ES2911447T3 - Resolución de derivados de diazaspiro[4.5]decano ópticamente activos - Google Patents
Resolución de derivados de diazaspiro[4.5]decano ópticamente activos Download PDFInfo
- Publication number
- ES2911447T3 ES2911447T3 ES17838138T ES17838138T ES2911447T3 ES 2911447 T3 ES2911447 T3 ES 2911447T3 ES 17838138 T ES17838138 T ES 17838138T ES 17838138 T ES17838138 T ES 17838138T ES 2911447 T3 ES2911447 T3 ES 2911447T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- mixture
- isomeric
- process according
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- YMANQWHUUFJQOW-UHFFFAOYSA-N 1,2-diazaspiro[4.5]decane Chemical class N1NCCC11CCCCC1 YMANQWHUUFJQOW-UHFFFAOYSA-N 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 165
- 239000000203 mixture Substances 0.000 claims abstract description 112
- 238000000034 method Methods 0.000 claims abstract description 70
- 150000003839 salts Chemical class 0.000 claims abstract description 47
- RGHNJXZEOKUKBD-QTBDOELSSA-N L-gulonic acid Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O RGHNJXZEOKUKBD-QTBDOELSSA-N 0.000 claims abstract description 36
- 239000011833 salt mixture Substances 0.000 claims abstract description 35
- ZFQRGFMVXLSLKZ-UHFFFAOYSA-N 2,2,7,7-tetramethyl-4a,5,8a,8b-tetrahydro-[1,3]dioxolo[3,4]furo[1,3-d][1,3]dioxine-3a-carboxylic acid;hydrate Chemical compound O.C12OC(C)(C)OCC2OC2(C(O)=O)C1OC(C)(C)O2 ZFQRGFMVXLSLKZ-UHFFFAOYSA-N 0.000 claims abstract description 31
- 239000002253 acid Substances 0.000 claims abstract description 27
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 14
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims abstract description 10
- 238000006243 chemical reaction Methods 0.000 claims description 69
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 42
- 239000002904 solvent Substances 0.000 claims description 37
- 230000008569 process Effects 0.000 claims description 34
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 30
- 239000002585 base Substances 0.000 claims description 22
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 17
- 239000012458 free base Substances 0.000 claims description 14
- 239000003960 organic solvent Substances 0.000 claims description 11
- 150000001299 aldehydes Chemical class 0.000 claims description 10
- 238000000746 purification Methods 0.000 claims description 10
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 9
- 239000007864 aqueous solution Substances 0.000 claims description 7
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzenecarboxaldehyde Natural products O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 claims description 7
- 239000004215 Carbon black (E152) Substances 0.000 claims description 6
- 229930195733 hydrocarbon Natural products 0.000 claims description 6
- 150000002430 hydrocarbons Chemical class 0.000 claims description 6
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 claims description 4
- HUMNYLRZRPPJDN-KWCOIAHCSA-N benzaldehyde Chemical group O=[11CH]C1=CC=CC=C1 HUMNYLRZRPPJDN-KWCOIAHCSA-N 0.000 claims 1
- 150000004682 monohydrates Chemical class 0.000 claims 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 abstract 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 26
- 239000000243 solution Substances 0.000 description 20
- 238000004128 high performance liquid chromatography Methods 0.000 description 19
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 15
- -1 for example Chemical compound 0.000 description 15
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- 239000012065 filter cake Substances 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 125000006239 protecting group Chemical group 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 239000000543 intermediate Substances 0.000 description 11
- 239000012074 organic phase Substances 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 10
- 238000005292 vacuum distillation Methods 0.000 description 10
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 238000005984 hydrogenation reaction Methods 0.000 description 9
- 239000011541 reaction mixture Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 8
- 238000010511 deprotection reaction Methods 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000000376 reactant Substances 0.000 description 8
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 238000005755 formation reaction Methods 0.000 description 6
- 125000000623 heterocyclic group Chemical group 0.000 description 6
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 6
- WXRWBESBLVCYGR-LBPRGKRZSA-N 8-o-tert-butyl 3-o-ethyl (3s)-2,8-diazaspiro[4.5]decane-3,8-dicarboxylate Chemical compound C1N[C@H](C(=O)OCC)CC21CCN(C(=O)OC(C)(C)C)CC2 WXRWBESBLVCYGR-LBPRGKRZSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 239000003651 drinking water Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 4
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 108010031944 Tryptophan Hydroxylase Proteins 0.000 description 4
- 102000005506 Tryptophan Hydroxylase Human genes 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 235000012206 bottled water Nutrition 0.000 description 4
- 125000005843 halogen group Chemical group 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- WHUQREWHTBAXRE-UHFFFAOYSA-N tert-butyl 4-(pyrrolidin-1-ylmethylidene)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1=CN1CCCC1 WHUQREWHTBAXRE-UHFFFAOYSA-N 0.000 description 4
- JYUQEWCJWDGCRX-UHFFFAOYSA-N tert-butyl 4-formylpiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(C=O)CC1 JYUQEWCJWDGCRX-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- PBKONEOXTCPAFI-UHFFFAOYSA-N 1,2,4-trichlorobenzene Chemical compound ClC1=CC=C(Cl)C(Cl)=C1 PBKONEOXTCPAFI-UHFFFAOYSA-N 0.000 description 3
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 3
- KDJGTMHOKBBFKF-NTISSMGPSA-N Cl.CCOC(=O)[C@@H]1CC2(CN1C(=O)OCc1ccccc1)CCNCC2 Chemical compound Cl.CCOC(=O)[C@@H]1CC2(CN1C(=O)OCc1ccccc1)CCNCC2 KDJGTMHOKBBFKF-NTISSMGPSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 3
- 239000003586 protic polar solvent Substances 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 239000010948 rhodium Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- UBOXGVDOUJQMTN-UHFFFAOYSA-N 1,1,2-trichloroethane Chemical compound ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N 1,3-Dimethylbenzene Natural products CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- XSRYJCWFFBJNSV-UHFFFAOYSA-N 2,8-diazaspiro[4.5]decane-3,8-dicarboxylic acid Chemical compound OC(=O)C1CC2(CN1)CCN(CC2)C(O)=O XSRYJCWFFBJNSV-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 2
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 2
- WXRWBESBLVCYGR-UHFFFAOYSA-N 8-o-tert-butyl 3-o-ethyl 2,8-diazaspiro[4.5]decane-3,8-dicarboxylate Chemical compound C1NC(C(=O)OCC)CC21CCN(C(=O)OC(C)(C)C)CC2 WXRWBESBLVCYGR-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- ZVDKZCQXXZLZPD-GORDUTHDSA-N C(/C(=N\O)/C(=O)O)Br Chemical compound C(/C(=N\O)/C(=O)O)Br ZVDKZCQXXZLZPD-GORDUTHDSA-N 0.000 description 2
- OYDQVRGPAKZTSR-INIZCTEOSA-N CCOC(=O)[C@@H]1CC2(CN1C(=O)OCc1ccccc1)CCNCC2 Chemical compound CCOC(=O)[C@@H]1CC2(CN1C(=O)OCc1ccccc1)CCNCC2 OYDQVRGPAKZTSR-INIZCTEOSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- GATVIKZLVQHOMN-UHFFFAOYSA-N Chlorodibromomethane Chemical compound ClC(Br)Br GATVIKZLVQHOMN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 229940126062 Compound A Drugs 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 2
- 101000892398 Homo sapiens Tryptophan 2,3-dioxygenase Proteins 0.000 description 2
- 101000851865 Homo sapiens Tryptophan 5-hydroxylase 2 Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 239000012901 Milli-Q water Substances 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 102100036474 Tryptophan 5-hydroxylase 2 Human genes 0.000 description 2
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 125000006242 amine protecting group Chemical group 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- TZCXTZWJZNENPQ-UHFFFAOYSA-L barium sulfate Chemical compound [Ba+2].[O-]S([O-])(=O)=O TZCXTZWJZNENPQ-UHFFFAOYSA-L 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- JPOXNPPZZKNXOV-UHFFFAOYSA-N bromochloromethane Chemical compound ClCBr JPOXNPPZZKNXOV-UHFFFAOYSA-N 0.000 description 2
- DIKBFYAXUHHXCS-UHFFFAOYSA-N bromoform Chemical compound BrC(Br)Br DIKBFYAXUHHXCS-UHFFFAOYSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N cycloheptane Chemical compound C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- ZQBFAOFFOQMSGJ-UHFFFAOYSA-N hexafluorobenzene Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1F ZQBFAOFFOQMSGJ-UHFFFAOYSA-N 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 229910052987 metal hydride Inorganic materials 0.000 description 2
- 150000004681 metal hydrides Chemical class 0.000 description 2
- 125000002496 methyl group Chemical class [H]C([H])([H])* 0.000 description 2
- UAEPNZWRGJTJPN-UHFFFAOYSA-N methylcyclohexane Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 2
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 2
- FBUKVWPVBMHYJY-UHFFFAOYSA-N nonanoic acid Chemical compound CCCCCCCCC(O)=O FBUKVWPVBMHYJY-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N o-dimethylbenzene Natural products CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 159000000001 potassium salts Chemical class 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 229940076279 serotonin Drugs 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- XSRYJCWFFBJNSV-ZETCQYMHSA-N (3S)-2,8-diazaspiro[4.5]decane-3,8-dicarboxylic acid Chemical compound C1N[C@H](C(=O)O)CC21CCN(C(O)=O)CC2 XSRYJCWFFBJNSV-ZETCQYMHSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- LZDKZFUFMNSQCJ-UHFFFAOYSA-N 1,2-diethoxyethane Chemical compound CCOCCOCC LZDKZFUFMNSQCJ-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- RRQYJINTUHWNHW-UHFFFAOYSA-N 1-ethoxy-2-(2-ethoxyethoxy)ethane Chemical compound CCOCCOCCOCC RRQYJINTUHWNHW-UHFFFAOYSA-N 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N 1H-imidazole Chemical compound C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- LFHLEABTNIQIQO-UHFFFAOYSA-N 1H-isoindole Chemical compound C1=CC=C2CN=CC2=C1 LFHLEABTNIQIQO-UHFFFAOYSA-N 0.000 description 1
- CJMUVMINGJFKIR-UHFFFAOYSA-N 2-(2-hydroxyethoxy)ethanol;methoxymethane Chemical compound COC.OCCOCCO CJMUVMINGJFKIR-UHFFFAOYSA-N 0.000 description 1
- SBASXUCJHJRPEV-UHFFFAOYSA-N 2-(2-methoxyethoxy)ethanol Chemical compound COCCOCCO SBASXUCJHJRPEV-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- ZFFBIQMNKOJDJE-UHFFFAOYSA-N 2-bromo-1,2-diphenylethanone Chemical compound C=1C=CC=CC=1C(Br)C(=O)C1=CC=CC=C1 ZFFBIQMNKOJDJE-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- GGDYAKVUZMZKRV-UHFFFAOYSA-N 2-fluoroethanol Chemical compound OCCF GGDYAKVUZMZKRV-UHFFFAOYSA-N 0.000 description 1
- MSXVEPNJUHWQHW-UHFFFAOYSA-N 2-methylbutan-2-ol Chemical compound CCC(C)(C)O MSXVEPNJUHWQHW-UHFFFAOYSA-N 0.000 description 1
- KIPMDPDAFINLIV-UHFFFAOYSA-N 2-nitroethanol Chemical compound OCC[N+]([O-])=O KIPMDPDAFINLIV-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OTLNPYWUJOZPPA-UHFFFAOYSA-N 4-nitrobenzoic acid Chemical compound OC(=O)C1=CC=C([N+]([O-])=O)C=C1 OTLNPYWUJOZPPA-UHFFFAOYSA-N 0.000 description 1
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 description 1
- IEYGNDKVLIUEOG-UHFFFAOYSA-N 9-O-tert-butyl 4-O-ethyl 1-hydroxy-2-oxa-3,9-diazaspiro[5.5]undec-3-ene-4,9-dicarboxylate Chemical compound OC1ON=C(CC11CCN(CC1)C(=O)OC(C)(C)C)C(=O)OCC IEYGNDKVLIUEOG-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- DOJXGHGHTWFZHK-UHFFFAOYSA-N Hexachloroacetone Chemical compound ClC(Cl)(Cl)C(=O)C(Cl)(Cl)Cl DOJXGHGHTWFZHK-UHFFFAOYSA-N 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 229910019020 PtO2 Inorganic materials 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 206010040108 Serotonin syndrome Diseases 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000003934 aromatic aldehydes Chemical class 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- FMWLUWPQPKEARP-UHFFFAOYSA-N bromodichloromethane Chemical compound ClC(Cl)Br FMWLUWPQPKEARP-UHFFFAOYSA-N 0.000 description 1
- 229950005228 bromoform Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical class CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- FJBFPHVGVWTDIP-UHFFFAOYSA-N dibromomethane Chemical compound BrCBr FJBFPHVGVWTDIP-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940019778 diethylene glycol diethyl ether Drugs 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 229940075557 diethylene glycol monoethyl ether Drugs 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- NKDDWNXOKDWJAK-UHFFFAOYSA-N dimethoxymethane Chemical compound COCOC NKDDWNXOKDWJAK-UHFFFAOYSA-N 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- ZKBUGUZPMZDNRT-QPJJXVBHSA-N ethyl (2z)-3-bromo-2-hydroxyiminopropanoate Chemical compound CCOC(=O)C(\CBr)=N\O ZKBUGUZPMZDNRT-QPJJXVBHSA-N 0.000 description 1
- VICYTAYPKBLQFB-UHFFFAOYSA-N ethyl 3-bromo-2-oxopropanoate Chemical compound CCOC(=O)C(=O)CBr VICYTAYPKBLQFB-UHFFFAOYSA-N 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- UZCGKGPEKUCDTF-UHFFFAOYSA-N fluazinam Chemical compound [O-][N+](=O)C1=CC(C(F)(F)F)=C(Cl)C([N+]([O-])=O)=C1NC1=NC=C(C(F)(F)F)C=C1Cl UZCGKGPEKUCDTF-UHFFFAOYSA-N 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 239000013505 freshwater Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000009904 heterogeneous catalytic hydrogenation reaction Methods 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- 238000009905 homogeneous catalytic hydrogenation reaction Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- ULYZAYCEDJDHCC-UHFFFAOYSA-N isopropyl chloride Chemical compound CC(C)Cl ULYZAYCEDJDHCC-UHFFFAOYSA-N 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- 229910000103 lithium hydride Inorganic materials 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- PYLWMHQQBFSUBP-UHFFFAOYSA-N monofluorobenzene Chemical compound FC1=CC=CC=C1 PYLWMHQQBFSUBP-UHFFFAOYSA-N 0.000 description 1
- 230000036651 mood Effects 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 210000003249 myenteric plexus Anatomy 0.000 description 1
- MBHINSULENHCMF-UHFFFAOYSA-N n,n-dimethylpropanamide Chemical compound CCC(=O)N(C)C MBHINSULENHCMF-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- JYVLIDXNZAXMDK-UHFFFAOYSA-N pentan-2-ol Chemical compound CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 210000004560 pineal gland Anatomy 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- LVTJOONKWUXEFR-FZRMHRINSA-N protoneodioscin Natural products O(C[C@@H](CC[C@]1(O)[C@H](C)[C@@H]2[C@]3(C)[C@H]([C@H]4[C@@H]([C@]5(C)C(=CC4)C[C@@H](O[C@@H]4[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@@H](O)[C@H](O[C@H]6[C@@H](O)[C@@H](O)[C@@H](O)[C@H](C)O6)[C@H](CO)O4)CC5)CC3)C[C@@H]2O1)C)[C@H]1[C@H](O)[C@H](O)[C@H](O)[C@@H](CO)O1 LVTJOONKWUXEFR-FZRMHRINSA-N 0.000 description 1
- 230000008433 psychological processes and functions Effects 0.000 description 1
- 238000011403 purification operation Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 235000019615 sensations Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- BAZAXWOYCMUHIX-UHFFFAOYSA-M sodium perchlorate Chemical compound [Na+].[O-]Cl(=O)(=O)=O BAZAXWOYCMUHIX-UHFFFAOYSA-M 0.000 description 1
- 229910001488 sodium perchlorate Inorganic materials 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- GETTZEONDQJALK-UHFFFAOYSA-N trifluorotoluene Substances FC(F)(F)C1=CC=CC=C1 GETTZEONDQJALK-UHFFFAOYSA-N 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000011995 wilkinson's catalyst Substances 0.000 description 1
- UTODFRQBVUVYOB-UHFFFAOYSA-P wilkinson's catalyst Chemical compound [Cl-].C1=CC=CC=C1P(C=1C=CC=CC=1)(C=1C=CC=CC=1)[Rh+](P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C=1C=CC=CC=1)P(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 UTODFRQBVUVYOB-UHFFFAOYSA-P 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Saccharide Compounds (AREA)
Abstract
Un procedimiento de aumento de la cantidad de una sal de ácido de un compuesto isomérico de fórmula I- (S): **(Ver fórmula)** en donde R1 es etilo y Pg1 es terc-butoxicarbonilo, en relación con una sal de ácido de una cantidad de un compuesto isomérico de fórmula I-(R): **(Ver fórmula)** en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R), comprendiendo el procedimiento: hacer reaccionar la mezcla de partida con ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, en presencia de un aldehído para formar una mezcla de sal de ácido que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos, en donde la mezcla de sal de ácido tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) presentes en la mezcla de partida.
Description
DESCRIPCIÓN
Resolución de derivados de diazaspiro[4.5]decano ópticamente activos
Campo de la invención
La presente invención se refiere a un procedimiento de preparación de productos intermedios útiles para la preparación de compuestos espirocíclicos que son inhibidores de la triptófano hidroxilasa (TPH), particularmente la isoforma 1 (TPH1), que son útiles en el tratamiento de enfermedades o trastornos asociados con serotonina periférica incluyendo, por ejemplo, enfermedades gastrointestinales, cardiovasculares, pulmonares, inflamatorias, metabólicas y de baja masa ósea, así como síndrome de serotonina, y cáncer.
Antecedentes
La serotonina (5-hidroxitriptamina, 5-HT) es un neurotransmisor que modula las funciones centrales y periféricas actuando sobre neuronas, músculo liso y otros tipos de células. La 5-HT está implicada en el control y la modulación de múltiples procesos fisiológicos y psicológicos. En el sistema nervioso central (SNC), la 5-HT regula el estado de ánimo, el apetito y otras funciones del comportamiento. En el sistema GI, la 5-HT desempeña un papel procinético general y es un mediador importante de la sensación (por ejemplo, náuseas y saciedad) entre el tracto G i y el cerebro. Se ha notificado que la desregulación del sistema de señalización periférico de 5-HT está implicada en la etiología de varias afecciones tales como osteoporosis, cáncer, enfermedades cardiovasculares, diabetes, aterosclerosis, así como enfermedades o trastornos gastrointestinales, pulmonares, inflamatorios y hepáticos.
Se han identificado dos isoformas de vertebrados de TPH, concretamente TPH1 y TPH2. TPH1 se expresa principalmente en la glándula pineal y tejidos no neuronales, tales como células enterocromafines (EC) ubicadas en el tracto gastrointestinal (GI). TPH2 (la forma dominante en el cerebro) se expresa exclusivamente en células neuronales, tales como células del rafe dorsal o del plexo mientérico. Los sistemas periféricos y centrales implicados en la biosíntesis de 5-HT están aislados, siendo 5-HT incapaz de cruzar la barrera hematoencefálica. Por lo tanto, los efectos farmacológicos de 5-HT pueden modularse por agentes que afectan a la TPH en la periferia, principalmente TPH1 en el intestino.
Informes recientes han descrito el desarrollo de nuevos inhibidores de TPH1 espirocíclicos útiles para reducir selectivamente los niveles de 5-HT intestinales como medio para tratar y prevenir enfermedades asociadas a 5-HT (véase, por ejemplo, la patente de Estados Unidos n.° 9.199.994). Los procedimientos de la presente invención son útiles para preparar inhibidores de TPH1 descritos en la patente de Estados Unidos n.° 9.199.994, tales como (S)-8-(2-amino-6-((R)-1-(5-cloro-[1,1'-bifenil]-2-il)-2,2,2-trifluoroetoxi)pirimidin-4-il)-2,8-diazaspiro[4.5]decano-3-carboxilato de etilo. En el documento EP2444402 se da a conocer un método para separar enantiómeros de 8-(2-amino-6-((R)-1-(5-cloro-[1,1'-bifenil]-2-il)-2,2,2-trifluoroetoxi)pirimidin-4-il)-2,8-diazaspiro[4.5]decano-3-carboxilato de etilo. Se da a conocer una separación adicional de enantiómeros en Bathori et al., Chemical Communications, vol. 51, n.° 26, páginas 5664-5667, 2015.
Sumario de la invención
La presente invención proporciona, entre otros, un procedimiento de aumento de la cantidad de un compuesto isomérico de fórmula I-(S):

en relación con una cantidad de un compuesto isomérico de fórmula I-(R):

en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R), comprendiendo el procedimiento:
hacer reaccionar la mezcla de partida con ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, en presencia de un aldehído para formar una mezcla de sal que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos,
en donde la mezcla de sal tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) presentes en la mezcla de partida, y en donde las variables constituyentes se definen en el presente documento.
La presente invención proporciona además una mezcla de compuestos isoméricos que tienen las fórmulas I-(S) e I-(R), en donde el exceso enantiomérico del compuesto isomérico de fórmula I-(S) es de aproximadamente el 90 % o mayor.
La presente invención proporciona además una sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos de fórmula I-(S) o fórmula I-(R).
Los detalles de una o más realizaciones de la invención se exponen en los dibujos adjuntos y la descripción a continuación. Otras características, objetos y ventajas de la invención serán evidentes a partir de la descripción y los dibujos, y de las reivindicaciones.
Descripción detallada de la invención
La presente solicitud proporciona, entre otros, un procedimiento de aumento de la cantidad de un compuesto isomérico de fórmula I-(S):

en relación con una cantidad de un compuesto isomérico de fórmula I-(R):

en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R), comprendiendo el procedimiento:
hacer reaccionar la mezcla de partida con ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, en presencia de un aldehído para formar una mezcla de sal que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos,
en donde la mezcla de sal tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) presentes en la mezcla de partida.
En algunas realizaciones, el ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, usado en la reacción es monohidrato de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico. En algunas realizaciones, la reacción se lleva a cabo con aproximadamente 1 equivalente molar del ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida.
En algunas realizaciones, el aldehído usado en la reacción es un aldehído aromático tal como benzaldehído. La cantidad de aldehído puede usarse en una cantidad catalítica con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida. En algunas realizaciones, la reacción se lleva a cabo con menos de 1 equivalente molar del aldehído con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida, por ejemplo, menos de 1 equivalente molar, menos de 0,8 equivalentes molares, menos de 0,6 equivalentes molares, menos de 0,4 equivalentes molares, menos de 0,2 equivalentes molares o menos de 0,1 equivalentes molares. En algunas realizaciones, la reacción se lleva a cabo con de aproximadamente 0,01 a aproximadamente 0,5 equivalentes molares del aldehído con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida, por ejemplo, de aproximadamente 0,01 a aproximadamente 0,5, de aproximadamente 0,01 a aproximadamente 0,4, de aproximadamente 0,01 a aproximadamente 0,3, de aproximadamente 0,01 a aproximadamente 0,2 o de aproximadamente 0,01 a aproximadamente 0,1 equivalentes molares. En realizaciones adicionales, la reacción puede llevarse a cabo, al menos en algún momento durante la reacción, a una temperatura elevada. En algunas realizaciones, la temperatura puede variar de aproximadamente 35 °C a aproximadamente 45 °C, de aproximadamente 30 °C a aproximadamente 40 °C, de aproximadamente 25 °C a aproximadamente 35 °C, de aproximadamente 20 °C a aproximadamente 30 °C o de aproximadamente 15 °C a aproximadamente 25 °C. También puede usarse un disolvente para llevar a cabo la reacción, tal como un disolvente orgánico (por ejemplo, un disolvente de éter tal como 2-metiltetrahidrofurano).
En algunas realizaciones, el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) es de aproximadamente el 75 % o más, aproximadamente el 80 % o más, aproximadamente el 85 % o más, aproximadamente el 90 % o más, aproximadamente el 95 % o más, aproximadamente el 97 % o más, aproximadamente el 98 % o más o aproximadamente el 99 % o más.
En algunas realizaciones, el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) puede oscilar entre aproximadamente el 75 % y aproximadamente el 99,9 %, aproximadamente el 80 % y aproximadamente el 99,9 %, aproximadamente el 85 % y aproximadamente el 99,9 %, aproximadamente el 90 % y aproximadamente el 99,9 %, aproximadamente el 95 % y aproximadamente el 99,9 %, aproximadamente el 96 % y aproximadamente el 99,9 %, aproximadamente el 97 % y aproximadamente el 99,9 %, aproximadamente el 98 % y aproximadamente el 99,9 %, aproximadamente el 99 % y aproximadamente el 99,9 %, o aproximadamente el 99,5 % y aproximadamente el 99,9 %.
En algunas realizaciones, el procedimiento comprende además purificar la mezcla de sal (por ejemplo, por medio de recristalización) para formar una mezcla de sal purificada que tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de las sales de ácido gulónico de los compuestos isoméricos antes de la purificación. La purificación puede llevarse a cabo en un disolvente tal como un disolvente orgánico (por ejemplo, un disolvente de éter tal como 2-metiltetrahidrofurano).
En algunas realizaciones, el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) después de la etapa de purificación es de aproximadamente el 90 % o más, aproximadamente el 95 % o más, aproximadamente el 96 % o más, aproximadamente el 97 % o más, aproximadamente el 98 % o más, o aproximadamente el 99 % o más.
En algunas realizaciones, el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) después de la etapa de purificación puede oscilar entre aproximadamente el 90 % y aproximadamente el 99,9 %, aproximadamente el 95 % y aproximadamente el 99,9 %, aproximadamente el 96 % y aproximadamente el 99,9 %, aproximadamente el 97 % y aproximadamente el 99,9 %, aproximadamente el 98 % y aproximadamente el 99,9 %, aproximadamente el 99 % y aproximadamente el 99,9 %, o aproximadamente el 99,5 % y aproximadamente el 99,9 %.
En algunas realizaciones, el procedimiento comprende además hacer reaccionar la mezcla de sal purificada con una base para formar una mezcla de base libre que comprende compuestos isoméricos que tienen la fórmula I-(S) y la fórmula I-(R):

En algunas realizaciones, la base usada en la reacción de formación de base libre es una base de metal alcalino tal como carbonato de sodio. La cantidad de base usada puede estar en un exceso molar con respecto a la cantidad combinada de sales de ácido gulónico en la mezcla de sal (por ejemplo, mayor de 1 equivalente molar con respecto a la cantidad de sales de ácido gulónico en la mezcla de sal), o en cualquier cantidad que sea suficiente para convertir las sales de ácido gluónico en compuestos de base libre. En algunas realizaciones, la cantidad de base usada en la reacción de formación de base libre es de aproximadamente 1,1a aproximadamente 100 equivalentes molares, de aproximadamente 1,1a aproximadamente 50 equivalentes molares, de aproximadamente 1,1a aproximadamente 25 equivalentes molares, de aproximadamente 1,1a aproximadamente 10 equivalentes molares o de aproximadamente 1,1a aproximadamente 5 equivalentes molares con respecto a la cantidad de sales de ácido gulónico en la mezcla de sal. En realizaciones adicionales, la base se proporciona como una disolución acuosa, tal como una disolución acuosa al 10 %, una disolución acuosa al 20 %, una disolución acuosa al 30 %, una disolución acuosa al 40 %, y similares. La reacción de formación de base libre puede llevarse a cabo adicionalmente, al menos en algún momento durante la reacción, a una temperatura elevada. En algunas realizaciones, la temperatura puede oscilar entre aproximadamente 10 °C y aproximadamente 30 °C, aproximadamente 15 °C y aproximadamente 25 °C o aproximadamente 15 °C y aproximadamente 20 °C. También puede usarse un disolvente para llevar a cabo la reacción de formación de base libre, tal como un disolvente orgánico que comprende un disolvente de éter (por ejemplo, un furano tal como 2-metiltetrahidrofurano) o disolvente de hidrocarburo (por ejemplo, tal como n-heptano), o una combinación de los mismos.
En algunas realizaciones, el exceso enantiomérico del compuesto isomérico de fórmula I-(S) en la mezcla de base libre es de aproximadamente el 90 % o más, aproximadamente el 95 % o más, aproximadamente el 97 % o más, aproximadamente el 98 % o más, aproximadamente el 99 % o más, o aproximadamente el 99,9 % o más.
En algunas realizaciones, el exceso enantiomérico del compuesto isomérico de fórmula I-(S) en la mezcla de base libre puede oscilar entre aproximadamente el 90 % y aproximadamente el 99,9 %, aproximadamente el 95 % y aproximadamente el 99,9 %, aproximadamente el 96 % y aproximadamente el 99,9 %, aproximadamente el 97 % y aproximadamente el 99,9 %, aproximadamente el 98 % y aproximadamente el 99,9 %, aproximadamente el 99 % y
aproximadamente el 99,9 %, o aproximadamente el 99,5 % y aproximadamente el 99,9 %.
Los procedimientos para preparar los compuestos y sales descritos en el presente documento pueden implicar la protección y desprotección de diversos grupos químicos (por ejemplo, protección y desprotección de grupos amina con un grupo protector de amino). La necesidad de protección y desprotección, y la selección de grupos protectores apropiados, puede determinarla fácilmente un experto en la técnica. Puede encontrarse la química de los grupos protectores, por ejemplo, en Wuts y Greene, Protective Groups in Organic Synthesis, 4a ed., John Wiley & Sons: Nueva Jersey, (2007). Los ajustes en los grupos protectores y los métodos de formación y escisión descritos en el presente documento pueden ajustarse según sea necesario en vista de los diversos sustituyentes. Pg1 es terc-butoxicarbonilo.
En algunas realizaciones, la mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) se prepara según un procedimiento que comprende hacer reaccionar un compuesto de fórmula II:

con gas hidrógeno en presencia de un catalizador de hidrogenación, en donde R1 es etilo y Pg1 es terc-butoxicarbonilo.
Como se usa en el presente documento, el término “catalizador de hidrogenación” se refiere a un metal (por ejemplo, paladio, níquel o rodio) adecuado para catalizar una reacción de hidrogenación (es decir, reacción de un compuesto con gas hidrógeno). Los catalizadores de hidrogenación de ejemplo incluyen, pero no se limitan a, paladio sobre carbono, catalizador de Lindlar (paladio depositado sobre carbonato de calcio o sulfato de bario), Ni Raney (por ejemplo, Ni Raney A5000), catalizador de Wilkinson, HRuCl(PPha)a, RhCl(PPh3)3, [Rh(COD)Cl]2, [Ir(COD)(PMePh2)2]+, [Rh(1,5-ciclooctadieno)(PPh3)2]+,PtO2 (catalizador de Adam), paladio sobre carbono, negro de paladio, y similares. Pueden encontrarse ejemplos adicionales de catalizadores de hidrogenación en Nishimura, Heterogeneous Catalytic Hidrogenation for Organic Synthesis, edición 1, Wiley (17 de abril de 2001) y Chaloner, Homogeneous Hydrogenation, edición 1, Springer Netherlands (6 de diciembre de 2010).
En algunas realizaciones, el catalizador de hidrogenación usado en la reacción es Ni Raney A5000. El catalizador de hidrogenación puede usarse en una cantidad catalítica con respecto a la cantidad del compuesto de fórmula II usado en la reacción. También puede usarse un disolvente para llevar a cabo la reacción de hidrogenación, tal como un disolvente orgánico que comprende un disolvente prótico (por ejemplo, etanol), o un disolvente de éter (por ejemplo, un disolvente de furano tal como tetrahidrofurano), o una combinación de los mismos. En realizaciones adicionales, la reacción puede llevarse a cabo, al menos en algún momento durante la reacción, a una temperatura elevada. En algunas realizaciones, la temperatura puede oscilar entre aproximadamente 30 °C y aproximadamente 45 °C, aproximadamente 30 °C y aproximadamente 40 °C o aproximadamente 35 °C y aproximadamente 40 °C.
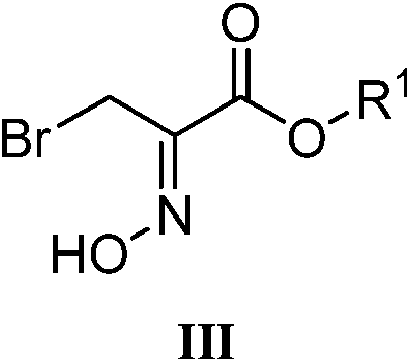
En algunas realizaciones, el compuesto de fórmula II se prepara según un procedimiento que comprende hacer reaccionar un compuesto de fórmula III:
con un compuesto de fórmula IV:

en presencia de una base, en donde R1 es etilo y Pg1 es terc-butoxicarbonilo.
En algunas realizaciones, la base usada en la reacción de los compuestos de fórmula III y IV es una base de amina tal como piridina, trietilamina o N,N-diisopropiletilamina. La cantidad de base usada puede ser un exceso molar con respecto a la cantidad del compuesto de fórmula IV. En algunas realizaciones, la cantidad de base usada puede oscilar entre aproximadamente 1,1 y aproximadamente 3 equivalentes molares, aproximadamente 1,1 y aproximadamente 2 equivalentes molares, aproximadamente 1,4 y aproximadamente 2 equivalentes molares o aproximadamente 1,4 y aproximadamente 1,8 equivalentes molares con respecto a 1 equivalente molar del compuesto de fórmula III. En algunas realizaciones, la reacción se lleva a cabo usando aproximadamente 1 equivalente molar del compuesto de fórmula III con respecto a 1 equivalente molar del compuesto de fórmula IV. En realizaciones adicionales, la reacción puede llevarse a cabo, al menos en algún momento durante la reacción, a una temperatura que es aproximadamente temperatura ambiente o inferior. En algunas realizaciones, la temperatura puede oscilar entre aproximadamente -10 °C y aproximadamente 25 °C, aproximadamente -10 °C y aproximadamente 20 °C, aproximadamente 0 °C y aproximadamente 20 °C, aproximadamente 0 °C y aproximadamente 15 °C o aproximadamente 10 °C y aproximadamente 15 °C. También puede usarse un disolvente para llevar a cabo la reacción, tal como un disolvente orgánico que comprende un disolvente de hidrocarburo (por ejemplo, tolueno) o un disolvente de éter (por ejemplo, un disolvente de furano tal como 2-metiltetrahidrofurano), o una combinación de los mismos.
En algunas realizaciones, el compuesto de fórmula III se prepara según un procedimiento que comprende hacer reaccionar un compuesto de fórmula V:

con hidroxilamina, o una sal de la misma, en donde R1 es etilo.
En algunas realizaciones, la hidroxilamina es una sal de hidroxilamina, tal como clorhidrato de hidroxilamina. La cantidad de la hidroxilamina, o sal de la misma, puede oscilar entre aproximadamente 1,1 y aproximadamente 2 equivalentes molares, aproximadamente 1,1 y aproximadamente 1,8 equivalentes molares, aproximadamente 1,1 y aproximadamente 1,6 equivalentes molares o aproximadamente 1,1 y aproximadamente 1,4 equivalentes molares basándose en 1 equivalente molar del compuesto de fórmula V. En realizaciones adicionales, la reacción puede llevarse a cabo, al menos en algún momento durante la reacción, a una temperatura que es aproximadamente temperatura ambiente o inferior. En algunas realizaciones, la temperatura puede oscilar entre aproximadamente 10 °C y aproximadamente 30 °C, aproximadamente 10 °C y aproximadamente 25 °C o aproximadamente 15 °C y aproximadamente 25 °C. También puede usarse un disolvente para llevar a cabo la reacción, tal como un disolvente de hidrocarburo (por ejemplo, tolueno) o un disolvente prótico (por ejemplo, agua), o una combinación de los mismos.
En algunas realizaciones, el compuesto de fórmula IV se prepara según un procedimiento que comprende hacer reaccionar un compuesto de fórmula VI:

La cantidad de pirrolidina usada puede ser un exceso molar con respecto a la cantidad del compuesto de fórmula VI usado. En algunas realizaciones, la cantidad de pirrolidina usada puede oscilar entre aproximadamente 1,1 y aproximadamente 3 equivalentes molares, aproximadamente 1,1 y aproximadamente 2 equivalentes molares o aproximadamente 1,1 y aproximadamente 1,8 equivalentes molares con respecto a 1 equivalente molar del compuesto de fórmula VI. También puede usarse un disolvente para llevar a cabo la reacción, tal como un disolvente orgánico (por ejemplo, un disolvente de hidrocarburo tal como tolueno). En realizaciones adicionales, la reacción puede llevarse a cabo, al menos en algún momento durante la reacción, a una temperatura elevada. En algunas realizaciones, la reacción puede llevarse a cabo a la temperatura de ebullición de los disolventes.
En algunas realizaciones, la presente solicitud proporciona un procedimiento de aumento de la cantidad de un compuesto isomérico de fórmula Ia-(S):

en relación con una cantidad de un compuesto isomérico de fórmula Ia-(R):

en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula Ia-(S) y fórmula Ia-(R), comprendiendo el procedimiento:
hacer reaccionar la mezcla de partida con monohidrato de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico en
presencia de benzaldehído para formar una mezcla de sal que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos, en donde la mezcla de sal tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula Ia-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula Ia-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas Ia-(S) y Ia-(R) presentes en la mezcla de partida;
recristalizar la mezcla de sal para formar una mezcla de sal purificada que tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula Ia-(S) en relación con la sal de ácido gulónico del compuesto isomérico de fórmula Ia-(R) en comparación con las cantidades relativas de las sales de ácido gulónico de los compuestos isoméricos antes de la purificación; y
hacer reaccionar la mezcla de sal purificada en presencia de carbonato de sodio para formar una mezcla de base libre que comprende compuestos isoméricos que tienen la fórmula Ia-(S) y la fórmula Ia-(R), en donde el exceso enantiomérico del compuesto isomérico de fórmula Ia-(S) en la mezcla de base libre es mayor de aproximadamente el 90 %.
La mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) puede prepararse según las realizaciones descritas anteriormente y también, por ejemplo, como se ilustra adicionalmente en el esquema 1.
Esquema 1.

Formula l-(S) Formula l-{R)
La mezcla de sal descrita en el presente documento que tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la sal de ácido gulónico del compuesto isomérico de fórmula I-(R) (es decir, una mezcla de sal enriquecida en el compuesto isomérico de fórmula I-(S)) puede prepararse según las realizaciones descritas anteriormente y también, por ejemplo, como se muestra a continuación en el esquema 2.
Esquema 2.

en donde R1 es etilo y Pg1 es terc-butoxicarbonilo.
Como se usa en el presente documento, el término “hacer reaccionar” se usa como se conoce en la técnica y generalmente se refiere a la reunión de reactivos químicos de tal manera que se permita su interacción a nivel molecular para lograr una transformación química o física. En algunas realizaciones, la reacción implica dos reactivos, en donde se usan uno o más equivalentes molares del segundo reactivo con respecto al primer reactivo. Las etapas de reacción de los procedimientos descritos en el presente documento pueden realizarse durante un tiempo y en condiciones adecuadas para preparar el producto identificado.
En algunas realizaciones, la preparación de compuestos o sales puede implicar la adición de ácidos o bases para afectar, por ejemplo, a la catálisis de una reacción deseada o a la formación de formas de sal tales como sales de adición de ácido (por ejemplo, formación de una sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico).
Ácidos de ejemplo pueden ser ácidos inorgánicos u orgánicos e incluyen, pero no se limitan a, ácidos fuertes y débiles. Algunos ejemplos de ácidos incluyen, pero no se limitan a, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido p-toluenosulfónico, ácido 4-nitrobenzoico, ácido metanosulfónico, ácido bencenosulfónico, ácido trifluoroacético, ácido nítrico, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, ácido acético, ácido propiónico, ácido butanoico, ácido benzoico, ácido tartárico, ácido pentanoico, ácido hexanoico, ácido heptanoico, ácido octanoico, ácido nonanoico y ácido decanoico.
Algunas bases de ejemplo incluyen, pero no se limitan a, carbonatos (por ejemplo, carbonato de sodio), bicarbonatos (por ejemplo, bicarbonato de sodio), hidróxidos (por ejemplo, hidróxido de sodio, hidróxido de potasio), alcóxidos, amidas metálicas, hidruros metálicos, dialquilamidas metálicas y arilaminas, en donde; los alcóxidos incluyen sales de litio, sodio y potasio de óxidos de metilo, etilo y terc-butilo; las amidas metálicas incluyen amida de sodio, amida de potasio y amida de litio; los hidruros metálicos incluyen hidruro de sodio, hidruro de potasio e hidruro de litio; y las dialquilamidas metálicas incluyen sales de litio, sodio y potasio de amidas sustituidas con metilo, etilo, n-propilo, ipropilo, n-butilo, t-butilo, trimetilsililo y ciclohexilo.
Todos los compuestos, y sales de los mismos, pueden encontrarse junto con otras sustancias tales como agua y disolventes (por ejemplo, hidratos y solvatos) o pueden estar aislados.
Como se usa en el presente documento, el término “enriquecido” se refiere a una cantidad aumentada de un compuesto o sal particular (por ejemplo, un compuesto (S)-isomérico o sal) en una mezcla en comparación con la cantidad del compuesto en la mezcla antes de enriquecerse. En algunas realizaciones, una mezcla puede enriquecerse en la cantidad de un primer compuesto isomérico o sal (por ejemplo, un compuesto (S)-isomérico o sal) en relación con un segundo compuesto isomérico o sal (por ejemplo, un compuesto (R)-isomérico) en comparación con la cantidad relativa de los compuestos isoméricos en una mezcla de partida (por ejemplo, antes de formar la mezcla enriquecida). Por ejemplo, una mezcla enriquecida en un compuesto isomérico o sal de fórmula I-(S) tiene una cantidad aumentada del compuesto isomérico de fórmula I-(S) en relación con el compuesto isomérico de fórmula I(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) en una mezcla de partida (por ejemplo, una mezcla racémica de los compuestos isoméricos de fórmulas I-(S) e I-(R)).
Las reacciones de los procedimientos descritos en el presente documento pueden llevarse a cabo en disolventes adecuados que pueden seleccionarse fácilmente por un experto en la técnica de síntesis orgánica. Los disolventes adecuados pueden ser sustancialmente no reactivos con los materiales de partida (reactantes), los productos intermedios o productos a las temperaturas a las que se llevan a cabo las reacciones, por ejemplo, temperaturas que pueden oscilar entre la temperatura de congelación del disolvente y la temperatura de ebullición del disolvente. Una reacción dada puede llevarse a cabo en un disolvente o una mezcla de más de un disolvente. Dependiendo de la etapa de reacción particular, pueden seleccionarse disolventes adecuados para una etapa de reacción particular. En algunas realizaciones, las reacciones pueden llevarse a cabo en ausencia de disolvente, tal como cuando al menos uno de los reactivos es un líquido o gas.
Los disolventes halogenados adecuados incluyen, pero no se limitan a, tetracloruro de carbono, bromodiclorometano, dibromoclorometano, bromoformo, cloroformo, bromoclorometano, dibromometano, cloruro de butilo, diclorometano, tetracloroetileno, tricloroetileno, 1,1,1-tricloroetano, 1,1,2-tricloroetano, 1,1-dicloroetano, 2-cloropropano, 1,1,1-trifluorotolueno, 1,2-dicloroetano, 1,2-dibromoetano, hexafluorobenceno, 1,2,4-triclorobenceno, 1,2-diclorobenceno, clorobenceno y fluorobenceno.
Los disolventes de éter adecuados incluyen, pero no se limitan a, dimetoximetano, tetrahidrofurano, 2-metiltetrahidrofurano, 1,3-dioxano, 1,4-dioxano, furano, dietil éter, dimetil éter de etilenglicol, dietil éter de etilenglicol, dimetil éter de dietilenglicol (diglima), dietil éter de dietilenglicol, dimetil éter de trietilenglicol, anisol y t-butil metil éter.
Los disolventes próticos adecuados incluyen, pero no se limitan a, agua, metanol, etanol, 2-nitroetanol, 2-fluoroetanol, 2,2,2-trifluoroetanol, etilenglicol, 1-propanol, 2-propanol, 2-metoxietanol, 1-butanol, 2-butanol, alcohol i-butílico, alcohol t-butílico, 2-etoxietanol, dietilenglicol, 1-, 2- o 3-pentanol, alcohol neopentílico, alcohol t-pentílico, monometil éter de dietilenglicol, monoetil éter de dietilenglicol, ciclohexanol, alcohol bencílico, fenol y glicerol.
Los disolventes apróticos adecuados incluyen, pero no se limitan a, tetrahidrofurano (THF), N,N-dimetilformamida (DMF), N,N-dimetilacetamida (DMA), 1,3-dimetil-3,4,5,6-tetrahidro-2(1H)-pirimidinona (DMPU), 1,3-dimetil-2-imidazolidinona (DMI), N-metilpirrolidinona (NMP), formamida, N-metilacetamida, N-metilformamida, acetonitrilo, dimetilsulfóxido, propionitrilo, formiato de etilo, acetato de metilo, hexacloroacetona, acetona, etil metil cetona, acetato de etilo, sulfolano, N,N-dimetilpropionamida, tetrametilurea, nitrometano, nitrobenceno y hexametilfosforamida.
Los disolventes de hidrocarburo adecuados incluyen, pero no se limitan a, benceno, ciclohexano, pentano, hexano, tolueno, cicloheptano, metilciclohexano, n-heptano, etilbenceno, m-, o- o p-xileno, octano, indano, nonano y naftaleno.
Las reacciones de los procedimientos descritos en el presente documento pueden llevarse a cabo en aire o bajo una atmósfera inerte (por ejemplo, atmósfera de nitrógeno o argón). Normalmente, las reacciones que contienen reactivos o productos que son sustancialmente reactivos con el aire pueden llevarse a cabo usando técnicas de síntesis sensibles al aire que conoce bien el experto en la técnica.
Tras llevar a cabo la preparación de compuestos y sales según los procedimientos descritos en el presente documento, pueden usarse las operaciones habituales de aislamiento y purificación, tales como concentración, filtración, extracción, extracción en fase sólida, recristalización, cromatografía, y similares, para aislar los productos deseados.
Las reacciones de los procedimientos descritos en el presente documento pueden llevarse a cabo a temperaturas apropiadas que puede determinarlas fácilmente el experto en la técnica. Las temperaturas de reacción dependerán de, por ejemplo, los puntos de fusión y ebullición de los reactivos y el disolvente, si está presente; la termodinámica de la reacción (por ejemplo, puede ser necesario llevar a cabo reacciones exotérmicas vigorosas a temperaturas reducidas); y la cinética de la reacción (por ejemplo, una alta barrera de energía de activación puede necesitar temperaturas elevadas). Por ejemplo, la expresión, “temperatura ambiente”, como se usa en el presente documento, se entiende en la técnica y se refiere en general a una temperatura (por ejemplo, una temperatura de reacción) que es aproximadamente la temperatura de la habitación en la que se lleva a cabo la reacción, por ejemplo, una temperatura de aproximadamente 20 °C a aproximadamente 30 °C.
Las reacciones pueden monitorizarse según cualquier método adecuado conocido en la técnica. Por ejemplo, la formación del producto puede monitorizarse por medios espectroscópicos, tales como espectroscopía de resonancia magnética nuclear (por ejemplo, 1H o 13C), espectroscopía infrarroja, espectrofotometría (por ejemplo, UV-visible), espectrometría de masas, o por métodos cromatográficos tales como cromatografía de líquidos de alto rendimiento (HPLC) o cromatografía en capa fina (TLC).
Compuestos y sales de la invención
La presente solicitud proporciona además una mezcla de compuestos isoméricos que tienen las fórmulas I-(S) e I-(R):

en donde el exceso enantiomérico del compuesto isomérico de fórmula I-(S) es de aproximadamente el 90 % o más, aproximadamente el 95 % o más, aproximadamente el 97 % o más, aproximadamente el 98 % o más, aproximadamente el 99 % o más, o aproximadamente el 99,9 % o más. R1 es etilo. Pg1 es terc-butoxicarbonilo. En algunas realizaciones, el exceso enantiomérico del compuesto isomérico de fórmula I-(S) puede oscilar entre aproximadamente el 90 % y aproximadamente el 99,9 %, aproximadamente el 95 % y aproximadamente el 99,9 %, aproximadamente el 96 % y aproximadamente el 99,9 %, aproximadamente el 97 % y aproximadamente el 99,9 %, aproximadamente el 98 % y aproximadamente el 99,9 %, aproximadamente el 99 % y aproximadamente el 99,9 %, o aproximadamente el 99,5 % y aproximadamente el 99,9 %.
En algunas realizaciones, la mezcla de compuestos isoméricos que tienen las fórmulas I-(S) e I-(R) se prepara según un procedimiento proporcionado en el presente documento, en donde la mezcla está enriquecida en el compuesto isomérico de fórmula I-(S).
La presente solicitud proporciona además una sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(S) o fórmula I-(R):

en donde
R1 es etilo. Pg1 es terc-butoxicarbonilo.
En algunas realizaciones, la sal es la sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(S).
En algunas realizaciones, la sal es la sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(R).
En algunas realizaciones, la sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(S) o fórmula I-(R) se prepara según un procedimiento proporcionado en el presente documento.
Los compuestos y sales de la invención también pueden incluir formas tautoméricas. Las formas tautoméricas resultan del intercambio de un enlace sencillo con un doble enlace adyacente junto con la migración concomitante de un protón. Las formas tautoméricas incluyen tautómeros prototrópicos que son estados de protonación isoméricos que tienen la misma fórmula empírica y carga total. Los tautómeros prototrópicos de ejemplo incluyen pares cetona-enol, pares
amida - ácido imídico, pares lactama - lactima, pares amida - ácido imídico, pares enamina - imina, y formas anulares donde un protón puede ocupar dos o más posiciones de un sistema heterocíclico, por ejemplo, 1H- y 3H-imidazol, 1H-, 2H- y 4H-1,2,4-triazol, 1H- y 2H-isoindol, y 1H- y 2H-pirazol.
El término “compuesto”, como se usa en el presente documento, pretende incluir todos los estereoisómeros, isómeros geométricos, tautómeros e isótopos de las estructuras representadas. Los compuestos y sales identificados en el presente documento por nombre o estructura como una forma tautomérica particular pretenden incluir otras formas tautoméricas a menos que se especifique lo contrario. Los compuestos y sales identificados en el presente documento por nombre o estructura sin especificar la configuración particular de un estereocentro pretenden abarcar todas las configuraciones posibles en el estereocentro. Por ejemplo, si un estereocentro particular en un compuesto de la invención podría ser R o S, pero el nombre o la estructura del compuesto no designa cuál es, entonces el estereocentro puede ser R o S.
Los compuestos y sales descritos en el presente documento pueden ser asimétricos (por ejemplo, teniendo uno o más estereocentros). Todos los estereoisómeros, tales como enantiómeros y diastereoisómeros, están previstos a menos que se indique lo contrario. Los compuestos y sales de la presente invención que contienen átomos de carbono sustituidos asimétricamente pueden aislarse en formas ópticamente activas o racémicas. Muchos isómeros geométricos de olefinas, dobles enlaces C=N, y similares también pueden estar presentes en los compuestos o sales descritos en el presente documento, y todos de tales isómeros estables se contemplan en la presente invención. Los isómeros geométricos cis y trans de los compuestos de la presente invención pueden aislarse como una mezcla de isómeros o como formas isoméricas separadas.
En algunas realizaciones, los compuestos o sales de la invención están sustancialmente aislados. Por “sustancialmente aislado” quiere decirse que el compuesto está al menos parcial o sustancialmente separado del entorno en el que se formó o detectó. La separación parcial puede incluir, por ejemplo, una composición enriquecida en los compuestos de la invención. La separación sustancial puede incluir composiciones que contienen al menos aproximadamente el 50 %, al menos aproximadamente el 60 %, al menos aproximadamente el 70 %, al menos aproximadamente el 80 %, al menos aproximadamente el 90 %, al menos aproximadamente el 95 %, al menos aproximadamente el 97 % o al menos aproximadamente el 99 % en peso de los compuestos o sales de la invención, o una sal de los mismos. Los métodos para aislar compuestos y sus sales son rutinarios en la técnica.
Los compuestos y sales de la invención también pueden incluir todos los isótopos de átomos que se encuentran en los productos intermedios o compuestos finales. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números másicos. Por ejemplo, los isótopos de hidrógeno incluyen tritio y deuterio.
Síntesis de inhibidores de TPH1
La mezcla de sal enriquecida en el compuesto isomérico de fórmula I-(S) descrito en el presente documento puede hacerse reaccionar adicionalmente, por ejemplo, para preparar productos intermedios útiles para la preparación de compuestos que son inhibidores de TPH1, como se muestra a continuación en el esquema 3. Por ejemplo, la mezcla de sal enriquecida en el compuesto isomérico de fórmula I-(S) se prepara según una o más realizaciones descritas en el presente documento (etapa 1), y la forma de base libre del compuesto isomérico de fórmula I-(S) se forma posteriormente (por ejemplo, por medio de reacción con carbonato de sodio) y se aísla (etapa 2). La amina libre del compuesto isomérico de fórmula I-(S) puede protegerse entonces usando condiciones de protección de amina convencionales como se muestra en la etapa 3, por ejemplo, reacción con Pg2-X en presencia de una base (por ejemplo, trimetilamina), en donde Pg2 es un grupo protector de amino (por ejemplo, terc-butoxicarbonilo, carbobenciloxilo, y similares) y X es halo (por ejemplo, Cl). La desprotección selectiva del grupo protector de amino Pg1 (etapa 4) forma el producto intermedio 1 deseado, un producto intermedio útil en la preparación de compuestos que son inhibidores de TPH1.
Esquema 3.

El producto intermedio 1 puede usarse en la preparación de compuestos inhibidores de TPH1, por ejemplo, como se muestra a continuación en el esquema 4, en donde R1 es alquilo C1-6, Pg2 es un grupo protector de amino y las variables W, X, Y, R2, R3, RA, RB, RC, RD y el anillo A son como se definen en la patente de Estados Unidos n.° 9.199.994, cuya divulgación se incorpora en el presente documento por referencia en su totalidad. Por ejemplo, el producto intermedio 1 (por ejemplo, (S)-2,8-diazaspiro[4.5]decano-2,3-dicarboxilato de 2-bencilo y 3-etilo) se añade a una disolución del compuesto A en un disolvente (por ejemplo, dioxano) en presencia de una base (por ejemplo, NaHCO3), y se calienta hasta reflujo para proporcionar un compuesto de fórmula C. En la etapa 2, el grupo Pg2 (por ejemplo, un grupo carbobenciloxilo (CBZ)) de fórmula C se elimina (por ejemplo, mediante reacción con yoduro de trimetilsililo (TMSI), un ácido fuerte o hidrogenación catalizada por metal de transición) para formar el compuesto inhibidor de TPH1 deseado.

El producto intermedio 1 del esquema 3 también puede usarse en la preparación de compuestos inhibidores de TPH1, por ejemplo, como se muestra a continuación en el esquema 5. Por ejemplo, el producto intermedio 1 (por ejemplo, (S)-2,8-diazaspiro[4.5]decano-2,3-dicarboxilato de 2-bencilo y 3-etilo) se añade a una disolución del compuesto A en un disolvente (por ejemplo, dioxano) en presencia de una base (por ejemplo, NaHCOa), y se calienta hasta reflujo para proporcionar un compuesto de fórmula B. La reacción posterior con ácido fenilborónico en condiciones convencionales de acoplamiento de arilarilo (por ejemplo, la reacción en presencia de un catalizador de paladio tal como PdCh(dppf)-CH2Cl2 en presencia de una base tal como KHCO3) proporciona el compuesto C. El grupo protector de grupo amino Pg2 (por ejemplo, un grupo carbobenciloxilo (CBZ)) de fórmula C se elimina entonces (por ejemplo, reacción con TMSI) para formar el compuesto inhibidor de TPH1 deseado de fórmula D.
Esquema 5.

Como se usa en el presente documento, el término “halo” se refiere a un átomo de halógeno seleccionado de F, Cl, I
y Br. En algunas realizaciones, el grupo halo es Cl.
Como se usa en el presente documento, el término “desprotección” se refiere a condiciones adecuadas para escindir un grupo protector de amina. En algunas realizaciones, la desprotección puede incluir la escisión de un grupo protector en presencia de un ácido fuerte, en presencia de una base fuerte, en presencia de un agente reductor o en presencia de un agente oxidante. Por ejemplo, la desprotección de un grupo protector de amina puede lograrse mediante métodos conocidos en la técnica para la eliminación de grupos protectores particulares para aminas, tales como los de Wuts y Greene, Protective Groups in Organic Synthesis, 4a ed., John Wiley & Sons: Nueva Jersey, páginas 696 887 (y, en particular, páginas 872-887) (2007). En algunas realizaciones, la desprotección comprende hacer reaccionar el compuesto protegido en condiciones ácidas (por ejemplo, ácido clorhídrico o ácido trifluoroacético).
Un experto en la técnica apreciará que los procedimientos descritos no son los medios exclusivos por los cuales pueden sintetizarse los compuestos y sales proporcionados en el presente documento y que está disponible un amplio repertorio de reacciones orgánicas de síntesis para su posible empleo en la síntesis de los compuestos proporcionados en el presente documento. El experto en la técnica sabe cómo seleccionar e implementar rutas de síntesis apropiadas. Pueden identificarse métodos de síntesis adecuados de materiales de partida, productos intermedios y productos mediante referencia a la bibliografía, incluyendo fuentes de referencia tales como: Advances in Heterocyclic Chemistry, vol. 1-107 (Elsevier, 1963-2012); Journal of Heterocyclic Chemistry vol. 1-49 (Journal of Heterocyclic Chemistry, 1964 2012); Carreira, et al. (Ed.) Science of Synthesis, vol. 1-48 (2001-2010) y Knowledge Updates KU2010/1-4; 2011/1-4; 2012/1-2 (Thieme, 2001-2012); Katritzky, et al. (Ed.) Comprehensive Organic Functional Group Transformations, (Pergamon Press, 1996); Katritzky et al. (Ed.); Comprehensive Organic Functional Group Transformations II (Elsevier, 2a edición, 2004); Katritzky et al. (Ed.), Comprehensive Heterocyclic Chemistry (Pergamon Press, 1984); Katritzky et al., Comprehensive Heterocyclic Chemistry II, (Pergamon Press, 1996); Smith et al., March s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6a ed. (Wiley, 2007); Trost et al. (Ed.), Comprehensive Organic Synthesis (Pergamon Press, 1991).
Ejemplos
La invención se describirá con mayor detalle por medio de ejemplos específicos. Los siguientes ejemplos se ofrecen con fines ilustrativos, y no pretenden limitar la invención de ninguna manera. Los expertos en la técnica reconocerán fácilmente una variedad de parámetros no críticos que pueden cambiarse o modificarse para producir esencialmente los mismos resultados.
El análisis por HPLC se realizó en una máquina Agilent 1100 con las siguientes condiciones: Columna: Altima C18, 150 mm de longitud, 3,1 mm de diámetro, 3 |im de tamaño de partícula. Fase móvil A: ácido fórmico al 0,1 % en agua mili-q. Fase móvil B: ácido fórmico al 0,1 % en acetonitrilo.
La pureza enantiomérica se determinó usando una de las siguientes condiciones:
Método de pureza enantiomérica A: Columna YMC Chiral Amylose-SA (250 mm de longitud, 4,6 mm de diámetro, 5 |im de tamaño de partícula) en una máquina de HPLC Agilent 1100 con n-heptano:isopropanol:etanol:dietilamina (80:10:10:0,1, % en v:v:v:v) como eluyente.
Método B de pureza enantiomérica: YMC Chiral NEA [NR30S05-2546WT] (250 mm de longitud, 4,6 mm de diámetro, 5 |im de tamaño de partícula) en una máquina Agilent 11100 HPLC con perclorato de sodio 150 mmol/l (pH 2,5) en agua mili-q y etanol como eluyente.
Ejemplo 1. 2,8-Diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo (mezcla isomérica) Etapa 1. (Z)-3-Bromo-2-(hidroxiimino)propanoato de etilo

Se cargó un reactor con hidroxilamina.HCl (13,4 kg, 192,8 mol, 1,25 eq.), agua potable (2,5 vol.) y tolueno (5 vol.). La mezcla se agitó y se enfrió hasta aproximadamente 15 °C. Se añadieron bromopiruvato de etilo (29,9 kg, 153,3 mol, 1,0 eq.) y tolueno (1,5 vol.) y la mezcla se agitó durante 16-20 horas entre 15-25 °C. Entonces se separaron las fases después de sedimentar durante al menos 15 minutos. La fase acuosa se retiró y la fase orgánica se mantuvo en el reactor. El reactor se cargó posteriormente con agua potable (0,5 vol.) y la mezcla resultante se agitó durante al menos 15 minutos. La fase acuosa se retiró, la fase orgánica se mantuvo en el reactor, y la extracción acuosa se realizó dos veces más. La fase orgánica se concentró usando destilación al vacío a 35-40 °C (~3,5 vol. eliminados; 3,6 volúmenes relativos restantes). El reactor se cargó posteriormente con n-heptano (3 vol.) y la disolución resultante se concentró usando destilación al vacío a 35-40 °C hasta que quedaron aproximadamente 3,6 volúmenes relativos. Se añadió nheptano adicional (3 vol.) y se concentró la disolución resultante usando destilación al vacío a 35-40 °C hasta que quedaron aproximadamente 3,6 volúmenes relativos. Entonces, la mezcla resultante se enfrió hasta aproximadamente 10 °C y se agitó durante aproximadamente 20-25 minutos. La mezcla se filtró y se retiraron las aguas madre resultantes. Se cargó el reactor con n-heptano (0,73 vol.) y se agitó durante al menos 5 minutos. La torta de filtro resultante se enjuagó con n-heptano y se secó durante al menos 5 minutos a temperatura ambiente. Se repitieron las etapas de enjuague y secado, momento en el cual la torta del filtro se secó durante entre 0,5-2,5 días a vacío y flujo de nitrógeno a temperatura ambiente. Pureza por HPLC: Lote 1: 93,21 % de área. Lote 2: 93,76 % de área. Pureza por 1H-RMN (dos lotes): Lote 1: 51,9 % en peso; lote 2: 93,7 % en peso.
Etapa 2. 4-(Pirrolidin-1-ilmetilen)piperidin-1-carboxilato de terc-butilo
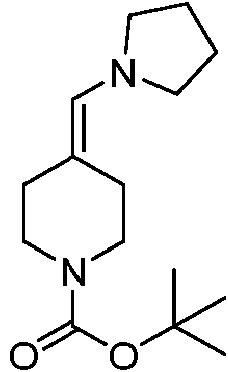
La reacción se cargó con N-Boc-piperidin-4-carbaldehído (18,2 kg, 85,3 mol, 1,0 eq.) y tolueno (16 vol.), y la mezcla se agitó hasta la disolución del N-Boc-piperidin-4-carbaldehído. Se añadieron pirrolidina (10,2 kg, 143,3 mol, 1,6 eq.) y tolueno adicional (0,5 vol.), y la mezcla resultante se calentó hasta reflujo (~111 °C) para eliminar el agua mediante destilación azeotrópica. La disolución resultante se concentró entonces usando destilación al vacío entre 35-40 °C hasta que quedaron 12,2 volúmenes relativos. Se añadió tolueno adicional (6 vol.), y se concentró la disolución resultante entre 35-40 °C hasta que quedaron 12,2 volúmenes relativos. La mezcla resultante se enfrió hasta aproximadamente 20 °C y se usó en la siguiente etapa sin purificación adicional. Lote 1: 84,1 % de área de 4-(pirrolidin-1 -ilmetilen)piperidin-1 -carboxilato de terc-butilo; 8,8 % de área de N-Boc-piperidin-4-carbaldehído; 7,0 % de área de pirrolidina. KF: <0,1 % en peso. Lote 2: 86,6 % de área de 4-(pirrolidin-1-ilmetilen)piperidin-1-carboxilato de terc-butilo; 6,3 % de área de N-Boc-piperidin-4-carbaldehído; 6,2 % área-% pirrolidina. KF: <0,1 % en peso.
Etapa 3. 1-Hidroxi-2-oxa-3,9-diazaspiro[5.5]undec-3-eno-4,9-dicarboxilato de 9-(terc-butilo) y 4-etilo

Se cargó un primer reactor con (Z)-3-bromo-2-(hidroxiimino)propanoato de etilo (del ejemplo 1, etapa 1; lote 1: 18,81 kg, 89,6 mol, 1,06 eq.; lote 2: 18,9 kg, 90,0 mol, 1,06 eq.) y 2-metiltetrahidrofurano (1,7-1,92 vol.) y la mezcla resultante se agitó y se enfrió hasta -5 °C. En un segundo reactor, una disolución de 4-(pirrolidin-1-ilmetilen)piperidin-1-carboxilato de terc-butilo en tolueno (del ejemplo 1, etapa 2; lote 1: 22,7 kg, 85,3 mol, 1,00 eq. lote 2: 22,7 kg, 85,3 mol, 1,00 eq.) se enfrió hasta -5 °C y se añadieron N,N'-diisopropiletilamina (lote 1: 17,4 kg, 134,2 mol, 1,6 eq. lote 2: 17,5 kg, 135,4 mol, 1,6 eq.) y tolueno (0,5 vol.). La disolución de (Z)-3-bromo-2-(hidroxiimino)propanoato y 2-metiltetrahidrofurano se añadió entonces al segundo reactor a lo largo de aproximadamente 1-2 horas mientras se
mantenía a una temperatura por debajo de 10 °C. El primer reactor se enjuagó entonces con 2-metiltetrahidrofurano adicional (0,22 vol.) que se añadió al segundo reactor. Tras completar la adición de la disolución de (Z)-3-bromo-2-(hidroxiimino)propanoato al segundo reactor, se calentó la mezcla resultante hasta aproximadamente 15 °C y se agitó durante aproximadamente 45 minutos. Se añadió disolución acuosa de ácido clorhídrico (HCl al 30% preparado a partir de 2,2 eq. de HCl y 3,5 vol. de agua potable) a la mezcla de reacción (lote 1: 24,2 kg, 199,1 mol, 2,3 eq.; lote 2: 23,9 kg, 196,7 mol, 2,3 eq.) y la mezcla resultante se calentó hasta 30 °C y se agitó durante aproximadamente 45 minutos. Las fases orgánica y acuosa se separaron después de permitir la sedimentación durante al menos 15 minutos y se retiró la fase acuosa. La fase orgánica se lavó con agua potable (1,0 vol.) y la mezcla se agitó durante al menos 5 minutos. Se separaron las fases después de permitir la sedimentación durante al menos 10 minutos y la fase acuosa se retiró y se extrajo adicionalmente con 2-metiltetrahidrofurano adicional (5 vol.). Se combinaron las fases orgánicas, se lavaron con una porción adicional de agua potable (1 volumen) y se retiró la fase acuosa. Las fases orgánicas combinadas resultantes se concentraron usando destilación al vacío entre 35-40 °C hasta que quedaron aproximadamente 5,4 volúmenes relativos, momento en el cual se añadió n-heptano (3 vol.) y la mezcla resultante se concentró a 35-40 °C hasta que quedaron aproximadamente 5,4 volúmenes relativos. Se repitió la adición de nheptano y destilación al vacío, momento en el cual se añadió n-heptano adicional (3 vol.) y la mezcla resultante se enfrió hasta aproximadamente 20 °C. Se filtró la mezcla y la torta de filtro resultante se lavó con n-heptano (2,57 vol.) y tolueno (0,14 vol.) (2x). La torta del filtro se secó, se lavó con una porción adicional de n-heptano (2,57 vol.) y se agitó. Luego se eliminó el n-heptano y la torta de filtro resultante se secó bajo nitrógeno durante entre 18-72 horas a temperatura ambiente y se usó en la siguiente etapa sin purificación adicional. Lote 1: Pureza por HPLC: 98,37 % de área. 1H-RMN: 91,8 % en peso. Lote 2: Pureza por HPLC: 99,52 % de área. 1H-RMN: 90,7 % en peso.
Etapa 4. 2,8-Diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo (mezcla isomérica)
Se cargó un reactor con 1-hidroxi-2-oxa-3,9-diazaspiro[5.5]undec-3-eno-4,9-dicarboxilato de 9-(terc-butilo) y 4-etilo (del ejemplo 1, etapa 3, 38,2 kg, 111,8 mol, 1,0 eq.), etanol (abs., 4 vol.) y tetrahidrofurano (4 vol.) y se agitó la mezcla. Se añadió entonces catalizador de esponja A5000 (28,9 kg) y el reactor se purgó varias veces con vacío y nitrógeno y luego se purgó con vacío e hidrógeno. Entonces, la mezcla resultante se calentó hasta aproximadamente 30 °C y el reactor se presurizó con hidrógeno a 4 ± 0,5 bar y la mezcla se agitó durante aproximadamente 16-22 horas a 30 ± 5 °C bajo hidrógeno. Entonces, el reactor se despresurizó y se cargó con nitrógeno. Entonces, el reactor se purgó con vacío e hidrógeno, se presurizó con hidrógeno a 4 ± 0,5 bar y la agitación continuó durante aproximadamente 46 horas a 30 ± 5 °C en hidrógeno. El reactor se despresurizó y la mezcla de reacción se filtró sobre un filtro de dicalita y se enjuagó con 2-metiltetrahidrofurano. Se recogió el filtrado, se lavó con 2-metiltetrahidrofurano adicional y se concentró a presión reducida entre 35-40 °C hasta que quedaron aproximadamente 3,75 volúmenes relativos. Se repitió el proceso de destilación dos veces adicionales, momento en el cual la mezcla resultante se enfrió hasta aproximadamente 20 °C y se usó el producto resultante sin purificación adicional. Pureza por HPLC: 78,8 % de área.
Ejemplo 2A. 2,8-Diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (mezcla isomérica enriquecida en el isómero (S))

Se cargó un reactor con monohidrato de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (32,65 kg, 111,5 mol, 1,0 eq.), 2-metiltetrahidrofurano (2 vol.) y benzaldehído (0,04 vol.). A continuación, se añadió a una disolución del 2,8-diazaspiro[4,5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo (mezcla isomérica del ejemplo 1) 2-metiltetrahidrofurano adicional (1 vol.). Se agitó la mezcla resultante y se calentó a 28 °C hasta que se formó una disolución. Entonces, la disolución se agitó durante al menos 30 minutos a 40 ± 3 °C. La mezcla resultante se enfrió entonces hasta 30 ± 3 °C y se agitó durante 2 horas adicionales. Entonces, la mezcla se sembró con (S)-2,8-diazaspiro[4,5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (2 g), y la mezcla resultante se agitó durante aproximadamente 14-15 horas a 30 ± 3 °C. La mezcla de reacción se enfrió entonces hasta aproximadamente 20 ± 3 °C durante 4 horas y luego se agitó durante 15 horas adicionales. La mezcla se filtró y el filtrado se separó para una reacción adicional. La torta de filtro resultante se lavó con 2-metiltetrahidrofurano (1 vol., 3x) y se secó durante 5 minutos entre cada adición de 2-metiltetrahidrofurano para proporcionar una mezcla de isómeros de 2,8-diazaspiro[4,5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico enriquecidos en el (S)-2,8-diazaspiro[4,5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico. Pureza por HPLC: 95,67 % de área. Pureza enantiomérica del isómero (S-): 78,55 % de área. El filtrado separado se concentró por separado entre 3540 °C hasta que quedaron 3,5 volúmenes relativos y la mezcla se enfrió hasta 27,5 °C. Entonces, la mezcla se sembró con (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2- ceto-L-gulónico y se agitó durante al menos 16 horas a 20 ± 3 °C. La mezcla resultante se filtró, se retiró el filtrado y la torta de filtro resultante se lavó con 2-metiltetrahidrofurano (0,57 vol.) y se secó (3x) para proporcionar una segunda cosecha de una mezcla de isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico enriquecidos en el (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico. Pureza por HPLC: 98,29 % de área; Pureza enantiomérica del isómero (S-) (método A): 93,60 % de área. Los volúmenes y las razones molares proporcionados son relativos a la mezcla isomérica de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3- etilo preparada en el ejemplo 1.
La mezcla obtenida de isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (23,2 kg, 39,5 mol, 1,0 eq.) se añadió entonces a un reactor con 2-metiltetrahidrofurano (16 vol.) y la mezcla se calentó hasta reflujo (aproximadamente 80 °C) hasta que los sólidos se disolvieron. Entonces, la mezcla se enfrió hasta 40 ± 3 °C durante aproximadamente 2 horas y se observó cristalización. Entonces, la mezcla se concentró entre 35-40 °C hasta que quedaron aproximadamente 9 volúmenes relativos. Entonces, la mezcla se enfrió hasta 20 ± 3 °C y la agitación continuó durante aproximadamente 2-3 horas. La mezcla resultante se filtró y se enjuagó con 2-metiltetrahidrofurano (1,4 vol., 2x con secado bajo nitrógeno durante 5 minutos entre cada lavado). Entonces se secó la torta de filtro bajo nitrógeno durante aproximadamente 14-16 horas para proporcionar la mezcla deseada de isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, acido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico enriquecidos en el (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico. Lote 1: Pureza por HPLC: 99,49 % de área; pureza enantiomérica del isómero (S-) (Método A): 93,98 % de área. Lote 2: Pureza por HPLC: >99,9 % de área; pureza enantiomérica del isómero (S-) (Método A): 95,22 % de área. Se realizó una segunda recristalización de los isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico que enriqueció adicionalmente la mezcla isomérica en el isómero (S-). Lote 1: Pureza por HPLC: 99,49 % de área; pureza enantiomérica del isómero (S-) (Método A): 99,02 % de área. Lote 2: Pureza por HPLC: 99,95 % de área; pureza enantiomérica del isómero (S-) (Método A): 99,48 % de área.
Ejemplo 2B. Preparación alternativa de (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico
Se disolvió 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo (1 g, 3,20 mmol) en THF (5 vol., 5 ml) a t.a. La disolución se trató con 1 equivalente molar de monohidrato de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico y entonces la temperatura se elevó lentamente hasta 40 °C. Después de 20 min, comenzó a formarse un precipitado a 40 °C, momento en el cual se añadió TBME (5 vol., 5 ml) a la mezcla, momento después del cual la reacción se enfrió lentamente hasta 5 °C a una velocidad de 1 °C/min. Después de este tiempo, el sólido formado se filtró y después se lavó con TBME frío. El sólido se secó al vacío para proporcionar (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (45 %, >99 % de ee) como un sólido cristalino adecuado para la siembra.
Ejemplo 3. (S)-2,8-Diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo

Se cargó un reactor con la mezcla enriquecida de isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(tercbutilo) y 3-etilo, ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico (ejemplo 2, 23,80 kg, 40,5 mol, 1,0 eq.), 2-metiltetrahidrofurano (2 vol.) y n-heptano (8 vol.), y la mezcla se agitó a 15 ± 3 °C. Se añadió carbonato de sodio (disolución al 10% en agua; 125,8 kg) a lo largo de 15 minutos mientras se mantenía la temperatura de la mezcla de reacción entre 15-20 °C y la mezcla resultante se agitó durante al menos 35 minutos. Se separaron las fases y se retiró la fase acuosa. A la fase orgánica restante se le añadió carbonato de sodio adicional (disolución al 10 % en agua; 27,4 kg) mientras se mantenía la temperatura de la mezcla de reacción entre 15-20 °C y la mezcla resultante se agitó durante al menos 35 minutos. Se separaron las fases, se retiró la fase acuosa y se concentró la fase orgánica hasta sequedad para proporcionar el compuesto del título. Pureza por HPLC: 97,65 % de área. Pureza enantiomérica del isómero (S-) (Método A): 98,89 % de área. Los volúmenes y las razones molares proporcionadas son relativos a los isómeros de 2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo, ácido 2,3:4,6-di-Oisopropiliden-2-ceto-L-gulónico preparados en el ejemplo 2A.
Ejemplo 4. (S)-2,8-Diazaspiro[4.5]decano-2,3,8-tricarboxilato de 2-bencilo, 8-(terc-butilo) y 3-etilo

Se cargó un reactor con (S)-2,8-diazaspiro[4.5]decano-3,8-dicarboxilato de 8-(terc-butilo) y 3-etilo (ejemplo 3, 12,6 kg, 32,2 mol, 1,0 eq.), carbonato de sodio (10 % en agua, 86,4 kg) y 2-metiltetrahidrofurano (0,55 vol. rel.), y la mezcla se enfrió hasta 0 ± 3 °C. Entonces se añadió una disolución de cloroformiato de bencilo (6,7 kg, 38,76 mol, 1,0 eq.) en 2-metiltetrahidrofurano (0,25 vol.) a lo largo de 40 minutos mientras se mantenía la temperatura de la mezcla de reacción entre -2-2 °C. Se usó 2-metiltetrahidrofurano adicional (0,25 vol.) para enjuagar la disolución residual de cloroformiato de bencilo en la mezcla de reacción, y la mezcla resultante se agitó durante 5-10 minutos a 0 ± 3 °C. Entonces, la mezcla se calentó hasta 30 ± 3 °C y se agitó durante 20-30 minutos adicionales. Entonces se separaron las fases y se eliminó la fase acuosa. La fase orgánica se lavó con una porción de agua potable (1 vol.) y la mezcla se calentó hasta 30 ± 3 °C y se agitó durante al menos 5 minutos. Entonces, se eliminó la fase acuosa y la fase orgánica restante se concentró usando destilación al vacío entre 35-40° hasta que quedaron aproximadamente 3 volúmenes relativos. Entonces se añadió etanol absoluto (3 vol.) y se concentró la mezcla resultante usando destilación al vacío entre 35 40° hasta que quedaron aproximadamente 3 volúmenes relativos. Se realizaron la adición de etanol y la destilación una segunda vez para proporcionar el producto del título. Pureza por HPLC: 65,58 % de área. También se observó el compuesto desprotegido, (S)-2,8-diazaspiro[4.5]decano-2,3-dicarboxilato de 2-bencilo y 3-etilo. La pureza por HPLC combinada del producto del título y el producto desprotegido con BOC fue del 80,45 % de área.
Ejemplo 5. Clorhidrato de (S)-2,8-diazaspiro[4.5]decano-2,3-dicarboxilato de 2-bencilo y 3-etilo

Se cargó un reactor con etanol absoluto (67 kg, 3,75 vol.) y se enfrió hasta aproximadamente 12,5 °C. Se añadió cloruro de acetilo (10,0 kg, 6,00 eq.) a lo largo de 55 minutos, mientras se mantenía la temperatura de la mezcla por debajo de 15 °C. Se añadió acetato de etilo (10,2 kg, 0,5 vol.) y se agitó la mezcla durante 15-25 minutos mientras se aumentaba la temperatura hasta 17 °C. A continuación, se añadió una disolución de (S)-2,8-diazaspiro[4.5]decano-2.3.8- tricarboxilato de 2-bencilo, 8-(terc-butilo) y 3-etilo en etanol (ejemplo 4, 17,3 kg de (S)-2,8-diazaspiro[4.5]decano-2.3.8- tricarboxilato de 2-bencilo, 8-(terc-butilo) y 3-etilo; disolución total de (S)-2,8-diazaspiro[4.5]decano-2,3,8-tricarboxilato de 2-bencilo, 8-(terc-butilo) y 3-etilo y etanol: 87 kg) a lo largo de 25 minutos mientras se mantenía la temperatura de la mezcla de reacción a aproximadamente 16 °C. Se añadió etanol absoluto adicional (0,5 vol.) y la mezcla de reacción se calentó hasta 30 ± 3 °C y se agitó durante aproximadamente 16 horas. Entonces, la mezcla se concentró usando destilación al vacío entre 35-40 °C hasta que quedaron aproximadamente 4 volúmenes relativos. Se añadió 2-metiltetrahidrofurano (3 vol.), y la mezcla resultante se concentró usando destilación al vacío entre 35 40 °C hasta que quedaron aproximadamente 4 volúmenes relativos. Se realizaron la adición de 2-metiltetrahidrofurano y destilación tres veces usando 3 vol. de 2-metiltetrahidrofurano y un tiempo final usando 2 vol. de 2-metiltetrahidrofurano. Entonces, la mezcla de reacción se calentó hasta aproximadamente 30-35 °C y se agitó durante aproximadamente 40 minutos. Entonces se añadió 2-metiltetrahidrofurano adicional (1,5 vol.) mientras se mantenía la temperatura a aproximadamente 29 °C. La mezcla se enfrió hasta 25 ± 3 °C a lo largo de un período de 1 h, después se agitó durante aproximadamente 18 horas. La mezcla se filtró y la torta del filtro se lavó con 2-metiltetrahidrofurano (1,0 vol.) y se agitó durante al menos 5 minutos. El lavado se repitió con el secado de la torta del filtro durante al menos 5 minutos entre lavados. La torta del filtro se secó entonces bajo flujo de nitrógeno durante aproximadamente 19 horas a temperatura ambiente para proporcionar el producto del título. Pureza por HPLC: 99,09 %. Pureza quiral (método B): 99,85 %. Exceso enantiomérico de isómero (S-) (Método B): > 98 %.
Se preparó una segunda cosecha del compuesto del título combinando los filtrados y los disolventes de lavado y se concentraron hasta aproximadamente 35 l, seguido de la adición de 35 l de metil terc-butil éter (MTBE) a lo largo de 45 minutos. Entonces, la mezcla se agitó durante 1 hora a 25 °C y se filtró. La torta de filtro resultante se lavó con MTBE adicional y se secó durante 17 horas para proporcionar una segunda cosecha del producto del título. Pureza por HPLC: 98,56 %. Pureza quiral (Método b ): 99,27 %. Exceso enantiomérico del isómero (S-) (Método B): >98 %.
Ejemplo 6. (S)-8-(2-Ammo-6-((R)-1-(5-doro-[1,1'-bifenM]-2-M)-2,2,2-tnfluoroetoxi)pmmidm-4-M)-2,8-diazaspiro[4.5]decano-3-carboxilato de etilo

Se preparó el compuesto del título a partir de clorhidrato de (S)-2,8-diazaspiro[4.5]decano-2,3-dicarboxilato de 2-bencilo y 3-etilo (ejemplo 5) según los procedimientos mostrados en el esquema 5 y en la patente de Estados Unidos n.° 9.199.994, cuya divulgación se incorpora en el presente documento por referencia en su totalidad. Se ha encontrado que el compuesto del título es un inhibidor de TPH1 según uno o más ensayos descritos en la patente de Estados Unidos n.° 9.199.994.
Claims (27)
- REIVINDICACIONESi. Un procedimiento de aumento de la cantidad de una sal de ácido de un compuesto isomérico de fórmula I-(S):
 en donde R1 es etilo y Pg1 es terc-butoxicarbonilo, en relación con una sal de ácido de una cantidad de un compuesto isomérico de fórmula I-(R):
en donde R1 es etilo y Pg1 es terc-butoxicarbonilo, en relación con una sal de ácido de una cantidad de un compuesto isomérico de fórmula I-(R): en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R), comprendiendo el procedimiento:hacer reaccionar la mezcla de partida con ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, en presencia de un aldehído para formar una mezcla de sal de ácido que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos,en donde la mezcla de sal de ácido tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) presentes en la mezcla de partida.
en una mezcla de partida que comprende ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R), comprendiendo el procedimiento:hacer reaccionar la mezcla de partida con ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, en presencia de un aldehído para formar una mezcla de sal de ácido que comprende sales de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico de los compuestos isoméricos,en donde la mezcla de sal de ácido tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la cantidad de sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de los compuestos isoméricos de fórmulas I-(S) e I-(R) presentes en la mezcla de partida. - 2. El procedimiento según la reivindicación 1, en donde el ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, es monohidrato de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico.
- 3. El procedimiento según la reivindicación 1 o 2, en donde la reacción se lleva a cabo con aproximadamente 1 equivalente molar del ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico, o un hidrato del mismo, con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida.
- 4. El procedimiento según una cualquiera de las reivindicaciones 1 a 3, en donde el aldehído es benzaldehído.
- 5. El procedimiento según una cualquiera de las reivindicaciones 1 a 4, en donde la reacción se lleva a cabo con menos de 1 equivalente molar del aldehido con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida.
- 6. El procedimiento según una cualquiera de las reivindicaciones 1 a 5, en donde la reacción se lleva a cabo con de aproximadamente 0,01 a aproximadamente 0,1 equivalentes molares del aldehído con respecto a la cantidad combinada de ambos compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de partida.
- 7. El procedimiento según una cualquiera de las reivindicaciones 1 a 6, en donde la reacción se lleva a cabo a una temperatura de aproximadamente 30 °C a aproximadamente 40 °C.
- 8. El procedimiento según una cualquiera de las reivindicaciones 1 a 7, en donde la reacción se lleva a cabo en presencia de un disolvente orgánico.
- 9. El procedimiento según una cualquiera de las reivindicaciones 1 a 7, en donde la reacción se lleva a cabo en presencia de un disolvente de éter.
- 10. El procedimiento según una cualquiera de las reivindicaciones 1 a 7, en donde la reacción se lleva a cabo en presencia de un disolvente orgánico que comprende 2-metiltetrahidrofurano.
- 11. El procedimiento según una cualquiera de las reivindicaciones 1 a 10, en donde el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en la mezcla de sal es del 75% o mayor.
- 12. El procedimiento según una cualquiera de las reivindicaciones 1 a 10, en donde el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en la mezcla de sal es del 90% o mayor.
- 13. El procedimiento según una cualquiera de las reivindicaciones 1 a 12, que comprende además recristalizar la mezcla de sal para formar una mezcla de sal purificada que tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) en relación con la sal de ácido gulónico del compuesto isomérico de fórmula I-(R) en comparación con las cantidades relativas de las sales de ácido gulónico de los compuestos isoméricos antes de la purificación.
- 14. El procedimiento según la reivindicación 13, en donde el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) es del 90% o mayor.
- 15. El procedimiento según la reivindicación 13, en donde el exceso enantiomérico de la sal de ácido gulónico del compuesto isomérico de fórmula I-(S) es del 95% o mayor.
- 16. El procedimiento según una cualquiera de las reivindicaciones 13 a 15, que comprende además hacer reaccionar la mezcla de sal purificada con una base para formar una mezcla de base libre que comprende compuestos isoméricos que tienen la fórmula I-(S) y la fórmula I-(R):

- 17. El procedimiento según la reivindicación 16, en donde la base está en forma de una disolución acuosa.
- 18. El procedimiento según la reivindicación 16, en donde la base es una disolución acuosa de carbonato de sodio.
- 19. El procedimiento según una cualquiera de las reivindicaciones 16 a 18, en donde la reacción de la mezcla de sal purificada se lleva a cabo con una cantidad en exceso molar de base con respecto a la cantidad combinada de ambas sales de ácido gulónico de los compuestos isoméricos de fórmula I-(S) y fórmula I-(R) en la mezcla de sal.
- 20. El procedimiento según una cualquiera de las reivindicaciones 16 a 19, en donde la reacción de la mezcla de sal purificada se lleva a cabo en presencia de un disolvente orgánico.
- 21. El procedimiento según una cualquiera de las reivindicaciones 16 a 19, en donde la reacción de la mezcla de sal purificada se lleva a cabo en presencia de un disolvente orgánico que comprende un disolvente de éter y un disolvente de hidrocarburo.
- 22. El procedimiento según una cualquiera de las reivindicaciones 16 a 19, en donde la reacción de la mezcla de sal purificada se lleva a cabo en presencia de un disolvente orgánico que comprende 2-metiltetrahidrofurano y n-heptano.
- 23. El procedimiento según una cualquiera de las reivindicaciones 16 a 22, en donde el exceso enantiomérico del compuesto isomérico de fórmula I-(S) en la mezcla de base libre es del 90% o mayor.
- 24. El procedimiento según una cualquiera de las reivindicaciones 16 a 22, en donde el exceso enantiomérico del compuesto isomérico de fórmula I-(S) en la mezcla de base libre es del 95% o mayor.
- 25. Una sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(S) o fórmula I-(R):
 en donde R1 es etilo y Pg1 es terc-butoxicarbonilo.
en donde R1 es etilo y Pg1 es terc-butoxicarbonilo. - 26. La sal según la reivindicación 25, que es la sal de ácido 2,3:4,6-di-O-isopropiliden-2-ceto-L-gulónico del compuesto isomérico de fórmula I-(S).
- 27. El procedimiento según una cualquiera de las reivindicaciones 1-24, que comprende además:recristalizar la mezcla de sal para formar una mezcla de sal purificada que tiene una cantidad aumentada de la sal de ácido gulónico del compuesto isomérico de fórmula Ia-(S) en relación con la sal de ácido gulónico del compuesto isomérico de fórmula Ia-(R) en comparación con las cantidades relativas de las sales de ácido gulónico de los compuestos isoméricos antes de la purificación; yhacer reaccionar la mezcla de sal purificada en presencia de carbonato de sodio para formar una mezcla de base libre que comprende compuestos isoméricos que tienen la fórmula Ia-(S) y la fórmula Ia-(R), en donde el exceso enantiomérico del compuesto isomérico de fórmula Ia-(S) en la mezcla de base libre es del 90% o mayor.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662419557P | 2016-11-09 | 2016-11-09 | |
| PCT/IB2017/001594 WO2018087602A1 (en) | 2016-11-09 | 2017-11-09 | Resolution of optically active diazaspiro[4.5]decane derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2911447T3 true ES2911447T3 (es) | 2022-05-19 |
Family
ID=61163735
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17838138T Active ES2911447T3 (es) | 2016-11-09 | 2017-11-09 | Resolución de derivados de diazaspiro[4.5]decano ópticamente activos |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20190263815A1 (es) |
| EP (1) | EP3538530B1 (es) |
| JP (2) | JP7229917B2 (es) |
| CN (2) | CN110177790B (es) |
| AR (1) | AR110150A1 (es) |
| CA (1) | CA3043442A1 (es) |
| DK (1) | DK3538530T3 (es) |
| ES (1) | ES2911447T3 (es) |
| TW (1) | TWI762527B (es) |
| WO (1) | WO2018087602A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210102887A (ko) * | 2018-11-14 | 2021-08-20 | 알타반트 사이언시스 게엠베하 | 말초 세로토닌과 관련된 질병 또는 장애를 치료하기 위한 트립토판 하이드록실라제 1 (tph1)의 결정질 스피로사이클릭 화합물 억제제 |
| CN111621492B (zh) * | 2020-06-15 | 2021-10-26 | 安徽红杉生物医药科技有限公司 | 内酰胺酶及其应用和酶法拆分制备(1r,4s)-文斯内酯的方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3682925A (en) * | 1969-01-30 | 1972-08-08 | Hoffmann La Roche | ({31 )-di-o-isopropylidene-2-keto-l-gulonates |
| US3904632A (en) * | 1972-05-08 | 1975-09-09 | Hoffmann La Roche | (')-di-o-isopropylidene-2-keto-l-gulonates |
| US4859771A (en) * | 1986-07-14 | 1989-08-22 | Merck & Co., Inc. | Process for resolution and racemization of amines with acidic α-hydrogens |
| US5030725A (en) * | 1990-02-06 | 1991-07-09 | Eli Lilly And Company | Method of resolving cis 3-amino-4-[2-(2-furyl)eth-1-yl]-1-methoxycarbonylmethyl-azetidin-2-one using (-)-DAG |
| CN1105723C (zh) | 1996-08-28 | 2003-04-16 | 普罗克特和甘保尔公司 | 螺环金属蛋白酶抑制剂 |
| DE19927412A1 (de) | 1999-06-16 | 2000-12-21 | Bayer Ag | Verfahren zur Enantiomerenanreicherung von cis-8-Benzyl-7,9-dioxo-2,8-diazabicyclo [4.3.0]nonan |
| EP2285769B1 (en) | 2008-06-02 | 2014-08-13 | Generics [UK] Limited | A process for the preparation of enantiomerically pure amines |
| TW201103540A (en) * | 2009-06-16 | 2011-02-01 | Ono Pharmaceutical Co | Compound having spiro-linked cyclic group and uses thereof |
| JP2014501771A (ja) * | 2011-01-07 | 2014-01-23 | ハー・ルンドベック・アクチエゼルスカベット | 4−((1r,3s)−6−クロロ−3−フェニル−インダン−1−イル)−1,2,2−トリメチル−ピペラジンと1−((1r,3s)−6−クロロ−3−フェニル−インダン,1−イル)−3,3−ジメチル−ピペラジンとを分割するための方法 |
| UA119247C2 (uk) | 2013-09-06 | 2019-05-27 | РОЙВЕНТ САЙЕНСИЗ ҐмбГ | Спіроциклічні сполуки як інгібітори триптофангідроксилази |
-
2017
- 2017-11-08 AR ARP170103102A patent/AR110150A1/es not_active Application Discontinuation
- 2017-11-09 CA CA3043442A patent/CA3043442A1/en active Pending
- 2017-11-09 WO PCT/IB2017/001594 patent/WO2018087602A1/en not_active Ceased
- 2017-11-09 CN CN201780069288.XA patent/CN110177790B/zh not_active Expired - Fee Related
- 2017-11-09 CN CN202211097232.4A patent/CN115536661A/zh active Pending
- 2017-11-09 DK DK17838138.0T patent/DK3538530T3/da active
- 2017-11-09 ES ES17838138T patent/ES2911447T3/es active Active
- 2017-11-09 EP EP17838138.0A patent/EP3538530B1/en active Active
- 2017-11-09 JP JP2019525887A patent/JP7229917B2/ja active Active
- 2017-11-09 TW TW106138813A patent/TWI762527B/zh not_active IP Right Cessation
-
2019
- 2019-05-09 US US16/407,375 patent/US20190263815A1/en not_active Abandoned
-
2021
- 2021-01-14 US US17/149,070 patent/US11708365B2/en active Active
-
2023
- 2023-02-15 JP JP2023021579A patent/JP2023078126A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| CN115536661A (zh) | 2022-12-30 |
| JP2023078126A (ja) | 2023-06-06 |
| CA3043442A1 (en) | 2018-05-17 |
| CN110177790A (zh) | 2019-08-27 |
| JP7229917B2 (ja) | 2023-02-28 |
| US20190263815A1 (en) | 2019-08-29 |
| US11708365B2 (en) | 2023-07-25 |
| WO2018087602A1 (en) | 2018-05-17 |
| TWI762527B (zh) | 2022-05-01 |
| DK3538530T3 (da) | 2022-03-28 |
| EP3538530A1 (en) | 2019-09-18 |
| JP2019534313A (ja) | 2019-11-28 |
| EP3538530B1 (en) | 2022-01-05 |
| CN110177790B (zh) | 2022-10-14 |
| US20210139481A1 (en) | 2021-05-13 |
| AR110150A1 (es) | 2019-02-27 |
| TW201829409A (zh) | 2018-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2882118T3 (es) | Procedimiento de síntesis de ruxolitinib | |
| ES2650915T3 (es) | Imidazopiridazinas amino-sustituidas | |
| ES2437755T3 (es) | Intermedios para derivados de tienopirazol que tienen actividad inhibitoria de PDE 7 | |
| JP2022520047A (ja) | 鏡像異性的に濃縮されたjak阻害剤を調製するための方法 | |
| JP2023078126A (ja) | 光学活性ジアザスピロ[4.5]デカン誘導体の分割 | |
| ES2817909T3 (es) | Procedimiento para la preparación de un intermedio de la sitagliptina mediante un método de reducción asimétrica | |
| US20240067652A1 (en) | Method of preparing indolinobenzodiazepine derivatives | |
| US20220363680A1 (en) | Processes for the synthesis of valbenazine | |
| ES2701251T3 (es) | Procedimiento de preparación de un intermediario para antagonistas de receptores opioides mu | |
| ES2718038T3 (es) | Impurezas diméricas de Apixaban y método de eliminación de las mismas | |
| ES2641901T3 (es) | Procedimiento para la fabricación de benzofuroquinolicinas sustituidas espirocíclicas | |
| TW202033521A (zh) | 製備囊泡單胺轉運蛋白2(vmat2)抑制劑之方法 | |
| ES2683125T3 (es) | Proceso para la preparación de piridinas 2,3-diaril-5-sustituidas y sus compuestos intermedios | |
| HK40084559A (en) | Resolution of optically active diazaspiro[4.5]decane derivatives | |
| HK40011685A (en) | Resolution of optically active diazaspiro[4.5]decane derivatives | |
| CN116143753B (zh) | Nlrp3抑制剂化合物 | |
| HK40011685B (en) | Resolution of optically active diazaspiro[4.5]decane derivatives | |
| WO2016071382A1 (en) | Synthesis of pi3k inhibitor and salts thereof | |
| RU2774253C2 (ru) | Синтез n-(гетероарил)-пирроло[3,2-d]пиримидин-2-аминов | |
| WO2026013121A1 (en) | Method for preparing fezolinetant | |
| ES2215922T3 (es) | Procedimiento para la preparacion de 9,10 dihidropirrolo(2,1-b)(1,3)benzotiacepinas aminosustituidas en la posicion 9'. | |
| KR20250052447A (ko) | Jak 키나아제 억제제의 전구체 약물 | |
| Kania et al. | Synthetic studies directed towards asmarines. | |
| JP2016132660A (ja) | 新規イミダゾピリミジンおよびその医薬用途 | |
| Woydziak et al. | Unusual tosyl transfer solvolysis reaction to 3‐n‐butyl‐4‐methyl‐5, 5‐di‐p‐toluenesulfonyl‐3‐pyrrolin‐2‐one |