ES2950450T3 - Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN - Google Patents
Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN Download PDFInfo
- Publication number
- ES2950450T3 ES2950450T3 ES18209821T ES18209821T ES2950450T3 ES 2950450 T3 ES2950450 T3 ES 2950450T3 ES 18209821 T ES18209821 T ES 18209821T ES 18209821 T ES18209821 T ES 18209821T ES 2950450 T3 ES2950450 T3 ES 2950450T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- pyridazin
- methyl
- tetramethylpiperidin
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 1,4-Disubstituted Pyridazine Chemical class 0.000 title claims description 130
- 230000007812 deficiency Effects 0.000 title claims description 12
- 238000000034 method Methods 0.000 title abstract description 152
- 150000001875 compounds Chemical class 0.000 claims abstract description 216
- 150000003839 salts Chemical class 0.000 claims abstract description 76
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 22
- 239000000203 mixture Substances 0.000 claims description 201
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 44
- 208000002320 spinal muscular atrophy Diseases 0.000 claims description 44
- 229910052757 nitrogen Inorganic materials 0.000 claims description 21
- 238000011282 treatment Methods 0.000 claims description 20
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 18
- 239000003814 drug Substances 0.000 claims description 16
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 125000001072 heteroaryl group Chemical group 0.000 claims description 10
- 125000005842 heteroatom Chemical group 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 10
- 208000003954 Spinal Muscular Atrophies of Childhood Diseases 0.000 claims description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims description 9
- 125000001424 substituent group Chemical group 0.000 claims description 8
- 208000033526 Proximal spinal muscular atrophy type 3 Diseases 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 201000004815 juvenile spinal muscular atrophy Diseases 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 5
- 230000001684 chronic effect Effects 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 208000032471 type 1 spinal muscular atrophy Diseases 0.000 claims description 3
- 208000032527 type III spinal muscular atrophy Diseases 0.000 claims description 3
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims 1
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims 1
- 125000004093 cyano group Chemical group *C#N 0.000 claims 1
- 125000006413 ring segment Chemical group 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 abstract description 11
- 230000001225 therapeutic effect Effects 0.000 abstract description 5
- 239000013543 active substance Substances 0.000 abstract 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 740
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 252
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 241
- 239000011541 reaction mixture Substances 0.000 description 161
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 144
- 239000000543 intermediate Substances 0.000 description 135
- 239000000243 solution Substances 0.000 description 133
- 239000007787 solid Substances 0.000 description 128
- 230000015572 biosynthetic process Effects 0.000 description 126
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 119
- 238000003786 synthesis reaction Methods 0.000 description 117
- 238000007429 general method Methods 0.000 description 116
- 238000005160 1H NMR spectroscopy Methods 0.000 description 111
- 235000019439 ethyl acetate Nutrition 0.000 description 109
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 107
- 239000000047 product Substances 0.000 description 101
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 90
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 86
- 238000006243 chemical reaction Methods 0.000 description 86
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 78
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical group C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 78
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 70
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 66
- 239000013058 crude material Substances 0.000 description 62
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 60
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 56
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 54
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 48
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 48
- 238000000746 purification Methods 0.000 description 48
- 239000002904 solvent Substances 0.000 description 48
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 47
- 238000006069 Suzuki reaction reaction Methods 0.000 description 47
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 46
- 239000012267 brine Substances 0.000 description 46
- 239000012043 crude product Substances 0.000 description 46
- 239000012044 organic layer Substances 0.000 description 46
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 46
- 229910021529 ammonia Inorganic materials 0.000 description 45
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 43
- 238000003818 flash chromatography Methods 0.000 description 42
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 40
- 238000005481 NMR spectroscopy Methods 0.000 description 39
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 39
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 38
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 37
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 37
- 239000000706 filtrate Substances 0.000 description 37
- 230000002829 reductive effect Effects 0.000 description 36
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 33
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 32
- 229910052938 sodium sulfate Inorganic materials 0.000 description 32
- 235000011152 sodium sulphate Nutrition 0.000 description 32
- 238000004128 high performance liquid chromatography Methods 0.000 description 31
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 30
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- 238000010511 deprotection reaction Methods 0.000 description 30
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 30
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 29
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 29
- 239000000460 chlorine Chemical group 0.000 description 26
- 238000002360 preparation method Methods 0.000 description 25
- JWAZRIHNYRIHIV-UHFFFAOYSA-N 2-naphthol Chemical compound C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 description 24
- 108090000623 proteins and genes Proteins 0.000 description 24
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 23
- 239000000463 material Substances 0.000 description 23
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 22
- 201000010099 disease Diseases 0.000 description 22
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 22
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 21
- 239000007864 aqueous solution Substances 0.000 description 21
- 239000000725 suspension Substances 0.000 description 21
- VIFJLLKYDCECIX-UHFFFAOYSA-N 6-chloro-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(Cl)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 VIFJLLKYDCECIX-UHFFFAOYSA-N 0.000 description 20
- 239000002253 acid Substances 0.000 description 20
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 20
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 20
- 239000003480 eluent Substances 0.000 description 19
- 102000004169 proteins and genes Human genes 0.000 description 19
- 239000011734 sodium Substances 0.000 description 19
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 18
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 18
- 239000003607 modifier Substances 0.000 description 18
- 239000003921 oil Substances 0.000 description 18
- 235000019198 oils Nutrition 0.000 description 18
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 18
- LETVJWLLIMJADE-UHFFFAOYSA-N pyridazin-3-amine Chemical compound NC1=CC=CN=N1 LETVJWLLIMJADE-UHFFFAOYSA-N 0.000 description 18
- 238000010898 silica gel chromatography Methods 0.000 description 18
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 17
- 239000000741 silica gel Substances 0.000 description 17
- 229910002027 silica gel Inorganic materials 0.000 description 17
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 16
- 239000007832 Na2SO4 Substances 0.000 description 16
- 238000004440 column chromatography Methods 0.000 description 16
- 229910052805 deuterium Inorganic materials 0.000 description 16
- 239000000284 extract Substances 0.000 description 16
- 238000002953 preparative HPLC Methods 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 14
- 101000617738 Homo sapiens Survival motor neuron protein Proteins 0.000 description 14
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 14
- 239000012074 organic phase Substances 0.000 description 14
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 13
- 102100021947 Survival motor neuron protein Human genes 0.000 description 13
- 239000000306 component Substances 0.000 description 13
- 238000001704 evaporation Methods 0.000 description 13
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 13
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 13
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- 230000008020 evaporation Effects 0.000 description 12
- 239000012458 free base Substances 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 12
- 229910000160 potassium phosphate Inorganic materials 0.000 description 12
- 235000011009 potassium phosphates Nutrition 0.000 description 12
- 239000000377 silicon dioxide Substances 0.000 description 12
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 11
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 11
- 230000001143 conditioned effect Effects 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 238000007327 hydrogenolysis reaction Methods 0.000 description 11
- 238000010992 reflux Methods 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical compound COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N acetic acid anhydride Natural products CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 10
- 239000004480 active ingredient Substances 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 238000001816 cooling Methods 0.000 description 10
- 238000010348 incorporation Methods 0.000 description 10
- 235000017557 sodium bicarbonate Nutrition 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 229910000029 sodium carbonate Inorganic materials 0.000 description 10
- 235000017550 sodium carbonate Nutrition 0.000 description 10
- 101150003085 Pdcl gene Proteins 0.000 description 9
- 125000000217 alkyl group Chemical group 0.000 description 9
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 9
- 239000003153 chemical reaction reagent Substances 0.000 description 9
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 9
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 9
- 235000019341 magnesium sulphate Nutrition 0.000 description 9
- 235000011056 potassium acetate Nutrition 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 125000006239 protecting group Chemical group 0.000 description 9
- 238000004007 reversed phase HPLC Methods 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 239000003826 tablet Substances 0.000 description 9
- FXPFRJGTQBVPMT-UHFFFAOYSA-N 3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=CC(O)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 FXPFRJGTQBVPMT-UHFFFAOYSA-N 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 8
- 238000004587 chromatography analysis Methods 0.000 description 8
- 208000035475 disorder Diseases 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 150000002430 hydrocarbons Chemical group 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- GUSWJGOYDXFJSI-UHFFFAOYSA-N 3,6-dichloropyridazine Chemical compound ClC1=CC=C(Cl)N=N1 GUSWJGOYDXFJSI-UHFFFAOYSA-N 0.000 description 7
- ZNTOQGWJGUIQSQ-UHFFFAOYSA-N 6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(O)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ZNTOQGWJGUIQSQ-UHFFFAOYSA-N 0.000 description 7
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 7
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 7
- 229910015845 BBr3 Inorganic materials 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- XBBXUQXQMUCNNJ-UHFFFAOYSA-N [3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound COC1=CC(OS(=O)(=O)C(F)(F)F)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 XBBXUQXQMUCNNJ-UHFFFAOYSA-N 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 7
- 125000002950 monocyclic group Chemical group 0.000 description 7
- 210000002161 motor neuron Anatomy 0.000 description 7
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 7
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 6
- QEIXVPUDYVNQBK-UHFFFAOYSA-N 3-chloro-6-(2-methoxy-4-pyrazol-1-ylphenyl)pyridazine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C1=CC=C(Cl)N=N1 QEIXVPUDYVNQBK-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- 101150081851 SMN1 gene Proteins 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 125000005843 halogen group Chemical group 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 239000003999 initiator Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 230000003287 optical effect Effects 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 229910052708 sodium Inorganic materials 0.000 description 6
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 5
- XUOPECWWIDEMRR-UHFFFAOYSA-N 3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound COC1=CC(O)=CC=C1B1OC(C)(C)C(C)(C)O1 XUOPECWWIDEMRR-UHFFFAOYSA-N 0.000 description 5
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 5
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- 229910000024 caesium carbonate Inorganic materials 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 238000006880 cross-coupling reaction Methods 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 125000000623 heterocyclic group Chemical group 0.000 description 5
- 150000003840 hydrochlorides Chemical class 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 230000000717 retained effect Effects 0.000 description 5
- 238000000926 separation method Methods 0.000 description 5
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 5
- KEZNMOUMHOZFRA-UHFFFAOYSA-N 1h-pyrazol-4-ylboronic acid Chemical compound OB(O)C=1C=NNC=1 KEZNMOUMHOZFRA-UHFFFAOYSA-N 0.000 description 4
- CRBIWPUGVLYXMC-UHFFFAOYSA-N 2-[[2-bromo-5-(5-methyl-1,3-oxazol-2-yl)-3-phenylmethoxyphenoxy]methoxy]ethyl-trimethylsilane Chemical compound O1C(C)=CN=C1C1=CC(OCOCC[Si](C)(C)C)=C(Br)C(OCC=2C=CC=CC=2)=C1 CRBIWPUGVLYXMC-UHFFFAOYSA-N 0.000 description 4
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 4
- ABLJWVJJULHGMK-UHFFFAOYSA-N 6-(4-bromo-2,6-dimethoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(Br)=CC(OC)=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 ABLJWVJJULHGMK-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 206010010356 Congenital anomaly Diseases 0.000 description 4
- XPDWGBQVDMORPB-UHFFFAOYSA-N Fluoroform Chemical compound FC(F)F XPDWGBQVDMORPB-UHFFFAOYSA-N 0.000 description 4
- 235000019502 Orange oil Nutrition 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 4
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 description 4
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 4
- 238000006795 borylation reaction Methods 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 238000010828 elution Methods 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- 230000000155 isotopic effect Effects 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- NCWXIIHXWQKDFT-UHFFFAOYSA-N methyl 4-bromo-3-hydroxy-5-phenylmethoxybenzoate Chemical compound COC(=O)C1=CC(O)=C(Br)C(OCC=2C=CC=CC=2)=C1 NCWXIIHXWQKDFT-UHFFFAOYSA-N 0.000 description 4
- 210000003205 muscle Anatomy 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 239000010502 orange oil Substances 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 4
- 230000005855 radiation Effects 0.000 description 4
- 239000012453 solvate Substances 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- NMPMHZMOPNJSAJ-UHFFFAOYSA-N tert-butyl-[3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy]-dimethylsilane Chemical compound COC1=CC(O[Si](C)(C)C(C)(C)C)=CC=C1B1OC(C)(C)C(C)(C)O1 NMPMHZMOPNJSAJ-UHFFFAOYSA-N 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- YMIIHJBTQLZXBV-QMMMGPOBSA-N (2s)-2-[tert-butyl(dimethyl)silyl]oxypropanal Chemical compound O=C[C@H](C)O[Si](C)(C)C(C)(C)C YMIIHJBTQLZXBV-QMMMGPOBSA-N 0.000 description 3
- ZEUFEQLEXFCKJR-UHFFFAOYSA-N (4-bromo-3-methoxyphenoxy)-tert-butyl-dimethylsilane Chemical compound COC1=CC(O[Si](C)(C)C(C)(C)C)=CC=C1Br ZEUFEQLEXFCKJR-UHFFFAOYSA-N 0.000 description 3
- UUNGMMOKFCBYHJ-UHFFFAOYSA-N (4-bromo-3-methoxyphenyl)hydrazine Chemical compound COC1=CC(NN)=CC=C1Br UUNGMMOKFCBYHJ-UHFFFAOYSA-N 0.000 description 3
- NLAPTVBPXBXLPN-UHFFFAOYSA-N (4-imidazol-1-yl-2-methoxyphenyl)boronic acid Chemical compound C1=C(B(O)O)C(OC)=CC(N2C=NC=C2)=C1 NLAPTVBPXBXLPN-UHFFFAOYSA-N 0.000 description 3
- CLIITJGOZJCJKU-UHFFFAOYSA-N 1-(4-bromo-3-methoxyphenyl)-3-nitropyrazole Chemical compound C1=C(Br)C(OC)=CC(N2N=C(C=C2)[N+]([O-])=O)=C1 CLIITJGOZJCJKU-UHFFFAOYSA-N 0.000 description 3
- BXGCNMWTCGUDLE-UHFFFAOYSA-N 1-(4-bromo-3-methoxyphenyl)imidazole Chemical compound C1=C(Br)C(OC)=CC(N2C=NC=C2)=C1 BXGCNMWTCGUDLE-UHFFFAOYSA-N 0.000 description 3
- JTXRERWJFTZFSO-UHFFFAOYSA-N 1-(4-bromo-3-methoxyphenyl)pyrazol-3-amine Chemical compound C1=C(Br)C(OC)=CC(N2N=C(N)C=C2)=C1 JTXRERWJFTZFSO-UHFFFAOYSA-N 0.000 description 3
- KPBBWGXWYGVRFC-UHFFFAOYSA-N 1-(4-bromo-3-methoxyphenyl)pyrazol-4-amine Chemical compound C1=C(Br)C(OC)=CC(N2N=CC(N)=C2)=C1 KPBBWGXWYGVRFC-UHFFFAOYSA-N 0.000 description 3
- MPRUKEFNWGKHFT-UHFFFAOYSA-N 1-(4-bromo-3-methoxyphenyl)pyrazole Chemical compound C1=C(Br)C(OC)=CC(N2N=CC=C2)=C1 MPRUKEFNWGKHFT-UHFFFAOYSA-N 0.000 description 3
- LHJDDHAMFMZGDH-UHFFFAOYSA-N 1-[3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazol-3-amine Chemical compound COC1=CC(N2N=C(N)C=C2)=CC=C1B1OC(C)(C)C(C)(C)O1 LHJDDHAMFMZGDH-UHFFFAOYSA-N 0.000 description 3
- YPIBYJATZVKUBP-UHFFFAOYSA-N 1-[3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazol-4-amine Chemical compound COC1=CC(N2N=CC(N)=C2)=CC=C1B1OC(C)(C)C(C)(C)O1 YPIBYJATZVKUBP-UHFFFAOYSA-N 0.000 description 3
- UNBQIMJREMUMCX-UHFFFAOYSA-N 1-[3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazole Chemical compound COC1=CC(N2N=CC=C2)=CC=C1B1OC(C)(C)C(C)(C)O1 UNBQIMJREMUMCX-UHFFFAOYSA-N 0.000 description 3
- PIPWNWZBTWYPJB-UHFFFAOYSA-N 1-bromo-4-iodo-2-methoxybenzene Chemical compound COC1=CC(I)=CC=C1Br PIPWNWZBTWYPJB-UHFFFAOYSA-N 0.000 description 3
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 3
- OTEMFHPLVRXVST-UHFFFAOYSA-N 2-(3-methoxy-4-propan-2-yloxyphenyl)ethanamine;hydrochloride Chemical compound Cl.COC1=CC(CCN)=CC=C1OC(C)C OTEMFHPLVRXVST-UHFFFAOYSA-N 0.000 description 3
- PQIIPMJJAHRHHZ-UHFFFAOYSA-N 2-(4-bromo-3-methoxy-5-phenylmethoxyphenyl)-5-methyl-1,3-oxazole Chemical compound BrC=1C(OC)=CC(C=2OC(C)=CN=2)=CC=1OCC1=CC=CC=C1 PQIIPMJJAHRHHZ-UHFFFAOYSA-N 0.000 description 3
- KLJZAIVAQHNLRW-UHFFFAOYSA-N 2-(6-chloropyridazin-3-yl)-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C1=CC=C(Cl)N=N1 KLJZAIVAQHNLRW-UHFFFAOYSA-N 0.000 description 3
- MMCZCFOMQLDRNW-UHFFFAOYSA-N 2-(6-piperidin-4-yloxypyridazin-3-yl)-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1CCNCC1 MMCZCFOMQLDRNW-UHFFFAOYSA-N 0.000 description 3
- NYKVTIHHAZZDOP-UHFFFAOYSA-N 2-bromo-5-(5-methyl-1,3-oxazol-2-yl)-3-phenylmethoxyphenol Chemical compound O1C(C)=CN=C1C1=CC(O)=C(Br)C(OCC=2C=CC=CC=2)=C1 NYKVTIHHAZZDOP-UHFFFAOYSA-N 0.000 description 3
- QEQKPUUULISNTI-UHFFFAOYSA-N 2-formamido-n-[2-(3-methoxy-4-propan-2-yloxyphenyl)ethyl]acetamide Chemical compound COC1=CC(CCNC(=O)CNC=O)=CC=C1OC(C)C QEQKPUUULISNTI-UHFFFAOYSA-N 0.000 description 3
- DMHAOTJKABKQIM-UHFFFAOYSA-N 3-(1-benzothiophen-2-yl)-6-chloropyridazine Chemical compound N1=NC(Cl)=CC=C1C1=CC2=CC=CC=C2S1 DMHAOTJKABKQIM-UHFFFAOYSA-N 0.000 description 3
- WICPHBMFEMZSJJ-UHFFFAOYSA-N 3-(2-methoxy-4-pyrazol-1-ylphenyl)-6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1CC1CC(C)(C)NC(C)(C)C1 WICPHBMFEMZSJJ-UHFFFAOYSA-N 0.000 description 3
- KMAVVCRXFBLEPK-UHFFFAOYSA-N 3-(2-methoxy-4-pyrazol-1-ylphenyl)-6-piperidin-4-yloxypyridazine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1CCNCC1 KMAVVCRXFBLEPK-UHFFFAOYSA-N 0.000 description 3
- UJNJJLIIJWOCCJ-UHFFFAOYSA-N 3-bromo-4-methoxy-n-[(1-methylpyrazol-4-yl)methyl]benzamide Chemical compound C1=C(Br)C(OC)=CC=C1C(=O)NCC1=CN(C)N=C1 UJNJJLIIJWOCCJ-UHFFFAOYSA-N 0.000 description 3
- AJUYTXBUCBBXKP-UHFFFAOYSA-N 3-chloro-6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazine Chemical compound C1C(C)(C)NC(C)(C)CC1CC1=CC=C(Cl)N=N1 AJUYTXBUCBBXKP-UHFFFAOYSA-N 0.000 description 3
- QVNKFWOWOWZRKB-UHFFFAOYSA-N 3-fluoro-4-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenol Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC(O)=CC=2)F)N=N1 QVNKFWOWOWZRKB-UHFFFAOYSA-N 0.000 description 3
- ZMKDLSXQKNJQCD-UHFFFAOYSA-N 3-methoxy-4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazin-3-yl]phenol Chemical compound COC1=CC(O)=CC=C1C(N=N1)=CC=C1CC1CC(C)(C)NC(C)(C)C1 ZMKDLSXQKNJQCD-UHFFFAOYSA-N 0.000 description 3
- OEISTHCMYDEJKZ-UHFFFAOYSA-N 4-[2-chloro-5-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-1h-pyrazole Chemical compound ClC=1C=C(B2OC(C)(C)C(C)(C)O2)C(OC)=CC=1C=1C=NNC=1 OEISTHCMYDEJKZ-UHFFFAOYSA-N 0.000 description 3
- UNEMEBDSEHTYDH-UHFFFAOYSA-N 4-bromo-3-methoxy-5-phenylmethoxybenzoic acid Chemical compound COC1=CC(C(O)=O)=CC(OCC=2C=CC=CC=2)=C1Br UNEMEBDSEHTYDH-UHFFFAOYSA-N 0.000 description 3
- UYDZUCNMZXCLJI-UHFFFAOYSA-N 4-bromo-3-methoxyphenol Chemical compound COC1=CC(O)=CC=C1Br UYDZUCNMZXCLJI-UHFFFAOYSA-N 0.000 description 3
- IEPXUEOXNHKTML-UHFFFAOYSA-N 4-bromo-3-phenylmethoxy-5-(2-trimethylsilylethoxymethoxy)benzoic acid Chemical compound C[Si](C)(C)CCOCOC1=CC(C(O)=O)=CC(OCC=2C=CC=CC=2)=C1Br IEPXUEOXNHKTML-UHFFFAOYSA-N 0.000 description 3
- FEWGGIQQEFOJMY-UHFFFAOYSA-N 4-bromo-3-phenylmethoxy-n-prop-2-ynyl-5-(2-trimethylsilylethoxymethoxy)benzamide Chemical compound C[Si](C)(C)CCOCOC1=CC(C(=O)NCC#C)=CC(OCC=2C=CC=CC=2)=C1Br FEWGGIQQEFOJMY-UHFFFAOYSA-N 0.000 description 3
- RDZLZTVEUNWIGI-UHFFFAOYSA-N 4-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound COC1=CC=C(O)C=C1B1OC(C)(C)C(C)(C)O1 RDZLZTVEUNWIGI-UHFFFAOYSA-N 0.000 description 3
- IYVIWFGZPOVASD-UHFFFAOYSA-N 4-methoxy-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=CC=C(O)C=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 IYVIWFGZPOVASD-UHFFFAOYSA-N 0.000 description 3
- RKQKQOUFFCTTHP-UHFFFAOYSA-N 5-(4-aminopyrazol-1-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(=CC=2)N2N=CC(N)=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 RKQKQOUFFCTTHP-UHFFFAOYSA-N 0.000 description 3
- XIPKSYAFRCAKRG-UHFFFAOYSA-N 5-bromo-3-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(Br)=CC=2O)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 XIPKSYAFRCAKRG-UHFFFAOYSA-N 0.000 description 3
- YBFXUHXZERVWLD-UHFFFAOYSA-N 6-(2-methoxy-6-phenylmethoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=NC=1C=1C(OC)=CC=CC=1OCC1=CC=CC=C1 YBFXUHXZERVWLD-UHFFFAOYSA-N 0.000 description 3
- JEDIADXYHHSDLC-UHFFFAOYSA-N 6-(4-bromo-2-fluoro-6-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(Br)=CC(F)=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 JEDIADXYHHSDLC-UHFFFAOYSA-N 0.000 description 3
- QMELFGFTFVSKDF-UHFFFAOYSA-N 6-(4-bromo-2-methoxy-6-phenylmethoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=NC=1C=1C(OC)=CC(Br)=CC=1OCC1=CC=CC=C1 QMELFGFTFVSKDF-UHFFFAOYSA-N 0.000 description 3
- GUEUELQNFNJYJO-UHFFFAOYSA-N 6-(4-chloro-2-fluorophenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC(Cl)=CC=2)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 GUEUELQNFNJYJO-UHFFFAOYSA-N 0.000 description 3
- SYGWNGZPDLGJKH-UHFFFAOYSA-N 6-(4-fluoro-2-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(F)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 SYGWNGZPDLGJKH-UHFFFAOYSA-N 0.000 description 3
- AMIPGOWGFJLCIG-UHFFFAOYSA-N 6-[4-phenylmethoxy-2-(trifluoromethoxy)phenyl]-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(OCC=3C=CC=CC=3)=CC=2)OC(F)(F)F)N=N1 AMIPGOWGFJLCIG-UHFFFAOYSA-N 0.000 description 3
- MMPZCASXVDLVJW-UHFFFAOYSA-N 6-[6-[methyl-(1,2,2,6,6-pentamethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalene-2-carbaldehyde Chemical compound C=1C=C(C=2C(=CC3=CC(C=O)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)N(C)C(C)(C)C1 MMPZCASXVDLVJW-UHFFFAOYSA-N 0.000 description 3
- YHRCPQNCRQLBGB-UHFFFAOYSA-N 6-bromo-7-phenylmethoxynaphthalen-2-ol Chemical compound C=1C2=CC(O)=CC=C2C=C(Br)C=1OCC1=CC=CC=C1 YHRCPQNCRQLBGB-UHFFFAOYSA-N 0.000 description 3
- PCEWEXKPLBHTKW-UHFFFAOYSA-N 6-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydroinden-1-one Chemical compound COC1=CC=2C(=O)CCC=2C=C1B1OC(C)(C)C(C)(C)O1 PCEWEXKPLBHTKW-UHFFFAOYSA-N 0.000 description 3
- PIEKJQDMRNPYAJ-UHFFFAOYSA-N 6-methoxy-5-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-2,3-dihydroinden-1-one Chemical compound COC1=CC=2C(=O)CCC=2C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 PIEKJQDMRNPYAJ-UHFFFAOYSA-N 0.000 description 3
- GIWDWVZVVVTMFD-UHFFFAOYSA-N 8-methoxy-5,6-dihydroimidazo[5,1-a]isoquinolin-9-ol Chemical compound C1CN2C=NC=C2C2=C1C=C(OC)C(O)=C2 GIWDWVZVVVTMFD-UHFFFAOYSA-N 0.000 description 3
- OVGPRJDCUGAYPD-UHFFFAOYSA-N 8-methoxy-9-propan-2-yloxy-5,6-dihydroimidazo[5,1-a]isoquinoline Chemical compound C1CN2C=NC=C2C2=C1C=C(OC)C(OC(C)C)=C2 OVGPRJDCUGAYPD-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 101150015954 SMN2 gene Proteins 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- RDEAKKBVKCVNOE-UHFFFAOYSA-N [1-(3-bromo-4-methoxyphenyl)pyrazol-3-yl]methanol Chemical compound C1=C(Br)C(OC)=CC=C1N1N=C(CO)C=C1 RDEAKKBVKCVNOE-UHFFFAOYSA-N 0.000 description 3
- JHRYZHFUBXMJTP-UHFFFAOYSA-N [3-hydroxy-5-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound COC1=CC(OS(=O)(=O)C(F)(F)F)=CC(O)=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 JHRYZHFUBXMJTP-UHFFFAOYSA-N 0.000 description 3
- WIBKLKSGWDVJBB-UHFFFAOYSA-N [3-methoxy-4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound COC1=CC(OS(=O)(=O)C(F)(F)F)=CC=C1C(N=N1)=CC=C1CC1CC(C)(C)NC(C)(C)C1 WIBKLKSGWDVJBB-UHFFFAOYSA-N 0.000 description 3
- FWFWDUFQLKABCM-UHFFFAOYSA-N [4-methoxy-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound COC1=CC=C(OS(=O)(=O)C(F)(F)F)C=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 FWFWDUFQLKABCM-UHFFFAOYSA-N 0.000 description 3
- CHCQIAAVRUVYIC-UHFFFAOYSA-N [4-phenylmethoxy-2-(trifluoromethoxy)phenyl]boronic acid Chemical compound C1=C(OC(F)(F)F)C(B(O)O)=CC=C1OCC1=CC=CC=C1 CHCQIAAVRUVYIC-UHFFFAOYSA-N 0.000 description 3
- WXVALKGUTHNNRO-UHFFFAOYSA-N [6-methoxy-2-methyl-1-(2-trimethylsilylethoxymethyl)benzimidazol-5-yl]boronic acid Chemical compound C1=C(B(O)O)C(OC)=CC2=C1N=C(C)N2COCC[Si](C)(C)C WXVALKGUTHNNRO-UHFFFAOYSA-N 0.000 description 3
- SPEUIVXLLWOEMJ-UHFFFAOYSA-N acetaldehyde dimethyl acetal Natural products COC(C)OC SPEUIVXLLWOEMJ-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- 230000031709 bromination Effects 0.000 description 3
- 238000005893 bromination reaction Methods 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- KOBAXFIJEBLKRZ-VIFPVBQESA-N ethyl (2s)-2-[tert-butyl(dimethyl)silyl]oxypropanoate Chemical compound CCOC(=O)[C@H](C)O[Si](C)(C)C(C)(C)C KOBAXFIJEBLKRZ-VIFPVBQESA-N 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 239000006260 foam Substances 0.000 description 3
- 239000011888 foil Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- BYYVXSILTNDSEQ-UHFFFAOYSA-N methyl 4-bromo-3-methoxy-5-phenylmethoxybenzoate Chemical compound COC(=O)C1=CC(OC)=C(Br)C(OCC=2C=CC=CC=2)=C1 BYYVXSILTNDSEQ-UHFFFAOYSA-N 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 230000001272 neurogenic effect Effects 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 125000006684 polyhaloalkyl group Polymers 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- JKANAVGODYYCQF-UHFFFAOYSA-N prop-2-yn-1-amine Chemical compound NCC#C JKANAVGODYYCQF-UHFFFAOYSA-N 0.000 description 3
- ORIHZIZPTZTNCU-YVMONPNESA-N salicylaldoxime Chemical compound O\N=C/C1=CC=CC=C1O ORIHZIZPTZTNCU-YVMONPNESA-N 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- NHWUVZVURBNCQD-UHFFFAOYSA-N tert-butyl 4-(2-fluoro-4-hydroxy-5-methoxyphenyl)pyrazole-1-carboxylate Chemical compound C1=C(O)C(OC)=CC(C2=CN(N=C2)C(=O)OC(C)(C)C)=C1F NHWUVZVURBNCQD-UHFFFAOYSA-N 0.000 description 3
- LMFLWOTXODZXBO-UHFFFAOYSA-N tert-butyl n-[2-(3-methoxy-4-propan-2-yloxyphenyl)ethyl]carbamate Chemical compound COC1=CC(CCNC(=O)OC(C)(C)C)=CC=C1OC(C)C LMFLWOTXODZXBO-UHFFFAOYSA-N 0.000 description 3
- LVVZUKZSPLLQLT-UHFFFAOYSA-N tert-butyl n-[2-(4-hydroxy-3-methoxyphenyl)ethyl]carbamate Chemical compound COC1=CC(CCNC(=O)OC(C)(C)C)=CC=C1O LVVZUKZSPLLQLT-UHFFFAOYSA-N 0.000 description 3
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 229910052723 transition metal Inorganic materials 0.000 description 3
- 150000003624 transition metals Chemical class 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- DKZRGRZRQAUPMR-UHFFFAOYSA-N (2-methoxy-6-phenylmethoxyphenyl)boronic acid Chemical compound COC1=CC=CC(OCC=2C=CC=CC=2)=C1B(O)O DKZRGRZRQAUPMR-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- NUPKSTHLQVYXPX-UHFFFAOYSA-N (8-methoxy-5,6-dihydroimidazo[5,1-a]isoquinolin-9-yl) trifluoromethanesulfonate Chemical compound C1CN2C=NC=C2C2=C1C=C(OC)C(OS(=O)(=O)C(F)(F)F)=C2 NUPKSTHLQVYXPX-UHFFFAOYSA-N 0.000 description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 2
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 description 2
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 2
- MLPAIIATLGUTEE-UHFFFAOYSA-N 1,3-thiazol-5-ol Chemical compound OC1=CN=CS1 MLPAIIATLGUTEE-UHFFFAOYSA-N 0.000 description 2
- LOZWAPSEEHRYPG-UHFFFAOYSA-N 1,4-dithiane Chemical compound C1CSCCS1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 2
- YLIHSEAYNQDGCV-UHFFFAOYSA-N 1-chloro-6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalene-2,7-diol Chemical compound C=1C=C(C=2C(=CC3=C(Cl)C(O)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 YLIHSEAYNQDGCV-UHFFFAOYSA-N 0.000 description 2
- UCNGGGYMLHAMJG-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(C)C=C1B1OC(C)(C)C(C)(C)O1 UCNGGGYMLHAMJG-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- ZILIWBGRMJXYTR-UHFFFAOYSA-N 1-phenylmethoxy-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoquinoline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C=CN=C2OCC=3C=CC=CC=3)C2=C1 ZILIWBGRMJXYTR-UHFFFAOYSA-N 0.000 description 2
- XLISNDFVWLZXMW-UHFFFAOYSA-N 2,2,6,6-tetramethyl-4-methylidenepiperidine Chemical compound CC1(C)CC(=C)CC(C)(C)N1 XLISNDFVWLZXMW-UHFFFAOYSA-N 0.000 description 2
- ONASENZNGCCTQH-UHFFFAOYSA-N 2,2,6,6-tetramethyl-4-methylidenepiperidine;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC1(C)CC(=C)CC(C)(C)N1 ONASENZNGCCTQH-UHFFFAOYSA-N 0.000 description 2
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 description 2
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 2
- LCEOAJWOZUSLAW-UHFFFAOYSA-N 2-(6-piperazin-1-ylpyridazin-3-yl)-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1N1CCNCC1 LCEOAJWOZUSLAW-UHFFFAOYSA-N 0.000 description 2
- FIKLWBXHHLBDME-UHFFFAOYSA-N 2-(6-piperidin-4-ylpyridazin-3-yl)-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1C1CCNCC1 FIKLWBXHHLBDME-UHFFFAOYSA-N 0.000 description 2
- KUQHEZJDFPHACI-UHFFFAOYSA-N 2-[(5-bromo-6-methoxy-2-methylbenzimidazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound C1=C(Br)C(OC)=CC2=C1N=C(C)N2COCC[Si](C)(C)C KUQHEZJDFPHACI-UHFFFAOYSA-N 0.000 description 2
- MGWPHVZBBRWTOC-UHFFFAOYSA-N 2-[6-(2,2-dimethylpiperidin-4-yl)oxypyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1CNC(C)(C)CC1OC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 MGWPHVZBBRWTOC-UHFFFAOYSA-N 0.000 description 2
- MOYDXTANPSDHTD-UHFFFAOYSA-N 2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-4-(1H-pyrazol-4-yl)phenol Chemical compound C=1C=C(C=2C(=CC=C(C=2)C2=CNN=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 MOYDXTANPSDHTD-UHFFFAOYSA-N 0.000 description 2
- RJVGWKITANOUKO-UHFFFAOYSA-N 2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)benzene-1,3-diol Chemical compound C=1C=C(C=2C(=CC(=CC=2O)C2=CNN=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 RJVGWKITANOUKO-UHFFFAOYSA-N 0.000 description 2
- IOOILPOCRHNWSR-UHFFFAOYSA-N 2-[[6-methoxy-2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzimidazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound COC1=CC=2N(COCC[Si](C)(C)C)C(C)=NC=2C=C1B1OC(C)(C)C(C)(C)O1 IOOILPOCRHNWSR-UHFFFAOYSA-N 0.000 description 2
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- YNFBMDWHEHETJW-UHFFFAOYSA-N 2-pyridin-2-yl-1h-benzimidazole Chemical compound N1=CC=CC=C1C1=NC2=CC=CC=C2N1 YNFBMDWHEHETJW-UHFFFAOYSA-N 0.000 description 2
- YEKKWGQNWUNPRI-UHFFFAOYSA-N 3-(2-fluoro-4-methoxyphenyl)-6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazine Chemical compound FC1=CC(OC)=CC=C1C(N=N1)=CC=C1OC1CC(C)(C)NC(C)(C)C1 YEKKWGQNWUNPRI-UHFFFAOYSA-N 0.000 description 2
- XUHQVVOQTYRUAQ-UHFFFAOYSA-N 3-(2-methoxy-4-pyrazol-1-ylphenyl)-6-pyrrolidin-3-yloxypyridazine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1CCNC1 XUHQVVOQTYRUAQ-UHFFFAOYSA-N 0.000 description 2
- QLACWJVHVUIAQK-UHFFFAOYSA-N 3-[2-methoxy-4-(1h-pyrazol-4-yl)phenyl]-6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazine Chemical compound COC1=CC(C2=CNN=C2)=CC=C1C(N=N1)=CC=C1CC1CC(C)(C)NC(C)(C)C1 QLACWJVHVUIAQK-UHFFFAOYSA-N 0.000 description 2
- ZWVAEWAYWBRRLZ-UHFFFAOYSA-N 3-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]naphthalene-2,7-diol Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC3=CC(O)=CC=C3C=2)O)N=N1 ZWVAEWAYWBRRLZ-UHFFFAOYSA-N 0.000 description 2
- GVDHMIXHGXIPNW-UHFFFAOYSA-N 3-[6-(piperidin-4-ylmethyl)pyridazin-3-yl]naphthalen-2-ol Chemical compound OC1=CC2=CC=CC=C2C=C1C(N=N1)=CC=C1CC1CCNCC1 GVDHMIXHGXIPNW-UHFFFAOYSA-N 0.000 description 2
- ANRDBHZBYHYNDL-UHFFFAOYSA-N 3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalene-2,7-diol Chemical compound C=1C=C(C=2C(=CC3=CC(O)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ANRDBHZBYHYNDL-UHFFFAOYSA-N 0.000 description 2
- PNVVUFYACYMHEY-UHFFFAOYSA-N 3-fluoro-5-(1-methylpyrazol-4-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(=CC=2O)C2=CN(C)N=C2)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 PNVVUFYACYMHEY-UHFFFAOYSA-N 0.000 description 2
- ABOVEBCKYNWNLM-UHFFFAOYSA-N 3-fluoro-5-(2-methoxypyridin-4-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=NC=CC(=C1)C1=CC(F)=C(C(O)=C1)C1=CC=C(N=N1)N(C)C1CC(C)(C)NC(C)(C)C1 ABOVEBCKYNWNLM-UHFFFAOYSA-N 0.000 description 2
- SNUFINWSPWMYDU-UHFFFAOYSA-N 3-methoxy-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound COC1=CC(C2=CNN=C2)=CC(O)=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 SNUFINWSPWMYDU-UHFFFAOYSA-N 0.000 description 2
- YKKWAPCXANUWLI-UHFFFAOYSA-N 3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzoic acid Chemical compound COC1=CC(C(O)=O)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 YKKWAPCXANUWLI-UHFFFAOYSA-N 0.000 description 2
- UGNZDAGQLDFKBI-UHFFFAOYSA-N 3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzonitrile Chemical compound COC1=CC(C#N)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 UGNZDAGQLDFKBI-UHFFFAOYSA-N 0.000 description 2
- KEGQTVHNBCOAFF-UHFFFAOYSA-N 3-methoxy-5-(1-methylpyrazol-4-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=CC(C2=CN(C)N=C2)=CC(O)=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 KEGQTVHNBCOAFF-UHFFFAOYSA-N 0.000 description 2
- UNIKTSOXRRFQII-UHFFFAOYSA-N 3-methoxy-5-(5-methyl-1,3-oxazol-2-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=CC(C=2OC(C)=CN=2)=CC(O)=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 UNIKTSOXRRFQII-UHFFFAOYSA-N 0.000 description 2
- IHCAPEFPRVYXMI-UHFFFAOYSA-N 3-phenylmethoxy-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isoquinoline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C=NC(OCC=2C=CC=CC=2)=C2)C2=C1 IHCAPEFPRVYXMI-UHFFFAOYSA-N 0.000 description 2
- JXAYZFQXXNLHKZ-UHFFFAOYSA-N 4,4,5,5-tetramethyl-2-[3-phenylmethoxy-6-(3-phenylmethoxypropoxy)naphthalen-2-yl]-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C(C(=CC1=C2)OCC=3C=CC=CC=3)=CC1=CC=C2OCCCOCC1=CC=CC=C1 JXAYZFQXXNLHKZ-UHFFFAOYSA-N 0.000 description 2
- BZCMZBGSVGHDKB-UHFFFAOYSA-N 4-(2-fluoro-6-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=CC(F)=C1C1=CC=NN=C1N(C)C1CC(C)(C)NC(C)(C)C1 BZCMZBGSVGHDKB-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- WWOBSLROTCFOPH-UHFFFAOYSA-N 4-(4-bromo-2-chloro-5-methoxyphenyl)-1h-pyrazole Chemical compound C1=C(Br)C(OC)=CC(C2=CNN=C2)=C1Cl WWOBSLROTCFOPH-UHFFFAOYSA-N 0.000 description 2
- JHXKBTFMEYOPEH-UHFFFAOYSA-N 4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-(trifluoromethoxy)phenol Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(O)=CC=2)OC(F)(F)F)N=N1 JHXKBTFMEYOPEH-UHFFFAOYSA-N 0.000 description 2
- CAQJOPZEWCGSMG-UHFFFAOYSA-N 4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-(trifluoromethoxy)phenol Chemical compound C=1C=C(C=2C(=CC(O)=CC=2)OC(F)(F)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 CAQJOPZEWCGSMG-UHFFFAOYSA-N 0.000 description 2
- RNHSYUZZAGJQDD-UHFFFAOYSA-N 4-bromo-6-(2-fluoro-6-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=CC(F)=C1C1=CC(Br)=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 RNHSYUZZAGJQDD-UHFFFAOYSA-N 0.000 description 2
- PMXSGPFPTQJFSV-UHFFFAOYSA-N 4-methoxy-n-[(1-methylpyrazol-4-yl)methyl]-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzamide Chemical compound COC1=CC=C(C(=O)NCC2=CN(C)N=C2)C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 PMXSGPFPTQJFSV-UHFFFAOYSA-N 0.000 description 2
- GZRHGDAVNTTXCB-UHFFFAOYSA-N 5-(1H-pyrazol-4-yl)-2-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-(trifluoromethoxy)phenol Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(=CC=2O)C2=CNN=C2)OC(F)(F)F)N=N1 GZRHGDAVNTTXCB-UHFFFAOYSA-N 0.000 description 2
- IUQMBINWIGHFMS-UHFFFAOYSA-N 5-(3-aminopyrazol-1-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(=CC=2)N2N=C(N)C=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 IUQMBINWIGHFMS-UHFFFAOYSA-N 0.000 description 2
- UOPZFPWKIZTDRV-UHFFFAOYSA-N 5-bromo-6-methoxy-2,3-dihydroinden-1-one Chemical compound C1=C(Br)C(OC)=CC2=C1CCC2=O UOPZFPWKIZTDRV-UHFFFAOYSA-N 0.000 description 2
- RNWGYGYYVMSQHF-UHFFFAOYSA-N 6-(1-benzothiophen-2-yl)-n-(1,2,2,6,6-pentamethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C1C(C)(C)N(C)C(C)(C)CC1NC1=CC=C(C=2SC3=CC=CC=C3C=2)N=N1 RNWGYGYYVMSQHF-UHFFFAOYSA-N 0.000 description 2
- DRMZKMYWQWJIDA-UHFFFAOYSA-N 6-(2,6-dimethoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=CC(OC)=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 DRMZKMYWQWJIDA-UHFFFAOYSA-N 0.000 description 2
- VOJKJGHVLGLPLE-UHFFFAOYSA-N 6-(6-ethenyl-3-phenylmethoxynaphthalen-2-yl)-n-methyl-n-(1,2,2,6,6-pentamethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC3=CC(C=C)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)N(C)C(C)(C)C1 VOJKJGHVLGLPLE-UHFFFAOYSA-N 0.000 description 2
- IICJUULRMBKDPB-UHFFFAOYSA-N 6-(6-methoxy-3-phenylmethoxynaphthalen-2-yl)-n-methyl-n-(1,2,2,6,6-pentamethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C2=CC(OC)=CC=C2C=C(C=2N=NC(=CC=2)N(C)C2CC(C)(C)N(C)C(C)(C)C2)C=1OCC1=CC=CC=C1 IICJUULRMBKDPB-UHFFFAOYSA-N 0.000 description 2
- NYKDGXJELNABPY-UHFFFAOYSA-N 6-(8-methoxy-5,6-dihydroimidazo[5,1-a]isoquinolin-9-yl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=2CCN3C=NC=C3C=2C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 NYKDGXJELNABPY-UHFFFAOYSA-N 0.000 description 2
- CONKBQPVFMXDOV-QHCPKHFHSA-N 6-[(5S)-5-[[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]methyl]-2-oxo-1,3-oxazolidin-3-yl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C[C@H]1CN(C(O1)=O)C1=CC2=C(NC(O2)=O)C=C1 CONKBQPVFMXDOV-QHCPKHFHSA-N 0.000 description 2
- GFGYTXQLYWJQPS-UHFFFAOYSA-N 6-[2,6-dimethoxy-4-(1h-pyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(C2=CNN=C2)=CC(OC)=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 GFGYTXQLYWJQPS-UHFFFAOYSA-N 0.000 description 2
- JVEWFEGMLHXNCA-UHFFFAOYSA-N 6-[2-methoxy-4-(1h-pyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(C2=CNN=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 JVEWFEGMLHXNCA-UHFFFAOYSA-N 0.000 description 2
- ZTYAGOCYJVKONG-UHFFFAOYSA-N 6-[2-methoxy-4-(5-methyl-1,3-oxazol-2-yl)-6-phenylmethoxyphenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=NC=1C=1C(OC)=CC(C=2OC(C)=CN=2)=CC=1OCC1=CC=CC=C1 ZTYAGOCYJVKONG-UHFFFAOYSA-N 0.000 description 2
- JCSGLZXJNZIEBY-UHFFFAOYSA-N 6-[4-(3-aminopyrazol-1-yl)-2-methoxyphenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(N2N=C(N)C=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 JCSGLZXJNZIEBY-UHFFFAOYSA-N 0.000 description 2
- WIMYXNRGSFKVLR-UHFFFAOYSA-N 6-[4-(4-aminopyrazol-1-yl)-2-methoxyphenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(N2N=CC(N)=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 WIMYXNRGSFKVLR-UHFFFAOYSA-N 0.000 description 2
- JDAHLFBDDBMJIO-UHFFFAOYSA-N 6-[4-fluoro-5-(1h-imidazol-5-yl)-2-methoxyphenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(F)=C(C=2N=CNC=2)C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 JDAHLFBDDBMJIO-UHFFFAOYSA-N 0.000 description 2
- ZUFBGECMKAHEED-UHFFFAOYSA-N 6-[6-(difluoromethyl)-3-phenylmethoxynaphthalen-2-yl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC3=CC(=CC=C3C=2)C(F)F)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ZUFBGECMKAHEED-UHFFFAOYSA-N 0.000 description 2
- DIPKZZHSVWPFQC-UHFFFAOYSA-N 6-bromo-3-phenylmethoxyisoquinoline Chemical compound C=1C2=CC(Br)=CC=C2C=NC=1OCC1=CC=CC=C1 DIPKZZHSVWPFQC-UHFFFAOYSA-N 0.000 description 2
- SSSMQFIHVLLJBE-UHFFFAOYSA-N 6-chloro-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(Cl)N=N1 SSSMQFIHVLLJBE-UHFFFAOYSA-N 0.000 description 2
- ADNBSVXKADLTAE-UHFFFAOYSA-N 7-(3-hydroxy-3-methylbutoxy)-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(OCCC(C)(C)O)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ADNBSVXKADLTAE-UHFFFAOYSA-N 0.000 description 2
- SNBAIZPWLNETCQ-UHFFFAOYSA-N 7-(3-hydroxypropoxy)-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(OCCCO)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 SNBAIZPWLNETCQ-UHFFFAOYSA-N 0.000 description 2
- HMXDQRRMUALXRR-UHFFFAOYSA-N 7-(3-methoxypropoxy)-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound OC1=CC2=CC(OCCCOC)=CC=C2C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 HMXDQRRMUALXRR-UHFFFAOYSA-N 0.000 description 2
- ZFMREKFGWZZCPD-UHFFFAOYSA-N 7-(difluoromethyl)-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(=CC=C3C=2)C(F)F)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ZFMREKFGWZZCPD-UHFFFAOYSA-N 0.000 description 2
- AZZWNVJXIUWPPS-UHFFFAOYSA-N 7-methoxy-3-[6-[methyl-(1,2,2,6,6-pentamethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound OC1=CC2=CC(OC)=CC=C2C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)N(C)C(C)(C)C1 AZZWNVJXIUWPPS-UHFFFAOYSA-N 0.000 description 2
- WYFYSTSNBJRGTC-UHFFFAOYSA-N 7-phenylmethoxy-6-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC3=CC(O)=CC=C3C=2)OCC=2C=CC=CC=2)N=N1 WYFYSTSNBJRGTC-UHFFFAOYSA-N 0.000 description 2
- CZTVOBCKOBFZEI-UHFFFAOYSA-N 8-methoxy-9-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5,6-dihydroimidazo[5,1-a]isoquinoline Chemical compound COC1=CC=2CCN3C=NC=C3C=2C=C1B1OC(C)(C)C(C)(C)O1 CZTVOBCKOBFZEI-UHFFFAOYSA-N 0.000 description 2
- OOZZFJILNSEBBF-UHFFFAOYSA-N 9-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5,6-dihydroimidazo[5,1-a]isoquinolin-8-ol Chemical compound C=1C=C(C=2C(=CC=3CCN4C=NC=C4C=3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 OOZZFJILNSEBBF-UHFFFAOYSA-N 0.000 description 2
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- VVJKKWFAADXIJK-UHFFFAOYSA-N Allylamine Chemical compound NCC=C VVJKKWFAADXIJK-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 102100032187 Androgen receptor Human genes 0.000 description 2
- 208000008037 Arthrogryposis Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- 208000029402 Bulbospinal muscular atrophy Diseases 0.000 description 2
- 206010068597 Bulbospinal muscular atrophy congenital Diseases 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- 229940126062 Compound A Drugs 0.000 description 2
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000007821 HATU Substances 0.000 description 2
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 2
- 101000775732 Homo sapiens Androgen receptor Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 208000027747 Kennedy disease Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- 208000032225 Proximal spinal muscular atrophy type 1 Diseases 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 206010038687 Respiratory distress Diseases 0.000 description 2
- 101150113275 Smn gene Proteins 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- JWUXJYZVKZKLTJ-UHFFFAOYSA-N Triacetonamine Chemical compound CC1(C)CC(=O)CC(C)(C)N1 JWUXJYZVKZKLTJ-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- 208000026481 Werdnig-Hoffmann disease Diseases 0.000 description 2
- 208000019291 X-linked disease Diseases 0.000 description 2
- UKSOJJRLFYFAKB-UHFFFAOYSA-N [1-(4-bromo-3-methoxyphenyl)pyrazol-4-yl]methanol Chemical compound C1=C(Br)C(OC)=CC(N2N=CC(CO)=C2)=C1 UKSOJJRLFYFAKB-UHFFFAOYSA-N 0.000 description 2
- NAZMTZCLBZQJNF-UHFFFAOYSA-N [2-fluoro-4-methoxy-5-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl]boronic acid Chemical compound COC1=CC(F)=C(B(O)O)C=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 NAZMTZCLBZQJNF-UHFFFAOYSA-N 0.000 description 2
- OHBYJTINNLFQIJ-UHFFFAOYSA-N [2-hydroxy-4-(5-methyl-1,3-oxazol-2-yl)-6-phenylmethoxyphenyl]boronic acid Chemical compound O1C(C)=CN=C1C1=CC(O)=C(B(O)O)C(OCC=2C=CC=CC=2)=C1 OHBYJTINNLFQIJ-UHFFFAOYSA-N 0.000 description 2
- TWHOHEIHZFMWLD-UHFFFAOYSA-N [2-methoxy-5-[(1-methylpyrazol-4-yl)methylcarbamoyl]phenyl]boronic acid Chemical compound C1=C(B(O)O)C(OC)=CC=C1C(=O)NCC1=CN(C)N=C1 TWHOHEIHZFMWLD-UHFFFAOYSA-N 0.000 description 2
- FGQYDZBIYZGFHE-UHFFFAOYSA-N [3-fluoro-4-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2)F)N=N1 FGQYDZBIYZGFHE-UHFFFAOYSA-N 0.000 description 2
- SKUKKAJYCUEXPH-UHFFFAOYSA-N [3-fluoro-5-hydroxy-4-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2O)F)N=N1 SKUKKAJYCUEXPH-UHFFFAOYSA-N 0.000 description 2
- SMTXSWTYHXXLPO-UHFFFAOYSA-N [3-hydroxy-4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(trifluoromethoxy)phenyl] trifluoromethanesulfonate Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2O)OC(F)(F)F)N=N1 SMTXSWTYHXXLPO-UHFFFAOYSA-N 0.000 description 2
- WLPSAHHHSZEEGB-UHFFFAOYSA-N [3-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(trifluoromethoxy)phenyl] trifluoromethanesulfonate Chemical compound C=1C=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2O)OC(F)(F)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 WLPSAHHHSZEEGB-UHFFFAOYSA-N 0.000 description 2
- HEXSOODLNUMCEE-UHFFFAOYSA-N [3-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl] trifluoromethanesulfonate Chemical compound C=1C=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 HEXSOODLNUMCEE-UHFFFAOYSA-N 0.000 description 2
- DASFMNLHSCXIBW-UHFFFAOYSA-N [4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-(trifluoromethoxy)phenyl] trifluoromethanesulfonate Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2)OC(F)(F)F)N=N1 DASFMNLHSCXIBW-UHFFFAOYSA-N 0.000 description 2
- ACAGFLBCLYSTJA-UHFFFAOYSA-N [4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-(trifluoromethoxy)phenyl] trifluoromethanesulfonate Chemical compound C=1C=C(C=2C(=CC(OS(=O)(=O)C(F)(F)F)=CC=2)OC(F)(F)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ACAGFLBCLYSTJA-UHFFFAOYSA-N 0.000 description 2
- WNXHEQQIBDAAAH-UHFFFAOYSA-N [7-hydroxy-6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-yl] trifluoromethanesulfonate Chemical compound C=1C=C(C=2C(=CC3=CC(OS(=O)(=O)C(F)(F)F)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 WNXHEQQIBDAAAH-UHFFFAOYSA-N 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000005605 benzo group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(I) oxide Inorganic materials [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 230000000875 corresponding effect Effects 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- 229940112669 cuprous oxide Drugs 0.000 description 2
- KRFJLUBVMFXRPN-UHFFFAOYSA-N cuprous oxide Chemical compound [O-2].[Cu+].[Cu+] KRFJLUBVMFXRPN-UHFFFAOYSA-N 0.000 description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 150000001975 deuterium Chemical group 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- YMWUJEATGCHHMB-DICFDUPASA-N dichloromethane-d2 Chemical compound [2H]C([2H])(Cl)Cl YMWUJEATGCHHMB-DICFDUPASA-N 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- 125000004982 dihaloalkyl group Chemical group 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 208000014720 distal hereditary motor neuropathy Diseases 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- LSEFCHWGJNHZNT-UHFFFAOYSA-M methyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(C)C1=CC=CC=C1 LSEFCHWGJNHZNT-UHFFFAOYSA-M 0.000 description 2
- 238000004452 microanalysis Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 125000006682 monohaloalkyl group Chemical group 0.000 description 2
- IYAIZRCBPGSZEB-UHFFFAOYSA-N n-methyl-6-[4-phenylmethoxy-2-(trifluoromethoxy)phenyl]-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC(OCC=3C=CC=CC=3)=CC=2)OC(F)(F)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 IYAIZRCBPGSZEB-UHFFFAOYSA-N 0.000 description 2
- YQILFOXWRJGMJD-UHFFFAOYSA-N naphthalene-2,7-diol Chemical compound C1=CC(O)=CC2=CC(O)=CC=C21.C1=CC(O)=CC2=CC(O)=CC=C21 YQILFOXWRJGMJD-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 238000004806 packaging method and process Methods 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229960005141 piperazine Drugs 0.000 description 2
- 229960005235 piperonyl butoxide Drugs 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 239000012041 precatalyst Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 208000022074 proximal spinal muscular atrophy Diseases 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 206010039722 scoliosis Diseases 0.000 description 2
- 238000002603 single-photon emission computed tomography Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical class [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 210000000278 spinal cord Anatomy 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000004808 supercritical fluid chromatography Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- YGOHRGAEXHUGRT-UHFFFAOYSA-N tert-butyl n-[3-(6-bromo-7-phenylmethoxynaphthalen-2-yl)oxypropyl]carbamate Chemical compound C=1C2=CC(OCCCNC(=O)OC(C)(C)C)=CC=C2C=C(Br)C=1OCC1=CC=CC=C1 YGOHRGAEXHUGRT-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- QIFVXSLMFCRLCB-UHFFFAOYSA-N (1-methyl-2-oxopyridin-4-yl)boronic acid Chemical compound CN1C=CC(B(O)O)=CC1=O QIFVXSLMFCRLCB-UHFFFAOYSA-N 0.000 description 1
- KFTRXTSNTQSGNE-UHFFFAOYSA-N (1-methylpyrazol-4-yl)methanamine Chemical compound CN1C=C(CN)C=N1 KFTRXTSNTQSGNE-UHFFFAOYSA-N 0.000 description 1
- SIFCHNIAAPMMKG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) acetate Chemical compound CC(=O)ON1C(=O)CCC1=O SIFCHNIAAPMMKG-UHFFFAOYSA-N 0.000 description 1
- BKWVXPCYDRURMK-UHFFFAOYSA-N (2,6-dimethoxyphenyl)boronic acid Chemical compound COC1=CC=CC(OC)=C1B(O)O BKWVXPCYDRURMK-UHFFFAOYSA-N 0.000 description 1
- XOVMDVZAWWQSDC-UHFFFAOYSA-N (2-fluoro-6-methoxyphenyl)boronic acid Chemical compound COC1=CC=CC(F)=C1B(O)O XOVMDVZAWWQSDC-UHFFFAOYSA-N 0.000 description 1
- DHQMUJSACXTPEA-UHFFFAOYSA-N (2-methoxypyridin-4-yl)boronic acid Chemical compound COC1=CC(B(O)O)=CC=N1 DHQMUJSACXTPEA-UHFFFAOYSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- RHNSMLZROCLQBJ-UHFFFAOYSA-N (3-methoxynaphthalen-2-yl)boronic acid Chemical compound C1=CC=C2C=C(B(O)O)C(OC)=CC2=C1 RHNSMLZROCLQBJ-UHFFFAOYSA-N 0.000 description 1
- YBNDRTRLXPEWKQ-UHFFFAOYSA-N (4-chloro-2-fluorophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Cl)C=C1F YBNDRTRLXPEWKQ-UHFFFAOYSA-N 0.000 description 1
- IFPQNCKVOQKEIU-UHFFFAOYSA-N (4-fluoro-2-phenylmethoxyphenyl)boronic acid Chemical compound OB(O)C1=CC=C(F)C=C1OCC1=CC=CC=C1 IFPQNCKVOQKEIU-UHFFFAOYSA-N 0.000 description 1
- GFTRRFZEDDIPRT-RXTYADHFSA-N (6s)-6-[(1s)-1-[tert-butyl(dimethyl)silyl]oxyethyl]-2,2-dimethylpiperidin-4-ol Chemical compound CC(C)(C)[Si](C)(C)O[C@@H](C)[C@@H]1CC(O)CC(C)(C)N1 GFTRRFZEDDIPRT-RXTYADHFSA-N 0.000 description 1
- STINCPOTZGHIKN-AAEUAGOBSA-N (6s)-6-[(1s)-1-[tert-butyl(dimethyl)silyl]oxyethyl]-2,2-dimethylpiperidin-4-one Chemical compound CC(C)(C)[Si](C)(C)O[C@@H](C)[C@@H]1CC(=O)CC(C)(C)N1 STINCPOTZGHIKN-AAEUAGOBSA-N 0.000 description 1
- LEUQTIHUFAPWJB-LRDDRELGSA-N (6s)-6-[(1s)-1-hydroxyethyl]-1-[(4-methoxyphenyl)methyl]-2,2-dimethylpiperidin-4-one Chemical compound C1=CC(OC)=CC=C1CN1C(C)(C)CC(=O)C[C@H]1[C@H](C)O LEUQTIHUFAPWJB-LRDDRELGSA-N 0.000 description 1
- WRZFLJFDZWZRMY-XPUUQOCRSA-N (6s)-6-[(1s)-1-hydroxyethyl]-2,2-dimethylpiperidin-4-one Chemical compound C[C@H](O)[C@@H]1CC(=O)CC(C)(C)N1 WRZFLJFDZWZRMY-XPUUQOCRSA-N 0.000 description 1
- IDFPQEHZYBXIFO-GFCCVEGCSA-N (R)-(4-fluoro-2-propylphenyl)-(1H-imidazol-2-yl)methanol Chemical compound CCCc1cc(F)ccc1[C@@H](O)c1ncc[nH]1 IDFPQEHZYBXIFO-GFCCVEGCSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- XHTYQFMRBQUCPX-UHFFFAOYSA-N 1,1,3,3-tetramethoxypropane Chemical compound COC(OC)CC(OC)OC XHTYQFMRBQUCPX-UHFFFAOYSA-N 0.000 description 1
- CGXOAAMIQPDTPE-UHFFFAOYSA-N 1,2,2,6,6-pentamethylpiperidin-4-amine Chemical compound CN1C(C)(C)CC(N)CC1(C)C CGXOAAMIQPDTPE-UHFFFAOYSA-N 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N 1,3,5-Me3C6H3 Natural products CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- IMLSAISZLJGWPP-UHFFFAOYSA-N 1,3-dithiolane Chemical compound C1CSCS1 IMLSAISZLJGWPP-UHFFFAOYSA-N 0.000 description 1
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 1
- UPSMYOUTOBWECX-UHFFFAOYSA-N 1-bromo-4-phenylmethoxy-2-(trifluoromethoxy)benzene Chemical compound C1=C(Br)C(OC(F)(F)F)=CC(OCC=2C=CC=CC=2)=C1 UPSMYOUTOBWECX-UHFFFAOYSA-N 0.000 description 1
- DPYMUHPZDKXNNI-UHFFFAOYSA-N 1-bromo-5-chloro-4-iodo-2-methoxybenzene Chemical compound COC1=CC(I)=C(Cl)C=C1Br DPYMUHPZDKXNNI-UHFFFAOYSA-N 0.000 description 1
- URLBZTBPPBPQEY-UHFFFAOYSA-N 1-bromo-6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=C(Br)C(O)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 URLBZTBPPBPQEY-UHFFFAOYSA-N 0.000 description 1
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 1
- NXBBLOPMUQCCKG-UHFFFAOYSA-N 1-chloro-6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=C(Cl)C(O)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 NXBBLOPMUQCCKG-UHFFFAOYSA-N 0.000 description 1
- AVJRGCKNVZTWQH-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-one Chemical compound O=C1N(C)C=CC(B2OC(C)(C)C(C)(C)O2)=C1 AVJRGCKNVZTWQH-UHFFFAOYSA-N 0.000 description 1
- HLNJFEXZDGURGZ-UHFFFAOYSA-M 1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1 HLNJFEXZDGURGZ-UHFFFAOYSA-M 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- CSHPTYYIGQDPOJ-UHFFFAOYSA-N 1h-pyrazol-4-amine;dihydrochloride Chemical compound Cl.Cl.NC=1C=NNC=1 CSHPTYYIGQDPOJ-UHFFFAOYSA-N 0.000 description 1
- UIEABCXJWANXFS-UHFFFAOYSA-N 1h-pyrazol-5-ylmethanol Chemical compound OCC=1C=CNN=1 UIEABCXJWANXFS-UHFFFAOYSA-N 0.000 description 1
- FTVFPPFZRRKJIH-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidin-4-amine Chemical compound CC1(C)CC(N)CC(C)(C)N1 FTVFPPFZRRKJIH-UHFFFAOYSA-N 0.000 description 1
- VDVUCLWJZJHFAV-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidin-4-ol Chemical compound CC1(C)CC(O)CC(C)(C)N1 VDVUCLWJZJHFAV-UHFFFAOYSA-N 0.000 description 1
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- UIBILPLZRULHTE-UHFFFAOYSA-N 2,2-dimethylpiperidin-4-ol Chemical compound CC1(C)CC(O)CCN1 UIBILPLZRULHTE-UHFFFAOYSA-N 0.000 description 1
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- IEMVEYRBXJCBBQ-UHFFFAOYSA-N 2,6-dimethylpiperidin-4-ol Chemical compound CC1CC(O)CC(C)N1 IEMVEYRBXJCBBQ-UHFFFAOYSA-N 0.000 description 1
- GDUVBKSVYWFWGV-UHFFFAOYSA-N 2-(1h-pyrazol-4-yl)phenol Chemical compound OC1=CC=CC=C1C1=CNN=C1 GDUVBKSVYWFWGV-UHFFFAOYSA-N 0.000 description 1
- HJGNAXXDNNYRNN-UHFFFAOYSA-N 2-(3-methoxy-4-propan-2-yloxyphenyl)ethanamine Chemical compound COC1=CC(CCN)=CC=C1OC(C)C HJGNAXXDNNYRNN-UHFFFAOYSA-N 0.000 description 1
- ULTRZADCHILMQC-UHFFFAOYSA-N 2-(4-bromo-3-ethoxy-5-phenylmethoxyphenyl)-5-methyl-1,3-oxazole Chemical compound BrC=1C(OCC)=CC(C=2OC(C)=CN=2)=CC=1OCC1=CC=CC=C1 ULTRZADCHILMQC-UHFFFAOYSA-N 0.000 description 1
- IYTUTWZXSBERDH-UHFFFAOYSA-N 2-(4-fluoro-2-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound COC1=CC(F)=CC=C1B1OC(C)(C)C(C)(C)O1 IYTUTWZXSBERDH-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- SXAMGRAIZSSWIH-UHFFFAOYSA-N 2-[3-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,2,4-oxadiazol-5-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NOC(=N1)CC(=O)N1CC2=C(CC1)NN=N2 SXAMGRAIZSSWIH-UHFFFAOYSA-N 0.000 description 1
- XXZCIYUJYUESMD-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-(morpholin-4-ylmethyl)pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)CN1CCOCC1 XXZCIYUJYUESMD-UHFFFAOYSA-N 0.000 description 1
- WWSJZGAPAVMETJ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-3-ethoxypyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C(=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2)OCC WWSJZGAPAVMETJ-UHFFFAOYSA-N 0.000 description 1
- FYELSNVLZVIGTI-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-5-ethylpyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1CC)CC(=O)N1CC2=C(CC1)NN=N2 FYELSNVLZVIGTI-UHFFFAOYSA-N 0.000 description 1
- WZFUQSJFWNHZHM-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)CC(=O)N1CC2=C(CC1)NN=N2 WZFUQSJFWNHZHM-UHFFFAOYSA-N 0.000 description 1
- ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 2-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]pyrazol-1-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C=1C=NN(C=1)CC(=O)N1CC2=C(CC1)NN=N2 ZRPAUEVGEGEPFQ-UHFFFAOYSA-N 0.000 description 1
- YJLUBHOZZTYQIP-UHFFFAOYSA-N 2-[5-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]-1,3,4-oxadiazol-2-yl]-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1=NN=C(O1)CC(=O)N1CC2=C(CC1)NN=N2 YJLUBHOZZTYQIP-UHFFFAOYSA-N 0.000 description 1
- SJWDXZKFWQQHMY-UHFFFAOYSA-N 2-[6-(1,2,2,6,6-pentamethylpiperidin-4-yl)oxypyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1C(C)(C)N(C)C(C)(C)CC1OC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 SJWDXZKFWQQHMY-UHFFFAOYSA-N 0.000 description 1
- FDDFUGAGCBKJRC-UHFFFAOYSA-N 2-[6-(1,7-diazaspiro[4.4]nonan-7-yl)pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1N(C1)CCC21CCCN2 FDDFUGAGCBKJRC-UHFFFAOYSA-N 0.000 description 1
- YYDKVSFEENAXCQ-UHFFFAOYSA-N 2-[6-(2,6-dimethylpiperidin-4-yl)oxypyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1C(C)NC(C)CC1OC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 YYDKVSFEENAXCQ-UHFFFAOYSA-N 0.000 description 1
- VFEQOBZBXSNQSK-UHFFFAOYSA-N 2-[6-(2,7-diazaspiro[3.5]nonan-7-yl)pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1N(CC1)CCC21CNC2 VFEQOBZBXSNQSK-UHFFFAOYSA-N 0.000 description 1
- RZKZALJPCPTXOQ-UHFFFAOYSA-N 2-[6-(2,9-diazaspiro[4.5]decan-2-yl)pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound OC1=CC(C2=CNN=C2)=CC=C1C(N=N1)=CC=C1N(C1)CCC21CCCNC2 RZKZALJPCPTXOQ-UHFFFAOYSA-N 0.000 description 1
- JPPCHSUPZIDLHA-UHFFFAOYSA-N 2-[6-(3-fluoropiperidin-4-yl)oxypyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1CCNCC1F JPPCHSUPZIDLHA-UHFFFAOYSA-N 0.000 description 1
- MHTZCIXQINDDRC-UHFFFAOYSA-N 2-[6-(7-methyl-2,7-diazaspiro[4.4]nonan-2-yl)pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1N(C)CCC11CN(C=2N=NC(=CC=2)C=2C(=CC(=CC=2)N2N=CC=C2)O)CC1 MHTZCIXQINDDRC-UHFFFAOYSA-N 0.000 description 1
- ZHCQCYLMVGYGFR-UHFFFAOYSA-N 2-[6-[(1,2,2,6,6-pentamethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1C(C)(C)N(C)C(C)(C)CC1NC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 ZHCQCYLMVGYGFR-UHFFFAOYSA-N 0.000 description 1
- ITTHQEAYIOMTMZ-ZFWWWQNUSA-N 2-[6-[(2S,4S)-2-methylpiperidin-4-yl]oxypyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1CN[C@@H](C)C[C@H]1OC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 ITTHQEAYIOMTMZ-ZFWWWQNUSA-N 0.000 description 1
- GNLRCCAIVNYVLW-CALCHBBNSA-N 2-[6-[(3aR,6aS)-2-(2-hydroxyethyl)-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrol-5-yl]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound C([C@@H]1CN(C[C@@H]1C1)CCO)N1C(N=N1)=CC=C1C(C(=C1)O)=CC=C1C=1C=NNC=1 GNLRCCAIVNYVLW-CALCHBBNSA-N 0.000 description 1
- IOOBNJURMOWBJW-OKILXGFUSA-N 2-[6-[(3aS,6aR)-2,3,3a,4,6,6a-hexahydro-1H-pyrrolo[3,4-c]pyrrol-5-yl]pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C=1C=C(C=2N=NC(=CC=2)N2C[C@H]3CNC[C@H]3C2)C(O)=CC=1N1C=CC=N1 IOOBNJURMOWBJW-OKILXGFUSA-N 0.000 description 1
- LKNUUOLYQLEBPT-IYBDPMFKSA-N 2-[6-[(3aS,6aR)-2-methyl-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrol-5-yl]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound C([C@@H]1CN(C[C@@H]1C1)C)N1C(N=N1)=CC=C1C(C(=C1)O)=CC=C1C=1C=NNC=1 LKNUUOLYQLEBPT-IYBDPMFKSA-N 0.000 description 1
- UWKSTVQYYZDJIB-UHFFFAOYSA-N 2-[6-[7-(2-hydroxyethyl)-2,7-diazaspiro[4.4]nonan-2-yl]pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C1N(CCO)CCC11CN(C=2N=NC(=CC=2)C=2C(=CC(=CC=2)N2N=CC=C2)O)CC1 UWKSTVQYYZDJIB-UHFFFAOYSA-N 0.000 description 1
- DYYYAFDPBKZELV-UHFFFAOYSA-N 2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-pyrazol-1-ylphenol Chemical compound C=1C=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 DYYYAFDPBKZELV-UHFFFAOYSA-N 0.000 description 1
- WXHLLJAMBQLULT-UHFFFAOYSA-N 2-[[6-[4-(2-hydroxyethyl)piperazin-1-yl]-2-methylpyrimidin-4-yl]amino]-n-(2-methyl-6-sulfanylphenyl)-1,3-thiazole-5-carboxamide;hydrate Chemical compound O.C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1S WXHLLJAMBQLULT-UHFFFAOYSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- BBVDXMFOLMWGNB-UHFFFAOYSA-N 2-bromo-1-iodo-3-methoxybenzene Chemical compound COC1=CC=CC(I)=C1Br BBVDXMFOLMWGNB-UHFFFAOYSA-N 0.000 description 1
- RCRYJYPWVPEYMN-UHFFFAOYSA-N 2-bromo-1-methoxy-3-phenylmethoxybenzene Chemical compound COC1=CC=CC(OCC=2C=CC=CC=2)=C1Br RCRYJYPWVPEYMN-UHFFFAOYSA-N 0.000 description 1
- KVKYADYEVUKNLZ-UHFFFAOYSA-N 2-bromo-3-phenylmethoxy-6-(3-phenylmethoxypropoxy)naphthalene Chemical compound C1=C2C=C(OCC=3C=CC=CC=3)C(Br)=CC2=CC=C1OCCCOCC1=CC=CC=C1 KVKYADYEVUKNLZ-UHFFFAOYSA-N 0.000 description 1
- FLKJSCUHRXTRPY-UHFFFAOYSA-N 2-bromo-3-phenylmethoxyphenol Chemical compound OC1=CC=CC(OCC=2C=CC=CC=2)=C1Br FLKJSCUHRXTRPY-UHFFFAOYSA-N 0.000 description 1
- YIQMMXVWWSWNGR-UHFFFAOYSA-N 2-bromo-4-iodo-1-methoxybenzene Chemical compound COC1=CC=C(I)C=C1Br YIQMMXVWWSWNGR-UHFFFAOYSA-N 0.000 description 1
- ABFPKTQEQNICFT-UHFFFAOYSA-M 2-chloro-1-methylpyridin-1-ium;iodide Chemical compound [I-].C[N+]1=CC=CC=C1Cl ABFPKTQEQNICFT-UHFFFAOYSA-M 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- CRGODOCVKOELQF-UHFFFAOYSA-N 2-methyl-4-[6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalen-2-yl]oxybutan-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(OCCC(C)(C)O)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 CRGODOCVKOELQF-UHFFFAOYSA-N 0.000 description 1
- JUSKPRNFXVJMED-UHFFFAOYSA-N 2-methyl-6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-1H-benzimidazol-5-ol Chemical compound C=1C=C(C=2C(=CC=3NC(C)=NC=3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 JUSKPRNFXVJMED-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- JZSAUQMXKHBZEO-UHFFFAOYSA-N 3,5-dichloropyridazine Chemical compound ClC1=CN=NC(Cl)=C1 JZSAUQMXKHBZEO-UHFFFAOYSA-N 0.000 description 1
- YVWATSXVUOJOIG-UHFFFAOYSA-N 3,6-dihydro-2h-pyran-4-ylboronic acid Chemical compound OB(O)C1=CCOCC1 YVWATSXVUOJOIG-UHFFFAOYSA-N 0.000 description 1
- PQTDPJMQCVPJEG-UHFFFAOYSA-N 3-(4-chloro-2-methoxyphenyl)-6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazine Chemical compound COC1=CC(Cl)=CC=C1C(N=N1)=CC=C1OC1CC(C)(C)NC(C)(C)C1 PQTDPJMQCVPJEG-UHFFFAOYSA-N 0.000 description 1
- CCHAEANBQMBWJU-UHFFFAOYSA-N 3-(5-methyl-1,3-oxazol-2-yl)-5-phenylmethoxyphenol Chemical compound O1C(C)=CN=C1C1=CC(O)=CC(OCC=2C=CC=CC=2)=C1 CCHAEANBQMBWJU-UHFFFAOYSA-N 0.000 description 1
- KYTVGRLIUBBZKZ-UHFFFAOYSA-N 3-(6-bromo-7-phenylmethoxynaphthalen-2-yl)oxypropylcarbamic acid Chemical compound C=1C2=CC(OCCCNC(=O)O)=CC=C2C=C(Br)C=1OCC1=CC=CC=C1 KYTVGRLIUBBZKZ-UHFFFAOYSA-N 0.000 description 1
- DPWQNNYQFIUBSP-GJZGRUSLSA-N 3-[(2s,6s)-2,6-dimethylpiperidin-4-yl]oxy-6-(2-methoxy-4-pyrazol-1-ylphenyl)pyridazine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1C[C@H](C)N[C@@H](C)C1 DPWQNNYQFIUBSP-GJZGRUSLSA-N 0.000 description 1
- DMMVXUFPDXGYRE-UHFFFAOYSA-N 3-[2-methoxy-4-(1h-pyrazol-4-yl)phenyl]-6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazine Chemical compound COC1=CC(C2=CNN=C2)=CC=C1C(N=N1)=CC=C1OC1CC(C)(C)NC(C)(C)C1 DMMVXUFPDXGYRE-UHFFFAOYSA-N 0.000 description 1
- DDHJEYOUTCNRTA-UHFFFAOYSA-N 3-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalene-2,7-diol Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC3=CC(O)=CC=C3C=2)O)N=N1 DDHJEYOUTCNRTA-UHFFFAOYSA-N 0.000 description 1
- XFUCCDHUVHJSHG-UHFFFAOYSA-N 3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-(piperidin-1-ylmethyl)naphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(CN4CCCCC4)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 XFUCCDHUVHJSHG-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- BBPZABXVRBFWGD-UHFFFAOYSA-N 3-bromo-4-methoxybenzoic acid Chemical compound COC1=CC=C(C(O)=O)C=C1Br BBPZABXVRBFWGD-UHFFFAOYSA-N 0.000 description 1
- NOJOUQQJSGRBMN-UHFFFAOYSA-N 3-bromo-4-methoxyphenol Chemical compound COC1=CC=C(O)C=C1Br NOJOUQQJSGRBMN-UHFFFAOYSA-N 0.000 description 1
- PSUXTZLDBVEZTD-UHFFFAOYSA-N 3-bromopropoxymethylbenzene Chemical compound BrCCCOCC1=CC=CC=C1 PSUXTZLDBVEZTD-UHFFFAOYSA-N 0.000 description 1
- QNZZWJGBBWNRHW-UHFFFAOYSA-N 3-chloro-6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazine Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(Cl)N=N1 QNZZWJGBBWNRHW-UHFFFAOYSA-N 0.000 description 1
- IBWYHNOFSKJKKY-UHFFFAOYSA-N 3-chloropyridazine Chemical compound ClC1=CC=CN=N1 IBWYHNOFSKJKKY-UHFFFAOYSA-N 0.000 description 1
- ZXEHOMBJSLISHU-UHFFFAOYSA-N 3-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound C=1C=C(C=2C(=CC(=CC=2O)C2=CNN=C2)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ZXEHOMBJSLISHU-UHFFFAOYSA-N 0.000 description 1
- MMOMXFQLECVNNA-UHFFFAOYSA-N 3-fluoro-5-(1H-pyrazol-4-yl)-2-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenol Chemical compound CC1(C)CC(CC(C)(C)N1)OC1=NN=C(C=C1)C1=C(F)C=C(C=C1O)C1=CNN=C1 MMOMXFQLECVNNA-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- WQJODUIHZFMFFC-UHFFFAOYSA-N 3-hydroxy-4-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzonitrile Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C(=CC(=CC=2)C#N)O)N=N1 WQJODUIHZFMFFC-UHFFFAOYSA-N 0.000 description 1
- QTUJFHJUHDTZFB-UHFFFAOYSA-N 3-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzonitrile Chemical compound C=1C=C(C=2C(=CC(=CC=2)C#N)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 QTUJFHJUHDTZFB-UHFFFAOYSA-N 0.000 description 1
- SYEBHTTYDRGCRC-UHFFFAOYSA-N 3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-n-prop-2-ynylbenzamide Chemical compound COC1=CC(C(=O)NCC#C)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 SYEBHTTYDRGCRC-UHFFFAOYSA-N 0.000 description 1
- UCRLUFAENGRCCZ-UHFFFAOYSA-N 3-methoxy-5-(5-methyl-1,3-oxazol-2-yl)-2-[6-[methyl-(2,2,6-trimethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound COC1=CC(C=2OC(C)=CN=2)=CC(O)=C1C(N=N1)=CC=C1N(C)C1CC(C)NC(C)(C)C1 UCRLUFAENGRCCZ-UHFFFAOYSA-N 0.000 description 1
- IDZWCHOIVBCPAS-UHFFFAOYSA-N 3-methyl-3-[6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-7-phenylmethoxynaphthalen-2-yl]oxybutan-1-ol Chemical compound C=1C=C(C=2C(=CC3=CC(OC(C)(C)CCO)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 IDZWCHOIVBCPAS-UHFFFAOYSA-N 0.000 description 1
- XPFCZYUVICHKDS-UHFFFAOYSA-N 3-methylbutane-1,3-diol Chemical compound CC(C)(O)CCO XPFCZYUVICHKDS-UHFFFAOYSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AWRIOTVUTPLWLF-UHFFFAOYSA-N 4-(2-aminoethyl)-2-methoxyphenol;hydron;chloride Chemical compound Cl.COC1=CC(CCN)=CC=C1O AWRIOTVUTPLWLF-UHFFFAOYSA-N 0.000 description 1
- FCXAYEPBCJFGPO-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole-1-carboxylic acid Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(C(O)=O)N=C1 FCXAYEPBCJFGPO-UHFFFAOYSA-N 0.000 description 1
- JRMKJOOJKCAEJK-UHFFFAOYSA-N 4-Hydroxymethylpyrazole Chemical compound OCC=1C=NNC=1 JRMKJOOJKCAEJK-UHFFFAOYSA-N 0.000 description 1
- NXOLRRXZUVZVHE-UHFFFAOYSA-N 4-[3-fluoro-5-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl]-1-methylpyridin-2-one Chemical compound CN(C1CC(C)(C)NC(C)(C)C1)c1ccc(nn1)-c1c(O)cc(cc1F)-c1ccn(C)c(=O)c1 NXOLRRXZUVZVHE-UHFFFAOYSA-N 0.000 description 1
- NAIXKVMLMVRZGO-UHFFFAOYSA-N 4-[3-fluoro-5-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl]-1H-pyridin-2-one Chemical compound CN(C1CC(C)(C)NC(C)(C)C1)C1=CC=C(N=N1)C1=C(F)C=C(C=C1O)C1=CC(=O)NC=C1 NAIXKVMLMVRZGO-UHFFFAOYSA-N 0.000 description 1
- UGIRVEPLXZTENQ-UHFFFAOYSA-N 4-[3-hydroxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(trifluoromethoxy)phenyl]-1-methylpyridin-2-one Chemical compound C=1C=C(C=2C(=CC(=CC=2O)C2=CC(=O)N(C)C=C2)OC(F)(F)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 UGIRVEPLXZTENQ-UHFFFAOYSA-N 0.000 description 1
- PMAXNUGIQISXHY-UHFFFAOYSA-N 4-[4-[6-(2,9-diazaspiro[4.5]decan-2-yl)pyridazin-3-yl]-3-hydroxyphenyl]-1-methylpyridin-2-one Chemical compound O=C1N(C)C=CC(C=2C=C(O)C(=CC=2)C=2N=NC(=CC=2)N2CC3(CC2)CNCCC3)=C1 PMAXNUGIQISXHY-UHFFFAOYSA-N 0.000 description 1
- ITWXOEQLYFYTJX-UHFFFAOYSA-N 4-borono-3-methoxybenzoic acid Chemical compound COC1=CC(C(O)=O)=CC=C1B(O)O ITWXOEQLYFYTJX-UHFFFAOYSA-N 0.000 description 1
- NUTRHYYFCDEALP-UHFFFAOYSA-N 4-bromo-3,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=C(Br)C(O)=C1 NUTRHYYFCDEALP-UHFFFAOYSA-N 0.000 description 1
- PGJZBAVEKVWHIO-UHFFFAOYSA-N 4-bromo-3-(trifluoromethoxy)phenol Chemical compound OC1=CC=C(Br)C(OC(F)(F)F)=C1 PGJZBAVEKVWHIO-UHFFFAOYSA-N 0.000 description 1
- RUTNWXBHRAIQSP-UHFFFAOYSA-N 4-bromo-3-methoxyaniline Chemical compound COC1=CC(N)=CC=C1Br RUTNWXBHRAIQSP-UHFFFAOYSA-N 0.000 description 1
- BJNIUDHOXUKBCY-UHFFFAOYSA-N 4-bromo-5-fluoro-2-methoxyphenol Chemical compound COC1=CC(Br)=C(F)C=C1O BJNIUDHOXUKBCY-UHFFFAOYSA-N 0.000 description 1
- SHYMDIOVKHXAQT-UHFFFAOYSA-N 4-chloro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound C=1C=C(C=2C(=CC(=C(Cl)C=2)C2=CNN=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 SHYMDIOVKHXAQT-UHFFFAOYSA-N 0.000 description 1
- MPZJVUYQTMFGPV-UHFFFAOYSA-N 4-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-5-(1H-pyrazol-4-yl)phenol Chemical compound C=1C=C(C=2C(=CC(=C(F)C=2)C2=CNN=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 MPZJVUYQTMFGPV-UHFFFAOYSA-N 0.000 description 1
- LVVXOUKKYWWJPD-UHFFFAOYSA-N 4-hydroxy-N-[(1-methylpyrazol-4-yl)methyl]-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]benzamide Chemical compound C=1C=C(C=2C(=CC=C(C=2)C(=O)NCC2=CN(C)N=C2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 LVVXOUKKYWWJPD-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical compound CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- STWTUEAWRAIWJG-UHFFFAOYSA-N 5-(1H-pyrazol-4-yl)-2-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenol Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC(=CC=2)C2=CNN=C2)O)N=N1 STWTUEAWRAIWJG-UHFFFAOYSA-N 0.000 description 1
- XJIMIVJABPKGIY-UHFFFAOYSA-N 5-(1H-pyrazol-4-yl)-2-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]phenol hydrochloride Chemical compound Cl.C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC(=CC=2)C2=CNN=C2)O)N=N1 XJIMIVJABPKGIY-UHFFFAOYSA-N 0.000 description 1
- SJIRMWMHWILMPD-UHFFFAOYSA-N 5-(1H-pyrazol-4-yl)-2-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazin-3-yl]phenol Chemical compound C1C(C)(C)NC(C)(C)CC1CC1=CC=C(C=2C(=CC(=CC=2)C2=CNN=C2)O)N=N1 SJIRMWMHWILMPD-UHFFFAOYSA-N 0.000 description 1
- NUYRCRJTWOXXJZ-UHFFFAOYSA-N 5-(5-methyl-1,3-oxazol-2-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-3-phenylmethoxyphenol Chemical compound C=1C=C(C=2C(=CC(=CC=2O)C=2OC(C)=CN=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 NUYRCRJTWOXXJZ-UHFFFAOYSA-N 0.000 description 1
- LWRBTOHOMHYHQA-UHFFFAOYSA-N 5-(5-methyl-1,3-oxazol-2-yl)-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(=CC=2)C=2OC(C)=CN=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 LWRBTOHOMHYHQA-UHFFFAOYSA-N 0.000 description 1
- FHZALEJIENDROK-UHFFFAOYSA-N 5-bromo-1h-imidazole Chemical compound BrC1=CN=CN1 FHZALEJIENDROK-UHFFFAOYSA-N 0.000 description 1
- LEVZYEAJEMIAEH-UHFFFAOYSA-N 5-chloro-2-[6-[(1,2,2,6,6-pentamethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C1C(C)(C)N(C)C(C)(C)CC1NC1=CC=C(C=2C(=CC(Cl)=CC=2)O)N=N1 LEVZYEAJEMIAEH-UHFFFAOYSA-N 0.000 description 1
- VGVJLODSYXCLIQ-UHFFFAOYSA-N 5-chloro-2-[6-[methyl-(1,2,2,6,6-pentamethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(Cl)=CC=2)O)N=NC=1N(C)C1CC(C)(C)N(C)C(C)(C)C1 VGVJLODSYXCLIQ-UHFFFAOYSA-N 0.000 description 1
- MAWZHKOLEMMOGH-UHFFFAOYSA-N 5-chloro-3-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(Cl)=CC=2O)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 MAWZHKOLEMMOGH-UHFFFAOYSA-N 0.000 description 1
- ALDHPMOLFIGMOK-UHFFFAOYSA-N 5-chloro-3-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol hydrochloride Chemical compound Cl.C=1C=C(C=2C(=CC(Cl)=CC=2O)F)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 ALDHPMOLFIGMOK-UHFFFAOYSA-N 0.000 description 1
- TVOYGKIUEVWKBY-UHFFFAOYSA-N 5-fluoro-2-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenol Chemical compound C=1C=C(C=2C(=CC(F)=CC=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 TVOYGKIUEVWKBY-UHFFFAOYSA-N 0.000 description 1
- MZRUFMBFIKGOAL-UHFFFAOYSA-N 5-nitro-1h-pyrazole Chemical compound [O-][N+](=O)C1=CC=NN1 MZRUFMBFIKGOAL-UHFFFAOYSA-N 0.000 description 1
- AXDNGXIRRGWVTJ-UHFFFAOYSA-N 5-pyrazol-1-yl-2-[6-(1,2,3,6-tetrahydropyridin-4-yl)pyridazin-3-yl]phenol Chemical compound OC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1C1=CCNCC1 AXDNGXIRRGWVTJ-UHFFFAOYSA-N 0.000 description 1
- JCXLDEBPXAHIOD-UHFFFAOYSA-N 5-pyrazol-1-yl-2-[6-[(2,2,6,6-tetramethylpiperidin-4-yl)methyl]pyridazin-3-yl]phenol Chemical compound C1C(C)(C)NC(C)(C)CC1CC1=CC=C(C=2C(=CC(=CC=2)N2N=CC=C2)O)N=N1 JCXLDEBPXAHIOD-UHFFFAOYSA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- HXFLZWAZSSPLCO-UHFFFAOYSA-N 6,6-dimethylbicyclo[3.1.1]heptyl Chemical group C1[C-]2C([CH2+])([CH2-])[C+]1CCC2 HXFLZWAZSSPLCO-UHFFFAOYSA-N 0.000 description 1
- MJELNGWHFBGDLA-UHFFFAOYSA-N 6-(2-methoxy-4-pyrazol-1-ylphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 MJELNGWHFBGDLA-UHFFFAOYSA-N 0.000 description 1
- YRGHMSWFPRXDJR-UHFFFAOYSA-N 6-(4-chloro-2-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(Cl)=CC=C1C1=CC=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=N1 YRGHMSWFPRXDJR-UHFFFAOYSA-N 0.000 description 1
- WSCQPDATNLSJBN-UHFFFAOYSA-N 6-(4-fluoro-2-phenylmethoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC(F)=CC=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 WSCQPDATNLSJBN-UHFFFAOYSA-N 0.000 description 1
- DIVZYKXFTVTRFD-UHFFFAOYSA-N 6-(4-imidazol-1-yl-2-methoxyphenyl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(N2C=NC=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 DIVZYKXFTVTRFD-UHFFFAOYSA-N 0.000 description 1
- QLKXBIVBZSPJQU-UHFFFAOYSA-N 6-(6-methoxy-3-phenylmethoxynaphthalen-2-yl)-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C2=CC(OC)=CC=C2C=C(C=2N=NC(=CC=2)N(C)C2CC(C)(C)NC(C)(C)C2)C=1OCC1=CC=CC=C1 QLKXBIVBZSPJQU-UHFFFAOYSA-N 0.000 description 1
- BXGCKOBKNKIBDC-UHFFFAOYSA-N 6-[2-fluoro-6-methoxy-4-(1-methylpyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(C2=CN(C)N=C2)=CC(F)=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 BXGCKOBKNKIBDC-UHFFFAOYSA-N 0.000 description 1
- ANKZJAFLHMSPBE-UHFFFAOYSA-N 6-[2-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(B2OC(C)(C)C(C)(C)O2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 ANKZJAFLHMSPBE-UHFFFAOYSA-N 0.000 description 1
- TUPPREIHMWBYND-UHFFFAOYSA-N 6-[2-methoxy-4-(5-methyl-1,3-oxazol-2-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC(C=2OC(C)=CN=2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 TUPPREIHMWBYND-UHFFFAOYSA-N 0.000 description 1
- WXDKMTVKFBVBCJ-UHFFFAOYSA-N 6-[2-methoxy-5-(1h-pyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=C(C2=CNN=C2)C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 WXDKMTVKFBVBCJ-UHFFFAOYSA-N 0.000 description 1
- RFAOZRYGIIUAPR-UHFFFAOYSA-N 6-[2-methoxy-6-phenylmethoxy-4-(1h-pyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(N(C)C2CC(C)(C)NC(C)(C)C2)N=NC=1C=1C(OC)=CC(C2=CNN=C2)=CC=1OCC1=CC=CC=C1 RFAOZRYGIIUAPR-UHFFFAOYSA-N 0.000 description 1
- PMCTWTPMZIAWNS-UHFFFAOYSA-N 6-[5-chloro-2-methoxy-4-(1h-pyrazol-4-yl)phenyl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound ClC=1C=C(C=2N=NC(=CC=2)N(C)C2CC(C)(C)NC(C)(C)C2)C(OC)=CC=1C=1C=NNC=1 PMCTWTPMZIAWNS-UHFFFAOYSA-N 0.000 description 1
- SIKBFGBUXXMZKR-UHFFFAOYSA-N 6-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C=1C=C(C=2C=C3C=CC(O)=CC3=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 SIKBFGBUXXMZKR-UHFFFAOYSA-N 0.000 description 1
- WLWAJESZYMOKQG-UHFFFAOYSA-N 6-[6-methoxy-2-methyl-1-(2-trimethylsilylethoxymethyl)benzimidazol-5-yl]-n-methyl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound COC1=CC=2N(COCC[Si](C)(C)C)C(C)=NC=2C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 WLWAJESZYMOKQG-UHFFFAOYSA-N 0.000 description 1
- MZXOHGOCNRWAPY-UHFFFAOYSA-N 6-bromo-2h-isoquinolin-3-one Chemical compound BrC1=CC=C2C=NC(O)=CC2=C1 MZXOHGOCNRWAPY-UHFFFAOYSA-N 0.000 description 1
- PTQMHGBFTYYQDX-UHFFFAOYSA-N 6-bromo-5-methoxy-2-methyl-1h-benzimidazole Chemical compound C1=C(Br)C(OC)=CC2=C1N=C(C)N2 PTQMHGBFTYYQDX-UHFFFAOYSA-N 0.000 description 1
- PEXFBIQTIZNHNP-UHFFFAOYSA-N 6-bromophthalazine Chemical compound C1=NN=CC2=CC(Br)=CC=C21 PEXFBIQTIZNHNP-UHFFFAOYSA-N 0.000 description 1
- KHRDKCVDWMPGNR-UHFFFAOYSA-N 6-hydroxy-5-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]-2,3-dihydroinden-1-one Chemical compound C=1C=C(C=2C(=CC=3C(=O)CCC=3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 KHRDKCVDWMPGNR-UHFFFAOYSA-N 0.000 description 1
- QYOLLMGXTMKUBP-UHFFFAOYSA-N 6-naphthalen-2-yl-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C1C(C)(C)NC(C)(C)CC1NC1=CC=C(C=2C=C3C=CC=CC3=CC=2)N=N1 QYOLLMGXTMKUBP-UHFFFAOYSA-N 0.000 description 1
- RPILYYNLROLJKA-UHFFFAOYSA-N 7-(2-morpholin-4-ylethoxy)-3-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]naphthalen-2-ol Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC3=CC(OCCN4CCOCC4)=CC=C3C=2)O)N=N1 RPILYYNLROLJKA-UHFFFAOYSA-N 0.000 description 1
- OGADZPJVAAZJTB-UHFFFAOYSA-N 7-(4-hydroxy-2-methylbutan-2-yl)oxy-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]naphthalen-2-ol Chemical compound C=1C=C(C=2C(=CC3=CC(OC(C)(C)CCO)=CC=C3C=2)O)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 OGADZPJVAAZJTB-UHFFFAOYSA-N 0.000 description 1
- WZVUPOQMVAEKQF-UHFFFAOYSA-N 7-bromo-1-phenylmethoxyisoquinoline Chemical compound C12=CC(Br)=CC=C2C=CN=C1OCC1=CC=CC=C1 WZVUPOQMVAEKQF-UHFFFAOYSA-N 0.000 description 1
- DSOKREQUHLPVFR-UHFFFAOYSA-N 7-bromo-2h-isoquinolin-1-one Chemical compound C1=CNC(=O)C2=CC(Br)=CC=C21 DSOKREQUHLPVFR-UHFFFAOYSA-N 0.000 description 1
- CYIGDJDFAMXXCZ-UHFFFAOYSA-N 7-phenylmethoxy-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)naphthalen-2-ol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC2=CC=C(O)C=C2C=C1OCC1=CC=CC=C1 CYIGDJDFAMXXCZ-UHFFFAOYSA-N 0.000 description 1
- FJLWZASCMYONKS-UHFFFAOYSA-N 7-phenylmethoxy-6-[6-(2,2,6,6-tetramethylpiperidin-4-yl)oxypyridazin-3-yl]naphthalen-2-ol Chemical compound C1C(C)(C)NC(C)(C)CC1OC1=CC=C(C=2C(=CC3=CC(O)=CC=C3C=2)OCC=2C=CC=CC=2)N=N1 FJLWZASCMYONKS-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- AMKGKYQBASDDJB-UHFFFAOYSA-N 9$l^{2}-borabicyclo[3.3.1]nonane Chemical compound C1CCC2CCCC1[B]2 AMKGKYQBASDDJB-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- VHIBOFWCGOAFJE-UHFFFAOYSA-N C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.Cl.[Fe+2] Chemical compound C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.C1=CC=C[C-]1P(C1=CC=CC=C1)C1=CC=CC=C1.Cl.Cl.[Fe+2] VHIBOFWCGOAFJE-UHFFFAOYSA-N 0.000 description 1
- UNBDBUKMDNRAQH-QCUKBLKESA-N COC1=CC=C(C\N=C\[C@H](C)O[Si](C)(C)C(C)(C)C)C=C1 Chemical compound COC1=CC=C(C\N=C\[C@H](C)O[Si](C)(C)C(C)(C)C)C=C1 UNBDBUKMDNRAQH-QCUKBLKESA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010011224 Cough Diseases 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010021118 Hypotonia Diseases 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 1
- 208000007379 Muscle Hypotonia Diseases 0.000 description 1
- 206010028289 Muscle atrophy Diseases 0.000 description 1
- 206010028293 Muscle contractions involuntary Diseases 0.000 description 1
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 1
- UGJBHEZMOKVTIM-UHFFFAOYSA-N N-formylglycine Chemical compound OC(=O)CNC=O UGJBHEZMOKVTIM-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000033952 Paralysis flaccid Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 208000033522 Proximal spinal muscular atrophy type 2 Diseases 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- 208000026214 Skeletal muscle atrophy Diseases 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920000147 Styrene maleic anhydride Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- AVRWEULSKHQETA-UHFFFAOYSA-N Thiophene-2 Chemical compound S1C=2CCCCCC=2C(C(=O)OC)=C1NC(=O)C1=C(F)C(F)=C(F)C(F)=C1F AVRWEULSKHQETA-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 239000003875 Wang resin Substances 0.000 description 1
- DATFJHHQYVQKJD-UHFFFAOYSA-N [1-[3-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazol-4-yl]methanol Chemical compound COC1=CC(N2N=CC(CO)=C2)=CC=C1B1OC(C)(C)C(C)(C)O1 DATFJHHQYVQKJD-UHFFFAOYSA-N 0.000 description 1
- HGVDVODSCOJZQN-UHFFFAOYSA-N [1-[3-methoxy-4-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl]pyrazol-4-yl]methanol Chemical compound COC1=CC(N2N=CC(CO)=C2)=CC=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 HGVDVODSCOJZQN-UHFFFAOYSA-N 0.000 description 1
- CSXUDLOCFCQFSV-UHFFFAOYSA-N [1-[4-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]pyrazol-3-yl]methanol Chemical compound COC1=CC=C(N2N=C(CO)C=C2)C=C1B1OC(C)(C)C(C)(C)O1 CSXUDLOCFCQFSV-UHFFFAOYSA-N 0.000 description 1
- ZTZYDMJFMFDYHU-UHFFFAOYSA-N [1-[4-methoxy-3-[6-[methyl-(2,2,6,6-tetramethylpiperidin-4-yl)amino]pyridazin-3-yl]phenyl]pyrazol-4-yl]methanol Chemical compound COC1=CC=C(N2N=CC(CO)=C2)C=C1C(N=N1)=CC=C1N(C)C1CC(C)(C)NC(C)(C)C1 ZTZYDMJFMFDYHU-UHFFFAOYSA-N 0.000 description 1
- JWOCIQZJFZPYNP-UHFFFAOYSA-N [2-methoxy-4-(5-methyl-1,3-oxazol-2-yl)-6-phenylmethoxyphenyl]boronic acid Chemical compound OB(O)C=1C(OC)=CC(C=2OC(C)=CN=2)=CC=1OCC1=CC=CC=C1 JWOCIQZJFZPYNP-UHFFFAOYSA-N 0.000 description 1
- NERFNHBZJXXFGY-UHFFFAOYSA-N [4-[(4-methylphenyl)methoxy]phenyl]methanol Chemical compound C1=CC(C)=CC=C1COC1=CC=C(CO)C=C1 NERFNHBZJXXFGY-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 239000000370 acceptor Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 125000005075 adamantyloxy group Chemical group C12(CC3CC(CC(C1)C3)C2)O* 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- HFEHLDPGIKPNKL-UHFFFAOYSA-N allyl iodide Chemical compound ICC=C HFEHLDPGIKPNKL-UHFFFAOYSA-N 0.000 description 1
- 125000005336 allyloxy group Chemical group 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 210000002226 anterior horn cell Anatomy 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 150000001499 aryl bromides Chemical class 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 230000035606 childbirth Effects 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- UBYDSYHGCPVBEQ-UHFFFAOYSA-N chloroform;propan-1-ol Chemical compound CCCO.ClC(Cl)Cl UBYDSYHGCPVBEQ-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- NMMPMZWIIQCZBA-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;2-phenylethanamine Chemical compound [Pd+]Cl.NCCC1=CC=CC=[C-]1.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 NMMPMZWIIQCZBA-UHFFFAOYSA-M 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 239000012045 crude solution Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000006612 decyloxy group Chemical group 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000036576 dermal application Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- 125000006003 dichloroethyl group Chemical group 0.000 description 1
- 125000004774 dichlorofluoromethyl group Chemical group FC(Cl)(Cl)* 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000006001 difluoroethyl group Chemical group 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 229940112141 dry powder inhaler Drugs 0.000 description 1
- 238000002567 electromyography Methods 0.000 description 1
- 230000003028 elevating effect Effects 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-L ethane-1,2-disulfonate Chemical compound [O-]S(=O)(=O)CCS([O-])(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-L 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- YFXCNIVBAVFOBX-UHFFFAOYSA-N ethenylboronic acid Chemical compound OB(O)C=C YFXCNIVBAVFOBX-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- LZCLXQDLBQLTDK-BYPYZUCNSA-N ethyl (2S)-lactate Chemical compound CCOC(=O)[C@H](C)O LZCLXQDLBQLTDK-BYPYZUCNSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001815 facial effect Effects 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 208000028331 flaccid paralysis Diseases 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000004886 head movement Effects 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 150000004050 homopiperazines Chemical class 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000003601 intercostal effect Effects 0.000 description 1
- 210000000876 intercostal muscle Anatomy 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- 229910052740 iodine Chemical group 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 238000010983 kinetics study Methods 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000003760 magnetic stirring Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- RQLPCVCAFJWECV-UHFFFAOYSA-N methyl 4-bromo-3,5-bis(phenylmethoxy)benzoate Chemical compound BrC=1C(OCC=2C=CC=CC=2)=CC(C(=O)OC)=CC=1OCC1=CC=CC=C1 RQLPCVCAFJWECV-UHFFFAOYSA-N 0.000 description 1
- UXSHRKILYILVSD-UHFFFAOYSA-N methyl 4-bromo-3,5-dihydroxybenzoate Chemical compound COC(=O)C1=CC(O)=C(Br)C(O)=C1 UXSHRKILYILVSD-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000020763 muscle atrophy Effects 0.000 description 1
- 238000001964 muscle biopsy Methods 0.000 description 1
- 230000004220 muscle function Effects 0.000 description 1
- 230000003387 muscular Effects 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- GMIARZNQLSJFCP-UHFFFAOYSA-N n,2,2,6,6-pentamethylpiperidin-4-amine Chemical compound CNC1CC(C)(C)NC(C)(C)C1 GMIARZNQLSJFCP-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- UABJRJJDKPUALY-UHFFFAOYSA-N n-methyl-6-(1-phenylmethoxyisoquinolin-7-yl)-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C=C3C(OCC=4C=CC=CC=4)=NC=CC3=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 UABJRJJDKPUALY-UHFFFAOYSA-N 0.000 description 1
- CLFMOTAMLFDNBG-UHFFFAOYSA-N n-methyl-6-[3-phenylmethoxy-6-(3-phenylmethoxypropoxy)naphthalen-2-yl]-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC3=CC(OCCCOCC=4C=CC=CC=4)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 CLFMOTAMLFDNBG-UHFFFAOYSA-N 0.000 description 1
- FSBBNRHWHSNXJO-UHFFFAOYSA-N n-methyl-6-[3-phenylmethoxy-6-(piperidin-1-ylmethyl)naphthalen-2-yl]-n-(2,2,6,6-tetramethylpiperidin-4-yl)pyridazin-3-amine Chemical compound C=1C=C(C=2C(=CC3=CC(CN4CCCCC4)=CC=C3C=2)OCC=2C=CC=CC=2)N=NC=1N(C)C1CC(C)(C)NC(C)(C)C1 FSBBNRHWHSNXJO-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MDWRQYBWVTXIIJ-UHFFFAOYSA-N naphthalen-2-yl trifluoromethanesulfonate Chemical compound C1=CC=CC2=CC(OS(=O)(=O)C(F)(F)F)=CC=C21 MDWRQYBWVTXIIJ-UHFFFAOYSA-N 0.000 description 1
- KPTRDYONBVUWPD-UHFFFAOYSA-N naphthalen-2-ylboronic acid Chemical compound C1=CC=CC2=CC(B(O)O)=CC=C21 KPTRDYONBVUWPD-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000002232 neuromuscular Effects 0.000 description 1
- 208000018360 neuromuscular disease Diseases 0.000 description 1
- 210000000715 neuromuscular junction Anatomy 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- YCIMNLLNPGFGHC-UHFFFAOYSA-N o-dihydroxy-benzene Natural products OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-M octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC([O-])=O QIQXTHQIDYTFRH-UHFFFAOYSA-M 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 239000012285 osmium tetroxide Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- OOFGXDQWDNJDIS-UHFFFAOYSA-N oxathiolane Chemical compound C1COSC1 OOFGXDQWDNJDIS-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- KSSNXJHPEFVKHY-UHFFFAOYSA-N phenol;hydrate Chemical compound O.OC1=CC=CC=C1 KSSNXJHPEFVKHY-UHFFFAOYSA-N 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- GRJHONXDTNBDTC-UHFFFAOYSA-N phenyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=CC=CC=C1 GRJHONXDTNBDTC-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- LWMPFIOTEAXAGV-UHFFFAOYSA-N piperidin-1-amine Chemical class NN1CCCCC1 LWMPFIOTEAXAGV-UHFFFAOYSA-N 0.000 description 1
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical class OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000002952 polymeric resin Substances 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 1
- 150000004892 pyridazines Chemical class 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 1
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 1
- QOTUIIJRVXKSJU-UHFFFAOYSA-N pyrrolidin-3-ylmethanol Chemical compound OCC1CCNC1 QOTUIIJRVXKSJU-UHFFFAOYSA-N 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 230000002787 reinforcement Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 1
- 229960001755 resorcinol Drugs 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 201000004193 respiratory failure Diseases 0.000 description 1
- 230000029058 respiratory gaseous exchange Effects 0.000 description 1
- 210000003019 respiratory muscle Anatomy 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 230000025185 skeletal muscle atrophy Effects 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- IBBSOVKTSBQQJO-UHFFFAOYSA-N tert-butyl 4-[2-fluoro-5-methoxy-4-(trifluoromethylsulfonyloxy)phenyl]pyrazole-1-carboxylate Chemical compound C1=C(OS(=O)(=O)C(F)(F)F)C(OC)=CC(C2=CN(N=C2)C(=O)OC(C)(C)C)=C1F IBBSOVKTSBQQJO-UHFFFAOYSA-N 0.000 description 1
- PDTZMULNKGUIEJ-UHFFFAOYSA-N tert-butyl 4-methylidenepiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(=C)CC1 PDTZMULNKGUIEJ-UHFFFAOYSA-N 0.000 description 1
- IOKGWQZQCNXXLD-UHFFFAOYSA-N tert-butyl n-(3-bromopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCBr IOKGWQZQCNXXLD-UHFFFAOYSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- BSJFXAFLSWDUPK-UHFFFAOYSA-N tert-butyl pyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC=N1 BSJFXAFLSWDUPK-UHFFFAOYSA-N 0.000 description 1
- RHLUYTCAIJXIPK-LIVGCHIRSA-N tert-butyl-[(1s)-1-[(2s)-4-[6-(2-methoxy-4-pyrazol-1-ylphenyl)pyridazin-3-yl]oxy-6,6-dimethylpiperidin-2-yl]ethoxy]-dimethylsilane Chemical compound COC1=CC(N2N=CC=C2)=CC=C1C(N=N1)=CC=C1OC1C[C@@H]([C@H](C)O[Si](C)(C)C(C)(C)C)NC(C)(C)C1 RHLUYTCAIJXIPK-LIVGCHIRSA-N 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- GZNAASVAJNXPPW-UHFFFAOYSA-M tin(4+) chloride dihydrate Chemical compound O.O.[Cl-].[Sn+4] GZNAASVAJNXPPW-UHFFFAOYSA-M 0.000 description 1
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Substances O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 1
- KJAMZCVTJDTESW-UHFFFAOYSA-N tiracizine Chemical compound C1CC2=CC=CC=C2N(C(=O)CN(C)C)C2=CC(NC(=O)OCC)=CC=C21 KJAMZCVTJDTESW-UHFFFAOYSA-N 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000037317 transdermal delivery Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000001228 trophic effect Effects 0.000 description 1
- 208000032521 type II spinal muscular atrophy Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/502—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with carbocyclic ring systems, e.g. cinnoline, phthalazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/20—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
La presente invención proporciona un compuesto de fórmula I o una sal farmacéuticamente aceptable del mismo; un método para fabricar los compuestos de la invención y sus usos terapéuticos. La presente invención proporciona además una combinación de agentes farmacológicamente activos y una composición farmacéutica. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN
Antecedentes de la invención
La atrofia muscular espinal (SMA, por sus siglas en inglés) proximal es un grupo heterogéneo desde un punto de vista clínico y heredado de trastornos neuromusculares caracterizados por la degeneración de las células del asta anterior de la médula espinal. Los pacientes padecen una debilidad simétrica de los músculos del tronco y las extremidades, estando las piernas más afectadas que los brazos y siendo los músculos proximales más débiles que los distales; los músculos del diafragma, faciales y oculares no se ven afectados. Existen tres formas de SMA con inicio en la infancia (tipos I, II y III) y una forma IV con inicio en la edad adulta catalogada de una manera relativamente reciente, todas las cuales se pueden distinguir en función de la edad de inicio y la gravedad de la evolución clínica evaluada mediante el examen clínico, biopsia muscular y electromiografía (EMG) (Munsat T L, Davies K E (1992)).
El tipo I (enfermedad de Werdnig-Hoffmann) es la forma más aguda y grave, con un inicio antes de los seis meses y fallecimiento normalmente antes de los dos años; los niños nunca son capaces de sentarse sin soporte. Los síntomas de la enfermedad pueden estar presentes en el útero, como reducción de los movimientos fetales; en el parto; o más a menudo, en los cuatro primeros meses de vida. Los niños afectados son especialmente hipotónicos, experimentan dificultades para alimentarse y respiración diafragmática, y a menudo están caracterizados por una debilidad general en los músculos intercostales y respiratorios accesorios. Los niños afectados nunca se sientan ni se ponen de pie y normalmente fallecen antes de la edad de 2; el fallecimiento se debe por lo general a una insuficiencia respiratoria.
El tipo II (forma crónica, intermedia) tiene un inicio entre los seis y los dieciocho meses de edad; son comunes las fasciculaciones musculares y los reflejos de los tendones se reducen progresivamente. Los niños son incapaces de ponerse de pie o caminar sin ayuda. Los problemas para alimentarse y tragar normalmente no están presentes en la SMA de tipo II, aunque en algunos pacientes puede ser necesaria una sonda nasogástrica. Por lo general, la mayoría de los pacientes desarrollan una escoliosis muscular progresiva que puede requerir corrección quirúrgica. Al igual que los pacientes con enfermedad de tipo I, la eliminación de las secreciones de la tráquea y la tos pueden volverse difíciles debido a una función bulbar deficiente y músculos intercostales débiles. Estos pacientes presentan una hipotonía profunda, parálisis fláccida simétrica y no controlan los movimientos de la cabeza.
El tipo III (enfermedad de Kugelberg-Welander o atrofia muscular espinal juvenil) es una forma crónica y leve, con un inicio después de la edad de 18 meses; se alcanzan los hitos motores de manera normal, y se puede conservar la deambulación hasta edades variables. Estos pacientes a menudo desarrollan escoliosis, y se observan con frecuencia síntomas de uso excesivo de las articulaciones, provocado generalmente por la debilidad. La esperanza de vida es casi normal, pero la calidad de vida está seriamente comprometida.
Los tipos I, II y III avanzan con el tiempo, acompañados por un deterioro del estado del paciente.
El tipo IV con inicio en la edad adulta está caracterizado por debilidad en la segunda o tercera década de vida, con una deficiencia motora moderada que no está acompañada por problemas respiratorios o nutricionales. La SMA adulta está caracterizada por un inicio gradual y una evolución muy lenta. Los músculos bulbares raramente están afectados en el tipo IV. No está claro que la SMA de tipo IV esté relacionada etiológicamente con las formas de tipo I-III.
Otras formas de atrofia muscular espinal incluyen la enfermedad ligada al cromosoma X, atrofia muscular espinal con dificultad respiratoria (SMARD, por sus siglas en inglés), atrofia muscular espinal y bulbar (enfermedad de Kennedy o atrofia muscular bulboespinal) y atrofia muscular espinal distal.
La SMA se debe a mutaciones en el gen de la supervivencia de neuronas motoras (SMN, por sus siglas en inglés), que existe en dos formas en los seres humanos (SMN1 y SMN2). La pérdida de SMN resulta nociva para las neuronas motoras y da como resultado una insuficiencia neuromuscular, un sello distintivo de la enfermedad Desde un punto de vista genético, SMA es una afección recesiva autosómica, provocada por la alteración del gen SMN1, ubicado en 5q13 (Lefebvre S., et al. (1995) Cell 80: 155-165). Más de un 98% de los pacientes con atrofia muscular espinal presentan una alteración homocigótica de SMN1 por deleción, reordenamiento o mutación. Sin embargo, todos estos pacientes conservan al menos una copia de SMN2.
A nivel genómico, solo se han detectado cinco nucleótidos que diferencian el gen SMN1 del gen SMN2. Además, los dos genes producen ARNm idénticos, excepto por un cambio de nucleótidos sinónimo en el exón 7, es decir, un cambio C ^ T seis pares de bases dentro del exón 7 en SMN2. Esta mutación modula la actividad de un potenciador del corte y empalme de exones (Lorson y Androphy (2000) Hum. Mol. Genet. 9:259-265). El resultado de este y los otros cambios de nucleótidos en las regiones intrónicas y promotoras es que la mayoría de los SMN2 son sometidos a corte y empalme de manera alternativa y sus transcritos carecen de los exones 3, 5 o 7. Por el contrario, el ARNm transcrito a partir del gen SMN1 es por lo general un ARNm completo donde únicamente una pequeña fracción de sus transcritos experimentan corte y empalme para eliminar el exón 3, 5 o 7 (Gennarelli et al. (1995) Biochem. Biophys. Res. Commun.
213:342-348; Jong et al. (2000) J. Neurol. Sci. 173:147-153). Todos los sujetos con SMA tienen al menos una, y por lo general, de dos a cuatro copias del gen SMN2, que codifica la misma proteína que SMN1; sin embargo, el gen SMN2 produce únicamente niveles muy bajos de la proteína SMN completa.
La proteína SMNA7 no es funcional y se cree que se degrada rápidamente. Aproximadamente un 10 % del pre-ARNm de SMN2 experimenta corte y empalme adecuado y posteriormente se traduce para producir la proteína SMN completa (FL-SMN), siendo el resto la copia SMNA7. La eficacia del corte y empalme de SMN2 puede depender de la gravedad de la enfermedad, y la producción de un transcrito completo de SMN2 podría variar entre un 10 % y un 50 %. Además, la presencia o ausencia del gen SMN1, del cual aproximadamente un 90 % se convierte en el producto génico y proteína FL-SMN, influencia la gravedad de SMA dependiendo de si puede o no compensar las copias truncadas SMNA7. Un nivel bajo de la proteína SMN permite el desarrollo embrionario pero no es suficiente para mantener la supervivencia de las neuronas motoras de la médula espinal.
La gravedad clínica de los pacientes con SMA se correlaciona inversamente con el número de genes SMN2 y con el nivel de proteína SMN funcional producida (Lorson C L, et al. (1999) PNAS; 96:6307-6311)(Vitali T. et al. (1999) Hum Mol Genet; 8:2525-2532)(Brahe C. (2000) Neuromusc. Disord.; 10:274-275)(Feldkotter M, et al. (2002) Am J Hum Genet; 70:358-368)(Lefebvre S, et al. (1997) Nature Genet; 16:265-269)(Coovert D D, et al. (1997) Hum Mol Genet; 6:1205-1214)(Patrizi A L, et al. (1999) Eur J Hum Genet; 7:301-309).
Las estrategias terapéuticas actuales para SMA se centran en su mayoría en elevar los niveles de proteína SMN (natural) completa, modular el corte y empalme hacia la inclusión del exón 7, estabilizar la proteína natural y, en menor grado, restaurar la función muscular en SMA al proporcionar refuerzo trófico o inhibir la atrofia muscular esquelética.
El mecanismo que da lugar a la pérdida de neuronas motoras y la atrofia muscular sigue siendo poco claro, aunque la disponibilidad de modelos en animales de la enfermedad está aumentando rápidamente el conocimiento en este campo (Frugier T, et al. (2000) Hum Mol. Genet. 9:849-58; Monani U R, et al. (2000) Hum Mol Genet 9:333-9; Hsieh-Li H M, et al. (2000) Nat Genet 24:66-70; Jablonka S, et al. (2000) Hum Mol. Genet. 9:341-6). Asimismo, la función de la proteína SMN se desconoce en parte y los estudios indican que puede estar implicada en el metabolismo del ARNm (Meister G, et al. (2002). Trends Cell Biol. 12:472-8; Pellizzoni L, et al. (2002). Science. 298: 1775-9), y probablemente en el transporte de proteínas/ARNm a las uniones neuromusculares (Ci-fuentes-Diaz C, et al. (2002) Hum Mol. Genet.
11: 1439-47; Chan Y B, et al. (2003) Hum Mol. Genet. 12:1367-76; McWhorter M L, et al. (2003) J. Cell Biol. 162:919-31; Rossoll W, et al. (2003) J. Cell Biol. 163:801-812).
Además de las SMA, se ha notificado por separado que en una subclase de artrogriposis múltiple congénica de tipo neurogénico (AMC congénita) está implicada la deleción del gen SMN1, lo que sugiere que un cierto grado de patología en los afectados se debe probablemente a niveles bajos de SMN de neuronas motoras (L. Burgien et al., (1996) J. Clin. Invest. 98(5):1130-32. La AMC congénita afecta a seres humanos y animales, por ejemplo, caballos, ganado, ovejas, cabras, cerdos, perros y gatos (M. Longeri et al., (2003) Genet. Sel. Evol. 35:S167-S175). Asimismo, se ha observado que el riesgo de desarrollar o la gravedad de la esclerosis lateral amiotrófica (ALS, por sus siglas en inglés) se correlaciona con niveles bajos de SMN de neuronas motoras.
Se carece de una cura de SMNA disponible en la actualidad y, por lo tanto, resultaría ventajoso proporcionar métodos novedosos para modular SMN con el fin de tratar a los aquejados con SMA, con AMC congénita neurogénica, ALS u otras afecciones relacionadas con la deficiencia de SMN. Resultaría más ventajoso aún proporcionar dianas farmacológicas novedosas que se puedan utilizar como la base para desarrollar agentes terapéuticos o diagnósticos eficaces para tales afecciones neuronales.
Compendio de la invención
Se siguen necesitando nuevos tratamientos y terapias para la atrofia muscular espinal. La invención proporciona compuestos, sales de estos, formulaciones farmacéuticas de estos y combinaciones de estos, compuestos que son moduladores de la atrofia muscular espinal. La invención proporciona además compuestos para su uso en el tratamiento, prevención o mejora de la atrofia muscular espinal, que comprende administrar a un sujeto que lo necesita una cantidad eficaz de un modulador de SMN (por ejemplo, un compuesto de la invención).
En la presente se describen varias realizaciones de la invención. Se reconocerá que se pueden combinar características especificadas en cada realización con otras características especificadas para proporcionar realizaciones adicionales.
En ciertos aspectos, los moduladores de SMN proporcionados en la presente son compuestos de Fórmula (I) y sales de estos:
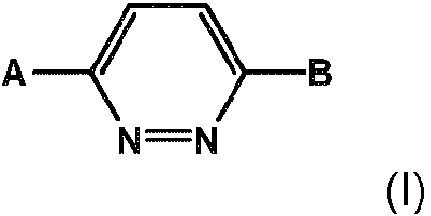
En otra realización, la invención proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con la definición de la fórmula (I), o subfórmulas de esta, y uno o más portadores farmacéuticamente aceptables.
En otra realización, la invención proporciona una combinación, en particular una combinación farmacéutica, que comprende una cantidad terapéuticamente eficaz del compuesto de acuerdo con la definición de la fórmula (I), o subfórmulas de esta, y uno o más coagentes terapéuticamente activos.
Otra realización de la invención es un método para modular la proteína SMN mediante la administración de un modulador de SMN. En otra realización, dicho modulador de SMN es capaz de aumentar uno o más de los niveles de FL-SMN o SMNA7. En otra realización más, dicho modulador de SMN es capaz de evitar que el exón 7 sea escindido del transcrito de SMN.
La presente invención se basa en el descubrimiento de que los moduladores de SMN de la invención (por ejemplo, compuestos de fórmula (I)) son capaces de modular las proteínas SMN, por ejemplo, mediante la activación del promotor de SMN, modulación del corte y empalme (por ejemplo, al evitar que el exón 7 sea eliminado por corte y empalme del gen SMN) y/o modulación de la estabilidad de la proteína SMN.
Descripción detallada de la invención
Como se ha mencionado anteriormente, la presente invención proporciona compuestos que modulan la actividad de SMN. Tales compuestos se pueden utilizar in vitro o in vivo para modular (preferentemente aumentar) la producción y actividad de SMN en diversos contextos.
En una primera realización, la invención proporciona compuestos de Fórmula (I) y sales farmacéuticamente aceptables de estos, que modulan la actividad de SMN. Los compuestos de Fórmula (I) están representados por la estructura:

En una segunda realización, la invención se refiere a un compuesto de la primera realización, o una sal de este, compuesto que está representado por la Fórmula (III):

donde Ri6 es un heteroarilo de 5 miembros que tiene un átomo de nitrógeno anular y 0 o 1 heteroátomos anulares adicionales seleccionados entre N, O o S, donde el heteroarilo está sustituido opcionalmente con alquilo C1-C4.
De acuerdo con la primera realización, la invención se refiere a un compuesto de fórmula (I), o una sal de este, donde B se selecciona del grupo que consiste en

donde X es O o N(Me), y R17 es hidrogeno o metilo.
En una tercera realización, la invención se refiere a un compuesto de acuerdo con la primera y segunda realizaciones, o una sal de este, donde X es -O-.
En una cuarta realización, la invención se refiere a un compuesto de acuerdo con las realizaciones anteriores, o una sal de este, donde R16 es:

En una quinta realización, la invención es un compuesto, o una sal de este, seleccionado del grupo que consiste en:
5-cloro-2-(6-(1,2,2,6,6-pentametilpiperidin-4-ilamino)piridazin-3-il)fenol;
2-(6-(piperidin-4-iloxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-(((2s,4R,6R)-2,6-dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-((-2,6-dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-(((2S,4S)-2-metilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-((3-fluoropiperidin-4-il)oxi)piridazin-3-il)-5-(1 H-pirazol-1 -il)fenol;
2-[6-(1,2,2,6,6-pentametilpiperidin-4-ilamino)piridazin-3-il]-5-pirazol-1 -ilfenol;
2-(6-(7-(2-hidroxietil)-2,7-diazaespiro[4.4]nonan-2-il)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-piperidin-4-ilpiridazin-3-il)-5-pirazol-1 -ilfenol;
2-(6-((3aR,6aS)-5-(2-hidroxietil)hexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol;
4-(3-hidroxi-4-(6-(5-metilhexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)fenil)-1-metilpiridin-2(1H)-ona;
4-(3-hidroxi-4-(6-((3aR,6aR)-1-metilhexahidropirrolo[3,4-¿]pirrol-5(1H)-il)piridazin-3-il)fenil)-1-metilpiridin-2(1H)-ona; 2-(6-(2,7-diazaespiro[4.5]decan-2-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol; y
4- (4-(6-(2,7-diazaespiro[4.5]decan-2-il)piridazin-3-il)-3-hidroxifenil)-1-metilpiridin-2(1 H)-ona.
En una sexta realización, la invención es un compuesto, o sal de este, seleccionado del grupo que consiste en: 2-[6-(7-metil-2,7-diazaespiro[4.4]non-2-il)piridazin-3-il]-5-pirazol-1 -ilfenol;
2-[6-(2,7-diazaespiro[3.5]non-7-il)piridazin-3-il]-5-pirazol-1 -ilfenol;
2-[6-(1,7-diazaespiro[4.4]non-7-il)piridazin-3-il]-5-pirazol-1 -ilfenol;
2-(6-((3aR,6aS)-hexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
5- pirazol-1-il-2-[6-(1,2,3,6-tetrahidropiridin-4-il)piridazin-3-il]fenol;
2-(6-((2,2-dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol;
2-(6-((3aR,6aS)-hexahidropirroloÍ3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol;
2-(6-((3aR,6as)-5-metilhexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol;
5-cloro-2-(6-(metil(1,2,2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)fenol; y
2-[6-(1,2,2,6,6-pentametilpiperidin-4-iloxi)piridazin-3-il]-5-pirazol-1 -ilfenol.
En una séptima realización, la invención se refiere a una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con una cualquiera de las realizaciones anteriores, o una sal farmacéuticamente aceptable de este, y uno o más portadores farmacéuticamente aceptables.
En una octava realización, la invención se refiere a una combinación que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con una cualquiera de la primera a sexta realizaciones, o una sal farmacéuticamente aceptable de este, y uno o más coagentes terapéuticamente activos.
En una novena realización, la invención se refiere a un compuesto de fórmula (I), o una sal de este, de acuerdo con una cualquiera de la primera a sexta realizaciones, para su uso en el tratamiento, prevención o mejora de una afección relacionada con la deficiencia de SMN, que comprende administrar a un sujeto que lo necesita una cantidad eficaz de un compuesto, o una sal de este, de una cualquiera de la primera a quinta realizaciones.
En una décima realización, la invención se refiere a un compuesto de fórmula (I), o una sal de este, para su uso como se describe en la novena realización, donde la afección relacionada con la deficiencia de SMN es la atrofia muscular espinal.
En una undécima realización, la invención se refiere a un compuesto de fórmula (I), o una sal de este, de acuerdo con una cualquiera de la primera a sexta realizaciones, o una sal farmacéuticamente aceptable de este, para su uso como un medicamento.
En una duodécima realización, la invención se refiere a un compuesto de fórmula (I), o una sal de este, de acuerdo con una cualquiera de la primera a sexta realizaciones, o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento de una afección relacionada con la deficiencia de SMN.
En una decimotercera realización, la invención se refiere al compuesto de fórmula (I), o una sal de este, de acuerdo con la duodécima realización, o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento de la atrofia muscular espinal.
En una decimocuarta realización, la invención se refiere al uso de un compuesto de fórmula (I), o una sal de este, de acuerdo con una cualquiera de la primera a sexta realizaciones, o una sal farmacéuticamente aceptable de este, en la fabricación de un medicamento para el tratamiento de la atrofia muscular espinal.
A fin de interpretar esta memoria descriptiva, se aplicarán las siguientes definiciones y, cuando proceda, los términos utilizados en singular también incluirán el plural y viceversa.
Tal como se utiliza en la presente, la expresión “modulador de SMN” incluye agentes, tales como los compuestos de la invención, como se definen en la presente, que poseen la capacidad de modular, por ejemplo, aumentar, los niveles de proteína SMN mediante al menos uno de los múltiples posibles mecanismos. Un conjunto no limitante de mecanismos incluyen la activación del promotor de SMN, modulación del corte y empalme (por ejemplo, evitar que el exón 7 se elimine por corte y empalme del gen SMN) y modulación de la estabilidad de la proteína SMN. Los moduladores de SMN pueden modular, por ejemplo, aumentar los niveles de FL-SMN y/o SMNA7 mediante cualquiera de dichos mecanismos, y/o pueden evitar que SMNA7 se degrade.
Tal como se utiliza en la presente, la expresión “compuestos de la invención” incluye, pero sin limitación, los compuestos de fórmula (I) como se definen en la presente.
Tal como se utiliza en la presente, la expresión “afecciones relacionadas con la deficiencia de SMN” incluye, sin carácter limitante, la atrofia muscular espinal (SMA), artrogriposis múltiple congénica de tipo neurogénico (AMC congénita) y esclerosis lateral amiotrófica (ALS).
Tal como se utiliza en la presente, la expresión “atrofia muscular espinal”, “SMA”, incluye las tres formas de SMA con inicio en la infancia: Tipo I (enfermedad de Werdnig-Hoffmann); tipo II (forma crónica intermedia), tipo III (enfermedad de Kugelberg-Welander o atrofia muscular espinal juvenil); tipo IV con inicio en la edad adulta; así como también otras formas de SMA, que incluyen la enfermedad ligada al cromosoma X, atrofia muscular espinal con dificultad respiratoria (SMARD), atrofia muscular espinal y bulbar (enfermedad de Kennedy o atrofia muscular bulboespinal) y atrofia muscular espinal distal.
A fin de interpretar esta memoria descriptiva, se aplicarán las siguientes definiciones y, cuando proceda, los términos utilizados en singular también incluirán el plural y viceversa.
Tal como se utiliza en la presente, la expresión “alquilo C1-10” se refiere a un resto hidrocarbonado ramificado o no ramificado totalmente saturado que tiene de 1 a 10 átomos de carbono. Los términos “alquilo C W y “alquilo C1-4” deben interpretarse en consecuencia. Los ejemplos representativos de alquilo C1-10 incluyen, sin carácter limitante,
metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, isobutilo, ferf-butilo, n-pentilo, isopentilo, neopentilo, n-hexilo, 3-metilhexilo, 2,2- dimetilpentilo, 2,3-dimetilpentilo, n-heptilo, n-octilo, n-nonilo y n-decilo.
Tal como se utiliza en la presente, la expresión “haloalquilo C1-10” se refiere a un grupo alquilo C1-10 como se define en la presente, donde al menos uno de los átomos de hidrógeno es reemplazado por un átomo halógeno. El grupo haloalquilo C1-10 puede ser monohaloalquilo C1-10, dihaloalquilo C1-10 o polihaloalquilo C1-10, incluido perhaloalquilo C1-10. Un monohaloalquilo C1-10 puede tener un yodo, bromo, cloro o fluoro en el grupo alquilo. Los grupos dihaloalquilo C1-10 y polihaloalquilo C1-10 pueden tener dos o más de los mismos átomos halógeno o una combinación de grupos halógenos diferentes en el alquilo. Normalmente, el grupo polihaloalquilo C1-10 contiene hasta 12, o 10, u 8, o 6, o 4, o 3, o 2 grupos halógenos. Los ejemplos no limitantes de haloalquilo C1-10 incluyen fluorometilo, difluorometilo, trifluorometilo, clorometilo, diclorometilo, triclorometilo, pentafluoroetilo, heptafluoropropilo, difluoroclorometilo, diclorofluorometilo, difluoroetilo, difluoropropilo, dicloroetilo y dicloropropilo. Un grupo perhaloalquilo C1-10 se refiere a un grupo alquilo C1-10 en el que todos los átomos de hidrógeno han sido reemplazados por átomos halógenos.
El término “arilo” se refiere a un grupo hidrocarbonado aromático que tiene 6-20 átomos de carbono en la porción anular. Normalmente, el arilo es un arilo monocíclico, bicíclico o tricíclico que tiene 6-20 átomos de carbono e incluye uno o más anillos aromáticos condensados a uno o más anillos hidrocarbonados no aromáticos. Los ejemplos no limitantes incluyen fenilo, naftilo o tetrahidronaftilo.
Tal como se utiliza en la presente, la expresión “alcoxi C1-10” se refiere a alquil C1-10-O-, donde el alquilo C1-10 es como se ha definido anteriormente. Los ejemplos representativos de alcoxi C1-10 incluyen, sin carácter limitante, metoxi, etoxi, propoxi, 2-propoxi, butoxi, fert-butoxi, pentiloxi, hexiloxi, heptiloxi, octiloxi y deciloxi.
Tal como se utiliza en la presente, el término “heterociclilo” o “heterociclo” se refiere a un anillo o sistema anular no aromático saturado o insaturado, que es un anillo monocíclico de 4, 5, 6 o 7 miembros que contiene 1, 2 o 3 heteroátomos seleccionados entre O, S y N, un sistema anular bicíclico de 7, 8, 9, 10, 11 o 12 miembros que contiene 1, 2, 3, 4 o 5 heteroátomos seleccionados entre O, S y N, o un sistema anular tricíclico de 10, 11, 12, 13, 14 o 15 miembros y que contiene 1, 2, 3, 4, 5, 6 o 7 heteroátomos seleccionados entre O, S y N, donde el N y S también se pueden oxidar opcionalmente a varios estados de oxidación. El grupo heterocíclico se puede unir mediante un heteroátomo o un átomo de carbono. El heterociclilo puede incluir anillos condensados o con puente, así como también anillos espirocíclicos. Los ejemplos de heterociclos incluyen tetrahidrofurano (THF), dihidrofurano, 1,4-dioxano, morfolina, 1,4-ditiano, piperazina, piperidina, 1,3-dioxolano, imidazolidina, imidazolina, pirrolina, pirrolidina, tetrahidropirano, dihidropirano, oxatiolano, ditiolano, 1,3-dioxano, 1,3-ditiano, oxatiano y tiomorfolina.
Tal como se utiliza en la presente, la expresión “cicloalquilo C3-12” se refiere a grupos hidrocarbonados saturados o insaturados monocíclicos, bicíclicos o tricíclicos de 3-12 átomos de carbono. La expresión “cicloalquilo C3-18” se refiere a un grupo hidrocarbonado totalmente saturado o insaturado monocíclico de 3-8 átomos de carbono. Los grupos hidrocarbonados monocíclicos de ejemplo incluyen, sin carácter limitante, ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo y ciclohexenilo. Los grupos hidrocarbonados biicíclicos de ejemplo incluyen bornilo, indilo, hexahidroindilo, tetrahidronaftilo, decahidronaftilo, biciclo[2.1.1]hexilo, biciclo[2.2.1]heptilo, biciclo[2.2.1]heptenilo, 6,6-dimetilbiciclo[3.1.1]heptilo, 2,6,6-trimetilbiciclo[3.1.1 ]heptilo, biciclo[2.2.2]octilo. Los grupos hidrocarbonados tricíclicos de ejemplo incluyen, por ejemplo, adamantilo.
Tal como se utiliza en la presente, la expresión “cicloalquiloxi C3-12” se refiere a cicloalquil C3-12-O-, donde cicloalquilo C3-12 se se ha definido anteriormente en la presente. Los ejemplos representativos de cicloalquiloxi C3-12 incluyen, sin carácter limitante, grupos monocíclicos tales como ciclopropoxi, ciclobutoxi, ciclopentiloxi, ciclopenteniloxi, ciclohexiloxi y ciclohexeniloxi y similares. Los grupos hidrocarbonados bicíclicos de ejemplo incluyen borniloxi, indiloxi, hexahidroindiloxi, tetrahidronaftiloxi, decahidronaftiloxi, biciclo[2.1.1 ]hexiloxi, biciclo[2.2.1]heptiloxi, biciclo[2.2.1]hepteniloxi, 6,6-dimetilbiciclo[3.1.1 ]heptiloxi, 2,6,6-trimetilbiciclo[3.1.1]heptiloxi, biciclo[2.2.2]octiloxi y similares. Los grupos hidrocarbonados tricíclicos de ejemplo incluyen, por ejemplo, adamantiloxi.
Tal como se utiliza en la presente, el término “ariloxi” se refiere tanto a un grupo -O-arilo como a uno -O-heteroarilo, donde arilo y heteroarilo se definen en la presente.
Tal como se utiliza en la presente, el término “heteroarilo” se refiere a un anillo aromático monocíclico de 5, 6 o 7 miembros que contiene 1,2, 3 o 4 heteroátomos seleccionados entre O, S y N, un sistema anular bicíclico condensado de 8, 9 o 10 miembros que contiene 1, 2, 3, 4 o 5 heteroátomos seleccionados entre O, S y N, o un sistema anular tricíclico condensado de 11, 12, 13 o 14 miembros que contiene 1,2, 3, 4, 5 o 6 heteroátomos seleccionados entre O, S y N, donde al menos uno de los anillos de los sistemas anulares bicíclico o tricíclicos es totalmente aromático. Los grupos heteroarilo típicos incluyen 2- o 3-tienilo, 2- o 3-furilo, 2- o 3-pirrolilo, 2-, 4- o 5-imidazolilo, 3-, 4- o 5-pirazolilo, 2-, 4- o 5-tiazolilo, 3-, 4-, o 5-isotiazolilo, 2-, 4-, o 5-oxazolilo, 3-, 4-, o 5-isoxazolilo, 3- o 5-1,2,4-triazolilo, 4- o 5-1,2,3-triazolilo, tetrazolilo, 2-, 3-, o 4-piridilo, 3- o 4-piridazinilo, 3-, 4-, o 5-pirazinilo, 2-pirazinilo, 2-, 4-, o 5-pirimidinilo, 1-, 2 , 3-, 5-, 6-, 7-, u 8-indolizi nilo, 1-, 3-, 4-, 5-, 6-, o 7-isoindolilo, 2-, 3-, 4-, 5-, 6-, o 7-indolilo, 2-, 3-, 4-, 5-, 6-, o 7-indazolilo, 2-, 4-, 5-, 6-, 7-, u 8- purinilo, 1-, 2-, 3-, 4-, 6-, 7-, 8-, o 9-quinolizinilo, 2-, 3-, 4-, 5-, 6-, 7-, u 8-quinoliilo, 1-, 3-, 4-, 5-, 6 , 7-, u 8-isoquinolinilo, 1-, 4-, 5-, 6-, 7-, u 8-ftalazinilo, 2-, 3-, 4-, 5-, o 6-naftiridinilo, 2-, 3-, 5-, 6-, 7-, u 8-quinazolinilo,
3- , 4-, 5-, 6-, 7-, u 8-cinnolinilo, 2-, 4-, 6-, o 7-pteridinilo, 1-, 2-, 3-, 4-, 5-, 6-, 7-, u 8-4aH-carbazolilo, 1-, 2-, 3-, 4-, 5-, 6 , 7-, u 8-carbazolilo, 1-, 3-, 4-, 5-, 6-, 7-, 8-, o 9-carbolinilo, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, o 10-fenantridinilo, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8-, o 9-acridinilo, 1-, 2-, 4-, 5-, 6-, 7-, 8-, o 9-perimidinilo, 2-, 3-, 4-, 5-, 6-, 8-, 9-, o 10-fenatrolinilo, 1-, 2-, 3-, 4- , 6-, 7-, 8-, o 9-fenazinilo, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, o 10-fenotiazinilo, 1-, 2-, 3-, 4-, 6-, 7-, 8-, 9-, o 10-fenoxazinilo, 2-, 3-, 4-, 5-, 6-, o 1-, 3-, 4-, 5-, 6-, 7-, 8-, 9-, o 10- bencisoquinolinilo, 2-, 3-, 4-, o tieno[2,3-b]furanilo, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10-, u 11-7H-pirazino[2,3-c]carbazolilo,2-, 3-, 5-, 6-, o 7-2H-furo[3,2-jb]-piranilo, 2-, 3-, 4-, 5-, 7-, u 8-5H-pirido[2,3-d]-o-oxazinilo, 1-, 3-, o 5-1H-pirazolo[4,3-d]-oxazolilo, 2-, 4-, o 54H-imidazo[4,5-d]tiazolilo, 3-, 5-, u 8-pirazino[2,3-d]piridazinilo, 2-, 3-, 5-, o 6- imidazo[2,1-jb]tiazolilo, 1-, 3-, 6-, 7-, 8-, o 9-furo[3,4-c]cinnolinilo, 1-, 2-, 3-, 4-, 5-, 6-, 8-, 9 , 10, u 11-4H-pirido[2,3-c]carbazolilo, 2-, 3-, 6-, o 7-imidazo[1,2-jb][1,2,4]triazinilo, 7-benzo[¿]tienilo, 2-, 4-, 5- , 6-, o 7-benzoxazolilo, 2-, 4-, 5-, 6-, o 7-bencimidazolilo, 2-, 4-, 4-, 5-, 6-, o 7-benzotiazolilo, 1-, 2-, 4-, 5-, 6-, 7-, 8-, o 9-benzoxapinilo, 2-, 4-, 5-, 6-, 7-, u 8-benzoxazinilo, 1-, 2-, 3-, 5-, 6-, 7-, 8-, 9-, 10-, u 11-1H-pirrolo[1,2-jb][2]benzazapinilo, 2-, 3-, 4-, 5-, 6-, 7-, u 8-quinolinilo, 1-, 3-, 4-, 5-, 6-, 7-, u 8-isoquinolinilo, 2-, 3-, 4-, 5-, 6-, o 7-indolilo, 2-, 3-, 4-, 5-, 6-, o 7-benzo[b]tienilo, 2-, 4-, 5-, 6-, o 7-benzoxazolilo, 2-, 4-, 5-, 6-, o 7-bencimidazolilo y 2-, 4-, 5-, 6-, o 7-benzotiazolilo.
Tal como se utilizan en la presente, el término “halógeno” o “halo” se refieren a fluoro, cloro, bromo y yodo.
Tal como se utiliza en la presente, el término “isómeros” se refiere a diferentes compuestos que tienen la misma fórmula molecular pero difieren en la exposición y configuración de los átomos. Asimismo, tal como se utiliza en la presente, la expresión “un isómero óptico” o “un estereoisómero” se refiere a cualquiera de las diversas configuraciones estereoisoméricas que pueden existir para un compuesto concreto de la presente invención, como se define en la presente, e incluye isómeros geométricos. Se entiende que un sustituyente se puede unir a un centro quiral de un átomo de carbono. Por lo tanto, la invención incluye enantiómeros, diastereómeros o racematos del compuesto de fórmula (I) como se define en la presente. Los “enantiómeros” son un par de estereoisómeros que no son imágenes especulares superponibles el uno del otro. Una mezcla 1:1 de un par de enantiómeros es una mezcla “racémica”. El término se utiliza para designar una mezcla racémica cuando sea apropiado. Los “diastereoisómeros” son estereoisómeros que tienen al menos dos átomos asimétricos, pero que no son imágenes especulares el uno del otro. La estereoquímica absoluta se especifica de acuerdo con el sistema de Cahn-Ingold-Prelog R-S. Cuando un compuesto es un enantiómero puro, la estereoquímica en cada carbono quiral se puede especificar mediante R o S. Los compuestos resueltos cuya configuración absoluta no se conoce se pueden designar (+) o (-) dependiendo de la dirección (dextro- o levorrotatoria) en la que rotan el plano de la luz polarizada en la longitud de onda de la línea D de sodio. Ciertos de los compuestos descritos en la presente contienen uno o más centros o ejes asimétricos y, por lo tanto, pueden dar lugar a enantiómeros, diastereómeros y otras formas estereoisoméricas que se puedan definir, en función de la estereoquímica absoluta, como (R)- o (S)-. Se pretende que la presente invención incluya todos estos posibles isómeros, incluidas mezcla racémicas, formas ópticamente puras y mezclas intermedias. Los isómeros (R) y (S) ópticamente activos se pueden preparar utilizando sintones quirales o reactivos quirales, o se pueden resolver utilizando técnicas convencionales. Si el compuesto contiene un doble enlace, el sustituyente puede tener la configuración E o Z. Si el compuesto contiene un cicloalquilo disustituido, el sustituyente cicloalquilo puede tener una configuración cis o trans. También se pretende que estén incluidas todas las formas tautoméricas.
Tal como se utilizan en la presente, los términos “sal” o “sales” se refieren a una sal de adición de ácido o adición de base de un compuesto de la invención. Las “sales” incluyen en particular “sales farmacéuticas aceptables”. La expresión “sales farmacéuticamente aceptables” se refiere a sales que retienen la eficacia y propiedades biológicas de los compuestos de esta invención y, que normalmente no son indeseables desde un punto de vista biológico ni de otro tipo. En muchos casos, los compuestos de la presente invención son capaces de formar sales de ácido y/o base en virtud de la presencia de grupos amino y/o carboxilo o grupos similares a estos.
Las sales de adición de ácido farmacéuticamente aceptables se pueden formar con ácidos inorgánicos y ácidos orgánicos, por ejemplo, sales de acetato, aspartato, benzoato, besilato, bromuro/bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, alcanforsulfato, cloruro/clorhidrato, clortofilonato, citrato, etanodisulfonato, fumarato, gluceptato, gluconato, glucuronato, hipurato, yodhidrato/yoduro, isetionato, lactato, lactobionato, laurilsulfato, malato, maleato, malonato, mandelato, mesilato, metilsulfato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/hidrogenofosfato/dihidrogenofosfato, poligalacturonato, propionato, estearato, succinato, sulfosalicilato, tartrato, tosilato y trifluoroacetato.
Los ácidos inorgánicos a partir de los que se pueden obtener sales incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares.
Los ácidos orgánicos a partir de los que se pueden obtener las sales incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido toluenosulfónico, ácido sulfosalicílico y similares. Se pueden formar sales de adición de base farmacéuticamente aceptables con bases inorgánicas y orgánicas.
Las bases inorgánicas a partir de las que se pueden obtener sales incluyen, por ejemplo, sales de amonio y metales de las columnas de I a XII de la tabla periódica. En ciertas realizaciones, las sales se obtienen a partir de sodio, potasio,
amonio, calcio, magnesio, hierro, plata, zinc y cobre; las sales particularmente adecuadas incluyen sales de amonio, potasio, sodio, calcio y magnesio.
Las bases orgánicas a partir de las que se pueden obtener sales incluyen, por ejemplo, aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas de origen natural, aminas cíclicas, resinas básicas de intercambio iónico y similares. Ciertas aminas orgánicas incluyen isopropilamina, benzatina, colinato, dietanolamina, dietilamina, lisina, meglumina, piperazina y trometamina.
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar a partir de un compuesto precursor, un resto básico o ácido, mediante métodos químicos convencionales. Por lo general, tales sales se pueden preparar haciendo reaccionar formas de ácido libre de estos compuestos con una cantidad estequiométrica de la base adecuada (tal como hidróxido, carbonato, bicarbonato de Na, Ca, Mg o K o similares) o haciendo reaccionar formas de base libre de estos compuestos con una cantidad estequiométrica del ácido adecuado. Tales reacciones se llevan a cabo normalmente en agua o en un disolvente orgánico o en una mezcla de los dos. Por lo general, cuando sea factible es deseable el uso de medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Se pueden consultar listas de sales adecuadas adicionales, por ejemplo, en “Remington's Pharmaceutical Sciences”, 20.a ed., Mack Publishing Company, Easton, Pa., (1985); y en “Handbook of Pharmaceutical Salts: Properties, Selection, and Use” de Stahl y Wermuth (Wiley-VCH, Weinheim, Alemania, 2002).
Se pretende que toda fórmula proporcionada en la presente también represente formas no marcadas así como formas marcadas con isótopos de los compuestos. Los compuestos marcados con isótopos tienen las estructuras mostradas en las fórmulas que se proporcionan en la presente excepto en que uno o más átomos son reemplazados por un átomo que tiene una masa atómica o número másico seleccionados. Los ejemplos de isótopos que se pueden incorporar en compuestos de la invención, como se definen en la presente, incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, tales como 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl, 125I, respectivamente. La invención incluye varios compuestos marcados con isótopos como se definen en la presente, por ejemplo, aquellos en los que están presentes isótopos radioactivos, tales como 3H, 13C y 14C. Tales compuestos marcados con isótopos son útiles en estudios metabólicos (con 14C), estudios de la cinética de reacción (con, por ejemplo, 2H o 3H), técnicas de detección u obtención de imágenes, tales como la tomografía por emisión de positrones (PET) o la tomografía computarizada de emisión monofotónica (SPECT), que incluyen ensayos de distribución tisular de fármacos o sustratos, o en el tratamiento radiactivo de pacientes. En particular, un 18F o compuesto marcado puede ser particularmente deseable para los estudios por PET o SPECT. Los compuestos marcados con isótopos de esta invención se pueden preparar por lo general llevando a cabo los procedimientos divulgados en los esquemas o en los ejemplos y preparaciones descritos más adelante utilizando un reactivo marcado con isótopos disponible en lugar de un reactivo no marcado con isótopos.
Además, la sustitución con isótopos más pesados, particularmente deuterio (es decir, 2H o D), puede proporcionar ciertas ventajas terapéuticas como resultado de una mayor estabilidad metabólica, por ejemplo, un aumento de la semivida in vivo o unos requisitos posológicos reducidos o una mejora en el índice terapéutico. Se entiende que el deuterio en este contexto se considera como un sustituyente de un compuesto de fórmula (I), como se define en la presente. La concentración de un isótopo más pesado de este tipo, específicamente deuterio, se puede definir por el factor de enriquecimiento isotópico. La expresión “factor de enriquecimiento isotópico”, tal como se utiliza en la presente, se refiere a la relación entre la abundancia isotópica y la abundancia natural de un isótopo especificado. Si se indica que un sustituyente en un compuesto de esta invención es deuterio, dicho compuesto tiene un factor de enriquecimiento isotópico para cada átomo de deuterio designado de al menos 3500 (incorporación de deuterio de un 52,5 % en cada átomo de deuterio designado), al menos 4000 (incorporación de deuterio de un 60 %), al menos 4500 (incorporación de deuterio de un 67,5 %), al menos 5000 (incorporación de deuterio de un 75 %), al menos 5500 (incorporación de deuterio de un 82,5 %), al menos 6000 (incorporación de deuterio de un 90 %), al menos 6333,3 (incorporación de deuterio de un 95 %), al menos 6466,7 (incorporación de deuterio de un 97 %), al menos 6600 (incorporación de deuterio de un 99 %) o al menos 6633,3 (incorporación de deuterio de un 99,5 %).
Los compuestos marcados con isótopos de fórmula (I), como se definen en la presente, generalmente se pueden preparar mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procesos análogos a los descritos en las Preparaciones y los Ejemplos adjuntos utilizando un reactivo marcado con isótopos apropiado en lugar del reactivo no marcado empleado previamente.
Los solvatos farmacéuticamente aceptables de acuerdo con la invención incluyen aquellos en los que el disolvente de cristalización pueda estar sustituido con isótopos, por ejemplo, D2O, d6-acetona, d6-DMSO.
Los compuestos de la invención, es decir, los compuestos de fórmula (I), como se define en la presente, que contienen grupos capaces de actuar como donantes y/o aceptores de enlaces de hidrógeno pueden ser capaces de formar cocristales con formadores de cocristales adecuados. Estos cocristales se pueden preparar a partir de compuestos de fórmula (I), como se define en la presente, mediante procedimientos de formación de cocristales conocidos. Tales procedimientos incluyen moler, calentar, cosublimar, cofundir o poner en contacto en solución compuestos de fórmula (I), como se define en la presente, con el formador de cocristales en condiciones de cristalización y aislar los cocristales
formados de esta manera. Los formadores de cocristales adecuados incluyen aquellos descritos en el documento WO 2004/078163. Así pues, la invención proporciona además cocristales que comprenden un compuesto de fórmula (I), como se define en la presente.
La expresión “una cantidad terapéuticamente eficaz” de un compuesto de la presente invención, como se define en la presente, se refiere a una cantidad de un compuesto de la presente invención que suscitará la respuesta biológica o médica de un sujeto, por ejemplo, reducción o inhibición de una actividad enzimática o proteica, o mejorará los síntomas, aliviará las afecciones, ralentizará o retrasará el avance de la enfermedad, o prevendrá una enfermedad, etc. En una realización no limitante, la expresión “una cantidad terapéuticamente eficaz” se refiere a la cantidad del compuesto de la presente invención, como se define en la presente, que, cuando se administra a un sujeto, es eficaz para (1) aliviar, inhibir, prevenir y/o mejorar al menos parcialmente una afección o un trastorno o una enfermedad (i) mediados por el gen o producto del gen de la supervivencia de neuronas motoras (SMN), o mediante la degradación de SMNA7, o mediante los niveles relativos de FL-SMN y SMNA7 (ii) asociados con la actividad de SMN, o (iii) caracterizados por la actividad (normal o anómala) de SMN; o (2) reducir o inhibir la actividad de SMN; o (3) reducir o inhibir la expresión de SMN1 o SMN2.
En otra realización no limitante, la expresión “una cantidad terapéuticamente eficaz” se refiere a la cantidad del compuesto de la presente invención, como se ha definido en la presente, que, cuando se administra a una célula o un tejido o un material biológico no celular o un medio es eficaz para reducir o inhibir al menos parcialmente la actividad de SMN; o reducir o inhibir al menos parcialmente la expresión de SMN, en ambos casos mediante la modulación de los niveles relativos de FL-SMN y SMNA7.
Las frases “cantidad terapéuticamente eficaz” y “cantidad eficaz” se utilizan en la presente para referirse a una cantidad suficiente para reducir en al menos aproximadamente un 15 por ciento, preferentemente, en al menos aproximadamente un 50 por ciento, más preferentemente en al menos un 90 por ciento, y de la manera más preferente prevenir un déficit significativo desde un punto de vista clínico en la actividad, función y respuesta del hospedador. Como alternativa, una cantidad terapéuticamente eficaz es suficiente para provocar una mejora en una afección/síntoma significativo desde un punto de vista clínico en el hospedador.
La cantidad eficaz puede variar dependiendo de factores tales como el tamaño y el peso del sujeto, el tipo de patología o el compuesto particular de la invención, como se define en la presente. Por ejemplo, la elección del compuesto de la invención puede afectar a lo que constituye una “cantidad eficaz”. Un experto en la técnica será capaz de estudiar los factores contenidos en la presente y realizar la determinación de lo que se refiere a la cantidad eficaz de los compuestos de la invención, como se definen en la presente, sin una experimentación excesiva.
El régimen de administración puede afectar lo que constituye una cantidad eficaz. El compuesto de la invención se puede administrar al sujeto antes o después del inicio de una afección relacionada con la deficiencia de SMN. Además, se pueden administrar varias dosificaciones divididas, así como también dosificaciones escalonadas, a diario o secuencialmente, o la dosis se puede infundir de manera continua o puede ser una inyección en embolada. Además las dosificaciones del (de los) compuesto(s) de la invención, como se definen(n) en la presente, se pueden aumentar o reducir proporcionalmente según indican las exigencias de la situación terapéutica o profiláctica.
Tal como se utiliza en la presente, el término “sujeto” se refiere a un animal. Normalmente, el animal es un mamífero. Un sujeto también se refiere a, por ejemplo, primates (por ejemplo, seres humanos, de sexo masculino o femenino), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, peces, pájaros y similares. En ciertas realizaciones, el sujeto es un primate. En otras realizaciones adicionales, el sujeto es un ser humano.
Tal como se utiliza en la presente, el término “inhibir”, “inhibición” o “que inhibe” se refiere a la reducción o supresión de una afección, síntoma, o trastorno o enfermedad dados, o una disminución significativa en la actividad de referencia de una actividad o proceso biológicos.
Tal como se utiliza en la presente, el término “tratar”, “que trata” o “tratamiento” de cualquier enfermedad o trastorno se refiere en una realización, a mejorar la enfermedad o trastorno (es decir, ralentizar o detener o reducir el desarrollo de la enfermedad o al menos uno de los síntomas clínicos de esta). En otra realización “tratar”, “que trata” o “tratamiento” se refiere a aliviar o mejorar al menos un parámetro físico incluidos aquellos que puede que no sean perceptibles para el paciente. En otra realización adicional, “tratar”, “que trata” o “tratamiento” se refiere a modular la enfermedad o trastorno, ya sea de modo físico (por ejemplo, mediante la estabilización de un síntoma perceptible), fisiológico (por ejemplo, mediante la estabilización de un parámetro físico) o ambos. En otra realización adicional, “tratar”, “que trata” o “tratamiento” se refiere a prevenir o retrasar el inicio o desarrollo o avance de la enfermedad o trastorno.
Tal como se utiliza en la presente, un sujeto “necesita” un tratamiento si tal sujeto se beneficiaría desde un punto de vista biológico, médico o en calidad de vida de tal tratamiento.
Tal como se utilizan en la presente, se ha de interpretar que el término “un”, “uno/a”, “el/la” y términos similares utilizados en el contexto de la presente invención (especialmente en el contexto de las reivindicaciones) abarcan tanto el singular como el plural a menos que se indique lo contrario en la presente o el contexto lo contradiga claramente.
Todos los métodos descritos en la presente se pueden llevar a cabo en cualquier orden adecuado a menos que se indique lo contrario en la presente o el contexto lo contradiga claramente. El uso de todos y cualquiera de los ejemplos, o lenguaje ilustrativo (por ejemplo, “tal como”) que se proporciona en la presente se pretende que simplemente ilustre mejor la invención y no impone una limitación en el alcance de la invención por lo demás reivindicada.
Cualquier átomo asimétrico (por ejemplo, de carbono o similares) del (de los) compuesto(s) de la presente invención como se define(n) en la presente, puede estar presente en la configuración racémica o enriquecida enantioméricamente, por ejemplo, la configuración (R), (S) o (R,S). En ciertas realizaciones, cada átomo asimétrico tiene un exceso enantiomérico de al menos un 50 %, un exceso enantiomérico de al menos un 60 %, un exceso enantiomérico de al menos un 70 %, un exceso enantiomérico de al menos un 80 %, un exceso enantiomérico de al menos un 90 %, un exceso enantiomérico de al menos un 95 % o un exceso enantiomérico de al menos un 99 % en la configuración (R) o (S). Los sustituyentes en átomos con enlaces insaturados pueden estar presentes, si es posible, en forma cis (Z) o trans (E).
Por consiguiente, tal como se utiliza en la presente, un compuesto de la presente invención, como se define en la presente, puede estar en forma de uno de los posibles isómeros, rotámeros, atropisómeros, tautómeros o mezclas de estos, por ejemplo, como racematos, isómeros ópticos (enantiómeros), diastereómeros, isómeros geométricos (cis o trans) sustancialmente puros o mezclas de estos.
Cualesquiera mezclas resultantes de isómeros se pueden separar, basándose en las diferencias fisicoquímicas de los constituyentes, en los racematos, diastereómeros, isómeros ópticos o geométricos sustancialmente puros, por ejemplo, mediante cromatografía y/o cristalización fraccionada.
Cualesquiera racematos resultantes de productos finales o intermedios se pueden resolver en los enantiómeros ópticos mediante métodos conocidos, por ejemplo, mediante la separación de las sales diastereoméricas de estos, obtenidas con un ácido o una base con actividad óptica, y liberando el compuesto ácido o básico con actividad óptica. En particular, se puede emplear, por tanto, un resto básico para resolver los compuestos de la presente invención, como se definen en la presente, en sus enantiómeros ópticos, por ejemplo, mediante la cristalización fraccionada de una sal formada con un ácido con actividad óptica, por ejemplo, ácido tartárico, ácido dibenzoiltartárico, ácido diacetiltartárico, ácido di-O,O'-p-toluoiltartárico, ácido mandélico, ácido málico o ácido canfor-10-sulfónico. Los productos racémicos también se pueden resolver mediante cromatografía quiral, por ejemplo, cromatografía de líquidos de alta resolución (HPLC) utilizando un adsorbente quiral.
Los compuestos de la presente invención, como se definen en la presente, se obtienen en forma libre o como una sal de estos.
Cuando están presentes tanto un grupo básico como un grupo ácido en la misma molécula, los compuestos de la presente invención, como se definen en la presente, también pueden formar sales internas, por ejemplo, moléculas zwitteriónicas.
Además, los compuestos de la presente invención, como se definen en la presente, incluidas sus sales, también se pueden obtener en forma de sus hidratos, o incluir otros disolventes utilizados para su cristalización. Los compuestos de la presente invención pueden formar de forma inherente o por diseño solvatos con disolventes farmacéuticamente aceptables (que incluye el agua); por lo tanto, se pretende que la invención abarque tanto las formas solvatadas como no solvatadas. El término “solvato” se refiere a un complejo molecular de un compuesto de la presente invención (que incluyen las sales farmacéuticas de este) con una o más moléculas de disolvente. Tales moléculas de disolvente son aquellas que se utilizan normalmente en el campo farmacéutico, las cuales se sabe que son inocuas para el receptor, por ejemplo, agua, etanol y similares. El término “hidrato” se refiere al complejo en el que la molécula de disolvente es agua.
Los compuestos de la presente invención, como se definen en la presente, que incluyen sales, hidratos y solvatos de estos, pueden formar de forma inherente o por diseño polimorfos.
La invención incluye además cualquier variante de los procesos de la presente, en la que un producto intermedio obtenible en cualquier etapa de estos se utiliza como material de partida y se llevan a cabo los pasos restantes, o en la que los materiales de partida se forman in situ en las condiciones de reacción, o en la que los componentes de reacción se utilizan en forma de sus sales o material ópticamente puro.
Los compuestos de la invención e intermedios también se pueden interconvertir entre sí de acuerdo con métodos conocidos generalmente por los expertos en la técnica.
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de la presente invención, como se definen en la presente, o una sal farmacéuticamente aceptable de este y un portador farmacéuticamente aceptable. La composición farmacéutica se puede formular para vías particulares de administración tales como la administración oral, administración parenteral y administración rectal, etc. Además, las composiciones farmacéuticas de la presente invención se pueden preparar en una forma sólida (que incluye, sin carácter limitante, cápsulas, comprimidos, pastillas, gránulos, polvos o supositorios) o en una forma líquida (que incluye, sin carácter limitante, soluciones, suspensiones o emulsiones). Las composiciones farmacéuticas se pueden someter a operaciones farmacéuticas convencionales tales como esterilización y/o pueden contener agentes tamponantes, agentes lubricantes o diluyentes inertes convencionales, así como también adyuvantes, tales como conservantes, estabilizantes, agentes humectantes, emulsionantes y tampones, etc.
Normalmente, las composiciones farmacéuticas son comprimidos o cápsulas de gelatina que comprenden el principio activo junto con
diluyentes, por ejemplo, lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa y/o glicina;
lubricantes, por ejemplo, sílice, talco, ácido esteárico, su sal de magnesio o de calcio y/o polietilenglicol; para comprimidos también
aglutinantes, por ejemplo, silicato de aluminio y magnesio, pasta de almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona; si se desea
desintegrantes, por ejemplo, almidones, agar, ácido algínico o su sal de sodio, o mezclas efervescentes; y/o
absorbentes, colorantes, sabores y edulcorantes.
Los comprimidos pueden tener ya sea un recubrimiento de tipo película o recubrimiento entérico de acuerdo con métodos conocidos en la técnica.
Las composiciones adecuadas para la administración oral incluyen una cantidad eficaz de un compuesto de la invención, como se define en la presente, en forma de comprimidos, pastillas para chupar, suspensiones acuosas u oleosas, polvos o gránulos dispersables, emulsión, cápsulas duras o blandas, o jarabes o elixires. Las composiciones destinadas al uso oral se preparan de acuerdo con cualquier método conocido en la técnica para la fabricación de composiciones farmacéuticas y tales composiciones pueden contener uno o más agentes seleccionados a partir del grupo que consiste en agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes con el fin de proporcionar preparados de sabor agradable y farmacéuticamente elegantes. Los comprimidos pueden contener el principio activo mezclado con excipientes farmacéuticamente aceptables atóxicos que son adecuados para la fabricación de comprimidos. Estos excipientes son, por ejemplo, diluyentes inertes, tales como carbonado de calcio, carbonato de sodio, lactosa, fosfato de calcio o fosfato de sodio; agentes granulantes y desintegrantes, por ejemplo, almidón de maíz o ácido algínico; agentes aglutinantes, por ejemplo, almidón, gelatina o goma arábiga; y agentes lubricantes, por ejemplo, estearato de magnesio, ácido esteárico o talco. Los comprimidos pueden estar no recubiertos o recubrirse mediante técnicas conocidas para retrasar la desintegración y la absorción en el tubo gastrointestinal, y de este modo proporcionar una acción sostenida durante un periodo más prolongado. Por ejemplo, se puede utilizar un material retardante tal como monoestearato de glicerilo o diestearato de glicerilo. Las formulaciones para el uso oral se pueden presentar como cápsulas de gelatina duras en las que el principio activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín, o como cápsulas de gelatina blandas en las que el principio activo se mezcla con agua o un medio oleoso, por ejemplo, aceite de cacahuete, parafina líquida o aceite de oliva.
Ciertas composiciones inyectables son soluciones o suspensiones isotónicas acuosas y los supositorios se preparan convenientemente a partir de emulsiones o suspensiones grasas. Dichas composiciones pueden estar esterilizadas y/o contener adyuvantes, tales como agentes conservantes, estabilizantes, humectantes o emulsionantes, promotores de la disolución, sales para regular la presión osmótica y/o tampones. Además, también pueden contener otras sustancias terapéuticamente valiosas. Dichas composiciones se preparan de acuerdo con métodos de mezcla, granulación o recubrimiento convencionales, respectivamente, y contienen aproximadamente un 0,1-75 % o contienen aproximadamente un 1-50 % del principio activo.
Las composiciones adecuadas para la aplicación transdérmica incluyen una cantidad eficaz de un compuesto de la invención con un portador adecuado. Los portadores adecuados para el suministro transdérmico incluyen disolventes absorbibles farmacológicamente aceptables para ayudar en el paso a través de la piel del hospedador. Por ejemplo, los dispositivos transdérmicos están en forma de un apósito que comprende un miembro de soporte, un depósito que contiene el compuesto opcionalmente con portadores, opcionalmente una barrera controladora de la velocidad para suministrar el compuesto a la piel del hospedador a una velocidad controlada y predeterminada durante un período de tiempo prolongado, y medios para asegurar el dispositivo a la piel.
Las composiciones adecuadas para la aplicación tópica, por ejemplo, a la piel y los ojos, incluyen suspensiones, soluciones acuosas, pomadas, cremas, geles o formulaciones pulverizables, por ejemplo, para el suministro en forma de aerosol o similares. Tales sistemas de suministro tópico serán apropiados en particular para la aplicación dérmica, por ejemplo, para el tratamiento del cáncer de piel, por ejemplo, para uso profiláctico en pulverizados, lociones, cremas solares y similares. Por lo tanto, son especialmente adecuados para su uso en formulaciones tópicas, incluidas las cosméticas, muy conocidas en la técnica. Estas pueden contener solubilizantes, estabilizantes, agentes potenciadores de la tonicidad, tampones y conservantes.
Tal como se utiliza en la presente, una aplicación tópica también se puede referir a la inhalación o a una aplicación intranasal. Se pueden suministrar convenientemente en forma de un polvo seco (ya sea solo, como una mezcla, por ejemplo, una mezcla seca con lactosa, o una partícula de componentes mezclados, por ejemplo, con fosfolípidos) a partir de un inhalador de polvo seco o una presentación de pulverizado en aerosol a partir de un recipiente presurizado, bomba, pulverizador, atomizador o nebulizador, con o sin el uso de un propulsor adecuado.
La presente invención proporciona además composiciones farmacéuticas y formas farmacéuticas anhidras que comprenden los compuestos de la presente invención como se definen en la presente, como principios activos, ya que el agua puede propiciar la degradación de ciertos compuestos.
Las composiciones farmacéuticas y formas farmacéuticas anhidras de la invención se pueden preparar utilizando ingredientes anhidros o con un contenido de humedad bajo y condiciones de humedad baja o humectación baja. Una composición farmacéutica anhidra se puede preparar y almacenar de modo que se mantenga su naturaleza anhidra. Por consiguiente, las composiciones anhidras se envasan utilizando materiales de los que se sabe que previenen la exposición al agua} de tal modo que se puedan incluir en kits de formulación adecuados. Los ejemplos de envases adecuados incluyen, sin carácter limitante, láminas de aluminio, plásticos, recipientes de dosis unitaria (por ejemplo, viales), envases blíster y envases con ampollas de papel de aluminio (strip packs) sellados herméticamente.
La invención proporciona además composiciones farmacéuticas y formas farmacéuticas que comprenden uno o más agentes que reducen la velocidad a la que se descompondrá el compuesto de la presente invención como principio activo. Tales agentes, que se denominan en la presente “estabilizantes”, incluyen, sin carácter limitante, antioxidantes tales como ácido ascórbico, tampones de pH o tampones salinos, etc.
Los compuestos de fórmula (I), como se definen en la presente, en forma libre o en forma de sal, presentan propiedades farmacológicas valiosas, por ejemplo, propiedades moduladoras de la producción de la proteína SMN completa, por ejemplo, como se indica en las pruebas in vitro e in vivo como las proporcionadas en las siguientes secciones, y, por tanto, están indicados para la terapia o para su uso como productos químicos de investigación, por ejemplo, como compuestos de referencia.
Por lo tanto, como una realización adicional, la presente invención proporciona el uso de un compuesto de fórmula (I), o una sal de este, como se define en la presente, para su uso en la terapia de una afección relacionada con la deficiencia de SMN. En una realización adicional, la terapia se selecciona a partir de una enfermedad que se puede tratar modulando la producción de la proteína SMN completa. En otra realización, la enfermedad se selecciona a partir de la lista mencionada anteriormente, convenientemente atrofia muscular espinal.
En otra realización, la invención proporciona un compuesto de fórmula (I), o una sal de este, como se define en la presente, para su uso en el tratamiento de una enfermedad que se trata modulando la producción de la proteína SMN completa que comprende la administración de una cantidad terapéuticamente aceptable de un compuesto de fórmula (I) o una sal de este a un paciente que necesita una terapia de este tipo. En una realización adicional, la enfermedad se selecciona a partir de la lista mencionada anteriormente, convenientemente atrofia muscular espinal.
Por lo tanto, como una realización adicional, la presente invención proporciona el uso de un compuesto de fórmula (I) o una sal de este para la fabricación de un medicamento. En una realización adicional, el medicamento es para el tratamiento de una enfermedad que se puede tratar mediante la modulación de la producción de la proteína s Mn . En otra realización, la enfermedad se selecciona a partir de la lista mencionada anteriormente, convenientemente atrofia muscular espinal.
La composición o combinación farmacéutica de la presente invención puede estar en una dosificación unitaria de aproximadamente 0,01-1000 mg de principio(s) activo(s) para un sujeto de aproximadamente .05-70 kg, o aproximadamente 1-20 kg, o aproximadamente 1-500 mg o aproximadamente 1-250 mg o aproximadamente 1-150 mg o aproximadamente 0,5-100 mg, o aproximadamente 0,01-1 mg o aproximadamente 0,01-0,1 mg o aproximadamente 1-50 mg de principios activos. La dosificación terapéuticamente eficaz de un compuesto, la composición farmacéutica o sus combinaciones depende de la especie del sujeto, el peso corporal, la edad y el estado del individuo, el trastorno o enfermedad que se esté tratando o su gravedad. Un médico, facultativo o veterinario experto puede determinar fácilmente la cantidad eficaz de cada uno de los principios activos necesaria para prevenir, tratar o inhibir el avance del trastorno o enfermedad.
Las propiedades posológicas citadas anteriormente se pueden demostrar en pruebas in vitro e in vivo utilizando convenientemente mamíferos, por ejemplo, ratones, ratas, perros, monos u órganos aislados, tejidos y preparados de estos. Los compuestos de la presente invención se pueden aplicar in vitro en forma de soluciones, por ejemplo, soluciones acuosas, e in vivo ya sea por vía enteral, parenteral, convenientemente intravenosa, por ejemplo, como una suspensión o en solución acuosa. La dosificación in vitro puede variar entre concentraciones de aproximadamente 10-3 molar y 10-9 molar. Una cantidad terapéuticamente eficaz in vivo puede variar dependiendo de la vía de administración, entre aproximadamente 0,1-500 mg/kg, o entre aproximadamente 1-100 mg/kg.
El compuesto de la presente invención se puede administrar ya sea de forma simultánea con, o antes o después de, uno o más agentes terapéuticos diferentes. El compuesto de la presente invención, como se define en la presente, se puede administrar por separado, por una vía de administración idéntica o diferente, o de forma conjunta en la misma composición farmacéutica que los otros agentes.
En una realización, la invención proporciona un producto que comprende un compuesto de fórmula (I) y al menos un agente terapéutico diferente como un preparado combinado para el uso simultáneo, separado o secuencial en terapia. En una realización, la terapia es para el tratamiento de una atrofia muscular espinal. Los productos proporcionados como un preparado combinado incluyen una composición que comprende el compuesto de fórmula (I), como se define en la presente, junto con el (los) otro(s) agente(s) terapéutico(s) en la misma composición farmacéutica, o el compuesto de fórmula (I), como se define en la presente, y el (los) otro(s) agente(s) terapéutico(s) en una forma separada, por ejemplo, en forma de un kit.
En una realización, la invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (I) y otro(s) agente(s) terapéutico(s). Opcionalmente, la composición farmacéutica puede comprender un portador farmacéuticamente aceptable, tal como se ha descrito anteriormente.
Se divulga un kit que comprende dos o más composiciones farmacéuticas separadas, donde al menos una de las cuales contiene un compuesto de fórmula (I), como se define en la presente. En una realización, el kit comprende medios para retener por separado dichas composiciones, tales como un recipiente, botella dividida o paquete de aluminio dividido. Un ejemplo de un kit de este tipo es un envase blíster, como el que se utiliza normalmente para el envasado de comprimidos, cápsulas y similares.
El kit se puede utilizar para administrar formas farmacéuticas diferentes, por ejemplo, orales y parenterales, para administrar las composiciones separadas en intervalos posológicos diferentes, o para titular las composiciones separadas unas frente a otras. Para contribuir al cumplimiento, el kit normalmente comprende instrucciones para su administración.
En las terapias combinadas de la invención, el compuesto de la invención y otro agente terapéutico pueden ser fabricados y/o formulados por los mismos fabricantes o por fabricantes diferentes. Es más, el compuesto de la invención y el otro agente terapéutico se pueden combinar en una terapia combinada: (i) antes de poner a disposición de los médicos el producto combinado (por ejemplo, en el caso de un kit que comprende el compuesto de la invención y el otro agente terapéutico); (ii) por el propio médico (o con las directrices del médico) poco antes de la administración; (iii) en el propio paciente, por ejemplo, durante la administración secuencial del compuesto de la invención y el otro agente terapéutico.
Se pretende que los siguientes ejemplos ilustren la invención y no se deben interpretar como que sean limitaciones de esta. Las temperaturas se proporcionan en grados Celsius. Si no se menciona lo contrario, todas las evaporaciones se realizan a presión reducida, normalmente entre aproximadamente 15 mm Hg y 100 mm Hg (= 20-133 mbar). La estructura de los productos finales, los intermedios y los materiales de partida se confirma mediante métodos analíticos estándar, por ejemplo, microanálisis y características espectroscópicas, por ejemplo, MS, IR, RMN. Las abreviaturas utilizadas son las convencionales en la técnica.
Todos los materiales de partida, componentes básicos, reactivos, ácidos, bases, agentes deshidratantes, disolventes y catalizadores utilizados para la síntesis de los compuestos de la presente invención se comercializan o se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la técnica (Houben-Weyl 4.a Ed. 1952, Methods of Organic Synthesis, Thieme, Tomo 21). Además, los compuestos de la presente invención se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la técnica como los que se muestran en los siguientes ejemplos.
Preparación de los compuestos
Se entiende que en la siguiente descripción, se permiten combinaciones de sustituyentes y/o variables de las fórmulas ilustradas únicamente si tales contribuciones dan como resultado compuestos estables.
También apreciarán los expertos en la técnica que en los procesos descritos a continuación puede ser necesario proteger con grupos protectores adecuados los grupos funcionales de los compuestos intermedios. Tales grupos
funcionales incluyen hidroxi, fenil, amino y ácido carboxílico. Los grupos protectores adecuados para hidroxi o fenol incluyen trialquilsililo o diarilalquilsililo (por ejemplo, í-butildimetilsililo, í-butildifeniisililo o trimetilsililo), tetrahidropiranilo, bencilo, bencilo sustituido, metilo y similares. Los grupos protectores adecuados para amino, amidino y guanidino incluyen t-butoxicarbonilo, benciloxicarbonilo y similares. Los grupos protectores adecuados para ácido carboxílico incluyen ésteres de alquilo, arilo o arilalquilo.
Los grupos protectores se pueden añadir o eliminar de acuerdo con técnicas estándar, que son muy conocidas por los expertos en la técnica y según como se describe en la presente. El uso de grupos protectores se describe con detalle en Green, T.W. y P.G.M. Wutz, Protective Groups in Organic Synthesis (1999), 3.a Ed., Wiley. El grupo protector también puede ser una resina polimérica, tal como una resina de Wang o una resina de cloruro de 2-clorotritilo. También apreciarán los expertos en la técnica que, aunque tales derivados protegidos de los compuestos de esta divulgación puede que no posean actividad farmacológica como tales, se pueden administrar a un sujeto y metabolizar posteriormente en el cuerpo para formar los compuestos de la invención que son farmacológicamente activos. Por ende, tales derivados se pueden describir como “profármacos”.
Los siguientes esquemas de reacción ilustran los métodos para preparar compuestos de los ejemplos y ejemplos de referencia. Se entiende que un experto en la técnica sería capaz de preparar estos compuestos mediante métodos similares o mediante métodos conocidos por un experto en la técnica. En general, los reactivos y componentes de partida se pueden obtener de proveedores tales como Sigma Aldrich, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI y Fluorochem USA, Strem, otros distribuidores comerciales, o sintetizar siguiendo referencias conocidas por los expertos en la técnica, o preparar como se describe en esta invención. A, B, X, R, R1, R2, R3, R4, son como se definen en la Memoria descriptiva a menos que se definan específicamente.
En general, los compuestos de piridazina de los ejemplos y ejemplos de referencia se pueden sintetizar siguiendo el procedimiento general descrito en el Esquema 1.
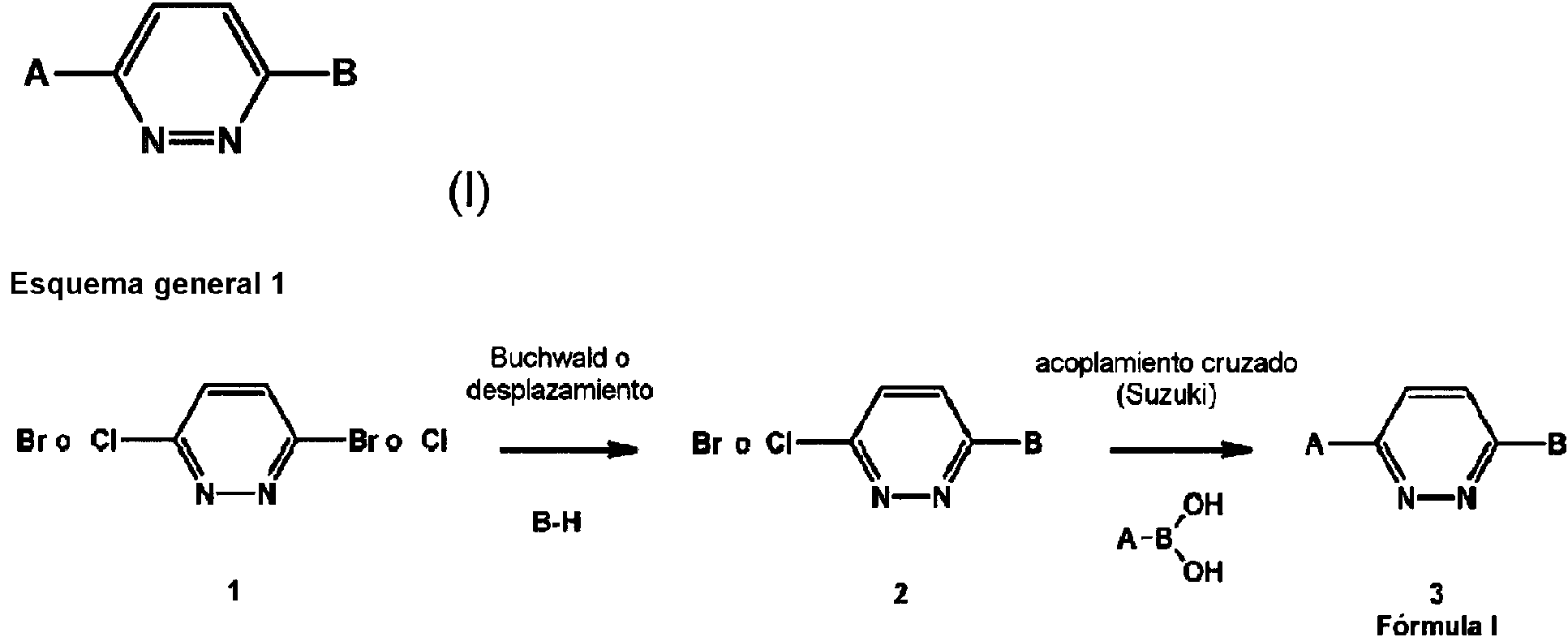
Los materiales de partida para el esquema de reacción anterior están comercializados o se pueden preparar de acuerdo con métodos conocidos por un experto en la técnica o mediante métodos divulgados en la presente. En general, los compuestos de los ejemplos y ejemplos de referencia se preparan en el Esquema 1 de reacción anterior de la siguiente manera:
La dihalopiridazina (1) reacciona en una reacción de desplazamiento o una reacción de acoplamiento cruzado mediada por metales (Buchwald) con un alcohol o una amina (B) para proporcionar el intermedio de tipo piridazina (2). La reacción de acoplamiento cruzada mediada por metales de transición, tal como una reacción de Suzuki, entre un compuesto de tipo haluro (2) y un compuesto arilo o heteroarilo sustituido A, tal como un ácido boronato o éster de boronato, proporciona el compuesto (3) de los ejemplos y ejemplos de referencia.
De una manera complementaria, los compuestos de los ejemplos y ejemplos de referencia se pueden sintetizar siguiendo el procedimiento general descrito en el Esquema 2.
Esquema general 2

Los materiales de partida para el esquema de reacción anterior están comercializados o se pueden preparar de acuerdo con métodos conocidos por un experto en la técnica o mediante métodos divulgados en la presente. En general, los compuestos de los ejemplos y ejemplos de referencia se preparan en el Esquema 2 de reacción anterior de la siguiente manera:
La dihalopiridazina (1) reacciona en una reacción de acoplamiento cruzado mediada por metales de transición, tal como una reacción de Suzuki, con un compuesto arilo o heteroarilo sustituido A , tal como un ácido o éster de boronato, para proporcionar el intermedio de tipo piridazina (4). El intermedio de tipo piridazina (4) reacciona mediante una reacción de desplazamiento con un alcohol o una amina (B) para proporcionar la piridazina (3) de los ejemplos y ejemplos de referencia.
Los compuestos de fórmula (I) también se pueden preparar siguiendo el procedimiento general descrito en el Esquema 3.
Esquema general 3

Los materiales de partida para el esquema de reacción anterior están comercializados o se pueden preparar de acuerdo con métodos conocidos por un experto en la técnica o mediante métodos divulgados en la presente. En general, los compuestos de los ejemplos y ejemplos de referencia se preparan en el Esquema 3 de reacción anterior de la siguiente manera:
La dihalopiridazina (1) reacciona en una reacción de acoplamiento cruzado mediada por metales de transición, tal como una reacción de Suzuki, con un compuesto arilo o heteroarilo sustituido A , tal como un ácido o éster de boronato, para proporcionar el intermedio de tipo piridazina (4). El intermedio de tipo piridazina (4) reacciona mediante una segunda reacción de acoplamiento cruzado mediada por metales, tal como una reacción de Suzuki, para proporcionar la piridazina (3) de los ejemplos y ejemplos de referencia de la invención.
Diversos grupos aromáticos A , tales como fenoles, naftilos, heteroarilo sustiuidos y similares, y diversos grupos amina o alcohol B, tales como aminopiperidinas, piperazinas, homopiperazinas, 4-hidroxipiperidinas, y similares, pueden seguir los Esquemas generales 1, 2 y 3 para proporcionar compuestos de los ejemplos y ejemplos de referencia. Se pueden necesitar estrategias de grupos protectores habituales para conseguir los compuestos finales de los ejemplos y ejemplos de referencia.
Todos los materiales de partida, componentes básicos, reactivos, ácidos, bases, agentes deshidratantes, disolventes, catalizadores y depuradores utilizados para sintetizar los compuestos de los ejemplos y ejemplos de referencia están comercializados o se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la técnica. Además, los compuestos de los ejemplos y ejemplos de referencia se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la técnica como los que se muestran en los siguientes ejemplos.
Se pretende que los siguientes ejemplos o ejemplos de referencia ilustren la invención y no se deben interpretar como que sean limitaciones de esta. Las temperaturas se proporcionan en grados centígrados. Si no se menciona lo contrario, todas las evaporaciones se realizan a presión reducida, preferentemente entre aproximadamente 15 mm Hg y 100 mm Hg (= 20-133 mbar). La estructura de los productos finales, los intermedios y los materiales de partida se confirma mediante métodos analíticos estándar, por ejemplo, microanálisis y características espectroscópicas, por
ejemplo, LCMS, MRN, CHN. Las abreviaturas utilizadas son las habituales en la técnica y se proporciona una lista de estas al final de la sección experimental.
PREPARACIÓN 1
Intermedio 1-1: Síntesis de 6-cloro-N-met¡l-N-(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)p¡r¡dazm-3-amma:

A 3,6-dicloropiridazina (4 g, 26,8 mmol) y N,2,2,6,6-pentametilpiperidin-4-amina (7,32 g, 43,0 mmol) en un matraz de fondo redondo de 300 mL se añadió butan-1-ol (67 mL) para obtener una solución incolora. La mezcla se calentó hasta 120 °C durante 72 h. Se eliminó el butan-1-ol utilizando un evaporador giratorio. El residuo se repartió entre agua y DCM, y la capa acuosa se extrajo opcionalmente con DCM. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron con MgSO4 y se concentraron al vacío. El material crudo negro se agitó en una pequeña cantidad de EtOAc durante toda la noche y el sólido blanquecino resultante se recogió para proporcionar el Intermed¡o 1-1 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (4,18 g, 14,78 mmol, 55,0 % de rendimiento). LCMS tR = 0,8 min (condición B), MS (M+1) = 283,5. 1H RMN (400 MHz, METANOL-d4) 5 ppm 7,40 (d, J=9,60 Hz, 1H), 7,14 (d, J=9,60 Hz, 1H), 4,96-5,13 (m, 1H), 2,93 (s, 3H), 1,59-1,68 (m, 2H), 1,51 (t, J=12,38 Hz, 2H), 1,20 (s, 6H), 1,33 (s, 6H).
PREPARACIÓN 2
Intermed¡o 1-2: Síntes¡s de 6-cloro-N-(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)p¡r¡dazm-3-amma

Una mezcla de 3,6-dicloropiridazina (6,26 g, 42 mmol) y 2,2,6,6-tetrametilpiperidin-4-amina (14,7 mL, 84 mmol) se agitó a 120 °C durante 1 h, sin disolvente. A esta mezcla cruda se añadió n-butanol (40 mL) y la reacción se agitó a 120 °C durante 1 h. La mezcla de reacción cruda se enfrió hasta la temperatura ambiente y se diluyó en agua y CH2O 2. La capa orgánica se secó con MgSO4, se filtró y se concentró. El material crudo se recristalizó en CH3CN para obtener el Intermed¡o 1-2 6-cloro-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (7,3 g) como un sólido blanquecino. LCMS tR = 1,10 min (condición B), MS (M+1) = 269,2. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,08 (d, J=9,3 Hz, 1H), 6,54 (d, J=9,3 Hz, 1H), 4,53 (d, J=7,6 Hz, 1H), 4,05-4,26 (m, 1H), 1,98 (dd, J=12,6, 3,8 Hz, 2H), 1,22 (s, 6H), 1,08 (s, 6H), 0,93 (t aparente, J=12,1 Hz, 2H).
PREPARACIÓN 3
Intermed¡o 1-3: Síntes¡s de 3-cloro-6-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡lox¡)p¡r¡daz¡na

A una solución de 2,2,6,6-tetrametilpiperidin-4-ol (106 mg, 0,67 mmol) en DMF (6,7 mL) se añadió NaH al 60 % p (35 mg, 0,87 mmol). La solución se agitó a TA durante 30 min, a continuación se añadió 3,6-dicloropiridazina (100 mg, 0,67 mmol) y la reacción se agitó durante 1 h. La mezcla de reacción cruda se diluyó en EtOAc. La capa orgánica se lavó con agua (5X), salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida para obtener el ¡ntermed¡o 1-3 3-cloro-6-(2,2,6,6-tetrametilpiperidin-4-iloxi)piridazina (135 mg). El material crudo se utilizó sin una purificación adicional. LCMS tR = 1,22 min (condición B); MS (M+1) = 270,2. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,53 (s, 1H), 7,37 (d, J=9,1 Hz, 1H), 6,91 (d, J=9,1 Hz, 1H), 5,68-5,78 (m, 1H), 2,20 (dd, J=12,4, 4,0 Hz, 2H), 1,32 (s, 6H), 1,27-1,29 (m, 2H), 1,20 (s, 6H).
PREPARACIÓN 4
Intermedio 1-4: Síntesis de 6-cloro-N-met¡l-N-(1,2,2,6,6-pentametilp¡per¡dm-4-¡l)μmdazm-3-amma

A una suspensión del Intermedio 1-56-doro-N-(1,2,2,6,6-pentametilp¡peridm-4-¡l)μmdazm-3-amma (4,0 g, 14,1 mmol) en DMF (140 mL) enfriada hasta 0 °C se añadió NaH al 60 % p (735 mg, 18,39 mmol) en porciones. La reacción se calentó hasta TA y se agitó durante 60 minutos. Después de 60 minutos, se añadió yoduro de metilo (0,88 mL, 14,1 mmol), la reacción se agitó durante 3 h más y a continuación se desactivó lentamente con agua a temperatura ambiente. La mezcla de reacción cruda se diluyó en EtOAc. La capa orgánica se lavó con agua (5X), salmuera, se secó con MgSO4 , se filtró y se concentró a presión reducida para obtener el Intermedio 1-4 6 -cloro-N-metil-N-(1,2,2,6,6-pentamet¡lp¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-am¡na (3,98 g). El material crudo se utilizó sin una purificación adicional. LCMS tR = 1,16 min (condición B), MS (M+1) = 297,0. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,10 (d, J=9,6 Hz, 1H), 6,69 (d, J=9,6 Hz, 1H), 4,79-5,00 (m, 1H), 2,86 (s, 3H), 2,22 (s, 3H), 1,61-1,73 (m, 2H), 1,48-1,57 (m, 2H), 1,14 (s, 6H), 1,10 (s, 6H).
Intermedio 1-5: Síntesis de 6-cloro-N-(1,2,2,6,6-pentametilpiperidm-4-N)piridazm-3-amma

Este material se preparó en dos lotes.
Lote 1: Una mezcla de 1,2,2,6,6-pentamet¡lp¡per¡d¡n-4-am¡na (15,1 g, 89,0 mmol) y 3,6-dicloropiridazina (6 , 6 g, 44,3 mmol) se calentó sin disolvente a 120 °C durante 30 min. El material crudo solidificó y se resuspendió en n-butanol (45 mL). La mezcla cruda se agitó a 120 °C durante 2 h más, a continuación se calentó hasta 16 0 °C durante 1 h, a continuación se enfrió y se combinó con el lote 2 para el tratamiento posterior.
Lote 2: Una mezcla de 1,2,2,6,6-pentamet¡lp¡per¡d¡n-4-am¡na (14,2 g, 83 mmol) y 3,6-dicloropiridazina (6,2 g, 41,6 mmol) en n-butanol (10 mL) se calentó a 120 °C durante 120 min. El material crudo solidificó, se resuspendió en nbutanol (15 mL) y se calentó a 120 °C durante 1 h. Este material crudo se combinó con el lote 1 para el tratamiento posterior y purificación. Se añadieron agua y CH2O 2 al material crudo combinado y la capa orgánica se separó, se lavó con agua y salmuera, se secó con MgSO4, se filtró y se concentró. El material crudo se recristalizó en CH3CN (400 mL) para obtener el Intermedio 1-5 6-cloro-N-(1,2,2,6,6-pentamet¡lp¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-am¡na (15,5 g, primera recolección). La recristalización se repitió utilizando CH3CN para obtener una 2.a recolección del Intermedio 1-5 6-cloro-N-(1,2,2,6,6-pentamet¡lp¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-am¡na (3,98 g). LCMS tR = 1,10 min (condición B); MS (M+1) = 283,0. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,15 (d, J=9,3 Hz, 1H), 6,65 (d, J=9,3 Hz, 1H), 4,70 (d, J=7,3 Hz, 1H), 4,08-4,26 (m, 1H), 2,31 (s, 3H), 1,89-2,09 (m, 2H), 1,42 (t aparente, J=12,1 Hz, 2H), 1,21 (s, 6H), 1,15 (s, 6H).
PREPARACIÓN 5
Intermedio 2-1: Síntesis de 3-cloro-6-(2-metoxi-4-(1H-pirazol-1-N)feml)piridazma:

Paso 1: (4-Bromo-3-metoxifenil)hidrazina
Se suspendió 4-bromo-3-metoxianilina (3,0 g, 14,85 mmol) en HCl concentrado (50 mL) y la mezcla se enfrió hasta 0 °C en un baño de hielo-agua. Se añadió muy lentamente una solución de nitrito de sodio (1,23 g, 17,82 mmol) en 10 mL a la mezcla de reacción. La mezcla se volvió amarilla y después marrón con un tono amarillo, lo que indicó diazotación. La sal de diazonio se mantuvo a 0 °C durante una hora y a continuación se añadió muy lentamente una solución de cloruro de estaño (II) dihidratado (10,05 g, 44,5 mmol) en HCl concentrado (20 mL) (cuidado, extremadamente exotérmica). La reacción se agitó durante 2 h a 0 °C y a continuación a TA durante toda la noche. La reacción se filtró y la masa retenida en el filtro se lavó con H2O fría para proporcionar (4-bromo-3-metoxifenil)hidrazina como un sólido tostado (3,1 g, MS: 218 [M+H+]).
Paso 2: 1-(4-Bromo-3-metoxifenil)-1H-pirazol
A una solución de (4-bromo-3-metoxifenil)hidrazina (62 g, 245 mmol) en etanol (310 mL) se añadió tetrametoxipropano (40,2 g, 245 mmol) durante unos pocos minutos y la mezcla se calentó hasta una temperatura interna de 70 °C. La mezcla se agitó a 70 °C durante 1,5 h y a continuación se enfrió lentamente hasta TA. Se eliminó el etanol al vacío y se añadió EtOAc para formar una suspensión espesa del residuo. El residuo se neutralizó con hidróxido de sodio acuoso 1 M (~700 mL) para provocar la precipitación. La mezcla bifásica se filtró y el filtrado se extrajo con EtOAc, se secó con sulfato de sodio y se concentró para proporcionar 30 g de 1-(4-bromo-3-metoxifenil)-1H-pirazol como un sólido negro (30 g, MS: 254 [M+H+].).
Paso 3: 1-(3-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol
Se añadieron 1-(4-bromo-3-metoxifenil)-1 H-pirazol (28,5 g, 113 mmol), bis(pinacolato)diboro (42,9 g, 169 mmol), carbonato de potasio (15,56 g, 113 mmol), y aducto de PdCl2(dppf).CH2Cl2 (9,20 g, 11,26 mmol) a un matraz de fondo redondo de 2 L, y después se añadió dioxano (700 mL). La mezcla de reacción se purgó con N2 y se agitó en N2 a una temp. interna de 84 °C durante toda la noche. La mezcla de reacción se filtró a través de un embudo con un filtro desechable y se concentró sobre gel de sílice. La mezcla se purificó utilizando cromatografía en columna (20 % de EtOAc en heptanos). Las fracciones deseadas se recogieron y concentraron para proporcionar 13,5 g de 1 -(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol.
Paso 4: 3-Cloro-6-(2-metoxi-4-( 1 H-pirazol-1-il)fenil)piridazina
Se introdujeron 3,6-dicloropiridizina (11,99 g, 80 mmol), 1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol (16,1 g, 53,6 mmol), carbonato de sodio (17,06 g, 161 mmol), y aducto de dicloruro de 1,1’-bis(difenilfosfino)ferroceno con diclorometano (3,07 g, 3,75 mmol) en un matraz de 3 bocas de fondo redondo de 250 mL equipado con una barrita de agitación magnética y entrada de nitrógeno. Se añadieron 1,4-dioxano (274 mL) y agua desionizada (46 mL) y en la mezcla de reacción se hizo vació y rellenó con nitrógeno tres veces. La mezcla de reacción se calentó en bloque calefactor de teflón hasta 85 °C durante 16 h. Después de eliminar el 1,4-dioxano al vacío, se añadió acetato de etilo para formar una suspensión espesa del residuo y se filtró a través de un embudo con una placa fritada de vidrio relleno de celite. El filtrado se concentró sobre gel de sílice y se purificó en una columna de gel de sílice de 330 g, eluyendo con un 10-35 % de acetato de etilo en heptanos. Las fracciones que contenían el producto se concentraron hasta un 10 % del volumen, se formó una suspensión espesa en acetato de etilo:heptanos 3:1 (100 mL), se agitó a TA durante 1 h y a continuación se filtró para proporcionar 7 g (46 %) de 3-cloro-6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazina como un sólido. 1H RMN (600 MHz, CLOROFORMO-d) 68,12 (dd, J=8,80, 11,30 Hz, 2H), 8,05 (d, J=1,88 Hz, 1 H), 7,80 (s, 1 H), 7,61 (d, J=1,25 Hz, 1 H), 7,54 (d, J=9,03 Hz, 1 H), 7,35 (dd, J=1,44, 8,22 Hz, 1 H), 6,55 (s, 1 H), 4,00 (s, 3H).
PREPARACIÓN 6
Intermedio 2-2: Síntesis de 2-(6-cloropiridazm-3-il)-5-(1H-pirazoM-il)fenol

Se añadió BCh (1 M en DCM, 91 mL, 91 mmol) a una solución de 3-cloro-6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazina (Intermedio 2-1, 8,6 g, 30 mmol) en DCM (150 mL) a 0 °C y la reacción se agitó durante 5 h a TA. Se añadió MeOH (50 mL) a la reacción a 0 °C, a continuación la reacción se calentó hasta TA y el disolvente se evaporó a presión reducida. El material crudo se trató con CH3CN caliente y a continuación se enfrió hasta 5 °C. La mezcla se filtró y se obtuvo el Intermedio 2-2 2-(6-cloropiridazin-3-il)-5-(1H-pirazol-1-il)fenol (7,6 g, 86 %) como un sólido amarillo. MS [M+H]: 273,2; 1H RMN (400 MHz, DMSO-d6) 6 11,95 (s, 1 H), 8,59 (d, J=2,0 Hz, 1 H), 8,49 (d, J=9,0 Hz, 1H), 8,09 (d,
J=8,5 Hz, 1H), 8,04 (d, J=9,0 Hz, 1H), 7,80 (d, J=2,0 Hz, 1H), 7,56 (d, J=2,0 Hz, 1H), 7,51 (dd, J=8,5, 2,0 Hz, 1H), 6,59 (t, J=2,0 Hz, 1H).
PREPARACIÓN 7
Intermedio 3-1: Síntesis de 7-(benciloxi)-6-(4,4,5,5-tetrametiM,3,2-dioxaborolan-2-il)naftalen-2-ol

Se añadieron bis(pinacolato)diboro (3,13 g, 12,33 mmol), KOAc (3,63 g, 37,0 mmol), y aducto de PdCl2(dppf).CH2Cl2 (0,504 g, 0,617 mmol) a un matraz de 250 mL que contenía 7-(benciloxi)-6-bromonaftalen-2-ol (2,03 g, 6,17 mmol). A continuación se añadió DMSO (30,8 mL) y se incorporó un condensador de reflujo. En la mezcla de reacción se hizo vacío, después se rellenó con N2 (2X) y después se calentó a 100 °C durante toda la noche. La mezcla de reacción se enfrió hasta la temperatura ambiente, se filtró a través de celite (embudo con filtro preempaquetado) utilizando EtOAc y se concentró al vacío para obtener un aceite crudo. La cromatografía ultrarrápida, eluyendo con un 5-30 % de EtOAc/heptano, proporcionó el producto, 7-(benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)naftalen-2-ol, (58 % de rendimiento) como un aceite incoloro. LCMS tR = 1,72 min (condición C), MS (M+1) = 377,6. 1H RMN (400 MHz, METANOL-d4) 5ppm 8,07 (s, 1H), 7,68 (t, J=8,91 Hz, 3H), 7,36-7,43 (m, 2H), 7,28-7,34 (m, 1H), 7,09 (s, 1H), 7,02 (d, J=2,51 Hz, 1H), 6,92 (dd, J=8,78, 2,51 Hz, 1H), 5,21 (s, 2H), 1,40 (s, 12H).
PREPARACIÓN 8
Intermedio 4-1: Síntesis de (6S)-6-((S)-1-(ferf-butNdimetilsMMoxi)etil)-2,2-dimetilpiperidm-4-ol

Paso 1. (S)-2-(tert-Butildimetilsililoxi)propanoato de etilo
A una solución de (S)-lactato de etilo (11,8 g, 100 mmol) en DMF (50 mL) se añadió imidazol (10,2 g, 150 mmol). La mezcla se enfrió en un baño de hielo y se añadió cloruro de tert-butildimetilsililo (15,8 g, 105 mmol) en tres porciones, con intervalos de 30 min entre cada adición. La mezcla de reacción se agitó durante toda la noche. La mezcla de reacción se diluyó con agua (30 mL) y se extrajo con Et2O (50 mL x 2). Los extractos combinados se lavaron con salmuera, se secaron con MgSO4 y se concentraron al vacío. El residuo se destiló al vacío (p.e. 70-78 °C, 0,5 mmHg) para proporcionar 22,07 g (95 %) de (S)-2-(tert-butildimetilsililoxi)propanoato de etilo: 1H RMN (400 MHz, CLOROFORMO-d) 54,32 (c, J=6,6 Hz, 1H), 4,11-4,25 (m, 2H), 1,40 (d, J=6,6 Hz, 3H), 1,29 (t, J=7,1 Hz, 3H), 0,93 (s, 9H), 0,12 (s, 3H), 0,09 (s, 3H).
Paso 2. (S)-2-(tert-Butildimetilsililoxi)propanal
A una solución de (S)-2-(tert-butildimetilsililoxi)propanoato de etilo (6,3 g, 27,1 mmol) en CH2O 2 (22 mL) se añadieron 54,2 mL de DIBAL (1,0 M en CH2Cl2 , 54,2 mmol) durante 20 min a -78 °C. Después de agitar durante 2 h a -78 °C, se añadió metanol (3 mL) a la solución a la misma temperatura. Se permitió que la mezcla se calentara hasta TA y se añadió tartrato de potasio y sodio acuoso (60 mL) a la solución. La mezcla resultante se agitó vigorosamente durante 3 h. La mezcla se extrajo con diclorometano (30 mL x 2), y los estratos combinados se lavaron con salmuera (20 mL), se secaron con sulfato de sodio y se concentraron al vacío. El residuo se destiló al vacío (p.e. 50-52 °C, 0,5 mmHg) para proporcionar 2,5 g (49 %) de (S)-2-(tert-butildimetilsililoxi)propanal: 1H RMN (400 MHz, CLOROFORMO-d) 59,59 (d, J=1,3 Hz, 1H), 4,07 (dq, J=6,9, 1,3 Hz, 1H), 1,26 (d, J=6,9 Hz, 3H), 0,93 (s, 9H), 0,08 (s, 3H), 0,07 (s, 3H).
Paso 3. (S,E)-N-(2-(tert-Butildimetilsililoxi)propilideno)-1-(4-metoxifenil)metanamina
A una solución de (S)-2-(tert-butildimetilsililoxi)propanal (2,07 g, 11,0 mmol) en diclorometano (60 mL) se añadió (4-metoxifenil)metanamina (1,51 g, 11,0 mmol) y MgSO4 (3,97 g, 33,0 mmol). Después de agitar durante toda la noche, la mezcla se filtró a través de celite y se lavó con diclorometano. El disolvente se eliminó a presión reducida tras lo
cual se obtuvieron 3,38 g (100 %) de (S,£)-N-(2-(tert-but¡ld¡met¡ls¡l¡lox¡)prop¡l¡deno)-1-(4-metox¡fen¡l)metanam¡na como un ace¡te amar¡llo pál¡do que se ut¡l¡zó en el s¡gu¡ente paso s¡n pur¡f¡cac¡ón: 1H RMN (400 MHz,CLOROFORMO-d) 5 7,56 (d, J=5,1 Hz, 1H), 7,09 (d, J=8,6 Hz, 2H), 6,80 (d, J=8,6 Hz, 2H), 4,44 (s, 2H), 4,24-4,34 (m, 1 H), 3,73 (s, 3H), 1,23 (d, J=6,6 Hz, 3H), 0,82 (s, 9H), 0,00 (s, 3H), -0,02 (s, 3H).
Paso 4. (S)-6-((S)-1-Hidroxietil)-1-(4-metoxibencil)-2,2-dimetilpiperidin-4-ona
A una soluc¡ón de (S,£)-N-(2-(terf-but¡ld¡met¡ls¡l¡lox¡)prop¡l¡deno)-1-(4-metox¡fen¡l)metanam¡na (3,38 g, 11 mmol) en d¡clorometano (90 mL) se añad¡eron TMSOTf (2,2 mL, 12,1 mmol) y tert-but¡ld¡met¡l(4-met¡lpenta-1,3-d¡en-2-¡lox¡)s¡lano (9,35 g, 44 mmol) a 0 °C. Después de ag¡tar durante 2 días a 0 °C, la mezcla de reacc¡ón se vert¡ó sobre una soluc¡ón de NaHCO3 ac. (100 mL), y después se extrajo con d¡clorometano (100 mL x 2). Las capas orgán¡cas comb¡nadas se secaron con MgSO4 y se evaporó el d¡solvente. El res¡duo se d¡solv¡ó en THF (60 mL) y a cont¡nuac¡ón se añad¡eron 41,8 mL de TBAF (1 M en THF, 41,8 mmol) a la soluc¡ón. La mezcla se ag¡tó durante 12 h, se desact¡vó con agua (100 mL), se extrajo con d¡clorometano (100 mL x 2) y los extractos comb¡nados se secaron con Na2SO4. Después de concentrar al vacío el d¡solvente, el producto se pur¡f¡có med¡ante cromatografía en columna (Et2O/Heptano) para obtener 1,3 g (41 %) de (S)-6-((S)-1-h¡drox¡et¡l)-1-(4-metox¡benc¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ona y 1,0 g (31 %) de (R)-6-((S)-1-h¡drox¡et¡l)-1-(4-metox¡benc¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ona.
(S)-6-((S)-1-Hidroxietil)-1-(4-metoxibencN)-2,2-dimetilpiperidm-4-ona: LCMS (m/z, MH+): 292,4; 1H RMN (400 MHz, CLOROFORMO-d) 57,13 (d, J=8,6 Hz, 2H), 6,74 (d, J=8,6 Hz, 2H), 3,84 (d, J=15,2 Hz, 1H), 3,63-3,70 (m, 1H), 3,65 (s, 3H), 3,56 (d, J=15,2 Hz, 1H), 3,36 (qd, J=8,5, 5,9 Hz, 1H), 2,93 (dt, J=8,2, 4,8 Hz, 1H), 2,40 (d, J=15,2 Hz, 1H), 2,29-2,36 (m, 1H), 2,08-2,17 (m, 2H), 1,17 (s, 3H), 1,14 (s, 3H), 0,81 (d, J=6,1 Hz, 3H).
(R)-6-((S)-1-Hidroxietil)-1-(4-metoxibencN)-2,2-dimetilpiperidm-4-ona: LCMS (m/z, MH+): 292,4; 1H RMN (400 MHz, CLOROFORMO-d) 57,30 (d, J=8,1 Hz, 2H), 6,84 (d, J=8,1 Hz, 2H), 4,09 (d, J=16,7 Hz, 1H), 3,82-3,92 (m, 1H), 3,74 (s, 3H), 3,26 (d, J=16,7 Hz, 1H), 2,71-2,78 (m, 1H), 2,55-2,66 (m, 2H), 2,23 (ddd, J=14,7, 4,5, 2,5 Hz, 1H), 2,15 (dd, J=13,6, 2,5 Hz, 1H), 1,70 (a., s, 1H), 1,29 (s, 3H), 1,00 (s, 3H), 0,89 (d, J=6,6 Hz, 3H).
Paso 5. (S)-6-((S)-1-Hidroxietil)-2,2-dimetilpiperidin-4-ona
A una soluc¡ón de (S)-6-((S)-1-h¡drox¡et¡l)-1-(4-metox¡benc¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ona (0,28 g, 0,96 mmol) en MeOH (40 mL) se añad¡ó ác¡do acét¡co (0,055 mL, 0,96 mmol) e h¡dróx¡do de palad¡o (0,13 g, 0,96 mmol). Después de desgas¡f¡car, la mezcla se ag¡tó durante toda la noche con h¡drógeno. La mezcla se f¡ltró a través de cel¡te, se lavó con MeOH (20 mL) y se concentró al vacío para obtener 0,11 g (67 %) de (S)-6-((S)-1-h¡drox¡et¡l)-2,2-d¡met¡lp¡ per¡d¡ n-4-ona que se ut¡l¡zó en el s¡gu¡ente paso s¡n pur¡f¡cac¡ón.
Paso 6. (S)-6-((S)-1-(tert-Butildimetilsililoxi)etil)-2,2-dimetilpiperidin-4-ona
A una soluc¡ón de (S)-6-((S)-1-h¡drox¡et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ona (0,11 g, 0,64 mmol) en DMF (3 mL) se añad¡eron ¡m¡dazol (0,13 g, 1,93 mmol) y TBSCl (0,14 g, 0,96 mmol). La mezcla se ag¡tó durante toda la noche. La mezcla de reacc¡ón se d¡luyó con agua (10 mL) y se extrajo con EtOAc (20 mL). El extracto se lavó con salmuera, se secó con Na2SO4 y se concentró al vacío. El res¡duo se pur¡f¡có med¡ante cromatografía en columna (EtOAc/Heptano) para proporc¡onar 65 mg (35 %) de (S)-6-((S)-1-(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡ per¡d¡ n-4-ona: 1H RMN (400 Mh z , CLOROFORMO-d) 53,99 (dq, J=6,3, 2,8 Hz, 1H), 2,96-3,04 (m, 1H), 2,21-2,32 (m, 3 H), 2,13 (d, J=13,1 Hz, 1H), 1,26 (s, 3H), 1,12 (d, J=6,1 Hz, 3H), 1,05 (s, 3H), 0,91 (s, 9H), 0,12 (s, 3H), 0,09 (s, 3H).
Paso 7. (6S)-6-((S)-1-(tert-Butildimetilsililoxi)etil)-2,2-dimetilpiperidin-4-ol
A una soluc¡ón de (S)-6-((S)-1 -(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ona (42 mg, 0,15 mmol) en MeOH (3 mL) se añad¡ó NaBH4 (5,6 mg, 0,15 mmol) a 0 °C. Después de que la mezcla se ag¡tara durante 1 h, la mezcla se d¡luyó con agua (5 mL) y se extrajo con EtOAc (20 mL). El extracto se lavó con salmuera, se secó con Na2SO4 y se concentró al vacío. La pur¡f¡cac¡ón med¡ante cromatografía en columna (EtOAc/Heptano) proporc¡onó 32 mg (76 %) de (6S)-6-((S)-1-(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ol (una mezcla 5:1 de d¡astereómeros): 1H RMN (400 MHz, CLOROFORMO-d) 54,17-4,27 (m, 1H), 3,71-3,78 (m, 1H), 2,92-3,02 (m, 1H), 1,56-1,67 (m, 2H), 1,30-1,37 (m, 2H), 1,25 (s, 3H), 1,07 (d, J=6,1 Hz, 3H), 1,02 (s, 3H), 0,83 (s, 9 H), 0,01 (s, 3H), 0,00 (s, 3H).
MÉTODO GENERAL 1-1
Procedimiento representativo para el acoplamiento cruzado de Suzuki (calentamiento convencional)
Se añad¡eron el éster borón¡co (2 equ¡valentes), Na2CO3 (3 equ¡valentes) y Pd(PPh3)4 (0,1 equ¡valentes) a un v¡al que contenía la clorop¡r¡daz¡na (1 equ¡valente). Se añad¡eron DME (0,2 M) y H2O (0,8 M) y en la mezcla de reacc¡ón se h¡zo vacío y se rellenó con N2 (2X). La reacc¡ón se calentó a 90 °C durante 18 h, se enfr¡ó hasta TA y a cont¡nuac¡ón se f¡ltró a través de cel¡te (embudo preempaquetado) con un lavado con MeOH. El f¡ltrado se ac¡d¡f¡có hasta pH 3 ut¡l¡zando HCl 1 M y a cont¡nuac¡ón se adsorb¡ó sobre una columna de SCX acond¡c¡onada con MeOH. La columna
se lavó varias veces (5-7 volúmenes de columna) con MeOH y a continuación se eluyó con NH3 2 N en MeOH. El residuo se purificó mediante cromatografía en gel de sílice para proporcionar el producto deseado.
MÉTODO GENERAL 1-2
Procedimiento representativo para el acoplamiento de Suzuki
Se añadió SiliaCat® DPP-Pd (0,05 equivalentes) a un vial apto para microondas que contenía una mezcla del intermedio de tipo cloropiridazina, tal como 1-1 (1 equivalente), ácido borónico (1,6 equivalentes) y Na2CO3 (3 equivalentes) en EtOH húmedo (0,2 M). La mezcla de reacción se selló, a continuación se calentó mediante irradiación de microondas a 130 °C durante 35 min. La mezcla de reacción se filtró a través de un pequeño tapón de celite con un lavado con MeOH/DCM y a continuación se concentró a sequedad al vacío. El residuo marrón resultante se repartió entre un 5 % de MeOH/DCM y una solución sat. ac. de NaHCO3. Después de la separación, la capa acuosa se extrajo con un 5 % de MeOH/DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con MgSO4, se filtraron y se concentraron a sequedad al vacío. El residuo resultante se disolvió en MeOH y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces con MeOH y a continuación se eluyó con NH3 3 N/MeOH. La evaporación del disolvente proporcionó el producto crudo. El producto crudo se disolvió en MeOH/DCM, a continuación se añadieron 3 g de siliabond® DMT (captador de paladio) y la mezcla se agitó a TA durante 1 h. La mezcla se filtró y a continuación se concentró a sequedad al vacío para proporcionar el producto deseado.
MÉTODO GENERAL 1-3
Procedimiento representativo para el acoplamiento de Suzuki
Se añadieron a un vial apto para microondas el intermedio de tipo cloropiridazina, tal como 1-1 (1 equivalente), reactivo de tipo ácido borónico (2 equivalentes) y Na2CO3 (3 equivalentes). A continuación se añadió Pd(PPh3)4 (0,1 equivalentes) a la mezcla de reacción y después se añadió DME (0,2 M) y H2O (0,8 M). La mezcla de reacción se selló, a continuación se hizo vacío, se rellenó con N2 (2X) y se calentó mediante irradiación de microondas a 125 °C durante 30 min. La mezcla de reacción se filtró a través de celite y se lavó con DCM. El filtrado resultante se lavó con una solución ac. de Na2CO31 M. La capa orgánica se secó con MgSO4, se filtró y se concentró para proporcionar el producto crudo. El producto crudo se purificó mediante cromatografía ultrarrápida para proporcionar el producto como un sólido blanquecino.
MÉTODO GENERAL 1-4
Procedimiento representativo para el acoplamiento de Suzuki
Se añadieron a un vial apto para microondas de 5 mL éster borónico (0,36 mmol), intermedio de tipo piridazina (0,36 mmol), Na2CO3 (1,08 mmol), DME (0,58 mL) y H2O (0,14 mL). El vial se desgasificó durante 5 min con N2 y a continuación se añadió el aducto PdCh(dppf)CH2Cl2 (29,4 mg, 0,04 mmol). La mezcla de reacción se calentó mediante irradiación de microondas a 120 °C durante 45 min. La mezcla cruda se diluyó con EtOAc, se filtró a través de celite y a continuación se concentró al vacío. La cromatografía ultrarrápida, eluyendo con un 0-100 % de EtOAc/heptano, proporcionó el producto.
MÉTODO GENERAL 2-1
Procedimiento representativo para la formación del éster de boronato.
Se añadieron bis(pinacolato)diboro (12,3 mmol), KOAc (37,0 mmol) y PdCh(dppf).CH2Cl2 (0,617 mmol) a un matraz de fondo redondo de 250 mL que contenía un bromuro de arilo (6,17 mmol). A continuación se añadió DMSO (31 mL) y se incorporó un condensador de reflujo. En la mezcla de reacción se hizo vacío, después se rellenó con N2 (2X) y después se calentó a 100 °C durante toda la noche. La mezcla de reacción se enfrió hasta TA, se filtró a través de celite (embudo con filtro preempaquetado) utilizando EtOAc y se concentró al vacío para obtener un aceite crudo. La cromatografía ultrarrápida, eluyendo con un 5-30 % de EtOAc/heptano, proporcionó el producto.
MÉTODO GENERAL 3-1
Procedimiento representativo para la desprotección de metoxi (tiofenol)
Se añadieron tiofenol (1 equivalente) y carbonato de potasio (1 equivalente) al sustrato de tipo metoxi en NMP (0,2 equivalentes) y la reacción se agitó a 190 °C durante 15 min en un reactor de microondas Biotage® Initiator. La mezcla de reacción se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (2 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). Después de la evaporación, el material se purificó mediante HPLC de fase inversa.
MÉTODO GENERAL 3-2
Procedimiento representativo para la desprotección de metoxi (BBr3)
El sustrato de tipo metoxi (1 equivalente) se disolvió en CH2CI2 (0,03 M) y se enfrió en un baño de hielo. Se añadió gota a gota una solución 1 M de BBr3 en CH2Cl2 (3 equivalentes). La mezcla de reacción cruda se agitó a TA durante toda la noche y a continuación se diluyó con CH2O 2 y agua. La capa orgánica se diluyó en EtOAc y se lavó con NaHCO3 ac. sat. (2X), agua, salmuera y se secó con Na2SO4. El producto crudo se purificó mediante HPLC, cromatografía en columna o recristalización.
MÉTODO GENERAL 3-3
Procedimiento representativo para la desprotección de metoxi (Lil, colidina)
A una solución del sustrato de tipo metoxi (1 equivalente) en 2,4,6-colidina (0,03 M, secada con MgSO4 y filtrada), se añadió Lil anhidro (9 equivalentes). La reacción se agitó a 170 °C durante 4 h, a continuación se enfrió y se diluyó con cantidades pequeñas de MeOH, EtOAc y H2O. La capa orgánica se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El producto crudo se purificó mediante HPLC.
MÉTODO GENERAL 4-1
Procedimiento representativo para la hidrogenólisis del grupo bencilo.
En un matraz de fondo redondo de 25 mL, se añadió Pd/C (1,6 mg, 0,016 mmol) o Pd(OH)2 (2,2 mg, 0,016 mmol) al sustrato protegido con bencilo (75 mg, 0,155 mmol) en EtOH (1,5 mL). Se añadió una solución de HCl 1 M (0,25 mL, 0,25 mmol) y en el recipiente de reacción se hizo vacío. Se burbujeó H2 a través de la solución durante 5 min y a continuación se agitó la reacción en H2 a TA. Después de 18 h, la mezcla de reacción se filtró a través de celite y se lavó con MeOH/DCM. El disolvente se eliminó al vacío y el material crudo se disolvió en MeOH que se purificó mediante HPLC preparativa (10-30 % de ACN/H2O con un 0,1 % de TFA). El residuo se disolvió en MeOH/DCM, se acidificó hasta aprox. pH 3 utilizando HCl 1 M y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces (3-4 volúmenes de columna) con MeOH y a continuación se eluyó con NH32 N/MeOH. La evaporación del disolvente proporcionó el producto deseado.
MÉTODO GENERAL 4-2
Procedimiento representativo para la hidrogenación
A un matraz de fondo redondo de 25 mL que contiene Pd/C al 10 % (0,026 mmol), se añadió el sustrato (0,52 mmol) en MeOH (2,5 mL). Se burbujeó H2 a través de la solución durante 5 min y a continuación se agitó la reacción en H2 a 55 psi a TA. Después de 18 h, la mezcla de reacción se filtró a través de celite y se lavó con MeOH. El disolvente se eliminó al vacío. El aceite resultante se disolvió en MeOH y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces (5-7 volúmenes de columna) con MeOH y a continuación se eluyó con NH32 N en MeOH para proporcionar el producto deseado.
MÉTODO GENERAL 5-1
Procedimiento repersentativo para la alquilación de fenol
Se añadió Cs2CO3 (1,489 mmol) a una solución del fenol (1,489 mmol) en acetona (15 mL) a TA. La mezcla de reacción se agitó durante 5 min y a continuación se añadió de una vez el bromuro (682 mg, 2,98 mmol), y después se añadió Nal (446 mg, 2,98 mmol). La mezcla de reacción se agitó a 60 °C durante toda la noche, se filtró con acetona y a continuación se concentró al vacío. El residuo resultante se repartió entre Et2O (60 mL) y agua (20 mL). Después de la separación, la capa orgánica se lavó con una solución ac. saturada de sulfito de sodio (20 mL), Na2CO3 2 M y salmuera. La capa orgánica se secó a continuación con MgSO4, se filtró y se concentró al vacío. La cromatografía ultrarrápida proporcionó el producto deseado.
MÉTODO GENERAL 6-1
Procedimiento representativo para una reacción SnAr
Se combinaron el Intermedio 2-2 (50 mg, 0,174 mmol), piperazino-1-carboxilato de ferf-butilo (59 mg, 0,314 mmol), DIPEA (0,06 mL, 0,349 mmol) y n-butanol (0,1 mL) en un vial de reacción de 4 mL y se calentó hasta 120 °C durante toda la noche. La reacción se enfrió hasta TA y se añadió EtOAc. El sólido blanco se filtró y se lavó con EtOAc, se disolvió en DCM y se lavó con H2O. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró al vacío
para proporcionar el producto deseado. La purificación mediante columna ultrarrápida o HPLC proporcionó el compuesto deseado.
Ejemplo de referencia 1-1: Síntesis de 6-(naftalen-2-¡l)-N-(2,2,6,6-tetrametilp¡per¡dm-4-¡l)μmdazm-3-amma

A un vial apto para microondas de 2 mL se añadieron el Intermedio 1-2 6-cloro-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (100 mg, 0,37 mmol), ácido naftalen-2-ilborónico (96 mg, 0,56 mmol), Na2CO3 (118 mg, 1,17 mmol), agua (0,25 mL), DME (1 mL) y pdCh(dppf).CH2Cl2 (30 mg, 0,037 mmol). El recipiente de reacción se selló y se calentó en un microondas a 120 °C durante 45 min. La mezcla cruda se diluyó con EtOAc, continuación la capa orgánica se lavó con agua, salmuera, se secó con MgSO4, se filtró y se concentró. El producto crudo se purificó mediante cromatografía en sílice para obtener 6-(naftalen-2-il)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (86 mg). LCMS Rt = 1,39 min (condición B); MS (M+1) = 361,3. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,30 (s, 1H), 8,12 (dd, J=8,6, 1,8 Hz, 1H), 7,86 (d, J=8,8 Hz, 1H), 7,76-7,84 (m, 2H), 7,67 (d, J=9,3 Hz, 1H), 7,37-7,48 (m, 2H), 6,69 (d, J=9,1 Hz, 1H), 4,82 (a. m, 1H), 4,31-4,51 (m, 2H), 2,07 (dd, J=12,9, 3,5 Hz, 2H), 1,35 (s, 6H), 1,22 (s, 6H).
L i i n m r r r n iliz n r imi n imil r l l E m l r f r n i 1-1





Ejemplo de referencia 1-22: Síntesis de N-metil-6-(ftalazm-6-il)-N-(2,2,6,6-tetrametilpipe
N
dm-4-il)piridazm-3-amina

Una mezcla de 6-bromoftalazina (0,11 g, 0,50 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (0,16 g, 0,58 mmol), 4,4,4',4l,5,5,5l,5l-octametil-2,2l-bi(1,3,2-dioxaborolano) (0,14 g, 0,55 mmol), acetato de potasio (0,15 g, 1,5 mmol) y [1,1-bis(difenilfosfino)ferroceno]dicloropaladio (II), complejo con diclorometano (0,04 g, 0,05 mmol) en dioxano (3 mL) se calentó a 80 °C durante 3 h. Se añadieron carbonato de potasio (0,20 g, 1,5 mmol) y agua (0,4 mL) y la mezcla se agitó durante 48 h a 90 °C. Después de enfriar, la reacción se purificó mediante extracción en fase sólida (SiliaBond ® carbonato, MeOH como eluyente). Después de la evaporación del disolvente a presión reducida, el material obtenido se purificó mediante HPLC preparativa de fase inversa utilizando de un 5 % a un 95 % de acetonitrilo en agua modificada con un 3 % de n-PrOH. LCMS: tR = 0,43 min [M+H] (método Q de LCMS); 377,245; 1H RMN (400 MHz, DMSO-d6) 69,74-9,62 (m, 2H), 8,75 (s, 1H), 8,74 (dd, J= 8,5, 2,0 Hz, 1H), 8,23 (d, J= 8,5 Hz, 1 H), 8,12 (d, J= 9,5 Hz, 1H), 7,19 (d, J= 9,5 Hz, 1H), 5,19 (tt, J= 12,0, 3,5 Hz, 1H), 2,98 (s, 3H), 1,56 (dd, J= 12,0, 3,5 Hz, 2H), 1,45 (t, J= 12,0 Hz, 2H), 1,27 (s, 6H), 1,10 (s, 6H).
Los siguientes compuestos se prepararon utilizando procedimientos similares al del Ejemplo de referencia 1-1 utilizando tecnolo ía de síntesis en fase de solución en aralelo de alta ca acidad.

Los siguientes compuestos finales se prepararon utilizando procedimientos similares al del Ejemplo de referencia 1 1, con desprotección posterior de metoxi como se resume en los MÉTODOS GENERALES 3-1 y 3-2 cuando sea a ro iado.




Ejemplo de referencia 4-1: Síntesis de 5-fluoro-2-{6-[metil(2,2,6,6-tetrametilpiperidm-4-N)ammo]piridazm-3-il}fenol

Paso 1: 6-(2-(Bendloxi)-4-fíuomfenil)-N-metil-N-(2,2,6,6-tetmmetilpipendin-4-il)pirídazin-3-amina
Se hicieron reaccionar el Intermedio 1-1 y ácido (2-(benciloxi)-4-fluorofenil)borónico de acuerdo con el MÉTODO GENERAL 1-1 para un acoplamiento de Suzuki. Se obtuvo un sólido tostado (94 % de rendimiento) después de purificación mediante SCX. No fue necesaria una cromatografía adicional. MS (M+1) = 449,2. 1H RMN (400 MHz, DMSO-d6) 5 ppm 7,79-7,67 (m, 2 H) 7,43-7,27 (m, 5 H) 7,10 (dd, J=11,37, 2,27 Hz, 1 H) 6,99 (d, J=9,60 Hz, 1 H) 6,88 (td, J=8,46, 2,27 Hz, 1 H) 5,20 (s, 2 H) 5,10-4,98 (m, 1 H) 2,90 (s, 3 H) 1,51 (dd, J=12,13, 3,54 Hz, 2 H) 1,41 (t, J=12,13 Hz, 2 H) 1,24 (s, 6 H) 1,11 (s, 1 H) 1,08 (s, 6 H)
Paso 2: 5-Fluoro-2-[6-[metil(2,2,6,6-tetrametilpiperidin-4-il)amino]piridazin-3-il]fenol
Siguiendo el MÉTODO GENERAL 4-1, se añadió Pd/C (10 % p, 47,0 mg, 0,044 mmol) a una solución de 6-(2-(benciloxi)-4-fluorofenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (198 mg, 0,441 mmol) en MeOH (2 mL)/EtOAc (2 mL) a TA. En la mezcla de reacción se hizo vacío y se rellenó con H2 (2X), a continuación se agitó en una atmósfera de H2 durante 4 h y se filtró a través de celite utilizando MeOH. El filtrado se concentró al vacío para proporcionar un aceite amarillo, que se redisolvió en DCM y se concentró al vacío para proporcionar el compuesto del título como un sólido amarillo (129 mg, 82 % de rendimiento). LCMS tR = 0,49 min (método Q de LCMS); MS (M+1) = 359,2. 1H RMN (400 MHz, DMSO-d6) 5 ppm 14,10 (s a, 1H), 8,11 (d, J=9,60 Hz, 1H), 7,83-7,92 (m, 1H), 7,32 (d, J=10,11 Hz, 1H), 6,67-6,77 (m, 2H), 4,83-4,98 (m, 1H), 2,95 (s, 3H), 1,54 (dd, J=12,13, 3,54 Hz, 2H), 1,43 (t, J=12,13 Hz, 2H), 1,25 (s, 6H), 1,09 (s, 6H).
Ejemplo de referencia 5-1: Síntesis de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-il)benzonitrilo

Paso 1: 3-Metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzonitrilo
A un matraz que contenía 6-(4-cloro-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (1,1 g, 2,88 mmol), Pd2(dba)3 (0,26 g, 0,29 mmol), dppf (0,32 g, 0,58 mmol), zinc en polvo (75 mg, 1,15 mmol) y cianuro de zinc (1,0 g, 8,63 mmol) se añadió DMA (8,99 mL). La reacción se agitó a 150 °C durante 18 h. La mezcla de reacción cruda se enfrió hasta TA, a continuación se diluyó con EtOAc y se filtró a través de celite. El filtrado se lavó con NaOH 1 M (4X), agua (6X) y salmuera. La capa orgánica se secó con Na2SO4, se filtró y se concentró a presión reducida. El producto crudo se purificó mediante cromatografía en columna para obtener 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzonitrilo (1,1 g). LCMS tR = 1,02 min (condición B); m S (M+1) = 380,4.
1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,09 (d, J=8,1 Hz, 1H), 7,80 (d, J=9,6 Hz, 1H), 7,38 (dd, J=7,8, 1,5 Hz, 1H), 7,21 (d, J=1,5 Hz, 1H), 6,81 (d, J=9,6 Hz, 1H), 5,12-5,28 (m, 1H), 3,90 (s, 3H), 3,00 (s, 3H), 1,71 (dd, J=12,5, 3,4 Hz, 2H), 1,42-1,51 (m, 2H), 1,38 (s, 6H), 1,23 (s, 6H).
Paso 2: 3-Hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzonitrilo
Siguiendo el MÉTODO GENERAL 3-3 para la desprotección de metoxi utilizando Lil y colidina, se preparó 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzonitrilo. LCMS tR = 0,52 min (método Q de LCMS); MS (M+1) = 366,2. RMN: 1H RMN (400 MHz, DMSO-d6) 5 ppm 8,27 (d, J=9,85 Hz, 1H), 8,08 (d, J=8,08 Hz, 1H), 7,28 7,46 (m, 3 H), 4,99 (s a, 1H), 2,97 (s, 3H), 1,37-1,60 (m, 4H), 1,25 (s, 6H), 1,08 (s, 6H).
Ejemplo de referencia 6-1: Síntesis de 1-alM-6-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)naftalen-2-ol

Paso 1: 6-(6-(Aliloxi)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpipendin-4-il)pindazin-3-amina:
A una solución de 6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (208 mg, 0,53 mmol) en DMF (5,3 mL) se añadió NaH al 60 % p (47 mg, 1,17 mmol) y después yoduro de alilo (54 |jL, 0,59 mmol). La reacción se agitó a TA durante 10 min. La mezcla de reacción cruda se diluyó con EtOAc. La capa orgánica se lavó con agua (5X) y salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El producto crudo se purificó mediante HPLC para obtener 6-(6-(aliloxi)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (85 mg). LCMS tR = 1,46 min (condición B); MS (M+1) = 431,1. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,37 (d, J=1,5 Hz, 1H), 8,20 (dd, J=8,6, 1,8 Hz, 1H), 7,84 (d, J=8,8 Hz, 1H), 7,83 (d, J=8,6 Hz, 1H), 7,79 (d, J=9,9 Hz, 1H), 7,20-7,25 (m, 1H), 7,19 (d, J=2,3 Hz, 1H), 6,92 (d, J=9,6 Hz, 1H), 6,09-6,23 (m, 1H), 5,51 (dd, J=17,2, 1,5 Hz, 1H), 5,36 (dd, J=10,5, 1,4 Hz, 1H), 5,16-5,31 (m, 1H), 4,66-4,74 (m, 2H), 3,03 (s, 3H), 1,76 (dd, J=12,5, 3,4 Hz, 2H), 1,45 1,53 (m, 2H), 1,41 (s, 6H), 1,24 (s, 6H).
Paso 2: 1-Alil-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol
Se calentó un matraz que contenía 6-(6-(aliloxi)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (40 mg, 0,09 mmol) en un baño de aceite precalentado a 220 °C durante 10 min. Después de 10 min, se permitió que el matraz se enfriara hasta TA. El producto crudo se purificó mediante HPLC (XBridge C8, H2O (0,1% de NH4OH ac. como modificador/CH3CN) para obtener 1 -alil-6-(6-(metil(2,2,6,6-tetrametilpiperidi n-4-il)amino)pi ridazin-3-il)naftalen-2-ol (25 mg). LCMS tR = 1,20 min (condición B); MS (M+1) = 431,0. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,40 (d, J=1,8 Hz, 1H), 8,21 (dd, J=9,0, 1,9 Hz, 1H), 8,01 (d, J=9,1 Hz, 1H), 7,71-7,86 (m, 2H), 7,17 (d, J=8,8 Hz, 1H), 6,92 (d, J=9,6 Hz, 1H), 6,03-6,21 (m, 1H), 5,18-5,39 (m, 1H), 5,15 (dd, J=6,9, 1,6 Hz, 1H), 5,11 (dd, J=13,9, 1,8 Hz, 1H), 3,88 (d, J=5,6 Hz, 2H), 3,03 (s, 3H), 1,76 (dd, J=12,4, 3,3 Hz, 2H), 1,43-1,57 (m, 2H), 1,41 (s, 6H), 1,25 (s, 6H).
Ejemplo de referencia 7-1: Síntesis de 6-(benzo[b]tiofen-2-il)-N-(1,2,2,6,6-pentametilpipendm-4-il)piridazm-3-amina

A 3,6-dicloropiridazina (4,18 g, 28,1 mmol) y Na2CO3 (8,93 g, 84 mmol) en DME (100 mL) y agua (25 mL) se añadió ácido benzo[b]tiofen-2-ilborónico (5 g, 28,1 mmol) y PdCh(dppf).CH2Cl2 (0,69 g, 0,84 mmol). En la reacción se hizo vacío y se purgó con N23X. La reacción se calentó a 85 °C durante 18 h y se enfrió hasta TA. El producto crudo se filtró a través de celite y se enjuagó con EtOAc y después CH2Cl2. El filtrado crudo se concentró al vacío y se diluyó en CH2O 2 y agua. La capa orgánica se separó y la emulsión restante se acidificó con HCl 1 M y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua, salmuera, se secaron con MgSO4, se filtraron y se concentraron. El producto crudo se purificó mediante cromatografía en gel de sílice. El producto se concentró parcialmente para proporcionar un precipitado que se filtró para obtener 3-(benzo[b]tiofen-2-il)-6-cloropiridazina (2,36 g) como un sólido amarillo claro. El filtrado se recristalizó en CH3CN para obtener 3-(benzo[b]tiofen-2-il)-6-cloropiridazina (0,46 g). LCMS tR = 1,57 minutos (condición B); (M+1) = 247,1.
Paso 2: 6-(Benzo[b]tiofen-2-il)-N-( 1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina
A una suspensión de 3-(benzo[b]tiofen-2-il)-6-cloropiridazina (100 mg, 0,41 mmol) en n-butanol (2 mL) en un vial apto para microondas 2 mL se añadió 1,2,2,6,6-pentametilpiperidin-4-amina (276 mg, 1,62 mmol) y DIPEA (0,14 mL, 0,81 mmol). El recipiente de reacción se selló y se calentó con irradiación de microondas durante 180 min a 180 °C. La mezcla de reacción cruda se diluyó en EtOAc. La capa orgánica se lavó con agua (5X), salmuera, se secó con MgSO4, se filtró y se concentró. El producto crudo se purificó mediante HPLC para obtener 25 mg de 6-(benzo[b]tiofen-2-il)-N-(1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina. LCMS tR = 0,54 min (método Q de LCMS); (M+1) = 381,1. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,73-7,82 (m, 1 H), 7,64-7,73 (m, 1 H), 7,54 (d, J=9,1 Hz, 1 H), 7,53 (s, 1H), 7,21 7,34 (m, 2 H), 6,56 (d, J=9,3 Hz, 1 H), 4,55 (d, J=7,8 Hz, 1 H), 4,14-4,32 (m, 1 H), 2,20 (s, 3 H), 1,93 (dd, J=12,4, 3,8 Hz, 2 H), 1,30 (t aparente, J=12,1 Hz, 2 H), 1,07 (s, 6 H), 1,10 (s, 6 H).
Ejemplo de referencia 8-1: Síntesis de N-al¡l-3-h¡drox¡-4-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡dm-4-il)amino)piridazin-3-il)benzamida

Paso 1: Ácido 3-metoxi-4-(6-(metil(2,2,6,6 tetrametilpiperidin-4-il)amino)piridazin-3-il)benzoico
A un vial apto para microondas se añadió 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermed¡o 1-1, 100 mg, 0,35 mmol), ácido 4-borono-3-metoxibenzoico (76 mg, 0,39 mmol), PdCl2(dppf).CH2Cl2 (29 mg, 0,035 mmol), Na2CO3 (112 mg, 1,06 mmol), DMF (2 mL) y agua (0,5 mL). El vial apto para microondas se selló y se calentó en un microondas a 120 °C durante 45 min. La mezcla de reacción cruda se enfrió y se diluyó en agua y éter. La capa acuosa se extrajo con éter. La emulsión resultante se diluyó en agua y NaOH 1 M. La capa acuosa se acidificó lentamente con HCl 1 M y se extrajo de nuevo con éter. La capa orgánica se concentró a presión reducida y el precipitado resultante se suspendió en 10 mL de MeOH y se filtró. El filtrado se concentró y utilizó sin una purificación adicional. LCMS tR = 0,63 min (condición B), MS (M+1) = 399,0.
Paso 2: Síntesis de N-alil-3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzamida
A una solución de ácido 3-metoxi-4-(6-(metil(2,2,6,6 tetrametilpiperidin-4-il)amino)piridazin-3-il)benzoico (0,354 mmol) en DMF (4 mL) y CH2Cl2 (4 mL) se añadieron TEA (0,40 mL, 2,83 mmol), alilamina (0,053 mL, 0,71 mmol), y HATU (202 mg, 0,53 mmol). Se permitió que la reacción cruda se agitara a TA durante toda la noche. La mezcla cruda se desactivó con NaHCO3 ac. sat. y se diluyó con CH2Cl2. La capa orgánica se lavó con agua (6X), salmuera, se secó con MgSO4, se filtró y se concentró. El producto crudo se purificó mediante HPLC para obtener N-alil-3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzamida (8,2 mg). LCMS tR = 1,08 min (condición B), Ms (M+1) = 438,1.
Paso 3: Siguiendo el MÉTODO GENERAL 3-2 para la desprotección de metoxi utilizando tribromuro de boro, se preparó N-alil-3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzamida. RMN: 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,14 (d, J=10,10 Hz, 1H), 7,85 (d, J=8,08 Hz, 1H), 7,42 (d, J=1,77 Hz, 1H), 7,39 (dd, J=8,21 Hz, 1,89 Hz, 1H), 7,31 (d, J=9,85 Hz, 1 H), 5,95 (ddt, J=17,18 Hz, 10,36 Hz, 5,43 Hz, 5,43 Hz, 1H), 5,25 (dq, J=17,18 Hz, 1,60 Hz, 1H), 5,15 (dq, J=10,33 Hz, 1,44 Hz, 1H), 5,11 (s a, 1H), 4,00 (dt, J=5,49 Hz, 1,55 Hz, 2H), 3,02 (s, 3H), 1,65-1,74 (m, 2H), 1,52-1,63 (m, 2H), 1,39 (s, 6H), 1,24 (s, 6H).
Ejemplo de referenc¡a 9-1: Síntes¡s de 2-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)ammo)p¡r¡dazm-3-¡l)-5-(1H-p¡razol-1-¡l)fenol

Paso 1: 6-(2-Metoxi-4-(1H-pirazol-1-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se acopló 1-(3-metoxi-4-(4,4,5,5-tetramet¡M,3,2-d¡oxaborolan-2-¡l)fen¡l)-1H-pirazol del Paso 3 del Intermedio 2-1, con el Intermedio 1-1 con métodos de acoplamiento de Suzuki estándar como se describe en el MÉTODO GENERAL 1 2 para proporcionar 6-(2-metox¡-4-(1H-p¡razol-1-¡l)fen¡l)-N-met¡l-N-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)μmdaz¡n-3-am¡na.
Paso 2: 2-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-1-il)fenol
Como se describe en el MÉTODO GENERAL 3-1, se añadió tiofenol (0,127 mL, 1,24 mmol) a un vial apto para microondas que contenía 6-(2-metox¡-4-(1H-p¡razol-1-¡l)fen¡l)-N-met¡l-N-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)μmdaz¡n-3-amina (520 mg, 1,24 mmol) y K2CO3 (171 mg, 1,24 mmol) en NMP (5 mL). Se hizo vacío en el vial de microondas y se rellenó con N2 (2X). La mezcla de reacción se calentó en el microondas a 190 °C durante 30 min. La mezcla de reacción se filtró a través de celite (embudo con filtro preempaquetado) y se lavó con MeOH. El filtrado se acidificó hasta pH 3 utilizando una solución acuosa de HCl 1 M y a continuación se adsorbió sobre una columna de SCX (10 g) acondicionada con metanol. La columna se lavó varias veces con metanol y a continuación se eluyó con una solución de amoniaco 2 N en metanol. El producto se recogió y concentró al vacío para proporcionar el producto crudo que se purificó mediante HPLC preparativa en condiciones básicas para proporcionar 2-(6-(metil(2,2,6,6-tetrametilpipendm^-il)ammo^mdazm^-N^-^H-pirazoM-Nfenol (198 mg, m S: 407,25 [M+H+], LC/Ms tR = 0,53 min (método Q de LCMS); 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,86 (d, J=2,02 Hz, 1H), 7,71 (d, J=10,11 Hz, 1H), 7,63 (d, J=1,52 Hz, 1H), 7,54 (d, J=8,59 Hz, 1H), 7,21-7,30 (m, 2H), 6,91 (d, J=9,60 Hz, 1H) 6,31-6,41 (m, 1H), 4,76-4,95 (m, 1H), 2,89-3,01 (m, 3H), 1,62 (dd, J=12,13, 3,54 Hz, 2H), 1,34 (s a, 2H), 1,27 (s, 6H), 1,10 (s, 6H).
Ejemplo de referencia 10-1: Síntesis de 5-(5-metiloxazol-2-il)-2-{6-[metil(2,2,6,6-tetrametilpiperidm-4-il)ammo]piridazm-3-il}fenol

Paso 1: 3-Metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-N-(prop-2-in-1-il)benzamida
A una solución cruda de ácido 3-metox¡-4-(6-(met¡l-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)benzo¡co (620 mg, 1,56 mmol) en DMF (10 mL) y CH2Cl2 (10 mL) se añadió propargilamina (129 mg, 2,33 mmol), DIPEA (0,82 mL, 4,67 mmol) y HATU (887 mg, 2,33 mmol). La reacción se agitó a Ta durante toda la noche y a continuación se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se concentró y se disolvió parcialmente en MeOH. El precipitado blanco resultante se filtró y el filtrado de MeOH se concentró. El filtrado se disolvió en MeOH y el precipitado resultante se filtró. El filtrado se concentró y purificó mediante HPLC (columna C18 sunfire, de un 20 % de CH3CN/H2O a un 100 % de CH3CN, 0,1 % de NH4OH ac. como modificador para obtener 3-metoxi-4-(6-(metil(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)-N-(prop-2-¡n-1-¡l)benzam¡da (190 mg). LCMS tR = 0,86 min (condición B); MS (M+1) = 436,3. 1H RMN (400 MHz, METANOL-d4) 5 ppm 7,82 (d, J=9,6 Hz, 1 H), 7,73 (d, J=8,1 Hz, 1 H), 7,58 (d, J=1,5 Hz, 1 H), 7,52 (dd, J=7,8, 1,5 Hz, 1 H), 7,10 (d, J=9,6 Hz, 1 H), 5,15-5,27 (m, 1H), 4,18 (d, J=2,3 Hz, 2 H), 3,93 (s, 3 H), 2,99 (s, 3 H), 2,63 (t, J=2,5 Hz, 1 H), 1,67 (dd, J=12,5, 3,4 Hz, 2 H), 1,54 (t, J=12,5 Hz, 2 H), 1,33 (s, 6 H), 1,20 (s, 6 H).
Paso 2: 6-(2-Metoxi-4-(5-metiloxazol-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A una solución de 3-metox¡-4-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)-N-(prop-2-¡n-1-¡l)benzam¡da (190 mg, 0,44 mmol) en dioxano (8 mL) se añadió NaH al 60 % p (52 mg, 1,31 mmol). La reacción se calentó a reflujo durante 5 h, a continuación se enfrió hasta TA y se diluyó en EtOAc y agua. La capa orgánica se lavó con agua, salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El material crudo se purificó mediante HPLC para obtener 6-(2-metox¡-4-(5-met¡loxazol-2-¡l)fen¡l)-N-met¡l-N-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-am¡na (106 mg). LCMS tR = 0,97 min (condición B); MS (M+1) = 436,3. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,08 (d, J=8,1 Hz, 1H), 7,85 (d, J=9,6 Hz, 1H), 7,72 (dd, J=8,1, 1,5 Hz, 1H), 7,65 (d, J=1,3 Hz, 1H), 6,88 (s, 1H), 6,82 (d, J=9,6 Hz, 1H), 5,20-5,39 (m, 1H), 3,96 (s, 3H), 2,99 (s, 3H), 2,43 (s, 3H), 1,74 (dd, J=12,4, 3,3 Hz, 2H), 1,47-1,59 (m, 2H), 1,43 (s, 6H), 1,31 (s, 6H).
Paso 3. 5-(5-Metiloxazol-2-il)-2[6-[metil(2,2,6,6-tetrametilpiperidin-4-il)amino]piridazin-3-i]fenol
A una solución de 6-(2-metox¡-4-(5-met¡loxazol-2-¡l)fen¡l)-N-met¡l-N-(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-am¡na (106 mg, 0,24 mmol) en 2,4,6 colidina (8 mL, secada con MgSO4, y filtrada) se añadió Lil anhidro (292 mg, 2,18 mmol). La reacción se agitó a 170 °C durante 4 h, a continuación se enfrió y se diluyó con cantidades pequeñas de MeOH y
EtOAc (150 mL) y H2O (30 mL). La capa orgánica se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró a presión reducida. El producto crudo se purificó mediante HPLC para proporcionar 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol (49 mg). LCMS tR = 0,55 min (método Q de LCMS); MS (M+1) = 422,3. 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,12 (d, J=9,9 Hz, 1H), 7,86 (d, J=8 , 8 Hz, 1H), 7,53 (dd, J=6,1, 1,8 Hz, 1H), 7,51 (s, 1H), 7,30 (d, J=10,1 Hz, 1H), 6,92 (d, J=1,0 Hz, 1H), 5,03-5,14 (m, 1H), 3,01 (s, 3H), 2,43 (d, J= 1,2 Hz, 3H), 1,68 (dd, J=12,6, 3,5 Hz, 2H), 1,56 (t, J=12,3 Hz, 2H), 1,37 (s, 6H), 1,21 (s, 6H).
Ejemplo de referencia 11-1: Síntesis de 5-((4-hidroximetil)-1H-pirazoM-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol

Paso 1. (1-(4-Bromo-3-metoxifenii)-1H-pirazoi-4-ii)metanoi
Una mezcla de 4-(hidroximetil)pirazol (500 mg, 5,10 mmol), salicilaldoxima (140 mg, 1,019 mmol), carbonato de cesio (4,98 g, 15,29 mmol), óxido cuproso (58,2 mg, 0,306 mmol), yodobromoanisol (1,59 g, 5,10 mmol) y N,N-dimetilformamida (10 mL) se combinó en un vial apto para microondas dotado de una entrada de N2 y una barrita de agitación magnética. La mezcla de reacción se agitó en una atmósfera de nitrógeno a 90 °C durante toda la noche. La mezcla de reacción se enfrió hasta TA, a continuación se filtró a través de celite y el filtrado se concentró al vacío. El material crudo se purificó mediante cromatografía en columna (de un 10 % a un 60 % de EtOAc en hepatanos) para obtener (1-(4-bromo-3-metoxifenil)-1H-pirazol-4-il)metanol (800 mg, MS: 285,3 [M+H+].)
Paso 2. (1-(3-Metoxi-4-(4,4,5,5-tetrametii-1,3,2-dioxaboroian-2-ii)fenii))-1H-pirazoi-4-ii)metanoi
A un vial apto para microondas se añadieron (1-(4-bromo-3-metoxifenil)-1H-pirazol-4-il)metanol (400 mg, 1,41 mmol), bis(pinacolato)diboro (538 mg, 2,12 mmol), acetato de potasio (415 mg, 4,24 mmol), PdCh(dppf) (103 mg, 0,14 mmol) y dppf (78 mg, 0,14 mmol), y después se añadió 1,4-dioxano ( 6 mL). La mezcla de reacción se purgó con N2 y se agitó en una atmósfera de N2 a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de un embudo con un filtro desechable y se concentró al vacío. La purificación mediante cromatografía en columna (de un 10 % a un 60 % de EtOAc en heptano) proporcionó (1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol-4-il)metanol (300 mg, MS: 331,2 [M+H+].).
Paso 3. (1-(3-Metoxi-4-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)fenii)-1H-pirazoi-4-ii)metanoi A un vial apto para microondas se añadieron (1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-4-il)metanol (87 mg, 0,26 mmol), Intermedio 1-1 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (74,5 mg, 0 , 2 6 mmol), fosfato de potasio (168 mg, 0,79 mmol), Pd2(dba)3 (12,06 mg, 0,01 mmol) y Sphos (10,82 mg, 0,03 mmol), y después se añadió 1,4-dioxano (5 mL)/H2O (1 mL). El vial se purgó con N2 durante 10 min y la mezcla de reacción se calentó a 100 °C en el microondas durante una hora. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar (1-(3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1H-pirazol-4-il)metanol (100 mg, MS: 451,4 [M+H+].).
Paso 4 5-(4-(Hidroximetii)-1H-pirazoi-1-ii)-2-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)fenoi Siguiendo el MÉTODO GENERAL 3-1 estándar para la desprotección de metoxi, se obtuvo el compuesto del título como un polvo amarillo pálido (30 mg). MS: 437,2 [M+H+], LCMS tR = 0,48 min (método Q de LCMS); 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,88 (s, 1H), 7,73 (d, J=10,11 Hz, 1H), 7,65 (s, 1H), 7,55 (d, J=8,08 Hz, 1 H), 7,20 7,27 (m, 2H), 6,93 (d, J=10,11 Hz, 1H), 4,86 (t, J=12,13 Hz, 1H), 4,61 (s, 2H), 2,91-2,98 (m, 3H), 1,62 (dd, J=12,38, 3,28 Hz, 2H), 1,32-1,37 (m, 2H), 1,28 (s, 6H), 1,11 (s, 6H).
Ejemplo de referencia 12-1: Síntesis de 5-(1H-imidazoM-N)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazin-3-il)fenol

Paso 1. 1-(4-Bromo-3-metoxifenil)-1H-imidazol
Una mezcla de 2-(2-piridil)bencimidazol (287 mg, 1,469 mmol), carbonato de cesio (5,98 g, 18,36 mmol), yoduro de cobre (I) (280 mg, 1,469 mmol) y DMF (5 mL) se combinó en un vial apto para microondas dotado de una entrada de N2 y una barrita de agitación magnética. La suspensión espesa se calentó a 60 °C durante 1 h, después se añadió imidazol (500 mg, 7,34 mmol) y 1-bromo-4-yodo-2-metoxibenceno (2,3 g, 7,34 mmol). La mezcla de reacción se calentó a 90 °C durante 2 días. La mezcla de reacción se filtró a través de celite, se lavó con EtOAc y se concentró al vacío. El material crudo se purificó mediante cromatografía en gel de sílice (de un 10 % a un 40 % de EtOAc en hepatanos) para obtener 1-(4-bromo-3-metoxifenil)-1H-imidazol (1,25 g, MS: 255,2 [M+H+])
Paso 2. Ácido (4-(1H-imidazol-1-il)-2-metoxifenil)borónico
A una solución agitada de 1-(4-bromo-3-metoxifenil)-1H-imidazol en THF (5 mL) se añadió n-butillitio 2,5 M en hexanos (0,348 ml, 0,869 mmol) gota a gota a -78 °C durante 15 min. Después de finalizar la adición, la solución de reacción se agitó a -78 °C durante 15 min, y se añadió borato de trimetilo (0,353 mL, 3,16 mmol). Se permitió que la reacción se calentara hasta TA y se siguió agitando durante toda la noche. La reacción se desactivó con una solución acuosa de HCl 1 M hasta pH 2, se diluyó con agua y se extrajo con DCM (3x). El producto permaneció en la solución acuosa que se concentró al vacío para obtener ácido (4-(1H-imidazol-1-il)-2-metoxifenil)borónico crudo (120 mg, MS: 219,2 [M+H+]).
Paso 3. 6-(4-( 1 H-imidazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadió ácido (4-(1H-imidazol-1-il)-2-metoxifenil)borónico (120 mg, 0,55 mmol), Intermedio 1-1 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (156 mg, 0,55 mmol), fosfato de potasio (351 mg, 1,65 mmol), Pd2(dba)3 (25,2 mg, 0,028 mmol), y SPhos (22,6 mg, 0,05 mmol), y después se añadió 1,4-dioxano (2 mL)/H2O (0,5 mL). El vial se purgó con N2 durante 10 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante 40 min. La mezcla de reacción se concentró al vacío y el pH se ajustó a 3 con una solución acuosa de HCl 1 M y a continuación se cargó en una columna de SCX. La columna se lavó con metanol y se eluyó con NH3 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 6-(4-(1 H-imidazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (90 mg, MS: 421,4 [M+H+]).
Paso 4: 5-(1H-imidazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol, se obtuvo 5-(1H-imidazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un polvo amarillo pálido (8 mg, MS: 407,2 [M+H+], LCMS tR = 0,40 min (método Q de LCMS); 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,82 (s a, 1H), 7,70 (d, J=10,11 Hz, 1H), 7,55 (d, J=8,59 Hz, 1H), 7,23 (s a, 1H), 7,12 (s a, 1H), 7,02 (d, J=2,02 Hz, 1H), 6,92 (d, J=10,11 Hz, 1H), 6,85 (dd, J=8,59, 2,02 Hz, 1H), 4,86-4,92 (m, 1H), 2,94 (s, 3H), 1,62 (dd, J=12,63, 3,54 Hz, 2H), 1,36 (s a, 2H), 1,28 (s, 6H), 1,11 (s, 6H).
Ejemplo de referencia 13-1: Síntesis de 5-(4-amino-1H-pirazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol

Paso 1: 1-(4-Bromo-3-metoxifenil)-1H-pirazol-4-amina
Una mezcla de diclorhidrato de pirazol-4-amina (0,75 g, 4,79 mmol), salicilaldoxima (0,131 g, 0,96 mmol), carbonato de cesio (4,69 g, 14,38 mmol), óxido cuproso (0,06 mg, 0,29 mmol), 1-bromo-4-yodo-2-metoxibenceno (1,5 g, 4,79 mmol) y N,N-dimetilformamida (5 mL) se combinó en un vial apto para microondas dotado de una entrada de N2 y una
barrita de agitación magnética. La mezcla de reacción se agitó en una atmósfera de nitrógeno a 90 °C durante toda la noche. Se permitió que la solución obtenida se enfriara hasta TA, a continuación se filtró a través de celite y el filtrado se concentró al vacío. El material crudo se purificó mediante cromatografía en gel de sílice (de un 10 % a un 60 % de EtOAc en hepatanos) para obtener 1-(4-bromo-3-metoxifenil)-1H-pirazol-4-amina (250 mg, MS: 270,2 [M+H+].)
Paso 2: 1-(3-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-4-amina
A un vial apto para microondas se añadieron 1-(4-bromo-3-metoxifenil)-1 H-pirazol-4-amina (250 mg, 0,93 mmol), bis(pinacolato)diboro (355 mg, 1,39 mmol), acetato de potasio (543 mg, 5,59 mmol), PdCl2(dppf) (68,20 mg, 0,09 mmol) y dppf (51,70 mg, 0,09 mmol), y después se añadió 1,4-dioxano (2 mL). La mezcla de reacción se purgó con N2 y se agitó con protección de N2 a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de un embudo con filtro desechable, se concentró al vacío y se purificó mediante cromatografía en columna (de un 10 % a un 60 % de EtOAc en heptano) para proporcionar 1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-4-amina (160 mg, MS: 316,2 [M+H+].).
Paso 3: 6-(4-(4-Amino-1H-pirazol-1-il)-2-metoxifeni)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadió 1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-4-amina (92 mg, 0,29 mmol), Intermedio 1-16-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (75 mg, 0,27 mmol), fosfato de potasio (169 mg, 0,78 mmol), Pd2(dba)3 (12,14 mg, 0,01 mmol), y SPhos (10,89 mg, 0,03 mmol), y después se añadió 1,4-dioxano (2 mL)/H2O (0,2 mL). El vial se purgó con N2 durante 10 minutos y la mezcla de reacción se calentó a 100 °C en un reactor de microondas durante una hora. La mezcla de reacción se concentró al vacío, a continuación el pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y a continuación se cargó en una columna de SCX. La columna se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 6-(4-(4-amino-1H-pirazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (100 mg, MS: 436,4 [M+H+].).
Paso 4: 5-(4-Amino-1H-pirazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Como se describe en el MÉTODO GENERAL 3-1, se añadió tiofenol (0,02 mL, 0,23 mmol) a un vial apto para microondas que contenía 6-(4-(4-amino-1H-pirazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (100 mg, 0,23 mmol) y K2CO3 (31,7 mg, 0,23 mmol) y Nm P (2 mL). Se hizo vacío en el vial de microondas y se rellenó con N2 (2X). La mezcla de reacción se calentó en un reactor de microondas a 190 °C durante 20 min, a continuación se filtró a través de celite (embudo con filtro preempaquetado) y se lavó con metanol. El filtrado se acidificó hasta pH 3 utilizando una solución acuosa de HCl 1 M y a continuación se adsorbió sobre una columna de SCX acondicionada con metanol. La columna se lavó varias veces con metanol y a continuación se eluyó con una solución de amoniaco 2 N en metanol. El eluyente se recogió, se concentró al vacío, a continuación se purificó mediante HPLC preparativa en condiciones básicas para obtener 5-(4-amino-1H-pirazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (50 mg, Ms : 422,26 [M+H+]; LCMS tR = 0,43 min (método Q de LCMS); 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,68 (d, J=10,11 Hz, 1H), 7,49 (d, J=8,59 Hz, 1H), 7,43 (s, 1H), 7,29 (s, 1H), 7,18 (dd, J=8,59, 2,53 Hz, 1H), 7,12 (d, J=2,02 Hz, 1H), 6,89 (d, J=9,60 Hz, 1H), 4,77-4,93 (m, 1H), 2,92 (s, 3H), 1,61 (dd, J=12,38, 3,28 Hz, 2H), 1,33 (t, J=12,38 Hz, 2H), 1,27 (s, 6H), 1,10 (s, 6H).
Ejemplo de referencia 14-1: Síntesis de 5-(4-ammo-1H-pirazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpipendm-4-il)amino)piridazin-3-il)

Paso 1: 3-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol
A un vial apto para microondas de 25 mL se añadió 4-bromo-3-metoxifenol (1,0 g, 4,93 mmol), bis(pinacolato)diboro (1,88 g, 7,39 mmol), acetato de potasio (2,41 g, 24,63 mmol), PdCh(dppf) (0,36 g, 0,49 mmol), dppf (0,27 g, 0,49 mmol) y 1,4-dioxano (10 mL). La solución de reacción se purgó con nitrógeno (3X) y se agitó a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de Celite y la masa retenida en el filtro se lavó con EtOAc. El filtrado se concentró al vacío para obtener un líquido marrón que se purificó mediante cromatografía en gel de sílice (10 %-50 % de EtOAc/Heptano) para proporcionar 3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (700 mg, MS: 251,4 [M+H+].).
Paso 2: 3-Metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
A un vial apto para microondas se añadió 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (500 mg, 2,0 mmol), Intermedio 1-1 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (565 mg, 2,0 mmol), fosfato de potasio (1,27 g, 6,0 mmol), Pd2(dba)3 (92 mg, 0,1 mmol), y SPhos (82 mg, 0,2 mmol), y después se añadió 1,4-dioxano (5 mL)/H2O (1 mL). El vial se purgó con N2 durante 10 min y la mezcla de reacción se calentó a 100 °C en un reactor de microondas durante una hora. La mezcla de reacción se concentró al vacío y el pH del material crudo se ajustó a 3 utilizando HCl acuoso 1 M y a continuación se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones del producto se recogieron y se secaron para proporcionar 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol que se utilizó sin una purificación adicional.
Paso 3: Trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo
A una solución de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (120 mg, 0,32 mmol) en DCM (2 mL) se añadió trietilamina (0,113 mL, 0,810 mmol) a TA. La mezcla de reacción se enfrió hasta 0 °C y después se añadió N-feniltrifluorometanosulfonimida (116 mg, 0,32 mmol). La mezcla de reacción se calentó hasta TA y se agitó durante 2 h. La reacción se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró para obtener el producto crudo, cuyo pH se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El producto crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones del producto se recogieron y se secaron para proporcionar trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (140 mg, MS: 503,4 [M+H+].).
Paso 4: 6-(2-Metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (60 mg, 0,12 mmol), ácido 1 H-pirazol-4-borónico (25,5 mg, 0,13 mmol), fosfato de potasio (76 mg, 0,36 mmol), Pd2(dba)3 (6 mg, 5,9 umol) y SPhos (5 mg, 0,012 mmol), y después se añadió 1,4-dioxano (1 mL)/H2O (0,2 mL). El vial se purgó con N2 durante 10 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante una hora. La mezcla de reacción se concentró al vacío y el pH del producto crudo se ajustó a 3 utilizando HCl acuoso 1 M y a continuación se cargó en una columna de SCX. El producto crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones con el producto se recogieron y secaron para proporcionar 6-(2-metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (MS: 421,4 [M+H+].).
Paso 5: 2-(6-(Metil(2!2!6!6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol, se obtuvo 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)fenol como un polvo amarillo pálido (8 mg, MS: 407,2 [M+H+]; LCMS tR = 0,48 min (método Q de LCMS); 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,83 (s, 2H), 7,75 (d, J=10,11 Hz, 1H), 7,51 (d, J=8,08 Hz, 1H), 7,13-7,17 (m, 1H), 7,01 (d, J=8,08 Hz, 1H), 6,93 (d, J=10,11 Hz, 1 H), 4,85 (t, J=12,38 Hz, 1H), 2,94 (s, 3H), 1,63 (dd, J=12,13, 3,03 Hz, 2H), 1,36 (t, J=12,38 Hz, 2H), 1,29 (s, 6H), 1,13 (s, 6H).
Ejemplo de referencia 15-1: Síntesis de 5-(3-ammopirazoM-N)-2-[6-[metil(2,2,6,6-tetrametilpiperidm-4-il)amino]piridazin-3-il}fenol

Paso 1: 1-(4-Bromo-3-metoxifenil)-3-nitro-1H-pirazol
Una mezcla de 1-bromo-4-yodo-2-metoxibenceno (3,99 g, 12,75 mmol), 3-nitro-1H-pirazol (1,730 g, 15,30 mmol), salicilaldoxima (0,350 g, 2,55 mmol), Cu2O (0,146 g, 1,020 mmol) y Cs2CO3 (6,23 g, 19,13 mmol) en DMF (13 mL) se desgasificó con N2 y se calentó a 95 °C durante toda la noche. Después de enfriar hasta TA, la mezcla se filtró a través de celite y se enjuagó con EtOAc. El filtrado se lavó con agua y salmuera. La solución orgánica se secó con Na2SO4, se filtró y se concentró al vacío. El residuo se suspendió en un 5 % de MeOH/DCM, y el precipitado se filtró, se enjuagó con un 5 % de MeOH/DCM y se secó para obtener 2,3 g de 1-(4-bromo-3-metoxifenil)-3-nitro-1 H-pirazol como un
sólido blanco. El filtrado del tratamiento anterior se purificó mediante cromatografía en gel de sílice (EtOAc/Heptano = de 10:90 a 50:50) para obtener 760 mg más de 1-(4-bromo-3-metoxifenil)-3-nitro-1H-pirazol como un sólido amarillo claro.
Paso 2: 1-(4-Bromo-3-metoxifenil)-1H-pirazol-3-amina
A una mezcla de 1-(4-bromo-3-metoxifenil)-3-nitro-1H-pirazol (2,3 g, 7,72 mmol) en DCM (24 mL) y ácido acético (6,18 mL, 108 mmol) se añadió zinc en polvo (2,52 g, 38,6 mmol) a 0 °C. La mezcla de reacción se agitó de 0 °C a RT durante toda la noche. La mezcla de reacción se filtró a través de celite, se enjuagó con EtOAc y se concentró al vacío. El residuo se purificó mediante cromatografía en sílice (EtOAc/heptano = de 10:90 a 50:50) para obtener una espuma blanca que se disolvió en 6 mL de tolueno, se concentró y se secó para obtener 1,9 g de 1-(4-bromo-3-metoxifenil)-1 H-pirazol-3-amina como un polvo blanco.
Paso 3. 1-(3-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) fenil)-1 H-pirazol-3-amina
Una mezcla de reacción desgasificada de 1-(4-bromo-3-metoxifenil)-1H-pirazol-3-amina (1 g, 3,73 mmol), bis(pinacolato)diboro (1,989 g, 7,83 mmol), Pd(dppf)Cl2 (0,273 g, 0,373 mmol), dppf (0,207 g, 0,373 mmol) y acetato de potasio (2,56 g, 26,1 mmol) en 1,4-dioxano (10 mL) se calentó a 88 °C durante toda la noche. Después de enfriar hasta TA, la mezcla se filtró a través de celite y se lavó con EtOAc. El filtrado se concentró y el residuo se purificó mediante cromatografía en gel de sílice (EtOAc/heptano = de 10:90 a 50:50, después de 50:50 a 60:40) para obtener 930 mg de 1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-3-amina como un sólido blanco.
Paso 4. 6-(4-(3-Amino-1H-pirazol-1-il)-2-metoxifeni)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Una mezcla de reacción desgasificada de 1-(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-3-amina (474 mg, 1,505 mmol), Intermedio 1-16-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (370 mg, 1,308 mmol), tetrakis(trifenilfosfina)paladio (0) (76 mg, 0,065 mmol) y NaHCO31 M (330 mg, 3,92 mmol) en 1,4-dioxano (7 mL) y agua (2,3 mL) se calentó a 100 °C durante 14 h. Después de enfriar hasta TA, la mezcla se filtró a través de celite, se lavó con EtOAc y el filtrado se concentró. El residuo se suspendido en MeOH, se acidificó hasta pH 2-3 utilizando HCl acuoso 1 M y se cargó sobre una columna de SCX de 10 g. La columna se lavó con MeOH y a continuación se eluyó con NH32 N en MeOH. Las fracciones recogidas se concentraron para obtener un aceite marrón claro, que se trató con éter y se concentró para obtener 570 mg de 6-(4-(3-amino-1H-pirazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido marrón claro.
Paso 5. 5-(3-Amino-1H-pirazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Una mezcla desgasificada de 6-(4-(3-amino-1H-pirazol-1-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (135 mg, 0,310 mmol), K2CO3 (42,8 mg, 0,310 mmol) y tiofenol (0,032 mL, 0,310 mmol) en NMP (2 mL) se calentó a 190 °C con irradiación de microondas durante 20 min. Después de la adición de 0,05 mmol de tiofenol y K2CO3 , la mezcla se calentó a 190 °C con irradiación de microondas durante otros 10 min. La mezcla se acidificó hasta pH 2-3 con una solución acuosa de HCl 1 M y a continuación se cargó sobre una columna de SCX de 5 g. La columna se lavó con MeOH y se eluyó con NH32 N en MeOH. Las fracciones recogidas se concentraron y disolvieron en una mezcla de MeOH y DMSO, a continuación se purificaron mediante HPLC preparativa para obtener 95 mg de 5-(3-amino-1H-pirazol-1-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un sólido amarillo oscuro. LCMS tp = 0,47 min (método Q de LCMS), MS (M+1) = 422,3, HRMS: 422,2672 [M+H+].
1H RMN (400 MHz, METANOL-d4) 5 ppm 8,08 (d, J=10,10 Hz, 1H), 7,98 (d, J=3,03 Hz, 1H), 7,80 (d, J=9,09 Hz, 1H), 7,31 (d, J=10,10 Hz, 1H), 7,15-7,22 (m, 2H), 5,91 (d, J=2,53 Hz, 1H), 5,06 (m, 1H), 3,01 (s, 3H), 1,70 (m, 2H), 1,51 1,65 (m, 2H), 1,39 (s, 6H), 1,24 (s, 6H).
Los siguientes compuestos finales se prepararon utilizando procedimientos similares a los de los Ejemplos de 9-1 a 1 -1 r f r n i l m n r l r m n n l i n^ MÉT D ENERALE .


Ejemplo 17-1: Síntesis de 2-(6-(p¡per¡dm-4-Mox¡)p¡r¡dazm-3-il)-5-(1H-p¡razoM-¡l)fenol

Paso 1. 4-((6-(2-Metoxi-4-(1H-pirazol-1-il)fenil)piridazin-3-il)oxi)piperidino-1-carboxilato de tert-butilo
Se añadió tert-butóxido de potasio (1,0 M en THF, 0,82 mL, 0,82 mmol) a 4-hidroxipiperidino-1-carboxilato de tertbutilo (0,17 g, 0,82 mmol) en THF (3 mL) a 0 °C y la mezcla se agitó durante 10 min a 0 °C. Se añadió el Intermedio 2-1 3-cloro-6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazina (0,13 g, 0,45 mmol) a la reacción a 0 °C y la mezcla se agitó durante 1 h a TA. Después de evaporar a presión reducida, el material crudo se purificó mediante cromatografía en sílice (de un 70 a un 100 % de EtOAc en heptano) para obtener 4-((6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazin-3-il)oxi)piperidino-1-carboxilato de tert-butilo (0,18 g, 89 %) como un sólido incoloro.
Paso 2: 3-(2-Metoxi-4-(1H-pirazol-1-il)fenil)-6-(piperidin-4-iloxi)piridazina
Se añadió ácido trifluoroacético (1 mL) a 4-((6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazin-3-il)oxi)piperidino-1 -carboxilato de tert-butilo (0,18 g, 0,40 mmol) en DCM (3 mL) a 0 °C. La reacción se agitó durante 1 h a TA. La mezcla de reacción se añadió a una solución acuosa de NaOH (1 M) y la fase acuosa se extrajo con cloroformo/propan-2-ol (3:1). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar 3-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)-6-(piperidin-4-iloxi)piridazino (0,14 g, 100 %) como un sólido incoloro.
Paso 3: 2-(6-(Piperidin-4-iloxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi con tiofenol, se trató 3-(2-metoxi-4-(1H-pirazol-1-il)fenil)-6-(piperidin-4-iloxi)piridazina (70 mg, 0,20 mmol) con tiofenol (27 mg, 0,24 mmol) y K2CO3 (25 mg, 0,18 mmol) en NMP (1,3 mL) durante 15 min a 190 °C. La purificación mediante HPLC (0,1 % de ácido trifluoroacético como modificador) proporcionó 2-(6-(piperidin-4-iloxi)piridazin-3-il)-5-(1 H-pirazol-1-il)fenol (12 mg, 18 %) como un sólido amarillo. LCMS tR = 0,50 min (método Q de LCMS); [M+H]: 338,16; 1H RMN (400 MHz, DMSO-d6) 6 13,01 (s a, 1H), 8,53 (d, J=2,5 Hz, 1H), 8,46 (s a, 2H), 8,45 (d, J=9,5 Hz, 1H), 7,50 (d, J=2,0 Hz, 1H), 7,47 (dd, J=8,5, 2,5 Hz, 2H), 7,44 (d, J=9,5 Hz, 1H), 6,51-6,62 (m, 1H), 5,51 (tt, J=7,5, 3,5 Hz, 1H), 3,33 (s a, 2H), 3,20 (s a, 2H), 2,18-2,31 (m, 2H), 2,01 (ddt, J=13,5, 8,5, 4,0 Hz, 2H).
Ejemplo 17-2: Síntesis de 2-(6-(((2S,4R,6R)-2,6-dimetilpipeNdm-4-N)oxi)pindazm-3-N)-5-(1H-pirazoM-N)fenol
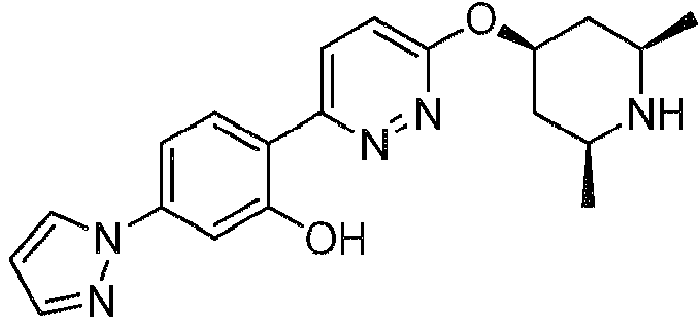
Paso 1: 3-(((2S,4R,6R)-2,6-Dimetilpiperidin-4-il)oxi)-6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazina
Se añadió tert-butóxido de potasio (1,0 M en THF, 0,72 mL, 0,72 mmol) a 2,6-dimetilpiperidin-4-ol (0,09 g, 0,66 mmol) en THF (3 mL) a 0 °C y la mezcla se agitó durante 10 min a 0 °C. Se añadió el Intermedio 2-1 3-cloro-6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazina (0,15 g, 0,51 mmol) a la reacción a 0 °C y la mezcla se agitó durante 1 h a TA. Se añadió agua y la fase acuosa se extrajo con cloroformo/propan-2-ol (3:1). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El material crudo se purificó mediante cromatografía en sílice (gel de sílice saturado con Et3N, de un 1 a un 15 % de MeOH en DCM) para obtener 3-(((2S,4R,6R)-2,6-dimetilpiperidin-4-il)oxi)-6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazina (59 mg, 31 %) como un sólido incoloro. También se aisló una mezcla racémica de 3-(((2R,6R)-2,6-dimetilpiperidin-4-il)oxi)-6-(2-metoxi-4-(1 H-pirazol-1-il)fenil)piridazina y 3-(((2S,6S)-2,6-dimetilpiperidin-4-il)oxi)-6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazina (91 mg, 47 %) como un sólido incoloro (intermedios para el Ejemplo 17-3, 17-4 más adelante).
Paso 2: 2-(6-(((2S,4R,6R)-2,6-Dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección con tiofenol, se trató 3-(((2S,4r,6R)-2,6-dimetilpiperidin-4-il)oxi)-6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazina (55 mg, 0,15 mmol) con tiofenol (0,11 M en NMP, 1,5 mL, 0,17
mmol) y K2CO3 (18 mg, 0,13 mmol) en NMP (1,5 mL) durante 15 min a 190 °C. Después de la purificación mediante HPLC (0,1 % de ácido trifluoroacético como modificador), en las fracciones que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (2 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). La evaporación a presión reducida proporcionó 2-(6-(((2S,4R,6R)-2,6-dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol (9 mg, 17 %) como un sólido amarillo. LCMS tR = 0,50 min (método Q de LCMS); [M+H]: 366,191; 1H RMN (400 MHz, DMSO-d6) 58,66 (s a, 1H), 8,53 (d, J=2,5 Hz, 1H), 8,45 (d, J=9,5 Hz, 1H), 8,12 (s a, 1H), 8,05 (d, J=8,5 Hz, 1H), 7,77 (d, J=2,0 Hz, 1H), 7,49 (d, J=2,0 Hz, 1H), 7,47 (dd, J=8,5, 2,0 Hz, 1H), 7,43 (d, J=9,5 Hz, 1H), 6,56 (dd, J=2,5, 2,0 Hz, 1H), 5-38-5,60 (m, 1H), 3,47 (s a, 2H), 2,45 (s a, 2H), 1,59 (c, J=12,0 Hz, 2H), 1,32 (d, J=6,5 Hz, 6H).
Ejemplo 17-3 y 17-4: Síntesis de 2-(6-((2,6-dimetilpiperidin-4-N)oxi)piridazin-3-N)-5-(1H-pirazoM-N)fenol, Enantiómero 1 y Enantiómero 2

La mezcla racémica aislada del Paso 1 del Ejemplo 17-2 se desprotegió siguiendo el MÉTODO GENERAL 3-1. El racemato (91 mg, 0,24 mmol) se trató con tiofenol (30 mg, 0,27 mmol) y K2CO3 (30 mg, 0,22 mmol) en NMP (1,6 mL) durante 15 min a 190 °C. Después de purificación mediante HPLC preparativa (0,1 % de ácido trifluoroacético como modificador), se generó la base libre en las fracciones deseadas mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (2 g, MeOH como eluyente y una solución 2 N de amoniaco en MeOH para liberar el material). Se aislaron el Enantiómero 1 y Enantiómero 2 mediante HPLC preparativa en fase normal (columna AD-H 4,6 x 250, 40 % de EtOH (dietilamina como modificador) en heptano). Se obtuvieron dos sólidos (5 mg (6 %) y 3 mg (3 %)). LCMS tR = 0,50 min (método Q de LCMS); [M+H]: 366,21; 1H RMN (400 MHz, DMSO-d6) 5 13,16 (s a, 1H), 8,52 (d, J=2,5 Hz, 1H), 8,40 (d, J=9,5 Hz, 1H), 8,03 (d, J=8,5 Hz, 1H), 7,75 (d, J=1,5 Hz, 1H), 7,47 (d, J=2,5 Hz, 1H), 7,45 (dd, J=8,5, 2,5 Hz, 1H), 7,35 (d, J=9,5 Hz, 1H), 6,54 (dd, J=2,5, 1,5 Hz, 1H), 5,52 (td, J=10,0, 5,0 Hz, 1H), 3,29-3,45 (m, 1H), 3,07 (dqd, J=9,5, 6,5, 3,0 Hz, 1H), 2,09-2,22 (m, 1H), 1,86-1,97 (m, 1H), 1,67 (ddd, J=12,0, 10,0, 5,0 Hz, 1H), 1,20-1,28 (m, 1H), 1,18 (d, J=7,0 Hz, 3H), 1,05 (d, J=6,5 Hz, 3H).
Ejemplo de referencia 17-5: Síntesis de 5-(1H-pirazoM-N)-2-(6-(pirroNdin-3-Noxi)piridazin-3-N)fenol

Paso 1. 3-((6-(2-Metoxi-4-(1H-pirazoi-1-ii)fenii)piridazin-3-ii)oxi)pirroiidino-1-carboxiiato de tert-butilo
Se añadió tert-butóxido de potasio (1,0 M en THF, 0,98 mL, 0,98 mmol) a 3-hidroxipirrolidino-1-carboxilato de tertbutilo (0,18 g, 0,98 mmol) en THF (3,3 mL) a 0 °C y la mezcla se agitó durante 10 min a 50 °C. Se añadió el Intermedio 2-1 3-cloro-6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazina (0,12 g, 0,40 mmol) a la reacción a 0 °C y la mezcla se agitó durante 2 h a TA. Se añadió agua (0,1 mL) y el disolvente se concentró a presión reducida para proporcionar (0,18 g, 100 %) de un sólido marrón.
Paso 2: 3-(2-Metoxi-4-(1H-pirazoi-1-ii)fenii)-6-(pirroiidin-3-iioxi)piridazina
Se añadió ácido trifluoroacético (1 mL) a 3-((6-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)piridazin-3-il)oxi)pirrolidino-1 -carboxilato de tert-butilo (0,18 g, 0,40 mmol) en DCM (3 mL) a 0 °C. La reacción se agitó durante 2 días a TA. La mezcla de reacción se añadió a una solución acuosa de NaOH (1 M) y la fase acuosa se extrajo con cloroformo/propan-2-ol (3:1). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar 3-(2-metoxi-4-(1 H-pirazol-1 -il)fenil)-6-(pirrolidin-3-iloxi)piridazina (0,14 g, 100 %) como un sólido marrón.
Paso 3: 5-(1H-Pirazoi-1-ii)-2-(6-(pirroiidin-3-iioxi)piridazin-3-ii)fenoi
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de fenol utilizando tiofenol, se trató 3-(2-metoxi-4-(1H-pirazol-1 -il)fenil)-6-(pirrolidin-3-iloxi)piridazina (91 mg, 0,24 mmol) con tiofenol (30 mg, 0,27 mmol) y K2CO3 (30 mg, 0,22 mmol) en NMP (1,6 mL) durante 15 min a 190 °C. Después de la purificación mediante HPLC (0,1 % de ácido trifluoroacético como modificador), en las fracciones que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (2 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). Se concentró el disolvente a presión reducida y se obtuvo un sólido marrón pálido (83 mg, 68 %). LCMS tR = 0,48 min (método Q de LCMS); [M+H]: 324,1457; 1H RMN (400 MHz, CLOROFORMO-d) 5 13,62 (s a, 1H), 7,97 (d, J=2,5 Hz, 1H), 7,97 (d, J=9,5 Hz, 1H), 7,74 (d, J=1,5 Hz, 1H), 7,70 (d, J=8,5 Hz, 1H), 7,39-7,43 (m, 1H), 7,38 (d, J=2,5 Hz, 1H), 7,12 (d, J=9,5 Hz, 1H), 6,44-6,51 (m, 1H), 5,74 (ddt, J=7,0, 5,0, 2,5 Hz, 1H), 3,24-3,31 (m, 2H), 3,17-3,24 (m, 1H), 3,02 (ddd, J=11,0, 8,5, 5,5 Hz, 1H), 2,20-2,33 (m, 1H), 2,02 2,11 (m, 1H).
Ejemplo 17-6 y 17-7: Síntesis de 2-(6-(((2S,4S)-2-met¡llp¡per¡dm-4-¡l)ox¡)p¡r¡dazm-3-¡l)-5-(1H-p¡razol-1-¡l)fenol, Enantiómero 1 y Enant¡ómero 2

Se añadió ferf-butóxido de potasio (1,0 M en THF, 1,4 mL, 1,4 mmol) a c/s-2-metilpiperidin-4-ol (0,10 g, 0,68 mmol) en THF (3 mL) y la mezcla se agitó durante 10 min a 50 °C. Se añadió el Intermed¡o 2-22-(6-cloropiridazin-3-il)-5-(1H-pirazol-1 -il)fenol (0,08 g, 0,28 mmol) a la reacción a 0 °C y la mezcla se agitó durante 2 días a TA. Se añadió ácido trifluoroacético (0,3 mL) y el disolvente se evaporó a presión reducida. El material crudo se purificó mediante HPLC preparativa (de un 5 a un 95 % de acetonitrilo en agua, 0,1 % de ácido trifluoroacético como modificador). Se aislaron el Enantiómero 1 y Enantiómero 2 mediante SFC preparativa. Se obtuvieron dos sólidos (10 mg (10 %) y (15 mg (16 %)).
Ejemplo 17-6, Enat¡ómero 1 LCMS tR = 0,48 min (método Q de LCMS); [M+H]: 352,1; 1H RMN (400 MHz, METANOL-d4) 58,28 (d, J=9,5 Hz, 1H), 8,23 (d, J=2,5 Hz, 1H), 7,93 (d, J=8,5 Hz, 1H), 7,73 (d, J=2,0 Hz, 1H), 7,40 (d, J=2,0 Hz, 1H), 7,37 (dd, J=8,5, 2,5 Hz, 1H), 7,26 (d, J=9,5 Hz, 1H), 6,50-6,56 (m, 1H), 5,31 (tt, J=11,0, 4,5 Hz, 1H), 3,20 (ddd, J=13,0, 4,5, 2,5 Hz, 1H), 2,90-2,97 (m, 1H), 2,84 (td, J=13,0, 2,5 Hz, 2H), 2,31 (dddq, J=17,0, 12,0, 4,5, 2,5z Hz, 2H), 1,63 (tdd, J=13,0, 11,0, 4,5 Hz, 1H), 1,32-1,38 (m, 1H), 1,20 (d, J=6,5 Hz, 3H)
Ejemplo 17-7, Enat¡ómero 2 LCMS tR = 0,48 min (método Q de LCMS); [M+H]: 352,1; 1H RMN (400 MHz, DMSO-d6) 5 13,2 (s a, 1H), 8,52 (d, J=2,5 Hz, 1H), 8,40 (d, J=9,5 Hz, 1H), 8,03 (d, J=8,5 Hz, 1H), 7,76 (d, J=1,5 Hz, 1H), 7,48 (d, J=2,0 Hz, 1H), 7,45 (dd, J=8,5, 2,0 Hz, 1H), 7,37 (d, J=9,5 Hz, 1H), 6,51-6,59 (m, 1H), 5,24 (tt, J=11,0, 4,5 Hz, 1H), 3,06 (ddd, J=12,0, 4,5, 2,5 Hz, 1H), 2,70-2,81 (m, 1H), 2,62-2,71 (m, 1H), 2,09-2,23 (m, 2H), 1,47 (qd, J=12,0, 4,5 Hz, 1H), 1,13-1,21 (m, 1H), 1,07 (d, J=6,0 Hz, 3H).
Ejemplo de referenc¡a 17-8 y 17-9: Síntes¡s de (5-(1H-p¡razol-1-¡l)-2-(6-(p¡rrol¡dm-3-¡lmetox¡)p¡ndazm-3-¡l)fenol, Enant¡ómero 1 y Enant¡ómero 2

Se añadió ferf-butóxido de potasio (1,0 M en THF, 0,7 mL, 0,7 mmol) a pirrolidin-3-ilmetanol (0,06 g, 0,64 mmol) en THF (1,4 mL) y la mezcla se agitó durante 10 min a 50 °C. Se añadió el Intermed¡o 2-22-(6-cloropiridazin-3-il)-5-(1H-pirazol-1 -il)fenol (0,05 g, 0,18 mmol) a la reacción a 0 °C y la mezcla se agitó durante 1 h a TA. El disolvente se evaporó a presión reducida. El material crudo se purificó mediante HPLC preparativa de fase inversa (de un 5 a un 95 % de acetonitrilo en agua, NH4OH 5 mM como modificador). Se aislaron el Enantiómero 1 y Enantiómero 2 mediante SFC preparativa. Se obtuvieron dos sólidos (11 mg (17 %) y 12 mg (20 %).
Ejemplo de referencia 17-8, Enatiómero 1 LCMS tR = 0,48 min (método Q de LCMS); [M+H]: 338,161; 1H RMN (400 MHz, METANOL-d4) 58,31 (d, J=9,5 Hz, 1H), 8,24 (d, J=2,5 Hz, 1H), 7,94 (d, J=8,5 Hz, 1H), 7,73 (d, J=2,0 Hz, 1H), 7,40 (d, J=2,0 Hz, 1H), 7,38 (dd, J=8,5, 2,0 Hz, 1H), 7,34 (d, J=9,5 Hz, 1H), 6,51-6,56 (m, 1H), 4,55-4,66 (m, 2H), 3,39 3,43 (m, 1H), 3,21-3,28 (m, 1H), 3,10-3,19 (m, 1H), 3,04 (dd, J=11,5, 7,0 Hz, 1H), 2,86 (hept, J=7,0 Hz, 1H), 2,20 (dtd, J=13,5, 8,1, 5,5 Hz, 1H), 1,84 (dq, J=13,5, 7,5 Hz, 1H).
Ejemplo de referencia 17-9, Enatiómero 2 LCMS tR = 0,47 min (método Q de LCMS); [M+H]: 338,161; 1H RMN (400 MHz, DMSO-d6) 513,0 (s a, 1H), 8,52 (d, J=2,5 Hz, 1H), 8,44 (d, J=9,5 Hz, 1H), 8,04 (d, J=8,5 Hz, 1H), 7,76 (d, J=2,0 Hz, 1H), 7,49 (d, J=2,0 Hz, 1H), 7,46 (dd, J=8,5, 2,0 Hz, 1H), 7,45 (d, J=9,5 Hz, 1H), 6,52-6,59 (m, 1H), 4,42-4,60 (m, 2H), 3,36 (dd, J=11,5, 8,0 Hz, 1H), 3,22-3,32 (m, 1H), 3,16 (m, 1H), 3,07 (dd, J=11,5, 7,0 Hz, 1H), 2,84 (pentet, J=7,5 Hz, 1H), 2,14 (dtd, J=13,0, 7,5, 5,5 Hz, 1H), 1,81 (dq, J=13,0, 7,5 Hz, 1H).
Los siguientes compuestos finales se prepararon utilizando procedimientos similares a los de los Ejemplos o ejemplos de referencia de 17-1 a 17-9, y los métodos generales que se resumen en la sección MÉTODOS GENÉRALES cuando sea a ro iado.

Ejemplo de referencia 17-13: Síntesis de la sal clorhidrato de 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol

Paso 1: 3-(4-Cloro-2-metoxifenil)-6-((2,2,6,6-tetrametilpiperidin-4-H)oxi)piridazina
Una mezcla de ácido 4-doro-2-metoxifenilborónico (1,12 g, 6,00 mmol), Intermedio 1-3 (1,94 g, 7,20 mmol), SiliaCat® DPP-Pd (1,15 g, 0,30 mmol) y carbonato de potasio en etanol/agua (10:1,33 mL) se calentó a reflujo durante 5 h. El disolvente se evaporó y el residuo marrón resultante se repartió entre DCM y una solución acuosa al 8 % de K2 CO3. Después de la separación, la capa acuosa se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron a sequedad al vacío. El material crudo se purificó mediante cromatografía ultrarrápida (amina 30 uM, -(CH2)3NH2, gel de sílice funcionalizado, de un 30 a un 100 % de Et2O en heptano) para obtener una mezcla (1,76 g) del producto deseado (75 %) y el Intermedio 1-3 (25 %). [M+H]: 376,3; 1H RMN (400 MHz, DMSO-d6) 67,91 (d, J=9,0 Hz, 1H), 7,71 (d, J=8,0 Hz, 1H), 7,27 (d, J=2,0 Hz, 1H), 7,12 7,20 (m, 2H), 5,68 (tt, J=11,0, 4,0 Hz, 1H), 3,85 (s, 3H), 1,99-2,11 (m, 2H), 1,20-1,30 (m, 8H), 1,10 (s, 6H).
Paso 2: 3-(2-Metoxi-4-(1H-pirazol-4-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina
Se añadió SiliaCat® DPP-Pd (0,37 g, 0,10 mmol) a un vial apto para microondas que contenía una mezcla de 3-(4-cloro-2-metoxifenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina (0,36 g, 0,96 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol (0,47 g, 2,4 mmol) y K2CO3 (0,53 g, 3,8 mmol) en etanol/agua (10:1,6,6 mL). La mezcla de reacción se selló, a continuación se calentó en un reactor de microondas a 160 °C durante 1 h. Después de enfriar, la reacción se purificó mediante extracción en fase sólida (5 g de SiliaBond® carbonato, MeOH como eluyente). El filtrado se concentró al vacío y el residuo resultante se purificó mediante cromatografía ultrarrápida (gel de sílice saturado con Et3N, de un 2 a un 25 % de MeOH en DCM) para obtener el producto deseado (0,08 mg, 21 %). [M+H]: 408,4; 1H RMN (400 MHz, DMSO-d6) 613,01 (s a, 1 H), 8,33 (s a, 1 H), 8,05 (s a, 1 H), 7,94 (d, J=9,5 Hz, 1 H), 7,70 (d, J=8,0 Hz, 1H), 7,38 (d, J=1,5 Hz, 1H), 7,34 (dd, J=8,0, 1,5 Hz, 1H), 7,14 (d, J=9,5 Hz, 1H), 5,57-5,74 (m, 1H), 3,90 (s, 3H), 2,00-2,14 (m, 2H), 1,17-1,31 (m, 8H), 1,11 (s, 6H).
Paso 3: 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol. Sal clorhidrato
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección utilizando tiofenol, se trató 3-(2-metoxi-4-(1 H-pirazol-4-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina (0,58 mg, 1,41 mmol) con tiofenol (0,15 mg, 1,36 mmol) y K2CO3 (0,23 mg, 1,64 mmol) en NMP (9 mL) durante 15 min a 190 °C. Después de la purificación (0,1 % de ácido trifluoroacético como modificador), en las fracciones deseadas se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (5 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se evaporó a presión reducida. El sólido beige resultante se disolvió en CH3CN/H2O/MeOH (6/1/6 mL), se añadió SiliaMetS® DMT (2,7 g, 1,4 mmol) y la mezcla se agitó durante 18 h. A continuación, la mezcla se filtró a través de un pequeño tapón de celite y el filtrado se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (5 g, MeOH como eluyente y una solución 2 N de amoniaco en MeOH para liberar el material). El disolvente se concentró al vacío y el sólido resultante se suspendió en CH3CN/H2O (4/1 mL). Se añadió HCl 4 N en 1,4-dioxano (4 equivalentes) y el disolvente se concentró al vacío para proporcionar un sólido amarillo (59 mg, 9 %). LCMS tR = 0,51 min (método Q de LCMS); [M+H]: 394,2225; 1H RMN (400 MHz, DMSO-d6) 69,10 (d, J=12,0 Hz, 1H), 8,49 (d, J=9,5 Hz, 1H), 8,36 (d, J=12,0 Hz, 1H), 8,16 (s, 1H), 7,94 (d, J=8,0 Hz, 1H), 7,47 (d, J=9,5 Hz, 1 H), 7,21-7,28 (m, 2H), 5,63-5,87 (m, 1H), 2,34 (dd, J=13,0, 4,0 Hz, 2H), 1,81 (dd, J=13,0, 10,5 Hz, 2H), 1,53 (s, 6H), 1,50 (s, 6H).
Ejemplo de referencia 18-1: Síntesis de la sal de HCl y 2-(6-piperazin-1-ilpiridazin-3-il)-5-pirazol-1-ilfenol

Paso 1 (N-arilación): 4-(6-(2-Metoxi-4-(1H-pirazol-1-il)fenil)piridazin-3-il)piperazino-1-carboxilato de tert-butilo Siguiendo el MÉTODO GENERAL 6-1 para SNAr, se combinaron el Intermedio 2-1 (50 mg, 0,174 mmol), piperazino-1-carboxilato de tert-butilo (59 mg, 0,314 mmol), DIPEA (0,06 mL, 0,349 mmol), y n-butanol (0,1 mL) en un vial de reacción de 4 mL y se calentaron a 120 °C durante toda la noche. La reacción se enfrió hasta TA y se añadió EtOAc. El sólido blanco que se formó se separó por filtración y se lavó con EtOAc, se disolvió en DCM y se lavó con H2O. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró al vacío para proporcionar el producto deseado. Paso 2: Se combinaron 4-(6-(2-metoxi-4-(1H-pirazol-1-il)fenil)piridazin-3-il)piperazino-1-carboxilato de tert-butilo (49,2 mg, 0,11 mmol), K2CO3 (15,6 mg, 0,11 mmol), tiofenol (17,2 uL, 0,17 mmol) y NMP (0,23 mL) en un vial apto para microondas de 2 mL y se calentaron en el microondas a 190 °C durante 0,5 h. La mezcla de reacción se enfrió hasta TA y se acidificó con ácido cítrico ac. al 5 %. Se añadió EtOAc y el producto precipitado resultante se aisló por filtración. Paso 3: El precipitado del Paso 2 se disolvió en HCl 4 M en 1,4-dioxano (2 mL), se agitó a TA durante 0,5 h y a continuación se concentró para proporcionar 2-(6-(piperazin-1-il)piridazin-3-il)-5-( 1 H-pirazol-1 -il)fenol, (11,0 mg, 0,03 mmol, 30 % rendimiento). LCMS tR = 0,46 min, M+1 = 323,5 (método Q de LCMS). 1H RMN (DMSO-d6) 59,32 (s a, 2H), 8,60 (d, J=2,3 Hz, 1H), 8,40 (d, J=10,0 Hz, 1H), 8,01 (d, J=8,8 Hz, 1H), 7,71-7,85 (m, 2H), 7,43-7,56 (m, 2H), 6,54-6,63 (m, 1H), 3,86-4,02 (m, 4H), 3,41-3,78 (m, 2H), 2,60-2,83 (m, 2H).
-





Ejemplo 19-1: Síntesis de 5-pirazol-1 -il-2-[6-(1,2,3,6-tetrahidropiridin-4-il)piridazin-3-M]fenol

Paso 1: Se preparó 4-(6-(2-metoxi-4-(1H-pirazol-1-il)feml)pindazin-3-il)-5,6-dihidropindmo-1(2H)-carboxilato de tertbutilo (151 mg, 0,35 mmol, 100 % de rendimiento) a partir del Intermedio 2-1 y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidropindino-1(2H)-carboxilato de tert-butilo utilizando el Método general 1-4 para el acoplamiento de Suzuki. Lc Ms tR = 1,58 min, M+1 = 434,8 (condición B).
Paso 2: Se preparó 5-(1H-p¡razol-1-¡l)-2-(6-(1,2,3,6-tetrah¡drop¡r¡d¡n-4-¡l)p¡ridaz¡n-3-¡l)fenol siguiendo el Método general 3-2 para la desprotecc¡ón con BBr3.
Se disolvió 4-(6-(2-metox¡-4-(1H-p¡razol-1-¡l)fen¡l)p¡r¡daz¡n-3-¡l)-5,6-d¡hidrop¡r¡d¡no-1(2H)-carbox¡lato de ferf-butilo (50 mg, 0,12 mmol) en DCM (0,6 mL). La solución se enfrió hasta -78 °C y se añadió gota a gota BBr31 M en DCM (0,6 mL). Se retiró la suspensión resultante del baño de hielo y se agitó a TA durante toda la noche. La reacción se desactivó con agua, se diluyó en MeOH y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces (5-7 volúmenes de columna) con MeOH y a continuación se eluyó con NH32 N en MeOH para proporcionar el producto deseado, 5-(1H-p¡razol-1-¡l)-2-(6-(1,2,3,6-tetrah¡drop¡r¡d¡n-4-il)p¡r¡daz¡n-3-¡l)fenol, (9,7 mg, 0,03 mmol, 25 % de rendimiento). LCMS tR = 0,49 min, M+1 = 320,1 (método Q de LCMS). 1H RMN (DMSO-d6) 8 8,64 (d, J=2,3 Hz, 1H), 8,52 (d, J=9,5 Hz, 1H), 8,19 (dd, J=9,0, 3,0 Hz, 2H), 7,80 (d, J=1,3 Hz, 1H), 7,43-7,61 (m, 2H), 6,89-7,03 (m, 1H), 6,52-6,66 (m, 1H), 3,56-3,73 (m, 2H), 3,02-3,15 (m, 2H), 2,66-2,74 (m, 2H).
Ejemplo 19-2: Síntesis de 2-(6-piperidin-4-ilpiridazin-3-il)-5-pirazol-1-ilfenol

A un matraz de fondo redondo que contenía Pd al 10 %/C (27,7 mg, 0,026 mmol), se añadió 5-pirazol-1-il-2-[6-(1,2,3,6-tetrahidrop¡r¡d¡n-4-¡l)p¡r¡daz¡n-3-¡l]fenol (Ejemplo 19-1) (166 mg, 0,52 mmol) en MeOH (2,5 mL). Se burbujeó H2 a través de la solución durante 5 min y a continuación se agitó la reacción en H2 a 55 psi a TA. Después de 18 h, la mezcla de reacción se filtró a través de celite y se lavó con MeOH. El disolvente se eliminó al vacío, el aceite resultante se disolvió en MeOH y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces (5-7 volúmenes de columna) con MeOH y a continuación se eluyó con NH32 N en MeOH para proporcionar el producto deseado, 2-(6-(p¡per¡d¡n-4-¡l)p¡r¡daz¡n-3-il)-5-(1H-p¡razol-1-¡l)fenol (19,8 mg, 0,06 mmol, 11 % de rendimiento). LCMS tR = 0,48 min, M+1 = 322,6 (método Q de LCMS). 1H RMN (DMSO-d6) 88,63 (d, J=2,5 Hz, 1H), 8,50 (d, J=9,3 Hz, 1H), 8,15 (d, J=8,5 Hz, 1H), 7,76-7,92 (m, 2H), 7,47-7,56 (m, 2H), 6,59 (dd, J=2,5, 1,8 Hz, 1H), 3,24 (s, 2H), 3,07-3,19 (m, 1H), 2,84 (td, J=12,2, 2,4 Hz, 2H), 2,00 (s, 2H), 1,77-1,92 (m, 2H).
L i i n m r r r n iliz n r imi n imil r l l E m l 1 -1 1 -2:


Ejemplo de referencia 20-1: Síntesis de 3-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)naftaleno-2,7-diol
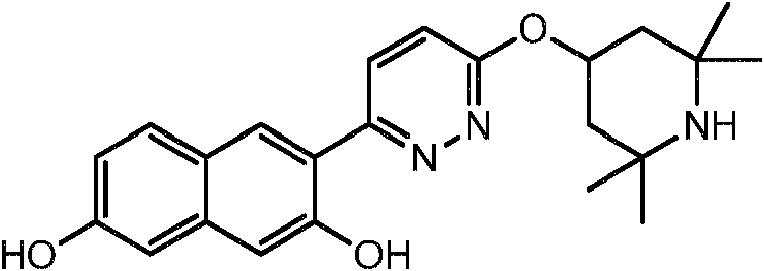
Paso 1: Siguiendo el procedimiento representativo MÉTODO GENERAL 1-1, acoplamiento cruzado de Suzuki, se hicieron reaccionar el Intermedio 3-1 y el Intermedio 1-3 para proporcionar 7-(benciloxi)-6-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)naftalen-2-ol (163 mg, 0,337 mmol). LCMS tR = 1,12 min, M+1 = 484,3 (condición B).
Paso 2: 3-(6-((2,2,6,6-Tetrametilpiperidin-4-il)oxi)piridazin-3-il)naftaleno-2,7-diol
(33 mg, 0,084 mmol, 54,1 % de rendimiento) se preparó a partir de 7-(bencNoxi)-6-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)naftalen-2-ol siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis. LCMS tR = 0,50 min (método Q de LCMS); MS (M+1) = 394,5. 1H RMN (400 MHz, DMSO-d6) 5 ppm 12,40 (s a, 1H), 9,84 (s, 1H), 8,47 (d, J=9,54 Hz, 1H), 8,43 (s, 1H) 7,74 (d, J=8,78 Hz, 1H), 7,40 (d, J=9,54 Hz, 1H), 7,09 (s, 1H), 6,87-6,95 (m, 2H), 5,62 5,73 (m, 1H), 2,11 (d, J=9,29 Hz, 2H), 1,39 (s a, 2H). 1,25 (s a, 6H), 1,12 (s a, 6H).
Ejemplo de referencia 20-2: Síntesis de 3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftaleno-2,7-diol

Paso 1: Siguiendo el procedimiento representativo MÉTODO GENERAL 1-1, acoplamiento cruzado de Suzuki, se hicieron reaccionar el Intermedio 3-1 y el Intermedio 1-1 para proporcionar 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (1,25 g, 2,36 mmol, 89 % de rendimiento). Lc Ms tR = 0,97 min, M 1 = 497,8 (condición C). 1H RMN (400 MHz, METANOL-d4) ó ppm 7,91 (s, 1H), 7,76 (d, J=9,54 Hz, 2H), 7,66 (d, J=8,78 Hz, 2H), 7,34-7,40 (m, 2H), 7,31 (s, 2H), 7,18-7,27 (m, 2H), 6,97-7,05 (m, 2H), 6,91 (dd, J=8,78, 2,26 Hz, 1H), 5,18 (m, 3H), 2,92 (s, 3H), 1,60-1,68 (m, 2H), 1,45-1,55 (m, 2H), 1,32 (s, 6H), 1,18 (s, 6H).
Paso 2: Se preparó 3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftaleno-2,7-diol (43 mg, 0,097 mmol, 48,2 % de rendimiento) a partir de 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol siguiendo el MéTo DO GENERAL 4-1 para la hidrogenólisis. LCMS tR = 0,48 min, M 1 = 407,2 (método Q de LCMS). 1H RMN (400 MHz, METANOL-d4) ó ppm 8,27 (d, J=10,04 Hz, 1H), 8,20 (s a, 3H), 7,71 (d, J=8,78 Hz, 1H), 7,35 (d, J=9,79 Hz, 1H), 7,05 (s, 1H), 6,85-6,94 (m, 2H), 5,27 (s a, 1H), 3,03 (s, 3H), 1,71-1,91 (m, 4H), 1,54 (s, 6H), 1,39 (s, 6H).
Ejemplo de referencia 20-3: Síntesis de 3-(6-((2,2,6,6-tetrametilpiperidm-4-N)amino)piridazm-3-N)naftaleno-2,7-diol
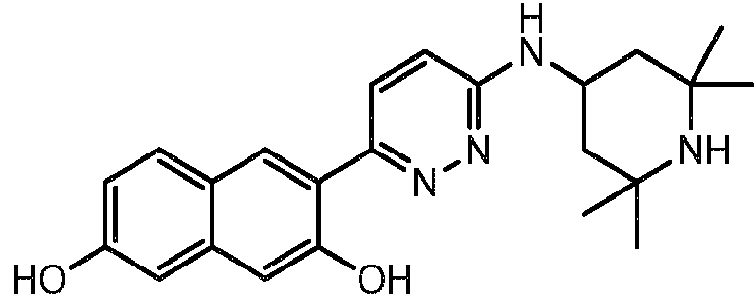
Paso 1: Siguiendo el procedimiento representativo MÉTODO GENERAL 1-1, acoplamiento cruzado de Suzuki, se hicieron reaccionar el Intermedio 3-1 y el Intermedio 1-2 para proporcionar 7-(benciloxi)-6-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (49 mg, 0,096 mmol, 52 % de rendimiento). LCMS tR = 1,16 min (condición C); MS (M+1) = 483,8.
Paso 2: Se preparó 3-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftaleno-2,7-diol (3 mg, 0,007 mmol, 38 % de rendimiento) a partir de 7-(benciloxi)-6-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol siguiendo el MÉTODO g En ERAL 4-1 para la hidrogenólisis. LCMS tR = 0,47 min (método Q de LCMS); MS (M+1) = 393,1. 1H RMN (400 MHz, METANOL-d4) ó ppm 8,42 (d, J=9,29 Hz, 1H), 8,16 (s, 1H), 7,79 (d, J=8,53 Hz, 1H), 7,64 (d, J=9,54 Hz, 1H), 7,15 (s, 1 H), 6,96-7,02 (m, 2H), 4,51 (t, J=11,92 Hz, 1H), 2,96-3,04 (m, 1H), 2,68 (s, 1H), 2,35 (dd, J=13,55, 3,01 Hz, 2H), 1,69-1,75 (m, 2H), 1,64 (s, 6H), 1,56 (s, 6H).
Ejemplo de referencia 20-4: Síntesis del éster ferf-butílico del ácido [3-(7-hidroxi-6-{6-[metil-(2,2,6,6-tetrametilpiperidin-4-il)amino]piridazin-3-il}naftalen-2-iloxi)propil]carbámico

Paso 1: (3-((7-(Benciloxi)-6-bromonaftalen-2-il)oxi)propil)carbamato de tert-butilo
Siguiendo el MÉTODO GENERAL 5-1 para la alquilación de fenol utilizando 7-(benciloxi)-6-bromonaftalen-2-ol (506,5 mg, 1,54 mmol) y 3-bromopropilcarbamato de tert-butilo (409 mg, 1,72 mmol) y después cromatografía en columna (eluyendo con un 3-80 % de EtOAc/heptano) se obtuvo (3-((7-(benciloxi)-6-bromonaftalen-2-il)oxi)propil)carbamato de tert-butilo como un sólido blanquecino (706 mg, 94 % de rendimiento). MS (M-Boc) = 388,3. 1H r Mn (400 MHz, CLOROFORMO-d) 6 ppm 7,99 (s, 1H), 7,59 (d, J=9,60 Hz, 1H), 7,54 (d, J=8,08 Hz, 2H), 7,42 (t, J=7,58 Hz, 2H), 7,32 7,37 (m, 1 H), 7,13 (s, 1 H), 7,05-7,00 (m, 2H), 5,26 (s, 2H), 4,68 (s a, 1 H), 4,14 (t, J=6,06 Hz, 2H), 3,37 (c, J=6,40 Hz, 2H), 2,09-2,01 (m, 2H), 1,47 (s, 9H).
Paso 2: (3-((7-(Benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando (3-((7-(benciloxi)-6-bromonaftalen-2-il)oxi)propil)carbamato de tert-butilo (659 mg, 1,36 mmol) se obtiene (3-((7-(benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo como un sólido blanquecino (662 mg, 87 % de rendimiento). MS (M-Bu) = 478,0. 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 8,16 (s, 1H), 7,65-7,72 (m, 3H), 7,39 (t, J=7,33 Hz, 2H), 7,29-7,34 (m, 1 H), 7,08 (s, 1 H), 7,04 (d, J=2,53 Hz, 1 H), 6,98 (dd, J=8,84, 2,27 Hz, 1H), 5,23 (s, 2H), 4,70 (s a, 1H), 4,16 (t, J=6,06 Hz, 2H), 3,38 (c, J=6,57 Hz, 2H), 2,00-2,10 (m, 2H), 1,47 (s, 9H), 1,41 (s, 12H).
Paso 3: (3-((7-(Benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo
Se hicieron reaccionar el Intermedio 1-1 (242,5 mg, 0,86 mmol) y (3-((7-(benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo (662 mg, 1,18 mmol) siguiendo el MÉTODO GENERAL 1-1 para el acoplamiento de Suzuki con las siguientes modificaciones: En lugar de purificación mediante SCX, el material crudo se purificó mediante cromatografía ultrarrápida (eluyendo con un 1-15 % de NH3 7 N en MeOH/DCM) para proporcionar un aceite amarillo. El aceite se lavó con Et2O y a continuación se concentró al vacío a sequedad para proporcionar (3-((7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo como un sólido amarillo pálido (492 mg, 83 % de rendimiento). MS (M+1) = 654,8.
Paso 4: (3-((7-Hidroxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo
Se añadió Pd/C (10 % p, 80 mg, 0,075 mmol) a una solución en EtOAc (5 mL)/MeOH (5 mL) de (3-((7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)propil)carbamato de tert-butilo (492 mg, 0,752 mmol) a TA. En la mezcla de reacción se hizo vacío y se rellenó con H2 (2X) y a continuación se agitó a TA en H2 (1 atm) durante 16 h. La mezcla de reacción se filtró a través de celite, se lavó con MeOH y a continuación se concentró el lavado al vacío a sequedad para proporcionar un sólido tostado. El sólido se disolvió en DCM y a continuación se adsorbió sobre una columna con amina unida a la sílice (columna de 10 g de Si-NH2 , marca Varian, Bond Elut NH2). A continuación, la columna se lavó con MeOH (150 mL) y a continuación el disolvente se concentró al vacío para proporcionar un aceite amarillo. El aceite se disolvió en Et2O y a continuación se concentró al vacío para proporcionar el compuesto del título como un sólido beige (386 mg, 91 % de rendimiento). MS (M+1) = 564,3; 1H RMN (400 MHz, DMSO-d6) 6 ppm 13,29 (s a, 1H), 8,34 (s, 1H), 8,28 (d, J=10,10 Hz, 1H), 7,76 (d, J=9,09 Hz, 1H), 7,34 (d, J=10,11 Hz, 1 H), 7,15 (s, 1H), 7,08 (d, J=2,53 Hz, 1H), 6,94 (dd, J=9,09, 2,53 Hz, 1H), 4,89-5,06 (m, 1H), 4,56 (s, 1H), 4,09 (t, J=6,32 Hz, 2H), 3,09-3,16 (m, 2H), 2,98 (s, 3H), 1,80-1,95 (m, 2H), 1,50-1,61 (m, 2H), 1,44 (t, J=12,38 Hz, 2H), 1,38 (s, 9H), 1,27 (s, 6H), 1,10 (s, 6H).
Ejemplo de referencia 20-5: Síntesis de 7-(3-ammopropoxi)-3-{6-[metil(2,2,6,6-tetrametilpiperidm-4-il)amino]piridazin-3-il}naftalen-2-ol

Se añadió TFA (1,5 ml, 19,47 mmol) a una solución del Ejemplo de referencia 20-4 (289,4 mg, 0,513 mmol) en DCM (6 ml) a TA. La mezcla de reacción se agitó a TA durante 30 min, se diluyó con MeOH y a continuación se adsorbió sobre una columna de SCX acondicionada con MeOH (10 g). La columna se lavó varias veces con MeOH y a continuación se eluyó con NH33 N en MeOH. La evaporación del disolvente proporcionó el compuesto del título como un sólido beige (178 mg, 75 % de rendimiento). Lc Ms tR = 0,44 min (método Q de LCMS); MS (M+1) = 464,2. 1H RMN (400 MHz, DMSO-d6) 6 ppm 8,35 (s, 1 H), 8,28 (d, J=9,60 Hz, 1 H), 7,77 (d, J=9,09 Hz, 1H), 7,36 (d, J=9,60 Hz,
1H), 7,16-7,18 (m, 1H), 7,11 (d, J=2,53 Hz, 1H), 6,96 (dd, J=8,84, 2,27 Hz, 1H), 4,91-5,04 (m, 1H), 4,56 (s, 1H), 4,17 (t, J=6,32 Hz, 2H), 2,98 (s, 3H), 2,88 (d, J=7,07 Hz, 2H), 1,90-2,05 (m, 2H), 1,46-1,62 (m, 4H), 1,30 (s, 6H), 1,14 (s, 6H).
Ejemplo de referencia 20-6: Síntesis de N-[3-(7-hidroxi-6-{6-[metil-(2,2,6,6-tetrametilpiperidm-4-il)ammo]piridazm-3-M}naftalen-2-Moxi)proμM]acetamida

Se añadió N-acetoxisuccinimida (98 mg, 0,624 mmol) a una solución del Ejemplo de referencia 20-5 (50 mg, 0,108 mmol) y TEA (0,747 mL, 5,39 mmol) en DMSO (2 mL) a TA. La mezcla de reacción se agitó TA durante 30 min y a continuación se concentró al vacío para eliminar la Et3 N en exceso. El residuo resultante se purificó mediante HPLC preparativa (eluyendo con un 5-80 % de MeCN/H2O con un 0,1 % de TFA como modificador). Las fracciones apropiadas que contenían el producto se combinaron y a continuación se adsorbieron sobre una columna de SCX acondicionada con MeOH (5 g, marca BSA Varian). La columna se lavó varias veces con MeOH y a continuación se eluyó con NH33 N en MeOH. La evaporación del disolvente proporcionó un aceite amarillo. Se añadió Et2O al aceite y a continuación se concentró a sequedad para proporcionar el compuesto del título como un sólido naranja amarillento (30 mg, 55 % de rendimiento). LCMS tR = 0,52 min (método Q de LCMS); MS (M+1) = 506,2. 1H RMN (400 MHz, DMSO-d6 ) 6 ppm 13,27 (s a, 1H), 8,35 (s, 1H), 8,28 (d, J=10,10 Hz, 1H), 7,77 (d, J=9,09 Hz, 2H), 7,35 (d, J=9,60 Hz, 1H), 7,17 (s, 1H), 7,09 (d, J=2,02 Hz, 1H), 6,95 (dd, J=9,09, 2,53 Hz, 1H), 4,92-5,04 (m, 1H), 4,11 (t, J=6,32 Hz, 2H), 3,20-3,28 (m, 2H), 2,98 (s, 3H), 1,87-1,96 (m, 2H), 1,82 (s, 3H), 1,55-1,63 (m, 2H), 1,44-1,55 (m, 2H), 1,30 (s, 6H), 1,14 (s, 6H).
Ejemplo de referencia 20-7: Síntesis de 7-(3-hidroxipropoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol

Siguiendo el MÉTODO GENERAL 5-1 para la alquilación de fenol, se añadió Cs2CO3 (485 mg, 1,489 mmol) a una solución de 7-(benciloxi)-6-bromonaftalen-2-ol (490 mg, 1,489 mmol) en acetona (14,90 mL) a TA. La mezcla de reacción se agitó durante 5 min y a continuación se añadió ((3-bromopropoxi)metil)benceno (682 mg, 2,98 mmol), y después se añadió Nal (446 mg, 2,98 mmol). La mezcla de reacción se calentó hasta 60 °C y se agitó durante toda la noche, a continuación se filtró, se lavó con acetona y se concentró al vacío. El residuo resultante se repartió entre Et2O (60 mL) y agua (20 mL). Después de la separación, la capa orgánica se lavó con una solución ac. saturada de sulfito de sodio (20 mL), Na2CO32 M y salmuera. La capa orgánica se secó a continuación con MgSO4 , se filtró y se concentró al vacío. La cromatografía en gel de sílice (10-60 % de EtOAc/heptano) proporcionó 3-(benciloxi)-6-(3-(benciloxi)propoxi)-2-bromonaftaleno (422 mg, 0,884 mmol, 60 % de rendimiento) como un sólido blanco (eluyendo con un 30 % de EtOAc). LCMS tR = 1,81 min (condición C); LCMS (M+1) = 479,9.
Paso 2: Se preparó 2-(3-(benciloxi)-6-(3-(benciloxi)propoxi)naftalen-2-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (355 mg, 0,677 mmol, 81 % de rendimiento) a partir del bromuro utilizando MÉTODO GENERAL 2-1 para la formación del éster de boronato. LCMS tR = 1,89 min (condición C); MS (M+1) = 525,3.
Paso 3:
Se preparó 6-(3-(benciloxi)-6-(3-(benciloxi)propoxi)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (59 mg, 0,091 mmol, 27 % de rendimiento) mediante una reacción de Suzuki a partir del éster borónico y el Intermedio 1-1 siguiendo el MÉTODO GENERAL 1-1. LCMS tR = 1,50 min (condición C); MS (M+1) = 645,3.
Paso 4: Se preparó 7-(3-hidroxipropoxi)-3-{6-[metil(2,2,6,6-tetrametilpiperidin-4-il)amino]piridazin-3-il}naftalen-2-ol (10 mg, 0,021 mmol, 23 % de rendimiento) a partir de 6-(3-(benciloxi)-6-(3-(benciloxi)propoxi)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina siguiendo el MÉTODO GENERAL 4-1 para la h¡drogenól¡s¡s. LCMS tR = 0,53 min (método Q de LCMS); MS (M+1) = 465,2. 1H RMN (400 MHz, DICLOROMETANO-d2) 5 ppm 7,97-8,08 (m, 2H), 7,63-7,72 (m, 1H), 7,19 (d, J=9,79 Hz, 1H), 7,04-7,12 (m, 1H), 7,01 (d, J=8,28 Hz, 1H), 6,89-6,97 (m, 1H), 4,99 (s a, 1H), 4,13-4,24 (m, 2H), 3,74-3,85 (m, 2H), 3,30-3,38 (m, 1H) 2,97 (s a, 3H), 2,12 (s a, 1H) 2,01-2,10 (m, 2H), 1,65-1,73 (m, 2H), 1,39-1,50 (m, 2H), 1,34 (s a, 6H), 1,17 (s a, 6H).
Ejemplo de referencia 20-8: Síntesis de 7-(3-metoxipropoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol

Se preparó 7-(3-metoxipropoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (15 mg, 0,031 mmol, 27 % de rendimiento) de una manera similar al Ejemplo de referencia 21-7. LCMS tR = 0,53 min (método Q de LCMS); MS (M+1) = 479,2. 1H RMN (400 MHz, DICLOROMETANO-d2) 5 ppm 8,01-8,07 (m, 2H), 7,69 (d, J=9,09 Hz, 2H), 7,20 (s, 1H), 7,09 (d, J=9,85 Hz, 1H), 7,00 (d, J=2,02 Hz, 1H), 6,95 (d, J=8,84 Hz, 1H), 5,42 (t, J=12,51 Hz, 1H), 4,15 (t, J=6,32 Hz, 2H), 3,58 (t, J=6,19 Hz, 2H), 3,35 (s, 3H), 3,01 (s, 3H), 1,94-2,14 (m, 4H), 1,77 (dd, J=13,39, 3,28 Hz, 2H), 1,58 (s, 6H), 1,51 (s, 6H).
Ejemplo de referencia 20-9: Síntesis de 7-(2-morfolmoetoxi)-3-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazm-3-il)naftalen-2-ol

Se preparó 7-(2-morfolinoetoxi)-3-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)naftalen-2-ol de manera similar al Ejemplo de referencia 21-7. LCMS tR = 0,43 min (método Q de LCMS); MS (M+1) = 507,3. 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,49 (d, J=9,79 Hz, 1H), 8,34 (s, 1H) 7,79 (d, J=9,03 Hz, 1H), 7,36 (d, J=9,54 Hz, 1H), 7,24 (s, 1H), 7,12 (d, J=2,26 Hz, 1H), 7,01 (dd, J=9,03, 2,51 Hz, 1H), 5,80-5,91 (m, 1H), 4,29 (t, J=5,40 Hz, 2H), 3,73-3,79 (m, 4H), 2,91 (t, J=5,40 Hz, 2H), 2,63-2,72 (m, 4H), 2,51 (dd, J=13,80, 4,02 Hz, 2H), 1,85 (dd, J=13,43, 10,92 Hz, 2H), 1,64 (s, 6H), 1,56 (s, 6H).
Ejemplo de referencia 21-1: Síntesis de 3-(6-(piperidin-4-ilmetil)piridazin-3-il)naftalen-2-ol

Paso 1: 4-((6-Cloropiridazin-3-il)metil)piperidino-1-carboxilato de tert-butilo
A un matraz que contenía 4-metilenopiperidino-1-carboxilato de tert-butilo (487 mg, 2,47 mmol) se añadió 9-borabiciclo[3.3.1]nonano 0,5 M en THF (5,4 mL, 2,72 mmol). La reacción se calentó a reflujo a 65 °C durante 1 h, a continuación se añadió a una suspensión desgasificada de 3,6-dicloropiridazina (368 mg, 2,47 mmol), K2CO3 (1,0 g, 7,41 mmol) y PdCl2(dppf)CH2Cl2 (101 mg, 0,12 mmol) en 1,4-dioxano (5,2 mL) y agua (0,88 mL). La mezcla de reacción resultante se calentó a 60 °C durante 3 h, a continuación se enfrió hasta TA y se diluyó con EtOAc. La suspensión se filtró a través de celite y se concentró al vacío. La cromatografía en gel de sílice, eluyendo con un 0-100 % de EtOAc/heptano, proporcionó el producto, 4-((6-cloropiridazin-3-il)metil)piperidino-1-carboxilato de tert-butilo (376 mg,
1,21 mmol, 49 % de rendimiento). 1H RMN (DMSO-d6) 6: 7,85 (d, J=8,8 Hz, 1H), 7,70 (d, J=8,8 Hz, 1H), 3,74-4,05 (m, 2H), 2,85 (d, J=7,3 Hz, 2H), 2,54-2,79 (m, 2H), 1,82-2,01 (m, 1H), 1,48-1,62 (m, 2H), 1,38 (s, 9H), 0,97-1,18 (m, 2H).
Paso 2: Se preparó 4-((6-(3-metoxinaftalen-2-il)piridazin-3-il)metil)piperidino-1-carboxilato de terí-butilo (63 mg, 0,145 mmol, 45 % de rendimiento) a partir de 4-((6-cloropiridazin-3-il)metil)piperidino-1-carboxilato de terí-butilo y ácido (3-metoxinaftalen-2-il)borónico utilizando el METODO GENERAL 1-4.
Paso 3: Se preparó 3-(6-(piperidin-4-ilmetil)piridazin-3-il)naftalen-2-ol (31 mg, 0,097 mmol, 67 % de rendimiento) a partir de 4-((6-(3-metoxinaftalen-2-il)piridazin-3-il)metil)piperidino-1-carboxilato de te/Y-butilo utilizando el MÉTODO GENERAL 3-2. LCMS tR = 0,53 min, M+1 = 320,2 (método Q de LCMS). 1H RMN (DMSO-d6) 6 8,62 (s, 1H), 8,51 (d, J=9,0 Hz, 1H), 7,93 (d, J=8,0 Hz, 1H), 7,82 (d, J=9,0 Hz, 1H), 7,76 (d, J=8,0 Hz, 1H), 7,48 (ddd, J=8,2, 6,9, 1,3 Hz, 1H), 7,29-7,40 (m, 2H), 2,85-2,99 (m, 4H), 2,41-2,48 (m, 2H), 1,84-1,99 (m, 1H), 1,48-1,65 (m, 2H), 1,10-1,27 (m, 2H).
Ejemplo de referencia 21-2: Síntesis de 5-(1H-pirazoM-N)-2-(6-((2,2,6,6-tetrametilpiperidin-4-N)metil)piridazin-3-il)fenol
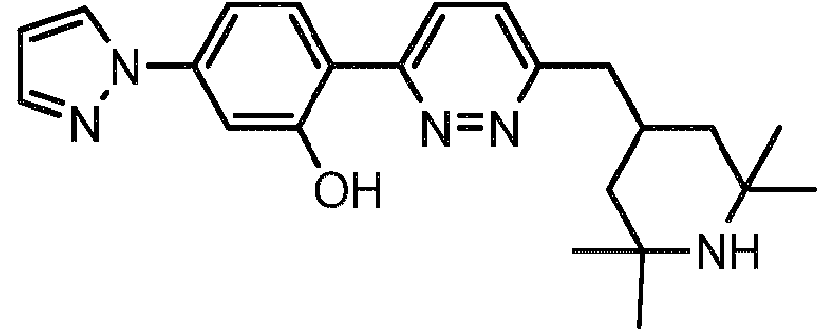
Paso 1: Trifluoroacetato de 2,2,6,6-tetrametil-4-metilenopiperidina
A un matraz de 100 mL que contenía bromuro de metiltrifenilfosfonio (5,2 g, 14,5 mmol) en éter (8 mL) enfriado hasta 0° C se añadió tert-butóxido de potasio (2,2 g, 19,3 mmol). La suspensión resultante se agitó durante 0,5 h a 0 °C y después se añadió gota a gota de 2,2,6,6-tetrametilpiperidin-4-ona (1,0 g, 6,44 mmol) en éter (5 mL). La mezcla resultante se agitó a TA durante toda la noche en cuyo momento se añadieron más tert-butóxido de potasio (0,72 g, 6,44 mmol) y bromuro de metiltrifenilfosfonio (1,7 g, 4,83 mmol). La reacción se agitó a TA durante 4 h, a continuación se enfrió hasta 0 °C y se desactivó con agua, se acidificó con una solución acuosa de HCl 1 M y se lavó con éter (3X). El pH de la mezcla acuosa se ajustó a 10 con NaOH 2 M y se extrajo con éter (3X). El extracto orgánicos se acidificó con ácido trifluoroacético, se secó con sulfato de sodio y se concentró al vacío para proporcionar como un aceite marrón el producto deseado, la sal trifluoroacetato de 2,2,6,6-tetrametil-4-metilenopiperidina (1,7 g, 4,46 mmol, 70 % de rendimiento), 1H RMN (DMSO-d6) 68,53 (s a, 2H), 5,00 (s, 2H), 2,20-2,30 (m, 4H), 1,33 (s, 12H).
Paso 2: 3-(2-Metoxi-4-(1H-pirazol-1-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina
A un matraz que contenía trifluoroacetato de 2,2,6,6-tetrametil-4-metilenopiperidina (250 mg, 0,936 mmol) se añadió 9-borabiciclo[3.3.1]nonano 0,5 M en THF (3,7 mL, 1,87 mmol). La reacción se calentó a reflujo a 65 °C durante 1 h, a continuación se añadió 9-borabiciclo[3.3.1]nonano 0,5 M en THF (0,75 mL, 0,375 mmol) y se continuó reflujo durante 1 h. Se añadió la mezcla resultante a una suspensión desgasificada del Intermedio 2-1 (268 mg, 0,936 mmol), K2CO3 (388 mg, 2,81 mmol) y PdCl2(dppf).CH2Cl2 (38 mg, 0,05 mmol) en 1,4-dioxano (2,7 mL) y agua (0,45 mL) y se calentó a 90 °C durante toda la noche, a continuación se enfrió hasta TA y se diluyó con EtOAc. La suspensión se filtró a través de celite y se concentró al vacío. El producto se adsorbió sobre una columna de SCX (5g) acondicionada con metanol. La columna se lavó varias veces con metanol y a continuación se eluyó con amoniaco 2 N en metanol. El producto crudo se recogió y se concentró al vacío para proporcionar el producto crudo. 3-(2-Metoxi-4-(1H-pirazol-1-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina. LCMS tR = 0,92 min, M+1 = 406,3.
Paso 3: Se preparó 5-(1 H-pirazol-1 -il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenol (56,1 mg, 0,143 mmol, 18 % de rendimiento) a partir de 3-(2-metoxi-4-(1H-pirazol-1-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina utilizando el MÉTODO GENERAL 3-1. LCMS tR = 0,52 min, M+1 = 392,3 (método Q de LCMS). 1H RMN (DMSO-d6) 68,64 (d, J=2,5 Hz, 1 H), 8,49 (d, J=9,0 Hz, 1 H), 8,17 (d, J=8,5 Hz, 1H), 7,87 (d, J=9,0 Hz, 1 H), 7,80 (d, J=1,8 Hz, 1H), 7,45-7,59 (m, 2H), 6,59 (dd, J=2,5, 1,8 Hz, 1H), 2,84 (d, J=7,0 Hz, 2H), 2,21-2,42 (m, 1H), 1,46 (d, J=11,8 Hz, 2H), 0,67-1,30 (m, 14H).
Ejemplo de referencia 22-1: Síntesis de 3-metoxi-2-(6-(metil(2,2,6-trimetilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol

Paso 1: 3-(Benciloxi)-4-bromo-5-hidroxibenzoato de metilo
Una mezcla de 4-bromo-3,5-dihidroxibenzoato de metilo (18,8 g, 76 mmol) y carbonato de potasio (5,26 g, 38,1 mmol) en DMF (190 mL) se añadió bromuro de bencilo (3,17 mL, 26,6 mmol). La mezcla se agitó durante toda la noche, se diluyó con 200 mL de agua y se acidificó hasta pH 1 mediante la adición lenta de ácido clorhídrico concentrado. La solución se extrajo con acetato de etilo/éter 1:1 (6X) y los extractos combinados se lavaron con agua (8X), bicarbonato de sodio saturado, salmuera, se secaron con sulfato de magnesio y se concentraron hasta obtener un sólido naranja. Los sólidos se suspendieron en DCM (200 mL) y se agitó durante toda la noche. Los sólidos (principalmente 4-bromo-3,5-dihidroxibenzoato sin reaccionar) se separaron por filtración y el filtrado se concentró hasta obtener un aceite naranja que se purificó mediante cromatografía en columna (80 g de gel de sílice, elución con DCM en heptano 2:1, y después se eluyó con DCM) para proporcionar 3-(benciloxi)-4-bromo-5-hidroxibenzoato de metilo (4,66 g). MS (M+1) = 337,0. 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,32-7,57 (m, 6H), 7,26 (d, J=1,52 Hz, 1H), 5,77 (s, 1H), 5,22 (s, 2H), 3,93 (s, 3H) así como también el 3,5-bis(benciloxi)-4-bromobenzoato de metilo dibencilado (1,8 g).
Paso 2: 3-(Benciloxi)-4-bromo-5-metoxibenzoato de metilo
Una mezcla de 3-(benciloxi)-4-bromo-5-hidroxibenzoato de metilo (3,69 g, 10,94 mmol) y carbonato de potasio (3,03 g, 21,98 mmol) en DMF (27 mL) se añadió yoduro de metilo (0,753 mL, 12,04 mmol). La mezcla se agitó durante toda la noche tras lo cual se diluyó con agua y se extrajo con acetato de etilo (4X). Los extractos combinados se lavaron con agua (8X), salmuera, se secaron con sulfato de magnesio y se concentraron para proporcionar 3-(benciloxi)-4-bromo-5-metoxibenzoato de metilo como un sólido blanco (3,72 g). MS (M+1) = 351,1; 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,31-7,59 (m, 7H), 5,24 (s, 2H), 3,99 (s, 3H), 3,95 (s, 3H).
Paso 3: Ácido 3-(benciloxi)-4-bromo-5-metoxibenzoico
A una solución de 3-(benciloxi)-4-bromo-5-metoxibenzoato de metilo (3,72 g, 10,59 mmol) en MeOH/THF 1:1 (50 mL) se añadió hidróxido de sodio acuoso (1 M, 53,0 mL, 53,0 mmol). Después de 10 minutos, los componentes volátiles se eliminaron a presión reducida y la solución se acidificó hasta pH 1 mediante la adición de ácido clorhídrico concentrado lo que dio como resultado la formación de un precipitado blanco expreso. La mezcla se extrajo con acetato de etilo (2X) y DCM (3X). Los extractos combinados se lavaron con salmuera, se secaron con sulfato de magnesio y se concentraron para proporcionar ácido 3-(benciloxi)-4-bromo-5-metoxibenzoico como un sólido blanco (3,41 g). MS (M-1) = 335,0. 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,21-7,49 (m, 7H), 5,16 (s, 2H), 3,91 (s, 3H).
Paso 4: 3-(Benciloxi)-4-bromo-5-metoxi-N-(prop-2-in-1-il)benzamida
A una suspensión de ácido 3-(benciloxi)-4-bromo-5-metoxibenzoico (2,0 g, 5,93 mmol) y 4 cotas de DMF en DCM (40 mL) se añadió lentamente cloruro de oxalilo (0,57 mL, 6,52 mmol). Después de tres horas se eliminó el disolvente y el residuo se redisolvió en DCM (10 mL). A esta solución se añadió lentamente una mezcla de propargilamina (0,46 mL, 7,12 mmol) y trietilamina (2,5 mL, 17,8 mmol) en DCM (2 mL). Después de 30 minutos, la solución se diluyó con éter, se lavó con agua (2X), ácido clorhídrico 1 M (2X), agua, bicarbonato de sodio saturado, salmuera, se secó con sulfato de magnesio y se concentró hasta obtener un sólido amarillo. El sólido se purificó disgregándolo en éter dietílico y se secó al vacío para proporcionar 3-(benciloxi)-4-bromo-5-metoxi-N-(prop-2-in-1-il)benzamida (1,88 g) como un sólido blanquecino. Ms = 374,0 (M+1).
Paso 5. 2-(3-(Benciloxi)-4-bromo-5-metoxifenil)-5-metiloxazol
A una solución de 3-(benciloxi)-4-bromo-5-metoxi-N-(prop-2-in-1-il)benzamida (0,455 g, 1,22 mmol) en dioxano (12 mL) se añadió hidruro de sodio (60 % p, 0,146 g, 3,65 mmol) y la mezcla se calentó a reflujo durante seis horas. La mezcla se enfrió hasta TA, se desactivó mediante la adición lenta de agua y se diluyó con acetato de etilo. La mezcla se lavó con agua, bicarbonato de sodio saturado, salmuera, se secó con sulfato de magnesio y se concentró. La cromatografía en columna ultrarrápida (12 g de sílice, 2 % de acetato de etilo en DCM) proporcionó 2-(3-(benciloxi)-4-bromo-5-metoxifenil)-5-metiloxazol (198 mg) como un sólido blanquecino. MS = 374 (M+1). 1H Rm N (400 MHz, CLOROFORMO-d) 6 ppm 7,55 (d, J=7,58 Hz, 2H), 7,43 (t, J=7,33 Hz, 2H), 7,32-7,39 (m, 2H), 7,27 (d, J=2,02 Hz, 1H), 6,89 (d, J=1,01 Hz, 1H), 5,27 (s, 2H), 4,02 (s, 3H), 2,44 (d, J=1,52 Hz, 3H).
Paso 6: Ácido (2-(benciloxi)-6-metoxi-4-(5-metiloxazol-2-H)fenil)borónico
A una solución agitada de 2-(3-(benciloxi)-4-bromo-5-metoxifenil)-5-metiloxazol (197 mg, 0,526 mmol) en THF (1,3 mL) enfriada hasta -78 °C se añadió n-butillitio (2,5 M en hexanos, 232 uL, 0,579 mmol). La solución se agitó durante 15 minutos, tras lo cual se añadió borato de trimetilo (235 uL, 2,11 mmol) y se permitió que la reacción se calentara lentamente hasta TA durante toda la noche. La reacción se desactivó mediante la adición de HCl 0,1 M y se diluyó con acetato de etilo, se lavó con agua, salmuera, se secó con sulfato de magnesio y se concentró. La cromatografía en columna ultrarrápida (12 g de sílice, 0-100 % de acetato de etilo en DCM durante 30 volúmenes de columna) proporcionó ácido (2-(benciloxi)-6-metoxi-4-(5-metiloxazol-2-il)fenil)borónico (63 mg) como una espuma blanca. MS = 340,1 (M+1). 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,28-7,46 (m, 5H), 7,25 (d, J=1,01 Hz, 1H), 7,08 (s a, 1H), 6,85 (d, J=1,01 Hz, 1H), 5,17 (s, 2H), 3,95 (s, 3H), 2,38 (d, J=1,52 Hz, 3H).
Paso 7: 6-(2-(Benciloxi)-6-metoxi-4-(5-metiloxazol-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Una mezcla de ácido (2-(benciloxi)-6-metoxi-4-(5-metiloxazol-2-il)fenil)borónico (63 mg, 0,186 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina Intermedio 1-1 (35 mg, 0,124 mmol), carbonato de sodio (39,4 mg, 0,371 mmol) en DME/agua 3:1 (825 uL) se desfasificó con una corriente seca de nitrógeno durante cinco minutos. Se añadió tetrakis(trifenilfosfina)paladio (0) (21 mg, 0,019 mmol) y la mezcla se calentó mediante radiación de microondas a 150 °C durante quince minutos. La reacción cruda se filtró, el filtrado se acidificó con HCl 2 M en MeOH y a continuación se concentró a sequedad. El residuo se redisolvió en metanol y se adsorbió sobre una columna de SCX acondicionada con MeOH. La columna se lavó varias veces (3-4 volúmenes de columna) con MeOH y a continuación se eluyó con amoniaco 3,5 N en MeOH. La evaporación del eluyente proporcionó el producto como una espuma marrón claro. La cromatografía en columna ultrarrápida (4 g de gel de sílice, gradiente de un 0-40 % de amoniaco 7 N en MeOH en DCM durante 30 volúmenes de columna) proporcionó 6-(2-(benciloxi)-6-metoxi-4-(5-metiloxazol-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido blanco. MS = 542,4 (M+1). 1H RMN (400 MHz, METANOL-d4) 6 ppm 7,45 (d, ^=1,01 Hz, 1H), 7,37 - 7,42 (m, 2H), 7,21-7,33 (m, 5H), 7,14 (d, J=9,60 Hz, 1H), 6,98 (d, J=1,01 Hz, 1H), 5,28 (m, 1H), 5,17 (s, 2H), 3,86 (s, 3H), 3,01 (s, 3H), 2,47 (d, J=1,01 Hz, 3H), 1,71 5 (dd, J= 12,1, 3,7 Hz, 2H), 1,62 (t, J=12,1Hz, 2H), 1,41 (s, 6H), 1,27 (s, 6H).
Paso 8. 3-Metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol
A una solución de 6-(2-(benciloxi)-6-metoxi-4-(5-metiloxazol-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (36 mg, 0,066 mmol) en acetato de etilo/MeOH 1:1 (1,3 mL) en una atmósfera de nitrógeno se añadió paladio sobre carbón (10 % de contenido de Pd, 7 mg, 6,6 umol). La atmósfera se reemplazó con hidrógeno (globo) y la mezcla se agitó rápidamente a TA durante toda la noche. La solución se diluyó con DCM y se filtró a través de celite. El filtrado se concentró hasta obtener un residuo amarillo y se acidificó con HCl en MeOH (producido mediante la adición dental de cloruro de ácido etilo (14 uL, 0,199 mmol) a 1 mL de MeOH). La solución se concentró al vacío, el residuo se redisolvió en MeOH y se cargó en una columna de SCX preacondicionada con MeOH. La columna se lavó con MeOH (20 mL) y se eluyó con amoniaco 3,5 N en MeOH (20 mL). La evaporación del eluyente proporcionó el producto como un residuo amarillo claro. La sonicación en éter dietílico dio como resultado la formación de un sólido amarillo claro. Se eliminó el disolvente a presión reducida para proporcionar 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol (22 mg). LCMS tR = 0,54 min (método Q de LCMS); MS = 352,3 (M+1). 1H RMN (400 MHz, METANOL-d4) 6 ppm 8,22 (d, J=10,11 Hz, 1H), 7,18-7,29 (m, 3H), 6,96 (d, J=1,01 Hz, 1 H), 5,07-5,20 (m, 1H), 3,98 (s, 3H), 3,03 (s, 3H), 2,46 (d, J=1,52 Hz, 3H), 1,73 (dd, J=12,5, 3,4 Hz, 2H), 1,62 (t, J=12,5 Hz, 2H), 1,42 (s, 6H), 1,26 (s, 6H).
Ejemplo de referencia 23-1: Síntesis de 2-(6-((6S)-6-((S)-1-hidroxietil)-2,2-dimetilpiperidm-4-Noxi)pi
N
dazm-3-N)-5-(1 H-pirazol-1 -il)fenol

Paso 1. 3-( (6S)-6-( (S)-1-(tert-Butildimetilsililoxi)etil)-2,2-dimetilpiperidin-4-iloxi)-6-( 2-metoxi-4-( 1H-pirazol-1-il)fenil)piridazina
A una solución del Intermedio 2-1 3-doro-6-(2-metoxi-4-(1H-p¡razoM-¡l)fen¡l)μmdazma (32 mg, 0,11 mmol) y el Intermedio de referencia 4-1 (6S)-6-((S)-1-(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-ol (Intermedio 4-1, 32 mg, 0,11 mmol) en DMF (1 mL) se añadió tert-butóxido de potasio (1 M en THF, 0,45 mL, 0,45 mmol) a 0 °C. La mezcla de reacción se agitó durante 1 h a TA. A continuación, la mezcla se desactivó con una solución saturada de cloruro de amonio (10 mL) y se extrajo con un 10 % de MeOH en diclorometano (20 mL). El extracto se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna (EtOAc/Heptano) para proporcionar 56 mg (94 %) de 3-((6S)-6-((S)-1-(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-¡lox¡)-6-(2-metox¡-4-(1H-p¡razol-1-il)fenil)piridazina: LCMS (m/z, MH+): 538,5; 1H RMN (400 MHz,CLOROFORMO-d) 58,05 (d, J=8,1 Hz, 1 H), 8,01 (d, J=2,5 Hz, 1 H), 7,97 (d, J=9,1 Hz, 1 H), 7,76 (d, J=1,5 Hz, 1 H), 7,55 (d, J=2,0 Hz, 1 H), 7,30 (dd, J=8,6, 2,0 Hz, 1 H), 6,94 (d, J=9,1 Hz, 1 H), 6,54-6,47 (m, 1 H), 5,79-5,87 (m, 1 H), 3,96 (s, 3 H), 3,83-3,93 (m, 1 H), 3,07-3,17 (m, 1 H), 2,13-2,22 (m, 1 H), 2,05-2,12 (m, 1 H), 1,43-1,57 (m, 2 H), 1,27 (s, 3 H), 1,16 (d, J=6,6 Hz, 3 H), 1,14 (s, 3 H), 0,90 (s, 9 H), 0,10 (s, 3 H), 0,08 (s, 3 H).
Paso 2. 2-(6-((6S)-6-((S)-1-Hidroxietil)-2,2-dimetilpiperidin-4-iloxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol
A una solución de 3-((6S)-6-((S)-1-(tert-but¡ld¡met¡ls¡l¡lox¡)et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-¡lox¡)-6-(2-metox¡-4-(1H-p¡razol-1-ilfenil)piridazina (56 mg, 0,1 mmol) en CH2O 2 (2 mL) se añadió BBr3 (1 M en heptano, 0,13 mL, 0,13 mmol) gota a gota a 0 °C. La mezcla se agitó durante toda la noche. La mezcla de reacción se desactivó con agua y a continuación se basificó con una solución saturada de NaHCO3. La capa orgánica se extrajo con un 10 % de MeOH en diclorometano (10 mL x 3). Los extractos combinados se secaron con Na2SO4 y se concentraron al vacío. La purificación mediante Hp Lc proporcionó 12 mg (28 %) de 2-(6-((6S)-6-((S)-1-h¡drox¡et¡l)-2,2-d¡met¡lp¡per¡d¡n-4-¡lox¡)p¡r¡daz¡n-3-¡l)-5-(1H-p¡razol-1-¡l)fenol: LCMS tR = 0,52 min (método Q de LCMS); MS = 410,3 (M+1); 1H RMN (400 MHz, METANOL-d4) 58,23 (d, J=9,6 Hz, 1H), 8,19 (d, J=2,0 Hz, 1H), 7,86 (d, J=8,6 Hz, 1H), 7,65 (d, J=2,0 Hz, 1H), 7,32-7,25 (m, 2H), 7,22 (d, J=9,6 Hz, 1H), 6,47-6,43 (m, 1H), 5,59-5,66 (m, 1H), 3,68-3,77 (m, 1H), 3,02-3,11 (m, 1H), 2,03-2,15 (m, 2H), 1,49-1,59 (m, 2H), 1,24 (s, 3H), 1,11 (d, J=6,4 Hz, 3H), 1,10 (s, 3H).
PREPARACIÓN 9
Intermedio 5-1: Síntesis de 7-(bencMoxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-N)naftalen-2-ol

Se preparó 7-(benc¡lox¡)-6-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)naftalen-2-ol (1,25 g, 2,366 mmol, 89 % de rendimiento) a partir de 7-(benc¡lox¡)-6-(4,4,5,5-tetramet¡l-1,3,2-d¡oxaborolan-2-¡l)naftalen-2-ol (1,45 g, 3,85 mmol) y el Intermedio 1-1 (0,75 g, 2,65 mmol) utilizando el Método general 1-1 para el acoplamiento de Suzuki. M+1 = 497,8. 1H RMN (400 MHz, METANOL-d4) 57,91 (s, 1H), 7,76 (d, J=9,54 Hz, 2H), 7,66 (d, J=8,78 Hz, 2H), 7,34 7,40 (m, 2H), 7,31 (s, 2H), 7,18-7,27 (m, 2H), 6,97-7,05 (m, 2H), 6,91 (dd, J=8,78, 2,26 Hz, 1H), 5,18 (m, 3H), 2,92 (s, 3H), 1,60-1,68 (m, 2H), 1,45-1,55 (m, 2H), 1,32 (s, 6H), 1,18 (s, 6H).
PREPARACIÓN 10
Intermedio 5-2: Síntesis de trifluorometanosulfonato de 7-(bencMoxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ilo

A una mezcla de reacción de 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (Intermedio 5-1, 3 g, 6,04 mmol) en DCM (30 mL) se añadió Et3N (2,10 mL, 15,10 mmol), y N-feniltrifluorometanosulfonimida (2,158 g, 6,04 mmol) en dos porciones. La mezcla se agitó a TA durante 3 h y a continuación se concentró al vacío. El residuo se cargó sobre dos columnas de SCX de 10 g, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron para obtener el Intermedio 5-2 como un sólido pálido (3,47 g, 91 % de rendimiento). MS (M+1) = 629,5.
PREPARACIÓN 11
Intermedio 5-3: Síntesis de trifluorometanosulfonato de 7-hidroxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ilo
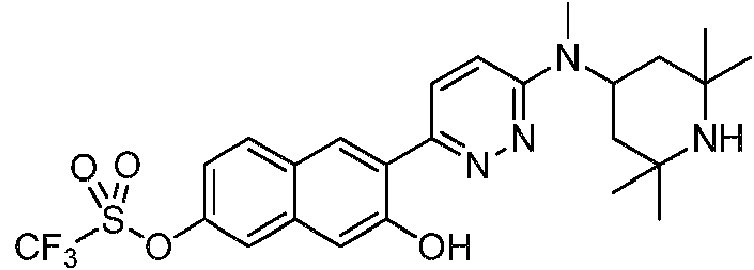
A una mezcla del Intermedio 5-2 (500 mg, 0,795 mmol) en DCM (4 mL) se añadió BBr3 (solución 1 M en DCM, 2,4 mL, 2,4 mmol) lentamente a -78 °C. La mezcla se agitó a -78°C durante 10 minutos, a continuación se calentó hasta TA y se agitó durante 1,5 horas. La reacción se desactivó con MeOH y se concentró. El residuo se cargó sobre una columna de SCX, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron para obtener el Intermedio 5-3 como un sólido amarillo claro (382 mg, 89 % de rendimiento). MS (M+1) = 539,3.
PREPARACIÓN 12
Intermedio 5-4: Síntesis de 6-(3-(benciloxi)-6-metoxinaftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina

A una mezcla de reacción de 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (Intermedio 5-1, 500 mg, 1,01 mmol) en DMF (3 mL) se añadió NaH al 60 % p (48,3 mg, 1,21 mmol) a 0°C. La mezcla se agitó durante 0,5 h y a continuación se añadió yoduro de metilo (0,063 mL, 1,01 mmol). La mezcla se agitó a TA durante 1,5 horas y a continuación se añadió otra porción de yoduro de metilo (0,063 mL, 1,01 mmol). La mezcla de reacción se agitó a TA durante toda la noche y a continuación se desactivó lentamente con agua. El residuo se repartió entre agua y DCM, y la capa acuosa se extrajo opcionalmente con DCM. Las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida en gel de sílice (0-20 % de NH3 1,5 M en MeOH/DCM) para obtener una mezcla del Intermedio 5-4 y 6-(3-(benciloxi)-6-metoxinaftalen-2-il)-N-metil-N-(1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina que se utilizó sin una purificación adicional (412 mg, 80 % de rendimiento). MS (M+1) = 511,5.
PREPARACIÓN 13
Intermedio 6-1: Síntesis de trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenilo

Paso 1: 6-(4-(Benciloxi)-2-(trifluorometoxi)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar el ácido (4-(benciloxi)-2-(trifluorometoxi)fenil)borónico (1,2 g, 61 % puro, 2,307 mmol) y el Intermedio 1-1 (246 mg, 0,871 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo el compuesto 6-(4-(benciloxi)-2-(trifluorometoxi)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido beige (405 mg, 90 % de rendimiento) después de purificación mediante cromatografía en columna ultrarrápida. MS (M+1) = 515,5.
Paso 2: 4-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol
A partir del compuesto 6-(4-(benciloxi)-2-(trifluorometoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (405 mg, 0,787 mmol), siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis del grupo bencilo, se obtuvo el compuesto 4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3(trifluorometoxi)fenol (335 mg, 100 % de rendimiento) después de purificación en columna de SCX. MS (M+1) = 425,3.
Paso 3: Trifluorometanosulfonato de 4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenilo
A una suspensión de 4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol (360 mg, 0,848 mmol) en DCM (5 mL) se añadió Et3N (0,296 mL, 2,120 mmol) y N-feniltrifluorometanosulfonimida (364 mg, 1,018 mmol). La mezcla se agitó a TA durante toda la noche. La mezcla de reacción se concentró. El residuo se cargó sobre una columna de SCX de 5 g, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y el producto crudo se purificó mediante cromatografía en columna ultrarrápida para obtener trifluorometanosulfonato de 4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenilo como un sólido beige 361 mg, 76 %). MS (M+1) = 557,5
Paso 4: Trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenilo
Una mezcla de trifluorometanosulfonato de 4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenilo (360 mg, 0,647 mmol), diacetato de yodobenceno (375 mg, 1,164 mmol) y Pd(OAc)2 (7,3 mg, 0,032 mmol) en AcOH (3,0 mL) y Ac2O (3,0 mL) se calentó a 60 °C durante toda la noche. La mezcla de reacción se enfrió hasta la temperatura ambiente y se concentró. El residuo se basificó con una solución acuosa de NaHCO3 y se extrajo con DCM. Las capas orgánicas combinadas se secaron con Na2SO4 y se concentraron. El material crudo se cargó sobre una columna de SCX, se lavó con MeOH y se diluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron. Se obtuvo el compuesto trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenilo como un sólido amarillo (110 mg, 28 %) después de la cromatografía en columna ultrarrápida y purificación mediante HPLC. MS (M+1) = 573,2.
PREPARACIÓN 14
Intermedio 6-2: Síntesis de trifluorometanosulfonato de 3-hidroxi-5-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo

Paso 1: 3-Metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol
A un recipiente resistente a la presión de 500 mL se añadió 4-bromo-3-metoxifenol (8,12 g, 40 mmol), bis(pinacolato)diboro (22,4 g, 88,0 mmol), acetato de potasio (27,4 g, 280 mmol), dppf (2,22 g, 4,00 mmol) y Pd(dppf)Cl2 (2,93 g, 4,00 mmol). Se añadió dioxano (120 mL) y la mezcla de reacción se purgó con nitrógeno durante 25 minutos. A continuación, la mezcla de reacción se selló y se agitó a 85 °C durante 20 h. La mezcla se diluyó con
acetato de etilo, se filtró a través de celite y se concentró hasta obtener un líquido marrón oscuro. El líquido se hizo pasar a través de un tapón de gel de sílice (60 g) eluyendo con heptano/acetato de etilo para proporcionar 3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol como un sólido blanco (4,0 g). 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,60 (d, J=8,1 Hz, 1H), 6,42 (dd, J=8,1,2,0 Hz, 1H), 6,33 (d, J=2,0 Hz, 1H), 3,64 (s, 3H), 1,35 (s, 12H).
Paso 2: 3-Metoxi-4-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)fenoi
A un vial apto para microondas de 25 mL se añadieron 3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (2,90 g, 10,8 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 2,55 g, 9,00 mmol), NaHCO3 (2,27 g, 27,0 mmol) y tetrakis(trifenilfosfina)paladio (0) (0,520 g, 0,450 mmol). Se añadieron dioxano (45 mL) y agua (15 mL) y la mezcla de reacción se purgó con nitrógeno durante 10 minutos. La mezcla de reacción se calentó con irradiación de microondas a 110 °C durante 16 h. Después de enfriar hasta TA, la mezcla se filtró a través de celite, lavando la almohadilla filtrante secuencialmente con acetato de etilo, DCM y MeOH. La concentración del filtrado proporcionó un sólido marrón que se purificó mediante cromatografía ultrarrápida (80 g de sílice, gradiente de un 0-20 % de amoniaco 2 M en MeOH en DCM) para proporcionar 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (1,6 g). MS (M+1) = 371,3.
Paso 3: Trifiuorometanosuifonato de 3-metoxi-4-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)feniio
Se añadió en porciones N-feniltrifluorometanosulfonimida (2,160 g, 6,05 mmol) una mezcla de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol y trietilamina (1,5 mL, 11 mmol) enfriada hasta 0°C. Se permitió que la mezcla se calentara hasta TA y se agitara durante 2 h. Se añadió una porción adicional de N-feniltrifluorometanosulfonimida (0,30 g, 0,86 mmol) y la mezcla se agitó a TA durante toda la noche. La solución se diluyó con una solución acuosa saturada de NaHCO3 y se extrajo con DCM (2x). Los extractos se concentraron y el residuo se purificó mediante cromatografía ultrarrápida (40 g de sílice, gradiente de un 0-25 % de MeOH en DCM) para proporcionar trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (1,8 g). MS (M+1) = 503,4. 1H RMN (400 MHz, METANOL-d4) 6 ppm 7,85 (d, J=9,6 Hz, 1 H), 7,78 (d, J=8,6 Hz, 1 H), 7,20 (d, J=9,6 Hz, 1H), 7,17 (d, J=2,5 Hz, 1H), 7,12 (dd, J=8,3, 2,3 Hz, 1H), 5,44-5,59 (m, 1H), 3,92 (s, 3H), 3,04 (s, 3H), 1,98 (m, J=8,1 Hz, 4H), 1,65 (s, 6H), 1,53 (s, 6H).
Paso 4: Trifiuorometanosuifonato de 3-hidroxi-5-metoxi-4-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)feniio
Una mezcla de trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (2,6 g, 5,17 mmol), Pd(OAc)2 (58 mg, 0,259 mmol) y diacetato de yodobenceno (2,33 g, 7,24 mmol) en ácido acético/anhídrido acético 1:1 (42 mL) se calentó a 50 °C durante 8 h. La mezcla se enfrió hasta TA y se concentró a presión reducida. La cromatografía ultrarrápida (elución con un 10-100 % de EtOH en DCM y después EtOH/amoniaco 7 N en MeOH 7:1) proporcionó una mezcla del compuesto del título y el acetato correspondiente (acetato de 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(((trifluorometil)sulfonil)oxi)fenilo). Después de la concentración, la mezcla se añadió metanol y se calentó a 70 °C durante 4 h. El disolvente se evaporó para proporcionar trifluorometanosulfonato de 3-hidroxi-5-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3- il)fenilo como un sólido de color tostado (1,25 g). MS (M+1) = 519,4. 1H RMN (400 MHz, METANOL-d4) 6 ppm 8,08 (d, J=10,1 Hz, 1 H), 7,23 (d, J=10,1 Hz, 1H), 6,57 (s, 2H), 5,16 (t, J=12,1 Hz, 1H), 3,89 (s, 3H), 3,02 (s, 3H), 1,69-1,77 (m, 2H), 1,56-1,67 (m, 2H), 1,41 (s, 6H), 1,27 (s, 6H).
PREPARACIÓN 15
Intermedio 7-1: Síntesis de 6-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-N)-2,3-dihidro-1 H-inden-1-ona

Paso 1: 6-Metoxi-5-(4,4,5,5-tetrametii-1,3,2-dioxaboroian-2-ii)-2,3-dihidro-1H-inden-1-ona
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 5-bromo-6-metoxi-2,3-dihidro-1 H-inden-1 -ona (1,0 mg, 4,15 mmol) se obtiene 6-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2,3-dihidro-1 H-inden-1 -ona (1,16 g) MS [M+H+] = 289,2.
Paso 2: 6-Metoxi-5-(6-(metii(2,2,6,6-tetrametiipiperidin-4-ii)amino)piridazin-3-ii)-2,3-dihidro-1H-inden-1-ona Siguiendo el MÉTODO GENERAL 1-4 para el acoplamiento de Suzuki utilizando 6-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2,3-dihidro-1H-inden-1-ona (300 mg, 1,06 mmol) y 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 611 mg, 2,12 mmol) se obtiene 6-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona (433 mg) MS [M+H+] = 409,7.
PREPARACIÓN 16
Intermedio 8-1: Síntesis de 5-bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)ammo)piridazm-3-i)fenol

Paso 1: (2-Fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se añadieron el Intermedio 1-1 (2,83 g, 10,0 mmol), ácido (2-fluoro-6-metoxifenil)borónico y K3PO4 (5,52 g, 26,0 mmol) a un vial apto para microondas. A continuación, se añadió a la mezcla el precatalizador de 2.a generación XPhos (0,32 g, 0,40 mmol) y después se añadió THF/agua 1:1 (50 mL). La mezcla de reacción se selló, se agitó a TA durante 4 h y a continuación se extrajo con CH2O 2 (2x). El material crudo se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (3 eq, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío para proporcionar el compuesto del título como un gel marrón (2,87 g, 77 %). [M+H]: 373,4; 1H RMN (400 MHz, DMSO) 57,43 (td, J= 8,5, 7,0 Hz, 1H), 7,36 (d, J= 9,5 Hz, 1H), 7,08 (d, J= 9,5 Hz, 1H), 6,98 (d, J= 8,5 Hz, 1H), 6,92 (td, J= 8,5, 1,0 Hz, 1H), 5,07 (s a, 1H), 3,74 (s, 3H), 3,31 (s, 3H), 1,47-1,55 (m, 2H), 1,33-1,48 (m, 2H), 1,24 (s, 6H), 1,09 (s, 6H).
Paso 2: 6-(4-Bromo-2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se calentaron (2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (1,83 g, 4,91 mmol), [Ir(COD)(OMe)]2 (0,16 g, 0,24 mmol), dtbpy (0,13 g, 0,24 mmol) y bis(pinacolato)diboro (1,87 g, 7,37 mmol) en dioxano (40 mL) a 80 °C durante toda la noche. Los componentes volátiles se eliminaron al vacío. Se añadieron EtOH (20 mL), H2O (20 mL) y CuBr2 (3,29g, 14,7 mmol). La mezcla se calentó a reflujo durante toda la noche. La mezcla de reacción se enfrió hasta TA y los componentes volátiles eliminaron al vacío. Se añadió una solución acuosa al 7 % de NH4OH y la fase acuosa se extrajo con DCM (3x). El producto se purificó a continuación mediante cromatografía en columna de gel de sílice (gradiente de un 1-10 % de amoniaco 7 N en MeOH, en CH2O 2) para obtener una mezcla inseparable del producto deseado (6-(4-bromo-2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina) y 4-bromo-6-(2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (1,26 g). [M+H]: 451,3.
Paso 3: 5-Bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Se calentaron 6-(4-bromo-2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (0,81 g, 1,80 mmol) y clorhidrato de piridina (1,65 g, 14,3 mmol) hasta 190 °C durante 45 min en un reactor de microondas Biotage® Initiator. La mezcla se diluyó en MeOH/DMSO y se purificó mediante HPLC preparativa de fase inversa (de un 5 a un 95 % de acetonitrilo en agua, 0 , 1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (4 eq, metanol como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío para proporcionar 5-bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un sólido beige (0,19 g, 23 %). [M+H]: 439,2; 1H Rm N (400 MHz, MeOD) 57,96 (d, J= 10,0 Hz, 1H), 7,27 (d, J= 10,0 Hz, 1H), 6,97 (t, J= 2,0 Hz, 1H), 6,89 (dd, J= 11,5, 2,0 Hz, 1H), 5,07-5,35 (m, 1H), 3,02 (s, 3H), 1,73 (dd, J= 12,5, 3,5 Hz, 2H), 1,62 (t, J= 12,5 Hz, 2H), 1,41 (s, 6H), 1,27 (s, 6H).
Preparación 17
Intermedio 9-1: Síntesis de trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo

Paso 1: (4-Bromo-3-metoxifenoxi)(tert-butil)dimetilsilano
Un matraz de fondo redondo de 5 L dotado de un agitador aéreo, termopar y entrada de N2 se añadieron 4-bromo-3-metoxifenol (254 g, 1251 mmol), DCM (2500 mL) y DIPEA (437 mL, 2502 mmol). La mezcla de reacción se enfrió en un baño de hielo y después se añadió tert-butilclorodimetilsilano (198 g, 1314 mmol). La mezcla de reacción se agitó a TA durante toda la noche y a continuación se diluyó con agua. La capa orgánica se separó, se secó con sulfato de sodio, se filtró y se concentró para proporcionar (4-bromo-3-metoxifenoxi)(tert-butil)dimetilsilano (472g, 1250 mmol, 100 % rendimiento). MS (M+1) = 319,2.
Paso 2: tert-Butil(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenoxi)dimetilsilano
Un matraz de fondo redondo de 500 mL que contenían (4-bromo-3-metoxifenoxi)(tert-butil)dimetilsilano (1,9 g, 6 mmol) y dioxano (60 mL), se añadieron 4,4,4',4',5,5,5',5'-octametil-2,2'-bi(1,3,2-dioxaborolano) (3,05 g, 12,00 mmol), acetato de potasio (2,35 g, 24,00 mmol), dppf (0,333 g, 0,600 mmol) y aducto de PdCh(dppf).CH2Cl2 (0,49 g, 0,600 mmol). En la reacción se hizo vacío, se rellenó con N2 dos veces y después se calentó a 90 °C durante toda la noche. A continuación, la reacción se diluyó con MeOH, se filtró a través de celite y se lavó con EtOAc. Después de la concentración al vacío, el residuo se purificó mediante cromatografía en gel de sílice utilizando EtOAc/Heptano (0 15 %) para proporcionar el sólido blanco tert-butil(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenoxi)dimetilsilano (1,6 g, 4,17 mmol, 70 % de rendimiento), m S (M+1) = 3 6 5 ,2.
Paso 3: 3-Metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
En un reactor encamisado de 20 L dotado de un condensador de reflujo, entrada de N2 , termopar y un agitador aéreo se introdujeron 2,7 L de dioxano y después tert-butil(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenoxi)dimetilsilano (290 g, 557 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 113 g, 398 mmol) y bicarbonato de sodio (100 g, 1194 mmol). Se añadieron 700 mL de agua, y después se añadió Pd(PPh3)4 (27,6 g, 23,88 mmol). La mezcla de reacción se calentó a 72 °C durante la noche. Después de enfriar hasta TA, las capas se separaron. La capa orgánica se concentró y el residuo se purificó mediante cromatografía en gel de sílice utilizando un 5 % de MeOH (que contenía un 1 % de TEA) en DCM para proporcionar el producto deseado, 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (83 g, 211 mmol, 53 % de rendimiento). MS = 371,4.
Paso 4: Trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo
Una mezcla de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (1,6 g, 4,32 mmol) y Et3N (1,50 mL, 10,8 mmol) en DCM (40 mL) se enfrió en un baño de hielo-agua y se añadió gota a gota N-feniltrifluorometanosulfonimida (2,16 g, 6,05 mmol). Después de agitar a TA durante 2 h, se añadieron otros 0,2 eq de N-feniltrifluorometanosulfonimida y la reacción se agitó durante toda la noche. La reacción se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se lavó con salmuera, se separó y se concentró y el residuo se purificó mediante cromatografía en columna utilizando MeOH/DCM (0-25 %) para proporcionar el Intermedio 9-1, trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (1,8 g, 3,58 mmol, 83 % de rendimiento), MS = 503,2.
Preparación 18
Intermedio 9-2: Síntesis de trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazin-3-il)fenilo

Un matraz de fondo redondo de 1 L dotado de una barrita de agitación magnética y una entrada de N2, se enfrió en un baño de hielo-agua y se añadieron trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (37,00 g, 6 8 , 6 mmol) y DCM (360 mL) Se añadió lentamente tribromuro de boro (1 M en DCM, 120 mL) mediante una jeringa. Después de agitar a TA durante 4 h, la reacción se desactivó con metanol y se agitó a TA durante 15 minutos. La mezcla de reacción se concentró para proporcionar un sólido vítreo pegajoso, que se calentó reflujo en HCl 1 M en MeOH (360 mL) durante toda la noche. Después de enfriar hasta TA, el material se concentró y el residuo se agitó en HCl 4 N en dioxano (18 mL) durante toda la noche. La filtración proporcionó la sal de HCl del producto deseado, trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo Intermedio 9-2 (36 g, 6 8 , 6 mmol, >100 % de rendimiento), MS (M+1) = 489,3. 1H RMN (DMSO-d6) 6 9,06 (d, J=11,6 Hz, 1H), 8,28 (d, J=10,1 Hz, 1H), 8,13 (d, J=12,1 Hz, 1H), 7,99 (d, J=8,1 Hz, 1H), 7,70 (d, J=9,1 Hz, 1H), 7,08-7,12 (m, 1H), 4,99 (s a., 1H), 3,03 (s, 3H), 2,03 (t, J=12,9 Hz, 2H), 1,80 (d, J=10,6 Hz, 2H), 1,54 (s, 6H), 1,48 (s, 6H).
Preparación 19
Intermedio 9-3: Síntesis de 6-(2-metoxi-4-(4,4,5,5-tetrametiM,3,2-dioxaborolan-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina

A un vial apto para microondas se añadieron trifluorometanosulfonato de 3-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (Intermedio 9-1, 4,0 g, 7,96 mmol), bis(pinacolato)diboro (4,45 g, 17,51 mmol), acetato de potasio (4,69 g, 47,8), PdCl2(dppf)CH2Cl2 (0,65 g, 0,79 mmol), dppf (0,44 g, 0,79 mmol) y 1,4-dioxano (10 mL). La solución se purgó con nitrógeno (3x) y se agitó a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con EtOAc. El filtrado se concentró al vacío para obtener un líquido marrón que se purificó mediante cromatografía en gel de sílice ( 10 %-60 % de EtOAc/Heptano) para proporcionar 6-(2-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (1,6 g, MS: 481,5 [M+H+]).
MÉTODO GENERAL 1-5
Procedimiento representativo para el acoplamiento de Suzuki
Se añadieron un intermedio de tipo cloropiridazina, tal como el Intermedio 1-1, (1 equivalente), reactivo de ácido borónico (1,2-1,5 equivalentes), Pd(PPh3)4 (0,1 equivalentes) y Na2CO3 o NaHCO3 (2,5-3 equivalentes) a un vial apto para microondas y después se añadió 1,4-dioxano y H2O (4:1). La mezcla de reacción se selló, a continuación se hizo vacío, se rellenó con N2 (4x) y se calentó mediante radiación de microondas a 120 °C duante 1 h. La mezcla de reacción se filtró a través de celite y se lavó con EtOAc o un 10 % de MeOH/DCM. El filtrado resultante se concentró y se acidificó hasta pH 3 utilizando una solución acuosa de HCl 1 M, a continuación se cargó en una columna de SCX, se lavó con MeOH y se eluyó con NH3 2 M MeOH. Las fracciones que contenían el producto se concentraron y se purificaron mediante cromatografía en columna ultrarrápida para proporcionar el producto deseado.
MÉTODO GENERAL 1-6
Procedimiento representativo para el acoplamiento de Suzuki
Se añadieron a un vial apto para microondas el sustrato de tipo halopiridazina (1 equivalente), reactivo de tipo ácido o éster borónico (2,5 equivalentes) y Na2CO3 (3 equivalentes). A continuación se añadió Pd(PPh3)4 (0,1 equivalentes) a la mezcla de reacción y después se añadió dioxano/agua (6/1, 0,1 M). La mezcla de reacción se selló y se calentó en un reactor de microondas Biotage® Initiator a 130 °C durante 1 h. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con metanol. El filtrado se concentró al vacío y el producto crudo se purificó mediante HPLC preparativa de fase inversa (0,1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (4 eq, metanol como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío y el sólido resultante se suspendió o disolvió en CH3CN/H2O (3/1 mL). Se añadió HCl acuoso 1 M (3 equivalentes) y el disolvente se concentró al vacío para proporcionar el compuesto deseado como la sal clorhidrato.
MÉTODO GENERAL 7-1
Procedimiento representativo para la borilación/bromación
En una mezcla de un intermedio de tipo fenilpiridazina con sustitución 2,6, tal como 6-(2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (1 equivalenet), bis(pinacolato)diboro (1,5 equivalentes), 4,4'-di-tertbutilbipiridina (dtbpy) (0,2 equivalentes) y [Ir(COD)(OMe)]2 (0,2 equivalentes) en 1,4-dioxanse hizo vacío, a continuación se rellenó con N2 (4x) y a continuación se calentó a 90 °C durante toda la noche. La mezcla de reacción se enfrió hasta TA y se concentró. Al residuo se añadieron CuBr2 (3 equivalentes), MeOH y agua (1:1). La mezcla se calentó a 85 °C durante toda la noche, a continuación se enfrió hasta TA, se diluyó con EtOAc, se filtró a través de celite y se lavó con EtOAc. El filtrado se lavó con salmuera, se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (MeOH/DCM) para proporcionar el producto deseado.
Ejemplo de referencia 24-1: Síntesis de 7-hidroxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-N)ammo)piridazm-3-il)-2-naftonitrilo

Una mezcla desgasificada de trifluorometanosulfonato de 7-hidroxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ilo (21 mg, 0,033 mmol), cianuro de zinc (5,00 mg, 0,043 mmol) y tetrakis(trifenilfosfina)paladio (0) (1,8 mg, 1,6 |jmol) en DMF (0,8 mL) se calentó a 120 °C con irradiación de microondas durante 1 h. La mezcla de reacción se filtró a través de celite, se lavó con EtOAc y se concentró. El residuo se cargó sobre una columna de SCX de 1 g, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron. El producto crudo se purificó mediante HPLC para obtener el compuesto del título como un sólido amarillo claro (6,9 mg, 50 % de rendimiento). LCMS tR = 0,55 min [Método Q], MS (M+1) = 416,3. 1H RMN (METANOL-d4) 5 8,26 (s, 1H), 8,16 (d, J=9,6 Hz, 1H), 8,04 (s, 1H), 7,86 (d, J=8 , 6 Hz, 1H), 7,32 (dd, J=8 ,6 , 1,5 Hz, 1H), 7,27 (s, 1H), 7,22 (d, J=9,6 Hz, 1H), 5,05 (t, J=11,9 Hz, 1H), 2,94 (s, 3H), 1,61 (dd, J=13,1, 3,5 Hz, 2H), 1,49 (t, J=12,4 Hz, 2H), 1,30 (s, 6H), 1,14 (s, 6H).
Ejemplo de referencia 24-2: Síntesis de 3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)ammo)piridazm-3-il)-7-(piperidin-1-ilmetil)naftalen-2-ol

Paso 1: 6-(3-(Bendloxi)-6-(piperídin-1-ilmetil)naftalen-2-il)-N-metil-N-(2,2,6,6-tetmmetilpiperidin-4-il)piridazin-3-amina
Una mezcla de reacción desgasificada del Intermedio 5-2 (125 mg, 0,199 mmol), potasio-1-trifluoroboratometilpiperidina (44,8 mg, 0,219 mmol), acetato de paladio (2,2 mg, 9,9 jmol), X-Phos (9,5 mg, 0,020 mmol) y Cs2CO3 (194 mg, 0,596 mmol) en THF (1 mL) y agua (0,1 mL) se calentó a 80 °C durante 25 h. La mezcla de reacción se filtró a través de élite y se lavó con EtOAc. El filtrado se concentró y se acidificó hasta pH 3 mediante la adición de HCl acuoso 1 M. El residuo se cargó sobre una columna de SCX, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y el producto crudo se purificó mediante HPLC para obtener el compuesto del título (34 mg). MS (M+1) = 578,7.
Paso 2: 3-(6-(Metil(2,2,6,6-tetmmetilpiperídin-4-il)amino)pirídazin-3-il)-7-(pipendin-1-ilmetil)naftalen-2-ol
Una mezcla purgada con H2 de 6-(3-(benciloxi)-6-(piperidin-1-ilmetil)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (34 mg, 0,059 mmol) y Pd al 10 % p/C (0,3 mg, 3 jmol) en MeOH (3 mL) y EtOAc (3 mL) se agitó en una atmósfera de H2 a TA durante toda la noche. La mezcla de reacción se filtró a través de celite, se enjuagó con MeOH y se concentró. El material crudo se purificó mediante HPLC para obtener el producto deseado como un sólido amarillo (15 mg, 52,3 % de rendimiento). LCMS tR = 0,45 min [Método Q], MS (M-1) = 486,4.
1H RMN (METANOL-d4) 5 8,16 (s, 1H), 8,12 (d, J=10,1 Hz, 1H), 7,69 (d, J=8 , 6 Hz, 1H), 7,46 (s, 1H), 7,15-7,22 (m,
2H), 7,14 (s, 1H), 4,98 (t, J=12,1 Hz, 1H), 3,49 (s, 2H), 2,89 (s, 3H), 2,37 (s a., 4H), 1,34-1,60 (m, 10H), 1,29 (s, 6H), 1,13 (s, 6H).
Ejemplo de referencia 24-3: Síntesis de 3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)-7-(pirrolidin-1-ilmetil)naftalen-2-ol

Una mezcla de reacción desgasificada del Intermedio 5-3 (50 mg, 0,093 mmol), potasio-1-trifluoroboratometilpirrolidina (26,6 mg, 0,139 mmol), acetato de paladio (1,0 mg, 4,6 |jmol), X-phos (4,4 mg, 9,3 |jmol) y Cs2COa (91 mg, 0,28 mmol) en THF (1 mL) y agua (0,1 mL) se calentó a 100 °C durante 1 h con irradiación de microondas. La mezcla de reacción se concentró, se acidificó hasta pH 3 mediante la adición de HCl acuoso 1 M y se cargó sobre una columna de SCX, a continuación se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y el material crudo se purificó mediante HPLC para obtener el producto deseado como un sólido blanco ( 6 mg, 13 % de rendimiento). MS (M+1) = 473,32. 1H RMN (METANOL-d4) 58,29 (s, 1H), 8,26 (d, J=10,1 Hz, 1H), 7,81 (d, J=8 , 6 Hz, 1H), 7,60 (s, 1H), 7,29-7,36 (m, 2H), 7,25 (s, 1H), 5,11 (t, J=12,4 Hz, 1H), 3,76 (s, 2H), 3,01 (s, 3H), 2,56-2,67 (m, 4H), 1,77-1,89 (m, 4H), 1,65-1,74 (m, 2H), 1,52-1,63 (m, 2H), 1,39 (s, 6H), 1,23 (s, 6H).
Ejemplo de referencia 24-4: Síntesis de 1-bromo-6-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)naftaleno-2,7-diol

Paso 1: 7-(Bendloxi)-1-bromo-6-(6-(metil(2,2,6,6-tetmmetilpiperídin-4-il)amino)pindazin-3-il)naftalen-2-ol
Una mezcla de 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (100 mg, 0,201 mmol) en DMF (1 mL) se añadió N-bromosuccinimida (39,4 mg, 0,221 mmol) a 0 °C. La mezcla de reacción se agitó a TA durante 1,5 h, a continuación se cargó sobre una columna de SCX de 2 g SCX, se lavó con MeOH, y se eluyó con NH3 2 M en MeOH. Las fracciones que contenían el producto se concentraron y el material crudo se purificó mediante cromatografía en columna de gel de sílice ultrarrápida para obtener el producto deseado como un sólido marrón (33,6 mg, 29 % de rendimiento). MS (M+1) = 577,3.
Paso 2: 3-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-7-(piperidin-1-ilmetil)naftalen-2-ol
Una mezcla de 7-(benciloxi)-1-bromo-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (29 mg, 0,050 mmol) en DCM (1 mL) se añadió lentamente BBr3 (solución 1 M en d Cm , 0,25 mL, 0,25 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 10 minutos, a continuación se calentó hasta la temperatura ambiente y se agitó a durante 2 h. La reacción se desactivó con MeOH y se concentró. El residuo se cargó sobre una columna de SCX de 1 g se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron para obtener el producto deseado como un sólido marrón claro (14 mg, 57 % de rendimiento). LCMS tR = 0,54 min [Método Q], MS (M+1) = 487,2. 1H RMN (METANOL-d4) 58,18-8,29 (m, 2H), 7,70 (d, J=8 , 6Hz, 1H), 7,50 (s, 1H), 7,33 (d, J=10,1 Hz, 1H), 6,99 (d, J=8 , 6 Hz, 1H), 5,16 (t, J=11,9 Hz, 1H), 3,03 (s, 3H), 1,72-1,80 (m, 2H), 1,61-1,70 (m, 2H), 1,45 (s, 6H), 1,29 (s, 6H).
Ejemplo de referencia 24-5: Síntesis de 1-cloro-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftaleno-2,7-diol
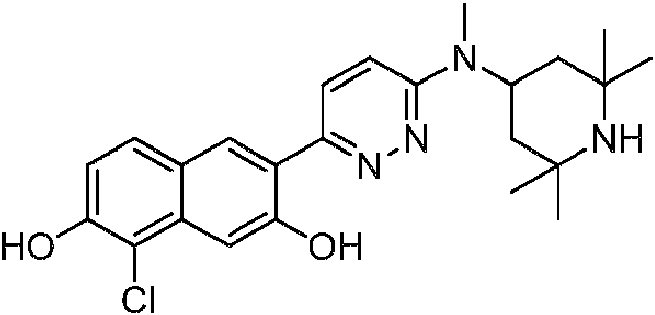
Paso 1: 7-(Bendloxi)-1-dom-6-(6-(metil(2,2,6,6-tetmmetilpipendin-4-il)amino)pindazin-3-il)naftalen-2-ol
Una mezcla de reacción de 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (100 mg, 0,201 mmol) en DMF (2 mL) se añadió N-clorosuccinimida (32,3 mg, 0,242 mmol) a temperatura ambiente. La mezcla de reacción se agitó durante toda la noche y a continuación se cargó sobre una columna de SCX, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y se purificaron mediante cromatografía en columna de gel de sílice ultrarrápida para obtener el producto del título como un producto sólido marrón claro (25 mg, 23 % de rendimiento). MS (M+1) = 531,6.
Paso 2: 1-Cloro-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftaleno-2,7-diol
Una mezcla de 7-(benciloxi)-1-cloro-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (25 mg, 0,047 mmol) en Dc M (1 mL) se añadió lentamente BBr3 (solución 1 M en DCM, 0,25 mL, 0,25 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 10 minutos, a continuación se calentó hasta TA y se agitó durante 1,5 h. La reacción se desactivó con MeOH y se concentró. El residuo se cargó sobre una columna de SCX de 1 g se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron para obtener el producto deseado como un sólido marrón claro (10 mg, 48,2 % de rendimiento). LCMS tR = 0,54 min [Método Q], MS (M+1) = 441,3.
1H RMN (METANOL-d4) 5 8,17-8,27 (m, 2H), 7,66 (d, J=9,1 Hz, 1H), 7,46 (s, 1H), 7,32 (d, J=10,1 Hz, 1H), 7,00 (d, J=9,1 Hz, 1H), 5,13 (t, J=11,9 Hz, 1H), 3,02 (s, 3H), 1,71-1,79 (m, 2H), 1,59-1,68 (m, 2H), 1,44 (s, 6H), 1,28 (s, 6H).
Ejemplo de referencia 24-6: Síntesis de 7-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)naftalen-2-ol

tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol a partir de Intermedio 5-4 (70 mg, 0,14 mmol). Se obtuvo un sólido blanco después de la purificación mediante HPLC (30 mg, 52 % de rendimiento). LCMS tR = 0,57 min [Método Q], MS (M+1) = 421,3. 1H RMN (METANOL-d4) 58,25 (d, J=9,6 Hz, 1H), 8,22 (s, 1H), 7,73 (d, J=8 , 6 Hz, 1H), 7,33 (d, J=9,6 Hz, 1H), 7,19 (s, 1H), 7,05 (d, J=2,5 Hz, 1H), 6,93 (dd, J=8 ,6 , 2,5 Hz, 1H), 5,10 (t, J=12,1 Hz, 1H), 3,90 (s, 3H), 3,02 (s, 3H), 1,68-1,76 (m, 2H), 1,55-1,65 (m, 2H), 1,41 (s, 6H), 1,25 (s, 6H).
Ejemplo de referencia 24-7: Síntesis de 7-metoxi-3-(6-(metil(1,2, 2,6,6-pentametilpiperidm-4-il)ammo)piridazm-3-il)naftalen-2-ol

Paso 1: 6-(3-(Bendbxi)-6-metoxinaftalen-2-il)-N-metil-N-(1,2,2,6,6-pentametilpipendin-4-il)pindazin-3-amina
A una mezcla del Intermedio 5-4 (400 mg, 0,783 mmol) en DMSO (1,5 mL) y agua (3 mL) se añadió formaldehído al 37 % p (0,087 mL, 1,2 mmol) y ácido fórmico (0,060 mL, 1,6 mmol). La mezcla se calentó a 120 °C con irradiación de microondas durante 2 0 min. La mezcla se calentó a 12 0 °C con irradiación de microondas durante 2 0 minutos dos veces más después de la adición de porciones adicionales de formaldehído (0,087 mL, 1,28 mmol) y ácido fórmico (0,060 ml, 1,6 mmol) cada vez. La mezcla de reacción se concentró y se cargó sobre una columna de SCX de 5 g se
lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y el producto crudo se purificó mediante cromatografía en columna ultrarrápida (0-20 % NH3 1,5 M en MeOH/DCM) para obtener el producto deseado como un sólido beige (340 mg, 83 % de rendimiento). MS (M+1) = 525,6.
Paso 2: 7-Metoxi-3-(6-(metil(1,2, 2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol
A partir del compuesto 6-(3-(benciloxi)-6-metoxinaftalen-2-il)-N-metil-N-(1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina (200 mg, 0,381 mmol), siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis del grupo bencilo, se obtuvo el compuesto 7-metoxi-3-(6-(metil(1,2,2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol como un sólido beige (150 mg, 91 % de rendimiento) después de purificación mediante cromatografía en columna ultrarrápida (0-15 % de NH3 1,5 M en MeOH/DCM). El producto se convirtió en la sal de HCl mediante la adición de HCl acuoso 4 M (0,2 mL, 2,3 equivalentes) y después se liofilizó. LCMS tR = 0,53 min [Método Q], MS (M+1) = 435,3. 1H RMN (METANOL-d4) 6 8,52 (d, J=10,1 Hz, 1H), 8,18 (s, 1H), 8,02 (d, J=9,6 Hz, 1H), 7,81 (d, J=9,1 Hz, 1H), 7,30 (s, 1H), 7,12 (d, J=2,5 Hz, 1H), 7,04 (dd, J=9,1, 2,5 Hz, 1H), 5,06 (t, J=11,4 Hz, 1H), 3,93 (s, 3H), 3,20 (s, 3H), 2,90 (s, 3H), 2,38 (t, J=13,1 Hz, 2H), 2,11 (dd, J=13,6, 3,0 Hz, 2H), 1,63 (s, 6H), 1,62 (s, 6H).
Ejemplo de referencia 24-8: Síntesis de 7-(3,6-dihidro-2H-piran-4-N)-3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazin-3-il)naftalen-2-ol

Una mezcla de reacción desgasificada del Intermedio 5-3 (100 mg, 0,186 mmol), éster pinacólico del ácido 3,6-dihidro-2H-piran-4-borónico (50,7 mg, 0,241 mmol) y tetrakis(trifenilfosfina)paladio (0) (10,73 mg, 9,28 μmol) en 1,4-dioxano (2 mL) y una solución ac. de Na2CO3 1 M (0,46 mL, 0,464 mmol) se hicieron reaccionar de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. El producto crudo se purificó mediante HPLC para obtener el compuesto del título como un sólido marrón claro (65 mg, 73 % de rendimiento). LCMS tR = 0,60 min [Método Q], MS (M+1) = 473,4. 1H RMN (METANOL-d4) 6 8,20-8,40 (m, 2H), 7,81 (d, J=8 , 6 Hz, 1H), 7,67 (s, 1H), 7,48 (dd, J=8 ,8 , 1,8 Hz, 1H), 7,35 (d, J=10,1 Hz, 1H), 7,29 (s, 1H), 6,37 (s a., 1H), 5,13 (t, J=12,0 Hz, 1H), 4,37 (d, J=2,5 Hz, 2H), 3,99 (t, J=5,6 Hz, 2H), 3,04 (s, 3H), 2,67 (s, 2H), 1,68-1,78 (m, 2H), 1,55-1, 66 (m, 2H), 1,41 (s, 6H), 1,25 (s, 6H).
Ejemplo de referencia 24-9: Síntesis de 3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piNdazm-3-N)-7-(tetrahidro-2H-piran-4-il)naftalen-2-ol

A partir del compuesto 7-(3,6-dihidro-2H-piran-4-il)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (200 mg, 0,381 mmol), siguiendo el MéTo Do GENERAL 4-1, se obtuvo el compuesto 3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-7-(tetrahidro-2H-piran-4-il)naftalen-2-ol como un sólido blanco después de purificación mediante HPLC (11 mg, 24 % de rendimiento). LCMS tR = 0,59 min [Método Q], MS (M+1) = 475,4. 1H RMN (METANOL-d4) 6 8,25-8,32 (m, 2H), 7,81 (d, J=8 , 6 Hz, 1H), 7,52 (s, 1H), 7,35 (d, J=10,1 Hz, 1H), 7,19-7,27 (m, 2H), 5,12 (t, J=12,9 Hz, 1H), 4,09 (dd, J=10,6, 3,0 Hz, 2H), 3,62 (td, J=11,2, 3,3 Hz, 2H), 3,04 (s, 3H), 2,94 (dt, J=10,5, 5,6 Hz, 1H), 1,81-1,97 (m, 4H), 1,68-1,76 (m, 2H), 1,55-1,65 (m, 2H), 1,41 (s, 6H), 1,25 (s, 6H).
Ejemplo de referencia 24-10: Síntesis de 7-(difluorometil)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol

Paso 1: 6-(3-(Benciloxi)-6-vinilnaftalen-2-il)-N-metil-N-(1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar el éter pinacólico del ácido vinilborónico (100 mg, 0,651 mmol) y el Intermedio 5-2 (315 mg, 0,501 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo un sólido beige (250 mg, 98 % de rendimiento) después de purificación mediante SCX. El material crudo se utilizó sin una purificación adicional. MS (M+1) = 507,5.
Paso 2: 7-(Benciloxi)-6-(6-(metil( 1,2,2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)-2-naftaldehído
A una mezcla de 6-(3-(benciloxi)-6-vinilnaftalen-2-il)-N-metil-N-(1,2,2,6,6-pentametilpiperidin-4-il)piridazin-3-amina (106 mg, 0,209 mmol) y tetróxido de osmio (solución acuosa al 4 % p, 0,080 mL, 0,013 mmol) en THF/agua 5:1 (60 mL) se añadió peryodato de sodio (112 mg, 0,523 mmol) a TA. La mezcla se agitó a TA durante toda la noche y a continuación se desactivó con una solución acuosa al 20 % de Na2S2O3. La mezcla cruda se basificó con una solución acuosa de NaHCO3 y se extrajo con DCM. La capa orgánica se secó con Na2SO4, se filtró y se concentró. El residuo se cargó sobre una columna de SCX de 2 g, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron para obtener 75 mg de un sólido beige que comprendió una mezcla 1:1 del compuesto del título y el acetal dimetílico correspondiente. La mezcla se disolvió en DCM (1,5 mL) y TFA (0,22 mL) y se agitó a TA durante 2 h. La mezcla se concentró, se basificó con una solución acuosa de NaHCO3 y se extrajo con DCM. La capa orgánica se secó con Na2SO4, se filtró y se concentró para obtener 7-(benciloxi)-6-(6-(metil(1,2,2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)-2-naftaldehído como un sólido marrón claro. El material crudo se utilizó sin una purificación adicional. MS (M+1) = 509,4.
Paso 3: 6-(3-(Benciloxi)-6-(difluorometil)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Una mezcla de 7-(benciloxi)-6-(6-(metil(1,2,2,6,6-pentametilpiperidin-4-il)amino)piridazin-3-il)-2-naftaldehído (75 mg, 0,147 mmol) en DCM (1,5 mL) se añadió trifluoruro de dietilaminoazufre (DAs T) (0,058 mL, 0,44 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 10 minutos, a continuación se calentó hasta TA y se agitó durante 2 días. La mezcla de reacción se desactivó con NaHCO3 acuoso a 0 °C y se extrajo con DCM. La capa orgánica se secó con Na2SO4, se filtró y se concentró. El producto crudo se purificó mediante HPLC para obtener el compuesto del título como un sólido blanco (20,6 mg, 26 % de rendimiento). MS (M+1) = 531,1.
Paso 4: 7-(Difluorometil)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol
A partir del compuesto 6-(3-(benciloxi)-6-(difluorometil)naftalen-2-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (15 mg, 0,028 mmol), siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis del grupo bencilo, se obtuvo el compuesto 7-(difluorometil)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol después de purificación mediante HPLC ( 6 mg, 50 % de rendimiento). LCMS tR = 0,60 min [Método Q], MS (M+1) = 441,3. 1H RMN (METANOL-d458,37 (s, 1H), 8,29 (d, J=9,6 Hz, 1H), 7,96 (d, J=8 , 6 Hz, 1H), 7,85 (s, 1H), 7,41 (d, J=8 , 6 Hz, 1H), 7,37 (s, 1H), 7,34(d, J=9,6 Hz, 1H), 6,87 (t, J=58,0 Hz, 1H), 5,23 (t, J=12,1 Hz, 1H), 3,03 (s, 3H), 1,74-1,83 (m, 2H), 1,64-1,74 (m, 2H), 1,47 (s, 6H), 1,28-1,37 (m, 6H).
Ejemplo de referencia 24-11 y 24-12: Síntesis de 7-((4-hidroxi-2-metilbutan-2-il)oxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-il)naftalen-2-ol y 7-(3-hidroxi-3-metilbutoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol

Paso 1: 3-((7-(Benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)-3-metilbutan-1-ol y 4-((7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-il)oxi)-2-metilbutan-2-ol Un matraz de fondo redondo de 50 mL se añadió 7-(benciloxi)-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (Ejemplo de referencia 20-2, Paso 1, 640 mg, 1,289 mmol) y 3-metilbutano-1,3-diol (550 μL, 5,15 mmol) en THF (8 , 6 mL) para obtener una solución tostada. Se añadieron trifenilfosfina (744 mg, 2,84 mmol) y DIAD (560 μl, 2,71 mmol) y la mezcla se agitó en nitrógeno a temperatura ambiente. Después de agitar durante toda la noche, la reacción se concentró a sequedad, a continuación se añadieron agua y EtOAc y se separó la capa orgánica. La capa orgánica se extrajo con HCl 0,2 N, a continuación se añadió NaHCO3 para neutralizar la capa acuosa que se extrajo a continuación con EtOAc (2x). Los extractos orgánicos combinados se secaron con MgSO4 , se filtraron y se concentraron al vacío. El material crudo (una mezcla ~1:1 de regioisómeros) se utilizó en el siguiente paso sin una purificación adicional.
Paso 2: Se prepararon 7-((4-hidroxi-2-metilbutan-2-il)oxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (43 mg, 0,097 mmol, 12 % de rendimiento, 2 pasos) y 7-(3-hidroxi-3-metilbutoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol (29 mg, 0,058 mmol, 9 % de rendimiento, 2 pasos) a partir de los aductos de bencilo siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis. La purificación y separación mediante HPLC preparativa (Waters Sunfire 30 mm ID x 50 mm, 0,1% de TFA, 25-50 % de ACN/H2O) proporcionó los productos regioisoméricos.
7-((4-Hidroxi-2-metilbutan-2-il)oxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol: LCMS tR = 0,52 min [Método Q], M+1 = 493,4. 1H RMN (400 MHz, CLOROFORMO-d) 6 8,04 (s, 1H), 8,00 (d, J=10,04 Hz, 1H), 7,68 (d, J=9,03 Hz, 1 H), 7,29 (s, 1H), 7,23-7,27 (m, 2H), 7,04 (d, J=10,04 Hz, 1 H), 6,98 (dd, J=2,26, 8,78 Hz, 1 H), 4,98 (s a., 1 H), 4,00 (t, J=5,90 Hz, 2H), 3,49 (s, 2H), 3,04 (s, 3H), 2,04 (t, J=5,90 Hz, 2H), 1,73 (dd, J=3,39, 12,42 Hz, 2H), 1,40-1,47 (m, 8 H), 1,38 (s, 6H), 1,21 (s, 6H).
7-(3-Hidroxi-3-metilbutoxi)-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)naftalen-2-ol: LCMS tR = 0,54 min [Método Q], M+1 = 493,4. 1H RMN (400 MHz, CLOROFORMO-d) 6 7,95-8,03 (m, 2H), 7,66 (d, J=8,78 Hz, 1H), 7,28 (s, 1H), 7,26 (s, 1H), 6,99-7,07 (m, 2H), 6,93 (dd, J=2,38, 8,91 Hz, 1H), 4,96 (s a., 1H), 4,31 (t, J=6,27 Hz, 2H), 3,04 (s, 3H), 2,07 (t, J=6,15 Hz, 2H), 1,72 (dd, J=3,39, 12,42 Hz, 2H), 1,38 (m, 8 H), 1,35 (s, 6H), 1,21 (s, 6H).
Ejemplo de referencia 25-1: Síntesis de 2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-N)-5-(1H-pirazol-4-il)benceno-1,3-diol

Paso 1: 6-(2,6-DimetoxifenH)-N-metH-N-(2,2,6,6-tetrametilpiperidin-4-H)piridazin-3-amina
Se hicieron reaccionar el Intermedio 1-1 (566 mg, 2,0 mmol) y ácido 2,6-dimetoxifenilborónico (437 mg, 2,4 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo 6-(2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido beige (375 mg, 49 % de rendimiento) después de la purificación mediante cromatografía en columna ultrarrápida. MS (M+1) = 385,4.
Paso 2: 6-(4-Bromo-2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar 6-(4-bromo-2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (240 mg, 0,624 mmol), bis(pinacolato)diboro (238 mg, 0,936 mmol), 4,4'-di-tert-butilbipiridino(dtbpy, 3,4 mg, 0,012 mmol) y [Ir(COD)(OMe)]2 (4,1 mg, 6,2 μmol) siguiendo el MÉTODO GENERAL 7-1 para la borilación/bromación, y se obtuvo 6-(4-bromo-2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido amarillo (176 mg, 60 % de rendimiento) después de la purificación mediante cromatografía en columna ultrarrápida y HPLC. MS (M+1) = 465,4.
Paso 3: 6-(2,6-Dimetoxi-4-( 1 H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar 6-(4-bromo-2,6-dimetoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (63 mg, 0,136 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (52,8 mg, 0,272 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo el compuesto del título como un sólido amarillo después de cromatografía en columna (43 mg, 70 % de rendimiento). MS (M+1) = 451,5.
Paso 4: 2-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)benceno-1,3-diol
A partir del compuesto 6-(2,6-dimetoxi-4-(1 H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (43 mg, 0,095mmol), siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol (22,1 mg, 0,2 mmol), se obtuvo el compuesto 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1 H-pirazol-4-il)benceno-1,3-diol como un sólido amarillo pálido (20 mg, 45 % de rendimiento) después de la purificación mediante HPLC. LCMS tR = 0,41 min [Método Q], MS (M+1) = 423,3. 1H RMN (METANOL-d4) 6 8,60 (d, J=10,1 Hz, 1H), 7,91 (s, 2H), 7,20 (d, J=10,1 Hz, 1H), 6 , 6 6 (s, 2H), 5,01 (t, J=12,4 Hz, 1H), 2,90-3,01 (m, 3H), 1,64-1,73 (m, 2H), 1,52-1,61 (m, 2H), 1,38 (s, 6H), 1,23 (s, 6H).
Ejemplo de referencia 25-2: Síntesis de 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpipendm-4-il)ammo)μmdazm-3-il)-5-(1 H-pirazol-4-il)fenol
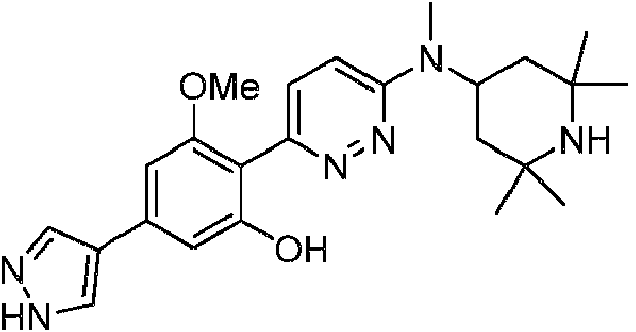
Paso 1: 1-(BencHoxi)-2-bromo-3-metoxibenceno
A una mezcla de 3-(benciloxi)-2-bromofenol (2,55 g, 9,14 mmol) en DMF ( 8 mL) se añadieron K2CO3 (1,894 g, 13,70 mmol) y Mel (0,63 mL, 10,05 mmol) a TA. La mezcla de reacción se agitó durante toda la noche, a continuación se desactivó con agua y se diluyó con EtOAc. La fase orgánica se lavó con agua (3x), salmuera, se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (EtOAc/Heptano) para proporcionar el compuesto del título como un aceite incoloro (2,66 g, 99 % de rendimiento). 1H RMN (CLOROFORMO-d) 6 7,49 (d, J=7,1 Hz, 2H), 7,36-7,43 (m, 2H), 7,30-7,35 (m, 1H), 7,20 (t, J=8,3 Hz, 1H), 6,56-6,65 (m, 2H), 5,18 (s, 2H), 3,92 (s, 3H).
Paso 2: Ácido (2-(benciloxi)-6-metoxifenil)borónico
A una mezcla de 1-(benciloxi)-2-bromo-3-metoxibenceno (2,66 g, 9,07 mmol) en THF (20 mL) se añadió butillitio (2,5 M en THF, 4 mL, 9,98 mmol) a -78 °C gota a gota durante 15 minutos. La mezcla se agitó a -78 °C durante 30 minutos y a continuación se añadió borato de trimetilo (4,0 mL, 36,3 mmol). Se permitió que la mezcla se calentara hasta TA y se agitó durante toda la noche. La mezcla se desactivó con HCl acuoso 1 M hasta pH 2 y se extrajo con DCM. La fase orgánica se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (EtOAc/Heptano) para proporcionar el producto deseado como un sólido blanco (1,07 g, 46 % de rendimiento). MS (M+1) = 259,4.
Paso 3: 6-(2-(Benciloxi)-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar el Intermedio 1 -1 (391 mg, 1,384 mmol) y ácido (2-(benciloxi)-6-metoxifenil)borónico (500 mg, 1,94 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo 6-(2-(benciloxi)-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido beige (358 mg, 56 % de rendimiento) después de la purificación mediante cromatografía en columna ultrarrápida. MS (M+1) = 461,5.
Paso 4: 6-(2-(Benciloxi)-4-bromo-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A partir del compuesto 6-(2-(benciloxi)-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (300 mg, 0,651 mmol), siguiendo el MÉTODO GENERAL 7-1 para la borilación/bromación, se obtuvo 6-(2-(benciloxi)-4-bromo-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina después de la purificación mediante cromatografía en columna (170 mg, 50 % pura, 24 % de rendimiento). MS (M+1) = 541,4.
Paso 5: 6-(2-(Benciloxi)-6-metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Sedición reaccionar 6-(2-(benciloxi)-4-bromo-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (129 mg, 50 % puro, 0,12 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (46,4 mg, 0,24 mmol) T acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo 6-(2-(benciloxi)-6-metoxi-4-(1 H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido amarillo (14 mg, 22 % de rendimiento) después de la purificación mediante HPLC. MS (M+1) = 527,4.
Paso 6: 3-Metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)fenol
A partir del compuesto 6-(2-(benciloxi)-6-metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (14 mg, 0,027 mmol), siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis del grupo bencilo, se obtuvo el compuesto 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)fenol después de la purificación mediante HPLC (4,2 mg, 35 % de rendimiento). LCMS tR = 0,43 min [Método Q], MS (M+1) = 437,4. 1H RMN (METANOL-d4) 6 8,23 (d, J=10,1 Hz, 1H), 8,01 (s a., 2H), 7,22 (d, J=10,1 Hz, 1H), 6,79 6,87 (m, 2H), 5,13 (t, J=12,4 Hz, 1H), 3,94 (s, 3H), 3,00 (s, 3H), 1,72-1,82 (m, 2H), 1,60-1,72 (m, 2H), 1,45 (s, 6H), 1,30 (s, 6H).
Ejemplo de referencia 25-3: Síntesis de 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpipendm-4-il)ammo)pindazm-3-il)-3-(trifluorometoxi)fenol

Paso 1: 4-(Benciloxi)-1-bmmo-2-(trifluommetoxi)benceno
Una mezcla de 4-bromo-3-(trifluorometoxi)fenol (2,97 g, 11,6 mmol) en DMF (10 mL) se añadieron Cs2CO3 (5,65 g, 17,3 mmol) y cloruro de bencilo (1,46 mL, 12,7 mmol) a TA. La mezcla de reacción se agitó durante toda la noche, a continuación se desactivó con agua y se diluyó con EtOAc. La fase orgánica se lavó con agua (3x), salmuera, se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (EtOAc/Heptano) para proporcionar el compuesto del título como un aceite incoloro (3,92 g, 98 % de rendimiento). 1H RMN (METANOL-d4) 6 7,51 (d, J=9,1 Hz, 1H), 7,40-7,45 (m, 4H), 7,34-7,40 (m, 1H), 6,97 (dd, J=2,5, 1,5 Hz, 1H), 6,82 (dd, J=9,1, 3,0 Hz, 1 H), 5,06 (s, 2H).
Paso 2: Ácido (4-(benciloxi)-2-(trifluorometoxi)fenil)borónico
A una mezcla de 4-(benciloxi)-1-bromo-2-(trifluorometoxi)benceno (1,8 g, 5,19 mmol) en THF (20 mL) se añadió butillitio (2,5 M en THF, 2,28 mL, 5,70 mmol) a -78 °C gota a gota durante 10 minutos. La mezcla de reacción se agitó a -78 °C durante 1 h y a continuación se añadió borato de trimetilo (1,73 mL, 15,6 mmol). La mezcla se calentó hasta la temperatura ambiente y se agitó durante toda la noche. La mezcla se concentró en éter dietílico tres veces y el residuo se secó a alto vacío para proporcionar el compuesto del título como un sólido gomoso (2,2 g, 61 % puro, 83 % de rendimiento). El material crudo se utilizó sin una purificación adicional. MS (M-1) = 311,3
Paso 3: 6-(4-(Benciloxi)-2-(trifluorometoxi)fenil)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se hicieron reaccionar el ácido (4-(benciloxi)-2-(trifluorometoxi)fenil)borónico (360 mg, 61 % puro, 0,692 mmol) y el Intermedio 1-2 (93 mg, 0,346 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki. Se obtuvo 6-(4-(benciloxi)-2-(trifluorometoxi)fenil)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido blanco (121 mg, 70 % de rendimiento) después de la purificación mediante cromatografía en columna ultrarrápida. MS (M+1) = 501,3.
Paso 4: 4-(6-((2,2,6,6-Tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol
A partir del compuesto 6-(4-(benciloxi)-2-(trifluorometoxi)fenil)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (120 mg, 0,24 mmol), siguiendo el MÉTODO GENERAL 4-1 para la hidrogenólisis del grupo bencilo, se obtuvo 4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol después de la purificación mediante columna de SCX (98 mg, 100 % de rendimiento). Ms (M+1) = 411,3.
Paso 5: Trifluorometanosulfonato de 4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenilo
A una suspensión de 4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol (98 mg, 0,239 mmol) en DCM (2 mL) se añadió Et3N (0,083 mL, 0,597 mmol) y N-feniltrifluorometanosulfonimida (172 mg, 0,48 mmol). Se añadió DMF (0,5 mL) para facilitar la disolución. La disolución se agitó a temperatura ambiente durante toda la noche y a continuación se concentró. El residuo se cargó sobre una columna de SCX de 2 g, se lavó con MeOH y se eluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron y el material crudo se purificó mediante cromatografía en columna ultrarrápida para obtener el compuesto del título como un sólido beige (89 mg, 69 %). MS (M+1) = 543,3.
Paso 6: Trífluorometanosulfonato de 3-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenilo
Una mezcla de trífluorometanosulfonato de 4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenilo ( 86 mg, 0,16 mmol), diacetato de yodobenceno (71,5 mg, 0,222 mmol) y Pd(OAc)2 (3,6 mg, 0,016 mmol) en AcoH (0,6 mL) y Ac2O (0,6 mL) se calentó a 75 °C durante 3 horas. A continuación, se calentó la mezcla a 80 °C durante toda la noche después de la adición de otros 40 mg de diacetato de yodobenceno. La mezcla de reacción se enfrió hasta la temperatura ambiente y se concentró. El residuo se cargó sobre una columna de SCX, se lavó con MeOH y se diluyó con NH32 M en MeOH. Las fracciones que contenían el producto se concentraron. El residuo se trató con NH3 7 M/MeOH y se agitó a 40 °C durante 4 h. El material crudo se purificó mediante cromatografía en columna ultrarrápida para obtener el compuesto del título como un sólido beige (32 mg, 36,1 %). MS (M+1) = 559,4.
Paso 7: 5-(1H-Pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol
Se hicieron reaccionar trifluorometanosulfonato de 3-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenilo (32 mg, 0,043 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (16,68 mg, 0,086 mmol) de acuerdo con el MÉTODO Ge NEr a L 1-5 para el acoplamiento de Suzuki. Se obtuvo 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-3-(trifluorometoxi)fenol como un sólido amarillo después de la purificación mediante HPLC. El producto se convirtió en la sal de HCl mediante la adición de HCl acuoso 1 M (0,1 mL, 2,3 equivalentes) y después se liofilizó (10 mg, 45 % de rendimiento). LCMS tR = 0,48 min [Método Q], MS (M+1) = 477,3. 1H RMN (METANOL-d4) 58,17 (s, 2H), 8,05 (d, J=9,6 Hz, 1H), 7,68 (d, J=9,6 Hz, 1H), 7,22-7,30 (m, 2H), 4,49 (t, J=12,1 Hz, 1H), 2,33 (dd, J=13,6, 3,5 Hz, 2H), 1,70 (t, J=12,9 Hz, 2H), 1,61 (s, 6H), 1,54 (s, 6H).
Ejemplo de referencia 25-4: Síntesis de 2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)-3-(trifluorometoxi)fenol

Se hicieron reaccionar el Intermedio 6-1 (40 mg, 0,070 mmol) y 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (29,1 mg, 0,140 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki, y se obtuvo 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)-3-(trifluorometoxi)fenol como un sólido blanco (7,5 mg, 21 % de rendimiento). LCMS tR = 0,52 min [Método Q], MS (m +1) = 505,4. 1H RMN (METANOL-d4 ) 58,04 (s, 1H), 7,85 (s, 1H), 7,79 (d, J=9,6 Hz, 1H), 7,25 (d, J=9,6 Hz, 1H), 7,11-7,17 (m, 1H), 7,07 (d, J=1,5 Hz, 1H), 5,24 (t, J=11,4 Hz, 1H), 3,94 (s, 3H), 3,02 (s, 3H), 1,72-1,82 (m, 2H), 1,60-1,72 (m, 2H), 1,44 (s, 6H), 1,29 (s, 6H).
Ejemplo de referencia 25-5: Síntesis de 2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazin-3-N)-5-(1H-pirazol-4-il)-3-(trifluorometoxi)fenol

Se hicieron reaccionar el Intermedio 6-1 (35 mg, 0,061 mmol) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (23,72 mg, 0,122 mmol) de acuerdo con el MÉTODO g ENe r Al 1-5 para el acoplamiento de Suzuki, y se obtuvo 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)-3-(trifluorometoxi)fenol como un sólido blanco (27 mg, 90 % de rendimiento). Lc Ms tR = 0,50 min [Método Q], m S (M+1) = 491,4. 1H RMN (METANOL-d4 ) 58,02 (s a., 2H), 7,79 (d, J=9,6 Hz, 1H), 7,25 (d, J=9,6 Hz, 1H), 7,18 (d, J=1,5 Hz, 1H), 7,10-7,13 (m, 1H), 5,26 (t, J=11,9 Hz, 1H), 3,01 (s, 3H), 1,72-1,84 (m, 2H), 1,61-1,72 (m, 2H), 1,45 (s, 6H), 1,30 (s, 6H).
Ejemplo de referencia 25-6: Síntesis de 4-(3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)ammo)piridazm-3-il)-5-(trifluorometoxi)fenil)-1 -metilpiridin-2(1 H)-ona
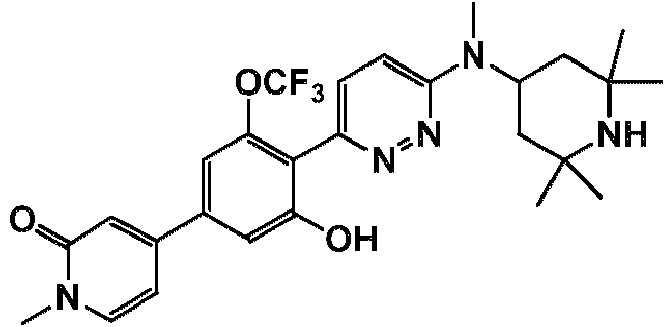
Se hicieron reaccionar el Intermedio 6-1 (40 mg, 0,070 mmol) y el éster pinacólico del ácido 1-metilpiridin-2-ona-4-borónico (32,8 mg, 0,140 mmol) de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento de Suzuki, y se obtuvo 4-(3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(trifluorometoxi)fenil)-1-metilpiridin-2(1H)-ona como un sólido blanco. El producto se convirtió en la sal de HCl mediante la adición de HCl 4 M en dioxano (0,1 mL, 5,7 equivalentes) y después se evaporó el disolvente (14,5 mg, 36 % de rendimiento). LCMS tR = 0,50 min [Método Q], MS (M+1) = 532,3. 1H RMN (METANOL-d4) 5 8,16-8,21 (m, 1H), 8,06-8,13 (m, 1H), 7,81 (d, J=7,1 Hz, 1H), 7,30 (s, 2H), 6,80 (d, J=2,0 Hz, 1H), 6,69 (dd, J=6 ,8 , 2,3 Hz, 1H), 5,02 (s a., 1H), 3,63 (s, 3H), 3,21 (s, 3H), 2,01-2,13 (m, 4H), 1,64 (s, 6H), 1,57 (s, 6H).
Ejemplo de referencia 26-1: Síntesis de 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)fenol
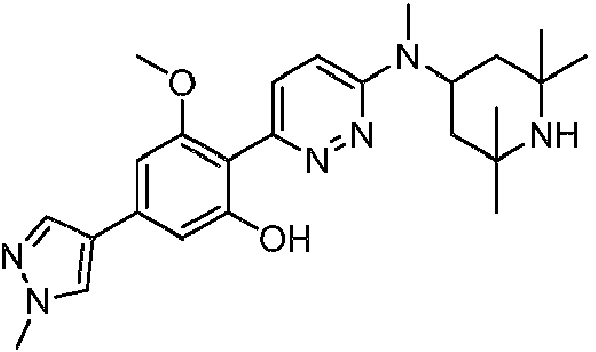
Una mezcla de trifluorometanosulfonato de 3-hidroxi-5-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (Intermedio 6-2, 100 mg, 0,193 mmol), 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (120 mg, 0,579 mmol) y carbonato de sodio (102 mg, 0,964 mmol) en DME/agua 3:1 (1,9 mL) se desgasificó con una corriente de nitrógeno seco durante 5 minutos. Se añadió tetrakis(trifenilfosfina)paladio (0) (16,7 mg, 0,014 mmol) y la mezcla se calentó con radiación de microondas a 90 °C durante una hora. La mezcla se repartió entre agua y diclorometano y la fase orgánica se acidificó con HCl en MeOH (4 equivalentes) y se concentró a sequedad. El material crudo se cargó sobre una columna de SCX (1 g, preacondicionada con MeOH), se lavó con MeOH y se eluyó con amoniaco 7 N en MeOH. El diluyente se concentró a sequedad y la purificación mediante cromatografía ultrarrápida (12 g de sílice, gradiente de un 1-12 % de amoniaco 7 N en MeOH, en DCM) proporcionó 3-metoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)fenol como un sólido amarillo claro (55 mg). LCMS tR = 0,44 min [Método Q], MS (M+1) = 451,5. 1H RMN (400 MHz, METANOL-d4) 58,25 (d, J = 10,11 Hz, 1H), 8,03 (s, 1H), 7,87 (s, 1H), 7,22 (d, J = 10,11 Hz, 1H), 6,74-6,85 (m, 2H), 5,05 (t, J = 12,38 Hz, 1H), 3,95 (s, 3H), 3,95 (s, 3H), 3,01 (s, 3H), 1,65-1,75 (m, 2H), 1,50-1,64 (m, 2H), 1,39 (s, 6H), 1,24 (s, 6H).
L i i n m r r r n n m n r imil r l E m l r f r n i 2 -1.


Ejemplo de referencia 27-1: Síntesis de 3-(bencMoxi)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)pindazm-3-il)-5-(5-metiloxazol-2-il)fenol

Paso 1: Ácido 3-(benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)benzoico
Se añadió cloruro de (2-(trimetilsilil)etoximetilo (SEM-Cl, 3,10 mL, 17,5 mmol) una mezcla de 3-(benciloxi)-4-bromo-5-hidroxibenzoato de metilo (Ejemplo de referencia 22-1 Paso 1, 5,36 g, 15,9 mmol) y carbonato de potasio (5,49 g, 39,7 mmol) en DMF (53,0 mL), y se permitió que la mezcla se agitaba a temperatura ambiente durante dos días. Se añadió una porción adicional de SEM-Cl (3,10 mL, 17,5 mmol) y la mezcla se agitó durante 4 horas más. La mezcla de reacción se repartió entre bicarbonato de sodio saturado y acetato de etilo/éter dietílico 1:1. La fase orgánica se lavó con agua (5x), salmuera, se secó con MgSO4 y se concentró hasta obtener un aceite naranja claro. El producto crudo se disolvió en tetrahidrofurano/metanol 2:1 (100 mL) y se añadió una solución acuosa de hidróxido de sodio (2,0 M, 63,6 mL, 127 mmol). La solución se agitó durante 1 hora tras lo cual se eliminaron los componentes volátiles mediante evaporación giratoria. La solución restante se acidificó hasta pH 3 mediante la adición lenta de ácido clorhídrico concentrado, se extrajo con diclorometano (1x) y a continuación con éter/acetato de etilo 1:1 (4x). Los extractos combinados se lavaron con salmuera, se secaron con MgSO4 y se concentraron hasta obtener un sólido. El producto crudo se purificó disgregándolo en hexano y se secó al vacío para proporcionar ácido 3-(benciloxi)-4-bromo
5-((2-(trimetilsilil)etoxi)metoxi)benzoico (6,36 g) como un sólido blanco. MS (M+1) = 453,4. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,28-7,59 (m, 7H) 5,36 (s, 2H) 5,18-5,28 (m, 2H) 3,75-3,90 (m, 2H) 0,92-1,03 (m, 2H) 0,00 (s, 9H).
Paso 2: 3-(Benciloxi)-4-bromo-N-(prop-2-inil)-5-((2-(trimetilsilil)etoxi)metoxi)benzamida
A una mezcla de ácido 3-(benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)benzoico (6,142 g, 13,55 mmol) y reactivo de Mukaiyama (yoduro de 2-cloro-1-metilpiridinio, 5,19 g, 20,3 mmol) en diclorometano (135 mL) se añadió trietilamina (7,55 mL, 54,2 mmol). La solución se agitó durante 10 minutos tras lo cual se añadió propargilamina (1,74 mL, 27,1 mmol). La solución se agitó durante toda la noche. La solución se diluyó con acetato de etilo/éter dietílico 1:1 y se lavó con agua, salmuera, se secó con MgSO4 y se concentró. El producto crudo se purificó mediante cromatografía ultrarrápida (40 g de gel de sílice, gradiente de acetato de etilo en diclorometano) para proporcionar 3-(benciloxi)-4-bromo-N-(prop-2-inil)-5-((2-(trimetilsilil)etoxi)metoxi)benzamida como un aceite naranja (6,49 g). MS (M+1) = 492,2. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,28-7,55 (m, 5H) 7,13 (dd, J=11,37, 1,77 Hz, 2H) 6,23 (s a., 1H) 5,34 (s, 2H) 5,20 (s, 2H) 4,22 (dd, J=5,05, 2,53 Hz, 2H) 3,81 (dd, J=9,09, 7,58 Hz, 2H) 2,28 (t, J=2,78 Hz, 1H) 0,91-1,00 (m, 2H) -0,03-0,03 (m, 9H).
Paso 3: 2-(3-(Benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)fenil)-5-metiloxazol
Se añadió hidruro de sodio (0,953 g, 39,7 mmol) a una solución de 3-(benciloxi)-4-bromo-N-(prop-2-inil)-5-((2-(trimetilsilil)etoxi)metoxi)benzamida (6,49 g, 13,2 mmol) en dioxano (100 mL) y la mezcla se calentó a reflujo durante toda la noche. La solución se enfrió hasta la temperatura ambiente y se desactivó mediante la adición lenta de NaHCO3 saturado. La solución se diluyó con acetato de etilo/éter dietílico y se lavó con agua (5x), NaHCO3 saturada, salmuera, se secó con sulfato de sodio y se concentró hasta obtener un líquido marrón espeso. La cromatografía ultrarrápida (80 g de gel de sílice, 5-40 % de EtOAc en heptano) proporcionó 2-(3-(benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)fenil)-5-metiloxazol como un aceite naranja (3,33 g). MS (M+1) = 492,21. 1H RMN (400 m Hz , CLOROFORMO-d) 5 ppm 7,30-7,58 (m, 7H), 6 , 86 (d, J=1,0 Hz, 1H), 5,40 (s, 2H), 5,25 (s, 2H), 3,80-3,92 (m, 2H), 2,41 (s, 3H), 0,95-1,04 (m, 2H), 0,02 (s, 10H).
Paso 4: Ácido (2-(benciloxi)-6-hidroxi-4-(5-metiloxazol-2-il)fenil)borónico
A una solución agitada de 2-(3-(benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)fenil)-5-metiloxazol (1,2 g, 2,447 mmol) en THF ( 6 mL) enfriada hasta -78 °C se añadió n-butillitio (2,5 M en heptano, 1,17 mL, 2,94 mmol) gota a gota. La solución se agitó durante 30 minutos tras lo cual se añadió borato de trimetilo (0,82 mL, 7,34 mmol) en una única porción. Se retiró el baño frío y se permitió que la solución se calentara hasta la temperatura ambiente durante dos horas. Se añadió HCl acuoso (0,1 M) y después acetato de etilo/éter dietílico 1:1. La solución se lavó con HCl 0,1 M, agua, salmuera, se secó con sulfato de magnesio y se concentró. Los sodio resultante se lavaron con DCM para proporcionar 105 mg de una mezcla 3:1 de ácido (2-(benciloxi)-6-hidroxi-4-(5-metiloxazol-2-il)fenil)borónico (MS (M+1 ) = 326,2) y 3-(benciloxi)-5-(5-metiloxazol-2-il)fenol (Ms (M+1 ) = 282,2) como un sólido blanquecino. Esta mezcla se utilizó sin una purificación adicional.
Paso 5: 3-(Bendloxi)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol
Una mezcla de 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 45 mg, 0,16 mmol), el ácido (2-(benciloxi)-6-hidroxi-4-(5-metiloxazol-2-il)fenil)borónico (103 mg, 0,239 mmol basándose en un 75 % de pureza), y carbonato de sodio (51 mg, 0,48 mmol) en DME/agua 3:1 se desgasificó con una corriente de nitrógeno seco durante cinco minutos. Se añadió tetrakis(trifenilfosfina)paladio (0) (18,39 mg, 0,016 mmol) y la mezcla se calentó con radiación de microondas a 140 °C durante 30 minutos. La mezcla se diluyó con diclorometano y se lavó con agua. La fase orgánica se acidificó con HCl en MeOH (3 equivalentes) y se concentró a sequedad. El material crudo se cargó sobre una columna de SCX (1 g, preacondicionada con MeOH), se lavó con MeOH y se eluyó con amoniaco 7 N en MeOH. El diluyente se concentró a sequedad y la purificación mediante cromatografía ultrarrápida (12 g de sílice, gradiente de un 2-12 % de amoniaco 7 N en MeOH, en DCM) proporcionó 3-(benciloxi)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol como un sólido amarillo claro. LCMS tR = 0,61 min [Método Q], MS (M+1) = 528,5. 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,18 (d, J=10,10 Hz, 1H), 7,28-7,51 (m, 6H), 7,23 (d, J=1,52 Hz, 1H), 7,13 (d, J=10,11 Hz, 1H), 6,95 (d, J=1,52 Hz, 1H), 5,25 (s, 2H), 5,14 (t, J=11,87 Hz, 1H), 2,99 (s, 3H), 2,45 (d, J=1,01 Hz, 3H), 1,66-1,75 (m, 2H), 1,53-1,64 (m, 2H), 1,40 (s, 6H), 1,25 (s, 6H).
Ejemplo de referncia 27-2: Síntesis de 3-etoxi-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)-5-(5-metiloxazol-2-il)fenol

Paso 1: 3-(Bendioxi)-2-bromo-5-(5-metiioxazoi-2-ii)fenoi
Se añadió ácido clorhídrico concentrado (3 mL) a una solución de 2-(3-(benciloxi)-4-bromo-5-((2-(trimetilsilil)etoxi)metoxi)fenil)-5-metiloxazol (0,60 g, 1,22 mmol) en THF ( 8 mL) y se agitó a temperatura ambiente durante tres horas. La solución se diluyó con agua y se extrajo con acetato de etilo/éter dietílico 1:1 (4x). Los extractos se lavaron con bicarbonato de sodio saturado, salmuera, se secaron con sulfato de magnesio y se concentraron hasta obtener un sólido. El sólido se purificó disgregándolo en heptano (2x) y se secó al vacío para proporcionar 3-(benciloxi)-2-bromo-5-(5-metiloxazol-2-il)fenol como un sólido blanquecino (412 mg). MS (M+1) = 360,2. 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,52 (d, J=7,07 Hz, 2H), 7,36-7,43 (m, 2H), 7,28-7,35 (m, 2H), 7,23 (d, J=1,52 Hz, 1 H), 6 , 86 (d, J=1,52 Hz, 1H), 5,24 (s, 2H), 4,21 (c, J=6,74 Hz, 2H), 2,41 (d, J=1,01 Hz, 3H), 1,50 (t, J=6,82 Hz, 3H).
Paso 2: 2-(3-(Benciioxi)-4-bromo-5-etoxifenii)-5-metiioxazoi
Se añadió yodoetano (111 uL, 1,37 mmol) a una mezcla de 3-(benciloxi)-2-bromo-5-(5-metiloxazol-2-il)fenol (412 mg, 1.14 mmol) y carbonato de potasio (632 mg, 4,58 mmol) en DMF (2,8 mL). Después de agitar durante dos horas, la solución se diluyó con acetato de etilo/éter dietílico 1:1, se lavó con agua (5x), salmuera, se secó con sulfato de magnesio y se concentró para proporcionar 2-(3-(benciloxi)-4-bromo-5-etoxifenil)-5-metiloxazol como un sólido cristalino blanco (421 mg). MS (M+1) = 388,2. 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 7,52 (d, J=7,07 Hz, 2H), 7,36-7,43 (m, 2H), 7,28-7,35 (m, 2H), 7,23 (d, J=1,52 Hz, 1H), 6 , 8 6 (d, J=1,52 Hz, 1H), 5,24 (s, 2H), 4,21 (c, J=6,74 Hz, 2H), 2,41 (d, J=1,01 Hz, 3H), 1,50 (t, J=6,82 Hz, 3H).
Paso 3: Ácido (2-(benciioxi)-6-etoxi-4-(5-metiioxazoi-2-ii)fenii)borónico
El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 6. MS (M+1) = 354,3. 1H RMN (400 MHz, METANOL-d4) 6 ppm 7,27-7,46 (m, 1H), 7,26 (s, 1H), 7,18 (s, 1H), 6,90 (d, J=1,0 Hz, 1H), 5.14 (s, 2H), 4,11 (c, J=7,1 Hz, 2H), 2,40 (d, J=1,0 Hz, 3H), 1,37 (t, J=6 , 8 Hz, 3H).
Paso 4: 6-(2-(Benciioxi)-6-etoxi-4-(5-metiioxazoi-2-ii)fenii)-N-metii-N-(2,2,6,6-tetrametiipiperidin-4-ii)piridazin-3-amina El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 7. MS (M+1) = 556,5
Paso 5: 3-Etoxi-2-(6-(metii(2,2,6-trimetiipiperidin-4-ii)amino)piridazin-3-ii)-5-(5-metiioxazoi-2-ii)fenoi.
El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 8. LCMS tR = 0,53 min [Método Q], MS (M+1) = 466,4. 1H RMN (400 MHz, METANOL-d4) 6 8,30 (d, J = 9,60 Hz, 1H), 7,26 (d, J = 9,60 Hz, 1 H), 7,20 (d, J = 4,04 Hz, 2H), 6,95 (s, 1H), 5,17 (s a., 1H), 4,23 (c, J = 6,91 Hz, 2H), 3,03 (s, 3H), 2,45 (s, 3H), 1,72-1,81 (m, 2H), 1,59-1,71 (m, 2H), 1,49 (t, J = 6,82 Hz, 3H), 1,44 (s, 6H), 1,29 (s, 6H).
Ejemplo de referencia 27-3: Síntesis de 3-(ciclopropNmetoxi)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazin-3-il)-5-(5-metiloxazol-2-il)fenol

Paso 1: 2-(3-(Benciioxi)-4-bromo-5-(ciciopropiimetoxi)fenii)-5-metiioxazoi
El compuesto del título se sintetizó a partir de 3-(benciloxi)-2-bromo-5-(5-metiloxazol-2-il)fenol (Ejemplo de referencia 27-2, Paso 1) y bromometil)ciclopropano de manera análoga del Ejemplo de referencia 27-2, Paso 2. MS (M+1) =
416.2. 1H RMN (400 MHz, CLOROFORMO-d) 57,23-7,52 (m, 7H), 6,83 (d, J = 1,01 Hz, 1H), 5,19 (s, 2H), 3,94 (d, J = 6,70 Hz, 2H), 2,36 (d, J = 1,01 Hz, 3H), 1,23 - 1,35 (m, 1H), 0,53-0,65 (m, 2H), 0,31-0,42 (m, 2H).
Paso 2: Ácido (2-(bendloxi)-6-(cidopropilmetoxi)-4-(5-metiloxazol-2-il)fenil)borónico
El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 6. MS (M+1) = 380.3. 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 7,30-7,50 (m, 8 H), 7,26 (s, 1H), 6,92 (d, J=1,0 Hz, 1H), 5,24 (s, 2H), 4,02 (d, J=7,1 Hz, 2H), 2,45 (d, J=1,0 Hz, 3H), 1,30 - 1,43 (m, 1H), 0,68-0,78 (m, 2H), 0,38-0,45 (m, 2H).
Paso 3: 6-(2-(Bendloxi)-6-(ddopropilmetoxi)-4-(5-metiloxazol-2-il)feniQ-N-metil-N-(2,2,6,6-tetrametilpipendin-4-il)piridazin-3-amina
El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 7. MS (M+1) = 582,5.
Paso 4: 3-(Cidopropilmetoxi)-2-(6-(metil(2,2,6,6-tetrametilpipendin-4-il)amino)pindazin-3-il)-5-(5-metioxazol-2-il)fenol
El compuesto del título se preparó de una manera análoga a la del Ejemplo de referencia 22-1, Paso 8. LCMS tR = 0,55 min [Método Q], MS (M-1) = 489,9. 1H RMN (400 MHz, METANOL-d4) 58,43 (d, J = 10,11 Hz, 1H), 7,25 (d, J = 10,11 Hz, 1H), 7,19 (d, J = 1,52 Hz, 1H), 7,14 (d, J = 1,52 Hz, 1H), 6,94 (d, J = 1,01 Hz, 1H), 5,13 (t, J = 12,13 Hz, 1H), 4,01 (d, J = 7,07 Hz, 2H), 3,02 (s, 3H), 2,45 (d, J = 1,01 Hz, 3H), 1,66 - 1,75 (m, 2H), 1,52-1,63 (m, 2H), 1,40 (s, 6H), 1,30-1,37 (m, 1H), 1,24 (s, 6H), 0,62-0,74 (m, 2H), 0,35-0,46 (m, 2H).
Ejemplo de referencia 28-1: Síntesis de 2-metil-5-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-il)-1H-benzo[d]imidazol-6-ol

Paso 1: 5-Bromo-6-metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol
A una mezcla de 5-bromo-6-metoxi-2-metil-1H-benzo[d]imidazol (400 mg, 1,659 mmol) en DMF (3 mL) se añadió NaH al 60 % p (80 mg, 1,991 mmol) a 0 °C. La mezcla se agitó desde 0 °C hasta la temperatura ambiente durante 0,5 hours y a continuación se añadió cloruro de 2-trimetilsililetioximetilo (SEMCl, 0,352 mL, 1,991 mmol) gota a gota. La mezcla de reacción se agitó a temperatura ambiente durante 2 horas, a continuación se desactivó con agua y se extrajo con DCM. La fase orgánica se secó con Na2SO4 y se concentró al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (10-100 % de EtOAc/Heptano y a continuación un 0-10 % de DCM/MeOH) para proporcionar el compuesto del título (310 mg, 50,3 % de rendimiento) como un aceite. MS (M+1) = 373,1.
Paso 2: 6-Metoxi-2-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol y ácido (6-metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol-5-il)borónico
Una mezcla de reacción desgasificada de 5-bromo-6-metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol (145 mg, 0,390 mmol), bis(pinacolato)diboro (218 mg, 0,859 mmol), Pd(dppf)Cl2 (31,9 mg, 0,039 mmol), dppf (21,7 mg, 0,039 mmol) y acetato de potasio (192 mg, 1,95 mmol) en dioxano (1,5 mL) se calentó a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de celite y se lavó con EtOAc. El filtrado se concentró hasta obtener un aceite marrón. El producto crudo se purificó mediante cromatografía en columna ultrarrápida (10-100 % de EtOAc/Heptano) para proporcionar una mezcla de 6-metoxi-2-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1-((2-(trimetilsilil)etoxi)metil)-1 H-benzo[d]imidazol y ácido (6 -metoxi-2 -metil-1 -((2 -(trimetilsilil)etoxi)metil)-1 H-benzo[d]imidazol-5-il)borónico (126,8 mg, 89,3 % de rendimiento total), que se utilizó en el siguiente paso sin una purificación adicional. MS (M+1) = 419,4 y 337,2, respectivamente.
Paso 3: 6-(6-Metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol-5-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
La mezcla del ácido (6-metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol-5-il)borónico y el éster pinacólico (78 mg, 0,139 mmol), y el Intermedio 1-1 (26 mg, 0,092 mmol) se hicieron reaccionar de acuerdo con el MÉTODO GENERAL 1-5 para el acoplamiento Suzuki. Se obtuvo 6-(6-metoxi-2-metil-1-((2-(trimetilsilil)etoxi)metil)-1H-benzo[d]imidazol-5-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina después de la purificación mediante cromatografía en columna ultrarrápida (44 mg, 89 % de rendimiento). MS (M+1) = 539,7.
Paso 4: 2-Metil-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-1H-benzo[d]imidazol-6-ol
A partir de 6-(6-metox¡-2-met¡l-1-((2-(tr¡met¡ls¡l¡l)etox¡)met¡l)-1H-benzo[d}¡m¡dazol-5-¡l)-N-meth¡l-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (44 mg, 0,082 mmol), s¡gu¡endo el METODO GENERAL 3.2 para la desprotecdón de metox¡ ut¡l¡zando BBr3 , se obtuvo 2-met¡l-5-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)am¡no)p¡r¡dazm-3-¡l)-1H-benzo[d]¡m¡dazol-6-ol como un sól¡do blanco después de la pur¡f¡cac¡ón med¡ante HPLC (16 mg, 50 % de rend¡m¡ento). LCMS tR = 0,40 m¡n [Método Q], MS (M+1) = 395,4. 1H RMN (METANOL-d4) 58,15 (d, J=10,1 Hz, 1H), 7,88 (s, 1H), 7,34 (d, J=9,6 Hz, 1H), 6,97 (s, 1H), 5,07 (t, J=11,9 Hz, 1H), 3,02 (s, 3H), 2,55 (s, 3H), 1,65-1,77 (m, 2H), 1,51-1,65 (m, 2H), 1,41 (s, 6H), 1,25 (s, 6H).
Ejemplo de referencia 29-1: Síntesis de 5-cloro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)ammo)piridazm-3-i)fenol

Se_ h¡c¡eron reacc¡onar el Intermedio 1-1 y 5-cloro-2-(4,4,5,5-tetramet¡l-1,3,2-d¡oxaborolan-2-¡l)fenol de acuerdo con el MÉTODO GENERAL 1-5 para el acoplam¡ento de Suzuk¡. Se obtuvo 5-doro-2-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)fenol como un sól¡do amar¡llo después de la pur¡f¡cac¡ón med¡ante HPLC. LCMS tR = 0,54 m¡n [Método Q], MS (M+1) = 375,2. 1H RMN (METANOL-d4) δ 8,16 (d, J=9,6 Hz, 1H), 7,74 (d, J=8 , 6 Hz, 1H), 7,45 (d, J=10,1 Hz, 1H), 6,91-7,05 (m, 2H), 5,28-5,44 (m, 1H), 3,06 (s, 3H), 1,88-2,05 (m, 4H), 1,65 (s, 6H), 1,52 (s, 6H).
Ejemplo de referencia 30-1: Síntesis de 5-(1H-pirazoM-N)-2-(6-((2,2,6,6-tetrametNpiperidin-4-N)ammo)piridazm-3-il)fenol

Paso 1: Se preparó 6-(2-metox¡-4-(1H-p¡razol-1-¡lfen¡l)-N-(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)p¡r¡dazm-3-amma (148 mg, 0,353 mmol, 48 % de rend¡m¡ento) s¡gu¡endo el MÉTODO GENERAL 1-4 para el acoplam¡ento de Suzuk¡ a part¡r de 1-(3-metox¡-4-(4,4,5,5-tetramet¡l-1,3,2-d¡oxaborolan-2-¡l)fen¡l-1H-p¡razol (Intermedio 2-1, Paso 3 , 447 mg, 1,49 mmol) y el Intermedio 1-2 (200 mg, 0,744 mmol). LCMS tR = 0,95 m¡n (cond¡c¡ón B de LCMS); MS (M+1) = 407,3. 1H RMN (400 MHz, CLOROFORMO-d) 57,97-8,06 (m, 2H), 7,73-7,82 (m, 2H), 7,52 (d, J=2,01 Hz, 1H), 7,24-7,31 (m, 1H), 6,64 (d, J=9,29 Hz, 1H), 6,47-6,54 (m, 1H), 4,49 (d, J=8,03 Hz, 1H), 4,27-4,42 (m, 1H), 3,95 (s, 3H), 2,12 (dd, J=3,76, 12,55 Hz, 2H), 1,33 (s, 6H), 1,17 (s, 6H), 1,03 (t, J=12,05 Hz, 2H).
Paso 2: 5-(1H-P¡razol-1-¡l)-2-(6-((2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)fenol
S¡gu¡endo el MÉTODO GENERAL 3-1 para la desprotecc¡ón de metox¡ ut¡l¡zando t¡ofenol, se obtuvo el compuesto del título como un polvo amar¡llo pál¡do ( 8 mg). LCm S tR = 0,48 m¡n [Método Q], MS (M+1) = 393,3. 1H RMN (400 MHz, METANOL-d4) 5 8,29 (d, J=2,51 Hz, 1H), 8,05 (d, J=9,79 Hz, 1H), 7,89 (d, J=8,53 Hz, 1H), 7,76 (d, J=1,51 Hz, 1H), 7,32-7,40 (m, 2H), 7,06 (d, J=9,79 Hz, 1H), 6,56 (t, J=2,13 Hz, 1H), 4,41-4,56 (m, 1H), 2,08 (dd, J=3,51, 12,80 Hz, 2H), 1,40 (s, 6H), 1,16-1,27 (m, 8H).
Ejemplo de referencia 30-2: Síntesis de 3-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)benzonitrilo

A un v¡al apto para m¡croondas se añad¡eron 3-metox¡-4-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)benzon¡tr¡lo (Ejemplo 5-1, Paso 1, 100 mg, 0,264 mmol) y clorh¡drato de p¡r¡d¡na (610 mg, 5,27 mmol), y la mezcla
se calentó a 150 °C durante 90 minutos en el microondas. La mezcla de reacción se disolvió en MeOH/DMSO y se purificó mediante HPLC preparativa (Waters Sunfire 30 mm ID x 50 mm, 0,1 % de TFA, 15-40 % de ACN/H2O) para proporcionar el compuesto del título como un producto minoritario (3 mg, 0,008 mmol). LCMS tR = 0,47 min (Método Q), MS (M+1) = 352,2. 1H RMN (400 MHz, CLOROFORMO-d) 57,81 (d, J=9,54 Hz, 1H), 7,65 (d, J=8,03 Hz, 1H), 7,34 (d, J=1,51 Hz, 1H), 7,18 (dd, J=1,76, 8,28 Hz, 1H), 6,87 (d, J=9,54 Hz, 1H), 4,83 (s a., 1H), 4,40 (d, J=7,53 Hz, 1H), 2,11 (dd, J=3,64, 12,67 Hz, 2H), 1,59 (td, J=7,72, 15,18 Hz, 1H), 1,33-1,43 (m, 7H), 1,25 (s a., 6H).
Ejemplo 31-1: Síntesis de 2-(6-((2,2-d¡met¡lp¡per¡dm-4-¡l)ox¡)p¡r¡dazm-3-¡l)-5-(1H-p¡razoM-¡l)fenol

Se añadió tert-butóxido de potasio (1,0 M en THF, 2,2 mL, 2,2 mmol) a 2,2-dimetilpiperidin-4-ol (0,22 g, 1, 66 mmol) en THF (2,2 mL) y DMF (0,6 mL) y la mezcla se agitó durante 10 minutos a 50 °C. Se añadió el Intermed¡o 2-2 (0,15 g, 0,55 mmol) a la reacción a 0 °C y la mezcla se agitó durante 4 h a TA. Se añadió una solución de bicarbonato de sodio y la fase acuosa se extrajo con cloroformo propanol 3:1 (2x). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El material crudo se purificó mediante HPLC preparativa (de un 10 a un 60 % de acetonitrilo en agua, 0,1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido tósico (5 eq, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). La evaporación a presión reducida proporcionó 2-(6-((2,2-dimetilpiperidin-4-il)oxi)piridazin-3-il)-5-(1H-pirazol-1-il)fenol como un sólido beige (0,11 g, 53 %). LCMS tR = 0,52 min [Método Q]; [M+H]: 366,2; 1H RMN (400 MHz, DMSO) 5 13,30 (s a, 1H), 8,60 (d, J= 2,5 Hz, 1H), 8,44 (d, J= 9,5 Hz, 1H), 8,07 (d, J= 8,5 Hz, 1H), 7,78 (d, J= 2,0 Hz, 1H), 7,50 (d, J= 2,0 Hz, 1H), 7,48 (dd, J= 8,5, 2,0 Hz, 1H), 7,40 (d, J= 9,5 Hz, 1H), 6,61-6,54 (m, 1H), 5,45 (td, J= 10,5, 5,0 Hz, 1H), 2,94-2,76 (m, 2H), 2,16-2,05 (m, 1H), 2,03-1,96 (m, 1H), 1,49-1,30 (m, 2H), 1,13 (s, 3H), 1,10 (s, 3H).
Ejemplo de referenc¡a 32-1: Síntes¡s de 2-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡dm-4-¡l)ammo)p¡r¡dazm-3-¡l)-4-(1H-p¡razol-4-¡l)fenol

Paso 1: 4-Metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 3-bromo-4-metoxifenol (1,0 g, 4,90 mmol) se obtiene 4-metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (1,23 mg) MS [M+H+] = 251,1.
Paso 2: 4-Metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
A un matraz apto para microondas se añadieron 4-metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (400 mg, 1,60 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermed¡o 1-1, 452 mg, 1,60 mmol), fosfato de potasio (1,4 g, 6,40 mmol), Pd2(dba)3 (146 mg, 0,16 mmol) y SPhos (65,7 mg, 0,16 mmol), y después se añadió 1,4-dioxano (4 mL)/H2O (0,8 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante 2 h. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 12 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (491 mg) MS [M+H+] = 371,2.
Paso 3: Trifluorometanosulfonato de 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo
A una solución de 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (490 mg, 1,32 mmol) en DCM ( 8 mL) se añadió trietilamina (0,461 mL, 3,31 mmol) a TA. La mezcla de reacción se enfrió hasta 0 °C, y después se añadió N-feniltrifluorometanosulfonimida (472 mg, 1,32 mmol). La mezcla de reacción se calentó hasta t A, se agitó durante 2 h y a continuación se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró para obtener el producto crudo, cuyo pH se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó sobre una columna de SCX. El producto crudo se lavó con
metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones del producto se recogieron y se secaron para proporcionar trifluorometanosulfonato de 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (665 mg) MS [M+H+] = 503,2.
Paso 4: 6-(2-Metoxi-5-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron trifluorometanosulfonato de 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (100 mg, 0,20 mmol), ácido 1H-pirazol-4-ilborónico (33,4 mg, 0,30 mmol), fosfato de potasio (127 mg, 0,60 mmol), Pd2(dba)3 (18,22 mg, 0,02 mmol) y SPhos (16,4 mg, 0,04 mmol), y después se añadió 1,4-dioxano (1,6 mL)/H2O (0,4 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante 1 hora. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 6-(2-metoxi-5-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina ( 6 8 mg) MS [M+H+] = 421,3.
Paso 5: 2-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-4-(1H-pirazol-4-il)fenol
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se obtuvo el compuesto del título. MS [M+H+] = 407,3, LCMS tR = 0,51 min [Método Q]; 1H RMN (400 MHz, DMSO-d6) 8 ppm 8,42 (d, J=9,79 Hz, 1H), 7,99-8,12 (m, 3H), 7,48 (d, J=7,78 Hz, 1H), 7,35 (d, J=9,79 Hz, 1H), 6,90 (d, J=8,28 Hz, 1H), 4,84-5,10 (m, 1H), 2,97 (s, 3 H), 1,49-1,58 (m, 2H), 1,37-1,49 (m, 2H), 1,23-1,28 (m, 7H), 1,09 (s, 6H).
Los siguientes compuestos se prepararon utilizando procedimientos similares al descrito en el Ejemplo de referencia 32-1, con desprotección posterior de metoxi como se resume en los MÉTODOS GENERALES 3-1 y 3-2 cuando sea a ro iado.


El siguiente compuesto se preparó utilizando un procedimiento similar al descrito en el Ejemplo de referencia 14-1, utilizando desprotección de metoxi como se resume en el MÉTODO GENERALE 3-2:

Ejemplo 34-1: Síntesis de 4-cloro-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-N)-5-(1H-pirazol-4-il)fenol

Paso 1: 4-(4-Bromo-2-cloro-5-metoxifenil)-1H-pirazol
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando ácido (1H-pirazol-4-il)borónico (161 mg, 1,44 mmol) y 1-bromo-5-cloro-4-yodo-2-metoxibenceno (500 mg, 1,44 mmol) se obtuvo 4-(4-bromo-2-doro-5-metoxifenil)-1H-pirazol (300 mg) MS [M+H+] = 286,8.
Paso 2: 4-(2-Cloro-5-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 4-(4-bromo-2-cloro-5-metoxifenil)-1H-pirazol (300 mg, 1,04 mmol) se obtuvo 4-(2-cloro-5-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol (178mg) MS [M+H+] = 335,2.
Paso 3: 6-(5-Cloro-2-metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando 4-(2-cloro-5-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol (118 mg, 0,35 mmol) y 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1,100 mg, 0,35 mmol) se obtuvo 6-(5-cloro-2-metoxi-4-(1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (138 mg) MS [M+H+] = 455,0.
Paso 4: 4-Cloro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-( 1H-pirazol-4-il)fenol Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 441,3, LCMS tR = 0,54 min [Método Q]; 1H RMN (400 MHz, DMSO-da) 5 ppm 13,72 (s a., 1H), 13,13 (s a., 1H), 8,29 (d, J=10,04 Hz, 1H), 8,04-8,26 (m, 2H), 8,01 (s, 1H), 7,35 (d, J=9,79 Hz, 1H), 7,20 (s, 1H), 4,81-5,23 (m, 1H), 2,95 (s, 3H), 1,36-1,63 (m, 4H), 0,96-1,32 (m, 12H).
Ejemplo de referencia 34-2: Síntesis de 4-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)ammo)piridazm-3-N)-5-(1 H-pirazol-4-il)fenol

Paso 1: 4-(2-Fluoro-4-hidroxi-5-metoxifenil)-1H-pirazol-1-carboxilato de tert-butilo
A un matraz de reacción se añadieron 4-bromo-5-fluoro-2-metoxifenol (500 mg, 2,26 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol-1-carboxilato de tert-butilo (998 mg, 3,39 mmol), fosfato de potasio (1,4 g, 6,79 mmol) y XPhos Paladaciclo (178 mg, 0,23 mmol), y después se añadió DMF (11 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 50 °C en el microondas durante 16 h. La mezcla de reacción se concentró al vacío y el producto crudo se purificó en gel de sílice para proporcionar 4-(2-fluoro-4-hidroxi-5-metoxifenil)-1H-pirazole-1-carboxilato de tert-butilo (700 mg) MS [M+H+]= 307,5.
Paso 2: 4-(2-Fluoro-5-metoxi-4-(((trifluorometil)sulfonil)oxi)fenil)-1H-pirazol-1-carboxilato de tert-butilo
A una solución de 4-(2-fluoro-4-hidroxi-5-metoxifenil)-1H-pirazol-1-carboxilato de tert-butilo (700 mg, 2,27 mmol) en DCM (11,4mL) se añadió trietilamina (01,27 mL, 9,08 mmol) a TA. La mezcla de reacción se enfrió hasta 0 °C, y después se añadió N-feniltrifluorometanosulfonimida (973 mg, 2,72 mmol). La reacción se calentó hasta TA y se agitó durante dos horas. La reacción se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró para obtener el producto crudo, que se purificó en gel de sílice para proporcionar 4-(2-fluoro-5-metoxi-4-(((trifluorometil)sulfonil)oxi)fenil)-1H-pirazol-1-carboxilato de tert-butilo (724mg) MS [M+H+-BOC] = 341,0.
Paso 3: 4-(2-Fluoro-5-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-1-carboxilato de tert-butilo
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 4-(2-fluoro-5-metoxi-4-(((trifluorometil)sulfonil)oxi)fenil)-1H-pirazol-1-carboxilato de tert-butilo (635 mg, 1,44 mmol) proporcionó el compuesto del título (170 mg). MS [M+H+] = 419,3.
Paso 4: 4-(2-Fluoro-5-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1H-pirazol-1-carboxilato de tert-butilo
A un matraz apto para microondas se añadieron 4-(2-fluoro-5-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol-1 -carboxilato de tert-butilo (98 mg, 0,23 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 66,3 mg, 0,23 mmol), fosfato de potasio (199 g, 0,94 mmol), Pd2(dba)3 (21,5 mg, 0,02 mmol) y SPhos (9,62 mg, 0,02 mmol), y después se añadió 1,4-dioxano (0,4 mL)/H2O (0,9 mL). El vial se purgó con N2 durante 5 minutos y a continuación, se calentó a 100 °C en el microondas durante una hora. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 4-(2-fluoro-5-metoxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1 H-pirazol-1-carboxilato de tert-butilo (126 mg) MS [M+H+] = 539,2.
Paso 5: 4-Fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1H-pirazol-4-il)fenol
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se obtuvo el compuesto del título. MS [M+H+] = 425,3, LCMS tR = 0,48 min [Método Q]; 1H RMN (400 MHz, DMSO-d6) 5 ppm 13,58 (s, 1H), 13,15 (s a., 1H), 7,90-8,39 (m, 3H), 7,83 (d, J=12,55 Hz, 1H), 7,24-7,41 (m, 2H), 4,80-5,17 (m, 1H), 2,95 (s, 3H), 1,38-1,56 (m, 4H), 1,25 (s, 6H), 1,09 (s, 6H).
Ejemplo de referencia 34-3: Síntesis de 5-fluoro-4-(1H-imidazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)fenol
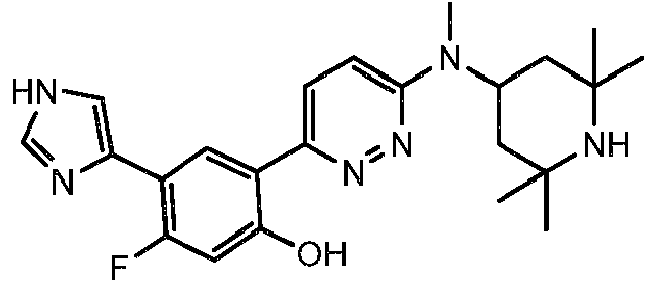
Paso 1: 6-(4-Fluoro-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron 2-(4-fluoro-2-metoxifenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (535 mg, 2,12 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 500 mg, 1,77 mmol), fosfato de potasio (1,5 g, 7,07 mmol), Pd2(dba)3 (162 mg, 0,18 mmol) y SPhos (72,6 mg, 0,18 mmol), y después se añadió 1,4-dioxano (3,7 mL)/H2O (0,7 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante una hora. La mezcla de reacción se concentró al vacío. Se ajustó el pH del material
crudo a 3 utilizando una solución acuosa de HCl 1 M, se cargó en una columna de SCX, se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 6-(4-fluoro-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (620 mg) MS [M+H+] = 373,3.
Paso 2: Ácido (2-fluoro-4-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)borónico
Siguiendo el MÉTODO GENERAL 7-1 para la borilación utilizando 6-(4-fluoro-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (100 mg, 0,23 mmol) se obtuvo ácido (2-fluoro-4-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)borónico (101 mg) MS [M+H+] = 417,3.
Paso 3: 6-(4-Fluoro-5-(1H-imidazol-4-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando (2-fluoro-4-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)borónico (50 mg, 0,12 mmol) y 4-bromo-1H-imidazol (35,3 mg, 0,24 mmol) se obtuvo 6-(4-fluoro-5-(1H-imidazol-4-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (52,7mg) MS [M+H+] = 439,3.
Paso 4: 5-Fluoro-4-(1H-imidazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 425,3, LCMS tR = 0,42 min [Método Q]; 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,18-8,27 (m, 2H), 8,12 (d, J=10,04 Hz, 1H), 7,49 (d, J=2,26 Hz, 1H), 7,32 (d, J=10,04 Hz, 1H), 6,73 (d, J=12,55 Hz, 1H), 5,23-5,43 (m, 1H), 2,95 (s, 3H), 1,84-1,95 (m, 4H), 1,56 (s, 6H), 1,42 (s, 6H).
Los siguientes compuestos finales se prepararon utilizando procedimientos similares al descrito en el Ejemplo de referencia 34-3:

Ejemplo 35-1: Síntesis de 6-hidroxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona

Paso 1: 6-Metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-H)-2,3-dihidro-1 H-inden-1-ona
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 5-bromo-6-metoxi-2,3-dihidro-1H-inden-1-ona (1,0 mg, 4,15 mmol) se obtuvo 6-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2,3-dihidro-1H-inden-1-ona (1,16 g) MS [M+H+] = 289,2.
Paso 2: 6-Metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona:
Intermedio 7-1
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando 6-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2,3-dihidro-1H-inden-1-ona (300 mg, 1,06 mmol) y 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 611 mg, 2,12 mmol) se obtuvo 6-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro- 1 H-inden-1 -ona (433 mg) MS [M+H+] = 409,7.
Paso 3: 6-Hidroxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 395,2, LCMS tR = 0,47 min [Método Q]; 1H RMN (400 MHz, DMSO-da) 5 ppm 13,50 (s, 1H), 8,29 (d, J=10,04 Hz, 1H), 8,10 (s, 1H), 7,37 (d, J=10,04 Hz, 1H), 7,08 (s, 1H), 4,86-5,27 (m, 1H), 3,02-3,11 (m, 2H), 2,97 (s, 3H), 2,62-2,72 (m, 2H), 1,37-1,59 (m, 4H), 1,25 (s, 6H), 1,09 (s, 6H).
Ejemplo de referencia 35-2: Síntesis de 6-(6-(metil(2,2,6,6-tetrametilpiperidm-4-N)amino)piridazm-3-N)-1,4-dihidroindeno[1,2-c]pirazol-7-ol

Paso 1: 6-(7-Metoxi-1,4-dihidroindeno[1,2-c]pirazol-6-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina A un matraz de fondo redondo de 100 ml que contenía tolueno (1,0 mL) se añadió 6-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona (Intermedio 7-1, 330 mg, 0,81 mmol), formiato de etilo (0,13 mL, 1,62 mmol), e hidruro de sodio (97 mg, 2,42 mmol). La mezcla de reacción se agitó a TA durante 16 h y a continuación se concentró al vacío. Se añadieron etanol (5,0 mL), ácido acético (0,51 mL, 8,89 mmol) e hidrazina hidratada (0,53 mL, 10,50 mmol). La mezcla se calentó a reflujo a 80 °C durante 3 h y se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 6-(7-metoxi-1,4-dihidroindeno[1,2-c]pirazol-6-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (75 mg) MS [M+H+] = 433,5.
Paso 2: 6-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-1,4-dihidroindeno[1,2-c]pirazol-7-ol Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se obtuvo el compuesto del título. MS [M+H+] = 419,2, LCMS tR = 0,47 min [Método Q]; 1H RMN (400 MHz, DMSO-de) 5 ppm 13,86 (s, 1H), 12,82 (s a., 1H), 8,24 (d, J=10,04 Hz, 1H), 8,02 (s, 1H), 7,66 (s, 1H), 7,37 (d, J=10,04 Hz, 1H), 7,13 (s, 1H), 4,78-5,12 (m, 1H), 3,60 (s, 2H), 2,96 (s, 3H), 1,36-1,64 (m, 4H), 1,18-1,35 (m, 7H), 1,09 (s a., 6H).
Ejemplo de referencia 35-3: Síntesis de la sal clorhidrato de la oxima de 6-hidroxi-5-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-N)-2,3-dihidro-1H-inden-1-ona

A un vial apto para microondas se añadieron 6-hidroxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona (Ejemplo de referencia 35-1,150 mg, 0,38 mmol), clorhidrato de hidroxilamina (264 mg, 3,80 mmol), piridina (0,25 mL, 3,04 mmol) y MeOH (1,0 mL). La mezcla resultante se agitó a TA durante 1,5 horas. La mezcla de reacción se acidificó con un exceso de ácido acético y se cargó sobre una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar el compuesto del título. MS [M+H+] = 410,2, LCMS tR = 0,48 min [Método Q]; 1H RMN (400 MHz, DMSO-d6) óppm 10,94-11,13 (m, 1H), 8,91-9,21 (m, 1H), 8,29 (d, J=10,04 Hz, 1H), 8,09-8,21 (m, 1H), 7,83 (s, 1H), 7,52-7,73 (m, 1H), 7,08 (s, 1H), 4,83-5,31 (m, 1H), 2,91-3,04 (m, 5H), 2,74-2,85 (m, 2H), 1,91 2,09 (m, 2H), 1,79 (d, J=10,79 Hz, 2H), 1,53 (s, 6H), 1,47 (s, 6H).
Ejemplo de referencia 35-4: Síntesis de 5-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)-2,3-dihidro-1H-indeno-1,6-diol

A un vial apto para microondas se añadieron 6-hidroxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1H-inden-1-ona (Ejemplo de referencia 35-1,50 mg, 0,13 mmol), borohidruro de sodio (9,59 mg, 0,25 mmol) y MeOH (1,5 mL). La suspensión resultante se agitó a TA durante dos horas. La mezcla de reacción se acidificó con un exceso de ácido acético y se cargó sobre una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar el compuesto del título. MS [M+H+] = 397,3, LCMS tR = 0,46 min [Método Q de LCMS]; 1H RMN (400 MHz, METANOL-d4 ) ó ppm 8,11 (d, J=9,54 Hz, 1H), 7,64 (s, 1H), 7,31 (d, J=9,54 Hz, 1H), 6,99 (s, 1H), 5,16 (t, J=6,53 Hz, 1H), 5,01-5,13 (m, 1H), 2,95-3,08 (m, 4H), 2,73-2,86 (m, 1H), 2,41-2,54 (m, 1H), 1,91 (s, 1H), 1,53-1,78 (m, 4H), 1,41 (s, 6H), 1,26 (s, 6H).
Ejemplo de referencia 35-5: Síntesis de la sal clorhidrato de 2-amino-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazm-3-il)-8H-mdeno[1,2-d]tiazol-5-ol

Paso 1: 5-Metoxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-8H-indeno[1,2-d]tiazol-2-amina
A un vial apto para microondas que contenía etanol (1,2 mL) se añadieron 6-metoxi-5-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-2,3-dihidro-1 H-inden-1 -ona (Intermedio 7-1, 100 mg, 0,25 mmol), tiourea (55,9 mL, 0,73 mmol) y yoduro (124 mg, 0,49 mmol). La mezcla de reacción se agitó a 100 °C durante 3 horas. La mezcla se diluyó con MeOH y se cargó sobre una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 5-metoxi-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-8H-indeno[1,2-d]tiazol-2-amina (114 mg) MS [M+H+] = 465,0.
Paso 2: 2-Amino-6-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-8H-indeno[1,2-d]tiazol-5-ol
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 451,2, LCMS tR = 0,46 min [Método Q]; 1H RMN (400 MHz, DMSO-d6) ó ppm 9,25-9,36 (m, 1H), 8,36 8,44 (m, 1H), 8,33 (d, J=9,85 Hz, 1H), 7,89 (s, 1H), 7,75-7,85 (m, 1H), 7,21 (s, 1H), 4,76-5,12 (m, 1H), 3,76 (s, 2H), 3,04 (s, 3H), 2,08 (t, J=12,88 Hz, 2H), 1,72-1,88 (m, 2H), 1,44-1,60 (m, 12H).
Ejemplo de referencia 35-6: Síntesis de la sal clorhidrato de 9-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5,6-dihidroimidazo[5,1-a]isoquinolin-8-ol

Paso 1: 4-Hidroxi-3-metoxifenetilcarbamato de tert-butilo
A un matraz de fondo redondo de 250 mL que contenía DCM ( 8 6 mL) se añadieron clorhidrato de 4-(2-aminoetil)-2-metoxifenol (3,5 g, 17,19 mmol), TEA (7,2 mL, 51,6 mmol) y Boc-anhídrido (3,94 mg, 18,04 mmol). La mezcla resultante se agitó a TA durante 18 horas, a continuación se diluyó con DCM, se lavó con H2O, HCl acuoso 1 N y salmuera, a continuación la capa orgánica se secó con sulfato de sodio y se concentró al vacío para proporcionar 4-hidroxi-3-metoxifenetilcarbamato de tert-butilo (4,59 g). 1H RMN (400 MHz, DMSO-d6) 5 ppm 8,71 (s, 1H), 6,85 (t, J=5,52 Hz, 1H), 6,72 (d, J=1,76 Hz, 1H), 6,63-6,68 (m, 1H), 6,55 (dd, J=8,03, 1,76 Hz, 1H), 3,74 (s, 3H), 3,01-3,13 (m, 2H), 2,53 2,62 (m, 2H), 1,37 (s, 9H).
Paso 2: 4-Isopropoxi-3-metoxifenetilcarbamato de tert-butilo
Un matraz de fondo redondo de 250 mL que contenía acetonitrilo (18,7 mL) se añadieron 4-hidroxi-3-metoxifenetilcarbamato de tert-butilo (1,0 g, 3,74 mmol), 2-bromopropano (0,51 g, 4,11 mmol) y carbonato de potasio (1,5 g, 11,22 mmol). La suspensión resultante se agitó a 65 °C durante 18 horas. Se realizó una segunda adición de 2- bromopropano (0,51 g, 4,11 mmol) y se siguió calentando durante otras 18 horas. La mezcla de reacción se concentró la reacción al vacío. El aceite resultante se disolvió en EtOAc y se lavó con H2O, bicarbonato de sodio acuoso saturado y salmuera, a continuación se secó con sulfato de sodio y se concentró al vacío. El producto crudo se purificó mediante cromatografía en gel de sílice (0-100 % de EtOAc en Heptano) para proporcionar 4-isopropoxi-3- metoxifenetilcarbamato de tert-butilo (0,81 g). 1H Rm N (400 MHz, CLOROF0RM0-d) 5 ppm
6,84 (d, J=8,03 Hz, 1H), 6,67-6,75 (m, 2H), 4,37-4,70 (m, 2H), 3,86 (s, 3H), 3,27-3,46 (m, 2H), 2,68-2,81 (m, 2H), 1,45 (s, 9H), 1,37 (d, J=6,02 Hz, 6H).
Paso 3: Clorhidrato de 2-(4-isopropoxi-3-metoxifenil)etanamina
En un matraz de fondo redondo de 50 mL se combinaron 4-isopropoxi-3-metoxifenetilcarbamato de tert-butilo (810 mg, 2,62 mmol) y HCl (4 M en 1,4-dioxano) (6,5 mL, 26,2 mmol). La suspensión se agitó a TA durante dos horas y a continuación se concentró al vacío para proporcionar clorhidrato de 2-(4-isopropoxi-3-metoxifenil)etanamina (643 mg) MS [M+H+] = 210,3.
Paso 4: 2-Formamido-N-(4-isopropoxi-3-metoxifenetil)acetamida
A clorhidrato de 2-(4-isopropoxi-3-metoxifenil)etanamina (487 mg, 2,33 mmol) se añadió MeOH y se cargó sobre una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 2-(4-isopropoxi-3-metoxifenil)etanamine como base libre. El material se disolvió en THF (23,3 mL) Ih se añadieron ácido 2-formamidoacético (360 mg, 3,49 mmol), DCC (528 mg, 2,56 mmol), HOBt (392 mg, 2,56 mmol) y NMM (1,02 mL, 9,31 mmol). La suspensión resultante se agitó a TA durante 3 horas, se diluyó con éter y se filtró a través de celite. El filtrado se purificó mediante cromatografía en gel de sílice (25 %-50 % de AcOH en DCM) para proporcionar 2-formamido-N-(4-isopropoxi-3-metoxifenetil)acetamida (650 mg) MS [M+H+] = 295,3.
Paso 5: 9-Isopropoxi-8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolina
A un matraz de fondo redondo de 50 mL se añadieron 2-formamido-N-(4-isopropoxi-3-metoxifenetil)acetamida (600 mg, 2,04 mmol) y después acetonitrilo (10,2 mL) y POCl3 (0,57 mL, 6,12 mmol). La mezcla de reacción se calentó hasta 80 °C durante una hora y a continuación se concentró al vacío. Al aceite resultante se añadieron H2O y carbonato de sodio acuoso saturado, y a continuación la solución se extrajo con EtOAc (2x). Los extractos orgánicos se combinaron, se secaron con sulfato de sodio y se concentraron al vacío. El producto crudo se purificó mediante cromatografía en gel de sílice (0-10 % de MeOH en DCM) para proporcionar 9-isopropoxi-8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolina ( 1 82 mg) MS [M+H+]= 259,2.
Paso 6: 8-Metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ol
A un matraz de fondo redondo de 50 mL se añadieron 9-isopropoxi-8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolina (180 mg, 0,70 mmol), cloroformo (13 mL) y ácido metanosulfónico (1,3 mL, 20,02 mmol). La mezcla resultante se calentó a 63 °C durante 2 h, a continuación se enfrió hasta TA y se concentró al vacío. Al aceite resultante se añadieron
H2O y carbonato de sodio saturado acuoso. La solución se extrajo con EtOAc (2x), y los extractos orgánicos se combinaron, se secaron con sulfato de sodio y se concentraron al vacío para proporcionar 8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ol (151 mg) MS [M+H+] = 217,4.
Paso 7: Trifluorometanosulfonato de 8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ilo
A una solución de 8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ol (196 mg, 2,27 mmol) en DCM (5,5 mL) se añadió trietilamina (0,38 mL, 2,72 mmol) a TA. La mezcla de reacción se enfrió hasta 0 °C, y después se añadió N-feniltrifluorometanosulfonimida (356 mg, 0,98 mmol). La mezcla de reacción se calentó hasta TA y se agitó durante 2 h. La reacción se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró para obtener el producto crudo, que se purificó en gel de sílice para proporcionar trifluorometanosulfonato de 8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ilo (316 mg) MS [M+H+] = 348,9.
Paso 8. 8-Metoxi-9-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidroimidazo[5,1-a]isoquinolina
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando trifluorometanosulfonato de 8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-ilo (316 mg, 1,24 mmol) se obtuvo 8-metoxi-9-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidroimidazo[5,1-a]isoquinolina (271 mg) MS [M+H+] = 327,4.
Paso 9: 6-(8-Metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando 8-metoxi-9-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5,6-dihidroimidazo[5,1-a]isoquinolina (104 mg, 0,32 mmol) y 6 -cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amins (Intermedio 1-1, 90 mg, 0,32 mmol), se obtuvo 6-(8-metoxi-5,6-dihidroimidazo[5,1-a]isoquinolin-9-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (140 mg) MS [M+H+] = 447,6.
Paso 10: 9-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5,6-dihidroimidazo[5,1-a]isoquinolin-8-ol Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se obtuvo el compuesto del título. MS [M+H+] = 433,3, LCMS tR = 0,40 min [Método Q]; 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,97 (d, J=1,26 Hz, 1H), 8,46 (d, J=10,04 Hz, 1H), 8,21 (s, 1H), 7,98 (d, J=1,25 Hz, 1H), 7,85 (d, J=10,04 Hz, 1H), 7,10 (s, 1H), 5,08-5,35 (m, 1H), 4,50 (t, J=6,65 Hz, 2H), 3,26-3,31 (m, 2H), 3,17 (s, 3H), 1,99-2,11 (m, 4H), 1,67 (s, 6H), 1,57 (s, 6H).
Ejemplo de referencia 36-1: Síntesis de 4-hidroxi-3-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)-N-((1-metil-1H-pirazol-4-il)metil)benzamida

Paso 1: 3-Bromo-4-metoxi-N-((1-metil-1H-pirazol-4-il)metil)benzamida
Un matraz de fondo redondo de 100 mL que contenía ácido 3-bromo-4-metoxibenzoico (500 mg, 2,16 mmol) y DCM (5 mL) se añadieron cloruro de oxalilo (0,23 mL, 2,60 mmol) y DMF (0,1 mL, 0,13 mmol). La mezcla de reacción se agitó a TA durante una hora y se concentró al vacío. Al aceite incoloro resultante se añadió DCM (2,5 mL) y se añadió a una mezcla de (1-metil-1H-pirazol-4-il)metanamina (241 mg, 2,16 mmol), TEA (0,60 mL, 4,33 mmol) y DCM (2,5 mL) a 0 °C. La mezcla de reacción se agitó a TA durante 0,5 horas y a continuación se concentró al vacío. El material crudo se purificó mediante cromatografía en gel de sílice (0-10 % de MeOH en DCM) para proporcionar 3-bromo-4-metoxi-N-((1-metil-1H-pirazol-4-il)metil)benzamida (702 mg) MS [M+H+] = 325,9.
Paso 2: Ácido (2-metoxi-5-(((1-metil-1H-pirazol-4-il)metil)carbamoil)fenil)borónico
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 3-bromo-4-metoxi-N-((1-metil-1H-pirazol-4-il)metil)benzamida (552 mg, 1,70 mmol) se obtuvo (2-metoxi-5-(((1-metil-1H-pirazol-4-il)metil)carbamoil)fenil)borónico (492 mg) MS [M+H+] = 290,1.
Paso 3: 4-Metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-N-((1-metil-1H-pirazol-4-il)metil)benzamida
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando ácido (2-metoxi-5-(((1-metil-1H-pirazol-4-il)metil)carbamoil)fenil)borónico (613 mg, 2,12 mmol) y 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 1-1, 300 mg, 1,06 mmol) se obtuvo 4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-N-((1-metil-1H-pirazol-4-il)metil)benzamida (522 mg) MS [M+H+] = 492,6.
Paso 4: 4-Hidroxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-N-((1-metil-1H-pirazol-4-il)metil)benzamida
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 478,3, LCMS tR = 0,46 min [Método Q]; 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,31 (d, J=2,26 Hz, 1H), 8,18 (d, J=10,04 Hz, 1H), 7,78 (dd, J=8 ,6 6 , 2,13 Hz, 1H), 7,61 (s, 1H), 7,50 (s, 1H), 7,36 (d, J=10,04 Hz, 1H), 7,01 (d, J=8,53 Hz, 1H), 5,06-5,27 (m, 1H), 4,45 (s, 2H), 3,87 (s, 3H), 3,03 (s, 3H), 1,68-1,77 (m, 2H), 1,54-1,67 (m, 2H), 1,42 (s, 6H), 1,27 (s, 6H).
Ejemplo de referencia 37-1: Síntesis de 4-(4-(hidroximetil)-1H-pirazoM-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)fenol
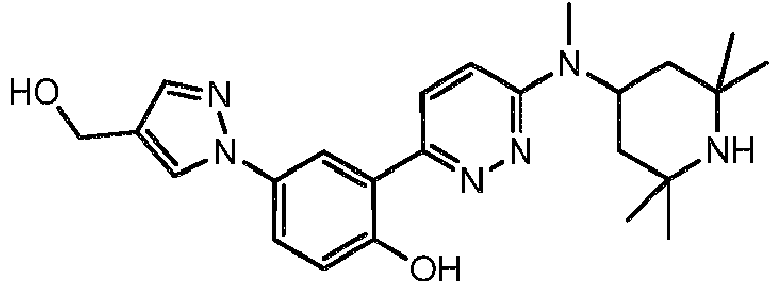
Paso 1: (1-(3-Bromo-4-metoxifenil)-1H-pirazol-3-il)metanol
A un matraz de fondo redondo de 100 mL se añadieron yoduro de cobre (I) (30,4 mg, 0,16 mmol), 2-(2-piridil)bencimidazol (31,2 mg, 0,16 mmol), carbonato de cesio (625 mg, 1,92 mmol) y DMF (5,3 mL). La mezcla de reacción se calentó hasta 60 °C durante 1 h, a continuación se añadieron (1H-pirazol-3-il)metanol (235 mg, 2,40 mmol) y 2-bromo-4-yodo-1-metoxibenceno (500 mg, 1,60 mmol) y la mezcla se calentó a 100 °C durante 18 horas. La reacción se enfrió se diluyó con EtOAc y se filtró a través de celite. El filtrado se concentró al vacío y se purificó mediante cromatografía en gel de sílice (0-100 % de EtOAc en Hepano) para proporcionar (1-(3-bromo-4-metoxifenil)-1 H-pirazol-3-il)metanol (353 mg) MS [M+2H+] = 285,0.
Paso 2: (1-(4-Metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil))-1H-pirazol-3-il)metanol
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando (1-(3-bromo-4-metoxifenil)-1H-pirazol-3-yl)metanol (353 mg, 1,25 mmol) se obtuvo (1-(4-metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1 H-pirazol-3-il)metanol (412 mg) MS [M+H+] = 331,2.
Paso 3: (1-(4-Metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1H-pirazol-4-il)metanol
A un vial apto para microondas se añadieron (1-(4-metoxi-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-1H-pirazol-3-il)metanol (254 mg, 0,77 mmol), 6-cloro-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina ( Intermedio 1-1, 145 mg, 0,51 mmol), fosfato de potasio (435 mg, 2,05 mmol), Pd2(dba)3 (46,9 mg, 0,05 mmol) y SPhos (21,1 mg, 0,05 mmol), y después se añadió 1,4-dioxano (1,3 mL)/H2O (0,3 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante 2 h. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 12 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar (1-(4-metoxi-3-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1H-pirazol-4-il)metanol (231 mg, MS [M+H+] = 451,3.
Paso 4: 2-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-4-(1H-pirazol-4-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 estándar para la desprotección de metoxi, se preparó el compuesto del título. MS [M+H+] = 437,3, LCMS tR = 0,50 min [Método Q]; 1H RMN (400 MHz, CLOROFORMO-d) 5 ppm 8,43 (d, J=0,50 Hz, 1H), 8,38 (d, J=10,04 Hz, 1H), 8,19 (d, J=2,76 Hz, 1H), 7,69 (dd, J=8,78, 2,51 Hz, 1H), 7,63 (s, 1H), 7,36 (d, J=10,04 Hz, 1H), 7,04 (d, J=8,78 Hz, 1H), 4,87-5,13 (m, 2H), 4,45 (d, J=5,27 Hz, 2H), 2,97 (s, 3H), 1,36-1,67 (m, 4H), 1,25 (s, 7H), 1,09 (s, 6H).
Ejemplo de referencia 38-1: Síntesis de 5-(1H-pirazol-4-N)-2-(6-((2,2,6,6-tetrametilpiperidin-4-N)metil)piridazin-3-il)fenol

Paso 1: 3-Cloro-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina
A un matraz de 50 mL se añadieron 2,2,2-trifluoroacetato de 2,2,6,6-tetrametil-4-metilenopiperidina (1,1 g, 4,12 mmol) y 9-BBN (0,5 M en THF) (16,5 mL, 8,23 mmol) y la mezcla de reacción se calentó a 65 °C durante 1 h. La reacción se enfrió hasta TA y se añadieron y calentaron a 60 °C durante toda la noche 3,5-dicloropiridazina (0,61 g, 4,12 mmol), K2CO3 (1,7 g, 12,35 mmol), y PdCl2(dppf).CH2Cl2 (0,17 g, 0,21 mmol) en 1,4-dioxano (8,5 mL)/H2O (1,7 mL). La mezcla de reacción se enfrió hasta TA, se diluyó con EtOAc, se filtró a través de celite y se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 12 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 3-cloro-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina (1,1 g) MS [M+H+] = 268,2.
Paso 2: 3-Metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 1-4 estándar para el acoplamiento de Suzuki utilizando tert-butil(3-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenoxi)dimetilsilano (408 mg, 1,12 mmol) y 3-cloro-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina (200 mg, 0,75 mmol) se obtiene 3-metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenol (165 mg) MS [M+H+] = 356,1.
Paso 3: Trifluorometanosulfonato de 3-metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenilo
A una solución de 3-metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenol (165 mg, 0,46 mmol) en DCM (2,8 mL) se añadió TEA (0,162 mL, 1,16 mmol) a TA. La mezcla de reacción se enfrió hasta 0 °C, y después se añadió N-feniltrifluorometanosulfonimida (174 mg, 0,49 mmol). La mezcla de reacción se calentó hasta TA y se agitó durante 2 h. La reacción se desactivó con una solución acuosa de bicarbonato de sodio y se extrajo con DCM. La capa orgánica se secó con sulfato de sodio, se filtró y se concentró al vacío. Se ajustó el pH del producto crudo a 3 utilizando una solución acuosa de HCl 1 M, se cargó en una columna de SCX, a continuación se lavó con metanol y se eluyó con amoniaco 2 N en metanol. Las fracciones del producto se recogieron y se secaron para proporcionar trifluorometanosulfonato de 3-metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenilo (160 mg) MS [M+H+] = 488,0.
Paso 4: 3-(2-Metoxi-4-(1H-pirazol-4-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina
A un vial apto para microondas se añadieron trifluorometanosulfonato de 3-metoxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenilo (160 mg, 0,33 mmol), ácido 1 H-pirazol-4-ilborónico (73,4 mg, 0,66 mmol), fosfato de potasio (209 mg, 0,99 mmol), Pd2(dba)3 (30,1 mg, 0,03 mmol) y SPhos (26,9 mg, 0,06 mmol), y después se añadió 1,4-dioxano (2,6 mL)/H2O (0,7 mL). El vial se purgó con N2 durante 5 minutos y la mezcla de reacción se calentó a 100 °C en el microondas durante 1 h. La mezcla de reacción se concentró al vacío. El pH del material crudo se ajustó a 3 utilizando una solución acuosa de HCl 1 M y se cargó en una columna de SCX. El material crudo se lavó con metanol y a continuación se eluyó con amoniaco 2 N en metanol. Las fracciones que contenían el producto se concentraron para proporcionar 3-(2-metoxi-4-(1 H-pirazol-4-il)fenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)metil)piridazina (133 mg) MS [M+H+] = 406,2.
Paso 5: 5-( 1 H-pirazol-4-il)-2-(6-((2, 2,6,6-tetrametilpiperidin-4-il)metil)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 3-2 estándar para la desprotección de metoxi, se obtuvo el compuesto del título. MS [M+H+] = 392,4, LCMS tR = 0,45 min [Método Q]; 1H RMN (400 MHz, DMSO-d6) 6 ppm 13,65 (s a., 1H), 13,05 (s a., 1H), 8,47 (d, J=9,03 Hz, 1H), 8,18 (s a., 2H), 8,01 (d, J=8,28 Hz, 1H), 7,84 (d, J=9,03 Hz, 1H), 7,24-7,33 (m, 2H), 2,83 (d, J=7,03 Hz, 2H), 2,20-2,37 (m, 1 H), 1,44 (dd, J=12,55, 2,76 Hz, 2H), 1,08 (s, 6H), 0,99 (s, 6H), 0,86 (t, J=12,42 Hz, 2H).
Ejemplo de referencia 39-1: Síntesis de 6-(3-(bencNoxi)isoquinoNn-6-N)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-N)piridazm-3-amina

Paso 1: 3-(Benciloxi)-6-bromoisoquinolina
A un matraz de fondo redondo de 25 mL que contenía 6-bromoisoquinolin-3-ol (500 mg, 2,23 mmol) se añadieron TEA (0,467 mL, 3,35 mmol) y bromuro de bencilo (0,319 mL, 2,68 mmol) en DMF (10 mL) para obtener una solución marrón. La reacción se calentó hasta 80 °C durante toda la noche. Después de enfriar hasta TA, la mezcla de reacción se concentró al vacío y se añadieron DCM y agua. A continuación, el agua se extrajo con DCM (2x), y las fracciones orgánicas combinadas se lavaron con salmuera, se secaron con MgSO4 , se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (10-30-50 % de EtOAc/Heptanos) para proporcionar el compuesto del título (132 mg, 0,420 mmol, 19 % de rendimiento). M+1 = 316,0. 1H RMN (400 Mh z , CLOROFORMO-d) 58,92 (s, 1H), 7,85 (d, J=1,25 Hz, 1H), 7,74 (d, J=8,78 Hz, 1H), 7,31-7,52 (m, 6H), 6,99 (s, 1H), 5,48 (s, 2H).
Paso 2: 3-(Benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)isoquinolina
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 3-(benciloxi)-6-bromoisoquinolina (125 mg, 0,398 mmol) se preparó el compuesto del título (115 mg, 0,398 mmol, 80 % de rendimiento). M+1 = 362,3. 1H RMN (400 MHz, CLOROFORMO-d) 5 8,98 (s, 1H), 8,20 (s, 1H), 7,87 (d, J=8,28 Hz, 1H), 7,72 (dd, J=8,28, 1,00 Hz, 1H), 7,50-7,54 (m, 2H), 7,36-7,42 (m, 2H), 7,30-7,35 (m, 1H), 7,12 (s, 1H), 5,48 (s, 2H), 1,40 (s, 12H).
Paso 3: 6-(3-(Bendbxi)isoquinolin-6-il)-N-metil-N-(2,2,6,6-tetmmetilpiperidin-4-il)piridazin-3-amina
El compuesto del título (38 mg, 0,077 mmol, 49 % de rendimiento) se preparó siguiendo el MÉTODO GENERAL 1-4 para el acoplamiento de Suzuki a partir de 3-(benciloxi)-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)isoquinolina (115 mg, 0,318 mmol) y el Intermedio 1-1 (45 mg, 0,159 mmol). LCMS tR = 0,61 min [Método Q]; MS (M+1) = 482,4. 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,95 (s, 1H), 8,21 (s, 1H), 8,01-8,09 (m, 2H), 7,96 (d, J=9,79 Hz, 1H), 7,49-7,54 (m, 2H), 7,36-7,42 (m, 2H), 7,30-7,35 (m, 1H), 7,23 (s, 1H), 7,16 (d, J=9,79 Hz, 1H), 5,40 (s, 2H), 5,28 (t, J=11,67 Hz, 1H), 3,00 (s, 3H), 1,67-1,73 (m, 2H), 1,56-1,65 (m, 2H), 1,41 (s, 6H), 1,26 (s, 6H).
Ejemplo de referencia 39-2: Síntesis de 6-(1-(bencMoxi)isoqumolm-7-il)-N-metil-N-(2,2,6,6-tetrametilpiperidm-4-il)piridazin-3-amina

Paso 1: 1-(Bendloxi)-7-bromoisoquinolina
A un matraz de fondo redondo de 25 mL que contenía 7-bromoisoquinolin-1-ol (500 mg, 2,23 mmol) se añadieron TEA (0,467 mL, 3,35 mmol) y bromuro de bencilo (0,319 mL, 2,68 mmol) en DMF (10 mL) para obtener una solución marrón. La reacción se calentó hasta 80 °C durante toda la noche. Después de enfriar hasta TA, la mezcla de reacción se concentró al vacío y se añadieron DCM y agua. La capa acuosa se extrajo con DCM (2x), y las fracciones orgánicas combinadas se lavaron con salmuera, se secaron con MgSO4 , se filtraron y se concentraron al vacío. El residuo se purificó mediante cromatografía en columna ultrarrápida (10-30-50 % de EtOAc/Heptanos) para proporcionar el compuesto del título (640 mg, 2,037 mmol, 91 % de rendimiento). M+1 = 316,0. 1H RMN (400 MHz, CLOROFORMO-d) 58,62 (d, J=2,01 Hz, 1H), 7,73 (dd, J=2,13, 8,41 Hz, 1H), 7,39 (d, J=8,28 Hz, 1H), 7,28-7,36 (m, 5H), 7,11 (d, J=7,53 Hz, 1H), 6,46 (d, J=7,28 Hz, 1H), 5,23 (s, 2H).
Paso 2: 1-(Benciloxi)-7-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)isoquinolina
Siguiendo el MÉTODO GENERAL 2-1 para la formación del éster de boronato utilizando 1-(benciloxi)-7-bromoisoquinolina (250 mg, 0,796 mmol) se obtuvo el compuesto del título (207 mg, 0,573 mmol, 72 % de
rendimiento). M+1 = 362,2. 1H RMN (400 MHz, CLOROFORMO-d) 6 8,96 (s, 1H), 8,00 (dd, J=7,91, 1,13 Hz, 1H), 7,47 (d, J=7,78 Hz, 1H), 7,27-7,34 (m, 5H), 7,11 (d, J=7,53 Hz, 1H), 6,46 (d, J=7,28 Hz, 1H), 5,23 (s, 2H), 1,36 (s, 12H). Paso 3: 6-( 1-(Benciloxi)isoquinolin-7-il)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
El compuesto del título (120 mg, 0,249 mmol, 94 % de rendimiento) se preparó siguiendo el MÉTODO GENERAL 1-4 para el acoplamiento de Suzuki a partir de 1-(benciloxi)-7-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)isoquinolina (144 mg, 0,398 mmol) e (Intermedio 1-1, 75 mg, 0,265 mmol). LCMS tR = 0,57 min [Método Q]; MS (M+1) = 482,4. 1H RMN (400 MHz, METANOL-d4) 6 ppm 8,81 (d, J=2,01 Hz, 1H), 8,39 (dd, J=8,41, 1, 88 Hz, 1H), 7,99 (d, J=9,79 Hz, 1H), 7,76 (d, J=8,53 Hz, 1H), 7,72-7,74 (m, 1H), 7,46 (d, J=7,28 Hz, 1H), 7,26-7,37 (m, 5H), 7,24 (d, J=9,79 Hz, 1H), 6,76 (d, J=7,53 Hz, 1H), 5,29 (s, 2H), 3,01 (s, 3H), 1,71-1,78 (m, 2H), 1,61-1,70 (m, 2H), 1,44 (s, 6H), 1,29 (s, 6H).
Ejemplo de referencia 40-1: Síntesis de la sal clorhidrato de 3-fluoro-5-(2-metoxipiridm-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
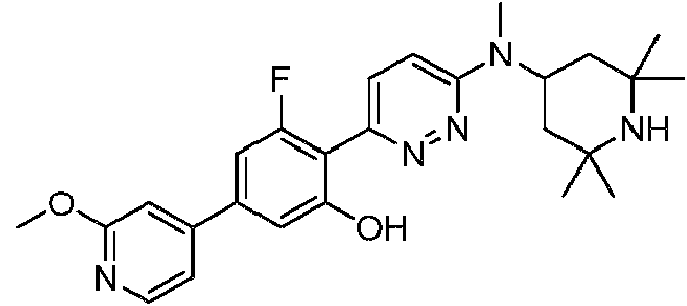
Siguiendo el MÉTODO GENERAL 1-6 para el acoplamiento cruzado de Suzuki, se hicieron reaccionar 5-bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (Intermedio 8-1) y ácido (2-metoxipiridin-4-il)borónico y el producto crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 % de CH3CN a un 30 % en H2O). Después de la formación de la sal, se obtuvo la sal clorhidrato de 3-fluoro-5-(2-metoxipiridin-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un sólido amarillo (5,7 mg). LCMS tR = 0,56 min [Método Q]; [M+H]: 466,4; 1H RMN (400 MHz, MeOD) 6 8,25 (d, J= 5,5 Hz, 1H), 8,20 (d, J= 10,0 Hz, 1H), 7,71 (d, J= 10,0 Hz, 1H), 7,30 (dd, J= 5,5, 1,5 Hz, 1H), 7,21 (dd, J= 12,0, 1,5 Hz, 1H), 7,21 (s, 1H), 7,13 (d, J= 1,0 Hz, 1H), 5,38 5,22 (m, 1H), 4,01 (s, 3H), 3,14 (s, 3H), 2,03 (d, J= 8,5 Hz, 4H), 1,67 (s, 6H), 1,56 (s, 6H).
Ejemplo de referencia 40-2: Síntesis de la sal clorhidrato de 4-(3-fluoro-5-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)piridin-2(1H)-ona

Se calentaron la sal clorhidrato de 3-fluoro-5-(2-metoxipiridin-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (Ejemplo de referencia 40-1, 10 mg, 0,02 mmol) y clorhidrato de piridina (50 mg, 0,43 mmol) a 170 °C durante 15 minutos en un reactor de microondas Biotage® Initiator. La mezcla se diluyó con MeOH/DMSO y se purificó mediante HPLC preparativa de fase inversa (de un 10 a un 45 % de acetonitrilo en agua, 0,1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (4 eq, metanol como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío y el sólido resultante se suspendió en CH3CN/H2O (3/1 mL). Se añadió HCl acuoso 1 M (3 equivalentes) y el disolvente se concentró al vacío para proporcionar el compuesto del título como un sólido amarillo (3 mg, 26 %). LCMS tR = 0,47 min [Método Q]; [M+H]: 452,3; 1H RMN (400 MHz, MeOD) 6 8,28 (d, J= 10,0 Hz, 1H), 8,07 (d, J= 10,0 Hz, 1H), 7,69 (d, J= 6,5 Hz, 1H), 7,25 (dd, J= 11,0, 1,5 Hz, 1H), 7,19 (s, 1H), 6,89 (s, 1H), 6,85 (dd, J= 6,5, 1,5 Hz, 1H), 5,07 (s a, 1H), 3,21 (s, 3H), 2,20-2,00 (m, 4H), 1, 66 (s, 6H), 1,59 (s, 6H).
Ejemplo de referencia 40-3: Síntesis de la sal clorhidrato de 4-(3-fluoro-5-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenil)-1-metilpiridin-2(1H)-ona

Siguiendo el MÉTODO GENERAL 1-6 para el acoplamiento cruzado de Suzuki, se hicieron reaccionar 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2(1H)-ona y 5-bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (Intermedio 8-1) y el producto crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 % de CH3CN a un 30 % en H2O). Después de la formación de la sal, se obtuvo la sal clorhidrato de 4-(3-fluoro-5-l^¡drox¡-4-(6-(met¡l(2,2,6,6-tetramet¡lp¡perid¡n-4-¡l)am¡no)p¡r¡daz¡n-3-¡l)fen¡l)-1-met¡lp¡^d¡n-2(1H)-ona como un sólido amarillo (10 mg, 12 %). LCMS tR = 0,49 min [Método Q]; [M+H]: 466,3; 1H RMN (400 MHz, MeOD) 8 8,22 (d, J= 10,0 Hz, 1H), 7,86-7,73 (m, 2H), 7,24-7,13 (m, 2H), 6,82 (d, J= 2,0 Hz, 1H), 6,72 (dd, J= 7,0, 2,0 Hz, 1H), 5,30-5,15 (m, 1H), 3,64 (s, 3H), 3,16 (s, 3H), 2,04 (d, J= 8,0 Hz, 4H), 1,67 (s, 6H), 1,57 (s, 6H).
Ejemplo de referencia 40-4: Síntesis de la sal clorhidrato de 5-(3-fluoro-5-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)feml)-1-metilpiridm-2(1H)-ona

Siguiendo el MÉTODO GENERAL 1-6 para el acoplamiento cruzado de Suzuki, se hicieron reaccionar 5-bromo-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol y 1-metil-5-(4,4,5,5-tetrametil-1,3,2-d¡oxaborolan-2-¡l)p¡rid¡n-2(1H)-ona y el producto crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 % de CH3CN a un 30 % en H2O). Después de la formación de la sal, se obtuvo la sal clorhidrato de 5-(3-fluoro-5-l^¡drox¡-4-(6-(met¡l(2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)am¡no)piridaz¡n-3-¡l)fen¡l)-1-met¡lp¡r¡d¡n-2(1H)-ona como un sólido amarillo (8,0 mg, 11 %). LCMS tR = 0,49 min [Método Q]; [M+H]: 466,3; 1H RMN (400 MHz, MeOD) δ 8,28 (d, J= 10,0 Hz, 1H), 8,08 (d, J= 10,0 Hz, 1H), 7,83 (d, J= 7,0 Hz, 1H), 7,23 (dd, J= 11,0, 1,5 Hz, 1H), 7,17 (s, 1H), 6,84 (d, J= 2,0 Hz, 1H), 6,75 (dd, J= 7,0, 2,0 Hz, 1H), 5,06 (s a, 1H), 3,66 (s, 3H), 3,21 (s, 3H), 2,19-2,01 (m, 4H), 1,66 (s, 6H), 1,59 (s, 6H).
Ejemplo de referencia 40-5: Síntesis de la sal clorhidrato de 3-fluoro-5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol

Paso 1: 3-(2-Fluoro-4-metoxifenil)-6-((2,2,6,6-tetrametilpiperídin-4-il)oxi)pirídazina
Se añadieron un vial apto para microondas el Intermedio 1-3 (2,14 g, 7,92 mmol), ácido (2-fluoro-4-metoxifem^borórnco (2,02 g, 11,9 mmol) y una solución acuosa 0,5 M de K3PO4 (32 mL, 16 mmol). Se añadió a la mezcla el precatalizador de 2.a generación XPhos (0,19 g, 0,24 mmol) y después se añadió THF (16 mL). La mezcla de reacción se selló, se agitó a TA durante 2 h y a continuación se extrajo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron al vacío para proporcionar 3-(2-fluoro-4-metox¡fen¡l)-6-((2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡)p¡r¡daz¡na como un sólido marrón (2,85 g, 90 %).
[M+H]: 360,3; 1H RMN (400 MHz, MeOD) 8 7,90 (dd, J= 9,5, 2,5 Hz, 1H), 7,81 (t, J= 9,0 Hz, 1H), 7,18 (d, J= 9,5 Hz, 1H), 6,94 (dd, J= 9,0, 2,5 Hz, 1H), 6 , 8 8 (dd, J= 13,0, 2,5 Hz, 1H), 5,79 (tt, J= 11,0, 4,0 Hz, 1H), 3,89 (s, 3H), 2,24 (dd, J= 12,0, 4,0 Hz, 2H), 1,43 (t, J= 12,0 Hz, 2H), 1,37 (s, 6H), 1,26 (s, 6H).
Paso 2: 3-Fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
Se disolvió 3-(2-fluoro-4-metoxifenil)-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina (1,08 g, 3,00 mmol) en CH2CI2 (15 mL) y se añadió gota a gota una solución 1 M de BBr3 en CH2CI2 (7,5 mL, 7,5 mmol). La mezcla de reacción se agitó a TA durante toda la noche y a continuación se diluyó con CH2O 2 y una solución acuosa tamponada de pH 4. La fase acuosa se lavó con cloroformo/propan-2-ol 3:1 (2x) y a continuación se basificó hasta pH 8 con NaHCO3 saturado. La fase acuosa se extrajo con cloroformo/propan-2-ol 3:1 (4x). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. Se obtuvo 3-fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol como un sólido marrón (0,59 g, 52 %).
[M+H]: 346,4; 1H RMN (400 MHz, MeOD) 57,88 (dd, J= 9,0, 2,2 Hz, 1H), 7,69 (t, J= 9,0 Hz, 1H), 7,16 (d, J= 9,0 Hz, 1H), 6,76 (dd, J= 9,0, 2,0 Hz, 1H), 6,65 (dd, J= 13,0, 2,0 Hz, 1H), 5,78 (tt, J= 11,0, 4,0 Hz, 1H), 2,26 (dd, J= 12,5, 4,0 Hz, 2H), 1,47 (t, J= 12,0 Hz, 2H), 1,39 (s, 6H), 1,29 (s, 6H).
Paso 3: Trifluorometanosulfonato de 3-fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo
Se mezclaron en un vial apto para microondas 3-fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol (0,70 g, 2,03 mmol), N-fenilbis(trifluorometanosulfonimida) (0,72 g, 2,03 mmol), K2CO3 (0,84 g, 6 , 0 8 mmol) y THF ( 10 mL). La mezcla de reacción se calentó hasta 120 °C durante 10 minutos en un reactor de microondas Biotage® Initiator. Los componentes volátiles se eliminaron al vacío. Se añadió una solución acuosa 1 M de NaOH y la fase acuosa se extrajo con DCM (2x). Las capas orgánicas combinadas se lavaron con salmuera, se secaron con Na2SO4 , se filtraron y se concentraron a sequedad al vacío. El material crudo se purificó mediante cromatografía en columna ultrarrápida utilizando gel de sílice (elución con gradiente de un 10-50 % de EtOAc/NH32 N en EtOH (3/1), en heptano) para obtener trifluorometanosulfonato de 3-fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo como un sólido gris-rojo (0,63 g, 66 %). [M+H]: 478,2; 1H RMN (400 MHz, MeOD) 5 8,09 (t, J= 8 , 6 Hz, 1H), 7,99 (dd, J= 9,2, 2,4 Hz, 1H), 7,50 (dd, J= 10,6, 2,4 Hz, 1H), 7,45 (dd, J= 8 ,6 , 2,4 Hz, 1H), 7,24 (d, J= 9,5 Hz, 1H), 5,82 (tt, J= 11,2, 4,2 Hz, 1H), 2,24 (dd, J= 12,6, 4,1 Hz, 2H), 1,43 (t, J= 11,7 Hz, 2H), 1,36 (s, 6H), 1,25 (s, 6H).
Paso 4: Trifluorometanosulfonato de 3-fluoro-5-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo
Se disolvieron trifluorometanosulfonato de 3-fluoro-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo (0,48 g, 1,01 mmol), PhI(OAc)2 (0,46 g, 1,41 mmol) y Pd(OAc)2 ( 68 mg, 0,10 mmol) en una mezcla de ácido acético (4 mL) y anhídrido acético (4 mL). La mezcla se agitó a 80 °C durante 3 h. La reacción cruda se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (5 eq, CH3CN como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío y se obtuvo trifluorometanosulfonato de 3-fluoro-5-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo como un sólido verde (0,50 g, 100 %). [M+H]: 494,3; 1H RMN (400 MHz, MeOD) 58,09 (t, J= 8,5 Hz, 1H), 7,99 (dd, J= 9,5, 2,5 Hz, 1H), 7,50 (dd, J= 10,5, 2,5 Hz, 1H), 7,45 (dd, J= 8,5, 2,5 Hz, 1H), 7,24 (d, J= 9,5 Hz, 1H), 5,82 (tt, J= 11,0, 4,0 Hz, 1H), 2,24 (dd, J= 12,5, 4,0 Hz, 2H), 1,43 (t, J= 12,0 Hz, 2H), 1,36 (s, 6H), 1,25 (s, 6H).
Paso 5: Sal clorhidrato de 3-fluoro-5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
Siguiendo el procedimiento representativo del MÉTODO GENERAL 1-6 para el acoplamiento cruzado de Suzuki, se hicieron reaccionar trifluorometanosulfonato de 3-fluoro-5-hidroxi-4-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenilo y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol-1-carboxilato y el producto crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 % de CH3CN a un 30 % en H2O). Después de la formación de la sal, se obtuvo la sal clorhidrato de 3-fluoro-5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol hidratado como un sólido amarillo (49 mg, 25 %). LCMS tR = 0,49 min; [M+H]: 412,3; 1H RMN (400 MHz, MeOD) 5 8,51 (d, J = 9,5 Hz, 1H), 8,26 (s, 2H), 7,87 (d, J= 9,5 Hz, 1H), 7,21 (dd, J= 12,0, 1,5 Hz, 1H), 7,16 (s, 1H), 5,77 (tt, J= 10,5, 4,0 Hz, 1H), 2,52 (dd, J= 14,0, 4,0 Hz, 2H), 1,93 (dd, J= 14,0, 10,5 Hz, 2H), 1,64 (s, 6H), 1,59 (s, 6H).
Ejemplo de referencia 40-6: Síntesis de la sal clorhidrato de 5-cloro-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol

Paso 1: 6-(4-Cloro-2-fluorofenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Se desgasificaron ácido (4-cloro-2-fluorofenil)borónico (4,29 g, 24,6 mmol), Intermedio 1-1 (6,63 g, 23,4 mmol) y Na2CO3 (7,45 g, 70,3 mmol) durante 10 minutos con N2 , y a continuación se añadió PdCl2(dppf)CH2Cl2 (0,96 g, 1,17 mmol). La mezcla de reacción se calentó a 90 °C durante 2 h. La mezcla de reacción se concentró al vacío y a
continuación se añadieron CH2CI2 y HCl 1 M. La fase acuosa se lavó con CH2CI2 y a continuación se basificó hasta pH 14 con una solución de NaOH 6 M. La fase acuosa se extrajo con CH2Cl2 (3x). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida para proporcionar 6-(4-cloro-2-fluorofenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina como un sólido marrón (8,49 mg, 95 %). [M+H]: 377,2; 1H RMN (400 MHz, MeOD) 6 7,87 (t, J= 8,5 Hz, 1H), 7,76 (dd, J= 9,5, 2,4 Hz, 1H), 7,32-7,40 (m, 2H), 7,17 (d, J= 9,5 Hz, 1H), 5,13-5,36 (m, 1H), 1,69 (dd, J= 12,5, 3,5 Hz, 2H), 1,57 (t, J= 12,5 Hz, 2H), 1,38 (s, 6H), 1,23 (s, 6H).
Paso 2: 5-Cloro-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol Sal clorhidrato.
Se disolvieron 6-(4-cloro-2-fluorofenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (50 mg, 0,13 mmol), PhI(OAc)2 (60 mg, 0,19 mmol) y Pd(OAc)2 (8,9 mg, 0,013 mmol) en una mezcla de ácido acético (0,6 mL) y anhídrido acético (0,6 mL). La mezcla de se agitó a 40 °C durante toda la noche. Se añadió una solución de tiosulfato de sodio saturado y la mezcla se agitó durante 8 días a TA. Se añadió una solución de carbonato de potasio y el pH de reacción se ajustó a 10. La fase acuosa se extrajo (2x) con diclorometano y metanol (9:1). Las fases orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El material crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 a un 30 % de acetonitrilo en agua, 0,1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (4 eq, CH3CN como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío y el sólido resultante se suspendió en CH3CN/H2O (3/1 mL). Se añadió HCl acuoso 1 M (3 equivalentes) y los componentes volátiles se concentraron al vacío para proporcionar la sal clorhidrato de 5-cloro-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un sólido amarillo (19 mg, 33 %). LCMS tR = 0,57 min [Método Q]; [M+H]: 393,2; 1H Rm N (400 MHz, MeOD) 6 8,21 (d, J= 10,0 Hz, 1H), 7,99 (d, J= 10,0 Hz, 1H), 6,99 (dd, J= 10,0, 2,0 Hz, 1H), 6,94 (t, J= 2,0 Hz, 1H), 5,09 (s a, 1H), 3,18 (s, 3H), 1,98-2,12 (m, 4H), 1,65 (s, 6H), 1,57 (s, 6H).
Ejemplo de referencia 40-7: Síntesis de la sal clorhidrato de 3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)piridazm-3-il)-5-(1H-pirazol-4-il)fenol

Se añadieron a un vial apto para microondas 5-cloro-3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (Ejemplo de referencia 40-6, 0,32 g, 0,82 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol-1-carboxilato de tert-butilo (1,00 g, 3,40 mmol) y Cs2CO3 (5,52 g, 26,0 mmol). A continuación, se añadió a la mezcla el precatalizador XPhos (60 g, 0,08 mmol) y después se añadieron dioxano (5 mL) y agua (0,9 mL). La mezcla de reacción se selló y agitó a 130 °C durante 2 h en un reactor de microondas Biotage® Initiator. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con metanol. El filtrado se concentró al vacío. El producto crudo se purificó mediante HPLC preparativa de fase inversa (de un 10 a un 45 % de acetonitrilo en agua, 0,1 % de ácido trifluoroacético como modificador). En las fracciones apropiadas que contenían el producto se generó la base libre mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (4 eq, metanol como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). El disolvente se concentró al vacío y el sólido resultante se disolvió en MeOH. Se añadió SiliaMetS® DMT ( 6 eq.) y la mezcla se agitó durante 18 h. El sólido se filtró y el filtrado se concentró al vacío. El sólido resultante se suspendió en CH3CN/H2O (6/2 mL). Se añadió HCl acuoso 1 M (3 equivalentes) y los componentes volátiles se concentraron al vacío para proporcionar la sal clorhidrato de 3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1 H-pirazol-4-il)fenol como un sólido amarillo (47 mg). LCMS tR = 0,48 min [Método Q]; [M+H]: 425,3; 1H RMN (400 MHz, MeOD) 6 8,25-8,32 (m, 1H), 8,14 (s, 2H), 8,04 (d, J= 10,0 Hz, 1H), 7,19 (dd, J= 11,5, 1,5 Hz, 1H), 7,11 (s, 1H), 5,10 (s a, 1H), 3,19 (s, 3H), 1,91-2,24 (m, 4H), 1,66 (s, 6H), 1,57 (s, 6H).
Ejemplo de referencia 40-8: Síntesis de la sal clorhidrato de 3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazin-3-il)-5-(1-metil-1 H-pirazol-4-il)fenol

Paso 1: 6-(2-Fluoro-6-metoxi-4-(1-metil-1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
Siguiendo el MÉTODO GENERAL 1-6 para el acoplamiento cruzado de Suzuki, se hicieron reaccionar una mezcla de 6-(4-bromo-2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina, 4-bromo-6-(2-fluoro-6-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 8-1, Paso 2 , 0,10 g, 0,22 mmol, cantidad total de los 2 regioisómeros) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol-1-carboxilato de tert-butilo (0,14 g, 0,67 mmol). El producto crudo se purificó mediante cromatografía en columna utilizando gel de sílice y una elución en gradiente de un 1-15 % de amoniaco 7 N en MeOH, en CH2O 2. Se obtuvo una mezcla inseparable del producto deseado (6-(2-fluoro-6-metoxi-4-(1-metil-1H-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6 tetra m eti lpiperidin-4-il)piridazin-3-amina) y 6-(2-fluoro-6-metoxifenil)-N-metil-4-(1-metiMH-pirazol-4-il)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina was como un sólido incoloro (80 mg, 80 %). [M+H]: 453,4.
Paso 2: Sal clorhidrato de 3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)fenol
Se disolvió una mezcla de 6-(2-fluoro-6-metoxi-4-(1-metiMH-pirazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina y 6-(2-fluoro-6-metoxifenil)-N-metil-4-(1-metiMH-pirazol-4-il)-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (0,03 g, 0,07 mmol, cantidad total de los 2 regioisómeros) en CH2O 2 (0,2 M). Se añadió rápidamente una solución 1 M de BBr3 en CH2Cl2 (0,3 mL, 3,5 mmol). La mezcla de reacción se agitó durante 3 h. Se añadió MeOH a la reacción a 0 1C y el disolvente se concentró a presión reducida. El material crudo se purificó mediante HPLC preparativa de fase inversa (de un 15 a un 45 % de acetonitrilo en agua, hidróxido de amonio 5 mM como modificador). El disolvente se concentró al vacío y el sólido resultante se suspendió en CH3CN/H2O (4/1 mL). Se añadió HCl acuoso 1 M (3 equivalentes) y los componentes volátiles se concentraron al vacío para proporcionar la sal clorhidrato de 3-fluoro-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-metil-1H-pirazol-4-il)fenol como un sólido amarillo (5 mg, 15 %). LCMS tR = 0,50 min [Método Q]; [M+H]: 439,4; 1H RMN (400 MHz, DMSO) 59,20 (d, J= 12,0 Hz, 1H), 8,29 (d, J= 12,0 Hz, 1H), 8,23 (s, 1H), 8,04 (d, J= 9,5 Hz, 1H), 7,92 (s, 1H), 7,79 (s a, 1H), 7,09 (dd, J= 12,0, 1,5 Hz, 1H), 7,04 (s, 1H), 4,92 (s a, 1H), 3,88 (s, 3H), 3,03 (s, 3H), 2,05 (t, J= 13,0 Hz, 2H), 1,81 (dd, J= 13,0, 3,5 Hz, 2H), 1,53 (s, 6H), 1,49 (s, 6H).
Ejemplo de referencia 41-1: Síntesis de 5-(5-metoxipiridm-3-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)amino)piridazm-3-il)fenol

Siguiendo el MÉTODO GENERAL 1-3 para el acoplamiento de Suzuki, se añadieron a un vial apto para microondas de 25 mL 3-metoxi-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridina (94 mg, 0,400 mmol), trifluorometanosulfonato de 3-hidroxi-4-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenilo (Intermedio 9-2, 105 mg, 0,2 mmol), bicarbonato de sodio (50,4 mg, 0,600 mmol) y Pd(PPh3)4 (11,56 mg, 10,00 |jmol), y después dioxano (2 mL) y agua (0,5 mL). La mezcla de reacción se purgó con N2 durante 10 minutos y se calentó en un microondas a 100 °C durante 1 h y a continuación se diluyó con EtOAc y se filtró a través de celite. El filtrado se acidificó hasta pH ~3 con HCl 1 N y se cargó sobre una columna de SCX. La columna se lavó con MeOH , y se eluyó con NH32 N en MeOH. Después de la concentración, el residuo se purificó mediante HPLC preparativa para proporcionar 5-(5-metoxipiridin-3-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (55 mg, 0 , 1 2 0 mmol, 60 % de rendimiento), LCMS: tR = 0,50 min [Método Q], MS (M+1) = 448,4; 1H RMN (METANOL-d4) 58,17 (d, J=5,1 Hz, 1H), 8,11 (d, J=9,1 Hz, 1H), 7,85 (d, J=7,1 Hz, 1H), 7,22-7,33 (m, 4H), 7,06 (s, 1H), 5,10 (m, 1H), 3,97 (s, 3H), 3,03 (s, 3H), 1,74 (dd, J=12,6, 3,0 Hz, 2H), 1,59 (t, J=12,4 Hz, 2H), 1,42 (s, 6H), 1,26 (s, 6H).
Los siguientes compuestos se prepararon utilizando procedimientos similares a los del Ejemplo de referencia 41-1 a partir de los Intermedios 1-1, 1-3 o el Ejemplo 32-1, Paso 4 , y métodos generales resumidos en la sección MÉTODOS GENERALES.




Ejemplo de referencia 42-1: Síntesis de 5-(1H-imidazol-2-il)-2-(6-(metil(2,2,6,6-tetrametilpipendm-4-il)amino)piridazin-3-il)fenol

Paso 1: 6-(4-(1H-Imidazol-2-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron 6-(2-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 9-3, 100 mg, 0,21 mmol), 2-bromo-1H-imidazol (61,2 mg, 0,42 mmol), Na2CO3 (44 mg, 0,42 mmol) y Pd(PPh3 )2Cl2 (14 mg, 0,02 mmol), y después DME (1 mL)/EtüH 0,25 mL)/(H2Ü (0,25 mL). El vial se purgó con N2 durante 10 min y la mezcla de reacción se calentó a 150 °C en un reactor de microondas durante 20 min. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con EtOAc. El filtrado se concentró al vacío para obtener el producto crudo que se purificó mediante cromatografía en gel de sílice (5 %-15 % de MeOH/DCM) para proporcionar 6-(4-(1H-imidazol-2-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (40 mg, MS: 421,3 [M+H+]).
Paso 2: 5-(1H-Imidazol-2-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol, se trató 6-(4-(1H-imidazol-2-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (40 mg, 0,1 mmol) con tiofenol (0,01 mL, 0,11 mmol) y K2CO3(13 mg, 0,1 mmol) en Nm P ( 2 mL) durante 30 min a 190 °C para proporcionar 5-(1H-imidazol-2-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol como un polvo amarillo pálido ( 6 mg). MS: 407,4 [M+H+]; LCMS tR = 0,40 min [Método Q]; 1H RMN (400 MHz, METANOL-d4 ) 5 ppm 8,00 (d, J=10,11 Hz, 1H), 7,68-7,78 (m, 1H), 7,32-7,41 (m, 2H), 7,19 (d, J=10,11 Hz, 1H), 7,05 (s, 2H), 4,96 (s a., 1H), 2,90 (s, 3H), 1,58 (dd, J=12,38, 3,28 Hz, 2H), 1,46 (t, J=12,38 Hz, 2H), 1,28 (s, 6H), 1,12 (s, 6H).
Ejemplo de referencia 42-2: Síntesis de 5-(1H-imidazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidm-4-il)ammo)pindazm-3-il)fenol

Paso 1: 4-Bromo-1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol
Una mezcla de 4-bromo-1H-imidazol (1,0 g, 6 , 8 mmol) en THF (15 mL) se añadió NaH (327 mg, 8,16 mmol) a 0 °C. La mezcla se agitó de 0 °C a TA durante 0,5 h. Se añadió gota a gota SEMCl (1,45 mL, 8,16 mmol) y la mezcla de reacción se agitó a TA durante 2 h. La mezcla de reacción se desactivó con agua y se extrajo con DCM. La capa orgánica se secó (Na2SO4), se filtró y se concentró al vacío para obtener el producto crudo que se purificó mediante cromatografía en gel de sílice (10-100 % EtOAc/Heptano, después un 0-15 % de MeOH/DCM) para proporcionar 4-bromo-1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol (1,36 g, MS: 279,3 [M+H+]).
Paso 2: 6-(2-Metoxi-4-(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron 6-(2-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 9-3, 50 mg, 0,10 mmol), 4-bromo-1-((2-(trimetilsilil)etoxi)metil)-1 H-imidazol (57,7 mg, 0,21 mmol), Na2CO3 (22 mg, 0,21 mmol) y Pd(PPh3)2Cl2 (7,3 mg, 0,01 mmol), y después DME (1 mL)/EtOH 0,25 mL)/(H2O (0,25 mL). El vial se purgó con N2 durante 10 minutos y la mezcla de reacción se calentó a 150 °C en un reactor de microondas durante 20 minutos. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con EtOAc. El filtrado se concentró al vacío para obtener el producto crudo que se purificó mediante cromatografía en gel de sílice (5 %-15 % de MeOH/DCM) para proporcionar 6-(2-metoxi-4-(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (50 mg). MS: 551,6 [M+H+]).
Paso 3: 2-(6-(Metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-4-il)fenol
Ssiguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol, se trató 6-(2-metoxi-4-(1-((2-(trimetilsilil)etoxi)metil)-1 H-imidazol-4-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (50 mg, 0,09 mmol) con tiophenol (0,01 mL, 0,11 mmol) y K2CO3 (12 mg, 0,09 mmol) en NMP (2 mL) durante 30 minutos a 190 °C para proporcionar 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-4-il)fenol y una impureza (50 mg, MS: 537,6 [M+H+]).
Paso 4: 5-(1H-imidazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
A un vial apto para microondas que contenía 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-4-il)fenol (mezcla del paso anterior, 50 mg, 0,09 mmol) en EtOH (1,0 mL)/DCM (1,0 mL) y HCl conc. (8,5 μL) se añadió BBr3 (0,46 mL, 0,46 mmol). El vial se purgó con N2 (2x) y la mezcla de reacción se calentó a 110 °C en un reactor de microondas durante 30 min. La mezcla de reacción se filtró a través de celite (embudo con filtro preempaquetado) con un lavado con MeOH. El filtrado se acidificó hasta pH 3 utilizando HCl 1 N y se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (1 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). Después de la evaporación, el material se purificó mediante HPLC de fase inversa para proporcionar 5-(1H-imidazol-4-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol (2 mg). MS: 407,4 [M+H+]. LCMStR = 0,40 min [Método Q]; 1H RMN (400 MHz, METANOL-d4) 6 ppm 8,01 (d, J=9,60 Hz, 1H), 7,57-7,74 (m, 2H), 7,39 (s, 1H), 7,14-7,30 (m, 3H), 4,98 (s a., 1H), 2,92 (s, 3H), 1,62 (d, J= 13,14 Hz, 2H), 1,52 (t, J=12,38 Hz, 2H), 1,32 (s, 6H), 1,16 (s, 6H).
Ejemplo de referencia 42-3: Síntesis de 5-(imidazo[1,2-a]pirazin-3-N)-2-(6-(metil(2,2,6,6-tetrametilpipendm-4-N)ammo)pindazm-3-N)fenol

Paso 1: 6-(4-(imidazo[1,2-a]pirazin-3-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina
A un vial apto para microondas se añadieron 6-(2-metoxi-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Intermedio 9-3, 50 mg, 0,1 mmol), 3-bromo-5,6,7,8-tetrahidroimidazo[1,2-a]pirazina (49,6 mg, 0,21 mmol), Na2CO3 (44 mg, 0,42 mmol) y Pd(PPh3)2Cl2 (7 mg, 0,01 mmol), y después DME (1 mL)/EtOH 0,25 mL)/(H2O (0,25 mL). El vial se purgó con N2 durante 10 minutos y la mezcla de reacción se calentó a 150 °C en un reactor de microondas durante 20 min. La mezcla de reacción se filtró a través de celite y la masa retenida en el filtro se lavó con EtOAc. El filtrado se concentró al vacío para obtener el producto crudo que se purificó mediante cromatografía en gel de sílice (5 %-15 % de MeOH/DCM) para proporcionar 6-(4-(imidazo[1,2-a]pirazin-3-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (49 mg). MS: 476,5 [M+H+]).
Paso 2: 5-(imidazo[1,2-a]pirazin-3-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol
Siguiendo el MÉTODO GENERAL 3-1 para la desprotección de metoxi utilizando tiofenol, se trató 6-(4-(imidazo[1,2-a]pirazin-3-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (49 mg, 0,1 mmol) con tiofenol (0,01 mL, 0,12 mmol) y K2CO3(14 mg, 0,11 mmol) en NMP (2 mL) durante 30 min a 190 °C para proporcionar 5-(imidazo[1,2-a]pirazin-3-il)-2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)fenol ( 8 mg). MS: 458,4 [M+H+]; LCMS tR = 0,47 min [Método Q]; 1H RMN (400 MHz, CLOROFORMO-d) 6 ppm 9,16 (s, 1H), 8,41 (d, J=5,05 Hz, 1H), 7,92-7,97 (m, 2H), 7,88 (d, J=10,11 Hz, 1H), 7,75 (d, J=8,08 Hz, 1H), 7,31 (s, 1H), 7,13 (s, 1H), 7,05 (d, J=9,60 Hz, 1H), 4,99 (s a., 1H), 3,05 (s, 3H), 1,72 (d, J=12,13 Hz, 2H), 1,46 (t, J=12,38 Hz, 2H), 1,39 (s, 6H), 1,23 (s, 6H).
Ejemplo de referencia 42-4: Síntesis de 2-(6-(metil(2,2,6,6-tetrametilpipendm-4-N)ammo)pindazm-3-N)-5-(5,6,7,8-tetrahidroimidazo[1,2-a]pirazin-3-N)fenol

A una solución de 6-(4-(imidazo[1,2-a]pirazin-3-il)-2-metoxifenil)-N-metil-N-(2,2,6,6-tetrametilpiperidin-4-il)piridazin-3-amina (Ejemplo de referencia 42-3, 50 mg, 0,1 mmol) en DCM (2 mL) se añadió una solución 1 M de BBr3 en DCM (0,52 mL) gota a gota a 78 °C. La mezcla de reacción cruda se calentó hasta TA y se agitó durante toda la noche. La reacción se desactivó con una solución ac. de NaHCO3 a 0 °C y se extrajo con DCM. La capa orgánica se secó con Na2SO4, se filtró y se concentró para obtener el producto crudo. El producto crudo se acidificó hasta pH 3 utilizando HCl 1 N y se purificó mediante captación y liberación utilizando SiliaBond® ácido propilsulfónico (1 g, MeOH como eluyente y una solución de amoniaco 2 N en MeOH para liberar el material). Después de la evaporación, el material se purificó mediante HPLC de fase inversa para proporcionar 2-(6-(metil(2,2,6,6-tetrametilpiperidin-4-il)amino)piridazin-3-il)-5-(5,6,7,8-tetrahidroimidazo[1,2-a]pirazin-3-il)fenol (4 mg). MS: 462,4 [M+H+]; LCMS tR = 0,36 min [Método Q]; 1H RMN (400 MHz, METANOL-ck) 6 ppm 8,03 (d, J=10,11 Hz, 1H), 7,74 (d, J=9,09 Hz, 1H), 7,22 (d, J=10,11 Hz, 1H), 6,96 (s, 2H), 6,99 (s, 1H), 5,02 (s a., 1H), 3,88-4,09 (m, 4H), 3,11 (t, J=5,31 Hz, 2H), 2,92 (s, 3H), 1,62 (d, J=12,63 Hz, 2H), 1,51 (t, J=12,38 Hz, 2H), 1,31 (s, 6H), 1,16 (s, 6H).
Los siguientes compuestos se prepararon utilizando procedimientos similares a los de los Ejemplos de referencia 42-1 42-4 l m n r l r m n n l i n MÉT D ENERALE .



Ejemplo 43-1: Síntesis de 2-(6-((3aR,6aS)-5-(2-hidroxietil)hexahidropirrolo[3,4-c]pirrol-2(1H)-N)piridazm-3-il)-5-(1H-pirazol-4-il)fenol

Paso 1: (3aR,6aS)-5-(6-Cloropiridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2(1H)-carboxilato de tert-butilo
A una solución de 3,6-dicloropiridazina (462 mg, 3,10 mmol) en n-butanol ( 8 mL) se añadieron DIPEA (1,354 mL, 7,75 mmol) y (3aR,6aS)-hexahidropirrolo[3,4-c]pirrol-2(1H)-carboxilato de tert-butilo (658 mg, 3,1 mmol). La mezcla de reacción se calentó a 120 °C durante 2 horas y a continuación se diluyó con DCM y agua. La capa orgánica se separó y se concentró hasta obtener un aceite amarronado, que se purificó mediante cromatografía en gel de sílice (0-25 % de EtOAc/DCM) para proporcionar (3aR,6aS)-5-(6-doropiridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2(1H)-carboxilato de tert-butilo (730 mg, 2,180 mmol, 70 % de rendimiento), Ms (M+1 ) = 325,2.
Paso 2: (3aR,6aS)-5-(6-(4-Cloro-2-hidroxifenil)piridazin-3-il)hexahidropyirrolo[3,4-c]pirrole-2(1H)-carboxilato de tertbutilo
La mezcla de reacción de (3aR,6aS)-5-(6-doropiridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2(1H)-carboxilato de tertbutilo (325 mg, 1 mmol), 5-cloro-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (280 mg, 1,100 mmol), carbonato de sodio (318 mg, 3,00 mmol) y PdCl2(dppf).CH2Cl2 (61,2 mg, 0,075 mmol) en dioxano (5 mL) y agua (5,00 mL) se desgasificó burbujeando N2 durante 10 minutos. Después de calentar a 90 °C durante toda la noche, la mezcla de reacción se filtró a través de celite y se lavó con EtOAc. El filtrado se concentró y el residuo se purificó mediante cromatografía en gel de sílice (0-10 % de MeOH/DCM) para proporcionar (3aR,6aS)-5-(6-(4-cloro-2-hidroxifenil)piridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2(1 H)-carboxilato de tert-butilo (180 mg, 0,432 mmol, 43,2 % de rendimiento) MS (M+1) = 417,0.
Paso 3: (3aR, 6aS)-5-(6-(2-Hidroxi-4-( 1 H-pirazol-4-il)feni)piridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2( 1 H)-carboxilato de tert-butilo
A un vial apto para microondas de 10 mL, se añadieron (3aR,6aS)-5-(6-(4-doro-2-hidroxifenil)piridazin-3-il)hexahidropirrolo[3,4-c]pirrol-2(1H)-carboxilato de tert-butilo (0,182 g, 0,437 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol-1-carboxilato de tert-butilo (0,385 g, 1,310 mmol), doro[2-(diddohexilfosfino)-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenil][2-(2-aminoetil)fenil]paladio (II) (0,035 g, 0,044 mmol) y Cs2CO3 (0,427 g, 1,310 mmol), y después se añadieron 1,4-dioxano (2 mL) y agua (0,5 mL). En la mezcla de reacción se hizo vacío, se rellenó con N2 dos veces y a continuación se calentó a 90 °C durante toda la noche. La mezcla de reacción se filtró a través de celite y se lavó con DMSO y MeOH. El filtrado se acidificó con HCl 1 N, se agitó a TA durante 3 horas y a continuación se extrajo con DCM. La capa acuosa se basificó con NH2 2 M en MeOH y se formó un precipitado amarronado, que se filtró y se lavó con DMSO para proporcionar un sólido gris 2-(6-((3aR,6aS)-hexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol (82 mg, 0,224 mmol, 51,2 % de rendimiento). MS (M+1) = 349,1. La solución de lavado con DMSO se concentró para proporcionar una solución en DMSO del (3aR,6aS)-5-(6-(2-hidroxi4- (1H-p¡razol-4-¡l)fenil)p¡r¡daz¡n-3-¡l)hexah¡drop¡rrolo[3,4-c]p¡rrole-2(1H)-carbox¡lato de tert-butilo (75 mg, 0,167 mmol, 38 % de rendimiento), MS (M+1) = 449,1.
Paso 4: 2-(6-((3aR, 6aS)-Hexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol
A una solución de (3aR,6aS)-5-(6-(2-h¡drox¡-4-(1H-p¡razol-4-¡l)fenil)p¡r¡daz¡n-3-¡l)hexah¡drop¡rrolo[3,4-c]p¡rrol-2(1H)-carboxilato de tert-butilo (75 mg, 0,167 mmol) en 1 mL de dioxano se añadió HCl 4 N en dioxanoe (1 mL, 4,00 mmol). La mezcla de reacción se agitó a TA durante toda la noche y a continuación se basificó con NH32 N en MeOH para formar un precipitado que se separó mediante centrifugación para proporcionar un sólido oscuro, 2-(6-((3aR,6aS)-hexahidropirrolo[3,4-c]pirrol-2(1 H)-il)piridazin-3-¡l)-5-(1 H-p¡razol-4-il)fenol (50 mg, 0,136 mmol, 82 % de rendimiento) MS(M+1) = 349,1.
Paso 5: 2-(6-((3aR,6aS)-5-(2-((tert-Butildimetilsilil)oxi)etil))hexahidropirrolo[3, 4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol
A una solución de 2-(6-((3aR,6aS)-hexahidropirrolo[3,4-c]pirrol-2(1H)-¡l)p¡r¡daz¡n-3-¡l)-5-(1H-p¡razol-4-¡l)fenol (31,4 mg, 0,09 mmol), 2-((tert-butildimet¡lsil¡l)ox¡)acetaldehído (47,1 mg, 0,270 mmol) y triacetoxihidroborato de sodio (57,2 mg, 0,270 mmol) en CH2Cl2 (2 mL), se añadió ácido acético (0,013 mL, 0,225 mmol). La mezcla de reacción se agitó a TA durante toda la noche, a continuación se desactivó con agua y se diluyó con DCM. La capa orgánica se acidificó con una solución de HCl 1 N hasta pH~3 y se filtró para separar los materiales insolubles. El filtrado se cargó sobre una columna de SCX, se lavó con MeOH y a continuación se eluyó con NH37 N en MeOH. La concentración proporcionó un sólido amarronado, 2-(6-((3aR,6aS)-5-(2-((terf-but¡ldimet¡ls¡l¡l)ox¡)et¡l)hexah¡drop¡rrolo[3,4-c]p¡rrol-2(1 H)-il)piridazin-3-¡l)-5-(1H-pirazol-4-il)fenol (23 mg, 0,020 mmol, 51 % de rendimiento), MS (M+1) = 507,1.
Paso 6: 2-(6-((3aR,6aS)-5-(2-Hiroxietil)hexahidropirrolo[3,4-c]pirrol-2(1H)-il)piridazin-3-il)-5-(1H-pirazol-4-il)fenol A una solución de 2-(6-((3aR,6aS)-5-(2-((terf-but¡ldimet¡ls¡l¡l)ox¡)et¡l)hexah¡drop¡rrolo[3,4-c]p¡rrol-2(1 H)-il)piridazin-3-il)-5- (1H-pirazol-4-il)fenol (20 mg, 0,039 mmol) en dioxano (2 mL) se añadió HCl 4 N en dioxano (1 mL, 4,00 mmol). La mezcla de reacción se agitó a TA durante toda la noche, a continuación se basificó con NH3 2 N en MeOH y se concentró. El producto crudo se purificó mediante HPLC preparativa para proporcionar un sólido blanquecino, 2-(6-((3aR,6aS)-5-(2-hidrox¡et¡l)hexahidrop¡rrolo[3,4-c]p¡rrol-2(1H)-¡l)p¡r¡daz¡n-3-¡l)-5-(1H-p¡razol-4-¡l)fenol (6,5 mg, 0,016 mmol, 41 % de rendimiento). LCMS tR = 0,87 min [Método Q]; MS (M+1) = 393,1. 1H RMN (400 MHz, METANOL-d4) 5 ppm 8,01 (d, J=9,60 Hz, 1 H), 7,93 (s, 2 H), 7,69 (d, J=8,08 Hz, 1 H), 7,08-7,19 (m, 3 H), 3,67-3,77 (m, 4 H), 3,56 (d, J= 11,12 Hz, 2 H), 3,11 (s a., 2 H), 2,97-3,05 (m, 2 H), 2,72 (t, J=5,81 Hz, 2 H), 2,63-2,70 (m, 2 H).
Los siguientes compuestos se prepararon utilizando procedimientos similares a los del Ejemplo 43-1, y los métodos enerales ue se resumen en la sección MÉTODOS GENERALES.


Ejemplo de referencia 44: Síntesis del Ejemplo de referencia 17-13 sal clorhidrato de 5-(1 W-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
Paso 1a: Preparación de 3-doro-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina
En un reactor de 30 L se introdujeron 3,6-dicloropiridazina (1 kg, 6,7 mol), 2,2,6,6-tetrametilpiperidin-4-ol (1,05 kg, 6,7
mol) y THF (5 L). La mezcla se agitó y se enfrió -5 °C. Se añadió lentamente tBuOK (1,13 kg, 10,1 mol) disuelto en
THF (10 L) al reactor mientras se mantenía la temperatura a -5-0 °C. La mezcla de reacción se volvió marrón oscuro durante la adición. Después de finalizar la adición, la mezcla se agitó durante 1 h a -5-0 °C, tras lo cual el análisis mediante HPLC mostró que la reacción había finalizado. Se añadió lentamente hielo-agua (1:1, 10 kg) para desactivar la reacción. La mezcla se concentró a presión reducida para eliminar la mayoría del THF. El residuo se extrajo con
EtOAc dos veces (10 L 5 L). La capa orgánica combinada se lavó con agua (10L * 3), y a continuación se concentró
a presión reducida para obtener un residuo negro. Se añadió éter de petróleo (25 L) a este residuo mientras se agitaba.
El sólido oscuro que se formó se separó por filtración. El filtrado amarillo pálido se concentró a presión reducida para obtener un sólido amarillo, que se secó a 50 °C al vacío para obtener 3-cloro-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina (1,3 kg, 4,8 mol), y se utilizó en el siguiente paso sin una purificación adicional. MS m/z 270,1 [M+H];
1H-RMN: (CDCla, 400 MHz) 57,36 (d, J= 9,2 Hz, 1H), 6,90 (d, J= 9,2 Hz, 1H), 5,74 (m, 1H), 2,20 (dd, 12,4 Hz, 2H), 1,30 (m, 14H).
Paso 1b: Preparación de 5-cloro-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol
En un reactor de 30 L se introdujeron 2-bromo-5-clorofenol (2,0 kg, 9,65 mol), B2Pin2 (2,7 kg, 10,6 mol), AcOK (1,9 kg,
19,3 mol) y 1,4-dioxano (15 L). La mezcla se agitó y se purgó con nitrógeno 3 veces. Se añadió PdCl2(dppf)-CH2Cl2
(100g, 0,12 mol) en nitrógeno y la mezcla se calentó hasta 75 °C (se puede retirar el baño de aceite en caso de una reacción muy exotérmica). La mezcla se calentó a 90 °C durante 16 horas, tras lo cual el análisis mediante HPLC
mostró que la reacción había finalizado. Después de enfriar hasta 35 °C, la mezcla se filtró a través de un lecho de
Celite. La solución de 5-cloro-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (1,22 kg, 4,8 mol) filtrada se utilizó en
el siguiente paso sin una purificación adicional. MS m/z 253,1 [M-H]; 1H-RMN: (CDCl3 , 400 MHz) 59,2 (a, 1H), 7,25
(d, J= 8 Hz, 1H), 6,64 (d, J= 1,6 Hz, 1H), 6,62 (dd, Jb= 8 Hz, Jb= 1,6 Hz, 1H), 1,05 (s, 12H).
Paso 2: Preparación de clorhidrato de 5-cloro-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
A la solución que contenía 5-cloro-2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenol (1,22 kg, 4,8 mol) del paso anterior se añadió 3-cloro-6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazina (1,17 kg, 4,4 mol). Se disolvió K3PO4-3 H2O (2,34 kg,
8,8 mol) en agua (5 L) y a continuación se añadió a la solución anterior. La mezcla se purgó con nitrógeno 3 veces.
Se añadió Pd(PPh3)4 (500 g, 0,42 mol) en el nitrógeno y la mezcla de reacción se calentó hasta reflujo a 89 °C durante
16 horas. Después de 16 horas, la HPLC mostró que la reacción había finalizado. Después de enfriar hasta ta temperatura ambiente, la mezcla se filtró a través de un lecho de celite y el filtrado se concentró a presión reducida.
Se añadieron CH2Cl2 (10 L * 3) y una solución al 10 % de K2CO3 (15 L) al residuo anterior. Las capas orgánicas se separaron y se combinaron, después de lavar con agua dos veces (10 L * 2) y concentrar a presión reducida para obtener un aceite amarillo. Se utilizó MTBE (10 L) para disolver el aceite amarillo. Se añadió llentamente con agitación
éter de petróleo (4 L). Precipitaron unos pocos sólidos oscuros y se separaron por filtración. El filtrado se concentró a presión reducida y se disolvió en en CH2Cl2 (20 L). Se añadió lentamente HCl 2 N (5 L) se formó una gran cantidad
de precipitado. Después de agitar durante 1 hora más, el sólido se recogió por filtración y se lavó con EtOAc (2 L). El sólido se secó al vacío a 50 °C para obtener clorhidrato de 5-cloro-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol (0,9 kg, 2,2 mol). MS m/z 362,0 [M+H]+; 1H-RMN: (DMSO-d6 , 400 MHz) 59,26 (d, J= 11,6 Hz, 1H), 8,49 (d,
J= 12 Hz, 1H), 8,38 (d, J= 9,6 Hz, 1H), 7,93 (d, J= 8,4 Hz, 1H), 7,44 (d, J= 9,6 Hz, 1H), 7,07 (d, J= 2 Hz, 1H), 7,02 (dd,
Ja= 2 Hz, Jb= 8,4 Hz, 1H), 5,73 (m, 1H), 2,31 (dd, Ja= 4 Hz, Jb= 13,2 Hz, 2H), 1,84 (dd, Ja= 11,6 Hz, Jb= 2 Hz, 2H), 1,51
(s, 6H), 1,49 (s, 6H).
Paso 3: Preparación de 5-(1H-pirazol-4-il)-2-(6-((2 ,2 ,6,6-tetram etilp iperid in-4-il)oxi)p iridazin-3-il)fenol
En un matraz de 1 L se introdujeron clorhidrato de 5-cloro-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
(9,2 g, 23 mmol), éster pinacólico del ácido N-Boc-pirazol-4-borónico (10,2 g, 35 mmol), Cs2CO3 (15 g, 46 mmol), 1,4-dioxane (100 mL) y agua (25 mL). La mezcla se purgó con nitrógeno 3 veces. Se añadieron X-Phos (0,88 g, 1,85
mmol) y Pd2dba3 (0,845 g, 0,922 mmol). La mezcla se purgó con nitrógeno 3 B es y a continuación se calentó a 80 °C
durante 3 h. Según el análisis mediante HPLC, la reacción había finalizado. Se añadió lentamente HCl al 37 % (10
mL) durante 20 min. Se añadieron etanol (100 mL) y H2O (200 mL) y la mezcla de reacción se calentó hasta 75-80 °C durante 16 horas. La mezcla de reacción se enfrió hasta 50-60 °C y los sólidos negros insolubles se filtraron. El filtrado
se enfrió hasta 30 °C y se añadió NaOH 2 N (50 mL) para basificar la solución hasta pH 8-9. El precipitado resultante
se agitó durante 30 min, a continuación se filtró y se secó al vacío a 50 °C para obtener 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol como un sólido amarillo. HRMS m/z 394,2239 [M+H]; 1H-RMN:
(DMSO-d6 , 400 MHz) 5 13,2 (a, 1H), 13,0 (a, 1H), 8,44 (d, J= 8 Hz, 1H), 8,14 (a, 2H), 7,93 (d, J= 8 Hz, 1H), 7,39 (d,
J= 12 Hz, 1H), 7,24 (d, J= 8 Hz, 2H), 5,64 (m, 1H), 2,10 (dd, Ja= 4 Hz, Jb= 12 Hz, 2H), 1,26-1,30 (m, J= 8 Hz, 2H), 1,23
(s, 6H), 1,10 (s, 6H). 13C-RMN: (DMSO-d6 , 100 MHz) 5 162,80, 158,55, 155,97, 136,13, 128,30, 127,71, 120,42, 120,01, 116,26, 115,13, 113,44, 71,32, 50,99, 43,20, 34,33, 29,14.
Paso 4: Preparación de clorhidrato de 5-(1H-pirazol-4-il)-2-(6-((2,2,6,6-tetrametilpiperidin-4-il)oxi)piridazin-3-il)fenol
A un matraz de 2 L se añadieron 5-(1H-p¡razol-4-¡l)-2-(6-((2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡)p¡r¡daz¡n-3-¡l)fenol (50 g, 127 mmol), HCl al 37 % (21 mL, 254 mmol), H2O (1 L) y EtOH (1 L). Tamb¡én se añad¡eron SMOPEX-234 (10 g, captador de Pd) y carbón act¡vo (10 g) La mezcla se calentó a reflujo (78 °C) durante 3 horas. Se perm¡t¡ó que la mezcla negra resultante se enfr¡ara hasta 60 °C y los agentes captadores de Pd se¡s separaron por f¡ltrac¡ón a 50-60 °C. El f¡ltrado se enfr¡ó hasta 15 °C gradualmente durante 1 h y se formó un prec¡p¡tado amar¡llo pál¡do. Después de 2 h, el sól¡do se recog¡ó por f¡ltrac¡ón, se lavó con EtOH (50 mL) y se secó al vacío a 50 °C para obtener clorh¡drato de 5-(1H-p¡razol-4-¡l)-2-(6-((2,2,6,6-tetramet¡lp¡per¡d¡n-4-¡l)ox¡)p¡r¡daz¡n-3-¡l)fenol (27,2 g, 63 mmol). HRMS m/z 394,2222 [M+H]; 1H-RMN: (DMSO-d6 , 400 MHz) 5 13,09 (a, 2H), 9,41 (d, J=12 Hz, 1H), 8,61 (d, J= 12 Hz, 1H), 8,49 (d, J= Hz, 1H), 8,15 (a, 2H), 7,95 (d, J= 8 Hz, 1H), 7,46 (d, J= 8 Hz, 1H), 7,26 (d, J= 8 Hz, 2H), 5,72 (m, 1H), 2,33 (dd, Ja= 3,2 Hz, Jb= 13,2 Hz, 2H), 1,86-1,92 (m, Ja= 8 Hz, 2H), 1,23 (s, 6H), 1,10 (s, 6H); 13C-RMN: (DMSO-d6 , 100 MHz) 5162,34, 158,51, 156,41, 136,29, 128,72, 127,94, 120,40, 119,95, 116,37, 115,18, 113,45, 67,94, 56,69, 29,23, 25,10; XRPD: 13,47505, 14,29462, 14,99017, 16,55045, 17,60726, 19,69314, 21,89296, 23,89703, 25,82989, 27,13969, 28,47844, 36,94252, 43,77528.
Cond¡c¡ones de LCMS:
Condición A:
Columna: Acqu¡ty BEH 1,7 mm 2,1 x 50 mm a 50 °C.
S¡stema neutro; Grad¡ente: de un 2 a un 98 % en 4,4 m¡n - flujo de 1 mL/m¡n; Eluyente A: agua acetato de amonio 3,75 mM 2 % de ACN; Eluyente B: aceton¡tr¡lo acetato de amon¡o 7,5 mM;
Condición B:
Columna: INERTSIL C8-3, 3 |jm x 33 mm x 3,0 mm a 40 °C
Tasa de flujo: 2 mL/m¡n
Fase móv¡l: A) HCOONH4 acuoso 5 mM, B) MeOH/CHaCN (1/1, v/v)
Grad¡ente: grad¡ente l¡neal desde un 5 % de A hasta un 95 % de B en 2 m¡n
Condición Q:
S¡stema de UPLC Acqu¡ty de Waters
BEH UPLC Acqu¡ty C18 1,7 um, 2,1 x 30 mm (parte n.°: 186002349) de Waters
Tasa de flujo: 1 mL/m¡n
Temperatura: 55 °C (temp. columna)
Compos¡c¡ones de las fases móv¡les:
A. 0,05 % de ác¡do fórm¡co en agua
B. 0,04 % de ác¡do fórm¡co en metanol
r n :

Abreviaturas:


Ejemplo biológico 1:
Se utilizó un ELISA SMN celular para medir los efectos de compuestos de bajo peso molecular sobre la elevación de la proteína SMN. Se sembraron células de una línea celular de mioblastos procedente del modelo en ratón SMNdelta7 ( cedido amablemente por Steve Burden, NYU) en una placa de 384 pocillos con una densidad de 3000 células/pocillo y se trataron con los compuestos durante 24 horas. Si prepararon las placas ELISA de captura recubriendo placas de 384 pocillos (Immulon 4HBX) con 0,5 ug/mL de mAb anti-SMN (BD Science, número de catálogo 610647) a 4 °C
durante toda la noche. Las placas se lavaron 5 veces con 110 uL de PBS-Tween (Tween-20 al 0,05 %, PBST), se bloquearon con 100 uL de BSA al 1 % en PBST durante 2 horas y se lavaron (5 veces) con 100 uL de PBST. Después de 24 horas de tratamiento con el compuesto las células se lisaron en un tampón RIPA modificado, en hielo durante 1 hora. A continuación, se añadieron 20 uL de lisado y 20 uL de BSA al 1 % a las placas ELISA de captura y se incubaron a 4 °C durante toda la noche. Las placas se lavaron (5 veces) con PBST y a continuación se incubaron con una disolución 1:100 de anticuerpo policlonal de conejo primario anti-SMN (Santa Cruz, número de catálogo SC-15320) a temperatura ambiente durante 1 hora y se lavaron posteriormente (5 veces) con 110 uL de PBST. Después de esto se añadió 1:100 el anticuerpo secundario de cabra anti-IgG de conejo-ligado a HRP (Cell Signaling, número de catálogo 7074). A continuación, las placas se lavaron con PBST y se incubaron con 40 uL de sustrato TMB (Cell Signaling, número de catálogo 7004L) a temperatura ambiente durante 1-10 minutos con agitación. La reacción se detuvo mediante la adición de 40 uL de solución de parada (Cell signaling, número de catálogo 7002L) y la absorción se midió 450 nm. Los datos se presentan como factor de activación respecto al control de DMSO y CE50.
Condición 1 del ensayo ELISA: intervalo de concentración del compuesto 20 nM - 10 uM; condición 2 del ensayo ELISA: concentración del compuesto 100 μM - 10 uM.
T l ivi : D n r n l E m l i l i 1 iliz n l n i i n 1 2 ELI A.



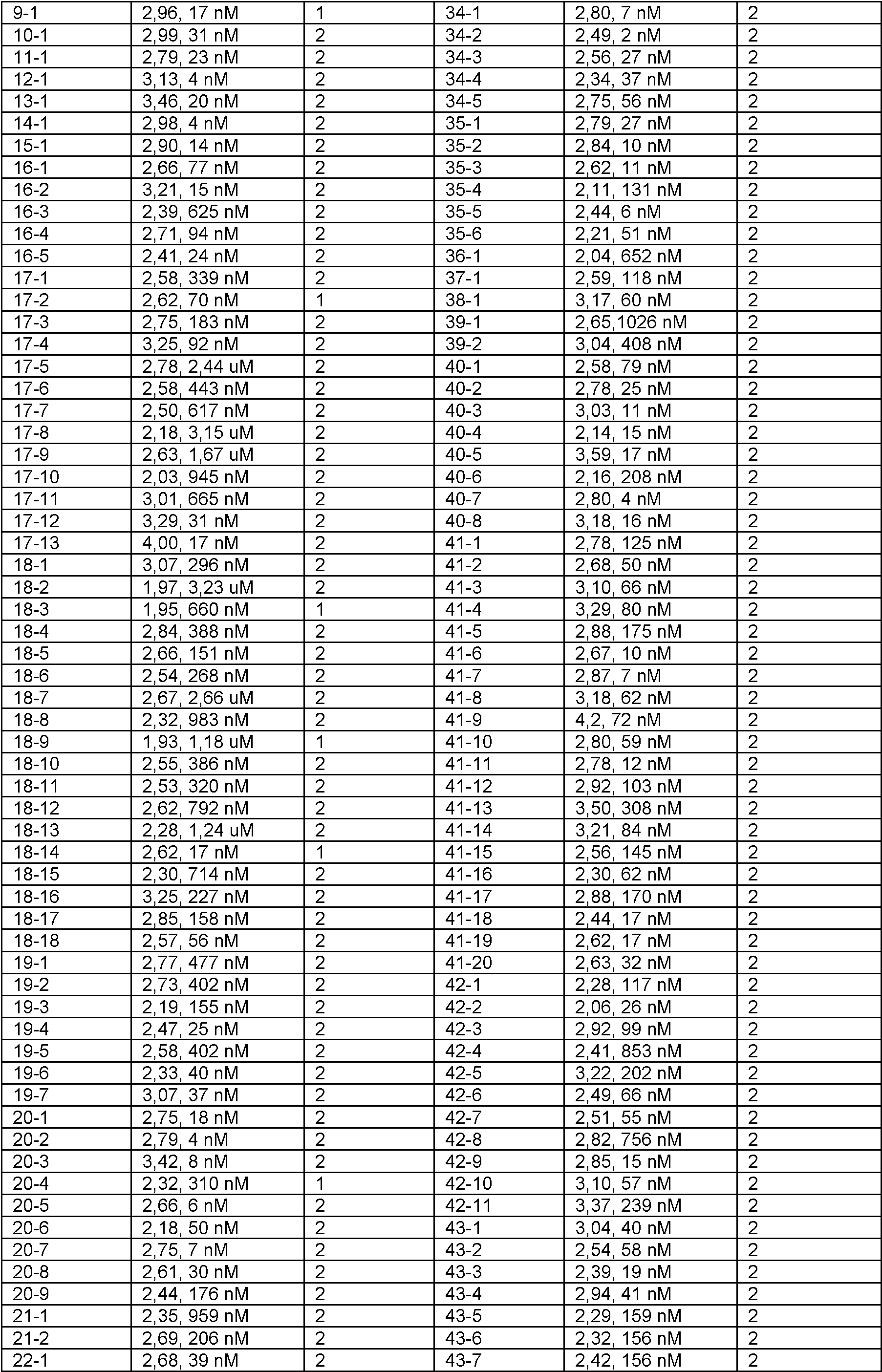
Claims (13)
1. Un compuesto o sal de este de acuerdo con la Fórmula (I)

A es 2-hidroxifenilo que está sustituido con 0, 1, 2 o 3 sustituyentes seleccionados independientemente entre alquilo C1-C4, haloalquilo C1-C4 alcoxi C1-C4, hidroxi, ciano, halógeno, amino, mono- y di-alquil C1-C4amino, heteroarilo y alquilo C1-C4 sustituido con hidroxi o amino, donde el heteroarilo tiene 5 o 6 átomos anulares, 1 o 2 heteroátomos anulares seleccionados entre N, O y S y está sustituido con 0, 1 o 2 sustituyentes seleccionados independientemente entre alquilo C1-C4, mono- y di-alquil C1-C4amino, hidroxialquil C1-C4amino, hidroxialquil C1-C4alquilo, heterociclo de 4-7 miembros-alquilo C1-C4, aminoalquilo C1-C4 y mono- y di-alquil C1-C4aminoalquilo C1-C4.
B se selecciona del grupo que consiste en

R17 es hidrógeno o metilo.
2. Un compuesto de acuerdo con la reivindicación 1 o una sal de este, cuyo compuesto está representado por la

donde R16 es un heteroarilo de 5 miembros que tiene un átomo de nitrógeno anular y 0 o 1 heteroátomos anulares adicionales seleccionados entre N, O o S, donde el heteroarilo está sustituido opcionalmente con alquilo C1-C4.
3. El compuesto, o sal de este, de acuerdo con la reivindicación 1, donde X es -O-:
4. Un compuesto o sal de este, de acuerdo con la reivindicación 2, donde R16 es:

5. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con una cualquiera de las reivindicaciones de 1 a 4, o una sal farmacéuticamente aceptable de este, y uno o más portadores farmacéuticamente aceptables.
6. Una composición que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con una cualquiera de las reivindicaciones de 1 a 4, o una sal farmacéuticamente aceptable de este, y uno o más coagentes terapéuticamente activos.
7. Un compuesto de acuerdo con una cualquiera de las reivindicaciones de 1 a 4, o una sal farmacéuticamente aceptable de este, para su uso como medicamento.
8. Un compuesto de acuerdo con una cualquiera de las reivindicaciones de 1 a 4, o una sal farmacéuticamente aceptable de este, para su uso en el tratamiento de una afección relacionada con la deficiencia de SMN.
9. Un compuesto para su uso de acuerdo con la reivindicación 8, o una sal farmacéuticamente aceptable de este, donde la afección relacionada con la deficiencia de SMN es atrofia muscular espinal.
10. El compuesto para su uso de acuerdo con la reivindicación 9, o sal farmacéuticamente aceptable de este, donde la atrofia muscular espinal es de tipo I (enfermedad de Werdnig-Hoffman).
11. El compuesto, o sal farmacéuticamente aceptable de este, donde la atrofia muscular espinal es de tipo II (forma intermedia crónica).
12. El compuesto para su uso de acuerdo con la reivindicación 9, o sal farmacéuticamente aceptable de este, donde la atrofia muscular espinal es de tipo III (enfermedad de Kugelberg-Welander o atrofia muscular espinal juvenil).
13. El compuesto para su uso de acuerdo con la reivindicación 9, o sal farmacéuticamente aceptable de este, donde la atrofia muscular espinal es de tipo IV con inicio en la edad adulta.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261682448P | 2012-08-13 | 2012-08-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2950450T3 true ES2950450T3 (es) | 2023-10-10 |
Family
ID=49029227
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13752768T Active ES2902198T3 (es) | 2012-08-13 | 2013-08-13 | Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN |
| ES18209821T Active ES2950450T3 (es) | 2012-08-13 | 2013-08-13 | Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13752768T Active ES2902198T3 (es) | 2012-08-13 | 2013-08-13 | Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN |
Country Status (40)
| Country | Link |
|---|---|
| US (6) | US8729263B2 (es) |
| EP (3) | EP4101849A1 (es) |
| JP (1) | JP6190883B2 (es) |
| KR (3) | KR20220029764A (es) |
| CN (1) | CN104583196B (es) |
| AP (1) | AP2015008242A0 (es) |
| AR (1) | AR092108A1 (es) |
| BR (1) | BR112015003004B1 (es) |
| CA (1) | CA2880273A1 (es) |
| CL (1) | CL2015000331A1 (es) |
| CR (1) | CR20150078A (es) |
| CU (1) | CU20150014A7 (es) |
| CY (1) | CY1124882T1 (es) |
| DK (1) | DK2885288T3 (es) |
| EA (1) | EA032005B1 (es) |
| EC (1) | ECSP15009862A (es) |
| ES (2) | ES2902198T3 (es) |
| GT (1) | GT201500030A (es) |
| HR (1) | HRP20211957T1 (es) |
| HU (1) | HUE057007T2 (es) |
| IL (1) | IL237067B (es) |
| JO (1) | JO3530B1 (es) |
| LT (1) | LT2885288T (es) |
| MA (1) | MA37834A1 (es) |
| MX (2) | MX375731B (es) |
| MY (2) | MY176488A (es) |
| NZ (1) | NZ704738A (es) |
| PE (2) | PE20151890A1 (es) |
| PH (1) | PH12015500236B1 (es) |
| PL (1) | PL2885288T3 (es) |
| PT (1) | PT2885288T (es) |
| RS (1) | RS62692B1 (es) |
| SG (2) | SG10201701123QA (es) |
| SI (1) | SI2885288T1 (es) |
| TN (1) | TN2015000038A1 (es) |
| TW (1) | TWI635083B (es) |
| UA (1) | UA114726C2 (es) |
| UY (1) | UY34974A (es) |
| WO (1) | WO2014028459A1 (es) |
| ZA (1) | ZA201500531B (es) |
Families Citing this family (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6069661B2 (ja) | 2010-06-24 | 2017-02-01 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 種々のアミロイドβペプチドアロフォームレベルの調節における組成物及びその使用 |
| US8729263B2 (en) | 2012-08-13 | 2014-05-20 | Novartis Ag | 1,4-disubstituted pyridazine analogs there of and methods for treating SMN-deficiency-related conditions |
| EP2968296B1 (en) | 2013-03-12 | 2020-09-02 | The Regents of the University of California | Gamma-secretase modulators |
| EP4105208A1 (en) * | 2013-07-31 | 2022-12-21 | Novartis AG | 1,4-disubstituted pyridazine derivatives and their use for treating smn-deficiency-related conditions |
| GB201410693D0 (en) | 2014-06-16 | 2014-07-30 | Univ Southampton | Splicing modulation |
| CN107109411B (zh) | 2014-10-03 | 2022-07-01 | 冷泉港实验室 | 核基因输出的定向增加 |
| US10472346B2 (en) | 2014-10-31 | 2019-11-12 | The General Hospital Corporation | Potent gamma-secretase modulators |
| EP3053577A1 (en) | 2015-02-09 | 2016-08-10 | F. Hoffmann-La Roche AG | Compounds for the treatment of cancer |
| EP3256126B1 (en) * | 2015-02-09 | 2024-03-27 | F. Hoffmann-La Roche AG | Compounds for the treatment of cancer |
| EP3359685B1 (en) | 2015-10-09 | 2026-01-28 | University Of Southampton | Modulation of gene expression for deregulated protein expression |
| EA201800367A1 (ru) * | 2015-12-10 | 2019-02-28 | ПиТиСи ТЕРАПЬЮТИКС, ИНК. | Способы лечения болезни хантингтона |
| US11096956B2 (en) | 2015-12-14 | 2021-08-24 | Stoke Therapeutics, Inc. | Antisense oligomers and uses thereof |
| EP3933041B1 (en) | 2015-12-14 | 2024-01-31 | Cold Spring Harbor Laboratory | Antisense oligomers for treatment of autosomal dominant retardation |
| MX2018009325A (es) * | 2016-02-01 | 2019-05-15 | Arrakis Therapeutics Inc | Compuestos y metodos para tratar enfermedades mediadas por el acido ribonucleico (arn). |
| MX2018010109A (es) * | 2016-02-23 | 2018-12-17 | Univ Indiana Res & Tech Corp | Terapias de combinacion para tratamiento de atrofia muscular espinal. |
| EP3634953B1 (en) | 2017-06-05 | 2024-01-03 | PTC Therapeutics, Inc. | Compounds for treating huntington's disease |
| JP2020523365A (ja) | 2017-06-14 | 2020-08-06 | ピーティーシー セラピューティクス,インコーポレーテッド | Rnaスプライシングを改変する方法 |
| CN111182898B (zh) | 2017-06-28 | 2024-04-16 | Ptc医疗公司 | 用于治疗亨廷顿氏病的方法 |
| MX2019015580A (es) | 2017-06-28 | 2020-07-28 | Ptc Therapeutics Inc | Metodos para tratar la enfermedad de huntington. |
| JP7312749B2 (ja) | 2017-08-04 | 2023-07-21 | スカイホーク・セラピューティクス・インコーポレーテッド | スプライシングをモジュレートする方法および組成物 |
| SG11202001590RA (en) | 2017-08-25 | 2020-03-30 | Stoke Therapeutics Inc | Antisense oligomers for treatment of conditions and diseases |
| GB2610100B (en) | 2017-10-23 | 2023-08-16 | Stoke Therapeutics Inc | Antisense oligomers for treatment of non-sense mediated RNA decay based conditions and diseases |
| RU2020112854A (ru) | 2017-11-30 | 2021-12-30 | Арракис Терапьютикс, Инк. | Фотозонды, связывающие нуклеиновые кислоты, и способы их применения |
| EP3728228A1 (en) | 2017-12-22 | 2020-10-28 | Ravenna Pharmaceuticals, Inc. | Aminopyridine derivatives as phosphatidylinositol phosphate kinase inhibitors |
| EA202092001A1 (ru) | 2018-03-27 | 2021-01-29 | ПиТиСи ТЕРАПЬЮТИКС, ИНК. | Соединения для лечения болезни гентингтона |
| JP2021521200A (ja) | 2018-04-10 | 2021-08-26 | スカイホーク・セラピューティクス・インコーポレーテッド | 癌の処置のための化合物 |
| US12060558B2 (en) | 2018-05-04 | 2024-08-13 | Stoke Therapeutics, Inc. | Methods and compositions for treatment of cholesteryl ester storage disease |
| HRP20240935T1 (hr) | 2018-06-27 | 2024-11-22 | Ptc Therapeutics, Inc. | Heterociklični i heteroaril spojevi za liječenje huntingtonove bolesti |
| WO2020005877A1 (en) | 2018-06-27 | 2020-01-02 | Ptc Therapeutics, Inc. | Heteroaryl compounds for treating huntington's disease |
| EP3814360B8 (en) | 2018-06-27 | 2024-11-06 | PTC Therapeutics, Inc. | Heteroaryl compounds for treating huntington's disease |
| WO2020051282A1 (en) | 2018-09-07 | 2020-03-12 | Teva Pharmaceuticals International Gmbh | Solid state forms of branaplam and their preparation |
| AU2019404930A1 (en) | 2018-12-21 | 2021-06-03 | Novartis Ag | Oral formulations of branaplam |
| JP7551629B2 (ja) | 2019-02-05 | 2024-09-17 | スカイホーク・セラピューティクス・インコーポレーテッド | スプライシングを調節するための方法および組成物 |
| WO2020163405A1 (en) * | 2019-02-05 | 2020-08-13 | Skyhawk Therapeutics, Inc. | Methods and compositions for modulating splicing |
| JP2022519311A (ja) * | 2019-02-05 | 2022-03-22 | スカイホーク・セラピューティクス・インコーポレーテッド | スプライシングを調節するための方法および組成物 |
| JP7603594B2 (ja) * | 2019-02-06 | 2024-12-20 | スカイホーク・セラピューティクス・インコーポレーテッド | スプライシングを調節するための方法および組成物 |
| JP7603595B2 (ja) * | 2019-02-06 | 2024-12-20 | スカイホーク・セラピューティクス・インコーポレーテッド | スプライシングを調節するための方法および組成物 |
| KR20210134003A (ko) | 2019-02-27 | 2021-11-08 | 스톡 테라퓨틱스, 인크. | 병태 및 질환의 치료를 위한 안티센스 올리고머 |
| WO2020190793A1 (en) * | 2019-03-15 | 2020-09-24 | Skyhawk Therapeutics, Inc. | Compositions and methods for correction of aberrant splicing |
| JP7649257B2 (ja) | 2019-05-13 | 2025-03-19 | ピーティーシー セラピューティクス, インコーポレイテッド | ハンチントン病を処置するための化合物 |
| UY38687A (es) * | 2019-05-17 | 2023-05-15 | Novartis Ag | Inhibidores del inflamasoma nlrp3, composiciones, combinaciones de los mismos y métodos para su uso |
| TW202112767A (zh) | 2019-06-17 | 2021-04-01 | 美商佩特拉製藥公司 | 作為磷脂酸肌醇磷酸激酶抑制劑之胺基吡啶衍生物 |
| WO2021014428A1 (en) | 2019-07-25 | 2021-01-28 | Novartis Ag | Regulatable expression systems |
| TW202131920A (zh) | 2019-11-01 | 2021-09-01 | 瑞士商諾華公司 | 剪接調節子用於減慢杭丁頓氏舞蹈症進展的治療之用途 |
| WO2021174176A1 (en) | 2020-02-28 | 2021-09-02 | Remix Therapeutics Inc. | Pyridazine dervatives for modulating nucleic acid splicing |
| MX2022010681A (es) | 2020-02-28 | 2023-03-21 | Remix Therapeutics Inc | Compuestos y metodos para modular el empalme. |
| WO2021174174A1 (en) | 2020-02-28 | 2021-09-02 | Remix Therapeutics Inc. | Thiophenyl derivatives useful for modulating nucleic acid splicing |
| CN115485025A (zh) | 2020-02-28 | 2022-12-16 | 雷密克斯医疗公司 | 用于调节剪接的化合物和方法 |
| WO2021207530A1 (en) | 2020-04-08 | 2021-10-14 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| MX2022012678A (es) | 2020-04-08 | 2023-01-11 | Remix Therapeutics Inc | Compuestos y metodos para modular el corte y empalme. |
| CN115768762A (zh) * | 2020-04-09 | 2023-03-07 | Ptc医疗公司 | 用于治疗亨廷顿氏病的化合物 |
| BR112022022889A2 (pt) | 2020-05-11 | 2023-04-04 | Stoke Therapeutics Inc | Oligômeros antissentido de opa1 para tratamento de condições e doenças |
| CA3182912A1 (en) | 2020-05-13 | 2021-11-18 | Chdi Foundation, Inc. | Htt modulators for treating huntington's disease |
| TW202216671A (zh) * | 2020-06-25 | 2022-05-01 | 瑞士商諾華公司 | 1,4—二取代的嗒𠯤化合物之製造方法 |
| MX2023000167A (es) | 2020-07-02 | 2023-05-03 | Remix Therapeutics Inc | Derivados de 5-[5-(piperidin-4-il)tieno[3,2-c]pirazol-2-il]indazol y compuestos relacionados como moduladores para el corte y empalme de ácidos nucleicos y para el tratamiento de enfermedades proliferativas. |
| TW202216710A (zh) | 2020-07-02 | 2022-05-01 | 美商雷密克斯醫療公司 | 調節剪接之化合物及方法 |
| JP2023535832A (ja) * | 2020-07-31 | 2023-08-21 | ラクティゲン セラピューティクス | Sarnaとmrna調節剤によるsmaの併用治療 |
| TW202304446A (zh) | 2021-03-29 | 2023-02-01 | 瑞士商諾華公司 | 剪接調節子用於減慢杭丁頓氏舞蹈症進展的治療之用途 |
| US20240400584A1 (en) | 2021-08-30 | 2024-12-05 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| US20240368163A1 (en) | 2021-08-30 | 2024-11-07 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| US20250333397A1 (en) | 2021-08-30 | 2025-10-30 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| WO2023034812A1 (en) | 2021-08-30 | 2023-03-09 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| CA3230256A1 (en) | 2021-08-30 | 2023-03-09 | Dominic Reynolds | Compounds and methods for modulating splicing |
| TW202330552A (zh) | 2021-10-13 | 2023-08-01 | 美商雷密克斯醫療公司 | 調節剪接之化合物及方法 |
| IL312078A (en) | 2021-10-13 | 2024-06-01 | Remix Therapeutics Inc | Compounds and methods for modulating nucleic acid splicing |
| MX2024005933A (es) | 2021-11-17 | 2024-08-06 | Chdi Foundation Inc | Moduladores de htt pata tratar la enfermedad de huntington. |
| AU2022389987A1 (en) * | 2021-11-22 | 2024-06-06 | Rgenta Therapeutics, Inc. | Heterocyclic substituted 1,3,4-thiadiazole and pyridazine compounds and methods of using the same |
| WO2023133229A2 (en) | 2022-01-05 | 2023-07-13 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| WO2023133217A1 (en) | 2022-01-05 | 2023-07-13 | Remix Therapeutics Inc. | 2-(indazol-5-yl)-6-(piperidin-4-yl)-1,7-naphthyridine derivatives and related compounds as modulators for splicing nucleic acids and for the treatment of proliferative diseases |
| TW202346305A (zh) | 2022-01-05 | 2023-12-01 | 美商雷密克斯醫療公司 | 用於調節剪切之化合物及方法 |
| CN118580191A (zh) * | 2022-01-28 | 2024-09-03 | 和记黄埔医药(上海)有限公司 | 用于制备咪唑并[1,2-b]哒嗪类化合物的中间体 |
| UY40374A (es) | 2022-08-03 | 2024-02-15 | Novartis Ag | Inhibidores de inflamasoma nlrp3 |
| WO2025247338A1 (zh) * | 2024-05-30 | 2025-12-04 | 纽欧申医药(上海)有限公司 | 哒嗪环类化合物、其药物组合物及其应用 |
Family Cites Families (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2516040C2 (de) | 1974-06-10 | 1984-12-20 | Dr. Karl Thomae Gmbh, 7950 Biberach | Benzimidazole, Verfahren zu deren Herstellung und diese enthaltende Arzneimittel |
| DE2427943C2 (de) | 1974-06-10 | 1984-08-02 | Dr. Karl Thomae Gmbh, 7950 Biberach | Benzimidazole, Verfahren zu deren Herstellung und diese enthaltende Arzneimittel |
| BR9408137A (pt) | 1993-11-24 | 1997-08-12 | Du Pont Merck Pharma | Composto éster de prodroga método de tratamento composição farmacêutica e método de inibição |
| HRP20020737A2 (en) | 2000-02-11 | 2005-10-31 | Vertex Pharmaceuticals Incorporated | Piperazine and piperidine derivatives for treatment or prevention of nruronal damage |
| EP1133993A1 (en) | 2000-03-10 | 2001-09-19 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Substances for the treatment of spinal muscular atrophy |
| EP1289993B9 (en) | 2000-06-07 | 2008-08-20 | Vertex Pharmaceuticals Incorporated | Caspase inhibitors and uses thereof |
| US6376508B1 (en) | 2000-12-13 | 2002-04-23 | Academia Sinica | Treatments for spinal muscular atrophy |
| WO2004078163A2 (en) | 2003-02-28 | 2004-09-16 | Transform Pharmaceuticals, Inc. | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| US6982259B2 (en) | 2002-04-30 | 2006-01-03 | Schering Aktiengesellschaft | N-heterocyclic derivatives as NOS inhibitors |
| IL166510A0 (en) | 2002-08-09 | 2006-01-15 | Nps Pharma Inc | 1,2,4"oxadiazole as modulators of metabotropic glutamate receptor-5 |
| MXPA05001592A (es) | 2002-08-09 | 2005-05-05 | Astrazeneca Ab | Oxadiazoles como moduladores de receptor-5 de glutamato metabotropico. |
| AU2003264018A1 (en) | 2002-08-09 | 2004-02-25 | Astrazeneca Ab | Compounds having an activity at metabotropic glutamate receptors |
| DK1562945T3 (da) * | 2002-11-11 | 2007-03-05 | Neurosearch As | Hidtil ukendte diazebicykliske biarylderivater |
| US20050075375A1 (en) | 2003-05-14 | 2005-04-07 | Anadys Pharmaceuticals, Inc. | Heterocyclic compounds for treating hepatitis C virus |
| EP1642898B1 (en) | 2003-06-27 | 2013-03-27 | Msd K.K. | Heteroaryloxy nitrogenous saturated heterocyclic derivative |
| US7666862B2 (en) | 2003-08-15 | 2010-02-23 | Merck & Co., Inc. | Mitotic Kinesin Inhibitors |
| US7399765B2 (en) * | 2003-09-19 | 2008-07-15 | Abbott Laboratories | Substituted diazabicycloalkane derivatives |
| US7655657B2 (en) * | 2003-12-22 | 2010-02-02 | Abbott Laboratories | Fused bicycloheterocycle substituted quinuclidine derivatives |
| WO2005077368A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077373A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| FR2868780B1 (fr) | 2004-04-13 | 2008-10-17 | Sanofi Synthelabo | Derives de la 1-amino-phthalazine, leur preparation et leur application en therapeutique |
| WO2006001958A2 (en) | 2004-05-21 | 2006-01-05 | Merck & Co., Inc. | Amino cyclopentyl heterocyclic and carbocyclic modulators of chemokine receptor activity |
| CA2571360C (en) | 2004-06-18 | 2014-11-25 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines useful in inducing cytokine biosynthesis in animals |
| US7829712B2 (en) | 2004-09-20 | 2010-11-09 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-CoA-desaturase |
| PL1637141T3 (pl) | 2004-09-21 | 2012-04-30 | Trobio Ab | Stabilizowana kompozycja proteazy zawierająca proteazę serynową, pochodne morfoliny i odwracalne inhibitory tej proteazy serynowej |
| EA012181B1 (ru) | 2004-10-18 | 2009-08-28 | Амген, Инк. | Соединения тиадиазола и их применение |
| EP1805183B1 (en) * | 2004-10-20 | 2010-12-29 | NeuroSearch A/S | Novel diazabicyclic aryl derivatives and their medical use |
| RU2499795C2 (ru) | 2005-07-04 | 2013-11-27 | Хай Пойнт Фармасьютикалс, ЛЛС | Антагонисты гистаминовых н3-рецепторов |
| CA2632642A1 (en) * | 2005-12-06 | 2007-06-14 | Neurosearch A/S | Novel diazabicyclic aryl derivatives and their medical use |
| TW200813051A (en) | 2006-05-08 | 2008-03-16 | Neurogen Corp | Substituted azaspiro derivatives |
| US20080247964A1 (en) * | 2006-05-08 | 2008-10-09 | Yuelian Xu | Substituted azaspiro derivatives |
| UA92083C2 (ru) * | 2006-05-19 | 2010-09-27 | Эбботт Леборетриз | Азабициклические алкановые производные, замещенные конденсированным бициклогетероциклом |
| WO2008058064A1 (en) | 2006-11-07 | 2008-05-15 | Lexicon Pharmaceuticals, Inc. | Amine-linked multicyclic compounds as inhibitors of the proline transporter |
| GB0704394D0 (en) | 2007-03-07 | 2007-04-11 | Senexis Ltd | Compounds |
| EP2014656A3 (en) * | 2007-06-11 | 2011-08-24 | High Point Pharmaceuticals, LLC | New heteocyclic h3 antagonists |
| BRPI0820171B8 (pt) | 2007-11-16 | 2021-05-25 | Rigel Pharmaceuticals Inc | compostos de carboxamida, sulfonamida e amina para distúrbios metabólicos, composição farmacêutica, e, uso dos mesmos |
| NZ720282A (en) | 2008-02-28 | 2017-12-22 | Vertex Pharma | Heteroaryl derivatives as cftr modulators |
| WO2009137503A1 (en) | 2008-05-05 | 2009-11-12 | Envivo Pharmaceuticals, Inc. | Hdac inhibitors and uses thereof |
| US20110142758A1 (en) | 2008-06-10 | 2011-06-16 | Neurosearch A/S | Indolyl-pyridazinyl-diazabicyclononane derivatives in labelled and unlabelled form and their use in diagnostic methods |
| GB0810902D0 (en) | 2008-06-13 | 2008-07-23 | Astex Therapeutics Ltd | New compounds |
| MX2010013876A (es) | 2008-06-20 | 2011-03-04 | Metabolex Inc | Agonistas de arilo grpr119 y sus usos . |
| EP2330894B8 (en) | 2008-09-03 | 2017-04-19 | BioMarin Pharmaceutical Inc. | Compositions including 6-aminohexanoic acid derivatives as hdac inhibitors |
| WO2010045306A2 (en) | 2008-10-16 | 2010-04-22 | Schering Corporation | Azine derivatives and methods of use thereof |
| KR101705158B1 (ko) | 2009-05-05 | 2017-02-09 | 다나-파버 캔서 인스티튜트 인크. | Egfr 억제제 및 질환 치료방법 |
| EP2475667A1 (en) | 2009-09-10 | 2012-07-18 | F. Hoffmann-La Roche AG | Inhibitors of jak |
| JO3250B1 (ar) | 2009-09-22 | 2018-09-16 | Novartis Ag | إستعمال منشطات مستقبل نيكوتينيك أسيتيل كولين ألفا 7 |
| US8673928B2 (en) | 2009-11-18 | 2014-03-18 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2011078143A1 (ja) | 2009-12-22 | 2011-06-30 | 塩野義製薬株式会社 | ピリミジン誘導体およびそれらを含有する医薬組成物 |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011130515A1 (en) * | 2010-04-14 | 2011-10-20 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Arylthiazolyl piperidines and related compounds as modulators of survival motor neuron (smn) protein production |
| AR081626A1 (es) * | 2010-04-23 | 2012-10-10 | Cytokinetics Inc | Compuestos amino-piridazinicos, composiciones farmaceuticas que los contienen y uso de los mismos para tratar trastornos musculares cardiacos y esqueleticos |
| WO2012022467A2 (en) | 2010-08-16 | 2012-02-23 | Santhera Pharmaceuticals (Schweiz) Ag | Novel benzoquinone derivatives and use thereof as modulators of mitochondrial function |
| WO2012127393A1 (en) | 2011-03-18 | 2012-09-27 | Novartis Ag | COMBINATIONS OF ALPHA 7 NICOTINIC ACETYLCHOLINE RECEPTOR ACTIVATORS AND mGluR5 ANTAGONISTS FOR USE IN DOPAMINE INDUCED DYSKINESIA IN PARKINSON'S DISEASE |
| UA113223C2 (xx) | 2012-08-13 | 2016-12-26 | Арилетинілпіримідини | |
| US8729263B2 (en) | 2012-08-13 | 2014-05-20 | Novartis Ag | 1,4-disubstituted pyridazine analogs there of and methods for treating SMN-deficiency-related conditions |
| US9604937B2 (en) | 2012-11-27 | 2017-03-28 | Thomas Helledays Stiftelse For Medicinsk Forskning | Pyrimidine-2,4-diamine derivatives for treatment of cancer |
| US9040712B2 (en) * | 2013-01-23 | 2015-05-26 | Novartis Ag | Thiadiazole analogs thereof and methods for treating SMN-deficiency-related-conditions |
| EP4105208A1 (en) | 2013-07-31 | 2022-12-21 | Novartis AG | 1,4-disubstituted pyridazine derivatives and their use for treating smn-deficiency-related conditions |
-
2013
- 2013-08-07 US US13/960,917 patent/US8729263B2/en active Active
- 2013-08-12 JO JOP/2013/0240A patent/JO3530B1/ar active
- 2013-08-12 TW TW102128893A patent/TWI635083B/zh not_active IP Right Cessation
- 2013-08-13 PT PT137527685T patent/PT2885288T/pt unknown
- 2013-08-13 EP EP21194692.6A patent/EP4101849A1/en active Pending
- 2013-08-13 EA EA201590371A patent/EA032005B1/ru not_active IP Right Cessation
- 2013-08-13 PE PE2015001214A patent/PE20151890A1/es unknown
- 2013-08-13 ES ES13752768T patent/ES2902198T3/es active Active
- 2013-08-13 MX MX2015001892A patent/MX375731B/es active IP Right Grant
- 2013-08-13 DK DK13752768.5T patent/DK2885288T3/da active
- 2013-08-13 SG SG10201701123QA patent/SG10201701123QA/en unknown
- 2013-08-13 CN CN201380042971.6A patent/CN104583196B/zh not_active Expired - Fee Related
- 2013-08-13 BR BR112015003004-1A patent/BR112015003004B1/pt not_active IP Right Cessation
- 2013-08-13 EP EP13752768.5A patent/EP2885288B1/en active Active
- 2013-08-13 MY MYPI2017000158A patent/MY176488A/en unknown
- 2013-08-13 MA MA37834A patent/MA37834A1/fr unknown
- 2013-08-13 UA UAA201500562A patent/UA114726C2/uk unknown
- 2013-08-13 SG SG11201500507UA patent/SG11201500507UA/en unknown
- 2013-08-13 JP JP2015527532A patent/JP6190883B2/ja not_active Expired - Fee Related
- 2013-08-13 WO PCT/US2013/054687 patent/WO2014028459A1/en not_active Ceased
- 2013-08-13 NZ NZ704738A patent/NZ704738A/en not_active IP Right Cessation
- 2013-08-13 HR HRP20211957TT patent/HRP20211957T1/hr unknown
- 2013-08-13 AR ARP130102863A patent/AR092108A1/es active IP Right Grant
- 2013-08-13 EP EP18209821.0A patent/EP3564233B1/en active Active
- 2013-08-13 UY UY0001034974A patent/UY34974A/es active IP Right Grant
- 2013-08-13 CA CA2880273A patent/CA2880273A1/en active Pending
- 2013-08-13 RS RS20211507A patent/RS62692B1/sr unknown
- 2013-08-13 MY MYPI2015000174A patent/MY174339A/en unknown
- 2013-08-13 KR KR1020227006110A patent/KR20220029764A/ko not_active Ceased
- 2013-08-13 HU HUE13752768A patent/HUE057007T2/hu unknown
- 2013-08-13 PL PL13752768T patent/PL2885288T3/pl unknown
- 2013-08-13 KR KR1020157006144A patent/KR102172911B1/ko not_active Expired - Fee Related
- 2013-08-13 KR KR1020207030949A patent/KR102368439B1/ko not_active Expired - Fee Related
- 2013-08-13 LT LTEPPCT/US2013/054687T patent/LT2885288T/lt unknown
- 2013-08-13 ES ES18209821T patent/ES2950450T3/es active Active
- 2013-08-13 PE PE2020000186A patent/PE20201165A1/es unknown
- 2013-08-13 SI SI201331954T patent/SI2885288T1/sl unknown
- 2013-08-13 AP AP2015008242A patent/AP2015008242A0/xx unknown
-
2014
- 2014-04-01 US US14/242,282 patent/US9545404B2/en active Active
-
2015
- 2015-01-23 ZA ZA2015/00531A patent/ZA201500531B/en unknown
- 2015-01-30 TN TNP2015000038A patent/TN2015000038A1/fr unknown
- 2015-02-02 IL IL237067A patent/IL237067B/en active IP Right Grant
- 2015-02-03 PH PH12015500236A patent/PH12015500236B1/en unknown
- 2015-02-11 MX MX2020010151A patent/MX2020010151A/es unknown
- 2015-02-11 CL CL2015000331A patent/CL2015000331A1/es unknown
- 2015-02-13 CU CUP2015000014A patent/CU20150014A7/es unknown
- 2015-02-13 GT GT201500030A patent/GT201500030A/es unknown
- 2015-02-13 CR CR20150078A patent/CR20150078A/es unknown
- 2015-03-13 EC ECIEPI20159862A patent/ECSP15009862A/es unknown
-
2016
- 2016-11-29 US US15/364,248 patent/US10195196B2/en active Active
-
2019
- 2019-01-02 US US16/237,907 patent/US10758533B2/en active Active
-
2020
- 2020-07-21 US US16/934,787 patent/US11229648B2/en active Active
-
2021
- 2021-12-13 US US17/644,045 patent/US20220387428A1/en not_active Abandoned
-
2022
- 2022-01-03 CY CY20221100002T patent/CY1124882T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2950450T3 (es) | Análogos de piridazina con disustitución 1,4 y métodos para tratar afecciones relacionadas con la deficiencia de SMN | |
| AU2016256728B2 (en) | Thiadiazole analogs thereof and methods for treating smn-deficiency-related-conditions | |
| AU2013302859B9 (en) | 1,4-disubstituted pyridazine analogs and methods for treating SMN-deficiency-related conditions | |
| HK40012123A (en) | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions | |
| EA040206B1 (ru) | Аналоги тиадиазола и способы лечения состояний, связанных с дефицитом smn | |
| HK1207862B (en) | 1,4-disubstituted pyridazine analogs and methods for treating smn-deficiency-related conditions |