ES2952989T3 - Compuestos heterocíclicos antibacterianos y su síntesis - Google Patents
Compuestos heterocíclicos antibacterianos y su síntesis Download PDFInfo
- Publication number
- ES2952989T3 ES2952989T3 ES18819475T ES18819475T ES2952989T3 ES 2952989 T3 ES2952989 T3 ES 2952989T3 ES 18819475 T ES18819475 T ES 18819475T ES 18819475 T ES18819475 T ES 18819475T ES 2952989 T3 ES2952989 T3 ES 2952989T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- hydrogen
- compound
- alkoxy
- cyano
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000003786 synthesis reaction Methods 0.000 title description 48
- 230000015572 biosynthetic process Effects 0.000 title description 41
- 230000000844 anti-bacterial effect Effects 0.000 title description 16
- 150000002391 heterocyclic compounds Chemical class 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 297
- -1 complexes Chemical class 0.000 claims abstract description 181
- 239000000203 mixture Substances 0.000 claims abstract description 181
- 150000003839 salts Chemical class 0.000 claims abstract description 110
- 239000012453 solvate Substances 0.000 claims abstract description 98
- 150000004677 hydrates Chemical class 0.000 claims abstract description 96
- 241000894006 Bacteria Species 0.000 claims abstract description 19
- 244000005700 microbiome Species 0.000 claims abstract description 6
- 241000233866 Fungi Species 0.000 claims abstract description 4
- 241000700605 Viruses Species 0.000 claims abstract description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 366
- 239000001257 hydrogen Substances 0.000 claims description 280
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 176
- 125000000217 alkyl group Chemical group 0.000 claims description 164
- 229910052731 fluorine Inorganic materials 0.000 claims description 155
- 229910052757 nitrogen Inorganic materials 0.000 claims description 152
- 229910052736 halogen Inorganic materials 0.000 claims description 145
- 150000002367 halogens Chemical class 0.000 claims description 145
- 125000003545 alkoxy group Chemical group 0.000 claims description 139
- 125000000623 heterocyclic group Chemical group 0.000 claims description 137
- 229910052760 oxygen Inorganic materials 0.000 claims description 125
- 229910052717 sulfur Inorganic materials 0.000 claims description 119
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 109
- 239000011737 fluorine Substances 0.000 claims description 107
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 106
- 229920006395 saturated elastomer Polymers 0.000 claims description 98
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 86
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 85
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 78
- 125000005842 heteroatom Chemical group 0.000 claims description 70
- 125000004452 carbocyclyl group Chemical group 0.000 claims description 67
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 58
- 125000001188 haloalkyl group Chemical group 0.000 claims description 58
- 125000006294 amino alkylene group Chemical group 0.000 claims description 55
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 53
- 238000000034 method Methods 0.000 claims description 50
- 239000000460 chlorine Substances 0.000 claims description 48
- 229910052801 chlorine Inorganic materials 0.000 claims description 46
- 125000003282 alkyl amino group Chemical group 0.000 claims description 40
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 37
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 33
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 claims description 33
- 125000005124 aminocycloalkyl group Chemical group 0.000 claims description 33
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 30
- 125000006310 cycloalkyl amino group Chemical group 0.000 claims description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 28
- HHVIBTZHLRERCL-UHFFFAOYSA-N sulfonyldimethane Chemical compound CS(C)(=O)=O HHVIBTZHLRERCL-UHFFFAOYSA-N 0.000 claims description 28
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 claims description 27
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 27
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 22
- 239000000741 silica gel Substances 0.000 claims description 21
- 229910002027 silica gel Inorganic materials 0.000 claims description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims description 19
- 238000004519 manufacturing process Methods 0.000 claims description 17
- 241000588697 Enterobacter cloacae Species 0.000 claims description 16
- 241000588724 Escherichia coli Species 0.000 claims description 16
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 claims description 16
- 239000003814 drug Substances 0.000 claims description 16
- 230000008569 process Effects 0.000 claims description 16
- 229940125782 compound 2 Drugs 0.000 claims description 14
- 238000011282 treatment Methods 0.000 claims description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 12
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 claims description 11
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 11
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 11
- 241000588923 Citrobacter Species 0.000 claims description 10
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 claims description 10
- 239000002904 solvent Substances 0.000 claims description 10
- 241000588919 Citrobacter freundii Species 0.000 claims description 9
- 241000194032 Enterococcus faecalis Species 0.000 claims description 9
- 241000588772 Morganella morganii Species 0.000 claims description 9
- 241000588770 Proteus mirabilis Species 0.000 claims description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- 229940076266 morganella morganii Drugs 0.000 claims description 9
- 241000588747 Klebsiella pneumoniae Species 0.000 claims description 8
- 241000191967 Staphylococcus aureus Species 0.000 claims description 8
- 229940032049 enterococcus faecalis Drugs 0.000 claims description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 8
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims description 7
- 241000588626 Acinetobacter baumannii Species 0.000 claims description 7
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims description 7
- 239000003638 chemical reducing agent Substances 0.000 claims description 7
- 125000006559 (C1-C3) alkylamino group Chemical group 0.000 claims description 6
- 241001148231 Acinetobacter haemolyticus Species 0.000 claims description 6
- 241000122230 Acinetobacter junii Species 0.000 claims description 6
- 241001135518 Acinetobacter lwoffii Species 0.000 claims description 6
- 208000035143 Bacterial infection Diseases 0.000 claims description 6
- 241000589513 Burkholderia cepacia Species 0.000 claims description 6
- 241001647372 Chlamydia pneumoniae Species 0.000 claims description 6
- 241000193403 Clostridium Species 0.000 claims description 6
- 241000194031 Enterococcus faecium Species 0.000 claims description 6
- 241000589989 Helicobacter Species 0.000 claims description 6
- 241000588915 Klebsiella aerogenes Species 0.000 claims description 6
- 241000588749 Klebsiella oxytoca Species 0.000 claims description 6
- 241000589242 Legionella pneumophila Species 0.000 claims description 6
- 201000009906 Meningitis Diseases 0.000 claims description 6
- 241000588655 Moraxella catarrhalis Species 0.000 claims description 6
- 241000588653 Neisseria Species 0.000 claims description 6
- 241000588652 Neisseria gonorrhoeae Species 0.000 claims description 6
- 241000588769 Proteus <enterobacteria> Species 0.000 claims description 6
- 241000589516 Pseudomonas Species 0.000 claims description 6
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 6
- 239000003054 catalyst Substances 0.000 claims description 6
- 229940092559 enterobacter aerogenes Drugs 0.000 claims description 6
- 229940115932 legionella pneumophila Drugs 0.000 claims description 6
- KKLUJZCDLWWLHI-LLVKDONJSA-N 6-[(5R)-5-[[2-(6-fluoro-4-methyl-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=NC2=CC=C(F)C(OCCNC[C@@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 KKLUJZCDLWWLHI-LLVKDONJSA-N 0.000 claims description 5
- NZZLMIOKPRNXCS-NSHDSACASA-N 6-[(5S)-5-[[2-[(3-chloro-5-methyl-6-oxo-1,5-naphthyridin-4-yl)oxy]ethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=CC2=NC=C(Cl)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 NZZLMIOKPRNXCS-NSHDSACASA-N 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 5
- 239000003463 adsorbent Substances 0.000 claims description 5
- 229940125904 compound 1 Drugs 0.000 claims description 5
- 229940125898 compound 5 Drugs 0.000 claims description 5
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 5
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims description 4
- IEXNRKDDQNSVJX-LBPRGKRZSA-N 6-[(5S)-5-[[2-(6-fluoro-2,4-dimethyl-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C(C)=NC2=CC=C(F)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 IEXNRKDDQNSVJX-LBPRGKRZSA-N 0.000 claims description 4
- KKLUJZCDLWWLHI-NSHDSACASA-N 6-[(5S)-5-[[2-(6-fluoro-4-methyl-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=NC2=CC=C(F)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 KKLUJZCDLWWLHI-NSHDSACASA-N 0.000 claims description 4
- DSXDPFVQKRFWCH-PYMCNQPYSA-N CC(COC1=C(F)C=CC2=C1N(C)C(=O)C=C2)NC[C@H]1CN(C(=O)O1)C1=CN=C2OCC(=O)NC2=N1 Chemical compound CC(COC1=C(F)C=CC2=C1N(C)C(=O)C=C2)NC[C@H]1CN(C(=O)O1)C1=CN=C2OCC(=O)NC2=N1 DSXDPFVQKRFWCH-PYMCNQPYSA-N 0.000 claims description 4
- 208000001572 Mycoplasma Pneumonia Diseases 0.000 claims description 4
- 201000008235 Mycoplasma pneumoniae pneumonia Diseases 0.000 claims description 4
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 125000002947 alkylene group Chemical group 0.000 claims description 4
- 229940125773 compound 10 Drugs 0.000 claims description 4
- 229940126214 compound 3 Drugs 0.000 claims description 4
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 4
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 claims description 4
- 239000012279 sodium borohydride Substances 0.000 claims description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 4
- NPVMLQWOADQASB-RXMQYKEDSA-N 6-[(5R)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound C1C(=O)NC2=C(O1)N=CC(=N2)N1C[C@@H](CN)OC1=O NPVMLQWOADQASB-RXMQYKEDSA-N 0.000 claims description 3
- CARDOQJNAKLKCT-ZDUSSCGKSA-N CN1C(=O)C=C(C)C2=C1C(OCCNC[C@H]1CN(C(=O)O1)C1=CN=C3OCC(=O)NC3=N1)=C(F)C=C2 Chemical compound CN1C(=O)C=C(C)C2=C1C(OCCNC[C@H]1CN(C(=O)O1)C1=CN=C3OCC(=O)NC3=N1)=C(F)C=C2 CARDOQJNAKLKCT-ZDUSSCGKSA-N 0.000 claims description 3
- YJDQUSLGYMRQEC-CYBMUJFWSA-N CN1C(=O)C=CC2=C1C(OCCNC[C@@H]1CN(C(=O)O1)C1=CN=C3OCC(=O)NC3=N1)=C(F)C=C2 Chemical compound CN1C(=O)C=CC2=C1C(OCCNC[C@@H]1CN(C(=O)O1)C1=CN=C3OCC(=O)NC3=N1)=C(F)C=C2 YJDQUSLGYMRQEC-CYBMUJFWSA-N 0.000 claims description 3
- 241000192125 Firmicutes Species 0.000 claims description 3
- 230000003115 biocidal effect Effects 0.000 claims description 3
- 229910052740 iodine Inorganic materials 0.000 claims description 3
- 229910017464 nitrogen compound Inorganic materials 0.000 claims description 3
- 150000002830 nitrogen compounds Chemical class 0.000 claims description 3
- 229910052763 palladium Inorganic materials 0.000 claims description 3
- 230000009467 reduction Effects 0.000 claims description 3
- BQXWFDSKBWMXMZ-NSHDSACASA-N (5S)-5-[[2-(6-fluoro-4-methyl-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-3-(3-oxo-4H-pyrazino[2,3-b][1,4]thiazin-6-yl)-1,3-oxazolidin-2-one Chemical compound CN1C(=O)C=NC2=CC=C(F)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4SCC(=O)NC4=N3)=C12 BQXWFDSKBWMXMZ-NSHDSACASA-N 0.000 claims description 2
- YJDQUSLGYMRQEC-ZDUSSCGKSA-N 6-[(5S)-5-[[2-(7-fluoro-1-methyl-2-oxoquinolin-8-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=CC2=C1C(OCCNC[C@H]1CN(C(=O)O1)C1=CN=C3OCC(=O)NC3=N1)=C(F)C=C2 YJDQUSLGYMRQEC-ZDUSSCGKSA-N 0.000 claims description 2
- KUWZQSKOJQPYQB-ZDUSSCGKSA-N 6-[(5S)-5-[[2-[(5-methyl-6-oxo-1,5-naphthyridin-4-yl)oxy]ethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrido[3,2-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=CC2=NC=CC(OCCNC[C@H]3CN(C(=O)O3)C3=NC4=C(OCC(=O)N4)C=C3)=C12 KUWZQSKOJQPYQB-ZDUSSCGKSA-N 0.000 claims description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 claims description 2
- 239000002808 molecular sieve Substances 0.000 claims description 2
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 claims description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 claims description 2
- 239000010457 zeolite Substances 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 41
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims 7
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 claims 2
- YKVLISOYHNSSQG-YFKPBYRVSA-N (5S)-5-(aminomethyl)-3-(3-oxo-4H-pyrazino[2,3-b][1,4]thiazin-6-yl)-1,3-oxazolidin-2-one Chemical compound NC[C@H]1CN(C(=O)O1)C1=CN=C2SCC(=O)NC2=N1 YKVLISOYHNSSQG-YFKPBYRVSA-N 0.000 claims 1
- BSCHIACBONPEOB-UHFFFAOYSA-N oxolane;hydrate Chemical compound O.C1CCOC1 BSCHIACBONPEOB-UHFFFAOYSA-N 0.000 claims 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 claims 1
- 230000002401 inhibitory effect Effects 0.000 abstract description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 239
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 157
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 152
- 239000011541 reaction mixture Substances 0.000 description 104
- 239000000243 solution Substances 0.000 description 88
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 79
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 77
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 75
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 70
- 239000007787 solid Substances 0.000 description 66
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 57
- 235000019439 ethyl acetate Nutrition 0.000 description 53
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 42
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 41
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 39
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 34
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 33
- 238000006243 chemical reaction Methods 0.000 description 33
- 239000012043 crude product Substances 0.000 description 33
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 33
- 238000005481 NMR spectroscopy Methods 0.000 description 31
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 30
- 229910052938 sodium sulfate Inorganic materials 0.000 description 30
- 235000011152 sodium sulphate Nutrition 0.000 description 30
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 29
- 239000012267 brine Substances 0.000 description 27
- 239000003208 petroleum Substances 0.000 description 27
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 27
- 239000012299 nitrogen atmosphere Substances 0.000 description 26
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- 238000000746 purification Methods 0.000 description 23
- 239000007832 Na2SO4 Substances 0.000 description 22
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 22
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- 238000004440 column chromatography Methods 0.000 description 21
- 239000000543 intermediate Substances 0.000 description 21
- 238000004128 high performance liquid chromatography Methods 0.000 description 20
- 239000012071 phase Substances 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- 238000005160 1H NMR spectroscopy Methods 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- 238000003756 stirring Methods 0.000 description 18
- 239000004480 active ingredient Substances 0.000 description 17
- 230000005764 inhibitory process Effects 0.000 description 17
- 239000010779 crude oil Substances 0.000 description 15
- 238000001990 intravenous administration Methods 0.000 description 15
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 15
- 230000001580 bacterial effect Effects 0.000 description 14
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 229940079593 drug Drugs 0.000 description 10
- 230000003287 optical effect Effects 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 10
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 10
- QSDYBYCXJOQJBI-UHFFFAOYSA-N 2-(7-fluoro-1-methyl-2-oxoquinolin-8-yl)oxyacetaldehyde Chemical compound CN1C(=O)C=CC2=C1C(OCC=O)=C(F)C=C2 QSDYBYCXJOQJBI-UHFFFAOYSA-N 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 239000004599 antimicrobial Substances 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 238000004809 thin layer chromatography Methods 0.000 description 9
- 239000000499 gel Substances 0.000 description 8
- AQIAIZBHFAKICS-UHFFFAOYSA-N methylaminomethyl Chemical compound [CH2]NC AQIAIZBHFAKICS-UHFFFAOYSA-N 0.000 description 8
- QHXLIQMGIGEHJP-UHFFFAOYSA-N picoline - borane complex Substances [B].CC1=CC=CC=N1 QHXLIQMGIGEHJP-UHFFFAOYSA-N 0.000 description 8
- 229960003010 sodium sulfate Drugs 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 7
- GSGNTVQLHGOEMB-UHFFFAOYSA-N 6-bromo-2-nitropyridin-3-ol Chemical compound OC1=CC=C(Br)N=C1[N+]([O-])=O GSGNTVQLHGOEMB-UHFFFAOYSA-N 0.000 description 7
- 108010054814 DNA Gyrase Proteins 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 6
- XXJGBENTLXFVFI-UHFFFAOYSA-N 1-amino-methylene Chemical compound N[CH2] XXJGBENTLXFVFI-UHFFFAOYSA-N 0.000 description 6
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 6
- GDVHYNCHZKTTSQ-UHFFFAOYSA-N 6-bromo-4h-pyrido[3,2-b][1,4]oxazin-3-one Chemical compound O1CC(=O)NC2=NC(Br)=CC=C21 GDVHYNCHZKTTSQ-UHFFFAOYSA-N 0.000 description 6
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 101710183280 Topoisomerase Proteins 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 239000007859 condensation product Substances 0.000 description 6
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- HZONUGDFRUXYSA-UHFFFAOYSA-N ethyl 2-(6-bromo-2-nitropyridin-3-yl)oxyacetate Chemical compound CCOC(=O)COC1=CC=C(Br)N=C1[N+]([O-])=O HZONUGDFRUXYSA-UHFFFAOYSA-N 0.000 description 6
- 150000002430 hydrocarbons Chemical group 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- LSYOFPBORRARMF-GSVOUGTGSA-N (5r)-5-(hydroxymethyl)-1,3-oxazolidin-2-one Chemical compound OC[C@H]1CNC(=O)O1 LSYOFPBORRARMF-GSVOUGTGSA-N 0.000 description 5
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 5
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 5
- QZNZGEODNPFQHQ-UHFFFAOYSA-N 3-bromo-4-fluoro-2-n-methylbenzene-1,2-diamine Chemical compound CNC1=C(N)C=CC(F)=C1Br QZNZGEODNPFQHQ-UHFFFAOYSA-N 0.000 description 5
- 108020004414 DNA Proteins 0.000 description 5
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 5
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 235000010323 ascorbic acid Nutrition 0.000 description 5
- 239000011668 ascorbic acid Substances 0.000 description 5
- 229960005070 ascorbic acid Drugs 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 235000003599 food sweetener Nutrition 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 239000002502 liposome Substances 0.000 description 5
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- 229960004793 sucrose Drugs 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- GYJXFTOULOIVHV-YFKPBYRVSA-N (5R)-5-(azidomethyl)-3-(3-oxo-4H-pyrazino[2,3-b][1,4]thiazin-6-yl)-1,3-oxazolidin-2-one Chemical compound [N-]=[N+]=NC[C@H]1CN(C(=O)O1)C1=CN=C2SCC(=O)NC2=N1 GYJXFTOULOIVHV-YFKPBYRVSA-N 0.000 description 4
- OJIXUMSJACCKAT-MRVPVSSYSA-N (5r)-5-[[tert-butyl(dimethyl)silyl]oxymethyl]-1,3-oxazolidin-2-one Chemical compound CC(C)(C)[Si](C)(C)OC[C@H]1CNC(=O)O1 OJIXUMSJACCKAT-MRVPVSSYSA-N 0.000 description 4
- HDWSBDUFYQCGBV-UHFFFAOYSA-N 1-methyl-5h-1,5-naphthyridine-2,8-dione Chemical compound C1=CN=C2C=CC(=O)N(C)C2=C1O HDWSBDUFYQCGBV-UHFFFAOYSA-N 0.000 description 4
- QOSWQSPJFYHYRG-UHFFFAOYSA-N 2-(6-fluoro-2,4-dimethyl-3-oxoquinoxalin-5-yl)oxyacetaldehyde Chemical compound FC=1C(=C2N(C(C(=NC2=CC=1)C)=O)C)OCC=O QOSWQSPJFYHYRG-UHFFFAOYSA-N 0.000 description 4
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 4
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 4
- LILXDMFJXYAKMK-UHFFFAOYSA-N 2-bromo-1,1-diethoxyethane Chemical compound CCOC(CBr)OCC LILXDMFJXYAKMK-UHFFFAOYSA-N 0.000 description 4
- IQCPYFIIMNLQNU-UHFFFAOYSA-N 2-bromo-1,3-difluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C(Br)=C1F IQCPYFIIMNLQNU-UHFFFAOYSA-N 0.000 description 4
- ABSFYSCEFALSQR-UHFFFAOYSA-N 2-bromo-3-fluoro-N-methyl-6-nitroaniline Chemical compound BrC1=C(NC)C(=CC=C1F)[N+](=O)[O-] ABSFYSCEFALSQR-UHFFFAOYSA-N 0.000 description 4
- XISPDFMAOWZEQG-UHFFFAOYSA-N 4h-1,4-oxazin-3-one Chemical compound O=C1COC=CN1 XISPDFMAOWZEQG-UHFFFAOYSA-N 0.000 description 4
- ZIGMNPBWRZELDV-SNVBAGLBSA-N 6-[(5R)-5-[[tert-butyl(dimethyl)silyl]oxymethyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound [Si](C)(C)(C(C)(C)C)OC[C@H]1CN(C(O1)=O)C1=NC2=C(OCC(N2)=O)N=C1 ZIGMNPBWRZELDV-SNVBAGLBSA-N 0.000 description 4
- XLHQCQPQTQQOOY-UHFFFAOYSA-N 7-fluoro-8-hydroxy-1-methylquinolin-2-one Chemical compound FC1=CC=C2C=CC(=O)N(C)C2=C1O XLHQCQPQTQQOOY-UHFFFAOYSA-N 0.000 description 4
- ADNVDQIYQRPQIN-UHFFFAOYSA-N 8-bromo-7-fluoro-1,4-dimethylquinolin-2-one Chemical compound CN1C(=O)C=C(C)C2=C1C(Br)=C(F)C=C2 ADNVDQIYQRPQIN-UHFFFAOYSA-N 0.000 description 4
- GHSXYAATYYDTOD-UHFFFAOYSA-N 8-bromo-7-fluoro-1-methyl-2-oxoquinoline-4-carboxylic acid Chemical compound CN1C(=O)C=C(C(O)=O)C2=C1C(Br)=C(F)C=C2 GHSXYAATYYDTOD-UHFFFAOYSA-N 0.000 description 4
- DZMRNYLHBHXWET-UHFFFAOYSA-N 8-bromo-7-fluoro-1-methylquinoxalin-2-one Chemical compound BrC=1C(=CC=C2N=CC(N(C=12)C)=O)F DZMRNYLHBHXWET-UHFFFAOYSA-N 0.000 description 4
- DRWFMWRHAAOVTP-UHFFFAOYSA-N 8-bromo-7-fluoro-4-(hydroxymethyl)-1-methylquinolin-2-one Chemical compound BrC=1C(=CC=C2C(=CC(N(C=12)C)=O)CO)F DRWFMWRHAAOVTP-UHFFFAOYSA-N 0.000 description 4
- JOFWNABEIODEPD-UHFFFAOYSA-N BrC=1C(=CC=C2C(=CC(N(C=12)C)=O)CCl)F Chemical compound BrC=1C(=CC=C2C(=CC(N(C=12)C)=O)CCl)F JOFWNABEIODEPD-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 108020003631 Kinetoplast DNA Proteins 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000003963 antioxidant agent Substances 0.000 description 4
- 235000006708 antioxidants Nutrition 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- UVUNBRCPBPDCLY-UHFFFAOYSA-N ethyl 2-[3-bromo-4-fluoro-2-(methylamino)anilino]acetate Chemical compound CCOC(=O)CNC1=C(NC)C(Br)=C(F)C=C1 UVUNBRCPBPDCLY-UHFFFAOYSA-N 0.000 description 4
- VASPVEJAFKLGAZ-UHFFFAOYSA-N ethyl 4-(2-bromo-3-fluoro-N-methylanilino)-2,4-dioxobutanoate Chemical compound BrC1=C(C=CC=C1F)N(C(CC(C(=O)OCC)=O)=O)C VASPVEJAFKLGAZ-UHFFFAOYSA-N 0.000 description 4
- 150000004675 formic acid derivatives Chemical class 0.000 description 4
- 230000036541 health Effects 0.000 description 4
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 150000002894 organic compounds Chemical class 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 235000015424 sodium Nutrition 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 3
- BBRMUSGOTQCSET-UHFFFAOYSA-N 3-chloro-6-methoxy-1h-1,5-naphthyridin-4-one Chemical compound N1C=C(Cl)C(=O)C2=NC(OC)=CC=C21 BBRMUSGOTQCSET-UHFFFAOYSA-N 0.000 description 3
- YRFBTUBTOVLVTQ-UHFFFAOYSA-N 7-fluoro-8-hydroxy-1,3-dimethylquinoxalin-2-one Chemical compound CN1C(=O)C(C)=NC2=CC=C(F)C(O)=C12 YRFBTUBTOVLVTQ-UHFFFAOYSA-N 0.000 description 3
- GYTKJXHIFHSJQN-UHFFFAOYSA-N 8-(2,2-diethoxyethoxy)-7-fluoro-1,3-dimethylquinoxalin-2-one Chemical compound CCOC(COC1=C(F)C=CC2=C1N(C)C(=O)C(C)=N2)OCC GYTKJXHIFHSJQN-UHFFFAOYSA-N 0.000 description 3
- WEWFYTPIDZTJAX-UHFFFAOYSA-N 8-bromo-7-fluoro-1,3-dimethylquinoxalin-2-one Chemical compound CN1C(=O)C(C)=NC2=C1C(Br)=C(F)C=C2 WEWFYTPIDZTJAX-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 3
- 108010041052 DNA Topoisomerase IV Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 102000004310 Ion Channels Human genes 0.000 description 3
- 108090000862 Ion Channels Proteins 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 3
- 239000006137 Luria-Bertani broth Substances 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- UJKSIVCMOGLESQ-UHFFFAOYSA-N N-(2-bromo-3-fluorophenyl)-N-methylacetamide Chemical compound BrC1=C(C=CC=C1F)N(C(C)=O)C UJKSIVCMOGLESQ-UHFFFAOYSA-N 0.000 description 3
- FIWILGQIZHDAQG-UHFFFAOYSA-N NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F Chemical compound NC1=C(C(=O)NCC2=CC=C(C=C2)OCC(F)(F)F)C=C(C(=N1)N)N1N=C(N=C1)C1(CC1)C(F)(F)F FIWILGQIZHDAQG-UHFFFAOYSA-N 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000007900 aqueous suspension Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- HEDRZPFGACZZDS-MICDWDOJSA-N deuterated chloroform Substances [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- SACNIGZYDTUHKB-UHFFFAOYSA-N ditert-butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C(C)(C)C)C(C)(C)C SACNIGZYDTUHKB-UHFFFAOYSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 229930195733 hydrocarbon Natural products 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 229940057995 liquid paraffin Drugs 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 150000007660 quinolones Chemical class 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 238000012453 sprague-dawley rat model Methods 0.000 description 3
- 239000012258 stirred mixture Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 2
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 2
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 description 2
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- NMPVEAUIHMEAQP-UHFFFAOYSA-N 2-Bromoacetaldehyde Chemical compound BrCC=O NMPVEAUIHMEAQP-UHFFFAOYSA-N 0.000 description 2
- JBCVVGVRIWGLJS-UHFFFAOYSA-N 2-[(5-methyl-6-oxo-1,5-naphthyridin-4-yl)oxy]acetaldehyde Chemical compound CN1C(=O)C=CC2=NC=CC(OCC=O)=C12 JBCVVGVRIWGLJS-UHFFFAOYSA-N 0.000 description 2
- HRZTZLCMURHWFY-UHFFFAOYSA-N 2-bromo-1,3-difluorobenzene Chemical compound FC1=CC=CC(F)=C1Br HRZTZLCMURHWFY-UHFFFAOYSA-N 0.000 description 2
- AFXKCBFBGDUFAM-UHFFFAOYSA-N 2-methylpropan-2-amine;hydrofluoride Chemical compound [F-].CC(C)(C)[NH3+] AFXKCBFBGDUFAM-UHFFFAOYSA-N 0.000 description 2
- BOLCKGGORFPPJC-UHFFFAOYSA-N 3-bromo-6-chloropyrazin-2-amine Chemical compound NC1=NC(Cl)=CN=C1Br BOLCKGGORFPPJC-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- TZKBVRDEOITLRB-UHFFFAOYSA-N 4-methyl-n-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1h-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2C=C3C=NNC3=NC=2)=C1 TZKBVRDEOITLRB-UHFFFAOYSA-N 0.000 description 2
- NPVMLQWOADQASB-YFKPBYRVSA-N 6-[(5S)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound O=C1NC2=NC(N3C[C@@H](OC3=O)CN)=CN=C2OC1 NPVMLQWOADQASB-YFKPBYRVSA-N 0.000 description 2
- BZTYGYDSIJOIJQ-UHFFFAOYSA-N 6-chloro-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound ClC1=CN=C2OCC(=O)NC2=N1 BZTYGYDSIJOIJQ-UHFFFAOYSA-N 0.000 description 2
- SSMACLPPRSQXHH-UHFFFAOYSA-N 6-methoxy-1h-1,5-naphthyridin-4-one Chemical compound N1C=CC(=O)C2=NC(OC)=CC=C21 SSMACLPPRSQXHH-UHFFFAOYSA-N 0.000 description 2
- TVGUNSQVENBUIF-UHFFFAOYSA-N 7-chloro-1-methyl-5h-1,5-naphthyridine-2,8-dione Chemical compound ClC1=CN=C2C=CC(=O)N(C)C2=C1O TVGUNSQVENBUIF-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 244000034356 Aframomum angustifolium Species 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 108010011485 Aspartame Proteins 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 102100033735 Bactericidal permeability-increasing protein Human genes 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 108010067770 Endopeptidase K Proteins 0.000 description 2
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 2
- 241001360526 Escherichia coli ATCC 25922 Species 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 101000871785 Homo sapiens Bactericidal permeability-increasing protein Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 241000202934 Mycoplasma pneumoniae Species 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 102000004257 Potassium Channel Human genes 0.000 description 2
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- WERKSKAQRVDLDW-ANOHMWSOSA-N [(2s,3r,4r,5r)-2,3,4,5,6-pentahydroxyhexyl] (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO WERKSKAQRVDLDW-ANOHMWSOSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 238000000246 agarose gel electrophoresis Methods 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 229960005475 antiinfective agent Drugs 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 2
- 239000000605 aspartame Substances 0.000 description 2
- 235000010357 aspartame Nutrition 0.000 description 2
- 229960003438 aspartame Drugs 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- UDSAIICHUKSCKT-UHFFFAOYSA-N bromophenol blue Chemical compound C1=C(Br)C(O)=C(Br)C=C1C1(C=2C=C(Br)C(O)=C(Br)C=2)C2=CC=CC=C2S(=O)(=O)O1 UDSAIICHUKSCKT-UHFFFAOYSA-N 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 238000001311 chemical methods and process Methods 0.000 description 2
- 229960003405 ciprofloxacin Drugs 0.000 description 2
- 229910001850 copernicium Inorganic materials 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 description 2
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 2
- 229960005542 ethidium bromide Drugs 0.000 description 2
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 2
- 230000036512 infertility Effects 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 229960002260 meropenem Drugs 0.000 description 2
- CTUAQTBUVLKNDJ-OBZXMJSBSA-N meropenem trihydrate Chemical compound O.O.O.C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](C(=O)N(C)C)C1 CTUAQTBUVLKNDJ-OBZXMJSBSA-N 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 239000002353 niosome Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Chemical group 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 108020001213 potassium channel Proteins 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000005180 public health Effects 0.000 description 2
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000008261 resistance mechanism Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 235000011069 sorbitan monooleate Nutrition 0.000 description 2
- 239000001593 sorbitan monooleate Substances 0.000 description 2
- 229940035049 sorbitan monooleate Drugs 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- 210000000434 stratum corneum Anatomy 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000007916 tablet composition Substances 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- JWKBFWULUAVNQC-AWEZNQCLSA-N (5S)-5-[[2-[7-fluoro-1-(2-hydroxyethyl)-2-oxoquinolin-8-yl]oxyethylamino]methyl]-3-(3-oxo-4H-pyrazino[2,3-b][1,4]thiazin-6-yl)-1,3-oxazolidin-2-one Chemical compound OCCN1C(=O)C=CC2=C1C(OCCNC[C@H]1CN(C(=O)O1)C1=CN=C3SCC(=O)NC3=N1)=C(F)C=C2 JWKBFWULUAVNQC-AWEZNQCLSA-N 0.000 description 1
- OMXHZUPGSIYJHV-KZUDCZAMSA-N (5S)-5-[[[3-(7-fluoro-1-methyl-2-oxoquinolin-8-yl)oxy-2-hydroxypropyl]amino]methyl]-3-(3-oxo-4H-pyrazino[2,3-b][1,4]thiazin-6-yl)-1,3-oxazolidin-2-one Chemical compound CN1C(=O)C=CC2=C1C(OCC(O)CNC[C@H]1CN(C(=O)O1)C1=CN=C3SCC(=O)NC3=N1)=C(F)C=C2 OMXHZUPGSIYJHV-KZUDCZAMSA-N 0.000 description 1
- LSYOFPBORRARMF-VKHMYHEASA-N (5s)-5-(hydroxymethyl)-1,3-oxazolidin-2-one Chemical compound OC[C@@H]1CNC(=O)O1 LSYOFPBORRARMF-VKHMYHEASA-N 0.000 description 1
- XUBOMFCQGDBHNK-JTQLQIEISA-N (S)-gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCN[C@@H](C)C1 XUBOMFCQGDBHNK-JTQLQIEISA-N 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N (e)-2-hydroxybut-2-enedioic acid Chemical compound OC(=O)\C=C(\O)C(O)=O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- CIISBYKBBMFLEZ-UHFFFAOYSA-N 1,2-oxazolidine Chemical compound C1CNOC1 CIISBYKBBMFLEZ-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- 125000004776 1-fluoroethyl group Chemical group [H]C([H])([H])C([H])(F)* 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- IYYJDNBUTYYVCV-UHFFFAOYSA-N 10-chloro-2,2-dimethyl-2,3-dihydro-5h-[1,4,2]oxazasilino[6,5,4-de]-1,5-naphthyridin-5-one Chemical compound ClC1=CN=C2C=CC(=O)N3C[Si](C)(C)OC1=C23 IYYJDNBUTYYVCV-UHFFFAOYSA-N 0.000 description 1
- MIARFAABFTZAEF-UHFFFAOYSA-N 2,2-dimethyl-2,3-dihydro-5h-[1,4,2]oxazasilino[6,5,4-de]-1,5-naphthyridin-5-one Chemical compound C1=CN=C2C=CC(=O)N3C[Si](C)(C)OC1=C23 MIARFAABFTZAEF-UHFFFAOYSA-N 0.000 description 1
- MGRFQXUMVXPLLR-UHFFFAOYSA-N 2-[(3-chloro-5-methyl-6-oxo-1,5-naphthyridin-4-yl)oxy]acetaldehyde Chemical compound CN1C(=O)C=CC2=NC=C(Cl)C(OCC=O)=C12 MGRFQXUMVXPLLR-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- XZRSXRUYZXBTGD-UHFFFAOYSA-N 2-bromo-3-fluoroaniline Chemical compound NC1=CC=CC(F)=C1Br XZRSXRUYZXBTGD-UHFFFAOYSA-N 0.000 description 1
- IVHKZCSZELZKSJ-UHFFFAOYSA-N 2-hydroxyethyl sulfonate Chemical compound OCCOS(=O)=O IVHKZCSZELZKSJ-UHFFFAOYSA-N 0.000 description 1
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- 238000004780 2D liquid chromatography Methods 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- QBPDSKPWYWIHGA-UHFFFAOYSA-N 3-hydroxy-2-nitropyridine Chemical compound OC1=CC=CN=C1[N+]([O-])=O QBPDSKPWYWIHGA-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- HVBSAKJJOYLTQU-UHFFFAOYSA-M 4-aminobenzenesulfonate Chemical compound NC1=CC=C(S([O-])(=O)=O)C=C1 HVBSAKJJOYLTQU-UHFFFAOYSA-M 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical compound O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 description 1
- QRXMUCSWCMTJGU-UHFFFAOYSA-N 5-bromo-4-chloro-3-indolyl phosphate Chemical compound C1=C(Br)C(Cl)=C2C(OP(O)(=O)O)=CNC2=C1 QRXMUCSWCMTJGU-UHFFFAOYSA-N 0.000 description 1
- JVIISELWCFVRTJ-YFKPBYRVSA-N 6-[(5R)-5-(azidomethyl)-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound [N-]=[N+]=NC[C@H]1CN(C(=O)O1)C1=CN=C2OCC(=O)NC2=N1 JVIISELWCFVRTJ-YFKPBYRVSA-N 0.000 description 1
- IYWLNGPBZLKFNL-RXMQYKEDSA-N 6-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound C1C(=O)NC2=C(O1)N=CC(=N2)N1C[C@@H](OC1=O)CO IYWLNGPBZLKFNL-RXMQYKEDSA-N 0.000 description 1
- IYFHGBJHRIRYIF-LBPRGKRZSA-N 6-[(5S)-5-[[2-(4-ethyl-6-fluoro-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CCN1C(=O)C=NC2=CC=C(F)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 IYFHGBJHRIRYIF-LBPRGKRZSA-N 0.000 description 1
- MXKGRWQRXOREGX-LBPRGKRZSA-N 6-[(5S)-5-[[2-(6-fluoro-4,8-dimethyl-3-oxoquinoxalin-5-yl)oxyethylamino]methyl]-2-oxo-1,3-oxazolidin-3-yl]-4H-pyrazino[2,3-b][1,4]oxazin-3-one Chemical compound CN1C(=O)C=NC2=C(C)C=C(F)C(OCCNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 MXKGRWQRXOREGX-LBPRGKRZSA-N 0.000 description 1
- XYYPQFQSSBCRMR-UHFFFAOYSA-N 6-chloro-4H-pyrazino[2,3-b][1,4]thiazin-3-one Chemical compound ClC1=CN=C2SCC(=O)NC2=N1 XYYPQFQSSBCRMR-UHFFFAOYSA-N 0.000 description 1
- RIWQBZCUHUKCCY-UHFFFAOYSA-N 8-bromo-7-fluoro-1-methyl-3,4-dihydroquinoxalin-2-one Chemical compound BrC=1C(=CC=C2NCC(N(C=12)C)=O)F RIWQBZCUHUKCCY-UHFFFAOYSA-N 0.000 description 1
- YIJIJGYAIZBDNW-UHFFFAOYSA-N 8-bromo-7-fluoro-1-methylquinolin-2-one Chemical compound FC1=CC=C2C=CC(=O)N(C)C2=C1Br YIJIJGYAIZBDNW-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241001232615 Acinetobacter baumannii ATCC 19606 = CIP 70.34 = JCM 6841 Species 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 229930183010 Amphotericin Natural products 0.000 description 1
- QGGFZZLFKABGNL-UHFFFAOYSA-N Amphotericin A Natural products OC1C(N)C(O)C(C)OC1OC1C=CC=CC=CC=CCCC=CC=CC(C)C(O)C(C)C(C)OC(=O)CC(O)CC(O)CCC(O)C(O)CC(O)CC(O)(CC(O)C2C(O)=O)OC2C1 QGGFZZLFKABGNL-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000006448 Buruli Ulcer Diseases 0.000 description 1
- 208000023081 Buruli ulcer disease Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 240000001546 Byrsonima crassifolia Species 0.000 description 1
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 1
- KQVXQEZHXHNJHR-KIYNQFGBSA-N CN1C(=O)C=NC2=CC=C(F)C(OCC(O)CNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 Chemical compound CN1C(=O)C=NC2=CC=C(F)C(OCC(O)CNC[C@H]3CN(C(=O)O3)C3=CN=C4OCC(=O)NC4=N3)=C12 KQVXQEZHXHNJHR-KIYNQFGBSA-N 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 241000943303 Enterococcus faecalis ATCC 29212 Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- 239000002211 L-ascorbic acid Substances 0.000 description 1
- 235000000069 L-ascorbic acid Nutrition 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 239000006142 Luria-Bertani Agar Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 206010066289 Mycobacterium ulcerans infection Diseases 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical class O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- WKDDRNSBRWANNC-UHFFFAOYSA-N Thienamycin Natural products C1C(SCCN)=C(C(O)=O)N2C(=O)C(C(O)C)C21 WKDDRNSBRWANNC-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- CTCBPRXHVPZNHB-VQFZJOCSSA-N [[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate;(2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O.C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O CTCBPRXHVPZNHB-VQFZJOCSSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- 229940009444 amphotericin Drugs 0.000 description 1
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 150000003975 aryl alkyl amines Chemical class 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005872 benzooxazolyl group Chemical group 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052792 caesium Inorganic materials 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229940041011 carbapenems Drugs 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 229960000484 ceftazidime Drugs 0.000 description 1
- NMVPEQXCMGEDNH-TZVUEUGBSA-N ceftazidime pentahydrate Chemical compound O.O.O.O.O.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC(C)(C)C(O)=O)C=2N=C(N)SC=2)CC=1C[N+]1=CC=CC=C1 NMVPEQXCMGEDNH-TZVUEUGBSA-N 0.000 description 1
- 229960004755 ceftriaxone Drugs 0.000 description 1
- VAAUVRVFOQPIGI-SPQHTLEESA-N ceftriaxone Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C(=O)NN1C VAAUVRVFOQPIGI-SPQHTLEESA-N 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- ITKVLPYNJQOCPW-UHFFFAOYSA-N chloro-(chloromethyl)-dimethylsilane Chemical compound C[Si](C)(Cl)CCl ITKVLPYNJQOCPW-UHFFFAOYSA-N 0.000 description 1
- BULLHNJGPPOUOX-UHFFFAOYSA-N chloroacetone Chemical compound CC(=O)CCl BULLHNJGPPOUOX-UHFFFAOYSA-N 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000005507 decahydroisoquinolyl group Chemical group 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N deuterated methanol Substances [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- WDRWZVWLVBXVOI-QTNFYWBSSA-L dipotassium;(2s)-2-aminopentanedioate Chemical compound [K+].[K+].[O-]C(=O)[C@@H](N)CCC([O-])=O WDRWZVWLVBXVOI-QTNFYWBSSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229960001484 edetic acid Drugs 0.000 description 1
- 230000007831 electrophysiology Effects 0.000 description 1
- 238000002001 electrophysiology Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960003276 erythromycin Drugs 0.000 description 1
- 229960000285 ethambutol Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- QUPDWYMUPZLYJZ-UHFFFAOYSA-N ethyl Chemical group C[CH2] QUPDWYMUPZLYJZ-UHFFFAOYSA-N 0.000 description 1
- ZANNOFHADGWOLI-UHFFFAOYSA-N ethyl 2-hydroxyacetate Chemical compound CCOC(=O)CO ZANNOFHADGWOLI-UHFFFAOYSA-N 0.000 description 1
- PVBRSNZAOAJRKO-UHFFFAOYSA-N ethyl 2-sulfanylacetate Chemical compound CCOC(=O)CS PVBRSNZAOAJRKO-UHFFFAOYSA-N 0.000 description 1
- 239000004403 ethyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010228 ethyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940117360 ethyl pyruvate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 201000006674 extrapulmonary tuberculosis Diseases 0.000 description 1
- 238000010265 fast atom bombardment Methods 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960003923 gatifloxacin Drugs 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- 244000000058 gram-negative pathogen Species 0.000 description 1
- 244000000059 gram-positive pathogen Species 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229960002182 imipenem Drugs 0.000 description 1
- GSOSVVULSKVSLQ-JJVRHELESA-N imipenem hydrate Chemical compound O.C1C(SCCNC=N)=C(C(O)=O)N2C(=O)[C@H]([C@H](O)C)[C@H]21 GSOSVVULSKVSLQ-JJVRHELESA-N 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- QWXYZCJEXYQNEI-OSZHWHEXSA-N intermediate I Chemical compound COC(=O)[C@@]1(C=O)[C@H]2CC=[N+](C\C2=C\C)CCc2c1[nH]c1ccccc21 QWXYZCJEXYQNEI-OSZHWHEXSA-N 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000003951 lactams Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 239000008263 liquid aerosol Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 229940041033 macrolides Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 238000007392 microtiter assay Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 235000013919 monopotassium glutamate Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 description 1
- 229960003702 moxifloxacin Drugs 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 150000002960 penicillins Chemical class 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 229960002292 piperacillin Drugs 0.000 description 1
- WCMIIGXFCMNQDS-IDYPWDAWSA-M piperacillin sodium Chemical compound [Na+].O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC=CC=1)C(=O)N[C@@H]1C(=O)N2[C@@H](C([O-])=O)C(C)(C)S[C@@H]21 WCMIIGXFCMNQDS-IDYPWDAWSA-M 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 230000003334 potential effect Effects 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 208000008128 pulmonary tuberculosis Diseases 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- ZHNFLHYOFXQIOW-LPYZJUEESA-N quinine sulfate dihydrate Chemical compound [H+].[H+].O.O.[O-]S([O-])(=O)=O.C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21.C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 ZHNFLHYOFXQIOW-LPYZJUEESA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 238000011125 single therapy Methods 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229940056729 sodium sulfate anhydrous Drugs 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005156 substituted alkylene group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 235000011044 succinic acid Nutrition 0.000 description 1
- 150000003444 succinic acids Chemical class 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-M sulfamate Chemical compound NS([O-])(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-M 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006089 thiamorpholinyl sulfoxide group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 238000001845 vibrational spectrum Methods 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Los compuestos de Fórmula I y Fórmula (B) se describen en el presente documento junto con sus estereoisómeros, sales, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos. Estos compuestos son útiles para matar o inhibir el crecimiento de un microorganismo seleccionado del grupo que consiste en bacterias, virus, hongos y protozoos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos heterocíclicos antibacterianos y su síntesis
Campo de la invención
La presente divulgación se refiere al campo de la química médica y más particularmente a la síntesis, caracterización y desarrollo de inhibidores de la topoisomerasa bacteriana efectivos contra especies bacterianas Gram-positivas y Gram-negativas con un amplio espectro de actividad antibacteriana. En particular, la presente divulgación se refiere a compuestos de Fórmula I, sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos y composiciones farmacéuticas que los contienen como ingrediente activo.
Antecedentes de la invención
Los agentes antimicrobianos son básicamente las sustancias que matan o inhiben el crecimiento de microorganismos. Estas sustancias pueden derivarse de fuentes naturales o pueden prepararse sintéticamente. Debido a su utilidad en el tratamiento de infecciones, los agentes antimicrobianos siguen siendo una preocupación seria para la Organización Mundial de la Salud, ya que forman componentes esenciales de los fármacos utilizados para la salud y el bienestar humano y animal (Bush, K., ASM News, 2004, 70, 282-287).
A pesar de los esfuerzos para proporcionar antimicrobianos mejorados en el mercado, las bacterias continúan evolucionando sobre estos agentes antimicrobianos recientemente desarrollados a través de su mecanismo de resistencia eficiente. La resistencia a los agentes antimicrobianos se ha convertido en un problema de salud pública mundial en los últimos años. El creciente impacto adverso en la salud pública, por lo tanto, sigue siendo la fuerza motriz para la innovación y el desarrollo de nuevos agentes antimicrobianos.
El mundo espera tener fármacos efectivos disponibles para cada enfermedad, pero la resistencia es una amenaza creciente para este régimen y, por lo tanto, es necesario desarrollar compuestos biológicamente activos que muestren una potente actividad antimicrobiana. En base a su diversidad estructural y actividad biológica de amplio espectro, los compuestos heterocíclicos podrían considerarse compuestos prometedores que podrían superar los problemas de resistencia existentes con los agentes antimicrobianos actuales.
Sumario de la invención
La presente divulgación proporciona un compuesto de Fórmula I

o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde que el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa
un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C i-6, alquilamino C i-6, alcoxi C i-6, haloalcoxi C i-6 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; n es 0 a 2; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
La presente divulgación se refiere además a un compuesto de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, para su uso en la destrucción o inhibición del crecimiento de un microorganismo seleccionado del grupo que consiste en bacterias, virus, hongos y protozoos. La presente divulgación proporciona un compuesto de Fórmula (B)

o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Y1 se selecciona de N o CR7 e Y2 es N; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
La presente divulgación se refiere además a un compuesto de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, para uso como medicamento.
La presente divulgación se refiere además a un compuesto de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, para su uso en la destrucción o inhibición del crecimiento de un microorganismo seleccionado del grupo que consiste en bacterias, virus, hongos y protozoos. La presente divulgación se refiere además a un compuesto de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, para su uso en el tratamiento de una infección bacteriana causada por una bacteria Gram-positiva, o una bacteria Gram-negativa seleccionada de Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis, Enterococcus faecium, Legionella pneumophila, Mycoplasma pneumoniae, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus houseri, Citrobacter freundii, Citrobacter kosari, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, Mycobacterium tuberculosis.
La presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo junto con un vehículo farmacéuticamente aceptable.
La presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo junto con un vehículo farmacéuticamente aceptable, y en combinación con al menos un antibiótico.
La presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas,
formas ópticamente activas y derivados farmacéuticamente activos de los mismos, comprendiendo dicho proceso hacer reaccionar compuestos de Fórmula (A) y compuestos de Fórmula (B) en presencia de al menos un agente reductor y un adsorbente para obtener los compuestos de Fórmula I.
En la presente memoria se describe un proceso de preparación de una composición que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, junto con un vehículo farmacéuticamente aceptable.
En la presente memoria se describe un proceso de preparación de una composición que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, junto con un vehículo farmacéuticamente aceptable, opcionalmente en combinación con una o más de otras composiciones farmacéuticas.
Estas y otras características, aspectos y ventajas del presente contenido se entenderán mejor con referencia a la siguiente descripción. Este resumen se proporciona para presentar una selección de conceptos de forma simplificada. Este resumen no pretende identificar las características clave o las características esenciales de la divulgación, ni pretende ser utilizado para limitar el alcance del contenido.
Descripción detallada
Los expertos en la técnica sabrán que la presente divulgación está sujeta a variaciones y modificaciones distintas de las descritas específicamente. Debe entenderse que la presente divulgación incluye todas esas variaciones y modificaciones. La divulgación también incluye todas las etapas, características, composiciones y compuestos a los que se hace referencia o se indica en esta memoria descriptiva, de forma individual o colectiva, y todas y cada una de las combinaciones de cualquiera o más de dichas etapas o características.
Definiciones
Por conveniencia, antes de una descripción adicional de la presente divulgación, aquí se recopilan ciertos términos empleados en la memoria descriptiva y ejemplos. Estas definiciones deben ser leídas a la luz del resto de la divulgación y entendidas por un experto en la técnica. Los términos utilizados en la presente memoria tienen los significados reconocidos y conocidos por los expertos en la técnica, sin embargo, por conveniencia y exhaustividad, los términos particulares y sus significados se establecen a continuación.
Los artículos "un", "una" y "el" se utilizan para referirse a uno o más de uno (es decir, al menos a uno) del objeto gramatical del artículo.
A lo largo de la descripción y las reivindicaciones que siguen, a menos que el contexto requiera lo contrario, la palabra "comprenden", y variaciones tales como "comprende" y "que comprende", se entenderá que implica la inclusión de un número entero o etapa o grupo de números enteros establecidos, pero no con exclusión de cualquier otro número entero o etapa o grupo de números enteros o etapas.
El término "que incluye" se utiliza para significar "que incluye, pero no se limita a". "Que incluye" y " que incluye, pero no se limita a" se usan indistintamente.
En las fórmulas estructurales proporcionadas en la presente memoria y a lo largo de la presente divulgación, se ha indicado el significado de los siguientes términos, a menos que se indique específicamente lo contrario.
En esta memoria descriptiva, el prefijo Cx-y como se usa en términos como alquilo Cx-y y similares (donde x e y son números enteros) indica el rango numérico de átomos de carbono que están presentes en el grupo; por ejemplo, alquilo C1-6 incluye alquilo C1 (metilo), alquilo C2 (etilo), alquilo C3 (propilo e isopropilo), y alquilo C4 (butilo, 1-metilpropilo, 2-metilpropilo y í-butilo). A menos que se indique específicamente, el átomo de enlace de un grupo puede ser cualquier átomo adecuado de ese grupo; por ejemplo, propilo incluye prop-1 -ilo y prop-2-ilo.
El término "alquilo" se refiere a una cadena hidrocarbonada saturada ramificada o no ramificada monoradical que tiene de 1 a 6 átomos de carbono. Este término se ejemplifica con grupos tales como n-butilo, iso-butilo, t-butilo, n-hexilo, n-decilo y similares. Los grupos pueden estar opcionalmente sustituidos.
El término "haloalquilo", como se usa en la presente memoria, se refiere a un grupo alquilo en donde uno o más átomos de hidrógeno están reemplazados por el mismo número de átomos de halógeno idénticos o diferentes. El término "haloalquilo" está ejemplificado por grupos tales como clorometilo, trifluorometilo, 1 -fluoroetilo, 2,2,2-trifluoroetilo y similares.
El término "alquileno" se refiere a una cadena hidrocarbonada saturada ramificada o no ramificada diradical que tiene de 1 a 6 átomos de carbono. Este término se ejemplifica con grupos tales como metileno, etileno, propileno, butileno, hexileno y similares. Los grupos pueden estar opcionalmente sustituidos. Los grupos alquileno sustituidos representativos incluyen alquilenos sustituidos con hidroxilo, alquileno sustituido con amino.
El término "alquenilo" se refiere a un monoradical de un grupo hidrocarbonado insaturado ramificado o no ramificado que tiene preferiblemente 2, 3, 4 , 5 o 6 átomos de carbono y que tiene 1,2 o 3 dobles enlaces (vinilo), preferiblemente 1 doble enlace. Los grupos pueden estar opcionalmente sustituidos.
El término "cicloalquilo" o "carbociclilo" se refiere a grupos carbocíclicos de 3 a 6 átomos de carbono que tienen un solo anillo cíclico o múltiples anillos condensados que pueden estar parcialmente insaturados. Dichos grupos cicloalquilo incluyen, a modo de ejemplo, estructuras de un solo anillo como ciclopropilo, ciclobutilo, ciclopentilo, ciclopentenilo, ciclohexilo, ciclohexenilo y similares, o estructuras de anillos múltiples o grupos carbocíclicos a los que se fusiona un grupo arilo, por ejemplo, indano, y similares. Los grupos pueden estar opcionalmente sustituidos.
Los términos "alcoxilo" o "alcoxi" se refieren a un grupo alquilo, como se define anteriormente, que tiene un radical oxígeno unido al mismo. Los grupos alcoxilo representativos incluyen metoxi, etoxi, propiloxi, terc-butoxi y similares. Un "éter" es dos hidrocarbonos unidos covalentemente por un oxígeno. En consecuencia, el sustituyente de un alquilo que convierte a ese alquilo en un éter es o se asemeja a un alcoxilo, tal como puede representarse por uno de -O alquilo, -O-alquenilo, -O-alquinilo.
"Halo" o "Halógeno", solo o en combinación con cualquier otro término, significa halógenos como cloro o cloro (Cl), flúor o flúor (F), bromo o bromo (Br) y yodo o yodo (I).
El término "heterociclilo" se refiere a un radical anular heterocíclico que puede estar opcionalmente sustituido con uno o más sustituyentes como se describe en la presente memoria más adelante en la divulgación. El radical del anillo heterociclilo puede unirse a la estructura principal en cualquier heteroátomo o átomo de carbono que dé como resultado la creación de una estructura estable. Además, el término "heterociclilo" se refiere a un radical anular estable de 3 a 7 miembros, que consiste en átomos de carbono y de uno a cinco heteroátomos seleccionados de nitrógeno, fósforo, oxígeno y azufre. Para los fines de esta divulgación, el radical anular heterocíclico puede ser un sistema anular monocíclico, bicíclico o tricíclico, y los átomos de nitrógeno, fósforo, carbono, oxígeno o azufre en el radical anular heterocíclico pueden oxidarse opcionalmente a varios estados de oxidación. Además, el átomo de nitrógeno puede estar opcionalmente cuaternizado; y el radical del anillo puede estar parcial o totalmente saturado. Los grupos heterociclilo preferidos incluyen, sin limitación, azetidinilo, acridinilo, benzodioxolilo, benzodioxanilo, benzofuranilo, carbazolilo, cinolinilo, dioxolanilo, indolizinilo, naftiridinilo, perhidroazepinilo, fenazinilo, fenotiazinilo, fenoxazinilo, ftalazinilo, piridilo, pteridinilo, purinilo, quinazolinilo, qunioxalinilo, quinolinilo, isoquinolinilo, tetrazolilo, imidazolilo, tetrahidroisoquinolinilo, piperidinilo, piperazinilo, homopiperazinilo, 2-oxoazepinilo, azepinilo, pirrolilo, 4-piperidonilo, pirrolidinilo, pirazinilo, pirimidinilo, piridazinilo, oxazolilo, oxazolinilo, triazolilo, indanilo, isoxazolilo, isoxazolidinilol, tiazolilo, tiazolinilo, tiazolidinilo, isotiazolilo, quinuclidinilo, isotiazolidinilo, indolilo, isoindolilo, indolinilo, isoindolinilo, octahidroindolilo, octahidroisoindolilo, quinolilo, isoquinolilo, decahidroisoquinolilo, bencimidazolilo, tiadiazolilo, benzopiranilo, benzotiazolilo, benzooxazolilo, tienilo, morfolinilo, tiomorfolinilo, sulfóxido de tiamorfolinilo, furilo, tetrahidrofurilo, tetrahidropiranilo, cromanilo e isocromanilo.
Como se usa en la presente memoria, se contempla que el término "sustituido" incluya todos los sustituyentes permisibles de los compuestos orgánicos. En un aspecto amplio, los sustituyentes permisibles incluyen sustituyentes acíclicos y cíclicos, ramificados y no ramificados, carbocíclicos y heterocíclicos, aromáticos y no aromáticos de compuestos orgánicos. Los sustituyentes ilustrativos incluyen, por ejemplo, los descritos en la presente memoria anteriormente. Los sustituyentes permisibles pueden ser uno o más e iguales o diferentes para compuestos orgánicos apropiados. Para los fines de esta divulgación, los heteroátomos como el nitrógeno pueden tener un sustituyente de hidrógeno y/o cualquier sustituyente permisible de los compuestos orgánicos descritos en la presente memoria que satisfagan las valencias de los heteroátomos.
El término "cantidad efectiva" significa una cantidad de un compuesto o composición que es suficiente para modificar significativa y positivamente los síntomas y/o afecciones a tratar (p. ej., proporcionar una respuesta clínica positiva). La cantidad efectiva de un ingrediente activo para usar en una composición farmacéutica variará con la afección particular que se esté tratando, la gravedad de la afección, la duración del tratamiento, la naturaleza de la terapia concurrente, el o los ingredientes activos particulares que se empleen, el o los excipientes/vehículos farmacéuticamente aceptables particulares utilizados, la vía de administración y factores similares dentro del conocimiento y la experiencia del médico tratante.
Los compuestos descritos en la presente memoria pueden contener uno o más centros quirales y/o dobles enlaces y, por lo tanto, pueden existir como estereoisómeros, como isómeros de doble enlace (es decir, isómeros geométricos), regioisómeros, enantiómeros o diastereómeros. En consecuencia, las estructuras químicas representadas en la presente memoria abarcan todos los enantiómeros y estereoisómeros posibles de los compuestos ilustrados o identificados, incluida la forma estereoisoméricamente pura (p. ej., geométricamente pura, enantioméricamente pura o diastereoméricamente pura) y mezclas enantioméricas y estereoisoméricas. Las mezclas enantioméricas y estereoisoméricas pueden resolverse en sus componentes enantiómeros o estereoisómeros utilizando técnicas de separación o técnicas de síntesis quiral bien conocidas por los expertos en la técnica. Los compuestos también pueden existir en varias formas tautoméricas que incluyen la forma enol, la forma ceto y mezclas de las mismas. En consecuencia, las estructuras químicas representadas en la presente memoria abarcan todas las formas tautoméricas posibles de los compuestos ilustrados o identificados.
El término "farmacéuticamente aceptable" se refiere a aquellos compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del buen criterio médico, adecuados para su uso en contacto con los tejidos de seres humanos y animales sin excesiva toxicidad, irritación, respuesta alérgica, u otro problema o complicación, acorde con una relación beneficio/riesgo razonable.
"Sal farmacéuticamente aceptable" abarca sales con un ácido o base farmacéuticamente aceptable. Los ácidos farmacéuticamente aceptables incluyen tanto ácidos inorgánicos, por ejemplo, ácido clorhídrico, sulfúrico, fosfórico, difosfórico, bromhídrico, yodhídrico y nítrico, como ácidos orgánicos, por ejemplo, ácido cítrico, fumárico, maleico, málico, mandélico, ascórbico, oxálico, succínico, tartárico, benzoico, acético, metanosulfónico, etanosulfónico, bencenosulfónico o p-toluenosulfónico. Las bases farmacéuticamente aceptables incluyen hidróxidos de metales alcalinos (p. ej., sodio o potasio) y metales alcalinotérreos (p. ej., calcio o magnesio), y bases orgánicas, por ejemplo, aminas de alquilo, aminas de arilalquilo y aminas heterocíclicas.
Los compuestos discutidos en la presente memoria en muchos casos pueden haber sido nombrados y/o verificados con ACD/Name de ACD/Labs® y/o Chemdraw de CambridgeSoft®.
El término "polimorfos" se refiere a formas cristalinas de la misma molécula, y diferentes polimorfos pueden tener diferentes propiedades físicas como, por ejemplo, temperaturas de fusión, calores de fusión, solubilidades, velocidades de disolución y/o espectros de vibración como resultado de la disposición o conformación de las moléculas en la red cristalina.
El término "solvato", como se usa en la presente memoria, se refiere a una forma cristalina de una sustancia que contiene disolvente.
El término "hidrato" se refiere a un solvato en donde el disolvente es agua.
Como se discutió en la sección de antecedentes, los mecanismos de resistencia efectivos de los microbios (especialmente las bacterias) representan una seria amenaza para la salud humana. Por lo tanto, es necesario desarrollar agentes antimicrobianos (antibacterianos) nuevos y eficientes como medida contra los adversarios que los microbios plantean a la salud humana a nivel mundial. La presente divulgación proporciona compuestos que pueden actuar como agentes antibacterianos eficaces que combaten tanto las cepas resistentes como las no resistentes.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I

o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde que el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-6, alquilamino C1-6, alcoxi C1-6, haloalcoxi C1-6 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6,
haloalcoxi C i-6, o alquilo C i-6; Zi se selecciona de O, S, NH o CH2; y Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Ri se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-5, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno , ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH y CH2; o Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5, o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es CR5; R5 se selecciona de hidrógeno, halógeno, ciano o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas
ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Ri se selecciona de alquilo C i-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, o anillo de heterociclilo saturado o insaturado de 3 7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (---) no representa un enlace; n es 0 a 2; Y1 se selecciona de N o CR7; Y2 es N; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH y CH2; o R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, cloro, flúor, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (-) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2; Y1 se selecciona de N o CR7; Y2 es CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 es alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 es alquilo C1-6, en donde alquilo C1-6 está además sustituido con 1 a 3 grupos seleccionados independientemente de halógeno, amino o hidroxilo; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6 y anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 es hidrógeno; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 es flúor; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 es cloro; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo
C i-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 e hidroxilo; R3 es hidrógeno; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 a 2; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 es hidroxilo; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 e hidroxilo; R3 es metilo; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 a 2; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6,
alquilamino C i-6, aminoalquileno C i-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 e hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (-) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-5 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-5, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-5, alquilamino C1-5, alcoxi C1-5, haloalcoxi C1-5 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n1 es 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-5, haloalquilo C1-5, haloalcoxi C1-5, o alquilo C1-5; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, C1-4 alcoxi, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-4, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-4, alquilamino C1-4, alcoxi C1-4, haloalcoxi C1-4 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6,
cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-4, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-4, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-4, alquilamino C1-4, alcoxi C1-4, haloalcoxi C1-4 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y el anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-4, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-4, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-4, alquilamino C1-4, alcoxi C1-4, haloalcoxi C1-4 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-4, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-4, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-4, alquilamino C1-4, alcoxi C1-4, haloalcoxi C1-4 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-5, alquenilo C2-5, cicloalquilo C3-5, alquilamino C1-5, aminoalquileno C1-5, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-5, alquenilo C2-5, cicloalquilo C3-5, alquilamino C1-5, aminoalquileno C1-5, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-5, alcoxi C1-5, haloalcoxi C1-5, cicloalquilo C3-5, cicloalquilamino C3-5, aminocicloalquilo C3-5, cicloalquilhidroxi C3-6, alquilamino C1-5, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-3, ciano
o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-3, hidroxilo o amino; X i es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-3, haloalquilo C1-3, haloalcoxi C1-3, o alquilo C1-3; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-3, haloalquilo C1-3, haloalcoxi C1-3, o alquilo C1-3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-3, alquilamino C1-3, alcoxi C1-3, haloalcoxi C1-3 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4, o anillo de carbociclilo o heterociclilo saturado de 4-7 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4 y carbociclilo o heterociclilo saturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, O-PO3H2, alquilo C1-4, alcoxi C1-4, haloalcoxi C1-4, cicloalquilo C3-5, cicloalquilamino C3-5, aminocicloalquilo C3-5, cicloalquilhidroxi C3-6, alquilamino C1-4, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-2, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-2, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-2, alquilamino C1-3, alcoxi C1-2, haloalcoxi C1-2 o COOH; o X3 es CH2 u O; y X3 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4, o anillo de carbociclilo o heterociclilo saturado de 4-6 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4 y carbociclilo o heterociclilo saturado de 4-6 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, O-PO3H2, alquilo C1-4, alcoxi C1-4, haloalcoxi C1-4, cicloalquilo C3-4, cicloalquilamino C3-5, aminocicloalquilo C3-5, cicloalquilhidroxi C3-6, alquilamino C1-4, o anillo de heterociclilo saturado o insaturado de 3-5 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1-2, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1-2, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (---) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-2, alquilamino C1-3, alcoxi C1-2, haloalcoxi C1-2 o COOH; o X3 es CH2 u O; y X3 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4, o anillo de carbociclilo o heterociclilo saturado de 4-7 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4 y carbociclilo o heterociclilo saturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, O-PO3H2, alquilo C1-4, alcoxi C1-4, haloalcoxi C1-4, cicloalquilo C3-5, cicloalquilamino C3-5, aminocicloalquilo C3-5, cicloalquilhidroxi C3-6, alquilamino C1-4, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C1, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C1, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1, o alquilo C1; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1, haloalquilo C1, haloalcoxi C1, o alquilo C1; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-2, alquilamino C1-3, alcoxi C1-2, haloalcoxi C1-2 o COOH; o X3 es CH2 u O; y X3 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se
selecciona de hidrógeno, halógeno, ciano, alcoxi C 1-2, haloalquilo C 1-2, haloalcoxi C 1-2, o alquilo C1-2; Z i se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo C1-4, haloalquilo C 1-4, cicloalquilo C3-4, alquilamino C1-4, o anillo de carbociclilo o heterociclilo saturado de 4-7 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1-4, haloalquilo C1-4, cicloalquilo C3-4, alquilamino C1-4 y carbociclilo o heterociclilo saturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, O-PO3H2, alquilo C 1-4, alcoxi C 1-4, haloalcoxi C 1-4, cicloalquilo C3-5, cicloalquilamino C3-5, aminocicloalquilo C3-5, cicloalquilhidroxi C3-6, alquilamino C 1-4, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R2 se selecciona de hidrógeno, flúor, alcoxi C 1-2, ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C 1-2, hidroxilo o amino; X 1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-2, haloalquilo C1-2, haloalcoxi C 1-2, o alquilo C1-2; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1-2, haloalquilo C1-2, haloalcoxi C1-2, o alquilo C1-2; X3 es N o CR6; y X4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C 1, alquilamino C 1-3, alcoxi C 1, haloalcoxi C1 o COOH; o X3 es CH2 u O; y X3 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y 1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1, o alquilo C1; Z 1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2FC3, CH2CHFCH3, CH2FC2CH3, CH2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2FC2CH2NH2,

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X 1 es N o CR4; R4 se selecciona de H, F, CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o CR6; y X4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH 3 )n H 2 , CH 2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y 1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de H, OH, alquilo C1-6 o F.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, CH2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2


R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X i es N o CR4; R4 se selecciona de H, F, CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o CR6; y X4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)n H2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y 1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, CH2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2,

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X 1 es N o CR4; R4 se selecciona de H, F, CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o CR6; y X4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)n H2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n1 es 0 o 1; Y 1 es CR7; R7 es H; Y2 es N; Z 1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, CH2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2,

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X 1 es N o CR4; R4 se selecciona de H, F, CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o CR6; y X4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)n H2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y 1 es CR7; Y2 es CR7; R7 es H; Z 1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, C2H5, o CH2CH2OH; R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X1 es N o
CR4; R4 se selecciona de H, F, CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o
CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, CH2NHCH3, OCH3, OCF3, y CH3; o X3 es CH2 u O; y X4 es CH2 cuan punteada (----) no representa un enlace; m es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, C n , OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, C H 2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2,

R2 se selecciona de H, F o Cl; R3 se selecciona de H, F, OCH3, OH o NH2; X1 es N o CR4; R4 se selecciona de H, F,
CN, OCH3, o CH3; X2 es N o CR5; Rs se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace;
n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, C H 2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2,

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H u OH; X1 es N o CR4; R4 se selecciona de H, F,
CN, OCH3, o CH3; X2 es N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace;
n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3,
CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, C H 2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X1 es CR4; R4 es H; X2 es
N o CR5; R5 se selecciona de H, F, CN, OCH3, CF3, OCF3, o CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada
( ) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, C H 2NHCH3, OCH3, O C F3, o
CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, C n , OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, C H 2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; X1 es CR4; R4 es H; X2 es
N; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la lín (----) no representa un enlace; n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH3, CH2CH3, CH(CH3)2, ciclopropilo, ciclobutilo, CH2CF3, CH2CHFCH3, CH2CF2CH3, C H 2CH(OH)CH3, CH2CH2OH, CH2CH2OCH3, CH2CH(OCH3)CH3, CH2CH2NH2, CH2CH2NHCH3, CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH2CHFCH2NH2, CH2CF2CH2NH2,

R2 se selecciona de H, F, Cl, OCH3, CN u OH; R3 se selecciona de H, F, OCH3, OH o NH2; Xi es CR4; R4 es H; X2 es
CR5; R5 es H o CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (— ) representa un enlace; R6 se selecciona de H, CN, COOH, CH2NH2, CH(CH3)NH2, CH2NHCH3, OCH3, OCF3, o CH3; o X3 es CH2 u O; y X4 e línea punteada (----) no representa un enlace; n1 es 0 o 1; Y1, e Y2 se seleccionan independientemente de N o CR7;
R7 se selecciona de H, F, CN, OCH3, o CH3; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de CH 3, C 2H5, o CH2CH2OH; R2 se selecciona de H, Cl o F; R3 se selecciona de H, CH3, y OH; X1 es CR4; R4 es H; X2 es N o CR5;
R5 es H o CH3; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 es H o CH3; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; n1 es 0 o 1; Y1 es CR7; Y2 es N o
C R 7; R7 es H; Z1 es O o S; y R8 es H.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R1 se selecciona de alquilo
C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H,
O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; y R9 se selecciona de hidrógeno, o alquilo C1-6.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 o hidroxilo.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde X1 es N o CR4; y R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde X2 es N o CR5; y R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; y R6 se selecciona de hidrógeno, ciano, alquilo C1-6, alquilamino
C1-6, alcoxi C1-6, haloalcoxi C1-6 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde m es 0 a 2.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Y1, e Y2 se seleccionan independientemente de N o CR7; y R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Z1 se selecciona de O, S, NH o CH2.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, que se selecciona de un grupo que consiste en:

(S)-6-(5-(((2-((5-metil-6-oxo-5 ,6-dihidro-1,5-naftiridin-4-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirido[3 ,2-b][1,4]oxazin-3(4H)-ona (Compuesto 1),

(S)-6-(5-(((2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidina-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 2),

(R)-6-(5-(((2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidina-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 3),

6-((5S)-5-(((1-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)propan-2-il)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 4),

(S)-6-(5-(((2-((7-fluoro-1,4-dimetil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 5),

(S)-6-(5-(((2-((3-cloro-5-metil-6-oxo-5 ,6-dihidro-1,5-naftiridin-4-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 6),

(S)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 7),

(R)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 8),

(S)-6-(5-(((2-((6-fluoro-2 ,4-dimetil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 9),

(S)-5-(((2-((6-fluoro-4-metil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-3-(3-oxo-3 ,4-dihidro-2H-pirazino[2 ,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Compuesto 10),
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, que se selecciona de un grupo que consiste en:

(S)-6-(5-(((2-((4-etil-6-fluoro-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 11),

(S)-6-(5-(((2-((6-fluoro-4 ,8-dimetil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 12),

6-((5S)-5-(((3-((6-fluoro-4-metil-3-oxo-3 ,4-dihidroquinoxalin-5-il)oxi)-2-hidroxipropil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Compuesto 13),

(S)-5-(((2-((7-fluoro-1-(2-hidroxietil)-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-3-(3-oxo-3 ,4-dihidro-2H-pirazino[2 ,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Compuesto 14), y

(5S)-5-(((3-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)-2-hidroxipropil)amino)metil)-3-(3-oxo-3 ,4-dihidro-2H-pirazino[2 ,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Compuesto 15)
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula (B) o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde

Y1 se selecciona de N o CR7 e Y2 es N; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2; y Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula (B) o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Y1 se selecciona de N o CR7 e Y2 es N; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, haloalquilo C1-4, haloalcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O, S, NH o CH2; y Rs se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula (B) o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, en donde Y1 se selecciona de N o CR7 e Y2 es N; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-4, o alquilo C1-4; Z1 se selecciona de O o S; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-4 o flúor.
Según una realización, la presente descripción se refiere a un compuesto de Fórmula (B) o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, que se selecciona de un grupo que consiste en:

(S)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Intermedio V)

(R)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2 ,3-b][1,4]oxazin-3(4H)-ona (Intermedio VI)

(S)-S-(aminometil)-3-(3-oxo-3 ,4-dihidro-2H-pirazino[2 ,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Intermedio VII).
Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, comprendiendo dicho proceso: (a) hacer reaccionar un compuesto de Fórmula (C) y un compuesto de Fórmula (D) en presencia de al menos un catalizador y al menos un disolvente para obtener un compuesto de Fórmula (E); (b) hacer reaccionar el compuesto de Fórmula (E) y al menos un compuesto de nitrógeno para obtener un compuesto de Fórmula (F); y (c) reducir el compuesto de Fórmula (F) para obtener un compuesto de Fórmula (B).

Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula (B) como se describe en la presente memoria, en donde al menos un catalizador se selecciona de un grupo que consiste en catalizador que contiene Pd, t-BuXPhos-Pd, Pd( OAc)2, y combinaciones de los mismos; el al menos un disolvente se selecciona del grupo que consiste en THF, tolueno, dioxano y combinaciones de los mismos, el al menos un compuesto nitrogenado es NaN3.
Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula (B) como se describe en la presente memoria, en donde la reducción del compuesto de Fórmula (F) para obtener un compuesto de Fórmula (B) se lleva a cabo en presencia de un agente reductor seleccionado de trifenilfosfina (Ph3P)/THF-H2O, o hidrógeno y paladio carbono (H2/Pd-C).
Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, comprendiendo dicho proceso hacer reaccionar compuestos de Fórmula (A) y compuestos de Fórmula (B) en presencia de al menos un agente reductor y un adsorbente para obtener los compuestos de Fórmula I, en donde R1 de Fórmula (A) se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X3 es N o C R 6; y X4 es C R 6 o alquilo C1-6 cuando la línea de puntos (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-6, alquilamino C1-6, alcoxi C1-6, haloalcoxi C1-6 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; y m es 0 a 2 ; R8 de Fórmula (B) se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2; y R1 de Fórmula I se selecciona de alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, di(alquil C1-6)amino, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-6, alquilamino C1-6, alcoxi C1-6, haloalcoxi C1-6, o COOH, en donde alquilo C1-6 y alquilamino C1-6 están opcionalmente sustituidos con uno o más grupos seleccionados de COOH, hidroxilo, amino o alquilo C1-6; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2 ; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor.

Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I como se describe en la presente memoria, en donde dicho proceso comprende un compuesto de Fórmula (A)

Ri de Fórmula (A) se selecciona de alquilo C i-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C1-6, alquenilo C2-6, cicloalquilo C3-6, alquilamino C1-6, aminoalquileno C1-6, y carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3H, O-PO3H2, COOR9, CONHR9, SO2NHR9, metilsulfona, alquilo C1-6, alcoxi C1-6, haloalcoxi C1-6, cicloalquilo C3-6, cicloalquilamino C3-6, aminocicloalquilo C3-6, cicloalquilhidroxi C3-6, alquilamino C1-6, di(alquil C1-6)amino, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C1-6; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C1-6 e hidroxilo; R3 se selecciona de hidrógeno, alquilo C1-6, flúor, alcoxi C1-6, hidroxilo o amino; X1 es N o CR4; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X2 es N o CR5; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; X3 es N o C R 6; y X4 es C R 6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C1-6, alquilamino C1-6, alcoxi C1-6, haloalcoxi C1-6 o COOH; o X3 es CH2 u O; y X4 es CH2 cuando la línea punteada (----) no representa un enlace; y m es 0 a 2.
Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I como se describe en la presente memoria, en donde dicho proceso comprende un compuesto de Fórmula (B)

donde R8 de Fórmula (B) se selecciona de hidrógeno, hidroxilo, alquilo C1-6 o flúor; Y1, e Y2 se seleccionan independientemente de N o CR7; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C1-6, haloalquilo C1-6, haloalcoxi C1-6, o alquilo C1-6; Z1 se selecciona de O, S, NH o CH2.
Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I como se describe en la presente memoria, en donde al menos un agente reductor se selecciona del grupo que consiste en borohidruro de sodio, cianoborohidruro de sodio, triacetoxiborohidruro de sodio y combinaciones de los mismos. Según una realización, la presente divulgación se refiere a un proceso de preparación de compuestos de Fórmula I como se describe en la presente memoria, en donde el adsorbente se selecciona del grupo que consiste en tamices moleculares, gel de sílice, zeolitas, sulfato de sodio anhidro, sulfato de magnesio anhidro, carbón activado y combinaciones de los mismos.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, para su uso como medicamento.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, para su uso como medicamento.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, para su uso en la
destrucción o inhibición del crecimiento de un microorganismo seleccionado del grupo que consiste en bacterias Grampositivas y Gram-negativas.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, para su uso en la destrucción o inhibición del crecimiento de bacterias Gram-positivas y Gram-negativas.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos del mismo, para su uso en el tratamiento de una infección bacteriana causada por una bacteria Gram-positiva o una bacteria Gram-negativa seleccionada de Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis Enterococcus faecium, Legionella pneumophila. Mycoplasma pneumoniae, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus houseri, Citrobacter freundii, Citrobacter kosari, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, Mycobacterium tuberculosis.
Según una realización, la presente divulgación se refiere a medicamentos que incluyen un compuesto de Fórmula I, o Fórmula (B), o una sal de adición del compuesto de Fórmula I o Fórmula (B), con un ácido o base farmacéuticamente aceptable. Estos medicamentos encuentran su uso en la terapéutica, especialmente en el tratamiento de infecciones bacterianas causadas por bacterias sensibles a fármacos y resistentes a fármacos, incluida la resistencia a las quinolonas pertenecientes a especies Gram positivas y Gram negativas seleccionadas de Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis Enterococcus faecium, Legionella pneumophila. Mycoplasma pneumonía, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus houseri, Citrobacter freundii, Citrobacter kosari, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, Mycobacterium tuberculosis.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y un derivado farmacéuticamente activo del mismo junto con un vehículo farmacéuticamente aceptable.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y un derivado farmacéuticamente activo del mismo junto con un vehículo farmacéuticamente aceptable, y en combinación con al menos un antibiótico.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que incluye un compuesto de Fórmula I o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y un derivado farmacéuticamente activo del mismo, y al menos un vehículo, diluyente o excipiente farmacéuticamente aceptable.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que incluye un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivado farmacéuticamente activo del mismo, para su uso en el tratamiento de una infección bacteriana causada por una bacteria Gram-positiva o Gram-negativa seleccionada de Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis, Enterococcus faecium, Legionella pneumophila. Mycoplasma pneumonia, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus houseri, Citrobacter freundii, Citrobacter kosari, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, Mycobacterium tuberculosis.
La infección bacteriana está causada por Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis, Enterococcus faecium, Legionella pneumophila. Mycoplasma pneumonia, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus
houserí, Citrobacter freundii, Citrobacter kosarí, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, o Mycobacterium tuberculosis.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y un derivado farmacéuticamente activo del mismo, junto con un vehículo farmacéuticamente aceptable, opcionalmente en combinación con una o más de otras composiciones farmacéuticas.
Según una realización, la presente divulgación se refiere a una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y un derivado farmacéuticamente activo del mismo, y un diluyente o vehículo farmacéuticamente aceptable.
Según una realización, la presente divulgación se refiere a una composición que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivado farmacéuticamente activo del mismo, y un vehículo.
La expresión "farmacéuticamente aceptable" incluye compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del buen criterio médico, adecuados para su uso en contacto con los tejidos de seres humanos y animales sin excesiva toxicidad, irritación, respuesta alérgica, u otro problema o complicación, acorde con una relación beneficio/riesgo razonable.
Los compuestos de Fórmula I o Fórmula (B) pueden formar sales ácidas o básicas estables farmacéuticamente aceptables, y en tales casos puede ser apropiada la administración de un compuesto como una sal. Los ejemplos de sales de adición de ácido incluyen acetato, adipato, ascorbato, benzoato, bencenosulfonato, bicarbonato, bisulfato, butirato, canforato, canforsulfonato, colina, citrato, ciclohexilsulfamato, dietilendiamina, etanosulfonato, fumarato, glutamato, glicolato, hemisulfato, 2-hidroxietilsulfonato, heptanoato , hexanoato, hidrocloruro, hidrobromuro, hidroyoduro, hidroximaleato, lactato, malato, maleato, metanosulfonato, meglumina, 2-naftalenosulfonato, nitrato, oxalato, pamoato, persulfato, fenilacetato, fosfato, difosfato, picrato, pivalato, propionato, quinato, salicilato, estearato, succinato, sulfamato, sulfanilato, sulfato, tartrato, tosilato (p-toluenosulfonato), trifluoroacetato y undecanoato. Los ejemplos de sales básicas incluyen sales de amonio; sales de metales alcalinos tales como sales de sodio, litio y potasio; sales de metales alcalinotérreos tales como sales de aluminio, calcio y magnesio; sales con bases orgánicas tales como sales de diciclohexilamina y N10 metil-D-glucamina; y sales con aminoácidos tales como arginina, lisina, ornitina, etc. Además, los grupos básicos que contienen nitrógeno pueden cuaternizarse con agentes tales como: haluros de alquilo inferior, tales como haluros de metilo, etilo, propilo y butilo; sulfatos de dialquilo tales como dimetilo, dietilo, dibutilo; sulfatos de diamilo; haluros de cadena larga tales como haluros de decilo, laurilo, miristilo y estearilo; haluros de arilalquilo tales como bromuro de bencilo y otros. Se prefieren las sales fisiológicamente aceptables no tóxicas, aunque pueden ser útiles otras sales, como para aislar o purificar el producto.
Las sales pueden formarse por medios convencionales, como, por ejemplo, haciendo reaccionar la forma de base libre del producto con uno o más equivalentes del ácido apropiado en un disolvente o medio en donde la sal sea insoluble, o en un disolvente como el agua, que se elimina in vacuo o mediante liofilización o intercambiando los aniones de una sal existente por otro anión en una resina de intercambio iónico adecuada.
Las composiciones de la divulgación pueden estar en una forma adecuada para uso oral (por ejemplo, como comprimidos, pastillas, cápsulas duras o blandas, suspensiones acuosas u oleosas, emulsiones, polvos o gránulos dispersables, jarabes o elixires), para uso tópico (por ejemplo, como cremas, ungüentos, geles o soluciones o suspensiones acuosas u oleosas), para administración por inhalación (por ejemplo, como un polvo finamente dividido o un aerosol líquido), para administración por insuflación (por ejemplo, como un polvo finamente dividido) o para administración parenteral (por ejemplo, como una solución acuosa u oleosa estéril para dosificación intravenosa, subcutánea, intramuscular o intramuscular o como un supositorio para dosificación rectal).
La presente divulgación se refiere a un proceso de preparación de una composición que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivado farmacéuticamente activo del mismo junto con un vehículo.
La presente divulgación se refiere a un proceso de preparación de una composición farmacéutica que comprende un compuesto de Fórmula I, o Fórmula (B), o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivado farmacéuticamente activo del mismo, junto con un vehículo farmacéuticamente aceptable, opcionalmente en combinación con una o más de otras composiciones farmacéuticas.
Las composiciones de la presente divulgación pueden obtenerse mediante procedimientos convencionales utilizando excipientes farmacéuticos convencionales bien conocidos en la técnica. Así, las composiciones destinadas a uso oral pueden contener, por ejemplo, uno o más agentes colorantes, edulcorantes, aromatizantes y/o conservantes.
Los excipientes farmacéuticamente aceptables adecuados para una formulación de comprimido incluyen, por ejemplo, diluyentes inertes tales como lactosa, carbonato de sodio, fosfato de calcio o carbonato de calcio; agentes de granulación y disgregación tales como almidón de maíz o ácido algénico; agentes aglutinantes tales como almidón; agentes lubricantes tales como estearato de magnesio, ácido esteárico o talco; agentes conservantes tales como phidroxibenzoato de etilo o propilo; y antioxidantes, tales como ácido ascórbico. Las formulaciones de comprimidos pueden estar sin recubrir o recubiertas para modificar su disgregación y la subsiguiente absorción del ingrediente activo dentro del tracto gastrointestinal, o para mejorar su estabilidad y/o apariencia, en cualquier caso, utilizando agentes de recubrimiento convencionales o procedimientos bien conocidos en la técnica.
Las composiciones para uso oral pueden estar en forma de cápsulas de gelatina dura en las que el ingrediente activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín, o como cápsulas de gelatina blanda en las que el ingrediente activo se mezcla con agua o aceite como aceite de cacahuete, parafina líquida o aceite de oliva.
Las suspensiones acuosas generalmente contienen el ingrediente activo en forma de polvo fino o en forma de partículas nano o micronizadas junto con uno o más agentes de suspensión, tales como carboximetilcelulosa sódica, metilcelulosa, hidroxipropilmetilcelulosa, alginato de sodio, polivinilpirrolidona, goma tragacanto y goma arábiga; agentes dispersantes o humectantes tales como lecitina o productos de condensación de un óxido de alquileno con ácidos grasos (por ejemplo, estearato de polioxetileno), o productos de condensación de óxido de etileno con alcoholes alifáticos de cadena larga, por ejemplo, heptadecaetilenoxietanol, o productos de condensación de óxido de etileno con ésteres parciales derivados a partir de ácidos grasos y un hexitol, como monooleato de polioxietilensorbitol, o productos de condensación de óxido de etileno con alcoholes alifáticos de cadena larga, por ejemplo, heptadecaetilenoxietanol, o productos de condensación de óxido de etileno con ésteres parciales derivados de ácidos grasos y un hexitol, como monooleato de polioxietilensorbitol, o productos de condensación de óxido de etileno con ésteres parciales derivados de ácidos grasos y anhídridos de hexitol, por ejemplo, monooleato de polietilensorbitán. Las suspensiones acuosas también pueden contener uno o más conservantes tales como p-hidroxibenzoato de etilo o propilo; antioxidantes como el ácido ascórbico); agentes colorantes; agentes aromatizantes; y/o agentes edulcorantes tales como sacarosa, sacarina o aspartamo.
Las suspensiones oleosas pueden formularse suspendiendo el ingrediente activo en un aceite vegetal tal como aceite de cacahuete, aceite de oliva, aceite de sésamo o aceite de coco o en un aceite mineral tal como parafina líquida. Las suspensiones oleosas también pueden contener un agente espesante como cera de abejas, parafina dura o alcohol cetílico. Se pueden añadir agentes edulcorantes como los expuestos anteriormente y agentes aromatizantes para proporcionar una preparación oral apetecible. Estas composiciones pueden conservarse mediante la adición de un antioxidante, comprendiendo el antioxidante ácido ascórbico.
Los polvos y gránulos dispersables adecuados para la preparación de una suspensión acuosa mediante la adición de agua contienen generalmente el ingrediente activo junto con un agente dispersante o humectante, un agente de suspensión y uno o más conservantes. Los agentes dispersantes o humectantes y los agentes de suspensión adecuados se ejemplifican mediante los ya mencionados anteriormente. También pueden estar presentes excipientes adicionales tales como agentes edulcorantes, aromatizantes y colorantes.
Las composiciones farmacéuticas de la divulgación también pueden estar en forma de emulsiones de aceite en agua. La fase oleosa puede ser un aceite vegetal, como el aceite de oliva o el aceite de cacahuete, o un aceite mineral, como, por ejemplo, la parafina líquida o una mezcla de cualquiera de ellos. Los agentes emulsionantes adecuados pueden ser, por ejemplo, gomas naturales como goma arábiga o goma tragacanto, fosfátidos naturales como soja, lecitina, ésteres o ésteres parciales derivados de ácidos grasos y anhídridos de hexitol (por ejemplo, monooleato de sorbitán) y productos de condensación de dichos ésteres parciales con óxido de etileno tales como monooleato de polioxietilensorbitán. Las emulsiones también pueden contener agentes edulcorantes, aromatizantes y conservantes. Los jarabes y elixires pueden formularse con agentes edulcorantes tales como glicerol, propilenglicol, sorbitol, aspartamo o sacarosa, y también pueden contener un agente demulcente, conservante, aromatizante y/o colorante. Las composiciones farmacéuticas también pueden estar en forma de una suspensión acuosa u oleosa inyectable estéril, que puede formularse según procedimientos conocidos utilizando uno o más de los agentes dispersantes o humectantes y agentes de suspensión apropiados, que se han mencionado anteriormente. Una preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico aceptable por vía parenteral, por ejemplo, una solución en 1,3-butanodiol.
Las composiciones para la administración por inhalación pueden estar en forma de un aerosol presurizado convencional dispuesto para dispensar el ingrediente activo como un aerosol que contiene gotitas sólidas o líquidas finamente divididas. Se pueden usar propulsores de aerosoles convencionales tales como hidrocarburos fluorados volátiles o hidrocarburos y el dispositivo de aerosol está convenientemente dispuesto para dispensar una cantidad medida de ingrediente activo.
Las composiciones para administración también se pueden formular como una preparación de liposomas. La preparación de liposomas puede comprender liposomas que penetran en las células de interés o en el estrato córneo
y se fusionan con la membrana celular, dando como resultado el suministro del contenido del liposoma a la célula. Otras formulaciones adecuadas pueden emplear niosomas. Los niosomas son vesículas lipídicas similares a los liposomas, con una membrana que consiste en gran parte en lípidos no iónicos, algunas formas de las cuales son efectivas para transportar compuestos a través del estrato córneo.
Las composiciones para administración también se pueden formular como una preparación de depósito, que se puede administrar por implantación o por inyección intramuscular. Las composiciones se pueden formular con material polimérico o hidrófobo adecuado (como una emulsión en un aceite aceptable), resinas de intercambio iónico o derivados escasamente solubles.
El compuesto de la presente divulgación también se puede administrar en formas de liberación sostenida o desde sistemas de administración de fármacos de liberación sostenida.
Para obtener más información sobre formulación, administración de fármacos y técnicas de procesamiento, se remite al lector a Remington’s Pharmaceutical Sciences (21a edición, 2005, Universidad de las ciencias en Filadelfia, Lippincott William & Wilkins)
La cantidad de ingrediente activo que se combina con uno o más excipientes para producir una sola forma de dosificación necesariamente variará dependiendo del huésped tratado y la vía particular de administración. Por ejemplo, una formulación destinada a la administración oral a seres humanos generalmente contendrá, por ejemplo, de 0,5 mg a 4 g de agente activo compuesto con una cantidad apropiada y conveniente de excipientes que puede variar de aproximadamente 5 a aproximadamente 98 por ciento en peso de la composición total. Las formas de unidades de dosificación contendrán generalmente aproximadamente 1 mg a aproximadamente 500 mg de un ingrediente activo. Para obtener más información sobre las vías de administración y los regímenes de dosificación, se remite al lector a Capítulo 25.3 en el Volumen 5 de Comprehensive Medicinal Chemistry (Corwin Hansch; Presidente del Consejo Editorial), Pergamon Press 1990 y Remington’s Pharmaceutical Sciences (2 ia edición, 2005, Universidad de las ciencias en Filadelfia, Lippincott William & Wilkins).
Como se indicó anteriormente, el tamaño de la dosis requerida para el tratamiento terapéutico o profiláctico de un estado de enfermedad particular variará necesariamente dependiendo del huésped tratado, la vía de administración y la gravedad de la enfermedad que se está tratando. Preferiblemente, se emplea una dosis diaria en el rango de 1-25 mg/kg. En consecuencia, la dosificación óptima puede ser determinada por el médico que está tratando a cualquier paciente en particular.
En cualquiera de las composiciones farmacéuticas, procesos, métodos, usos, medicamentos y características de fabricación mencionados en la presente memoria, también se aplica cualquiera de los aspectos alternativos de los compuestos de la divulgación descrita en la presente memoria.
Los compuestos divulgados en la presente memoria pueden aplicarse como terapia única o pueden implicar, además de un compuesto de la divulgación, una o más de otras sustancias y/o tratamientos. Dicho tratamiento conjunto puede lograrse mediante la administración simultánea, secuencial o separada de los componentes individuales del tratamiento. Cuando la administración sea secuencial o separada, el retraso en la administración del segundo componente no debe ser tal que se pierda el efecto beneficioso de la combinación. Las clases y sustancias adecuadas se pueden seleccionar de una o más de las siguientes: i) otros agentes antibacterianos, por ejemplo, macrólidos, p. ej., eritromicina, azitromicina o claritromicina; quinolonas, p. ej., ciprofloxacina o levofloxacina; p lactamas, p. ej., penicilinas, p. ej., amoxicilina o piperacilina; cefalosporinas, p. ej., ceftriaxona o ceftazidima; carbapenemos, p. ej., meropenem o imipenem, etc.; aminoglucósidos, p. ej., gentamicina o tobramicina; u oxazolidinonas; y/o ii) agentes antiinfecciosos, por ejemplo, un triazol antifúngico, p. ej., o anfotericina; y/o iii) productos terapéuticos proteicos biológicos, por ejemplo, anticuerpos, citocinas, productos proteicos bactericidas/que aumentan la permeabilidad (BPI); y/o iv) uno o más agentes antibacterianos útiles en el tratamiento de Mycobacterium tuberculosis tales como uno o más de rifampicina, isoniazida, pirizinamida, etambutol, quinolonas, p. ej., moxifloxacina o gatifloxacina, estreptomicina y/o v) inhibidores de la bomba de expulsión.
Según una realización, la presente divulgación se refiere a un compuesto de Fórmula I, o Fórmula (B), o una sal farmacéuticamente aceptable del mismo y un agente quimioterapéutico seleccionado de: i) uno o más agentes antibacterianos adicionales; y/o ii) uno o más agentes antiinfecciosos; y/o iii) productos terapéuticos proteicos biológicos, por ejemplo, anticuerpos, citocinas, productos proteicos bactericidas/que aumentan la permeabilidad (BPI); iv) uno o más agentes antibacterianos útiles en el tratamiento de tuberculosis pulmonar, tuberculosis extrapulmonar, infecciones por avium, úlceras de buruli y/o v) uno o más inhibidores de la bomba de expulsión.
Si no están disponibles comercialmente, los materiales de partida necesarios para los procedimientos como los descritos en la presente memoria se pueden hacer mediante procedimientos que se seleccionan de técnicas químicas orgánicas estándar, técnicas que son análogas a la síntesis de compuestos estructuralmente similares conocidos, o técnicas que son análogas al procedimiento o los procedimientos descritos en los Ejemplos.
Se observa que muchos de los materiales de partida para los métodos sintéticos que se describen en la presente memoria están disponibles comercialmente y/o ampliamente informados en la literatura científica o podrían fabricarse a partir de compuestos disponibles comercialmente usando adaptaciones de procesos informados en la literatura
científica. Se remite además al lector a Advanced Organic Chemistry, 5a edición, por Jerry March y Michael Smith, publicado por John Wiley & Sons 2001, para obtener una orientación general sobre las condiciones de reacción y los reactivos.
También se apreciará que en algunas de las reacciones mencionadas en la presente memoria puede ser necesario/deseable proteger cualquier grupo sensible en los compuestos. Los casos en los que es necesaria o deseable la protección son conocidos por los expertos en la técnica, así como los métodos adecuados para dicha protección. Se pueden usar grupos protectores convencionales según la práctica estándar (para una ilustración, véase T. W. Greene, Protective Groups in Organic Synthesis, publicado por John Wiley and Sons, 1991) y como se ha descrito anteriormente en la presente memoria.
Ejemplos
Abreviaturas
A PCI- ionización química a presión atmosférica;
ATP- trifosfato de adenosina;
BSA- albúmina de suero bovino;
C D Cl3- cloroformo deuterado;
CLSI- Instituto de Estándares Clínicos y de Laboratorio;
DCM- diclorometano;
DMAP- 4-dimetilaminopiridina;
DMF- dimetilformamida;
DMSO- dimetilsulfóxido;
DTT- ditiotretol;
EtOAc- acetato de etilo;
EDTA- ácido etilendiaminotetracético;
HPLC- cromatografía líquida de alta presión;
LC/M S- cromatografía de líquidos/espectrometría de masas;
MPLC- cromatografía líquida de media presión;
MeOD- metanol deuterado, es decir, D3COD;
MeOH- metanol;
MS- espectroscopia de masas; ESP (o ES) - electropulverización; EI - impacto de electrones;
MTBD- N-metil-1,5,7-triazabiciclo[4.4.0]dec-5-eno;
RMN- espectroscopía por resonancia magnética nuclear;
DO- densidad óptica;
S D S- dodecilsulfato de sodio;
S TEB - tampón sacarosa tris EDTA;
TA E- tampón EDTA de ácido trisacético;
TH F- tetrahidrofurano;
TFA- ácido trifluoroacético;
h- hora(s);
min- minuto(s);
d- día(s);
v/v- relación volumen/volumen;
Boc- t-butoxicarbonilo;
Cbz- benciloxicarbonilo;
Bz- benzαlo;
TLC - cromatografía de capa fina;
atm- presión atmosférica;
rt- temperatura ambiente;
mg- miligramo;
ng- nanogramo;
g- gramo;
qL- microlitro;
mL- mililitro;
L- litro;
qM- micromolar;
mM- milimolar;
nm- nanómetro
Consideraciones Generales
Si no están disponibles comercialmente, los materiales de partida necesarios para los procedimientos como los descritos en la presente memoria pueden prepararse mediante procedimientos que se seleccionan de técnicas químicas orgánicas estándar, técnicas que son análogas a la síntesis de compuestos estructuralmente similares conocidos, o técnicas que son análogas. al procedimiento descrito o los procedimientos descritos en los Ejemplos. Se observa que muchos de los materiales de partida para los métodos sintéticos descritos en la presente memoria están disponibles comercialmente y/o ampliamente informados en la literatura científica o podrían fabricarse a partir de compuestos disponibles comercialmente usando adaptaciones de procesos informados en la literatura científica. Se remite además al lector Advanced Organic Synthesis, 5a Edición, por Jerry March y Michael Smith, publicado por John Wiley & Sons 2001, para obtener orientación general sobre las condiciones de reacción y los reactivos.
También se apreciará que en algunas de las reacciones mencionadas en la presente memoria puede ser necesario/deseable proteger cualquier grupo sensible en los compuestos. Los casos en los que es necesaria o deseable la protección son conocidos por los expertos en la técnica, así como los métodos adecuados para dicha protección. Se pueden usar grupos protectores convencionales según la práctica estándar (para una ilustración, véase T. W. Greene, Protective Groups in Organic Synthesis, publicado por John Wiley and Sons, 1991) y como se ha descrito anteriormente en la presente memoria.
Los siguientes ejemplos proporcionan detalles sobre la síntesis, actividades y aplicaciones de los compuestos de la presente divulgación. Debe entenderse que lo siguiente es solo representativo y que la invención no está limitada por los detalles expuestos en estos ejemplos.
Material y métodos
Las evaporaciones se llevaron a cabo por evaporación rotatoria in vacuo y los procedimientos de elaboración se llevaron a cabo después de la eliminación de los sólidos residuales por filtración; las temperaturas se expresan en °C ; las operaciones se llevaron a cabo a temperatura ambiente, es decir, típicamente en el rango de 18 a 26 °C y sin exclusión de aire, a menos que se indique lo contrario, o a menos que el experto en la técnica trabajara bajo una atmósfera inerte; se usó cromatografía en columna (mediante el procedimiento ultrarrápido) para purificar los compuestos y se realizó en gel de sílice Merck Kiesel (Art. 9385) a menos que se indique lo contrario; en general, el curso de las reacciones se siguió por TLC, HPLC o LC/MS y los tiempos de reacción se dan solo como ilustración; los rendimientos se dan solo a modo de ilustración y no son necesariamente los máximos alcanzables; la estructura de los productos finales de la invención fue generalmente confirmada por RMN y técnicas de espectro de masas. Los espectros de resonancia magnética de protones se determinaron generalmente en DMSO d6, a menos que se indique lo contrario, usando un espectrómetro Bruker DRX 300 o un espectrómetro Bruker DRX-400, operando a una intensidad de campo de 300 MHz o 400 MHz, respectivamente. En los casos en los que el espectro de RMN es
complejo, solo se notifican las señales de diagnóstico. Los desplazamientos químicos se informan en partes por millón campo abajo del tetrametilsilano como estándar externo (escala *) y las multiplicidades de los picos se muestran así: s, singlete; d, doblete; dd, doblete de dobletes; dt, doblete de tripletes; dm, doblete de multipletes; t, triplete, m, multiplete; br, amplio. Los datos espectrales de masas de bombardeo con átomos rápidos (FAB) generalmente se obtuvieron utilizando un espectrómetro Platform (suministrado por Micromass) ejecutado en electropulverización y, según fuera apropiado , se recopilaron datos de iones positivos o de iones negativos o utilizando el sistema LC/MS Agilent serie 1100 equipado con Sedex. 75ELSD y, según fuera apropiado, se recopilaron datos de iones positivos o datos de iones negativos. El ion principal de masa más baja se informa para moléculas en las que la división de isótopos da como resultado múltiples picos espectrales de masa (por ejemplo, cuando hay cloro presente). La HPLC de fase inversa se llevó a cabo usando YMC Pack ODS AQ (100 x 20 mm DI, tamaño de partícula 5 Á, tamaño de poro 12 nm) en instrumentos Agilent; cada intermedio se purificó al estándar requerido para la etapa subsiguiente y se caracterizó con suficiente detalle para confirmar que la estructura asignada era correcta; la pureza se evaluó mediante HPLC, TLC o RMN y la identidad se determinó mediante espectroscopia infrarroja (IR), espectroscopia de masas o espectroscopia RMN, según fuera apropiado.
Ejemplo 1
Procedimiento general para la síntesis de compuestos de Fórmula I

La presente invención proporciona un proceso para preparar compuestos de Fórmula I y sales farmacéuticamente aceptables de los mismos; implicando el proceso hacer reaccionar un compuesto de Fórmula (A) y un derivado de amina de Fórmula (B) en condiciones de aminación reductora, en donde n =0 o 1; Rs es H.
Procedimiento general para preparar los compuestos de Fórmula (A)
Se llevó a cabo la hidroxilación catalizada por paladio de halo arenos tales como los de Fórmula (D) para obtener los compuestos de Fórmula (C). Además, los compuestos de Fórmula (C) se trataron con reactivo de acetal de haloalquilo (p. ej., BrCH2(CH2)n1CH(0CH2CH3)2) para obtener los compuestos de Fórmula (A).

Los compuestos de Fórmula la y Fórmula Ib en donde Z1 es O; m=0 ; R3 es H u OH; y Rs es H; pueden prepararse haciendo reaccionar compuestos de Fórmula (E) con compuestos de Fórmula (G) o (F) como se muestra en el Esquema 1.
Esquema 1

Los compuestos de Fórmula (E) se pueden preparar a partir de compuestos de Fórmula (H) y compuestos de Fórmula I como se resume en el Esquema 2. El acoplamiento de Buchwald catalizado por paladio de compuestos de Fórmula (H) con Fórmula (I) en condiciones de reacción óptimas proporcionó los compuestos de Fórmula (J). Otros compuestos de Fórmula (J) se convirtieron en compuestos de Fórmula (K) a través de una reacción de azidación y reducción de la funcionalidad azida proporcionando los compuestos de Fórmula (E).

Ejemplo 2
S íntesis de intermedios
S íntesis de 2-((5-m etil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)acetaldehído (Intermedio I)

Etapa 1 : S íntesis de 2,2-dim etil-2,3-dihidro-5H-[1,4,2]oxazasilino[6,5,4-de][1,5]naftiridin-5-ona (Ia)
A una suspensión agitada de NaH (0,34 g, 8,5 mmoles) en DMF seca se le añadió 6-metoxi-1,5-naftiridin-4-ol (Combi-Blocks, 1 g, 5,6 mmoles) en porciones a 0 °C. La mezcla de reacción se dejó agitar a rt durante 1 h. A esto se le añadió cloro (clorometil) dimetil) silano (1,29 g, 9,09 mmoles) a temperatura ambiente y la mezcla de reacción se calentó hasta 100 °C durante 16 h. La mezcla de reacción se concentró y el producto bruto se coevaporó con tolueno para obtener el producto bruto Ia (0,5 g) como un sólido blanquecino y el material se usó como tal para la etapa siguiente sin purificación adicional. LC-M S Calc. para C 1 1 H12 N2 O2 SK 232,31; Obs.: 233.1[M++H].
Etapa 2: Síntesis de 8-hidroxi-1-metil-1,5-naftiridin-2(1H)-ona (Ib)
A una solución agitada de Ia (0,5 g, 2 ,15 mmoles) en dioxano seco y MeOH (2:1, 16 mL) se añadió C sF (0,98 g, 6,46 mmoles) y la mezcla resultante se calentó hasta 80 °C durante 12 h. La mezcla de reacción se enfrió hasta rt y el disolvente se evaporó, el residuo se neutralizó con HCl 1,5 N. El producto orgánico se extrajo con EtOAc:MeOH (9:1, 50 mL) y se lavó con solución de salmuera (20 mL). La capa orgánica se secó sobre Na2SO4 y se concentró para obtener el producto crudo Ib (0,3 gm) como un sólido blanquecino. El crudo se usó como tal para la siguiente etapa sin purificación adicional. LC-MS Calc. para C 9H8 N2 O2 : 176,18; Obs.: 177,1 [M++H]; 1H RMN (400 MHz, DMSO-D6): 5 7,95-7,90 (m, 2H), 6,91 (d, j= 9.6 Hz, 1H), 6,59 (s, 1H), 3,96 (s, 3H), 3 ,12 (s, 1H).
Etapa 3: Síntesis de 8-(2,2-dietoxietoxi)-1-metil-1,5-naftiridin-2(1H)-ona (Ic)
A una solución agitada de Ib (0,3 g, 17,04 mmoles) en DMSO seco (6 mL) se añadió C s 2 CO3 (0,66 g, 2,04 mmoles) seguido de la adición de dietilacetal de bromoacetaldehído (0,47 g, 2,38 mmoles). La mezcla resultante se calentó hasta 90 °C durante 16 h. La mezcla de reacción se diluyó con EtOAc (50 mL) y se lavó con agua (2x 50 mL). La capa orgánica se secó sobre Na2SO4 y se concentró para obtener el producto crudo. El producto crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 de malla, 30 % de EtOAc en éter de petróleo) para producir Ic como un líquido viscoso amarillo (0,2 gm). LC-MS Calc. para C 15 H20 N2 O4 : 292,34; Obs.: 293,2 [M++H].
Etapa 4: Síntesis de 2-((5-metil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)acetaldehído (I)
Una solución de Ic (0,2 g, 0,68 mmoles) en diclorometano (3 mL), se enfrió hasta 0 °C y se añadió ácido trifluoroacético (1 mL). La mezcla de reacción se dejó en agitación a rt durante 30 min. La mezcla de reacción se concentró y el residuo se diluyó con DCM (5 mL) y se neutralizó usando solución de NaHCO3 al 10 % . La capa orgánica se separó, se secó sobre sulfato de sodio y se concentró para obtener el producto crudo I (0,1 gm). El material crudo utilizado se utilizó para la síntesis del Compuesto 1 sin purificación adicional. LC-MS Calc. para C 1 1 H 10 N2 O3 : 218 ,21; Obs.: 219,1 [M++H].
Síntesis de 2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)acetaldehído (Intermedio II)

Etapa 1: Síntesis de 7-fluoro-8-hidroxi-1-metilquinolin-2(1H)-ona (IIa)
A una mezcla de 8-bromo-7-fluoro-1-metilquinolin-2(1H)-ona (CAS:1002108-91-9, 1 g, 3,9 mmoles) en 1,4-dioxano (20 mL) se purgó N2 durante 10 min. A esto se añadió Pd2 (dba)3 (0,071 g, 0,078 mmoles) y t-Butilxfos-Pd-G3 (0,123 g, 0,156 mmoles). Después de 5 minutos bajo desgasificación, se añadió KOH (0,43 g, 7,8 mmoles) disuelto en H2 O (5 mL) a la mezcla de reacción. La mezcla resultante se agitó a 100 °C durante 2 horas. La mezcla de reacción se enfrió y acidificó con HCl diluido 1,5 N y se extrajo con acetato de etilo (4 x 125 mL). La capa orgánica se lavó con salmuera (2 x 100 mL), se secó sobre Na2SO4 y se concentró a presión reducida. El crudo se trituró con éter de petróleo para producir el compuesto puro IIa (0,5 g). LC-MS Calc. para C 10 H8 FNO2 : 193,18; Obs.: 194.2, [M++H]; 1H RMN (400 MHz, DMSO-D6): 5 10,02 (s, 1 H), 7,79 (d, J = 9,30 Hz, 1 H), 7 ,11 -7 ,14 (m, 2H), 6,52 (d, J = 9,30 Hz, 1 H), 3,89 (s, 3H).
Etapa 2: Síntesis de 8-(2,2-dietoxietoxi)-7-fluoro-1-metilquinolin-2(1H)-ona (IIb)
A una mezcla de IIa (0,5 g, 2,59 mmoles) en DMSO (20 mL) se añadió Cs2CO3 (2,52 g, 7,77 mmoles) y bromoacetaldehído dietilacetal (0,765 g, 3,88 mmoles). La mezcla resultante se agitó a 90 °C durante 12 horas. La mezcla de reacción se enfrió y se inactivó con agua extraída con acetato de etilo (4 x 125 mL). La capa orgánica se lavó con salmuera (2 x 100 mL), se secó sobre Na2SO4 y se concentró a presión reducida. El crudo se purificó por cromatografía en columna usando gel de sílice (230 - 400 de malla) eluyendo con 10 - 15 % de acetato de etilo en éter de petróleo para producir el compuesto puro IIb (0,5 gm). LC-MS Calc. para C 16 H20 FNO4 : 309,34; Obs.: 310 ,1, [M++H].
Etapa 3: Síntesis de 2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)acetaldehído (II)
A una mezcla de IIb (0,3 g, 0,96 mmoles) en (3 ml de DCM) se enfrió hasta 0 °C y se añadió TFA (4 mL). La mezcla de reacción se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se enfrió y se inactivó con una solución de bicarbonato de sodio al 10 % extraída con DCM. La capa orgánica se lavó con salmuera (2 x 100 mL), se secó sobre Na2 SO4 y se concentró a presión reducida. El crudo se trituró con éter de petróleo para producir II crudo (0,2 gm) y como tal utilizado para la siguiente etapa sin purificación adicional. 1H RMN (400 MHz, CDCl3): 5 9,93 (s, 1 H), 7,58 (d, J = 9,52 Hz, 1 H), 7,27-7,28 (m, 1 H), 6,68 (d, J = 9,40 Hz, 1 H), 4,66 (s, 1 H), 3,98 ( s, 3H).
Síntesis de 6-bromo-2H-pirido[3,2-b][1,4]oxazin-3(4H)-ona (Intermedio III)

Etapa 1: Síntesis de 6-bromo-2-nitropiridin-3-ol (IIIa)
A una solución de 2-nitropiridin-3-ol (20 g, 0,142 moles) en DMF (400 mL), se añadió en porciones N-bromosuccinimida (32,52 g, 0,187 moles) durante un período de 5 horas a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 12 h. Una vez completada la reacción, la mezcla de reacción se concentró in vacuo. El residuo se recogió en éter y la mezcla se agitó durante 30 min. El precipitado se eliminó por filtración y el filtrado se concentró in vacuo para obtener 6-bromo-2-nitropiridin-3-ol, IIIa (40 g, 46 % ) como una mezcla de compuestos mono y di bromo. La LCMS del crudo mostró un 46 % del compuesto monobromo esperado, este material se usó como tal para la siguiente etapa sin purificación adicional. LC-MS Calc. para C5H3BrN2O3: 218,99; Obs.: 219,2.
Etapa 2: Síntesis de 2-((6-bromo-2-nitropiridin-3-il)oxi)acetato de etilo (IIIb)
A la solución de 6-bromo-2-nitropiridin-3-ol, IIIa (40 g, 0,182 moles) en acetona (400 mL), enfriada hasta 0 °C, se añadió carbonato de potasio (50,41 g, 0,365 moles) y se agitó durante 5 min. Luego, se añadió lentamente bromoacetato de etilo (39,7 g, 0,237 moles) y se sometió a reflujo a 65 °C durante 12 h. Una vez completada la reacción, se filtró y el filtrado se concentró in vacuo. El crudo se diluyó con agua y se extrajo con acetato de etilo (2 x 600 mL). Las capas orgánicas combinadas se lavaron con solución de salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron in vacuo. Se purificó por cromatografía en columna sobre gel de sílice con elución en gradiente de 20-22 % de acetato de etilo en éter de petróleo para obtener 2-((6-bromo-2-nitropiridin-3-il)oxi)acetato de etilo, IIIb (21 g, 75,32 % ) como un sólido amarillo pálido. lC-MS Calc. para CgHgBrN2O5: 305,38; Obs.: 306,2.
Etapa 3: Síntesis de 6-bromo-2H-pirido[3,2-b][1,4]oxazin-3(4H)-ona (III)
A una solución agitada de 2-((6-bromo-2-nitropiridin-3-il)oxi)acetato de etilo, IIIb (21 g, 0,0687 moles) en ácido acético glacial (400 mL), se añadió polvo de hierro (1,1,51 g, 0,2063 moles) y se calentó hasta 100 °C durante 6 horas. Una vez completada la reacción, la mezcla de reacción se filtró a través de un lecho de celite utilizando acetato de etilo, metanol al 10 % y se concentró in vacuo. Se lavó con metanol para obtener 6-bromo-2H-pirido[3,2-b][1,4]oxazin-3(4H)-ona pura, III (12 g, 76,28 % ) . LC-MS Calc. para C/HaBrNgOg: 229,03; Obs.: 230,2.
Síntesis de 6-(5-(am¡nomet¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡rido[3.2-b][1.4]oxaz¡n-3(4H)-ona (Intermedio IV)

Etapa 1: (R)-5-(((terc-butildimetilsilil)oxi)metil)oxazolidin-2-ona (IVa)
A una solución agitada de TBDM S-Cl (38,46 g, 0,256 moles), imidazol (23,2 g, 0,341 moles), DMAP (2,08 g, 0,017 moles) en DMF (200 mL), enfriada hasta 0 °C , se añadió (R)-5-(hidroximetil)oxazolidin-2-ona (CAS: 97859-49 9, 20 g, 0,1709 mmoles) en DMF (25 mL) y se agitó a 25 °C durante 2 h. Una vez completada la reacción, la mezcla de reacción se inactivó con agua y se extrajo con acetato de etilo. La capa orgánica separada se lavó con agua, solución de salmuera, se secó sobre Na2SO4, se filtró y se concentró a presión reducida. Se purificó por cromatografía en columna sobre gel de sílice (230-400 de malla, 20-25 % de acetato de etilo en éter de petróleo) para obtener IVa (32 g, 81% ). LC-MS Calc. para C 10 H2 1 NO3 SK 231,37; Obs.: 232,1; 1H RMN (400 MHz, CDC13): d5.11 (s, 1H), 4,72 4,66 (m, 1 H), 3,86-3,82 (m, 1 H), 3,79-3,76 (m, 1 H), 3,66-3,62 (m, 1 H), 3,58-3,55 (m, 1 H) , 0,91 (s, 9H), 0,11 (s, 6H).
Etapa 2: (R)-6-(5-(((terc-but¡ld¡met¡ls¡l¡l)ox¡)met¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-3(4H)-ona (IVb)
A una solución agitada de IVa (32 g, 0,139 moles) y III (31,7 g, 0,139 moles) en 1,4-dioxano seco (50 mL), se añadieron complejo de cloruro de t-butil-X-Phos mesilo (5,5 g, 0,0069 moles) y terc-butóxido de sodio (19,94 g, 0,207 moles) y se desgasificó durante 20 min. Luego, se calentó en tubo sellado a 100 °C durante 16 h. Una vez completada la reacción, la mezcla de reacción se concentró a presión reducida. Se purificó por cromatografía en columna sobre gel de sílice (230-400 de malla, 25-30 % de acetato de etilo en éter de petróleo) para proporcionar IVb (45,6 g, 86 % ). LC-MS Calc. para C^^aN sO aSi: 379,49; Obs.: 380,0; 1H RMN (400 MHz, DMSO-d6): 57,60 (d, J = 8,68 Hz, 1 H), 7,43 (d, J = 8,68 Hz, 1 H), 4,77-4,73 (m, 1H), 4,67 (s, 2H), 4,15-4,10 (m, 1H), 3,93-3,89 (m, 3H), 0,79 (s, 9H), 0,04 (s, 6H).
Etapa 3: (R)-6-(5-(H¡drox¡met¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-3(4H)-ona (IVc)
A una solución agitada de IVb (45 g, 0 ,118 moles) en THF (250 mL), enfriada hasta 0 °C , se añadió gota a gota fluoruro de terc-butilamonio (1 M en THF) (296 mL, 0,296 moles) y se agitó a 25 °C durante 3 horas. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, se obtuvo un sólido, se filtró y se secó al vacío para obtener un sólido blanco de IVc (29 g, 92 % ). LC-MS Calc. para C 1 1 H 1 1 N3 O5 : 265,23; Obs.: 265,9; 1H RMN (400 MHz, DMSO-d6):
5 11,22 (s, 1 H), 7,60 (d, J = 8,80 Hz, 1H), 7,42 (d, J = 8,40 Hz, 1H), 5,21 (bs, 1H), 4,70-4,66 (m, 1H), 4,60 (s, 2H), 4,12-4,07 (m, 1H), 3,92-3,88 (m, 1H), 3,69-3,65 (m, 1H), 3,54-3,34 (m, 1H).
Etapa 4: metanosulfonato de (R)-(2-oxo-3-(3-oxo-3,4-d¡h¡dro-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-6-¡l)oxazol¡d¡n-5-¡l)met¡lo (IVd)
A una solución agitada de IVc (29 g, 0,109 moles) en DMF seca (300 mL), enfriada hasta 0 °C , se añadieron trietilamina (45,7 mL, 0,328 moles) y cloruro de mesilo (17 mL, 0,218 moles) y se agitó a 25 °C durante 2 h. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, se obtuvo un sólido, se filtró , se secó al vacío para obtener un sólido blanco de IVd (30 g, 80 % ). LC-MS Calc. para C12H13Ne3O7S: 343,31; obs.: 344,0; 1H RmN (400 MHz, DMSO-d6): 5 11,27 (s, 1 H), 7,61 (d, J = 8,40 Hz, 1 H), 7,46 (d, J = 8,80 Hz, 1 H), 5,00 (bs, 1 H), 4,63 (s, 2H), 4,63-4,51 (m, 2H), 4,25-4,20 (m, 1H), 3,90-3,85 (m, 1H), 3,35 (s, 3H).
Etapa 5: (R)-6-(5-(az¡domet¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-3(4H)-ona (IVe)
A una solución agitada de IVd (30 g, 0,087 moles) en DMF (300 mL), enfriada hasta 0 °C , se añadió azida de sodio (17 g, 0,262 moles) y se calentó a 60 °C durante 3 h. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, se obtuvo un sólido, se filtró y se secó in vacuo obtener un sólido blanco de IVe (22 g, 87 % ) . LC-MS Calc. para C 1 1 H 10 N6O4 : 290,24; Obs.: 290,9; 1H RMN (400 MHz, DMSO-d6): 5 11,26 (s, 1H), 7,61 (d, J = 8,40 Hz, 1H), 7,45 (d, J = 8,80 Hz, 1 H), 4,88 (bs, 1H), 4,63 (s, 2H), 4,16 (t, J = 9,60 Hz, 1H), 3,70-3,84 (m, 3H).
Etapa 6: (S)-6-(5-(am¡nomet¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-3(4H)-ona (IV)
A una solución agitada de IVe (22 g, 0,075 moles) en THF:MeOH ( 1 :1) (400 mL), se añadió paladio al 10 % sobre carbono (7 g) y se agitó a 25 °C bajo H2 durante 4 h. Una vez completada la reacción, la mezcla de reacción se filtró a través de un lecho de celite usando THF y MeOH y se concentró a presión reducida para obtener IV (15 g, 75 % ).
LC-MS Calc. para C 1 1 H 12 N4O4 : 264,24; Obs.: 265,1.
Pur¡f¡cac¡ón del Intermed¡o IV: Protección Boc: A una solución agitada de IV crudo (15 g, 0,056 moles) en 1,4 dioxano: agua (1:1, 200 mL) se le añadió Na2CO3 (12 g, 0 ,113 moles) seguido de la adición de (Boc)2 O (25 g, 0 ,113 moles) a 0 °C y se dejó en agitación a rt durante 12 h. La mezcla de reacción se diluyó con EtOAc (250 mL) y se lavó con agua (2 x 250 mL). Las capas orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre sulfato de sodio y se evaporaron a presión reducida para obtener el crudo. El crudo se purificó mediante cromatografía en columna (230/400 de malla, DCM al 4 % en MeOH) para obtener IV con la protección Boc deseada como sólido blanco (12 g, 58 % ) . LC-MS Calc. para C 16 H20 N4O6 : 364,36; Obs.: 265,1; 1H RMN (400 MHz, DMSO-D6): 5 11,23 (s, 1 H), 7,61 (d, 1 H, J=8,8 Hz), 7,45 (d, 1H, J=8,8 Hz), 7,24 (m, 1H), 4,71 (m, 1H), 4,60 (s, 2H), 4,16-4,11 (m, 1H), 3,84 3,80 (m, 1 H), 3,25 (m, 2H),1,36 (s, 9H).
El IV protegido con Boc (12 g, 0,033 mmoles) recogió en 1,4 dioxano (60 mL) y se le añadió HCl 4 M en dioxano (120 mL) a 0 °C. La mezcla resultante se agitó a rt durante 3 h. La mezcla de reacción se concentró para obtener la sal de HCl de amina cruda. El crudo se disolvió en MeOH/DCM seco (200 mL) y se neutralizó con resina, se filtró y se concentró para producir la amina pura IV como un sólido blanquecino (8 g, 92 % ). LC-MS Calc. para C 11 H 12 N4 O4 : 264,24; Obs.: 265,1 [M+H]; 1H RMN (400 MHz, DMSO-D6): 5 11,05 (brs, 1H), 7,62 (d, 1H, J=8,8 Hz), 7,45 (d, 1H, J=8,8 Hz), 7,05 (brs, 2H), 4,83 (m, 1H), 4,62(s, 2H), 4,23-4,20 (m, 1H), 3,87-3,82 (m, 1H), 3,19-3,10 (m, 2H).
Síntes¡s de 6-cloro-2H-p¡raz¡no[2,3-b][1,4]oxaz¡n-3(4H)-ona (Intermed¡o Va)

A una solución agitada de 3-bromo-6-cloropirazin-2-amina (75 g, 0,3598 moles) en 1,4-dioxano (1.500 mL) a temperatura ambiente en atmósfera de nitrógeno se añadió terc-butóxido de sodio (110,65 g, 1,1514 moles) y se agitó durante 30 minutos. Luego, se añadió gota a gota glicolato de etilo (112 ,37 g, 1,0794 moles) durante un período de 30 minutos a temperatura ambiente. La mezcla resultante se calentó hasta 100 °C y se agitó durante 2 horas. El progreso de la reacción se monitorizó por TLC.
Después de eso, la mezcla de reacción se enfrió hasta temperatura ambiente y se concentró a presión reducida para eliminar el dioxano. El residuo obtenido se diluyó con agua (750 mL) y se neutralizó con HCl (1,5 N). El sólido precipitado se retiró por filtración y se secó al vacío para obtener el compuesto Va como un sólido blanquecino. Rendimiento: 60 g, 89,9 % ; LC-MS Calc. para C 6H4CLN 3 O2 , 185,57, Observado 184,0 (M-1H); 1H RMN (400 MHz, DMSO-d6): 5 11,86 (s, 1 H), 7,87 (s, 1 H), 4,90 (s, 2H).
Síntesis de (S)-6-(5-(aminometil)-2-oxooxazoMdin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona, (Intermedio V)

Etapa 1: (R)-6-(5-(((terc-butildimetilsilil)oxi)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Vb)
A una solución agitada de Va (2,5 g, 13,51 mmoles) y IIIa (3,43 g, 14,86 mmoles) en 1,4-dioxano seco (40 mL), se añadieron complejo de t-butil-X-Phos cloruro de mesilo (0,53 g, 0,67 mmoles) y terc-butóxido de sodio (1,94 g, 20,27 mmoles) y se desgasificó durante 20 minutos Luego, se calentó en tubo sellado a 100 °C durante 16 h. Una vez completada la reacción, la mezcla de reacción se concentró a presión reducida. Se purificó por cromatografía en columna sobre gel de sílice (230-400 de malla, 25-30 % de acetato de etilo en éter de petróleo) para proporcionar Vb (3 g, 59%). LC-MS Calc. para C1sH24N4O5Si, 380,48, observado 381,1 (M+1H); 1H RMN (400 MHz, DMSO-d6): 5 11,61 (s, 1 H), 8,37 - 8,35 (m, 1H), 4,85 - 4,79 (m, 3H), 4 ,12 - 4,06 (m, 1H), 3,89 - 3,74 (m, 3H), 0,84 - 0,71 (m, 9H), 0,03 -0,00 (m, 6H).
Etapa 2: (R)-6-(5-(hidroximetil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Vc)
A una solución agitada de Vb (3 g, 7,89 mmoles) en THF (30 mL), enfriada hasta 0 °C , se añadió gota a gota fluoruro de terc-butilamonio (1 M en THF) (15,8 mL, 15,78 mmoles) y se agitó a temperatura ambiente durante 3 horas. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, se extrajo con acetato de etilo, se secó sobre sulfato de sodio y se evaporó. El producto crudo se purificó mediante cromatografía en columna sobre gel de sílice (230-400 de malla, 50-50 % de acetato de etilo en éter de petróleo) para producir Vc (1,5 g, 71 % ) . LC-MS Calc. para C 10 H 10 N4O5 , 266,21, Observado 267,1 (M+1H); 1H RMN (400 MHz, DMSO-d6): 5 11,62 (s, 1H), 8,38 (s, 1H), 5,23 -5,07 (m, 2H), 5,03 (s, 1 H), 4,86 - 4,73 (m, 1H), 4,10 - 3,86 (m, 2H), 3,70 - 3,48 (m, 2H).
Ruta alternativa para la síntesis de Vc

A una solución agitada de Va (40,0 g, 0,215 moles) y (R)-5-(hidroximetil)oxazolidin-2-ona (CAS: 97859-49-9, 28,0 g, 0,237 moles) en 1,4-dioxano (600 mL) se añadió terc-butóxido de sodio (31,08 g, 0,323 moles) a temperatura ambiente. La mezcla resultante se desgasificó con una corriente de nitrógeno y se le añadió t-butil-X-Phos Paladaciclo (8,56 g, 0,0107 moles) a temperatura ambiente. A continuación, la mezcla resultante se calentó hasta 100 °C y se agitó durante 3 horas. Posteriormente, la mezcla de reacción se enfrió hasta temperatura ambiente, se concentró in vacuo. El residuo obtenido se diluyó con agua (500 mL), se neutralizó con HCl 1,5 N (pH-7). El sólido precipitado se retiró por filtración y se lavó con éter dietílico, se secó in vacuo para obtener el compuesto Vc como sólido marrón. Rendimiento: 55,0 g (crudo), 95,9 % .
Etapa 3: metanosulfonato de (R)-(2-oxo-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]oxazin-6-il)oxazolidin-5-il)metilo (Vd)
A una solución agitada de Vc (1,5 g, 5,63 mmoles) en DMF seca (15 mL), enfriada hasta 0 °C, se añadieron trietilamina (2,3 mL, 16,91 mmoles) y cloruro de mesilo (0,69 mL, 8,45 mmoles) y se agitó a 25 °C durante 2 h. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, el sólido resultante se filtró, se lavó con éter de petróleo y se secó al vacío para producir un sólido marrón de Vd (1,2 g, 63 % ). LC-MS Calc. para C 1 1 H 12 N4O7 S, 344,30, Observado 345,0 (M+1H); 1H RMN (400 MHz, DMSO-d6): 5 11,67 (s, 1H), 8,38 (s, 1H), 5,06 - 5,04 (m, 1H), 4,87 (s, 2H), 4,57 -4,54 (m, 2H), 4,23 - 4,20 (m, 1 H), 3,86 - 3,82 (m, 1 H), 3,28 (s, 3H), 3,25 - 3,23 (m, 1 H).
Etapa 4: (R)-6-(5-(azidometil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Ve)
A una solución agitada de Vd (1,2 g, 3,48 mmoles) en DMF (12 mL), enfriada hasta 0 °C , se añadió azida de sodio (0,56 g, 8,72 mmoles) y se calentó a 65 °C durante 3 h. Una vez completada la reacción, la mezcla de reacción se inactivó con agua, el sólido obtenido se filtró, se lavó con éter de petróleo y se secó para proporcionar el sólido marrón Ve (0,7 g, 70 % ) . LC-MS Calc. para C 10 H9N7 O4 , 291,23, Observado 290,1 (M-1H); 1H RMN (400 MHz, DMSO-d6): 5 11,66 (s, 1 H), 4,94 - 4,86 (m, 3H), 4,18 - 4,13 (m, 1H), 3,81 - 3,75 (m, 3H).
Etapa 5 (S)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (V)
A una solución agitada de Ve (0,7 g, 2,40 mmoles) en THF:MeOH ( 1 :1) (40 mL) se añadió PPh3 (1,9 g, 7,21 mmoles) a temperatura ambiente. La mezcla de reacción se calentó a 70 °C durante 3 h. Una vez completada la reacción por TLC, la mezcla de reacción se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (2 x 100 mL) 2 veces. Además, la capa acuosa se concentró y se secó para proporcionar V (0,3 g, 47 % ) . LC-MS Calc. para C 10 H 1 1 N5 O4 , 265,23, Observado 264,1 (M-1H); 1H RMN (400 MHz, DMSO-d6): 8,38 (s, 1H), 4,85 (s, 2H), 4,69 - 4,67 (m, 1 H), 4,11 - 4,06 (m, 1H), 3,88 - 3,84 (m, 1H), 3,17 (s, 1H), 2,91 - 2,83 (m, 2H).
Síntesis de (R)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Intermedio VI)

El intermedio VI se sintetizó utilizando esquemas y procedimientos análogos al Intermedio V que implica (S)-5-(hidroximetil)oxazolidin-2-ona (CAS: 97859-49-9) y 6-cloro-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Va) como materiales de partida. LC-MS Calc. para C 10 H 1 1 N5 O4, 265,23, Observado 264,2 (M-1H); 1H r Mn (400 MHz, DMSO-d6): 8,36 (s, 1H), 4,84 (s, 2H), 4,69 - 4,65 (m, 1H), 4 ,12 - 4,06 (m, 1H), 3,89 - 3,84 (m, 1H), 3,16 (s, 1H), 2,92 - 2,83 (m, 2H).
Síntesis de 6-cloro-2H-pirazino[2,3-b][1,4]tiazin-3(4H)-ona, VIIa

A una solución agitada de 3-bromo-6-cloropirazin-2-amina I (10 g, 0,0479 moles) en 1,4-dioxano (1,5 L) a temperatura ambiente en atmósfera de nitrógeno se añadió terc-butóxido de sodio (14,75 g, 0,1535 moles) y se agitó durante 30 minutos. Luego, se añadió gota a gota tioglicolato de etilo (11,53 g, 0,0959 moles) durante un período de 30 minutos a temperatura ambiente. La mezcla resultante se calentó hasta 100 °C y se agitó durante 2 horas. El progreso de la reacción se monitorizó por TLC.
Después de eso, la mezcla de reacción se enfrió hasta temperatura ambiente y se concentró a presión reducida para eliminar el dioxano. El residuo obtenido se diluyó con agua (750 mL) y se neutralizó usando HCl (1,5 N). El sólido precipitado se retiró por filtración y se secó para obtener el compuesto VlIa como un sólido blanquecino. Rendimiento: 6 g, 62,5 % ; LC_MS: Calc. para C6H4CIN30s : 201,63; Obs.: 199,9 [M-1H]. 1H-RMN (400 MHz, DMSO-da): 5 11,54 (s, 1 H), 8,25 (s, 1 H), 3,83 (s, 2H).
Síntesis de (S)-5-(aminometil)-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Intermedio VII)

Etapa 1: (R)-5-(mdroximetil)-3-(3-oxo-3,4-dimdro-2H-pirazino[2,3-b][1,4]tiazm-6-N)oxazohdm-2-ona (VIIb)
A una solución agitada del compuesto VIIa (6 g, 0,0297 moles) y (R)-5-(hidroximetil)oxazolidin-2-ona (CAS: 97859-49 9, 3,83 g, 0,0327 moles) en 1,4-dioxano (100 mL) se añadió terc-butóxido de sodio (4,29 g, 0,0446 moles) a temperatura ambiente. La mezcla resultante se desgasificó con una corriente de nitrógeno durante 10 minutos. Luego, se añadió t-butil-X-Phos Paladaciclo (1,18 g, 0,0014 moles) a temperatura ambiente y se desgasificó de nuevo con nitrógeno durante 5 minutos. A continuación, la mezcla resultante se calentó hasta 100 °C y se agitó durante 5 horas. Posteriormente, la mezcla de reacción se enfrió hasta temperatura ambiente y se concentró in vacuo. El residuo obtenido se diluyó con agua (50 mL), se neutralizó con HCl acuoso (1,5 N, pH~7). El sólido precipitado se filtró y se lavó con éter dietílico, se secó bajo vacuo para obtener compuesto VIIb como sólido marrón. Rendimiento: 4 g (crudo), que se tomó para la siguiente etapa sin más purificación. LC_MS: Calc. para C 10 H 10 N4O4S: 282,27; Obs.: 283,2 [M+H]+.1H RMN (300 MHz, DMSO-da): 511.24 (s, 1 H), 8,24 (s, 1 H), 5,27-5,24 (m, 1 H), 4,80-4,76 (m, 1 H), 4,09 (t, J = 9,36 Hz, 1 H), 3,91 -3,87 (m, 1H) ), 3,78-3,68 (m, 3H), 3,60-3,57 (m, 1 H).
Etapa 2: metanosulfonato de (R)-(2-oxo-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-5-il)metilo (VIIb)
A una solución agitada de VIIa (4 g, 0,0141 moles) en DMF seca (40 mL) a 0 °C en atmósfera de nitrógeno se añadieron trietilamina (5,5 mL, 0,0425 moles) y cloruro de mesilo (2,43, 0,0215 moles) sucesivamente. A continuación, la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 2 h. Después de eso, la mezcla de reacción se inactivó con agua, el sólido formado se filtró, se lavó con éter de petróleo y se secó para obtener el compuesto. VIIb como un sólido marrón (3 g, crudo), que se tomó para la siguiente etapa sin más purificación. LC_MS: Calc. para C 1 1 H 12 N4O6S 2 : 360,36; Obs.: 361,00 [M+H]+.
Etapa 3: (R)-5-(azidometil)-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (VIIc)
A una solución agitada de VIIb (3,0 g, 0,0083 moles) en DMF (30 mL) a 0 °C en atmósfera de nitrógeno se añadió azida sódica (2,16 g, 0,033 moles). Luego, la mezcla de reacción se calentó hasta 65 °C y se agitó durante 3 h. Después de eso, la mezcla de reacción se inactivó con agua, el sólido obtenido se filtró, se lavó con éter de petróleo y se secó para obtener el compuesto. VIIc como un sólido marrón (1,7 g, 66,66 % ). LC_MS: Calc. para C 10 H9N7 O3 S: 307,29; Obs.: 307,9 [M+H]+.
Etapa 4: (S)-5-(am¡nomet¡l)-3-(3-oxo-3,4-dih¡dro-2H-p¡raz¡no[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (VII)
A una solución agitada del compuesto VIIc (1,7 g, 0,0055 moles) en una mezcla de THF: H2 O (1:1) (80 mL) bajo atmósfera de nitrógeno se añadió PPh3 (4,34 g, 0,0165 moles) a temperatura ambiente. La mezcla de reacción se calentó hasta 70 °C y se agitó durante 3 horas. Una vez completada la reacción por TLC, la mezcla de reacción se enfrió hasta temperatura ambiente y se extrajo con acetato de etilo (2 x 100 mL). La capa acuosa se separó y se concentró in vacuo para obtener el compuesto VII (0,9 g, 58,06 % ) . LC_MS: Calc. para C 10 H 1 1 N5 O3 S: 281,29; Obs.: 282,1 [M+H]+.
Síntesis de 2-(6-Fluoro-4-metil-3-oxo-3,4-d¡h¡droquinoxal¡n-5-¡l)acetaldehído, (Intermedio VIII)

Etapa 1: 2-Bromo-1,3-difluoro-4-nitrobenceno (VIIIa)
A una solución agitada de 2-bromo-1,3-difluorobenceno (CAS 64248-56-2, 10,0 g, 51,8 mmoles) en H2 SO4 (30 mL) a 0 °C bajo atmósfera de nitrógeno se añadió HNO3 (65 % , 25 mL) gota a gota. La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 1 hora. Después de eso, la mezcla de reacción se vertió sobre hielo y se agitó vigorosamente durante 5 minutos. La suspensión resultante se extrajo con CH 2 Cl2 (4 x 125 mL), se lavó con salmuera (200 mL), se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto VIIIa (crudo). El producto crudo obtenido se llevó a la siguiente etapa sin más purificación. Rendimiento: 9,00 g, 72,99 % . 1H-RMN (400 MHz, DMSO-cfe): 58,34 - 8,29 (m, 1 H), 7,56 - 7,51 (m, 1 H).
Etapa 2: 2-Bromo-3-fluoro-N-metil-6-nitroan¡l¡na (VIIIb)
A una solución agitada del compuesto VIIIa (9,00 g, 37,8 mmoles) y DIPEA (13,3 mL, 75,6 mmoles) en THF (90 mL) a temperatura ambiente en atmósfera de nitrógeno se añadió metilamina (2M en THF, 37,8 mL, 75,6 mmoles). La mezcla resultante se calentó hasta 60 °C y se agitó durante 3 horas. Después de eso, la mezcla de reacción se concentró a presión reducida y el residuo obtenido se purificó por cromatografía en columna usando gel de sílice (60 120 de malla) eluyendo con éter de petróleo para dar el compuesto VIIIb como sólido de color amarillo pálido. Rendimiento: 7,70 g, 81,74 % . 1H-RMN (400 MHz, DMSO-cfe): 57,91 - 7,87 (m, 1 H), 6,80 - 6,76 (m, 2H), 2,75 (s, 3H).
Etapa 3: 6-Bromo-5-fluoro-N1-metilbenceno-1,2-diamina (VIIIc)
A una solución agitada del compuesto VIIIb (7,70 g, 30,9 mmoles) en una mezcla de metanol/agua (400 mL, 3:1) se añadieron sucesivamente cloruro de amonio (8,20 g, 154,6 mmoles) y polvo de hierro (6,90 g, 123,7 mmoles). La mezcla resultante se calentó a reflujo durante 16 horas. Después de eso, la mezcla de reacción se filtró hasta el material sólido y el filtrado se concentró a presión reducida. El residuo obtenido se diluyó con agua y se extrajo con acetato de etilo (2 x 300 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El residuo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con acetato de etilo al 15-20 % en éter de petróleo para obtener el compuesto VIIIc como sólido de color marrón pálido. Rendimiento: 3,50 g, 51,69 % ; 1H RMN (400 MHz, DMSO-de): 56,70 (t, J= 8,6 Hz, 1 H), 6,61 -6,59 (m, 1 H), 4,82 (brs, 2H), 3,92 (brs, 1H), 2,61 (s, 3H).
Etapa 4: (3-Bromo-4-fluoro-2-(metilamino)fenil)glicinato de etilo (VIIId)
A una solución agitada del compuesto VIIIc (3,50 g, 16,0 mmoles) en DMF (70 mL) a 0 °C bajo atmósfera de nitrógeno se añadieron K2 CO3 (3,30 g, 23,9 mmoles) y bromoacetato de etilo (2,10 mL, 19,2 mmoles) sucesivamente. La mezcla resultante se calentó hasta 75 °C y se agitó durante 1 hora. Una vez completada, la mezcla de reacción se inactivó con agua helada (50 mL) y se extrajo con acetato de etilo (4 x 100 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con 10 -15 % de acetato de etilo en éter de petróleo para obtener el compuesto VIIId. Rendimiento: 3,50 g, 71,86 % . 1H-RMN (400 MHz, DMSO-de): 56,84 (t, J= 8,6 Hz, 1H), 6,41 - 6,38 (m, 1H), 5,39 - 5,37 (m, 1H), 4,16 - 4,08 (m, 1H), 4,01 - 3,94 (m, 1H), 2,59 (d, J = 5,7 Hz, 3H), 1,18 (t, J = 7,0 Hz, 3H).
Etapa 5: 8-Bromo-7-fluoro-1-met¡l-3,4-d¡h¡droquinoxal¡n-2(1H)-ona (VlIIe)
A una solución agitada del compuesto VIIId (3,50 g, 11,5 mmoles) en 1,4-dioxano (70 mL) a 0 °C en atmósfera de nitrógeno se añadió NaH (dispersión al 60 % en aceite, 0,14 g, 3,44 mmoles). La mezcla resultante se calentó hasta 100 °C y se agitó durante 2 horas. Después de eso, la mezcla de reacción se inactivó con agua helada (50 mL) y se extrajo con acetato de etilo (3 x 100 mL). La fase orgánica combinada se lavó con salmuera (100 mL), se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con 25-30 % de acetato de etilo en éter de petróleo para obtener el compuesto VlIIe como un sólido blanquecino. Rendimiento: 2,70 g, 90,91 % . 1H-RMN (400 MHz, DMSO-de): 56,99 -6,94 (m, 1 H), 6,88 - 6,84 (m, 1H), 6,24 (s, 1H), 3,60 (s, 2H), 3,35 (s, 3H).
Etapa 6: 8-Bromo-7-fluoro-1-metilquinoxalin-2(1H)-ona (VIIIf)
A una solución agitada del compuesto VlIIe (2,7 g, 10,42 mmoles) en 1,4-dioxano (30 mL) a temperatura ambiente en atmósfera de nitrógeno se añadió MnO2 (5,44 g, 62,54 mmoles) de una vez. La mezcla resultante se calentó hasta 100 °C y se agitó durante 2 horas. Después de eso, la mezcla de reacción se enfrió hasta temperatura ambiente, se filtró a través de una almohadilla de celite y se lavó a fondo con acetato de etilo. El filtrado combinado se concentró a presión reducida para obtener el compuesto VIIIf (crudo) como un sólido marrón pálido, que se usó para la siguiente etapa sin ninguna purificación adicional. Rendimiento: 2,1 g (crudo). 1H-RMN (30o MHz, DMSO-de): 58,24 (s, 1H), 7,91 - 7,86 (m, 1 H), 7,46-7,40 (m, 1 H), 3,91 (s, 3H).
Etapa 7: 7-Fluoro-8-h¡drox¡-1-met¡lqu¡noxal¡n-2(1H)-ona (VIIIg)
Una solución agitada del compuesto VIIIf (2,1 g, 8,17 mmoles) en una mezcla de 1,4-dioxano (25 mL) y agua (15 mL) a temperatura ambiente se desgasificó con una corriente de nitrógeno durante 15 minutos. Luego, se añadieron sucesivamente t-BuXPhos Paladaciclo (0,19 g, 0,24 mmoles) y tris(dibencilidenacetona)dipaladio (0) (0,15 g, 0,16 mmoles). La mezcla resultante se desgasificó nuevamente con una corriente de nitrógeno durante 10 minutos. Luego, se añadió KOH (0,91 g, 16,34 mmoles) a la mezcla de reacción en atmósfera de nitrógeno. A continuación, la mezcla resultante se calentó hasta 100 °C y se agitó durante 16 horas.
Después de eso, la mezcla de reacción se enfrió hasta 0 °C , se inactivó con agua y se lavó con acetato de etilo (2 x 50 mL). A continuación, la fase acuosa se acidificó con HCl 1,5 (5 mL) y se extrajo con acetato de etilo (3 x 50 mL). La fase orgánica combinada se lavó con solución de salmuera (50 mL), se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto VIIIg (crudo) como un sólido marrón pálido. El producto crudo se llevó a la siguiente etapa sin más purificación. Rendimiento: 1,1 g, 69,62 % . LC_MS: Calc. para C 9H7 FN2 O2 194,17; Obs.: 194,9 [M+H]+.
Etapa 8: 8-(2,2-D¡etox¡etox¡)-7-fluoro-1-met¡lqu¡noxal¡n-2(1H)-ona (VIIIh)
A una solución agitada del compuesto VIIIg (1,1 g, 5,67 mmoles) en dimetilsulfóxido (11 mL) a temperatura ambiente en atmósfera de nitrógeno se añadieron sucesivamente carbonato de cesio (2,57 g, 7,93 mmoles) y bromoacetaldehído dietil acetal (1,4 mL, 8,50 mmoles). La mezcla resultante se calentó hasta 90 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se diluyó con agua (20 mL) y se extrajo con acetato de etilo (3 x 25 mL). La fase orgánica combinada se lavó con solución de salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna utilizando gel de sílice (230-400 de malla) eluyendo con acetato de etilo al 15 % en éter de petróleo para obtener el compuesto VIIIh como un sólido marrón pálido. Rendimiento: 1,2 g, 70,58 % . LC_MS: Calc. para C 15 H 19 FN2 O4310,33; Obs.: 311,2 [M+H]+.
Etapa 9: 2-((6-Fluoro-4-met¡l-3-oxo-3,4-d¡h¡droqu¡noxal¡n-5-¡l)ox¡)acetaldehído (VIII)
A una solución agitada del compuesto VIIIh (1,2 g, 3,86 mmoles) en diclorometano (15 mL) a 0 °C en atmósfera de nitrógeno se añadió gota a gota ácido trifluoroacético (6 mL, 5 volúmenes). La mezcla resultante se dejó con agitación a temperatura ambiente durante 2 horas. Después de eso, la mezcla de reacción se concentró, se basificó con solución al 10 % de NaHCO3, se extrajo con diclorometano (3 x 50 mL), se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto VIII (crudo) como un sólido marrón. El producto crudo se usó como tal para la siguiente etapa sin purificación adicional. Rendimiento: 0,6 g. LC_MS: Calc. para C 1 1 H9 FN2 O3236,2; Obs. 237,0 [M+H]+.
Síntesis de 2-(7-fluoro-1,4-d¡met¡l-2-oxo-1,2-d¡h¡droqu¡nol¡n-8-¡l)acetaldehído, (Intermedio IX)

A una solución agitada de 2-bromo-3-fluoroanilina (25,0 g, 131,57 mmoles) en diclorometano (500 mL) a 0 °C bajo nitrógeno se añadió anhídrido acético (18,65 mL, 197,36 mmoles). La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 16 horas. Después de eso, la mezcla de reacción se diluyó con agua y se extrajo con diclorometano (2 x 500 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró in vacuo para obtener el compuesto IXa (crudo), que se tomó como tal para la siguiente etapa sin más purificación. Rendimiento: 25 g, 81,91 % . LC-MS Calc. para CsH7BrFNO, 232,05; Obs.; 232,0 [M+]; 1H-RMN (300 MHz, DMSO-de): 5 9,57 (s, 1H), 7,49-7,34 (m, 2H), 7,20-7,14 (m, 1H), 2,09 (s, 3H).
Etapa 2: N-(2-Bromo-3-fluorofenil)-N-metilacetamida (IXb)
A una solución agitada del compuesto IXa (25,0 g, 107,73 mmoles) en THF seco (500 mL) en atmósfera de nitrógeno, se añadió hidruro de sodio (al 60 % en aceite mineral, 5,38 g y 134,66 mmoles) y se agitó durante 30 minutos. Luego, se añadió sulfato de dimetilo (13,79 mL, 145,44 mmoles) a la mezcla de reacción a 0 °C . La mezcla resultante se calentó hasta temperatura ambiente y se agitó a temperatura ambiente durante 3 horas. Después de eso, la mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo (2 x 500 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre sulfato de sodio y se concentró in vacuo para obtener el compuesto IXb como un sólido blanco, que se usó como tal para la siguiente etapa sin más purificación. Rendimiento: 20 g, 75,47 % . LC-MS Calc. para CgHgBrFNO 246,08; Obs.: 248,0; [M++2H]; 1H RMN (300 MHz, DMSO-cfe): 57,58-7,32 (m, 3H), 3,05 (s, 3H), 1,67 (s, 3H).
Etapa 3: 4-((2-Bromo-3-fluorofenil)(metil)amino)-2,4-dioxobutanoato de etilo (IXc)
A una solución agitada del compuesto IXb (20,0 g, 81,95 mmoles) en etóxido de sodio (al 20 % en etanol, 55 mL, 163,90 mmoles) a temperatura ambiente en atmósfera de nitrógeno se añadió oxalato de dietilo (47,89 g, 327,81 mmoles). A continuación, la mezcla resultante se calentó a 80 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se evaporó por completo a presión reducida para obtener el compuesto crudo, que se purificó adicionalmente por cromatografía en columna sobre gel de sílice (230-400 de malla, EtOAc al 40 % en éter de petróleo) para obtener el compuesto. IXc como un aceite incoloro. Rendimiento: 15,0 g, 53,32 % . LC-MS Calc. para C13HaBrFNO4346,15; Obs.: 347,0 [M++H]; 1H RMN (400 MHz, DMSO- de): 57,63-7,48 (m, 3H), 5,37 (s, 1H), 4,18-4,13 (m, 2H), 3,22 (s, 3H), 1,30 -1,15 (m, 3H).
Etapa 4: Ácido 8-bromo-7-fluoro-1-metil-2-oxo-1,2-dihidroquinolina-4-carboxílico (IXd)
Una solución del compuesto IXc (23,0 g, 66,44 mmoles) en H2 SO4 conc. (230 mL) se calentó hasta 90 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se inactivó con agua helada y se extrajo con acetato de etilo (2 x 500 mL), se lavó con agua, salmuera, se secó sobre sulfato de sodio y se concentró in vacuo. El producto crudo obtenido se trituró adicionalmente con éter dietílico, se filtró y se secó para obtener el compuesto IXd como un sólido blanco, que se usó como tal para la siguiente etapa sin más purificación. Rendimiento: 15 g, 75,26 % . LC-MS Calc. para CnH/BrFNOa 300,08; Obs.: 300,0; [M+]; 1H RMN (400 MHz, DMSO- de): 5 14,22 (s, br, 1H), 8,25-8,22 (m, 1H), 7,40-7,35 (m, 1H), 6,95 (s, 1H), 3,81 (s, 3H).
Etapa 5: 8-Bromo-7-fluoro-4-(hidroximetil)-1-metilquinolin-2(1H)-ona (IXe)
A una solución agitada del compuesto IXd (12,0 g, 40,00 mmoles) en THF (120 mL) a 0 °C en atmósfera de nitrógeno, se añadieron sucesivamente trietilamina (7,23 mL, 52,00 mmoles) y cloroformiato de isobutilo (6,55 g, 48 mmoles) y se agitó durante 1 hora. Luego, se añadieron NaBH4 (3,80 g, 100,00 mmoles) en porciones y metanol (10 mL) a la mezcla de reacción y se continuó agitando durante 10 minutos. Después de eso, la mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo (2 x 500 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre sulfato de sodio y se concentró in vacuo. El producto crudo obtenido se purificó por cromatografía en columna sobre
gel de sílice (230-400 de malla, EtOAc al 40 % en éter de petróleo) para obtener el compuesto IXe como un sólido blanco. Rendimiento: 6,5 g, 56,82 % . LC-MS Calc. para CnHgBrFNO2 , 286,10; Obs.: 288,0 [M++2H];1H RMN (400 MHz, DMSO-cfe): 5 7,82-7,78 (m, 1H), 7,34-7,30 (m, 1H), 6,68 (s, 1H), 5,61-5,58 (m, 1H), 4,74-4,72 (m, 2H), 3,80 (s, 3H).
Etapa 6: 8-Bromo-4-(clorometil)-7-fluoro-1-met¡lqu¡nol¡n-2(1H)-ona (IXf)
A una solución agitada del compuesto IXe (3,0 g, 10,48 mmoles) en diclorometano (60 mL) a 0 °C en atmósfera de nitrógeno, se añadió gota a gota cloruro de tionilo (2,34 mL, 31,46 mmoles). A continuación, la mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 5 horas. Después de eso, la mezcla de reacción se inactivó con solución acuosa al 10 % de NaHCO3 y se extrajo con diclorometano (2 x 100 mL). El extracto orgánico combinado se lavó con salmuera, se secó sobre sulfato de sodio y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con EtOAc al 20 % en éter de petróleo) para obtener el compuesto IXf como un sólido blanquecino. Rendimiento: 1,5 g, 47,16 % . LC-MS Calc. para CnHsBrClFNO, 304,54; Obs.:306,0 [M++2H];1H-RMN (300 MHz, DMSO-cfe): 57,98-7,93 (m, 1 H), 7,44-7,39 (m, 1 H), 6,87 (s, 1H), 5,04 (s, 2H), 3,81 (s, 3H).
Etapa 7: 8-Bromo-7-fluoro-1,4-dimetilqu¡nol¡n-2(1H)-ona (IXg)
A una solución agitada del compuesto IXf (1,5 g, 4,93 mmoles) en DMF (30 mL) a 0°C bajo atmósfera de nitrógeno se añadió NaBH4 (93 mg, 2,46 mmoles). La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 3 horas. Después de eso, la mezcla de reacción se enfrió hasta 0 °C , se inactivó con agua (60 mL) y se extrajo con acetato de etilo (2 x 100 mL). La fase orgánica combinada se lavó con salmuera, se secó sobre sulfato de sodio y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) 20 % de acetato de etilo en éter de petróleo) para obtener el compuesto IXg como un sólido blanquecino. Rendimiento: 0,7 g, 53,84 % . LC-MS Calc. para CnHgBrFNO, 270,10; Obs.: 272,0 [M++2H];1H RMN (400 MHz, DMSO-cfe): 57,83 (m, 1 H), 7,31 (m, 1 H), 6,55 (s, 1 H), 3,78 (s, 3H), 2,40 (s, 3H).
Etapa 8: 7-Fluoro-8-h¡drox¡-1,4-d¡met¡lqu¡nol¡n-2(1H)-ona (IXh)
Una solución agitada del compuesto IXg (1,0 g, 3,7037 mmoles) en una mezcla de 1,4-dioxano (15 mL) y agua (5 mL) a temperatura ambiente se desgasificó con una corriente de nitrógeno durante 15 minutos. Luego, se añadieron sucesivamente t-BuXPhos Paladaciclo (90 mg, 0 ,1111 mmoles) y tris(dibencilidenacetona)dipaladio (0) (68 mg, 0,074 mmoles). La mezcla resultante se desgasificó nuevamente con una corriente de nitrógeno durante 10 minutos. Luego, se añadió KOH (0,415 g, 7,4074 mmoles) a la mezcla de reacción bajo atmósfera de nitrógeno a temperatura ambiente. A continuación, la mezcla resultante se calentó hasta 100 °C y se agitó durante 16 horas.
Después de eso, la mezcla de reacción se enfrió hasta 0 °C , se inactivó con agua y se lavó con acetato de etilo (2 x 50 mL). A continuación, la fase acuosa separada se acidificó con HCl (1,5 N, 5 mL) y se extrajo con acetato de etilo (3 x 50 mL). La fase orgánica combinada se lavó con solución de salmuera (50 mL), se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto IXh (crudo) como un sólido marrón pálido, que se tomó para la siguiente etapa sin ninguna purificación adicional. Rendimiento: 0,5 g, 65,86 % . LC_MS: Calc. para C 1 1 H 10 FNO2 207,2; Obs.: 208,1 [M+H]+.
Etapa 9: 8-(2,2-D¡etox¡etox¡)-7-fluoro-1,4-d¡met¡lqu¡nol¡n-2(1H)-ona (IX¡)
A una solución agitada del compuesto IXh (0,5 g, 2,41 mmoles) en sulfóxido de dimetilo (10 mL) a temperatura ambiente bajo atmósfera de nitrógeno, se añadieron carbonato de cesio (1,1 g, 3,38 mmoles) y acetal dietílico de bromoacetaldehído (0,55 mL, 3,62 mmoles). Luego, la mezcla de reacción se calentó hasta 90 °C . y se agitó durante 16 horas. Después de eso, la mezcla de reacción se diluyó con agua (20 mL) y se extrajo con acetato de etilo (3 x 25 mL). La fase orgánica combinada se lavó con solución de salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna utilizando gel de sílice (230-400 de malla) eluyendo con acetato de etilo al 15 % en éter de petróleo para obtener el compuesto IX¡ como un sólido marrón pálido. Rendimiento: 0,5 g, 64,1 % . LC_MS: Calc. para C 17 H22 FNO4323,36; Obs.: 324,1 [M+H] .
Etapa 10: 2-((7-Fluoro-1,4-d¡met¡l-2-oxo-1,2-d¡h¡droqu¡nol¡n-8-¡l)ox¡)acetaldehído (IX)
A una solución agitada del compuesto IX¡ (0,5 g, 1,54 mmoles) en diclorometano (10 mL) a 0 °C bajo atmósfera de nitrógeno se añadió gota a gota ácido trifluoroacético (5 mL, 10 volúmenes). La mezcla resultante se dejó con agitación a temperatura ambiente durante 2 horas. Después de eso, la mezcla de reacción se concentró, se basificó con solución al 10 % de NaHCO3, se extrajo con diclorometano (3 x 50 mL). El extracto orgánico combinado se secó sobre Na2SO4 anhidro y se concentró in vacuo para obtener el compuesto IX (crudo) como un sólido marrón, que se usó para la siguiente etapa sin más purificación. Rendimiento: 0,25 g. LC_MS: Calc. para C 13 H 12 FNO3249,24; Obs. 250,1 [M+H]+; 1H-RMN (300 MHz, DMSO-cfe) : 59,67 (s, 1 H), 7,60-7,55 (m, 1 H), 7,29-7,22 (m, 1 H), 6,53 (s, 1 H), 4,88 (s, 2H), 3,79 (s, 3H), 2,49 (s, 3H).
Síntes¡s de 7-Fluoro-1-met¡l-8-(2-oxopropox¡)qu¡nol¡n-2(1H)-ona (Intermed¡o X)

A una solución agitada del compuesto lia (0,6 g, 3,10 mmoles) en DMF (6 mL) a temperatura ambiente en atmósfera de nitrógeno, se añadieron sucesivamente carbonato de potasio (0,65 g, 4,66 mmoles) y cloroacetona (0,46 g, 4,96 mmoles). La mezcla de reacción se calentó luego hasta temperatura ambiente y se agitó durante 16 horas. Después de eso, la mezcla de reacción se diluyó con agua (20 mL) y se extrajo con acetato de etilo (3 x 25 mL). La fase orgánica combinada se lavó con solución de salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con acetato de etilo al 10 % en éter de petróleo para obtener el compuesto X como un sólido marrón. Rendimiento: 0,26 g, 33,76 % . LC_MS: Calc. para C 13 H12 FNO3249,24; Obs.:250,1 [M+H]+.
Síntesis de 2-((6-fluoro-2,4-dimetil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)acetaldehído, (Intermedio XI)

Etapa 1: 8-Bromo-7-fluoro-1,3-dimetilquinoxalin-2(1H)-ona (XIa)
A una solución agitada del compuesto VIIIc (3,00 g, 13,7 mmoles) en etanol (15 mL) a temperatura ambiente en atmósfera de nitrógeno se añadieron piruvato de etilo (1,52 mL, 13,7 mmoles) y ácido acético (15 mL) sucesivamente. La mezcla resultante se calentó a reflujo y se agitó durante 16 horas. Después de eso, la mezcla de reacción se concentró a presión reducida. El residuo obtenido se diluyó con agua (100 mL) y se extrajo con acetato de etilo (2 x 50 mL). La fase orgánica combinada se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se evaporó in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (230-400 de malla) eluyendo con 20-30 % de acetato de etilo en éter de petróleo para obtener el compuesto XIa como un sólido blanquecino. Rendimiento: 2,30 g, 61,99 % ; 1H-RMN (400 MHz, DMSO-d6) : 57,82-7,78 (m, 1H), 7,41-7,37 (m, 1H), 3,91 (s, 3H), 2,42 (s, 3H).
Etapa 2: 7-Fluoro-8-hidroxi-1,3-dimetilquinoxalin-2(1 H)-ona (XIb)
Una solución agitada del compuesto XIa (2,1 g, 7,74 mmoles) en una mezcla de 1,4-dioxano (25 mL) y agua (15 mL) a temperatura ambiente se desgasificó con una corriente de nitrógeno durante 15 minutos. Luego, se añadieron sucesivamente t-BuXPhos Paladaciclo (0,18454 g, 0,234 mmoles) y tris (dibencilidenacetona)dipaladio (0) (0,141 g, 0,16 mmoles). La mezcla resultante se desgasificó nuevamente con una corriente de nitrógeno durante 10 minutos. Luego, se añadió KOH (0,867 g, 15,48 mmoles) a la mezcla de reacción en atmósfera de nitrógeno. A continuación, la mezcla resultante se calentó hasta 100 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se enfrió hasta 0 °C , se inactivó con agua y se lavó con acetato de etilo (2 x 50 mL). A continuación, la fase acuosa se acidificó con HCl acuoso (1,5 N, 5 mL) y se extrajo con acetato de etilo (3 x 50 mL). La fase orgánica combinada se lavó con solución de salmuera (50 mL), se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto XIb (crudo) como un sólido marrón pálido, que se tomó para la siguiente etapa sin ninguna purificación adicional. Rendimiento: 1,0 g, 62,5 % . LC_MS: Calc. para C 10 H9FN2 O2208,19; Obs.: 209,0 [M+H]+.
Etapa 3: 8-(2,2-Dietoxietoxi)-7-fluoro-1,3-dimetilquinoxalin-2(1 H)-ona (XIc)
A una solución agitada del compuesto XIb (1,0 g, 4,807 mmoles) en sulfóxido de dimetilo (10 mL) a temperatura ambiente en atmósfera de nitrógeno, se añadieron sucesivamente carbonato de cesio (2,34 g, 7,21 mmoles) y dietil
acetal de bromoacetaldehído (1,2 mL, 7,69 mmoles). La mezcla resultante se calentó hasta 90 °C . y se agitó durante 16 horas. Después de eso, la mezcla de reacción se diluyó con agua (20 mL) y se extrajo con acetato de etilo (3 x 25 mL). La fase orgánica combinada se lavó con solución de salmuera, se secó sobre Na2SO4 y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna utilizando gel de sílice (230-400 de malla) eluyendo con acetato de etilo al 15 % en éter de petróleo para obtener el compuesto XIc como un sólido marrón pálido. Rendimiento: 1,1 g, 73,33 % . LC_MS: Calc. para C 16 H2 1 FN2 O4324,35; Obs.: 324,9 [M+H]+.
Etapa 4: 2-((6-Fluoro-2,4-dimetil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)acetaldehído (XI)
A una solución agitada del compuesto XIc (1,1 g, 3,39 mmoles) en diclorometano (15 mL) a 0 °C en atmósfera de nitrógeno se añadió gota a gota ácido trifluoroacético (5,5 mL, 5 volúmenes). La mezcla resultante se dejó con agitación a temperatura ambiente durante 2 horas. Después de eso, la mezcla de reacción se concentró, se basificó con una solución al 10 % de NaHCO3 y se extrajo con diclorometano (3 x 50 mL). La fase orgánica combinada se secó sobre Na2SO4 y se concentró in vacuo para obtener el compuesto XI (crudo) como un sólido marrón, que se usó como tal para la siguiente etapa sin purificación adicional. Rendimiento: 0,4 g. LC_MS: Calc. para C 12 H 1 1 FN2 O3 250,23; Obs.
250,9 [M+H]+.
Síntesis de 2-((3-Cloro-5-metil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)acetaldehído, (Intermedio XII)

Etapa 1: 3-Cloro-6-metoxi-1,5-naftiridin-4-ol (XIIa)
A una solución agitada de 6-metoxi-1,5-naftiridin-4-ol (10,0 g, 56,76 mmoles) en ácido acético glacial (150 mL) a temperatura ambiente en atmósfera de nitrógeno se añadió N-clorosuccinimida (8,48 g, 63,57 mmoles). La mezcla de reacción se calentó hasta 60 °C y se agitó durante 3 horas. Después de eso, la mezcla de reacción se enfrió hasta temperatura ambiente y el precipitado sólido se filtró, se lavó con n-hexano y se secó en vacuo para obtener el compuesto XIIa como un sólido blanco, que se usó como tal para la siguiente etapa sin ninguna purificación adicional. Rendimiento: 10,0 g (crudo), 85,03 % . LC_MS Calc. para C 9H7 CLN 2 O2 , 210,62; Obs: 211,0 [M++H]; 1H-RMN (300 MHz, DMSO-de): 5 8,40 (s, 1H) 7,99 (d, J = 11,60 Hz, 1H), 7,20 (d, J = 12,0 Hz, 1H), 3,95 (s, 3H), 3,33 (brs, 1H).
Etapa 2: 10-Cloro-2,2-dimetil-2,3-dihidro-5H-[1,4,2]oxazasilino[6,5,4-de][1,5]naftiridin-5-ona (XIIb)
A una solución agitada del compuesto XIIa (2,0 g, 9,49 mmoles) en DMF (60 mL) a 0 °C en atmósfera de nitrógeno, se añadió hidruro de sodio (0,57 g, 14,24 mmoles, al 60 % disperso en aceite mineral). La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 1 hora. Luego, se añadió cloro (clorometil)dimetilsilano (2,02 mL, 15,19 mmoles) a la mezcla de reacción a temperatura ambiente y se dejó agitar durante otra 1,5 hora. Luego, la mezcla de reacción se calentó hasta 100 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se inactivó con metanol (1 mL) y se concentró por completo in vacuo. El producto crudo obtenido se trituró con éter dietílico, se filtró y se secó en vacuo para obtener el compuesto XIIb como un sólido naranja pálido, que se usó como tal para la siguiente etapa sin ninguna purificación. Rendimiento: 3,0 g (crudo). LC_MS Calc. para C n H n C lN 2 O2 Si, 266,76; Obs: 266,8; 1H-RMN (400 MHz, DMSO- de): 58,45 (s, 1 H) 7,88 (d, J = 9,60 Hz, 1 H), 6,89 (d, J = 9,60 Hz, 1 H), 3,62 (s, 2H), 0,47 (s, 6H).
Etapa 3: 7-Cloro-8-hidroxi-1-metil-1,5-naftiridin-2(1H)-ona (XIIc)
A una solución agitada del compuesto XlIb (3,0 g, 11,27 mmoles) en una mezcla de 1,4-dioxano/metanol (113 mL, 2:1) a temperatura ambiente en atmósfera de nitrógeno se añadió fluoruro de cesio (5,13 g, 33,82 mmoles). A continuación, la mezcla de reacción se calentó hasta 80 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se concentró in vacuo. El residuo obtenido se disolvió en agua (5 mL) y se neutralizó con HCl 1,5 N (pH ajustado ~6-7). El precipitado sólido se filtró y se secó en vacuo para obtener el compuesto Xllc como un sólido blanquecino, que se usó como tal para la siguiente etapa sin ninguna purificación. Rendimiento: 2,0 g (crudo). LC_MS Calc. para C 9 H7 CLN 2 O2 , 210,62; Obs.; 211.1 ; [M++H]; 1H-RMN (400 MHz, DMSO-de): 5 12,53 (brs, 1H), 8,29 (s, 1H), 7,69 (d, J = 9,20 Hz, 1 H), 6,84 (d, J = 9,60 Hz, 1 H), 3,98 (s, 3H).
Etapa 4: 7-Cloro-8-(2,2-d¡etox¡etox¡)-1-metil-1,5-naft¡r¡d¡n-2(1H)-ona (Xlld)
A una solución agitada del compuesto Xllc (0,5 g, 2,37 mmoles) en DMSO (5 mL) a temperatura ambiente en atmósfera de nitrógeno se añadieron bromoacetaldehído dietil acetal (0,45 mL, 2,85 mmoles) y carbonato de cesio (1,16 g, 3,56 mmoles) sucesivamente. Luego, la mezcla de reacción se calentó hasta 100 °C y se agitó durante 16 horas. Después de eso, la mezcla de reacción se inactivó con agua y se extrajo con acetato de etilo (3 x 50 mL). La fase orgánica combinada se lavó con una solución de salmuera (50 mL), se secó sobre sulfato de sodio anhidro y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna usando gel de sílice (60-120 de malla) eluyendo con acetato de etilo al 30 % en éter de petróleo para obtener el compuesto Xlld como un sólido marrón. Rendimiento: 0,3 g, 38,34 % . LC_MS Calc. para C 15 H 19 CLN 2 O4 , 326,78; Obs.; 327,1; [M+ H]; 1H-RMN (400 MHz, DMSO- de): 58.34 (s, 1H), 8.08 (d, J = 10,00 Hz, 1H), 6,86 (d, J = 10,40 Hz, 1H), 4,74 (s, 1H), 4,41 (d, J = 4,80 Hz, 2H), 3,95 (s, 3H), 3,66-3,60 (m, 2H), 3,47-3,40 (m, 2H), 1,03-1,00 (m, 6H).
Etapa 5: 2-((3-Cloro-5-met¡l-6-oxo-5,6-d¡h¡dro-1,5-naft¡r¡d¡n-4-¡l)ox¡)acetaldehído (Xll)
A una solución agitada del compuesto 5 (0,3 g, 0,92 mmoles) en diclorometano (3 mL) a 0 °C bajo atmósfera de nitrógeno se añadieron ácido trifluoroacético (3 mL). A continuación, la mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 3 horas. Después de eso, la mezcla de reacción se inactivó con solución al 10 % de NaHCO3 y se extrajo con acetato de etilo (3 x 20 mL). La fase orgánica combinada se lavó con una solución de salmuera (20 mL), se secó sobre sulfato de sodio anhidro y se concentró in vacuo para obtener el compuesto 6 como un sólido marrón pálido, que se usó como tal para la siguiente etapa sin ninguna purificación. Rendimiento: 0,2 g (crudo). LC_MS Calc. para CnH gCl^O a, 252,65; Obs: 250,8 [M+-H]; 1H-RMN (300 MHz, DMSO-de): 5 9,62 (s, 1H), 8,28-8,24 (m, 1H), 8,02 (d, J = 14,0 Hz, 1H), 6,86-6,82 (m, 1H), 4,20-4,02 (m, 2H), 3,93 (s, 3H)
Además, los compuestos de Fórmula I se prepararon utilizando los intermedios anteriores.
Síntes¡s de (S)-6-(5-(((2-((5-met¡l-6-oxo-5,6-d¡h¡dro-1,5-naft¡r¡d¡n-4-¡l)ox¡)et¡l)am¡no)met¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡r¡do[3,2-b][1,4]oxaz¡n-3(4H)-ona (Compuesto 1)

A una mezcla de aldehido, l (0,1 g, 0,45 mmoles) y amina lV (0,12 g, 0,45 mmoles) en metanol seco (10 mL) y diclorometano seco (10 mL) se añadió AcOH (0,10 mL) y se dejó con agitación durante 16 horas a temperatura ambiente. A esto se añadió resina de cianoborohidruro (0,34 g, 0,68 mmoles) y se agitó durante otros 15 minutos a temperatura ambiente. La mezcla de reacción se filtró y el filtrado se concentró para obtener el crudo. El crudo se purificó por HPLC preparativa para dar el producto puro (Compuesto 1) como sal de formiato (sólido blanquecino, 13 mg, 6%). LC-MS Calc. para C 22 H22 N6O6 : 466,45; Obs.: 467,2 [M++H]; 1H RMN (400 MHz, DMSO-D6): 5 11,16 (s, 1 H), 8,34 (m, 1 H), 7,83 (d, J = 8,4 Hz, 1 H), 7,57 (d, J = 8,4, Hz, 1 H), 7,38 (d, J = 9 Hz, 1 H ), 7,21 (m, 1 H), 6,82 (d, J = 9,8 Hz, 1 H), 4,76 (brs, 1H), 4,62 (s, 2H), 4,23 (m, 2H), 4 ,13 -4 ,11 (m, 1H), 3,87 (m, 1H), 3,80 (s, 3H) ,3,08 (m,2H), 2,96 (m,3H).
Síntes¡s de (S)-6-(5-(((2-((7-fluoro-1-met¡l-2-oxo-1,2-d¡h¡droqu¡nol¡n-8-¡l)ox¡)et¡l)am¡no)met¡l)-2-oxooxazol¡d¡n-3-¡l)-2H-p¡raz¡no[2,3-b][1,4]oxaz¡n-3(4H)-ona (Compuesto 2)

A una mezcla de aldehido II (0,2 g, 0,85 mmoles) y amina VI (0,22 g, 0,85 mmoles) en MeOH seco (10 mL) y DCM (10 mL), se añadió AcOH (0,25 mL) y se dejó con agitación durante 16 h. A esto se añadió resina de cianoborohidruro (2,05 % en mmoles, 0,63 g, 1,27 mmoles) y se agitó durante 5 min. La mezcla de reacción se filtró y el filtrado se concentró para obtener el producto crudo. El crudo se purificó por el método de HPLC preparativa en fase inversa para proporcionar el Compuesto 2 como sólido blanquecino (sal de formiato, 50 mg). LC-MS Calc. para C 22 H2 1 FN6 O6 : 484,44; Obs.: 485,1; [M++H]; 1H RMN (400 MHz, DMSO-Da): 5 8,39 (s, 1H), 8,23 (s, 1H), 7,85 (d, J = 9,44 Hz, 1H), 7,49-7,50 (m, 1H), 7,22 (t, J = 10,08 Hz, 1H), 6,56 (d, J = 9,36 Hz, 1H), 4,79-4,81 (m, 3H), 4,03-4,04 (m, 3H), 3,83 3,85 (m, 5H), 2,93-2,94 (m, 4H).
Síntesis de (R)-6-(5-(((2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 3)

A una mezcla de compuestos II (0,5 g, 1,88 mmoles) y compuesto VI (0,443 g, 1,88 mmoles) en una mezcla de MeOH seco (20 mL) y CH 2 C l2 (20 mL) a temperatura ambiente en atmósfera de nitrógeno se añadió AcOH (0,5 mL) seguido de complejo de 2-picolina borano (2,05 % en mmoles) (0,120 g, 1,13 mmoles). La mezcla de reacción se continuó agitando a temperatura ambiente durante 1 hora. La mezcla de reacción se inactivó con HCOOH al 1 % en agua y se concentró in vacuo para obtener el crudo. El crudo obtenido se purificó por cromatografía en columna eluyendo con 8 10 % de metanol en diclorometano. El producto puro obtenido se trituró adicionalmente con éter dietílico para producir el Compuesto 3 como un sólido blanquecino (sal de formiato). Rendimiento: 0,2 g, 21,95 % . LC-MS Calc. para 22 H2 1 FN6O6 : 484,44; Obs.: 485,1; [M++H]; 1H RMN (400 MHz, DMSO-cfe) : 5 11,63 (S,1H), 8,39 (s, 1H), 8,14 (s, 1H), 7,87 (d, J = 9,44 Hz, 1H), 7,54-7,51 (m, 1H), 7,24 (t, J = 10,08 Hz, 1H), 6,57 (d, J = 9,20 Hz, 1H), 4,86-4,81 (m, 3H), 4 ,17-4 ,11 (m, 3H), 3,88-3,83 (m, 4H), 3,33-3,11 (m , 4H). HPLC Pureza = 96,21 % , Columna: X-Bridge C8 (50 x 4,6) mm, 3,5 gm, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: TFA al 0,1 % en acetonitrilo.
Síntesis de 6-((5S)-5-(((1-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)propan-2-il)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 4)

A una mezcla agitada del compuesto X (260 mg, 1,0 mmol) y compuesto V (275 mg, 1,0 mmol) en una mezcla de MeOH seco/diclorometano (30 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno se añadieron AcOH (0,6 mL) y complejo de 2-picolina-borano (110 mg, 1,0 mmol) sucesivamente. La mezcla de reacción se continuó agitando a temperatura ambiente durante 16 horas. Después de eso, la mezcla de reacción se inactivó con HCOOH al 0,1 % en agua y se concentró a presión reducida. El producto crudo obtenido se purificó adicionalmente por HPLC PREP para obtener el Compuesto 4 como un sólido blanquecino. Rendimiento: 100 mg, 50,20 % . LC_MS Calc. para C 23 H23 FN6O6 , 498,47; Obs.: 499,1 [M++H].1H RMN (300 MHz, DMSO-cfe) : 5 11,60 (brs, 1H), 8,38-8,35 (m, 1H), 7,85 7,82 (d, J = 9,3 Hz, 1 H), 7,52-7,47 (t, J = 6,6 Hz, 1 H), 7,24-7,18 (t, J = 9,3 Hz, 1 H), 6,57-6,53 (d, J = 9,3 Hz, 1 H), 4,84 (brs, 3H), 4,15-4,09 (m, 1H), 3,95-3,85 (m, 2H), 3,80 (s, 3H), 3,19-3,04 (m, 4H), 1,17 -1,14 (d, 3H); HPLC Pureza = 99,25 % (Columna HPLC: xBridge C8 (50 x 4,6) mm, 3,5 gm, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: TFA al 0,1 % en acetonitrilo.
Síntesis de (S)-6-(5-(((2-((7-fluoro-1,4-dimetil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 5)

A una mezcla agitada del compuesto IX (250 mg, 1,00 mmol) y compuesto V (280 mg, 1,00 mmol) en una mezcla de MeOH seco/diclorometano (20 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno, se añadieron sucesivamente AcOH (0,5 mL) y complejo de 2-picolina-borano (64 mg, 0,6024 mmoles). La mezcla de reacción se continuó agitando a temperatura ambiente durante 1 hora. Después de eso, la mezcla de reacción se inactivó con HCOOH al 0,1 % en agua y se concentró a presión reducida. El producto crudo obtenido se purificó adicionalmente por HPLC PREP para obtener el Compuesto 5 como un sólido blanquecino. Rendimiento: 50 mg, 10,0 % . LC_MS Calc. para C 24 H25 FN6O6 , 512,50; Obs.: 513,1 [M++H]; 1H-RMN (400 MHz, DMSO-cfe) : 5 11,65 (brs, 1H), 8,39 (s, 1H), 7,62-7,58 (m, 1H), 7,31-7,26 (t, J = 10,0 Hz, 1H), 6,54 (d, J = 5,2 Hz, 1H), 4,87-4,83 (m, 3H), 4,24-4,17 (m, 2H), 3,83 (s, 3H), 3,78-3,74 (m, 1H), 2,98 (brs, 2H), 2,50 (s, 3H), 2,06 (brs, 2H); HPLC Pureza = 96,24 % (Columna HPLC: Atlantis dC18 (250*4,6) mm 5 μm, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: Acetonitrilo.
Síntesis de (S)-6-(5-(((2-((3-cloro-5-metil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 6)

A una solución agitada del compuesto XII (0,2 g, 0,79 mmoles) y compuesto V (0.232 g, 0,87 mmoles) en una mezcla de MeOH seco/diclorometano (8 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno, se añadió AcOH (0,2 mL) y se dejó con agitación durante 10 min. Luego, se añadió complejo de 2-picolina borano (0,051 g, 0,477 mmoles) a 0 °C . A continuación, la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 3 horas. Después de eso, la mezcla de reacción se inactivó con ácido fórmico al 1 % en agua y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por HPLC PREP (fase inversa) para obtener el Compuesto 6 como un sólido blanco (sal de formiato). Rendimiento: 0,010 g. LC_MS Calc. para C21H2üCIN7O6, 501,88; Obs.: 502,2 [M++H].
Síntesis de ((S)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 7)

A una solución agitada del compuesto VII (0,15 g, 0,635 mmoles) y compuesto V (0,17 g, 0,635 mmoles) en una mezcla de metanol/diclorometano seco (40 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno se añadieron AcOH (0,3 mL) y Pic-BH3 (32 mg, 0,38 mmoles). La mezcla resultante se continuó agitando a temperatura ambiente
durante 1 hora. El progreso de la reacción se monitorizó por TLC. Después de eso, la mezcla de reacción se inactivo con agua (3 mL) y se concentró a presión reducida. El producto crudo obtenido se purificó adicionalmente por cromatografía en columna utilizando gel de sílice (230-400 de malla) eluyendo con metanol al 5 % en DCM para obtener el compuesto 7a como un sólido marrón. Rendimiento: 0,12 g, 38,7 % . LC_MS: Calc. para C 2 1 H22 FN7 O6 487,45; Obs.: 486,3 [M-H]+.
A una solución agitada del compuesto 7a (0,12 g, 0,2462 mmoles) en 1,4-dioxano (4 mL) a temperatura ambiente bajo atmósfera de nitrógeno se añadió MnO2 (12 mg, 0,1478 mmoles) de una vez. La mezcla resultante se calentó a 100 °C y se agitó durante 2 horas. Después de eso, la mezcla de reacción se enfrió hasta temperatura ambiente, se filtró a través de una almohadilla de celite y se lavó minuciosamente con acetato de etilo y DCM (1:1). El filtrado combinado se concentró a presión reducida para obtener el compuesto crudo como un sólido marrón pálido, que se purificó más por HPLC PREP para obtener el compuesto Compuesto 7 como sal de ácido trifluoroacético: Polvo amorfo blanquecino; Rendimiento: 50 mg, 42 % .
LC_MS: Calc. para C21H20FN7O6 485,43; Obs.: 486,1 [M+H]+; 1H-RMN (400 MHz, DMSO-de) : 5 11,68 (s, 1 H), 9,32 (bs, 2H), 8,40 (s, 1 H), 8,23 (s, 1H), 7,71 - 7,68 (m, 1H), 7,44-7,39 (t, J = 9,32 Hz, 1H), 5,13 (bs, 1H), 4,89 (s, 2H), 4,35 4,34 (m, 2H), 4,26-4,24 (m, 1H), 3,86 (s, 3H), 3,84- 3,82 (m, 1H), 3,55 - 3,51 (m, 4H). LC_MS: Calc. para C21H20FN 7O6485,43; Obs.: 486,1 [M+H]+; HPLC: 1,95 min; 98,77 % ; Columna de HPLC: X-Bridge C8 (50X4,6) mm, 3,5 gm, Fase móvil A: TFA al 0,1 % en H2O, Fase móvil B: Acetonitrilo.
Síntesis de ((R)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 8)

A una solución agitada del compuesto VIII (0,250 g, 1,06 mmoles) y compuesto VI (0,280 g, 1,06 mmoles) en una mezcla de MeOH seco/diclorometano (20 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno, se añadió AcOH (0,5 mL) y se dejó con agitación durante 10 min. La mezcla de reacción se enfrió hasta 0 °C y se le añadió complejo de 2-picolina borano (0,067 g, 0,63 mmoles). A continuación, la mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 3 horas. Después de eso, la mezcla de reacción se inactivó con agua y se concentró in vacuo. El producto crudo obtenido se purificó por cromatografía en columna usando gel de sílice (60-120 de malla) eluyendo con 4 % de metanol en diclorometano para obtener el compuesto 8a. Rendimiento: 0,150 g, 30,91 % ; LC_MS Calc. para C 2 1 H22 FN7 O6 , 487,45; Obs.: 487,8 [M++H].
A una solución agitada del compuesto 8a (0,150 g, 0,307 mmoles) en 1,4-dioxano (3 mL) a temperatura ambiente bajo atmósfera de nitrógeno se añadió MnO2 (0,192 g, 2 ,15 mmoles). Luego, la mezcla de reacción se calentó hasta 100 °C y se agitó durante 2 horas. Después de eso, la mezcla de reacción se filtró a través de celite, se lavó con diclorometano/metanol (100 mL, 1:1) y se concentró in vacuo. El producto crudo obtenido se purificó por HPLC PREP (fase inversa) para obtener el Compuesto 8 como sal de formiato; Polvo amorfo blanco; Rendimiento: 0,020 g, 13,29 % .
LC_MS Calc. para C 2 1 H20 FN7 O6 , 485,43; Obs.: 483,9 [M+-H]; 1H-RMN (400 MHz, DMSO- de): 58,36 (s, 2H), 8,15 (s, 1 H), 7,61-7,58 (m, 1H), 7,34-7,30 (m, 1H), 4,84 (s, 2H), 4,81-4,78 (m, 1H), 4,12-4,4,07 (m, 3H), 3,86-3,82 (m, 4H), 3,00-2,97 (m, 2H), 2,94-2,89 (m, 2 H). Hp L c Pureza = 91,42 % , Columna: X-Bridge C8 (50 x 4,6) mm, 3,5 gm, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: Acetonitrilo.
Síntesis de (S)-6-(5-(((2-((6-fluoro-2,4-dimetil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 9)

A una mezcla agitada del compuesto XI (0,2 g, 0,8 mmoles) y compuesto V (210 mg, 0,8 mmoles) en una mezcla de MeOH seco/diclorometano (30 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno se añadieron AcOH (0,4 mL) y complejo de 2-picolina-borano (51 mg, 0,48 mmoles). La mezcla de reacción se continuó agitando a temperatura ambiente durante 1 hora. Después de eso, la mezcla de reacción se inactivó con HCOOH al 0,1 % en agua y se concentró a presión reducida. El producto crudo obtenido se purificó adicionalmente por HPLC P R E P para obtener el Compuesto 9 como un sólido blanquecino como una sal de ácido trifluoroacético. Rendimiento: 50 mg, 12,82 % . LC_MS Calc. para C 22 H22 FN7 O6 , 499,46; Obs.: 500,0 [M++H]; 1H RMN (400 MHz, DMSO-cfe): 5 11,69 (brs, 1 H), 9,32 (brs, 2H), 8,40 (s, 1H) 7,62-7,58 (m, 1H), 7,39-7,34 (m, 1H), 5 ,16 -5 ,12 (m, 1H), 4,89 (s, 2H), 4,36-4,24 (m, 2H), 3,87 (s, 3H), 3,85-3,83 (m, 2H), 3,57-3,52 (m, 4H), 2,33 (s, 3H); HPLC Pureza = 97,03 % (Columna HPLC: X-Bridge C8 (50 x 4,6) mm, 3,5 p m, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: Acetonitrilo.
Síntesis de (S)-5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroqumoxaMn-5-N)oxi)etil)ammo)metil)-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Compuesto 10)

A una solución agitada del compuesto VIII (0,150 g, 0,63 mmoles) y compuesto VII (0,179 g, 0,63 mmoles) en una mezcla de MeOH seco/diclorometano (12 mL, 1:1) a temperatura ambiente en atmósfera de nitrógeno, se añadió AcOH (0,3 mL) y se dejó con agitación durante 10 min. Luego, se añadió complejo de 2-picolina borano (0,040 g, 0,38 mmoles) a 0 °C . La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 3 horas. Después de eso, la mezcla de reacción se inactivó con agua y se concentró in vacuo. El producto crudo obtenido se purificó por cromatografía en columna usando gel de sílice (60-120 de malla) eluyendo con 5 % de metanol en diclorometano para obtener el compuesto 10a. Rendimiento: 0,1 g, 31,65 % ; LC_MS Calc. para C 2 1 H22 FN7 O5 S 503,51; Obs.: 504,1 [M++H].
A una solución agitada del compuesto 10a (0,1 g, 0,198 mmoles) en 1,4-dioxano (2 mL) a temperatura ambiente en atmósfera de nitrógeno, se añadió dióxido de manganeso (0,124 g, 1,39 mmoles). Luego, la mezcla de reacción se calentó hasta 100 °C y se agitó durante 2 horas. Después de eso, la mezcla de reacción se filtró a través de una almohadilla de celite, se lavó con diclorometano/metanol (1:1, 100 mL) y se concentró in vacuo. El producto crudo obtenido se purificó adicionalmente por HPLC PREP (fase inversa) para obtener el Compuesto 10 como sal de formiato. Polvo amorfo blanco: Rendimiento: 0,020 g, 20,14 % . LC_MS Calc. para C 21 H20 FN7 O5 S 501,49; Obs.: 499,8 [M+-H]; 1H RMN (400 MHz, DMSO-cfe): 5 11,96 (brs, 1H), 8,78 (s, 1H), 8,30 (bs, 1H), 8,15 (s, 1H), 7,61-7,57 (m, 1H), 7,34-7,29 (m, 1H), 4,84-4,80 (m, 1H), 4,12-4,4,07 (m, 3H), 3,86-3,82 (m, 4H), 3,76 (s, 2H), 2,98-2,91 (m, 4H); HPLC Pureza = 99,55 % , Columna: X-Bridge C8 (50 x 4,6) mm, 3,5 μm, Fase móvil A: TFA al 0,1 % en agua, Fase móvil B: Acetonitrilo.
Ejemplo 3
Actividad biológica
Actividad antibacteriana:
Los compuestos de Fórmula I son de interés debido a sus potentes efectos antibacterianos. La capacidad de los compuestos de la presente divulgación como se divulga en la presente memoria para lograr un efecto antibacteriano puede evaluarse con respecto a su capacidad para inhibir el crecimiento de especies bacterianas como Escherichia coli A TCC 25922, Staphylococcus aureus A TCC 29213, Klebsiella pneumoniae A TCC 13883, Acinetobacter baumannii A TCC 19606, Pseudomonas aeruginosa A TCC 27853, Enterococcus faecalis A TCC 29212, Enterococcus faecalis A TCC 29212, Proteus mirabilis A TCC 43071, Enterobacter cloacae A TCC 13047, Citrobacter freundii A TCC 43864 y Morganella morganii A TCC 25830 utilizando un ensayo basado en el siguiente protocolo de concentración inhibidora mínima (MIC).
Las bacterias de ensayo se cultivaron en Caldo de Luria Bertani (HIMEDIA M1245), se disolvieron 25 gramos del polvo en 1.000 mL de agua destilada y se esterilizaron en autoclave a una presión de 100 kPa (15 lb) (121 °C) durante 20 minutos. La esterilidad del medio se comprobó incubando a 37 °C durante un periodo de 48 h.
Los cultivos bacterianos que se almacenaron como preparaciones madre en glicerol a -80 °C se subcultivaron en placas de agar LB para obtener colonias aisladas. Se cultivó una única colonia de cada cepa en caldo LB. Los cultivos se incubaron a 37 °C , 200 rpm hasta que alcanzaron una densidad óptica (DO a 600 nm) de 0,8 a 1. Este cultivo en fase logarítmica se diluyó en caldo LB hasta un número de células de 5-8x10A5 CFU/mL para ser utilizado como inóculo para experimentos de MIC. Los compuestos de ensayo se disolvieron en sulfóxido de dimetilo (DMSO) hasta una concentración madre de 4 mg/ml. Se preparó una serie de diluciones dobles de esta preparación madre en DMSO en una placa de microtitulación de fondo en V de 96 pocillos de las filas A a H. Se transfirió un volumen de 3 pL de estas diluciones a una placa de ensayo de microtitulación de fondo plano de 96 pocillos. Se incluyeron controles para monitorizar los efectos del DMSO y la esterilidad de los medios. Cada pocillo se inoculó con 150 pL del cultivo diluido anterior. Las placas se incubaron a 37 °C toda la noche en una incubadora humidificada. A la mañana siguiente, se leyeron las placas utilizando un espectrofotómetro a una longitud de onda de 600 nM.
La concentración inhibidora mínima (MIC) se define como la concentración más baja de fármaco que contiene un pocillo que muestra una inhibición del 90 % del crecimiento bacteriano. La actividad antibacteriana (MIC) determinada contra patógenos representativos Gram-positivos (Staphylococcus aureus ATCC 29213, Enterococcus faecalis ATCC 29212) y Gram-negativos (Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 13883, Acinetobacter baumannii ATCC 19606, Pseudomonas aeruginosa ATCC 27853, Proteus mirabilis ATCC 43071, Enterobacter cloacae ATCC 13047, Citrobacter freundii ATCC 43864, Morganella morganii ATCC 25830) se informaron en la Tabla 1. Los compuestos ejemplificados que pertenecen a la Fórmula I demostraron una potente actividad antibacteriana contra patógenos tanto Gram-positivos como Gram-negativos.
Tabla 1 Estudios MIC en Medios LB


S.au: S. aureus A TCC 29213; E.fa: E. faecalis A TCC 29212; E.co: E. coli ATCC 25922; P.ae: P. aurigenosa A TCC 27853; K.pn. K. pneumoniae A TCC 13883; A.ba: A. baumannii A TCC 19606; C.fr: Citrobacter freundii ATCC 43864; E.cl: Enterobacter cloacae ATCC 13047; M.mo: Morganella morganii ATCC 25830; P.mi; Proteus mirabilis ATCC 43071
Ensayo de inhibición enzimática: Determinación del valor CI50 contra el superenrollamiento por girasa de E. co li y decatenación por Topo IV de E. co li
Los compuestos de Fórmula I o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas, formas ópticamente activas y derivados farmacéuticamente activos de los mismos, se evaluaron para su uso en la eliminación o inhibición del crecimiento de bacterias Grampositivas y Gram-negativas a través de la inhibición de las topoisomerasas bacterianas de Tipo II, a saber, la ADN girasa y la Topo IV. La presente divulgación también proporciona evidencia para el tratamiento de infecciones causadas por bacterias tanto Gram-positivas como Gram-negativas a través de la inhibición de las topoisomerasas bacterianas usando la ADN girasa y enzimas Topo IV de E. coli.
Procedimiento para el ensayo del superenrollamiento por ADN girasa de E. co li
El superenrollamiento por girasa de E. coli y su inhibición se ensayaron usando un kit procurado por Inpiralis (K0001) y el protocolo (PMID: 2172086) se adaptó con las modificaciones necesarias. Los compuestos a ensayar se incubaron durante 10 min con 2,5 nM de ADN girasa de E. coli en una reacción de 30 pl de volumen y DMSO al 3,2 % . A continuación, las reacciones se iniciaron con la adición de 60 ng de ADN del plásmido pBR322 relajado y continuaron durante 45 min a 37 °C . La mezcla de reacción contenía Tris.HCl 35 mM (pH 7,5), KCl 24 mM, espermidina 1,8 mM, MgCl24 mM, DTT 2 mM, glicerol al 6,5 % (p/v), 0,1 mg/mL de BSA y ATP 1 mM. A continuación, la reacción se detuvo mediante la adición de 0,75 pL de proteinasa K (20 mg/mL) y 3 pL de SD S al 2 % y se incubó adicionalmente a 37 °C durante 30 min. A esto le siguió la adición de 4 pL de St EB (sacarosa al 40 % (p/v), Tris-HCl 100 mM, pH 8, EDTA 1 mM, 0,5 mg/ml de azul de bromofenol) y las formas superenrolladas/relajadas del ADN plasmídico se separaron por electroforesis en gel de agarosa. Los geles de agarosa al 1 % se corrieron durante 3 h a 4 V/cm en 1 x TAE (Tris 40 mM, ácido acético 20 mM, EDTA 1 mM). Para visualizar el ADN, los geles se tiñeron durante 10 min con 0,7 μg/mL de bromuro de etidio y el exceso de colorante se eliminó mediante varios lavados con agua. Los valores de CI50 se determinaron cuantificando el ADN superenrollado y relajado en cada una de las reacciones a partir de una imagen del gel mediante un método densitométrico utilizando el software Quantity One (Bio-rad).
Procedimiento para el ensayo de decatenación por topoisomerasa IV de E. co li
La actividad de decatenación de la topoisomerasa IV de E. coli y su inhibición se ensayaron utilizando un kit procurado por Inpiralis (D4002) y el protocolo del kit se adaptó con las modificaciones necesarias similares a los ensayos de superenrollamiento por girasa. Los compuestos a ensayar se incubaron durante 10 minutos con 5 nM de topoisomerasa IV de E. coli en un volumen de reacción de 30 pl y DMSO al 3,2 % . Las reacciones se iniciaron con la adición de 60 ng de kADN y continuaron durante 40 min a 37 °C . La mezcla de reacción final contiene Tris-HCl 40 mM (pH 7,6), glutamato de potasio 100 mM, acetato de magnesio 10 mM, DTT 10 mM, ATP 1 mM y 50 μg/ml de albúmina. Las reacciones se detuvieron mediante la adición de 0,75 pL de proteinasa K (20 mg/mL) y 3 pL de SD S al 2 % y se incubaron a 37 °C durante 30 min. A esto le siguió la adición de 4 pL de STEB (sacarosa al 40 % (p/v), Tris-HCl 100 mM pH 8, EDTA 1 mM, 0,5 mg/ml de azul de bromofenol) y las formas de kADN/minicírculos se separaron por electroforesis en gel de agarosa. Los geles de agarosa al 1 % se corrieron durante 3 h a 4 V/cm en 1X TAE (Tris 40 mM, ácido acético 20 mM, EDTA 1 mM). Para visualizar el ADN, los geles se tiñeron durante 10 min con 0,7 μg/mL de bromuro de etidio y el exceso de colorante se eliminó mediante varios lavados con agua. Las CI50 se determinaron cuantificando la banda de ADN de cinetoplasto (kADN) dentro del pocillo del gel y los minicírculos decatenados que migran en el gel en cada una de las reacciones a partir de una imagen del gel mediante un método densitométrico utilizando el software Quantity One (Bio-rad).
Los compuestos representativos de Fórmula I se evaluaron frente a la ADN girasa y enzima Topo IV de E. coli utilizando un ensayo de superenrollamiento basado en gel para la inhibición de la girasa y un ensayo de decatenación para la inhibición de Topo IV. Los resultados de las Topo isomerasas bacterianas de Tipo II (Girasa y Topo IV) se presentan en la Tabla 2 a continuación.
Los resultados presentados en la Tabla 2 indican que los compuestos de Fórmula I ejercen actividad antibacteriana a través de la inhibición de la actividad de la topoisomerasa bacteriana de tipo II y significa el modo dual de inhibición de las topoisomerasas bacterianas (Girasa y Topo IV) para la actividad antibacteriana observada de los compuestos.
Tabla 2 Evaluación de compuestos de Fórmula I frente a ADN girasa y enzima Topo IV de
E. co li

Se encontró que los valores de CI50 para la mayoría de los compuestos pertenecientes a la Fórmula I eran <0,250 tanto para la ADN girasa de E. coli como para la Topo IV de E. coli. Por otro lado, se encontró que los valores de CI50 para el Compuesto 2 eran <0,05 para ambas enzimas. Esto indica que los compuestos de Fórmula I ejercen su actividad antibacteriana a través de la inhibición de las topoisomerasas bacterianas (enzimas Girasa y Topo IV) dentro de la célula bacteriana.
Con el fin de ensayar la capacidad de los compuestos para retener la actividad antibacteriana contra cepas clínicas de bacterias, se llevaron a cabo estudios de susceptibilidad antibacteriana (determinación de MIC50 y MIC90) para un compuesto representativo (Compuesto 2) de la serie utilizando cepas clínicas de cinco especies de bacterias Gramnegativas (E. coli, P. aurigenosa, K. pneumoniae , A. baumannii, E. cloacae) según las pautas estándar del CLSI y los resultados se presentan en la Tabla 3 a continuación. Los fármacos estándar ciprofloxacino y meropenem se utilizaron como controles positivos en el estudio.
Tabla 3: Estudios de susceptibilidad (determinación de MIC50 y MIC90)
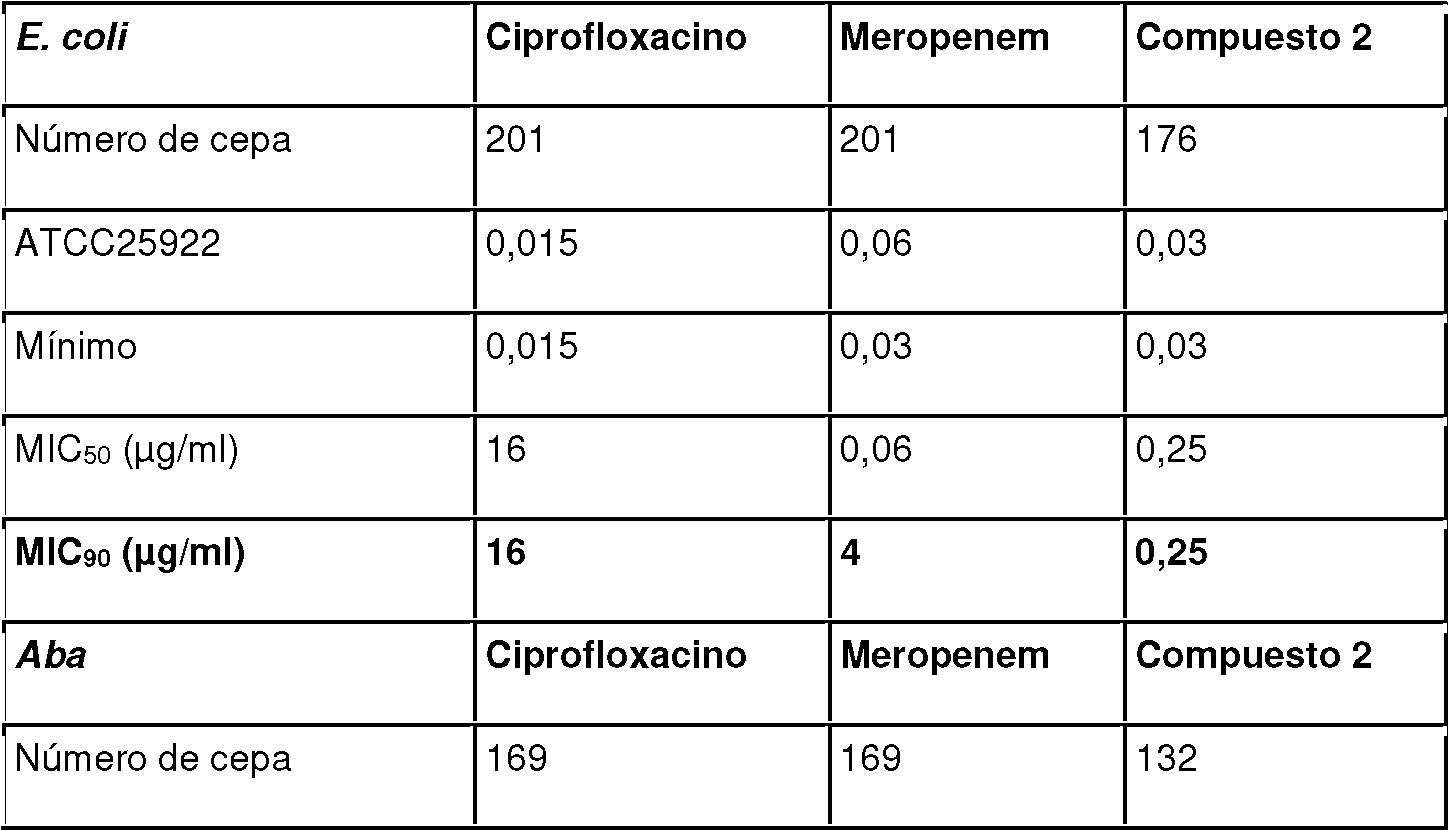

Los estudios de susceptibilidad antibacteriana ilustrados en la Tabla 3 indican que los compuestos de Fórmula I funcionan contra cepas clínicas de especies bacterianas gram-negativas tanto sensibles como resistentes a fármacos y retienen la actividad antibacteriana. Los valores de MIC90 del Compuesto 2 tienen un rango de 0,25 a 1 μg/ml para 5 especies bacterianas y se encuentra que es superior en comparación con los fármacos estándar utilizados en el estudio.
Ensayo de inhibición de hERG
Para ensayar si los compuestos de la presente divulgación tienen algún riesgo de seguridad al inhibir el canal iónico cardíaco, particularmente el canal de potasio (IKr, hERG), los compuestos se ensayaron usando ensayos electrofisiológicos para evaluar su actividad potencial en el canal iónico hERG. Los compuestos se ensayaron para determinar la inhibición del canal de K+ del gen relacionado con el éter humano (hERG) usando electrofisiología automatizada QPatch HTX. Se generaron curvas de concentración-respuesta de 6 puntos utilizando diluciones en
serie triples a partir de una concentración de ensayo final máxima de 300 pM y los resultados se presentan en la tabla 4.
Los compuestos de Fórmula I se solubilizaron a 100 mM en DMSO antes de la dilución en HBPS a 300 pM. Se generaron curvas de concentración-respuesta de 6 puntos utilizando diluciones en serie de 3,16 veces de la concentración de ensayo superior.
Procedimiento:
Los registros electrofisiológicos se realizaron a partir de una línea celular de ovario de hámster chino que expresaba de manera estable el canal de potasio hERG de longitud completa. Las corrientes iónicas de células individuales se midieron en una configuración de pinza de parche de células enteras a temperatura ambiente (21 -23 ° C) utilizando la plataforma QPatch HTX (Sophion). La solución intracelular contenía (mM): 120 KF, 20 KCl, 10 EGTA, 10 H EPES y se tamponó a pH 7,3. La solución extracelular (solución salina fisiológica tamponada con H EPES, HBPS) contenía (mM): 145 NaCl, 4 KCl, 2 C aC l2 , 1 MgCl2 , 10 HEPES, 10 glucosa, tamponada a pH 7,4. Las células se pinzaron a un potencial de retención de -80 mV. Las células se escalonaron a 20 mV durante 2s y luego a -40 mV durante 3s antes de volver al potencial de retención. Este barrido se repitió 10 veces a intervalos de 10s. Las corrientes de hERG se midieron desde el escalón de cola y se referenciaron a la corriente de retención. A continuación, los compuestos se incubaron durante 2 minutos antes de una segunda medición de la corriente del canal iónico usando un tren de pulsos idéntico.
Tabla 4: Valores de CI50 para hERG

Formulación intravenosa del Compuesto 2 para estudios farmacocinéticos (PK)
El Compuesto 2 se formuló en una solución de ácido L-ascórbico al 10 % en agua para lograr la solubilidad deseable para la vía de administración intravenosa y se ajustó a pH 4 con NaOH 1 N.
Procedimiento: Se pesó la cantidad apropiada del Compuesto 2 para ser ensayado y se disolvió en 1 mL de la solución de ácido ascórbico al 10 % . (Agitar con vórtex durante unos segundos si el compuesto no se disuelve instantáneamente). Se sometió a ultrasonidos la solución del compuesto a 37 °C durante 5 minutos usando un sonicador de baño para obtener una solución visualmente clara. El pH de la solución preparada anteriormente se ajustó a pH ~ 4 con solución de NaOH 1 N (p/v) con sonicación (formulación final pH ~ 4). Los detalles de la solubilidad de la Formulación del Compuesto 2 se dan en la Tabla 5.
Tabla 5: Solubilidad de la formulación IV

Se observó que la formulación IV preparada del Compuesto 2 era estable a temperatura ambiente durante más de 24 horas.
Estudios farmacocinéticos (PK)
in vivo
en ratas
Los estudios farmacocinéticos en ratas se llevaron a cabo en ratas Sprague-Dawley (SD) para estimar el aclaramiento plasmático, el volumen de distribución, la semivida terminal y la biodisponibilidad oral del Compuesto 2 después de 1 h de infusión intravenosa (IV) y sonda oral.
El Compuesto 2 exhibió un aclaramiento moderado, un volumen de distribución bajo y una semivida moderada y una buena biodisponibilidad oral en ratas SD. Se observó un aumento proporcional a la dosis en AUC y Cmáx durante la infusión IV y la dosificación oral del Compuesto 2 a dosis de 5, 10, 30 y 100 mg/kg en ratas SD. Este estudio sugiere que el Compuesto 2 tiene un perfil farmacocinético deseable para mantener los niveles en sangre del producto parental por encima de las MIC para demostrar la eficacia en modelos de infección en ratas mediante la administración de infusión IV.
Procedimiento: El objetivo de este estudio fue investigar el perfil farmacocinético del Ejemplo 12, después de dosis únicas ascendentes mediante infusión intravenosa (IV) a velocidad constante durante 1 h, en ratas macho Sprague Dawley. El estudio se realizó utilizando el siguiente diseño de estudio (n=3/grupo). Para la dosificación oral, las ratas se dosificaron por vía oral mediante una aguja de sonda forzada. El volumen de dosis requerido de las formulaciones de ensayo apropiadas se tomó en una jeringa graduada y se administró lentamente. Se usaron animales en ayunas durante la noche para la dosificación PO y se proporcionó alimento 4 h después de la dosificación. El diseño experimental farmacocinético para el Compuesto 2 se tabula en la Tabla 6 a continuación:
Tabla 6: Diseño experimental farmacocinético para el Compuesto 2 pág 106

Se utilizó muestreo de sangre en serie para la recogida de sangre. Se recogieron muestras de sangre antes de la dosis, 0,25, 0,5 h (durante la infusión), 1 h (final de la infusión) y 0,033, 0,25, 0,5, 1, 2, 4, 8 y 24 h después de la infusión. En cada punto de tiempo, se recogieron aproximadamente 100 pL de sangre de la vena yugular en un tubo de microcentrífuga etiquetado que contenía solución de K2 EDTA 200 mM (20 pL por mL de sangre) y el volumen equivalente de solución salina heparinizada se reemplazó después de la recogida de la muestra. Las muestras de sangre se procesaron para obtener las muestras de plasma dentro de los 30 min del tiempo de muestreo programado. Todas las muestras de plasma se almacenaron por debajo de -60 °C hasta el bioanálisis.
Las muestras de plasma se analizaron para determinar el Compuesto 2 utilizando un método de LC-MS/MS adecuado para su propósito con un límite inferior de cuantificación (LLOQ) de 8,1 ng/mL. Los parámetros farmacocinéticos del Compuesto 2 se calcularon utilizando la herramienta de análisis no compartimental del software validado Phoenix® WinNonlin® (versión 6.4) con método lineal ascendente y descendente para estimar AUC.
Las ratas Sprague Dawley macho (8-12 semanas de edad, con un peso de 280 ± 20 g en el momento de la dosificación) utilizadas en el estudio se obtuvieron de los laboratorios de Invigo Research, EE. UU. La solución anestésica (Ketamina y xilazina) se preparó mezclando 2 mL de Ketamina (50 mg/mL) con 0,5 mL de Xilazina (20 mg/mL). Las ratas fueron anestesiadas con una solución de ketamina y xilaxina por vía intraperitoneal a una dosis de 1 mL/kg. Se canularon las venas yugular y femoral de las ratas, y el estudio se realizó 48 h después de la canulación. Todos los animales se mantuvieron en ayunas durante la noche antes de la administración de la dosis y se les proporcionó alimento 4 h después de la administración de la dosis. Todos los animales recibieron agua ad libitum durante el período de estudio. El perfil farmacocinético IV y oral del Compuesto 2 se presenta en la Tabla 7.
Tabla 7: Perfil farmacocinético del Compuesto 2

*AUC de PK de la infusión IV 5 mg/kg utilizada para calcular la biodisponibilidad oral
Eficacia in vivo del Compuesto 2 en modelos de infección en rata: eficacia in vivo en el modelo de E. c o liy K. pneum oniae en muslo de rata:
El Compuesto 2 se ensayó en un modelo de infección en el muslo de rata después de la infusión intravenosa del compuesto a dosis de 100 mg/kg una vez, 30 mg/kg una vez al día durante un período de 1 h para evaluar su eficacia. Este estudio se realizó siguiendo todas las prácticas éticas establecidas en las directrices para el cuidado de los animales (Número de registro No. 1852/PO/RcN16/CPCSEA). El estudio fue aprobado por el Comité de Ética Animal Institucional (IAEC) del centro de ensayo. La formulación utilizada fue ácido L-ascórbico al 10 % en agua MilliQ fresca (p/v) con pH ajustado a ~4,0 con NaOH 1 N. El Día -4 (4 días antes de la fecha deseada de infección), se administró a cada rata una única inyección intraperitoneal de ciclofosfamida equivalente a 150 mg/kg y se devolvió a su jaula. El día -1 (un día antes de la infección) cada rata recibió una dosis equivalente a 100 mg/kg de ciclofosfamida. Este procedimiento aseguró que los animales serían neutropénicos el día 0. El día de la infección, el cultivo nocturno de los microorganismos apropiados [E. coli [ATCC25922]/ A. baumannii [ATCC19606]/ K. pneumoniae [ATCC13883] se ajustó a 1 DO [igual a ~ 109 CFU/mL], se centrifugó y las células se sedimentaron. Las células sedimentadas se suspendieron en solución salina normal estéril para obtener 107 CFU/ml y se usaron para la infección. El inóculo se diluyó diez veces en serie en caldo CSD B estéril y 0,05 mL de seis diluciones se sembraron en placas de agar CSDA para determinar el recuento viable (CFU/ml) del inóculo. Todos los animales se dividieron en diferentes grupos según lo especificado en el diseño experimental para cada microorganismo. Todas las infecciones se realizaron en una cabina de seguridad biológica, con la debida protección del personal. La infección se realizó mediante la inyección de 0,2 mL de inóculo [aproximadamente 1 x 107 CFU/ml en caldo] del microorganismo apropiado usando una jeringa y aguja de 1,0 mL, post-lateralmente en el muslo derecho del animal [aproximadamente 2 x 106 CFU/muslo]. Esto fue seguido de agitación/mezcla suave del inóculo entre dos animales para una distribución uniforme.
Dos horas después de la infección, a los animales de los grupos 4, 5 y 6 se les administró por vía intravenosa el Compuesto 2, como una infusión de velocidad constante (duración de la infusión 1 h), bajo anestesia de Ketamina 60 mg/kg IP Xilazina 10 mg/kg IP, en un volumen de dosis de 10 mL/kg, a razón de 0,16 mL/min. Los niveles de dosis del Compuesto 2 fueron 10, 30 y 100 mg/kg. El ciprofloxacino [10 mg/kg] y el vehículo [10 % de ácido L-ascórbico en agua fresca MilliQ (p/v) con ajuste de pH [a pH ~ 4,0] con solución de hidróxido de sodio 1N (p/v)] se dosificaron por vía intravenosa como una única dosis en bolo. La duración total del estudio fue de 10 h.
Los animales se sacrificaron 10 h después de la infección y se recogieron tejidos del muslo para enumerar el recuento de CFU bacterianas. Los músculos del muslo se extirparon asépticamente, se pesaron y se colocaron en 1 mL de caldo CSD B estéril y se homogeneizaron (Omni Tip (manual de 220 V)). Se prepararon diluciones seriadas de diez veces de los homogeneizados de muslo en caldo de lactosa estéril y se sembraron 0,05 mL de cuatro diluciones para cada muslo en placas de agar CSDA. Las colonias bacterianas se enumeraron después de una incubación durante la noche a 370C. Las densidades bacterianas se estimaron como Log10CFU/gramo de muslo. Se estimó la Media ± SD de Log10CFU/gramo de muslo en cada grupo. Las diferencias significativas entre las medias de los grupos y el control se analizarán mediante ANOVA unidireccional, seguido de una prueba de comparación múltiple de Dunnett, utilizando Graphpad Prism con niveles de confianza del 95 % . Se consideró significativo un valor de p < 0,05. Los resultados del estudio de eficacia se presentan en la Tabla 8.
Tabla 8: Eficacia del Compuesto 2 frente a E. co li [A TC C 25922] en un modelo de infección de muslo neutropénica en rata


Tabla 9: Eficacia del Compuesto 2 contra
K. pneumoniae [ATCC13883]
en un modelo de infección de muslo neutropénica en rata

*(P<0,05) Significativamente diferente del control de infección PI de 2 h;
#(P<0,05) Significativamente diferente del control de infección PI de 10 h.
El Ejemplo 2 mostró una eficacia bactericida dependiente de la dosis significativa con respecto al control de 2 horas después de la infección (PI) a 3, 10 y 30 mg/kg, y la eficacia fue comparable al fármaco estándar ciprofloxacino a una dosis similar (10 mg/kg).
Eficacia in vivo en el modelo de infección por
E. c o li
del tracto urinario (UTI) en ratas:
El propósito de este estudio es evaluar la eficacia del Compuesto 2 contra E. coli [ATCC25922] después de dosis de dosis única de infusión intravenosa de 3, 10 y 30 mg/kg en un modelo en rata de infección del tracto urinario.
Procedimiento
Antes del inicio del proceso de infección, todos los animales fueron divididos en diferentes grupos. Todas las jaulas de animales agrupadas se llevaron a una sala de procedimientos, cerca de una cabina de seguridad biológica. Todas
las infecciones se realizaron en una cabina de seguridad biológica, con la debida protección del personal. Los animales se anestesiaron mediante una inyección intraperitoneal de un cóctel de ketamina y xilazina (60+10 mg/kg i.p.). Una vez que los animales estuvieron en un plano de anestesia lo suficientemente profundo como se monitorizó mediante el reflejo del pedal, se afeitó la pared abdominal de cada rata con maquinillas eléctricas y se limpió la piel con povidona yodada al 10 % . Después de una incisión de 1,5 a 2 cm en la pared abdominal inferior, los músculos de la pared abdominal se separaron con disección roma. Se aisló y expuso la vejiga urinaria, se extrajo la orina del interior de la vejiga y se inyectaron en la vejiga 0,1 mL de solución salina estéril o cultivo bacteriano de E. coli (aproximadamente 1 x 108CFU/animal). Después de la reposición de la vejiga a su ubicación original, se aproximaron los músculos abdominales mediante sutura y se cerró la piel. Las heridas se limpiaron con povidona yodada al 10 % .
El vehículo de formulación IV utilizado fue ácido L-ascórbico al 10 % en agua MilliQ fresca (p/v) con pH ajustado a 4,0 con solución de hidróxido de sodio 1N (p/v), y el volumen de la dosis fue de 10 mL/kg. El meropenem se preparó en agua MilliQ y el pH de la solución se ajustó a 4,5 usando HCl. Cuatro horas después de la infección, se dosificó a los animales por vía intravenosa, como dosis únicas (para los compuestos de ensayo), como una infusión de velocidad constante, bajo anestesia con 60 mg/kg de ketamina IP 10 mg/kg de xilazina IP, a un volumen de dosis de 10 mL/kg, a la velocidad de 0,03 mL/min. Los niveles de dosis de los compuestos de ensayo fueron 3, 10 y 30 mg/kg. El meropenem se administró como una dosis única en bolo a un volumen de dosis de 5 mL/kg.
Todos los animales fueron sacrificados a las 24 h después de la infección, como se especifica en el diseño experimental, mediante una sobredosis de CO2 en una cámara de exposición apropiada. Los animales del grupo 1 se sacrificaron a las 4 h después de la infección.
Los animales sacrificados se sumergieron en etanol al 70 % para la descontaminación de la superficie. Los órganos se extrajeron asépticamente; se cortó la vejiga cerca de la uretra y se extirparon los riñones mediante disección roma para evitar el sangrado. La vejiga y cada riñón por separado se homogeneizaron en PBS. Las CFU por mililitro de homogeneizado de vejiga y riñón se determinaron después de 18 a 24 h de incubación a 37 °C . Se enumeró el número de bacterias por órgano y los resultados del estudio se presentan en las Tablas 9 y 10.
Tabla 10: Eficacia del Compuesto 2 contra
E. co li
[ATCC25922] en modelo de rata con infección del tracto urinario - riñones

*(P<0,05) significativamente diferente del control de infección PI de 4 h;
#(P<0,05) significativamente diferente del control de infección PI de 24 h.
Tabla 11: Eficacia del Compuesto 2 contra E. co li [ATCC25922] en modelo de rata con infección del tracto urinario - vejiga
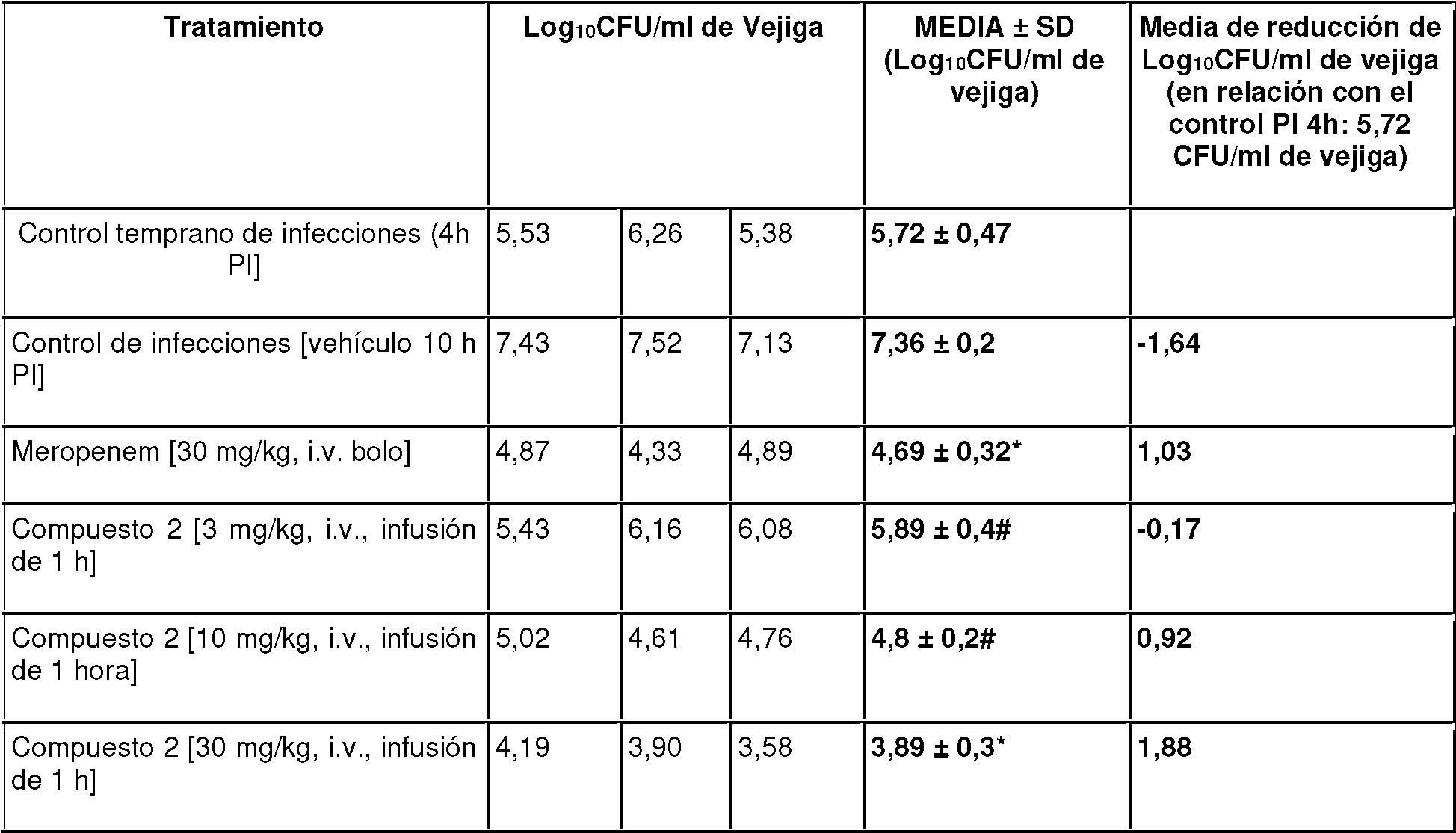
*(P<0,05) significativamente diferente del control de infección PI de 4 h;
# (P<0,05) significativamente diferente del control de infección PI de 24 h.
El Compuesto 2 mostró un efecto bactericida dependiente de la dosis significativo con respecto al control de PI de 4 h a 10 y 30 mg/kg y fue bacteriostático a 3 mg/kg en comparación con el control de PI de 4 h y la eficacia fue mejor que el fármaco estándar meropenem a dosis similar (30mg/kg).
Eficacia in vivo en el modelo de pulmón de rata con P. aeruginosa:
El propósito de este estudio fue evaluar la eficacia del Compuesto 2 contra P. aeruginosa [ATCC27853], después de dosis de dosis únicas de infusión intravenosa de 10, 30 y 100 mg/kg en un modelo de infección pulmonar neutropénica en ratas.
Procedimiento
Antes del inicio del proceso de infección, todos los animales fueron divididos en diferentes grupos. Todas las jaulas de animales agrupadas se llevaron a una sala de procedimientos, cerca de una cabina de seguridad biológica. Todas las infecciones se realizaron en una cabina de seguridad biológica, con la debida protección del personal. Los animales se colocaron en una cámara de inducción y se indujo la anestesia exponiendo a los animales a isoflurano al 3-5 % en un flujo de oxígeno ajustado a aproximadamente (~) 1 litro por minuto (LPM). Una vez que los animales se encontraban en un plano de anestesia lo suficientemente profundo como se monitorizó mediante el reflejo del pedal, se extrajeron e infectaron (2). La infección se inició instilando 0,07 mL (que contenía ~1x109 CFU/ml) del inóculo; 35 gl en cada fosa nasal del animal anestesiado usando una pipeta de 100 gl (~7X107CFU/animal). Se siguió un mezclado suave del inóculo entre dos animales para una distribución uniforme.
El vehículo de formulación IV utilizado para el Compuesto 2 fue ácido L-ascórbico al 10 % en agua MilliQ fresca (p/v) con pH ajustado a ~4,0 con solución de hidróxido de sodio 1N (p/v), y el volumen de la dosis fue de 10 mL/kg. El meropenem se formuló en solución salina. Cuatro horas después de la infección, los animales recibieron dosis intravenosas, en dosis únicas, por infusión, con anestesia de 60 mg/kg de ketamina IP 10 mg/kg de xilazina IP, a un volumen de dosis de 10 mL/kg, a una velocidad constante de 0,03 mL/mín. Los niveles de dosis del. Compuesto 2 de ensayo fueron 10, 30 y 100 mg/kg.
Todos los animales en grupos fueron sacrificados a las 24 h después de la infección, como se especifica en el diseño experimental, mediante una sobredosis de CO2 en una cámara de exposición apropiada. Los animales del grupo 1 se sacrificaron 4 h después de la infección. Los animales sacrificados se sumergieron en etanol al 70 % para la descontaminación de la superficie. Todo el pulmón se aisló asépticamente, se pesó y se colocó en 1 mL de caldo
CSD B estéril y se homogeneizó (Omni Tip (manual de 220 V)). Se prepararon diluciones seriadas de diez veces de los homogeneizados de pulmón en caldo CSD estéril y se sembraron 0,05 mL de cuatro diluciones para cada tejido en placas de agar CSDA. Las colonias bacterianas se enumeraron después de una noche de incubación a 370C. Las densidades bacterianas se estimaron como Log10 CFU/g de pulmón. Se estimó la Media ± SD de Log10 CFU/g de pulmón en cada grupo. Las diferencias significativas entre las medias de los grupos y el control se analizaron mediante ANOVA unidireccional, seguido de una prueba de comparación múltiple de Dunnett, utilizando Graphpad Prism con niveles de confianza del 95 % . Se consideró significativo un valor de p <0,05 y los resultados del estudio se presentan en la Tabla 10.
Tabla 12: Eficacia del Compuesto 2 contra P. aeruginosa [ATCC27853] en un modelo de infección pulmonar neutropénica en rata

Análisis de datos: Anova unidireccional seguido de la prueba de comparación múltiple de Dunnett; *(P<0,05) significativamente diferente del control de infección PI de 4h. # (P<0,05) significativamente diferente del control de infección PI de 24 h.
El Compuesto 2 mostró una eficacia significativa a dosis de 30 mg/kg y 100 mg/kg con respecto al control temprano de la infección y la eficacia fue comparable al fármaco estándar meropenem a una dosis similar (30 mg/kg).
VENTAJA
Los ejemplos de implementación mencionados anteriormente tal como se describen en este contenido y su equivalente tienen muchas ventajas, incluidas las que se describen.
Los compuestos de la presente divulgación muestran una alta actividad antibacteriana contra varios patógenos, incluidas bacterias Gram-positivas y Gram-negativas a través de la inhibición de la topoisomerasa bacteriana a través de un mecanismo novedoso.
Los compuestos de la presente divulgación demuestran un alto grado de selectividad contra el canal hERG (canal de potasio cardíaco) y pueden carecer de cardiotoxicidad en animales y seres humanos.
La ejemplificación representativa de la presente divulgación demuestra un perfil farmacocinético deseable en ratas y eficaz en varios modelos de infección en ratas, lo que confirma la prueba in vivo del principio en animales a través de la inhibición de la topoisomerasa bacteriana.
Aunque el contenido se ha descrito con considerable detalle con referencia a ciertas realizaciones del mismo, son posibles otras realizaciones. Como tal, el alcance de la invención no debe limitarse a la descripción de las realizaciones contenidas en la presente memoria.
Claims (18)
1. Un compuesto de Fórmula I

o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo,
en donde
R 1 se selecciona de alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3 H, O-PO3 H2 , COO R 9 , CONHR 9 , SO2 NHR9 , metilsulfona, alquilo C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 , cicloalquilo C 3 -6 , cicloalquilamino C 3 -6 , aminocicloalquilo C 3 -6 , cicloalquilhidroxi C 3 -6 , alquilamino C 1 -6 , o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S;
R9 se selecciona de hidrógeno, o alquilo C 1 - 6 ; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C 1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C 1 -6 , flúor, alcoxi C 1 -6 , hidroxilo o amino;
X 1 es N o C R 4 ;
R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ;
X 2 es N o C R 5 ;
R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; X 3 es N o CR6; y X 4 es CR6 cuando la línea punteada (----) representa un enlace;
R6 se selecciona de hidrógeno, ciano, alquilo C 1 -6 , alquilamino C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 o COOH; o
X3 es CH 2 u O; y X 4 es CH 2 cuando la línea punteada (----) no representa un enlace;
n1 es 0 a 2;
Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ;
R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ;
Z 1 se selecciona de O, S, NH o CH 2 ; y
R8 se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor.
2. El compuesto según se reivindica en la reivindicación 1, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo,
en donde
R1 se selecciona de alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1 -6 , alquenilo
C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3 H, O-PO3 H2 , COO R 9 , CONHR9 , SO2 NHR9 , metilsulfona, alquilo C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 , cicloalquilo C 3 -6 , cicloalquilamino C 3 -6 , aminocicloalquilo C 3 -6 , cicloalquilhidroxi C 3 -6 , alquilamino C 1 -6 o di(alquil C 1 -6 )amino, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C 1 - 6 ; R2 se selecciona de hidrógeno, flúor, cloro, alcoxi C 1 -4 , ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C 1 - 4 , hidroxilo o amino; X 1 es N o C R 4 ; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 - 4 , haloalquilo C 1 -4 , haloalcoxi C 1 -4 , o alquilo C 1 - 4 ; X 2 es N o C R 5 ; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -4 , haloalquilo C 1 - 4 , haloalcoxi C 1 -4 , o alquilo C 1 - 4 ; X 3 es N o CR6; y X 4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C 1 - 4 , alquilamino C 1 -4 , alcoxi C 1 -4 , haloalcoxi C 1 -4 y COOH; o X 3 es CH 2 u O; y X4 es CH 2 cuando la línea punteada (----) no representa un enlace; n es 0 a 2; Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 - 4 , haloalquilo C 1 -4 , haloalcoxi C 1 -4 , o alquilo C 1 - 4 ; Z 1 se selecciona de O, S, NH o CH 2 ; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor.
3. El compuesto según se reivindica en la reivindicación 1, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo,
en donde
R 1 se selecciona de alquilo C 1 -4 , haloalquilo C 1 -4 , cicloalquilo C 3 -4 , alquilamino C 1 -4 , o anillo de carbociclilo o heterociclilo saturado de 4-7 miembros, en donde el anillo de heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1 - 4 , haloalquilo C 1 - 4 , cicloalquilo C 3 -4 , alquilamino C 1 -4 y el anillo de carbociclilo o heterociclilo saturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, O-PO3 H2 , alquilo C 1 -4 , alcoxi C 1 -4 , haloalcoxi C 1 -4 , cicloalquilo C 3 -5 , cicloalquilamino C 3 -5 , aminocicloalquilo C 3 - 5 , cicloalquilhidroxi C 3 -6 , alquilamino C 1 -4 , di(alquil C1-4)amino, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R2 se selecciona de hidrógeno, flúor, alcoxi C 1 - 2 , ciano o hidroxilo; R3 se selecciona de hidrógeno, flúor, alcoxi C 1 - 2 , hidroxilo o amino; X 1 es N o C R 4 ; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 - 2 , haloalquilo C 1 - 2 , haloalcoxi C 1 - 2 , o alquilo C 1 - 2 ; X 2 es N o C R 5 ; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 - 2 , haloalquilo C 1 - 2 , haloalcoxi C 1 - 2 , o alquilo C 1 - 2 ; X 3 es N o CR6; y X4 es CH cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C 1 - 2 , alquilamino C 1 - 3 , alcoxi C 1 - 2 , haloalcoxi C 1 -2 o COOH; o X 3 es CH 2 u O; y X 3 es CH 2 cuando la línea punteada (----) no representa un enlace; n es 0 o 1; Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ;
R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 - 2 , haloalquilo C 1 - 2 , haloalcoxi C 1 - 2 , o alquilo C 1 - 2 ;
Z 1 se selecciona de O, S, NH o CH 2 ; y
R8 se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor.
4. El compuesto según se reivindica en la reivindicación 1, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo,
en donde
R 1 se selecciona de CH 3 , CH 2 CH 3 , CH(CH3)2, ciclopropilo, ciclobutilo, CH 2 C F 3 , CH 2 C H FC H 3 , CH 2 C F 2 CH 3 , CH2CH(OH)CH3, CH 2 CH 2 OH, CH 2 CH 2 OCH 3 , CH2CH(OCH3)CH3, CH 2 CH 2 NH2 , CH 2 CH 2 NHCH3 , CH2CH(NH2)CH3, CH2CH2N(CH3)2, CH 2 C H FC H 2 NH2 , CH 2 C F 2 CH 2 NH2 , CH 2 CH 2 OPO3 H2 , CH2C(CH3)2OH, CH2C(CH3)2OPO3H2,

R2 se selecciona de H, F, Cl, OCH 3 , CN u OH; R3 se selecciona de H, F, O CH 3 , OH o NH2 ;
Xi es N o C R 4 ;
R4 se selecciona de H, F, CN, O CH 3 , o CH 3 ; X 2 es N o C R 5 ; R5 se selecciona de H, F, CN, OCH 3 , C F 3 , O C F 3 , o CH 3 ; X 3 es N o CR6; y X4 es CH cuando la línea punteada (----) representa un enlace;
R6 se selecciona de H, CN, COOH, CH 2 NH2 , CH(CH3)NH2, CH 2 NHCH3 , OCH 3 , O C F 3 , o CH 3 ; o
X 3 es CH 2 u O; y X4 es CH 2 cuando la línea punteada (----) no representa un enlace;
n1 es 0 o 1;
Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ;
R7 se selecciona de H, F, CN, OCH 3 , o CH 3 ;
Z 1 se selecciona de O, S, NH o CH 2 ; y
R8 se selecciona de H, OH, alquilo C 1 -6 o F.
5. El compuesto según se reivindica en la reivindicación 1, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo, que se selecciona del grupo que consiste en:

(S)-6-(5-(((2-((5-metil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirido[3,2-b][1,4]oxazin-3(4H)-ona (Compuesto 1),

(S)-6-(5-(((2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 2),

(R)-6-(5-(((2-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 3),

6-((5S)-5-(((1-((7-fluoro-1-metil-2-oxo-1,2-dihidroquinolin-8-il)oxi)propan-2-il)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 4),

(S)-6-(5-(((2-((7-fluoro-1,4-dimetil-2-oxo-1,2-dihidroquinolin-8-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 5),

(S)-6-(5-(((2-((3-cloro-5-metil-6-oxo-5,6-dihidro-1,5-naftiridin-4-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 6),

(S)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidina-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 7),

(R)-6-(5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidina-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 8),

(S)-6-(5-(((2-((6-fluoro-2,4-dimetil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Compuesto 9), y

(S)-5-(((2-((6-fluoro-4-metil-3-oxo-3,4-dihidroquinoxalin-5-il)oxi)etil)amino)metil)-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Compuesto 10),
6. Un compuesto de Fórmula (B)

o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo,
en donde
Y 1 se selecciona de N o C R 7 ;
Y 2 es N;
R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ;
Z 1 se selecciona de O, S, NH o CH 2 ; y
Rs se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor.
7. El compuesto según se reivindica en la reivindicación 6, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo, que se selecciona del grupo que consiste en:

(S)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,41oxazn-3(4H)-ona (Intermedio V)

(R)-6-(5-(aminometil)-2-oxooxazolidin-3-il)-2H-pirazino[2,3-b][1,4]oxazin-3(4H)-ona (Intermedio VI)

(S)-5-(aminometil)-3-(3-oxo-3,4-dihidro-2H-pirazino[2,3-b][1,4]tiazin-6-il)oxazolidin-2-ona (Intermedio VII)
8. Un proceso de preparación de compuestos de Fórmula (B) según se reivindica en una cualquiera de las reivindicaciones 6 o 7, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas de los mismos, comprendiendo dicho proceso:
a) hacer reaccionar un compuesto de Fórmula (C) y un compuesto de Fórmula (D) en presencia de al menos un catalizador y al menos un disolvente para obtener un compuesto de Fórmula (E);
b) hacer reaccionar el compuesto de Fórmula (E) y al menos un compuesto de nitrógeno para obtener un compuesto de Fórmula (F); y
c) reducir el compuesto de Fórmula (F) para obtener un compuesto de Fórmula (B).

9. El proceso según se reivindica en la reivindicación 8, en donde al menos un catalizador se selecciona de un grupo que consiste en catalizador que contiene Pd, t-BuXPhos-Pd, Pd(OAc)2 , y combinaciones de los mismos; el al menos un disolvente se selecciona del grupo que consiste en THF, tolueno, dioxano y combinaciones de los mismos, el al menos un compuesto de nitrógeno es NaN3.
10. El proceso según se reivindica en la reivindicación 8, en donde la reducción del compuesto de Fórmula (F) para obtener un compuesto de Fórmula (B) se lleva a cabo en presencia de un agente reductor seleccionado de trifenilfosfina [(Ph3P)/THF-H2O], o hidrógeno y paladio sobre carbón (H2/Pd-C).
11. Un proceso de preparación de compuestos de Fórmula I según se reivindica en una cualquiera de las reivindicaciones 1 - 5, o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas de los mismos, comprendiendo dicho proceso hacer reaccionar un compuesto de Fórmula (A) y un compuesto de Fórmula (B)

en presencia de al menos un agente reductor y un adsorbente para obtener los compuestos de Fórmula I, en donde R 1 de Fórmula (A) se selecciona de alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3 H, O -PO3 H2 , COO R 9 , CONHR9 , SO2 NHR9 , metilsulfona, alquilo C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 , cicloalquilo C 3 -6 , cicloalquilamino C 3 -6 , cicloalquilhidroxi C 3 -6 , aminocicloalquilo C 3 -6 , alquilamino C 1 -6 , o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C 1 - 6 ; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C 1-6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C 1 -6 , flúor, alcoxi C 1 -6 , hidroxilo o amino; X 1 es N o C R 4 ; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; X 2 es N o C R 5 ; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; X 3 es N o CR6; y X 4 es CR6 o alquilo C 1-6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C 1 -6 , alquilamino C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 o COOH; o X3 es CH 2 u O; y X 4 es CH 2 cuando la línea punteada (----) no representa un enlace; y m es 0 a 2; R8 de Fórmula (B) se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor; Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; Z 1 se selecciona de O, S, NH o CH 2 ; y R 1 de Fórmula I se selecciona de alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , o anillo carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros, en donde el anillo heterociclilo está opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S, en donde alquilo C 1 -6 , alquenilo C 2 -6 , cicloalquilo C 3 -6 , alquilamino C 1 -6 , aminoalquileno C 1 -6 , y un anillo de carbociclilo o heterociclilo saturado o insaturado de 4-7 miembros están opcionalmente sustituidos con 1 a 3 grupos seleccionados independientemente de halógeno, amino, hidroxilo, SO3 H, O-PO3 H2 , COO R 9 , CONHR9 , SO2 NHR9 , metilsulfona, alquilo C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 , cicloalquilo C 3 -6 , cicloalquilamino C 3 -6 , aminocicloalquilo C 3 -6 , cicloalquilhidroxi C 3 -6 , alquilamino C 1 -6 , di(alquil C 1 -6 )amino, o anillo de heterociclilo saturado o insaturado de 3-7 miembros opcionalmente sustituido con hasta tres heteroátomos seleccionados independientemente de O, N o S; R9 se selecciona de hidrógeno, o alquilo C 1 - 6 ; R2 se selecciona de hidrógeno, flúor, cloro, ciano, alcoxi C 1 -6 o hidroxilo; R3 se selecciona de hidrógeno, alquilo C 1 -6 , flúor, alcoxi C 1 -6 , hidroxilo o amino; X 1 es N o C R 4 ; R4 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; X 2 es N o C R 5 ; R5 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; X3 es N o CR6; y X 4 es CR6 cuando la línea punteada (----) representa un enlace; R6 se selecciona de hidrógeno, ciano, alquilo C 1 -6 , alquilamino C 1 -6 , alcoxi C 1 -6 , haloalcoxi C 1 -6 , o COOH, en donde alquilo C 1-6 y alquilamino C 1-6 están opcionalmente sustituidos con uno o más grupos seleccionados de COOH, hidroxilo, amino o alquilo C 1 - 6 ; o X 3 es CH 2 u O; y X 4 es CH 2 cuando la línea punteada (----) no representa un enlace; m es 0 a 2; Y 1 , e Y 2 se seleccionan independientemente de N o C R 7 ; R7 se selecciona de hidrógeno, halógeno, ciano, alcoxi C 1 -6 , haloalquilo C 1 -6 , haloalcoxi C 1 -6 , o alquilo C 1 - 6 ; Z 1 se selecciona de O, S, NH o CH 2 ; y R8 se selecciona de hidrógeno, hidroxilo, alquilo C 1 -6 o flúor.
12. El proceso según se reivindica en la reivindicación 11, en donde el al menos un agente reductor se selecciona del grupo que consiste en borohidruro de sodio, cianoborohidruro de sodio, triacetoxiborohidruro de sodio y combinaciones de los mismos.
13. El proceso según se reivindica en la reivindicación 11, en donde el adsorbente se selecciona del grupo que consiste en tamices moleculares, gel de sílice, zeolitas, sulfato de sodio anhidro, sulfato de magnesio anhidro, carbón activado y combinaciones de los mismos.
14. Un compuesto según se reivindica en una cualquiera de las reivindicaciones 1 a 5 o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo para su uso como medicamento.
15. Un compuesto según se reivindica en una cualquiera de las reivindicaciones 1 a 5 o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas
ópticamente activas del mismo para su uso para matar o inhibir el crecimiento de un microorganismo seleccionado del grupo que consiste en bacterias, virus, hongos y protozoos.
16. Un compuesto según se reivindica en una cualquiera de las reivindicaciones 1 a 5 o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo, para uso en el tratamiento de una infección bacteriana causada por un bacteria Grampositiva o una bacteria Gram-negativa seleccionada de Escherichia coli, Pseudomonas aurigenosa, Klebsiella pneumoniae, Acinetobacter baumannii, Enterobacter cloacae, Staphylococcus aureus, Enterococcus faecalis, Enterococcus faecium, Legionella pneumophila. Mycoplasma pneumonía, Acinetobacter haemolyticus, Acinetobacter junii, Acinetobacter lwoffi, Burkholderia cepacia, Chlamydophila pneumoniae, Clostridium difficili, Enterobacter aerogenes, Enterobacter cloacae, Moraxella catarrhalis, Neisseria gonorrhoeae, Neisseria meningitides, Proteus mirabilis, Proteus houseri, Citrobacter freundii, Citrobacter kosari, Citrobacter barakii, Seratia marcescens, Klebsiella oxytoca, Morganella morganii, Helicobacter pyroli, Mycobacterium tuberculosis.
17. Una composición farmacéutica que comprende un compuesto de Fórmula I, según se reivindica en cualquiera de las reivindicaciones 1 a 5 o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo junto con un vehículo farmacéuticamente aceptable.
18. Una composición farmacéutica que comprende un compuesto de Fórmula I según se reivindica en cualquiera de las reivindicaciones 1 a 5 o sus estereoisómeros, sales farmacéuticamente aceptables, complejos, hidratos, solvatos, tautómeros, polimorfos, mezclas racémicas y formas ópticamente activas del mismo junto con un vehículo farmacéuticamente aceptable, y en combinación con al menos un antibiótico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201741042876 | 2017-11-29 | ||
| PCT/IN2018/050799 WO2019106693A1 (en) | 2017-11-29 | 2018-11-29 | Anti-bacterial heterocyclic compounds and their synthesis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2952989T3 true ES2952989T3 (es) | 2023-11-07 |
Family
ID=64665590
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18819475T Active ES2952989T3 (es) | 2017-11-29 | 2018-11-29 | Compuestos heterocíclicos antibacterianos y su síntesis |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11401277B2 (es) |
| EP (1) | EP3717490B1 (es) |
| JP (1) | JP7390728B2 (es) |
| CN (1) | CN111527093B (es) |
| ES (1) | ES2952989T3 (es) |
| WO (1) | WO2019106693A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4558125A1 (en) * | 2022-07-19 | 2025-05-28 | BUGWORKS Research India Pvt Ltd | An antibacterial parenteral formulation and methods thereof |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2186550B2 (es) * | 2001-06-27 | 2003-11-16 | Vita Lab | Nuevos derivados de oxazolidinonas como antibacterianos. |
| CA2515269A1 (en) * | 2003-02-07 | 2004-08-19 | Warner-Lambert Company Llc | Oxazolidinone derivatives n-substituted by a bicyclic ring, for use as antibacterial agents |
| JP2009539807A (ja) * | 2006-06-09 | 2009-11-19 | グラクソ グループ リミテッド | 抗菌薬としての置換1−メチル−1h−キノリン−2−オンおよび1−メチル−1h−1,5−ナフチリジン−2−オン |
| CL2008001002A1 (es) * | 2007-04-11 | 2008-10-17 | Actelion Pharmaceuticals Ltd | Compuestos derivados de oxazolidinona; composicion farmaceutica que comprende a dichos compuestos; y su uso para preparar un medicamento para tratar una infeccion bacteriana. |
| CL2008001003A1 (es) * | 2007-04-11 | 2008-10-17 | Actelion Pharmaceuticals Ltd | Compuestos derivados de oxazolidinona; composicion farmaceutica que comprende a dichos compuestos; y su uso para preparar un medicamento para tratar una infeccion bacteriana. |
| ES2411930T3 (es) * | 2008-02-20 | 2013-07-09 | Actelion Pharmaceuticals Ltd. | Compuestos antibióticos azatricíclicos |
| KR101592046B1 (ko) * | 2008-02-22 | 2016-02-05 | 액테리온 파마슈티칼 리미티드 | 옥사졸리디논 유도체 |
| DK2344495T3 (en) * | 2008-10-07 | 2015-02-02 | Actelion Pharmaceuticals Ltd | TRICYCLIC OXAZOLIDINON ANTIBIOTIC COMPOUNDS |
| RU2014108919A (ru) * | 2011-08-11 | 2015-09-20 | Актелион Фармасьютиклз Лтд | Хиназолин-2,4-дионовые производные |
| SG11201401710PA (en) * | 2011-11-08 | 2014-09-26 | Actelion Pharmaceuticals Ltd | 2-oxo-oxazolidin-3,5-diyl antibiotic derivatives |
| KR20150143720A (ko) * | 2013-04-16 | 2015-12-23 | 액테리온 파마슈티칼 리미티드 | 항균성 이방향족 유도체 |
| PT3634968T (pt) * | 2017-06-08 | 2023-04-14 | Bugworks Res Inc | Compostos heterocíclicos úteis como agentes anti-bacterianos e método para a sua produção |
| RS64146B1 (sr) * | 2018-03-28 | 2023-05-31 | Bugworks Research Inc | Oksazolidinonska antibiotska jedinjenja i postupak dobijanja |
-
2018
- 2018-11-29 CN CN201880076740.XA patent/CN111527093B/zh active Active
- 2018-11-29 ES ES18819475T patent/ES2952989T3/es active Active
- 2018-11-29 EP EP18819475.7A patent/EP3717490B1/en active Active
- 2018-11-29 WO PCT/IN2018/050799 patent/WO2019106693A1/en not_active Ceased
- 2018-11-29 US US16/763,222 patent/US11401277B2/en active Active
- 2018-11-29 JP JP2020547482A patent/JP7390728B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US11401277B2 (en) | 2022-08-02 |
| CN111527093B (zh) | 2023-06-09 |
| JP7390728B2 (ja) | 2023-12-04 |
| JP2021504489A (ja) | 2021-02-15 |
| EP3717490A1 (en) | 2020-10-07 |
| EP3717490B1 (en) | 2023-05-24 |
| WO2019106693A1 (en) | 2019-06-06 |
| US20210070772A1 (en) | 2021-03-11 |
| CN111527093A (zh) | 2020-08-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA3203205A1 (en) | Pharmaceutical combinations of sos1 inhibitors for treating and/or preventing cancer | |
| ES2942761T3 (es) | Compuestos heterocíclicos útiles como agentes antibacterianos y método para la producción de los mismos | |
| US10711011B2 (en) | Substituted oxazolidines as anti-bacterial agents | |
| EP1984369A2 (en) | Nitroimidazole compounds | |
| ES2952989T3 (es) | Compuestos heterocíclicos antibacterianos y su síntesis | |
| ES2942552T3 (es) | Compuestos antibióticos de oxazolidinona y procedimiento de preparación | |
| CN116419753B (zh) | 作为bcl-2抑制剂的杂环化合物 | |
| JP2024529132A (ja) | 抗生物質ピラジノチアジン誘導体及びその調製方法 | |
| WO2002102792A1 (en) | Novel heterocyclic antibacterial compounds | |
| TW202345792A (zh) | 苯并噻吩化合物 | |
| HK40013941A (en) | 2(1h)-quinolinone derivative |