ES2943011T3 - Procedimiento de preparación del compuesto imidazol dicetona deuterado - Google Patents
Procedimiento de preparación del compuesto imidazol dicetona deuterado Download PDFInfo
- Publication number
- ES2943011T3 ES2943011T3 ES16919556T ES16919556T ES2943011T3 ES 2943011 T3 ES2943011 T3 ES 2943011T3 ES 16919556 T ES16919556 T ES 16919556T ES 16919556 T ES16919556 T ES 16919556T ES 2943011 T3 ES2943011 T3 ES 2943011T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- compounds
- reaction
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 30
- -1 deuterated imidazole diketone compound Chemical class 0.000 title claims abstract description 7
- 238000002360 preparation method Methods 0.000 title description 10
- 150000001875 compounds Chemical class 0.000 claims abstract description 48
- 239000002904 solvent Substances 0.000 claims abstract description 10
- 238000004519 manufacturing process Methods 0.000 claims abstract description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 5
- 238000006482 condensation reaction Methods 0.000 claims abstract description 5
- 238000006467 substitution reaction Methods 0.000 claims abstract description 5
- 238000005886 esterification reaction Methods 0.000 claims abstract description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 27
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 claims description 15
- 239000000203 mixture Substances 0.000 claims description 14
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 12
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 11
- FFDGPVCHZBVARC-UHFFFAOYSA-N N,N-dimethylglycine Chemical compound CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 claims description 8
- 239000003795 chemical substances by application Substances 0.000 claims description 8
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 8
- 229940011051 isopropyl acetate Drugs 0.000 claims description 8
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims description 8
- 239000002585 base Substances 0.000 claims description 7
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 6
- 229910052805 deuterium Inorganic materials 0.000 claims description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 150000008044 alkali metal hydroxides Chemical group 0.000 claims description 4
- 108700003601 dimethylglycine Proteins 0.000 claims description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 4
- 229940078490 n,n-dimethylglycine Drugs 0.000 claims description 4
- IVRIRQXJSNCSPQ-UHFFFAOYSA-N propan-2-yl carbonochloridate Chemical compound CC(C)OC(Cl)=O IVRIRQXJSNCSPQ-UHFFFAOYSA-N 0.000 claims description 4
- 238000007363 ring formation reaction Methods 0.000 claims description 4
- 239000003153 chemical reaction reagent Substances 0.000 claims description 3
- 239000007858 starting material Substances 0.000 claims description 3
- 239000007821 HATU Substances 0.000 claims description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 150000001408 amides Chemical class 0.000 claims description 2
- 230000032050 esterification Effects 0.000 claims description 2
- 229910052739 hydrogen Inorganic materials 0.000 claims description 2
- 239000001257 hydrogen Substances 0.000 claims description 2
- 229910001854 alkali hydroxide Inorganic materials 0.000 claims 1
- 239000003054 catalyst Substances 0.000 claims 1
- 239000002699 waste material Substances 0.000 abstract description 2
- 239000002994 raw material Substances 0.000 abstract 2
- 238000006352 cycloaddition reaction Methods 0.000 abstract 1
- 238000007257 deesterification reaction Methods 0.000 abstract 1
- 230000007613 environmental effect Effects 0.000 abstract 1
- 238000004904 shortening Methods 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 206010060862 Prostate cancer Diseases 0.000 description 10
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 229910001873 dinitrogen Inorganic materials 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 3
- TYXKOMAQTWRDCR-UHFFFAOYSA-N 4-isothiocyanato-2-(trifluoromethyl)benzonitrile Chemical compound FC(F)(F)C1=CC(N=C=S)=CC=C1C#N TYXKOMAQTWRDCR-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- 229960000997 bicalutamide Drugs 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 2
- GVMQERXYJLHNIZ-UHFFFAOYSA-N 4-(2-carboxypropan-2-ylamino)-2-fluorobenzoic acid Chemical compound OC(=O)C(C)(C)NC1=CC=C(C(O)=O)C(F)=C1 GVMQERXYJLHNIZ-UHFFFAOYSA-N 0.000 description 2
- BAJCFNRLEJHPTQ-FIBGUPNXSA-N 4-bromo-2-fluoro-N-(trideuteriomethyl)benzamide Chemical compound BrC1=CC(=C(C(=O)NC([2H])([2H])[2H])C=C1)F BAJCFNRLEJHPTQ-FIBGUPNXSA-N 0.000 description 2
- ZQQSRVPOAHYHEL-UHFFFAOYSA-N 4-bromo-2-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(Br)C=C1F ZQQSRVPOAHYHEL-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 239000003098 androgen Substances 0.000 description 2
- 239000007810 chemical reaction solvent Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229960004671 enzalutamide Drugs 0.000 description 2
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical class [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 2
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- NSTREUWFTAOOKS-UHFFFAOYSA-N 2-fluorobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1F NSTREUWFTAOOKS-UHFFFAOYSA-N 0.000 description 1
- MWFMGBPGAXYFAR-UHFFFAOYSA-N 2-hydroxy-2-methylpropanenitrile Chemical compound CC(C)(O)C#N MWFMGBPGAXYFAR-UHFFFAOYSA-N 0.000 description 1
- PMDYLCUKSLBUHO-UHFFFAOYSA-N 4-amino-2-(trifluoromethyl)benzonitrile Chemical compound NC1=CC=C(C#N)C(C(F)(F)F)=C1 PMDYLCUKSLBUHO-UHFFFAOYSA-N 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000003837 Second Primary Neoplasms Diseases 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 235000008242 dietary patterns Nutrition 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 150000001468 imidazolidinediones Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- LRDFRRGEGBBSRN-UHFFFAOYSA-N isobutyronitrile Chemical compound CC(C)C#N LRDFRRGEGBBSRN-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 210000005001 male reproductive tract Anatomy 0.000 description 1
- 208000010658 metastatic prostate carcinoma Diseases 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- ZWZVWGITAAIFPS-UHFFFAOYSA-N thiophosgene Chemical compound ClC(Cl)=S ZWZVWGITAAIFPS-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/86—Oxygen and sulfur atoms, e.g. thiohydantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Se describe un método para preparar un compuesto de imidazol dicetona deuterado, que comprende los siguientes pasos: (1) usar un compuesto de fórmula (I) y un compuesto de fórmula (II) como materias primas para obtener un compuesto de fórmula (III) por medio de de una reacción de sustitución; (2) llevar a cabo una reacción de esterificación de los grupos carboxilo en el compuesto de fórmula (III) para obtener un compuesto de fórmula (IV); (3) llevar a cabo una reacción de cicloadición del compuesto de fórmula (IV) y un compuesto de fórmula (V) para obtener un compuesto de fórmula (VI); (4) llevar a cabo una reacción de desesterificación del compuesto de fórmula (VI) para obtener un compuesto de fórmula (VII); y (5) usando el compuesto de fórmula (VII) y un compuesto de fórmula (VIII) como materia prima, mediante una reacción de condensación de formación de amida, para obtener el compuesto de imidazol dicetona deuterado representado por la fórmula (IX). En comparación con los métodos existentes, el método de la presente invención es más seguro, consume menos solvente y reduce los desechos, minimizando así los impactos ambientales, acorta el ciclo de producción y aumenta la producción y el rendimiento total y, por lo tanto, tiene amplias perspectivas de mercado. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Procedimiento de preparación del compuesto imidazol dicetona deuterado
Campo técnico
La presente invención se refiere al campo de la síntesis medicinal, y particularmente a un procedimiento de preparación de compuestos de imidazol dicetona deuterados.
Antecedentes de la técnica
El cáncer de próstata (CaP) es un tumor maligno frecuente en el aparato genital masculino, y la aparición de esta enfermedad aumenta con la edad, cuya tasa de incidencia presenta una evidente disparidad regional, siendo más elevada en regiones de Europa y América. El CaP es el segundo cáncer que causa la muerte masculina, sólo por detrás del cáncer de pulmón. En el pasado, en el espectro tumoral de nuestro país, el CaP pertenecía a las enfermedades menores y no se le prestaba suficiente atención. Con el desarrollo social y el progreso de nuestro país, el envejecimiento de la sociedad, la urbanización de la población humana, la occidentalización del patrón dietético y el avance de la tecnología de detección, la tasa de incidencia del cáncer de próstata en nuestro país presenta una evidente tendencia al alza. La encuesta realizada por el segundo hospital adscrito a la universidad de medicina de Tianjin y el grupo cooperativo de diagnóstico y tratamiento del cáncer de próstata de Tianjin en 2011 mostró que la tasa de incidencia del cáncer de próstata en la ciudad de Tianjin estaba aumentando rápidamente, y la tasa de incidencia del cáncer de próstata aumentó cuatro veces durante los últimos 20 años. Los pacientes con cáncer de próstata ya han ocupado el 13,4% de los pacientes hospitalizados por tumores en urología, y el cáncer de próstata se convierte en un tumor común desde el pasado cáncer raro. En todo el país, la tasa de incidencia del cáncer de próstata presenta la misma tendencia.
El andrógeno es una proteína reguladora de la transcripción inversa dependiente de ligando, con un peso molecular de 1,1 x 105 Dalton. Los andrógenos desempeñan un papel muy importante en la causa del cáncer de próstata y durante su progresión, así como en enfermedades relacionadas con las hormonas masculinas como el acné, la lipsotrichia masculina, etc.
El procedimiento de tratamiento habitual para el cáncer de próstata es la cirugía o los antagonistas androgénicos como la bicalutamida. Sin embargo, tras 2-4 años de tratamiento, los pacientes pueden desarrollar resistencia al fármaco, mientras que la bicalutamida tiene además un efecto adverso de irritación de la proliferación del cáncer, por lo que los pacientes deben dejar de utilizarla. En la actualidad, ya se han desarrollado compuestos con los mismos puntos diana de unión que la bicalutamida, junto con otros fármacos comercializados para el tratamiento del cáncer de próstata metastásico, como la patente CN201280052853.9.
En el que el siguiente compuesto tiene características farmacológicas mejoradas:

Sin embargo, en la ruta sintética divulgada en la patente CN201280052853.9 se utilizaron isotiocianato e isobutironitrilo para el acoplamiento. La principal deficiencia de este procedimiento es que sólo se obtiene un 11% de rendimiento del producto objetivo en la etapa final. Esto da como resultado un rendimiento total de sólo el 3,5% de esta ruta, comenzando con el material de partida disponible comercialmente. Además, la purificación de cada intermediario requiere una laboriosa cromatografía en columna que requiere mucho tiempo, y el tiempo total de producción es mayor, lo que no es deseable para la producción industrial.
El documento CN 104 803 918 A divulga un procedimiento de preparación de enzalutamida, que comprende las siguientes etapas: añadir un compuesto con fórmula F a un disolvente; hacer reaccionar con una fuente de metilamina; y después de la reacción, recoger el compuesto con fórmula G de los productos de reacción, a fin de obtener la enzalutamida - en donde la fórmula general de la reacción es como sigue:

El documento EP 2 792 674 A1 divulga compuestos de imidazolidinediona de fórmula (I) (como se muestra a continuación), procesos para su preparación, usos y composiciones farmacéuticas de los mismos.

Por lo tanto, para mejorar la eficiencia de la producción y reducir el coste de producción, es necesario mejorar el procedimiento sintético.
Contenido de la invención
Con el fin de resolver los problemas anteriores, la presente invención proporciona un procedimiento de preparación de compuestos de imidazol dicetona deuterados, e incluye las siguientes etapas:

en las que Ri y R2 se seleccionan independientemente entre alquilos C1-C4 ;
R3 , R4 y R5 se seleccionan entre hidrógeno y deuterio, en el que al menos uno de ellos se selecciona entre deuterio;
(1) utilización de un compuesto de fórmula (I) y compuestos de fórmula (II) como material de partida, en el que los compuestos de fórmula (III) se obtienen mediante una reacción de sustitución;
(2) Los compuestos de fórmula (IV) se preparan por esterificación del grupo carboxilo en compuestos de fórmula (III), en los que R se selecciona entre alquilos C1-C6 ;
(3) La ciclización de compuestos de fórmula (IV) y compuesto de fórmula (V) proporciona compuestos de fórmula (VI);
(4) Los compuestos de fórmula (VI) se desesterifican para producir compuestos de fórmula (VII);
(5) se hacen reaccionar compuestos de fórmula (VII) y compuestos de fórmula (VIII), en los que los compuestos de imidazol dicetona deuterados de fórmula (IX) se obtienen mediante una reacción de condensación que forma un enlace amida.
Preferentemente R1 y R2 son ambos metilo.
Preferentemente, R3 , R4 y R5 son todos deuterio.
Preferentemente, R es metilo.
Preferentemente, en la ciclización de la etapa (3), los disolventes son la mezcla de dimetilsulfóxido y acetato de isopropilo.
Preferentemente, la proporción en volumen de dimetilsulfóxido y acetato de isopropilo es de 1:2.
Preferentemente, en la etapa (4), dicha reacción se lleva a cabo en presencia de base, y la base se selecciona entre los hidróxidos de metales alcalinos.
Preferentemente, dichos hidróxidos alcalinos se seleccionan entre LiOH, KOH y NaOH, y más preferentemente LiOH.
Preferentemente, en la etapa (5), dicha reacción de condensación de amida se lleva a cabo en presencia de agente de condensación, y el agente de condensación se selecciona de clorocarbonato de isopropilo, N,N'-carbonildiimidazol, o 2-(7-oxibenzotriazol)-N,N,N',N'-tetrametilurea hexafluorofosfato (HATU).
Preferentemente, en la etapa (1), dicha reacción de sustitución se lleva a cabo en medio alcalino en presencia de Cu, CuI y N,N-dimetilglicina.
Preferentemente, en la etapa (2), dicho reactivo de metilesterificación para el grupo carboxilo es yoduro de metilo. Dichos alquilos C1-C4 denotan alquilos C1, C2, C3, C4 , es decir, alquilos de cadena recta o cadena ramificada que tienen de 1 a 4 átomos de carbono, como metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc-butilo, sec-butilo, etc. En comparación con los procedimientos de la técnica anterior, la presente invención presenta las siguientes ventajas evidentes:
(1) El procedimiento de la presente invención puede realizar el rendimiento total del 40%, mucho mejor que el rendimiento total del 3,5% del procedimiento actual.
(2) Para el procedimiento de la presente invención, los procedimientos son sencillos y no requieren cromatografía en columna, por lo que pueden utilizarse medios sencillos de precipitación o cristalización para la purificación de los productos.
(3) El procedimiento de la presente invención evita el uso del reactivo extremadamente tóxico acetona cianohidrina, y es más ecológico y seguro.
En la presente invención, las abreviaturas se enumeran en la siguiente Tabla:


Siguiendo ejemplos específicos de dichas realizaciones, se ilustra con más detalle el contenido anterior de la presente invención. Sin embargo, no debe interpretarse que el ámbito del objeto anterior de la presente invención se limite únicamente a los siguientes ejemplos.
Ejemplos
Ejemplo 1 Preparación del ácido 4-(1-carboxil-1-metil-etilamino)-2-fluoro-benzoico

Al hervidor de reacción se introdujo gas nitrógeno, al que se añadieron N,N-dimetilformamida (20 L), agua (2 L), ácido 4-bromo-2-fluorobenzoico (2,0 kg), 2-metilalanina (2,82 kg), N,N-dimetilglicina (474 g), carbonato potásico (6,31 kg), polvo de cobre (116 g) y CuI (348 g). Bajo la protección del nitrógeno, la mezcla se agitó a 110 °C durante 16 horas. La solución de reacción se enfrió a temperatura ambiente y, a continuación, se añadió agua helada (30 L). El pH de la solución se ajustó a 4-5 añadiendo gota a gota ácido clorhídrico helado 6N, y se extrajo con acetato de etilo dos veces (20L*2). La fase orgánica se combinó y se lavó con agua (5L) una vez. La fase orgánica se concentró hasta sequedad, a la que se añadió diclorometano (15 L) para transformar en pulpa, después se filtró, se secó al vacío a 40 °C, para proporcionar el compuesto objetivo 1,797 kg, con un rendimiento del 81,7%.
Ejemplo 2 Preparación del éster metílico del ácido 2-fluoro-4-[(1-metoxil-2-metiM-oxo-2-propil)amino]benzoico

Al hervidor de reacción se añadieron N,N-dimetilformamida (7 L), ácido 4-(1-carboxil-1-metil-etilamino)-2-fluorobenzoico (750 g) y carbonato potásico (6,31 kg). A continuación, se añadió yoduro de metilo (945 g) y la mezcla se agitó durante toda la noche. A la solución de reacción, se añadió gota a gota agua (20 L), y se agitó para la cristalización, se filtró, se secó al vacío a 50 °C durante 16 horas, para proporcionar el compuesto objetivo 787 g, con un rendimiento del 93,9%.
1HNMR (DMSO, 400 MHz): 1,49 (6H, s), 3,64 (3H, s), 3,75 (3H, s), 6,16 (1H, dd, J = 2,1, 14,5 Hz), 6,31 (1H, dd, J = 2,1, 8,8 Hz), 7,16 (1H, s), 7,63 (1H, t, J= 8,8 Hz).
empo reparac n e s er me co e c o - - -cano- - r uorome em - , - me - -oxo- - o--imidazolidinil}-2-fluorobenzoico

Al hervidor de reacción se añadieron éster metílico del ácido 2-fluoro-4-[(1-metil-2-metil-1-oxo-2-propil)amino]benzoico (1,70 kg), 4-isotiocianato-2-(trifluorometil)benzonitrilo (2,88 kg), dimetilsulfóxido (1,7 L) y acetato de isopropilo (3,4 L). Bajo la protección del nitrógeno, la mezcla se agitó a 83 °C durante 40 horas. La solución de reacción se concentró a presión reducida hasta que no se evaporó disolvente, después se añadió gota a gota metanol (8,5 L), y se agitó a 0-5 °C para cristalización, filtrándose. La torta de filtración se lavó con metanol y se secó al vacío a 50 °C para proporcionar el compuesto objetivo 2,3 kg, con un rendimiento del 78,2%.
1HNMR (DMSO, 400 MHz): 1.57 (6H, s), 3,91 (3H, s), 7,43 (1H, dd, J = 1,8, 8,3 Hz), 7,53 (1H, dd, J= 1,8, 11,2 Hz), 8,08-8,12 (2H, m), 8,30 (1H, d, J = 1,5 Hz), 8,42 (1H, d, J= 8,2 Hz).
De acuerdo con el procedimiento anterior, los inventores investigaron los disolventes de reacción, y los resultados se muestran en la Tabla 1:
Tabla 1 Investigación de disolventes de reacción

Puede demostrarse que cuando el disolvente es la mezcla de dimetilsulfóxido y acetato de iso-propilo, el rendimiento es mayor.
Ejemplo 4 Preparación del ácido 4-{3-[4-ciano-3-(trifluorometil)fenil]-5,5-dimetil-4-oxo-2-tio-1-imidazolidinil}-2-fluorobenzoico

Al hervidor de reacción se introdujo gas nitrógeno, al que se añadieron 4-{3-[4-ciano-3-(trifluorometil)fenil]-5,5-dimetil-4-oxo-2-tio-1-imidazolidinil}-2-fluorobenzoico (1,70 kg) y tetrahidrofurano (3,4 L), y la mezcla se agitó hasta que la solución fue clara. Se añadió gota a gota una solución acuosa de hidróxido de litio (hidróxido de litio monohidratado 0,46 kg agua 3,4 L). A la temperatura de 40 °C, la mezcla se incubó durante 1 h. La mezcla se enfrió a la temperatura, y se añadió agua (3,4 L), después se ajustó el pH a 1-2 utilizando salmuera. Se añadió
acetato de etilo (13,6 L) para la extracción. La fase orgánica se lavó con agua una vez, se secó con sulfato sódico anhidro, se filtró y se concentró a presión reducida hasta unos 3,4 L de disolvente restante. Se añadió n-heptano (10,2 L) y se agitó para cristalización 1 h, después se filtró y se secó al vacío para proporcionar el compuesto objetivo 1,31 kg, con un rendimiento del 79,4%.
1HNMR (DMSO, 400 MHz): 1,57 (6H, s), 7,39 (1H, dd, J= 1,6, 8,3 Hz), 7,48 (1H, dd, J= 1,6, 11,0 Hz), 8,06-8,12 (2H, m), 8,32 (1H, d, J= 1,2 Hz), 8,42 (1H, d, J= 8,2 Hz), 13,58 (1H, brd).
De acuerdo con el procedimiento anterior, los inventores investigaron las bases, y los resultados se muestran en la Tabla 2:
Tabla 2 Investigación de bases

Se puede comprobar que el carbonato de potasio y el carbonato de potasio básicamente no reaccionaron, y cuando la base se selecciona de los hidróxidos de metales alcalinos, el rendimiento es mayor. Entre otras cosas, el rendimiento es mayor con LiOH, y la pureza también es mayor, por lo que es preferible el LiOH.
Ejemplo 5 Preparación de 4-(3-(4-ciano-3-(trifluorometil)piridin-3-il)-5,5-dimetil-4-oxo-2-tioxoimidazolidin-1-il)-2-fluoro-N-trideuterado-metilbenzamida

Al hervidor de reacción se añadieron ácido 4- f 3-[4-ciano-3-(trifluorometil)fenil]-5,5-dimetil-4-oxo-2-tio-1-imidazolidinil}-2-fluorobenzoico (1,30 kg) y diclorometano (13 L), y la mezcla se agitó para su disolución. Se añadió N,N'-carbonildiimidazol (0,7 kg) por tandas, y se agitó durante 2h. Se añadieron trietilamina (1,2 L) y clorhidrato de metilamina deuterada (302 g), y se agitó durante 4 horas a temperatura ambiente. La solución de reacción se lavó sucesivamente con solución acuosa de hidróxido sódico 1 N (13 L), ácido clorhídrico 1 N (13 L) y agua (6,5 L). La fase orgánica se secó con sulfato sódico anhidro y se concentró a presión reducida para precipitar sólo sólidos. Se añadieron gota a gota metil terc-butil éter (3,9 L) y n-heptano (3,9 L) sucesivamente, se agitó, precipitó durante 1 h y se filtró, para obtener productos brutos. Los productos brutos se disolvieron en alcohol anhidro (13 L) bajo calentamiento, y a la temperatura de 0-5 °C, la solución se agitó y cristalizó durante 2 h, filtrándose a continuación. A la temperatura de 50 °C, el cristal se secó al vacío durante 8 horas, para proporcionar el compuesto objetivo como sólido blanco (1,09 kg), con un rendimiento del 80,7%. La pureza fue del 99,8% por HPLC.
1HNMR (DMSO, 400MHz): 1,57 (6H, s), 7,36 (1H, dd, J = 1,2, 8,2 Hz), 7,46 (1H, dd, J = 1,2, 10,7 Hz), 7,82 (1H, t, J = 8,2 Hz), 8,11 (1H, d, J = 8,2 Hz), 8,32 (1H, s), 8,42 (1H, d, J = 8,2 Hz), 8,46 (1H, s).
De acuerdo con el procedimiento anterior, los inventores investigaron los agentes de condensación, y los resultados se muestran en la Tabla 3:
Tabla 3 Investigación de agentes de condensación

Puede demostrarse que cuando se utiliza clorocarbonato de isopropilo o CDI como agente de condensación, el rendimiento es mayor. Entre ellos, el clorocarbonato de isopropilo necesita reducir la temperatura para formar el anhídrido mezclado, por lo que la operación tiene un alto estándar para el equipo. Considerando globalmente, el agente de condensación es preferentemente CDI.
Ejemplo 6 (no forma parte de la invención)

(1) Preparación de 4-isotiocianato-2-(trifluorometil)benzonitrilo

Al hervidor de reacción se introdujo gas nitrógeno, al que se añadieron 4-ciano-3-(trifluorometil)fenilamina (200 g), nheptano (450 mL) y agua (500 mL), seguido de agitación para producir una suspensión. Se añadió gota a gota tiofosgeno (148 g). La mezcla se agitó a 40 °C durante 16 horas y luego se dejó reposar para la separación de líquidos. La fase acuosa se extrajo una vez con n-heptano (500 mL) y se combinó la fase orgánica. El disolvente se eliminó mediante concentración a presión reducida, seguida de destilación a presión reducida para proporcionar el compuesto objetivo (220 g), con un rendimiento del 89,8%.
1HNMR (DMSO, 400 MHz): 7,52 (1H, dd, J= 1,7, 8,3), 7,60 (1H, d, J= 1,7 Hz), 7,87 (1H, d, J = 8,3 Hz).
(2) Preparación de 4-bromo-2-fluoro-N-trideuterometilbenzamida

Al hervidor de reacción, se añadieron ácido 4-bromo-2-fluorobenzoico (50 g) y diclorometano (500 mL), después se agitó. Se añadió N,N-carbonildiimidazol (73,9 g) por tandas y se agitó durante 2 h. Se añadieron trietilamina (95,5 mL) y clorhidrato de metilamina deuterada (30,8 g) y se agitó durante 4 horas. La solución de reacción se lavó secuencialmente con solución acuosa de hidróxido sódico 1 N (500 mL), ácido clorhídrico 1 N (500 mL) y agua (250 mL). La fase orgánica se secó con sulfato sódico anhidro, se filtró, se concentró a presión reducida para obtener los sólidos blanquecinos, que se secaron 8 horas para proporcionar el compuesto objetivo como sólidos blanquecinos (44,0 g), con un rendimiento del 82,2%.
(3) Preparación del ácido 2-(3-fluoro-4-(trideuterometilformamil)fenilamino)-2-metilpropiónico

Al hervidor de reacción se introdujo gas nitrógeno, al que se añadieron N,N-dimetilformamida (680 mL), agua (70 mL), 4-bromo-2-fluoro-N-trideuterometilbenzamida (150 g), 2-metilalanina (199,8 g), N,N-dimetilglicina (33,3 g), carbonato potásico (446,1 g), polvo de cobre (8,3 g) y CuI (24,6 g). Bajo la protección del nitrógeno, la mezcla se agitó a 110 °C durante 16 horas. La solución de reacción se enfrió a temperatura ambiente y, a continuación, se añadió agua (1,8 L). Las impurezas se extrajeron con acetato de etilo. El pH de la solución se ajustó a 3-4 con ácido cítrico, se cristalizó durante 1 hora a 5 °C, se filtró y se secó al vacío para obtener el compuesto objetivo (100 g), con un rendimiento del 60,9%.
(4) éster metílico del ácido 2-(3-fluoro-4-(trideuterometilformamil)fenilamino)-2-metilpropiónico

Al hervidor de reacción se añadieron N,N-dimetilformamida (630 mL), ácido 2-(3-fluoro-4-(trideuterometilformamil)fenilamino)-2-metilpropiónico (90 g), agua (2,2 mL) y carbonato potásico (58,7 g). A continuación, se añadió yoduro de metilo (26,5 mL) y la mezcla se agitó a 40 °C durante 3 horas. A la solución de reacción, se añadió ácido acético (6,4 mL), y se agitó a 60 °C durante 1 hora. Se añadió gota a gota agua (1,35 L), se enfrió a temperatura ambiente, se agitó para la cristalización, se filtró, se secó al vacío, para proporcionar el compuesto objetivo 91 g, con un rendimiento del 95,2%.
(5) 4-(3-(4-ciano-3-(trifluorometil)piridin-3-il)-5,5-dimetil-4-oxo-2-tioxoimidazolidin-1-il)-2-fluoro-N-trideuterado-metilbenzamida

Al hervidor de reacción se introdujo gas nitrógeno, al que se añadió éster metílico del ácido 2-(3-fluoro-4-(trideuterometilformamil)fenilamino)-2-metilpropiónico (27,1 g), 4-isotiocianato-2-(trifluorometil)benzonitrilo (45,6 g), dimetilsulfóxido (27,1 mL) y acetato de isopropilo (54,2 mL). Bajo la protección del nitrógeno, la mezcla se agitó a 83 °C durante 24 horas. La solución de reacción se concentró a presión reducida hasta que no se evaporó disolvente, después se añadió metanol (135,5 mL) gota a gota, y se agitó a 0-5 °C durante 1 hora para cristalización, y se filtró para obtener el producto bruto. El producto bruto se disolvió en alcohol anhidro (250 mL) bajo calentamiento y se cristalizó a 0-5 °C durante 1 hora, se filtró y la torta de filtración se secó al vacío a 50 °C para proporcionar el compuesto objetivo 34,5 g, con un rendimiento del 73,9%.
En resumen, en comparación con los procedimientos de la técnica anterior, el procedimiento según la presente invención es más seguro, consume menos disolventes, minimiza los residuos y el efecto sobre el medio ambiente, acorta el ciclo de producción y mejora la capacidad y el rendimiento total del procedimiento, con una amplia perspectiva de mercado.
Claims (10)
1. Un procedimiento de preparación de compuestos de imidazol dicetona deuterados, caracterizado porque comprende las siguientes etapas:
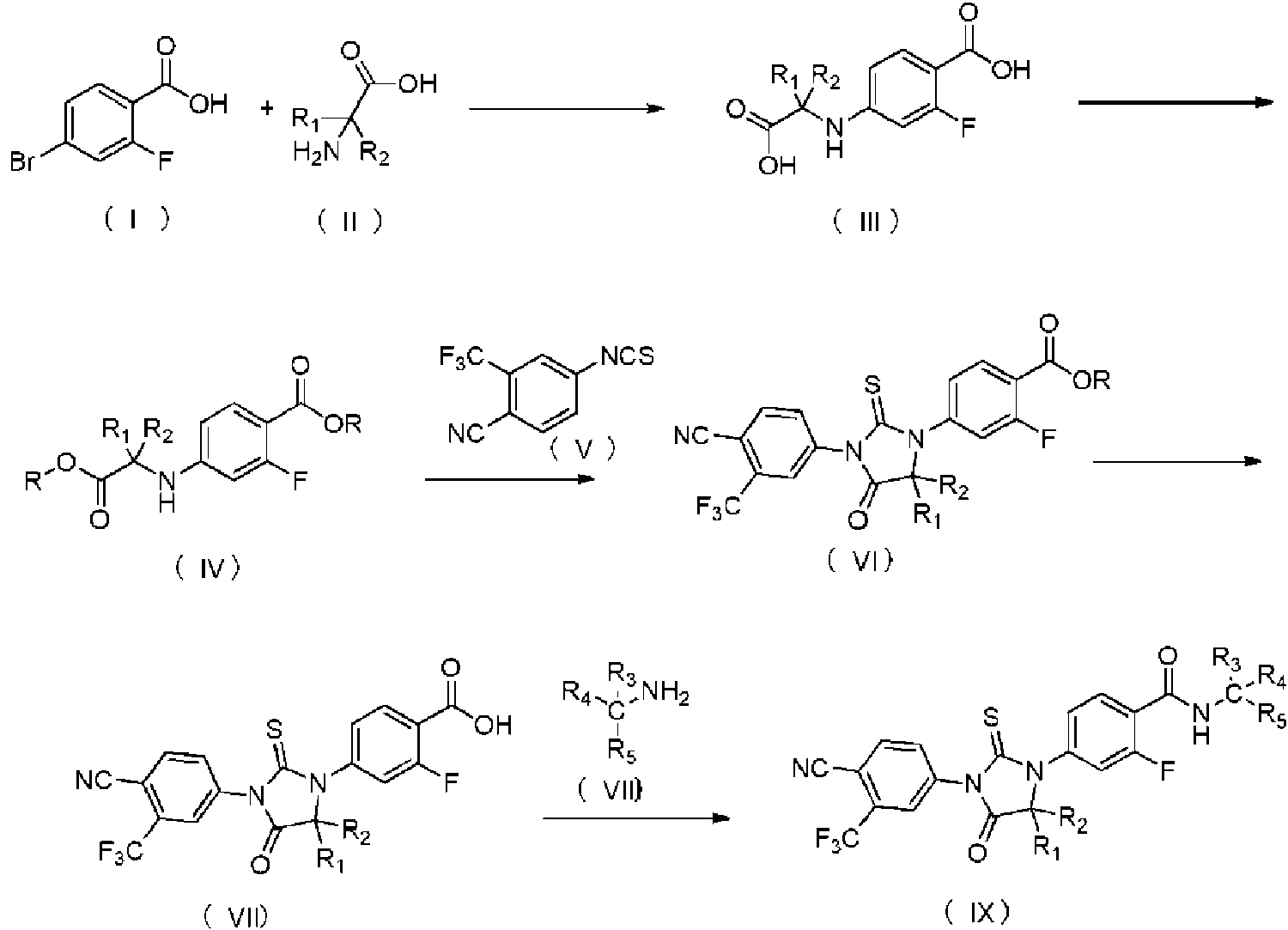
en las que R1 y R2 se seleccionan independientemente entre alquilos C1-C4 ;
R3 , R4 y R5 se seleccionan entre hidrógeno y deuterio, siendo al menos uno de ellos deuterio;
(1) utilización de un compuesto de fórmula (I) y compuestos de fórmula (II) como material de partida, en el que los compuestos de fórmula (III) se obtienen mediante una reacción de sustitución; (2) Los compuestos de fórmula (IV) se preparan por esterificación del grupo carboxilo en compuestos de fórmula (III), en los que R se selecciona entre alquilos C1-C6 ;
(3) La ciclización de compuestos de fórmula (IV) y compuesto de fórmula (V) proporciona compuestos de fórmula (VI);
(4) Los compuestos de fórmula (VI) se desesterifican para producir compuestos de fórmula (VII); (5) se hacen reaccionar compuestos de fórmula (VII) y compuestos de fórmula (VIII), en el que los compuestos de imidazol dicetona deuterados de fórmula (IX) se obtienen mediante una reacción de condensación que forma un enlace amida.
2. El procedimiento según la reivindicación 1, caracterizado porque R1 y R2 son ambos metilo; y/o R3, R4 y R5 son todos deuterio; y/o R es metilo.
3. El procedimiento según la reivindicación 1 ó 2, caracterizado porque en la etapa (1), la temperatura de reacción está comprendida entre 40 y 120 °C; y/o
en la etapa (2), la temperatura de reacción está comprendida entre -10 y 60 °C; y/o
en la etapa (3), la temperatura de reacción está comprendida entre 40 y 90 °C; y/o
en la etapa (4), la temperatura de reacción está comprendida entre -10 y 70 °C; y/o en la etapa (5), la temperatura de reacción está comprendida entre -10 y 40 °C.
4. El procedimiento según la reivindicación 1 o 2, caracterizado porque en la ciclización de la etapa (3), los disolventes son dimetilsulfóxido o la mezcla de dimetilsulfóxido y acetato de isopropilo, en la que la relación de volumen de dimetilsulfóxido y acetato de isopropilo está en el intervalo de 50:1 a 1:10.
5. El procedimiento según la reivindicación 4, caracterizado porque la relación de volumen de dimetilsulfóxido y acetato de isopropilo es 1:2.
6. El procedimiento según la reivindicación 1 o 2, caracterizado porque en la etapa (4), dicha reacción se lleva a cabo en presencia de una base, y la base se selecciona entre los hidróxidos de metales alcalinos.
7. El procedimiento según la reivindicación 6, caracterizado porque dichos hidróxidos alcalinos se seleccionan del grupo que consiste en LiOH, KOH y NaOH, preferentemente LiOH.
8. El procedimiento según la reivindicación 1 o 2, caracterizado porque en la etapa (5), dicha reacción de condensación de amida se lleva a cabo en presencia de un agente de condensación, y el agente de condensación se selecciona del grupo que consiste en clorocarbonato de isopropilo, N,N'-carbonildiimidazol y 2-(7-oxibenzotriazol)-N,N,N',N'-tetrametilurea hexafluorofosfato (HATU).
9. El procedimiento según la reivindicación 1 o 2, caracterizado porque en la etapa (1), dicha reacción de sustitución se lleva a cabo en medio alcalino, utilizando Cu, CuI, y N,N-dimetilglicina como catalizador.
10. El procedimiento según la reivindicación 1 o 2, caracterizado porque en la etapa (2), dicho reactivo de metilesterificación para el grupo carboxilo es yoduro de metilo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201610901502.0A CN107954936B (zh) | 2016-10-17 | 2016-10-17 | 一种制备氘代咪唑二酮类化合物的方法 |
| PCT/CN2016/110978 WO2018072300A1 (zh) | 2016-10-17 | 2016-12-20 | 一种制备氘代咪唑二酮类化合物的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2943011T3 true ES2943011T3 (es) | 2023-06-08 |
Family
ID=61954120
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16919556T Active ES2943011T3 (es) | 2016-10-17 | 2016-12-20 | Procedimiento de preparación del compuesto imidazol dicetona deuterado |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20200087261A1 (es) |
| EP (1) | EP3527556B1 (es) |
| JP (1) | JP7241682B2 (es) |
| CN (1) | CN107954936B (es) |
| AU (1) | AU2016426847B2 (es) |
| CA (1) | CA3040785C (es) |
| DK (1) | DK3527556T3 (es) |
| ES (1) | ES2943011T3 (es) |
| FI (1) | FI3527556T3 (es) |
| HU (1) | HUE061557T2 (es) |
| PL (1) | PL3527556T3 (es) |
| PT (1) | PT3527556T (es) |
| TW (1) | TWI728077B (es) |
| WO (1) | WO2018072300A1 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109180511A (zh) * | 2018-08-22 | 2019-01-11 | 辽宁东科药业有限公司 | 一种盐酸丁卡因的制备方法 |
| CN114621109B (zh) * | 2020-12-14 | 2024-04-26 | 成都苑东生物制药股份有限公司 | 一种阿帕他胺的合成方法及其中间体 |
| CN113698310B (zh) * | 2021-08-20 | 2023-03-17 | 江西金丰药业有限公司 | 一种恩杂鲁胺双酯中间体的制备方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| LT2656842T (lt) * | 2006-03-27 | 2016-09-26 | The Regents Of The University Of California | Androgeno receptoriaus moduliatorius, skirtas prostatos vėžio ir su androgeno receptoriumi susijusių ligų gydymui |
| NZ594741A (en) * | 2009-02-24 | 2014-03-28 | Medivation Prostate Therapeutics Inc | Specific diarylhydantoin and diarylthiohydantoin compounds |
| CN101817787B (zh) * | 2009-02-26 | 2013-07-24 | 童友之 | 抗前列腺癌的雄性激素受体拮抗剂 |
| PL2538785T3 (pl) * | 2010-02-24 | 2018-08-31 | Medivation Prostate Therapeutics Llc | Sposoby syntezy zwiazków diarylotiohydantoinowych i diarylohydantoinowych |
| CN103159680A (zh) * | 2011-12-14 | 2013-06-19 | 爱美尼迪药物有限公司 | 咪唑二酮类化合物及其用途 |
| JP6469092B2 (ja) * | 2013-05-29 | 2019-02-13 | ヒノバ ファーマシューティカルズ インコーポレイテッド | イミダゾリジンジオン化合物及び薬物組成物 |
| CN104341352A (zh) | 2013-08-09 | 2015-02-11 | 南京衡杰生物科技有限公司 | 作为雄激素受体拮抗剂的二芳基乙内酰脲化合物及其应用 |
| CN104803918B (zh) * | 2014-01-26 | 2017-11-10 | 上海医药工业研究院 | 恩杂鲁胺的制备方法 |
| CN104016924B (zh) * | 2014-06-16 | 2016-04-13 | 上海鼎雅药物化学科技有限公司 | 一种“一锅法”合成恩杂鲁胺的方法 |
| RU2557235C1 (ru) * | 2014-07-08 | 2015-07-20 | Александр Васильевич Иващенко | Замещенные 2-тиоксо-имидазолидин-4-оны и их спироаналоги, противораковый активный компонент, фармацевтическая композиция, лекарственный препарат, способ лечения рака простаты |
-
2016
- 2016-10-17 CN CN201610901502.0A patent/CN107954936B/zh active Active
- 2016-12-20 PL PL16919556.7T patent/PL3527556T3/pl unknown
- 2016-12-20 HU HUE16919556A patent/HUE061557T2/hu unknown
- 2016-12-20 US US16/342,912 patent/US20200087261A1/en not_active Abandoned
- 2016-12-20 DK DK16919556.7T patent/DK3527556T3/da active
- 2016-12-20 JP JP2019520547A patent/JP7241682B2/ja active Active
- 2016-12-20 CA CA3040785A patent/CA3040785C/en active Active
- 2016-12-20 EP EP16919556.7A patent/EP3527556B1/en active Active
- 2016-12-20 PT PT169195567T patent/PT3527556T/pt unknown
- 2016-12-20 WO PCT/CN2016/110978 patent/WO2018072300A1/zh not_active Ceased
- 2016-12-20 ES ES16919556T patent/ES2943011T3/es active Active
- 2016-12-20 FI FIEP16919556.7T patent/FI3527556T3/fi active
- 2016-12-20 AU AU2016426847A patent/AU2016426847B2/en active Active
-
2017
- 2017-03-24 TW TW106109895A patent/TWI728077B/zh active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2019532075A (ja) | 2019-11-07 |
| TWI728077B (zh) | 2021-05-21 |
| AU2016426847B2 (en) | 2020-11-26 |
| HUE061557T2 (hu) | 2023-07-28 |
| CN107954936A (zh) | 2018-04-24 |
| CA3040785A1 (en) | 2018-04-26 |
| DK3527556T3 (da) | 2023-05-01 |
| AU2016426847A1 (en) | 2019-06-06 |
| TW201815768A (zh) | 2018-05-01 |
| FI3527556T3 (fi) | 2023-04-25 |
| EP3527556A4 (en) | 2020-04-29 |
| JP7241682B2 (ja) | 2023-03-17 |
| PL3527556T3 (pl) | 2023-08-14 |
| US20200087261A1 (en) | 2020-03-19 |
| PT3527556T (pt) | 2023-05-02 |
| EP3527556A1 (en) | 2019-08-21 |
| CN107954936B (zh) | 2021-03-19 |
| EP3527556B1 (en) | 2023-01-25 |
| WO2018072300A1 (zh) | 2018-04-26 |
| CA3040785C (en) | 2021-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111423452A (zh) | 瑞卢戈利的中间体及其制备方法和应用 | |
| CN100402523C (zh) | 杂环化合物及以其为有效成分的抗肿瘤药 | |
| CN103694238B (zh) | No供体型苦参碱衍生物、其制备方法及医药用途 | |
| ES2943011T3 (es) | Procedimiento de preparación del compuesto imidazol dicetona deuterado | |
| CN106632193A (zh) | 一种白杨素氨基酸衍生物的制备方法 | |
| ES2879294T3 (es) | Formas polimórficas de Belinostat y procesos para la preparación de las mismas | |
| CN102408437A (zh) | 一种阿扑西林的制备方法 | |
| CN110229168A (zh) | 11,20-二羰基济源冬凌草甲素及其l-氨基酸-14-酯三氟乙酸盐 | |
| CN117186013B (zh) | 一种以松香酸为疏水基团的蛋白降解剂及其制备方法、药物组合物和应用 | |
| WO2009035168A1 (ja) | 4-カルバモイル-5-ヒドロキシ-イミダゾール誘導体のスルホン酸塩化合物 | |
| CN103030631B (zh) | 用于制备嘧啶二酮类dpp-iv抑制剂的化合物 | |
| CN102219811B (zh) | Ca-4衍生物、其制法及其医药用途 | |
| CN103922992B (zh) | 一种抗癌活性吲哚酮衍生物、合成方法及其用途 | |
| CN104817568A (zh) | 5,6-双脱氢去甲斑蝥醇衍生物及其抗肿瘤应用 | |
| CN101768075A (zh) | 15-羰基甜菊醇衍生物和盐、制备方法及用途 | |
| JP2008308690A (ja) | ポリ(エチレングリコール)機能性誘導体およびその製造方法 | |
| CN117843638A (zh) | 基于rsl3诱导gpx4蛋白降解的双功能分子化合物的制备与应用 | |
| CN104557916B (zh) | 2‑取代β‑咔啉类化合物及其用于制备预防或治疗肿瘤药物的应用 | |
| CN114560845B (zh) | 喹啉化合物的晶型ɑ及其制备方法和应用 | |
| CN110698533A (zh) | 一种熊果酸吲哚醌基类衍生物及其制备方法和应用 | |
| CN120398997A (zh) | 一种具有HIF-1α蛋白降解活性的化合物及其合成方法与应用 | |
| CN102659895A (zh) | 甾体生物碱盐酸盐及其制备方法和用途 | |
| CN121202872A (zh) | 一种帕博西尼衍生物及其制备方法和应用、药物组合物及其应用 | |
| CN116283817A (zh) | 一类no供体型hdac抑制剂、组合物及其用途 | |
| CN121135703A (zh) | 一类3,5-二芳基-1,2,4-噁二唑类衍生物及其制备方法和应用 |