ES2953190T3 - Proteínas de unión a BCMA (CD269/TNFRSF17) - Google Patents
Proteínas de unión a BCMA (CD269/TNFRSF17) Download PDFInfo
- Publication number
- ES2953190T3 ES2953190T3 ES18163942T ES18163942T ES2953190T3 ES 2953190 T3 ES2953190 T3 ES 2953190T3 ES 18163942 T ES18163942 T ES 18163942T ES 18163942 T ES18163942 T ES 18163942T ES 2953190 T3 ES2953190 T3 ES 2953190T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- bcma
- cells
- cell
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2878—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the NGF-receptor/TNF-receptor superfamily, e.g. CD27, CD30, CD40, CD95
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
- A61K47/6817—Toxins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/567—Framework region [FR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/72—Increased effector function due to an Fc-modification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Toxicology (AREA)
- Cell Biology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
La presente invención se refiere a proteínas de unión a antígenos y fragmentos de las mismas que se unen específicamente al antígeno de maduración de células B (BCMA), particularmente al BCMA humano (hBCMA) y que inhiben la unión de BAFF y APRIL al receptor de BCMA. Además se describen composiciones farmacéuticas, métodos de detección y tratamiento médico. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Proteínas de unión a BCMA (CD269/TNFRSF17)
Campo de la invención
La presente invención se refiere a proteínas de unión a antígeno y fragmentos de las mismas que se unen específicamente a antígeno de maduración de linfocitos B (BCMA) y en particular BCMA humano (hBCMA).
La presente invención también se refiere a procedimientos para tratar enfermedades o trastornos con dichos fragmentos de unión a antígeno, composiciones farmacéuticas que comprenden dichos fragmentos de unión a antígenos y procedimientos de preparación. Otras realizaciones de la presente invención resultarán evidentes a partir de la descripción posterior.
Antecedentes de la invención
BCMA (CD269 o TNFRSF17) es un miembro de la superfamilia de receptores de TNF. Es un receptor de membrana integral no glucosilado para los ligandos BAFF y APRIL. Los ligandos de BCMA también puede unirse a receptores adicionales: TACI (activador transmembrana y modulador de calcio y agente de interacción con el ligando de ciclofilina), que se une a APRIL y BAFF; así como BAFF-R (receptor BAFF o BR3), que muestra afinidad restringida pero alta por BAFF. Juntos, estos receptores y sus ligandos correspondientes regulan diferentes aspectos de la inmunidad humoral, desarrollo de linfocitos B y homeostasis.
La expresión de BCMA está típicamente restringida al linaje de linfocitos B y se ha indicado que aumenta en diferenciación de linfocitos B terminales. BCMA se expresa por blastos de plasma humano, células plasmáticas de amígdalas, bazo y médula ósea, pero también por linfocitos B de memoria de las amígdalas y por linfocitos B de centro germinal, que tienen un fenotipo bajo de TACI-BAFFR (Darce y col, 2007). BCMA está prácticamente ausente en linfocitos B de memoria y vírgenes (Novak y col, 2004a y b). El antígeno BCMA se expresa en la superficie celular de modo que es accesible para el anticuerpo, pero también se expresa en el Golgi. Como se sugiere por su perfil de expresión, la señalización de BCMA, típicamente ligada con supervivencia y proliferación de linfocitos B, es importante en las etapas tardías de diferenciación de linfocitos B, así como la supervivencia de células plasmáticas de médula ósea de larga vida (O'Connor y col, 2004) y plasmoblastos (Avery y col, 2003). Además, como BCMA se une a APRIL con alta afinidad, se sugiere que el eje de señalización BCMA-APRIL predomina en las etapas tardías de diferenciación de linfocitos B, siendo quizás la interacción fisiológicamente más relevante.
El mieloma múltiple (MM) es un tumor maligno de linfocitos B clonal que se produce en múltiples sitios dentro de la médula ósea antes de propagarse a la circulación, de novo, o como una progresión a partir de gammapatía monoclonal de importancia indeterminada (MGUS). Se caracteriza habitualmente por aumentos de actividad de osteoclastos y paraproteínas, así como hipercalcemia, citopenia, disfunción renal, hiperviscosidad y neuropatía periférica. También son habituales reducciones tanto en los niveles de anticuerpo normal como en los números de neutrófilos, lo que conduce a una susceptibilidad a infección con riesgo para la vida. Se ha implicado a BCMA en el crecimiento y supervivencia de líneas celulares de mieloma in vitro (Novak y col, 2004a y b; Moreaux y col, 2004).
Se ha indicado que la expresión de BCMA (tanto transcrito como proteína) se correlaciona con progresión de enfermedad en MM. Usando micromatrices Affymetrix, se demostró que los genes TACI y BCMA se sobreexpresaron en células de mieloma múltiple (MMC) en comparación con sus homólogos normales (Moreaux y col, 2004). Se ha usado análisis de expresión génica para comparar células de mieloma humanas con células plasmáticas purificadas de pacientes con MGUS y de médula ósea normal así como con células de tumor primario de leucemias de linaje de linfocitos B (Bellucci y col, 2005). El gen de BCMA se expresó en gran medida en todas las muestras de mieloma. Aunque las células plasmáticas purificadas de pacientes con MGUS tuvieron expresión más baja de BCMA, no hubo diferencia significativa en comparación con la expresión hallada en células plasmáticas normales o células de mieloma. Por el contrario, la expresión de BCMA fue significativamente más baja en leucemia linfocítica crónica de linfocitos B (CLL), leucemia linfocítica aguda pre-B (ALL) y ALL de linfocitos T (T-ALL).
Los modelos de ratón que sobre-expresan de forma transgénica BAFF o APRIL tienen un aumento significativo en linfomas de linfocitos B (Batten y col, 2004 - BAFF; Planelles y col, 2004 - APRIL). En los seres humanos, se ha detectado exceso de BAFF y APRIL en los sueros y micro-ambientes de pacientes con varios tumores malignos de linfocitos B, así como otros trastornos de linfocitos B.
Se ha mostrado que un anticuerpo SG1 anti-BCMA IgG1 bloquea la activación de NFkB dependiente de APRIL. Este anticuerpo aparentemente demostró citotoxicidad contra células de mieloma múltiple tanto como anticuerpo desnudo como conjugados anticuerpo-fármaco con mono metil auristatina F (MMFA). Se logró ADCC mejorada (100 veces) mediante quimerización con una región Fc humana que comprende mutaciones que mejoran la unión de FcyRIIIA (Ryan Maureen et al: "Antibody targeting of B-cell maturation antigen on malignant plasma cell", Modecular Cancer
Therapeutics, American Association of Cáncer Research, US vol. 6, n.° 11, 1° de noviembre de 2001 (01-11-2001), páginas 3009-3018, ISSN: 1535-7163) También se han dado a conocer previamente IgG1 anti-BMCA quimérica (humanizada), incluyendo sus versiones afucosiladas, y se supone que tienen actividad ADCC (documento WO 2010/104949 A2).
Breve Descripción de las Figuras
Figura 1: Ensayo de unión de FMAT - Figura que muestra los resultados del ensayo de FMAT con respecto a unión del anticuerpo CA8 con células HEK293 que expresan BCMA humano y de cynomolgus. El CA8 quimérico humano se une bien a células que expresan BCMA humano y de cynomolgus.
Figura 2: Ensayo de unión de ELISA - Figura que muestra los resultados de ELISA con respecto a anticuerpos CA8 que se unen a proteínas recombinantes de BCMA humanas y de cynomolgus. Esta muestra claramente que los anticuerpos CA8 quiméricos humanos se unen igualmente a proteínas BCMA humanas y de cynomolgus.
Figura 3: Ensayo de unión BiaCore - Figura que muestra la unión de CA8 con proteínas BCMA-Fc, TACI-Fc y BAFF-R-Fc en el experimento de Biacore. El anticuerpo quimérico CA8 no se une a proteínas TACI o BAFF-R.
Figura 4: Ensayo de unión a células - Figura que muestra unión de S307118G03, S3222110D07, S332121F02 y S332126E04 murinos con células de mieloma múltiple H929 y S3322110D07, S332121F02 y S332126E04 con las células ARH77 transfectadas con BCMA como se determinó por FACS.
La línea celular de mieloma múltiple H929 o células transfectantes que expresaban BCMA ARH77-hBCMA 10B5 se tiñeron con anticuerpos murinos anti-BCMA (histograma sólido) o control de isotipo de IgG2a murino (histogramas abiertos). Las células se analizaron por FACS para detectar anticuerpo unido a las células.
Figura 5: Ensayo de unión a células - Figura que muestra unión de CA8 quimérico con un panel de líneas celulares de mieloma múltiple como se determinó por FACS. La unión con H929, OPM-2, JJN-3 y U266 se ensayó por citometría de flujo y los valores de intensidad de fluorescencia media (MFI) se midieron para determinar la unión. Se usó Synagis como un control de isotipo irrelevante.
Figura 6: Ensayo de unión a células - Figura que muestra curvas de unión de variantes de CA8 humanizadas con células ARH77 transfectadas con BCMA (A) y células de mieloma múltiple H929 (B) como se determinó por FACS. Se ensayaron las variantes humanizadas, J6M0, J6M1, J6M2, J9M0, J9M1 y J9M2 por citometría de flujo y los valores de intensidad de fluorescencia media (IFM) se midieron para determinar la unión en comparación con la quimera de CA8.
Figura 7: Ensayos de neutralización de ligando -
(A y B) Figura que muestra la capacidad de CA8 y J6M0 para neutralizar la unión de BAFF o APRIL recombinante con BCMA recombinante con el que se ha revestido una placa de ELISA. Los valores de DO se usaron para calcular la inhibición mediada por anticuerpo de la señal máxima conseguida por el ligando relevante solo que se une a BCMA recombinante. Se presentan datos como porcentaje de inhibición de la señal máxima. Los anticuerpos ensayados fueron CA8 quimérico y CA8 humanizado versión J6M0 en forma tanto de tipo silvestre como afucosilado (Potelligent).
(A) Neutralización de unión del ligando BAFF, (B) Neutralización de unión de ligando APRIL.
(C) Figura que muestra la capacidad del anticuerpo de BCMA J6M0 en la inhibición de fosforilación inducida por APRIL o BAFF de NFkappaB en células H929. Las células H-929 se lavaron 3 veces para retirar cualquier sBCMA y se resuspendieron en medio sin suero. Se añadió anticuerpo J6M0 potelligent a una placa de 96 pocillos para proporcionar una concentración de pocillo final hasta 100 μg/ml junto con ligando BAFF o APRIL para proporcionar una concentración de pocillo final de 0,6 o 0,2 μg/ml respectivamente. Las células H-929 se sembraron después a 7,5 x 104 células/pocillo en medio sin suero. 30 minutos después, las células se lisaron y se midieron los niveles de NFkappaB fosforilado usando un ensayo MSD de pNFkappaB. Lector de MSD 502819. Estos son datos de un experimento independiente. Cada punto de datos es la media/DT de dos repeticiones.
Figura 8: Ensayo de ADCC - Figura que muestra la actividad de ADCC de CA8 quimérico y CA8 desfucosilado (potenciado con Fc) con células diana que expresan BCMA.
Se incubaron células NK humanas con células diana transfectadas con BCMA ARH7710b5 marcadas con europio en presencia de diversas concentraciones de anticuerpo. Se midió la liberación de europio de las células diana y se calculó la lisis específica. (A) Curvas de respuesta a dosis de ADCC de CA8 quimérico en comparación con control de isotipo. (B) Curvas de respuesta a dosis de ADCC para CA8 quimérico y CA8 quimérico desfucosilado (potenciado con Fc), frente a la línea celular que expresa BCMA ARH7710b5.
Figura 9: Ensayo de ADCC - Figura que muestra el ensayo de ADCC en anticuerpos humanizados CA8 usando células diana que expresan ARH77 BCMa .
Se incubaron PBMC humanas con células diana transfectadas con BCMA ARH77 marcado con europio en presencia de una serie de concentraciones de las series J5, J6, J7, J8 o J9 de anticuerpos CA8 humanizados. Se midió la liberación de europio de las células diana y se calculó la lisis específica. Se muestran los valores de CE50 en gg/ml.
Figura 10: Ensayo de ADCC - Figura que muestra actividad ADCC de H3L0 quimérico, S332121F02 (A), S3322110D07 (B) S307118G03 (C) y S307118G03 humanizado (D) frente a células diana ARH7710B5 con células NK purificadas como células efectoras. Las células diana NK humanas se incubaron con células diana transfectadas con BCMA ARH77 10b5 marcadas con europio en presencia de diversas concentraciones de anticuerpo. Se midió la liberación de europio de las células diana y se calculó la lisis específica.
Figura 11: Curvas de respuesta a dosis de ensayo de viabilidad - Figura que muestra las curvas de respuesta a dosis en un ensayo de viabilidad celular con respecto al anticuerpo CA8 quimérico, conjugados de anticuerpo-fármaco CA8 quimérico-vcMMAE y CA8 quimérico-mcMMAF en líneas celulares de mieloma múltiple humanas (A) NCI-H929 (B) U266-B1 (C) JJN3 y (D) OPM2. Se añadió anticuerpo a las células y se midió el número de células viables después de 96 horas usando CelltiterGlo. Los puntos de datos representan la media de mediciones de CellTiterGlo por triplicado. Las barras de error representan el error típico.
Figura 12: Impacto de anticuerpo quimérico CA8 en el ciclo celular.
(A) Histogramas del ciclo celular de células NCI-H929 tratadas con CA8 quimérico no conjugado, ADC de CA8 quimérico-vcMMAE o ADC de CA8 quimérico- mcMMAF a 50 ng/ml para los puntos temporales indicados. Se usó Paclitaxel (100 nM) como un control positivo para detención del ciclo celular en G2/M y muerte celular. Se usó IgG1 humana de control como un control negativo. Se llevó a cabo análisis del ciclo celular en los tiempos mostrados en los gráficos. (B) Cuantificación de la población de células con ADN 4N indicativa de detención en G2/M y (C) población de células con ADN sub-2N indicativo de muerte celular para cada uno de los tratamientos indicados. Las células se sembraron en placas de 12 pocillos (2 x 105 células por pocillo en 1 ml de RPMI FBS 10 %). Se añadió anticuerpo o ADC 6 horas después de la siembra de células.
Figura 13: Impacto de CA8 quimérico en fosfo-histona H3.
El tratamiento con CA8 ADC quimérico da como resultado aumento de la tinción de fosfo-histona H3 de células NCI-H929.
(A, B) representaciones de puntos de células teñidas con yoduro de propidio para medir el contenido de ADN (FL3-H), eje x, y anticuerpo anti-fosfo-histona H3 (Thr11) (FL1-H), eje y, después del tratamiento con IgG de control (A) o CA8 quimérico-mcMMAF (B). (C) Cuantificación de células NCI-H929 positivas para fosfo histona H3 después de un tratamiento de 48 horas con las concentraciones indicadas de ADC de CA8 quimérico. Se usó Paclitaxel (100 nM) como un control positivo para la detención mitótica y se usó IgG 1 quimérica de control como un control negativo. Las células se sembraron en placas de 12 pocillos (2 x 105 células por pocillo en 1 ml de RPMI FBS 10 %). Se añadió anticuerpo o ADC 6 horas después de la siembra de células.
Figura 14: Impacto de CA8 quimérico en Anexina-V.
El tratamiento con ADC CA8 quimérico da como resultado aumento de la tinción de Anexina-V de células NCI-H929.
(A) Histogramas de Anexina-V-FITC (FL1-H; paneles superiores) y tinción con yoduro de propidio de células vivas (FL3-H; paneles inferiores) después del tratamiento con concentraciones crecientes de ADC de CA8 quiméricos. (B) Cuantificación de células NCI-H929 positivas para Anexina-V después de un tratamiento de 96 horas con las concentraciones indicadas de ADC de CA8 quimérico. Se usó Paclitaxel (100 nM) como un control positivo para apoptosis y se usó IgG 1 quimérica de control como un control negativo. Las células se sembraron en placas de 12 pocillos (2 x 105 células por pocillo en 1 ml de RPMI FBS 10 %). Se añadió anticuerpo o ADC 6 horas después de la siembra de células.
Figura 15: Curvas de respuesta a dosis de ensayo de viabilidad - Figura que muestra curvas de respuesta a dosis para el no conjugado (desnudo) y conjugados de anticuerpo-fármaco de vcMMAE y mcMMAF de anticuerpos CA8 quimérico o J6M0 humanizado. Se ensayaron conjugados de fármacos con anticuerpos frente a las líneas celulares de mieloma múltiple humano NCI-H929 y OPM2.
Figura 16: Curvas de respuesta a dosis de ensayo de viabilidad - Figura que muestra las curvas de respuesta a dosis para los anticuerpos no conjugados, conjugados de anticuerpo-fármaco con vcMMAE y mcMMAF de anticuerpos murinos anti-BCMA S332121F02, S322110D07, S332126E04 y S307118G03 en líneas celulares de mieloma múltiple
humano NCI-H929 y U266 -B1.
Figura 17: Actividad ADCC de moléculas ADC J6M0 - Figura que muestra ensayo de ADCC en anticuerpos J6M0 usando células diana que expresan BCMA ARH77. Se incubaron PBMC humanas con células diana transfectadas con BCMA ARH77 marcadas con europio en presencia de una serie de concentraciones de anticuerpos de BCMA J6M0 WT y Potelligent conjugados con Mm AE, MMAF o se controló la liberación de europio no conjugado en el lector de multimarcador Victor 21420.
Figura 18 Curvas de respuesta a dosis de ADCC de CA8 J6M0 Potelligent frente a un panel de 5 líneas de mieloma múltiple - Se incubaron PBMC humanas con células diana de mieloma múltiple en presencia de diversas concentraciones de anticuerpo CA8 J6M0 Potelligent a una relación E:T de 50:1 durante 18 horas. El porcentaje de células diana que permanecen en la mezcla de efector más diana se midió después por FACS usando un anticuerpo anti-CD138 marcado con fluorescencia para detectar las células diana y se calculó el porcentaje de citotoxicidad. A) Curvas de respuesta a dosis ejemplares para CA8 J6M0 Potelligent frente a las cinco líneas celulares de mieloma múltiple ensayadas. Cada punto de datos es de un valor de ensayo sencillo.
Figura 19 Efecto de aumento de dosis de J6M0 y J6M0 conjugado con fármaco en el crecimiento y establecimiento de células NCI-H929 en ratones CB.17 SCID. Volúmenes tumorales calculados de tumores de NCI-H929 en ratones CB17 SCID después de dosificación intraperitoneal dos veces por semana de 50 ó 100 |ug de J6M0 anti-BCMA o control de isotipo de IgG1 no conjugado o conjugado con MMAE o MMAF durante 2 semanas. Los puntos de datos representan volumen tumoral medio de n = 5 por grupo.
Figura 20 - Determinación de niveles de BCMA soluble en suero de voluntarios sanos y pacientes con mieloma.
Se recogieron muestras de suero de pacientes con MM. Las muestras fueron de una diversidad de etapas (enfermedad progresiva, remisión, recaída, nuevo diagnóstico y otros). Las muestras mostradas en la figura son las de suero diluido 1/500 antes del ensayo.
Se usó un kit de ELISA de tipo sándwich de BCMA/TNFRSF17 humano de R & D Systems que mide los niveles de BCMA humano soluble para detectar BCMA siguiendo el protocolo convencional proporcionado con el kit.
Sumario de la Invención
La invención se define por las reivindicaciones adjuntas.
Cualquier referencia en la descripción al procedimiento de tratamiento se refiere a los compuestos, composiciones farmacéuticas y medicamentos de la presente invención para su uso en un procedimiento para el tratamiento del cuerpo humano (o animal) mediante terapia (o para diagnóstico).
Descripción Detallada de la Invención
La presente invención proporciona un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) que comprende una región variable de cadena pesada de SEC ID N°. 23 y una región variable de cadena ligera de SEC ID N°. 31. Se proporciona además un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) que comprende una cadena pesada de SEC ID N°. 55 y una cadena ligera de SEC ID N°. 63. En un aspecto, el anticuerpo anti-antígeno de maduración de linfocitos B (CD269) de la invención tiene unión potenciada a Fcy RIIIA o tiene función efectora mediada por Fcy RIIIA potenciada, por ejemplo en un aspecto de la invención tal como se proporciona en el presente documento el anticuerpo está desfucosilado.
Se dan a conocer en el presente documento proteínas de unión a antígeno que se unen a dianas unidas a membrana y siendo la proteína de unión a antígeno capaz de internalización. En una realización adicional se proporciona un inmunoconjugado que comprende la proteína de unión a antígeno de la presente invención y un agente citotóxico. En una realización adicional la proteína de unión a antígeno tiene función efectora ADCC, por ejemplo la proteína de unión a antígeno tiene función efectora ADCC potenciada.
En una realización tal se proporcionan proteínas de unión a antígeno o fragmentos de las mismas que se unen específicamente a BCMA, por ejemplo que se unen específicamente a BCMA humano (hBCMA) y que inhiben la unión de BAFF y/o APRIL con el receptor de Bc MA.
En una realización adicional las proteínas de unión a antígeno o fragmentos se unen específicamente a BCMA e inhibe la unión de BAFF y/o APRIL con BCMA teniendo las proteínas de unión a antígeno o fragmentos de las mismas la capacidad para unirse a Fcy RIIIA y mediar en funciones efectoras mediadas por FcgRIIIA, o tener función efectora mediada por Fcy RIIIA potenciada. En una realización de la invención como se proporciona en el presente documento las proteínas de unión a antígeno son capaces de internalización.
En un aspecto de la invención se proporciona una proteína de unión a antígeno de acuerdo con la invención como se describe en el presente documento que se une a BCMA no unido a membrana, por ejemplo a BCMA del suero.
En un aspecto de la invención, la proteína de unión a antígeno tiene función efectora potenciada. En otro aspecto, la proteína de unión a antígeno está conjugada con un agente citotóxico. Aún en una realización adicional, la proteína de unión a antígeno tiene tanto función efectora potenciada como está conjugada con un agente citotóxico.
Las proteínas de unión a antígeno de la presente invención pueden comprender regiones variables de cadena pesada y regiones variables de cadena ligera de la invención que pueden tomar el formato de la estructura de un anticuerpo natural o fragmento funcional o equivalente del mismo. Una proteína de unión a antígeno de la invención puede comprender por lo tanto las regiones VH de la invención con el formato de un anticuerpo de longitud completa, un fragmento (Fab’)2, un fragmento Fab o equivalente de los mismos (tales como scFv, bi- tri- o tetracuerpos, Tandab, etc.), cuando se emparejan con una cadena ligera apropiada. El anticuerpo puede ser un IgG1, IgG2, IgG3, o IgG4; o IgM; IgA, IgE o IgD o una variante modificada de los mismos. El dominio constante de la cadena pesada de anticuerpo puede seleccionarse en consecuencia. El dominio constante de cadena ligera puede ser un dominio constante kappa o lambda. Además, la proteína de unión a antígeno puede comprender modificaciones de todas las clases, por ejemplo dímeros de IgG, mutantes de Fc que ya no se unen a receptores de Fc o median en la unión de C1q. La proteína de unión a antígeno también puede ser un anticuerpo quimérico del tipo descrito en el documento WO86/01533 que comprende una reacción de unión a antígeno y una región no de inmunoglobulina.
La región constante se selecciona de acuerdo con cualquier funcionalidad requerida, por ejemplo una IgG1 puede demostrar capacidad lítica a través de unión a complemento y/o mediará en ADCC (citotoxicidad celular dependiente de anticuerpo).
En un aspecto la proteína de unión a antígeno es un anticuerpo o fragmento de unión a antígeno del mismo que comprende una o más CDR de acuerdo con la invención descrita en el presente documento, o uno o ambos de los dominios variables de cadena pesada o ligera de acuerdo con la invención descrita en el presente documento. En una realización la proteína de unión a antígeno se une a BCMA de primate. En una realización tal la proteína de unión a antígeno se une adicionalmente a BCMA de primate no humano, por ejemplo BCMA de macaco cynomolgus.
En otro aspecto, la proteína de unión a antígeno se selecciona del grupo que consiste en un dAb, Fab, Fab’, F(ab’)2, Fv, diacuerpo, triacuerpo, tetracuerpo, minianticuerpo y un minicuerpo.
En un aspecto de la presente invención la proteína de unión a antígeno es un anticuerpo humanizado o quimérico, en un aspecto adicional el anticuerpo es humanizado.
En un aspecto el anticuerpo es un anticuerpo monoclonal.
En otro aspecto la proteína de unión a antígeno se une a BCMA humano con alta afinidad, por ejemplo cuando se mide por Biacore la proteína de unión a antígeno se une a BCMA humano con una afinidad de 20 nM o menos, una afinidad de 15 nM o menos, una afinidad de 5 nM o menos, una afinidad de 1.000 μM o menos, una afinidad de 500 pM o menos, una afinidad de 400 μM o menos o 300 μM o menos o por ejemplo aproximadamente 120 pM. En una realización adicional la proteína de unión a antígeno se une a BCMA humano cuando se mide por Biacore de entre aproximadamente 100 pM y aproximadamente 500 pM, entre aproximadamente 100 pM y aproximadamente 400 pM o entre aproximadamente 100 pM y aproximadamente 300 pM. En una realización de la presente invención la proteína de unión a antígeno se une a BCMA con una afinidad de menos de 150 pM.
En una realización tal, esto se mide por Biacore, por ejemplo como se expone en el Ejemplo 4.
En otro aspecto la proteína de unión a antígeno se une a BCMA humano y neutraliza la unión de los ligandos BAFF y/o APRIL con el receptor de BCMA en un ensayo de neutralización celular en el que la proteína de unión a antígeno tiene una CI50 de entre aproximadamente 1 nM y aproximadamente 500 nM, entre aproximadamente 1 nM y aproximadamente 100 nM, entre aproximadamente 1 nM y aproximadamente 50 nM, entre aproximadamente 1 nM y aproximadamente 25 nM o entre aproximadamente 5 nM y aproximadamente 15 nM. En una realización adicional de la presente invención la proteína de unión a antígeno se une a BCMA y neutraliza BCMA en un ensayo de neutralización celular en el que la proteína de unión a antígeno tiene una CI5 de aproximadamente 10 nM.
En una realización tal, esto se mide por un ensayo de neutralización celular, por ejemplo como se expone en el Ejemplo 4.6.
Las proteínas de unión a antígeno, por ejemplo anticuerpos de la presente invención pueden producirse por transfección de una célula huésped con un vector de expresión que comprende la secuencia codificante para la proteína de unión a antígeno de la invención. Un vector de expresión o plásmido recombinante se produce colocando
estas secuencias codificantes para la proteína de unión a antígeno en asociación operativa con secuencias de control reguladoras convencionales capaces de controlar la replicación y expresión en, y/o secreción de, una célula huésped. Las secuencias reguladoras incluyen secuencias promotoras, por ejemplo, promotor de CMV y secuencias señal que pueden derivar de otros anticuerpos conocidos. De forma similar, puede producirse un segundo vector de expresión que tenga una secuencia de ADN que codifique una cadena pesada o ligera de proteína de unión a antígeno complementaria. En ciertas realizaciones este segundo vector de expresión es idéntico al primero excepto en lo que se refiere a las secuencias codificantes y marcadores seleccionables, para asegurar tanto como sea posible que cada cadena polipeptídica se exprese de forma funcional. Como alternativa, las secuencias codificantes de cadena pesada y ligera para la proteína de un antígeno pueden residir en un vector sencillo.
Una célula huésped seleccionada se co-transfecta por técnicas convencionales tanto con el primer como con el segundo vector (o simplemente se transfectan por un vector sencillo) para crear la célula huésped transfectada de la invención que comprende las cadenas ligeras y pesadas recombinantes o sintéticas. La célula transfectada se cultiva después por técnicas convencionales para producir la proteína de unión a antígeno modificada por ingeniería genética de la invención. La proteína de unión a antígeno que incluye la asociación de la cadena ligera y/o cadena pesada recombinante se explora a partir de cultivo por ensayo apropiado, tal como ELISA o RIA. Pueden emplearse técnicas convencionales similares para construir otras proteínas de unión a antígeno.
Pueden seleccionarse vectores adecuados para las etapas de clonación y subclonación empleadas en los procedimientos y construcción de las composiciones de la presente invención por un experto en la materia. Por ejemplo, puede usarse la serie de pUC convencional de vectores de clonación. Un vector, pUC19 está disponible en el mercado de proveedores, tales como Amersham (Buckinghamshire, Reino Unido) o Pharmacia (Uppsala, Suecia). Adicionalmente, cualquier vector que sea capaz de replicar fácilmente, tenga una abundancia de sitios de clonación y genes seleccionables (por ejemplo, resistencia a antibióticos) y se manipule fácilmente puede usarse para clonación. Por lo tanto, la selección del vector de clonación no es un factor limitante en la presente invención.
Los vectores de expresión también pueden caracterizarse por genes adecuados para amplificar la expresión de las secuencias de ADN heterólogas, por ejemplo, el gen de dihidrofolato reductasa de mamífero (DHFR). Otras secuencias de vector incluyen una secuencia señal de poli A, tal como de hormona de crecimiento bovina (BGH) y la secuencia promotora de betaglobina (betaglopro). Los receptores de expresión útiles en el presente documento pueden sintetizarse por técnicas bien conocidas por los expertos en la materia.
Los componentes de tales vectores, por ejemplo, replicones, genes de selección, potenciadores, promotores, secuencias señal y similares, pueden obtenerse de fuentes comerciales o naturales o sintetizarse por procedimientos conocidos para su uso en la dirección de la expresión y/o secreción del producto del ADN recombinante en un huésped seleccionado. Otros vectores de expresión apropiados de los que se conocen numerosos tipos en la técnica para expresión de mamíferos, bacteriana, de insectos, de levadura y fúngica también pueden seleccionarse para este fin.
La presente invención también abarca una línea celular transfectada con un plásmido recombinante que contiene las secuencias codificantes de las proteínas de unión a antígeno de la presente invención. Las células huésped útiles para la clonación y otras manipulaciones de estos vectores de clonación también son convencionales. Sin embargo, pueden usarse células de diversas cepas de E. coli para la replicación de los receptores de clonación y otras etapas en la construcción de proteínas de unión a antígeno de la presente invención.
Las células huésped o líneas celulares adecuadas para la expresión de las proteínas de unión a antígeno de la invención incluyen células de mamífero tales como NS0, Sp2/0, CHO (por ejemplo DG44), COS, HEK, un fibroblasto (por ejemplo 3T3) y células de mieloma, por ejemplo puede expresarse en una célula de mieloma o CHO. Pueden usarse células humanas, permitiendo de este modo que la molécula se modifique con patrones de glucosilación humanos. Como alternativa, pueden emplearse otras líneas celulares eucariotas. Se conocen en la técnica la selección de células huésped de mamífero adecuadas y procedimientos para transformación, cultivo, amplificación, exploración y producción de productos y purificación. Véase, por ejemplo, Sambrook y col., mencionado anteriormente.
Las células bacterianas puede demostrar ser útiles como células huésped adecuadas para la expresión de los Fab recombinantes u otras realizaciones de la presente invención (véase, por ejemplo, Plückthun, A., Immunol. Rev., 130: 151-188 (1992)). Sin embargo, debido a la tendencia de las proteínas expresadas en células bacterianas a estar en una forma desplegada o plegada de forma inapropiada o en una forma no glucosilada, cualquier Fab recombinante producido en una célula bacteriana tendría que explorarse con respecto a retención de capacidad de unión a antígeno. Si la molécula expresada por la célula bacteriana se produjo en una forma plegada de forma apropiada, esa célula bacteriana sería un huésped deseable, o en realizaciones alternativas la molécula puede expresarse en el huésped bacteriano y después volver a plegarse posteriormente. Por ejemplo, diversas cepas de E. coli usadas para expresión se conocen bien como células huésped en el campo de la biotecnología. También pueden emplearse diversas cepas de B. subtilis, Streptomyces, otros bacilos y similares en este procedimiento.
Cuando se desee, también están disponibles cepas de células de levadura conocidas por los expertos en la materia
como células huésped, así como células de insecto, por ejemplo Drosophila y lepidópteros, y sistemas de expresión virales. Véase, por ejemplo Miller y col., Genetic Engineering, 8: 277-298, Plenum Press (1986) y referencias citadas en la misma.
Los procedimientos generales por los que los vectores pueden construirse, los procedimientos de transfección requeridos para producir las células huésped de la invención y los procedimientos de cultivo necesarios para producir la proteína de unión a antígeno de la invención de dicha célula huésped pueden ser todos técnicas convencionales. Típicamente, el procedimiento de cultivo de la presente invención es un procedimiento de cultivo sin suero, habitualmente cultivando células sin suero en suspensión. De forma similar, una vez producidas, las proteínas de unión a antígeno de la invención pueden purificarse de los contenidos de cultivo celular de acuerdo con procedimientos convencionales de la técnica, incluyendo precipitación con peróxido de amonio, columnas de afinidad, cromatografía en columna, electroforesis en gel y similares. Tales técnicas están dentro de la experiencia de la técnica y no limitan la presente invención. Por ejemplo, se describen preparaciones de anticuerpos alterados en los documentos WO 99/58679 y WO 96/16990.
Otro procedimiento más de expresión de las proteínas de unión a antígeno puede utilizar expresión en un animal transgénico, tal como se describe en la Patente de Estados Unidos N° 4.873.316. Esto se refiere a un sistema de expresión que usa el promotor de caseína de los animales que cuando se incorpora de forma transgénica en un mamífero permite a la hembra producir la proteína recombinante deseada en su leche.
En una realización adicional de la invención se proporciona un procedimiento para producir un anticuerpo de la invención comprendiendo dicho procedimiento la etapa de cultivar una célula huésped transformada o transfectada con un vector que codifica la cadena ligera y/o pesada del anticuerpo de la invención y recuperar el anticuerpo producido de este modo.
De acuerdo con la presente invención se proporciona un procedimiento para producir un anticuerpo anti-BCMA de la presente invención que se una a, y neutralice la actividad de BCMA humano comprendiendo dicho procedimiento las etapas de:
proporcionar un primer vector que codifica una cadena pesada del anticuerpo;
proporcionar un segundo vector que codifica una cadena ligera del anticuerpo;
transformar una célula huésped de mamífero (por ejemplo CHO) con dichos primer y segundo vectores;
cultivar la célula huésped de la etapa (c) en condiciones que conduzcan a la secreción del anticuerpo de dicha célula huésped en dicho medio de cultivo;
recuperar el anticuerpo secretado de la etapa (d).
Una vez expresado por el procedimiento deseado, el anticuerpo se examina después con respecto a actividad in vitro mediante el uso de un ensayo apropiado. Se emplean formatos de ensayo de ELISA convencionales en la actualidad para evaluar unión cualitativa y cuantitativa del anticuerpo con BCMA. Adicionalmente, también pueden usarse otros ensayos in vitro para verificar la eficacia de neutralización antes de estudios clínicos humanos posteriores realizados para evaluar la persistencia del anticuerpo en el cuerpo a pesar de los mecanismos de eliminación habituales.
La dosis y duración del tratamiento se refiere a la duración relativa de las moléculas de la presente invención en la circulación humana y pueden ajustarse por un experto en la materia dependiendo de la afección que se trate y la salud general del paciente. Se prevé que la dosificación repetida (por ejemplo una vez a la semana, una vez cada dos semanas o una vez cada tres semanas) durante un periodo de tiempo extendido (por ejemplo de cuatro a seis meses) puede requerirse para conseguir máxima eficacia terapéutica.
En una realización de la presente invención se proporciona una célula huésped recombinante transformada, transfectada o transducida que comprende al menos un casete de expresión, por ejemplo en la que el casete de expresión comprende un polinucleótido que codifica una cadena pesada de una proteína de unión a antígeno de acuerdo con la invención descrita en el presente documento y comprende adicionalmente un polinucleótido que codifica una cadena ligera de una proteína de unión a antígeno de acuerdo con la invención descrita en el presente documento o en la que hay dos casetes de expresión y el primero codifica la cadena ligera y el segundo codifica la cadena pesada. Por ejemplo en una realización el primer casete de expresión comprende un polinucleótido que codifica una cadena pesada de una proteína de unión a antígeno que comprende una región constante o fragmento de unión a antígeno de la misma que está ligada a una región constante de acuerdo con la invención descrita en el presente documento y comprende adicionalmente un segundo casete que comprende un polinucleótido que codifica una cadena ligera de una proteína de unión a antígeno que comprende una región constante o fragmento de unión a antígeno de la misma que se liga a una región constante de acuerdo con la invención descrita en el presente
documento, por ejemplo el primer casete de expresión comprende un polinucleótido que codifica una cadena pesada seleccionada de SEC ID N°: 56, y un segundo casete de expresión que comprende un polinucleótido que codifica una cadena ligera seleccionada de SEC ID N°: 64.
En otra realización de la invención se proporciona una célula huésped transformada de forma estable que comprende un vector que comprende uno o más casetes de expresión que codifican una cadena pesada y/o una cadena ligera del anticuerpo que comprende una región constante o fragmento de unión a antígeno de la misma que está ligado a una región constante como se describe en el presente documento. Por ejemplo tales células huésped pueden comprender un primer vector que codifica la cadena ligera y un segundo vector que codifica la cadena pesada, por ejemplo el primer vector codifica una cadena pesada seleccionada de SEC ID N°: 55, y un segundo vector que codifica una cadena ligera, por ejemplo la cadena ligera de SEC ID N°: 63. En un ejemplo tal el primer vector codifica una cadena pesada seleccionada de SEC ID N°: 55 y un segundo vector que codifica una cadena ligera, por ejemplo la cadena ligera de SEC ID N°: 63.
En otra realización de la presente invención se proporciona una célula huésped de acuerdo con la invención descrita en el presente documento siendo la célula eucariota, por ejemplo siendo la célula de mamífero. Los ejemplos de tales líneas celulares incluyen CHO o NS0.
En otra realización de la presente invención se proporciona un procedimiento para la producción de un anticuerpo que comprende una región constante o fragmento de unión a antígeno de la misma que está ligada con una región constante de acuerdo con la invención descrita en el presente documento comprendiendo dicho procedimiento la etapa de cultivar una célula huésped en un medio de cultivo, por ejemplo medio de cultivo sin suero.
En otra realización de la presente invención se proporciona un procedimiento de acuerdo con la invención descrita en el presente documento en el que dicho anticuerpo se purifica adicionalmente al menos a 95 % o más (por ejemplo 98 % o más) con respecto a dicho medio de cultivo sin suero que contiene anticuerpos.
En otra realización más se proporciona una composición farmacéutica que comprende una proteína de unión a antígeno y un vehículo farmacéuticamente aceptable.
En otra realización de la presente invención se proporciona un equipo de herramientas que comprende la composición de acuerdo con la invención descrita en el presente documento junto con instrucciones para su uso.
El modo de administración del agente terapéutico de la invención puede ser cualquier vía adecuada que suministra el agente al huésped. Las proteínas de unión a antígeno y composiciones farmacéuticas de la presente invención son particularmente útiles para administración parenteral, es decir, por vía subcutánea (s.c.), vía intratecal, vía intraperitoneal, vía intramuscular (i.m.) o vía intravenosa (i.v.). En una realización tal las proteínas de unión a antígeno de la presente invención se administran por vía intravenosa o vía subcutánea.
Los agentes terapéuticos de la invención pueden prepararse como composiciones farmacéuticas que contienen una cantidad eficaz de la proteína de unión a antígeno de la invención como un principio activo en un vehículo farmacéuticamente aceptable. En una realización el agente profiláctico de la invención es una suspensión o solución acuosa que contiene la proteína de unión a antígeno en una forma lista para inyección. En una realización la suspensión o solución se tampona a pH fisiológico. En una realización las composiciones para administración parenteral comprenderán una solución de la proteína de unión a antígeno de la invención o un cóctel de la misma disuelto en un vehículo farmacéuticamente aceptable. En una realización el vehículo es un vehículo acuoso. Puede emplearse una diversidad de vehículos acuosos, por ejemplo, solución salina 0,9 %, glicina 0,3 % y similares. Estas soluciones pueden hacerse estériles y generalmente sin materia en partículas. Estas soluciones pueden esterilizarse por técnicas de esterilización convencionales bien conocidas (por ejemplo, filtración). Las composiciones pueden contener sustancias adyuvantes farmacéuticamente aceptables según se requiera para aproximar las condiciones fisiológicas tales como ajuste de pH y agentes tamponantes, etc. La concentración de la proteína de unión a antígeno de la invención en dicha formulación farmacéutica puede variar ampliamente, es decir, de menos de aproximadamente 0,5 %, habitualmente a o al menos aproximadamente a 1 %, hasta tanto como aproximadamente 15 ó 20 % en peso y se seleccionará principalmente basándose en volúmenes de fluido, viscosidades, etc., de acuerdo con el modo particular de administración seleccionado.
Por lo tanto, una composición farmacéutica de la invención para infusión intravenosa puede prepararse para contener aproximadamente 250 ml de solución de Ringer estéril y de aproximadamente 1 a aproximadamente 30 o de 5 mg a aproximadamente 25 mg de una proteína de unión a antígeno de la invención por ml de solución de Ringer. Se conocen bien o resultarán evidentes para los expertos en la materia procedimientos concretos para preparar composiciones administrables por vía parenteral y se describen en más detalle en, por ejemplo, Remington’s Pharmaceutical Science, 15a ed., Mack Publishing Company, Easton, Pennsylvania. Para la preparación de formulaciones de proteína de unión a antígeno administrables por vía intravenosa de la invención véase Lasmar U y Parkins D “The formulation of Biopharmaceutical products”, Pharma. Sci.Tech.today, páginas 129-137, Vol.3 (3 de abril de 2000); Wang, W
“Instability, stabilisation and formulation of liquid protein pharmaceuticals”, Int. J. Pharm 185 (1999) 129-188; Stability of Protein Pharmaceuticals Part A and B ed Ahern T. J., Manning M.C., Nueva York, NY: Plenum Press (1992); Akers, M. J. “Excipient-Drug interactions in Parenteral Formulations”, J. Pharm Sci 91 (2002) 2283-2300; Imamura, K y col “Effects of types of sugar on stabilization of Protein in the dried state”, J Pharm Sci 92 (2003) 266-274; Izutsu, Kkojima, S. “Excipient crystalinity and its protein-structure-stabilizing effect during freeze-drying”, J Pharm. Pharmacol, 54 (2002) 1033-1039; Johnson, R, “Mannitol-sucrose mixtures-versatile formulations for protein peroxidise 19g19n”, J. Pharm. Sci, 91 (2002) 914-922; y Ha, E Wang W, Wang Y.j. “Peroxide formation in polysorbate 80 and protein stability”, J. Pharm Sci, 91,2252-2264, (2002).
En una realización, el agente terapéutico de la invención, cuando está en una preparación farmacéutica, está presente en formas de dosis unitarias. La dosis terapéuticamente eficaz apropiada se determinará fácilmente por los expertos en la materia. Las dosis adecuadas pueden calcularse para pacientes de acuerdo con su peso, por ejemplo las dosis adecuadas pueden estar en el intervalo de aproximadamente 0,1 a aproximadamente 20 mg/kg, por ejemplo de aproximadamente 1 a aproximadamente 20 mg/kg, por ejemplo de aproximadamente 10 a aproximadamente 20 mg/kg o por ejemplo de aproximadamente 1 a aproximadamente 15 mg/kg, por ejemplo de aproximadamente 10 a aproximadamente 15 mg/kg. Para tratar de forma eficaz afecciones tales como mieloma Múltiple, SLE o IPT en un ser humano, las dosis adecuadas pueden estar dentro del intervalo de aproximadamente 0,1 a aproximadamente 1000 mg, por ejemplo de aproximadamente 0,1 a aproximadamente 500 mg, por ejemplo de aproximadamente 500 mg, por ejemplo de aproximadamente 0,1 a aproximadamente 100 mg, de aproximadamente 0,1 a aproximadamente 80 mg, de aproximadamente de 0,1 a aproximadamente 60 mg, de aproximadamente 0,1 a aproximadamente 40 mg, por ejemplo de aproximadamente 1 a aproximadamente 100 mg, o de aproximadamente 1 a aproximadamente 50 mg, de una proteína de unión a antígeno de la presente invención, que puede administrarse por vía parenteral, por ejemplo por vía subcutánea, vía intravenosa o intramuscular. Dicha dosis puede, si es necesario, repetirse a intervalos de tiempo apropiados seleccionados según sea apropiado por un médico.
Las proteínas de unión a antígeno descritas en el presente documento pueden liofilizarse para almacenamiento y reconstituirse en un vehículo adecuado antes de su uso. Se ha mostrado que esta técnica es eficaz con inmunoglobulinas convencionales y pueden emplearse técnicas de reconstitución y peroxidación conocidas en la técnica.
En otro aspecto de la invención se proporciona una proteína de unión a antígeno como se describe en el presente documento para su uso en un medicamento.
En un aspecto de la presente invención se proporciona una proteína de unión a antígeno de acuerdo con la invención como se describe en el presente documento para su uso en el tratamiento de artritis reumatoide, Diabetes Mellitus de Tipo 1, esclerosis múltiple o psoriasis comprendiendo dicho procedimiento la etapa de administrar a dicho paciente una cantidad terapéuticamente eficaz de la proteína de unión a antígeno como se ha descrito en el presente documento.
En una realización de la presente invención, se proporcionan procedimientos para tratar cáncer en un ser humano que comprenden administrar a dicho ser humano una proteína de unión a antígeno que se une específicamente a BCMa . En algunos casos la proteína de unión a antígeno es parte de un inmunoconjugado.
En otro aspecto de la presente invención se proporciona una proteína de unión a antígeno de acuerdo con la invención como se describe en el presente documento para su uso en el tratamiento de una enfermedad mediada por linfocitos B o mediada por células plasmáticas o enfermedad o trastorno mediado por anticuerpos seleccionado de Mieloma Múltiple (MM), leucemia linfocítica crónica (CLL), mieloma múltiple no secretor, Mieloma Múltiple Indolente, gammapatía monoclonal de importancia indeterminada (MGUS), plasmacitoma solitario (Hueso, Extramedular), linfoma linfoplasmacítico (LPL), macroglobulinemia de Waldenstrom, leucemia de células plasmáticas, amiloidosis Primaria (AL), enfermedad de cadena pesada, lupus eritematoso sistémico (SLE), síndrome de POEMS / mieloma osteosclerótico, crioglobulinemia de Tipo I y II, enfermedad de deposición de cadena ligera, síndrome de Goodpasture, púrpura trombocitopénica idiopática (ITP), glomerulonefritis aguda, Pénfigo y trastornos penfigoides, Epidermólisis ampollosa adquirida; o cualquier leucemia de linfocitos B de Linfoma No de Hodgkin o linfoma de Hodgkin (HL) con expresión de BCMA o cualquier enfermedad en la que los pacientes desarrollen anticuerpos neutralizadores para terapia de reemplazo de proteína recombinante en la que dicho procedimiento comprende la etapa de administrar a dicho paciente una cantidad terapéuticamente eficaz de la proteína de unión a antígeno como se describe en el presente documento.
Los trastornos de linfocitos B pueden dividirse en defectos de desarrollo de linfocitos B/producción de inmunoglobulinas (inmunodeficiencias) y proliferación excesiva/incontrolada (linfomas, leucemias). Como se usa en el presente documento, trastornos de linfocitos B se refiere a ambos tipos de enfermedades, y se proporcionan procedimientos para tratar trastornos de linfocitos B con una proteína de unión a antígeno.
En un aspecto particular, la enfermedad o trastorno se selecciona del grupo que consiste en Mieloma Múltiple (MM),
Leucemia Linfocítica Crónica (CLL), Plasmacitoma solitario (Hueso, Extramedular), Macroglobulinemia de Waldenstrom.
En un aspecto de la presente invención la enfermedad es Mieloma Múltiple, Mieloma Múltiple Indolente (SMM) o Plasmacitoma solitario (Hueso, Extramedular).
En un aspecto de la presente invención la enfermedad es Mieloma Múltiple.
En un aspecto de la presente invención la enfermedad es  lupus eritematoso sistémico (SLE).
lupus eritematoso sistémico (SLE).
En un aspecto de la presente invención la enfermedad es púrpura trombocitopénica idiopática (ITP).
También se proporciona uso de la proteína de unión a antígeno como se describe en el presente documento en la preparación de un medicamento para el tratamiento de enfermedades y trastornos como se describe en el presente documento.
En otro aspecto de la divulgación se proporciona el uso de la proteína de unión a antígeno como se describe en el presente documento para su uso en el tratamiento o profilaxis de una enfermedad o trastorno mediado por anticuerpo o mediado por células plasmáticas seleccionado de artritis reumatoide, Diabetes Mellitus de Tipo 1, esclerosis múltiple o psoriasis.
En otro aspecto de la invención se proporciona el uso de la proteína de unión a antígeno como se describe en el presente documento para su uso en el tratamiento o profilaxis de una enfermedad o trastorno mediada por anticuerpo o mediada por células plasmáticas seleccionada de Mieloma Múltiple (MM), leucemia linfocítica crónica (CLL), gammapatía monoclonal de importancia indeterminada (MGUS), Mieloma Múltiple Indolente (SMM), plasmacitoma solitario (Hueso, Extramedular), Macroglobulinemia de Waldenstrom, Amiloidosis Primaria (AL), enfermedad de cadena pesada, lupus eritematoso sistémico (SLE), síndrome de POEMS / mieloma osteosclerótico, crioglobulinemia de Tipo I y II, enfermedad de deposición de cadena ligera, síndrome de Goodpasture, púrpura trombocitopénica idiopática (ITP), glomerulonefritis aguda, Pénfigo y trastornos penfigoides y Epidermólisis ampollosa adquirida, cualquier Linfoma No de Hodgkin y leucemia con expresión de BCMA o cualquier enfermedad en la que los pacientes desarrollen anticuerpos neutralizadores para terapia de reemplazo de proteína recombinante en la que dicho procedimiento comprende la etapa de administrar a dicho paciente una cantidad terapéuticamente eficaz de la proteína de unión a antígeno como se describe en el presente documento.
En un aspecto, la invención proporciona una composición farmacéutica que comprende una proteína de unión a antígeno de la presente invención o un fragmento funcional de la misma y un vehículo farmacéuticamente aceptable para tratamiento o profilaxis de artritis reumatoide, Diabetes Mellitus de Tipo I, esclerosos múltiple o psoriasis, o una enfermedad o trastorno mediado por anticuerpos o mediado por células plasmáticas seleccionado de Mieloma Múltiple (MM), leucemia linfocítica crónica (CLL), gammapatía monoclonal de importancia indeterminada (MGUS), Mieloma Múltiple Indolente (SMM), plasmacitoma solitario (Hueso, Extramedular), Macroglobulinemia de Waldenstrom, Amiloidosis Primaria (AL), enfermedad de cadena pesada, lupus eritematoso sistémico (SLE), síndrome de POEMS / mieloma osteosclerótico, crioglobulinemia de Tipo I y II, enfermedad de deposición de cadena ligera, síndrome de Goodpasture, púrpura trombocitopénica idiopática (ITP), glomerulonefritis aguda, Pénfigo y trastornos Penfigoides y Epidermólisis ampollosa adquirida, cualquier Linfoma No de Hodgkin y leucemia con expresión de BCMA o cualquier enfermedad en la que los pacientes desarrollen anticuerpos neutralizadores para terapia de reemplazo de proteína recombinante en la que dicho procedimiento comprende la etapa de administrar a dicho paciente una cantidad terapéuticamente eficaz de la proteína de unión a antígeno descrita en el presente documento.
En otra realización de la presente invención se proporciona un procedimiento para tratar a un paciente humano aquejado de artritis reumatoide, Diabetes Mellitus de Tipo I, esclerosos múltiple o psoriasis, o una enfermedad o trastorno mediado por anticuerpos o mediado por células plasmáticas comprendiendo dicho procedimiento la etapa de administrar una cantidad terapéuticamente eficaz de la proteína de unión a antígeno de acuerdo con la invención como se describe en el presente documento, por ejemplo se proporciona un procedimiento para tratar a un paciente humano aquejado de una enfermedad o trastorno mediada por anticuerpo o mediado por células plasmáticas seleccionado de Mieloma Múltiple (MM), leucemia linfocítica crónica (CLL), gammapatía monoclonal de importancia indeterminada (MGUS), Mieloma Múltiple Indolente (SMM), plasmacitoma solitario (Hueso, Extramedular), Macroglobulinemia de Waldenstrom, Amiloidosis Primaria (AL), enfermedad de cadena pesada, lupus eritematoso sistémico (SLE), síndrome de POEMS / mieloma osteosclerótico, crioglobulinemia de Tipo I y II, enfermedad de deposición de cadena ligera, síndrome de Goodpasture, púrpura trombocitopénica idiopática (ITP), glomerulonefritis aguda, Pénfigo y trastornos Penfigoides y Epidermólisis ampollosa adquirida, cualquier Linfoma No de Hodgkin y leucemia con expresión de BCMA o cualquier enfermedad en la que los pacientes desarrollen anticuerpos neutralizadores para terapia de reemplazo de proteína recombinante en la que dicho procedimiento comprende la etapa de administrar una composición farmacéutica que comprende una proteína de unión a antígeno de acuerdo con la invención en el presente documento en combinación con un vehículo farmacéuticamente aceptable.
En una realización adicional se proporciona un procedimiento para tratar a un paciente humano aquejado de Mieloma Múltiple (MM).
Definiciones
Como se usa en el presente documento, los términos “cáncer”, “neoplasia” y “tumor” se usan de forma intercambiable y, en la forma singular o plural, se refieren a células que han experimentado una transformación maligna que las hace patológicas para el organismo huésped. Las células cancerosas primarias pueden distinguirse fácilmente de células no cancerosas por técnicas bien establecidas, particularmente examen histológico. La definición de la célula cancerosa, como se usa en el presente documento, incluye no solamente una célula cancerosa primaria, sino cualquier célula derivada de un ancestro de célula cancerosa. Esto incluye células cancerosas metastatizadas, y cultivos in vitro y líneas celulares derivadas de células cancerosas. Cuando se hace referencia a un tipo de cáncer que normalmente se manifiesta como un tumor sólido, un tumor “clínicamente detectable” es uno que es detectable basándose en la masa tumoral; por ejemplo, por procedimientos tales como exploración de tomografía computarizada (CT), imagen de resonancia magnética (MRI), rayos X, ultrasonidos o palpación en examen físico, y/o que es detectable debido a la expresión de uno o más antígenos específicos de cáncer en una muestra que puede obtenerse de un paciente. Los tumores pueden ser un cáncer hematopoyético (o hematológico o relacionado con la sangre), por ejemplo, cánceres derivados de células sanguíneas o células inmunes, que pueden denominarse “tumores líquidos”. Los ejemplos específicos de afecciones clínicas basadas en tumores hematológicos incluyen leucemias tales como leucemia mielocítica crónica, leucemia mielocítica aguda, leucemia linfocítica crónica y leucemia linfocítica aguda; tumores malignos de células plasmáticas tales como mieloma múltiple, MGUS y macroglobulinemia de Waldenstrom; linfomas tales como linfoma no de Hodgkin, linfoma de Hodgkin y similares.
El cáncer puede ser cualquier cáncer en el que esté presente un número anómalo de blastocitos o proliferación de células no deseadas o que se diagnostique como un cáncer hematológico, incluyendo tumores malignos tanto linfoides como mieloides. Los tumores malignos mieloides incluyen, pero sin limitación, leucemia mieloide (o mielocítica, mielógena o mieloblástica) aguda (indiferenciada o diferenciada), leucemia promieloide (o promielocítica, promielógena o promieloblástica) aguda, leucemia mielomonocítica (o mielomonoblástica) aguda, leucemia monocítica (o monoblástica) aguda, eritroleucemia y leucemia megacariocítica (o megacarioblástica). Estas leucemias pueden denominarse juntas leucemia mieloide (o mielocítica o mielógena) aguda (AML). Los tumores malignos mieloides también incluyen trastornos mieloproliferativos (MPD) que incluyen, pero sin limitación, leucemia mielógena (o mieloide) crónica (CML), leucemia mielomonocítica crónica (CMMl), trombocitopenia (o trombocitosis) esencial y policitemia vera (PCV). Los tumores malignos mieloides también incluyen mielodisplasia (o síndrome mielodisplásico o MDS), que puede denominarse anemia refractaria (RA), anemia refractaria con exceso de blastos (RAEB) y anemia refractaria con exceso de blastos en transformación (RAEBT); así como mielofibrosis (MFS) con o sin metaplasia mieloide agnogénica.
Los cánceres hematopoyéticos también incluyen tumores malignos linfoides, que pueden afectar a los ganglios linfáticos, bazos, médula ósea, sangre periférica y/o sitios extranodales. Los cánceres linfoides incluyen tumores malignos de linfocitos B, que incluyen, pero sin limitación, linfomas no de Hodgkin de linfocitos B (B-NHL). Los B-NHL puede ser indolentes (o de grado bajo), de grado intermedio (o agresivos) o de grado alto (muy agresivos). Los linfomas de linfocitos B indolentes incluyen linfoma folicular (FL); linfoma linfocítico pequeño (SLL); linfoma de zona marginal (MZL) incluyendo MZL nodal, MZL extranodal, MZL esplénico y MZL esplénico con linfocitos vellosos; linfoma linfoplasmacítico (LPL); y linfoma de tejido linfoide asociado a mucosa (MALT o zona margina extranodal). Los B-NHL de grado intermedio incluyen linfoma de células de manto (MCL) con o sin implicación leucémica, linfoma de células grandes difuso (DLBCL), linfoma de células grandes folicular (o grado 3 o grado 3B), y linfoma mediastinal primario (PML). Los B-NHL de grado alto incluyen linfoma de Burkitt (BL), linfoma de tipo Burkitt, linfoma de células pequeñas no escindidas (SNCCL) y linfoma linfoblástico. Otros B-NHL incluyen linfoma inmunoblástico (o immunocitoma), linfoma de efusión primaria, linfomas asociados con VIH (o relacionados con SIDA) y linfoma o trastorno linfoproliferativo postrasplante (PTLD). Los tumores malignos de linfocitos B también incluyen, pero sin limitación, leucemia linfocítica crónica (CLL), leucemia prolinfocítica (PLL), macroglobulinemia de Waldenstrom (WM), tricoleucemia (HCL), leucemia de linfocitos granulares grandes (LGL), leucemia linfoide (o linfocítica o linfoblástica) aguda y enfermedad de Castleman. El NHL también puede incluir linfomas no de Hodgkin de linfocitos T (T-NHL), que incluyen, pero sin limitación, linfoma no de Hodgkin de linfocitos T no especificado de otro modo (NOS), linfoma de linfocitos T periférico (PTCL), linfoma de células grandes anaplásicas (ALCL), trastorno linfoide angioinmunoblástico (AILD), linfoma de linfocitos T / linfocitos citolíticos naturales (NK) nasal, linfoma gamma/delta, linfoma de linfocitos T cutáneo, micosis fungoide y síndrome de Sezary.
Los cánceres hematopoyéticos también incluyen linfoma (o enfermedad) de Hodgkin incluyendo linfoma de Hodgkin clásico, linfoma de Hodgkin esclerosante nodular, linfoma de Hodgkin de celularidad mixta, linfoma de Hodgkin con linfocitos predominantes (LP), linfoma de Hodgkin LP nodular y linfoma de Hodgkin empobrecido con respecto a linfocitos. Los cánceres hematopoyéticos también incluyen enfermedades de células plasmáticas o cánceres tales como mieloma múltiple (MM), incluyendo MM indolente, gammapatía monoclonal de importancia indeterminada (o
desconocida o poco clara) (MGUS), plasmacitoma (hueso, extramedular), linfoma linfoplasmacítico (LPL), Macroglobulinemia de Waldenstrom, leucemia de células plasmáticas y amiloidosis primaria (AL). Los cánceres hematopoyéticos también pueden incluir otros cánceres de células hematopoyéticas adicionales, incluyendo leucocitos polimorfonucleares (o neutrófilos), basófilos, eosinófilos, células dendríticas, plaquetas, eritrocitos y linfocitos citolíticos naturales. Los tejidos que incluyen células hematopoyéticas indicadas en el presente documento como “tejidos celulares hematopoyéticos” incluyen médula ósea, sangre periférica, timo y tejidos linfoides periféricos, tales como bazo, ganglios linfáticos, tejidos linfoides asociados con mucosa (tales como los tejidos linfoides asociados con intestino), amígdalas, placas de Peyer y apéndice, y tejidos linfoides asociados con otra mucosa, por ejemplo, los revestimientos bronquiales.
La expresión “proteína de unión a antígeno” como se usa en el presente documento se refiere a anticuerpos, fragmentos de anticuerpo y otras construcciones proteicas que son capaces de unirse a y neutralizar BCMA humano.
Los términos Fv, Fc, Fd, Fab, o F(ab)2 se usan con sus significados convencionales (véase, por ejemplo, Harlow y col., Antibodies A Laboratory Manual, Cold Spring Harbor Laboratory, (1988)).
El término “anticuerpo” se usa en el presente documento en el sentido más amplio y específicamente abarca anticuerpos monoclonales (incluyendo anticuerpos monoclonales de longitud completa), anticuerpos policlonales, anticuerpos multiespecíficos (por ejemplo anticuerpos biespecíficos).
La expresión “anticuerpo monoclonal” como se usa en el presente documento se refiere a un anticuerpo obtenido de una población de anticuerpos sustancialmente homogéneos, es decir los anticuerpos individuales que comprenden la población son idénticos excepto por posibles mutaciones de origen natural que pueden estar presentes en cantidades menores. Los anticuerpos monoclonales son altamente específicos que se dirigen contra un sitio de unión antigénico sencillo. Además, a diferencia de las preparaciones de anticuerpo policlonal que típicamente incluyen diferentes anticuerpos dirigidos contra diferentes determinantes (epítopos), cada anticuerpo monoclonal se dirige contra un determinante sencillo en el antígeno.
Un “anticuerpo quimérico” se refiere a un tipo de anticuerpo obtenido por ingeniería genética en el que una parte de la cadena pesada y/o ligera es idéntica a u homóloga de secuencias correspondientes en anticuerpos derivadas de una clase o subclase de anticuerpo donador particular, mientras que el resto de la cadena o cadenas es idéntico a, u homólogo de secuencias correspondientes en anticuerpos derivados de otra especie o que pertenecen a otra clase o subclase de anticuerpo, así como fragmentos de tales anticuerpos, siempre que muestren la actividad biológica deseada (Patente de Estados Unidos N°4.816.567y Morrison y col. Proc. Nati. Acad. Sci. USA 81:6851-6855) (1984)).
Un “anticuerpo humanizado” se refiere a un tipo de anticuerpo obtenido por ingeniería genética que tiene sus CDR derivadas de una inmunoglobulina donadora no humana, derivando las partes derivadas de inmunoglobulina restantes de la molécula de una, o más, inmunoglobulinas humanas. Además, los restos de soporte del marco pueden alterarse para conservar la afinidad de unión (véase, por ejemplo, Queen y col., Proc. Natl Acad Sci USA, 86: 10029-10032 (1989), Hodgson y col., Bio/Technology, 9: 421 (1991)). Un anticuerpo aceptor humano adecuado puede ser uno seleccionado de una base de datos convencional, por ejemplo la base de datos KABAT®, base de datos de Los Alamos y base de datos Swiss Protein, por homología con las secuencias de nucleótidos y aminoácidos del anticuerpo donador. Un anticuerpo humano caracterizado por una homología con las regiones marco del anticuerpo donador (basándose en aminoácidos) puede ser adecuado para proporcionar una región constante de cadena pesada y/o una región marco variable de cadena pesada para inserción de las CDR donadoras. Un anticuerpo aceptor adecuado capaz de donar regiones marco constantes o variables de cadena ligera puede seleccionarse de una manera similar. Debería observarse que no se requiere que las cadenas pesada y ligera del anticuerpo aceptor se originen del mismo anticuerpo aceptor. La técnica anterior describe varios modos de producir tales anticuerpos humanizados, véase por ejemplo los documentos EP-A-0239400 y EP-A-054951.
Para ácidos nucleicos, la expresión “identidad sustancial” indica que dos ácidos nucleicos, o secuencias designadas de los mismos, cuando se alinean y comparan de forma óptima, son idénticos, con inserciones o deleciones de nucleótidos apropiadas, en al menos aproximadamente 80 % de los nucleótidos, al menos aproximadamente 90 % a aproximadamente 95 %, o al menos aproximadamente 98 % a aproximadamente 99,5 % de los nucleótidos. Como alternativa, existe identidad sustancial cuando los segmentos hibridan en condiciones de hibridación selectivas, con el complemento de la cadena.
“Identidad” significa, para polinucleótidos y polipéptidos, según sea el caso, la comparación calculada usando un algoritmo proporcionado en (1) y (2) a continuación:
(1) Se calcula la identidad para polinucleótidos multiplicando el número total de nucleótidos en una secuencia dada por el número entero que define el porcentaje de identidad dividido por 100 y después se resta ese producto de dicho número total de nucleótidos en dicha secuencia, o:
nn ≤ xn - (xn • y),
en la que nn es el número de alteraciones de nucleótidos, xn es el número total de nucleótidos en una secuencia dada, y es 0,95 para 95 %, 0,97 para 97 % o 1,00 para 100 % y • es el símbolo para el operador de multiplicación, y en la que cualquier número no entero producto de xn y se redondea hacia abajo al número entero más cercano antes de restarlo de xn. Las alteraciones de una secuencia polinucleotídica que codifica un polipéptido pueden crear mutaciones sin sentido, de sentido erróneo o de desplazamiento de la fase en esta secuencia codificante y de este modo alterar el polipéptido codificado por el polinucleótido tras dichas alteraciones.
(2) Se calcula la identidad para polipéptidos multiplicando el número total de aminoácidos por el número entero que define el porcentaje de identidad dividido por 100 y después se resta ese producto de dicho número total de aminoácidos, o:
na ≤ xa - (xa • y),
en la que na es el número de alteraciones de aminoácidos, xa es el número total de aminoácidos en la secuencia, y es 0,95 para 95 %, 0,97 para 97 % o 1,00 para 100 % y • es el símbolo para el operador de multiplicación, y en la que cualquier producto no entero de xa y se redondea hacia abajo al número entero más cercano antes de restarlo de xa.
Para secuencias de nucleótidos y aminoácidos, el término “idéntico” indica el grado de identidad entre dos secuencias de aminoácidos o ácidos nucleicos cuando se alinean de forma óptima y se comparan con inserciones o deleciones apropiadas.
“Aislado” significa alterado “por la mano del hombre” de su estado natural, se ha cambiado o retirado de su ambiente original, o ambos. Por ejemplo, un polinucleótido o un polipéptido presente de forma natural en un organismo vivo no está “aislado”, pero el mismo polinucleótido o polipéptido separado de los materiales coexistentes de su estado natural está “aislado”, incluyendo pero sin limitación cuando dicho polinucleótido o polipéptido se introduce de nuevo en una célula, incluso si la célula es de la misma especie o tipo que de la que se separó el polinucleótido o polipéptido.
A lo largo de la presente memoria descriptiva y las reivindicaciones adjuntas la expresión “que comprende” y “comprende” incorpora “que consiste en” y “consiste en”. Es decir, estas palabras pretenden transmitir la posible inclusión de otros elementos o números enteros no enumerados de forma específica, cuando el contexto lo permita.
La expresión “se une específicamente” como se usa a lo largo de la presente memoria descriptiva en relación con proteínas de unión a antígeno de la invención significa que la proteína de unión a antígeno se une a BCMA humano (hBCMA) sin unión o con unión insignificante con otras proteínas humanas. La expresión sin embargo no excluye el hecho de que las proteínas de unión a antígeno de la invención pueden también reaccionar de forma cruzada con otras formas de BCMA, por ejemplo BCMA de primates. Por ejemplo en una realización la proteína de unión a antígeno no se une a TACI o BAFF-R.
El término “inhibe” como se usa a lo largo de la presente memoria descriptiva en relación con proteínas de unión a antígeno de la invención significa que la actividad biológica de BCMA se reduce en presencia de las proteínas de unión a antígeno de la presente invención en comparación con la actividad de BCMA en ausencia de tales proteínas de unión a antígeno. La inhibición puede deberse pero sin limitación a uno o más de bloquear unión de ligando, evitar que el ligando active el receptor y/o regular de forma negativa el BCMA. “Inhibe” también puede referirse a una proteína de unión a antígeno que se une a BCMA y provoca apoptosis celular o ADCC. Los anticuerpos de la invención pueden neutralizar la actividad de los ligandos de BCMA BAFf y/o APRIL que se unen a BCMA. Los niveles de neutralización pueden medirse de varias maneras, por ejemplo mediante el uso de los ensayos como se exponen en los ejemplos posteriores, por ejemplo en 4.4 en un ensayo de señalización de NFk B de células H929. Los ligandos de BCMA BAFF y APRIL son capaces de inducir señalización de NFk B y acontecimientos cadena abajo después de la unión a BCMA. La neutralización de BCMA en este ensayo se mide evaluando la capacidad de los anticuerpos monoclonales anti-BCMA para inhibir la inducción de NFk B conducida por BAFF o APRIL.
Si un anticuerpo o fragmento de unión a antígeno del mismo es capaz de neutralización entonces esto es indicativo de inhibición de la interacción entre BAFF o APRIL humanos y BCMa . Los anticuerpos que se considera que tienen actividad neutralizadora frente a BCMA humano tendrían una CI50 de menos de 30 microgramos/ml, menos de 20 microgramos/ml, menos de 10 microgramos/ml, menos de 5 microgramos/ml, menos de 1 microgramo/ml o menos de 0,1 microgramos/ml en el ensayo de estimulación de H929 como se expone en el Ejemplo 4.4
Las “CDR” se definen como las secuencias de aminoácidos de región determinante de complementariedad de un anticuerpo que son los dominios hipervariables de cadenas pesadas y ligeras de inmunoglobulina. Hay tres CDR (o regiones CDR) de cadena pesada y tres de cadena ligera en la parte variable de una inmunoglobulina. Por lo tanto, las “CDR” como se usan en el presente documento pueden referirse a las tres CDR de cadena pesada o las tres CDR de cadena ligera (o todas las CDR tanto de cadena pesada como de cadena ligera, si es apropiado).
Las CDR proporcionan la mayoría de los restos de contacto para la unión del anticuerpo con el antígeno o epítopo. Las CDR de interés de la presente invención derivan de secuencias de cadena pesada y ligera variable de anticuerpo donador, e incluyen análogos de las CDR de origen natural, compartiendo o conservando también dichos análogos la misma especificidad de unión a antígeno y/o capacidad de neutralización que el anticuerpo donador del que derivaron.
Las secuencias de CDR de anticuerpos pueden determinarse por el sistema de numeración de Kabat (Kabat y col; (Sequences of proteins of Immunological Interest NIH, 1987), como alternativa pueden determinarse usando el sistema de numeración de Chothia (Al-Lazikani y col., (1997) JMB 273,927-948), el procedimiento de definición del contacto (MacCallum R. M., y Martin A.C.R. y Thornton J. M, (1996), Journal of Molecular Biology, 262 (5), 732-745) o cualquier otro procedimiento establecido para enumerar los restos en un anticuerpo y determinar las CDR conocidas por los expertos en la materia.
Otras convenciones de numeración para secuencias de CDR disponibles para un experto en la materia incluyen procedimientos de “AbM” (Universidad de Bath) y de “contacto” (Escuela Universitaria de Londres). Puede determinarse que la región solapante mínima usando al menos dos de los procedimientos de Kabat, Chothia, AbM y contacto proporciona la “unidad de unión mínima”. La unidad de unión mínima puede ser una subporción de una CDR.
La Tabla A a continuación representa una definición usando cada convención de numeración para cada CDR o unidad de unión. El esquema de numeración de Kabat se usa en la Tabla X para enumerar la secuencia de aminoácidos de dominio variable. Debería observarse que algunas de las definiciones de CDR pueden variar dependiendo de la publicación individual usada.
Tabla A

A lo largo de la presente memoria descriptiva, los restos de aminoácidos en secuencias de anticuerpos se numeran de acuerdo con el esquema de Kabat. De forma similar, los términos “CDR”, “CDRL1”, “CDRL2”, “CDRL3”, “CDRH1”, “CDRH2”, “CDRH3” siguen el sistema de numeración de Kabat como se expone en Kabat y col; Sequences of proteins of Immunological Interest NIH, 1987.
El término “variante” se refiere a al menos uno, dos o tres cambios de aminoácidos en la secuencia. Estos cambios de aminoácidos pueden ser deleción, sustitución o adición pero son preferentemente sustitución. En una realización tal las sustituciones son sustituciones conservativas.
En una realización alternativa la secuencia variante contiene al menos una sustitución conservando a la vez la canónica de la proteína de unión a antígeno.
Las regiones determinantes de complementariedad (CDR) L1, L2, L3, H1 y H2 tienden a mostrar estructuralmente una de un numero finito de conformaciones de cadena principal. La clase de estructura canónica particular de una CDR se define tanto por la longitud de la CDR como por el empaquetamiento del bucle, determinado por restos localizados en posiciones claves tanto en las CDR como en las regiones marco (restos de determinación estructural o SDR). Martin y Thornton (1996; J Mol Biol 263: 800-815) han generado un procedimiento automático para definir los moldes canónicos de “restos clave”. Se usa análisis de grupos para definir las clases canónicas para conjuntos de CDR y se identifican después los moldes canónicos analizando restos hidrófobos enterrados, de unión a hidrógeno y por ejemplo glicinas conservadas. Las CDR de las secuencias de anticuerpos pueden asignarse a clases canónicas comparando las secuencias con los moldes de restos clave y puntuando cada molde usando matrices de identidad o similitud.
Los términos “VH” y “VL” se usan en el presente documento para referirse al dominio variable de cadena pesada y el dominio variable de cadena ligera respectivamente de un anticuerpo.
Como se usa en el presente documento el término “dominio” se refiere a una estructura proteica plegada que tiene estructura terciaria independiente del resto de la proteína. En general, los dominios son responsables de propiedades funcionales discretas de proteínas y en muchos casos pueden añadirse, retirarse o transferirse a otras proteínas sin
pérdida de función del resto de la proteína y/o del dominio. Un “dominio variable sencillo de anticuerpo” es un dominio polipeptídico plegado que comprende secuencias características de dominios variables de anticuerpo. Incluye por lo tanto dominios variables de anticuerpo completo y dominios variables modificados, por ejemplo, en los que uno o más bucles se han reemplazado por secuencias que no son características de dominios variables de anticuerpo o dominios variables de anticuerpo que se han truncado o comprenden extensiones C terminales o N terminales, así como fragmentos plegados de dominios variables que conservan al menos la actividad de unión y especificidad del dominio de longitud completa.
La frase “dominio variable sencillo de inmunoglobulina” se refiere a un dominio variable de anticuerpo (VH, VHH, VL) que se une específicamente a un antígeno o epítopo independientemente de un domino o región V diferente. Un dominio variable sencillo de inmunoglobulina puede estar presente en un formato (por ejemplo, homo o heteromultímero) con otras regiones variables o dominios variables diferentes en el que las otras regiones o dominios no se requieren para unión a antígeno por el dominio variable de inmunoglobulina sencillo (es decir, en el que el dominio variable sencillo de inmunoglobulina se une a antígeno independientemente de los dominios variables adicionales). Un “anticuerpo de dominio” o “dAb” es el mismo que un “dominio variable sencillo de inmunoglobulina” que es capaz de unirse a un antígeno como se usa el término en el presente documento. Un domino variable sencillo de inmunoglobulina puede ser un dominio variable de anticuerpo humano, pero también incluye dominios variables de anticuerpo sencillo de otras especies tales como dAb de roedor (por ejemplo, como se desvela en el documento WO 00/29004), tiburón nodriza y VHH de Camélido. Los VHH de Camélido son polipéptidos de dominio variable sencillo de inmunoglobulina que derivan de especies que incluyen camello, llama, alpaca, dromedario y guanaco, que producen anticuerpos de cadena pesada desprovistos de forma natural de cadenas ligeras. Tales dominios VHH pueden humanizarse de acuerdo con técnicas convencionales disponibles en la materia, y aún se considera que tales dominios son “anticuerpos de dominio” de acuerdo con la invención. Como se usa en el presente documento “VH” incluye dominios VHH de camélido. NARV son otro tipo de dominios variables sencillos de inmunoglobulina que se identificaron en peces cartilaginosos incluyendo el tiburón nodriza. Estos dominios también se conocen como región variable de Nuevo Receptor de Antígenos (habitualmente abreviado a V(NAR) o NARV). Para detalles adicionales véase Mol. Immunol. 44, 656-665 (2006) y documento US20050043519A.
La expresión “dominio de unión a epítopo” se refiere a un dominio que se une específicamente a un antígeno o epítopo independientemente de un domino o región V diferente, este puede ser un anticuerpo de dominio (dAb), por ejemplo un dominio variable sencillo de inmunoglobulina de humano, camélido o tiburón o puede ser un dominio que sea un derivado de un armazón seleccionado del grupo que consiste en CTLA-4 (Evibody); lipocalina; moléculas derivadas de Proteína A tales como dominio Z de Proteína A (Affibody, SpA), dominio A (Avimer/Maxibody); proteína de choque Térmico tales como GroEl y GroES; peroxidasa μg (transcuerpo); proteína de repetición de anquirina (DARPin); aptámero peptídico; dominio de lectinas de tipo C (Tetranectina); cristalina y humana y ubiquitina humana (afilinas); dominios PDZ; dominios de tipo toxina kunitz de escorpión de inhibidores de proteasa humanos; y fibronectina (adnectina); que se ha sometido a ingeniería proteica para obtener unión a un ligando distinto del ligando natural.
CTLA-4 (Antígeno asociado con Linfocitos T Citotóxicos 4) es un receptor de la familia CD28 expresado principalmente en linfocitos T CD4+. Su dominio extracelular tiene un pliegue de Ig de tipo dominio variable. Los bucles correspondientes a CDR de anticuerpos pueden sustituirse con secuencia heteróloga para conferir diferentes propiedades de unión. Las moléculas CTLA-4 modificadas por ingeniería genética para tener especificidades de unión diferentes también se conocen como Evibodies. Para detalles adicionales véase Journal of Immunological Methods 248 (1-2), 31-45 (2001)
Las lipocalinas son una familia de proteínas extracelulares que transportan moléculas hidrófobas pequeñas tales como esteroides, bilinas, retinoides y lípidos. Tienen una estructura secundaria de lámina p rígida con varios bucles en el extremo abierto de la estructura cónica que puede modificarse por ingeniería genética para unirse a diferentes antígenos diana. Las anticalinas son de entre 160 y 180 aminoácidos de tamaño, y se derivan de lipocalinas. Para detalles adicionales véase Biochim Biophys Acta 1482: 337-350 (2000), documento US7250297B1 y documento US20070224633
Un affibody es un armazón derivado de Proteína A de Staphylococcus aureus que puede modificarse por ingeniería genética para unirse a antígeno. El dominio consiste en un haz de tres hélices de aproximadamente 58 aminoácidos. Se han generado bibliotecas por selección aleatoria de restos superficiales. Para detalles adicionales véase Protein Eng. Des. Sel. 17, 455-462 (2004) y documento EP1641818A1
Los avímeros son proteínas multidominio derivadas de la familia del armazón de dominio A. Los dominios nativos de aproximadamente 35 aminoácidos adoptan una estructura con enlaces disulfuro definida. Se genera diversidad por la redistribución de la variación natural mostrada por la familia de dominios A. Para detalles adicionales véase Nature Biotechnology 23(12), 1556 - 1561 (2005) y Expert Opinion on Investigational Drugs 16(6), 909-917 (junio de 2007)
Una Transferrina es una glicoproteína de transporte de suero monomérica. Las transferrinas pueden modificarse por ingeniería genética para unirse a diferentes antígenos diana por inserción de secuencias peptídicas en un bucle de
superficie permisivo. Los ejemplos de armazones de transferrinas modificas por ingeniería genética incluyen el transcuerpo. Para detalles adicionales véase J. Biol. Chem 274, 24066-24073 (1999).
Las Proteínas de Repetición de Anquirina Diseñadas (DARPin) derivan de Anquirina que es una familia de proteínas que median en la unión de proteínas de membrana integrales con el citoesqueleto. Una repetición de anquirina sencilla es un motivo de 33 restos que consiste en dos hélices a y un giro p. Pueden modificarse por ingeniería genética para unirse a diferentes antígenos diana seleccionando de forma aleatoria restos en la primera hélice a y un giro p de cada repetición. Su interfaz de unión puede aumentarse aumentando el número de módulos (un procedimiento para maduración de afinidad). Para detalles adicionales véase J. Mol. Biol. 332, 489-503 (2003), PNa S 100(4), 1700-1705 (2003) y J. Mol. Biol. 369, 1015-1028 (2007) y documento US20040132028A1.
La fibronectina es un armazón que puede modificarse por ingeniería genética para unirse a un antígeno. La adnectina consiste en una cadena principal de la secuencia de aminoácidos natural del 10° dominio de las 15 unidades repetidas de fibronectina humana de tipo III (FN3). Tres bucles en un extremo del sándwich p pueden modificarse por ingeniería genética para permitir que una adnectina reconozca específicamente una diana terapéutica de interés. Para detalles adicionales véase Protein Eng. Des. Sel. 18, 435-444 (2005), documentos US20080139791, WO2005056764 y US6818418B1.
Los aptámeros peptídicos son moléculas de reconocimiento combinatorias que consisten en una proteína de armazón constante, típicamente tiorredoxina (TrxA) que contiene un bucle peptídico variable restringido insertado en el sitio activo. Para detalles adicionales véase Expert Opin. Biol. Ther. 5, 783-797 (2005).
Los microcuerpos derivan de microproteínas de origen natural de 25-50 aminoácidos de longitud que contienen 3-4 puentes de cisteína, los ejemplos de microproteínas incluyen KalataB1 y conotoxina y notinas. Las microproteínas tienen un bucle que puede modificarse por ingeniería genética para incluir hasta 25 aminoácidos sin afectar al plegamiento global de la microproteína. Para detalles adicionales de dominios de notina modificados por ingeniería genética, véase documento WO2008098796.
Otros dominios de unión a epítopo incluyen proteínas que se han usado como un armazón para modificar por ingeniería genética diferentes propiedades de unión de antígeno diana, incluyen cristalina y humana y ubiquitina humana (afilinas), dominios de tipo kunitz de inhibidores de proteasa humana, dominios PDZ de la proteína de unión a Ras AF-6, toxinas de escorpión (caribdotoxina), dominio de lectinas de tipo C (tetranectinas) se revisan en el Capítulo 7 - Non-Antibody Scaffolds de Handbook of Therapeutic Antibodies (2007, editado por Stefan Dubel) y Protein Science 15:14-27 (2006). Los dominios de unión de epítopos de la presente invención podrían derivar de cualquiera de estos dominios proteicos alternativos.
Como se usa en el presente documento, la expresión “sitio de unión a antígeno” se refiere a un sitio en una proteína que es capaz de unirse específicamente a antígeno, esto puede ser un dominio sencillo, por ejemplo un dominio de unión a epítopo, o pueden ser dominios emparejados VH/VL como se encuentran en un anticuerpo convencional. En algunas realizaciones de la invención los dominios Fv de cadena sencilla (ScFv) pueden proporcionar sitios de unión a antígeno.
Los términos “mAbdAb” y dAbmAb” se usan en el presente documento para referirse a propiedades de unión a antígeno de la presente invención. Los dos términos pueden usarse de forma intercambiable y se pretende que tengan el mismo significado como se usan en el presente documento.
La expresión “proteína de unión a antígeno” como se usa en el presente documento se refiere a anticuerpos, fragmentos de anticuerpo por ejemplo un anticuerpo de dominio (dAb), ScFv, Fab, Fab2, y otras construcciones proteicas. Las moléculas de unión a antígeno pueden comprender al menos un dominio variable de Ig, por ejemplo anticuerpos, anticuerpos de dominio (dAb), Fab, Fab’, F(ab’)2, Fv, ScFv, diacuerpos, mAbdAb, affibodies, anticuerpos heteroconjugados o anticuerpos biespecíficos. En una realización la molécula de unión a antígeno es un anticuerpo. En otra realización la molécula de unión a antígeno es un dAb, es decir un dominio variable sencillo de inmunoglobulina tal como un VH, VHH o VL que se une específicamente a un antígeno o epítopo independientemente de un domino o región V diferente. Las moléculas de unión a antígeno pueden ser capaces de unirse a dos dianas, es decir pueden ser proteínas de dirección doble. Las moléculas de unión a antígeno pueden ser una combinación de anticuerpos y fragmentos de unión a antígeno tales como por ejemplo, uno o más anticuerpos de dominio y/o uno o más ScFv ligados a un anticuerpo monoclonal. Las moléculas de unión a antígeno también pueden comprender un dominio no de Ig por ejemplo un dominio que sea un derivado de un armazón seleccionado del grupo que consiste en CTLA-4 (Evibody); lipocalina; moléculas derivadas de Proteína A tales como dominio Z de Proteína A (Affibody, SpA), dominio A (Avimer/Maxibody); proteínas de choque Térmico tales como GroEl y GroES; peroxidasa μg (transcuerpo); proteína de repetición de anquirina (DARPin); aptámero peptídico; dominio de lectina de tipo C (Tetranectina); cristalina y humana y ubiquitina humana (afilinas); dominios PDZ; dominios de tipo toxina kunitz de escorpión de inhibidores de proteasa humana; y fibronectina (adnectina); que se ha sometido a ingeniería proteica para obtener unión a OSM. Como se usa en el presente documento “proteína de unión a antígeno” será capaz de antagonizar y/o neutralizar OSM
humano. Además, una proteína de unión a antígeno puede inhibir y/o bloquear la actividad de OSM uniéndose a OSM y evitando que un ligando natural se una y/o active el receptor gp130.
La expresión “Función Efectora” como se usa en el presente documento pretende referirse a uno o más de actividad citotóxica mediada por células dependiente de anticuerpo (ADCC), respuestas mediadas por actividad citotóxica dependiente de complemento (CDC), fagocitosis mediada por Fc y reciclaje de anticuerpos mediante el receptor FcRn. Para anticuerpos de IgG, las funcionalidades efectoras que incluyen ADCC y ADCP están mediadas por interacción de la región constante de cadena pesada con una familia de receptores de Fcy presentes en la superficie de células inmunes. En seres humanos estos incluyen FcyRI (CD64), FcyRIi (CD32) y FcyRIII (CD16). La interacción entre la proteína de unión a antígeno unida al antígeno y la formación de complejos Fc/ Fcy induce una serie de efectos incluyendo citotoxicidad, activación de células inmunes, fagocitosis y liberación de citocinas inflamatorias.
Se cree que la interacción entre la región constante de una proteína de unión a antígeno y diversos receptores de Fc (FcR) median en las funciones efectoras de la proteína de unión a antígeno. Efectos biológicos significativos pueden ser una consecuencia de la funcionalidad efectora, en particular, citotoxicidad celular dependiente de anticuerpo (ADCC), fijación de complemento (citotoxicidad dependiente de complemento o CDC) y semivida/eliminación de la proteína de unión a antígeno. Habitualmente, la capacidad para mediar en la función efectora requiere unión de la proteína de unión a antígeno con un antígeno y no todas las proteínas de unión a antígeno mediarán en cada función efectora.
La función efectora puede medirse de varias maneras incluyendo por ejemplo mediante unión del FcyRIII con linfocíticos Citolíticos Naturales o mediante FcyRI con monocitos/macrófagos para medir con respecto a función efectora ADCC. Por ejemplo una proteína de unión a antígeno de la presente invención puede evaluarse con respecto a función efectora ADCC en un ensayo de linfocitos Citolíticos Naturales. Pueden encontrarse ejemplos de tales ensayos en Shields y col, 2001 The Journal of Biological Chemistry, Vol. 276, p 6591-6604; Chappel y col, 1993 The Journal of Biological Chemistry, Vol 268, p 25124-25131; Lazar y col, 2006 p Na S, 103; 4005-4010.
Los ejemplos de ensayos para determinar función CDC incluyen los descritos en 1995 J Imm Meth 184: 29-38.
Algunos isotipos de regiones constantes humanas, en particular isotipos de IgG4 e IgG2, esencialmente carecen de las funciones de a) activación de complemento por la ruta clásica; y b) citotoxicidad celular dependiente de anticuerpo. Pueden llevarse a cabo diversas modificaciones de la región constante de cadena pesada de proteínas de unión a antígeno dependiendo de la propiedad efectora deseada. Se ha descrito de forma separada que las regiones constantes de IgG1 que contienen mutaciones específicas reducen la unión a receptores de Fc y por lo tanto reducen ADCC y CDC (Duncan y col. Nature 1988, 332; 563-564; Lund y col. J. Immunol. 1991, 147; 2657-2662; Chappel y col. PNAS 1991,88; 9036-9040; Burton y Woof, Adv. Immunol. 1992, 51; 1 -84; Morgan y col., Immunology 1995, 86; 319-324; Hezareh y col., J. Virol. 2001,75 (24); 12161-12168).
En una realización de la presente invención se proporciona una proteína de unión a antígeno que comprende una región constante de modo que la proteína de unión a antígeno tiene ADCC reducida y/o activación del complemento o funcionalidad efectora. En una realización tal la región constante de cadena pesada puede comprender una región constante deshabilitada de forma natural de isotipo IgG2 o IgG4 o una región constante de IgG1 mutada. Se describen ejemplos de modificaciones adecuadas en el documento EP0307434. Un ejemplo comprende las sustituciones de restos de alanina en las posiciones 235 y 237 (numeración de índice de EU).
También se ha descrito que las regiones constantes de IgG1 humana que contienen mutaciones específicas o glucosilación alterada en el resto Asn297 potencian la unión con receptores de Fc. En algunos casos también se ha mostrado que estas mutaciones potencian ADCC y CDC (Lazar y col. PNAS 2006, 103; 4005-4010; Shields y col. J Biol Chem 2001,276; 6591-6604; Nechansky y col. Mol Immunol, 2007, 44; 1815-1817).
En una realización de la presente invención, tales mutaciones están en una o más de las posiciones seleccionadas de 239, 332 y 330 (IgG1), o las posiciones equivalentes en otros isotipos de IgG. Los ejemplos de mutaciones adecuadas son S239D, I332E y A330L. En una realización la proteína de unión a antígeno de la invención descrita en el presente documento se muta en las posiciones 239 y 332, por ejemplo S239D e I332E o en una realización adicional está mutada en tres o más posiciones seleccionadas de 239, 332 y 330, por ejemplo S239D, I332E y A330L (numeración de índice de EU).
En una realización alternativa de la presente invención, se proporciona una proteína de unión a antígeno que comprende una región constante de cadena pesada con un perfil de glucosilación alterado de modo que la proteína de unión a antígeno tiene función efectora potenciada. Por ejemplo, teniendo la proteína de unión a antígeno ADCC potenciada o CDC potenciada o teniendo función efectora tanto ADCC como CDC potenciada. Se describen ejemplos de metodologías adecuadas para producir proteínas de unión a antígeno con un perfil de glucosilación alterado en los documentos WO2003011878, WO2006014679 y EP1229125, todos los cuales pueden aplicarse a las proteínas de unión a antígeno de la presente invención.
La presente invención también proporciona un procedimiento para la producción de una proteína de unión a antígeno de acuerdo con la invención que comprende las etapas de:
a) cultivar una célula huésped recombinante que comprende un vector de expresión que comprende el ácido nucleico aislado como se describe en el presente documento, en la que el gen FUT8 que codifica alfa-1,6-fucosiltransferasa se ha inactivado en la célula huésped recombinante; y
b) recuperar la proteína de unión a antígeno.
Tales procedimientos para la producción de proteínas de unión a antígeno pueden realizarse, por ejemplo, usando el sistema de tecnología POTELLIGENT™ disponible de BioWa, Inc. (Princeton, NJ) en el que CHOK1SV que carecen de una copia funcional del gen FUT8 producen anticuerpos monoclonales que tienen actividad de citotoxicidad mediada por células dependiente de anticuerpo (ADCC) potenciada que aumenta en relación con un anticuerpo monoclonal idéntico producido en una célula con un gen FUT8 funcional. Se describen aspectos del sistema de tecnología POTELLIGENT™ en los documentos US7214775, US6946292, WO0061739 y WO0231240 todos los cuales se incorporan en el presente documento por referencia. Los expertos habituales en la materia también reconocerán otros sistemas apropiados.
En una realización de la presente invención se proporciona una proteína de unión a antígeno que comprende una región constante de cadena pesada quimérica, por ejemplo una proteína de unión a antígeno que comprende una región constante de cadena pesada quimérica con al menos un dominio CH2 de IgG3 de modo que la proteína de unión a antígeno tenga función efectora potenciada, por ejemplo en la que tiene ADCC potenciada o CDC potenciada, o funciones ADCC y CDC potenciadas. En una realización tal, la proteína de unión a antígeno puede comprender un domino CH2 de IgG3 o ambos dominios CH2 pueden ser de IgG3.
También se proporciona un procedimiento para producir una proteína de unión a antígeno de acuerdo con la invención que comprende las etapas de:
a) cultivar una célula huésped recombinante que comprende un vector de expresión que comprende un ácido nucleico aislado como se ha descrito en el presente documento en la que el vector de expresión comprende una secuencia de ácido nucleico que codifica un dominio Fc que tiene restos de aminoácidos de dominios Fc tanto de IgG1 como de IgG3; y
b) recuperar la proteína de unión a antígeno.
Tales procedimientos para la producción de proteínas de unión a antígeno pueden realizarse, por ejemplo, usando el sistema de tecnología COMPLEGENT™ disponible de BioWa, Inc. (Princeton, NJ) y Kyowa Hakko Kogyo (ahora, Kyowa Hakko Kirin Co., Ltd.) Co., Ltd. en el que una célula huésped recombinante que comprende un vector de expresión en el que se expresa una secuencia de ácido nucleico que codifica un domino Fc quimérico que tiene restos de aminoácidos de dominio de Fc tanto de IgG1 como de IgG3 para producir una proteína de unión a antígeno que tiene actividad de citotoxicidad dependiente de complemento (CDC) potenciada que aumenta en relación con una proteína de unión a antígeno idéntica en lo demás que carece de un domino de Fc quimérico tal. Se describen aspectos del sistema de tecnología COMPLEGENT™ en los documentos WO2007011041 y US20070148165 cada uno de los cuales se incorpora en el presente documento por referencia. En una realización alternativa la actividad CDC puede aumentarse introduciendo mutaciones específicas de secuencia en la región Fc de una cadena IgG. Los expertos en la materia también reconocerán otros sistemas apropiados.
Resultará evidente para los expertos en la materia que tales modificaciones pueden no solamente usarse solas sino que pueden usarse en combinación entre sí para potenciar adicionalmente la función efectora.
En una realización tal de la presente invención se proporciona una proteína de unión a antígeno que comprende una región constante de cadena pesada que comprende una región constante de cadena pesada mutada y quimérica por ejemplo comprendiendo una proteína de unión a antígeno al menos un dominio CH2 de IgG3 y un dominio CH2 de IgG1, en la que el dominio c H2 de IgG 1 tiene una o más mutaciones en posiciones seleccionadas de 239, 332 y 330 (por ejemplo las mutaciones pueden seleccionarse de S239D, I332E y A330L) de modo que la proteína de unión a antígeno tiene función efectora potenciada, por ejemplo en la que tiene una o más de las siguientes funciones, ADCC potenciada o CDC potenciada, por ejemplo en la que tiene ADCC potenciada y CDC potenciada. En una realización el dominio CH2 de IgG1 tiene las mutaciones S239D e I332E.
En una realización alternativa de la presente invención se proporciona una proteína de unión a antígeno que comprende una región constante de cadena pesada quimérica y que tiene un perfil de glucosilación alterado. En una realización tal la región constante de cadena pesada comprende al menos un dominio CH2 de IgG3 y un dominio CH2 de IgG1 y tiene un perfil de glucosilación alterado de modo que la relación de fucosa y manosa es 0,8:3 o menos, por ejemplo siendo la proteína de unión a antígeno desfucosilada de modo que dicha proteína de unión a antígeno tenga
una función efectora potenciada en comparación con una proteína de unión a antígeno equivalente con una región constante de cadena pesada de inmunoglobulina que carece de dichas mutaciones y perfil de glucosilación alterado, por ejemplo en el que tiene una o más de las siguientes funciones, ADCC potenciada o CDC potenciada, por ejemplo en la que tiene ADCC potenciada y CDC potenciada.
En una realización alternativa la proteína de unión a antígeno tiene al menos un dominio CH2 de IgG3 y al menos un dominio constante de cadena pesada de IgG1 estando ambos dominios CH2 de IgG mutados de acuerdo con las limitaciones descritas en el presente documento.
En un aspecto de la invención se proporciona un procedimiento para producir una proteína de unión a antígeno de acuerdo con la invención descrita en el presente documento que comprende las etapas de:
a) cultivar una célula huésped recombinante que contenga un vector de expresión que contenga un ácido nucleico aislado como se describe en el presente documento, comprendiendo adicionalmente dicho vector de expresión una secuencia de ácido nucleico de Fc que codifica un dominio Fc quimérico que tiene restos de aminoácidos de dominio Fc tanto de IgG1 como de IgG3 y en la que el gen FUT8 que codifica alfa-1,6-fucosiltransferasa se ha inactivado en la célula huésped recombinante; y
b) recuperar la proteína de unión a antígeno.
Tales procedimientos para la producción de proteínas de unión a antígeno pueden realizarse, por ejemplo, usando el sistema de tecnología ACCRETAMAB™ disponible de BioWa, Inc. (Princeton, NJ) que combina los sistemas de tecnología POTELLIGENT™ y COMPLEGENT™ para producir una proteína de unión a antígeno que tenga actividad potenciada tanto ADCC como CDC que aumenta en relación con un anticuerpo monoclonal idéntico en lo demás, que carezca de un dominio Fc quimérico y que tenga fucosa en el oligosacáridos.
En otra realización más de la presente invención se proporciona una proteína de unión a antígeno que comprende una región constante de cadena pesada mutada y quimérica teniendo dicha proteína de unión a antígeno un perfil de glucosilación alterado de modo que la proteína de unión a antígeno tiene función efectora potenciada, por ejemplo teniendo una o más de las siguientes funciones, ADCC potenciada o CDC potenciada. En una realización las mutaciones se seleccionan de las posiciones 239, 332 y 330, por ejemplo las mutaciones se seleccionan de S239D, I332E y A330L. En una realización adicional la región constante de cadena pesada comprende al menos un dominio CH2 de IgG3 y un domino CH2 de IgG1. En una realización la región constante de cadena pesada tiene un perfil de glucosilación alterado de modo que la relación de fucosa y manosa es de 0,8:3 o menos, por ejemplo la proteína de unión a antígeno está desfucosilada, de modo que dicha proteína de unión a antígeno tiene una función efectora potenciada en comparación con una proteína de unión a antígeno no quimérica equivalente o con una región constante de cadena pesada de inmunoglobulina que carece de dichas mutaciones y perfil de glucosilación alterado.
Inmunoconjugados
También se proporciona un inmunoconjugado (denominado de forma intercambiable “conjugados anticuerpo-fármaco” o “ADC”) que comprende una proteína de unión a antígeno de acuerdo con la invención como se describe en el presente documento incluyendo, pero sin limitación, un anticuerpo conjugado con uno o más agentes citotóxicos, tales como un agente quimioterapéutico, un fármaco, un agente inhibidor del crecimiento, una toxina (por ejemplo, una toxina proteica, una toxina enzimáticamente activa de origen bacteriano, fúngico, vegetal o animal, o fragmentos de la misma) o un isótopo radioactivo (es decir, un radioconjugado).
Se han usado inmunoconjugados para el suministro local de agentes citotóxicos, es decir, fármacos que matan o inhiben el crecimiento o proliferación de células, en el tratamiento de cáncer (Lambert, J. (2005) Curr. Opinion in Pharmacology 5: 543-549; Wu y col. (2005) Nature Biotechnology 23(9):1137-1146; Payne, G. (2003) i 3:207-212; Syrigos y Epenetos (1999) Anticancer Research 19: 605-614; Niculescu-Duvaz y Springer (1997) Adv. Drug Deliv. Rev. 26:151-172; Patente de Estados Unidos N° 4.975.278). Los inmunoconjugados posibilitan el suministro dirigido de un resto farmacológico a un tumor, y la acumulación intracelular en el mismo, en el que la administración sistémica de fármacos no conjugados puede dar como resultado niveles inaceptables de toxicidad para células normales así como para las células tumorales que se busca eliminar (Baldwin y col., Lancet (Mar. 15, 1986) pp. 603-05; Thorpe (1985) "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review," en Monoclonal Antibodies '84: Biological And Clinical Applications (A. Pinchera y col., eds) pp. 475-506. Se ha indicado que tanto los anticuerpos policlonales como los anticuerpos monoclonales son útiles en estas estrategias (Rowland y col., (1986) Cancer Immunol. Immunother. 21: 183-87). Los fármacos usados en estos procedimientos incluyen daunomicina, doxorrubicina, metotrexato y vindesina (Rowland y col., (1986) mencionado anteriormente). Las toxinas usadas en conjugados de anticuerpo-toxina incluyen toxinas bacterianas tales como toxina diftérica, toxinas vegetales tales como ricina, toxinas de moléculas pequeñas tales como geldanamicina (Mandler y col (2000) J. Nat. Cancer Inst. 92(19): 1573-1581; Mandler y col (2000) Bioorganic & Med. Chem. Letters 10: 1025-1028; Mandler y col (2002) Bioconjugate Chem. 13: 786-791), maitansinoides (documento EP 1391213; Liu y col., (1996) Proc. Natl. Acad. Sci. USA 93: 8618-8623), y
caliqueamicina (Lode y col (1998) Cáncer Res. 58: 2928; Hinman y col (1993) Cáncer Res. 53: 3336-3342).
En una realización, la presente invención incluye inmunoconjugados que tienen la siguiente estructura general:
ABP - ((Engarce)n - Ctx)m
En la que ABP es una proteína de unión a antígeno
El enlazador está ausente o es cualquier engarce escindible o no escindible descrito en el presente documento
Ctx es cualquier agente citotóxico descrito en el presente documento
n es 0, 1,2 o 3 y
m es 1,2, 3, 4, 5, 6, 7, 8, 9 o 10.
Se representan ejemplos de anticuerpos ligados con un engarce MC con auristatinas tales como MMAE y MMAF en la siguiente estructura:

En ciertas realizaciones, un inmunoconjugado comprende una proteína de unión a antígeno, incluyendo pero sin limitación, un anticuerpo y un agente quimioterapéutico u otra toxina. Se describen en el presente documento agentes quimioterapéuticos útiles en la generación de inmunoconjugados. Las toxinas enzimáticamente activas y fragmentos de los mismos que pueden usarse incluyen cadena A de difteria, fragmentos activos que no se unen de la toxina diftérica, cadena A de exotoxina (de Pseudomonas aeruginosa), cadena A de ricina, cadena A de abrina, cadena A de modeccina, alfa-sarcina, proteínas de Aleurites fordii, proteínas de diantina, proteínas de Phytolacca americana, curcina, crotina, inhibidor de Saponaria officinalis, gelonina, mitogelina, restrictocina, fenomicina, neomicina y los tricotecenos. Véase, por ejemplo, documento WO 93/21232 publicado el 28 de octubre de 1993. Está disponible una diversidad de radionúclidos para la producción de anticuerpos radioconjugados. Los ejemplos incluyen 211At- 212Bi, 131I, 131In, 90Y y 186Re.
Las proteínas de unión a antígeno de la presente invención también pueden conjugarse con una o más toxinas, incluyendo, pero sin limitación, una caliqueamicina, maitansinoides, dolastatinas, aurostatinas, un tricoteceno y CC1065, y los derivados de estas toxinas que tienen actividad tóxica. Los agentes citotóxicos adecuados incluyen, pero sin limitación, una auristatina incluyendo dovalina-valina-dolaisoleunina-dolaproina-fenilalanina (MMAF) y monometil auristatina E (MMAE) así como formas estéricas de MMAE, un agente de unión al surco menor del ADN, un agente alquilante del surco menor del ADN, una enediina, una lexitropsina, una duocarmicina, un taxano, incluyendo paclitaxel y docetaxel, una puromicina, una dolastatina, un maitansinoide y un alcaloide de la vinca. Los agentes citotóxicos específicos incluyen topotecan, morfolino-doxorrubicina, rizoxina, cianomorfolino-doxorrubicina, dolastatina-10, equinomicina, combretastatina, caliqueamicina, maitansina, DM-1, DM-4, netropsina. Otros agentes
citotóxicos adecuados incluyen agentes antitubulina, tales como una auristatina, un alcaloide de la vinca, una podofilotoxina, un taxano, un derivado de bacatina, una criptofisina, un maitansinoide, una combretastatina o una dolastatina. Los agentes antitubulina incluyen dimetilvalina-valina-dolaisoleucina-dolaproina-fenilalanina-pfenilendiamina (AFP), MMAF, MMAE, auristatina E, vincristina, vinblastina, vindesina, vinorelbina, VP-16, camptotecina, paclitaxel, docetaxel, epotilona A, epotilona B, nocodazol, colchicinas, colcimida, estramustina, cemadotina, discodermolida, maitansina, DM-1, DM-4 o eleuterobina.
Se produjeron conjugados de fármaco con anticuerpo conjugando el agente antitubulina de moléculas pequeñas monometil auristatina E (MMAE) o monometil auristatina F (MMAF) con los anticuerpos. En el caso de MMAE el engarce consiste en una maleimida sensible a tiol, un espaciador de caproilo, el dipéptido valina-citrulina y paminobenciloxicarbonilo, un grupo de fragmentación autodegradante. En el caso de MMAf se usa un engarce de maleimidocaproilo resistente a proteasa. El procedimiento de conjugación conduce a heterogeneidad en la unión de fármaco-anticuerpo, variando tanto el número de fármacos unido a cada molécula de anticuerpo (relación molar [MR]), como el sito de unión. La especie más prevalente es el material con una MR = 4; menos prevalentes son materiales con MR de 0, 2, 6 y 8. La m R de fármaco y anticuerpo media global es aproximadamente 4.
Producción de inmunoconjugados
Los puntos de unión son cisteínas producidas por reducción leve de los disulfuros intercatenarios del anticuerpo que se lleva a cabo mientras los anticuerpos se inmovilizan en resina de afinidad de Proteína G (permitiendo de este modo el uso de grandes excesos de reactivos sin purificaciones intermedias). Mientras esté inmovilizado, un gran exceso de TCEP reducirá completamente los disulfuros intercatenarios pero no tiene impacto sobre la unión del anticuerpo con la resina.
El número de tioles por anticuerpo generados por este procedimiento depende de la fuente e isotipo de los anticuerpos. Por ejemplo, los IgG1 humanos (y quiméricos ratón-humano) tienen 4 disulfuros reducibles y por lo tanto generan 8 tioles tras su reducción completa, mientras que los IgG1 murinos tienen 5 disulfuros reducibles y producen 10 tioles. Si se desean ADC con la máxima carga farmacológica (por ejemplo, 10 fármacos por anticuerpo para las IgG1 murinas), entonces puede añadirse simplemente el engarce de maleimido-fármaco a los anticuerpos inmovilizados en exceso suficiente para asegurar la conjugación completa. Sin embargo, también pueden prepararse ADC con menos fármacos por anticuerpo a partir de anticuerpos completamente reducidos incluyendo un agente de protección biológicamente inerte tal como N-etilmaleimida (NEM) que ocupa algunos de los tioles disponibles en el anticuerpo. Cuando se añade engarce de maleimido-fármaco y el agente de protección simultáneamente al anticuerpo completamente reducido y en gran exceso (al menos 3 veces), los dos electrófilos de maleimida compiten por el número limitante de tioles disponibles. De esta manera, la carga farmacológica se determina por las tasas de reacción de tiol relativas del engarce-fármaco y agente de protección, y de este modo puede considerarse que están bajo control cinético. Las tasas de reacción relativas de engarces de maleimido-fármaco varían significativamente y de este modo la relación molar de fármaco-engarce con NEM presente en una mezcla de reacción deben determinarse de forma empírica para llegar a un panel de ADC con un nivel deseado de carga farmacológica. La fracción molar de los engarces farmacológicos SGD-1006 (vcMMAE) y SGD-1269 (mcMMAF) en mezclas de NEM que producen ADC con aproximadamente 4 fármacos por anticuerpo se resumen en la Tabla 2 para isotipos de IgG humano y murino habituales.
Auristatinas y dolastatinas
En algunas realizaciones, el inmunoconjugado comprende una proteína de unión a antígeno o anticuerpo conjugado con dolastatinas o análogos peptídicos de dolastatinas y derivados, las auristatinas (Patentes de Estados Unidos N° 5.635.483; 5.780.588). Se ha mostrado que las dolastatinas y auristatinas interfieren con dinámica de microtúbulos, hidrólisis de GTP y división nuclear y celular (Woyke y col. (2001) Antimicrob. Agents and Chemother. 45(12): 3580 3584) y tienen actividad antineoplásica (Patente de Estados Unidos N° 5.663.149) y antifúngica (Pettit y col. (1998) Antimicrob. Agents Chemother. 42: 2961 -2965). El resto farmacológico de dolastatina o auristatina (que son derivados pentapeptídicos de dolastatinas) puede unirse al anticuerpo a través del extremo N (amino) terminal o el C (carboxilo) terminal del resto farmacológico peptídico (documento WO 02/088172).
Las realizaciones de auristatina ejemplares incluyen los restos farmacológicos de monometil auristatina ligados en extremo N terminal DE y DF, desvelados en “Monomethylvaline Compounds Capable of Conjugation to Ligands,” Patente de Estados Unidos N° 7.498.298, la divulgación de la cual se incorpora expresamente por referencia en su totalidad. Como se usa en el presente documento, la abreviatura “MMAE” se refiere a monometil auristatina E. Como se usa en el presente documento la abreviatura “MMAF” se refiere a dovalina-valina-dolaisoleucina-dilaproinafenilalanina.
Típicamente, pueden prepararse restos farmacológicos basados en péptidos formando un enlace peptídico entre dos o más aminoácidos y/o fragmentos peptídicos. Tales enlaces peptídicos pueden prepararse, por ejemplo, de acuerdo con el procedimiento de síntesis de fase líquida (véase E. Schroder y K. Lubke, “The Peptides,” volumen 1, pp 76-136,
1965, Academic Press) que se conoce bien en el campo de la química peptídica. Los restos farmacológicos de auristatina/dolastatina pueden prepararse de acuerdo con los procedimientos de la Patente de Estados Unidos N° 5.635.483; Patente de Estados Unidos N° 5.780.588; Pettit y col. (1989) J. Am. Chem. Soc. 111: 5463-5465; Pettit y col. (1998) Anti-Cancer Drug Design 13:243-277; Pettit, G. R., y col. Synthesis, 1996, 719-725; y Pettit y col. (1996) J. Chem. Soc. Perkin Trans. 15:859-863. Véase también Doronina (2003) Nat Biotechnol 21(7): 778-784; “Monomethylvaline Compounds Capable of Conjugation to Ligands”, Patente de Estados Unidos N° 7.498.298, presentada el 5 de noviembre de 2004, que se incorpora por la presente por referencia en su totalidad (que desvela, por ejemplo, engarces y procedimientos para preparar compuestos de monometil valina tales como MMAE y MMAF conjugados con engarces). Se desvelan compuestos biológicamente activos orgánicos que actúan como agentes citotóxicos, específicamente pentapéptidos, en las Patentes de Estados Unidos N° 6.884.869; 7.498.298; 7.098.308; 7.256.257; y 7.423.116. Se describen anticuerpos monoclonales ligados con MMAE y MMAF así como diversos derivados de auristatinas y procedimientos para prepararlos en la Patente de Estados Unidos N° 7.964.566.
Los ejemplos de auristatinas incluyen MMAE y MMAF, las estructuras de las cuales se muestran a continuación:

Maitansina y maitansinoides
Los maitansinoides son inhibidores mitóticos que actúan inhibiendo la polimerización de tubulinas. Se aisló en primer lugar maitansina del arbusto Africano oriental Maytenus serrata (Patente de Estados Unidos N° 3.896.111). Posteriormente, se descubrió que ciertos microbios también producen maitansinoides, tales como maitansinol y ésteres de maitansinol C-3 (Patente de Estados Unidos N° 4.151.042). Pueden prepararse fármacos maitansinoides altamente citotóxicos a partir de precursores de ansamitocina producidos por fermentación de microorganismos tales como Actinosinema. Se describen procedimientos para aislar ansamitocinas en la Patente de Estados Unidos N° 6.573.074. Se desvelan maitansinol sintético y derivados y análogos del mismo, por ejemplo, en las Patentes de Estados Unidos N° 4.137.230; 4.248.870; 4.256.746; 4.260.608; 4.265.814; 4.294.757; 4.307.016; 4.308.268; 4.308.269; 4.309.428; 4.313.946; 4.315.929; 4.317.821; 4.322.348; 4.331.598; 4.361.650; 4.364.866; 4.424.219; 4.450.254; 4.362.663; y 4.371.533.
Se preparan conjugados de anticuerpos-maitansinoide ligando químicamente un anticuerpo con una molécula de maitansinoide sin reducir significativamente la actividad biológica del anticuerpo ni de la molécula de maitansinoide. Véase, por ejemplo, Patente de Estados Unidos N° 5.208.020. Una media de 3-4 moléculas de maitansinoide conjugadas por molécula de anticuerpo ha mostrado eficacia en la potenciación de citotoxicidad de células diana sin afectar de forma negativa a la función o solubilidad del anticuerpo, incluso aunque se esperaría que una molécula de toxina/anticuerpo potenciara la citotoxicidad frente al uso de anticuerpo desnudo. Los maitansinoides se conocen bien en la técnica y pueden sintetizarse por técnicas conocidas o aislarse a partir de fuentes naturales. Se desvelan maitansinoides adecuados, por ejemplo, en la Patente de Estados Unidos N° 5.208.020 y en las otras patentes y publicaciones no de patente a las que se ha hecho referencia anteriormente en el presente documento. Los maitansinoides son maitansinol y análogos de maitansinol modificados en el anillo aromático o en otras posiciones de la molécula de maitansinol, tales como diversos ésteres de maitansinol. Se desvelan procedimientos para preparar maitansinoides para engarce con anticuerpos en las Patentes de Estados Unidos N° 6.570.024 y 6.884.874.
Caliqueamicina
La familia de caliqueamicina de antibióticos es capaz de producir roturas de ADN bicatenarias a concentraciones subpicomolares. Para la preparación de conjugados de la familia de caliqueamicina véase Patentes de Estados Unidos N° 5.712.374, 5.714.586, 5.739.116, 5.767.285, 5.770.701, 5.770.710, 5.773.001, 5.877.296 (todas de American Cyanamid Company). Los análogos estructurales de caliqueamicina que pueden usarse incluyen, pero sin limitación, gamma.11, alfa.2I, .alfa.3I, N-acetil-gamma.11, PSAG y zeta.I1 (Hinman y col., Cancer Research 53: 3336-3342 (1993), Lode y col., Cancer Research 58: 2925-2928 (1998) y las patentes de Estados Unidos anteriormente mencionadas de American Cyanamid). Otro fármaco antitumoral con el que puede conjugarse el anticuerpo es QFA que es un antifolato. Tanto caliqueamicina como QFA tienen sitios intracelulares de acción y no cruzan fácilmente la membrana plasmática. Por lo tanto, la captación celular de estos agentes a través de internalización mediada por anticuerpo potencia en gran medida sus efectos citotóxicos.
Otros agentes citotóxicos
Otros agentes antitumorales que pueden conjugarse con los anticuerpos incluyen BCNU, estreptozocina, vincristina y 5-fluorouracilo, la familia de agentes conocida colectivamente como complejo LL-E33288 descrita en las Patentes de Estados Unidos N° 5.053.394, 5.770.710, así como esperamicinasa (Patente de Estados Unidos N° 5.877.296).
Las toxinas enzimáticamente activas y fragmentos de las mismas que pueden usarse incluyen cadena A de difteria, fragmentos activos sin unión de toxina diftérica, cadena A de exotoxina (de Pseudomonas aeruginosa), cadena A de ricina, cadena A de abrina, cadena A de modeccina, alfa-sarcina, proteínas de Aleurites fordii, proteínas de diantina, proteínas de Phytolacca americana (PAPI, PAPII y PAP-S), inhibidor de Momordica charantia, curcina, crotina, inhibidor de Saponaria officinalis, gelonina, mitogelina, restrictocina, fenomicina, neomicina y los tricotecenos. Véase, por ejemplo, documento WO 93/21232 publicado el 28 de octubre de 1993.
La presente invención contempla adicionalmente un inmunoconjugado formado entre un anticuerpo y un compuesto con actividad nucleolítica (por ejemplo, una ribonucleasa o una endonucleasa de ADN tal como una desoxirribonucleasa; DNasa).
Para destrucción selectiva del tumor, el anticuerpo puede comprender un átomo altamente radiactivo. Está disponible una diversidad de isótopos radiactivos para la producción de anticuerpos radioconjugados. Los ejemplos incluyen At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32, Pb212 e isótopos radiactivos de Lu. Cuando el conjugado se usa para detección, este puede comprender un átomo radiactivo para estudios escintigráficos, por ejemplo tc99m o I123, o un marcador de espín para imágenes de resonancia magnética nuclear (RMN) (también conocida como imagen de resonancia magnética, mri), tal como yodo-123 de nuevo, yodo-131, indio-111, flúor-19, carbono-13, nitrógeno-15, oxígeno-17, gadolinio, manganeso o hierro.
Los marcadores radiactivos o de otro tipo pueden incorporarse en el conjugado de maneras conocidas. Por ejemplo, el péptido puede biosintetizarse o puede sintetizarse por síntesis de aminoácidos químicos usando precursores de aminoácidos adecuados que implican, por ejemplo, flúor 19 en lugar de hidrógeno. Pueden unirse marcadores tales como tc99m o I123, Re186, Re188 e In111 mediante un resto de cisteína en el péptido. Puede unirse Itrio-90 mediante un resto de lisina. Puede usarse el procedimiento de IODOGEN (Fraker y col. (1978) Biochem. Biophys. Res. Commun.
80: 49-57) para incorporar yodo-123. “Monoclonal Antibodies in Immunoscintigraphy” (Chatal, CRC Press 1989) describe otros procedimientos en detalle.
Preparación de ADC
En los conjugados de anticuerpo con fármaco, el anticuerpo puede conjugarse directamente con el agente citotóxico o mediante un engarce. Los engarces adecuados incluyen, por ejemplo, engarces escindibles y no escindibles. Un engarce escindible es típicamente susceptible de escisión en condiciones intracelulares. Los engarces escindibles adecuados incluyen, por ejemplo, un engarce peptídico escindible por una proteasa intracelular, tal como proteasa lisosomal o una proteasa endosomal. En realizaciones ejemplares, el engarce puede ser un engarce dipeptídico, tal como un engarce valina-citrulina (val-cit) o fenilalanina-lisina (phe-lys). Otros engarces adecuados incluyen engarces hidrolizables a un pH de menos de 5,5 tales como un engarce de hidrazona. Los engarces escindibles adecuados adicionales incluyen engarces de disulfuro.
Bristol-Myers Squibb ha descrito conjugados de fármacos antitumorales escindibles por enzima lisosomal particulares. Véase, por ejemplo, Patente de Estados Unidos N° 6.214.345. Seattle Genetics ha publicado la solicitud de Patente de Estados Unidos 2003/0096743 y solicitud de Patente de Estados Unidos 2003/0130189, que describen paminobenciléteres en agentes de suministro de fármacos. Los engarces descritos en estas solicitudes se limitan a composiciones de aminobenciléter.
Pueden prepararse conjugados de la proteína de unión a antígeno y el agente citotóxico usando una diversidad de agentes de acoplamiento de proteínas bifuncionales tales como N-succinimidil-3-(2-piridilditio)propionato (SPDP), succinimidil-4-(N-maleimidometil) ciclohexano-1-carboxilato (SMCC), iminotiolano (IT), derivados bifuncionales de
imidoésteres (tales como dimetil adipimidato HCl), ásteres activos (tales como disuccinimidil suberato), aldehidos (tales como glutaraldehído), compuestos de bis-azido (tales como bis(p-azidobenzoil) hexanodiamina), derivados de bis-diazonio (tales como bis-(p-diazoniobenzoil)-etilendiamina), diisocianatos (tales como tolueno 2,6-diisocianato), y compuestos de flúor bis-activo (tales como 1,5-difluoro-2,4-dinitrobenceno).
Adicionalmente el engarce puede estar compuesto de uno o más componentes de engarce. Los componentes de engarce ejemplares incluyen 6-maleimidocaproilo (“MC”), maleimidopropanoil (“MP”), valina-citrulina (“val-cit”), alanina-fenilalanina (“ala-phe”), p-aminobenciloxicarbonilo (“PAB”), N-Succinimidil 4-(2-piridiltio)pentanoato (“SPP”), N-Succinimidil 4-(N-maleimidometil)ciclohexano-1 carboxilato (“SMCC”), y N-Succinimidil (4-yodoacetil)aminobenzoato (“SIAB”). Se conocen en la técnica componentes de engarce adicionales y algunos se describen en el presente documento. Véase también “Monomethylvaline Compounds Capable of Conjugation to Ligands,” Patente de Estados Unidos N° US 7.498.298, presentada el 5 de noviembre de 2004.
Los engarces también pueden comprender aminoácidos y/o análogos de aminoácidos. Los componentes de engarce aminoacidicos incluyen un dipéptido, un tripéptido, un tetrapéptido o un pentapéptido. Los dipéptidos ejemplares incluyen: valina-citrulina (vc o val-cit), alanina-fenilalanina (af o ala-phe). Los tripéptidos ejemplares incluyen: glicinavalina-citrulina (gly-val-cit) y glicina-glicina-glicina (gly-gly-gly). Los restos de aminoácidos que comprenden un componente de engarce aminoacidico incluyen los de origen natural, asi como aminoácidos menores y análogos de aminoácidos de origen no natural, tales como citrulina. Pueden diseñarse componentes de engarce aminoacidicos y optimizarse en su selectividad para escisión enzimática por una enzima particular, por ejemplo, una proteasa asociada a tumor, catepsina B, C y D o una proteasa de plasmita.
Las proteinas de unión a antigeno y anticuerpos pueden hacerse reactivos para conjugación con reactivos de engarce. Los grupos nucleófilos en anticuerpos incluyen, pero sin limitación: (i) grupos de amina N-terminal, (ii) grupos de amina de cadena lateral, por ejemplo, lisina, (iii) grupos de tiol de cadena lateral, por ejemplo, cisteina y (iv) grupos amino o hidroxilo de azúcares en los que el anticuerpo está glucosilado. Los grupos de amina, tiol e hidroxilo son nucleófilos y capaces de reaccionar para formar enlaces covalentes con grupos electrófilos en restos de engarce y reactivos de engarce incluyendo: (i) ésteres activos tales como ésteres de n Hs , ésteres de HOBt, haloformatos y haluros ácidos; (ii) alquil y bencil haluros tales como haloacetamidas; (iii) grupos de aldehidos, cetonas, carboxilo y maleimida. Ciertos anticuerpos tienen disulfuros intercatenarios reducibles, es decir puentes de cisteina. Los anticuerpos pueden hacerse reactivos para conjugación con reactivos de engarce por tratamiento con un agente reductor tal como DTT (ditiotreitol). Cada puente de cisteina formará por lo tanto, teóricamente, dos nucleófilos de tiol reactivos. Pueden introducirse grupos nucleófilos adicionales en anticuerpos a través de la reacción de lisinas con 2-iminotiolano (reactivo de Traut) dando como resultado conversión de una amina en un tiol. Pueden introducirse grupos de tiol reactivos en el anticuerpo (o fragmento del mismo) introduciendo uno, dos, tres, cuatro o más restos de cisteina (por ejemplo, preparando anticuerpos mutantes que comprenden uno o más restos de aminoácido cisteina no nativos).
Las proteinas de unión a antigenos y anticuerpos también pueden modificarse para introducir restos electrófilos, que pueden reaccionar con sustituyentes nucleófilos en el reactivo o fármaco de engarce. Los azúcares de anticuerpos glucosilados pueden oxidarse, por ejemplo, con reactivos oxidantes de peryodato, para formar aldehido o grupos de cetona que pueden reaccionar con el grupo amina de reactivos de engarce o restos farmacológicos. Los grupos base de Schiff de imina resultantes pueden formar un engarce estable, o pueden reducirse, por ejemplo, por reactivos de borohidruro para formar engarces de amina estables. En una realización, la reacción de la parte de carbohidrato de un anticuerpo glucosilado con galactosa oxidasa o metaperyodato de sodio puede producir grupos carbonilo (aldehido y cetona) en la proteina que pueden reaccionar con grupos apropiados en el fármaco (Hermanson, Bioconjugate Techniques). En otra realización, las proteinas que contienen restos de serina o treonina N terminales pueden reaccionar con metaperyodato sódico, dando como resultado producción de un aldehido en lugar del primer aminoácido (Geoghegan & Stroh, (1992) Bioconjugate Chem. 3: 138-146; Patente de Estados Unidos N° 5.362.852). Tales aldehidos pueden reaccionar con un resto farmacológico o nucleófilo de engarce.
Los grupos nucleófilos en un resto farmacológico incluyen, pero sin limitación: amina, tiol, hidroxilo, hidracida, oxima, hidracina, tiosemicarbazona, carboxilato de hidracina y grupos de arilhidracida capaces de reaccionar para formar enlaces covalentes con grupos electrófilos en restos de engarce y reactivos de engarce que incluyen: (i) ésteres activos tales como ésteres de NHS, ésteres de HOBt, haloformatos y haluros ácidos; (ii) alquil y bencil haluros tales como haloacetamidas; (iii) grupos aldehidos, cetonas, carboxilo y maleimida.
En algunas realizaciones, el engarce es escindible por un agente de escisión que está presente en el ambiente intracelular (por ejemplo, dentro de un lisosoma, endosoma o caveola). El engarce puede ser, por ejemplo, un engarce de peptidilo que se escinde por una enzima peptidasa o proteasa intracelular, incluyendo, pero sin limitación, una proteasa lisosomal o endosomal. Tipicamente, el engarzador de peptidilo es de al menos dos aminoácidos de longitud o al menos tres aminoácidos de longitud. Los agentes de escisión pueden incluir catepsina B y D y plasmina, de todos los cuales se sabe que hidrolizan derivados farmacológicos dipeptidicos dando como resultado la liberación de fármaco activo dentro de células diana (véase, por ejemplo, Dubowchik y Walker, 1999, Pharm. Therapeutics 83: 67 123). Los engarzadores de peptidilo pueden ser escindibles por enzimas que están presentes en las células. Por
ejemplo, puede usarse un engarce de peptidilo que es escindible por la proteasa dependiente de tiol catepsina B, que se expresa en gran medida en tejido canceroso (por ejemplo, un engarce de Phe-Leu o de Gly-Phe-Leu-Gly (SEC ID Ns: 50)). Otros engarces tales se describen, por ejemplo, en la Patente de Estados Unidos N° 6.214.345. En realizaciones específicas, el engarce de peptidilo escindible por una proteasa intracelular es un engarce Val-Cit o un engarce Phe-Lys (véase, por ejemplo Patente de Estados Unidos N° 6.214.345, que describe la síntesis de doxorrubicina con el engarce val-cit). Una ventaja de usar liberación proteolítica intracelular del agente terapéutico es que el agente se atenúa típicamente cuando se conjuga y las estabilidades en suero de los conjugados son típicamente altas.
En otras realizaciones, el engarzador escindible es sensible a pH, es decir, sensible a hidrólisis a ciertos valores de pH. Típicamente, el engarce sensible a pH es hidrolizable en condiciones ácidas. Por ejemplo, puede usarse un engarce lábil para ácido que es hidrolizable en el lisosoma (por ejemplo, una hidrazona, semicarbazona, tiosemicarbazona, amida cis-aconítica, ortoéster, acetal, cetal o similares). (Véase, por ejemplo, Patente de Estados Unidos N° 5.122.368; 5.824.805; 5.622.929; Dubowchik y Walker, 1999, Pharm. Therapeutics 83: 67-123; Neville y col., 1989, Biol. Chem. 264: 14653-14661). Tales engarces son relativamente estables en condiciones de pH neutro, tales como las de la sangre, pero son inestables a pH por debajo de 5,5 ó 5,0, el pH aproximado del lisosoma. En ciertas realizaciones, el engarce hidrolizable es un engarce de tioéter (tal como, por ejemplo, un tioéter unido al agente terapéutico mediante un enlace de acilhidrazona (véase, por ejemplo, Patente de Estados Unidos N° 5.622.929)).
En otras realizaciones más, el engarce es escindible en condiciones reductoras (por ejemplo, un engarce disulfuro). Se conoce una diversidad de engarces disulfuro en la técnica, incluyendo, por ejemplo, los que pueden formarse usando SATA (N-succinimidil-5-acetiltioacetato), SPDP (N-succinimidil-3-(2-piridilditio)propionato), SPDB (N-succinimidil-3-(2-piridilditio)butirato) y SMPT (N-succinimidil-oxicarbonil-alfa-metil-alfa-(2-piridil-ditio)tolueno), SPDB y SMPT (Véase, por ejemplo, Thorpe y col., 1987, Cancer Res. 47: 5924-5931; Wawrzynczak y col., In Immunoconjugates: Antibody Conjugates in Radioimagery and Therapy of Cancer (C. W. Vogel ed., Oxford U. Press, 1987. Véase también Patente de Estados Unidos N° 4.880.935.))
En otras realizaciones específicas más, el engarce es un engarce de malonato (Johnson y col., 1995, Anticancer Res.
15: 1387-93), un engarce de maleimidobenzoilo (Lau y col., 1995, Bioorg-Med-Chem. 3(10): 1299-1304), o un análogo de 3’-N-amida (Lau y col., 1995, Bioorg-Med-Chem. 3(10): 1305-12).
Típicamente, el engarce no es sustancialmente sensible al ambiente extracelular. Como se usa en el presente documento, “no sustancialmente sensible al ambiente extracelular”, en el contexto de un engarce, significa que no más de aproximadamente 20 %, típicamente no más de aproximadamente 15 %, más típicamente no más de aproximadamente 10 %, incluso más típicamente no más de aproximadamente 5 %, no más de aproximadamente 3 % o no más de aproximadamente 1 % de los engarces, en una muestra de ADC o derivado de ADC, se escinden cuando el ADC o derivado de ADC está presente en un ambiente extracelular (por ejemplo, en plasma). Si un engarce no es sustancialmente sensible al ambiente extracelular puede determinarse, por ejemplo, incubando de forma independiente con plasma tanto (a) el ADC o derivado de ADC (la “muestra de ADC”) como (b) una cantidad molar igual de anticuerpo no conjugado o agente terapéutico (la “muestra de control”) durante un periodo de tiempo predeterminado (por ejemplo, 2, 4, 8, 16 ó 24 horas) y comparando después la cantidad de anticuerpo no conjugado o agente terapéutico presente en la muestra de ADC con la presente en muestra de control, como se mide, por ejemplo, por cromatografía líquida de alto rendimiento.
En otras realizaciones no mutuamente exclusivas, el engarce promueve la internalización celular. En ciertas realizaciones, el engarce promueve la internalización celular cuando se conjuga con el agente terapéutico (es decir, en el medio del resto de agente terapéutico-engarce del ADC o derivado de ADC como se describe en el presente documento). En otras realizaciones más, el engarce promueve la internalización celular cuando se conjuga tanto con el agente terapéutico como con la proteína de unión a antígeno o anticuerpo derivado del mismo (es decir, en el medio del ADC o derivado de ADC como se describe en el presente documento).
Una diversidad de engarces que pueden usarse con las presentes composiciones y procedimientos se describen en el documento WO 2004010957 titulado “Drug Conjugates and Their Use for T reating Cancer, An Autoimmune Disease or an Infectious Disease” presentado el 31 de julio de 2003 y la Solicitud Provisional de Estados Unidos N° 60/400.403, titulada “Drug Conjugates and their use for treating cancer, an autoimmune disease or an infectious disease”, presentada el 31 de julio de 2002.
Como alternativa, puede prepararse una proteína de fusión que comprende la proteína de unión a antígeno y agente citotóxico, por ejemplo, por técnicas recombinantes o síntesis peptídica. La longitud del ADN puede comprender regiones respectivas que codifican las dos partes del conjugado adyacentes entre sí o separadas por una región que codifica un péptido de engarce que no destruye las propiedades deseadas del conjugado.
En otra realización más, el anticuerpo puede conjugarse con un “receptor” (tal como estreptavidina) para su utilización en pre-dirección de tumor en la que el conjugado de anticuerpo-receptor se administra al paciente, seguido de retirada
de conjugado no unido de la circulación usando un agente de eliminación y la administración de un “ligando” (por ejemplo, avidina) que se conjuga con un agente citotóxico (por ejemplo, un radionucleótido).
La expresión “anticuerpo No Humano o fragmento de anticuerpo del mismo” como se usa en el presente documento pretende referirse a anticuerpos o fragmentos de los mismos que se originan en cualquier especie distinta de seres humanos, en la que humano incluye anticuerpos quiméricos.
La expresión “anticuerpo donador” se refiere a un anticuerpo (monoclonal y/o recombinante) que contribuye a las secuencias de aminoácidos de sus dominios variables, CDR, u otros fragmentos funcionales o análogos de los mismos para un primer compañero de inmunoglobulina, para proporcionar la región codificante de inmunoglobulina alterada y anticuerpo alterado expresado resultante con la especificidad antigénica y actividad neutralizadora característica del anticuerpo donador.
La expresión “anticuerpo aceptor” se refiere a un anticuerpo (monoclonal y/o recombinante) heterólogo para el anticuerpo donador, que aporta todas (o cualquier parte, pero preferentemente todas) las secuencias de aminoácidos que codifican sus regiones marco de cadena pesada y/o ligera, y/o sus regiones constantes de cadena pesada y/o ligera para el primer compañero de inmunoglobulina. El anticuerpo humano es el anticuerpo aceptor.
La expresión “secuencia aceptora humana” como se usa en el presente documento pretende referirse a un marco de un anticuerpo o fragmento de anticuerpo del mismo que comprende la secuencia de aminoácidos de un marco VH o VL derivado de un anticuerpo humano o fragmento de anticuerpo del mismo o un marco de secuencia consenso humano en el que pueden incorporarse CDR de una especie no humana.
El término “incorporación” de las CDR o regiones hipervariables como se usa en el presente documento abarca cualquier medio por el que las CDR no humanas se sitúan con el marco aceptor humano. Se apreciará que esto puede conseguirse de diversas formas, por ejemplo, pueden generarse ácidos nucleicos que codifiquen la secuencia de aminoácidos deseada mutando ácidos nucleicos que codifican la secuencia de dominio variable no humana de modo que los restos marco de la misma cambien a restos del marco aceptor humano o mutando ácido nucleico que codifica la secuencia de dominio variable humana de modo que las CDR cambian a restos no humanos, o sintetizando ácidos nucleicos que codifican la secuencia deseada. En una realización la secuencia final se genera in silico.
La presente invención se ilustra por los siguientes ejemplos.
Ejemplos
Ejemplo 1 Generación y selección de anticuerpos monoclonales
1.1 Estrategias de inmunización
El mAb anti BCMA humano murino parental CA8 se identificó a partir de hibridomas derivados de ratones inmunizados con BCMA humano de longitud completa. Se inmunizó un ratón BALB/c i.p. con 25 μg de proteína recombinante (rBCMA) combinada con CFA. El ratón se estimuló tres veces a intervalos de un mes con 25 μg de proteína rBCMA de longitud completa 10 μg de emulsión estable de monofosforil lípido A (MPL-SE) (Corixa Corporation, Seattle, WA) y se le proporcionó un refuerzo pre-fusión de 30 μg de proteína rBCMA i.v. 3 días antes de la fusión. Se generaron hibridomas y se clonaron usando el kit de clonación de hibridoma ClonaCell-HY (StemCell Technologies, Vancouver, BC) o usando un procedimiento convencional. En el procedimiento convencional, los linfocitos B de los bazos de los animales inmunizados se fusionaron con células de mieloma Sp2/0 en presencia de PEG (Sigma-Aldrich, St. Louis, MO). Después de recuperación durante una noche, las células fusionadas se sembraron a dilución limitante en placas de 96 pocillos y se sometieron a selección de hipoxantina-aminopterina-timidina. Los sobrenadantes de cultivo de hibridoma se examinaron con respecto a la presencia de anticuerpos anti-BCMA por ELISA y citometría de flujo.
El mAb anti BCMA humano murino parental S307118G03 se identificó a partir de hibridomas derivados de ratones SJL inmunizados con quimera de TNFRSF17-Fc/BCMA humano recombinante (R&D 193-Fc) usando el procedimiento de RIMMS (múltiples sitios de inmunización rápida). El día 0, se emulsionaron 5 μg de proteína por ratón en adyuvante AS02a en 2 sitios en la espalda (sobre las caderas y sobre los hombros) y subyacentes a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 6 y el día 11 se inyectaron 2,5 μg de proteína por ratón en adyuvante RIBI subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 14 se sacrificaron los animales. Los ganglios linfáticos y el bazo se escindieron, se rompieron y se realizó una fusión de células somáticas inducida por PEG1500 usando una relación 3:1 con célula de mieloma de ratón X63 AG8653.GFP.Bcl-2.11 (BioCat 112754; R17209/58). La fusión se sembró en 10 placas de 96 pocillos y se exploró directamente a partir de estas.
El mAb anti BCMA humano murino parental S336105A07 se identificó a partir de hibridomas derivados de inmunizaciones idénticas. Los ganglios linfáticos y el bazo se escindieron el día 14, se rompieron y se realizó una electrofusión de Cytopulse usando una relación 1:1 con células de mieloma de ratón X63 AG8 653.GFP.Bcl-2.11
(BioCat 112754; R17209/58). La fusión se sembró en omnitrays que contenía medio semisólido antes de seleccionar en 10 placas de 96 pocillos y se exploró directamente a partir de estas 5 días después.
Los mAb parentales murinos anti BCMA humano S332121F02 y 332126E04 se identificaron a partir de hibridomas derivados de ratones SJL inmunizados con fusión de Fc recombinante del dominio extracelular de BCMA humano (4 53) BCMA usando el procedimiento de RIMMS (inmunización rápida). El día 0, se emulsionaron 5 qg de proteína por ratón en adyuvante AS02a en 2 sitios en la espalda (sobre las caderas y sobre los hombros) y subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 6 se inyectaron 5 qg de proteína BMCA-Fc de cynomolgus recombinante por ratón en adyuvante RIBI subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 11 se inyectaron 2,5 qg de BCMA-Fc recombinante humano y 2,5 qg de BCMA-Fc de cynomolgus recombinante por ratón en adyuvante RIBI subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 14 los animales se sacrificaron y las células se trataron como para S307118G03.
El mAb parental murino anti BCMA humano S322110D07 se identificó a partir de hibridomas derivados de ratones SJL inmunizados con fusión de Fc recombinante del dominio extracelular de BCMA humano (4-53) en complejo con April humano recombinante (R&D 5860-AP/CF) premezclado a relación molar 1:1. Los ratones se inmunizaron i.p. con 5 qg de complejo April/ BCMA-Fc de Cynomolgus en PBS, se suspendió en adyuvante RIBI, 100 ql de dosis por ratón y se reforzaron tres veces a intervalos de 3-4 semanas con 2,5 qg de complejo April/ BCMA-Fc de Cynomolgus en PBS, se suspendió en adyuvante RIBI, se inyectaron 100 ql de dosis por ratón por vía intraperitoneal y se proporcionó un refuerzo pre-fusión del mismo inmunógeno 1 día antes de la fusión y se trató como para S307118G03.
El mAb anti BCMA humano murino parental S335115G01 y S335122F05 se identificaron a partir de hibridomas derivados de ratones SJL inmunizados con una mezcla de fusión de Fc recombinante del dominio extracelular de BCMA humano (4-53) y fusión de Fc recombinante del domino extracelular de BCMA (4-52) de cynomolgus usando el procedimiento de RIMMs (múltiples sitios de inmunización rápida). El día 0, se emulsionaron 2,5 qg de cada proteína por ratón en adyuvante AS02a y se inyectaron en 2 sitios en la espalda (sobre las caderas y sobre los hombros) y subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 6 y el día 11 se inyectaron 2,5 qg de cada proteína por ratón en adyuvante subyacente a los ganglios linfáticos principales en 4 sitios en la parte frontal. El día 14 los animales se sacrificaron. Los ganglios linfáticos y de bazo se escindieron, se rompieron y se realizó una electrofusión de Cytopulse usando una relación 1:1 con células de mieloma de ratón X63 AG8 653.GFP.Bcl-2.11 (BioCat 112754; R17209/58). La fusión se sembró en omnitrays que contenían medio semisólido antes de seleccionar en 32 placas de 96 pocillos y se exploró directamente a partir de estos 5 días después.
Ejemplo 2 Humanización.
2.1 Clonación de regiones variables de hibridoma de CA8
Se extrajo ARN total de células de hibridoma de CA8, se generó después secuencia de ADNc de dominio variable pesado y ligero por transcripción inversa y reacción en cadena de la polimerasa (RT-PCR). El cebador directo para RT-PCR fue una mezcla de cebadores degenerados específicos para secuencias líder de gen de inmunoglobulina murina y el cebador inverso fue específico para las regiones constantes de anticuerpo. Se usaron en este caso cebadores inversos específicos para IgG1, IgG2a e IgG2b puesto que el isotipo era desconocido. Para diseñar los cebadores, se generaron alineamientos de múltiples secuencias de ADN de las secuencias líder de los genes de VH y Vk de ratón.
2.2 Clonación de CA8 quimérico
Las construcciones de expresión de ADN que codifican el anticuerpo quimérico se prepararon de novo por acumulación de oligonucleótidos solapantes que incluían sitios de restricción para clonar en vectores de expresión de mamífero así como una secuencia señal humana. Se introdujeron sitios de restricción HindIII y Spel para enmarcar el dominio VH que contenía la secuencia señal para clonar en vectores de expresión de mamíferos que contenían la región constante humana y1. Se introdujeron sitios de restricción HindIII y BsiWI para enmarcar el dominio VL que contenía la secuencia señal para clonar en el vector de expresión de mamífero que contenía la región constante kappa humana.
2.3 Clonación de las variantes de CA8 humanizadas
Las construcciones de expresión de ADN que codificaban las variantes de anticuerpo humanizado se prepararon de novo por acumulación de oligonucleótidos solapantes que incluían sitios de restricción para clonar en vectores de expresión de mamífero así como una secuencia señal humana. Se introdujeron sitios de restricción HindIII y SpeI para enmarcar el dominio VH que contenía la secuencia señal para clonar en vectores de expresión de mamífero que contenían la región constante y1 humana. Se introdujeron sitios de restricción HindIII y BsiWI para enmarcar el dominio VL que contenía la secuencia señal para clonar en el vector de expresión de mamífero que contenía la región constante kappa humana.
2.4 Expresión de los anticuerpos CA8 recombinantes (incluyendo cuantificación de anticuerpos)
Se cotransfectaron de forma transitoria células HEK 2936E con plásmido de expresión que codificaban las cadenas pesada y ligera respectivamente y se expresaron a pequeña escala para producir anticuerpo. Los anticuerpos se cuantificaron por ELISA. Se revistieron placas de ELISA con IgG anti humano (Sigma I3382) a 1 mg/ml y se bloquearon con solución de bloqueo (BSA al 4 % en solución salina tamponada con Tris). Se añadieron diversas diluciones de los sobrenadantes de cultivo tisular y la placa se incubó durante 1 hora a temperatura ambiente. También se añadieron diluciones de un anticuerpo convencional conocido a la placa. La placas se lavó en TBST y se detectó la unión mediante la adición de un anticuerpo de cadena ligera kappa antihumano marcado con peroxidasa (Sigma A7164) a una dilución de 1/1000 en solución de bloqueo. La placa se incubó durante 1 hora a temperatura ambiente antes de lavar en TBST. La placa se reveló por adición de sustrato OPD (Sigma P9187) y el revelado del color se detuvo mediante la adición de H2SO42 M. La absorbancia se midió a 490 nm y se representó una curva patrón usando datos para las diluciones convencionales conocidas. La curva patrón se usó para estimar la concentración de anticuerpo en los sobrenadantes de cultivo tisular. Se purificaron preparaciones de anticuerpo a mayor escala usando proteína A y las concentraciones se midieron usando un Nanodrop (Thermo Scientific).
Tabla 1. Diseño de variantes humanizadas pesadas y ligeras variables de CA8

2.5 Producción de anticuerpos desfucosilados
Para generar anticuerpos desfucosilados las cadenas pesada y ligera se cotransfectaron respectivamente en células CHO DG44 MS705 BioWa y se expresaron a escala para producir anticuerpo. Brevemente, se linealizaron 30 μg de ADN durante una noche con Not1, el ADN se precipitó con etanol y se volvió a disolver en tampón TE. A partir del cultivo, se obtuvieron 2,4 X 107 células BioWa DG44 y se lavaron en 14 ml de PBS-sacarosa caliente. Las células se centrifugaron y el sedimento se resuspendió en 1,6 ml de PBS-sacarosa. La mitad (0,8 ml) de las células anteriormente mencionadas, suspendidas en PBS-sacarosa, se añadieron a una cubeta BioRad con los 30 μg del ADN linealizado (en 50 μl de tampón TE). Se programó un GenePulser BioRad a 380V con una capacitación de 25 pF y la cubeta se introdujo para electroporación. Los 850 pl resultantes de células electroporadas y ADN se añadieron a (80 ml de) medio SFM512 caliente (que incluía rojo fenol, 2 X HT (nucleósidos), glutamax y suplemento Gibco 4). Finalmente, los 80 ml resultantes de suspensión celular se transfirieron (150 μl/pocillo) a cada pocillo de una de 4 placas de 96 pocillos. Después de 48 horas, el medio se cambió a nucleósido libre retirando aproximadamente 130 pl de medio acondicionado y reemplazándolo con 150 pl de medio SFM512 de selección nuevo (que incluía rojo fenol y glutamax). Cada 3-4 días, se retiraron 130-150 pl de medio acondicionado y se reemplazó con medio de selección nuevo. Los pocillos se controlaron con respecto a cambio de color y se ensayaron con respecto a concentración de IgG como se ha analizado anteriormente.
2.6 Anticuerpos adicionales. Clonación de regiones variables de hibridoma
Se extrajo ARN total de células de hibridoma S307118G03, S332121F02, S332126E04, S322110D07, S336105A07, S335115G01 y S335122F05. Se generó después secuencia de ADNc de dominio variable pesado y ligero por transcripción inversa y reacción en cadena de la polimerasa (RT-PCR). El cebador directo para RT-PCR fue una mezcla de cebadores degenerados específicos para secuencias líder de gen de inmunoglobulina murina y el cebador
inverso fue específico para las regiones constantes de anticuerpo, en este caso isotipo IgG2a. Se diseñaron cebadores basándose en una estrategia descrita por Jones y Bendig (Bio/Technology 9: 88, 1991). Se llevó a cabo RT-PCR para ambas secuencias de región V para permitir verificación posterior de las secuencias de región V correctas. Se obtuvieron datos de secuencia de ADN para los productos de región V generados por la RT-PCR.
2.7 Anticuerpos adicionales. Clonación de las quimeras
Las construcciones de expresión de ADN que codificaban los anticuerpos quiméricos se prepararon de novo por clonación de PCR infusion advantage (Clonetech) de los productos de PCR del gen V en vectores de expresión de mamíferos. Este procedimiento de clonación permitió la fusión de las regiones variables murinas con regiones constantes de cadena L kappa y cadena H de IgG1 humana.
2.8 S307118G03. Clonación de las variantes humanizadas
La clonación se llevó a cabo como en el párrafo 2.3.
2.9 S307118G03. Expresión de los anticuerpos recombinantes
Se cotransfectaron de forma transitoria plásmidos de expresión que codificaban las cadenas pesadas y ligeras relevantes (enumeradas en la Tabla 8 posterior) en células HEK 293 6E y se expresaron a pequeña escala para producir anticuerpo. Los anticuerpos se purificaron por Proteína A de los sobrenadantes y se cuantificaron usando el espectrofotómetro Nanodrop.
Ejemplo 3 Conjugación de anticuerpos con vcMMAE y mcMMAF para formar conjugados de fármaco con anticuerpo (ADC)
Tabla B Estructuras químicas de engarces farmacológicos
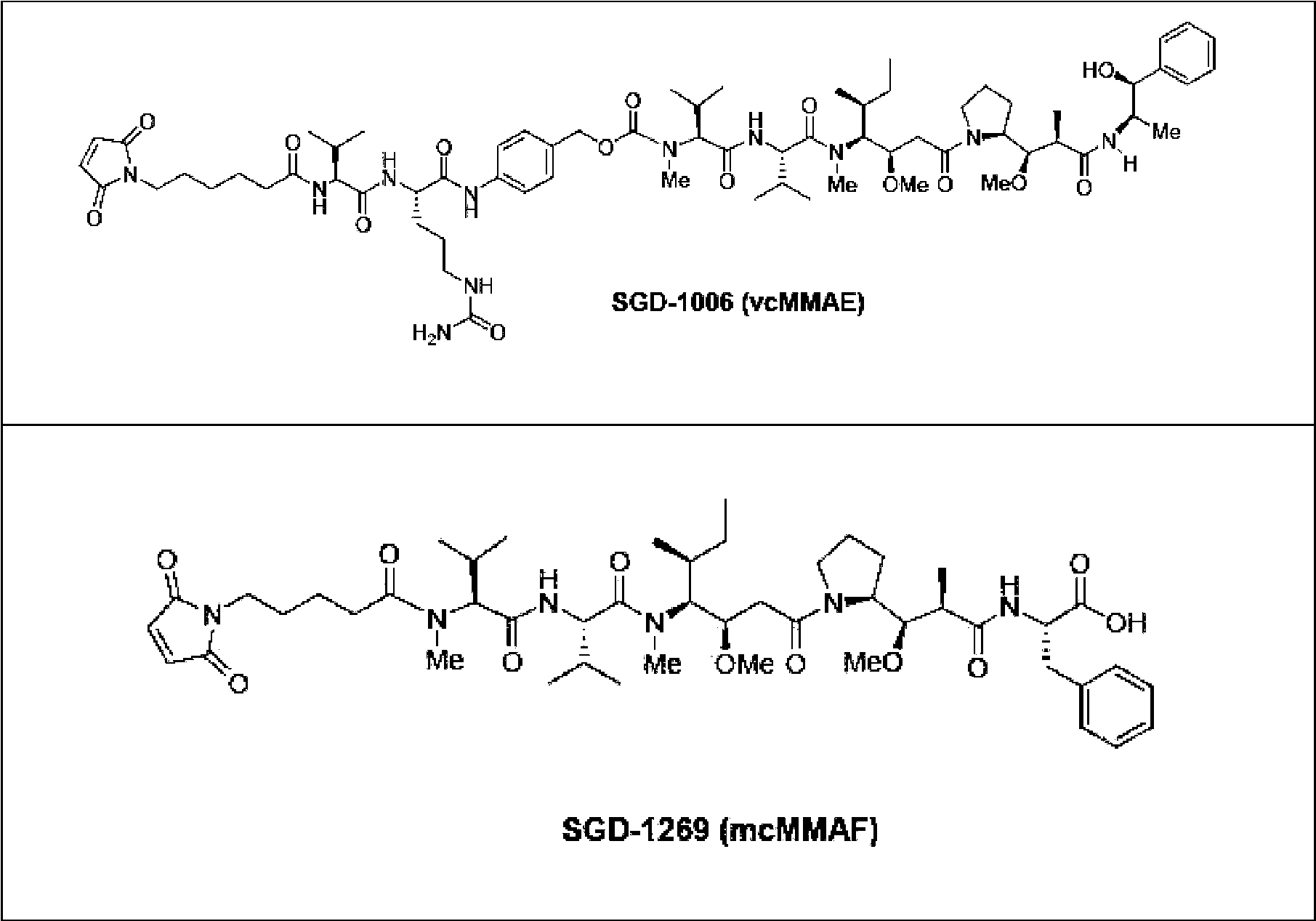
Se añadió suspensión de resina de Gammabind más Proteína G (GE Healthcare) (75 μl) a cada pocillo de una placa de filtrado de pocillo profundo (capacidad de 2 ml). Los anticuerpos para conjugar se agruparon por especie e isotipo y se transfirieron hasta 0,5 mg de cada anticuerpo a cada pocillo de la placa. Cada anticuerpo se transfirió a dos pocillos separados para facilitar la preparación de dos conjugados, con los engarces farmacológicos SGD-1006 y
SGD-1269. La placa de filtrado se agitó después a 1200 RPM durante 2 horas a 5 °C para unir los anticuerpos de la resina. La placa de filtrado se centrifugó después a 500 x g durante 3 minutos para asegurar la sedimentación de todos los fluidos y resina al fondo de cada pocillo.
Los anticuerpos unidos se redujeron después añadiendo 500 gl de TCEP 10 mM en KPO4100 mM, NaCl 150 mM, pH 7, EDTA 1mM y agitando durante 30 minutos a 22 °C. Después de la reducción, la placa se centrifugó de nuevo para retirar la solución de TCEP y posteriormente se lavó con PBS EDTA 1mM, 1 ml por pocillo. La solución de lavado se retiró por centrifugación y el procedimiento se repitió 3 veces para un total de 4 lavados. Los anticuerpos unidos o reducidos se conjugaron después usando una mezcla de n Em y engarce farmacológico preparado de acuerdo con las fracciones molares indicadas en la Tabla 2.
Tabla 2.

* también para quiméricos de IgG 1 murino / humano
Se prepararon de este modo mezclas separadas de NEM y engarce farmacológico para cada especie / isotipo de anticuerpo usando soluciones madre de DMSO 10 mM de sGd -1006, SGD-1269 (Véase Tabla B) y NEM. Cuando se mezcla a la relación apropiada la concentración de maleimida total fue por lo tanto aún 10 mM, y este valor se usó para calcular el volumen de solución de maleimida para añadir a cada pocillo. Por ejemplo para una IgG 1 murina con 5 disulfuros reducibles (10 tioles disponibles cuando se reduce) 0,5 mg de anticuerpo a 150 kDa es 3,33 nmoles correspondientes a 33,3 nmoles de tiol. Un exceso de 3 veces es por lo tanto 100 nmoles de maleimida total o 10 gl de la mezcla de NEM /engarce farmacológico 10 mM. Para el conjugado SGD-1269 esta mezcla se prepararía después con 5,86 gl de SGD-1269 y 4,14 gl de NEM. La mezcla de maleimida se diluiría después en 500 gl de PBS antes de adición al anticuerpo reducido inmovilizado. En la práctica, puesto que se conjugaron múltiples anticuerpos de cada isotipo de forma simultánea se preparó una solución mixta sencilla de SGD-1269 / NEM para cada isotipo multiplicando el número de pocillos que contenían el isotipo por 10 gl por pocillo, diluyendo después en un volumen de PBS igual a 500 gl multiplicado por el número de pocillos. De forma similar se preparó un total de ocho mezclas de engarce farmacológico / NEM, cuatro con SGD-1006 y cuatro con SGD-1269, y se diluyeron en PBS. Estas mezclas se añadieron después a los anticuerpos reducidos (500 gl por pocillo) y la placa se agitó durante 30 minutos a 22 °C. La placa se centrifugó después como anteriormente para retirar la solución de reacción en exceso y posteriormente se lavó 4 veces con PBS como anteriormente.
Los ADC unidos se eluyeron después añadiendo 200 gl de glicina 50 mM pH 2,5 a cada pocillo y agitando la placa durante 3 minutos a 1200 RPM. En agitación se añadieron 20 gl de tampón de neutralización (fosfato potásico 1 M, pH 7,4, NaCl 500 mM, Tween-200,2 %) a cada pocillo de una placa de recogida de 1 ml. Los ADC se eluyeron después a la placa de recogida centrifugando a 1500 x g durante 6 minutos. La placa de recogida se agitó después brevemente para asegurar la mezcla completa del tampón de neutralización.
La concentración de cada ADC se determinó después con un lector de placa de absorbancia transfiriendo las soluciones a una placa de ensayo de UV (Costar modelo 3635, Corning) y midiendo la densidad óptica a 280 nm. Se usó un coeficiente de extinción de IgG medio de 1,45 ml mg-1 cm-1 para proporcionar una estimación adecuada de la concentración de ADC por todo el panel. Para confirmar la conjugación exitosa, se usó un procedimiento de HPLC de proteína de fase inversa (descrito posteriormente) para estimar la carga farmacológica de los controles de isotipo. Para la placa que contenía las variantes de humanización de CA8 se usó este procedimiento para estimar la carga de todos los ADC directamente.
El procedimiento de cromatografía de proteína de fase inversa para determinar carga farmacológica emplea la fase estacionaria polimérica PLRP-S (Agilent Technologies). Puesto que los anticuerpos se redujeron completamente durante el procedimiento de conjugación todas las subunidades de anticuerpo eluyen de la columna como cadenas polipeptídicas sencillas permitiendo que se evalúen de forma separada las subpoblaciones de especies de cadena ligera y pesada con diversos niveles de carga farmacológica. Por lo tanto, el análisis de estos datos permite el cálculo de la carga farmacológica de cadena ligera media y la carga farmacológica de cadena pesada media como factores independientes que pueden después combinarse para determinar la carga farmacológica de anticuerpo media con el conocimiento básico de que cada anticuerpo está comprendido por dos cadenas pesadas y dos ligeras. Las condiciones cromatográficas fueron como sigue: Una columna PRLP-S, 1000 Á, 50 x 2,1 mm, tamaño de partícula 8
gm (Agilent Technologies) con agua TFA 0,05 % como fase móvil A y acetonitrilo TFA 0,01 % como fase móvil B; elución con un gradiente lineal de B 27 % a B 42 % en 12,5 minutos.
Los anticuerpos anti-BCMA se conjugaron con SGD-1006 y SGD-1269 en tres lotes separados durante un periodo de siete meses. En el primer lote se conjugó un total de 29 anticuerpos (dando como resultado 58 ADC). La carga farmacológica de cada control de isotipo determinada por cromatografía de PLRP y los datos se resumen en la Tabla 3.
Tabla 3.

Para el segundo lote se conjugaron 25 anticuerpos adicionales (dando como resultado 50 ADC). La carga farmacológica de cada control de isotipo se determinó de nuevo por cromatografía de PLRP y los datos se resumen en la Tabla 4.
Tabla 4.

En el tercer lote se conjugaron 30 anticuerpos (dando como resultado 60 ADC), incluyendo 13 variantes humanizadas de CA8. En este lote final, la carga farmacológica de todos los ADC se determinó y se resumen en los siguientes mapas de dos placas (Tabla 5 y 6).
Tabla 5.

Tabla 6.

Se indica la carga farmacológica media y el % de CV para cada serie de isotipo en la parte de abajo. Se observó una variabilidad atípicamente grande de la carga farmacológica para los ADC SGD-1269 preparados con anticuerpos mIgG2b; la razón para esto no está clara. Además, los anticuerpos CA8 potenciados con Fc produjeron niveles de carga farmacológica algo inferiores a los de las otras variantes humanas de CA8; para abordar esto, se conjugó CA8 con Fc potenciado adicional en una reacción de fase de solución para alcanzar mejor la carga farmacológica conseguida para los otros anticuerpos.
Ejemplo 4. Datos de unión
4.1 Ensayo de unión de FMAT para mostrar unión de CA8 quimérico con células que expresan BCMA humano o de cynomolgus.
Se recuperaron células HEK293 humanas transfectadas crioconservadas, con BCMA de cynomolgus y con transfección simulada de almacenamiento en LN2. Se prepararon pocillos de ensayo con anticuerpo CA8 quimérico humano, a una serie de diferentes concentraciones, mezclado con células HEK293 con BCMA humano, HEK293 con BCMA de cynomolgus y con transfección simulada respectivamente. Se añadió conjugado secundario de Azul FMAT anti-IgG humana para detección de CA8 quimérico humano. Las placas de ensayo se dejaron durante un mínimo de 90 minutos antes de que el resultado se leyera en el lector de placas ABI8200 (FMAT).
Esto mostró que el anticuerpo CA8 en forma quimérica se une bien a proteínas BCMA tanto humanas como de cynomolgus expresadas en células HEK293.
Los resultados se muestran en la Figura 1.
4.2 Experimento de ELISA que muestra unión de CA8 quimérico con proteína BCMA recombinante
Se ensayaron anticuerpos CA8 quiméricos con respecto a unión con BCMA humano y BCMA de cynomolgus expresado como fusiones de Fc. Se revistieron placas de ELISA con Fc-BCMA humano y Fc-BCMA de cynomolgus y las placas se bloquearon usando BSA para reducir la unión no específica. Se añadieron anticuerpos quiméricos CA8 en un intervalo de concentración de 5 μg/ml a 0,1 μg/ml a las placas de ELISA revestidas con BCMA humano y de cynomolgus. Cualquier anticuerpo CA8 quimérico humano unido se detectó usando anticuerpo secundario conjugado con HRP anti-IgG humana según fuera apropiado. Se añadió sustrato de HRP (TMB) para desarrollar el ELISA. Este mostró que el anticuerpo CA8 se une a BCMA de cynomolgus y humano recombinante en un ensayo de ELISA.
Los resultados se muestran en la Figura 2.
4.3 Experimento de Biacore para mostrar la unión de anticuerpo CA8 con proteínas BCMA y TACI para determinar la reactividad cruzada con proteína TACI.
Se inyectó anticuerpo quimérico CA8 y se capturó en proteína A (se usó una microplaca sensora derivatizada con proteína A). Se bloqueó la unión de proteína A residual con una inyección de una alta concentración de solución de IgG humana. Se ensayaron después soluciones de BCMA-Fc, TACI-Fc o BAFF-R-Fc con respecto a unión con el anticuerpo. Las tres proteínas se inyectaron en secuencia y se midieron los acontecimientos de unión. La superficie se regeneró entre las inyecciones de cada proteína.
Se analizaron sensogramas en el programa Biaevaluation. Se realizó resta de la referencia doble para retirar el ruido del instrumento y cualquier unión no específica de las curvas del sensograma.
Esto mostró que CA8 era específico con respecto a unión para unión con BCMA y no con TACI y BAFFR.
La unión del anticuerpo CA8 con BCMA-Fc, TACI-Fc y BAFF-R-Fc se representó como se muestra en la Figura 3.
4.4 Unión de células y datos de neutralización
4.4.1 Unión de anticuerpos murinos anti BCMA con células de mieloma múltiple y células que expresan BCMA.
Se tiñeron células de la línea celular de mieloma múltiple H929 y transfectantes que expresaban ARH77-hBCMA 10B5 BCMA con S332211 D07, S3332121F02 o S332126E04 murino o con el control de isotipo murino a 5 μg/ml. Se tiñó la línea celular de mieloma múltiple H929 con S307118G03 murino. Las células se incubaron durante 20 minutos a temperatura ambiente (TA) y después se lavaron con tampón FACS (PBS BSA 0,5 % azida sódica 0,1 %) para retirar anticuerpo no unido. Las células se incubaron con un anticuerpo anti-IgG de ratón marcado con PE secundario durante 15 minutos a TA y después se lavaron con tampón FACS para retirar anticuerpo no unido. Las células se analizaron por FACS para detectar anticuerpo unido a las células.
Los resultados (Figura 4) mostraron que los 4 anticuerpos murinos se unieron a la línea celular de mieloma múltiple H929 y los tres anticuerpos ensayados en células transfectadas con BCMA ARH77 se unieron a estos.
4.4.2 Curva de unión de CA8 quimérico con células de mieloma múltiple como se determinó por FACS
Se usó un panel de líneas celulares de mieloma múltiple para determinar la unión de CA8 quimérico. Las líneas celulares H929, OPM-2, JJN-3 y U266 se tiñeron con CA8 quimérico o anticuerpo irrelevante (Synagis) a diversas concentraciones durante 20 minutos a TA. Las células se lavaron después con tampón FACS (PBS BSA 0,5 % azida sódica al 0,1 %) para retirar anticuerpo no unido. Las células se incubaron con un anticuerpo anti-IgG humano marcado con PE secundario durante 15 minutos a TA y después se lavaron con tampón FACS para retirar el anticuerpo no unido. Las células se analizaron por FACS y se midieron los valores de intensidad de fluorescencia media (IFM) para determinar la unión.
Los resultados mostraron que CA8 quimérico se unió a líneas celulares de mieloma múltiple H929, OPM-2, JJN-3 y U266 de una manera dependiente de dosis (Figura 5).
4.4.3 Unión de CA8 humanizado con células transfectadas con BCMA según se determinó por FACS
Se tiñeron células transfectantes que expresaban BCMA ARH77-hBCMA 10B5 o células H929 con CA8 quimérico o variantes humanizadas de CA8 designadas J6M0, J6M1, J6M2, J9M0, J9M1, J9M2 a diversas concentraciones durante 20 minutos a TA. Las células se lavaron después con tampón FACS (PBS BSA 0,5 % azida sódica 0,1 %) para retirar el anticuerpo no unido. Las células se incubaron con un anticuerpo anti-IgG humano marcado con PE secundario durante 15 minutos a TA y después se lavaron con tampón de FACS para retirar anticuerpo no unido. Las células se analizaron por FACS y se midieron los valores de intensidad de fluorescencia media (IFM) para determinar la unión.
Los resultados mostraron que CA8 quimérico y todos los anticuerpos ensayados aparte de J9M2 se unieron a células transfectantes que expresaban BCMa ARH77-hBCMA 10B5 y células H929 de una manera dependiente de dosis (Figura 6).
4.5 Demostración de la capacidad de CA8 y la versión humanizada J6M0 para neutralizar la unión de BAFF o APRIL con BCMA recombinante.
El objetivo de este ensayo fue evaluar la capacidad del anticuerpo CA8, y su versión humanizada J6M0, tanto en forma de tipo silvestre como afucosilada (Potelligent), a diversas concentraciones para neutralizar la capacidad de unión de uno de los ligandos de BCMA, Ba Ff o APRIL.
Se revistieron durante una noche placas de fondo plano de 96 pocillos con solución de 1 μg/ml de BCMA humana recombinante Fc 4-53 en PBS. Después de una etapa de lavado usando TWEEN200,05 %, las placas se bloquearon con solución de Albúmina de Suero Bovino 2 % en PBS durante 1 hora a temperatura ambiente. Las placas se lavaron como anteriormente y se añadieron 40 pl de cada anticuerpo (IgG murino, c A8 murino y CA8 quimérico) comenzando a 10 μg/ml, valorado a 1 en 2 por duplicado a los pocillos relevantes y se incubó durante 1 hora a temperatura ambiente. Se añadieron 40 pl de BSA 2 % a los pocillos de control relevantes. Se añadieron 10 pl de BAFF humano recombinante (2149-BF/CF, R&D Systems) o APRIL humano recombinante (5860-AP/CF, R&D Systems) a 30 ng/ml y 750 ng/ml, respectivamente, dando una concentración final de 6 ng/ml y 150 ng/ml respectivamente en cada pocillo. Se añadió un volumen equivalente de BSA 2 % a los pocillos de control relevantes. Se permitió que las placas se incubaran durante 2 horas a temperatura ambiente, después de lo cual se lavaron como anteriormente. Se añadió ligando antihumano biotinilado (BAFF BAF124 o APRIL BAF884, R&D Systems) a los pocillos relevantes a 50 ng/ml y se incubó durante 1 hora. Después de una etapa de lavado, se añadieron 50 pl de una dilución 1:4000 de Estreptavidina-HRP (Amersham RPN4401) a cada pocillo y se incubó durante 30 minutos a temperatura ambiente. El proceso de lavado se repitió de nuevo seguido de la adición de 100 pl de solución de sustrato de Tetrametilbencidina (T8665, Sigma) a cada pocillo. Las placas se incubaron durante 20-25 minutos a temperatura ambiente, envueltas en papel de aluminio. La reacción se detuvo con la adición de 100 pl de H2SO4 1 M. Se determinó la densidad óptica a 450 nm usando lector Spectromax. Véase Figura 7A y B.
En un ensayo basado en placas para neutralización de unión de BAFF o APRIL con BCMA, los valores de CE50 calculados para CA8 quimérico fueron 0,695 μg/ml y 0,773 μg/ml respectivamente. Los valores para el J6M0 humanizado fueron 0,776 ng/ml y 0,630 ng/ml. Los valores para la versión de J6M0 potelligent fueron 0,748 y 0,616 ng/ml respectivamente.
4.6 Efecto de CA8 quimerizado y anticuerpo de BCMA J6M0 humanizado en fosforilación inducida por BAFF o APRIL de NFk B en células H929.
En un conjunto de experimentos, se sembraron células H-929 a 75.000 células/pocillo en una placa de 96 pocillos en medio sin suero. El anticuerpo CA8 quimérico se añadió 24 horas después para proporcionar concentraciones finales en los pocillos de hasta 200 μg/ml. Diez minutos después, se añadió ligando BAFF o APRIL a las células para proporcionar concentraciones finales en los pocillos de 0,6 ó 0,3 μg/ml respectivamente. Después de 30 minutos las células se lisaron y se midieron los niveles de NfkappaB fosforilado usando un ensayo de pNFkappaB MSD.
El anticuerpo de BCMA quimérico CA8 neutralizó la señalización de células NfkappaB inducida por BAFF y APRIL en células H-929. Fue particularmente potente en la neutralización de señalización de células NfkappaB inducida por BAFF en este tipo celular con una CI50 media de 10 nM, en comparación con 257 nM para señalización de células Nfkappap inducida por APRIL.
Datos promediados para 2 experimentos
Las CI50 fueron 10 nM para neutralización de Nfkappap inducida por BAFF y 257 nM para neutralización de Nfkappap inducida por APRIL (media de 2 experimentos independientes) y se muestran en la Tabla 7.
Tabla 7

Se llevó a cabo un conjunto de experimentos adicional para dirigirse a entender por qué hubo una discrepancia tal
entre la potencia de neutralización de APRIL y BAFF en el sistema basado en células. Después del descubrimiento de la forma soluble de BCMA el diseño experimental se cambió para incluir una etapa en la que las células H929 se lavaban antes del ensayo para reducir la interferencia de la unión del anticuerpo con BCMA soluble. Las células H-929 se lavaron 3 veces para retirar cualquier sBCMA y se resuspendieron en medio sin suero. Se añadió anticuerpo J6M0 potelligent a una placa de 96 pocillos para proporcionar concentraciones finales en los pocillos de hasta 100 μg/ml junto con ligando BAFF o APRIL para proporcionar una concentración final de los pocillos de 0,6 ó 0,2 μg/ml. Las células H-929 se sembraron después a 7,5 x 104 células/pocillo en medio sin suero. 30 minutos después las células se lisaron y los niveles de NFkappaB fosforilado se midieron usando un ensayo de pNFkappaB MSD. Estos son datos de un experimento. Cada punto de datos es la medida/dt de dos repeticiones. Los datos de este experimentos se muestran en la Figura 7c. Las CI50 para inhibición de señalización por BAFF y APRIL se determinaron como 0,91 μg/ml y 2,43 μg/ml respectivamente.
4.7 Análisis de ProteOn de construcciones humanizadas y quiméricas de CA8 anti-BCMA
La exploración inicial de variantes quiméricas y humanizadas de CA8 se llevó a cabo en el ProteOn XPR36 (Biorad). El procedimiento fue como sigue: se inmovilizó Proteína A en una microplaca de GLC (Biorad, Cat N°: 276-5011) por acoplamiento de amina primaria, se capturaron después variantes de CA8 en este superficie y se pasó BCMA humano recombinante (materiales internos o comerciales US Biological, B0410 (solamente ciclo 2)) a 256, 64, 16, 4, 1 nM con una inyección de 0 nM (es decir, solamente tampón) usada para doble referencia de las curvas de unión, el tampón usado es el tampón HBS-EP. Se usó NaOH 50 mM para regenerar la superficie de captura. Los datos se ajustaron al modelo 1:1 usando el software de análisis inherente al ProteOn XPR36. El ciclo 1 corresponde a la primera exploración de variantes de CA8 humanizadas (serie J0 a J5) y el ciclo 2, a la segunda exploración de variantes de CA8 humanizadas (serie J5 a J9). Ambos ciclos se llevaron a cabo a 25 °C.
Los datos obtenidos del ciclo 1 se exponen en la Tabla 8 y los datos del ciclo 2 se exponen en la Tabla 9. Varias moléculas en el Ciclo 2 (Tabla 09) no dieron valores de afinidad medibles por ProteOn, esto se debió a que la velocidad de disociación estaba más allá de la sensibilidad de la máquina en este ensayo. Esto no indica sin embargo, que todas estas moléculas se unan estrechamente a BCMA humano recombinante. Del Ciclo 1 los datos indican que algunas construcciones no mostraron ninguna unión con BCMA de cynomolgus recombinante.
Tabla 8: Análisis cinéticos de Ciclo 1 de moléculas anti-BCMA frente a BCMA Humano Recombinante


Tabla 9. Ciclo 2. Análisis cinéticos de moléculas anti-BCMA frente a BCMA Humano Recombinante
Para los anticuerpos J8M0, J9M0, J8M1, J9M2, J7M2, J5M0, J7M1, J7M0, J8M2, J9M1, J5M2, J5M1 la velocidad de disociación estuvo más allá de la sensibilidad del ensayo, por lo tanto no se muestran datos.

4.8. Análisis de BIAcore de construcciones humanizadas y quiméricas de CA8 anti-BCMA (serie J7 a J9)
Se inmovilizó proteína A en una microplaca CM5 (GE Healthcare, Cat Ns: BR-1005-30) por acoplamiento de amina primaria y esta superficie se usó después para capturar las moléculas de anticuerpo. Se usó BCMA humano recombinante (US Biological, B0410) como analito a 256 nM, 64 nM, 16 nM, 4 nM y 1 nM. La regeneración de la superficie de captura se llevó a cabo usando NaOH 50 mM. Se tomaron dobles referencias de todas las curvas de unión con una inyección de tampón (es decir, 0 nM) y los datos se ajustaron a los del uso del modelo 1:1 inherente al software de evaluación T100. El ciclo se llevó a cabo a 37 °C, usando HBS-EP como el tampón de ejecución.
Los resultados mostraron que las moléculas ensayadas con la excepción de J9M2 se unen a BCMA humano recombinante, con afinidad similar a la molécula quimérica. Los datos generados de este experimento se presentan en la tabla 10.
Tabla 10. Análisis cinético de moléculas humanizadas anti-BCMA frente a BCMA humano recombinante


4.9 Análisis de BIAcore de construcciones quiméricas y humanizadas de CA8 anti-BCMA J6M0 y J9M0
Se inmovilizó proteína A en una microplaca CM5 (GE Healthcare, Cat N°: BR-1005-30) por acoplamiento de amina primaria y esta superficie se usó después para capturar las moléculas de anticuerpo. Se usó BCMA humano recombinante (US Biological, B0410) como analito a 256 nM, 64 nM, 16 nM, 4 nM y 1 nM. Se llevó a cabo regeneración de la superficie de captura usando NaOH 50 mM. Se tomaron doble referencias de todas las curvas de unión con una inyección de tampón (es decir, 0 nM). Los datos se ajustaron a los del uso de modelo 1:1 inherente a software de evaluación T100. El ciclo se llevó a cabo a 25 °C y 37 °C para el experimento 1 y solamente 37 °C para el experimento 2 usando HBS-EP como el tampón de ejecución.
Ambos ciclos identificaron J9M0 como la mejor molécula con respecto a la afinidad global por BCMA humano. Los datos generados de este experimento se presentan en la tabla 11.
Tabla 11 Análisis cinéticos de moléculas humanizadas anti-BCMA frente a BCMA humano

4.10 Análisis de ProteOn de nuevas construcciones quiméricas anti-BCMA
La exploración inicial de las nuevas variantes quiméricas del segundo lote de hibridomas se llevó a cabo en el ProteOn
XPR36 (Biorad). El procedimiento fue como sigue; la Proteína A se inmovilizó en una microplaca GLM (Biorad, Cat Ns: 176-5012) por acoplamiento de amina primaria, se capturaron después variantes anti-BCMA en esta superficie y se pasó BCMA humano recombinante (material interno) a 256, 64, 16, 4, 1 nM con una inyección 0 nM (es decir, tampón solamente) usada para realizar doble referencia de las curvas de unión, el tampón usado es el tampón HBS-EP. Se llevó a cabo regeneración de la superficie de captura usando NaOH 50 nM. Los datos se ajustaron al modelo 1:1 usando el software de análisis inherente al ProteOn XPR36. El ciclo se llevó a cabo a 25 °C.
Se presentan datos generados de este experimento en la tabla 12.
Tabla 12: Análisis cinéticos de moléculas humanizadas anti-BCMA frente a BCMA humano.

Ejemplo 5 Ensayos de muerte celular
5.1 Potencias de ADCC de CA8 quimérico y versión de CA8 quimérico desfucosilado en células ARH77 que expresan BCMA
Se incubaron linfocitos citolíticos naturales (NK) humanos con células diana transfectadas con BCMA ARH77A marcadas con europio (10B5) en presencia de diversas concentraciones de anticuerpo a una relación E:T de 5:1 durante 2 horas. Se midió la liberación de europio de las células diana y se calculó la lisis específica.
Resultado: CA8 quimérico y CA8 quimérico desfucosilado destruyeron células diana que expresaban BCMA mediante ADCC. El anticuerpo quimérico desfucosilado mostró actividad ADCC más potente, como se midió por un mayor porcentaje de lisis conseguido con todas las células diana ensayadas y una CE50 diez veces más baja en la línea celular diana que expresaba BCMA alto 10B5, en comparación con el anticuerpo quimérico precursor. Véase Figura 8A y 8B.
5.2. Actividad ADCC de anticuerpos humanizados CA8 usando células diana que expresan BCMA ARH77 y PBMC como efectores
Se incubaron PBMC humanas con células diana transfectadas con BCMA ARH77 marcadas con europio (10B5) en presencia de diversas concentraciones de versiones humanizadas de anticuerpo CA8 (5 gg/ml a 0,005 gg/ml) a una relación E:T de 5:1 durante 2 horas. Se midió la liberación de europio de las células diana y se calculó la lisis específica. Resultado:
Todas las series J5, J6, J7 J8 y J9 de variantes humanizadas de CA8 mostraron actividad ADCC contra la línea celular que expresaba BCMA con alto ARH7710B5 de una manera dependiente de dosis. La ADCC estuvo a un nivel similar al hallado en los experimentos usando molécula de CA8 quimérica. Véase Figura 9.
5.3. Potencias de ADCC de S322110F02, S322110D07 y S307118G03 quiméricos y S307118G03 humanizado H3L0 frente a células ARH77 10B5 que expresan BCMA con linfocitos NK purificados como células efectoras
Se incubaron células diana citolíticas naturales (NK) humanas con células diana transfectadas con BCMA ARH77 marcadas con europio (10B5) en presencia de diversas concentraciones de anticuerpo a una relación de E:T de 5:1 durante 2 horas. Se midió la liberación de europio de las células diana y se calculó la lisis específica.
Resultado: los 4 anticuerpos ensayados mostraron actividad ADCC frente a células ARH7710B5. Véase Figura 10.
5.4. Actividad de Conjugado Anticuerpo-Fármaco (ADC) de ADC de CA8 Quimérico
Medición de actividad ADC del anticuerpo CA8 quimérico, conjugados de fármaco con anticuerpo CA8 quiméricomcMMAF y conjugados de fármaco con anticuerpo CA8 quimérico-vcMMAE frente a líneas celulares de mieloma múltiple humano.
Se trataron líneas celulares de mieloma múltiple con conjugados de fármaco-anticuerpo CA8 quimérico para determinar las concentraciones de ADC requeridas para inhibición de crecimiento y muerte.
Los conjugados de fármaco con anticuerpos ensayados se añadieron a pocilios que contenían células de mieloma múltiple a concentraciones que variaban de 1 gg/ml a 5 ng/ml. Las placas se incubaron a 37 °C durante 96 horas momento en el cual se cuantificaron las células viables usando Cell titre Glo. El anticuerpo CA8 quimérico no conjugado no mostró actividad inhibidora del crecimiento significativa a las concentraciones de anticuerpo que se ensayaron. El conjugado de anticuerpo-fármaco CA8 quimérico-mcMMAF mostró mayor actividad inhibidora del crecimiento que el conjugado de anticuerpo-fármaco CA8 quimérico-vcMMAE en las 4 de las líneas celulares de mieloma múltiple que se ensayaron. Véase Figura 11 y Tabla 13.
Tabla 13 Valores de CI50 representados en ng/ml para los conjugados de anticuerpo-fármaco CA8 quimérico-vcMMAE y CA8 quimérico-mcMMAF en 4 líneas celulares de mieloma múltiple.

5.5. Medición de la actividad de detención del ciclo celular de anticuerpo CA8 quimérico, conjugados de fármaco con anticuerpo CA8 quimérico-mcMMAF y conjugados de fármaco con anticuerpo CA8 quimérico-vcMMAE frente a línea celular de mieloma múltiple humano H929.
Para determinar el mecanismo por el que los Conjugados de Fármaco con Anticuerpo CA8 quimérico (ADC) provocan inhibición de crecimiento en células de mieloma múltiple, el ciclo celular de células NCl-H929 se controló midiendo el contenido de ADN celular a través de tinción de yoduro de propidio de células fijadas en múltiples puntos temporales después de tratamiento con anticuerpo CA8 quimérico y ADC de CA8 quimérico.
A la concentración de ADC de CA8 quimérico ensayada (50 ng/ml), el ADC CA8 quimérico-mcMMAF provocó detención del ciclo celular en G2/M significativa (contenido de ADN 4N) que alcanzó un pico a las 48 horas. En los puntos temporales posteriores de 48, 72 y 96 horas, el tratamiento con el ADC de CA8 quimérico-mcMMAF dio como resultado acumulación de una población celular con contenido de ADN por debajo de 2N, lo que es representativo de muerte celular. A la concentración ensayada de 50 ng/ml el ADC de CA8 quimérico-vcMMAE no tuvo efecto significativo en la detención del ciclo celular en G2/M o acumulación sub-G1. Véase Figura 12.
5.6. Tinción con Fosfo-Histona-H3 (Thr11) como un marcador para conjugado de fármaco con anticuerpo CA8 quimérico-mcMMAF y conjugado de fármaco con anticuerpo CA8 quimérico-vcMMAE indujo detención mitótica.
Para determinar si la acumulación de células con contenido de ADN 4N es un resultado específico de la detención mitótica inducida por ADC con CA8 quimérico se tiñeron células NCI-H929 con un anticuerpo anti-fosfo-Histona H3 después del tratamiento con concentraciones crecientes de CA8 quimérico no conjugado, CA8 quimérico-vcMMAE o CA8 quimérico-mcMMAF durante 48 horas.
El tratamiento con ADC de CA8 quimérico dio como resultado una acumulación dependiente de dosis de células NCI-H929 que se tiñeron positivas para peróxido-Histona H3 (Thr11), un marcador específico de células mitóticas. El ADC de CA8 quimérico-mcMMAF provocó la acumulación de células positivas para peróxido-Histona H3 a concentraciones más bajas que el ADC de CA8 quimérico-vcMMAE. Véase Figura 13.
5.7 Medición de apoptosis en células NCI-H929 en respuesta a ADC de CA8 quimérico por tinción con respecto a Anexina-V.
Para determinar si la acumulación de células con contenido de ADN sub-2N es un resultado específico de apoptosis inducida por los ADC de CA8 quimérico, se tiñeron células NCI-H929 con un anticuerpo anti-Anexina V después del tratamiento con concentraciones crecientes de CA8 quimérico no conjugado, CA8 quimérico-vcMMAE o CA8 quimérico-mcMMAF durante 48 horas. El tratamiento con ADC de CA8 quimérico dio como resultado una acumulación dependiente de dosis de células NCI-H929 que se tiñeron positivas con respecto a Anexina-V, un marcador específico de apoptosis. El ADC de CA8 quimérico-mcMMAF provocó la acumulación de células positivas para Anexina-V a concentraciones más bajas que el ADC de CA8 quimérico-vcMMAE. Véase Figura 14.
5.8 Actividad de Conjugado de Anticuerpo-Fármaco (ADC) de variantes humanizadas de conjugados de anticuerpo anti-BCMA CA8-fármaco
Las células se sembraron en placas de 96 pocillos (4.000 células por pocillo en 100 gl de RPMI FBS 10 %).
Se añadió anticuerpo desnudo o ADC 6 horas después de la siembra de las células y las placas se incubaron durante 144 horas. Se midió la inhibición del crecimiento en presencia de los anticuerpos o ADC a las 144 horas usando Cell Titre glo. Los puntos de datos representan la media de mediciones de CellTiterGlo por triplicado. Las barras de error representan el error típico.
Se trataron líneas celulares de mieloma múltiple NCI-H929 y OPM2 con conjugados de anticuerpo anti-BCMA CA8 humanizado-fármaco para determinar las concentraciones de ADC requeridas para inhibición del crecimiento y muerte. Las formas conjugadas de anticuerpo-fármaco con mcMMAF y vcMMAE de estos anticuerpos mostraron actividad inhibidora del crecimiento significativa comparable con la hallada con la quimera de CA8. La variante J6M0 mostró mayor potencia que la quimera y los datos se muestran en la Figura 15 en células H929 y células OPM2. El conjugado de anticuerpo-fármaco con mcMMAF mostró mayor actividad inhibidora del crecimiento que el conjugado de anticuerpo-fármaco con vcMMAE para todos los anticuerpos en ambas líneas celulares ensayadas. Se muestran resultados para todas las variantes humanizadas en la Tabla 14.
Tabla 14. Valores de CI50 representados en ng/ml para los conjugados de anticuerpo anti BCMA-fármaco en células NCI-H929 y U266-B1

5.9 Actividad de conjugado anticuerpo-fármaco (ADC) de otros conjugados de fármaco-anticuerpo anti-BCMA murino
Se sembraron células en placas de 96 pocillos (4.000 células por pocillo en 100 μl de RPMI FBS 10 %).
Se añadió anticuerpo o ADC 6 horas después de la siembra de las células y las placas se incubaron durante 144 horas. Se midió la inhibición del crecimiento en presencia de los ADC a las 144 horas usando CellTiterGlo. Se muestra la media de mediciones de CellTiterGlo por triplicado. Las Tablas 15a y 15b son de experimentos llevados a cabo a diferentes tiempos en diferentes series de anticuerpos. Se usaron las líneas celulares de mieloma múltiple NCI-H929 y U266-B1 para los anticuerpos en la Tabla 15a.
Las formas conjugadas de anticuerpo-fármaco con mcMMAF y vcMMAE de los anticuerpos murinos S322110D07, S332121F02 y S332136E04 mostraron actividad inhibidora del crecimiento significativa. El conjugado de anticuerpofármaco con mcMMAF mostró mayor actividad inhibidora del crecimiento que el conjugado de anticuerpo-fármaco con vcMMAE en todos los anticuerpos anti-BCMA murinos ensayados en los que se vio actividad. Se muestran las cifras
de CI50 en la Tabla 15a. Véase la Figura 16 para curvas de respuesta a dosis para esos tres anticuerpos y también S107118G03. Las barras de error representan el error típico. Las células NCI-H929, U266-B1, JJN3 y o Pm 2 para anticuerpos en la Tabla 15b se trataron con una serie diferente de conjugados de fármaco-anticuerpo anti-BCMA murino para determinar las concentraciones de ADC requeridas para inhibición del crecimiento y muerte. Se muestran las cifras de CI50 en la Tabla 15b. Los 5 anticuerpos mostrados en la tabla tuvieron actividad ADC significativa.
Tabla 15a. Valores de CI50 representados en ng/ml para los conjugados de fármaco-anticuerpo anti-BCMA en células NCl-H929 y U266-B1

g
Tabla 15b Valores de CI50 representados en ng/ml para los conjugados de fármaco-anticuerpo anti BCMA en células NCI-H929, U266-B1, JJN3 y OPM2

5.10 Potencia de ADCC de J6M0 afucosilado conjugado (Potelligent)
Se ensayó J6M0 afucosilado conjugado con MMAE o MMAF en ensayos de ADCC usando transfectantes de BCMA para asegurar que su actividad ADCC no se viera comprometida por la conjugación. Se incubaron células ARH77-10B5 marcadas con europio con diversos anticuerpos de BMCA Potelligent y J6M0 WT a concentraciones de hasta 10000 ng/ml durante 30 minutos antes de la adición de PBMC (relación de PBMC: células diana 50:1). Dos horas después se tomó una muestra de una alícuota de medio celular y se mezcló con solución de potenciación. Después de 30 minutos en un agitador de placas, se controló la liberación de europio en el lector de multimarcador Victor 2 1420. Los puntos de datos representan medias de los valores por triplicado. Estos datos son representativos de los 2 experimentos.
No hubo diferencias significativas en la potencia de ADCC entre las formas no conjugada y ADC de J6M0 Potelligent. En el mismo experimento se incluyó una versión de tipo silvestre de J6M0 para mostrar la comparación de la potencia con la versión afucosilada. Como se esperaba, la desfucosilación dio como resultado una CE50 menor y una lisis máxima mayor. No se observó lisis con la forma deshabilitada de Fc de J6M0 (Figura 17).
5.11 Potencia de ADCC de J6M0 afucosilado en líneas celulares MM
Se incubaron PBMC humanas con células diana de mieloma múltiple a una relación de E:T de 50:1 en presencia de diversas concentraciones de J6M0 afucosilado (Potelligent). El porcentaje de células diana restantes en la mezcla de efector células diana después de 18 horas se midió por FACS usando un anticuerpo anti-CD138 marcado con fluorescencia para detectar las células diana y se calculó el porcentaje de lisis. Éste es representativo de varios experimentos.
El anticuerpo J6M0 Potelligent mostró actividad ADCC contra las cinco líneas celulares diana de mieloma múltiple ensayadas. Fue importante ensayar esto puesto que se llevaron a cabo estudios anteriores usando células transfectadas. Los resultados se muestran en la Figura 18. Se muestra el conjunto de datos completo con múltiples donadores en la Tabla 16. Las potencias estuvieron todas en un intervalo similar al hallado con los transfectantes. La actividad ADCC no estaba directamente relacionada con la expresión de superficie de BCMA en estas líneas celulares.
Tabla 16. Valores de CE50 generados en 13 ensayos independientes usando 11 donares (designados A-K) a lo largo de las cinco líneas celulares de mieloma múltiple.

Ejemplo 6. Datos de xenotrasplante
6.1. Se ensayaron xenotrasplantes murinos de líneas celulares MM humanas para asegurar que la potencia de anticuerpo detectada in vitro también podía demostrarse in vivo. La línea celular seleccionada para estudios de xenotrasplantes fue NCI-H929 que es sensible a muerte por ADC y ADCC in vitro. Se llevaron a cabo estudios en ratones Cb .17 SCID inmunocomprometidos que carecían de linfocitos T y B pero que mantenían linfocitos NK para posibilitar la actividad ADCC. Sin embargo debería observarse que aunque IgG1 humana puede entrar en contacto con receptores Fc murinos, la potenciación de Potelligent no mejora la afinidad como lo hace con receptores de Fc humanos.
6.2. Impacto de J6M0 no conjugado y conjugado con MMAE o MMAF en crecimiento de tumor de NCI-H929
Para analizar de forma independiente las actividades tanto de ADCC como de ADC de J6M0 los inventores ensayaron el anticuerpo J6M0 en presencia y ausencia de conjugación con MMAF o MMAE. Ensayando el J6M0 no conjugado, cualquier efecto antitumoral podría atribuirse a alguna combinación de ADCC y actividad inhibidora funcional.
Los ratones con tumores NCI-H929 que habían alcanzado un volumen de 200 mm3 de media se trataron con un control
de IgG1 humana o el anticuerpo J6M0 (no conjugado, MMAE o MMAF) dos veces por semana a una dosis de 50 pg o 100 μg, durante 2 semanas. Los resultados de este estudio muestran que una dosis de 100 μg del conjugado J6M0-MMAF dio como resultado eliminación de tumores en los ratones que habían completado la dosificación. Los ratones J6M0-MMAF se mantuvieron durante 40 días después de la última dosis sin que se produjera recurrencia del tumor. Estos resultados de este experimento demuestran que la conjugación con MMAF había aumentado la actividad antitumoral tanto frente a anticuerpo J6M0 no conjugado como frente a conjugado J6M0-MMAE. Véase Figura 19.
Ejemplo 7. Evaluación de Niveles de BCMA Soluble de Suero de Paciente con MM
7.1 No se sabe en la actualidad si BCMA está presente de forma extracelular y puede detectarse en la sangre. En este trabajo, los inventores determinaron el nivel en suero de BCMA humano de pacientes con MM. Las muestras de suero de 54 pacientes con discrasia de células plasmáticas y MM y 20 muestras de control normales se analizaron por ELISA. Se obtuvo la Aprobación para Sujetos Humanos de la Junta de Evaluación Institucional Occidental.
7.2 Evaluación de los niveles de BCMA Humano en Suero
Se recogió sangre de pacientes y controles normales en la clínica, en tubos de recogida de suero. Las muestras de pacientes con MM fueron de una diversidad de etapas (enfermedad progresiva, remisión, recaída, nuevo diagnóstico y otras). Las muestras sanguíneas se centrifugaron a 10.000 rpm durante 10 minutos y el suero se transfirió a tubos de plástico de microcentrífuga estériles.
Se usó un kit de ELISA de BCMA humano/TNFRSF17 de R&D Systems (N° de catalogo DY193E) que mide los niveles de BCMA humano soluble para detectar BCMA siguiendo el protocolo convencional proporcionado con el kit.
Brevemente, se revistieron microplacas de 96 pocillos con 100 pl por pocillo de anticuerpo de captura y se incubó durante una noche a 4 °C. Las placas se lavaron tres veces con tampón de lavado (Tween 200,05 % en PBS, pH 7,2) y se bloqueó con 300 pl de BSA 1 % en PBS a temperatura ambiente durante 2 horas. Las placas se lavaron tres veces con tampón de lavado. Se añadieron 100 pl de muestra de suero o patrón a cada pocillo y se incubó durante 2 horas a temperatura ambiente. Las placas se lavaron tres veces con tampón de lavado y después se añadieron 100 pl del anticuerpo de detección a cada pocillo y se incubaron durante 2 horas a temperatura ambiente. Se añadieron 100 pl de estreptavidina-HRP en cada pocillo después de lavar las placas tres veces y se incubaron en una habitación oscura durante 20 minutos. Las placas se lavaron tres veces y se añadieron 50 pl de solución de parada y después se determinó por lector de microplacas con longitud de onda de 570 nm.
Se llevó a cabo una serie de ensayos para determinar el factor de dilución en suero apropiado para los niveles de BCMA que estaban presentes. Se descubrió que un factor de dilución de 1:500 era adecuado para la mayoría de las muestras y es el factor de dilución usado en los datos mostrados en la Figura 20. El conjunto de datos completo se muestra en la Tabla 17.
Las muestras de suero de pacientes y de control normal diluidas y procesadas por triplicado tuvieron niveles de BCMA determinados. Los niveles en suero de BCMA estaban significativamente elevados en los sueros de pacientes de MM en comparación con controles normales en este estudio. Cuando el subconjunto de enfermedad se dividió adicionalmente hubo una tendencia hacia niveles en suero elevados de BCMA en los sueros de pacientes con MM en progreso en comparación con los de remisión. Este es el primer informe que identifica BCMA en suero en cualquier enfermedad humana y sugiere que estos niveles pueden ser un nuevo biomarcador para controlar el estado de enfermedad y la respuesta terapéutica de pacientes con MM y para otros pacientes con enfermedades mediadas por células plasmáticas.
Tabla 17. Cifras que representan la concentración en suero de BCMA soluble en ng/ml calculadas a partir de muestras diluidas a 1/50, 1/500 y 1/5000. Los P valores se calcularon usando el Ensayo de T de una cola y los valores a 95 % de significación están debajo de la tabla.


P-Valores (Ensayo de T de una cola, 95 % de Significación)
~ 1 -500 Sencillo
Normal frente a Progresivo: p = 0,0010*
Progresivo frente a Remisión: p = 0,0146*
~ 1 -500 por triplicado
Normal frente a Progresivo: p = 0,0004*
Progresivo frente a Remisión: p = 0,091*
~ 1 -50 Ensayo 1
Normal frente a Progresivo: p = 0,0171*
Progresivo frente a Remisión: p = 0,0777*
~ 1 -50 Ensayo 2
Normal frente a Progresivo: p = 0,0184*
Progresivo frente a Remisión: p = 0,0876
* muestra la significación
Sumario de Secuencias (Tabla C)





Claims (21)
1. Un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) que comprende una región variable de cadena pesada de SEC ID N°. 23 y una región variable de cadena ligera de SEC ID N°. 31.
2. Un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) de acuerdo con la reivindicación 1 que comprende una cadena pesada de SEC ID N°. 55 y una cadena ligera de SEC ID N°. 63.
3. Un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) de acuerdo con cualquier reivindicación anterior en el que el anticuerpo es un anticuerpo monoclonal.
4. Un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) de acuerdo con cualquier reivindicación anterior en el que el anticuerpo tiene unión potenciada a FcyRIIIA o tiene función efectora mediada por FcyRIIIA potenciada.
5. Un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) de acuerdo con la reivindicación 4 en el que el anticuerpo está desfucosilado.
6. Secuencias de ácido nucleico que codifican ambas de las secuencias variables de cadena pesada y secuencias variables de cadena ligera del anticuerpo de cualquier reivindicación anterior.
7. Secuencias de ácido nucleico que comprenden SEC ID N°. 56 que codifica la cadena pesada; y SEC ID N°.
64 que codifica la cadena ligera del anticuerpo de cualquier reivindicación anterior.
8. Un vector de expresión que comprende las secuencias de ácido nucleico de acuerdo con la reivindicación 6 o la reivindicación 7.
9. Una célula huésped transfectada o transformada recombinante que comprende las secuencias de ácido nucleico de acuerdo con la reivindicación 6 o la reivindicación 7, o el vector de expresión de acuerdo con la reivindicación 8 en la que la célula huésped comprende ambas secuencias de ácido nucleico que codifican la cadena pesada y la cadena ligera.
10. Un procedimiento para la producción de un anticuerpo, comprendiendo el procedimiento cultivar la célula huésped de acuerdo con la reivindicación 9 en condiciones adecuadas para la expresión de dichas secuencias de ácido nucleico o vector(es), por lo cual se produce el anticuerpo.
11. Un anticuerpo como se produce por el procedimiento de la reivindicación 10.
12. Un inmunoconjugado que comprende el anticuerpo de una cualquiera de las reivindicaciones 1 a 5 o la reivindicación 11 y un agente citotóxico.
13. Un inmunoconjugado de acuerdo con la reivindicación 12 en el que el agente citotóxico es monometil auristatina E (MMAE) o monometil auristatina F (MMAF).
14. Un inmunoconjugado de la reivindicación 13 en el que el agente citotóxico es monometil auristatina F (MMAF).
15. Un inmunoconjugado de una cualquiera de las reivindicaciones 12 a 14 en el que el anticuerpo se une al agente citotóxico a través de un engarce.
16. Un inmunoconjugado de la reivindicación 15, en el que el engarce se selecciona de 6-maleimidocaprαlo (MC), maleimido propanoílo (MP), valina-citrulina (val-cit), alanina-fenilalanina (ala-phe), paminobenciloxicarbonilo (PAB), N-Succinimidil 4-(2-piridiltio)pentanoato (SPP), N-succinimidil 4-(N-maleimidometil)ciclohexano-1 carboxilato (SMCC), y N-Succinimidil (4-yodo-acetil) aminobenzoato (SIAB).
17. Un inmunoconjugado de la reivindicación 16 en el que el engarce es 6-maleimidocaprαlo (MC).
18. Un inmunoconjugado que comprende un anticuerpo anti-antígeno de maduración de linfocitos B (CD269) y un agente citotóxico en el que el anticuerpo tiene unión potenciada a FcyRIIIA o tiene función efectora mediada por FcyRIIIA potenciada y comprende la secuencia de aminoácidos de cadena pesada de acuerdo con la SEC ID N°. 55 y la secuencia de aminoácidos de cadena ligera de acuerdo con la reivindicación SEC ID N°. 63 y en el que el anticuerpo está desfucosilado y en el que el agente citotóxico es monometil auristatina F (MMAF) y en el que dicho MMAF se une a dicho anticuerpo a través de un engarce 6-maleimidocaprαlo (MC).
19. Una composición farmacéutica que comprende el anticuerpo de acuerdo con una cualquiera de las reivindicaciones 1 a 5 o la reivindicación 11 del inmunoconjugado de acuerdo con una cualquiera de las reivindicaciones 12 a 18 y un vehículo farmacéuticamente aceptable.
20. Un anticuerpo de acuerdo con una cualquiera de las reivindicaciones 1 a 5 y 11, un inmunoconjugado de acuerdo con una cualquiera de las reivindicaciones 12 a 18, o una composición farmacéutica de acuerdo con la reivindicación 19 para su uso en el tratamiento de un paciente humano aquejado de una enfermedad o trastorno de linfocitos B seleccionado de Mieloma Múltiple (MM), leucemia linfocítica crónica (CLL), Mieloma múltiple no secretor, Mieloma múltiple latente, gammapatía monoclonal de importancia indeterminada (MGUS), plasmacitoma solitario (Hueso, Extramedular), linfoma linfoplasmático (LPL), Macroglobulinemia de Waldenstrom, leucemia de células plasmáticas, Amiloidosis Primaria (AL), enfermedad de cadena pesada, lupus eritematoso sistémico (SLE), síndrome de POEMS / mieloma osteosclerótico, crioglobulinemia de Tipo I y II, enfermedad de deposición de cadena ligera, síndrome de Goodpasture, púrpura trombocitopénica idiopática (ITP), glomerulonefritis aguda, Pénfigo y trastornos Penfigoides y Epidermólisis ampollosa adquirida, o cualquier leucemia de linfocitos B linfoma no Hodgkin o linfoma de Hodgkin.
21. El anticuerpo, inmunoconjugado o composición farmacéutica para su uso de acuerdo con la reivindicación 20 en el que la enfermedad de linfocitos B es mieloma múltiple (MM).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161490732P | 2011-05-27 | 2011-05-27 | |
| US201261647196P | 2012-05-15 | 2012-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2953190T3 true ES2953190T3 (es) | 2023-11-08 |
Family
ID=46148885
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18163942T Active ES2953190T3 (es) | 2011-05-27 | 2012-05-24 | Proteínas de unión a BCMA (CD269/TNFRSF17) |
Country Status (40)
| Country | Link |
|---|---|
| US (1) | US20140105915A1 (es) |
| EP (4) | EP3415531B1 (es) |
| JP (7) | JP6263467B2 (es) |
| KR (1) | KR101972446B1 (es) |
| CN (2) | CN103562225B (es) |
| AR (1) | AR086579A1 (es) |
| AU (1) | AU2012264890C1 (es) |
| BR (1) | BR112013028779B8 (es) |
| CA (1) | CA2833820C (es) |
| CL (2) | CL2013003373A1 (es) |
| CO (1) | CO6811809A2 (es) |
| CR (1) | CR20130624A (es) |
| CY (2) | CY2023019I2 (es) |
| DK (1) | DK3415531T5 (es) |
| DO (1) | DOP2013000280A (es) |
| EA (2) | EA201790330A1 (es) |
| ES (1) | ES2953190T3 (es) |
| FI (2) | FI3415531T3 (es) |
| FR (1) | FR23C1032I2 (es) |
| HR (1) | HRP20231066T1 (es) |
| HU (2) | HUE063461T2 (es) |
| IL (2) | IL228784B (es) |
| LT (2) | LT3415531T (es) |
| LU (1) | LUC00314I2 (es) |
| MA (1) | MA35208B1 (es) |
| MX (1) | MX351069B (es) |
| MY (1) | MY177970A (es) |
| NL (1) | NL301241I2 (es) |
| PE (1) | PE20141045A1 (es) |
| PH (1) | PH12013502421A1 (es) |
| PL (1) | PL3415531T3 (es) |
| PT (1) | PT3415531T (es) |
| RS (1) | RS64791B1 (es) |
| SG (1) | SG194176A1 (es) |
| SI (1) | SI3415531T1 (es) |
| SM (1) | SMT202300400T1 (es) |
| TW (2) | TWI609883B (es) |
| UY (1) | UY34101A (es) |
| WO (1) | WO2012163805A1 (es) |
| ZA (1) | ZA201308635B (es) |
Families Citing this family (248)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100447244C (zh) | 1999-08-17 | 2008-12-31 | 比奥根艾迪克Ma公司 | Baff受体(bcma),一种免疫调节剂 |
| KR101972446B1 (ko) * | 2011-05-27 | 2019-04-25 | 글락소 그룹 리미티드 | Bcma(cd269/tnfrsf17)결합 단백질 |
| UA112434C2 (uk) | 2011-05-27 | 2016-09-12 | Ґлаксо Ґруп Лімітед | Антигензв'язувальний білок, який специфічно зв'язується з всма |
| TWI679212B (zh) | 2011-11-15 | 2019-12-11 | 美商安進股份有限公司 | 針對bcma之e3以及cd3的結合分子 |
| RU2766608C2 (ru) | 2012-04-11 | 2022-03-15 | Дзе Юнайтед Стейтс Оф Америка, Эз Репрезентед Бай Дзе Секретари, Департмент Оф Хелс Энд Хьюман Сёрвисез | Химерные антигенные рецепторы, нацеленные на антиген созревания b-клеток |
| WO2014068079A1 (en) | 2012-11-01 | 2014-05-08 | Max-Delbrück-Centrum für Molekulare Medizin | An antibody that binds cd269 (bcma) suitable for use in the treatment of plasma cell diseases such as multiple myeloma and autoimmune diseases |
| US9243058B2 (en) | 2012-12-07 | 2016-01-26 | Amgen, Inc. | BCMA antigen binding proteins |
| TW201425336A (zh) * | 2012-12-07 | 2014-07-01 | Amgen Inc | Bcma抗原結合蛋白質 |
| EP2762496A1 (en) * | 2013-02-05 | 2014-08-06 | EngMab AG | Method for the selection of antibodies against BCMA |
| WO2014122143A1 (en) | 2013-02-05 | 2014-08-14 | Engmab Ag | Method for the selection of antibodies against bcma |
| EP2762497A1 (en) * | 2013-02-05 | 2014-08-06 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA |
| WO2014124280A1 (en) * | 2013-02-08 | 2014-08-14 | Institute For Myeloma & Bone Cancer Research | Improved diagnostic, prognostic, and monitoring methods for multiple myeloma, chronic lymphocytic leukemia, and b-cell non-hodgkin lymphoma |
| AR095374A1 (es) * | 2013-03-15 | 2015-10-14 | Amgen Res Munich Gmbh | Moléculas de unión para bcma y cd3 |
| GB2513405A (en) | 2013-04-26 | 2014-10-29 | Adc Biotechnology Ltd | Method of synthesising ADCs using affinity resins |
| DK3019240T3 (da) | 2013-07-09 | 2024-06-03 | Annexon Inc | Anti-komplementfaktor C1Q antistoffer og anvendelser deraf |
| BR112016018100A2 (pt) | 2014-02-07 | 2018-02-20 | Univ Mcmaster | acoplador antigênico de células t trifuncional, métodos e usos do mesmo |
| CA2939411A1 (en) | 2014-02-27 | 2015-09-03 | Ucl Business Plc | Ligand |
| US20170335281A1 (en) | 2014-03-15 | 2017-11-23 | Novartis Ag | Treatment of cancer using chimeric antigen receptor |
| HUE049514T2 (hu) | 2014-04-25 | 2020-09-28 | Bluebird Bio Inc | Javított eljárások adoptív sejtterápiák kialakítására |
| KR102526945B1 (ko) * | 2014-04-30 | 2023-04-27 | 막스-델부뤽-센트럼 퓌어 몰레쿨라레 메디친 인 데어 헬름홀츠-게마인샤프트 | Cd269에 대한 인간화 항체 |
| SI3151672T1 (sl) | 2014-06-06 | 2021-03-31 | Bluebird Bio, Inc. | Izboljšani T-celični sestavki |
| BR112017001183A2 (pt) | 2014-07-21 | 2017-11-28 | Novartis Ag | tratamento de câncer usando receptor de antígeno quimérico anti-bcma humanizado |
| SG10201913782UA (en) | 2014-07-21 | 2020-03-30 | Novartis Ag | Treatment of cancer using a cll-1 chimeric antigen receptor |
| JP2017528433A (ja) | 2014-07-21 | 2017-09-28 | ノバルティス アーゲー | 低い免疫増強用量のmTOR阻害剤とCARの組み合わせ |
| ES2878449T3 (es) | 2014-07-24 | 2021-11-18 | 2Seventy Bio Inc | Receptores antigénicos quiméricos de BCMA |
| EP2982692A1 (en) | 2014-08-04 | 2016-02-10 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA |
| JP6839074B2 (ja) | 2014-09-17 | 2021-03-03 | ノバルティス アーゲー | 養子免疫療法のためのキメラ受容体での細胞毒性細胞のターゲティング |
| GB201419185D0 (en) * | 2014-10-28 | 2014-12-10 | Adc Biotechnology Ltd | Method of synthesising ADCs using affinity resin |
| AU2015339012B2 (en) | 2014-10-31 | 2020-11-05 | Abbvie Biotherapeutics Inc. | Anti-CS1 antibodies and antibody drug conjugates |
| EP3023437A1 (en) | 2014-11-20 | 2016-05-25 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA |
| EP3029068A1 (en) | 2014-12-03 | 2016-06-08 | EngMab AG | Bispecific antibodies against CD3epsilon and BCMA for use in the treatment of diseases |
| HRP20191873T1 (hr) | 2014-12-12 | 2020-01-24 | Bluebird Bio, Inc. | Kimerni antigenski receptori |
| WO2016126608A1 (en) | 2015-02-02 | 2016-08-11 | Novartis Ag | Car-expressing cells against multiple tumor antigens and uses thereof |
| US10294304B2 (en) * | 2015-04-13 | 2019-05-21 | Pfizer Inc. | Chimeric antigen receptors targeting B-cell maturation antigen |
| TWI703159B (zh) * | 2015-04-13 | 2020-09-01 | 美商輝瑞股份有限公司 | Bcma特異性治療性抗體及其用途 |
| EP3286211A1 (en) | 2015-04-23 | 2018-02-28 | Novartis AG | Treatment of cancer using chimeric antigen receptor and protein kinase a blocker |
| US9708412B2 (en) | 2015-05-21 | 2017-07-18 | Harpoon Therapeutics, Inc. | Trispecific binding proteins and methods of use |
| SI3331910T1 (sl) * | 2015-08-03 | 2020-07-31 | Engmab Sarl | Monoklonska protitelesa proti humani antigen dozorevanja limfocitov B (BCMA) |
| US11667691B2 (en) | 2015-08-07 | 2023-06-06 | Novartis Ag | Treatment of cancer using chimeric CD3 receptor proteins |
| WO2017072716A1 (en) * | 2015-10-30 | 2017-05-04 | Glaxosmithkline Intellectual Property Development Limited | Prognostic method |
| CN108473575B (zh) * | 2015-11-13 | 2022-04-19 | 美国卫生和人力服务部 | 抗-bcma多肽和蛋白质 |
| FI3380620T3 (fi) | 2015-11-23 | 2024-08-01 | Novartis Ag | Optimoituja lentiviruksen siirtovektoreita ja niiden käyttötapoja |
| EP3380518B1 (en) | 2015-11-24 | 2026-03-25 | Annexon, Inc. | Anti-complement factor c1q fab fragments and uses thereof |
| FI3380522T3 (fi) * | 2015-11-25 | 2024-01-16 | Visterra Inc | Vasta-ainemolekyylejä april-proteiinille ja niiden käyttöjä |
| JP2019505476A (ja) | 2015-12-01 | 2019-02-28 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 組合せ処置およびその方法 |
| US11479755B2 (en) | 2015-12-07 | 2022-10-25 | 2Seventy Bio, Inc. | T cell compositions |
| RU2018127657A (ru) | 2015-12-30 | 2020-01-31 | Новартис Аг | Виды терапии на основе иммуноэффекторных клеток с улучшенной эффективностью |
| ES2898329T3 (es) | 2016-01-12 | 2022-03-07 | Oncotracker Inc | Métodos mejorados para supervisar el estado inmunitario de un sujeto |
| CN108778329B (zh) * | 2016-02-17 | 2022-09-16 | 西雅图基因公司 | Bcma抗体和其用以治疗癌症和免疫病症的用途 |
| KR20180118175A (ko) | 2016-03-04 | 2018-10-30 | 노파르티스 아게 | 다중 키메라 항원 수용체 (car) 분자를 발현하는 세포 및 그에 따른 용도 |
| EP3432924A1 (en) | 2016-03-23 | 2019-01-30 | Novartis AG | Cell secreted minibodies and uses thereof |
| CA3019650C (en) | 2016-04-01 | 2023-07-25 | Kite Pharma, Inc. | Chimeric receptors and methods of use thereof |
| ES2891578T3 (es) | 2016-04-01 | 2022-01-28 | Kite Pharma Inc | Antígeno quimérico y receptores de células T y métodos de uso |
| TWI795133B (zh) | 2016-04-01 | 2023-03-01 | 美商凱特製藥公司 | Bcma結合分子類及彼等之用途 |
| CN109715808A (zh) | 2016-04-15 | 2019-05-03 | 诺华股份有限公司 | 用于选择性蛋白质表达的组合物和方法 |
| EP3493844A4 (en) | 2016-05-20 | 2021-03-24 | Harpoon Therapeutics Inc. | SINGLE DOMAIN SERUM ALBUMIN BINDING PROTEIN |
| US11623958B2 (en) | 2016-05-20 | 2023-04-11 | Harpoon Therapeutics, Inc. | Single chain variable fragment CD3 binding proteins |
| KR20250175345A (ko) | 2016-06-21 | 2025-12-16 | 테네오바이오, 인코포레이티드 | Cd3 결합 항체 |
| BR112019002035A2 (pt) | 2016-08-01 | 2019-05-14 | Novartis Ag | tratamento de câncer usando um receptor de antígeno quimérico em combinação com um inibidor de uma molécula pró-macrófago m2 |
| IL322083A (en) | 2016-08-24 | 2025-09-01 | Teneobio Inc | Non-human transgenic animals that produce modified heavy chain antibodies only |
| MA46236A (fr) * | 2016-09-14 | 2019-07-24 | Janssen Biotech Inc | Récepteurs antigéniques chimériques comprenant des domaines de la fibronectine de type iii spécifiques du bcma, et utilisations correspondantes |
| PE20241349A1 (es) | 2016-09-14 | 2024-07-03 | Teneobio Inc | Anticuerpos de union a cd3 |
| JP7267914B2 (ja) | 2016-11-02 | 2023-05-02 | エンクマフ エスアーエールエル | Bcma及びcd3に対する二重特異性抗体、及び多発性骨髄腫を治療するために併用して使用される免疫療法薬 |
| WO2018085690A1 (en) | 2016-11-04 | 2018-05-11 | Bluebird Bio, Inc. | Anti-bcma car t cell compositions |
| EP3548055A4 (en) | 2016-12-02 | 2020-08-19 | University of Southern California | SYNTHETIC IMMUNE RECEPTORS AND THEIR PROCESSES FOR USE |
| WO2018111340A1 (en) | 2016-12-16 | 2018-06-21 | Novartis Ag | Methods for determining potency and proliferative function of chimeric antigen receptor (car)-t cells |
| IL317134A (en) * | 2016-12-21 | 2025-01-01 | Teneobio Inc | An antibody containing only heavy chains that binds a human B-cell maturation antigen, a pharmaceutical composition containing the same, its use in the treatment of B-cell disorders and a method for its preparation |
| WO2018133877A1 (zh) * | 2017-01-23 | 2018-07-26 | 科济生物医药(上海)有限公司 | 靶向bcma的抗体及其应用 |
| ES2912408T3 (es) | 2017-01-26 | 2022-05-25 | Novartis Ag | Composiciones de CD28 y métodos para terapia con receptores quiméricos para antígenos |
| CN110582509A (zh) | 2017-01-31 | 2019-12-17 | 诺华股份有限公司 | 使用具有多特异性的嵌合t细胞受体蛋白治疗癌症 |
| MX2019009552A (es) | 2017-02-17 | 2019-10-02 | Hutchinson Fred Cancer Res | Terapias de combinacion para el tratamiento de canceres relacionados con el antigeno de maduracion de celulas b (bcma) y trastornos autoinmunitarios. |
| WO2018160731A1 (en) | 2017-02-28 | 2018-09-07 | Novartis Ag | Shp inhibitor compositions and uses for chimeric antigen receptor therapy |
| WO2018158349A1 (en) | 2017-02-28 | 2018-09-07 | Affimed Gmbh | Tandem diabody for cd16a-directed nk-cell engagement |
| US11535668B2 (en) | 2017-02-28 | 2022-12-27 | Harpoon Therapeutics, Inc. | Inducible monovalent antigen binding protein |
| CN108624549B (zh) * | 2017-03-23 | 2020-11-20 | 上海奥浦迈生物科技有限公司 | 一种cho dg44培养基及其应用 |
| EP3615055A1 (en) | 2017-04-28 | 2020-03-04 | Novartis AG | Cells expressing a bcma-targeting chimeric antigen receptor, and combination therapy with a gamma secretase inhibitor |
| US20200179511A1 (en) | 2017-04-28 | 2020-06-11 | Novartis Ag | Bcma-targeting agent, and combination therapy with a gamma secretase inhibitor |
| EP3621994A4 (en) | 2017-05-12 | 2020-12-30 | Harpoon Therapeutics, Inc. | MESOTHELIN BINDING PROTEINS |
| US11166985B2 (en) | 2017-05-12 | 2021-11-09 | Crispr Therapeutics Ag | Materials and methods for engineering cells and uses thereof in immuno-oncology |
| MX2019013514A (es) | 2017-05-12 | 2020-01-20 | Crispr Therapeutics Ag | Materiales y metodos para modificar celulas por ingenieria genetica y usos de los mismos en inmunooncologia. |
| US11635435B2 (en) | 2017-06-13 | 2023-04-25 | Oncotracker, Inc. | Diagnostic, prognostic, and monitoring methods for solid tumor cancers |
| JP2020523383A (ja) * | 2017-06-14 | 2020-08-06 | ダナ−ファーバー キャンサー インスティテュート,インコーポレイテッド | B細胞成熟抗原(bcma)指向性ナノ粒子 |
| WO2018229715A1 (en) | 2017-06-16 | 2018-12-20 | Novartis Ag | Compositions comprising anti-cd32b antibodies and methods of use thereof |
| CN110945026B (zh) * | 2017-06-20 | 2024-03-19 | 特纳奥尼股份有限公司 | 仅有重链的抗bcma抗体 |
| KR20250007003A (ko) | 2017-06-20 | 2025-01-13 | 테네오바이오, 인코포레이티드 | 항-bcma 중쇄-단독 항체 |
| MA51447A (fr) | 2017-08-01 | 2020-06-10 | Medimmune Llc | Conjugué anticorps monoclonal-médicament dirigé contre bcma |
| EP3692066A2 (en) | 2017-09-14 | 2020-08-12 | GlaxoSmithKline Intellectual Property Development Limited | Combination treatment for cancer |
| JP2020533382A (ja) * | 2017-09-14 | 2020-11-19 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 癌の組合せ治療 |
| JP2020533383A (ja) * | 2017-09-14 | 2020-11-19 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | 癌の組合せ治療 |
| CN111201243B (zh) * | 2017-09-29 | 2023-08-11 | 财团法人牧岩生命科学研究所 | 对bcma具有高亲和力的抗bcma抗体和包含其的用于治疗癌症的药物组合物 |
| US12065498B2 (en) * | 2017-09-29 | 2024-08-20 | Cell Design Labs, Inc. | Methods of making bispecific anti-CD307E and anti-BCMA chimeric antigen receptors and uses of the same |
| CN111479925B (zh) | 2017-10-12 | 2024-03-08 | 麦克马斯特大学 | 具有y182t突变的t细胞-抗原偶联物及其方法和用途 |
| IL315737A (en) | 2017-10-13 | 2024-11-01 | Harpoon Therapeutics Inc | B-cell maturation antigen-binding proteins |
| MX2020003915A (es) | 2017-10-13 | 2020-10-08 | Harpoon Therapeutics Inc | Proteinas trispecificas y metodos de uso. |
| AU2018351050B2 (en) | 2017-10-18 | 2025-09-18 | Novartis Ag | Compositions and methods for selective protein degradation |
| WO2019081983A1 (en) | 2017-10-25 | 2019-05-02 | Novartis Ag | CD32B TARGETING ANTIBODIES AND METHODS OF USE |
| US20210179709A1 (en) | 2017-10-31 | 2021-06-17 | Novartis Ag | Anti-car compositions and methods |
| SG11202003501XA (en) | 2017-11-01 | 2020-05-28 | Juno Therapeutics Inc | Antibodies and chimeric antigen receptors specific for b-cell maturation antigen |
| PT3703750T (pt) | 2017-11-01 | 2025-01-17 | Memorial Sloan Kettering Cancer Center | Recetores de antigénio quimérico específicos para o antigénio de maturação das células b e polinucleótidos codificantes |
| AU2018358004A1 (en) | 2017-11-01 | 2020-05-28 | Hummingbird Bioscience Pte. Ltd. | CD47 antigen-binding molecules |
| GB201720426D0 (en) * | 2017-12-07 | 2018-01-24 | Hummingbird Bioscience Pte Ltd | CD47 and BCMA antigen-binding molecules |
| MX2020004568A (es) | 2017-11-06 | 2020-10-05 | Juno Therapeutics Inc | Combinación de una terapia celular y un inhibidor de gamma secretasa. |
| CN111787938A (zh) | 2017-11-15 | 2020-10-16 | 诺华股份有限公司 | 靶向bcma的嵌合抗原受体、靶向cd19的嵌合抗原受体及组合疗法 |
| US20200371091A1 (en) | 2017-11-30 | 2020-11-26 | Novartis Ag | Bcma-targeting chimeric antigen receptor, and uses thereof |
| WO2019133799A1 (en) * | 2017-12-29 | 2019-07-04 | University Of Florida Research Foundation | Monoclonal antibodies targeting microtubule-binding domain of tau protein |
| US12539308B2 (en) | 2018-01-08 | 2026-02-03 | The Trustees Of The University Of Pennsylvania | Immune-enhancing RNAs for combination with chimeric antigen receptor therapy |
| AU2019215031C1 (en) | 2018-01-31 | 2026-02-26 | Novartis Ag | Combination therapy using a chimeric antigen receptor |
| PT3674328T (pt) * | 2018-02-01 | 2024-03-14 | Innovent Biologics Suzhou Co Ltd | Recetor de antigénio quimérico (car) que se liga a bcma e utilizações do mesmo |
| US11807663B2 (en) * | 2018-02-01 | 2023-11-07 | Innovent Biologics (Suzhou) Co., Ltd. | Fully humanized anti-B cell maturation antigen (BCMA) single-chain antibody and use thereof |
| US20200399383A1 (en) | 2018-02-13 | 2020-12-24 | Novartis Ag | Chimeric antigen receptor therapy in combination with il-15r and il15 |
| BR112020017053A2 (pt) | 2018-02-21 | 2020-12-15 | Celgene Corporation | Anticorpos que se ligam ao bcma e usos dos mesmos |
| WO2019195535A1 (en) * | 2018-04-05 | 2019-10-10 | Novartis Ag | Trispecific binding molecules against cancers and uses thereof |
| CN116836297A (zh) * | 2018-04-12 | 2023-10-03 | 上海赛比曼生物科技有限公司 | 靶向bcma的嵌合抗原受体及其制法和应用 |
| CA3095373A1 (en) | 2018-04-13 | 2019-10-17 | Affimed Gmbh | Nk cell engaging antibody fusion constructs |
| WO2019210153A1 (en) | 2018-04-27 | 2019-10-31 | Novartis Ag | Car t cell therapies with enhanced efficacy |
| EP3788369A1 (en) | 2018-05-01 | 2021-03-10 | Novartis Ag | Biomarkers for evaluating car-t cells to predict clinical outcome |
| MX2020012028A (es) | 2018-05-11 | 2021-03-29 | Crispr Therapeutics Ag | Metodos y composiciones para tratar el cancer. |
| EP3801769A1 (en) | 2018-05-25 | 2021-04-14 | Novartis AG | Combination therapy with chimeric antigen receptor (car) therapies |
| KR102870868B1 (ko) | 2018-06-01 | 2025-10-15 | 노파르티스 아게 | Bcma에 대한 결합 분자 및 이의 용도 |
| WO2019237035A1 (en) | 2018-06-08 | 2019-12-12 | Intellia Therapeutics, Inc. | Compositions and methods for immunooncology |
| TWI890660B (zh) | 2018-06-13 | 2025-07-21 | 瑞士商諾華公司 | Bcma 嵌合抗原受體及其用途 |
| AR116109A1 (es) | 2018-07-10 | 2021-03-31 | Novartis Ag | Derivados de 3-(5-amino-1-oxoisoindolin-2-il)piperidina-2,6-diona y usos de los mismos |
| US11110123B2 (en) | 2018-07-17 | 2021-09-07 | Triumvira Immunologics Usa, Inc. | T cell-antigen coupler with various construct optimizations |
| TWI838389B (zh) | 2018-07-19 | 2024-04-11 | 美商再生元醫藥公司 | 雙特異性抗-BCMAx抗-CD3抗體及其用途 |
| MD3823665T2 (ro) | 2018-07-19 | 2024-05-31 | Regeneron Pharma | Receptori antigenci chimerici cu specificitate BCMA și utilizările acestora |
| KR102835308B1 (ko) | 2018-08-08 | 2025-07-21 | 드래곤플라이 쎄라퓨틱스, 인크. | Nkg2d, cd16 및 종양 관련 항원에 결합하는 단백질 |
| EP3833392A4 (en) | 2018-08-08 | 2022-05-18 | Dragonfly Therapeutics, Inc. | MULTISPECIFIC BINDING PROTEINS FOR BINDING CD33, NKG2D AND CD16 AND METHODS OF USE |
| EP4635978A2 (en) | 2018-08-31 | 2025-10-22 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| EP3844265A2 (en) | 2018-08-31 | 2021-07-07 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| US12195544B2 (en) | 2018-09-21 | 2025-01-14 | Harpoon Therapeutics, Inc. | EGFR binding proteins and methods of use |
| US10815311B2 (en) | 2018-09-25 | 2020-10-27 | Harpoon Therapeutics, Inc. | DLL3 binding proteins and methods of use |
| MA53732A (fr) * | 2018-09-28 | 2022-01-05 | Amgen Inc | Anticorps dirigés contre bcma soluble |
| US12116415B2 (en) | 2018-10-09 | 2024-10-15 | Single Cell Technology, Inc. | Anti-BCMA antibodies |
| US20220003772A1 (en) | 2018-10-31 | 2022-01-06 | Glaxosmithkline Intellectual Property Development Limited | Methods of treating cancer |
| MA54078A (fr) | 2018-11-01 | 2021-09-15 | Juno Therapeutics Inc | Méthodes pour le traitement au moyen de récepteurs antigéniques chimériques spécifiques de l'antigene de maturation des lymphocytes b |
| JP7410143B2 (ja) * | 2018-11-01 | 2024-01-09 | 山▲東▼新▲時▼代▲薬▼▲業▼有限公司 | 二重特異性抗体及びその用途 |
| KR20210106437A (ko) | 2018-12-20 | 2021-08-30 | 노파르티스 아게 | 3-(1-옥소이소인돌린-2-일)피페리딘-2,6-디온 유도체를 포함하는 투약 요법 및 약학적 조합물 |
| BR112021014662A2 (pt) * | 2019-02-01 | 2021-09-21 | Glaxosmithkline Intellectual Property Development Limited | Tratamentos de combinação para o câncer que compreendem belantamab mafodotin e um anticorpo anti ox40 e usos e métodos dos mesmos |
| CN113490528B (zh) | 2019-02-15 | 2024-12-03 | 诺华股份有限公司 | 3-(1-氧代-5-(哌啶-4-基)异吲哚啉-2-基)哌啶-2,6-二酮衍生物及其用途 |
| CA3123519A1 (en) | 2019-02-15 | 2020-08-20 | Novartis Ag | Substituted 3-(1-oxoisoindolin-2-yl)piperidine-2,6-dione derivatives and uses thereof |
| US20220152150A1 (en) | 2019-02-25 | 2022-05-19 | Novartis Ag | Mesoporous silica particles compositions for viral delivery |
| EP3942025A1 (en) | 2019-03-21 | 2022-01-26 | Novartis AG | Car-t cell therapies with enhanced efficacy |
| WO2020206330A1 (en) | 2019-04-05 | 2020-10-08 | Teneobio, Inc. | Heavy chain antibodies binding to psma |
| EP3953455A1 (en) | 2019-04-12 | 2022-02-16 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| EP3959320A1 (en) | 2019-04-24 | 2022-03-02 | Novartis AG | Compositions and methods for selective protein degradation |
| MX2021013359A (es) | 2019-04-30 | 2022-01-31 | Crispr Therapeutics Ag | Terapia de celulas alogénicas de neoplasias malignas de células b usando células t modificadas genéticamente dirigidas a cd19. |
| US11162079B2 (en) | 2019-05-10 | 2021-11-02 | The Regents Of The University Of California | Blood type O Rh-hypo-immunogenic pluripotent cells |
| KR20220024106A (ko) | 2019-05-20 | 2022-03-03 | 노파르티스 아게 | Mcl-1 억제제 항체-약물 접합체 및 사용 방법 |
| JP7489407B2 (ja) | 2019-05-21 | 2024-05-23 | ノバルティス アーゲー | Cd19結合分子及びその使用 |
| JP2022533671A (ja) * | 2019-05-22 | 2022-07-25 | ザ ボード オブ トラスティーズ オブ ザ レランド スタンフォード ジュニア ユニバーシティー | 薬物複合体およびその使用方法 |
| SG11202113008YA (en) * | 2019-05-31 | 2021-12-30 | Medimmune Llc | Combination therapy |
| CR20210622A (es) | 2019-06-14 | 2022-06-27 | Teneobio Inc | Anticuerpos multiespecíficos de cadena pesada que se unen a cd22 y cd3 |
| MX2022001065A (es) | 2019-07-30 | 2022-02-14 | Shanghai Hansoh Biomedical Co Ltd | Anticuerpo anti-bcma, fragmento de union al antigeno y uso medico del mismo. |
| CN120204384A (zh) | 2019-08-06 | 2025-06-27 | 葛兰素史密斯克莱知识产权发展有限公司 | 生物药物组合物和相关方法 |
| CN112409482B (zh) * | 2019-08-20 | 2022-08-26 | 杭州尚健生物技术有限公司 | Bcma抗体 |
| EP4023673A4 (en) | 2019-10-10 | 2023-03-15 | Suzhou Qin Pharmaceuticals Co., Ltd. | ANTI-BCMA HUMANIZED MONOCLONAL ANTIBODY WITH HUMAN-MONKEY CROSS-REACTIVITY |
| JP7773466B2 (ja) * | 2019-11-26 | 2025-11-19 | シャンハイ エピムアブ バイオセラピューティクス カンパニー リミテッド | Cd3およびbcmaに対する抗体およびそれらから作製した二重特異性結合タンパク質 |
| IL293215A (en) | 2019-11-26 | 2022-07-01 | Novartis Ag | Chimeric antigen receptors that bind bcma and cd19 and their uses |
| BR112022011902A2 (pt) | 2019-12-20 | 2022-09-06 | Novartis Ag | Terapias de combinação |
| WO2021136323A1 (en) * | 2020-01-03 | 2021-07-08 | Salubris (Chengdu) Biotech Co., Ltd. | Antibodies binding bcma and uses thereof |
| WO2021136308A1 (en) * | 2020-01-03 | 2021-07-08 | Biosion Inc. | Antibodies binding bcma and uses thereof |
| CN111234020B (zh) * | 2020-01-23 | 2020-10-23 | 和铂医药(苏州)有限公司 | 一种bcma结合蛋白及其制备方法和应用 |
| JP2023512023A (ja) | 2020-01-28 | 2023-03-23 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッド | 併用療法及びその使用及び方法 |
| CN113248611B (zh) * | 2020-02-13 | 2026-02-06 | 上海泰槿生物技术有限公司 | 抗bcma抗体、其药物组合物及应用 |
| EP4110376A2 (en) | 2020-02-27 | 2023-01-04 | Novartis AG | Methods of making chimeric antigen receptor-expressing cells |
| CN115397460A (zh) | 2020-02-27 | 2022-11-25 | 诺华股份有限公司 | 制备表达嵌合抗原受体的细胞的方法 |
| JP2023524875A (ja) | 2020-05-11 | 2023-06-13 | ヤンセン バイオテツク,インコーポレーテツド | 多発性骨髄腫を治療するための方法 |
| AU2021284401A1 (en) * | 2020-06-05 | 2023-01-05 | Eisai R&D Management Co., Ltd. | Anti-BCMA antibody-drug conjugates and methods of use |
| JP2023529211A (ja) | 2020-06-11 | 2023-07-07 | ノバルティス アーゲー | Zbtb32阻害剤及びその使用 |
| CN113817056B (zh) * | 2020-06-18 | 2022-11-11 | 重庆精准生物技术有限公司 | 一种靶向cd70的单链抗体及其应用 |
| KR20230027056A (ko) | 2020-06-23 | 2023-02-27 | 노파르티스 아게 | 3-(1-옥소이소인돌린-2-일)피페리딘-2,6-디온 유도체를 포함하는 투약 요법 |
| JP7819176B2 (ja) | 2020-08-03 | 2026-02-24 | ノバルティス アーゲー | ヘテロアリール置換3-(1-オキソイソインドリン-2-イル)ピペリジン-2,6-ジオン誘導体及びその使用 |
| MX2023002107A (es) | 2020-08-21 | 2023-03-15 | Novartis Ag | Composiciones y metodos para la generacion in vivo de celulas que expresan car. |
| US20230374147A1 (en) | 2020-11-02 | 2023-11-23 | Hummingbird Bioscience Pte. Ltd. | Bcma/taci antigen-binding molecules |
| AU2021386367A1 (en) | 2020-11-24 | 2023-06-22 | Les Laboratoires Servier | Bcl-xl inhibitor antibody-drug conjugates and methods of use thereof |
| JP2024501971A (ja) | 2020-12-31 | 2024-01-17 | サナ バイオテクノロジー,インコーポレイテッド | Car-t活性を調節するための方法及び組成物 |
| US20220251241A1 (en) * | 2021-02-08 | 2022-08-11 | Ngm Biopharmaceuticals, Inc. | Htra1-binding agents and methods of use thereof |
| EP4301774A4 (en) | 2021-03-03 | 2025-08-13 | Dragonfly Therapeutics Inc | METHODS OF TREATING CANCER USING MULTI-SPECIFIC BINDING PROTEINS THAT BIND TO NKG2D, CD16, AND A TUMOR-ASSOCIATED ANTIGEN |
| CN115109156B (zh) * | 2021-03-22 | 2024-03-08 | 浙江纳米抗体技术中心有限公司 | 一种靶向bcma的纳米抗体及其应用 |
| TW202304979A (zh) | 2021-04-07 | 2023-02-01 | 瑞士商諾華公司 | 抗TGFβ抗體及其他治療劑用於治療增殖性疾病之用途 |
| WO2022229853A1 (en) | 2021-04-27 | 2022-11-03 | Novartis Ag | Viral vector production system |
| BR112023024231A2 (pt) | 2021-05-19 | 2024-01-30 | Sana Biotechnology Inc | Células t primárias rhd negativas hipoimunogênicas |
| US20240226164A1 (en) | 2021-05-27 | 2024-07-11 | Sana Biotechnology, Inc. | Hypoimmunogenic cells comprising engineered hla-e or hla-g |
| CN117396513A (zh) | 2021-05-28 | 2024-01-12 | 葛兰素史密斯克莱知识产权发展有限公司 | 治疗癌症的联合疗法 |
| AU2022283819A1 (en) | 2021-06-01 | 2024-01-04 | Triumvira Immunologics Usa, Inc. | Claudin 18.2 t cell-antigen couplers and uses thereof |
| CN115521381B (zh) | 2021-06-24 | 2024-10-29 | 益科思特(北京)医药科技发展有限公司 | 结合bcma和cd3的双特异性抗体及其制备方法与应用 |
| US11453723B1 (en) | 2021-06-25 | 2022-09-27 | Mcmaster University | BCMA T cell-antigen couplers and uses thereof |
| US20240316102A1 (en) | 2021-07-01 | 2024-09-26 | Indapta Therapeutics, Inc. | Engineered natural killer (nk) cells and related methods |
| KR20240046319A (ko) | 2021-07-14 | 2024-04-08 | 사나 바이오테크놀로지, 인크. | 저면역원성 세포에서의 y 염색체-연결 항원의 변경된 발현 |
| TW202323822A (zh) | 2021-08-03 | 2023-06-16 | 英商葛蘭素史密斯克藍智慧財產發展有限公司 | 生藥組合物及穩定同位素標記肽之圖譜定位方法 |
| EP4381081A1 (en) | 2021-08-04 | 2024-06-12 | Sana Biotechnology, Inc. | Use of cd4-targeted viral vectors |
| AU2022327174A1 (en) | 2021-08-11 | 2024-02-15 | Sana Biotechnology, Inc. | Inducible systems for altering gene expression in hypoimmunogenic cells |
| KR20240053675A (ko) * | 2021-08-11 | 2024-04-24 | 악소 바이오파마슈티컬 인코포레이티드 | sBCMA 변이체 및 이의 FC 융합 단백질을 이용한 IgA, IgM 및/또는 IgG의 생산 감소 방법 |
| EP4388000A1 (en) | 2021-08-20 | 2024-06-26 | Novartis AG | Methods of making chimeric antigen receptor?expressing cells |
| CA3233953A1 (en) | 2021-10-05 | 2023-04-13 | Matthew Bruce | Combination therapies for treating cancer |
| US20250313861A1 (en) | 2021-10-22 | 2025-10-09 | Sana Biotechnology, Inc. | Methods of engineering allogeneic t cells with a transgene in a tcr locus and associated compositions and methods |
| MX2024005392A (es) | 2021-11-03 | 2024-08-06 | Janssen Biotech Inc | Métodos para tratar cánceres y potenciar la eficacia de anticuerpos biespecíficos para bcmaxcd3. |
| WO2023081754A1 (en) * | 2021-11-04 | 2023-05-11 | Dana-Farber Cancer Institute, Inc. | Developing inducible cluster chimeric antigen receptor (ccar) constructs |
| WO2023104138A1 (zh) * | 2021-12-09 | 2023-06-15 | 江苏先声药业有限公司 | Bcma抗体及其应用 |
| US12486322B2 (en) | 2021-12-13 | 2025-12-02 | Annexon, Inc. | Anti-complement factor C1q antibodies with single binding arms and uses thereof |
| US20250059239A1 (en) | 2021-12-17 | 2025-02-20 | Sana Biotechnology, Inc. | Modified paramyxoviridae fusion glycoproteins |
| WO2023115041A1 (en) | 2021-12-17 | 2023-06-22 | Sana Biotechnology, Inc. | Modified paramyxoviridae attachment glycoproteins |
| CN119072319A (zh) | 2021-12-23 | 2024-12-03 | 萨那生物技术股份有限公司 | 用于治疗自身免疫性疾病的嵌合抗原受体(car)t细胞及相关方法 |
| EP4463135A2 (en) | 2022-01-10 | 2024-11-20 | Sana Biotechnology, Inc. | Methods of ex vivo dosing and administration of lipid particles or viral vectors and related systems and uses |
| US20260053939A1 (en) | 2022-01-25 | 2026-02-26 | Glaxosmithkline Intellectual Property Development Limited | Combination Therapy for Cancer |
| EP4472646A1 (en) | 2022-02-01 | 2024-12-11 | Sana Biotechnology, Inc. | Cd3-targeted lentiviral vectors and uses thereof |
| WO2023150647A1 (en) | 2022-02-02 | 2023-08-10 | Sana Biotechnology, Inc. | Methods of repeat dosing and administration of lipid particles or viral vectors and related systems and uses |
| EP4479416A1 (en) | 2022-02-17 | 2024-12-25 | Sana Biotechnology, Inc. | Engineered cd47 proteins and uses thereof |
| WO2023178357A1 (en) | 2022-03-18 | 2023-09-21 | Evolveimmune Therapeutics, Inc. | Bispecific antibody fusion molecules and methods of use thereof |
| WO2023193015A1 (en) | 2022-04-01 | 2023-10-05 | Sana Biotechnology, Inc. | Cytokine receptor agonist and viral vector combination therapies |
| WO2023215725A1 (en) | 2022-05-02 | 2023-11-09 | Fred Hutchinson Cancer Center | Compositions and methods for cellular immunotherapy |
| WO2023214325A1 (en) | 2022-05-05 | 2023-11-09 | Novartis Ag | Pyrazolopyrimidine derivatives and uses thereof as tet2 inhibitors |
| CA3253069A1 (en) * | 2022-05-31 | 2023-12-07 | Oncopeptides Innovation 1 Ab | NEW POLYPEPTIDES |
| JP2025525439A (ja) | 2022-06-30 | 2025-08-05 | インダプタ セラピューティクス インコーポレイテッド | 操作されたナチュラルキラー(nk)細胞と抗体療法との組み合わせおよび関連する方法 |
| WO2024026377A1 (en) | 2022-07-27 | 2024-02-01 | Sana Biotechnology, Inc. | Methods of transduction using a viral vector and inhibitors of antiviral restriction factors |
| WO2024064838A1 (en) | 2022-09-21 | 2024-03-28 | Sana Biotechnology, Inc. | Lipid particles comprising variant paramyxovirus attachment glycoproteins and uses thereof |
| WO2024081820A1 (en) | 2022-10-13 | 2024-04-18 | Sana Biotechnology, Inc. | Viral particles targeting hematopoietic stem cells |
| AU2023369684A1 (en) | 2022-10-26 | 2025-04-17 | Novartis Ag | Lentiviral formulations |
| WO2024119157A1 (en) | 2022-12-02 | 2024-06-06 | Sana Biotechnology, Inc. | Lipid particles with cofusogens and methods of producing and using the same |
| CN120303298A (zh) | 2022-12-05 | 2025-07-11 | 葛兰素史密斯克莱知识产权发展有限公司 | 使用b细胞成熟抗原拮抗剂的治疗方法 |
| WO2024179465A1 (zh) | 2023-02-27 | 2024-09-06 | 上海君赛生物科技有限公司 | 表达膜结合细胞因子的肿瘤浸润淋巴细胞 |
| KR20250131823A (ko) | 2023-03-31 | 2025-09-03 | 아벨제타 인크. | Cd20 및 bcma를 표적화하는 이중특이적 키메라 항원 수용체 |
| EP4698666A1 (en) | 2023-04-18 | 2026-02-25 | Sana Biotechnology, Inc. | Universal protein g fusogens and adapter systems thereof and related lipid particles and uses |
| WO2024220560A1 (en) | 2023-04-18 | 2024-10-24 | Sana Biotechnology, Inc. | Engineered protein g fusogens and related lipid particles and methods thereof |
| WO2024220598A2 (en) | 2023-04-18 | 2024-10-24 | Sana Biotechnology, Inc. | Lentiviral vectors with two or more genomes |
| EP4716750A1 (en) | 2023-05-23 | 2026-04-01 | Sana Biotechnology, Inc. | Tandem fusogens and related lipid particles |
| PE20252789A1 (es) | 2023-05-24 | 2025-12-22 | Kumquat Biosciences Inc | Compuestos heterociclicos y usos de estos |
| AU2024279278A1 (en) | 2023-05-31 | 2025-12-18 | Capstan Therapeutics, Inc. | Lipid nanoparticle formulations and compositions |
| TW202502811A (zh) | 2023-06-01 | 2025-01-16 | 瑞士商赫孚孟拉羅股份公司 | 與bcma特異性結合之免疫刺激性抗原結合分子 |
| WO2024246083A1 (en) | 2023-06-01 | 2024-12-05 | F. Hoffmann-La Roche Ag | Bispecific antibodies targeting bcma and cd28 |
| WO2025006799A1 (en) | 2023-06-27 | 2025-01-02 | Capstan Therapeutics, Inc. | Extracorporeal and ex vivo engineering of select cell populations from peripheral blood |
| WO2025043172A1 (en) | 2023-08-23 | 2025-02-27 | Sana Biotechnology, Inc. | Modified cd47 proteins and their uses |
| WO2025054202A1 (en) | 2023-09-05 | 2025-03-13 | Sana Biotechnology, Inc. | Method of screening a sample comprising a transgene with a unique barcode |
| AU2024345039A1 (en) | 2023-09-20 | 2026-03-19 | Evolveimmune Therapeutics, Inc. | Multispecific antibodies that bind cd3 and cd2 and methods of use thereof |
| JP2025059072A (ja) * | 2023-09-28 | 2025-04-09 | 沛爾生技醫藥股▲分▼有限公司 | Nkp30膜貫通ドメインを含む単離核酸分子及びこれを含むキメラ抗原受容体 |
| WO2025076113A1 (en) | 2023-10-05 | 2025-04-10 | Capstan Therapeutics, Inc. | Ionizable cationic lipids with conserved spacing and lipid nanoparticles |
| US20250127728A1 (en) | 2023-10-05 | 2025-04-24 | Capstan Therapeutics, Inc. | Constrained Ionizable Cationic Lipids and Lipid Nanoparticles |
| US20250144234A1 (en) | 2023-11-02 | 2025-05-08 | Capstan Therapeutics Inc. | RNA for In vivo Transfection with Increased Expression |
| WO2025101820A1 (en) | 2023-11-08 | 2025-05-15 | Fred Hutchinson Cancer Center | Compositions and methods for cellular immunotherapy |
| US20250332281A2 (en) | 2023-12-15 | 2025-10-30 | Capstan Therapeutics, Inc. | Humanized anti-cd8 antibodies and uses thereof |
| WO2025151838A1 (en) | 2024-01-12 | 2025-07-17 | Sana Biotechnology, Inc. | Safety switches to control in vitro and in vivo proliferation of cell therapy products |
| WO2025171388A1 (en) | 2024-02-09 | 2025-08-14 | Dispatch Biotherapeutics, Inc. | Engineered cancer antigens with modified domains and related methods and uses |
| WO2025171383A2 (en) | 2024-02-09 | 2025-08-14 | Dispatch Biotherapeutics, Inc. | Engineered cancer antigens and related methods and uses |
| US20250302763A1 (en) | 2024-02-22 | 2025-10-02 | Capstan Therapeutics, Inc. | Immune engineering amplification |
| WO2025184529A1 (en) | 2024-03-01 | 2025-09-04 | Sana Biotechnology, Inc. | Viral particles with fusogen display and related compositions and methods |
| WO2025217454A2 (en) | 2024-04-11 | 2025-10-16 | Capstan Therapeutics, Inc. | Ionizable cationic lipids and lipid nanoparticles |
| WO2025217452A1 (en) | 2024-04-11 | 2025-10-16 | Capstan Therapeutics, Inc. | Constrained ionizable cationic lipids and lipid nanoparticles |
| WO2026006767A1 (en) | 2024-06-28 | 2026-01-02 | Dispatch Biotherapeutics, Inc. | Tethered il-9/il-9r and related engineered cells and methods |
Family Cites Families (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3896111A (en) | 1973-02-20 | 1975-07-22 | Research Corp | Ansa macrolides |
| US4151042A (en) | 1977-03-31 | 1979-04-24 | Takeda Chemical Industries, Ltd. | Method for producing maytansinol and its derivatives |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| US4265814A (en) | 1978-03-24 | 1981-05-05 | Takeda Chemical Industries | Matansinol 3-n-hexadecanoate |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| JPS5562090A (en) | 1978-10-27 | 1980-05-10 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS5566585A (en) | 1978-11-14 | 1980-05-20 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| JPS55164687A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55102583A (en) | 1979-01-31 | 1980-08-05 | Takeda Chem Ind Ltd | 20-acyloxy-20-demethylmaytansinoid compound |
| JPS55162791A (en) | 1979-06-05 | 1980-12-18 | Takeda Chem Ind Ltd | Antibiotic c-15003pnd and its preparation |
| JPS55164685A (en) | 1979-06-08 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55164686A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4309428A (en) | 1979-07-30 | 1982-01-05 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| JPS5645483A (en) | 1979-09-19 | 1981-04-25 | Takeda Chem Ind Ltd | C-15003phm and its preparation |
| EP0028683A1 (en) | 1979-09-21 | 1981-05-20 | Takeda Chemical Industries, Ltd. | Antibiotic C-15003 PHO and production thereof |
| JPS5645485A (en) | 1979-09-21 | 1981-04-25 | Takeda Chem Ind Ltd | Production of c-15003pnd |
| WO1982001188A1 (en) | 1980-10-08 | 1982-04-15 | Takeda Chemical Industries Ltd | 4,5-deoxymaytansinoide compounds and process for preparing same |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| JPS57106673A (en) | 1980-12-24 | 1982-07-02 | Chugai Pharmaceut Co Ltd | Dibenzo(b,f)(1,4)oxazepin derivative |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| JPS57192389A (en) | 1981-05-20 | 1982-11-26 | Takeda Chem Ind Ltd | Novel maytansinoid |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| GB8422238D0 (en) | 1984-09-03 | 1984-10-10 | Neuberger M S | Chimeric proteins |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US4880935A (en) | 1986-07-11 | 1989-11-14 | Icrf (Patents) Limited | Heterobifunctional linking agents derived from N-succinimido-dithio-alpha methyl-methylene-benzoates |
| JP3101690B2 (ja) | 1987-03-18 | 2000-10-23 | エス・ビィ・2・インコーポレイテッド | 変性抗体の、または変性抗体に関する改良 |
| US4873316A (en) | 1987-06-23 | 1989-10-10 | Biogen, Inc. | Isolation of exogenous recombinant proteins from the milk of transgenic mammals |
| US4975278A (en) | 1988-02-26 | 1990-12-04 | Bristol-Myers Company | Antibody-enzyme conjugates in combination with prodrugs for the delivery of cytotoxic agents to tumor cells |
| US5770701A (en) | 1987-10-30 | 1998-06-23 | American Cyanamid Company | Process for preparing targeted forms of methyltrithio antitumor agents |
| US5053394A (en) | 1988-09-21 | 1991-10-01 | American Cyanamid Company | Targeted forms of methyltrithio antitumor agents |
| US5606040A (en) | 1987-10-30 | 1997-02-25 | American Cyanamid Company | Antitumor and antibacterial substituted disulfide derivatives prepared from compounds possessing a methyl-trithio group |
| IL106992A (en) | 1988-02-11 | 1994-06-24 | Bristol Myers Squibb Co | Acylhydrazone derivatives of anthracycline and methods for their preparation |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5362852A (en) | 1991-09-27 | 1994-11-08 | Pfizer Inc. | Modified peptide derivatives conjugated at 2-hydroxyethylamine moieties |
| US5622929A (en) | 1992-01-23 | 1997-04-22 | Bristol-Myers Squibb Company | Thioether conjugates |
| ZA932522B (en) | 1992-04-10 | 1993-12-20 | Res Dev Foundation | Immunotoxins directed against c-erbB-2(HER/neu) related surface antigens |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US5773001A (en) | 1994-06-03 | 1998-06-30 | American Cyanamid Company | Conjugates of methyltrithio antitumor agents and intermediates for their synthesis |
| GB9424449D0 (en) | 1994-12-02 | 1995-01-18 | Wellcome Found | Antibodies |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| US5714586A (en) | 1995-06-07 | 1998-02-03 | American Cyanamid Company | Methods for the preparation of monomeric calicheamicin derivative/carrier conjugates |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| DK0871490T3 (da) | 1995-12-22 | 2003-07-07 | Bristol Myers Squibb Co | Forgrenede hydrazonlinkere |
| DE19742706B4 (de) | 1997-09-26 | 2013-07-25 | Pieris Proteolab Ag | Lipocalinmuteine |
| GB9809839D0 (en) | 1998-05-09 | 1998-07-08 | Glaxo Group Ltd | Antibody |
| IL127127A0 (en) | 1998-11-18 | 1999-09-22 | Peptor Ltd | Small functional units of antibody heavy chain variable regions |
| US7115396B2 (en) | 1998-12-10 | 2006-10-03 | Compound Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| US6818418B1 (en) | 1998-12-10 | 2004-11-16 | Compound Therapeutics, Inc. | Protein scaffolds for antibody mimics and other binding proteins |
| CA2704600C (en) | 1999-04-09 | 2016-10-25 | Kyowa Kirin Co., Ltd. | A method for producing antibodies with increased adcc activity |
| CN101371923B (zh) * | 1999-08-17 | 2013-04-17 | 比奥根艾迪克Ma公司 | Baff受体(bcma),一种免疫调节剂 |
| CN100447244C (zh) * | 1999-08-17 | 2008-12-31 | 比奥根艾迪克Ma公司 | Baff受体(bcma),一种免疫调节剂 |
| EP1229125A4 (en) | 1999-10-19 | 2005-06-01 | Kyowa Hakko Kogyo Kk | PROCESS FOR PREPARING A POLYPEPTIDE |
| US6573074B2 (en) | 2000-04-12 | 2003-06-03 | Smithkline Beecham Plc | Methods for ansamitocin production |
| EP1332209B1 (en) | 2000-09-08 | 2009-11-11 | Universität Zürich | Collections of repeat proteins comprising repeat modules |
| AR030612A1 (es) | 2000-09-12 | 2003-08-27 | Smithkline Beecham Corp | Procedimiento e intermedios |
| US6946292B2 (en) | 2000-10-06 | 2005-09-20 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions with increased antibody dependent cytotoxic activity |
| US6374470B1 (en) | 2000-10-06 | 2002-04-23 | Milliken & Company | Face plate for spun-like textured yarn |
| AU2002220265A1 (en) * | 2000-11-03 | 2002-05-15 | University Of Vermont And State Agricultural College | Compositions for inhibiting grb7 |
| WO2002066516A2 (en) * | 2001-02-20 | 2002-08-29 | Zymogenetics, Inc. | Antibodies that bind both bcma and taci |
| US7256257B2 (en) | 2001-04-30 | 2007-08-14 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| NZ571596A (en) | 2001-08-03 | 2010-11-26 | Glycart Biotechnology Ag | Antibody glycosylation variants having increased antibody-dependent cellular cytotoxicity |
| PT1419179E (pt) | 2001-08-10 | 2010-03-16 | Univ Aberdeen | Domínios de ligação de antigenes |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| US20080267965A1 (en) * | 2002-02-21 | 2008-10-30 | Kalled Susan L | Use of Bcma as an Immunoregulatory Agent |
| DK1545613T3 (da) | 2002-07-31 | 2011-11-14 | Seattle Genetics Inc | Auristatinkonjugater og deres anvendelse til behandling af cancer, en autoimmun sygdom eller en infektiøs sygdom |
| EP1391213A1 (en) | 2002-08-21 | 2004-02-25 | Boehringer Ingelheim International GmbH | Compositions and methods for treating cancer using maytansinoid CD44 antibody immunoconjugates and chemotherapeutic agents |
| CA2531238C (en) | 2003-07-04 | 2015-02-24 | Affibody Ab | Polypeptides having binding affinity for her2 |
| WO2005019255A1 (en) | 2003-08-25 | 2005-03-03 | Pieris Proteolab Ag | Muteins of tear lipocalin |
| EP2478912B1 (en) | 2003-11-06 | 2016-08-31 | Seattle Genetics, Inc. | Auristatin conjugates with anti-HER2 or anti-CD22 antibodies and their use in therapy |
| EP1711196A4 (en) | 2003-12-05 | 2011-09-14 | Bristol Myers Squibb Co | INHIBITORS OF TYPE-2 VASCULAR ENDOTHELIAL GROWTH FACTOR RECEPTORS |
| US20070249530A1 (en) * | 2004-01-29 | 2007-10-25 | Genentech, Inc. | Bcma Polypeptides and Uses Thereof |
| AU2005269759A1 (en) | 2004-07-21 | 2006-02-09 | Glycofi, Inc. | Immunoglobulins comprising predominantly a GlcNAc2Man3GlcNAc2 glycoform |
| US7947805B2 (en) * | 2004-12-23 | 2011-05-24 | Merck Serono S.A. | BCMA polypeptides and uses thereof |
| WO2007011041A1 (ja) | 2005-07-22 | 2007-01-25 | Kyowa Hakko Kogyo Co., Ltd. | 遺伝子組換え抗体組成物 |
| US7923538B2 (en) | 2005-07-22 | 2011-04-12 | Kyowa Hakko Kirin Co., Ltd | Recombinant antibody composition |
| WO2007149932A2 (en) * | 2006-06-22 | 2007-12-27 | Genentech, Inc. | Methods and compositions for targeting hepsin |
| WO2008025747A1 (en) * | 2006-08-28 | 2008-03-06 | Ares Trading S.A. | Process for the purification of fc-fusion proteins |
| EP1958957A1 (en) | 2007-02-16 | 2008-08-20 | NascaCell Technologies AG | Polypeptide comprising a knottin protein moiety |
| KR20100097737A (ko) * | 2008-01-15 | 2010-09-03 | 에프. 호프만-라 로슈 아게 | Ccr5에 대한 어푸코실화된 항체 및 이의 용도 |
| MX341884B (es) * | 2009-03-10 | 2016-09-07 | Biogen Ma Inc | Anticuerpos anti-antigeno de maduracion de celulas b (bcma). |
| WO2011108008A2 (en) * | 2010-03-04 | 2011-09-09 | Transgene Biotek Ltd. | Antibody for targeted induction of apoptosis, cdc and adcc mediated killing of cancer cells, tbl-cln1 |
| EP3974453A3 (en) * | 2010-11-16 | 2022-08-03 | Amgen Inc. | Agents and methods for treating diseases that correlate with bcma expression |
| KR101972446B1 (ko) * | 2011-05-27 | 2019-04-25 | 글락소 그룹 리미티드 | Bcma(cd269/tnfrsf17)결합 단백질 |
| UA112434C2 (uk) * | 2011-05-27 | 2016-09-12 | Ґлаксо Ґруп Лімітед | Антигензв'язувальний білок, який специфічно зв'язується з всма |
-
2012
- 2012-05-24 KR KR1020137034803A patent/KR101972446B1/ko active Active
- 2012-05-24 EP EP18163942.8A patent/EP3415531B1/en active Active
- 2012-05-24 SI SI201232040T patent/SI3415531T1/sl unknown
- 2012-05-24 AU AU2012264890A patent/AU2012264890C1/en active Active
- 2012-05-24 LT LTEP18163942.8T patent/LT3415531T/lt unknown
- 2012-05-24 CN CN201280025793.1A patent/CN103562225B/zh active Active
- 2012-05-24 SG SG2013076138A patent/SG194176A1/en unknown
- 2012-05-24 CA CA2833820A patent/CA2833820C/en active Active
- 2012-05-24 RS RS20231039A patent/RS64791B1/sr unknown
- 2012-05-24 WO PCT/EP2012/059762 patent/WO2012163805A1/en not_active Ceased
- 2012-05-24 US US14/122,391 patent/US20140105915A1/en not_active Abandoned
- 2012-05-24 DK DK18163942.8T patent/DK3415531T5/da active
- 2012-05-24 JP JP2014511888A patent/JP6263467B2/ja active Active
- 2012-05-24 PT PT181639428T patent/PT3415531T/pt unknown
- 2012-05-24 EA EA201790330A patent/EA201790330A1/ru unknown
- 2012-05-24 FI FIEP18163942.8T patent/FI3415531T3/fi active
- 2012-05-24 EP EP23183917.6A patent/EP4338754A3/en active Pending
- 2012-05-24 SM SM20230400T patent/SMT202300400T1/it unknown
- 2012-05-24 HU HUE18163942A patent/HUE063461T2/hu unknown
- 2012-05-24 MX MX2013013942A patent/MX351069B/es active IP Right Grant
- 2012-05-24 EP EP20153215.7A patent/EP3693394A1/en not_active Withdrawn
- 2012-05-24 HR HRP20231066TT patent/HRP20231066T1/hr unknown
- 2012-05-24 EA EA201391457A patent/EA028220B1/ru not_active IP Right Cessation
- 2012-05-24 ES ES18163942T patent/ES2953190T3/es active Active
- 2012-05-24 PE PE2013002545A patent/PE20141045A1/es active IP Right Grant
- 2012-05-24 CN CN201610808775.0A patent/CN106279418A/zh active Pending
- 2012-05-24 PH PH1/2013/502421A patent/PH12013502421A1/en unknown
- 2012-05-24 MY MYPI2013702251A patent/MY177970A/en unknown
- 2012-05-24 BR BR112013028779A patent/BR112013028779B8/pt active IP Right Grant
- 2012-05-24 EP EP12723206.4A patent/EP2714737A1/en not_active Withdrawn
- 2012-05-24 PL PL18163942.8T patent/PL3415531T3/pl unknown
- 2012-05-25 TW TW101118855A patent/TWI609883B/zh active
- 2012-05-25 TW TW106128318A patent/TWI644924B/zh active
- 2012-05-25 UY UY0001034101A patent/UY34101A/es not_active Application Discontinuation
- 2012-05-28 AR ARP120101871A patent/AR086579A1/es active IP Right Grant
-
2013
- 2013-10-08 IL IL228784A patent/IL228784B/en active IP Right Grant
- 2013-11-18 ZA ZA2013/08635A patent/ZA201308635B/en unknown
- 2013-11-19 CO CO13271946A patent/CO6811809A2/es active IP Right Grant
- 2013-11-25 CL CL2013003373A patent/CL2013003373A1/es unknown
- 2013-11-26 DO DO2013000280A patent/DOP2013000280A/es unknown
- 2013-11-27 CR CR20130624A patent/CR20130624A/es unknown
- 2013-12-25 MA MA36613A patent/MA35208B1/fr unknown
-
2016
- 2016-10-12 CL CL2016002585A patent/CL2016002585A1/es unknown
-
2017
- 2017-10-25 IL IL255253A patent/IL255253A0/en unknown
- 2017-12-18 JP JP2017241606A patent/JP2018087190A/ja active Pending
-
2019
- 2019-04-22 JP JP2019080646A patent/JP7018910B2/ja active Active
-
2021
- 2021-11-19 JP JP2021188586A patent/JP7260621B2/ja active Active
-
2023
- 2023-04-06 JP JP2023062129A patent/JP7475515B2/ja active Active
- 2023-09-07 CY CY2023019C patent/CY2023019I2/el unknown
- 2023-09-07 LU LU00314C patent/LUC00314I2/fr unknown
- 2023-09-07 FR FR23C1032C patent/FR23C1032I2/fr active Active
- 2023-09-07 FI FIC20230029C patent/FIC20230029I1/fi unknown
- 2023-09-07 CY CY20231100477T patent/CY1126300T1/el unknown
- 2023-09-07 NL NL301241C patent/NL301241I2/nl unknown
- 2023-09-08 LT LTPA2023524C patent/LTPA2023524I1/lt unknown
- 2023-10-27 HU HUS2300027C patent/HUS2300027I1/hu unknown
-
2024
- 2024-04-16 JP JP2024066216A patent/JP7756194B2/ja active Active
-
2025
- 2025-10-06 JP JP2025168003A patent/JP2026010000A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7756194B2 (ja) | Bcma(cd269/tnfrsf17)結合タンパク質 | |
| US12466893B2 (en) | Antigen binding proteins | |
| ES2719548T3 (es) | Anticuerpos humanizados para liv-1 y uso de los mismos para tratar el cáncer | |
| TWI649334B (zh) | 抗體 | |
| TW202330036A (zh) | 抗體-藥物結合物之製造方法 | |
| CN104936617A (zh) | Cd33抗体及其在治疗癌症中的用途 | |
| JP2016503067A (ja) | 抗ntb−a抗体ならびに関連する組成物および方法 | |
| CN110352074B (zh) | 用于偶联的半胱氨酸突变抗体 | |
| KR20240099178A (ko) | 항 cd37 항체-약물 콘주게이트 | |
| HK40102023A (en) | Antigen binding proteins | |
| HK40022680A (en) | Antigen binding proteins | |
| NZ616433B2 (en) | Bcma (cd269/tnfrsf17) -binding proteins |