ES2910378T3 - Copolímeros en bloque no iónicos y composiciones farmacéuticas derivadas de los mismos - Google Patents
Copolímeros en bloque no iónicos y composiciones farmacéuticas derivadas de los mismos Download PDFInfo
- Publication number
- ES2910378T3 ES2910378T3 ES18777331T ES18777331T ES2910378T3 ES 2910378 T3 ES2910378 T3 ES 2910378T3 ES 18777331 T ES18777331 T ES 18777331T ES 18777331 T ES18777331 T ES 18777331T ES 2910378 T3 ES2910378 T3 ES 2910378T3
- Authority
- ES
- Spain
- Prior art keywords
- pvp
- pla
- molecular weight
- block copolymers
- average molecular
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F226/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen
- C08F226/06—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen by a heterocyclic ring containing nitrogen
- C08F226/10—N-Vinyl-pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/045—Hydroxy compounds, e.g. alcohols; Salts thereof, e.g. alcoholates
- A61K31/05—Phenols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/192—Carboxylic acids, e.g. valproic acid having aromatic groups, e.g. sulindac, 2-aryl-propionic acids, ethacrynic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5192—Processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F299/00—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers
- C08F299/02—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates
- C08F299/04—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates from polyesters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
- C08G63/08—Lactones or lactides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/68—Polyesters containing atoms other than carbon, hydrogen and oxygen
- C08G63/688—Polyesters containing atoms other than carbon, hydrogen and oxygen containing sulfur
- C08G63/6882—Polyesters containing atoms other than carbon, hydrogen and oxygen containing sulfur derived from hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L39/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a single or double bond to nitrogen or by a heterocyclic ring containing nitrogen; Compositions of derivatives of such polymers
- C08L39/04—Homopolymers or copolymers of monomers containing heterocyclic rings having nitrogen as ring member
- C08L39/06—Homopolymers or copolymers of N-vinyl-pyrrolidones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/04—Polyesters derived from hydroxycarboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L87/00—Compositions of unspecified macromolecular compounds, obtained otherwise than by polymerisation reactions only involving unsaturated carbon-to-carbon bonds
- C08L87/005—Block or graft polymers not provided for in groups C08L1/00 - C08L85/04
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/12—Copolymers
- C08G2261/126—Copolymers block
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Dispersion Chemistry (AREA)
- Pain & Pain Management (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biomedical Technology (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Una composición farmacéutica seca que comprende copolímeros en bloque de PVP-PLA como se define en la Fórmula I: **(Ver fórmula)** donde: x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C, n es, en promedio, de 20 y 40, y m es, en promedio, de 10 y 40, en donde los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da; en donde el peso molecular promedio en número se mide mediante resonancia magnética nuclear (RMN) de protones, en asociación molecular con al menos un ingrediente farmacéutico activo (IFA).
Description
DESCRIPCIÓN
Copolímeros en bloque no iónicos y composiciones farmacéuticas derivadas de los mismos
Referencia cruzada a solicitudes relacionadas
Esta solicitud reivindica el beneficio de prioridad de la solicitud de patente provisional de EE.UU. n° 62/479.582 titulada NON-IONIC BLOCK COPOLYMERS AND PHARMACEUTICAL COMPOSITIONS DERIVED THEREFROM, presentada el 31 de marzo de 2017, y de la solicitud del Reino Unido n° 1705287.9, titulada NON-IONIC BLOCK COPOLYMERS AND PHARMACEUTICAL COMPOSITIONS DERIVED THEREFROM presentada el 31 de marzo de 2017.
Campo
La presente descripción se refiere en general a vehículos para ingredientes farmacéuticos activos (IFA). Más particularmente, la presente descripción se refiere a copolímeros en bloque para la administración micelar de IFA.
Antecedentes
PVP-PLA es un copolímero en bloque no iónico de polivinilpirrolidona (PVP) y poli(D,L lactida) (PLA). (Polímero 53, 2012, 5743-5753). La tecnología de PVP-PLA se puede utilizar para la formulación de ingredientes farmacéuticos activos (IFA) para su administración a personas que los necesiten, y es particularmente útil para la administración de IFA de baja solubilidad en agua o iFa que son insolubles en agua. La tecnología tiene la capacidad de formar micelas independientemente del pH cuando la concentración de polímero en el agua está por encima de la concentración micelar crítica (CMC) y atrapar los IFA dentro de sus núcleos en el proceso. Tales micelas se rompen cuando la concentración de polímero disminuye por debajo de la CMC. Siguiendo este principio, las micelas de PVP-PLA cargadas con IFA comenzarán a liberar su contenido de IFA tras la dilución, por ejemplo, después de la inyección en la sangre; sin embargo, la CMC de la formulación de PVP-PLA/IFA puede variar según la naturaleza del fármaco atrapado.
Aunque PVP-PLA es valioso para la administración de fármacos, la tecnología sigue siendo un desafío, por ejemplo, debido a uno o más de: métodos de síntesis ineficientes y no rentables; limitaciones en la capacidad de cargar diferentes IFA; o limitaciones a la cantidad de IFA que se puede cargar.
Por lo tanto, sigue existiendo la necesidad de copolímeros en bloque de PVP-PLA que puedan fabricarse de manera eficiente. Sigue existiendo la necesidad de tecnologías de administración de fármacos con PVP-PLA flexibles con la capacidad de cargar los IFA respectivos en una concentración suficientemente alta y/o cargar una variedad de IFA. Sigue existiendo la necesidad de especies de copolímeros en bloque de PVP-PLA que tengan la capacidad de cargar IFA en una concentración suficientemente alta para que la administración de la formulación de PVP-PLA IFA sea lo menos frecuente posible.
Resumen
Un objetivo de la presente descripción es evitar o mitigar al menos una desventaja de los enfoques anteriores.
En un primer aspecto, la presente descripción proporciona copolímeros en bloque de PVP-PLA como se define en la Fórmula I:

donde x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C, n es, en promedio, entre 20 y 40, y m es, en promedio, entre 10 y 40, en donde los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da.
En otro aspecto, se proporciona un sistema de administración de nanovehículos que comprende micelas formadas por los copolímeros en bloque de PVP-PLA.
En otro aspecto, se proporciona una composición farmacéutica seca que comprende los copolímeros en bloque de PVP-PLA en asociación molecular con al menos un ingrediente farmacéutico activo (IFA).
En otro aspecto, se proporciona una composición farmacéutica líquida que comprende nanopartículas formadas por copolímeros en bloque de PVP-PLA y que comprende al menos un ingrediente farmacéutico activo (IFA).
En otro aspecto, se proporciona un método para administrar al menos un ingrediente farmacéutico activo (IFA) a un
sujeto que lo necesite, que comprende administrar la composición farmacéutica seca al sujeto.
En otro aspecto, se proporciona un método para administrar al menos un ingrediente farmacéutico activo (IFA) a un sujeto que lo necesite, que comprende administrar la composición farmacéutica líquida al sujeto.
En otro aspecto, se proporciona la composición farmacéutica seca o la composición farmacéutica líquida para el uso en la administración de al menos un ingrediente farmacéutico activo (IFA) a un sujeto.
En otro aspecto, se proporciona un kit que comprende la composición farmacéutica seca o la composición farmacéutica líquida, e instrucciones para su uso en la administración de al menos un ingrediente farmacéutico activo (IFA) a un sujeto. En un aspecto, los copolímeros en bloque descritos anteriormente pueden encontrar aplicación en la mitigación de los efectos hemolíticos.
En un aspecto, los copolímeros en bloque descritos anteriormente pueden encontrar aplicación en la mitigación de la actividad hemolítica de una molécula de carga.
En otro aspecto, se proporciona un método para preparar copolímeros en bloque de PVP-PLA como se define en la Fórmula I:

donde x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C, n es, en promedio, entre 20 y 40, m es, en promedio, entre 10 y 40, y los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da, comprendiendo el método: iniciar la polimerización de D,L-Lactida a partir del alcohol iniciador x para formar poli(ácido láctico) (PLA), añadir un xantato al PLA para formar un macroiniciador de PLA y polimerizar la NVP, mediante polimerización controlada, sobre el macroiniciador de PLA para formar el compuesto de copolímero en bloque de fórmula I.
Otros aspectos y características de la presente descripción resultarán evidentes para los expertos en la técnica tras la revisión de la siguiente descripción de realizaciones específicas junto con las figuras adjuntas.
Breve descripción de los dibujos
A continuación se describirán realizaciones de la presente descripción, únicamente a modo de ejemplo, con referencia a las figuras adjuntas.
La Fig. 1 representa la distribución de tamaños de las micelas obtenidas de tres lotes diferentes de polímero de PVP-PLA.
La Fig. 2 representa los patrones de XRPD para flurbiprofeno (línea superior) y su mezcla física con PVP-PLA (línea inferior).
La Fig. 3 representa el patrón de XRPD de la torta liofilizada de flurbiprofeno que contiene una cantidad similar de IFA que la mezcla física que se muestra en la Figura 2.
La Fig. 4 muestra imágenes, con el panel A que muestra la imagen de la torta liofilizada y la disolución obtenida después de la reconstitución de la torta con agua para inyección a una concentración de flurbiprofeno de 50 mg/mL, y el panel B que muestra una vista más cercana de la disolución del panel A.
La Fig. 5 representa la distribución de tamaños de partículas de una formulación de flurbiprofeno reconstituida a 50 mg/mL.
La Fig. 6 representa el patrón de XRPD del producto de FLU en el momento inicial y después de T=3 meses de almacenamiento en ambas condiciones.
La Fig. 7 representa distribuciones de tamaños de partículas de formulaciones de flurbiprofeno preparadas utilizando los polímeros WB-4 y WB-7.
La Fig. 8 representa los patrones de XRPD para el IFA de paracetamol y la torta liofilizada del producto farmacéutico.
La Fig. 9 representa una imagen de la torta liofilizada de paracetamol y la disolución obtenida después de la reconstitución de la torta con agua para inyección a la concentración de paracetamol de 50 mg/mL.
La Fig. 10 representa la distribución de tamaños de partículas para la formulación de APAP reconstituida a 50 mg/mL.
La Fig. 11 representa distribuciones de tamaños de partículas para formulaciones de paracetamol preparadas usando dos polímeros diferentes WB-4 y WB-7 y reconstituidas a 50 mg/mL.
La Fig. 12 representa una imagen de la torta liofilizada de celecoxib y la disolución obtenida después de la reconstitución de la torta con agua para inyección a la concentración de CEL de 25 mg/mL.
La Fig. 13 muestra la distribución de tamaños de partículas para la formulación de celecoxib reconstituida a 25 mg/mL.
La Fig. 14 muestra una fotografía de formulaciones de PPF preparadas con muestras de polímeros WB-4 a WB-8 antes de la filtración y la liofilización.
La Fig. 15 representa la distribución de tamaño promedio en volumen de las partículas en formulaciones de PPF y polímeros WB-4 a WB-8 antes de la filtración y la liofilización.
La Fig. 16 muestra que la distribución de tamaños de partículas para las formulaciones de PPF preparadas usando los polímeros WB-4, WB-5 y WB-6 es monomodal y que el tamaño promedio en volumen de las micelas varía de aprox. 15 a 150 nm.
La Fig. 17 representa perfiles de tiempo-concentración de FLU f en ratas después de una dosis IV de 2,5 (línea inferior), 10 (línea media) o 30 mg/kg (línea superior) de PPI-1501.
La Fig. 18 muestra perfiles comparativos de tiempo-concentración en plasma de FLU después de la administración IV de una disolución de FLU de 2,5 mg/kg (redibujada de la Referencia 1; línea inferior) y después de la administración IV de PPI-1501 a 2,5 mg/kg (línea superior).
La Fig. 19 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU dosificado como PPI 1501 a la dosis IV de 2,5 mg/kg.
La Fig. 20 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU administrado como PPI 1501 a la dosis IV de 10 mg/kg.
La Fig. 21 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU administrado como PPI 1501 a la dosis IV de 30 mg/kg.
La Fig. 22 representa el ajuste del modelo de dos compartimentos de la concentración plasmática de FLU administrado como una disolución simple a la dosis IV de 2,5 mg/kg.
La Fig. 23 representa una simulación comparativa de la concentración de FLU humana después de una dosis IV única de 100 mg de FLU en disolución frente a la formulación de PPI 1501 y FA. La línea horizontal correspondiente a la concentración de 4 mg/L representa el nivel umbral de recurrencia del dolor.
La Fig. 24 representa una simulación comparativa de la concentración de FLU humana después de una dosis IV bid de 100 mg de FLU en disolución frente a la formulación de PPI 1501 y FA. La línea horizontal correspondiente a la concentración de 4 mg/L representa el nivel umbral de recurrencia del dolor.
La Fig. 25 representa una simulación comparativa de la concentración de FLU humana después de una dosis IV cada 6 h de 50 mg de FLU en disolución frente a la formulación de PPI 1501 y FA. La línea horizontal azul correspondiente a la concentración de 4 mg/L representa el nivel umbral de reaparición del dolor.
La Fig. 26 muestra el porcentaje de hemólisis medido para el flurbiprofeno sin formular y su formulación, ambos a la misma concentración de 5 mg/mL.
La Fig. 27 muestra el porcentaje de hemólisis para diferentes concentraciones de fármaco en formulaciones de flurbiprofeno, celecoxib y paracetamol.
Descripción detallada
Copolímeros en bloque de PVP-PLA
En un aspecto, se proporcionan copolímeros en bloque de PVP-PLA como se define en la Fórmula I:

donde: x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C, n es, en promedio, entre 20 y 40, y m es, en promedio, entre 10 y 40, en donde los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da.
Por "alcohol iniciador" se entiende una especie que tiene un grupo hidroxilo capaz de servir de sustrato para la polimerización, en este caso de poli(D,L lactida) (PLA). Se entenderá que la referencia a un "alcohol iniciador" en el contexto de la estructura anterior pretende significar la forma reaccionada del mismo.
Todos los puntos de ebullición a los que se hace referencia en la presente memoria son a presión estándar. El punto de ebullición del alcohol iniciador se puede seleccionar para conseguir la eficacia de polimerización deseada sin una evaporación problemática del alcohol iniciador.
En una realización, el alcohol iniciador tiene un punto de ebullición superior a 150 °C. En una realización, el alcohol iniciador tiene un punto de ebullición superior a 160 °C. En una realización, el alcohol iniciador tiene un punto de ebullición superior a 170 °C. En una realización, el alcohol iniciador tiene un punto de ebullición superior a 180 °C. En una realización, el alcohol iniciador tiene un punto de ebullición superior a 190 °C. En una realización, el alcohol iniciador tiene un punto de ebullición superior a 200 °C.
En una realización, el alcohol iniciador se selecciona del grupo que consiste en: 1-hexanol; 1-heptanol; éter monoetílico de dietilenglicol; éter monometílico de dietilenglicol; éter monometílico de trietilenglicol; éter monometílico de tetraetilenglicol; monometiléteres de oligoetilenglicol de fórmula II

donde a > 5;
éteres monoetílicos de oligoetilenglicol de fórmula III

donde b > 1;
y mezclas de los mismos.
En una realización, x es éter monoetílico de dietilenglicol (DEGMEE).
El "peso molecular promedio numérico" (Mn) se entenderá como la media aritmética ordinaria o promedio de las masas moleculares de las macromoléculas individuales. Se determina midiendo la masa molecular de n moléculas de polímero, sumando las masas y dividiendo por n:

El peso molecular promedio numérico de un polímero se puede determinar, por ejemplo, mediante cromatografía de permeación en gel, viscosimetría, p. ej. a través de la ecuación de Mark-Houwink, métodos coligativos como la osmometría de presión de vapor, la determinación del grupo final o la RMN de protones.
En una realización, los copolímeros en bloque pueden formar nanopartículas con al menos un ingrediente farmacéutico activo (IFA), en el que las nanopartículas son adecuadas para la administración a un sujeto. La formación de
nanopartículas puede solubilizar el IFA.
Por "nanopartículas" se entiende las partículas que tienen un tamaño de menos de 200 nm. En algunos casos, las nanopartículas pueden tener un tamaño inferior a 100 nm. Las nanopartículas pueden dimensionarse para evitar o reducir la excreción renal. Por ejemplo, las nanopartículas pueden tener un tamaño superior a 15 nm. Las nanopartículas a las que se hace referencia en la presente memoria pueden ser micelas.
Por "micela" se entenderá un autoensamblaje supramolecular compuesto por moléculas que se disponen en forma generalmente esférica en disoluciones acuosas. La formación de una micela es una respuesta a la naturaleza anfipática de los copolímeros en bloque de PVP-PLA, que contienen tanto regiones hidrófilas (grupos PLA) como regiones hidrófobas (grupos PVP). Una micela típica en disolución acuosa se forma con las regiones hidrófilas del polímero en contacto con el disolvente circundante, secuestrando las regiones hidrófobas en el centro de la micela. En la presente memoria, el término "micela" o "micelar" también se puede usar para referirse a la estructura de una forma seca de una composición coloidal previamente líquida de nanopartículas micelares, en la que algunos elementos de una estructura micelar se retienen en forma seca, o en la que la forma seca reforma fácilmente las micelas tras la hidratación.
Las moléculas, incluidos los IFA, se pueden solubilizar mediante la formación de nanopartículas que comprenden la molécula o el IFA. Las nanopartículas pueden ser una nanodispersión.
Por "nanodispersión" se entiende una dispersión de nanopartículas en un medio.
Por "adecuado para la administración a un sujeto" significa que las nanopartículas o una suspensión de nanopartículas posee propiedades que las hacen adecuadas para su administración a un sujeto, p. ej. para cumplir con los requisitos de seguridad. La suspensión de nanopartículas puede, por ejemplo, cumplir con los requisitos establecidos en USP 788, que entre otros establece limitaciones sobre la cantidad de partículas permitidas en el material destinado a la administración parenteral. Se considera que se cumplen estos requisitos si el número de partículas por recipiente (de inyección) es inferior a 6000 para partículas de 10 micras o más, y menos de 600 para partículas iguales a 25 micras o más.
En algunas realizaciones, la suspensión de nanopartículas puede ser esencialmente clara. En algunas realizaciones, la suspensión puede estar libre de partículas visibles. "Esencialmente clara" generalmente significa que está libre de partículas visibles. En algunas realizaciones, la suspensión puede estar libre de partículas subvisibles.
En algunas realizaciones, la suspensión de nanopartículas puede tener una transmitancia óptica indicativa de la claridad general de una disolución. La transmitancia óptica puede, por ejemplo, ser una DO650 de más del 70%, 80%, 90%, 95%, 96%, 97% o 98%, según los requisitos.
En alguna realización, los copolímeros en bloque tienen un Mn de menos de 12.000 Da, 11.000 Da, 10.000 Da, 9.000 Da, 8000 Da, 7000 Da o 6000 Da. En alguna realización, los copolímeros en bloque tienen un Mn de menos de 7000 Da. En alguna realización, los copolímeros en bloque tienen un Mn de más de 4000 Da, 5000 Da o 6000 Da.
Grupo 1
En una realización, los copolímeros en bloque son capaces de formar nanopartículas con al menos un ingrediente farmacéutico activo.
Por "capaz de formar nanopartículas" se entiende que los copolímeros en bloque forman espontáneamente nanopartículas que comprenden los copolímeros en bloque y el IFA cuando se mezclan en condiciones adecuadas. Las nanopartículas pueden ser estables, lo que significa que persisten como nanopartículas (sujetas a equilibrio) en una disolución acuosa durante un período de tiempo determinado. Por ejemplo, las nanopartículas pueden ser estables durante al menos un día. Las nanopartículas pueden ser estables durante al menos dos días. Las nanopartículas pueden ser estables durante al menos tres días. La estabilidad puede estar indicada por la falta de formación de partículas o sedimentos visibles indeseables. La formación de nanopartículas puede solubilizar el IFA.
En una realización, los copolímeros en bloque son capaces de formar nanopartículas con al menos flurbiprofeno (FLU). En una realización, n es, en promedio, de 20 a 28. En una realización, n es, en promedio, de 21 a 27. En una realización, m es, en promedio, de 11 a 37. En una realización, m es, en promedio, de 12 a 36. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 3000 Da a 6600 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 3100 Da a 6500 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1400 Da a 2900 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1500 Da a 2800 Da.
Cuando se hace referencia a una característica del "Bloque de PLA" en la presente memoria, se entenderá que esto se refiere al componente de poli(ácido láctico) de los polímeros. Este valor se puede obtener, por ejemplo, utilizando los valores de Mn calculados por RMN de protones en la Tabla 20 para PLA-PEOX y restando el peso molecular de PEOX (121,2) y el del alcohol iniciador utilizado (134,2).
En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 1200 Da a 4200 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 1300 Da a 4100 Da.
Cuando se hace referencia a una característica del "Bloque de PVP", se entenderá que esto se refiere al componente de polivinilpirrolidona de los polímeros. Este valor se puede obtener, por ejemplo, usando los valores de Mn calculados por RMN de protones en la Tabla 20 para PVP.
En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de aproximadamente 0,3 a aproximadamente 1,4. En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de aproximadamente 0,4 a aproximadamente 1,3.
En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden definirse mediante algunos o todos los parámetros establecidos para los polímeros del Grupo 1 en la Tabla 20. En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden poseer algunas o todas las características de los polímeros WB-DMAP, WB-1, WB-2, WB-3, WB-4, WB-5, WB-6, WB-7 o WB-8 de la Tabla 20.
Grupo 2
En una realización, los copolímeros en bloque de PVP-PLA son capaces de formar nanopartículas con al menos dos ingredientes farmacéuticos activos (IFA) diferentes.
Por "capaz de formar nanopartículas con al menos dos IFA diferentes" se entenderá que el copolímero en bloque puede al menos formar nanopartículas con al menos dos IFA diferentes de forma independiente. En algunas realizaciones, las nanopartículas pueden formar nanopartículas con una mezcla de al menos dos IFA diferentes.
En una realización, los al menos dos IFA diferentes comprenden flurbiprofeno y celecoxib. En una realización, n es, en promedio, de 21,5 a 28. En una realización, n es, en promedio, de 22 a 27. En algunas realizaciones, n puede ser, en promedio, aproximadamente 24. En una realización, m es, en promedio, de 18 a 37. En una realización, m es, en promedio, de 19 a 36. En algunas realizaciones, m puede ser, en promedio, aproximadamente 29. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4600 Da a 6600 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4700 Da a 6500 Da. En algunas realizaciones, el peso molecular promedio en número puede ser de aproximadamente 5700 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1600 Da a 2900 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1700 Da a 2800 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 1900 Da a 4200 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 2000 Da a 4100 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de p La y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,3 a 1,4. En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,4 a 1,3. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en peso (Mw) de PLA en los copolímeros en bloque del 35% al 60%, basado en el peso total del polímero. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en peso (Mw) de PVP en los copolímeros en bloque del 40% al 65%, basado en el peso total del polímero. En algunas realizaciones, el Mw promedio de PLA es de alrededor del 45% y el Mw promedio de PVP es de alrededor del 55%.
"Peso molecular promedio en peso" (Mw) se entenderá como otra forma de describir la masa molar de un polímero, donde las moléculas más grandes tienen una contribución mayor que una molécula más pequeña. La masa molar promedio en masa se calcula mediante:

donde Ni es el número de moléculas de masa molecular Mi. La masa molecular promedio en masa se puede determinar, por ejemplo, mediante dispersión de luz estática, dispersión de neutrones de ángulo pequeño, dispersión de rayos X y velocidad de sedimentación, cromatografía de permeación de gel (GPC) relativa o cromatografía de exclusión por tamaño (SEC).
En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden definirse mediante algunos o todos los parámetros establecidos para los polímeros del Grupo 2 en la Tabla 20. En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden poseer algunas o todas las características de los polímeros WB-1, WB-2, WB-3, WB-4, WB-5, WB-6 o WB-8 de la Tabla 20.
Grupo 3
En una realización, los copolímeros en bloque son capaces de formar nanopartículas con al menos tres ingredientes farmacéuticos activos (IFA) diferentes.
Por "capaz de formar nanopartículas con al menos tres IFA diferentes" se entenderá que el copolímero en bloque puede al menos formar nanopartículas con al menos tres IFA diferentes de forma independiente. En algunas realizaciones, las nanopartículas pueden formar nanopartículas con una mezcla de al menos tres IFA diferentes.
En una realización, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En una realización, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol. En una realización, n es, en promedio, de 21,5 a 28. En una realización, n es, en promedio, de 22 a 27. En algunas realizaciones, n puede ser, en promedio, aproximadamente 24. En una realización, m es, en promedio, de 25 a 37. En una realización, m es, en promedio, de 26 a 36. En algunas realizaciones, m puede ser, en promedio, aproximadamente 31. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4800 Da a 6600 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4900 Da a 6500 Da. En algunas realizaciones, el peso molecular promedio en número es de aproximadamente 5900 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1600 Da a 2900 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 1700 Da a 2800 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 2700 Da a 4200 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 2800 Da a 4100 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,3 a 1,0. En una realización, los copolímeros en bloque de PVP-PLA tienen una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,4 a 0,9. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en peso (Mw) de PLA en los copolímeros en bloque del 35% al 60%, basado en el peso total del polímero. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en peso (Mw) de PVP en los copolímeros en bloque del 40% al 65%, basado en el peso total del polímero. En algunas realizaciones, el Mw promedio de PLA es de alrededor del 45% y el Mw promedio de PVP es de alrededor del 55%.
En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden definirse mediante algunos o todos los parámetros establecidos para los polímeros del Grupo 3 en la Tabla 20. En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden poseer algunas o todas las características de los polímeros WB-1, WB-2, WB-3, WB-4, WB-5 o WB-6.
Grupo 4
En una realización, los copolímeros en bloque pueden ser un subconjunto de aquellos capaces de formar nanopartículas con al menos tres ingredientes farmacéuticos activos (IFA) diferentes.
En una realización, n es, en promedio, de 24 a 28. En una realización, n es, en promedio, de 25 a 27. En algunas realizaciones, n puede ser, en promedio, aproximadamente 26. En una realización, m es, en promedio, de 25 a 33. En una realización, m es, en promedio, de 26 a 32. En algunas realizaciones, m puede ser, en promedio, aproximadamente 29. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 5400 Da a 6600 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 5500 Da a 6500 Da. En algunas realizaciones, el peso molecular promedio en número es de aproximadamente 6000 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 2200 Da a 2900 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PLA de 2300 Da a 2800 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 2700 Da a 3700 Da. En una realización, los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número para el bloque de PVP de 2800 Da a 3600 Da. En una realización, la proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) es de 0,5 a 1,0. En una realización, la proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) es de 0,6 a 0,9. En una realización, el peso molecular promedio en peso (Mw) de PLA en los copolímeros en bloque es del 35 % al 50 %, basado en el peso total del polímero. En una realización, el peso molecular promedio en peso (Mw) de PVP en los copolímeros en bloque es del 50 % al 65 %, basado en el peso total del polímero. En algunas realizaciones, el Mw promedio de PLA es de alrededor del 45% y el Mw promedio de PVP es de alrededor del 55%.
En algunas realizaciones, los copolímeros en bloque de PVP-PLA pueden definirse por algunos o todos los parámetros establecidos para los polímeros del Grupo 4 en la Tabla 20.
Grupos 1 a 4
En las realizaciones anteriores de los Grupos 1 a 4 y anteriores, el peso molecular promedio en número puede medirse mediante resonancia magnética nuclear (RMN) de protones.
En las realizaciones anteriores de los Grupos 1 a 4 y anteriores, el peso molecular promedio en peso puede medirse mediante análisis termogravimétrico (ATG).
En una realización, los copolímeros en bloque de PVP-PLA tienen un índice de polidispersidad (PDI) de <1,8. En una realización, los copolímeros en bloque de PVP-PLA tienen un índice de polidispersidad (PDI) de <1,6. En una realización, los copolímeros en bloque de PVP-PLA tienen un índice de polidispersidad (PDI) de <1,5. En una realización, el índice de polidispersidad es <1,4. En una realización, el índice de polidispersidad es <1,3. En una realización, el índice de polidispersidad es <1,2. En una realización, el índice de polidispersidad es <1,1. En una realización, el PDI se mide mediante cromatografía de permeación en gel con dispersión de luz (GPC-LS).
Sistema de administración de nanovehículos
En un aspecto, se proporciona un sistema de administración de nanovehículos que comprende micelas formadas por los copolímeros en bloque de PVP-PLA descritos anteriormente. "Sistema de administración de nanovehículos" significa nanopartículas formadas a partir de los copolímeros en bloque descritos anteriormente. Las nanopartículas pueden consistir en los copolímeros en bloque descritos anteriormente en algunas realizaciones. El sistema de administración de nanovehículos puede ser útil para solubilizar otra molécula, que puede ser de baja solubilidad o hidrofóbica. El sistema de administración de nanovehículos puede ser útil para solubilizar uno o más IFA, como se describe en la presente memoria.
Formulación Farmacéutica Seca
En un aspecto, se proporciona una composición farmacéutica seca que comprende los copolímeros en bloque de PVP-PLA descritos anteriormente en asociación molecular con al menos un ingrediente farmacéutico activo (IFA).
Por "composición farmacéutica seca" se entiende una formulación preparada secando (p. ej., eliminando el disolvente) de una mezcla del IFA y los copolímeros en bloque para formar una mezcla íntima del IFA y los copolímeros en bloque. En la presente memoria, se entenderá que "seco" significa "sustancialmente seco", e indica que al menos alrededor del 90%, al menos alrededor del 95%, 96%, 97%, 98%, 99% o 99,9% del disolvente se ha eliminado durante el proceso de secado. La composición farmacéutica seca puede estar en forma de una torta de polvo. El término "polvo" se refiere a un material particulado sustancialmente seco, de flujo libre, que tiene una alta densidad aparente. Los polvos secados por aspersión típicamente tienen una densidad aparente en el intervalo de alrededor de 0,05 - 1,00 g/cm3, más típicamente entre alrededor de 0,2 - 0,5 g/cm3. Ventajosamente, los polvos son adecuados para su incorporación en varias formas farmacéuticas no intravenosas, incluidos, entre otros, los comprimidos, incluidos los comprimidos de desintegración rápida, comprimidos oblongos, cápsulas, bolsitas, disoluciones, suspensiones, cremas, geles, ungüentos, pesarios, supositorios, enemas, gotas, aerosoles o inhaladores de polvo seco, y similares. El término "torta", en comparación con un polvo, se refiere a un material sin partículas que no fluye y que tiene una baja densidad aparente, típicamente en el intervalo de aproximadamente 0,0001 - 0,05 g/cm3. De acuerdo con los métodos descritos en la presente memoria, se puede formar una torta, por ejemplo, como resultado de la liofilización.
"Asociación molecular" significa que al menos una parte del IFA está en contacto íntimo con el segmento hidrofóbico de los copolímeros en bloque de PVP-PLA.
Las composiciones farmacéuticas sólidas pueden estar en forma de, o formularse como, diversas formas farmacéuticas, en algunas realizaciones. Los ejemplos incluyen, pero sin limitación, comprimidos, comprimidos oblongos, cápsulas, formulaciones en bolsitas, películas, pastillas, goma de mascar, pastas, ungüentos, aerosoles, inhaladores de aerosol, inhaladores de polvo seco, supositorios, pesarios, etc.
"Ingrediente farmacéutico activo (IFA)" se refiere a un agente que tiene un efecto terapéutico o promotor de la salud cuando se administra a un ser humano o un animal, por ejemplo, un agente capaz de tratar o prevenir una enfermedad o afección. Los ejemplos de agentes terapéuticos incluyen, pero sin limitación, fármacos, profármacos, vitaminas y suplementos.
Los IFA contemplados en la presente memoria incluyen, por ejemplo, compuestos individuales de baja solubilidad como se define en la presente memoria, que incluyen aquellos fármacos categorizados como "ligeramente solubles", "muy ligeramente solubles", "prácticamente insolubles" e "insolubles" en USP 24, págs. 2254-2298; y aquellos fármacos clasificados por requerir 100 mL o más de agua para disolver 1 g del fármaco, como se enumera en USP 24, págs. 2299-2304.
Los compuestos ejemplares incluyen, sin limitación; compuestos de las siguientes clases: abortivos, inhibidores de la ECA, agonistas adrenérgicos alfa y beta, bloqueadores adrenérgicos alfa y beta, supresores adrenocorticales, hormonas adrenocorticotrópicas, disuasivos del alcohol, inhibidores de la aldosa reductasa, antagonistas de la aldosterona, anabólicos, analgésicos (incluidos narcóticos y analgésicos no narcóticos), anestésicos, andrógenos, antagonistas de los
receptores de la angiotensina II, anoréxicos, antiácidos, antihelmínticos, agentes antiacné, antialérgicos, agentes antialopecia, antiamebianos, antiandrógenos, agentes antianginosos, antiarrítmicos, antiarterioscleróticos, agentes antiartríticos/antirreumáticos, antiasmáticos, antibacterianos, antibacterianos adyuvantes, anticolinérgicos, anticoagulantes, anticonvulsivos, antidepresivos, antidiabéticos, agentes antidiarreicos, antidiuréticos, antídotos de venenos, antidiscinéticos, antieccemáticos, antieméticos, antiestrógenos, antifibróticos, antiflatulentos, antifúngicos, agentes antiglaucoma, antigonadotropinas, agentes antigotosos, antihistamínicos, antihiperactivos, antihiperlipoproteinémicos, antihiperfosfatémicos, antihipertensivos, agentes antihipertiroideos, antihipotensivos, agentes antihipotiroideos, antiinflamatorios, antipalúdicos, antimaníacos, antimetahemoglobinémicos, agentes antimigrañosos, antimuscarínicos, antimicobacterianos, agentes antineoplásicos y adjuntos, antineutropénicos, antiosteoporóticos, antipagéticos, agentes antiparkinsonianos, agentes antifeocromocitoma, agentes antipneumocystis, agentes antihipertrofia prostática, antiprotozoarios, antipruriginosos, antipsoriáticos, antipsicóticos, antipiréticos, antirickettsiales, antiseborreicos, antisépticos/desinfectantes, antiespasmódicos, antisifilíticos, antitrombocitémicos, antitrombóticos, antitusivos, antiulcerativos, antiurolíticos, antivenenos, agentes antivirales, ansiolíticos, inhibidores de la aromatasa, antagonistas de las benzodiacepinas, inhibidores de la resorción ósea, agentes bradicárdicos, antagonistas de la bradicinina, broncodilatadores, bloqueadores de los canales de calcio, reguladores del calcio, inhibidores de la anhidrasa carbónica, cardiotónicos, antagonistas de CCK, agentes quelantes, agentes colelitolíticos, coleréticos, colinérgicos, inhibidores de la colinesterasa, reactivadores de la colinesterasa, estimulantes del SNC, anticonceptivos, agentes de desbridamiento, descongestionantes, despigmentantes, supresores de la dermatitis herpetiforme, auxiliares digestivos, diuréticos, agonistas de los receptores de dopamina, antagonistas de los receptores de dopamina, ectoparasiticidas, eméticos, inhibidores de encefalinasa, enzimas, cofactores enzimáticos, estrógenos, expectorantes, antagonistas de los receptores de fibrinógeno, suplementos de flúor, estimulantes de la secreción gástrica y pancreática, citoprotectores gástricos, inhibidores de la bomba de protones gástrica, inhibidores de la secreción gástrica, gastroprocinéticos, glucocorticoides, inhibidores de la a-glucosidasa, principios estimulantes de las gónadas, inhibidores de la hormona del crecimiento, factores liberadores de la hormona del crecimiento, estimulantes del crecimiento, hematínicos, hematopoyéticos, hemolíticos, hemostáticos, antagonistas de la heparina, inductores de enzimas hepáticas, hepatoprotectores, antagonistas del receptor de histamina H2, inhibidores de la proteasa del VIH, inhibidores de la HMG CoA reductasa, inmunomoduladores, inmunosupresores, sensibilizadores de la insulina, resinas de intercambio iónico, queratolíticos, hormonas estimulantes de la lactancia, laxantes/catárticos, antagonistas de los leucotrienos, agonistas de la LH-RH, lipotrópicos, inhibidores de la 5-lipoxigenasa, supresores del lupus eritematoso, inhibidores de la metaloproteinasa de la matriz, mineralocorticoides, mióticos, inhibidores de la monoaminooxidasa, mucolíticos, relajantes musculares, midriáticos, antagonistas de narcóticos, neuroprotectores, nootrópicos, hormonas ováricas, oxitócicos, inhibidores de la pepsina, agentes de pigmentación, expansores del volumen plasmático, activadores/abridores de los canales de potasio, progestágenos, inhibidores de la prolactina, prostaglandinas, inhibidores de proteasas, radiofármacos, inhibidores de la 5a-reductasa, estimulantes respiratorios, inhibidores de la transcriptasa inversa, sedantes/hipnóticos, serénicos, inhibidores de la recaptación de serotonina y noradrenalina, agonistas de los receptores de serotonina, antagonistas de los receptores de serotonina, inhibidores de la recaptación de serotonina, análogos de somatostatina, trombolíticos, antagonistas del receptor de tromboxano A2, hormonas tiroideas, hormonas tirotrópicas, tocolíticos, inhibidores de la topoisomerasa I y II, uricosúricos, vasomoduladores, incluidos los vasodilatadores y vasoconstrictores, vasoprotectores, inhibidores de la xantina oxidasa y combinaciones de los mismos.
Los ejemplos de IFA incluyen, sin limitación, paracetamol, acetohexamida, ácido acetilsalicílico, alclofenaco, alopurinol, atropina, benztiazida, carprofeno, celecoxib, clordiazepóxido, clorpromazina, clonidina, codeína, fosfato de codeína, sulfato de codeína, deracoxib, diacereína, diclofenaco, diltiazem, estradiol, etodolaco, etopósido, etoricoxib, fenbufeno, fendofenac, fenprofeno, fentiazac, flurbiprofeno, griseofulvina, haloperidol, ibuprofeno, indometacina, indoprofeno, ketoprofeno, lorazepam, acetato de medroxiprogesterona, megestrol, metoxsaleno, metilprednisona, morfina, sulfato de morfina, naproxeno, nicergolina, nifedipina, niflumic, oxaprozina, oxazepam, oxifenbutazona, paclitaxel, palperidona, fenindiona, fenobarbital, piroxicam, pirprofeno, prednisolona, prednisona, procaína, progesterona, propofol, pirimetamina, risperidona, rofecoxib, sulfadiazina, sulfamerazina, sulfisoxazol, sulindac, suprofen, temazepam, ácido tiaprofénico, tilomisol, tolmético, valdecoxib y ziprasidona.
Otros ejemplos de IFA incluyen, sin limitación, acenocumarol, acetildigitoxina, anetol, anileridina, benzocaína, benzonatato, betametasona, acetato de betametasona, valerato de betametasona, bisacodilo, bromodifenhidramina, butamben, clorambucilo, cloranfenicol, clordiazepóxido, clorobutanol, clorocresol, clorpromazina, palmitato de clindamicina, clioquinol, acetato de cortisona, clorhidrato de ciclizina, clorhidrato de ciproheptadina, demeclociclina, diazepam, dibucaína, digitoxina, mesilato de dihidroergotamina, dimetisterona, disulfiram, docusato de calcio, docusato de sodio, dihidrogesterona, enalaprilato, tartrato de ergotamina, eritromicina, estolato de eritromicina, pivalato de flumetasona, acetónido de fluocinolona, fluorometolona, enantato de flufenazina, flurandrenolida, guaifenesina, halazona, hidrocortisona, levotiroxina sódica, metilclotiazida, miconazol, nitrato de miconazol, nitrofurazona, nitromersol, oxazepam, pentazocina, pentobarbital, primidona, sulfato de quinina, estanozolol, nitrato de sulconazol, sulfadimetoxina, sulfaetidol, sulfametizol, sulfametoxazol, sulfapiridina, testosterona, triazolam, triclorometiazida y trioxsaleno.
En una realización, la composición farmacéutica seca es reconstituible en agua o una disolución acuosa en nanopartículas formadas por los copolímeros en bloque de PVP-PLA y que comprenden al menos un IFA. Las nanopartículas pueden ser micelas.
En una realización, la composición farmacéutica seca se seca por congelación o se seca por pulverización. La
composición también se puede "secar en lecho", es decir, secar en un lecho fluidizado.
En una realización, la composición farmacéutica seca es amorfa.
"Amorfo" significa que el IFA se mantiene en un estado generalmente desordenado, es decir, no cristalino.
En una realización, la composición farmacéutica seca es estable durante al menos seis meses a 40 °C.
Por "estable", en el contexto de una composición seca, se entiende que la composición seca puede almacenarse durante un período de tiempo después del cual todavía es reconstituible en forma de nanopartículas estables.
Ese período de tiempo puede ser, p. ej. 1 mes, 2 meses, 3 meses, 4 meses, 5 meses o 6 meses. La temperatura a la que es estable puede ser, por ejemplo, la temperatura ambiente, por ejemplo alrededor de 25 °C. También puede ser 40 °C. La composición puede ser estable durante este período a una humedad relativa del 75%. En algunas realizaciones, las composiciones son estables durante 6 meses a 40 °C y 75% de humedad relativa. Las nanopartículas estables así reconstituidas, en algunas realizaciones, son estables durante al menos tres días. La composición seca puede ser estable durante 3 meses a 25 °C. La composición seca puede ser estable durante 6 meses a 25 °C. La composición seca puede ser estable durante 3 meses a 40 °C. La composición seca puede ser estable durante 6 meses a 60 °C. La composición seca puede ser estable durante 3 meses a 25 °C.
"Nivel de carga de fármacos" (NCF), tal como se hace referencia en la presente memoria, significa la proporción en peso del fármaco con respecto a la suma del peso del fármaco y del polímero en la formulación.
En una realización, la composición farmacéutica seca tiene un NCF de al menos un 10 % p/p del al menos un IFA. En una realización, la composición farmacéutica seca tiene un NCF de al menos un 20 % p/p del al menos un IFA. En una realización, la composición farmacéutica seca tiene un NCF de al menos un 30 % p/p del al menos un IFA. El NCF se puede seleccionar de acuerdo con el IFA y de acuerdo con los requisitos, y siempre que se cumplan los requisitos para la administración. El NCF puede ser del 5 %, 7,5 %, 10 %, 12,5 %, 15 %, 17,5 %, 20 %, 22,5 %, 25 %, 27,5 %, 30 % o superior al 30 %.
En una realización, el al menos un IFA es hidrofóbico.
Como se usa en la presente memoria, "hidrofóbico" significa sustancialmente inmiscible con un medio acuoso. En una realización, el al menos un IFA puede ser insoluble o escasamente soluble. En una realización, el al menos un IFA tiene una solubilidad en agua de 0 g/L a 33 g/L. El IFA puede ser insoluble o ligeramente soluble (es decir, 0 g/L a 10 g/L). En una realización, el al menos un IFA tiene una solubilidad en agua de 0 g/L a 14 g/L. El IFA puede ser escasamente soluble. En una realización, el IFA tiene una solubilidad en agua de 10 g/L a 33 g/L.
En una realización, el al menos un IFA se selecciona de la clase II y clase IV del Sistema de Clasificación Biofarmacéutica (BCS).
En una realización, el al menos un IFA comprende un analgésico.
Por "analgésico" se entiende cualquier miembro del grupo de fármacos utilizados para lograr la reducción o el alivio del dolor. Algunos ejemplos incluyen paracetamol, medicamentos antiinflamatorios no esteroideos (AINE), inhibidores de la COX-2, opioides, etc.
En una realización, el analgésico comprende un fármaco antiinflamatorio no esteroideo (AINE).
"AINE" se refiere a un fármaco no narcótico que proporciona efectos analgésicos (reductores del dolor) y antipiréticos (reductores de la fiebre) y, en dosis más altas, efectos antiinflamatorios. Algunos ejemplos incluyen aspirina, ibuprofeno y naproxeno.
En una realización, el AINE comprende flurbiprofeno.
En una realización, el AINE comprende un inhibidor de la COX-2.
Por "inhibidor COX-2" se entiende un fármaco que inhibe selectivamente la enzima ciclooxigenasa-2. En una realización, el inhibidor de COX-2 comprende celecoxib.
En una realización, el analgésico comprende paracetamol.
En una realización, el al menos un IFA comprende un anestésico.
Por "anestésico" se entiende uno de los fármacos utilizados para prevenir el dolor, p. ej. durante la cirugía. Los ejemplos no limitantes de anestésicos no opioides incluyen barbitúricos (p. ej., amobarbital, metohexital, tiamilal, tiopental), benzodiacepinas (p. ej., diazepam, lorazepam, midazolam), etomidato, ketamina y propofol. Los ejemplos no limitantes de anestésicos opioides incluyen alfentanilo, fentanilo, remifentanilo, sufentanilo, buprenorfina, butorfanol, diacetilmorfina, hidromorfona, levorfanol, meperidina, metadona, morfina, nalbufina, oxicodona, oximorfona
y pentazocina.
En una realización, el anestésico comprende propofol.
En una realización, la composición farmacéutica seca se puede administrar por vía oral.
En una realización, la composición farmacéutica seca es reconstituible en agua para formar un líquido esencialmente claro que comprende nanopartículas formadas por los copolímeros en bloque de PVP-PLA y que comprende al menos un IFA.
En una realización, el líquido esencialmente claro comprende al menos 20 g/L de al menos un IFA. En una realización, el líquido esencialmente claro comprende al menos 30 g/L de al menos un IFA. En una realización, el líquido esencialmente claro comprende al menos 40 g/ L de al menos un IFA. En una realización, el líquido esencialmente claro comprende al menos 50 g/L de al menos un IFA.
En una realización, la composición farmacéutica seca proporciona un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 4 horas. En una realización, la composición farmacéutica seca proporciona un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 6 horas. En una realización, la composición farmacéutica seca proporciona un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 8 horas. En una realización, la composición farmacéutica seca proporciona un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 12 horas.
En algunas realizaciones, la composición farmacéutica seca comprende al menos dos IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 2. En algunas realizaciones, los al menos dos IFA comprenden flurbiprofeno y celecoxib.
En algunas realizaciones, la composición farmacéutica seca comprende al menos tres IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 3. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
En algunas realizaciones, la composición farmacéutica seca comprende al menos tres IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 4. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
Composición farmacéutica líquida
En un aspecto, se proporciona una composición farmacéutica líquida que comprende nanopartículas formadas por los copolímeros en bloque de PVP-PLA descritos anteriormente y que comprende al menos un ingrediente farmacéutico activo (IFA).
En una realización, la composición farmacéutica líquida es un líquido esencialmente clara.
En una realización, el líquido esencialmente claro se puede filtrar a través de un filtro de esterilización. Un "filtro de esterilización" se entenderá como un filtro que consigue la filtración necesaria para cumplir los requisitos de esterilidad. Por ejemplo, dicho filtro puede tener un tamaño de poro de 0,45 micras o 0,2 micras.
En una realización, el líquido esencialmente claro se administra por vía parenteral. En una realización, el líquido esencialmente claro es inyectable. En una realización, el líquido esencialmente claro se puede administrar por vía intravenosa. En una realización, el líquido esencialmente claro se puede administrar por infusión. El líquido esencialmente claro puede administrarse por vía parenteral, intravenosa, por infusión, intraocular, intratecal, intramuscular, intraperitoneal o intraespinal.
En una realización, la composición farmacéutica líquida es estable durante al menos tres días a 25 °C.
En una realización, la composición farmacéutica líquida proporciona un nivel plasmático farmacéuticamente efectivo de al menos un IFA en 2 minutos. La composición farmacéutica líquida puede proporcionar un nivel plasmático farmacéuticamente eficaz en 1 minuto, 2 minutos, 5 minutos, 10 minutos, 20 minutos o 30 minutos.
Un "nivel plasmático farmacéuticamente eficaz" se refiere a una cantidad de IFA que, cuando está en plasma, es suficiente para lograr la eficacia terapéutica deseada. Este nivel puede variar, por ejemplo, según el IFA, la enfermedad, el trastorno y/o los síntomas de la enfermedad o trastorno, la gravedad de la enfermedad, el trastorno y/o los síntomas de la enfermedad o el trastorno, la edad, el peso y/o la salud del paciente a tratar.
En una realización, se mantiene un nivel plasmático farmacéuticamente eficaz del IFA durante al menos 4 horas. En una realización, se mantiene un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 6 horas. En una realización, se mantiene un nivel plasmático farmacéuticamente efectivo de al menos un IFA durante al menos 8 horas. En una realización, se mantiene un nivel plasmático farmacéuticamente efectivo de al menos un IFA
durante al menos 12 horas.
En una realización, la composición farmacéutica líquida comprende además un diluyente, excipiente o vehículo farmacéuticamente aceptable. "Farmacéuticamente aceptable" se refiere a aditivos que no son tóxicos cuando se administran a un paciente en una cantidad suficiente para proporcionar un efecto terapéutico del IFA y que no destruyen la actividad biológica del al menos un IFA. "Diluyente, excipiente o vehículo" se entenderá como un aditivo que no tiene sustancialmente actividad farmacológica propia.
En una realización, el al menos un IFA es hidrofóbico.
En una realización, el al menos un IFA puede ser insoluble o escasamente soluble. En una realización, el al menos un IFA tiene una solubilidad en agua de 0 g/L a 33 g/L. El IFA puede ser insoluble o ligeramente soluble (es decir, 0 g/L a 10 g/L). En una realización, el IFA tiene una solubilidad en agua de 0 g/L a 14 g/L . El IFA puede ser escasamente soluble. En una realización, el IFA tiene una solubilidad en agua de 10 g/L a 33 g/L.
En una realización, el al menos un IFA se selecciona de la clase II y clase IV del Sistema de Clasificación Biofarmacéutica (BCS).
En una realización, el al menos un IFA comprende un analgésico.
En una realización, el analgésico comprende un fármaco antiinflamatorio no esteroideo (AINE).
En una realización, el AINE comprende flurbiprofeno.
En una realización, el AINE comprende un inhibidor de la COX-2.
En una realización, el inhibidor de COX-2 comprende celecoxib.
En una realización, el analgésico comprende paracetamol.
En una realización, el al menos un IFA comprende un anestésico.
En una realización, el anestésico comprende propofol.
En una realización, la composición farmacéutica líquida comprende al menos 20 g/L de al menos un IFA. En una realización, la composición farmacéutica líquida comprende al menos 30 g/L de al menos un IFA. En una realización, la composición farmacéutica líquida comprende al menos 40 g/L de al menos un IFA. En una realización, la composición farmacéutica líquida comprende al menos 50 g/L de al menos un IFA.
En algunas realizaciones, la composición farmacéutica líquida comprende al menos dos IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 2. En algunas realizaciones, los al menos dos IFA comprenden flurbiprofeno y celecoxib.
En algunas realizaciones, la composición farmacéutica líquida comprende al menos tres IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 3. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
En algunas realizaciones, la composición farmacéutica líquida comprende al menos tres IFA, en donde los copolímeros en bloque son como se definieron anteriormente en el Grupo 4. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
Hemólisis
En un aspecto, los copolímeros en bloque descritos anteriormente pueden encontrar aplicación en la mitigación de los efectos hemolíticos.
En un aspecto, los copolímeros en bloque descritos anteriormente pueden encontrar aplicación en la mitigación de la actividad hemolítica de una molécula de carga. La carga puede ser una molécula pequeña o grande, como un IFA. Por "actividad hemolítica" o "efectos hemolíticos" se entenderá la propensión de una molécula o formulación particular a causar hemólisis, es decir, lisis de glóbulos rojos (RBC). Estos pueden ser glóbulos rojos de mamíferos, por ejemplo, glóbulos rojos humanos. Se puede determinar la propensión a causar hemólisis in vitro, por ejemplo, usando glóbulos rojos derivados de sangre, por ejemplo, derivados de sangre humana completa. Una prueba in vitro ejemplar es el método seguido en el Ejemplo 5. Por ejemplo, los glóbulos rojos suspendidos aislados de sangre humana completa se pueden mezclar con moléculas de carga sin formular o formulaciones de prueba que comprenden la molécula de carga en una concentración final dada con respecto a la molécula de carga incubada a 37 °C durante 60 minutos y centrifugar para eliminar los glóbulos rojos intactos. A continuación, se puede medir la absorbancia a 540 nm y compararla con las muestras de control, p. ej. para 0 y 100% de hemólisis. La prueba in vitro puede ser una establecida
para proporcionar una indicación del potencial hemolítico de la molécula de carga o el potencial hemoprotector de la formulación in vivo.
Cuando se mencione la mitigación de la hemólisis para una molécula de carga formulada en particular (es decir, formulada con copolímero en bloque de PVP-PLA), se entenderá que esto significa que existe un grado de protección proporcionado por la formulación (el potencial hemoprotector) a los glóbulos rojos contra la hemólisis inducida por la molécula de carga cuya protección puede expresarse como un porcentaje de la causada por la molécula de carga en un estado sin formular o formulado convencionalmente. Por ejemplo, el grado de hemoprotección proporcionado a los glóbulos rojos por una formulación de FLU con copolímero en bloque de PVP-PLA frente a la actividad hemolítica de
FLU sin formular o formulado convencionalmente puede calcularse como:
100% - ((porcentaje de hemólisis observado para la formulación de FLU y PVP-PLA) (porcentaje de hemólisis observado para la formulación de FLU no formulado o convencional) x 100 %).
Así, en un escenario de ejemplo en el que FLU no formulado a una concentración final de 5 g/L provoque un 90 % de hemólisis y FLU con PVP-PLa provoque un 7 % (es decir, similar a la del control negativo), el grado de protección sería del 100 % -(7/90 x 100%) = 92%.
Por "estado sin formular" se entenderá la molécula en ausencia de los copolímeros en bloque de PVP-PLA descritos en la presente memoria. Igualmente, "formulado convencionalmente" se entenderá como alguna otra formulación de la molécula que no incluye los copolímeros en bloque de PVP-PLA descritos en la presente memoria.
En una realización, se proporciona un uso de los copolímeros en bloque descritos anteriormente para mitigar la actividad hemolítica de una molécula de carga, como un IFA. En una realización, se proporciona un uso de los copolímeros en bloque descritos anteriormente para mitigar el potencial hemolítico de una molécula de carga, tal como un IFA, a niveles de control negativo. En algunas realizaciones, se entenderá que los niveles de control negativo son el grado de hemólisis provocado por la adición de la disolución de control negativo. Un ejemplo es el control negativo descrito en el Ejemplo 5. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo
1. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 2. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 3. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 4. En una realización, el grado de protección, como se definió anteriormente, es de al menos el 50%. En una realización, el grado de protección es de al menos el 60%. En una realización, el grado de protección es de al menos el 70%. En una realización, el grado de protección es de al menos el 80%. En una realización, el grado de protección es de al menos el 90%. En una realización, el grado de protección es de al menos del 92%. En una realización, el grado de protección es de al menos del 95%.
En una realización, se proporciona un copolímero en bloque, como se describe anteriormente, para el uso en la mitigación de la actividad hemolítica de una molécula de carga, como un IFA. En una realización, se proporciona un copolímero en bloque para mitigar el potencial hemolítico de una molécula de carga, como un IFA, a niveles de control negativo. En algunas realizaciones, se entenderá que los niveles de control negativo son el grado de hemólisis provocado por la adición de la disolución de control negativo. Un ejemplo es el control negativo descrito en el Ejemplo
5. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 1. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 2. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 3. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 4. En una realización, el grado de protección, como se definió anteriormente, es de al menos el 50%. En una realización, el grado de protección es de al menos el 60%. En una realización, el grado de protección es de al menos el 70%. En una realización, el grado de protección es de al menos el 80%. En una realización, el grado de protección es de al menos el 90%. En una realización, el grado de protección es de al menos del 92%. En una realización, el grado de protección es de al menos del 95%.
En una realización, se proporciona un método para mitigar la actividad hemolítica de una molécula de carga, tal como un IFA, y el método comprende las etapas de: mezclar los copolímeros en bloque descritos anteriormente con el IFA para formar una composición farmacéutica líquida, exhibiendo dicha composición farmacéutica líquida una actividad hemolítica reducida o nula en comparación con el IFA en un estado sin formular. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 1. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 2. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 3. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 4. En una realización, el grado de protección, como se definió anteriormente, es de al menos el 50%. En una realización, el grado de protección es de al menos el 60%. En una realización, el grado de protección es de al menos el 70%. En una realización, el grado de protección es de al menos el 80%. En una realización, el grado de protección es de al menos el 90%. En una realización, el grado de protección es de al menos del 92%. En una realización, el grado de protección es de al menos del 95%.
En una realización, la composición farmacéutica seca descrita anteriormente exhibe efectos hemolíticos mitigados para el IFA, cuando se reconstituye para formar una composición farmacéutica líquida. En una realización, la composición farmacéutica seca descrita anteriormente muestra una actividad hemolítica mitigada para el IFA, cuando se reconstituye para formar una composición farmacéutica líquida, p. ej. en comparación con el IFA solo. En una
realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 1. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 2. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 3. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 4. En una realización, el grado de protección, como se definió anteriormente, es de al menos el 50%. En una realización, el grado de protección es de al menos el 60%. En una realización, el grado de protección es de al menos el 70%. En una realización, el grado de protección es de al menos el 80%. En una realización, el grado de protección es de al menos el 90%. En una realización, el grado de protección es de al menos del 92%. En una realización, el grado de protección es de al menos del 95%.
En una realización, la formulación líquida descrita anteriormente exhibe una actividad hemolítica mitigada para el IFA. En una realización, la formulación líquida descrita anteriormente muestra una actividad hemolítica mitigada para el IFA, p. ej. en comparación con el IFA solo. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 1. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 2. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 3. En una realización, los copolímeros en bloque pueden ser como se definieron para el Grupo 4. En una realización, el grado de protección, como se definió anteriormente, es de al menos el 50%. En una realización, el grado de protección es de al menos el 60%. En una realización, el grado de protección es de al menos el 70%. En una realización, el grado de protección es de al menos el 80%. En una realización, el grado de protección es de al menos el 90%. En una realización, el grado de protección es de al menos del 92%. En una realización, el grado de protección es de al menos del 95%.
En una realización de lo anterior, el IFA comprende celecoxib. En algunas de estas realizaciones, el NCF de celecoxib es del 5 % al 15 %. En una realización, el NCF de celecoxib es de alrededor del 10 %.
En una realización de lo anterior, el IFA comprende paracetamol. En algunas de estas realizaciones, el NCF del paracetamol es del 13 % al 23 %. En una realización, el NCF del paracetamol es de alrededor del 18%.
En lo anterior, el IFA puede ser uno que provoque hemólisis en un estado sin formular o formulado convencionalmente. En algunas realizaciones, el nivel de hemólisis provocado por el IFA puede ser un nivel que impida su administración a un sujeto. El IFA puede ser uno del que se sabe que causa hemólisis, p. ej. cuando se administra (por ejemplo, por vía parenteral) a un sujeto en un estado sin formular. El IFA puede ser un iFa que, en su estado sin formular, provoque más del 10 % de hemólisis de glóbulos rojos en la prueba expuesta en el Ejemplo 5. Se entenderá que un iFa que "cause hemólisis" puede hacerlo a un nivel deseado de administración en un estado sin formular o formulado convencionalmente, y no necesariamente en absoluto (o en la misma medida) a otros niveles en un estado sin formular o formulado convencionalmente. Un ejemplo de IFA que provoca hemólisis en un estado sin formular es el flurbiprofeno.
En algunas realizaciones de lo anterior, el IFA es flurbiprofeno. En algunas de estas realizaciones, el NCF del flurbiprofeno es del 15 al 25%. En una realización, el NCF del flurbiprofeno es de aproximadamente el 20 %.
Método de formación de composiciones farmacéuticas
En un aspecto, se proporciona un método para formar una composición farmacéutica que comprende mezclar los copolímeros en bloque descritos anteriormente con al menos un IFA para formar nanopartículas que comprenden al menos un IFA.
En una realización, el al menos un IFA comprende al menos dos IFA, y los copolímeros en bloque son como se definieron anteriormente en el Grupo 2. En algunas realizaciones, los al menos dos IFA comprenden flurbiprofeno y celecoxib.
En una realización, el al menos un IFA comprende al menos tres IFA, y los copolímeros en bloque son como se definieron anteriormente en el Grupo 3. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol. En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
En una realización, el al menos un IFA comprende al menos tres IFA, y los copolímeros en bloque son como se definieron anteriormente en el Grupo 4. En algunas realizaciones, los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol En algunas realizaciones, los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
En una realización, los métodos descritos anteriormente pueden comprender una etapa de secado de la composición farmacéutica.
En algunas realizaciones que involucran una pluralidad de IFA, la pluralidad de IFA se puede mezclar junto con los copolímeros.
Alternativamente, el método puede comprender mezclar por separado cada IFA con una cantidad del copolímero en bloque antes de combinarlos. Estas realizaciones pueden comprender una etapa adicional de incubar las dos poblaciones de nanopartículas así formadas juntas durante un tiempo suficiente para que tenga lugar un intercambio de IFA a medida que se equilibra la mezcla.
Métodos de administración, usos y kits
En un aspecto, se proporciona un método para administrar al menos un ingrediente farmacéutico activo (IFA) a un sujeto que lo necesita, que comprende administrar al sujeto la composición farmacéutica seca descrita anteriormente. En una realización, la administración es por vía oral.
En un aspecto, se proporciona un método para administrar al menos un ingrediente farmacéutico activo (IFA) a un sujeto que lo necesita, que comprende administrar al sujeto la composición farmacéutica líquida descrita anteriormente. En una realización, dicha administración comprende inyectar, administrar por vía parenteral, administrar por vía intravenosa, infundir, administrar por vía intraocular, administrar por vía intratecal, administrar por vía intramuscular, administrar por vía intraperitoneal o administrar por vía intraespinal.
En una realización, el nivel plasmático farmacéuticamente eficaz se mantiene durante al menos 4 horas. En una realización, el nivel plasmático farmacéuticamente eficaz se mantiene durante al menos 6 horas. En una realización, el nivel plasmático farmacéuticamente eficaz se mantiene durante al menos 8 horas. En una realización, el nivel plasmático farmacéuticamente eficaz se mantiene durante al menos 12 horas.
En una realización, el sujeto tiene dolor y el al menos un IFA comprende un analgésico. En una realización, el analgésico comprende un AINE. En una realización, el AINE comprende flurbiprofeno. En una realización, el AINE comprende celecoxib.
En una realización, el analgésico comprende paracetamol.
En una realización, el sujeto necesita anestesia y al menos un IFA es un anestésico. En una realización, el anestésico es propofol.
En un aspecto, se proporciona la composición farmacéutica seca descrita anteriormente o la composición farmacéutica líquida descrita anteriormente para el uso en la administración de al menos un ingrediente farmacéutico activo (IFA) a un sujeto.
En una realización, la composición es para el uso en el tratamiento del dolor en el sujeto, en el que al menos un IFA comprende un analgésico.
En una realización, el analgésico comprende un AINE.
En una realización, el AINE comprende flurbiprofeno.
En una realización, el AINE comprende celecoxib.
En una realización, el analgésico comprende paracetamol.
En una realización, la composición se utiliza para proporcionar anestesia al sujeto, en la que el IFA comprende un anestésico.
En una realización, el anestésico es propofol.
En un aspecto, se proporciona un kit que comprende la composición farmacéutica seca descrita anteriormente o la composición farmacéutica líquida descrita anteriormente, e instrucciones de uso en la administración de al menos un ingrediente farmacéutico activo (IFA) a un sujeto.
En un aspecto, se proporciona un kit que comprende la composición farmacéutica seca descrita anteriormente o la composición farmacéutica líquida descrita anteriormente, e instrucciones de uso en la administración de al menos un ingrediente farmacéutico activo (IFA) para el tratamiento del dolor en un sujeto.
En una realización, el al menos un IFA comprende flurbiprofeno, celecoxib o paracetamol.
En un aspecto, se proporciona un kit que comprende la composición farmacéutica seca descrita anteriormente o la composición farmacéutica líquida descrita anteriormente, e instrucciones de uso en la administración de al menos un ingrediente farmacéutico activo (IFA) para proporcionar anestesia a un sujeto.
En una realización, el al menos un IFA comprende propofol.
Métodos de producción
En otro aspecto, se proporciona un método para preparar copolímeros en bloque de PVP-PLA como se define en la Fórmula I:
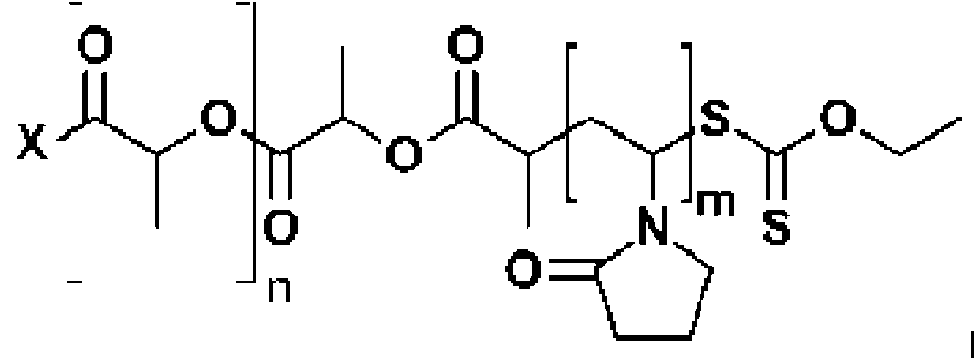
donde: x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C, n es, en promedio, entre 20 y 40, y m es, en promedio, entre 10 y 40, en donde los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da, comprendiendo el método: iniciar la polimerización de D,L-Lactida a partir del alcohol iniciador x para formar poli(ácido láctico) (PLA), añadir un xantato al PLA para formar un macroiniciador de PLA, y polimerizar la NVP, mediante polimerización controlada, sobre el macroiniciador de PLA para formar el compuesto de copolímero en bloque de fórmula I.
En una realización, la polimerización controlada es una polimerización por radicales controlada.
En una realización, el copolímero en bloque de PVP-PLA es como se define en la presente memoria.
En una realización, el copolímero en bloque de PVP-PLA es como se definió anteriormente en el Grupo 1.
En una realización, el copolímero en bloque de PVP-PLA es como se definió anteriormente en el Grupo 2.
En una realización, el copolímero en bloque de PVP-PLA es como se definió anteriormente en el Grupo 3.
En una realización, el copolímero en bloque de PVP-PLA es como se definió anteriormente en el Grupo 4.
En una realización, la polimerización de la D,L-lactida es mediante una polimerización por coordinación-inserción organocatalítica. En una realización, el catalizador para la polimerización de D,L-lactida es 4-dimetilaminopiridina (DMAP). En una realización, la polimerización de la D,L-lactida tiene lugar a una temperatura de al menos 60 °C a 90 °C. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 24 horas. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 48 horas. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 60 horas.
En una realización, la polimerización de la D,L-lactida es mediante una polimerización por coordinación-inserción organocatalítica. En una realización, el catalizador para la polimerización de la D,L-lactida es octoato estañoso (Sn(Oct)2). En una realización, la polimerización de la D,L-lactida tiene lugar a una temperatura de al menos 135 °C a 160 °C. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 24 horas. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 48 horas. En una realización, la polimerización de la D,L-lactida tiene lugar durante al menos 60 horas.
En una realización, el catalizador para la polimerización de la NVP es azobisisobutironitrilo (AIBN). En una realización, la polimerización de la NVP tiene lugar a una temperatura de al menos 70 °C a 90 °C. En una realización, la polimerización de la NVP tiene lugar durante al menos 24 horas. En una realización, la polimerización de la NVP tiene lugar durante al menos 48 horas. En una realización, la polimerización de la NVP tiene lugar durante al menos 60 horas.
En una realización, el método comprende además eliminar el xantato.
En una realización, el método comprende además añadir un resto funcional.
Por "resto funcional" se entiende una modificación o adición molecular que proporciona una actividad o reactividad funcional deseada. El "resto funcional" puede ser un "resto selectivo" diseñado para dirigir las nanopartículas resultantes formadas a partir de los copolímeros en bloque hacia un tipo de célula particular, p. ej. seleccionando como objetivo una proteína o receptor particular de la superficie celular. La adición de un resto funcional puede dar como resultado nanopartículas funcionalizadas o decoradas. Por ejemplo, los restos funcionales pueden incluir un colorante, un marcador o un grupo funcional terminal.
En una realización, el resto funcional comprende un resto selectivo.
"Resto selectivo" se refiere a cualquier compuesto químico, incluyendo, por ejemplo, un péptido o ácido nucleico, que imparte una función de selección del objetivo a las micelas formadas por los copolímeros en bloque a las que está unido. Los restos selectivos pueden tener, por ejemplo, afinidad por proteínas o receptores de la superficie celular particulares. Esto puede ser característico de un órgano o tipo de célula en particular. En una realización, el resto selectivo comprende albúmina, folato o un aptámero. Por "aptámero" se entiende moléculas de ADN o ARN
monocatenario (ssADN o ssARN) que pueden unirse a un objetivo preseleccionado, como una proteína o un péptido, con alta afinidad y especificidad.
Ejemplos
Introducción a los ejemplos 1 y 2
La tecnología de PVP-PLA se puede utilizar para la formulación de fármacos, p. ej. con una solubilidad en agua limitante de la dosis. La tecnología tiene la capacidad de formar micelas independientemente del pH cuando la concentración de polímero en agua está por encima de la concentración micelar crítica (CMC). Las micelas se rompen cuando la concentración de polímero disminuye por debajo de la CMC. Siguiendo este principio, una formulación que comprenda micelas de PVP-PLA reconstituidas comenzará a liberar su contenido de fármaco, por ejemplo, tras la inyección en la sangre. La tecnología de PVP-PLA sigue siendo un desafío debido a una síntesis y polidispersión ineficientes, y a la falta de plataformas de administración flexibles que tengan la capacidad de formular una variedad de medicamentos y/o atraparlos a una concentración suficientemente alta (es decir, el nivel de carga de fármacos o NCF).
Se buscaron nuevos copolímeros de PVP-PLA o métodos de producción que proporcionaran propiedades físicas o químicas relevantes, rendimiento deseado, eficiencia de síntesis, baja polidispersidad, facilidad de producción, flexibilidad deseada en la formulación de una gama de fármacos y/o alto n Cf . Se utilizaron dos enfoques principales para intentar mejorar la síntesis del copolímero en bloque de PVP-PLA.
En el primer enfoque, el PVP-PLA se preparó utilizando una polivinilpirrolidona con un grupo hidroxilo terminal (denominado "PVP-OH") como macroiniciador seguido de (i) una polimerización por coordinación-inserción organometálica (Esquema 1A), (ii) una polimerización aniónica (Esquema 1B), o (iii) una polimerización por coordinación-inserción organocatalítica (ver Esquema 1C) de la D,L-lactida [1]. Este enfoque se denominará posteriormente "Enfoque de PVP-OH". El Esquema 1 muestra cada ruta de síntesis para un tipo de catalizador, un disolvente y una temperatura específica. Sin embargo, cada ruta de síntesis se probó en dos o tres disolventes diferentes, utilizando dos tipos de catalizadores y a dos temperaturas diferentes. En total se realizaron de 8 a 12 síntesis por ruta.

Esquema 1
En el segundo enfoque, el PVP-PLA se produjo a partir de la preparación de la poli(D,L-lactida) con un grupo hidroxilo terminal (denominado "PLA-OH"), y el PLA-OH se usó como un agente de transferencia de macrocadena para polimerizar la N-vinil-2-pirrolidinona (NVP). La preparación del PLA-OH se realizó después de (i) una polimerización por coordinación-inserción organocatalítica (ver Esquema 2A-1) o (ii) una polimerización por coordinación-inserción organocatalítica (ver Esquemas 2B-1 y 2C-1). El PLA-OH obtenido se convirtió en un agente de transferencia de macrocadena mediante la sustitución del grupo hidroxilo terminal por un grupo xantato de O-etilo. La sustitución del grupo hidroxilo requirió dos etapas, como se ilustra en el Esquema 2. Después de la sustitución, el agente de transferencia de macrocadena resultante de PLA se usó para polimerizar la NVP (véanse los Esquemas 2A-4, 2B-4 y 2C-4). Este segundo enfoque se denominará posteriormente "Enfoque PLA-OH".

Esquema 2
Ejemplo 1: El enfoque de PVP-OH
Se obtuvo PVP-PLA utilizando PVP-OH como macroiniciador para la polimerización de D,L-lactida. El PVP-OH utilizado durante este proyecto es un polímero comercial de Polysciences (Número de catálogo 16693, Número de lote 652063 y Mw 2500). Sin embargo, también se puede producir PVP-OH mediante una polimerización radical de N-vinil-2-pirrolidinona (NVP) en presencia de 2-isopropoxietanol (IPE) o mercaptoetanol o disulfuros funcionalizados como agente de transferencia de cadena (ATC) [2, 3]. Independientemente del ATC, la mayoría de las cadenas de PVP obtenidas tenían uno o dos agentes de transferencia de cadena como grupo terminal. El polímero obtenido fue una mezcla de OH-PVP-OH, PVP-OH y PVP en diferentes proporciones. Sin embargo, la proporción de PVP-OH puede aumentar según el tipo de agente de transferencia de cadena utilizado y las condiciones experimentales de la polimerización. La síntesis de PVP-OH con un peso molecular y un índice de polidispersidad (PDI) controlados sigue siendo un desafío.
Polimerización por coordinación-inserción organometálica de PVP-PLA
La polimerización por coordinación-inserción organometálica de PVP-PLA se realizó en presencia de dilaurato de dibutilestaño (DBTDl ) u óxido de dibutilestaño (DBTO) como catalizador. El Esquema 3 muestra la ruta de síntesis para la preparación de PVP-PLA usando DBTDL como catalizador en diferentes condiciones experimentales y disolventes.

Esquema 3
Procedimiento de la síntesis
Se disolvió una cantidad requerida de PVP-OH (previamente secada usando tolueno como disolvente) en N-Metil-2-pirrolidona (NMP) y luego se añadió a la mezcla una cierta cantidad de dilaurato de dibutilestaño. Después de la dispersión del DBTDL, se añadió a la mezcla la proporción requerida de D,L-lactida (el compuesto se seca previamente después de la purificación por recristalización en acetato de etilo). La disolución de la mezcla de reacción obtenida se agitó en una atmósfera de argón durante 1 hora y luego se puso a 130 °C y se mantuvo en una atmósfera de argón durante otras 4 horas. La polimerización se dejó durante 20 horas más y luego se enfrió a temperatura ambiente antes de la precipitación en éter dietílico. Se obtuvo un producto sólido blanco después de la precipitación.
La disolución enfriada se añadió gota a gota a una cierta cantidad de éter dietílico y luego el producto se recogió mediante filtración. El producto obtenido se lavó con tres porciones de éter dietílico antes de secarlo durante la noche a 40 °C. La Tabla 1 muestra la cantidad de materias primas utilizadas para la preparación de PVP-PLA en presencia de DTBL como catalizador y NMP como disolvente.
Tabla 1

Otras condiciones experimentales
La síntesis de PVP-PLA mediante polimerización por coordinación-inserción organometálica se repitió en varias condiciones experimentales. La Tabla 2 ilustra todas las condiciones y la cantidad de materias primas utilizadas para generar el copolímero en bloque de PVP-PLA.
Tabla 2

Resultados
Independientemente de las condiciones experimentales utilizadas, se obtuvo un producto muy viscoso para cada caso. Sin embargo, los resultados de las pruebas o de diferentes técnicas muestran que la D,L-lactida no reaccionó con la PVP-OH como se esperaba. Sin limitarse por la teoría, la proporción limitada de PVP-OH puede ser la causa principal de esos resultados. Como se mencionó anteriormente, es un desafío obtener PVP-OH en una proporción mayor al 50% [3]. El producto obtenido suele ser una mezcla de HO-PVP-OH, PVP-OH y PVP. Para comprender los resultados obtenidos, se decidió repetir la síntesis con DBTDL y DBTO en NMP a 130 °C y 120 °C, respectivamente. Sin embargo, la PVP-OH se reemplazó por un éter monometílico de poli(etilenglicol) (MePEG-OH) comercial de Sigma Aldrich. El MePEG-OH (número de catálogo 202509, número de lote MKBS5550V y Mw 2000) contiene más del 90 % de un grupo hidroxilo terminal en comparación con el 50 % de PVP-OH. Los resultados obtenidos con MePEG-OH fueron mucho mejores que con PVP-OH excepto en el caso de MePEG-OH - la D,L-lactida reaccionó con MePEG-OH pero el rendimiento fue inferior al 25%. Además, el Mn fue mucho más pequeño de lo esperado. Según estos resultados, la polimerización por coordinación-inserción organometálica de PVP-OH no fue un método lo suficientemente bueno para preparar PVP-PLA para el desarrollo rentable de productos farmacéuticos potentes que son comercializables.
Polimerización aniónica de PVP-PLA
La polimerización aniónica de PVP-PLA se realizó en presencia de hidruro de sodio (NaH) o hidruro de potasio (KH) como catalizador. El Esquema 4 muestra la ruta de síntesis resultante para la preparación de PVP-PLA utilizando NaH como catalizador en diferentes condiciones experimentales.

Esquema 4
Procedimiento de la síntesis
Se disolvió una cantidad requerida de PVP-OH en N-metil-2-pirrolidona (NMP) y luego se añadió una cierta cantidad de NaH a la mezcla. Después de la dispersión del NaH, se añadió a la mezcla la proporción requerida de D,L-lactida (previamente secada y purificada por recristalización en acetato de etilo). La mezcla obtenida se agitó bajo una atmósfera de nitrógeno durante 1 hora y luego se puso a 130 °C y se mantuvo bajo una atmósfera de nitrógeno durante otras 4 horas. La polimerización se dejó durante 20 horas y luego se enfrió a temperatura ambiente antes de la precipitación en éter dietílico.
La disolución enfriada se añadió gota a gota a una cantidad de éter dietílico y luego el producto se recogió mediante filtración. El producto obtenido se lavó con tres porciones de éter dietílico antes de secarlo durante la noche a 40 °C. La Tabla 3 muestra las cantidades de materias primas utilizadas para la preparación de PVP-PLA en presencia de NaH como catalizador y NMP como disolvente.
Tabla 3

Otras condiciones experimentales
Como se realizó en el método de síntesis anterior, la síntesis de PVP-PLA por polimerización aniónica se repitió en varias condiciones experimentales. La Tabla 4 ilustra todas las condiciones y las cantidades de materias primas utilizadas para generar el PVP-PLA.
Tabla 4


Resultados
Independientemente de las condiciones experimentales utilizadas, se obtuvo en cada caso un sólido blanco. Los resultados de diferentes técnicas muestran que la D,L-Lactida reaccionó parcialmente con la PVP-OH solo en el caso de la síntesis con NaH en THF a temperatura ambiente. El rendimiento obtenido para esta síntesis estuvo siempre por debajo del 30%. Estos resultados sugieren que la sal sódica de PVP-OH no es tan eficiente como un macroiniciador, ya que se esperaba un rendimiento del 50 % en lugar del 30 % [3]. El método también condujo a la producción de una proporción muy pequeña de un homopolímero de PLA-OH, pero suficiente para generar una distribución bimodal de tamaños de micelas. La distribución bimodal de tamaños de micelas no es adecuada para las formulaciones parenterales, especialmente si uno de los tamaños obtenidos es mayor de 500 nm. Además, sea cual sea el proceso de purificación utilizado, es difícil obtener únicamente PVP-PLA con el peso molecular esperado. Por ello, el producto final contiene, en distintas proporciones, cuatro polímeros (PVP-PLA, PLA-OH, HO-PVP-OH y PVP). El producto principal sigue siendo PVP-PLA, pero este método no permitió el control del peso molecular. El Mw es mucho más pequeño de lo esperado. En base a los resultados señalados anteriormente, la polimerización aniónica tampoco fue un método suficientemente bueno para preparar PVP-PLA para el desarrollo rentable de productos farmacéuticos potentes que son comercializables.
Polimerización por coordinación-inserción organocatalítica de PVP-PLA
Se intentó el método de polimerización por coordinación-inserción organocatalítica para producir el PVP-PLA en presencia de cloruro de 1,3-dimesitilimidazolinio (DMIC) como catalizador. Se sabe que este tipo de compuesto de carbeno N-heterocíclico produce una polimerización viva por apertura del anillo (ROP). Sin embargo, el compuesto DMIC nunca se ha utilizado en la polimerización de PLA cuando se utiliza PVP-OH como macroiniciador. Además, DMIC no está disponible comercialmente y debe prepararse. La preparación de DMIC requiere tres etapas y cada etapa condujo a un rendimiento superior al 85%. Los Esquemas 5 a 7 ilustran las rutas de síntesis para la preparación del DMIC. Las Tablas 5 a 7 muestran la cantidad de materias primas utilizadas para cada etapa.
Etapa 1: Síntesis de DMEDI
El Esquema 5 muestra la ruta de síntesis de la primera etapa de la preparación de DMEDI.
i-PrOH, H20

2,4,6-trimetilanilina Glioxal

Esquema 5
Tabla 5: Materias primas para la síntesis de DMEDI

Etapa 2: Síntesis de DMEDADH
El Esquema 6 representa la ruta de síntesis de la segunda etapa de la preparación de DMEDADH.

Esquema 6
Tabla 6: Materias primas para la síntesis de DMEDADH.

Etapa 3: Síntesis de DMIC
El Esquema 7 representa la ruta de síntesis de la tercera etapa que condujo a la preparación de DMIC.

Esquema 7
Tabla 7: Materias Primas para la Síntesis de DMIC.

Procedimiento de la síntesis
El procedimiento para la preparación de DMIC ha sido descrito por Delaude et al. [4]. Como se mencionó anteriormente, DMIC no se ha utilizado en la preparación de PVP-PLA.
Síntesis de PVP-PLA usando DMIC como catalizador
La preparación de PVP-PLA usando DMIC como catalizador y THF o DMSO como disolvente se presenta en el Esquema 8.

Esquema 8
La preparación de PVP-PLA en presencia de DMIC comenzó con la disolución de una cantidad de PVP-OH en THF o DMSO y luego se añadió a la mezcla una cantidad de DMIC. Después de la disolución del DMIC, se añadió a la mezcla la proporción requerida de D,L-lactida (previamente secada y purificada por recristalización en acetato de etilo). La mezcla obtenida se agitó en una atmósfera de argón durante 1 hora y luego se puso a 130 °C y se mantuvo en una atmósfera de argón durante otras 4 horas. La polimerización se dejó durante 20 horas más y luego se enfrió a temperatura ambiente antes de la precipitación en éter dietílico.
La disolución enfriada se añadió gota a gota a una cantidad de éter dietílico y luego el producto se recogió mediante filtración. El producto obtenido se lavó con tres porciones de éter dietílico antes de secarlo durante la noche a 40 °C. La Tabla 8 muestra la cantidad de materias primas utilizadas para la preparación de PVP-PLA en presencia de DTBL como catalizador y NMP como disolvente.
Tabla 8: Materia prima para la síntesis de PVP-PLA en presencia de DMIC

Tabla 9: Materia prima para la síntesis de PVP-PLA en presencia de DMIC

Resultados
Los datos obtenidos de diferentes técnicas muestran que la D,L-Lactida reaccionó parcialmente con la PVP-OH en ambos disolventes. Sin embargo, el rendimiento de cada síntesis fue de alrededor del 25%. Este método parecía prometedor y su optimización podría conducir a un mayor rendimiento. A pesar del bajo rendimiento, este método condujo a un mejor control del peso molecular. Los resultados obtenidos sugieren que la polimerización por coordinación-inserción organocatalítica podría ser un buen método para preparar PVP-PLA para el desarrollo rentable de productos farmacéuticos potentes que sean comercializables.
Ejemplo 2: El enfoque de PLA-OH
Como se ilustra en el Esquema 2, el PVP-PLA se obtuvo utilizando D,L-Lactida como materia prima para generar PLA-OH mediante una polimerización por coordinación-inserción organocatalítica (véase el Esquema 2A-1) o mediante una polimerización por coordinación-inserción organometálica (véanse los Esquemas 2B-1 y 2C-1). Por lo tanto, el PLA-OH obtenido se convirtió luego en un agente de transferencia de macrocadena al sustituir el grupo hidroxilo terminal con un grupo xantato de O-etilo. El nuevo agente de transferencia de macrocadena obtenido se utilizó para iniciar la polimerización de NVP. La D,L-lactida utilizada durante este proyecto fue de Sigma Aldrich (número de catálogo 303143, número de lote STBF0369V y Mw 144,13) y se secó y purificó mediante recristalización en acetato de etilo antes de utilizarse.
La idea detrás del enfoque de PLA-OH era aprovechar el conocimiento adquirido de la falta de éxito con el enfoque de PVP-OH para generar un PVP-PLA con un alto rendimiento y una alta pureza. Es deseable evitar tener un material
con tres homopolímeros diferentes remanentes antes de la siguiente etapa de polimerización, como se observa en el caso de la preparación de PVP-OH. Por esa razón, PLA-OH se preparó primero evitando usar el método de polimerización aniónica. Por esa razón, se seleccionaron dos métodos diferentes de coordinación-inserción para probar la táctica del enfoque de PLA-OH.
Polimerización por coordinación-inserción organocatalítica de PLA-OH
La polimerización por coordinación-inserción organocatalítica de PLA-OH se realizó en presencia de 4-dimetilaminopiridina (DMAP) como catalizador y alcohol isopropílico (IPA) como iniciador. El Esquema 9 muestra la ruta sintética para la preparación de PLA-OH en NMP a 80 °C.

Esquema 9
Procedimiento de la síntesis
PLA-OH se sintetiza mediante una polimerización de apertura de anillo (ROP) de D,L-lactida utilizando una cantidad de IPA como iniciador y DMAP como catalizador. El procedimiento comenzó poniendo la D,L-lactida (recristalizada en acetato de etilo antes de usarla) en un matraz redondo seco bajo una atmósfera de argón. Se añadieron NMP e IPA, seguido de la adición de DMAP en el matraz de reacción. La mezcla de reacción se calentó a 80 °C durante 24 horas y se mantuvo bajo una atmósfera de argón durante ese período de tiempo. Después de enfriar a temperatura ambiente, el producto bruto obtenido se disolvió en DCM y se precipitó en hexanos. El polímero precipitado se recogió y se disolvió de nuevo en DCM seguido de otra precipitación en hexanos. Finalmente, el PLA-OH recogido se secó a vacío a 40 °C durante 48 horas. La Tabla 10 ilustra la cantidad de reactivos utilizados para la síntesis de PLA-OH.
Tabla 10: Materias primas utilizadas para la síntesis de PLA-OH en presencia de DMAP

Resultados
Los datos obtenidos de la preparación de PLA-OH muestran que la D,L-Lactida polimerizó en presencia de DMAP e IPA. El rendimiento obtenido para la síntesis fue cercano al 60%. En cuanto a DMIC, este método parece prometedor y su optimización podría conducir a un mayor rendimiento. A pesar del bajo rendimiento, este método también condujo a un mejor control del peso molecular. Los resultados obtenidos muestran que la polimerización por coordinación-inserción organocatalítica de PLA-OH puede generar productos farmacéuticos potentes, económicos y comercializables.
Polimerización por coordinación-inserción organometálica de PLA-OH
Se utilizaron dos métodos de polimerización por coordinación-inserción organometálica para producir el PLA-OH. Para cada método se utilizó un catalizador diferente. Los Esquemas 10 y 11 muestran la ruta de síntesis en presencia de DBTDL o Sn(Oct)2 como catalizador. Como se ilustra, la otra diferencia principal entre las dos síntesis es que se usó NMP como disolvente para la síntesis con DBTDL, mientras que la realizada en presencia de Sn(Oct)2 es a granel. La temperatura requerida para ambas síntesis fue muy cercana (145 °C y 150 °C, respectivamente). Las Tablas 11 y 12 ilustran la cantidad de reactivos necesarios para cada polimerización.
El Esquema 10 muestra la ruta de síntesis para la preparación de PLA-OH usando DBTDL como catalizador en NMP a 145 °C.

Esquema 10
Tabla 11: Materias primas utilizadas para la síntesis de PLA-OH en presencia de DBTDL

El Esquema 11 muestra la ruta de síntesis para la preparación de PLA-OH a granel a 145 °C y utilizando Sn(Oct)2 como catalizador.

Esquema 11
Tabla 12: Materias primas utilizadas para la síntesis de PLA-OH en presencia de Sn(Oct)2

Procedimiento de la síntesis
El PLA-OH se sintetizó a través de la ROP de D,L-lactida utilizando en ese caso éter monoetílico de dietilenglicol (DEGMEE) como iniciador y DBTDL o Sn(Oct)2 como catalizador. Para la síntesis en presencia de Sn(Oct)2 , el procedimiento comenzó poniendo la D,L-lactida (recristalizada en acetato de etilo antes de su uso) en un matraz redondo seco bajo una atmósfera de argón. Se añadieron DEGMEE y Sn(Oct)2 al matraz. Luego, la mezcla de reacción se calentó a 150 °C durante 24 horas. Después de enfriar a temperatura ambiente, el producto bruto obtenido se disolvió en cierta cantidad de DCM y se precipitó en hexanos. El polímero precipitado se recogió mediante filtración y luego se volvió a disolver en DCM, seguido de precipitación en hexanos una vez más. Finalmente, el PLA-OH recogido se secó a vacío a 40 °C durante 48 horas.
Resultados
Los resultados muestran que D,L-lactida se polimerizó usando ambos métodos de polimerización por coordinacióninserción organometálica. La Tabla 13 ilustra los resultados seleccionados obtenidos de ambos métodos de síntesis, así como el resultado de la polimerización por coordinación-inserción organocatalítica en presencia de DMAP.
Tabla 13: Resultados obtenidos de los tres métodos de preparación de PLA-OH.

Como se ilustra, las tres síntesis condujeron a un peso molecular comparable (Mn), y esos valores están dentro del intervalo esperado de valores de Mn, que es entre 1500 y 2500. Sin embargo, los rendimientos obtenidos con cada método son muy diferentes. La Tabla 13 también ilustra que la síntesis en presencia de DBTDL requirió 72 horas para producir un rendimiento del 62 % mientras que la síntesis con Sn(Oct)2 condujo a un rendimiento del 94% en 24 horas. En el caso de la síntesis en presencia de d Ma P, el rendimiento obtenido es similar al método de DBTDL, excepto que en este caso solo se requirieron 24 hr. Los tres métodos parecen muy prometedores, pero se han seleccionado los métodos con DMAP y Sn(Oct)2 para un mayor desarrollo y optimización preliminar. La selección se hizo estrictamente basándose en el rendimiento obtenido sin ninguna optimización y las condiciones experimentales, especialmente el tiempo requerido para producir el rendimiento obtenido.
Síntesis de PVP-PLA
El PLA-OH obtenido del DMAP y Sn(Oct)2 se utilizó para producir PVP-PLA. Ambos PLA-OH se convirtieron en un agente de transferencia de macrocadena mediante la sustitución del grupo hidroxilo terminal por un grupo xantato de O-etilo. La sustitución del grupo hidroxilo requirió dos etapas, como se ilustra en los Esquemas 12 y 13. Después de la sustitución, el agente de transferencia de macrocadena resultante de PLA se usó para polimerizar la NVP como se indica en el Esquema 14.
Preparación de PLA-Br
Se disolvió PLA-OH en THF (destilado antes de su uso) en un matraz de fondo redondo seco con trietilamina mientras se agitaba en una atmósfera de argón. La mezcla de reacción se enfrió en un baño de hielo. Después del enfriamiento, se añadió gota a gota bromuro de 2-bromopropionilo desde un matraz de adición. A continuación, la mezcla de reacción se agitó durante 72 horas a temperatura ambiente. La sal de trimetilamina precipitada se eliminó mediante filtración y el filtrado se evaporó hasta sequedad. El producto sólido se disolvió en diclorometano (DCM) y se lavó metódicamente con una disolución saturada de bicarbonato de sodio (4 x 200 mL). La capa orgánica se lavó con agua (4 x 300 mL), se secó sobre sulfato de sodio anhidro y se filtró. El filtrado se concentró mediante evaporación y se precipitó en hexanos y se secó a vacío a 40 °C durante 48 horas. La Tabla 14 y el Esquema 12 ilustran, respectivamente, la cantidad de reactivos utilizados y la ruta de síntesis de la preparación de PLA-Br.
Tabla 14: Cantidad de cada compuesto utilizado para la preparación de PLA-Br.

El Esquema 12 representa una ruta de síntesis para la preparación de PLA-Br utilizando el PLA-OH obtenido previamente.

Esquema 12
Incorporación de O-etil xantato de potasio para producir PLA-POEX
El PLA-Br preparado se mezcló con etilxantogenato de potasio (previamente preparado y secado a vacío a 40 °C) en
un matraz seco de fondo redondo, se añadió DCM seco para dispersar ambos sólidos y la mezcla se purgó con argón durante 2 h. En otro matraz seco de fondo redondo, se mezcló piridina con DCM mientras se agitaba bajo argón también durante 2 horas. La disolución de DCM se añadió a la mezcla de reacción de PLA-Br y se mantuvo con agitación continua y argón durante 1 h. Luego, la mezcla de reacción se agitó a temperatura ambiente durante 48 horas. La disolución de reacción se lavó consecutivamente con disolución saturada de cloruro de amonio (4 x 200 mL) y disolución saturada de bicarbonato de sodio (4 x 200 mL). La capa orgánica se lavó con más agua (4 x 200 mL), se secó sobre sulfato de sodio anhidro y se filtró. El filtrado se concentró mediante evaporación y se precipitó en hexanos y se secó a vacío a 40 °C durante 48 horas. La Tabla 15 y el Esquema 13 ilustran, respectivamente, la cantidad de reactivos utilizados y la ruta de síntesis de PLA-POEX.
Tabla 15: Cantidad de cada compuesto utilizado para la preparación de PLA-POEX.

El Esquema 13 representa una ruta de síntesis para la preparación de PLA-POEX usando el PLA-Br obtenido previamente.

Esquema 13
Preparación de PVP-PLA
En un matraz de fondo redondo seco, se disolvió PLA-POEX (previamente secado a vacío a 40 °C) en THF (frasco anhidro recién destilado o recién abierto). A esta disolución se le añadieron y disolvieron NVP y AIBN. La disolución homogénea se agitó y desgasificó con argón durante 2 horas. El matraz de reacción se puso en un baño de aceite precalentado a 80 °C durante 48 horas. Después de este tiempo, la PVP-PLA en bruto se disolvió en DCM, se precipitó en hexanos y se secó a vacío a 40 °C durante la noche. El polímero se purificó adicionalmente mediante la disolución repetida en DCM y se precipitó en hexanos por segunda vez, y finalmente se secó a vacío a 40 °C durante 48 horas. La Tabla 16 y el Esquema 14 ilustran, respectivamente, la cantidad de reactivos utilizados y el esquema de síntesis de PVP-PLA.
Tabla 16: Cantidad de cada materia prima utilizada para la elaboración de PVP-PLA

El Esquema 14 representa una ruta de síntesis para la preparación de PVP-PLA utilizando el PLA-POEX obtenido previamente.

Esquema 14
Características de los PVP-PLA obtenidos
Se prepararon cuatro lotes de PVP-PLA, uno de PLA-OH con DMAP como catalizador y los otros tres de PLA-OH con Sn(Oct)2 como catalizador. Los cuatro lotes se caracterizaron utilizando múltiples técnicas. El objetivo era confirmar las propiedades según los requisitos y también mostrar la reproducibilidad y robustez del método.
La Tabla 17 ilustra el rendimiento obtenido para cada síntesis. Las tres síntesis con Sn(Oct)2 condujeron a un rendimiento y eficiencia mucho mejores que la síntesis con DMAP.
Tabla 17: Rendimiento y eficiencia obtenidos para la preparación de PVP-PLA

Tras la validación del peso molecular mediante técnicas de GPC-LS y RMN de protones, se aplicó el análisis termogravimétrico (ATG), así como análisis elemental (AE) y RMN de protones para determinar el contenido de PLA de cada polímero.
La Tabla 18 ilustra los resultados obtenidos de cada método. Como era de esperar a partir de los resultados del peso molecular, el contenido de PLA estuvo entre el 35 y el 45 %. Los resultados de las técnicas de RMN de protones y AE muestran nuevamente la reproducibilidad del método de Sn(Oct)2.
Tabla 18: Valor del contenido de PLA obtenido a partir de ATG, AE y RMN de protones.

Propiedades del polímero en disolución
Para establecer la capacidad de cada polímero de formar micelas, se preparó una disolución de polímero a una concentración de 90 mg/mL en tampón fosfato 100 mM a 25 °C y luego se midió la distribución de tamaños de cada polímero. La Figura 1 muestra el perfil de distribución de tamaños obtenido para cada polímero. Una vez más, la distribución de tamaños confirma la reproducibilidad del método de Sn(Oct)2. Sin embargo, la distribución de tamaños observada para el polímero WB-DMAp es completamente diferente (más de 2 veces menor). Este resultado no sorprende, dado que el peso molecular obtenido para este polímero es un 35% inferior al valor promedio de los polímeros WB-1, WB-2 y WB-3. Los cuatro polímeros muestran una distribución de tamaños unimodal. Se requirió purificación adicional para obtener esta distribución unimodal.
La Figura 1 representa la distribución de tamaños de las micelas obtenidas de tres lotes diferentes del nuevo polímero PVP-PLA.
La Tabla 19 muestra el valor del tamaño y el valor de PDI medidos para los cuatro polímeros. Como se muestra, los polímeros del método de Sn(Oct)2 condujeron a tamaños equivalentes. La concentración crítica de micelas (CMC) es otra propiedad física medida, y los resultados obtenidos para todos los polímeros del método de Sn(Oct)2 también son equivalentes. Los resultados son muy diferentes del método de DMAP.
Tabla 19: Tamaño de partículas obtenido y CMC para cada polímero.

Aquí, la CMC en disolución de PBS 10 mM a temperatura ambiente se determinó a partir de los espectros de excitación de pireno (lem = 390 nm), y se expresó como una proporción de intensidad de excitación a 337 y 333 nm.
Síntesis de polímeros adicionales
Dado el éxito con el método de Sn(Oct)2, se prepararon cinco polímeros adicionales, WB-4 a WB-8 utilizando el enfoque de PLA-OH con Sn(Oct)2 como catalizador. La Tabla 20 enumera las características de los polímeros producidos mediante el enfoque de PLA-OH. En la presente memoria, cabe señalar que Mn se determinó mediante resonancia magnética nuclear de protones (RMN) en DMSO deuterado a temperatura ambiente, y mediante cromatografía de permeación en gel con detector de dispersión de luz (GPC-LS). Los valores de GPC-LS se determinaron en DMF-LiBr (10 mM) a una concentración de polímero de 5 mg/mL y utilizando un patrón de poliestireno con un Mn de 30k y un PDI de 1,008 para calibrar el sistema. Se midió el valor dn/dc requerido para cada polímero y la temperatura del sistema se fijó a 35 °C. Los valores porcentuales bajo "ATG" se determinaron a partir de los valores de Mw determinados mediante el análisis termogravimétrico.
La Tabla 20 enumera las propiedades de varios polímeros producidos. Cabe señalar que los valores de PLA:PVP se calculan utilizando el valor de Mn de PLA-POEX obtenido de la RMN de protones, del cual se han sustraído la masa de PEOX (121,2) y del alcohol iniciador (134,2).




Discusión de los ejemplos 1 y 2
De todas las síntesis y caracterizaciones realizadas y presentadas, el enfoque de PLA-OH es el más prometedor, siendo inesperadamente más eficiente que los enfoques equivalentes de PVP-OH.
De los tres métodos ilustrados para el enfoque ventajoso PLA-OH, el método de Sn(Oct)2 dio el rendimiento total más alto para la preparación de PVP-PLA (66% en comparación con 25% para DMAP). Este método también ha mostrado muy buena reproducibilidad y robustez. Incluso sin ninguna optimización adicional, se logró un buen control del peso molecular y PDI con el método de Sn(Oct)2. Por todas esas razones, se seleccionó el método de Sn(Oct)2 y los polímeros producidos con este método para su posterior estudio y optimización.
Ejemplo 3: formulaciones farmacéuticas
Introducción
El control del dolor es una carga importante para los pacientes, los servicios de salud y la sociedad. Por ejemplo, se realizan más de 100 millones de cirugías solo en los EE. UU. cada año, y el 80 % de los pacientes experimentan dolor posoperatorio de intensidad moderada que requiere una intervención analgésica.
En más del 50% de los casos, los pacientes reciben un fármaco opioide administrado por vía intravenosa para combatir el dolor. Los segundos agentes más utilizados para tratar el dolor posoperatorio son los antiinflamatorios no esteroideos (AINE) y el paracetamol; estos medicamentos se pueden usar solos para tratar el dolor moderado y como parte de un enfoque multimodal para complementar y reducir la dosis y la frecuencia de la analgesia opioide concomitante cuando se haya demostrado que son efectivos. Sin embargo, los fármacos AINE y el paracetamol sufren un inconveniente común en su uso como analgésicos intravenosos: el de la escasa solubilidad junto con, en muchos casos, una potencia relativamente baja. Los intentos anteriores para superar este inconveniente han sido variados.
Por ejemplo, la alta dosis requerida (hasta 4000 mg/día) y la baja solubilidad del paracetamol se han evitado con el desarrollo de productos como Ofirmev™ (Mallinckrodt) y Caldolor (Cumberland Pharmaceuticals). Estas son formulaciones de gran volumen y baja concentración que deben infundirse lentamente al paciente. Si bien estas disoluciones de baja concentración brindan beneficios, el paciente debe recibir una nueva vía intravenosa para administrar el producto (lo que genera inconvenientes, molestias y costos de tratamiento) y la infusión lenta da como resultado una Cmax más baja y una Tmax más tardía para el producto que las que se podrían lograr si se administrase una dosis intravenosa en bolo o de presión lenta. Estos productos deben administrarse cada 4-6 horas
Por otra parte, Pfizer desarrolló una nueva entidad química de profármaco soluble en agua de su potente inhibidor de COX-2 valdecoxib (Bextra™), llamado Dynastat® (parecoxib), para superar la insolubilidad intrínseca de valdecoxib. El producto debe administrarse cada 4-6 hr. Si bien este es un excelente analgésico posoperatorio, los costos y el tiempo necesarios para desarrollar nuevas entidades químicas, junto con la variabilidad inherente de paciente a paciente que resulta de los profármacos, no hacen que esta sea una opción rentable. El tiempo necesario para la hidrólisis y activación del profármaco también puede retrasar el inicio del analgésico para la primera y más importante administración.
Los productos como Toradol™ (ketorolaco para inyección) y Dynastat™ (diclofenaco para inyección) utilizan enfoques de formulación novedosos para superar la insolubilidad intrínseca de los AINE. Así, Toradol emplea disolventes orgánicos (no beneficiosos para el paciente) en los que el ketorolaco es soluble, mientras que Dyloject emplea hidroxipropil beta-ciclodextrina como agente solubilizante. Desafortunadamente, a pesar de estos enfoques, tanto Toradol como Dyloject sufren problemas de inestabilidad y han sido objeto de numerosas retiradas de los productos y del mercado; la escasez de suministros es común; de nuevo, estos productos deben administrarse cada 4-6 horas.
Se seleccionaron fármacos para el control del dolor de baja solubilidad (incluidos fluriboprofeno, celecoxib y paracetamol) como ejemplos para la formulación con los copolímeros en bloque descritos en la presente memoria. Paralelamente, se seleccionó el propofol anestésico hidrofóbico no opioide como ejemplo adicional para la formulación.
Métodos y resultados
Formulaciones de flurbiprofeno
Formulación de flurbiprofeno con polímero WB-2
La formulación de flurbiprofeno (denominada "Flu") se preparó como sigue. Se disolvieron 12,8 g del copolímero en bloque de PVP-PLA WB-2 y 3,2 g de flurbiprofeno en 16 mL de etanol a temperatura ambiente durante aproximadamente 15 minutos para proporcionar una disolución final con una concentración de 200 mg/mL de flurbiprofeno y de 800 mg/mL de PVP-PLA, respectivamente. A esta disolución se le añadieron gota a gota 16 mL de agua a una velocidad de aproximadamente 2 mL/min con agitación vigorosa utilizando una barra agitadora. A esta mezcla se le añadieron 13 mL de una disolución acuosa de NaOH 1 N con agitación para llevar el pH a aproximadamente 6,9. A continuación, se añadieron 260 mL de agua a la mezcla durante aprox. 25 min con agitación vigorosa seguido de la adición de 12,8 mL de tampón de fosfato de sodio 100 mM de pH 7,0. A continuación, la mezcla se mantuvo con agitación magnética durante aproximadamente 10 min. A continuación, la disolución se concentró
hasta aproximadamente el 65 % de su peso inicial a presión reducida en un aparato Büchi Collegiate Rotavapor® equipado con una trampa de disolvente con hielo seco y una bomba de vacío Heidolph Rotavac Valve. La temperatura del baño de agua se mantuvo a 30-35 °C. A continuación, la mezcla se diluyó con agua para obtener una concentración final de flurbiprofeno de 12,5 mg/mL y se filtró a través de filtros Nylon Target2 de 0,2 pm (Thermo Scientific). La disolución resultante tuvo un contenido de etanol de aprox. 0,2% (p/p). A continuación, la formulación filtrada se transfirió a viales de vidrio de 10 mL en alícuotas de 4 mL, correspondientes a 50 mg de flurbiprofeno. Los viales que contenían la mezcla se liofilizaron utilizando un liofilizador VirTis Genesis 25EL. La composición de las tortas liofilizadas de flurbiprofeno resultantes se muestra en la Tabla 21.
Tabla 21

En consecuencia, se logró un nivel de carga de fármacos (NCF) de casi el 20% (p/p).
Se registraron los patrones de difracción de rayos X en polvo (XRPD) para el IFA flurbiprofeno, una mezcla física de flurbiprofeno y copolímero en bloque de PVP-PLA WB-2 que contenía un 20 % (p/p) de IFA, y para la torta liofilizada de flurbiprofeno. Las mediciones de XRPD para flurbiprofeno y la mezcla física se realizaron en un difractómetro Bruker D8 Discover, mientras que el patrón de difracción para la torta liofilizada se registró en un instrumento Bruker D8 Advanced. Ambos instrumentos estaban equipados con una fuente de rayos X de Ka1 de cobre (A= 1,54 A) y utilizaron la geometría Bragg-Brentano 0-20. La Figura 2 muestra los patrones de XRPD para flurbiprofeno y su mezcla física con PVP-PLA. En ambos casos, la presencia de picos agudos bien definidos confirma la naturaleza cristalina del IFA.
La Figura 3 muestra el patrón de XRPD de la torta liofilizada de flurbiprofeno que contiene la misma cantidad de IFA que la mezcla física que se muestra en la Figura 2. Sin embargo, en este caso, no se observaron los picos agudos de flurbiprofeno cristalino. Esto confirma que en la formulación sólida liofilizada de flurbiprofeno, el IFA está presente en estado amorfo.
La Figura 4, panel A muestra la imagen de la torta liofilizada y la disolución obtenida tras la reconstitución de la torta con agua para inyección a una concentración de flurbiprofeno de 50 mg/mL. La torta muestra una fina estructura esponjosa sin agrietarse ni colapsarse. La inspección más detallada del líquido reconstituido (Figura 4, panel B) confirma la claridad de la disolución y la ausencia de partículas sólidas visibles.
Las tortas liofilizadas se reconstituyeron en agua para inyección en menos de 2 minutos para proporcionar disoluciones claras y sin partículas con una concentración de flurbiprofeno de 50 mg/mL. El pH de las disoluciones reconstituidas estuvo en el intervalo de 7,2 a 7,4 medido utilizando un medidor de pH Accumet AP61 equipado con un electrodo de combinación de cuerpo epoxi relleno de gel. La transmitancia óptica se determinó en cubetas de poliestireno desechables de 1 cm en un espectrómetro Agilent Cary UV-Vis-NIR 5000. Las medidas se realizaron a 650 nm y a temperatura ambiente usando una cubeta vacía como blanco. Las mezclas reconstituidas tuvieron una transmitancia óptica entre el 99% y 102%. La osmolaridad de las muestras reconstituidas se midió con un Micro-Osmómetro 3300 de depresión del punto de congelación (Advanced Instruments) y estuvo en el intervalo de 320 a 340 mOsm/kg. El tamaño medio Z de las micelas y su distribución de tamaños se determinó a 25 °C mediante dispersión de luz dinámica usando un aparato Malvern Zetasizer Nano ZS equipado con un láser de He-Ne de 10 mW que funcionó a 633 nm. El tamaño de partícula promedio Z y su polidispersidad variaron de 31,5 a 31,8 nm y de 0,24 a 0,28, respectivamente.
La Figura 5 muestra que la distribución de tamaños de partículas para la formulación de flurbiprofeno reconstituida a 50 mg/mL es unimodal y que el tamaño promedio en volumen de las micelas varía de aprox. 8 a aprox. 100 nm
La estabilidad de las tortas liofilizadas de flurbiprofeno preparadas con el copolímero en bloque de PVP-PLA WB-2 se controló a 25 °C/65 % de HR y 40 °C/75 % de HR. La Figura 6 muestra el patrón de XRPD del producto farmacéutico a tiempo cero y después de T=3 meses de almacenamiento en ambas condiciones. La ausencia de picos agudos bien resueltos de flurbiprofeno cristalino confirma que el IFA puede conservar su estado amorfo al menos durante 3 meses en ambas condiciones. Estos datos se han ampliado a T=6 meses, en los que la ausencia de picos nítidos bien resueltos de flurbiprofeno cristalino volvió a confirmar que el IFA puede conservar su estado amorfo al menos durante 6 meses en ambas condiciones (datos no mostrados).
Las características de las tortas reconstituidas (flurbiprofeno = 50 mg/mL) en las diferentes etapas de evaluación de la estabilidad se muestran en la Tabla 22. No se observaron cambios significativos tras 6 meses de almacenamiento tanto a 25 como a 40 °C, lo que confirma la estabilidad del producto liofilizado.
Tabla 22

Formulaciones de flurbiprofeno con los polímeros WB-4 y WB-7
Dado el éxito con WB-2, se intentaron formulaciones de FLU con los polímeros WB-4 y WB-7. Los copolímeros en bloque de PVP-PLA WB-4 o WB-7 se disolvieron cada uno junto con flurbiprofeno en 2,75 mL de etanol con agitación magnética en un vaso de precipitados de vidrio a temperatura ambiente durante aproximadamente 15 minutos para dar a la disolución una concentración de 200 y 800 mg/mL de flurbiprofeno y copolímero en bloque de PVP-PLA, respectivamente. A ambas disoluciones se les añadieron gota a gota 2,75 mL de agua durante aprox. 1 min y con agitación vigorosa utilizando una barra de agitación magnética y una placa de agitación. A continuación, se añadió una disolución acuosa de NaOH 1 N con agitación para llevar el pH cerca de la neutralidad (6,2-6,8). Finalmente, se añadió agua a la mezcla con agitación vigorosa, seguido de la adición de 2,2 mL de tampón de fosfato de sodio 100 mM, pH 7,0. A continuación, las mezclas se mantuvieron con agitación magnética durante aproximadamente 10 min. A continuación, ambas disoluciones se concentraron hasta aproximadamente el 60 % de su peso inicial a presión reducida en un aparato Büchi Collegiate Rotavapor® equipado con una trampa de disolvente de hielo seco y una bomba de vacío Heidolph Rotavac Valve. La temperatura del baño de agua se mantuvo a 30-35 °C. A continuación, las disoluciones claras resultantes se diluyeron con agua para obtener una concentración final de flurbiprofeno de 12,5 mg/mL y se filtraron a través de filtros Nylon Target2 de 0,2 pm (Thermo Scientific). Las formulaciones filtradas se transfirieron como alícuotas de 4 mL (correspondientes a 50 mg de flurbiprofeno) a viales de vidrio de 10 mL. Los viales llenos se liofilizaron usando un liofilizador VirTis Genesis 25EL. En la Tabla 23 se muestran las cantidades de los productos utilizados para la preparación de las formulaciones de flurbiprofeno con los polímeros WB-4 o WB-7. La composición de las tortas liofilizadas de flurbiprofeno resultantes preparadas con los polímeros WB-4 o WB-7 se muestra en la Tabla 24 y la Tabla 25, respectivamente.
Las tortas liofilizadas se reconstituyeron en agua para inyección en menos de 1 min para proporcionar disoluciones claras y sin partículas con una concentración de flurbiprofeno de 50 mg/mL. El pH de las disoluciones reconstituidas para las formulaciones obtenidas con ambos polímeros estuvo en el intervalo de 7,2 a 7,4 medido usando un medidor de pH Accumet AP61 equipado con un electrodo de combinación de cuerpo epoxi relleno de gel. La osmolalidad de las muestras reconstituidas se midió con un Micro-Osmómetro 3300 de depresión del punto de congelación (Advanced Instruments) y estuvo en el intervalo de 380 a 420 mOsm/kg. La transmitancia óptica se determinó en cubetas de poliestireno desechables de 1 cm usando un espectrómetro Agilent Cary UV-Vis-NIR 5000. Las medidas se realizaron a 650 nm y a temperatura ambiente usando una cubeta vacía como blanco. Las disoluciones reconstituidas preparadas a partir de las tortas que contenían el polímero WB-4 mostraron una transmitancia óptica entre el 98 % y el 100 %. Las disoluciones de las muestras preparadas con el polímero WB-7 tienen una transmitancia ligeramente menor, en el intervalo de 84-85%. El tamaño medio Z de las micelas y su distribución de tamaños se determinó a 25 °C mediante dispersión de luz dinámica usando un aparato Malvern Zetasizer Nano ZS equipado con un láser de He-Ne de 10 mW que funcionó a 633 nm. El tamaño de partícula promedio Z para ambas formulaciones reconstituidas de las tortas que contenían WB-4 y WB-7 fue similar e igual a 43,9 y 43,6 nm, respectivamente. La Figura 7 muestra que las distribuciones de tamaños de partículas para las formulaciones preparadas utilizando dos muestras de polímero diferentes WB-4 y WB-7 y reconstituidas a 50 mg/mL tienen una forma similar, con un tamaño promedio en volumen de las micelas de aprox. 10 a aprox. 200 nm. Los valores de los parámetros determinados en la caracterización de las formulaciones de flurbiprofeno reconstituidas se muestran en la Tabla 26.
Tabla 23

Tabla 24

Tabla 25

Tabla 26

Formulaciones de flurbiprofeno con otros polímeros de PLA-OH
Dados los éxitos anteriores, se probó la capacidad de otros polímeros derivados del método de PLA-OH para formular FLU, es decir, los polímeros WB-DMAP, WB-1, WB-3, WB-5, WB-6 y WB-8. Se descubrió que todos los polímeros eran capaces de formular FLU, logrando disoluciones claras adecuadas que tenían un NCF adecuado.
Formulaciones de acetaminofeno
Formulación de paracetamol con polímero WB-2 (sin antioxidante)
La formulación de paracetamol (APAP) se preparó como sigue. Se disolvieron 4,40 g de APAP en 29 mL de etanol en un vaso de precipitados con agitación a temperatura ambiente durante aproximadamente 10 minutos, y después la adición de 20,05 g de copolímero en bloque WB-4 y 10 minutos adicionales de agitación. A esta disolución se le añadieron gota a gota 200 mL de agua a una velocidad de aproximadamente 10 mL/min y con agitación vigorosa utilizando una barra agitadora. A esta mezcla se añadieron 0,38 mL de una disolución acuosa de NaOH 1 N con agitación para llevar el pH a aproximadamente 7,1. A continuación, se añadieron 200 mL de agua a la mezcla durante aprox. 20 min con agitación vigorosa seguido de la adición de 12,2 mL de disolución acuosa de manitol a una concentración de 100 mg/mL. La agitación continuó durante 10 minutos más. A continuación, la disolución se concentró durante aprox. 2 h hasta el 42 % de su peso inicial a presión reducida en un aparato Büchi Collegiate Rotavapor® equipado con una trampa de disolvente de hielo seco y una bomba de vacío Heidolph Rotavac Valve. La temperatura del baño de agua se mantuvo a 30-35 °C. A la disolución concentrada se añadieron 4,9 mL de tampón de fosfato de sodio 100 mM, pH 7,0. A continuación, la mezcla se diluyó con 29 mL de agua para obtener una concentración final de APAP de 20 mg/mL y se filtró a través de filtros de Nylon Target2 de 0,2 pm (Thermo Scientific). A continuación, la formulación filtrada se transfirió a viales de vidrio de 10 mL en alícuotas de 5 mL, correspondientes a 100 mg de APAP. Los viales que contenían la formulación se liofilizaron utilizando un liofilizador VirTis Genesis 25EL. La composición de las tortas liofilizadas de paracetamol resultantes se muestra en la Tabla 27.
Tabla 27

En consecuencia, se logró un NCF de más del 17%.
Se registraron los patrones de difracción de rayos X en polvo (XRPD) para el IFA de paracetamol y para la torta liofilizada de paracetamol. Las mediciones de XRPD se registraron en un instrumento Bruker D8 Advanced equipado con fuente de rayos X de Ka1 de cobre (A= 1,54 A) y se utilizó la geometría Bragg-Brentano 0-20. La Figura 8 muestra los patrones de XRPD para el IFA de paracetamol y la torta liofilizada del producto farmacéutico. No se detectó la presencia de picos agudos característicos del paracetamol cristalino, en el caso de la formulación APAP no se observó, lo que confirma que en las tortas sólidas liofilizadas el IFA se encuentra presente en estado amorfo.
La Figura 9 muestra la imagen de la torta liofilizada y la disolución obtenida después de la reconstitución de la torta con agua para inyección a la concentración de paracetamol de 50 mg/mL. La torta muestra una estructura fina similar a una esponja sin grietas ni colapso. La inspección minuciosa del líquido reconstituido (Figura 9b) confirma la claridad de la disolución y la ausencia de partículas sólidas visibles.
Las tortas liofilizadas se disuelven por completo en agua en aprox. 2-3 min para obtener disoluciones claras y sin partículas con una concentración de paracetamol de 50 mg/mL. El pH de las disoluciones reconstituidas estaba en el intervalo de 7,5 a 7,7 medido usando un medidor de pH Accumet AP61 equipado con un electrodo de combinación de cuerpo epoxi relleno de gel. La transmitancia óptica se determinó en cubetas de poliestireno desechables de 1 cm en un espectrómetro Agilent Cary UV-Vis-NIR 5000. Las medidas se realizaron a 650 nm y a temperatura ambiente usando una cubeta vacía como blanco. Las mezclas reconstituidas tuvieron una transmitancia óptica de alrededor del 86%. La osmolalidad de las muestras reconstituidas se midió con un Micro-Osmómetro 3300 de depresión del punto de congelación (Advanced Instruments), y estuvo en el intervalo de 250 a 265 mOsm/kg. El tamaño medio Z de las micelas y su distribución de tamaños se determinó a 25 °C mediante dispersión de luz dinámica usando un aparato Malvern Zetasizer Nano ZS equipado con un láser de He-Ne de 10 mW que funcionó a 633 nm. El tamaño de partícula
promedio Z y su polidispersidad variaron de 48 a 51 nm y de 0,19 a 0,22, respectivamente. La Figura 10 muestra que la distribución de tamaños de partículas para la formulación de APAP reconstituida a 50 mg/mL es monomodal y que el tamaño promedio en volumen de las micelas varía de aprox. 15 a aprox. 200 nm.
Formulaciones de paracetamol con polímeros WB-4 y WB-7
Dado el éxito anterior, las formulaciones de APAP con los polímeros WB-4 y WB-7 se prepararon de la siguiente manera. Se disolvió APAP en 3 mL de etanol en un vaso de precipitados de vidrio con agitación a temperatura ambiente durante aproximadamente 5 minutos, y después la adición del copolímero en bloque WB-4 o WB-7 y agitación adicional de 10 minutos. A estas disoluciones se les añadió gota a gota 40 mL de agua a una velocidad de aproximadamente 10 mL/min y con agitación vigorosa. A continuación, se añadió una disolución acuosa de NaOH 0,1 N para llevar el pH a aproximadamente 7,2, seguido de la adición de 40 mL de agua durante aprox. 10 minutos. En la siguiente etapa, se añadieron a ambas disoluciones 1,25 mL de disolución acuosa de manitol a una concentración de 100 mg/mL y se continuó agitando durante 10 min más. Ambas formulaciones se concentraron posteriormente durante aprox. 30-40 min al 20-40% de su peso inicial a presión reducida en un aparato Büchi Collegiate Rotavapor® equipado con una trampa de disolvente de hielo seco y una bomba de vacío Heidolph Rotavac Valve. La temperatura del baño de agua se mantuvo a 40-45 °C. A cada una de estas disoluciones concentradas se añadieron 0,5 mL de tampón de fosfato de sodio 100 mM, pH 7,0. A continuación, las mezclas se diluyeron con agua para obtener una concentración final de APAP de 20 mg/mL. Las disoluciones se filtraron a través de filtros Nylon Target2 de 0,2 pm (Thermo Scientific). A continuación, las formulaciones filtradas se transfirieron a viales de liofilización de vidrio de 10 mL en alícuotas de 5 mL, correspondientes a 100 mg de APAP. Los viales que contenían las formulaciones se liofilizaron usando un liofilizador VirTis Genesis 25EL. En la Tabla 28 se enumeran los productos utilizados para preparar formulaciones reconstituidas sólidas de paracetamol que contienen polímeros WB-4 o WB-7. Las composiciones de las tortas liofilizadas resultantes se muestran en las Tablas 29 y 30, respectivamente.
Las tortas liofilizadas de formulación preparadas con el polímero WB-4 se reconstituyeron en agua para inyección en menos de 3 minutos para proporcionar disoluciones claras y sin partículas de paracetamol con una concentración de 50 mg/mL. Por el contrario, la reconstitución de las tortas preparadas con el polímero WB-7 fue mucho más rápida (menos de 1 min), pero las disoluciones resultantes mostraron una turbidez significativa en este caso. Esta diferencia está respaldada por mediciones de transmitancia óptica. Las formulaciones reconstituidas que contienen el polímero WB-4 se caracterizan por una transmitancia óptica aprox. del 95%. Las disoluciones de las formulaciones preparadas con el polímero WB-7 tienen una transmitancia significativamente menor, en el intervalo del 40-42%. Mientras que el tamaño de partícula promedio Z para las formulaciones reconstituidas de las tortas de APAP/WB-4 fue de 44,3 nm, el tamaño de las partículas en la formulación reconstituida de APAP/WB-7 fue mucho mayor e igual a 218 nm. La Figura 11 muestra que las distribuciones de tamaños de partículas para las formulaciones preparadas usando dos polímeros diferentes WB-4 y WB-7 y reconstituidas a 50 mg/mL tienen diferentes formas, con el tamaño promedio en volumen de las micelas de aprox. 10 a aprox. 200 nm. Esto contrasta fuertemente con el comportamiento de las formulaciones de flurbiprofeno preparadas con estos dos polímeros. El pH de las disoluciones reconstituidas para las formulaciones obtenidas con ambos polímeros fue similar, y en el intervalo de 7,20-7,25. La osmolalidad de las muestras reconstituidas también fue similar para ambos polímeros y estuvo comprendida en el intervalo de 271-281 mOsm/kg. Los valores de los parámetros determinados en la caracterización de las formulaciones de APAP reconstituidas se muestran en la Tabla 31.
Tabla 28

Tabla 29

Tabla 30

Tabla 31

En consecuencia, se lograron NCF de casi el 15%.
WB-7 no pudo formular APAP de manera adecuada para su administración debido a la presencia de partículas visibles y subvisibles.
Con WB-4, se consideró que la disolución resultante poseía propiedades adecuadas para la administración. Otros experimentos indicaron que WB-4 podría formular APAP a un NCF del 18 % (90 % de transmisión óptica), 20 % (84 % de transmisión óptica) y 25 % (78 % de transmisión óptica).
Formulaciones de paracetamol con otros polímeros
Dado el éxito anterior con WB-4, se intentaron otras formulaciones de prueba de APAP basadas en los protocolos anteriores con los polímeros WB-5, WB-6 y WB-8 (datos no mostrados). En resumen, se consideró que los polímeros WB-5 y WB-6 fueron capaces de producir nanodispersiones de APAP que fueron adecuadas para cumplir los requisitos de administración.
F o rm u la c io n e s d e c e le c o x ib
Formulaciones de celecoxib con polímero WB-4
La formulación de celecoxib (CEL) se preparó como sigue. Se disolvieron 450 mg de copolímero en bloque WB-4 en 1 mL de agua para inyección con agitación magnética durante aprox. 10 minutos. A la disolución de polímero, se añadieron 0,4 mL de disolución acuosa de NaOH 0,1 N con agitación para llevar el pH a aproximadamente 7,5. A continuación, se añadieron a la disolución 0,25 mL de tampón de fosfato de sodio 100 mM, pH 7,0, seguido de la adición de 1,25 mL de disolución acuosa de manitol a una concentración de 100 mg/mL. A continuación, se disolvieron 50 mg de celecoxib en 1 mL de etanol en un vial de vidrio con agitación magnética a temperatura ambiente y se añadieron gota a gota a la disolución de polímero durante aprox. 1 min. La disolución clara resultante se enfrió en un baño de hielo y se colocó en una cámara fría a 6 °C con agitación durante aprox. 20 minutos. Después de sacar la muestra de la cámara fría, se añadieron 1,10 mL de agua a la formulación. A continuación, la disolución se concentró durante aprox. 30 min al 25% de su peso inicial a presión reducida en un aparato Büchi Collegiate Rotavapor® equipado con una trampa de disolvente de hielo seco y una bomba de vacío Heidolph Rotavac Valve. La temperatura del baño de agua se mantuvo a 30-35 °C. A la disolución concentrada se le añadieron 3,64 mL de agua para obtener la formulación de concentración final de CEL de 10 mg/mL. A continuación, la formulación se filtró a través de filtros Nylon Target2 de 0,2 pm (Thermo Scientific). La formulación filtrada se transfirió a un vial de vidrio de 10 mL y se liofilizó usando un liofilizador VirTis Genesis 25EL. La composición de las tortas liofilizadas de celecoxib resultantes se muestra en la Tabla 32.
Tabla 32

En consecuencia, se logró un NCF de casi el 8%.
La Figura 12 muestra una imagen de la torta liofilizada y la disolución obtenida después de la reconstitución de la torta con agua para inyección a la concentración de CEL de 25 mg/mL. La torta muestra una estructura fina similar a una esponja sin agrietarse ni colapsarse. La inspección minuciosa del líquido reconstituido (Figura 12b) confirma la claridad de la disolución y la ausencia de partículas sólidas visibles.
Las tortas liofilizadas se disuelven en agua en menos de 1 min para proporcionar disoluciones claras y sin partículas con una concentración de celecoxib de 25 mg/mL. El pH de las disoluciones reconstituidas estuvo en el intervalo de 6,8 a 7,2 medido usando un medidor de pH Accumet AP61 equipado con un electrodo de combinación de cuerpo epoxi relleno de gel. La transmitancia óptica se determinó en cubetas de poliestireno desechables de 1 cm en un espectrómetro Agilent Cary UV-Vis-NIR 5000. Las medidas se realizaron a 650 nm y a temperatura ambiente usando una cubeta vacía como blanco. Las mezclas reconstituidas tuvieron una transmitancia óptica de alrededor del 92%. La osmolalidad de las muestras reconstituidas se midió con un Micro-Osmómetro 3300 de depresión del punto de congelación (Advanced Instruments) y estuvo en el intervalo de 420 a 450 mOsm/kg. El tamaño medio Z de las micelas y su distribución de tamaños se determinó a 25 °C mediante dispersión de luz dinámica usando un aparato Malvern Zetasizer Nano ZS equipado con un láser de He-Ne de 10 mW que funcionó a 633 nm. El tamaño de partícula promedio Z y su polidispersidad variaron de 47 a 52 nm y de 0,38 a 0,45, respectivamente. La Figura 13 muestra que la distribución de tamaños de partículas para la formulación de celecoxib reconstituida a 25 mg/mL es monomodal y que el tamaño promedio en volumen de las micelas varía de aprox. 10 a aprox. 200 nm.
Formulaciones de celecoxib con otros polímeros
Dado el éxito anterior con WB-4, se intentaron otras formulaciones de prueba de CEL con polímeros WB-5 a WB-8 basándose en los protocolos anteriores (datos no mostrados). En resumen, se consideró que los polímeros WB-5, WB-6 y WB-8 fueron capaces de producir nanodispersiones de CEL que fueron adecuadas para cumplir los requisitos de administración.
F o rm u la c io n e s d e P ro p o fo l
Las formulaciones de propofol (PPF) se prepararon como sigue. Se disolvieron 900 mg de polímero de PVP-PLA (WB-4, WB-5, WB-6, WB-7 y WB-8) en 7,5 mL de tampón fosfato 100 mM pH 7,0 en un vial de vidrio con agitación a temperatura ambiente durante aproximadamente 10 minutos, y después la adición de 100 mg (107 pL) de PPF. Las disoluciones se mantuvieron con agitación vigorosa a temperatura ambiente durante aprox. 16 h, seguido de la adición de 2,5 mL de agua. La Figura 1 muestra las imágenes de las formulaciones antes de la filtración. La distribución de tamaños de las partículas obtenidas antes de la filtración se muestra en la Figura 2. Las formulaciones preparadas con los polímeros WB-4, WB-5 y WB-6 son disoluciones homogéneas y claras. Muestran una distribución monomodal de tamaños de partículas con un tamaño promedio Z de 33 a 44 nm. Estas formulaciones se filtraron posteriormente a través de filtros Nylon Target2 de 0,2 pm (Thermo Scientific), se transfirieron a viales de vidrio de 5 mL en alícuotas de 2 mL y se liofilizaron usando un liofilizador VirTis Genesis 25EL. Por el contrario, las muestras preparadas con los polímeros WB-7 y WB-8 muestran una distribución de tamaños bimodal y un tamaño promedio z de aprox. 120 nm, no se pueden filtrar con filtros de 0,2 pm y no se analizaron más.
La Figura 14 muestra una fotografía de las formulaciones de PPF preparadas con muestras de los polímeros WB-4 a WB-8 antes de la filtración y la liofilización, con WB-4 a WB-6 produciendo disoluciones relativamente claras.
La Figura 15 representa la distribución del tamaño promedio en volumen de partículas en las formulaciones de PPF y polímeros WB-4 a WB-8 antes de la filtración y la liofilización.
Las tortas liofilizadas (20 mg de PPF) se reconstituyeron con agua en menos de 1 min después de agitar suavemente para obtener disoluciones claras y sin partículas con una concentración de PPF de 10 mg/mL. La Tabla 2 muestra las propiedades fisicoquímicas de las formulaciones reconstituidas. El pH de las disoluciones reconstituidas fue de 7,02 7,03 medido con un medidor de pH Accumet AP61 equipado con un electrodo de combinación de cuerpo epoxi relleno de gel. La transmitancia óptica se determinó en cubetas de poliestireno desechables de 1 cm en un espectrómetro Agilent Cary UV-Vis-NIR 5000. Las medidas se realizaron a 650 nm y a temperatura ambiente usando una cubeta vacía como blanco. Las mezclas reconstituidas tuvieron una transmitancia óptica aprox. entre el 77 y 83%. La osmolalidad de las muestras reconstituidas se midió con un Micro-Osmómetro 3300 de depresión del punto de congelación (Advanced Instruments) y estuvo en el intervalo de 188 a 223 mOsm/kg. El tamaño promedio Z de las micelas y su distribución de tamaños se determinó a 25 °C mediante dispersión de luz dinámica utilizando un aparato Malvern Zetasizer Nano ZS equipado con un láser de He-Ne de 10 mW que funcionó a 633 nm. El tamaño de partícula promedio Z varió de 43,5 a 58,7 nm.
La Figura 16 muestra que la distribución de tamaños de partículas para las formulaciones de PPF preparadas usando los polímeros WB-4, WB-5 y WB-6 es monomodal, y que el tamaño promedio en volumen de las micelas varía de aprox. 15 a 150 nm.
La Tabla 33 describe la caracterización fisicoquímica de las formulaciones de PPF preparadas con muestras de polímeros WB-4, WB-5 y WB-6 y reconstituidas a 10 mg/mL.
Tabla 33
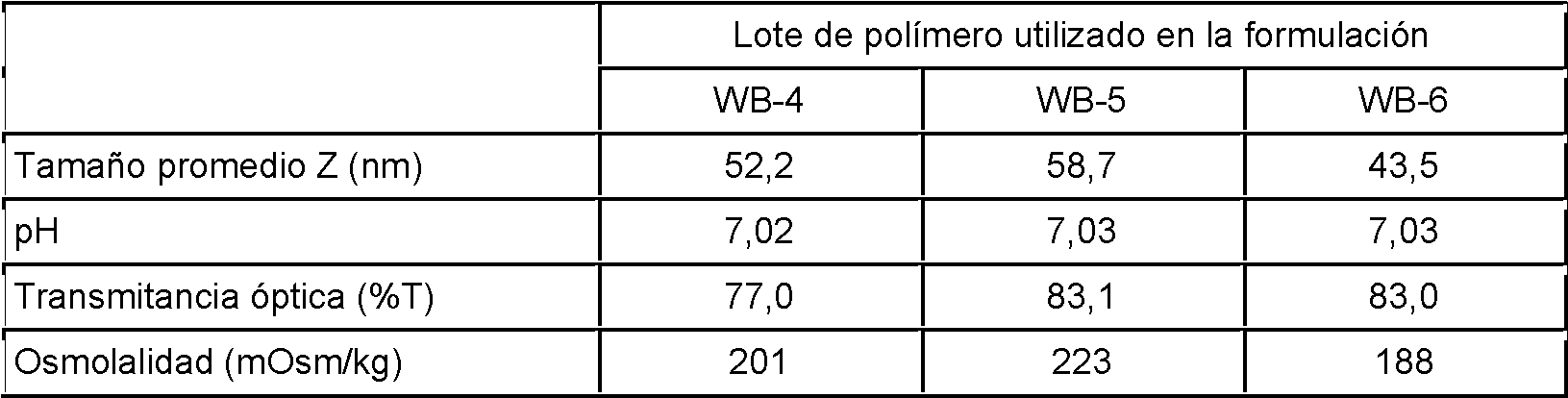
Las tres formulaciones eran adecuadas para la administración.
Resumen
La Tabla 34 resume brevemente los resultados de la formulación, indicando qué polímeros se formularon con éxito (S), qué IFA y cuáles no (N).
Tabla 34

Discusión
Los copolímeros en bloque de PVP-PLA descritos en la presente memoria se han utilizado para aumentar la solubilidad en agua de una diversidad de moléculas farmacológicas miles de veces y para producir productos sólidos que se reconstituyen rápidamente en disolución acuosa para generar líquidos de baja viscosidad y muy alta concentración. Además, estas formulaciones se han liofilizado para optimizar su estabilidad tanto a temperatura ambiente como a temperaturas elevadas, como podría ocurrir de vez en cuando. Estos copolímeros pueden lograr un NCF y concentraciones en disolución que son adecuadas para la administración.
Los polímeros se pueden clasificar en términos de su capacidad para formular IFA de la siguiente manera.
Se ha demostrado que los polímeros del Grupo 1 formulan eficazmente al menos un IFA, es decir, FLU como una composición micelar adecuada para la administración a un sujeto. Estos incluyen WB-1 a WB-8. WB-DMAP también puede incluirse en este grupo.
Se ha demostrado que los polímeros del Grupo 2 formulan eficazmente al menos dos IFA, es decir, FLU y CEL. En este grupo se incluyen WB-4 a WB-6 y WB-8. WB-1 a WB-3 no se probaron, pero deberían incluirse debido a sus propiedades similares.
Se ha demostrado que los polímeros del Grupo 3 formulan eficazmente al menos tres IFA. De hecho, también formulan los cuatro que se probaron, es decir, FLU, CEL, APAP y PPF. WB-4 A WB-6 están incluidos en este grupo. Nuevamente, WB-1 a WB- también se incluyen debido a propiedades similares.
Se puede pensar en el polímero del Grupo 4 como un subconjunto del Grupo 3 que ha estado sujeto a las pruebas más rigurosas e incluye WB-4 a WB-6.
Estos grupos se enumeran en la Tabla 20, en la que se han enumerado los valores mínimo, máximo y promedio para varios parámetros para los grupos. Las tendencias son evidentes en estos datos, con intervalos reducidos o diferentes valores promedio en los Grupos 1 a 4 que indican aumentos inesperados en la flexibilidad. Los parámetros de diseño podrían seleccionarse fácilmente dentro de estos intervalos para generar polímeros que tengan las características deseadas. La selección de las variables objetivo para los copolímeros en bloque dependerá de los requisitos, p. ej. si se desea seleccionar un copolímero en bloque en función de la capacidad de solubilizar un IFA específico, o si se prefiere una plataforma de administración de fármacos flexible.
Ejemplo 4: Estudio Farmacocinético
Introducción
El presente estudio examinó la farmacocinética (FC) del flurbiprofeno (FLU) formulado con nanotecnología micelar SmartCelle (PPI-1501) después de la administración intravenosa (IV) a ratas como se describe en el Informe JRF: Estudio farmacocinético de dosis única de formulación de flurbiprofeno a través de la ruta intravenosa en ratas Wistar; JRF International, septiembre de 2016. La farmacocinética de FLU tal como se presenta en el informe se ajustó utilizando modelos no compartimentales y de dos compartimentos para evaluar la bondad del ajuste.
Los resultados ajustados obtenidos en el estudio JRF se compararon después con los de la bibliografía, generados después de administrar una disolución IV de FLU simple de baja concentración no micelar a ratas. Esto se llevó a cabo para determinar las posibles diferencias en la farmacocinética de FLU inducida por la formulación micelar.
Luego, ambos conjuntos de datos de roedores se extrapolaron usando los parámetros FC de FLU humanos publicados para estimar la FC de FLU después de la administración IV de PPI 1501 o FLU sin formular a humanos.
Finalmente, la farmacocinética predicha de FLU después de la administración de PPI-1501 a humanos se comparó con la generada por la administración IV del profármaco de FLU flurbiprofeno axetilo (FA) a humanos. A continuación, se evaluó el impacto de la formulación micelar en la farmacodinámica de FLU en comparación con una disolución simple o el profármaco.
Métodos
Según el protocolo de JRF, se administró PPI 1501 reconstituido a ratas Wistar mediante inyección intravenosa durante 15 segundos a través de la vena yugular en 3 dosis: 2,5 mg/kg, 10 mg/kg y 30 mg/kg. Estas dosis se seleccionaron para producir exposiciones en ratas que abarcaban las dosis terapéuticas humanas potenciales, teniendo en cuenta las diferencias en la tasa de eliminación entre ratas y humanos.
Se recogieron muestras de sangre de 3 ratas en los siguientes puntos de tiempo: 2, 15, 30 minutos y 1, 2, 4, 6, 12 y 24 horas. Se tomaron muestras de cada rata 2 veces de la vena yugular.
Resultados
Variabilidad
Los perfiles de tiempo-concentración de FLU generados por la formulación PPI 1501 se resumen en las Tablas 35-37 y la Figura 17.
Tabla 35: Perfil de tiempo-concentración de FLU en ratas después de una dosis IV de 2,5 mg/kg de PPI-1501

Tabla 36: Perfil de tiempo-concentración de FLU en ratas después de una dosis IV de 10 mg/kg de PPI-1501.

Tabla 37: Perfil de tiempo-concentración de FLU en ratas después de una dosis IV de 30 mg/kg de PPI-1501.

La propia Figura 17 representa los perfiles de tiempo-concentración de FLU en ratas después de una dosis IV de 2,5 (línea inferior), 10 (línea media) o 30 mg/kg (línea superior) de PPI-1501.
La variabilidad entre animales (CV) para la mayoría de los puntos de tiempo estuvo dentro del 20%. Solo se observaron mayores grados de variación en puntos de tiempo posteriores cuando los niveles de FLU estaban más cerca de los niveles de cuantificación del bioensayo.
Linealidad
Los datos se analizaron primero usando un modelo estándar no compartimental, los resultados se resumen en la Tabla 38.
Tabla 38. Parámetros FC IV de FLU de PPI 1501 basados en el análisis no compartimental estándar (ANC)

Las concentraciones plasmáticas normalizadas por dosis a tiempo 0 (C0) y el ABCinf a la dosis de 10 mg/kg fueron el 93 % y el 80 %, respectivamente, de los observados con la dosis de 2,5 mg/kg, mientras que los valores de estos parámetros para la dosis de 30 mg/kg fueron solo del 63 % de los observados con la dosis de 2,5 mg/kg. Estos datos sugieren una linealidad de dosis aceptable a las dosis entre 10 mg/kg y 2,5 mg/kg, pero una reducción en la exposición al aumentar la dosis.
Por esta razón, las comparaciones con los datos de la disolución intravenosa de roedores y las extrapolaciones a los resultados potenciales en humanos que se muestran más adelante en este informe se realizaron solo con los datos de 2,5 mg/kg en ratas. Como la dosis humana prevista de PPI-1501 es de 100 mg BID (ver más abajo), 2,5 mg/kg representa el intervalo superior del posible régimen de tratamiento (175 mg para un individuo de 70 kg).
Después se compararon los resultados del estudio JRF con los valores de la bibliografía para la FC de FLU
administrado como una disolución IV generados por Park y Kim (1) y Knihinicki et al. (2); la comparación se muestra en la Tabla 39.
Tabla 39: Diferencias de los parámetros farmacocinéticos de ANC entre FLU en PPI 1501 frete a las disoluciones a una dosis IV de 2,5 mg/kg

Como se puede ver, los resultados FC de roedores obtenidos por separado para una disolución simple de FLU de dosis baja por Park y Kim o Knihinicki fueron similares para todos los parámetros medidos (diferencias de 0,72 a 1,17), lo que indica que un meta-análisis entre estudios, aunque no proporciona datos comparativos absolutos, es aceptable para identificar las diferencias significativas entre la administración de FLU en formas micelares y no micelares. Se destacan los parámetros en los que se producen diferencias considerables entre la disolución simple y la administración micelar.
Así, al comparar los valores de la disolución simple con los obtenidos con la formulación micelar, se evidencian una serie de diferencias;
1. La vida media (t1/2) de FLU administrado mediante PPI-1501 es considerablemente mayor que cuando se administra como una disolución.
2. El tiempo medio de residencia (MRT) de FLU administrado mediante PPI-1501 es mayor que cuando se administra como una disolución.
3. La velocidad de aclaramiento (CL) de FLU administrado mediante PPI-1501 es considerablemente más lenta que cuando se administra como una disolución.
4. Lo que es más importante, la exposición de las ratas a FLU (ABCinf) es considerablemente mayor cuando se administra FLU en forma micelar.
La Figura 18 muestra perfiles comparativos de tiempo-concentración en plasma de FLU después de la dosificación IV de una disolución de FLU de 2,5 mg/kg (redibujada de la Referencia 1; línea inferior) y después de la dosificación IV de PPI-1501 a 2,5 mg/kg (línea superior). Los resultados durante el período de prueba destacan la eliminación más rápida de FLU cuando se administra como una disolución y también la Cmax más baja que se logra cuando se administra FLU en forma micelar. Obsérvese la eliminación/redistribución inicial más rápida de FLU cuando se administra como una disolución simple. En conclusión, la administración en forma micelar parece aumentar la exposición de las ratas a FLU.
Análisis utilizando un modelo de dos compartimentos
Si bien el ANC se empleó anteriormente para permitir la comparación de los datos de roedores generados con PPI-1501 con los datos de disolución simple de la bibliografía, los informes más recientes indican que FLU tanto en la clínica como en los roedores se ajusta mejor usando un modelo de 2 compartimentos (2-8). Las Figuras 18-20, por lo tanto, presentan los datos de JRF ajustados utilizando un modelo de 2 compartimentos.
La Figura 19 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU dosificado como PPI 1501 a la dosis IV de 2,5 mg/kg.
La Figura 20 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU dosificado como PPI 1501 a la dosis IV de 10 mg/kg.
La Figura 21 representa el ajuste del modelo de dos compartimentos del perfil de concentración plasmática de FLU dosificado como PPI 1501 a la dosis IV de 30 mg/kg.
La Tabla 40 muestra los parámetros FC que se generan.
Tabla 40: Análisis de datos farmacocinéticos de dos compartimentos basado en los datos de la Tabla 21
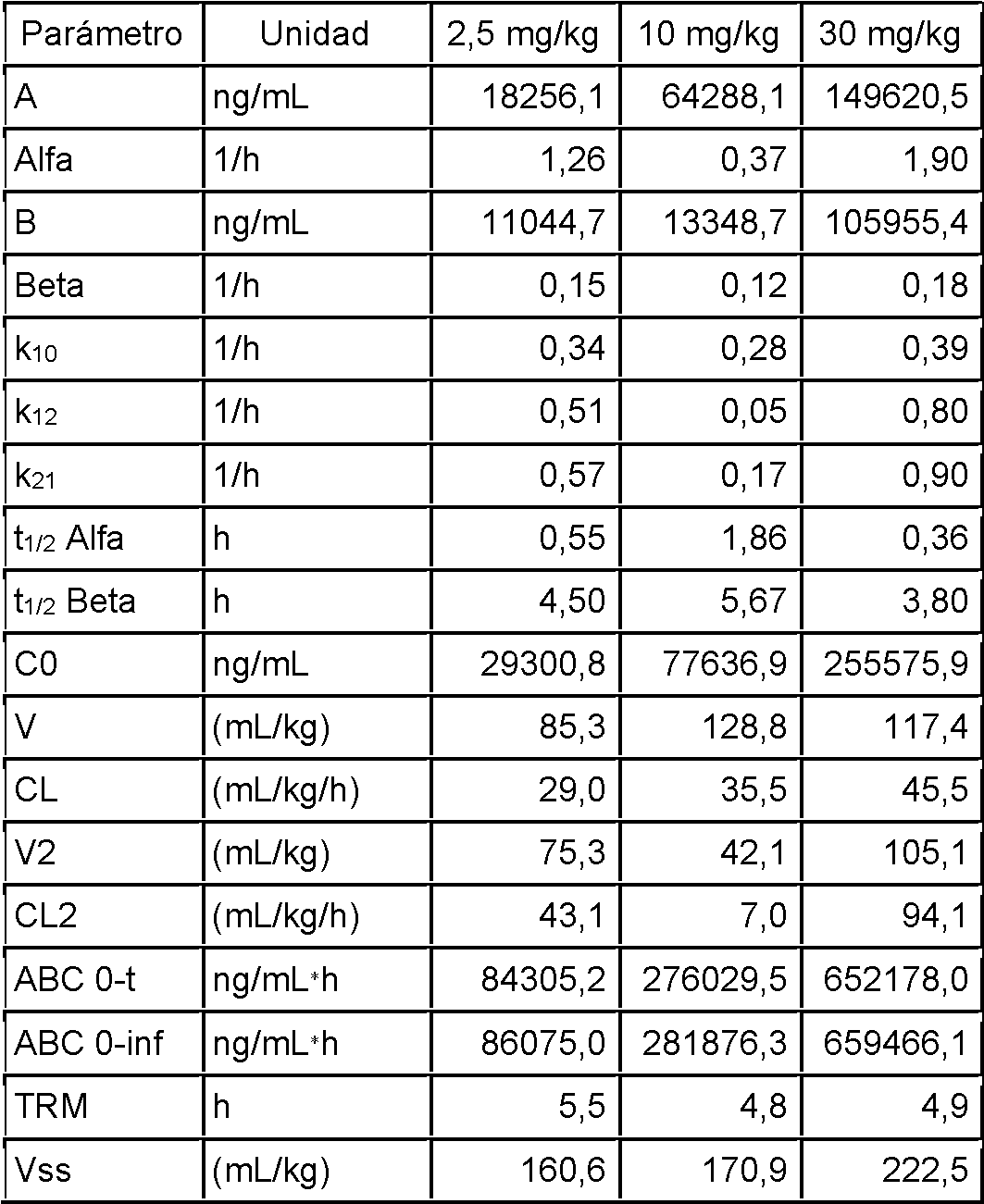
Con fines comparativos, los datos de farmacocinética de ratas para una disolución de FLU generados por Park y Kim (1) también se ajustaron al modelo de 2 compartimentos (Figura 22 y Tabla 41).
La Figura 22 representa el ajuste del modelo de dos compartimentos de la concentración plasmática de FLU dosificado como una disolución simple a la dosis IV de 2,5 mg/kg. Obsérvese la fase de redistribución inicial más pronunciada y rápida de la disolución de FLU en comparación con FLU de PPI-1501.
La comparación del perfil IV entre FLU en PPI 1501 y FLU en disolución reveló una concentración inicial más baja y una fase de distribución alfa más lenta para el producto micelar, probablemente debido a la liberación retardada de FLU de las micelas de PPI 1501 en comparación con la disolución simple. Por el contrario, la constante de eliminación de FLU formulada como PPI-1501 en la fase beta fue más lenta que cuando se administró FLU como una disolución. Estos valores, a su vez, dieron lugar a una V más alta (en un 70 %) pero a una V2 más baja (en un 39 %) para FLU de PPI 1501 y un CL y CL2 un 40 % y un 92 % más bajos para la formulación micelar.
Tabla 41: Parámetros farmacocinéticos estimados de 2 compartimentos de FLU en disolución (2,5 mg/kg) y diferencias de los valores de los parámetros entre FLU en PPI 1501 (2,5 mg/kg)


Los parámetros en las celdas de los cuadros sombreados son los más críticos para influir en el perfil de tiempoconcentración de la Figura 22. Los resultados son similares a aquellos cuando se utilizó el análisis no compartimental.
Extrapolaciones al rendimiento de FC y FD humanos
Suponiendo que el perfil FC en plasma de FLU mediado por PPI 1501 IV en la rata es traducible a humanos, es decir, que las mismas proporciones de diferencias de parámetros FC para FLU en PPI 1501 observadas en la rata se aplican a humanos, entonces se pueden estimar los parámetros FC IV humanos para FLU en PPI 1501. Esto puede lograrse mediante la simple multiplicación de las proporciones de diferencias proporcionadas en la Tabla 41 (última columna) para obtener de nuevo los valores comparativos de los parámetros FC humanos de FLU en PPI 1501 frente a los obtenidos de la inyección de la disolución simple; dichos datos también pueden compararse con los datos obtenidos al inyectar a humanos el profármaco FA (Tabla 42).
Los parámetros FC IV humanos de FLU en disolución se estimaron en función de los parámetros FC orales de FLU y su biodisponibilidad oral estimada (informe del proyecto 1, Duan, 2015, Tabla 42, referencia 3-8). A partir de la Tabla 10, está claro que el MRT estimado después de la dosis IV de FLU en PPI 1501 es >2 veces el de FLU en disolución y la dosis IV de FA (Tabla 42), atribuible al aumento de V y la disminución de CL.
Tabla 42: Parámetros FC IV humanos de 2 compartimentos estimados de FLU en PPI 1501 en humanos, suponiendo que la misma proporción de diferencia de valores críticos para 2 compartimentos observados en ratas son aplicables a humanos

Estos valores de parámetros compartimentales se usaron además para simular los perfiles de tiempo-concentración IV de FLU en disolución frente a los del PPI 1501 y el profármaco FA administrado IV. (Figuras 23-25).
La Figura 23 representa la simulación comparativa de la concentración humana de FLU después de una dosis única IV de 100 mg de FLU en disolución (la línea superior en el tiempo 0) frente a la formulación de PPI 1501 (la línea superior a las 24 h) y FA (la línea inferior). Aquí, una concentración de 4 mg/l representa el nivel umbral de reaparición del dolor.
La Figura 24 representa una simulación comparativa de la concentración de FLU humana después de una dosis IV BID de 100 mg de FLU en disolución (la línea superior en el tiempo 0) frente a la formulación de PPI 1501 (la línea superior a las 24 h) y FA (la línea inferior). La línea horizontal corresponde a una concentración de 4 mg/L y representa el nivel umbral de recurrencia del dolor.
La Figura 25 representa una simulación comparativa de la concentración de FLU humana después de una dosis IV cada 6 h de 50 mg de FLU en disolución (la línea superior en el tiempo 0) frente a la formulación de PPI 1501 (la línea superior a las 24 h) y FA (la línea inferior). La línea horizontal correspondiente a la concentración de 4 mg/L representa el nivel umbral de recurrencia del dolor.
Como se muestra en la Figura 23, la concentración plasmática de FLU fue máxima después de la administración IV de 100 mg del fármaco como una disolución simple; la concentración plasmática máxima más baja se mostró cuando se administraron 100 mg de FLU en PPI 1501; en todos los casos, sin embargo, la Cmax fue mayor que la supuesta concentración terapéutica mínima de FLU de 4 mg/L. Sin embargo, la formulación de PPI 1501 entregó las concentraciones plasmáticas más altas después de 4 horas y mantuvo las concentraciones plasmáticas >4 mg/L durante más de 14,9 horas. En comparación, después de la misma dosis IV de FA, la duración de las concentraciones plasmáticas > 4 mg/L fue solo de 5,5 horas, casi 3 veces más corta (Figura 23); la inyección de FA también dio como resultado la proporción más alta de Cmax/C24 en plasma, un parámetro estrechamente relacionado con el perfil de seguridad de FLU (Tabla 43). El FLU de PPI-1501 exhibió la proporción más baja.
Se observaron resultados similares después de una dosis IV BID de 100 mg, o 4 veces al día con 50 mg de FLU en disolución frente a la formulación de PPI 1501 y FA dosificado por vía IV (Figuras 24 y 25). En particular, las concentraciones plasmáticas de FLU permanecieron >4 mg/L durante 23,2 horas y >24 horas después de la dosis IV de FLU en la formulación de PPI 1501 únicamente, en comparación con las 12,5 horas en ambas dosis después de las dosis IV de FA (Tabla 43).
Tabla 43: Parámetros farmacocinéticos comparativos de FLU en disolución y en PPI 1501 frente a los de FA después de diferentes dosis IV

En conclusión, el presente estudio demostró que FLU en PPI 1501 en ratas mostró un ABC superior tras la administración que el logrado por FLU en disolución o FA, principalmente debido a la eliminación reducida del fármaco. Suponiendo que las mismas diferencias de parámetros de 2 compartimentos sean aplicables de manera similar a los humanos, la dosis de 100 mg de FLU cada día, o bid, o cada 6 horas de 50 mg puede tener la ventaja de lograr concentraciones plasmáticas > 4 mg/L (un umbral de concentración efectiva) para 14,9 y >23,2 horas, ~3 o 2 veces mejor que la dosis IV de FA. Además, la proporción Cmax/C24 reducida puede representar otro beneficio para el perfil de seguridad.
Ejemplo 5: Protección contra la hemolisis
Introducción
Se estudió el efecto hemoprotector de las formulaciones de IFA que comprendían los copolímeros en bloque de PVP-PLA.
Métodos
Los copolímeros en bloque de PVP-PLA analizados procedían de otro lote sintetizado, en el que n=27 (valor medio), m=35 (valor medio), Mn(PLA)=2075, Mn(PVP)=3980 y Mn(PLA-PVP) = 6055 (determinado mediante RMN). El %PLA fue el 44% y el %PVP fue 56 (los dos últimos valores determinados mediante ATG). En consecuencia, se entendería que el polímero está dentro de los parámetros más amplios de los Grupos 1 a 3, tal como se definen en la presente memoria.
Para probar la capacidad de las formulaciones de mitigar el potencial hemolítico del IFA, se realizaron experimentos de hemólisis in vitro de la siguiente manera. Se aislaron glóbulos rojos humanos (RBC) mediante centrifugación de sangre completa recogida en un tubo Vacutainer que contenía EDTA. Los RBC se lavaron tres veces en disolución salina normal y finalmente se diluyeron en PBS 9:1 a pH 7,4. Las formulaciones liofilizadas se reconstituyeron en agua y luego se diluyeron a las concentraciones deseadas con PBS. La suspensión de glóbulos rojos se mezcló con las formulaciones para generar una concentración final de IFA de 0,05 mg/mL a 5 mg/mL (se entiende que una concentración de 5 mg/mL de IFA en la sangre de un ser humano es mucho mayor que la que podría usarse terapéuticamente), se incubó a 37 °C durante 60 min y se centrifugó para eliminar los glóbulos rojos intactos. Se diluyeron 0,1 mL de sobrenadante con 0,75 mL de PBS y se transfirieron a una cubeta. La absorbancia A de dicha disolución se determinó a 540 nm usando un espectrómetro Agilent Cary UV-Vis-NIR 5000. Se prepararon muestras de control para el 0 y 100 % de hemólisis incubando glóbulos rojos con PBS y 1 % de Triton X-100, respectivamente. El porcentaje de hemólisis se expresa como 100x(A-A0)/(A100-Ac), donde A0 y A100 es la absorbancia del control negativo y positivo, respectivamente.
Resultados
La Figura 26 muestra el porcentaje de hemólisis medido para el flurbiprofeno sin formular y la formulación de PVP-PLA/FLU (NCF=20 %) a la misma concentración de 5 mg/mL. Los datos representan un único experimento realizado por triplicado ± desviación estándar. Los valores de absorbancia medidos a 540 nm fueron los siguientes: 0,10±0,01 (control de 0% de hemólisis), 0,58±0,03 (control de 100% de hemólisis), 0,12±0,01 (FLU/PVP-PLA), 0,54±0,03 (FLU). El flurbiprofeno (FLU) sin formular muestra una actividad hemolítica significativa, lo que resulta en un alto valor de 92,1 ± 17,2 % de hemólisis. Por el contrario, cuando FLU queda atrapado en los vehículos de PVP-PLA (FLU/PVP-PLA), esencialmente no se observa hemólisis (% de hemólisis = 3,1 ± 0,6 %), lo que muestra un fuerte efecto protector de las micelas poliméricas sobre el fármaco encapsulado.
La Figura 27 muestra el porcentaje de hemólisis para diferentes concentraciones de fármaco en formulaciones de flurbiprofeno (FLU), celecoxib (CEL) y paracetamol (APAP). Los puntos de datos representan experimentos individuales realizados por triplicado ± desviación estándar. Esencialmente no se observó hemólisis (% de hemólisis <10%) para las formulaciones de celecoxib y paracetamol en todo el intervalo de concentraciones estudiado, es decir, para las concentraciones <25 mg/mL y 50 mg/mL, para CEL y APAP, respectivamente.
Discusión
En todos los casos, las micelas de PVP-PLA mostraron la capacidad de mitigar la actividad hemolítica de los IFA hacia los glóbulos rojos, estableciendo así un efecto protector. Se ha demostrado que un IFA como Flu, APAP y Cel induce la hemólisis en los glóbulos rojos humanos a diversas concentraciones y en diversos grados. En la presente memoria se ha demostrado que las micelas, tales como las micelas de PVP-PLA, mediante la encapsulación del IFA, poseen un efecto hemoprotector mediante el cual mitigan la actividad hemolítica de IFA. Esto se confirma de manera más marcada mediante los experimentos con flurbiprofeno. Esta capacidad hemoprotectora proporciona una característica de seguridad importante para la administración de IFA, por ejemplo, a través de la ruta parenteral, donde las concentraciones iniciales de IFA pueden ser muy altas.
Ejemplo 6: Discusión general
En la presente memoria se han descrito nuevos copolímeros en bloque de PVP-PLA, junto con nuevos métodos de producción que demuestran una sorprendente eficiencia de síntesis.
Estos copolímeros se han utilizado para aumentar la solubilidad en agua de las moléculas farmacológicas miles de veces para producir nuevas formulaciones secas y líquidas adecuadas para la administración.
Se han observado tendencias en las propiedades de los copolímeros, de modo que los polímeros podrían diseñarse para, p. ej. un IFA en particular, o para lograr una plataforma de administración de medicamentos más flexible.
Estas nuevas formulaciones se han liofilizado para optimizar su estabilidad tanto a temperatura ambiente como a temperaturas elevadas, como podría ocurrir de vez en cuando. Los productos sólidos así producidos se reconstituyen rápidamente en disolución acuosa para generar líquidos de muy alta concentración y baja viscosidad.
Estos líquidos, cuando se inyectan en animales, generan concentraciones sanguíneas muy altas de su fármaco atrapado. Por lo tanto, la naturaleza de la nueva especie de PVP-PLA hace que los fármacos insolubles sean solubles en disolución acuosa, los mantiene en forma amorfa tanto en forma líquida como sólida, los estabiliza y los administra rápidamente y en una alta concentración a la sangre después de la inyección intravenosa.
Estas propiedades son ideales, por ejemplo, para productos para el dolor posoperatorio, que generan analgesia de forma rápida y fiable. Los polímeros de PVP-PLA prolongan ventajosamente el tiempo de residencia en la sangre y, por lo tanto, mantienen la analgesia durante más tiempo. Se ha demostrado que algunos polímeros mantienen niveles efectivos de IFA en la sangre durante al menos 12 horas. Esto puede significar menos inyecciones para un sujeto y menos recurrencia del dolor (dolor irruptivo).
Lo que es más importante, tales productos pueden proporcionar al paciente analgésicos efectivos, seguros, prácticos y de poca incomodidad con la oportunidad de reemplazar o reducir significativamente la necesidad de analgésicos opioides con sus muchos inconvenientes. Los copolímeros se pueden usar, por ejemplo, para administración oral o parenteral. Los copolímeros en bloque descritos se sintetizaron utilizando un proceso novedoso que produjo productos de alta pureza con rendimientos inesperados para este tipo de sistema. Las características de los copolímeros así sintetizados permiten atrapar una variedad de fármacos insolubles a una alta concentración y convertirlos en amorfos para permitir una liberación rápida in vivo una vez administrados.
En la descripción anterior, con fines explicativos, se exponen numerosos detalles para proporcionar una comprensión completa de las realizaciones. Sin embargo, será evidente para un experto en la técnica que estos detalles específicos no son necesarios. En otros casos, las estructuras y los circuitos eléctricos bien conocidos se muestran en forma de diagrama de bloques para no dificultar la comprensión. Por ejemplo, no se proporcionan detalles específicos sobre si las realizaciones descritas en la presente memoria se implementan como una rutina de software, circuito de hardware, firmware o una combinación de los mismos.
Las realizaciones descritas anteriormente pretenden ser solo ejemplos. Los expertos en la técnica pueden efectuar alteraciones, modificaciones y variaciones en las realizaciones particulares. El alcance de las reivindicaciones no debe estar limitado por las realizaciones particulares establecidas en la presente memoria, sino que debe interpretarse de manera coherente con la memoria descriptiva en su conjunto.
Referencias
1. Kazunari Masutani, Yoshiharu Kimura, PLA Synthesis. From the Monomer to the Polymer, capítulo 1, The Royal Society of Chemistry, 2015, páginas 1-36.
2. Ann-Christine Albertsson, Indra K. Verma, Recent Development in Ring Opening Polymerization of Lactones for Biomedical Applications, Biomacromolecules, 2003, 4, páginas 1466-1486.
3. Irene Bartolozzi, Roberto Solaro, Etienne Schacht, Emo Chiellini, Hydroxyl end-capped macromers of N-vinyl-2-pyrrolidinone as precursors of amphiphilic block copolymers, European Polymer Journal, 2007, 43, páginas 4628-4638.
4. Lionel Delaude, Morgan Hans, Somenath Chowdhury, Microwave-Assisted Synthesis of the 1,3-Dimesitylimidazolinium Chloride, Org. Synth., 2010, 87, páginas 77-87.
5. Lei Xiong, Hong Wei Jiang, Affiliated with South China University of Technology, Di Zhen Wang, Synthesis, characterization and degradation of poly(dl-lactide)-block-polyvinylpyrrolidone-block-poly(dl-lactide) copolymers, Journal of Polymer Research, 2009, 16, páginas 191-197.
6. KM Park, CK Kim. (1999) Preparation and evaluation of flurbiprofen-loaded microemulsion for parenteral delivery. Intl J Pharmaceutics, 181: 173-179.
7. RD Knihinicki, RO Day, GG Graham, KM Williams. (1990) Stereoselective disposition of ibuprofen and flurbiprofen in rats. Chirality, 2: 134-140.
8. Szpunar, G. J., Albert, K. S., Bole, G. G., Dreyfus, J. N., Lockwood, G. F. y Wagner, J. G. (1987), Pharmacokinetics of flurbiprofen in man. I. Area/dose relationships. Biopharm. Drug Dispos., 8: 273-283.
9. Cefali, E. A., Poynor, W. J., Sica, D. y Cox, S. (1991), Pharmacokinetic Comparison of Flurbiprofen in End-Stage Renal Disease Subjects and Subjects with Normal Renal Function. Journal of Clinical Pharma, 31: 808-814.
10. Taburet, A. M., Singlas, E., Glass, R. C., Thomas, F. y Leutenegger, E. (1995), Pharmacokinetic comparison of oral and local action transcutaneous flurbiprofen in healthy volunteers. Journal of Clinical Pharmacy and Therapeutics, 20: 101-107.
11. Qayyum, A., Najmi, M. H. y Farooqi, Z-R. (2011) Determination of Pharmacokinetics of Flurbiprofen in Pakistani Population Using Modified HPLC Method. J. Chromatogr. Sci. 49: 108-113.
12. Kaiser DG, Brooks CD, Lomen PL. (1986) Pharmacokinetics of flurbiprofen. Am. J. Med. 80(3A):10-5.
13. Kumpulainen E, Valitalo P, Kokki M, Lehtonen M, Hooker A, Ranta VP, Kokki H. (2010) Plasma and cerebrospinal fluid pharmacokinetics of flurbiprofen in children. Br J Clin Pharmacol. 70:557-66 5
Claims (16)
1. Una composición farmacéutica seca que comprende copolímeros en bloque de PVP-PLA como se define en la Fórmula I:

donde:
x es un alcohol iniciador que tiene un punto de ebullición superior a 145 °C,
n es, en promedio, de 20 y 40, y
m es, en promedio, de 10 y 40,
en donde los copolímeros en bloque tienen un peso molecular promedio en número (Mn) de al menos 3000 Da; en donde el peso molecular promedio en número se mide mediante resonancia magnética nuclear (RMN) de protones, en asociación molecular con al menos un ingrediente farmacéutico activo (IFA).
2. La composición farmacéutica seca de la reivindicación 1, en la que el punto de ebullición del alcohol iniciador es superior a 200 °C.
3. La composición farmacéutica seca de la reivindicación 1, en la que el alcohol iniciador se selecciona del grupo que consiste en:
1-hexanol;
1-heptanol;
éter monoetílico de dietilenglicol;
éter monometílico de dietilenglicol;
éter monometílico de trietilenglicol;
éter monometílico de tetraetilenglicol;
monometiléteres de oligoetilenglicol de fórmula II

donde a > 5;
éteres monoetílicos de oligoetilenglicol de fórmula III

donde b > 1; y mezclas de los mismos.
4. La composición farmacéutica seca de la reivindicación 1, en la que x es éter monoetílico de dietilenglicol (DEGMEE).
5. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 4, que es reconstituible en agua o una disolución acuosa hasta nanopartículas formadas por los copolímeros en bloque de PVP-PLA y que comprenden
al menos un IFA.
6. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 5, que está liofiliza o secada por pulverización.
7. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 6, que es amorfa.
8. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 7, que es estable durante al menos seis meses a 40 °C.
9. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 8, que tiene un nivel de carga de fármacos (NCF) de al menos un 5 % p/p del al menos un IFA, al menos un 10 % p/p del al menos un IFA, al menos un 20 % p/p del al menos un IFA, o al menos el 30 % p/p del al menos un IFA.
10. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 9, en la que al menos un IFA es hidrofóbico, opcionalmente en la que al menos un IFA tiene una solubilidad en agua de 0,005 g/L a 30 g/L, 0,005 g/L a 14 g/L, o de 10 g/L a 30 g/L.
11. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 10, en la que al menos un IFA comprende un analgésico o al menos un anestésico,
donde opcionalmente:
- el analgésico comprende un fármaco antiinflamatorio no esteroideo (AINE), en el que opcionalmente el AINE comprende flurbiprofeno, celecoxib o paracetamol; y/o
- el al menos un anestésico comprende propofol.
12. La composición farmacéutica seca de una cualquiera de las reivindicaciones 1 a 11, que es reconstituible en agua para formar un líquido esencialmente claro que comprende nanopartículas formadas por los copolímeros en bloque de PVP-PLA y que comprende al menos un IFA.
13. La composición farmacéutica seca de la reivindicación 12, en la que el líquido esencialmente claro comprende al menos 20 g/L del IFA, al menos 30 g/L del IFA, al menos 40 g/L del IFA o al menos 50 g/L del IFA.
14. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 13, en la que:
- n es, en promedio, de 21,5 a 28,
- m es, en promedio, de 18 a 37,
- los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4600 Da a 6600 Da, - el peso molecular promedio en número del bloque de PLA es de 1600 Da a 2900 Da,
- el peso molecular promedio en número del bloque de PVP es de 1900 Da a 4200 Da,
- una proporción del peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,3 a 1,4,
- el peso molecular promedio en peso (Mw) del PLA en los copolímeros en bloque es del 35 % al 60 %, basado en el peso total del polímero, y
- el peso molecular promedio en peso (Mw) de la PVP en los copolímeros en bloque es del 40 % al 65 %, basado en el peso total del polímero;
en donde el peso molecular promedio en número se mide mediante resonancia magnética nuclear de protones (RMN) y el peso molecular promedio en peso se mide mediante análisis termogravimétrico (ATG);
opcionalmente en donde la composición farmacéutica seca comprende al menos dos IFA,
opcionalmente, en donde al menos dos IFA comprenden flurbiprofeno y celecoxib.
15. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 13, en la que:
- n es, en promedio, de 21,5 a 28, opcionalmente de 24 a 28,
- m es, en promedio, de 25 a 37, opcionalmente de 25 a 33,
- los copolímeros en bloque de PVP-PLA tienen un peso molecular promedio en número de 4800 Da a 6600 Da, opcionalmente de 5400 Da a 6600 Da,
- el peso molecular promedio en número del bloque de PLA es de 1600 Da a 2900 Da, opcionalmente de 2200 Da a 2900 Da,
- el peso molecular promedio en número del bloque de PVP es de 2700 Da a 4200 Da, opcionalmente de 2700 Da a 3700 Da,
- una proporción entre el peso molecular promedio en número del bloque de PLA y el peso molecular promedio en número del bloque de PVP (PLA:PVP) de 0,3 a 1,0, opcionalmente de 0,5 a 1,0,
- el peso molecular promedio en peso (Mw) del PLA en los copolímeros en bloque es del 35 % al 60 %, opcionalmente del 35 % al 50 %, basado en el peso total del polímero, y
- el peso molecular promedio en peso (Mw) de PVP en los copolímeros en bloque es del 40 % al 65 %, opcionalmente del 50 % al 65 % basado en el peso total del polímero;
en donde el peso molecular promedio en número se mide mediante resonancia magnética nuclear de protones (RMN) y el peso molecular promedio en peso se mide mediante análisis termogravimétrico (ATG);
opcionalmente, en donde la composición farmacéutica seca comprende al menos tres IFA, opcionalmente, en donde los al menos tres IFA diferentes comprenden al menos tres de flurbiprofeno, celecoxib, paracetamol y propofol,
opcionalmente en donde los al menos tres IFA diferentes comprenden los cuatro flurbiprofeno, celecoxib, paracetamol y propofol.
16. La composición farmacéutica seca de cualquiera de las reivindicaciones 1 a 15 para el uso en un tratamiento, opcionalmente para el uso en el tratamiento del dolor, en donde el al menos un IFA comprende un analgésico.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762479582P | 2017-03-31 | 2017-03-31 | |
| GB1705287.9A GB2561009B (en) | 2017-03-31 | 2017-03-31 | Non-ionic PVP-PLA block copolymers and pharmaceutical compositions derived therefrom |
| PCT/CA2018/050399 WO2018176158A1 (en) | 2017-03-31 | 2018-03-29 | Non-ionic block copolymers and pharmaceutical compositions derived therefrom |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2910378T3 true ES2910378T3 (es) | 2022-05-12 |
Family
ID=58682539
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18777331T Active ES2910378T3 (es) | 2017-03-31 | 2018-03-29 | Copolímeros en bloque no iónicos y composiciones farmacéuticas derivadas de los mismos |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US11266741B2 (es) |
| EP (2) | EP4008735A1 (es) |
| ES (1) | ES2910378T3 (es) |
| GB (1) | GB2561009B (es) |
| PT (1) | PT3601392T (es) |
| WO (1) | WO2018176158A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20240016762A1 (en) * | 2020-08-21 | 2024-01-18 | Altus Formulation lnc. | Dual analgesic/anti-inflammatory compositions comprising cb2 receptor agonists, combinations, and methods of use thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7262253B2 (en) * | 2003-12-02 | 2007-08-28 | Labopharm, Inc. | Process for the preparation of amphiphilic poly (N-vinyl-2-pyrrolidone) block copolymers |
| WO2011130834A1 (en) * | 2010-04-23 | 2011-10-27 | Labopharm Inc. | Non-intravenous dosage form comprising solid formulation of liquid biologically active agent and uses thereof |
-
2017
- 2017-03-31 GB GB1705287.9A patent/GB2561009B/en active Active
-
2018
- 2018-03-29 EP EP22154117.0A patent/EP4008735A1/en active Pending
- 2018-03-29 US US16/497,925 patent/US11266741B2/en active Active
- 2018-03-29 WO PCT/CA2018/050399 patent/WO2018176158A1/en not_active Ceased
- 2018-03-29 EP EP18777331.2A patent/EP3601392B1/en active Active
- 2018-03-29 PT PT187773312T patent/PT3601392T/pt unknown
- 2018-03-29 ES ES18777331T patent/ES2910378T3/es active Active
-
2022
- 2022-02-02 US US17/591,241 patent/US12186398B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US11266741B2 (en) | 2022-03-08 |
| US12186398B2 (en) | 2025-01-07 |
| US20210322556A1 (en) | 2021-10-21 |
| US20220152208A1 (en) | 2022-05-19 |
| EP3601392A4 (en) | 2020-12-02 |
| PT3601392T (pt) | 2022-04-14 |
| GB2561009A (en) | 2018-10-03 |
| GB2561009B (en) | 2020-05-13 |
| EP4008735A1 (en) | 2022-06-08 |
| WO2018176158A1 (en) | 2018-10-04 |
| EP3601392B1 (en) | 2022-03-09 |
| CA3054981A1 (en) | 2018-10-04 |
| EP3601392A1 (en) | 2020-02-05 |
| GB201705287D0 (en) | 2017-05-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Attia et al. | The effect of kinetic stability on biodistribution and anti-tumor efficacy of drug-loaded biodegradable polymeric micelles | |
| ES2271039T3 (es) | Composiciones que comprenden micelas polimericas. | |
| Roberge et al. | BEPO®: bioresorbable diblock mPEG-PDLLA and triblock PDLLA-PEG-PDLLA based in situ forming depots with flexible drug delivery kinetics modulation | |
| ES2531679T3 (es) | Composiciones para la administración de liberación controlada de péptidos | |
| AU2009331026B2 (en) | Preparation method of polymeric micelles composition containing a poorly water-soluble drug | |
| Cao et al. | Unified therapeutic‐prophylactic vaccine demonstrated with a postoperative filler gel to prevent tumor recurrence and metastasis | |
| BRPI0821616B1 (pt) | Composição micelar de copolímero de bloco anfifílico contendo taxano e método para preparação da mesma | |
| Nagahama et al. | Self-assembling polymer micelle/clay nanodisk/doxorubicin hybrid injectable gels for safe and efficient focal treatment of cancer | |
| Wei et al. | Folate-decorated PEG–PLGA nanoparticles with silica shells for capecitabine controlled and targeted delivery | |
| Sotoudegan et al. | Nimodipine-loaded Pluronic® block copolymer micelles: preparation, characterization, in-vitro and in-vivo studies | |
| CN101155844A (zh) | 聚乙二醇-聚缩醛和聚乙二醇-聚缩醛-聚原酸酯接枝共聚物和药物组合物 | |
| Li et al. | Photodynamic therapy-mediated remote control of chemotherapy toward synergistic anticancer treatment | |
| CN101820919A (zh) | 用于局部药物输送的可注射聚合物-脂质共混物 | |
| US10143689B2 (en) | SN-38 loaded iron crosslinked micelle and methods thereof | |
| ES2910378T3 (es) | Copolímeros en bloque no iónicos y composiciones farmacéuticas derivadas de los mismos | |
| Nguyen et al. | Investigation of the properties of chitosan–Pluronic based nanogel drug delivery systems utilizing various Pluronic types | |
| Zhu et al. | Self‐assembled micelles prepared from poly (D, L‐lactide‐co‐glycolide)‐poly (ethylene glycol) block copolymers for sustained release of valsartan | |
| WO2008033497A1 (en) | Nanostructured smart gel for time release drug delivery | |
| EP2415463B1 (en) | Particulate medicinal composition | |
| Salimi et al. | Polymeric micelle as a new carrier in Oral drug delivery systems | |
| CA3054981C (en) | Non-ionic block copolymers and pharmaceutical compositions derived therefrom | |
| ES2734675T3 (es) | Formulación de comprimido dispersable en agua que contiene deferasirox | |
| CN111635508B (zh) | 一种侧链含氨基聚乙二醇-脂肪族饱和聚酯嵌段共聚物及其应用 | |
| Jeetah et al. | Dual encapsulation and controlled delivery of anti-TB drugs from PEG-block-poly (ester-ether) nanomicelles | |
| KR102539414B1 (ko) | 저분자량의 양친성 블록 공중합체를 포함하는 나노입자의 제조 방법 |