ES2909709T3 - Inhibidores del receptor 2 activado por proteasa - Google Patents
Inhibidores del receptor 2 activado por proteasa Download PDFInfo
- Publication number
- ES2909709T3 ES2909709T3 ES17725905T ES17725905T ES2909709T3 ES 2909709 T3 ES2909709 T3 ES 2909709T3 ES 17725905 T ES17725905 T ES 17725905T ES 17725905 T ES17725905 T ES 17725905T ES 2909709 T3 ES2909709 T3 ES 2909709T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- mmol
- methanol
- fluoro
- imidazol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 108010070503 PAR-2 Receptor Proteins 0.000 title description 59
- 239000003112 inhibitor Substances 0.000 title description 2
- 102000018402 Protease-activated receptor 2 Human genes 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 320
- 150000003839 salts Chemical class 0.000 claims abstract description 65
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 55
- 125000003118 aryl group Chemical group 0.000 claims abstract description 51
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 47
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 40
- 125000004429 atom Chemical group 0.000 claims abstract description 39
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims abstract description 37
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 36
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims abstract description 34
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims abstract description 34
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 30
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 27
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims abstract description 24
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 23
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 21
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 20
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 20
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 19
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 19
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 19
- 125000005843 halogen group Chemical group 0.000 claims abstract description 17
- 125000006714 (C3-C10) heterocyclyl group Chemical group 0.000 claims abstract description 16
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 13
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 13
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 11
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims abstract description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 56
- 239000008194 pharmaceutical composition Substances 0.000 claims description 35
- 208000002193 Pain Diseases 0.000 claims description 33
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 33
- 201000010099 disease Diseases 0.000 claims description 32
- 208000035475 disorder Diseases 0.000 claims description 24
- 230000000694 effects Effects 0.000 claims description 24
- 239000003937 drug carrier Substances 0.000 claims description 14
- 201000004624 Dermatitis Diseases 0.000 claims description 13
- 230000004054 inflammatory process Effects 0.000 claims description 12
- 206010061218 Inflammation Diseases 0.000 claims description 11
- 206010009887 colitis Diseases 0.000 claims description 11
- 208000036110 Neuroinflammatory disease Diseases 0.000 claims description 10
- 208000003251 Pruritus Diseases 0.000 claims description 10
- 230000007803 itching Effects 0.000 claims description 10
- 208000037883 airway inflammation Diseases 0.000 claims description 9
- 230000001404 mediated effect Effects 0.000 claims description 7
- 125000002950 monocyclic group Chemical group 0.000 claims description 7
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 3
- 101000603877 Homo sapiens Nuclear receptor subfamily 1 group I member 2 Proteins 0.000 claims 1
- 101001098560 Homo sapiens Proteinase-activated receptor 2 Proteins 0.000 claims 1
- 101000713170 Homo sapiens Solute carrier family 52, riboflavin transporter, member 1 Proteins 0.000 claims 1
- 102100036863 Solute carrier family 52, riboflavin transporter, member 1 Human genes 0.000 claims 1
- -1 wherein said C6- 10 Chemical group 0.000 abstract description 70
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 268
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 231
- 239000000203 mixture Chemical class 0.000 description 200
- 238000000034 method Methods 0.000 description 163
- 235000019439 ethyl acetate Nutrition 0.000 description 133
- 229910052739 hydrogen Inorganic materials 0.000 description 132
- 239000007787 solid Substances 0.000 description 117
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 110
- 238000004128 high performance liquid chromatography Methods 0.000 description 106
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 98
- 238000005160 1H NMR spectroscopy Methods 0.000 description 96
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 96
- 125000000217 alkyl group Chemical group 0.000 description 94
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 94
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 89
- 239000000243 solution Substances 0.000 description 89
- 239000000543 intermediate Substances 0.000 description 77
- 239000012043 crude product Substances 0.000 description 76
- 239000012299 nitrogen atmosphere Substances 0.000 description 72
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 70
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 69
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 66
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 66
- 239000002253 acid Substances 0.000 description 61
- 239000011541 reaction mixture Substances 0.000 description 59
- 102000032628 PAR-2 Receptor Human genes 0.000 description 58
- 238000003818 flash chromatography Methods 0.000 description 54
- 230000002829 reductive effect Effects 0.000 description 52
- 238000010828 elution Methods 0.000 description 51
- 239000012044 organic layer Substances 0.000 description 50
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 49
- 239000002904 solvent Substances 0.000 description 48
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 47
- 239000007832 Na2SO4 Substances 0.000 description 46
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 46
- 229910052938 sodium sulfate Inorganic materials 0.000 description 46
- 235000011152 sodium sulphate Nutrition 0.000 description 46
- 239000000377 silicon dioxide Substances 0.000 description 43
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 40
- 239000003208 petroleum Substances 0.000 description 40
- 239000012071 phase Substances 0.000 description 38
- 125000001309 chloro group Chemical group Cl* 0.000 description 32
- 125000001424 substituent group Chemical group 0.000 description 32
- 238000011282 treatment Methods 0.000 description 32
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 31
- 239000003814 drug Substances 0.000 description 28
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 26
- 239000003921 oil Substances 0.000 description 26
- 235000019198 oils Nutrition 0.000 description 26
- 239000000047 product Substances 0.000 description 26
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 23
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 23
- 125000001072 heteroaryl group Chemical group 0.000 description 22
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 21
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 20
- 235000019341 magnesium sulphate Nutrition 0.000 description 20
- 239000004480 active ingredient Substances 0.000 description 19
- 125000003545 alkoxy group Chemical group 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 19
- 229940079593 drug Drugs 0.000 description 19
- 235000019441 ethanol Nutrition 0.000 description 19
- 125000001183 hydrocarbyl group Chemical group 0.000 description 19
- 239000012074 organic phase Substances 0.000 description 19
- 239000000651 prodrug Substances 0.000 description 19
- 229940002612 prodrug Drugs 0.000 description 19
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 18
- 239000008346 aqueous phase Substances 0.000 description 18
- 239000012267 brine Substances 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 17
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 17
- 238000004296 chiral HPLC Methods 0.000 description 17
- SUBYSXRGWSURGY-UHFFFAOYSA-N 2-(imidazol-1-ylmethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCN1C=CN=C1 SUBYSXRGWSURGY-UHFFFAOYSA-N 0.000 description 16
- 125000001246 bromo group Chemical group Br* 0.000 description 16
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 16
- 239000003480 eluent Substances 0.000 description 16
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 16
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 15
- 125000003342 alkenyl group Chemical group 0.000 description 15
- 125000004093 cyano group Chemical group *C#N 0.000 description 15
- 238000001514 detection method Methods 0.000 description 15
- 238000009472 formulation Methods 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 229920006395 saturated elastomer Polymers 0.000 description 15
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 15
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 14
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- 239000001257 hydrogen Substances 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 239000000463 material Substances 0.000 description 14
- 238000010898 silica gel chromatography Methods 0.000 description 14
- 238000006467 substitution reaction Methods 0.000 description 14
- 108090000765 processed proteins & peptides Proteins 0.000 description 13
- 102000004196 processed proteins & peptides Human genes 0.000 description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 12
- 229910003827 NRaRb Inorganic materials 0.000 description 12
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 230000003281 allosteric effect Effects 0.000 description 12
- 125000000539 amino acid group Chemical group 0.000 description 12
- 150000001413 amino acids Chemical group 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 150000002367 halogens Chemical group 0.000 description 12
- 208000004296 neuralgia Diseases 0.000 description 12
- 208000021722 neuropathic pain Diseases 0.000 description 12
- 201000008482 osteoarthritis Diseases 0.000 description 12
- 229920001184 polypeptide Polymers 0.000 description 12
- 229910052701 rubidium Inorganic materials 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 11
- 235000019253 formic acid Nutrition 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 239000012453 solvate Substances 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- ZOMATQMEHRJKLO-UHFFFAOYSA-N 1h-imidazol-2-ylmethanol Chemical compound OCC1=NC=CN1 ZOMATQMEHRJKLO-UHFFFAOYSA-N 0.000 description 10
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 10
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 10
- QNKYCVGXFPFZFZ-UHFFFAOYSA-N [7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran-4-yl]-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC=C(C=2C(=COC=21)CC(F)(F)F)C(O)C=1NC=CN=1 QNKYCVGXFPFZFZ-UHFFFAOYSA-N 0.000 description 10
- 239000005557 antagonist Substances 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 125000004122 cyclic group Chemical group 0.000 description 10
- 239000002552 dosage form Substances 0.000 description 10
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 10
- 239000000546 pharmaceutical excipient Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 238000004808 supercritical fluid chromatography Methods 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- QDAZIAFLGNFROW-UHFFFAOYSA-N (4-fluoro-1-methylindol-7-yl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=C2C=CN(C2=C(C=C1)C(O)C=1NC=CN=1)C QDAZIAFLGNFROW-UHFFFAOYSA-N 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- 125000000392 cycloalkenyl group Chemical group 0.000 description 9
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 9
- 150000003384 small molecules Chemical class 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 8
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 8
- 125000000304 alkynyl group Chemical group 0.000 description 8
- 239000012298 atmosphere Substances 0.000 description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 8
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 8
- 201000006417 multiple sclerosis Diseases 0.000 description 8
- 238000003419 tautomerization reaction Methods 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 7
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 7
- 229920002472 Starch Polymers 0.000 description 7
- 238000010521 absorption reaction Methods 0.000 description 7
- 230000008901 benefit Effects 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000002207 metabolite Substances 0.000 description 7
- 238000002953 preparative HPLC Methods 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 description 6
- OPZDXMCOWFPQPE-UHFFFAOYSA-N 2-bromo-4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C(Br)=C1 OPZDXMCOWFPQPE-UHFFFAOYSA-N 0.000 description 6
- FXJNBIBSXXPDGG-UHFFFAOYSA-N 3-ethyl-8-fluoroindolizine-5-carbaldehyde Chemical compound C(C)C1=CC=C2C(=CC=C(N12)C=O)F FXJNBIBSXXPDGG-UHFFFAOYSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000003963 antioxidant agent Substances 0.000 description 6
- 235000006708 antioxidants Nutrition 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 125000002619 bicyclic group Chemical group 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 235000011187 glycerol Nutrition 0.000 description 6
- 230000036541 health Effects 0.000 description 6
- 150000004677 hydrates Chemical class 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 238000012746 preparative thin layer chromatography Methods 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 229960004063 propylene glycol Drugs 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- XEGXZUHQEVUODL-UHFFFAOYSA-N (3,4-difluoro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound CCCc1c(F)c(F)ccc1C(O)c1ncc[nH]1 XEGXZUHQEVUODL-UHFFFAOYSA-N 0.000 description 5
- CQJNUZJADLSZGY-UHFFFAOYSA-N (3-ethyl-7-fluoro-1-benzothiophen-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C=1C2=C(SC=1)C(=CC=C2C(O)C=1NC=CN=1)F CQJNUZJADLSZGY-UHFFFAOYSA-N 0.000 description 5
- WNTPTQLSIKUWGF-UHFFFAOYSA-N (3-ethyl-8-fluoroindolizin-5-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C1=CC=C2C(=CC=C(N12)C(O)C=1NC=CN=1)F WNTPTQLSIKUWGF-UHFFFAOYSA-N 0.000 description 5
- IDFPQEHZYBXIFO-GFCCVEGCSA-N (R)-(4-fluoro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound CCCc1cc(F)ccc1[C@@H](O)c1ncc[nH]1 IDFPQEHZYBXIFO-GFCCVEGCSA-N 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 5
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 5
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 5
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 5
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 5
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 5
- 101001098529 Homo sapiens Proteinase-activated receptor 1 Proteins 0.000 description 5
- 101000713169 Homo sapiens Solute carrier family 52, riboflavin transporter, member 2 Proteins 0.000 description 5
- 206010065390 Inflammatory pain Diseases 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 102100037136 Proteinase-activated receptor 1 Human genes 0.000 description 5
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- ZOVRXKJGEDLEOO-UHFFFAOYSA-N [2-(2,5-dihydropyrrol-1-yl)-4-fluorophenyl]-(1H-imidazol-2-yl)methanol Chemical compound N1(CC=CC1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 ZOVRXKJGEDLEOO-UHFFFAOYSA-N 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 230000000996 additive effect Effects 0.000 description 5
- 125000004414 alkyl thio group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 230000008485 antagonism Effects 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 5
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 5
- 229910052805 deuterium Inorganic materials 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 5
- 231100000252 nontoxic Toxicity 0.000 description 5
- 230000003000 nontoxic effect Effects 0.000 description 5
- 235000015320 potassium carbonate Nutrition 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 235000010356 sorbitol Nutrition 0.000 description 5
- 239000000600 sorbitol Substances 0.000 description 5
- 239000008107 starch Substances 0.000 description 5
- 229940032147 starch Drugs 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 239000000454 talc Substances 0.000 description 5
- 235000012222 talc Nutrition 0.000 description 5
- 229910052623 talc Inorganic materials 0.000 description 5
- 239000003643 water by type Substances 0.000 description 5
- KSARXAFMALPZDK-UHFFFAOYSA-N (3-ethyl-7-fluoro-1-benzofuran-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C1=COC2=C1C(=CC=C2F)C(O)C=1NC=CN=1 KSARXAFMALPZDK-UHFFFAOYSA-N 0.000 description 4
- MZZXWDJEZGHGFN-UHFFFAOYSA-N (3-ethyl-8-fluoroindolizin-5-yl)methanol Chemical compound C(C)C1=CC=C2C(=CC=C(N12)CO)F MZZXWDJEZGHGFN-UHFFFAOYSA-N 0.000 description 4
- SUUQCDYQOMBVTO-UHFFFAOYSA-N (4-fluoro-2-pyrrolidin-1-ylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)N1CCCC1 SUUQCDYQOMBVTO-UHFFFAOYSA-N 0.000 description 4
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 4
- WZGMPEWKMWAHJV-UHFFFAOYSA-N FC1=CC(=C(C=O)C=C1)N1CCCC1 Chemical compound FC1=CC(=C(C=O)C=C1)N1CCCC1 WZGMPEWKMWAHJV-UHFFFAOYSA-N 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- 101000613565 Homo sapiens PRKC apoptosis WT1 regulator protein Proteins 0.000 description 4
- 101001135199 Homo sapiens Partitioning defective 3 homolog Proteins 0.000 description 4
- 101001098557 Homo sapiens Proteinase-activated receptor 3 Proteins 0.000 description 4
- 101001113471 Homo sapiens Proteinase-activated receptor 4 Proteins 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 229910002666 PdCl2 Inorganic materials 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 229940118430 Protease-activated receptor-2 antagonist Drugs 0.000 description 4
- 102000002020 Protease-activated receptors Human genes 0.000 description 4
- 108050009310 Protease-activated receptors Proteins 0.000 description 4
- 102100037133 Proteinase-activated receptor 3 Human genes 0.000 description 4
- 102100023710 Proteinase-activated receptor 4 Human genes 0.000 description 4
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 4
- OTOXQRVCQXMWOS-UHFFFAOYSA-N [2-(cyclopenten-1-yl)-4-fluorophenyl]-(1H-imidazol-2-yl)methanol Chemical compound C1(=CCCC1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 OTOXQRVCQXMWOS-UHFFFAOYSA-N 0.000 description 4
- IWNABUVVTVUARB-UHFFFAOYSA-N [4-fluoro-2-(1,3-oxazol-4-yl)phenyl]-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)C=1N=COC=1 IWNABUVVTVUARB-UHFFFAOYSA-N 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 235000010443 alginic acid Nutrition 0.000 description 4
- 229920000615 alginic acid Polymers 0.000 description 4
- 150000004781 alginic acids Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 4
- 229910021538 borax Inorganic materials 0.000 description 4
- VQFAIAKCILWQPZ-UHFFFAOYSA-N bromoacetone Chemical compound CC(=O)CBr VQFAIAKCILWQPZ-UHFFFAOYSA-N 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 208000007118 chronic progressive multiple sclerosis Diseases 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- 208000037765 diseases and disorders Diseases 0.000 description 4
- 238000010494 dissociation reaction Methods 0.000 description 4
- 230000005593 dissociations Effects 0.000 description 4
- 239000003995 emulsifying agent Substances 0.000 description 4
- 150000002170 ethers Chemical class 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 230000002757 inflammatory effect Effects 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 235000019359 magnesium stearate Nutrition 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- 201000001119 neuropathy Diseases 0.000 description 4
- 230000007823 neuropathy Effects 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- 239000004006 olive oil Substances 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 150000002894 organic compounds Chemical class 0.000 description 4
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- JAQOMSTTXPGKTN-UHFFFAOYSA-N propylboronic acid Chemical compound CCCB(O)O JAQOMSTTXPGKTN-UHFFFAOYSA-N 0.000 description 4
- 206010039073 rheumatoid arthritis Diseases 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 235000010339 sodium tetraborate Nutrition 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 description 4
- 239000001993 wax Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- BFCJTVKXAJFHRK-UHFFFAOYSA-N (2-cyclopentyl-4-fluorophenyl)-(1H-imidazol-2-yl)methanol Chemical compound C1(CCCC1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 BFCJTVKXAJFHRK-UHFFFAOYSA-N 0.000 description 3
- SDERHZQTCLJMQS-UHFFFAOYSA-N (3-chloro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound ClC=1C(=C(C=CC=1)C(O)C=1NC=CN=1)CCC SDERHZQTCLJMQS-UHFFFAOYSA-N 0.000 description 3
- YEMHFNLAQRARIZ-UHFFFAOYSA-N (3-cyclopropyl-7-fluoro-1-benzofuran-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound C1(CC1)C1=COC2=C1C(=CC=C2F)C(O)C=1NC=CN=1 YEMHFNLAQRARIZ-UHFFFAOYSA-N 0.000 description 3
- SCJIAJSZIDORJD-UHFFFAOYSA-N (3-ethyl-7-fluoro-1-benzofuran-4-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C(C)C1=COC2=C1C(=CC=C2F)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C SCJIAJSZIDORJD-UHFFFAOYSA-N 0.000 description 3
- DHWDWJVAUUPMPE-UHFFFAOYSA-N (3-ethyl-8-fluoroindolizin-5-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C(C)C1=CC=C2C(=CC=C(N12)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)F DHWDWJVAUUPMPE-UHFFFAOYSA-N 0.000 description 3
- IDFPQEHZYBXIFO-UHFFFAOYSA-N (4-fluoro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)CCC IDFPQEHZYBXIFO-UHFFFAOYSA-N 0.000 description 3
- LYHAQSRVLVVYAF-UHFFFAOYSA-N (4-fluoro-2-propylphenyl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)CCC LYHAQSRVLVVYAF-UHFFFAOYSA-N 0.000 description 3
- OUOXWUZKAAFUAG-UHFFFAOYSA-N (4-fluorodibenzofuran-1-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC=C(C2=C1OC1=C2C=CC=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C OUOXWUZKAAFUAG-UHFFFAOYSA-N 0.000 description 3
- AVBIITJVIKUGON-UHFFFAOYSA-N (5-fluoro-8-formylnaphthalen-1-yl) trifluoromethanesulfonate Chemical compound FC(S(=O)(=O)OC1=CC=CC2=C(C=CC(=C12)C=O)F)(F)F AVBIITJVIKUGON-UHFFFAOYSA-N 0.000 description 3
- NNQBAGFIYBBAFL-UHFFFAOYSA-N 1-(diethoxymethyl)imidazole Chemical compound CCOC(OCC)N1C=CN=C1 NNQBAGFIYBBAFL-UHFFFAOYSA-N 0.000 description 3
- UONVQIIPSNCTPM-UHFFFAOYSA-N 1-bromo-4-fluorodibenzofuran Chemical compound FC1=CC=C(Br)C2=C1OC1=C2C=CC=C1 UONVQIIPSNCTPM-UHFFFAOYSA-N 0.000 description 3
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 3
- WHCXAOCMHZXMSF-UHFFFAOYSA-N 2,3-dihydro-1H-pyrrolo[1,2-a]indol-8-yl-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C[Si](C)(C)CCOCN1C=CN=C1C(O)C1=C2N3CCCC3=CC2=CC=C1 WHCXAOCMHZXMSF-UHFFFAOYSA-N 0.000 description 3
- NHARSUQSOKDUNB-UHFFFAOYSA-N 2,3-dihydro-1H-pyrrolo[1,2-a]indol-8-ylmethanol Chemical compound OCC1=C2N3CCCC3=CC2=CC=C1 NHARSUQSOKDUNB-UHFFFAOYSA-N 0.000 description 3
- GJTNBJJSKKRCQZ-UHFFFAOYSA-N 2,3-dihydro-1H-pyrrolo[1,2-a]indole-8-carbaldehyde Chemical compound O=CC1=C2N3CCCC3=CC2=CC=C1 GJTNBJJSKKRCQZ-UHFFFAOYSA-N 0.000 description 3
- HNWZAXTVADNFNX-UHFFFAOYSA-N 2,3-dihydro-1H-pyrrolo[1,2-a]indole-8-carboxylic acid Chemical compound OC(=O)C1=C2N3CCCC3=CC2=CC=C1 HNWZAXTVADNFNX-UHFFFAOYSA-N 0.000 description 3
- VUWNFSDDDVZPAN-UHFFFAOYSA-N 3-chloro-2-pyrazol-1-ylbenzoic acid Chemical compound ClC=1C(=C(C(=O)O)C=CC=1)N1N=CC=C1 VUWNFSDDDVZPAN-UHFFFAOYSA-N 0.000 description 3
- QDIOUOIFUWIGKR-UHFFFAOYSA-N 3-chloro-N-methoxy-N-methyl-2-pyrazol-1-ylbenzamide Chemical compound ClC=1C(=C(C(=O)N(C)OC)C=CC=1)N1N=CC=C1 QDIOUOIFUWIGKR-UHFFFAOYSA-N 0.000 description 3
- CXQMCAFKBDMJLQ-UHFFFAOYSA-N 3-ethyl-7-fluoro-1-benzofuran-4-carbaldehyde Chemical compound C(C)C1=COC=2C1=C(C=CC=2F)C=O CXQMCAFKBDMJLQ-UHFFFAOYSA-N 0.000 description 3
- OFQLKGRRTLHHFP-UHFFFAOYSA-N 4-(2-bromo-5-fluorophenyl)-1,3-oxazole Chemical compound BrC1=C(C=C(C=C1)F)C=1N=COC=1 OFQLKGRRTLHHFP-UHFFFAOYSA-N 0.000 description 3
- ZZAGPQOFAOWZIC-UHFFFAOYSA-N 4-(5-bromo-2-fluorophenyl)butanoic acid Chemical compound OC(=O)CCCC1=CC(Br)=CC=C1F ZZAGPQOFAOWZIC-UHFFFAOYSA-N 0.000 description 3
- PLLZMPBNDYFPIE-UHFFFAOYSA-N 4-bromo-3-(bromomethyl)-7-fluoro-1-benzofuran Chemical compound BrC1=CC=C(C2=C1C(=CO2)CBr)F PLLZMPBNDYFPIE-UHFFFAOYSA-N 0.000 description 3
- YUDUEAJKLNFBJV-UHFFFAOYSA-N 4-bromo-3-ethyl-7-fluoro-1-benzofuran Chemical compound BrC1=CC=C(C2=C1C(=CO2)CC)F YUDUEAJKLNFBJV-UHFFFAOYSA-N 0.000 description 3
- GRVDFALMWASOCV-UHFFFAOYSA-N 4-bromo-7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran Chemical compound BrC1=CC=C(C2=C1C(=CO2)CC(F)(F)F)F GRVDFALMWASOCV-UHFFFAOYSA-N 0.000 description 3
- PESURLRFOORKLG-UHFFFAOYSA-N 4-bromo-7-fluoro-3-methyl-1-benzofuran Chemical compound BrC1=CC=C(C2=C1C(=CO2)C)F PESURLRFOORKLG-UHFFFAOYSA-N 0.000 description 3
- AOKVWJLEQVSXMW-UHFFFAOYSA-N 4-fluoro-1h-indole-7-carbaldehyde Chemical compound FC1=CC=C(C=O)C2=C1C=CN2 AOKVWJLEQVSXMW-UHFFFAOYSA-N 0.000 description 3
- PRLOWNASKFQQTM-UHFFFAOYSA-N 4-fluoro-2-(1,3-oxazol-4-yl)benzaldehyde Chemical compound FC1=CC(=C(C=O)C=C1)C=1N=COC=1 PRLOWNASKFQQTM-UHFFFAOYSA-N 0.000 description 3
- DSGISHNJDSOHOL-UHFFFAOYSA-N 4-fluoro-2-(1,3-thiazol-4-yl)benzaldehyde Chemical compound FC1=CC(=C(C=O)C=C1)C=1N=CSC=1 DSGISHNJDSOHOL-UHFFFAOYSA-N 0.000 description 3
- VWGPYMZQHRBCBP-UHFFFAOYSA-N 4-fluoro-2-propylbenzaldehyde Chemical compound CCCc1cc(F)ccc1C=O VWGPYMZQHRBCBP-UHFFFAOYSA-N 0.000 description 3
- AASFCLBJHSQKGB-UHFFFAOYSA-N 4-fluoro-8-hydroxynaphthalene-1-carbaldehyde Chemical compound FC1=CC=C(C2=C(C=CC=C12)O)C=O AASFCLBJHSQKGB-UHFFFAOYSA-N 0.000 description 3
- ONPZJOOQXFPZGM-UHFFFAOYSA-N 4-fluoro-8-phenylmethoxynaphthalene-1-carbaldehyde Chemical compound C(C1=CC=CC=C1)OC=1C=CC=C2C(=CC=C(C=12)C=O)F ONPZJOOQXFPZGM-UHFFFAOYSA-N 0.000 description 3
- SCUMKZZUXCNAAF-UHFFFAOYSA-N 4-fluorodibenzofuran-1-carbaldehyde Chemical compound FC1=CC=C(C2=C1OC1=C2C=CC=C1)C=O SCUMKZZUXCNAAF-UHFFFAOYSA-N 0.000 description 3
- VXNXSGLEDHVMQC-UHFFFAOYSA-N 7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran-4-carbaldehyde Chemical compound FC=1C=CC(=C2C(=COC2=1)CC(F)(F)F)C=O VXNXSGLEDHVMQC-UHFFFAOYSA-N 0.000 description 3
- HHFBDRUPPURJPW-UHFFFAOYSA-N 8-bromo-5-fluoro-3,4-dihydro-2H-naphthalen-1-one Chemical compound FC1=CC=C(Br)C2=C1CCCC2=O HHFBDRUPPURJPW-UHFFFAOYSA-N 0.000 description 3
- UFLYDNJASUTAFT-UHFFFAOYSA-N 8-bromo-5-fluoronaphthalen-1-ol Chemical compound C1=CC(Br)=C2C(O)=CC=CC2=C1F UFLYDNJASUTAFT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 241000416162 Astragalus gummifer Species 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- QPSBMLXJDPBDGV-UHFFFAOYSA-N CC(=O)COC1=C(F)C=CC(Br)=C1 Chemical compound CC(=O)COC1=C(F)C=CC(Br)=C1 QPSBMLXJDPBDGV-UHFFFAOYSA-N 0.000 description 3
- FDQLEBKCRLSPBP-UHFFFAOYSA-N CCC(=O)COC1=C(F)C=CC(Br)=C1 Chemical compound CCC(=O)COC1=C(F)C=CC(Br)=C1 FDQLEBKCRLSPBP-UHFFFAOYSA-N 0.000 description 3
- IDFPQEHZYBXIFO-LBPRGKRZSA-N CCCC1=C(C=CC(F)=C1)[C@H](O)C1=NC=CN1 Chemical compound CCCC1=C(C=CC(F)=C1)[C@H](O)C1=NC=CN1 IDFPQEHZYBXIFO-LBPRGKRZSA-N 0.000 description 3
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 208000004454 Hyperalgesia Diseases 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical group [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 3
- JZBFSNNIIJRJCC-UHFFFAOYSA-N [2-(cyclopenten-1-yl)phenyl]-(1H-imidazol-2-yl)methanol Chemical compound C1(=CCCC1)C1=C(C=CC=C1)C(O)C=1NC=CN=1 JZBFSNNIIJRJCC-UHFFFAOYSA-N 0.000 description 3
- NBONXCQDLGCRAH-UHFFFAOYSA-N [4-fluoro-2-(1,3-oxazol-4-yl)phenyl]methanol Chemical compound FC1=CC(=C(C=C1)CO)C=1N=COC=1 NBONXCQDLGCRAH-UHFFFAOYSA-N 0.000 description 3
- NUHLOPDVFBYSIY-UHFFFAOYSA-N [4-fluoro-2-(1,3-thiazol-4-yl)phenyl]-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)C=1N=CSC=1 NUHLOPDVFBYSIY-UHFFFAOYSA-N 0.000 description 3
- BOTOZRGVQPCYPM-NSCUHMNNSA-N [4-fluoro-2-[(E)-prop-1-enyl]phenyl]-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)\C=C\C BOTOZRGVQPCYPM-NSCUHMNNSA-N 0.000 description 3
- BOTOZRGVQPCYPM-IHWYPQMZSA-N [4-fluoro-2-[(Z)-prop-1-enyl]phenyl]-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)\C=C/C BOTOZRGVQPCYPM-IHWYPQMZSA-N 0.000 description 3
- AMFDONZGNGFNEX-UHFFFAOYSA-N [7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran-4-yl]-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC=C(C=2C(=COC=21)CC(F)(F)F)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C AMFDONZGNGFNEX-UHFFFAOYSA-N 0.000 description 3
- VZTPJESDEHPOGX-UHFFFAOYSA-N [7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran-4-yl]methanol Chemical compound FC1=CC=C(C=2C(=COC=21)CC(F)(F)F)CO VZTPJESDEHPOGX-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 239000000783 alginic acid Substances 0.000 description 3
- 229960001126 alginic acid Drugs 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 3
- 125000003368 amide group Chemical group 0.000 description 3
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 229960005070 ascorbic acid Drugs 0.000 description 3
- 239000011668 ascorbic acid Substances 0.000 description 3
- 208000006673 asthma Diseases 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 235000010445 lecithin Nutrition 0.000 description 3
- 239000000787 lecithin Substances 0.000 description 3
- 229940067606 lecithin Drugs 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- FLRRZZBKDDEUBU-UHFFFAOYSA-N methyl 2-(3-hydroxypropyl)-1H-indole-7-carboxylate Chemical compound OCCCC=1NC2=C(C=CC=C2C=1)C(=O)OC FLRRZZBKDDEUBU-UHFFFAOYSA-N 0.000 description 3
- SHOWRISMEMBQHK-UHFFFAOYSA-N methyl 2-[3-(4-methylphenyl)sulfonyloxypropyl]-1H-indole-7-carboxylate Chemical compound S(=O)(=O)(C1=CC=C(C)C=C1)OCCCC=1NC2=C(C=CC=C2C=1)C(=O)OC SHOWRISMEMBQHK-UHFFFAOYSA-N 0.000 description 3
- ULYHNOAMYHEXLE-UHFFFAOYSA-N methyl 2-amino-5-bromo-3-(5-hydroxypent-1-ynyl)benzoate Chemical compound NC1=C(C(=O)OC)C=C(C=C1C#CCCCO)Br ULYHNOAMYHEXLE-UHFFFAOYSA-N 0.000 description 3
- SHIJOOINYUMOLY-UHFFFAOYSA-N methyl 3-ethyl-8-fluoroindolizine-5-carboxylate Chemical compound C(C)C1=CC=C2C(=CC=C(N12)C(=O)OC)F SHIJOOINYUMOLY-UHFFFAOYSA-N 0.000 description 3
- BRQFCUJIHJLNML-UHFFFAOYSA-N methyl 4-fluoro-2-(1,3-oxazol-4-yl)benzoate Chemical compound FC1=CC(=C(C(=O)OC)C=C1)C=1N=COC=1 BRQFCUJIHJLNML-UHFFFAOYSA-N 0.000 description 3
- VQPGHNYFGRSWFG-UHFFFAOYSA-N methyl 5-bromo-2-(3-hydroxypropyl)-1H-indole-7-carboxylate Chemical compound BrC=1C=C2C=C(NC2=C(C=1)C(=O)OC)CCCO VQPGHNYFGRSWFG-UHFFFAOYSA-N 0.000 description 3
- NKBXAVGOIJIPHJ-UHFFFAOYSA-N methyl 7-fluoro-3-(2,2,2-trifluoroethyl)-1-benzofuran-4-carboxylate Chemical compound FC=1C=CC(=C2C(=COC2=1)CC(F)(F)F)C(=O)OC NKBXAVGOIJIPHJ-UHFFFAOYSA-N 0.000 description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 3
- 125000006574 non-aromatic ring group Chemical group 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000006072 paste Substances 0.000 description 3
- 208000033808 peripheral neuropathy Diseases 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- ACVYVLVWPXVTIT-UHFFFAOYSA-M phosphinate Chemical compound [O-][PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-M 0.000 description 3
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 3
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 3
- 230000004962 physiological condition Effects 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 125000003367 polycyclic group Chemical group 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 3
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 3
- 239000011698 potassium fluoride Substances 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000000159 protein binding assay Methods 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 3
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 3
- 235000012239 silicon dioxide Nutrition 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 125000000547 substituted alkyl group Chemical group 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 3
- 150000007970 thio esters Chemical class 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 235000010487 tragacanth Nutrition 0.000 description 3
- 239000000196 tragacanth Substances 0.000 description 3
- 229940116362 tragacanth Drugs 0.000 description 3
- 229910052722 tritium Chemical group 0.000 description 3
- 238000000825 ultraviolet detection Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- OYGNFQIMXBCASK-UHFFFAOYSA-N (2-cyclobutyl-4-fluorophenyl)-(1H-imidazol-2-yl)methanol Chemical compound C1(CCC1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 OYGNFQIMXBCASK-UHFFFAOYSA-N 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- WBRVTPPTAVKUPS-UHFFFAOYSA-N (3-chloro-2-pyrazol-1-ylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound ClC=1C(=C(C=CC=1)C(O)C=1NC=CN=1)N1N=CC=C1 WBRVTPPTAVKUPS-UHFFFAOYSA-N 0.000 description 2
- NWTVOCBCRNXJHG-UHFFFAOYSA-N (3-chloro-2-pyrazol-1-ylphenyl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound ClC=1C(=C(C=CC=1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)N1N=CC=C1 NWTVOCBCRNXJHG-UHFFFAOYSA-N 0.000 description 2
- LEOFZXSUICVEKE-UHFFFAOYSA-N (3-chloro-8-fluoroindolizin-5-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C[Si](C)(C)CCOCN1C=CN=C1C(O)C1=CC=C(F)C2=CC=C(Cl)N12 LEOFZXSUICVEKE-UHFFFAOYSA-N 0.000 description 2
- WKGQVKYNRGBYEO-UHFFFAOYSA-N (3-chloro-8-fluoroindolizin-5-yl)methanol Chemical compound ClC1=CC=C2C(=CC=C(N12)CO)F WKGQVKYNRGBYEO-UHFFFAOYSA-N 0.000 description 2
- MKHYWEHKOFHAEH-UHFFFAOYSA-N (3-cyclopropyl-8-fluoroindolizin-5-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C1(CC1)C1=CC=C2C(=CC=C(N12)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)F MKHYWEHKOFHAEH-UHFFFAOYSA-N 0.000 description 2
- UKMKXTXTLURTNA-UHFFFAOYSA-N (4-fluoro-2-pyrrolidin-1-ylphenyl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)N1CCCC1 UKMKXTXTLURTNA-UHFFFAOYSA-N 0.000 description 2
- CBYQPTIOTHIEKC-UHFFFAOYSA-N (4-fluoro-8-methylnaphthalen-1-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=CC=C(C2=C(C=CC=C12)C)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C CBYQPTIOTHIEKC-UHFFFAOYSA-N 0.000 description 2
- DGECFFLSWXKTPD-UHFFFAOYSA-N (4-fluorodibenzofuran-1-yl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC=C(C2=C1OC1=C2C=CC=C1)C(O)C=1NC=CN=1 DGECFFLSWXKTPD-UHFFFAOYSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 2
- VASPISDTJISIDL-UHFFFAOYSA-N 1,3-difluoro-2-(2-iodophenoxy)benzene Chemical compound FC1=C(C(=CC=C1)F)OC1=C(C=CC=C1)I VASPISDTJISIDL-UHFFFAOYSA-N 0.000 description 2
- KUURTEAOMQZIHM-UHFFFAOYSA-N 1,3-difluoro-2-(2-nitrophenoxy)benzene Chemical compound [O-][N+](=O)C1=CC=CC=C1OC1=C(F)C=CC=C1F KUURTEAOMQZIHM-UHFFFAOYSA-N 0.000 description 2
- CCXQVBSQUQCEEO-UHFFFAOYSA-N 1-bromobutan-2-one Chemical compound CCC(=O)CBr CCXQVBSQUQCEEO-UHFFFAOYSA-N 0.000 description 2
- IBXNCJKFFQIKKY-UHFFFAOYSA-N 1-pentyne Chemical compound CCCC#C IBXNCJKFFQIKKY-UHFFFAOYSA-N 0.000 description 2
- HDSWCGPCUGNIIP-UHFFFAOYSA-N 2,3-dihydro-1H-pyrrolo[1,2-a]indol-8-yl(1H-imidazol-2-yl)methanol Chemical compound OC(C1=NC=CN1)C1=C2N3CCCC3=CC2=CC=C1 HDSWCGPCUGNIIP-UHFFFAOYSA-N 0.000 description 2
- SICCCLXKABFYKB-UHFFFAOYSA-N 2-(2,6-difluorophenoxy)aniline Chemical compound NC1=CC=CC=C1OC1=C(F)C=CC=C1F SICCCLXKABFYKB-UHFFFAOYSA-N 0.000 description 2
- JFTZVYKESKQING-UHFFFAOYSA-N 2-(cyclopenten-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CCCC1 JFTZVYKESKQING-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- PZWNNLKICUALRY-UHFFFAOYSA-N 3-(3-bromo-6-fluoropyridin-2-yl)prop-2-yn-1-ol Chemical compound BrC=1C(=NC(=CC=1)F)C#CCO PZWNNLKICUALRY-UHFFFAOYSA-N 0.000 description 2
- JBUYFJDUOIWCCE-UHFFFAOYSA-N 3-(3-bromo-6-fluoropyridin-2-yl)prop-2-ynyl methanesulfonate Chemical compound CS(=O)(=O)OCC#CC1=NC(=CC=C1Br)F JBUYFJDUOIWCCE-UHFFFAOYSA-N 0.000 description 2
- HUUACIALCVAHKF-UHFFFAOYSA-N 3-(6-aminopyridin-2-yl)prop-2-yn-1-ol Chemical compound NC1=CC=CC(=N1)C#CCO HUUACIALCVAHKF-UHFFFAOYSA-N 0.000 description 2
- HOSOLMRHAWETDC-UHFFFAOYSA-N 3-chloro-2-pyrazol-1-ylbenzaldehyde Chemical compound ClC=1C(=C(C=O)C=CC=1)N1N=CC=C1 HOSOLMRHAWETDC-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- RSWRJJLJDRAQDP-UHFFFAOYSA-N 4-bromo-1-fluoro-5-phenylmethoxynaphthalene Chemical compound C(C1=CC=CC=C1)OC1=C2C(=CC=C(C2=CC=C1)F)Br RSWRJJLJDRAQDP-UHFFFAOYSA-N 0.000 description 2
- FDRAHNNVATYADT-UHFFFAOYSA-N 4-fluoro-1-methylindole-7-carbaldehyde Chemical compound C1=CC(C=O)=C2N(C)C=CC2=C1F FDRAHNNVATYADT-UHFFFAOYSA-N 0.000 description 2
- XYBJAZYMWVJNCY-UHFFFAOYSA-N 4-fluoro-8-methylnaphthalene-1-carbaldehyde Chemical compound FC1=CC=C(C2=C(C=CC=C12)C)C=O XYBJAZYMWVJNCY-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- YPTHSYKJDRMAJY-UHFFFAOYSA-N 5-bromo-2-fluorophenol Chemical compound OC1=CC(Br)=CC=C1F YPTHSYKJDRMAJY-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- KSZUALMWWVCIEF-UHFFFAOYSA-N 8-bromo-1-ethyl-5-fluoroindolizine Chemical compound BrC1=CC=C(N2C=CC(=C12)CC)F KSZUALMWWVCIEF-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 239000005695 Ammonium acetate Substances 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 208000008035 Back Pain Diseases 0.000 description 2
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- GRLMBZPNUUXBMF-UHFFFAOYSA-N CCC(=C=C)C1=NC(F)=CC=C1Br Chemical compound CCC(=C=C)C1=NC(F)=CC=C1Br GRLMBZPNUUXBMF-UHFFFAOYSA-N 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 208000000094 Chronic Pain Diseases 0.000 description 2
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 description 2
- 229940126062 Compound A Drugs 0.000 description 2
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 2
- 208000001640 Fibromyalgia Diseases 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 229910010084 LiAlH4 Inorganic materials 0.000 description 2
- 208000008930 Low Back Pain Diseases 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 2
- 208000001089 Multiple system atrophy Diseases 0.000 description 2
- 101100189356 Mus musculus Papolb gene Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 101100406879 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) par-2 gene Proteins 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- 208000004983 Phantom Limb Diseases 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 208000007400 Relapsing-Remitting Multiple Sclerosis Diseases 0.000 description 2
- 244000000231 Sesamum indicum Species 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 102000003566 TRPV1 Human genes 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 208000008548 Tension-Type Headache Diseases 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 101150016206 Trpv1 gene Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- QVGJZIZAGQAGCH-UHFFFAOYSA-N [2-(2,5-dihydropyrrol-1-yl)-4-fluorophenyl]-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound N1(CC=CC1)C1=C(C=CC(=C1)F)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C QVGJZIZAGQAGCH-UHFFFAOYSA-N 0.000 description 2
- NBTGMLXUWXTKKI-UHFFFAOYSA-N [2-(azetidin-1-yl)-4-fluorophenyl]-(1H-imidazol-2-yl)methanol Chemical compound N1(CCC1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 NBTGMLXUWXTKKI-UHFFFAOYSA-N 0.000 description 2
- DOWRULCVGBSQAS-UHFFFAOYSA-N [4-fluoro-2-(1,3-oxazol-4-yl)phenyl]-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)C=1N=COC=1 DOWRULCVGBSQAS-UHFFFAOYSA-N 0.000 description 2
- GMGFDPMZWLQZJC-UHFFFAOYSA-N [4-fluoro-2-(1,3-thiazol-4-yl)phenyl]-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)C=1N=CSC=1 GMGFDPMZWLQZJC-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 208000005298 acute pain Diseases 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 230000029936 alkylation Effects 0.000 description 2
- 238000005804 alkylation reaction Methods 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 235000010210 aluminium Nutrition 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001409 amidines Chemical class 0.000 description 2
- 125000004103 aminoalkyl group Chemical group 0.000 description 2
- 235000019257 ammonium acetate Nutrition 0.000 description 2
- 229940043376 ammonium acetate Drugs 0.000 description 2
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000008135 aqueous vehicle Substances 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 125000004452 carbocyclyl group Chemical group 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 201000005795 chronic inflammatory demyelinating polyneuritis Diseases 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 208000018631 connective tissue disease Diseases 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 229940097362 cyclodextrins Drugs 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 230000001804 emulsifying effect Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- IZLKBNIPMWRBPE-UHFFFAOYSA-N ethyl 3-ethyl-8-fluoroindolizine-5-carboxylate Chemical compound C(C)C1=CC=C2C(=CC=C(N12)C(=O)OCC)F IZLKBNIPMWRBPE-UHFFFAOYSA-N 0.000 description 2
- AMBXIDNDSAORHZ-UHFFFAOYSA-N ethyl 5-fluoro-6-pent-1-ynylpyridine-2-carboxylate Chemical compound FC=1C=CC(=NC=1C#CCCC)C(=O)OCC AMBXIDNDSAORHZ-UHFFFAOYSA-N 0.000 description 2
- CURXEPHOFMUDQR-UHFFFAOYSA-N ethyl 6-bromo-5-fluoropyridine-2-carboxylate Chemical compound CCOC(=O)C1=CC=C(F)C(Br)=N1 CURXEPHOFMUDQR-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 229940093471 ethyl oleate Drugs 0.000 description 2
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 229940012356 eye drops Drugs 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 125000001207 fluorophenyl group Chemical group 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 239000006260 foam Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000010255 intramuscular injection Methods 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 150000003951 lactams Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- AWIJRPNMLHPLNC-UHFFFAOYSA-N methanethioic s-acid Chemical compound SC=O AWIJRPNMLHPLNC-UHFFFAOYSA-N 0.000 description 2
- GYGVCXXNIAZBTE-UHFFFAOYSA-N methyl 5-fluoro-6-pent-1-ynylpyridine-2-carboxylate Chemical compound FC=1C=CC(=NC=1C#CCCC)C(=O)OC GYGVCXXNIAZBTE-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000002808 molecular sieve Substances 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 201000005518 mononeuropathy Diseases 0.000 description 2
- 210000004897 n-terminal region Anatomy 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000002687 nonaqueous vehicle Substances 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 239000006174 pH buffer Substances 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 210000000578 peripheral nerve Anatomy 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Chemical group O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 235000011007 phosphoric acid Nutrition 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229920000137 polyphosphoric acid Polymers 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 206010063401 primary progressive multiple sclerosis Diseases 0.000 description 2
- 230000000135 prohibitive effect Effects 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000006337 proteolytic cleavage Effects 0.000 description 2
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 201000008628 secondary progressive multiple sclerosis Diseases 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000004328 sodium tetraborate Substances 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 208000020431 spinal cord injury Diseases 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 229960004274 stearic acid Drugs 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- FKHIFSZMMVMEQY-UHFFFAOYSA-N talc Chemical compound [Mg+2].[O-][Si]([O-])=O FKHIFSZMMVMEQY-UHFFFAOYSA-N 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- ZXFVPQKCMGUYTI-UHFFFAOYSA-N tert-butyl 4-(5-bromo-2-fluorophenyl)butanoate Chemical compound BrC=1C=CC(=C(C=1)CCCC(=O)OC(C)(C)C)F ZXFVPQKCMGUYTI-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- DUYAAUVXQSMXQP-UHFFFAOYSA-M thioacetate Chemical compound CC([S-])=O DUYAAUVXQSMXQP-UHFFFAOYSA-M 0.000 description 2
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 239000003440 toxic substance Substances 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 230000008733 trauma Effects 0.000 description 2
- YYQKQPYPLADFMK-UHFFFAOYSA-N tributyl(1,3-thiazol-4-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CSC=N1 YYQKQPYPLADFMK-UHFFFAOYSA-N 0.000 description 2
- AVBGNFCMKJOFIN-UHFFFAOYSA-N triethylammonium acetate Chemical compound CC(O)=O.CCN(CC)CC AVBGNFCMKJOFIN-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- NKVUXPXCCHUPEK-UHFFFAOYSA-N (1-ethyl-4-fluoroindol-7-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)N1C=CC2=C(C=CC(=C12)C(O)C=1NC=CN=1)F NKVUXPXCCHUPEK-UHFFFAOYSA-N 0.000 description 1
- OQYFMLPZOYSJIC-UHFFFAOYSA-N (1-ethyl-4-fluoroindol-7-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C(C)N1C=CC2=C(C=CC(=C12)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)F OQYFMLPZOYSJIC-UHFFFAOYSA-N 0.000 description 1
- MBLOLINBNOUHFB-UHFFFAOYSA-N (1-ethyl-5-fluoroindolizin-8-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C=1C=CN2C(=CC=C(C=12)C(O)C=1NC=CN=1)F MBLOLINBNOUHFB-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- KXSQRPMICQOFOY-UHFFFAOYSA-N (3,4-difluoro-2-pyrazol-1-ylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC=1C(=C(C=CC=1F)C(O)C=1NC=CN=1)N1N=CC=C1 KXSQRPMICQOFOY-UHFFFAOYSA-N 0.000 description 1
- JEUQUHDWHVGXER-UHFFFAOYSA-N (3-chloro-8-fluoroindolizin-5-yl)-(1H-imidazol-2-yl)methanol Chemical compound ClC1=CC=C2C(=CC=C(N12)C(O)C=1NC=CN=1)F JEUQUHDWHVGXER-UHFFFAOYSA-N 0.000 description 1
- CVYXMLRDJQJNCC-UHFFFAOYSA-N (3-cyclopropyl-8-fluoroindolizin-5-yl)-(1H-imidazol-2-yl)methanol Chemical compound C1(CC1)C1=CC=C2C(=CC=C(N12)C(O)C=1NC=CN=1)F CVYXMLRDJQJNCC-UHFFFAOYSA-N 0.000 description 1
- VTFLXHJGYLBZOO-UHFFFAOYSA-N (4-fluoro-1-methylindol-7-yl)-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound FC1=C2C=CN(C2=C(C=C1)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C)C VTFLXHJGYLBZOO-UHFFFAOYSA-N 0.000 description 1
- ZLMJYEFZTYHDRJ-UHFFFAOYSA-N (4-fluoro-2-pyrazol-1-ylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)C(O)C=1NC=CN=1)N1N=CC=C1 ZLMJYEFZTYHDRJ-UHFFFAOYSA-N 0.000 description 1
- JJYQEWONVVLREO-UHFFFAOYSA-N (4-fluoro-8-methylnaphthalen-1-yl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC=C(C2=C(C=CC=C12)C)C(O)C=1NC=CN=1 JJYQEWONVVLREO-UHFFFAOYSA-N 0.000 description 1
- QFSGSWIIBVNWEC-UHFFFAOYSA-N (7-fluoro-3-methyl-1-benzofuran-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC=C(C=2C(=COC=21)C)C(O)C=1NC=CN=1 QFSGSWIIBVNWEC-UHFFFAOYSA-N 0.000 description 1
- VTMCSOXBNFEORE-UHFFFAOYSA-N (8-fluoro-3-methylindolizin-5-yl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC=C(N2C(=CC=C12)C)C(O)C=1NC=CN=1 VTMCSOXBNFEORE-UHFFFAOYSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- KSARXAFMALPZDK-GFCCVEGCSA-N (R)-(3-ethyl-7-fluoro-1-benzofuran-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C1=COC2=C1C(=CC=C2F)[C@@H](O)C=1NC=CN=1 KSARXAFMALPZDK-GFCCVEGCSA-N 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N (R)-alpha-Tocopherol Natural products OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- XEGXZUHQEVUODL-LBPRGKRZSA-N (S)-(3,4-difluoro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC=1C(=C(C=CC=1F)[C@H](O)C=1NC=CN=1)CCC XEGXZUHQEVUODL-LBPRGKRZSA-N 0.000 description 1
- KSARXAFMALPZDK-LBPRGKRZSA-N (S)-(3-ethyl-7-fluoro-1-benzofuran-4-yl)-(1H-imidazol-2-yl)methanol Chemical compound C(C)C1=COC2=C1C(=CC=C2F)[C@H](O)C=1NC=CN=1 KSARXAFMALPZDK-LBPRGKRZSA-N 0.000 description 1
- SUUQCDYQOMBVTO-ZDUSSCGKSA-N (S)-(4-fluoro-2-pyrrolidin-1-ylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound FC1=CC(=C(C=C1)[C@H](O)C=1NC=CN=1)N1CCCC1 SUUQCDYQOMBVTO-ZDUSSCGKSA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- GLDXZAUMBBIJNJ-UHFFFAOYSA-N 1,3-difluoro-2-(3-iodophenoxy)benzene Chemical compound Fc1cccc(F)c1Oc1cccc(I)c1 GLDXZAUMBBIJNJ-UHFFFAOYSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- QMOFBUHLIBXFFY-UHFFFAOYSA-N 1-ethyl-4-fluoroindole-7-carbaldehyde Chemical compound CCN1C=CC2=C(F)C=CC(C=O)=C12 QMOFBUHLIBXFFY-UHFFFAOYSA-N 0.000 description 1
- VZCRLPWSYHSFDT-UHFFFAOYSA-N 1-ethyl-5-fluoroindolizine-8-carbaldehyde Chemical compound CCC1=C2N(C=C1)C(F)=CC=C2C=O VZCRLPWSYHSFDT-UHFFFAOYSA-N 0.000 description 1
- PWKNBLFSJAVFAB-UHFFFAOYSA-N 1-fluoro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1F PWKNBLFSJAVFAB-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- JRHPOFJADXHYBR-UHFFFAOYSA-N 1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CNC1CCCCC1NC JRHPOFJADXHYBR-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical class OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 1
- CKKOVFGIBXCEIJ-UHFFFAOYSA-N 2,6-difluorophenol Chemical compound OC1=C(F)C=CC=C1F CKKOVFGIBXCEIJ-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 1
- NYXFGTZHLKBZIL-UHFFFAOYSA-N 2-bromo-1-(2-bromo-5-fluorophenyl)ethanone Chemical compound FC1=CC=C(Br)C(C(=O)CBr)=C1 NYXFGTZHLKBZIL-UHFFFAOYSA-N 0.000 description 1
- WCCCDMWRBVVYCQ-UHFFFAOYSA-N 2-bromo-1-cyclopropylethanone Chemical compound BrCC(=O)C1CC1 WCCCDMWRBVVYCQ-UHFFFAOYSA-N 0.000 description 1
- LBTJGMXVLOOKBO-UHFFFAOYSA-N 2-bromo-3,4-difluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C(Br)=C1F LBTJGMXVLOOKBO-UHFFFAOYSA-N 0.000 description 1
- SPVFFPDJYPMULI-UHFFFAOYSA-N 2-bromo-3,4-difluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(F)=C1Br SPVFFPDJYPMULI-UHFFFAOYSA-N 0.000 description 1
- VEJPLARLFWIYNM-UHFFFAOYSA-N 2-bromo-3-chlorobenzaldehyde Chemical compound ClC1=CC=CC(C=O)=C1Br VEJPLARLFWIYNM-UHFFFAOYSA-N 0.000 description 1
- SITHNMNGOHVILG-UHFFFAOYSA-N 2-bromo-3-chlorobenzoic acid Chemical compound OC(=O)C1=CC=CC(Cl)=C1Br SITHNMNGOHVILG-UHFFFAOYSA-N 0.000 description 1
- NDOPHXWIAZIXPR-UHFFFAOYSA-N 2-bromobenzaldehyde Chemical compound BrC1=CC=CC=C1C=O NDOPHXWIAZIXPR-UHFFFAOYSA-N 0.000 description 1
- IKCLCGXPQILATA-UHFFFAOYSA-N 2-chlorobenzoic acid Chemical class OC(=O)C1=CC=CC=C1Cl IKCLCGXPQILATA-UHFFFAOYSA-N 0.000 description 1
- JBVSBLLOZVDAAZ-UHFFFAOYSA-N 2-diazonio-1-[(2-methylpropan-2-yl)oxy]ethenolate Chemical compound CC(C)(C)OC([O-])=C[N+]#N JBVSBLLOZVDAAZ-UHFFFAOYSA-N 0.000 description 1
- AFENDNXGAFYKQO-UHFFFAOYSA-N 2-hydroxybutyric acid Chemical class CCC(O)C(O)=O AFENDNXGAFYKQO-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- SDXAWLJRERMRKF-UHFFFAOYSA-N 3,5-dimethyl-1h-pyrazole Chemical class CC=1C=C(C)NN=1 SDXAWLJRERMRKF-UHFFFAOYSA-N 0.000 description 1
- CKGBPLNCKGNHPG-UHFFFAOYSA-N 3-(2,6-difluorophenoxy)aniline Chemical compound FC1=C(OC=2C=C(N)C=CC=2)C(=CC=C1)F CKGBPLNCKGNHPG-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- VCBPQSPQALVDDA-UHFFFAOYSA-N 3-chloro-8-fluoroindolizine-5-carbaldehyde Chemical compound ClC1=CC=C2C(=CC=C(N12)C=O)F VCBPQSPQALVDDA-UHFFFAOYSA-N 0.000 description 1
- JVQIKJMSUIMUDI-UHFFFAOYSA-N 3-pyrroline Chemical compound C1NCC=C1 JVQIKJMSUIMUDI-UHFFFAOYSA-N 0.000 description 1
- QSVDFJNXDKTKTJ-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-indene Chemical compound C1CCCC2=C1CC=C2 QSVDFJNXDKTKTJ-UHFFFAOYSA-N 0.000 description 1
- RSXICCZXXOLXIG-UHFFFAOYSA-N 4-bromo-2-ethynyl-1-fluorobenzene Chemical compound FC1=CC=C(Br)C=C1C#C RSXICCZXXOLXIG-UHFFFAOYSA-N 0.000 description 1
- SHSYFORYQKPNSF-UHFFFAOYSA-N 5-bromo-2-fluorobenzenethiol Chemical compound FC1=CC=C(Br)C=C1S SHSYFORYQKPNSF-UHFFFAOYSA-N 0.000 description 1
- QMLPBIBVTKYXKR-UHFFFAOYSA-N 5-bromo-6-[3-[tert-butyl(dimethyl)silyl]oxyprop-1-ynyl]pyridin-2-amine Chemical compound BrC=1C=CC(=NC=1C#CCO[Si](C)(C)C(C)(C)C)N QMLPBIBVTKYXKR-UHFFFAOYSA-N 0.000 description 1
- 108010031646 5-isoxazoyl-cyclohexylalanyl-isoleucyl-(3-(aminomethyl)phenyl)-(4-(aminomethyl)piperidin-1-yl)methanone Proteins 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- GSJJXNDSWVHYMU-UHFFFAOYSA-N 6-[3-[tert-butyl(dimethyl)silyl]oxyprop-1-ynyl]pyridin-2-amine Chemical compound [Si](C)(C)(C(C)(C)C)OCC#CC1=CC=CC(=N1)N GSJJXNDSWVHYMU-UHFFFAOYSA-N 0.000 description 1
- UVDJYBXHNFJTME-UHFFFAOYSA-N 6-bromo-5-fluoropyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(F)C(Br)=N1 UVDJYBXHNFJTME-UHFFFAOYSA-N 0.000 description 1
- BKLJUYPLUWUEOQ-UHFFFAOYSA-N 6-bromopyridin-2-amine Chemical compound NC1=CC=CC(Br)=N1 BKLJUYPLUWUEOQ-UHFFFAOYSA-N 0.000 description 1
- PRXUTIXCOBFMMM-UHFFFAOYSA-N 7-bromo-4-fluoro-1h-indole Chemical compound FC1=CC=C(Br)C2=C1C=CN2 PRXUTIXCOBFMMM-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 101100478290 Arabidopsis thaliana SR30 gene Proteins 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 206010009094 Chronic paroxysmal hemicrania Diseases 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 208000006561 Cluster Headache Diseases 0.000 description 1
- 206010009895 Colitis ischaemic Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000013586 Complex regional pain syndrome type 1 Diseases 0.000 description 1
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 1
- VMQMZMRVKUZKQL-UHFFFAOYSA-N Cu+ Chemical compound [Cu+] VMQMZMRVKUZKQL-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-YMDCURPLSA-N D-galactopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-YMDCURPLSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 206010012442 Dermatitis contact Diseases 0.000 description 1
- 206010012455 Dermatitis exfoliative Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 208000005171 Dysmenorrhea Diseases 0.000 description 1
- 206010013935 Dysmenorrhoea Diseases 0.000 description 1
- 206010014201 Eczema nummular Diseases 0.000 description 1
- 206010058838 Enterocolitis infectious Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 101100322888 Escherichia coli (strain K12) metL gene Proteins 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- 101150095928 F2rl1 gene Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 206010016952 Food poisoning Diseases 0.000 description 1
- 208000019331 Foodborne disease Diseases 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 208000035154 Hyperesthesia Diseases 0.000 description 1
- 108060005987 Kallikrein Proteins 0.000 description 1
- 102000001399 Kallikrein Human genes 0.000 description 1
- 208000003892 Kartagener syndrome Diseases 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 229930182821 L-proline Natural products 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 108090000543 Ligand-Gated Ion Channels Proteins 0.000 description 1
- 102000004086 Ligand-Gated Ion Channels Human genes 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 206010027918 Mononeuropathy multiplex Diseases 0.000 description 1
- 208000023178 Musculoskeletal disease Diseases 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- ILUJQPXNXACGAN-UHFFFAOYSA-N O-methylsalicylic acid Chemical class COC1=CC=CC=C1C(O)=O ILUJQPXNXACGAN-UHFFFAOYSA-N 0.000 description 1
- 229910004727 OSO3H Inorganic materials 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 208000007542 Paresis Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000009675 Perioral Dermatitis Diseases 0.000 description 1
- 206010056238 Phantom pain Diseases 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- IGVPBCZDHMIOJH-UHFFFAOYSA-N Phenyl butyrate Chemical class CCCC(=O)OC1=CC=CC=C1 IGVPBCZDHMIOJH-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 206010036105 Polyneuropathy Diseases 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 206010037779 Radiculopathy Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 201000001947 Reflex Sympathetic Dystrophy Diseases 0.000 description 1
- 206010038419 Renal colic Diseases 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 206010039793 Seborrhoeic dermatitis Diseases 0.000 description 1
- HOWDUIVVWDUEED-WAUHAFJUSA-N Ser-Leu-Ile-Gly-Lys-Val-Amide Chemical compound OC[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(N)=O HOWDUIVVWDUEED-WAUHAFJUSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 206010041955 Stasis dermatitis Diseases 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 208000000491 Tendinopathy Diseases 0.000 description 1
- 206010043255 Tendonitis Diseases 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108060005989 Tryptase Proteins 0.000 description 1
- 102000001400 Tryptase Human genes 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000000697 Vocal Cord Dysfunction Diseases 0.000 description 1
- 208000013154 Vocal cord disease Diseases 0.000 description 1
- 208000035562 Wallenberg syndrome Diseases 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- CBMCZKMIOZYAHS-NSCUHMNNSA-N [(e)-prop-1-enyl]boronic acid Chemical compound C\C=C\B(O)O CBMCZKMIOZYAHS-NSCUHMNNSA-N 0.000 description 1
- CBMCZKMIOZYAHS-IHWYPQMZSA-N [(z)-prop-1-enyl]boronic acid Chemical compound C\C=C/B(O)O CBMCZKMIOZYAHS-IHWYPQMZSA-N 0.000 description 1
- ZQKHRAXLCGDJDL-UHFFFAOYSA-N [1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C[Si](C)(C)CCOCN1C=CN=C1CO ZQKHRAXLCGDJDL-UHFFFAOYSA-N 0.000 description 1
- XGXCVNUHLXARIE-UHFFFAOYSA-N [2-(1-bicyclo[3.1.0]hexanyl)-4-fluorophenyl]-(1H-imidazol-2-yl)methanol Chemical compound C12(CCCC2C1)C1=C(C=CC(=C1)F)C(O)C=1NC=CN=1 XGXCVNUHLXARIE-UHFFFAOYSA-N 0.000 description 1
- ZKHBCXGLSKRDPO-UHFFFAOYSA-N [2-(1-bicyclo[3.1.0]hexanyl)-4-fluorophenyl]-[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C12(CCCC2C1)C1=C(C=CC(=C1)F)C(O)C=1N(C=CN=1)COCC[Si](C)(C)C ZKHBCXGLSKRDPO-UHFFFAOYSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- GPWHDDKQSYOYBF-UHFFFAOYSA-N ac1l2u0q Chemical compound Br[Br-]Br GPWHDDKQSYOYBF-UHFFFAOYSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000008484 agonism Effects 0.000 description 1
- 150000001335 aliphatic alkanes Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 206010053552 allodynia Diseases 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 210000001742 aqueous humor Anatomy 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 230000002567 autonomic effect Effects 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 150000001539 azetidines Chemical class 0.000 description 1
- 210000004082 barrier epithelial cell Anatomy 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960004217 benzyl alcohol Drugs 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- JBFDZEJAJZJORO-UHFFFAOYSA-N bicyclo[4.1.0]hept-3-ene Chemical compound C1C=CCC2CC21 JBFDZEJAJZJORO-UHFFFAOYSA-N 0.000 description 1
- DCRRIOWFXXDTHV-UHFFFAOYSA-N bicyclo[4.2.0]oct-3-ene Chemical compound C1C=CCC2CCC21 DCRRIOWFXXDTHV-UHFFFAOYSA-N 0.000 description 1
- RPZUBXWEQBPUJR-UHFFFAOYSA-N bicyclo[4.2.0]octane Chemical compound C1CCCC2CCC21 RPZUBXWEQBPUJR-UHFFFAOYSA-N 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 125000001626 borono group Chemical group [H]OB([*])O[H] 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000005884 carbocyclylalkyl group Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 201000008240 chemical colitis Diseases 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 150000005827 chlorofluoro hydrocarbons Chemical class 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 208000018912 cluster headache syndrome Diseases 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- SFJMFSWCBVEHBA-UHFFFAOYSA-M copper(i)-thiophene-2-carboxylate Chemical compound [Cu+].[O-]C(=O)C1=CC=CS1 SFJMFSWCBVEHBA-UHFFFAOYSA-M 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 208000012790 cranial neuralgia Diseases 0.000 description 1
- 201000010549 croup Diseases 0.000 description 1
- 150000001925 cycloalkenes Chemical group 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 201000003146 cystitis Diseases 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000005534 decanoate group Chemical class 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 229920006237 degradable polymer Polymers 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 125000005131 dialkylammonium group Chemical group 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- HQWPLXHWEZZGKY-UHFFFAOYSA-N diethylzinc Chemical compound CC[Zn]CC HQWPLXHWEZZGKY-UHFFFAOYSA-N 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- UQGFMSUEHSUPRD-UHFFFAOYSA-N disodium;3,7-dioxido-2,4,6,8,9-pentaoxa-1,3,5,7-tetraborabicyclo[3.3.1]nonane Chemical compound [Na+].[Na+].O1B([O-])OB2OB([O-])OB1O2 UQGFMSUEHSUPRD-UHFFFAOYSA-N 0.000 description 1
- AJFXNBUVIBKWBT-UHFFFAOYSA-N disodium;boric acid;hydrogen borate Chemical compound [Na+].[Na+].OB(O)O.OB(O)O.OB(O)O.OB([O-])[O-] AJFXNBUVIBKWBT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000001667 episodic effect Effects 0.000 description 1
- 230000004890 epithelial barrier function Effects 0.000 description 1
- OLAMWIPURJGSKE-UHFFFAOYSA-N et2o diethylether Chemical compound CCOCC.CCOCC OLAMWIPURJGSKE-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- HBTUTSUYMCYGKN-UHFFFAOYSA-N ethyl 3-chloro-8-fluoroindolizine-5-carboxylate Chemical compound ClC1=CC=C2C(=CC=C(N12)C(=O)OCC)F HBTUTSUYMCYGKN-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 208000004526 exfoliative dermatitis Diseases 0.000 description 1
- 239000006277 exogenous ligand Substances 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical class CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000004404 heteroalkyl group Chemical group 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical class OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 125000003037 imidazol-2-yl group Chemical group [H]N1C([*])=NC([H])=C1[H] 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 208000027139 infectious colitis Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000000266 injurious effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 201000008222 ischemic colitis Diseases 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical class CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000010983 kinetics study Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 229960000448 lactic acid Drugs 0.000 description 1
- 208000004343 lateral medullary syndrome Diseases 0.000 description 1
- 229940033355 lauric acid Drugs 0.000 description 1
- 238000011542 limb amputation Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 238000000622 liquid--liquid extraction Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- QBZXOWQOWPHHRA-UHFFFAOYSA-N lithium;ethane Chemical compound [Li+].[CH2-]C QBZXOWQOWPHHRA-UHFFFAOYSA-N 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000011418 maintenance treatment Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- NYKVKANRUGAWAL-UHFFFAOYSA-N methyl 2-amino-5-bromo-3-iodobenzoate Chemical compound COC(=O)C1=CC(Br)=CC(I)=C1N NYKVKANRUGAWAL-UHFFFAOYSA-N 0.000 description 1
- AYMMPTHLFMQVFH-UHFFFAOYSA-N methyl 6-bromo-5-fluoropyridine-2-carboxylate Chemical compound COC(=O)C1=CC=C(F)C(Br)=N1 AYMMPTHLFMQVFH-UHFFFAOYSA-N 0.000 description 1
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical class COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 208000008275 microscopic colitis Diseases 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 210000002200 mouth mucosa Anatomy 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- FGNGTWFJQFTFGN-UHFFFAOYSA-N n,n,n',n'-tetramethylethane-1,2-diamine Chemical compound CN(C)CCN(C)C.CN(C)CCN(C)C FGNGTWFJQFTFGN-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical class C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 208000020469 nerve plexus disease Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 230000000474 nursing effect Effects 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical class CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229940042125 oral ointment Drugs 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229940098695 palmitic acid Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 208000007777 paroxysmal Hemicrania Diseases 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- CRWVOXFUXPYTRK-UHFFFAOYSA-N pent-4-yn-1-ol Chemical compound OCCCC#C CRWVOXFUXPYTRK-UHFFFAOYSA-N 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical class CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 125000005498 phthalate group Chemical class 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-M picolinate Chemical compound [O-]C(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-M 0.000 description 1
- 201000006380 plexopathy Diseases 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 230000007824 polyneuropathy Effects 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 201000009266 primary ciliary dyskinesia Diseases 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- TVDSBUOJIPERQY-UHFFFAOYSA-N prop-2-yn-1-ol Chemical compound OCC#C TVDSBUOJIPERQY-UHFFFAOYSA-N 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical class CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229940095574 propionic acid Drugs 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-N propynoic acid Chemical class OC(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-N 0.000 description 1
- 230000004844 protein turnover Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-CDYZYAPPSA-N pyridin-2-amine Chemical compound NC1=CC=CC=[15N]1 ICSNLGPSRYBMBD-CDYZYAPPSA-N 0.000 description 1
- ILVXOBCQQYKLDS-UHFFFAOYSA-N pyridine N-oxide Chemical compound [O-][N+]1=CC=CC=C1 ILVXOBCQQYKLDS-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical class OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 1
- 208000008742 seborrheic dermatitis Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000009991 second messenger activation Effects 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 108010054243 seryl-leucyl-isoleucyl-glycyl-lysyl-valinamide Proteins 0.000 description 1
- 150000004666 short chain fatty acids Chemical class 0.000 description 1
- 235000021391 short chain fatty acids Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 229940100996 sodium bisulfate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229940001482 sodium sulfite Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical class OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000004426 substituted alkynyl group Chemical group 0.000 description 1
- 150000003900 succinic acid esters Chemical class 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 201000004415 tendinitis Diseases 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 150000005621 tetraalkylammonium salts Chemical class 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000005208 trialkylammonium group Chemical group 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 1
- 206010044652 trigeminal neuralgia Diseases 0.000 description 1
- ZNEOHLHCKGUAEB-UHFFFAOYSA-N trimethylphenylammonium Chemical compound C[N+](C)(C)C1=CC=CC=C1 ZNEOHLHCKGUAEB-UHFFFAOYSA-N 0.000 description 1
- PRXNKYBFWAWBNZ-UHFFFAOYSA-N trimethylphenylammonium tribromide Chemical compound Br[Br-]Br.C[N+](C)(C)C1=CC=CC=C1 PRXNKYBFWAWBNZ-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BSVBQGMMJUBVOD-UHFFFAOYSA-N trisodium borate Chemical compound [Na+].[Na+].[Na+].[O-]B([O-])[O-] BSVBQGMMJUBVOD-UHFFFAOYSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 238000001195 ultra high performance liquid chromatography Methods 0.000 description 1
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 230000001457 vasomotor Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 208000009935 visceral pain Diseases 0.000 description 1
- 210000004127 vitreous body Anatomy 0.000 description 1
- 210000001260 vocal cord Anatomy 0.000 description 1
- ZBGXUVOIWDMMJE-QHNZEKIYSA-N vorapaxar Chemical group C(/[C@@H]1[C@H]2[C@H](C(O[C@@H]2C)=O)C[C@H]2[C@H]1CC[C@H](C2)NC(=O)OCC)=C\C(N=C1)=CC=C1C1=CC=CC(F)=C1 ZBGXUVOIWDMMJE-QHNZEKIYSA-N 0.000 description 1
- 229960005044 vorapaxar Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- GDJZZWYLFXAGFH-UHFFFAOYSA-M xylenesulfonate group Chemical group C1(C(C=CC=C1)C)(C)S(=O)(=O)[O-] GDJZZWYLFXAGFH-UHFFFAOYSA-M 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 235000016804 zinc Nutrition 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto, en el que el compuesto es un compuesto de fórmula (I): **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, en la que: R3 es halógeno; X es N o CH; Y es C o N; y Z es C o N, en la que Y y Z no son ambos N, y (1) cuando tanto Z como Y son C, el enlace " " es un doble enlace, y R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo, **(Ver fórmula)** F, Cl, Br, OH, metoxi y CN; o (2) cuando uno de Z y Y es N, el otro es C, el enlace " " es un enlace sencillo, y R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo, **(Ver fórmula)** F, Cl, Br, OH, metoxi y CN.
Description
DESCRIPCIÓN
Inhibidores del receptor 2 activado por proteasa
Campo de la invención
La presente solicitud se refiere al tratamiento o prevención de afecciones o trastornos relacionados con el dolor, inflamación musculoesquelética tal como la osteoartritis, trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis, colitis y afecciones relacionadas con compuestos de imidazoles sustituidos, composiciones que los contienen y métodos para usarlos. Cualquier referencia a la solicitud que se relacione con, proporcione o tenga realizaciones que sean métodos de tratamiento debe entenderse como un compuesto o compuestos para su uso.
Antecedentes
Se cree que las acciones del receptor 2 activado por proteasa (PAR2) están involucradas en dolor, inflamación musculoesquelética, trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis, colitis y afecciones relacionadas. Los receptores activados por proteasa se componen de cuatro miembros de la familia (PAR1, PAR2, PAR3 y PAR4), que son receptores acoplados a proteína G activados por una escisión proteolítica de la región del extremo terminal N del receptor (Ramachandran, Hollenberg et al., Nature Reviews Drug Discovery, 2012, 11: 69 86). Tras la escisión proteolítica, el receptor se activa para la activación del segundo mensajero y las respuestas celulares. Se sabe que muchas enzimas escinden la región del extremo terminal N de los receptores PAR para iniciar este proceso, tal como la trombina, la tripsina, la triptasa de mastocitos, las calicreínas y enzimas relacionadas en las cascadas inflamatorias y de coagulación. Se sabe que otros ligandos activan el receptor, tal como péptidos cortos tales como SLIGKV-NH2 (SEQ ID NO.: 2), SLIGLR-NH2 (SEQ ID NO.: 3) o ligandos de moléculas pequeñas tales como GB-110 (Fairlie et al., Journal of Medicinal Chemistry, 2013, 56, 7477-7497).
Se ha demostrado que la expresión de PAR2 aumenta en el revestimiento sinovial, los condrocitos y los tejidos en la artritis reumatoide humana y en modelos animales de artritis (Amiable, N. et al., Bone, 44, 1143-1150). PAR2 también potencia la señalización a través de canales tales como TRPV1 (Dai, et al., Journal of Innate Immunology, 2010, 2, 495-504), un canal iónico dependiente de ligando implicado en el dolor inflamatorio. También se sabe que la señalización de PAR2 sensibiliza a TRPV1 in vivo, lo que produce hiperalgesia térmica.
Se sabe que la inhibición de los receptores PAR2 es responsable de las vías de señalización inflamatorias. En ratones que carecen del receptor PAR2, hay un inicio tardío de la inflamación (Linder et al., Journal of Immunology, 2000, 165, 6504-6510). Otros estudios de desactivación de PAR2 en roedores han demostrado que PAR2 juega un papel importante en la fisiopatología de muchas enfermedades como el dolor, la inflamación musculoesquelética como la osteoartritis, los trastornos neuroinflamatorios, la inflamación de las vías respiratorias, la picazón, la dermatitis, la colitis y otras afecciones relacionadas (Fairlie et al., Journal of Medicinal Chemistry, 2013, 56, 7477-749). También se ha demostrado que los antagonistas del receptor PAR2, tal como GB88, bloquean las respuestas inflamatorias in vivo, tal como el modelo de artritis inducida por colágeno en ratas (Fairlie et al., FASEB Journal, 2012, 26, 2877-2887). Yau M-K et al (J. Med. Chem. 2013, 56, 19, 7477-7497) divulga moduladores de PAR2 conocidos.
Por lo tanto, se cree que los antagonistas de PAR2 probablemente proporcionen beneficios a numerosas personas y tengan el potencial de aliviar el dolor y las afecciones relacionadas con la inflamación.
Resumen de la solicitud
La invención está definida por las reivindicaciones. La invención proporciona un compuesto, en el que el compuesto es un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en la que:
R3 es halógeno;
X es N o CH;
Y es C o N; y
Z es C o N, en la que Y y Z no son ambos N, y
(1) cuando tanto Z como Y son C, el enlace " ------" es un doble enlace, y
R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo
está opcionalmente fusionado con un arilo Ca-io, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,

F, Cl, Br, OH, metoxi y CN; o
(2) cuando uno de Z y Y es N, el otro es C, el enlace es un enlace sencillo, y R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,
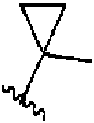
F, Cl, Br, OH, metoxi y CN;
o el compuesto es un compuesto seleccionado del grupo que consiste en:

y sales farmacéuticamente aceptables de los mismos.
Esta solicitud proporciona un compuesto de fórmula (I),

o una sal farmacéuticamente aceptable del mismo, en la que:
R3 es -H o halógeno (tal como -F);
X es N o CH;
Y es C o N; y
Z es C o N, en la que Y y Z no son ambos N, y
(1) cuando tanto Z como Y son C, el enlace " r r r r " es un doble enlace, y
R1 se selecciona de alquilo (tal como alquilo C1-12-, alquilo C2-12-, o alquilo C2-6-, por ejemplo, etilo o propilo); alquenilo (tal como alquenilo C2-12- o alquenilo C2-6-, por ejemplo,

cicloalquilo (tal como cicloalquilo C3-10- o cicloalquilo C3-6-, por ejemplo, ciclobutilo, ciclopentilo o

); cicloalquenilo (tal como cicloalquenilo C3-10- o cicloalquenilo C3-6-, por ejemplo,

); arilo (tal como arilo C6-10-); heterociclilo (tal como heterociclilo de 3 a 10 miembros o heterociclilo de 3 a 6 miembros, por ejemplo,

en la que n se selecciona de 0-3); y heteroarilo (tal como heteroarilo de 5 a 10 miembros, por ejemplo,

en la que n se selecciona de 0-3); en la que R1 está sustituido con 0-3 R, en la que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo o -CF3); cicloalquilo; halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en la que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
R2 es -H o -halógeno (tal como -F o -Cl); o
R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o -CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; o
(2) cuando uno de Z y Y es N y el otro es C, el enlace
" es un enlace sencillo, y R1 y R2se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o -CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; - N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb; en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
siempre que cuando R1 es metilo y X es CH, R2 y R3 no son ambos -H. En algunas realizaciones de lo anterior, cuando R1 es metilo y X es CH, R3 es -F.
En ciertas realizaciones, R3 es -H. En ciertas realizaciones, R3 es -F.
En ciertas realizaciones, X es CH. En ciertas realizaciones, X es N.
En ciertas realizaciones, tanto Z como Y son C y el enlace
" es un doble enlace. En ciertas realizaciones, tanto Z como Y son C, el enlace "
" es un doble enlace y el compuesto se puede representar por la fórmula (I-A):

o una sal farmacéuticamente aceptable del mismo, en la que:
X es N o CH;
R1 se selecciona de alquilo (tal como alquilo C1-12-, alquilo C2-12-, o alquilo C2-6-, por ejemplo, etilo o propilo); alquenilo (tal como alquenilo C2-12-, o alquenilo C2-6-, por ejemplo,

); cicloalquilo (tal como cicloalquilo C3-10 o cicloalquilo C3-6-, por ejemplo, ciclobutilo, ciclopentilo, o

); cicloalquenilo (tal como cicloalquenilo C3-10- o cicloalquenilo C3-6-, por ejemplo,

); arilo (tal como arilo C6-10-); heterociclilo (tal como heterociclilo de 3 a 10 miembros o heterociclilo de 3 a 6 miembros, por ejemplo,

en el que n se selecciona de 0-3); y heteroarilo (tal como heteroarilo de 5 a 10 miembros, por ejemplo,

en el que n se selecciona de 0-3); en el que R1 está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo o -CF3); cicloalquilo; halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -c N; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -c(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
R2 es -H o -halógeno (tal como -F o -Cl); o
R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o - CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; y
R3 es -H o halógeno (tal como -F);
siempre que cuando R1 es metilo y X es CH, R2 y R3 no son ambos -H. En algunas realizaciones de lo anterior, cuando R1 es metilo y X es CH, R3 es -F.
En determinadas realizaciones en las que uno de Z y Y es N y el otro es C, el compuesto se puede representar mediante la fórmula (I-B):

o una sal farmacéuticamente aceptable del mismo, en la que:
X es N o CH;
R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o - CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; y
R3 es -H o halógeno (tal como -F).
En ciertas realizaciones, Y es C y Z es N y el enlace
nnnn” es
" es un enlace sencillo. En ciertas realizaciones, Y es N y Z es C y el enlace "
" es un enlace sencillo.
En otro aspecto, la presente solicitud proporciona un compuesto que se une a un sitio alostérico de un polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que el compuesto interactúa con uno o más residuos de aminoácidos seleccionados del grupo que consiste en Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto se une a un sitio alostérico de un polipéptido pAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131 e His 135 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto se une a un sitio alostérico de un polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas realizaciones, dicho compuesto es un antagonista de PAR2. En algunas realizaciones, dicho compuesto es una molécula pequeña (por ejemplo, un antagonista de molécula pequeña). En ciertas realizaciones de lo anterior, el compuesto tiene una estructura de fórmula (I), (I-A) o (I-B), o una sal farmacéuticamente aceptable del mismo.
En otro aspecto, la presente solicitud proporciona un compuesto que se une a un sitio alostérico del polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que dicho compuesto tiene una vida media disociativa objetivo (t1/2) de más de 10 minutos. En algunas realizaciones, el compuesto se une a un sitio alostérico del polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el
que dicho compuesto tiene una vida media disociativa objetivo (ti/2) seleccionada entre más de 3000 minutos, 10 a 3000 minutos, 10 a 2000 minutos, 10 a 1000 minutos, 10 a 900 minutos, 10 a 800 minutos, 10 a 700 minutos, 10 a 600 minutos, 10 a 500 minutos, 10 a 400 minutos, 10 a 300 minutos, 10 a 200 minutos, 10 a 100 minutos, 10 a 50 minutos, 10 a 40 minutos, 10 a 30 minutos y 10 a 20 minutos. En algunas realizaciones, dicho compuesto tiene una vida media disociativa objetivo de 10 a 200 minutos. En algunas realizaciones de lo anterior, el compuesto se une a un sitio alostérico de un polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que el compuesto interactúa con uno o más residuos de aminoácidos seleccionados del grupo que consiste en Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131 e His 135 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En determinadas realizaciones, la vida media disociativa se determina en un ensayo de disociación de FLPR, un ensayo de resonancia de plasmones superficiales o un ensayo de unión radioquímica. En algunas realizaciones, dicho compuesto es un antagonista de PAR2. En algunas realizaciones, dicho compuesto es una molécula pequeña (por ejemplo, un antagonista de molécula pequeña). En algunas realizaciones de lo anterior, el compuesto tiene una estructura de fórmula (I), (I-A) o (I-B), o una sal farmacéuticamente aceptable del mismo.
En determinadas realizaciones, esta solicitud proporciona una composición farmacéutica que comprende al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo. Las composiciones farmacéuticas como se describen en el presente documento pueden comprender además un excipiente farmacéuticamente aceptable. En determinadas realizaciones, esta solicitud también proporciona un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo, o una composición que comprende cualquiera de los anteriores para su uso como medicamento.
En otro aspecto, esta solicitud proporciona métodos para tratar una enfermedad o trastorno mediado por la actividad de PAR2, tal como los descritos en el presente documento, que comprenden administrar a un sujeto que necesita dicho tratamiento, tal como un paciente, una cantidad eficaz de al menos un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo en una dosis, frecuencia y duración para proporcionar un efecto beneficioso para el sujeto.
En ciertas realizaciones, esta solicitud proporciona el uso de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición que comprende cualquiera de los anteriores en la preparación de un medicamento para el tratamiento de enfermedades o trastornos regulados por la actividad de PAR2, y el uso de tales compuestos y sales para el tratamiento de tales enfermedades y trastornos.
En determinadas realizaciones, esta solicitud proporciona un método para tratar una enfermedad o trastorno en un sujeto, tal como un paciente, que comprende modular PAR2, en el que la modulación de PAR2 comprende administrar al sujeto al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición que comprende cualquiera de los anteriores, en una dosis, con una frecuencia y por una duración para proporcionar un efecto beneficioso al sujeto paciente. En ciertas realizaciones de este tipo, la enfermedad o trastorno se selecciona del grupo que consiste en dolor, inflamación musculoesquelética (tal como osteoartritis), trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis o colitis. En ciertas realizaciones, la enfermedad o trastorno es osteoartritis.
En determinadas realizaciones, esta solicitud proporciona un método para modular la actividad de PAR2, que comprende poner en contacto una célula que comprende PAR2 con una cantidad eficaz de al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición que comprende cualquiera de los anteriores.
En determinadas realizaciones, esta solicitud describe un método para modular la actividad de un receptor PAR2, que comprende poner en contacto una célula que comprende el receptor PAR2 con una cantidad eficaz de al menos un compuesto descrito en el presente documento o una sal farmacéuticamente aceptable del mismo, y/o con al menos un compuesto o composición farmacéutica como se describe en el presente documento. En ciertas realizaciones de lo anterior, el contacto es in vitro, ex vivo, o in vivo.
Las realizaciones, características y ventajas adicionales de la solicitud serán evidentes a partir de la siguiente descripción detallada ya través de la práctica de las realizaciones descritas en esta solicitud.
Descripción detallada de las figuras
La Figura 1 muestra la estructura cristalina de PAR1 unida a Vorapaxar en comparación con la estructura cristalina de PAR2 unida al compuesto del Ejemplo 3.
La Figura 2 muestra la estructura cristalina de PAR2 en complejo con el compuesto del Ejemplo 3.
La Figura 3 muestra las interacciones Proteína-ligando con el compuesto del Ejemplo 3 con base en la estructura cristalina de PAR2.
Descripción detallada
La invención está definida por las reivindicaciones. La invención proporciona un compuesto, en el que el compuesto es un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en la que:
R3 es halógeno;
X es N o CH;
Y es C o N; y
Z es C o N, en el que Y y Z no son ambos N, y
i “ ” es un d
(1) cuando tanto Z como Y son C, el enlace ' ° s a omos " es un doble enlace, y R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,

F, Cl, Br, OH, metoxi y CN; o
md es un s
(2) cuando uno de Z y Y es N, el otro es C, el enlace "rrr-. están unidos a fo m es un en|ace sencillo, y R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,

F, Cl, Br, OH, metoxi y CN;
o el compuesto es un compuesto seleccionado del grupo que consiste en:

y sales farmacéuticamente aceptables de los mismos.
La presente solicitud proporciona un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en la que:
R3 es -H o halógeno (tal como -F);
X es N o CH;
Y es C o N; y
Z es C o N, en el que Y y Z no son ambos N, y
(1) cuando tanto Z como Y son C, el enlace
d “ ” es un
alquilo C1-12-, (
" es un doble enlace, y
R1 se selecciona de alquilo (tal como alquilo C1-12-, alquilo C2-12-, o alquilo C2-6-, por ejemplo, etilo o propilo); alquenilo (tal como alquenilo C2-12- o alquenilo C2-6-, por ejemplo,

); cicloalquilo (tal como cicloalquilo C3-10- o cicloalquilo C3-6-, por ejemplo, ciclobutilo, ciclopentilo o

); cicloalquenilo (tal como cicloalquenilo C3-10- o cicloalquenilo C3-6-, por ejemplo,

); arilo (tal como arilo C6-10-); heterociclilo (tal como heterociclilo de 3 a 10 miembros o heterociclilo de 3 a 6 miembros, por ejemplo,

en el que n se selecciona de 0-3); y heteroarilo (tal como heteroarilo de 5 a 10 miembros o heteroarilo de 5 a 6 miembros, por ejemplo,
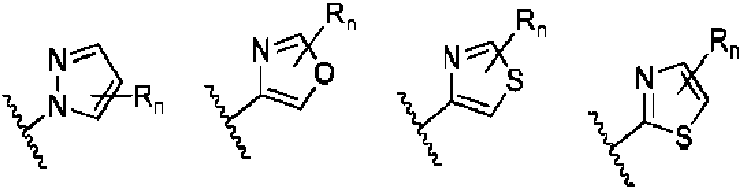
en el que n se selecciona de 0-3); en el que R1 está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo o -CF3); cicloalquilo; halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; - N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
R2 es -H o -halógeno (tal como -F o -Cl); o
R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o -CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo;
- N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; o
(2) cuando uno de Z y Y es N y el otro es C, el enlace " ------
" es un enlace sencillo, y R1 y R2se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o -CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; - N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; siempre que cuando R1 es metilo y X es CH, R2 y R3 no son ambos -H. En ciertas realizaciones, cuando R1 es metilo y X es CH, R3 es -F.
En ciertas realizaciones, X es CH. En ciertas realizaciones, X es N.
En ciertas realizaciones, R3 es -H. En ciertas realizaciones, R3 es -F.
En ciertas realizaciones, tanto Z como Y son C y el enlace " ------
id “ ” es.
enlace doble,;
" es un doble enlace. En ciertas realizaciones, tanto Z como Y son C, el enlace " ------e C y el bor
id “ ” es un d<
" es un doble enlace, y el compuesto se puede representar por la fórmula (I-A):

o una sal farmacéuticamente aceptable del mismo, en la que:
X es N o CH;
R1 se selecciona de alquilo (tal como alquilo C1-12-, alquilo C2-12-, o alquilo C2-6-, por ejemplo, etilo o propilo); alquenilo (tal como alquenilo C2-12-, o alquenilo C2-6-, por ejemplo,

); cicloalquilo (tal como cicloalquilo C3-10- o cicloalquilo C3-6-, por ejemplo, ciclobutilo, ciclopentilo o

); cicloalquenilo (tal como cicloalquenilo C3-10- o cicloalquenilo C3-6-, por ejemplo,

); arilo (tal como arilo Ce-io-); heterociclilo (tal como heterociclilo de 3 a 10 miembros o heterociclilo de 3 a 6 miembros, por ejemplo,

en el que n se selecciona de 0-3); y heteroarilo (tal como heteroarilo de 5 a 10 miembros, por ejemplo,

en el que n se selecciona de 0-3); en el que R1 está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo o -CF3); cicloalquilo; halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
R2 es -H o -halógeno (tal como -F o -Cl); o
R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o - CF3); cicloalquilo (por ejemplo, ciclopropilo o
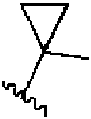
); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo; y
R3 es -H o halógeno (tal como -F);
siempre que cuando R1 es metilo y X es CH, R2 y R3 no son ambos -H. En algunas realizaciones de lo anterior, cuando R1 es metilo y X es CH, R3 es -F.
En ciertas realizaciones, X es CH. En ciertas realizaciones, X es N.
En ciertas realizaciones, R3 es -H. En ciertas realizaciones, R3 es -F.
En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es alquilo C1-12 que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es alquilo C2-12 que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es alquilo C2-6 que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es un alquilo C3-6 lineal o ramificado que está opcionalmente sustituido con 0-3 R. En algunas de las realizaciones anteriores, R1 no está sustituido. En algunas realizaciones, R1 es etilo o propilo. En algunas realizaciones, R1 es propilo.
En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es alquenilo C2-12 que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es alquenilo C2-6 que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es un alquenilo C3-6 lineal o ramificado que está opcionalmente sustituido con 0-3 R. En algunas de las realizaciones anteriores, R1 no está sustituido. En algunas realizaciones, R1 es

En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es cicloalquilo C3-10- que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es cicloalquilo C3-6- que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 se selecciona de ciclobutilo, ciclopentilo y

en el que R1 está opcionalmente sustituido con 0-3 R. En algunas de las realizaciones anteriores, R1 no está sustituido.
En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es cicloalquenilo C3-10- que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es cicloalquenilo C3-6- que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es

que está opcionalmente sustituido con 0-3 R. En algunas de las realizaciones anteriores, R1 no está sustituido.
En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es heterociclilo de 3 a 10 miembros que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es heterociclilo de 3 a 6 miembros que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 se selecciona de:

en el que n es 0-3. En algunas de las realizaciones anteriores, R1 no está sustituido. En algunas de las realizaciones anteriores, cada ocurrencia de R se selecciona independientemente de: alquilo C1-3 (tal como -Me) y halógeno (tal como -F). En algunas realizaciones, R1 se selecciona de:

En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 es heteroarilo de 5 a 10 miembros que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 es heteroarilo de 5 o 6 miembros que está opcionalmente sustituido con 0-3 R. En algunas realizaciones, R1 se selecciona de:

En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R2 es -H o -halógeno. En algunas realizaciones, R2 es -H. En algunas realizaciones, R2 es -F o -Cl.
En ciertas realizaciones de compuestos de fórmula (I) o (I-A), R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R. En algunas de las realizaciones anteriores, la estructura
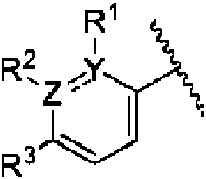
de fórmula (I) o la estructura

de fórmula (I-A) se selecciona de:

en el que R5 es -H o alquilo C1-3-, n se selecciona de 0-3. En algunas de estas realizaciones, R5 es -Me o -Et. En algunas de las realizaciones anteriores, cada ocurrencia de R se selecciona independientemente de: alquilo C1-4, cicloalquilo C3-6 y halo. En algunas de las realizaciones anteriores, cada ocurrencia de R se selecciona independientemente de: metilo, etilo, propilo, isopropilo, -CH2CF3, -CF3, ciclopropilo,

-F, -Cl y -Br, por ejemplo, metilo, etilo, -CH2CF3, ciclopropilo y

En algunas realizaciones, la estructura

de fórmula (I) o la estructura

de fórmula (I-A) se selecciona de:

En determinadas realizaciones en las que uno de Z y Y es N y el otro es C, el compuesto se puede representar mediante la fórmula (I-B):

o una sal farmacéuticamente aceptable del mismo, en la que:
X es N o CH;
R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de alquilo, tal como alquilo C1-4 (por ejemplo, metilo, etilo, propilo, isopropilo, -CH2CF3 o - CF3); cicloalquilo (por ejemplo, ciclopropilo o

); halo, tal como -F, -Cl o -Br (por ejemplo, -Cl); -OH; alcoxi, tal como metoxi; -CN; -NRaRb, -N(Ra)C(O) alquilo; -N(Ra)CO2alquilo; -N(Ra)SO2alquilo; -C(O)alquilo; -CO2H; -CO2alquilo; -CONRaRb; -SO2alquilo; y SO2NRaRb en el que Ra y Rb son independientemente para cada ocurrencia H o alquilo;
R3 es -H o halógeno (tal como -F).
En ciertas realizaciones, Y es C y Z es N y el enlace
nd “ ...... ” es un s
un enlace sencillo
" es un enlace sencillo. En ciertas realizaciones, Y es N y Z es C y el enlace
y el enlace 1
nd “ -----” es un s
a í n r o a l i ^ a n i A n
" es un enlace sencillo.
En ciertas realizaciones, X es CH. En ciertas realizaciones, X es N.
En ciertas realizaciones, R3 es -H. En ciertas realizaciones, R3 es -F.
En ciertas realizaciones de compuestos de fórmula (I) o (I-B), la estructura

se selecciona de:

en el que n se selecciona de 0-3. En algunas de estas realizaciones, cada ocurrencia de R se selecciona independientemente de: alquilo C1-4, cicloalquilo C3-6 y halo. En algunas realizaciones, cada ocurrencia de R se selecciona independientemente de: metilo, etilo, propilo, isopropilo, -CH2CF3, -CF3, ciclopropilo,

-F, -Cl y -Br, tales como metilo, etilo, isopropilo, ciclopropilo y -Cl. En algunas realizaciones, la estructura
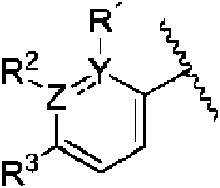
de fórmula (I) o (I-B) se selecciona de:

En ciertas realizaciones, el compuesto de la presente solicitud se selecciona de:

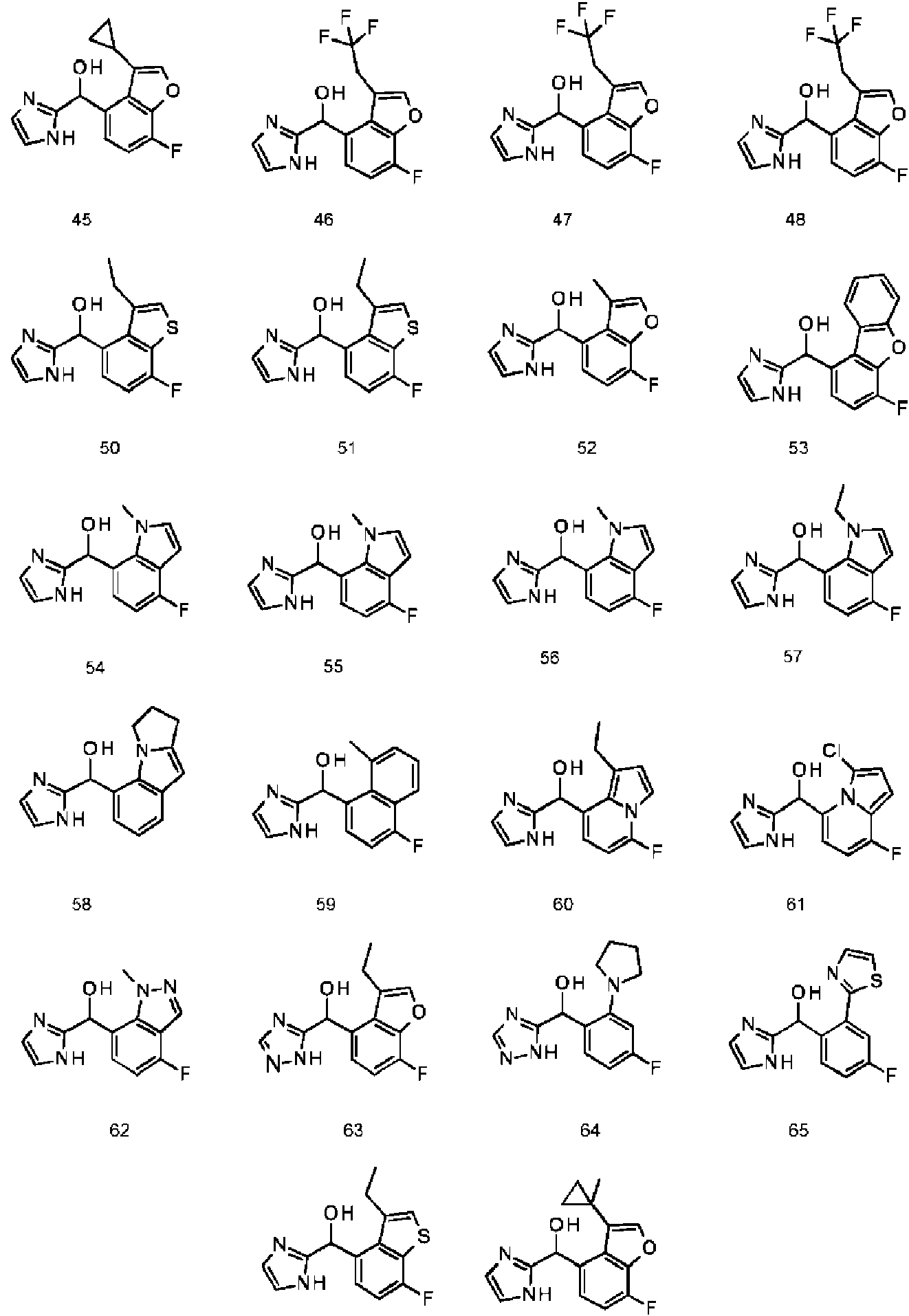
, y sales farmacéuticamente aceptables de los mismos.
Un ejemplo de un compuesto de la memoria descriptiva es:
(4-Fluoro-2-propilfenil)(1H-imidazol-2-il)metanol;
(R)-(4-fluoro-2-propilfeml)(1H-imidazol-2-il)metanol;
(s)-(4-Fluoro-2-propilfenil)(1H-imidazol-2-il)metanol;
(2-Ciclopentenilfenil)(1H-imidazol-2-il)metanol;
(2-Ciclopentenil-4-fluorofenil)(1H-imidazol-2-il)metanol;
(2-Ciclopentil-4-fluorofenil)(1H-imidazol-2-il)metanol;
(2-Ciclobutil-4-fluorofenil)(1H-imidazol-2-il)metanol;
(E)-(4-Fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol;
(z)-(4-Fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol;
(3,4-Difluoro-2-propilfenil)(1H-imidazol-2-il)metanol;
(R)-(3,4-Difluoro-2-propilfeml)(1H-imidazol-2-il)metanol;
(s)-(3,4-Difluoro-2-propilfenil)(1H-imidazol-2-il)metanol;
(3-Cloro-2-propilfenil)(1H-imidazol-2-il)metanol;
(4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-imidazol-2-il)metanol;
(R)-(4-Fluoro-2-(pirrolidin-1-il)feml)(1H-imidazol-2-il)metanol;
(S)-(4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-imidazol-2-il)metanol;
(2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol;
(R)-(2-(2,5-d¡h¡dro-1H-p¡rrol-1-¡l)-4-fluorofen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il)metano1;
(2-(Azet¡d¡n-1-¡l)-4-fluorofen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-Cloro-2-(1H-p¡razol-1-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3,4-D¡fluoro-2-(lH-p¡razol-1-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-(1H-p¡razol-1-¡l)fen¡l)(lH-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-(3-fluoro-3-met¡lazet¡d¡n-1-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-(3-met¡lazet¡d¡n-1-¡l)fen¡l)(lH-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-((S)-3-fluorop¡rrol¡d¡n-1-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-((R)-3-fluorop¡rrol¡d¡n-1-¡l)fen¡l)(lH-¡m¡dazol-2-¡l)metanol;
(2-(3,3-D¡fluorop¡rrol¡d¡n-1-¡l)-4-fluorofen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-((R)-3-met¡lp¡rrol¡d¡n-1-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(2-(3-Azab¡c¡clo[3.1.0]hexan-3-¡l)-4-fluorofen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(2-(B¡c¡clo[3.1.0]hexan-1-¡l)-4-fluorofen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-(oxazol-4-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-2-(t¡azol-4-¡l)fen¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-Et¡l-8-fluoro¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(R)-(3-Et¡l-8-fluoro¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(3-Et¡l-8-fluoro¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(8-Fluoro-3-met¡l¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(8-Fluoro-3-¡soprop¡l¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-C¡cloprop¡l-8-fluoro¡ndol¡z¡n-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-Et¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(R)-(3-Et¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(3-Et¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-C¡cloprop¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(R) -(3-C¡cloprop¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(S) -(3-C¡cloprop¡l-7-fluorobenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(7-Fluoro-3-(2,2,2-tr¡fluoroet¡l)benzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(R)-(7-Fluoro-3-(2,2,2-tr¡fluoroet¡l)benzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(7-Fluoro-3-(2,2,2-tr¡fluoroet¡l)benzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-Et¡l-7-fluorobenzo[b]t¡ofen-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(R)-(3-Et¡l-7-fluorobenzo[b]t¡ofen-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(3-Et¡l-7-fluorobenzo[b]t¡ofen-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(7-Fluoro-3-met¡lbenzofuran-4-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluorod¡benzo[b,d]furan-1-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-1-met¡l-1H-¡ndol-7-¡l)(lH-¡m¡dazol-2-¡l)metanol;
(R)-(4-Fluoro-1-met¡l-1H-¡ndol-7-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(s)-(4-Fluoro-1-met¡l-1H-¡ndol-7-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(l-Et¡l-4-fluoro-1H-¡ndol-7-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(2,3-D¡h¡dro-1H-p¡rrolo[1,2-a]¡ndol-5-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-8-met¡lnaftalen-1-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(l-Et¡l-5-fluoro-¡ndol¡z¡n-8-¡l)-(lH-¡m¡dazol-2-¡l)metanol;
(3-Cloro-8-fluoro-¡ndol¡z¡n-5-¡l)-(1H-¡m¡dazol-2-¡l)metanol;
(4-Fluoro-1-met¡l-1H-¡ndazol-7-¡l)(1H-¡m¡dazol-2-¡l)metanol;
(3-Et¡l-7-fluorobenzofuran-4-¡l)(lH-1,2,4-tr¡azol-5-¡l)metanol;
(4-Fluoro-2-(p¡rrol¡d¡n-1-¡l)fen¡l)(1H-1,2,4-tr¡azol-5-¡l)metanol;
(4-Fluoro-2-(t¡azol-2-¡l)fen¡l)(lH-¡m¡dazol-2-¡l)metanol;
[7-Fluoro-3-(1-met¡lc¡cloprop¡l)benzofuran-4-¡l]-(1H-¡m¡dazol-2-¡l)metanol;
o una sal farmacéut¡camente aceptable del m¡smo.
En otro aspecto, la presente sol¡c¡tud proporc¡ona un compuesto que se une a un s¡t¡o alostér¡co de un pol¡pépt¡do PAR2 que comprende una secuenc¡a de am¡noác¡dos expuesta en la SEQ ID NO.: 1, en el que el compuesto ¡nteractúa con uno o más res¡duos de am¡noác¡dos selecc¡onados del grupo que cons¡ste en Asp 228, Lys 131, H¡s 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas real¡zac¡ones, el compuesto se une a un s¡t¡o alostér¡co de un pol¡pépt¡do pAR2 que comprende una secuenc¡a de am¡noác¡dos expuesta en la SEQ ID NO.: 1, en el que el compuesto ¡nteractúa con los res¡duos de am¡noác¡dos Asp 228, Lys 131 e H¡s 135 de la SEQ ID NO.: 1. En algunas real¡zac¡ones, el compuesto se une a un s¡t¡o alostér¡co de un pol¡pépt¡do PAR2 que comprende una secuenc¡a de am¡noác¡dos expuesta en la SEQ ID NO.: 1, en el que el compuesto ¡nteractúa con los res¡duos de am¡noác¡dos Asp 228, Lys 131, H¡s 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas real¡zac¡ones, d¡cho compuesto es un antagon¡sta de PAR2. En algunas real¡zac¡ones, d¡cho compuesto es una molécula pequeña (por ejemplo, un antagon¡sta de molécula pequeña). En c¡ertas real¡zac¡ones de lo anter¡or, el compuesto t¡ene una estructura de fórmula (I), (I-A) o (I-B), o una sal farmacéut¡camente aceptable del m¡smo.
Se ha demostrado que los compuestos que actúan sobre su objetivo con largos tiempos medios de residencia en el objetivo tienen ventajas potenciales tales como una larga duración de la actividad y selectividad del objetivo. Esto se puede medir en enzimas y proteínas utilizando métodos como la espectroscopia de resonancia de plasmones superficiales, ensayos de unión radioquímica o deducirse de estudios dependientes del tiempo en un ensayo bioquímico. Los métodos cinéticos se pueden utilizar para determinar la constante de asociación (Kasociación), la constante de disociación (Kdisociación) y el valor de Kd para un compuesto dado (Copeland et al., Nat. Rev. Drug Disc.
2006, 5, 730-739). Estas mediciones cinéticas se pueden informar como tiempo de residencia (t = 1/kdisociación), o como la vida media disociativa (t-i/2 = 0.693/kdisociación). Se cree que el valor del tiempo de residencia o la vida media disociativa necesaria para obtener una actividad de larga duración (por ejemplo, efecto PD) varía de un objetivo a otro y generalmente está influenciado por los factores ambientales circundantes, tal como las concentraciones locales de ligandos competidores (tal como agonistas), la tasa de renovación de proteínas, así como la distribución y localización del compuesto. Los compuestos con esta propiedad también se describen a veces como que tienen cinéticas de disociación lenta.
En otro aspecto, la presente solicitud proporciona un compuesto que se une a un sitio alostérico del polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que dicho compuesto tiene una vida media disociativa objetivo (t-io) de más de 10 minutos. En algunas realizaciones, la presente solicitud proporciona un compuesto que se une a un sitio alostérico del polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que dicho compuesto tiene una vida media disociativa objetivo (ti/2) seleccionada entre más de 3000 minutos, 10 a 3000 minutos, 10 a 2000 minutos, 10 a 1000 minutos, 10 a 900 minutos, 10 a 800 minutos, 10 a 700 minutos, 10 a 600 minutos, 10 a 500 minutos, 10 a 400 minutos, 10 a 300 minutos, 10 a 200 minutos, 10 a 100 minutos, 10 a 50 minutos, 10 a 40 minutos, 10 a 30 minutos y 10 a 20 minutos. En algunas realizaciones, dicho compuesto tiene una vida media disociativa objetivo de 10 a 200 minutos. En algunas realizaciones de lo anterior, el compuesto se une a un sitio alostérico de un polipéptido PAR2 que comprende una secuencia de aminoácidos expuesta en la SEQ ID NO.: 1, en el que el compuesto interactúa con uno o más residuos de aminoácidos seleccionados del grupo que consiste en Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131 e His 135 de la SEQ ID NO.: 1. En algunas realizaciones, el compuesto interactúa con los residuos de aminoácidos Asp 228, Lys 131, His 135 y Tyr 82 de la SEQ ID NO.: 1. En determinadas realizaciones, la vida media disociativa se determina en un ensayo de disociación de FLPR, un ensayo de resonancia de plasmones superficiales o un ensayo de unión radioquímica. En algunas realizaciones, dicho compuesto es un antagonista de PAR2. En algunas realizaciones, dicho compuesto es una molécula pequeña (por ejemplo, un antagonista de molécula pequeña). En algunas realizaciones de lo anterior, el compuesto tiene una estructura de fórmula (I), (I-A) o (I-B), o una sal farmacéuticamente aceptable del mismo.
En determinadas realizaciones, esta solicitud se refiere a una composición farmacéutica que comprende (a) un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo; y (b) un excipiente farmacéuticamente aceptable.
En determinadas realizaciones, esta solicitud se refiere a un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica que comprende cualquiera de los anteriores, para su uso como medicamento.
En determinadas realizaciones, esta solicitud se refiere a un método para tratar una enfermedad o trastorno mediado por la actividad de PAR2, que comprende administrar a un sujeto que necesita dicho tratamiento una cantidad eficaz de al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica que comprende cualquiera de los anteriores. En ciertas realizaciones de este tipo, la enfermedad o trastorno mediado por la actividad de PAR2 se selecciona del grupo que consiste en dolor, inflamación musculoesquelética (tal como osteoartritis), trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis o colitis. En ciertas realizaciones de este tipo, la enfermedad o trastorno mediado por la actividad de PAR2 es osteoartritis.
En ciertas realizaciones, esta solicitud se relaciona con el uso de un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica de cualquiera de los anteriores, en la preparación de un medicamento para el tratamiento de enfermedades o trastornos regulados por la actividad de PAR2, y el uso de tales compuestos para el tratamiento de tales enfermedades y trastornos. En ciertas realizaciones de este tipo, la enfermedad o trastorno se selecciona del grupo que consiste en dolor, inflamación musculoesquelética (tal como osteoartritis), trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis o colitis. En ciertas realizaciones de este tipo, la enfermedad o trastorno mediado por la actividad de PAR2 es osteoartritis.
En determinadas realizaciones, esta solicitud se refiere a un método para modular (por ejemplo, inhibir) la actividad de un receptor PAR2, que comprende poner en contacto una célula que comprende el PAR2 con una cantidad eficaz de al menos un compuesto descrito en el presente documento, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica de cualquiera de los anteriores. En ciertas realizaciones de este tipo, el contacto es in vitro, ex vivo, o in vivo.
En ciertas realizaciones, esta solicitud se refiere a un método para tratar una enfermedad o trastorno en un paciente que lo necesita, que comprende administrar un compuesto descrito en el presente documento, o una sal
farmacéuticamente aceptable del mismo, o una composición farmacéutica de cualquiera de los anteriores, en el que el la enfermedad o trastorno se selecciona del grupo que consiste en dolor, inflamación musculoesquelética (tal como osteoartritis), trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis o colitis. En ciertas realizaciones de este tipo, la enfermedad o trastorno mediado por la actividad de PAR2 es osteoartritis.
Los expertos en la técnica reconocerán que las especies enumeradas o ilustradas en el presente documento no son exhaustivas, y que también se pueden seleccionar especies adicionales dentro del alcance de estos términos definidos.
La solicitud también incluye profármacos, sales, solvatos, tales como hidratos, farmacéuticamente aceptables, de los compuestos descritos en el presente documento, preferiblemente de los descritos anteriormente y de los compuestos específicos ejemplificados en el presente documento, y composiciones farmacéuticas que comprenden dichos profármacos, sales o solvatos, tales como hidratos y métodos de uso de tales sales o hidratos.
La presente solicitud también se refiere a los metabolitos farmacéuticamente activos de los compuestos descritos en el presente documento y los usos de tales metabolitos en los métodos de la solicitud.
Definiciones
Las definiciones establecidas en esta solicitud pretenden aclarar los términos utilizados en esta solicitud.
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que comúnmente entiende un experto en la técnica a la que pertenece esta solicitud. Si una definición establecida en esta sección es contraria o incompatible con una definición establecida en una patente, solicitud u otra publicación, prevalecerá la definición establecida en esta sección. Aunque cualquier método y material similar o equivalente a los descritos en el presente documento también puede usarse en la práctica o prueba de las realizaciones en la presente solicitud, ahora se describen los métodos y materiales preferidos.
Para proporcionar una descripción más concisa, algunas de las expresiones cuantitativas dadas en el presente documento no están calificadas con el término "aproximadamente". Se entiende que, ya sea que el término "aproximadamente" se use explícitamente o no, cada cantidad dada en el presente documento se refiere al valor real dado, y también se refiere a la aproximación a dicho valor dado que razonablemente se inferiría basado en la habilidad ordinaria en la técnica, incluyendo equivalentes y aproximaciones debido a las condiciones experimentales y/o de medición para dicho valor dado. Siempre que se dé un rendimiento en porcentaje, dicho rendimiento se refiere a una masa de la entidad para la que se da el rendimiento con respecto a la cantidad máxima de la misma entidad que podría obtenerse en las condiciones estequiométricas particulares. Las concentraciones que se dan como porcentajes se refieren a proporciones en masa, a menos que se indique lo contrario.
Salvo que se indique lo contrario, los métodos y técnicas de las presentes realizaciones se realizan generalmente de acuerdo con métodos convencionales bien conocidos en la técnica y como se describe en varias referencias generales y más específicas que se citan y analizan a lo largo de la presente memoria descriptiva. Véase, por ejemplo, Loudon, Organic Chemistry, cuarta edición, Nueva York: Oxford University Press, 2002, páginas 360-361, 1084-1085; Smith y March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, quinta edición, Wiley-Interscience, 2001.
La nomenclatura utilizada en el presente documento para nombrar los compuestos en cuestión se ilustra en los Ejemplos del presente documento. Esta nomenclatura generalmente se ha derivado usando el software ChemBioDraw Ultra comercialmente disponible (Cambridgesoft/Perkin Elmer), Versión 12.0.
Debe entenderse que la presente descripción no se limita a las realizaciones particulares descritas, ya que tales pueden, por supuesto, variar. También debe entenderse que la terminología utilizada en el presente documento tiene el propósito de describir realizaciones particulares únicamente, y no pretende ser limitativa, ya que el alcance de la presente solicitud estará limitado únicamente por las reivindicaciones adjuntas.
Se aprecia que ciertas características de la solicitud, que se describen, para mayor claridad, en el contexto de realizaciones separadas, también pueden proporcionarse en combinación en una sola realización. Por el contrario, diversas características de la solicitud, que se describen, por brevedad, en el contexto de una sola realización, también se pueden proporcionar por separado o en cualquier subcombinación adecuada. Todas las combinaciones de las realizaciones pertenecientes a los grupos químicos representados por las variables están específicamente abarcadas por la presente solicitud y se divulgan en el presente documento como si todas y cada una de las combinaciones se divulgaran individual y explícitamente, en la medida en que dichas combinaciones abarcan compuestos que son compuestos estables. (es decir, compuestos que se pueden aislar, caracterizar y probar por su actividad biológica). Además, todas las subcombinaciones de los grupos químicos enumerados en las realizaciones que describen tales variables también están específicamente abarcadas por la presente solicitud y se divulgan en el presente documento como si todas y cada una de tales subcombinaciones de grupos químicos se revelaran individual y explícitamente en el presente documento.
Cualquier fórmula representada en el presente documento pretende representar un compuesto de esa fórmula estructural, así como ciertas variaciones o formas. Por ejemplo, una fórmula proporcionada en el presente documento pretende incluir una forma racémica, o uno o más isómeros enantioméricos, diastereoméricos o geométricos, o formas tautoméricas, o una mezcla de las mismas. Además, cualquier fórmula proporcionada en el presente documento pretende referirse también a un solvato, tal como un hidrato, solvato o polimorfo de dicho compuesto, o una mezcla de los mismos. Cualquier fórmula proporcionada en el presente documento pretende referirse a formas físicas amorfas y/o cristalinas del compuesto. Los compuestos descritos en el presente documento pueden ser analíticamente puros o una mezcla en la que el compuesto comprende al menos el 50 %, al menos el 70 %, al menos el 80 %, al menos el 90 %, al menos el 95 % o al menos el 98 % en peso de la mezcla.
Además, cuando las características o aspectos de las realizaciones de esta solicitud se describen en términos de grupos Markush, los expertos en la materia reconocerán que las realizaciones descritas en el presente documento también se describen en términos de cualquier miembro individual o subgrupo de miembros del grupo Markush. Por ejemplo, si X se describe como seleccionado del grupo que consiste en bromo, cloro y yodo, las reivindicaciones de que X es bromo y las reivindicaciones de que X es bromo y cloro se describen completamente.
El término "en el presente documento" se refiere a la solicitud completa.
Como se usa en el presente documento, las formas singulares "un", "uno, una" y "el, la" incluyen referentes plurales a menos que el contexto dicte claramente lo contrario. Se observa además que las reivindicaciones pueden redactarse para excluir cualquier elemento opcional. Como tal, esta declaración pretende servir como base antecedente para el uso de terminología exclusiva como "solamente", "únicamente" y similares en relación con la mención de los elementos reivindicados, o el uso de una limitación "negativa".
Como se usa en el presente documento, los términos "que incluye", "que contiene" y "que comprende" se usan en su sentido abierto y no limitativo.
Como se usa en el presente documento, "sujeto" (tal como en el sujeto del tratamiento) se refiere tanto a mamíferos como a no mamíferos. Los mamíferos incluyen, por ejemplo, seres humanos; primates no humanos, por ejemplo, simios y monos; y no primates, por ejemplo ratones, ratas, conejos, perros, gatos, vacas, caballos, ovejas y cabras. Los no mamíferos incluyen, por ejemplo, gusanos, peces y pájaros. En algunas realizaciones, el sujeto es un ser humano.
"Sustancialmente", tal como se usa el término en el presente documento, se refiere a estar completamente o casi completamente; por ejemplo, una composición que está "sustancialmente libre" de un componente no tiene el componente o contiene una cantidad en trazas tal que cualquier propiedad funcional relevante de la composición no se ve afectada por la presencia de la cantidad en trazas, o un compuesto es "sustancialmente puro" si solo existen trazas insignificantes de impurezas presentes.
El término "acilo" se reconoce en la técnica y se refiere a un grupo representado por la fórmula general hidrocarbilC(O)-, preferiblemente alquilC(O)-.
El término "acilamino" se reconoce en la técnica y se refiere a un grupo amino sustituido con un grupo acilo y puede representarse, por ejemplo, mediante la fórmula hidrocarbilC(O)NH-.
El término "aciloxi" se reconoce en la técnica y se refiere a un grupo representado por la fórmula general hidrocarbilC(O)O-, preferiblemente alquilC(O)O-.
El término "alcoxi" se refiere a un grupo alquilo, preferiblemente un grupo alquilo inferior, que tiene un oxígeno unido al mismo. Los grupos alcoxi representativos incluyen metoxi, etoxi, propoxi, tert-butoxi y similares.
El término "alcoxialquilo" se refiere a un grupo alquilo sustituido con un grupo alcoxi y puede estar representado por la fórmula general alquil-O-alquilo.
El término "alquenilo", tal como se usa en el presente documento, se refiere a un grupo alifático que contiene al menos un doble enlace y pretende incluir tanto "alquenilos no sustituidos" como "alquenilos sustituidos", el último de los cuales se refiere a fracciones alquenilo que tienen sustituyentes que reemplazan un hidrógeno en uno o más carbonos del grupo alquenilo. Dichos sustituyentes pueden aparecer en uno o más carbonos que están incluidos o no en uno o más dobles enlaces. Además, dichos sustituyentes incluyen todos los contemplados para los grupos alquilo, tal como se analiza a continuación, excepto cuando la estabilidad es prohibitiva. Por ejemplo, se contempla la sustitución de grupos alquenilo por uno o más grupos alquilo, carbociclilo, arilo, heterociclilo o heteroarilo.
El término "alquinilo", tal como se usa en el presente documento, se refiere a un grupo alifático que contiene al menos un triple enlace y pretende incluir tanto "alquinilos no sustituidos" como "alquinilos sustituidos", el último de los cuales se refiere a fracciones alquinilo que tienen sustituyentes que reemplazan uno o más hidrógenos en uno o más carbonos del grupo alquinilo. Dichos sustituyentes pueden ocurrir en uno o más carbonos que están incluidos o no en uno o más enlaces triples. Además, tales sustituyentes incluyen todos los contemplados para los grupos alquilo, tal como se
discutió anteriormente, excepto en el que la estabilidad es prohibitiva. Por ejemplo, se contempla la sustitución de grupos alquinilo por uno o más grupos alquilo, carbociclilo, arilo, heterociclilo o heteroarilo.
Un grupo "alquilo" o "alcano" es un hidrocarburo no aromático de cadena lineal o ramificado que está completamente saturado. Típicamente, un grupo alquilo de cadena lineal o ramificada tiene de 1 a 20 átomos de carbono, tal como de 1 a 12 átomos de carbono, preferiblemente de 1 a aproximadamente 10, más preferiblemente de 1 a 4, a menos que se defina de otra manera. Los ejemplos de grupos alquilo de cadena lineal y ramificada incluyen metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, tert-butilo, pentilo, isopentilo, tert-pentilo, hexilo, isohexilo, pentilo y octilo. Un grupo alquilo de cadena lineal o ramificada C1-C6 también se denomina grupo "alquilo inferior".
Además, el término "alquilo" (o "alquilo inferior"), tal como se utiliza en la memoria descriptiva, los ejemplos y las reivindicaciones, pretende incluir tanto "alquilos no sustituidos" como "alquilos sustituidos", el último de los cuales se refiere a fracciones alquilo que tienen sustituyentes que reemplazan a un hidrógeno o más hidrógenos en uno o más carbonos de la cadena hidrocarbonada. Dichos sustituyentes, si no se especifica lo contrario, pueden incluir, por ejemplo, un halógeno, un hidroxilo, un carbonilo (tal como un carboxilo, un alcoxicarbonilo, un formilo o un acilo), un tiocarbonilo (tal como un tioéster, un tioacetato o un tioformiato), un alcoxilo, un fosforilo, un fosfato, un fosfonato, un fosfinato, un amino, un amido, una amidina, una imina, un ciano, un nitro, un azido, un sulfhidrilo, un alquiltio, un sulfato, un sulfonato, un sulfamoilo, un sulfonamido, un sulfonilo, un heterociclilo, un aralquilo o una fracción aromática o heteroaromática. Los expertos en la técnica entenderán que las fracciones sustituidas en la cadena hidrocarbonada pueden estar sustituidos ellos mismos, si es apropiado. Por ejemplo, los sustituyentes de un alquilo sustituido pueden incluir formas sustituidas y no sustituidas de grupos amino, azido, imino, amido, fosforilo (incluidos fosfonato y fosfinato), sulfonilo (incluidos sulfato, sulfonamido, sulfamoilo y sulfonato) y sililo, así como éteres, alquiltios, carbonilos (incluyendo cetonas, aldehídos, carboxilatos y ésteres), -CF3, -CN y similares. A continuación se describen ejemplos de alquilos sustituidos. Los cicloalquilos se pueden sustituir adicionalmente con alquilos, alquenilos, alcoxis, alquiltios, aminoalquilos, alquilos sustituidos con carbonilo, -CF3, -CN y similares.
El término "(ÁTOMO)i-j" con j > i, cuando se usa junto con una fracción química, tal como acilo, aciloxi, alquilo, alquenilo, alquinilo o alcoxi, se entiende que incluye grupos que contienen átomos de i a j (incluidos i y j). Por ejemplo, el término "alquilo Cx-y" se refiere a grupos hidrocarbonados saturados sustituidos o no sustituidos, incluidos grupos alquilo de cadena lineal y alquilo de cadena ramificada que contienen de xa y carbonos en la cadena, incluidos grupos haloalquilo tales como trifluorometilo y 2,2,2-trifluoroetilo, etc. Alquilo C0 se refiere a un átomo de hidrógeno en el que el grupo está en una posición terminal, un enlace si es interno. Del mismo modo, por ejemplo, cicloalquilo C3-6 se refiere a un cicloalquilo como se define en el presente documento que tiene de 3 a 6 átomos de carbono en el anillo. Los términos "alquenilo C2-y" y "alquinilo C2-y" se refieren a grupos alifáticos insaturados sustituidos o no sustituidos análogos en longitud y posible sustitución con los alquilos descritos anteriormente, pero que contienen al menos un doble o triple enlace respectivamente.
El término "alquilamino", tal como se usa en el presente documento, se refiere a un grupo amino sustituido con al menos un grupo alquilo.
El término "alquiltio", tal como se usa en el presente documento, se refiere a un grupo tiol sustituido con un grupo alquilo y puede representarse por la fórmula general alquilS-.
El término "hidrocarbilo", tal como se usa en el presente documento, se refiere a un grupo que está unido a través de un átomo de carbono que no tiene un sustituyente =O o =S, y normalmente tiene al menos un enlace carbonohidrógeno y una cadena principalmente de carbono, pero puede incluir opcionalmente heteroátomos. Por lo tanto, los grupos tales como metilo, etoxietilo, 2-piridilo y trifluorometilo se consideran hidrocarbilos para los fines de esta solicitud, pero sustituyentes tales como acetilo (que tiene un sustituyente =O en el carbono de enlace) y etoxi (que está unido a través de oxígeno, no de carbono) no lo son. Los grupos hidrocarbilo incluyen, pero no se limitan a, arilo, heteroarilo, carbociclo, heterociclilo, alquilo, alquenilo, alquinilo y combinaciones de los mismos.
Los términos "amina" y "amino" se reconocen en la técnica y se refieren tanto a aminas sustituidas como no sustituidas y sus sales. Por ejemplo, una fracción que puede ser representada por

en el que cada R30 representa independientemente un hidrógeno o un grupo hidrocarbilo, o dos R30 se toman junto con el átomo de N al que están unidos para completar un heterociclo que tiene de 4 a 8 átomos en la estructura del anillo.
El término "aminoalquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo amino.
El término "amida", tal como se usa en el presente documento, se refiere a un grupo:

en el que cada R30 representa independientemente un hidrógeno o un grupo hidrocarbilo, o dos R30 se toman junto con el átomo de N al que están unidos para completar un heterociclo que tiene de 4 a 8 átomos en la estructura del anillo.
El término "carbamato" está reconocido en la técnica y se refiere a un grupo

en el que R29 y R30 representan independientemente hidrógeno o un grupo hidrocarbilo, tal como un grupo alquilo, o R29 y R30 tomados junto con el átomo o a átomos que intervienen completan un heterociclo que tiene de 4 a 8 átomos en la estructura del anillo.
El término "halógeno" o "haluro" representa cloro, flúor, bromo o yodo. El término "halo" representa flúor, cloro, bromo o yodo.
El término "haloalquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo con uno o más sustituyentes halo, o uno, dos o tres sustituyentes halo. Los ejemplos de grupos haloalquilo incluyen -CF3, -CH2F, -CHF2, -CH2Br, -CH2CF3, y -CH2CH2F.
El término "heteroátomo", tal como se usa en el presente documento, se refiere a un átomo de cualquier elemento que no sea carbono o hidrógeno. Los ejemplos de heteroátomos incluyen, pero no se limitan a, nitrógeno, oxígeno y azufre.
El término "heteroalquilo", tal como se usa en el presente documento, se refiere a una cadena saturada o insaturada de átomos de carbono y al menos un heteroátomo, en la que no hay dos heteroátomos adyacentes.
El término "arilo", tal como se usa en el presente documento, incluye anillos aromáticos monocíclicos sustituidos o no sustituidos en los que cada átomo del anillo es carbono. Preferiblemente, el anillo es un anillo de 5 a 7 miembros, más preferiblemente un anillo de 6 miembros. El término "arilo" también incluye sistemas de anillos policíclicos que tienen dos o más anillos cíclicos en los que dos o más carbonos son comunes a dos anillos contiguos en los que al menos uno de los anillos es aromático, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos. Los grupos arilo incluyen benceno, naftaleno, fenantreno, fenol, anilina y similares.
El término "aralquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo arilo.
Un grupo "aroilo", tal como se usa el término en el presente documento, se refiere a un grupo arilo unido a través de un grupo carbonilo exocíclico, tal como un grupo benzoilo.
El término "heteroarilo", tal como se usa en el presente documento, incluye un sistema de anillo aromático monocíclico sustituido o no sustituido, preferiblemente anillos aromáticos de 5 a 7 miembros, más preferiblemente anillos de 5 a 6 miembros, cuyas estructuras de anillo incluyen al menos un heteroátomo, preferiblemente uno a cuatro heteroátomos, más preferiblemente de uno a dos heteroátomos. Por ejemplo, un heteroarilo de 5 miembros es furano, tiofeno, pirrol, oxazol, isoxazol, tiazol, isotiazol, pirazol, imidazol, oxadiazol, tiadiazol, triazol o tetrazol. En otro ejemplo, un heteroarilo de 6 miembros es piridina, pirazina, pirimidina, piridazina o triazina. El término "heteroarilo" también incluye sistemas de anillos "policíclicos" sustituidos o no sustituidos que tienen dos o más anillos cíclicos en los que dos o más carbonos son comunes a dos anillos contiguos en los que al menos uno de los anillos es heteroaromático, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos.
Los ejemplos ilustrativos de grupos heteroarilo incluyen, pero no se limitan a, las siguientes entidades, en forma de fracciones correctamente unidas:

El término "heteroaralquilo" o "hetaralquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo heteroarilo.
Un grupo "heteroaroilo", tal como se usa el término en el presente documento, se refiere a un grupo heteroarilo unido a través de un grupo carbonilo exocíclico, análogo a un grupo benzoilo pero en el que el anillo fenilo del grupo benzoilo se reemplaza por un grupo heteroarilo.
Los términos "heterociclilo", "heterociclo" y "heterocíclico", tal como se usan en el presente documento, se refieren a estructuras de anillos no aromáticos sustituidos o no sustituidos, preferiblemente anillos de 3 a 10 miembros, más preferiblemente anillos de 3 a 7 miembros, cuyas estructuras de anillo incluyen al menos un heteroátomo, preferiblemente de uno a cuatro heteroátomos, más preferiblemente uno o dos heteroátomos. Los términos "heterociclilo" y "heterocíclico" también incluyen sistemas de anillos policíclicos sustituidos o no sustituidos que tienen dos o más anillos cíclicos en los que dos o más carbonos son comunes a dos anillos contiguos en los que al menos uno de los anillos es heterocíclico, por ejemplo, los otros anillos cíclicos pueden ser cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos. Los grupos heterociclilo incluyen, por ejemplo, piperidina, piperazina, pirrolidina, morfolina, lactonas, lactamas y similares, tales como por ejemplo, piperidina, piperazina, pirrolidina, morfolina, lactonas, lactamas, azetidinas y similares.
El término "heterociclilalquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo heterociclo que está opcionalmente sustituido.
Los términos "carbociclo" y "carbocíclico", tal como se usan en el presente documento, se refieren a un anillo saturado o insaturado en el que cada átomo del anillo es carbono. El término carbociclo incluye tanto carbociclos aromáticos como carbociclos no aromáticos. Los carbociclos no aromáticos incluyen tanto anillos de cicloalcano, en los que todos los átomos de carbono están saturados, como anillos de cicloalqueno, que contienen al menos un doble enlace. Carbociclo incluye anillos monocíclicos de 5-7 miembros y bicíclicos de 8-12 miembros. Cada anillo de un carbociclo bicíclico puede seleccionarse de anillos saturados, insaturados y aromáticos. El carbociclo incluye moléculas bicíclicas en las que uno, dos o tres o más átomos se comparten entre los dos anillos. El término "carbociclo fusionado" se refiere a un carbociclo bicíclico en el que cada uno de los anillos comparte dos átomos adyacentes con el otro anillo. Cada anillo de un carbociclo fusionado puede seleccionarse de anillos aromáticos, insaturados y saturados. En un ejemplo de realización, un anillo aromático, por ejemplo, fenilo, puede fusionarse con un anillo saturado o insaturado, por ejemplo, ciclohexano, ciclopentano o ciclohexeno. Cualquier combinación de anillos bicíclicos saturados, insaturados y aromáticos, según lo permita la valencia, se incluye en la definición de carbocíclico. Los ejemplos de "carbociclos" incluyen ciclopentano, ciclohexano, biciclo[2.2.1]heptano, 1,5-ciclooctadieno, 1,2,3,4-tetrahidronaftaleno, biciclo[4.2.0]oct-3-eno, naftaleno y adamantano. Los ejemplos de carbociclos fusionados incluyen decalina, naftaleno, 1,2,3,4-tetrahidronaftaleno, biciclo[4.2.0]octano, 4,5,6,7-tetrahidro-1H-indeno y biciclo[4.1.0]hept-3-eno. Los "carbociclos" pueden estar sustituidos en una cualquiera o más posiciones capaces de portar un átomo de hidrógeno.
Un grupo "cicloalquilo", tal como se usa en el presente documento, se refiere a un hidrocarburo cíclico sustituido o no sustituido que está completamente saturado. “Cicloalquilo” incluye anillos monocíclicos y bicíclicos sustituidos o no sustituidos. Típicamente, un grupo cicloalquilo monocíclico tiene de 3 a aproximadamente 10 átomos de carbono, más típicamente de 3 a 8 átomos de carbono a menos que se defina de otra manera. Dicho grupo cicloalquilo monocíclico puede estar sustituido o no sustituido. El segundo anillo de un cicloalquilo bicíclico se puede seleccionar de anillos saturados, insaturados y aromáticos que están sustituidos o sin sustituir. Cicloalquilo incluye moléculas bicíclicas sustituidas o no sustituidas en las que uno, dos o tres o más átomos se comparten entre los dos anillos. El término "cicloalquilo fusionado" se refiere a un cicloalquilo bicíclico sustituido o no sustituido en el que cada uno de los anillos comparte dos átomos adyacentes con el otro anillo. El segundo anillo de un cicloalquilo bicíclico condensado se puede seleccionar de anillos saturados, insaturados y aromáticos.
El término "carbociclilalquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo carbociclo.
Un grupo "cicloalquenilo", tal como se usa en el presente documento, se refiere a un hidrocarburo cíclico que contiene uno o más dobles enlaces. Un grupo "cicloalquinilo" es un hidrocarburo cíclico que contiene uno o más enlaces triples.
Los términos "policiclilo", "policiclo" y "policíclico", tal como se usan en el presente documento, se refieren a dos o más anillos (por ejemplo, cicloalquilos, cicloalquenilos, cicloalquinilos, arilos, heteroarilos y/o heterociclilos) en los que dos o más átomos son comunes a dos anillos adyacentes, por ejemplo, los anillos son "anillos fusionados". Cada uno de
los anillos del policiclo puede estar sustituido o no sustituido. En ciertas realizaciones, cada anillo del policiclo contiene de 3 a 10 átomos en el anillo, preferiblemente de 5 a 7.
El término "carbonato" está reconocido en la técnica y se refiere a un grupo -OCO2-R30, en el que R30 representa un grupo hidrocarbilo.
El término "carboxi", como se usa en el presente documento, se refiere a un grupo representado por la fórmula -CO2H
El término "éster", como se usa en el presente documento, se refiere a un grupo -C(O)OR30 en el que R30 representa un grupo hidrocarbilo.
El término "éter", como se usa en el presente documento, se refiere a un grupo hidrocarbilo unido a través de un oxígeno a otro grupo hidrocarbilo. Por consiguiente, un sustituyente éter de un grupo hidrocarbilo puede ser hidrocarbil-O-. Los éteres pueden ser simétricos o asimétricos. Los ejemplos de éteres incluyen, pero no se limitan a, heterociclo-O-heterociclo y aril-O-heterociclo. Los éteres incluyen grupos "alcoxialquilo", que pueden estar representados por la fórmula general alquil-O-alquilo.
El término "sulfato" está reconocido en la técnica y se refiere al grupo -OSO3H, o una sal farmacéuticamente aceptable del mismo.
El término "sulfonamida" se reconoce en la técnica y se refiere al grupo representado por las fórmulas generales

en las que R29 y R30 representan independientemente hidrógeno o hidrocarbilo, tal como alquilo, o R29 y R30 tomados junto con el átomo o átomos intervinientes completan un heterociclo que tiene de 4 a 8 átomos en la estructura del anillo.
El término "sulfóxido" se reconoce en la técnica y se refiere al grupo -S(O)-R30, en el que R30 representa un hidrocarbilo.
El término "sulfonato" se reconoce en la técnica y se refiere al grupo SO3H, o una sal farmacéuticamente aceptable del mismo.
El término "sulfona" está reconocido en la técnica y se refiere al grupo -S(O)2-R30, en el que R30 representa un hidrocarbilo.
El término "tioalquilo", tal como se usa en el presente documento, se refiere a un grupo alquilo sustituido con un grupo tiol.
El término "tioéster", tal como se usa en el presente documento, se refiere a un grupo -C(O)SR30 o -SC(O)R30 en el que R30 representa un hidrocarbilo.
El término "tioéter", tal como se usa en el presente documento, es equivalente a un éter, en el que el oxígeno se reemplaza con azufre.
El término "urea" está reconocido en la técnica y puede representarse mediante la fórmula general

en la que R29 y R30 representan independientemente hidrógeno o un hidrocarbilo, tal como alquilo, o cualquier ocurrencia de R29 tomada junto con R30 y el átomo o átomos intervinientes completan un heterociclo que tiene de 4 a 8 átomos en la estructura del anillo.
El término "sustituido", como se usa en el presente documento, se refiere a fracciones que tienen sustituyentes que reemplazan uno o más hidrógenos en uno o más carbonos de la cadena. Se entenderá que "sustitución" o "sustituido con" incluye la condición implícita de que tal sustitución está de acuerdo con la valencia permitida del átomo sustituido y el sustituyente, y que la sustitución da como resultado un compuesto estable, por ejemplo, que no experimenta transformación espontánea tal como por reordenamiento, ciclación, eliminación, etc. Como se usa en el presente documento, se contempla que el término "sustituido" incluya todos los sustituyentes permisibles de compuestos orgánicos. En un aspecto amplio, los sustituyentes permisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Los sustituyentes permitidos pueden ser uno o más e iguales o diferentes para compuestos orgánicos apropiados. Para los fines de esta solicitud, los heteroátomos como el nitrógeno pueden tener sustituyentes de hidrógeno y/o cualquier
sustituyente permisible de los compuestos orgánicos descritos en el presente documento que satisfagan las valencias de los heteroátomos. En algunas realizaciones, "sustituido" significa que el grupo o fracción especificado tiene uno, dos o tres sustituyentes. En otras realizaciones, "sustituido" significa que el grupo o fracción especificado tiene uno o dos sustituyentes. En aún otras realizaciones, "sustituido" se refiere al grupo o fracción especificado que tiene un sustituyente.
Los sustituyentes pueden incluir cualquier sustituyente descrito en el presente documento, por ejemplo, un alquilo inferior (tal como alquilo C1-6, por ejemplo, -metilo, -etilo y -propilo), un halógeno, un hidroxilo, un carbonilo (tal como un carboxilo, un alcoxicarbonilo, un formilo o un acilo), un tiocarbonilo (tal como un tioéster, un tioacetato, o un tioformiato), un alcoxilo, un fosforilo, un fosfato, un fosfonato, un fosfinato, un amino, un amido, una amidina, una imina, un ciano, un nitro, un azido, un sulfhidrilo, un alquiltio, un sulfato, un sulfonato, un sulfamoilo, un sulfonamido, un sulfonilo, un heterociclilo, un aralquilo o una fracción aromática o heteroaromática. Los expertos en la técnica entenderán que los propios sustituyentes pueden sustituirse, si es apropiado.
Eliminado
El término "opcionalmente sustituido", tal como se usa en el presente documento, significa que la sustitución es opcional y, por lo tanto, es posible que el átomo o fracción designada no esté sustituida.
Cualquier disustituyente al que se hace referencia en el presente documento pretende abarcar las diversas posibilidades de unión cuando se permite más de una de tales posibilidades. Por ejemplo, la referencia al disustituyente -A-B-, en el que A t B, se refiere en el presente documento a dicho disustituyente con A unido a un primer miembro sustituido y B unido a un segundo miembro sustituido, y también se refiere a tal disustituyente con A unido al segundo miembro sustituido y B unido al primer miembro sustituido.
"Grupo protector", tal como se usa en el presente documento, se refiere a un grupo de átomos que, cuando se unen a un grupo funcional reactivo en una molécula, enmascaran, reducen o previenen la reactividad del grupo funcional. Normalmente, un grupo protector puede eliminarse selectivamente según se desee durante el curso de una síntesis. Se pueden encontrar ejemplos de grupos protectores en Greene y Wuts, Protective Groups in Organic Chemistry, 3.a edición, 1999, John Wiley & Sons, NY y Harrison et al., Compendium of Synthetic Organic Methods, vols. 1-8, 1971 1996, John Wiley & Sons, Nueva York. Los grupos protectores de nitrógeno representativos incluyen, pero no se limitan a, formilo, acetilo, trifluoroacetilo, bencilo, benciloxicarbonilo ("CBZ"), tert-butoxicarbonilo ("Boc"), trimetilsililo ("TMS"), 2-trimetilsilil-etanosulfonilo ("TES"), tritilo y grupos tritilo sustituidos, aliloxicarbonilo, 9-fluorenilmetiloxicarbonilo ("FMOC"), nitro-veratriloxicarbonilo ("NVOC") y similares. Los grupos protectores de hidroxilo representativos incluyen, pero no se limitan a, aquellos en los que el grupo hidroxilo está acilado (esterificado) o alquilado, tal como éteres de bencilo y tritilo, así como éteres de alquilo, éteres de tetrahidropiranilo, éteres de trialquilsililo (por ejemplo, grupos TMS o TIPS) éteres de glicol, tales como derivados de etilenglicol y propilenglicol y éteres de alilo.
El término "farmacéuticamente aceptable" se emplea en el presente documento para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del buen juicio médico, adecuados para usar en contacto con los tejidos de seres humanos y animales sin excesiva toxicidad, irritación, respuesta alérgica u otro problema o complicación, acorde con una proporción razonable de beneficio/riesgo.
Una "sal farmacéuticamente aceptable" pretende significar una sal de un ácido libre o una base de un compuesto representado en el presente documento que no es tóxico, es biológicamente tolerable o bien biológicamente adecuado para su administración al sujeto. Véase, en general, S.M. Berge, et al., "Pharmaceutical Salts", J. Pharm. Sci., 1977, 66, 1-19. Las sales farmacéuticamente aceptables preferidas son aquellas que son farmacológicamente eficaces y adecuadas para el contacto con los tejidos de los sujetos sin una toxicidad, irritación o respuesta alérgica indebidas. Un compuesto descrito en el presente documento puede poseer un grupo suficientemente ácido, un grupo suficientemente básico, ambos tipos de grupos funcionales o más de uno de cada tipo y, en consecuencia, reaccionar con varias bases inorgánicas u orgánicas y ácidos inorgánicos y orgánicos para formar una sal farmacéuticamente aceptable.
Para un compuesto descrito en el presente documento que contiene un grupo básico, tal como una amina, se puede preparar una sal farmacéuticamente aceptable mediante cualquier método adecuado disponible en la técnica, por ejemplo, tratamiento de la base libre con un ácido inorgánico, tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido sulfámico, ácido nítrico, ácido bórico, ácido fosfórico y similares, o con un ácido orgánico, tal como ácido acético, ácido fenilacético, ácido propiónico, ácido esteárico, ácido láctico, ácido ascórbico, ácido maleico, ácido hidroximaleico, ácido isetiónico, ácido succínico, ácido valérico, ácido fumárico, ácido malónico, ácido pirúvico, ácido oxálico, ácido glicólico, ácido salicílico, ácido oleico, ácido palmítico, ácido láurico, un ácido piranosidilo, tal como ácido glucurónico o ácido galacturónico, un ácido alfa-hidroxi, tal como ácido mandélico, ácido cítrico o ácido tartárico, un aminoácido, tal como ácido aspártico o ácido glutámico, un ácido aromático, tal como ácido benzoico, ácido 2-acetoxibenzoico, ácido naftoico o ácido cinámico, un ácido sulfónico, tal como ácido lauril sulfónico, ácido ptoluenosulfónico, ácido metanosulfónico o ácido etanosulfónico, o cualquier mezcla compatible de ácidos como los que se dan como ejemplos en el presente documento, y cualquier otro ácido y mezcla de los mismos que se consideren equivalentes o sustitutos aceptables a la luz del nivel ordinario de conocimiento en esta tecnología.
Para un compuesto descrito en el presente documento que contiene un grupo ácido, tal como un grupo de ácido carboxílico, las sales de adición de base se pueden preparar mediante cualquier método adecuado disponible en la técnica, por ejemplo, el tratamiento de dicho compuesto con una cantidad suficiente de la base deseada, ya sea puro o en un disolvente inerte adecuado. Los ejemplos de sales de adición de bases farmacéuticamente aceptables incluyen, pero no se limitan a, sal de litio, sodio, potasio, calcio, amonio, zinc o magnesio, u otras sales metálicas; sales orgánicas de amino, tales como sales de alquil, dialquil, trialquil o tetraalquilamonio.
Otros ejemplos de sales farmacéuticamente aceptables incluyen, pero no se limitan a, camsilato, sulfatos, pirosulfatos, bisulfatos, sulfitos, bisulfitos, fosfatos, monohidrogenofosfatos, dihidrogenofosfatos, metafosfatos, pirofosfatos, cloruros, bromuros, yoduros, acetatos, propionatos, decanoatos, caprilatos, acrilatos, formiatos, isobutiratos, caproatos, heptanoatos, propiolatos, oxalatos, malonatos, succinatos, suberatos, sebacatos, fumaratos, maleatos, butino-1,4-dioatos, hexino-1,6-dioatos, benzoatos, clorobenzoatos, metilbenzoatos, dinitrobenzoatos, hidroxibenzoatos, metoxibenzoatos, ftalatos, sulfonatos, metilsulfonatos, propilsulfonatos, besilatos, xilenosulfonatos, naftaleno-1-sulfonatos, naftaleno-2-sulfonatos, fenilacetatos, fenilpropionatos, fenilbutiratos, citratos, lactatos, yhidroxibutiratos, glicolatos, tartratos y mandelatos. Las listas de otras sales farmacéuticamente aceptables adecuadas se encuentran en Remington's Pharmaceutical Sciences, 17a edición, Mack Publishing Company, Easton, Pensilvania, 1985.
Las formas neutras de los compuestos se regeneran preferiblemente poniendo en contacto la sal con una base o un ácido y aislando el compuesto original de la manera convencional. La forma original del compuesto difiere de las diversas formas de sal en ciertas propiedades físicas, tal como la solubilidad en disolventes polares, pero por lo demás las sales son equivalentes a la forma original del compuesto para los fines de la presente solicitud.
El término "profármaco" pretende abarcar compuestos que, en condiciones fisiológicas, se convierten en los agentes terapéuticamente activos de la presente solicitud, por ejemplo, un compuesto descrito en el presente documento. Un método común para preparar un profármaco es incluir una o más fracciones seleccionadas que se hidrolizan en condiciones fisiológicas para producir la molécula deseada. En determinadas realizaciones, el profármaco se convierte mediante una actividad enzimática del animal huésped. Por ejemplo, la reductasa podría reducir un profármaco con un grupo nitro en un anillo aromático para generar el grupo amino deseado del compuesto activo correspondiente in vivo. En otro ejemplo, los grupos funcionales tales como un hidroxilo, carbonato o ácido carboxílico en el compuesto original se presentan como un éster, que podría escindirse mediante esterasas. Además, los grupos amino en los compuestos originales se presentan en, pero no se limitan a carbamato, formas N-alquiladas o N-aciladas (Simplicio et al., "Prodrugs for Amines", Molecules, (2008), 13:519-547). En ciertas realizaciones, algunos o todos los compuestos descritos en el presente documento en una formulación representada anteriormente pueden reemplazarse con el profármaco adecuado correspondiente.
Un "profármaco farmacéuticamente aceptable" es un profármaco que no es tóxico, es biológicamente tolerable y, por lo demás, biológicamente adecuado para su administración al sujeto. Los procedimientos ilustrativos para la selección y preparación de derivados de profármacos adecuados se describen, por ejemplo, en "Design of Prodrugs", H. Bundgaard, Elsevier, 1985.
Un "metabolito farmacéuticamente activo" o "metabolito" se refiere a un producto farmacológicamente activo del metabolismo/modificación bioquímica de un compuesto descrito en el presente documento, por ejemplo, un compuesto de Fórmula (I), (I-A), (I-B), o una sal del mismo, bajo condiciones fisiológicas, por ejemplo, a través de cierta ruta enzimática. Por ejemplo, un metabolito oxidativo se forma por oxidación del compuesto original durante el metabolismo, tal como la oxidación de un anillo de piridina a N-óxido de piridina. En otro ejemplo, se forma un metabolito oxidativo por desmetilación de un grupo metoxi para obtener como resultado un grupo hidroxilo.
Los profármacos y los metabolitos activos de un compuesto pueden determinarse utilizando técnicas rutinarias conocidas o disponibles en la técnica. Véase por ejemplo, Bertolini et al., J. Med. Chem. 1997, 40, 2011-2016; Shan et al., J. Pharm. Sci. 1997, 86 (7), 765-767; Bagshawe, Drug Dev. Res. 1995, 34, 220-230; Bodor, Adv. Drug Res.
1984, 13, 255-331; Bundgaard, Design of Prodrugs (Elsevier Press, 1985); y Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991).
Los compuestos de la presente solicitud, tales como los compuestos de fórmulas (I), (I-A) y (I-B), también pueden existir como varios "solvatos" o "hidratos". Un "hidrato" es un compuesto que existe en una composición con moléculas de agua. La composición puede incluir agua en cantidades estequiométricas, tal como un monohidrato o un dihidrato, o puede incluir agua en cantidades aleatorias. Un "solvato" es una composición similar excepto que un disolvente distinto del agua, tal como con metanol, etanol, dimetilformamida, éter dietílico y similares reemplaza el agua. Por ejemplo, el metanol o el etanol pueden formar un "alcoholato", que nuevamente puede ser estequiométrico o no estequiométrico. También se pueden preparar mezclas de tales solvatos o hidratos. La fuente de tal solvato o hidrato puede ser del disolvente de cristalización, inherente al disolvente de preparación o cristalización, o adventicio a tal disolvente.
Los compuestos de la solicitud, incluyendo sus sales y profármacos farmacéuticamente aceptables, pueden existir como diversos polimorfos, pseudopolimorfos o en estado amorfo. El término "polimorfo", tal como se usa en el presente documento, se refiere a diferentes formas cristalinas del mismo compuesto y otras formas moleculares en estado
sólido que incluyen pseudopolimorfos, tales como hidratos, solvatos o sales del mismo compuesto. Diferentes polimorfos cristalinos tienen diferentes estructuras cristalinas debido a un diferente empaquetamiento de moléculas en la red, tal como resultado de cambios de temperatura, presión o variaciones en el proceso de cristalización. Los polimorfos difieren entre sí en sus propiedades físicas, tal como las características de difracción de rayos X, la estabilidad, los puntos de fusión, la solubilidad o las velocidades de disolución en ciertos disolventes. Por lo tanto, las formas polimórficas cristalinas son aspectos importantes en el desarrollo de formas de dosificación adecuadas en la industria farmacéutica.
La presente solicitud abarca además compuestos aislados de acuerdo con la fórmula (I), (I-A) o (I-B). El término "compuesto aislado" se refiere a una preparación de un compuesto de fórmula (I), (I-A) o (I-B), o una mezcla de compuestos de acuerdo con la fórmula (I), (I-A) o (I-B), en la que el compuesto aislado ha sido separado de los reactivos usados, y/o los subproductos formados, en la síntesis del compuesto o compuestos. "Aislado" no significa que la preparación sea técnicamente pura (homogénea), si no que es lo suficientemente pura para formar compuestos en una forma en la que se puede usar terapéuticamente. Preferiblemente, un "compuesto aislado" se refiere a una preparación de un compuesto de fórmula (I), (I-A) o (I-B) o una mezcla de compuestos de acuerdo con la fórmula (I), (I-A) o (I-B), que contiene el mencionado compuesto o mezcla de compuestos de acuerdo con la fórmula (I), (I-A) o (I B) en una cantidad de al menos el 10 por ciento en peso del peso total. Preferiblemente, la preparación contiene el compuesto mencionado o la mezcla de compuestos en una cantidad de al menos el 50% en peso del peso total; más preferiblemente al menos el 80% en peso del peso total; y lo más preferiblemente al menos el 90%, al menos el 95% o al menos el 98% en peso del peso total de la preparación.
Los compuestos de la solicitud y los intermedios pueden aislarse de sus mezclas de reacción y purificarse mediante técnicas estándar tales como filtración, extracción líquido-líquido, extracción en fase sólida, destilación, recristalización o cromatografía, incluida la cromatografía en columna ultrarrápida o HPLC.
Isomerismo y tautomerismo en compuestos descritos
Tautomerismo
Dentro de la presente solicitud, debe entenderse que un compuesto descrito en el presente documento o una sal del mismo puede exhibir el fenómeno de tautomerismo por el cual dos compuestos químicos que son capaces de interconvertirse fácilmente al intercambiar un átomo de hidrógeno entre dos átomos, cualquiera de los cuales forma un enlace covalente. Dado que los compuestos tautoméricos existen en equilibrio móvil entre sí, pueden considerarse formas isoméricas diferentes del mismo compuesto. Debe entenderse que los dibujos de fórmulas dentro de esta memoria descriptiva pueden representar solo una de las posibles formas tautoméricas. Sin embargo, también debe entenderse que la solicitud abarca cualquier forma tautomérica y no debe limitarse simplemente a cualquier forma tautomérica utilizada en los dibujos de fórmulas. Los dibujos de fórmulas dentro de esta memoria descriptiva pueden representar solo una de las formas tautoméricas posibles y debe entenderse que la memoria descriptiva abarca todas las formas tautoméricas posibles de los compuestos dibujados, no solo aquellas formas que ha sido conveniente mostrar gráficamente en el presente documento. Por ejemplo, el tautomerismo puede ser exhibido por un grupo pirazolilo unido como lo indica la línea ondulada. Si bien ambos sustituyentes se denominarían grupo 4-pirazolilo, es evidente que un átomo de nitrógeno diferente tiene el átomo de hidrógeno en cada estructura.

Tal tautomerismo también puede ocurrir con pirazoles sustituidos tales como 3-metilo, 5-metilo o 3,5-dimetilpirazoles y similares. Otro ejemplo de tautomerismo es el tautomerismo amido-imido (lactama-lactima cuando es cíclico), tal como se ve en los compuestos heterocíclicos que tienen un átomo de oxígeno en el anillo adyacente a un átomo de nitrógeno en el anillo. Por ejemplo, el equilibrio:

es un ejemplo de tautomerismo. En consecuencia, se pretende que una estructura representada en el presente documento como un tautómero también incluya el otro tautómero.
Isomería óptica
Se entenderá que cuando los compuestos de la presente solicitud contienen uno o más centros quirales, los compuestos pueden existir y pueden aislarse como formas enantioméricas o diastereoisómeras puras o como mezclas racémicas. Por lo tanto, la presente solicitud incluye todos los posibles enantiómeros, diastereómeros, racematos en sus formas puras o mezclas de los mismos, y sales de los mismos, de los compuestos de la solicitud.
Los isómeros resultantes de la presencia de un centro quiral comprenden un par de isómeros no superponibles que se denominan "enantiómeros". Los enantiómeros individuales de un compuesto puro son ópticamente activos, es decir,
son capaces de rotar el plano de la luz polarizada plana. Los enantiómeros individuales se designan de acuerdo con el sistema de Cahn-Ingold-Prelog. La prioridad de los sustituyentes se clasifica en función de los pesos atómicos, un peso atómico más alto, determinado por el procedimiento sistemático, tiene una clasificación de prioridad más alta. Una vez que se determina la clasificación de prioridad de los cuatro grupos, la molécula se orienta de modo que el grupo de clasificación más baja apunte lejos del espectador. Entonces, si el orden de rango descendente de los otros grupos procede en el sentido de las agujas del reloj, la molécula se designa (R) y si el rango descendente de los otros grupos procede en sentido contrario a las agujas del reloj, la molécula se designa (S). En el ejemplo del Esquema 14, en la clasificación de Cahn-Ingold-Prelog es A > B > C > D. El átomo de clasificación más bajo, D, está orientado lejos del observador.

Configuración (R) Configuración (S)
En ciertas realizaciones, la preparación terapéutica puede enriquecerse para proporcionar predominantemente un enantiómero de un compuesto (por ejemplo, de fórmula (I), (I-A) o (I-B)). Una mezcla enantioméricamente enriquecida puede comprender, por ejemplo, al menos 60 por ciento en moles de un enantiómero, o más preferiblemente al menos 75, 90, 95 o incluso 99 por ciento en moles. En ciertas realizaciones, un compuesto de la solicitud puede tener más del 30 % de ee, 40 % de ee, 50 % de ee, 60 % de ee, 70 % de ee, 80 % de ee, 90 % de ee, o incluso 95 % o más de ee. En ciertas realizaciones, el compuesto enriquecido en un enantiómero está sustancialmente libre del otro enantiómero, en el que sustancialmente libre significa que la sustancia en cuestión constituye menos del 10 %, o menos del 5 %, o menos del 4 %, o menos del 3 %, o menos del 2%, o menos del 1% en comparación con la cantidad del otro enantiómero, por ejemplo, en la composición o mezcla de compuestos. Por ejemplo, si una composición o mezcla de compuestos contiene 98 gramos de un primer enantiómero y 2 gramos de un segundo enantiómero, se diría que contiene 98 por ciento en moles del primer enantiómero y solo 2% del segundo enantiómero.
En ciertas realizaciones, los compuestos de la solicitud pueden tener más de un estereocentro. En ciertas realizaciones de este tipo, los compuestos de la solicitud pueden estar enriquecidos en uno o más diastereómeros. Por ejemplo, un compuesto de la solicitud puede tener más del 30 % de de, 40 % de de, 50 % de de, 60 % de de, 70 % de de, 80 % de de, 90 % de de, o incluso 95 % o más de de.
Los isómeros ópticos aislados pueden purificarse a partir de mezclas racémicas mediante técnicas de separación quiral bien conocidas, tales como, pero no se limitan a, cromatografía de fase normal e inversa y cristalización. De acuerdo con uno de tales métodos, una mezcla racémica de un compuesto de la solicitud, o un intermedio quiral del mismo, se separa usando una sal quiral o se lleva a cabo en una columna ChiralCel OD. La columna se opera de acuerdo con las instrucciones del fabricante.
Los isómeros ópticos aislados (compuestos enantioméricamente puros) también se pueden preparar mediante el uso de compuestos intermedios quirales o catalizadores en la síntesis. Cuando se usa un compuesto intermedio quiral sintético, el centro óptico (centro quiral) puede conservarse sin racemización durante el resto del procedimiento preparativo, tal como es bien conocido en la técnica. El catalizador quiral se puede usar para impartir al menos algún grado de pureza enantiomérica a los productos de reacciones catalizadas por el catalizador quiral. Y, en algunos casos, los compuestos que tienen al menos algún grado de enriquecimiento enantiomérico se pueden obtener mediante procesos físicos tales como la cristalización selectiva de sales o complejos formados con adyuvantes quirales.
Puede existir una variedad de compuestos en la presente solicitud en formas geométricas o estereoisómeras particulares. La presente solicitud tiene en cuenta todos estos compuestos, incluidos los tautómeros, los isómeros cis y trans, los enantiómeros R y S, los diastereómeros, los isómeros (D), los isómeros (L), las mezclas racémicas de los mismos y otras mezclas de los mismos, como los cubiertos dentro del alcance de esta solicitud. Todas las formas tautoméricas están abarcadas en la presente solicitud. Pueden estar presentes átomos de carbono asimétricos adicionales en un sustituyente tal como un grupo alquilo. Todos estos isómeros, así como sus mezclas, están destinados a ser incluidos en esta solicitud, a menos que se indique específicamente la estereoquímica o la forma isomérica.
Isomería rotacional
Se entiende que debido a las propiedades químicas (es decir, resonancia que otorga cierto carácter de doble enlace al enlace CN) de rotación restringida al rededor el enlace de la unión amida (tal como se ilustra a continuación), es posible observar especies de rotámeros separadas e incluso, en algunas circunstancias, aislar tales especies (ver a continuación). Se entiende además que ciertos elementos estructurales, incluido el volumen estérico o los sustituyentes en el nitrógeno de la amida, pueden mejorar la estabilidad de un rotámero hasta el punto de que un compuesto puede aislarse y existir indefinidamente como un único rotámero estable. Por lo tanto, la presente solicitud
incluye cualquier posible rotámero estable de fórmula (I) que sea biológicamente activo en el tratamiento del cáncer u otros estados patológicos proliferativos.

Regioisomerismo
Los compuestos preferidos de la presente solicitud tienen una disposición espacial particular de los sustituyentes en los anillos aromáticos, que están relacionados con la relación estructura-actividad demostrada por la clase de compuestos. A menudo, dicha disposición de la sustitución se denota mediante un sistema de numeración; sin embargo, los sistemas de numeración a menudo no son consistentes entre diferentes sistemas de anillos. En los sistemas aromáticos de seis miembros, las disposiciones espaciales se especifican mediante la nomenclatura común
"para" para sustitución 1,4, "meta" para sustitución 1,3 y "orto" para sustitución 1,2, tal como se muestra a continuación.

Marcación isotópica en compuestos descritos
La presente solicitud incluye además todos los compuestos marcados con isótopos farmacéuticamente aceptables
[por ejemplo, de fórmula (I), (I-A) o (I-B)]. Un compuesto "isotópicamente" o "radiomarcado" es un compuesto en el que uno o más átomos se reemplazan o sustituyen por un átomo que tiene una masa atómica o un número de masa diferente de la masa atómica o el número de masa que normalmente se encuentra en la naturaleza (es decir, de origen natural). Por ejemplo, en ciertas realizaciones, en compuestos [por ejemplo, de fórmula (I), (I-A) o (I-B)], los átomos de hidrógeno se reemplazan o sustituyen por uno o más átomos de deuterio o tritio (por ejemplo, átomos de hidrógeno en un alquilo C1-6 o un alcoxi C1-6 se reemplazan con deuterio, tal como d3-metoxi o 1,1,2,2-d4-3-metilbutilo).
Ciertos compuestos marcados isotópicamente [por ejemplo, compuestos de fórmula (I), (I-A) o (I-B)], por ejemplo, los que incorporan un isótopo radiactivo, son útiles en estudios de distribución de fármacos y/o sustratos en tejidos. Los isótopos radiactivos tritio, es decir, 3H, y carbono 14, es decir, 14C, son particularmente útiles para este propósito en vista de su facilidad de incorporación y rápidos medios de detección.
Dichos compuestos marcados isotópicamente son útiles en estudios metabólicos (preferiblemente con 14C), estudios de cinética de reacción (con, por ejemplo, 2H o 3H), técnicas de detección o formación de imágenes [como la tomografía por emisión de positrones (PET) o la tomografía computarizada por emisión de fotón único (Sp Ec T)], incluidos los ensayos de distribución de fármacos o sustratos en tejidos, o en el tratamiento radiactivo de pacientes. Además, la sustitución con isótopos más pesados como el deuterio (es decir, 2H) puede proporcionar ciertas ventajas terapéuticas derivadas de una mayor estabilidad metabólica, por ejemplo, mayor vida media in vivo o requisitos de dosificación reducida.
Sustitución con isótopos emisores de positrones, tal como 11C, 18F, 15O, y 13N, puede ser útil en los estudios de tomografía por emisión de positrones (PET) para examinar la ocupación del receptor del sustrato.
Los compuestos marcados con isótopos [por ejemplo, de fórmula (I), (I-A) o (I-B)] o sus profármacos correspondientes generalmente se pueden preparar mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procesos análogos a los descritos en los ejemplos adjuntos usando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado empleado anteriormente. Los isótopos adecuados que pueden incorporarse en los compuestos de la presente solicitud incluyen, pero no se limitan a, isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor, cloro y yodo, tales como 2H (también escrito como D para deuterio), 3H (también escrito como T para tritio), 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 75Br, 31P y 32P.
Los compuestos marcados isotópicamente de esta solicitud y sus profármacos generalmente se pueden preparar llevando a cabo los procedimientos divulgados en los esquemas o en los ejemplos y preparaciones descritos a continuación sustituyendo un reactivo marcado isotópicamente fácilmente disponible por un reactivo no marcado isotópicamente.
En diversas realizaciones, el compuesto o conjunto de compuestos, tal como los que se usan en los métodos de la invención, puede ser uno cualquiera de cualquiera de las combinaciones y/o subcombinaciones de las realizaciones enumeradas anteriormente.
Composiciones farmacéuticas
Las composiciones y métodos de la presente solicitud se pueden utilizar para tratar a un sujeto, tal como un mamífero, por ejemplo, un humano o un mamífero no humano, que lo necesite. Cuando se administra a un animal, tal como un ser humano, la composición o el compuesto se administra preferiblemente como una composición farmacéutica que comprende, por ejemplo, un compuesto de la solicitud y un vehículo farmacéuticamente aceptable. En ciertas realizaciones, la solicitud se refiere a una composición farmacéutica que comprende como ingrediente activo una cantidad terapéuticamente eficaz de un compuesto descrito en el presente documento, o una de sus sales, solvatos o profármacos farmacéuticamente aceptables, en asociación con al menos un vehículo, excipiente o diluyente farmacéuticamente aceptable.
El término "vehículo farmacéuticamente aceptable", tal como se usa en el presente documento, se refiere a un material, composición o vehículo farmacéuticamente aceptable, tal como un relleno líquido o sólido, diluyente, excipiente, disolvente o material de encapsulación, que puede actuar, por ejemplo, para estabilizar, aumentar solubilidad o para aumentar la absorción de un compuesto tal como un compuesto de la solicitud. Cada vehículo debe ser "aceptable" en el sentido de ser compatible con los demás ingredientes de la formulación y no perjudicial para el paciente.
Los vehículos farmacéuticamente aceptables son bien conocidos en la técnica. Por ejemplo, algunos ejemplos de materiales que pueden servir como vehículos farmacéuticamente aceptables incluyen, pero no se limitan a: (1) azúcares, tales como lactosa, glucosa, sacarosa o dextrano; (2) almidones, tales como almidón de maíz y almidón de patata; (3) celulosa y sus derivados, tales como carboximetilcelulosa de sodio, etilcelulosa y acetato de celulosa; (4) tragacanto en polvo; (5) malta; (6) gelatina; (7) talco; (8) excipientes, tal como manteca de cacao y ceras para supositorios; (9) aceites, tales como aceite de cacahuete, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; (10) glicoles, tales como glicerol o propilenglicol; (11) polioles, tales como glicerina, sorbitol, manitol y polietilenglicol; (12) ésteres, tales como oleato de etilo y laurato de etilo; (13) agar; (14) agentes tamponantes, tales como hidróxido de magnesio e hidróxido de aluminio; (15) ácido algínico; (16) agua libre de pirógenos; (17) solución salina isotónica; (18) solución de Ringer; (19) alcohol etílico; (20) soluciones tampón de fosfato; (21) antioxidantes, tales como ácido ascórbico o glutatión; y (22) otras sustancias compatibles no tóxicas empleadas en formulaciones farmacéuticas, tales como agentes quelantes, proteínas de bajo peso molecular u otros estabilizantes o excipientes.
La elección de un vehículo farmacéuticamente aceptable, incluido un agente fisiológicamente aceptable, depende, por ejemplo, de la vía de administración de la composición. La composición farmacéutica puede ser un sistema de administración de fármacos autoemulsionante o automicroemulsionante. La composición farmacéutica también puede ser un liposoma u otra matriz polimérica, que se puede haber incorporado en ella. Los liposomas, por ejemplo, que comprenden fosfolípidos u otros lípidos, son vehículos no tóxicos, fisiológicamente aceptables y metabolizables que son relativamente simples de preparar y administrar.
Agentes humectantes, emulsionantes y lubricantes, tales como lauril sulfato de sodio y estearato de magnesio, así como agentes colorantes, agentes de liberación, agentes de recubrimiento, agentes edulcorantes, aromatizantes y perfumantes, conservantes y antioxidantes también pueden estar presentes en las composiciones. Dichas composiciones líquidas pueden contener opcionalmente: excipientes farmacéuticamente aceptables tales como agentes de suspensión (por ejemplo, sorbitol, metilcelulosa, alginato de sodio, gelatina, hidroxietilcelulosa, carboximetilcelulosa, gel de estearato de aluminio y similares); vehículos no acuosos, por ejemplo, aceite (por ejemplo, aceite de almendras o aceite de coco fraccionado), propilenglicol, alcohol etílico o agua; conservantes (por ejemplo, p-hidroxibenzoato de metilo o propilo o ácido sórbico); agentes humectantes tales como lecitina; y, si se desea, agentes saborizantes o colorantes.
Los ejemplos de antioxidantes farmacéuticamente aceptables incluyen: (1) antioxidantes solubles en agua, tales como ácido ascórbico, clorhidrato de cisteína, bisulfato de sodio, metabisulfito de sodio, sulfito de sodio y similares; (2) antioxidantes solubles en aceite, tales como palmitato de ascorbilo, hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), lecitina, galato de propilo, alfa-tocoferol y similares; y (3) agentes quelantes de metales, tales como ácido cítrico, ácido etilendiaminotetraacético (EDTA), sorbitol, ácido tartárico, ácido fosfórico y similares.
Una composición farmacéutica se puede administrar a un sujeto por cualquiera de varias vías de administración que incluyen, pero no se limitan a, por ejemplo, por vía oral [por ejemplo, pociones como en soluciones o suspensiones acuosas o no acuosas, comprimidos, píldoras, cápsulas (incluyendo cápsulas para espolvorear y cápsulas de gelatina), bolos, polvos, gránulos, pastas para aplicar en la lengua]; absorción a través de la mucosa oral (por ejemplo, sublingualmente); por vía anal, rectal o vaginal (por ejemplo, tal como un pesario, crema o espuma); por vía parenteral (incluyendo por vía intramuscular, intravenosa, subcutánea o intratecal como, por ejemplo, una solución o suspensión estéril); por vía nasal; intraperitoneal; subcutáneamente; transdérmica (por ejemplo, tal como un parche aplicado a la piel); y tópicamente (por ejemplo, tal como crema, ungüento o spray aplicado a la piel, o como gotas para los ojos). La composición o compuesto también puede formularse para inhalación. En ciertas realizaciones, la composición o el compuesto pueden disolverse o suspenderse simplemente en agua estéril. Los detalles de las vías de administración apropiadas y las composiciones adecuadas para las mismas se pueden encontrar en, por ejemplo, las patentes de los Estados Unidos Nos. 6,110,973; 5,763,493; 5,731,000; 5,541,231; 5,427,798; 5,358,970 y 4,172,896, así como en las patentes allí citadas. Las composiciones estériles también están contempladas por la solicitud, incluidas las
composiciones que están de acuerdo con las reglamentaciones nacionales y locales que rigen tales composiciones. Preferiblemente, las composiciones se formulan para administración intravenosa u oral.
Para la administración oral, los compuestos de la solicitud se pueden proporcionar en forma sólida, tal como comprimidos, píldoras, grageas, polvos, gránulos o cápsulas, o como una solución, emulsión o suspensión. Para preparar las composiciones orales, el ingrediente activo se mezcla con uno o más vehículos farmacéuticamente aceptables, tales como citrato de sodio o fosfato dicálcico, y/o cualquiera de los siguientes: (1) rellenos o extensores, tales como almidones, lactosa, sacarosa, glucosa, manitol y/o ácido silícico; (2) aglutinantes, tales como, por ejemplo, carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidona, sacarosa y/o goma arábiga; (3) humectantes, tales como glicerol; (4) agentes desintegrantes, tales como agar-agar, carbonato de calcio, almidón de patata o tapioca, ácido algínico, ciertos silicatos y carbonato de sodio; (5) agentes retardantes de la solución, tales como parafina; (6) aceleradores de absorción, tales como compuestos de amonio cuaternario; (7) agentes humectantes, tales como, por ejemplo, alcohol cetílico y monoestearato de glicerol; (8) absorbentes, tales como caolín y arcilla de bentonita; (9) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, laurilsulfato de sodio y mezclas de los mismos; (10) agentes complejantes, tales como ciclodextrinas modificadas y no modificadas; (11) agentes colorantes; (12) agentes emulsionantes y de suspensión, tales como alcoholes isoestearílicos etoxilados, polioxietilén sorbitol, ésteres de sorbitán, celulosa microcristalina, metahidróxido de aluminio, bentonita, agar y tragacanto; y (13) otras sustancias compatibles no tóxicas empleadas en formulaciones farmacéuticas, tales como, sin limitación, agentes tamponantes, agentes perfumantes y conservantes, agentes edulcorantes, agentes saborizantes.
Los comprimidos orales pueden fabricarse por compresión o moldeo, opcionalmente con uno o más ingredientes accesorios, tales como diluyentes, agentes desintegrantes, agentes aglutinantes, agentes lubricantes, agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes. Los rellenos inertes adecuadas incluyen carbonato de sodio y calcio, fosfato de sodio y calcio, lactosa, almidón, azúcar, glucosa, metilcelulosa, estearato de magnesio, manitol, sorbitol y similares. Los ejemplos de excipientes orales líquidos incluyen etanol, glicerol, agua y similares. El almidón, la polivinilpirrolidona (PVP), el glicolato sódico de almidón, la celulosa microcristalina y el ácido algínico son ejemplos de agentes desintegrantes. Los agentes aglutinantes pueden incluir hidroxipropilmetilcelulosa, almidón y gelatina. El agente lubricante, si está presente, puede ser estearato de magnesio, ácido esteárico o talco. Si se desea, los comprimidos se pueden recubrir con un material tal como monoestearato de glicerilo o diestearato de glicerilo para retrasar la absorción en el tracto gastrointestinal, o se pueden recubrir con un recubrimiento entérico.
Otras formas de dosificación sólidas de las composiciones farmacéuticas, tal como grageas, cápsulas (incluidas las cápsulas para espolvorear y las cápsulas de gelatina), píldoras y gránulos, se pueden marcar o preparar opcionalmente con recubrimientos y cubiertas, tal como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de formulación farmacéutica. Por ejemplo, para preparar cápsulas de gelatina dura, el ingrediente o ingredientes activos pueden mezclarse con un diluyente sólido, semisólido o líquido. Las cápsulas de gelatina blanda se pueden preparar mezclando el ingrediente activo con agua, aceite tal como el aceite de cacahuete o aceite de oliva, parafina líquida, una mezcla de mono y diglicéridos de ácidos grasos de cadena corta, polietilenglicol 400 o propilenglicol.
Las composiciones farmacéuticas también pueden formularse para proporcionar una liberación lenta o controlada del ingrediente activo utilizando, por ejemplo, hidroxipropilmetilcelulosa en proporciones variables para proporcionar el perfil de liberación deseado, otras matrices poliméricas, liposomas y/o microesferas. Pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro que retiene bacterias, o incorporando agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse en agua estéril, o algún otro medio inyectable estéril inmediatamente antes de su uso. Estas composiciones también pueden contener opcionalmente agentes opacificantes y pueden ser de una composición que liberen el o los ingredientes activos solo, o preferentemente, en una cierta porción del tracto gastrointestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que se pueden usar incluyen sustancias poliméricas y ceras. El principio activo también puede estar en forma microencapsulada, si procede, con uno o varios de los excipientes anteriormente descritos.
Las formas de dosificación líquidas útiles para la administración oral incluyen emulsiones farmacéuticamente aceptables, liofilizados para reconstitución, microemulsiones, soluciones, suspensiones, jarabes y elixires, o pueden liofilizarse o presentarse como un producto seco para reconstitución con agua u otro vehículo adecuado antes de su uso. Además del ingrediente activo, las formas de dosificación líquidas pueden contener diluyentes inertes comúnmente utilizados en la técnica, como, por ejemplo, agua u otros disolventes, ciclodextrinas y derivados de las mismas, agentes solubilizantes y emulsionantes, tales como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, aceites (en particular, aceites de semilla de algodón, cacahuete, maíz, germen, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurílico, polietilenglicoles y ésteres de ácidos grasos de sorbitán y mezclas de los mismos.
Además, las formulaciones de las composiciones farmacéuticas para administración en la boca pueden presentarse como un enjuague bucal, un aerosol oral o un ungüento oral.
Las frases "administración parenteral" y "administrado por vía parenteral", tal como se usan en el presente documento, significan modos de administración distintos de la administración enteral y tópica, generalmente por inyección, e incluyen, sin limitación inyección e infusión intravenosa, intramuscular, intraarterial, intratecal, intracapsular, intraorbital, intracardiaca, intradérmica intranasal, intraperitoneal, transtraqueal, subcutánea, subcuticular, intraarticular, subcapsular, subaracnoidea, intraespinal e intraesternal.
Para uso parenteral, los agentes de la aplicación pueden proporcionarse en soluciones o suspensiones acuosas estériles, tamponadas a un pH e isotonicidad apropiados o en un aceite aceptable por vía parenteral. Los vehículos acuosos adecuados incluyen solución de Ringer y cloruro de sodio isotónico. Dichas formas pueden presentarse en forma de dosis unitaria, tal como ampollas o dispositivos de inyección desechables, en formas de dosis múltiples, tal como viales de los que se puede extraer la dosis adecuada, o en forma sólida o concentrada previamente que puede reconstituirse en una formulación inyectable estéril, tal como soluciones o dispersiones justo antes de su uso, que pueden contener antioxidantes, tampones, bacteriostáticos, solutos que hacen que la formulación sea isotónica con la sangre del receptor previsto o agentes de suspensión o espesantes. Las dosis de infusión ilustrativas oscilan entre aproximadamente 1 y 1000 pg/kg/minuto de agente mezclado con un vehículo farmacéutico durante un período que oscila entre varios minutos y varios días.
Los ejemplos de vehículos acuosos y no acuosos adecuados que pueden emplearse en las composiciones farmacéuticas de la solicitud incluyen agua, etanol, polioles (tales como glicerol, propilenglicol, polietilenglicol y similares) y mezclas adecuadas de los mismos, aceites vegetales, tales como aceite de oliva y ésteres orgánicos inyectables, tales como el oleato de etilo. La fluidez adecuada se puede mantener, por ejemplo, mediante el uso de materiales de recubrimiento, tales como la lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de las dispersiones y mediante el uso de tensioactivos.
Estas composiciones también pueden contener adyuvantes tales como conservantes, humectantes, emulsionantes y dispersantes. La prevención de la acción de los microorganismos se puede asegurar mediante la inclusión de varios agentes antibacterianos y antifúngicos, por ejemplo, parabeno, clorobutanol, ácido fenol sórbico y similares. También puede ser deseable incluir agentes isotónicos, tales como azúcares, cloruro de sodio y similares en las composiciones.
En algunos casos, para prolongar el efecto de un fármaco, es deseable retardar la absorción del fármaco por inyección subcutánea o intramuscular. Esto se puede lograr mediante el uso de una suspensión líquida de material cristalino o amorfo que tiene poca solubilidad en agua. La velocidad de absorción del fármaco depende entonces de su velocidad de disolución, que, a su vez, puede depender del tamaño del cristal y la forma cristalina. Alternativamente, la absorción retardada de una forma de fármaco administrada por vía parenteral se logra disolviendo o suspendiendo el fármaco en un vehículo oleoso.
Las formas de depósito inyectables se fabrican formando matrices microencapsuladas de los compuestos en cuestión en polímeros biodegradables tales como poliláctido-poliglicólido. Dependiendo de la proporción de fármaco a polímero y de la naturaleza del polímero particular empleado, se puede controlar la velocidad de liberación del fármaco. Los ejemplos de otros polímeros biodegradables incluyen poli(ortoésteres) y poli(anhídridos). Las formulaciones inyectables de depósito también se preparan atrapando el fármaco en liposomas o microemulsiones que son compatibles con el tejido corporal.
En una realización preferida, cuando dichas composiciones farmacéuticas son para administración humana, particularmente para vías de administración invasivas (es decir, vías, tal como inyección o implantación, que eluden el transporte o la difusión a través de una barrera epitelial), la solución acuosa está libre de pirógenos o sustancialmente libre de pirógenos. Los excipientes se pueden elegir, por ejemplo, para efectuar la liberación retardada de un agente o para dirigirse selectivamente a una o más células, tejidos u órganos.
Para la administración rectal, vaginal o uretral, las formulaciones de las composiciones farmacéuticas pueden presentarse como un supositorio, que puede prepararse mezclando uno o más compuestos activos con uno o más excipientes o vehículos no irritantes adecuados que comprenden, por ejemplo, manteca de cacao, polietilenglicol , una cera de supositorio o un salicilato, y que es sólido a temperatura ambiente, pero líquido a temperatura corporal y, por lo tanto, se derretirá en el recto o en la cavidad vaginal y liberará el compuesto activo. Las formulaciones que son adecuadas para la administración vaginal también incluyen pesarios, tampones, cremas, geles, pastas, espumas o formulaciones en aerosol que contienen los vehículos que se sabe que son apropiados en la técnica.
Para aplicaciones tópicas o administración transdérmica, los compuestos activos de la presente solicitud se pueden mezclar en condiciones estériles con un vehículo farmacéuticamente aceptable y con cualquier conservante, tampón, excipiente o propelente, tales como grasas animales y vegetales, aceites, ceras, parafinas, almidón, tragacanto, derivados de celulosa, polietilenglicoles, siliconas, bentonitas, ácido silícico, talco y óxido de zinc, o mezclas de los mismos. Las formas de dosificación para administración tópica incluyen polvos, aerosoles, ungüentos, pastas, cremas, lociones, geles, soluciones, parches e inhalantes. Los compuestos activos pueden mezclarse con un vehículo farmacéutico a una concentración de aproximadamente 0.1% a aproximadamente 10% de fármaco por vehículo.
Los polvos y aerosoles pueden contener, además de un principio activo, excipientes tales como lactosa, talco, ácido silícico, hidróxido de aluminio, silicatos de calcio y poliamida en polvo, o mezclas de estas sustancias. Los aerosoles
pueden contener además propelentes habituales, tales como clorofluorohidrocarburos e hidrocarburos volátiles no sustituidos, tales como butano y propano.
Los parches transdérmicos tienen la ventaja adicional de proporcionar un suministro controlado de un compuesto de la presente solicitud al cuerpo. Dichas formas de dosificación pueden prepararse disolviendo o dispersando el compuesto activo en el medio adecuado. También se pueden usar potenciadores de la absorción para aumentar el flujo del compuesto a través de la piel. La velocidad de dicho flujo se puede controlar proporcionando una membrana de control de la velocidad o dispersando el compuesto en una matriz polimérica o gel.
Las formulaciones oftálmicas, los ungüentos para los ojos, los polvos, las soluciones y similares también se contemplan dentro del alcance de esta solicitud. Los ejemplos de formulaciones oftálmicas se describen en las publicaciones de patente de los Estados Unidos Nos. 2005/0080056, 2005/0059744, 2005/0031697 y 2005/004074 y la patente de los Estados Unidos No. 6,583,124. Si se desea, las formulaciones oftálmicas líquidas tienen propiedades similares a las de los fluidos lagrimales, el humor acuoso o el humor vítreo o son compatibles con tales fluidos. Una vía de administración preferida es la administración local (por ejemplo, administración tópica, tal como gotas para los ojos o administración a través de un implante).
Alternativa o adicionalmente, las composiciones se pueden formular para la administración a través de un catéter, cánula intraluminal, cable u otro dispositivo intraluminal. El suministro a través de dichos dispositivos puede ser especialmente útil para el suministro a la vejiga, la uretra, el uréter, el recto o el intestino.
Los métodos de introducción también pueden proporcionarse mediante dispositivos recargables o biodegradables. Se han desarrollado y probado varios dispositivos poliméricos de liberación lenta in vivo en los últimos años para la administración controlada de fármacos, incluidos los compuestos biofarmacéuticos proteináceos. Se puede usar una variedad de polímeros biocompatibles (incluidos los hidrogeles), incluidos los polímeros biodegradables y no degradables, para formar un implante para la liberación sostenida de un compuesto en un sitio objetivo particular.
Un médico o veterinario que tenga una experiencia normal en la técnica puede determinar y prescribir fácilmente la cantidad terapéuticamente eficaz de la composición farmacéutica requerida. El término "cantidad terapéuticamente eficaz" o "dosis", o "dosificación", como se usa en el presente documento, se refiere a una cantidad o dosis suficiente para producir generalmente el beneficio terapéutico deseado o una cantidad suficiente para modular la actividad biológica del receptor objetivo en sujetos que necesitan tal tratamiento.
Las cantidades o dosis efectivas de los compuestos de la solicitud pueden determinarse mediante métodos de rutina, tales como modelado, escalada de dosis o ensayos clínicos, teniendo en cuenta factores de rutina. Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas pueden variar. En general, una dosis diaria adecuada de un compuesto activo utilizado en las composiciones y métodos de la solicitud será la cantidad del compuesto que sea la dosis más baja eficaz para producir un efecto terapéutico.
El nivel de dosificación seleccionado dependerá de una variedad de factores que incluyen la actividad del compuesto particular o combinación de compuestos empleados, o las sales, solvatos y profármacos del mismo, la vía de administración, el tiempo de administración, la velocidad de excreción del compuesto o compuestos particulares empleados, la duración del tratamiento, otras fármacos, compuestos y/o materiales usados en combinación con un compuesto o compuestos particulares empleados, edad, sexo, peso, condición, salud general, historial médico previo del paciente siendo tratado, y la preferencia y experiencia del médico o veterinario a cargo, y factores tales bien conocidos en las artes médicas.
Por ejemplo, al elegir un régimen para un sujeto, tal como un paciente, frecuentemente puede ser necesario comenzar con una dosis más alta y cuando la condición está bajo control reducir la dosis. En otro ejemplo, también es posible comenzar con una dosificación de la composición farmacéutica del compuesto a niveles inferiores a los necesarios para lograr el efecto terapéutico deseado y aumentar gradualmente la dosificación hasta lograr el efecto deseado.
Los compuestos de la solicitud son efectivos en un amplio rango de dosificación. Por ejemplo, en el tratamiento de humanos adultos, se pueden usar dosis de aproximadamente 0.05 a aproximadamente 5000 mg, preferiblemente de aproximadamente 1 a aproximadamente 2000 mg, y más preferiblemente entre aproximadamente 2 y aproximadamente 2000 mg por día. Una dosificación típica es de aproximadamente 10 mg a aproximadamente 1000 mg por día, o de 25 a 200 mg por día, o de 50 a 100 mg por día, o menos de 100 mg por día.
En algunas realizaciones, los compuestos de la solicitud se administran en forma de dosificación unitaria que incluye desde aproximadamente 0.05 mg hasta aproximadamente 1000 mg de ingrediente activo junto con un vehículo farmacéuticamente aceptable por unidad de dosificación. En otras realizaciones, una forma de dosificación unitaria incluye de aproximadamente 10 a aproximadamente 200 mg de ingrediente activo. En otras realizaciones, las formas de dosificación adecuadas para la administración oral, nasal, pulmonar o transdérmica incluyen desde aproximadamente 125 |jg hasta aproximadamente 1250 mg, preferiblemente desde aproximadamente 250 |jg hasta aproximadamente 500 mg, y más preferiblemente desde aproximadamente 2.5 mg hasta aproximadamente 250 mg, de los compuestos mezclados con un vehículo o diluyente farmacéuticamente aceptable. Los métodos para determinar la eficacia y la dosificación son conocidos por los expertos en la técnica (Isselbacher et al.. (1996) Principles of Internal Medicine Harrison, decimotercera ed., 1814-1882).
Las formas de dosificación se pueden administrar diariamente o más de una vez al día, tal como dos o tres veces al día. Alternativamente, las formas de dosificación se pueden administrar con menos frecuencia que diariamente, tal como cada dos días, o semanalmente, si el médico que las prescribe lo considera aconsejable. Se puede administrar una dosis mayor mediante múltiples administraciones del agente. En algunas realizaciones, las formas de dosificación se administran una, dos o tres veces al día. En realizaciones preferidas, el compuesto activo se administrará una vez al día. Una vez que se ha producido una mejoría de la enfermedad del paciente, se puede ajustar la dosis para el tratamiento de mantenimiento. Por ejemplo, la dosificación o la frecuencia de administración, o ambas, pueden reducirse en función de los síntomas, hasta un nivel en el que se mantenga el efecto terapéutico o profiláctico deseado. Por supuesto, si los síntomas se han aliviado a un nivel apropiado, el tratamiento puede suspenderse. Sin embargo, los pacientes pueden requerir un tratamiento intermitente a largo plazo ante cualquier recurrencia de los síntomas. Los pacientes también pueden requerir un tratamiento crónico a largo plazo.
Métodos y usos
En diversas realizaciones, los compuestos de la solicitud se pueden usar para modular, tal como activar (agonista) o bloquear la activación de (antagonista), un receptor PAR2. Por consiguiente, en varias realizaciones, la solicitud proporciona un método para modular un receptor PAR2 que comprende poner en contacto el receptor con una cantidad o concentración eficaz de un compuesto de la solicitud. En diversas realizaciones, el compuesto de la solicitud es un antagonista de un receptor PAR2. En varias realizaciones, el contacto puede tener lugar in vivo dentro de los tejidos de un paciente, tal como un paciente humano. En diversas realizaciones, la modulación de un receptor PAR2, por ejemplo, el antagonismo de PAR2, por un compuesto de la solicitud puede usarse para tratar una enfermedad o trastorno en un paciente, tal como se describe en el presente documento.
En varias realizaciones, la solicitud proporciona un método para tratar una enfermedad o trastorno en un paciente en el que la modulación de un receptor pAR2 es una indicación médica, que comprende administrar al sujeto, tal como un paciente, un compuesto de la solicitud en una dosis, a una frecuencia y duración para proporcionar un efecto beneficioso para el sujeto. La modulación, tal como agonismo o antagonismo, de un receptor PAR2 puede estar médicamente indicada en el tratamiento de una enfermedad o trastorno en el que el receptor PAR2 desempeña un papel metabólico o regulador. Ciertas condiciones de este tipo pueden tratarse mediante la modulación selectiva de un receptor PAR2, mientras que los otros receptores activados por proteasa, tales como PAR1, PAR3 y PAR4, no se ven afectados por la administración del compuesto de la solicitud en la dosis proporcionada. En diversas realizaciones, los compuestos de la solicitud pueden ser antagonistas de PAR2, y algunos de ellos son antagonistas selectivos de PAR2 con respecto a otros receptores activados por proteasa tales como PAR1, PAR3 y PAR4. Por "selectivo" se entiende que un receptor es modulado a concentraciones del compuesto al menos 10 veces más bajas que las concentraciones a las que el receptor comparativo es modulado por ese compuesto. En realizaciones adicionales, el compuesto de la solicitud puede modular adicionalmente otros tipos o clases de receptores activados por proteasa tales como PAR1, PAR3 y PAR4.
En varias realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de una enfermedad o trastorno en un paciente. Por ejemplo, un compuesto de la solicitud se puede usar en la preparación de un medicamento para administrar a un paciente que padece una enfermedad o trastorno. Más específicamente, la enfermedad o trastorno puede comprender dolor, inflamación musculoesquelética, trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis, colitis y afecciones relacionadas.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de un dolor, que incluye, pero no se limitan a, dolor agudo, dolor crónico, dolor inflamatorio y neuropático. El dolor puede ser crónico, alodinia (la percepción del dolor de un estímulo normalmente inocuo), hiperalgesia (una respuesta exagerada a cualquier estímulo de dolor dado) y una expansión del campo receptivo (es decir, el área que es "dolorosa" cuando se aplica un estímulo), dolor fantasma o dolor inflamatorio. Los tipos de dolor agudo comprenden, pero no se limitan a, dolor asociado con daño tisular, dolor posoperatorio, dolor después de un traumatismo, dolor causado por quemaduras, dolor causado por infección local o sistémica, dolor visceral asociado con enfermedades que comprenden: pancreatitis, cistitis intestinal, dismenorrea, síndrome del intestino irritable, enfermedad de Crohn, cólico ureteral e infarto de miocardio. Además, el término "dolor" comprende dolor asociado con trastornos del SNC que comprenden: esclerosis múltiple, lesión de la médula espinal, lesión cerebral traumática, enfermedad de Parkinson y accidente cerebrovascular. En algunas realizaciones, "dolor" se refiere a tipos de dolor crónico que comprenden dolor de cabeza (por ejemplo, trastornos de migraña, dolor de cabeza de tipo episódico y tensión crónica, dolor de cabeza de tipo tensión, dolor de cabeza en racimo y hemicránea paroxística crónica), dolor lumbar, dolor por cáncer, dolor de osteoartritis y dolor neuropático, pero no se limita a los mismos. El dolor inflamatorio (dolor en respuesta a una lesión tisular y el proceso inflamatorio resultante) como se define en el presente documento se refiere al dolor inflamatorio asociado con enfermedades que comprenden enfermedades del tejido conectivo, artritis reumatoide, lupus eritematoso sistémico, esclerosis múltiple y artritis, pero no se limita a ellas. El dolor neuropático (dolor resultante del daño a los nervios periféricos o al propio sistema nervioso central) incluye afecciones que comprenden, pero no se limitan a, neuropatías metabólicas (por ejemplo, neuropatía diabética), neuralgia postherpética, neuralgia del trigémino, neuralgia craneal, dolor neuropático después de accidente cerebro vascular, dolor neuropático asociado a la esclerosis múltiple, dolor neuropático asociado a VIH/SIDA, dolor neuropático asociado al cáncer, dolor neuropático asociado al túnel carpiano, dolor neuropático asociado a lesiones de la médula espinal, síndrome de dolor regional complejo, dolor neuropático asociado a la fibromialgia, distrofia simpática refleja, síndrome del miembro fantasma o
nervio periférico o traumatismo de la médula espinal, transección nerviosa incluida la cirugía, amputación de extremidades y dolor del muñón, dolor causado por los efectos secundarios de las terapias contra el cáncer y el SIDA, dolor neuropático posquirúrgico, dolor asociado a neuropatía tal como en neuropatía idiopática o postraumática, y dolor neuropático causado por enfermedad de tejido conectivo tal como artritis reumatoide, síndrome de Wallenberg, lupus eritematoso sistémico, esclerosis múltiple o poliarteritis nodosa. La neuropatía se puede clasificar como radiculopatía, mononeuropatía, mononeuropatía múltiple, polineuropatía o plexopatía.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de un trastorno inflamatorio musculoesquelético, que incluye, pero no se limitan a, dolor lumbar, fibromialgia, gota, osteoartritis, artritis reumatoide y tendinitis.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de un trastorno neuroinflamatorio, que incluye, pero no se limitan a, esclerosis múltiple (MS), que incluye, pero no se limitan a, por ejemplo, esclerosis múltiple con recaída y remisión (RRMS), esclerosis múltiple progresiva secundaria (SPMS) y esclerosis múltiple progresiva primaria (PPMS); enfermedad de Parkinson; atrofia multisistémica (MSA); degeneración corticobasal; paresia supranuclear progresiva; síndrome de Guillain-Barré (GBS); y polineuropatía desmielinizante inflamatoria crónica (CIDP).
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de un trastorno inflamatorio de las vías respiratorias, que incluye, pero no se limitan a, síndrome de Kartagener, asma (tal como asma difícil o asma persistente grave), disfunción de las cuerdas vocales (tal como cierre incontrolado de las cuerdas vocales al respirar), crup espasmódico, enfermedad vasomotora refleja y trastornos autonómicos.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento del picazón.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de dermatitis, que incluye, pero no se limitan a, dermatitis de contacto, dermatitis atópica, dermatitis seborreica, dermatitis numular, dermatitis exfoliativa, dermatitis crónica, dermatitis por estasis y dermatitis perioral.
En algunas realizaciones, la solicitud proporciona un uso de un compuesto de la solicitud para el tratamiento de la colitis, que incluye, pero no se limitan a, enfermedad inflamatoria intestinal (IBD), colitis (enfermedad de Crohn o colitis ulcerosa), colitis microscópica, colitis química, colitis isquémica, colitis infecciosa (intoxicación alimentaria causada por infecciones e infecciones causadas por parásitos o bacterias).
Se cree que el antagonismo de PAR2, en particular, está médicamente indicado para el tratamiento de las condiciones mencionadas anteriormente. Por antagonismo se entiende bloquear un receptor, en este caso un receptor PAR2, sin provocar que transduzca una señal. Es decir, el antagonismo da como resultado el bloqueo de un ligando endógeno o exógeno para que no active o provoque el antagonismo del receptor.
Está dentro de la experiencia habitual evaluar la eficacia de cualquier compuesto descrito y reivindicado en el presente documento en la modulación del receptor PAR2 y en los diversos ensayos celulares usando los procedimientos descritos anteriormente o encontrados en la literatura científica. Por consiguiente, el experto en la materia puede preparar y evaluar cualquiera de los compuestos reivindicados sin experimentación indebida.
Cualquier compuesto que se descubra que es un modulador, agonista o antagonista efectivo, también puede probarse en modelos animales y en estudios clínicos humanos usando la habilidad y experiencia del investigador para guiar la selección de dosis y regímenes de tratamiento.
En ciertas realizaciones, la solicitud comprende un método para llevar a cabo un negocio farmacéutico, mediante la determinación de una formulación y dosificación apropiadas de un compuesto de la solicitud para tratar o prevenir cualquiera de las enfermedades o afecciones que se describen en el presente documento, realizando perfiles terapéuticos de formulaciones identificadas para la eficacia y toxicidad en animales, y proporcionar una red de distribución para vender una preparación identificada como que tiene un perfil terapéutico aceptable. En ciertas realizaciones, el método incluye además proporcionar un grupo de ventas para comercializar la preparación a los proveedores de atención médica.
En ciertas realizaciones, la solicitud se refiere a un método para llevar a cabo un negocio farmacéutico mediante la determinación de una formulación y dosificación apropiadas de un compuesto de la solicitud para tratar o prevenir cualquiera de las enfermedades o afecciones descritas en el presente documento, y otorgar licencia a un tercero, los derechos para un mayor desarrollo y venta de la formulación.
El término "proveedores de atención médica" se refiere a individuos u organizaciones que brindan servicios de atención médica a una persona, comunidad, etc. Los ejemplos de "proveedores de atención médica" incluyen médicos, hospitales, comunidades de jubilados de atención continua, centros de enfermería especializada, centros de atención subaguda, clínicas, clínicas para múltiples especialidades, centros ambulatorios independientes, agencias de salud en el hogar y de HMO.
Combinaciones de fármacos
Los compuestos de la presente solicitud pueden usarse en composiciones o métodos farmacéuticos en combinación con uno o más ingredientes activos adicionales en el tratamiento de las enfermedades y trastornos descritos en el presente documento. Otros ingredientes activos adicionales incluyen otros compuestos farmacéuticos o agentes que mitigan los efectos adversos de las terapias para los objetivos de la enfermedad previstos. Tales combinaciones pueden servir para aumentar la eficacia, mejorar otros síntomas de enfermedad, disminuir uno o más efectos secundarios o disminuir la dosis requerida de un compuesto de la invención. En ciertas realizaciones, dicha combinación proporciona un efecto aditivo, en el que un efecto aditivo se refiere a la suma de cada uno de los efectos de la administración individual del compuesto de la solicitud y uno o más agentes terapéuticos adicionales. En otras realizaciones, dicha combinación proporciona un efecto sinérgico, en el que el efecto terapéutico supera la suma de cada uno de los efectos de la administración individual del compuesto de la solicitud y uno o más agentes terapéuticos adicionales.
Los ingredientes activos adicionales pueden administrarse en una composición farmacéutica separada de un compuesto de la presente solicitud o pueden incluirse con un compuesto de la presente solicitud en una única composición farmacéutica. Los ingredientes activos adicionales pueden administrarse simultáneamente, antes o después de la administración de un compuesto de la presente solicitud. Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas pueden variar para obtener una cantidad del ingrediente activo que sea eficaz para lograr la respuesta terapéutica deseada para un sujeto en particular, tal como un paciente, composición y modo de administración sin ser tóxico para el sujeto.
Los agentes de combinación incluyen ingredientes activos adicionales que se sabe o se descubre que son efectivos en el tratamiento de las enfermedades y trastornos descritos en el presente documento, incluidos los activos contra otro objetivo asociado con la enfermedad. Por ejemplo, las composiciones y formulaciones de la solicitud, así como los métodos de tratamiento, pueden comprender además otros fármacos o productos farmacéuticos, por ejemplo, otros agentes activos útiles para tratar o paliar las enfermedades objetivo o los síntomas o afecciones relacionados. Las composiciones farmacéuticas de cualquier compuesto descrito en el presente documento pueden comprender adicionalmente uno o más de dichos agentes activos, y los métodos de tratamiento pueden comprender adicionalmente la administración de una cantidad eficaz de uno o más de dichos agentes activos.
Ejemplos
Los siguientes ejemplos se ofrecen para ilustrar pero no para limitar la solicitud. Un experto en la técnica reconocerá que las siguientes reacciones y esquemas de síntesis pueden modificarse eligiendo materiales de partida y reactivos adecuados para acceder a otros compuestos de la solicitud, o una de sus sales farmacéuticamente aceptables.
Protocolos de síntesis
A continuación se describirán ejemplos de entidades químicas útiles en métodos de la solicitud con referencia a esquemas de síntesis ilustrativos para su preparación general y los ejemplos específicos que siguen. Los expertos reconocerán que, para obtener los diversos compuestos de este documento, los materiales de partida pueden seleccionarse adecuadamente de modo que las sustituciones deseadas finalmente se lleven a cabo a través del esquema de reacción con o sin protección, según sea apropiado, para producir el producto deseado. Alternativamente, puede ser necesario o deseable emplear, en lugar del sustituyente finalmente deseado, un grupo adecuado que pueda utilizarse en el esquema de reacción y reemplazarse según corresponda con el sustituyente deseado. Además, un experto en la técnica reconocerá que las transformaciones que se muestran en los siguientes esquemas se pueden realizar en cualquier orden que sea compatible con la funcionalidad de los grupos adheridos particulares. Cada una de las reacciones representadas en los esquemas generales se lleva a cabo preferiblemente a una temperatura de aproximadamente 0 °C a la temperatura de reflujo del disolvente orgánico utilizado. A menos que se especifique lo contrario, las variables son como se definieron anteriormente en referencia a la fórmula (I), (I-A) o (I-B). Los compuestos marcados isotópicamente como se describe en el presente documento se preparan de acuerdo con los métodos descritos a continuación, usando materiales de partida adecuadamente marcados. Dichos materiales generalmente están disponibles a través de proveedores comerciales de reactivos químicos radiomarcados.
Términos y abreviaturas:
ACN acetonitrilo;
Ac. acuoso;
Atm presión atmosférica;
Boc f-butoxicarbonilo;
Bórax tetraborato disódico o borato de sodio o tetraborato de sodio;
Cbz benciloxicarbonilo;
CDI 1,1-carbonildiimidazol;
dba dibencilidenacetona;
DCM diclorometano;
DEA dietilamina;
DIBAL-H hidruro de diisobutilaluminio;
DIPEA diisopropiletilamina;
EMD 1,2-dimetoxietano;
DMF N,N-dimetilformamida;
DMSO dimetilsulfóxido;
Et2O éter dietílico;
EtOAc acetato de etilo;
EtOH etanol;
eq. o equiv. equivalente;
h hora(s);
HATU hexafluorofosfato de 2-(7-aza-1H-benzotriazol-1-il)-1,1,3,3-tetrametiluronio;
HBTU hexafluorofosfato de 0-benzotriazol-N,N,N',N'-tetrametiluronio
HPLC cromatografía líquida de alto rendimiento;
LCMS cromatografía líquida con espectrometría de masas;
LDA diisopropilamida de litio;
LiHMDS bis(trimetilsilil)amida de litio;
MeOH metanol;
min minuto(s);
MS espectrometría de masas;
MW microonda(s);
NH4OAc acetato de amonio;
RMN resonancia magnética nuclear;
ox oxidación;
Psi libras por pulgada cuadrada;
cuant. cuantitativo;
RCM metátesis de cierre de anillo;
ta temperatura ambiente;
sat. saturado;
SFC cromatografía de fluidos supercríticos;
T3P anhídrido propilfosfónico;
TFA ácido trifluoroacético;
THF tetrahidrofurano;
TLC cromatografía de capa fina;
TMEDA tetrametiletilendiamina;
UPLC cromatografía líquida de ultra alto rendimiento.
Condiciones de LC-MS:
Método A: La LC-MS se llevo a cabo utilizando un LCMS-2020 de Shimadzu. Detección UV = 190-400 nm, espectrometría de masas = ESI. La columna utilizada fue Ascentis express C18, 2.1 mm * 50 mm, 2.7 pm a 40 °C. El caudal fue de 1 ml/min utilizando un gradiente de disolvente de 5 a 100 % de B durante 1.8 minutos, en el que A = agua (TFA al 0.05 %) y B = acetonitrilo (TFA al 0.05 %). Donde se indique, con designación base o modificada para eluyentes o condiciones básicas, las fases móviles se alcalinizaron y se reemplazaron con A= agua NH4HCO36.5 mM a pH 10 en A, y B = acetonitrilo (sin aditivo).
Método B: La UPLC-MS se llevó a cabo utilizando un UPLC Acquity de Waters y un espectrómetro de masas SQD de Waters. Detección UV = 210-400 nm, espectrómetro de masas = ESI con conmutación positiva/negativa y voltaje de cono = 10 V. La columna utilizada fue HSS T3 Acquity de Waters, 1.8 pm, 2.1 * 30 mm, a una temperatura de 30 °C. El caudal fue de 1 ml/min usando un gradiente de disolventes de 2 a 98 % de B durante 1.5 minutos (tiempo total del proceso con equilibrio de vuelta a las condiciones iniciales de 2 min), en el que A = ácido fórmico al 0.1 % en agua y B = ácido fórmico al 0.1 % en acetonitrilo.
Método C: Los cromatogramas analíticos de HPLC se realizaron utilizando un instrumento de la serie 1100 de Agilent. Los espectros de masas se registraron con un detector Micromass ZQ de Waters a 100 °C. El espectrómetro de masas estaba equipado con una fuente de iones de electroaspersión (ESI) operado en modo de iones positivos y configurado para escanear entre m/z 150-750 con un tiempo de escaneo de 0.3 s. Los productos y los compuestos intermedios se analizaron mediante HPLC/MS en un Gemini (5.0 mM, 2.10 * 30 mm) utilizando un gradiente de tampón de pH alto del 5 % al 100 % de ACN en H2O ((N ^^C O aa l 0.03 %/NH4OH al 0.375%) durante 3.0 min a razón de 1.8 ml/min durante un ciclo de 3.5 min y en un Kinetex EVO (5.0 mM, 2.10 * 50 mm) usando un gradiente de tampón de pH bajo del 5 % al 100 % de ACN en H2O (HCOOH al 0.05 %) durante 2.8 min a razón de 2.2 ml/min durante un ciclo de 3.5 min.
Método D: Instrumentos: Acquity clase H de Waters, arreglo de fotodiodos, detector de SQ; columna: BEH C18, 1.7 micras, 2.1 * 50 mm; gradiente [tiempo (min)/disolvente B en A (%)]: 0.00/5, 0.40/5, 0.8/35, 1.20/55, 2.50/100, 3.30/100, 4.00/5; disolventes: disolvente A = acetato de amonio 5 mM y ácido fórmico al 0.1 % en H2O; disolvente B = ácido fórmico al 0.1% en MeCN; volumen de inyección 2 pl; detección UV de 200 a 400 nm; detección de masa 100 a 1200 AMU (electroaspersión ve); columna a temperatura ambiente; caudal 0.5 ml/min.
Espectroscopia de RMN de 1H: Los desplazamientos químicos se reportan en partes por millón a partir de un estándar de tetrametilsilano.
Esquema general de síntesis
Los compuestos de fórmula (I), (I-A) o (I-B) de la solicitud pueden prepararse siguiendo el siguiente esquema general de síntesis:
Procedimiento general 1:

Acoplamiento general tipo Suzuki (a a b, en el que Y = B(OH)2 o B(OR)2: se añadió 2-bromo-4-fluorobenzaldehído (5 g, 24.63 mmol) al ácido propilborónico (3.25 g, 36.94 mmol), PdCh(dppf) (1.802 g, 2.46 mmol) y CS2CO3 (16.05 g, 49.26 mmol) en dioxano (40 ml) y agua (10 ml), luego se calentó a 80 °C bajo atmósfera de nitrógeno. La suspensión resultante se agitó a 80 °C durante 12 horas. La mezcla de reacción se diluyó con EtOAc (50 ml) y se lavó secuencialmente con salmuera saturada (20 ml x 3). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para producir un producto crudo. El producto crudo se purifica mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 5 % de EtOAc en éter de petróleo.
Acoplamiento general de tipo Buchwald (a a b, en el que Y = NH): se añadió PdOAc2 (553 mg, 2.46 mmol) a pirrolidina (2.59 g, 36.9 mmol), BINAP (4.6 g, 7.4 mmol), CS2CO3 (16.05 g, 4.93 mmol) y 2-bromo-4-fluorobenzaldehído (5 g, 24.63 mmol) en tolueno (50 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a 120 °C durante 1 hora. La mezcla de reacción se filtró a través de Celite. El disolvente se eliminó a presión reducida. El producto crudo se purifica mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1 % de EtOAc en éter de petróleo.
Acoplamiento general tipo Stille (a a b, en el que Y = SnBu3): se añadió 4-(tributilestanil)tiazol (1198 mg, 3.20 mmol) a una solución agitada de 2-bromo-4-fluorobenzaldehído (500 mg, 2.46 mmol) y cloruro de bis(trifenilfosfina)paladio(II) (104 mg, 0.15 mmol ) en DMF (10 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a 100 °C durante 16 horas. La solución se lavó con salmuera y KF. La capa acuosa se extrae con EtOAc (2 x 20 ml). El disolvente se eliminó a presión reducida para proporcionar el producto crudo. El producto crudo se purifica mediante cromatografía ultrarrápida sobre sílice con EtOAc/éter de petróleo (1:30).
Alquilación general de aldehído (b a c): A la solución de 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (1.355 g, 6.83 mmol) en THF (30 ml) a -78 °C se le añade n-butil-litio (5.05 ml, 8.07 mmol ) gota a gota. La mezcla de reacción se agitó a -78 °C durante 30 min, luego se agrega 4-fluoro-2-(pirrolidin-1-il)benzaldehído (1.2 g, 6.21 mmol). La reacción se calentó a temperatura ambiente y se agitó a temperatura ambiente durante 1 h. A continuación, la reacción se detiene con solución sat. De NH4O y se extrae con EtOAc (3 * 60 ml). La capa orgánica se combina, se lavó con agua (60 ml), salmuera (60 ml), se secó sobre MgSO4, se filtró y se concentra para obtener un sólido blanquecino.
Estrategias generales de desprotección:
Método 1: A una solución de (4-fluoro-2-propilfenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (11.6 g, 31.82 mmol) en DCM (50 ml) se añadió TFA (24.52 ml, 318.22 mmol). La solución resultante se somete a reflujo durante 3 horas y se evaporó. El residuo se vuelve a disolver en DCM (50 ml) y la solución se trata con NaHCO3 saturado.
Método 2: En un matraz con forma de guisante de 125 ml se añadió (2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (3 g, 7.70 mmol) y HCl en dioxano (50 ml, 200 mmol) para obtener una solución incolora. La mezcla resultante se agitó a temperatura ambiente durante 30 horas. El disolvente se eliminó a presión reducida. La mezcla de reacción se apaga con NaHCO3 saturado (150 ml), se extrae con EtOAc (3 * 125 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar un sólido amarillo. El precipitado se lavó con EtOAc (20 ml) y se secó al vacío.
Método 3: Se añade cloruro de hidrógeno/EtOAc (10 ml) a (4-fluoro-2-(oxazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (200 mg, 0.51 mmol) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 36 horas. El disolvente se eliminó a presión reducida. El producto crudo se purifica por
HPLC preparativa (columna Xselect CSH Fluoro fenilo OBD, de sílice 5 ^m, diámetro de 19 mm, longitud de 100 mm), utilizando mezclas polares decrecientes de agua (que contienen ácido fórmico al 0.05%) y MeCN como eluyentes.
Método 4: Método combinado de alquilación/desprotección en un solo recipiente: se añadió n-BuLi 2.50 M en THF (11.8 ml, 29.5 mmol) a una solución enfriada de 1-(dietoximetil)imidazol (5.02 g, 29.5 mmol) en THF (75 ml) a -45 °C, y la mezcla se agitó durante 15 min. La mezcla se enfría a -78 °C y se añadió gota a gota una solución de 3-etil-8-fluoro-indolizina-5-carbaldehído (2.82 g, 14.8 mmol) en THF (25 ml). La mezcla se agitó durante 30 min a -78 °C y luego se calentó a 0 °C durante 1 h. La mezcla se diluyó con HCl 0.1 M (50.0 ml) y EtOAc (50.0 ml) a 0 °C. La mezcla se agitó a 23 °C durante 15 min. Se añade más EtOAc (50.0 ml) y se separan las fases. La fase orgánica se extrae con HCl 0.1 M (3 x 50.0 ml) y las fases acuosas combinadas se diluyó cuidadosamente con solución saturada de NaHCO3 (50.0 ml). La fase acuosa w se extrae con EtOAc (3 x 200 ml) y las fases orgánicas combinadas se lavaron con salmuera (200 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida.
Método 5: se añadió percloroestanano (130 mg, 0.50 mmol) a (3-ciclopropil-8-fluoroindolizin-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (200 mg, 0.50 mmol) en DCM (4 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 30 horas. El disolvente se eliminó a presión reducida y se vuelve a disolver en MeOH (2 ml). La mezcla de reacción se neutraliza con NH3.H2O a pH=7~8. El producto crudo se purifica por HPLC preparativa (columna Xselect CSH Prep C18 OBD, de sílice 5 ^m, diámetro de 19 mm, longitud de 100 mm), utilizando como eluyentes mezclas polares decrecientemente de agua (con NH4HCO3 al 0.05%) y MeCN.
Método 6: Se añade gota a gota cloruro de hidrógeno en dioxano (73.0 ml, 291.91 mmol) a (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (5.7 g, 14.60 mmol) en 1,4-dioxano (20 ml) en baño de hielo bajo atmósfera de nitrógeno. La temperatura se incrementa a temperatura ambiente de forma natural. La solución resultante se agitó a 40 °C durante la noche. El disolvente se eliminó bajo presión reducida para obtener un producto crudo. El producto crudo se alcaliniza con NH37 M en MeOH, y se purifica mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 30 % de MeCN en agua.
Procedimiento general 2:

Los aldehídos heterocíclicos se sintetizan y reaccionan de manera análoga al procedimiento general 1 utilizando un grupo de protección similar y procedimientos de desprotección.
Ejemplo 1. (4-fluoro-2-propilfenil)(1H-imidazol-2-il)metanol

A una solución de (4-fluoro-2-propilfenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (11.6 g, 31.82 mmol) en DCM (50 ml) se le añadió t Fa (24.52 ml, 318.22 mmol). La solución resultante se sometió a reflujo durante 3 horas y se evaporó. El residuo se volvió a disolver en DCM (50 ml) y la solución se trató con NaHCO3 saturado. El precipitado se filtró y se secó al vacío para obtener (4-fluoro-2-propilfenil)(1H-imidazol-2-il)metanol (5.88 g, 79 %) como un sólido blancuzco. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 11.82 (br. s., 1 H) 7.51 (dd, J = 8.57, 6.31 Hz, 1 H) 6.59 -7.09 (m,
4 H) 6.00 (d, J = 4.33 Hz, 1 H) 5.90 (d, J = 4.33 Hz, 1 H) 2.63 - 2.84 (m, 1 H) 2.52 - 2.62 (m, 1 H) 1.31 - 1.63 (m, 2 H) 0.90 (t, J = 7.25 Hz, 3 H). LC-MS (Método B): m/z (ES+), [M+H]+ = 235; TFA, HPLC tR = 0.57 min.
Compuesto intermedio C: (4-fluoro-2-propilfenil)(1 -((2-(trimetilsiMl)etoxi)metil)-1H-imidazol-2-M)metanol
A una solución agitada de 4-fluoro-2-propilbenzaldehído (302 mg, 1.82 mmol) en THF (5 ml) a -78 °C, se le añadió una mezcla de 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (400 mg, 2.02 mmol), n-BuLi (258 mg, 4.03 mmol) en THF (5 ml) durante un período de 1 minuto bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 1 hora. La temperatura se aumentó a temperatura ambiente durante un período de varias horas. La mezcla de reacción se inactivó con NH4Cl saturado (5 ml), se extrajo con DCM (3 * 10 ml). La capa orgánica se secó sobre Na2SO4, se filtro y se evaporó para obtener un aceite amarillo. El residuo se purificó por TLC preparativa (EtOAc:éter de petróleo = 1:2), para producir (4-fluoro-2-propilfenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (C, 400 mg, 54.4 %) como un aceite anaranjado. RMN de 1H (300 MHz, CDCl3-cf) 5 ppm 7.21 (dd, J = 8.57, 5.93 Hz, 1 H) 7.05 (dd, J = 16.48, 1.41 Hz, 2 H) 6.79 - 6.98 (m, 2 H) 6.16 (s, 1 H) 4.90 - 5.15 (m, 2 H) 3.20 - 3.45 (m, 2 H) 2.60 - 2.87 (m, 2 H) 1.46 - 1.75 (m, 2 H) 1.00 (t, J = 7.25 Hz, 3 H) 0.72 - 0.89 (m, 2 H) -0.09 - 0.03 (m, 9 H). LC-MS (Método A): m/z (ES+), [M+H]+ = 365; ácido, HPLC tR = 1.062 min.
Compuesto intermedio B: 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol
Se enfrió hidruro de sodio (0.529 g, 22.03 mmol) en THF (50 ml) a 0 °C durante un período de 10 minutos bajo atmósfera de nitrógeno. A esto se le añadió lentamente imidazol (1 g, 14.69 mmol) en t Hf (30 ml) bajo atmósfera de nitrógeno. La temperatura se aumentó gradualmente hasta la temperatura ambiente. La mezcla resultante se agitó a 25 °C durante 30 minutos y luego se añadió lentamente a la mezcla (2-(clorometoxi)etil)trimetilsilano (3.67 g, 22.03 mmol) a 0 °C durante un período de 1 minuto bajo atmósfera de nitrógeno. La temperatura se aumentó gradualmente hasta la temperatura ambiente. La mezcla resultante se agitó a 25 °C durante 14 horas. La mezcla se concentró y se diluyó con EtOAc (200 ml) y se lavó secuencialmente con agua (150 ml). La capa orgánica se secó sobre Na2SO4, se filtro y se evaporo para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida C18 ultrarrápida con un gradiente de elución de 0 a 50 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (B, 2.91 g, 100 %) como un aceite blanco que solidificó al reposar. RMN de 1H (DMSO-d6, 300 MHz) 5 -0.05 - 0.05 (9H, d, J = 6.9 Hz), 0.83 - 0.97 (2H, m), 3.52 -3.64 (2H, m), 5.54 - 5.61 (2H, s), 7.70 - 7.78 (1H, s), 7.84 - 7.91 (1H, s), 9.25 - 9.31 (1H, s). LC-MS (Método A): m/z (ES+), [M+H]+ = 199; HPLC tR = 1.177 min.
Compuesto intermedio A: 4-Fluoro-2-propilbenzaldehído
Se añadió 2-bromo-4-fluorobenzaldehído (5 g, 24.63 mmol) a ácido propilborónico (3.25 g, 36.94 mmol), PdCl2(dppf) (1.802 g, 2.46 mmol) y CS2CO3 (16.05 g, 49.26 mmol) en dioxano (40 ml) y agua (10 ml), luego se calentó a 80°C bajo atmósfera de nitrógeno. La suspensión resultante se agitó a 80 °C durante 12 horas. La mezcla de reacción se diluyó con EtOAc (50 ml) y se lavó secuencialmente con salmuera saturada (20 ml x 3). La capa orgánica se seco sobre Na2SO4, se filtro y se evaporo para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 5 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 4-fluoro-2-propilbenzaldehído (A, 3.10 g, 76 %) como un líquido amarillo pálido. RMN de 1H (400 MHz, DMSO-d6) 5 ppm 0.93 (3 H, t, J = 7.3), 1.58 (2 H, m), 2.96 - 3.05 (2 H, m), 7.21 - 7.32 (2 H, m), 7.93 (1 H, dd, J = 8.2, 6.3), 10.20 (1 H, s).
Ejemplo 2 y 3. (R)-(4-fluoro-2-propilfenil)(1H-imidazol-2-il)metanol (2) y (S)-(4-Fluoro-2-propilfenil)(1H-imidazol-2-il) metanol (3).

La mezcla racémica del Ejemplo 1 se separó mediante SFC quiral utilizando una columna Chiralpak AD-H (30 * 250 mm, 5 ^m). La fase móvil A era CO2 supercrítico y la fase móvil B era isopropanol que contenía dimetilamina al 0.1%. Se ejecutó un gradiente de hasta 15 % de B a razón de 2.8 ml/min y se aislaron dos picos para obtener (R)-(4-fluoro-2-propilfenil)(1H-imidazol-2-il)metanol (Pico 1 a los 3.25 min, 2.490 g, 33.4 % y > 99 de ee, Ejemplo 2) y (s)-(4-fluoro-2-propilfenil)(1H-imidazol-2-il)metanol (Pico 2 a los 4.54 min, 2.480 g, 33.3 % y >99 % de ee, Ejemplo 3) como sólidos blancos. La estereoquímica absoluta se asignó con en base una estructura cristalina del compuesto con receptor PAR2.
Ejemplo 2. Datos analíticos: RMN de 1H (300 MHz, DMSO-d6) 5 ppm 11.83 (br. s., 1 H) 7.51 (dd, J = 8.48, 6.22 Hz, 1 H) 6.61 - 7.06 (m, 4 H) 6.00 (br. s., 1 H) 5.90 (s, 1 H) 2.64 - 2.78 (m, 1 H) 2.52 - 2.62 (m, 1 H) 1.35 - 1.63 (m, 2 H) 0.90 (t, J = 7.35 Hz, 3 H). LC-MS (Método A): [M H]+ = 235; HPLC tR = 0.57 min. SFC quiral tR = 3.25 min.
Ejemplo 3. Datos analíticos: RMN de 1H (300 MHz, DMSO-d6) 5 ppm 11.83 (br. s., 1 H) 7.51 (dd, J = 8.57, 6.31 Hz, 1 H) 6.65 - 7.05 (m, 4 H) 5.81 - 6.22 (m, 2 H) 2.64 - 2.78 ( m, 1 H) 2.53 - 2.62 (m, 1 H) 1.35 - 1.62 (m, 2 H) 0.90 (t, J = 7.35 Hz, 3 H). LC-MS (Método A): [M H]+ = 235; HPLC tp = 0.57 min. SFC quiral tp = 4.54 minutos
Ejemplo 4. (2-ciclopentenilfenil)(1H-imidazol-2-il)metanol.

Ejemplo 4
Se preparo (2-ciclopentenilfenil)(1H-imidazol-2-il)metanol se preparó de manera análoga al Ejemplo 1, utilizando 2-(ciclopent-1-en-1-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano y 2-bromo-benzaldehído. RMN de 1H (400 MHz, DMSO): 5 ppm 1.89 - 1.97 (m, 2H), 2.50 - 2.56 (m, 2H), 2.67 - 3.32 (m, 2H), 5.94 - 6.03 (m, 3H), 6.74 - 6.76 (d, 1H), 6.97 (s, 1H), 7.15-7.24 (m, 3H), 7.53- 7.55 (m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 241; base, HPLC tp = 1.420 min. Ejemplo 5. (2-ciclopentenil-4-fluorofenil)(1H-imidazol-2-il)metanol

Ejemplo 5
Se preparó (2-ciclopentenil-4-fluorofenil)(1H-imidazol-2-il)metanol de manera análoga al Ejemplo 1 usando 2-(ciclopent-1-en-1-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano para proporcionar un sólido blanco (68 mg, 68%). RMN de 1H (400 MHz, DMSO): 5 ppm 1.87- 1.96 (m, 2H), 2.32-2.49 (m, 2H), 2.66-2.71 (m, 2H), 5.75 (s, 1H), 5.90-5.91 (d , 1H), 5.99 - 6.06 (m, 1H), 6.75 (s, 1H), 6.97 - 6.99 (m, 2H), 7.00 - 7.09 (m, 1H), 7.54 - 7.58 (m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 259; base, HPLC tp = 2.185 min.
Ejemplo 6. (2-ciclopentil-4-fluorofenil)(1H-imidazol-2-il)metanol

Ejemplo 5 Ejemplo 6
Una solución de (2-(ciclopent-1-en-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol (254 mg, 0.98 mmol) en MeOH (10 ml) se agitó bajo una atmósfera de hidrógeno (5 atm) y 70 °C durante 12 horas. La mezcla de reacción se filtró a través de Celite. El disolvente se eliminó a presión reducida. El residuo se purificó por TLC preparativa (DCM: MeOH = 10: 1), para proporcionar (2-ciclopentil-4-fluorofenil)(1H-imidazol-2-il)metanol (48.6 mg, 18.99 %) como un sólido blanco. RMN de 1H (400 MHz, DMSO): 5 ppm 1.40- 1.61 (m, 4H), 1.65- 1.75 (m, 3H), 1.88- 1.91 (m, 1H), 3.32-3.40 (m, 1H), 5.97 (s , 1H), 6.05 (s, 1H), 6.75 (s, 1H), 6.94- 7.03 (m, 3H), 7.46- 7.48 (m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 261; base, HPLC tp = 2.286 min.
Ejemplo 7. (2-ciclobutil-4-fluorofenil)(1H-imidazol-2-il)metanol.
Se preparó (2-cidobutil-4-fluorofenil)(1H-imidazol-2-il)metanol de manera análoga al Ejemplo 1 usando bromuro de cidobutilcinc(N) para proporcionar un sólido blanco (55 mg, 61%). RMN de 1H (300 MHz, DMSO-d6) 5 ppm 1.76 (1 H, d, J = 9.2 Hz), 1.81 - 1.96 (1 H, m), 1.93 -2.12 (3 H, m), 2.30 (1 H, d, J = 10.7 Hz), 3.79 (1 H, q, J = 8.9 Hz), 5.84 (1 H, d, J = 4.3 Hz), 6.00 (1 H, d, J = 4.4 Hz), 6.74 (1 H , s), 6.94 - 7.03 (2 H, m), 7.07 (1 H, dd, J = 10.9, 2.7 Hz), 7.48 (1 H, dd, J = 8.6, 6.3 Hz), 11.80 (1 H, s ). LC-MS (Método A): m/z (ES+), [M H]+ = 247; tR = 0.79 min.
Ejemplo 8. (E)-(4-Fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol
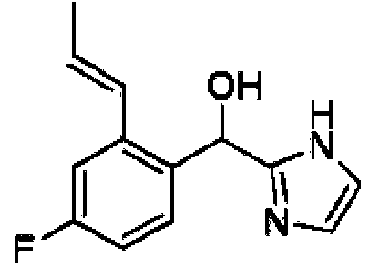
Ejemplo 8
Se preparó (E)-(4-fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol de manera análoga usando el ácido (E)-prop-1-enilborónico. RMN de 1H (400 MHz, DMSO-d6) 51.84 (dd, J = 1.5, 6.5 Hz, 3H), 5.93 (d, J = 4.0 Hz, 1H), 6.11 (d, J = 4.0 Hz, 1H), 6.17 - 6.26 (m, 1H), 6.80 - 6.84 (m, 2H), 6.97 (br. s, 1H), 7.03 (td, J = 3.0, 8.5 Hz, 1H), 7.22 (dd, J = 2.5, 11.0 Hz, 1H), 7.46 (dd, J = 6.5, 8.5 Hz, 1H), 11.91 (br. s, 1H). LCMS (Método D): m/z 233 (M H)+ (ES+), tR = 1.53 min.
Ejemplo 9. (Z)-(4-Fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol

Ejemplo 9
Se preparó (Z)-(4-fluoro-2-(prop-1-enil)fenil)(1H-imidazol-2-il)metanol de forma análoga usando ácido (Z)-prop-1-enilborónico. RMN de 1H (500 MHz, MeOD) 57.53 (dd, J = 8.6, 5.9 Hz, 1H), 7.01 (td, J = 8.6, 2.7 Hz, 1H), 6.95 (s, 2H), 6.89 (dd, J = 9.7, 2.7 Hz, 1H), 6.47 (d, J = 11.1 Hz, 1H), 5.95 (s, 1H), 5.83 (dq, J = 11.5, 7.0 Hz, 1H), 1.62 (dd, J = 7.0, 1.6 Hz, 3H). LC-MS (Método C): m/z (ES+), [M-H2O+H]+ = 215.3.
Ejemplo 10. (3,4-difluoro-2-propilfenil)(1H-imidazol-2-il)metanol

Ejemplo 10
Se preparó (3,4-difluoro-2-propilfenil)(1H-imidazol-2-il)metanol de manera análoga al Ejemplo 1 usando 2-bromo-3,4-difluorobenzaldehído y ácido propilborónico (750 mg, 60%). LC-MS (Método A): m/z (ES+), [M H]+ = 253; HPLC tR = 1.416 min.
Ejemplo 11. Pico 1, Enantiómero puro de (3,4-difluoro-2-propilfenil)(1H-imidazol-2-il)metanol

Ejemplo 11, Enantiómero puro, Pico 1
El material racémico del ejemplo 10 se sometió a SFC quiral utilizando EnantioCel-C1, (21.2 * 250 mm) con una fase móvil A: CO2, fase móvil B: EtOH (IPAal 0.1%) en una proporción de 80:20. Caudal: 40 ml/min. RMN de 1H (400 MHz, Metanol-d4) 5 -0.15 - 0.20 (3 H, s), 0.93 - 0.98 (2 H, t, J = 7.3), 1.30 -1.51 (2 H, dtd, J = 30.2, 16.4, 15.1, 6.7), 2.65 -2.76 (2 H, m), 5.99 - 6.04 (1 H, s), 6.94 - 7.17 (3 H, m), 7.26 - 7.34 (1 H, m). SFC quiral tR = 4.1 min.
Ejemplo 12. Pico 2, enantiómero puro (3,4-difluoro-2-propilfenil)(1H-imidazol-2-il)metanol.

Ejemplo 12, Enantiomero puro, Pico 2
El material racémico del Ejemplo 10 se sometió a SFC quiral utilizando EnantioCel-CI, (21.2 * 250 mm) con una fase móvil A: CO2, fase móvil B: EtOH (IPA al 0.1%) en una proporción de 80:20. Caudal: 40 ml/min. RMN de 1H (400 MHz, metanol-d4) 50.91 - 1.00 (3 H, t, J = 7.3), 1.29 - 1.52 (2 H, m), 2.65 - 2.76 (2 H, m), 5.99 - 6.04 (1 H, s), 6.97 - 7.02 (2 H, s), 7.05 - 7.17 (1 H, m), 7.26 - 7.34 (1 H, m). SFC quiral tR = 5.5 min.
Ejemplo 13. (3-cloro-2-propilfenil)(1H-imidazol-2-il)metanol.

Ejemplo 13
Se preparó (3-cloro-2-propilfenil)(1H-imidazol-2-il)metanol de manera análoga al Ejemplo 1 utilizando 2-bromo-3-clorobenzaldehído y ácido propilborónico. RMN de 1H (300 MHz, DMSO-d6) 50.93 (3 H, t, J = 7.3), 1.35 (2 H, m), 2.68 (1 H, dt, J = 13.1,6.5), 2.76 - 2.93 (1 H, m), 5.93 (1 H, d, J = 4.4), 6.16 (1 H, d, J = 4.3), 6.75 (1 H, s), 6.98 (1 H, s), 7.22 (1 H, d, J = 7.8), 7.29 -7.34 (1 H, m), 7.50 (1 H, dd, J = 7.7, 1.3), 11.90 (1 H, s). LC-MS (Método A): m/z (ES+), base [M+H]+ = 251; HPLC tR = 1.53 min.
Ejemplo 14. (4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-imidazol-2-il)metanol

A la solución de (4-fluoro-2-(pirrolidin-1-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (2.2 g, 5.62 mmol) en DCM (30 ml) se le añadió TFA (43.3 ml, 561.86 mmol). La reacción se agitó a temperatura ambiente durante 6 horas. Se eliminó el TFA. El residuo se repartió entre EtOAc (30 ml) y NaHCO3 saturado (30 ml). La capa orgánica se recogió, se lavo con agua (30 ml), salmuera (30 ml), se seco sobre MgSO4, se filtro y se concentró. El residuo se purificó mediante CombiFlash (DCM/MeOH 10:1, columna de 80 g) para obtener un sólido blanquecino. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 1.75 - 1.96 (m, 4 H) 3.04 -3.29 (m, 4 H) 5.79 -5.95 (m, 1 H) 6.00 -6.12 (m, 1 H) 6.53 -7.14 (m, 4 H)7.18 -7.47 (m, 1 H) 11.67 - 11.96 (m, 1 H). LC-MS (Método B): m/z (ES+), [M H]+ = 262; TFA, HPLC tR = 0.54 minutos
Compuesto intermedio C: (4-fluoro-2-(pirrolidin-1 -il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol
A la solución de 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (1.355 g, 6.83 mmol) en THF (30 ml) a -78 oC se le añadió n-butil-litio (5.05 ml, 8.07 mmol) gota a gota. La mezcla de reacción se agitó a -78 oC durante 30 min y luego se añadió 4-fluoro-2-(pirrolidin-1-il)benzaldehído (1.2 g, 6.21 mmol). La reacción se calentó a temperatura ambiente y se agitó a
temperatura ambiente durante 1 hora. A continuación, la reacción se inactivó con una solución de NH4CI saturada y se extrajo con EtOAc (3 * 60 ml). La capa orgánica se combinó, se lavo con agua (60 ml), salmuera (60 ml), se seco sobre MgSO4, se filtro y se concentró para obtener un sólido blanquecino. RMN de 1H (300 MHz, DMSO-d6) 60.00 (s, 10 H) 0.72 - 1.02 (m, 2 H) 1.71 -1.94 (m, 4 H) 2.80 - 3.14 (m, 4 H) 3.44 - 3.59 (m, 2 H) 5.10 - 5.27 (m, 1 H) 5.34 - 5.38 (m, 1 H) 5.46 - 5.61 (m, 1 H) 5.74 - 5.91 (m, 1 H) 6.04 - 6.24 (m, 1 H) 6.60 - 6.84 (m, 3 H) 7.16 - 7.31 (m, 1 H) 7.42 -7.61 (m, 1 H). LC-MS (Método B): m/z (ES+), [M H]+ = 392; TFA, HPLC tR = 0.98 min.
Compuesto intermedio B: Véase el Ejemplo 1
Compuesto intermedio C: 4-Fluoro-2-(pirrolidin-1 -il)benzaldehído
Se añadió PdOAC2 (553 mg, 2.46 mmol) a pirrolidina (2.59 g, 36.9 mmol), BINAP (4.6 g, 7.4 mmol), CS2CO3 (16.05 g, 4.93 mmol) y 2-bromo-4-fluorobenzaldehído (5 g, 24.63 mmol) en tolueno (50 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a 120 °C durante 1 hora. La mezcla de reacción se filtró a través de Celite. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1% de EtOAc en éter de petróleo. Las fracciones puras se evaporaron hasta sequedad para proporcionar 4-fluoro-2-(pirrolidin-1-il)benzaldehído (3.6 g, 76 %) como un sólido blanco. RMN de 1H (CDCh, 400 MHz) 6 10.03 (s, 1H), 7.76 - 7.72 (m, 1H), 6.57 -6.53 (m, 2H), 3.41 - 3.36 (t, 4H), 2.07 -2.00 (t, 4H). m/z (ES+), [M H]+ = 194.0; ácido, HPLC tR = 0.925 min.
Ejemplo 15. Isómero 1, enantiómero puro (4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-imidazol-2-il)metanol.
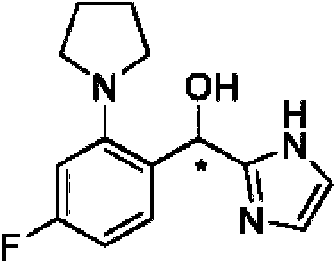
Ejemplo 15: Enantiómero puro, Isómero 1
La mezcla racémica del Ejemplo 14 se separó por HPLC quiral para obtener el Pico 1 y el Pico 2 como sólidos blancos. RMN de 1H (300 MHz, DMSO-d6) 6 ppm 1.75 -2.01 (m, 4 H) 2.97 - 3.33 (m, 9 H) 5.69 - 5.95 (m, 1 H) 5.99 -6.09 (m, 1 H) 6.48 -6.80 (m, 2 H) 6.78 -7.00 (m, 2 H) 7.17 -7.47 (m, 1 H) 11.65 -12.13(m , 1 H). LC-MS (Método B): [M H]+ = 262; TFA, HPLC tR = 0.55 min. La pureza quiral se determinó utilizando una columna Lux Amylose-2 (4.6 * 250 mm, 5 |jm). La fase móvil A era hexano y la fase móvil B era una mezcla de metanol:isopropanol 1:1. A esto se añadió un aditivo de dietilamina al 0.1%. La columna se procesó a razón de 1 ml/min con detección a 220 nM. El pico 1 eluyó a los 6.12 min >98 % de ee.
Ejemplo 16. Isómero 2, enantiómero puro (4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-imidazol-2-il)metanol.

Ejemplo 16: Enantiómero puro, Isómero 2
La mezcla racémica del Ejemplo 14 se separó por HPLC quiral para obtener el Pico 1 y el Pico 2 como sólidos blancos. RMN de 1H (300 MHz, DMSO-d6) 6 ppm 1.72 -2.05 (m, 5 H) 2.98 - 3.32 (m, 7 H) 5.67 - 5.96 (m, 1 H) 5.98 -6.16 (m, 1 H) 6.52 -6.79 (m, 3 H) 6.73 -7.03 (m, 2 H) 7.17 -7.38 (m, 1 H) 11.67 -12.12 (m, 1 H). LC-MS (Método B): [M H]+ = 262; TFA, HPLC tR = 0.55 min La pureza quiral se determinó utilizando una columna Lux Amylose-2 (4.6 * 250 mm, 5 u). La fase móvil A era hexano y la fase móvil B era una mezcla de metanol:isopropanol 1:1. A esto se añadió un aditivo de dietilamina al 0.1%. La columna se procesó a razón de 1 ml/min con detección a 220 nm. El pico 2 eluyó a los 7.29 min con >98 % de ee.
Ejemplo 17. (2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol
En un matraz con forma de guisante de 125 ml se añadió (2-(2,5-dihidro-1H-pirrol-1-M)-4-fluorofenil)(1 -((2-(trimetilsilil)etoxi)metM)-1H- imidazol-2-il)metanol (3 g, 7.70 mmol) y HCl en dioxano (50 ml, 200 mmol) para obtener una solución incolora. La mezcla resultante se agitó a temperatura ambiente durante 30 horas. El disolvente se eliminó a presión reducida. La mezcla de reacción se inactivó con NaHCO3 saturado (150 ml), se extrajo con EtOAc (3 * 125 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido amarillo. El precipitado se lavó con EtOAc (20 ml) y se secó al vacío para producir (2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il) metanol (1.00 g, 50.1 %) como un sólido blanco, que se usó sin más purificación. RMN de 1H (300 MHz, MeOD) 5 ppm 4.06 (4 H, s), 5.86 (2 H, s), 6.25 (1 H, s), 6.60 (1 H, td), 6.74 (1 H, dd), 6.94 (2 H, s), 7.20 (1 H, dd). LC-MS (Método A): m/z (ES+), [M H]+ = 260; ácido, HPLC tp = 0.592 min.
Se preparó (2-(2,5-Dihidro-1H-pirrol-1 -il)-4-fluorofenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol de manera análoga al Ejemplo 14, usando 2,5-dihidro-1H-pirrol.
Ejemplo 18. Isómero 1, enantiómero puro (2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol

Ejemplo 18: Enantiómero puro, Isómero 1
El Ejemplo 17 se purificó mediante HPLC quiral preparativa (columna: CHIRALPAK AD-H SFC, 5 * 25 cm, 5 |jm; fase móvil A: CO2 :6%0, fase móvil B: EtOH(IpA al 0.1%)-40%; Caudal: 160 ml/min; Detección a 215 nm. RMN de 1H (300 MHz, MeOD) 5 ppm 4.12 (4 H, s), 5.92 (2 H, s), 6.31 (1 H, s), 6.66 (1 H, td), 6.80 (1 H, dd), 7.00 (2 H, s), 7.26 (1 H, dd). LC-MS (Método A): m/z (ES+), [M H]+ = 260; ácido, HPLC tp = 1.306 min. SFC quiral tp = 5.45 min (pico 1) > 98% de ee.
Ejemplo 19. Isómero 2, enantiómero puro (2-(2,5-dihidro-1H-pirrol-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol

Ejemplo 19: Enantiómero puro, Isómero 2
El Ejemplo 17 se purificó mediante HPLC quiral preparativa (columna: CHIRALPAK AD-H SFC, 5 * 25 cm, 5 jm ; fase móvil A: CO2 :6%0, fase móvil B: EtOH(IPA al 0.1%)-40%; Caudal: 160 ml/min; Detección a 215 nm. RMN de 1H (300 MHz, MeOD) 5 ppm 4.12 (3 H, s), 5.92 (2 H, s), 6.31 (1 H, s), 6.66 (1 H, td), 6.80 (1 H, dd), 7.01 (2 H, s), 7.26 (1 H, dd). LC-MS (Método A): m/z (ES+), [M H]+ = 260; ácido, HPLC tp = 1.30 min SFC quiral tp = 7.60 min (pico 2) > 98% de ee.
Ejemplo 20. (2-(Azetidin-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol

Se empleó un procedimiento análogo al del Ejemplo 14 usando azetidina para proporcionar (2-(azetidin-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol (17 mg, 12%). p MN de 1H (300 MHz, CDCh) 5 ppm 7.17 (dd, J = 7.08, 8.59 Hz, 1H), 6.91 (s, 2H), 6.47 (dt, J = 2.55, 8.45 Hz, 1H), 6.18 (dd, J = 2.64, 12.09 Hz, 1H), 5.78 - 5.91 (m, 2H), 3.78 - 4.06 (m, 4H), 2.05 - 2.29 (m, 2H), 2.05 - 2.29 (m, 2H), 2.20 (quin, J =7.27 Hz, 2H). LC-MS (Método B): [M H]+ = 248; TFA, HPLC tp = 0.35 min.
Ejemplo 21. (3-cloro-2-(1H-pirazol-1-il)fenil)(1H-imidazol-2-il)metanol

Se añadió ácido 2,2,2-trifluoroacético (2.5 ml, 32.45 mmol) a (3-doro-2-(1H-pirazoM-il)fenil)(1-((2-(trimetilsNil) etoxi)metil)-1H-imidazol-2-il)metanol (200 mg, 0.49 mmol) en DCM (5 ml). La solución resultante se agitó a ta durante 18 horas. El disolvente se eliminó a presión reducida. La mezcla de reacción se neutralizó con NaHCO3 saturado, se extrajo con EtOAc (3 x 5 ml), se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 5 a 70 % de MeCN en agua. Las fracciones puras se evaporaron hasta sequedad para proporcionar (3-cloro-2-(1H-pirazol-1-il)fenil)(1H-imidazol-2-il)metanol (45 mg, 33 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 5 ppm 5.24 - 5.26 (m, 1H), 6.21 - 6.22 (m, 1H), 6.42 - 6.43 (m, 1H), 6.86(s, 2H), 7.52 - 7.60 (m, 2H), 7.65 - 7.68 (m, 2H), 7.73 (s, 1H), 11.86 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 275.1; ácido, HPLC tp = 0.477 min.
Compuesto intermedio D, (3-cloro-2-(1H-pirazol-1-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol

Se añadió lentamente n-butil-litio (0.774 ml, 1.94 mmol) a 1 -((2-(trimetilsilil)etoxi)metil)-1H-imidazol (384 mg, 1.94 mmol) en THF (6 ml) a -78 °C bajo atmósfera de nitrógeno. Media hora más tarde, se añadió 3-cloro-2-(1H-pirazol-1-il)benzaldehído (200 mg, 0.97 mmol). La solución resultante se agitó a -78 °C durante 1.5 horas. La mezcla de reacción se inactivó con NH4O saturado (5 ml), se extrajo con EtOAc (3 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un líquido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 5 a 70 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (3-cloro-2-(1H-pirazol-1-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (380 mg, 97 %) como un líquido blanco. RMN de 1H (400 MHz, DMSO) 5 -0.05 -0.02 (m, 24H), 0.70 - 0.78 (m, 1H), 0.81 - 0.85 (m, 4H), 3.18 - 3.24 (m, 2H), 3.43 - 3.47 (m, 3H), 5.01 - 5.03 (m, 1H), 5.27 - 5.29 (m, 1H), 5.32 (s, 3H), 5.52 - 5.54 (m, 1H), 6.23 -6.24 (m, 1H), 6.29 (s, 1H), 6.72 (s, 1H), 6.93 (s, 2H), 7.14 (s, 1H), 7.26 (s, 2H), 7.59 - 7.60 (m, 2H), 7.67 (s, 1H), 7.77 (s, 2H), 7.81 -7.82 (m, 1H).
Compuesto intermedio C, 3-cloro-2-(1H-pirazol-1-il)benzaldehído

Se añadió lentamente DIBAL-H (3.39 ml, 3.39 mmol) a 3-cloro-N-metoxi-N-metil-2-(1H-pirazol-1-il)benzamida (600 mg, 2.26 mmol) en THF (10 ml) a -78°C bajo atmósfera de nitrógeno. La solución resultante se agitó a -40 °C durante 2 horas. La mezcla de reacción se vertió en agua (10 ml), se extrajo con EtOAc (3 x 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante
cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron hasta sequedad para proporcionar 3-doro-2-(1H-pirazol-1-il)benzaldehído (345 mg, 73.9 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 56.63 - 6.64 (m, 1H), 7.66 - 7.75 (m, 1H), 7.84 - 7.91 (m, 2H), 8.01 - 8.04 (m, 1H), 8.24 - 8.25 (m, 1H), 9.21 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 207; ácido, HPLC tp = 0.788 min.
Compuesto intermedio B, 3-cloro-N-metoxi-N-metil-2-(1H-pirazol-1-il)benzamida

Se añadió lentamente DIEA (0.941 ml, 5.39 mmol) a ácido 3-cloro-2-(1H-pirazol-1-il)benzoico (200 mg, 0.90 mmol), HATU (512 mg, 1.35 mmol) y N, Clorhidrato de O-dimetilhidroxilamina (219 mg, 2.25 mmol) en DMF (5 ml) a 0 °C durante un período de 6 minutos bajo atmósfera de nitrógeno. La temperatura se aumentó a temperatura ambiente de forma natural. La solución resultante se agitó a ta por 2 horas. La mezcla de reacción se diluyó con agua, se extrajo con EtOAc (3 x 10 ml), se lavó con salmuera saturada (15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 50 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 3-cloro-N-metoxi-N-metil-2-(1H-pirazol-1-il)benzamida (210 mg, 88 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 52.95 - 3.01 (m, 3H), 3.45 (m, 3H), 6.46 (s, 1H), 7.51 - 7.60 (m, 2H), 7.68 - 7.77 (m, 2H), 7.91 -7.95 (m, 3H). LC-MS (Método A): m/z (ES+), [M H]+ = 266; ácido, HPLC tp = 0.717 min.
Compuesto intermedio A, ácido 3-cloro-2-(1H-pirazol-1-il)benzoico

Se añadieron ácido 2-bromo-3-clorobenzoico (500 mg, 2.12 mmol), 1H-pirazol (145 mg, 2.12 mmol), N1,N2-dimetilciclohexano-1,2-diamina (15.1 mg, 0.11 mmol), yoduro de cobre (I) (60.7 mg, 0.32 mmol), K2CO3 (734 mg, 5.31 mmol) y L-prolina (73.3 mg, 0.64 mmol) a DMSO (6 ml) a 25 °C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 100 °C durante 1 hora. La mezcla de reacción se diluyó con agua (10 ml), se extrajo con EtOAc (2 x 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un líquido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir ácido 3-cloro-2-(1H-pirazol-1-il)benzoico (250 mg, 52.8 %) como un aceite blanco. RMN de 1H (400 MHz, DMSO) 5 ppm 6.46 (s, 1H), 7.41 - 7.50 (m, 1H), 7.58 - 7.62 (m, 1H), 7.66 - 7.67 (m, 1H), 7.75 - 7.77 (m, 1H), 7.81 - 7.83 (m, 1H), 7.96 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 222.9; ácido, HPLC tp = 0.54 min.
Ejemplo 22. (3,4-difluoro-2-(1H-pirazol-1-il)fenil)(1H-imidazol-2-il)metanol

El Ejemplo 22 se preparó de manera análoga al Ejemplo 21, usando ácido 2-bromo-3,4-difluorobenzoico y 1H-pirazol. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 5.47 - 5.49 (m, 1H), 6.27 - 6.29 (m, 1H), 6.48 - 6.50 (m, 1H), 6.73 (s, 1H), 6.96 (s, 1H), 7.49 - 7.54 (m, 1H), 7.60 - 7.68 (m, 1H), 7.78 - 7.79 (m, 1H), 7.93 - 7.94 (m, 1H) 11.91 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 277; ácido, HPLC tp = 0.656 min.
Ejemplo 23. (4-Fluoro-2-(1H-pirazol-1-il)fenil(1H-imidazol-2-il)metanol

Ejemplo 23
El Ejemplo 23 se preparó de manera análoga al Ejemplo 21, usando éster de ácido 2-bromo-4-fluorobenzoico y 1H-pirazol. (En este caso, la reducción de DIBAL-H resultante en el éster condujo al alcohol y no al aldehído. Se necesitó una etapa adicional para oxidar el alcohol bencílico al aldehído usando 20 equivalentes de MnO2 en DCM.) RMN de 1H (400 MHz, DMSO-d6) 6 ppm 5.80 (1 H, s), 6.53 (1 H, t), 6.89 (2 H, d), 7.25 - 7.35 (2 H, m), 7.69 (1 H , dd), 7.78 (1 H, d), 8.41 (1 H, d), 12.05 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 259; HPLC tR = 0.609 min.
Los siguientes ejemplos se prepararon por rutas análogas al Ejemplo 14, usando métodos de separación quiral como se indica cuando se emplean:


Ejemplo 31. (2-(Biciclo[3.1.0]hexan-1-il)-4-fluorofenil)(1H-imidazol-2-il)metanol

Ejemplo 31
A la solución de (2-(biciclo[3.1.0]hexan-1-il)-4-fluorofenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (43 mg, 0.11 mmol) en DCM (3 ml) se le añadió TFA (0.823 ml, 10.68 mmol). La reacción se agitó a temperatura ambiente durante 6 horas. Se eliminó el TFA. El residuo se repartió entre EtOAc (30 ml) y solución saturada de NaHCO3 (30 ml). La capa orgánica se recogió, se lavó con agua (30 ml), salmuera (30 ml), se secó sobre MgSO4, se filtró y concentró. El residuo se purificó mediante CombiFlash (DCM/MeOH 10:1, columna de 24 g) para obtener un sólido blanco. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 0.43 - 0.93 (m, 2 H) 1.13 - 1.46 (m, 1 H) 1.53 - 2.19 (m, 6 H) 5.78 -6.00 (m, 1 H) 6.07 - 6.25 (m, 1 H) 6.62 - 7.14 (m, 4 H) 6.93 - 6.95 (m, 1 H) 7.37 - 7.61 (m, 1 H) 11.74 - 12.03 (m, 1 H). LC-MS (Método B): m/z (ES+), [M H]+ = 273; ácido, HPLC tR = 0.65 min.
Una solución de dietilzinc (1.153 ml, 1.15 mmol) en DCM (2.5 ml) se enfrió a -40 °C. A continuación, se añadió gota a gota TFA (0.089 ml, 1.15 mmol). Después de 20 min más, se añadió gota a gota diyodometano (0.093 ml, 1.15 mmol). Después de 20 minutos adicionales, se añadió (2-(ciclopent-1-en-1-il)-4-fluorofenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (compuesto intermedio del Ejemplo 5, 80 mg, 0.21 mmol). La reacción se calentó a temperatura ambiente. Después de 30 min adicionales, la reacción se detuvo con NH4Cl acuoso saturado (20 ml) y las capas se separaron. La capa acuosa se extrajo con DCM (3 * 20 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4, se filtraron y concentraron. El producto crudo se purificó con CombiFlash (acetato de etilo en hexanos) para producir un producto como un aceite amarillo.
Ejemplo 32. (4-Fluoro-2-(oxazol-4-il)fenil)(1H-imidazol-2-il)metanol
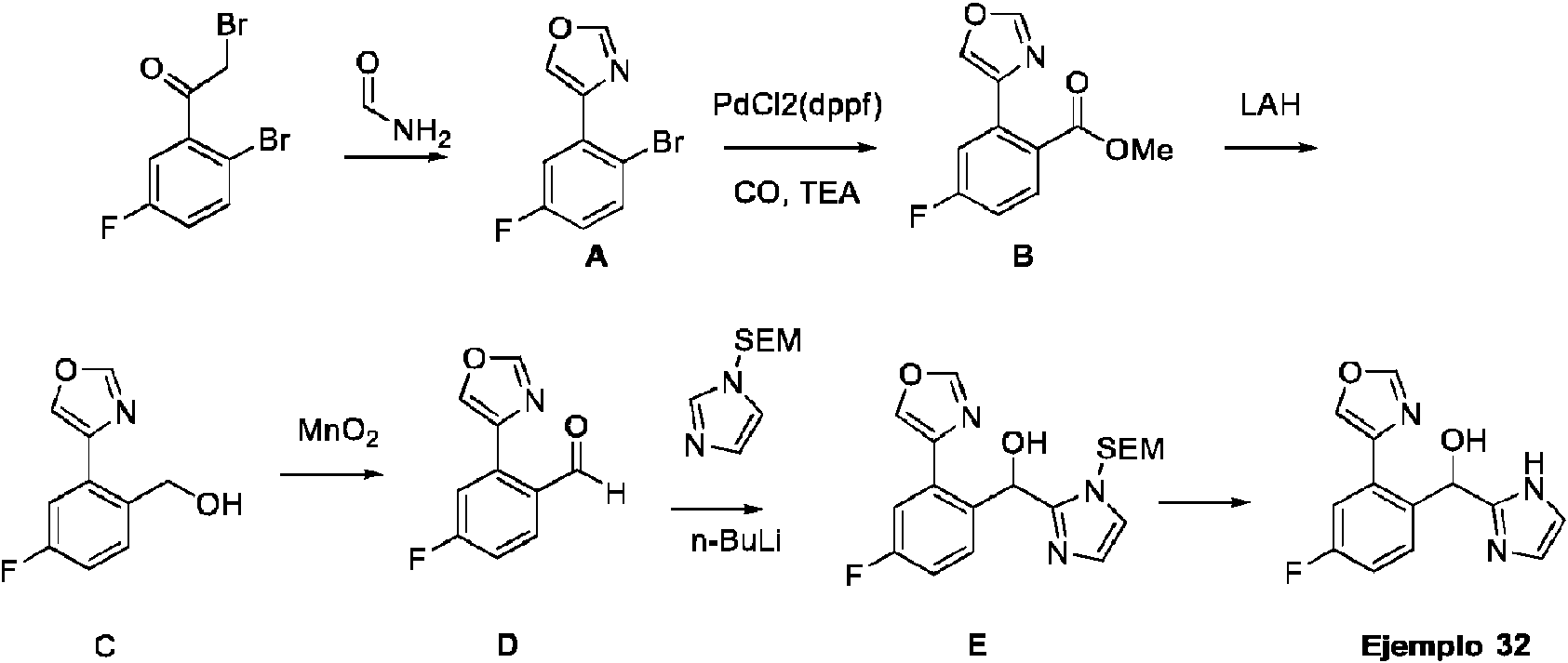
Se añadió cloruro de hidrógeno/EtOAc (10 ml) a (4-fluoro-2-(oxazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (200 mg, 0.51 mmol) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 36 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó por HPLC preparativa (columna Xselect CSH Fluoro fenilo OBD, sílice de 5 |jm, 19 mm de diámetro, 100 mm de longitud), usando mezclas decrecientemente polares de agua (que contenía ácido fórmico al 0.05%) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluoro-2-(oxazol-4-il)fenil)(1H-imidazol-2-il)metanol (100 mg, 75 %) como un sólido blanco. RMN de 1H (400 MHz, DMSO) 5 ppm 6.05 (1
H, s), 6.91 (2 H, s), 7.24 (1 H, td), 7.46 (1 H, dd), 7.71 (1 H, dd), 8.26 (1 H, s), 8.56 (1 H, d), 8.78 (1 H, d). LC-MS (Método A): m/z (ES+), [M H]+ = 259; base, HPLC tR = 1.50 min.
Compuesto intermedio E, (4-Fluoro-2-(oxazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol
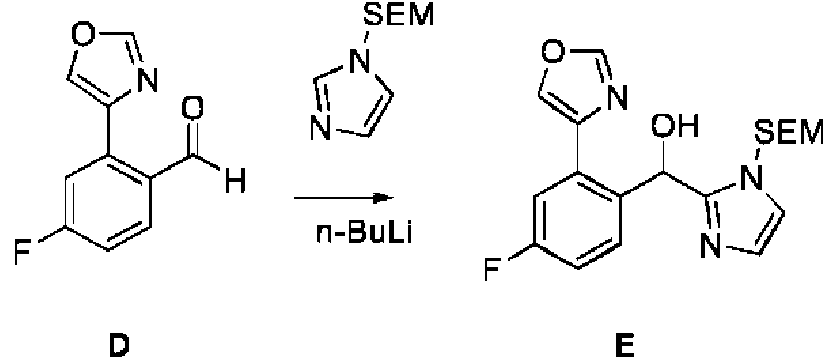
Se añadió gota a gota butil-litio (349 mg, 5.45 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (865 mg, 4.36 mmol) en THF (10 ml) enfriado a -78 °C en nitrógeno. La mezcla resultante se agitó a -78 °C durante 1 hora. Se añadió 4-fluoro-2-(oxazol-4-il)benzaldehído (417 mg, 2.08 mmol) en THF (10 ml). La mezcla resultante se agitó a -78 °C durante 30 minutos.
La mezcla de reacción se inactivó con NH4Cl (10 ml), se extrajo con EtOAc (2 * 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco.
El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (4-fluoro-2-(oxazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (500 mg, 58 %) como un sólido blanco. RMN de 1H 8 ppm -0.07 (3 H, m), 0.83 (15 H, m), 1.18 (5 H, m), 1.36 (1 H, dd), 1.55 (1 H, dd), 1.92 (1 H, s), 2.00 (4 H, s), 3.46 (15 H, m), 4.04 (3 H, q), 5.32 (12 H, m), 5.55 (3 H, m), 5.79 (1 H, m), 6.19 (4 H, m), 6.75 (3 H, m), 6.94 (4 H, m), 7.25 (11 H, m), 7.42 (4 H, m), 7.75 (7 H, m), 7.90 (2 H, d), 8.48 (2 H, d), 11.97(1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 390.10; ácido, HPLC tR= 1.285 min.
Compuesto intermedio D: 4-Fluoro-2-(oxazol-4-il)benzaldehído

Se añadió óxido de manganeso (IV) (7201 mg, 82.83 mmol) a (4-fluoro-2-(oxazol-4-il)fenil)metanol (400 mg, 2.07 mmol) en DCM (10 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 24 horas. La mezcla se filtró a través de una capa de Celite para producir 4-fluoro-2-(oxazol-4-il)benzaldehído (300 mg, 76 %) como un sólido blanco. RMN de 1H 8 ppm 0.85 (2 H, m), 1.24 (1 H, s), 1.57 (9 H, d), 5.42 (1 H, t), 5.55 (2 H, s), 5.69 (3 H, q), 7.44 (7 H, m), 7.66 (12 H, m), 7.94 (7 H, m), 8.34 (1 H, t), 8.64 (3 H, d), 8.76 (4 H, d), 10.18 (1 H, d), 10.36 (4 H, d). LC-MS (Método A): m/z (ES+), [M H]+ = 192.1; ácido, HPLC tR = 0.998 min.
Compuesto intermedio C: (4-fluoro-2-(oxazol-4-il)fenil)metanol

Se añadió LiAlH4 (64.3 mg, 1.70 mmol) a 4-fluoro-2-(oxazol-4-il)benzoato de metilo (250 mg, 1.13 mmol) en THF (5 ml) enfriado a -20 °C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a -20 °C durante 30 minutos. La mezcla de reacción se inactivó con agua (1 ml), se extrajo con EtOAc (2 x 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar (4-fluoro-2-(oxazol-4-il)fenil)metanol (120 mg, 55.0 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 193.95; ácido, HPLC tR = 0.684 min.
Compuesto intermedio B : 4-fluoro-2-(oxazol-4-il)benzoato de metilo

Se añadió CO (exceso) a PdCl2(dppf) (159 mg, 0.22 mmol), TEA (0.806 ml, 5.78 mmol) y 4-(2-bromo-5-fluorofenil)oxazol (350 mg, 1.45 mmol) en MeOH (5 ml). La mezcla resultante se agitó a 130 °C durante 20 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 4-fluoro-2-(oxazol-4-il)benzoato de metilo (251 mg, 78 %) como un sólido blanco. RMN de 1H 5 ppm (400 MHz, CDCls) 0.88 (5 H, dd), 0.98 (1 H, m), 1.29 (9 H, m), 1.46 (5 H, m), 1.85 (5 H, s), 2.07 (2 H, s), 2.55 (7 H, s), 2.74 (1 H, s), 3.13 (5 H, s), 3.49 (1 H, d), 3.90 (20 H, d), 4.15 (2 H, q), 7.14 (11 H, m), 7.31 (3 H, td), 7.53 (4 H, dd), 7.91 (12 H, m), 8.08 (5 H, d). LC-MS (Método A): m/z (ES+), [M H]+ = 222.1; ácido, HPLC tR = 1.024 min.
Compuesto intermedio A, 4-(2-bromo-5-fluorofenil)oxazol

Se añadió 2-bromo-1-(2-bromo-5-fluorofenil)etanona (1 g, 3.38 mmol) a formamida (0.152 g, 3.38 mmol) bajo atmósfera de nitrógeno y se selló en un tubo de microondas. La reacción se calentó a 120 °C durante 2 días en el reactor de microondas y se enfrió a temperatura ambiente. La mezcla de reacción se diluyó con EtOAc (25 ml) y se lavó con salmuera saturada (15 ml x 3). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 4 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 4-(2-bromo-5-fluorofenil)oxazol (0.30 g, 36%) como un sólido amarillo pálido.
Ejemplo 33. (4-Fluoro-2-(tiazol-4-il)fenil)(1H-imidazol-2-il)metanol

Se añadió cloruro de hidrógeno (exceso) en EtOAc a (4-fluoro-2-(tiazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (100 mg, 0.25 mmol) en EtOAc (6 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 14 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó por HPLC preparativa (columna XSelect CSH Prep C18 OBD, sílice de 5 ^m, 19 mm de diámetro, 150 mm de longitud), utilizando mezclas decrecientemente polares de agua (que contenía ácido fórmico al 0.05%) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluoro-2-(tiazol-4-il)fenil)(1H-imidazol-2-il)metanol (29.0 mg, 34.2%) como un sólido blanco. RMN de 1H (300 m Hz , CD3OD) 56.21 -6.22 (m, 1H), 7.13 - 7.25(m, 3H), 7.37 -7.41(m, 1H), 7.58 - 7.62(m, 1H), 7.84 (s, 1H), 8.33(s, 1.5H), 9.11 - 9.12(m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 276.2; ácido, HPLC tR = 1.207 min.
Compuesto intermedio B, (4-fluoro-2-(tiazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol

Se añadió n-butil-litio (0.386 ml, 0.97 mmol) a una solución agitada de 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (191 mg, 0.97 mmol) en THF (10 ml) a -78 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 1 hora. Se añadió una solución de 4-fluoro-2-(tiazol-4-il)benzaldehído (100 mg, 0.48 mmol) en THF (3 ml) a una solución agitada a -78 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 2 horas. La mezcla de reacción se inactivó con NH4Cl saturado (10 ml), se extrajo con EtOAc (3 x 20 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un aceite amarillo. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 70 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (4-fluoro-2-(tiazol-4-il)fenil)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il) metanol (100 mg, 51 %) como un aceite amarillo. LC-m S (Método A): m/z (ES+), [M H]+ = 406.2; ácido, HPLC tR = 0.721 min.
Compuesto intermedio A, 4-fluoro-2-(tiazol-4-il)benzaldehído

Se añadió 4-(tributilestanil)tiazol (1198 mg, 3.20 mmol) a una solución agitada de 2-bromo-4-fluorobenzaldehído (500 mg, 2.46 mmol) y cloruro de bis(trifenilfosfina)paladio(II) (104 mg, 0.15 mmol) en DMF (10 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a 100 °C durante 16 horas. La solución se lavó con salmuera y KF. La capa acuosa se extrajo con EtOAc (2 x 20 ml). El disolvente se eliminó a presión reducida para proporcionar el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice con EtOAc/éter de petróleo (1:30). Las fracciones puras se evaporaron hasta sequedad para proporcionar 4-fluoro-2-(tiazol-4-il)benzaldehído (150 mg, 29.4 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 208; ácido, HPLC tR = 0.802 min.
Ejemplo 34. (3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol

Se añadió HCl (0.125 ml, 4.11 mmol) a (3-etil-8-fluoroindolizin-5-il)(1-((2-(trimetilsilil)etoxi)metil)- 1H-imidazol-2-il) metanol (160 mg, 0.41 mmol) en EtOAc (5 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 30 horas. El disolvente se eliminó a presión reducida y se volvió a disolver en MeOH (2 ml). La mezcla de reacción se neutralizó con NH3.H2O a pH = 7~8. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para proporcionar (3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol (90 mg, 85 %) como un sólido blanquecino. RMN de 1H (400 MHz, DMSO) 5 ppm 1.29 (3 H, t), 2.52 (1 H, s), 3.20 (1 H, dq), 6.42 (4 H, m),
6.61 (2 H, h), 6.85 (1 H, s), 7.10 (2 H, m), 12.08 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 260.1; base, HPLC tR = 2.528 min.
Compuesto intermedio E, (3-etil-8-fluoroindolizin-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol

Se añadió butil-litio (317 mg, 4.94 mmol) en porciones a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (840 mg, 4.24 mmol) en THF (5 ml) enfriado a -78 °C durante un período de 5 minutos bajo atmósfera de nitrógeno. La mezcla resultante se agitó a -78 °C durante 30 minutos. Se añadió en porciones 3-etil-8-fluoroindolizina-5-carbaldehído (270 mg, 1.41 mmol) en THF (5 ml) a la mezcla resultante bajo atmósfera de nitrógeno. La mezcla resultante se agitó a -78 °C durante 30 minutos. La mezcla de reacción se inactivó con NH4Cl (5 ml), se extrajo con EtOAc (2 * 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para producir (3-etil-8-fluoroindolizin-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (400 mg, 72.7 %) como un aceite incoloro. RMN de 1H (400 Mh z , CDCh) 8 -0.06 (2 H, d), 0.08 (m), 0.71 (qdd), 1.29 (t), 1.48 (t), 2.07 (1 H, s), 3.38 (m), 4.15 (q), 5.10 (m), 5.81 (dd), 6.11 (dd), 6.63 (s), 6.73 (q), 7.10 (dd). LC-MS (Método A): m/z (ES+), [M H]+ = 390.15; ácido, HPLC tR = 1.585 min.
Compuesto intermedio D, 3-etil-8-fluoroindolizina-5-carbaldehído

Se añadió óxido de manganeso (IV) (4500 mg, 51.76 mmol) al compuesto C (~400 mg) y DCM (2 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 48 horas. La mezcla de reacción se filtró a través de Celite. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1% de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 3-etil-8-fluoroindolizina-5-carbaldehído (160 mg, 32.3 %) como un aceite amarillo. RMN de 1H (300 MHz, CDCla-d) 89.93 (s, 1H), 7.47 - 7.36 (m, 1H), 6.99 (d, J = 4.4 Hz, 1H), 6.82 (d, J = 4.4 Hz, 1H), 6.57 - 6.44 (m, 1H), 2.99 (q, J = 7.6 Hz, 2H), 1.33 (t, J = 9.4, 8.4 Hz, 3H). LC-MS (Método A): m/z (ES+), [M H]+ = 191.95; ácido, HPLC tR = 1.004 min.
Compuesto intermedio C, (3-etil-8-fluoroindolizin-5-il)metanol

Se añadió lentamente hidruro de litio y aluminio (172 mg, 4.52 mmol) a 3-etil-8-fluoroindolizina-5-carboxilato de metilo (500 mg, 2.26 mmol) en THF (8 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 0 °C durante 1 hora. La mezcla de reacción se inactivó con agua (5 ml), se extrajo con EtOAc (3 * 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 60 % de MeCN en agua. Las fracciones puras se evaporaron hasta sequedad para proporcionar (3-etil-8-fluoroindolizin-5-il)metanol (260 mg, 59.5 %) como un sólido gris. RMN de 1H (400 MHz, CDCla) 8 ppm 6.73 - 6.63 (m, 2H), 6.44 (dd, J = 7.4, 5.6 Hz, 1H), 6.24 (dd, J = 9.9, 7.4 Hz, 1H), 4.94 (s, 2H), 3.30 (q, J = 7.4 Hz, 2H), 1.42 (t, J = 7.4 Hz, 3H). LC-MS (Método A): m/z (ES+), [M H]+ = 194.05; ácido, HPLC tR = 1.498 min.
Compuesto intermedio B, 3-etil-8-fluoroindolizina-5-carboxilato de metilo

Se añadió cloruro de cobre (I) (0.483 g, 4.88 mmol) a 5-fluoro-6-(pent-1-in-1-M)picolinato de metilo (1.08 g, 4.88 mmol) en trietilamina (1.000 ml) y DMA ( 3.5 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 120 °C durante 16 horas. La mezcla de reacción se diluyó con EtOAc (25 ml) y se lavó secuencialmente con agua (15 ml x 3), salmuera saturada (15 ml x 2) y NH4Cl saturado (10 ml x 2). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 0 % de EtOAc en éter de petróleo. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 60 % de MeCN en agua. Las fracciones puras se evaporaron hasta sequedad para proporcionar 3-etil-8-fluoroindolizina-5-carboxilato de metilo (0.550 g, 50.9 %) como un sólido amarillo. RMN de 1H (400 MHz, CDCl3) 5 ppm 7.56 (d, J = 7.0 Hz, 1H), 7.17 (dd, J = 7.8, 5.5 Hz, 2H), 6.87 (d, J = 4.2 Hz, 2H), 6.75 (d, J = 4.1 Hz, 2H), 6.61 (s, 1H), 6.46 (td, J = 7.1,5.1 Hz, 1H), 6.41 -6.29 (m, 3H), 3.99 (s, 5H), 2.85 (q, J = 7.5 Hz, 2H), 2.76 - 2.66 (m, 4H), 1.41 (t, J = 7.5 Hz, 3H), 1.31 (t, J = 7.4 Hz, 6H). LC-MS (Método A): m/z (ES+), [M H]+ = 222.0; ácido, HPLC tR= 1.072 min.
Compuesto intermedio A, 5-fluoro-6-(pent-1-inil) picolinato de metilo

Se añadió pent-1-ino (0.524 g, 7.69 mmol) y cloruro de bis(trifenilfosfina)paladio(II) (0.450 g, 0.64 mmol) a yoduro de cobre(I) (0.122 g, 0.64 mmol), TEA (1.787 ml, 12.82 mmol) y 6-bromo-5-fluoropicolinato de metilo (1.5 g, 6.41 mmol) en acetonitrilo (20 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 80 °C durante 3 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 5-fluoro-6-(pent-1-in-1-il)picolinato de metilo (1.28 g, 90 %) como un sólido naranja. RMN de 1H (400 MHz, CDCb) 5 ppm 8.20 - 8.06 (m, 2H), 7.52 (dd, J = 8.6, 8.0 Hz, 2H), 4.14 (q, J = 7.1 Hz, 1H), 4.01 (s, 7H), 2.50 (td, J = 7.1, 1.0 Hz, 4H), 2.07 (s, 1H), 1.71 (h, J = 7.3 Hz, 4H), 1.28 (t, J = 7.1 Hz, 1H), 1.09 (t, J = 7.4 Hz, 6H). LC-MS (Método A): m/z (ES+), [M H]+ = 222.2; ácido, HPLC tR = 1.140 min.
Ejemplo 35. Enantiómero puro, isómero 1 de (3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol

Ejemplo 35, Enantiómero puro, Isómero 1
El producto se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con EtOH al 10 % en heptano (modificado con DEA al 0.1) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir P1-(3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol (60.0 mg, 37.5 %) como un sólido marrón (Ejemplo 35) y P2-(3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol (60.0 mg, 37.5 %) como un sólido marrón (Ejemplo 36). RMN de 1H (400 MHz, DMSO) 5 1.32 (3 H, t), 3.22 (1 H, dt), 6.21 (1 H, s), 6.46 (2 H, m), 6.65 (2 H, m), 6.77 (2 H, s), 7.20 (2 H, s), 13.20 (1 H, s). m/z (ES+), [M H]+ = 260.4; ácido, HPLC tR = 2.478 min. La HPLC quiral analítica se completó utilizando la columna AmChemteq ACI Am-1, 0.46 x 15 cm, 5 ^m. La fase móvil A era de hexanos (con DEA al 0.1%) y la fase móvil B era etanol (mezcla de A:B 70/30). Detección UV-VIS a 220 nm. Flujo = 1.0 ml/min. tR = 2.95 min; Pureza del 99%.
Ejemplo 36. Enantiómero puro, isómero 2 de (3-etil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol

Ejemplo 36, Enantiomero puro, Isómero 2
Véase el Ejemplo 35. RMN de 1H (300 MHz, DMSO) 5 ppm 1.28 (3 H, t), 3.19 (2 H, qd), 6.44 (4 H, m), 6.60 (2 H, m), 6.99 (2 H, s), 12.21 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 260.3; ácido, HPLC tR = 2.213 min. HPLC quiral usando los métodos descritos en el Ejemplo 35. tR = 3.76 minutos; Pureza del 99% de ee.
Síntesis alternativa al ejemplo 34, 35 y 36. (3-etil-8-fluoroindolizin-5-il)-(1H-imidazol-2-il)metanol y enantiómeros puros
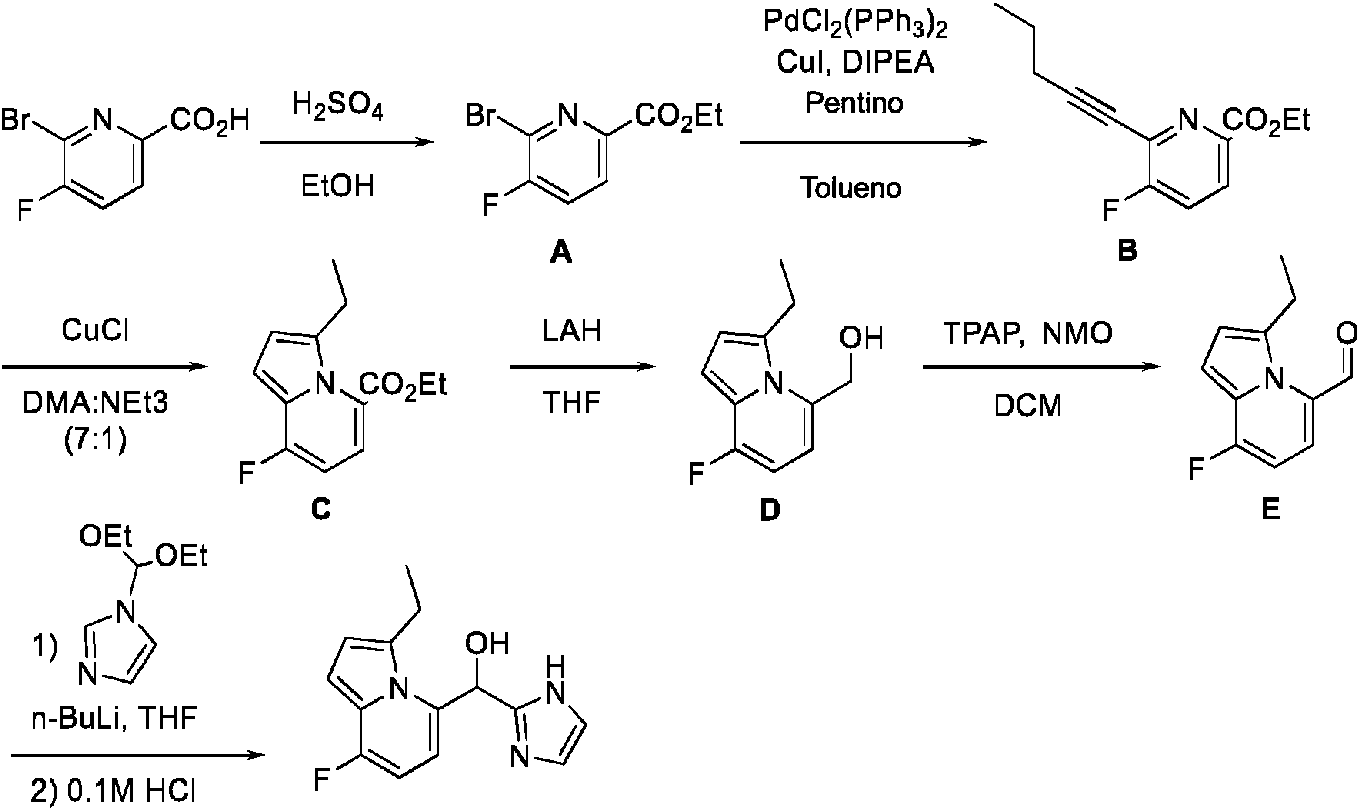
Ejemplo 34, mezcla racémica
Se añadió n-BuLi 2.50 M en THF (11.8 ml, 29.5 mmol) a una solución enfriada de 1-(dietoximetil)imidazol (5.02 g, 29.5 mmol) en THF (75 ml) a -45 °C, y la mezcla se agitó durante 15 m. La mezcla se enfrió a -78 °C y se añadió gota a gota una solución de 3-etil-8-fluoroindolizina-5-carbaldehído (2.82 g, 14.8 mmol) en THF (25 ml). La mezcla se agitó durante 30 min a -78 °C y luego se calentó a 0 °C durante 1 h. La mezcla se diluyó con HCl 0.1 M (50.0 ml) y EtOAc (50.0 ml) a 0 °C. La mezcla se agitó a 23 °C durante 15 min. Se añadió más EtOAc (50.0 ml) y se separaron las fases. La fase orgánica se extrajo con HCl 0.1 M (3 x 50.0 ml) y las fases acuosas combinadas se diluyeron cuidadosamente con NaHCO3 saturado (50.0 ml). La fase acuosa se extrajo con EtOAc (3 x 200 ml) y las fases orgánicas combinadas se lavaron con salmuera (200 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El compuesto era idéntico al material producido por la otra vía descrita y podía purificarse mediante cromatografía SFC quiral para proporcionar enantiómeros puros como se describió previamente.
Síntesis alternativa Ejemplo 34, compuesto intermedio E. 3-Etil-8-fluoroindolizina-5-carbaldehído
Se añadió NMO (8.23 g, 70.3 mmol) a una mezcla de (3-etil-8-fluoroindolizin-5-il)metanol (6.79 g, 35.1 mmol) y tamices moleculares de 4 Á en DCM (100 ml). La mezcla se agitó a 23 °C durante 30 min. La mezcla se enfrió a 0 °C y se añadió TPAP (1.23 g, 3.51 mmol) en porciones. La mezcla se calentó a 23 °C durante 1 h y se filtró a través de un lecho de Florisil. El filtrado se concentró a presión reducida y el producto se purificó por cromatografía en gel de sílice (cartucho de 120 g) eluyendo con hexano y EtOAc (0-30 %) para proporcionar el compuesto del título como un sólido (2.82 g, 42 %). RMN de 1H (500 MHz, CDCls) 59.90 (s, 1H), 7.38 (dd, J = 7.9, 5.6 Hz, 1H), 6.96 (d, J = 4.2 Hz, 1H), 6.79 (d, J = 4.2 Hz, 1H), 6.47 (dd, J = 9.3, 7.9 Hz, 1H), 2.96 (q, J = 7.4 Hz, 2H), 1.31 (t, J = 7.4 Hz, 3H); LC-MS (Método C) m/z (ES+), Sin ionización. tR = 1.76 min.
Síntesis alternativa Ejemplo 34, Compuesto intermedio D. (3-Etil-8-fluoroindolizin-5-il)metanol
Se añadió LAH 2 M en THF (20.5 ml, 40.9 mmol) a una solución enfriada de 3-etil-8-fluoroindolizina-5-carboxilato de etilo (9.63 g, 40.9 mmol) en THF (90.0 ml) bajo atmósfera de nitrógeno a 0 °C. La mezcla se agitó a 0 °C durante 15 min y luego se diluyó con acetona (10.0 ml) y solución sat. solución de tartrato de K/Na (100 ml). Se añadió agua (100 ml) y el producto se extrajo de la fase acuosa con EtOAc (3 x 200 ml). Las fases orgánicas combinadas se lavaron con salmuera (200 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 120 g) con hexano y EtOAc (5-60 %) para proporcionar el
compuesto del título como un sólido (6.79 g, 86 %). RMN de 1H (500 MHz, CDCI3) 56.68 (d, J = 4.1 Hz, 1H), 6.64 (d, J = 4.1 Hz, 1H), 6.41 (dd, J = 7.4, 5.6 Hz, 1H), 6.22 (dd, J = 9.8, 7.4 Hz, 1H), 4.91 (d, J = 6.2 Hz, 2H), 3.27 (q, J = 7.4 Hz, 2H), 1.68 (t, J = 6.2 Hz, 1H), 1.40 (t, J = 7.4 Hz, 3H). LC-MS (Método C) m/z (ES+), [M H]+ = 193.8. HPLC tR = 1.51 min.
Síntesis alternativa Ejemplo 34, Compuesto intermedio C. 3-etil-8-fluoroindolizina-5-carboxilato de etilo
Se añadió una solución de 5-fluoro-6-pent-1-inil-piridin-2-carboxilato de etilo (14.5 g, 61.6 mmol) en DMA desgasificada (193 ml) a CuCl (3.05 g, 30.8 mmol) bajo atmósfera de nitrógeno. NEt3 desgasificada (27.5 ml, 197 mmol) y la mezcla se agitó a 130 °C durante 20 h. La mezcla se enfrió y se filtró a través de Celite, lavando con EtOAc. El filtrado se concentró a presión reducida. Se añadió agua (400 ml) al residuo y la fase acuosa se extrajo con EtOAc (3 x 500 ml). Las fases orgánicas combinadas se lavaron con salmuera (500 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 220 g) con hexano y EtOAc (0-20 %) para proporcionar el compuesto del título como un aceite (9.63 g, 66 %). RMN de 1H (500 MHz, CDCh) 5 7.14 (dd, J = 7.7, 5.5 Hz, 1H), 6.83 (d, J = 4.2 Hz, 1H), 6.72 (d, J = 4.2 Hz, 1H), 6.33 (dd, J = 9.7, 7.8 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 2.71 (q, J = 7.4 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.28 (t, J = 7.4 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 235.9. HPLC tR = 2.18 m.
Síntesis alternativa Ejemplo 34, Compuesto intermedio B. 5-fluoro-6-pent-1-inil-piridin-2-carboxilato de etilo
Se añadió 1-pentino (12.1 ml, 123 mmol) a una mezcla de 6-bromo-5-fluoro-piridin-2-carboxilato de etilo (20.4 g, 82.0 mmol), Cul (2.34 g, 12.3 mmol) y PdCk(PPh3)2 (5.76 g, 8.20 mmol) en tolueno desgasificado (500 ml) a 23 °C bajo atmósfera de nitrógeno. Se añadió DIPEA (28.6 ml, 164 mmol) y la mezcla se agitó en la oscuridad (cubierta con papel de aluminio) durante 18 h. La mezcla se diluyó con una solución saturada de NaHCO3 (400 ml). La fase acuosa se extrajo con EtOAc (3 * 400 ml) y las fases orgánicas combinadas se lavaron con salmuera (400 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 330 g) con hexano y EtOAc (0-50 %) para proporcionar el compuesto del título como un aceite (16.8 g, 87 %). RMN de 1H (500 MHz, CDCh) 58.05 (dd, J = 8.6, 4.0 Hz, 1H), 7.54 - 7.43 (m, 1H), 4.45 (q, J = 7.1 Hz, 2H), 2.47 (td, J = 7.1,0.7 Hz, 2H), 1.68 (h, J = 7.3 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H), 1.06 (t, J = 7.4 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 235.9. HPLC tR = 1.92 m.
Síntesis alternativa Ejemplo 34, Compuesto intermedio A. 6-bromo-5-fluoro-piridin-2-carboxilato de etilo
Se disolvió ácido 6-bromo-5-fluoro-piridin-2-carboxílico (24.0 g, 109 mmol) en una mezcla de EtOH (400 ml) y H2SO4 concentrado (17.5 ml, 328 milimoles). La mezcla se calentó a 90 °C durante 2.5 h. Después de enfriar a 0 °C, se añadió lentamente una solución de NaOH 3.50 M (90.0 ml, 315 mmol) con agitación vigorosa (pH ajustado a aproximadamente 4), seguido de una disolución saturada de NaHCO3 hasta que el pH fue de aprox. 8. La suspensión se filtró a través de una capa de Celite y el filtrado se concentró a presión reducida. El residuo se suspendió en agua fría (300 ml) y se agitó a 0 °C durante 5 min. La mezcla se filtró, se lavó con agua y el sólido se secó al vacío para proporcionar el compuesto del título como un sólido (20.4 g, 75%). RMN de 1H (500 MHz, CDCh) 58.12 (dd, J = 8.3, 3.6 Hz, 1H), 7.53 (dd, J = 8.3, 7.0 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 247.8. HPLC tR = 1.74 m.
Ejemplo 37. (8-Fluoro-3-metilindolizin-5-il)(1H-imidazol-2-il)metanol

Ejemplo 37
Preparado de manera análoga al Ejemplo 34. RMN de 1H (300 MHz, DMSO-c6) 5 ppm 12.08 (s, 1H), 7.08 (s, 1H), 6.86 (s, 1H), 6.53 (t, J = 3.7 Hz, 2H), 6.47 - 6.35 (m, 2H), 6.30 (d, J = 7.1 Hz, 1H), 2.71 (s, 3H). LC-MS (Método A): m/z (ES+), [M H]+ = 246.2; álcali, HPLC tR = 1.357 min.
Ejemplo 38. (8-Fluoro-3-isoproilindolizin-5-il)(1H-imidazol-2-il)metanol
El Ejemplo 38 se preparó de manera análoga al Ejemplo 34. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 12.12 (s, 1H), 7.10 (s, 1H), 6.86 (s, 1H), 6.74 (d, J = 4.2 Hz, 1H), 6.63 (d, J = 4.2 Hz, 1H), 6.56 -6.26 (m, 4H), 3.77 (p, J = 6.6 Hz, 1H), 1.35 - 1.21 (m, 6H). LC-m S (Método A modificado a condiciones básicas): m/z (ES+), [M H]+ = 274.2; álcali, HPLC tR= 1.559 min.
Ejemplo 39. (3-Cicloorooil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol

El Ejemplo 38 se preparó de manera análoga al Ejemplo 34. Se realizaron las siguientes modificaciones para la etapa final de desprotección. Se añadió percloroestanano (130 mg, 0.50 mmol) a (3-ciclopropil-8-fluoroindolizin-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (200 mg, 0.50 mmol) en DCM (4 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 30 horas. El disolvente se eliminó a presión reducida y se volvió a disolver en MeOH (2 ml). La mezcla de reacción se neutralizó con NH3.H2O a pH=7~8. El producto crudo se purificó por HPLC preparativa (columna XSelect CSH Prep C18 OBD, sílice de 5 pm, 19 mm de diámetro, 100 mm de longitud), utilizando mezclas decrecientemente polares de agua (que contiene NH4HCO3 al 0.05%) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (3-ciclopropil-8-fluoroindolizin-5-il)(1H-imidazol-2-il)metanol (10.00 mg, 7.40 %) como un sólido blanquecino. RMN de 1H (400 MHz, MeOD) 5 ppm 0.78 (1 H, tt), 0.91 (1 H, tt), 1.03 (2 H, s), 1.31 (1 H, s), 2.42 (1 H, ddd), 5.11 (5 H, s), 6.27 (2 H, m), 6.54 (2 H, m), 7.08 (2 H, s), 7.37 (1 H, s). LC-MS (Método A, modificado a condiciones básicas): m/z (ES+), [M H]+ = 272.0; base, HPLC tR = 1.511 min.
Ejemplo 40. (3-Etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol.
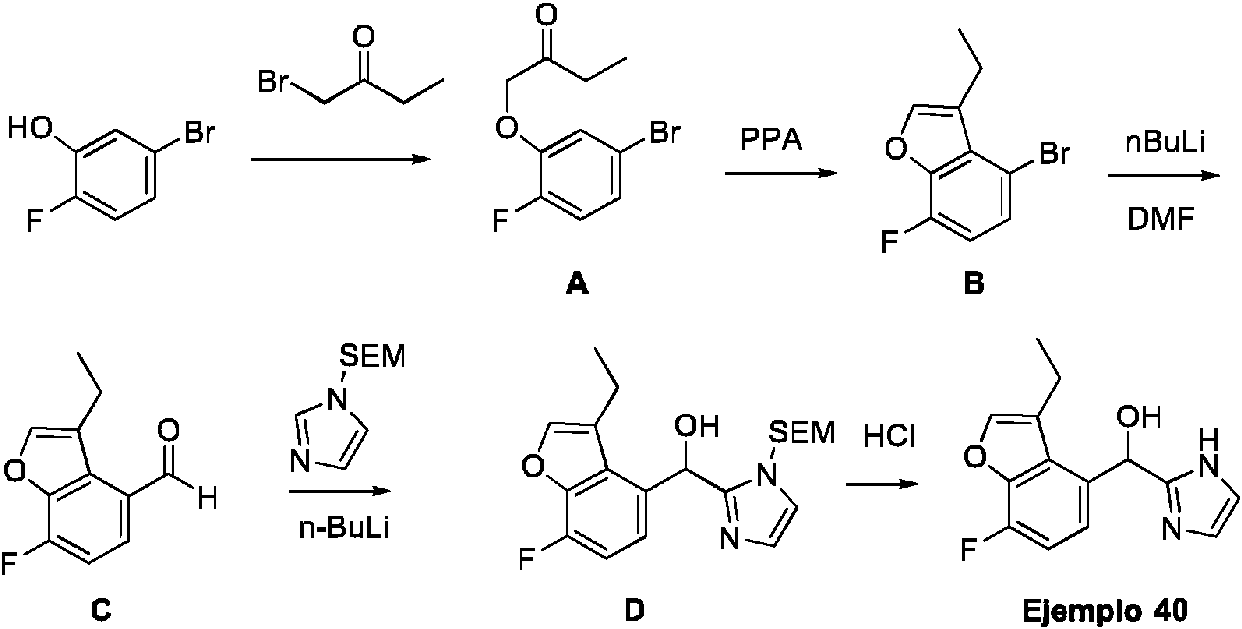
Se añadió gota a gota cloruro de hidrógeno en dioxano (73.0 ml, 291.91 mmol) a (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (5.7 g, 14.60 mmol) en 1,4-dioxano (20 ml) en baño de hielo bajo atmósfera de nitrógeno. La temperatura se aumentó a temperatura ambiente de forma natural. La solución resultante se agitó a 40 °C durante la noche. El disolvente se eliminó a presión reducida para obtener un producto crudo. El producto crudo se alcalinizó con NH37 M en MeOH, y se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 30 % de MeCN en agua. Las fracciones puras se evaporaron hasta sequedad para proporcionar (3-etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol (2.500 g, 65.8 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 1.11 -1.29 (3 H, t, J = 7.4), 2.63 - 2.98 (2 H, m), 6.18 - 6.32 (2 H, m), 6.84 - 6.93 (2 H, s), 7.12 - 7.22 (1 H, dd, J = 10.7, 8.5), 7.23 -7.33 (1 H, dd, J = 8.5, 4.7), 7.77 -7.84 (1 H, s), 12.01 - 12.13 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 261.4; ácido, HPLC tR = 1.539 min.
Compuesto intermedio D, (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol
Se añadió gota a gota butil-litio (13.05 ml, 32.62 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (7.06 g, 35.59 mmol), en THF (150 ml) enfriado a -70 °C durante un período de 5 minutos bajo atmósfera de nitrógeno. Se añadió gota a gota 3-etil-7-fluorobenzofuran-4-carbaldehído (5.7 g, 29.66 mmol) en THF (15 ml) después de agitar la reacción durante 1.5 horas a -70 °C. La reacción fue incompleta por lo que la mezcla se dejó en agitación durante 1 hora a -70 °C. La mezcla de reacción se inactivó con NH4Cl saturado (200 ml), se extrajo con EtOAc (3 * 200 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un aceite amarillo. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 50 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il) metanol (5.72 g, 49.4 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO-d6 ) 6 ppm -0.07 -0.01 (9H, s), 0.69 -0.94 (2H, dddd, J = 35.8, 13.8, 9.7, 6.9), 1.06 - 1.18 (3H, t, J = 7.4), 2.22 -2.42 (1H, m), 2.52 -2.67 (1H, m), 3.36 -3.56 (2H, ddq, J = 9.5, 6.7, 3.0), 5.26 - 5.36 (1 H, d, J = 10.7), 5.60 - 5.71 (1 H, d, J = 10.7), 6.18 - 6.25 (1 H, d, J = 6.0), 6.29 -6.38 (1 H, d, J = 6.0), 6.69 -6.76 (1 H, d, J = 1.1), 7.12 -7.22 (1 H, dd, J = 10.8, 8.5), 7.27-7.38 (2 H, m), 7.73 -7.79 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 391.4; ácido, HPLC tR = 2.323 min.
Compuesto intermedio C, 3-etil-7-fluorobenzofurano-4-carbaldehído

Se añadió lentamente DMF (0.637 ml, 8.23 mmol) a 4-bromo-3-etil-7-fluorobenzofurano (1 g, 4.11 mmol) y butil-litio (2.468 ml, 6.17 mmol) en THF (20 ml) a -78 °C durante un período de 1 hora bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 1 hora. La mezcla de reacción se inactivó con NH4Cl saturado (20 ml), se extrajo con EtOAc (2 * 25 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 20 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron hasta sequedad para proporcionar 3-etil-7-fluorobenzofuran-4-carbaldehído (0.570 g, 72.1 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 193; ácido, HPLC tR = 1.652 min.
Compuesto intermedio B, 4-bromo-3-etil-7-fluorobenzofurano
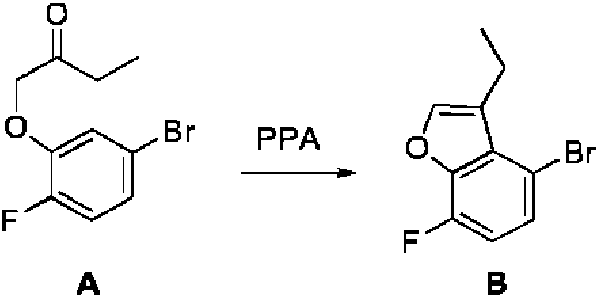
Se añadió ácido polifosfórico (11.76 g, 49.03 mmol) a 1-(5-bromo-2-fluorofenoxi)butan-2-ona (6.4 g, 24.51 mmol) en tolueno (50 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 120 °C durante 14 horas. La mezcla de reacción se vertió en agua (50 ml), se extrajo con EtOAc (2 x 75 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un líquido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1% de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 4-bromo-3-etil-7-fluorobenzofurano (2.20 g, 36.9 %) como un sólido blanco que se usó tal cual sin purificación adicional.
Compuesto intermedio A, 1-(5-bromo-2-fluorofenoxi)butan-2-ona

Se añadió 1-bromobutan-2-ona (26.1 g, 172.78 mmol) a 5-bromo-2-fluorofenol (30 g, 157.07 mmol) y K2CO3 (54.3 g, 392.67 mmol) en MeCN (500 ml) a 25 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. La solución resultante se agitó a 60°C durante 3 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 1-(5-bromo-2-fluorofenoxi)butan-2-ona (31.0 g, 76 %) como un sólido blanco. RMN de 1H (300 MHz, metanol-d4) 6 ppm 1.02 -1.14 (t, J = 7.3 Hz, 3H), 2.54 - 2.67 (q, J = 7.3 Hz,
2H), 4.78 -4.85 (s, 2H), 7.00 -7.13 (m, 2H), 7.14 -7.22 (dd, J = 7.6, 2.0 Hz, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = desconocido; ácido, HPLC tR = 1.574 min.
Ejemplo 41. Enantiómero puro, Pico 2, (R)-(3-etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol

Ejemplo 41
El Ejemplo 40 se purificó mediante HPLC quiral preparativa en una CHIRALPAK AD-H SFC, 5 * 25 cm, 5 pm con MeOH al 30 % en otro disolvente (modificado con IPAmina al 0.1 %) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (3-etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il) metanol (0.850 g, 38.2 %) como un sólido blanco. fase móvil A: CO2; fase móvil B: MeOH (IPAmina al 0.1%) (proporción 70:30). Caudal: 170 ml/min; 254 nm ; Pico 2 tR = 5.66 min. RMN de 1H (300 MHz, Metanol-d4) 6 ppm 1.20 - 1.37 (3 H, t, J = 7.4), 2.62 - 2.86 (2 H, m), 6.31 - 6.38 (1 H, s), 6.95 - 7.08 (3 H, m), 7.08 - 7.18 (1 H, dd, J = 8.4, 4.5), 7.55 - 7.62 (1 H, t, J = 1.4). LC-MS (Método A): m/z (ES+), [M H]+ = 260.95; ácido, HPLC tR = 1.263 min.
Ejemplo 42. Enantiómero puro, Pico 1, (S)-(3-etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol

El Ejemplo 40 se purificó mediante HPLC quiral preparativa en una CHIRALPAK AD-H SFC, 5 * 25 cm, 5 pm con MeOH al 30 % en otro disolvente (modificado con IPAmina al 0.1 %) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (3-etil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il) metanol (0.850 g, 38.2 %) como un sólido blanco. Fase móvil A: CO2; fase móvil B: MeOH (iPAmina al 0.1%) (proporción 70:30). Caudal: 170 ml/min; 254 nm; Pico 1, tR = 4.29 min. Estereoquímica absoluta establecida por la estructura cristalina de rayos X en la proteína Par2. RMN de 1H (300 MHz, DMSO-d6) 6 ppm 1.17 -1.29 (3 H, t, J 7.4), 2.64 -2.98 (2 H, m), 6.18 -6.25 (1 H, s), 6.85 -6.92 (2 H, s), 7.10 -7.34 (2 H, m), 7.77 -7.83 (1 H, s), 8.11 -8.17 (1 H, s), 11.81 -12.88 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 260.95; ácido, HPLC tR = 1.263 min.
Ejemplo 43. (3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol

Ejemplo 43
El Ejemplo 43 se preparó de manera análoga al Ejemplo 40, utilizando 2-bromo-1-ciclopropiletanona. RMN de1H (400 MHz, DMSO-c/6) 6 ppm 0.55 (1 H, q), 0.64 (1 H, m), 0.78 (2 H, m), 1.98 (1 H, s), 6.79 (1 H, d), 7.11 (1 H, m), 7.28 (2 H, m), 7.60 (2 H, s), 7.93 (1 H, d), 14.23 (2 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 273.1; base, HPLC tR = 2.552 min.
Ejemplo 44. Enantiómero puro, Isómero 1, (3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol

Enantiómero puro, Isómero 1, Ejemplo 44
El Ejemplo 43 se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con IPA al 10% en hexano (modificado con DEA al 0.1) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir P1-(3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol (40.0 mg, 40.0 %, Ejemplo 44) y P2-(3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol (40.0 mg, 40.0 %, Ejemplo 45) como un sólido blanquecino. RMN de 1H (400 MHz, DMSO-d6) 5 ppm 0.51 (1 H, q), 0.81 (3 H, m), 2.16 (1 H, p), 6.26 (1 H, d), 6.62 (1 H, d), 6.76 (1 H, s), 7.00 (1 H, s), 7.18 (1 H, dd), 7.33 (1 H, dd), 7.79 (1 H, d), 11.96 (1 H, s). lC-MS (Método A, modificado a condiciones básicas): m/z (ES+), [M H]+ = 273.2; base, HPLC tR = 1.814 min. Métodos analíticos de HPLC quirales: Columna ODH, (0.46 x 10 cm, 5 ^M), fase móvil A: Hexanos, fase móvil B: etanol (proporción 90:10), detección a 254 nm. tR = 3.75.
Ejemplo 45. Enantiómero puro, Isómero 2, (3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol

Enantiómero puro, Isómero 2, Ejemplo 45
El Ejemplo 45 se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con IPA al 10% en hexano (modificado con DEA al 0.1) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir P1-(3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol (40.0 mg, 40.0 %, Ejemplo 45) y P2-(3-ciclopropil-7-fluorobenzofuran-4-il)(1H-imidazol-2-il)metanol (40.0 mg, 40.0 %, Ejemplo 45) como un sólido blanquecino. RMN de 1H (400 MHz, DMSO-d6) 5 ppm 0.53 (1 H, m), 0.80 (3 H, m), 2.15 (1 H, q), 6.26 (1 H, d), 6.62 (1 H, d), 6.76 (1 H, s), 7.00 (1 H, s), 7.18 (1 H, dd), 7.33 (1 H, dd), 7.79 (1 H, d), 11.96 (1 H, s). lC-MS (Método A, modificado a condiciones básicas): m/z (ES+), [M H]+ = 273.2; base, HPLC tR = 1.903 min. Métodos analíticos de HPLC quirales: Columna ODH, (0.46 x 10 cm, 5 ^M), fase móvil A: Hexanos, fase móvil B: etanol (relación 90:10), detección a 254 nm. tR = 5.43.
Ejemplo 46, (7-Fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol
Se añadió EtOAc/HCl (2 ml) a (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (100 mg, 0.22 mmol) a ta bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 16 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó por HPLC preparativa (columna XSelect CSH Prep C18 OBD, sílice de 5 |jm, 19 mm de diámetro, 150 mm de longitud), usando mezclas decrecientemente polares de agua (que contenía ácido fórmico al 0.1%) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (20.00 mg, 24.30 %) como un sólido blanco. RMN de 1H (400 MHz, metanol-d4) 83.43 - 3.61 (1 H, m), 3.91 (1 H, dtd), 6.28 (1 H, s), 7.06 - 7.18 (4 H, m), 7.85 - 7.93 (1 H, m), 8.46 (1 H, s). LC-MS (Método A): m/z (ES+), [M ] = 315.2; ácido, HPLC tR = 2.21 min. RMN de F (400 MHz, Metanol-d4, 23 °C) 8 -140.3 (1F),-67.2 (3F).
Compuesto intermedio H, (7-Fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il) metanol.

Se añadió gota a gota BuLi (0.471 ml, 1.18 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (234 mg, 1.18 mmol) en THF (5 ml) enfriado a -78 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 1 hora. Se añadió gota a gota 7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carbaldehído (145 mg, 0.59 mmol) en THF (1 ml). La solución resultante se agitó a -78 °C durante otra hora. La mezcla de reacción se inactivó con NH4Cl saturado (10 ml), se extrajo con EtOAc (3 * 5 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un residuo incoloro. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (7-fluoro-3-(2,2,2-trifluoroetil) benzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2 -il)metanol (138 mg, 52.7 %) como una goma incolora. LC-MS (Método A): m/z (ES+), [M H]+ = 445.2; ácido, HPLC tR = 0.82 min.
Compuesto intermedio G, 7-Fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carbaldehído

Se añadió dióxido de manganeso (10.51 mg, 0.12 mmol) a una mezcla agitada de (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)metanol (30 mg, 0.12 mmol) en DCM (5 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 16 horas. La mezcla de reacción se filtró a través de Celite. El disolvente se eliminó a presión reducida. Esto dio como resultado 7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carbaldehído (20.00 mg, 67.2 %) como un sólido blanco. RMN de 1H (300 MHz, CDCh) 8 ppm 4.14 (2 H, qd), 7.27 (1 H, dd), 7.75 - 7.88 (2 H, m), 10.08 (1 H, s).
Compuesto intermedio F, (7-Fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)metanol

Se añadió LiAlH4 (5.50 mg, 0.14 mmol) a 7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carboxilato de metilo (40 mg, 0.14 mmol) en THF (5 ml) enfriado a -20 °C bajo atmósfera de nitrógeno. La solución resultante se calentó a 0 °C y se agitó a 0 °C durante 30 minutos. La mezcla de reacción se inactivó con NH4Cl saturado (5 ml), se extrajo con EtOAc (2 * 5 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar (7-fluoro-3-(2,2,2-trifluoroetil) benzofuran-4-il)metanol (35.0 mg, 97 %) como un sólido amarillo claro.
Compuesto intermedio E, 7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carboxilato de metilo

Se agitaron 4-bromo-7-fluoro-3-(2,2,2-trifluoroetil)benzofurano (100 mg, 0.34 mmol), aducto PdCl2(dppf)-CH2Cl2 (275 mg, 0.34 mmol) y Et3N (0.047 ml, 0.34 mmol) en MeOH (10 ml) bajo una atmósfera de monóxido de carbono a 40 atm y 130 °C durante 16 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1% de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-carboxilato de metilo (40.0 mg, 43.0 %) como un sólido blanco. RMN de 1H (300 MHz, CDCh) 8 ppm 3.88 - 4.06 (5 H, m), 7.11 (1 H, dd), 7.77 (1 H, s), 7.89 (1 H, dd).
Compuesto intermedio D, 4-bromo-7-fluoro-3-(2,2,2-trifluoroetil)benzofurano

Se añadió trimetil(trifluorometil)silano (942 mg, 6.62 mmol) a ((tiofeno-2-carbonil)oxi)cobre (211 mg, 1.10 mmol), fluoruro de potasio (385 mg, 6.62 mmol) y 4-bromo-3-(bromometil)-7-fluorobenzofurano (340 mg, 1.10 mmol) en THF (10 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 75 °C durante 4 horas. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (2 x 20 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un residuo naranja. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 1% de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 4-bromo-7-fluoro-3-(2,2,2-trifluoroetil)benzofurano (120 mg, 36.6 %) como un líquido incoloro. RMN de 1H (300 MHz, CDCla) 8 ppm 4.13 (2 H, q), 6.98 (1 H, dd), 7.36 (1 H, dd), 7.72 (1 H, q).
Compuesto intermedio C, 4-Bromo-3-(bromometil)-7-fluorobenzofurano
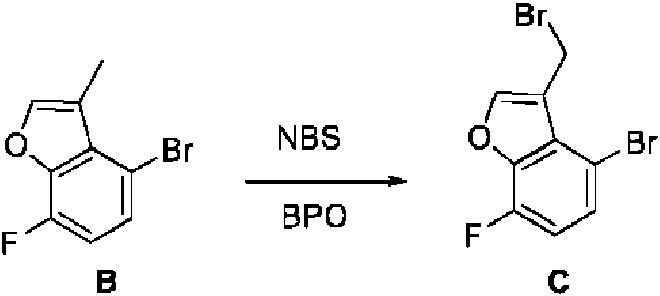
Se añadió NBS (31.1 mg, 0.17 mmol) a 4-bromo-7-fluoro-3-metilbenzofurano (40 mg, 0.17 mmol) y peroxianhídrido benzoico (42.3 mg, 0.17 mmol) en CCl4 (2 ml) a 25 °C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 85 °C durante 16 horas. El residuo se purificó por TLC preparativa (éter de petróleo:EtOAc = 50:1), para proporcionar 4-bromo-3-(bromometil)-7-fluorobenzofurano (20.00 mg, 37.2 %) como un sólido amarillo.
Compuesto intermedio B, 4-Bromo-7-fluoro-3-metilbenzofurano
Se añadió ácido polifosfórico (5 ml, 14.17 mmol) a 1-(5-bromo-2-fluorofenoxi)propan-2-ona (3.5 g, 14.17 mmol) a 25°C bajo atmósfera de nitrógeno. La solución resultante se agitó a 110 °C durante 16 horas. La mezcla de reacción se inactivó con agua (25 ml), se extrajo con EtOAc (3 x 100 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para producir un residuo oscuro. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 5 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron hasta sequedad para producir 4-bromo-7-fluoro-3-metilbenzofurano (1.370 g, 42.2 %) como un sólido blanco. RMN de 1H (400 MHz, CDCls) 5 ppm 2.45 (3 H, d), 6.91 (1 H, dd), 7.29 (1 H, dd), 7.48 (1 H, q).
Compuesto intermedio A, 1-(5-Bromo-2-fluorofenoxi)propan-2-ona

Se añadió gota a gota 1-bromopropan-2-ona (8.61 g, 62.83 mmol) a 5-bromo-2-fluorofenol (10 g, 52.36 mmol) y K2CO3 (10.85 g, 78.53 mmol) en acetona (200 ml) a 60°C durante un período de 30 minutos bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 60 °C durante 2 horas. La mezcla de reacción se filtró a través de Celite. La mezcla de reacción se diluyó con EtOAc (300 ml) y se lavó con agua (200 ml x 3). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 1-(5-bromo-2-fluorofenoxi)propan-2-ona (10.90 g, 84 %) como un sólido blanco. RMN de 1H (400 MHz, DMSO-CÍ6) 57.34 - 7.09 (m, 3H), 5.00 (s, 2H), 2.16 (s, 3H).
Ejemplo 47. Enantiómero puro, isómero 1, (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol

Ejemplo 47, Enantiómero puro, Isómero 1
El producto crudo (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (200 mg, 0.64 mmol) se purificó mediante HpLC quiral preparativa en una columna Chiralpak IB, eluyendo isocráticamente con EtOH al 30% en heptano (modificado con dietilamina) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (50 mg, 25 % ) Pico 1 como un sólido blanco y (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (40 mg, 20 %) Pico 2 como un sólido blanco. RMN de 1H (400 MHz, DMSO-d6) 5 ppm 3.98 - 4.26 (2 H, m), 6.19 (1 H, d), 6.48 (1 H, d), 6.80 (1 H, s), 7.02 (1 H, s), 7.21 - 7.39 (2 H, m), 8.09 (1 H, s), 11.99 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 315.3; ácido, Hp Lc tR = 1.44 min. HPLC analítica quiral. Columna Chiral Amylose-SA, 0.46 x 15 cm, 5 ^M, fase móvil A, hexanos (IPA al 0.2 %), fase móvil B, etanol (70:30). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 2.71 min.
Ejemplo 48, Enantiómero puro, isómero 2, (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol

Ejemplo 48, Enantiómero puro, Isómero 2
El producto crudo (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (200 mg, 0.64 mmol) se purificó mediante HpLC quiral preparativa en una columna Chiralpak IB, eluyendo isocráticamente con EtOH al 30% en heptano (modificado con dietilamina) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para producir (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (50 mg, 25 % ) Pico 1 como un sólido blanco y (7-fluoro-3-(2,2,2-trifluoroetil)benzofuran-4-il)(1H-imidazol-2-il)metanol (40 mg, 20 %) Pico 2 como un sólido blanco. RMN de 1H (400 MHz, DMSO-CÍ6) 5 ppm 4.03 - 4.21 (2 H, m), 6.19 (1 H, d), 6.48 (1 H, d), 6.80 (1 H, s), 7.02 (1 H, s), 7.21 - 7.39 (2 H, m), 8.09 (1 H, s), 11.99 (1 H, s). LC-MS (Método A): m/z (ES+),
[M H]+ = ácido, HPLC tR = 1.99 min. HPLC analítica quiral. Columna Chiral Amylose-SA, 0.46 * 15 cm, 5 |jm, fase móvil A, hexanos (IPA al 0.2 %), fase móvil B, etanol (70:30). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 4.44 min.
Ejemplo 49, (3-etil-7-fluorobenzo[b]tiofen-4-il)(1H-imidazol-2-il)metanol

Ejemplo 49
El Ejemplo 49 se preparó de forma análoga al Ejemplo 40 utilizando 5-bromo-2-fluorobencenotiol y 1-bromobutan-2-ona). LC-MS (Método A): m/z (ES+), [M H]+ = 277; ácido, HPLC tR = 1.487 min.
Ejemplo 50. Enantiómero puro, isómero 1, (3-etil-7-fluorobenzo[b]tiofen-4-il)(1H-imidazol-2-il)metanol

Ejemplo 50, Enantiómero puro, Isómero 1
El Ejemplo 49 se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con EtOH al 6% en heptano (modificado con TEAA) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (3-etil-7-fluorobenzo[b]tiofen-4-il)(1H-imidazol-2-il)metanol (44.0 mg, 32.4 %) como un sólido blanco (Ejemplo 51, pico 1). RMN de 1H (400 MHz, DMSO) 8 1.29 - 1.33 (m, 3H), 3.06 - 3.22 (m, 2H), 6.27 - 6.28 (m, 1H), 6.52 - 6.53 (m, 1H), 6.89 (s, 2H), 7.19 - 7.23 (m, 1H), 7.49 - 7.52 (m, 2H), 11.97 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 277; ácido, HPLC tR = 1.484 min. HPLC analítica quiral. Columna ODH, 0.46 * 10 cm, 5 jm , fase móvil A, hexanos (0.1 A), fase móvil B, etanol (85:15). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 2.59 min.
Ejemplo 51. Enantiómero puro, isómero 2, (3-etil-7-fluorobenzo[b]tiofen-4-il)(1H-imidazol-2-il)metanol

Ejemplo 51, Enantiómero puro, Isómero 2
El Ejemplo 49 se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con EtOH al 6% en heptano (modificado con TEAA) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (3-etil-7-fluorobenzo[b]tiofen-4-il)(1H-imidazol-2-il)metanol (44.0 mg, 32.4 %) como un sólido blanco (Ejemplo 51, pico 2). RMN de 1H (400 Mh z , DMSO) 8 ppm 1.30 - 1.33 (m, 3H), 3.09 -3.24 (m, 2H), 6.27 -6.28 (m, 1H), 6.52 -6.53 (m, 1H), 6.89 (s, 2H), 7.19 -7.23 (m, 1H), 7.49 -7.52 (m, 2H), 11.97 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 277; ácido, HPLC tR = 1.487 min. HPLC analítica quiral. Columna ODH, 0.46 * 10 cm, 5 jm , fase móvil A, hexanos (0.1 A), fase móvil B, etanol (85:15). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 3.96 min.
Ejemplo 52. (7-Fluoro-3-metilbenzofuran-4-il)(1H-imidazol-2-il)metanol.

Ejemplo 52
El Ejemplo 52 se preparó de forma análoga al Ejemplo 40, usando 1-bromopropan-2-ona. RMN de 1H (400MHz, DMSO-d6) 5 ppm 2.26 (s,3H) , 6.27 - 6.24 (s, 2H), 6.84 - 6.95 (m, 2H), 7.13 - 7.18 (m, 1H), 7.25 - 7.28 (m, 1H), 7.80 (s, 1H), 8.21 (s, 0.5H).
Ejemplo 53. (4-fluorodibenzo[b,d]furan-1-il)(1H-imidazol-2-il)metanol

Se añadió HCl/EtOAc (10 ml) a (4-fluorodibenzo[b,d]furan-1-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (300 mg, 0.73 mmol). La mezcla resultante se agitó a temperatura ambiente durante 12 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó por Hp Lc preparativa (columna XSelect CSH Prep C18 OBD, sílice de 5 pm, 19 mm de diámetro, 150 mm de longitud), usando mezclas decrecientemente polares de agua (que contenía ácido fórmico al 0.1 %) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluorodibenzo[b,d]furan-1-il)(1H-imidazol-2-il)metanol (130 mg, 50.7 %) como un sólido blanco. RMN de 1H (300 MHz, metanol-d4) 5 ppm 6.55 (s, 1H), 7.16 (d, 2H), 7.34 (dd, 3H), 7.54 (t, 1H), 7.67 (d, 1H), 7.98 (d, 1H), 8.22 (s, 1.5H). LC-MS (método A): m/z (ES+), [M H]+ = 283; base, HPLC tR = 1.46 min.
Compuesto intermedio E, (4-Fluorodibenzo[b,d]furan-1-il(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol

Se añadió gota a gota BuLi (747 pl, 1.87 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (370 mg, 1.87 mmol) en THF (10 ml) a -78 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. Se añadió 4-fluorodibenzo[b,d]furan-1-carbaldehído (200 mg, 0.93 mmol) en (1 ml). La mezcla resultante se agitó a -78 °C durante 1 hora. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 x 10 ml), la capa orgánica se evaporó. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de agua en MeCN. Las fracciones puras se evaporaron a sequedad para producir (4-fluorodibenzo[b,d]furan-1-il)(1-((2-(trimetilsilil)etoxi)metil)-1 H-imidazol-2 -il)metanol (320 mg, 83 %) como un aceite de color amarillo pálido. LC-Ms (Método A): m/z (ES+), [M H]+ = 413; base, HPLC tR = 1.543 min.
Compuesto intermedio D, 4-Fluorodibenzo[b,d]furan-1-carbaldehído

Se añadió gota a gota BuLi (1.132 ml, 2.83 mmol) a 1-bromo-4-fluorodibenzo[b,d]furano (500 mg, 1.89 mmol) en THF (10 ml) a -78 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. Se añadió DMF (551 mg, 7.54 mmol). La mezcla resultante se agitó a -78 °C durante 1 hora. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 x 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y evaporó. El producto crudo se purificó
mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 2 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para producir 4-fluorodibenzo[b,d]furan-1-carbaldehído (360 mg, 89 %) como un sólido blanco.
Compuesto intermedio C, 1-Bromo-4-fluorodibenzo[b,d]furano

Se anadió gota a gota BuLi (0.482 ml, 1.20 mmol) a 1,3-difluoro-2-(2-yodofenoxi)benceno (100 mg, 0.30 mmol) en THF (10 ml) a -78 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. La mezcla resultante se dejó calentar a 0°C y se agitó durante 30 min. Después de enfriar a -78°C, se añadió gota a gota dibromuro de etileno (85 mg, 0.45 mmol) y se continuó agitando a baja temperatura durante 30 minutos más, luego se calentó a ta. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y evaporó. El residuo se purificó por TLC preparativa (éter de petróleo), para proporcionar 1-bromo-4-fluorodibenzo[b,d]furano (55.0 mg, 68.9 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO-d6 ) 6 ppm 7.43 -7.72 (m, 4H), 7.83 - 7.90 (m, 1H), 8.47 (d, 1H).
Compuesto intermedio B, 1,3-difluoro-2-(3-yodofenoxi)benceno

Se anadió gota a gota nitrito de sodio (206 mg, 2.94 mmol) en agua (5 ml) a 2-(2,6-difluorofenoxi)anilina (500 mg, 2.26 mmol) en HCl (6781 pl, 6.78 mmol) a 0 °C. La mezcla resultante se agitó a 0 °C durante 1 hora. Se añadió gota a gota yoduro de sodio (847 mg, 5.65 mmol) en agua (5 ml). Después de 0.5 h, la mezcla se calentó a 100 °C durante 1 h. Se enfrió a ta y se extrajo con EtOAc (10 ml * 3). La capa orgánica combinada se lavó con Na2S2O3.5 H2O (30 ml) y se secó con Na2SO4. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 2 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron hasta sequedad para producir 1,3-difluoro-2-(2-yodofenoxi)benceno (530 mg, 70.6 %) como un sólido blanco. RMN de 1H (400 MHz, DMSO-d6 ) 6 ppm 6.68 (dq, 1H), 6.92 (td, 1H), 7.28 - 7.47 (m, 4H), 7.91 (dd, 1H).
Compuesto intermedio A, 3-(2,6-difluorofenoxi)anilina

Se anadió 1-fluoro-2-nitrobenceno (6.51 g, 46.12 mmol) a 2,6-difluorofenol (5 g, 38.43 mmol) y K2CO3 (15.94 g, 115.30 mmol) en DMF (50 ml) a 25°C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 100 °C durante 12 horas. La mezcla de reacción se inactivó con agua (50 ml), se extrajo con EtOAc (3 * 50 ml), la capa orgánica se lavó con agua (100 ml * 3) y se secó sobre Na2SO4, se filtró y evaporó. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para producir 1,3-difluoro-2-(2-nitrofenoxi)benceno (9.50 g, 98 %) como un sólido amarillo. Se añadió Pd/C (4.02 g, 3.78 mmol) a 1,3-difluoro-2-(2-nitrofenoxi)benceno (9.5 g, 37.82 mmol) en etanol (100 ml) a 25 °C bajo atmósfera de hidrógeno. La mezcla resultante se agitó a temperatura ambiente durante 2 horas. La mezcla se filtró a través de una almohadilla de Celite. El disolvente se eliminó a presión reducida. Se usó 2-(2,6-difluorofenoxi)anilina (7.50 g, 90 %) como un aceite marrón en la siguiente etapa directamente sin purificación adicional. RMN de 1H (300 MHz, DMSO-d6 ) 6 ppm 5.09 (s, 2H), 6.32 - 6.48 (m, 2H), 6.70 - 6.85 (m, 2H), 7.18 - 7.40 (m, 3H).
E je m p lo 54. (4 -F lu o ro -1 -m e til-1 H -in d o l-7 - il) (1 H -im id a zo l-2 - il)m e ta n o l
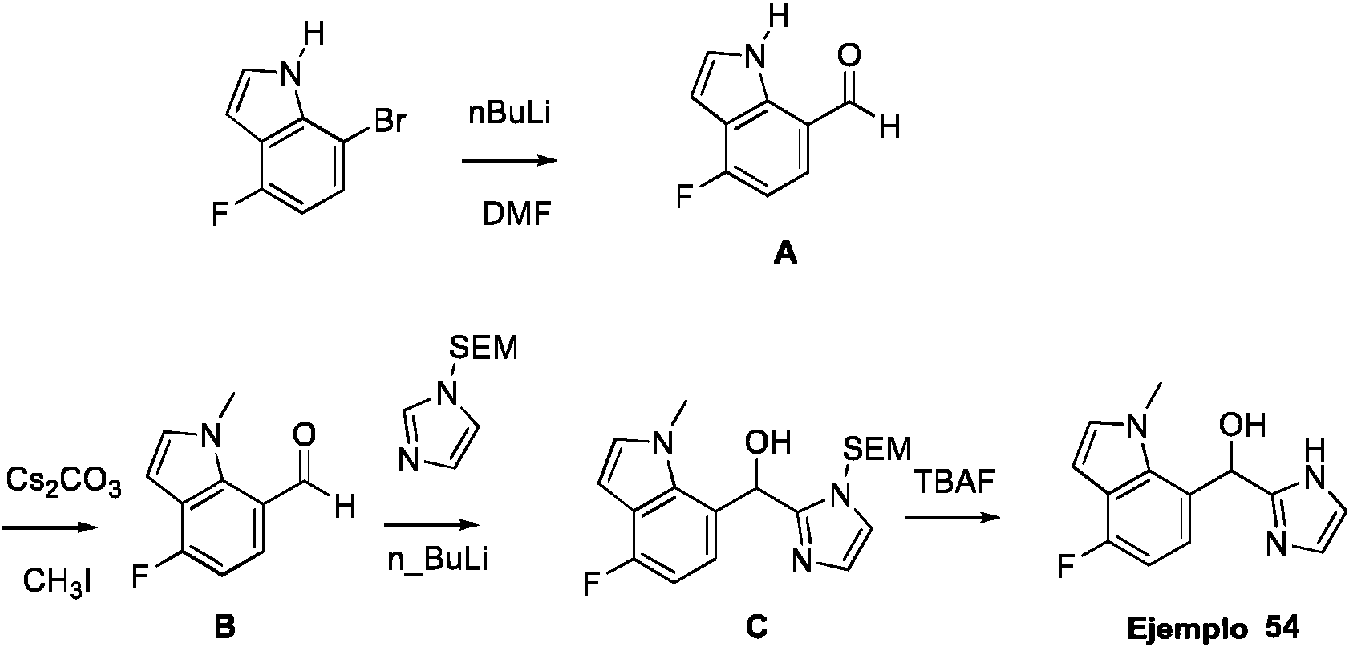
Se añadió lentamente TBAF (0.799 ml, 0.80 mmol) a (4-fluoro-1-metil-1H-indol-7-il)(1-((2-(trimetMsMil)etoxi)metil)-1H-imidazol-2- il)metanol (200 mg, 0.53 mmol) en THF (7 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a 50 °C durante 6 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 70 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para proporcionar (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (36.9 mg, 28.2 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 5 ppm 4.00 (s, 3H), 6.28 - 6.29 (m, 1H), 6.41 - 6.43 (m, 1H), 6.46 - 6.47 (m, 1H), 6.71 - 6.75 (m, 1H), 6.92 - 7.00 (m, 2H), 7.02 - 7.04 (m, 1H), 7.27 (s, 1H), 11.96 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 246; ácido, HPLC tR = 0.544 min.
Compuesto intermedio C, (4-Fluoro-1-metil-1H-indol-7-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol
Se añadió lentamente n-butil-litio (1.255 ml, 3.14 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (467 mg, 2.35 mmol) en THF (8 ml) a -78° C bajo atmósfera de nitrógeno. Una hora más tarde, se añadió 1-etil-4-fluoro-1H-indol-7-carbaldehído (300 mg, 1.57 mmol). La solución resultante se agitó a -78 °C durante 3 horas. La mezcla de reacción se inactivó con NH4Cl saturado (5 ml), se extrajo con EtOAc (2 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 100 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir (1-etil-4-fluoro-1H-indol-7-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (525 mg, 86 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 5 ppm -0.04 -0.00 (m, 12H), 0.81 -0.85 (m, 2H), 1.16 -1.18 (m, 1H), 1.20- 1.26 (m, 2H), 1.99 (s, 1H), 3.35 -3.47 (m, 3H), 4.19 (s, 1H), 4.32 (s, 1H), 5.25 -5.28 (m, 1H), 5.58 - 5.60 (m, 1H), 6.25 - 6.27 (m, 1H), 6.40 - 6.41 (m, 1H), 5.52 - 5.53 (m, 1H), 6.71 - 6.76 (m, 1H), 6.80 (s, 1H), 7.02 -7.05 (m, 1H), 7.31 - 7.34 (m, 2H). LC-MS (Método A): m/z (ES+), [M H]+ = 390.1; ácido, HPLC tR = 0.58 min.
Compuesto intermedio B, 4-fluoro-1-metil-1H-indol-7-carbaldehído
Se añadió Cs2CO3 (1997 mg, 6.13 mmol) a 4-fluoro-1H-indol-7-carbaldehído (500 mg, 3.06 mmol) en acetonitrilo (8 ml) calentado a 70 °C bajo atmósfera de nitrógeno. Media hora más tarde, se enfrió a ta y se añadió yodometano (870 mg, 6.13 mmol). La mezcla resultante se agitó a 70 °C durante 2 horas. La mezcla de reacción se filtró, el disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 4-fluoro-1-metil-1H-indol-7-carbaldehído (337 mg, 62.1 %) como un líquido blanco. RMN de 1H (300 MHz, DMSO) 5 ppm 4.08 -4.11 (m, 3H), 6.66 -6.67 (m, 1H), 7.01 - 7.07 (m, 1H), 7.49 (s, 1H), 7.80 - 7.83 (m, 1H), 10.32 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 177.95; ácido, HPLC tR = 1.407 min.
Compuesto intermedio A, 4-Fluoro-1H-indol-7-carbaldehído
Se añadió lentamente n-butil-litio (14.39 ml, 35.98 mmol) a 7-bromo-4-fluoro-1H-indol (2.2 g, 10.28 mmol) en THF (30 ml) a -78 °C bajo atmósfera de nitrógeno, se agitó durante 15 minutos. Luego se calentó lentamente a 5°C, se agitó durante 30 minutos. Se enfrió a -78°C, se añadió DMF (3.98 ml, 51.39 mmol), se calentó lentamente a 25°C, se agitó durante 2 horas. La mezcla de reacción se inactivó con agua (50 ml), se extrajo con Et2O (3 * 50 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 4-fluoro-1H-indol-7-carbaldehído (1.370 g, 82 %) como un sólido blanco. RMN de 1H (300 MHz, DMSO) 5 ppm 6.64 - 6.66 (m, 1H), 7.02 - 7.08 (m, 1H), 7.44 - 7.46 (m, 1H), 7.79 - 7.84 (m, 1H), 10.13 (s, 1H), 11.90 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 164; ácido, HPLC tR = 1.052 min.
Ejemplo 55. Enantiómero puro, isómero 1, (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol

Ejemplo 55, Enantiómero puro, Isómero 1
El Ejemplo 54, (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.22 g, 0.90 mmol), se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA, eluyendo isocráticamente con heptano al 10% en IPA (modificado con EtaN) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.090 g, 40.9 %, Pico 1, Ejemplo 55) como un sólido blanco y (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.110 g, 50.0 %, Pico 2 , Ejemplo 56) como un sólido blanco. RMN de 1H (300 MHz, CDCla) 8 ppm 3.75 (s, 3H), 6.44 (s, 1H), 6.56 (d, 1H), 6.67 (dt, 1H), 6.88 - 6.96 (m, 3H), 6.96 - 7.07 (m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 246; ácido, HPLC tR = 1.26 min. HPLC analítica quiral. CHIRALPAK AS-3, 0.46 * 5 cm, 5 |j M, fase móvil A, hexanos (0.2 A), fase móvil B, alcohol isopropílico (90:10). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 2.72 min.
Ejemplo 56. Enantiómero puro, isómero 2, (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol
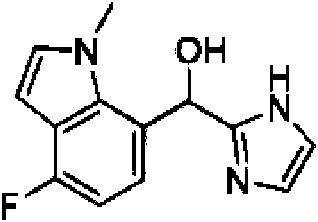
Ejemplo 56, Enantiómero puro, Isómero 2
El Ejemplo 54, (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.22 g, 0.90 mmol), se purificó mediante HPLC quiral preparativa en una columna Chiralpak IA , eluyendo isocráticamente con heptano al 10% en IPA (modificado con EtaN) como eluyente. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.090 g, 40.9 %, Pico 1, Ejemplo 55) como un sólido blanco y (4-fluoro-1-metil-1H-indol-7-il)(1H-imidazol-2-il)metanol (0.110 g, 50.0 %, Pico 2 , Ejemplo 56) como un blanco sólido. RMN de 1H (300 MHz, CDCla) 8 ppm 3.73 (s, 3H), 6.45 (s, 1H), 6.56 (d, 1H), 6.68 (dt, 1H), 6.86 -6.96 (m, 3H), 6.96 - 7.08 (m, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 246; ácido, HPLC tR = 1.26 min. HPLC analítica quiral. CHIRALPAK AS-3, 0.46 * 5 cm, 5 jm , fase móvil A, hexanos (0.2 % DEA), fase móvil B, alcohol isopropílico (90:10). Caudal 1.0 ml/min, detección UV-vis 254 nm. tR = 4.82 min.
Ejemplo 57. (1-etil-4-fluoro-1H-indol-7-il)(1H-imidazol-2-il)metanol

Ejemplo 57
El Ejemplo 55 se preparó de manera análoga al Ejemplo 54. RMN de 1H (300 MHz, DMSO) 8 ppm 1.22 -1.29 (m, 3H), 4.37 -4.46 (m, 1H), 4.55 -4.67(m, 1H), 6.27 (s, 2H), 6.50 -6.51(m, 1H), 6.71 -6.78 (m, 2H), 7.01 -7.08 (m, 2H), 7.35 - 7.36 (m, 1H), 11.93 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 259.9; ácido, HPLC tR = 1.375 min.
Ejemplo 58. (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)(1H-imidazol-2-il)metanol
Se añadió (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (120 mg , 0.31 mmol) a HCl/EtOAc (5 ml, 64.90 mmol) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 8 horas. La mezcla se evaporó para obtener el producto crudo como un sólido púrpura. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)(1H-imidazol-2-il) metanol (35.0 mg, 38.6 %) como un sólido púrpura. Rm N de 1H (300 MHz, DMSO) 8 ppm: 2.54 - 2.59 (2H, m), 2.94 -2.99 (2H, m) 3.96 -4.04 (1H, m), 4.32 -4.40 (1H, m), 6.25 (1H, s), 6.60 (1H, s), 6.23 (1H, s), 6.94 -6.99 (1H, m), 7.51 - 7.54 (1H, m), 7.62 (2H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 254,4; ácido, HPLC tR = 1.30 min.

Compuesto intermedio H, (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il) metanol

Se añadió gota a gota BuLi (112 mg, 1.75 mmol) a 1 -((2-(trimetilsilil)etoxi)metil)-1H-imidazol (209 mg, 1.05 mmol) en THF (10 ml) a -78 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a -78 °C durante 1 hora. Se añadió gota a gota 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carbaldehído (130 mg, 0.70 mmol). La solución resultante se agitó a -78 °C durante 2 horas. La mezcla de reacción se inactivó con NH4G saturado (10 ml), se extrajo con EtOAc (2 * 15 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un residuo incoloro. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)(1-((2-(trimetilsilil)etoxi)metil)-1 H-imidazol- 2 -il)metanol (150 mg, 55.7 %) como una goma incolora. lC-MS (Método A): m/z (ES+), [M H]+ = 384; ácido, HPLC tR = 0.84 min.
Compuesto intermedio G, 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carbaldehído

Se añadió dióxido de manganeso (65.0 mg, 0.75 mmol) a (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)metanol (140 mg, 0.75 mmol) en DCM (10 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 16 horas. El sólido se filtró. El filtrado se evaporó para proporcionar 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carbaldehído (130 mg, 94 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 186.0; ácido, HPLC tR = 0.97 min. Compuesto intermedio F, (2,3-dihidro-1H-pirrolo[1,2-a]indol-5-il)metanol

Se añadió LÍAIH4 (11.32 mg, 0.30 mmol) al ácido 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carboxílico (60 mg, 0.30 mmol) en THF (5 ml) enfriado a 0 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se inactivó con NH4Cl saturado (10 ml), se extrajo con EtOAc (2 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para producir (2,3-dihidro-1H-pirrolo[1,2-a]indol-5- il) metanol (42.0 mg, 75 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 188.2; ácido, HPLC tR = 0.81 min.
Compuesto intermedio E, ácido 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carboxílico
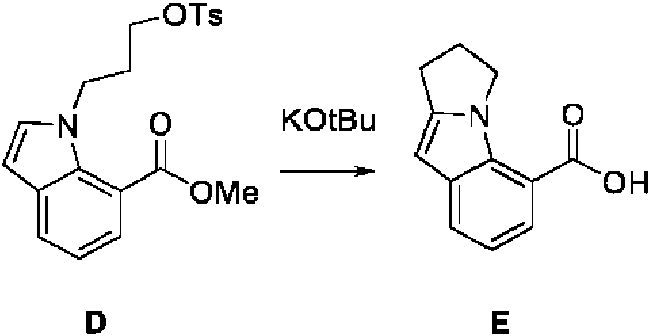
Se añadió 2-metilpropan-2-olato de potasio (46.3 mg, 0.41 mmol) a 2-(3-(tosiloxi)propil)-1H-indol-7-carboxilato de metilo (160 mg, 0.41 mmol) en BuOH (5 ml) a 25°C bajo atmósfera de nitrógeno. La solución resultante se agitó a 100 °C durante 30 minutos. La mezcla de reacción se acidificó con HCl 2M. La mezcla de reacción se diluyó con EtOAc (20 ml) y se lavó secuencialmente con salmuera saturada (10 ml). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar ácido 2,3-dihidro-1H-pirrolo[1,2-a]indol-5-carboxílico (62.0 mg, 74.6 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M H]+ = 202.2; ácido, HPLC tR = 0.86 min.
Compuesto intermedio D, 2-(3-(tosiloxi)propil)-1H-indol-7-carboxilato de metilo

Se añadió cloruro de 4-metilbenceno-1-sulfonilo (147 mg, 0.77 mmol) a 2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo (180 mg, 0.77 mmol) y Et3N (0.108 ml, 0.77 mmol) en DCM (10 ml) a 25 °C bajo atmósfera de nitrógeno. La solución resultante se agitó a temperatura ambiente durante 4 horas. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 50 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron hasta sequedad para proporcionar 2-(3-(tosiloxi)propil)-1H-indol-7-carboxilato de metilo (260 mg, 87 %) como un sólido blanco. LC-MS (Método A): m/z (ES+), [M h ]+ = 388.0; ácido, HPLC tR = 1.09 min.
Compuesto intermedio C, 2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo
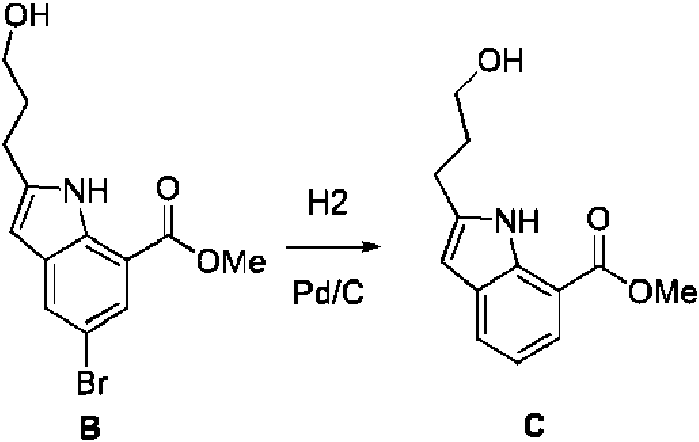
Pd/C (157 mg, 0.15 mmol) y 5-bromo-2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo (230 mg, 0.74 mmol) en MeOH (10 ml) se agitó en una atmósfera de nitrógeno a 1 atm y ta durante 4 horas. Se eliminó el disolvente. El
producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 80 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para producir 2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo (100 mg, 58.2 %) como una goma incolora. RMN de 1H (300 MHz, DMSO) 8 ppm: 1.17 (2H, m), 2.85 (2H, m), 3.48 (2H, m), 3.92(3H, s), 4.59 (1H, m), 6.28 (1H, m), 7.05 (1H, m), 7.66 -7.73 (2H, m), 10.85 (1H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 234.2; ácido, HPLC tR = 0.83 min.
Compuesto intermedio B, 5-bromo-2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo

Se añadió NaAuCl4.2 H2O (49.3 mg, 0.13 mmol) a 2-amino-5-bromo-3-(5-hidroxipent-1-in-1-il)benzoato de metilo (400 mg, 1.28 mmol) en etanol (2 ml) a 25°C. La solución resultante se agitó a 25 °C durante 16 horas. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 80 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar 5-bromo-2-(3-hidroxipropil)-1H-indol-7-carboxilato de metilo (230 mg, 57.5 %) como un sólido naranja. LC-MS (Método A): m/z (ES+), [M H]+ = 312.0; ácido, HPLC tR= 1.11 minutos
Compuesto intermedio A, 2-amino-5-bromo-3-(5-hidroxipent-1-inil)benzoato de metilo

Se suspendieron pent-4-in-1-ol (35.4 mg, 0.42 mmol), 2-amino-5-bromo-3-yodobenzoato de metilo (100 mg, 0.28 mmol), PdCl2(PPha)2 (45.9 mg, 0.06 mmol), DIEA (0.098 ml, 0.56 mmol) y yoduro de cobre (I) (10.70 mg, 0.06 mmol) se suspendieron en THF (2 ml) bajo atmósfera de nitrógeno. La reacción se agitó a temperatura ambiente durante 2 horas. Se eliminó el disolvente. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 80 % de EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para producir 2-amino-5-bromo-3-(5-hidroxipent-1-in-1-il)benzoato de metilo (80 mg, 91 %) como un sólido marrón (80 mg, 91 % ). RMN de 1H (300 MHz, CDCh-d6 ) 8 ppm 1.67 - 1.76 (2H,m), 2.50 -2.57 (2H, m), 3.50 -3.82 (2H, m), 3.82 (3H, s), 4.58 (1H, tr), 6.76 (2H, brs), 7.52 (1H, d), 7.79 (1H, d). LC-MS (Método A): m/z (ES+), [M H]+ = 312.2; ácido, HPLC tR= 1.54 min.
E je m p lo 59, (4 -F lu o ro -8 -m e tiln a fta le n -1 - il) (1 H -im id a z o l-2 - il)m e ta n o l

Se añadió HCl/EtOAc (15 ml) a (4-fluoro-8-metilnaftalen-1-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (250 mg , 0.65 mmol) a ta. La mezcla resultante se agitó a temperatura ambiente durante 12 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó por HPLC preparativa (columna XSelect CSH Prep C18 OBD, sílice de 5 ^m, 19 mm de diámetro, 150 mm de longitud), utilizando mezclas decrecientemente polares de agua (que contiene ácido fórmico al 0.1 %) y MeCN como eluyentes. Las fracciones que contenían el compuesto deseado se evaporaron a sequedad para proporcionar (4-fluoro-8-metilnaftalen-1-il)(1H-imidazol-2-il)metanol (70.0 mg, 35.7 %) como un sólido blanco. RMN de 1H (400 MHz, metanol-d4) 5 ppm 3.01 (3 H, s), 7.08 (1 H, s), 7.12 - 7.25 (3 H, m), 7.34 (1 H, t), 7.45 - 7.54 (2 H, m), 8.07 (1 H, dt), 8.33 (1 H, s). LC-MS (Método A): m/z (ES+), [M H]+ = 257; base, HPLC tR = 1.411 min.
Compuesto intermedio J, (4-fluoro-8-metilnaftalen-1-il)(1-((2-(trimetilsilil)etoxi)metil-1H-imidazol-2-il)metanol

Se añadió gota a gota BuLi (0.638 ml, 1.59 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (316 mg, 1.59 mmol) en THF (10 ml) a -78 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. Se añadió 4-fluoro-8-metil-1-naftaldehído (150 mg, 0.80 mmol) a -78°C. La mezcla resultante se agitó a -78 °C durante 1 hora. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y evaporó. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 0 a 100 % de agua en MeCN. Las fracciones puras se evaporaron a sequedad para producir (4-fluoro-8-metilnaftalen-1-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (270 mg, 88 %) como un sólido marrón. LC-MS (Método A): m/z (ES+), [M H]+ = 387; ácido, HPLC tR = 0.888 min.
C o m p u e s to in te rm e d io I, 4 -F lu o ro -8 -m e til-1 -n a fta ld e h íd o

Se añadió el aducto PdCh(dppf)-CH2Cl2 (25.3 mg, 0.03 mmol) a trifluorometanosulfonato de 5-fluoro-8-formilnaftalen-1-ilo (50 mg, 0.16 mmol), trimetilboroxina (97 mg, 0.78 mmol) y K2CO3 (64.3 mg, 0.47 mmol) en DME (2 ml) y agua (1 ml) bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 85 °C durante 2 horas. La mezcla de reacción se diluyó con EtOAc (10 ml) y se lavó con agua (5 ml x 3). La capa orgánica se secó sobre Na2SO4, se filtró y evaporó. El residuo se purificó por TLC preparativa (EtOAc:éter de petróleo = 1:10), para producir 4-fluoro-8-metil-1-naftaldehído (20.00 mg, 68.5 %) como un sólido amarillo pálido.
Compuesto intermedio H, trifluorometanosulfonato de 5-fluoro-8-formilnaftalen-1-ilo

Se añadió Tf2O (1.237 ml, 7.32 mmol) a 4-fluoro-8-hidroxi-1-naftaldehído (1.16 g, 6.10 mmol) y piridina (0.740 ml, 9.15 mmol) en DCM (20 ml) a 0 °C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con DCM (3 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó. El producto crudo se purificó mediante cromatografía ultrarrápida en sílice, gradiente de elución de 0 a 2 %. EtOAc en éter de petróleo. Las fracciones puras se evaporaron a sequedad para proporcionar trifluorometanosulfonato de 5-fluoro-8-formilnaftalen-1-ilo (1.100 g, 56.0 %) como un sólido naranja. RMN de 1H (300 MHz, DMSO-d6) 5 ppm 7.73 (1 H, m), 7.92 (1 H, m), 7.99 - 8.09 (1 H, m), 8.24 (1 H, m), 8.37 (1 H, m), 10.69(1 H, s).
Compuesto intermedio G, 4-Fluoro-8-hidroxi-1-naftaldehído

Se añadió gota a gota tricloruro de boro (2.283 ml, 2.28 mmol) a 8-(benciloxi)-4-fluoro-1-naftaldehído (320 mg, 1.14 mmol) en DCM (10 ml) a 0°C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a 0 °C durante 50 minutos. La mezcla de reacción se inactivó con HCl 2 M (5 ml), se extrajo con DCM (3 * 5 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 2 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para producir 4 fluoro-8-hidroxi-1-naftaldehído (200 mg, 92 %) como un sólido naranja.
Compuesto intermedio F, 8-(Benciloxi)-4-fluoro-1-naftaldehído

Se añadió gota a gota BuLi (49.3 mg, 0.77 mmol) a 5-(benciloxi)-4-bromo-1-fluoronaftaleno (170 mg, 0.51 mmol) en THF (10 ml) a -78 °C durante un período de 0.5 horas bajo atmósfera de nitrógeno. Se añadió DMF (0.159 ml, 2.05 mmol). La solución resultante se agitó a -78 °C durante 1 hora. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 * 5 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar 8-(benciloxi)-4-fluoro-1-naftaldehído (130 mg, 90 %) como un sólido amarillo. El producto se usó en la siguiente etapa directamente sin purificación adicional.
C o m p u e s to in te rm ed io E, 5 -(B e n c ilo x i)-4 -b ro m o -1 -flu o ro n a fta le n o

Se añadió gota a gota alfa-bromotolueno (180 mg, 1.05 mmol) a 8-bromo-5-fluoronaftalen-1-ol (230 mg, 0.95 mmol) y CS2CO3 (622 mg, 1.91 mmol) en acetona (5 ml) a 25°C bajo atmósfera de nitrógeno. La mezcla resultante se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se inactivó con agua (10 ml), se extrajo con EtOAc (3 * 5 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido amarillo pálido. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para producir 5-(benciloxi)-4-bromo-1-fluoronaftaleno (170 mg, 53.8 %) como un sólido amarillo pálido.
Compuesto intermedio D, 8-Bromo-5-fluoronaftalen-1-ol
tnbromuro de


C D
Se añadió lentamente tribromuro de trimetilfenilamonio (155 mg, 0.41 mmol) a 8-bromo-5-fluoro-3,4-dihidronaftalen-1(2H)-ona (100 mg, 0.41 mmol) en THF (5 ml) a 25 °C. La mezcla resultante se agitó a temperatura ambiente durante 45 minutos. La mezcla de reacción se inactivó con agua (5 ml), se extrajo con EtOAc (3 * 5 ml), la capa orgánica se lavó con agua (20 ml) y se secó sobre Na2SO4, se filtró y se evaporó para obtener un aceite de color amarillo pálido. El residuo se disolvió en DMF (5 ml) y se añadieron bromuro de litio (71.5 mg, 0.82 mmol) y carbonato de litio (97 mg, 1.32 mmol). La suspensión se calentó a 130 °C durante 1 h, se enfrió a ta y el sólido se eliminó por filtración (EtOAc (20 ml), el filtrado se lavó con agua (3 * 10 ml) y se secó con Na2SO4. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 5 % de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para producir 8-bromo-5-fluoronaftalen-1-ol (63.0 mg, 63.5 %) como un sólido amarillo pálido.
Compuesto intermedio C, 8-Bromo-5-fluoro-3,4-dihidronaftalen-1(2H)-ona

Se añadió PPA (20 ml, 10.72 mmol) a ácido 4-(5-bromo-2-fluorofenil)butanoico (2.8 g, 10.72 mmol). La mezcla resultante se agitó a 120 °C durante 2 horas. La mezcla de reacción se vertió en agua con hielo (100 ml), se extrajo con EtOAc (3 x 30 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para producir un sólido amarillo pálido. El residuo se purificó por TLC preparativa (EtOAc:éter de petróleo = 1:20), para proporcionar 8-bromo-5-fluoro-3,4-dihidronaftalen-1(2H)-ona (0.50 g, 19.1 %) como un sólido naranja.
Compuesto intermedio B, ácido 4-(5-Bromo-2-fluorofenil)butanoico
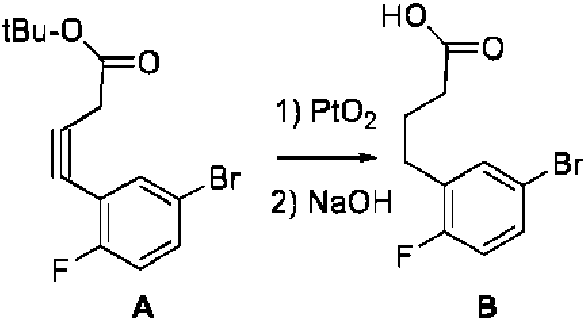
Se añadió óxido de platino (IV) (1.08 mg, 4.79 pmol) a 4-(5-bromo-2-fluorofenil)but-3-inoato de tert-butilo (50 mg, 0.16 mmol), hidróxido de amonio (0.056 mg, 1.60 pmol ) en MeOH (5 ml) a 25 °C bajo atmósfera de hidrógeno. La mezcla resultante se agitó a temperatura ambiente durante 12 horas. La mezcla se filtró a través de una almohadilla de Celite. El disolvente se eliminó a presión reducida. Se usó 4-(5-bromo-2-fluorofenil)butanoato de tert-butilo (45.0 mg, 89 %)
como un aceite de color amarillo pálido en la siguiente etapa directamente sin purificación adicional. Se añadió NaOH (10 ml, 10.00 mmol) a 4-(5-bromo-2-fluorofenil)butanoato de tert-butilo (140 mg, 0.44 mmol) en MeOH (5 ml) a 25 °C. La mezcla resultante se agitó a 50 °C durante 4 horas. La mezcla de reacción se ajustó a pH = 3-4 con HCl 2 M. La mezcla de reacción se extrajo con EtOAc (10 ml x 2). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para proporcionar ácido 4-(5-bromo-2-fluorofenil)butanoico (100 mg, 87 %) como un aceite amarillo.
Compuesto intermedio A, 4-(5-bromo-2-fluorofenil)but-3-inoato de tert-butilo

Se añadió yoduro de cobre (I) (7.18 mg, 0.04 mmol) a 4-bromo-2-etinil-1-fluorobenceno (150 mg, 0.75 mmol) y 2-diazoacetato de tert-butilo (107 mg, 0.75 mmol) en acetonitrilo (10 ml) a temperatura ambiente. La mezcla resultante se agitó a temperatura ambiente durante 1 hora. La mezcla de reacción se diluyó con EtOAc (25 ml) y se lavó secuencialmente con agua (10 ml x 3), salmuera saturada (10 ml x 2). La capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 100 a 1% de éter de petróleo en EtOAc. Las fracciones puras se evaporaron a sequedad para proporcionar 4-(5-bromo-2-fluorofenil)but-3-inoato de tert-butilo (100 mg, 42.4 %) como un aceite amarillo.
Ejemplo 60. (1-etil-5-fluoro-indolizin-8-il)-(1H-imidazol-2-il)metanol
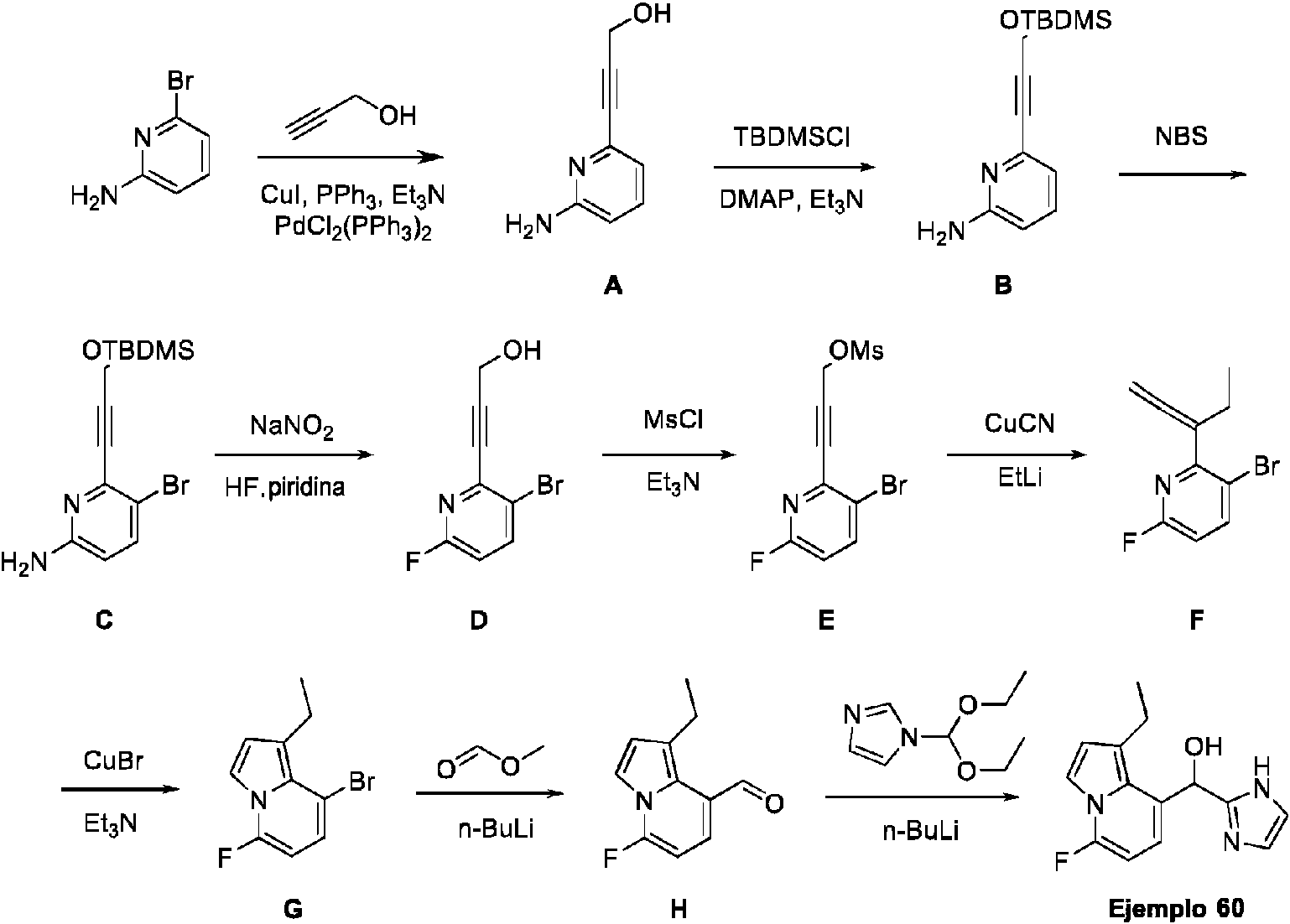
Se añadió n-BuLi (0.0960 ml, 0.241 mmol, solución 2.50 M en hexanos) a una mezcla de 1-(dietoximetil)imidazol (41.0 mg, 0.241 mmol) en THF (0.500 ml) a -45 °C. La mezcla se agitó durante 15 m y luego se enfrió a -60°C. Se añadió una solución de 1-etil-5-fluoro-indolizina-8-carbaldehído (23.0 mg, 0.120 mmol) en THF (0.500 ml). La mezcla se agitó durante 10 minutos a -60 °C y luego se calentó a 0 °C durante 1 hora. La mezcla se diluyó con HCl 0.100 M y EtOAc y la fase orgánica se extrajo con HCl 0.100 M (4 x 10.0 ml). Las fases acuosas combinadas se diluyeron cuidadosamente con NaHCO3 saturado y la mezcla acuosa se extrajo con EtOAc (4 x 10.0 ml). Las fases orgánicas combinadas se secaron (MgSO4) y se concentró a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con DCM y MeOH (0 - 10 %) para proporcionar el compuesto del título como un sólido (12.2 mg, 38 %). RMN de 1H (500 MHz, MeOD) 5 ppm 1.17 (t, J = 7.5 Hz, 3H), 2.81 (ddd, J = 15.0, 7.5, 3.0 Hz, 2H), 6.15 (dd, J = 7.4, 6.1 Hz, 1H), 6.33 (s, 1H), 6.62 (t, J = 6.8 Hz, 1H), 6.73 (d, J = 2.9 Hz, 1H), 7.00 (s, 2H), 7.37 (d, J = 2.8 Hz, 1H); LC-MS (Método C): m/z (ES+), [M H]+ = 260.27; HPLC tR = 2.10 m.
C o m p u e s to in te rm ed io H, 1 -e til-5 -flu o ro -in d o liz in a -8 -ca rb a ld e h íd o
Se añadió n-BuLi (0.061 ml, 0.152 mmol, solución 2.50 M en hexanos) a una solución enfriada de 8-bromo-1-etil-5-fluoro-indolizina (35.0 mg, 0.145 mmol) en THF (1.40 ml) a - 78°C. La solución resultante se agitó durante 20 min a -78 °C y se añadió formiato de metilo (0.0180 ml, 0.289 mmol). La mezcla se agitó durante 10 min a -78 °C y luego se calentó a 23 °C. La mezcla se diluyó con agua y la fase acuosa se extrajo con EtOAc (3 x 10.0 ml). Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 - 60 %) para proporcionar el compuesto del título como un sólido (16.0 mg, 58 %). RMN de 1H (500 MHz, CDCk) 8 1.29 (t, J = 5.5 Hz, 3H), 3.09 (q, J = 7.5 Hz, 2H), 6.23 (dd, J = 7.5, 5.4 Hz, 1H), 6.86 (d, J = 2.8 Hz, 1H), 7.39 (dd, J = 7.5, 6.2 Hz, 1H), 7.42 (d, J = 2.9 Hz, 1H), 10.14 (s, 1H); LC-MS (Método C): m/z (ES+), [M H]+ 192.21; (A05), HPLC tR = 2.69 min.
Compuesto intermedio G, 8-Bromo-1-etil-5-fluoro-indolizina
Se añadieron CuBr (7.90 mg, 0.0560 mmol) y Et3N (0.0260 ml, 0.186 mmol) a una mezcla de 3-bromo-2-(1-etilpropa-1,2-dienil)-6-fluoropiridina (45.0 mg, 0.186 mmol) en DMA (2.10 ml). La mezcla se agitó durante 5 horas a 130 °C con protección de la luz. La mezcla se enfrió a 23 °C y se diluyó con NH4Cl saturado. La fase acuosa se extrajo con EtOAc (3 x 10 ml) y las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 - 40 %) para proporcionar el compuesto del título como un aceite (36.0 mg, 80 %). RMN de 1H (500 MHz, CDCk) 8 1.29 (t, J = 7.5 Hz, 3H), 3.13 (q, J = 7.5 Hz, 2H), 5.94 (dd, J = 7.6, 5.6 Hz, 1H), 6.75 (d, J = 2.8 Hz, 1H), 6.78 (dd, J = 7.6, 5.5 Hz, 1H), 7.29 (d, J = 2.8 Hz, 1H); m/z: sin ionización.
Compuesto intermedio F, 3-Bromo-2-(1-etilpropa-1,2-dienil)-6-fluoro-piridina
Una suspensión de CuCN (317 mg, 3.54 mmol) en THF seco (7.00 ml) se enfrió a -50°C bajo atmósfera de nitrógeno. A esto se le añadió EtLi (7.08 ml, 3.54 mmol, 0.500 M en benceno) y la suspensión se agitó durante 30 min a -50 °C. Se transfirió con una cánula una solución enfriada de metanosulfonato de 3-(3-bromo-6-fluoro-2-piridil)prop-2-inilo (436 mg, 1.42 mmol) en THF seco (7.00 ml) a -50 °C. La mezcla se agitó durante 2 horas a -78 °C y se calentó a 23 °C. La mezcla se diluyó con una mezcla 10:1 de NH4Cl y NH4OH (15.0 ml). La fase acuosa se extrajo con EtOAc (3 x 15.0 ml) y las fases orgánicas combinadas se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 - 40 %) para proporcionar el compuesto del título como un aceite (217 mg, 63 %). RMN de 1H (500 MHz, CDCls) 8 1.01 (t, J = 7.4 Hz, 3H), 2.40 (ddq, J = 10.9, 7.3, 3.5 Hz, 2H), 4.99 (t, J = 3.5 Hz, 2H), 6.62 (dd, J = 8.5, 3.8 Hz, 1H), 7.84 (dd, J = 8.5, 7.3 Hz, 1H); LC-MS (Método C): m/z (ES+), [M H]+ = 242.12, 244.17; HPLC tR = 2.83 m.
Compuesto intermedio E, metanosulfonato de 3-(3-bromo-6-fluoro-2-piridil)prop-2-inilo
Se añadieron DIPEA (0.315 ml, 1.81 mmol) y cloruro de metanosulfonilo (0.140 ml, 1.81 mmol) a una mezcla de 3-(3-bromo-6-fluoro-2-piridil)prop-2-in-1-ol (104 mg, 0.452 mmol) en DCM (1.90 ml) a 0°C bajo atmósfera de nitrógeno. La mezcla se agitó a 23 °C durante 1 h y luego se diluyó con HCl 1 N acuoso. La fase acuosa se extrajo con EtOAc (3 x 10.0 ml) y las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 - 60 %) para proporcionar el compuesto del título como un aceite (108 mg, 77 %). Rm N de 1H (500 MHz, CDCla) 83.24 (s, 3H), 5.17 (s, 2H), 6.91 (dd, J = 8.7, 3.4 Hz, 1H), 8.00 (dd, J = 8.7, 6.9 Hz, 1H); LC-MS (Método C): m/z (ES+), [M H]+ = 308.09, 310.10; HPLC tR = 2.17 m.
Compuesto intermedio D, 3-(3-Bromo-6-fluoro-2-piridil) prop-2-in-1-ol
Se añadió NaNO2 (63.1 mg, 0.914 mmol) a una solución de (dimetil)silil]oxiprop-1-inil]piridin-2-amina (208 mg, 0.609 mmol) en HF.piridina (0.900 ml) a -10 °C. La mezcla se agitó durante 1 hora a -10°C y luego se diluyó con agua. La fase acuosa se extrajo con DCM (3 x 10 ml) y las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 - 60 %) para proporcionar el compuesto del título como un sólido (104.0 mg, 74 %). RMN de 1H (500 MHz, CDCls) 82.07 (t, J = 6.4 Hz, 1H), 4.59 (d, J = 6.3 Hz, 2H), 6.85 (dd, J = 8.6 , 3.4 Hz, 1H), 7.97 (dd, J = 8.6 , 7.0 Hz, 1H); LC-MS (Método C): m/z (ES+), [M H]+ = 230.15, 232.15; HPLC tR = 1.59 min.
Compuesto intermedio C, 5-Bromo-6-[3-[tert-butil(dimetil)silil]oxiprop-1-inil]piridin-2-amina
Se añadió NBS (20.7 mg, 0.117 mmol) a una solución de 6-[3-[tert-butil(dimetil)silil]oxiprop-1-inil]piridin-2-amina (30.0 mg, 0.114 mmol) en MeOH (0.350 ml) a 0°C. La mezcla se agitó durante 20 min a 23 °C y luego se diluyó con agua. La fase acuosa se extrajo con DCM (3 x 10 ml) y las fases orgánicas combinadas se lavaron con salmuera (50.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida para proporcionar el compuesto del título como un sólido (208 mg, 100%). El producto se usó en la siguiente etapa sin purificación adicional. RMN de 1H (500 MHz, CDCls) 80.19 (s, 6 H), 0.94 (s, 9H), 4.60 (s, 4H), 6.36 (d, J= 8.7 Hz, 1H), 7.55 Hz, 1H); LC-MS (Método C): m/z (ES+), [M H]+ = 341.20, 343.21; HPLC tR = 3.00 m.
C o m p u e s to in te rm e d io B, 6 -[3 -[te rt-b u til(d im e til)s ilM ]o x ip ro p -1 -in il]p ir id in -2 -a m in a
Se añadieron Et3N (0.223 ml, 1.604 mmol), cloruro de terc-butildimetilsililo (174.6 mg, 1.158 mmol) y 4-dimetilaminopiridina (10.9 mg, 0.0890 mmol) a una mezcla de 3-(6-amino-2-piridil)prop-2-in-1-ol (132 mg, 0.891 mmol) en DCM (7.20 ml) a 0°C bajo atmósfera de nitrógeno. La mezcla se agitó a 23 °C durante 6 h y luego se diluyó con agua. La fase acuosa se extrajo con DCM (3 * 10.0 ml) y las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con hexanos y EtOAc (0 -100 %) para proporcionar el compuesto del título como un sólido (168.0 mg, 72 %). RMN de 1H (500 MHz, CDCh) 50.14 (s, 6 H), 0.91 (s, 9H), 4.52 (s, 2H), 4.55 (s, 2H), 6.43 (dd, J = 8.3, 0.6 Hz, 1H), 6.80 (dd, J = 7.4, 0.6 Hz, 1H), 7.35 (dd, J = 8.3, 7.4 Hz, 1H); m/z (ES+), [M H]+ = 263.34; (B05), HPLC tR = 2.84 min.
Compuesto intermedio A, 3-(6-Amino-2-piridil)prop-2-in-1-ol
Se añadió alcohol propargílico (0.101 ml, 1.73 mmol) a una mezcla de PdCl2(PPh3)2 (40.6 mg, 0.0580 mmol), HPP3 (33.4 mg, 0.127 mmol), Cul (22.0 mg, 0.115 mmol) y 2-amino-6-bromopiridina (200.0 mg, 1.16 mmol) en THF (4.20 ml) y Et3N (0.242 ml, 1.73 mmol). La mezcla se agitó durante 2 días a 23 °C. La mezcla se diluyó con NH4Q saturado y la fase acuosa se extrajo con EtOAc (2 x 10.0 ml). Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice, eluyendo con DCM y MeOH (0 - 10 %) para proporcionar el compuesto del título como un sólido (112 mg, 65 %). RMN de 1H (500 MHz, MeOD) 54.38 (s, 2H), 6.53 (d, J = 8.4 Hz, 1H), 6.72 (d, J = 7.2 Hz, 1H), 7.40 (dd, J = 8.4, 7.3 Hz, 1H); LC-MS (Método C) m/z (ES+), [M H]+ = 149.14; HPLC tR = 0.56 min.
Ejemplo 61. (3-cloro-8-fluoro-indolizin-5-il)-(1H-imidazol-2-il)metanol

Se añadió 1.00 de TBAF 1 M en THF (0.260 ml, 0.260 mmol) a una mezcla de (3-cloro-8-fluoro-indolizin-5-il)-[1-(2-trimetilsililetoximetil)imidazol-2-il]metanol (34.0 mg, 0.0900 mmol) en THF (2.50 ml) a 23 °C bajo atmósfera de nitrógeno. La mezcla se calentó a reflujo durante 3 h y se diluyó con NaHCO3 saturado (10.0 ml). La fase acuosa se extrajo con EtOAc (3 * 25.0 ml) y las fases orgánicas combinadas se lavaron con salmuera (25.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 12 g) con hexano y EtOAc (0 - 100 %) para proporcionar el compuesto del título como un sólido (11.0 mg, 48 %). RMN de 1H (500 MHz, MeOD) 57.14 (s, 1H), 7.06 (s, 2H), 6.72 (d, J = 4.3 Hz, 1H), 6.69 (d, J = 4.3 Hz, 1H), 6.42 (d, J = 9.2 Hz, 2H). LC-MS (Método C): m/z (ES+), [M H]+ = 266.0. HPLC tR = 2.23 m.
Compuesto intermedio F. (3-cloro-8-fluoro-indolizin-5-il)-[1-(2-trimetilsililetoximetil)imidazol-2-il]metanol
Se añadió n-BuLi 2.50 M en THF (0.260 ml, 0.640 mmol) a una mezcla de 2-(imidazol-1-ilmetoxi)etil-trimetilsilano (127 mg, 0.640 mmol) en THF (4.00 ml) a -78 °C bajo atmósfera de nitrógeno. La mezcla se agitó a -78 °C durante 1 hora y se añadió gota a gota a una solución enfriada de 3-cloro-8-fluoro-indolizina-5-carbaldehído (97.0 mg, 0.490 mmol) en THF (2 ml) a -78 °C La mezcla se agitó a -78 °C durante 20 min y se calentó a 0 °C durante 20 min. La mezcla se diluyó con NH4G (10.0 ml). La fase acuosa se extrajo con EtOAc (3 x 25.0 ml) y las fases orgánicas combinadas se lavaron con salmuera (50.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 24 g) con hexano y EtOAc (0 - 70 %) para proporcionar el compuesto del título como un sólido (36.0 mg, 19 %). RMN de 1H (500 MHz, CDCh) 57.19 (s, 1h ), 7.05 (d, J = 2.9 Hz, 2H), 6.72 - 6.66 (m, 2H), 6.36 -6.24 (m, 2H), 5.27 (q, J = 10.8 Hz, 2H), 3.95 (s, 1H), 3.56 - 3.35 (m, 2H), 0.79 (ddd, J = 10.3, 6.4, 5.4 Hz, 3H), -0.04 (s, 9H). LC-MS (Método C): m/z (ES+), [M H]+ = 396.0. HPLC tR = 3.05 min.
C o m p u e s to in te rm ed io E. 3 -C lo ro -8 -flu o ro - in d o liz in a -5 -ca rb a ld e h íd o
Se añadieron tamices moleculares de 4 A a una mezcla de NMO (144 mg, 1.23 mmol) y (3-cloro-8-fluoro-indolizin-5-il)metanol (123 mg, 0.620 mmol) en DCM (5.00 ml) a 23° C bajo atmósfera de nitrógeno. La mezcla se agitó durante 20 min. Se añadió TPAP (10.8 mg, 0.0300 mmol) y la mezcla se agitó durante 10 min y luego se filtró a través de una capa de Florisil. El filtrado se concentró a presión reducida y el producto se purificó por cromatografía en gel de sílice (cartucho de 24 g) con hexano y EtOAc (0 - 30 %) para proporcionar el compuesto del título como un sólido (97.0 mg; 80 %). RMN de 1H (500 MHz, CDCh) 8 10.86 (d, J = 0.5 Hz, 1H), 7.47 (dd, J = 8.0, 5.6 Hz, 1H), 6.91 (d, J = 4.4 Hz, 1H), 6.84 (d, J = 4.4 Hz, 1H), 6.51 (dd, J = 9.3, 8.0 Hz, 1H). LC-MS (Método C): m/z (ES+), [M H]+ = 198.1. HPLC tR = 2.67 min.
Compuesto intermedio D. (3-Cloro-8-fluoro-indolizin-5-il)metanol
Se añadió LAH 1.00 M en THF (0.620 ml, 0.620 mmol) a una mezcla de 3-cloro-8-fluoro-indolizina-5-carboxilato de etilo (150 mg, 0.620 mmol) en THF (10.0 ml) a 0 °C bajo atmósfera de nitrógeno. La mezcla se agitó a 23 °C durante 20 min y luego se diluyó con agua (1.00 ml), NaOH 1 M (2.00 ml) y agua (1.00 ml). La mezcla se filtró a través de Celite y el sólido se lavó con EtOAc (10.0 ml). El filtrado se concentró a presión reducida para proporcionar el compuesto del título como un sólido (123 mg; 99%). RMN de 1H (500 MHz, CDCla) 86.72 (d, J = 4.3 Hz, 1H), 6.68 (d, J = 4.3 Hz, 1H), 6.52 (dd, J = 7.6, 5.4 Hz, 1H), 6.33 (dd, J = 9.8, 7.6 Hz, 1H), 5.16 (s, 2H). LC-MS (Método C): m/z (ES+), [M H]+ = 200.2. tR = 2.53 min.
Compuesto intermedio C. 3-cloro-8-fluoro-indolizina-5-carboxilato de etilo
Se añadió gota a gota una solución de NCS (419 mg, 3.14 mmol) en DCM (10.0 ml) a una mezcla de 8-fluoroindolizina-5-carboxilato de etilo (591 mg, 2.85 mmol) en DCM (20.0 ml) a 0° C bajo atmósfera de nitrógeno. La mezcla se agitó en la oscuridad a 0 °C durante 3.5 h y luego se diluyó con Na2S2O3 1 M (50.0 ml). La fase acuosa se extrajo con DCM (3 x 50.0 ml) y las fases orgánicas combinadas se lavaron con salmuera (50.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 40 g) con hexano (100 %) para proporcionar el compuesto del título como un sólido (656 mg, 95 %). RMN de 1H (500 MHz, CDCta) 87.02 (dd, J = 7.7, 5.2 Hz, 1H), 6.82 (d, J = 4.3 Hz, 1H), 6.79 (d, J = 4.4 Hz, 1H), 6.40 (dd, J = 9.7, 7.7 Hz, 1H), 4.45 (q, J = 7.2 Hz, 2H), 1.41 (t, J = 7.2 Hz, 4H). LC-MS.
(Método C): m/z (ES+), [M H]+ = 242.0. HPLC tR = 3.04 m.
Compuesto intermedio B. 8-fluoroindolizina-5-carboxilato de etilo
Se añadió CuCl (0.760 g, 7.67 mmol) a una solución de 5-fluoro-6-prop-1-inil-piridin-2-carboxilato de etilo (3.18 g, 15.4 mmol) en una mezcla 7:1 de DMA desgasificada (48.0 ml) y NeT3 (6.85 ml, 49.1 mmol) a 23 °C bajo atmósfera de nitrógeno. La mezcla se agitó a 130 °C durante 17 h y se filtró a través de Celite. El filtrado se diluyó con EtOAc (250 ml) y agua (250 ml). La fase acuosa se extrajo con EtOAc (3 x 250 ml) y las fases orgánicas combinadas se lavaron con salmuera (250 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 80 g) con hexano y EtOAc (0 - 20 %) para proporcionar el compuesto del título como un sólido (1.68 g, 53 %). RMN de 1H (500 MHz, CDCh) 88.74 (ddd, J = 3.9, 2.8, 1.4 Hz, 1H), 7.61 (dd, J = 8.0, 5.5 Hz, 1H), 6.91 (dd, J= 4.1, 2.8 Hz, 1H), 6.83 (dd, J = 4.1, 1.4 Hz, 1H), 6.40 (dd, J = 9.7, 8.0 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 1.96 m.
Compuesto intermedio A. 5-fluoro-6-prop-1-inil-piridin-2-carboxilato de etilo
Se añadieron DIPEA (8.37 ml, 48.1 mmol) y TBAF 1 M en THF (28.8 ml, 28.8 mmol) a una mezcla de 6-bromo-5-fluoro-piridin-2-carboxilato de etilo (5.96 g, 24.0 mmol), 1-(trimetilsilil)-1 -propino (4.27 ml, 28.8 mmol), Cul (0.690 g, 3.60 mmol) y PdCl2(PPh3)2 (169 g, 2.40 mmol) en tolueno desgasificado (225 ml) a 23 °C bajo atmósfera de nitrógeno. La mezcla se agitó durante 16 h y luego se diluyó con Et2O (250 ml) y NH4Cl saturado (150 ml). La fase acuosa se extrajo con Et2O (3 x 250 ml), y las fases orgánicas combinadas se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 120 g) con hexano y EtOAc (0 - 30 %) para proporcionar el compuesto del título como un sólido (3.21 g, 65 %). RMN de 1H (500 MHz, CDCh) 8 8.06 (dd, J = 8.6 , 4.0 Hz, 1H), 7.54 -7.42 (m, 1H), 4.46 (q, J = 7.1 Hz, 2H), 2.14 (d, J = 0.9 Hz, 3H), 1.42 (t, J = 7.1 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 208.1. HPLC tR = 2.60 min.
Ejemplo 62. (4-Fluoro-1-metil-1H-indazol-7-il)(1H-imidazol-2-il)metanol

Se añadió n-BuLi 2.50 M en THF (0.260 ml, 0.640 mmol) a una mezcla de 1-(dietoximetil)imidazol (109 mg, 0.640 mmol) en THF (1.00 ml) a -45 °C y la mezcla se agitó durante 15 m manteniendo la temperatura entre -45 °C y -40 °C. La mezcla se enfrió a -60 °C y se añadió una solución de 4-fluoro-1-metil-indazol-7-carbaldehído (57.0 mg, 0.320 mmol) en THF (1.00 ml). La mezcla se agitó durante 10 min a -60 °C y se calentó a 0 °C durante 1 h. La mezcla se diluyó con HCl 0.25 M (5.00 ml) y EtOAc (5.00 ml) a 0 °C y se agitó a 23 °C durante 10 min. La fase orgánica se extrajo con HCl 0.250 M (3 x 10.0 ml) y las fases acuosas combinadas se diluyeron con NaHCO3 saturado (20.0 ml) y se extrajo con EtOAc (3 x 50.0 ml). Las fases orgánicas combinadas se lavaron con salmuera (50.0 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 4 g) eluyendo con DCM y MeOH ( 0 - 10 %) para proporcionar el compuesto del título como un sólido (4.35 mg, 6 %): RMN de 1H (500 MHz, DMSO) 5 12.10 (s, 1H), 8.13 (s, 1H), 7.29 (dd, J = 8.0, 5.3 Hz, 1H), 6.94 (br, 1H), 6.86 (dd, J = 9.8, 8.0 Hz, 2H), 6.51 (d, J = 4.5 Hz, 1H), 6.35 (d, J = 4.5 Hz, 1H), 4.11 (s, 3H). LC-MS (Método C): m/z (ES ), [MH2OH] = 229.0. HPLC tR = 1.26 min.
Compuesto intermedio D.4-Fluoro-1-metil-indazol-7-carbaldehído
Una solución de (4-fluoro-1-metil-indazol-7-il)metanol (50.0 mg, 0.280 mmol) en CHCl3 (1.00 ml) se añadió gota a gota a una mezcla de MnO2 (121 mg, 1.39 mmol) en CHCl3 (1.00 ml) a 0 °C. La mezcla se sometió a reflujo durante 3.5 h y se filtró a través de una capa de Celite. El sólido se lavó con DCM y el filtrado se concentró a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 4 g) eluyendo con hexano y EtOAc (0 - 30 %) para proporcionar el compuesto del título como un sólido (40.0 mg, 81 %): r Mn de 1H (500 MHz, CDCl3) 5 ppm 10.09 (s, 1H), 8.15 (s, 1H), 7.86 (dd, J = 8.1, 5.1 Hz, 1H), 6.92 (dd, J = 8.8 , 8.1 Hz, 1H), 4.50 (s, 3H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 1.58 min.
Compuesto intermedio C. (4-Fluoro-1-metil-indazol-7-il)metanol
Se añadió LAH 2.00 M en THF (0.220 ml, 0.440 mmol) a una mezcla de 4-fluoro-1-metil-indazol-7-carboxilato de etilo (98.0 mg, 0.440 mmol) en THF (2.50 ml) a 0 °C bajo atmósfera de nitrógeno. La mezcla se calentó a 23 °C y se agitó durante 20 min. La mezcla se diluyó con acetona (1.00 ml) y tartrato de Na/K saturado (10.0 ml) a 0 °C y se agitó durante 5 min. La fase acuosa se extrajo con EtOAc (3 x 20.0 ml) y las fases orgánicas combinadas se secaron sobre MgSO4, se filtraron y concentraron a presión reducida para proporcionar el compuesto del título como un sólido (78.0 mg, 98%). RMN de 1H (500 MHz, CDCl3) 58.04 (s, 1H), 7.19 (dd, J = 7.8, 5.0 Hz, 1H), 6.69 (dd, J = 9.5, 7.8 Hz, 1H), 4.98 (d, J = 5.4 Hz, 2H), 4.39 (s, 3H), 1.74 (t, J = 5.8 Hz, 1H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 1.31 min.
Compuesto intermedio B. 4-fluoro-1-metil-indazol-7-carboxilato de etilo
Se añadió yodometano (20.1 pl, 0.320 mmol) a una suspensión de Cs2CO3 (105 mg, 0.320 mmol) y 4-fluoro-1H-indazol-7-carboxilato de etilo (56.0 mg, 0.270 mmol) en d Mf (1.00 ml), y la mezcla se agitó a 23 °C durante 2.5 h. Se añadió agua (10.0 ml) y la fase acuosa se extrajo con EtOAc (3 x 10.0 ml). Las fases orgánicas combinadas se lavaron con salmuera (20.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 12 g) con hexano y EtOAc (0 - 50 %) para proporcionar el compuesto del título como un sólido (36 mg, 60 %). RMN de 1H (500 MHz, CDCl3) 58.11 (s, 1H), 7.94 (dd, J = 8.2, 5.2 Hz, 1H), 6.77 (dd, J = 8.9, 8.2 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 4.28 (s, 3H), 1.43 (t, J = 7.1 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 222.9. HPLC tR = 1.98 m.
Compuesto intermedio A. 4-fluoro-1H-indazol-7-carboxilato de etilo
Se añadieron 2-amino-4-fluoro-3-metil-benzoato de etilo (505 mg, 2.56 mmol) y KOAc (131 mg, 1.33 mmol) a tolueno anhidro (15.0 ml) bajo atmósfera de nitrógeno. La mezcla se calentó a reflujo y se añadió AC2O (0.730 ml, 7.68 mmol). La mezcla se agitó a reflujo durante 10 min y se añadió nitrito de isopentilo (0.570 ml, 4.23 mmol) durante 1 h. Se continuó agitando a reflujo durante 15 h. La mezcla se enfrió y se filtró. El filtrado se concentró a presión reducida y el residuo se lavó con hexanos, agua y finalmente con hexanos para proporcionar el compuesto del título como un sólido
(347 mg, 65%). RMN de 1H (500 MHz, CDCla) 511.30 (s, 1H), 8.20 (d, J = 1.7 Hz, 1H), 8.07 (dd, J = 8.2, 4.8 Hz, 1H), 6.87 (dd, J = 9.4, 8.2 Hz, 1H), 4.48 (q, J = 7.1 Hz, 2H), 1.47 (t, J = 7.1 Hz, 3H). LC-MS (Método C): m/z (ES+), [M H]+ = 208.9. HPLC tR = 1.86 min.
Ejemplo 63. (3-Etil-7-fluorobenzofuran-4-il)(1H-1,2,4-triazol-5-il)metanol

Se añadió cloruro de hidrógeno en EtOAc (exceso) a (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol-5- il)metanol (150 mg, 0.38 mmol) en EtOAc (10 ml) bajo atmósfera de nitrógeno. La solución resultante se agitó a ta durante 16 horas. El disolvente se eliminó a presión reducida. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 5 a 80 % de MeCN en agua. Las fracciones puras se evaporaron a sequedad para proporcionar (3-etil-7-fluorobenzofuran-4-il)(1H-1,2,4-triazol-5-il)metanol (30.0 mg, 30.0 %) como un sólido blanco. RMN de 1H (400 MHz, CD3OD) 5 ppm 1.26 - 1.35 (m, 3H), 2.05 (s, 0.1H), 2.76 - 2.94 (m, 2H), 6.46 (s, 1H), 7.03 -7.07 (m, 1H), 7.20 -7.23 (m, 1H), 7.63 (s, 1H), 8.17 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 262.3; ácido, HPLC tR = 1.560 min.
Compuesto intermedio B. (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol-5-il)metanol

Se añadió 3-etil-7-fluorobenzofuran-4-carbaldehído (450 mg, 2.34 mmol) a 1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol (933 mg, 4.68 mmol) y n-butil-litio (1.967 ml, 4.92 mmol) en THF (10 ml) a -78°C durante un período de 1 hora bajo atmósfera de nitrógeno. La mezcla resultante se agitó a -78 °C durante 30 minutos. La mezcla de reacción se inactivó con NH4Cl saturado (5 ml), se extrajo con EtOAc (2 * 10 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un líquido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida C18, gradiente de elución de 5 a MeCN al 100 % en agua. Las fracciones puras se evaporaron a sequedad para producir (3-etil-7-fluorobenzofuran-4-il)(1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol-5-il)metanol (150 mg, 16.36 %) como un líquido blanco. RMN de 1H (400 MHz, DMSO-d6) 5 ppm 8.66 (s, 1H), 7.82 (d, J = 1.4 Hz, 1H), 7.36 (dd, J = 8.4, 4.5 Hz, 1H), 7.22 (dd, J = 10.8, 8.4 Hz, 1H), 6.56 (d, J = 5.8 Hz, 1H), 6.41 (d, J = 5.8 Hz, 1H), 5.65 (d, J = 10.8 Hz, 1H), 5.43 (d, J = 10.8 Hz, 1H), 3.55 - 3.38 (m, 2H), 3.18 (d, J = 5.2 Hz, 1H), 2.58 (ddt, J = 15.7, 8.4, 6.8 Hz, 1H), 2.41 (ddt, J = 14.6, 8.9, 7.3 Hz, 1H), 1.15 (t, J = 7.4Hz, 3H), 0.81 (dddd, J = 45.9, 13.7, 10.6, 6.1 Hz, 2H). LC-MS (Método A): m/z (ES+), [M H]+ = 392; ácido, HPLC tR = 0.987 min.
Compuesto intermedio A. 1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol

Se añadió gota a gota SEM-Cl (6.93 ml, 39.09 mmol) a 1H-1,2,4-triazol (3 g, 43.44 mmol) y NaH (2.085 g, 52.12 mmol) en THF (100 ml) a 0°C durante un período de 20 minutos bajo atmósfera de nitrógeno. La mezcla resultante se agitó a ta durante 16 horas. La mezcla de reacción se inactivó con hielo (25 ml), se extrajo con EtOAc (2 x 25 ml), la capa orgánica se secó sobre Na2SO4, se filtró y se evaporó para obtener un sólido blanco. El producto crudo se purificó mediante cromatografía ultrarrápida sobre sílice, gradiente de elución de 0 a 10 % de MeOH en DCM. Las fracciones puras se evaporaron a sequedad para proporcionar 1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol (2.00 g, 23.1 %) como
un sólido blanco. RMN de 1H (300 MHz, DMSO-d3) 5 ppm 8.68 (s, 1H), 5.43 (s, 1H), 3.56 - 3.43 (m, 1H), 0.92 - 0.77 (m, 1H).
Ejemplo 64. (4-Fluoro-2-(pirrolidin-1-il)fenil)(1H-1,2,4-triazol-5-il)metanol

El Ejemplo 64 se preparó de manera análoga al Ejemplo 63 usando 4-fluoro-2-(pirrolidin-1-il)benzaldehído y 1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,4-triazol. RMN de 1H (300 MHz, CDCb) 5 ppm 8.01 (s, 1H), 7.24 - 7.33 (m, 1H), 6.94 (dd, J = 2.54, 10.64 Hz, 1H), 6.84 (dt, J = 2.64, 8.19 Hz, 1H), 6.26 (s, 1H), 3.00 -3.28 (m, 4H), 1.92 -2.10(m , 4H). LC-MS (Método B): LC-MS (Método A): m/z (ES+), [M H]+ = 263; ácido, HPLC tR = 0.43 min.
Ejemplo 65. (4-Fluoro-2-(tiazol-2-il)fenil)(1H-imidazol-2-il)metanol

Ejemplo 65
Preparado de manera análoga al Ejemplo 33. RMN de 1H (400 MHz, CD3DO) 5 ppm 6.45 - 6.50 (m, 1H), 7.17 - 7.23 (m, 2H), 7.32 - 7.37 (m, 1H), 7.53 - 7.56 (m, 1H), 7.76 - 7.77 (m, 2H), 7.96 - 7.97 (m, 1H), 8.35 (s, 1H). LC-MS (Método A): m/z (ES+), [M H]+ = 276.3; ácido, HPLC tR = 1.312 min.
Ejemplo 66. [7-Fluoro-3-(1-metilciclopropil)benzofuran-4-il]-(1H-imidazol-2-il)metanol
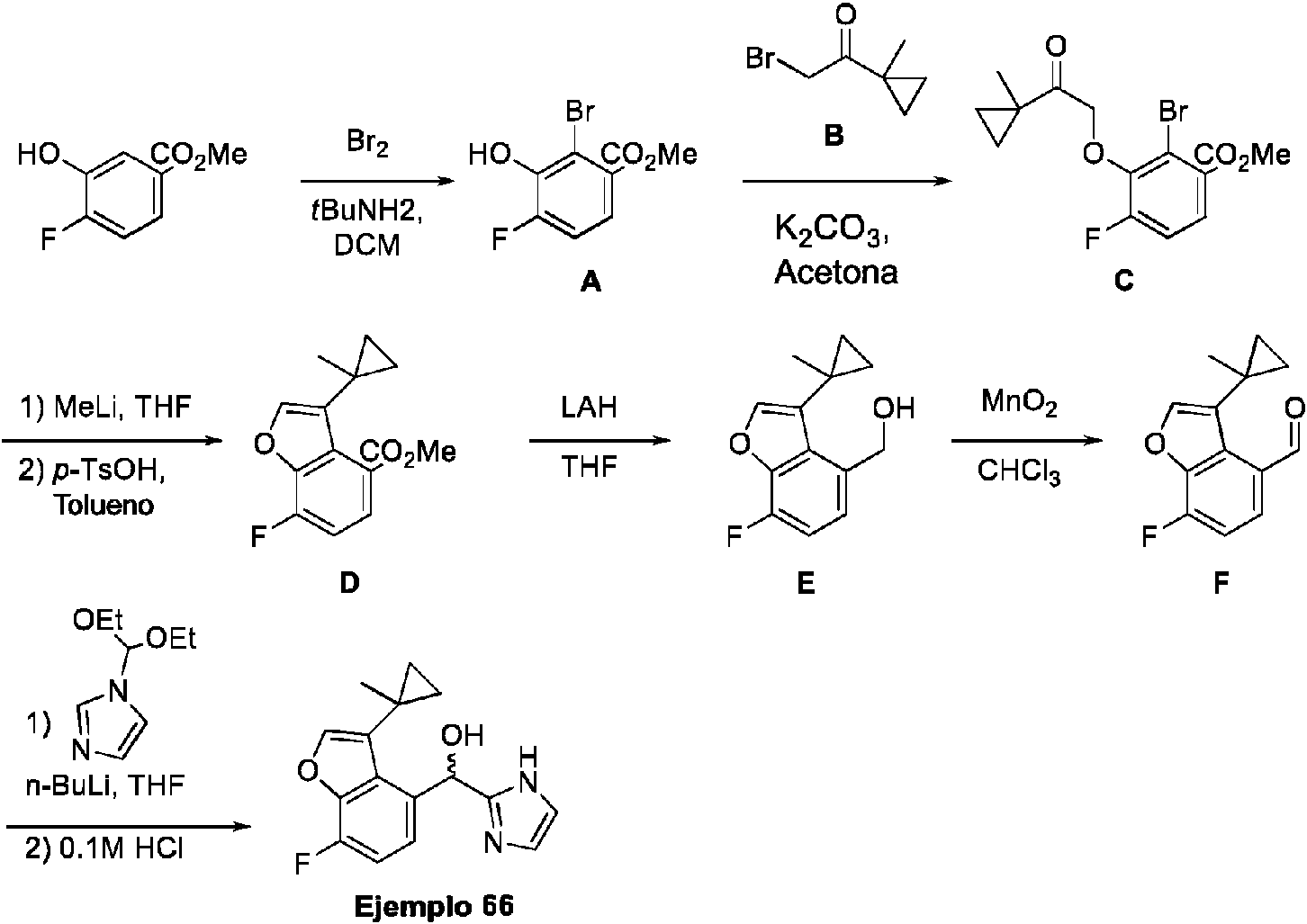
Se enfrió 1-(dietoximetil)imidazol (81.9 mg, 0.480 mmol) en THF (1.00 ml) a -45 °C y se añadió gota a gota n-BuLi 2.5 M en THF (0.190 ml, 0.480 mmol). La mezcla se agitó a -45 °C durante 20 min. La mezcla se enfrió a -60 °C y se añadió gota a gota una solución de 7-fluoro-3-(1-metilciclopropil)benzofuran-4-carbaldehído (50.0 mg, 0.230 mmol) en THF (1.00 ml). La solución se calentó a 0 °C durante 1 h. Se añadió HCl 0.1 M (5.00 ml) y la mezcla se calentó a 23 °C y se agitó durante 10 min. Se añadió EtOAc (5.00 ml). La fase orgánica se extrajo con HCl 0.1 M (2 x 5.00 ml) y las fases acuosas combinadas se diluyeron con NaHCO3 saturado (10.0 ml). La mezcla acuosa se extrajo con EtOAc (3
x 25.0 ml) y las fases orgánicas combinadas se lavaron con salmuera (25.0 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El producto se purificó mediante un HPLC de Waters (Gemini NX, 150 X 30, 5 micras, columna C-18), eluyendo isocráticamente con una mezcla de agua y MeCN (50%), que contenía 0.1 % de (NH4)2CO3 para proporcionar el compuesto del título como un sólido (37 mg, 56%). RMN de 1H (500 MHz, DMSO) 8 12.03 (s, 1H), 7.94 (s, 1H), 7.28 (dd, J = 8.5, 4.6 Hz, 1H), 7.19 (dd, J = 10.7, 8.5 Hz, 1H), 7.02 (s, 1H), 6.73 (s, 1H), 6.68 (d, J = 4.2 Hz, 1H), 6.13 (d, J = 4.2 Hz, 1H), 1.42 (s, 3H), 1.01 -0.87 (m, 1H), 0.87 -0.79 (m, 1H), 0.79 -0.72 (m, 1H), 0.72 - 0.59 (m, 1H). LC-MS (Método C): m/z (ES+), [M H]+ = 287.1. HPLC tR = 1.57 m.
Compuesto intermedio F. 7-Fluoro-3-(1-metilciclopropil)benzofuran-4-carbaldehído
Se añadió MnO2 (114 mg, 1.31 mmol) a una mezcla de [7-fluoro-3-(1-metilciclopropil)benzofuran-4-il]metanol (72.0 mg, 0.330 mmol) en CHCh (2.00 ml) a 23 °C bajo atmósfera de nitrógeno. La mezcla se sometió a reflujo durante 18 h y se filtró a través de una capa de Celite. El sólido se lavó con DCM y el filtrado se concentró a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 12 g) eluyendo con hexano y EtOAc (0-15 %) para proporcionar el compuesto del título como un sólido (52.0 mg, 73 %). Rm N de 1H (500 MHz, CDCh) 8 10.87 (d, J = 0.6 Hz, 1H), 7.95 (dd, J = 8.6 , 4.6 Hz, 1H), 7.67 (s, 1H), 7.20 - 7.06 (m, 1H), 1.45 (s, 3H), 0.99 - 0.89 (m, 2H), 0.89 - 0.79 (m, 2H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 2.54 m.
Compuesto intermedio E. [7-Fluoro-3-(1-metilciclopropil)benzofuran-4-il]metanol
Se añadió LAH 1.00 M en THF (0.370 ml, 0.370 mmol) a una solución enfriada de 7-fluoro-3-(1-metilciclopropil) benzofuran-4-carboxilato de metilo (92.0 mg, 0.370 mmol) en THF (1.00 ml) a 0 °C bajo atmósfera de nitrógeno. La mezcla se agitó a 0 °C durante 15 min y se diluyó con acetona (0.500 ml) y solución saturada de tartrato de K/Na (10.0 ml). Se añadió agua (10.0 ml) y la fase acuosa se extrajo con EtOAc (3 x 25.0 ml). Las fases orgánicas combinadas se lavaron con salmuera (30.0 ml), se secaron (MgSO4), se filtraron y concentraron a presión reducida para proporcionar el compuesto del título como un aceite (72.0 mg; 88%). Rm N de 1H (500 MHz, CDCh) 8 7.51 (s, 1H), 7.29 (dd, J = 8.3, 4.4 Hz, 1H), 7.03 (dd, J = 10.3, 8.3 Hz, 1H), 5.17 (s, 2H), 1.71 (br, 1H), 1.44 (s, 3H), 0.91 - 0.83 (m, 2H), 0.79 - 0.72 (m, 2H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 2.29 m.
Compuesto intermedio D. 7-fluoro-3-(1-metilciclopropil)benzofuran-4-carboxilato de metilo
Se añadió gota a gota MeLi 1.60 M en Et2O (0.700 ml, 1.11 mmol) a una solución enfriada de 2-bromo-4-fluoro-3-[2-(1-metilciclopropil)-2-oxo-etoxi]benzoato de metilo (320 mg, 0.930 mmol) en THF (92.0 ml) a -78 °C. La mezcla se agitó a -78 °C durante 30 min. Se añadió MeLi adicional 1.60 M en Et2O (0.280 ml, 0.440 mmol) a -78 °C. La mezcla se calentó a 0 °C y se diluyó con NH4Cl saturado (30 ml). La mezcla se concentró parcialmente a presión reducida para eliminar el t Hf y se añadió agua (20.0 ml). La fase acuosa se extrajo con EtOAc (3 * 50.0 ml) y las fases orgánicas combinadas se lavaron con salmuera (50.0 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El residuo se disolvió en tolueno (9.00 ml) y se añadieron tamices moleculares de 4 A y monohidrato de p-TSA (44.1 mg, 0.230 mmol). La mezcla se calentó a reflujo durante 45 min. Después de enfriar a temperatura ambiente, la mezcla se filtró y el filtrado se concentró a presión reducida. El producto se purificó por cromatografía en gel de sílice (cartucho de 12 g), eluyendo con hexano y EtOAc ( 0 - 15 %) para proporcionar el compuesto del título como un aceite (99.0 mg, 43 %). RMN de 1H (500 MHz, CDCh) 87.70 (dd, J = 8.5, 4.6 Hz, 1H), 7.61 (s, 1H), 7.04 (dd, J = 9.8, 8.5 Hz, 1H), 3.96 (s, 3H), 1.38 (s, 3H), 0.88 - 0.76 (m, 2H), 0.74 - 0.66 (m, 2H). LC-MS (Método C): m/z (ES+), Sin ionización. Hp Lc tR = 2.60 m.
Compuesto intermedio C. 2-bromo-4-fluoro-3-[2-(1-metilciclopropil)-2-oxo-etoxi]benzoato de metilo
Se añadieron 2-bromo-1-(1-metilciclopropil)etanona (206 mg, 1.16 mmol) y K2CO3 (210 mg, 1.52 mmol) a una solución de 2-bromo-4-fluoro-3-hidroxibenzoato de metilo (252 mg, 1.01 mmol) en acetona (5.00 ml). La mezcla se calentó a 60 °C durante 18 h. La mezcla se filtró a través de Celite y el sólido se lavó con EtOAc. El filtrado se concentró a presión reducida. El producto se purificó mediante cromatografía en gel de sílice (cartucho de 25 g), eluyendo con EtOAc y hexanos (0 - 25 %) para proporcionar el compuesto del título como un aceite (0.332 g, 95 %). RMN de 1H (500 MHz, CDCla) 8 7.52 (dd, J = 8.7, 5.3 Hz, 1H), 7.09 (dd, J = 10.2, 8.7 Hz, 1H), 4.95 (d, J = 1.1 Hz, 2H), 3.92 (s, 3H), 1.39 (s, 3H), 1.38 - 1.35 (m, 2H), 0.81 - 0.77 (m, 2H). LC-MS (Método C): m/z (ES+), [M H]+ = 345.0. HPLC tR = 2.31 min.
Compuesto intermedio B. 2-Bromo-1-(1-metilciclopropil)etanona
Se añadió gota a gota una solución de bromo (0.780 ml, 15.3 mmol) en DCM (7.50 ml) a una solución enfriada de metil 1-metilciclopropilcetona (1.68 ml, 15.3 mmol) en MeOH (12.0 ml) a 0 °C. La mezcla se agitó durante 1 h y se vertió sobre hielo. La fase acuosa se extrajo con DCM (4 * 20.0 ml) y las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y concentraron a presión reducida para proporcionar el compuesto del título (2.55 g, 94 %) como un aceite. RMN de 1H (500 MHz, CDCh) 84.01 (s, 2H), 1.45 (d, J = 0.6 Hz, 3H), 1.37 - 1.32 (m, 2H), 0.86 - 0.82 (m, 2H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 2.02 min.
C o m p u e s to in te rm e d io A. 2 -b ro m o -4 -flu o ro -3 -h id ro x i-b e n z o a to de m etilo
Se añadió gota a gota una solución de bromo (0.510 ml, 10.0 mmol) en DCM (5.00 ml) a una solución enfriada de tercbutilamina (2.10 ml, 20.0 mmol) en DCM (20.0 ml) a -78 °C. La mezcla se agitó a -78 °C cubierta con papel de aluminio durante 1 hora. Se añadió gota a gota una solución de 4-fluoro-3-hidroxi-benzoato de metilo (1.70 g, 10.0 mmol) en DCM (20.0 ml) a -78 °C. La mezcla se agitó a -78 °C, se cubrió con papel de aluminio y se calentó lentamente a 23 °C durante 18 h. La mezcla se filtró y se añadió HCl 1 N (30.0 ml) al filtrado. La fase acuosa se extrajo con DCM (2 x 25.0 ml). Las fases orgánicas combinadas se lavaron con salmuera (30.0 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida. El residuo se diluyó con HCl 1 N (25.0 ml) y la fase acuosa se extrajo con DCM (3 * 25.0 ml). Las fases orgánicas combinadas se lavaron con salmuera (25.0 ml), se secaron sobre MgSO4, se filtraron y concentraron a presión reducida para proporcionar el compuesto del título como un sólido (1.45 g; 58%). RMN de 1H (500 MHz, CDCla) 87.49 (dd, J = 8.7, 5.4 Hz, 1H), 7.18 - 7.05 (m, 1H), 5.90 (d, J = 1.7 Hz, 1H), 3.93 (s, 3H). LC-MS (Método C): m/z (ES+), Sin ionización. HPLC tR = 1.89 m.
Métodos de ensayo:
Ensayo de IP-One:
Protocolo para probar compuestos con respecto a la actividad antagonista de PAR2 utilizando un ensayo IP-One HTRF
La producción de monofosfato de inositol (IP1) se midió en células 1321N1 que expresan de forma estable PAR2 humano utilizando un kit de ensayo IP-One HTRF (Cisbio). Se añadieron 80 nl de compuesto en DMSO al 100 % en placas blancas de 384 pocillos de pequeño volumen usando un Echo 555 (Labcyte) y se incubaron durante 30 min a 37 °C con 4 pl de células (15,000 células por pocillo) en tampón de estimulación (HBSS con HEPES 20 mM, pH 7.4). La producción de IP1 se inició mediante la adición de 4 pl de SLIGRL-NH2 140 pM (SEQ ID NO.: 4) en tampón de estimulación suplementado con LiCl 100 mM. Después de 60 min a 37 °C, las células se lisaron y las concentraciones de IP1 se detectaron de acuerdo con el protocolo del fabricante (Cisbio). Los datos se normalizaron a concentraciones de IP1 usando una curva estándar de IP1 de acuerdo con el protocolo del fabricante (Cisbio).
Ensayo de FLPR activado con tripsina:
Protocolo para probar compuestos con respecto a la actividad antagonista de PAR2 usando un ensayo de movilización de Ca2+
La movilización de calcio se midió utilizando células 1321N1 que expresan de forma estable PAR2 humano. Las células se sembraron en placas de 384 pocillos a razón de 4,000 células por pocillo en 20 pl de DMEM con Glutamax suplementado con FBS al 10 % y se incubaron durante 18-24 h a 37 °C, 5% de CO2 y 95 % de humedad. Las células se cargaron con 20 pl de colorante de calcio Fluo-8 NW (Cat: 36316, AAT Bioquest) y se mantuvo a 37 °C durante 30 min antes de agregar 10 pl del compuesto preparado en HEPES 20 mM pH 7.4, HBSS, DMSO al 2.5 %, BSA al 0.1 % y Vorapaxar 6 pM (para bloquear la actividad inducida por tripsina de PARI ). La adición se realizó con un FLIPRTETRA (cabezal de 384 pocillos, Molecular Devices) y la respuesta se leyó simultáneamente para detectar cualquier actividad agonista. Las células se incubaron durante 30 min a temperatura ambiente con los compuestos y luego se evaluó la actividad antagonista mediante la adición de 10 pl de tripsina 234 nM preparada en HEPES 20 mM pH 7.4, HBSS y BSA al 0.1 % y detección simultánea de la movilización de calcio utilizando el FLIPRTETRA.
Tabla 1. Datos del ensa o IP-One FLPR tripsina



Estudios de unión
En algunas realizaciones, la solicitud proporciona compuestos que se unen a un sitio distinto del sitio de PARI Vorapaxar utilizando técnicas de cristalografía en una forma mutante del receptor PAR2.
La estructura de PAR1 en complejo con Vorapaxar mostró que el ligando se une en un bolsillo periférico cercano a la superficie extracelular de PAR1 (Zhang, C. et al. Nature 2012, 492, 387-392. Mientras que Vorapaxar exhibe una alta selectividad frente a PAR2, se conservan casi todos los residuos implicados en las interacciones. Los estudios mutacionales han demostrado que Glu260 y Asp256 del bucle extracelular 2 (ECL2) son importantes para unir el péptido activador. Ambos residuos se conservan en PAR2 (Glu232 y Asp228). Por lo tanto, se cree que el bolsillo de Vorapaxar es el sitio ortostérico para los receptores relacionados activados por proteasa.
Una estructura cristalina con el Ejemplo 3 en complejo con PAR2 reveló que el ligando se une en un nuevo bolsillo alostérico, a 8 A del sitio de unión de Vorapaxar identificado en la estructura PARI (Figura 1). El bolsillo está completamente enterrado en el receptor (Figura 2), lo que es coherente con las largas tasas de disociación de los compuestos de esta serie. El ligando está situado en una parte relativamente rígida de la proteína cerca del puente disulfuro entre la transmembrana 3 (TM3) y ECL2. Hace interacciones polares con Asp228, Tyr82 y His135 (Figura 3). Según los datos estructurales, los bolsillos alostéricos y ortostéricos en PAR2 están claramente separados. Ninguno de los ligandos probados en la serie del Ejemplo 3 ha demostrado actividad contra PARI (ensayo tipo p-arrestina por DiscoverX, Ejemplo 3 IC50 > 50 j M, Ejemplo 35 > 50 j M).
Ensayo de tasa de disociación de FLPR: Protocolo para probar compuestos con respecto al tiempo medio de residencia de PAR2
El ensayo de FLPR descrito anteriormente utilizó la misma línea celular y los mismos protocolos de manejo de células, pero se modificó de las siguientes maneras: Las células se incubaron con el compuesto en diferentes momentos (60 min, 120 min, 180 min y 240 min), momento en el que se lavaron las células. Tinte de calcio Fluo-8 NW (Cat: 36316, AAT Bioquest) similar al protocolo descrito anteriormente, una hora antes de la adición del agonista. El agonista peptídico SLIGLR-NH2 (e Cbü) se usó en lugar de tripsina.
Tabla 2. Datos del ensa o de la tasa de disociación de FLPR

Claims (12)
1. Un compuesto, en el que el compuesto es un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del 
R3 es halógeno;
X es N o CH;
Y es C o N; y
Z es C o N, en la que Y y Z no son ambos N, y
(1) cuando tanto Z como Y son C, el enlace " " es un doble enlace, y R1 y R2 pueden tomarse junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,

F, Cl, Br, OH, metoxi y CN; o
(2) cuando uno de Z y Y es N, el otro es C, el enlace " 77= " es un enlace sencillo, y R1 y R2 se toman junto con los átomos a los que están unidos para formar un anillo monocíclico aromático o no aromático de 3 a 10 miembros que tiene 0-3 heteroátomos o grupos de heteroátomos seleccionados independientemente de N, NH, O, S, SO y SO2, en el que dicho anillo está sustituido con 0-3 R, y en el que dicho anillo está opcionalmente fusionado con un arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros, en el que dicho arilo C6-10, heteroarilo de 5 a 10 miembros, cicloalquilo C3-10, o un heterociclilo de 3 a 10 miembros está sustituido con 0-3 R, en el que cada ocurrencia de R se selecciona independientemente de metilo, etilo, propilo, isopropilo, CH2CF3, CF3, ciclopropilo,

F, Cl, Br, OH, metoxi y CN.
2. Un compuesto que se selecciona del grupo que consiste en:


y sales farmacéuticamente aceptables de los mismos.
3. El compuesto de acuerdo con la reivindicación 1, en el que la estructura
se selecciona de:

en el que R5 es H o alquilo C1-3, y n es 0-3.
4. El compuesto de acuerdo con la reivindicación 1, en el que la estructura

se selecciona de:

5. El compuesto de acuerdo con la reivindicación 1, en el que la estructura

se selecciona de:

6. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 o 3-5, en el que X es CH.
7. El compuesto de acuerdo con una cualquiera de las reivindicaciones 1 o 3-6, en el que R3 es -F.
8. El compuesto de la reivindicación 1 o la reivindicación 2, en el que el compuesto se selecciona de:


y sales farmacéuticamente aceptables de los mismos.
9. El compuesto de la reivindicación 8 , en el que el compuesto se selecciona de:

y sales farmacéuticamente aceptables de los mismos.
10. Una composición farmacéutica que comprende (a) un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-9; y (b) un excipiente farmacéuticamente aceptable.
11. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-9 o una composición farmacéutica de acuerdo con la reivindicación 10 para uso en el tratamiento de una enfermedad o trastorno mediado por la actividad de PAR2.
12. El compuesto para uso de acuerdo con la reivindicación 11, en el que la enfermedad o trastorno es dolor, inflamación musculoesquelética, trastornos neuroinflamatorios, inflamación de las vías respiratorias, picazón, dermatitis o colitis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662335496P | 2016-05-12 | 2016-05-12 | |
| PCT/EP2017/061409 WO2017194716A1 (en) | 2016-05-12 | 2017-05-11 | Inhibitors of protease-activated receptor-2 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2909709T3 true ES2909709T3 (es) | 2022-05-10 |
Family
ID=58772863
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17725905T Active ES2909709T3 (es) | 2016-05-12 | 2017-05-11 | Inhibidores del receptor 2 activado por proteasa |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US10550089B2 (es) |
| EP (1) | EP3455212B1 (es) |
| DK (1) | DK3455212T3 (es) |
| ES (1) | ES2909709T3 (es) |
| WO (1) | WO2017194716A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017194716A1 (en) | 2016-05-12 | 2017-11-16 | Astrazeneca Ab | Inhibitors of protease-activated receptor-2 |
| US20240066027A1 (en) | 2020-12-03 | 2024-02-29 | Domain Therapeutics | Novel par-2 inhibitors |
| KR20250057699A (ko) | 2022-06-03 | 2025-04-29 | 도메인 테라퓨틱스 | 신규한 par-2 억제제 |
| WO2025172573A1 (en) | 2024-02-16 | 2025-08-21 | Domain Therapeutics | Combinations of par2 inhibitors and immune checkpoint inhibitors for the treatment of cancer |
| WO2025191185A1 (en) | 2024-03-15 | 2025-09-18 | Domain Therapeutics | Azine-based compounds as par-2 inhibitors and therapeutic uses thereof |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11209375A (ja) * | 1998-01-23 | 1999-08-03 | Nippon Bayer Agrochem Co Ltd | 複素環置換アルコール誘導体及び農園芸用殺菌剤 |
| GB9929674D0 (en) * | 1999-12-15 | 2000-02-09 | Novartis Res Found | Protease-activated receptor (PAR) modulator assays |
| US20060104944A1 (en) * | 2004-11-18 | 2006-05-18 | Mousa Shaker A | Activators and inhibitors of protease activated receptor2 (PAR2) and methods of use |
| US7592362B2 (en) * | 2006-01-19 | 2009-09-22 | Pfizer Limited | Substituted imidazoles |
| CA2637312A1 (en) * | 2006-01-27 | 2007-08-02 | F. Hoffmann-La Roche Ag | Use of substituted 2-imidazole of imidazoline derivatives |
| US20100041712A1 (en) * | 2008-08-18 | 2010-02-18 | Pfizer Inc. | Substituted imidazole combinations |
| EP3351641A1 (en) * | 2011-04-08 | 2018-07-25 | Tufts Medical Center, Inc. | Pepducin design and use |
| HK1223096A1 (zh) * | 2013-09-25 | 2017-07-21 | Vertex Pharmaceuticals Incorporated | 用作par-2信號通路抑制劑的咪唑並噠嗪 |
| WO2017083618A1 (en) * | 2015-11-13 | 2017-05-18 | Oasis Pharmaceuticals, LLC | Protease-activated receptor-2 modulators |
| WO2017194716A1 (en) | 2016-05-12 | 2017-11-16 | Astrazeneca Ab | Inhibitors of protease-activated receptor-2 |
-
2017
- 2017-05-11 WO PCT/EP2017/061409 patent/WO2017194716A1/en not_active Ceased
- 2017-05-11 DK DK17725905.8T patent/DK3455212T3/da active
- 2017-05-11 ES ES17725905T patent/ES2909709T3/es active Active
- 2017-05-11 US US16/300,641 patent/US10550089B2/en active Active
- 2017-05-11 EP EP17725905.8A patent/EP3455212B1/en active Active
-
2019
- 2019-12-27 US US16/728,814 patent/US10995072B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017194716A1 (en) | 2017-11-16 |
| DK3455212T3 (da) | 2022-03-28 |
| US20200140392A1 (en) | 2020-05-07 |
| US20190144394A1 (en) | 2019-05-16 |
| EP3455212A1 (en) | 2019-03-20 |
| US10550089B2 (en) | 2020-02-04 |
| US10995072B2 (en) | 2021-05-04 |
| EP3455212B1 (en) | 2022-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN113811300B (zh) | Tead转录因子的新型小分子抑制剂 | |
| ES2855135T3 (es) | Amidas heterocíclicas como inhibidores de quinasa | |
| ES2909709T3 (es) | Inhibidores del receptor 2 activado por proteasa | |
| ES2780848T3 (es) | Análogos de tiadiazol y su uso para tratar enfermedades asociadas con una deficiencia de neuronas motoras SMN | |
| ES2620446T3 (es) | Nuevo compuesto heterocíclico bicíclico | |
| ES2641478T3 (es) | Derivados de 3-acetilamino-1-(fenil-heteroaril-aminocarbonil o fenil-heteroaril-carbonilamino)benceno para el tratamiento de trastornos hiperproliferativos | |
| ES2525581T3 (es) | Derivados de N-sulfonilbenzamida útiles como inhibidores del canal de sodio dependiente de voltaje | |
| ES2582576T3 (es) | Derivados de ciclohexilamida y su uso como antagonistas del receptor de CRF-1 | |
| CN111406054B (zh) | 作为组蛋白脱乙酰基酶6抑制剂的1,2,4-噁二唑衍生物 | |
| EA016301B1 (ru) | Пирролопиримидины и их применение | |
| ES2446356T3 (es) | N-fenil-1-(4-piridinil)-1H-pirazol-3-aminas sustituidas | |
| BR112021000332A2 (pt) | Compostos químicos | |
| WO2015038778A1 (en) | SUBSTITUTED UREA EIF2α KINASE ACTIVATORS | |
| ES2923790T3 (es) | Piperidinas halosustituidas como moduladores de receptores de orexina | |
| EP4396176A1 (en) | Indole compounds and uses thereof in the treatement of cystic fibrosis | |
| CA2901766A1 (en) | N-(2-cyano heterocyclyl)pyrazolo pyridones as janus kinase inhibitors | |
| ES2872775T3 (es) | Compuestos de pirropirimidina como inhibidores de MNK | |
| EP4066895A1 (en) | Compound having lysophosphatidic acid receptor agonistic activity and pharmaceutical use of said compound | |
| US20250320189A1 (en) | New derivatives for treating trpm3 mediated disorders | |
| JP6770962B2 (ja) | チアゾール誘導体の製造方法 | |
| US20250346569A1 (en) | New derivatives for treating trpm3 mediated disorders | |
| MX2015002367A (es) | Derivados de diazepinona utiles para el tratamiento del sindrome x fragil, enfermedad de parkinson o enfermedad de reflujo. | |
| BR112021018303B1 (pt) | Composto inibidor de pequenas moléculas de fatores de transcrição de tead ou sal farmaceuticamente aceitável deste, composição farmacêutica e uso destes | |
| TW202412782A (zh) | 用於治療trpm3介導之病症的吲唑衍生物 | |
| EP4532469A1 (en) | Spleen tyrosine kinase inhibitors and methods of use thereof |