ES2889775T3 - Proceso mejorado para la síntesis del fármaco-enlazador vc-seco - Google Patents
Proceso mejorado para la síntesis del fármaco-enlazador vc-seco Download PDFInfo
- Publication number
- ES2889775T3 ES2889775T3 ES18811180T ES18811180T ES2889775T3 ES 2889775 T3 ES2889775 T3 ES 2889775T3 ES 18811180 T ES18811180 T ES 18811180T ES 18811180 T ES18811180 T ES 18811180T ES 2889775 T3 ES2889775 T3 ES 2889775T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- antibody
- duba
- drug
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 43
- 230000008569 process Effects 0.000 title claims description 38
- 238000003786 synthesis reaction Methods 0.000 title claims description 13
- 230000015572 biosynthetic process Effects 0.000 title claims description 11
- 150000001875 compounds Chemical class 0.000 claims abstract description 63
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 20
- RFQYSAASDBNNDZ-UCGHAGIGSA-N [(1s)-1-(chloromethyl)-3-[6-[(4-hydroxybenzoyl)amino]imidazo[1,2-a]pyridine-2-carbonyl]-9-methyl-1,2-dihydrobenzo[e]indol-5-yl] n-[2-[[4-[[(2s)-5-(carbamoylamino)-2-[[(2s)-2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxycarbonylamino]-3-methylbutanoyl]amino]pe Chemical compound N([C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=O)C(=O)NC=1C=CC(COC(=O)N(C)CCN(CCOCCO)C(=O)OC=2C3=CC=CC(C)=C3C=3[C@H](CCl)CN(C=3C=2)C(=O)C=2N=C3C=CC(NC(=O)C=4C=CC(O)=CC=4)=CN3C=2)=CC=1)C(=O)OCCOCCN1C(=O)C=CC1=O RFQYSAASDBNNDZ-UCGHAGIGSA-N 0.000 claims description 17
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 15
- 229910000041 hydrogen chloride Inorganic materials 0.000 claims description 15
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 14
- 229940049595 antibody-drug conjugate Drugs 0.000 claims description 14
- 239000003814 drug Substances 0.000 claims description 13
- 229940079593 drug Drugs 0.000 claims description 13
- 238000006243 chemical reaction Methods 0.000 claims description 12
- 239000000427 antigen Substances 0.000 claims description 11
- 108091007433 antigens Proteins 0.000 claims description 11
- 102000036639 antigens Human genes 0.000 claims description 11
- 239000000611 antibody drug conjugate Substances 0.000 claims description 10
- 230000021615 conjugation Effects 0.000 claims description 8
- 229960000575 trastuzumab Drugs 0.000 claims description 6
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 claims description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 claims description 5
- 229940043279 diisopropylamine Drugs 0.000 claims description 4
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Substances CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 claims description 4
- 239000012634 fragment Substances 0.000 claims description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 21
- 239000002904 solvent Substances 0.000 description 17
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- 239000000203 mixture Substances 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 12
- 238000004519 manufacturing process Methods 0.000 description 11
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 10
- VQQZUHVNVDPTLO-SSEXGKCCSA-N tert-butyl N-[2-[[(1S)-1-(chloromethyl)-3-[6-[[4-(methoxymethoxy)benzoyl]amino]imidazo[1,2-a]pyridine-2-carbonyl]-9-methyl-1,2-dihydrobenzo[e]indol-5-yl]oxycarbonyl-[2-(2-hydroxyethoxy)ethyl]amino]ethyl]-N-methylcarbamate Chemical compound COCOc1ccc(cc1)C(=O)Nc1ccc2nc(cn2c1)C(=O)N1C[C@@H](CCl)c2c1cc(OC(=O)N(CCOCCO)CCN(C)C(=O)OC(C)(C)C)c1cccc(C)c21 VQQZUHVNVDPTLO-SSEXGKCCSA-N 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 235000018417 cysteine Nutrition 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- -1 EGFL Proteins 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N dichloromethane Natural products ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical group ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000002019 disulfides Chemical class 0.000 description 4
- 229960005501 duocarmycin Drugs 0.000 description 4
- 229930184221 duocarmycin Natural products 0.000 description 4
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 4
- 0 CC(C)(C)OC(N(C)CCN(CCOCCO)C(Oc1c(cccc2C)c2c([C@](CCl)CN2C(c3c[n](cc(cc4)NC(c(cc5)ccc5OC*)=O)c4n3)=O)c2c1)=O)=O Chemical compound CC(C)(C)OC(N(C)CCN(CCOCCO)C(Oc1c(cccc2C)c2c([C@](CCl)CN2C(c3c[n](cc(cc4)NC(c(cc5)ccc5OC*)=O)c4n3)=O)c2c1)=O)=O 0.000 description 3
- 229960005532 CC-1065 Drugs 0.000 description 3
- 102000003735 Mesothelin Human genes 0.000 description 3
- 108090000015 Mesothelin Proteins 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 102100030859 Tissue factor Human genes 0.000 description 3
- 102100033579 Trophoblast glycoprotein Human genes 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000000010 aprotic solvent Substances 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 239000012830 cancer therapeutic Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 229940127089 cytotoxic agent Drugs 0.000 description 3
- 239000002254 cytotoxic agent Substances 0.000 description 3
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 3
- 239000004210 ether based solvent Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 150000003573 thiols Chemical class 0.000 description 3
- 238000011282 treatment Methods 0.000 description 3
- YCGQPIRMLGEWMW-UHFFFAOYSA-N 1-[1-butyl-4-(3-methoxyphenyl)-2-oxo-1,8-naphthyridin-3-yl]-3-[4-[(dimethylamino)methyl]-2,6-di(propan-2-yl)phenyl]urea;hydrochloride Chemical compound Cl.CC(C)C=1C=C(CN(C)C)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OC)=C1 YCGQPIRMLGEWMW-UHFFFAOYSA-N 0.000 description 2
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 2
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 2
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 102100032912 CD44 antigen Human genes 0.000 description 2
- 102100025221 CD70 antigen Human genes 0.000 description 2
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 102100036466 Delta-like protein 3 Human genes 0.000 description 2
- AZVARJHZBXHUSO-UHFFFAOYSA-N Duocarmycin A Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3CC4CC44C5=C(C(C=C43)=O)NC(C5=O)(C)C(=O)OC)=CC2=C1 AZVARJHZBXHUSO-UHFFFAOYSA-N 0.000 description 2
- VQNATVDKACXKTF-UHFFFAOYSA-N Duocarmycin SA Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C(C64CC6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-UHFFFAOYSA-N 0.000 description 2
- 102000017930 EDNRB Human genes 0.000 description 2
- 102000012804 EPCAM Human genes 0.000 description 2
- 101150084967 EPCAM gene Proteins 0.000 description 2
- 101710194572 Endothelin receptor type B Proteins 0.000 description 2
- 102000000820 Enterotoxin Receptors Human genes 0.000 description 2
- 108010001687 Enterotoxin Receptors Proteins 0.000 description 2
- 102100031507 Fc receptor-like protein 5 Human genes 0.000 description 2
- 101710120217 Fc receptor-like protein 5 Proteins 0.000 description 2
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 2
- 102100035139 Folate receptor alpha Human genes 0.000 description 2
- 230000005526 G1 to G0 transition Effects 0.000 description 2
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 2
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 2
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 2
- 101000928513 Homo sapiens Delta-like protein 3 Proteins 0.000 description 2
- 101001023230 Homo sapiens Folate receptor alpha Proteins 0.000 description 2
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 2
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 2
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 2
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 2
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 2
- 101000897042 Homo sapiens Nucleotide pyrophosphatase Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 101000904724 Homo sapiens Transmembrane glycoprotein NMB Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 2
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 2
- 102100034256 Mucin-1 Human genes 0.000 description 2
- 102100023123 Mucin-16 Human genes 0.000 description 2
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 2
- 102100035486 Nectin-4 Human genes 0.000 description 2
- 101710043865 Nectin-4 Proteins 0.000 description 2
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 2
- 102100021969 Nucleotide pyrophosphatase Human genes 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- 102100035721 Syndecan-1 Human genes 0.000 description 2
- 101150057140 TACSTD1 gene Proteins 0.000 description 2
- 108010000499 Thromboplastin Proteins 0.000 description 2
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- 102100027212 Tumor-associated calcium signal transducer 2 Human genes 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 239000003610 charcoal Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 150000001945 cysteines Chemical class 0.000 description 2
- 229940127276 delta-like ligand 3 Drugs 0.000 description 2
- 229960005519 duocarmycin A Drugs 0.000 description 2
- 229960005510 duocarmycin SA Drugs 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 235000019439 ethyl acetate Nutrition 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- AZVARJHZBXHUSO-DZQVEHCYSA-N methyl (1R,4R,12S)-4-methyl-3,7-dioxo-10-(5,6,7-trimethoxy-1H-indole-2-carbonyl)-5,10-diazatetracyclo[7.4.0.01,12.02,6]trideca-2(6),8-diene-4-carboxylate Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)N[C@@](C5=O)(C)C(=O)OC)=CC2=C1 AZVARJHZBXHUSO-DZQVEHCYSA-N 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 239000002516 radical scavenger Substances 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- RZZQCIBGHFVPJC-UHFFFAOYSA-N tert-butyl n-[2-[2-(2-hydroxyethoxy)ethylamino]ethyl]-n-methylcarbamate Chemical compound CC(C)(C)OC(=O)N(C)CCNCCOCCO RZZQCIBGHFVPJC-UHFFFAOYSA-N 0.000 description 2
- ZGYICYBLPGRURT-UHFFFAOYSA-N tri(propan-2-yl)silicon Chemical compound CC(C)[Si](C(C)C)C(C)C ZGYICYBLPGRURT-UHFFFAOYSA-N 0.000 description 2
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 1
- 102100023990 60S ribosomal protein L17 Human genes 0.000 description 1
- 210000002925 A-like Anatomy 0.000 description 1
- 102000000074 ADP-ribosyl Cyclase Human genes 0.000 description 1
- 108010080394 ADP-ribosyl Cyclase Proteins 0.000 description 1
- 102000004145 Annexin A1 Human genes 0.000 description 1
- 108090000663 Annexin A1 Proteins 0.000 description 1
- 101100208111 Arabidopsis thaliana TRX5 gene Proteins 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 1
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 1
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 1
- 102100026094 C-type lectin domain family 12 member A Human genes 0.000 description 1
- 101710188619 C-type lectin domain family 12 member A Proteins 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 102100036369 Carbonic anhydrase 6 Human genes 0.000 description 1
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 1
- 102100039496 Choline transporter-like protein 4 Human genes 0.000 description 1
- SBJKKFFYIZUCET-JLAZNSOCSA-N Dehydro-L-ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(=O)C1=O SBJKKFFYIZUCET-JLAZNSOCSA-N 0.000 description 1
- SBJKKFFYIZUCET-UHFFFAOYSA-N Dehydroascorbic acid Natural products OCC(O)C1OC(=O)C(=O)C1=O SBJKKFFYIZUCET-UHFFFAOYSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 230000010777 Disulfide Reduction Effects 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 108091008794 FGF receptors Proteins 0.000 description 1
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 1
- 102000010451 Folate receptor alpha Human genes 0.000 description 1
- 108050001931 Folate receptor alpha Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102100022623 Hepatocyte growth factor receptor Human genes 0.000 description 1
- 101000690301 Homo sapiens Aldo-keto reductase family 1 member C4 Proteins 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000714525 Homo sapiens Carbonic anhydrase 6 Proteins 0.000 description 1
- 101000910338 Homo sapiens Carbonic anhydrase 9 Proteins 0.000 description 1
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101000606465 Homo sapiens Inactive tyrosine-protein kinase 7 Proteins 0.000 description 1
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101001116548 Homo sapiens Protein CBFA2T1 Proteins 0.000 description 1
- 101100420560 Homo sapiens SLC39A6 gene Proteins 0.000 description 1
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 1
- 101000635804 Homo sapiens Tissue factor Proteins 0.000 description 1
- 101000658574 Homo sapiens Transmembrane 4 L6 family member 1 Proteins 0.000 description 1
- 101000955999 Homo sapiens V-set domain-containing T-cell activation inhibitor 1 Proteins 0.000 description 1
- 102100039813 Inactive tyrosine-protein kinase 7 Human genes 0.000 description 1
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 1
- 102000000646 Interleukin-3 Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- 101710098610 Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 1
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 1
- 108010089836 Proto-Oncogene Proteins c-met Proteins 0.000 description 1
- 108091007561 SLC44A4 Proteins 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 108090000058 Syndecan-1 Proteins 0.000 description 1
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 1
- 101150117918 Tacstd2 gene Proteins 0.000 description 1
- 102100034902 Transmembrane 4 L6 family member 1 Human genes 0.000 description 1
- 101710190034 Trophoblast glycoprotein Proteins 0.000 description 1
- 241000722921 Tulipa gesneriana Species 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 102100023144 Zinc transporter ZIP6 Human genes 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 229950002903 bivatuzumab Drugs 0.000 description 1
- 229960003008 blinatumomab Drugs 0.000 description 1
- 229960000455 brentuximab vedotin Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 101150027734 cript gene Proteins 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 235000020960 dehydroascorbic acid Nutrition 0.000 description 1
- 239000011615 dehydroascorbic acid Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 229950009760 epratuzumab Drugs 0.000 description 1
- 229950009569 etaracizumab Drugs 0.000 description 1
- 229950009929 farletuzumab Drugs 0.000 description 1
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 102000006815 folate receptor Human genes 0.000 description 1
- 108020005243 folate receptor Proteins 0.000 description 1
- 229950002140 futuximab Drugs 0.000 description 1
- 229950000918 glembatumumab Drugs 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- 102000054751 human RUNX1T1 Human genes 0.000 description 1
- 229940076264 interleukin-3 Drugs 0.000 description 1
- 229950010939 iratumumab Drugs 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229950007752 isatuximab Drugs 0.000 description 1
- 230000000503 lectinlike effect Effects 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229950003734 milatuzumab Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 210000003061 neural cell Anatomy 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 229940126618 pankomab Drugs 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 238000009522 phase III clinical trial Methods 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229950007463 rovalpituzumab Drugs 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000002653 sulfanylmethyl group Chemical group [H]SC([H])([H])[*] 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 101150047061 tag-72 gene Proteins 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 229950009027 trastuzumab duocarmazine Drugs 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 229950000302 vadastuximab Drugs 0.000 description 1
- 229950006959 vorsetuzumab Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Cell Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Endocrinology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicinal Preparation (AREA)
- Saccharide Compounds (AREA)
Abstract
Un compuesto de Fórmula (II) **(Ver fórmula)**
Description
DESCRIPCIÓN
Proceso mejorado para la síntesis del fármaco-enlazador vc-seco
Campo de la invención
La presente invención se refiere a un proceso mejorado para la síntesis del fármaco-enlazador vc-seco-DUBA y sus intermediarios, así como al uso de dicho proceso mejorado en un proceso para preparar un conjugado anticuerpofármaco que comprende el fármaco-enlazador vc-seco-DUBA.
Antecedentes de la presente invención
Las duocarmicinas son miembros de una familia de antibióticos antitumorales que incluyen duocarmicina A, duocarmicina SA y CC-1065. Son conocidos por sus potentes propiedades antitumorales, pero normalmente no se usan solos debido a su toxicidad extremadamente alta. Actualmente, se exploran las duocarmicinas como fármacos citotóxicos en conjugados anticuerpo-fármaco (ADC).
Los ADC tienen el potencial de abordar la gran necesidad insatisfecha de nuevos tratamientos efectivos en el cáncer al dirigir el fármaco citotóxico altamente potente específicamente a las células cancerosas, lo que mejora así la eficacia mientras se reducen los efectos secundarios tóxicos sistémicos potenciales del fármaco de molécula pequeña.
Uno de los aspectos clave para el éxito comercial futuro de los ADC es un proceso para la síntesis del fármaco citotóxico y la construcción del fármaco-enlazador correspondiente, en el que se une un resto enlazador al fármaco citotóxi
Fármaco-enlazador vc-seco-DUBA de Fórmula (I)

descrito por primera vez en el documento WO2011/133039 como compuesto 18b en la pág. 210, 11. 21-27, es un ejemplo de un análogo de CC-1065 altamente potente. El ADC de vc-seco-DUBA con el anticuerpo anti-HER2 trastuzumab, es decir, SYD985 o (vic-) trastuzumab duocarmazina, se usó con éxito en varios estudios preclínicos (M.M.C. van der Lee y otros, Molecular Cancer Therapeutics, 2015, 14(3), 692-703; J. Black y otros, Molecular Cancer Therapeutics, 2016, 15 (8), 1900-1909) y ensayos clínicos de fase I (ClinicalTrials.gov NCT02277717). Actualmente, el tratamiento con SYD985 se compara directamente con el tratamiento elegido por el médico en un ensayo clínico de fase III en pacientes con cáncer de mama localmente avanzado o metastásico h ER2 positivo (TULIP; ClinicalTrials.gov NCT03262935).
La síntesis del fármaco-enlazador vc-seco-DUBA se describe en el documento WO2011/133039 como un proceso de cuatro etapas. La preparación de vc-seco-DUBA al seguir este proceso a escala de laboratorio de 50-100 mg proporcionó el fármaco-enlazador con un rendimiento global de sólo el 21-25 %. Las dos últimas etapas, es decir, las etapas 3 y 4 de este proceso, son cruciales para el rendimiento global de vc-seco-DUBA del proceso total, al mostrar un rendimiento combinado de sólo aproximadamente el 50 %. A escala industrial, el rendimiento de este proceso de cuatro etapas será aún menor.
Por tanto, existe la necesidad de un proceso mejorado para preparar vc-seco-DUBA. En particular, existe la necesidad de un proceso que sea eficiente en términos de rendimiento y pureza química, rentable en términos de reactivos y condiciones de reacción, y que sea adecuado para la producción a escala industrial.
Breve descripción de la presente invención
La presente invención se refiere a un proceso mejorado para la síntesis del fármaco-enlazador vc-seco-DUBA y sus intermediarios con condiciones de proceso que son adecuadas para la producción a escala industrial, y que proporciona el producto vc-seco -DUBA deseado con un rendimiento mejorado.
En un primer aspecto, la presente invención se refiere a un compuesto de Fórmula (II)

En un segundo aspecto, la invención proporciona un proceso que comprende hacer reaccionar un compuesto de Fórmula (III)

con cloruro de hidrógeno en 1,4-dioxano para formar un compuesto de Fórmula (II).
En un tercer aspecto, la invención se refiere al uso de un compuesto de Fórmula (II) en un proceso para preparar vcseco-DUBA de Fórmula (I)

En un cuarto aspecto, la invención se refiere al uso del proceso para la fabricación de vc-seco-DUBA en un proceso para la fabricación de un vc-seco -DUBA que contiene el conjugado de anticuerpo-fármaco.
Descripción detallada de la presente invención
Las duocarmicinas son una clase de toxinas relacionadas estructuralmente que se aislaron por primera vez de un caldo de cultivo de especies de Streptomyces. Son miembros de una familia de antibióticos antitumorales que incluyen duocarmicina A, duocarmicina SA y CC-1065. Las duocarmicinas se unen al surco menor del ADN y posteriormente causan una alquilación irreversible del ADN. Esto altera la arquitectura del ácido nucleico, lo que eventualmente conduce a la muerte de las células tumorales.
El documento WO2011/133039 describe específicamente el fármaco-enlazador vc-seco-DUBA altamente potente de Fórmula (I) (compuesto 18b en la pág. 210, 11. 21-27) que comprende un derivado de duocarmicina de c C-1065

La presente invención se refiere a un proceso mejorado para la producción de vc-seco-DUBA con un rendimiento sorprendentemente alto y que puede aplicarse con éxito a escala industrial.
La síntesis química del fármaco-enlazador vc-seco-DUBA en el Ejemplo 10 del documento WO2011/133039 se describe como un proceso de cuatro etapas

en donde PNP-Cl es cloroformiato de 4-nitrofenilo, Et3N es trietilamina, Boc es terc-butiloxicarbonilo, TFA es ácido trifluoroacético, CHCb es cloroformo y DMF es W,W-dimetilformamida.
En una escala de laboratorio de 50-100 mg, este proceso de cuatro etapas muestra un rendimiento general de solo 21-25 %. A escala industrial este rendimiento será significativamente menor.
El bajo rendimiento global de este proceso de cuatro etapas puede atribuirse en gran parte al bajo rendimiento combinado de sólo aproximadamente el 50 % a escala de laboratorio de las dos últimas etapas, es decir, las etapas 3 y 4. Los presentes inventores descubrieron sorprendentemente que un procedimiento modificado, que implicaba cambiar el reactivo ácido en la etapa 3, producía un nuevo intermediario que podía aislarse mediante cristalización. Inesperadamente, se encontró que este procedimiento modificado de la etapa 3 y el uso del nuevo intermediario resultó en un rendimiento considerablemente mayor de vc-seco-DUBA.
Normalmente, se introduce una etapa de cristalización en una síntesis química cuando es necesario aumentar la pureza del producto. Sin embargo, la introducción de dicha etapa suele reducir el rendimiento de dicho producto, ya que queda una cantidad considerable de producto en las aguas madres. Sorprendentemente, los presentes inventores encontraron que la introducción de una etapa de cristalización en la síntesis de vc-seco-DUBA como se describe en la presente descripción anteriormente, que conduce al nuevo intermediario de Fórmula (II)

no sólo condujo a un aumento en la pureza (de 94-96 % a >99,0 %), sino que también mostró un aumento inesperado y significativo en el rendimiento de vc-seco-DUBA (de 53 % a -79 %).
Por tanto, en una modalidad, la presente invención se refiere a un compuesto de Fórmula (II).
En una segunda modalidad, la presente invención se refiere a un proceso para preparar un compuesto de Fórmula ( II) que comprende hacer reaccionar un compuesto de Fórmula (III)

con cloruro de hidrógeno en 1,4-dioxano para formar el compuesto de Fórmula (II). Normalmente, el compuesto de Fórmula (III) se hace reaccionar con cloruro de hidrógeno al 10-20 % en masa en 1,4-dioxano. Preferiblemente, el compuesto de Fórmula (III) se hace reaccionar con 12-18 % de cloruro de hidrógeno en 1,4-dioxano, más preferiblemente con 15 % de cloruro de hidrógeno en 1,4-dioxano. Normalmente, la relación de masa del compuesto de Fórmula (III):HCl en 1,4-dioxano varía de 1:0,5 a 1:25. Preferiblemente, la relación de masa del compuesto de Fórmula (III):HCl en 1,4-dioxano varía de 1:1 a 1:10. Más preferiblemente de 1:5 a 1:10.
Normalmente, la cantidad de cloruro de hidrógeno es un exceso molar de la cantidad del compuesto de Fórmula (III). Preferiblemente, la cantidad de cloruro de hidrógeno es al menos 2 equivalentes molares de la cantidad del compuesto de Fórmula (III), más preferiblemente de 2 a 50 equivalentes.
Preferiblemente, dicha reacción tiene lugar en presencia de un removedor, tal como triisopropilsilano en agua y/o metanol. Dicha agua y/o metanol pueden estar presentes en una cantidad inferior al 25 % en masa de la masa total de disolvente, preferiblemente menos del 15 %, más preferiblemente menos del 10 %.
El compuesto de Fórmula (III) se puede preparar, como se describe, por ejemplo, en R.C. Elgersma y otros, Molecular Cáncer Therapeutics, 2015, 12(6), 1813-1835, mediante la reacción de un compuesto de Fórmula (IV)

con cloroformiato de 4-nitrofenilo para formar un compuesto de Fórmula (V)

seguido de hacer reaccionar el compuesto de Fórmula (V) con un compuesto de Fórmula (VI)

en presencia de 1-hidroxibenzotriazol hidratado para formar el compuesto de Fórmula (III).
Normalmente, la reacción del compuesto de Fórmula (IV) con cloroformiato de 4-nitrofenilo se realiza a una temperatura de 0 a 20 °C. Preferiblemente, la temperatura es de 0 a 10 °C, más preferiblemente de 0 a 6 °C, incluso más preferiblemente de 2 a 6 °C, más preferiblemente de 3 a 5 °C.
Los disolventes adecuados para usar en la preparación del compuesto de Fórmula (V) son, sin limitación, disolventes orgánicos, preferiblemente disolventes apróticos, más preferiblemente disolventes apróticos polares. Los disolventes preferidos son disolventes de éter, disolventes de amida o mezclas de los mismos. Los disolventes particularmente preferidos son tetrahidrofurano (THF), W,W-dimetilacetamida (DMA) o mezclas de los mismos. Lo más preferido es una mezcla de THF y DMA.
Las bases adecuadas para usar en la preparación del compuesto de Fórmula (V) son bases orgánicas, por ejemplo, aminas terciarias. Una base particularmente adecuada es Et3N.
Normalmente, la reacción del compuesto de Fórmula (V) con el compuesto de Fórmula (VI) se realiza a una temperatura de 0 a 20 °C. Preferiblemente, la temperatura es de 0 a 10 °C, más preferiblemente de 4 a 10 °C.
Los disolventes adecuados para usar en la preparación del compuesto de Fórmula (III) son, sin limitación, disolventes orgánicos, preferiblemente disolventes apróticos, disolventes polares o mezclas de los mismos. Los disolventes preferidos son disolventes de éter, disolventes de amida o mezclas de los mismos. Los disolventes particularmente preferidos son THF, DMA o mezclas de los mismos. Lo más preferido es una mezcla de THF y DMA.
El compuesto de Fórmula (IV) puede producirse mediante, o de forma análoga a, cualquier proceso adecuado conocido en la técnica anterior, por ejemplo, el proceso descrito en el Ejemplo 6a del documento WO2015/185142.
En otra modalidad, la presente invención se refiere al uso de un compuesto de Fórmula (II) para preparar vc-seco-DUBA.
En otra modalidad más, la presente invención se refiere a un proceso para preparar vc-seco-DUBA en el que un compuesto de Fórmula (II) se hace reaccionar con un compuesto de Fórmula (VII)

Inesperadamente, el rendimiento de vc-seco-DUBA aumentó aún más cuando el compuesto de Fórmula (II) se hizo reaccionar con el compuesto de Fórmula (VII) mediante el uso de W,W-diisopropilamina (DIPEA) como base en lugar de trietilamina (Et3N), que se usó en el Ejemplo 10 del documento WO2011/133039. Normalmente, la relación molar del compuesto de Fórmula (II):DIPEA varía de 1:1 a 1:15. Preferiblemente, la relación varía de 1:1 a 1:10, más preferiblemente de 1:2 a 1:7, incluso más preferiblemente de 1:3 a 1:5, más preferiblemente la relación es aproximadamente 1:4.
Normalmente, la reacción del compuesto de Fórmula (II) con el compuesto de Fórmula (VII) se realiza a una temperatura de 0 a 20 °C. Preferiblemente, la temperatura es de 0 a 10 °C, más preferiblemente de 0 a 5 °C.
Los disolventes adecuados para usar en la reacción de Fórmula (II) con el compuesto de Fórmula (VII) para preparar vc-seco-DUBA son, sin limitación, disolventes orgánicos, preferiblemente disolventes apróticos, más preferiblemente disolventes apróticos polares. Los disolventes preferidos son disolventes de éter, disolventes de amida o mezclas de los mismos. Los disolventes particularmente preferidos son THF, DMA, W,W-dimetilformamida (DMF) o mezclas de los mismos. El más preferido es DMA.
En una modalidad preferida, el proceso se realiza en presencia de hidrato de 1-hidroxibenzotriazol. Normalmente, la relación molar del compuesto de Fórmula (II): hidrato de 1-hidroxibenzotriazol varía de 1:1 a 1:10. Preferiblemente, la relación varía de 1:1 a 1:7, más preferiblemente de 1:2 a 1:5, incluso más preferiblemente de 1:2 a 1:3, más preferiblemente la relación es aproximadamente 1:2,5.
La presente invención se refiere adicionalmente a un proceso para la preparación de un ADC vc-seco-DUBA de Fórmula (VIII)
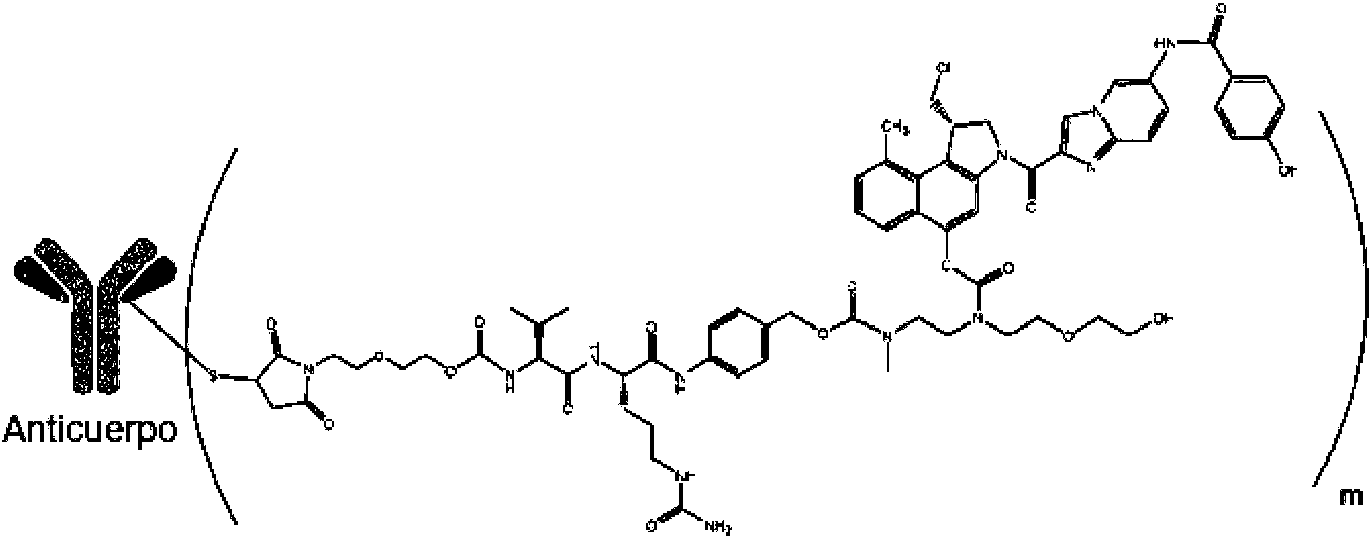
en donde el compuesto fármaco-enlazador vc-seco-DUBA se prepara con el proceso de acuerdo con la invención como se describe en la presente descripción anteriormente.
m representa una relación promedio de fármaco a anticuerpo (DAR) de 1 a 8, preferiblemente de 1 a 6, más preferiblemente de 1 a 4.
En el contexto de la presente invención, cualquier anticuerpo, en particular cualquier anticuerpo que se sepa que tiene actividad terapéutica o cualquier anticuerpo conocido en la técnica de los ADC, o cualquier fragmento de unión a antígeno del mismo, por ejemplo, un fragmento F(ab')2 o Fab', un anticuerpo monocatenario (sc), un scFv, un anticuerpo de dominio único (sd), un diacuerpo o un minicuerpo, se pueden usar para la conjugación (de tipo salvaje o específico de sitio) de vc-seco-DUBA. Los anticuerpos pueden ser de cualquier isotipo, tales como los anticuerpos
IgG, IgA o IgM. Preferiblemente, el anticuerpo es un anticuerpo IgG, más preferiblemente un anticuerpo IgGi o IgG2. Los anticuerpos pueden ser quiméricos, humanizados o humanos. Preferiblemente, los anticuerpos son humanizados. Incluso más preferiblemente, el anticuerpo es un anticuerpo IgG humanizado o humano, más preferiblemente un anticuerpo monoclonal IgG1 (mAb) humanizado o humano. Preferiblemente, dicho anticuerpo tiene cadenas ligeras k (kappa), es decir, un anticuerpo IgG1-K humanizado o humano.
En los anticuerpos humanizados, las regiones determinantes de complementariedad de unión a antígeno (CDR) en las regiones variables de la cadena pesada (HC) y la cadena ligera (LC) se derivan de anticuerpos de una especie no humana, comúnmente ratón, rata o conejo. Estas CDR no humanas se pueden colocar dentro de un marco humano (región marco (FR) FR1, FR2, FR3 y FR4) de las regiones variables de HC y LC. Los aminoácidos seleccionados en las FR humanas pueden intercambiarse por los correspondientes aminoácidos originales de especies no humanas, por ejemplo, para mejorar la afinidad de unión, mientras se conserva una baja inmunogenicidad. Alternativamente, los marcos no humanos se retienen y los aminoácidos seleccionados de las FR de especies no humanas pueden intercambiarse por sus correspondientes aminoácidos humanos para reducir la inmunogenicidad, mientras se conserva la afinidad de unión del anticuerpo. Las regiones variables así humanizadas se combinan con regiones constantes humanas.
Estos anticuerpos se pueden producir de forma recombinante, sintética o mediante otros métodos adecuados conocidos en la técnica.
Normalmente, el anticuerpo es un anticuerpo monoespecífico (es decir, específico para un antígeno; tal antígeno puede ser común entre especies o tener secuencias de aminoácidos similares entre especies) o biespecífico (es decir, específico para dos antígenos diferentes de una especie) que comprende al menos una región variable HC y LC que se une a una diana seleccionada del grupo que consiste en anexina A1, B7H4, CA6, CA9, CA15-3, CA19-9, CA27-29, CA125, CA242 (antígeno canceroso 242), CCR2, CCR5, CD2, CD19, CD20, CD22, CD30 (factor de necrosis tumoral 8), CD33, CD37, CD38 (ADP ribosa hidrolasa cíclica), CD40, CD44, CD47 (proteína asociada a integrina), CD56 (molécula de adhesión de células neurales), CD70, CD74, CD79, CD115 (receptor del factor estimulante de colonias 1), CD123 (receptor de interleucina-3), CD138 (Sindecán 1), CD203c (ENPP3), CD303, CD333, CEA, CEACAM, CLCA-1 (molécula tipo lectina similar a C-1), CLL-1, c-MET (receptor del factor de crecimiento de hepatocitos), Cripto, DLL3, EGFL, EGFR, EPCAM, EPh (por ejemplo, EphA2 o EPhB3), ETBR (receptor de endotelina tipo B), FAP, FcRL5 (proteína similar al receptor Fc 5, CD307), FGFR (por ejemplo, FGFR3), FOLR1 (receptor de folato alfa), GCC (guanilil ciclasa C), GPNMB, HER2, HMW-MAA (antígeno asociado a melanoma de alto peso molecular), integrina a (por ejemplo, avp3 y avp5), IGF1R, TM4SF1 (o antígeno L6), carbohidrato similar a Lewis A, Lewis X, Lewis Y (CD174), LIV1, mesotelina (MSLN), MN (CA9), MUC1, MUC16, NaPi2b, Nectina-4, PD-1, PD-L1, PSMA, PTK7, SLC44A4, STEAP-1, antígeno 5T4 (o TPb G, glicoproteína trofoblasto), TF (factor tisular, tromboplastina, CD142), TF-Ag, Tag72, TNFR, TROP2 (transductor de señal de calcio 2 asociado a tumor), VEGFR y VLA.
Los ejemplos de anticuerpos adecuados incluyen blinatumomab (CD19), epratuzumab (CD22), iratumumab y brentuximab (CD30), vadastuximab (CD33), tetulumab (CD37), isatuximab (CD38), bivatuzumab (CD44), lorvotuzumab (CD56), vorsetuzumab (CD70), milatuzumab (CD74), polatuzumab (CD79), rovalpituzumab (DLL3), futuximab (EGFR), oportuzumab (EPCAM), farletuzumab (FOLR1), glembatumumab (GPNMB), trastuzumab y pertuzumab (HER2), etaracizumab (integrina), anetumab (mesotelina), pankomab (MUC1), enfortumab (Nectina-4) y H8, A1 y A3 (antígeno 5T4).
La conjugación del fármaco-enlazador vc-seco-DUBA con el anticuerpo puede realizarse como se describe, por ejemplo, en los documentos WO2011/133039, WO2015/177360 y WO2017/137628.
Los ADC de tipo salvaje se producen al conjugar el fármaco-enlazador con el anticuerpo a través de los tioles libres de las cadenas laterales de las cisteínas generadas mediante la reducción de los enlaces disulfuro intercatenarios. La fabricación implica la reducción parcial de los disulfuros intercatenarios expuestos al disolvente seguida de la modificación de los tioles resultantes con fármacos-enlazadores que contienen maleimida. La estrategia de unión de cisteína resulta en un máximo de dos fármacos por disulfuro reducido. La mayoría de las moléculas de IgG humanas tienen cuatro enlaces disulfuro expuestos al disolvente, por lo que es posible un intervalo de cero a ocho fármacos por anticuerpo. El número exacto de fármacos por anticuerpo se determina mediante el grado de reducción de disulfuro y el número de equivalentes molares de fármaco-enlazador usados en la reacción de conjugación siguiente. La reducción completa de los cuatro enlaces disulfuro da una construcción homogénea con ocho fármacos por anticuerpo, mientras que una reducción parcial normalmente resulta en una mezcla heterogénea con cero, dos, cuatro, seis u ocho fármacos por anticuerpo.
Los ADC específicos de sitio se producen mediante la conjugación del fármaco-enlazador al anticuerpo a través de las cadenas laterales de residuos de cisteína modificados genéticamente en posiciones adecuadas del anticuerpo mutado. Las cisteínas modificadas genéticamente suelen estar tapadas por otros tioles, como la cisteína o el glutatión, para formar disulfuros. Estos residuos tapados deben destaparse antes de que se produzca la unión del fármaco. La unión del fármaco a los residuos modificados genéticamente se logra mediante la reducción tanto de los disulfuros intercatenarios nativos como mutantes, luego al reoxidar las cisteínas intercatenarias nativas mediante el uso de un oxidante suave tal como CuSO4 o ácido deshidroascórbico, seguido de la conjugación estándar de la cisteína
modificada genéticamente sin tapar con un fármaco-enlazador, o mediante el uso de agentes reductores suaves que reducen los disulfuros mutantes a una velocidad mayor que los enlaces disulfuro intercatenarios, seguido de la conjugación estándar de la cisteína modificada genéticamente sin tapar con un fármaco-enlazador. En condiciones óptimas, se unirán dos fármacos por anticuerpo (es decir, la relación de fármaco a anticuerpo, DAR, es 2) (si una cisteína se modifica genéticamente en la cadena pesada o la cadena ligera del mAb).
En una modalidad preferida, el anticuerpo a usar de acuerdo con la presente invención es un anticuerpo anti-HER2, incluso más preferido el anticuerpo anti-HER2 trastuzumab.
En una modalidad particular, la presente invención se refiere a un proceso para la preparación de un ADC trastuzumab vc-seco-DUBA de Fórmula (IX)

en donde el compuesto fármaco-enlazador vc-seco-DUBA se prepara con el proceso de acuerdo con la invención como se describe en la presente descripción anteriormente. 2,6-2,9 representa un DAR promedio de 2,6-2,9.
Ejemplos
Ejemplo 1 - Preparación de metilCBI-azaindol-benzamida-MOM-Boc-etilendiamina-D (4)

Se hizo reaccionar metilCBI-azaindol-benzamida-MOM (1) (1,0 g, 1,75 mmol) con cloroformiato de 4-nitrofenilo (PNP-Cl) (0,43 g, 2,12 mmol) en una mezcla de tetrahidrofurano (THF) (4,5 g) y W,W-dimetilacetamida (DMA) (3,0 g) en presencia de trietilamina (Et3N) (0,55 g, 4,94 mmol) durante aproximadamente 1,5 horas a una temperatura de 0 °C y se dejó calentar hasta 6 °C. Se obtuvo una suspensión que comprendía metilCBI-azaindol-benzamida-MOM-PNP (2).
En la segundo etapa, se disolvió (2-((2-(2-hidroxietoxi)etil) amino)etil)(metil)-carbamato de tere- butilo (3) (0,58 g, 2,19 mmol) en DMA (1,7 g) y se añadió hidrato de 1-hidroxibenzotriazol (HOBt) (0,35 g, 2,28 mmol). Esta solución obtenida se hizo reaccionar con la suspensión durante 1,5 horas a una temperatura de 4 °C y se dejó calentar hasta 10 °C.
Una vez completada la reacción, se añadió acetato de etilo (EtOAc) (8,8 g) a la mezcla de reacción y la solución se lavó con salmuera (11,3 g), solución saturada de bicarbonato de sodio (3,8 g) y nuevamente con salmuera (3,8 g). La capa orgánica se separó y se purificó mediante filtración con carbón. El disolvente se evaporó en un evaporador
rotatorio de vacío. La metilCBI-azaindol-benzamida-MOM-Boc-etilendiamina-D (4) obtenida se disolvió en acetona (20 g) y, finalmente, se purificó de nuevo mediante filtración con carbón.
El producto crudo se purificó mediante cromatografía en columna de gel de sílice, al eluir con una fase móvil -DCM:MeOH = 97:3 a 94:6. Las fracciones de producto combinadas se concentraron y se secaron al vacío para producir metilCBI-azaindol-benzamida-MOM-Boc-etilendiamina-D (4) (1,27 g, 1,48 mmol; 84 % de rendimiento, 93,82 % de pureza).
Ejemplo 2 - Preparación de vc-seco-DUBA
Preparación de clorhidrato de metilCBI-azaindol-benzamida-etilendiamina-D (5)

Los grupos metoximetilo (MOM) y terc- butiloxicarbonilo (Boc) de metilCBI-azaindol-benzamida-MOM-Bocetilendiamina-D (4) (1,27 g, 1,48 mmol) se eliminaron con cloruro de hidrógeno (HCl) al 15 % en 1,4-dioxano (7,5 g) en presencia de un removedor (triisopropilsilano (0,63 g), agua (0,4 g) y metanol (0,3 g)). El clorhidrato de metilCBI-azaindol-benzamida-etilendiamina-D (5) cristalizó a partir de la solución de reacción como un sólido amarillo.
El sólido amarillo obtenido se filtró, se lavó con acetona y se secó sobre el filtro mediante el uso de nitrógeno y vacío y proporcionó un producto puro (5) (1,0 g, 1,33 mmol; 90 % de rendimiento, > 90 % de pureza).
Preparación de vc-seco-DUBA

Se hizo reaccionar clorhidrato de metilCBI-azaindol-benzamida-etilendiamina-D (5) (1,0 g, 1,33 mmol) durante 1,5 horas en la oscuridad a una temperatura de 0 °C y se dejó calentar hasta 5 °C con maleimida-OEG2-val-cit-PABA-PNP (6) (0,98 g, 1,29 mmol) en DMA (17,8 g) en presencia de W,W-diisopropilamina (DIPEA) (0,65 g, 5,10 mmol) y HOBt (0,47 g, 3,16 mmol). La mezcla de reacción se añadió gota a gota a agua (201,1 g) a una temperatura de 23 a 25 °C (50 a 60 min) y se obtuvo un precipitado de producto crudo vc-seco-DUBA. Después de 30 min de agitación, el producto crudo precipitado se filtró en un filtro de presión. La torta del filtro se lavó a fondo con agua y se secó en el filtro al vacío y con una ligera corriente de nitrógeno.
El producto crudo vc-seco-DUBA se sometió primero a cromatografía ultrarrápida a baja presión (fase estacionaria -gel de sílice de 0,040 a 0,063 mm; fase móvil - diclorometano:metanol = 90:10). Las fracciones que cumplen (con pureza UPLC-IN de vc-seco-DUBA > 90 %) se recogieron en un matraz, se filtraron y se evaporaron. Se realizó una purificación adicional mediante cromatografía preparativa (fase estacionaria - gel de sílice de 0,015 a 0,040 mm; fase móvil - diclorometano:metanol = 90:10 a 85:15). Las fracciones que cumplen (con pureza UPLC-IN de vc-seco-DUBA > 90 %) se recogieron en un matraz y el disolvente se cambió a DMA. La concentración se realizó a una temperatura máxima de 25 °C. Las soluciones concentradas se combinaron, se filtraron mediante un filtro de 0,2 pm y se añadieron a agua para precipitar vc-seco-DUBA puro como un polvo amarillo fino (rendimiento: 35-45 %; pureza: > 99,0 %).
El producto se filtró, se lavó con agua y se secó en el filtro mediante el uso de nitrógeno y vacío a una temperatura máxima de 25 °C.
Ejemplo comparativo - Preparación de vc-seco-DUBA
La síntesis de vc-seco-DUBA se realiza al seguir el procedimiento descrito en el Ejemplo 10 del documento WO2011/133039.

Etapa 1
MetilCBI-azaindol-benzamida-MOM-Boc-etilendiamina-D (4) (0,1 mmol) se suspendió en cloroformo (CHCb) (6 ml) y se enfrió en hielo. Se añadieron 2 ml de ácido (ácido trifluoroacético (TFA) o HCl al 15 % en 1,4 dioxano (7,5 g)) y la mezcla se agitó durante 3 h. Después, la mezcla se concentró al vacío.
Etapa 2
El residuo se disolvió en W,W-dimetilformamida (DMF) (4 ml), la solución se enfrió en hielo y se añadieron maleimida-OEG2 val-cit-PABA-PNP (6) (0,13 mmol) y la base (1 mmol, Et3N o DIPEA). La mezcla se agitó durante 2 horas, se concentró al vacío y el residuo se purificó mediante cromatografía en columna (SO 2 , diclorometano:metanol, 1:0 a
El procedimiento anterior se realizó mediante el uso de HCl en 1,4-dioxano o el ácido TFA de la técnica anterior para eliminar los grupos MOM y Boc de metilCBI-azaindol-benzamida-MOM-Boc-etilendiamina-D (4) en la primera etapa y mediante el uso ya sea de DIPEA o la base Et3N de la técnica anterior para facilitar la reacción de acoplamiento de clorhidrato de metilCBI-azaindol-benzamida-etilendiamina-D (5) y maleimida-OEG2-val-cit-PABA-PNP (6) en la etapa 2 en orden de determinar la influencia de la elección del ácido y la base en la eficiencia de la preparación de vc-seco-DUBA.
La siguiente tabla muestra el rendimiento de vc-seco-DUBA.

El uso de HCl en 1,4-dioxano en lugar de TFA en la etapa 1 resultó en un aumento del 25,8 % en el rendimiento global de vc-seco-DUBA determinado mediante HPLC. El uso de DIPEA en lugar de Et3N en la etapa 2 resultó en una disminución del 23,6 % en el rendimiento global de vc-seco-DUBA determinado mediante HPLC. Sin embargo, el uso de HCl en 1,4-dioxano en la etapa 1 y DIPEA en la etapa 2 resultó en un aumento del 29,8 % en el rendimiento global de vc-seco-DUBA determinado mediante HPLC.
Claims (1)
- REIVINDICACIONESUn compuesto de Fórmula (II)
 Un proceso que comprende hacer reaccionar un compuesto de Fórmula (III)
Un proceso que comprende hacer reaccionar un compuesto de Fórmula (III) con cloruro de hidrógeno en 1,4-dioxano para formar el compuesto de Fórmula (II) de acuerdo con la reivindicación 1.El uso del compuesto de Fórmula (II) de acuerdo con la reivindicación 1 para preparar vc-seco-DUBA de Fórmula (I)
con cloruro de hidrógeno en 1,4-dioxano para formar el compuesto de Fórmula (II) de acuerdo con la reivindicación 1.El uso del compuesto de Fórmula (II) de acuerdo con la reivindicación 1 para preparar vc-seco-DUBA de Fórmula (I) Un proceso para la síntesis de vc-seco-DUBA de Fórmula (I)
Un proceso para la síntesis de vc-seco-DUBA de Fórmula (I) que comprende hacer reaccionar el compuesto de Formula (II) de acuerdo con la reivindicación 1 con un compuesto de Fórmula (VII)
que comprende hacer reaccionar el compuesto de Formula (II) de acuerdo con la reivindicación 1 con un compuesto de Fórmula (VII) para formar el compuesto de Fórmula (I).5. El proceso de acuerdo con la reivindicación 4, en donde la reacción del compuesto de Fórmula (II) y el compuesto de Fórmula (VII) se realiza en presencia de W,W-diisopropilamina.6. El proceso de acuerdo con la reivindicación 4, en donde la reacción del compuesto de Fórmula (II) y el compuesto de Fórmula (VII) se realiza en W,W-dimet¡lacetamida en presencia de W,W-diisopropilamina e hidrato de 1-hidroxibenzotriazol.7. El proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 6, en donde el compuesto de Fórmula (II) se prepara al hacer reaccionar un compuesto de Fórmula (III)
para formar el compuesto de Fórmula (I).5. El proceso de acuerdo con la reivindicación 4, en donde la reacción del compuesto de Fórmula (II) y el compuesto de Fórmula (VII) se realiza en presencia de W,W-diisopropilamina.6. El proceso de acuerdo con la reivindicación 4, en donde la reacción del compuesto de Fórmula (II) y el compuesto de Fórmula (VII) se realiza en W,W-dimet¡lacetamida en presencia de W,W-diisopropilamina e hidrato de 1-hidroxibenzotriazol.7. El proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 6, en donde el compuesto de Fórmula (II) se prepara al hacer reaccionar un compuesto de Fórmula (III) con cloruro de hidrógeno en 1,4-dioxano para formar el compuesto de Fórmula (II).8. El proceso de acuerdo con la reivindicación 7, en donde el compuesto de Fórmula (III) se prepara al hacer reaccionar un compuesto de Fórmula (IV)
con cloruro de hidrógeno en 1,4-dioxano para formar el compuesto de Fórmula (II).8. El proceso de acuerdo con la reivindicación 7, en donde el compuesto de Fórmula (III) se prepara al hacer reaccionar un compuesto de Fórmula (IV) con cloroformiato de 4-nitrofenilo para formar un compuesto de Fórmula (V)
con cloroformiato de 4-nitrofenilo para formar un compuesto de Fórmula (V) seguido de hacer reaccionar el compuesto de Fórmula (V) con un compuesto de Fórmula (VI)
seguido de hacer reaccionar el compuesto de Fórmula (V) con un compuesto de Fórmula (VI) en presencia de 1-hidroxibenzotriazol hidratado para formar el compuesto de Fórmula (III).9. Un proceso para la síntesis de un conjugado anticuerpo-fármaco de Fórmula (VIII)
en presencia de 1-hidroxibenzotriazol hidratado para formar el compuesto de Fórmula (III).9. Un proceso para la síntesis de un conjugado anticuerpo-fármaco de Fórmula (VIII) que comprende el proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 8 para formar el compuesto de Fórmula (I), seguido de la conjugación del compuesto de Fórmula (I) con un anticuerpo o un fragmento de unión a antígeno del mismo,en donde Anticuerpo es un anticuerpo o un fragmento de unión a antígeno del mismo y m representa una relación promedio de fármaco a anticuerpo de 1 a 8, preferiblemente de 1 a 6, más preferiblemente de 1 a 4.10. El proceso de acuerdo con la reivindicación 9, en donde el compuesto de Fórmula (I) se conjuga con el anticuerpo anti-HER2 trastuzumab.11. Un proceso para la síntesis de un conjugado anticuerpo-fármaco de Fórmula (IX)
que comprende el proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 8 para formar el compuesto de Fórmula (I), seguido de la conjugación del compuesto de Fórmula (I) con un anticuerpo o un fragmento de unión a antígeno del mismo,en donde Anticuerpo es un anticuerpo o un fragmento de unión a antígeno del mismo y m representa una relación promedio de fármaco a anticuerpo de 1 a 8, preferiblemente de 1 a 6, más preferiblemente de 1 a 4.10. El proceso de acuerdo con la reivindicación 9, en donde el compuesto de Fórmula (I) se conjuga con el anticuerpo anti-HER2 trastuzumab.11. Un proceso para la síntesis de un conjugado anticuerpo-fármaco de Fórmula (IX) que comprende el proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 8 para formar el compuesto de Fórmula (I), seguido de la conjugación del compuesto de Fórmula (I) con el anticuerpo anti-HER2 trastuzumab, en donde 2,6-2,9 representa una relación promedio de fármaco a anticuerpo de 2,6 a 2,9.
que comprende el proceso de acuerdo con una cualquiera de las reivindicaciones 4 a 8 para formar el compuesto de Fórmula (I), seguido de la conjugación del compuesto de Fórmula (I) con el anticuerpo anti-HER2 trastuzumab, en donde 2,6-2,9 representa una relación promedio de fármaco a anticuerpo de 2,6 a 2,9.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17203457 | 2017-11-24 | ||
| PCT/EP2018/082199 WO2019101850A1 (en) | 2017-11-24 | 2018-11-22 | Improved process for the synthesis of linker-drug vc-seco-duba |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2889775T3 true ES2889775T3 (es) | 2022-01-13 |
Family
ID=60452506
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18811180T Active ES2889775T3 (es) | 2017-11-24 | 2018-11-22 | Proceso mejorado para la síntesis del fármaco-enlazador vc-seco |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US11633492B2 (es) |
| EP (1) | EP3713939B1 (es) |
| JP (1) | JP7279042B2 (es) |
| KR (1) | KR102728434B1 (es) |
| CN (1) | CN111556870B (es) |
| AU (1) | AU2018371966B2 (es) |
| CY (1) | CY1124480T1 (es) |
| DK (1) | DK3713939T3 (es) |
| ES (1) | ES2889775T3 (es) |
| HR (1) | HRP20211435T1 (es) |
| HU (1) | HUE055305T2 (es) |
| LT (1) | LT3713939T (es) |
| PL (1) | PL3713939T3 (es) |
| PT (1) | PT3713939T (es) |
| WO (1) | WO2019101850A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4175672A1 (en) * | 2020-07-06 | 2023-05-10 | Byondis B.V. | Antifolate linker-drugs and antibody-drug conjugates |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7714016B2 (en) | 2005-04-08 | 2010-05-11 | Medarex, Inc. | Cytotoxic compounds and conjugates with cleavable substrates |
| CN102695545B (zh) | 2009-09-08 | 2016-08-17 | 特色疗法股份有限公司 | 包括酶可剪切的酮改性的阿片样物质药物前体以及其可任选的抑制剂的组合物 |
| PT3056203T (pt) | 2010-04-21 | 2018-02-15 | Syntarga Bv | Conjugados de análogos de cc-1065 e ligantes bifuncionais |
| SG11201609372UA (en) * | 2014-05-22 | 2016-12-29 | Synthon Biopharmaceuticals Bv | Site-specific conjugation of linker drugs to antibodies and resulting adcs |
| US9890159B2 (en) | 2014-06-05 | 2018-02-13 | Synthon Biopharmaceuticals B.V. | Process for making duocarmycin prodrugs |
| JP2015199772A (ja) | 2015-06-25 | 2015-11-12 | シグネーチャー セラピューティクス,インク.Signature Therapeutics, Inc. | 酵素切断可能なオピオイドプロドラッグとそのインヒビターとを含んでなる組成物 |
| CA3026139C (en) | 2016-02-12 | 2023-09-12 | Synthon Biopharmaceuticals B.V. | Selective reduction of cysteine-engineered antibodies |
| CA3044898C (en) | 2016-11-25 | 2022-04-05 | Mabwell (shanghai) Bioscience Co., Ltd. | Di-substituted maleic amide linker for antibody-drug conjugating and preparation method and use thereof |
-
2018
- 2018-11-22 PT PT188111801T patent/PT3713939T/pt unknown
- 2018-11-22 ES ES18811180T patent/ES2889775T3/es active Active
- 2018-11-22 KR KR1020207018101A patent/KR102728434B1/ko active Active
- 2018-11-22 HR HRP20211435TT patent/HRP20211435T1/hr unknown
- 2018-11-22 JP JP2020528269A patent/JP7279042B2/ja active Active
- 2018-11-22 US US16/766,150 patent/US11633492B2/en active Active
- 2018-11-22 DK DK18811180.1T patent/DK3713939T3/da active
- 2018-11-22 WO PCT/EP2018/082199 patent/WO2019101850A1/en not_active Ceased
- 2018-11-22 HU HUE18811180A patent/HUE055305T2/hu unknown
- 2018-11-22 CN CN201880075808.2A patent/CN111556870B/zh active Active
- 2018-11-22 LT LTEPPCT/EP2018/082199T patent/LT3713939T/lt unknown
- 2018-11-22 EP EP18811180.1A patent/EP3713939B1/en active Active
- 2018-11-22 AU AU2018371966A patent/AU2018371966B2/en active Active
- 2018-11-22 PL PL18811180T patent/PL3713939T3/pl unknown
-
2021
- 2021-09-06 CY CY20211100786T patent/CY1124480T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CY1124480T1 (el) | 2022-07-22 |
| US20200368362A1 (en) | 2020-11-26 |
| JP2021504351A (ja) | 2021-02-15 |
| HUE055305T2 (hu) | 2021-11-29 |
| KR102728434B1 (ko) | 2024-11-08 |
| CN111556870B (zh) | 2023-09-29 |
| AU2018371966A1 (en) | 2020-06-18 |
| US11633492B2 (en) | 2023-04-25 |
| CN111556870A (zh) | 2020-08-18 |
| CA3083169A1 (en) | 2019-05-31 |
| PL3713939T3 (pl) | 2021-12-13 |
| RU2020120838A3 (es) | 2022-03-03 |
| WO2019101850A1 (en) | 2019-05-31 |
| KR20200091431A (ko) | 2020-07-30 |
| LT3713939T (lt) | 2021-10-25 |
| JP7279042B2 (ja) | 2023-05-22 |
| AU2018371966B2 (en) | 2021-02-25 |
| EP3713939B1 (en) | 2021-07-21 |
| HRP20211435T1 (hr) | 2022-01-07 |
| RU2020120838A (ru) | 2021-12-24 |
| PT3713939T (pt) | 2021-09-16 |
| EP3713939A1 (en) | 2020-09-30 |
| DK3713939T3 (da) | 2021-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI819474B (zh) | 細胞毒性苯并二氮呯衍生物 | |
| JP2022512057A (ja) | 抗体薬物複合体のためのリンカーおよびその使用 | |
| JP2023532380A (ja) | 二成分毒素を担った抗体薬物複合体及びその応用 | |
| CA3208591A1 (en) | Anti-her2 antibody-drug conjugates and uses thereof | |
| JP7811217B2 (ja) | 抗her2抗体-免疫アゴニストコンジュゲート及びその使用 | |
| TW202506191A (zh) | 包含可切割連接子的軛合物 | |
| KR102858995B1 (ko) | 설포말레이미드계 링커 및 상응하는 컨쥬게이트 | |
| CN119095625A (zh) | 抗体-药物偶联物 | |
| JP2025533983A (ja) | リンカー-ペイロード化合物、複合体、及びその用途 | |
| ES2889775T3 (es) | Proceso mejorado para la síntesis del fármaco-enlazador vc-seco | |
| CN119156232A (zh) | 抗体-药物偶联物前体及其合成中间体 | |
| US20230149558A1 (en) | Duocarmycin derivative and use thereof | |
| CA3083169C (en) | Improved process for the synthesis of linker-drug vc-seco-duba | |
| WO2024024935A1 (ja) | 抗腫瘍効果を有する抗体薬物複合体の新規製造方法 | |
| CA3236102A1 (en) | Pyridazinedione-based heterobicyclic covalent linkers and methods and applications thereof | |
| RU2777160C2 (ru) | Улучшенный способ синтеза линкер-лекарственного средства vc-seco-duba | |
| WO2026007511A1 (en) | Drug conjugates, their preparation methods and uses | |
| RU2815199C2 (ru) | Линкеры на основе сульфомалеимида и соответствующие конъюгаты | |
| WO2026044149A2 (en) | Synthesis of drug-linker combinations for antibody-drug conjugates | |
| WO2024248944A1 (en) | Topoisomerase inhibitors, methods of making, and methods of use thereof | |
| CN120957758A (zh) | 大环化合物及其制备方法和用途 | |
| CN120957759A (zh) | 具有抗肿瘤作用的化合物及其制备方法和用途 | |
| KR20190143246A (ko) | 신규한 링커 화합물 및 이를 포함하는 위치 특이적인 항체-약물 결합체 화합물 |