción. La solución resultante se agitó durante una noche a temperatura ambiente, a continuación se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (2:1) como eluyente. El producto impuro (~150 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001 (SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (28 % CH3CN asciende al 43 % en 7 minutos, asciende al 100 % en 2 minutos, desciende al 28 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 62 mg (29 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 453 (M+H)+. 1H RMN (400 MHz, CD3OD) ó 7,75-7,67 (m, 2H), 7,49-7,34 (m, 5H), 4,89-4,80 (m, 1H), 3,75-3,72 (m, 1H), 3,64-3,59 (m, 1H), 3,27-3,23 (m, 1H), 3,05-2,90 (m, 2H), 2,83 (c, 2H), 2,40 y 2,30 (2 singletes, rotámeros de amida, A C H 3, 3H), 2,27-1,97 (m, 6H), 1,90-1,70 (m, 4H), 1,40 (t, 3H).
O (COCI)2 o
< 0 ^'OH DCM, DMF ° ^ A ' q r |
Compuesto 246.1. C loruro de 2-metoxiacetilo. A una solución de ácido 2-metoxiacético (2,00 g, 22,2 mmol, 1,00 equiv) en diclorometano (20 ml) se le añadió gota a gota (COCl)2 (5,65 g, 2,00 equiv) en W,A/-dimetilformamida (0,1 ml) (se observó desprendimiento de gas). La reacción se agitó durante 1 h a 40 °C. La mezcla resultante se concentró a continuación a presión reducida para dar 2,10 g (70 %) de cloruro de 2-metoxiacetilo como un aceite de color amarillo.

Compuesto 246.2. 1-Bromo-3-metoxipropan-2-ona. A una solución de TMSCHN2 (2 M en hexano) (16 ml, 2,00 equiv) en diclorometano (40 ml) se le añadió cloruro de 2-metoxiacetilo (compuesto 246.1, 2,10 g, 15,5 mmol, 1,00 equiv, 80 %) gota a gota a 0 °C. Después de agitar durante 20 minutos, se añadió HBr (48 %, 2 ml) a la reacción. La solución resultante se agitó durante 30 minutos a 25 °C. La mezcla se lavó con 1 x 20 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. Esto dio como resultado 2,30 g (71 %) de 1-bromo-3-metoxipropan-2-ona como un aceite de color amarillo.

Compuesto 246.3. 4-CidobutN-5-(5-(metoximetM)-1H-imidazol-2-M)-2-metNbenzoato de metilo. A un matraz de fondo redondo, se le añadió una solución de 1-bromo-3-metoxipropan-2-ona (compuesto 246.2, 505 mg, 1,81 mmol, 1,00 equiv, 60%) en CH3CN (16 ml). Se añadieron clorhidrato de 5-carbamimidoil-4-ciclobutil-2-metilbenzoato de metilo (compuesto 245.1, 450 mg, 1,59 mmol, 1,00 equiv) y carbonato potásico (554 mg, 3,61 mmol, 3,00 equiv, 90 %) a la reacción. La solución resultante se agitó durante una noche a 80 °C en nitrógeno. Después de enfriar a temperatura ambiente, la reacción se inactivó cuidadosamente mediante la adición de agua. La mezcla resultante se extrajo con 2 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:2) como eluyente para proporcionar 170 mg (24%) de 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoato de metilo como un sólido de color blanco.

Compuesto 246.4. Ácido 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoico. Una solución de 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoato de metilo (compuesto 246.3, 150 mg, 0,430 mmol, 1,00 equiv, 90%) e hidróxido sódico acuoso (76,0 mg, 1,90 mmol, 4,00 equiv en 2 ml de agua) en metanol (4 ml) se agitó durante 2 horas a 60 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se eliminó a presión reducida. El valor del pH de la fase acuosa restante se ajustó a ~4 con cloruro de hidrógeno (ac., 4 M). La mezcla resultante se extrajo con 2 x 50 ml de acetato de etilo y las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron al vacío. Esto dio como resultado 120 mg (84%) de ácido 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-

Compuesto 246. 4-(1-(4-CidobutN-5-(5-(metoximetM)-1H-imidazol-2-M)-2-metNbenzoM)piperidm-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoico (compuesto 246.4, 50,0 mg, 0,150 mmol, 1,00 equiv, 90%) en W,A/-dimetilformamida (3 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 41,0 mg, 0,170 mmol, 1,10 equiv), EDCHCl (64,0 mg, 0,330 mmol, 2,00 equiv), y 4-dimetilaminopiridina (41,0 mg, 0,340 mmol, 2,00 equiv) a la reacción. La solución resultante se agitó durante 3 h a 25 °C, a continuación se inactivó con agua, y se extrajo con 2 x 30 ml de acetato de etilo. Las capas orgánicas combinadas se concentraron al vacío. El producto impuro (~50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (26,0 % de CH3Cn , asciende al 42,0 % en 7 minutos, asciende al 100,0 % en 2 minutos, desciende al 26,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 18 mg (25 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 469 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,73-7,64 (m, 3H), 7,52-7,32 (m, 4H), ~4,9 (1H parcialmente oscurecido por pico de agua), 4,60 (s, 2H), 3,81-3,68 (m, 1H), 3,68-3,52 (m, 1H), 3,46 (s, 3H), 3,35-3,22 (m, 1H parcialmente oscurecido por pico de disolvente metanol), 3,09-2,95 (m, 2H), 2,52 y 2,41 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,16-1,97 (m, 6H), 1,94-1,58 (m, 4H).

Compuesto 247. 4-(1-(4-CidobutN-5-(5-(metoximetM)-1H-imidazol-2-M)-2-metNbenzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 246), usando clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2, sal HCl) en lugar de clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5). m/z (ES+) 487
(M+H)+. 1H RMN (400 MHz, CD3OD): 57,79-7,67 (m, 2H), 7,66 (d, 3H), 7,53-7,39 (m, 2H), 4,99-4,93 (m, 1H), 4,85 (s, 2H), 3,76-3,56 (m, 3H), 3,50 (s, 3H), 2,42 y 2,34 (2 singletes, rotámeros de amida, A C H 3, 3H), 2,27-1,97 (m, 8H), 1,92-1,81 (m, 2H), 1,35-1,26 (m, 1H.

Compuesto 248. 4-(1-(4-CidobutN-5-(5-(metoximetM)-4-metiMH-imidazol-2-M)-2-metNbenzoN)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 246). m/z (ES+) 501 (M+H)+. 1H RMN (400 m Hz , CD3OD): 57,79 7,77 (m, 2H), 7,66 (d, 2H), 7,52-7,38 (m, 2H), 4,87-4,80 (m, 1H), 4,55 (s, 2H), 3,78-3,74 (m, 1H), 3,62-3,56 (m, 2H), 3,50 (s, 3H), 3,44-3,32 (m, 1H), 2,52 y 2,42 (2 singletes, rotámeros de amida, A C H 3, 3H), 2,44 (s, 3H), 2,11-1,99 (m, 8H), 1,93-1,83 (m, 2H).

Compuesto 249. 4-(1-(4-CidobutM-5-(4,5-dimetiMH-imidazol-2-M)-2-metMbenzoM)piperidm-4-M)benzomtrMo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 246). m/z (ES+) 453 (M+H)+. 1H RMN (400 MHz, CD3OD): 57,68 (d, 2H), 7,51 (d, 2H), 7,31 (d, 1H), 7,32 y 7,30 (2 singletes, rotámeros de amida, ArH, 1H), 4,90-4,88 (m, 1H), 3,96-3,87 (m, 1H), 3,70-3,62 (m, 1H), 3,32-3,29 (m, 1H), 3,02 (t, 2H), 2,44 y 2,33 (2 singletes, rotámeros de amida, A C H 3, 3H), 2,20 (s, 6H), 2,18-1,90 (m, 7H), 1,88 1,78 (m, 3H).

Compuesto 250. 4-(1-(4-Ciclobutil-2-metil-5-(5-metil-1H-imidazol-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 246). m/z (ES+) 439 (M+H)+. 1H RMN (400 MHz, CD3OD): 57,68 (d, 2H), 7,49-7,39 (m, 4H), 7,36 y 7,34 (2 singletes, rotámeros de amida, ArH, 1H), 4,90-4,86 (m, 1H), 3,75-3,70 (m, 1H), 3,63-3,55 (m, 1H), 3,33-3,24 (m, 1H), 3,04-2,99 (m, 2H), 2,51 y 2,41 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,44 (s, 3H), 2,28-1,96 (m, 6H), 1,89-1,81 (m, 3H), 1,77-1,74 (m, 1H).

Compuesto 251. 4-(1-(4-Ciclobutil-2-metil-5-(5-(trifluorometil)-1H-imidazol-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4- 1- 4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2Compuesto 252. 4-(1-(5-(5-Cloro-1H-imidazol-2-il)-4-ciclobutil-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 4-(1-(4-ciclobutil-5-(5-(metoximetil)-1H-imidazol-2-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 246). m/z (ES+) 459 (M+H)+.
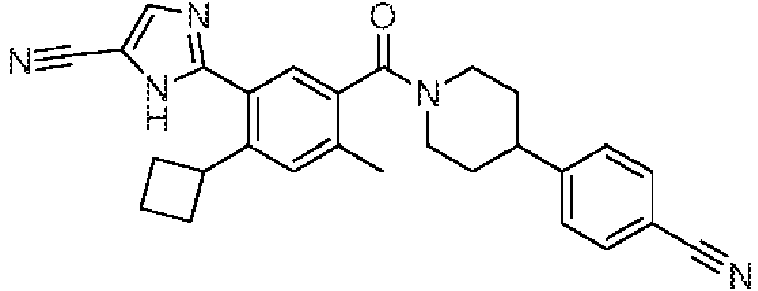
Compuesto 253. 2-(5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilfenil)-1H-imidazol-5-carbonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados

Compuesto 254. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-N,4-dimetilbenzamida. A una solución de ácido 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilbenzoico (compuesto 234, 50 mg, 0,124 mmol) en DMF (2 ml) se le añadieron Ed CI (36 mg, 0,186 mmol), HOBt (10 mg, 0,5 mmol), diisopropiletilamina (54 mg, 0,434 mmol), y metilamina (125 ul, 2,5 M en THF). La mezcla de reacción se agitó durante 12 horas a temperatura ambiente y se inactivó con NaHCO3 acuoso saturado (50 ml). Después de la extracción con acetato de etilo (2 x 50 ml), las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron, y se concentraron al vacío. La purificación mediante cromatografía ultrarrápida en SiO2 con acetato de etilo respecto a acetato de etilo/metanol = 98/2 produjo 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-N,4-dimetilbenzamida (28,3 mg, 55% de rendimiento) como un sólido de color blanco. m/z (ES+) 416 (M+H)+. 1H RMN (400 MHz, DMSO-da): 88,13 (c, J = 4,5 Hz, 1H), 7,77 (d, J = 8,3 Hz, 2H), 7,55-7,45 (m, 2H), 7,26 (s ancho, 1H), 7,12 y 6,99 (2 singletes, rotámeros de amida, Ar-H, 1H), 4,70 (d ancho, J = 11,9 Hz, 1H), 3,86-3,72 (m, 1H), 3,50-3,35 (m, 1H), 3,13 (t con estructura fina, J = 12,3 Hz, 1H), 2,99-2,78 (m, 2H), 2,72 (d, J = 4,6 Hz, 3H), 2,39-2,17 (m, 5H , 2,17-1,98 m, 2H , 1,98-1,81 m, 2H , 1,81-1,35 (m, 4H).

Compuesto 255. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-N-(2-metoxietil)-4-metilbenzamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-N,4-dimetilbenzamida (compuesto 254). m/z (ES+) 460 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 88,27 (t, 1H), 7,78 (d, 2H), 7,49 (d, 2H), 7,26 (s, 1H), 7,12 y 6,98 (2 singletes, rotámeros de amida, 1H), 4,70 (d, 1H), 3,85-3,75 (m, 1H), 3,44 (t, 3H), 3,39-3,33 (m, 2H), 3,30 (s, 3H), 3,19 - 3,03 (m, 1H), 3,02-2,71 (m, 2H), 2,32 y 2,26 (2 singletes, rotámeros de amida, ArCH, 3H), 2,38-2,17 (m, 2H), 2,15 2,00 (m, 2H), 1,98 -1,81 (m, 2H), 1,83 -1,47 (m, 4H).

Compuesto 256. 5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-ciclobutil-N-etil-4-metilbenzamida. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de 5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-N,4-dimetilbenzamida (compuesto 254). m/z (ES+) 430 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 88,21 (t, 1H), 7,78 (d, 2H), 7,56-7,45 (m, 2H), 7,31- 7,20 (m, 1H), 7,11 y 6,98 (2 singletes, rotámeros de amida, 1H), 4,74-4,65 (m, 1H), 3,85-3,74 (m, 1H), 3,50-3,37 (m, 1H), 3,22 (c, 2H), 3,20 - 3,05 (m, 2H), 2,32 y 2,22 (2 singletes, rotámeros de amida, ArCH3, 3H), 3,01 -2,76 (m, 2H), 2,38-2,17 (m, 2H), 2,18 1,97 (m, 2H), 1,98-1,84 (m, 2H), 1,81-1,38 (m, 3H), 1,10 (t, 3H).
Compuesto 257.1. Cloruro de (S)-tetrahidrofuran-2-carbonilo. A un matraz de fondo redondo, se le añadió una solución de ácido (S)-tetrahidrofuran-2-carboxílico (4,64 g, 40,0 mmol, 1,00 equiv) en diclorometano (25 ml). Se añadieron W,A/-dimetilformamida (0,05 ml, 0,05 equiv) y (COCl)2 (5,2 ml, 1.50 equiv) gota a gota a la reacción. La solución resultante se agitó durante 1 h a 25 °C, a continuación se concentró al vacío y para dar 5,00 g (93%) de cloruro de (S)-tetrahidrofuran-2-carbonilo como un aceite de color amarillo.

Compuesto 257.2. (S)-2-Bromo-1-(tetrahidrofuran-2-il)etanona. A un matraz de fondo redondo de tres bocas de 250 ml, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se le añadió una solución de (diazometil)trimetilsilano (20 ml, 2 M en hexano) en éter (150 ml). Una solución de (S)-cloruro de tetrahidrofuran-2-carbonilo (compuesto 257.1, 5,00 g, 37,2 mmol, 1,00 equiv) en éter/DCM (25/10 ml) se añadió gota a gota a 0 °C y se agitó durante 20 minutos a 0 °C. Se añadió gota a gota bromuro de hidrógeno (48 %) (8 ml, 1,50 equiv). La solución resultante se agitó durante 30 minutos a 10 °C, a continuación se lavó con 2 x 100 ml de agua y 1 x 50 ml de solución saturada de cloruro sódico. La mezcla se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50) como eluyente para proporcionar 2,50 g (35%) de (S)-2-bromo-1-(tetrahidrofuran-2-il)etanona como un aceite de color amarillo.

Compuesto 257.3. 2,4-Dimetil-5-(5-(tetrahidrofuran-2-il)-1H-imidazol-2-il)benzoato de (S)-metilo. A un matraz de fondo redondo, se le añadió clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5, 1,30 g) en CH3CN (30 ml). Se añadieron (S)-2-bromo-1-(tetrahidrofuran-2-il)etanona (compuesto 257.2, 1,92 g, 9,95 mmol) y carbonato potásico (4,20 g, 30,4 mmol) a la reacción. La solución resultante se agitó durante 3 días a 75 °C en nitrógeno. Después de enfriar a temperatura ambiente, los sólidos se retiraron por filtración. El filtrado se concentró al vacío y se diluyó con 30 ml de H2O. La fase acuosa se extrajo con 3 x 50 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y a continuación se concentraron al vacío. La mezcla se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:2 ) como eluyente para dar 400 mg (25 %) de 2,4-dimetil-5-(5-(tetrahidrofuran- 2-il)-1H-imidazol-2-il)benzoato de (S)-metilo como un sólido de color amarillo claro.
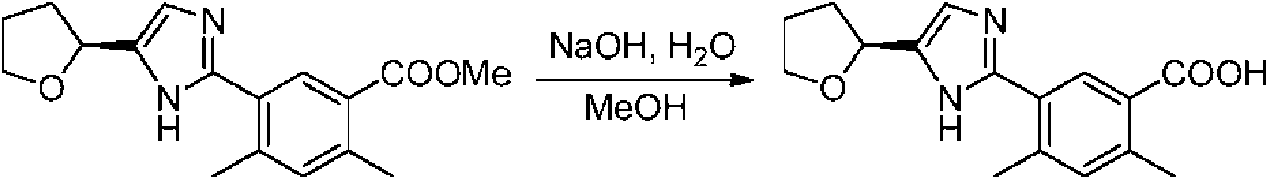
Compuesto 257.4. Ácido (S)-2,4-dimetil-5-(5-(tetrahidrofuran-2-il)-1H-imidazol-2-il)benzoico. A un matraz de fondo redondo se le añadió una solución de 2,4-dimetil-5-(5-(tetrahidrofuran-2-il)-1H-imidazol-2-il)benzoato de (S)-metilo (compuesto 257.3, 400 mg , 1,33 mmol, 1,00 equiv) e hidróxido sódico (300 mg, 7,50 mmol, 5,63 equiv) en una mezcla disolvente de metanol y H2O (20/10 ml). La solución resultante se agitó durante 2 h a 70 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. La capa acuosa residual se lavó con 2 x 50 ml de acetato de etilo. El pH de la capa acuosa restante se ajustó a 5-6 con cloruro de hidrógeno (ac., 6 M), a continuación se extrajo con 3 x 50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. Esto dio como resultado 340 mg (90 %) de 2,4-dimetil-5-(5-(tetrahidrofuran-2-¡l)-1H-im¡dazol-2-¡l benzoato de S -metilo como un sólido de color amarillo.
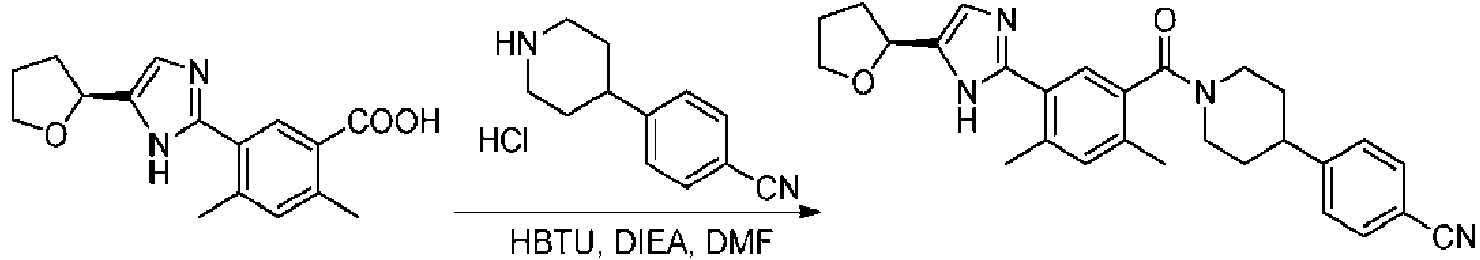
Compuesto 257. (S)-4-(1-(2,4-DimetN-5-(5-(tetrahidrofuran-2-N)-1H-imidazol-2-N)benzoN)piperidm-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 2,4-dimetil-5-(5-(tetrahidrofuran-2-il)-1H-imidazol-2-il)benzoato de (S)-metilo (compuesto 257.4, 143 mg, 0,500 mmol, 1,00 equiv) en W,W-dimetilformamida (10 ml). Se añadió HBTU (285 mg, 0,750 mmol, 1,50 equiv) a la reacción, y se agitó durante 30 minutos a 25 °C. A esto se le añadió clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 133 mg, 0,600 mmol, 1,20 equiv) y DIEA (390 mg, 3,02 mmol, 6,04 equiv) gota a gota. La solución resultante se agitó durante 30 minutos a 25 °C, a continuación se inactivo con 20 ml de agua. La mezcla se extrajo con 3 x 25 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con metanol/acetato de etilo (1:30) como eluyente. El producto (~100 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones [(1# Pre-HPLC-001 (SHIMADZU): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (35 % CH3CN
asciende al 52 % en 8 minutos, asciende al 100 % en 1 minuto, desciende al 35 % en 1 minuto); detector, Waters 2489 254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 34,6 mg (15%) del compuesto del título como un sólido de color blanco. m/z ES+) 455 (M+H)+.

Compuesto 258.1. Ácido 5-(5-etil-4H-1,2,4-triazol-3-il)-4-fluoro-2-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 152.8 y usando ácido 4-fluoro-2-metilbenzoico en lugar de ácido 4-bromo-2-metilbenzoico y propionohidrazida en lu ar de acetohidrazida.

Compuesto 258.2. 4-(1-(5-(5-Etil-4H-1,2,4-triazol-3-il)-4-fluoro-2-metilbenzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de ácido 5-(5-etil-4H-1,2,4-triazol-3-il)-4-fluoro-2-metilbenzoico (compuesto 258.1, 125 mg, 0,500 mmol, 1,00 equiv) en DMF (10 ml). Se añadieron EDCHCl (143 mg, 0,750 mmol, 1,50 equiv), clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 122 mg, 0,550 mmol, 1,10 equiv), y 4-dimetilaminopiridina (183 mg, 1,50 mmol, 3,00 equiv) por partes. La solución resultante se agitó durante 1 h a 40 °C en un baño de aceite, a continuación se inactivó con 50 ml de NH4Cl (ac. sat.). La mezcla resultante se extrajo con 40 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 3 x 30 ml de NH4Cl (ac. sat.), se secaron sobre sulfato sódico anhidro, y se concentraron al vacío. Esto dio como resultado 180 mg (86 %) de 4-(1-(5-(5-etil-4H-

Compuesto 258. 4-(1-(4-(Azetidin-1-il)-5-(5-etil-4H-1,2,4-triazol-3-il)-2-metilbenzoil)piperidin-4-il)benzonitrilo. A un tubo sellado de 10 ml, se le añadió una solución de 4-(1-[[5-(5-etil-4H-1,2,4-triazol-3-il)-4-fluoro-2-metilfenil]carbonil]piperidin-4-il)benzonitrilo (compuesto 258.2, 83,5 mg, 0,200 mmol, 1,00 equiv) en 1,4-dioxano (5 ml). A la mezcla de reacción se le añadió por partes clorhidrato de azetinina (93,0 mg, 0,990 mmol, 5,00 equiv). A la mezcla anterior también se le añadió carbonato potásico (276 mg, 2,00 mmol, 10,0 equiv). La solución resultante se agitó durante una noche a 105 °C detrás de una pantalla protectora contra explosiones, a continuación se enfrió ata. La reacción se inactivó con 20 ml de agua. La mezcla se extrajo con 30 ml de acetato de etilo y la capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El producto (~80 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 50 ml de NH4CO3 y CH3CN (41,0 % CH3CN asciende al 43,0 % en 8 minutos, asciende al 100,0 % en 2 minutos, desciende al 41,0 % en 2 minutos); detector, Waters 2489254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 18,0 mg (20 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 455 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,68 (d, J = 6,3 Hz, 2H), 7,48 (d, J =5,7 Hz, 2H), 7,24 & 7,13 (2 singletes, rotámeros de amida, Ar-H, 1H), 6,52 (s, 1H), 4,91-7,77 (m, 1H parcialmente oscurecido por pico de agua), 3,81-3,67 (m, 1H), 3,68 (t, J = 5,6 Hz, 4H), 3,32-3,18 (m, 1H), 3,05-1,91

Compuesto 259. 4-(1-(5-(5-Etil-4H-1,2,4-triazol-3-il)-2-metil-4-(metiltio)benzoil)piperidin-4-il)benzonitrilo. A un matraz de fondo redondo, se le añadió una solución de 4-(1-(5-(5-etil-4H-1,2,4-triazol-3-il)-4-fluoro-2-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 258.2, 20 mg, 0,050 mmol, 1,0 equiv) en W,W-etilformamida (3 ml). A la reacción se le añadió tiometóxido sódico (70 mg, 0,10 mmol, 2,0 equiv). La mezcla resultante se agitó durante 15 h a 110 °C en un baño de aceite, y a continuación se enfrió a temperatura ambiente y se inactivó con 100 ml de agua. La mezcla se extrajo con 50 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2 x 50 ml de
solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El producto (50 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (37% CH3CN asciende al 49% en 7 minutos, asciende al 100% en 3 minutos, desciende al 37% en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 4 mg (19 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 446 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,69 (d, J = 8,4 Hz, 2H), 7,61-7,43 (m, 3H), 7,35 (s ancho, 1H), ~4,85 (1H parcialmente oscurecido por pico de agua), 3,77-3,60 (m, 1H), ~3,3 (1H parcialmente oscurecido por pico de disolvente metanol), 3,08-2,94 (m, 2H), 2,91 (c, J = 7,7 Hz, 2H), 2,51 (s, 3H), 2,47 y 2,37 (2 singletes, rotámeros de amida, Ar-CHs, 3H), 2,10-1,97 (m, 1H), 1,88-1,55 (m, 3H), 1,40 (t, 3H).
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 5-(4-(4-cianofenil i eridin-1-carbonil -2-ciclobutil-N 4-dimetilbenzamida com uesto 254 :

(continuación)

Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida

(continuación)

(continuación)
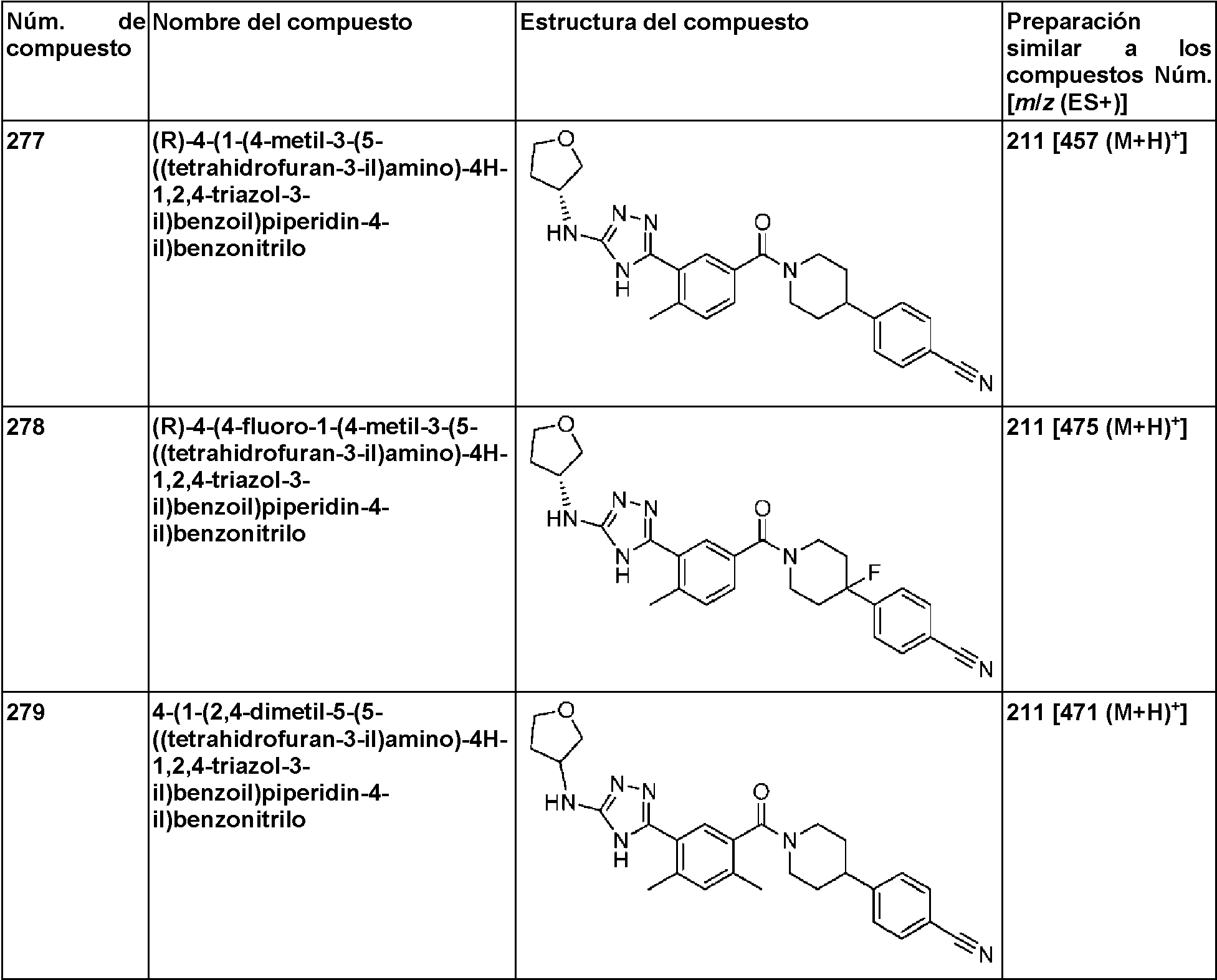
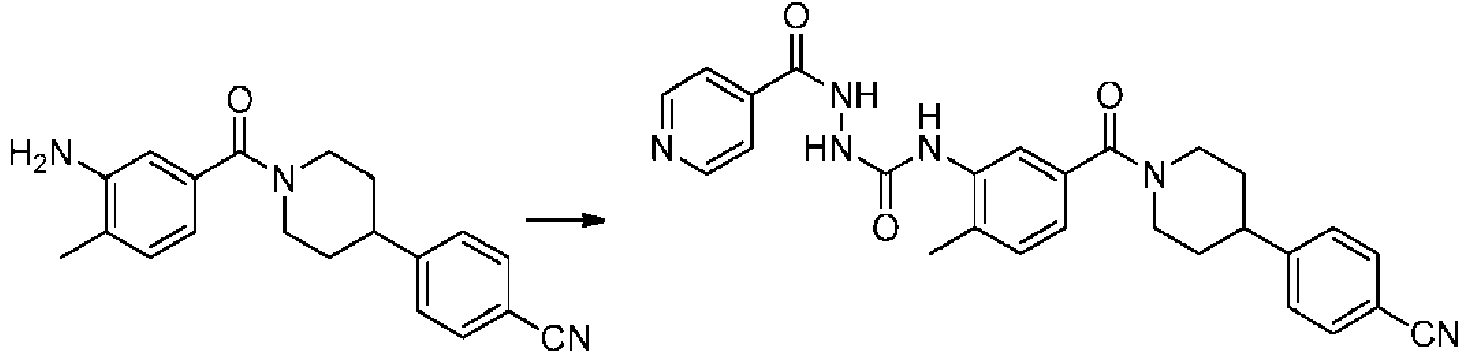
Compuesto 280.1. N-(5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilfenil)-2-isonicotinoilhidrazincarboxamida.
Una solución de 4-(1-(3-amino-4-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 121.1, 0,1 g, 0,31 mmol) y trietilamina (0,09 ml, 0,62 mmol) en DCM (5 ml) se añadió a una solución de fosgeno (20 % en tolueno, 0,31 ml, 0,62 mmol) en DCM (5 ml) a 0°C. Después la mezcla de reacción resultante se agitó a temperatura ambiente durante 1,5 horas, todos los disolventes se retiraron a presión reducida. El residuo se secó a alto vacío durante 30 minutos y a continuación se disolvió de nuevo en EtOH (5 ml). Se añadió isonicotinohidrazida (0,05 g, 0,34 mmol). La mezcla se calentó a 80 °C durante una noche. El etanol se retiró y el residuo se purificó utilizando TLC preparativa (MeOH al 10 % en DCM) para dar el producto como un sólido de color marrón claro. Rendimiento: 0,14 g, 93%. m/z (ES+) 483,2 (M+H)+.

Compuesto 280. 4-(1-(4-Metil-3-(5-(piridin-4-il)-1,3,4-oxadiazol-2-il)benzoil)piperidin-4-il)benzonitrilo A una solución de N-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilfenil)-2-isonicotinoilhidrazincarboxamida (compuesto 280.1, 0,14 g, 0,29 mmol) y PPh3 (0,09 g, 0,35 mmol) en DCM (10 ml) se le añadió trietilamina (0,061 ml, 0,43 mmol) seguido de CCl4 (0,08 ml, 0,87 mmol). La mezcla se calentó a reflujo durante 3 horas. TLC y LCMS mostraron que la reacción estaba completa. La mezcla se enfrió a temperatura ambiente, se diluyó con DCM (100 ml), y se lavó con agua (20 ml). La capa orgánica se secó con Na2SO4, se concentró y se purificó usando TLC preparativa (MeOH al 5% en CH2Cl2) para dar 54 mg (42%) de un sólido de color blanco. m/z (ES+) 465,0 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 810,00 (ancho 1H), 8,77 (d, 2H), 7,92 (s, 1H), 7,81-7,73 (m, 4H), 7,50 (d, 2H), 7,33 (d, 1H), 7,14 (d, 1H), 4,64 (m, 1h 

Compuesto 281.1. N-(5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilfenil)-1H-imidazol-1-carbotioamida. A una solución de 4-(1-(3-amino-4-metilbenzoil)piperidin-4-il)benzonitrilo (121.1, 0,1 g, 0,32 mmol) en DMF (3 ml), se le añadió 1,1'-tio-CDI (0,056 g, 0,32 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas. LCMS mostró que la reacción estaba completa. La mezcla se llevó a la siguiente etapa en un solo recipiente sin tratamiento final o

Compuesto 281.2. 1-(4-Aminopiridin-3-il)-3-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilfenil)tiourea. A una solución de N-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilfenil)-1H-imidazol-1-carbotioamida (281.1) en DMF continuada desde la etapa anterior, se le añadió piridin-3,4-diamina (0,034, 0,32 mmol). La mezcla se agitó a temperatura ambiente durante 3 horas. LCMS mostró que la reacción estaba completa. La reacción se llevó a la

Compuesto 281. 4-(1-(3-((1H-Imidazo[4,5-c]piridin-2-il)amino)-4-metilbenzoil)piperidin-4-il)benzonitrilo. A una solución de 1-(4-aminopiridin-3-il)-3-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilfenil)tiourea (281.2) en DMF continuada desde la etapa anterior, se le añadió EDCI (0,12 g, 0,64 mmol). La mezcla se agitó a temperatura ambiente durante una noche. El DMF se retiró a alto vacío y el residuo se purificó usando TLC preparativa (MeOH al 5% en EtOAc) para dar 76 mg (56% durante 3 etapas) de un polvo de color blanco. m/z (ES+) 437 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 88,84 (ancho, 1H), 8,48 (s, 1H), 8,40 (s, 1H), 8,08 (d, 1H), 7,78(d, 2H), 7,53 (d, 2H), 7,29 (d, 2H), 7,05 (d, 
Compuesto 282.1. N-((5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilfenil)carbamotioil)isobutiramida. En un matraz de fondo redondo, se colocó una solución de NH4SCN (72 mg, 0,95 mmol, 2,00 equiv) en acetona (10 ml). Se añadió una solución de cloruro de 2-metilpropanoílo (50 mg, 0,47 mmol, 1,00 equiv) en acetona (5 ml) gota a gota a
25 °C y la reacción se agitó durante una noche a 40 °C en un baño de aceite. A esto se le añadió 4-(1-(3-amino-4-metilbenzoil)piperidin-4-il)benzonitrilo (compuesto 121.1, 150 mg, 0,42 mmol, 1,00 equiv) a 25°C. La solución resultante se agitó durante 2 h a 25 °C, a continuación se concentró a presión reducida. El residuo se diluyó con 100 ml de acetato de etilo. La capa orgánica se lavó con 2x30 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (20:1) como eluyente para proporcionar 150 mg (64 %) del compuesto del título como un aceite de color marrón

Compuesto 282. 4-(1-(3-((5-Isopropil-4H-1,2,4-triazol-3-il)amino)-4-metilbenzoil)piperidin-4-il)benzonitrilo. En un tubo sellado de 10 ml se colocó una solución de N-((5-(4-(4-cianofenil)piperidin-1-carbonil)-2-metilfenil)carbamotioil)isobutiramida (compuesto 282.1, 200 mg, 0,40 mmol, 1,00 equiv, 90 %) en etanol (3 ml). Se añadieron NH2NH2HCl (234 mg, 2,23 mmol, 5,00 equiv) y carbonato potásico (185 mg, 1,34 mmol, 3,00 equiv) a la reacción. La solución resultante se agitó durante una noche a 80 °C en un baño de aceite detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se repartió entre acetato de etilo y agua. La fase orgánica se lavó con solución saturada de cloruro sódico, se secó (Na2SO4) y se concentró. El producto impuro (~50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-006(Waters)): Columna, SunFire Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,05 % de TFA y CH3CN (se mantiene un 5,0 % de CH3CN en 2 minutos, asciende al 30,0 % en 1 minuto, asciende al 60,0 % en 12 minutos, asciende al 100,0 % en 1 minuto); Detector, UV 254/220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 10,3 mg (6 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 429 (M+H)+.
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación de los compuestos 280, 121, 281 y 282:

(continuación)

(continuación)

Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación de los compuestos 26, 43, 48, 50, 51,64 y 80:

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)
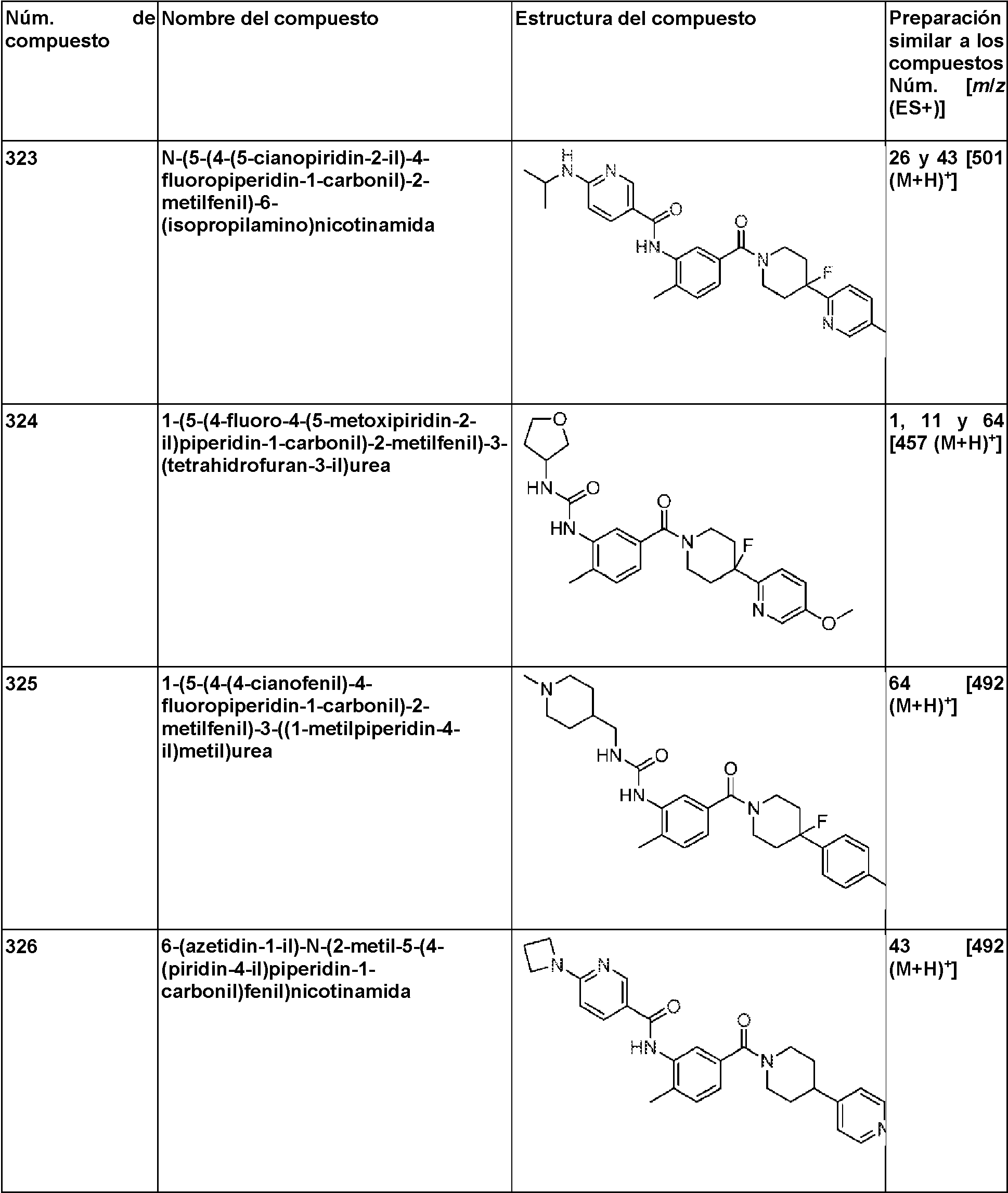
(continuación)

(continuación)

(continuación)

(continuación)

(continuación)


Compuesto 346.1. Ácido 2-(1-bencil-4-hidroxipiperidin-4-il)-5-bromobenzoico. A una solución agitada de ácido 2,5-dibromobenzoico (27,8 g, 100 mmol, 1,00 equiv) en THF/ Et2O (450/50 ml) en nitrógeno a -78 °C, se le añadió gota a gota n-BuLi (2,5 M) (88 ml, 2,20 equiv). Después de 2 h a -78 °C, se añadió 1-bencilpiperidin-4-ona (26,5 g, 140 mmol, 1,40 equiv). La solución resultante se agitó durante 0,5 h a -78 °C, y a continuación se calentó a temperatura ambiente, y se agitó durante una noche. La reacción se inactivó añadiendo cuidadosamente 200 ml de agua. El pH de la mezcla se ajustó a 2~3 con cloruro de hidrógeno (ac., 2 M). La fase acuosa se extrajo con 3x500 ml de acetato de etilo y 3x500 ml de tetrahidrofurano. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 38,9 g (impuros) del compuesto del título como un sólido de color amarillo.

Compuesto 346.2. 1'-Bencil-5-bromo-3H-espiro[isobenzofuran-1,4'-piperidin]-3-ona. En un matraz de fondo redondo, se colocó una solución del compuesto 346.1 impuro (38,9 g, 100 mmol, 1,00 equiv) en tetrahidrofurano (800 ml). A esto se le añadió ácido sulfúrico (8 ml) gota a gota. La solución resultante se agitó durante una noche a reflujo en un baño de aceite. El valor de pH de la solución se ajustó lentamente a 10 con LiOH (ac. sat.). La fase acuosa se extrajo con 3 x 500 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y a continuación se concentraron al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:8—1:5) como eluyente para proporcionar 8,00 g (22%) del compuesto del título como un sólido de color blanco.

Compuesto 346.3.1'-Bencil-5-bromo-3H-espiro[isobenzofuran-1,4'-piperidina]. A una solución del compuesto 346.2 (10,0 g, 26,9 mmol, 1,00 equiv) en tetrahidrofurano (150 ml) en nitrógeno a -10 °C se le añadió gota a gota BH3-THF (135 ml, 5,00 equiv). La solución resultante se agitó durante 30 minutos a temperatura ambiente y a continuación se calentó a temperatura de reflujo durante una noche. Después de enfriar a -10 °C, se añadió cloruro de hidrógeno (ac., 6 M, 60 ml) gota a gota a -10 °C. La solución resultante se calentó a reflujo durante 5 h en un baño de aceite. La mezcla de reacción se enfrió y el pH de la solución se ajustó a 10 con hidróxido potásico (ac., 1 M). La fase acuosa resultante se extrajo con 2x200 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2x300 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro y se

Compuesto 346.4. 1'-Bencil-3H-espiro[isobenzofuran-1,4'-piperidin]-5-carbonitrilo. En un matraz de fondo redondo de tres bocas, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución del compuesto 346.3 (12,0 g, 33,6 mmol, 1,00 equiv) en N,N-dimetilformamida (150 ml). Se añadieron Zn(CN)2 (4,50 g, 38,5 mmol, 1,14 equiv) y Pd(PPh3)4 (4,00 g) a la reacción. La solución resultante se agitó durante una noche a 90 °C en un baño de aceite. La mezcla de reacción se enfrió a ta, a continuación se inactivó con 200 ml de FeSO4 (ac., sat.) y se diluyó con acetato de etilo. La mezcla resultante se agitó vigorosamente después se filtró a través de celite y se lavó con FeSO41 M, agua y acetato de etilo. Las capas se separaron y la fase acuosa se extrajo con 2 x 200 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2x100 ml de cloruro sódico (ac. sat.), se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con éter de petróleo/acetato de etilo (10:1—5:1) como eluyente para dar 9,12 g (89 %) del compuesto del título como un aceite de color amarillo.

Compuesto 346.5. 3H-Espiro[isobenzofuran-1,4'-piperidin]-5-carbonitrilo. A una solución de 346.4 (9,12 g, 30,0 mmol, 1,00 equiv) en DCE (150 ml) a 0°C, se le añadió gota a gota cloroformiato de 1-cloroetilo (8,52 g, 60,0 mmol, 2,00 equiv). Después de agitar a temperatura ambiente durante 30 minutos, se añadió trietilamina (9,09 g, 3,00 equiv) cuidadosamente a la mezcla. La solución resultante se calentó a reflujo durante 2 h en un baño de aceite, a continuación se concentró a presión reducida. El residuo se disolvió en 100 ml de metanol y a continuación se calentó a reflujo durante 1 h en un baño de aceite. La mezcla resultante se concentró a presión reducida y el residuo se recogió en agua (100 ml). El pH de la mezcla se ajustó a 10 con hidróxido sódico (acuoso, 1 M). La fase acuosa se extrajo con 3x200 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2 x 100 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/metanol (100:0 a 3:1) como eluyente para

Compuesto 346. N-(5-(5-Ciano-3H-espiro[isobenzofuran-1,4'-pipeNd m ]-1'-NcarbonM)-2-metNfem l-6-(pirroMdm -1-il)nicotinamida. El compuesto del título se preparó usando procedimientos similares a los usados para la

Compuesto 347.1. 4-(4-cianofenil)-2-oxopiperidin-1-carboxilato de terc-butilo. En un matraz de fondo redondo, se colocó una solución de 4-(4-cianofenil)piperidin-1-carboxilato de terc-butilo (compuesto 1.4, 515 mg, 1,80 mmol, 1,00 equiv) en acetato de etilo (5 ml). Se añadió cuidadosamente una solución de N a O (963 mg, 4,50 mmol, 2,50 equiv) en agua (5 ml) y RuCh (74,7 mg, 0,36 mmol, 0,20 equiv). La mezcla resultante se agitó durante una noche a temperatura ambiente. Los sólidos se retiraron por filtración y el filtrado se lavó con 2x20 ml de agua, se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 432 mg (80%) del compuesto del título como un aceite de color amarillo claro.
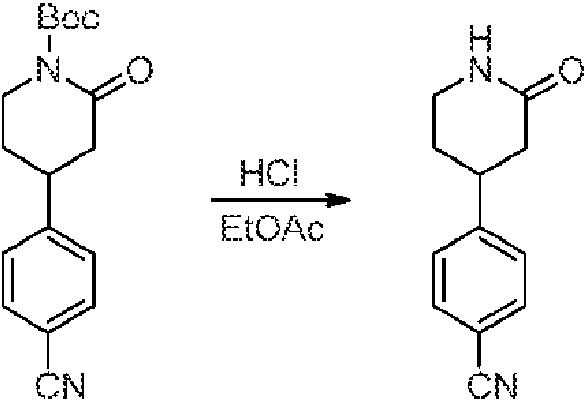
Compuesto 347.2. 4-(2-Oxopiperidin-4-il)benzonitrilo. En un matraz de fondo redondo, se colocó una solución del compuesto 347.1 (432 mg, 1,44 mmol, 1,00 equiv) en acetato de etilo (10 ml). Se burbujeó cloruro de hidrógeno gaseoso a través de la solución y la mezcla resultante se agitó durante 0,5 h a temperatura ambiente. Los sólidos se recogieron por filtración, a continuación se disolvieron en 50 ml de acetato de etilo. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 254 mg (88%) del compuesto del título como un
sólido de color blanco.

Compuesto 347.3. 4-(6-Metoxi-2,3,4,5-tetrahidropiridin-4-il)benzonitrilo. En un matraz de fondo redondo, se colocó una solución de 4-(2-oxopiperidin-4-il)benzonitrilo (347.2, 220 mg, 1,10 mmol, 1,00 equiv) en diclorometano (10 ml). Se añadió lentamente tetrafluoroborato de trimetiloxonio (244,2 mg, 1,65 mmol, 1,50 equiv) y la mezcla resultante se agitó durante 2 h a temperatura ambiente. El pH se ajustó cuidadosamente a 8 con bicarbonato sódico (ac.). La capa orgánica se lavó con 2x20 ml de H2O, se secó sobre sulfato sódico anhidro y se concentró a presión reducida para dar 212 mg (90 %) del compuesto del título como un sólido de color blanco.

Compuesto 347.4. 3-Amino-4-metilbenzohidrazida. En un matraz de fondo redondo, se colocó una solución de 3-amino-4-metilbenzoato de metilo (6,60 g, 40,0 mmol, 1,00 equiv) en etanol (100 ml). A la reacción se le añadió hidrato de hidrazina (10,0 g, 200 mmol, 5,00 equiv). La solución resultante se agitó durante 2 h a 100 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se repartió entre agua y acetato de etilo (20 ml). La capa acuosa se extrajo con 4x20 ml de acetato de etilo y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro, se concentraron a presión reducida y se secaron a alto vacío para dar 4,60 g (70 %) del compuesto del título como un sólido de color marrón.

Compuesto 347.5. 4-(3-(3-Amino-4-metilfenil)-5,6,7,8-tetrahidro-[1,2,4]triazolo[4,3-a]piridin-7-il)benzonitrilo. A una solución de 4-(6-metoxi-2,3,4,5-tetrahidropiridin-4-il) benzonitrilo (compuesto 347.3, 5,00 g, 23,36 mmol, 1,00 equiv) en 1,2-diclorobenceno (100 ml), se le añadido 3-amino-4-metilbenzohidrazida (compuesto 347.4, 4,63 g, 28,0 mmol, 1,20 equiv). La solución resultante se agitó durante una noche a 150 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla resultante se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (1:0-100:1) como eluyente para proporcionar 2,40 g (31 %) del compuesto del título como un sólido de color blanco.

Compuesto 347. N-(5-(7-(4-Cianofeml)-5,6,7,8-tetrahidro-[1,2,4]triazolo[4,3-a]pmdm-3-M)-2-metNfenM)-6-(isopropilam ino)nicotinamida. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 43 y usando el compuesto 347.5 en lugar del compuesto 42.2. m/z (ES+) 492 (M+H)+.

Compuesto 348.1. Ácido 2-hidroxi-6-metil-5-nitronicotínico. A una mezcla agitada de ácido 2-hidrox¡-6-metilnicotínico (14,0 g, 91,5 mmol, 1,00 equiv) en ácido sulfúrico (conc.) (140 ml) a 0 °C se le añadió gota a gota una solución de HNO3 (12,0 g, 190 mmol, 2,00 equiv) en ácido sulfúrico (conc.) (10 ml). La mezcla resultante se agitó a
continuación durante 2 h a 90 °C, se enfrió a temperatura ambiente y a continuación se inactivó con 250 ml de agua con hielo. Los sólidos resultantes se recogieron por filtración y se secaron en un horno a presión reducida para dar 15,8 g (87 %) del compuesto del título como un sólido de color amarillo

Compuesto 348.2. 2-Cloro-6-metil-5-nitronicotinato de metilo. A una solución de ácido 2-hidroxi-6-metil-5-nitronicotínico (compuesto 348.1, 15,0 g, 75,8 mmol, 1,00 equiv) en clorobenceno (150 ml) se le añadió cuidadosamente tricloruro de fosforilo (45,0 g, 296 mmol, 4,00 equiv) y N,N-dimetilformamida (1,5 ml, 0,10 equiv). La mezcla resultante se agitó durante 2 h a 135 °C, y a continuación se concentró a presión reducida. El residuo se disolvió en 20 ml de DCM. A esto se añadió gota a gota metanol (20 ml). La solución resultante se agitó durante 3 h a 25 °C, y a continuación se concentró a presión reducida. Al residuo se añadieron cuidadosamente 100 ml de agua y el pH de la mezcla resultante se ajustó lentamente a 8 con bicarbonato sódico (ac.). La fase acuosa se extrajo con 2x200 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 17,6 g (impuros) del compuesto del título como un sólido de color amarillo.

Compuesto 348.3. 5-Amino-6-metilnicotinato de metilo. Un matraz de fondo redondo, que contenía una solución de 2-cloro-6-metil-5-nitronicotinato de metilo (12,8 g, 55,5 mmol, 1,00 equiv) en metanol (120 ml) se purgó con nitrógeno. Se añadieron trietilamina (15,0 g, 149 mmol, 2,68 equiv) y paladio sobre carbono (1,30 g). Después de purgar adicionalmente el matraz con nitrógeno, la atmósfera se cambió a hidrógeno y la solución resultante se agitó durante 2 días a temperatura ambiente a presión atmosférica. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:50-1:5) como eluyente para proporcionar 3,50 g (38 %) del compuesto del título como un sólido de color amarillo.

Compuesto 348.4. Ácido 5-amino-6-metilnicotínico. En un matraz de fondo redondo, se colocó una solución de 5-amino-6-metilnicotinato de metilo (5,00 g, 30,1 mmol, 1,00 equiv) e hidróxido sódico (20 g) en MeOH/H2O (80/200 ml). La solución resultante se calentó a reflujo durante una noche. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El pH de la fase acuosa restante se ajustó a 4 con cloruro de hidrógeno acuoso (2 M). La mezcla resultante se concentró a continuación a presión reducida para dar 5,00 g (impuros) del compuesto del título como un sólido de color amarillo.
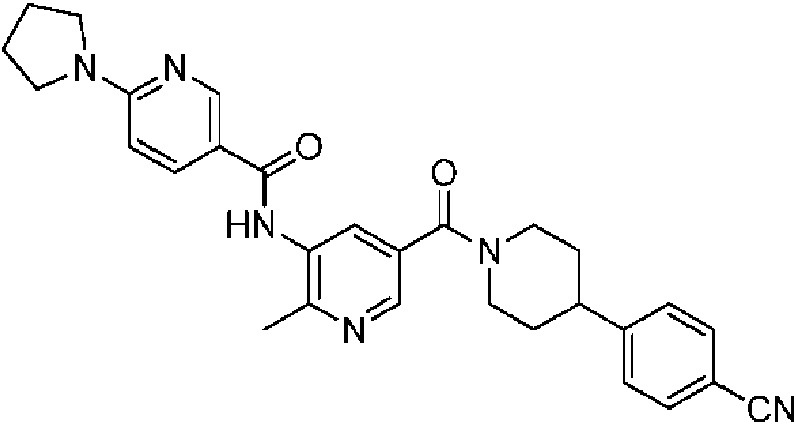
Compuesto 348. N-(5-(4-(4-Cianofenil)piperidin-1-carbonil)-2-metilpiridin-3-il)-6-(pirrolidin-1-il)nicotinamida. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 43 y usando los compuestos 348.4 y 1.5 en lugar de ácido 5-amino-2,4-dimetilbenzoico y el compuesto 11.2, respectivamente, miz (ES+) 495 ÍM+HÍ+.

Compuesto 349.1.1-Óxido de 2-bromo-5-metilpiridina. En un matraz de fondo redondo, se colocó una solución de 2-bromo-5-metilpiridina (10,0 g, 58,1 mmol, 1,00 equiv) en diclorometano (250 ml). Se añadió mCPBA (15,0 g, 86,9 mmol, 1,50 equiv) en varios lotes a temperatura ambiente. La solución resultante se agitó durante una noche a 30 °C, a continuación se diluyó con 50 ml de hidróxido sódico 2 N (ac.). El valor pH de la solución se ajustó a 10 con hidróxido
sódico 2 N (ac.). La fase acuosa se extrajo con 3x100 ml de diclorometano y las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron a presión reducida para dar 11,0 g (91 %) de 2-bromo-5-metilpiridin-1 -io-1-olato como un sólido de color amarillo.

Compuesto 349.2. 1-Óxido de 2-bromo-5-metil-4-nitropiridina. En un matraz de fondo redondo, se colocó HNO3 (15 ml) y ácido sulfúrico (20 ml). A esto se le añadió el compuesto 349.1 (11,0 g, 52,7 mmol, 1,00 equiv, 90%) en varios lotes a temperatura ambiente. La mezcla resultante se agitó a continuación durante una noche a 100 °C, a continuación se enfrió a temperatura ambiente y se inactivó con 50 ml de agua con hielo. El pH se ajustó lentamente a 2-3 con hidróxido sódico (ac. 2 M) y la mezcla resultante se extrajo con 3x50 ml de diclorometano. Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:2 ) como eluyente para proporcionar 3,00 g (23 %) del compuesto del título como un sólido de color amarillo.

Compuesto 349.3. 4-Amino-5-metilpicolinato de metilo. En un autoclave de 250 ml (30 atm, ADVERTENCIA: llevar a cabo detrás de una pantalla protectora contra explosiones, purgado y mantenido con una atmósfera inerte de nitrógeno, se colocó una solución de clorhidrato de 1-óxido de 2-bromo-5-metil-4-nitropiridina (compuesto 349.2, 3,00 g, 12,9 mmol, 1,00 equiv) en metanol (120 ml). Se añadieron trietilamina (2,60 g, 25,7 mmol, 2,00 equiv) y Pd(dppf)Ch (600 mg, 0,82 mmol, 0,06 equiv) a la reacción. El autoclave se purgó y la mezcla se agitó a continuación durante una noche a 30 atm de CO (g) a 90 °C. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo:metanol (20:1) como eluyente para proporcionar 1,50 g (67 %) 4-amino-5-metilpiridin-2-carboxilato de metilo como un sólido de color amarillo.
. 
Compuesto 349.4. Ácido 4-amino-5-metilpicolínico. Una solución de 4-amino-5-met¡lp¡col¡nato de metilo (compuesto 349.3, 1,50 g, 8,12 mmol, 1,00 equiv, 90 %) e hidróxido sódico 5 N (ac., 15 ml) en metanol (15 ml) se agitó durante una noche a temperatura ambiente, a continuación se concentró a presión reducida. El residuo se diluyó con 20 ml de H2O y el pH se ajustó a 3-4 con cloruro de hidrógeno acuoso 2 M. El precipitado resultante se recogió por

Compuesto 349. N-(2-(4-(4-Cianofenil)piperidin-1-carbonil)-5-metilpiridin-4-il)-6-(isopropilamino)nicotinamida. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 43 y usando los compuestos 349.4 y 1.5 en lugar de ácido 5-amino-2,4-dimetilbenzoico y el compuesto 11.2, respectivamente, miz (ES+) 483 (M+H .

Compuesto 350.1.1-Óxido de 2-bromo-3-metilpiridina. El compuesto del título se preparó (8,00 g 85 %) usando un procedimiento similar al usado para la preparación del compuesto 349,1 y usando 2-bromo-3-metilpiridina (8,60 g) en lugar de 2-bromo-5-metilp¡r¡d¡na.

Compuesto 350.2. 6-Bromo-5-metilpicolinonitrilo. A una solución de 1-óxido de 2-bromo-3-metilpiridina
(compuesto 350.1, 5,65 g, 30,1 mmol, 1,00 equiv) en acetonitrilo (50 ml) se le añadieron trietilamina (6,10 g, 60,3 mmol, 2,00 equiv) y TMSCN (8,90 g, 3,00 equiv). La solución resultante se calentó a reflujo y se agitó durante una noche en un baño de aceite, a continuación se enfrió a temperatura ambiente y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para dar 2,00 g (34 %) del compuesto del título como un sólido de color amarillo



Compuesto 350.3. 6-Bromo-5-metilpicolinim idamida. En un tubo sellado de 50 ml, se colocó una solución de 6-bromo-5-metilpicolinonitrilo (compuesto 350.2, 900 mg, 4,57 mmol, 1,00 equiv) en metanol (40 ml). Se burbujeó NH3 (g) a través de la solución y la solución resultante se agitó durante una noche a 95 °C en un baño de aceite detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la mezcla se concentró a

Compuesto 350.4. 6-Amino-5-metilpicolinamida. En un tubo sellado de 50 ml, se colocó una solución de 6-bromo-5-metilpicolinimidamida (compuesto 350.3, 800 mg, 3,74 mmol, 1,00 equiv) y CuSO4 (80 mg) en NH3H2O (40 ml). La mezcla resultante se agitó durante una noche a 80 °C detrás de una pantalla protectora contra explosiones. Después de enfriar a temperatura ambiente, la mezcla se extrajo a continuación con 2x50 ml de acetato de etilo. Las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y se concentraron a presión reducida para dar 750 mg (impuros) del compuesto del título como un sólido de color marrón.

Compuesto 350.5. C lorhidrato de ácido 6-amino-5-metilpicolínico. En un matraz de fondo redondo, se colocó una solución de 6-amino-5-metilpicolinamida (compuesto 350.4, 500 mg, 3,31 mmol, 1,00 equiv) en cloruro de hidrógeno (conc., 15 ml). La solución resultante se calentó a reflujo durante una noche en un baño de aceite, a continuación se

Compuesto 350. N-(6-(4-(4-Cianofenil)piperidin-1-carbonil)-3-metilpiridin-2-il)-6-(isopropilamino)nicotinamida. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 43 y usando los compuestos 350.5 y 1.5 en lugar de ácido 5-amino-2,4-dimetilbenzoico y el compuesto 11.2, respectivamente, miz (ES+) 483 (M+H .

Compuesto 351.1. 5-Nitro-1H-pirazol-3-carboxilato de metilo. En un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución de ácido 5-nitro-1H-pirazol-3-carboxílico (5,00 g, 31,9 mmol, 1,00 equiv) en metanol (150 ml). A esto se añadió cloruro de tionilo (5,60 g, 47,8 mmol, 1,50 equiv) gota a gota a 0 °C. La solución resultante se agitó durante 18 h a 30 °C en un baño de aceite, a continuación se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:30-1:10) como eluyente para proporcionar 5,00 g (78 %) del compuesto del título como un sólido de color marrón.
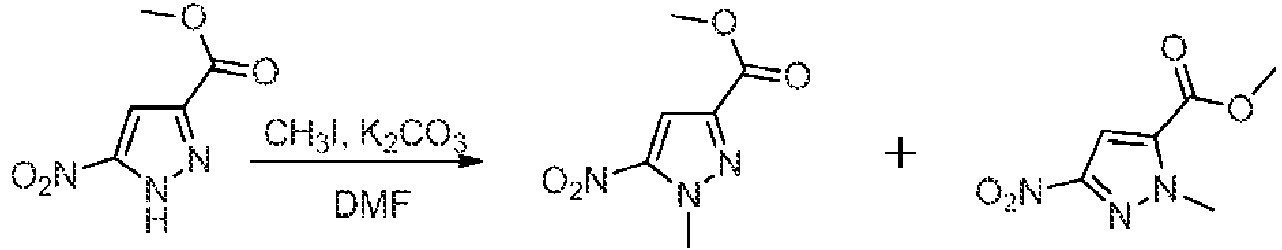
Compuesto 351.2 y 351.3. 1-Metil-5-nitro-1H-pirazol-3-carboxilato de metilo y 1-metM-3-nitro-1H-pirazol-5-carboxilato de metilo. En un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución de 5-nitro-1H-pirazol-3-carboxilato de metilo (3,00 g, 17,5 mmol, 1,00 equiv) en N,N-dimetilformamida (30 ml). Se añadieron carbonato potásico (4,84 g, 35,0 mmol, 2,00 equiv) y CH3I (2,98 g, 21,0 mmol, 1,20 equiv) a 0°C. La solución resultante se agitó durante 2 h a 40°C en un baño de aceite. Después de enfriar a temperatura ambiente, los sólidos se retiraron por filtración, seguida de la adición de 120 ml de agua con hielo. La mezcla se extrajo con 3x100 ml de acetato de etilo, y las capas orgánicas combinadas se secaron sobre sulfato sódico anhidro y a continuación se concentraron al vacío. El producto impuro se purificó mediante recristalización a partir de éter. Esto dio como resultado 1,10 g (34%) de 1-metil-3-nitro-1H-pirazol-5-carboxilato de metilo como un sólido de color blanco. m/z (ES+) 186(M+H)+. 1H-RMN, (300 MHz, DMSO-da, ppm): □ 7,57 (s, 1H), 4,22 (s, 3H), 3,91 (s, 3H). El filtrado se concentró al vacío y el producto impuro (2 g) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (10 % CH3CN asciende al 56,5 % en 10 minutos); Detector, uv254 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 470 mg (15 %) de 1-metil-5-nitro-1H-pirazol-3-carboxilato de metilo como un sólido de color blanco. m/z (ES+) 186 (M+H)+. 1H-r Mn , (300 MHz, DMSO-d6, ppm): □ □7,64 (1H, s), 4,24 (3H, s), 3,87 (
Compuesto 351.4. 5-Amino-1-metil-1H-pirazol-3-carboxilato de metilo. A una solución de 1-metil-5-nitro-1H-pirazol-3-carboxilato de metilo ((351.2, 0,48 g, 2,60 mmol) en MeOH (15 ml) en nitrógeno, se le añadió paladio sobre carbono (10 %, 0,25 g). El matraz se desgasificó adicionalmente con nitrógeno y se llenó con H2 a través de un globo. La mezcla se agitó a temperatura ambiente durante 1,5 horas. Después de purgar el sistema con nitrógeno, la mezcla de reacción se filtró a través de una torta de Celite y se concentró para dar 363 mg (90 %) de un sólido de color blanco. m/z (ES+) 156 (M+H)+.

Compuesto 351.5. 5-(3-isobutilureido)-1-metil-1H-pirazol-3-carboxilato de metilo. A una solución de 5-amino-1-metil-1H-pirazol-3-carboxilato de metilo (compuesto 351.4, 0,088 g, 0,57 mmol) en THF (5 ml), se le añadió trifosgeno (0,084 g, 0,28 mmol) seguido de DIEA(0,2 ml, 1,14 mmol). Después de que la mezcla se agitó a temperatura ambiente durante 2 horas, se añadió isobutilamina (0,225 ml, 2,28 mmol). La mezcla resultante se agitó a temperatura ambiente durante una noche. La mezcla se repartió entre agua y EtOAc. La capa de EtOAc se lavó con solución acuosa de NaH2PO41 M, seguida de solución saturada de cloruro sódico, se secó con Na2SO4, y se concentró para dar 120 mg (83 %) de un sólido de color amarillo claro, m/z ES+ 255 M+H .

Compuesto 351.6. Ácido 5-(3-isobutilureido)-1-metil-1H-pirazol-3-carboxílico A una solución de 5-(3-isobutilureido)-1-metil-1H-pirazol-3-carboxilato de metilo (compuesto 351.5, 0,12 g , 0.47 mmol) en MeOH (5 ml), se le añadió LiOH 1 M en H2O (1,42 ml, 1,42 mmol). La mezcla se agitó a temperatura ambiente durante 5 horas. TLC mostró que la reacción estaba completa. La mezcla se acidificó a pH 3-4 a 0 °C y a continuación se extrajo con EcOAc (2 x 50 ml). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron para dar 110 mg (100 %) de un aceite transparente. m/z (ES+) 241 (M+H)+.

Compuesto 351. 1-(3-(4-(4-Cianofenil)piperidin-1-carbonil)-1-metil-1H-pirazol-5-il)-3-isobutilurea Una solución de ácido 5-(3-isobutilureido)-1-metil-1H-pirazol-3-carboxílico (compuesto 351.6, 0,11 g, 0,46 mmol), sa1HCl de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 0,097 g, 0,46 mmol), EDCI (0,090 g, 0,51 mmol), HOBT (0,065 g, 0,51 mmol, con el 20 % de H2O) y DlEA (0,22 ml, 1,38 mmol) en DMF (5 ml) se agitó a temperatura ambiente durante una noche. A continuación se diluyó la mezcla de reacción con 50 ml de acetato de etilo y se lavó con 2x20 ml de solución saturada de cloruro sódico. La fase orgánica se secó sobre sulfato sódico anhidro y se concentró a presión reducida. El residuo se purificó usando TLC preparativa y se reveló usando acetato de etilo neto para dar 25 mg del compuesto del título como un sólido de color blanquecino m/z (ES+) 409 (M+H)+. 1H RMN (400 MHz, DMSO-da) 88,46 (s, 1H), 7,76 (d, 2H), 7,49 (d, 2H), 6,48 (t, 1H), 6,37 (s, 1H), 4,84 (d, 1H), 4,63 (d, 1H), 3,65 (s, 3H), 3,14 (m, 1H), 2,92 (t, 3H), 2,79 (m, 2H), 1,85 (m, 1H), 1,67 (m, 1H), 1,57 (m, 2H), 0,87 (d, 6H).
Los compuestos en la siguiente tabla se prepararon usando manipulaciones químicas estándar, materiales de partida disponibles fácilmente y procedimientos similares a los usados para la preparación de los compuestos 346, 347, 348, 349 350 351:

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)
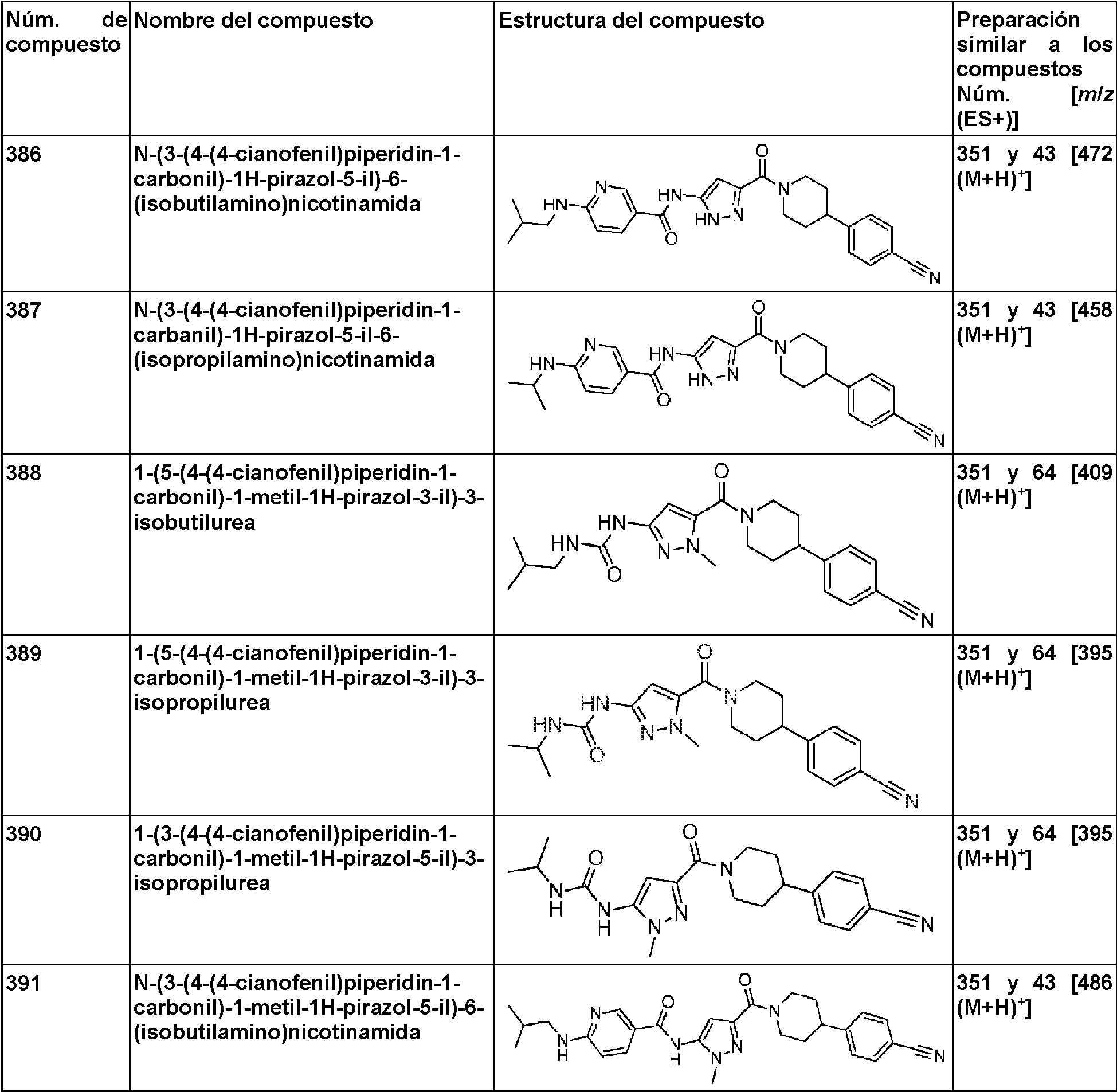
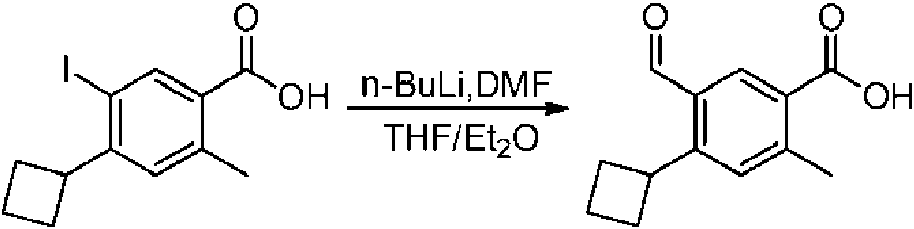
Compuesto 392.1. Ácido 4-ciclobutil-5-formil-2-metilbenzoico. En un matraz de fondo redondo de tres bocas, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución de ácido 4-ciclobutil-5-yodo-2-metilbenzoico (compuesto 230.1, 5,00 g, 12,7 mmol, 1,00 equiv, 80%) en una mezcla disolvente de tetrahidrofurano y Et2O (50/50 ml). A esto le siguió la adición de butillitio (15 ml, 2,50 equiv, 95 %) gota a gota con agitación a -78 °C. A esto se le añadió N,N-dimetilformamida (2,50 g, 32,5 mmol, 2,00 equiv). La solución resultante se agitó durante 1 h a -78 °C y a continuación se inactivó cuidadosamente mediante adición lenta de 50 ml de NH4Cl (ac.). El pH se ajustó a 1-2 con cloruro de hidrógeno (6 M). La solución resultante se diluyó con 100 ml de acetato de etilo, a continuación se lavó con 4x50 ml de solución saturada de cloruro sódico. La capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:1) como eluyente para proporcionar 1,62 g (41 %) de ácido 4-ciclobutil-5-formil-2-metilbenzoico como un sólido de color blanco.

Compuesto 392.2. 4-(1-(4-CidobutN-5-formN-2-metNbenzoM)piperidm-4-N)benzomtrMo. En un matraz de fondo redondo, se colocó una solución de ácido 4-cidobutil-5-formil-2-metilbenzoico (compuesto 392.1, 490 mg, 1,57 mmol, 1,00 equiv, 70%) en N,N-dimetilformamida (8 ml). Se añadieron el compuesto 1.5 (500 mg, 1,57 mmol, 1,00 equiv), EDCHCl (860 mg, 4,26 mmol, 2,00 equiv, 95%), y 4-dimetilaminopiridina (550 mg, 4,28 mmol, 2,00 equiv, 95%) a la mezcla de reacción. La solución resultante se agitó durante una noche a temperatura ambiente, a continuación se diluyó con 30 ml de acetato de etilo. La mezcla resultante se lavó con 4x30 ml de solución saturada de cloruro sódico, a continuación se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:2) como eluyente para dar 560 mg

Compuesto 392. 4-(1-(4-CidobutN-5-formN-2-metNbenzoM)piperidm-4-N)benzomtrMo. En un matraz de fondo redondo, se colocó una solución de 4-[1-[(4-ciclobutil-5-formil-2-metilfenil)carbonil]piperidin-4-il]benzonitrilo (392.2, 300 mg, 0,74 mmol, 1,00 equiv, 95%) en N,N-dimetilformamida (10 ml). Se añadieron 3-bromodihidro-2H-piran-4(3H)-ona (compuesto 1.10.1, 210 mg, 1,17 mmol, 1,00 equiv), amoniaco (82 mg, 0,59 mmol, 3,00 equiv, 25% ac.), y NH4OAc (270 mg, 2,81 mmol, 4,50 equiv, 80 %) y la mezcla resultante se agitó durante una noche a 130 °C en atmósfera de nitrógeno. Después de enfriar a temperatura ambiente, la mezcla se diluyó a continuación con 50 ml de acetato de etilo, se lavó con 4x50 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo como eluyente. El producto impuro (~80 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (26 % CH3CN asciende al 41 % en 7 minutos, asciende al 100 % en 3 minutos, desciende al 26 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 24,6 mg (7 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 481 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,67 (d, J =7,8 Hz, 2H), 4,47 (d, J = 8,1 Hz, 2H), 7,33 (s ancho, 1H), 7,26 y 7,15 (2 singletes, rotámeros de amida, Ar-H, 1H), ~4,9 (1H parcialmente oscurecido por pico de agua), 4,68 (s, 2H), 4,06-3,88 (m, 3H), 3,73-3,58 (m, 1H), 3,33-3,18 (m, 1H), 3,07-2,92 (m, 2H), 2,83-2,73 (m, 2H), 2,44 & 2,34 (2 singletes, rotámeros de amida, Ar-CH3, 3H), 2,25-1,91 (m, 6H), 1,91-1,50 (m, 4H). 1H RMN (400 MHz, CDCh): 8 7,64 (m, 2H), 7,34 (m, 2H), 7,13-6,93 (m, 2H), 4,99 (m, 1H), 4,75 (s, 2H), 3,98 (m, 2H), 3,73 (m, 1H), 3,53 (m, 1H), 3,31-2,83 (m, 5H), 2,41 (m, 1H), 2,37-1,61 (m, 11H), 1,44 (m, 1H).
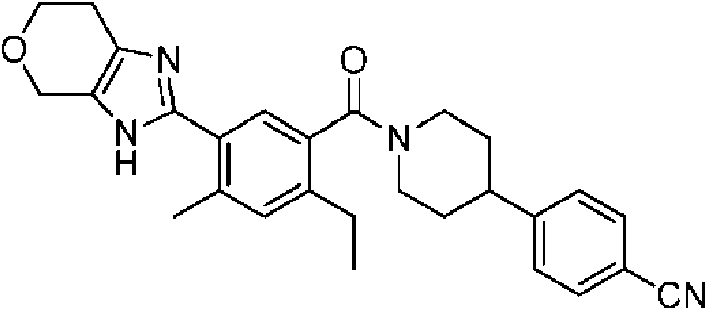
Compuesto 393. 4-(1-(2-etN-4-metM-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 pero se usó el compuesto 211.2 en lugar del compuesto 2.2. m/z (ES+) 455 (
Compuesto 394. 4-(1-(4-etil-2-metil-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 393 pero se usó el compuesto 152.1 en lugar de 2-bromo
4-metilbenzoato de metilo. m/z(ES+ 455 M+H .

Compuesto 395. 4-(1-(4-EtN-2-metN-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 394 pero se usó sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. miz (ES+) 473 (
Compuesto 396. 4-(1-(2,4-DietN-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoM)-4-fluoropiperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 excepto que se usaron los compuestos 204.3 y 11.2 en lugar de los compuestos 2.2 y 1.5, respectivamente, miz (ES+) 487 (M+H)+.

Compuesto 397. 4-(1-(5-(5-AcetN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]pmdm-2-M)-2,4-dietNbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 excepto que se usó el compuesto 204.3 en lugar del compuesto 2.2. miz (ES+) 510
Compuesto 398.4-(1-(5-(5-Acetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-4-etil-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos

Compuesto 399. 4-(1-(5-(5-Acetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-4-etil-2-metilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1,2 y 394 y usando sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. miz ES+ 514 M+H .

Compuesto 400.1. 3-Metildihidro-2H-piran-4(3H)-ona. Un matraz de fondo redondo de tres bocas de 1 l se purgó y se mantuvo con una atmósfera de nitrógeno y una solución de LDA (2 M en THF, 132 ml, 1,20 equiv) en tetrahidrofurano (300 ml). La mezcla se enfrió a -78 °C, a continuación, se añadió una solución de dihidro-2H-piran4(3H)-ona (22,0 g, 220 mmol, 1,00 equiv) en hexametilfosforamida (40 ml, 230 mmol, 1,05 equiv) gota a gota seguida de la adición gota a gota de yoduro de metilo (34 ml, 550 mmol, 2,5 equivalentes) a -78 °C. La solución resultante se agitó a -78 °C durante 5 minutos, a continuación a 25 °C durante 5 minutos. La reacción se inactivó cuidadosamente con NH4Cl acuoso saturado (80 ml) y se extrajo con éter (2 x 100 ml). Las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice con PE/Et2O (5:1) como eluyente para obtener el compuesto del título como un aceite de color amarillo (6,00 g, 24%).


Compuesto 400.2. 3-Bromo-5-metildihidro-2H-piran-4(3W)-ona. Un matraz de fondo redondo de tres bocas de 250 ml se purgó y se mantuvo con una atmósfera de nitrógeno. Se añadieron LDA (2,0 M en THF) (6 ml, 12 mmol, 1,20 equiv) y tetrahidrofurano (30 ml). La solución se enfrió a -78 °C y a continuación se añadió TMSCl (7 ml, 55 mmol) gota a gota y se agitó a -78 °C durante 5 minutos. Una solución de 3-metildihidro-2H-piran-4(3H)ona (compuesto 400.1, I , 14 g, 9,99 mmol, 1,00 equiv) en tetrahidrofurano (20 ml) se añadió gota a gota y la mezcla resultante se agitó a -78 °C durante 10 minutos. La mezcla se inactivó cuidadosamente con una mezcla de trietilamina (15 ml) y NaHCO3 acuoso saturado (100 ml). La fase acuosa se extrajo con éter (2 x 50 ml), y las fases orgánicas combinadas se lavaron con ácido cítrico acuoso (3 x 100 ml), se secaron (K2CO3), se filtraron y se concentraron al vacío para obtener el éter silílico de enol intermedio, como un aceite incoloro. El éter silílico de enol se disolvió en tetrahidrofurano (20 ml) y el sistema se purgó con nitrógeno. La mezcla se enfrió a 0 °C, y a continuación se añadió W-bromosuccinimida (1,95 g, I I , 0 mmol, 1,10 equiv) por partes. La mezcla resultante se agitó a 25 °C durante 1 h, a continuación se inactivó con NaHCO3 acuoso saturado (30 ml). La fase acuosa se extrajo con éter (2 x 30 ml) y las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice con PE/Et20 (5:1) como elu ente para obtener el compuesto del título como un aceite incoloro (1,00 g, 52 %).

Compuesto 400.3. Ácido 2-etil-4-metil-5-(7-metil-3,4,6,7-tetrahidropirano[3,4-c(]imidazol-2-il)benzoico. En un matraz de fondo redondo, se colocó una solución de 3-bromo-5-metildihidro-2H-piran-4(3H)ona (compuesto 400.2, 800 mg, 4,14 mmol, 1,6 equiv) en W,W-dimetilformamida (5 ml). Se añadieron hidróxido de amonio (NH3 al 25 % en H2O) (530 mg, 7,8 mmol, 3,0 equiv), NH4OAc (904 mg, 11,7 mmol, 4,50 equiv) y ácido 2-etil-5-formil-4-metilbenzoico (compuesto 211.4, 500 mg, 2,60 mmol, 1,00 equiv) al matraz. La mezcla resultante se agitó a 130 °C durante 2 h, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con diclorometano/metanol

Compuesto 400. 4-(1-(2-Etil-4-metil-5-(7-metM-3,4,6,7-tetrahidropirano[3,4-c(]imidazol-2-il)benzoil)piperidm-4-il)benzonitrilo. En un matraz de fondo redondo, se colocó una solución de ácido 2-etil-4-metil-5-(7-metil-3,4,6,7-tetrahidropirano[3,4- d ]imidazol-2-il)benzoico (compuesto 400.3, 30 mg, 0,10 mmol, 1,0 equiv) en DMF/DCM (5/5 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 50 mg, 0,22 mmol, 2,2 equiv), EDC (40 mg, 2,0 equiv), y 4-dimetilaminopiridina (28 mg, 2,0 equiv). La solución resultante se agitó durante una noche a 25 °C, a continuación se inactivó con agua (10 ml). La mezcla se extrajo con acetato de etilo (3 x 5 ml) y las capas orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (2:1) como eluyente para obtener el compuesto del título como un

Compuesto 401. 4-(1-(4-Ciclopropil-2-metil-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 392 pero se usó el compuesto 142.2 en lugar del compuesto 152.3. miz (ES+) 467 (M+H)+.

Compuesto 402. 4-(1-(4-CidopropM-2-etM-5-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-M)benzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 392 pero se usó el compuesto 226.5 en lugar del compuesto 230.1. m lz {ES+) 481 (M+H)+.

Compuesto 403. 2-(5-(4-(4-Cianofeml)piperidm-1-carbonM)-2-cidopropM-4-metMfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 pero se usaron el compuesto 142.2 y dicarbonato de dimetilo/DIEA en lugar del compuesto 2.2 y anhídrido acético, respectivamente. miz (ES+) 524 (M+H)+.

Compuesto 404. 4-(1-(5-(5-AcetN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)4-ddopropN-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación de los compuestos 1 y 2 pero usando el compuesto 142.2 en lugar del compuesto 2.2. miz ES+ 508 M+H .

Compuesto 405. 4-(1-(4-Ciclobutil-2-metil-5-(3,4,6,7-tetrahidropirano [3,4-d]imidazol-2-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 392 excepto que se usó sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 499 (M+H)+. 1H RMN (300 MHz, CD3OD) 67,97 (m, 2H), 7,97 (m, 2H), 7,67-7,53 (m, 2H), 4,80 (s, 2H), 4,10 (m, 2H), 3,75 (m, 1H), 3,55 (m, 2H), 3,27 (m, 1H), 2,90 (m, 2H), 2,53 y 2,43 (2 singletes, rotámeros de amida, ArCH3, 3H , 2,38-1,80 m, 10H.

Compuesto 406.1. Ácido 5-(5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-4-ciclobutil-2-metilbenzoico. A una solución de ácido 4-ciclobutil-5-formil-2-metilbenzoico (compuesto 392.1, 2,00 g, 9,16 mmol, 1,00 equiv) en N,N-dimetilformamida (15 ml), se le añadieron 3-bromo-4-oxopiperidin-1-carboxilato de terc-butilo (9,00 g, 16,2 mmol, 2,00 equiv), hidróxido de amonio (3,84 g, 27,4 mmol, 3,00 equiv, 25%), y NH4OAc (3,18 g, 41,3 mmol, 4,50 equiv). La mezcla resultante se agitó durante una noche a 130 °C en nitrógeno. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 50 ml de acetato de etilo, se lavó con 3x30 ml de solución saturada de cloruro
sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con diclorometano/metanol (20:1) como eluyente para proporcionar 2,00 g (27 %) del compuesto del título como un sólido de color marrón

Compuesto 406.2. 2-(5-(4-(4-cianofenM)piperidm-1-carboml)-2-ddobutM-4-metMfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de terc-butilo. A una solución de ácido 5-(5-(terc-butoxicarbonil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c] piridin-2-il)-4-ciclobutil-2-metilbenzoico (compuesto 406.1, 1,00 g, 1,22 mmol, 1,00 equiv, 50%) en N,N-dimetilformamida (5 ml), se le añadieron sal clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 270 mg, 1,21 mmol, 1,00 equiv), EDCHCl (460 mg, 2,40 mmol, 2,00 equiv), y 4-dimetilaminopiridina (296 mg, 2,42 mmol, 2,00 equiv). La mezcla resultante se agitó a 25 °C durante una noche. La mezcla se diluyó a continuación con 30 ml de acetato de etilo, se lavó con 3x30 ml de solución saturada de cloruro sódico, se secó sobre sulfato de sodio anhidro, y se concentró a presión reducida para dar 890 mg (63 %) del compuesto del título como un sólido de color marrón.

Compuesto 406.3.4-(1-(4-CidobutM-2-metM-5-(4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-M)benzoM)piperidm-4-il)benzonitrilo. Una solución de 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2-ciclobutil-4-metilfenil)-6,7-dihidro-3H-imidazo[4,5-c ]piridin-5(4H)-carboxilato de terc-butilo (406.2, 600 mg, 0,620 mmol, 1,00 equiv, 60 %) en diclorometano (3 ml) y ácido trifluoroacético (1,5 ml) se agitó durante 3 horas a 25 °C, a continuación se diluyó con 5 ml de ácido clorhídrico (3 mol/l) y 10 ml de agua. La mezcla resultante se lavó con 3x20 ml de acetato de etilo y después el pH se ajustó a ~9 con hidróxido sódico (ac., 3 M). La mezcla resultante se extrajo con 4x20 ml de diclorometano. Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron a presión reducida para dar 140 mg (37 %) del

Compuesto 406. 4-(1-(4-CiclobutN-2-metN-5-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-il)benzoil)piperidin-4-il)benzonitrilo. A una solución de 4-(1-(4-ciclobutil-2-metil-5-(4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)benzoil) piperidin-4-il)benzonitrilo (406.3, 190 mg, 0,30 mmol, 1,00 equiv, 75 %) en tetrahidrofurano (10 ml), se le añadieron NaBH(OAc)3 (252 mg, 1,13 mmol, 3,00 equiv, 95 %) y HCHO ( 37 %) (2,2 ml, 2,00 equiv). La mezcla resultante se agitó durante 2 h a 40 °C en un baño de aceite, y a continuación se concentró a presión reducida. El residuo se diluyó con diclorometano y se lavó con bicarbonato sódico acuoso saturado y solución saturada de cloruro sódico. La capa orgánica se secó (Na2SO4) y se concentró. El producto impuro (~80 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (36 % CH3CN asciende al 48 % en 8 minutos, asciende al 100 % en 1 minuto, desciende al 36 % en 1,5 minutos); detector, Waters 2489254 y 220 nm]. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 23,4 mg (16 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 494 (M+H)+. 1H RMN (300 MHz, CD3OD): ó 7,69 (d, J = 6,0 Hz, 2H), 7,48 (d, J = 6,0 Hz, 2H), 7,37-7,31 (m, 1H), 7,26 y 7,15 (2 singletes, rotámeros de amida, Ar-H, 1h ), ~4,9 (1H parcialmente oscurecido por pico de agua), 4,02-3,90 (m, 1H), 3,73-3,57 (m, 1H), 3,57 (s, 2H), 3,34 3,21 (m, 1H), 3,06-2,94 (m, 2H), 2,90-2,77 (m, 4H), 2,54 (s, 3H), 2,45 y 2,34 (2 singletes, rotámeros de amida, ArCH3,
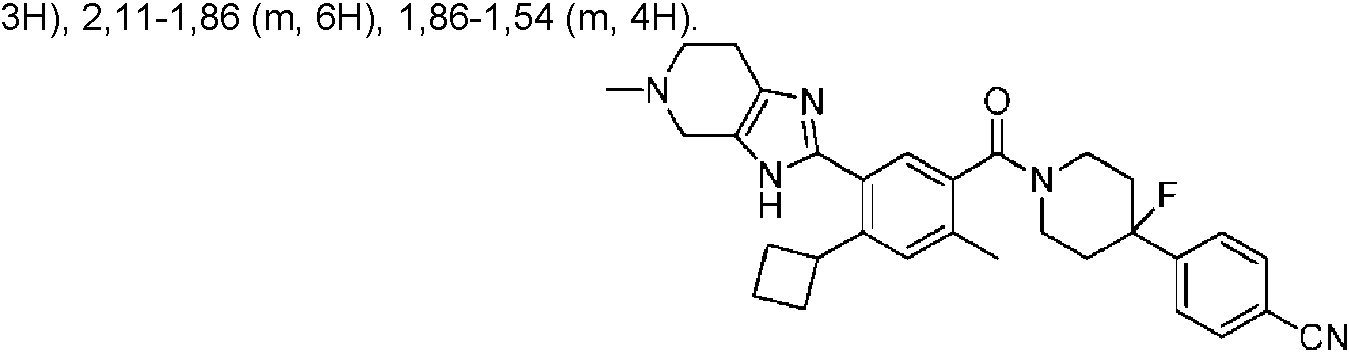
Compuesto 407. 4-(1-(4-CiclobutN-2-metN-5-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)benzoM)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y
procedimientos similares a los usados para la preparación del compuesto 406 excepto que se usó sa1HCl del

Compuesto 408.4-(1-(4-MetN-3-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 406 excepto que se usó ácido 3-yodo-4-metilbenzoico en luaar del compuesto 230.1. m/z (ES+) 440 (M+H)+.

Compuesto 409. 4-(1-(4-CidobutN-2-metN-5-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-il)benzoil)piperidin-4-il)benzonitrilo. A una solución de 4-(1-(4-ciclobutil-2-metil-5-(4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin -4-il)benzonitrilo (406.3, 180 mg, 0,300 mmol, 1,00 equiv, 80%) en N,N-dimetilformamida (5 ml) se le añadieron dicarbonato de dimetilo (176 mg, 1,31 mmol, 5,00 equiv) y DIEA (169 mg , 1,31 mmol, 5,00 equiv). La solución resultante se agitó durante una noche a 25 °C y a continuación se inactivo con 20 ml de metanol. La mezcla resultante se concentró a presión reducida y el producto impuro se purificó por HPLC preparativa usando las siguientes condiciones (1#-Pre-HPLC-001(SHIMADzU)): columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (28 % de CH3Cn , asciende al 43 % en 8 minutos, asciende al 100 % en 3 minutos, desciende al 28 % en 2 minutos); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 29,9 mg (19 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 538 (M+H)+. 1H RMN (400 MHz, CD3OD): 87,70 (m, 2H), 7,50 7,35 (m, 4H), 4,90 (m, 1H), 4,70 (s, 2H), 4,00 (m, 2H), 3,80 (m, 4H), 3,60 (m, 1H), 3,25 (m, 1H), 3,05 (m, 2H), 2,85 (m, 2H), 2,50 y 2,40 (2 singletes, rotámeros de amida, ArCH3, 3H), 2,16-2,00 (m, 6H), 1,86-1,64 (m, 4H).

Compuesto 410. 2-(5-(4-(4-Cianofeml)-4-fluoropiperidm-1-carboml)-2-ddobutN-4-metNfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usó el compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 556 (M+H)+. 1H RMN (400 MHz, CD3OD) 87,78 (m, 2H), 7,67 (m, 2H), 7,35-7,20 (m, 2H), 4,80 (m, 1H), 4,55 (s, 2H), 4,00 (m, 1H), 4,84 (m, 2H), 3,77 (s, 3H), 3,60 (m, 1H), 3,28 (m, 1H), 2,80 (s, 2H), 2,47 y 2,36 (2 singletes, rotámeros de amida, ArCHa, 3H), 2,30-1,80 (m, 9H), 1,62 (m, 1H), 1,32 (m, 1H).
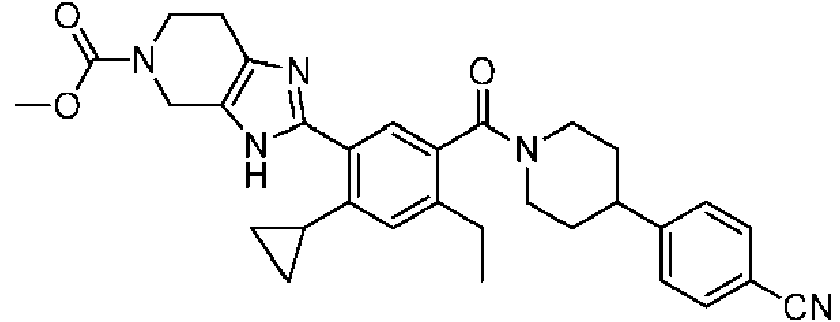
Compuesto 411. 2-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2-ddopropN-4-etNfeml)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usó el compuesto 226.5 en lugar del compuesto 230.1. m/z (ES+) 538 (M+H)+.

Compuesto 412. 4-(1-(4-CiclopropN-2-etN-5-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]pmdm-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 406 excepto que se usó el compuesto 226.5 en lugar del compuesto 230.1. m/z ES+ 494 M+H .
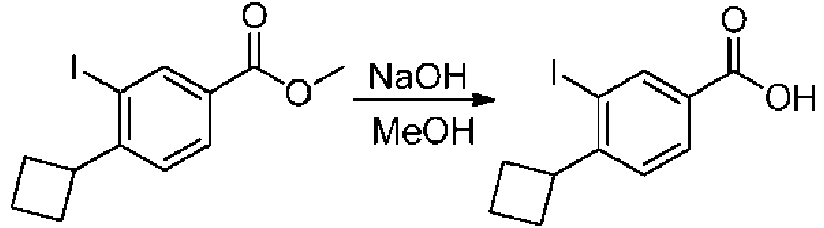
Compuesto 413.1. Ácido 4-ciclobutil-3-yodobenzoico. Una solución del compuesto 168.2 (11,0 g, 34,8 mmol, 1,00 equiv) e hidróxido sódico (4.00 g, 100 mmol, 3,00 equiv) en metanol (~100 ml) se agitó a 50 °C durante una noche. Después de enfriar a temperatura ambiente, la mezcla se concentró a sequedad. El residuo se recogió en agua (50 ml) equiv, y se lavó con acetato de etilo. El pH de la capa acuosa se ajustó a 3-4 con cloruro de hidrógeno acuoso 6 M. El precipitado resultante se recogió por filtración y se secó para dar 8,60 g (82%) del compuesto del título como un sólido de color blanco.

Compuesto 413. 4-(1-(4-CidobutM-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-M)benzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 392 excepto que se usó el compuesto 413.1 en lugar del compuesto 230.1. m/z (ES+) 477 (M+H)+. 1H RMN (300 MHz, CD3OD) ó 7,76-7,65 (m, 5H), 7,48 (m, 2H), 5,00 (m, 1H), 4,80 (s, 2H), 4,10 (m, 2H), 3,80 (m, 2H), 3,45 (m, 1H), 3,04 (m, 2H), 2,90 (m, 2H), 2,18-1,95 (m, 6H), 1,85-1,77 (m, 4H).

Compuesto 414. 4-(1-(4-Ciclobutil-3-(3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)benzoil) -4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 413 excepto que se usó sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 485 (M+H)+. 1H RMN (300 MHz, CD3OD) ó 7,82-7,77 (m, 3H), 7,68-7,65 (m, 4H),

Compuesto 415. 4-(1-(4-CidobutN-3-(7-metN-3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoM)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 414 excepto que se usó 3-metildihidro-2H-piran-4(3H)-ona en lugar de dihidro-2H-piran-4(3H)-ona. m/z (ES+) 499 (M+H)+.
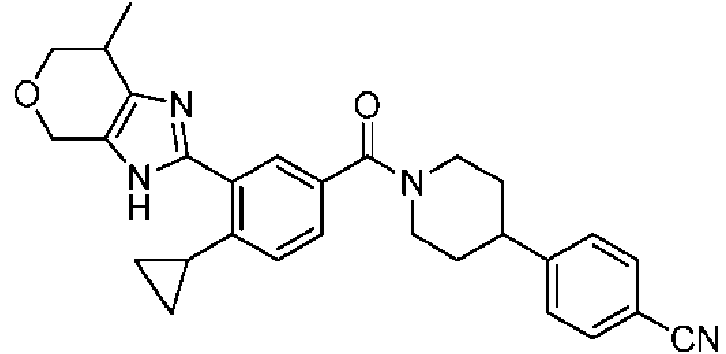
Compuesto 416. 4-(1-(4-CidopropN-3-(7-metM-3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 413 excepto que se usaron bromo(cidopropil)magnesio y 3-metildihidro-2H-piran-4(3H)-ona en lugar de bromo(ciclobutil)magnesio y dihidro-2H-piran-4(3H)-ona, respectivamente, miz (ES+) 467 (M+H .

Compuesto 417. 4-(1-(4-CidobutN-3-(5-metM-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 406 excepto que se usó el compuesto 413.1 en lugar del compuesto 230.1. m/z (ES+) 480 (M+H)+. 1H RMN (300 MHz, CD3OD) 67,72-7,63 (m, 4H), 7,56 (s, 1H), 7,49 (m, 2H), 4,82 (m, 1H), 4,56 (s, 2H), 3,89 (m, 2H), 3,80 (m, 2H), 3,08 (m, 2H), 3,04 (s, 3H), 2,99 (m, 2H), 2,18-1,66 (m, 11H).

Compuesto 418. 4-(1-(4-CidobutN-3-(5-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)benzoM)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 417 excepto que se usó sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 498 (M+H)+. 1H RMN (300 MHz, CD3OD) 67,79 (m, 2H), 7,74 7,59 (m, 5H), 4,73 (s ancho, 1H), 4,54 (s ancho, 2H), 3,94 (m, 2H), 3,76 (s ancho, 2H), 3,61 (s ancho, 1H), 3,20 (s

Compuesto 419. 2-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2-cidobutNfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usó el compuesto 413.1 en lugar del compuesto 230.1. m/z ES+ 524 M+H .

Compuesto 420. 2-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carbonM)-2-ddobutNfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usaron los compuestos 413.1 y 11.2 en lugar de los compuestos 230.1 y 1.5, respectivamente. m/z (ES+) 542 (M+H).

Compuesto 421. 2-(5-(4-(4-Cianofeml)piperidm-1-carboml)-2-ddobutMfenM)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de etilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usó el compuesto 413.1 y dicarbonato de dietilo en lugar del compuesto 230.1 y dicarbonato de dimetilo, respectivamente. m/z (ES+) 538 (M+H).

Compuesto 422. 2-(5-(4-(4-Cianofeml)piperidm-1-carbonM)-2-ddopropNfeml)-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 421 excepto que se usaron bromo(ciclopropil)magnesio y dicarbonato de dimetildietilo en lugar de bromo(ciclobutil)magnesio y dicarbonato de

Compuesto 423. 4-(1-(2,4-Dimetil-5-(5-(oxetan-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4-il)benzonitrilo. Una mezcla de 4-(1-(2,4-dimetil-5-(4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4- il)benzonitrilo (compuesto 2.9, 62 mg), oxetan-3-ona (18 jl), cianotrihidroborato sódico (18 mg), y ácido acético (80 j l ) en MeOH/THF (1:1 v/v) se agitó durante 16 horas. La reacción se diluyó a continuación con diclorometano, se lavó con solución saturada de bicarbonato sódico seguida de solución saturada de cloruro sódico. La capa orgánica se secó (MgSO4) y se concentró. El residuo se purificó por TLC preparativa al 8 % de MeOH en DCM para dar 42 mg de sólido de color blanco (60%). m/z (ES+) 496 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 87,63 (d, 2H), 7,32 (d, 2H), 7,27 y 7,20 (2 singlete, rotámeros de amida, 1H), 7,02 (s, 1H), 4,96 (s, 1H), 4,74 (m, 4H), 3,88 - 3,77 (m, 1H), 3,63 (d, 1H), 3,49 (d, 2H), 3,08 (t, 1H), 2,97 - 2,62 (m, 6H), 2,45 (d, 3H), 2,31 y 2,22 (2 singletes, rotámeros de amida, 3H), 2,11 -1,40 (m, 4H).

Compuesto 424. 4-(1-(5-(5-Cidopropil-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridin-2-il)2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 423. m/z (ES+) 480 (M+H)+. □1H RMN (400 MHz, Cloroformo-d) 810,58, 7,61 (d, 2H), 7,33 (d, 2H), 7,23 y 7,15 (2 singletes, rotámeros de amida, 1H), 6,97 (s, 1H), 4,93 (s, 1H), 3,80 - 3,66 (m, 2H), 3,64 - 3,49 (m, 1H), 3,02 (m, 3H), 2,91 - 2,64 (m, 4H), 2,41 (d, 3H), 2,28 y 2,18 (2 singletes, rotámeros de amida, 3H), 2,07 -1,86 (m, 2H), 1,84 - 1,37 (m, 2H), 0,66 - 0,40 (m, 4H).

Compuesto 425. 4-(1-(2,4-DimetN-5-(5-metN-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]pmdm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 406 pero usando el compuesto 2.9 en lugar del compuesto 406.3. m/z (ES+) 454 (M+H)+.

Compuesto 426. 4-(1-(5-(5-EtM-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridm-2-M)-2,4-dimetMbenzoM)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 423. m/z (Es ) 468 (M+H)+. 1H RMN (400 MHz, Cloroformod) 87,63 (d, 2H), 7,40-7,19 (m, 3H), 7,04 (s, 1H), 4,95 (s, 1H), 3,75-3,51 (m, 3H), 3,09 (m, 1H), 2 ,95-2,77 (m, 6H), 2,71 (m, 2H), 2,46 (s, 3H), 2,30 y 2,21 (2 singletes, rotámeros de amida, 3H), 2,12 -1,89 (m, 1H), 1,85-1,43 (m, 3H), 1,22 (m, 3H).

Compuesto 427. 4-(1-(5-(5-(2-FluoroetN)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]pmdm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. Una mezcla de 4-(1-(2,4-dimetil-5-(4,5,6,7-tetrahidro-IH-imidazo[4,5-c]piridin-2-il)benzoil)piperidin-4- il)benzonitrilo (compuesto 2.9, 88 mg), 1-bromo-2-fluoroetano (150 pl), yoduro potásico (50 mg), y trietilamina (140 pl) en DMF (2 ml) se agitó durante 48 horas. La mezcla de reacción se diluyó a continuación con diclorometano y se lavó con solución saturada de cloruro sódico. La capa orgánica se secó (MgSO4) y se concentró. El residuo se purificó mediante TLC preparativa usando el 8 % de MeOH en DCM para dar 51 mg de un sólido de color blanco (52 %). m/z (ES+) 486 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 7,63 (d, 2H), 7,32 (d, 2H), 7,28 y 7,20 (2 singletes, rotámeros de amida, 1H), 7,00 (s, 1H), 4,93 (m, 1H), 4,67 (dt, Ji=47,6 Hz, J2=4,9 Hz, 2H), 3,71 (d, 2H), 3,62 (d, 1H), 3,03-2,71 (m, 8H), 2,43 (d, 3H), 2,29 y 2,19 (2 singletes, rotámeros de amida, 3H), 2,08 - 1,89 (m, 2H), 1,85 - 1,49 (m, 2H), 3,18 - 3,05 (m, 1H).

Compuesto 428. 4-(4-Fluoro-1-(5-(5-(2-fluoroetM)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]pmdm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 427 pero usando sa1HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) 504 (M+H)+. 1H RMN (400 MHz, Cloroformo-d) 87,70 (d, 2H),
7,50 (d, 2H), 7,28 y 7,21 (2 singletes, rotámeros de amida, 1H), 7,00 (s, 1H), 4,92 - 4,77 (m, 1H), 4,66 (dt, Ji=47,7 Hz, J2=4,8 Hz, 2H), 3,68 (d, 2H), 3,58 - 3,35 (m, 2H), 3,33-3,11 (m, 1H), 2,95 (dt, 4H), 2,76 (d, 2H), 2,42 (s, 3H), 2,28 y 2,20 (2 singletes, rotámeros de amida, 3H , 2,16 -1,72 m, 4H .

Compuesto 429.1. 4-Etil-5-oxoazepan-1,4-dicarboxilato de 1-terc-butilo. A una solución de 4-oxopiperidin-1-carboxilato de terc-butilo (20,0 g, 100 mmol, 1,00 equiv) en éter (60 ml) -30 °C, se le añadió gota a gota una solución de BF3.Et2O (16,0 ml, 1,30 equiv) en éter (20 ml). Después de agitar 30 minutos a -30 °C, una solución de 2-diazoacetato de etilo (16,0 g, 140 mmol, 1,30 equiv) en éter (20 ml) se añadió gota a gota a la reacción a -30 °C. La solución resultante se agitó durante 1 h a -30 °C, a continuación se calentó a 25 °C y se agitó durante 2 h. La reacción se inactivó a continuación con 100 ml de carbonato potásico acuoso al 30 %. La mezcla resultante se extrajo con 2x250 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 2x50 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó mediante cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1/10) como eluyente para proporcionar 19,0 g (66 %) del compuesto del título como un aceite de color amarillo claro.

Compuesto 429.2. 4-Oxoazepan-1-carboxilato de terc-butilo. A una solución de 4-etil-5-oxoazepan-1,4-dicarboxilato de 1-terc-butilo (429.1, 19,0 g, 66,6 mmol, 1,00 equiv) en 1,4-dioxano (190 ml) se le añadió gota a gota hidróxido sódico (4,00 g , 100 mmol, 1,50 equiv) en agua (100 ml). La mezcla resultante se agitó a temperatura ambiente durante una noche. El pH se ajustó a continuación a 4-5 con cloruro de hidrógeno (ac. 3 M) y la solución resultante se extrajo con 2x50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 2x10 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo impuro se purificó mediante cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:3) como eluyente para proporcionar 11,0 g (77 % del compuesto del título como un aceite de color amarillo.

Compuesto 429.3. 4-Bromo-5-oxoazepan-1-carboxilato de terc-butilo. A una solución de 4-oxoazepan-1-carboxilato de terc-butilo (429.2, 11,0 g, 51,6 mmol, 1,00 equiv) en cloroformo (220 ml) 0 °C se le añadió gota a gota una solución de Br2 (12,4 g, 77,6 mmol, 1,50 equiv) en cloroformo (110 ml). La mezcla resultante se agitó a temperatura ambiente durante una noche. Los sólidos formados se recogieron por filtración y se recogieron en 200 ml de diclorometano. Se añadieron Et3N (12,2 g) y (Boc)2O (8,70 g, 40,3 mmol, 1,00 equiv) a la mezcla a 0 °C. La solución resultante se agitó durante 3 h a temperatura ambiente, y a continuación se concentró a presión. El residuo impuro se purificó mediante cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:10) como eluyente para dar
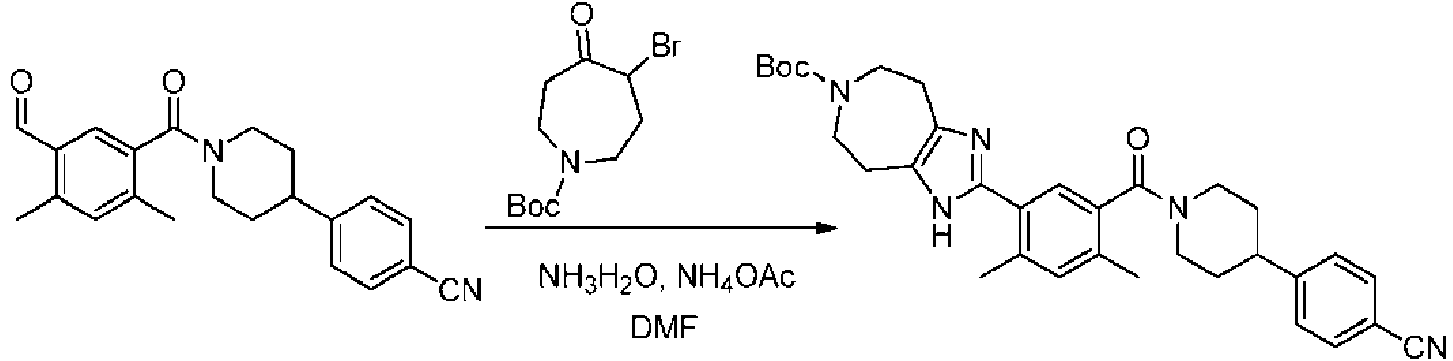
Compuesto 429.4. 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dimetilfenil)-4,5,7,8-tetrahidroimidazo[4,5-d]azepin-6(1H)-carboxilato de terc-butilo. A una solución de 4-bromo-5-oxoazepan-1-carboxilato de terc-butilo (compuesto 429.3, 844 mg, 2,89 mmol, 2,00 equiv) en N,N-dimetilformamida (15 ml), se le añadieron compuesto 16.4 (500 mg, 1,44 mmol, 1,00 equiv), NH4OH (606 mg, 4,33 mmol, 3,00 equiv, 25 %), y NH4OAc (500 mg, 6,49 mmol, 4,50 equiv). La mezcla resultante se agitó durante una noche a 130 °C en nitrógeno. Después de enfriar a temperatura ambiente, se añadieron 30 ml de acetato de etilo. La mezcla resultante se lavó con 3x20 ml de solución saturada de
cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo como eluyente para dar 343 mg (43 %) del compuesto del título como un sólido de color marrón.

Compuesto 429.5. Trifluoroacetato de 4-(1-(5-(1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. A una solución del compuesto 429.4, (550 mg, 0,990 mmol, 1,00 equiv, en DCM (3 ml) se le añadió ácido trifluoroacético (1,3 ml). Después de agitar a 25 °C durante 4 h, la mezcla se concentró a presión reducida y se secó para dar 400 mg (71 %) del compuesto del título como un sólido de color marrón.

Compuesto 429. 4-(1-(5-(6-Acetil,4,5,6,7,8-hexahidroimidazo[4,5-d]azepin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 excepto que se usó el compuesto 429.5 en lugar del compuesto 2.9. m/z (ES+) 496 (M+H)+. 1H RMN (300 MHz, CD3OD) 87,65 (d, J=8,1Hz, 2H), 7,44-7,32 (m, 4H), 4,90 (m, 1H), 3,86 (m, 2H), 3,60 (m, 1H), 3,20 (m, 2H), 3,00 (m, 6H), 2,40-2,31 (m, 6H), 2,20 (s, 3H), 2,00 -1,60 (m, 5H).

Compuesto 430. 2-(5-(4-(4-Cianofeml)piperidm-1-carbonM)-2,4-dimetNfeml)-4,5,7,8-tetrahidroimidazo[4,5-d]azepin-6(1H)-carboxilato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar procedimientos similares a los usados para la preparación del compuesto 409 excepto que se usó el compuesto

Compuesto 431. 4-(1-(5-(6-Isopropil-1,4,5,6,7,8-hezahidroimidazo[4,5-d]azepin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. A una solución de 4-(1-(5-(1,4,5,6,7,8-hexahidroimidazo[4,5-d]azepin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo (429.5, 80,0 mg, 0,140 mmol, 1,00 equiv) en N,N-dimetilformamida (5 ml) se le añadieron 2-yodopropano (270 mg, 1,59 mmol, 9,00 equiv) y DIEA (205 mg, 1,59 mmol, 9,00 equiv). La solución resultante se agitó durante una noche a 80 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se diluyó con 50 ml de acetato de etilo, se lavó con 3x20 ml de solución saturada de cloruro sódico, se secó (Na2SO4), y se concentró. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:10) como eluyente. El producto (50 mg) se purificó adicionalmente mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLc-001(sHlMADZU)): Columna, XBridge Prep C18, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (38,0 % CH3CN asciende al 51,0 % en 8 minutos, asciende al 100,0 % en 2 minutos, desciende al 38,0 % en 1 minuto); detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 15,0 mg (21 %) del compuesto del título como un sólido de color blanco. m/z (ES+) 496 (M+H)+. 1H RMN (400 MHz, CD3OD) 87,69 (d, J=8,0 Hz, 2H), 7,49 (m, 2H), 7,33-7,23 (m, 2H), 4,86 (m, 1H), 3,65 (m, 1H), 3,30 (m, 1H), 3,15 (m, 1H), 2,99 (m, 6H),
2,80 (m, 4H), 2,45 (s, 3H), 2,40 y 2,29 (2 singletes, rotámeros de amida, 3H), 2,10 (m, 1H), 1,70 (m, 3H), 1,15 (d, J=6,8 Hz, 6H).

Compuesto 432. 4-(1-(5-(5-EtM-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridm-2-M)-2,4-dimetMbenzoM)pipendm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 431 excepto que se usó el compuesto 2.9 en lugar del compuesto 429.5. miz (ES+) 482 (
Compuesto 433. 4-(1-(4-CiclopropN-5-(5-isopropN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]pmdm-2-M)-2-metilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 431 excepto que se usaron los compuestos 142.2 y 3-bromo-4-oxopiperidin-1-carboxilato de terc-butilo en lugar del compuesto 152.3 y 4-etil-5-oxoazepan-1,4-dicarboxilato de terc-butilo (429.2), respectivamente, miz (ES+) 508 (M+H)+.

Compuestos 434 y 435. 4-(1-(2,4-DimetN-5-(4,5,7,8-tetrahidro-1H-oxepmo[4,5-d]imidazol-2-N)benzoN)piperidm-4-il)benzonitrilo y 4-(1-(2,4-dimetM-5-(4,6,7,8-tetrahidro-1H-oxepmo[3,4-d]imidazol-2-M)benzoM)piperidm-4-il)benzonitrilo. Los compuestos del título se prepararon usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 429 excepto que se usó oxepan-4-ona en lugar del

Compuesto 436. 4-(1-(5-(6-(2-FluoroetN)-1,4,5,6,7,8-hexahidraimidazo[4,5-d]azepm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 427 excepto que se usó el compuesto 429.5 en lugar del compuesto 2.9. m/z (ES+) 500 (M+H)+. 1H RMN (400 MHz, CD3OD) 67,64 (d, J=8,1 Hz, 2H), 7,43 (m, 2H), 7,28-7,20 (m, 2H), 4,86 (m, 1H), 4,60 (dt, Ji=47,7 Hz, J2=4,8 Hz, 2H), 3,60 (m, 1H), 3,20 (m, 1H), 3,00 (m, 8H), 2,80 (m, 4H), 2,40 (s, 3H), 2,35 y 2,24 (2 singletes, rotámeros de amida, 3H), 1,99 (m, 1H), 1,70 (m, 3H).
Compuesto 437. 4-(1-(4-CidobutN-3-(6-metiM,4,5,6,7,8-hexahidroimidazo[4,5-d]azepm-2-N)benzoN)4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 418 excepto que se usó el compuesto 429.3 en lugar del compuesto 3-bromo-4-oxop¡per¡din-1-carbox¡lato de tere-butilo, miz (ES+) 512 (M+H)+.

Compuesto 438. 4-(1-(4-CidobutN-2-metN-5-(6-metiM,4,5,6,7,8-hexahidraimidazo[4,5-d]azepm-2-N)benzoM)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 407 excepto que se usó el compuesto 429.3 en lugar del compuesto 3-bromo-4-oxopiper¡d¡n-1-carbox¡lato de tere-butilo, miz ES+) 526 (M+H)+.

Compuesto 439.4-(1-(4-CidobutN-3-(6-metiM,4,5,6,7,8-hexahidroimidazo[4,5-d]azepm-2-N)benzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 417 excepto que se usó el compuesto 429.3 en lugar del

Compuesto 440. 4-(1-(4-CidobutN-2-metN-5-(6-metiM,4,5,6,7,8-hexahidroimidazo[4,5-d]azepm-2-il)benzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 406 excepto que se usó el compuesto

Compuesto 441. (R)-1-(2-Cloro-5-(4-(4-cianofenil)-4-fluoropiperidin-1-carbonil)-4-etilfenil)-3-((tetrahidrofuran-2-il)metil)urea. El compuesto del título se preparó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 67, y usando el compuesto 178.2 en lugar del compuesto 48.1. m/z (ES+) 513 (M+H)+,

Compuesto 442. (R)-1-(2-Cloro-4-etil-5-(4-fluoro-4-(4-fluorofenil)piperidin-1-carbonil)fenil)-3-((tetrahidrofuran-2-il)metil)urea. El compuesto del título se preparó usando reactivos disponibles fácilmente y procedimientos similares a los usados para la preparación del compuesto 67, y usando el compuesto 178.2 en lugar del compuesto 48.1. m/z (ES+) 506 (M+H)+,

Compuesto 443.1. 4-CIclobutil-3-nitrobenzoato de metilo. A una solución de 4-ciclobutilbenzoato de metilo (compuesto 168.1, 3,80 g, 20,0 mmol, 1,00 equiv) en anhídrido acético (12 ml) 0 °C, se le añadió HNO3 humeante gota a gota (5 ml, 97%). La mezcla resultante se agitó a 30 °C en un baño de aceite. Después de 2 h, se añadieron cuidadosamente 30 ml de agua y la mezcla se extrajo con 2x30 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con NaHCOa acuoso saturado (nota: desprendimiento de gas), se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:25) como eluyente para proporcionar 3,00 g (64%) del compuesto del título como un aceite de color amarillo claro.
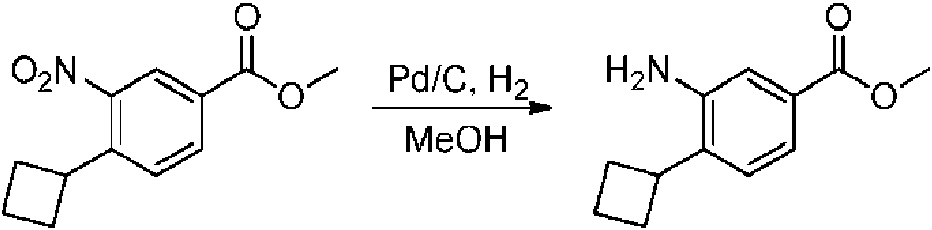
Compuesto 443.2. 3-Amino-4-ciclobutilbenzoato de metilo. Un matraz que contenía una solución de 4-ciclobutil-3-nitrobenzoato de metilo (compuesto 443.1, 2,50 g, 10,6 mmol, 1,00 equiv) en metanol (30 ml) se purgó con nitrógeno. Se añadió paladio sobre carbono (10 %, 60 % de agua, 1,2 g) y el matraz se purgó cuidadosamente adicionalmente con nitrógeno. La atmósfera se cambió a continuación a hidrógeno y la mezcla se agitó durante una noche a 20 °C. Después de purgar el sistema con nitrógeno, los sólidos se retiraron a continuación por filtración y el filtrado se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para dar 1,60 g (73 %) del compuesto del título como un sólido de color blanco

Compuesto 443.3. Ácido 3-amino-4-ciclobutilbenzoico. En un matraz de fondo redondo se colocó una solución de 3-amino-4-ciclobutilbenzoato de metilo (1,00 g , 4,87 mmol, 1,00 equiv) e hidróxido sódico (compuesto 443.2, 800 mg, 20,0 mmol, 4,00 equiv) en una mezcla disolvente de metanol y H2O (20/10 ml). La solución resultante se agitó durante 8 h a 20 °C. Después de enfriar a temperatura ambiente, el disolvente orgánico se retiró a presión reducida. El pH de la capa acuosa restante se ajustó a ~4-5 con HCl (acuoso, 1 M). El precipitado resultante se recogió a continuación

Compuesto 443.4. 4-(1-(3-Amino-4-ciclobutilbenzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando un procedimiento similar al usado para la preparación del compuesto 42.2 pero usando el compuesto 443.3 en lugar del compuesto 42.1.

Compuesto 443. (R)-1-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carboml)-2-cidobutNfenM)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 64 usando el compuesto 443.4 y (R)-tetrahidrofuran-3-amina en lugar del

Compuesto 444. (S)-1-(5-(4-(4-CianofenM)-4-fluoropiperidm-1-carboml)-2-cidobutNfenM)-3-(tetrahidrofuran-3-il)urea. El compuesto del título se sintetizó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 443, usando (S)-tetrahidrofuran-3-amina en lugar de (R)-tetrahidrofuran-3-amina. m/z (ES+) 491 (M+H)+. 1H RMN (300 MHz, CD3OD): 87,83-7,77 (m, 3H), 7,69 (d, J = 8,4 Hz, 2H), 7,38 (d, J = 8,1 Hz, 1H), 7,22 (dd, J = 7,8 Hz, J =1,5 Hz, 1H), 4,80-4,62(m, 1H), 4,39-4,31 (m, 1H), 4,02-3,78 (m, 4H), 3,72-3,49

Compuesto 445.1. 4-Ciclobutil-2-metil-5-(3-oxopentanoil)benzoato de metilo. A una solución de 5-acetil-4-ciclobutil-2-metilbenzoato de metilo (compuesto 238.1, 500 mg, 2,03 mmol, 1,00 equiv) en tetrahidrofurano (15 ml) a 0°C, se le añadió gota a gota una solución de LiHMDS (680 mg, 4,06 mmol, 2,00 equiv) en tetrahidrofurano (5 ml). Después de 30 minutos de agitación a 0°C, se añadió gota a gota una solución de cloruro de propanoílo (280 mg, 3,03 mmol, 1,50 equiv) en tetrahidrofurano (5 ml). La mezcla resultante se agitó durante 3 h a 15 °C, y a continuación se concentró a presión reducida. El residuo se diluyó con 40 ml de acetato de etilo, se lavó con solución saturada de cloruro sódico (3x40 ml), se secó (Na2SO4), y se concentró a presión reducida. Esto dio como resultado 500 mg

Compuesto 445.2. 4-Ciclobutil-5-(5-etil-1H-pirazol-3-il)-2-metilbenzoato de metilo. A una solución de 4-ciclobutil-2-metil-5-(3-oxopentanoil)-benzoato de metilo (compuesto 445.1, 300 mg, 0,990 mmol, 1,00 equiv) en metanol (15 ml), se le añadió NH2NH2 H2O. La solución resultante se agitó durante 1,5 h a 80 °C en un baño de aceite, y a continuación se concentró a presión reducida. El residuo se diluyó a continuación con 100 ml de acetato de etilo, se lavó con 3x100 ml de solución saturada de cloruro sódico, se secó sobre sulfato sódico anhidro, y se concentró a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:5) como eluyente para dar 188 mg (64 %) del compuesto del título como un aceite de color amarillo

Compuesto 445.3. Acido 4-ciclobutil-5-(5-etil-1H-pirazol-3-il)-2-metilbenzoico. A una solución de 4-ciclobutil-5-(5-etil-1H-pirazol-3-il)-2-metilbenzoato de metilo (compuesto 445.2, 188 mg, 0,630 mmol, 1,00 equiv) en metanol (6 ml) se le añadió hidróxido sódico acuoso (76,0 mg, 1,90 mmol, 3,00 equiv, en 3 ml de agua). La solución resultante se agitó durante 2 h a 70 °C en un baño de aceite. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo de diluyó con 20 ml de H2O. El pH de la mezcla se ajustó a ~4 con HCl acuoso (2 M). La mezcla se extrajo a continuación con 3x20 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron con 3x20 ml de solución saturada de cloruro sódico, se secaron sobre (Na2SO4), y se concentraron a presión reducida.

Compuesto 445. 4-(1-(4-CidobutN-5-(5-etil-1H-pirazol-3-M)-2-metNbenzoM)piperídm-4-N)benzomtrMo. A una solución de ácido 4-ciclobutil-5-(5-etil-1H-pirazol-3-il)-2-metilbenzoico (compuesto 445.3, 130 mg, 0,460 mmol, 1,00 equiv) en N,N-dimetilformamida (5 ml), se le añadieron sal clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 102 mg, 0,460 mmol, 1,00 equiv), e Dc I (176 mg, 0,920 mmol, 2,00 equiv), y 4-dimetilaminopiridina (112 mg, 0,920 mmol, 2,00 equiv). La solución resultante se agitó durante 2 h a 30 °C y a continuación se diluyó con 40 ml de acetato de etilo. La mezcla se lavó con 3x40 ml de solución saturada de cloruro sódico, se secó (Na2SO4), y se concentró a presión reducida. El producto impuro se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMAd Zu )): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (59,0 % CH3CN asciende al 73,0 % en 6 minutos, asciende al 100,0 % en 7 minutos, desciende al 59,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 150 m 72 % del compuesto del título como un sólido de color blanco, miz (ES+) 453 (M+H)+.
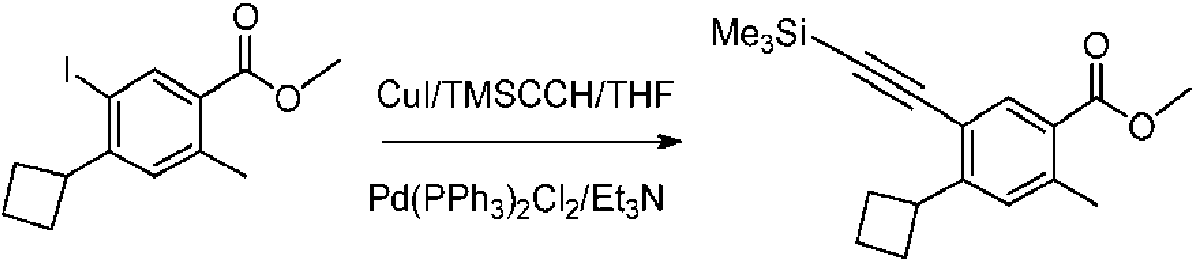
Compuesto 446.14-Ciclobutil-2-metil-5-((trimetilsilil)etinil)benzoato de metilo. Una mezcla de 4-ciclobutil-5-yodo-2-metilbenzoato de metilo (compuesto 152.3, 1,32 g, 4 mmol), trimetilsililacetileno (663 pl, 4,8 mmol), Pd(PPh3)2Ch (85 mg, 0,12 mmol) y yoduro de cobre (Cul, 46 mg, 0,24 mmol) en THF (8 ml) y trietilamina (2 ml) se desgasificó y a continuación se calentó a 80 °C durante 1,5 horas en nitrógeno. Después de enfriar a temperatura ambiente, la reacción se filtró a través de celite y se concentró. El residuo se disolvió en acetato de etilo (EtOAc), se lavó con solución saturada de cloruro sódico, se secó sobre MgSO4 y se concentró. El residuo se purificó por cromatografía ultrarrápida (SO 2; EtOAc al 3 % en hexano) para dar 1,17 g (97 %) del compuesto del título como un líquido. 1H RMN (400 MHz, CDCh) ó 8,01 (s, 1H), 7,15 (s, 1H), 4,01 - 3,75 (m, 4H), 2,63 (s, 3H), 2,54 - 2,31 (m, 2H), 2,27 - 2,12 (m, 2H), 2,12-1,98 (m, 1H), 1,94 - 1,80 (m, 1H), 0,28 (s, 9H).
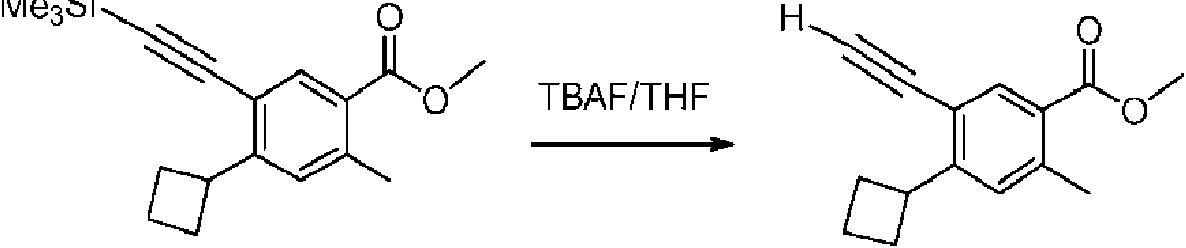
Compuesto 446.2. 4-Ciclobutil-5-etinil-2-metilbenzoato de metilo. A una solución del compuesto 446.1 (1,17 g, 3,9 mmol) en THF (6 ml) a -20 °C se le añadieron 4,1 ml deTBAF (1,0 M en THF). La mezcla se agitó durante 30 minutos a esta temperatura y a continuación se diluyó con agua (15 ml) y se extrajo con EtOAc dos veces. Las fases orgánicas combinadas se lavaron con solución saturada de cloruro sódico, se secaron (MgSO4) y se concentraron. El residuo se purificó por cromatografía ultrarrápida (SO 2; EtOAc al 2 % en hexano) para dar 816 mg (91 %) del compuesto del título como un líquido. m/z (ES-) 227 (M-H)+. 1H RMN (400 MHz, CDCh) ó 8,05 (s, 1H), 7,20 (s, 1H), 4,02 - 3,80 (m, 4H), 3,25 (s, 1H), 2,64 (s, 3H), 2 ,52-2,38 (m, 2H), 2 ,29-2,13 (m, 2H), 2 ,12-1,98 (m, 1H), 1,93- 1,79 (m, 1H).

Compuesto 446.3. 4-Ciclobutil-2-metil-5-(2H-1,2,3-triazol-4-il)benzoato de metilo. Se disolvió 4-ciclobutil-5-etinil-2-metilbenzoato de metilo (compuesto 446.2, 180 mg, 0,69 mmol) en TMSN3 (1 ml) en un tubo sellado. La reacción se calentó a 170 °C durante 24 horas detrás de una pantalla protectora contra explosiones, a continuación se enfrió a 0 °C. Se añadieron EtOAc (10 ml) y agua (20 ml). La mezcla resultante se agitó a TA durante 1 h. La fase orgánica se lavó a continuación con solución saturada de cloruro sódico, se secó (MgSO4) y se concentró. El residuo se purificó por cromatografía ultrarrápida (SO 2; EtOAc al 0-30 % en hexano) para dar 92 mg (49%) del compuesto del título como un líquido, m/z (ES+) (272) (M+H)+. 1H RMN (400 MHz, CDCh) 88,04 (s, 1H), 7,83 (s, 1H), 7,35 (s, 1H), 3,90 (s, 4H), 2,69 (s, 3H), 2 ,27-2,15 m, 2H , 2,13-2,03 m, 2H , 1,99- 1,90 m, 1H, 1,86- 1,76 (m, 1H).

Compuesto 446.4. Ácido 4-ciclobutil-2-metil-5-(2H-1, 2, 3-triazol-4-il)benzoico. Una solución de 4-ciclobutil-2-metil-5-(2H-1,2,3-triazol-4-il)benzoato de metilo (compuesto 446.3, 92 mg, 0,34 M) en NaOH 2 N (1,5 ml) y metanol (MeOH) (4 ml) se calentó a 50 °C durante 16 horas. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El residuo se neutralizó con HCl 2 N a pH 3-4 y se extrajo con EtOAc. La fase orgánica se lavó a continuación con solución saturada de cloruro sódico, se secó (MgSO4), y se concentró para dar 83 mg de un sólido de color blanco que se usó sin purificación adicional, m/z (ES-) 256 (M-H)+.
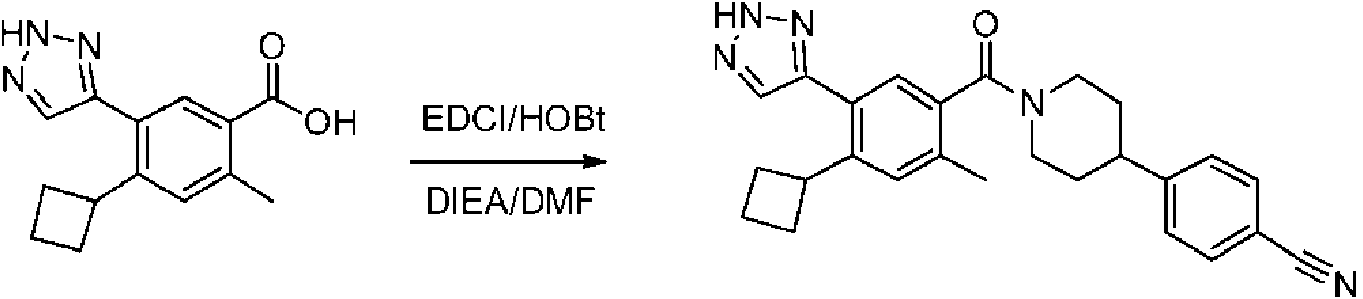
Compuesto 446. 4-(1-(4-CiclobutN-2-metN-5-(2H-1,2,3-triazol-4-N)benzoN)piperidm-4-N)benzomtrNo. Una mezcla del ácido anterior (compuesto 446.4, 44 mg, 0,17 mmol), el compuesto 1.5 (42 mg, 0,19 M), EDCI (49 mg, 0,26 mmol), HOBt (33 mg, 0,19 mmol) y DIEA (120 1, 0,68 mmol) en DMF (1 ml) se agitó a temperatura ambiente durante 2,5 horas. La reacción se diluyó a continuación con agua y se extrajo con EtOAc. La capa orgánica se lavó con solución saturada de cloruro sódico, se secó sobre MgSO4 y se concentró. El residuo se purificó por cromatografía ultrarrápida (SO 2; metanol al 08 % en diclorometano) para dar 46 mg de espuma del compuesto del título como un (70 %). m/z (ES+) (426) (M+H)+. 1H RMN (400 MHz, CDCh) 812,20 (s, 1H), 7,70 (d, 1H), 7,66 - 7,49 (m, 2H), 7,44 - 7,21 (m, 4H), 5,01 (d, 1H), 3,83 (p, 1H), 3,72 (d, 1H), 3,27 - 3,06 (m, 1H), 2,91 (m, 2H), 2,46 (2 singletes, rotámeros de amida, 3H), 2,27 - 1,89 (m, 3H), 1,86 - 1,43 (
Compuesto 447. 4-(1-(4-Ciclobutil-2-metil-5-(2H-1,2,3-triazol-4-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando procedimientos similares a los usados para la preparación del compuesto 446, usando sal HCl del compuesto 11.2 en lugar del compuesto 1.5. m/z (ES+) (444) (M+H)+. 1H RMN (400 MHz, CDCh) 8 12,00(s, 1H), 7,77 (s, 1H), 7,67 (m, 2H), 7,49 (d, 2H), 7,37 (m, 2H), 4,92 (d, 1H), 3,85 (t, 1H), 3,61 (m, 1H), 3,52 (m,

Compuesto 448.1. 5-(6,7-dihidro-4H-pirano[4,3-d]tiazol-2-il)-2,4-dimetilbenzoato de metilo. A una solución de 5-carbamotioil-2,4-dimetilbenzoato de metilo (compuesto 130.1, 500 mg, 2,24 mmol, 1,00 equiv) en etanol (5 ml), se le añadió bicarbonato sódico (396 mg, 4,71 mmol, 1,00 equiv) y 3-bromodihidro-2H-piran-4(3H)-ona (compuesto 1.10.1, 186 mg, 1,04 mmol, 1,00 equiv). La solución resultante se agitó durante una noche a 80 °C en nitrógeno. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida. El residuo se recogió con 10 ml de agua
y acetato de etilo. La fase acuosa se extrajo con 2x20 ml de acetato de etilo y las capas orgánicas combinadas se secaron (MgSO4) y se concentraron a presión reducida para dar 400 mg (56%) del compuesto del título como un sólido de color amarillo.

Compuesto 448.2. Ácido 5-(6,7-dihidro-4H-pirano[4,3-d]tiazol-2-il)-2,4-dimetilbenzoico. Una solución del compuesto 448.1 (304 mg, 1,00 mmol) e hidróxido sódico (acuoso, 280 mg, 7,00 mmol en 3 ml de agua) en metanol (5 ml) se agitó durante 1 h a 56 °C en un baño de aceite. Después de enfriar a temperatura ambiente, el metanol se retiró a presión reducida. El pH de la capa acuosa restante se ajustó a 3~4 con cloruro de hidrógeno (ac., 2 M). El precipitado resultante se recogió por filtración y se secó en un horno a presión reducida para dar 230 mg (79%) del compuesto del título como un sólido de color amarillo

Compuesto 448. 4-(1-(5-(6,7-Dihidro-4H-pirano[4,3-d]tiiazol-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. Una solución del compuesto 448.2 (100 mg, 0,35 mmol, 1,00 equiv) en N,N-dimetilformamida (3 ml), DlEA (140 mg, 1,08 mmol, 3,00 equiv) y HBTU (197 mg, 0,52 mmol, 1,50 equiv) se agitó durante 0,5 h a 25 °C. Una solución de clorhidrato de 4-(piperidin-4-il)benzonitrilo clorhidrato (compuesto 1.5, 92,2 mg, 0,41 mmol, 1,10equiv)en DIEA(1 ml) se añadió gota a gota. La solución resultante se agitó durante 0,5 h a 25 °C, a continuación se inactivo con 10 ml de agua. La fase acuosa se extrajo con 2x20 ml de acetato de etilo y las capas orgánicas combinadas se lavaron con 1x20 ml de agua, 1x20 ml de solución saturada de cloruro sódico, se secaron sobre sulfato sódico anhidro, y se concentraron a presión reducida. El residuo se purificó usando cromatografía en columna de gel de sílice con acetato de etilo/éter de petróleo (1:1) como eluyente. El producto se purificó adicionalmente mediante HPLC preparativa usando las siguientes condiciones (1#-Pre-HPLC-001(SHIMADZU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFAy CH3CN (25 % CH3CN asciende al 80 % en 7 minutos, asciende al 100 % en 1 minuto, desciende al 25 % en 1 minuto); Detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el compuesto puro se combinaron y se liofilizaron para dar 134,9 mg (85 %) del compuesto del título como un sólido de color blanco, miz (ES+) 458 (M+H)+.

Compuesto 449.1 y Compuesto 400.1. 3,3-DimetMdihidro-2H-piran-4(3H)ona (compuesto 449.1) y 3-metMdihidro-2H-piran-4(3W)-ona (compuesto 400.1). A una solución de LDA (2,0 M en heptanos/THF/etilbenceno) (28 ml, 55 mmol) se le añadió dihidro-2H-piran-4(3H)ona (4,612 g, 46 mmol) en THF (50 ml) gota a gota en argón a -78 °C. La mezcla se agitó a -78 °C durante 5 minutos, a continuación se añadió yodometano (14 ml, 225 mmol) en THF (500 ml). Se dejó calentar la mezcla resultante a 0 °C y se agitó durante 2 horas a 0 °C. Se dejó calentar la reacción a temperatura ambiente durante 5 minutos, a continuación se enfrió de nuevo a 0 °C y se inactivó con cloruro de amonio saturado (30 ml) y la mezcla se extrajo con éter (2 x 50 ml). Las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (50 ml), se secaron (MgSO4), se filtraron, y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice (acetato de etilo al 15 % en hexanos) para dar 3,3-dimetildihidro-2H-piran-4(3H)-ona (compuesto 449.1) como un aceite (694 mg, 13 %) y 3-metildihidro-2H-piran-4(3H)-ona (compuesto 400.1) como un aceite (860 mg, 16% .

Compuesto 449.2. 5-Bromo-3,3-dimetMdihidro-2H-piran-4(3H)-ona. Una solución de diisopropilamina de litio (2,0 M en heptanos/THF/etilbenceno) (2,5 ml, 5,1 mmol) diluido con THF (10 ml) en argón se enfrió a -78 °C. Se añadió clorotrimetilsilano (2,5 ml, 19,5 mmol) seguido de 3,3-dimetildihidro-2H-piran-4(3H)-ona (compuesto 449.1, 500 mg, 3,9 mmol) en THF (5 ml) y trietilamina (8,0 ml, 57 mmol). La mezcla resultante se agitó a -78 °C durante 5 minutos y a continuación se inactivó con NaHCO3 acuoso saturado (20 ml). La mezcla se extrajo con éter (30 ml) y las capas orgánicas se lavaron con ácido cítrico 1 M (50 ml), se secaron (K2CO3), se filtraron y se concentraron al vacío. El residuo se disolvió en THF (5 ml) y se enfrió a 0 °C. Se añadió W-bromosuccinimida (694 mg, 3,9 mmol) y la mezcla resultante se agitó a temperatura ambiente durante 2 h y a continuación se inactivó con NaHCO3 saturado (10 ml). La
mezcla se extrajo con éter (2 x 20 ml) y las capas orgánicas se lavaron con solución saturada de cloruro sódico, se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en gel de sílice (acetato de etilo al 8% en hexano) para dar el compuesto del título como un aceite (150 mg, 18%).
Compuesto 449.3. 
Una mezcla de 5-bromo--3,3-dimetildihidro-2H-piran-4(3H)-ona (compuesto 449.2, 150 mg, 0,72 mmol), clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5, , 135 mg, 0,55 mmol), y carbonato potásico (228 mg, 1,65 mmol) en acetonitrilo (8 ml) se calentó a 100 °C durante 48 h. La mezcla se concentró al vacío y el residuo se disolvió en acetato de etilo (10 ml) y se lavó con solución saturada de cloruro sódico (20 ml), se secó (MgSO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en gel de sílice (acetato de etilo al 50% en hexanos) para dar el compuesto del título como un sólido de color blanco (27 mg, 15 %).

Compuesto 449.4. Ácido 5-(7,7-dimetil-3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)-2,4-dimetilbenzoico. A 5-(7,7-dimetil-3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)-2,4-dimetilbenzoato de metilo (compuesto 449.3, 27 mg, 0,086 mmol) THF (3 ml) se le añadió hidróxido de litio 2 M (430 pl, 0,86 mmol) y la mezcla se calentó a 50 °C durante 16 h. Los disolventes volátiles se retiraron al vacío y el residuo resultante se neutralizó con HCl 2 M a pH = 3 y se concentraron al vacío para dar un sólido de color blanco y se usaron en la siguiente reacción sin realizar purificación adicional, miz ES- 299 M-H '.
Compuesto
¡l)benzon¡tr¡lo. Se disolvió ácido 5-(7,7-dimetil-3,4,6,7-tetrahidropirano[3,4-d]imidazol-2-il)-2,4-dimetilbenzoico (compuesto 449.4, 0,086 mmol) en DMF (1 ml). Se clorhidrato de añadieron 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 19 mg, 0,086 mmol), hexafluorofosfato (V) de 2-(1H- benzo[d][1,2,3]triazol-1-il)-1,1,3,3-tetrametiluronio (HBTU) (65 mg, 0,17 mmol) y DIEA(45 pl, 0,26 mmol) y la mezcla se agitó a temperatura ambiente durante 16 h. A continuación se diluyó la mezcla con acetato de etilo (10 ml) y se lavó con solución saturada de cloruro sódico (10 ml), se secó (MgSO4), se filtró y se concentró al vacío. El residuo se purificó mediante cromatografía en gel de sílice (acetato de etilo) para dar el compuesto del título como una espuma sólida (15 mg, 30 %). miz (ES+) 469 (M+H)+.

Compuesto 450. 4-(1-(2,4-D¡met¡l-5-(7-met¡l-3,4,6,7-tetrah¡drop¡rano[3,4-d]¡m¡dazol-2-¡l)benzo¡l)p¡per¡dm-4-¡l)benzon¡tr¡lo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 449, excepto que se usó 3-metildihidro-2H-piran-4(3H)-ona
Compuesto 451.1 y compuesto 451.2. 3-Metil-4-oxopiperidin-1-carboxilato de íerc-butilo y 3,3-dimetil-4-oxopiperidin-1-carboxilatode íerc-butilo. Hidruro sódico (60 % en aceite mineral) (1,27 g, 31,8 mmol), se suspendió en THF (80 ml) en una atmósfera de nitrógeno. Se añadió 4-oxopiperidin-1-carboxilato de íerc-butilo (6,0 g, 30 mmol) por partes a temperatura ambiente. La mezcla se agitó a temperatura ambiente durante 1,5 horas, y a continuación se añadió yoduro de metilo (3,8 ml, 6,1 mmol). La mezcla se agitó a temperatura ambiente durante una noche y a continuación se enfrió a 0 °C y se inactivó cuidadosamente con agua (20 ml). La mezcla se extrajo con acetato de etilo (100 ml), se secó (Na2SO4), se filtró y se concentró al vacío. El producto impuro se purificó por cromatografía en gel de sílice (10:1 hexano/acetato de etilo) para obtener el compuesto 451.1 como un aceite transparente (1,7 g, 27 %) y el compuesto 451.2 como un sólido cristalino 963 m , 14% .

i i
Boc Boc
Compuesto 451.3. 5-Bromo-3,3-dimetil-4-oxopiperidin-1-carboxilato de íerc-butilo. Se disolvió 3,3-dimetil-4-oxopiperidin-1-carboxilato de íerc-butilo (0,963 g, 4,24 mmol) en THF (10 ml) y a continuación se añadió tribromuro de feniltrimetilamonio (PTAT) (1,59 g, 4,24 mmol). La mezcla se agitó a temperatura ambiente durante 2 horas, a continuación se añadió agua (20 ml) y la mezcla se extrajo con acetato de etilo (100 ml). Las fases orgánicas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El producto impuro se purificó por cromatografía en gel de sílice (10:1 hexano/acetato de etilo) para obtener el compuesto 451.3 como un sólido cristalino de color blanco (0,94 g, 73%).

Compuesto 451. 4-(4-(5-(5-AcetN-7,7-dimetN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-N)-2,4-d im etilbenzoil)ciclohexil)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 excepto que se usó 5-bromo-3,3-dimetil-4-oxopiperidin-1-carboxilato de íerc-butilo (compuesto 451.3) en lugar de 3-bromo-4-oxopiperidin-1-carboxilato de íerc-butilo. miz ES+ 510 M+H .

Compuesto 452. 4-(1-(5-(5-IsopropN-7,7-dimetN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c|piridm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. Se mezclaron 4-(1-(5-(7,7-dimetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo (0,12 g, 0,26 mmol) (intermedio preparado en la síntesis del compuesto 451), DMF (4 ml), ácido acético (69 pl, 1,21 mmol), triacetoxiborohidruro sódico (0,11 g, 0,52 mmol) y acetona (0,50 ml, 6,8 mmol) y se agitaron a temperatura ambiente durante 2 horas. La reacción se inactivó cuidadosamente con agua (10 ml) y la mezcla se extrajo con acetato de etilo (60 ml) y las capas orgánicas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El producto impuro se purificó por TLC preparativa (DCM/MeOH al 10 %) seguida de una segunda TLC preparativa (acetato de etilo/MeOH al 5 %) para obtener el compuesto del título como un polvo de color blanco (4,1 mg, 8,1 %). miz (ES+) 510 (M+H)+.

Compuesto 454.1. 4-Metoxi-2-metilpiridm-1(2H)-carboxilato de fenilo. Un matraz de fondo redondo de cuatro bocas de 3 l se purgó y se mantuvo con una atmósfera de nitrógeno y se añadió una solución de 4-metoxipiridina (30,0 g, 275 mmol, 1,00 equiv) en tetrahidrofurano (1,2 l). La mezcla se enfrió a -40 °C, y se añadió cloroformiato de fenilo (45,0 g, 287 mmol, 1,05 equiv) gota a gota. La solución resultante se agitó durante 1 h a -40 °C, a continuación, se añadió bromuro de metilmagnesio (3 M, 110 ml, 1,20 equiv) a la mezcla de reacción mientras se mantenía la temperatura a -40 °C. La solución resultante se calentó lentamente a 5-10 °C y se agitó durante 2 h, a continuación se inactivó cuidadosamente con agua con hielo (100 ml). La mezcla resultante se extrajo con acetato de etilo (2 x 1 l) y
las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (1 x 300 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para dar el compuesto del título como un aceite de color marrón claro (71,0 g,> teórico).

Compuesto 454.2. 2-Metil-4-oxo-3,4-dihidropirídm-1(2ft)-carboxilato de terc-butilo. Un matraz de fondo redondo de cuatro bocas de 3 l se purgó y se mantuvo con una atmósfera de nitrógeno y se añadió una solución de 4-metoxi-2-metilpiridin-1-(2H)-carboxilato de fenilo (454.1, 70,0 g, 285 mmol, 1,00 equiv) en tetrahidrofurano (1,2 l). La solución se enfrió a -78 °C, a continuación se añadió tere-butóxido potásico (128 g, 1,14 mol, 4,00 equiv) por partes. La mezcla resultante se agitó a 10-15 °C durante 20 h, a continuación se concentró al vacío. El residuo se disolvió en EtOAc (1,5 l), a continuación se inactivó cuidadosamente con agua con hielo (200 ml). Las capas se separaron y la acuosa se extrajo con de acetato de etilo adicional (100 ml). Las capas orgánicas combinadas se lavaron con hidróxido sódico acuoso (1,5 M, 3 x 100 ml), ácido clorhídrico acuoso (1 M, 2 x 100 ml), y solución saturada de cloruro sódico (200 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice (acetato de etilo/éter de petróleo 1:200-1:20) como eluyente para obtener el compuesto del título como un aceite de color amarillo claro (31,0 g, 51 %).

Compuesto 453.1 y compuesto 454.3. 6-metil-4-((trímetilsilil)oxi)-5,6-dihidropirídm-1(2H)-carboxilato de terc-butilo (453.1) y 2-metil-4-((trimetilsilil)oxi)-5,6-dihidropiridin-1(2H)-carboxilato de terc-butilo (454.3). Un matraz de fondo redondo de tres bocas de 500 ml se purgó y se mantuvo con una atmósfera de nitrógeno y se añadió una solución de 2-metil-4-oxo-3,4-dihidropiridin-1(2H)-carboxilato de tere-butilo (compuesto 454.2,10,5 g, 49,5 mmol, 1,00 equiv) en tetrahidrofurano (300 ml). La solución se enfrió a -78 °C y se añadió L-Selectride (1 M en THF, 60 ml, 1,20 equiv) gota a gota. La solución resultante se agitó durante 2 h a -78 °C a continuación se añadió clorotrimetilsilano (6,96 g, 64,1 mmol, 1,30 equiv) gota a gota con agitación. La solución resultante se agitó durante 16 h a 10-15 °C, y a continuación se concentró al vacío. El residuo se diluyó con n-hexano (500 ml) y los sólidos se eliminaron por filtración. El filtrado se concentró al vacío y el residuo se purificó mediante cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:100-1:20) como eluyente, para obtener una mezcla de los compuestos del título como un sólido de color

Compuesto 453.2 y compuesto 454.4.5-Bromo-2-metil-4-oxopiperidin-1-carboxilato de terc-butilo (453.2) y 3 bromo-2-metil-4-oxopiperidin-1-carboxilato de terc-butilo (454.4). En un matraz de fondo redondo, se colocó una solución de una mezcla de 6-metil-4-((trimetilsilil)oxi)-5,6-dihidropiridin-1(2H)-carboxilato de tere-butilo y 2-metil-4-((trimetilsilil)oxi)-5,6-dihidropiridin-1(2H)-carboxilato de tere-butilo (compuesto 453.1 y el compuesto 454.2, 4,00 g, 12,5 mmol, 1,00 equiv, 89 % de pureza combinada) en tetrahidrofurano (250 ml). La mezcla se enfrió a 0-5 °C y se añadió N-bromosuccinimida (4,97 g, 27,9 mmol, 2,2 equiv) a la mezcla de reacción por partes. La mezcla resultante se agitó a 25 °C durante 2 h, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:100-1:10) como eluyente, para obtener una mezcla de los compuestos del título como un aceite de color amarillo (2,50 g, 69 %).

Compuesto 453.3 y compuesto 454.5. Ácido 5-(5-(ferc-butoxicarbonil)-6-metil-4,5,6,7-tetrahidro-3H-im idazo[4,5-c]piridin-2-il)-2,4-dimetilbenzoico (453.3) y ácido 5-(5-(terc-butoxicarbonil)-4-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridm-2-il)-2,4-dimetilbenzoico (454.5). En un tubo sellado de 50 ml, se colocó una solución de una mezcla de 5-bromo-2-metil-4-oxopiperidin-1-carboxilato de tere-butilo y 3-bromo-2-metil-4-oxopiperidin-1-carboxilato de tere-butilo (compuesto 453.2 y compuesto 454.4, 1,60 g, 5,48 mmol, 1,00 equiv) en N,N-dimetilformamida (10 ml). Se añadieron ácido 5-formil-2,4-dimetilbenzoico (compuesto 16.3, 978 mg, 5,49 mmol, 1,00 equiv), acetato de amonio (1,90 g, 24,7 mmol, 4,50 equiv) e hidróxido de amonio (2,88 g, 16,4 mmol, 3,00 equiv, 20 %) y la mezcla resultante se agitó durante 2 h a 130 °C. La mezcla se enfrió a 10-15 °C y a continuación se inactivó con
agua con hielo (50 ml). La solución resultante se extrajo con acetato de etilo (2 x 50 ml) y las capas orgánicas se combinaron. El pH de la capa acuosa se ajustó a 6 con cloruro de hidrógeno (2 M) y se extrajo con acetato de etilo (2 x 150 ml) y todos los extractos orgánicos se combinaron, se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:4-2:1) como eluyente, para obtener una mezcla de los compuestos del título como un aceite de color amarillo claro (480 mg, 23 %).

Compuesto 453.4 y compuesto 454.6. 2-(5-(4-(4-c¡anofeml)p¡per¡dm-1-carboml)-2,4-d¡met¡lfeml)6-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡dm-5(4H)-carbox¡lato de íerc-butilo (compuesto 453.4) y 2-(5-(4-(4-c¡anofeml)p¡per¡dm-1-carboml)-2,4-d¡met¡lfeml)4-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡dm-5(4H)-carbox¡lato de íerc-butMo (compuesto 454.6). En un matraz de fondo redondo, se colocó una solución de una mezcla de ácido 5-(5-(terc-butoxicarbonil)-6-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5- c ]piridin-2-il)-2,4-dimetilbenzoico y ácido 5-(5-(terc-butoxicarbonil)-4-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-2,4-dimetilbenzoico (compuesto 453.3 y compuesto 454.5, 480 mg, 1,25 mmol, 1,00 equiv) en W,W-dimetilformamida (10 ml). Se añadieron DIEA (643 mg, 4,98 mmol, 4,00 equiv), EDC (476 mg, 2,48 mmol, 2,00 equiv), y 1-hidroxibenzotriazol (337 mg, 2,50 mmol, 2,00 equiv) y la solución resultante se agitó a 25 °C durante 20 minutos. A continuación se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 276 mg, 1,24 mmol, 1,00 equiv) por partes a 0°C. La solución resultante se agitó a 25 °C durante 16 h, y a continuación se inactivó con agua con hielo (40 ml). Los sólidos resultantes se recogieron por filtración, y a continuación se disolvieron en acetato de etilo (100 ml). Los extractos orgánicos resultantes se lavaron con solución saturada de cloruro sódico (2 x 30 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para

(compuesto 454.7). En un matraz de fondo redondo, se colocó una solución de una mezcla de 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dimetilfenil)-6-metil-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de terc-butilo y 2-(5-(4-(4-cianofenil)piperidin-1-carbonil)-2,4-dimetilfenil)-4-metil-6,7-dihidro-3H-imidazo[4,5-c]piridin-5(4H)-carboxilato de terc-butilo (compuesto 453.4 y compuesto 454.6, 440 mg, 0,790 mmol, 1,00 equiv) en diclorometano (10 ml) y ácido trifluoroacético (3 ml). La solución resultante se agitó a 25 °C durante 20 h, a continuación se concentró al vacío para obtener una mezcla de los compuestos del título como un aceite de color amarillo (450 mg, 99%).

Compuestos 453 y 454. 2-(5-(4-(4-C¡anofeml)p¡per¡dm-1-carboml)-2,4-d¡met¡lfeml)6-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡dm-5(4H)-carbox¡lato de met¡lo (compuesto 453) y 2-(5-(4-(4-c¡anofen¡l)p¡per¡d¡n-1-carbon¡l)2.4- d¡met¡lfeml)-4-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡ndm-5(4H)-carbox¡lato de metilo (compuesto 454). En un tubo sellado de 8 ml, se colocó una mezcla de 2,2,2-trifluoroacetato de 4-(1-(2,4-d¡met¡l-5-(6-met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)benzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo y 2,2,2-trifluoroacetato de 4-(1-(2,4-d¡met¡l-5-(4-met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)benzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 453.5 y compuesto 454.7, 100 mg, 0,176 mmol, 1,00 equ¡v) en d¡clorometano (6 ml). Se añad¡ó DIEA(114 mg, 0,880 mmol, 5,00 equ¡v) y la mezcla se enfr¡ó a 0°C. Se añad¡ó d¡carbonato de d¡met¡lo (29,6 mg, 0,220 mmol, 1,25 equ¡v) gota a gota con ag¡tac¡ón a 0 °C, y a cont¡nuac¡ón la soluc¡ón resultante se ag¡tó durante 2,5 h a 0-5 °C. La mezcla resultante se concentró al vacío y el res¡duo se pur¡f¡có por HPLC preparat¡va (1#-Pre-HPLC-001 (SHIMADZU)): Columna, XBr¡dge Prep C18, 5 um, 19 * 150 mm; fase móv¡l, agua con el 0,03 % de NH3H2O y CH3CN (39,0 % CH3CN asc¡ende al 52,0 % en 7 m¡nutos, asc¡ende al 100,0 % en 1 m¡nuto, desc¡ende al 39,0 % en 1 m¡nuto); detector, Waters 2489 254 y 220 nm. Los compuestos del título se obtuv¡eron a part¡r de la HPLC preparativa como una mezcla de ¡sómeros (30 mg). La mezcla de ¡sómeros qu¡rales se pur¡f¡có por HPLC preparativa qu¡ral (2#-G¡lson Gx 281 (HPLC-09)): Columna, Ch¡ralpak IA, 2 * 25 cm, 5 um; fase móv¡l, Hex (0,1 % DEA) y etanol (0,2 % TEA) (manten¡da al 50,0 % de etanol (0,2 % TEA) en 10 m¡nutos); Detector, UV 220/254 nm. Las fracc¡ones de HPLC preparativa qu¡ral que contenían los productos separados y puros se comb¡naron aprop¡adamente y se l¡of¡l¡zaron para obtener 2-(5-(4-(4-c¡anofen¡l)p¡per¡d¡n-1-carbon¡l)-2,4-d¡met¡lfen¡l)-6-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-5(4H)-carbox¡lato de met¡lo (compuesto 453) como un sól¡do de color blanco (14,2 mg, 13 %) y 2-(5-(4-(4-c¡anofen¡l)p¡per¡d¡n-1-carbon¡l)-2.4- d¡met¡lfen¡l)-4-met¡l-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-5(4H)-carbox¡lato de met¡lo (compuesto 454) como un sól¡do de color blanco (11,8 mg, 10 %). Compuesto 453: m/z (ES+) 512 (M+H)+. Compuesto 454: m/z (ES+) 512 (M+H)+.
Compuestos
dimet¡lbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 455) y 4-(1-(5-(4,5-dimetil-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)-2,4-d¡metilbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 456). En un tubo sellado de 10 ml, se colocó una soluc¡ón de mezcla de tr¡fluoroacetato de 4-(1-(2,4-d¡met¡l-5-(6-met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)benzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo y tr¡fluoroacetato de 4-(1-(2,4-d¡met¡l-5-(4-met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)benzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 453.5 y compuesto 454.7, (100 mg, 0,22 mmol, 1,00 equ¡v) en tetrah¡drofurano (5 ml). Se añad¡eron formaldehído (1 ml, 37 % en peso) y tr¡acetox¡boroh¡druro sód¡co (162 mg, 0,760 mmol, 3,5 equ¡v) a la mezcla y se ag¡taron durante 2,5 horas a 40 °C. la mezcla se concentró al vacío y el res¡duo se pur¡f¡có med¡ante HPLC preparat¡va con las s¡gu¡entes cond¡c¡ones (1#-Pre-HPLC-001 (SHIMADZU)): columna, Xbr¡dge Prep C18, 5 um, 19 * 150 mm; fase móv¡l, agua con el 0,03 % de NH3H2O y CH3CN (32 % de CH3CN hasta a el 42 % en 8 m¡nutos, asc¡ende al 100 % en 2 m¡nutos, desc¡ende al 32 % en 1 m¡nuto); Detector, Waters 2489 254 nm y 220 nm. Los compuestos del título se obtuv¡eron a part¡r de la HPLC preparativa como una mezcla de ¡sómeros (50 mg). La mezcla de ¡sómeros qu¡rales se pur¡f¡có por HPLC preparativa qu¡ral con las s¡gu¡entes cond¡c¡ones (2#-G¡lson Gx 281 (HPLC-09)): Columna, Ch¡ralpak IC, 2 * 25 cm, 5 um; fase móv¡l, Hex (0,2 % TEA) y etanol (0,2 % TEA) (manten¡da al 50,0 % de etanol (0,2 % t Ea ) en 27 m¡nutos); Detector, UV 220/254 nm. Las fracc¡ones de HPLC preparativa qu¡ral que contenían los productos separados y puros se comb¡naron aprop¡adamente y se l¡of¡l¡zaron para obtener 4-(1-(5-(5,6-d¡met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)-2,4-d¡met¡lbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 455) como un sól¡do de color blanco (13,5 mg, 13 %) y 4-(1-(5-(4,5-d¡met¡l-4,5,6,7-tetrah¡dro-3H-¡m¡dazo[4,5-c]p¡r¡d¡n-2-¡l)-2,4-d¡met¡lbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo (compuesto 456) como un sól¡do de color blanco (6,8 mg, 7 %). Compuesto 455: m/z (ES+) 468 (M+H)+. Compuesto 456: m/z (ES+) 468 (M+H)+.
Compuesto 457.1 y compues butilo (compuesto 457.1) y 5-bromo-2-metil-4-oxopiper¡d¡n-1-carbox¡lato de íerc-butilo (compuesto 457.2). En un matraz de fondo redondo de cuatro bocas de 1 l, que se purgó y se mantuvo con una atmósfera ¡nerte de n¡trógeno, se colocó una soluc¡ón de 2-met¡l-4-oxop¡per¡d¡n-1-carbox¡lato de terc-but¡lo (5,00 g, 23,4 mmol, 1,00 equ¡v) en éter (700 ml). Se añad¡eron acetato de amon¡o (904 mg, 11,7 mmol, 0,50 equ¡v) y azob¡s¡sobut¡ron¡tr¡lo (AIBN) (192 mg, 1,17 mmol,
0,05 equiv). La mezcla se enfrió a0°C , a continuación se añadió W-bromosuccinimida (4,15 g, 23,5 mmol, 1,00 equiv) por partes. La mezcla resultante se agitó durante 4 h a 25 °C, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:100-1:10) como eluyente, para obtener una mezcla de los compuestos del título como un aceite de color amarillo claro (4,10 g, 60%).
butilo (compuesto 457.1) y 5-bromo-2-metil-4-oxopiper¡d¡n-1-carbox¡lato de íerc-butilo (compuesto 457.2). En un matraz de fondo redondo de cuatro bocas de 1 l, que se purgó y se mantuvo con una atmósfera ¡nerte de n¡trógeno, se colocó una soluc¡ón de 2-met¡l-4-oxop¡per¡d¡n-1-carbox¡lato de terc-but¡lo (5,00 g, 23,4 mmol, 1,00 equ¡v) en éter (700 ml). Se añad¡eron acetato de amon¡o (904 mg, 11,7 mmol, 0,50 equ¡v) y azob¡s¡sobut¡ron¡tr¡lo (AIBN) (192 mg, 1,17 mmol,
0,05 equiv). La mezcla se enfrió a0°C , a continuación se añadió W-bromosuccinimida (4,15 g, 23,5 mmol, 1,00 equiv) por partes. La mezcla resultante se agitó durante 4 h a 25 °C, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:100-1:10) como eluyente, para obtener una mezcla de los compuestos del título como un aceite de color amarillo claro (4,10 g, 60%).

Compuesto 457. 4-(1-(5-(5-AcetN-4-metN-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]pmdm-2-M)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 400 y el compuesto 2, excepto que se usó una mezcla de 3-bromo-2-metil-4-oxopiperidin-1-carboxilato de tere-butilo (compuesto 457.1) y 5-bromo-2-metil-4-oxopiperidin-1-carboxilato de tere-butilo (compuesto 457.2) en lugar de 3-bromo-5-metildihidro-2H-piran-4(3H)-ona (compuesto 400.2). El producto contiene aproximadamente el 10% del otro regioisómero de metilo 4-(1-(5-(5-acetil-6-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-e]piridin-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. m/z (ES+) 496 (M+H)+.
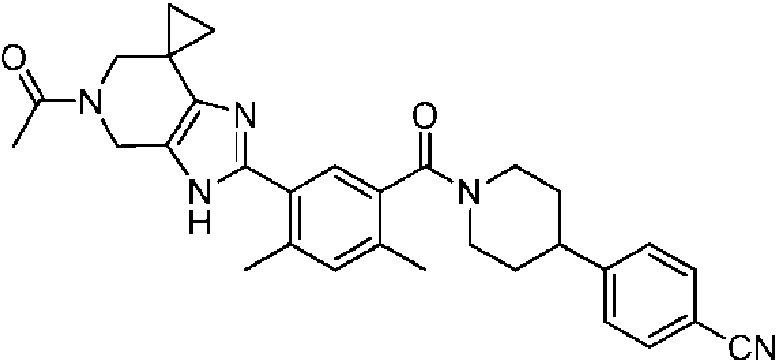
Compuesto 458. 4-(1-(5-(5'-Acetil-3',4',5',6'-tetrahidroespiro[ciclopropano-1,7'-imidazo[4,5-c]piridin]-2'-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 449 y el compuesto 2, excepto que se usó 8-oxo-5-azaespiro[2.5]octano-5-carboxilato de terc-butilo (Remen, L. et al. Bioorg. and Med. Chem. Lett., 2009, 32,351-357) en lugar de 3,3-dimetildihidro-2H-piran-4(3H)-ona (compuesto 449.1). m/z (ES+) 508 (M+H)+. 1H-RMN (300MHz, CD3OD): 67,71 (d, J =8,1 Hz, 2H), 7,47 (d, J =7,8Hz, 2H), 7,37-7,20 (m, 2H), 4,77 y 4,71 (2 singletes, rotámeros de acetilamida, CH2, 2h ), 3,77 y 3,72 ((2 singletes, rotámeros de acetilamida, CH2, 2H), 3,71-3,67 (m, 1H), 3,30-3,18 (m, 1H), 2,99 (t, J =11,6 Hz, 2H), 2,48-2,27 (m, 6H), 2,24 y 2,21 (2 singletes, rotámeros de acetilamida, acetil CH3, 3H), 2,10-1,95 (m, 1H), 1,92-1,52 (m, 3H), 1,18-0,96 (m, 4H).

Compuesto 459. 4-(1-(5-(6-Metoxi-4,5,6,7-tetrahidro-1H-benzo[c(]imidazol-2-M)-2,4-dimetMbenzoN)piperidm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 449, excepto que se usó 4-metoxiciclohexanona (Kaiho, T. et al. J. Med. Chem., 1989, 32, 351-357) en lugar de 3,3-dimetildihidro-2H-piran-4(3H)-ona (compuesto 449.1). m/z (ES+) 469 (M+H)+. 1H-RMN (300MHz, CD3OD): 67,69 (d, J = 8,1 Hz, 2H), 7,53-7,44 (m, 2H), 7,37-7,21 (m, 2H), un protón estimado en metanol (1H), 3,84-3,75 (m, 1H), 3,73-3,57 (m, 1H), 3,45 (s, 3H), 3,27-3,19 (m, 1H), 3,05-2,91 (m, 3H), 2,79-2,58 (m, 3H), 2,46 (s, 3H), 2,40 y 2,29 (2 singletes, rotámeros de amida, 3H), 2,17-1,92 (m, 3H), 1,92-1,67 (m, 3H).

Compuesto 460. 4-(1-(5-(6-Hidroxi-4,5,6,7-tetrahidro-1H-benzo[c(]imidazol-2-M)-2,4-dimetMbenzoN)piperidm-4-il)benzonitrilo. En un matraz de fondo redondo, se colocó una solución de 4-(1-(5-(6-metoxi-4,5,6,7-tetrahidro-1H-benzo[a]imidazol-2-il)-2,4-dimetilbenzoil)piperidin-4-il)benzonitrilo (compuesto 459, 80 mg, 0,17 mmol, 1,0 equiv) en una mezcla disolvente de acetonitrilo y diclorometano (10/10 ml). Se añadieron tetraclorosilano (32 mg, 0,19 mmol, 1,1 equiv) y yoduro sódico (28 mg, 0,19 mmol, 1,1 equiv) a la reacción y la mezcla resultante se agitó durante una
noche a 20 °C. La reacción se inactivó con bicarbonato sódico acuoso (10 ml) y la fase acuosa se extrajo con acetato de etilo ( 3x10 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron al vacío y el residuo se purificó por cromatografía en gel de sílice con acetato de etilo como eluyente para obtener el compuesto del título como un sólido de color amarillo 7,1 m , 9 % . miz ES+ 455 M+H .

Compuesto 461.1. 4-Metoxihepta-1,6-dieno. En un matraz de fondo redondo, se colocó una solución de hepta-1,6-dien-4-ol (2,00 g, 17,8 mmol, 1,00 equiv) y yodometano (5,00 g, 35,2 mmol, 2,00 equiv) en tetrahidrofurano (30 ml). La solución se enfrió a 0°C y se añadió hidruro sódico (1,00 g, 25,0 mmol, 1,50 equiv, 60% en aceite mineral) a la reacción por partes. La mezcla resultante se agitó durante una noche a temperatura ambiente, a continuación se inactivó cuidadosamente con agua (5 ml) y se diluyó con éter (30 ml). Los extractos orgánicos resultantes se lavaron con solución saturada de cloruro sódico (2 x 20 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener el compuesto del título como un aceite incoloro 200 84 %.

Compuesto 461.2. 4-Metoxiciclopent-1-eno. En un matraz de fondo redondo, que se purgó y se mantuvo con una atmósfera inerte de nitrógeno, se colocó una solución de 4-metoxihepta-1,6-dieno (compuesto 461.1, 200 mg, 1,43 mmol, 1,00 equiv, 90 %) en diclorometano (25 ml). Se añadió catalizador de Grubbs II (55 mg, 0,06 mmol, 0,04 equiv) y la solución resultante se agitó durante una noche a temperatura ambiente. La mezcla se concentró al vacío para obtener el compuesto del título como un aceite incoloro (100 mg, 57%).

Compuesto 461.3. 2-Bromo-4-metoxiciclopentanol. En un matraz de fondo redondo, se añadió una solución de 4-metoxiciclopent-1-eno (compuesto 461.2, 1,00 g, 8,66 mmol, 1,00 equiv, 85 %) en diclorometano (100 ml) y una solución de W-bromosuccinimida (2,00 g, 11,3 mmol, 1,00 equiv) en agua (20 ml). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas y a continuación la mezcla resultante se lavó con agua (20 ml), se secó (Na2SO4), se filtró y se concentró al vacío para obtener el compuesto del título como un aceite incoloro (1,00 g , 36 %).

Compuesto 461.4. 2-Bromo-4-metoxiciclopentanona. En un matraz de fondo redondo, se colocó una solución de 2-bromo-4-metoxiciclopentanol (compuesto 461.3, 1,00 g, 3,08 mmol, 1,00 equiv, 60 %) en diclorometano (100 ml). Se añadió peryodinano de Dess-Martin (2,00 g, 4,72 mmol, 1,10 equiv) por partes y la mezcla se agitó durante una noche a temperatura ambiente. La mezcla se diluyó a continuación con agua (20 ml) y se inactivó con Na2S2O4 (4 g). La capa acuosa se extrajo con diclorometano (100 ml) y las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (30 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener el compuesto del título como un aceite de color marrón claro (800 mg, 81 %).

Compuesto 461.5. 5-(3a-Hidroxi-5-metoxi-1,3a,4,5,6,6a-hexahidrociclopenta[G(]imidazol-2-M)-2,4-dimetilbenzoato de metilo. En un matraz de fondo redondo, se colocó una solución de 2-bromo-4-metoxiciclopentanona (compuesto 461.4, 600 mg, 1,86 mmol, 1,00 equiv, 60 %) en ACN (15 ml). Se añadieron clorhidrato de 5-carbamimidoil-2,4-dimetilbenzoato de metilo (compuesto 2.5, 320 mg) y carbonato potásico (430 mg, 3,11 mmol) y la mezcla se agitó durante una noche a 80 °C. La mezcla se concentró al vacío y el residuo se diluyó con acetato de etilo (50 ml) y a continuación se lavó con solución saturada de cloruro sódico (2 x 20 ml), se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con diclorometano/metanol (10/1) como eluyente para obtener el compuesto del título como un aceite de color marrón (200 mg, 27 %).
Compuesto 461.6.
un matraz de fondo redondo, se colocó una solución de 5-(3a-hidroxi-5-metoxi-1,3a,4,5,6,6ahexahidrocidopenta[d]imidazol-2-il)-2,4-dimetilbenzoato de metilo (compuesto 461.5, 200 mg, 0,38 mmol, 1,00 equiv, 60 %) en W,W-dimetilformamida (ml). Se añadió ácido p-toluenosulfónico (20 mg, 0,12 mmol, 0,18 equiv) y la mezcla resultante se agitó durante una noche a 80 °C, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con diclorometano/metanol (10/1) como eluyente para obtener el compuesto del título como un aceite de color marrón (100 mg, 66%).

Compuesto 461.7. Ácido 5-(5-metoxi-1,4,5,6-tetrahidrociclopenta[c(]imidazol-2-il)-2,4-dimetilbenzoico. En un matraz de fondo redondo, se colocó una solución de 5-(5-metoxi-1,4,5,6-tetrahidrociclopenta[d]imidazol-2-il)-2,4-dimetilbenzoato de metilo (compuesto 461.6, 100 mg, 0,270 mmol, 1,00 equiv, 80%) en metanol (3 ml). Se añadió una solución de hidróxido sódico (67,0 mg, 1,68 mmol, 5,00 equiv) en agua (3 ml) y la mezcla resultante se agitó durante una noche a 70 °C, a continuación se concentró al vacío. El residuo se diluyó con agua (3 ml) y el pH de la solución se ajustó a 2-3 con cloruro de hidrógeno acuoso (12 M). La mezcla se extrajo acetato de etilo ( 3x 10 ml), y las capas orgánicas combinadas se concentraron al vacío para obtener el compuesto del título como un sólido de

¡l)benzon¡tr¡lo. En un matraz de fondo redondo, se colocó una solución de ácido 5-(5-metoxi-1,4,5,6-tetrahidrociclopenta[d]imidazol-2-il)-2,4-dimetilbenzoico (compuesto 461.7, 60 mg, 0,17 mmol, 1,00 equiv, 80 %) en W,W-dimetilformamida (4 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 46 mg, 0,20 mmol, 1,00 equiv), 4-dimetilaminopiridina (52 mg, 0,43 mmol, 2,00 equiv), y EDCHCl (80 mg, 0,42 mmol, 2,00 equiv) y la mezcla se agitó durante una noche a temperatura ambiente. La solución resultante se diluyó con acetato de etilo (30 ml) y se lavó con solución saturada de cloruro sódico ( 3 x 10 ml), se secó (Na2SO4), se filtró, y se concentró al vacío. El residuo (40 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHIMAZDU)): Columna, SunFire Prep C18, 19 * 150 mm 5 um; fase móvil, agua con el 0,05 % de TFA y CH3CN (15,0 % CH3CN asciende al 55,0 % en 7 minutos, asciende al 100,0 % en 1 minuto, desciende al 15,0 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el producto puro se combinaron y se liofilizaron para obtener el compuesto del título como un sólido de color blanco 8,9 m , 11 %). miz (ES+) 455 (M+H)+.

Compuesto 462.1.5-Bromo-2,2-dimetildihidro-2H-piran-4(3H)-ona. En un matraz de fondo redondo, se colocó una solución de 2,2-dimetiloxan-4-ona (1,00 g, 7,80 mmol, 1,00 equiv) en éter (20 ml). Se añadió W-bromosuccinimida (1,50 g, 25,5 mmol, 3,26 equiv) por partes, seguida de la adición de acetato de amonio (60,0 mg, 0,78 mmol, 0,10 equiv). La mezcla resultante se agitó durante una noche a 25 °C, a continuación se diluyó con acetato de etilo (20 ml). La mezcla resultante se lavó con solución saturada de cloruro sódico (2 x 40 ml), se secó (Na2SO4), se filtró, y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:30) como eluyente para obtener el compuesto del título como un aceite de color amarillo (508 mg, 31 %).
Compuesto 462.
il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 461, excepto que se usó 5-bromo-2,2-dimetildihidro-2H-piran-4(3H)-ona (compuesto 462.1) en lugar de 2bromo-4-metoxicilopentanona (compuesto 461.4). m/z (ES+) 469 (M+H)+.

Compuesto 463.1. Trimetil((4-(trimetilsilil)but-3-in-1-il)oxi)silano. En un matraz de fondo redondo de tres bocas de 1 l, se colocó una solución de but-3-in-1-ol (20,0 g, 285 mmol, 1,00 equiv) en tetrahidrofurano (300 ml) y la mezcla se purgó con nitrógeno. La mezcla se enfrió a -78 °C, a continuación se añadió n-butillitio (2,5 M en THF) (270 ml, 2,40 equiv) gota a gota, seguido de la adición de clorotrimetilsilano (67,9 g, 625 mmol, 2,20 equiv). La mezcla resultante se agitó a continuación durante 1 h a 25 °C, a continuación se inactivó cuidadosamente con bicarbonato sódico acuoso (250 ml). La fase acuosa se extrajo con éter (3 x 100 ml) y las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó por cromatografía en gel de sílice con éter/éter de petróleo (1:10-1:1) como eluyente para obtener el compuesto del título como un aceite incoloro (10,0 g, 16 %).

Compuesto 463.2. Tr¡met¡l((4-(tr¡met¡ls¡l¡l)but-3-en-1-¡l)ox¡)s¡lano. En un matraz de fondo redondo, se colocó una solución del compuesto 463.1, 2,00 g, 7,46 mmol, 1,00 equiv, 80%) en hexano (30 ml). El sistema se purgó con nitrógeno. Se añadieron quinolina (0,1 ml, 0,10 equiv) y reactivo de Lindlar (contaminado con Pb) (0,2 g, 0,10 equiv, 5 %) a la mezcla. La mezcla resultante se hidrogenó durante una noche a temperatura ambiente a presión atmosférica de hidrógeno. Después de la finalización de la reacción, el sistema se purgó con nitrógeno y los sólidos se retiraron por filtración. El filtrado se concentró al vacío y el residuo se purificó por cromatografía en gel de sílice con hexano/éter (20:1) como eluyente para obtener el compuesto del título como un aceite incoloro (1,50 g, 74 %).

Compuesto 463.3. 6-Metil-3,6-dihidro-2H-pirano. En un matraz de fondo redondo, se colocó una mezcla de trimetil((4-(trimetilsilil)but-3-en-1-il)oxi)silano (compuesto 463.2, 1,50 g, 6,93 mmol, 1,00 equiv), acetaldehído (900 mg , 20,4 mmol, 3,00 equiv), y cloruro de indio (III) (1,50 g, 1,00 equiv) en diclorometano (15 ml). La mezcla resultante se agitó durante una noche a 25 °C, a continuación se diluyó con DCM (50 ml). Los extractos orgánicos resultantes se lavaron con solución saturada de cloruro sódico (3 x 20 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener el compuesto del título como un aceite de color marrón 0,600 , 88 %).
Compuesto 463.4 y compues -ona (compuesto 463.4) y 3-bromo-2-met¡ld¡h¡dro-2H-p¡ran-4(3H)-ona (compuesto 463.5). En un matraz de fondo redondo, se colocó una solución de ácido o-yodobezoico (IBX) (3,42 g, 6,11 mmol, 2,00 equiv) en DMSO (12 ml). La mezcla se agitó durante 30 minutos a 25 °C, a continuación se añadió gota a gota una solución de 6-metil-3,6-dihidro-2H-pirano (compuesto 463.3, 600 mg, 4,28 mmol, 1,00 equiv, 70 %) en diclorometano (30 ml). La mezcla se enfrió a 0-5 °C, y a continuación se añadió W-bromosuccinimida (1,20 g, 6,74 mmol, 1,10 equiv) por partes. La solución resultante se agitó durante una noche a 25 °C., a continuación los sólidos se retiraron con filtración. El filtrado se diluyó con diclorometano (50 ml) y se lavó con solución saturada de cloruro sódico (3 x 20 ml). La mezcla se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:20) como eluyente para obtener una mezcla de los compuestos del título como un aceite de color marrón (600 mg, 73 %).
-ona (compuesto 463.4) y 3-bromo-2-met¡ld¡h¡dro-2H-p¡ran-4(3H)-ona (compuesto 463.5). En un matraz de fondo redondo, se colocó una solución de ácido o-yodobezoico (IBX) (3,42 g, 6,11 mmol, 2,00 equiv) en DMSO (12 ml). La mezcla se agitó durante 30 minutos a 25 °C, a continuación se añadió gota a gota una solución de 6-metil-3,6-dihidro-2H-pirano (compuesto 463.3, 600 mg, 4,28 mmol, 1,00 equiv, 70 %) en diclorometano (30 ml). La mezcla se enfrió a 0-5 °C, y a continuación se añadió W-bromosuccinimida (1,20 g, 6,74 mmol, 1,10 equiv) por partes. La solución resultante se agitó durante una noche a 25 °C., a continuación los sólidos se retiraron con filtración. El filtrado se diluyó con diclorometano (50 ml) y se lavó con solución saturada de cloruro sódico (3 x 20 ml). La mezcla se secó (Na2SO4), se filtró y se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:20) como eluyente para obtener una mezcla de los compuestos del título como un aceite de color marrón (600 mg, 73 %).
Compuestos 463. 
il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 461, excepto que se usó una mezcla de 4-bromo-2-metildihidro-2H-piran-3(4H)-ona (compuesto 463.4) y 3-bromo-2-metildihidro-2H-piran-4(3H)-ona (compuesto 463.5) en lugar de 2-bromo-4-metoxic¡clopentanona (compuesto 461.4). miz (ES+) 455 (M+H)+.

Compuesto 464.1. 4-(Al¡lox¡)pent-1-eno. Un matraz de fondo redondo de cuatro bocas de 500 ml se purgó y se mantuvo con una atmósfera inerte de nitrógeno, a continuación, se añadió una suspensión de hidruro sódico (14,0 g, 350 mmol, 2,01 equiv, 60%) en W,W-dimetilformamida (100 ml). La mezcla se enfrió a 0 °C, a continuación se añadió una solución de pent-4-en-2-ol (15,0 g, 174 mmol, 1,00 equiv) en W,W-dimetilformamida (100 ml) gota a gota y la mezcla se agitó durante 20 minutos a 0 °C. La mezcla se enfrió a -20 °C y se añadió una solución de 3-bromoprop-1-eno (20,9 g, 172 mmol, 0,99 equiv) en W,W-dimetilformamida (100 ml). La mezcla resultante se dejó calentar a temperatura ambiente y se agitó durante una noche, a continuación se inactivó cuidadosamente con H2O (500 ml). La capa acuosa se extrajo con acetato de etilo (4 x 100 ml) y las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (2 x 200 ml), se secaron (Na2¿O4), se filtraron y se concentraron al vacío para obtener el compuesto del título como un aceite de color amarillo (13,3 g, 61 %).

Compuesto 464.2. 2-Met¡l-3,6-d¡h¡dro-2H-p¡rano. Un matraz de fondo redondo de cuatro bocas de 500 ml se purgó y se mantuvo con una atmósfera inerte de nitrógeno, a continuación, se añadió una solución de 4-(aliloxi)pent-1-eno (compuesto 464.1, 3,00 g, 23,8 mmol, 1,00 equiv) en DCE (200 ml). Se añadió catalizador de Grubbs (810 mg, 0,950 mmol, 0,04 equiv) y la mezcla se agitó a 60 °C durante 4 h. A continuación, la mezcla se concentró al vacío a 20 °C para obtener el compuesto del título como un aceite incoloro 200 86 %.

Compuesto 464.3 y 464.4. 4-Bromo-6-met¡ltetrah¡dro-2H-p¡ran-3-ol y 5-bromo-2-met¡ltetrah¡dro-2H-p¡ran-4-ol. En un matraz de fondo redondo, se colocó una solución de metil-3,6-dihidro-2H-pirano (compuesto 464.2, 2,00 g, 20,4 mmol, 1,00 equiv) en una mezcla de tetrahidrofurano y H2O (20/20 ml). Se añadió A/-bromosuccinimida (3,60 g, 20,3 mmol, 1,00 equiv) y la mezcla resultante se agitó a temperatura ambiente durante una noche. La mezcla se extrajo con diclorometano (2 x 20 ml) y las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (2 x 20 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener una mezcla de los compuestos del título como un aceite de color amarillo 1,10 , impuro .

Compuesto 464.5 y 464.6. 4-Bromo-6-met¡ld¡h¡dro-2H-p¡ran-3(4H)-ona y 5-bromo-2-met¡ld¡h¡dro-2H-p¡ran-4(3H)-ona. En un matraz de fondo redondo, se colocó una mezcla de 4-bromo-6-metiltetrahidro-2H-piran-3-ol (compuesto 464.3) y 5-bromo-2-metiltetrahidro-2H-piran-4-ol (compuesto 464.4) (1,10 g, 5,64 mmol, 1,00 equiv) como una solución en diclorometano (30 ml). Se añadió peryodinano de Dess-Martin (2,90 g, 6,84 mmol, 1,21 equiv) y la solución resultante se agitó durante una noche a temperatura ambiente, a continuación se inactivó con agua (20 ml). La capa acuosa se extrajo con diclorometano (2 x 20 ml) y las capas orgánicas combinadas se lavaron con solución saturada de cloruro sódico (2 x 50 ml), se secaron (Na2SO4), se filtraron y se concentraron al vacío para obtener una mezcla de los compuestos del título como un aceite de color amarillo (0,7 g).

Compuesto 464.7. 2,4-D¡met¡l-5-(6-met¡l-3,4,6,7-tetrah¡drop¡rano[3,4-c(]¡m¡dazol-2-¡l)benzoato de metilo. En un matraz de fondo redondo, se colocó una mezcla de 4-bromo-6-metild¡h¡dro-2H-p¡ran-3(4H)-ona (compuesto 464.5) y 5-bromo-2-metild¡h¡dro-2H-piran-4(3H)-ona (compuesto 464.6) (700 mg, 3,63 mmol) como una solución en acetonitrilo (20 ml). Se añadieron clorhidrato de 5-carbam¡m¡do¡l-2,4-d¡met¡lbenzoato de metilo (compuesto 2.5, 750 mg) y carbonato potásico (1,00 g, 7,25 mmol) y la mezcla se agitó durante una noche a 75 °C en nitrógeno. La mezcla se enfrió y los sólidos se eliminaron por filtración. El filtrado se concentró al vacío y el residuo se purificó por cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:1) como eluyente para obtener el compuesto del título como un

Compuesto 464.8. Ácido 2,4-dimetil-5-(6-metil-3,4,6,7-tetrahidropirano[3,4-c/]imidazol-2-il)benzoico. En un matraz de fondo redondo, se colocó una solución de 2,4-d¡met¡l-5-(6-met¡l-3,4,6,7-tetrah¡drop¡rano[3,4- d ]¡midazol-2-¡l)benzoato de metilo (compuesto 464.7, 100 mg, 0,330 mmol, 1,00 equiv) en metanol (10 ml). Se añadió una solución de hidróxido de litio (76 mg, 3,17 mmol, 10,0 equiv) en agua (10 ml) y la solución resultante se agitó durante 4 h a temperatura ambiente. La mezcla se concentró al vacío y se añadió HCl acuoso a continuación hasta que el pH fue de 5-6. La mezcla se ajustó a pH 5-6 con HCl acuoso y a continuación la mezcla resultante se concentró al vacío. Se añadió MeOH (5 ml) al residuo y los sólidos se retiraron por filtración. El filtrado se concentró al vacío para obtener el compuesto del título como un sólido de color amarillo (60 mg, 63 %).
Compues
¡l)benzon¡tr¡lo. En un matraz de fondo redondo, se colocó una solución de ácido 2,4-dimetil-5-[6-met¡l-3H,4H,6H,7H-p¡rano[3,4-d]¡m¡dazol-2-¡l]benzo¡co (50 mg, 0,17 mmol, 1,00 equiv) en W,W-dimet¡lformamida (10 ml). Se añadieron ED CHcl (67 mg, 0,35 mmol, 2,00 equiv), 4-dimetilaminopiridina (64 mg, 0,52 mmol, 3,00 equiv), y clorhidrato de 4-(piper¡d¡n-4-il)benzon¡tr¡lo (compuesto 1.5, 39 mg, 0,18 mmol, 1,00 equiv) y la solución se agitó durante 2 h a temperatura ambiente. La reacción se inactivó con agua (20 ml) y la fase acuosa se extrajo con acetato de etilo (2 x 20 ml). Las fases orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en gel de sílice con acetato de etilo/éter de petróleo (1:1) como eluyente. El producto impuro (20 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(SHlMADZU)): Columna, XBridge Prep Phenyl, 5 um, 19 * 150 mm; fase móvil, agua con el 0,03 % de NH3H2O y CH3CN (30 % CH3CN asciende al 60 % en 9 minutos, asciende al 100 % en 1 minuto, desciende al 30 % en 1 minuto); detector, Waters 2489 254 y 220 nm. Las fracciones que contenían el producto puro se combinaron y se liofilizaron para obtener el compuesto del título como un sólido de color blanco 8,0 m , 10% . miz (ES+) 455 (M+H)+.
Compuesto 465.
tetrah¡droc¡clopenta[c(]¡m¡dazol-5-carbomtr¡lo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 461, excepto que se usó ciclopent-3-enocarbonitrilo (Johnson, C. R. et al. J. Org. Chem, 1969, 34, 860-864) en lugar de 4-metoxiciclopent-1-eno (compuesto 461.2). m/z (ES+) 450 (M+H)+.

Compuesto 466.1. Dimetanosulfonato de Tetrahidrofuran-3,4-diilo. En un matraz de fondo redondo, se colocó una solución de tetrahidrofuran-3,4-diol (500 mg, 4,80 mmol, 1,00 equiv) y trietilamina (1,45 g, 14,3 mmol, 3,00 equiv) en diclorometano (8 ml). La mezcla se enfrió a 0-5 °C y se añadió una solución de cloruro de metanosulfonilo (1,40 g, 12,2 mmol, 2,50 equiv) en diclorometano (2 ml) gota a gota. La solución se agitó a temperatura ambiente durante 2 h, y a continuación se diluyó con diclorometano (100 ml). La solución se lavó con NH4Cl acuoso (2x 30 ml), a continuación solución saturada de cloruro sódico (30 ml), se secó (Na2SO4), se filtró y se concentró al vacío para obtener el compuesto del título como un sólido de color marrón 1,00 , 72 %.

Compuesto 466.2. 3,4-Diazidotetrahidrofurano. En un matraz de fondo redondo, se colocó una mezcla de dimetanosulfonato de tetrahidrofuran-3,4-diilo (compuesto 466.1, 2,00 g, 7,30 mmol, 1,00 equiv, 95%) y azida sódica (4,00 g, 61,5 mmol, 8,00 equiv) en W,A/-dimetilformamida (20 ml). La mezcla resultante se agitó durante una noche a 100 °C detrás de una pantalla protectora contra explosiones, a continuación se enfrió y se diluyó con éter (100 ml). Las mezcla se lavó con solución saturada de cloruro sódico (5 x 20 ml), se secó (Na2SO4), se filtró y se concentró al vacío para obtener el compuesto del título como un aceite incoloro 1,00 , 80 %).

Compuesto 466.3. Tetrahidrofuran-3,4-diamina. En un matraz de fondo redondo, se colocó una solución de 3,4-diazidotetrahidrofurano (compuesto 466.2, 900 mg, 5,26 mmol, 1,00 equiv, 90 %) en etanol (10 ml). El sistema se purgó con nitrógeno y se añadió paladio sobre carbono (10 % en peso de Pd) (900 mg). Después de purgar adicionalmente el sistema con nitrógeno, la atmósfera se cambió a hidrógeno y la suspensión resultante se agitó durante una noche a temperatura ambiente en una atmósfera de hidrógeno. Después de purgar el sistema con nitrógeno, los sólidos se retiraron por filtración y el filtrado se concentró al vacío para obtener el compuesto del título como un aceite incoloro (600 mg
Compuesto 466.4. Clorhidrato de 5-(imino(metoxi)metil)-2,4-dimetilbenzoato de metilo. En un matraz de fondo redondo de tres bocas de 50 ml, se colocó una solución de 5-ciano-2,4-dimetilbenzoato de metilo (compuesto 2.3, 900 mg, 4,28 mmol, 1,00 equiv, 90 %) en metanol (20 ml). Se introdujo cloruro de hidrógeno gaseoso por borboteo a través de la solución durante 0,5 h. La mezcla de reacción se transfirió a un tubo sellado de 30 ml y se agitó durante una noche a temperatura ambiente. La mezcla se concentró al vacío y el residuo se diluyó con acetato de etilo (30 ml) y se extrajo con agua (10 ml). La fase acuosa se concentró al vacío para obtener el compuesto del título como un sólido

Compuesto 466.5. 2,4-DimetN-5-(3a,4,6,6a-tetrahidro-1H-furo[3,4-G(]imidazol-2-N)benzoato de metilo. En un matraz de fondo redondo, se colocó una solución de tetrahidrofuran-3,4-diamina (compuesto 466.3, 140 mg, 1,17 mmol, 1,00 equiv, 85 %) en etanol (6 ml). Se añadieron clorhidrato de 5-(imino(metoxi)metil)-2,4-dimetilbenzoato de metilo (compuesto 466.4, 300 mg, 0,930 mmol, 1,00 equiv) y trietilamina (140 mg, 1,38 mmol, 1,00 equiv) y la solución resultante se agitó durante una noche a 80 °C, a continuación se concentró al vacío. El residuo se purificó por cromatografía en gel de sílice con diclorometano/metanol (10/1) como eluyente para obtener el compuesto del título como un aceite de color marrón (120 mg, 34 %).

Compuesto 466.5. Acido 2,4-dimetil-5-(3a,4,6,6a-tetrahidro-1H-furo[3,4-d]imidazol-2-il)benzoico. En un matraz de fondo redondo, se colocó una solución de 2,4-dimetil-5-(3a,4,6,6a-tetrahidro-1H-furo[3,4- d]imidazol-2-il)benzoato de metilo (compuesto 466.5, 100 mg, 0,330 mmol, 1,00 equiv, 90 %) e hidróxido sódico (73 mg, 1,82 mmol, 5,00 equiv)
en metanol/H2O (3/3 ml). La solución resultante se agitó durante una noche a 70 ° C , a continuación se concentró al vacío. El residuo se diluyó con H2O (5 ml) y el pH de la solución se ajustó a 2-3 con cloruro de hidrógeno (12 N), a continuación se extrajo con acetato de etilo ( 3 x 10 ml). Las capas orgánicas combinadas se concentraron al vacío para obtener el compuesto del título como un sólido de color amarillo (80,0 mg, 84 %).

Compuesto 466. 4-(1-(2,4-D¡met¡l-5-(-3a,4,6,6a-tetrah¡dro-1H-furo[3,4-d]¡m¡dazol-2-¡l)benzo¡l)p¡per¡dm-4-il)benzonitrilo. En un matraz de fondo alrededor, se colocó una solución de ácido 2,4-dimetil-5-(3a,4,6,6a-tetrahidro-1Hfuro[3,4-d]imidazol-2-il)benzoico (compuesto 466.5, 80,0 mg, 0,250 mmol, 1,00 equiv, 80%) en N,N-dimetilformamida (4 ml). Se añadieron clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5, 82,0 mg, 0,350 mmol, 1,20 equiv), 4-dimetilaminopiridina (76,0 mg, 0,620 mmol, 2,00 equiv), y EDCHCl (116 mg, 0,610 mmol, 2,00 equiv) y la solución resultante se agitó durante una noche a temperatura ambiente. La mezcla se diluyó con acetato de etilo (30 ml) y se lavó con solución saturada de cloruro sódico (3 x 15 ml), se secó (Na2SO4), se filtró, y se concentró al vacío. El producto impuro (50 mg) se purificó mediante HPLC preparativa con las siguientes condiciones (1#-Pre-HPLC-001(Sh IMADZU)): Columna, Xbridge Prep C18, 5 um 19 * 150 mm; fase móvil, agua con el 0,05 % de TFA y CH3CN (15 % CH3CN asciende al 50 % en 9 minutos, asciende al 100 % en 1 minuto, desciende al 15 % en 1 minuto); Detector, Waters 2489254 y 220 nm. Las fracciones que contenían el producto puro se combinaron y se liofilizaron para obtener el compuesto del título como un sólido de color blanco (13 mg, 12 %). m/z (ES+) 429 (M+H)+. 1H-RMN (300MHz, CD3OD): ó 7,70 (d, 2H), 7,53-7,36 (m, 4H), 5,09 (s, 2H), 4,17 (d, J =10,8 Hz, 2H), 3,77 (d, J =10,5 Hz, 2H), 3,60-3,44 (m, 1H), ~3,3 (m, parcialmente solapado con agua, 1H), 3,10-2,92 (m, 2H), 2,48 (s, 3H), 2,45 y 2,35 (2

Compuesto 467. 4-(1-(4-Fluoro-2-met¡l-5-(3,4,6,7-tetrah¡drop¡rano[3,4-c(]¡m¡dazol-2-¡l)benzo¡l)p¡per¡dm-4-¡l)benzon¡tr¡lo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 1 y el compuesto 2 excepto que se usó ácido 4-fluoro-2-metilbenzoico en lugar de ácido 2,4-dimetilbenzoico. m/z ES+ 445 M+H .
Compuesto 468.  -cloro-2-met¡lbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 excepto que se usó ácido 4-
-cloro-2-met¡lbenzo¡l)p¡per¡d¡n-4-¡l)benzon¡tr¡lo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 excepto que se usó ácido 4-

Compuesto 469. 1-(2-(2-Cloro-5-(4-(4-fluorofeml)p¡per¡dm-1-carboml)-4-met¡lfeml)-6,7-d¡h¡dro-3H-¡m¡dazo[4,5-c]p¡r¡dm-5(4H)-¡l)etanona. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 y el compuesto 468. m/z (ES+) 495 (M+H)+.

Compuesto 470.1. 2-Bromo-4-fluorobenzoato de metilo. En un matraz de fondo redondo, se colocó una solución de ácido 2-bromo-4-fluorobenzoico (21,8 g, 99,5 mmol, 1,00 equiv) en una mezcla disolvente de ácido sulfúrico (20 ml) y metanol (20 ml). La solución resultante se agitó durante 5 h a 85 °C, a continuación se enfrió y se concentró al vacío. El residuo se diluyó con acetato de etilo (200 ml) y se lavó con solución saturada de cloruro sódico (200 ml), a continuación NaHCO3 acuoso (100 ml Nota: desprendimiento de gas), se secó (Na2SO4), se filtró y se concentró al vacío para obtener el compuesto del título como un aceite de color amarillo claro 22,0 g, 95 %).

Compuesto 470.1. 2-Etil-4-fluorobenzoato de metilo. El compuesto del título (aceite incoloro, 14,5 g, 93%) se preparó usando un procedimiento similar al usado para la preparación del compuesto 48.1 y usando 2-bromo-4-

Compuesto 470.4-(1-(2-Et¡l-4-fluoro-5-(3,4,6,7-tetrah¡drop¡rano[3,4-c(]¡m¡dazol-2-¡l)benzo¡l)-4-fluorop¡per¡dm-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 1 y el compuesto 2, excepto que se usó 2-etil-4-fluorobenzoato de metilo (compuesto 470.1) en lugar de ácido 2,4-dimetilbenzoico, y se usó clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2) en lugar de clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5). m/z (ES+) 477 (M+H)+.

Compuesto 471.1. Ácido 4-cloro-5-yodo-2-metilbenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2,1 excepto que se usó ácido 4-cloro-2-metilbenzoico en lugar de ácido 2,4-dimetilbenzoico.

Compuesto 471.2. Ácido 4-cloro-5-formil-2-metilbenzo¡co. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 392.2 excepto que se usó ácido 4-cloro-5-yodo-2-metilbenzoico (compuesto 471.1) en lugar de ácido 4-ciclobutil-5-yodo-2-metilbenzoico (compuesto 392.1.
Compuesto 471. 4-(1-(4-Cloro-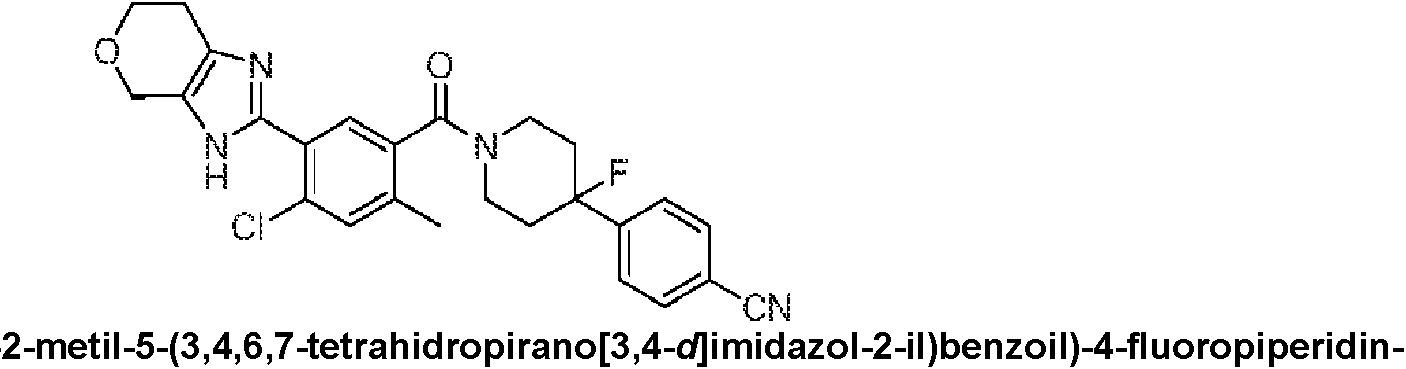
4-¡l)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 400, excepto que se usó ácido 4-cloro-5-formil-2-metilbenzoico (compuesto 471.2) en lugar de ácido 2-etil-5-formil-4-metilbenzoico (compuesto 211.4), se usó 3-bromodihidro-2H-piran-4(3H)-ona en lugar de 3-bromo-5-metildihidro-2H- piran-4(3H)-ona (compuesto 400.2) y se usó clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2) en lugar de clorhidrato de 4-(piperidin-4-¡IJbenzonitrilo (compuesto 1.5). m/z (ES+) 479 (M+H .

Compuesto 472.1. 4-Cloro-2-etil-5-yodobenzoato de metilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 211.2
excepto que se usó 4-cloro-2-etilbenzoato de metilo (compuesto 178.2) en lugar de 2-etil-4-metilbenzoato de metilo (compuesto 48.1).

Compuesto 472.2. Ácido 4-cloro-2-etil-5-yodobenzoico. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 211.3 excepto que se usó 4-cloro-2-etil-5-yodobenzoato de metilo (compuesto 472.1) en lugar de 2-etil-5-yodo-4-metilbenzoato de metilo (compuesto 211.2).
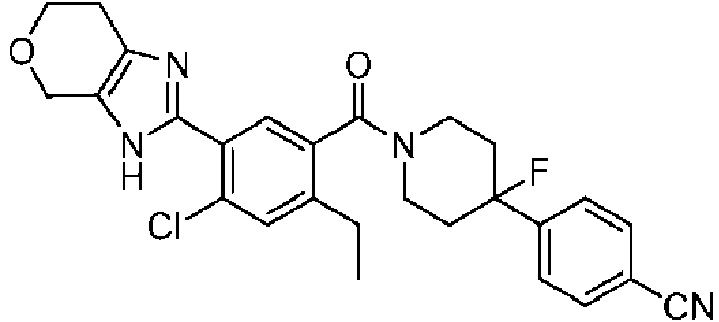
Compuesto 472. 4-(1-(4-Cloro-2-etil-5-(3,4,6,7-tetrahidropirano[3,4-c(]imidazol-2-il)benzoil)-4-fluoropiperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 400, excepto que se usó ácido 4-cloro-2-etil-5-yodobenzoico (compuesto 472.2) en lugar de ácido 2-etil-5-formil-4-metilbenzoico (211.4), se usó 3-bromodihidro-2H-piran-4(3H)-ona en lugar de 3-bromo-5-metildihidro-2H- piran-4(3H)-ona (compuesto 400.2) y se usó clorhidrato de 4-(4-fluoropiperidin-4-il)benzonitrilo (compuesto 11.2) en lugar de clorhidrato de 4-(piperidin-4-il)benzonitrilo (compuesto 1.5). m/z (ES+) 493 (M+H)+.

Compuesto 473. 4-(1-(5-(5-Acetil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridin-2-il)-4-cloro-2-etilbenzoil)piperidin-4-il)benzonitrilo. El compuesto del título se preparó usando manipulaciones químicas estándar y procedimientos similares a los usados para la preparación del compuesto 2 excepto que se usó 4-cloro-2-etil-5-yodobenzoato de metilo (compuesto 472.1) en lugar de 5-yodo-2,4-dimetilbenzoato de metilo (compuesto 2.2). mlz (ES+) 516 (M+H)+. 1H-RMN (300MHz, CD3OD): ó 7,73-7,54 (m, 4H), 7,53-7,42 (m, 2H), 4,75 y 4,70 (2 singletes, rotámeros de acetilamida, CH2, 2H), 4,02-3,88 (m, 2H), 3,68-3,52 (m, 1H), 3,32-3,20 (m, 1H), 3,07-2,88 (m, 3H), 2,88-2,58 (m, 3H), 2,25 y 2,22 (2 singletes, rotámeros de acetilamida, acetil CH3, 3H), 2,11-1,98 (m, 1H), 1,93-1,53 (m, 3H), 1,39-1,22 (m, 3H).
Pueden encontrarse compuestos ejemplares adicionales en la tabla 1. Los ejemplos que no caen dentro del alcance de las reivindicaciones son solo para fines ilustrativos.
Ejemplo 1- Inhibición de FASN mediante compuestos de la presente descripción
Determinación de la actividad bioquímica de FASN: La enzima FASN se aisló a partir de células SKBr3. SKBr3 es una línea celular de cáncer de mama humana con altos niveles de expresión de FASN. Se estima que FASN comprende aproximadamente el 25% de las proteínas citosólicas en esta línea celular. Las células SKBR3 se homogeneizaron en un homogeneizador Dounce y a continuación se centrifugan durante 15 minutos a 4 °C para retirar la materia particulada. A continuación se analizó el contenido de proteína del sobrenadante, se diluyó hasta la concentración apropiada, y se usó para medir la actividad de FASN. La presencia de FASN se confirmó por análisis de Western blot. Un procedimiento similar para el aislamiento de FASN a partir de células SKBr3 se describe en Teresa, P. et al. (Clin. Cancer Res. 2009; 15(24), 7608-7615).
La actividad de FASN del extracto de células SKBr3 se determinó midiendo la oxidación de NADPH o la cantidad de coenzima A (CoA) que contiene tiol liberada durante la reacción de ácido graso sintasa. El colorante CPM (7-dietilamino-3-(4'-maleimidil-fenil)-4-metilcoumarina) contiene un grupo reactivo tiol que incrementa su emisión de fluorescencia al reaccionar con grupo sulfhidrilo de CoA. Las actividades bioquímicas mostradas en la tabla 1 se determinaron usando la medición de fluorescencia de liberación de CoA mediante un procedimiento descrito en Chung C.C. et al.(Assay and Drug Development Technologies, 2008, 6(3), 361-374).
Ejemplo 2 - Actividad antiviral
La actividad antiviral de la estructura (I-Z) se evaluó usando el sistema de replicón VHClb:

El replicón se construyó usando la línea celular ET (luc-ubi-neo/ET), una línea celular de hepatoma humano Huh7 que alberga un replicón de VHC con un reportero de luciferasa (Luc) estable y tres mutaciones adaptativas al cultivo celular (Pietschmann, y col (2002) J. Virol. 76:4008-4021). El ensayo de evaluación antiviral del replicón de VHC examinó los efectos de los compuestos a seis concentraciones semilogarítmicas. Se incluyó interferón alfa-2b humano en cada serie como compuesto de control positivo. Se sembraron cultivos sub-confluentes de la línea celular ET en placas de 96 pocillos que estaban dedicadas al análisis de los números de células (citotoxicidad) o la actividad antiviral y al día siguiente se añadieron fármacos a los pocillos apropiados. Las células se procesaron 72 h más tarde cuando las células aún eran sub-confluentes. Se determinaron los valores de CE50 (concentraciones que inhibían el replicón en un 50 % y un 90 %, respectivamente), CI50 (concentración que disminuye la viabilidad celular en un 50 %) e ES (índice selectivo: CI50/CE50). Los niveles de replicón de ARN de VHC se ensayaron como actividad Luc derivada de replicón de ARN de VHC o como ARN de VHC mediante TaqMan RT-PCR. Se usaron dos procedimientos para estimar los recuentos celulares (citotoxicidad). Cuando se empleó el sistema de ensayo Luc, se usó el ensayo de proliferación celular colorimétrico CytoTox-1 (Promega) para estimar el número de células, mientras que los niveles de ARN ribosómico (ARNr) determinados a través de TaqMan RT-PCR se usaron como una indicación del número de células en el ensayo basado en ARN. Un resumen de los resultados se enumera a continuación en la tabla 2.
Tabla 2

Ejemplo 3 - La inhibición de FASN se correlaciona con la inhibición de VHC
Las actividades antivirales de 15 compuestos de la presente descripción (los números se correlacionan con los compuestos de la Tabla 1) se midieron usando el sistema de replicón de VHC. Se estableció la línea celular de replicón 1b (replicón VHC 1b/Luc-Neo (1b Con1 con el gen Firefly integrado)) siguiendo procedimientos publicados (Lohmann et a/.(1999) Science 285(5424):110-113, Lohmann et al. (2001) J. Virol. 75(3): 1437-1449 yd Qi et al. (2009) Antiviral Res. 81(2): 166-173) usando Huh7 mediante selección con G418. El replicón se ensambló usando fragmentos de genes sintéticos. La línea GT1b tiene PV-EKT y alberga 3 mutaciones adaptativas E1202G(NS3), T1280I(NS3), K1846T(NS4B) y la cadena principal es Con1. El medio de cultivo fue:
a) DMEM suplementado con FbS al 10%, G418 (250 ^g/ml), estreptomicina (100 ^g/ml)/penicilina (100 U/ml), L-glutamina (100*), NEAA (100*)
b) Medios preparados de la siguiente manera:
i) 500 ml de medio DMEM (Gibco, Núm. de catálogo 11960-077)
ii) 57 ml de Suero Fetal Bovino (Gibco, Núm. de catálogo 16140-071)
iii) 5,7 ml de Penicilina-estreptomicina (Gibco, Núm. de catálogo 15140-122)
iv) 5,7 ml de MEM sin aminoácidos esenciales (Gibco, Núm. de catálogo 111140-050)
v) 5,7 ml de L-glutamina (Gibco, Núm. de catálogo 125030-081)
vi) 574,1 ml de medio 2,87 ml de 50 mg/ml de G418 [final 0,25 mg/ml] (Gibco, Núm. de catálogo 10131-027) Se disolvieron compuestos en DMSO para proporcionar una solución madre 10 mM o se usaron a partir de soluciones madre de DMSO. Se diluyeron compuestos para generar diluciones sucesivas semi-log de 10 puntos (3,16 veces) para ensayo en placas de 384 pocillos (PP de 384 pocillos cualificada para Echo (Labcyte Núm. de Cat. P-05525)) más DMSO por duplicado. Este experimento se repitió tres veces en tres días diferentes.
Las células se recogieron cuando la confluencia alcanzó el 90 %-100 %. Las concentraciones celulares se ajustaron a 8x104 células/ml y se añadieron a microplacas de ensayo blancas de 384 pocillos (tratadas con cultivo tisular -Greiner Núm. de Ca. 781080) hasta alcanzar una densidad celular final de 2.000 células/pocillo. Las placas se incubaron en CO2 al 5 % y a 37 °C durante 72 horas.
Después de 72 horas de incubación, se prepararon reactivo Bright-Glo Luciferase (Promega, Núm. de cat. e2650) y Cell Titer Flo (Promega, Núm. de cat G6080/1/2) y se almacenaron en la oscuridad, mientras se equilibraban a temperatura ambiente. Las células tratadas se equilibraron del mismo modo a temperatura ambiente. Se añadieron 10 ^l de Cell Titer Flo a cada pocillo de células tratadas con el compuesto y se incubaron en placas de microvaloración durante aprox. 0,5 horas. La viabilidad celular se midió usando un lector Envision (disponible de Perkin Elmer) para estimar la citotoxicidad. Se añadieron 30 ^l de sustrato de luciferasa de luciérnaga a cada pocillo y la
quimioluminiscencia se midió como un indicador de la medida de la replicación del VHC.
La actividad anti-replicón (% de inhibición) se calcula usando la ecuación:
í Comv — Controls
% de Inhibición = (1 — --------------- ; 1x100
V DMSO — Control)
La citotoxicidad se calcula usando la ecuación:
¡ Comp — Fondos
% de Citotoxicidad = (1 — ------V DMSO — F--o--n- — 1x100
do)
Se determinó que había una correlación entre la potencia de la inhibición de FASN y la actividad antiviral tal como se ilustra en tabla 3 a continuación y en la figura 1. Se observa que ninguno de los compuestos causó citotoxicidad significativa.
Tabla 3
Molécula CI50 bioquímica (pM) CE50 antiviral (pM)
1 0,230 0,425
2 0,065 0,192
12 0,370 1,003
14 0,263 0,260
20 0,022 0,011
27 0,107 0,153
43 0,110 0,154
55 0,035 0,034
58 0,025 0,078
67 0,090 0,270
68 0,100 0,301
70 0,037 0,099
73 0,040 0,117
152 0,052 0,072
343 0,600 0,624
Ejemplo - bidores de FASN conservan la actividad contra mutantes de VHC que otorgan resistencia a agentes antivirales de acción directa
- bidores de FASN conservan la actividad contra mutantes de VHC que otorgan resistencia a agentes antivirales de acción directa
Uno de los principales desafíos en el tratamiento de la hepatitis C es la rápida aparición de resistencia en respuesta a agentes antivirales de acción directa. La resistencia normalmente se produce cuando el virus genera una mutación puntual que soporta funciones virales esenciales, pero impide que los agentes antivirales se unan. Tres inhibidores de FASN (compuestos 55, 20 y 70) se ensayaron para determinar su capacidad para inhibir mutantes de VHC que otorgan resistencia a los agentes antivirales representativos. Cada uno de estos mutantes se introdujo en una construcción GT1b basada en una cadena principal Con1 que contiene el gen PVIRES-Luciferase Ubi-Neo y que alberga 1 mutación adaptativa (S2204I). (Lohmann et al. (1999) Science 285(5424):110-113, Lohmann et al. (2001) J. Virol. 75(3): 1437 1449 yd Qi et al. (2009) Antiviral Res. 81(2):166-173). Las actividades antivirales se midieron mediante el procedimiento descrito en el ejemplo 3.
Las mutaciones estudiadas se muestran en la tabla 4 a continuación.
Tabla 4 Mutaciones estudiadas

Un inhibidor alostérico de NS4B conocido (Compuesto A), un inhibidor de NS5A conocido (Compuesto B), un inhibidor de NS5B no nucleósido (Compuesto C), un inhibidor de proteasa NS3/NS4A conocido (Compuesto D) y un inhibidor de NS5B nucleósido conocido (Compuesto E) se ensayaron en paralelo con los inhibidores de FASN de la presente descripción para confirmar el rendimiento de las mutaciones de resistencia.
Las CE50 antivirales para los diversos compuestos contra el panel de mutantes, junto con el desplazamiento relativo en CE50 respecto al replicón de tipo silvestre GT1b se muestran a continuación. La variación de ensayo normal es ± 3-4 veces. Los desplazamientos de CE50 fuera de este intervalo implican resistencia y se indican en negrita. Los 3 inhibidores de FASN conservan actividad en todo el panel de mutantes, mientras que los agentes antivirales de acción directa presentan resistencia contra mutaciones en sus respectivos sitios de unión.
Tabla 5 CE50 antivirales

T l D l z mi n n v n E n r l i ilv r
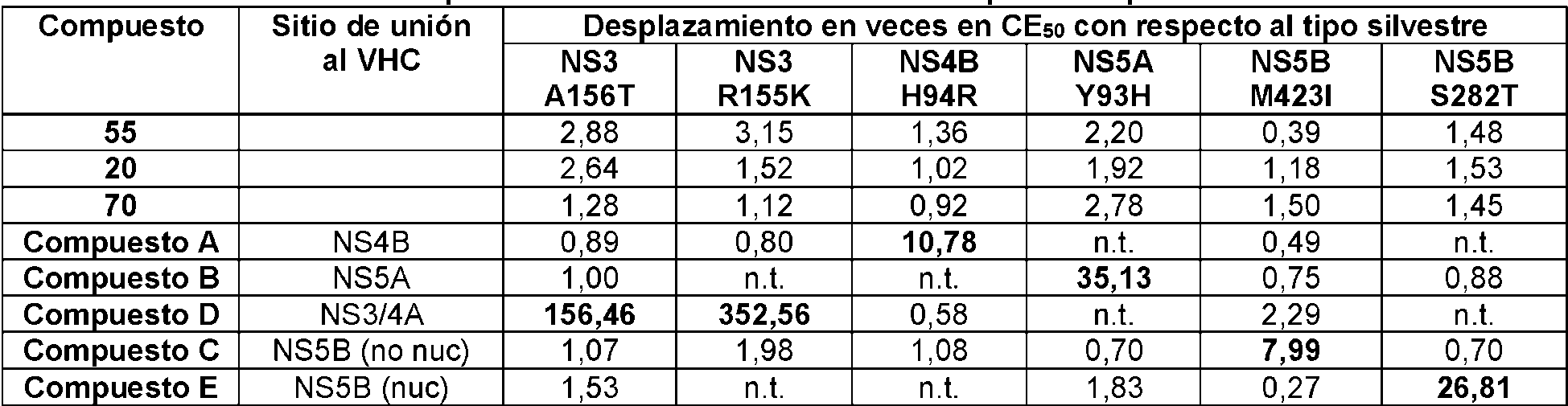
Ejemplo 5 - Inhibidores de FASN útiles en terapias de combinación
Este ejemplo describe la actividad antiviral y la citotoxicidad in vitro del compuesto de estructura (V-K) en combinación con IFN-a, Ribavirina, Compuestos B, C, D E contra una línea celular de replicón GT1 b de VHC.


Materiales:
Virus: El plásmido del replicón GTb1 se ensambló usando fragmentos de genes sintéticos. El genoma del replicón contiene segmentos del gen PVIRES-Luciferase Ubi-Neo y alberga 1 mutación adaptativa (S22041), y la cadena principal es Con1. La línea celular de replicón GT1b se estableció mediante los siguientes procedimientos publicados. Medio y reactivos: La tabla 7 a continuación proporciona detalles respecto a los reactivos del medio de cultivo usados en este ejemplo.
Tabla 7. Lista de reactivos del medio de cultivo

Instrumentos analíticos: Se usaron los siguientes instrumentos analíticos para realizar los ensayos de este ejemplo:
POD-810
Topcount(PE)
Envision (PE)
Multidrop (Thermo)
Procedimientos:
Preparación de placas del compuesto para ensayo de compuestos individuales: Los compuestos se suministraron como polvos secos y se reconstituyeron en DMSO para generar soluciones madre. El sistema POD-810 se usó para generar diluciones sucesivas semi-log de 10 puntos (3,16 veces) para el ensayo en placas de 96 pocillos. Las concentraciones de ensayo más altas se detallan para cada compuesto en la Tabla 8.
Protocolo de ensayo (compuestos individuales): Cada compuesto se ensayó con diluciones sucesivas de 3,16 veces (semi-log) de 10 concentraciones más DMSO por duplicado. Se recogieron las células GT1b de replicón de VHC y se ajustaron a una concentración celular de 8E 04 células/ml. Se usó un Multidrop para sembrar 100 pl/pocillo en 96 microplacas de ensayo para alcanzar una densidad celular final de 8000 células/pocillo. Las placas se incubaron en CO2 al 5 %, a 37 °C durante 72 horas.
Al final de la incubación de 72 horas, se midieron la actividad antiviral y la citotoxicidad. Se prepararon el reactivo Bright-Glo Luiferase y Cell Titer Flo y se almacenaron en la oscuridad, mientras que se equilibraban a temperatura ambiente. Las placas de células se dejaron equilibrar a temperatura ambiente también. Se usó un Multidrop para añadir 20 pl de Cell Titer Flo a cada pocillo de células tratadas con el compuesto y libres de compuesto. Las placas se incuban durante 1 hora, y la viabilidad celular se mide en un lector Envision para el cálculo de la citotoxicidad. Cincuenta microlitros de sustrato de luciferasa de luciérnaga se añaden a cada pocillo, se incuban durante 2 minutos, y la quimioluminiscencia se mide para el cálculo de CE50.
La actividad anti-replicón (% de inhibición) se calculó usando la siguiente ecuación:
% de Inhibición = [ l — ( (Compuesto — Fondo)/(DMSO — Fondo))x 100].
Compuestos de ensayo y configuración del ensayo para estudios de combinación de dos compuestos: Las soluciones madre de DMSO de los compuestos usados en el ensayo de un compuesto individual también se usaron en este análisis. Se generaron matrices de dilución de combinación mediante POD-810 en las microplacas de ensayo de 96 pocillos. El sistema POD-810 se usó para generar diluciones sucesivas de 2 veces, de 7 puntos en un formato de matriz. La concentración máxima ensayada para cada compuesto se detalla a continuación.
Tabla 8. Actividades esperadas y concentraciones superiores de compuestos ensayados en estudios con a ente único de combinación
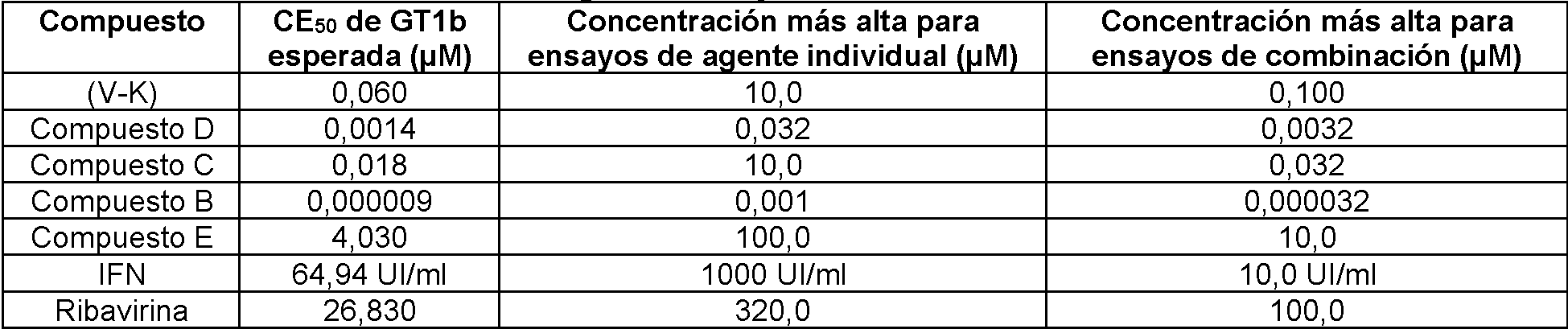
El compuesto de Estructura (V-K) se ensayó en solitario y en combinación con compuestos detallados en la Tabla 9. Cada compuesto también se ensayó en solitario como un único agente.
T l . m in i n m r v l i n
in v i r .

Configuración del ensayo (combinaciones de dos fármacos): Cada compuesto se ensayó con diluciones sucesivas de 2 veces para 7 concentraciones en formato de matriz más cada fármaco en solitario. Se recogieron las células GT1b de replicón de VHC y se ajustaron a una concentración celular de 8E 04 células/ml. Se usó un Multidrop para sembrar 100 pl en microplacas de ensayo de 96 pocillos para alcanzar una densidad celular final de 8000 células/pocillo. Las placas se incubaron en CO2 al 5%, a 37 °C durante 72 horas.
Al final de la incubación de 72 horas, se midieron la actividad antiviral y la citotoxicidad. Se prepararon el reactivo Bright-Glo Luiferase y Cell Titer Flo y se almacenaron en la oscuridad, mientras que se les dejaba equilibrarse a temperatura ambiente. Las placas de células se dejaron equilibrar a temperatura ambiente también. Se usó un Multidrop para añadir 20 pl de Cell Titer Flo a cada pocillo de células tratadas con el compuesto y libres de compuesto. Las placas se incubaron durante 1 hora, y la viabilidad celular se midió en un lector Envision para el cálculo de la citotoxicidad. A continuación se retiró el líquido de las placas, después de lo cual se añadieron 50 pl de PBS y 50 pl de solución de sustrato de luciferasa de luciérnaga a cada pocillo, después de un período de incubación de 2 minutos,
se midió la quimioluminiscencia (para el cálculo de la replicación del VHC). Los datos se analizaron utilizando MacSynergy™ II.
Resultados del ensayo:
Actividad y citotoxicidad de los compuestos. Los valores de CE50 y CC50 se resumen a continuación en la Tabla 10.
Tabla 10. CE50 CC50 de cada com uesto de ensa o

Efecto de combinación. El efecto de la combinación de los pares de compuestos se calculó usando MacSynergy™ II y los resultados se resumen en la Tabla 11 a continuación.

Conclusiones
Los factores Z de los pares de compuestos resumidos en la Tabla 12 indican que la calidad del ensayo es mejor que el estándar de CC.

Los valores de CE50 de los compuestos individuales en la matriz de combinación (resumidos en la Tabla 13) son coherentes con los datos de CE50 obtenidos para inhibición de compuesto individual Tabla 10.
Tabla 13. Resumen de CE50 de dosis individual en combinación de com uestos

Se demostró que el compuesto de estructura (V-K) tiene actividad antiviral aditiva sin incrementar la citotoxicidad en combinación con agentes que representan diversos mecanismos. Estos resultados se resumen en la Tabla 14 a continuación.
Tabla 14. Resumen de mecanismos antivirales que son aditivos con el compuesto de estructura (V-K). La
expresión "antiviral de acción directa" ("AAD") se refiere a un compuesto que se une a e inhibe una proteína vir l n l r n r ín l h .

IFN-a y RBV representan el actual procedimiento recomendado para el tratamiento de la infección por hepatitis C, y los inhibidores de la proteasa del VHC Telapreviry Boceprivir han sido aprobados recientemente. La actividad antiviral aditiva y la falta de citotoxicidad mejorada en combinación con IFN-a y RBV sugieren, además, que los compuestos de esta invención interferirán en los procesos críticos del huésped, tales como la defensa celular (IFN-a) o la biosíntesis de nucleótidos de guanidina (RBV). Por lo tanto, los compuestos de esta invención, tales como el compuesto de estructura (V-K) deben ser terapéuticamente útiles si se administran en regímenes de combinación con el procedimiento recomendado actual. Por otra parte, las actividades antivirales aditivas observadas con el compuesto B, el compuesto C, y el compuesto E sugieren que las moléculas de esta invención, tales como el compuesto de estructura (V-K) pueden combinarse productivamente con agentes actualmente en desarrollo que se dirigen a mecanismos más recientes (por ejemplo, inhibidores de NS5A y NS5B).
Ejemplo 6 - Actividad anti-tumoral - Ensayo de citotoxicidad multiplexado
Las células se cultivaron en RPMI 1640, FBS al 10%, L-alanil-L-glutamina 2 mM, piruvato de Na 1 mM o un medio especial en una atmósfera humidificada con el 5 % de CO2 a 37 °C. Las células se sembraron en placas de 384 pocillos y se incubaron en una atmósfera humidificada con el 5% de CO2 a 37 °C. Los compuestos se añadieron 24 horas después de la siembra de las células. Al mismo tiempo, se generó una placa de células no tratadas a tiempo cero. Después de un período de incubación de 72 horas, las células se fijaron y se tiñeron con anticuerpos marcados fluorescentemente y colorante nuclear para permitir la visualización de los núcleos, las células apoptóticas y las células mitóticas. Se detectaron células apoptóticas usando un anticuerpo anti-caspasa-3 activa. Se detectaron células mitóticas usando un anticuerpo anti fosfo-histona-3.
Los compuestos se diluyeron en serie en incrementos semi-log (3,16 veces) y se ensayaron en 10 concentraciones en una concentración final de ensayo del 0,1 % de DMSO a partir de la concentración de ensayo más alta especificada en el capítulo información de la muestra. Se llevó a cabo microscopía de fluorescencia automatizada usando un analizador IN Cell Analyzer 1000 de GE Healthcare, y las imágenes se recogieron con un objetivo 4X.
Se adquirieron imágenes en formato tiff de doce bits usando el InCell Analyzer 10003.2 y se analizaron con el software Developer Toolbox 1.6. Los valores de CE50 y CI50 se calcularon usando regresión no lineal para ajustar los datos a un modelo de respuesta a la dosis de Un Sitio de 4 parámetros, 4 puntos, sigmoidea, donde : y (ajuste) = A [(B -A)/(1 ((C/x) A D))]. Los cálculos del ajuste de curva, CE50/CI50 y la generación del informe se realizan usando un software basado en el motor de reducción datos personalizado MathIQ based (AIM).
El ensayo de citotoxicidad multiplexada usa una técnica de análisis basado en imágenes de células, donde las células se fijaron y se tiñeron con anticuerpos marcados fluorescentemente y colorante nuclear para visualizar los núcleos, y las células y apoptóticas. Se detectan células apoptóticas usando un anticuerpo anti-caspasa-3 activa. Se detectan células mitóticas usando un anticuerpo anti fosfo-histona-3.
La proliferación celular se mide por la intensidad de la señal del colorante nuclear incorporado. La salida ensayo de proliferación celular se denomina el recuento de células relativo. Para determinar la conclusión de proliferación celular, la salida de datos de proliferación celular se transforma en porcentaje de control (PDC) mediante la siguiente fórmula: PDC = recuento de células relativo (pocillos de compuesto)/recuento de células relativo (pocillos de vehículo) * 100 La placa no tratada a tiempo cero se usa para determinar el número de duplicaciones en el periodo de ensayo de 72 horas: Número de duplicaciones en 72 horas = LN[Número de células (conclusión a las 72 h) *Número de células (tiempo cero)]/LN(2). La salida de cada biomarcador es el incremento en veces respecto al fondo de vehículo normalizado al recuento de células relativo en cada pocillo.
El marcados caspasa-3 activada etiqueta células desde fase temprana a tardía de apoptosis. La salida se muestra como el incremento en veces de células apoptóticas respecto al fondo de vehículo normalizado al recuento de células relativo en cada pocillo. Las concentraciones de compuesto de ensayo que causan una inducción de 5 veces en la señal de la caspasa-3 indican la inducción de apoptosis significativa. Los pocillos con concentraciones más altas que el recuento celular relativo CI95 se eliminan del análisis de inducción de caspasa3.
El marcador fosfo-histona-3 etiqueta células mitóticas. La salida se muestra como el incremento en veces de inducción de células mitóticas respecto al fondo de vehículo normalizado al recuento de células relativo en cada pocillo. Cuando la inducción en veces de señal de células mitóticas respecto al fondo es ~1, no hay "ningún efecto" sobre el ciclo celular. Un incremento de dos o más veces en la señal de fosfo-histona-3 respecto al fondo de vehículo indica la inducción significativa por el compuesto de ensayo de bloqueo mitótico.
Una disminución de dos o más veces en la señal de fosfo-histona-3 puede indicar bloqueo de G1/S solo cuando los niveles de citotoxicidad están por debajo del recuento celular relativo medido CI95. Cuando se observa una disminución de 2 o más veces en la señal de fosfo-histona-3 a concentraciones más altas que el recuento celular relativo CI95, la
disminución de los recuentos de células mitóticas se debe muy probablemente a un efecto de citotoxicidad más general en lugar de un verdadero bloqueo de fase G1/S. Los pocillos con concentraciones más altas que el recuento celular relativo CI95 se eliminan del análisis de inducción de fosfo-histona-3.
La proliferación celular medida mediante recuento de células relativo fueron los criterios de respuesta positiva Apoptosis:
Un incremento de >5 veces en señal de caspasa-3 activada indica una respuesta apoptótica
Mitosis:
Un incremento de >2 veces en fosfo-histona-3 indica bloqueo mitótico
Una disminución de <2 veces en fosfo-histona-3 indica bloqueo de G1/S
Tabla 15. Resultados

Aunque en el presente documento se han mostrado y descrito aspectos preferidos de la presente invención, será obvio para los expertos en la técnica que dichos aspectos se proporcionan a modo de ejemplo solamente. Numerosas variaciones, cambios y sustituciones se les ocurrirán a los expertos en la técnica sin alejarse de la invención. Debe entenderse que diversas alternativas a los aspectos de la invención descrita en este documento se pueden emplear en la puesta en práctica de la invención. Se pretende que las siguientes reivindicaciones definan el alcance de la invención y que los procedimientos y estructuras dentro del alcance de estas reivindicaciones y sus equivalentes estén cubiertos por el mismo.
Claims (1)
- REIVINDICACIONES1. Un compuesto de Fórmula X:
 o una sal farmacéuticamente aceptable del mismo, donde:R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) donde:el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está sustituido opcionalmente por uno o más halógenos;cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH o halógeno;L3 es C(R60)2, O o NR50;cada R60 es independientemente H, -OH, -CN, -Ot-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado), o -C(O)-N(R601)2 en donde:t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;cada R50 es independientemente H, -C(O)-Ot-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C3-C5 cíclico), -alquilo C3-C5 cíclico conteniendo opcionalmente un heteroátomo de oxígeno o nitrógeno, -C(O)-N(R501)2, o alquilo C1-C4 lineal o ramificado, en donde:t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;n es 1, 2 o 3;m es 1 o 2;R21 es H, halógeno, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 donde el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno;R22 es H, halógeno, alquilo C1-C2;cada R26 es independientemente -OH, -CN, halógeno, alquilo C1-C4 lineal o ramificado, alquilo C1-C4)t-Ot-cicloalquilo C3-C5, -(alquilo C1-C4)t-O-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C1-C4), o -C(O)-N(R501)2 en donde t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;s es 0, 1 o 2;cada R601 y R501 es independientemente H o alquilo C1-C4 lineal o ramificado; yen donde dos de R26, R60, R50, R501 y R601 se unen opcionalmente para formar un anillo en donde los dos de R26, R60, R50, R501 y R601 pueden ser dos R26, dos R60, dos R50, dos R501 o dos R601.2. El compuesto de acuerdo con la reivindicación 1 en donde R21 es halógeno, alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C5.3. Compuesto de acuerdo con la reivindicación 1 o 2, en donde (i) R3 es H o halógeno; o (ii) R3 es H o F.4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-3, en donde (i) R1 es -CN o haloalquilo C1-C2; o (ii) R1 es -CN; o (iii) R1 es -CF3.5. El compuesto de acuerdo con una las reivindicaciones 1-4, en donde (i) n es 1; o (ii) n es 2.6. El compuesto de una cualquiera de las reivindicaciones 1-5, en donde (i) m es 1; o (ii) m es 2.7. El compuesto de una cualquiera de las reivindicaciones 1-6, en donde (i), R21 es alquilo C1-C2 o cicloalquilo C3-C5 y R22 es alquilo C1-C2, o (ii) R21 es cicloalquilo C3-C5 y R22 es alquilo C1-C2.8. El compuesto de una cualquiera de las reivindicaciones 1-4 en donde(i) n es 2, m es 1, L3 es -N-C(O)-O-(alquilo C1-C2); o (ii) L3 es NR50; R50 es alquilo C1-C2; R21 es ciclobutilo; R22 es H o metilo; R3 es H; R1 es -CN; m es 2 y n es 1 o 2; o (iii) n es 2, m es 1, L3 es O y s es 0.9. El compuesto de una cualquiera de las reivindicaciones 1-6, en donde (i) R22 es H, metilo o etilo; o (ii) R22 es metilo; o (iii) R22 es H.10. El compuesto de la reivindicación 1, en donde (i) R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4, R22 es metilo, n es 2 y L3 es NR50 en donde R50 es metilo o etilo; o(ii) R1 es -c N, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4, R22 es metilo, n es 2 y L3 es O.11. El compuesto de la reivindicación 1, que tiene una fórmula seleccionada del grupo que consiste en:
o una sal farmacéuticamente aceptable del mismo, donde:R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) donde:el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está sustituido opcionalmente por uno o más halógenos;cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH o halógeno;L3 es C(R60)2, O o NR50;cada R60 es independientemente H, -OH, -CN, -Ot-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado), o -C(O)-N(R601)2 en donde:t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;cada R50 es independientemente H, -C(O)-Ot-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C3-C5 cíclico), -alquilo C3-C5 cíclico conteniendo opcionalmente un heteroátomo de oxígeno o nitrógeno, -C(O)-N(R501)2, o alquilo C1-C4 lineal o ramificado, en donde:t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;n es 1, 2 o 3;m es 1 o 2;R21 es H, halógeno, alquilo C1-C4 lineal o ramificado, cicloalquilo C3-C5 donde el cicloalquilo C3-C5 opcionalmente incluye un heteroátomo de oxígeno o nitrógeno;R22 es H, halógeno, alquilo C1-C2;cada R26 es independientemente -OH, -CN, halógeno, alquilo C1-C4 lineal o ramificado, alquilo C1-C4)t-Ot-cicloalquilo C3-C5, -(alquilo C1-C4)t-O-(alquilo C1-C4 lineal o ramificado), -C(O)-Ot-(alquilo C1-C4), o -C(O)-N(R501)2 en donde t es 0 o 1, yel cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno;s es 0, 1 o 2;cada R601 y R501 es independientemente H o alquilo C1-C4 lineal o ramificado; yen donde dos de R26, R60, R50, R501 y R601 se unen opcionalmente para formar un anillo en donde los dos de R26, R60, R50, R501 y R601 pueden ser dos R26, dos R60, dos R50, dos R501 o dos R601.2. El compuesto de acuerdo con la reivindicación 1 en donde R21 es halógeno, alquilo C1-C4 lineal o ramificado o cicloalquilo C3-C5.3. Compuesto de acuerdo con la reivindicación 1 o 2, en donde (i) R3 es H o halógeno; o (ii) R3 es H o F.4. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1-3, en donde (i) R1 es -CN o haloalquilo C1-C2; o (ii) R1 es -CN; o (iii) R1 es -CF3.5. El compuesto de acuerdo con una las reivindicaciones 1-4, en donde (i) n es 1; o (ii) n es 2.6. El compuesto de una cualquiera de las reivindicaciones 1-5, en donde (i) m es 1; o (ii) m es 2.7. El compuesto de una cualquiera de las reivindicaciones 1-6, en donde (i), R21 es alquilo C1-C2 o cicloalquilo C3-C5 y R22 es alquilo C1-C2, o (ii) R21 es cicloalquilo C3-C5 y R22 es alquilo C1-C2.8. El compuesto de una cualquiera de las reivindicaciones 1-4 en donde(i) n es 2, m es 1, L3 es -N-C(O)-O-(alquilo C1-C2); o (ii) L3 es NR50; R50 es alquilo C1-C2; R21 es ciclobutilo; R22 es H o metilo; R3 es H; R1 es -CN; m es 2 y n es 1 o 2; o (iii) n es 2, m es 1, L3 es O y s es 0.9. El compuesto de una cualquiera de las reivindicaciones 1-6, en donde (i) R22 es H, metilo o etilo; o (ii) R22 es metilo; o (iii) R22 es H.10. El compuesto de la reivindicación 1, en donde (i) R1 es -CN, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4, R22 es metilo, n es 2 y L3 es NR50 en donde R50 es metilo o etilo; o(ii) R1 es -c N, cada R2 es H, R3 es H o F, R21 es cicloalquilo C3-C4, R22 es metilo, n es 2 y L3 es O.11. El compuesto de la reivindicación 1, que tiene una fórmula seleccionada del grupo que consiste en: 12. Un compuesto que tiene la Fórmula (Vl-J):
12. Un compuesto que tiene la Fórmula (Vl-J): o una sal del mismo farmacéuticamente aceptable, en donde:R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) en donde:el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH, o halógeno;R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;R22 es H, halógeno, o alquilo C1-C2;R35 es -C(O)-NHR351; yR351 es alquilo C1-C6 lineal o ramificado, cicloalquilo, heterociclilo, arilo o heteroarilo;en donde opcionalmente el compuesto tiene una fórmula seleccionada del grupo que consiste en:
o una sal del mismo farmacéuticamente aceptable, en donde:R1 es H, -CN, halógeno, alquilo C1-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 lineal o ramificado) en donde:el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH, o halógeno;R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;R22 es H, halógeno, o alquilo C1-C2;R35 es -C(O)-NHR351; yR351 es alquilo C1-C6 lineal o ramificado, cicloalquilo, heterociclilo, arilo o heteroarilo;en donde opcionalmente el compuesto tiene una fórmula seleccionada del grupo que consiste en: y13. Un compuesto que tiene la Fórmula (XI):
y13. Un compuesto que tiene la Fórmula (XI): o una sal del mismo farmacéuticamente aceptable, en donde:R1 es H, -CN, halógeno, alquilo Ci-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), o -O-(alquilo Ci-C4 lineal o ramificado) en donde:el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH, o halógeno;R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;R22 es H, halógeno, alquilo C1-C2; yR351 es alquilo C1-C2 o C2-O-(alquilo C1 o C2);En donde opcionalmente el compuesto tiene una fórmula seleccionada del grupo que consiste en:
o una sal del mismo farmacéuticamente aceptable, en donde:R1 es H, -CN, halógeno, alquilo Ci-C4 lineal o ramificado, -O-(cicloalquilo C3-C5), o -O-(alquilo Ci-C4 lineal o ramificado) en donde:el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; ycuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;cada R2 es independientemente H, halógeno o alquilo C1-C4 lineal o ramificado;R3 es H, -OH, o halógeno;R21 es ciclobutilo, azetidin-1-ilo, o ciclopropilo;R22 es H, halógeno, alquilo C1-C2; yR351 es alquilo C1-C2 o C2-O-(alquilo C1 o C2);En donde opcionalmente el compuesto tiene una fórmula seleccionada del grupo que consiste en: 13 y un vehículo, excipiente o diluyente farmacéuticamente aceptable.15. Una cantidad terapéuticamente efectiva de uno cualquiera de los compuestos de las reivindicaciones 1-13 para uso en el tratamiento dea) una infección vírica en un sujeto, en donde opcionalmente la infección viral comprende infección por hepatitis C; b) una afección caracterizada por la desregulación de una función sintasa de ácido graso en un sujeto;c) cáncer en un sujeto, en donde opcionalmente el cáncer es cáncer de mama.
13 y un vehículo, excipiente o diluyente farmacéuticamente aceptable.15. Una cantidad terapéuticamente efectiva de uno cualquiera de los compuestos de las reivindicaciones 1-13 para uso en el tratamiento dea) una infección vírica en un sujeto, en donde opcionalmente la infección viral comprende infección por hepatitis C; b) una afección caracterizada por la desregulación de una función sintasa de ácido graso en un sujeto;c) cáncer en un sujeto, en donde opcionalmente el cáncer es cáncer de mama.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161450482P | 2011-03-08 | 2011-03-08 | |
| US201161450561P | 2011-03-08 | 2011-03-08 | |
| US201161508611P | 2011-07-16 | 2011-07-16 | |
| US201261585642P | 2012-01-11 | 2012-01-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2883274T3 true ES2883274T3 (es) | 2021-12-07 |
Family
ID=48221725
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17153778T Active ES2883274T3 (es) | 2011-03-08 | 2012-03-08 | Moduladores heterocíclicos de la síntesis de lípidos |
| ES12754969.9T Active ES2624131T3 (es) | 2011-03-08 | 2012-03-08 | Moduladores heterocíclicos de la síntesis de lípidos |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12754969.9T Active ES2624131T3 (es) | 2011-03-08 | 2012-03-08 | Moduladores heterocíclicos de la síntesis de lípidos |
Country Status (21)
| Country | Link |
|---|---|
| EP (2) | EP3190108B1 (es) |
| JP (3) | JP5973473B2 (es) |
| KR (2) | KR101930106B1 (es) |
| CN (2) | CN105001225B (es) |
| AU (1) | AU2012225390B2 (es) |
| BR (3) | BR112013022761B8 (es) |
| CA (1) | CA2829082C (es) |
| DK (1) | DK2683244T3 (es) |
| EA (1) | EA031193B1 (es) |
| ES (2) | ES2883274T3 (es) |
| HK (1) | HK1216310A1 (es) |
| HR (1) | HRP20170500T1 (es) |
| HU (1) | HUE032459T2 (es) |
| IL (2) | IL228276A (es) |
| LT (1) | LT2683244T (es) |
| MX (2) | MX369388B (es) |
| PL (1) | PL2683244T3 (es) |
| PT (1) | PT2683244T (es) |
| SI (1) | SI2683244T1 (es) |
| WO (1) | WO2012122391A1 (es) |
| ZA (1) | ZA201306655B (es) |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8871790B2 (en) | 2011-03-08 | 2014-10-28 | 3-V Biosciences, Inc. | Heterocyclic modulators of lipid synthesis |
| US20170119786A1 (en) | 2011-03-08 | 2017-05-04 | 3-V Biosciences, Inc. | Heterocyclic modulators of lipid synthesis |
| US9624173B2 (en) | 2011-03-08 | 2017-04-18 | 3-V Biosciences, Inc. | Heterocyclic modulators of lipid synthesis |
| AU2013286894B2 (en) | 2012-07-03 | 2017-12-07 | Sagimet Biosciences Inc. | Heterocyclic modulators of lipid synthesis |
| AR092211A1 (es) * | 2012-09-24 | 2015-04-08 | Merck Patent Ges Mit Beschränkter Haftung | Derivados de hidropirrolopirrol |
| SG11201504740UA (en) | 2012-12-27 | 2015-07-30 | Univ Drexel | Novel antiviral agents against hbv infection |
| BR112015019208A2 (pt) * | 2013-02-14 | 2017-07-18 | Galderma Res & Dev | processo de síntese |
| MX2015011198A (es) * | 2013-03-06 | 2015-11-30 | Hoffmann La Roche | Compuestos antivirales. |
| PH12015502097B1 (en) | 2013-03-13 | 2023-02-03 | Forma Therapeutics Inc | Novel compounds and compositions for inhibition of fasn |
| EP3083583B1 (en) | 2013-12-20 | 2020-11-18 | Sagimet Biosciences Inc. | Heterocyclic modulators of lipid synthesis and combinations thereof |
| EP3092232B1 (en) * | 2014-01-07 | 2022-03-09 | Sagimet Biosciences Inc. | Heterocyclic modulators of lipid synthesis for use against cancer and viral infections |
| CN104774204A (zh) * | 2014-01-15 | 2015-07-15 | 鲁南贝特制药有限公司 | 一种制备奈韦拉平及其中间体的方法 |
| WO2015199206A1 (ja) * | 2014-06-27 | 2015-12-30 | 塩野義製薬株式会社 | Trpv4阻害活性を有する6員環誘導体 |
| JP6691909B2 (ja) * | 2014-08-15 | 2020-05-13 | サギメット バイオサイエンシーズ インコーポレイテッド | 薬物耐性癌の治療で用いるための脂肪酸シンターゼ阻害剤 |
| KR20170129151A (ko) * | 2015-03-19 | 2017-11-24 | 3-브이 바이오사이언시스, 인코포레이티드 | 지질 합성의 복소환식 조절제 |
| CN105175277B (zh) * | 2015-05-18 | 2018-04-03 | 中山大学肿瘤防治中心 | 一种3‑磷酸甘油醛脱氢酶的抑制剂及其制备方法和应用 |
| WO2016205590A1 (en) | 2015-06-18 | 2016-12-22 | Cephalon, Inc. | Substituted 4-benzyl and 4-benzoyl piperidine derivatives |
| EA201890086A1 (ru) | 2015-06-18 | 2018-06-29 | Сефалон, Инк. | 1,4-замещенные производные пиперидина |
| CN115089595A (zh) * | 2015-09-04 | 2022-09-23 | 波利摩迪股份公司 | 用于治疗人乳头状瘤病毒皮肤感染的含乙酰水杨酸的组合物和医疗装置 |
| KR102644934B1 (ko) * | 2016-11-11 | 2024-03-08 | 새지메트 바이오사이언시스, 인코포레이티드 | 지질 합성의 헤테로사이클릭 조절인자 |
| CN107043347B (zh) * | 2016-11-18 | 2019-07-02 | 泰州禾益新材料科技有限公司 | 一种阿加曲班中间体(2r,4r)-4-甲基哌啶-2-甲酸乙酯的合成方法 |
| CN109212056B (zh) * | 2018-08-19 | 2021-05-07 | 丁立平 | 一种测定水源水中三种痕量单卤代甲基苯甲酸的气相色谱-质谱联用法 |
| TWI767148B (zh) | 2018-10-10 | 2022-06-11 | 美商弗瑪治療公司 | 抑制脂肪酸合成酶(fasn) |
| US10793554B2 (en) | 2018-10-29 | 2020-10-06 | Forma Therapeutics, Inc. | Solid forms of 4-(2-fluoro-4-(1-methyl-1H-benzo[d]imidazol-5-yl)benzoyl)piperazin-1-yl)(1-hydroxycyclopropyl)methanone |
| WO2021229533A1 (en) * | 2020-05-14 | 2021-11-18 | Palani Llc | A fatty acid based composition for treatment and/or prevention of enveloped-virus related infections |
| CN114469945A (zh) * | 2020-10-28 | 2022-05-13 | 歌礼生物科技(杭州)有限公司 | 脂肪酸合酶抑制剂的用途和药物组合物 |
| CN113584044A (zh) * | 2021-08-28 | 2021-11-02 | 青岛农业大学 | 一种牙鲆socs3基因及其真核表达载体、表达系统和异源表达方法 |
| EP4594313A1 (en) * | 2022-10-01 | 2025-08-06 | Assia Chemical Industries Ltd. | Solid state forms of denifanstat |
| WO2024240108A1 (zh) * | 2023-05-19 | 2024-11-28 | 上海海和药物研究开发股份有限公司 | 一种芳基脲类parp1抑制剂、其制备方法及用途 |
| CN119080742A (zh) * | 2023-06-05 | 2024-12-06 | 甘莱制药有限公司 | 脂质合成的杂环调节剂的工业化制备 |
| AU2024298839A1 (en) | 2023-07-21 | 2026-03-05 | Sagimet Biosciences Inc. | Pharmaceutical composition containing heterocyclic compound |
| WO2025086054A1 (zh) * | 2023-10-23 | 2025-05-01 | 歌礼生物科技(杭州)有限公司 | 一种包含苯甲腈衍生物的药物组合物 |
| WO2025120168A1 (en) | 2023-12-07 | 2025-06-12 | Institut National De Recherche Pour L'agriculture, L'alimentation Et L'environnement (Inrae) | Sulfonamide derivatives and use thereof in the treatment of cryptosporidium infections |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3625214A (en) | 1970-05-18 | 1971-12-07 | Alza Corp | Drug-delivery device |
| US4906474A (en) | 1983-03-22 | 1990-03-06 | Massachusetts Institute Of Technology | Bioerodible polyanhydrides for controlled drug delivery |
| US4789734A (en) | 1985-08-06 | 1988-12-06 | La Jolla Cancer Research Foundation | Vitronectin specific cell receptor derived from mammalian mesenchymal tissue |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| JP2876058B2 (ja) | 1986-08-18 | 1999-03-31 | エミスフィア・テクノロジーズ・インコーポレイテッド | 薬物送達システム |
| US6024983A (en) | 1986-10-24 | 2000-02-15 | Southern Research Institute | Composition for delivering bioactive agents for immune response and its preparation |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US5001139A (en) | 1987-06-12 | 1991-03-19 | American Cyanamid Company | Enchancers for the transdermal flux of nivadipine |
| US4992445A (en) | 1987-06-12 | 1991-02-12 | American Cyanamid Co. | Transdermal delivery of pharmaceuticals |
| US4897268A (en) | 1987-08-03 | 1990-01-30 | Southern Research Institute | Drug delivery system and method of making the same |
| US5766573A (en) | 1988-12-06 | 1998-06-16 | Riker Laboratories, Inc. | Medicinal aerosol formulations |
| US5190029A (en) | 1991-02-14 | 1993-03-02 | Virginia Commonwealth University | Formulation for delivery of drugs by metered dose inhalers with reduced or no chlorofluorocarbon content |
| US5376359A (en) | 1992-07-07 | 1994-12-27 | Glaxo, Inc. | Method of stabilizing aerosol formulations |
| US5928647A (en) | 1993-01-11 | 1999-07-27 | Dana-Farber Cancer Institute | Inducing cytotoxic T lymphocyte responses |
| KR20080028964A (ko) * | 2005-06-27 | 2008-04-02 | 엑셀리시스, 인코포레이티드 | 피라졸계 lxr 변조제 |
| ATE452635T1 (de) * | 2005-12-01 | 2010-01-15 | Hoffmann La Roche | Heteroaryl-substituierte piperidin-derivate als l-cpt1-hemmer |
| US7994222B2 (en) * | 2006-09-05 | 2011-08-09 | Bipar Sciences, Inc. | Monitoring of the inhibition of fatty acid synthesis by iodo-nitrobenzamide compounds |
| WO2008059214A1 (en) * | 2006-11-13 | 2008-05-22 | Astrazeneca Ab | Bisamlde derivatives and use thereof as fatty acid synthase inhibitors |
| TW200831092A (en) * | 2006-12-21 | 2008-08-01 | Astrazeneca Ab | Therapeutic agents |
| TW200833663A (en) * | 2006-12-21 | 2008-08-16 | Astrazeneca Ab | Therapeutic agents |
| WO2008075077A1 (en) * | 2006-12-21 | 2008-06-26 | Astrazeneca Ab | Piperidine derivatives for the treatment of obesity |
-
2012
- 2012-03-08 ES ES17153778T patent/ES2883274T3/es active Active
- 2012-03-08 ES ES12754969.9T patent/ES2624131T3/es active Active
- 2012-03-08 PT PT127549699T patent/PT2683244T/pt unknown
- 2012-03-08 EP EP17153778.0A patent/EP3190108B1/en active Active
- 2012-03-08 CA CA2829082A patent/CA2829082C/en active Active
- 2012-03-08 WO PCT/US2012/028309 patent/WO2012122391A1/en not_active Ceased
- 2012-03-08 KR KR1020137026499A patent/KR101930106B1/ko active Active
- 2012-03-08 EP EP12754969.9A patent/EP2683244B1/en active Active
- 2012-03-08 LT LTEP12754969.9T patent/LT2683244T/lt unknown
- 2012-03-08 BR BR112013022761A patent/BR112013022761B8/pt active IP Right Grant
- 2012-03-08 KR KR1020187017297A patent/KR101968058B1/ko active Active
- 2012-03-08 BR BR122015005121-5A patent/BR122015005121B1/pt active IP Right Grant
- 2012-03-08 HR HRP20170500TT patent/HRP20170500T1/hr unknown
- 2012-03-08 CN CN201510354353.6A patent/CN105001225B/zh active Active
- 2012-03-08 EA EA201391290A patent/EA031193B1/ru unknown
- 2012-03-08 MX MX2016009579A patent/MX369388B/es unknown
- 2012-03-08 JP JP2013557870A patent/JP5973473B2/ja active Active
- 2012-03-08 DK DK12754969.9T patent/DK2683244T3/en active
- 2012-03-08 CN CN201280020832.9A patent/CN103561576B/zh active Active
- 2012-03-08 BR BR122015005122-3A patent/BR122015005122B1/pt active IP Right Grant
- 2012-03-08 PL PL12754969T patent/PL2683244T3/pl unknown
- 2012-03-08 HU HUE12754969A patent/HUE032459T2/en unknown
- 2012-03-08 SI SI201230912A patent/SI2683244T1/sl unknown
- 2012-03-08 AU AU2012225390A patent/AU2012225390B2/en active Active
- 2012-03-08 MX MX2013010170A patent/MX342287B/es active IP Right Grant
-
2013
- 2013-09-03 IL IL228276A patent/IL228276A/en active IP Right Grant
- 2013-09-04 ZA ZA2013/06655A patent/ZA201306655B/en unknown
-
2016
- 2016-02-03 JP JP2016018611A patent/JP2016074742A/ja active Pending
- 2016-02-03 JP JP2016018610A patent/JP6061437B2/ja active Active
- 2016-04-11 HK HK16104111.8A patent/HK1216310A1/zh unknown
- 2016-09-13 IL IL247804A patent/IL247804B/en active IP Right Grant
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2883274T3 (es) | Moduladores heterocíclicos de la síntesis de lípidos | |
| US9809591B2 (en) | Heterocyclic modulators of lipid synthesis | |
| US20230405016A1 (en) | Heterocyclic modulators of lipid synthesis | |
| KR102644934B1 (ko) | 지질 합성의 헤테로사이클릭 조절인자 | |
| HK1194615B (en) | Heterocyclic modulators of lipid synthesis | |
| HK1194615A (en) | Heterocyclic modulators of lipid synthesis |