ES2942329T3 - Régimen de dosificación de vaborbactam y meropenem para el tratamiento de infecciones bacterianas en sujetos con función renal reducida - Google Patents
Régimen de dosificación de vaborbactam y meropenem para el tratamiento de infecciones bacterianas en sujetos con función renal reducida Download PDFInfo
- Publication number
- ES2942329T3 ES2942329T3 ES16783764T ES16783764T ES2942329T3 ES 2942329 T3 ES2942329 T3 ES 2942329T3 ES 16783764 T ES16783764 T ES 16783764T ES 16783764 T ES16783764 T ES 16783764T ES 2942329 T3 ES2942329 T3 ES 2942329T3
- Authority
- ES
- Spain
- Prior art keywords
- meropenem
- compound
- pharmaceutically acceptable
- acceptable salt
- administered
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
En el presente documento se describen métodos para tratar o mejorar una infección bacteriana que comprende administrar una composición que comprende un Compuesto I de éster de ácido borónico cíclico en combinación con meropenem. En algunas realizaciones, la infección bacteriana es una infección del tracto respiratorio inferior. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Régimen de dosificación de vaborbactam y meropenem para el tratamiento de infecciones bacterianas en sujetos con función renal reducida
Antecedentes
Campo
Las realizaciones de la presente solicitud se refieren a compuestos antimicrobianos, composiciones, su uso y preparación como agentes terapéuticos.
Los antibióticos han sido herramientas efectivas en el tratamiento de enfermedades infecciosas durante el último medio siglo. Desde el desarrollo de la terapia antibiótica hasta finales de la década de 1980, el control de las infecciones bacterianas fue casi total en los países desarrollados. Sin embargo, ante la presión del uso de antibióticos, se han generalizado múltiples mecanismos de resistencia que amenazan la utilidad clínica de la terapia antibacteriana. El aumento de cepas resistentes a los antibióticos ha sido especialmente común en los grandes hospitales y centros asistenciales. Las consecuencias del aumento de cepas resistentes incluyen una mayor morbilidad y mortalidad, una hospitalización más prolongada de los pacientes y un incremento de los costes de tratamiento.
Diversas bacterias han desarrollado enzimas desactivadoras de p-lactámicos, a saber, p-lactamasas, que contrarrestan la eficacia de los diversos p-lactámicos. las p-lactamasas se pueden agrupar en 4 clases en base a sus secuencias de aminoácidos, a saber, las clases A, B, C y D de Ambler. Las enzimas de las clases A, C y D incluyen p-lactamasas de serina de sitio activo, y las enzimas de la clase B, que se encuentran con menos frecuencia, son Zn-dependientes. Estas enzimas catalizan la degradación química de los antibióticos p-lactámicos, volviéndolos inactivos. Algunas p-lactamasas se pueden transferir entre diferentes cepas y especies bacterianas. La rápida propagación de la resistencia bacteriana y la evolución de cepas multirresistentes limitan gravemente las opciones de tratamiento con p-lactámicos disponibles.
El aumento de cepas bacterianas que expresan p-lactamasa de clase D, tales como Acinetobacter baumannii, se ha convertido en una amenaza emergente multirresistente. Las cepas de A. baumannii expresan p-lactamasas de clase A, C y D. Las p-lactamasas de clase D, tales como las de las familias OXA, son especialmente efectivas para destruir los antibióticos p-lactámicos de tipo carbapenem, por ejemplo, el imipenem, el componente activo de los carbapenems del Primaxin® de Merck (Montefour, K.; et al. Crit. Care Nurse 2008, 28, 15; Perez, F. et al. Expert Rev. Anti Infect. Ther. 2008, 6, 269; Bou, G.; Martinez-Beltran, J. Antimicrob. Agents Chemother. 2000, 40, 428. 2006, 50, 2280; Bou, G. et al., J. Antimicrob. Agents Chemother. 2000, 44, 1556). Esto ha impuesto una amenaza acuciante al uso efectiva de los medicamentos de esa categoría para tratar y prevenir las infecciones bacterianas. De hecho, el número de p-lactamasas de serina catalogadas ha pasado de menos de diez en la década de 1970 a más de 300 variantes. Estas cuestiones propiciaron el desarrollo de cinco “generaciones” de cefalosporinas. Cuando se introdujeron inicialmente en la práctica clínica, las cefalosporinas de espectro extendido resistían la hidrólisis por las p-lactamasas de clase A prevalentes, TEM-1 y SHV-1. Sin embargo, el desarrollo de cepas resistentes por la evolución de sustituciones de un solo aminoácido en TEM-1 y SHV-1 dio lugar a la aparición del fenotipo de plactamasa de espectro extendido (ESBL).
Recientemente se han desarrollado nuevas p-lactamasas que hidrolizan la clase carbapenem de antimicrobianos, incluidos imipenem, biapenem, doripenem, meropenem y ertapenem, así como otros antibióticos p-lactámicos. Estas carbapenemasas pertenecen a las clases moleculares A, B y D. Las carbapenemasas de clase A del tipo KPC predominan en Klebsiella pneumoniae, pero ahora también se han descrito en otras Enterobacteriaceae, Pseudomonas aeruginosa y Acinetobacter baumannii. La carbapenemasa KPC se describió por primera vez en 1996 en Carolina del Norte, pero desde entonces se ha diseminado ampliamente por Estados Unidos. Ha sido especialmente problemática en la zona de Nueva York, donde se han notificado diversos casos de propagación en hospitales importantes y de morbilidad en pacientes. Estas enzimas también se han notificado recientemente en Francia, Grecia, Suecia, Reino Unido, y recientemente se ha notificado un brote en Alemania. El tratamiento de cepas resistentes con carbapenems se puede asociar a malos resultados.
Otro mecanismo de resistencia mediada por p-lactamasa a los carbapenems implica la combinación de mecanismos de permeabilidad o eflujo combinados con la hiperproducción de beta-lactamasas. Un ejemplo es la pérdida de una porina combinada con la hiperproducción de betalactamasa ampC, que provoca resistencia al imipenem en Pseudomonas aeruginosa. La sobreexpresión de la bomba de eflujo combinada con la hiperproducción de la plactamasa ampC también puede provocar resistencia a un carbapenem tal como el meropenem.
Debido a que hay tres clases moleculares principales de p-lactamasas en base a serina, y cada una de estas clases contiene un número significativo de variantes de p-lactamasas, es poco probable que la inhibición de una o un pequeño número de p-lactamasas tenga valor terapéutico. Los inhibidores de p-lactamasas heredados son en gran medida inefectivos contra, al menos, las carbapenemasas de clase A, contra las cefalosporinasas de clase C mediadas por cromosomas y plásmidos y contra muchas de las oxacilinasas de clase D. Por lo tanto, es necesario mejorar el tratamiento combinado con inhibidores de la p-lactamasa.
Documentos citados
Griffith D et al., 54ta Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); Washington, DC, EE.UU, (2014) desvela “A Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of the Beta-lactamase inhibitor RPX7009 Alone, Meropenem Alone, and both in Combination (Carbavance) TID for 7 days in Healthy Adult Subjecf.
Kuti J L et al., J. of Critical Care (2010) 25, 155 a 156 desvela “Optimal cefepime and meropenem dosing for ventilator-associated pneumonia patients with reduced renal function: An update to our clinical pathway”.
Yagi Yusuke et al., J. Clin. Pharm. (2014) 36, 648 a 656 desvela “Outcome evaluation of an intervention to improve the effective and safe use of meropenem".
Sumario
La presente invención se define en las reivindicaciones adjuntas.
Algunas realizaciones descritas en la presente memoria se refieren al Compuesto I o a una sal aceptable para uso farmacéutico del mismo para su uso en un procedimiento para tratar o mejorar una infección bacteriana en un sujeto que padece una función renal reducida en combinación con meropenem:

en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran después de una sesión de diálisis y en el que:
(i) el sujeto tiene un nivel de aclaramiento de creatinina igual o superior a 30 ml/min e inferior a 50 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g y la cantidad de meropenem es de aproximadamente 1,0 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 8 horas (q8h); o,
(ii) el sujeto tiene un nivel de aclaramiento de creatinina igual o superior a 20 ml/min e inferior a 30 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g y la cantidad de meropenem es de aproximadamente 1,0 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 12 horas (q12h); o bien
(iii) el sujeto tiene un aclaramiento de creatinina igual o superior a 10 ml/min e inferior a 20 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 0,5 g y la cantidad de meropenem es de aproximadamente 0,5 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 12 horas (q12h); o bien
(iv) el sujeto tiene un aclaramiento de creatinina inferior a 10 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 0,5 g y la cantidad de meropenem es de aproximadamente 0,5 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 24 horas (q24h).
En algunas realizaciones, el sujeto padece una infección del tracto respiratorio inferior.
En algunas realizaciones, el procedimiento además comprende la administración de un medicamento adicional seleccionado entre un agente antibacteriano, un agente antifúngico, un agente antiviral, un agente antiinflamatorio o un agente antialérgico.
En algunas realizaciones, el sujeto tratado por el procedimiento descrito anteriormente es un mamífero. En algunas realizaciones adicionales, el sujeto es un ser humano.
Breve descripción de las figuras
La FIG. 1 es un gráfico que representa el perfil de concentración plasmática (mg/l) de varias dosis del Compuesto I en función del tiempo tras una única infusión IV en sujetos sanos en el estudio desvelado en el Ejemplo 1.
La FIG. 2 es un gráfico que representa La dosis del Compuesto I frente al AUC (hs*mg/l) tras dosis únicas o múltiples en sujetos sanos en el estudio desvelado en el Ejemplo 1.
La FIG. 3 es un gráfico que representa el perfil de concentración plasmática (mg/l) del Compuesto I solo y en combinación con meropenem tras infusiones de 3 horas en sujetos adultos sanos en el estudio desvelado en el Ejemplo 2.
La FIG. 4 es un gráfico que representa el perfil de concentración plasmática (mg/l) de meropenem solo y en combinación con el Compuesto I tras infusiones de 3 horas en sujetos adultos sanos en el estudio desvelado en el Ejemplo 2.
La FlG. 5 es un gráfico que representa el perfil de concentración plasmática (mg/l) del Compuesto I solo y en combinación con meropenem tras una dosis única y 7 días de dosificación TID (es decir, tres veces al día) por medio de infusiones de 3 horas en sujetos adultos sanos en el estudio desvelado en el Ejemplo 3.
La FIG. 6 es un gráfico que representa el perfil de concentración plasmática (mg/l) de meropenem solo y en combinación con el Compuesto I después de una dosis única y 7 días de dosificación TID (es decir, tres veces al día) por medio de infusiones de 3 horas en sujetos adultos sanos en el estudio desvelado en el Ejemplo 3.
La FIG. 7 es un gráfico que representa el perfil de concentración plasmática (mg/l) de 2 g del Compuesto I solo y en combinación con 2 g de meropenem tras dosis únicas y múltiples por infusión de 3 horas en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 8 es un gráfico que representa el perfil de concentración plasmática (mg/l) de 2 g del Compuesto I solo y en combinación con 2 g de meropenem tras dosis únicas y múltiples por infusión de 1 horas en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 9 es un gráfico que representa el perfil de concentración plasmática media (mg/l) del Compuesto I tras infusiones de 1 hora o 3 horas de 2 g de Compuesto I en combinación con 2 g de meropenem en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 10 es un gráfico que representa el perfil de concentración plasmática (mg/l) de 2 g de meropenem solo y en combinación con 2 g del Compuesto I tras dosis única y múltiple por infusión de 3 horas en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 11 es un gráfico que representa el perfil de concentración plasmática (mg/l) de 2 g de meropenem solo y en combinación con 2 g del Compuesto I tras dosis única y múltiple por infusión de 1 horas en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 12 es un gráfico que representa el perfil de concentración plasmática media (mg/l) de meropenem I tras infusiones de 1 o 3 horas de 2 g de Compuesto I en combinación con 2 g de meropenem en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 13 es un gráfico que representa el perfil de concentración plasmática (mg/l) de meropenem lactama abierta después de una infusión de 1 hora de 2 g de meropenem solo y en combinación con 2 g del Compuesto I en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 14 es un gráfico que representa el perfil de concentración plasmática media (mg/l) de meropenem lactama abierta tras infusiones de 1 hora o 3 horas de 2 g de meropenem en combinación con 2 g de Compuesto I en sujetos sanos en el estudio desvelado en el Ejemplo 4.
La FIG. 15 es un gráfico que representa el aclaramiento de la combinación de 1 g de Compuesto I y 1 g de meropenem de acuerdo con el aclaramiento de creatinina en sujetos con diversos grados de insuficiencia renal en el estudio desvelado en el Ejemplo 5.
La FIG. 16 es un gráfico que representa el perfil de concentración plasmática media (pg/ml) de meropenem antes y después del inicio de la tercera infusión de meropenem 2 g durante 3 horas en el estudio desvelado en el Ejemplo 6.
La FIG. 17 es un gráfico que representa el perfil de concentración plasmática media (pg/ml) del Compuesto I antes y después del inicio de la tercera infusión de 2 g de Compuesto I durante 3 horas en el estudio desvelado en el Ejemplo 6.
La FIG. 18 es un gráfico que representa el perfil de concentración media en plasma y líquido de revestimiento epitelial (ELF) (pg/ml) de meropenem en el momento de la broncoscopia con lavado broncoalveolar (BAL) (dosis de meropenem de 2 g infundida durante 3 horas) en el estudio desvelado en el Ejemplo 6.
La FIG. 19 es un gráfico que representa el perfil de concentración media en plasma y líquido de revestimiento epitelial (ELF) (pg/ml) del Compuesto I en el momento de la broncoscopia con lavado broncoalveolar (BAL) (dosis de 2 g de Compuesto I infundida durante 3 horas) en el estudio desvelado en el Ejemplo 6.
La FIG. 20 es un gráfico que representa el perfil de concentración plasmática media (pg/ml) del Compuesto I y meropenem antes y después del inicio de la tercera infusión de meropenem 2 g/Compuesto I 2 g durante 3 horas en el estudio desvelado en el Ejemplo 6.
La FIG. 21 es un gráfico que representa el perfil de concentración media de líquido de revestimiento epitelial (ELF) (pg/ml) del Compuesto I y meropenem en el momento de la broncoscopia con lavado broncoalveolar (BAL) (dosis de meropenem de 2 g infundida durante 3 horas) en el estudio desvelado en el Ejemplo 6.
La FIG. 22 es un gráfico que representa la actividad de 1 g meropenem /1 g Compuesto I administrado por infusión de 3 horas cada 8 horas contra ciertas cepas de K. pneumoniae Resistente a Carbapenem en un Modelo de Fibra Hueca in vitro.
La FIG. 23 es un gráfico que representa la actividad de 1 g meropenem /1 g Compuesto I administrado por infusión de 3 horas cada 8 horas contra ciertas cepas de K. pneumoniae Resistente a Carbapenem en un Modelo de Fibra Hueca in vitro.
La FIG. 24 es un gráfico que representa la actividad de 2 g meropenem /1 g Compuesto I administrado por infusión de 3 horas cada 8 horas contra ciertas cepas de K. pneumoniae Resistente a Carbapenem en un Modelo de Fibra Hueca in vitro.
La FIG.25 es un gráfico que representa la actividad de 1 g de meropenem /1 g de Compuesto I administrado por infusión de 3 horas cada 8 horas contra ciertas cepas de P aeruginosa en un Modelo de Fibra Hueca in vitro.
La FIG. 26 es un gráfico que muestra los perfiles farmacocinéticos representativos de 2 g de meropenem y 2 g del Compuesto I administrados cada 8 horas por medio de infusión de 3 horas en un Modelo de Fibra Hueca in vitro.
La FIG.27 es un gráfico que representa la actividad de 2 g de meropenem administrados cada 8 horas por medio de infusión de 3 horas contra determinadas cepas de P. aeruginosa en un Modelo de Fibra Hueca in vitro.
La FIG. 28 es un gráfico que representa la actividad de 2 g de meropenem /1 g de Compuesto I administrado por infusión de 3 horas cada 8 horas contra ciertas cepas de P aeruginosa en un Modelo de Fibra Hueca in vitro. Descripción detallada de las realizaciones
Definiciones
El término “agente” o “agente de ensayo” incluye cualquier sustancia, molécula, elemento, compuesto, entidad o una combinación de los mismos. Incluye, pero no se limita a, por ejemplo, proteínas, polipéptidos, péptidos o miméticos, pequeñas moléculas orgánicas, polisacáridos, polinucleótidos y similares. Puede ser un producto natural, un compuesto sintético o un compuesto químico, o una combinación de dos o más sustancias. A menos que se especifique lo contrario, los términos “agente”, “sustancia” y “compuesto” se usan en la presente memoria indistintamente.
El término “mamífero” se usa en su sentido biológico habitual. Por lo tanto, incluye específicamente a los seres humanos, el ganado vacuno, los caballos, los perros, los gatos, las ratas y los ratones, pero también a muchas otras especies.
El término “infección microbiana” se refiere a la invasión del organismo huésped, ya sea vertebrado, invertebrado, pez, planta, ave o mamífero, por microbios patógenos. Incluye el crecimiento excesivo de microbios que normalmente están presentes en el cuerpo de un mamífero u otro organismo. En términos más generales, una infección microbiana puede ser cualquier situación en la que la presencia de una población o poblaciones microbianas sea perjudicial para un mamífero huésped. Por lo tanto, un mamífero “sufre” una infección microbiana cuando un número excesivo de una población microbiana está presente en o sobre el cuerpo de un mamífero, o cuando los efectos de la presencia de una población o poblaciones microbianas está dañando las células u otros tejidos de un mamífero. En concreto, esta descripción se aplica a una infección bacteriana. Cabe señalar que los compuestos de las realizaciones preferentes también son útiles para tratar el crecimiento microbiano o la contaminación de cultivos celulares u otros medios, o superficies u objetos inanimados, y nada de lo en la presente memoria expuesto debe limitar las realizaciones preferentes únicamente al tratamiento de organismos superiores, excepto cuando así se especifique explícitamente en las reivindicaciones.
El término “portador aceptable para uso farmacéutico” o “excipiente aceptable para uso farmacéutico” incluye todos y cada uno de los disolventes, medios de dispersión, revestimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardadores de la absorción y similares. El uso de tales medios y agentes para sustancias activas para uso farmacéutico es muy conocido en la técnica. Salvo en la medida en que algún medio o agente convencional sea incompatible con el principio activo, se contempla su uso en las composiciones terapéuticas. También se pueden incorporar compuestos activos complementarios a las composiciones. Además, se pueden incluir diversos adyuvantes tales como los que se usan habitualmente en la técnica. Estos y otros compuestos de este tipo se describen en la bibliografía, por ejemplo, en el Merck Index, Merck & Company, Rahway, NJ. Las consideraciones para la inclusión de diversos componentes en las composiciones farmacéuticas se describen, por ejemplo, en Gilman et al. (Eds.) (1990); Goodman y Gilman: The Pharmacological Basis of Therapeutics, 8va Ed., Pergamon Press.
El término “sal aceptable para uso farmacéutico” se refiere a sales que conservan la eficacia biológica y las propiedades de los compuestos de las realizaciones preferentes y que no son biológicamente o de otro modo indeseables. En muchos casos, los compuestos de las realizaciones preferentes son capaces de formar sales ácidas y/o básicas en virtud de la presencia de grupos amino y/o carboxilo o grupos similares a los mismos. Se pueden formar sales de adición ácida aceptables para uso farmacéutico con ácidos inorgánicos y ácidos orgánicos. Los ácidos inorgánicos de los que se pueden derivar sales incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares. Los ácidos orgánicos de los que se pueden derivar sales incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido pirúvico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico, ácido salicílico y similares. Se pueden formar sales de adición de bases aceptables para uso farmacéutico con bases inorgánicas y orgánicas. Las bases inorgánicas de las que se pueden derivar sales incluyen, por ejemplo, sodio, potasio, litio, amonio, calcio, magnesio, hierro, zinc, cobre, manganeso, aluminio y similares; particularmente preferentes son las sales de amonio, potasio, sodio, calcio y magnesio. Las bases orgánicas de las que se pueden derivar sales incluyen, por ejemplo, aminas primarias, secundarias y terciarias, aminas sustituidas, incluidas las aminas sustituidas naturales, aminas cíclicas, resinas básicas de intercambio iónico y similares, específicamente tales como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina y etanolamina. Muchas sales de este tipo son conocidas en la técnica, como se describe en el documento WO 87/05297, Johnston et al., publicado el 11 de septiembre de 1987.
El término “solvato” se refiere al compuesto formado por la interacción de un disolvente y un EPI, un metabolito o una sal del mismo. Los solvatos adecuados son solvatos aceptables para uso farmacéutico, incluidos los hidratos. El término “sujeto”, como se usa en la presente memoria, significa un ser humano o un mamífero no humano, por ejemplo, un perro, un gato, un ratón, una rata, una vaca, una oveja, un cerdo, una cabra, un primate no humano o un ave, por ejemplo, un pollo, así como cualquier otro vertebrado o invertebrado.
Un efecto terapéutico alivia, hasta cierto punto, uno o más de los síntomas de la infección, e incluye curar una infección. El término “curación” significa que se eliminan los síntomas de la infección activa, incluida la eliminación del exceso de miembros de microbios viables de los implicados en la infección. Sin embargo, pueden existir ciertos efectos a largo plazo o permanentes de la infección incluso después de obtener la curación (tales como daños extensos en los tejidos).
Los términos “tratar”, “tratamiento” o “en tratamiento”, como se usa en la presente memoria, se refiere a la administración de una composición farmacéutica con fines profilácticos y/o terapéuticos. El término “tratamiento profiláctico” hace referencia al tratamiento de un paciente que aún no está infectado, pero que es susceptible de contraer una infección concreta o corre el riesgo de contraerla, por medio del cual el tratamiento reduce la probabilidad de que el paciente desarrolle una infección. El término “tratamiento terapéutico” se refiere a la administración de un tratamiento a un paciente que ya padece una infección.
Procedimientos de Tratamiento
La información técnica que se expone a continuación puede, en algunos aspectos, ir más allá del alcance de la invención reivindicada en la presente memoria, que se define por medio de las reivindicaciones adjuntas. La información técnica adicional se proporciona para situar la invención real en un contexto técnico más amplio y para ilustrar posibles avances técnicos relacionados.
Algunas realizaciones descritas en la presente memoria se relacionan con el Compuesto I o una sal aceptable para uso farmacéutico del mismo para su uso en un procedimiento para tratar una infección bacteriana en combinación con meropenem, que comprende administrar una cantidad efectiva del Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem a un sujeto que lo necesite:

(Compuesto I)
en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g a aproximadamente 3,0 g y la cantidad de meropenem es de aproximadamente 1,0 g a aproximadamente 3,0 g.
En algunas realizaciones, la cantidad del Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 2,0 g. En algunas realizaciones, la cantidad de meropenem es de aproximadamente 2,0 g. En algunas realizaciones, la cantidad tanto del Compuesto I o la sal aceptable para uso farmacéutico del mismo como de meropenem es de aproximadamente 2,0 g.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran al menos una vez al día. En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem se administran 3 veces al día. En algunas otras realizaciones, la dosis diaria del Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 6,0 g y en las que la dosis diaria de meropenem es de aproximadamente 6,0 g.
En algunas realizaciones, la administración es por infusión intravenosa. En algunas realizaciones, la infusión intravenosa se completa en aproximadamente 1 a 5 horas. En algunas otras realizaciones, la infusión intravenosa se completa es de aproximadamente 3 horas.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra antes o después del meropenem. En algunas otras realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se encuentran en una única forma farmacéutica. En algunas realizaciones, la forma de dosificación única además comprende un excipiente, diluyente o portador aceptable para uso farmacéutico.
Sujetos con Función Renal Reducida
Algunas realizaciones descritas en la presente memoria se refieren al Compuesto I o a una sal aceptable para uso farmacéutico del mismo para su uso en un procedimiento para tratar una infección bacteriana en combinación con
meropenem, que comprende seleccionar para el tratamiento a un sujeto que necesita tratamiento para una infección bacteriana y que padece una función renal reducida; administrar una cantidad efectiva del Compuesto I o de una sal aceptable para uso farmacéutico del mismo y meropenem a dicho sujeto. En algunas realizaciones, dicho sujeto tiene un aclaramiento de creatinina de > 30 ml/min y < 50 ml/min. En algunas realizaciones, dicho sujeto tiene un aclaramiento de creatinina de > 20 ml/min y < 3o ml/min. En algunas realizaciones, dicho sujeto tiene un aclaramiento de creatinina de > 10 ml/min y < 20 ml/min. En algunas realizaciones, dicho sujeto tiene un aclaramiento de creatinina < 10 ml/min. En algunas realizaciones, la infección bacteriana es una infección del tracto respiratorio inferior.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en un intervalo de dosis de aproximadamente 250 mg a aproximadamente 2,0 g. En otras realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en una dosis de aproximadamente 500 mg a aproximadamente 1,0 g. En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en una dosis de aproximadamente 1,0 g. En otras realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en una dosis de aproximadamente 500 mg. En algunas realizaciones, meropenem se administra en un intervalo de dosis de aproximadamente 250 mg a aproximadamente 2,0 g. En otras realizaciones, meropenem se administra en una dosis de aproximadamente 500 mg a aproximadamente 1,0 g. En algunas realizaciones, meropenem se administra en una dosis de aproximadamente 1,0 g. En otras realizaciones, meropenem se administra en una dosis de aproximadamente 500 mg. En algunas otras realizaciones, tanto el Compuesto I o la sal aceptable para uso farmacéutico del mismo como el meropenem se administran en una dosis de aproximadamente 1,0 g. En algunas otras realizaciones, tanto el Compuesto I o la sal aceptable para uso farmacéutico del mismo como el meropenem se administran en una dosis de aproximadamente 500 mg.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem se administran al menos una vez al día (cada 24 horas). En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem se administran 2 veces al día (cada 12 horas). En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem se administran 3 veces al día (cada 8 horas). En algunas realizaciones, la dosis diaria del Compuesto I o de la sal aceptable para uso farmacéutico del mismo es de aproximadamente 3,0 g, y la dosis diaria de meropenem es de aproximadamente 3,0 g. En algunas realizaciones, la dosis diaria del Compuesto I o de la sal aceptable para uso farmacéutico del mismo es de aproximadamente 2,0 g, y la dosis diaria de meropenem es de aproximadamente 2,0 g. En algunas realizaciones, la dosis diaria del Compuesto I o de la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g, y la dosis diaria de meropenem es de aproximadamente 1,0 g. En algunas realizaciones, la dosis diaria del Compuesto I o de la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g y en las que la dosis diaria de meropenem es de aproximadamente 1,0 g. En otras realizaciones, la dosis diaria del Compuesto I o de la sal aceptable para uso farmacéutico del mismo es de aproximadamente 500 mg y en las que la dosis diaria de meropenem es de aproximadamente 500 mg.
En algunas realizaciones, la administración es por infusión intravenosa. En algunas realizaciones, la infusión intravenosa se completa en aproximadamente 1 a 5 horas. En algunas otras realizaciones, la infusión intravenosa se completa en aproximadamente 3 horas.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra antes o después del meropenem. En algunas otras realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se encuentran en una única forma farmacéutica. En algunas realizaciones, la forma de dosificación única además comprende un excipiente, diluyente o portador aceptable para uso farmacéutico.
Sujetos con Infección del Tracto Respiratorio Inferior
Algunas realizaciones descritas en la presente memoria se refieren al Compuesto I o una sal aceptable para uso farmacéutico del mismo para su uso en un procedimiento de tratamiento o mejora de una infección del tracto respiratorio inferior en combinación con meropenem, que comprende la administración de una cantidad efectiva del Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem a un sujeto que lo necesite.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en un intervalo de dosis de aproximadamente 250 mg a aproximadamente 5,0 g. En algunas realizaciones más, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra en un intervalo de dosis de aproximadamente 1,0 g a aproximadamente 3,0 g. En algunas realizaciones más, la cantidad de Compuesto I es de aproximadamente 2,0 g. En algunas realizaciones, meropenem se administra en un intervalo de dosis de aproximadamente 250 mg a aproximadamente 5,0 g. En algunas realizaciones adicionales, meropenem se administra en un intervalo de dosis de aproximadamente 1,0 g a aproximadamente 3,0 g. En algunas realizaciones adicionales, la cantidad de meropenem es de aproximadamente 2,0 g. En algunas realizaciones, tanto el Compuesto I o la sal aceptable para uso farmacéutico del mismo como meropenem se administran en una dosis de aproximadamente 2,0 g.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran al menos una vez al día. En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem se administran 3 veces al día. En algunas realizaciones, la dosis diaria del Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 3,0 g a aproximadamente 6,0 g y en las que la dosis diaria de meropenem es de aproximadamente 3,0 g a aproximadamente 6,0 g. En algunas realizaciones más, la dosis diaria del Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 6,0 g y en las que la dosis diaria de meropenem es de aproximadamente 6,0 g.
En algunas realizaciones, la administración es por infusión intravenosa. En algunas realizaciones, la infusión intravenosa se completa en aproximadamente 1 a 5 horas. En algunas otras realizaciones, la infusión intravenosa se completa en aproximadamente 3 horas.
En algunas realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra antes o después del meropenem. En algunas otras realizaciones, el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se encuentran en una única forma farmacéutica. En algunas realizaciones, la forma de dosificación única además comprende un excipiente, diluyente o portador aceptable para uso farmacéutico.
En cualquiera de las realizaciones de los procedimientos en la presente memoria descritos, el procedimiento puede comprender además la administración de un medicamento adicional seleccionado entre un agente antibacteriano, un agente antifúngico, un agente antiviral, un agente antiinflamatorio o un agente antialérgico.
En algunas realizaciones, el sujeto tratado por el procedimiento descrito anteriormente es un mamífero. En algunas realizaciones adicionales, el sujeto es un ser humano.
En cualquiera de las realizaciones de los procedimientos en la presente memoria descritos, el tratamiento es para la infección causada por Enterobacteriaceae resistentes a carbapenemes.
Indicaciones
Las composiciones que comprenden el Compuesto I y un compuesto carbapenem meropenem descrito en la presente memoria se pueden usar para tratar infecciones bacterianas. Las infecciones bacterianas que se pueden tratar con una combinación de Compuesto I y meropenem pueden comprender un amplio espectro de bacterias. Los organismos ejemplo incluyen bacterias grampositivas, bacterias gramnegativas, bacterias aerobias y anaerobias, tales como Staphylococcus, Lactobacillus, Streptococcus, Sarcina, Escherichia, Enterobacter, Klebsiella, Pseudomonas, Acinetobacter, Mycobacterium, Proteus, Campylobacter, Citrobacter, Nisseria, Baccillus, Bacteroides, Peptococcus, Clostridium, Salmonella, Shigella, Serratia, Haemophilus, Brucella y otros organismos.
Más ejemplos de infecciones bacterianas incluyen Pseudomonas aeruginosa, Pseudomonas fluorescens, Pseudomonas acidovorans, Pseudomonas alcaligenes, Pseudomonas putida, Stenotrophomonas maltophilia, Burkholderia cepacia, Aeromonas hydrophilia, Escherichia coli, Citrobacter freundii, Salmonella typhimurium, Salmonella typhi, Salmonella paratyphi, Salmonella enteritidis, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiella oxytoca, Serratia marcescens, Francisella tularensis, Morganella morganii, Proteus mirabilis, Proteus vulgaris, Providencia alcalifaciens, Providencia rettgeri, Providencia stuartii, Acinetobacter baumannii, Acinetobacter calcoaceticus, Acinetobacter haemolyticus, Yersinia enterocolitica, Yersinia pestis, Yersinia pseudotuberculosis, Yersinia intermedia, Bordetella pertussis, Bordetella parapertussis, Bordetella bronchiseptica, Haemophilus influenzae, Haemophilus parainfluenzae, Haemophilus haemolyticus, Haemophilus parahaemolyticus, Haemophilus ducreyi, Pasteurella multocida, Pasteurella haemolytica, Branhamella catarrhalis, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni, Campylobacter coli, Borrelia burgdorferi, Vibrio cholerae, Vibrio parahaemolyticus, Legionella pneumophila, Listeria monocytogenes, Neisseria gonorrhoeae, Neisseria meningitidis, Kingella, Moraxella, Gardnerella vaginalis, Bacteroides fragilis, Bacteroides distasonis, el grupo de homología de Bacteroides 3452A, Bacteroides vulgatus, Bacteroides ovalus, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides eggerthii, Bacteroides splanchnicus, Clostridium difficile, Mycobacterium tuberculosis, Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium leprae, Corynebacterium diphtheriae, Corynebacterium ulcerans, Streptococcus pneumoniae, Streptococcus agalactiae, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, Staphylococcus intermedius, Staphylococcus hyicus subsp. hyicus, Staphylococcus haemolyticus, Staphylococcus hominis o Staphylococcus saccharolyticus.
En algunas realizaciones, la infección está causada por una bacteria seleccionada entre Pseudomonas aeruginosa, Pseudomonas fluorescens, Stenotrophomonas maltophilia, Escherichia coli, Citrobacter freundii, Salmonella typhimurium, Salmonella typhi, Salmonella paratyphi, Salmonella enteritidis, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiella oxytoca, Serratia marcescens, Acinetobacter calcoaceticus, Acinetobacter haemolyticus, Yersinia enterocolitica, Yersinia pestis, Yersinia pseudotuberculosis, Yersinia intermedia, Haemophilus influenzae, Haemophilus parainfluenzae, Haemophilus haemolyticus, Haemophilus parahaemolyticus, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni, Campylobacter coli, Vibrio cholerae, Vibrio parahaemolyticus, Legionella pneumophila, Listeria
monocytogenes, Neisseria gonorrhoeae, Neisseria meningitidis, Moraxella, Bacteroides fragilis, Bacteroides vulgatus, Bacteroides ovalus, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides eggerthii o Bacteroides splanchnicus.
En algunas realizaciones, la infección bacteriana es una infección gramnegativa. En algunas realizaciones, la infección bacteriana es una infección del tracto respiratorio inferior. En algunas realizaciones, la infección bacteriana es causada por Pseudomonas aeruginosa. En algunas realizaciones, la infección bacteriana es causada por Klebsiella pneumonia.
Compuestos Antibacterianos
El Compuesto I tiene las estructuras que se muestran a continuación:

En algunas realizaciones, debido al fácil intercambio de ésteres de boro, el Compuesto I se puede convertir o existir en equilibrio con formas alternativas. Por consiguiente, en algunas realizaciones, el Compuesto I puede existir en combinación con una o más de estas formas. Por ejemplo, el Compuesto I puede existir en combinación con una o más formas de cadena abierta (Fórmula la), forma dimérica (Fórmula Ib), forma dimérica cíclica (Fórmula Ic), forma trimérica (Fórmula Id), forma trimérica cíclica (Fórmula Ie), y similares. El Compuesto I y su enantiómero, diastereoisómero o tautómero, o sal aceptable para uso farmacéutico se describe en la Patente de los Estados Unidos Núm. 8.680.136.

El meropenem es un antibiótico inyectable de espectro ultra-amplio usado para tratar una amplia variedad de infecciones. Es un p-lactámico y pertenece al subgrupo de los carbapenemas. Tiene la estructura que se muestra a continuación:

Algunas realizaciones incluyen el Compuesto I para su uso en procedimientos para tratar o prevenir una infección bacteriana que comprenden administrar a un sujeto que lo necesita, una cantidad efectiva del Compuesto I y meropenem, en el que el Compuesto I puede estar en cualquiera de las formas descritas anteriormente o una combinación de las mismas.
Algunas realizaciones además comprenden la administración de un medicamento adicional, ya sea en una composición separada o en la misma composición. En algunas realizaciones, el medicamento adicional incluye un agente antibacteriano, un agente antifúngico, un agente antivírico, un agente antiinflamatorio o un agente antialérgico. En algunas realizaciones, el medicamento adicional comprende un agente antibacteriano tal como una (3-lactama) adicional.
En algunas realizaciones, la p-lactama adicional incluye Amoxicilina, Ampicilina (Pivampicilina, Hetacilina, Bacampicilina, Metampicilina, Talampicilina), Epicilina, Carbenicilina (Carindacilina), Ticarcilina, Temocilina, Azlocilina, Piperacilina, Mezlocilina, Mecillinam (Pivmecillinam), Sulbenicilina, Bencilpenicilina (G), Clometocilina, Benzatina bencilpenicilina, Procaína bencilpenicilina, Azidocilina, Penamecilina, Fenoximetilpenicilina (V), Propicilina, Fenoximetilpenicilina benzatina, Feneticilina, Cloxacilina (Dicloxacilina, Flucloxacilina), Oxacilina, Meticilina, Nafcilina, Faropenem, Biapenem, Doripenem, Ertapenem, Imipenem, Panipenem, Tomopenem, Razupenem, Tebipenem, Sulopenem, Cefazolina, Cefacetrilo, Cefadroxil, Cefalexina, Cefaloglicina, Cefalonio, Cefaloridina, Cefalotina, Cefapirina, Cefatrizina, Cefazedona, Cefazaflur, Cefradina, Cefroxadina, Ceftezol, Cefaclor, Cefamandole, Cefminox, Cefonicid, Ceforanida, Cefotiam, Cefprozil, Cefbuperazona, Cefuroxima, Cefuzonam, Cefoxitina, Cefotetán, Cefmetazol, Loracarbef, Cefixima, Ceftazidima, Ceftriaxona, Cefcapeno, Cefdaloxima, Cefdinir, Cefditoren, Cefetamet, Cefmenoxima, Cefodizima, Cefoperazona, Cefotaxima, Cefpimizol, Cefpiramida, Cefpodoxima, Cefsulodina, Cefteram, Ceftibuten, Ceftioleno, Ceftizoxima, Flomoxef, Latamoxef, Cefepim, Cefozopran, Cefpirom, Cefquinom, Ceftobiprol, Ceftarolina, Ceftolozano, CXA-101, RWJ-54428, MC-04,546, ME1036, BAL30072, SYN 2416, Ceftiofur, Cefquinom, Cefovecin, Aztreonam, Tigemonam, Carumonam, RWJ-442831, RWJ-333441, RWJ-333442, S649266, GSK3342830 y AIC 499.
En algunas realizaciones, el p-lactámico adicional incluye Ceftazidima, Doripenem, Ertapenem, Imipenem o Panipenem.
Algunas realizaciones incluyen una composición farmacéutica que comprende una cantidad efectiva para uso terapéutico de cualquiera de los compuestos anteriores y un excipiente aceptable para uso farmacéutico.
Administración y composiciones farmacéuticas
Algunas realizaciones incluyen composiciones farmacéuticas que comprenden: (a) una cantidad segura y efectiva para uso terapéutico del Compuesto I, o su correspondiente enantiómero, diastereoisómero o tautómero, o sal aceptable para uso farmacéutico; (b) meropenem, y (c) un soporte aceptable para uso farmacéutico.
El Compuesto I y el meropenem se administran a una dosis efectiva para uso terapéutico, por ejemplo, una dosis suficiente para proporcionar tratamiento para los estados de enfermedad descritos anteriormente. En algunas realizaciones, una dosis única de Compuesto I y meropenem puede oscilar entre aproximadamente 250 mg y aproximadamente 5000 mg o entre aproximadamente 1000 mg y aproximadamente 3000 mg. En algunas realizaciones, el Compuesto I y el meropenem se pueden administrar al menos una vez al día, por ejemplo de 1 a 5 veces al día.
La administración de las composiciones farmacéuticas descritas en la presente memoria se puede llevar a cabo a través de cualquiera de los modos de administración aceptados para los agentes que tienen utilidades similares, que incluyen, pero sin limitarse a ello, por vía oral, sublingual, bucal, subcutánea, intravenosa, intranasal, tópica, transdérmica, intradérmica, intraperitoneal, intramuscular, intrapulmonar, vaginal, rectal o intraocular. Las administraciones intravenosas, orales y parenterales son habituales en el tratamiento de las indicaciones que son objeto de las realizaciones preferentes.
El Compuesto I y el meropenem se pueden formular en composiciones farmacéuticas para su uso en el tratamiento de estas afecciones. Se usan técnicas estándar de formulación farmacéutica, tales como las desveladas en Remington's The Science and Practice of Pharmacy, 21st Ed., Lippincott Williams & Wilkins (2005).
Además del Compuesto I y el meropenem, algunas realizaciones incluyen composiciones que contienen un portador aceptable para uso farmacéutico. El término “portador aceptable para uso farmacéutico”, como se usa en la presente memoria, significa uno o más diluyentes de carga sólidos o líquidos compatibles o sustancias encapsulantes, que son adecuados para la administración a un mamífero. El término “compatible”, como se usa en la presente memoria, significa que los componentes de la composición se pueden mezclar con el compuesto en cuestión, y entre sí, de forma que no se produzcan interacciones que reduzcan sustancialmente la eficacia farmacéutica de la composición en situaciones de uso ordinario. Los portadores aceptables para uso farmacéutico deben, por supuesto, ser de una pureza suficientemente alta y de una toxicidad suficientemente baja para hacerlos adecuados para su administración preferentemente a un animal, preferentemente mamífero, que esté siendo tratado.
Algunos ejemplos de sustancias que pueden servir como portadores aceptables para uso farmacéutico o componentes de los mismos son los azúcares, tales como la lactosa, la glucosa y la sacarosa; los almidones, tales como el almidón de maíz y la fécula de patata; la celulosa y sus derivados, tales como la carboximetilcelulosa sódica, la etilcelulosa y la metilcelulosa; el tragacanto en polvo; la malta; la gelatina; el talco; los lubricantes sólidos, tales como el ácido esteárico y el estearato de magnesio; el sulfato de calcio; aceites vegetales, tales como el aceite de cacahuete, el aceite de semilla de algodón, el aceite de sésamo, el aceite de oliva, el aceite de maíz y el aceite de teobroma; polioles, tales como el propilenglicol, la glicerina, el sorbitol, el manitol y el polietilenglicol; ácido algínico emulgentes, tales como el TWEENS; agentes humectantes, tales como el lauril sulfato de sodio; agentes colorantes; agentes aromatizantes; agentes para la elaboración de comprimidos, estabilizadores; antioxidantes; conservantes; agua libre de pirógenos; solución salina isotónica; y soluciones tampón de fosfato.
La elección de un portador aceptable para uso farmacéutico que se usará junto con la combinación viene determinada básicamente por la forma en que se administrará la combinación.
Las composiciones descritas en la presente memoria se suministran preferentemente en forma de dosificación unitaria. Como se usa en la presente memoria, una “forma de dosificación unitaria” es una composición que contiene una cantidad de un compuesto o una composición que es adecuada para su administración a un animal, preferentemente un sujeto mamífero, en una sola dosis, de acuerdo con la buena práctica médica. Sin embargo, la preparación de una forma de dosificación única o unitaria no implica que la forma de dosificación se administre una vez al día o una vez por curso de terapia. Dichas formas de dosificación se contemplan para ser administradas una, dos, tres o más veces al día y pueden ser administradas como infusión durante un período de tiempo (por ejemplo, de aproximadamente 30 minutos a aproximadamente 2 a 6 horas), o administradas como infusión continua, y pueden ser administradas más de una vez durante un curso de terapia, aunque no se excluye específicamente una única administración. Los expertos reconocerán que la formulación no contempla específicamente todo el curso de la terapia y tales decisiones se dejan para los expertos en la técnica del tratamiento en lugar de la formulación.
Las composiciones útiles de acuerdo con lo descrito anteriormente pueden estar en cualquiera de una variedad de formas adecuadas para una variedad de vías de administración, por ejemplo, para la administración oral, sublingual, bucal, nasal, rectal, tópica (que incluyen transdérmica e intradérmica), ocular, intracerebral, intracraneal, intratecal, intraarterial, intravenosa, intramuscular u otras vías parentales de administración. Los expertos apreciarán que las composiciones orales y nasales incluyen composiciones que se administran por inhalación, y que se elaboran mediante el uso de las metodologías disponibles. Dependiendo de la vía particular de administración deseada, se puede usar una variedad de portadores aceptables para uso farmacéutico muy conocidos en la técnica. Los portadores aceptables para uso farmacéutico incluyen, por ejemplo, cargas sólidas o líquidas, diluyentes, hidrótropos, agentes tensioactivos y sustancias encapsulantes. Se pueden incluir materiales farmacéuticamente activos opcionales, que no interfieran sustancialmente con la actividad del compuesto o la composición. La cantidad de portador empleada junto con el compuesto o la composición es suficiente para proporcionar una cantidad práctica de material para la administración por dosis unitaria del compuesto. Las técnicas y composiciones para hacer formas de dosificación útiles en los procedimientos descritos en la presente memoria se describen en las siguientes referencias, todas ellas incorporadas por referencia a la presente memoria: Modern Pharmaceutics, 4ta edición, Capítulos 9 y 10 (Banker & Rhodes, editores, 2002) Lieberman et al., Pharmaceutical Dosage Forms: Tablets (1989); y Ansel, Introduction to Pharmaceutical Dosage Forms, 8va Edición (2004). En algunas realizaciones, las composiciones farmacéuticas se administran por vía intravenosa. En algunas realizaciones, las composiciones farmacéuticas se administran por vía oral. En algunas otras realizaciones, las composiciones farmacéuticas se administran por vía intraperitoneal.
Se pueden usar varias formas de dosificación oral, que incluyen formas sólidas tales como comprimidos, cápsulas (por ejemplo, cápsula de gel líquido y cápsula de gel sólido), gránulos y polvos a granel. Estas formas orales comprenden una cantidad segura y efectiva, por lo general de al menos aproximadamente 5%, con un máximo de aproximadamente 90%, del compuesto. Los comprimidos pueden ser comprimidos, triturados, con revestimiento entérico, con revestimiento de azúcar, con revestimiento de película o comprimidos múltiples, que contienen aglutinantes adecuados, lubricantes, diluyentes, agentes desintegrantes, agentes colorantes, agentes aromatizantes, agentes inductores de flujo y agentes fundentes. Las formas de dosificación oral líquida incluyen soluciones acuosas, emulsiones, suspensiones, soluciones y/o suspensiones reconstituidas a partir de gránulos no efervescentes, y preparados efervescentes reconstituidos a partir de gránulos efervescentes, que contienen disolventes adecuados, conservantes, agentes emulsionantes, agentes de suspensión, diluyentes, edulcorantes, agentes fundentes, agentes colorantes y agentes aromatizantes.
Los portadores aceptables para uso farmacéutico adecuados para la preparación de formas de dosificación unitarias para la administración peroral son muy conocidos en la técnica. Los comprimidos suelen incluir adyuvantes convencionales compatibles farmacéuticamente como diluyentes inertes, tales como carbonato de calcio, carbonato de sodio, manitol, lactosa y celulosa; aglutinantes tales como almidón, gelatina y sacarosa; disgregantes tales como almidón, ácido algínico y croscarmelosa; lubricantes tales como estearato de magnesio, ácido esteárico y talco. Para mejorar las características de fluidez de la mezcla de polvos se pueden usar glidantes tales como el dióxido de silicio. Se pueden añadir agentes colorantes, tales como los colorantes FD&C, para mejorar el aspecto. Los edulcorantes y los agentes aromatizantes, tales como el aspartamo, la sacarina, el mentol, la menta, la sacarosa y los sabores frutales, son adyuvantes útiles para los comprimidos masticables. Las cápsulas comprenden típicamente
uno o más diluyentes sólidos desvelados anteriormente. La selección de los componentes del portador depende de consideraciones secundarias, como el sabor, el coste y la estabilidad en el estante, que no son críticas, y pueden ser fácilmente llevadas a cabo por los expertos en la técnica.
Las composiciones perorales también incluyen soluciones líquidas, emulsiones, suspensiones y similares. Los portadores aceptables para uso farmacéutico adecuados para la preparación de dichas composiciones son muy conocidos en la técnica. Los componentes típicos de los soportes para jarabes, elixires, emulsiones y suspensiones son el etanol, el glicerol, el propilenglicol, el polietilenglicol, la sacarosa líquida, el sorbitol y el agua. Para una suspensión, los agentes de suspensión típicos incluyen la metilcelulosa, la carboximetilcelulosa sódica, el AVICEL RC-591, el tragacanto y el alginato de sodio; los agentes humectantes típicos incluyen la lecitina y el polisorbato 80; y los conservantes típicos incluyen el metilparabeno y el benzoato de sodio. Las composiciones líquidas perorales también pueden contener uno o más componentes, tales como los edulcorantes, los agentes aromatizantes y los colorantes desvelados anteriormente.
Dichas composiciones también pueden ser revestidas por procedimientos convencionales, típicamente con revestimientos dependientes del pH o del tiempo, de forma que la composición en cuestión sea liberada en el tracto gastrointestinal en la proximidad de la aplicación tópica deseada, o en varios momentos para extender la acción deseada. Tales formas de dosificación incluyen típicamente, pero no se limitan a, uno o más de ftalato de acetato de celulosa, ftalato de polivinilacetato, ftalato de hidroxipropilmetilcelulosa, etilcelulosa, revestimientos de Eudragit, ceras y goma laca.
Las composiciones en la presente memoria descritas pueden incluir opcionalmente otros activos farmacológicos. Otras composiciones útiles para lograr la administración sistémica de los compuestos en cuestión incluyen formas de dosificación sublinguales, bucales y nasales. Dichas composiciones suelen incluir una o más sustancias de relleno solubles, tales como la sacarosa, el sorbitol y el manitol, y aglutinantes tales como la acacia, la celulosa microcristalina, la carboximetilcelulosa y la hidroxipropilmetilcelulosa. También se pueden incluir los glidificantes, lubricantes, edulcorantes, colorantes, antioxidantes y aromatizantes desvelados anteriormente.
Una composición líquida, formulada para uso oftálmico tópico, está formulada de forma que pueda ser administrada tópicamente en el ojo. La comodidad se debería maximizar en la medida de lo posible, aunque a veces las consideraciones de formulación (por ejemplo, la estabilidad del fármaco) pueden requerir una comodidad menos óptima. En el caso de que no se pueda maximizar la comodidad, el líquido se puede formular de forma que sea tolerable para el paciente para su uso tópico oftálmico. Además, un líquido oftálmico aceptable puede ya sea estar envasado para un solo uso o contener un conservante para evitar la contaminación en múltiples usos.
Para la aplicación oftálmica, las soluciones o medicamentos se preparan a menudo mediante el uso de una solución salina fisiológica como portador principal. Las soluciones oftálmicas se pueden mantener preferentemente a un pH cómodo con un sistema tampón adecuado. Las formulaciones también pueden contener conservantes, estabilizadores y tensioactivos convencionales, aceptables para uso farmacéutico.
Los conservantes que se pueden usar en las composiciones farmacéuticas en la presente memoria desveladas incluyen, pero no se limitan a, cloruro de benzalconio, PHMB, clorobutanol, timerosal, fenilmercúrico, acetato y nitrato fenilmercúrico. Un tensioactivo útil es, por ejemplo, el Tween 80. Asimismo, en las preparaciones oftálmicas en la presente memoria desveladas se pueden usar diversos portadores útiles. Estos portadores incluyen, entre otros, alcohol polivinílico, povidona, hidroxipropilmetilcelulosa, poloxámeros, carboximetilcelulosa, hidroxietilcelulosa y agua purificada.
Se pueden añadir ajustadores de tonicidad de acuerdo con lo necesario o conveniente. Incluyen, pero no se limitan a, sales, particularmente cloruro de sodio, cloruro de potasio, manitol y glicerina, o cualquier otro ajustador de tonicidad adecuado y oftálmicamente aceptable.
Se pueden usar diversos tampones y medios para ajustar el pH, a condición de que la preparación resultante sea oftalmológicamente aceptable. Para muchas composiciones, el pH estará entre 4 y 9. En consecuencia, los tampones incluyen tampones de acetato, tampones de citrato, tampones de fosfato y tampones de borato. Se pueden usar ácidos o bases para ajustar el pH de estas formulaciones de acuerdo con lo necesario.
En un sentido similar, un antioxidante oftálmicamente aceptables incluye, pero no se limitan a, metabisulfito de sodio, tiosulfato de sodio, acetilcisteína, hidroxianisol butilado e hidroxitolueno butilado.
Otros componentes excipientes que se pueden incluir en los preparados oftálmicos son los agentes quelantes. Un agente quelante útil es el edetato de disodio (EDTA), aunque también se pueden usar otros agentes quelantes en lugar de éste o junto con él.
Para uso tópico, se emplean cremas, ungüentos, geles, soluciones o suspensiones, etc., que contienen la composición en la presente memoria desvelada. Las formulaciones tópicas pueden estar compuestas generalmente por un portador farmacéutico, un co-disolvente, un emulsionante, un potenciador de la penetración, un sistema de conservación y un emoliente.
Para la administración intravenosa, las composiciones en la presente memoria descritas se pueden disolver o dispersar en un diluyente aceptable para uso farmacéutico, tal como una solución salina o dextrosa. Se pueden incluir excipientes adecuados para lograr el pH deseado, que incluyen pero no limitándose a NaOH, carbonato de sodio, acetato de sodio, HCl y ácido cítrico. En varias realizaciones, el pH de la composición final oscila entre 2 y 8, o preferentemente entre 4 y 7. Los excipientes antioxidantes pueden incluir bisulfito de sodio, bisulfito de sodio de acetona, formaldehido de sodio, sulfoxilato, tiourea y EDTA. Otros ejemplos no limitantes de excipientes adecuados que se encuentran en la composición intravenosa final pueden ser los fosfatos de sodio o de potasio, el ácido cítrico, el ácido tartárico, la gelatina y los hidratos de carbono tales como la dextrosa, el manitol y el dextrano. Otros excipientes aceptables se describen en Powell, et al., Compendium of Excipients for Parenteral Formulations, PDA J Pharm Sci and Tech 1998, 52238 a 311 y Nema et al., Excipients and Their Role in Approved Injectable Products: Usage and Future Directions, PDA J Pharm Sci and Tech 2011, 65 287 a 332. También se pueden incluir agentes antimicrobianos para conseguir una solución bacteriostática o fungistática, entre los que se incluyen, pero sin limitarse a ellos, el nitrato fenilmercúrico, el timerosal, el cloruro de bencetonio, el cloruro de benzalconio, el fenol, el cresol y el clorobutanol.
La composición resultante se puede infundir en el paciente durante un período de tiempo. En diversas realizaciones, el tiempo de infusión oscila entre 5 minutos y una infusión continua, entre 10 minutos y 8 horas, entre 30 minutos y 4 horas, y entre 1 hora y 3 horas. En una realización, el fármaco se infunde durante un período de 3 horas. La infusión se puede repetir en el intervalo de dosis deseado, que puede incluir, por ejemplo, 6 horas, 8 horas, 12 horas o 24 horas.
Las composiciones para la administración intravenosa se pueden proporcionar a los cuidadores en forma de uno más sólidos que se reconstituyen con un diluyente adecuado tales como agua estéril, solución salina o dextrosa en agua poco antes de la administración. Las soluciones concentradas reconstituidas se pueden diluir en soluciones parenterales que tienen un volumen de aproximadamente 25 a aproximadamente 1000 ml, de aproximadamente 30 a aproximadamente 500 ml, o de aproximadamente 50 a aproximadamente 250 ml. En otras realizaciones, las composiciones se suministran en solución lista para ser administrada por vía parenteral. En otras realizaciones, las composiciones se proporcionan en una solución que se diluye aún más antes de la administración. En las realizaciones que incluyen la administración de una combinación de un compuesto descrito en la presente memoria y otro agente, la combinación se puede proporcionar a los cuidadores como una mezcla, o los cuidadores pueden mezclar los dos agentes antes de la administración, o los dos agentes se pueden administrar por separado.
La dosis real de los compuestos activos en la presente memoria descritos depende del compuesto específico y de la afección a tratar; la selección de la dosis adecuada está dentro de los conocimientos de los expertos.
Kits para Administración Intravenosa
Algunas realizaciones incluyen un kit que comprende el Compuesto I y un agente antibacteriano carbapenem Meropenem. En algunas realizaciones, los kits se usan para administración intravenosa.
En una realización, ambos componentes se suministran en un único recipiente estéril. En el caso de los sólidos para reconstitución, los agentes se pueden premezclar y añadir al recipiente simultáneamente o se pueden introducir en polvo seco en el recipiente en dos etapas separadas. En algunas realizaciones, los sólidos son productos cristalinos estériles. En otra realización, los sólidos son liófilos. En una realización, ambos componentes se liofilizan juntos. Otros ejemplos no limitantes de excipientes adecuados que se encuentran en la composición intravenosa final pueden ser los fosfatos de sodio o de potasio, el ácido cítrico, el ácido tartárico, la gelatina y los hidratos de carbono tales como la dextrosa, el manitol y el dextrano. Una realización incluye sólidos no estériles que se irradian antes o después de introducirlos en el recipiente.
En el caso de un líquido, los agentes se pueden disolver o dispersar en un diluyente listo para su administración. En otra realización, la solución o dispersión se puede diluir aún más antes de su administración. Algunas realizaciones incluyen el suministro del líquido en una bolsa intravenosa. El líquido se puede congelar para mejorar su estabilidad. En una realización, el recipiente incluye otros ingredientes tales como un ajustador de pH, un agente solubilizante o un agente dispersante. Los ejemplos no limitantes de ajustadores del pH incluyen NaOH, carbonato de sodio, acetato de sodio, HCl y ácido cítrico.
En una realización alternativa, los dos componentes se pueden suministrar en recipientes separados. Cada recipiente puede incluir un sólido, una solución o una dispersión. En tales realizaciones, los dos recipientes se pueden suministrar en un único envase o por separado. En una realización, el compuesto descrito en la presente memoria se proporciona como una solución mientras que el agente adicional (por ejemplo, agente antibacteriano) se proporciona como un sólido listo para la reconstitución. En una de estas realizaciones, la solución del compuesto descrito en la presente memoria se usa como diluyente para reconstituir el otro agente.
En algunas realizaciones, el kit puede comprender uno o más medicamentos adicionales seleccionados entre un agente antibacteriano, un agente antifúngico, un agente antiviral, un agente antiinflamatorio o un agente antialérgico. Los medicamentos adicionales se pueden preparar del mismo modo que se ha descrito anteriormente.
Ejemplos
Los siguientes ejemplos, incluidos los experimentos y resultados obtenidos, se proporcionan únicamente con fines ilustrativos y no se deben interpretar como limitativos de la presente solicitud.
Ejemplo 1
El Ejemplo 1 proporciona un resumen de un estudio clínico de la seguridad, tolerabilidad y farmacocinética del inhibidor de betalactamasa Compuesto I en sujetos adultos sanos.
Procedimientos: Se reclutaron 56 sujetos sanos en una de las 7 cohortes de 8 sujetos cada una en la fase de dosis única ascendente (250 mg, 500 mg, 750 mg, 1000 mg, 1250 mg, 1500 mg y 2000 mg). A continuación, se incluyó a otros 32 sujetos en una de las 4 cohortes de la fase de dosis múltiples (250 mg, 1000 mg, 1500 mg y 2000 mg, administrados q8h durante 7 días). Dentro de cada cohorte, los sujetos fueron asignados aleatoriamente al Compuesto I (n = 6) o a placebo salino normal (n = 2). Todas las infusiones se administraron durante 3 horas. Se obtuvieron muestras de plasma y orina tras dosis únicas o múltiples y se analizó su contenido en Compuesto I por medio de un procedimiento validado de HPLC/MS.
Resultados: La Tabla 1 resume la farmacocinética media del Compuesto I en diferentes dosis. El perfil de concentración del Compuesto I en función del tiempo tras una única fusión IV y el perfil AUC del Compuesto I en función de la dosis se ilustran en las FIGs. 1 y 2 respectivamente.

Las concentraciones máximas para el Compuesto I se alcanzaron al final de la infusión de 3 horas. La exposición al Compuesto I (Cmáx y AUC) aumentó de forma proporcional a la dosis tras la administración de dosis únicas y múltiples (véanse las FIGs. 1 y 2). No hubo indicios de acumulación con dosis múltiples, lo que concuerda con la semivida terminal observada (< 2 horas). Tanto el volumen de distribución como el aclaramiento plasmático fueron independientes de la dosis. Se midieron altas concentraciones de las FIGs. 1 y 2 en la orina. La recuperación urinaria fue del 80% o superior durante 48 horas en todos los grupos de dosis.
Ningún sujeto interrumpió el estudio debido a eventos adversos (EA) y no se observaron eventos adversos graves (EAG). Los EA fueron similares entre los sujetos tratados con el Compuesto I y los tratados con placebo, sin evidencia de aumento de la incidencia o gravedad de los EA con el aumento de la dosis, y todos los EA fueron leves o moderados.
Conclusión: El Compuesto I fue seguro y bien tolerado en todas las dosis probadas. El AUC y la Cmáx aumentaron proporcionalmente independientemente de la dosis.
Ejemplo 2
El Ejemplo 2 proporciona un resumen de un estudio clínico de la seguridad, tolerabilidad y farmacocinética del inhibidor de betalactamasa Compuesto I solo, meropenem solo y la combinación de ambos en sujetos adultos sanos.
Procedimiento: Se inscribieron 80 sujetos sanos en 1 de 5 cohortes en la fase de dosis única ascendente (250 mg, 1000 mg, 1500 mg y 2000 mg de Compuesto I en combinación con 1 o 2 g de meropenem). Dentro de cada cohorte, se administró a los sujetos dosis únicas del Compuesto I o de meropenem el día 1, y del Compuesto I o de meropenem el día 3. La combinación de ambos fármacos se administró el día 7. Todos los fármacos se infundieron durante 3 horas. Se obtuvieron muestras de plasma y orina y se analizaron por medio de procedimientos HPLC/MS validados. La farmacocinética del Compuesto I solo y en combinación con meropenem tras infusiones de 3 horas en sujetos adultos sanos y la farmacocinética del meropenem solo y en combinación con el Compuesto I tras infusiones de 3 horas en sujetos adultos sanos se ilustran en las FIGs. 1 y 2 respectivamente.
Resultados: Los parámetros farmacocinéticos, derivados por medio de procedimientos no compartimentales, para cada fármaco solo y en combinación del Compuesto I y meropenem se muestran a continuación en las Tablas 2 y 3. La Tabla 2 resume los parámetros farmacocinéticos del Compuesto I tras la administración de una dosis única del Compuesto I solo o en combinación con meropenem en infusiones de 3 horas a voluntarios sanos (los datos son la media ± desviación estándar). La Tabla 3 resume los parámetros farmacocinéticos de meropenem tras la administración de una dosis única de meropenem solo o en combinación con el Compuesto I en infusiones de 3 horas a voluntarios sanos (los datos son la media ± desviación estándar).


Las concentraciones máximas del Compuesto I y del meropenem se alcanzaron al final de las infusiones de 3 horas. Las exposiciones al Compuesto I y al meropenem (Cmáx y AUC) aumentaron proporcionalmente con la dosis. Los parámetros PK del Compuesto I y meropenem tras una dosis única sola o en combinación no muestran cambios importantes en las propiedades PK de ninguno de los dos fármacos (Tablas 2 y 3). La PK de meropenem solo y en combinación con el Compuesto I observada en este estudio concuerda con la bibliografía publicada. Véase, por ejemplo, Lodise T.P. et al., “Penetration of meropenem into epithelial lining fluid of patients with ventilator-associated pneumonía", Antimicrob Agents Chemother. 2011; 55 (4):1606 a 1610 y Kuti J. L. et al., “Use of Monte Carlo simulation to design an optimized pharmacodynamics dosing strategy for meropenem,’’ J. Clin. Pharmacol. 2003; 43 (10): 1116 a 23.
La Tabla 4 resume los eventos adversos (EA) emergentes del tratamiento observados en > 3 sujetos que recibieron la combinación del Compuesto I y meropenem. Ningún sujeto interrumpió el tratamiento debido a EA y no se observaron EAG. No hubo pruebas de que aumentara el número o la gravedad de los EA con el aumento de la dosis de cualquiera de los fármacos solos o en combinación, y todos los EA fueron de gravedad leve o moderada.

Conclusión: El Compuesto I solo y en combinación con 1 o 2 g de meropenem fue seguro y bien tolerado a todas las dosis probadas. El AUC y la Cmáx aumentaron proporcionalmente con la dosis y los parámetros farmacocinéticos del Compuesto I y del meropenem son similares. No hubo efectos del meropenem o del Compuesto I sobre la PK del otro agente.
Ejemplo 3
El Ejemplo 3 proporciona un resumen de un estudio clínico de la seguridad, tolerabilidad y farmacocinética del inhibidor de betalactamasa Compuesto I solo, meropenem solo y la combinación de ambos tras 7 días de TID (tres veces al día) en sujetos adultos sanos.
Procedimiento: Se inscribieron 80 sujetos sanos en 1 de 5 cohortes en la fase de dosis única ascendente (250 mg, 1000 mg, 1500 mg y 2000 mg de Compuesto I en combinación con 1 o 2 g de meropenem). Dentro de cada cohorte, a los sujetos se les administró el Compuesto I o meropenem el día 1, posteriormente se les cruzó al Compuesto I o meropenem el día 3, posteriormente se les administró tanto el Compuesto I como meropenem en combinación el día 7 seguido por 7 días de dosificación TID. Todas las infusiones se administraron durante 3 horas. Se obtuvieron muestras intensivas de plasma y orina PK después de la dosificación y se analizaron por medio de procedimientos HPLC/MS validados. La farmacocinética plasmática del Compuesto I solo y en combinación con meropenem tras una y 7 días de dosificación TID por medio de infusiones de 3 horas en sujetos sanos y la farmacocinética plasmática del meropenem solo y en combinación con el Compuesto I tras una y 7 días de dosificación TID por medio de infusiones de 3 horas en sujetos sanos se ilustran en las FIGs.5 y 6 respectivamente.
Resultados: Los parámetros farmacocinéticos, derivados mediante el uso de procedimientos no compartimentales, para cada fármaco solo y en combinación en las cohortes de Compuesto I/meropenem 1 g/1 g y 2 g/ 2 g se muestran en las Tablas 5 y 6 a continuación. La Tabla 5 resume los parámetros farmacocinéticos del Compuesto I (media ± desviación estándar) tras una dosis única sola (única) y una dosis única (primera) seguida por 7 días de dosificación TID (última) del Compuesto I administrado en combinación con meropenem como infusiones de 3 horas a sujetos sanos. La Tabla 6 resume los parámetros farmacocinéticos de meropenem (media ± desviación estándar) tras una dosis única sola (única) y una dosis única (primera) seguida por 7 días de dosificación TID (última) de meropenem administrado en combinación con el Compuesto I como infusiones de 3 horas a sujetos sanos.


Las concentraciones máximas del Compuesto I y del meropenem se alcanzaron al final de las infusiones de 3 horas. La exposición al Compuesto I y al meropenem (Cmáx y AUC) aumentó proporcionalmente a la dosis. Los parámetros PK del Compuesto I y meropenem solos o en combinación no muestran cambios importantes en las propiedades PK de ninguno de los dos fármacos (véanse las Tablas 5 y 6). No se observó acumulación ni del Compuesto I ni del meropenem tras 7 días de dosificación TID. La PK de meropenem solo y en combinación con el Compuesto I observada en este estudio concuerda con la bibliografía publicada.
La Tabla 7 resume el número (%) de sujetos con al menos un EA emergente del tratamiento y el número de eventos adversos durante la fase de dosis múltiples. Un sujeto que recibió meropenem 1 g/Compuesto I 2 g lo interrumpió prematuramente debido a un EA de tromboflebitis. Todos los EA, excepto 2, fueron de gravedad leve o moderada. Sólo se observaron náuseas leves en los sujetos que recibieron meropenem 2 g, solo o en combinación. No hay pruebas de que la adición del Compuesto I cambiara el perfil EA del meropenem.

Conclusión: El Compuesto I solo y en combinación con 1 g o 2 g de meropenem fue seguro y bien tolerado a todas las dosis probadas, sin evidencia de que el perfil de seguridad del meropenem se modificara por la adición del Compuesto I. No se observó acumulación ni del Compuesto I ni del meropenem tras 7 días de dosificación TID. No se observaron efectos del meropenem sobre la farmacocinética del Compuesto I ni viceversa.
Ejemplo 4
El Ejemplo 4 proporciona un resumen de un estudio preliminar de la farmacocinética de la combinación del Compuesto I (2 g) y meropenem (2 g) en sujetos adultos sanos por medio de una infusión de 1 o 3 horas.
Resultados: La farmacocinética del Compuesto I tras infusiones de 3 horas o 1 hora (2 g de Compuesto I solo y en combinación con 2 g de meropenem) en sujetos sanos se ilustra en las FIGs. 7 y 8 respectivamente. La farmacocinética media del Compuesto I tras infusiones de 1 o 3 horas de 2 g de Compuesto I en combinación con 2 g de meropenem en sujetos sanos se resume en la FIG. 9. Con respecto al Compuesto I, no se observaron efectos del meropenem sobre la farmacocinética del Compuesto I con ninguna de las dos velocidades de infusión. Además, no hay un efecto significativo de la velocidad de infusión sobre la exposición al Compuesto I (p = 0,18).
La farmacocinética del meropenem tras infusiones de 3 horas o 1 hora (2 g de meropenem solo y en combinación con 2 g del Compuesto I) en sujetos sanos se ilustra en las FIGs. 10 y 11 respectivamente. La farmacocinética media de meropenem tras infusiones de 1 o 3 horas de 2 g de meropenem en combinación con 2 g del Compuesto I en sujetos sanos se resume en la FIG. 12. La farmacocinética del meropenem lactama abierta tras infusiones de 1 hora de 2 g solo y en combinación con 2 g de Compuesto I y la farmacocinética media del meropenem lactama abierta tras infusiones de 1 o 3 horas de 2 g de meropenem en combinación con 2 g de Compuesto I se ilustran en las FIGs. 13 y 14.
Para el meropenem, no se observaron efectos del Compuesto I sobre la farmacocinética del meropenem con ninguna de las tasas de infusión. La exposición al meropenem (AUC) tras una infusión de 3 horas de 2 g de meropenem es coherente con la bibliografía publicada. Se produjo un aumento de la exposición al meropenem (AUC) con la infusión de 1 hora en comparación con la infusión de 3 horas. La exposición a meropenem (AUC) tras una infusión de 1 hora de 2 g de meropenem es aproximadamente un 48% mayor que la observada tras una infusión de 3 horas de 2 g de meropenem (211 frente a 142 mg*h/L). El aclaramiento ajustado al peso (Cl) de meropenem tras una infusión de 1 hora de 2 g de meropenem es aproximadamente un 25% más lento que el observado tras una infusión de 3 horas (0,14 frente a 0,19 l/h/kg; p = 0,015). Las posibles razones de la diferencia observada en el aclaramiento ajustado al peso de meropenem se pueden deber a un aclaramiento renal saturable a dosis de 2 g debido a una Cmáx elevada o a que una infusión más prolongada reduce la “dosis” debido a la degradación (la apertura del anillo p-lactámico da lugar a la formación de meropenem lactama abierta).
Ejemplo 5
El Ejemplo 5 proporciona un resumen de un estudio abierto de la seguridad y farmacocinética de la combinación del Compuesto I y meropenem en sujetos con función renal reducida, que incluyen pacientes con hemodiálisis estándar. Se evaluó la seguridad y farmacocinética de una dosis IV única de 1 g de meropenem más 1 g del Compuesto I, infundida durante 3 horas. Se inscribieron 41 sujetos en 5 grupos en función de su grado de insuficiencia renal. Las cinco cohortes incluían: pacientes con función renal normal (CrCl > 90 ml/min), insuficiencia renal leve (CrCl 60 a 89ml/min), insuficiencia renal moderada (CrCl 30 a < 60ml/min), insuficiencia renal grave (CrCl < 30ml/min, y pacientes con enfermedad renal terminal que requieren hemodiálisis. No se estudiaron los pacientes en tratamiento renal sustitutivo diferente de la hemodiálisis estándar (incluida la hemofiltración venovenosa continua, la hemodiálisis venovenosa continua y el tratamiento renal sustitutivo continuo).
La FIG. 15 muestra la relación entre la TFG estimada y el aclaramiento plasmático de meropenem o el Compuesto I. El aclaramiento plasmático de ambos fármacos se mantuvo similar en todo el intervalo de la función renal, como evidencian la agrupación de valores y el descenso lineal del aclaramiento con la disminución de la función renal. La eliminación de meropenem y del Compuesto I durante la hemodiálisis se estudió en 9 pacientes con insuficiencia renal grave en hemodiálisis crónica. Los pacientes recibieron una dosis única de meropenem 1 g/Compuesto I 1 g, seguida por una sesión de hemodiálisis. Tanto el meropenem como el Compuesto I se eliminaron del plasma por medio de hemodiálisis. Estos datos indican que se deben administrar dosis de mantenimiento de cada fármaco (ajustadas al grado de función renal endógena subyacente) después de una sesión de diálisis.
Determinación de la combinación de dosis de Compuesto I/meropenem en pacientes con insuficiencia renal El ajuste de la dosis de acuerdo con el grado de insuficiencia renal se determinó por medio del análisis de las estimaciones de la farmacocinética de cada sujeto y la determinación de las exposiciones de acuerdo con los posibles regímenes de dosificación de meropenem o del Compuesto I. El objetivo era mantener las exposiciones (como AUC) en todo el espectro de la función renal lo más consistentes posible. A la vista de los análisis PK-PD en modelos no clínicos que muestran que el AUC está vinculado a la eficacia del Compuesto I, el AUC fue el controlador apropiado de la eficacia de este agente. Dado que T > MIC es el índice PK-PD importante de
meropenem, se evaluaron diferentes intervalos de dosificación para asegurar que el T > MICpunto de ruptura estuviera por encima de los valores umbral (T > MIC > 40%) para la eficacia. A efectos de este análisis, el punto de ruptura de susceptibilidad previsto para el meropenem en base a la dosis de 2 gramos y la infusión de 3 horas fue de 8 pg/ml. Se consideró fármaco libre tanto para el meropenem como para el Compuesto I (unión a proteínas plasmáticas del 6% y 33%, respectivamente).
Meropenem
La Tabla A muestra el AUC de meropenem medido en cada paciente y los índices PK-PD para tres posibles regímenes de dosificación en cada paciente de acuerdo con la PK de meropenem medida en cada sujeto. Se identificaron los regímenes de dosificación de meropenem para cada uno de los estratos de función renal que cumplirían o alcanzarían las exposiciones objetivo (T > MIC de al menos el 40%) en todos los sujetos (véanse las celdas sombreadas).
La Tabla A resume el Análisis de diferentes regímenes de dosificación de meropenem por sujetos individuales. El objetivo PK-PD para meropenem es un T > MIC de al menos el 40% del intervalo de dosis cuando la MIC es de 8 pg/ml. El sombreado en los diferentes grupos de aclaramiento de creatinina denota el régimen de dosificación de meropenem recomendado.
Tabla A.

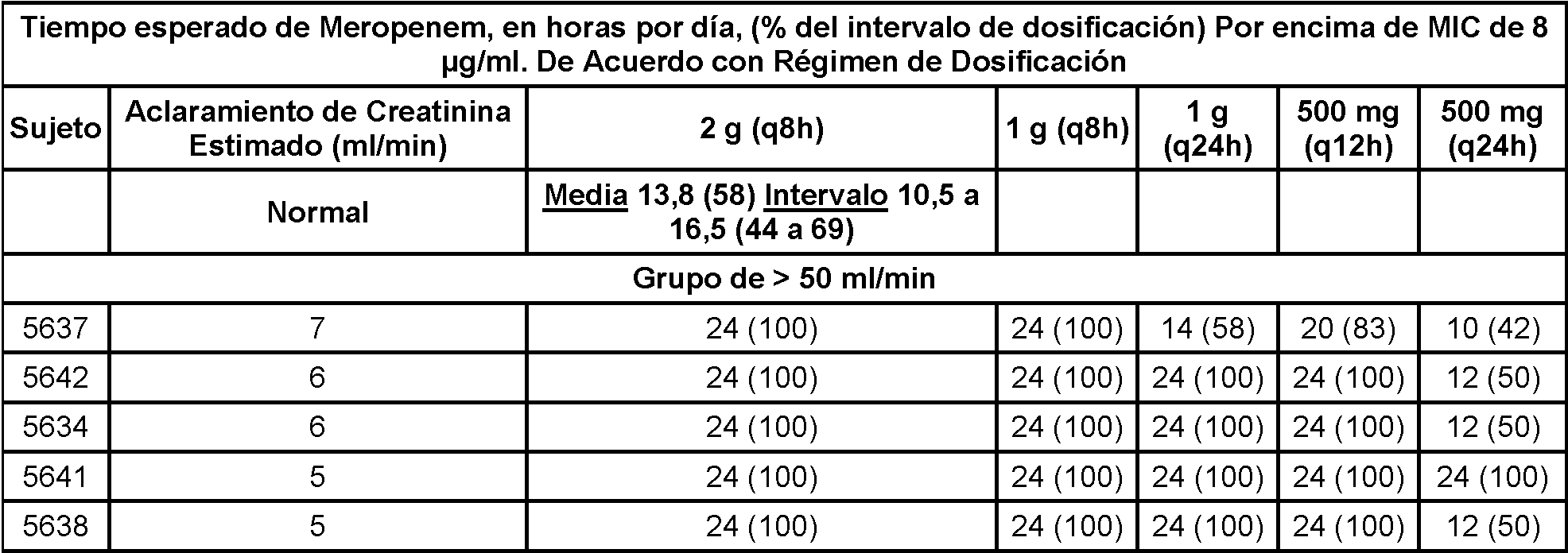
Compuesto I
La Tabla B muestra el AUC del Compuesto I medido en cada paciente y el AUC de 24 hs para tres regímenes de dosificación potenciales de acuerdo con el aclaramiento medido del Compuesto I en cada sujeto. Dado que el AUC es la métrica PK objetivo y que el aclaramiento del Compuesto I se mantuvo cercano al aclaramiento del meropenem, las dosis unitarias y de 24 hs se mantuvieron en una relación 1:1 en todo el intervalo de función renal.
Consideraciones para sujetos con aclaramiento de creatinina < 10 ml/min
Como se muestra en la FIG. 15, a medida que el aclaramiento de creatinina desciende por debajo de 10 ml/min, el aclaramiento no renal de meropenem asume una mayor proporción del aclaramiento total. Por el contrario, el Compuesto I no tiene un aclaramiento no renal medible. Por lo tanto, para mantener una relación de dosis 1:1 que proporcione exposiciones terapéuticas de cada componente y evitar la acumulación del Compuesto I, los pacientes con un aclaramiento de creatinina <10 ml/min deben recibir hemodiálisis cada 3 días aproximadamente (es decir, dos veces por semana).
La Tabla B proporciona un resumen del análisis de diferentes regímenes de dosificación del Compuesto I por sujetos individuales inscritos en el estudio. El sombreado en los diferentes grupos de aclaramiento de creatinina denota el régimen de dosificación de meropenem recomendado.
TABLA B. COMPUESTO I ESPERADO FÁRMACO LIBRE 24 Hs AUC (MG*HR/L)


Con base en el análisis anterior, los regímenes de dosificación de la Combinación de Compuesto I/meropenem de la Tabla C se pueden usar en sujetos con función renal alterada.
TABLA C: DOSIFICACIÓN DE LA COMBINACIÓN DE COMPUESTO I/MEROPENEM de acuerdo con LA
FUNCIÓN RENAL

Se concluye que el ajuste de la dosis para la función renal se puede basar en el meropenem o en el Compuesto I, dado que ambos fármacos se ven afectados de forma similar a medida que disminuye la función renal. En sujetos con un aclaramiento de creatinina igual o superior a 50 ml/min, no es necesario ajustar la dosis. Se puede usar la dosis estándar de 2 g de compuesto 1/2 g de meropenem TID (cada 8 horas). En sujetos con un aclaramiento de creatinina igual o superior a 30 ml/min e inferior a 50 ml/min, se puede usar una dosis reducida de 1 g de Compuesto I/1 g de meropenem TID (cada 8 horas) y seguir consiguiendo los efectos deseados. Para sujetos con un
aclaramiento de creatinina igual o superior a 20 ml/min e inferior a 30 ml/min, se puede usar una dosis reducida de 1 g de Compuesto I/1 g de meropenem administrado cada 12 horas. En sujetos con un aclaramiento de creatinina igual o superior a 10 ml/min e inferior a 20 ml/min, se puede usar una dosis reducida de 500 mg de Compuesto I/500 mg de meropenem administrados cada 12 horas. En sujetos con un aclaramiento de creatinina inferior a 10 ml/min, se puede usar una dosis reducida de 500 mg de Compuesto I/500 mg de meropenem cada 24 horas.
Ejemplo 6
El Ejemplo 6 proporciona un resumen de un estudio clínico aleatorizado y abierto que evalúa las concentraciones plasmáticas, de líquido de revestimiento epitelial (ELF) y de macrófagos alveolares (AM) de la combinación de 2 g de Compuesto I/ 2 g de meropenem (“la Combinación”) en sujetos adultos sanos.
Para las infecciones del tracto respiratorio inferior, se ha defendido que el líquido de revestimiento epitelial (ELF) y los macrófagos alveolares (AM) son sitios de infección importantes para los patógenos extracelulares e intracelulares comunes, respectivamente. Se necesitan estudios con broncoscopia y lavado broncoalveolar (BAL), que puedan evaluar de forma fiable la penetración intrapulmonar de los antibióticos en la ELF y la AM. Los objetivos primarios de este estudio farmacocinético son determinar y comparar las concentraciones plasmáticas, ELF y Am del Compuesto I y meropenem administrados tras múltiples dosis intravenosas (2 g de meropenem/ 2 g de Compuesto I administrados q8h durante 3 dosis) en sujetos adultos sanos de ambos sexos. Un objetivo secundario de este estudio fue evaluar la seguridad y tolerabilidad de la administración intravenosa de la Combinación en sujetos adultos sanos.
Procedimientos de Análisis Farmacocinético
Diseño del estudio y sujetos. Se incluyó en este análisis farmacocinético a un total de veinticinco (n = 25) sujetos de ambos sexos que cumplían los criterios de inclusión en el estudio y completaron todas las fases del estudio farmacocinético. Cada sujeto recibió la Combinación (2 g de meropenem / 2 g de Compuesto I) administrada cada 8 horas durante un total de tres dosis bajo observación directa en el lugar del estudio. Se recolectaron muestras de sangre para medir las concentraciones del fármaco en plasma antes (tiempo 0), y a las 1,5, 2,95, 3,083, 3,25, 3,5, 4, 6 y 8 horas después del inicio de una infusión intravenosa de 3 horas de la tercera dosis combinada. A cada sujeto se le llevó a cabo una única broncoscopia estandarizada con BAL programada en un intervalo de tiempo tras la última dosis de la Combinación, como se indica en la siguiente tabla:

La urea se ha usado comúnmente como marcador endógeno para estimar el volumen aparente de la ELF. Se obtuvieron muestras de sangre para determinar las concentraciones plasmáticas de urea justo antes de la broncoscopia programada. Se obtuvieron alícuotas de BAL para determinar las concentraciones de urea en BAL y el recuento celular con diferencial. El procedimiento estandarizado de broncoscopia con BAL para la recolección de muestras intrapulmonares se ha descrito previamente en las referencias que figuran a continuación.
Ensayos de Fármaco y Urea. Los procedimientos de preparación de muestras y los ensayos para las concentraciones de meropenem, Compuesto I y meropenem lactama abierta en plasma, ELF y AM se llevaron a cabo con una cromatografía líquida de alto rendimiento con detección por espectrometría de masas en MicroConstants, Inc., San Diego, CA (Informes MC14B-0013, MC14B-0015, MC14I-011 y MC14I-0012). Las concentraciones de urea en plasma y BAL se llevaron a cabo con un procedimiento en base a microplacas con una solución cromogénica de O-ftalaldehído en MicroConstants, Inc., San Diego, CA.
Cálculos Farmacocinéticos de las Concentraciones Plasmáticas. Se usaron procedimientos no compartimentales para generar parámetros farmacocinéticos para el meropenem, el Compuesto I y el meropenem lactama abierta en plasma. La concentración plasmática máxima (Cmáx) y el tiempo hasta la Cmáx (Tmáx) se leyeron a partir del perfil de concentración plasmática-tiempo observado tras el inicio de la perfusión intravenosa de la tercera dosis de la Combinación. El área bajo la curva de concentración plasmática-tiempo durante 8 horas (AUC0-8) después de la tercera dosis se calculó con la regla lineal-log trapezoidal (WinNonlin®, versión 6.3, Pharsight Corporation, Cary, Carolina del Norte). La constante de velocidad de eliminación (p) se determinó por medio de regresión no lineal por mínimos cuadrados. La semivida de eliminación (t-io) se calculó por medio de la división de p entre el logaritmo natural de dos. Para el meropenem y el Compuesto I, se calcularon el aclaramiento aparente (CL) y los términos de volumen de distribución (Vss) con las ecuaciones no compartimentales estándar incluidas en el programa WinNonlin®.
Cálculos de Volumen de ELF y Concentraciones de Antibiótico en ELF y AM Los cálculos de volumen de ELF y concentraciones de fármaco en ELF y AM fueron llevados a cabo con sobrenadante de BAL y células pulmonares
(alveolares) (“granulo celular”) de aspirados recuperados de la 2da, 3ra y4ta instilaciones (BAL2). La concentración de fármaco (ABXELF) en el líquido de revestimiento epitelial ELF) se determinó del siguiente modo:
A B X e l f = A B X b a l x ( V b a l / V e l f )
donde ABX bal es la concentración medida de meropenem, Compuesto I o meropenem lactama abierta en el líquido BAL, V bal es el volumen de líquido BAL aspirado, y V elf es el volumen de e Lf muestreado por el BAL. V elf se deriva de lo siguiente:
Velf = V bal x
UreaBAL / Ureap
donde UreaBAL es la concentración de urea en el líquido BAL y UreaP es la concentración de urea en plasma.
La concentración del fármaco (ABXAM) en las células alveolares (CA) se determinó de la siguiente manera:
ABXam = ABXm / Vac
donde ABXM es la concentración medida de meropenem, Compuesto I o meropenem lactama abierta en la suspensión celular de 1 ml, y VAC es el volumen de células alveolares en la suspensión celular de 1 ml. Se llevó a cabo un recuento celular diferencial para determinar el número de macrófagos presentes. En los cálculos del volumen de células alveolares en la suspensión de gránulos se usó un volumen medio de células macrofágicas de 2,42 |jl/106 células.
Las proporciones de concentración de ELF y AM con respecto a las concentraciones plasmáticas simultáneas se calcularon para cada sujeto y se resumieron para cada grupo en cada tiempo de muestreo. Las concentraciones medias y medianas de meropenem y Compuesto I de los tiempos de muestreo broncopulmonar (por ejemplo, 1,5, 3,25, 4, 6 y 8 horas) se usaron para estimar elAUC0-8 del plasma, ELF y AM. El tiempo de muestreo de 8 horas también se usó como valor a tiempo cero para determinar el término de área del plasma, ELF y AM. El AUC0-8 para cada matriz se determinó con el procedimiento lineal trapezoidal. Se calculó la proporción de AUC0-8 de e Lf a plasma y de AM a plasma.
Resultados
Veintiséis (26) sujetos adultos sanos se inscribieron en este estudio. Un sujeto abandonó el estudio debido a eventos adversos y no se llevaron a cabo las fases farmacocinéticas (por ejemplo, la recolección de muestras de sangre para medir las concentraciones del fármaco en plasma y una broncoscopia con BAL a la hora de muestreo prevista [4 horas]). Las características de los 25 sujetos del estudio que recibieron la Combinación durante tres dosis y completaron todas las fases del estudio farmacocinético se recolectan en la Tabla 8.
Las concentraciones plasmáticas medias (±DE) de meropenem antes y después del inicio de la infusión intravenosa de la tercera dosis de la Combinación se muestran en la FIG. 16. La media (± DE) de la Cmáx y el AUCo-8 para las concentraciones plasmáticas de meropenem fueron 58,2 ± 10,8 ^g/ml y 185,5 ± 33,6 ^g-h/ml, respectivamente. Los parámetros farmacocinéticos medios (±DE) de meropenem en plasma se resumen en la Tabla 9. Las concentraciones plasmáticas medias (±DE) del Compuesto I antes y después del inicio de la infusión intravenosa de la tercera dosis de la Combinación se muestran en la FIG. 17. La media (± DE) de la Cmáx y el AUCo-8 para las concentraciones plasmáticas del Compuesto I fueron 59,0 ± 8,4 ^g/ml y 204,2 ± 34,6 (^g-h/ml, respectivamente. Los parámetros farmacocinéticos medios (±DE) del Compuesto I en plasma se resumen en la Tabla 10.
Las concentraciones medias (±DE) de meropenem en plasma y ELF en los tiempos de muestreo broncopulmonar se ilustran en la FIG. 18. Las concentraciones medias de meropenem en plasma y ELF oscilaron entre 1,36 y 41,2 ^g/ml y 2,51 y 28,3 (^g/ml, respectivamente. Las concentraciones medias (±DE) de meropenem después de la última dosis en plasma, ELF y a M en los cinco tiempos de muestreo broncopulmonar se reportan en la Tabla 11. Las concentraciones de meropenem en las células alveolares estaban por debajo del límite cuantificable en todas las muestras.
Las concentraciones medias (±DE) del Compuesto I en plasma, ELF, y AM en los tiempos de muestreo broncopulmonar se ilustran en la FIG. 19. Las concentraciones medias del Compuesto I en plasma y ELF oscilaron entre 2,74 y 51,1 ^g/ml y 2,61 y 26,1 (^g/ml, respectivamente. Las FIGs. 20 y 21 ilustran la magnitud y el curso temporal similares de las concentraciones de meropenem y del Compuesto I en plasma y ELF. Las concentraciones medias (±DE) del Compuesto I tras la última dosis en plasma, ELF y AM en los cinco momentos de muestreo broncopulmonar se recolectan en la Tabla 12. Las concentraciones del Compuesto I en los macrófagos alveolares fueron medibles en todas las muestras y oscilaron entre 1,26 y 93,9 ^g/ml.
Las proporciones medias (±DE) de ELF a las concentraciones plasmáticas simultáneas para meropenem se reportan en la Tabla 13. Las proporciones medias de ELF a concentraciones plasmáticas simultáneas para meropenem durante el período de 8 horas tras la administración del fármaco oscilaron entre 0,525 y 2,13. Los valores de AUC0-8 en base a las concentraciones medias y medianas de ELF fueron de 111,7 y 102,4 ^g-h/ml, respectivamente. La proporción entre las concentraciones de ELF y las concentraciones plasmáticas totales de meropenem en base a los
valores medios y medianos de AUCo-8 fue de 0,63 y 0,58, respectivamente. Las proporciones entre las concentraciones plasmáticas de ELF y de meropenem no unido (unión a proteínas = 2%) en base a los valores medios y medianos de AUC0-8 fueron de 0,65 y 0,59, respectivamente.
Las proporciones medias (±DE) de ELF y AM a las concentraciones plasmáticas simultáneas para el Compuesto I se informan en la Tabla 14. Las proporciones medias de ELF y AM respecto a la concentración plasmática simultánea para el Compuesto I durante el período de 8 horas tras la administración del fármaco oscilaron entre 0,45 y 1,01 y entre 0,062 y 2,58, respectivamente. Los valores de AUC0-8 en base a las concentraciones medias y medianas de ELF fueron de 105,1 y 96,7 pg-hs/ml, respectivamente. La proporción entre las concentraciones de ELF y las concentraciones plasmáticas totales del Compuesto I, en base a los valores medio y mediano del AUC0-8, fue de 0,53 y 0,48, respectivamente. Las proporciones entre las concentraciones plasmáticas de ELF y de Compuesto I no unido (unión a proteínas = 33%) en base a los valores medios y medianos de AUC0-8 fueron de 0,79 y 0,72, respectivamente.
Sumario
La Combinación (2 g de meropenem / 2 g de Compuesto I) administrada cada 8 horas, como infusiones IV de 3 horas, logró un curso temporal y una magnitud similares de las concentraciones de meropenem y Compuesto I en plasma y ELF. La penetración intrapulmonar de meropenem y del Compuesto I, en base a los valores AUC0-8 de las concentraciones plasmáticas de ELF y total, fue de aproximadamente 63% y 53%, respectivamente. Cuando se consideraron las concentraciones plasmáticas no unidas, la penetración fue del 65% y del 79% para el meropenem y el Compuesto I, respectivamente. Los resultados de este estudio apoyan la exploración de la combinación meropenem 2 g/Compuesto I 2 g como posible agente antimicrobiano para el tratamiento de infecciones bacterianas del tracto respiratorio inferior causadas por patógenos susceptibles.
Las concentraciones de meropenem en las células alveolares estaban por debajo del límite cuantificable para todas las muestras. Por el contrario, las concentraciones del Compuesto I fueron medibles en todas las muestras de células alveolares y las concentraciones de AM oscilaron entre 1,26 y 93,9 pg/ml. Vale la pena señalar que dos sujetos del tiempo de muestreo de 6 horas tuvieron las concentraciones más altas reportadas del Compuesto I en a M (35,4 y 93,9 (pg/ml) lo que consecuentemente infló la proporción media de AM a la concentración plasmática (2,58 ± 3,57, Tabla 14). Ambos sujetos tenían concentraciones extremadamente altas de glóbulos rojos en su líquido BAL (176.000 y 226.250 células/mm3) lo que puede haber contribuido a tan altas mediciones de concentraciones de AM.
La proporción de la exposición sistemática de meropenem lactama abierta a meropenem fue de aproximadamente 11% y 15% de acuerdo con la comparación de la concentración plasmática máxima y los valores de AUC0-8, respectivamente. Las concentraciones medias en la ELF de meropenem lactama abierta oscilaron entre sólo 1,81 y 2,69 pg/ml durante las primeras 6 horas tras la administración de meropenem, y todas las concentraciones en la ELF de meropenem lactama abierta estaban por debajo del límite cuantificable en el tiempo de muestreo de 8 horas. Sólo tres concentraciones AM de meropenem lactama abierta fueron medibles y oscilaron entre 1,91 y 8,46 pg/ml.
Conte et al. administraron meropenem a una dosis de 500 mg, 1 gramo o 2 gramos cada 8 horas, como infusiones IV de 30 minutos, para un total de cuatro dosis. Las concentraciones medias de ELF de meropenem a la 1, 2, 3, 5 y 8 horas fueron de 5,3, 2,7, 1,9, 0,7 y 0,2 pg/ml para la dosis de 500 mg y de 7,7, 4,0, 1,7, 0,8 y 0,03 pg/ml para la dosis de 1 gramo. Las relaciones entre las concentraciones de ELF y las concentraciones plasmáticas totales en los momentos de muestreo oscilaron entre 0,49 y 2,3 para la dosis de 500 mg y entre 0,32 y 0,53 para la dosis de 1 gramo. La penetración intrapulmonar de meropenem en base a los valores de AUC0-8 de las concentraciones plasmáticas de ELF y total fue de aproximadamente 43% y 28% para las dosis de 500 mg y 1 gramo, respectivamente. Para la dosis de 2 gramos, las concentraciones medias de ELF de meropenem y los índices de penetración en los tiempos de muestreo de 1 y 3 horas fueron de 2,9 y 2,8 pg/ml, y de 0,05 y 0,22, respectivamente. Para la dosis de 2 gramos, el número de observaciones fue limitado (n = 8) y no fue posible calcular el valor AUC0-8 para ELF.
Los hallazgos de meropenem en este estudio no son directamente comparables con los de Conte et al. debido a las diferencias en el diseño del estudio. En este estudio se evaluó una dosis de 2 gramos de meropenem administrada como infusión prolongada de 3 horas y en combinación con el Compuesto I. Además, este estudio incluyó una recolección más amplia de concentraciones de ELF (n = 30) durante el intervalo de dosificación de 8 horas, lo que permitió una estimación exacta del valor AUC0-8. Concentraciones medias más elevadas de meropenem en plasma y ELF tras la administración de 2 gramos con infusiones prolongadas (intervalo: 1,36 a 41,2 pg/ml y 2,51 a 28,3 pg/ml, respectivamente). También es posible que las infusiones más prolongadas de carbapenems puedan proporcionar una mayor penetración en la ELF, como se ha informado anteriormente para el biapenem (Kikuchi et al.). Las proporciones medias de ELF a concentraciones plasmáticas simultáneas para meropenem durante el período de 8 horas oscilaron entre 0,525 y 2,13. Los valores AUC0-8 en base a las concentraciones medias y medianas de ELF fueron 111,7 y 102,4 (pg-h/ml, respectivamente. La proporción entre las concentraciones de ELF y las concentraciones plasmáticas totales de meropenem en base a los valores medios y medianos de AUC0-8 fue de 0,63 y 0,58, respectivamente. Estos datos respaldan la realización de nuevos estudios sobre la combinación del Compuesto I/meropenem para el tratamiento de infecciones pulmonares.
TABLA 8. CARACTERÍSTICAS DE LOS SUJETOS DEL ESTUDIO QUE RECIBIERON LA COMBINACIÓN CADA 8
HORAS DURANTE 3 DOSIS
Tiempo de Sexo Edad Altura Peso IMC Recuento Total de Células Macrófagos Muestreo de BAL (años) (cm) (kilogramos) (kg/m2) en el Líquido BAL (mm3) (%) 1,5 horas 5 M 32 ± 9 181 ± 7 83,2 ± 5,5 25,5 ± 3,3 114±46 89 ± 7 3,25 horas 3 M, 2 F 80,5 ± 11,9 26,6 ± 1,6 92 ± 52 83 ± 13 4 horas 5 M 40 ± 9 179 ± 10 80,5 ± 13,0 25,2 ± 2,3 173 ± 80 91 ± 4 6 horas 3 M, 2 F 43 ± 8 169 ± 9 80,9 ± 8,3 28,5 ± 0,7 197 ± 186 80 ± 10 8 horas 3 M, 2 F 40 ± 12 168 ± 5 76,2 ± 9,2 26,9 ± 2,1 130 ± 76 85 ± 8 Los datos se expresan como media ± DE excepto para el sexo
M = varones; F = mujeres
IMC = índice de masa corporal = peso [kg] (altura [m])2
TABLA 9. PARAMETROS FARMACOCINETICOS NO COMPARTIMENTALES EN PLASMA DE MEROPENEM 2 G
CADA 8 HORAS DURANTE 3 DOSIS
Cmáx. (Hg / ml) Tmáx (horas) AUCo-8 (Hg/ml) t i /2 (horas) Vss (litros) CL (L/h) Todos los Sujetos3 58,2 ± 10,8 2 ± 0 ,5 ± 33,6 1 ± 015 163 ± 2,6 111 ± 2,1 Grupo de Muestreo BAL de 1,5 hsb 56,9 ± 19,3 2 ± 001 167.,8 ± 41,7 0,98 ± 0 175 ± 2,5 12,5 ± 2,8 Grupo de Muestreo BAL de 3,25 hsb 57,9 ± 7,5 3 ± 007 ,8 ± 29 ,7 1,04 ± 013 167 ± 2,6 111 ± 1,8 Grupo de Muestreo BAL de 4 hsb 59,6 ± 7,4 2 ± 0 196.,2 ± 33,5 1,07 ± 015 153 ± 2,1 10,5 ± 1,9 Grupo de Muestreo BAL de 6 hsb 59,4 ± 11,5  197.,4 ± ,7 1,12 ± 0 161 ± 3,4 10,4 ± 2,0 Grupo de Muestreo BAL de 8 hsb 57,3 ± 9,0 2
197.,4 ± ,7 1,12 ± 0 161 ± 3,4 10,4 ± 2,0 Grupo de Muestreo BAL de 8 hsb 57,3 ± 9,0 2  ,4 ± ,7 0,96 ± 013 156 ± 2,8 112 ± 1,9 Los datos se expresan como media ± DE.
,4 ± ,7 0,96 ± 013 156 ± 2,8 112 ± 1,9 Los datos se expresan como media ± DE.
a. 25 sujetos por estimación de parámetro
b. 5 sujetos por estimación de parámetro
TABLA 10. PARAMETROS FARMACOCINETICOS NO COMPARTIMENTALES EN PLASMA DEL COMPUESTO I 2
G CADA 8 HORAS DURANTE 3 DOSIS
Cmáx. (Hg/ml) Tmáx (horas) AUCo-8 (Hg/ml) t i /2 (horas) Vss (litros) CL (L/h) Todos los Sujetosa ,0 ± 84 2 ± 0 ,2 ± 34,6 1,27 ± 0,21 176 ± 2,6 10,1 ± 1,9 Grupo de Muestreo BAL de 1,5 hsb 56, 1 ± 13,0 2 ± 001  ± ,6 1,18 ± 0 186 ± 2,3 11,3 ± 2,6 Grupo de Muestreo BAL de 3,25 hsb ,7 ± 58 3 ± 007 ,5 ± 32,2 1,26 ± 0,23 173 ± 2,3 9,7 ± 1,6 Grupo de Muestreo BAL de 4 hsb
± ,6 1,18 ± 0 186 ± 2,3 11,3 ± 2,6 Grupo de Muestreo BAL de 3,25 hsb ,7 ± 58 3 ± 007 ,5 ± 32,2 1,26 ± 0,23 173 ± 2,3 9,7 ± 1,6 Grupo de Muestreo BAL de 4 hsb  ± 57 2 ± 0 213,,7 ± 35,4 1,34 ± 0,26 169 ± 0,9 9,5 ± 1,3 Grupo de Muestreo BAL de 6 hsb
± 57 2 ± 0 213,,7 ± 35,4 1,34 ± 0,26 169 ± 0,9 9,5 ± 1,3 Grupo de Muestreo BAL de 6 hsb  9 7 3 ± 007 ,8 ± 33,7 1,37 ± 0,27 181 ± 3,9 9,5 ± 1,6 Grupo de Muestreo BAL de 8 hsb ,9 ± 88 2 ± 0, 197,,5 ± 36,6 1,18 ± 0,16 170 ± 3,3 10,4 ± 2,0 Los datos se expresan como media ± DE.
9 7 3 ± 007 ,8 ± 33,7 1,37 ± 0,27 181 ± 3,9 9,5 ± 1,6 Grupo de Muestreo BAL de 8 hsb ,9 ± 88 2 ± 0, 197,,5 ± 36,6 1,18 ± 0,16 170 ± 3,3 10,4 ± 2,0 Los datos se expresan como media ± DE.
a. 25 sujetos por estimación de parámetro
b. 5 sujetos por estimación de parámetro
TABLA 11. CONCENTRACIONES DE MEROPENEM EN PLASMA, ELF Y AM EN EL MOMENTO DE LA
BRONCOSCOPIA Y BAL
Tiempo de Muestreo de BAL Plasma (Hg/ml) ELF (Hg/ml) AM (Hg/ml) 1,5 horas 41,2 ± 5,0 21,4 ± 4,0 BQL 3,25 horas 47,7 ± 7,3 28,3 ± 6,7 BQL 4 horas 23,8 ± 4,3 16,1 ± 4,8 BQL 6 horas 7,24 ± 2,79 7,52 ± 5,29 BQL 8 horas 1,36 ± 0,51 2,51 ± 1,13 BQL Los datos se expresan como media ± DE
5 sujetos por período de muestreo
Tiempo de Muestreo de BAL Plasma (pg/ml) ELF (pg/ml) AM (pg/ml) BQL = por debajo del límite cuantificable
TABLA 12. CONCENTRACIONES DEL COMPUESTO I EN PLASMA, ELF Y AM EN EL MOMENTO DE LA
BRONCOSCOPIA Y BAL
Tiempo de Muestreo de BAL Plasma (pg/ml) ELF (pg/ml) AM (pg/ml) 1,5 horas 42,1 ± 5,0 18,6 ± 3,8 2,71 ± 1,44 3,25 horas 51,1 ± 6,8 26,1 ± 1,1 8,79 ± 9,43 4 horas 28,2 ± 5,3 15,7 ± 3,4 5,51 ± 3,15 6 horas 10,8 ± 2,8 8,03 ± 5,80 27,6 ± 39,6 8 horas 2,74 ± 1,12 2,61 ± 1,35 4,40 ± 4,10 Los datos se expresan como media ± DE
5 sujetos por período de muestreo
TABLA 13. RELACION ENTRE LAS CONCENTRACIONES PLASMATICAS DE ELF Y LAS CONCENTRACIONES
PLASMÁTICAS TOTALES DE MEROPENEM
Tiempo de Muestreo de BAL ELF a Plasma
1,5 horas 0,525 ± 0,107
3,25 horas 0,590 ± 0,079
4 horas 0,705 ± 0,302
6 horas 1,037 ± 0,475
8 horas 2,133 ± 1,366
Los datos se expresan como media ± DE
5 sujetos por período de muestreo
TABLA 14. RELACIÓN ENTRE ELF Y AM Y LAS CONCENTRACIONES PLASMÁTICAS TOTALES DEL
COMPUESTO I
Tiempo de Muestreo de BAL ELF a Plasma AM a Plasma 1,5 horas 0,450 ± 0,123 0,062 ± 0,029 3,25 horas 0,508 ± 0,096 0,165 ± 0,163 4 horas 0,570 ± 0,159 0,191 ± 0,101 6 horas 0,705 ± 0,329 2,58 ± 3,57 8 horas 1,009 ± 0,391 1,603 ± 1,103 Los datos se expresan como media ± DE
5 sujetos por período de muestreo
Ejemplo 7
El Ejemplo 7 proporciona un resumen de un estudio de Modelo de Fibra Hueca de los perfiles farmacocinéticos de la combinación del Compuesto I y meropenem en dos regímenes de dosificación diferentes (2 g de meropenem/2 g de Compuesto I y 1 g de meropenem/1 g de Compuesto I) administrados cada 8 horas por infusión de 3 horas. La combinación es muy activa frente a patógenos gramnegativos, que incluyen Enterobacteriaceae K. pneumonía y P aeruginosa productoras de KPC y resistentes a los carbapenemes. El objetivo de este estudio fue demostrar la eficacia de meropenem en combinación con el Compuesto I contra aislados clínicos de P aeruginosa mediante el uso de exposiciones humanas simuladas en un modelo in vitro de fibra hueca. La simulación farmacocinética se basó en los datos del estudio clínico desvelado en el Ejemplo 2.
Procedimientos: Se analizaron tres cepas de P aeruginosa. Las concentraciones inhibitorias mínimas (MIC) se determinaron por medio de ensayo de microdilución en caldo mediante el uso de los procedimientos de referencia CLSI y se muestran en la Tabla D.

Modelo PK-PD in v ito Se usaron seis cartuchos de fibra hueca de tamaño medio (FiberCell Systems) por experimento. Para cada experimento se usaron tres cepas estudiadas por duplicado. Se inocularon células en fase logarítmica y se incubaron durante 2 horas antes del inicio del tratamiento para alcanzar aproximadamente 108 UFC/ml. Los parámetros PK objetivo se enumeran en las Tablas E y F. Las exposiciones se basaron en las bibliografías publicadas desveladas en el Ejemplo 2. Se recolectaron muestras del compartimento central para determinar las concentraciones de fármaco durante un período de 32 horas y se analizaron por medio de un procedimiento LC-MS/MS.


Se usaron cepas de Enterobacteriaceae productoras de Klebsiella pneumoniae carbapenemasa (KPC) con MIC de meropenem solo de 8 a 512 pg/ml y con meropenem/Compuesto I (en el que el Compuesto I se administró a una concentración fija de 8 pg/ml con meropenem, la MIC de meropenem oscila entre < 0,06 y 8 pg/ml), así como cepas de P aeruginosa con MIC de meropenem y meropenem/Compuesto I de 2 a 8 pg/ml.
Resultados:
La exposición de la combinación del régimen de dosificación de 1 g de meropenem y 1 g de Compuesto I se asoció con la eliminación efectiva y sin rebrote a las 32 horas de cepas de K. pneumonia productoras de KPC con meropenem solo (la MIC oscila entre 8 y 64 pg/ml) y con la combinación de meropenem y Compuesto I (donde el Compuesto I se administró a la concentración fija de 4 pg/ml con meropenem, la MIC de meropenem oscila entre < 0,06 y 2 pg/ml) (véase la FIG. 22 y la FIG. 23). Diversos clones de las cepas KP1061, KP1087, KP1004 y KP1074 que sobrevivieron a las 32 horas fueron sometidos a pruebas de susceptibilidad al meropenem y a la combinación meropenem/Compuesto I, y resultaron ser indistinguibles de las cepas preexpuestas.
Por otro lado, se observó una menor mortalidad para la cepa KP1099 con meropenem solo (la MIC es de 128 pg/ml) y la combinación de meropenem y el Compuesto I (cuando se administró el Compuesto I a la concentración fija de 4 pg/ml, la MIC de meropenem se redujo a 4 pg/ml). Véase la FIG. 23. El rebrote se observó a las 16 horas del inicio del tratamiento. Cuando se investigaron las colonias de KP1099 que sobrevivieron a la exposición a tres dosis de 1 g de meropenem/1 g de Compuesto I, su susceptibilidad a meropenem/Compuesto I se redujo entre 16 y 32 veces, lo que indica selección de resistencia en condiciones de exposición inadecuada.
Es importante destacar que la exposición a un régimen de dosificación de 2 g de meropenem/2 g de Compuesto I se asoció con una destrucción eficiente y ningún desarrollo de rebrote/resistencia mediante el uso de cepas con meropenem solo y meropenem/Compuesto I. Para la cepa KP1094, la MIC para meropenem solo fue tan alta como 512 pg/ml. Sin embargo, cuando el Compuesto I se administró a la concentración fija de 8 pg/ml con meropenem, la MIC observada de meropenem se redujo a 8 pg/ml (véase la FIG. 24).
La exposición del régimen de dosificación de 1 g de meropenem/1 g de Compuesto I resultó en una muerte efectiva y ningún rebrote a las 32 horas debido al desarrollo de resistencia para la cepa de P aeruginosa PAM3210 con meropenem y meropenem/Compuesto I (cuando el Compuesto I se administró a la concentración fija de 4 pg/ml u 8
|jg/ml, la MIC de meropenem sigue siendo 2 |jg/ml. Sin embargo, se produjo un rebrote y desarrollo de resistencia en las cepas PAM3353 y PAM3377 con una MIC de 8 jg/ml para meropenem (véase la FIG. 25).
Para la eficacia de las exposiciones humanas simuladas de meropenem en comparación con la combinación del Compuesto I 2 g/meropenem 2 g contra Pseudomonas aeruginosa en el modelo de fibra hueca in vitro, se observó que el modelo simulaba efectivamente las exposiciones humanas tanto de meropenem como del Compuesto I. (véase la FIG. 26). La actividad antibacteriana del meropenem en el modelo se muestra en la FIG.27. Meropenem 2 g q8h por infusión de 3 horas produjo más de 4 logs de eliminación bacteriana contra la cepa con una MIC de 2 mg/l, casi 4 logs de eliminación contra la cepa con una MIC de 4 a 8 mg/l. Se desarrolló resistencia en la cepa con una MIC de 8 mg/l. La actividad antibacteriana de la combinación de 2 g de meropenem/2 g del Compuesto I en el modelo se muestra en la FIG. 28. La combinación produjo más de 4 logs de eliminación bacteriana contra todas las cepas probadas, sin rebrotes ni desarrollo de resistencias durante el período de prueba de 32 horas. El régimen de dosificación de 2 g de meropenem/2 g de Compuesto I fue eficaz contra las tres cepas. No se identificaron mutantes resistentes entre las bacterias supervivientes (véase la FIG. 28). Los resultados se resumen en la Tabla G a continuación.

En conclusión, los estudios PK/PD en modelos in vitro de infecciones demuestran que las exposiciones humanas de la combinación 2 g/2 g de meropenem/Compuesto I se asocian con una amplia eliminación de patógenos diana y prevención de la resistencia para las cepas con Compuesto I a 8 jg/ml fijo y MIC de meropenem menor o igual a 8 jg/ml. Además, la combinación de dosis 2 g/2 g redujo las exposiciones asociadas al desarrollo de resistencias. Además, la combinación del Compuesto I 2 g/meropenem 2 g administrado cada 8 horas por infusión de tres horas fue altamente eficaz en este modelo in vitro contra cepas de P. aeruginosa con MIC tan altas como 8 mg/l, sin recrecimiento ni desarrollo de resistencia en el transcurso del estudio de 32 horas. Meropenem 2 g q8h por infusión de 3 horas fue efectivo contra 2 de las 3 cepas, pero se desarrolló resistencia en la tercera cepa con una MIC de 8 mg/l.
Claims (8)
1. Un Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para su uso en un procedimiento de tratamiento o mejora de una infección bacteriana en un sujeto que padece una función renal reducida:

en el que:
el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran después de una sesión de diálisis y en el que:
(i) el sujeto tiene un nivel de aclaramiento de creatinina igual o superior a 30 ml/min e inferior a 50 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g y la cantidad de meropenem es de aproximadamente 1,0 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 8 horas (q8h); o, (ii) el sujeto tiene un nivel de aclaramiento de creatinina igual o superior a 20 ml/min e inferior a 30 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 1,0 g y la cantidad de meropenem es de aproximadamente 1,0 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 12 horas (q12h); o bien
(iii) el sujeto tiene un aclaramiento de creatinina igual o superior a 10 ml/min e inferior a 20 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 0,5 g y la cantidad de meropenem es de aproximadamente 0,5 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 12 horas (q12h); o bien
(iv) el sujeto tiene un aclaramiento de creatinina inferior a 10 ml/min, y en el que la cantidad de Compuesto I o la sal aceptable para uso farmacéutico del mismo es de aproximadamente 0,5 g y la cantidad de meropenem es de aproximadamente 0,5 g, y en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y el meropenem se administran cada 24 horas (q24h).
2. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para uso de acuerdo con la reivindicación 1, en el que la composición se administra por medio de infusión intravenosa; y opcionalmente, en el que la infusión intravenosa se completa en aproximadamente 3 horas.
3. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para su uso de acuerdo con cualquiera de las reivindicaciones 1 a 2, en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo se administra antes o después del meropenem.
4. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para uso de acuerdo con cualquiera de las reivindicaciones 1 a 3, en el que el Compuesto I o la sal aceptable para uso farmacéutico del mismo y meropenem son:
(i) formulado en una composición farmacéutica; o
(ii) en una forma farmacéutica única, opcionalmente, en la que la forma farmacéutica única además comprende un excipiente, diluyente o portador aceptable para uso farmacéutico.
5. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para uso de acuerdo con cualquiera de las reivindicaciones 1 a 4, en el que dicho procedimiento además comprende administrar un medicamento adicional seleccionado entre un agente antibacteriano, un agente antifúngico, un agente antiviral, un agente antiinflamatorio o un agente antialérgico.
6. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para uso de acuerdo con cualquiera de las reivindicaciones 1 a 5, en el que el sujeto padece una infección del tracto respiratorio inferior.
7. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para su uso de acuerdo con cualquiera de las reivindicaciones 1 a 6, en el que la infección está causada por enterobacteriaceae resistente a carbapenemes.
8. El Compuesto I o una sal aceptable para uso farmacéutico del mismo y meropenem para su uso de acuerdo con la reivindicación 7, en el que la infección bacteriana está causada por Klebsiella pneumoniae, Escherichia coli o Enterobacter cloacae, o combinaciones de las mismas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562152668P | 2015-04-24 | 2015-04-24 | |
| PCT/US2016/028437 WO2016172208A1 (en) | 2015-04-24 | 2016-04-20 | Methods of treating bacterial infections |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2942329T3 true ES2942329T3 (es) | 2023-05-31 |
Family
ID=57144340
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16783764T Active ES2942329T3 (es) | 2015-04-24 | 2016-04-20 | Régimen de dosificación de vaborbactam y meropenem para el tratamiento de infecciones bacterianas en sujetos con función renal reducida |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US20160339045A1 (es) |
| EP (1) | EP3285776B1 (es) |
| JP (2) | JP6945452B2 (es) |
| CN (2) | CN112755039A (es) |
| AU (1) | AU2016252555A1 (es) |
| CA (1) | CA2982911C (es) |
| ES (1) | ES2942329T3 (es) |
| WO (1) | WO2016172208A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2807546C (en) | 2010-08-10 | 2022-08-23 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| KR20150103269A (ko) | 2013-01-04 | 2015-09-09 | 렘펙스 파머수티클스 인코퍼레이티드 | 보론산 유도체 및 그의 치료적 용도 |
| EP3139930B1 (en) | 2014-05-05 | 2024-08-14 | Melinta Therapeutics, Inc. | Salts and polymorphs of cyclic boronic acid ester derivatives and therapeutic uses thereof |
| ES2750805T3 (es) | 2014-05-05 | 2020-03-27 | Rempex Pharmaceuticals Inc | Síntesis de sales de boronato y usos de las mismas |
| AU2015264418A1 (en) | 2014-05-19 | 2016-11-10 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| WO2016003929A1 (en) | 2014-07-01 | 2016-01-07 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| US10662205B2 (en) | 2014-11-18 | 2020-05-26 | Qpex Biopharma, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| US20180051041A1 (en) | 2015-03-17 | 2018-02-22 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| MX389349B (es) | 2016-06-30 | 2025-03-20 | Qpex Biopharma Inc | Derivados de ácido borónico y usos terapéuticos de los mismos. |
| JP7436206B2 (ja) * | 2017-01-09 | 2024-02-21 | レンペックス・ファーマシューティカルズ・インコーポレイテッド | 細菌感染症を処置する方法 |
| US11376237B2 (en) * | 2017-10-03 | 2022-07-05 | Melinta Subsidiary Corp. | Methods of treating bacterial infections |
| US11286270B2 (en) | 2017-10-11 | 2022-03-29 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis thereof |
| EP3781576B1 (en) | 2018-04-20 | 2024-06-12 | Qpex Biopharma, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| AU2021266715A1 (en) | 2020-05-05 | 2022-11-17 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis, polymorphic forms, and therapeutic uses thereof |
| CN116098993B (zh) * | 2022-11-17 | 2024-10-25 | 中国农业科学院特产研究所 | 一种狐、貉肺炎三联灭活疫苗及其制备方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4783443A (en) | 1986-03-03 | 1988-11-08 | The University Of Chicago | Amino acyl cephalosporin derivatives |
| CA2807546C (en) | 2010-08-10 | 2022-08-23 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| US10561675B2 (en) * | 2012-06-06 | 2020-02-18 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
-
2016
- 2016-04-20 WO PCT/US2016/028437 patent/WO2016172208A1/en not_active Ceased
- 2016-04-20 US US15/134,198 patent/US20160339045A1/en not_active Abandoned
- 2016-04-20 ES ES16783764T patent/ES2942329T3/es active Active
- 2016-04-20 AU AU2016252555A patent/AU2016252555A1/en not_active Abandoned
- 2016-04-20 CA CA2982911A patent/CA2982911C/en active Active
- 2016-04-20 EP EP16783764.0A patent/EP3285776B1/en active Active
- 2016-04-20 CN CN202110100198.0A patent/CN112755039A/zh active Pending
- 2016-04-20 JP JP2017555494A patent/JP6945452B2/ja active Active
- 2016-04-20 CN CN201680023884.XA patent/CN107530364A/zh active Pending
-
2018
- 2018-07-06 US US16/029,388 patent/US20190105337A1/en not_active Abandoned
-
2021
- 2021-06-18 JP JP2021101472A patent/JP7245289B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2982911C (en) | 2023-10-03 |
| US20190105337A1 (en) | 2019-04-11 |
| WO2016172208A1 (en) | 2016-10-27 |
| JP2018513188A (ja) | 2018-05-24 |
| EP3285776B1 (en) | 2022-12-21 |
| CN112755039A (zh) | 2021-05-07 |
| EP3285776A1 (en) | 2018-02-28 |
| CN107530364A (zh) | 2018-01-02 |
| EP3285776A4 (en) | 2019-01-23 |
| US20160339045A1 (en) | 2016-11-24 |
| JP2021143196A (ja) | 2021-09-24 |
| HK1249432A1 (en) | 2018-11-02 |
| JP7245289B2 (ja) | 2023-03-23 |
| CA2982911A1 (en) | 2016-10-27 |
| AU2016252555A1 (en) | 2017-11-09 |
| JP6945452B2 (ja) | 2021-10-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2942329T3 (es) | Régimen de dosificación de vaborbactam y meropenem para el tratamiento de infecciones bacterianas en sujetos con función renal reducida | |
| US11007206B2 (en) | Cyclic boronic acid ester derivatives and therapeutic uses thereof | |
| ES2985626T3 (es) | Sales y polimorfos de derivados cíclicos de éster de ácido borónico y usos terapéuticos de los mismos | |
| ES3026983T3 (en) | Ceftolozane antibiotic compositions | |
| EP3849562B1 (en) | Combination compositions comprising a beta-lactamase inhibitor | |
| ES3036963T3 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| ES2979266T3 (es) | Procedimientos de tratamiento de infecciones bacterianas con una combinación de vaborbactam y meropenem | |
| HK40051777A (en) | Methods of treating bacterial infections | |
| HK1249432B (en) | Dosage regimen of vaborbactam and meropenem for treating bacterial infections in subjects with reduced renal function | |
| WO2014071155A1 (en) | Therapeutic uses of tigemonam and carumonam | |
| HK40097685A (en) | Methods of treating bacterial infections with vaborbactam and meropenem | |
| WO2024128238A1 (ja) | 細菌感染症の治療剤 | |
| HK40018408B (en) | Methods of treating bacterial infections with a combination of vaborbactam and meropenem | |
| HK40018408A (en) | Methods of treating bacterial infections with a combination of vaborbactam and meropenem |