ES2951064T3 - Procedimiento de preparación de 2-amino-5-hidroxipropiofenona - Google Patents
Procedimiento de preparación de 2-amino-5-hidroxipropiofenona Download PDFInfo
- Publication number
- ES2951064T3 ES2951064T3 ES20741510T ES20741510T ES2951064T3 ES 2951064 T3 ES2951064 T3 ES 2951064T3 ES 20741510 T ES20741510 T ES 20741510T ES 20741510 T ES20741510 T ES 20741510T ES 2951064 T3 ES2951064 T3 ES 2951064T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- acid
- chloride
- sodium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C221/00—Preparation of compounds containing amino groups and doubly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C225/00—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones
- C07C225/22—Compounds containing amino groups and doubly—bound oxygen atoms bound to the same carbon skeleton, at least one of the doubly—bound oxygen atoms not being part of a —CHO group, e.g. amino ketones having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/08—Preparation of nitro compounds by substitution of hydrogen atoms by nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/45—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by at least one doubly—bound oxygen atom, not being part of a —CHO group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C68/00—Preparation of esters of carbonic or haloformic acids
- C07C68/02—Preparation of esters of carbonic or haloformic acids from phosgene or haloformates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/96—Esters of carbonic or haloformic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La presente invención se refiere a un proceso para la preparación de 2-amino-5-hidroxipropiofenona, un intermedio clave para la síntesis de análogos de camptotecina, incluida la 7-etil-10-hidroxicamptotecina (SN-38). (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Procedimiento de preparación de 2-amino-5-hidroxipropiofenona
Campo de la invención
La presente invención se refiere en general a un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona, un intermedio clave en la síntesis de análogos de camptotecina que incluyen 7-etil-10-hidroxicamptotecina (SN-38).
Antecedentes de la invención
La 2-amino-5-hidroxipropiofenona es un intermedio importante útil en la preparación de 7-etil-10-hidroxicamptotecina (SN-38), que es un intermedio crucial en la preparación de una clase de agente antineoplásico irinotecán {(4S)-4,11-dietil-4-hidroxi-9-[(4-piperidinopiperidino)carboniloxi]-1 H-pirano[3',4':6,7]indolizino[1,2-b]quinolina-3,14(4H,12H)diona}.
El irinotecán se usa más comúnmente para tratar el carcinoma metastásico. Cuando se administra a un paciente, el irinotecán se metaboliza a un metabolito más activo, 7-etil-10-hidroxicamptotecina (SN-38). La fórmula estructural de estos compuestos se representa de la siguiente manera:
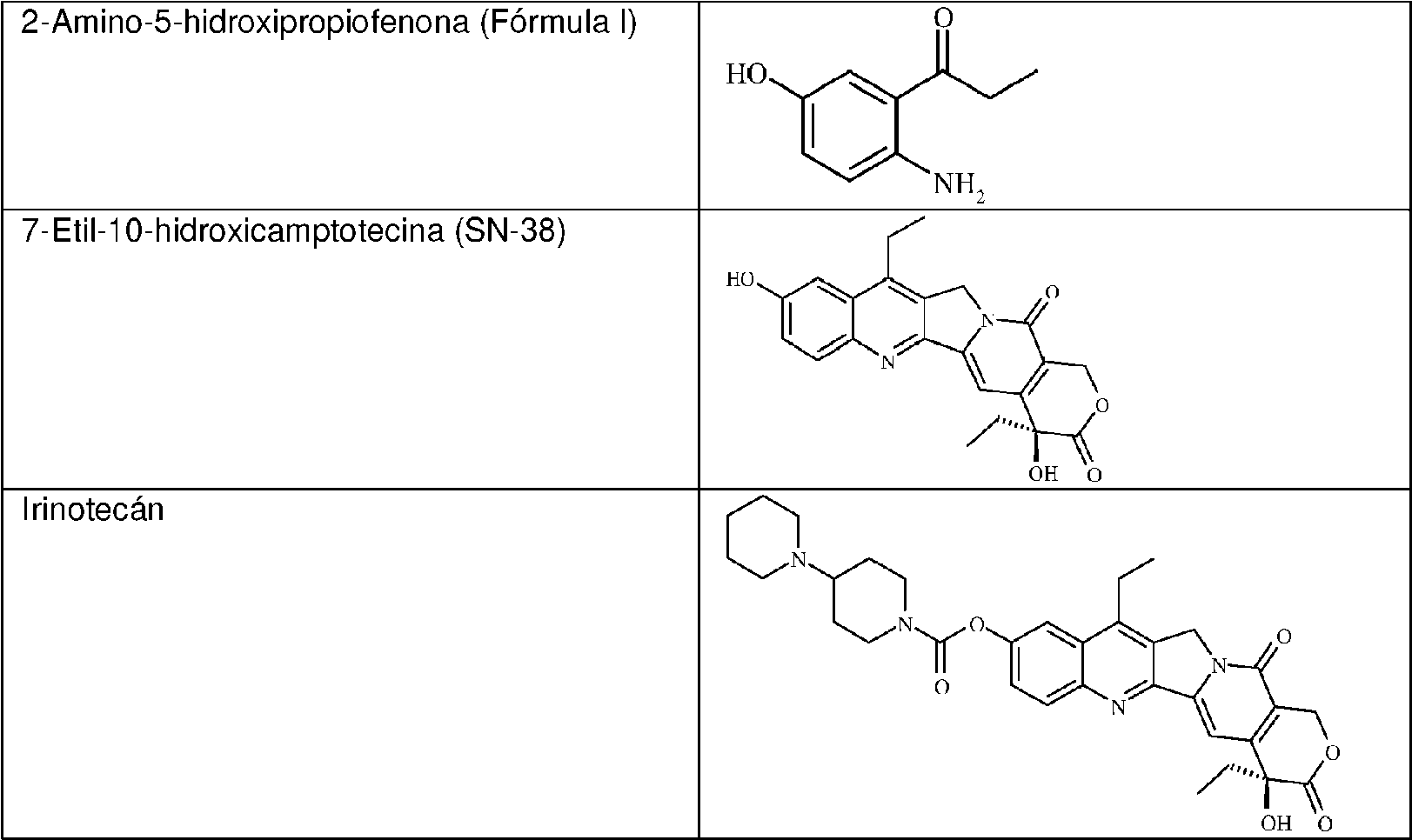
En la técnica se conocen varios métodos para la síntesis de 7-etil-10-hidroxicamptotecina (SN-38) y su posterior conversión en irinotecán. Uno de los mejores métodos conocidos hasta ahora es sintetizar los dos intermedios clave, que son 2-amino-5-hidroxipropiofenona de fórmula I y cetonas tricíclicas intermedias y la posterior condensación de estos dos compuestos mediante la reacción de Friedlander para dar 7-etil-10-hidroxicamptotecina (SN-38) y luego convertir SN-38 en irinotecán (Referencia: Patente de EE. UU. No. 6.121.451) y el procedimiento representado esquemáticamente de la siguiente manera:

La 2-amino-5-hidroxipropiofenona de Fórmula I es uno de los intermedios clave en la preparación de 7-etil-10-hidroxicamptotecina (SN-38). Hasta la fecha, muchas publicaciones han descrito la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I y todas estas referencias tienen varios inconvenientes, que incluyen una baja selectividad de reacción, implican reacciones peligrosas y obtienen menos rendimientos con baja pureza. La literatura reportada sobre la preparación del compuesto de Fórmula I se describe a continuación: Patente de EE. UU. No.
7.126.000 ("la patente '000") describe un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I a partir de 5-hidroxi-2-nitrobenzaldehído o de 5-benciloxi-2-nitrobenzaldehído. El procedimiento descrito en la patente '000 se representa esquemáticamente de la siguiente manera:

El procedimiento descrito bajo la patente '000 implica el uso de 5-hidroxi-2-nitrobenzaldehído o su derivado protegido como material de partida y la posterior conversión de este intermedio en el compuesto de Fórmula I implica muchas etapas de reacción tediosos, por ejemplo, el uso de reacciones de Grignard altamente sensibles y la reducción de la cadena de alqueno. El uso del compuesto aldehido como material de partida es considerablemente costoso y la formación de impurezas durante la etapa de la reacción de Grignard y la reducción final hace que el procedimiento sea tedioso y antieconómico.
La Patente de EE. UU. No. 7.608.740 ("la patente '740") describe un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I a partir de 3-fluoropropiofenona. El procedimiento descrito en la patente '740 se representa esquemáticamente como sigue:

El procedimiento descrito bajo la patente '740 implica principalmente la nitración de 5-fluoropropiofenona utilizando ácido nítrico. Mientras se realiza la reacción de nitración, siempre existe la posibilidad de sustitución del grupo nitro en las posiciones orto y/o para del anillo en lugar de un solo grupo para según sea necesario y esto conduce a la contaminación del producto con compuestos sustituidos en para y, por lo tanto, se requieren purificaciones cromatográficas engorrosas para purificar el producto.
La Patente China número CN101723841B ("la patente '841") describe un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I a partir de 3-cloropropiofenona. El procedimiento descrito en la patente '841 se representa esquemáticamente como sigue:

De nuevo, el procedimiento descrito en la publicación '841 adolece de la formación de regioisómeros/isómeros posicionales durante la reacción de nitración.
Xiao-Dong Xiong et. al. en Organic Preparations and Procedures International, 41, 423-427, 2009 describe un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I mediante la conversión del grupo aldehído en propiofenona. Este procedimiento tiene desventajas como que el aldehído intermedio en sí mismo es más costoso e implica varias etapas para convertirlo en un grupo propionilo. El procedimiento descrito por Xiao-Dong Xiong et. al. se representa esquemáticamente como sigue:

Aurore et al. en Bioorganic & Medicinal Chemistry Letters Volumen 14, Número 9, páginas 2363-2365, 2004 (la referencia 'Aurore) describe un procedimiento para la preparación de 2-Amino-5-hidroxipropiofenona de Fórmula I por nitración de 3'-hidroxipropiofenona. El procedimiento descrito por Aurore et al. se representa esquemáticamente como sigue:

El procedimiento descrito bajo Aurore et. al. implica la nitración de 5-hidroxipropiofenona utilizando ácido nítrico. Este procedimiento tiene la desventaja de que implica la formación de regioisómeros/isómeros posicionales no deseados en la reacción de nitración por sustitución del grupo nitro en las posiciones orto y para en lugar de la misma reacción solo en la posición para. La eliminación de isómeros posicionales mediante el uso de purificaciones normales siempre es una tarea desafiante, ya que están estrechamente asociados entre sí por su factor de retención. Por lo tanto, requiere múltiples purificaciones en columna e incluso entonces no hay garantía de obtener el compuesto de alta pureza requerido para el uso farmacéutico.
En base a los inconvenientes mencionados anteriormente, existe una necesidad vital de desarrollar un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que sea fácilmente susceptible de producción a gran escala.
Por lo tanto, los presentes inventores se centraron en investigar cómo simplificar el procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I que evite los inconvenientes antes mencionados, procedimiento que implica principalmente la nitración regioselectiva para evitar/reducir la formación de regioisómeros/isómeros posicionales, evitando así las engorrosas purificaciones en columna y hace que el procedimiento sea viable para la fabricación a gran escala.
Sumario de la invención
Por consiguiente, la presente invención proporciona un procedimiento mejorado para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, un compuesto intermedio útil en la preparación de 7-etil-10-hidroxicamptotecina (SN-38).
De acuerdo con una realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, y

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado;
b) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que comprende:
a) Hacer reaccionar un compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, en donde "PG" representa un grupo protector de hidroxilo adecuado; y
b) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoilo, p- metoxibencilo, t-butildimetilsililo, tbutildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde "PG" representa un grupo protector de hidroxilo adecuado,

b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, y

c) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde "PG" representa un grupo protector de hidroxilo adecuado,
b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, y
c) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, tbutildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde "PG" representa un grupo protector de hidroxilo adecuado,

b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV,

c) Desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V, y

d) Reducir el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hldroxlpropiofenona de Fórmula I; (o)
e) Reducir el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI y

f) Desproteger el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde "PG" representa un grupo protector de hidroxilo adecuado,
b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV,
c) Desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V, y
d) Reducir el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I; (o)
e) Reducir el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI y
f) Desproteger el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que comprende:
a) Hacer reaccionar un compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, en donde "PG" representa un grupo protector de hidroxilo adecuado,
b) Desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V, y
c) Reducir el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I; (o)
d) Reducir el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI y
e) Desproteger el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula III,

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula III, en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula III, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IIIa.

De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IIIb.

De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IIIc.
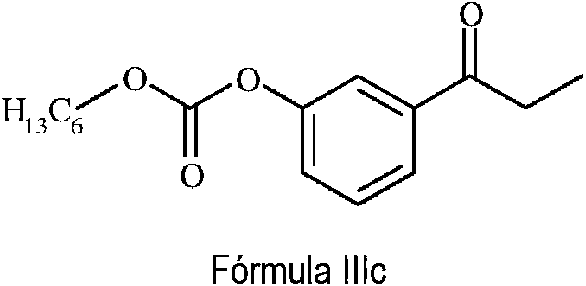
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IV:

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IV; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano,
alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butiisilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IV, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación del compuesto de Fórmula IV, que comprende hacer reaccionar un compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV;

en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, tbutildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo. De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IVa.

De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IVb.

De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula IVc.

De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula VI.

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula VI; en donde el grupo
“PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula VI, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de un compuesto de Fórmula VI, que comprende reducir un compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI; en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de irinotecán, que comprende: preparar un compuesto de Fórmula I de acuerdo con los procedimientos descritos anteriormente y convertir el compuesto de Fórmula I en 7-etil-10-hidroxicamptotecina (SN-38) y posteriormente en irinotecán.
Descripción detallada de la invención
La presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, un compuesto intermedio útil en la preparación de 7-etil-10-hidroxicamptotecina (SN-38).
De acuerdo con una realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula III, en donde "PG" es un grupo protector de hidroxilo adecuado, con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, y

b) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde “PG” representa un grupo protector de hidroxilo adecuado,

b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV, y

d) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I.
En otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde “PG” representa un grupo protector de hidroxilo adecuado,

b) Hacer reaccionar el compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV,

c) Desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V, y

d) Reducir el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I; (o)
e) Reducir el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI, y

f) Desproteger el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I.
Los materiales de partida, un compuesto de Fórmula II es bien conocido en la técnica y se puede producir mediante métodos conocidos en la técnica reconocidos por el químico orgánico con experiencia ordinaria en la técnica.
La presente invención se refiere a un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que involucra principalmente la nitración regioselectiva al crear un impedimento estérico en la 3'-hidroxipropiofenona mediante la protección del grupo hidroxilo, lo que ventajosamente evita la formación de regioisómeros/isómeros posicionales, haciendo así que el procedimiento esté libre de tediosas etapas de purificación y/o cristalización en columna, etapas de procedimiento que se consideran en la técnica anterior para separar regioisómeros/isómeros posicionales no deseados. El procedimiento regioselectivo de la presente invención es más adecuado para aplicaciones comerciales con mayor regioselectividad y evita los problemas asociados con los procedimientos reportados.
Protección del grupo hidroxi
La etapa a) del procedimiento anterior implica la protección del grupo hidroxi de un compuesto de Fórmula II con un agente formador de grupos protectores adecuado en presencia de una base y en un disolvente para obtener un compuesto de Fórmula III; en donde "PG" es un grupo protector adecuado.
El grupo protector de hidroxilo adecuado incluye, pero no se limita a, formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, tbutildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo. Otros grupos protectores de hidroxilo que son bien conocidos en la técnica también pueden considerarse como grupos protectores de hidroxilo de la invención, descritos específicamente en detalle en Protecting Groups in Organic Synthesis, Theodora W. Greene y Peter G. M. Wuts, 3a edición, 1999, publicado por John Wiley and Son, Inc.; preferiblemente el grupo protector es formiato de tricloroetilo, formiato de etilo y formiato de hexilo.
El agente formador de grupos protectores de hidroxilo adecuado incluye, pero no se limita a, cloroformiato de tricloroetilo, cloroformiato de metilo, cloroformiato de etilo, cloroformiato de hexilo, cloroformiato de isobutilo, cloroformiato de bencilo, cloruro de metoximetilo, cloruro de metoxietoximetilo, tetrahidropiranil éter, bromuro de alilo, cloruro de metoxitritilo, cloruro de metiltiometilo, bromuro de bencilo, bromuro de benzoílo, cloruro de p-metoxibencilo, cloruro de t-butildimetilsililo, cloruro de t-butildifenilsililo, cloruro de triisopropilsililo, cloruro de di-terc-butilsilileno, cloruro de tetraisopropildisiloxanilideno, cloruro de pivaloílo y cloruro de benzoílo; preferiblemente cloroformiato de tricloroetilo, cloroformiato de etilo o cloroformiato de hexilo.
La base adecuada usada en este documento en la etapa a) se selecciona de una base inorgánica o una orgánica; además, la base inorgánica adecuada se selecciona, pero no se limita a, de hidróxidos de metales alcalinos tales como hidróxido de sodio, hidróxido de potasio y similares; hidruros de metales alcalinos tales como hidruro de sodio, hidruro de potasio y similares; amidas de metales alcalinos tales como amida de sodio, amida de potasio, amida de litio y similares; alcóxidos de metales alcalinos tales como metóxido de sodio, etóxido de sodio, t-butóxido de sodio, t-butóxido de potasio y similares; carbonatos de metales alcalinos tales como carbonato de sodio, carbonato de potasio, carbonato de cesio y similares; bicarbonatos de metales alcalinos tales como bicarbonato de sodio, bicarbonato de potasio y similares; fosfatos
tales como fosfato sódico, fosfato trisódico, fosfato potásico, fosfato tripotásico y similares; carbonato de amonio; y la base orgánica se selecciona de, pero no se limita a, trietilamina, tri-n-propilamina, tri-n-butilamina, metildibutilamina, diisopropilamina, diisopropiletilamina, diciclohexilamina, metildiciclohexilamina, etildiisopropilamina, N,N-dietildiciclohexilamina, piridina, dimetilamino-4-piridina, N-metilpiperidina, N-etilpiperidina, N-etilpiperidina, N-butilpiperidina, 1,2-dimetilpiperidina y similares; o mezclas de los mismos; preferiblemente hidróxido de sodio, hidruro de sodio, amida de sodio, t-butóxido de potasio o carbonato de cesio; más preferiblemente hidróxido de sodio.
El disolvente adecuado usado en este documento en la etapa a) se selecciona, pero no se limita a, de cetonas que incluyen, pero no se limitan a, acetona, metil etil cetona, metil isobutil cetona, dietil cetona y similares; los nitrilos incluyen, pero no se limitan a, acetonitrilo, propionitrilo y similares; los éteres incluyen, pero no se limitan a, éter dietílico, tetrahidrofurano, metiltetrahidrofurano, 1,4-dioxano y similares; los disolventes apróticos polares incluyen, pero no se limitan a, dimetilacetamida, dimetilformamida, dimetilsulfóxido, N-metilpirrolidona y similares; los disolventes halogenados incluyen, pero no se limitan a, cloruro de metileno, cloruro de etileno, cloroformo y similares; los hidrocarburos aromáticos incluyen, pero no se limitan a, tolueno, xileno y similares; agua o mezclas de los mismos; preferiblemente, el disolvente adecuado es acetonitrilo, tetrahidrofurano, dimetilformamida, cloruro de metileno, agua o mezclas de los mismos; más preferiblemente cloruro de metileno, agua o mezclas de los mismos.
La reacción de un compuesto de Fórmula II con un grupo protector de hidroxilo adecuado se lleva a cabo a una temperatura de aproximadamente -10 °C a la temperatura de reflujo; preferiblemente a una temperatura de aproximadamente 0 °C a aproximadamente 80 °C.
Una vez finalizada la reacción de protección, el compuesto resultante de Fórmula III se puede aislar como una forma sólida para la conversión en las etapas posteriores o se puede continuar sin aislar el mismo. Si implica aislamiento, el aislamiento puede llevarse a cabo por filtración directa del sólido precipitado o por extracción de la masa de reacción con un disolvente orgánico inmiscible en agua adecuado; preferiblemente acetato de etilo, cloruro de metileno y similares; más preferiblemente cloruro de metileno. Luego, la capa orgánica que contiene el producto se puede separar y el compuesto de Fórmula III se puede aislar mediante precipitación, evaporación y, opcionalmente, el producto resultante se puede secar adicionalmente.
En otra realización, el compuesto de Fórmula III se representa específicamente como un compuesto de Fórmula IIIa, Fórmula IIIb o Fórmula IIIc.

Reacción de nitración
Hasta la fecha, todos los procedimientos de nitración reportados en la técnica implican la formación de regioisómeros/isómeros posicionales no deseados en la reacción de nitración por sustitución del grupo nitro en las posiciones orto y para en lugar de la misma reacción solo en la posición para. Esto se debe a la presencia de un grupo hidroxilo libre y, como resultado, de la falta de impedimento estérico en las posiciones orto y para, la posibilidad de nitración en ambas posiciones es muy favorable. La nitración en múltiples posiciones en lugar de solo en la requerida posición para crea una carga innecesaria para el procedimiento a escala comercial, ya que las impurezas regioisómeras formadas son difíciles de eliminar del producto requerido mediante las técnicas de purificación normales debido a la baja diferencia de polaridad de los regioisómeros. Por lo tanto, requiere múltiples purificaciones en columna e incluso entonces no hay garantía de obtener el compuesto de alta pureza requerido para el uso farmacéutico.
Para superar las dificultades asociadas con la técnica, los inventores de la presente invención han encontrado sorprendentemente que crear impedimento estérico protegiendo el grupo hidroxilo libre vecino en el anillo arilo con un grupo protector adecuado que evite la nitración en la posición orto no deseada y favorece la solo requerida posición para y, por lo tanto, como resultado la formación de regioisómeros no deseados se reduce en gran medida, lo que hace que el procedimiento actual esté libre de la tediosa purificación en columna y/o las múltiples etapas de cristalización para separar los isómeros no deseados y hacer que el procedimiento tenga un rendimiento más alto y el producto sea más puro.
La etapa de nitración b) del procedimiento anterior implica la reacción del compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV; en donde "PG" es un grupo protector adecuado.
El reactivo de nitración adecuado puede seleccionarse del grupo que comprende mezcla de nitración (una mezcla de ácido nítrico y ácido sulfúrico), nitrato de sodio, nitrato de potasio, nitrato de calcio, nitrato cúprico y similares y mezclas de los mismos; preferiblemente mezcla de nitración o nitrato de potasio.
La reacción de Fórmula III con un reactivo de nitración adecuado se puede realizar ventajosamente en un disolvente adecuado. El disolvente adecuado incluye, pero no se limita a, hidrocarburos halogenados, hidrocarburos aromáticos, amidas, nitrilos, ácidos y mezclas de los mismos. Los hidrocarburos halogenados incluyen, pero no se limitan a, cloruro de metileno, cloruro de etileno, cloroformo y similares; los hidrocarburos aromáticos incluyen, pero no se limitan a, tolueno, xileno y similares; las amidas incluyen, pero no se limitan a, dimetilformamida, dimetilacetamida, dimetilsulfóxido, N-metilpirrolidinona y similares; los nitrilos incluyen, pero no se limitan a, acetonitrilo, propionitrilo y similares; preferiblemente cloruro de metileno.
La etapa de nitración puede llevarse a cabo opcionalmente en presencia de un ácido seleccionado del grupo que comprende ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido perclórico, ácido fosfórico, ácido acético, anhídrido acético, ácido trifluoroacético, ácido tricloroacético, ácido metanosulfónico y similares y mezclas de los mismos; preferiblemente ácido sulfúrico.
La reacción del compuesto de Fórmula III con un reactivo de nitración adecuado se lleva a cabo a una temperatura de aproximadamente -25 °C a aproximadamente la temperatura de reflujo; Preferiblemente, la reacción se lleva a cabo entre aproximadamente -15 °C y aproximadamente 15 °C.
El compuesto resultante de Fórmula IV se puede aislar como una forma sólida para su conversión en las etapas posteriores o se puede continuar sin aislarlo. Si implica aislamiento, el aislamiento puede llevarse a cabo por filtración directa del sólido precipitado o por extracción de la masa de reacción con un disolvente orgánico inmiscible en agua adecuado; preferiblemente acetato de etilo, cloruro de metileno y similares; más preferiblemente cloruro de metileno. Luego, la capa orgánica que contiene el producto se puede separar y el compuesto de Fórmula IV se puede aislar mediante precipitación, evaporación y, opcionalmente, el producto resultante se puede secar adicionalmente.
En otra realización, el compuesto de Fórmula IV se representa específicamente como un compuesto de Fórmula IVa, Fórmula IVb o Fórmula IVc.

De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I, que comprende: convertir el compuesto de Fórmula IV obtenido anteriormente en 2-amino-5-hidroxipropiofenona de Fórmula I implicando desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V y reducir luego el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I, (o) reducir primero el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI y desproteger luego el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I.
Reacción de desprotección
El procedimiento precedente de la etapa de desprotección implica la desprotección del grupo protector hidroxi del compuesto de Fórmula IV o de Fórmula VI en presencia de un agente desprotector adecuado a una temperatura de aproximadamente 0 °C a aproximadamente la temperatura de reflujo; preferiblemente, la reacción se lleva a cabo entre aproximadamente 25 °C y aproximadamente 50 °C.
El agente desprotector adecuado usado en este documento para desproteger el compuesto de Fórmula IV o el de Fórmula VI se puede seleccionar de una base adecuada o un ácido adecuado. La base adecuada incluye, pero no se limita a, bases inorgánicas seleccionadas de hidróxidos de metales alcalinos tales como hidróxido de litio, hidróxido de sodio, hidróxido de potasio y similares; alcóxidos de metales alcalinos tales como metóxido de sodio, etóxido de sodio, terc-butóxido de sodio, terc-butóxido de potasio y similares; carbonatos de metales alcalinos tales como carbonato de sodio, carbonato de potasio, carbonato de cesio y similares; bicarbonatos de metales alcalinos tales como bicarbonato de sodio, bicarbonato de potasio y similares; y bases orgánicas seleccionadas del grupo que consiste en trietilamina, isopropiletilamina, diisopropilamina, diisopropiletilamina, N-metilmorfolina, piperidina, piridina y similares y mezclas de los mismos. El ácido adecuado incluye, pero no se limita a, ácido clorhídrico, ácido bromhídrico, tribromuro de boro, yoduro de trimetilsililo, bromuro de zinc, cloruro de titanio (IV), 2,3-dicloro-5,6-dicianobenzoquinona y similares y mezclas de estos; preferiblemente hidróxido de sodio, carbonato de potasio, ácido clorhídrico y mezclas de los mismos; más preferiblemente carbonato de potasio.
La reacción de desprotección se lleva a cabo ventajosamente en un disolvente adecuado. El disolvente adecuado incluye, pero no se limita a, alcoholes, hidrocarburos halogenados, amidas, nitrilos, ácidos y mezclas de estos. Los
alcoholes incluyen, pero no se limitan a, metanol, etanol, isopropanol, n-butanol y similares; los hidrocarburos halogenados incluyen, pero no se limitan a, cloruro de metileno, cloruro de etileno, cloroformo y similares; las amidas incluyen, pero no se limitan a, dimetilformamida, dimetilacetamida, dimetilsulfóxido, N-metilpirrolidinona y similares; los nitrilos incluyen, pero no se limitan a, acetonitrilo, propionitrilo y similares; los ácidos incluyen, pero no se limitan a, ácido fórmico, ácido acético y similares y mezclas de los mismos; preferiblemente metanol, etanol, dimetilformamida y mezclas de los mismos; más preferiblemente metanol.
Una vez completada la reacción de desprotección, el compuesto resultante de Fórmula V o Fórmula VI se puede aislar como una forma sólida para su conversión en las etapas posteriores o se puede continuar sin aislar el mismo. Si implica aislamiento, el aislamiento puede llevarse a cabo por filtración directa del sólido precipitado o por extracción de la masa de reacción con un disolvente orgánico inmiscible en agua adecuado; preferiblemente acetato de etilo, cloruro de metileno y similares; preferiblemente cloruro de metileno. Luego, la capa orgánica que contiene el producto puede separarse y el compuesto de Fórmula V o Fórmula VI puede aislarse mediante precipitación, evaporación y, opcionalmente, el producto resultante puede secarse adicionalmente.
Reducción del grupo nitro
El procedimiento anterior de la etapa de reducción del grupo nitro implica la reducción del compuesto de Fórmula IV o del de Fórmula V en presencia de un agente reductor adecuado a una temperatura de aproximadamente 25 °C a aproximadamente la temperatura de reflujo; preferiblemente, la reacción se lleva a cabo entre aproximadamente 25 °C y aproximadamente 80° C.
El agente reductor adecuado para la reducción del grupo nitro se selecciona del grupo que comprende hierro en HCl, hierro/NH4Cl, SnCl2 , ditionito de sodio, hidrosulfito de sodio, cloruro de estaño (11), cloruro de titanio (111), zinc/NH4Cl, Zn/hidracina hidrato, hierro/hidracina hidrato, níquel Raney y similares y mezclas de los mismos. La etapa de reducción se puede llevar a cabo en combinación con hidrógeno gaseoso; preferiblemente ditionito de sodio, hierro/NH4Cl o níquel Raney/ hidrógeno gaseoso; más preferiblemente ditionito de sodio.
La reacción de reducción se lleva a cabo ventajosamente en un disolvente adecuado. El disolvente adecuado incluye, pero no se limita a, alcoholes, hidrocarburos halogenados, nitrilos, agua y mezclas de los mismos. Los alcoholes incluyen, pero no se limitan a, metanol, etanol, isopropanol, n-butanol y similares; los hidrocarburos halogenados incluyen, pero no se limitan a, cloruro de metileno, cloruro de etileno y similares; los nitrilos incluyen, pero no se limitan a, acetonitrilo, propionitrilo y similares; agua y sus mezclas; preferiblemente metanol, etanol, agua y mezclas de los mismos; más preferiblemente agua.
Una vez completada la reacción de reducción, el compuesto resultante de Fórmula I puede aislarse como un sólido mediante filtración directa de la masa de reacción o puede aislarse extrayendo la masa de reacción con un disolvente orgánico inmiscible en agua adecuado; preferiblemente acetato de etilo, cloruro de metileno y similares. Luego, la capa orgánica que contiene el producto se puede separar y el compuesto de Fórmula I se puede aislar mediante precipitación, evaporación y el producto resultante se puede secar adicionalmente opcionalmente.
La presente invención proporciona 2-amino-5-hidroxipropiofenona de Fórmula I, obtenida mediante el procedimiento descrito en el presente documento, que tiene una pureza de al menos aproximadamente 95 %, medida por HPLC, preferiblemente al menos aproximadamente 97 % medida por HPLC, y más preferiblemente al menos aproximadamente 99%, medida por HPLC; y menos que 5% de otro regioisómero de Fórmula I' y/o Fórmula I", preferiblemente menos que 2% medido por HPLC.

La referencia de Aurore describe la reacción de nitración en un compuesto con el grupo hidroxi no protegido da como resultado un compuesto de Fórmula IV que contiene aproximadamente 60% de impurezas regioisómeras de Fórmula I' y I". Por el contrario, el procedimiento descrito en el presente documento llega a un compuesto de Fórmula IV, que puede estar involucrada en la protección del grupo hidroxilo para mejorar la selectividad de la reacción; en consecuencia, el compuesto de Fórmula IV se obtiene con una pureza de más que 95% y limita considerablemente la formación de impurezas regioisómeras no deseadas de Fórmula I' y I" a menos que aproximadamente 5%, por lo que se evitan engorrosas purificaciones en columna y/o cristalizaciones múltiples.
La preparación comparativa del compuesto de Fórmula IV usando el procedimiento de Aurore produjo el compuesto de Fórmula IV que tenía un nivel sustancialmente mayor de impurezas regioisómeras de Fórmula I' y I" que el presente procedimiento. Los resultados obtenidos a partir del procedimiento de Aurore y el presente procedimiento se resumen
en la Tabla I, como se muestra a continuación en la sección de Ejemplos, donde los valores se expresan como porcentaje en peso (% p/p) determinado por HPLC.
En otra realización, la presente invención proporciona un compuesto de Fórmula III,
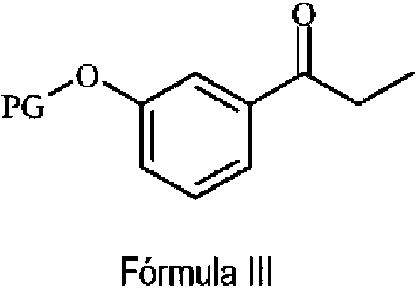
en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
En otra realización, la presente invención proporciona un compuesto de Fórmula III,

en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, tbutildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo. De acuerdo con otra realización, la presente invención proporciona un compuesto de Fórmula III, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
En otra realización, la presente invención proporciona un compuesto de Fórmula IIIa.

En otra realización, la presente invención proporciona un compuesto de Fórmula IIIb.

En otra realización, la presente invención proporciona un compuesto de Fórmula IIIc.

En otra realización, la presente invención proporciona un compuesto de Fórmula IV:

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
En otra realización, la presente invención proporciona un compuesto de Fórmula IV.

en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, tbutildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo. En otra realización, la presente invención proporciona un compuesto de Fórmula IV, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
En otra realización, la presente invención proporciona un compuesto de Fórmula IVa.

En otra realización, la presente invención proporciona un compuesto de Fórmula IVb.

En otra realización, la presente invención proporciona un compuesto de Fórmula IVc.

En otra realización, la presente invención proporciona un compuesto de Fórmula VI.

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado.
En otra realización, la presente invención proporciona un compuesto de Fórmula VI.

en donde el grupo “PG” se selecciona del grupo que comprende formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, tbutildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo. En otra realización, la presente invención proporciona un compuesto de Fórmula VI, en donde el grupo “PG” se selecciona de formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
De acuerdo con otra realización, la presente invención proporciona un procedimiento para la preparación de irinotecán, que comprende: preparar un compuesto de Fórmula I de acuerdo con los procedimientos descritos anteriormente y convertir el compuesto de Fórmula I en 7-etil-10-hidroxicamptotecina (SN-38) y posteriormente en irinotecán.
El compuesto de Fórmula I preparado por el procedimiento de la presente invención se puede utilizar como intermedio en la preparación de 7-etil-10-hidroxicamptotecina (SN-38) y posteriormente convertirlo en irinotecán, por los métodos conocidos en la técnica reconocidos por un químico orgánico con conocimientos ordinarios en la técnica.
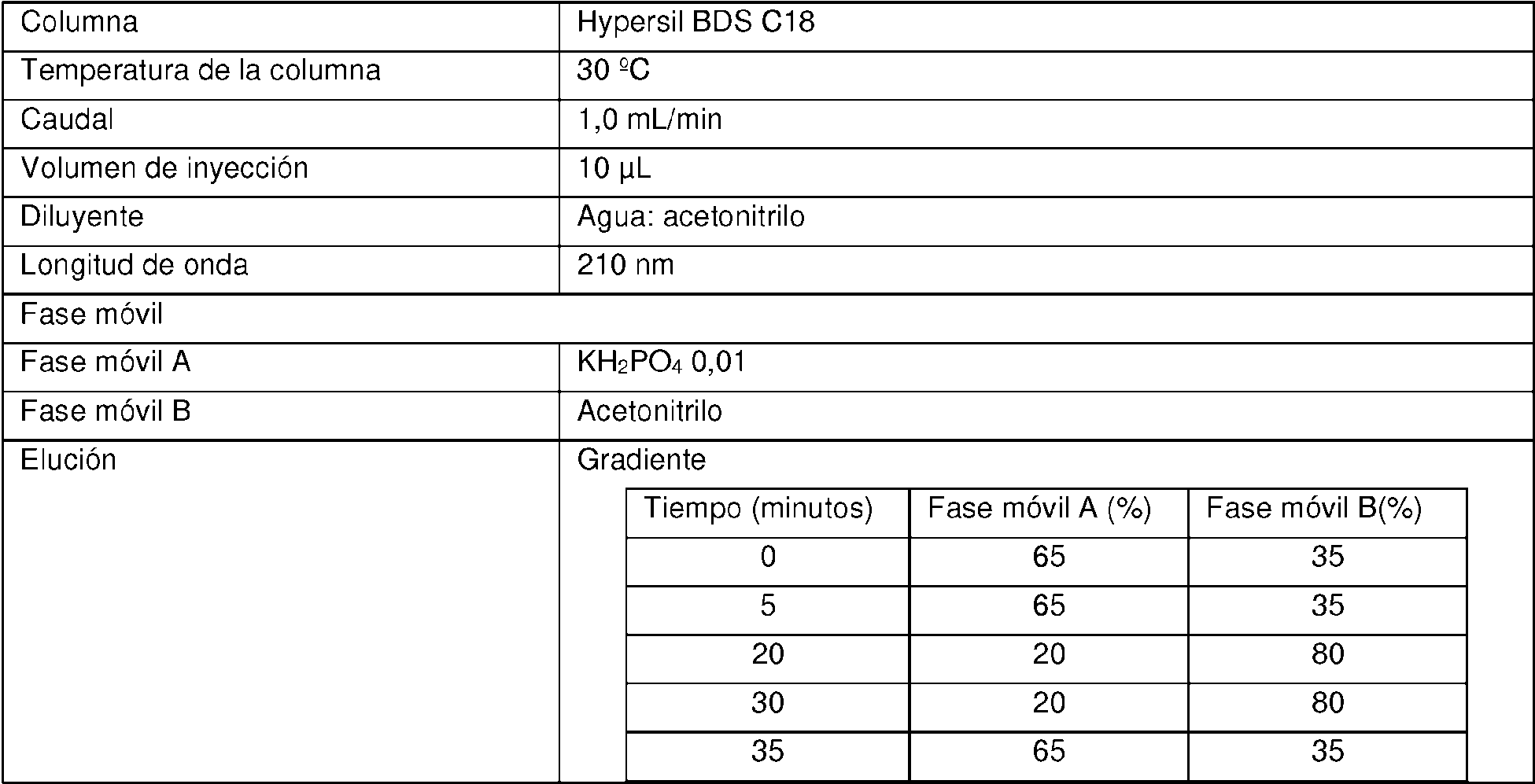
La presente invención proporciona 2-amino-5-hidroxipropiofenona de Fórmula I y sus intermedios, obtenidos mediante el procedimiento anterior, analizados utilizando cromatografía de líquidos de alta resolución ("HPLC") con las condiciones que se describen a continuación:
Ejemplos
Ejemplo 1
Preparación de un compuesto de Fórmula IlIa (PG: cloroformiato de 2,2,2-tricloroetilo)
En un matraz de fondo redondo de 2 litros equipado con un agitador mecánico y un termómetro se cargaron agua (700 mL) y 3-hidroxipropiofenona (140 g) a una temperatura de 25 °C a 30 °C. La masa de reacción se enfrió a una temperatura de 0 °C a 10 °C y se añadió una solución de NaOH (39,1 g de NaOH disueltos en 140 mL de agua). Se agitó la masa de reacción durante 10 min a la misma temperatura y se añadió cloroformiato de 2,2,2-tricloroetilo (237,3 g) a la masa de reacción mientras se mantenía la temperatura por debajo de 10 °C. La masa de reacción se agitó durante 2 horas a una temperatura a una temperatura de 0 °C a 10 °C. Una vez completada la reacción, se filtró el sólido y se lavó la torta con hexano (300 mL) y se secó el material húmedo al vacío para obtener 224 g de Fórmula III. 1H-RMN (DMSO): 5 = 1,09 (t, 3H), 3,07 (q, 2H), 5,08 (s, 2H) 7,57-7,96 (m, 4H); Pureza por HPLC: 99,5%.
Ejemplo 2
Preparación de un compuesto de Fórmula IVa (PG: cloroformiato de 2,2,2-tricloroetilo)
En un matraz de fondo redondo de 1 litro equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IIIa del Ejemplo-1 (50 g) y cloruro de metileno (500 mL) a una temperatura de 25°C a 30°C. A la masa de reacción se añadió sulfato sódico anhidro (25 g) a la misma temperatura. La masa de reacción se enfrió a una temperatura de 5 °C a 10 °C y se añadió ácido sulfúrico (125 mL). La masa de reacción se enfrió a -1 °C y se añadió nitrato potásico (38,7 g) en porciones a la misma temperatura. La masa de reacción se agitó durante 60 min a 5-10°C. Una vez finalizada la reacción, la masa de reacción se vertió en agua (1000 mL) previamente enfriada. Se separó la capa orgánica y la capa acuosa se extrajo con cloruro de metileno (400 mL). Luego, la capa orgánica combinada se lavó con una solución de NaOH 2 M (500 mL) y una solución de cloruro de sodio al 10% (500 mL). La capa orgánica combinada se destiló completamente al vacío para proporcionar 47,3 g de Fórmula IV. 1H-RMN (DMSO): 5 = 1,12 (t, 3H), 2,91 (q, 2H), 5,10 (s, 2H), 7,72 (d, J=2,4 Hz, 1H), 7,77 (dd, 1H), 8,29 (d, J = 9Hz, 1H); Pureza por HPLC: 98%.
Ejemplo 3
Preparación del compuesto de Fórmula V
En un matraz de fondo redondo de 1 litro equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IVa (46 g) del Ejemplo 2, carbonato de potasio (25 g) y metanol (500 mL) a una temperatura de 25 °C a 30 °C y se agitó la masa de reacción durante 60 min a la misma temperatura. Una vez finalizada la reacción, se filtraron las sales y se cargó el filtrado con agua (500 mL). La capa acuosa se lavó con cloruro de metileno (250 mL). El pH de la masa se ajustó a 3,5 usando una solución de ácido clorhídrico. Luego, la masa de reacción se extrajo con cloruro de metileno (1 x 500 mL y 1 x 250 mL). La capa orgánica combinada se lavó con agua (250 mL). La capa orgánica se destiló completamente al vacío y el residuo se eliminó con hexano (100 mL). Se cargó tolueno (50 mL) al material bruto y se agitó durante 30 min a una temperatura de 25 °C a 30 °C. El sólido se filtró para dar 20 g del compuesto de Fórmula V.1H-RMN (DMSO): 5 = 1,10 (t, 3H), 2,77 (q, 2H), 6,81 (d, J=2,4 Hz, 1H), 6,99 (dd, 1H), 8,10 (d, J = 9 Hz, 1H) 11,37 (brs, 1H); Pureza por HPLC: 97,5%.
Ejemplo 4
Preparación del compuesto de Fórmula I
En un matraz de fondo redondo de 500 mL equipado con un agitador mecánico y un termómetro se cargaron agua (125 mL), ditionito de sodio (17,8 g) y carbonato de sodio (8,7 g) a una temperatura de 25 °C a 30 °C y se agitó durante 10 min a la misma temperatura. La masa de reacción se enfrió a una temperatura de 0 °C a 5 °C y se añadió una solución del compuesto de Fórmula V (5 g disueltos en 10 mL de metanol) a la misma temperatura. La masa de reacción se dejó entre 25 °C y 30 °C y se agitó durante 1 hora. El sólido se filtró y se lavó con agua para dar 3,6 g de 2-amino 5-hidroxipropiofenona de Fórmula I. 1H-RMN (DMSO): 5 = 1,05 (t, 3H), 2,87 (q, 2H), 6,16 (s, 2H), 6,45 (d, 1H), 6,81 (dd, 1H), 7,11 (d, J=9 Hz, 1H), 8,65 (s, 1H); Pureza por HPLC: 97,5%.
Ejemplo 5
Preparación de un compuesto de Fórmula IIIb (PG: cloroformiato de etilo)
En un matraz de fondo redondo de 500 mL equipado con un agitador mecánico y un termómetro se cargaron agua (100 mL) y 3-hidroxipropiofenona (20 g) a una temperatura de 25 °C a 30 °C. La masa de reacción se enfrió a una temperatura de 0 °C a 10 °C y se añadió una solución de NaOH (9,6 g de NaOH disueltos en 20 mL de agua). Se agitó la masa de reacción durante 10 min a la misma temperatura y se añadió cloroformiato de etilo (17,4 g) a la masa de reacción manteniendo la temperatura por debajo de 10 °C. Una vez finalizada la reacción, la masa de reacción se extrajo con cloruro de metileno (150 mL) y la fase orgánica se lavó con NaOH al 10% (100 mL). La capa orgánica resultante se destiló completamente al vacío por debajo de 50 °C. Se cargaron 500 mL de hexano, se mantuvo la
masa durante la noche a 2-8 °C. El sólido se filtró y se lavó la torta con hexano frío (50 mL) y se secó el material húmedo al vacío para obtener 15,5 g de carbonato de etilo de 3-hidroxipropiofenona.
Ejemplo 6
Preparación de un compuesto de Fórmula IVb (PG: cloroformiato de etilo)
En un matraz de fondo redondo de 250 mL equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IIIb del Ejemplo-5 (5 g) y cloruro de metileno (50 mL) a una temperatura de 25 °C a 30 °C bajo atmósfera de nitrógeno. A la masa de reacción se añadió sulfato sódico anhidro (2,5 g) a la misma temperatura. La masa de reacción se enfrió a una temperatura de 5 °C a 10 °C y se añadió ácido sulfúrico (18,3 mL) y nitrato de potasio (5,7 g) en porciones por debajo de 10 °C. La masa de reacción se agitó durante 60 min a 0-10 °C. Una vez finalizada la reacción se vertió la masa de reacción en agua (200 mL) previamente enfriada. Se separó la capa orgánica y la capa acuosa se extrajo con cloruro de metileno (50 mL). Luego, la capa orgánica combinada se lavó con una solución de NaOH 2 M (100 mL) y una solución de cloruro de sodio al 10% (100 mL). La capa orgánica combinada se destiló completamente al vacío para proporcionar 2,4 g de compuesto de Fórmula IVb.
Ejemplo 7
Preparación del compuesto de Fórmula V
En un matraz de fondo redondo de 100 mL equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IVb (2 g) del Ejemplo 6, carbonato de potasio (1 g) y metanol (10 mL) a una temperatura de 25 °C a 30 °C y la masa de reacción se agitó durante 60 min a la misma temperatura. Una vez finalizada la reacción se filtraron las sales y se cargó el filtrado con agua (50 mL). La capa acuosa se lavó con cloruro de metileno (50 mL). El pH de la masa se ajustó a 1,2 usando una solución de ácido clorhídrico. Luego, la masa de reacción se extrajo con cloruro de metileno (3 x 50 mL). La capa orgánica se destiló completamente al vacío para proporcionar 1,4 g de compuesto de Fórmula V.
Ejemplo 8
Preparación de un compuesto de Fórmula IIIc (PG: cloroformiato de hexilo)
En un matraz de fondo redondo de 500 mL equipado con un agitador mecánico y un termómetro se cargaron agua (120 mL) y 3-hidroxipropiofenona (20 g) a una temperatura de 25 °C a 30 °C. La masa de reacción se enfrió a una temperatura de 0 °C a 10 °C y se añadió una solución de NaOH (9,6 g de NaOH disueltos en 20 mL de agua). Se agitó la masa de reacción durante 10 min a la misma temperatura y se añadió cloroformiato de hexilo (26,3 g) a la masa de reacción manteniendo la temperatura por debajo de 10 °C. Una vez finalizada la reacción, la masa de reacción se extrajo con cloruro de metileno (150 mL) y la fase orgánica se lavó con NaOH al 10% (100 mL). La capa orgánica resultante se destiló completamente al vacío por debajo de 50 °C para obtener 34,8 g de carbonato de hexilo de 3-hidroxipropiofenona.
Ejemplo 9
Preparación del compuesto de Fórmula IVc (PG: cloroformiato de hexilo)
En un matraz de fondo redondo de 250 mL equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IIIc del Ejemplo-8 (5 g) y cloruro de metileno (50 mL) a una temperatura de 25 °C a 30 °C bajo atmósfera de nitrógeno. A la masa de reacción se añadió sulfato sódico anhidro (5 g) a la misma temperatura. La masa de reacción se enfrió una temperatura de 5°C a 10°C y se añadió ácido sulfúrico (15 mL) y nitrato de potasio (4,6 g) en porciones por debajo de 10 °C. La masa de reacción se agitó durante 2 horas a 5-10 °C. Una vez finalizada la reacción, se vertió la masa de reacción en agua (200 mL) previamente enfriada. Se separó la capa orgánica y la capa acuosa se extrajo con cloruro de metileno (100 mL). Luego, la capa orgánica combinada se lavó con una solución de NaOH 2 M (100 mL) y una solución de cloruro de sodio al 10% (100 mL). La capa orgánica combinada se destiló completamente al vacío para proporcionar 5,3 g del compuesto de Fórmula IV.
Ejemplo 10
Preparación del compuesto de Fórmula V
En un matraz de fondo redondo de 100 mL equipado con un agitador mecánico y un termómetro se cargaron el compuesto de Fórmula IVc (5 g) del Ejemplo 9, carbonato de potasio (3 g) y metanol (50 mL) a una temperatura de 25 °C a 30 °C y se agitó la masa de reacción durante 60 min a la misma temperatura. Una vez finalizada la reacción, se filtraron las sales y se cargó el filtrado con agua (50 mL). La capa acuosa se lavó con cloruro de metileno (50 mL). El pH de la masa se ajustó a 1,5 usando una solución de ácido clorhídrico. Luego, la masa de reacción se extrajo con cloruro de metileno (3 x 50 mL). La capa orgánica se destiló completamente al vacío para proporcionar 1,2 g de Fórmula V.
Ejemplo 11
Preparación del compuesto de Fórmula I (PG: cloroformiato de 2,2,2-tricloroetilo)
En un matraz de fondo redondo de 2 litros equipado con un agitador mecánico y un termómetro se cargaron hidróxido de sodio (41,5 g), agua (600 mL) y 3-hidroxipropiofenona (120 g) a una temperatura de 25 °C a 30 °C y se dejó agitar durante 10-15 min a la misma temperatura. La masa de reacción se cargó con dicloruro de metileno (480 mL) y se dejó enfriar a una temperatura de 5-15 °C, se cargaron bromuro de tetrabutilamonio (0,6 g) y cloroformiato de 2,2,2-tricloroetilo (203 g) a 5-15 °C y se dejó en agitación durante 30-40 min a la misma temperatura. Una vez finalizada la reacción, se separaron las capas orgánica y acuosa y la capa orgánica se lavó con agua (2 x 240 mL) y con una solución de cloruro de sodio (24 g disueltos en 240 mL de agua). La capa orgánica se separó y se concentró al vacío por debajo de 40 °C y se aisló el compuesto de Fórmula Illa sólido mediante filtraciones de suspensiones en heptano.
En un matraz de fondo redondo de 2 litros equipado con un agitador mecánico y un termómetro se cargaron el sólido húmedo obtenido anteriormente (Fórmula Illa) y dicloruro de metileno (672 mL) a 25-30°C y la masa de reacción se dejó enfriar a una temperatura de -20 a -25 °C. A la masa de reacción se añadió ácido sulfúrico (389,8 mL) y ácido nítrico fumante (130 g) a una temperatura de -20 a -25 °C durante 30 min. Una vez finalizada la reacción, se separó la capa de ácido sulfúrico y la masa de reacción se dejó a 25-30°C. La masa de reacción se lavó con agua (672 mL) y la capa orgánica se concentró al vacío por debajo de 45 °C para obtener el compuesto de Fórmula IVa.
En un matraz de fondo redondo de 2 litros equipado con un agitador mecánico y un termómetro se cargaron el sólido húmedo obtenido anteriormente (Fórmula IVa) y metanol (448 mL) a 25-30 °C y se agitó durante 15-30 min a la misma temperatura. A la masa de reacción se añadió carbonato potásico (47,5 g) y se agitó durante 1 hora a 25-30 °C. La masa de reacción se añadió a una solución preparada por separado de carbonato de sodio (182,3 g) y ditionato de sodio (357 g) en agua (5,6 litros) a 5 °C y se dejó agitar durante 1 hora a 25-30 °C. Los sólidos precipitados se filtraron y lavaron con agua (224 mL). El sólido húmedo se disolvió en acetato de etilo (2,6 litros) a 25-30 °C y se lavó con agua (2 x 560 mL). La capa orgánica se separó y se concentró completamente al vacío por debajo de 45 °C para obtener el compuesto del título y se secó al vacío a 75 °C durante 7-8 h para proporcionar 57,6 g del compuesto del título. Pureza HPLC: 99,9%.
Los siguientes ejemplos comparativos se llevaron a cabo siguiendo la enseñanza de Aurore et al. en Bioorganic & Medicinal Chemistry Letters Volumen 14, Número 9, páginas 2363-2365, 2004.
Ejemplo comparativo
Preparación del compuesto de Fórmula V a partir de 3'-hidroxipropiofenona
En un matraz de fondo redondo de 500 mL equipado con un agitador mecánico y un termómetro se cargó ácido acético (400 mL) a 25-35 °C y la masa de reacción se calentó a 65-70 °C. La masa de reacción se cargó con ácido nítrico al 65-75% (50 mL) a la misma temperatura. Una vez finalizada la reacción, la masa de reacción se enfrió a 25-35°C. La masa de reacción se añadió lentamente a agua enfriada y se extrajo con acetato de etilo (2 x 800 mL). La capa orgánica combinada se destiló al vacío por debajo de 50 °C para obtener el compuesto bruto. El compuesto bruto obtenido se purificó por purificación en columna de gel de sílice utilizando como eluyente acetato de etilo/hexeno. Las fracciones puras se concentraron al vacío por debajo de 50 °C para obtener 41 g del compuesto del título.
Tabla 1

Claims (15)
1. Un procedimiento para la preparación de 2-amino-5-hidroxipropiofenona de Fórmula I:

que comprende:
a) Hacer reaccionar un compuesto de Fórmula III con un reactivo de nitración adecuado para obtener un compuesto de Fórmula IV,

en donde el grupo “PG” representa un grupo protector de hidroxilo adecuado; y
b) Convertir el compuesto de Fórmula IV en 2-amino-5-hidroxipropiofenona de Fórmula I.
2. El procedimiento según la reivindicación 1, en donde el "grupo protector de hidroxilo adecuado" se selecciona de formiato de tricloroetilo, formiato de metilo, formiato de etilo, formiato de hexilo, formiato de isobutilo, formiato de bencilo, metoximetilo, metoxietoximetilo, tetrahidropiranilo, tetrahidrofurano, alilo, metoxitritilo, metiltiometilo, bencilo, benzoílo, p-metoxibencilo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo, di-terc-butilsilileno, tetraisopropildisiloxanilideno, pivaloílo y benzoílo, y preferiblemente es formiato de tricloroetilo, formiato de etilo o formiato de hexilo.
3. El procedimiento según la reivindicación 1, en donde el reactivo de nitración se selecciona de una mezcla de nitración (una mezcla de ácido nítrico y ácido sulfúrico), nitrato de sodio, nitrato de potasio, nitrato de calcio, nitrato cúprico y mezclas de los mismos, y preferiblemente es una mezcla de nitración o nitrato de potasio.
4. El procedimiento según la reivindicación 1, en donde la etapa (a) se lleva a cabo en un disolvente adecuado y opcionalmente en presencia de un ácido, en donde el disolvente se selecciona de cloruro de metileno, cloruro de etileno, cloroformo, tolueno, xileno, dimetilformamida, dimetilacetamida, dimetilsulfóxido, N-metilpirrolidinona, acetonitrilo, propionitrilo y mezclas de los mismos, y en donde el ácido se selecciona de ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido perclórico, ácido fosfórico, ácido acético, anhídrido acético, ácido trifluoroacético, ácido tricloroacético, ácido metanosulfónico y mezclas de los mismos.
5. Un procedimiento según la reivindicación 1, en donde el compuesto de Fórmula III se obtiene haciendo reaccionar un compuesto de Fórmula II con un agente protector de hidroxilo adecuado para obtener un compuesto de Fórmula III; en donde "PG" representa un grupo protector de hidroxilo adecuado.

6. Un procedimiento según la reivindicación 5, en donde la etapa (b) comprende
c) Desproteger el compuesto de Fórmula IV con un agente desprotector adecuado para obtener un compuesto de Fórmula V, y

d) Reducir el compuesto de Fórmula V con un agente reductor adecuado para obtener 2-amino-5-hldroxlpropiofenona de Fórmula I.
7. Un procedimiento según la reivindicación 5, en donde la etapa (b) comprende
e) Reducir el compuesto de Fórmula IV con un agente reductor adecuado para obtener un compuesto de Fórmula VI y

f) Desproteger el compuesto de Fórmula VI con un agente desprotector adecuado para obtener 2-amino-5-hidroxipropiofenona de Fórmula I.
8. El procedimiento según las reivindicaciones 5, 6 ó 7, en donde el agente protector de hidroxilo adecuado se selecciona de cloroformiato de tricloroetilo, cloroformiato de metilo, cloroformiato de etilo, cloroformiato de hexilo, cloroformiato de isobutilo, cloroformiato de bencilo, cloruro de metoximetilo, cloruro de metoxietoximetilo, tetrahidropiranil éter, bromuro de alilo, cloruro de metoxitritilo, cloruro de metiltiometilo, bromuro de bencilo, bromuro de benzoílo, cloruro de p-metoxibencilo, cloruro de t-butildimetilsililo, cloruro de t-butildifenilsililo, cloruro de triisopropilsililo, cloruro de di-terc-butilsilileno, cloruro de tetraisopropildisiloxanilideno, cloruro de pivaloílo y cloruro de benzoílo, y preferiblemente es cloroformiato de tricloroetilo, cloroformiato de etilo o cloroformiato de hexilo.
9. El procedimiento según las reivindicaciones 5, 6 ó 7, en donde la etapa (a) se lleva a cabo en presencia de una base adecuada y un disolvente adecuado, en donde la base se selecciona de hidróxido de sodio, hidróxido de potasio, hidruro de sodio, hidruro de potasio, amida de sodio, amida de potasio, amida de litio, metóxido de sodio, etóxido de sodio, t-butóxido de sodio, t-butóxido de potasio, carbonato de sodio, carbonato de potasio, carbonato de cesio, bicarbonato de sodio, bicarbonato de potasio, fosfato de sodio, fosfato trisódico, fosfato de potasio, fosfato tripotásico, carbonato de amonio, trietilamina, tri-n-propilamina, tri-n-butilamina, metildibutilamina, diisopropilamina, diisopropiletilamina, diciclohexilamina, metildiciclohexilamina, etildiisopropilamina, N,N-dietildiciclohexilamina, piridina, dimetilamino-4-piridina, N-metilpiperidina, N-etilpiperidina, N-etilpiperidina, N-butilpiperidina, 1,2-dimetilpiperidina o mezclas de las mismas, y en donde el disolvente se selecciona de acetona, metil etil cetona, metil isobutil cetona, dietil cetona, acetonitrilo, propionitrilo, éter dietílico, tetrahidrofurano, metiltetrahidrofurano, 1,4-dioxano, dimetilacetamida, dimetilformamida, dimetilsulfóxido, N-metilpirrolidona, cloruro de metileno, cloruro de etileno, cloroformo, tolueno, xileno, agua o mezclas de los mismos.
10. El procedimiento según las reivindicaciones 5, 6 ó 7, en donde la etapa (a) se lleva a cabo en un disolvente adecuado opcionalmente en presencia de un ácido, en donde el disolvente se selecciona de cloruro de metileno, cloruro de etileno, cloroformo, tolueno, xileno, dimetilformamida, dimetilacetamida, dimetilsulfóxido, N-metilpirrolidinona, acetonitrilo, propionitrilo y mezclas de los mismos y/o donde el ácido se selecciona de ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido perclórico, ácido fosfórico, ácido acético, anhídrido acético, ácido trifluoroacético, ácido tricloroacético, ácido metanosulfónico y mezclas de los mismos.
11. El procedimiento según la reivindicación 10, en donde el disolvente es cloruro de metileno y el ácido es ácido sulfúrico.
12. El procedimiento según las reivindicaciones 6 ó 7, en donde el agente desprotector adecuado utilizado en la etapa (c) o la etapa (f) se selecciona de hidróxido de litio, hidróxido de sodio, hidróxido de potasio, metóxido de sodio, etóxido de sodio, terc-butóxido de sodio, terc-butóxido de potasio, carbonato de sodio, carbonato de potasio, carbonato de cesio, bicarbonato de sodio, bicarbonato de potasio, trietilamina, isopropiletilamina, diisopropilamina, diisopropiletilamina, N-metilmorfolina, piperidina, piridina, ácido clorhídrico, ácido bromhídrico, tribromuro de boro, yoduro de trimetilsililo, bromuro de zinc, cloruro de titanio (IV), 2,3-dicloro-5,6-dicianobenzoquinona y mezclas de los mismos, y preferiblemente es carbonato de potasio.
13. El procedimiento según las reivindicaciones 6 ó 7, en donde la etapa (c) y la etapa (f) se llevan a cabo en un disolvente adecuado; en donde el disolvente se selecciona de metanol, etanol, isopropanol, n-butanol, cloruro de metileno, cloruro de etileno, cloroformo, dimetilformamida, dimetilacetamida, dimetilsulfóxido, N-metilpirrolidinona, acetonitrilo, propionitrilo, ácido fórmico, ácido acético y mezclas de los mismos, y preferiblemente es metanol.
14. El procedimiento según las reivindicaciones 6 ó 7, en donde el agente reductor adecuado usado en la etapa (d) o la etapa (e) se selecciona del grupo que comprende hierro en HCl, hierro/NH4Cl, SnCl2 , ditionito de sodio, hidrosulfito de sodio, cloruro de estaño (II), cloruro de titanio (III), zinc/NH4Cl, zinc/hidrazina hidrato, hierro/hidrazina hidrato, níquel Raney y mezclas de los mismos, y preferiblemente es ditionito de sodio, y/o en donde la etapa (d) y la etapa (e) se llevan a cabo en un disolvente adecuado, en donde el disolvente se selecciona de metanol, etanol, isopropanol, nbutanol, cloruro de metileno, cloruro de etileno, acetonitrilo, propionitrilo, agua y mezclas de los mismos, y preferiblemente es agua.
15. El procedimiento según una cualquiera de las reivindicaciones precedentes, que comprende además convertir el compuesto de Fórmula I en 7-etil-10-hidroxicamptotecina (SN-38) y posteriormente en irinotecán.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201941001793 | 2019-01-15 | ||
| PCT/IB2020/050256 WO2020148641A1 (en) | 2019-01-15 | 2020-01-14 | Process for preparation of 2-amino-5-hydroxy propiophenone |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2951064T3 true ES2951064T3 (es) | 2023-10-17 |
Family
ID=71613360
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20741510T Active ES2951064T3 (es) | 2019-01-15 | 2020-01-14 | Procedimiento de preparación de 2-amino-5-hidroxipropiofenona |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11434196B2 (es) |
| EP (1) | EP3911660B1 (es) |
| ES (1) | ES2951064T3 (es) |
| WO (1) | WO2020148641A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115387113A (zh) * | 2022-07-28 | 2022-11-25 | 郑喜才 | 一种远红外除臭面料及其制备方法 |
| CN117924092B (zh) * | 2023-12-28 | 2025-03-04 | 棓诺(苏州)新材料有限公司 | 一种2-烯丙基-4-硝基苯酚的制备方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB711905A (en) * | 1950-05-12 | 1954-07-14 | Erwin Bunm | 1-(úÝ-hydroxyphenyl)-1-hydroxy-2-aralkylaminopropanes |
| TW438775B (en) | 1995-04-07 | 2001-06-07 | Pharmacia & Upjohn Co Llc | Novel intermediates and process for the manufacture of camptothecin derivatives (CPT-11) and related compounds |
| FR2785284B1 (fr) * | 1998-11-02 | 2000-12-01 | Galderma Res & Dev | Analogues de la vitamine d |
| AR035684A1 (es) * | 2001-02-21 | 2004-06-23 | Yakult Honsha Kk | Procedimiento para preparar 2'-amino-5'-hidroxipropiofenona, uso de la misma para la preparacion de analogos de camptotecina, procedimiento para prepararlos, compuestos intermediarios, procedimiento para preparar una cetona triciclica utilizada en la sintesis de analogos de camptotecina |
| US7608740B2 (en) * | 2005-08-03 | 2009-10-27 | Avra Laboratories Pvt. Ltd | Method of synthesizing key intermediates for the production of camptothecin derivatives |
| CN101362701A (zh) * | 2008-09-12 | 2009-02-11 | 复旦大学 | 2-氨基-5-羟基苯丙酮的制备方法 |
| CN101723841B (zh) * | 2009-12-04 | 2012-03-07 | 华东师范大学 | 2-氨基-5-烷氧基苯丙酮的制备方法 |
| CN104803861B (zh) * | 2014-01-27 | 2017-05-24 | 上海博邦医药科技有限公司 | 一种合成盐酸他喷他多的方法 |
| CN108794737B (zh) * | 2018-06-26 | 2019-07-02 | 中国科学院长春应用化学研究所 | 具有紫外光响应功能的封端改性聚乙二醇交联剂及制法和含该交联剂的水凝胶敷料及制法 |
-
2020
- 2020-01-14 EP EP20741510.0A patent/EP3911660B1/en active Active
- 2020-01-14 WO PCT/IB2020/050256 patent/WO2020148641A1/en not_active Ceased
- 2020-01-14 ES ES20741510T patent/ES2951064T3/es active Active
- 2020-01-14 US US17/419,378 patent/US11434196B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020148641A1 (en) | 2020-07-23 |
| US11434196B2 (en) | 2022-09-06 |
| US20220081388A1 (en) | 2022-03-17 |
| EP3911660B1 (en) | 2023-05-10 |
| EP3911660A4 (en) | 2022-03-30 |
| EP3911660A1 (en) | 2021-11-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3867231B1 (en) | Novel processes | |
| US20140058107A1 (en) | Apixaban preparation process | |
| JPS59130288A (ja) | 置換5H↓−ピリミド〔5.4↓−b〕インド−ル、その製法およびこれを含有する精神作用を有する医薬製剤 | |
| KR102266680B1 (ko) | 벨리노스테트의 다형태 및 이의 제조 방법 | |
| ES2951064T3 (es) | Procedimiento de preparación de 2-amino-5-hidroxipropiofenona | |
| JP2021119142A (ja) | キサンチンをベースとする化合物の調製方法 | |
| TWI808068B (zh) | 製備3-經取代5-胺基-6H-噻唑并[4,5-d]嘧啶-2,7-二酮化合物之方法 | |
| WO2009083258A2 (en) | Solid and crystalline dutasteride and processes for preparation thereof | |
| ES2795227T3 (es) | Procedimiento para producir un derivado de aminopirrolidina | |
| CN114031623B (zh) | 一种c14位氨基取代粉防己碱衍生物及其制备和应用 | |
| WO2022022613A1 (zh) | 一种三氮唑并[1,5-a]吡嗪制备方法及其应用 | |
| ES2741505T3 (es) | Procedimiento para la preparación de acetato de abiraterona y sus productos intermedios | |
| EP1791539A1 (en) | Process to prepare camptothecin derivatives | |
| JP5133704B2 (ja) | 製造方法 | |
| CN101880285B (zh) | 一种烯丙基取代喜树碱化合物的合成方法 | |
| US20080275241A1 (en) | Polymorphic Forms of Dolasetron Base and Processes of Preparing Dolasetron Base, Its Polymorphic Forms and Salt Thereof | |
| JP7416842B2 (ja) | 縮合多環式化合物の調製方法 | |
| WO2020020190A1 (zh) | 一种喹啉衍生物的合成方法 | |
| ES2232309A1 (es) | Procedimiento simplificado para la obtencion de gatifloxacino. | |
| TWI844822B (zh) | 稠合多環化合物之合成方法 | |
| CN113429333B (zh) | 一种加雷沙星中间体的合成方法 | |
| JP2021523187A (ja) | リナグリプチンおよびその塩の製造のための中間体およびプロセス | |
| WO2023013757A1 (ja) | アルキルシリルオキシ置換ベンジル化合物の製造方法 | |
| KR20230160303A (ko) | 5-{5-클로로-2-[(3s)-3-[(모르폴린-4-일)메틸]-3,4-디하이드로이소퀴놀린-2(1h)-카르보닐]페닐}-1,2-디메틸-1h-피롤-3-카르복실산 유도체의 신규한 합성 방법 및 약학적 화합물의 생산을 위한 이의 용도 | |
| WO2012032531A1 (en) | Process for the manufacture of irinotecan hydrochloride by total synthesis |