ES2926912T3 - Heterodímeros de ácido glutámico - Google Patents
Heterodímeros de ácido glutámico Download PDFInfo
- Publication number
- ES2926912T3 ES2926912T3 ES19214298T ES19214298T ES2926912T3 ES 2926912 T3 ES2926912 T3 ES 2926912T3 ES 19214298 T ES19214298 T ES 19214298T ES 19214298 T ES19214298 T ES 19214298T ES 2926912 T3 ES2926912 T3 ES 2926912T3
- Authority
- ES
- Spain
- Prior art keywords
- mip
- ureido
- acid
- pentyl
- carboxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/06—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton
- C07C275/16—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an acyclic and saturated carbon skeleton being further substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/385—Haptens or antigens, bound to carriers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0455—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/001—Acyclic or carbocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/04—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms
- C07C275/20—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an unsaturated carbon skeleton
- C07C275/24—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/28—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C275/30—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton being further substituted by halogen atoms, or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/38—Radicals substituted by singly-bound nitrogen atoms having only hydrogen or hydrocarbon radicals attached to the substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F1/00—Compounds containing elements of Groups 1 or 11 of the Periodic Table
- C07F1/005—Compounds containing elements of Groups 1 or 11 of the Periodic Table without C-Metal linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F13/00—Compounds containing elements of Groups 7 or 17 of the Periodic Table
- C07F13/005—Compounds without a metal-carbon linkage
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Epidemiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Psychiatry (AREA)
- Physical Education & Sports Medicine (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
- Medicinal Preparation (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Nuclear Medicine (AREA)
Abstract
Compuestos de Fórmula (Ia) donde R es un arilo C6-C12 sustituido o no sustituido, un heteroarilo C6-C12 sustituido o no sustituido, un alquilo C1-C6 sustituido o no sustituido o -NR'R',Q es C(O), O, NR' , S, S(O)2, C(O)2(CH2)pY es C(O), O, NR', S, S(O)2, C(O)2(CH2)pZ es H o C1 -alquilo C4, R' es H, C(O), S(O)2, C(O)2, un arilo C6-C12 sustituido o no sustituido, un heteroarilo C6-C12 sustituido o no sustituido o un alquilo C1-C6 sustituido o no sustituido, cuando sustituidos, arilo, heteroarilo y alquilo están sustituidos con halógeno, C6-C12heteroarilo, -NR'R' o COOZ, que tienen propiedades diagnósticas y terapéuticas, tales como el tratamiento y manejo del cáncer de próstata y otras enfermedades relacionadas con la inhibición de la NAALADasa. Los radiomarcadores se pueden incorporar a la estructura a través de una variedad de grupos prostéticos unidos a la cadena lateral del aminoácido X a través de un enlace de carbono o heteroátomo. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Heterodímeros de ácido glutámico
Antecedentes de la invención
Al menos 1 millón de hombres sufren de cáncer de próstata y se estima que la enfermedad afectará a uno de cada seis hombres estadounidenses entre las edades de 60 y 80 años. Cada año se diagnostican más de 300000 nuevos casos de cáncer de próstata. El cáncer de próstata afectará a uno de cada seis hombres en los Estados Unidos, y la mortalidad por la enfermedad es superada solo por el cáncer de pulmón. Se estima que actualmente se gastan 2 mil millones de dólares en todo el mundo en cirugía, radiación, terapia con fármacos y tratamientos mínimamente invasivos, mil millones de dólares del gasto en los EE. UU. Actualmente no existe una terapia efectiva para el cáncer de próstata recurrente, metastásico e independiente de andrógenos. Actualmente se necesitan nuevos agentes que permitan una visualización rápida del cáncer de próstata y un direccionamiento específico para permitir radioterapia. La dipeptidasa ácida ligada a alfa N-acetilada (NAALADasa), también conocida como glutamato carboxipeptidasa II (GCPII), es una neuropeptidasa que escinde N-acetilaspartil-glutamato (NAAG) en N-acetilaspartato y glutamato en el sistema nervioso, ver más abajo, que representa la escisión hidrolítica de NAAG por NAALDasa a través del intermedio tetraédrico. La enzima es una proteína de tipo II de la clase cocatalítica de metalopeptidasas, que contiene dos átomos de zinc en el sitio activo.

Independientemente de su caracterización en el sistema nervioso, se demostró que una forma de NAALADasa se expresaba a altos niveles en adenocarcinomas prostáticos humanos y se denominó antígeno de membrana específico de próstata (PSMA). Se sabe que el gen NAALADasa/PSMA produce múltiples formas de corte y empalme de ARNm y, según la evidencia inmunohistoquímica previa, se ha asumido que el cerebro y la próstata humanos expresan diferentes isoformas de la enzima.
El antígeno de membrana específico de próstata (PSMA) humano, también conocido como folato hidrolasa I (FOLH1), es una glicoproteína transmembrana de tipo II de 750 aminoácidos que se expresa principalmente en el epitelio prostático humano normal, pero se regula positivamente en el cáncer de próstata, incluido el metastásico. El PSMA es una exopeptidasa única con reactividad hacia los folatos poli-gamma-glutamados, capaz de eliminar secuencialmente los poli-gamma-glutamil terminales. Dado que el PSMA se expresa en prácticamente todos los cánceres de próstata y su expresión aumenta aún más en los carcinomas poco diferenciados, metastásicos y refractarios a las hormonas, es un objetivo muy atractivo para la imagenología y la terapia de la próstata. El desarrollo de ligandos que interactúen con el PSMA y transporten los radionúclidos apropiados puede proporcionar una opción de direccionamiento prometedora y novedosa para la detección, el tratamiento y el manejo del cáncer de próstata.
La forma radioinmunoconjugada del anticuerpo monoclonal (mAb) 7E11 anti-PSMA, conocida como exploración PROSTASCINT, se utiliza actualmente para diagnosticar la metástasis y la recurrencia del cáncer de próstata. Los primeros resultados prometedores de varios ensayos de Fase I y II han utilizado el PSMA como diana terapéutica. PROSTASCINT se dirige al dominio intracelular de PSMA y se cree que se une principalmente a las porciones necróticas del tumor de próstata. 14 Más recientemente, se han desarrollado anticuerpos monoclonales que se unen al dominio extracelular de PSMA y se han radiomarcado y se ha demostrado que se acumulan en modelos de tumores de próstata positivos para PSMA en animales.
Si bien los anticuerpos monoclonales son prometedores para la detección y el tratamiento de tumores, ha habido éxitos clínicos limitados fuera del linfoma debido a su baja permeabilidad en los tumores sólidos. Los miméticos de bajo peso molecular, con mayor permeabilidad en tumores sólidos, tendrán una clara ventaja en la obtención de un alto porcentaje por gramo y un alto porcentaje de unión específica.
Kozikowski y otros (2004) J. Med. Chem. 47: 1729-1738 describen la síntesis de inhibidores a base de urea como sondas de sitio activo de glutamato carboxipeptidasa II y su eficacia como analgésicos.
Foss y otros (2005) Clinical Cancer Research 11(11): 4022-4028 describen varios ligandos de molécula pequeña radiomarcados para el antígeno de membrana específico de próstata y su aplicación a la obtención de imágenes in vivo en modelos experimentales de cáncer de próstata.
Resumen de la invención
Un aspecto de la presente invención se refiere a un kit que comprende: (i) un compuesto que comprende un resto de unión a glutamato-urea-lisina-PSMA conjugado con un resto quelante de metales, y (ii) un radionúclido.
En una modalidad preferida del kit de la invención, el radionúclido se selecciona de tecnecio-99m, renio-186, renio-188 o combinaciones de los mismos.
La presente invención también se refiere al uso del kit de la invención para la producción de un compuesto de unión a PSMA, que comprende la etapa de quelar el radionúclido (ii) con el resto quelante de metales del compuesto (i).
Los restos químicos adecuadas, las definiciones de los restos químicos, los excipientes y los métodos y modos de administración pueden encontrarse en las Solicitudes publicadas en EE. UU. Núms. 2004/0054190 y 2004/0002478 y Solicitudes Internacionales Nos. WO 02/22627 y WO 03/060523.
Breve descripción de los dibujos
Las Figuras 1A-1C son cromatogramas de HPLC respectivamente de la coinyección de TC-99m-glu-urea-DpK (Tc-99m-MIP 1008), el análogo de renio y los complejos de diéster de renio.
Las Figuras 2A-2D muestran la estabilidad del complejo Tc-99m de Glu-urea-DpK (Tc-99m-MIP 1008) a 37 °C en PBS respectivamente pH 7,2, cisteína 0,1 M en PbS, DTPA 0,1 M en PBS y 5 % de suero de ratón en PBS durante 6 horas.
Las Figuras 3A-3B son cromatogramas de HPLC respectivos de N-[N-[(S)-1,3-dicarboxipropil]carbamoil]-S-3-yodo-L-tirosina (1-131 DCIT) reacción cruda Figura 3A, arriba, purificado a las 2 horas, Figura 3B, centro y a los 2 días Figura 3C, abajo.
Las Figuras 4A-4D son cromatogramas de radio-HPLC de I-131 MIP 1072 purificado; a los 3 días en A) DMSO. B) Genstisato al 3 %-ascorbato al 6 %/ácido ascórbico, C) PBS, pH = 7,2, D) etanol al 10 % en solución salina a 37 °C. Como se muestra arriba, el 1-131-1072 (pico 12 minutos) permaneció estable a lo largo de los experimentos.
La Figura 5 muestra 1-123 DCIT unido específicamente a células LnCap y no a células PC3 (conjunto de la izquierda) como es evidente por los recuentos desplazables por compuesto no radiomarcado (conjunto del medio) o PMPA (conjunto de la derecha) en células LnCap. Los histogramas muestran la media ± s Em , cada experimento se realizó por duplicado.
La Figura 6 es un análisis de Scatchard en un ensayo de unión celular de PSMA con ácido 2-{3-[1-carboxi-2-(4-hidroxi-fenil)-etil]-ureido}-pentanodioico (DCT) frío.
La Figura 7 muestra evaluaciones biológicas de compuestos seleccionados en las células LNCaP positivas para PSMA.
La Figura 8 muestra las evaluaciones biológicas de los compuestos principales en las células LNCaP positivas para PSMA.
La Figura 9 muestra el análisis de Scatchard en el ensayo de unión celular de PSMA con MIP1072.
La Figura 10 muestra la internalización de I-131-MIP1072.
Las Figuras 11A y 11B muestran respectivamente la estabilidad de I-131 MIP-1072 frente a DCT y fenacetina en microsomas de rata durante 60 minutos.
La Figura 12 muestra la biodistribución tisular de 1-131 MIP1072 en ratones con tumores de xenoinjerto.
La Figura 13 muestra la inhibición de la actividad de NAALADasa a partir de lisados celulares de LNCaP. La Figura 14 muestra la inhibición de la actividad de NAALADasa a partir de lisados celulares de LNCaP. La Figura 15 muestra la inhibición de la actividad de NAALADasa a partir de lisados celulares de LNCaP. La Figura 16 muestra la capacidad de los compuestos de ensayo para inhibir la unión de un inhibidor de NAALADasa conocido, 131I-DCIT, a PSMA en células LNCaP. Las células se incubaron con varias concentraciones de compuestos de prueba y 131I-DCIT durante 1 hora, luego se lavaron y contaron para determinar los valores de IC50.
La Figura 17 es un análisis de unión directa de MIP-1072. Se incubó 123I-MIP-1072 (3 nM, >1000 mCi/pmol) con células LNCaP positivas para PSMA o células PC3 negativas para PSMA (300000 células/pocillo), tanto en ausencia como en presencia de MIP-1072 10 pM o 10 pM no radiactivo o de un inhibidor específico de PSMA (PMPA).
La Figura 18 muestra el análisis de unión por saturación de 123 I-MIP-1072 en células LNCaP.
La Figura 19 muestra la internalización de 123I-MIP-1072.
La Figura 20 muestra la captación de 123I-MIP-1072 en ratones portadores de xenoinjerto LNCaP. Se evaluó la biodistribución tisular de 123I-MIP-1072 (2 pCi/ratón, >1000 mCi/pmol) en tejidos seleccionados de ratones desnudos portadores de tumores LNCaP (PSMA positivos). Los resultados se expresan como el porcentaje de la dosis inyectada por gramo de los tejidos seleccionados (%ID/g).
Figura 21 Captación de 123I-MIP-1072 en ratones portadores de xenoinjertos LNCaP y PC3. Se evaluó la biodistribución tisular de 123I-MIP-1072 (2 pCi/ratón, >1000 mCi/pmol) en tejidos seleccionados de ratones desnudos portadores de tumores LNCaP (PSMA positivo) y PC3 (PSMA negativo) con (Bloqueado) o sin (Normal) pretratamiento con 50 mg/kg de PMPA.
Descripción detallada de la invención
Definiciones
"Sal farmacéuticamente aceptable" se refiere a aquellas sales que retienen la eficacia biológica y las propiedades de las bases libres y que se obtienen por reacción con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido ptoluenosulfónico, ácido salicílico y similares.
"Alquilo" se refiere a un hidrocarburo alifático saturado de cadena lineal, ramificada o cíclica. Los grupos alquilo típicos incluyen metilo, etilo, propilo, isopropilo, butilo, isobutilo, butilo terciario, pentilo, hexilo y similares. El grupo alquilo puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en hidroxilo, ciano o alcoxi. Cuando el grupo alquilo es un sustituyente R', es un alquilo inferior de 1 a 6 carbonos, con mayor preferencia de 1 a 4 carbonos.
"Arilo" se refiere a un grupo aromático que tiene al menos un anillo que tiene un sistema de electrones pi conjugado e incluye grupos arilo carbocíclico, arilo heterocíclico y biarilo. El grupo arilo puede estar opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en halógeno, trihalometilo, hidroxilo, SH, OH, NO2, amina, tioéter, ciano, alcoxi, alquilo y amino. Los ejemplos de grupos arilo incluyen grupos fenilo, naftilo y antracilo. Se prefieren los grupos fenilo y fenilo sustituido.
"Heteroarilo" se refiere a un grupo arilo que tiene de 1 a 3 heteroátomos como átomos del anillo, y el resto de los átomos del anillo son carbonos. Los heteroátomos incluyen oxígeno, azufre y nitrógeno. Así, los grupos arilo heterocíclicos incluyen furanilo, tienilo, piridilo, pirrolilo, pirrolo alquilo N-inferior, pirimidilo, pirazinilo, imidazolilo y similares.
Síntesis
Todas las reacciones se llevaron a cabo en cristalería seca bajo una atmósfera de argón a menos que se indique de otra manera. Las reacciones se purificaron por cromatografía en columna, a presión media mediante el uso de un Biotage SP4 o por cromatografía líquida preparativa de alta presión.
Se registró 1H NMR en un instrumento Bruker de 400 MHz. Los espectros se reportan como ppm 8 y se refieren a las resonancias del solvente en CDCh, DMSO-cfó o metanol-d4. Todos los disolventes se adquirieron de Sigma-Aldrich. Los reactivos se adquirieron de Sigma Aldrich, Bachem, Akaal, Fisher, Alfa Aesar, Acros y Anaspec. Se usan las siguientes abreviaturas cloruro de metileno (DCM), acetato de etilo (EA), hexanos (Hex), dicloroetano (DCE), dimetilformamida (DMF), ácido trifluoroacético (TFA), tetrahidrofurano (THF), carbonildiimidazol (CDI), dimetilaminopiridina (DMAP), trietilamina (TEA), trifluorometanosulfonato de metilo (MeOTf), ácido (S)-2-amino-6-(bis-piridin-2-ilmetil-amino)-hexanoico (dpK), ácido glutámico (Glu), diisopropiletilamina (DIEA), benciloxicarbonilo (CBZ).
Síntesis de intermediarios
Todos los siguientes compuestos se prepararon con rendimientos generales que oscilaban entre el 20 y el 40 % siguiendo la ruta que se muestra en el Esquema 1. La primera etapa, realizada a 0 °C en condiciones inertes, usó el éster di-f-butílico del ácido glutámico con CDI en presencia de una base para formar el derivado 2 intermediario Gluurea-imidazol. Este intermediario se activó con MeOTf en condiciones básicas para producir el imidazol 3 metilado, que en condiciones inertes reaccionó fácilmente con las aminas. Los grupos protectores del éster terc-butílico se eliminaron mediante el uso de TFA al 20 % en DCM durante 1 a 4 horas a temperatura ambiente. Una vez completada la desprotección, las reacciones se concentraron en un evaporador rotatorio o se secaron con nitrógeno y se purificaron en una columna de sílice o se recristalizaron. Los productos finales se probaron in vitro e in vivo.

Esquema 1. Via general para la síntesis de compuestos de PSMA
Éster di-terc-butílico del ácido L-(S)-2-[(imidazol-1-carbonil)-amino]-pentanodioico (2)
A una suspensión de clorhidrato de glutamato de di-í-butilo (15,0 g, 51 mmol) en DCM (150 mL) enfriada hasta 0 °C se le añadió TEA (18 mL) y DMAP (250 mg). Después de agitar durante 5 minutos, se añadió CDI (9,0 g, 56 mmol) y la reacción se agitó durante toda la noche con calentamiento hasta temperatura ambiente. La reacción se diluyó con DCM (150 mL) y se lavó con bicarbonato de sodio saturado (60 mL), agua (2 X 100 mL) y salmuera (100 mL). La capa orgánica se secó sobre sulfato sódico y se concentró para dar el producto crudo como un semisólido, que solidificó lentamente al reposar. El material bruto se trituró con hexano/acetato de etilo para dar un sólido blanco que se filtró, se lavó con hexano (100 mL) y se secó para dar el producto deseado (15,9 g, 45 mmol, 88 %) como un sólido blanco. 1H NMR (400 MHz, DMSO-^) 57,63 (s, 1H), 7,00 (br, 2H), 6,31 (d, 1H), 4,02 (m, 1H), 2,19 (m, 2H), 1,86 (m, 1H), 1,67 (m, 1H), 1,39 (s, 9H), 1,38 (s, 9H). SGAS m / z; 354 (M+H)+.

Alternativamente, los análogos pueden prepararse a través del isocianato generado in situ mediante el uso de trifosgeno. Este enfoque puede lograrse mediante la activación del residuo de glutamato y el acoplamiento con un residuo de lisina (ruta A) o mediante la activación del residuo de lisina y el acoplamiento con el glutamato (ruta B) como se muestra en el esquema 2 más abajo.

Éster di-terc-butílico del ácido L-(S,S)-2-[3-(5-Benciloxicarbonilamino-1-terc-butoxicarbonil-pentil-ureido)-pentanodioico (3)
Ruta A. En un matraz de fondo redondo, se combinaron 1,8 mL de TEA (13,2 mmol) con 1,8 gramos (6 mmol) de clorhidrato del éster di-terc-butílico del ácido L-glutámico en 20 mL de DCM. Esta solución se añade gota a gota durante 45 minutos a una solución de 10 mL de DCM y trifosgeno (0,7 g, 2,2 mmol) a 0 °C. Después de agitar durante 30 min adicionales, se añadió en una porción una solución de H-lis-(Z)-O-t-butil éster HCl (2,2 g, 6 mmol) que contenía TEA (1,8 mL, 13 mmol) en 15 mL de DCM. La solución se agitó durante 1 hora. La reacción se concentró, se diluyó con 50 mL de acetato de etilo, se lavó con NaHSO42N (2x 50 mL), salmuera (50 mL) y se secó sobre sulfato de sodio para producir un aceite amarillo. Purificación por cromatografía en columna para proporcionar el producto deseado como un aceite transparente que al reposar solidifica en un sólido blanco (1,9 g, 54 %).
Ruta B. En un matraz de fondo redondo, se suspende trifosgeno (2,9 g, 10 mmol) en DCM (50 mL) y se agita a 0 °C. A la solución de trifosgeno se añadió gota a gota una solución de base libre de H-lisina(Z) (9,1 g, 27 mmol) y DIEA (10,4 mL, 60 mmol) DCM (50 mL) durante 2,5 horas. Después de 2,5 horas, se añadió en una porción una solución de clorhidrato de éster di-terc-butílico del ácido L-glutámico (8 g, 27 mmol) que contenía DIEA (10,4 mL, 60 mmol) DCM (50 mL) y se dejó agitar durante 45 minutos. La reacción se concentró hasta sequedad, se diluyó con 150 mL de acetato de etilo, se lavó con NaHSO42 N (2 x 200 mL), salmuera (150 mL) y se secó sobre sulfato de sodio para producir un aceite amarillo. Este aceite se purificó por cromatografía en columna (SiO2) para proporcionar el producto deseado como un aceite transparente que al reposar solidifica en un sólido blanco (12,0 g, 72 %). (400 MHz, CDCls) 57,34 (m, 5H), 5,33-5,28 (m, 3H), 5,08 (d, J= 7,4 Hz, 2H), 4,38-4,29 (m, 2H), 3,15 (m, 2H), 2,32-2,01 (m, 2H), 1,90-1,50 (m, 8H), 1,43-1,40 (m, 27H, t-Bu's). ESMS m/z: 622 (M+H)+.

Éster di-terc-butílico del ácido 2-[3-(5-amino-1-terc-butoxicarbonil-pentil)-ureido]-pentanodioico (4). A una solución de éster di-terc-butílico del ácido 2-(3-(5-benciloxicarbonilamino-1-terc-butoxicarbonil-pentil)-ureido]-pentanodioico (630 mg, 1,0 mmol) en etanol (20 mL) se añadió formiato de amonio (630 mg, 10 eqv) seguido de Pd-C al 10 % y la suspensión se dejó reposar con agitación ocasional durante toda la noche hasta que se completó. La reacción se filtró a través de celite y se concentró para dar el producto deseado (479 mg, 98 %) como un sólido ceroso. 1H NMR (400 MHz, CDCls) 57,15-6,0 (bm, 4H, NH's), 4,29 (m, 2H), 3,02 (m, 2H), 2,33 (m, 2H), 2,06-1,47 (m, 8H), 1,45-1,40
(m, 27H, t-Bu's). ESMS m/z: 488 (M+H)+.

Síntesis de los compuestos modelo núcleo de nudo Glu-Urea-Glu.
En esta serie, se incorpora un nudo en la cadena lateral del ácido glutámico o lisina antes de la conjugación para formar el dímero de urea. En el ejemplo más abajo, el ácido carboxílico de cadena lateral de uno de los ácidos glutámicos se modifica en un nudo para agregar un quelante, átomo o grupo funcional que es o contiene un radionúclido (Esquema 4).

Éster di-terc-butílico del ácido 2-{3-[3-(4-amino-butilcarbamoil)-1-metoxicarbonil-propil]-ureido}-pentanodioico (28)
A una solución de N-BOC Ácido glutámico a-metil éster BOC-Glu(OH)-Ome (960 mg, 3,7 mmol) en DMF (6 mL) enfriada hasta 0 °C se añadió EDC (845 mg, 1,3 eqv) y TEA (1,3 mL). Después de agitar durante 10 minutos, se añadió la sal clorhidrato de N-CBZ-1,4-diaminobutano de diamina monoprotegida (1 g, 3,8 mmol) y la reacción se dejó en agitación durante toda la noche con calentamiento hasta temperatura ambiente. La reacción en bruto se diluyó con EA (100 mL) y se lavó con agua (30 mL), solución acuosa al 5 % de Ácido cítrico (30 mL), bicarbonato de sodio saturado (30 mL), agua (30 mL) y salmuera (30 mL). La capa orgánica se secó sobre sulfato de sodio y se concentró para proporcionar el producto bruto como un jarabe espeso (2,1 g). Al jarabe obtenido se le añadió HCl 4 N en dioxano (10 mL) y la reacción se agitó a temperatura ambiente durante 3 h. La concentración proporcionó un sólido ceroso (1,8 g) como la sal de clorhidrato. La sal se acopló al éster di-terc-butílico del ácido L-(S)-2-[(imidazol-1-carbonil)-amino]-pentanodioico activado (2) como se describe en las secciones experimentales anteriores para proporcionar el dímero x protegido (1,9 g). Este material se suspendió en EtOH absoluto (20 mL), se añadió formato de amonio en exceso (5 g), seguido de Pd(OH)2 al 20 % sobre carbón (100 mg) y la suspensión se agitó muy suavemente durante toda la noche para efectuar la escisión del grupo de protección CBZ. La filtración a través de celite y la concentración proporcionaron la amina libre deseada (1,4 g, 2,7 mmol, 73 %, 4 etapas). 1H NMR (400 MHz, CDCls) 58,41 (br, 2H), 7,369br, 1H), 6,44 (bs, 1H), 6,37 (bs, 1H), 4,37-4,29 (m, 2H), 3,71 (s, 3H), 3,20 1,50 (m, 16H), 1,45 (s, 9H), 1,43 (s, 9H). ESMS m/z: 517 (M+H)+.

Ácido Re(CO)3 -2-(3-{3-[4-(Bis-piridin-2-ilmetil-amino)-butilcarbamoil]-1-carboxi-propil}-ureido)-pentanodioico[Br] (29) (MIP-1100)
El intermediario protegido se preparó mediante aminación reductora mediante el uso de piridina-2-carboxaldehído como se describió anteriormente. El tratamiento con LiOH 2 M en MeOH efectuó la hidrólisis del éster metílico. Se eliminó el metanol y se añadió un exceso de DCM:TFA (1:1) y la reacción se agitó a temperatura ambiente durante toda la noche. El material bruto se convirtió en el conjugado de renio deseado siguiendo el procedimiento descrito anteriormente. La HPLC preparativa proporcionó la molécula deseada (9,5 mg, 16 %). 1H Nm R (400 MHz, DMSO-CÍ6) 5 8,78 (m, 2H), 8,31 (br, 1H), 7,95 (m, 2H), 7,59 (m, 2H), 7,39 (m, 2H), 6,60-6.33 (m, 2H), 4,89 (m, 4H), 4,00 (m, 1H), 3,76 (m, 1H), 3,20-1,2 (m, 16H) (3 CO2H no visto). ESMS 842 (M-H)+.

Síntesis de los compuestos modelo de núcleo de Glu-urea-X-bencil-lisina.
Los siguientes compuestos se prepararon todos con rendimientos generales que oscilaban entre el 20 y el 40 % mediante el uso de la ruta representada en el Esquema 3. La Glu-urea-lisina desprotegida en Z se mezcló con el aldehido apropiado (0,9 equivalentes) a temperatura ambiente durante una hora para formar el intermediario de base 10chiff. La base de 11chiff se redujo mediante el uso de 1 equivalente de triacetoxiborohidruro de sodio. Los compuestos se desprotegieron mediante el uso de TFA al 50 % en DCM durante 1 hora a temperatura ambiente. Una vez completadas, las reacciones se concentraron en un evaporador rotatorio o se secaron con nitrógeno y se extrajeron mediante el uso de cloruro de metileno y agua. La capa de agua se evaporó hasta sequedad para proporcionar el producto desprotegido con un rendimiento del 40-80 %.

X = Ha uro
Esquema 3. Vía general para la síntesis de compuestos de PSMA halogenados.
4-trimetilestananil-benzaldehído (5). A una solución de 4-yodobenzaldehído (1,92 g, 8,27 mmol) en dioxano seco (60 mL) se añadió hexametilditina (4,1 mL, 19,8 mmol) seguido de Pd(Ph3P)Cl2 (150 mg) y la mezcla de reacción se calentó durante 3 h a reflujo hasta que se consideró completo. La reacción se filtró a través de celite y se purificó por cromatografía en columna mediante el uso de hexanos/acetato de etilo (9/1) como eluyente para proporcionar (2,24 g, 98 %) como un aceite transparente. 1H NMR (400 MHz, CDCl3) 59,97 (s, 1H), 7,81 (d, J= 7,8 Hz, 2H), 7,72 (d, J= 7,8 Hz, 2H), 0,29 (s, 9H). ESMS m/z: 268 (Sn-grupo).

Éster di-terc-butílico del ácido 2-{3-[1-terc-butoxicarbonil-5-(4-trimetilestananil-bencilamino)-pentil]-ureido}-pentanodioico (6). A una solución de éster di-terc-butílico del ácido 2-[3-(5-amino-1-terc-butoxicarbonil-pentil)-ureido]-pentanodioico (150 mg, 0,31 mmol) en DCE (10 mL) se añadió 4 -trimetilestananil-benzaldehído (82 mg, 0,31 mmol) seguido de triacetoxiborohidruro de sodio (98 mg, 0,47 mmol) y la reacción se agitó durante toda la noche a 40 °C. La reacción se concentró y se purificó por cromatografía en columna mediante el uso de hexanos/acetato de etilo como eluyente para proporcionar el producto deseado (88 mg, 38 %) como un jarabe espeso que comienza a solidificarse al reposar. 1H NMR (400 MHz, DMSO-cf6) 57,48 (d, J = 7,4 Hz, 2H), 7,30 (d, J= 7,4 Hz, 2H), 6,27 (m, 2H, NH's), 3,96 (m, 4H), 2,74 (bm, 2H), 2,21 (m, 2H), 1,87 (m, 2H), 1,65-1,19 (m, 7H), 1,35 (m, 27H, í-Bu's), 0,23 (s, 9H). ESMS m/z: 742 (Sn-grupo).

Ácido (S, S)-2-[3-(5-Amino-1-carboxi-pentil)-ureido]- pentanodioico (7) (MIP 1033) El mismo procedimiento experimental que se muestra en el esquema 1 produjo un 8 % de éster di-terc-butílico del ácido 2-[3-(5-benciloxicarbonilamino-1-terc-butoxicarbonil-pentil)-ureido]-pentanodioico. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente y se purificó por HPLC para proporcionar el producto deseado. 1H NMR (éster tri-t-butílico de amina Z-protegida) (400 MHz, CDCls,) 5 12,2 (s, 3H), 6,4 (s, 2H), 4,15 (m, 2H), 3,45 (m, 1H), 2,75 (bs, 1H), 2,2 (m, 4H), 1,90 (m, 2H), 1,65 (m, 2H), 1,50 (s, 2H), 1,35 (m, 2H). ESMS m/z: 622 (M-H)+.

Ácido (S)-2-(3,3-bis-piridin-2-ilmetil-ureido)-pentanodioico (8) (MIP 1025).
El mismo procedimiento experimental que en la síntesis general produjo 0,65 g, 48 % del éster di-terc-butílico del ácido 2-(3,3-bis-piridin-2-ilmetil-ureido)-pentanodioico. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente y se purificó por HPLC para proporcionar el producto deseado. 1H NMR (400 MHz, DMSO-cfe) 5, 12,0 (bs, 2H), 8,68 (d, 2H), 8,00 (m, 2H), 7,41 (d, 4H), 7,14 (d, 1H), 4,73 (d, 4H), 3,96 (s, 1H), 2,18 (m, 2H), 1,80 (m, 2H).

Ácido (S,S)-2-{3-[3-(Bis-piridin-2-ilmetil-amino)-1-carboxi-propil]-ureido}-pentanodioico (9) (MIP 1028). El mismo procedimiento experimental que en la síntesis general del esquema 1 produjo 0,16 g, 35 % de éster di-terc-butílico del ácido 2-{3-[3-(Bis-piridin-2-ilmetil-amino)-1-carboxi-propil]-ureido}-pentanodioico. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente y se purificó por HPLC para proporcionar el producto
deseado. 1H NMR (400 MHz, DMSO-cfe) 512,4 (br, 2H), 9,37 (s, 1H), 8,52 (d, 2H), 7,80 (t, 2H), 7,14 (dd, 4H), 6,45 (m, 2H), 4,49 (br, 4H), 4,12 (s, 1H), 4,05 (s, 1H), 3,21 (m, 2H), 2,24 (m, 2H), 1,80 (m, 2H), 1,40 (m, 2H). ESMS m/z: (diethyl ester) 429 (M)+, 451 (M+Na).

Ácido (S,S)-2-{3-[5-(Bis-piridin-2-ilmetil-amino)-1-carboxi-pentil]-ureido}-pentanodioico (10) (MIP 1008). El mismo procedimiento experimental que en la síntesis general produjo 0,09 g, 12 % de éster di-terc-butílico del ácido 2-{3-[5-(Bis-piridin-2-ilmetil-amino)-1-carboxi-pentil]-ureido}-pentanodioico. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente y se purificó por HPLC para proporcionar el producto deseado. 1H NMR (400 MHz, DMSO-CÍ6) 5 12,7 (s, 2H), 8,97 (s, 1H), 8,65 (dd, 2H), 7,91 (dd, 2H), 7,45 (m, 4H), 6,44 (d, 1H), 6,28 (d, 1H), 4,45 (br, 4H), 4,10 (m, 2H), 3,15 (br, 2H), 2,60 (m, 2H), 2,25 (m, 2H), 1,90 (m, 2H), 1,78 (m, 2H), 1,45 (m, 2H).

Ácido (S)-2-{3-[1-carboxi-2-(4-yodo-fenil)-etil]-ureido}-pentanodioico (11) (MIP-1034). El mismo procedimiento experimental que en la síntesis general, produjo 0,038 g, 5 % de éster di-terc-butílico del ácido 2-{3-[1-Carboxy-2-(4-yodo-fenil)-etil]-ureido}-pentanodioico. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente. 1H NMR (400 MHz, DMSO-^) 5 12,40 (s, 3H), 7,65 (dd, 2H), 7,05 (dd, 2H), 6,30 (m, 2H), 4,25 (s, 1H), 4,05 (s, 1H), 2,90 (m, 2H), 2,2 (m, 2H), 1,80 (m, 2H). ESMS m/z: 429 (M)+, 451 (M+Na).

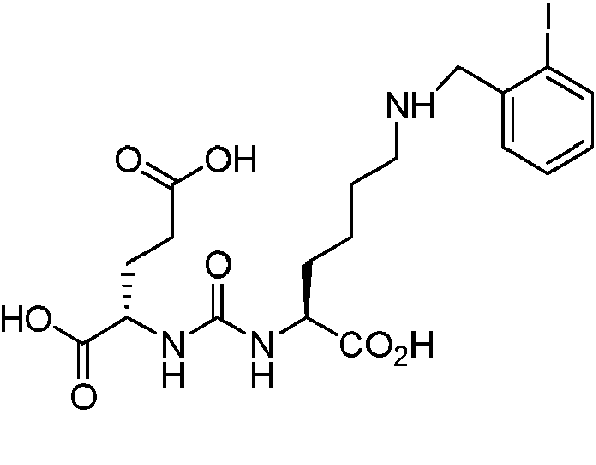
Ácido (S,S)-2-{3-[1-Carboxy-5-(2-yodo-bencilamino)-pentil]-ureido}-pentanodioico (12) (MIP 1035). El mismo procedimiento general, mediante el uso del éster di t -butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente (5,5 mg, 66 %). 1H NMR (400 MHz, DMSO-^) 512,4 (s, 3H), 8,8 (s, 1H), 7,94 (m, 1H), 7,5 (m, 1H), 7,16 (t, 1H), 6,38 (m, 2H), 4,15 (m, 5H), 3,06 (s, 2H), 2,85 (s, 1H), 2,2 (m, 2H), 1,90 (m, 1H), 1,70 (m, 2H), 1,50 (s, 2H), 1,35 (m, 2H). ESMS m/z: 536 (M+H)+.
(S,S)-2-{3-[1-Carboxy-5-(3-yodo-bencilamino)-pentil]-ureido}-pentanodioico (13) (MIP 1089). El mismo procedimiento general, mediante el uso del éster di-f-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente (4,1 mg, 53 %). 1H NMR (400 MHz, DMSO-^) 512,4 (s, 3H), 8,7 (s, 2H), 7,9 (s, 1H), 7,8 (d, 1H), 7,44 (d, 1H), 7,22 (t, 1H), 6,25 (s, 2H), 4,09 (m, 5H), 2,89 (s, 1H), 2,75 (s, 1H), 2,2 (d, 2H), 1,90 (m, 2H), 1,65 (m, 2H), 1,40 (m, 2H).

(S,S)-2-{3-[1-Carboxy-5-(4-yodo-bencilamino)-pentil]-ureido}-pentanodioico (14) (MIP 1072). El mismo procedimiento general, mediante el uso del éster di f -butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente (12 mg, 66 %). 1H NMR (400 MHz, DMSO-^) 512,4 (bs, 3H), 8,8 (br, 1H), 7,8 (d, 2H), 7,27 (d, 2H), 6,35 (br, 2H), 4,1 (m, 4H), 2,89 (m, 2H), 2,2 (d, 2H), 1,90 (m, 2H), 1,65 (m, 4H), 1,35 (m, 2H). ESMS m/z: 536 (M+H)+.

(S,S)-2-{3-[1-Carboxy-5-(4-fluoro-bencilamino)-pentil]-ureido}-pentanodioico (15) (MIP 1090). El mismo procedimiento general, mediante el uso del éster di f -butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente. 1H NMR (400 MHz, DMSO-^) 5 12,4 (br, 3H), 8,7 (br, 1H), 7,5 (m, 2H), 7,3 (m, 2H), 6,35 (m, 2H), 4,1 (m, 4H), 2,9 (m, 2H), 2,2 (d, 2H), 1,90 (m, 2H), 1,60 (m, 4H), 1,35 (m, 2H). ESMS m/z: 428 (M+H)+, 450 (M+ Na).

(S,S)-2-{3-[1-Carboxy-5-(4-bromo-bencilamino)-pentil]-ureido}-pentanodioico (16) (MIP 1094). El mismo procedimiento general, mediante el uso del éster di f -butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. 1HNMR (éster tri f-butílico) (400 MHz, CDCl3) 57,52 (d, 2h), 7,32 (d, 2H), 6,28 (m, 2H), 3,98 (m, 2H), 2,55 (t, 2H), 2,48 (t, 2H), 2,22 (m, 2H), 1,85 (m, 2H), 1,62 (m, 2H), 1,45 (m, 2H), 1,37 (s, 27H), 1,28 (m, 2H) ESMS m/z: 642 (M+H)+. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente. eSm S m/z: 474 (M+H)+.

Ácido (S,S)-2-{3-[1-Carboxy-5-(4-iodo-benzoylamino)-pentyl]-ureido}-pentanodioico (17) (MIP 1044). El mismo procedimiento general, mediante el uso del éster di t -butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente. 1H NMR (400 MHz, DMSO-cfe) 512,4 (s, 3H), 8,45 (s, 1H), 7,8 (dd, 2H), 7,6 (dd, 2H), 6,3 (s, 2H), 5,75 (s, 1H), 4,1 (m, 4H), 3,2 (s, 2H), 2,25 (d, 2H), 1,90 (m, 1H), 1,65 (m, 2H), 1,4 (m, 2H).

Ácido 2-{3-[1-carboxi-5-(4-yodo-bencenosulfonilamino)-pentil]-ureido}-pentanodioico (18). (MIP 1097).
En un matraz de fondo redondo, se suspendió éster di t-butílico del ácido 2-[3-(5-amino-1-carboxi-pentil)-ureido]-pentanodioico (300 mg, 0,62 mmol) en agua (10 mL) y se añadieron 1,4 dioxano (10 mL) y TEA (1,75 mL, 1,25 mmol) seguido de cloruro de 4-yodo-bencenosulfonilo y la mezcla se agitó durante toda la noche a 50 °C. La mezcla de reacción se evaporó hasta sequedad, se recogió en DCM y se sometió a cromatografía sobre gel de sílice para proporcionar el producto deseado (375 mg, 80 %) como un aceite transparente. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente seguido de purificación por HPLC para proporcionar el producto deseado MIP-1097 como un sólido más blanco (270 gramos, 90 % de rendimiento). 1H NMR (400 MHz, DMSO-CÍ6) 57,97 (d, 2H), 7,68 (t, 1H), 7,53(d, 2H), 6,35 (dd, 2H), 4,10 (m, 1H), 4,00 (m, 1H), 2,65 (m, 2H), 2,22 (m, 2H), 1,9 (m, 1H), 1,7 (m, 1H), 1,55 (m, 1H), 1,45 (m, 1H), 1,35 (m, 2H), 1,25 (m, 2H), (3 CO2H no visto). ESMS m/z: 565 (M+H)+.

Ácido 2-(3-{1-carboxi-5-[3-(4-yodo-fenil)-ureido]-pentil}-ureido)-pentanodioico (19) (MIP 1095)
En un matraz de fondo redondo, se disuelve isocianato de 4-yodo-fenilo (100 mg, 0,41 mmol) en DCM (10 mL) que contiene TEA (0,057 mL, 0,4 mmol). Se añadió éster di t-butílico del ácido 2-[3-(5-amino-1-carboxi-pentil)-ureido]-pentanodioico (200 mg, 0,41 mmol) y se agitó durante 3 horas. La mezcla de reacción se evaporó hasta sequedad y la mezcla bruta se recogió en metanol (5 mL). La adición gota a gota de agua (20 mL) produjo un precipitado blanco que se recogió y se lavó con agua (20 mL) y se secó para producir el éster tri-terc-butílico deseado como un sólido blanco que se desprotegió directamente mediante el uso del método descrito anteriormente para producir el producto deseado (158 mg, 53 %) como un sólido blanco. 1H NMR (400 MHz, DMSO-cf6) 58,51 (s, 1H), 7,5 (d, 2H), 7,22 (d, 2H), 6,3 (t, 2H), 6,16 (t, 1H), 4,05 (m, 2H), 3,05 (m, 2H), 2,24 (m, 2H), 1,9 (m, 1H), 1,68 (m, 2H), 1,52 (m, 1H), 1,38 (m, 2H), 1,28 (m, 2H), (3 CO2H no visto). ESMS m/z: 565 (M+H)+.

Síntesis de Glu-Urea-P-Fenil Glicinas
Ácido (±)3-amino-3-(3-yodo-fenil)-propiónico (20).
Se suspendieron ácido malónico (2,2 g, 21,5 mmol) y 3-yodobenzaldehído (5 g, 21,5 mmol) en etanol (50 mL) y se añadió acetato de amonio (1,66 g, 21,5 mmol) y la reacción se calentó a reflujo durante toda la noche. La reacción se enfrió hasta temperatura ambiente, se filtró y se lavó con etanol seguido de éter y se secó para proporcionar el producto (3,4 g, 11,6 mmol, 54 %) como un sólido blanco. 1H NMR (400 MHz, DMSO-^) 57,80 (s, 1H), 7,64 (dd, J = 7,8 Hz, 1H), 7,42 (dd, J = 7,6 Hz, 1H), 7,16 (dd, J = 7,8 Hz, 1H), 7,14 (dd, J = 7,6 Hz, 1H), 4,21 (m, 1H), 2,36 (m, 2H).

Éster metílico del ácido (±)-3-Amino-3-(3-yodo-fenil)- propiónico (21).
A una suspensión de ácido (±)3-amino-3-(3-yodo-fenil)-propiónico (3,1 g, 10,6 mmol) en metanol se le añadió cloruro de tionilo (0,95 mL, 12,7 mmol) y la reacción se agitó a temperatura ambiente durante toda la noche. La concentración seguida de trituración con éter da un sólido blanco. El sólido se filtra, se lava con éter y se seca para proporcionar el producto deseado (3,5 g, 10 mmol, 95 %) como un sólido blanco. 1H NMR (400 MHz, DMSO-cf6) 5 8,79 (br, 2H), 8,01 (s, 1H), 7,74 (d, J = 8,1 Hz, 1H). 7,57 (d, J = 7,8 Hz, 1H), 7,21 (dd, J = 8,1,7,8 Hz, 1H), 4,56 (br, 1H), 3,54 (s, 3H), 3,23-3,17 (m, 1H), 3,04-2,98 (m, 1H).

(S, R) y éster di-terc-butílico del ácido (S, S)-2-{3-[1-(3-Iodo-fenil)-2-metoxicarbonil-etil]-ureido}-pentanodioico (22). Se disolvió éster di-terc-butílico del ácido 2-[(imidazol-1-carbonil)-amino]-pentanodioico (370 mg, 1,05 mmol) en DCE (10 mL) y se enfrió hasta 0 °C. Se añadió MeoTf (142 ^L, 1,25 mmol) y se dejó que la reacción prosiguiera durante 20 min. Se añadió éster metílico del ácido (±)3-amino-3-(3-yodo-fenil)-propiónico (356 mg, 1,045 mmol) y la reacción se dejó calentar hasta temperatura ambiente y luego se calentó hasta 55 °C y se agitó durante toda la noche. La reacción se diluyó con DCM (50 mL) y se lavó con agua (30 mL), 5 % de Ácido cítrico (30 mL), bicarbonato de sodio saturado (30 mL), agua (30 mL) y salmuera (30 mL). La capa orgánica se secó sobre sulfato de sodio y se concentró para proporcionar el producto bruto. El producto se purificó por cromatografía en columna para proporcionar el producto deseado (303 mg, 0,51 mmol, 49 %) como una espuma blanca. 1H NMR (400 MHz, CDCh) 57,66 (s, 1H), 7,57 (d, J = 7,6 Hz, 1H), 7,29 (s, 1H), 7,07-7,02 (m, 1H), 5,74 (br, 1H), 5,17 (br, 2H), 4,30 (m, 1H), 3,63 (s, 1.5H), 3,62 (s 1.5H), 2,88-2,76 (m, 2H), 2,38-2,24 (m, 2H), 2,10-2,00 (m, 1H), 1,90-1,80 (m, 1H), 1,46 (s, 9H), 1,44 (s, 9H).

(S, R) y ácido (S, S)-2-{3-[2-carboxi-1-(3-yodo-fenil)-etil]-ureido}-pentanodioico (23).
En una solución de éster di-terc-butílico del ácido (±)2-{3-[1-(3-yodo-fenil)-2-metoxicarbonil-etil]-ureido}-pentanodioico se disolvió (289 mg, 0,49 mmol) en metanol (3 mL) y LiOH 2 M (0,5 mL) y la reacción se agitó a temperatura ambiente durante toda la noche.
La reacción se diluyó con agua (20 mL) y la capa orgánica se extrajo con acetato de etilo (2 X 20 mL) y luego se acidificó con HCl 1 N a pH ~ 2. La capa acuosa se extrajo con acetato de etilo (3 x 20 mL), se secó sobre sulfato de sodio y se concentró para proporcionar el producto bruto (206 mg, 0,36 mmol, 73 %) como un sólido blanco. Al material bruto se le añadió dCm (2 mL) seguido de TFA (2 mL) y la reacción se agitó a temperatura ambiente durante toda la noche. La concentración seguida de recristalización desde acetato de etilo proporcionó el producto deseado (22 mg, 0,047 mmol, 10 %) como un sólido blanco. 1H NMR (400 MHz, DMSO-cfe) 512,39 (br, 3H), 7,64 (br, 1H), 7,56 (m, 1H), 7,30 (bm, 1H), 7,10 (bm, 1H), 6,72 (bm, 1H), 6,34 (bm, 1H), 4,94 (br, 1H), 4,03 (bm, 1H), 2,64 (br, 2H), 2,20 (br, 2H), 1,86 (br, 1H), 1,71 (br, 1H). ESMS m/z: 463 (M-H)+.

(S,S)-2-{3-[1-Carboxy-5-(2-cloro-bencilamino)-pentil]-ureido}-pentanodioico (7) (MIP-1137). El mismo procedimiento general que se muestra en el Esquema 1, mediante el uso de éster di-í-butílico del ácido 2-[3-(5-Amino-1-carboxipentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (100 mg, 45 %) como un sólido blanquecino. 1H NMR (400 MHz, DMSO-cfe) 59,0 (br, 3H), 7,63 (d, 1H), 7,2 (m, 2H), 7,15 (d, 1H), 6,30 (d, 2H), 4,1 (m, 4H), 2,9 (br, 2H), 2,2 (m, 2H), 1,90 (m, 2H), 1,60 (m, 4H), 1,35 (m, 2H). ESMS m/z: 444 (M+H)+.

(S,S)-2-{3-[1-Carboxy-5-(3-cloro-bencilamino)-pentil]-ureido}-pentanodioico (8) (MIP 1131). El mismo procedimiento general que se muestra en el Esquema 1, mediante el uso de éster di í-butílico del ácido 2-[3-(5-Amino-1-carboxipentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos previamente para proporcionar el producto deseado (200 mg, 90 %) como un sólido blanquecino. 1H NMR (400 MHz, DMSO-d6) 58,9 (br, 3H), 7,6 (s, H), 7,43 (m, 3H), 6,39 (br, 2H), 4,1 (m, 4H), 2,9 (br, 2H), 2,2 (m, 2H), 1,90 (m, 2H), 1,60 (m, 4H), 1,35 (m, 2H). ESMS m/z: 444 (M+H)+.

(S,S)-2-{3-[1-Carboxy-5-(4-cloro-bencilamino)-pentil]-ureido}-pentanodioico (9) (MIP 1135). El mismo procedimiento general que se muestra en el Esquema 1, mediante el uso de éster di í-butílico del ácido 2-[3-(5-Amino-1-carboxipentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (10 mg, 66 %) como un sólido blanquecino. ESMS m/z: 444 (M+H)+.

Ácido (S)-2-(3-((R)-5-(bencilamino)-1-carboxipentil)ureido)pentanodioico (10). (MIP-1106). El mismo procedimiento general que se muestra en el Esquema 1, mediante el uso de éster di f-butílico del ácido 2-[3-(5-Amino-1-carboxipentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (5 mg, 47 %) como un sólido blanquecino. ESMS m/z: 410 (M+H)+.
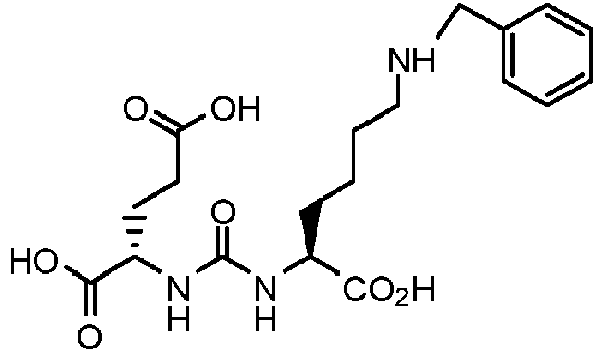
Ácido 2-(3-{1-carboxi-5-[3-(fenil)-ureido]-pentil}-ureido)-pentanodioico (11) (MIP 1111).
En un matraz de fondo redondo se disolvió isocianato de fenilo (100 mg, 0,84 mmol) en DCM (10 mL) éster di fbutílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico (409 mg, 0,84 mmol) y se agitó durante 3 horas. La mezcla de reacción se evaporó hasta sequedad y la mezcla bruta se purificó mediante cromatografía en columna ultrarrápida 2:1 de hexanos/acetato de etilo para proporcionar el éster terc-butílico como un sólido blanco que se desprotegió mediante el uso de TFA/CH2Cl2 para obtener el producto deseado. 1H NMR (400 MHz, DMSO-cfe) 5 12,5 (s, 3H), 8,54 (s, 1H), 7,40 (dd, 2H), 7,26 (dd, 2H), 6,30 (t, 2H), 6,17 (t, 1H), 4,05 (m, 2H), 3,05 (m, 2H), 2,44 (m, 2H), 1,90 (m, 1H), 1,68 (m, 2H) 1,52 (m, 1H). 1,40 (m, 2H). 1,29 (m, 2H). ESMS m/z: 439 (M+H)+.

Ácido 2-(3-{1-carboxi-5-[3-(4-bromo-fenil)-ureido]-pentil}-ureido)-pentanodioico (12) (MIP 1129)
En un matraz de fondo redondo, se disolvió isocianato de 4-bromo-fenilo (100 mg, 0,50 mmol) en DCM (10 mL). Se añadió éster di f-butílico del ácido 2-[3-(5-amino-1-carboxi-pentil)-ureido]-pentanodioico (246 mg, 0,50 mmol) y se agitó durante 3 horas. La mezcla de reacción se evaporó hasta sequedad y la mezcla bruta se purificó mediante cromatografía en columna ultrarrápida 2:1 de hexanos/acetato de etilo para proporcionar el éster terc-butílico como un sólido blanco que se desprotegió mediante el uso de TFA/CH2Cl2 para obtener el producto deseado.
1H NMR (400 MHz, DMSO-cfe) 512,5 (s, 3H), 8,55 (s, 1H), 7,35 (d, 4H), 6,30 (t, 2H), 6,18 (t, 1H), 4,08 (m, 2H), 3,05 (m, 2H), 2,22 (m, 2H), 1,90 (m, 1H), 1,68 (m, 2H), 1,52 (m, 1H), 1,40 (m, 2H), 1,30 (m, 2H). ESMS m/z: 518 (M+H)+.

Ácido 2-(3-{1-carboxi-5-[3-(4-doro-fenil)-ureido]-pentil}-ureido)-pentanodioico (13) (MIP 1110)
En un matraz de fondo redondo, se disolvió isocianato de 4-cloro-fenilo (100 mg, 0,65 mmol) en DCM (10 mL), se añadió ácido 2-[3-(5-amino-1-carboxi-pentil)-ureido]-pentanodioico, éster di f-butílico (318 mg, 0,65 mmol) y se agitó durante 3 horas. La mezcla de reacción se evaporó hasta sequedad y la mezcla bruta se purificó mediante cromatografía en columna ultrarrápida 2:1 de hexanos/acetato de etilo para proporcionar el éster terc-butílico como un sólido blanco (470 mg, 96 %) que se desprotegió mediante el uso de TFA/CH2Cl2 que proporcionó el producto deseado
1H NMR (400 MHz, DMSO-^) 512,5 (s, 3H), 8,35 (s, 1H), 7,40 (dd, 2H), 7,19 (dd, 2H), 6,30 (t, 2H), 6,10 (t, 1H), 4,08 (m, 2H), 3,05 (m, 2H), 2,32 (m, 2H), 1,90 (m, 1H), 1,68 (m, 2H), 1,52 (m, 1H), 1,40 (m, 2H), 1,30 (m, 2H). ESMS m/z: 474 (M+H)+.

Ácido (S)-2-(3-((R)-1-carboxi-5-(22iridin22ne-1-ilmetilamino)pentil)ureido)pentanodioico. (14) (MIP-1108). El mismo procedimiento general que se muestra en el Esquema A, mediante el uso de éster di f -butílico del ácido 2-[3-(5 -Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos previamente para proporcionar el producto deseado (51 mg, 70 %) como un sólido blanquecino. 1H NMR (400 MHz, DMSO-cfe) 58,9 (br, 3H), 7,95 (m, 5H), 7,6 (m, 2H), 6,35 (br, 2H), 4,1 (m, 4H), 2,9 (br, 2H), 2,55 (m, 2H), 2,25 (m, 2H), 1,70 (m, 4H), 1,3 (m, 2H). ESMS m/z: 460 (M+H)+.

Ácido 2-(3-{1-carboxi-5-[3-(3-yodo-bencil)-ureido]-pentil}-ureido)-pentanodioico (15)
(MIP-1101). El mismo procedimiento general que se muestra en el Esquema 2, mediante el uso de éster di f-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado. ESMS m/z: 579 (M+H)+.

Ácido (19S,23S)-2-(4-yodobencil)-1-(4-yodofenil)-13,21-dioxo-2,14,20,22-tetraazapentacosano-19,23,25-tricarboxílico (16) (MIP-1130). El mismo procedimiento general que se muestra en el Esquema A, mediante el uso de éster di t -butílico del ácido 2-[3-(5 - Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos previamente para producir el producto deseado (8,3 mg, 10 %) como un sólido blanquecino. 1H NMR (400 MHz, d Ms O-cí6) 57,8 (d), 7,3 (d), 6,3 (dd), 4,25 (br), 4,05 (m), 2,97 (m), 2,85 (br), 2,22 (m), 2,05 (m), ,90 (m), 1,64 (m), 1,48 (m), 1,35 (m), 1,2 (m). ESMS m/z: 936 (M+H)+.

Experimentación general con Renio:
Los complejos de renio de los inhibidores de SAAC se aíslan convenientemente de las reacciones del precursor fácilmente disponible [Net4]2[Re(CO)3Br3] con el inhibidor de SAAC. Dado que los conjuntos de donantes provistos por el terminal SAAC están bien documentados como quelantes efectivos para el núcleo {M(CO)3}+1 y han sido diseñados para adoptar la disposición facial requerida sobre el sitio del metal, las preparaciones de los complejos no fueron excepcionales.
El sistema {Re(I)(CO)3}+ siguió una química de reacción similar a la del núcleo de tricarbonilo Tc-99m. El uso de [Net4]2[ReBr3(CO)3], como material de partida, facilitó la formación del núcleo fac-{Re(CO)3(L)3}. El [Net4]2[ReBr3(CO)3] se derivó fácilmente del [ReBr(CO)5]. La síntesis de los complejos Re(I) se realizó haciendo reaccionar [Net4]2[ReBr3(CO)3] con el ligando TEC apropiado en una proporción de 1:1,2 en 10 mL de metanol. La reacción se dejó calentar a 80 °C durante 4 horas. Después de enfriar, todos los siguientes productos de reacción se purificaron mediante el uso de una pequeña columna de sílice con rendimientos que oscilaban entre el 10 y el 30 %.

Glu-urea-Lys-PEG2-ReDP:
Ácido [Re(CO)3{(17R,21S)-11,19-dioxo-1-(24iridin-2-il)-2-(24iridin-2-ilmetil)-5,8-dioxa-2,12,18,20-tetraazatricosano-17,21,23-tricarboxílico}][Br]. (17) (MIP-1133). El compuesto de dipiridilo PEG2, ácido (17R,21S)-11,19-dioxo-1-(24iridin-2-il)-2-(24iridin-2-ilmetil)-5,8-dioxa-2,12,18,20 -tetraazatricosano-17,21,23-tricarboxílico se preparó mediante el mismo procedimiento general que se muestra en el Esquema 1, mediante el uso del éster di f-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El complejo de éster de renio se preparó mediante el mismo procedimiento que se describe en el experimento general de renio. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (2 mg, 20 %) como un sólido blanquecino. 1H NMR (400 MHz, DMSO-da) 58,8 (d), 8,00 (dd), 7,55 (d), 7,42 (dd), 6,45 (s), 3,95 (m), 3,4-3,6 (m), 2,45 (m), 1,25 (m), 1,1 (m), 0,8 (m). ESMS m/z: 931 (M+H)+.
Glu-urea-Lys-PEG4-ReDP:
Ácido[Re(CO)s{(23R,27S)-17,25-dioxo-1-(piridin-2-il)-2-(piridin-2-ilmetil)-5,8,11,14-tetraoxa-2,18,24,26-tetraazanonacosano-23,27,29-tricarboxílico}][Br]. (18) (KM11-200). El compuesto de dipiridilo PEG4 ácido (23R,27S)-17,25-dioxo-1-(piridin-2-il)-2-(piridin-2-ilmetil)-5,8,11,14-tetraoxa-2,18,24,26-tetraazanonacosano-23,27,29-tricarboxílico se preparó mediante el mismo procedimiento general que se muestra en el Esquema A, mediante el uso del éster di f-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El complejo de éster de renio se preparó mediante el mismo procedimiento que se describe en el experimento general de renio. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado. (5,1 mg, 29,6%) como un sólido blanco. ESMS m/z: 1019 (M+H)+.
Glu-urea-Lys-PEG8-ReDP:
Ácido [Re(CO)s{(35R,39S)-29,37-dioxo-1-(25iridin-2-il)-2-(25iridin-2-ilmetil)-5,8,11,14,17,20,23,26-octaoxa-2,30,36,38-tetraazahentetracontano-35,39,41-tricarboxílico}][Br]. (19) (MIP-1132). El compuesto de dipiridilo PEG8, ácido (35R,39S)-29,37-dioxo-1-(piridin-2-il)-2-(piridin-2-ilmetil)-5,8,11,14,17,20,23,26-octaoxa-2,30,36,38-tetraazahentetracontane-35,39,41-tricarboxílico se preparó mediante el mismo procedimiento general que se muestra en el Esquema A, mediante el uso del éster di f-butílico del ácido 2-[3-(5-Amino -1-carboxi-pentil)-ureido]-pentanodioico. El complejo de éster de renio se preparó mediante el mismo procedimiento que se describe en el experimento general de renio. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (8,0 mg, 30,4 %) como un sólido blanco. ESMS m/z: 1195 (M+H)+.
Glu-urea-Lys-C11PAMA-Re:
Ácido [Re(CO)3{(19R,23S)-13,21-dioxo-2-(25iridin-2-ilmetil)-2,14,20,22-tetraazapentacosano-1,19,23,25-tetracarboxílico}] (20) (MIP-1109). Se preparó el compuesto C11-PAMA, ácido (19R,23S)-13,21-dioxo-2-(25iridin-2-ilmetil)-2,14,20,22-tetraazapentacosano-1,19,23,25-tetracarboxílico mediante el mismo procedimiento general que se muestra en el Esquema A, mediante el uso del éster di f-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El complejo de éster de renio se preparó mediante el mismo procedimiento que se describe en el experimento general de renio. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente para proporcionar el producto deseado (3,0 mg, 75 %) como un sólido blanquecino. ESMS m/z: 922 (M+H)+.

X = I, Br, Cl, F, H
La Tabla 1 más abajo es un resumen de los inhibidores de PSMA sintetizados investigados.
Tabla 1

Tabla 1. Resumen de los datos de unión celular in vitro de los derivados de Glu-Urea-Lis adicionales o reensayados. Análogos de p-aminoácidos
Análogos de p-aminoácidos de MIP-1072, MIP-1095, MIP-1027 específicamente, pero es muy conveniente la extensión a otros análogos tales como los conjugados de tecnecio, así como otros análogos de halógeno. Nosotros no tenemos nuevos ejemplos para apoyar esta afirmación en este momento.
Análogos de p-aminoácido MIP-1072

Las propiedades del tecnecio y del renio, metales del Grupo VII, son muy similares debido a su relación periódica. Se anticipó que los metales demostrarían una química de reacción similar, que suele ser el caso de la química de tricarbonilo, nitrógeno y tiazol de estos dos metales. Asimismo, debido a su tamaño similar que estabiliza la configuración de espín apareado los electrones d6 de M(I), el perhenato y el pertechnetato tienen comportamientos de reacción muy similares. La síntesis de los TEC de renio nos permitió una ruta fácil para caracterizar estructuralmente los productos. La relación periódica entre Tc y Re indica que los radiofármacos Tc-99m pueden diseñarse mediante modelación de complejos análogos de renio.
Algunos de los nuevos compuestos se sintetizaron con cantidades macroscópicas de renio para su caracterización mediante métodos convencionales, incluida la espectrometría de masas y la espectrometría de 1H y 13C NMR. Después de la purificación, los complejos de renio sintetizados se pasaron por una columna de HpLC para la
purificación y la identificación de los tiempos de retención para compararlos con los productos de reacción de Tc. Los complejos de renio-TEC también se cristalizaron.
Los complejos de renio de los inhibidores de SAAC se aíslan convenientemente de las reacciones de los precursores fácilmente disponibles {Re(CO)3(H2O)3}+1 y [Net4]2[Re(CO)3Br3] con el inhibidor de SAAC. Dado que los conjuntos de donantes provistos por el terminal SAAC están bien documentados como quelantes efectivos para el núcleo {M(CO)3}+1 y han sido diseñados para adoptar la disposición facial requerida sobre el sitio del metal, las preparaciones de los complejos no fueron excepcionales.
Experimentación General
El sistema {Re(I)(CO)3}+ siguió una química de reacción similar a la del núcleo de tricarbonilo Tc-99m. El uso de [Net4]2[ReBr3(CO)3], como material de partida, facilitó la formación del núcleo fac-{Re(CO)3(L)3}. El [Net4]2[ReBr3(CO)3] se derivó fácilmente del [ReBr(CO)5]. La síntesis de los complejos Re(I) se realizó haciendo reaccionar [Net4]2[ReBr3(CO)3] con el ligando TEC apropiado en una proporción de 1:1,2 en 10 mL de metanol. La reacción se dejó calentar a 80 °C durante 4 horas. Después de enfriar, todos los siguientes productos de reacción se purificaron mediante el uso de una pequeña columna de sílice con rendimientos que oscilaban entre el 10 y el 30 %. Éster [Re(CO)3(2-{3-[3-(Bis-piridin-2-ilmetil-amino)-1-carboxi-propil]-ureido}-pentanodietilo)] [Br] (24). 1H NMR (400 MHz, DMSO-CÍ6) 58,65 (dd, 2H), 7,85 (dd, 2H), 7,7 (dd, 4H), 7,25 (dd, 2H), 6,42 (dd, 1H), 6,0 (dd, 1H), 4,5 (m, 2H), 4,16 (m, 2H), 3,80 (m, 4H), 2,45 (m, 2H), 2,0 (dd, 2H), 1,5 (m, 4H), 1,25 (m, 6H). ESMS m/z: 812-815.

Ácido [Re(CO)3(ácido 2-{3-[5-(Bis-piridin-2-ilmetil-amino)-1-carboxi-pentil]-ureido}-pentanodioico)] [Br] (25) (MIP 1029). 1H NMR (400 MHz, DMSO-^) 512,6 (s, 2H), 8,91 (s, 1H), 8,63 (dd, 2H), 7,85 (dd, 2H), 7,75 (dd, 4H), 7,3 (dd, 2H), 6,44 (d, H), 6,28 (d, 1H), 4,45 (s, 2H), 4,10 (m, 2H), 3,15 (s, 1H), 2,60 (m, 2H), 2,25 (m, 2H), 1,90 (m, 1H), 1,78 (m, 2H), 1,45 (m, 2H). ESMS m/z: 770-774.

Ácido 2-{3-[1-carboxi-5-(carboximetil-piridin-2-ilmetil-amino)-pentil]-ureido}-pentanodioico (26). El mismo procedimiento general, mediante el uso del éster di í-butílico del ácido 2-[3-(5-Amino-1-carboxi-pentil)-ureido]-pentanodioico previamente preparado y protegido. El compuesto se desprotegió mediante el uso de los métodos descritos anteriormente (2,2 mg, 65 %). 1H NMR (400 MHz, DMSO-CÍ6) 58,65 (d, 1H), 7,91 (dd, 1H), 7,56 (d, 1H), 7,45 (dd, 1H), 6,31 (m, 2H), 4,34 (s, 2H), 4,08 (m, 4H), 3,10 (m, 2H), 2,24 (m, 2H), 1,95 (m, 1H), 1,68 (m, 4H), 1,5 (m, 1H), 1,22 (m, 2H). ESMS m/z: 469 (M+H)+.
M+1469.
Ácido [Re(CO)3(2-{3-[1-carboxi-5-(carboximetil-piridin-2-ilmetil-amino)-pentil]-ureido}-pentanodioico)] (27). 1H NMR (400 MHz, DMSO-CÍ6) 58,75 (d, 1H), 8,13 (dd, 1H), 7,69 (d, 1H), 7,57 (dd, 1H), 6,45 (m, 2H), 4,75 (m, 1H), 4,50 (m, 1H), 4,20 (m, 2H), 3,61 (m, 4H), 3,15 (m, 2H), 2,38 (m, 1H), 2,0 (m, 2H), 1,75 (m, 4H), 1,62 (m, 1H), 1,25 (m, 2H).
ESMS m/z 779-782 (M+2Na)+.

Síntesis de análogos de Glu-Urea-Lis(N -bencil-X) (3).
Los compuestos de la estructura general 3 se prepararon con rendimientos generales que oscilaban entre el 20 y el 40 % mediante el uso de la ruta general representada en el Esquema A. El intermediario sintético clave (1) se hizo reaccionar con el aldehído apropiado a temperatura ambiente durante una hora para formar la base intermedia 30iridi. La base 30iridi no se aisló, pero se redujo in situ con triacetoxiborohidruro de sodio. Los grupos protectores de éster t-butílico se eliminaron mediante el uso de TFA al 50 % en DCM durante 1 hora a temperatura ambiente. Una vez completada la desprotección, las reacciones se concentraron en un evaporador rotatorio y se purificaron mediante Hp Lc o cromatografía ultrarrápida para proporcionar los productos deseados (3) con un rendimiento del 40-80 %.

Síntesis de análogos de Glu-Urea-Ureido(Fenil-X)
Los siguientes compuestos de la estructura general 8 se prepararon con rendimientos generales que oscilaban entre el 20 y el 60 % mediante la ruta representada en el Esquema B. El intermediario sintético clave (4) se hizo reaccionar con el fenilisocianato apropiado a temperatura ambiente para proporcionar los intermediarios protegidos deseados (5) en buenos rendimientos. Los grupos protectores de éster t-butílico se eliminaron en presencia de TFA al 50 % en DCM durante 1 hora a temperatura ambiente. Una vez completadas, las reacciones se concentraron en un evaporador rotatorio y se purificaron por HPLC o por recristalización para proporcionar los productos deseados (6) con un rendimiento del 40-90 %.

Preparación y Caracterización de los Complejos Radiomarcados
Etiquetado de tecnecio-99m
La preparación de los complejos marcados con 99mTc se logró mediante la adición de 100 |jl de una solución que contenía [99m Tc(CO)3(H2O)3]+ a 500 j l de soluciones 10-4 M del inhibidor-SAAC. Las mezclas se calentaron a 70 °C durante 30 min. Se analizó la pureza radioquímica de los productos mediante HPLC de fase inversa.
La estabilidad de los compuestos radiomarcados en solución y en suero se determinó en función del tiempo y de las condiciones de solución. Específicamente, después del radiomarcaje y el aislamiento, el producto se almacenó a temperatura ambiente durante 6 h, después de lo cual se realizó un análisis de HPLC para verificar el grado de retención de la etiqueta, así como la posible degradación del producto. Se analizó la reformación de TcO4" y la presencia del material reducido TcO2.
Para ayudar a predecir la estabilidad in vivo, se realizaron retos de ligando. Específicamente, las estabilidades de los complejos de 99mTc se investigaron mediante incubación de los complejos purificados por HPLC en suero de ratón al 5 % a temperatura ambiente y 37 °C. La capacidad de los ligandos competidores, como la cisteína y el DTPA, para extraer Tc-99m de los complejos se estudió mediante incubación de los complejos purificados con soluciones (PBS pH 7,2) que contenían ligandos competidores en concentraciones finales de 0,1 M.
Los resultados de los estudios de competencia de marcaje no demostraron degradación de los complejos de Tc-99m hasta las 6 horas en el suero o en el estudio de ligandos competidores. Los resultados de la incubación a 37 °C después de 6 horas se muestran en la Figura 2.
Yodaciones de DCT
La preparación del compuesto marcado con yodo-131 N-[N-[(S)-1,3-dicarboxipropil]carbamoil]-S-3-yodo-L-tirosina (l-131-DClT) se logró mediante la adición de 100 ul de [1 -l3 l] Nal en NaOH 0,1 N a una solución de PBS (pH 7,2) que contiene DCT (1 mg/mL) en un tubo lodogen™ (Fisher Scientific, Pierce). La mezcla se agitó durante 3 minutos y se almacenó a temperatura ambiente durante 20 minutos.
La estabilidad del compuesto radiomarcado en solución se determinó en función del tiempo. Específicamente, después del radiomarcaje y aislamiento, el producto se almacenó a temperatura ambiente durante 48 horas, después de lo cual se realizó un análisis por HPLC para comprobar el grado de retención del marcador, así como la posible degradación del producto. Se analizó la reformación de Nal y la presencia de los yodatos reducidos. Los resultados del estudio de estabilidad del marcaje no demostraron una degradación significativa del I-131 DCIT durante 2 días a temperatura ambiente. Los resultados del estudio se muestran en la Figura 3.
La preparación del ácido 2-{3-[1-carboxi-5-(4-yodo-benzoilamino)-pentil]-ureido}-pentanodioico marcado con yodo-131 (M31-MIP 1072) se logró mediante la adición de 100 ul de [I-131] Nal en NaOH 0,1 N con 30 j l de metanol con ácido acético al 0,5 % a una solución de PBS (pH 7,2) que contiene MIP 1072 (1 mg/mL) en un TUBO IODOGEN (Fisher Scientific). La mezcla se agitó durante 3 minutos y se almacenó a temperatura ambiente durante 20 minutos. La estabilidad del compuesto radiomarcado en solución se determinó en función del tiempo. Específicamente, después del radiomarcaje y el aislamiento, el producto se almacenó a 37 °C durante 3 días, luego de lo cual se realizó un análisis por HPLC para verificar el grado de retención de la etiqueta, así como la posible degradación del producto. Se analizó la reformación de Nal y la presencia de los yodatos reducidos. Los resultados del estudio de estabilidad de marcaje no demostraron una degradación significativa del l-131 1072 a los 3 días a temperatura ambiente en DMSO, etanol al 10 %/solución salina, PBS pH 7,2 y ascorbato al 6 %/solución de ácido gentísico al 3 %. Los resultados del estudio se muestran en la Figura 4.
Caracterización biológica de conjugados SAAC-urea-glutamato
Los conjugados SAAC-urea-Glu recién preparados se seleccionaron en un ensayo de unión a células de cáncer de próstata humano mediante el uso de células PSMA positivas, células LnCap, y PSMA negativas, células PC3. Los compuestos que demuestren captación específica o unión a células positivas para PSMA se estudiarán para la localización de tumores in vivo.
Ensayos de detección en frío in vitro versus 1-131 DClT. Las células de cáncer de próstata humano LNCaP y PC3 se obtuvieron de American Type Culture Collection, Rockville, MD. Las células LNCaP se mantuvieron en medio RPMl-1640 suplementado con suero fetal bovino (FBS) al 10%. Las células PC3 se cultivaron en medio F12K suplementado con FBS al 10%. La unión del compuesto radiomarcado y la competencia con derivados fríos a las células LNCaP y PC-3 se realizó de acuerdo con los métodos de Tang y otros (Tang, H.; Brown, M.; Ye, Y.; Huang, G.; Zhang, Y.; Wang, Y.; Zhai, H.; Chen, X.; Shen, T.Y.; Tenniswood, M., Prostate targeting ligands based on N-acetylated alpha-linked acidic dipeptidase, Biochem. Biophys. Res. Commun. 2003, 307, 8-14) con las modificaciones apropiadas. Las células se sembraron en placas de 12 pocillos a aproximadamente 4 x 105 células/pocillo y se incubaron durante 48 horas en una incubadora humidificada a 37 °C/5 % de dióxido de carbono antes de la adición del compuesto. Cada conjugado único de SAAC-urea-Glu se preparó y se diluyó en medio de
cultivo celular sin suero que contenía albúmina de suero bovino (BSA) al 0,5 % en combinación con I-131 DCIT 3 nM (inhibidor conocido). La unión total se determinó mediante incubación de 1-131 DCIT sin compuesto de prueba. Las placas se incubaron a temperatura ambiente durante 1 hora. Las células se retiraron de las placas mediante pipeteo suave y se transfirieron a tubos eppendorff. Las muestras se microcentrifugaron durante 15 segundos a 10K x g. El medio se aspiró y el sedimento se lavó dos veces mediante dispersión en medio de ensayo fresco seguido de microcentrifugación. La unión celular de I-131 DCIT se determinó mediante conteo del sedimento celular en un contador gamma automatizado. La unión no específica se determinó como los recuentos asociados con las células después de incubar con compuesto no radiomarcado 2 uM o ácido 2-fosfonometil-pentanodioico (PMPA). Los compuestos de control se representan a continuación.

Los dos compuestos clave para los ensayos de unión se muestran arriba: el I-DCIT (Kozikowski y otros) y el ácido 2-fosfonometil-pentanodioico (PMPA -derecha), un potente inhibidor con ICso = 6 nM.
(ii) Cribado de dosis in vitro.
I-131 DCIT se unió específicamente a las células LnCap y no a las células PC3, como es evidente por los recuentos desplazables por el compuesto no radiomarcado o PMPa solo en células LnCap (Figura 5). Las constantes de unión se determinaron mediante incubación de células LnCap con diversas cantidades de DCIT no radiomarcado en presencia de una cantidad constante de I-131 DCIT y mediante división por la actividad específica de cada solución para determinar el número de fmoles del compuesto unido (Figura 6). Se determinó que Kd era 264 nM y Bmáx era 254 fmoles. Los compuestos MIP-1008 y MIP-1033 que a 2 uM competían con I-131 DCIT por unirse a células LnCap, se volvieron a analizar a varias dosis para determinar los valores de IC-50 (Figuras 7 y 8). Mientras que MIP-1072, MIP-1095 y MIP-1097 mostraron valores de IC50 <50 nm, los compuestos MIP-1008 y MiP-1033 exhibieron IC-50 de 98 nM y 497 nM, respectivamente. Los compuestos MIP-1025, MIP-1028 y MIP-1029 no compitieron por la unión (Tabla 1).
Con el fin de confirmar los resultados del análisis de Scatchard de la figura 7 que indica la internalización de MIP-1072 en células LNCaP, se monitoreó la tasa de captación de MIP-1072 en células LNCaP. Cada pocillo se dosificó con 100 nM de MIP-1072 (2 uCi/pocillo) a 4 °C y 37 °C. La unión a PSMA alcanzó el equilibrio después de 15 min como lo demuestra la meseta en la curva a 4 °C. Las células incubadas a 37 °C continuaron la internalización de MIP-1072 después de alcanzar el equilibrio. Este resultado, Figura 10, confirma el Scatchard e indica que MIP-1072 está realmente internalizado.
(iii) Ensayo experimental de microsomas
Se añadieron microsomas de hígado de rata macho agrupados (1 mg/mL, BD Biosciences), sistema de regeneración NADPH (NADP 1,3 mM, glucosa 6-fosfato 3,3 mM y glucosa 6-fosfato deshidrogenasa 0,4 U/mL, BD Biosciences) y compuesto de prueba (MIP -1072 50 pM, DCT 50 pM y fenacetina 100 j M) a tampón de fosfato de potasio 0,1 M (pH 7,4) con el fin de controlar la degradación catastrófica de los compuestos de prueba. La mezcla se incubó a 37 °C y en el tiempo indicado (0, 15, 60 min) se detuvo la reacción mediante la adición de un volumen igual de metanol enfriado con hielo (500 j L). A continuación, la suspensión resultante se centrifugó a 21 000xG durante 10 min y el sobrenadante se recogió y se inyectó en un LCMS modelo MSD SL de Agilent mediante el uso de una proporción de agua:acetonitrilo 95:5 (con ácido fórmico al 0,1 %) a una proporción de agua:acetonitrilo 40:60 gradiente (con ácido fórmico al 0,1 %) y monitoreo del ion original solo en modo de ion único. Los resultados, que se muestran en las Figuras 11A y 11B, se expresan como degradación del ion original con respecto al punto de tiempo de 0 min.
La estabilidad de MIP-1072 se evaluó mediante el uso de microsomas de hígado de rata. MIP-1072 (50 j M) y fenacetina (100 j M) se incubaron con microsomas de hígado de rata a 37 °C durante el tiempo indicado. La fenacetina se usó como sustancia de control que se sabe que se metaboliza. MIP-1072 no fue degradado por los microsomas de hígado de rata durante el período de incubación. Sin embargo, la fenacetina se degradó en un 22 % después de una incubación de 60 min.
El compuesto principal, MIP 1072, se marcó con I-131 para estudios de distribución en tejidos en ratones con tumores implantados LNCaP (PSMA positivo) y PC3 (PSMA negativo). El compuesto se radiomarcó por la ruta que se muestra más abajo.

Los resultados de la biodistribución tisular coincidieron con los datos in vitro y demostraron una captación significativa en los tumores LNCaP (PSMA positivos). Los resultados también mostraron un alto grado de especificidad con muy poca actividad en los tumores PC3 (PSMA negativos). Más abajo se muestra un gráfico que representa la distribución de ratones (Figura 12).
La evaluación biológica mediante el uso de N-[N-[(S)-1,3-dicarboxipropil]carbamoil]-S-3-yodo-L-tirosina (I-131-DCIT) versus complejos "fríos" demostró ser una primera exploración rápida, seguida de curvas de dosis para determinar valores precisos de ICso. La serie principal de compuestos que exhibieron valores IC50 < 50 nM. Los datos in vivo de la serie principal demostraron una alta afinidad, con un 3 % de ID/g acumulada en los tumores LNCaP, y una alta especificidad con una proporción de LNCaP a PC3 superior a 15 a 1.
Protocolo de lisis celular LNCaP
2 matraces T75 confluentes
Lavar las células de la placa al pipetear hacia arriba y hacia abajo con los medios.
Lavar con sacarosa 0,32 M, volver a centrifugar
Volver a suspender el sedimento celular en 1 mL de Tris-HCl 50 mM, pH 7,4, Triton X-100 al 0,5 %.
Centrifugar a 14000 rpm durante 1 min para precipitar los núcleos
Retirar el sobrenadante y dividir en alícuotas de 50 ul
Almacenar a -80 °C
Ensayo de proteínas:
Bio-Rad Proteína Estándar II - 1,44 mg/mL
Dado que se usa detergente en la etapa de lisis, preparar el reactivo de trabajo A' mediante adición de 20 ul de reactivo S a cada 1 mL de reactivo A que se necesitará para la ejecución. (Si se forma un precipitado, calentar y agitar)
Preparar 5 diluciones de proteínas - 0, 0,2, 0,4, 0,8, 1,6 mg/mL
Preparar también diluciones 1/10, 1/100 y 1/1000 de lo desconocido.
Combinar 25 ^l de estándar/desconocido, 100 ^l de A', 800 ^l de reactivo B por duplicado. Mezclar
Después de ~15 min, medir la absorbancia a 750 nM
Ensayo de NAALADasa:
Tampón Rxn: Tris-HCl 50 mM, pH 7,4, CoCl220 mM, NaCl 32 mM
Preparar NAAG frío (solución madre 100 mM) diluir 1/100 en tampón Rxn para 1 mM
Combinar 600 ul de tampón y lisado de células LNCaP (200 ^g) Preincubar a 37 °C durante 3 min
Preincubar el tampón Rxn y el lisado de células LNCaP durante 3 min a 37 °C
Agregar 6 ^l de NAAG 1 mM (para una concentración final de 1 ^M) enriquecido con 1 000000 CPM de 3H-NAAG (100 ^l de NAAG 1 mM 10 ^l de 3H-NAAG (10 ^Ci)). Para competencia agregar PMPA.
Incubar durante 30 minutos
En el momento indicado, detener la reacción y retirar 100 ul de la mezcla de reacción y agregar un volumen igual de KH2PO40,25 M enfriado con hielo, pH 4,3 para detener la reacción.
Aplicar la mitad de la mezcla a una columna de intercambio catiónico AG 50W-X4 de 250 mg (malla 200-400, forma H+, hinche la resina con DI H2O antes de usar). Guardar la otra mitad para contar.
Lavar la columna con 500 ^L 1:1 Tampón de reacción /0.25M KH2PO4
Eluir con KCl 3M (1,5 mL)
Contar 100 ul de la carga, elución y reacción (diluida 1:6) para minimizar la inactivación
Notas:
Tiempo = 0 valores de control se restarán de los puntos de tiempo experimentales
Resultados expresados como pmol de 3H-glutamato formado/min/mg de proteína
Grant dice que se aumente solo 10 minutos para garantizar la linealidad, aunque Luthi-Carter, y otros (J Pharm Exp
Therap 1998 286(2)) dice que 2 horas todavía no hacen efecto sobre la linealidad y menos del 20 % del sustrato consumido
Tratamientos Terapéuticos
Los compuestos de la presente pueden usarse para inhibir la NAALADasa para tratamientos terapéuticos. Las enfermedades que podrían ser receptivas al tratamiento con NAALADasa incluyen neuropatía diabética sensorial y dolorosa, daño neuronal y cáncer de próstata, esquizofrenia, cáncer colorrectal, inflamación, esclerosis lateral amiotrófica o neuropatía diabética. Los presentes compuestos también pueden usarse como analgésicos. Puede encontrarse orientación para el modelado de tales tratamientos terapéuticos en Goodman & Gilman's The Pharmacological Basis of Therapeutics, McGraw Hill, 10 edition, 2001, Pharmaceutical Preformulation and Formulation: A Practical Guide from Candidate Drug Selection to Commercial Dosage Form, CRC, 2001 y Handbook of Pharmaceutical Excipients, AphA Publications, 5 edition, 2005.
Unión competitiva de análogos (Figura 16)
La capacidad de los análogos no radiactivos para competir con 131I-DCIT para unirse a PSMA se probó en la línea celular de cáncer de próstata humano positivo para PSMA, células LNCaP. Se incubaron células LNCaP (300 000 células/pocillo) durante 1 hora con [131I]-DCIT 3 nM en presencia de MIP-1072 1-10 000 nM en medio RPMI-1640 suplementado con albúmina sérica bovina al 0,5 %, luego se lavaron y se contaron en un contador gamma. Todos los documentos citados en esta especificación, incluidas las solicitudes de patentes, se incorporan por referencia en su totalidad.
Unión directa e internalización de MIP-1072
Se examinó la unión directa de 123I-MIP-1072 a células de cáncer de próstata (Figura 17). Las células LNCaP, o la línea celular negativa para PSMA, células PC3, se incubaron en medio RPMI-1640 suplementado con albúmina de suero bovino al 0,5 % durante 1 hora con 123I-MIP-10723 nM solo, o en presencia de MIP-1072 sin marcar 10 pM, o 10 pM de ácido 2-(fosfonometil)-pentanodioico (PMPA), un inhibidor de la NAALADasa estructuralmente no relacionado. Las células se lavaron y se contaron en un contador gamma.
La constante de afinidad (Kd) de MIP-1072 se determinó mediante análisis de unión por saturación (Figura 18). Las células LNCaP se incubaron durante 1 hora con 131I-MIP-1072 30-100 000 pM en HBS (Hepes 50 mM, pH 7,5, cloruro de sodio al 0,9 %) a 4 °C o 37 °C en ausencia o presencia de 10 pM de MIP-1072 sin marcar (para determinar la unión no específica). A continuación, se lavaron las células y se midió la cantidad de radiactividad en un contador gamma. La unión específica se calculó como la diferencia entre la unión total y la unión no específica. La constante de afinidad (Kd) de la interacción de MIP-1072 con PSMA en células LNCaP se determinó mediante un análisis de unión por saturación realizado mediante la titulación de 123I-MIP-1072 (3 pM-1000 nM) en presencia y ausencia de un exceso de MIP-1072 no radiomarcado (10 pM). Se determinó una Kd de 4,8 nM y Bmáx de 1490 fmoles/106 células a 4 °C mediante análisis de regresión no lineal mediante el uso del software Graph Pad Prism (Figura 18). Kd no fue significativamente diferente a 37 °C, 8,1 nM. Bmáx, sin embargo, fue mayor a 37 °C que a 4 °C; 1490 vs. 4400 fmol/106 células, respectivamente, lo que indica la internalización de MIP-1072. Los resultados de más abajo son representativos de dos análisis independientes.
La capacidad de MIP-1072 para internalizarse en células LNCaP se confirmó mediante un ensayo de lavado con ácido (Figura 19). Las células LNCaP se incubaron en HBS con 100 nM de 123I-MIP-1072 durante 0-2 horas a 4 y 37 °C. En el momento indicado se retiró el medio y las células se incubaron en tampón ácido suave (glicina 50 mM, NaCl 150 mM, pH 3,0) a 4 °C durante 5 minutos. Después de la breve incubación, las células se centrifugaron a 20000 x g durante 5 minutos. El sobrenadante y el sedimento celular se contaron en un contador gamma. Con el fin de confirmar los resultados del análisis de unión de saturación que indica la internalización de MIP-1072 en células LNCaP, nosotros monitoreamos la tasa de captación de MIP-1072 en células LNCaP. Cada pocillo se dosificó con 100 nM de MIP-1072 (2 uCi/pocillo) a 4 °C y 37 °C. La unión a PSMA alcanzó el equilibrio después de 15 min como lo demuestra la meseta en la curva a 4 °C. Las células incubadas a 37 °C continuaron la internalización de MIP-1072 después de alcanzar el equilibrio. Los resultados muestran un aumento dependiente del tiempo e insensible al ácido en la radiactividad asociada con el sedimento a 37 °C, pero no a 4 °C, lo que indica que el 123I-MIP-1072 se internaliza a 37 °C, pero no a 4 °C (Figura 19).
Captación tumoral y distribución tisular de 123I-MIP-1072
Se realizó un análisis cuantitativo de la distribución tisular de 123I-MIP-1072 en grupos separados de ratones macho NCr Nude_/' que portaban xenoinjertos LNCaP positivos para PSMA (aproximadamente 100-200 mm3) administrado a través de la vena de la cola como una inyección en bolo (aproximadamente 2 pCi/ratón) en un volumen constante de 0,05 mL. Los animales (n=5/punto de tiempo) fueron sacrificados por asfixia con dióxido de carbono a las 0,25, 1, 2, 4, 8 y 24 horas después de la inyección. Los tejidos (sangre, corazón, pulmones, hígado, bazo, riñones, glándulas suprarrenales, estómago, intestino grueso y delgado (con su contenido), testículos, músculo esquelético, hueso,
cerebro, tejido adiposo y tumor) se diseccionaron, se extirparon, se pesaron húmedos, se transfirieron a tubos de plástico y se contaron en un contador y automatizado (LKB Modelo 1282, Wallac Oy, Finlandia). Para comparar la captación de 123I-MIP-1072 en tumores LNCaP versus tumores PC3, y para demostrar que el compuesto estaba en el mecanismo a través de la competencia con ácido 2-(fosfonometil)-pentanodioico (PMPA), se pretrataron algunos ratones que portaban xenoinjertos LNCaP o PC3 con 50 mg/kg de PMPA 5 minutos antes de la inyección con 123I-MIP-1072 y los tejidos seleccionados se recogieron 1 hora después de la inyección. La captación y exposición de MIP-1072 fue mayor en el xenoinjerto de riñón y LNCaP que expresa altos niveles de PSMA. La absorción máxima en el riñón fue de 158 ± 46 % ID/g a las 2 horas y el xenoinjerto de LNCaP fue de 17 ± 6 % ID/g a 1 hora (Figura 20). La captación en estos tejidos diana fue rápida, mientras que el lavado fue más lento en el xenoinjerto LNCaP. Se demostró que 123I-MIP-1072 estaba en el mecanismo in vivo como lo evidenció la localización en tumores LNCaP que expresan PSMA, pero no en tumores PC3 que no expresan PSMA (Figura 21). Además, tanto el tumor como los riñones se bloquearon al tratar previamente a los ratones con PMPA, un potente inhibidor de PSMA.
Claims (3)
1. Un kit que comprende: (i) un compuesto que comprende un resto de unión a PSMA de glutamato-urea-lisina conjugado con un resto quelante de metales, y (ii) un radionúclido.
2. El kit de la reivindicación 1, en donde el radionúclido se selecciona de tecnecio-99m, renio-186, renio-188 o combinaciones de los mismos.
3. Uso del kit de la reivindicación 1 o la reivindicación 2 para la producción de un compuesto de unión a PSMA, que comprende la etapa de quelar el radionúclido (ii) con el resto quelante de metales del compuesto (i).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85749006P | 2006-11-08 | 2006-11-08 | |
| US87867807P | 2007-01-05 | 2007-01-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2926912T3 true ES2926912T3 (es) | 2022-10-31 |
Family
ID=39365345
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18150494T Active ES2847275T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
| ES15168948.6T Active ES2684322T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
| ES07844938.6T Active ES2547481T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
| ES19214298T Active ES2926912T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18150494T Active ES2847275T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
| ES15168948.6T Active ES2684322T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
| ES07844938.6T Active ES2547481T3 (es) | 2006-11-08 | 2007-11-07 | Heterodímeros de ácido glutámico |
Country Status (16)
| Country | Link |
|---|---|
| US (11) | US20080193381A1 (es) |
| EP (5) | EP3335736B1 (es) |
| JP (2) | JP5606737B2 (es) |
| CN (2) | CN103922998A (es) |
| BR (1) | BRPI0718700B8 (es) |
| CA (1) | CA2669127C (es) |
| CY (1) | CY1116610T1 (es) |
| DK (1) | DK2097111T3 (es) |
| ES (4) | ES2847275T3 (es) |
| HK (1) | HK1200164A1 (es) |
| HU (3) | HUE026216T2 (es) |
| PL (3) | PL2097111T3 (es) |
| PT (2) | PT3699162T (es) |
| SI (1) | SI2097111T1 (es) |
| TW (1) | TWI492761B (es) |
| WO (1) | WO2008058192A2 (es) |
Families Citing this family (93)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6528499B1 (en) | 2000-04-27 | 2003-03-04 | Georgetown University | Ligands for metabotropic glutamate receptors and inhibitors of NAALADase |
| US9492400B2 (en) | 2004-11-04 | 2016-11-15 | Massachusetts Institute Of Technology | Coated controlled release polymer particles as efficient oral delivery vehicles for biopharmaceuticals |
| EP1745802A1 (en) * | 2005-07-20 | 2007-01-24 | Kreatech Biotechnology B.V. | Method of conjugating therapeutic compounds to cell targeting moieties via metal complexes |
| US9267937B2 (en) | 2005-12-15 | 2016-02-23 | Massachusetts Institute Of Technology | System for screening particles |
| ES2776100T3 (es) | 2006-03-31 | 2020-07-29 | Massachusetts Inst Technology | Sistema para el suministro dirigido de agentes terapéuticos |
| WO2007150030A2 (en) | 2006-06-23 | 2007-12-27 | Massachusetts Institute Of Technology | Microfluidic synthesis of organic nanoparticles |
| ES2847275T3 (es) | 2006-11-08 | 2021-08-02 | Molecular Insight Pharm Inc | Heterodímeros de ácido glutámico |
| US9217129B2 (en) | 2007-02-09 | 2015-12-22 | Massachusetts Institute Of Technology | Oscillating cell culture bioreactor |
| EP2136788B1 (en) * | 2007-03-30 | 2011-10-26 | Bind Biosciences, Inc. | Cancer cell targeting using nanoparticles |
| WO2008124632A1 (en) | 2007-04-04 | 2008-10-16 | Massachusetts Institute Of Technology | Amphiphilic compound assisted nanoparticles for targeted delivery |
| CA2926573C (en) | 2007-06-26 | 2018-08-28 | The Johns Hopkins University | Labeled inhibitors of prostate specific membrane antigen (psma), biological evaluation, and use as imaging agents |
| EP4464384A3 (en) | 2007-08-17 | 2025-01-08 | Purdue Research Foundation | Psma binding ligand-linker conjugates and methods for using |
| EP2630966B1 (en) | 2007-10-12 | 2017-04-19 | Massachusetts Institute of Technology | Vaccine nanotechnology |
| CA2711678A1 (en) | 2008-01-09 | 2009-07-16 | Molecular Insight Pharmaceuticals, Inc. | Inhibitors of carbonic anhydrase ix |
| JP2012501965A (ja) * | 2008-06-16 | 2012-01-26 | バインド バイオサイエンシズ インコーポレイテッド | 薬剤を装填したポリマーナノ粒子及びその製造方法と使用方法 |
| WO2010005740A2 (en) * | 2008-06-16 | 2010-01-14 | Bind Biosciences, Inc. | Methods for the preparation of targeting agent functionalized diblock copolymers for use in fabrication of therapeutic targeted nanoparticles |
| US8613951B2 (en) * | 2008-06-16 | 2013-12-24 | Bind Therapeutics, Inc. | Therapeutic polymeric nanoparticles with mTor inhibitors and methods of making and using same |
| ES2721850T3 (es) | 2008-06-16 | 2019-08-05 | Pfizer | Nanopartículas poliméricas terapéuticas que comprenden alcaloides vinca y procedimientos de fabricación y uso de las mismas |
| RU2494096C2 (ru) * | 2008-08-01 | 2013-09-27 | Дзе Джонс Хопкинс Юниверсити | Агенты, связывающиеся с psma, и их применение |
| US8591905B2 (en) | 2008-10-12 | 2013-11-26 | The Brigham And Women's Hospital, Inc. | Nicotine immunonanotherapeutics |
| US8277812B2 (en) | 2008-10-12 | 2012-10-02 | Massachusetts Institute Of Technology | Immunonanotherapeutics that provide IgG humoral response without T-cell antigen |
| WO2010065906A2 (en) | 2008-12-05 | 2010-06-10 | Molecular Insight Pharmaceuticals, Inc. | Ca-ix specific radiopharmaceuticals for the treatment and imaging of cancer |
| WO2010065899A2 (en) | 2008-12-05 | 2010-06-10 | Molecular Insight Pharmaceuticals, Inc. | Technetium-and rhenium-bis(heteroaryl)complexes and methods of use thereof |
| TW201034691A (en) * | 2008-12-05 | 2010-10-01 | Molecular Insight Pharm Inc | Technetium-and rhenium-bis(heteroaryl) complexes and methods of use thereof |
| WO2010068866A2 (en) * | 2008-12-12 | 2010-06-17 | Bind Biosciences | Therapeutic particles suitable for parenteral administration and methods of making and using same |
| US20100216804A1 (en) * | 2008-12-15 | 2010-08-26 | Zale Stephen E | Long Circulating Nanoparticles for Sustained Release of Therapeutic Agents |
| AU2010249719A1 (en) | 2009-05-19 | 2012-05-31 | Aic Blab Company | Composite current collector and methods therefor |
| AU2010260195B2 (en) | 2009-06-15 | 2014-11-20 | Molecular Insight Pharmaceuticals, Inc. | Process for production of heterodimers of glutamic acid |
| US12083191B2 (en) | 2009-10-19 | 2024-09-10 | Case Western Reserve University | Composition and methods for imaging cells |
| EP2495043A4 (en) * | 2009-10-30 | 2014-04-23 | Sumitomo Osaka Cement Co Ltd | EXHAUST GAS CLEANER FOR A COMBUSTION ENGINE |
| US8357401B2 (en) | 2009-12-11 | 2013-01-22 | Bind Biosciences, Inc. | Stable formulations for lyophilizing therapeutic particles |
| ES2780156T3 (es) | 2009-12-15 | 2020-08-24 | Pfizer | Composiciones terapéuticas de nanopartículas poliméricas con alta temperatura de transición vítrea o copolímeros de alto peso molecular |
| EP2338892A1 (en) | 2009-12-18 | 2011-06-29 | Bayer Schering Pharma Aktiengesellschaft | Prostate specific membrane antigen inhibitors |
| US9951324B2 (en) | 2010-02-25 | 2018-04-24 | Purdue Research Foundation | PSMA binding ligand-linker conjugates and methods for using |
| JP2011206049A (ja) | 2010-03-08 | 2011-10-20 | Sumio Sugano | 壊死マーカー及びその用途 |
| US10363309B2 (en) | 2011-02-04 | 2019-07-30 | Case Western Reserve University | Targeted nanoparticle conjugates |
| WO2013022797A1 (en) * | 2011-08-05 | 2013-02-14 | Molecular Insight Pharmaceuticals | Radiolabeled prostate specific membrane antigen inhibitors |
| EP2800471A4 (en) | 2012-01-06 | 2015-11-04 | Molecular Insight Pharm Inc | METAL COMPLEX OF POLY (CARBOXYL) AMINE CONTAINING LIGANDS HAVING AFFINITY FOR CARBON IX ANHYDRASE |
| MX356097B (es) | 2012-09-17 | 2018-05-14 | Pfizer Inc Star | Proceso para la preparacion de nanoparticulas terapeuticas. |
| BR112015011118B1 (pt) * | 2012-11-15 | 2022-12-13 | Endocyte, Inc | Conjugado; composição farmacêutica; e uso de um conjugado |
| EP3300746B1 (en) * | 2013-01-14 | 2019-05-15 | Molecular Insight Pharmaceuticals, Inc. | Triazine based radiopharmaceuticals and radioimaging agents |
| EP2958596B1 (en) | 2013-02-15 | 2019-12-04 | Case Western Reserve University | Psma ligands and uses thereof |
| US11975074B2 (en) | 2013-02-15 | 2024-05-07 | Case Western Reserve University | Photodynamic therapy composition |
| US12465651B2 (en) | 2013-02-15 | 2025-11-11 | Case Western Reserve University | Photodynamic therapy composition |
| US10207005B2 (en) | 2013-02-15 | 2019-02-19 | Case Western Reserve University | Photodynamic therapy composition |
| US10434194B2 (en) | 2013-06-20 | 2019-10-08 | Case Western Reserve University | PSMA targeted nanobubbles for diagnostic and therapeutic applications |
| WO2015057692A1 (en) * | 2013-10-14 | 2015-04-23 | The Johns Hopkins University | Prostate-specific membrane antigen-targeted photosensitizers for photodynamic therapy |
| LT4095130T (lt) | 2013-10-18 | 2024-04-25 | Novartis Ag | Žymėti prostatos specifinio membranos antigeno (psma) inhibitoriai, jų naudojimas kaip vizualizavimo medžiagų ir farmacinių medžiagų prostatos vėžiui gydyti |
| US20150110814A1 (en) * | 2013-10-18 | 2015-04-23 | Psma Development Company, Llc | Combination therapies with psma ligand conjugates |
| JP6908964B2 (ja) | 2013-10-18 | 2021-07-28 | ピーエスエムエー ディベロップメント カンパニー,エルエルシー | Psmaリガンドコンジュゲートによる併用療法 |
| BR112016010927A2 (pt) | 2013-11-14 | 2017-08-08 | Endocyte Inc | Conjugado de fórmula |
| WO2015073896A2 (en) | 2013-11-15 | 2015-05-21 | Psma Development Company, Llc | Biomarkers for psma targeted therapy for prostate cancer |
| MA39734B1 (fr) | 2014-03-14 | 2019-07-31 | Pfizer | Nanoparticules thérapeutiques comportant un agent thérapeutique, et leurs procédés de fabrication et d'utilisation |
| EP3183236B1 (en) | 2014-08-24 | 2022-04-20 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften | Method for the production of 18f-labeled active esters and their application exemplified by the preparation of a psma-specific pet-tracer |
| AU2015315465B2 (en) | 2014-09-08 | 2019-08-29 | Molecular Insight Pharmaceuticals, Inc. | Organ protection in PSMA-targeted radionuclide therapy of prostate cancer |
| US10736974B2 (en) | 2014-10-22 | 2020-08-11 | The Johns Hopkins University | Scaffolds and multifunctional intermediates for imaging PSMA and cancer therapy |
| US10188759B2 (en) | 2015-01-07 | 2019-01-29 | Endocyte, Inc. | Conjugates for imaging |
| WO2016112382A2 (en) * | 2015-01-09 | 2016-07-14 | The Johns Hopkins University | Intranasal administration of glutamate carboxypeptidase (gcp-ii) inhibitors |
| AU2016333334B2 (en) | 2015-09-30 | 2019-07-25 | Deutsches Krebsforschungszentrum | 18F-tagged inhibitors of prostate specific membrane antigen (PSMA) and their use as imaging agents for prostate cancer |
| KR101639599B1 (ko) | 2015-11-09 | 2016-07-14 | 서울대학교산학협력단 | 펩타이드 싸이오우레아 유도체, 이를 포함하는 방사성 동위원소 표지 화합물 및 이를 유효 성분으로 함유하는 전립선암 치료 또는 진단용 약학적 조성물 |
| WO2017117687A1 (en) | 2016-01-10 | 2017-07-13 | British Columbia Cancer Agency Branch | 18/19f-labelled compounds which target the prostate specific membrane antigen |
| WO2017184231A1 (en) | 2016-04-20 | 2017-10-26 | Dow Corning Corporation | Lithium alkylsiliconate composition, coating, and method of making same |
| FI127538B (en) * | 2016-06-22 | 2018-08-31 | Dextech Medical Ab | MODIFIED Dextran Conjugates |
| CN109843339B (zh) * | 2016-06-23 | 2023-07-07 | 康奈尔大学 | 影响肿瘤杀伤的双重靶向构建体 |
| US10806806B2 (en) | 2016-06-23 | 2020-10-20 | Cornell University | Trifunctional constructs with tunable pharmacokinetics useful in imaging and anti-tumor therapies |
| EP3493856B1 (en) * | 2016-06-28 | 2023-08-09 | Cornell University | 18f-labeled triazole containing psma inhibitors |
| IL315062A (en) | 2017-04-05 | 2024-10-01 | Univ Cornell | Trifunctional constructs with tunable pharmacokinetics useful in imaging and anti-tumor therapies |
| JP7755371B2 (ja) * | 2017-06-09 | 2025-10-16 | アヴェンセル ユーロップ ゲー・エム・ベー・ハー | 共通キメラ抗原受容体発現免疫細胞のためのターゲティングモジュールならびに癌、感染および自己免疫障害の処置における使用 |
| SG11202005511VA (en) | 2017-12-13 | 2020-07-29 | Sciencons AS | Complex comprising a psma-targeting compound linked to a lead or thorium radionuclide |
| KR20250126870A (ko) | 2018-02-06 | 2025-08-25 | 더 존스 홉킨스 유니버시티 | 암 방사선 치료를 위한 psma 표적 방사성 할로겐화 우레아-폴리아미노카복실레이트 |
| WO2019183633A1 (en) | 2018-03-23 | 2019-09-26 | Case Western Reserve Univeristy | Psma targeted conjugate compounds and uses thereof |
| US12208102B2 (en) | 2018-04-17 | 2025-01-28 | Endocyte, Inc. | Methods of treating cancer |
| EP3853213A4 (en) * | 2018-09-21 | 2022-07-06 | Endocyte, Inc. | Shielding agents and their use |
| CN113164450A (zh) * | 2018-10-11 | 2021-07-23 | 普罗热尼奇制药公司 | 用于治疗转移性前列腺癌的组合治疗 |
| RU2697519C1 (ru) * | 2018-10-15 | 2019-08-15 | Общество с ограниченной ответственностью "Изварино Фарма" | Средство пептидной природы, включающее псма-связывающий лиганд на основе производного мочевины, способ его получения и применение для получения конъюгата с лекарственным и диагностическим агентом |
| KR102403970B1 (ko) * | 2018-12-27 | 2022-05-31 | (주)퓨쳐켐 | 카르복시산이 도입된 psma-표적 화합물 및 그의 용도 |
| WO2020150740A1 (en) | 2019-01-18 | 2020-07-23 | Case Western Reserve University | Psma ligand targeted compounds and uses thereof |
| EP3917626B1 (en) * | 2019-01-30 | 2023-05-31 | Technische Universität München | Psma binding dual mode radiotracer and therapeutic |
| WO2020210909A1 (en) | 2019-04-17 | 2020-10-22 | Provincial Health Services Authority | Novel radiolabelled compounds for diagnosis or treatment of prostate-specific membrane antigen-expressing cancer |
| CN114096264B (zh) | 2019-05-20 | 2025-03-14 | 因多塞特股份有限公司 | 制备psma缀合物的方法 |
| CN114401947B (zh) | 2019-06-21 | 2024-11-29 | 省卫生服务机构 | 靶向前列腺特异性膜抗原的放射性标记化合物 |
| KR102269315B1 (ko) | 2019-10-24 | 2021-06-24 | 서울대학교산학협력단 | 전립선 암의 영상 또는 치료를 위한 동위원소 표지 화합물 |
| AU2020387750B2 (en) * | 2019-11-21 | 2021-10-21 | Ferronova Pty Ltd | Magnetic tracer compositions |
| JP2023519915A (ja) * | 2020-03-26 | 2023-05-15 | ラミレス-フォート,マリグダリア,カレス | 紫外線照射治療 |
| WO2021202376A1 (en) | 2020-03-30 | 2021-10-07 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Method for blocking uptake of prostate-specific membrane antigen (psma)-targeted radionuclides by exocrine organs |
| CN111548305B (zh) * | 2020-05-12 | 2021-08-31 | 北京师范大学 | 一种可用于靶向psma的喹啉类化合物及其制备方法 |
| US12521435B2 (en) | 2020-07-03 | 2026-01-13 | Case Western Reserve University | Photodynamic therapy composition |
| CN112209970B (zh) * | 2020-10-21 | 2021-10-29 | 北京师范大学 | 一种锝-99m标记含异腈的谷氨酸-脲衍生物的制备方法和应用 |
| EP4244216A1 (en) | 2020-11-12 | 2023-09-20 | ABX Advanced Biochemical Compounds GmbH | Ligands of prostate specific membrane antigen (psma) containing heteroaromatic linker building blocks |
| CA3237590A1 (en) | 2021-11-09 | 2023-05-08 | James Basilion | Psma targeted conjugate compounds and uses thereof |
| AU2024208501A1 (en) | 2023-01-10 | 2025-07-24 | Sun Pharma Advanced Research Company Limited | Ligand-drug conjugates |
| AU2024317528A1 (en) | 2023-07-31 | 2026-03-05 | Curium Us Llc | [177 lu] lutetium-psma it composition and dosimetry, kit, method of making, and method of using thereof |
| WO2025184495A1 (en) * | 2024-02-29 | 2025-09-04 | Shuttle Pharmaceuticals Holdings, Inc. | Boron containing psma ligand for use in the treatment of cancer |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3527789A (en) * | 1967-10-06 | 1970-09-08 | Shell Oil Co | Production of poly(lower)alkyl alkenepolycarboxylates |
| JP2608550B2 (ja) * | 1986-10-17 | 1997-05-07 | 株式会社 片山化学工業研究所 | 軟水ボイラの防食処理方法 |
| US4888136A (en) * | 1988-05-02 | 1989-12-19 | Witco Corporation | New flame retardant compositions of matter and cellulosic products containing same |
| TW353663B (en) * | 1991-04-06 | 1999-03-01 | Hoechst Ag | Process for the preparation of phosphorus-containing L-amino acids, their derivatives and intermediates for this process |
| IT1270260B (it) * | 1994-06-21 | 1997-04-29 | Zambon Spa | Derivati dell'acido fosfonico ad attivita' inibitrice delle metallopeptidasi |
| US5662885A (en) | 1994-07-22 | 1997-09-02 | Resolution Pharmaceuticals Inc. | Peptide derived radionuclide chelators |
| US6011021A (en) | 1996-06-17 | 2000-01-04 | Guilford Pharmaceuticals Inc. | Methods of cancer treatment using naaladase inhibitors |
| US5795877A (en) * | 1996-12-31 | 1998-08-18 | Guilford Pharmaceuticals Inc. | Inhibitors of NAALADase enzyme activity |
| US5672592A (en) * | 1996-06-17 | 1997-09-30 | Guilford Pharmaceuticals Inc. | Certain phosphonomethyl-pentanedioic acid derivatives thereof |
| US5902817A (en) | 1997-04-09 | 1999-05-11 | Guilford Pharmaceuticals Inc. | Certain sulfoxide and sulfone derivatives |
| US6046180A (en) | 1996-06-17 | 2000-04-04 | Guilford Pharmaceuticals Inc. | NAALADase inhibitors |
| US5824662A (en) * | 1996-09-27 | 1998-10-20 | Guilford Pharmaceuticals Inc. | Treatment of global and focal ischemia using naaladase inhibitors |
| US6071965A (en) * | 1996-06-17 | 2000-06-06 | Guilford Pharmaceuticals Inc. | Phosphinic alkanoic acid derivatives |
| US6025344A (en) | 1996-06-17 | 2000-02-15 | Guilford Pharmaceuticals Inc. | Certain dioic acid derivatives useful as NAALADase inhibitors |
| PL332413A1 (en) | 1996-09-27 | 1999-09-13 | Guilford Pharm Inc | Compositions containing inhibitors of naaladase as well as methods of treating glutamatic anomaly and influencing neuronic functions among animals |
| US5962521A (en) | 1997-04-04 | 1999-10-05 | Guilford Pharmaceuticals Inc. | Hydroxamic acid derivatives |
| ZA983930B (en) | 1997-05-14 | 1999-11-08 | Lilly Co Eli | Excitatory amino acid receptor modulators. |
| US6528499B1 (en) | 2000-04-27 | 2003-03-04 | Georgetown University | Ligands for metabotropic glutamate receptors and inhibitors of NAALADase |
| CA2367787C (en) * | 1999-04-28 | 2011-07-26 | Alan P. Kozikowski | Ligands for metabotropic glutamate receptors |
| US6228888B1 (en) | 1999-07-01 | 2001-05-08 | Guilford Pharmaceuticals Inc. | Methods for treating anxiety, anxiety disorders and memory impairment using naaladase inhibitors |
| AU2003205077A1 (en) | 2002-01-10 | 2003-07-30 | Johns Hopkins University | Imaging agents and methods of imaging naaladase of psma |
| JP4846199B2 (ja) * | 2002-03-11 | 2011-12-28 | モレキュラ インサイト ファーマシューティカルズ インコーポレイテッド | テクネチウム−ジビリジン複合体、およびその使用法 |
| CA2504935C (en) * | 2002-11-07 | 2012-04-10 | Newsouth Innovations Pty Limited | Induction of the mitochondrial permeability transition |
| JP2007525452A (ja) * | 2003-04-15 | 2007-09-06 | マリンクロッド・インコーポレイテッド | Reおよびtcトリカルボニル錯体のための配位子を含有する二官能性三座ピラゾリル |
| US7682601B2 (en) * | 2003-04-15 | 2010-03-23 | Mallinckrodt Inc. | Bifunctional tridentate pyrazolyl containing ligands for re and tc tricarbonyl complexes |
| US7741510B2 (en) * | 2005-01-13 | 2010-06-22 | E. I. Du Pont De Nemours And Company | Rheology control agents |
| US20060155021A1 (en) * | 2005-01-13 | 2006-07-13 | Lenges Christian P | Coating compositions containing rheology control agents |
| WO2006093991A1 (en) * | 2005-03-02 | 2006-09-08 | The Cleveland Clinic Foundation | Compounds which bind psma and uses thereof |
| ES2847275T3 (es) * | 2006-11-08 | 2021-08-02 | Molecular Insight Pharm Inc | Heterodímeros de ácido glutámico |
| CA2926573C (en) * | 2007-06-26 | 2018-08-28 | The Johns Hopkins University | Labeled inhibitors of prostate specific membrane antigen (psma), biological evaluation, and use as imaging agents |
| EP4464384A3 (en) * | 2007-08-17 | 2025-01-08 | Purdue Research Foundation | Psma binding ligand-linker conjugates and methods for using |
| EP3222617B1 (en) | 2009-03-19 | 2022-07-06 | The Johns Hopkins University | Psma-targeting compounds and uses thereof |
| AU2010260195B2 (en) * | 2009-06-15 | 2014-11-20 | Molecular Insight Pharmaceuticals, Inc. | Process for production of heterodimers of glutamic acid |
| WO2013022797A1 (en) | 2011-08-05 | 2013-02-14 | Molecular Insight Pharmaceuticals | Radiolabeled prostate specific membrane antigen inhibitors |
| LT4095130T (lt) | 2013-10-18 | 2024-04-25 | Novartis Ag | Žymėti prostatos specifinio membranos antigeno (psma) inhibitoriai, jų naudojimas kaip vizualizavimo medžiagų ir farmacinių medžiagų prostatos vėžiui gydyti |
| AU2015315465B2 (en) | 2014-09-08 | 2019-08-29 | Molecular Insight Pharmaceuticals, Inc. | Organ protection in PSMA-targeted radionuclide therapy of prostate cancer |
-
2007
- 2007-11-07 ES ES18150494T patent/ES2847275T3/es active Active
- 2007-11-07 ES ES15168948.6T patent/ES2684322T3/es active Active
- 2007-11-07 HU HUE07844938A patent/HUE026216T2/en unknown
- 2007-11-07 EP EP18150494.5A patent/EP3335736B1/en not_active Revoked
- 2007-11-07 HU HUE19214298A patent/HUE059645T2/hu unknown
- 2007-11-07 PL PL07844938T patent/PL2097111T3/pl unknown
- 2007-11-07 ES ES07844938.6T patent/ES2547481T3/es active Active
- 2007-11-07 SI SI200731686T patent/SI2097111T1/sl unknown
- 2007-11-07 CN CN201410158114.9A patent/CN103922998A/zh active Pending
- 2007-11-07 DK DK07844938.6T patent/DK2097111T3/en active
- 2007-11-07 ES ES19214298T patent/ES2926912T3/es active Active
- 2007-11-07 EP EP07844938.6A patent/EP2097111B1/en active Active
- 2007-11-07 TW TW096142019A patent/TWI492761B/zh not_active IP Right Cessation
- 2007-11-07 PT PT192142982T patent/PT3699162T/pt unknown
- 2007-11-07 PL PL15168948T patent/PL2942065T3/pl unknown
- 2007-11-07 JP JP2009536459A patent/JP5606737B2/ja active Active
- 2007-11-07 CA CA2669127A patent/CA2669127C/en active Active
- 2007-11-07 PT PT78449386T patent/PT2097111E/pt unknown
- 2007-11-07 CN CN200780049490.2A patent/CN101778910B/zh active Active
- 2007-11-07 EP EP21208033.7A patent/EP4023624A1/en not_active Withdrawn
- 2007-11-07 WO PCT/US2007/083934 patent/WO2008058192A2/en not_active Ceased
- 2007-11-07 US US11/936,659 patent/US20080193381A1/en active Granted
- 2007-11-07 HU HUE15168948A patent/HUE039199T2/hu unknown
- 2007-11-07 EP EP15168948.6A patent/EP2942065B1/en active Active
- 2007-11-07 BR BRPI0718700A patent/BRPI0718700B8/pt active IP Right Grant
- 2007-11-07 PL PL19214298.2T patent/PL3699162T3/pl unknown
- 2007-11-07 EP EP19214298.2A patent/EP3699162B1/en not_active Revoked
-
2011
- 2011-10-12 US US13/271,549 patent/US9309193B2/en active Active
- 2011-11-11 US US13/294,677 patent/US8487129B2/en active Active
-
2014
- 2014-08-27 JP JP2014172732A patent/JP5936659B2/ja active Active
-
2015
- 2015-01-15 HK HK15100503.3A patent/HK1200164A1/xx unknown
- 2015-08-13 CY CY20151100716T patent/CY1116610T1/el unknown
-
2016
- 2016-03-14 US US15/068,841 patent/US10640461B2/en active Active
- 2016-10-28 US US15/337,867 patent/US9878980B2/en active Active
-
2017
- 2017-10-26 US US15/794,539 patent/US10131627B2/en active Active
-
2018
- 2018-10-10 US US16/156,289 patent/US10647666B2/en active Active
-
2020
- 2020-05-08 US US16/870,288 patent/US20210070695A1/en not_active Abandoned
- 2020-11-16 US US17/099,440 patent/US20210070696A1/en not_active Abandoned
-
2021
- 2021-04-23 US US17/238,535 patent/US20210246103A1/en not_active Abandoned
-
2022
- 2022-11-07 US US17/982,182 patent/US12534429B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2926912T3 (es) | Heterodímeros de ácido glutámico | |
| AU2007316391B2 (en) | Heterodimers of glutamic acid | |
| HK40036808A (en) | Heterodimers of glutamic acid | |
| HK40036808B (en) | Heterodimers of glutamic acid | |
| HK1257260B (en) | Heterodimers of glutamic acid |