ES2907651T3 - Enlazadores covalentes en conjugados de anticuerpo-fármaco y métodos para obtener y usar los mismos - Google Patents
Enlazadores covalentes en conjugados de anticuerpo-fármaco y métodos para obtener y usar los mismos Download PDFInfo
- Publication number
- ES2907651T3 ES2907651T3 ES16837634T ES16837634T ES2907651T3 ES 2907651 T3 ES2907651 T3 ES 2907651T3 ES 16837634 T ES16837634 T ES 16837634T ES 16837634 T ES16837634 T ES 16837634T ES 2907651 T3 ES2907651 T3 ES 2907651T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- antibody
- mixture
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68033—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a maytansine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0205—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-(X)3-C(=0)-, e.g. statine or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06052—Val-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/02—Linear peptides containing at least one abnormal peptide link
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Cell Biology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Reproductive Health (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un enlazador-agente activo o una sal farmacéuticamente aceptable del mismo, en el que el enlazador-agente activo es **(Ver fórmula)** o **(Ver fórmula)**
Description
DESCRIPCIÓN
Enlazadores covalentes en conjugados de anticuerpo-fármaco y métodos para obtener y usar los mismos
REFERENCIA CRUZADA A LA SOLICITUD RELACIONADA
La presente solicitud reivindica el beneficio de la Solicitud Provisional U.S. Serie No. 62/205.121, presentada el 14 de agosto de 2016.
ANTECEDENTES DE LA INVENCIÓN
Los conjugados de anticuerpo-fármaco (ADC), una clase de terapias dirigidas en rápido crecimiento, representan un nuevo enfoque prometedor para mejorar tanto la selectividad como la actividad citotóxica de los fármacos contra el cáncer. Un ejemplo de fármaco de tipo ADC que se ha aprobado para uso terapéutico en los EE.UU. es brentuximab vedotin (ADCETRIS®), un anticuerpo quimérico anti-CD30 conjugado con monometil auristatina E usado en el tratamiento del linfoma anaplásico de células grandes y el linfoma de Hodgkin.
Un método convencional empleado en el diseño de conjugados de anticuerpo-fármaco (ADC) implica el acoplamiento de moléculas de fármaco a grupos tiol de cadenas de anticuerpos a través de un resto enlazante. Los grupos tiol libres se obtienen como resultado de la ruptura de los enlaces de disulfuro intercatenarios de cisteína de un anticuerpo a través de una reacción de reducción. Un anticuerpo típico contiene 4 enlaces de disulfuro intercatenarios (2 entre las cadenas pesadas y 2 entre las cadenas pesada y ligera). Estos disulfuros intercatenarios se pueden reducir selectivamente con ditiotreitol, tris(2-carboxietil)fosfina, u otros agentes reductores suaves, para dar como resultado 8 grupos sulfhidrilo reactivos para la conjugación. Este método puede enlazar hasta ocho moléculas de fármaco a un anticuerpo determinado.
Debido al hecho de que se rompen al menos dos enlaces de disulfuro, los ADC diseñados con este principio son inestables una vez que entran en circulación, y de este modo se acortará la vida media de los ADC. Como resultado, el desarrollo reciente en el diseño y la síntesis de ADC adopta un enfoque diferente, a saber, aquel que se basa en conectar covalentemente dos grupos tiol mediante un agente de acoplamiento, estableciendo así puentes de tiol entre las dos cadenas pesadas y entre las cadenas pesada y ligera de un anticuerpo dado. Los esfuerzos de investigación actuales que exploran este enfoque se centran principalmente en diseñar la estructura de un agente de acoplamiento que no solo tiene las funcionalidades para unir dos grupos tiol de forma covalente, sino que también incluye los componentes necesarios para facilitar actividades biológicas específicas.
Estudios anteriores en el campo han usado bis-maleimidas para reaccionar con los dos grupos tiol resultantes de un enlace de disulfuro roto. El acoplamiento covalente entre maleimidas y tioles es una reacción clásica de conversión de alquenos. Las reacciones de formación puentes de tiol estudiadas más recientemente también se basan en este principio, implicando las reacciones ejemplares a maleimidas, bis-maleimidas, y maleimidas con sustituyentes de halógeno. Sin embargo, hasta la fecha, las composiciones de enlazadores formadores de puentes de tiol se limitan únicamente a compuestos basados en maleimidas, y a menudo, no se especifican para aplicaciones en los ADC dirigidos a tumores. Por ejemplo, los métodos previamente descritos de acoplamiento covalente con tiol que implican el uso de compuestos basados en maleimidas similares no especificaron sus aplicaciones en el acoplamiento con agentes activos tales como moléculas farmacéuticas, proteínas, o polipéptidos dirigidos contra tumores (véase, por ejemplo, la Publicación de Solicitud de Patente PCT No. WO 2013132268). Otros métodos que describieron específicamente aplicaciones en ADC dirigidos a tumores que usan compuestos basados en maleimidas no emplearon el mecanismo de formación de puentes de tiol covalente en el que un enlazador reacciona simultáneamente con dos grupos tiol, como lo proporciona la presente invención (véase, por ejemplo, Publicación de Solicitud la Patente China No. CN 103933575).
El documento WO2015004400A1 describe conjugados de anticuerpo-fármaco y el uso de los mismos en terapia, en particular en terapia anticancerosa o antiinflamatoria.
El documento WO2013085925A1 describe conjugados de anticuerpo-fármaco de tipo anticuerpo-citotoxina y compuestos relacionados, tales como conjugados de enlazador-citotoxina, y los enlazadores usados para obtenerlos, análogos de tubulisina, y síntesis de intermedios; composiciones.
El documento CN103933575A describe un enlazador tridentado y su uso, que puede usarse para obtener conjugados de anticuerpo-fármaco.
BREVE SUMARIO DE LA INVENCIÓN
La presente invención proporciona enlazadores novedosos y ventajosos para acoplar un anticuerpo a otro compuesto. Además de los enlazadores cuya síntesis estructural y el uso de los mismos se describen aquí, la presente invención también proporciona conjugados de anticuerpo/agente activo, y aplicaciones de los mismos, como se usan en, por ejemplo, aplicaciones de conjugados de anticuerpo-fármaco.
Algunas realizaciones proporcionan un enlazador-agente activo o una de sus sales farmacéuticamente aceptables, en las que el enlazador-agente activo es

Algunas realizaciones proporcionan un conjugado de fármaco-anticuerpo o una sal farmacéuticamente aceptable del mismo, en las que el conjugado de anticuerpo-fármaco es

en el que A es un anticuerpo o un fragmento de anticuerpo.
Como se usa aquí, la referencia a “nulo” significa que la entidad no está presente.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra un esquema del puente de tiol covalente establecido por un enlazador que reacciona con dos grupos tiol vecinos de un enlace de disulfuro roto o reducido de anticuerpo.
La Figura 2 es una imagen SDS-PAGE de un ADC anti-Her-2 ejemplar, que comprende un anticuerpo monoclonal anti-Her-2 conjugado con una citotoxina preparada usando Mc-VC-PAB-MMAE.
La Figura 3 muestra un cromatograma HIC-HPLC de MSL-C31.
La Figura 4 muestra los resultados de SDS-PAGE en condiciones reducidas de MSL-C75.
La Figura 5 muestra un cromatograma HIC-HPLC de MSL-C75.
La Figura 6 muestra el resultado de SDS-PAGE en condiciones reducidas para a) anticuerpo desnudo, y b) CD59-C78.
La Figura 7 muestra un cromatograma HIC-HPLC de CD59-C78.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
En la siguiente descripción detallada, se hace referencia a los dibujos adjuntos, que representan realizaciones ejemplares, no limitativas y no exhaustivas de la invención. Estas realizaciones se describen con suficiente detalle para permitir que los expertos en la técnica pongan en práctica la invención.
La presente invención proporciona moléculas enlazadoras novedosas y ventajosas capaces de acoplarse covalentemente a dos tioles libres de un anticuerpo a través de un extremo del enlazador, y unirse a un agente activo en otro extremo del enlazador. También se proporcionan aquí conjugados de anticuerpo/agente activo, incluyendo, por ejemplo, conjugados de anticuerpo-fármaco (ADC).
Definiciones
Como se usa aquí, las abreviaturas de compuestos orgánicos comunes se definen a continuación:
BOC Terc-butoxicarbonilo
Fmoc 9-Fluorenilmetoxicarbonilo
°C Temperatura en grados centígrados
DIPEA Diisopropiletilamina
DMF N,N’-dimetilformamida
T.A. Temperatura ambiente
EtOH Etanol
h Hora
Et3N trietilamina
HOBt N-hidroxibenzotriazol
Prep-HPLC Cromatografía de líquidos de altas prestaciones preparativa
NaHCO3 Bicarbonato de sodio
DCC Diciclohexilcarbodiimida
MeCN Acetonitrilo
EDC 1-Etil-3-(3-dimetilaminopropil)carbodiimida
DIC N,N’-diisopropilcarbodiimida
DEA Dietilamina
K2CO3 Carbonato de potasio
M Mol/litro
ml Mililitro
MgSO4 Sulfato de magnesio
THF Tetrahidrofurano
CH2Cl2 Diclorometano
NaCl Cloruro de sodio
NaSO4 Sulfato de sodio
HCl Ácido clorhídrico
LC-MS Cromatografía de líquidos-espectrometría de masas
CHCl3 Triclorometano
NaAc Acetato de sodio
Ac2O Anhídrido acético
TFA Ácido trifluoroacético
NaOH Hidróxido de sodio
La expresión “agente activo”, como se usa según la presente invención, incluye cualquier sustancia natural o sintética que tiene un efecto fisiológico cuando se administra a un animal. El agente activo se puede usar según la invención para tratar, por ejemplo, animales de sangre caliente, particularmente mamíferos, incluyendo seres humanos, animales veterinarios, y animales de granja. El agente activo puede actuar o visualizarse en una diana deseada dentro o sobre el cuerpo del animal, incluyendo el tejido tumoral.
Ejemplos no limitantes de “agentes activos” son fármacos que actúan en sitios sinápticos y sitios de unión de neuroefectores; analgésicos generales y locales; hipnóticos y sedantes; fármacos para el tratamiento de trastornos psiquiátricos tales como depresión y esquizofrenia; antiepilépticos y anticonvulsivos; medicamentos para el tratamiento de la enfermedad de Parkinson y de Huntington, envejecimiento y enfermedad de Alzheimer; antagonistas de aminoácidos excitadores, factores neurotróficos y agentes neurorregenerativos; factores tróficos; fármacos destinados al tratamiento de traumatismos o accidentes cerebrovasculares del sistema nervioso central (SNC); fármacos para el tratamiento de la adicción y el abuso de drogas; fármacos para el tratamiento de infecciones bacterianas, virales y/o microbianas, tales como gripe, VIH, herpes, varicela, y similares; autacoides y fármacos antiinflamatorios; agentes quimioterapéuticos para infecciones parasitarias y enfermedades causadas por microbios; agentes inmunosupresores; fármacos contra el cáncer; hormonas y antagonistas de hormonas; metales pesados y antagonistas de metales pesados; antagonistas para agentes tóxicos no metálicos; agentes citostáticos; agentes de visualización y otras sustancias de diagnóstico; agentes inmunoactivos e inmunorreactivos; transmisores y sus respectivos agonistas de receptores y antagonistas de receptores, sus respectivos precursores y metabolitos; inhibidores de transportadores; antibióticos; antiespasmódicos; antihistamínicos; antieméticos; relajantes; estimulantes; oligonucleótidos sentido y antisentido; dilatadores cerebrales; psicotrópicos; antimaníacos; dilatadores y constrictores vasculares; antihipertensivos; fármacos para el tratamiento de la migraña; agentes hiperglucémicos e hipoglucémicos; minerales y agentes nutricionales; fármacos contra la obesidad; anabólicos; antiasmáticos; y mezclas de los mismos.
Un “anticuerpo”, también conocido como una inmunoglobulina, es una proteína grande en forma de Y usada por el sistema inmunitario para identificar y neutralizar entidades extrañas tales como bacterias y virus. Un anticuerpo tiene cuatro cadenas polipeptídicas, dos cadenas pesadas idénticas y dos cadenas ligeras idénticas conectadas por enlaces de disulfuro de cisteína.
Un “anticuerpo monoclonal” es un anticuerpo monoespecífico en el que todas las moléculas de anticuerpo son idénticas debido a que se obtienen por células inmunitarias idénticas que son clones de una única célula madre. Los anticuerpos monoclonales se pueden preparar fusionando células de mieloma con células de bazo de un ratón (o células B de un conejo) que ha sido inmunizado con el antígeno deseado, y después purificando los hibridomas resultantes mediante técnicas tales como la purificación por afinidad. Los anticuerpos monoclonales recombinantes se pueden preparar en virus o células de levadura en lugar de en ratones, a través de tecnologías que incluyen la clonación de repertorio o la presentación en fagos/levaduras, la clonación de segmentos de genes de inmunoglobulina para crear bibliotecas de anticuerpos con secuencias de aminoácidos ligeramente diferentes a partir de las cuales se pueden obtener anticuerpos con especificidades deseadas. Los anticuerpos resultantes pueden prepararse a gran escala mediante fermentación.
Los anticuerpos “quiméricos” o “humanizados” son anticuerpos que contienen una combinación de las secuencias de ADN original (normalmente de ratón) y humano que se usan en el proceso recombinante, tales como aquellos en los que el ADN de ratón que codifica la porción de unión de un anticuerpo monoclonal se fusiona con ADN productor de anticuerpo humano para producir un anticuerpo monoclonal parcialmente humano y parcialmente de ratón. Los anticuerpos completamente humanizados se producen usando ratones transgénicos (manipulados para producir anticuerpos humanos) o bibliotecas de presentación de fagos.
Un “enlazador” es un resto con dos extremos reactivos, uno para unirse o asociarse de otro modo con un resto biológico o un fragmento del mismo, tal como un anticuerpo (o un fragmento del mismo), y el otro para la conjugación con un agente activo tal como una citotoxina.
Una “citotoxina” es una entidad que, cuando está en presencia de una célula, tal como una célula cancerosa, es tóxica o induce cambios funcionales clave en esa célula.
Como se usa aquí, “alquilo” se refiere a una cadena hidrocarbonada lineal o ramificada que está completamente saturada (es decir, no contiene enlaces dobles o triples). El grupo alquilo puede tener 1 a 9 átomos de carbono (siempre que aparezca aquí, un intervalo numérico tal como “1 a 9” se refiere a cada número entero en el intervalo dado; por ejemplo, “1 a 9 átomos de carbono” significa que el grupo alquilo puede comprender 1 átomo de carbono, 2 átomos de carbono, 3 átomos de carbono, etc., hasta 9 átomos de carbono inclusive, aunque la presente definición también cubre la aparición del término “alquilo” en el que no se designa un intervalo numérico). El grupo alquilo también puede ser un alquilo de tamaño medio que tiene 1 a 9 átomos de carbono. Los grupos alquilo típicos incluyen, pero de ninguna manera se limitan a, metilo, etilo, propilo, isopropilo, butilo, isobutilo, butilo terciario, pentilo, hexilo, y similares.
Como se usa aquí, “alquenilo” se refiere a una cadena hidrocarbonada lineal o ramificada que contiene uno o más dobles enlaces. El grupo alquenilo puede tener 2 a 9 átomos de carbono, aunque la presente definición también cubre la aparición del término “alquenilo” en el que no se designa intervalo numérico. El grupo alquenilo también puede ser un alquenilo de tamaño medio que tiene 2 a 9 átomos de carbono. El grupo alquenilo también podría ser un alquenilo inferior que tiene 2 a 4 átomos de carbono. El grupo alquenilo se puede designar como “alquenilo de C2-4” o designaciones similares. Sólo a modo de ejemplo, “alquenilo de C2-4” indica que hay dos a cuatro átomos de carbono en la cadena de alquenilo, es decir, la cadena de alquenilo se selecciona de etenilo, propen-1 -ilo, propen-2-ilo, propen-3-ilo, buten-1-ilo, buten-2-ilo, buten-3-ilo, buten-4-ilo, 1-metil-propen-1-ilo, 2-metil-propen-1-ilo, 1 -etil-eten-1 -ilo, 2-metil-propen-3-ilo, buta-1,3-dienilo, buta-1,2-dienilo, y buta-1,2-dien-4-ilo. Los grupos alquenilo típicos incluyen, pero de ningún modo se limitan a, etenilo, propenilo, butenilo, pentenilo, y hexenilo, y similares.
Como se usa aquí, “alquinilo” se refiere a una cadena hidrocarbonada lineal o ramificada que comprende uno o más enlaces triples. El grupo alquinilo puede tener 2 a 9 átomos de carbono, aunque la presente definición también cubre la aparición del término “alquinilo” en el que no se designa un intervalo numérico. El grupo alquinilo también puede ser un alquinilo de tamaño medio que tiene 2 a 9 átomos de carbono. El grupo alquinilo también podría ser un alquinilo inferior que tiene 2 a 4 átomos de carbono. El grupo alquinilo se puede designar como “alquinilo de C2-4” o designaciones similares. Solo a modo de ejemplo, “alquinilo de C2-4” indica que hay dos a cuatro átomos de carbono en la cadena de alquinilo, es decir, la cadena de alquinilo se selecciona de etinilo, propin-1 -ilo, propin-2-ilo, butin-1 -ilo, butin-3-ilo, butin-4-ilo, y 2-butinilo. Los grupos alquinilo típicos incluyen, pero de ningún modo se limitan a, etinilo, propinilo, butinilo, pentinilo, y hexinilo, y similares.
El término “aromático” se refiere a un anillo o sistema anular que tiene un sistema de electrones p conjugados, e incluye tanto grupos aromáticos carbocíclicos (por ejemplo, fenilo) como heterocíclicos (por ejemplo, piridina). El término incluye grupos monocíclicos o policíclicos de anillos condensados (es decir, anillos que comparten pares de átomos adyacentes), siempre que todo el sistema anular sea aromático.
Como se usa aquí, “cicloalquilo” significa un anillo o sistema anular de carbociclilo completamente saturado. Los ejemplos incluyen, pero no se limitan a, ciclopropilo, ciclobutilo, ciclopentilo, y ciclohexilo.
Como se usa aquí, “heteroarilo” se refiere a un anillo o sistema anular aromático (es decir, dos o más anillos condensados que comparten dos átomos adyacentes) que comprende uno o más heteroátomos, es decir, un elemento distinto del carbono, incluyendo, pero sin limitarse a, nitrógeno, oxígeno, y azufre, en el esqueleto del anillo. Cuando el heteroarilo es un sistema anular, todos los anillos del sistema son aromáticos. El grupo heteroarilo puede tener 5 18 miembros anulares (es decir, el número de átomos que forman el esqueleto del anillo, incluyendo los átomos de carbono y los heteroátomos), aunque la presente definición también cubre la aparición del término “heteroarilo” en el que no se designa un intervalo numérico. Los ejemplos de anillos heteroarílicos incluyen, pero no se limitan a, furilo, tienilo, ftalazinilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, isoxazolilo, isotiazolilo, triazolilo, tiadiazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, triazinilo, quinolinilo, isoquinolinilo, bencimidazolilo, benzoxazolilo, benzotiazolilo, indolilo, isoindolilo, y benzotienilo.
Como se usa aquí, “heterociclilo” significa un anillo o sistema anular cíclico no aromático que contiene al menos un heteroátomo en el esqueleto del anillo. Los heterociclilos se pueden unir entre sí de forma condensada, puenteada, o espiroconectada. Los heterociclilos pueden tener cualquier grado de saturación, siempre que al menos un anillo en el sistema anular no sea aromático. El o los heteroátomos pueden estar presentes en un anillo aromático o no aromático en el sistema anular. El grupo heterociclilo puede tener 3 a 20 miembros anulares (es decir, el número de átomos que forman el esqueleto del anillo, incluyendo los átomos de carbono y los heteroátomos), aunque la presente definición también cubre la aparición del término “heterociclilo” en el que no se designa un intervalo numérico. El grupo heterociclilo también puede ser un heterociclilo de tamaño medio que tiene 3 a 10 miembros anulares. El grupo heterociclilo también podría ser un heterociclilo que tiene 3 a 6 miembros anulares. Los ejemplos de anillos
heterociclílicos incluyen, pero no se limitan a, azepinilo, acridinilo, carbazolilo, cinolinilo, dioxolanilo, imidazolinilo, imidazolidinilo, morfolinilo, oxiranilo, oxepanilo, tiepanilo, piperidinilo, piperazinilo, dioxopiperazinilo, pirrolidinilo, pirrolidonilo, pirrolidionilo, 4-piperidonilo, pirazolinilo, pirazolidinilo, 1,3-dioxinilo, 1,3-dioxanilo, 1,4-dioxinilo, 1,4-dioxanilo, 1,3-oxatianilo, 1,4-oxatiinilo, 1,4-oxatianilo, 2H-1,2-oxazinilo, trioxanilo, hexahidro-1,3,5-triazinilo, 1,3 dioxolilo, 1,3-dioxolanilo, 1,3-ditiolilo, 1,3-ditiolanilo, isoxazolinilo, isoxazolidinilo, oxazolinilo, oxazolidinilo, oxazolidinonilo, tiazolinilo, tiazolidinilo, 1,3-oxatiolanilo, indolinilo, isoindolinilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidrotiofenilo, tetrahidrotiopiranilo, tetrahidro-1,4-tiazinilo, tiamorfolinilo, dihidrobenzofuranilo, bencimidazolidinilo, y tetrahidroquinolina.
Un “(heterociclil)alquilo” es un grupo heterociclilo conectado, como sustituyente, a través de un grupo alquileno. Los ejemplos incluyen, pero no se limitan a, imidazolinilmetilo e indoliniletilo.
Enlazador/agente activo
En un aspecto, la presente invención proporciona un enlazador-agente activo o una de sus sales farmacéuticamente aceptables, en el que el enlazador-agente activo es

En otro aspecto, la presente invención proporciona un conjugado de anticuerpo-fármaco o una de sus sales farmacéuticamente aceptables, en la que el conjugado de anticuerpo-fármaco es

o
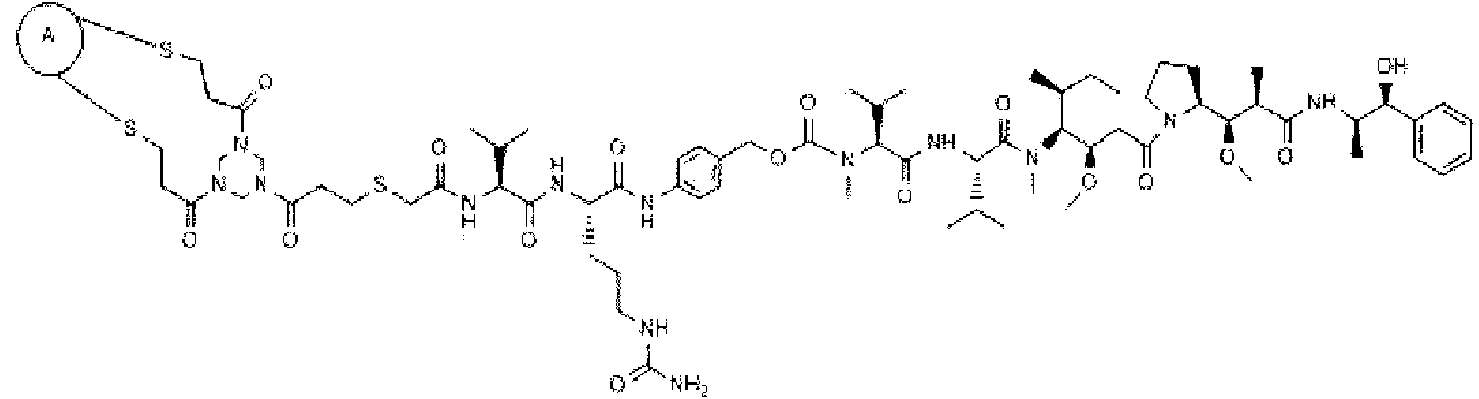
en el que A es un anticuerpo o un fragmento de anticuerpo.
En otro aspecto, la presente invención proporciona un conjugado de anticuerpo-fármaco para uso en un método para tratar una condición patológica en un sujeto, en el que dicho método comprende administrarlo a un sujeto que necesita dicho tratamiento, y en particular el sujeto es un ser humano.
En otro aspecto, A es un anticuerpo, o un fragmento del mismo, que se une a HER2, y la condición patológica es un cáncer de mama.
En otro aspecto, dicha condición patológica se selecciona de cáncer, trastornos inflamatorios, trastornos autoinmunes, trastornos del sistema nervioso, y trastornos cardiovasculares,
preferiblemente el cáncer se selecciona de carcinoma, linfoma, blastoma, sarcoma, y leucemia o neoplasias malignas linfoides,
más preferiblemente, el cáncer es un cáncer positivo para HER2, y en particular cáncer de mama.
EJEMPLOS
Los siguientes son ejemplos que ilustran los procedimientos para poner en práctica la invención. Estos ejemplos no deben interpretarse como limitativos.
EJEMPLO I SÍNTESIS DE CONJUGADOS DE ENLAZADOR-AGENTE ACTIVO
EJEMPLO I-1 SÍNTESIS DEL COMPUESTO 9 (no según la presente invención)

i) Et3N, MeOH, T.A., 2 h; ii) NaH, THF, T.A., 5 h; iii) Ácido clorhídrico, 1,4-dioxano, T.A., 16 h; iv) NaHCO3, H2O/THF, T.A., 3 h; v) HOBt, DIC, DIPEA, DMF, T.A., 12 h; vi) Et3N, CH2Cl2, T.A., 2 h.
Síntesis de compuesto 2
Una disolución de 1,3-diamino-2-propanol (3,15 g, 0,035 moles) y Et3N (4,85 ml, 0,035 moles) en metanol (120 ml) se calentó a 45°C. A la disolución, se añadió gota a gota lentamente (Boc)2O (17,05 g, 0,078 moles) en metanol (80 ml). La disolución de reacción se agitó a 45°C durante 30 min. Después agitar adicionalmente a temperatura ambiente durante 1,5 h, el disolvente se eliminó a presión reducida. El producto bruto se extrajo con éter dietílico (200 ml x 3) y se secó sobre sulfato de sodio para producir el compuesto 2 (9,94 g, 97,8%) como un polvo blanco. LC-MS m/z (ES+), 291,19 (M+H)+.
Síntesis de compuesto 3
Se añadió bromoacetato de terc-butilo (5,41 ml, 33,5 mmoles) a una disolución del compuesto 2 (3,89 g, 13,4 mmoles) en THF seco (40 ml) a temperatura ambiente. A la disolución, se añadió posteriormente hidruro de sodio (dispersión al 60% en aceite mineral, 2,42 g, 60,5 mmoles). La mezcla se filtró después de 5 horas. El filtrado se evaporó, y el residuo se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 10:1 a 5:1) para dar el producto 3 (3,96 g, 73,1%) como un sólido blanco. LC-MS m/z (ES+), 405,26 (M+H)+.
Síntesis de compuesto 4
Se añadieron 5 ml de ácido clorhídrico a una disolución agitada del compuesto 3 (1,0 g, 2,5 mmoles) en 1,4-dioxano (10 ml) a temperatura ambiente. La reacción se completó después de 16 horas. Después, el disolvente se eliminó para dar un producto bruto blanco (384,2 mg, 69,8%). El producto 4 se pudo usar en la siguiente etapa sin purificación. LC-MS m/z (ES+), 221,05 (M+H)+.
Síntesis de compuesto 5
Se añadió cloruro de acriloílo (267 ml, 3,3 mmoles) a una disolución del compuesto 4 (242,1 mg, 1,1 mmoles) en disolución saturada de bicarbonato de sodio/THF (v/v = 1:1,20 ml) a 0°C. La mezcla resultante se agitó vigorosamente a 0°C. Después de 10 minutos, se dejó que la disolución alcanzara la temperatura ambiente y se hizo reaccionar durante 3 horas. La mezcla se acidificó con ácido clorhídrico para ajustar el pH <4. La mezcla se extrajo con acetato de etilo (50 ml x 2). Las capas orgánicas se combinaron y se lavaron con disolución saturada de cloruro de sodio (40 ml), se secaron sobre sulfato de magnesio anhidro, se filtraron, y se eliminó el disolvente. El residuo sólido se purificó por prep-HPLC para dar el producto 5 como un polvo gris (196,0 mg, 69,6%). LC-MS m/z (ES+), 257,12 (M+H)+.
Síntesis de compuesto 7
A una disolución agitada del compuesto 5 (25,6 mg, 0,10 mmoles) y DIPEA (16,5 ml, 0,10 mmoles) en DMF seco (6 ml) a 0°C se añadió HOBt (14,9 mg, 0,11 mmoles) y DIC (13,9 mg, 0,11 mmoles). Después de 15 minutos, el compuesto 6 (20,5 mg, 0,09 mmoles) se añadió a la disolución. La mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante la noche. La mezcla se diluyó con agua (10 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, y el disolvente se eliminó a vacío. El producto bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 3:1) para producir el compuesto 7 como un sólido blanco (24,3 mg, 62,6%). LC-m S m/z (ES+), 467,22 (M+H)+.
Síntesis de compuesto 9
El compuesto 7 (140,1 mg, 0,3 mmoles) se disolvió en 5 ml de CH2CL y se enfrió a 0°C en un baño de hielo. Se añadieron Et3N (1 mg, 0,01 mmoles) y el compuesto 8 (73,7 mg, 0,1 mmoles) a la mezcla resultante, y se dejó agitar durante 30 minutos. La disolución se dejó calentar a temperatura ambiente y se agitó durante otras 1,5 horas. El disolvente se eliminó a presión reducida, y el residuo sólido se purificó por pre-HPLC para dar el compuesto 9 como un polvo blanco (90 mg, 74,8%). LC-MS m/z (ES+), 1204,49 (M+H)+.
EJEMPLO I-2 SÍNTESIS DEL COMPUESTO 17 (no según la presente invención)

i) EtaN, CH2CI2, T.A., 5 h; ii) EtaN, THF, T.A., 4 h; iii) CH2CI2, T.A., 12 h; iv) TFA, CH2CI2, T.A., 3 h; v) K2CO3, FbO/EtOAc, T.A., 5 h; vi) HOBt, DIC, DIPEA, DMF, T.A., 24 h.
Síntesis de compuesto 11
El compuesto 10 (263,3 mg, 1,15 mmoles) se disolvió en CH2Cl2 (15 ml), Et3N (5,9 ml, 0,04 mmoles), y el compuesto 6 (300,1 mg, 0,39 mmoles) se añadió a la mezcla resultante a 0°C. La disolución se dejó calentar a temperatura ambiente y se mantuvo durante 5 horas. El disolvente se eliminó a vacío, y el producto bruto se purificó por prep-HPLC para producir el compuesto 11 (193,2 mg, 49,7%) como un sólido blanco. LC-MS m/z (ES+), 994,43 (M+H)+.
Síntesis de compuesto 13
A una mezcla de compuesto 12 (0,45 g, 4,2 moles) y Et3N (8 ml, 0,06 moles) en THF (15 ml), se añadió gota a gota a 0°C una disolución de 2-(terc-butoxicarboniloxiimino)-2-fenilacetonitrilo (2,1 g, 8,3 moles) en THF (30 ml). Después de la adición completa, la disolución se dejó calentar a temperatura ambiente y se dejó agitar durante 4 horas. La mezcla de reacción se concentró a aceite a presión reducida, y se añadió CH2CL (50 ml). La mezcla se lavó con hidróxido de sodio (5%, 30 ml) y salmuera (30 ml). La capa orgánica se secó sobre sulfato de sodio y se concentró a presión reducida. El producto bruto se purificó por cromatografía en columna (gel de sílice, MeOH:CH2Cl2 = 1:10, v/v) para dar compuesto 13 como un aceite amarillo (803,1 mg, 60,7%). LC-MS m/z (ES+), 304,22 (M+H)+.
Síntesis de compuesto 14
Se añadió anhídrido succínico (265,2 mg, 2,65 mmoles) a una disolución del compuesto 13 (800,1 mg, 2,65 mmoles) en CH2Cl2 (10 ml). La mezcla se agitó a temperatura ambiente durante la noche, y después se concentró hasta un aceite a presión reducida. El producto bruto se purificó por cromatografía en columna (gel de sílice, MeOH:CH2Cl2 = 1:8, v/v) para dar compuesto 14 (506,5 mg 47,6%) como un aceite gris. LC-MS m/z (ES+), 404,24 (M+H)+.
Síntesis de compuesto 15
El compuesto 14 (503,5 mg, 1,25 mmoles) se disolvió en CH2CL (10 ml) junto con ácido trifluoroacético (2 ml). La disolución se dejó reaccionar durante 3 horas a temperatura ambiente. El disolvente se eliminó a presión reducida para producir el compuesto 15 como un aceite gris (190,2 mg, 75,1%) sin más purificación. LC-MS m/z (ES+), 204,13 (M+H)+.
Síntesis de compuesto 16
A una mezcla de carbonato de potasio (68,3 mg, 0,5 mmoles) en agua (5 ml) y compuesto 15 (67,2 mg, 0,33 mmoles) en acetato de etilo (10 ml), se añadió gota a gota una disolución de cloruro de acriloílo (53,6 ml, 0,67 mmoles) en acetato de etilo (10 ml) a 0i) 2C. La mezcla resultante se agitó a 0°C en un baño de hielo durante 10 minutos, y después se dejó reaccionar a temperatura ambiente durante 5 horas. La mezcla se acidificó con ácido clorhídrico para ajustar el pH<5. La mezcla se extrajo con acetato de etilo (20 ml x 2). Las capas orgánicas se combinaron y se lavaron con disolución saturada de cloruro de sodio (20 ml), se secaron sobre sulfato de magnesio anhidro, se filtraron, y se eliminó el disolvente. El residuo sólido se purificó por cromatografía en columna (MeOH:CH2Cl2 = 10:1) para dar producto 16 como un aceite amarillo (63,1 mg, 61,7%). LC-MS m/z (ES+), 312,15 (M+H)+.
Síntesis de compuesto 17
El compuesto 16 (22,2 mg, 0,07 mmoles), HOBt (9,7 mg, 0,07 mmoles) y DIC (11 ml, 0,07 mmoles) se disolvieron en 8 ml de DMF y se enfriaron a 0°C en un baño de hielo. Posteriormente, se añadieron compuesto 11 (59,1 mg, 0,06 mmoles) y DIPEA (12,4 ml, 0,07 mmoles) a la mezcla. La disolución se dejó calentar a temperatura ambiente, y se dejó reaccionar durante 24 horas. La mezcla resultante se concentró y se purificó por prep-HPLC para producir el compuesto 17 (11,3 mg, 39,5%) como un sólido blanco. LC-MS m/z (Es ), 1287,56 (M+H)+.
EJEMPLO I-3 SÍNTESIS DEL COMPUESTO 25 (no según la presente invención)

i) HOBt, DIPEA, Piridina, DMF, T.A., 32 h; ii) DEA, DMF, T.A., 2 h; iii) EtaN, CH2CL, T.A., 4 h; iv) EtaN, CH2Cl2, T.A., 12 h; v) EDCI, HOBt, DIPEA, CH2Cl2, T.A., 24 h.
Síntesis de compuesto 20
A una disolución agitada de 18 (191,6 mg, 0,25 mmoles) en DMF seca (15 ml) a 0°C bajo nitrógeno, se añadió HOBt (33,8 mg, 0,25 mmoles) y compuesto 19 (197,3 mg, 0,26 mmoles). Después de 15 minutos, se añadieron piridina (3 ml) y DIPEA (52,4 ml, 0,30 mmoles), y se hicieron reaccionar durante 30 minutos a 0°C. Después, la mezcla de reacción
se dejó calentar a temperatura ambiente y se agitó durante 3 horas. La mezcla se repuso con DIPEA (52,4 pl, 0,30 mmoles), y se hizo reaccionar durante 32 horas. Una vez completada la reacción, se eliminó el disolvente a presión reducida. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 20:1) para dar el compuesto 20 (126,4 mg, 37,5%) como un sólido blanco. LC-MS m/z (ES+), 1344,77 (M+H)+.
Síntesis de compuesto 21
Se añadió dietilamina (4 ml, 38,8 mmoles) a una disolución del compuesto 20 (126,4 mg, 0,1 mmoles) en DMF (8 ml) a temperatura ambiente. Después de agitar durante 2 horas, el disolvente se eliminó a presión reducida para dar un producto bruto 21 (104,1 mg, 99%). El producto se pudo usar en la siguiente etapa sin purificación. LC-MS m/z (ES+), 1123,72 (M+H)+.
Síntesis de compuesto 23
Una disolución de cloruro de acriloílo (7,31 ml, 90,00 mmoles) en CH2CL seco (50 ml) se añadió gota a gota a una mezcla del compuesto 22 (2,64 g, 20,00 mmoles) y Et3N (16,60 ml; 120,00 mmoles) en CH2Cl2 seco (60 ml) a 0°C bajo argón. Se dejó que la mezcla alcanzara la temperatura ambiente y se hizo reaccionar durante 4 horas. Después, la mezcla se trató con disolución saturada de bicarbonato de sodio (150 ml) y salmuera (150 ml), y se extrajo con CH2CL (150 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, y el disolvente se eliminó a vacío. El producto bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 10:1) para producir el compuesto 23 como un sólido blanco (4,88 g, 83,0%). LC-MS m/z (Es+), 295,10 [M+H]+.
Síntesis de compuesto 24
A una disolución de compuesto 23 (444,1 mg, 1,5 mmoles) y Et3N (0,23 ml, 1,66 mmoles) en CH2Cl2 (30 ml), se añadió gota a gota en un baño de hielo ácido mercaptoacético (0,23 ml, 3,32 mmoles) en CH2CL (5 ml). Después de la adición completa, la disolución se dejó calentar a temperatura ambiente y se dejó agitar durante la noche. El disolvente se eliminó a vacío, y el residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 60:1-30:1) para dar el compuesto 24 como un aceite amarillo (1,72 g, 36,87%). LC-MS m/z (ES-), 385,10 (M-H)-.
Síntesis de compuesto 25
El compuesto 24 (22,2 mg, 0,057 mmoles), HOBt (9,3 mg, 0,069 mmoles) y EDCI (16,5 mg, 0,086 mmoles) se disolvieron en 5,0 ml de CH2Cl2. Posteriormente, se añadieron compuesto 21 (64,5 mg, 0,063 mmoles) y DIPEA (20,3 pl, 0,104 mmoles) a la mezcla. La disolución se dejó calentar a temperatura ambiente y se dejó reaccionar durante 24 horas. La disolución resultante se separó por prep-HPLC para dar el compuesto 25 como un sólido blanco (25,3 mg, 29,7%). LC-MS m/z (ES+), 1479,80 (M+H)+.
EJEMPLO I-4 SÍNTESIS DEL COMPUESTO 31

i) HOBt, DIPEA, Piridina, DMF, T.A., 24 h; ii) DEA, DMF, T.A., 2,5 h; iii) EtaN, CH2CI2, T.A., 12 h; iv) DIC, HOBt, DIPEA, DMF, T.A., 24 h.
Síntesis de compuesto 27
A una disolución de 18 (194,2 mg, 0,25 mmoles) en DMF seca (15 ml) a 0°C bajo nitrógeno, se añadieron HOBt (34,2 mg, 0,25 mmoles) y compuesto 26 (200,3 mg, 0,28 mmoles). Después de agitar durante 15 minutos, se añadieron piridina (3 ml) y DIPEA (53,1 ml, 0,32 mmoles), y se dejó reaccionar durante 30 minutos a 0°C. Después, la mezcla de reacción se dejó agitar a temperatura ambiente durante 3 horas. La mezcla se repuso con DIPEA (53,1 ml, 0,32 mmoles) y se hizo reaccionar durante otras 24 horas. Una vez completada la reacción, la mezcla se concentró a vacío. El producto bruto se purificó por cromatografía en columna para dar el compuesto 27 (126,2 mg, 37,0%) como un sólido blanco. LC-MS m/z (ES+), 1398,75 (M+H)+.
Síntesis de compuesto 28
Se añadió dietilamina (4 ml) a una disolución del compuesto 27 (126,2 mg, 0,09 mmoles) en DMF (8 ml) a temperatura ambiente. Después de agitar durante 2,5 horas, el disolvente se eliminó a presión reducida para dar un producto bruto 28 (73,3 mg, 68,9%) que pudo usarse sin purificación. LC-MS m/z (ES+), 1176,68 (M+H)+.
Síntesis de compuesto 30
A una disolución de compuesto 29 (1,87 g, 7,5 mmoles) y Et3N (104 ml, 0,75 mmoles) en CH2Cl2 (40 ml), se añadió gota a gota a 0°C ácido mercaptoacético (103,9 ml, 1,5 mmoles) en CH2Cl2 (10 ml). Una vez completada la adición, la disolución se dejó calentar a temperatura ambiente y se dejó agitar durante la noche. El disolvente se eliminó a vacío, y el residuo se purificó por cromatografía en columna para dar el compuesto 30 como un sólido blanco (1,72 g, 36,87%). LC-MS m/z (ES-), 342,11 (M-H)-.
Síntesis de compuesto 31
El compuesto 30 (22,2 mg, 0,065 mmoles), HOBt (8,8 mg, 0,065 mmoles) y DIC (11 ml, 0,065 mmoles) se añadieron a DMF (8 ml), y se enfriaron a 0°C en un baño de hielo. Se añadieron compuesto 28 (63,7 mg, 0,054 mmoles) y DIPEA (12,4 ml, 0,054 mmoles) a la disolución. La mezcla se dejó calentar a temperatura ambiente y se dejó reaccionar durante 24 horas. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 60:1-30:1) para producir compuesto 31 como un sólido blanco (11,3 mg, 39,5%). LC-MS m/z (ES+), 1499,78 (M+H)+.
EJEMPLO I-5 SÍNTESIS DEL COMPUESTO 36 (no según la presente invención)

i) K2CO3, EtOAc/H2O, T.A., 2 h; ii) EDCI, DMF, T.A., 12 h; iii) DIPEA, DMF, T.A., 12 h; iv) EDCI, HOBt, DIPEA, DMF, T.A., 24 h.
Síntesis de compuesto 33
Ácido 2,4-diaminobenzoico 32 (501,8 mg, 3,3 mmoles) se dispersó en acetato de etilo (20 ml), y se añadió una disolución de carbonato de potasio (9,0 g, 66 mmoles) en agua (20 ml). A la mezcla, se añadió cuidadosamente cloruro de acriloílo (1 ml, 13 mmoles), lo que dio como resultado la formación de un precipitado marrón claro. A continuación, se permitió que la mezcla de reacción alcanzara la temperatura ambiente, lo que condujo a la disolución del precipitado. Después de 2 horas, la reacción se completó. La capa orgánica se descartó, y la capa acuosa se acidificó con ácido clorhídrico al 5%, apareciendo un precipitado a pH<4. El precipitado se filtró, se lavó con éter de petróleo, y se diluyó con acetato de etilo. Después, la capa orgánica se secó sobre sulfato de magnesio anhidro, se filtró, y se eliminó a vacío. El residuo sólido se suspendió en agua, se filtró, y se lavó a fondo con éter dietílico para dar el producto 33 (670,2 mg, 78%) como un polvo gris. LC-MS m/z (ES+), 261,09 (M+H)+.
Síntesis de compuesto 34
El compuesto 33 (260,1 mg, 1,0 mmoles) y EDCI (210,9 mg, 1,1 mmoles), N-hidroxisuccinimida (126,6 mg, 1,1 mmoles) se disolvieron en DMF (14 ml). La disolución se agitó durante la noche a temperatura ambiente. Una vez completada la reacción, se eliminó el disolvente a presión reducida. El residuo sólido se purificó por cromatografía en columna con CH2Cl2/MeOH (10:1) como eluyente para dar el producto 34 como un sólido marrón claro (273 mg, 76,4%). LC-MS m/z (ES+), 358,11 (M+H)+.
Síntesis de compuesto 35
A una disolución de ácido 4-aminobutírico (82,4 mg, 0,8 mmoles) y DIPEA (489 pl, 2,8 mmoles) en DMF (10 ml), se añadió a 0°C en un baño de hielo una disolución del compuesto 34 (260,7 mg, 0,73 mmoles) en DMF (10 ml). La mezcla se agitó durante 10 minutos a 0°C, y después se agitó a temperatura ambiente durante la noche. El disolvente se eliminó a vacío cuando se completó la reacción. El residuo sólido se purificó por cromatografía en columna (CH2Cl2:MeOH = 10:1, v/v) para producir el compuesto 35 (192,3 mg, 76,2%). LC-MS m/z (ES+), 346,14 (M+H)+.
Síntesis de compuesto 36
HOBt (14,9 mg, 0,11 mmoles), EDCI (21,1 mg, 0,11 mmoles) y DIPEA (19,2 pl, 0,11 mmoles) se añadieron a una disolución agitada de 35 (34,5 mg, 0,1 mmoles) en DMF seca (10 ml) a 0°C. Después de 15 minutos, se añadió el compuesto 21 (112,3 mg, 0,1 mmoles). La mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante 24 horas. A la mezcla se añadió agua (15 ml), y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, se filtraron, y se eliminaron a vacío. El producto bruto se purificó por cromatografía en columna (CH2CL:MeOH = 10:1, v/v) para producir el compuesto 36 como un sólido blanco (56,5 mg, 39%). LC-MS m/z (ES+), 1450,84 (M+H)+.
EJEMPLO I-6 SÍNTESIS DEL COMPUESTO 43 (no según la presente invención)
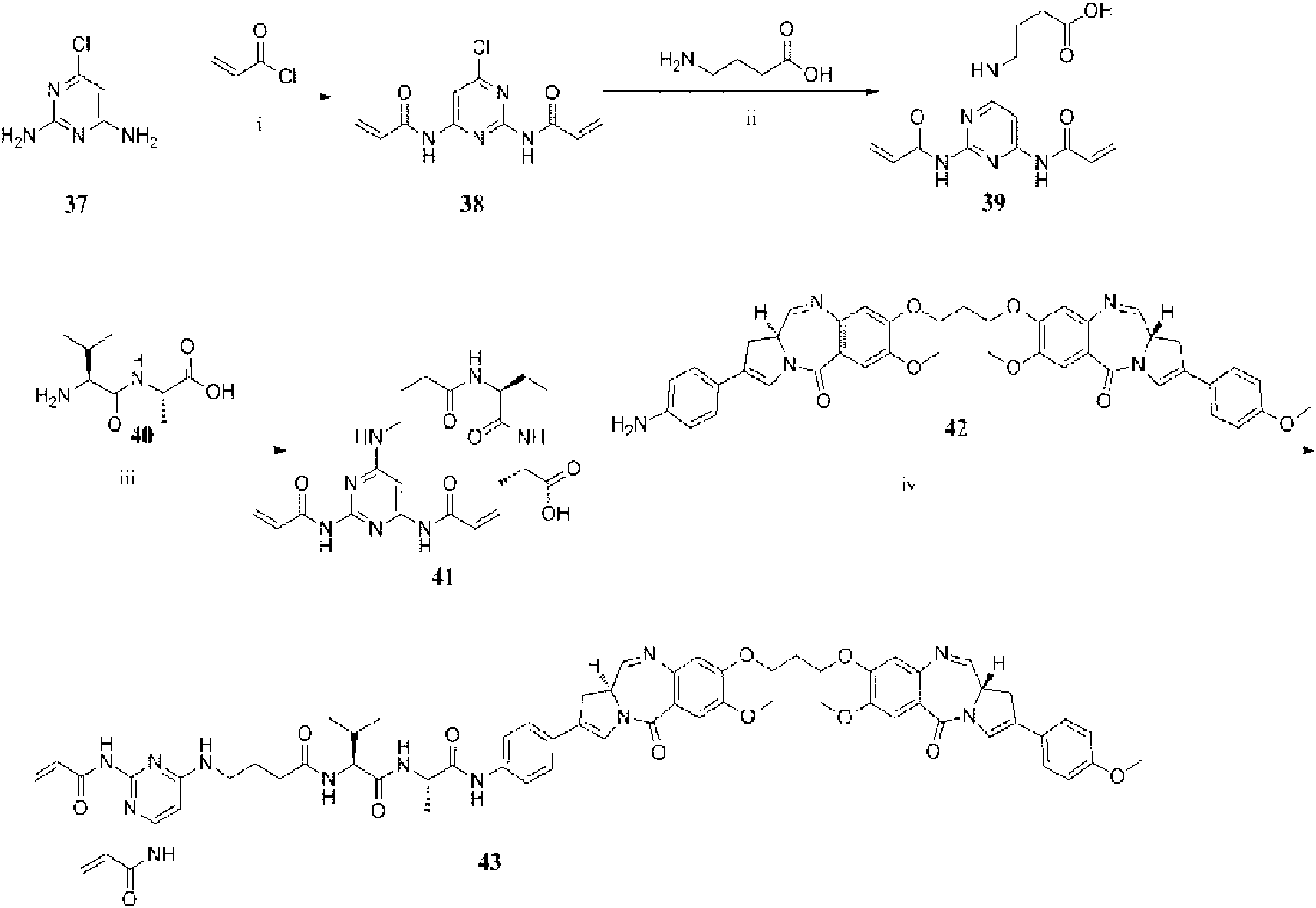
i) DMAP, CH2Cl2, T.A., 1 h; ii) NaOH, EtOH/H2O, 80°C, 2 h; iii) EDCI, HOBt, DIPEA, DMF, T.A., 18 h; (iv) EEDQ, CH2Cl2, MeOH, T.A., 18 h.
Síntesis de compuesto 38
En un matraz de fondo redondo de 100 ml se colocó una disolución del compuesto 37 (1,05 g, 6,9 mmoles) y DMAP (1,94 g, 15,9 mmoles) en CH2CL (20 ml), se añadió gota a gota cloruro de acriloílo (1,38 g, 15,2 mmoles) con agitación a 0°C. La reacción se agitó durante 1 hora a temperatura ambiente. La reacción se inactivó mediante la adición de agua (50 ml), y se extrajo con CH2Cl2 (20 ml x 3). La fase orgánica se combinó, se lavó con disolución saturada de bicarbonato de sodio (30 ml), y salmuera (30 ml), se secó sobre sulfato de sodio anhidro, y se filtró. El filtrado se concentró a presión reducida para dar el compuesto 38 (1,51 g, 85,8%) como un semisólido amarillo claro. LC-MS m/z (ES+), 253,04 (M+H)+.
Síntesis de compuesto 39
El compuesto 38 (1,56 g, 5,94 mmoles) y ácido 4-aminobutanoico (673,0 mg, 6,53 mmoles) se disolvieron en EtOH (15 ml) junto con hidróxido de sodio (713,3 mg, 17,82 mmoles) en agua (15 ml). La mezcla de reacción se agitó durante 2 horas a 80°C. Después de enfriar a temperatura ambiente, el disolvente se concentró a presión reducida. La mezcla se acidificó a pH<7 con ácido clorhídrico (0,5 N). El sólido se recogió por filtración, se lavó con agua (10 ml x 2), y se secó para producir el compuesto 39 (490,2 mg, 25,8%) como un sólido blanquecino. LC-MS m/z (ES+), 320,03 (M+H)+.
Síntesis de compuesto 41
El compuesto 39 (230,1 mg, 0,72 mmoles), HOBt (116,7 mg, 0,86 mmoles), EDCI (151,8 mg, 0,79 mmoles) y DIPEA (279,2 mg, 2,2 mmoles) se añadieron a DMF (20 ml) y se agitaron a 0°C en un baño de hielo. Después de 1 hora, se añadió compuesto 40 (149,1 mg, 0,79 mmoles) a la disolución. La mezcla se dejó calentar a temperatura ambiente y se dejó reaccionar durante 18 horas. La mezcla de reacción se diluyó con salmuera (50 ml), se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas se combinaron y se secaron con sulfato de sodio anhidro, se filtraron, y se concentraron. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 200/1 ~10/1, v/v) para producir compuesto 41 como un sólido amarillo (97,3 mg, 27,6%). LC-MS m/z (ES+), 490,23 (M+H)+.
Síntesis de compuesto 43
Una disolución de compuesto 41 (90,1 mg, 0,18 mmoles), EEDQ (54,6 mg, 0,22 mmoles) en CH2Cl2 anhidro (7 ml) y MeOH anhidro (0,07 ml) se agitó durante 1 hora a temperatura ambiente. Posteriormente, se añadió compuesto 42 (133,5 mg, 0,184 mmoles). La mezcla de reacción se agitó durante 18 horas más. La mezcla de reacción se concentró a vacío, y se purificó por prep-HPLC para producir el compuesto 43 (12,3 mg, 5,6%) como un sólido blanco. LC-MS m/z (ES+), 1197,51 (M+H)+.
EJEMPLO I-7 SÍNTESIS DEL COMPUESTO 49 (no según la presente invención)

i) HCl(g), MeOH, 0°C, 2 h; ii) piridina, 100°C, 2 h; iii) NaOH, THF/H2O, T.A., 4 h; iv) EDCI, HOBt, DIPEA, DMF, T.A., 18 h; v) EEDQ, CH2Cl2/MeOH, T.A., 18 h.
Síntesis de compuesto 45
A una disolución de compuesto 44 (1,13 g, 6,53 mmoles) en metanol (30 ml), se introdujo ácido clorhídrico (gas) con agitación a 0°C. La reacción se agitó durante 2 horas a 0°C. La mezcla se concentró, y el residuo se disolvió en agua (50 ml). La mezcla se ajustó a 8 con bicarbonato de sodio saturado, se extrajo con acetato de etilo (20 ml x 3). La capa orgánica se combinó y se secó con sulfato de sodio anhidro, y se filtró. El filtrado se concentró para dar el compuesto 45 (710,1 mg, 65,1%) como un sólido amarillo. LC-MS m/z (ES+), 168,07 (M+H)+.
Síntesis de compuesto 46
Se añadió gota a gota cloruro de acriloílo (947,1 mg, 10,47 mmoles) a una disolución del compuesto 45 (700,5 mg, 4,19 mmoles) en piridina (20 ml) con agitación a 0°C en un baño de hielo. La mezcla de reacción se agitó durante 2 horas a 100°C. Después de enfriar a temperatura ambiente, la mezcla de reacción se concentró a presión reducida. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 200/1 ~ 10/1, v/v) para obtener el compuesto 46 (520,4 mg, 45,2%) como un sólido amarillo. LC-MS m/z (ES+), 276,09 (M+H)+.
Síntesis de compuesto 47
Una mezcla de compuesto 46 (510,2 mg, 1,85 mmoles), hidróxido de sodio (370,6 mg, 9,26 mmoles), THF (10 ml) y agua (10 ml) se agitó durante 4 horas a temperatura ambiente. Una vez completada la reacción, la mezcla se
concentró, y el pH se ajustó a 7 con ácido clorhídrico (1N). El compuesto 47 (360,2 mg, 74,1%) se recogió por filtración como un sólido blanquecino. LC-MS m/z (ES+), 262,07 (M+H)+.
Síntesis de compuesto 48
El compuesto 47 (120,5 mg, 0,5 mmoles), HOBt (4,7 mg, 0,6 mmoles), EDCI (96,8 mg, 0,5 mmoles) y DIPEA (178,1 mg, 1,4 mmoles) se añadieron a DMF (10 ml) y se agitaron a 0°C en un baño de hielo. Después de 1 hora, se añadió compuesto 40 (95,1 mg, 0,5 mmoles) a la disolución. La mezcla se dejó calentar a temperatura ambiente y se dejó reaccionar durante 18 horas. La mezcla de reacción se diluyó con salmuera (20 ml), se extrajo con acetato de etilo (10 ml x 3). Las capas orgánicas se combinaron y se secaron con sulfato de sodio anhidro, se filtraron, y se concentraron. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 100/1 ~20/1, v/v) para producir compuesto 41 como un sólido blanquecino (71 mg, 35,8%). LC-MS m/z (ES+), 432,07 (M+H)+.
Síntesis de compuesto 49
El compuesto 48 (60,2 mg, 0,14 mmoles), EEDQ (41,3 mg, 0,17 mmoles) se disolvieron en CH2Cl2 anhidro (6 ml) y MeOH anhidro (0,06 ml), y se dejaron agitar durante 1 hora a temperatura ambiente. Se añadió compuesto 42 (100,9 mg, 0,14 mmoles) a la disolución. La mezcla de reacción se agitó durante 18 horas adicionales. La mezcla de reacción se concentró a presión reducida, y se purificó por prep-HPLC para producir el compuesto 49 (12,4 mg, 7,61%) como un sólido blanco. LC-MS m/z (ES+), 1139,45 (M+H)+.
EJEMPLO I-8 SÍNTESIS DEL COMPUESTO 55 (no según la presente invención)

i) EtaN, MeOH, T.A., 3 h; ii) Na2COs, THF/H2O, T.A., 3 h; iii) HOBt, DIC, DIPEA, DMF, T.A., 12 h; iv) TFA, CH2Cl2, T.A., 3 h; v) EtsN, CH2Cl2, T.A., 2 h; vi)
Síntesis de compuesto 50
Se añadió Et3N (332,7 pl, 2,4 mmoles) a una disolución del compuesto 4 (440,0 mg, 2 mmoles) en MeOH (10 ml) a 45°C. Después, se añadió una disolución de (Boc)2O (436,2 mg, 2 mmoles) en MeOH (10 ml). La mezcla resultante se agitó vigorosamente a 45°C durante 30 minutos, seguido de agitación a temperatura ambiente durante 2 horas. Una vez eliminado el disolvente, el residuo se lavó con ácido clorhídrico (5%, 10 ml), disolución saturada de cloruro de sodio (15 ml), y se extrajo con acetato de etilo (10 ml x 3). La capa orgánica se secó sobre sulfato de magnesio
anhidro, se filtró, y se eliminó a vacío. El residuo sólido se recristalizó en éter de petróleo/acetato de etilo (10 ml, v:v = 2:1) para dar el compuesto 50 (375,4 mg, 75,6%) como un polvo blanco. LC-MS m/z (ES+), 249,15 (M+H)+.
Síntesis de compuesto 51
Se añadió cloruro de acriloílo (121 ml, 1,5 mmoles) a una disolución del compuesto 50 (248,0 mg, 1,0 mmoles) en disolución saturada de bicarbonato de sodio/THF (v/v = 1:1,20 ml) a 0°C. La mezcla resultante se agitó vigorosamente a 0°C durante 10 minutos. Se dejó que la mezcla alcanzara la temperatura ambiente, y se hizo reaccionar durante 3 horas. La mezcla se acidificó con ácido clorhídrico para ajustar el pH<4. La mezcla se extrajo mediante acetato de etilo (20 ml x 3). Las capas orgánicas se combinaron y se lavaron con disolución saturada de cloruro de sodio (30 ml), se secaron sobre sulfato de magnesio anhidro, se filtraron, y se eliminó el disolvente. El residuo sólido se purificó por prep-HPLC para dar el producto 51 (176,1 mg, 58,3%) como un aceite gris. LC-MS m/z (ES+), 303,16 (M+H)+.
Síntesis de compuesto 52
El compuesto 51 (30,2 mg, 0,1 mmoles), HOBt (14,7 mg, 0,11 mmoles) y DIC (13,9 mg, 0,11 mmoles) se disolvieron en DMF seca (6 ml) y se enfriaron a 0°C en un baño de hielo. Después de 15 minutos, se añadieron compuesto 6 (20,5 mg, 0,09 mmoles) y DIPEA (19,2 ml, 0,11 mmoles) a la mezcla. La disolución se dejó calentar a temperatura ambiente, y se agitó durante la noche. La mezcla se diluyó con agua (10 ml) y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se secaron sobre sulfato de magnesio anhidro, y el disolvente se eliminó a vacío. El producto bruto se purificó por cromatografía en columna (CH2Cl2:MeOH = 20:1, v/v) para producir el compuesto 52 como un sólido blanco (28,2 mg, 60,7%). LC-MS m/z (ES+), 513,26 (M+H)+.
Síntesis de compuesto 53
Una disolución de compuesto 52 (28,2 mg, 0,06 mmoles), ácido trifluoroacético (1,0 ml) en CH2Cl2 (5 ml) se agitó durante 3 horas a temperatura ambiente. El disolvente se eliminó a presión reducida para producir el compuesto 53 (20,4 mg, 88,2%) como un sólido gris que pudo usarse en la siguiente etapa sin más purificación. LC-MS m/z (ES+), 413,21 (M+H)+.
Síntesis de compuesto 54
El compuesto 53 (41,2 mg, 0,1 mmoles) se disolvió en 10 ml de DMF y se enfrió a 0°C en un baño de hielo. Et3N (6,9 ml, 0,05 mmoles) y el compuesto 8 (73,7 mg, 0,1 mmoles) se añadieron gota a gota a la disolución, y se dejó agitar durante 30 minutos. La disolución se dejó calentar a temperatura ambiente, y se agitó durante otras 5 horas. La mezcla se concentró a presión reducida, y el residuo se purificó por pre-HPLC para dar el compuesto 54 (73 mg, 63,6%) como un polvo blanco. LC-MS m/z (ES+), 1148,47 (M+H)+.
Síntesis de compuesto 55
Se añadió bromuro de bromoacetilo (150,0 mg, 0,75 mmoles) a una disolución del compuesto 54 (573,7 mg, 0,5 mmoles) en DMF (10 ml) a 0°C. La mezcla resultante se agitó a 0°C durante 10 minutos, y después se dejó agitar a temperatura ambiente durante otras 3 horas. A la mezcla se añadió agua (20 ml), y se extrajo mediante acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se concentraron, y el residuo se purificó por pre-HPLC para dar el compuesto 55 (421 mg, 66,4%) como un polvo amarillo. LC-MS m/z (ES+), 1270,38 (M+H)+.
EJEMPLO I-9 SÍNTESIS DEL COMPUESTO 59 (no según la presente invención)
i) K2CO3, H2O/EtOAc, T.A., 5 h; ii) CHCI3, Reflujo, 12 h; iii) AC2O, NaAc, 100°C, 2 h; iv) HOBt, DIC, DIPEA, DMF, T.A., 24 h.
Síntesis de compuesto 56
A una mezcla de carbonato de potasio (51,2 mg, 0,37 mmoles) en agua (5 ml) y compuesto 15 (50,0 mg, 0,25 mmoles) en acetato de etilo (10 ml), se añadió gota a gota lentamente a 0°C una disolución de cloruro de acriloílo (16 ml, 0,2 mmoles) en acetato de etilo (8 ml). Una vez completada la adición, la mezcla resultante se agitó a 0°C en un baño de hielo durante 10 minutos adicionales. Después se dejó que la mezcla alcanzara la temperatura ambiente, y se hizo reaccionar durante 5 horas. La mezcla se acidificó con ácido clorhídrico para ajustar el pH<5. La mezcla se extrajo con acetato de etilo (15 ml x 3). Las capas orgánicas combinadas se lavaron con disolución saturada de cloruro de sodio (15 ml), se secaron sobre sulfato de magnesio anhidro, se filtraron, y se eliminó el disolvente. El residuo sólido se purificó por cromatografía en columna (MeOH:CH2Cl2 = 8:1) para dar producto 56 (26,2 mg, 40,1%) como un aceite gris. LC-MS m/z (ES+), 258,14 (M+H)+.
Síntesis de compuesto 57
El compuesto 56 (26,0 mg, 0,10 mmoles) se agitó en CHCl3 (10 ml), se añadió anhídrido maleico (28,1 mg, 0,11 mmoles) a la mezcla. La reacción se calentó a reflujo durante 12 horas. Después de enfriar a temperatura ambiente, el producto sólido precipitado se filtró y se lavó con CHCl3 (30 ml x 3) para producir el compuesto 57 (20,1 mg, 59,1%) como un sólido blanco. LC-MS m/z (ES+), 356,14 (M+H)+.
Síntesis de compuesto 58
Una disolución del compuesto 57 (20,1 mg, 0,054 mmoles) y acetato de sodio (6,3 mg, 0,047 mmoles) en anhídrido acético (10 ml) se calentó a 100°C durante 2 horas. Una vez completada la reacción, la disolución de reacción se vertió lentamente en hielo picado con agitación, y se añadió agua con hielo. El sólido precipitó después de 1 hora de agitación. El residuo sólido se filtró y se lavó con agua con hielo (15 ml x 3) para dar el compuesto 58 (9,1 mg, 46,1%) como un sólido blanco. LC-MS m/z (ES+), 338,13 (M+H)+.
Síntesis de compuesto 59
El compuesto 58 (8,1 mg, 0,02 mmoles), HOBt (3,25 mg, 0,02 mmoles) y DIC (3,7 ml, 0,02 mmoles) se disolvieron en 5 ml de DMF y se enfriaron a 0°C en un baño de hielo. Posteriormente, se añadieron compuesto 11 (20,1 mg, 0,02 mmoles) y DIPEA (4,1 ml, 0,02 mmoles) a la mezcla. La disolución se dejó calentar a temperatura ambiente, y se dejó reaccionar durante 24 horas. La mezcla resultante se concentró y se purificó por prep-HPLC para producir el compuesto 59 (9,3 mg, 34,9%) como un sólido blanco. LC-MS m/z (ES+), 1313,53 (M+H)+.
EJEMPLO I-10 SÍNTESIS DEL COMPUESTO 64 (no según la presente invención)

i) DMF, 80°C, 6 h; ii) K2CO3, EtOAc/H2O, T.A., 5 h; iii) Ac2O, NaAc, 90°C, 3 h; iv) HOBt, DIC, DIPEA, DMF, T.A., 24 h.
Síntesis de compuesto 60
A una disolución de compuesto 32 (140,0 mg, 0,9 mmoles) en DMF (6 ml), se añadió gota a gota anhídrido maleico (89,8 mg, 0,9 mmoles). La mezcla se agitó a 60°C durante 6 horas, y después se concentró hasta un aceite a presión reducida. El compuesto 60 (sólido blanco, 209,1 mg, 90,7%) se obtuvo por recristalización en acetato de etilo (5 ml). LC-MS m/z (ES+), (M+H)+ 251,06.
Síntesis de compuesto 61
A una mezcla de carbonato de potasio (201,3 mg, 1,4 mmoles) en agua (15 ml) y compuesto 60 (209,1 mg, 0,9 mmoles) en acetato de etilo (20 ml), se añadió gota a gota a 0°C una disolución de cloruro de acriloílo (90 ml, 1,1 mmoles) en acetato de etilo (10 ml). Una vez completada la adición, la mezcla se dejó calentar a temperatura ambiente, y se hizo reaccionar durante 5 horas. La mezcla se acidificó con ácido clorhídrico para ajustar el pH<5. La mezcla se extrajo con acetato de etilo (20 ml x 2). Las capas orgánicas se combinaron y se lavaron con disolución saturada de cloruro de sodio (20 ml), se secaron sobre sulfato de magnesio anhidro, y se filtraron. El disolvente se eliminó a vacío para dar compuesto 61 (201,0 mg, 82,7%) como un sólido gris que pudo usarse en la siguiente etapa sin más purificación. LC-MS m/z (ES+), (M+H)+ 304,07.
Síntesis de compuesto 62
A una disolución de compuesto bruto 61 (201,0 mg, 0,7 mmoles) en anhídrido acético (30 ml), se añadió acetato de sodio (60,5 mg, 0,7 mmoles). La mezcla se agitó a 90°C durante 3 horas. La mezcla de reacción se vertió en agua con hielo, y se agitó durante 30 min y se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se concentraron y se purificaron por prep-HPLC para dar el compuesto 62 (92,2 mg, 48,6%) como un sólido blanco. LC-MS m/z (ES+), (M+H)+ 287,06.
Síntesis de compuesto 64
El compuesto 62 (57,0 mg, 0,2 mmoles), HOBt (27,2 mg, 0,2 mmoles), DIC (29,3 ml, 0,2 mmoles) y DIPEA (0,2 mmoles, 33,1 ml) se añadieron a DMF (6 ml). Después de 30 minutos, se añadió a 0°C el compuesto 63 (109,8 mg, 0,15 mmoles). La mezcla se dejó agitar a temperatura ambiente durante 24 horas. La disolución se concentró y se purificó por prep-HPLC para dar el compuesto 64 (94,8 mg, 47,6%) como un sólido blanco. LC-MS m/z (ES+), 1000,53 (M+H)+.
EJEMPLO I-11 SÍNTESIS DEL COMPUESTO 69 (no según la presente invención)

i) DMF, Reflujo, 12 h; ii) Ac2O, NaAc, Reflujo, 6 h; iii) DMF, 80°C, 12 h; iv) HOBt, DIC, DIPEA, NMP, T.A., 36 h.
Síntesis de compuesto 66
A una mezcla de compuesto 65 (145,0 mg, 1,0 mmoles) en DMF (6 ml), se añadió gota a gota lentamente anhídrido maleico (89,8 mg, 0,9 mmoles). La mezcla se calentó a reflujo durante 12 horas. Después de enfriar a temperatura ambiente, el producto sólido precipitado se filtró y se lavó con CH2Cl2 (20 ml x 3) para producir el compuesto 66 (110,2 mg, 45,1%) como un sólido blanco. LC-MS m/z (ES+), 244,02 (M+H)+.
Síntesis de compuesto 67
Una mezcla de compuesto 66 (110,2 mg, 0,5 mmoles) y acetato de sodio (14,2 mg, 0,2 mmoles) en anhídrido acético (10 ml) se calentó a reflujo durante 6 horas. Una vez completada la reacción, la mezcla de reacción se vertió lentamente en hielo picado con agitación, y se añadió agua con hielo. El residuo sólido precipitó después de 1 hora de agitación. El residuo sólido se filtró y se lavó con agua con hielo (10 ml x 3) para dar el compuesto 67 (87,3 mg, 85,7%) como un sólido blanco. LC-MS m/z (ES+), 226,01 (M+H)+.
Síntesis de compuesto 68
Se añadió anhídrido succínico (38,8 mg, 0,4 mmoles) a una mezcla del compuesto 67 (87,3 mg, 0,4 mmoles) en DMF (6 ml). La mezcla se agitó a 80°C durante la noche, y después se concentró hasta un aceite a presión reducida. El producto bruto se purificó por prep-HPLC para dar el compuesto 68 (45,0 mg, 35,7%) como un sólido blanco. LC-MS m/z (ES+), 326,02 (M+H)+.
Síntesis de compuesto 69
El compuesto 68 (45,0 mg, 0,14 mmoles), HOBt (19,0 mg, 0,14 mmoles), DIC (20,5 ml, 0,14 mmoles) y DIPEA (23,2 ml, 0,14 mmoles) se añadieron a DMF (10 ml) y se agitaron a 0°C en un baño de hielo durante 1 hora. Se añadió compuesto 63 (76,9 mg, 0,11 mmoles) a la disolución. La mezcla se dejó calentar a temperatura ambiente, y se dejó reaccionar durante 36 horas. La mezcla de reacción se purificó por prep-HPLC para producir el compuesto 69 (64,3 mg, 44,3%) como un sólido blanco. LC-MS m/z (ES+), 1039,49 (M+H)+.
EJEMPLO I-12 SÍNTESIS DEL COMPUESTO 74 (no según la presente invención)

i) Et3N, n-butanol, 90°C, 18 h, ii) Et3N, CH2Cl2, T.A., 18 h, iii) EDCI, HOBt, DIPEA, DMF, T.A., 18 h, iv) EEDQ, CH2G 2, MeOH, T.A., 18 h.
Síntesis de compuesto 71
El compuesto 69 (1,0 g, 6,10 mmoles) y ácido 4-aminobutanoico (691,9 mg, 6,71 mmoles) y trietilamina (3,09 g, 30,5 mmoles) se disolvieron en n-butanol (30 ml). La mezcla de reacción se agitó durante 18 horas a 90°C. Después de enfriar a temperatura ambiente, el disolvente se concentró a presión reducida. El residuo se diluyó con agua (100 ml), y se extrajo con acetato de etilo (30 ml x 3). La fase orgánica se combinó y se secó sobre sulfato de sodio anhidro, se filtró, y se concentró. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 200/1 ~10/1, v/v) para producir compuesto 71 como un sólido amarillo (482,3 mg, 34,3%). LC-MS m/z (ES+), 231,06 (M+H)+.
Síntesis de compuesto 72
Se añadió gota a gota cloruro de acriloílo (947,2 mg, 10,47 mmoles) a una disolución del compuesto 71 (450,5 mg, 1,95 mmoles) y TEA (590,8 mg, 5,85 mmoles) en CH2G 2 (20 ml) con agitación a 0°C en un baño de hielo. La mezcla de reacción se agitó durante 18 horas a temperatura ambiente. La reacción se inactivó mediante la adición de agua (50 ml), y se extrajo con CH2Cl2 (20 ml x 3). La capa orgánica se combinó, se lavó con salmuera (30 ml), se secó sobre sulfato de sodio anhidro, se filtró, y se concentró para producir el compuesto 72 (255,4 mg, 46%) como un sólido amarillo. LC-MS m/z (ES+), 285,07 (M+H)+-
Síntesis de compuesto 73
El compuesto 3 (155,7 mg, 0,547 mmoles), HOBt (88,7 mg, 0,656 mmoles), EDCI (115,4 mg, 0,602 mmoles) y DIPEA (212,1 mg, 1,64 mmoles) se añadieron a DMF (10 ml) y se agitaron a 0°C en un baño de hielo durante 1 hora. Se añadió compuesto 40 (113,3 mg, 0,602 mmoles) a la disolución. La mezcla se dejó calentar a temperatura ambiente, y se dejó reaccionar durante 18 horas. La mezcla de reacción se diluyó con salmuera (30 ml), se extrajo con CH2G 2 (10 ml x 3). La capa orgánica se combinó y se secó sobre sulfato de sodio anhidro, se filtró, y se concentró. El residuo se purificó por cromatografía en columna (MeOH:CH2Cl2 = 200/1 ~10/1, v/v) para producir compuesto 73 como un sólido amarillo (90,2 mg, 36,2%). LC-MS m/z (ES+), 455,17 (M+H)+.
Síntesis de compuesto 74
Una disolución de compuesto 73 (85,4 mg, 0,19 mmoles), EEDQ (55,4 mg, 0,23 mmoles) en CH2Cl2 anhidro (7 ml) y MeOH anhidro (0,07 ml) se agitó durante 1 hora a temperatura ambiente. Posteriormente, se añadió compuesto 42 (135,7 mg, 0,19 mmoles). La mezcla de reacción se agitó durante 18 horas más. La mezcla de reacción se concentró a vacío, y se purificó por prep-HPLC para producir el compuesto 74 (19,1 mg, 6,7%) como un sólido blanco. LC-MS m/z (ES+), 1162,45 (M+H)+.
Tabla 1 Otros conjugados de enlazador-agente activo


EJEMPLO II PREPARACIÓN DE CONJUGADOS DE ANTICUERPO-FÁRMACO
Procedimiento general de conjugación
DTPA (1 mM) y 3,0-5,0 equiv. de TCEP (10 mM) se añadieron al anticuerpo (10 mg/ml) en un tubo de PCR, y se mezclaron durante 1 hora para reducir los enlaces de disulfuro en el anticuerpo. Se añadieron a las mezclas de reacción diversas cantidades del conjugado de enlazador-agente activo, y se agitaron durante la noche usando un mezclador de vórtice. Todas las reacciones se llevaron a cabo a temperatura ambiente. Los productos resultantes se analizaron mediante electroforesis en gel (SDS-PAGE) y HIC (cromatografía de interacción hidrófoba)-HPLC.
El conjugado de anticuerpo-fármaco se analizó mediante SDS-PAGE en condiciones reducidas: las muestras mezcladas con amortiguador se cargaron en geles de archilamida al 8%-15%, y se tomaron fotografías de gel con un sistema de imágenes UVP BioDoc-it.
Las bandas (>50 kDa) aparecieron en los carriles 2 y 3, lo que sugiere que la conjugación del puente de tiol fue efectiva (H, L). Las bandas a 75 kDa representan la parte compuesta por una cadena pesada y una ligera que estaban unidas por un par de tioles puenteados (LH). Dos cadenas pesadas estaban unidas por uno o dos pares de tioles puenteados, que formaban las bandas a 100 kDa (HH). Las bandas a 125 kDa representan la parte en la que solo no se formó un puente entre una cadena pesada y una ligera (LHH). Por último, las bandas a 150 kDa revelaron la formación de un anticuerpo totalmente puenteado (LHHL).
El conjugado de anticuerpo-fármaco se analizó mediante HIC-HPLC en las siguientes condiciones: columna de HPLC: columna TSK-GEL Butyl-NPR 4,6*35 mm; amortiguador A: 75% de fosfato de sodio (20 mM, pH 7,0), 25% de isopropanol (v/v); amortiguador B: fosfato de sodio (20 mM, pH 7,0), sulfato de amonio (1,5 mM); caudal: 1,0 ml/min.
Evaluación in vitro de conjugados de anticuerpo-fármaco
El primer día, una línea de células tumorales de líneas celulares de cáncer de ovario humano, tal como OVCAR3, se sembró en placas a 5000 células en 100 ml de medio de cultivo en una placa de cultivo de 96 pocillos (Cellstar). Las células se incubaron durante la noche a 37°C en una incubadora de CO2 con agua saturada. El segundo día, dos ADC conjugados con tiol covalente de MSL-31 y MSL-75 se diluyeron en serie, y los conjugados se añadieron a la placa de 96 pocillos que contenía células OVCAR3, 100 ml por pocillo. El conjugado inicial fue de 100000 ng/ml, y se diluyó hasta 0,001 ng/ml. Las células OVCAR3 con ADC añadido se incubaron a 37°C durante 72 horas. A continuación, se midió la viabilidad de la célula tumoral tratada con ADC conjugado con tiol covalente usando un kit de viabilidad celular, Cell Counting Kit-8 (DOJINDO, CK04), según el protocolo del fabricante, en un lector de placas (BIO-RAD iMarkSpectraMax L de Molecular device). El valor de lC50, 50% de inhibición del crecimiento celular, se calculó usando un software de ajuste de curvas, Graphpad.
EJEMPLO II-1 PREPARACIÓN DE MSL-C31
DTPA (1 mM) y 5,0 equiv. de TCEP (10 mM) se añadieron a anti-MSL (10 mg/ml) en un tubo de PCR. La mezcla se agitó con un mezclador de vórtice durante 1 hora para reducir los enlaces de disulfuro en el anticuerpo. Posteriormente, se añadió 25% de DMSO a las mezclas de reacción. El compuesto 31 (5,0 equiv., 10 mM) se añadió a las mezclas de reacción y se agitó durante la noche usando un mezclador de vórtice. Todas las reacciones se llevaron a cabo a temperatura ambiente. Una vez finalizada la reacción, la mezcla se cargó en el tubo de ultrafiltración centrífuga Millipore de 4 ml, y se lavó tres veces con PBS, y después se recogieron las mezclas de reacción. La Figura 2 muestra que los productos (el carril derecho) de la conjugación covalente de al menos un par de grupos sulfhidrilo migrarán de forma más lenta que los de la cadena pesada (H) o ligera (L) sola (mostrado en el carril izquierdo). La conjugación covalente entre las cadenas pesada y ligera produjo la banda de 75 kDa (HL); la conjugación covalente de dos cadenas pesadas produjo la banda de 100 kDa (HH). Hay 3 picos en el cromatograma de HIC-HPLC, que representan 3, 4 y 5 de DAR, respectivamente. La DAR del MSL-C31 resultante fue 3,60 basado en los resultados de HIC-HPLC (Figura 3).
La Figura 2 muestra el resultado de SDS-PAGE en condiciones reducidas para a) anticuerpo desnudo, b) MSL-C31, el producto del conjugado de tiol covalente.
La Figura 3 muestra un cromatograma de HIC-HPLC de MSL-C31.
EJEMPLO II-2 PREPARACIÓN DE MSL-C75
DTPA (1 mM) y 5,0 equiv. de TCEP (10 mM) se añadieron a anti-MSL (10 mg/ml) en un tubo de PCR. La mezcla se agitó con un mezclador de vórtice durante 1 hora para reducir los enlaces de disulfuro en el anticuerpo. Posteriormente, se añadió 25% de DMSO a las mezclas de reacción. Se añadieron 5,0 equiv. del compuesto 75 (5,0 equiv., 10 mM) a las mezclas de reacción, y se agitó durante la noche usando un mezclador de vórtice. Todas las reacciones se llevaron a cabo a temperatura ambiente. Una vez finalizada la reacción, la mezcla se cargó en el tubo de ultrafiltración centrífuga Millipore de 4 ml, y se lavó tres veces con PBS, y después se recogieron las mezclas de reacción. Las muestras se sometieron a análisis por electroforesis. Los resultados de SDS-PAGE en condiciones reducidas se presentaron en la Figura 4. El carril izquierdo era anticuerpo desnudo, y el carril derecho era conjugado covalente de MSL-C75. La DAR del MSL-C75 resultante fue 3,87 basado en HIC-HPLC.
La Figura 4 muestra los resultados de SDS-PAGE en condiciones reducidas de MSL-C75.
La Figura 5 muestra un cromatograma de HIC-HPLC de MSL-C75.
La Tabla 2 muestra los valores de IC50 de dos muestras: ADC-C31 y ADC-C75, analizadas en células de cáncer de ovario humano.

EJEMPLO II-3 PREPARACIÓN DE CD59-C78 (no según la presente invención)
DTPA (1 mM) y 5,0 equiv. de TCEP (10 mM) se añadieron a anti-CD59 (10 mg/ml) en un tubo de PCR. La mezcla se agitó con un mezclador de vórtice durante 1 hora para reducir los enlaces de disulfuro en el anticuerpo. Posteriormente, se añadió 25% de DMSO a las mezclas de reacción. Se añadió compuesto 78 (5,0 equiv., 10 mM) a las mezclas de reacción, y se agitó durante la noche usando un mezclador de vórtice. Todas las reacciones se llevaron a cabo a temperatura ambiente. Una vez finalizada la reacción, la mezcla se cargó en el tubo de ultrafiltración centrífuga Millipore de 4 ml, y se lavó tres veces con PBS. La DAR del CD59-C78 resultante fue 3,66 basado en HIC-HPLC.
La Figura 6 muestra el resultado de SDS-PAGE en condiciones reducidas para a) anticuerpo desnudo, b) CD59-C78.
La Figura 7 muestra un cromatograma de HIC-HPLC de CD59-C78.
EJEMPLO II-4 PREPARACIÓN DE CONJUGADO DE HER2-C75
DTPA (1 mM) y 5,0 equiv. de TCEP (10 mM) se añadieron a anti-Her2 (10 mg/ml) en un tubo de PCR. La mezcla se agitó con un mezclador de vórtice durante 1 hora para reducir los enlaces de disulfuro en el anticuerpo. Posteriormente, se añadió 25% de DMSO a las mezclas de reacción. Se añadió compuesto 75 (5,0 equiv., 10 mM) a las mezclas de reacción, y se agitó durante la noche usando un mezclador de vórtice. Todas las reacciones se llevaron a cabo a temperatura ambiente. Una vez finalizada la reacción, la mezcla se cargó en el tubo de ultrafiltración centrífuga Millipore de 4 ml, y se lavó tres veces con PBS.
Estudio de estabilidad del conjugado de HER2-C75
Para determinar las tasas de degradación de los ADC de la conjugación de tiol covalente frente a la conjugación de tiol normal, se usaron tres muestras de ADC preparadas mediante los procedimientos anteriores para el ELISA para el estudio de estabilidad. El plan de implementación de ELISA es el siguiente: en primer lugar, Her2-ECD (100 ng) se recubrió en cada pocillo de una placa de 96 pocillos; en segundo lugar, sangre periférica humana tratada con Anti-Her2-Mc-VC-PAB-MMAE (de conjugación de tiol normal), Anti-Her2-Di-Mc-VC-PAB-MMAE (conjugación de bismaleimida-tiol) y Anti-Her2-PY-QJ2-ADC (HER2-C75, de conjugación de tiol covalente de esta invención) se incubó en el pocillo revestido para permitir la unión a Her-2 ECD. Por último, la cantidad de ADC anti-her2 unidos al antígeno Her2-ECD se mide mediante la unión de IgG antihumana conjugada con estreptavidina/peroxidasa de rábano picante, que después cataliza la conversión del sustrato cromogénico o-fenilendiamina (OPD) en un producto coloreado. Los resultados muestran que durante 3 días de incubación con las sangres periféricas, la tasa de degradación de ADC con la conjugación de tiol normal fue 26,7%, la del 2° ADC con conjugación de bis-maleimida-tiol fue 18,9%, y la del 3er ADC de esta invención fue 5,1%. La tasa de degradación de ADC con el enlazador covalente de esta invención es la más baja entre los tres ADC, como se muestra en la Tabla 3 a continuación.
Tabla 3. Sumario de las tasas de degradación del conjugado de anti-Her2-fármaco en sangre humana

EJEMPLO III - Actividad de los conjugados de anticuerpo-fármaco
Los conjugados de anticuerpo-fármaco de la presente invención se pueden usar, en una realización, para dianizar específicamente células tumorales y unirse a antígenos asociados a tumores en la superficie de las células tumorales. El complejo de ADC-antígeno entrará en la célula tumoral por endocitosis y liberará su carga útil después de que el endosoma del complejo de ADC-antígeno se fusione con los lisosomas. Las cargas útiles liberadas inhiben las funciones clave de una célula tumoral y/o incluso exterminan las células tumorales. Si las cargas útiles se liberan en la superficie de la célula o cerca de las células tumorales, la célula tumoral también se puede destruir de manera espectadora.
Así, la invención proporciona diversos complejos de proteína-fármaco que inhiben el crecimiento de células tumorales y/o su diferenciación. Estos compuestos, armados con diversos tipos de cargas útiles activas, se pueden usar para tratar neoplasias, trastornos autoinmunes e inflamación, incluso las diferenciaciones de un tumor o una célula madre.
EJEMPLO III-1 - Actividad contra tumores
En una realización, o la invención objeto, los conjugados de anticuerpo-fármaco (ADC) combinan el direccionamiento único de los mAb con la capacidad de eliminación del cáncer de los fármacos citotóxicos, y permiten que los ADC sean agentes potentes para la eliminación de tumores. Además, el uso de conjugación de tiol covalente según la presente invención aumenta la estabilidad de los ADC, limitando la descomposición del ADC en la circulación sanguínea, reduciendo así su toxicidad. Este ejemplo proporciona datos que muestran el tratamiento del cáncer humano, usando ADC preparados como se describe anteriormente, en células de cáncer de mama humano implantadas en ratones atímicos (MDA-MB-231).
Para determinar la función biológica de diversos ADC con conjugados covalentes o normales, se ha conjugado mAb anti-Her-2 con tiol con conjugación normal o se ha conjugado con tiol covalente con el mismo enlazador y citotoxina. A los ratones atímicos se les implantó una línea celular de cáncer humano (MDA-MB-231), y los ADC (0, 2,5, 5,0 y 10 mg/kg) se administraron mediante inyección intraperitoneal cuando el volumen del tumor alcanzó alrededor de 100 milímetros cúbicos. Los volúmenes de los tumores se monitorizaron cada 3 días, y los animales se sacrificaron al final de los 21 días, y los tumores se diseccionaron y se pesaron.
La Tabla 4 a continuación proporciona una comparación de la eficacia entre ADC normal frente a ADC de conjugación de tiol covalente administrado en ratones atímicos implantados con células de cáncer de mama humano.
Tabla 4. ADC conjugado con tiol covalente inhibe el crecimiento de células tumorales humanas en el estudio con ratones atímicos

REFERENCIAS
1. Yao Xuejing, Jing Jiang, Xin Wang, Changjiang Huang Dong Li, Kuan Xie, Qiaoyu Xu, Hongwen Li, Zhuanglin Li, Liguang Lou and Jianmin Fang, 2015 “A novel humanized anti-her2 antibody conjugated with MMAE exerts potent anti-tumor activity”, Breast Cancer Research & Treatment, 153(1): 123-133.
2. An Deqiang et a l, 20140723
Claims (5)
1. Un enlazador-agente activo o una sal farmacéuticamente aceptable del mismo, en el que el enlazador-agente activo es

2. Un conjugado de anticuerpo-fármaco o una sal farmacéuticamente aceptable del mismo, en el que el conjugado de anticuerpo-fármaco es

en el que A es un anticuerpo o un fragmento de anticuerpo.
3. Un conjugado de anticuerpo-fármaco de la reivindicación 2 para uso en un método para tratar una condición patológica en un sujeto, en el que dicho método comprende una administración a un sujeto que necesita de tal tratamiento, y en particular el sujeto es un ser humano.
4. Un conjugado de anticuerpo-fármaco de la reivindicación 3 para uso en un método para tratar una condición patológica en un sujeto, en el que A es un anticuerpo, o un fragmento del mismo, que se une a HER2, y la condición patológica es un cáncer de mama.
5. El conjugado de anticuerpo-fármaco para uso según la reivindicación 3, en el que dicha condición patológica se selecciona de cáncer, trastornos inflamatorios, trastornos autoinmunes, trastornos del sistema nervioso, y trastornos cardiovasculares,
preferiblemente el cáncer se selecciona de carcinoma, linfoma, blastoma, sarcoma, y leucemia o neoplasias malignas linfoides,
más preferiblemente, el cáncer es un cáncer positivo para HER2, y en particular, cáncer de mama.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562205121P | 2015-08-14 | 2015-08-14 | |
| PCT/US2016/046987 WO2017031034A2 (en) | 2015-08-14 | 2016-08-15 | Covalent linkers in antibody-drug conjugates and methods of making and using the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2907651T3 true ES2907651T3 (es) | 2022-04-25 |
Family
ID=58052236
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16837634T Active ES2907651T3 (es) | 2015-08-14 | 2016-08-15 | Enlazadores covalentes en conjugados de anticuerpo-fármaco y métodos para obtener y usar los mismos |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US10537644B2 (es) |
| EP (2) | EP3981433A1 (es) |
| JP (1) | JP6714687B2 (es) |
| KR (1) | KR102097876B1 (es) |
| CN (3) | CN107921030B (es) |
| AU (1) | AU2016308539B2 (es) |
| CA (1) | CA2994853C (es) |
| DK (1) | DK3334462T3 (es) |
| ES (1) | ES2907651T3 (es) |
| HU (1) | HUE057906T2 (es) |
| PL (1) | PL3334462T3 (es) |
| PT (1) | PT3334462T (es) |
| RU (1) | RU2698727C9 (es) |
| TW (1) | TWI715611B (es) |
| WO (1) | WO2017031034A2 (es) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2784603T3 (es) | 2015-06-02 | 2020-09-29 | Novo Nordisk As | Insulinas con extensiones recombinantes polares |
| WO2017055582A1 (en) | 2015-10-01 | 2017-04-06 | Novo Nordisk A/S | Protein conjugates |
| US10864279B2 (en) * | 2016-12-16 | 2020-12-15 | Industrial Technology Research Institute | Linker-drug and antibody-drug conjugate (ADC) employing the same |
| CN106946926A (zh) * | 2017-02-27 | 2017-07-14 | 西北大学 | 一种具有多齿氨羧类二聚体螯合剂及其制备方法、应用和分离介质 |
| JP7152408B2 (ja) | 2017-03-10 | 2022-10-12 | キアペグ ファーマシューティカルズ アクチエボラグ | 遊離可能コンジュゲート |
| MA49339A (fr) * | 2017-04-05 | 2020-02-12 | Novo Nordisk As | Conjugués insuline-fc à extension oligomère |
| CN109232464B (zh) * | 2017-07-10 | 2022-11-15 | 上海新理念生物医药科技有限公司 | 噁二唑型连接子及其应用 |
| CN120248019B (zh) * | 2017-12-15 | 2026-02-03 | 四川科伦博泰生物医药股份有限公司 | 生物活性物偶联物及其制备方法和用途 |
| WO2019157772A1 (zh) * | 2018-02-13 | 2019-08-22 | 和元生物技术(上海)股份有限公司 | 抗trailr2抗体-毒素-偶联物及其在抗肿瘤治疗中的药物用途 |
| WO2019171358A1 (en) | 2018-03-09 | 2019-09-12 | Quiapeg Pharmaceuticals Ab | Releasable antibody conjugates |
| CN110507824A (zh) * | 2018-05-21 | 2019-11-29 | 荣昌生物制药(烟台)有限公司 | 一种抗间皮素抗体及其抗体药物缀合物 |
| CN109602915A (zh) * | 2018-07-03 | 2019-04-12 | 烟台迈百瑞国际生物医药有限公司 | 抗体-t2毒素缀合物及其用途 |
| WO2020053815A1 (en) | 2018-09-12 | 2020-03-19 | Quiapeg Pharmaceuticals Ab | Releasable glp-1 conjugates |
| CA3106493C (en) * | 2018-12-17 | 2024-01-09 | Remegen, Co., Ltd. | A linker for antibody-drug conjugates and its use |
| CA3085001C (en) * | 2019-05-20 | 2021-10-05 | Mabplex International, Ltd. | One-pot process for preparing intermediate of antibody-drug conjugate |
| RU2745738C1 (ru) * | 2019-05-20 | 2021-03-31 | МэбПлекс Интернэшнл Ко., Лтд. | Однореакторный способ получения промежуточного продукта конъюгата антитело-лекарственное средство |
| FR3096259B1 (fr) * | 2019-05-20 | 2023-12-15 | Mc Saf | conjugués anticorps-médicament et leur utilisation en thérapie |
| CN111620927B (zh) * | 2019-05-20 | 2022-11-11 | 烟台迈百瑞国际生物医药股份有限公司 | 一种抗体药物偶联物中间体的一锅法制备工艺 |
| FR3101541B1 (fr) * | 2019-10-04 | 2021-10-22 | Mc Saf | conjugués anticorps-médicament et leur utilisation en thérapie |
| WO2022022649A1 (zh) * | 2020-07-30 | 2022-02-03 | 成都科岭源医药技术有限公司 | 一种双药链接组装单元及双药靶向接头-药物偶联物 |
| CN115105607B (zh) * | 2021-03-22 | 2024-12-20 | 成都科岭源医药技术有限公司 | 一种用于adc的双药-接头的制备方法及其用途 |
| EP4596536A1 (en) | 2022-09-30 | 2025-08-06 | Shanghai de Novo Pharmatech Co., Ltd. | Benzazepine derivative, conjugate containing same, and use thereof |
| WO2025003392A1 (en) * | 2023-06-28 | 2025-01-02 | Université De Tours | Bioconjugation systems and uses thereof |
| CN119431181A (zh) * | 2023-08-04 | 2025-02-14 | 泰诚思(上海)生物医药有限公司 | 双联烯连接子、连接子与药物缀合物、抗体偶联药物及其用途 |
| TW202543679A (zh) * | 2023-12-29 | 2025-11-16 | 大陸商杭州尚健生物技術有限公司 | 一種免疫綴合物及其用途 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2615887A (en) * | 1952-10-28 | Reaction products of nitriles | ||
| US20030215395A1 (en) | 2002-05-14 | 2003-11-20 | Lei Yu | Controllably degradable polymeric biomolecule or drug carrier and method of synthesizing said carrier |
| US7282590B2 (en) * | 2004-02-12 | 2007-10-16 | The Research Foundation Of State University Of New York | Drug conjugates |
| US6998978B2 (en) | 2004-04-29 | 2006-02-14 | International Business Machines Corporation | Method and apparatus for responding to medical alerts |
| KR101200133B1 (ko) * | 2004-06-01 | 2012-11-13 | 제넨테크, 인크. | 항체 약물 접합체 및 방법 |
| US7947839B2 (en) * | 2004-12-01 | 2011-05-24 | Genentech, Inc. | Heterocyclic-substituted bis-1,8 naphthalimide compounds, antibody drug conjugates, and methods of use |
| WO2009148554A1 (en) * | 2008-05-30 | 2009-12-10 | Siemens Medical Solutions Usa, Inc. | Achievement of high therapeutic index through molecular imaging guided targeted drug treatment |
| MX2014006739A (es) | 2011-12-05 | 2015-06-05 | Igenica Biotherapeutics Inc | Conjugados de anticuerpo-farmaco y compuestos relacionados, composiciones , y metodos. |
| EP2822597A1 (en) * | 2012-03-09 | 2015-01-14 | UCL Business Plc. | Chemical modification of antibodies |
| WO2013185117A1 (en) * | 2012-06-07 | 2013-12-12 | Ambrx, Inc. | Prostate-specific membrane antigen antibody drug conjugates |
| CN103933575B (zh) | 2013-01-23 | 2017-09-29 | 上海新理念生物医药科技有限公司 | 一种三齿型连接子及其应用 |
| FR3008408B1 (fr) * | 2013-07-11 | 2018-03-09 | Mc Saf | Nouveaux conjugues anticorps-medicament et leur utilisation en therapie |
| JP6393324B2 (ja) * | 2013-10-30 | 2018-09-19 | デンツプライ シロナ インコーポレーテッド | 1,3,5−トリアクリロイル−ヘキサヒドロ−1,3,5−トリアジン部分を含有する重合性モノマー、これらを製造する方法、およびこれらを含有する歯科用組成物 |
| CA2950155C (en) * | 2014-06-02 | 2021-11-23 | Regeneron Pharmaceuticals, Inc. | Biologically active molecule conjugates, reagents and methods of manufacture, and therapeutic uses |
| WO2016064749A2 (en) * | 2014-10-20 | 2016-04-28 | Igenica Biotherapeutics, Inc. | Novel antibody-drug conjugates and related compounds, compositions, and methods of use |
-
2016
- 2016-08-15 CN CN201680046806.1A patent/CN107921030B/zh active Active
- 2016-08-15 PT PT168376341T patent/PT3334462T/pt unknown
- 2016-08-15 WO PCT/US2016/046987 patent/WO2017031034A2/en not_active Ceased
- 2016-08-15 CN CN202210509207.6A patent/CN114939174B/zh active Active
- 2016-08-15 CN CN202410947073.5A patent/CN118743763A/zh active Pending
- 2016-08-15 KR KR1020187003685A patent/KR102097876B1/ko active Active
- 2016-08-15 US US15/524,487 patent/US10537644B2/en active Active
- 2016-08-15 RU RU2018108613A patent/RU2698727C9/ru active
- 2016-08-15 EP EP21206552.8A patent/EP3981433A1/en active Pending
- 2016-08-15 ES ES16837634T patent/ES2907651T3/es active Active
- 2016-08-15 JP JP2018505740A patent/JP6714687B2/ja active Active
- 2016-08-15 TW TW105126166A patent/TWI715611B/zh active
- 2016-08-15 EP EP16837634.1A patent/EP3334462B8/en active Active
- 2016-08-15 HU HUE16837634A patent/HUE057906T2/hu unknown
- 2016-08-15 AU AU2016308539A patent/AU2016308539B2/en active Active
- 2016-08-15 CA CA2994853A patent/CA2994853C/en active Active
- 2016-08-15 PL PL16837634T patent/PL3334462T3/pl unknown
- 2016-08-15 DK DK16837634.1T patent/DK3334462T3/da active
-
2018
- 2018-02-14 US US15/896,428 patent/US10772965B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2907651T3 (es) | Enlazadores covalentes en conjugados de anticuerpo-fármaco y métodos para obtener y usar los mismos | |
| ES2994375T3 (en) | Hydrophilic drug-linker compounds | |
| AU2018327171B2 (en) | Improved method for producing antibody-drug conjugate | |
| ES2747386T3 (es) | Dímeros de benzodiacepina unidos por heteroarileno, conjugados de los mismos y métodos de preparación y uso | |
| JP2021532162A (ja) | トル様受容体7(TLR7)アゴニストとしての1H−ピラゾロ[4,3−d]ピリミジン化合物ならびにその方法および使用 | |
| JP2020531472A (ja) | 免疫刺激因子トル様受容体7(tlr7)アゴニストとしての、6−アミノ−7,9−ジヒドロ−8h−プリン−8−オン誘導体 | |
| JP2020531468A (ja) | 免疫刺激因子トル様受容体7(tlr7)アゴニストとしての6−アミノ―7,9−ジヒドロ−8h−プリン−8−オン誘導体 | |
| JP2020531466A (ja) | 免疫刺激物質トル様受容体7(tlr7)アゴニストとしての6−アミノ−7,9−ジヒドロ−8h−プリン−8−オン誘導体 | |
| JP2020531469A (ja) | 三環式基を有するトル様受容体7(tlr7)アゴニスト、そのコンジュゲート、ならびにそれらの方法および使用 | |
| AU2011307236A1 (en) | Thiazolidine linker for the conjugation of drugs to antibodies | |
| JP2021536430A (ja) | リンカー、それを含む抗体薬物コンジュゲート、およびその使用 | |
| KR20190039570A (ko) | 세코-시클로프로파피롤로인돌 화합물, 그의 항체-약물 접합체, 및 제조 및 사용 방법 | |
| CN116036303A (zh) | 一种抗体-药物偶联物及其制备方法和应用 | |
| ES2889775T3 (es) | Proceso mejorado para la síntesis del fármaco-enlazador vc-seco | |
| HK40066766A (en) | Covalent linkers in antibody-drug conjugates and methods of making and using the same | |
| HK1251182B (en) | Covalent linkers in antibody-drug conjugates and methods of making and using the same | |
| JP7667870B2 (ja) | Dna毒性二量体及びその複合体 | |
| CN120285217A (zh) | 双特异性抗体-奥瑞他汀类药物偶联物及其用途 | |
| BR112018002033B1 (pt) | Agente ligante-ativo, conjugado de anticorpo-fármaco e seus usos | |
| HK40120190A (zh) | 含毛兰素的抗体药物偶联物及其制备方法和应用 |