ES2953834T3 - Derivados novedosos de ftalazinona y su procedimiento de fabricación - Google Patents
Derivados novedosos de ftalazinona y su procedimiento de fabricación Download PDFInfo
- Publication number
- ES2953834T3 ES2953834T3 ES14843941T ES14843941T ES2953834T3 ES 2953834 T3 ES2953834 T3 ES 2953834T3 ES 14843941 T ES14843941 T ES 14843941T ES 14843941 T ES14843941 T ES 14843941T ES 2953834 T3 ES2953834 T3 ES 2953834T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- fluoro
- carbonyl
- phthalazin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- IJAPPYDYQCXOEF-UHFFFAOYSA-N phthalazin-1(2H)-one Chemical class C1=CC=C2C(=O)NN=CC2=C1 IJAPPYDYQCXOEF-UHFFFAOYSA-N 0.000 title abstract description 96
- 238000004519 manufacturing process Methods 0.000 title description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 13
- 150000001875 compounds Chemical class 0.000 claims description 951
- -1 methylene, carbonyl Chemical group 0.000 claims description 190
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 84
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 61
- 206010028980 Neoplasm Diseases 0.000 claims description 37
- 239000000203 mixture Substances 0.000 claims description 34
- 239000001257 hydrogen Substances 0.000 claims description 32
- 229910052739 hydrogen Inorganic materials 0.000 claims description 32
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 32
- 150000003839 salts Chemical class 0.000 claims description 25
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 19
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 16
- 125000000217 alkyl group Chemical group 0.000 claims description 16
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical group CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 claims description 14
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 13
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 13
- 206010006187 Breast cancer Diseases 0.000 claims description 10
- 208000026310 Breast neoplasm Diseases 0.000 claims description 10
- 125000003118 aryl group Chemical group 0.000 claims description 10
- 108090000623 proteins and genes Proteins 0.000 claims description 10
- 102000036365 BRCA1 Human genes 0.000 claims description 9
- 230000007547 defect Effects 0.000 claims description 8
- 125000005843 halogen group Chemical group 0.000 claims description 8
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 claims description 7
- 101150072950 BRCA1 gene Proteins 0.000 claims description 7
- 101150008921 Brca2 gene Proteins 0.000 claims description 7
- 125000000623 heterocyclic group Chemical group 0.000 claims description 7
- 108700020463 BRCA1 Proteins 0.000 claims description 6
- 108700020462 BRCA2 Proteins 0.000 claims description 6
- 102000052609 BRCA2 Human genes 0.000 claims description 6
- QKIUAMUSENSFQQ-UHFFFAOYSA-N dimethylazanide Chemical group C[N-]C QKIUAMUSENSFQQ-UHFFFAOYSA-N 0.000 claims description 6
- 125000004494 ethyl ester group Chemical group 0.000 claims description 6
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 6
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 6
- JFCUZMQKURGQOK-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C JFCUZMQKURGQOK-UHFFFAOYSA-N 0.000 claims description 5
- 101001077417 Gallus gallus Potassium voltage-gated channel subfamily H member 6 Proteins 0.000 claims description 5
- 101001010792 Homo sapiens Transcriptional regulator ERG Proteins 0.000 claims description 5
- 206010033128 Ovarian cancer Diseases 0.000 claims description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 5
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 5
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 claims description 5
- 206010060862 Prostate cancer Diseases 0.000 claims description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 125000001153 fluoro group Chemical group F* 0.000 claims description 5
- 230000004927 fusion Effects 0.000 claims description 5
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 5
- 201000002528 pancreatic cancer Diseases 0.000 claims description 5
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 5
- YNBQAYKYNYRCCA-UHFFFAOYSA-N venadaparib Chemical compound C1(CC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F YNBQAYKYNYRCCA-UHFFFAOYSA-N 0.000 claims description 5
- XESQDDBWEOEUDO-UHFFFAOYSA-N C1(CCC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XESQDDBWEOEUDO-UHFFFAOYSA-N 0.000 claims description 4
- YCAPBAODPGKNAI-UHFFFAOYSA-N C1(CCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F YCAPBAODPGKNAI-UHFFFAOYSA-N 0.000 claims description 4
- IGINPWSVDNTKOK-UHFFFAOYSA-N Cl.C1(CC1)CNC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CC1)CNC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F IGINPWSVDNTKOK-UHFFFAOYSA-N 0.000 claims description 4
- FSVSACOGQQIXAF-UHFFFAOYSA-N Cl.C1(CC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F FSVSACOGQQIXAF-UHFFFAOYSA-N 0.000 claims description 4
- UBSLSGKRSOQQKE-UHFFFAOYSA-N Cl.C1(CCC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CCC1)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F UBSLSGKRSOQQKE-UHFFFAOYSA-N 0.000 claims description 4
- HQGFJTPHAXRFJN-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)C HQGFJTPHAXRFJN-UHFFFAOYSA-N 0.000 claims description 4
- KKSZOZGTFLMQIZ-UHFFFAOYSA-N C1(CC1)CNC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)CNC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F KKSZOZGTFLMQIZ-UHFFFAOYSA-N 0.000 claims description 3
- GKUXCCVQBLJTMD-MRXNPFEDSA-N C1(CC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F GKUXCCVQBLJTMD-MRXNPFEDSA-N 0.000 claims description 3
- XZUMNWIZCNUMAS-GOSISDBHSA-N C1(CCCC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCCC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XZUMNWIZCNUMAS-GOSISDBHSA-N 0.000 claims description 3
- PNQARVCRZIKCHP-UHFFFAOYSA-N Cl.C1(CC1)CNCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CC1)CNCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F PNQARVCRZIKCHP-UHFFFAOYSA-N 0.000 claims description 3
- HPHLEONXLLWYKF-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNC(C)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNC(C)C HPHLEONXLLWYKF-UHFFFAOYSA-N 0.000 claims description 3
- KTJXLDNKRDIIKT-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C KTJXLDNKRDIIKT-UHFFFAOYSA-N 0.000 claims description 3
- LWCQZYNHAVNPAH-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNC(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNC(C)C LWCQZYNHAVNPAH-UHFFFAOYSA-N 0.000 claims description 3
- DSBIAGOLCXYHPB-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)C DSBIAGOLCXYHPB-UHFFFAOYSA-N 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 208000020816 lung neoplasm Diseases 0.000 claims description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 2
- OSXBCQJXDXZASP-UHFFFAOYSA-N Cl.C1(CCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F OSXBCQJXDXZASP-UHFFFAOYSA-N 0.000 claims description 2
- 206010009944 Colon cancer Diseases 0.000 claims description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 2
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 2
- OOGCIODHIGNQIQ-UHFFFAOYSA-N 4-[[4-fluoro-3-[3-[[1-(trifluoromethyl)cyclopropyl]amino]azetidine-1-carbonyl]phenyl]methyl]-2h-phthalazin-1-one Chemical compound FC1=CC=C(CC=2C3=CC=CC=C3C(=O)NN=2)C=C1C(=O)N(C1)CC1NC1(C(F)(F)F)CC1 OOGCIODHIGNQIQ-UHFFFAOYSA-N 0.000 claims 2
- DKJMZXNQISEHFU-UHFFFAOYSA-N C1(CC1)CNCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)CNCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F DKJMZXNQISEHFU-UHFFFAOYSA-N 0.000 claims 1
- 238000002360 preparation method Methods 0.000 abstract description 350
- 230000000694 effects Effects 0.000 abstract description 9
- 230000002401 inhibitory effect Effects 0.000 abstract description 8
- 102000004190 Enzymes Human genes 0.000 abstract description 3
- 108090000790 Enzymes Proteins 0.000 abstract description 3
- 229910052799 carbon Inorganic materials 0.000 description 486
- 238000000034 method Methods 0.000 description 330
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 250
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 185
- 238000005160 1H NMR spectroscopy Methods 0.000 description 174
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 155
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 153
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 120
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 110
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 87
- 125000005605 benzo group Chemical group 0.000 description 77
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 73
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 73
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 70
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 67
- AJSXBKSREJIQBJ-UHFFFAOYSA-N NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F AJSXBKSREJIQBJ-UHFFFAOYSA-N 0.000 description 59
- 229940086542 triethylamine Drugs 0.000 description 51
- 239000000243 solution Substances 0.000 description 47
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 39
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 38
- XZUFXXPSLGVLFC-UHFFFAOYSA-N 2-fluoro-5-formylbenzoic acid Chemical compound OC(=O)C1=CC(C=O)=CC=C1F XZUFXXPSLGVLFC-UHFFFAOYSA-N 0.000 description 36
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 35
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 32
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 31
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 30
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 30
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 29
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 26
- MBABOKRGFJTBAE-UHFFFAOYSA-N methyl methanesulfonate Chemical compound COS(C)(=O)=O MBABOKRGFJTBAE-UHFFFAOYSA-N 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- 238000006243 chemical reaction Methods 0.000 description 23
- KEKUNQAVGWOYDW-UHFFFAOYSA-N 3-dimethoxyphosphoryl-3h-2-benzofuran-1-one Chemical compound C1=CC=C2C(P(=O)(OC)OC)OC(=O)C2=C1 KEKUNQAVGWOYDW-UHFFFAOYSA-N 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 201000011510 cancer Diseases 0.000 description 20
- 239000012044 organic layer Substances 0.000 description 20
- 239000002253 acid Substances 0.000 description 19
- 238000004587 chromatography analysis Methods 0.000 description 19
- 239000000377 silicon dioxide Substances 0.000 description 19
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 18
- ISYHEXMNTJYNPN-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)=O Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)=O ISYHEXMNTJYNPN-UHFFFAOYSA-N 0.000 description 16
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 description 16
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 description 16
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 16
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 15
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- 150000001408 amides Chemical class 0.000 description 12
- 238000005859 coupling reaction Methods 0.000 description 12
- 238000009472 formulation Methods 0.000 description 12
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 12
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 11
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 11
- 239000011734 sodium Substances 0.000 description 11
- 108010064218 Poly (ADP-Ribose) Polymerase-1 Proteins 0.000 description 10
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 10
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 238000006772 olefination reaction Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 235000015424 sodium Nutrition 0.000 description 8
- 229940083542 sodium Drugs 0.000 description 8
- 229910052708 sodium Inorganic materials 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- IFKPLJWIEQBPGG-QGZVFWFLSA-N (5s)-6-(dimethylamino)-5-methyl-4,4-diphenylhexan-3-one Chemical compound C=1C=CC=CC=1C([C@H](C)CN(C)C)(C(=O)CC)C1=CC=CC=C1 IFKPLJWIEQBPGG-QGZVFWFLSA-N 0.000 description 7
- 101100063435 Caenorhabditis elegans din-1 gene Proteins 0.000 description 7
- 230000008878 coupling Effects 0.000 description 7
- BSEXNZMHLUMQKR-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1.NC(=O)C1CC1 BSEXNZMHLUMQKR-UHFFFAOYSA-N 0.000 description 7
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 6
- 102100037664 Poly [ADP-ribose] polymerase tankyrase-1 Human genes 0.000 description 6
- 101710129670 Poly [ADP-ribose] polymerase tankyrase-1 Proteins 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 238000003570 cell viability assay Methods 0.000 description 6
- 230000001413 cellular effect Effects 0.000 description 6
- 238000010168 coupling process Methods 0.000 description 6
- 239000003814 drug Substances 0.000 description 6
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- YNTJKQDWYXUTLZ-UHFFFAOYSA-N 2-(3-chlorophenoxy)propanoic acid Chemical compound OC(=O)C(C)OC1=CC=CC(Cl)=C1 YNTJKQDWYXUTLZ-UHFFFAOYSA-N 0.000 description 5
- QRHLIWLOPSLNCU-UHFFFAOYSA-N 2h-phthalazin-1-one;hydrochloride Chemical compound Cl.C1=CC=C2C(=O)NN=CC2=C1 QRHLIWLOPSLNCU-UHFFFAOYSA-N 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 5
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 5
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- SHQSVMDWKBRBGB-UHFFFAOYSA-N cyclobutanone Chemical compound O=C1CCC1 SHQSVMDWKBRBGB-UHFFFAOYSA-N 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 230000008439 repair process Effects 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 4
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 4
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 239000012661 PARP inhibitor Substances 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 4
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 4
- 102100037171 Protein JTB Human genes 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- 238000006482 condensation reaction Methods 0.000 description 4
- 239000007884 disintegrant Substances 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- NVHXMNNCHAFIHX-UHFFFAOYSA-N pyridazin-3-amine Chemical compound NC1=CC=C=N[N]1 NVHXMNNCHAFIHX-UHFFFAOYSA-N 0.000 description 4
- IQHXABCGSFAKPN-UHFFFAOYSA-N pyrrolidine-3-carboxamide Chemical compound NC(=O)C1CCNC1 IQHXABCGSFAKPN-UHFFFAOYSA-N 0.000 description 4
- 238000006268 reductive amination reaction Methods 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- GQLSEYOOXBRDFZ-UHFFFAOYSA-N N-formylnornicotine Natural products O=CN1CCCC1C1=CC=CN=C1 GQLSEYOOXBRDFZ-UHFFFAOYSA-N 0.000 description 3
- IDRGFNPZDVBSSE-UHFFFAOYSA-N OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F Chemical compound OCCN1CCN(CC1)c1ccc(Nc2ncc3cccc(-c4cccc(NC(=O)C=C)c4)c3n2)c(F)c1F IDRGFNPZDVBSSE-UHFFFAOYSA-N 0.000 description 3
- 239000012828 PI3K inhibitor Substances 0.000 description 3
- 102100023652 Poly [ADP-ribose] polymerase 2 Human genes 0.000 description 3
- 101710144590 Poly [ADP-ribose] polymerase 2 Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 3
- 229930195731 calicheamicin Natural products 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000005889 cellular cytotoxicity Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 238000002784 cytotoxicity assay Methods 0.000 description 3
- 231100000263 cytotoxicity test Toxicity 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- DHMTURDWPRKSOA-RUZDIDTESA-N lonafarnib Chemical compound C1CN(C(=O)N)CCC1CC(=O)N1CCC([C@@H]2C3=C(Br)C=C(Cl)C=C3CCC3=CC(Br)=CN=C32)CC1 DHMTURDWPRKSOA-RUZDIDTESA-N 0.000 description 3
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- QJZUKDFHGGYHMC-UHFFFAOYSA-N pyridine-3-carbaldehyde Chemical compound O=CC1=CC=CN=C1 QJZUKDFHGGYHMC-UHFFFAOYSA-N 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 230000005783 single-strand break Effects 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- 229960000241 vandetanib Drugs 0.000 description 3
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 3
- BZMMRNKDONDVIB-UHFFFAOYSA-N (1-ethoxycyclopropyl)oxy-trimethylsilane Chemical compound CCOC1(O[Si](C)(C)C)CC1 BZMMRNKDONDVIB-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- KDSNLYIMUZNERS-UHFFFAOYSA-N 2-methylpropanamine Chemical compound CC(C)CN KDSNLYIMUZNERS-UHFFFAOYSA-N 0.000 description 2
- SYBYTAAJFKOIEJ-UHFFFAOYSA-N 3-Methylbutan-2-one Chemical compound CC(C)C(C)=O SYBYTAAJFKOIEJ-UHFFFAOYSA-N 0.000 description 2
- YGHRJJRRZDOVPD-UHFFFAOYSA-N 3-methylbutanal Chemical compound CC(C)CC=O YGHRJJRRZDOVPD-UHFFFAOYSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- PWJFNRJRHXWEPT-UHFFFAOYSA-N ADP ribose Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OCC(O)C(O)C(O)C=O)C(O)C1O PWJFNRJRHXWEPT-UHFFFAOYSA-N 0.000 description 2
- SRNWOUGRCWSEMX-KEOHHSTQSA-N ADP-beta-D-ribose Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=CN=C(C=2N=C1)N)OP(O)(=O)OP(O)(=O)OC[C@H]1O[C@@H](O)[C@H](O)[C@@H]1O SRNWOUGRCWSEMX-KEOHHSTQSA-N 0.000 description 2
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 206010071980 BRCA1 gene mutation Diseases 0.000 description 2
- 108091007743 BRCA1/2 Proteins 0.000 description 2
- 206010071981 BRCA2 gene mutation Diseases 0.000 description 2
- YHPXMAFYEGPKGP-UHFFFAOYSA-N C1(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F YHPXMAFYEGPKGP-UHFFFAOYSA-N 0.000 description 2
- HDYQOBLXSWUURO-SFHVURJKSA-N CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C Chemical compound CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C HDYQOBLXSWUURO-SFHVURJKSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- HBVWVOGUSFGHDW-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC#C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC#C HBVWVOGUSFGHDW-UHFFFAOYSA-N 0.000 description 2
- LPZFTMHNYRWIGW-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC(C)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC(C)C LPZFTMHNYRWIGW-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 230000005778 DNA damage Effects 0.000 description 2
- 231100000277 DNA damage Toxicity 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XFSQIPZQQQUUHN-UHFFFAOYSA-N FC1(CN(C1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F Chemical compound FC1(CN(C1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F XFSQIPZQQQUUHN-UHFFFAOYSA-N 0.000 description 2
- HJNZDMWQMFANKE-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O HJNZDMWQMFANKE-UHFFFAOYSA-N 0.000 description 2
- XWBHCBHHEYSFFJ-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O XWBHCBHHEYSFFJ-UHFFFAOYSA-N 0.000 description 2
- AHKRWJRFSSUHKJ-QGZVFWFLSA-N FC1=C(C(=O)N2C[C@@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O AHKRWJRFSSUHKJ-QGZVFWFLSA-N 0.000 description 2
- JGWRANKDUONGFX-CQSZACIVSA-N FC1=C(C(=O)N2C[C@@H](CC2)N2C(NCC2=O)=O)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)N2C(NCC2=O)=O)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O JGWRANKDUONGFX-CQSZACIVSA-N 0.000 description 2
- XWJWZYKNJCCVKV-GOSISDBHSA-N FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C(=O)N(C)C)C=CC=N2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C(=O)N(C)C)C=CC=N2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O XWJWZYKNJCCVKV-GOSISDBHSA-N 0.000 description 2
- DCOSLQSNCCDQHQ-KRWDZBQOSA-N FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O DCOSLQSNCCDQHQ-KRWDZBQOSA-N 0.000 description 2
- AHKRWJRFSSUHKJ-KRWDZBQOSA-N FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O AHKRWJRFSSUHKJ-KRWDZBQOSA-N 0.000 description 2
- TVZJPABYJKMMBN-UHFFFAOYSA-N FC1=C(C=C(C=C2/OC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F Chemical compound FC1=C(C=C(C=C2/OC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F TVZJPABYJKMMBN-UHFFFAOYSA-N 0.000 description 2
- HGZQLWMEMSGRKZ-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC#C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC#C HGZQLWMEMSGRKZ-UHFFFAOYSA-N 0.000 description 2
- FLGYFLGQOQVVJN-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CNCC(C)C FLGYFLGQOQVVJN-UHFFFAOYSA-N 0.000 description 2
- CLBLPGAIABHVGV-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CC)CC Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CC)CC CLBLPGAIABHVGV-UHFFFAOYSA-N 0.000 description 2
- FEXCXBJVIQMDFJ-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC=1N=NC=CC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC=1N=NC=CC1 FEXCXBJVIQMDFJ-UHFFFAOYSA-N 0.000 description 2
- DTQVFJOGSJBHGN-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(C)(C)C DTQVFJOGSJBHGN-UHFFFAOYSA-N 0.000 description 2
- JIMFHZIXARVSJA-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCCC(F)(F)F Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCCC(F)(F)F JIMFHZIXARVSJA-UHFFFAOYSA-N 0.000 description 2
- SLLINAVMZWCBGT-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)O Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)O SLLINAVMZWCBGT-UHFFFAOYSA-N 0.000 description 2
- HRGJMHDPVJPZCT-GOSISDBHSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N1CCCCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N1CCCCC1 HRGJMHDPVJPZCT-GOSISDBHSA-N 0.000 description 2
- WOBNJYNYIRZXGO-CYBMUJFWSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)O Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)O WOBNJYNYIRZXGO-CYBMUJFWSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 108010033040 Histones Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229940124647 MEK inhibitor Drugs 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001328813 Methles Species 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- NMKURCNRNJLZQC-MRXNPFEDSA-N N1(CCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound N1(CCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F NMKURCNRNJLZQC-MRXNPFEDSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 150000003973 alkyl amines Chemical class 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 2
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- KZZKOVLJUKWSKX-UHFFFAOYSA-N cyclobutanamine Chemical compound NC1CCC1 KZZKOVLJUKWSKX-UHFFFAOYSA-N 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- JMYVMOUINOAAPA-UHFFFAOYSA-N cyclopropanecarbaldehyde Chemical compound O=CC1CC1 JMYVMOUINOAAPA-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000001952 enzyme assay Methods 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 2
- 235000008191 folinic acid Nutrition 0.000 description 2
- 239000011672 folinic acid Substances 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000007902 hard capsule Substances 0.000 description 2
- XLSMFKSTNGKWQX-UHFFFAOYSA-N hydroxyacetone Chemical compound CC(=O)CO XLSMFKSTNGKWQX-UHFFFAOYSA-N 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 2
- 229960001691 leucovorin Drugs 0.000 description 2
- 239000012669 liquid formulation Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 2
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- XNLICIUVMPYHGG-UHFFFAOYSA-N pentan-2-one Chemical compound CCCC(C)=O XNLICIUVMPYHGG-UHFFFAOYSA-N 0.000 description 2
- FDPIMTJIUBPUKL-UHFFFAOYSA-N pentan-3-one Chemical compound CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 2
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N pentanal Chemical compound CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229920003124 powdered cellulose Polymers 0.000 description 2
- 235000019814 powdered cellulose Nutrition 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 230000000754 repressing effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 239000008109 sodium starch glycolate Substances 0.000 description 2
- 229920003109 sodium starch glycolate Polymers 0.000 description 2
- 229940079832 sodium starch glycolate Drugs 0.000 description 2
- 239000007901 soft capsule Substances 0.000 description 2
- 229960003787 sorafenib Drugs 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- PVYJZLYGTZKPJE-UHFFFAOYSA-N streptonigrin Chemical compound C=1C=C2C(=O)C(OC)=C(N)C(=O)C2=NC=1C(C=1N)=NC(C(O)=O)=C(C)C=1C1=CC=C(OC)C(OC)=C1O PVYJZLYGTZKPJE-UHFFFAOYSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- OBMKZINZPBARIK-UHFFFAOYSA-N (1-aminocyclopropyl)methanol Chemical compound OCC1(N)CC1 OBMKZINZPBARIK-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- NXMXETCTWNXSFG-SCSAIBSYSA-N (2r)-1-methoxypropan-2-amine Chemical compound COC[C@@H](C)N NXMXETCTWNXSFG-SCSAIBSYSA-N 0.000 description 1
- DXSUORGKJZADET-RXMQYKEDSA-N (2r)-3,3-dimethylbutan-2-amine Chemical compound C[C@@H](N)C(C)(C)C DXSUORGKJZADET-RXMQYKEDSA-N 0.000 description 1
- JOZZAIIGWFLONA-RXMQYKEDSA-N (2r)-3-methylbutan-2-amine Chemical compound CC(C)[C@@H](C)N JOZZAIIGWFLONA-RXMQYKEDSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- NXMXETCTWNXSFG-BYPYZUCNSA-N (2s)-1-methoxypropan-2-amine Chemical compound COC[C@H](C)N NXMXETCTWNXSFG-BYPYZUCNSA-N 0.000 description 1
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 1
- FLWWDYNPWOSLEO-HQVZTVAUSA-N (2s)-2-[[4-[1-(2-amino-4-oxo-1h-pteridin-6-yl)ethyl-methylamino]benzoyl]amino]pentanedioic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1C(C)N(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FLWWDYNPWOSLEO-HQVZTVAUSA-N 0.000 description 1
- NECZZOFFLFZNHL-XVGZVFJZSA-N (2s)-2-amino-5-[[(2r)-3-[2-[bis[bis(2-chloroethyl)amino]-oxidophosphaniumyl]oxyethylsulfonyl]-1-[[(r)-carboxy(phenyl)methyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)P(=O)(N(CCCl)CCCl)OCCS(=O)(=O)C[C@H](NC(=O)CC[C@H](N)C(O)=O)C(=O)N[C@@H](C(O)=O)C1=CC=CC=C1 NECZZOFFLFZNHL-XVGZVFJZSA-N 0.000 description 1
- DXSUORGKJZADET-YFKPBYRVSA-N (2s)-3,3-dimethylbutan-2-amine Chemical compound C[C@H](N)C(C)(C)C DXSUORGKJZADET-YFKPBYRVSA-N 0.000 description 1
- JOZZAIIGWFLONA-YFKPBYRVSA-N (2s)-3-methylbutan-2-amine Chemical compound CC(C)[C@H](C)N JOZZAIIGWFLONA-YFKPBYRVSA-N 0.000 description 1
- PJRSUKFWFKUDTH-JWDJOUOUSA-N (2s)-6-amino-2-[[2-[[(2s)-2-[[(2s,3s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[(2-aminoacetyl)amino]-4-methylsulfanylbutanoyl]amino]propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]propanoyl]amino]acetyl]amino]propanoyl Chemical compound CSCC[C@H](NC(=O)CN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(N)=O PJRSUKFWFKUDTH-JWDJOUOUSA-N 0.000 description 1
- CGMTUJFWROPELF-YPAAEMCBSA-N (3E,5S)-5-[(2S)-butan-2-yl]-3-(1-hydroxyethylidene)pyrrolidine-2,4-dione Chemical compound CC[C@H](C)[C@@H]1NC(=O)\C(=C(/C)O)C1=O CGMTUJFWROPELF-YPAAEMCBSA-N 0.000 description 1
- ULLMNWIRHQTTIE-MRVPVSSYSA-N (3R)-N-(cyclopropylmethyl)pyrrolidine-3-carboxamide Chemical compound C1(CC1)CNC(=O)[C@H]1CNCC1 ULLMNWIRHQTTIE-MRVPVSSYSA-N 0.000 description 1
- AAFUQJGCJNFFRT-SNVBAGLBSA-N (3r)-n-phenylpyrrolidin-3-amine Chemical compound C1NCC[C@H]1NC1=CC=CC=C1 AAFUQJGCJNFFRT-SNVBAGLBSA-N 0.000 description 1
- MAYZWDRUFKUGGP-VIFPVBQESA-N (3s)-1-[5-tert-butyl-3-[(1-methyltetrazol-5-yl)methyl]triazolo[4,5-d]pyrimidin-7-yl]pyrrolidin-3-ol Chemical compound CN1N=NN=C1CN1C2=NC(C(C)(C)C)=NC(N3C[C@@H](O)CC3)=C2N=N1 MAYZWDRUFKUGGP-VIFPVBQESA-N 0.000 description 1
- XRBSKUSTLXISAB-XVVDYKMHSA-N (5r,6r,7r,8r)-8-hydroxy-7-(hydroxymethyl)-5-(3,4,5-trimethoxyphenyl)-5,6,7,8-tetrahydrobenzo[f][1,3]benzodioxole-6-carboxylic acid Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H](CO)[C@@H]2C(O)=O)=C1 XRBSKUSTLXISAB-XVVDYKMHSA-N 0.000 description 1
- XRBSKUSTLXISAB-UHFFFAOYSA-N (7R,7'R,8R,8'R)-form-Podophyllic acid Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C(CO)C2C(O)=O)=C1 XRBSKUSTLXISAB-UHFFFAOYSA-N 0.000 description 1
- INAUWOVKEZHHDM-PEDBPRJASA-N (7s,9s)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1 INAUWOVKEZHHDM-PEDBPRJASA-N 0.000 description 1
- NOPNWHSMQOXAEI-PUCKCBAPSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4-(2,3-dihydropyrrol-1-yl)-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCC=C1 NOPNWHSMQOXAEI-PUCKCBAPSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- AGNGYMCLFWQVGX-AGFFZDDWSA-N (e)-1-[(2s)-2-amino-2-carboxyethoxy]-2-diazonioethenolate Chemical compound OC(=O)[C@@H](N)CO\C([O-])=C\[N+]#N AGNGYMCLFWQVGX-AGFFZDDWSA-N 0.000 description 1
- ZGYIXVSQHOKQRZ-COIATFDQSA-N (e)-n-[4-[3-chloro-4-(pyridin-2-ylmethoxy)anilino]-3-cyano-7-[(3s)-oxolan-3-yl]oxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound N#CC1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZGYIXVSQHOKQRZ-COIATFDQSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 125000005871 1,3-benzodioxolyl group Chemical group 0.000 description 1
- FONKWHRXTPJODV-DNQXCXABSA-N 1,3-bis[2-[(8s)-8-(chloromethyl)-4-hydroxy-1-methyl-7,8-dihydro-3h-pyrrolo[3,2-e]indole-6-carbonyl]-1h-indol-5-yl]urea Chemical compound C1([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C4=CC(O)=C5NC=C(C5=C4[C@H](CCl)C3)C)=C2C=C(O)C2=C1C(C)=CN2 FONKWHRXTPJODV-DNQXCXABSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 1
- OQBCJXUAQQMTRW-UHFFFAOYSA-N 1-(1-methylcyclopropyl)ethanone Chemical compound CC(=O)C1(C)CC1 OQBCJXUAQQMTRW-UHFFFAOYSA-N 0.000 description 1
- APWRZPQBPCAXFP-UHFFFAOYSA-N 1-(1-oxo-2H-isoquinolin-5-yl)-5-(trifluoromethyl)-N-[2-(trifluoromethyl)pyridin-4-yl]pyrazole-4-carboxamide Chemical compound O=C1NC=CC2=C(C=CC=C12)N1N=CC(=C1C(F)(F)F)C(=O)NC1=CC(=NC=C1)C(F)(F)F APWRZPQBPCAXFP-UHFFFAOYSA-N 0.000 description 1
- WICRKCABAQJKSY-UHFFFAOYSA-N 1-(aminomethyl)cyclopropane-1-carbonitrile Chemical compound NCC1(C#N)CC1 WICRKCABAQJKSY-UHFFFAOYSA-N 0.000 description 1
- SQGAMWPLHIDBLX-UHFFFAOYSA-N 1-(azetidin-3-yl)azetidine Chemical compound C1CCN1C1CNC1 SQGAMWPLHIDBLX-UHFFFAOYSA-N 0.000 description 1
- KWXIMSDLQSTXBC-UHFFFAOYSA-N 1-(methoxymethyl)cyclopropan-1-amine Chemical compound COCC1(N)CC1 KWXIMSDLQSTXBC-UHFFFAOYSA-N 0.000 description 1
- FBWOFVFUKXZXKT-UHFFFAOYSA-N 1-(trifluoromethyl)cyclopropan-1-amine Chemical compound FC(F)(F)C1(N)CC1 FBWOFVFUKXZXKT-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- HVCFCNAITDHQFX-UHFFFAOYSA-N 1-cyclopropylethanone Chemical compound CC(=O)C1CC1 HVCFCNAITDHQFX-UHFFFAOYSA-N 0.000 description 1
- SOWNWZQOFXEMNE-SSDOTTSWSA-N 1-ethyl-3-[(3R)-pyrrolidin-3-yl]imidazolidine-2,4-dione Chemical compound C(C)N1C(N(C(C1)=O)[C@H]1CNCC1)=O SOWNWZQOFXEMNE-SSDOTTSWSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- VSRXAWSAKJABKW-UHFFFAOYSA-N 1-methylcyclopropan-1-amine Chemical compound CC1(N)CC1 VSRXAWSAKJABKW-UHFFFAOYSA-N 0.000 description 1
- FTGZMZBYOHMEPS-UHFFFAOYSA-N 2,2-dimethylcyclopentan-1-one Chemical compound CC1(C)CCCC1=O FTGZMZBYOHMEPS-UHFFFAOYSA-N 0.000 description 1
- BTOTXLJHDSNXMW-POYBYMJQSA-N 2,3-dideoxyuridine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(=O)NC(=O)C=C1 BTOTXLJHDSNXMW-POYBYMJQSA-N 0.000 description 1
- BOMZMNZEXMAQQW-UHFFFAOYSA-N 2,5,11-trimethyl-6h-pyrido[4,3-b]carbazol-2-ium-9-ol;acetate Chemical compound CC([O-])=O.C[N+]1=CC=C2C(C)=C(NC=3C4=CC(O)=CC=3)C4=C(C)C2=C1 BOMZMNZEXMAQQW-UHFFFAOYSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 1
- CCFQICSWPMSZEA-UHFFFAOYSA-N 2-[[1-[2-fluoro-5-[(4-oxo-3H-phthalazin-1-yl)methyl]benzoyl]azetidin-3-yl]amino]cyclopentene-1-carboxylic acid Chemical compound OC(=O)C1=C(CCC1)NC1CN(C1)C(=O)c1cc(Cc2n[nH]c(=O)c3ccccc23)ccc1F CCFQICSWPMSZEA-UHFFFAOYSA-N 0.000 description 1
- QCXJFISCRQIYID-IAEPZHFASA-N 2-amino-1-n-[(3s,6s,7r,10s,16s)-3-[(2s)-butan-2-yl]-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-10-propan-2-yl-8-oxa-1,4,11,14-tetrazabicyclo[14.3.0]nonadecan-6-yl]-4,6-dimethyl-3-oxo-9-n-[(3s,6s,7r,10s,16s)-7,11,14-trimethyl-2,5,9,12,15-pentaoxo-3,10-di(propa Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N=C2C(C(=O)N[C@@H]3C(=O)N[C@H](C(N4CCC[C@H]4C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]3C)=O)[C@@H](C)CC)=C(N)C(=O)C(C)=C2O2)C2=C(C)C=C1 QCXJFISCRQIYID-IAEPZHFASA-N 0.000 description 1
- QINPEPAQOBZPOF-UHFFFAOYSA-N 2-amino-n-[3-[[3-(2-chloro-5-methoxyanilino)quinoxalin-2-yl]sulfamoyl]phenyl]-2-methylpropanamide Chemical compound COC1=CC=C(Cl)C(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=C(NC(=O)C(C)(C)N)C=CC=2)=C1 QINPEPAQOBZPOF-UHFFFAOYSA-N 0.000 description 1
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 1
- WNZQDUSMALZDQF-UHFFFAOYSA-N 2-benzofuran-1(3H)-one Chemical compound C1=CC=C2C(=O)OCC2=C1 WNZQDUSMALZDQF-UHFFFAOYSA-N 0.000 description 1
- TUHIBIVYQLRGME-UHFFFAOYSA-N 2-cyclopropylacetaldehyde Chemical compound O=CCC1CC1 TUHIBIVYQLRGME-UHFFFAOYSA-N 0.000 description 1
- VXDHQYLFEYUMFY-UHFFFAOYSA-N 2-methylprop-2-en-1-amine Chemical compound CC(=C)CN VXDHQYLFEYUMFY-UHFFFAOYSA-N 0.000 description 1
- LGEXGKUJMFHVSY-UHFFFAOYSA-N 2-n,4-n,6-n-trimethyl-1,3,5-triazine-2,4,6-triamine Chemical compound CNC1=NC(NC)=NC(NC)=N1 LGEXGKUJMFHVSY-UHFFFAOYSA-N 0.000 description 1
- CTRPRMNBTVRDFH-UHFFFAOYSA-N 2-n-methyl-1,3,5-triazine-2,4,6-triamine Chemical class CNC1=NC(N)=NC(N)=N1 CTRPRMNBTVRDFH-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- GSSXLFACIJSBOM-UHFFFAOYSA-N 2h-pyran-2-ol Chemical compound OC1OC=CC=C1 GSSXLFACIJSBOM-UHFFFAOYSA-N 0.000 description 1
- YIMDLWDNDGKDTJ-QLKYHASDSA-N 3'-deamino-3'-(3-cyanomorpholin-4-yl)doxorubicin Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1C#N YIMDLWDNDGKDTJ-QLKYHASDSA-N 0.000 description 1
- FNYHFEKPIFKMGC-UHFFFAOYSA-N 3,3-difluoro-2H-azete Chemical compound FC1(CN=C1)F FNYHFEKPIFKMGC-UHFFFAOYSA-N 0.000 description 1
- KVTUSMPNLUCCQO-UHFFFAOYSA-N 3,3-difluoropyrrolidine Chemical compound FC1(F)CCNC1 KVTUSMPNLUCCQO-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- PWMYMKOUNYTVQN-UHFFFAOYSA-N 3-(8,8-diethyl-2-aza-8-germaspiro[4.5]decan-2-yl)-n,n-dimethylpropan-1-amine Chemical compound C1C[Ge](CC)(CC)CCC11CN(CCCN(C)C)CC1 PWMYMKOUNYTVQN-UHFFFAOYSA-N 0.000 description 1
- XZBYEADXFLEAHO-UHFFFAOYSA-N 3-(azetidin-3-yl)-5,5-dimethylimidazolidine-2,4-dione Chemical compound N1CC(C1)N1C(NC(C1=O)(C)C)=O XZBYEADXFLEAHO-UHFFFAOYSA-N 0.000 description 1
- AZEVUKHOQBCVKK-CQSZACIVSA-N 3-[(3R)-3-[tert-butyl(dimethyl)silyl]oxypyrrolidine-1-carbonyl]-4-fluorobenzaldehyde Chemical compound C(C)(C)(C)[Si](O[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)(C)C AZEVUKHOQBCVKK-CQSZACIVSA-N 0.000 description 1
- SYEGSOIORRSJIP-RXMQYKEDSA-N 3-[(3R)-pyrrolidin-3-yl]imidazolidine-2,4-dione Chemical compound N1C[C@@H](CC1)N1C(NCC1=O)=O SYEGSOIORRSJIP-RXMQYKEDSA-N 0.000 description 1
- AZEVUKHOQBCVKK-AWEZNQCLSA-N 3-[(3S)-3-[tert-butyl(dimethyl)silyl]oxypyrrolidine-1-carbonyl]-4-fluorobenzaldehyde Chemical compound C(C)(C)(C)[Si](O[C@@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)(C)C AZEVUKHOQBCVKK-AWEZNQCLSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 1
- RDEINJOCAVHFTC-UHFFFAOYSA-N 3-[3-[[tert-butyl(dimethyl)silyl]oxymethyl]azetidine-1-carbonyl]-4-fluorobenzaldehyde Chemical compound C(C)(C)(C)[Si](OCC1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)(C)C RDEINJOCAVHFTC-UHFFFAOYSA-N 0.000 description 1
- OISBARLBVPDUPG-UHFFFAOYSA-N 3-[3-[tert-butyl(dimethyl)silyl]oxyazetidine-1-carbonyl]-4-fluorobenzaldehyde Chemical compound C(C)(C)(C)[Si](OC1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)(C)C OISBARLBVPDUPG-UHFFFAOYSA-N 0.000 description 1
- FNVOFDGAASRDQY-UHFFFAOYSA-N 3-amino-2,2-dimethylpropan-1-ol Chemical compound NCC(C)(C)CO FNVOFDGAASRDQY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- SEPQTYODOKLVSB-UHFFFAOYSA-N 3-methylbut-2-enal Chemical compound CC(C)=CC=O SEPQTYODOKLVSB-UHFFFAOYSA-N 0.000 description 1
- GZPHSAQLYPIAIN-UHFFFAOYSA-N 3-pyridinecarbonitrile Chemical compound N#CC1=CC=CN=C1 GZPHSAQLYPIAIN-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- SCJCDNUXDWFVFI-UHFFFAOYSA-N 4,4,4-trifluorobutanal Chemical compound FC(F)(F)CCC=O SCJCDNUXDWFVFI-UHFFFAOYSA-N 0.000 description 1
- XNXGGVZXBSADFN-UHFFFAOYSA-N 4-[[3-[3-(dicyclopropylamino)azetidine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one Chemical compound C1(CC1)N(C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC=1F)C1CC1 XNXGGVZXBSADFN-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- QBLLOOKBLTTXHB-UHFFFAOYSA-N 4-fluoropiperidine Chemical compound FC1CCNCC1 QBLLOOKBLTTXHB-UHFFFAOYSA-N 0.000 description 1
- UZOVYGYOLBIAJR-UHFFFAOYSA-N 4-isocyanato-4'-methyldiphenylmethane Chemical compound C1=CC(C)=CC=C1CC1=CC=C(N=C=O)C=C1 UZOVYGYOLBIAJR-UHFFFAOYSA-N 0.000 description 1
- HQQTZCPKNZVLFF-UHFFFAOYSA-N 4h-1,2-benzoxazin-3-one Chemical compound C1=CC=C2ONC(=O)CC2=C1 HQQTZCPKNZVLFF-UHFFFAOYSA-N 0.000 description 1
- IRPVABHDSJVBNZ-RTHVDDQRSA-N 5-[1-(cyclopropylmethyl)-5-[(1R,5S)-3-(oxetan-3-yl)-3-azabicyclo[3.1.0]hexan-6-yl]pyrazol-3-yl]-3-(trifluoromethyl)pyridin-2-amine Chemical compound C1=C(C(F)(F)F)C(N)=NC=C1C1=NN(CC2CC2)C(C2[C@@H]3CN(C[C@@H]32)C2COC2)=C1 IRPVABHDSJVBNZ-RTHVDDQRSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- SCPSQJAXCIJGSE-SSDOTTSWSA-N 5-bromo-N-[(3R)-pyrrolidin-3-yl]pyrimidin-2-amine Chemical compound BrC=1C=NC(=NC=1)N[C@H]1CNCC1 SCPSQJAXCIJGSE-SSDOTTSWSA-N 0.000 description 1
- WYXSYVWAUAUWLD-SHUUEZRQSA-N 6-azauridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=N1 WYXSYVWAUAUWLD-SHUUEZRQSA-N 0.000 description 1
- XQJWCWHEDIFYRV-SSDOTTSWSA-N 6-chloro-n-methyl-n-[(3r)-pyrrolidin-3-yl]pyridazin-3-amine Chemical compound C=1C=C(Cl)N=NC=1N(C)[C@@H]1CCNC1 XQJWCWHEDIFYRV-SSDOTTSWSA-N 0.000 description 1
- 229960005538 6-diazo-5-oxo-L-norleucine Drugs 0.000 description 1
- YCWQAMGASJSUIP-YFKPBYRVSA-N 6-diazo-5-oxo-L-norleucine Chemical compound OC(=O)[C@@H](N)CCC(=O)C=[N+]=[N-] YCWQAMGASJSUIP-YFKPBYRVSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 description 1
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 description 1
- 230000005730 ADP ribosylation Effects 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- CEIZFXOZIQNICU-UHFFFAOYSA-N Alternaria alternata Crofton-weed toxin Natural products CCC(C)C1NC(=O)C(C(C)=O)=C1O CEIZFXOZIQNICU-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 238000011729 BALB/c nude mouse Methods 0.000 description 1
- 108700040618 BRCA1 Genes Proteins 0.000 description 1
- 108700010154 BRCA2 Genes Proteins 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- CQUKBGPEALMHRZ-GFCCVEGCSA-N BrC=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F Chemical compound BrC=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F CQUKBGPEALMHRZ-GFCCVEGCSA-N 0.000 description 1
- LVWHZKAXMXKQSW-MRXNPFEDSA-N BrC=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound BrC=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F LVWHZKAXMXKQSW-MRXNPFEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- MBABCNBNDNGODA-LTGLSHGVSA-N Bullatacin Natural products O=C1C(C[C@H](O)CCCCCCCCCC[C@@H](O)[C@@H]2O[C@@H]([C@@H]3O[C@H]([C@@H](O)CCCCCCCCCC)CC3)CC2)=C[C@H](C)O1 MBABCNBNDNGODA-LTGLSHGVSA-N 0.000 description 1
- KGGVWMAPBXIMEM-JQFCFGFHSA-N Bullatacinone Natural products O=C(C[C@H]1C(=O)O[C@H](CCCCCCCCCC[C@H](O)[C@@H]2O[C@@H]([C@@H]3O[C@@H]([C@@H](O)CCCCCCCCCC)CC3)CC2)C1)C KGGVWMAPBXIMEM-JQFCFGFHSA-N 0.000 description 1
- KGGVWMAPBXIMEM-ZRTAFWODSA-N Bullatacinone Chemical compound O1[C@@H]([C@@H](O)CCCCCCCCCC)CC[C@@H]1[C@@H]1O[C@@H]([C@H](O)CCCCCCCCCC[C@H]2OC(=O)[C@H](CC(C)=O)C2)CC1 KGGVWMAPBXIMEM-ZRTAFWODSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- JPRNSLMDGWQPLV-GOSISDBHSA-N C(#C)C=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C(#C)C=1C=NC(=NC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F JPRNSLMDGWQPLV-GOSISDBHSA-N 0.000 description 1
- LXCYEDZDACEINB-UHFFFAOYSA-N C(C)(C)(C)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C(C)(C)(C)NCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F LXCYEDZDACEINB-UHFFFAOYSA-N 0.000 description 1
- ACDSSLQUKSPVEN-GOSISDBHSA-N C(C)(C)(C)OC(=O)N1C[C@@H](CC1)NC1CN(C1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O Chemical compound C(C)(C)(C)OC(=O)N1C[C@@H](CC1)NC1CN(C1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O ACDSSLQUKSPVEN-GOSISDBHSA-N 0.000 description 1
- GRBALCNGXPQUPF-UHFFFAOYSA-N C(C)(C)(C)[Si](OC1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](OC1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C GRBALCNGXPQUPF-UHFFFAOYSA-N 0.000 description 1
- LXGHACDPJGFZMA-UHFFFAOYSA-N C(C)(C)(C)[Si](OC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](OC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C LXGHACDPJGFZMA-UHFFFAOYSA-N 0.000 description 1
- PTPTYCIYSVBKPL-UHFFFAOYSA-N C(C)(C)(C)[Si](OCC1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](OCC1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C PTPTYCIYSVBKPL-UHFFFAOYSA-N 0.000 description 1
- HYZYOCLFCXWBOI-UHFFFAOYSA-N C(C)(C)(C)[Si](OCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](OCC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C HYZYOCLFCXWBOI-UHFFFAOYSA-N 0.000 description 1
- LEBDBNNCXQRUPW-SFHVURJKSA-N C(C)(C)(C)[Si](O[C@@H]1CN(CC1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](O[C@@H]1CN(CC1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C LEBDBNNCXQRUPW-SFHVURJKSA-N 0.000 description 1
- FRXGTVKFRIQJSA-SFHVURJKSA-N C(C)(C)(C)[Si](O[C@@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](O[C@@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C FRXGTVKFRIQJSA-SFHVURJKSA-N 0.000 description 1
- LEBDBNNCXQRUPW-GOSISDBHSA-N C(C)(C)(C)[Si](O[C@H]1CN(CC1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](O[C@H]1CN(CC1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)(C)C LEBDBNNCXQRUPW-GOSISDBHSA-N 0.000 description 1
- FRXGTVKFRIQJSA-GOSISDBHSA-N C(C)(C)(C)[Si](O[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C Chemical compound C(C)(C)(C)[Si](O[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)(C)C FRXGTVKFRIQJSA-GOSISDBHSA-N 0.000 description 1
- INVHQLOJOLGBBZ-UHFFFAOYSA-N C(C)(CC)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C(C)(CC)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F INVHQLOJOLGBBZ-UHFFFAOYSA-N 0.000 description 1
- PAVQLIVVGKTZLY-GFCCVEGCSA-N C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)=O Chemical compound C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)=O PAVQLIVVGKTZLY-GFCCVEGCSA-N 0.000 description 1
- COAQVLDCQQJJAL-MRXNPFEDSA-N C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)C=C1/OC(C2=CC=CC=C12)=O)F)=O)=O Chemical compound C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)C=C1/OC(C2=CC=CC=C12)=O)F)=O)=O COAQVLDCQQJJAL-MRXNPFEDSA-N 0.000 description 1
- KKRRUBURPYTBBN-MRXNPFEDSA-N C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O)=O Chemical compound C(C)N1C(N(C(C1)=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O)=O KKRRUBURPYTBBN-MRXNPFEDSA-N 0.000 description 1
- WPTYACPKDLPKFN-UHFFFAOYSA-N C(C1=CC=CC=C1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C(C1=CC=CC=C1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F WPTYACPKDLPKFN-UHFFFAOYSA-N 0.000 description 1
- IETXZSCCTUWGCI-UHFFFAOYSA-N C(C1=CC=CC=C1)OC(NC1CN(C1)C(C1=C(C=CC(=C1)C=C1/OC(C2=CC=CC=C12)=O)F)=O)=O Chemical compound C(C1=CC=CC=C1)OC(NC1CN(C1)C(C1=C(C=CC(=C1)C=C1/OC(C2=CC=CC=C12)=O)F)=O)=O IETXZSCCTUWGCI-UHFFFAOYSA-N 0.000 description 1
- ARTKTMHHFVWWPV-QGZVFWFLSA-N C1(CC1)CNC(=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O Chemical compound C1(CC1)CNC(=O)[C@H]1CN(CC1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O ARTKTMHHFVWWPV-QGZVFWFLSA-N 0.000 description 1
- XGKMGCPQORHNIC-UHFFFAOYSA-N C1(CC1)N(C)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)N(C)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XGKMGCPQORHNIC-UHFFFAOYSA-N 0.000 description 1
- XJRSFIGBYPAQJH-UHFFFAOYSA-N C1(CC1)N(CC)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)N(CC)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XJRSFIGBYPAQJH-UHFFFAOYSA-N 0.000 description 1
- YDYWRTNDZPFULS-UHFFFAOYSA-N C1(CC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F YDYWRTNDZPFULS-UHFFFAOYSA-N 0.000 description 1
- XVUOMQCEJSMKLO-UHFFFAOYSA-N C1(CCC1)N(C1CN(C1)C(=O)C=1C=C(C=O)C=CC=1F)C1=NC=CC=N1 Chemical compound C1(CCC1)N(C1CN(C1)C(=O)C=1C=C(C=O)C=CC=1F)C1=NC=CC=N1 XVUOMQCEJSMKLO-UHFFFAOYSA-N 0.000 description 1
- ULPUEPLXFXQABL-QGZVFWFLSA-N C1(CCC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCC1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F ULPUEPLXFXQABL-QGZVFWFLSA-N 0.000 description 1
- YVKPGDKGLJISLZ-UHFFFAOYSA-N C1(CCCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound C1(CCCCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F YVKPGDKGLJISLZ-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- PXASOIWCJBGNDV-UHFFFAOYSA-N CC1(C(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C Chemical compound CC1(C(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C PXASOIWCJBGNDV-UHFFFAOYSA-N 0.000 description 1
- ANRVJYJCLZLPEH-UHFFFAOYSA-N CC1(NC(N(C1=O)C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)=O)C Chemical compound CC1(NC(N(C1=O)C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)=O)C ANRVJYJCLZLPEH-UHFFFAOYSA-N 0.000 description 1
- SHMCGLCCBFKAQR-SFHVURJKSA-N CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)C Chemical compound CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(C=C2/OC(C3=CC=CC=C23)=O)C=CC1F)C SHMCGLCCBFKAQR-SFHVURJKSA-N 0.000 description 1
- LMGUJBCWYQDOCZ-AWEZNQCLSA-N CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)C Chemical compound CN([C@@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)C LMGUJBCWYQDOCZ-AWEZNQCLSA-N 0.000 description 1
- HDYQOBLXSWUURO-GOSISDBHSA-N CN([C@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C Chemical compound CN([C@H]1CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C HDYQOBLXSWUURO-GOSISDBHSA-N 0.000 description 1
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- PTHCMJGKKRQCBF-UHFFFAOYSA-N Cellulose, microcrystalline Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC)C(CO)O1 PTHCMJGKKRQCBF-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- XCDXSSFOJZZGQC-UHFFFAOYSA-N Chlornaphazine Chemical compound C1=CC=CC2=CC(N(CCCl)CCCl)=CC=C21 XCDXSSFOJZZGQC-UHFFFAOYSA-N 0.000 description 1
- MKQWTWSXVILIKJ-LXGUWJNJSA-N Chlorozotocin Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](C=O)NC(=O)N(N=O)CCCl MKQWTWSXVILIKJ-LXGUWJNJSA-N 0.000 description 1
- 208000031404 Chromosome Aberrations Diseases 0.000 description 1
- 101150065749 Churc1 gene Proteins 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- IITPWJYQPFSALK-UHFFFAOYSA-N Cl.C(CCC)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C(CCC)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F IITPWJYQPFSALK-UHFFFAOYSA-N 0.000 description 1
- SKJNBPGSVZCBIA-UHFFFAOYSA-N Cl.C1(CC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F SKJNBPGSVZCBIA-UHFFFAOYSA-N 0.000 description 1
- MTIYJGHLCMNFOE-UHFFFAOYSA-N Cl.C1(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.C1(CCC1)NC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F MTIYJGHLCMNFOE-UHFFFAOYSA-N 0.000 description 1
- DCHCKRLBIVHWDS-UHFFFAOYSA-N Cl.FC1=C(C(=O)N2CC(C2)NC2(CCC2)C#N)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound Cl.FC1=C(C(=O)N2CC(C2)NC2(CCC2)C#N)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O DCHCKRLBIVHWDS-UHFFFAOYSA-N 0.000 description 1
- IGOCVQDNRHAQJO-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CN1CCCC1 Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CN1CCCC1 IGOCVQDNRHAQJO-UHFFFAOYSA-N 0.000 description 1
- JFHRMJGWXVUYJU-UHFFFAOYSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CO)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CO)C JFHRMJGWXVUYJU-UHFFFAOYSA-N 0.000 description 1
- NQZLGLWQJLPYHH-PFEQFJNWSA-N Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@@H](COC)C Chemical compound Cl.FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@@H](COC)C NQZLGLWQJLPYHH-PFEQFJNWSA-N 0.000 description 1
- MBAYZFBQZBQXSC-UHFFFAOYSA-N Cl.N1(CCC1)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.N1(CCC1)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F MBAYZFBQZBQXSC-UHFFFAOYSA-N 0.000 description 1
- GYAWDZQAHLZBAE-UHFFFAOYSA-N Cl.N1(CCC1)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.N1(CCC1)CC1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F GYAWDZQAHLZBAE-UHFFFAOYSA-N 0.000 description 1
- FDJKSTSBXSIUGA-PKLMIRHRSA-N Cl.N1(CCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound Cl.N1(CCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F FDJKSTSBXSIUGA-PKLMIRHRSA-N 0.000 description 1
- LNUOPXGERWHVGE-UHFFFAOYSA-N ClC1=CC=C(N=N1)N(C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)C Chemical compound ClC1=CC=C(N=N1)N(C1CN(C1)C(=O)C=1C=C(C=O)C=CC1F)C LNUOPXGERWHVGE-UHFFFAOYSA-N 0.000 description 1
- LDQKAUFAHSJDQL-UHFFFAOYSA-N ClC1=CC=C(N=N1)N(C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C Chemical compound ClC1=CC=C(N=N1)N(C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)C LDQKAUFAHSJDQL-UHFFFAOYSA-N 0.000 description 1
- GKRVIHBKCFHKBK-GFCCVEGCSA-N ClC1=CC=C(N=N1)N([C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)C Chemical compound ClC1=CC=C(N=N1)N([C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F)C GKRVIHBKCFHKBK-GFCCVEGCSA-N 0.000 description 1
- GBSVZMJQKHYSKB-UHFFFAOYSA-N ClC1=CC=C(N=N1)NC1CN(C1)C(=O)C=1C=C(C=O)C=CC1F Chemical compound ClC1=CC=C(N=N1)NC1CN(C1)C(=O)C=1C=C(C=O)C=CC1F GBSVZMJQKHYSKB-UHFFFAOYSA-N 0.000 description 1
- QCHVOOFOYMSFJU-OAHLLOKOSA-N ClC1=CC=C(N=N1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound ClC1=CC=C(N=N1)N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F QCHVOOFOYMSFJU-OAHLLOKOSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- AUGQEEXBDZWUJY-ZLJUKNTDSA-N Diacetoxyscirpenol Chemical compound C([C@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@@H]1C=C(C)CC[C@@]13COC(=O)C)O2 AUGQEEXBDZWUJY-ZLJUKNTDSA-N 0.000 description 1
- AUGQEEXBDZWUJY-UHFFFAOYSA-N Diacetoxyscirpenol Natural products CC(=O)OCC12CCC(C)=CC1OC1C(O)C(OC(C)=O)C2(C)C11CO1 AUGQEEXBDZWUJY-UHFFFAOYSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 239000012594 Earle’s Balanced Salt Solution Substances 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- XOBROCLBNQEWGP-UHFFFAOYSA-N FC1(CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F Chemical compound FC1(CN(CC1)C(=O)C1CN(C1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F XOBROCLBNQEWGP-UHFFFAOYSA-N 0.000 description 1
- CUYPYCSNFQRPCE-UHFFFAOYSA-N FC1(CN(CC1)C(=O)C1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F Chemical compound FC1(CN(CC1)C(=O)C1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F)F CUYPYCSNFQRPCE-UHFFFAOYSA-N 0.000 description 1
- OUVGLFVQEUHVNU-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)C=O Chemical compound FC1=C(C(=O)N2CC(C2)N(C(=O)C2CC2)C)C=C(C=C1)C=O OUVGLFVQEUHVNU-UHFFFAOYSA-N 0.000 description 1
- GQSXADLYCJAFJO-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)N2C(NC(C2=O)(C)C)=O)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)N2C(NC(C2=O)(C)C)=O)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O GQSXADLYCJAFJO-UHFFFAOYSA-N 0.000 description 1
- WBZBIEKLPNTZSF-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)N2C(NC(C2=O)(C)C)=O)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)N2C(NC(C2=O)(C)C)=O)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O WBZBIEKLPNTZSF-UHFFFAOYSA-N 0.000 description 1
- PPMMTDXCIFLGBU-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O PPMMTDXCIFLGBU-UHFFFAOYSA-N 0.000 description 1
- SVYJHOXVESXHTF-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)NC(C#N)C(C)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)NC(C#N)C(C)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O SVYJHOXVESXHTF-UHFFFAOYSA-N 0.000 description 1
- JEDMBNWTLRDEMV-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)NC(C#N)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)NC(C#N)C)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O JEDMBNWTLRDEMV-UHFFFAOYSA-N 0.000 description 1
- ACACWIKBWUYZEX-UHFFFAOYSA-N FC1=C(C(=O)N2CC(C2)NC(C#N)CC)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2CC(C2)NC(C#N)CC)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O ACACWIKBWUYZEX-UHFFFAOYSA-N 0.000 description 1
- DCOSLQSNCCDQHQ-QGZVFWFLSA-N FC1=C(C(=O)N2C[C@@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O DCOSLQSNCCDQHQ-QGZVFWFLSA-N 0.000 description 1
- WLQOIDALVJIKDU-CQSZACIVSA-N FC1=C(C(=O)N2C[C@@H](CC2)N2C(NCC2=O)=O)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)N2C(NCC2=O)=O)C=C(C=C1)C=C1/OC(C2=CC=CC=C12)=O WLQOIDALVJIKDU-CQSZACIVSA-N 0.000 description 1
- UBTRRYSXYVURPM-MRXNPFEDSA-N FC1=C(C(=O)N2C[C@@H](CC2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O UBTRRYSXYVURPM-MRXNPFEDSA-N 0.000 description 1
- PKSXZWVOMAYFBX-GOSISDBHSA-N FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C#N)C=CC=N2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C#N)C=CC=N2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O PKSXZWVOMAYFBX-GOSISDBHSA-N 0.000 description 1
- HXBWCZPIYYCDAA-CQSZACIVSA-N FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C(=O)N(C)C)C=CC=N2)C=C(C=C1)C=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)NC2=C(C(=O)N(C)C)C=CC=N2)C=C(C=C1)C=O HXBWCZPIYYCDAA-CQSZACIVSA-N 0.000 description 1
- CDIPREVYHWXGFY-QGZVFWFLSA-N FC1=C(C(=O)N2C[C@@H](CC2)NC2=NC=C(C=N2)C#N)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@@H](CC2)NC2=NC=C(C=N2)C#N)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O CDIPREVYHWXGFY-QGZVFWFLSA-N 0.000 description 1
- NMKKMKYCPZNGBT-ZDUSSCGKSA-N FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=O Chemical compound FC1=C(C(=O)N2C[C@H](CC2)N(C(=O)C2CC2)C)C=C(C=C1)C=O NMKKMKYCPZNGBT-ZDUSSCGKSA-N 0.000 description 1
- UBTRRYSXYVURPM-INIZCTEOSA-N FC1=C(C(=O)N2C[C@H](CC2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O Chemical compound FC1=C(C(=O)N2C[C@H](CC2)NC(=O)C2CC2)C=C(C=C1)CC1=NNC(C2=CC=CC=C12)=O UBTRRYSXYVURPM-INIZCTEOSA-N 0.000 description 1
- OLEGXIQBBUTEOE-UHFFFAOYSA-N FC1=C(C=C(C=C2/OC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C Chemical compound FC1=C(C=C(C=C2/OC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C OLEGXIQBBUTEOE-UHFFFAOYSA-N 0.000 description 1
- MGPUDPFBVIIFEC-UHFFFAOYSA-N FC1=C(C=C(C=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F Chemical compound FC1=C(C=C(C=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F MGPUDPFBVIIFEC-UHFFFAOYSA-N 0.000 description 1
- WZYXXJPBQJQXOD-UHFFFAOYSA-N FC1=C(C=C(C=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C Chemical compound FC1=C(C=C(C=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C WZYXXJPBQJQXOD-UHFFFAOYSA-N 0.000 description 1
- FFRZAANCQSDBSN-GFCCVEGCSA-N FC1=C(C=C(C=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=N1 Chemical compound FC1=C(C=C(C=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=N1 FFRZAANCQSDBSN-GFCCVEGCSA-N 0.000 description 1
- ZBKGAURWXUKYNI-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCC(CC1)F ZBKGAURWXUKYNI-UHFFFAOYSA-N 0.000 description 1
- CSTDWBPBBLWRIH-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)C(=O)N1CCCC1 CSTDWBPBBLWRIH-UHFFFAOYSA-N 0.000 description 1
- OESOOLTWQMDYHK-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CN1CCCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CN1CCCC1 OESOOLTWQMDYHK-UHFFFAOYSA-N 0.000 description 1
- FCBDBZPMBINKMN-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CO Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)CO FCBDBZPMBINKMN-UHFFFAOYSA-N 0.000 description 1
- YKTQVVOHKGWOGM-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)C YKTQVVOHKGWOGM-UHFFFAOYSA-N 0.000 description 1
- DKBIZLOYXMFSDU-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)CC Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N(C1=NC=CC=N1)CC DKBIZLOYXMFSDU-UHFFFAOYSA-N 0.000 description 1
- ZWGWTNXHGBSGOA-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N1CCCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N1CCCC1 ZWGWTNXHGBSGOA-UHFFFAOYSA-N 0.000 description 1
- ITJJIWONXAEHMW-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N1CCCCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N1CCCCC1 ITJJIWONXAEHMW-UHFFFAOYSA-N 0.000 description 1
- AAXQZBTZUAUBNO-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(C)C(C)C AAXQZBTZUAUBNO-UHFFFAOYSA-N 0.000 description 1
- LUHMKJUXMVIJHV-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CO)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC(CO)C LUHMKJUXMVIJHV-UHFFFAOYSA-N 0.000 description 1
- GWHPOEISAJOCTO-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1(CC1)CO Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1(CC1)CO GWHPOEISAJOCTO-UHFFFAOYSA-N 0.000 description 1
- BTZPBGWUPOBAQU-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1(CC1)COC Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1(CC1)COC BTZPBGWUPOBAQU-UHFFFAOYSA-N 0.000 description 1
- APUAGCHOXIDOQO-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1=NC=CC=N1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NC1=NC=CC=N1 APUAGCHOXIDOQO-UHFFFAOYSA-N 0.000 description 1
- DYBUUZMRIQXGKY-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(CO)(C)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(CO)(C)C DYBUUZMRIQXGKY-UHFFFAOYSA-N 0.000 description 1
- BZMOFWVBTOTEIH-UHFFFAOYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(F)(F)F Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)NCC(F)(F)F BZMOFWVBTOTEIH-UHFFFAOYSA-N 0.000 description 1
- YLODFAUXEWZAFV-HNNXBMFYSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@@H]1CNCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@@H]1CNCC1 YLODFAUXEWZAFV-HNNXBMFYSA-N 0.000 description 1
- YLODFAUXEWZAFV-OAHLLOKOSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@H]1CNCC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1CC(C1)N[C@H]1CNCC1 YLODFAUXEWZAFV-OAHLLOKOSA-N 0.000 description 1
- AJLKUMKMDMUPFE-QGZVFWFLSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N(C=1N=NC=CC1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N(C=1N=NC=CC1)C AJLKUMKMDMUPFE-QGZVFWFLSA-N 0.000 description 1
- VHHWGQUFEYBJGI-HXUWFJFHSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N(CC1=NC=CC=C1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)N(CC1=NC=CC=C1)C VHHWGQUFEYBJGI-HXUWFJFHSA-N 0.000 description 1
- IBXWHRBKCFOINO-HXUWFJFHSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=CC=C(C=C1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=CC=C(C=C1)C IBXWHRBKCFOINO-HXUWFJFHSA-N 0.000 description 1
- HTWRNLOZWWVLKT-QGZVFWFLSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=C(C=C1)C(F)(F)F Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=C(C=C1)C(F)(F)F HTWRNLOZWWVLKT-QGZVFWFLSA-N 0.000 description 1
- VFJOAEODASGSQP-QGZVFWFLSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=C1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=C1 VFJOAEODASGSQP-QGZVFWFLSA-N 0.000 description 1
- VIBFYTPXQPVJIJ-MRXNPFEDSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=N1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC1=NC=CC=N1 VIBFYTPXQPVJIJ-MRXNPFEDSA-N 0.000 description 1
- JRDMOEXZJKOIMR-MRXNPFEDSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC=1N=NC=CC1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC=1N=NC=CC1 JRDMOEXZJKOIMR-MRXNPFEDSA-N 0.000 description 1
- VBZXVPZPBZMLOW-OAHLLOKOSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC=1SC=CN1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@@H](CC1)NC=1SC=CN1 VBZXVPZPBZMLOW-OAHLLOKOSA-N 0.000 description 1
- ICVGAQIBCKXPAP-KRWDZBQOSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C1=NC=CC=N1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C1=NC=CC=N1)C ICVGAQIBCKXPAP-KRWDZBQOSA-N 0.000 description 1
- VONJGTHLELFXOE-SFHVURJKSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C1=NC=CC=N1)CC Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C1=NC=CC=N1)CC VONJGTHLELFXOE-SFHVURJKSA-N 0.000 description 1
- AJLKUMKMDMUPFE-KRWDZBQOSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C=1N=NC=CC1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(C=1N=NC=CC1)C AJLKUMKMDMUPFE-KRWDZBQOSA-N 0.000 description 1
- ARYYJPZZAXETBK-FQEVSTJZSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(CC1=CC=NC=C1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(CC1=CC=NC=C1)C ARYYJPZZAXETBK-FQEVSTJZSA-N 0.000 description 1
- VHHWGQUFEYBJGI-FQEVSTJZSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(CC1=NC=CC=C1)C Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)N(CC1=NC=CC=C1)C VHHWGQUFEYBJGI-FQEVSTJZSA-N 0.000 description 1
- VIBFYTPXQPVJIJ-INIZCTEOSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)NC1=NC=CC=N1 Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)NC1=NC=CC=N1 VIBFYTPXQPVJIJ-INIZCTEOSA-N 0.000 description 1
- WOBNJYNYIRZXGO-ZDUSSCGKSA-N FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)O Chemical compound FC1=C(C=C(CC2=NNC(C3=CC=CC=C23)=O)C=C1)C(=O)N1C[C@H](CC1)O WOBNJYNYIRZXGO-ZDUSSCGKSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- MPJKWIXIYCLVCU-UHFFFAOYSA-N Folinic acid Natural products NC1=NC2=C(N(C=O)C(CNc3ccc(cc3)C(=O)NC(CCC(=O)O)CC(=O)O)CN2)C(=O)N1 MPJKWIXIYCLVCU-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000006947 Histones Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 241000544038 Luronium Species 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229920002774 Maltodextrin Polymers 0.000 description 1
- 239000005913 Maltodextrin Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- VJRAUFKOOPNFIQ-UHFFFAOYSA-N Marcellomycin Natural products C12=C(O)C=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C=C2C(C(=O)OC)C(CC)(O)CC1OC(OC1C)CC(N(C)C)C1OC(OC1C)CC(O)C1OC1CC(O)C(O)C(C)O1 VJRAUFKOOPNFIQ-UHFFFAOYSA-N 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- IVDYZAAPOLNZKG-KWHRADDSSA-N Mepitiostane Chemical compound O([C@@H]1[C@]2(CC[C@@H]3[C@@]4(C)C[C@H]5S[C@H]5C[C@@H]4CC[C@H]3[C@@H]2CC1)C)C1(OC)CCCC1 IVDYZAAPOLNZKG-KWHRADDSSA-N 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Mitomycin E Natural products O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- MTHLDFHTYSVAHD-SECBINFHSA-N N,N-dimethyl-2-[[(3R)-pyrrolidin-3-yl]amino]pyridine-3-carboxamide Chemical compound CN(C(C1=C(N=CC=C1)N[C@H]1CNCC1)=O)C MTHLDFHTYSVAHD-SECBINFHSA-N 0.000 description 1
- MZMYAEZGBJNORO-UHFFFAOYSA-N N-(azetidin-3-yl)-N-cyclobutylpyrimidin-2-amine Chemical compound N1CC(C1)N(C1=NC=CC=N1)C1CCC1 MZMYAEZGBJNORO-UHFFFAOYSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- IDQSXIWJCYXUQF-QMMMGPOBSA-N N-methyl-N-[(3S)-pyrrolidin-3-yl]cyclopropanecarboxamide Chemical compound CN(C(=O)C1CC1)[C@@H]1CNCC1 IDQSXIWJCYXUQF-QMMMGPOBSA-N 0.000 description 1
- VTTFHGJVTCQWLY-QGZVFWFLSA-N N1(CCCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound N1(CCCC1)[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F VTTFHGJVTCQWLY-QGZVFWFLSA-N 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-O NAD(+) Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-O 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- XFCOVBLNDZJVES-ZDUSSCGKSA-N N[C@@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound N[C@@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XFCOVBLNDZJVES-ZDUSSCGKSA-N 0.000 description 1
- XFCOVBLNDZJVES-CYBMUJFWSA-N N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F Chemical compound N[C@H]1CN(CC1)C(=O)C=1C=C(CC2=NNC(C3=CC=CC=C23)=O)C=CC1F XFCOVBLNDZJVES-CYBMUJFWSA-N 0.000 description 1
- 229910052779 Neodymium Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- SYNHCENRCUAUNM-UHFFFAOYSA-N Nitrogen mustard N-oxide hydrochloride Chemical compound Cl.ClCC[N+]([O-])(C)CCCl SYNHCENRCUAUNM-UHFFFAOYSA-N 0.000 description 1
- KGTDRFCXGRULNK-UHFFFAOYSA-N Nogalamycin Natural products COC1C(OC)(C)C(OC)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=C4C5(C)OC(C(C(C5O)N(C)C)O)OC4=C3C3=O)=C3C=C2C(C(=O)OC)C(C)(O)C1 KGTDRFCXGRULNK-UHFFFAOYSA-N 0.000 description 1
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 1
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 1
- KUWMJTUSKBDLPX-SNVBAGLBSA-N O=C1N(C(CN1)=O)[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F Chemical compound O=C1N(C(CN1)=O)[C@H]1CN(CC1)C(=O)C=1C=C(C=O)C=CC1F KUWMJTUSKBDLPX-SNVBAGLBSA-N 0.000 description 1
- 229930187135 Olivomycin Natural products 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- VREZDOWOLGNDPW-ALTGWBOUSA-N Pancratistatin Chemical compound C1=C2[C@H]3[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)[C@@H]3NC(=O)C2=C(O)C2=C1OCO2 VREZDOWOLGNDPW-ALTGWBOUSA-N 0.000 description 1
- VREZDOWOLGNDPW-MYVCAWNPSA-N Pancratistatin Natural products O=C1N[C@H]2[C@H](O)[C@H](O)[C@H](O)[C@H](O)[C@@H]2c2c1c(O)c1OCOc1c2 VREZDOWOLGNDPW-MYVCAWNPSA-N 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 102100038239 Protein Churchill Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 1
- NSFWWJIQIKBZMJ-YKNYLIOZSA-N Roridin A Chemical compound C([C@]12[C@]3(C)[C@H]4C[C@H]1O[C@@H]1C=C(C)CC[C@@]13COC(=O)[C@@H](O)[C@H](C)CCO[C@H](\C=C\C=C/C(=O)O4)[C@H](O)C)O2 NSFWWJIQIKBZMJ-YKNYLIOZSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- BXFOFFBJRFZBQZ-QYWOHJEZSA-N T-2 toxin Chemical compound C([C@@]12[C@]3(C)[C@H](OC(C)=O)[C@@H](O)[C@H]1O[C@H]1[C@]3(COC(C)=O)C[C@@H](C(=C1)C)OC(=O)CC(C)C)O2 BXFOFFBJRFZBQZ-QYWOHJEZSA-N 0.000 description 1
- 108700011582 TER 286 Proteins 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- CGMTUJFWROPELF-UHFFFAOYSA-N Tenuazonic acid Natural products CCC(C)C1NC(=O)C(=C(C)/O)C1=O CGMTUJFWROPELF-UHFFFAOYSA-N 0.000 description 1
- UMILHIMHKXVDGH-UHFFFAOYSA-N Triethylene glycol diglycidyl ether Chemical compound C1OC1COCCOCCOCCOCC1CO1 UMILHIMHKXVDGH-UHFFFAOYSA-N 0.000 description 1
- FYAMXEPQQLNQDM-UHFFFAOYSA-N Tris(1-aziridinyl)phosphine oxide Chemical compound C1CN1P(N1CC1)(=O)N1CC1 FYAMXEPQQLNQDM-UHFFFAOYSA-N 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- ZYVSOIYQKUDENJ-ASUJBHBQSA-N [(2R,3R,4R,6R)-6-[[(6S,7S)-6-[(2S,4R,5R,6R)-4-[(2R,4R,5R,6R)-4-[(2S,4S,5S,6S)-5-acetyloxy-4-hydroxy-4,6-dimethyloxan-2-yl]oxy-5-hydroxy-6-methyloxan-2-yl]oxy-5-hydroxy-6-methyloxan-2-yl]oxy-7-[(3S,4R)-3,4-dihydroxy-1-methoxy-2-oxopentyl]-4,10-dihydroxy-3-methyl-5-oxo-7,8-dihydro-6H-anthracen-2-yl]oxy]-4-[(2R,4R,5R,6R)-4-hydroxy-5-methoxy-6-methyloxan-2-yl]oxy-2-methyloxan-3-yl] acetate Chemical class COC([C@@H]1Cc2cc3cc(O[C@@H]4C[C@@H](O[C@@H]5C[C@@H](O)[C@@H](OC)[C@@H](C)O5)[C@H](OC(C)=O)[C@@H](C)O4)c(C)c(O)c3c(O)c2C(=O)[C@H]1O[C@H]1C[C@@H](O[C@@H]2C[C@@H](O[C@H]3C[C@](C)(O)[C@@H](OC(C)=O)[C@H](C)O3)[C@H](O)[C@@H](C)O2)[C@H](O)[C@@H](C)O1)C(=O)[C@@H](O)[C@@H](C)O ZYVSOIYQKUDENJ-ASUJBHBQSA-N 0.000 description 1
- IFJUINDAXYAPTO-UUBSBJJBSA-N [(8r,9s,13s,14s,17s)-17-[2-[4-[4-[bis(2-chloroethyl)amino]phenyl]butanoyloxy]acetyl]oxy-13-methyl-6,7,8,9,11,12,14,15,16,17-decahydrocyclopenta[a]phenanthren-3-yl] benzoate Chemical compound C([C@@H]1[C@@H](C2=CC=3)CC[C@]4([C@H]1CC[C@@H]4OC(=O)COC(=O)CCCC=1C=CC(=CC=1)N(CCCl)CCCl)C)CC2=CC=3OC(=O)C1=CC=CC=C1 IFJUINDAXYAPTO-UUBSBJJBSA-N 0.000 description 1
- XZSRRNFBEIOBDA-CFNBKWCHSA-N [2-[(2s,4s)-4-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-2,5,12-trihydroxy-7-methoxy-6,11-dioxo-3,4-dihydro-1h-tetracen-2-yl]-2-oxoethyl] 2,2-diethoxyacetate Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)C(OCC)OCC)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 XZSRRNFBEIOBDA-CFNBKWCHSA-N 0.000 description 1
- 229940028652 abraxane Drugs 0.000 description 1
- ZOZKYEHVNDEUCO-XUTVFYLZSA-N aceglatone Chemical compound O1C(=O)[C@H](OC(C)=O)[C@@H]2OC(=O)[C@@H](OC(=O)C)[C@@H]21 ZOZKYEHVNDEUCO-XUTVFYLZSA-N 0.000 description 1
- 229950002684 aceglatone Drugs 0.000 description 1
- WJGAPUXHSQQWQF-UHFFFAOYSA-N acetic acid;hydrochloride Chemical compound Cl.CC(O)=O WJGAPUXHSQQWQF-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 229930188522 aclacinomycin Natural products 0.000 description 1
- LJZPVWKMAYDYAS-QKKPTTNWSA-N aclacinomycin T Chemical class O([C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)[C@H]1C[C@H](N(C)C)[C@H](O)[C@H](C)O1 LJZPVWKMAYDYAS-QKKPTTNWSA-N 0.000 description 1
- 229930183665 actinomycin Natural products 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 229950004955 adozelesin Drugs 0.000 description 1
- BYRVKDUQDLJUBX-JJCDCTGGSA-N adozelesin Chemical compound C1=CC=C2OC(C(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)NC=C5C)=CC2=C1 BYRVKDUQDLJUBX-JJCDCTGGSA-N 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 229940061720 alpha hydroxy acid Drugs 0.000 description 1
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- 229960002749 aminolevulinic acid Drugs 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- BBDAGFIXKZCXAH-CCXZUQQUSA-N ancitabine Chemical compound N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 BBDAGFIXKZCXAH-CCXZUQQUSA-N 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045686 antimetabolites antineoplastic purine analogs Drugs 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 229940045713 antineoplastic alkylating drug ethylene imines Drugs 0.000 description 1
- 229940045688 antineoplastic antimetabolites pyrimidine analogues Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 150000008209 arabinosides Chemical class 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229950011321 azaserine Drugs 0.000 description 1
- BTRRCAGMXNRVFD-UHFFFAOYSA-N azetidin-3-yl-(4-fluoropiperidin-1-yl)methanone Chemical compound N1CC(C1)C(=O)N1CCC(CC1)F BTRRCAGMXNRVFD-UHFFFAOYSA-N 0.000 description 1
- OTQLOBVJJPWUNE-UHFFFAOYSA-N azetidin-3-ylmethoxy-tert-butyl-dimethylsilane Chemical compound C(C)(C)(C)[Si](OCC1CNC1)(C)C OTQLOBVJJPWUNE-UHFFFAOYSA-N 0.000 description 1
- ULMHPBVREZOAQD-UHFFFAOYSA-N azetidin-3-yloxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OC1CNC1 ULMHPBVREZOAQD-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- ZWAJDQXQVFJJTE-MRXNPFEDSA-N benzyl N-[(3R)-1-(2-fluoro-5-formylbenzoyl)pyrrolidin-3-yl]carbamate Chemical compound C(C1=CC=CC=C1)OC(N[C@H]1CN(CC1)C(C1=C(C=CC(=C1)C=O)F)=O)=O ZWAJDQXQVFJJTE-MRXNPFEDSA-N 0.000 description 1
- IPVFSDVNBSIMRM-HXUWFJFHSA-N benzyl N-[(3R)-1-[2-fluoro-5-[(4-oxo-3H-phthalazin-1-yl)methyl]benzoyl]pyrrolidin-3-yl]carbamate Chemical compound C(C1=CC=CC=C1)OC(N[C@H]1CN(CC1)C(C1=C(C=CC(=C1)CC1=NNC(C2=CC=CC=C12)=O)F)=O)=O IPVFSDVNBSIMRM-HXUWFJFHSA-N 0.000 description 1
- XHTQWZFEUYPKGP-FQEVSTJZSA-N benzyl N-[(3S)-1-[2-fluoro-5-[(Z)-(3-oxo-2-benzofuran-1-ylidene)methyl]benzoyl]pyrrolidin-3-yl]carbamate Chemical compound C(C1=CC=CC=C1)OC(N[C@@H]1CN(CC1)C(C1=C(C=CC(=C1)C=C1/OC(C2=CC=CC=C12)=O)F)=O)=O XHTQWZFEUYPKGP-FQEVSTJZSA-N 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 description 1
- UWOOBFRZDNDUQB-UHFFFAOYSA-N benzyl n-(azetidin-3-yl)carbamate Chemical compound C=1C=CC=CC=1COC(=O)NC1CNC1 UWOOBFRZDNDUQB-UHFFFAOYSA-N 0.000 description 1
- DSOICHFMGRBFCM-LLVKDONJSA-N benzyl n-[(3r)-pyrrolidin-3-yl]carbamate Chemical compound C=1C=CC=CC=1COC(=O)N[C@@H]1CCNC1 DSOICHFMGRBFCM-LLVKDONJSA-N 0.000 description 1
- DSOICHFMGRBFCM-NSHDSACASA-N benzyl n-[(3s)-pyrrolidin-3-yl]carbamate Chemical compound C=1C=CC=CC=1COC(=O)N[C@H]1CCNC1 DSOICHFMGRBFCM-NSHDSACASA-N 0.000 description 1
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 229950006844 bizelesin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical class N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229960005520 bryostatin Drugs 0.000 description 1
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 1
- MUIWQCKLQMOUAT-AKUNNTHJSA-N bryostatin 20 Natural products COC(=O)C=C1C[C@@]2(C)C[C@]3(O)O[C@](C)(C[C@@H](O)CC(=O)O[C@](C)(C[C@@]4(C)O[C@](O)(CC5=CC(=O)O[C@]45C)C(C)(C)C=C[C@@](C)(C1)O2)[C@@H](C)O)C[C@H](OC(=O)C(C)(C)C)C3(C)C MUIWQCKLQMOUAT-AKUNNTHJSA-N 0.000 description 1
- MBABCNBNDNGODA-LUVUIASKSA-N bullatacin Chemical compound O1[C@@H]([C@@H](O)CCCCCCCCCC)CC[C@@H]1[C@@H]1O[C@@H]([C@H](O)CCCCCCCCCC[C@@H](O)CC=2C(O[C@@H](C)C=2)=O)CC1 MBABCNBNDNGODA-LUVUIASKSA-N 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- XSBPYGDBXQXSCU-UHFFFAOYSA-N but-3-yn-1-amine Chemical compound NCCC#C XSBPYGDBXQXSCU-UHFFFAOYSA-N 0.000 description 1
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108700002839 cactinomycin Proteins 0.000 description 1
- 229950009908 cactinomycin Drugs 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- IVFYLRMMHVYGJH-PVPPCFLZSA-N calusterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-PVPPCFLZSA-N 0.000 description 1
- 229950009823 calusterone Drugs 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- BBZDXMBRAFTCAA-AREMUKBSSA-N carzelesin Chemical compound C1=2NC=C(C)C=2C([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)C3=CC4=CC=C(C=C4O3)N(CC)CC)=C2C=C1OC(=O)NC1=CC=CC=C1 BBZDXMBRAFTCAA-AREMUKBSSA-N 0.000 description 1
- 229950007509 carzelesin Drugs 0.000 description 1
- 108010047060 carzinophilin Proteins 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 235000015218 chewing gum Nutrition 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229950008249 chlornaphazine Drugs 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960001480 chlorozotocin Drugs 0.000 description 1
- 231100000005 chromosome aberration Toxicity 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cis-cyclohexene Natural products C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960002286 clodronic acid Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960002271 cobimetinib Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- LMKPHJYTFHAGHK-UHFFFAOYSA-N cyclodrine Chemical compound C1CCCC1(O)C(C(=O)OCCN(CC)CC)C1=CC=CC=C1 LMKPHJYTFHAGHK-UHFFFAOYSA-N 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- UVJHQYIOXKWHFD-UHFFFAOYSA-N cyclohexa-1,4-diene Chemical compound C1C=CCC=C1 UVJHQYIOXKWHFD-UHFFFAOYSA-N 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- VELDYOPRLMJFIK-UHFFFAOYSA-N cyclopentanecarbaldehyde Chemical compound O=CC1CCCC1 VELDYOPRLMJFIK-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- IGSKHXTUVXSOMB-UHFFFAOYSA-N cyclopropylmethanamine Chemical compound NCC1CC1 IGSKHXTUVXSOMB-UHFFFAOYSA-N 0.000 description 1
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229950006418 dactolisib Drugs 0.000 description 1
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 229960005052 demecolcine Drugs 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- 229950003913 detorubicin Drugs 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960002086 dextran Drugs 0.000 description 1
- 229940096516 dextrates Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 1
- 229950002389 diaziquone Drugs 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- 229940095079 dicalcium phosphate anhydrous Drugs 0.000 description 1
- BIPUHAHGLJKIPK-UHFFFAOYSA-N dicyclopropylmethanone Chemical compound C1CC1C(=O)C1CC1 BIPUHAHGLJKIPK-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 230000005782 double-strand break Effects 0.000 description 1
- NOTIQUSPUUHHEH-UXOVVSIBSA-N dromostanolone propionate Chemical compound C([C@@H]1CC2)C(=O)[C@H](C)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](OC(=O)CC)[C@@]2(C)CC1 NOTIQUSPUUHHEH-UXOVVSIBSA-N 0.000 description 1
- 229950004683 drostanolone propionate Drugs 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- FSIRXIHZBIXHKT-MHTVFEQDSA-N edatrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CC(CC)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FSIRXIHZBIXHKT-MHTVFEQDSA-N 0.000 description 1
- 229950006700 edatrexate Drugs 0.000 description 1
- 230000000235 effect on cancer Effects 0.000 description 1
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical compound NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- XOPYFXBZMVTEJF-UHFFFAOYSA-N eleutherobin Natural products C1=CC2(OC)OC1(C)C(OC(=O)C=CC=1N=CN(C)C=1)CC(C(=CCC1C(C)C)C)C1C=C2COC1OCC(O)C(O)C1OC(C)=O XOPYFXBZMVTEJF-UHFFFAOYSA-N 0.000 description 1
- XOPYFXBZMVTEJF-PDACKIITSA-N eleutherobin Chemical compound C(/[C@H]1[C@H](C(=CC[C@@H]1C(C)C)C)C[C@@H]([C@@]1(C)O[C@@]2(C=C1)OC)OC(=O)\C=C\C=1N=CN(C)C=1)=C2\CO[C@@H]1OC[C@@H](O)[C@@H](O)[C@@H]1OC(C)=O XOPYFXBZMVTEJF-PDACKIITSA-N 0.000 description 1
- 229950000549 elliptinium acetate Drugs 0.000 description 1
- 229940120655 eloxatin Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- JOZGNYDSEBIJDH-UHFFFAOYSA-N eniluracil Chemical compound O=C1NC=C(C#C)C(=O)N1 JOZGNYDSEBIJDH-UHFFFAOYSA-N 0.000 description 1
- 229950010213 eniluracil Drugs 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 208000002854 epidermolysis bullosa simplex superficialis Diseases 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- ITSGNOIFAJAQHJ-BMFNZSJVSA-N esorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)C[C@H](C)O1 ITSGNOIFAJAQHJ-BMFNZSJVSA-N 0.000 description 1
- 229950002017 esorubicin Drugs 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 229940087861 faslodex Drugs 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- 229940083124 ganglion-blocking antiadrenergic secondary and tertiary amines Drugs 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 230000007614 genetic variation Effects 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229930182478 glucoside Natural products 0.000 description 1
- 150000008131 glucosides Chemical class 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229940028286 ibadronate Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 229950001750 lonafarnib Drugs 0.000 description 1
- YROQEQPFUCPDCP-UHFFFAOYSA-N losoxantrone Chemical compound OCCNCCN1N=C2C3=CC=CC(O)=C3C(=O)C3=C2C1=CC=C3NCCNCCO YROQEQPFUCPDCP-UHFFFAOYSA-N 0.000 description 1
- 229950008745 losoxantrone Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940035034 maltodextrin Drugs 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- MQXVYODZCMMZEM-ZYUZMQFOSA-N mannomustine Chemical compound ClCCNC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CNCCCl MQXVYODZCMMZEM-ZYUZMQFOSA-N 0.000 description 1
- 229950008612 mannomustine Drugs 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229950009246 mepitiostane Drugs 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- VJRAUFKOOPNFIQ-TVEKBUMESA-N methyl (1r,2r,4s)-4-[(2r,4s,5s,6s)-5-[(2s,4s,5s,6s)-5-[(2s,4s,5s,6s)-4,5-dihydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyloxan-2-yl]oxy-4-(dimethylamino)-6-methyloxan-2-yl]oxy-2-ethyl-2,5,7,10-tetrahydroxy-6,11-dioxo-3,4-dihydro-1h-tetracene-1-carboxylat Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=C(O)C=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1C[C@H](O)[C@H](O)[C@H](C)O1 VJRAUFKOOPNFIQ-TVEKBUMESA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- HRHKSTOGXBBQCB-VFWICMBZSA-N methylmitomycin Chemical compound O=C1C(N)=C(C)C(=O)C2=C1[C@@H](COC(N)=O)[C@@]1(OC)[C@H]3N(C)[C@H]3CN12 HRHKSTOGXBBQCB-VFWICMBZSA-N 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 229960003539 mitoguazone Drugs 0.000 description 1
- MXWHMTNPTTVWDM-NXOFHUPFSA-N mitoguazone Chemical compound NC(N)=N\N=C(/C)\C=N\N=C(N)N MXWHMTNPTTVWDM-NXOFHUPFSA-N 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 230000003232 mucoadhesive effect Effects 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- JKMOISGOVOSNMI-UHFFFAOYSA-N n-(azetidin-3-yl)-6-chloro-n-methylpyridazin-3-amine Chemical compound C=1C=C(Cl)N=NC=1N(C)C1CNC1 JKMOISGOVOSNMI-UHFFFAOYSA-N 0.000 description 1
- RSEOLOUMBPVXRT-UHFFFAOYSA-N n-(azetidin-3-yl)-n-methylcyclopropanecarboxamide Chemical compound C1CC1C(=O)N(C)C1CNC1 RSEOLOUMBPVXRT-UHFFFAOYSA-N 0.000 description 1
- YDGWWNRXHXZWPS-SSDOTTSWSA-N n-[(3r)-pyrrolidin-3-yl]pyrimidin-2-amine Chemical compound C1NCC[C@H]1NC1=NC=CC=N1 YDGWWNRXHXZWPS-SSDOTTSWSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 description 1
- 229950010159 nemorubicin Drugs 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- KGTDRFCXGRULNK-JYOBTZKQSA-N nogalamycin Chemical compound CO[C@@H]1[C@@](OC)(C)[C@@H](OC)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=C4[C@@]5(C)O[C@H]([C@H]([C@@H]([C@H]5O)N(C)C)O)OC4=C3C3=O)=C3C=C2[C@@H](C(=O)OC)[C@@](C)(O)C1 KGTDRFCXGRULNK-JYOBTZKQSA-N 0.000 description 1
- 229950009266 nogalamycin Drugs 0.000 description 1
- KPMKEVXVVHNIEY-RITPCOANSA-N norcamphor Chemical compound C1C[C@@H]2C(=O)C[C@H]1C2 KPMKEVXVVHNIEY-RITPCOANSA-N 0.000 description 1
- 108020004017 nuclear receptors Proteins 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229960000572 olaparib Drugs 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- CZDBNBLGZNWKMC-MWQNXGTOSA-N olivomycin Chemical class O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1)O[C@H]1O[C@@H](C)[C@H](O)[C@@H](OC2O[C@@H](C)[C@H](O)[C@@H](O)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@H](O)[C@H](OC)[C@H](C)O1 CZDBNBLGZNWKMC-MWQNXGTOSA-N 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- VREZDOWOLGNDPW-UHFFFAOYSA-N pancratistatine Natural products C1=C2C3C(O)C(O)C(O)C(O)C3NC(=O)C2=C(O)C2=C1OCO2 VREZDOWOLGNDPW-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 108010021753 peptide-Gly-Leu-amide Proteins 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 238000005502 peroxidation Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NUKCGLDCWQXYOQ-UHFFFAOYSA-N piposulfan Chemical compound CS(=O)(=O)OCCC(=O)N1CCN(C(=O)CCOS(C)(=O)=O)CC1 NUKCGLDCWQXYOQ-UHFFFAOYSA-N 0.000 description 1
- 229950001100 piposulfan Drugs 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229950004406 porfiromycin Drugs 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 102000003998 progesterone receptors Human genes 0.000 description 1
- 108090000468 progesterone receptors Proteins 0.000 description 1
- JKANAVGODYYCQF-UHFFFAOYSA-N prop-2-yn-1-amine Chemical compound NCC#C JKANAVGODYYCQF-UHFFFAOYSA-N 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- WOLQREOUPKZMEX-UHFFFAOYSA-N pteroyltriglutamic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(=O)NC(CCC(=O)NC(CCC(O)=O)C(O)=O)C(O)=O)C(O)=O)C=C1 WOLQREOUPKZMEX-UHFFFAOYSA-N 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- BGUWFUQJCDRPTL-UHFFFAOYSA-N pyridine-4-carbaldehyde Chemical compound O=CC1=CC=NC=C1 BGUWFUQJCDRPTL-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 229940099538 rapamune Drugs 0.000 description 1
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 description 1
- 229960000460 razoxane Drugs 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 description 1
- MBABCNBNDNGODA-WPZDJQSSSA-N rolliniastatin 1 Natural products O1[C@@H]([C@@H](O)CCCCCCCCCC)CC[C@H]1[C@H]1O[C@@H]([C@H](O)CCCCCCCCCC[C@@H](O)CC=2C(O[C@@H](C)C=2)=O)CC1 MBABCNBNDNGODA-WPZDJQSSSA-N 0.000 description 1
- IMUQLZLGWJSVMV-UOBFQKKOSA-N roridin A Natural products CC(O)C1OCCC(C)C(O)C(=O)OCC2CC(=CC3OC4CC(OC(=O)C=C/C=C/1)C(C)(C23)C45CO5)C IMUQLZLGWJSVMV-UOBFQKKOSA-N 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- XIIOFHFUYBLOLW-UHFFFAOYSA-N selpercatinib Chemical compound OC(COC=1C=C(C=2N(C=1)N=CC=2C#N)C=1C=NC(=CC=1)N1CC2N(C(C1)C2)CC=1C=NC(=CC=1)OC)(C)C XIIOFHFUYBLOLW-UHFFFAOYSA-N 0.000 description 1
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 229960003885 sodium benzoate Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000008137 solubility enhancer Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 229950006315 spirogermanium Drugs 0.000 description 1
- ICXJVZHDZFXYQC-UHFFFAOYSA-N spongistatin 1 Natural products OC1C(O2)(O)CC(O)C(C)C2CCCC=CC(O2)CC(O)CC2(O2)CC(OC)CC2CC(=O)C(C)C(OC(C)=O)C(C)C(=C)CC(O2)CC(C)(O)CC2(O2)CC(OC(C)=O)CC2CC(=O)OC2C(O)C(CC(=C)CC(O)C=CC(Cl)=C)OC1C2C ICXJVZHDZFXYQC-UHFFFAOYSA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- CMIBWIAICVBURI-SSDOTTSWSA-N tert-butyl (3r)-3-aminopyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@@H](N)C1 CMIBWIAICVBURI-SSDOTTSWSA-N 0.000 description 1
- CMIBWIAICVBURI-ZETCQYMHSA-N tert-butyl (3s)-3-aminopyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@H](N)C1 CMIBWIAICVBURI-ZETCQYMHSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- SDLGKSBUCUJCRU-SECBINFHSA-N tert-butyl-dimethyl-[(3r)-pyrrolidin-3-yl]oxysilane Chemical compound CC(C)(C)[Si](C)(C)O[C@@H]1CCNC1 SDLGKSBUCUJCRU-SECBINFHSA-N 0.000 description 1
- SDLGKSBUCUJCRU-VIFPVBQESA-N tert-butyl-dimethyl-[(3s)-pyrrolidin-3-yl]oxysilane Chemical compound CC(C)(C)[Si](C)(C)O[C@H]1CCNC1 SDLGKSBUCUJCRU-VIFPVBQESA-N 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 230000009974 thixotropic effect Effects 0.000 description 1
- YFTWHEBLORWGNI-UHFFFAOYSA-N tiamiprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC(N)=NC2=C1NC=N2 YFTWHEBLORWGNI-UHFFFAOYSA-N 0.000 description 1
- 229950011457 tiamiprine Drugs 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- PXSOHRWMIRDKMP-UHFFFAOYSA-N triaziquone Chemical compound O=C1C(N2CC2)=C(N2CC2)C(=O)C=C1N1CC1 PXSOHRWMIRDKMP-UHFFFAOYSA-N 0.000 description 1
- 229960004560 triaziquone Drugs 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 235000019731 tricalcium phosphate Nutrition 0.000 description 1
- 229930013292 trichothecene Natural products 0.000 description 1
- 150000003327 trichothecene derivatives Chemical class 0.000 description 1
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- HDZZVAMISRMYHH-LITAXDCLSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@@H](CO)[C@H](O)[C@H]1O HDZZVAMISRMYHH-LITAXDCLSA-N 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- LLDWLPRYLVPDTG-UHFFFAOYSA-N vatalanib succinate Chemical compound OC(=O)CCC(O)=O.C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 LLDWLPRYLVPDTG-UHFFFAOYSA-N 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/502—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with carbocyclic ring systems, e.g. cinnoline, phthalazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
Abstract
La presente invención se refiere a derivados de ftalazinona, incluidas composiciones farmacéuticas y a la preparación de derivados de ftalazinona. Y más particularmente, la presente invención proporcionó una composición farmacéutica de derivados de ftalazinona para inhibir la actividad de la enzima poli(ADP-ribosido) polimerasa. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados novedosos de ftalazinona y su procedimiento de fabricación
Campo técnico
La presente invención se refiere a derivados de ftalazinona, isómeros, una sal farmacéuticamente aceptable de los mismos y a su estudio para usos medicinales.
Antecedentes de la técnica
El interés en inhibidores de poli ADP ribosa polimerasa (PARP) está aumentando de acuerdo con la detección de una alta tasa de mutación del gen BRCA1 o BRCA2 debido al reciente cáncer de mama y ovario genético. En general, con las mutaciones genéticas BRCA el riesgo de cáncer de mama es 5 veces más alto, y uno de ellos en particular, la probabilidad de un riesgo considerable para TNBC (cáncer de mama triple negativo) se incrementa. TNBC representa el 15 % de pacientes con cáncer de mama está relacionado con las carencias de receptores de estrógeno, receptores de progesterona. Como no ha habido ningún tratamiento especial hasta ahora, el objeto de TNBC contiene potencial de mercado importante.
La PARP es una de las enzimas reparadoras para rompimientos de ADN de doble cadena dañado. En caso de inhibición de PARP, el daño de una cadena consecutivamente genera defectos en ADN de doble cadena. En este punto, los defectos de doble cadena pueden ser recuperados por el complejo de la proteína BRCA. Así en general, aun cuando una de las rutas de reparación de ADN no se trabaja, la mayoría de la célula puede estar viva. Pero los pacientes de cáncer que hereditariamente pierden la trayectoria de reparación del complejo de proteína BRCA por la mutación de BRCA1/BRCA2 pueden aumentar la dependencia en la trayectoria de Pa Rp de reparación de ADN. Especialmente, como la posibilidad de defectos en la replicación del ADN en el caso de célula de cáncer es superior a la célula normal, la célula cancerosa tiene mayor dependencia en la trayectoria de PARP que la célula normal. En otras palabras, los inhibidores de PARP fundamentalmente bloquean el sistema de reparación de la célula cancerosa después de la apoptosis de células cancerosas.
Hasta la fecha, 18 miembros de la familia de PARP han sido identificados y caracterizados, con PARP-1 que es el más ampliamente estudiado y PARP-2 que es su pariente más cercano. A pesar del gran número de enzimas en esta familia, PARP-1 representa > 90 % de la ADP-ribosilación dentro de la célula. Debido a la homología estructural entre PARP-1 y PARP-2, más inhibidores de PARP-1 también inhiben PARP-2, La enzima PARP-1 es una proteína de 113 kDa con tres principales dominios estructurales, un dominio de unión a ADN con dos dedos de zinc, un dominio catalítico de 55 kDa, que utiliza nicotinamida adenina dinucleótido (NAD+) como un sustrato para construir polímeros de ADP-ribosa en las histonas y otras proteínas receptoras nucleares que incluyen el dominio de automodificación de PARP-1 por sí mismo. Es publicado y aceptado generalmente que la actividad catalítica de PARP-1 es estimulada por el daño del ADN causado por la peroxidación, irradiación y químicos que dañan el ADN, los agentes quimioterapéuticos. Con este fin, la enzima PARP-1 se une al ADN dañado y estimula la polimerización de ADP-ribosa que resulta en el desenrollo del ADN de histonas y que expone el ADN dañado para reparación. Por consiguiente, PARP-1 se asocia con el mantenimiento y reparación de ADN.
El cáncer de mama TNBC está asociado con mutaciones del gen BRCA1 y BRCA2. El papel central del gen BRCA es la recuperación de la ruptura de doble cadena (OSD) a través de la recombinación homóloga (HR). La inhibición de PARP-1 conducirá a un aumento en rupturas de cadena sencilla (SSB), la preponderancia de estas SSB conducirá eventualmente a DSB incrementado. El aumento de DSB en pacientes con cáncer de mutación del gen BRCA1/BRCA2 en la presencia de tipos de células deficientes HR conduce a aberraciones cromosómicas e inestabilidad del genoma que resulta en muerte celular.
Un inhibidor de PARP convencionalmente conocido Olaparib (WO2002036576, WO2003093261, US2004876080, US2005059663) se desarrolla para el tratamiento del cáncer, tal como, específicamente, el cáncer de estómago, cáncer de ovario, cáncer de mama.
Las siguientes cuatro patentes se publican con el grupo fenilo modificado de la estructura de ftalazinona (WO2007138351, WO2007138355, WO2009063244 y WO2009112832).
Desde 2011 las compañías farmacéuticas China y la India publicaron varias patentes con derivados modificados de Olaparib (WO2012019426A1, WO2012019427A1, WO2012019430A1, WO2012071684A1, WO2012072033A1 y WO2012014221).
Como agentes anticancerígenos los inhibidores de PARP, han ido progresado con respecto a la anterior literatura clínica publicada, tiene un nuevo mecanismo de acción para el tratamiento del cáncer. Los inhibidores de PARP se desarrollan como primer objeto para la medicina personalizada basada en mutación genética personal así que la atención mundial se centra. Los inhibidores de Pa Rp se ha reportado por exhibir en particular un efecto significativo sobre el cáncer causado por mutaciones genéticas en BRCA1/2, y la presente invención con nuevo mecanismo para
el tratamiento de pacientes con cáncer con variación genética en genes BRCA1/2 se espera que abra un nuevo capítulo.
Divulgación de la invención
Problema técnico
Un objeto de la presente invención es proporcionar un derivado de ftalazinona novedoso.
Además, el objeto de la presente invención es proporcionar un uso médico para el tratamiento útil de enfermedades mejoradas por la inhibición de PARP, o cánceres causados por defecto genérico del gen de fusión de BRCA1, BRCA2 y ERG.
Sin embargo, los objetos técnicos que se lograrán en la presente invención no se limitan a los mencionados anteriormente y los expertos en la materia pueden entender claramente otros objetos a partir de la siguiente descripción.
Solución al Problema
Para resolver el problema antes descrito, la presente invención proporciona un compuesto representado por la Fórmula I, racémico, enantiómero, diastereómero del mismo, o una sal farmacéuticamente aceptable del mismo.
[Fórmula I]

en donde
n es 1 o 2,
R es 
En donde, cuando R es

m es 0, 1 o 2,
L es metileno, carbonilo, NRc2CO, NRc3, CONRc4 o
CH2NRc5, en donde Rc2, Rc3, Rc4 y Rc5 cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6 o cicloalquilo C3-8,
RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o cicloalquilo C3-8,
RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etil éster, dimetilamina, metilo, metoximetilo o propargilo,
Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos,
en donde, cuando R es

p y q cada uno es independientemente de 1 o 2
W es CRd1Rd2 o NRd3, en donde Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno, fluoro o metilo) en donde, cuando R es

R1, R2 y R3 cada uno es independientemente hidrógeno, metilo o etilo.
La presente invención proporciona una composición farmacéutica para el tratamiento de cánceres que comprende el compuesto representado por la Fórmula I, racémico, enantiómero, diastereómero del mismo o sal farmacéuticamente aceptable del mismo.
Se divulga también un método de preparación del compuesto representado por la Fórmula I, racémico, enantiómero, diastereómero del mismo o sal farmacéuticamente aceptable del mismo, que no pertenece a la materia objeto de la invención.
Efectos ventajosos de la invención
Los compuestos de la presente invención son muy activos en la represión de PARP y según sus composiciones farmacéuticas se espera que sean útiles para aplicaciones terapéuticas que son mejoradas por la supresión de la actividad de PARP y cáncer con gen de fusión BRCA1, BRCA2 y ERG mutado en mono o combinación de tratamiento con radiación y con quimioterapia.
Mejor modo para llevar a cabo la invención
A continuación en el presente documento la presente invención se describirá en detalle.
La presente invención proporciona un compuesto representado por la Fórmula I, racémico, enantiómero, diastereómero del mismo, o una sal farmacéuticamente aceptable del mismo.
[Fórmula I]

en donde,
n es 1 o 2,
R es

En donde, cuando R es

m es 0, 1 o 2,
L es metileno, carbonilo, NRc2CO, NRc3, CONRc4 o
CH2NRc5, en donde Rc2, Rc3, Rc4 y Rc5 cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6, cicloalquilo C3-8,
RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o cicloalquilo C3-8,
RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etil éster, dimetilamina, metilo, metoximetilo o propargilo,
Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos,
en donde, cuando R es

p y q cada uno es independientemente de 1 o 2,
W es CRd1Rd2 o NRd3, Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno, fluoro o metilo),
en donde, cuando R es

R1, R2 y R3 cada uno es independientemente hidrógeno o alquilo C1-6 ,
En la presente invención, el compuesto de Fórmula I preferentemente se selecciona de i) a vi) descritos a continuación:
i) En el caso de que R sea, L es metileno, carbonilo, NRc2CO, NRc3,

CONRc4o Ch^NR05, en donde Rc2, Rc3, Rc4y Rc5cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6 o cicloalquilo C3-8), RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o ciclopropilo, RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etiléster, dimetilamina, metilo, metoximetilo o propargilo, Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos.
ii) En el caso de que R sea, p y q cada uno es independientemente de 1 a 2, W es CRd1Rd2

o NRd3, en donde Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno o metilo).
iii) En el caso de que R sea, R1, R2 y R3 cada uno es independientemente hidrógeno, metilo o

etilo.
iv) En el caso de que R sea, L es metileno o carbonilo, Z es

v) En el caso de que R sea, L es NRc2CO,

en donde Rc2 es hidrógeno, metilo, etilo o propilo)j, Z es

vii) En el caso de que R sea

Algunos ejemplos particularmente preferidos del compuesto de Fórmula I de acuerdo a la presente invención comprenden los siguientes:
(R) -4-(3-(3-aminopirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-1)
(S) -4-(3-(3-aminopirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-2)
4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-3)
(R) -4-(4-fluoro-3-(3-hidroxipirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-4)
(S) -4-(4-fluoro-3-(3-hidroxipirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-5)
4-(4-fluoro-3-(3-hidroxiazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-6)
4-(4-fluoro-3-(3-(hidroximetil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-7)
(R) -N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)ciclopropanocarboxamida; (I-8) N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)ciclopropanocarboxamida; (I-9)
(S) -N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)ciclopropanocarboxamida; (I-10) (R) -N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida; (I-11)
N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)-N-metilciclopropanocarboxamida; (I-12) (S) -N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida; (I-13)
3- (1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)-5,5-dimetilimidazolidin-2,4-diona; (I-14) (R)-3-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)imidazolidin-2,4-diona; (I-15)
(R)-1-etil-3-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)imidazolidin-2,4-diona; (I-16) 4- (4-fluoro-3-(3-(4-fluoropiperidin-1-carbonil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-17)
4-(3-(3-(3,3-difluoroazetidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-18)
4-(3-(3-(3,3-difluoropirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-19)
4-(3-(3-(3,3-difluoropirrolidin-1-carbonil)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-20)
(R)-N-(ciclopropilmetil)-1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-carboxamida; (I-21) 4-(4-fluoro-3-(3-(pirrolidin-1-carbonil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-22)
(R) -4-(3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-23) (S) -4-(3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-24)
4-(3-(3-(cidobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-25)
4-(3-(3-(ddopropNamino)azetidin-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-26)
4-(3-(3-(ddopentNamino)azetidin-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-27)
4-(3-(3-(cidohexilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-28)
(R)-4-(3-(3-(cidopropilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-29)
(R)-4-(3-(3-(ddobutNamino)pirroNdin-1-carbomi)-4-fluorobendi)ftaiazin-1(2H)-ona; (I-30)
(R)-4-(3-(3-(cidopentilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-31)
4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-32)
4-(3-(3-((ddopropNmetil)amino)azetidin-1-carbomi)-4-fluorobendi)ftaiazin-1(2H)-ona; (I-33)
4-(3-(3-(bis(cidopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-34)
4-(4-fiuoro-3-(3-(isobutNammo)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-35)
4-(4-fluoro-3-(3-((1-hidroxipropan-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-36)
4-(4-fluoro-3-(3-(neopentilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-37)
4-(3-(3-((2,2-dimetilddopentN)amino)azetidin-1-carbomi)-4-fluorobendi)ftaiazin-1(2H)-ona; (I-38)
2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)cidopent-1-enocarboxilato de etilo; (I-39)
4-(4-fluoro-3-(3-(pentan-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-40)
4-(4-fluoro-3-(3-((3-metilbutan-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-41)
4-(3-(3-((1-ddopropNetil)amino)azetidin-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-42)
4-(3-(3-(biddo[2,2,1]heptan-2-iiamino)azetidin-1-carbomi)-4-fluorobendi)ftaiazin-1(2H)-ona; (I-43)
4-(3-(3-(sec-butilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-44)
4-(3-(3-((dicidopropNmetil)amino)azetidin-1-carbonN)-4-fluorobencN)ftaiazin-1(2H)-ona; (I-45)
4-(4-fluoro-3-(3-((4-metilpentan-2-N)amino)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-46)
4-(4-fluoro-3-(3-((3-hidroxibutan-2-N)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-47)
4-(4-fiuoro-3-(3-(pentan-2-Namino)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-48)
4-(4-fluoro-3-(3-((l-(1-metilcidopropN)etil)amino)azetidin-1-carbonN)bencii)ftaiazin-1(2H)-ona; (I-49)
4-(4-fluoro-3-(3-((3,3,3-trifluoro-2-metilpropN)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-50)
4-(3-(3-(aNiamino)azetidin-1-carbonN)-4-fiuorobencN)ftaiazin-1(2H)-ona; (I-51)
4-(4-fiuoro-3-(3-(isopentNamino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-52)
4-(3-(3-(butNamino)azetidin-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-53)
4-(4-fluoro-3-(3-((3-metilbut-2-en-1-N)amino)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-54)
4-(3-(3-((cidopentNmetil)amino)azetidin-1-carbonN)-4-fluorobencN)ftaiazin-1(2H)-ona; (I-55)
4-(4-fluoro-3-(3-((4,4,4-tnfiuorobutN)ammo)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-56)
4-(4-fiuoro-3-(3-(pentNamino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-57)
4-(3-(3-((2-ddopropNetil)amino)azetidin-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-58)
4-(4-fiuoro-3-(3-(propNamino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-59)
4-(4-fluoro-3-(3-(metil(piridin-4-Nmetil)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-60)
(R)-4-(4-fluoro-3-(3-(metil(piridin-4-Nmetil)amino)pirroNdin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (l-61)
(s)-4-(4-fluoro-3-(3-(metil(piridin-4-Nmetil)amino)pirroNdin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-62)
(s)-4-(4-fluoro-3-(3-(metil(pindin-2-Nmetil)amino)pirroNdin-1-carbomi)bendi)ftaiazin-l(2H)-ona; (l-63)
(R) -4-(4-fluoro-3-(3-(metil(piridin-2-Nmetil)amino)pirroNdin-1-carbonN)bencN)ftaiazin-l(2H)-ona; (l-64)
4-(4-fluoro-3-(3-(metil(piridin-2-Nmetil)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-65)
4-(4-fluoro-3-(3-(metil(pindin-3-Nmetil)amino)azetidin-1-carbomi)bendi)ftaiazin-l(2H)-ona; (l-66)
(S) -4-(4-fluoro-3-(3-(metil(piridin-3-Nmetil)amino)pirroNdin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (l-67)
(R) -4-(4-fluoro-3-(3-(metil(pindin-3-Nmetil)amino)pirroNdin-1-carbomi)bendi)ftaiazin-l(2H)-ona; (l-68)
4-(3-(3-(cidopropN(metil)amino)azetidin-1-carbonN)-4-fluorobencN)ftaiazin-1(2H)-ona; (I-69)
4-(3-(3-(cidopropN(etil)amino)azetidin-1-carbonN)-4-fiuorobencN)ftaiazin-1(2H)-ona; (I-70)
4-(3-(3-(cidobutN(metil)amino)azetidin-1-carbonii)-4-fluorobencN)ftaiazin-1(2H)-ona; (I-71)
4-(3-(3-(cidopentN(prop-2-in-1-N)amino)azetidin-1-carbonN)-4-fiuorobencN)ftaiazin-1(2H)-ona; (I-72)
4-(3-(3-(3,3-difluoropirroNdin-1-N)azetidin-1-carbomi)-4-fluorobendi)ftaiazin-1(2H)-ona; (I-73)
4-(4-fiuoro-3-(3-(4-fiuoropipendin-1-N)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-74)
4-(3-([1,3'-biazetidina]-1-carbomi)-4-fiuorobendi)ftaiazin-1(2H)-ona; (I-75)
4-(4-fiuoro-3-(3-(pirroNdin-1-N)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-76)
4-(4-fiuoro-3-(3-(piperidin-1-N)azetidin-1-carbonN)bencN)ftaiazin-l(2H)-ona; (l-77)
4-(4-fiuoro-3-(3-(4-metilpiperazin-1-N)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-78)
4-(4-fiuoro-3-(3-(fenNamino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-79)
4-(4-fluoro-3-(3-((1-(trifiuorometil)cidopropN)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-80)
4-(4-fiuoro-3-(3-(prop-2-in-1-Namino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-81)
(S) -4-(4-fiuoro-3-(3-((1-metoxipropan-2-N)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-82)
(R)-4-(4-fluoro-3-(3-((l-metoxipropan-2-N)amino)azetidin-1-carbomi)bendi)ftaiazin-l(2H)-ona; (l-83)
4-(4-fiuoro-3-(3-((1-(hidroximetil)cidopropN)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-84)
4-(4-fiuoro-3-(3-((1-metilcidopropN)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-85)
(R)-4-(3-(3-((3,3-dimetilbutan-2-N)amino)azetidin-1-carboml)-4-fluorobendl)ftaiazin-1(2H)-ona; (I-86)
(s)-4-(3-(3-((3,3-dimetilbutan-2-N)amino)azetidin-1-carbonN)-4-fluorobencN)ftaiazin-1(2H)-ona; (I-87)
(R)-4-(4-fluoro-3-(3-((3-metilbutan-2-N)amino)azetidin-1-carbomi)bendi)ftaiazin-1(2H)-ona; (I-88)
(s)-4-(4-fiuoro-3-(3-((3-metilbutan-2-N)amino)azetidin-1-carbonN)bencN)ftaiazin-1(2H)-ona; (I-89)
4-(4-fluoro-3-(3-((1-(metoximetil)cidopropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-90)
4-(3-(3-(but-3-¡n-1-¡lam¡no)azet¡d¡n-1-carboml)-4-fluorobendl)ftalaz¡n-1(2H)-ona; (I-91)
4-(4-fluoro-3-(3-((2-met¡lal¡l)am¡no)azet¡d¡n-1-carboml)bendl)ftalaz¡n-1(2H)-ona; (I-92)
4-(4-fluoro-3-(3-((3-hidroxi-2,2-dimetilpropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-93)
1-(((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)metil)cidopropanocarbonitrilo; (I-94)
4-(4-fluoro-3-(3-((2,2,2-trifluoroetil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-95)
(R) -4-(4-fluoro-3-(3-(pirrolidin-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-96)
(s)-4-(4-fluoro-3-(3-(pirrolidin-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-97)
1-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)cidopentanocarbonitrilo; (I-98) 1- ((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)cidobutanocarbonitrilo; (I-99) 2- ((l-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)propanonitrilo; (I-100)
2-((l-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)butanonitrilo; (I-101)
2-((l-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)-3-metilbutanonitrilo; (I-102) 2-ddoprop¡l-2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)aceton¡trilo; (I-103) 4-(4-fluoro-3-(3-((1-(trifluorometil)cidobutil)amino)azetidin-1-carbonil)bencil)ftalazin-l(2H)-ona; (I-104)
(S) -4-(4-fluoro-3-(3-(metil(pirimidin-2-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-105)
(S)-4-(4-fluoro-3-(3-(etil(pirimidin-2-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-106)
4-(4-fluoro-3-(3-(metil(pirimidin-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-107)
4-(4-fluoro-3-(3-(etil(pirimidin-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-108)
4-(4-fluoro-3-(3-(pirimidin-2-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-109)
(S)-4-(4-fluoro-3-(3-(pirimidin-2-ilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-110)
(s)-4-(3-(3-((6-doropiridazin-3-il)(metil)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-111) (s)-4-(3-(3-((6-doropiridazin-3-il)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-112)
(s)-4-(4-fluoro-3-(3-(metil(piridazin-3-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-1l3)
4-(3-(3-((6-doropiridazin-3-il)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-114)
4-(3-(3-((6-doropiridazin-3-il)(metil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-115)
4-(3-(3-(cidobutil(pirimidin-2-il)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-1l6)
4-(4-fluoro-3-(3-(piridazin-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; ( l- l l7 )
(R)-4-(3-(3-((6-dorop¡ndaz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carboml)-4-fluorobendl)ftalaz¡n-1(2H)-ona; (I-118)
(R)-4-(3-(3-((6-doropiridazin-3-il)(metil)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-119) (R)-4-(4-fluoro-3-(3-(pirimidin-2-ilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-120)
(R)-4-(3-(3-(et¡l(p¡nm¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carboml)-4-fiuorobendl)ftalaz¡n-1(2H)-ona; (I-121)
(R)-4-(4-fluoro-3-(3-(piridazin-3-ilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-122)
(R)-4-(4-fluoro-3-(3-(metil(piridazin-3-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-123)
(R)-4-(4-fluoro-3-(3-(tiazol-2-ilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-124)
(R)-4-(3-(3-((5-etinilpirimidin-2-il)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-125)
(R)-2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)amino)pirimidin-5-carbonitrilo; (I-126)
(R)-2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)amino)nicotinonitrilo; (I-127) (R)-4-(4-fluoro-3-(3-(piridin-2-ilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-128)
(R)-2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)amino)-N,N-dimetilnicotinamida; (I-129)
(R)-4-(3-(3-((5-bromopirimidin-2-il)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-130)
(R)-4-(4-fluoro-3-(3-((5-(trifluorometil)piridin-2-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-131) (R)-4-(4-fluoro-3-(3-((4-(trifluorometil)piridin-2-il)amino)pirrolidin-1-carbonil)bencil)ftalazin-l(2H)-ona; (l-132) 4-(3-(3-(bencilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-133)
4-(4-fluoro-3-(3-((3,3,3-trifluoropropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-134)
(R)-4-(3-(3-(azetidin-1-il)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-l35)
(R)-4-(3-([1,3'-bipirrolidina]-1'-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-136)
(R)-4-(4-fluoro-3-(3-(piperidin-1-il)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-137)
(R)-4-(4-fluoro-3-(3-(p-tolilamino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-138)
(R)-4-(4-fluoro-3-(3-(femlammo)p¡rrol¡d¡n-1-carboml)bendl)ftalaz¡n-1(2H)-ona; (I-139)
4-(4-fluoro-3-(3-(pirrolidin-1-ilmetil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-l40)
4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-l(2H)-ona; (l-141)
4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-142)
4-(3-(3-((c¡doprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-143)
4-(4-fluoro-3-(3-((¡soprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-144)
4-(3-(3-(((c¡doprop¡lmet¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-145)
4-(4-fluoro-3-(3-((¡sobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-146)
4-(3-(3-((terc-but¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-147)
4-(3-(3-((c¡dobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-l(2H)-ona; (l-148)
4-(4-fluoro-3-(3-((prop-2-¡n-1-¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-149)
4-(4-fluoro-3-(3-((fen¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-150)
1-((((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l)am¡no)met¡l)ddopropanocarbomtrilo; (I-151)
1-(((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)metil)amino)cidopropanocarbonitrilo; (I-152)
4-(3-(3-((cidopropil(prop-2-in-1-il)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-153) 4-(3-(3-((ciclopropil(metil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-154)
4-(3-(3-((ciclopropil(etil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-155)
4-(3-(3-(cidobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-156)
4-(3-(3-(cidopropilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-157)
4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-158)
4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-159)
4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-160)
4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-161)
4-(4-fluoro-3-(3-((1-hidroxipropan-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-162) 4-(3-(3-(butilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-163)
4-(3-([1,3'-biazetidina]-1'-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-164)
(R)-4-(4-fluoro-3-(3-((1-metoxipropan-2-il)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-165) 1-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)ciclobutanocarbonitrilo clorhidrato; (I-166)
(R)-4-(3-(3-(azetidin-1-il)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-167)
4-(4-fluoro-3-(3-(pirrolidin-1-ilmetil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-168)
4-(4-fluoro-3-(3-(piperidin-1-ilmetil)azetidin-1-carbonil)bencil)ftalazin-l(2H)-ona clorhidrato; (l-169)
4-(3-(3-(azetidin-1-ilmetil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-170)
4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-171)
4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-172)
4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-173) 4-(4-fluoro-3-(3-((isobutilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-174)
4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-175) y
4-(4-fluoro-3-(3-((prop-2-in-1-ilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-176).
En la presente invención, "alquilamina C1-4" es una hidrocarbonil amina saturada con cadenas lineales o ramificadas de 1~4 átomos de carbono. Alquilaminas ejemplares incluyen, pero sin limitación, metilamina, etilamina, propilamina, butilamina, 1 metiletilamina, dietilamina o dimetilamina.
En la presente invención, "alquilo C1-6" es una hidrocarbonil amina saturada con cadenas lineales o ramificadas de 1 6 átomos de carbono. El alquilo ejemplar incluye, pero sin limitación, metilo, etilo, propilo, butilo, pentilo, hexilo, 1-metiletilo, 1 -metilpropilo, 2-metilpropilo, 1,1 -dimetiletilo, 1 -metilbutilo, 1,1-dimetilpropilo, 1 -metilpentilo o 1,1-dimetilbutilo.
En la presente invención, "alcoxi C1-6" es un grupo OR con R como se ha definido anteriormente. El alcoxi ejemplar con 1-6 átomos de carbono incluye, pero sin limitación, metoxi, etoxi, propoxi, butoxi, 1-metiletoxi, 1,1 -dimetiletoxi, 1-metilpropoxi, 2-metilpropoxi o ciclopropilmetoxi.
En la presente invención, "haloalquilo C1-6" pretende ser un radical alquilo C1-6 que tiene uno o más átomos de hidrógeno reemplazados por un átomo de halógeno definido anteriormente. El haloalquilo ejemplar incluye, pero sin limitación, difluorometilo o trifluorometilo.
En la presente invención, "halo" pretende ser un átomo de bromo, flúor o cloro.
En la presente invención, “alquenilo C2-6” pretende ser una cadena de hidrocarbonilo lineal o ramificada de 2-6 átomos de carbono y al menos un doble enlace carbono-carbono. Los alquenilos tienen una configuración de doble enlace "cis" o "trans" y "E" o "Z". Alquenilo ejemplar incluye, pero sin limitación, crotilo (-CH2CH=CHCH3),vinilo (-CH=CH2) o alilo (-CH2CH=CH2).
En la presente invención, “alquinilo C2-6” pretende ser un radical hidrocarbonilo lineal o ramificado con 2-6 átomos de carbono y al menos un triple enlace carbono-carbono. El alquinilo ejemplar incluye, pero sin limitación, etinilo (-CECH) o propargilo (-CH2CECH).
En la presente invención, "cicloalquilo C3-8" pretende ser un anillo hidrocarbonilo saturado con 3-8 átomos de carbono. El cicloalquilo ejemplar incluye, pero sin limitación, ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo.
En la presente invención, "cicloalquenilo C3-8" pretende ser un anillo hidrocarbonilo insaturado con 3-8 átomos de carbono y al menos un doble enlace carbono-carbono. Los alquenilos tienen una configuración de doble enlace "cis" o "trans" y "E" o "Z". El cicloalquenilo ejemplar incluye, pero sin limitación, ciclopropenilo, ciclobutenilo, ciclopentenilo, ciclohexenilo, 1,3-ciclohexadieno, 1,4-ciclohexadieno, cicloheptenilo, ciclooctenilo o 1,5-ciclooctadieno.
En la presente invención, "ciclo aromático C6-10" pretender ser un radical hidrocarbonilo aromático con 6-10 átomos de carbono. El anillo aromático ejemplar incluye, pero sin limitación, fenilo o naftilo.
En la presente invención, "heterociclo" pretender ser un anillo mono-tricíclico de hidrocarbonilo saturado o parcialmente insaturado con al menos un átomo de nitrógeno. Los mono heterociclos ejemplares con 5-6 átomos incluyen, pero sin limitación, pirrolidinilo, piperidinilo, pirollilo, pirazolilo, imidazolilo, oxazolilo, isoxazolilo, triazolilo, tiazolilo, piridilo, piridazinilo, pirimidinilo, pirazinilo y triazinilo. También, el anillo aromático bicíclico ejemplar incluye, pero sin limitación, benzotiazolilo, benzoxazolilo, benzoxazinona, benzoxadiazolilo, 1,3-benzodioxolilo, benzofurilo, benzopirazinilo, indolilo, indazolilo, bencimidazolilo, benzopiranilo, pirolopiridanilo, furopiridinilo o imidazotiazolilo.
El término “farmacéuticamente aceptable”, como se usa aquí, cuando se hace referencia a un componente de una composición farmacéutica significa que el componente, cuando se administra a un animal, no tiene excesivos efectos adversos tal como excesiva toxicidad, irritación o reacción alérgica en consonancia con una relación beneficio/riesgo razonable.
El término "tratamiento" como se usa aquí cubre cualquier tratamiento de una enfermedad en un mamífero, particularmente un humano, e incluye la inhibición de la enfermedad, es decir, deteniendo su desarrollo; o aliviando la enfermedad, es decir, causando la regresión de la enfermedad y/o sus síntomas o condiciones y detener la progresión de la enfermedad.
El término "cantidad terapéuticamente efectiva" significa una cantidad de un compuesto de la presente invención que mejora, atenúa o elimina una enfermedad o condición particular o previene o retrasa la aparición de una enfermedad o condición particular. En el caso de cáncer, la cantidad terapéuticamente efectiva del fármaco puede reducir el número de células cancerígenas; reducir el tamaño del tumor; inhibir (es decir, detener en cierta medida y preferentemente parar) la infiltración de la célula cancerígena en órganos periféricos; inhibir (es decir, detener en cierta medida y preferentemente parar) la metástasis de tumor; inhibir, en cierta medida, el crecimiento del tumor; y/o aliviar en cierta medida una o más de los síntomas asociados con el cáncer. En la medida que el fármaco puede prevenir el crecimiento y/o eliminar las células cancerígenas existentes, puede ser citostático o citotóxico.
Los compuestos de la invención pueden contener centros asimétricos o quirales, y por lo tanto existen en diferentes formas estereoisoméricas. Se pretende que todas las formas estereoisoméricas de los compuestos de la invención, incluyendo pero no limitadas a, diastereómeros, enantiómeros y atropisómeros, así como sus mezclas tal como mezclas racémicas, forman parte de la presente invención. Un estereoisómero específico también puede ser conocido como un enantiómero, y una mezcla de dichos isómeros se denomina frecuentemente una mezcla enantiomérica. Una mezcla 50:50 de enantiómeros es referida como una mezcla racémica o un racemato.
"Diastereómero" se refiere a un estereoisómero con dos o más centros de quiralidad y cuyas moléculas no son imágenes de espejo uno del otro. Los diastereómeros tienen diferentes propiedades físicas, por ejemplo, puntos de fusión, puntos de ebullición, propiedades espectrales y reactividades. Las mezclas de diastereómeros pueden separarse bajo procedimientos analíticos de alta resolución tales como electroforesis y cromatografía.
"Enantiómeros" se refiere a dos estereoisómeros de un compuesto que con imágenes en el espejo no superponibles uno del otro.
La expresión “sal farmacéuticamente aceptable” como se usa aquí, se refiere a sales orgánicas o inorgánicas farmacéuticamente aceptables de un compuesto de la invención. Las sales ejemplares incluyen, pero sin limitación, sales de sulfato, citrato, acetato, oxalato, cloruro, bromuro, yoduro, nitrato, bisulfato, fosfato, fosfato ácido, isonicotinato, lactato, salicilato, citrato, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, metanosulfonato "mesilato", etanosulfonato, bencenosulfonato, p-toluenosulfonato, y pamoato (es decir, 1,1'-metileno-bis-(2-hidroxi-3-naftoato)). Una sal farmacéuticamente aceptable puede implicar la inclusión de otra molécula tal como un ion de acetato, un ion de succinato u otro contraión. El contraión puede ser cualquier radical orgánico o inorgánico que estabiliza la carga en el compuesto de origen. Además, una sal farmacéuticamente aceptable puede tener más de un átomo cargado en su estructura. Los casos donde múltiples átomos cargados son parte de la sal farmacéuticamente aceptable pueden tener múltiples contraiones. Por lo tanto, una sal farmacéuticamente aceptable puede tener uno o más átomos cargados y/o uno o más contraiones.
Si el compuesto de la invención es una base, la sal farmacéuticamente aceptable deseada puede prepararse por cualquier método adecuado disponible en la técnica, por ejemplo, el tratamiento de la base libre con un ácido inorgánico, tal como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido metanosulfónico, ácido fosfórico y lo similar, o con un ácido orgánico, tal como ácido acético, ácido maleico, ácido succínico, ácido mandélico, ácido fumárico, ácido malónico, ácido pirúvico, ácido oxálico, ácido glicólico, ácido salicílico, un ácido piranosidilo, tal como ácido glucurónico o ácido galacturónico, un ácido alfa hidroxi, tal como el ácido cítrico o ácido tartárico, un aminoácido, tal como ácido aspártico o ácido glutámico, un ácido aromático, tal como ácido benzoico o ácido cinámico, un ácido sulfónico, tal como el ácido p-toluenosulfónico o ácido etanosulfónico o similares.
Si el compuesto de la invención es un ácido, la sal farmacéuticamente aceptable deseada puede prepararse por cualquier método adecuado, por ejemplo, el tratamiento del ácido libre con una base inorgánica u orgánica, tal como
una amina (primaria, secundaria o terciaria), un hidróxido de metal alcalino o hidróxido del metal alcalinotérreo o similares. Los ejemplos ilustrativos de sales adecuadas incluyen, pero sin limitación, sales orgánicas derivadas de aminoácidos tales como glicina y arginina, amoníaco, aminas primarias, secundarias y terciarias y aminas cíclicas, tal como piperidina, morfolina y piperazina, y sales inorgánicas derivadas de sodio, calcio, potasio, magnesio, manganeso, hierro, cobre, zinc, aluminio y litio.
También se divulga un método de preparación del compuesto representado por la Fórmula I o una sal farmacéuticamente aprobada del mismo, que no pertenece a la materia objeto de la invención.
Se muestra a continuación un método de preparación.
[Esquema 
El compuesto de Fórmula I de la presente invención, como se muestra en el esquema 1, se puede preparar mediante una serie de etapas a partir del compuesto de Fórmula 2.
n y R, ilustrados en el esquema 1, se definen de la siguiente manera:
En donde,
n es 1 o 2;
R es

En donde, cuando R es

m es 0, 1 o 2.
L es metileno, carbonilo, NRc2CO, NRc3, CONRc4 o CH2NRc5, en donde Rc2, Rc3, Rc4 y Rc5 cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6 o cicloalquilo C3-8,
RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o cicloalquilo C3-8,
RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etil éster, dimetilamina, metilo, metoximetilo o propargilo,
Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos,
en donde, cuando R es

p y q cada uno es independientemente de 1 o 2,
W es CRd1Rd2 o NRd3, en donde Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno, fluoro o metilo, en donde, cuando R es

R1, R2 y R3 cada uno es independientemente hidrógeno, metilo o etilo.
El método de preparación de la Fórmula I comprende:
La preparación de un compuesto de Fórmula 4 a partir de un compuesto de Fórmula 2 y 3 que reacciona con la reacción de acoplamiento de amida (etapa 1)
La preparación de un compuesto de Fórmula 6 a partir de un compuesto de Fórmula 4 y 5 que reacciona con la reacción de olefinación (etapa 2)
La preparación de un compuesto de Fórmula I a partir de un compuesto de Fórmula 6 y la hidrazina monohidratada que ha reaccionado con la reacción de condensación (etapa 3)
Cada etapa del método de preparación anterior se describe con más detalle a continuación.
i) El compuesto de Fórmula 4 puede prepararse a partir de la Fórmula 2 por acoplamiento de amida en la etapa 1 anterior. Se lleva a cabo una reacción de acoplamiento de amida con clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida (EDCI) y 4-(dimetilamino)piridina (DMAP) o hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HBTU) y N,N-diisopropil etilamina (DIPEA). Los ejemplos de disolventes útiles en la reacción incluyen cloroformo o dimetilformamida. La reacción se calienta a 20~35 °C por durante 1~30 horas, con el fin de obtener el compuesto de Fórmula 4.
El ejemplo de preparación del compuesto de Fórmula 4 a partir del compuesto de Fórmula 2 y 3 por acoplamiento de amida en la etapa 1 anterior se ilustra a continuación.

ii) El compuesto de Fórmula 6 puede prepararse a partir de la Fórmula 4 por olefinación en la etapa 2 anterior. Una reacción olefinación se lleva a cabo con la Fórmula 5 en condición básica. Los ejemplos de disolvente útil en la reacción incluye THF. La reacción se enfría a 0 °C durante 1~5 horas, con el fin de obtener el compuesto de Fórmula 6.
El ejemplo de preparación del compuesto de Fórmula 6 a partir del compuesto de Fórmula 4 y 5 por olefinación en la etapa 2 anterior se ilustra a continuación.

iii) El compuesto de Fórmula I se puede preparar a partir de la Fórmula 6 por condensación en la etapa 3 anterior. Una reacción de condensación se lleva a cabo con monohidrato de hidracina. El ejemplo de disolvente útil en la reacción incluye agua. La reacción se calienta a 30~70 °C durante 20 horas, con el fin de obtener el compuesto de Fórmula I.
El ejemplo de preparación del compuesto de Fórmula I a partir del compuesto de Fórmula 6 por condensación en la etapa 3 anterior se ilustra a continuación.

En otro aspecto, la presente invención proporciona otro método de preparación del compuesto representado por la Fórmula I o una sal farmacéuticamente aprobada del mismo.
Además, se muestra a continuación otro método de preparación.

El compuesto de Fórmula I de la presente invención, como se muestra en el esquema 2, se puede preparar mediante una serie de etapas a partir del compuesto de Fórmula I.
n, R4 y R, ilustrados en El esquema 2, se definen a continuación: En donde
n es 1 o 2 ;
R4 y R5 cada uno es independientemente terc-butildimetilsiloxilo (

) terc-butildimetilsiloximetilo (

) bencilcarbamato, amina, CH2OH o hidroxilo;
R es

en donde, cuando R es

m es 0, 1 o 2,
L es metileno, carbonilo, NRc2CO, NRc3, CONRc4 o CH2NRc5, en donde Rc2, Rc3, Rc4 y Rc5 cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6 o cicloalquilo C3-8,
RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o cicloalquilo C3-8,
RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etil éster, dimetilamina, metilo, metoximetilo o propargilo,
Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos,
en donde, cuando R es

p y q cada uno es independientemente de 1 o 2,
W es CRd1Rd2 o NRd3, en donde Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno, fluoro o metilo, en donde, cuando R es
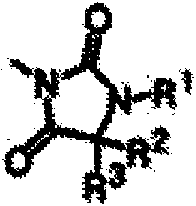
R1, R2 y R3 cada uno es independientemente hidrógeno, metilo o etilo.
Otro método de preparación de la Fórmula I comprende
La preparación de un compuesto de Fórmula 8 a partir a partir de un compuesto de Fórmula 7 y 3 que ha reaccionado con la reacción de acoplamiento de amida (etapa 1)
La preparación de un compuesto de Fórmula 9 a partir de un compuesto de Fórmula 8 y 5 que ha reaccionado con la reacción de olefinación (etapa 2)
La preparación de un compuesto de Fórmula 10 a partir a partir de un compuesto de Fórmula 9 y monohidrato de hidrazina que ha reaccionado con la reacción de condensación (etapa 3)
La preparación de un compuesto de Fórmula 11 a partir de un compuesto de Fórmula 10 que ha reaccionado con carboxibencilo o terc-butildimetilsiloxilo (

) de una reacción de desprotección (etapa 4)
La preparación de un compuesto de Fórmula I a partir a partir de un compuesto de Fórmula 11 que ha reaccionado con el acoplamiento de amida, la sustitución y la reacción de aminación reductora (etapa 5)
Cada etapa del método de preparación anterior se describe con más detalle a continuación.
i) El compuesto de Fórmula 8 puede prepararse a partir de la Fórmula 7 por acoplamiento de amida en la etapa 1 anterior. Se lleva a cabo una reacción de acoplamiento de amida con clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida (EDCI) y 4-(dimetilamino)piridina (DMAP) o hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HBTU) y N,N-diisopropil etilamina (DIPEA). Los ejemplos de disolventes útiles en la reacción incluyen cloroformo o dimetilformamida. La reacción se calienta a 20~35 °C durante 1~30 horas, con el fin de obtener el compuesto de Fórmula 8.
El ejemplo de preparación del compuesto de Fórmula 8 a partir del compuesto de Fórmula 7 y 3 por acoplamiento de amida en la etapa 1 anterior se ilustra a continuación.

ii) El compuesto de Fórmula 9 puede prepararse a partir de la Fórmula 8 por olefinación en la etapa 2 anterior. Una reacción olefinación se lleva a cabo con la Fórmula 5 en condición básica. Los ejemplos de disolvente útil en la reacción incluye THF. La reacción se enfría a 0 °C durante 1~5 horas, con el fin de obtener el compuesto de Fórmula 9.
El ejemplo de preparación del compuesto de Fórmula 9 a partir del compuesto de Fórmula 8 y 5 por olefinación en la etapa 2 anterior se ilustra a continuación.

iii) El compuesto de Fórmula 10 se puede preparar a partir de la Fórmula 9 por condensación en la etapa 3 anterior. Una reacción de condensación se lleva a cabo con monohidrato de hidrazina. El ejemplo de disolvente útil en la reacción incluye agua. La reacción se calienta a 30~70 °C durante 20 horas, con el fin de obtener el compuesto de la Fórmula 10.
El ejemplo de preparación del compuesto de Fórmula 10 a partir del compuesto de Fórmula 9 por condensación en la etapa 3 anterior se ilustra a continuación.

iv) El compuesto de Fórmula 11 se puede preparar de la Fórmula 10 por desprotección de carboxibencilo o tercbutiloxicarbonilo (

) en la etapa 4 anterior. Una reacción de desprotección de carboxibencilo o terc-butildimetilsiloxilo (

) se realiza con Pd/C en atmósfera de N2 o solución 1 M de fluoruro de tetra-n-butilamonio en THF (TBAF), para obtener así el compuesto de Fórmula 11.
El ejemplo de preparación del compuesto de Fórmula 11 a partir del compuesto de Fórmula 10 por condensación en la etapa 4 anterior se ilustra a continuación.
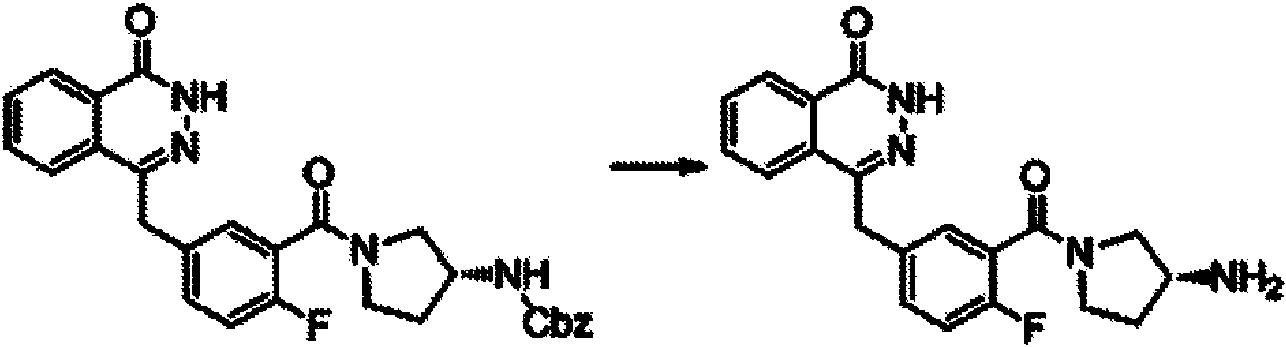
v) El compuesto de Fórmula I se puede preparar a partir de la Fórmula 11 por acoplamiento de amida, sustitución y la reacción de aminación reductora.
Una etapa 5 del esquema 2 se describe en más detalle a continuación.
En la etapa 5 del método de preparación, el compuesto de Fórmula I se puede preparar con el acoplamiento de amida, sustitución y aminación reductora.
En la reacción de acoplamiento de amida anterior, la reacción se lleva a cabo con clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida (EDCI) y 4-(dimetilamino)piridina (DMAP) o hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HBTU) y N,N-diisopropil etilamina (DIPEA). Los ejemplos de disolventes útiles en la reacción incluyen cloroformo o dimetilformamida para obtener el compuesto de Fórmula I. Un ejemplo se ilustra a continuación.

Además, en la etapa 5, el compuesto de Fórmula I se puede preparar con sustitución.
En la reacción de sustitución anterior, el compuesto de mesilato está preparado del compuesto de alcohol por la reacción con cloruro de metanosulfonilo en diclorometano. Y luego, el grupo funcional deseado se puede introducir por la reacción del compuesto mesilato con (En el esquema anterior, p, q, m, W, Z, RX y RY, son

los mismos a lo definido en la Fórmula I). Un ejemplo de preparación del compuesto de Fórmula I forma el compuesto de Fórmula 8 por sustitución en el presente invención se ilustra a continuación (la sustitución Rc6 se introduce con K2CO3 y Rc6-I).

Además, en la reacción de aminación reductora anterior, el compuesto de Fórmula I puede prepararse a partir de

(En el esquema anterior, m, Z, RX y RY, son los mismos a los definidos en la Fórmula I). La reacción se lleva a cabo
con el triacetoxiborohidruro de sodio y ácido acético por toda la noche. Los ejemplos de disolvente útil en la reacción incluyen cloroformo o diclorometano. Un ejemplo de preparación del compuesto de Fórmula I se ilustra a continuación (la sustitución de Rc4 se introduce con K2CO3 y Rc4- en dimetilformamida).

Además, la presente invención proporciona una composición farmacéutica para el tratamiento de cánceres que comprende el compuesto de Fórmula I, su racémico, enantiómero, diastereoisómero, o su sal farmacéuticamente aceptable. Los cánceres pueden deberse a la actividad de PARP, o defecto genérico del gen de fusión de BRCA1, BRCA2 y ERG. Los cánceres ejemplares incluyen, pero sin limitación cáncer de mama, cáncer de ovario, cáncer de páncreas, cáncer gástrico, cáncer de pulmón, cáncer colorrectal, tumor cerebral, cáncer de próstata y un sarcoma de Ewing. Como se utiliza aquí, el término "defecto genérico" pretende ser una mutación del gen, deficiencia del gen o defecto de la expresión génica, pero sin limitación esto.
Se divulga también un método para tratar cánceres en un sujeto que lo necesite, que no pertenece a la materia objeto de la invención, que comprende administrar una cantidad efectiva de la composición farmacéutica al sujeto. La dosificación de la composición farmacéutica de la presente invención puede variar dependiendo del peso del paciente, edad, género, condición física, dieta, tiempo y modo de administración, tasas de excreción y la severidad de la enfermedad. Los mamíferos (incluyendo humanos) son deseables para el individuo sin límite.
Los compuestos de la invención destinados para el uso farmacéutico pueden ser administrados como un sólido o líquido, tal como comprimido, cápsula, solución o suspensión. Las composiciones farmacéuticas adecuadas para el suministro de compuestos de la presente invención y los métodos para su preparación serán aparentes para aquellos con experiencia en la técnica. Dichas composiciones y métodos para su preparación pueden encontrarse, por ejemplo, en Remingtons Pharmaceutical Sciences, 19a edición (Mack Publishing Company, 1995).
La presente invención proporciona compuestos, específicamente, activos en la inhibición de la actividad de PARP. Los compuestos de la invención pueden usarse en el tratamiento del cáncer mediante la inhibición de PARP. Los ejemplares incluyen cáncer de pulmón, cáncer gastrointestinal, cáncer de próstata, cáncer de útero o especialmente cáncer de mama, o cáncer de ovario.
Los compuestos de la invención pueden combinarse en una formulación de combinación farmacéutica, o régimen de dosificación como terapia de combinación, con un segundo compuesto que tiene propiedades anti-cancerígenas o por lo menos dos clases de ingredientes activos farmacéuticos.
Los ejemplos de agentes incluyen: oxaliplatino (ELOXATIN®, Sanofi), bortezomib (VELCADE®, Millennium Pharm.), sutent (SUNITINIB®, SU11248, Pfizer), letrozol (FEMARA®, Novartis), mesilato de imatinib (GLEEVEC®, Novartis), XL-518 (Mek inhibitor, Exelixis, WO 2007/044515), ARRY- 8 8 6 (inhibidor Mek, AZD6244, Array BioPharma, Astra Zeneca), SF 1126 (inhibidor PI3K, Semafore Pharmaceuticals), BEZ 235 (inhibidor PI3K, Novartis), XL-147 (inhibidor PI3K, Exelixis), PTK787/ZK 222584 (Novartis), f ulvestrant (FASLODEX®, AstraZeneca), leucovorin (ácido folinico), rapamicina (sirolimus, RAPAMUNE®, Wyeth), lapatinib (t Yk ERB ®, GSK572016, Glaxo Smith Kline), lonafarnib (SARASAR®, SCH 66336, Schering Plough), sorafenib (NEXAVAR®, BAY43-9006, Bayer Labs), gefitinib (IRESSA®, AstraZeneca), irinotecan (CAMPTOSAR®, CPT-11, Pfizer), tipifarnib (ZARNESt Ra ®, Johnson & Johnson), ABRAXANE® (libre de cremofor), formulaciones de nanoparticula diseñada de albúmina de paclitaxel (American Pharmaceutical Partners, Schaumberg, II), vandetanib (rINN, ZD6474, ZACTIMA® AstraZeneca), cloranmbucil, AG1478, AG1571 (SU 5271; SUGEN), temsirolimus (TORiSe L®, Wyeth), pazopanib (GlaxoSmithKline), canfosfamida (TELCYTA ®, Telik), tiotepa y ciclofosfamida (CYTOXAN®, NEOSAR ®); sulfonatos de alquilo tal como busulfan, improsulfan y piposulfan; aziridinas tal como benzodopa, carboquone, meturedopa y uredopa; etileniminas y metilamelaminas incluyendo altretamina, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida y trimetilomelamina; acetogeninas (especialmente b ullatacin y b ullatacinona); una camptotecina (incluyendo el topotecán análogo sintético); briostatina; callistatina; CC-1065 (que incluye sus análogos sintéticos adozelesina, carzelesina y bizelesina); criptoficinas (particularmente criptoficina 1 y criptoficina 8 ); dolastatina; duocarmicina (incluyendo los análogos sintéticos, KN-2189 y CB1-TM1); eleuterobina; pancratistatina; una sarcodictiina; espongistatina; mostazas de nitrógeno tal como clorambucilo, clornafazina, clorofosfamida, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalán, novembicina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo; nitrosoureas tal como carmustina, clorozotocina, fotemustina, lomustina, nimustina y ranimnustina; antibióticos tal como antibióticos de enediina (por ejemplo, caliqueamicina, caliqueamicina gamma II, caliqueamicina omega II (Angew Chem. Intl. Ed. Engl. (1994) 33:183-186); dinemicina, dinemicina A; bifosfonatos, tal como clodronato; una esperamicina; así como cromóforo de neocarzinostatina y cromóforos antibióticos de endiina cromoproteina relacionados), aclacinomisinas, actinomicina, autramicina, azaserina, bleomicinas, cactinomicina,
carabicina, caminomicina, carzinofilina, cromomicinis, dactinomicina, daunorrubicina, detorubicina, 6-diazo-5-oxo-L-norleucina, morfolino-doxorrubicina, cianomorfolino-doxorrubicina, 2-pirrolino-doxorrubicina y deoxidoxorubicina), epirubicina, esorubicina, idarrubicina, nemorubicina, marcellomicina, mitomicinas tales como mitomicina C, ácido micofenólico, nogalamicina, olivomicinas, peplomicina, porfiromicina, puromicina, quelamicina, rodorubicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatin, zorubicina; anti-metabolitos tal como metotrexato y 5-fluorouracilo (5-FU); análogos del ácido fólico tal como denopterina, metotrexato, pteropterina, trimetrexato; análogos de purina tal como fludarabina, 6-mercaptopurina, tiamiprina, tioguanina; análogos de pirimidina tal como ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, dideoxiuridina, doxifluridina, enocitabina, floxiridina; andrógenos tal como calusterona, dromostanolona propionato, epitiostanol, mepitiostano, testolactona; anti-adrenales tal como aminoglutetimida, mitotano, trilostano; proveedor de ácido fólico tal como ácido frolínico; aceglatona; aldofosfamida glucósido; ácido aminolev ulínico; eniluracilo; amsacrina; bestrabucilo; bisantreno; edatraxato; defofamina; demecolcina; diaziquona; elformitina; acetato de eliptinio; una epotilona; etoglucida; nitrato de galio; hidroxiurea; lentinano; lonidainina; maitansinoides tal como maitansina y ansamitocinas; mitoguazona; mitoxantrona; mopidanmol; nitraerina; pentostatina; fenamet; pirarubicina; losoxantrona; ácido podofílinico; 2-etilhidrazida; procarbazina; complejo de polisacárido PSK (JHS Natural Products, Eugene, Oreg); razoxano; rizoxina; sizofurano; espirogermanio; ácido tenuazonico; triaziquona; 2,2',2"-triclorotrietilamina; tricotecenos (especialmente toxina T-2, verracurina A, roridina A y anguidina); uretan; vindesina; dacarbazina; mannomustina; mitobronitol; mitolactol; pipobroman; gacitosina; arabinosida ("Ara-C"); ciclofosfamida; tiotepa; 6-tioguanina; mercaptopurina; metotrexato; análogos de platino tal como cisplatino y carboplatino; vinblastina; etopósido (VP-16); ifosfamida; mitoxantrona; vincristina; vinorelbina (NAVELBINE®); Novantrona; tenipósido; edatrexato; daunomicina; aminopterina; capecitabina (XELODA®, Roche); ibandronato; CPT-11; inhibidor de topoisomerasa RFS 2000; difluorometilornitina (DMFo ); retinoides tal como el ácido retinoico; y sales farmacéuticamente aceptables, ácidos y derivados de cualquiera de los anteriores.
Por ejemplo, los compuestos de la invención, especialmente de Fórmula I o una sal farmacéuticamente aceptable pueden administrarse simultáneamente, gradualmente, o de forma individual con al menos uno de los agentes terapéuticos.
Administración oral
Los compuestos de la invención pueden ser administrados oralmente. La administración oral puede implicar deglución, para que el compuesto entre en el tracto gastrointestinal y/o administración bucal, lingual o sublingual, por la que el compuesto entra en el torrente sanguíneo directamente desde la boca.
Las formulaciones adecuadas para la administración oral incluyen tapones sólidos, microparticulados sólidos, semisólidos y líquidos (incluyendo múltiples fases o sistemas dispersados) tal como comprimidos; cápsulas blandas o duras que contiene multi- o nano-particulados, líquidos, emulsiones o polvos; pastillas (que incluyen relleno líquido); gomas de mascar; geles; formas de dosificación de dispersión rápida; lms; óvulos; aerosoles; y parches bucales/mucoadhesivos. Las formulaciones líquidas (que incluyen múltiples fases y sistemas dispersos) incluyen emulsiones, suspensiones, soluciones, jarabes y elixires. Dichas formulaciones pueden presentarse como llers en cápsulas blandas o duras (hechas, por ejemplo, de gelatina o hidroxipropilmetilcelulosa) y típicamente comprenden un portador, por ejemplo, agua, etanol, polietilenglicol, propilenglicol, metilcelulosa, o un aceite adecuado, y uno o más agentes emulsionantes y/o agentes de suspensión. Las formulaciones líquidas se pueden también preparar por la reconstitución de un sólido, por ejemplo, de una bolsita.
Los compuestos de la invención pueden utilizarse también en las formas de dosificación de rápida disolución, de rápida desintegración tal como las descritas en Expert Opinion in Therapeutic Patents, 11(6), 981-986 por Liang and Chen (2001).
La porción de liberación inmediata puede comprender un desintegrante. Los ejemplos de desintegrantes incluyen glicolato sódico de almidón, carboximetilcelulosa de sodio, carboximetilcelulosa de calcio, croscarmelosa de sodio, crospovidona, polivinilpirrolidona, metil celulosa, celulosa microcristalina, celulosa en polvo, hidroxipropilcelulosa sustituida con alquilo inferior, polacrilina de potasio, almidón, almidón pregelatinizado, alginato de sodio y mezclas de los mismos. Generalmente, el desintegrante que comprende de 1 % e peso a 80 % en peso, preferentemente de 5 % en peso a 60 % en peso de la capa.
Los ejemplos de materiales de matriz, rellenos o diluyentes incluyen lactosa, manitol, xilitol, dextrosa, sacarosa, sorbitol, azúcar compresible, celulosa microcristalina, celulosa en polvo, almidón, almidón pregelatinizado, dextratos, dextrano, dextrina, dextrosa, maltodextrina, carbonato de calcio, fosfato dibásico de calcio, fosfato tribásico de calcio, sulfato de calcio, carbonato de magnesio, óxido de magnesio, poloxámeros, óxido de polietileno, hidroxipropil metil celulosa y mezclas de los mismos.
Al preparar las formas de dosificación que incorporan las composiciones de la invención, los compuestos también pueden ser mezclados con excipientes convencionales tales como aglutinantes,
que incluyen gelatina, almidón pregelatinizado y lo similar; lubricantes, tal como aceite vegetal hidrogenado, ácido esteárico y lo similar; diluyentes, tal como lactosa, manosa y sacarosa; desintegrantes, tal como
carboximetilcelulosa y sodio almidón glicolato; agentes de suspensión, tal como povidona, alcohol de polivinilo, y lo similar; absorbantes, tal como dióxido de silicio; conservadores, tal como metilparabeno, propilparabeno y benzoato de sodio; agentes tensoactivos; tal como lauril sulfato de sodio, polisorbato 80 y lo similar; saborizantes; y edulcorantes. Si están presentes, los agentes tensoactivos podrían comprender de 0,2 % en peso a 5 % en peso, y los absorbantes comprenderían de 0,2 % en peso a 1 % en peso. Otros excipientes incluyen uno o más de: antioxidantes, colorantes, agentes saborizantes, conservantes y agentes de enmascaramiento de sabor.
Las mezclas de comprimidos o porciones de mezclas de pueden ser alternativamente granulados húmedos, secos, o fundidos, de congelamiento por fusión o extruido antes de formar el comprimido. La formulación final puede comprender una o más capas y puede estar recubierta o no recubierta; incluso puede ser encapsulada.
La formulación de comprimidos se discute en “Pharmaceutical Dosage Forms: Tablets, Vol. 1", por H. Lieberman and L. Lachman, Marcel Dekker, Nueva York, Nueva York, 1980 (ISBN 0-8247-6918-X).
Las formulaciones sólidas para administración oral se pueden formular para ser de liberación inmediata y/o modificada. Las formulaciones de liberación modificada incluyen liberación retardada, sostenida, pulsada, controlada, dirigida y programada.
Administración parenteral
Los compuestos de la invención pueden también administrarse directamente en el torrente sanguíneo, en el músculo o en el órgano interno. Los medios adecuados para administración parenteral incluyen intravenosa, intraarterial, intraperitoneal, intratecal, intraventricular, intrauretral, intraesternal, intracraneal, intramuscular y subcutánea.
Los dispositivos adecuados para administración parenteral incluyen inyectores de aguja (incluyendo microaguja), inyectores sin aguja y técnicas de infusión. Un ejemplo de una inyección libre de aguja es Powderject™.
Las formulaciones parenterales son generalmente soluciones acuosas que pueden contener excipientes tales como sales, carbohidratos y agentes reguladores de pH (preferentemente, a un pH de 3 a 9), pero, para algunas aplicaciones, pueden ser formuladas adecuadamente como una solución no acuosa estéril o como una forma en polvo, forma seca para ser utilizada conjuntamente con un vehículo disponible tal como agua estéril libre de pirógeno. La preparación de formulaciones parenterales bajo condiciones estériles, por ejemplo, por liofilización, pueden ser fácilmente realizadas utilizando técnicas farmacéuticas estándar bien conocidas por aquellos con experiencia en la técnica.
Una forma de dosificación apropiada tal como la combinación con intensificador de solubilidad puede aumentar la solubilidad del compuesto de Fórmula I usado en solución no oral.
Las formulaciones para administración parenteral pueden ser formuladas para ser de liberación inmediata y/o modificada/controlada. Las formulaciones de liberación controlada/modificada incluyen liberación retardada, sostenida, pulsada, controlada, dirigida y programada. Así los compuestos de la invención pueden formularse como un sólido, semisólido o líquido tixotrópico para administración como un depósito implantado que proporciona liberación modificada del compuesto activo. Los ejemplos de dichas formulaciones incluyen stents recubiertos de fármaco y microesferas de PGLA.
Administración local
Los compuestos de la invención puedan ser administrados tópicamente a la piel o mucosa, es decir, dérmicamente o transdérmicamente. Las formulaciones típicas para este propósito incluyen geles, hidrogeles, lociones, soluciones, cremas, ungüentos, polvos, aderezos, espumas, películas, parches cutáneos, obleas, implantes, esponjas, fibras, vendas y microemulsiones. Puede usarse también liposomas.
Dosis
En pacientes humanos, la dosis diaria precisa administrada depende de varios factores tal como la edad, sexo, peso y condición del paciente a tratarse. La cantidad de dosis puede ser seleccionada dentro de los límites del objeto que logra el efecto del tratamiento sin efectos adversos graves o perjudiciales.
Por ejemplo, la dosificación del compuesto de la invención puede administrarse en una cantidad efectiva que varía de 0,05 a 1000 mg diarios en pacientes. Los siguientes niveles de dosificación y otros niveles de dosificación aquí son para el sujeto humano promedio que tiene un intervalo de peso de aproximadamente 65 a 70kg. La persona con experiencia fácilmente será capaz de determinar los niveles de dosificación necesaria para un sujeto cuyo peso cae fuera de este intervalo, tal como niños y ancianos.
La presente invención explicar, pero sin limitación, en detalle a través de los siguientes ejemplos y ejemplos
experimentales.
rEiemplos!
<Ejemplo 1>
(R) -4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-1)
Etapa 1: Preparación de (R)-bencilo (1-(2-fluoro-5-formilbenzoil)pirrolidin-3-il)carbamato
Ácido 2-fluoro-5-formilbenzoico (220 mg, 0,91 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 384 mg, 1,01 mmol) y N,N-diisopropil etilamina (DIPEA, 0,52 ml, 2,98 mmol) se añadió a una solución de (R)-bencilo pirrolidin-3-ilcarbamato (200 mg, 0,91 mmol) en DMF (5 ml) y se agitó durante 12 horas. La mezcla de reacción se concentró al vacío, se añadió diclorometano y lavó con NH4Cl(ac) y agua. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto intermedio (R)-bencilo (1-(2-fluoro-5-formilbenzoil)pirrolidin-3-il)carbamato (229 mg, 68 %).
Etapa 2: Preparación de (R.Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)carbamato
El compuesto intermedio (etapa 1) (236 mg, 0,62 mmol) y trietilamina (0,12 ml, 0,81 mmol) se añadió a una solución de dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (220 mg, 0,91 mmol) en THF (1,7 ml) y se agitó durante 5 horas a 0 °C. La mezcla de reacción se concentró al vacío entonces el residuo blanco formó suspensión en agua durante 30 minutos, filtró, lavó con agua, hexano y éter, y se secó para producir el compuesto intermedio (R,Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)carbamato (164 mg, 59 %).
Etapa 3: Preparación de (R)-bencilo (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)carbamato Monohidrato de hidrazina (19 ul, 0,38 mmol) se añadió a una suspensión del compuesto intermedio (etapa 2) (164 mg, 0,35 mmol) en agua (1,5 ml) y agitado durante 2 horas a 40 °C. La reacción se enfrió a temperatura ambiente y se concentró al vacío. Se añadió agua a la mezcla de reacción y el producto se extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron usando cromatografía en sílice para dar (R)-bencilo (1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)carbamato (100 mg, 47 %).
Etapa 4: Preparación de (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona
El compuesto intermedio (etapa 3) (100 mg, 0,20 mmol) y Pd/C 10% en peso (10 mg) en metanol (30 ml) fue hidrogenado a la atmósfera por 6 horas. La mezcla de reacción se filtró, evaporó al vacío y purificó mediante cromatografía en sílice para dar el compuesto del título (63 mg, 85 %).
1H-RMN (DMSO, 400 MHz): 8 12,60 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,50-7,44 (m, 1H), 7,40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,67 (m, 1H), 3,59-3,33 (m, 3H), 3,29-3,06 (m, 3H).
<Ejemplo 2>
(S) -4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-2)
Etapa 1: Preparación de (S)-bencilo (1-(2-fluoro-5-form¡lbenzoil)p¡rrol¡d¡n-3-¡l)carbamato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, (S)-bencilpirrolidin-3-ilcarbamato (150 mg, 0,68 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (114 mg, 0,68 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 335 mg, 0,88 mmol) y N,N-diisopropil etil amina (DIPEA, 0,24 ml, 1,36 mmol) para producir el compuesto intermedio (S)-bencilo (1-(2-fluoro-5-formilbenzoil)pirrolidin-3-il)carbamato (156 mg, 62 %).
Etapa 2: Preparación de (S,Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)carbamato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). Por lo tanto, este compuesto intermedio (etapa 1) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (102 mg, 0,42 mmol) y trietil amina (88 ul, 0,63 mmol) para producir el compuesto intermedio (S, Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)carbamato (117 mg, 57%).
Etapa 3: Preparación de (S)-bencilo (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)carbamato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). Por lo tanto, este compuesto intermedio (etapa 2) reaccionó con monohidrato de hidrazina (24 ul, 0,48 mmol) para dar el compuesto intermedio (S)-bencilo (1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)carbamato (58 mg, 48 %).
Etapa 4: Preparación de (S)-4-(3-(3-am¡nop¡rrol¡din-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 4). Así, este compuesto intermedio (etapa 3) (58 mg, 0,12 mmol) reaccionó con Pd/C 10 % en peso (6 mg) para dar el compuesto del título (39 mg, 88 %).
1H-RMN (DMSO, 400 MHz): 8 12,60 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,50-7,44 (m, 1H), 7,40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,67 (m, 1H), 3,59-3,33 (m, 3H), 3,29-3,06 (m, 3H).
<Ejemplo 3>
4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-3)
Etapa 1 : Preparación de bencilo (1-(2-fluoro-5-formilbenzo¡l)azet¡d¡n-3-¡l)carbamato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, bencilo azetidin-3-ilcarbamato (500 mg, 2,42 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (408 mg, 2,42 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 1,20g, 3,15 mmol) y N,N-diisopropil etilamina (DIPEA, 0,84 ml, 4,85 mmol) para dar el compuesto intermedio bencilo (1-(2-fluoro-5-formilbenzoil)azetidin-3-il)carbamato (604 mg, 70 %).
Etapa 2: Preparación de (Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)carbamato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(604 mg, 1,69 mmol) reaccionó con dimetilo (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (410 mg, 1,69 mmol) y trietilamina (0,35 ml, 2,54 mmol) para producir el compuesto intermedio (Z)-bencilo (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)carbamato (497 mg, 62 %).
Etapa 3: Preparación de bencilo (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalazin-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)carbamato Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). Por lo tanto, este compuesto intermedio (etapa 2) (497 mg, 1,05 mmol) reaccionó con monohidrato de hidrazina (0,1 ml, 2,1 mmol) para producir el compuesto bencilo intermedio (1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)carbamato (261 mg, 51 %).
Etapa 4: Preparación de 4-(3-(3-am¡noazet¡din-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 4). Así, este compuesto intermedio (etapa 3) (261 mg, 0,54 mmol) reaccionó con Pd/C 10 % en peso (30 mg) para dar el compuesto del título (174 mg, 91 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,27-8,25 (m, 1H), 7,97 (d, 1H), 7,91-7,81 (m, 2H), 7,48-7,44 (m, 1H), 7,41-7,39 (m, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,15-4,10 (m, 2H), 3,66-3,59 (m, 3H).
<Ejemplo 4>
(R)-4-(4-fluoro-3-(3-hidroxipirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-4)
Etapa 1: Preparación de (R)-3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrolid¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, (R)-3-((tercbutildimetilsilil)oxi)pirrolidina (300 mg, 1,49 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (250 mg, 1,49 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 734 mg, 1,93 mmol) y N,N-diisopropil etilamina (DIPEA, 0,52 ml, 2,97 mmol) para producir el compuesto intermedio (R)-3-(3-((tercbutildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobenzaldehído (377 mg, 72%).
Etapa 2: Preparación de (R.Z)-3-(3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). Por lo tanto, este
compuesto intermedio (etapa 1) (377 mg, 1,07 mmol) fue reaccionado con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (260 mg, 1,07 mmol) y trietilamina (0,22 ml, 1,61 mmol) para producir el compuesto intermedio (R,Z)-3-(3-(3-((terc-butildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona (300 mg, 60 %).
Etapa 3: Preparación de ((R)-4-(3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). Por lo tanto, este compuesto intermedio (etapa 2) (300 mg, 0,64 mmol) reaccionó con monohidrato de hidrazina (63 ul, 1,28 mmol) para producir el compuesto intermedio (R)-4-(3-(3-((terc-butildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (148 mg, 48 %).
Etapa 4: Preparación de (R)-4-(4-fluoro-3-(3-h¡drox¡p¡rrol¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
El compuesto intermedio (etapa 3) (148 mg, 0,30 mmol) en THF (3 ml) enfriado a 0 °C se añadió una solución de fluoruro de tetra-n-butilamonio en THF (TBAF, 0,60 ml, 0,60 mmol), y la mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se concentró al vacío, se añadió diclorometano y se lavó con NH4Cl (ac) saturado y agua. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron mediante cromatografía en sílice para dar el compuesto del título (101 mg, 92 %).
1H-RMN (DMSO, 400 MHz): 8 12,61(s, 1H), 8,25 (d, 1H), 7,98 (d, 1H), 7,87 (t, 1H), 7,82 (t, 1H), 7,51-7,45 (m, 1H), 7.40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,65 (m, 2H), 3,59-3,28 (m, 5H).
<Ejemplo 5>
(S)-4-(4-fluoro-3-(3-h¡drox¡p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-5)
Etapa 1: Preparación de (S)-3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrolid¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, (S)-3-((tercbutildimetilsilil)oxi)pirrolidina (300 mg, 1,49 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (250 mg, 1,49 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 734 mg, 1,93 mmol) y N,N-diisopropil etilamina (DIPEA, 0,52ml, 2,97 mmol) para producir el compuesto intermedio (S)-3-(3-((tercbutildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobenzaldehído (377 mg, 72%).
Etapa 2: Preparación de (S.Z)-3-(3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrol¡d¡n-1-carbonil)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(377 mg, 1,07 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (260 mg, 1,07 mmol) y trietilamina (0,22 ml, 1,61 mmol) para producir el compuesto intermedio (S,Z)-3-(3-(3-((terc-butildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona (300 mg, 60 %).
Etapa 3: Preparación de (S)-4-(3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). De este modo, este compuesto intermedio (etapa 2) (300 mg, 0,64 mmol) reaccionó con hidrazina monohidratada (63 ul, 1,28 mmol) para producir el compuesto intermedio (S)-4-(3-(3-((terc-butildimetilsilil)oxi)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (148 mg, 48 %).
Etapa 4: Preparación de (S)-4-(4-fluoro-3-(3-h¡drox¡p¡rrol¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 4 (etapa 4). De este modo, este compuesto intermedio (etapa 3)(144 mg, 0,30 mmol) reaccionó con una solución 1 M de fluoruro de tetra-n-butilamonio en THF (TBAF, 0,60 ml, 0,60 mmol) para producir el compuesto del título (101 mg, 92 %).
1H-RMN (DMSO, 400 MHz): 8 12,61(s, 1H), 8,25 (d, 1H), 7,98 (d, 1H), 7,87 (t, 1H), 7,82 (t, 1H), 7,51-7,45 (m, 1H), 7.40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,65 (m, 2H), 3,59-3,28 (m, 5H).
<Ejemplo 6>
4-(4-fluoro-3-(3-h¡drox¡azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-6)
Etapa 1: Preparación de 3-(3-((terc-but¡ld¡met¡ls¡l¡l)oxi)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, 3-((tercbutildimetilsilil)oxi)azetidina (300 mg, 1,60 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (269 mg, 1,60 mmol),
O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 789 mg, 2,08mmol) y N,N-diisopropil etilamina (DIPEA, 0,56 ml, 3,20 mmol) para producir el compuesto intermedio 3-(3-((terc-butildimetilsilil)oxi)azetidin-1-carbonil)-4-fluorobenzaldehído (378 mg, 70 %).
Etapa 2: Preparación de (Z)-3-(3-(3-((terc-butildimetilsilil)oxi)azetidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(378mg, 1,12 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (271 mg, 1,12 mmol) y trietilamina (0,23 ml, 1,68 mmol) para producir el compuesto intermedio (Z)-3-(3-(3-((terc-butildimetilsilil)oxi)azetidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona (284 mg, 56 %).
Etapa 3: Preparación de 4-(3-(3-((terc-but¡ld¡met¡ls¡l¡l)ox¡)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(284 mg, 0,62 mmol) reaccionó con hidrazina monohidratada (61 ul, 1,26 mmol) para producir el compuesto intermedio 4-(3-(3-((terc-butildimetilsilil)oxi)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (149 mg, 51 %).
Etapa 4: Preparación de 4-(4-fluoro-3-(3-h¡drox¡azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 4 (etapa 4). De este modo, este compuesto intermedio (etapa 3)(149 mg, 0,32 mmol) reaccionó con una solución 1 M de fluoruro de tetra-n-butilamonio en THF (TBAF, 0,64 ml, 0,64 mmol) para producir el compuesto del título (98 mg, 92 %).
1H-RMN (DMSO, 400 MHz): 8 12,60 (s, 1H), 8,27-8,26 (m, 1H), 7,96 (d, 1H), 7,91-7,81 (m, 2H), 7,48-7,45 (m, 1H), 7.41- 7,39 (m, 1H), 7,20 (t, 1H), 4,33 (s, 2H), 4,15-4,11 (m, 3H), 3,66-3,58 (m, 2H).
<Ejemplo 7>
4-(4-fluoro-3-(3-(h¡drox¡met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-7)
Etapa 1: Preparación de 3-(3-(((terc-but¡ld¡met¡ls¡l¡l)oxi)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 1). De este modo, 3-(((tercbutildimetilsilil)oxi)metil)azetidina (300 mg, 1,60 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (269 mg, 1,60 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 789 mg, 2,08 mmol) y N,N-diisopropil etilamina (DIPEA, 0,56 ml, 3,20 mmol) para producir el compuesto intermedio 3-(3-(((tercbutildimetilsilil)oxi)metil)azetidin-1-carbonil)-4-fluorobenzaldehído (378 mg, 72 %).
Etapa 2: Preparación______ de______ (Z)-3-(3-(3-(((terc-butildimetilsilil)oxi)metil)azetidin-1-carbonil)-4-fluorobencil¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(378 mg, 1,12 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (271 mg, 1,12 mmol) y trietilamina (0,23 ml, 1,68 mmol) para producir el compuesto intermedio (Z)-3-(3-(3-(((terc-butildimetilsilil)oxi)metil)azetidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona (284 mg, 56 %). Etapa 3: Preparación de 4-(3-(3-(((terc-but¡ld¡met¡ls¡l¡l)ox¡)met¡l)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 1 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(284 mg, 0,63 mmol) reaccionó con hidrazina monohidratada (61 ul, 1,25 mmol) para producir el compuesto intermedio 4-(3-(3-(((terc-butildimetilsilil)oxi)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (149 mg, 51 %).
Etapa 4: Preparación de 4-(4-fluoro-3-(3-(h¡drox¡met¡l)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 4 (etapa 4). De este modo, este compuesto intermedio (etapa 3)(149 mg, 0,32 mmol) reaccionó con una solución 1 M de fluoruro de tetra-n-butilamonio en THF (TBAF, 0,64 ml, 0,64 mmol) para producir el compuesto del título (98 mg, 92 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,28-8,26 (m, 1H), 7,97 (d, 1H), 7,92-7,81 (m, 2H), 7,49-7,45 (m, 1H), 7.42- 7,39 (m, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,15-4,11 (m, 3H), 3,66-3,58 (m, 2H), 3,47 (m, 2H).
<Ejemplo 8>
(R) -N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da: (I-8) Etapa 1: Preparación de (R)-N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)metil)benzo¡l)p¡rrol¡d¡n-3-il)ciclopropanocarboxam¡da
Ác¡do ciclopropanocarboxílico (20 ul. 0.28 mmol). 1-et¡l-3-(3-dimet¡lam¡noprop¡l)carbod¡¡m¡da clorhidrato (EDCI. 66 mg, 0.42 mmol) y 4-(dimet¡lam¡no)pir¡d¡na (DMAP. 68 mg. 0.56 mmol) se añadió a una solución de (R)-4-(3-(3-aminop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(100 mg. 0.28 mmol) en diclorometano (1.5 ml) y se agitó durante 12 horas. La mezcla de reacción se concentró al vacío. se añadió diclorometano y lavó con NH4Cl (ac) saturado y agua. Las capas orgánicas combinadas se secaron sobre MgSO4. filtraron. evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto del título (83 mg.68 %).
1H-RMN (CDCl3.400 MHz): 8 10.36 (d.1H). 8.46 (m.1H). 7.79 (m.2H). 7.71 (m.1H). 7.35 (m.1H). 7.32 (m.1H). 7.04 (c.1H). 5.89 (m.1H). 4.60 (c.0.3H). 4.45 (c.0.7H). 4.26 (d.2H). 3.82 (m.1H). 3.65 (m.1H). 3.51 (m.1H). 3.40 (m.1H). 3.18 (m.1H). 2.28 (m.2H). 1.61 (m.1H). 0.98 (m.1H). 0.96 (m.2H). 0.74 (m.2H).
<Ejemplo 9>
N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)c¡clopropanocarboxam¡da: (I-9) Etapa 1: Preparación_____ de_____ N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalazin-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-il)c¡clopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 8 (etapa 1). De este modo. 4-(3-(3-aminoazet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg. 0.28 mmol) reaccionó con ácido ciclopropanocarboxilico (20 ul. 0.28 mmol). 1-etil-3-(3-d¡met¡lam¡noprop¡l)carbod¡¡mida clorhidrato (EDCI. 66 mg. 0.42 mmol) y 4-(dimet¡lam¡no)pir¡d¡na (DMAP. 68 mg. 0.56 mmol) para producir el compuesto del título (78 mg. 62 %). 1H-RMN (MeOD. 400 MHz): 88.39 (dd. 1H). 7.94 (dd. 1H). 7.85-7.80 (m. 2H). 7.52-7.45 (m. 2H). 7.17-7.12 (m. 1H).
4.63-4.53 (m. 1H). 4.44-4.37 (m. 3H). 4.30-4.21 (m. 1H). 4.01-3.89 (m. 2H). 1.59-1.52 (m. 1H). 0.92-0.78 (m. 4H). <Ejemplo 10>
(S) -N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxam¡da: (I-10) Etapa 1: Preparación  (S)-N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzoil)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxamida
(S)-N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzoil)p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 8 (etapa 1). De este modo. (S)-4-(3-(3-aminop¡rrol¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona (ejemplo 2)(100 mg. 0.28 mmol) reaccionó con ácido ciclopropanocarboxilico (20 ul. 0.28 mmol). 1-etil-3-(3-d¡met¡lam¡noprop¡l)carbod¡¡mida clorhidrato (EDCI. 66 mg. 0.42 mmol) y 4-(dimet¡lam¡no)pir¡d¡na (DMAP. 68 mg. 0.56 mmol) para producir el compuesto del título (77 mg. 65 %). 1H-RMN (CDCh.400 MHz): 89.55 (s. 0.6H). 9.52 (s. 0.6H). 8.46 (d. 1H). 7.82-7.71 (m. 3H). 7.38-7.26 (m. 2H). 7.04 (c.
1H). 5.75 (m. 1H). 4.60 (m. 0.4H). 4.42 (m. 0.6H). 4.28 (s. 0.8H). 4.27 (s. 1.2H). 3.92-3.64 (m. 2.4H). 3.56-3.33 (m.
1H). 3.17 (dd. 0.6H). 2.33-2.16 (m. 1H). 1.93 (m. 1H). 1.32 (m. 1H). 0.96 (m. 2H). 0.75 (m. 2H).
<Ejemplo 11>
(R)-N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1 -¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)-N-met¡lc¡clopropanocarboxam¡da: (I-11)
Etapa 1: Preparación de (R)-N-(1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡din-3-¡l)-N-met¡lc¡clopropanocarboxam¡da
Ácido 2-fluoro-5-formilbenzoico (350 mg. 2.08 mmol). O-(benzotriazol-1-¡l)-N,N,N'.N'-tetrametiluron¡o hexafluorofosfato (HBTU. 1.02g. 2.79 mmol) y N,N-diisopropil etilamina (DlPEA. 0.72 ml. 4.16 mmol) se añadió a una solución de (R)-N-metil-N-(p¡rrol¡d¡n-3-¡l)c¡clopropanocarboxamida (350 mg. 2.08 mmol) en DMF(5 ml) y se agitó durante 12 horas. La mezcla de reacción se concentró al vacío. se añadió diclorometano y lavó con NH4Cl (ac) saturado y agua. Las capas orgánicas combinadas se secaron sobre MgSO4. filtraron. evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto intermedio (R)-N-(1-(2-fluoro-5-formilbenzo¡l)p¡rrolid¡n-3-¡l)-N-metilciclopropanocarboxamida (470 mg.72 %).(R)-bencilo (1-(2-fluoro-5-formilbenzo¡l)p¡rrolid¡n-3-¡l)carbamato (229 mg.68 %).
Etapa 2: Preparación de (R.Z)-N-(1-(2-fluoro-5-((3-oxo¡sobenzofuran-1(3H)-¡l¡den)met¡l)benzoil)p¡rrol¡d¡n-3-¡l)-N-metilciclopropanocarboxamida
Trietilamina (0,38 ml, 2,21 mmol) se añadió en gotas a una solución del compuesto intermedio (etapa 1)(470 mg, 1,48 mmol) y dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (357 mg, 1,48 mmol) en THF (1,7ml) y se agitó durante 5 horas a 0°C. La mezcla de reacción se concentró al vacío y luego el residuo blanco formó suspensión en agua por 30 minutos, filtró, lavó con agua, hexano y éter y se secó para dar el compuesto intermedio (R,Z)-N-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida (398 mg, 62 %).
Etapa 3: Preparación de (R)-N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida
Hidrazina monohidratada (90 ul, 1,83 mmol) se añadió a una suspensión del compuesto intermedio (etapa 2) (398 mg, 0,92 mmol) en etanol (1,5 ml) y agitar a 40 °C durante 2 horas. La reacción se enfrió a temperatura ambiente y se concentró al vacío. Se añadió agua a la mezcla de reacción y el producto se extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto del título (184 mg, 45 %).
1H-RMN (CDCla,400 MHz): 85,35 (c, 0,7H), 4,78 (c, 0,3H), 3,65 (m, 1H), 3,51 (m, 1H), 3,40 (m, 1H), 3,01 (s, 3H), 2,28 (m, 2H), 1,61 (m, 1H), 0,98 (m, 1H), 0,96 (m, 2H), 0,74 (m, 2H).
<Ejemplo 12>
N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)-N-met¡lc¡clopropanocarboxam¡da: (I-121
Etapa 1: Preparación de N-(1-(2-fluoro-5-form¡lbenzo¡l)azet¡din-3-¡l)-N-met¡lc¡clopropanocarboxam¡da
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, N-(azetidin-3-il)-N-metilciclopropanocarboxamida (200 mg, 1,30 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (218 mg, 1,30 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 640 mg, 1,68 mmol) y N,N-diisopropil etilamina (DIPEA, 0,45 ml, 2,59 mmol) para producir el compuesto intermedio N-(1-(2-fluoro-5-formilbenzoil)azetidin-3-il)-N-metilciclopropanocarboxamida (256 mg, 65 %).
Etapa 2: Preparación de (Z)-N-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)-N-metilciclopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(256 mg, 0,84 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (204 mg, 0,84 mmol) y trietilamina (0,17 ml, 1,26 mmol) para producir el compuesto intermedio (Z)-N-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)-N-metilciclopropanocarboxamida (202 mg, 57 %).
Etapa 3: Preparación de N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)-N-metilciclopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(202 mg, 0,48 mmol) reaccionó con hidrazina monohidratada (46 ul, 0,96 mmol) para producir el compuesto del título (106 mg, 51 %).
1H-RMN (MeOD, 400 MHz): 88,39 (dd, 1H), 7,94 (dd, 1H), 7,85-7,80 (m, 2H), 7,52-7,45 (m, 2H), 7,17-7,12 (m, 1H), 4,63-4,53 (m, 1H), 4,44-4,37 (m, 3H), 4,30-4,21 (m, 1H), 4,01-3,89 (m, 2H), 3,26 (s, 3H), 1,59-1,52 (m, 1H), 0,92-0,78 (m, 4H).
<Ejemplo 13>
(S)-N-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)-N-met¡lc¡clopropanocarboxam¡da: (I-13)
Etapa 1: Preparación de (S)-N-(1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡din-3-¡l)-N-met¡lc¡clopropanocarboxam¡da
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (S)-N-metil-N-(pirrolidin-3-il)ciclopropanocarboxamida (350 mg, 2,08 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (349 mg, 2,08 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 1,02 mg, 2,70 mmol) y N,N-diisopropil etilamina (DIPEA, 0,72 ml, 4,16 mmol) para producir el compuesto intermedio (S)-N-(1-(2-fluoro-5-formilbenzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida (470 mg, 71 %).
Etapa 2: Preparación de (S,Z)-N-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(470 mg, 1,48 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (357 mg, 1,48 mmol) y trietilamina (0,31 ml, 2,12 mmol) para producir el compuesto intermedio (S,Z)-N-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida (397 mg, 62 %).
Etapa 3: Preparación de (S)-N-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)-N-metilciclopropanocarboxamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(397 mg, 0,92 mmol) reaccionó con hidrazina monohidratada (90 ul, 1,83 mmol) para producir el compuesto del título (185 mg, 45 %).
1H-RMN (CDCla, 400 MHz): 810,13 (s, 0,5H), 9,97 (s, 0,5H), 8,46 (s, 1H), 7,81-7,70 (m, 3H), 7,37 (m, 1H), 7,30 (m, 1H), 7,03 (c, 1H), 5,34 (m, 0,5H), 5,13 (m, 0,5H), 4,27 (d, 2H), 3,88 (m, 0,5H), 3,78 (m, 0,5H), 3,66-3,34 (m, 3H), 3,20 3,14 (m, 3,5H), 2,98-2,82 (m, 0,5H), 2,20-2,06 (m, 1H), 1,78-1,65 (m, 1H), 1,06-0,92 (m, 2H), 0,81 (m, 2H).
<Ejemplo 14>
3-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)-5.5-d¡met¡l¡m¡dazol¡d¡n-2.4-d¡ona: (I-14)
Etapa 1: Preparación de 3-(3-(4.4-d¡met¡l-2.5-d¡oxo¡m¡dazol¡d¡n-1-¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, 3-(azetidin-3-il)-5,5-dimetilimidazolidin-2,4-diona (150 mg, 0,82 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (137 mg, 0,82 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 403 mg, 1,06 mmol) y N,N-diisopropil etilamina (DIPEA, 0,29 ml, 1,63 mmol) para producir el compuesto intermedio 3-(3-(4,4-dimetil-2,5-dioxoimidazolidin-1-il)azetidin-1-carbonil)-4-fluorobenzaldehído (152 mg, 56 %).
Etapa 2: Preparación de (Z)-3-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)-5,5-d¡met¡l¡m¡dazol¡d¡n-2,4-d¡ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(152 mg, 0,45 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (111 mg, 0,45 mmol) y trietilamina (96 ul, 0,68 mmol) para producir el compuesto intermedio (Z)-3-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)azetidin-3-il)-5,5-dimetilimidazolidin-2,4-diona (89 mg, 43 %). Etapa 3: Preparación de 3-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)-5,5-d¡met¡l¡m¡dazol¡d¡n-2,4-d¡ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(89 mg, 0,19 mmol) reaccionó con hidrazina monohidratada (20 ul, 0,39 mmol) para producir el compuesto del título (47 mg, 51 %).
1H-RMN (MeOD, 400 MHz): 88,36-8,35 (m, 1H), 7,92 (t, 1H), 7,98 (d, 1H), 7,90-7,82 (m, 2H), 7,48-7,43 (m, 2H), 7,22 (t, 1H), 4,34 (s, 2H), 4,07-4,03 (m, 1H), 3,94 (t, 1H), 3,85-3,76 (m, 2H), 3,27-3,23 (m, 1H), 1,32 (s, 6H).
<Ejemplo 15>
(R)-3-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)¡m¡dazol¡d¡n-2.4-d¡ona: (I-15) Etapa 1: Preparación de (R)-3-(3-(2.5-d¡oxo¡m¡dazol¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-3-(pirrolidin-3-il)imidazolidin-2,4-diona (150 mg, 0,88 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (149 mg, 0,88 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 437 mg, 1,15 mmol) y N,N-diisopropil etilamina (DIPEA, 0,31 ml, 1,77 mmol) para producir el compuesto intermedio (R)-3-(3-(2,5-dioxoimidazolidin-1-il)pirrolidin-1-carbonil)-4-fluorobenzaldehído (158 mg, 56 %).
Etapa 2 : Preparación de (R,Z)-3-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-¡l)¡m¡dazol¡d¡n-2,4-d¡ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(158 mg, 0,50 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1
il)fosfonato (120 mg, 0,50 mmol) y trietilamina (58 ul, 0,42 mmol) para producir el compuesto intermedio (R,Z)-3-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)imidazolidin-2,4-diona (93 mg, 43 %).
Etapa 3 : Preparación de (R)-3-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)imidazolidin-2,4-diona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(93 mg, 0,21 mmol) reaccionó con hidrazina monohidratada (21 ul, 0,43 mmol) para producir el compuesto del título (54 mg, 56 %).
1H-RMN (MeOD, 400 MHz): 88,35-8,32 (m, 1H), 7,91 (t, 1H), 7,86-7,77 (m, 2H), 7,47-7,38 (m, 2H), 7,16-7,10 (m, 1H), 4,80-4,58 (m, 1H), 4,35 (d, 2H), 3,98-3,94 (m, 2H), 3,68-3,60 (m, 1H), 3,51-3,45 (m, 2H), 2,50-2,32 (m, 1H), 2,24-2,12 (m, 2H).
<Ejemplo 16>
(R)-1-et¡l-3-(1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)¡m¡dazol¡d¡n-2.4-d¡ona: (I-161
Etapa 1: Preparación de (R)-3-(3-(3-et¡l-2.5-d¡oxo¡m¡dazol¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-1-etil-3-(pirrolidin-3-il)imidazolidin-2,4-diona (200 mg, 1,01 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (170 mg, 1,01 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 500 mg, 1,31 mmol) y N,N-diisopropil etilamina (DIPEA, 0,35 ml, 2,02 mmol) para producir el compuesto intermedio (R)-3-(3-(3-etil-2,5-dioxoimidazolidin-1-il)pirrolidin-1-carbonil)-4-fluorobenzaldehído (197 mg, 56 %).
Etapa 2: Preparación de (R,Z)-1-etil-3-(1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-¡l)¡m¡dazol¡d¡n-2,4-d¡ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(197 mg, 0,56 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (137 mg, 0,56 mmol) y trietilamina (0,12 ml, 0,85 mmol) para producir el compuesto intermedio (R,Z)-1 -etil-3- (1-(2-fluoro-5-((3-oxoisobenzofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)imidazolidin-2,4-diona (134 mg, 51 %).
Etapa 3: Preparación de (R)-1-etil-3-(1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-¡l)¡m¡dazol¡d¡n-2,4-d¡ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(134 mg, 0,29 mmol) reaccionó con hidrazina monohidratada (28 ul, 0,58 mmol) para producir el compuesto del título (66 mg, 48 %).
1H-RMN (MeOD, 400 MHz): 88,37-8,34 (m, 1H), 7,92 (t, 1H), 7,87-7,80 (m, 2H), 7,49-7,43 (m, 1H), 7,41-7,39 (m, 1H), 7,17-7,11 (m, 1H), 4,78-4,59 (m, 1H), 4,37 (d, 2H), 3,94 (s, 2H), 3,87 (d, 2H), 3,84-3,79 (m, 1H), 3,67-3,60 (m, 1H), 3,47-3,35 (m, 2H), 2,59-2,44 (m, 1H), 2,19 (d, 2H), 1,20-1,13 (m, 3H).
<Ejemplo 17>
4- (4-fluoro-3-(3-(4-fluorop¡per¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-17)
Etapa 1: Preparación de 4-fluoro-3-(3-(4-fluorop¡per¡din-1-carbon¡l)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, azetidin-3-il(4-fluoropiperidin-1-il)metanona (200 mg, 1,07 mmol) reaccionó con ácido 2-fluoro-5-formil benzoico (180 mg, 1,07 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 529 mg, 1,40 mmol) y N,N-diisopropil etilamina (DIPEA, 0,37 ml, 2,14 mmol) para producir el compuesto intermedio 4-fluoro-3-(3-(4-fluoropiperidin-1-carbonil)azetidin-1-carbonil)benzaldehído (216 mg, 60 %).
Etapa 2: Preparación de (Z)-3-(4-fluoro-3-(3-(4-fluoropiperidin-1-carbonil)azetidin-1-carbonil)benciliden)isobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(261 mg, 0,64 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (156 mg, 0,64 mmol) y trietilamina (0,13 ml, 0,96 mmol) para producir el compuesto intermedio (Z)-3-(4-fluoro-3-(3-(4-fluoropiperidin-1-carbonil)azetidin-1-carbonil)benciliden)isobenzofuran-1(3H)-ona (140 mg, 48 %).
Etapa 3: Preparación de 4-(4-fluoro-3-(3-(4-fluorop¡per¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, este compuesto ¡ntermed¡o (etapa 2)(140 mg, 0,31 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (30 ul, 0,62 mmol) para produc¡r el compuesto del título (76 mg, 53 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,95 (d, 1H), 7,79-7,90 (m, 2H), 7,45-7,60 (m, 2H), 7,15 (t, 1H), 4,37 (s, 2H), 4,32 (d, 1H), 4,15-4,27 (m, 3H), 3,70-3,88 (m, 2H), 3,41-3,62 (m, 2H), 3,27-3,36 (m, 2H), 1,76-1,94 (m, 4H). <Ejemplo 18>
4-(3-(3-(3.3-difluoroazetidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-18)
Etapa 1: Preparac¡ón de 3-(3-(3.3-d¡fluoroazet¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, azet¡d¡n-3- ¡l(3,3-d¡fluoroazet¡d¡n-1-¡l)metanona (200 mg, 1,14 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l benzo¡co (190 mg, 1,14 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 560 mg, 1,47 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,41 ml, 2,27 mmol) para produc¡r el compuesto ¡ntermed¡o 3-(3-(3,3-d¡fluoroazet¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (222 mg, 60 %).
Etapa 2: Preparac¡ón______ de______ (Z)-3-(3-(3-(3,3-d¡fluoroazet¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(222 mg, 0,68 mmol) reacc¡onó con d¡met¡l (3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (164 mg, 0,68 mmol) y tr¡et¡lam¡na (0,14 ml, 1,02 mmol) para produc¡r el ¡ntermed¡o (Z)-3-(3-(3-(3,3-d¡fluoroazet¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona (145 mg, 48 %).
Etapa 3: Preparac¡ón de 4-(3-(3-(3.3-d¡fluoroazet¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, este compuesto ¡ntermed¡o (etapa 2)(145 mg, 0,33 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (31 ul, 0,65 mmol) para produc¡r el compuesto del título (79 mg, 53 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,94 (d, 1H), 7,79-7,89 (m, 2H), 7,46-7,58 (m, 2H), 7,14 (t, 1H), 4,46-4,64 (m, 2H), 4,25-4,40 (m, 5H), 4,05-4,23 (m, 3H), 3,51-3,59 (m, 1H).
<Ejemplo 19>
4- (3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-19) Etapa 1: Preparac¡ón de 3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, azet¡d¡n-3-¡l(3,3-d¡fluorop¡rrol¡d¡n-1-¡l)metanona (200 mg, 1,05 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l benzo¡co (176 mg, 1,05 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 518 mg, 1,05 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,37 ml, 1,37 mmol) para produc¡r el compuesto ¡ntermed¡o 3-(3-(3,3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (214 mg, 60 %).
Etapa 2: Preparac¡ón______ de______ (Z)-3-(3-(3-(3,3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(214 mg, 0,63 mmol) reacc¡onó con d¡met¡l (3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (152 mg, 0,63 mmol) y tr¡et¡lam¡na (0,13 ml, 0,94 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(3-(3-(3,3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona (138 mg, 48 %). Etapa 3: Preparac¡ón de 4-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, este compuesto ¡ntermed¡o (etapa 2)(138mg, 0,31 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (30 ul, 0,62 mmol) para produc¡r el compuesto del título (75mg, 53 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,95 (d, 1Hz), 7,80-7,90 (m, 2H), 7,45-7,54 (m, 2H), 7,15 (t, 1H), 4,38 (s, 2H), 4,29-4,36 (m, 2H), 4,16-4,28 (m, 3H), 3,58-3,85 (m, 4H), 2,36-2,51 (m, 2H).
<Ejemplo 20>
4-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-20) Etapa 1: Preparación de 3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo. (3,3-d¡fluorop¡rrol¡d¡n-1-¡l)(p¡rrol¡d¡n-3-¡l)metanona (210 mg. 1.03 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l benzo¡co (172 mg. 1.03 mmol). O-(benzotr¡azol-1-¡l)-N,N,N'.N'-tetramet¡luron¡o hexafluorofosfato (HBTU. 507 mg. 1,33 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA. 0.36 ml. 2.05 mmol) para produc¡r el compuesto ¡ntermed¡o 3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (218 mg. 60 %).
Etapa 2: Preparac¡ón______ de______ (Z)-3-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo. este compuesto ¡ntermed¡o (etapa 1)(218 mg. 0.62 mmol) reacc¡onó con d¡met¡l (3-oxo-1.3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (149 mg. 0.62 mmol) y tr¡et¡lam¡na (0.13 ml. 0.93 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona (139 mg. 48 %). Etapa 3: Preparac¡ón de 4-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-carbon¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo. este compuesto ¡ntermed¡o (etapa 2)(139 mg. 0.30 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (29 ul. 0.59 mmol) para produc¡r el compuesto del título (76 mg. 53 %).
1H-RMN (DMSO. 400 MHz): 812.60 (s. 1H). 8.26 (d. 1H). 7.98 (d. 1H). 7.90 (t. 1H). 7.86-7.80 (m. 1H). 7.46-7.37 (m.
2H). 7.22 (t. 1H). 4.33 (s. 2H). 4.08-3.98 (m. 1H). 3.79-3.67 (m. 2H). 3.64-3.42 (m. 3H). 3.32-3.16 (m. 2H). 2.49-2.32 (m. 2H). 2.23-2.04 (m. 1H). 1.97-1.87 (m. 1H). 1.18 (t. 1H).
<Ejemplo 21>
(R)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-carboxam¡da: (I-21)
Etapa 1: Preparac¡ón de (R)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-carboxam¡da
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo. (R)-N-(c¡cloprop¡lmet¡l)p¡rrol¡d¡n-3-carboxam¡da (300 mg. 1.78 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l benzo¡co (299 mg.
1.78 mmol). O-(benzotr¡azol-1-¡l)-N,N,N'.N'-tetramet¡luron¡o hexafluorofosfato (HBTU. 879 mg. 2.32 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA. 0.63 ml. 3.57 mmol) para produc¡r el compuesto ¡ntermed¡o (R)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-carboxam¡da (266 mg. 47 %).
Etapa 2: Preparac¡ón______de______(R.Z)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-((3-oxo¡sobenzofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-carboxam¡da
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo. este compuesto ¡ntermed¡o (etapa 1)(266 mg. 0.84 mmol) reacc¡onó con d¡met¡l (3-oxo-1.3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (203 mg. 0.84 mmol) y tr¡et¡lam¡na (0.18 ml. 1.26 mmol) para produc¡r el compuesto ¡ntermed¡o (R.Z)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-((3-oxo¡sobenzofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-carboxam¡da (186 mg.
51 %).
Etapa 3: Preparac¡ón de (R)-N-(c¡cloprop¡lmet¡l)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-carboxam¡da
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo. este compuesto ¡ntermed¡o (etapa 2)(186 mg. 0.43 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (42 ul. 0.85 mmol) para produc¡r el compuesto del título (84 mg. 44 %).
1H-RMN (MeOD. 400 MHz): 88.36-8.34 (m. 1H). 7.93 (t. 1H). 7.88-7.78 (m. 2H). 7.47-7.39 (m. 2H). 7.14 (t. 1H). 4.37 (s. 2H). 3.82-3.53 (m. 2H). 3.49-3.33 (m. 2H). 3.12-2.96 (m. 3H). 2.22-1.96 (m. 2H). 0.99-0.88 (m. 1H). 0.52-0.44 (m.
2H). 2.24-2.02 (m. 2H).
<Ejemplo 22>
4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-22)
Etapa 1: Preparación de 4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, azet¡d¡n-3-¡l(p¡rrol¡d¡n-1-¡l)metanona (200 mg, 1,3o mmol) fue el ác¡do 2-fluoro-5-form¡l benzo¡co (218 mg, 1,30 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 639 mg, 1,69 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,45 ml, 2,59 mmol) para produc¡r el ¡ntermed¡o 4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benzaldehído (244 mg, 62 %).
Etapa 2: Preparac¡ón de (Z)-3-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(244 mg, 0,80 mmol) reacc¡onó con d¡met¡l (3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (194 mg, 0,80 mmol) y tr¡et¡lam¡na (0,17 ml, 1,21 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona (172 mg, 51 %).
Etapa 3: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, este compuesto ¡ntermed¡o (etapa 2)(172 mg, 0,42 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (40 ul, 0,82 mmol) para produc¡r el compuesto del título (97 mg, 55 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 8,37 (m, 1H), 7,24 (d, 1H), 7,80-7,90 (m, 2H), 7,49 (m, 2H), 7,14 (t, 1H), 4,37 (s, 2H), 4,32 (t, 1H), 4,22 (m, 3H), 3,71 (m, 1H), 3,44 (m, 2H), 3,30-3,44 (m, 2H), 1,86-1,97(m, 4H).
<Ejemplo 23>
(R) -4-(3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-23) Etapa 1: Preparac¡ón de (R)-3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-azet¡d¡n-3-¡l(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-¡l)metanona (200 mg, 1,05 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l benzo¡co (176 mg, 2,05 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 515 mg, 1,36 mmol) y N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,52 ml, 2,98 mmol) para produc¡r el compuesto ¡ntermed¡o (R)-3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (150 mg, 42 %).
Etapa 2: Preparac¡ón de (R,Z)-3-(3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(150 mg, 0,44 mmol) reacc¡onó con d¡met¡l (3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (106 mg, 0,44 mmol) y tr¡et¡lam¡na (92 ul, 0,65 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sobenzofuran-1(3H)-ona (102 mg, 51 %). Etapa 3: Preparac¡ón de (R)-4-(3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, este compuesto ¡ntermed¡o (etapa 2)(102mg, 0,22mmol) reacc¡onó con h¡draz¡na monoh¡dratada (21 ul, 0,45mmol) para produc¡r el compuesto del título (53mg, 50 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,94-7,92 (m, 1H), 7,89-7,80 (m, 2H), 7,56-7,39 (m, 2H), 7,16 (t, 1H), 4,36 (s, 2H), 4,20-4,03 (m, 2H), 3,96-3,80 (m, 2H), 3,67 (m, 1H), 3,44 (t, 2H), 3,39-3,32 (m, 2H), 2,98-2,90 (m, 1H), 2,10 (s, 6H), 1,82-1,80 (m, 2H).
<Ejemplo 24>
(S) -4-(3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-24) Etapa 1: Preparac¡ón de (S)-3-(3-(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (S)-azet¡d¡n-3-¡l(3-(d¡met¡lam¡no)p¡rrol¡d¡n-1-¡l)metanona (200 mg, 1,05 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡l
benzoico (176 mg, 2,05 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 515 mg, 1,36 mmol) y N,N-diisopropil etilamina (DIPEA, 0,52 ml, 2,98 mmol) para producir el compuesto intermedio (S)-3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobenzaldehído (150 mg, 42 %).
Etapa 2: Preparación de (S,Z)-3-(3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(150 mg, 0,44 mmol) reaccionó con dimetil (3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (106 mg, 0,44 mmol) y trietilamina (92 ul, 0,65 mmol) para producir el compuesto intermedio (S,Z)-3-(3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobenciliden)isobenzofuran-1(3H)-ona (102 mg, 51 %). Etapa 3: Preparación de (S)-4-(3-(3-(3-(dimetilamino)pirrolidin-1-carbonil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, este compuesto intermedio (etapa 2)(102 mg, 0,22 mmol) reaccionó con hidrazina monohidratada (21 ul, 0,45 mmol) para producir el compuesto del título (53 mg, 50 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,94-7,92 (m, 1H), 7,89-7,80 (m, 2H), 7,56-7,39 (m, 2H), 7,16 (t, 1H), 4,36 (s, 2H), 4,20-4,03 (m, 2H), 3,96-3,80 (m, 2H), 3,67 (m, 1H), 3,44 (t, 2H), 3,39-3,32 (m, 2H), 2,98-2,90 (m, 1H), 2,10 (s, 6H), 1,82-1,80 (m, 2H).
<Ejemplo 25>
4-(3-(3-(ciclobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-25)
Etapa 1: Preparación de 4-(3-(3-(c¡clobut¡lam¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Ciclobutanona (42 ul, 0,57 mmol) se añadió a una solución de 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3)(100 mg, 0,28 mmol) en 1,2-dicloroetano (2ml) y se agitó durante 30min luego ácido acético (32 ul, 0,56 mmol) y triacetoxiborohidruro (118 mg, 0,56 mmol) se añadió a la mezcla de reacción a 0°C y se agitó durante 12 horas. La mezcla de reacción se concentró en el vacío, añadió 2N NaOH(ac) y extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto del título (82 mg, 72 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,50-7,21 (m, 2H), 7,21 (t, 1H), 4,33 (s, 2H), 4,10 (t, 1H), 3,97 (t, 1H), 3,67-3,51 (m, 3H), 3,08 (m, 1H), 2,76 (m, 1H), 1,99 (t, 2H), 1,69 1,46 (m, 4H).
<Ejemplo 26>
4-(3-(3-(ciclopropilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-26)
Etapa 1 : Preparación de 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con (1-etoxiciclopropoxi)trimetilsilano (51 ul, 0,57 mmol) y triacetoxiborohidruro de sodio (248 mg, 1,17 mmol) para producir el compuesto del título (152 mg, 68 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,93 (d, 1H), 7,79-7,89 (m, 2H), 7,48-7,54 (m, 1H), 7,42 (d, 1H), 7,15 (t, 1H), 4,00-4,05 (m, 2H), 3,75-3,80 (m, 2H), 3,34-3,39 (m, 1H), 3,12 (s, 2H), 1,32-1,36 (m, 1H), 0,64-0,72 (m, 2H), 0,41 0,46 (m, 2H).
<Ejemplo 27>
4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-27)
Etapa 1: Preparación de4-(3-(3-(c¡clopent¡lam¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con ciclopentanona (51 ul, 0,57 mmol) y triacetoxiborohidruro de sodio (248 mg, 1,178 mmol) para producir el compuesto del título (240 mg, 66 %).
1H-RMN (DMSO, 400 MHz): 5 12,60 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,44-7,50 (m, 1H), 7.40 (d, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,13 (t,.1H), 4,00 (t, 1H), 3,54-3,70 (m, 3H), 2,82-2,91 (m, 1H), 1,52-1,67 (m, 4H), 1,43 (sa, 2H), 1,10-1,27 (m, 2H).
<Ejemplo 28>
4-(3-(3-(ciclohexilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-28)
Etapa 1: Preparación de 4-(3-(3-(c¡clohex¡lam¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con c¡clohexanona (59 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (203 mg, 82 %).
1H-RMN (DMSO, 400 MHz): 512,61 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,91-7,81 (m, 2H), 7,49-7,45 (m, 1H), 7,41-7,39 (m, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,16-3,98 (m, 2H), 3,67-3,58 (m, 3H), 2,33-2,26 (m, 1H), 1,72-1,61 (m, 4H), 1,52 (d, 1H), 1,23-1,06 (m, 3H), 1,00-0,90 (m, 2H).
<Ejemplo 29>
(R)-4-(3-(3-(ciclopropilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-29)
Etapa 1: Preparac¡ón de (R)-4-(3-(3-(c¡cloprop¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(200 mg, 0,54 mmol) reacc¡onó con (1-etox¡c¡clopropox¡)tr¡met¡ls¡lano (49 ul, 0,54 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (142 mg, 65 %).
1H-RMN (DMSO, 400 MHz): 5 12,60 (s, 1H), 8,27-8,25 (m, 1H), 7,97 (d, 1H), 7,89-7,82 (m, 2H), 7,44-7,33 (m, 2H), 7,22 (t, 1H), 4,32 (s, 2H), 3,29-3,23 (m, 2H), 3,15-2,99 (m, 1H), 2,06-1,73 (m, 3H), 1,24 (s, 1H), 0,39-0,13 (m, 4H). <Ejemplo 30>
(R)-4-(3-(3-(ciclobutilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-30)
Etapa 1 : Preparac¡ón de (R)-4-(3-(3-(c¡clobut¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(200 mg, 0,54 mmol) reacc¡onó con c¡clobutanona (41 ul, 0,54 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (163 mg, 72 %).
1H-RMN (DMSO, 400 MHz): 5 12,60 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,44-7,50 (m, 1H), 7.40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,67 (m, 1H), 3,59-3,33 (m, 3H), 3,29-3,06 (m, 3H), 2,76 (m, 1H), 1,99 (t, 2H), 1,69-1,46(m, 4H).
<Ejemplo 31>
(R)-4-(3-(3-(ciclopentilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-31)
Etapa 1: Preparac¡ón de (R)-4-(3-(3-(c¡clopent¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(200 mg, 0,54 mmol) reacc¡onó con c¡clopentanona (48 ul, 0,54 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (190 mg, 81 %).
1H-RMN (DMSO, 400 MHz): 5 12,60 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,44-7,50 (m, 1H), 7.40 (d, 1H), 7,21 (t, 1H), 4,37 (s, 2H), 3,79-3,67 (m, 1H), 3,59-3,33 (m, 3H), 3,29-3,06 (m, 3H), 2,82-2,91 (m, 1H), 1,52-1,67 (m, 4H), 1,43 (sa, 2H), 1,10-1,27 (m, 2H).
<Ejemplo 32>
4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-32)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(¡soprop¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con acetona (42 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (123 mg, 55 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,27-8,25 (m, 1H), 7,97 (d, 1H), 7,91-7,81 (m, 2H), 7,48-7,44 (m, 1H), 7,41-7,39 (m, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,15-4,10 (m, 2H), 3,66-3,59 (m, 3H), 3,17 (d, 1H), 2,68-2,61 (m, 1H), 0,92-0,83 (m, 6H).
<Ejemplo 33>
4-(3-(3-((c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-33)
Etapa 1: Preparac¡ón de 4-(3-(3-((c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con c¡clopropanocarbaldehído (42 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (153 mg, 66 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,28-7,97 (m, 2H), 7,91-7,81 (m, 2H), 7,48-7,40 (m, 2H), 7,22 (m, 1H), 4,33 (s, 2H), 4,13-3,98 (m, 2H), 3,70-3,56 (m, 3H), 2,29 (m, 2H), 0,83-0,72 (m, 1H), 0,37-0,32 (m, 2H), 0,06 (m, 2H).
<Ejemplo 34>
4-(3-(3-(b¡s(c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-34)
Etapa 1: Preparac¡ón de 4-(3-(3-(b¡s(c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con c¡clopropanocarbaldehído (84 ul, 1,14 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (116 mg, 43 %).
1H-RMN (DMSO, 400 MHz): 812,57 (s, 1H), 8,23 (m, 1H), 7,97-7,77 (m, 3H), 7,47-7,36 (m, 2H), 7,18 (m, 1H), 4,29 (s, 2H), 3,99 (m, 1H), 3,88-3,58 (m, 4H), 2,38-2,31 (m, 4H), 0,77 (m, 2H), 0,38 (s, 2H), 0,36 (s, 2H), 0,01 (s, 4H).
<Ejemplo 35>
4-(4-fluoro-3-(3-(¡sobut¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-35)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(¡sobut¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con ¡sobut¡raldehído (52 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (194 mg, 83%).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,51-7,49 (m, 1H), 7,45-7,43 (m, 1H), 7,15 (t, 1H), 4,38 (s, 2H), 4,28-4,26 (m, 1H), 4,16-4,12 (m, 1H), 3,88-3,84 (m, 1H), 3,79-3,75 (m, 1H), 3,66 3,64 (m, 1H), 2,31-2,28 (m, 2H), 1,71-1,68 (m, 1H), 0,91 (dd, 6H).
<Ejemplo 36>
4-(4-fluoro-3-(3-((1-h¡drox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-36)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((1-h¡drox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 1-h¡drox¡propan-2-ona (39 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (82 mg, 35 %).
1H-RMN (MeOD, 400 MHz): 58,36-8,34 (m, 1H), 7,94-7,92 (m, 1H), 7,88-7,81 (m, 2H), 7,50-7,49 (m, 1H), 7,46-7,44 (m, 1H), 7,16-7,11 (m, 1H), 4,36 (s, 2H), 4,35-4,33 (m, 1H), 4,20-4,17 (m, 1H), 3,90-3,3,83 (m, 3H), 3,46-3,43 (m, 1H), 3,39-3,36 (m, 1H), 2,77-2,75 (m, 1H), 1,95 (s, 1H), 1,02 (c, 3H).
<Ejemplo 37>
4-(4-fluoro-3-(3-(neopentilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-37)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(neopent¡lam¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con p¡valaldehído (62 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (185 mg, 77 %).
1H-RMN (MeOD, 400 MHz): 58,34 (m, 1H), 7,92-7,78 (m, 3H), 7,50-7,43 (m, 2H), 7,13 (t, 1H), 4,35 (s, 2H), 4,29-4,10 (m, 2H), 3,88-3,75 (m, 2H), 3,62 (m, 1H), 2,23 (m, 2H), 0,91 (s, 9H).
<Ejemplo 38>
4-(3-(3-((2.2-dimetilciclopentil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-38)
Etapa 1: Preparac¡ón de 4-(3-(3-((2.2-d¡met¡lc¡clopent¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 2,2-d¡met¡lc¡clopentanona (72 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (99 mg, 39 %).
1H-RMN (MeOD, 400 MHz): 58,35 (m, 1H), 7,93-7,79 (m, 3H), 7,51-7,42 (m, 2H), 7,14 (t, 1H), 4,36(s, 2H), 4,30-4,10 (m, 2H), 3,90-3,68 (m, 3H), 2,43 (m, 1H), 1,95-1,82 (m, 1H), 1,68-1,28 (m, 5H), 1,01 (d, 3H), 0,84 (s, 3H).
<Ejemplo 39>
Etilo____________ 2-((1-(2-fluoro-5-((4-oxo-3.4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)ciclopent-1-enocarboxilato; (I-39)
Etapa 1: Preparac¡ón de etilo 2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)c¡clopent-1-enocarbox¡lato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 2-oxoc¡clopentanocarbox¡lato de et¡lo (85 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (64 mg, 23 %).
1H-RMN (CDCla,400 MHz): 511,44 (s, 1H), 8,48 (m, 1H), 7,80-7,73 (m, 3H), 7,55 (m, 1H), 7,34-7,28 (m, 1H), 7,01 (t, 1H), 4,50 (t, 1H), 4,39-4,27 (m, 4H), 4,15 (m, 2H), 4,01 (m, 2H), 2,52-2,38 (m, 4H), 1,86-1,80 (m, 2H), 1,28 (t, 3H). <Ejemplo 40>
4-(4-fluoro-3-(3-(pentan-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-40)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(pentan-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con pentan-3-ona (48 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (46 mg, 19 %).
1H-RMN (CDCla,400 MHz): 511,32 (s a, 1H), 8,47 (m, 1H), 7,77-7,72 (m, 3H), 7,51 (m, 1H), 7,32-7,31 (m, 1H), 7,00 (t, 1H), 4,39 (m, 1H), 4,29 (s, 2H), 4,17 (m, 1H), 3,85-3,74 (m, 3H), 2,36 (m, 1H), 2,06 (s a, 1H), 1,38 (m, 4H), 0,87 (dd, 6H).
<Ejemplo 41>
4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-41) Etapa 1: Preparación de 4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 3-met¡lbutan-2-ona (61 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (125 mg, 52%).
1H-RMN (CDCla, 400 MHz): 8 11,22 (s, 1H), 8,50-8,45 (m, 1H), 7,79-7,71 (m, 3H), 7,52-7,50 (m, 1H), 7,33-7,29 (m, 1H), 7,01 (t, 1H), 4,38 (m, 1H), 4,29 (s, 2H), 4,21-4,13 (m, 1H), 3,86-3,72 (m, 3H), 2,50-2,40 (m, 1H), 1,66-1,55 (m, 1H), 0,94-0,84 (m, 9H).
<Ejemplo 42>
4-(3-(3-((1-c¡cloprop¡let¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-42)
Etapa 1: Preparac¡ón de 4-(3-(3-((1-c¡cloprop¡let¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 1-c¡cloprop¡letanona (56 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (239 mg, 43 %).
1H-RMN (CDCla, 400 MHz): 8 11,18 (s, 1H), 8,49-8,47 (m, 1H), 7,79-7,72 (m, 3H), 7,52-7,50 (m, 1H), 7,33-7,29 (m, 1H), 7,01 (t, 1H), 4,40 (m, 1H), 4,29 (s, 2H), 3,79 (m, 1H), 1,93 (s a, 1H), 1,83 (m, 1H), 1,14 (m, 3H), 0,70-0,40 (m, 3H), 0,15-0,04 (m, 2H).
<Ejemplo 43>
4-(3-(3-(b¡c¡clor2.2.11heptan-2-¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I -43)
Etapa 1: Preparac¡ón de 4-(3-(3-(b¡c¡clo[2.2.11heptan-2-¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con norcanfor (62 mg, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (48 mg, 19 %).
1H-RMN (MeOD, 400 MHz): 88,37 (d, 1H), 7,92 (d, 1H), 7,82-7,88 (m, 2H), 7,48-7,50 (m, 1H), 7,41-7,43 (m, 1H), 7,14 (t, 1H), 4,38 (s, 2H), 4,28-4,37 (m, 1H), 4,09-4,26 (m, 1H), 3,70-3,91 (m, 2H), 3,61-3,69 (m, 1H), 2,88-2,95 (m, 1H), 2,14-2,23 (m, 2H), 1,73-1,94 (m, 2H), 1,48-1,71 (m, 2H), 1,22-1,40 (m, 4H).
<Ejemplo 44>
4-(3-(3-(sec-but¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-44)
Etapa 1: Preparac¡ón de 4-(3-(3-(sec-but¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con butan-2-ona (51 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (74 mg, 32 %).
1H-RMN (CDCh, 400 MHz): 8 10,58 (s, 1H), 8,49-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,52-7,49 (dd, 1H), 7,33-7,30 (m, 1H), 7,01 (t, 1H), 4,44-4,35 (m, 1H), 4,28 (s, 2H), 4,22-4,14 (m, 1H), 3,86-3,73 (m, 3H), 2,59-2,52 (m, 1H), 1,48-1,24 (m, 3H), 1,02-0,98 (m, 3H), 0,91-0,86 (m, 3H).
<Ejemplo 45>
4-(3-(3-((d¡c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I -45)
Etapa 1: Preparac¡ón de 4-(3-(3-((d¡c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con
diciclopropilmetanona (43 ul, 0,57 mmol) y triacetoxiborohidruro de sodio (247 mg, 1,17 mmol) para producir el compuesto del título (28 mg, 11 %).
1H-RMN (CDCla, 400 MHz): 8 10,63 (s, 1H), 8,50-8,45 (m, 1H), 7,87-7,71 (m, 3H), 7,52-7,50 (m, 1H), 7,33-7,31 (m, 1H), 7,01 (t, 1H), 4,41-4,37 (m, 1H), 4,28 (s, 2H), 4,20-4,16 (m, 1H), 3,99-3,93 (m, 1H), 3,89-3,76 (m, 2H), 1,74 (s a, 1H), 1,08 (t, 1H), 0,85-0,77 (m, 2H), 0,54-0,42 (m, 4H), 0,24-0,18 (m, 2H), 0,08-0,04 (m, 2H).
<Ejemplo 46>
4-(4-fluoro-3-(3-((4-met¡lpentan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I -46)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-((4-met¡lpentan-2-¡l)amino)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con 4-metilpentan-2-ona (57 mg, 0,57 mmol) y triacetoxiborohidruro de sodio (247 mg, 1,17 mmol) para producir el compuesto del título (104 mg, 42 %).
1H-RMN (MeOD, 400 MHz): 88,37-8,35 (m, 1H), 7,95-7,92 (m, 1H), 7,87-7,82 (m, 2H), 7,51-7,49 (m, 1H), 7,45-7,43 (m, 1H), 7,16 (c, 1H), 4,37 (s, 2H), 4,34-4,32 (m, 1H), 4,19-4,17 (m, 1H), 3,84-3,82 (m, 3H), 2,78-2,76 (m, 1H), 1,71 1,68 (m, 1H), 1,32-1,27 (m, 2H), 1,03-0,99 (m, 3H), 0,92-0,86 (m, 6H).
<Ejemplo 47>
4-(4-fluoro-3-(3-((3-h¡drox¡butan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I -47)
Etapa 1 : Preparación de 4-(4-fluoro-3-(3-((3-h¡drox¡butan-2-¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con 3-hidroxibutan-2-ona (49 ul, 0,57 mmol) y triacetoxiborohidruro de sodio (247 mg, 1,17 mmol) para producir el compuesto del título (60 mg, 25 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,36 (m, 1H), 7,94-7,92 (m, 1H), 7,88-7,80 (m, 2H), 7,52-7,49 (m, 1H), 7,46-7,44 (m, 1H), 7,16-7,11 (m, 1H), 4,37 (s, 2H), 4,35-4,32 (m, 1H), 4,19-4,16 (m, 1H), 3,91-3,89 (m, 3H), 2,04-1,99 (m, 2H), 1,15-1,12 (m, 3H), 0,12-0,95 (m, 3H).
<Ejemplo 48>
4-(4-fluoro-3-(3-(pentan-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-48)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(pentan-2-¡lam¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con pentan-2-ona (49 mg, 0,57 mmol) y triacetoxiborohidruro de sodio (247 mg, 1,17 mmol) para producir el compuesto del título (113 mg, 51 %).
1H-RMN (CDCla, 400 MHz): 810,55 (s, 1H), 8,49-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,50 (d, 1H), 7,33-7,29 (m, 1H), 7,01 (t, 1H), 4,42-4,36 (m, 1H), 4,21-4,11 (m, 1H), 3,85-3,72 (m, 3H), 2,66-2,59 (m, 1H), 1,40-1,25 (m, 5H), 1,02-0,98 (m, 3H), 0,92-0,88 (m, 3H).
<Ejemplo 49>
4-(4-fluoro-3-(3-((1-(1-met¡lc¡cloprop¡l)et¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-49) Etapa 1: Preparación de 4-(4-fluoro-3-(3-((1-(1-met¡lc¡cloprop¡l)et¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reaccionó con 1-(1-metilciclopropil)etanona (63 ul, 0,57 mmol) y triacetoxiborohidruro de sodio (247 mg, 1,17 mmol) para producir el compuesto del título (37 mg, 16 %).
1H-RMN (CDCh, 400 MHz): 8 10,22 (s, 1H), 8,48-8,45 (m, 1H), 7,78-7,71 (m, 3H), 7,50-7,49 (m, 1H), 7,33-7,29 (m, 1H), 7,02 (t, 1H), 4,40-4,34 (m, 1H), 4,27 (s, 2H), 4,15 (t, 1H), 3,84-3,73 (m, 3H), 1,85-1,79 (m, 1H), 1,41 (s a, 1H), 1,10-1,07 (dd, 3H), 0,97-0,95 (d, 3H).
<Ejemplo 50>
4-(4-fluoro-3-(3-((3.3.3-tr¡fluoro-2-met¡lprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-50) Etapa 1: Preparación de 4-(4-fluoro-3-(3-((3.3.3-tr¡fluoro-2-met¡lprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo. 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0.57 mmol) reacc¡onó con 3.3.3-tr¡fluoro-2-met¡lpropanal(0.1 ul. 0.57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg. 1.17 mmol) para produc¡r el compuesto del título (137 mg. 52 %).
1H-RMN (MeOD. 400 MHz): 88.26 (d. 1H). 7.72-7.84 (m. 3H). 7.33-7.41 (m. 2H). 7.04 (t. 1H). 4.28 (s. 2H). 4.18 (t.
1H). 4.02-4.05 (m. 1H). 3.71-3.76 (m. 1H). 3.63-3.67 (m. 1H). 3.54-3.58 (m. 1H). 2.65-2.70 (m. 1H). 2.32-2.38 (m. 2H).
1.05-1.07 (m. 3H).
<Ejemplo 51>
4-(3-(3-(al¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-51)
Etapa 1: Preparac¡ón de 4-(3-(3-(al¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo. 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg. 0.57 mmol) reacc¡onó con acr¡laldehído (38 ul. 0.57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg. 1.17 mmol) para produc¡r el compuesto del título (147 mg. 66 %).
1H-RMN (CDCla. 400 MHz): 8 10.02 (s. 1H). 8.40-8.38 (m. 1H). 7.72-7.64 (m. 3H). 7.44-7.42 (m. 1H). 7.25-7.22 (m.
1H). 6.97-6.93 (m. 1H). 5.79 (m. 1H). 5.13-5.03 (m. 2H). 4.30-4.27 (m. 1H). 4.20 (s. 2H). 4.10-4.08 (m. 1H). 3.83-3.79 (m. 1H). 3.72-3.65 (m. 2H). 3.14 (d. 2H).
<Ejemplo 52>
4-(4-fluoro-3-(3-(¡sopent¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-52)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(¡sopent¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo. 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg. 0.57 mmol) reacc¡onó con 3-met¡lbutanal(61 ul. 0.57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg. 1.17 mmol) para produc¡r el compuesto del título (190 mg. 79 %).
1H-RMN (CDCla. 400 MHz): 8 10.14 (s. 1H). 8.47-8.45 (m. 1H). 7.79-7.71 (m. 3H). 7.50 (m. 1H). 7.33-7.29 (m. 1H).
7.02 (t. 1H). 4.36 (t. 1H). 4.27 (s. 2H). 4.18 (t. 1H). 3.88-3.68 (m. 3H). 2.56 (m. 2H). 1.62 (m. 1H). 1.51 (s a. 1H). 1.38 1.35 (m. 2H). 0.90-0.88 (dd. 6H).
<Ejemplo 53>
4-(3-(3-(but¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-53)
Etapa 1: Preparac¡ón de 4-(3-(3-(but¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo. 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg. 0.57 mmol) reacc¡onó con but¡raldehído (51 ul. 0.57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg. 1.17 mmol) para produc¡r el compuesto del título (198 mg. 85 %).
1H-RMN (CDCh. 400 MHz): 8 10.28 (s. 1H). 8.40-8.38 (m. 1H). 7.72-7.66 (m. 1H). 7.44-7.41 (m. 1H). 7.24-7.22 (m.
1H). 6.97-6.92 (m. 1H). 4.30-4.27 (m. 1H). 4.20 (s. 2H). 4.11-4.09 (m. 1H). 3.81-3.79 (m. 1H). 3.72-3.70 (m. 1H). 3.64 3.62 (m. 1H). 2.50-2.46 (m. 2H). 2.02 (s. 1H). 1.40-1.37(m. 2H). 1.30-1.24 (m. 2H). 0.86-0.80 (m. 5H).
<Ejemplo 54>
4-(4-fluoro-3-(3-((3-met¡lbut-2-en-1-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-54)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-((3-met¡lbut-2-en-1-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (20o mg, 0,57 mmol) reacc¡onó con 3-met¡lbut-2-enal(55 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (146 mg, 61 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,79-7,94 (m, 3H), 7,43-7,51 (m, 2H), 7,13 (t, 1H), 5,19 (sa, 1H), 4,37 (s, 2H), 4,26 (t, 1H), 4,13 (t, 1H), 3,83-3,87 (m, 1H), 3,74-3,78 (m, 1H), 3,63-3,66 (m, 1H), 3,11 (d, 2H), 1,71 (s, 3H), 1,64 (s, 3H).
<Ejemplo 55>
4-(3-(3-((c¡clopent¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-55)
Etapa 1: Preparac¡ón de 4-(3-(3-((c¡clopent¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con c¡clopentanocarbaldehído (61 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (178 mg, 72 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,36 (m, 1H), 7,94-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,51-7,43 (m, 2H), 7,17-7,12 (m, 1H), 4,38 (s, 2H), 4,29-4,28 (m,.1H), 4,15-4,13 (m, 1H), 3,88-3,87 (m, 1H), 3,79-3,77 (m, 1H), 3,66-3,64 (m, 1H), 2,45-2,42 (m, 2H), 1,98-1,95 (m, 1H), 1,81-1,79 (m, 2H), 1,65-1,56 (m, 4H), 1,18-1,16 (m, 2H).
<Ejemplo 56>
4-(4-fluoro-3-(3-((4.4.4-tr¡fluorobut¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-56)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((4.4.4-tr¡fluorobut¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 4,4,4-tr¡fluorobutanal(72 mg, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (111 mg, 42%).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,95-7,92 (m, 1H), 7,89-7,82 (m, 2H), 7,52-7,50 (m, 1H), 7,45-7,43 (m, 1H), 7,17-7,12 (m, 1H), 4,37 (s, 2H), 4,30-4,28 (m, 1H), 4,16-4,14 (m, 1H), 3,86-3,83 (m, 1H), 3,78-3,76 (m, 1H), 3.68- 3,66 (m, 1H), 2,58-2,55 (m, 2H), 2,24-2,17 (m, 2H), 1,74-1,68 (m, 2H).
<Ejemplo 57>
4-(4-fluoro-3-(3-(pent¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-57)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(pent¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con pentanal (61 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (200 mg, 83 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,94-7,92 (m, 1H), 7,89-7,82 (m, 2H), 7,51-7,49 (m, 1H), 7,45-7,43 (m, 1H), 7,17-7,12 (m, 1H), 4,37 (s, 2H), 4,30-4,28 (m, 1H), 4,17-4,15 (m, 1H), 3,88-3,86 (m, 1H), 3,80-3,78 (m, 1H), 3.69- 3,67 (m, 1H), 2,53-2,49 (m, 2H), 1,51-1,47 (m, 2H), 1,35-1,29 (m, 4H), 0,91 (t, 3H).
<Ejemplo 58>
4-(3-(3-((2-c¡cloprop¡let¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-58)
Etapa 1: Preparac¡ón de 4-(3-(3-((2-c¡cloprop¡let¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con 2-c¡cloprop¡lacetaldehído (48 mg, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el
compuesto del título (240 mg, 69 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,36 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,52-7,50 (m, 1H), 7,45-7,43 (m, 1H), 7,17-7,15 (m, 1H), 4,38 (s, 2H), 4,30-4,29 (m, 1H), 4,15-4,14 (m, 1H), 3,87-3,86 (m, 1H), 3,78-3,77 (m, 1H), 3,68-3,66 (m, 1H), 2,60-2,56 (m, 2H), 1,39-1,34 (m, 2H), 0,72-0,71 (m, 1H), 0,46-0,42 (m, 2H), 0,06-0,04 (m, 2H). <Ejemplo 59>
4-(4-fluoro-3-(3-(prop¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-59)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(prop¡lamino)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 25 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 0,57 mmol) reacc¡onó con prop¡onaldehído (42 ul, 0,57 mmol) y tr¡acetox¡boroh¡druro de sod¡o (247 mg, 1,17 mmol) para produc¡r el compuesto del título (191 mg, 85%).
1H-RMN (MeOD, 400 MHz): 8 10,39 (s, 1H), 8,47-8,45 (m, 1H), 7,80-7,72 (m, 3H), 7,51-7,48 (m, 1H), 7,33-7,29 (m, 1H), 7,04-6,99 (m, 1H), 4,39-4,34 (m, 1H), 4,27 (s, 2H), 4,19-4,17 (m, 1H), 3,89-3,85 (m, 1H), 3,81-3,77 (m, 1H), 3,71 3,70 (m, 1H), 2,55-2,50 (m, 2H), 1,52-1,47 (m, 2H), 0,94-0,91 (m, 3H).
<Ejemplo 60>
4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-4-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-60)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Ison¡cot¡naldehído (26 ul, 0,28 mmol) se añad¡ó a una soluc¡ón de 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg, 0,28 mmol) en 1,2-d¡cloroetano (1,1ml) y se ag¡tó durante 30m¡n luego ác¡do acét¡co (31 ul, 0,54 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) se añad¡ó a la mezcla de reacc¡ón a 0°C y se ag¡tó durante 12 horas. La mezcla de reacc¡ón se concentró en el vacío, añad¡ó 2N NaOH(ac) y extrajo en d¡clorometano. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4, f¡ltraron, evaporaron al vacío y pur¡f¡caron usando cromatografía en síl¡ce para produc¡r el compuesto ¡ntermed¡o 4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (108 mg,87 %).
Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-4-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona K2CO3(51 mg,0,48 mmol) y yodometano (33 ul,0,48 mmol) se añad¡eron a una soluc¡ón de el compuesto ¡ntermed¡o (etapa 1)(108 mg,0,24 mmol) en DMF(1,5 ml) y se ag¡tó durante 3 horas. La mezcla de reacc¡ón se concentró al vacío, se añad¡ó d¡clorometano y lavó con NH4Cl (ac) saturado y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4 , f¡ltraron, evaporaron al vacío y pur¡f¡caron usando cromatografía en síl¡ce para produc¡r el compuesto del título (99 mg,91 %).
1H-RMN (MeOD, 400 MHz): 88,47 (d, 2H), 8,36 (d, 2H), 7,94 (d, 1H), 7,78-7,91 (m, 2H), 7,40-7,55 (m, 3H), 7,15 (t, 1H), 4,38 (s, 2H), 4,18-4,25 (m, 1H), 3,97-4,14 (m, 2H), 3,80-3,90 (m, 1H), 3,42-3,54 (m, 3H), 2,08(s, 3H).
<Ejemplo 61>
(R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-61) Etapa 1: Preparac¡ón de (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(100 mg, 0,28 mmol) reacc¡onó con ¡son¡cot¡naldehído (26 ul, 0,28 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (106 mg, 83 %). Etapa 2: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(106 mg, 0,23 mmol) reacc¡onó con K2COa(51 mg, 0,48 mmol) y yodometano (33 ul,0,48 mmol) para produc¡r el compuesto del título (98 mg, 91 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 8,51-8,47 (m, 2H), 8,26 (t, 1H), 7,97 (m, 1H), 7,92-7,78 (m, 2H), 7,45-7,33 (m, 3H), 7,24 (m, 2H), 4,32 (s, 2H), 3,76 (m, 1H), 3,51 (m, 3H), 3,30 (s, 2H), 3,08 (m, 1H), 1,92 (s, 3H), 1,81(m, 2H), 1,19 (m, 1H).
<Ejemplo 62>
(S)-4-(4-fluoro-3-(3-(metil(piridin-4-ilmetil)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-62) Etapa 1: Preparación de (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (S)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 2)(100 mg, 0,23 mmol) reacc¡onó con ¡son¡cot¡naldehído (33 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (106 mg, 83 %)). Etapa 2: Preparac¡ón de (S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-4-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(106 mg, 0,24 mmol) reacc¡onó con K2CÜ3(51 mg, 0,48 mmol) y yodometano (33 ul,0,48 mmol) para produc¡r el compuesto del título (98 mg, 91 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 8,51-8,47 (m, 2H), 8,26 (t, 1H), 7,97 (m, 1H), 7,92-7,78 (m, 2H), 7,45-7,33 (m, 3H), 7,24 (m, 2H), 4,32 (s, 2H), 3,76 (m, 1H), 3,51 (m, 3H), 3,30 (s, 2H), 3,08 (m, 1H), 1,92 (s, 3H), 1,81(m, 2H), 1,19 (m, 1H).
<Ejemplo 63>
(S)-4-(4-fluoro-3-(3-(metil(piridin-2-ilmetil)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-63) Etapa 1: Preparac¡ón de (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (S)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 2)(100 mg, 0,23 mmol) reacc¡onó con p¡col¡naldehído (33 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (85 mg, 81 %). Etapa 2: Preparac¡ón de (S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(85 mg, 0,19 mmol) reacc¡onó con K2CO3(51 mg,0,37 mmol) y yodometano (23 ul,0,37 mmol) para produc¡r el compuesto del título (79 mg, 88 %).
1H-RMN (MeOD, 400 MHz): 810,82 (s, 0,4H), 10,40 (s, 0,6H), 8,62 (dd, 1H), 8,46 (m, 1H), 7,75 (m, 3H), 7,52 (m, 1H), 7.37 (m, 2H), 7,17 (m, 1H), 7,0 (m, 1H), 4,15 (s, 2H), 3,96 (m, 1H), 3,89 (m, 1H), 3,74 (d, 1H), 3,63 (m, 2H), 3,46 (m, 1H), 3,41 (m, 1H), 2,21 (s, 2H), 1,93 (m, 3H).
<Ejemplo 64>
(R)-4-(4-fluoro-3-(3-(metil(piridin-2-ilmetil)amino)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-64) Etapa 1: Preparac¡ón de (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(100 mg, 0,23 mmol) reacc¡onó con p¡col¡naldehído (33 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (85 mg, 81 %). Etapa 2: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-2-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(85 mg, 0,19 mmol) reacc¡onó con K2CO3(51 mg, 0,37 mmol) y yodometano (23 ul,0,37 mmol) para produc¡r el compuesto del título (79 mg, 88 %).
1H-RMN (MeOD, 400 MHz): 810,82 (s, 0,4H), 10,40 (s, 0,6H), 8,62 (dd, 1H), 8,46 (m, 1H), 7,75 (m, 3H), 7,52 (m, 1H), 7.37 (m, 2H), 7,17 (m, 1H), 7,0 (m, 1H), 4,15 (s, 2H), 3,96 (m, 1H), 3,89 (m, 1H), 3,74 (d, 1H), 3,63 (m, 2H), 3,46 (m, 1H), 3,41 (m, 1H), 2,21 (s, 2H), 1,93 (m, 3H).
<Ejemplo 65>
4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-2-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-65)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg, 0,28 mmol) reacc¡onó con p¡col¡naldehído (26 ul, 0,28 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o 4-(4-fluoro-3-(3-((p¡r¡d¡n-2-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (108 mg, 87 %). Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-2-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(108mg, 0,24 mmol) reacc¡onó con K2CO3(51 mg, 0,48 mmol) y yodometano (33 ul,0,48 mmol) para produc¡r el compuesto del título (99mg, 91 %).
1H-RMN (MeOD, 400 MHz): 88,47 (d, 1H), 8,35 (d, 1H), 7,94 (d, 1H), 7,78-7,89 (m, 3H), 7,43-7,54 (m, 3H), 7,30-7,35 (m, 1H), 7,14 (t, 1H), 4,38 (s, 2H), 4,17-4,22 (m, 1H), 3,95-4,06 (m, 2H), 3,87-3,94 (m, 1H), 3,57 (s, 2H), 3,44-3,51 (m, 1H), 2,18 (s, 3H).
<Ejemplo 66>
4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1
(2H)-ona; (I-66)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg, 0,28 mmol) reacc¡onó con n¡cot¡naldehído (26 ul, 0,28 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o 4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (108 mg, 87 %). Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(108 mg, 0,24 mmol) reacc¡onó con K2CO3(51 mg, 0,48 mmol) y yodometano (33 ul, 0,48 mmol) para produc¡r el compuesto del título (99 mg, 91 %).
1H-RMN (MeOD, 400 MHz): 88,43-8,51 (m, 2H), 8,36 (d, 1H), 7,94 (d, 1H), 7,79-7,90 (m, 3H), 7,48-7,53 (m, 1H), 7,39 7,48 (m, 2H), 7,15 (t, 1H), 4,38 (s, 2H), 4,18-4,25 (m, 1H), 3,96-4,10 (m, 2H), 3,89-3,96 (m, 1H), 3,39-3,56 (m, 2H), 2,06 (s, 3H).
<Ejemplo 67>
(S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-67) Etapa 1: Preparac¡ón de (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (S)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 2)(100 mg, 0,23 mmol) reacc¡onó con n¡cot¡naldehído (26 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (S)-4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (86 mg, 82 %). Etapa 2: Preparac¡ón de (S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(86 mg, 0,19 mmol) reacc¡onó con K2CO3(52 mg, 0,38 mmol) y yodometano (24 ul, 0,38 mmol) para produc¡r el compuesto del título (98 mg, 91 %).
1H-RMN (MeOD, 400 MHz): 810,36 (s, 0,4H), 10,01 (s, 0,6H), 8,46 (m, 3H), 7,73 (m, 3H), 7,40 (m, 1H), 7,38 (m, 3H), 7,04 (m, 1H), 4,27 (s, 2H), 3,89 (m, 1H), 3,67 (m, 3H), 3,47 (m, 3H), 3,12 (m, 1H), 2,13 (m, 5H), 1,87 (m, 1H).
<Ejemplo 68>
(R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-68) Etapa 1: Preparación de (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, (R)-4-(3-(3-am¡nop¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 1)(100 mg, 0,23 mmol) reacc¡onó con n¡cot¡naldehído (26 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o (R)-4-(4-fluoro-3-(3-((p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (86 mg, 82 %). Etapa 2: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡d¡n-3-¡lmet¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(86 mg, 0,19 mmol) reacc¡onó con K2CO3(52 mg, 0,38 mmol) y yodometano (24 ul, 0,38 mmol) para produc¡r el compuesto del título (98 mg, 91 %).
1H-RMN (MeOD, 400 MHz): 810,36 (s, 0,4H), 10,01 (s, 0,6H), 8,46 (m, 3H), 7,73 (m, 3H), 7,40 (m, 1H), 7,38 (m, 3H), 7,04 (m, 1H), 4,27 (s, 2H), 3,89 (m, 1H), 3,67 (m, 3H), 3,47 (m, 3H), 3,12 (m, 1H), 2,13 (m, 3H), 1,87 (m, 1H).
<Ejemplo 69>
4-(3-(3-(c¡cloprop¡l(met¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-69)
Etapa 1: Preparac¡ón de 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg, 0,23 mmol) reacc¡onó con (1-etox¡c¡clopropox¡)tr¡met¡ls¡lano (46 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto ¡ntermed¡o 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (58 mg, 71 %). Etapa 2: Preparac¡ón de 4-(3-(3-(c¡cloprop¡l(met¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(58mg, 0,16mmol) reacc¡onó con K2CO3(44mg, 0,32mmol) y yodometano (33 ul, 0,32mmol) para produc¡r el compuesto del título (62mg, 95 %).
1H-RMN (MeOD, 400 MHz): 88,32-8,39 (d, 1H), 7,89-7,95 (d, 1H), 7,78-7,88 (m, 2H), 7,46-7,53 (m, 1H), 7,39-7,45 (m, 1H), 7,14 (t, 1H), 4,37 (s, 2H), 4,14-4,22 (m, 1H), 4,05-4,13 (m, 2H), 3,98-4,03 (m, 2H), 3,54-3,63 (m, 1H), 2,26 (s, 3H), 0,34-0,56 (m, 4H).
<Ejemplo 70>
4-(3-(3-(c¡cloprop¡l(et¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-70)
Etapa 1: Preparac¡ón de 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 60(etapa 1) para produc¡r el compuesto ¡ntermed¡o 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (58 mg, 71 %).
Etapa 2: Preparac¡ón de 4-(3-(3-(c¡cloprop¡l(et¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 2). De este modo, este compuesto ¡ntermed¡o (etapa 1)(58 mg, 0,16 mmol) reacc¡onó con K2CO3(44 mg, 0,32 mmol) y yodoetano (33 ul, 0,32 mmol) para produc¡r el compuesto del título (58 mg,91 %).
1H-RMN (MeOD, 400 MHz): 88,32-8,40 (m, 1H), 7,90-7,95 (m, 1H), 7,77-7,89 (m, 2H), 7,46-7,54 (m, 1H), 7,30-7,45 (m, 1H), 7,14 (t, 1H), 4,36 (s, 2H), 3,95-4,24 (m, 4H), 3,73-3,84 (m, 1H), 2,60-2,71 (m, 2H), 1,68-1,78 (m, 1H), 1,19 1,26 (m, 1H), 1,01-1,10 (m, 3H), 0,35-0,58 (m, 4H).
<Ejemplo 71>
4-(3-(3-(c¡clobut¡l(met¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-71)
Etapa 1: Preparac¡ón de 4-(3-(3-(c¡clobut¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 60 (etapa 1). De este modo, 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3)(100 mg, 0,23 mmol) reacc¡onó con c¡clobutanona (17 ul, 0,23 mmol) y tr¡acetox¡boroh¡druro de sod¡o (114 mg, 0,54 mmol) para produc¡r el compuesto
intermedio 4-(3-(3-(cidobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (84 mg, 90 %).
Etapa 2: Preparación de 4-(3-(3-(c¡clobut¡l(met¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 60 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(84 mg, 0,21 mmol) reaccionó con K2CO3(57 mg, 0,41 mmol) y yodometano (25 ul, 0,41 mmol) para producir el compuesto del título (82 mg, 93 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,93 (d, 1H), 7,79-7,89 (m, 2H), 7,48-7,54 (m, 1H), 7,42 (d, 1H), 7,15 (t, 1H), 4,37 (s, 2H), 4,08-4,18 (m, 1H), 3,98-4,04 (m, 1H), 3,89-3,99 (m, 2H), 3,35-3,43 (m, 1H), 2,80-2,90 (m, 1H), 2,06 (s, 3H), 1,81-1,97 (m, 4H), 1,59-1,72 (m, 2H).
<Ejemplo 72>
4-(3-(3-(c¡clopent¡l(prop-2-¡n-1-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-72) Etapa 1: Preparación de 4-(3-(3-(c¡clopent¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 60 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3)(100 mg, 0,23 mmol) reaccionó con ciclopentanona (20 ul, 0,23 mmol) y triacetoxiborohidruro de sodio (114 mg, 0,54 mmol) para producir el compuesto intermedio 4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (74 mg, 77 %).
Etapa 2: Preparación de 4-(3-(3-(c¡clopent¡l(prop-2-¡n-1-¡l)am¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 60 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(74 mg, 0,18 mmol) reaccionó con K2CO3(49 mg, 0,35 mmol) y solución 1 N de bromuro de propargilo en tolueno (0,35 ml, 0,35 mmol) para producir el compuesto del título (67 mg,81 %).
1H-RMN (CDCla,400MHz): 810,32 (s, 1H), 8,48-8,46 (m, 1H), 7,80-7,71 (m, 3H), 7,51-7,48 (m, 1H), 7,33-7,29 (m, 1H), 7,01 (t, 1H), 4,29-4,25 (m, 3H), 4,12-3,99 (m, 3H), 3,81-3,74 (m, 1H), 3,52-3,39 (m, 2H), 2,96-2,89 (m, 1H), 2,16 (m, 1H), 1,83-1,51 (m, 6H), 1,41-1,30 (m, 2H)
<Ejemplo 73>
4-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-73)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
4-(4-fluoro-3-(3-hidroxiazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (ejemplo 6)(200 mg, 0,57 mmol) se disolvió en diclorometano (2,5ml) y 1,4-dioxano (1ml). Periodinano Dess-Martin (DMP, 484 mg, 1,14 mmol) se añadió a 0°C, después de agitar a temperatura ambiente durante 12 horas. La mezcla de reacción se añadió a diclorometano y lavó con hidróxido de sodio acuoso y agua. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (162 mg, 81 %).
Etapa 2: Preparación de 4-(3-(3-(3.3-d¡fluorop¡rrol¡d¡n-1-¡l)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona 3,3-difluoropirrolidina (43 ul, 0,46 mmol) se añadió a una solución de el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) en 1,2-dicloroetano/metanol (2 ml/1 ml) y se agitó durante 30 min luego ácido acético (31 ul, 0,54 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) se añadió a la mezcla de reacción a 0°C y se agitó durante 12 horas. La mezcla de reacción se concentró en el vacío, añadió 2N NaOH(ac) y extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron al vacío y purificaron usando cromatografía en sílice para producir el compuesto del título (134 mg, 66 %).
1H-RMN (DMSO, 400 MHz): 812,62 (s, 1H), 8,26 (d, 1H), 7,99 (d, 1H), 7,90 (t, 1H), 7,84 (t, 1H), 7,45 (d, 2H), 7,23 (t, 1H), 4,33 (s, 2H), 4,11 (m, 1H), 4,07-0,03 (m, 1H), 3,96 (t, 1H), 3,89-3,85 (m, 1H), 3,81-3,78 (m, 1H), 2,90 (t, 2H), 2,69 2,65 (m, 2H), 2,30-2,19 (m, 2H).
<Ejemplo 74>
4-(4-fluoro-3-(3-(4-fluorop¡per¡d¡n-1-¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-74)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto
intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(4-fluorop¡per¡d¡n-1-il)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 4-fluoropiperidina (43 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (145 mg, 72 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 8,26 (d, 1H), 7,99 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,48-7,42 (m, 2H), 7,22 (t, 1H), 4,78-4,60 (m, 1H), 4,33 (s, 2H), 4,05-4,01 (m, 1H), 3,91 (t, 1H), 3,81-3,77 (m, 1H), 3,74-3,70 (m, 1H), 2,41 2,31 (m, 2H), 2,25-2,12 (m, 2H), 1,91-1,77 (m, 2H), 1,75-1,64 (m, 2H).
<Ejemplo 75>
4-(3-(ri.3'-biazetidinal-1'-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-75)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(3-([1.3'-b¡azet¡d¡na1-1'-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con azetidina (28 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (113 mg, 74 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 8,26 (d, 1H), 7,78 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,45 (m, 2H), 7,22 (t, 1H), 4,33 (d, 2H), 4,11 (m, 1H), 3,97 (t, 1H), 3,87 (t, 1H), 3,68 (m, 1H), 3,60 (t, 1H), 3,10 (m, 4H), 1,95 (t, 2H).
<Ejemplo 76>
4-(4-fluoro-3-(3-(pirrolidin-1-il)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-76)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(p¡rrol¡din-1-¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con pirrolidina (38 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (159 mg, 85 %).
1H-RMN (MeOD, 400 MHz): 88,37-8,35 (m, 1H), 7,94 (d, 1H), 7,98 (d, 1H), 7,92-7,82 (m, 2H), 7,48-7,42 (m, 2H), 7,22 (t, 1H), 4,34 (s, 2H), 4,07-4,03 (m, 1H), 3,94 (t, 1H), 3,85-3,81 (m, 1H), 3,76-3,73 (m, 1H), 3,27-3,23 (m, 1H), 2,38 (d, 4H), 1,69 (s, 4H).
<Ejemplo 77>
4-(4-fluoro-3-(3-(piperidin-1-il)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-77)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(p¡per¡din-1-¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con piperidina (43 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (162 mg, 85 %).
1H-RMN (DMSO, 400 MHz): 8 12,62 (s, 1H), 8,25 (d, 1H), 8,01 (d, 1H), 7,88 (t, 1H), 7,82 (t, 1H), 7,48-7,39 (m, 2H), 7,21 (t, 1H), 4,33 (s, 2H), 4,10-4,08 (m, 1H), 3,95 (t, 1H), 3,85 (t, 1H), 3,69-3,65 (m, 1H), 3,61-3,55 (m, 1H), 2,42 (t,
4H), 1,55-1,49 (m, 6H).
<Ejemplo 78>
4-(4-fluoro-3-(3-(4-met¡lp¡peraz¡n-1-¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-78)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(4-met¡lp¡peraz¡n-1-¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 1-metilpiperazina (50 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (90 mg, 45 %).
1H-RMN (MeOD, 400 MHz): 88,37-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,81 (m, 2H), 7,57-7,40 (m, 2H), 7,15 (t, 1H), 4,38 (s, 2H), 4,21-4,02 (m, 2H), 3,97-3,82 (m, 2H), 3,65 (m, 1H), 2,94-2,80 (m, 6H), 2,59 (s, 3H).
<Ejemplo 79>
4-(4-fluoro-3-(3-(fen¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-79)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(fen¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con anilina (42 ul, 0,46 mmol), epóxido de titanio (IV)(Ti[OCH2(CH3)4], 96 ml, 46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,0 mmol) para producir el compuesto del título (43 mg, 22 %).
1H-RMN (DMSO, 400 MHz): 812,60 (s, 1H), 8,25 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,82 (t, 1H), 7,41 (m, 1H), 7,33 (m, 1H), 7,23 (t, 1H), 7,06 (t, 2H), 6,59 (d, 2H), 6,51 (t, 1H), 4,38 (m, 1H), 4,33 (s, 2H), 4,28-4,19(m, 2H), 3,80 (dd, 1H), 3,70 (m, 1H).
<Ejemplo 80>
4-(4-fluoro-3-(3-((1-(tr¡fluoromet¡l)c¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-80) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((1-(tr¡fluoromet¡l)c¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 1-(tr¡fluoromet¡l)c¡cloprop¡lam¡na (74 mg, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (51 mg, 24 %). 1H-RMN (MeOD, 400 MHz): 88,25 (d, 1H), 7,83(d, 1H), 7,71-7,76 (m, 2H), 7,33-7,39(m, 1H), 7,31-7,40 (m, 1H), 7,03 (t, 1H), 4,25 (s, 2H), 4,20-4,26 (m, 1H), 4,01-4,06(m, 1H), 3,66-3,80(m, 3H), 0,97-0,99(m, 2H), 0,81-0,83 (m, 2H). <Ejemplo 81>
4-(4-fluoro-3-(3-(prop-2-¡n-1-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-81)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto
intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-(prop-2-¡n-1-¡lam¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con propargilamina (29 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (134 mg, 66 %).
1H-RMN (CDCl3 , 400 MHz): 8 10,40 (s, 1H), 8,48-8,46 (m, 1H), 7,79-7,71 (m, 3H), 7,52-7,50 (m, 1H), 7,32-7,30 (m, 1H), 7,02 (t, 1H), 4,43-4,38 (m, 1H), 4,28 (s, 2H), 4,23-4,20 (m, 1H), 4,00-3,93 (m, 1H), 3,88-3,85 (m, 2H), 3,42 (c, 2H), 2,21 (m, 1H).
<Ejemplo 82>
(S)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-82) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (S)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (S)-1-metoxipropano-2-amina (48 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (125 mg, 64 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,52-7,50 (m, 1H), 7,45-7,43 (m, 1H), 7,14 (t, 1H), 4,38 (s, 2H), 4,12-4,10 (m, 1H), 4,18-4,15 (m, 1H), 3,84-3,77 (m, 3H), 3,22-3,19 (m, 1H), 2,89 2,87 (m, 1H), 2,03 (s, 3H), 1,00 (t, 3H).
<Ejemplo 83>
(R)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-83) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (R)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (R)-1-metoxipropano-2-amina (48 ul, 0,4 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (125 mg, 64 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,52-7,50 (m, 1H), 7,45-7,43 (m, 1H), 7,14 (t, 1H), 4,38 (s, 2H), 4,12-4,10 (m, 1H), 4,18-4,15 (m, 1H), 3,84-3,77 (m, 3H), 3,22-3,19 (m, 1H), 2,89 2,87 (m, 1H), 2,03 (s, 3H), 1,00 (t, 3H).
<Ejemplo 84>
4-(4-fluoro-3-(3-((1-(h¡drox¡met¡l)c¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-84) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((1-(hidroximetil)ciclopropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (1-aminociclopropil)metanol(40 mg, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (84 mg, 43 %).
1H-RMN (MeOD, 400 MHz): 58,35-8,34 (m, 1H), 7,95-7,82 (m, 3H), 7,51-7,44 (m, 2H), 7,13 (t, 1H), 4,37-4,33 (m, 3H), 4,19-4,17 (m, 1H), 3,90-3,86 (m, 3H), 1,29-1,27 (m, 1H), 0,57-0,54 (m, 2H), 0,52-0,50 (m, 2H).
<Ejemplo 85>
4-(4-fluoro-3-(3-((1-met¡lc¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-85)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((1-met¡lc¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 1-metilciclopropanamina (43 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (35 mg, 19 %).
1H-RMN (MeOD, 400 MHz): 58,36 (d, 1H), 7,93(d, 1H), 7,80-7,86(m, 2H), 7,42-7,52(m, 2H), 7,14(t, 1H), 4,32-4,37(m, 3H), 4,16(t, 1H), 3,78-3,88(m, 3H), 1,29(s, 3H), 0,51-0,57(m, 2H, 0,33-0,39(m, 2H).
<Ejemplo 86>
(R) -4-(3-(3-((3.3-d¡met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-86) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (R)-4-(3-(3-((3,3-d¡met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (R)-3,3-dimetilbutan-2-amina (55 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (110 mg, 55 %).
1H-RMN (MeOD, 400 MHz): 58,39(d, 1H), 7,87-8,38(m, 3H), 7,43-7,53(m, 2H), 7,17(t, 1H), 4,41(s, 2H), 4,28-4,40(m, 1H), 4,09-4,18(m, 1H), 3,71-3,90(m, 3H), 2,17-2,27(m, 1H), 0,94-0,98(m, 12H).
<Ejemplo 87>
(S) -4-(3-(3-((3.3-d¡met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-87) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (S)-4-(3-(3-((3,3-d¡met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (S)-3,3-dimetilbutan-2-amina (55 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (110 mg, 55 %).
1H-RMN (CDCla, 400 MHz): 510,28 (d, 1H), 8,48-8,46 (m, 1H), 7,79-7,70 (m, 3H), 7,50 (d, 1H), 7,32-7,28 (m, 1H), 7,01 (t, 1H), 4,43-4,32 (m, 1H), 4,27 (s, 2H), 4,21-4,09 (m, 1H), 3,84-3,70 (m, 3H), 2,26-2,14 (m, 1H), 0,96-0,90 (m, 3H), 0,88-0,86 (dd, 9H).
<Ejemplo 88>
(R)-4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-88)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto
intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (R)-4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (R)-3-metilbutan-2-amina (53 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (113 mg, 58 %).
1H-RMN (MeOD, 400 MHz): 88,39 (d, 1H), 7,83-8,38 (m, 3H), 7,44-7,52 (m, 2H), 7,17 (t, 1H), 4,63 (sa, 2H), 4,40 (s, 2H), 3,76-3,86 (m, 3H), 2,44-2,46 (m, 1H), 1,64-1,67 (m, 1H), 0,86-0,96 (m, 9H).
<Ejemplo 89>
(S)-4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-89)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de (S)-4-(4-fluoro-3-(3-((3-met¡lbutan-2-¡l)am¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, este compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con (S)-3-metilbutan-2-amina (53 ul, 0,46 mmol) y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (113 mg, 58 %).
1H-RMN (CDCla,400MHz): 810,61 (s, 1H), 8,48-8,46 (m, 1H), 7,80-7,71 (m, 3H), 7,53-7,47 (m, 1H), 7,32-7,30 (m, 1H), 7,01 (t, 1H), 4,38 (m, 1H), 4,28 (s, 2H), 4,15 (m, 1H), 3,84-3,71 (m, 3H), 2,45 (m, 1H), 1,59 (m, 1H), 0,94-0,83 (m, 9H).
<Ejemplo 90>
4-(4-fluoro-3-(3-((1-(metox¡met¡l)c¡cloprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-90) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((1-(metoximetil)ciclopropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 1-(metoximetil)ciclopropanamina (46 mg, 0,46 mmol), y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (78 mg, 39 %) 1H-RMN (MeOD, 400 MHz): 88,37-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,50-7,43 (m, 2H), 7,16-7,13 (m, 1H), 4,37 (s, 2H), 7,36-7,33 (m, 1H), 7,15-7,14 (m, 1H), 3,87-3,82 (m, 3H), 3,31 (s, 3H), 1,29 (s a, 2H), 0,60-0,59 (m, 2H), 0,53-0,51 (m, 2H).
<Ejemplo 91>
4-(3-(3-(but-3-¡n-1-¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-91)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(3-(3-(but-3-¡n-1-¡lam¡no)azet¡din-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con but-3-in-1-amina (38 ul, 0,46 mmol), y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (145 mg, 78 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,35 (m, 1H), 7,94-7,82 (m, 3H), 7,52-7,43 (m, 2H), 7,17-7,14 (m, 1H), 4,37 (s, 2H), 4,32-4,29 (m, 1H), 4,15-4,13 (m, 1H), 3,88-3,69 (m, 3H), 2,69-2,66 (m, 2H), 2,34-2,31 (m, 3H).
<Ejemplo 92>
4-(4-fluoro-3-(3-((2-met¡lal¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-92)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((2-met¡lal¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 2-metilprop-2-en-1-amina (35 ul, 0,46 mmol), y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (83 mg, 45 %).
1H-RMN (MeOD, 400 MHz): 88,38 (d, 1H), 8,36 (d, 1H), 7,81-7,95 (m, 2H), 7,44-7,50 (m, 1H), 7,11-7,17 (m, 1H), 4,38 (sa, 2H), 4,26-4,30 (m, 1H), 4,11-4,13 (m, 1H), 3,85-3,88 (m, 1H), 3,76-3,78 (m, 1H), 3,66-3,69 (m, 1H), 3,34 (s, 2H), 3,08 (s, 2H), 1,25 (s, 3H).
<Ejemplo 93>
4-(4-fluoro-3-(3-((3-h¡drox¡-2.2-d¡met¡lprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-93) Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((3-l'l¡drox¡-2,2-d¡met¡lprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 3-amino-2,2-dimetilpropan-1-ol(47 mg, 0,46 mmol), y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (24 mg, 12%).
1H-RMN (CDCla, 400 MHz): 811,53 (s a, 1H), 8,46 (m, 1H), 7,78-7,72 (m, 3H), 7,51-7,49 (m, 1H), 7,34-7,29 (m, 1H), 7,01 (t, 1H), 4,36-4,32 (m, 3H), 4,18-4,10 (m, 1H), 3,90-3,78 (m, 2H), 3,67 (m, 1H), 2,53-2,45 (m, 5H), 0,93 (s, 3H), 0,91 (s, 3H).
<Ejemplo 94>
1-(((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no) met¡l)c¡clopropanocarbon¡tr¡lo; (I-94)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación_____de_____1-(((1-(2-fluoro-5-((4-oxo-3,4-d¡l'l¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-iDamino^etiDciclopropanocarbonitrilo
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(162 mg, 0,46 mmol) reaccionó con 1-(am¡nomet¡l)c¡clopropancarbon¡tr¡lo (44 mg, 0,46 mmol), y triacetoxiborohidruro de sodio (227 mg, 1,07 mmol) para producir el compuesto del título (91 mg, 46 %). 1H-RMN (MeOD, 400 MHz): 88,38 (dd, 1H), 8,00 (d, 1H), 7,83-7,92 (m, 2H), 7,54-7,62 (m, 2H), 7,14 (t, 1H), 4,41 (s, 2H), 4,28-4,36 (m, 1H), 4,14-4,18 (m, 1H), 3,71-3,81 (m, 3H), 1,35 (s, 4H).
<Ejemplo 95>
4-(4-fluoro-3-(3-((2.2.2-tr¡fluoroet¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-95)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg, 162 %).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((2.2.2-tr¡fluoroet¡l)am¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 73 (etapa 2). De este modo. el compuesto intermedio (etapa 1)(162 mg. 0.46 mmol) reaccionó con 2.2.2-trifluoroetanoamina (36 ul. 0.46 mmol). y triacetoxiborohidruro de sodio (227 mg. 1.07 mmol) para producir el compuesto del título (200 mg. 60 %).
1H-RMN (MeOD. 400 MHz): 88.38-8.35 (m. 1H). 7.95-7.92 (m. 1H). 7.89-7.82 (m. 2H). 7.52-7.50 (m. 1H). 7.45-7.43 (m. 1H). 7.17-7.12 (m. 1H). 4.37 (s. 2H). 4.30-4.28 (m. 1H). 4.16-4.14 (m. 1H). 3.86-3.83 (m. 1H). 3.78-3.76 (m. 1H).
3.68-3.66 (m. 1H). 2.90-2.83 (m. 2H).
<Ejemplo 96>
(R) -4-(4-fluoro-3-(3-(p¡rrol¡d¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-96)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg. 162 %).
Etapa 2: Preparación de (R)-terc-butil-3-((1-(2-fluoro-5-((4-oxo-3.4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-¡l)amino)p¡rrol¡d¡n-1-carbox¡lato
El compuesto intermedio (etapa 1. 162 mg. 0.46 mmol) se añadió a una solución de (R)-terc-butil-3-aminopirrolidin-1-carboxilato (25 ul. 0.46 mmol) en diclorometano (3ml). metanol(1ml) y se agitó durante 30min luego ácido acético (36 ul. 0.69 mmol) y triacetoxiborohidruro de sodio (144 mg. 0.69 mmol) se añadió a la mezcla de reacción a 0°C y se agitó durante 12 horas. La mezcla de reacción se concentró en vacuo. se añadió 2N NaOH(ac) y extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4. filtraron. evaporaron in vacuo y purificaron usando cromatografía en sílice para producir (R)-terc-butil-3-((1-(2-fluoro-5-((4-oxo-3.4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-il)amino)pirrolidin-1-carboxilato (122 mg. 43 %).
Etapa 3: Preparación de (R)-4-(4-fluoro-3-(3-(p¡rrol¡d¡n-3-¡lam¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona El compuesto intermedio (etapa 2)(122 mg. 0.20 mmol) en diclorometano (4ml) enfriado a 0°C se añadió una solución 1.0 N HCl (0.40 ml. 0.40 mmol). y la mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se concentró in vacuo. añadió diclorometano y lavó con 2N NaOH(ac) y agua. Las capas orgánicas combinadas se secaron sobre MgSO4. filtraron. evaporaron in vacuo y purificaron usando cromatografía en sílice para producir el compuesto del título (76 mg. 90 %).
1H-RMN (MeOD. 400 MHz): 88.38-8.36 (m. 1H). 7.95-7.83 (m. 3H). 7.54 (m. 1H). 7.44-7.42 (m. 1H). 7.25 (t. 1H). 4.38 (s. 2H). 4.35-4.33 (m. 1H). 4.19-4.17 (m. 1H). 3.78-3.77 (m. 1H). 3.74-3.71 (m. 2H). 3.36-3.21 (m. 2H). 3.17-3.08 (m.
2H). 2.09-1.99 (m. 1H). 1.82-1.79 (m. 1H). 1.28 (s. 1H).
<Ejemplo 97>
(S) -4-(4-fluoro-3-(3-(p¡rrol¡d¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-97)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-oxoazetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró usando el procedimiento descrito para el ejemplo 73(etapa 1) para producir el compuesto intermedio 4-(4-fluoro-3-(3-oxoazetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (81 mg. 162 %).
Etapa 2: Preparación de (S)-terc-butil-3-((1-(2-fluoro-5-((4-oxo-3.4-dihidroftalazin-1-il)metil)benzoil)azetidne-3-¡l)amino)p¡rrol¡d¡n-1-carbox¡lato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 96 (etapa 2). De este modo. el compuesto intermedio (etapa 1)(162 mg. 0.46 mmol) reaccionó con (S)-terc-butil-3-aminopirrolidin-1-carboxilato (25 ul. 0.46 mmol). y triacetoxiborohidruro de sodio (144 mg. 0.69 mmol) para producir el compuesto del título (122 mg.
43 %).
Etapa 3: Preparación de (S)-4-(4-fluoro-3-(3-(p¡rrol¡d¡n-3-¡lam¡no)azetid¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 96 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(122 mg, 0,20 mmol) reaccionó con solución HCl 1,0 N(0,40 ml, 0,40 mmol)) para producir el compuesto del título (76 mg, 90 %).
1H-RMN (MeOD, 400 MHz): 88,38-8,36 (m, 1H), 7,95-7,83 (m, 3H), 7,54 (m, 1H), 7,44-7,42 (m, 1H), 7,25 (t, 1H), 4,38 (s, 2H), 4,35-4,33 (m, 1H), 4,19-4,17 (m, 1H), 3,78-3,77 (m, 1H), 3,74-3,71 (m, 2H), 3,36-3,21 (m, 2H), 3,17-3,08 (m, 2H), 2,09-1,99 (m, 1H), 1,82-1,79 (m, 1H), 1,28 (s, 1H).
<Ejemplo 98>
1-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)c¡clopentanocarbon¡tr¡lo: (I-98]
Etapa 1: Preparación_____ de____ 1-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-¡l)amino)c¡clopentanocarbon¡tr¡lo
Ciclopentanona (79 ul, 1,17 mmol) se añadió a una solución de 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (200 mg, 1,17 mmol) en 1,2-dicloroetano (6 ml) y se agitó durante 30 min luego ácido acético (67 ul, 1,17 mmol) y cianuro de trimetilsililo (0,30 ml, 2,34 mmol) se añadió a la mezcla de reacción a 0°C y se agitó durante 12 horas. La mezcla de reacción se concentró in vacuo, se añadió 2N NaOH(ac) y extrajo en diclorometano. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron in vacuo y purificaron usando cromatografía en sílice para producir el compuesto del título (328 mg, 63 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,93-7,79 (m, 3H), 7,51-7,43 (m, 2H), 7,14 (t, 1H), 4,43-4,39 (m, 1H), 4,36 (s, 2H), 4,25 (m, 1H), 3,96-3,91 (m, 3H), 2,10-2,02 (m, 2H), 1,86-1,76 (m, 6H).
<Ejemplo 99>
1- ((1-(2-fluoro-5-((4-oxo-3.4-d ¡h ¡ droftalaz ¡ n-1-¡ l)met¡l)benzo¡ l)azet¡ d ¡n-3-¡l)am ¡no)c ¡ clobutanocarbon ¡tr¡ lo; (I-99) Etapa 1: Preparación_____ de_____1-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-¡l)amino)c¡clobutanocarbon¡tr¡lo
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 1,17 mmol) reaccionó con ciclobutanona (87 ul, 1,17 mmol), cianuro de tetrametilsililo (0,30 ml, 2,34 mmol) para producir el compuesto del título (293 mg, 58 %).
1H-RMN (MeOD, 400 MHz): 88,35 (m, 1H), 7,92-7,78 (m, 3H), 7,50-7,43 (m, 2H), 7,13 (t, 1H), 4,41-4,39 (m, 1H), 4,36 (s, 2H), 4,23 (m, 1H), 3,98-3,92 (m, 2H), 3,85-3,78(m, 1H), 2,48-2,41 (m, 2H), 2,20-2,02 (m, 2H).
<Ejemplo 100>
2- ((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)propanon¡tr¡lo: (I-100) Etapa 1: Preparación_____ de_____2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)azetidin-3-¡l)amino)propanon¡tr¡lo
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3) (200 mg, 1,17 mmol) reaccionó con acetaldehído (70 ul, 1,17 mmol), cianuro de tetrametilsililo (0,30 ml, 2,34 mmol) para producir el compuesto del título (336 mg, 71 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,94-7,79 (m, 3H), 7,50-7,44 (m, 2H), 7,14 (t, 1H), 4,41-4,17 (m, 4H), 4,00 3,93 (m, 1H), 3,89-3,66 (m, 3H), 1,42 (d, 3H).
<Ejemplo 101 >
2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)butanon¡tr¡lo: (I-101) Etapa 1: Preparación de 2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)butanon¡tr¡lo Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 3, 200 mg, 1,17 mmol) reaccionó con propionaldehído (84 ul, 1,17 mmol), cianuro de tetrametilsililo (0,30 ml, 2,34 mmol) para producir el compuesto del
título (294 mg, 60 %).
1H-RMN (MeOD, 400 MHz): 88,35 (m, 1H), 7,94-7,80 (m, 3H), 7,49-7,44 (m, 2H), 7,14 (t, 1H), 4,40-4,16 (m, 4H), 4,00 3,91 (m, 1H), 3,88-3,76 (m, 2H), 3,61-3,50 (m, 1H), 1,79-1,68 (m, 2H), 1,08-1,01 (m, 3H).
<Ejemplo 102>
2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)-3-met¡lbutanon¡tr¡lo: (I-102) Etapa 1: Preparación de 2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡din-3-¡l)am¡no)-3-metilbutanonitrilo
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 1,17 mmol) reaccionó con isobutilaldehído (0,1 ml, 1,17 mmol), cianuro de tetrametilsililo (0,30 ml, 2,34 mmol) para producir el compuesto del título (294 mg, 58 %).
1H-RMN (MeOD, 400 MHz): 88,34 (m, 1H), 7,92-7,78 (m, 3H), 7,49-7,44 (m, 2H), 7,13 (t, 1H), 4,39-4,15 (m, 4H), 4,02 3,96 (m, 1H), 3,88-3,76 (m, 2H), 3,46-3,39 (dd, 1H), 1,93 (m, 1H), 1,04-0,87 (m, 6H).
<Ejemplo 103>
2-c¡cloprop¡l-2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)aceton¡tr¡lo: (I-103)
Etapa 1: Preparación de 2-c¡cloprop¡l-2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)amino)aceton¡tr¡lo
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 1,17 mmol) reaccionó con ciclopropancabaldehído (79 ul, 1,17 mmol), cianuro de tetrametilsililo (0,30 ml, 2,34 mmol) para producir el compuesto del título (409 mg, 81 %).
1H-RMN (MeOD, 400 MHz): 88,35 (m, 1H), 7,93-7,78 (m, 3H), 7,50-7,44 (m, 2H), 7,13 (t, 1H), 4,40-4,17 (m, 4H), 4,01 3,79 (m, 3H), 3,42-3,37 (dd, 1H), 1,17 (m, 1H), 0,65-0,42 (m, 4H).
<Ejemplo 104>
4-(4-fluoro-3-(3-((1-(tr¡fluoromet¡l)c¡clobut¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-104) Etapa 1: Preparación de 4-(4-fluoro-3-(3-((1-(tr¡fluoromet¡l)c¡clobut¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 98 (etapa 1). De este modo, 4-(3-(3-aminoazet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (ejemplo 3) (200 mg, 1,17 mmol) reaccionó con ciclobutanona (79 ul, 1,17 mmol), trifluorometiltrimetilsilano (0,30 ml, 2,34 mmol) para producir el compuesto del título (328 mg, 63 %).
1H-RMN (MeOD, 400 MHz): 88,26 (d, 1H), 7,72-7,84 (m, 3H), 7,34-7,41 (m, 2H), 7,04 (t, 1H), 7,71-7,76 (m, 2H), 7,33 7,39 (m, 1H), 7,31-7,40 (m, 1H), 7,03 (t, 1H), 4,25 (s, 2H), 4,20-4,26 (m, 1H), 4,01-4,06 (m, 1H), 3,66-3,80 (m, 3H), 0,97-0,99 (m, 2H), 0,81-0,83 (m, 2H).
<Ejemplo 105>
(S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-105)
Etapa 1: Preparación de (S)-4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)amino)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (S)-N-met¡l-N-(pirrol¡d¡n-3-¡l)p¡r¡m¡d¡n-2-am¡na (200 mg, 1,12 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (188 mg, 1,12 mmol), O-(benzotriazol-1-¡l)-N,N,N',N'-tetrametiluron¡o hexafluorofosfato (HBTU, 553 mg, 1,46 mmol), N,N-diisopropil etilamina (DIPEA, 0,40 ml, 2,24 mmol) para producir (S)-4-fluoro-3-(3-(met¡l(pir¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbonil)benzaldehído (154 mg, 43 %).
Etapa 2: Preparación_______ de_______ (S.Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡np)p¡rrol¡d¡n-1carbonil)benc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(154 mg, 0,47 mmol) reaccionó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-il)fosfonato (114 mg, 0,47 mmol), trietilamina (98 ul, 0,47 mmol) para producir el compuesto intermedio (S,Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡np)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona (107 mg, 51 %).
Etapa 3: Preparación de (S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)1:talaz¡n-1(2H)-ona) Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(107 mg, 0,24 mmol) reaccionó con hidrazina monohidratada (23 ul, 0,48 mmol) para producir el compuesto del título (55 mg, 50 %).
1H-RMN (CDCl3 , 400 MHz): 89,96 (d, 1H), 8,45 (m, 1H), 8,43 (dd, 2H), 7,75 (m, 3H), 7,39 (m, 1H), 7,31 (m, 1H), 7,02 (m, 1H), 6,52 (c, 1H), 5,53 (m, 1H), 4,26 (d, 2H), 3,93 (m, 1H), 3,66 (m, 1H), 3,44 (m, 1H), 3,27 (m, 1H), 3,10 (s, 1,3H), 3,04 (s, 1,7H), 2,21 (m, 1H), 2,06 (m, 1H).
<Ejemplo 106>
(S)-4-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-106)
Etapa 1: Preparación de (S)-4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (S)-N-etil-N^pirrolidin^-i^pirimidin^-amina (200 mg, 1,12 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (175 mg, 1,04 mmol), O-(benzotr¡azol-1-¡l)-N,N,N,N-tetramet¡luron¡o hexafluorofosfato (HBTU, 512 mg, 1,35 mmol), N,N-diisopropil etilamina (DIPEA, 0,36 ml, 2,08 mmol) para producir (S)-4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbonil)benzaldehído (149 mg, 42 %).
Etapa 2: Preparación de (S,Z)-3-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(149 mg, 0,43 mmol) fue d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (105 mg, 0,43 mmol), trietilamina (92 ul, 0,66 mmol) para producir (S,Z)-3-(4-fluoro-3-(3-(etil(pirimidin-2-il)amino)pirrolidin-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona (102 mg, 51 %).
Etapa 3: Preparación de (S)-4-(3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)1:talaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(102 mg, 0,22 mmol) reaccionó con hidrazina monohidratada (21 ul, 0,45 mmol) para producir el compuesto del título (53 mg, 50 %).
1H-RMN (CDCla, 400 MHz): 810,63 (d, 1H), 8,49 (m, 1H), 8,32 (dd, 2H), 7,73 (m, 3H), 7,39 (m, 1H), 7,28 (m, 1H), 7,02 (m, 1H), 6,50 (m, 1H), 5,13 (dt, 1H), 4,29 (d, 2H), 3,95 (m, 1H), 3,58 (m, 2H), 3,46 (m, 2H), 3,27 (m, 1H), 2,23 (m, 1H), 2,09 (m, 1H), 1,93 (dt, 3H).
<Ejemplo 107>
4-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-107)
Etapa 1: Preparación de 4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, N-(azetidin^-i^-N-metilpirimidin^-amina (200 mg, 1,12 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (204 mg, 1,22 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, HBTU, 600 mg, 1,58 mmol), N,N-diisopropil etilamina (DIPEA, 0,42 ml, 2,44 mmol) para producir 4-fluoro-3-(3-(metil(pirimidin-2-il)amino)azetidin-1-carbonil)benzaldehído (149 mg, 42 %).
Etapa 2: Preparación de (Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(202 mg, 0,65 mmol) reaccionó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-il)fosfonato (156 mg, 0,65 mmol), trietilamina (0,13 ml, 0,96 mmol) para producir el compuesto intermedio (Z)-3-(4fluoro-3-(3-(metil(pirimidin-2-il)amino)azetidin-1-carbonil)benciliden)isobenzofuran-1(3H)-ona (169 mg, 61 %).
Etapa 3: Preparación de 4-(4-fluoro-3-(3-(met¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(169 mg, 0,39 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (38 ul, 0,79 mmol) para produc¡r el compuesto del título (89 mg, 51 %).
1H-RMN (CDCla, 400 MHz): 88,53 (dd, 1H), 8,31 (d, 2H), 7,80-7,70 (m, 3H), 7,54 (dd, 1H), 7,34-7,30 (m, 1H), 7,10 7,03 (m, 1H), 6,55 (t, 1H), 5,48-5,41 (m, 1H), 4,52-4,45 (m, 1H), 4,39-4,27 (m, 4H), 4,23-4,12 (m, 1H), 3,21 (s, 3H).
<Ejemplo 108>
4-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-108)
Etapa 1: Preparac¡ón de 4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, N-(azet¡d¡n-3-¡l)-N-et¡lp¡r¡m¡d¡n-2-am¡na (200 mg, 1,12 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (188 mg, 1,12 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 553 mg, 1,45 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,39 ml, 2,24 mmol) para produc¡r 4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benzaldehído (195 mg, 53 %).
Etapa 2: Preparac¡ón de (Z)-3-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(195 mg, 0,59 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (144 mg, 0,59 mmol), tr¡et¡lam¡na (0,12 ml, 0,89 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l) benc¡l¡den)¡sobenzofuran-1(3H)-ona (161 mg, 61 %).
Etapa 3: Preparac¡ón de 4-(4-fluoro-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(161 mg, 0,36 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (36 ul, 0,73 mmol) para produc¡r el compuesto del título (84 mg, 51 %).
1H-RMN (CDCla, 400 MHz): 88,53 (dd, 1H), 8,31 (d, 2H), 7,80-7,70 (m, 3H), 7,54 (dd, 1H), 7,34-7,30 (m, 1H), 7,10 7,03 (m, 1H), 6,55 (t, 1H), 5,09-5,01 (m, 1H), 4,55-4,46 (m, 1H), 4,44-4,28 (m, 4H), 4,20-4,13 (m, 1H), 3,82-3,63 (m, 2H), 1,18 (t, 3H).
<Ejemplo 109>
4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-109)
Etapa 1: Preparac¡ón de 4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, N-(azet¡d¡n-3-¡l)-p¡r¡m¡d¡n-2-am¡na (220 mg, 1,46 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (246 mg, 1,46 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 722 mg, 1,90 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,51 ml, 2,93 mmol) para produc¡r 4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benzaldehído (233 mg, 53 %).
Etapa 2: Preparac¡ón de (Z)-3-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(233 mg, 0,78 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (188 mg, 0,78 mmol), tr¡et¡lam¡na (0,16 ml, 1,16 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (197 mg, 61 %).
Etapa 3: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(197 mg, 0,47 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (46 ul, 0,95 mmol) para produc¡r el compuesto del título (104 mg, 51 %).
1H-RMN (DMSO, 400 MHz): 57,45 (d, 2H), 7,41 (d, 1H), 7,14 (d, 1H), 7,06-6,96 (m, 3H), 6,63-6,62 (m, 2H), 6,40-6,35 (m, 1H), 5,80 (t, 1H), 3,78-3,70 (m, 1H), 3,49-3,44 (m, 3H), 3,40-3,36 (m, 1H), 3,12-3,08 (m, 1H), 3,05-3,01 (m, 1H).
<Ejemplo 110>
(S)-4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-110)
Etapa 1: Preparación de (S)-4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lamino)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (S)-N-(p¡rrol¡d¡n-3-¡l)p¡r¡m¡d¡n-2-am¡na (300 mg, 1,82 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (307 mg, 1,82 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 900 mg, 2,38 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,64 ml, 3,65 mmol) para produc¡r (S)-4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (252 mg, 44 %).
Etapa 2: Preparac¡ón de (S.Z)-3-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(252 mg, 0,80 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (195 mg, 0,80 mmol), tr¡et¡lam¡na (0,17 ml, 1,20 mmol) para produc¡r el compuesto ¡ntermed¡o (S,Z)-3-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (138 mg, 40 %).
Etapa 3: Preparac¡ón de (S)-4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(138 mg, 0,32 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (32 ul, 0,65 mmol) para produc¡r el compuesto del título (73 mg, 51 %).
1H-RMN (CDCla, 400 MHz): 510,74 (d, 1H), 8,46 (m, 1H), 8,32 (dd, 2H), 7,73 (m, 3H), 7,40 (m, 1H), 7,28 (m, 1H), 7,01 (c, 1H), 6,60 (m, 1H), 5,60 (m, 1H), 4,60 (m, 1H), 4,27 (d, 2H), 4,03 (dd, 0,4H), 3,84 (m, 0,6H), 3,73 (m, 1H), 3,49 (m, 1H), 3,41 (m, 1H), 3,25 (m, 1H), 2,28 (m, 1H), 1,95 (m, 1H).
<Ejemplo 111>
(S)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)
ftalaz¡n-1(2H)-ona; (I-111)
Etapa 1: Preparac¡ón de (S)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (S)-6-cloro-N-met¡l-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (120 mg, 0,71 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (118 mg, 0,71 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 348 mg, 0,91 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,25 ml, 1,41 mmol) para produc¡r (S)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (115 mg, 45 %).
Etapa 2: Preparac¡ón_____ de_____ (S,Z)-3-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(115 mg, 0,31 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (77 mg, 0,31 mmol), tr¡et¡lam¡na (66 ul, 0,48 mmol) para produc¡r el compuesto ¡ntermed¡o (S,Z)-3-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona (85 mg, 56 %).
Etapa 3: Preparac¡ón de (S)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(85 mg, 0,18 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (17 ul, 0,36 mmol) para produc¡r el compuesto del título (46 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 510,68 (d, 1H), 8,45 (m, 1H), 7,76 (m, 3H), 7,37 (m, 1H), 7,28 (m, 1H), 6,83 (d, 1H), 7,05 (c, 1H), 6,83 (c, 1H), 5,57 (m, 0,4H), 5,39 (m, 0,6H), 4,29 (d, 2H), 3,92 (m, 1H), 3,66 (m, 2H), 3,50 (m, 1H), 3,26 (m, 1H), 2,91 (s, 1,5H), 2,96 (s, 1,5H), 2,24 (m, 2H).
<Ejemplo 112>
(S)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-112) Etapa 1: Preparación de (S)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (S)-6-cloro-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,01 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (169 mg, 1,01 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 496 mg, 1,31 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,35 ml, 2,01 mmol) para produc¡r (S)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (158 mg, 45 %).
Etapa 2: Preparac¡ón de (S,Z)-3-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(158 mg, 0,45 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (109 mg, 0,45 mmol), tr¡et¡lam¡na (95 ul, 0,68 mmol) para produc¡r el compuesto ¡ntermed¡o (S,Z)-3-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona (117 mg, 56 %).
Etapa 3: Preparac¡ón de (S)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(117 mg, 0,25 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (25 ul, 0,50 mmol) para produc¡r el compuesto del título (64 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,83 (d, 1H), 8,41 (m, 1H), 7,72 (m, 3H), 7,35 (m, 1H), 7,02 (m, 2H), 6,67 (dd, 1H), 5,37 (m, 1H), 4,57 (m, 1H), 4,27 (dd, 2H), 3,77 (m, 1H), 3,43 (m, 2H), 3,23 (m, 2H), 2,02 (m, 1H).
<Ejemplo 113>
(S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-113)
Etapa 1: Preparac¡ón de (S)-4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (S)-N-met¡l-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,22 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (204 mg, 1,22 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 600 mg, 1,58 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA,.42 ml, 2,43 mmol) para produc¡r (S)-4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (172 mg, 45 %).
Etapa 2: Preparac¡ón de (S,Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(172 mg, 0,55 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (132 mg, 0,55 mmol), tr¡et¡lam¡na (0,12 ml, 0,55 mmol) para produc¡r el compuesto ¡ntermed¡o (S,Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (132 mg, 56 %).
Etapa 3: Preparac¡ón de (S)-4-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(132 mg, 0,31 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (30 ul, 0,61 mmol) para produc¡r el compuesto del título (71 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,68 (d, 1H), 8,6 (m, 1H), 8,45 (m, 1H), 7,75 (m, 3H), 7,37 (m, 2H), 7,20 (m, 1H), 7,04 (m, 1H), 6,67 (dd, 1H), 4,65 (m, 2H), 4,27 (d, 2H), 3,91 (m, 2H), 3,48 (m, 1H), 3,22 (m, 1H), 2,40 (m, 1H), 2,02 (m, 1H).
<Ejemplo 114>
4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-114)
Etapa 1: Preparac¡ón de 3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, N
(azetidin-3-il)-6-doropiridazin-3-amina (200 mg, 1,08 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (182 mg, 1,08 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 534 mg, 1,40 mmol), N,N-diisopropil etilamina (DIPEA, 0,38 ml, 2,17 mmol) para producir 3-(3-((6-cloropiridazin-3-il)amino)azetidin-1-carbonil)-4-fluorobenzaldehído (184 mg, 51 %).
Etapa 2: Preparación de (Z)-3-(3-(3-((6-cloropiridazin-3-il)amino)azetidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(184 mg, 0,55 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (133 mg, 0,55 mmol), trietilamina (0,12 ml, 0,83 mmol) para producir el compuesto intermedio (Z)-3-(3-(3-((6-cloropiridazin-3-il)amino)azetidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona (146 mg, 59 %).
Etapa 3: Preparación de 4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(146mg, 0,33mmol) reaccionó con hidrazina monohidratada (32 ul, 0,66mmol) para producir el compuesto del título (82mg, 54 %).
1H-RMN (CDCla, 400 MHz): 8 10,16 (s, 1H), 8,47-8,42 (m, 1H), 7,79-7,67 (m, 4H), 7,47 (dd, 1H), 7,35-7,31 (m, 1H), 7.18 (d, 1H), 7,04-6,99 (m, 1H), 6,72 (d, 1H), 5,29-5,25 (m, 1H), 4,87-4,73 (m, 1H), 4,65-4,59 (m, 1H), 4,47-4,43 (m, 1H), 4,31-4,26 (m, 3H), 4,09-4,05 (m, 1H), 3,92-3,88 (m, 1H).
<Ejemplo 115>
4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)
ftalazin-1(2H)-ona; (I-115)
Etapa 1: Preparación de 3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)amino)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, N-(azetidin-3-il)-6-cloro-N-metilpiridazin-3-amina (200 mg, 1,00 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (169 mg, 1,00 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 496 mg, 1,31 mmol), N,N-diisopropil etilamina (DIPEA, 0,35 ml, 2,01 mmol) para producir 3-(3-((6-cloropiridazin-3-il)(metil)amino)azetidin-1-carbonil)-4-fluorobenzaldehído (179 mg, 51 %).
Etapa 2: Preparación de (Z)-3-(3-(3-((6-cloropiridazin-3-il)(metil)amino)azetidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(179 mg, 0,51 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (124 mg, 0,51 mmol), trietilamina (0,11 ml, 0,77 mmol) para producir el compuesto intermedio (Z)-3-(3-(3-((6-cloropiridazin-3-il)(metil)amino)azetidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona (140 mg, 59 %).
Etapa 3: Preparación de 4-(3-(3-((6-cloropiridazin-3-il)(metil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(140mg, 0,30mmol) reaccionó con hidrazina monohidratada (30 ul, 0,60mmol) para producir el compuesto del título (78mg, 54 %).
1H-RMN (CDCla, 400 MHz): 8 10,16 (s, 1H), 8,47-8,42 (m, 1H), 7,79-7,67 (m, 4H), 7,47 (dd, 1H), 7,35-7,31 (m, 1H), 7.18 (d, 1H), 7,04-6,99 (m, 1H), 6,72 (d, 1H), 5,21-5,15 (m, 1H), 4,87-4,73 (m, 1H), 4,65-4,59 (m, 1H), 4,47-4,43 (m, 1H), 4,31-4,26 (m, 3H), 4,09-4,05 (m, 1H), 3,92-3,88 (m, 1H), 3,06 (s, 3H).
<Ejemplo 116>
4-(3-(3-(c¡clobut¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1
(2H)-ona; (I-116)
Etapa 1: Preparación de 3-(3-(c¡clobut¡l(p¡r¡m¡d¡n-2-¡l)amino)azet¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, N-(azetidin-3-il)-N-ciclobutilpirimidin-2-amina (500 mg, 2,44 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (411 mg, 2,44 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 1,2g, 3,18 mmol), N,N-diisopropil etilamina (DIPEA, 0,86 ml, 4,89 mmol) para producir 3-(3-(ciclobutil(pirimidin-2-il)amino)azetidin-1-carbonil)4-fluorobenzaldehído (442 mg, 51 %).
Etapa 2: Preparación de (Z)-3-(3-(3-(c¡clobut¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(442 mg, 1,24 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (302 mg, 1,24 mmol), tr¡et¡lam¡na (0,26 ml, 1,87 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(3-(3-(c¡clobut¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona (288 mg, 49 %).
Etapa 3: Preparac¡ón de 4-(3-(3-(c¡clobut¡l(p¡r¡m¡d¡n-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(288 mg, 0,62 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (60 ul, 1,22 mmol) para produc¡r el compuesto del título (124 mg, 42 %).
1H-RMN (CDCla, 400 MHz): 88,46-8,44 (m, 1H), 8,28 (d, 2H), 7,75-7,72 (m, 3H), 7,52-7,49 (m, 1H), 7,36-7,28 (m, 1H), 7,04-6,99 (m, 1H), 6,55 (t, 1H), 4,97-4,90 (m, 1H), 4,82-4,74 (m, 1H), 4,66-4,62 (m, 1H), 4,47-4,43 (m, 1H), 4,31-4,26 (m, 3H), 4,17-4,13 (m, 1H), 2,25-2,01 (m, 5H).
<Ejemplo 117>
4-(4-fluoro-3-(3-(piridazin-3-ilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-117)
Etapa 1: Preparac¡ón de 4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, N-(azet¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,33 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (224 mg, 1,33 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 656 mg, 1,73 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,46 ml, 2,66 mmol) para produc¡r 4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benzaldehído (203 mg, 51 %).
Etapa 2: Preparac¡ón de (Z)-3-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(203 mg, 0,68 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (164 mg, 0,68 mmol), tr¡et¡lam¡na (0,14 ml, 1,02 mmol) para produc¡r el compuesto ¡ntermed¡o (Z)-3-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (172 mg, 61 %).
Etapa 3: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(172 mg, 0,41 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (40 ul, 0,82 mmol) para produc¡r el compuesto del título (93 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 8 10,86 (s, 1H), 8,58-8,57 (m, 1H), 8,44 (d, 1H), 7,78-7,69 (m, 3H), 7,48-7,46 (m, 1H), 7,35-7,31 (m, 1H), 7,18-7,15 (m, 1H), 7,00 (t, 1H), 6,70 (d, 1H), 5,68 (sa, 1H), 4,81-4,74 (m, 1H), 4,61-4,42 (m, 2H), 4,26 (s, 2H), 4,11-4,07 (m, 1H), 3,93-3,90 (m, 1H).
<Ejemplo 118>
(R)-4-(3-(3-((6-cloropiridazin-3-il)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-118) Etapa 1: Preparac¡ón de (R)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-6-cloro-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,00 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (169 mg, 1,00 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 496 mg, 1,31 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,35 ml, 2,01 mmol) para produc¡r (R)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (179 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-3-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(179 mg, 0,51 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1il)fosfonato (124 mg, 0,51 mmol), trietilamina (0,1 ml, 0,77 mmol) para producir el compuesto intermedio (R,Z)-3-(3-(3-((6-doropiridazin-3-il)amino)pirrolidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona (146 mg, 61 %).
Etapa 3: Preparación de (R)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(146mg, 0,31 mmol) reaccionó con hidrazina monohidratada (30 ul, 0,31mmol) para producir el compuesto del título (78mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,85 (s, 0,6H), 10,61 (s, 0,4H), 8,43 (m, 1H), 7,79-7,66 (m, 3,5H), 7,32 (m, 1,5H), 7,15 6,98 (m, 2H), 6,69 (d, 0,4H), 6,58 (d, 0,6H), 5,37 (t, 1H), 4,65 (m, 0,4H), 4,58 (m, 0,6H), 4,26 (d, 2H), 3,99-3,83 (m, 1H), 3,79-3,66 (m, 2H), 3,43 (m, 0,5H), 3,25 (dd, 0,5H), 2,32 (m, 1H), 2,04 (m, 1H).
<Ejemplo 119>
(R)-4-(3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)
ftalazin-1(2H)-ona; (I-119)
Etapa 1: Preparación de (R)-3-(3-((6-clorop¡r¡daz¡n-3-¡l)(met¡l)am¡no)pirrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-6-cloro-N-metil-N-(pirrolidin-3-il)piridazin-3-amina (200 mg, 0,94 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (158 mg, 0,94 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 463 mg, 1,22 mmol), N,N-diisopropil etilamina (DIPEA, 0,33 ml, 1,88 mmol) para producir (R)-3-(3-((6-cloropiridazin-3-il)(metil)amino)pirrolidin-1-carbonil)-4-fluorobenzaldehído (174 mg, 51 %).
Etapa 2: Preparación_____ de_____ (R,Z)-3-(3-(3-((6-cloropiridazin-3-il)metil)amino)pirrolidin-1-carbonil)-4-fluorobencil¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(174 mg, 0,48 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (116 mg, 0,48 mmol), trietilamina (0,1 ml, 0,72 mmol) para producir el compuesto intermedio (R,Z)-3-(3-(3-((6-cloropiridazin-3-il)metil)amino)pirrolidin-1-carbonil)-4-fluorobenciliden)isofuran-1(3H)-ona (140 mg, 61 %).
Etapa 3: Preparación de (R)-4-(3-(3-((6-cloropiridazin-3-il)(metil)amino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(140 mg, 0,29 mmol) reaccionó con hidrazina monohidratada (29 ul, 0,59 mmol) para producir el compuesto del título (74 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,42 (s, 0,6H), 10,29 (s, 0,4H), 8,46 (m, 1H), 7,81-7,70 (m, 3H), 7,38 (m, 1H), 7,34-7,24 (m, 1H), 7,22 (d, 1H), 7,03 (m, 1H), 6,82 (dd, 1H), 5,58 (m, 0,5H), 5,41 (m, 0,5H), 4,29 (s, 0,8H), 4,28 (s, 1,2H), 3,92 (m, 1H), 3,74-3,61 (m, 2H), 3,44 (m, 0,5H), 3,26 (dd, 0,5H), 2,99 (s, 1,2H), 2,94 (s, 1,8H), 2,32-2,04 (m, 2H).
<Ejemplo 120>
(R)-4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-120)
Etapa 1: Preparación de (R)-4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lamino)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-N-(pirrolidin-3-il)pirimidin-2-amina (200 mg, 1,22 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (205 mg, 1,22 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 600 mg, 1,58 mmol), N,N-diisopropil etilamina (DIPEA, 0,42 ml, 2,44 mmol) para producir (R)-4-fluoro-3-(3-(pirimidin-2-ilamino)pirrolidin-1-carbonil)benzaldehído (195 mg, 51 %).
Etapa 2: Preparación de (R.Z)-3-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(195 mg, 0,62 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (150 mg, 0,62 mmol), trietilamina (0,13 ml, 0,94 mmol) para producir el compuesto intermedio (R,Z)-3-(4-fluoro-3-(3-(pirimidin-2-ilamino)pirrolidin-1-carbonil)benciliden)isofuran-1(3H)-ona (163 mg, 61 %).
Etapa 3: Preparación de (R)-4-(4-fluoro-3-(3-(p¡r¡m¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(163 mg, 0,38 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (37 ul, 0,76 mmol) para produc¡r el compuesto del título (88 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,73 (s, 0,6H), 10,51 (s, 0,4H), 8,42 (s, 1H), 8,30 (dd, 2H), 7,78-7,68 (m, 3H), 7,41 (m, 1H), 7,28 (m, 1H), 7,03 (c, 1H), 6,59 (m, 1H), 5,57 (dd, 1H), 4,63 (m, 0,4H), 4,53 (m, 0,6H), 4,27 (s, 0,8H), 4,26 (s, 1,2H), 4,02 (dd, 0,4H), 3,88-3,68 (m, 1,6H), 3,62 (dd, 0,4H), 3,52-3,40 (m, 1H), 3,26 (d, 0,6H), 2,37-2,24 (m, 1H), 2,08 1,95 (m, 1H).
<Ejemplo 121 >
(R)-4-(3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1
(2H)-ona; (I-121)
Etapa 1: Preparac¡ón de (R)-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-et¡l-N-(p¡rrol¡d¡n-3-¡l)p¡r¡m¡d¡n-2-am¡na (220 mg, 1,14 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (158 mg, 0,94 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 564 mg, 1,49 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,40 ml, 2,29 mmol) para produc¡r (R)-3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (200 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-3-(3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(200 mg, 0,58 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (141 mg, 0,58 mmol), tr¡et¡lam¡na (0,12 ml, 0,87 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona (163 mg, 61 %).
Etapa 3: Preparac¡ón de (R)-4-(3-(3-(et¡l(p¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(163 mg, 0,36 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (34 ul, 0,72 mmol) para produc¡r el compuesto del título (87 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,43 (s, 0,5H), 10,38 (s, 0,5H), 8,47 (m, 1H), 8,30 (m, 2H), 7,81-7,68 (m, 3H), 7,39 (dd, 1H), 7,28 (m, 1H), 7,03 (m, 1H), 6,51 (m, 1H), 5,37-5,18 (m, 1H), 4,29 (s, 0,8H), 4,27 (s, 1,2H), 4,02-3,90 (m, 1H), 3,67 3,53 (m, 2H), 3,52-3,37 (m, 2,5H), 3,27 (t, 0,5H), 2,22 (m, 1H), 2,13 (m, 1H), 1,22 (t, 1,5H), 1,16 (t, 1,5H).
<Ejemplo 122>
(R)-4-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-122)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,34 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (225 mg, 1,34 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 660 mg, 1,74 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,47 ml, 2,68 mmol) para produc¡r (R)-4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (214 mg, 51 %).
Etapa 2: Preparac¡ón de (R.Z)-3-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(214 mg, 0,68 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (165 mg, 0,68 mmol), tr¡et¡lam¡na (0,14 ml, 1,02 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (179 mg, 61 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(p¡r¡daz¡n-3-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(179 mg, 0,42 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (41 ul, 0,84 mmol) para
producir el compuesto del título (97 mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,88 (s, 0,6H), 10,49 (s, 0,4H), 8,58 (m, 1H), 8,44 (m, 1H), 7,74 (m, 3H), 7,39-7,25 (m, 2H), 7,17 (m, 1H), 7,02 (m, 1H), 6,71 (d, 0,4H), 6,63 (d, 0,6H), 5,30 (m, 0,4H), 5,00 (m, 0,6H), 4,28 (s, 0,8H), 4,26 (s, 1,2H), 4,02 (m, 0,6H), 3,87 (m, 1,4H), 3,79-3,64 (m, 1H), 3,52-3,42 (m, 0,5H), 3,25 (m, 0,5H), 2,43-2,26 (m, 1H), 2,07 (m, 1H).
<Ejemplo 123>
(R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1
(2H)-ona; (I-123)
Etapa 1: Preparación de (R)-4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)amino)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-met¡l-N-(p¡rrol¡d¡n-3-¡l)p¡r¡daz¡n-3-am¡na (200 mg, 1,12 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (188 mg, 1,12 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 553 mg, 1,46 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,39 ml, 2,24 mmol) para produc¡r (R)-4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (187 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(187 mg, 0,57 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (138 mg, 0,57 mmol), tr¡et¡lam¡na (0,12 ml, 0,86 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (155 mg, 61 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(met¡l(p¡r¡daz¡n-3-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(155 mg, 0,35 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (34 ul, 0,70 mmol) para produc¡r el compuesto del título (83mg, 52 %).
1H-RMN (CDCla, 400 MHz): 810,70 (s, 0,6H), 10,25 (s, 0,4H), 8,61 (m, 1H), 8,46 (m, 1H), 7,81-7,70 (m, 3H), 7,39 (m, 1H), 7,33-7,21 (m, 2H), 7,08-6,97 (m, 1H), 6,81 (m, 1H), 5,74 (m, 0,4H), 5,52 (m, 0,6H), 4,29 (s, 0,8H), 4,27 (s, 1,2H), 3,92 (m, 1H), 3,75-3,63 (m, 2H), 3,52-3,39 (m, 0,4H), 3,24 (dd, 0,6H), 2,99 (s, 1,2H), 2,94 (s, 1,8H), 2,25 (m, 1H), 2,08 (m, 1H).
<Ejemplo 124>
(R)-4-(4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-124)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-(p¡rrol¡d¡n-3-¡l)t¡azol-2-am¡na (200 mg, 1,18 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (198 mg, 1,18 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 582 mg, 1,54 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,42 ml, 2,36 mmol) para produc¡r (R)-4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (170 mg, 45 %).
Etapa 2: Preparac¡ón de (R.Z)-3-(4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(170 mg, 0,53 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (128 mg, 0,53 mmol), tr¡et¡lam¡na (0,11 ml, 0,80 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (125 mg, 54 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(t¡azol-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(125 mg, 0,29 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (28 ul, 0,57 mmol) para produc¡r el compuesto del título (67 mg, 52 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,85 (m, 3H), 7,39 (m, 2H), 7,15 (m, 1H), 6,96 (dd, 1H), 6,58 (dd, 1H), 4,41
(m, 2H), 3,71 (m, 3H), 3,40 (m, 1H), 3,21 (m, 1H), 2,29 (m, 1H), 2,04 (m, 1H).
<Ejemplo 125>
(R)-4-(3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-125) Etapa 1: Preparación de (R)-3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)pirrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-5-et¡n¡l-N-(p¡rrol¡d¡n-3-¡l)p¡r¡m¡d¡n-2-am¡na (230 mg, 1,22 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (205 mg, 1,22 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 602 mg, 1,59 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,43 ml, 2,44 mmol) para produc¡r (R)-3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (210 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-3-(3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 2)(210 mg, 0,62 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (150 mg, 0,62 mmol), tr¡et¡lam¡na (0,13 ml, 0,93 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l¡den)¡sofuran-1(3H)-ona (164 mg, 58 %).
Etapa 3: Preparac¡ón de (R)-4-(3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(164 mg, 0,36 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (36 ul, 0,72 mmol) para produc¡r el compuesto del título (103mg, 61 %).
1H-RMN (CDCla, 400 MHz): 8 10,10-.93 (dd, 1H), 8,41-8,38 (m, 3H), 7,78-7,68 (m, 3H), 7,41-7,37 (m, 1H), 7,32-7,27 (m, 1H), 7,04-7,02 (m, 1H), 5,48-5,44 (m, 1H), 4,66-4,51 (m, 1H), 4,27 (s, 2H), 4,03 (m, 0,5H), 3,85-3,39 (m, 3H), 3,28 3,26 (m, 0,5H), 3,19 (m, 2H), 2,40-2,23 (m, 1H), 2,17-1,93 (m, 1H).
<Ejemplo 126>
(R)-2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo: (I-126)
Etapa 1: Preparac¡ón de (R)-2-((1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-2-(p¡rrol¡d¡n-3-¡lam¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo (230 mg, 1,22 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (205 mg, 1,22 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 602 mg, 1,59 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,43 ml, 2,44 mmol) para produc¡r (R)-3-(3-((5-et¡n¡lp¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído (210 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-2-((1-(2-fluoro-5-((3-oxo¡sofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(210 mg, 0,63mol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (150 mg, 0,63 mmol), tr¡et¡lam¡na (0,13 ml, 0,93 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-2-((1-(2-fluoro-5-((3-oxo¡sofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo (164 mg, 58 %).
Etapa 3: Preparac¡ón de (R)-2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)p¡r¡m¡d¡n-5-carbon¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(164 mg, 0,36 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (164 mg, 0,36 mmol) para produc¡r el compuesto del título (103 mg, 61 %).
1H-RMN (CDCla, 400 MHz): 89,93-9,81 (m, 1H), 8,50-8,43 (m, 2H), 7,79-7,62 (m, 3H), 7,41-7,27 (m, 3H), 7,07-6,99 (m, 1H), 5,83-5,76 (m, 1H), 4,71-4,55 (m, 1H), 4,27 (d, 2H), 4,05-4,00 (m, 0,5H), 3,88-3,62 (m, 2H), 3,52-3,44 (m, 1H), 3,29-3,25 (m, 0,5H), 2,42-2,28 (m, 1H), 2,09-1,96 (m, 1H).
<Ejemplo 127>
(R)-2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrolid¡n-3-¡l)
amino)nicotinonitrilo; (I-127)
Etapa 1: Preparación de (R)-2-((1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)n¡cot¡non¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-2-(p¡rrol¡d¡n-3-¡lam¡no)n¡cot¡non¡tr¡lo (300 mg, 1,59 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (205 mg, 1,22 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 785 mg, 2,07 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,55 ml, 3,18 mmol) para produc¡r (R)-2-((1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)n¡cot¡non¡tr¡lo (275 mg, 51 %).
Etapa 2: Preparac¡ón de (R,Z)-2-((1-(2-fluoro-5-((3-oxo¡sofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)n¡cot¡non¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(275 mg, 0,81 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (196 mg, 0,81 mmol), tr¡et¡lam¡na (1,7 ml, 1,22 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-2-((1-(2-fluoro-5-((3-oxo¡sofuran-1(3H)-¡l¡den)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)n¡cot¡non¡tr¡lo (155 mg, 42 %).
Etapa 3: Preparac¡ón de (R)-2-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)n¡cot¡non¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(155mg, 0,34mmol) reacc¡onó con h¡draz¡na monoh¡dratada (34 ul, 0,68mmol) para produc¡r el compuesto del título (78 mg, 49 %).
1H-RMN (DMSO, 400 MHz): 812,59 (d, 1H), 8,28 (m, 2H), 7,85 (m, 4H), 7,38 (m, 2H), 7,22 (m, 2H), 6,72 (m, 1H), 4,57 (m, 1H), 4,32 (d, 2H), 3,75 (m, 1H), 3,46 (m, 2H), 3,21 (m, 1H), 2,10 (m, 2H).
<Ejemplo 128>
(R)-4-(4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalazin-1(2H)-ona; (I-128)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-(p¡rrol¡d¡n-3-¡l)p¡r¡d¡n-2-am¡na (250 mg, 0,92 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (154 mg, 0,92 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 453 mg, 1,19 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,32 ml, 1,83 mmol) para produc¡r (R)-4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (178 mg, 62 %).
Etapa 2: Preparac¡ón de (R.Z)-3-(4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(178 mg, 0,57 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (138 mg, 0,57 mmol), tr¡et¡lam¡na (0,12 ml, 0,85 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (144 mg, 59 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(p¡r¡d¡n-2-¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(144 mg, 0,34 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (33 ul, 0,67 mmol) para produc¡r el compuesto del título (91 mg, 61 %).
1H-RMN (MeOD, 400 MHz): 88,35-8,31(m, 1H), 7,95-7,93(m, 1H), 7,88-7,81(m, 2H), 7,76-7,73(m, 1H), 7,45-7,39(m, 3H), 7,34-7,32(m, 1H), 7,15-7,08(m, 2H), 4,67-4,30 (m, 1H), 4,38-4,31 (d, 2H), 3,98-3,12 (m, 5H), 2,35-2,19 (m, 1H), 2,06-1,88 (m, 1H).
<Ejemplo 129>
(R)-2-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrolid¡n-3-¡l)
amino)-N,N-dimetilnicotinamida; (I-129)
Etapa 1: Preparac¡ón de (R)-2-((1-(2-fluoro-5-form¡lbenzo¡l)p¡rrol¡d¡n-3-¡l)am¡no)-N,N-d¡met¡ln¡cot¡nam¡da
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N,N
dimetil-2-(pirrolidin-3-ilamino)nicotinamida (320 mg, 1,36 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (300 mg, 1,36 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 673 mg, 1,77 mmol), N,N-diisopropil etilamina (DIPEA, 0,48 ml, 2,73 mmol) para producir (R)-2-((1-(2-fluoro-5-formilbenzoil)pirrolidin-3-il)amino)-N,N-dimetilnicotinamida (220 mg, 42 %).
Etapa 2: Preparación de (R,Z)-2-((1-(2-fluoro-5-((3-oxoisofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)amino)-N,N-dimetilnicotinamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(220 mg, 0,57 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (139 mg, 0,57 mmol), trietilamina (0,2 ml, 0,86 mmol) para producir el compuesto intermedio (R,Z)-2-((1-(2-fluoro-5-((3-oxoisofuran-1(3H)-iliden)metil)benzoil)pirrolidin-3-il)amino)-N,N-dimetilnicotinamida (109 mg, 38 %). Etapa 3: Preparación de (R)-2-((1-(2-fluoro-5-((4-oxo-3,4-dihidroftalazin-1-il)metil)benzoil)pirrolidin-3-il)amino)-N,N-dimetilnicotinamida
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(109 mg, 0,22 mmol) reaccionó con hidrazina monohidratada (22 ul, 0,44 mmol) para producir el compuesto del título (39 mg, 35 %).
1H-RMN (MeOD, 400 MHz): 88,36 (m, 1H), 7,88 (m, 4H), 7,44 (m, 3H), 7,13 (m, 1H), 6,69 (m, 1H), 4,56 (m, 1H), 4,36 (d, 2H), 3,85 (m, 1H), 3,55 (m, 2H), 3,21 (m, 1H), 3,02 (s, 6H), 2,30 (m, 1H), 2,07 (m, 1H).
<Ejemplo 130>
(R)-4-(3-(3-((5-bromop¡r¡m¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)
ftalazin-1(2H)-ona; (I-130)
Etapa 1: Preparación de (R)-3-(3-((5-bromop¡r¡m¡d¡n-2-¡l)am¡no)pirrol¡d¡n-1-carbon¡l)-4-fluorobenzaldehído
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-5-bromo-N-(pirrolidin-3-il)pirimidin-2-amina (320 mg, 1,32 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (221 mg, 1,32 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 649 mg, 1,71 mmol), N,N-diisopropil etilamina (DIPEA, 0,46 ml, 2,63 mmol) para producir (R)-3-(3-((5-bromopirimidin-2-il)amino)pirrolidin-1-carbonil)-4-fluorobenzaldehído (274 mg, 53 %).
Etapa 2: Preparación de (R,Z)-3-(4-fluoro-3-(3-((5-bromopirimidin-2-il)aminopirrolidin-1-carbonil)benciliden)isofuran-1(3H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 2). De este modo, el compuesto intermedio (etapa 1)(274 mg, 0,70 mmol) reaccionó con dimetil(3-oxo-1,3-dihidroisobenzofuran-1-il)fosfonato (169 mg, 0,70 mmol), trietilamina (0,15 ml, 1,04 mmol) para producir el compuesto intermedio (R,Z)-3-(4-fluoro-3-(3-((5-bromopirimidin-2-il)aminopirrolidin-1-carbonil)benciliden)isofuran-1(3H)-ona (217 mg, 61 %).
Etapa 3: Preparación de (R)-4-(4-fluoro-3-(3-((5-bromopirimidin-2-il)aminopirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 3). De este modo, el compuesto intermedio (etapa 2)(217 mg, 0,43 mmol) reaccionó con hidrazina monohidratada (45 ul, 0,86 mmol) para producir el compuesto del título (131 mg, 59 %).
1H-RMN (CDCla, 400 MHz): 89,93-9,81 (m, 1H), 8,50-8,43 (m, 2H), 7,79-7,62 (m, 3H), 7,41-7,27 (m, 3H), 7,07-6,99 (m, 1H), 5,83-5,76 (m, 1H), 4,71-4,55 (m, 1H), 4,27 (d, 2H), 4,05-4,00 (m, 0,5H), 3,88-3,62 (m, 2H), 3,52-3,44 (m, 1H), 3,29-3,25 (m, 0,5H), 2,42-2,28 (m, 1H), 2,09-1,96 (m, 1H).
<Ejemplo 131 >
(R)-4-(4-fluoro-3-(3-((5-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)
ftalaz¡n-1(2H)-ona; (I-131)
Etapa 1: Preparación de (R)-4-fluoro-3-(3-((5-(tr¡fluoromet¡lp¡r¡d¡n-2-¡l)aminop¡rrol¡d¡n-1-carbon¡l)benzaldehído Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 11 (etapa 1). De este modo, (R)-N-(pirrolidin-3-il)-5-(trifluorometil-2-amina (200 mg, 0,86 mmol) reaccionó con ácido 2-fluoro-5-formilbenzoico (145 mg, 0,86 mmol), O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio hexafluorofosfato (HBTU, 426 mg, 1,12 mmol), N,N-diisopropil etilamina (DIPEA, 0,31 ml, 1,72 mmol) para producir (R)-4-fluoro-3-(3-((5-(trifluorometilpiridin-2il)aminopirrolidin-1-carbonil)benzaldehído (174 mg, 53 %).
Etapa 2: Preparación______de______(R.Z)-3-(4-fluoro-3-(3-((5-(tr¡fluoromet¡lp¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbonil)benc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(174 mg. 0.46 mmol) reacc¡onó con d¡met¡l(3-oxo-1.3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (111 mg. 0.46 mmol). tr¡et¡lam¡na (96 ul. 0.69 mmol) para produc¡r el compuesto ¡ntermed¡o R.Z)-3-(4-fluoro-3-(3-((5-(tr¡fluoromet¡lp¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (139 mg. 61 %). Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-((5-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo. el compuesto ¡ntermed¡o (etapa 2)(139mg. 0.28mmol) reacc¡onó con h¡draz¡na monoh¡dratada (28 ul. 0.56mmol) para produc¡r el compuesto del título (85mg. 59 %).
1H-RMN (DMSO. 400 MHz): 812.59 (d. 1H). 8.29 (m. 2H). 7.85 (m. 3H). 7.62 (m. 2H). 7.39 (m. 2H). 7.20 (m. 1H). 6.58 (m. 1H). 4.38 (m. 1H). 4.32 (d. 2H). 3.75 (m. 1H). 3.47 (m. 2H). 3.21 (m. 1H). 2.20 (m. 1H). 1.92 (m. 1H).
<Ejemplo 132>
(R)-4-(4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)
ftalazin-1(2H)-ona; (I-132)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡np)p¡rrol¡d¡n-1-carbon¡l)benzaldehído Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo. (R)-N-(p¡rrol¡d¡n-3-¡l)-4-(tr¡fluoromet¡l)p¡r¡d¡n-2-am¡na (200 mg. 0.86 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (145 mg. 0.86 mmol). O-(benzotr¡azol-1-¡l)-N,N,N'.N'-tetramet¡luron¡o hexafluorofosfato (HBTU. 426 mg. 1.12 mmol). N,N-d¡¡soprop¡l et¡lam¡na (DIPEA. 0.31 ml. 1.72 mmol) para produc¡r (R)-4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡np)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (174 mg. 53 %).
Etapa 2: Preparac¡ón______de_____ (R.Z)-3-(4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(174 mg. 0.46 mmol) reacc¡onó con d¡met¡l(3-oxo-1.3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (111 mg. 0.46 mmol). tr¡et¡lam¡na (96 ul. 0.69 mmol) para produc¡r el compuesto ¡ntermed¡o (R.Z)-3-(4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sofuran-1(3H)-ona (139 mg. 61 %). Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-((4-(tr¡fluoromet¡l)p¡r¡d¡n-2-¡l)am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo. el compuesto ¡ntermed¡o (etapa 2)(139mg. 0.28mmol) reacc¡onó con h¡draz¡na monoh¡dratada (28 ul. 0.56mmol) para produc¡r el compuesto del título (85mg. 59 %).
1H-RMN (DMSO. 400 MHz): 812.59 (d. 1H). 8.29 (m. 2H). 7.85 (m. 3H). 7.62 (m. 2H). 7.39 (m. 2H). 7.20 (m. 1H). 6.58 (m. 1H). 4.38 (m. 1H). 4.32 (d. 2H). 3.75 (m. 1H). 3.47 (m. 2H). 3.21 (m. 1H). 2.20 (m. 1H). 1.92 (m. 1H).
<Ejemplo 133>
4-(3-(3-(benc¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona; (I-133)
Etapa 1: Preparac¡ón de 4-(3-(3-(benc¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Bromuro de benc¡lo (0.10 ml. 0.86 mmol) y K2CO3(119 mg. 0.86 mmol) se añad¡ó a una suspens¡ón del ejemplo 3 producto (150 mg. 0.43 mmol) en d¡met¡lformam¡da (DMF. 8ml) y se ag¡tó durante 8 horas a 80°C. La reacc¡ón de enfr¡ó a temperatura amb¡ente y se concentró al vacío. añad¡ó d¡clorometano y se lavó una soluc¡ón de b¡carbonato de sod¡o y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4. f¡ltraron. evaporaron ¡n vacuo y pur¡f¡caron usando cromatografía en síl¡ce para produc¡r el compuesto del título (86 mg. 45 %).
1H-RMN (CDCla. 400 MHz): 8 10.17 (s. 1H). 8.47-8.46 (m. 1H). 7.82-7.72 (m. 3H). 7.54-7.50 (m. 1H). 7.37-7.31 (m.
5H). 7.06-7.01 (m. 1H). 4.37-4.32 (m. 1H). 4.29 (s. 2H). 4.19-4.16 (m. 1H). 3.89-3.73 (m. 5H).
<Ejemplo 134>
4-(4-fluoro-3-(3-((3.3.3-trifluoropropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-134)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-((3.3.3-tr¡fluoroprop¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 133 (etapa 1). De este modo. 4-(3-(3-am¡noazet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (150 mg. 0.43 mmol) reacc¡onó con 3-bromo-1.1.1-tr¡fluoroprapan(0,25 ml. 0.86 mmol). K2CO3(119 mg. 0.86 mmol) para produc¡r el compuesto del título (98 mg.51 %).
1H-RMN (MeOD. 400 MHz): 88.38-8.35 (m. 1H). 7.95-7.92 (m. 1H). 7.89-7.82 (m. 2H). 7.52-7.50 (m. 1H). 7.45-7.43 (m. 1H). 7.17-7.12 (m. 1H). 4.37 (s. 2H). 4.30-4.28 (m. 1H). 4.16-4.14 (m. 1H). 3.86-3.83 (m. 1H). 3.78-3.76 (m. 1H).
3.68-3.66 (m. 1H). 2.48-2.42 (m. 2H). 1.92-1.85 (m. 2H).
<Ejemplo 135>
(R)-4-(3-(3-(azetidin-1-il)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-135)
Etapa 1: Preparac¡ón de (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡lmetansulfonato El compuesto ¡ntermed¡o del ejemplo 5 (S)-4-(4-fluoro-3-(3-h¡drox¡p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (539 mg. 1.25 mmol) en d¡clorometano (7ml) enfr¡ado a 0°C se añad¡ó tr¡et¡lam¡na (0.1 ml. 1.50 mmol). cloruro de metanosulfon¡lo (MsCl. 0.11 ml. 1.38 mmol). y la mezcla se ag¡tó a temperatura amb¡ente durante 3 horas. La mezcla de reacc¡ón se concentró ¡n vacuo. añad¡ó d¡clorometano y lavó con b¡carbonato de sod¡o acuoso y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4. f¡ltraron. evaporaron ¡n vacuo para produc¡r (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡lmetanosulfonato (434 mg. 78 %).
Etapa 2: Preparac¡ón de (R)-4-(3-(3-(azet¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
El compuesto ¡ntermed¡o (etapa 1)(434 mg. 0.97 mmol) en d¡met¡lformam¡da (DMF. 5ml) se añad¡ó azet¡d¡na (0.20 ml.
2.93 mmol). LhCO3(216 mg. 2.93 mmol) y la mezcla se ag¡tó a 80C durante 16 horas. La mezcla de reacc¡ón se concentró ¡n vacuo. añad¡ó d¡clorometano y lavó con cloruro de amon¡o acuoso y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4. f¡ltraron. evaporaron ¡n vacuo y pur¡f¡caron usando cromatografía en síl¡ce para produc¡r el compuesto del título (117 mg.45 %).
1H-RMN (DMSO. 400 MHz): 812.61 (s. 1H). 8.26 (d. 1H). 7.96 (t. 1H). 7.91-7.80(m. 2H). 7.46-7.35 (m. 1H). 7.25-7.18 (m. 1H). 4.33 (s. 2H). 3.73-3.58 (m. 1H). 3.44-3.39 (m. 1H). 3.27-3.19 (m. 1H). 3.13-3.08 (m. 1H). 2.98 (t. 1H). 2.86 (d.
1H). 2.77-2.61 (m. 1H). 2.16 (t. 2H). 2.03 (t. 2H). 1.77-1.62 (m. 2H).
<Ejemplo 136>
(R)-4-(3-(r1.3'-b¡p¡rrol¡d¡na1-1'-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-136)
Etapa 1: Preparac¡ón de (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡lmetansulfonato El compuesto ¡ntermed¡o (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 135(etapa 1) con un rend¡m¡ento de 78 %(434mg).
Etapa 2: Preparac¡ón de (R)-4-(3-([1.3'-b¡p¡rrol¡d¡na1-1'-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 135 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(434 mg. 0.97 mmol) reacc¡onó con p¡rrol¡d¡na (0.22 ml. 2.93 mmol). LhCO3(216 mg.
2.93 mmol) para produc¡r el compuesto del título (130 mg.32 %).
1H-RMN (MeOD. 400 MHz): 88.35 (d. 1H). 7.93 (d. 1H). 7.90-7.79 (m. 2H). 7.54-7.44 (m. 1H). 7.42-7.37 (m. 1H). 7.18 7.11 (m. 1H). 4.37 (s. 2H). 3.85-3.74 (m. 1H). 2.65 (t. 2H). 2.57-2.50 (m. 1H). 2.40 (t. 1H). 2.23-2.07 (m. 1H). 1.97-1.90 (m. 1H). 1.85 (m. 2H). 1.76 (m. 2H).
<Ejemplo 137>
(R)-4-(4-fluoro-3-(3-(piperidin-1-il)pirrolidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-137)
Etapa 1: Preparac¡ón de (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡lmetansulfonato El compuesto ¡ntermed¡o (S)-1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)p¡rrol¡d¡n-3-¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 135(etapa 1) con un rend¡m¡ento de 78 %(434mg).
Etapa 2: Preparación de (R)-4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 135 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(434 mg, 0,97 mmol) reacc¡onó con p¡per¡d¡na (0,10 ml, 2,93 mmol), L¡2CO3(216 mg, 2,93 mmol) para produc¡r el compuesto del título (88 mg,21 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 8,26 (d, 1H), 7,97 (d, 1H), 7,91-7,80 (m, 2H), 7,46-7,39 (m, 1H), 7,38-7,35 (m, 1H), 7,25-7,19 (m, 1H), 4,32 (s, 2H), 3,75-3,59 (m, 1H), 3,39-3,36 (m, 0,5H), 3,29-3,12 (m, 2H), 2,97 (t, 0,5H), 2,83 2,71 (m, 1H), 2,42-2,28 (m, 3H), 2,15-1,98 (m, 2H), 1,73-1,63 (m, 1H), 1,51-1,29 (m, 6H).
<Ejemplo 138>
(R)-4-(4-fluoro-3-(3-(p-tol¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-138)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-(p-tol¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-4-fluoro-3-(3-(p-tol¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (200 mg, 1,13 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (190 mg, 1,13 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 559 mg, 1,48 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,40 ml, 2,27 mmol) para produc¡r (R)-4-fluoro-3-(3-(ptolyam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (230 mg, 62 %).
Etapa 2: Preparac¡ón de (R.Z)-3-(4-fluoro-3-(3-(p-tol¡am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(230 mg, 0,70 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (170 mg, 0,70 mmol), tr¡et¡lam¡na (0,15 ml, 1,10 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(p-tol¡am¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona (190 mg, 61 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(p-tol¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(190 mg, 0,43 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (42 ul, 0,86 mmol) para produc¡r el compuesto del título (129 mg, 66 %).
1H-RMN (DMSO, 400 MHz): 812,59 (d, 1H), 8,26 (m, 1H), 7,85 (m, 3H), 7,60 (m, 1H), 7,36 (m, 1H), 7,20 (m, 1H), 6,88 (dd, 2H), 6,48 (dd, 2H), 5,60 (m, 1H), 4,32 (d, 2H), 3,91 (m, 2H), 3,52 (m, 2H), 3,11 (m, 1H), 2,14 (m, 1H), 2,12 (s, 3H), 1,90 (m, 1H).
<Ejemplo 139>
(R)-4-(4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-139)
Etapa 1: Preparac¡ón de (R)-4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 1). De este modo, (R)-N-fen¡lp¡rrol¡d¡n-3-am¡na (200 mg, 1,23 mmol) reacc¡onó con ác¡do 2-fluoro-5-form¡lbenzo¡co (207 mg, 1,23 mmol), O-(benzotr¡azol-1-¡l)-N,N,N',N'-tetramet¡luron¡o hexafluorofosfato (HBTU, 607 mg, 1,60 mmol), N,N-d¡¡soprop¡l et¡lam¡na (DIPEA, 0,43 ml, 2,46 mmol) para produc¡r (R)-4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benzaldehído (238 mg, 62 %).
Etapa 2: Preparac¡ón de (R.Z)-3-(4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(238 mg, 0,76 mmol) reacc¡onó con d¡met¡l(3-oxo-1,3-d¡h¡dro¡sobenzofuran-1-¡l)fosfonato (185 mg, 0,76 mmol), tr¡et¡lam¡na (0,16 ml, 1,14 mmol) para produc¡r el compuesto ¡ntermed¡o (R,Z)-3-(4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l¡den)¡sobenzofuran-1(3H)-ona (200 mg, 61 %).
Etapa 3: Preparac¡ón de (R)-4-(4-fluoro-3-(3-(fen¡lam¡no)p¡rrol¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 11 (etapa 3). De este modo, el compuesto ¡ntermed¡o (etapa 2)(200mg, 0,47 mmol) reacc¡onó con h¡draz¡na monoh¡dratada (46 ul, 0,94mmol) para produc¡r el compuesto del título (136mg, 66 %).
1H-RMN (MeOD, 400 MHz): 88,39-8,34 (m, 1H), 7,97-7,70 (m, 3H), 7,50-7,33 (m, 2H), 7,19-7,07 (m, 3H), 6,70-6,57(m,
3H), 4,40 (s, 1H), 4,33 (s, 1H), 4,17-4,05 (m, 1H), 3,95-3,79 (m, 1H), 3,72-3,47 (m, 2H), 3,41-3,16 (m, 1H), 2,37-2,20 (m, 1H), 2,09-1,91 (m, 1H).
<Ejemplo 140>
4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-140)
Etapa 1: Preparación de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)metil)benzo¡l)azet¡d¡n-3-¡l)met¡lmetansulfonato El compuesto ¡ntermed¡o (ejemplo 7) 4-(4-fluoro-3-(3-(h¡drox¡met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (460 mg, 1,25 mmol) en d¡clorometano (7ml) enfr¡ado a 0°C se añad¡ó tr¡et¡lam¡na (0,1 ml, 1,50 mmol), cloruro de metanosulfon¡lo (MsCl, 0,11 ml, 1,38 mmol), y la mezcla se ag¡tó a temperatura amb¡ente durante 3 horas. La mezcla de reacc¡ón se concentró ¡n vacuo, añad¡ó d¡clorometano y lavó con b¡carbonato de sod¡o acuoso y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4, f¡ltraron, evaporaron ¡n vacuo para produc¡r (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡lmetansulfonato (417 mg, 75 %).
Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
El compuesto ¡ntermed¡o (etapa 1)( 417 mg, 0,94 mmol) en 1,4-d¡oxano (5 ml) se añad¡ó p¡rrol¡d¡na (0,27 ml, 4,00 mmol) y la mezcla se ag¡tó a 80°C durante 16 horas. La mezcla de reacc¡ón se concentró ¡n vacuo, añad¡ó d¡clorometano y lavó con cloruro de amon¡o acuoso y agua. Las capas orgán¡cas comb¡nadas se secaron sobre MgSO4 , f¡ltraron, evaporaron ¡n vacuo y pur¡f¡caron usando cromatografía en síl¡ce para produc¡r el compuesto del título (138 mg,35 %).
1H-RMN (CDCla, 400 MHz): 89,85 (s, 1H), 8,46-8,44 (m, 1H), 7,78-7,70 (m, 3H), 7,49-7,47 (m, 1H), 7,32-7,27 (m, 1H), 7,03-6,98 (m, 1H), 4,29-4,24 (m, 3H), 4,14-4,10 (t, 1H), 3,85-3,77 (m, 2H), 2,91-2,64 (m, 3H), 2,48 (s, 4H), 1,77 (s, 4H).
<Ejemplo 141 >
4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona: (I-141)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con p¡per¡d¡na (0,44 ml, 4,00 mmol) para produc¡r el compuesto del título (138 mg, 35 %).
1H-RMN (CDCla, 400 MHz): 89,94 (s, 1H), 8,46 (m, 1H), 7,78-7,70 (m, 3H), 7,50-7,48 (m, 1H), 7,31-7,27 (m, 1H), 7,01 (t, 1H), 4,27-4,23 (t, 1H), 4,26 (s, 2H), 7,13-4,09 (t, 1H), 3,83-3,70 (m, 2H), 2,88-2,84 (m, 1H), 2,57-2,55 (m, 2H), 2,32 (s, 4H), 1,54 (s, 4H), 1,42 (s, 2H).
<Ejemplo 142>
4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona: (I-142)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con azet¡d¡na (0,28 ml, 4,00 mmol) para produc¡r compuesto del título (154 mg, 41 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,94 (d, 1H), 7,84 (m, 2H), 7,50 (m, 2H), 7,13 (t, 1H), 4,38 (s, 2H), 4,19 (t, 1H), 4,09 (m, 1H), 3,74 (m, 2H), 3,30 (m, 4H), 2,69 (m, 3H), 2,14 (t, 2H).
<Ejemplo 143>
4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-143)
Etapa 1: Preparación de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(3-(3-((c¡cloprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con c¡cloprop¡lam¡na (0.28 ml. 4.00 mmol) para produc¡r el compuesto del título (206 mg. 54 %).
1H-RMN (CDCla. 400 MHz): 8 10.03 (s. 1H). 8.47-8.45 (m. 1H). 7.80-7.71 (m. 3H). 7.52-7.48 (m. 1H). 7.32-7.28 (m.
1H). 7.01 (t. 1H). 4.27 (s. 2H). 4.26-4.22 (m. 1H). 4.10 (t. 1H). 3.85-3.81 (m. 1H). 3.73-3.70 (m. 1H). 2.99-2.88 (m. 2H).
2.81-2.72 (m. 1H). 2.07 (m. 1H). 1.57 (s a. 1H). 0.45-0.41 (m. 2H). 0.29-0.26 (m. 2H).
<Ejemplo 144>
4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-144)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-((¡soprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con ¡soprop¡lam¡na (0.34 ml. 4.00 mmol) para produc¡r el compuesto del título (234 mg. 61 %).
1H-RMN (CDCla. 400 MHz): 810.20 (s a. 1H). 8.47-8.45 (m. 1H). 7.80-7.70 (m. 3H). 7.51-7.49 (dd. 1H). 7.32-7.28 (m.
1H). 7.01 (t. 1H). 4.27 (s. 2H). 4.28-4.23 (m. 1H). 4.12 (t. 1H). 3.84-3.81 (dd. 1H). 3.75-3.71 (dd. 1H). 2.88-2.69 (m.
4H). 1.05-1.03 (dd. 6H).
<Ejemplo 145>
4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1
(2H)-ona; (I-145)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(3-(3-(((c¡cloprop¡lmet¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con c¡cloprop¡lmet¡lam¡na (0.34 ml. 4.00 mmol) para produc¡r el compuesto del título (189 mg. 48 %).
1H-RMN (CDCh. 400 MHz): 810.36 (s a. 1H). 8.47-8.45 (m. 1H). 7.80-7.71 (m. 3H). 7.51-7.49 (m. 1H). 7.32-7.28 (m.
1H). 7.01 (t. 1H). 4.27-4.24 (m. 3H). 4.13 (t. 1H). 3.86-3.75 (m. 2H). 2.94-2.75 (m. 3H). 2.48-2.46 (dd. 2H). 1.61 (s a.
1H). 0.95-0.91 (m. 1H). 0.51-0.46 (m. 2H). 0.12-0.09 (m. 2H).
<Ejemplo 146>
4-(4-fluoro-3-(3-((isobutilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-146)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg).
Etapa 2: Preparación de 4-(4-fluoro-3-(3-((¡sobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con 2-met¡lpropan-1-am¡na (0,40 ml, 4,00 mmol) para produc¡r el compuesto del título (207 mg, 52 %).
1H-RMN (CDCl3 , 400 MHz): 810,69 (s a, 1H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,50-7,48 (m, 1H), 7,32-7,28 (m, 1H), 7,00 (t, 1H), 4,27-4,23 (m, 3H), 4,14 (t, 1H), 3,86-3,78 (m, 2H), 2,93-2,78 (m, 3H), 2,45 (s, 1H), 2,44 (s, 1H), 1,81 1,71 (m, 1H), 0,91 (s, 3H), 0,89 (s, 3H).
<Ejemplo 147>
4-(3-(3-((terc-butilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-147)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(3-(3-((terc-but¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con 2-met¡lpropan-2-am¡na (0,42 ml, 4,00 mmol) para produc¡r el compuesto del título (71 mg, 18 %).
1H-RMN (CDCla, 400 MHz): 810,49 (s a, 1H), 8,47-8,45 (m, 1H), 7,80-7,72 (m, 3H), 7,52-7,48 (m, 1H), 7,31-7,28 (m, 1H), 7,01 (t, 1H), 4,27-4,22 (m, 3H), 4,14 (t, 1H), 2,90-2,75 (m, 3H), 1,14 (s, 9H).
<Ejemplo 148>
4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona; (I-148)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(3-(3-((c¡clobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con c¡clobut¡lam¡na (0,43 ml, 4,00 mmol) para produc¡r el compuesto del título (138 mg, 35 %).
1H-RMN (CDCla, 400 MHz): 810,33 (s a, 1H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,50-7,48 (m, 1H), 7,31-7,28 (m, 1H), 7,00 (t, 1H), 4,27-4,22 (m, 3H), 4,11 (t, 1H), 3,83-3,72 (m, 2H), 3,23 (m, 1H), 2,83-2,72 (m, 3H), 2,23-2,17 (m, 2H), 1,71-1,62 (m, 5H).
<Ejemplo 149>
4-(4-fluoro-3-(3-((prop-2-in-1-ilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona; (I-149)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de4-(4-fluoro-3-(3-((prop-2-¡n-1-¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo, el compuesto ¡ntermed¡o (etapa 1)(417 mg, 0,94 mmol) reacc¡onó con proparg¡lam¡na (0,27 ml, 4,00 mmol) para produc¡r el compuesto del título (163 mg, 43 %).
1H-RMN (CDCh, 400 MHz): 8 10,61 (s, 1H), 8,48-8,46 (m, 1H), 7,80-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32-7,28 (m, 1H), 7,01 (t, 1H), 4,28-4,24 (m, 3H), 4,12 (t, 1H), 3,88-3,76 (m, 2H), 3,43 (m, 2H), 3,00-2,87 (m, 2H), 2,78-2,72 (m, 1H), 2,27-2,23 (m, 1H), 1,45 (s a, 1H).
<Ejemplo 150>
4-(4-fluoro-3-(3-((fen¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona; (I-150)
Etapa 1: Preparación de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 4-(4-fluoro-3-(3-((fen¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con an¡l¡na (0.36 ml. 4.00 mmol) para produc¡r compuesto del título (45 mg. 11 %).
1H-RMN (CDCla. 400 MHz): 8 11.25 (s. 1H). 8.48-8.46 (m. 1H). 7.77-7.70 (m. 3H). 7.51-7.49 (dd. 1H). 7.33-7.29 (m.
1H). 7.17 (t. 2H). 7.00 (t. 1H). 6.72 (t. 1H). 6.59 (m. 2H). 4.31-4.27 (m. 3H). 4.17-4.09 (m. 1H). 3.39-3.78 (m. 2H). 3.41 3.31 (m. 2H). 2.95-2.89 (m. 1H).
<Ejemplo 151 >
1-((((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l) am¡no)met¡l)c¡clopropanocarbon¡tr¡lo; (I-151)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón de 1-((((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l)am¡no)met¡l)c¡clopropanocarbon¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con 1-(am¡nomet¡l)c¡clopropancarbon¡tr¡lo (384 mg.
4.00 mmol) para produc¡r el compuesto del título (121 mg. 29 %).
1H-RMN (MeOD. 400 MHz): 88.36(d. 1H). 7.82-7.95(m. 3H). 7.44-7.46(m. 2H). 7.13(t. 1H). 4.37(s. 2H). 4.21(t. 1H).
4.10(t. 1H). 3.74-3.85(m. 2H). 2.80-2.87(m. 3H). 2.68-2.70(m. 2H). 1.20 (s. 2H). 0.95(s. 2H).
<Ejemplo 152>
1-(((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l) am¡no)c¡clopropanocarbon¡tr¡lo; (I-152)
Etapa 1: Preparac¡ón de (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato El compuesto ¡ntermed¡o (1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l metansulfonato se elaboró usando el proced¡m¡ento descr¡to para el ejemplo 140(etapa 1) con un rend¡m¡ento de 75 %(417mg). Etapa 2: Preparac¡ón_____de_____1-(((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)met¡l)am¡no)c¡clopropanocarbon¡tr¡lo
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 140 (etapa 2). De este modo. el compuesto ¡ntermed¡o (etapa 1)(417 mg. 0.94 mmol) reacc¡onó con 1-am¡noc¡clopropancarbon¡tr¡lo (474 mg. 4.00 mmol) para produc¡r compuesto del título (44 mg. 11 %).
1H-RMN (CDCla. 400 MHz): 8 10.39 (s. 1H). 8.48-8.46 (m. 1H). 7.77-7.72 (m. 3H). 7.53-7.49 (m. 1H). 7.35-7.29 (m.
1H). 7.02 (t. 1H). 4.28 (s. 2H). 4.27-4.09 (m. 2H). 3.86-3.69 (m. 2H). 2.75 (m. 1H). 1.90 (m. 1H). 1.64 (s. 2H). 1.23 (m.
2H). 1.00 (m. 2H).
<Ejemplo 153>
4-(3-(3-((c¡cloprop¡l(prop-2-¡n-1-¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)
ftalaz¡n-1(2H)-ona; (I-153)
Etapa 1: Preparac¡ón de 4-(3-(3-((c¡cloprop¡l(prop-2-¡n-1-¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n
1(2H)-ona
4-(3-(3-((cidopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (ejemplo 144)(126 mg, 0,52 mmol) en dimetilformamida (DMF, 3ml) se añadió Na2CO3(63 mg, 1,56 mmol), 3-bromoprop-1-in (0,16 ml, 2,60 mmol) y la mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla de reacción se concentró in vacuo, añadió diclorometano y lavó con cloruro de amonio acuoso y agua. Las capas orgánicas combinadas se secaron sobre MgSO4 , filtraron, evaporaron in vacuo y purificaron usando cromatografía en sílice para producir el compuesto del título (190 mg,82 %).
1H-RMN (CDCl3 , 400 MHz): 8 11,08 (s, 1H), 8,49-8,47 (m, 1H), 7,79-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32-7,29 (m, 1H), 7,01 (t, 1H), 4,29 (s, 2H), 4,26-4,05 (m, 2H), 3,83-3,68 (m, 2H), 3,40 (m, 2H), 2,96-2,87 (m, 3H), 2,23 (m, 1H),
1,98-1,95 (m, 1H), 0,53-0,48 (m, 2H), 0,34-0,31 (m, 2H).
<Ejemplo 154>
4-(3-(3-((ciclopropil(metil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1
(2H)-ona; (I-154)
Etapa 1: Preparación de 4-(3-(3-((c¡cloprop¡l(met¡l)am¡no)met¡l)azet¡din-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 153 (etapa 1). De este modo, el compuesto intermedio (ejemplo 144) 4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)-ftalazin-1(2H)-ona (150 mg, 0,34 mmol) reaccionó con Na2CO3(78 mg, 0,74 mmol) y yoduro de metilo (46 ul, 0,74 mmol) para producir el compuesto del título (121 mg,85 %).
1H-RMN (CDCla, 400 MHz): 8 10,91 (s, 1H), 8,49-8,47 (m, 1H), 7,79-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32-7,28 (m, 1H), 7,00 (t, 1H), 4,28 (s, 2H), 4,26-4,04 (m, 2H), 3,81-3,66 (m, 2H), 2,96-2,87 (m, 1H), 2,81-2,74 (m, 3H), 1H), 0,49-0,41 (m, 2H), 0,34-0,26 (m, 2H).
<Ejemplo 155>
4-(3-(3-((ciclopropil(etil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1
(2H)-ona; (I-155)
Etapa 1: Preparación de 4-(3-(3-((c¡cloprop¡l(et¡l)am¡no)met¡l)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 153 (etapa 1). De este modo, el compuesto intermedio (ejemplo 144) 4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)-ftalazin-1(2H)-ona (150 mg, 0,34 mmol) reaccionó con Na2CO3(78 mg, 0,74 mmol) y yoduro de etilo (49 ul, 0,74 mmol) para producir el compuesto del título (113 mg,77 %).
1H-RMN (CDCla, 400 MHz): 8 10,60 (s, 1H), 8,48-8,46 (m, 1H), 7,79-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32-7,28 (m,
1H), 7,01 (t, 1H), 4,28 (s, 2H), 4,26-4,04 (m, 2H), 3,81-3,66 (m, 2H), 2,96-2,87 (m, 1H), 2,81-2,74 (m, 2H), 2,65-2,60
(m, 2H), 1,64 (m, 1H), 1,03 (m, 1H), 0,49-0,41 (m, 2H), 0,34-0,26 (m, 2H).
<Ejemplo 156>
4-(3-(3-(ciclobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-156)
Etapa 1: Preparación de 4-(3-(3-(c¡clobut¡lam¡no)azetid¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona clorhidrato
El compuesto intermedio (ejemplo 25) 4-(3-(3-(ciclobutilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona
(100 mg, 0,25 mmol) en diclorometano (3ml) enfriado a 0°C se añadió solución 1 N de ácido clorhídrico (0,50 ml, 0,50 mmol). Después de agitar a temperatura ambiente durante 1 hora, los disolventes se evaporaron para producir el compuesto del título (102 mg, 93 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 9,23 (d, 2H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,50
7,21 (m, 2H), 7,21 (t, 1H), 4,33 (s, 2H), 4,10 (t, 1H), 3,97 (t, 1H), 3,67-3,51 (m, 3H), 3,08 (m, 1H), 2,76 (m, 1H), 1,99 (t,
2H), 1,69-1,46(m, 4H).
<Ejemplo 157>
4-(3-(3-(ciclopropilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-157)
Etapa 1: Preparación de 4-(3-(3-(c¡cloprop¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 26) 4-(3-(3-(ciclopropilciclopropilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (100 mg, 0,25 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,50 ml, 0,50 mmol) para producir el compuesto del título (97 mg, 91 %).
1H-RMN (MeOD, 400 MHz): 88,36 (d, 1H), 7,93 (d, 1H), 7,79-7,89 (m, 2H), 7,48-7,54 (m, 1H), 7,42 (d, 1H), 7,15 (t, 1H), 4,00-4,05 (m, 2H), 3,75-3,80 (m, 2H), 3,34-3,39 (m, 1H), 3,12 (s, 2H), 1,32-1,36 (m, 1H), 0,64-0,72 (m, 2H), 0,41 0,46 (m, 2H).
<Ejemplo 158>
4-(3-(3-(c¡clopent¡lam¡no)azet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-158)
Etapa 1: Preparación de 4-(3-(3-(c¡clopent¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 27) 4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (117 mg, 0,24 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,48 ml, 0,48 mmol) para producir el compuesto del título (99 mg, 90 %).
1H-RMN (DMSO, 400 MHz): 8 12,60 (s, 1H), 9,23 (d, 2H), 8,26 (d, 1H), 7,97 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,44 7,50 (m, 1H), 7,40 (d, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,13 (t,.1H), 4,00 (t, 1H), 3,54-3,70 (m, 3H), 2,82-2,91 (m, 1H), 1,52-1,67 (m, 4H), 1,43 (sa, 2H), 1,10-1,27 (m, 2H).
<Ejemplo 159>
4-(4-fluoro-3-(3-(¡soprop¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-159)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(¡soprop¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorhidrato Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 32) 4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (100 mg, 0,25 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,50 ml, 0,50 mmol) para producir el compuesto del título (102 mg, 95 %).
1H-RMN (DMSO, 400 MHz): 812,61 (s, 1H), 9,23 (d, 2H), 8,27-8,25 (m, 1H), 7,97 (d, 1H), 7,91-7,81 (m, 2H), 7,48-7,44 (m, 1H), 7,41-7,39 (m, 1H), 7,21 (t, 1H), 4,33 (s, 2H), 4,15-4,10 (m, 2H), 3,66-3,59 (m, 3H), 3,17 (d, 1H), 2,68-2,61 (m, 1H), 0,92-0,83 (m, 6H).
<Ejemplo 160>
4-(3-(3-((c¡cloprop¡lmet¡l)am¡no)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona clorhidrato; (I-160)
Etapa 1: Preparación de 4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-onaclorhidrato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 33) 4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (100 mg, 0,25 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,50 ml, 0,50 mmol) para producir el compuesto del título (98 mg, 89 %).
1H-RMN (DMSO, 400 MHz): 8 12,61 (s, 1H), 9,23 (d, 2H), 8,28-7,97 (m, 2H), 7,91-7,81 (m, 2H), 7,48-7,40 (m, 2H), 7,22 (m, 1H), 4,33 (s, 2H), 4,13-3,98 (m, 2H), 3,70-3,56 (m, 3H), 2,29 (m, 2H), 0,83-0,72 (m, 1H), 0,37-0,32 (m, 2H), 0,06 (m, 2H).
<Ejemplo 161 >
4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-161)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-(¡sobut¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorhidrato Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 35) 4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (100 mg, 0,24 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,48 ml, 0,48 mmol) para producir el compuesto del título (101 mg, 95 %).
1H-RMN (MeOD, 400 MHz): 58 ,38 -8 ,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,51-7,49 (m, 1H), 7,45-7,43 (m, 1H), 7,15 (t, 1H), 4,38 (s, 2H), 4,28-4,26 (m, 1H), 4,16-4,12 (m, 1H), 3,88-3,84 (m, 1H), 3,79-3,75 (m, 1H), 3,66 3,64 (m, 1H), 2,31-2,28 (m, 2H), 1,71-1,68 (m, 1H), 0,91 (dd, 6H).
<Ejemplo 162>
4-(4-fluoro-3-(3-((1-h¡drox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1
(2H)-ona clorhidrato; (I-162)
Etapa 1: Preparación de 4-(4-fluoro-3-(3-((1-h¡drox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorhidrato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 36) 4-(4-fluoro-3-(3-((1-h¡drox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,24 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,48 ml, 0,48 mmol) para produc¡r el compuesto del título (91 mg, 85 %).
1H-RMN (MeOD, 400 MHz): 58,36-8,34 (m, 1H), 7,94-7,92 (m, 1H), 7,88-7,81 (m, 2H), 7,50-7,49 (m, 1H), 7,46-7,44 (m, 1H), 7,16-7,11 (m, 1H), 4,36 (s, 2H), 4,35-4,33 (m, 1H), 4,20-4,17 (m, 1H), 3,90-3,3,83 (m, 3H), 3,46-3,43 (m, 1H), 3,39-3,36 (m, 1H), 2,77-2,75 (m, 1H), 1,95 (s, 1H), 1,02 (c, 3H).
<Ejemplo 163>
4-(3-(3-(but¡lam¡no)azet¡din-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-163)
Etapa 1: Preparac¡ón de 4-(3-(3-(but¡lam¡no)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 53) 4-(3-(3-(but¡lam¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,24 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,48 ml, 0,48 mmol) para produc¡r el compuesto del título (104 mg, 98 %).
1H-RMN (CDCla, 400 MHz): 5 10,28 (s, 1H), 9,19 (d, 2H), 8,40-8,38 (m, 1H), 7,72-7,66 (m, 1H), 7,44-7,41 (m, 1H), 7,24-7,22 (m, 1H), 6,97-6,92 (m, 1H), 4,30-4,27 (m, 1H), 4,20 (s, 2H), 4,11-4,09 (m, 1H), 3,81-3,79 (m, 1H), 3,72-3,70 (m, 1H), 3,64-3,62 (m, 1H), 2,50-2,46 (m, 2H), 2,02 (s, 1H), 1,40-1,37(m, 2H), 1,30-1,24 (m, 2H), 0,86-0,80 (m, 5H).
<Ejemplo 164>
4-(3-(r1.3'-b¡azet¡d¡na1-1'-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-ona clorhidrato; (I-164)
Etapa 1: Preparac¡ón de 4-(3-([1.3'-b¡azet¡d¡na1-1'-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 75) 4-(3-([1,3'-b¡azet¡d¡na]-1'-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,25 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,50 ml, 0,50 mmol) para produc¡r el compuesto del título (88 mg, 82 %).
1H-RMN (DMSO, 400 MHz): 512,61 (s, 1H), 9,23 (d, 2H), 8,26 (d, 1H), 7,78 (d, 1H), 7,89 (t, 1H), 7,83 (t, 1H), 7,45 (m, 2H), 7,22 (t, 1H), 4,33 (d, 2H), 4,11 (m, 1H), 3,97 (t, 1H), 3,87 (t, 1H), 3,68 (m, 1H), 3,60 (t, 1H), 3,10 (m, 4H), 1,95 (t, 2H).
<Ejemplo 165>
(R)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-165)
Etapa 1: Preparac¡ón de (R)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 83) (R)-4-(4-fluoro-3-(3-((1-metox¡propan-2-¡l)am¡no)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (117 mg, 0,23 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,46 ml, 0,46 mmol) para produc¡r el compuesto del título (93 mg, 88 %).
1H-RMN (MeOD, 400 MHz): 58,38-8,35 (m, 1H), 7,95-7,93 (m, 1H), 7,89-7,82 (m, 2H), 7,52-7,50 (m, 1H), 7,45-7,43
(m, 1H), 7,14 (t, 1H), 4,38 (s, 2H), 4,12-4,10 (m, 1H), 4,18-4,15 (m, 1H), 3,84-3,77 (m, 3H), 3,22-3,19 (m, 1H), 2,89 2,87 (m, 1H), 2,03 (s, 3H), 1,00 (t, 3H).
<Ejemplo 166>
1-((1-(2-fluoro-5-((4-oxo-3.4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)amino)c¡clobutanocarbon¡tr¡lo clorhidrato; (I-166)
Etapa 1: Preparación_____ de_____ 1-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)c¡clobutanocarbonitr¡loclorh¡drato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 99) 1-((1-(2-fluoro-5-((4-oxo-3,4-d¡h¡droftalaz¡n-1-¡l)met¡l)benzo¡l)azet¡d¡n-3-¡l)am¡no)c¡clobutanocarbon¡tr¡lo (100 mg, 0,23 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,46 ml, 0,46 mmol) para produc¡r el compuesto del título (86 mg, 79 %).
1H-RMN (MeOD, 400 MHz): 68,35 (m, 1H), 7,92-7,78 (m, 3H), 7,50-7,43 (m, 2H), 7,13 (t, 1H), 4,41-4,39 (m, 1H), 4,36 (s, 2H), 4,23 (m, 1H), 3,98-3,92 (m, 2H), 3,85-3,78(m, 1H), 2,48-2,41 (m, 2H), 2,20-2,02(m, 2H).
<Ejemplo 167>
(R)-4-(3-(3-(azet¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-167)
Etapa 1: Preparac¡ón de (R)-4-(3-(3-(azet¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 135) (R)-4-(3-(3-(azet¡d¡n-1-¡l)p¡rrol¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,24 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,48 ml, 0,48 mmol) para produc¡r el compuesto del título (99 mg, 93 %).
1H-RMN (DMSO, 400 MHz): 6 12,61 (s, 1H), 9,23 (d, 2H), 8,26 (d, 1H), 7,96 (t, 1H), 7,91-7,80(m, 2H), 7,46-7,35 (m, 1H), 7,25-7,18 (m, 1H), 4,33 (s, 2H), 3,73-3,58 (m, 1H), 3,44-3,39 (m, 1H), 3,27-3,19 (m, 1H), 3,13-3,08 (m, 1H), 2,98 (t, 1H), 2,86 (d, 1H), 2,77-2,61 (m, 1H), 2,16 (t, 2H), 2,03 (t, 2H), 1,77-1,62 (m, 2H).
<Ejemplo 168>
4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-168) Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡rrol¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 140) -(4-fluoro-3-(3-(p¡rrol¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,23 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,46 ml, 0,46 mmol) para produc¡r el compuesto del título (96 mg, 91 %).
1H-RMN (CDCla, 400 MHz): 69,85 (s, 1H), 9,19 (d, 2H), 8,46-8,44 (m, 1H), 7,78-7,70 (m, 3H), 7,49-7,47 (m, 1H), 7,32 7,27 (m, 1H), 7,03-6,98 (m, 1H), 4,29-4,24 (m, 3H), 4,14-4,10 (t, 1H), 3,85-3,77 (m, 2H), 2,91-2,64 (m, 3H), 2,48 (s, 4H), 1,77 (s, 4H).
<Ejemplo 169>
4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-169)
Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 142) 4-(4-fluoro-3-(3-(p¡per¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,23 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,46 ml, 0,46 mmol) para produc¡r el compuesto del título (105 mg, 97%).
1H-RMN (CDCla, 400 MHz): 69,94 (s, 1H), 9,19 (d, 2H), 8,46 (m, 1H), 7,78-7,70 (m, 3H), 7,50-7,48 (m, 1H), 7,31-7,27 (m, 1H), 7,01 (t, 1H), 4,27-4,23 (t, 1H), 4,26 (s, 2H), 7,13-4,09 (t, 1H), 3,83-3,70 (m, 2H), 2,88-2,84 (m, 1H), 2,57-2,55 (m, 2H), 2,32 (s, 4H), 1,54 (s, 4H), 1,42 (s,2H).
<Ejemplo 170>
4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbonil)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-170) Etapa 1: Preparación de 4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 143) 4-(3-(3-(azet¡d¡n-1-¡lmet¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,24 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,48 ml, 0,48 mmol) para produc¡r el compuesto del título (101 mg, 95%).
1H-RMN (400 MHz, MeOD): 88,36 (d, 1H), 7,94 (d, 1H), 7,84 (m, 2H), 7,50 (m, 2H), 7,13 (t, 1H), 4,38 (s, 2H), 4,19 (t, 1H), 4,09 (m, 1H), 3,74 (m, 2H), 3,30 (m, 4H), 2,69 (m, 3H), 2,14 (t, 2H).
<Ejemplo 171>
4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-171)
Etapa 1: Preparac¡ón de 4-(3-(3-((c¡cloprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 25) 4-(3-(3-((c¡cloprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,25 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídrico (0,50 ml, 0,50 mmol) para produc¡r el compuesto del título (99 mg, 91 %).
1H-RMN (CDCla, 400 MHz): 8 10,03 (s, 1H), 9,18 (d, 2H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,52-7,48 (m, 1H), 7.32- 7,28 (m, 1H), 7,01 (t, 1H), 4,27 (s, 2H), 4,26-4,22 (m, 1H), 4,10 (t, 1H), 3,85-3,81 (m, 1H), 3,73-3,70 (m, 1H), 2,99 2,88 (m, 2H), 2,81-2,72 (m, 1H), 2,07 (m, 1H), 1,57 (s a, 1H), 0,45-0,41 (m, 2H), 0,29-0,26 (m, 2H).
<Ejemplo 172>
4-(4-fluoro-3-(3-((¡soprop¡lam¡no)met¡l)azet¡d¡n-1-carbonil)benc¡l)ftalaz¡n-1(2H)-ona clorhidrato; (I-172) Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((¡soprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 145) 4-(4-fluoro-3-(3-((¡soprop¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,24 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,48 ml, 0,48 mmol) para produc¡r el compuesto del título (95 mg, 89 %).
1H-RMN (CDCla, 400 MHz): 810,20 (s a, 1H), 9,19 (d, 2H), 8,47-8,45 (m, 1H), 7,80-7,70 (m, 3H), 7,51-7,49 (dd, 1H), 7.32- 7,28 (m, 1H), 7,01 (t, 1H), 4,27 (s, 2H), 4,28-4,23 (m, 1H), 4,12 (t, 1H), 3,84-3,81 (dd, 1H), 3,75-3,71 (dd, 1H), 2,88-2,69 (m, 4H), 1,05-1,03 (dd, 6H).
<Ejemplo 173>
4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1
(2H)-ona clorhidrato; (I-173)
Etapa 1: Preparac¡ón de 4-(3-(3-(((c¡cloprop¡lmet¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró ut¡l¡zando el proced¡m¡ento descr¡to para el ejemplo 156 (etapa 1). De este modo, el compuesto ¡ntermed¡o (ejemplo 146) 4-(3-(3-(((c¡cloprop¡lmet¡l)am¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobenc¡l)ftalaz¡n-1(2H)-ona (100 mg, 0,23 mmol) reacc¡onó con soluc¡ón 1 N de ác¡do clorhídr¡co (0,46 ml, 0,46 mmol) para produc¡r el compuesto del título (94 mg, 90 %).
1H-RMN (CDCh, 400 MHz): 810,36 (s a, 1H), 9,18 (d, 2H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32-7,28 (m, 1H), 7,01 (t, 1H), 4,27-4,24 (m, 3H), 4,13 (t, 1H), 3,86-3,75 (m, 2H), 2,94-2,75 (m, 3H), 2,48-2,46 (dd, 2H), 1,61 (s a, 1H), 0,95-0,91 (m, 1H), 0,51-0,46 (m, 2H), 0,12-0,09 (m, 2H).
<Ejemplo 174>
4-(4-fluoro-3-(3-((isobutilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-174) Etapa 1: Preparac¡ón de 4-(4-fluoro-3-(3-((¡sobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)benc¡l)ftalaz¡n-1(2H)-onaclorh¡drato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 147) 4-(4-fluoro-3-(3-((isobutilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (100 mg, 0,23 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,46 ml, 0,46 mmol) para producir el compuesto del título (101 mg, 93 %).
1H-RMN (CDCla, 400 MHz): 810,69 (s a, 1H), 9,20 (d, 2H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,50-7,48 (m, 1H), 7,32-7,28 (m, 1H), 7,00 (t, 1H), 4,27-4,23 (m, 3H), 4,14 (t, 1H), 3,86-3,78 (m, 2H), 2,93-2,78 (m, 3H), 2,45 (s, 1H), 2,44 (s, 1H), 1,81-1,71 (m, 1H), 0,91 (s, 3H), 0,89 (s, 3H).
<Ejemplo 175>
4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; (I-175) Etapa 1: Preparación de 4-(3-(3-((c¡clobut¡lam¡no)met¡l)azet¡d¡n-1-carbon¡l)-4-fluorobencil)ftalaz¡n-1(2H)-onaclorh¡drato Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 149) 4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona (100 mg, 0,24 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,48 ml, 0,48 mmol) para producir el compuesto del título (99 mg, 91 %).
1H-RMN (CDCla, 400 MHz): 810,33 (s a, 1H), 9,19 (d, 2H), 8,47-8,45 (m, 1H), 7,80-7,71 (m, 3H), 7,50-7,48 (m, 1H), 7,31-7,28 (m, 1H), 7,00 (t, 1H), 4,27-4,22 (m, 3H), 4,11 (t, 1H), 3,83-3,72 (m, 2H), 3,23 (m, 1H), 2,83-2,72 (m, 3H), 2,23-2,17 (m, 2H), 1,71-1,62 (m, 5H).
<Ejemplo 176>
4-(4-fluoro-3-(3-((prop-2-in-1-ilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato; (I-176) Etapa 1: Preparación de 4-(4-fluoro-3-(3-((prop-2-in-1-ilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-onaclorhidrato
Este compuesto se elaboró utilizando el procedimiento descrito para el ejemplo 156 (etapa 1). De este modo, el compuesto intermedio (ejemplo 150) 4-(4-fluoro-3-(3-((prop-2-in-1-ilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona (100 mg, 0,25 mmol) reaccionó con solución 1 N de ácido clorhídrico (0,50 ml, 0,50 mmol) para producir el compuesto del título (96 mg, 88 %).
1H-RMN (CDCls,400 MHz): 810,61 (s, 1H), 9,19 (d, 2H), 8,48-8,46 (m, 1H), 7,80-7,71 (m, 3H), 7,51-7,49 (m, 1H), 7,32 7,28 (m, 1H), 7,01 (t, 1H), 4,28-4,24 (m, 3H), 4,12 (t, 1H), 3,88-3,76 (m, 2H), 3,43 (m, 2H), 3,00-2,87 (m, 2H), 2,78 2,72 (m, 1H), 2,27-2,23 (m, 1H), 1,45 (s a, 1H).
<Experimento 1> Prueba in vitro de actividad antitumoral
Para evaluar la prueba in vitro, los compuestos inventados (de I-1 a I-155) se determinaron como sigue y se muestran en las tablas 1,2, 3, 4 y 5.
1. Determinación de niveles de PAR celular
El ensayo de PAR celular se realizó para medir la actividad inhibidora de PARP celular de los compuestos de la presente invención. Las células cancerosas cervicales humanas Hela cultivadas en medio esencial mínimo con sales balanceadas de Earle (MEM/EBSS) que contiene 10% de FBS se sembraron en placas de cultivo celular de 96 pocillos, y se incubaron por 1 día a 37 °C en atmósfera de 5 % CO2. Después de que los compuestos inventados diluidos de manera serial en DMSO se añadieron a cada pocillo, y el daño de ADN se provocó por la adición de solución de H2O2 en H2O. Se determinaron los niveles de pAr celular y EC50 se calculó utilizando un anticuerpo PAR y solución de detección.
Tabla 1
T 11

continuación

Intervalo A: EC50 ^ 5 nM
Intervalo B: 5 nM < EC50 ^ 50 nM
Intervalo C: 50 nM < EC50^ 300 nM
Intervalo D: 300 nM < EC50^ 1.000 nM
Intervalo E: EC50> 1.000 nM
De acuerdo con la tabla 1 anterior, los compuestos de la presente invención demostraron potente actividad inhibitoria de PARP celular en ensayo de PAR celular.
2. Ensayo de viabilidad celular (MDA-MB-436)
El ensayo de viabilidad celular se realizó mediante el uso de la línea celular MDA-MB-436 para medir la citotoxicidad de los compuestos en la presente invención. La línea celular de cáncer de mama MDA-MB-436 se sembró en placas de 96 pocillos en 37 °C bajo atmosfera de 5 % CO2 usando DMEM/F12 (Gibco) que contiene 10 % de FBS (Hyclone). Después de 24 horas de incubación, las células se trataron con los compuestos inventados a diferentes concentraciones durante 6 días. El medio se cambió cada 3 días. 6 días después, se utilizó un reactivo de fluorescencia para determinar los valores de IC50.
Tabla 2
T l 21

Intervalo A: IC50 ^ 5 nM
Intervalo B: 5 nM < IC50 ^ 50 nM
Intervalo C: 50 nM < IC50 ^ 300 nM
Intervalo D: 300 nM < IC50 ^ 1.000 nM
Intervalo E: IC50 > 1.000 nM
De acuerdo con la tabla 2 anterior, los compuestos de la presente invención demostraron potente inhibición del crecimiento de células cancerosas en el ensayo de citotoxicidad celular usando la línea celular de cáncer de mama MDA-MB-436.
3. Ensayo de viabilidad celular (Capan-1)
El ensayo de viabilidad celular se realizó utilizando la línea celular Capan-1 para medir la citotoxicidad de los compuestos de la presente invención. La línea celular de cáncer de páncreas Capan-1 se sembró en placas de 96 pocillos a 37 °C en atmósfera de 5 % CO2 usando IMDM (Sigma) que contiene 10 % de FBS (Hyclone). Después de 24 horas de incubación, las células se trataron con los compuestos inventados a diferentes concentraciones durante 10 días. Después del día 7 de incubación, el medio se cambió. 10 días después, se utilizó un reactivo de fluorescencia para determinar los valores de IC50.
Tabla 3
T l 1

Intervalo A: IC50 ^ 5 nM
Intervalo B: 5 nM < IC50 ^ 50 nM
Intervalo C: 50 nM < IC50 ^ 300 nM
Intervalo D: 300 nM < IC50 ^ 1.000 nM
Intervalo E: IC50 > 1.000 nM
De acuerdo con la tabla 3 anterior, los compuestos de la presente invención demostraron potente inhibición del crecimiento de células cancerosas en ensayo de citotoxicidad celular utilizando la línea celular de cáncer de páncreas Capan-1.
4. Ensayo de viabilidad celular (LNCap)
El ensayo de viabilidad celular se realizó mediante el uso de la línea celular LNCaP para medir la citotoxicidad de los compuestos de la presente invención. La línea celular de cáncer de próstata LNCaP se sembró en placas de 96 pocillos a 37 °C en atmósfera de 5 % CO2 usando RPMI-1640 (Hyclone) que contiene 10 % de FBS (Hyclone). Después de 24 horas de incubación, las células se trataron con los compuestos inventados a diferentes concentraciones durante 11 días. El medio se cambió cada 3 a 4 días. 11 días después, se utilizó un reactivo de fluorescencia para determinar los valores de IC50.
Tabla 4
T l 41

continuación

Intervalo A: IC50 ^ 5 nM
Intervalo B: 5 nM < IC50 ^ 50 nM
Intervalo C: 50 nM < IC50 ^ 300 nM
Intervalo D: 300 nM < IC50 ^ 1.000 nM
Intervalo E: IC50 > 1.000 nM
De acuerdo con la tabla 4 anterior, los compuestos de la presente invención demostraron potente inhibición del crecimiento de células cancerosas en el ensayo de citotoxicidad celular utilizando la línea celular de cáncer de próstata LNCaP.
5. Ensayo de la enzima tankirasa-1
El ensayo de la enzima tankirasa-1 se realizó para determinar actividad inhibidora de la tankirasa-1 de compuestos de la presente invención. La reacción enzimática se realizó cubriendo la placa con sustratos de tankirasa-1, y luego se lavó la placa. Los compuestos de la presente diluidos en serie en DMSO se añadieron al regulador de pH de reacción que activa tankirasa-1 y se incubaron durante 1 hora a temperatura ambiente. Después de parar la reacción, se lavó la placa, y estreptavidina-HRP se añadió a cada pocillo. IC50 se calculó añadiendo sustrato quimioluminiscente y se leyó inmediatamente la muestra en un lector (Biotek) de microplaca.
Tabla 5
T l 1

Intervalo A: IC50 ^ 5 nM
Intervalo B: 5 nM < IC50 ^ 50 nM
Intervalo C: 50 nM < IC50 ^ 300 nM
Intervalo D: 300 nM < IC50 ^ 1.000 nM
Intervalo E: IC50 >1.000 nM
De acuerdo con la tabla 5 anterior, los compuestos de la presente invención exhibieron fuerte actividad inhibidora contra tankirasa-1.
<Experimento 2> Prueba in vivo para la actividad antitumoral
Para evaluar la eficacia antitumoral in vivo, los compuestos inventados (I-80~171) se determinaron como un resultado que sigue.
Las células cancerosas de páncreas humano Capan-1 se establecieron como xenoinjertos subcutáneos por inyección de 8 X 106 células en los flancos de ratones desnudos Balb/c hembra adultos. Los ratones con tumores establecidos (160-250mm3) se seleccionaron para el estudio (n = 6/grupo de tratamiento). Los compuestos de prueba se formularon en DW y se administraron por vía oral (po) en una dosis de 30~200 mg/kg. El vehículo solo se administró para los grupos de control. Los animales se dosificaron 5 días por semana (de lunes a viernes) durante 3 semanas
consecutivas.
Los animales se pesaron y el tamaño del tumor se determinó dos veces por semana por las medidas del calibrador y se calcularon los volúmenes del tumor (volumen = [/ x w2]/2(mm3), donde l y w se refieren a las dimensiones más grandes y más pequeñas en cada medición). Se calcularon los volúmenes del tumor medios de cada grupo. El cambio en el volumen medio del tumor tratado se dividió por el cambio en el volumen del tumor de control medio, multiplicado por 100 y sustraer del 100 % para dar la inhibición del crecimiento tumoral para cada grupo.
Tabla 6
T l

Como puede observarse en la tabla 6, los compuestos seleccionados mostraron inhibición del crecimiento tumoral útil.
<Experimento 3> Farmacocinéticas de ratón
Para evaluar la prueba farmacocinética, los compuestos inventados (de I-25 a I-1171) se determinan como sigue. Se recolectaron muestras de sangre en 15, 30, 60, 120, 240, 480, 1140 minutos. La cuantificación es utilizando el método LC-EM/EM específico para el compuesto seleccionado. Los parámetros farmacocinéticos se calcularon utilizando el software de análisis compartimentado WinNonLinnon.
Tabla 7
T l 7

Como puede observarse en la tabla 7, los compuestos seleccionados mostraron farmacocinéticas significativas en ratones machos Balb/c.
<Experimento 4> Prueba de solubilidad
Para evaluar la solubilidad, los compuestos inventados (I-159, I-161 e I-171) se determinaron como sigue. Los compuestos se disolvieron en agua (1 ml, 10 ml y 100 ml) a 25 °C a presión atmosférica.
Tabla 8
T l 1

Como puede observarse en la tabla 8, los compuestos inventados mostraron significativa solubilidad.
Se entenderá que la invención descrita y definida en esta especificación se extiende a todas las combinaciones alternativas de dos o más de las características individuales mencionadas o evidentes del texto o dibujos. Todas estas diferentes combinaciones constituyen diversos aspectos alternativos de la invención.
Aplicabilidad industrial
Los compuestos de la presente invención son muy activos en la represión de PARP y según sus composiciones farmacéuticas se espera que sean útiles para aplicaciones terapéuticas que son mejoradas por la supresión de la actividad de PARP y cáncer con gen de fusión BRCA1, BRCA2 y ERG mutado en mono o combinación de tratamiento con radiación y con quimioterapia.
Claims (26)
1. Un compuesto representado por la Fórmula I, una mezcla racémica del mismo, un enantiómero, un diastereómero del mismo, o una sal farmacéuticamente aceptable del mismo
[Fórmula I]

en la presente Fórmula I,
n es 1 o 2,
R es

en donde, cuando R es
m es 0, 1 o 2,
L es metileno, carbonilo, NRc2CO, NRc3, CONRc4 o CH2NRc5, en donde Rc2, Rc3, Rc4 y Rc5 cada uno es independientemente hidrógeno, alquilo C1-6, alquinilo C2-6 o cicloalquilo C3-8,
RX es hidrógeno, ciano, hidroxilo, trifluorometilo, metilo, etilo o ciclopropilo,
RY es hidrógeno, dimetilamida, ciano, hidroxilo, trifluorometilo, halo, etil éster, dimetilamina, metilo, metoximetilo o propargilo,
Z es alquilo C1-6, metoxialquilo C1-6, alquinilo C2-6, cicloalquilo C3-8, ciclo aromático C6-10 o heterociclo de 3-8 miembros que tiene 1-3 nitrógenos,
en donde, cuando R es

p y q cada uno es independientemente de
W es CRd1Rd2 o NRd3, en donde Rd1, Rd2 y Rd3 cada uno es independientemente hidrógeno, fluoro o metilo, en donde, cuando R es

R1, R2 y R3 cada uno es independientemente hidrógeno, metilo o etilo.
2. El compuesto de acuerdo con la reivindicación 1,
en donde L es NRc2CO en donde Rc2 es hidrógeno, metilo, etilo o propilo,
Z es

3. El compuesto de acuerdo con la reivindicación 1,
en donde R es

4. El compuesto de acuerdo con la reivindicación 1, en donde el compuesto se selecciona del grupo que consiste en los siguientes compuestos:
4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
(R)-4-(3-(3-(ciclopropilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
(R)-4-(3-(3-(ciclopentilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona;
4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencill)ftalazin-1(2H)-ona;
4-(4-fluoro-3-(3-((1-(trifluorometil)ciclopropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona;
4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona;
4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1 (2H)-ona;
4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona;
4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato;
4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato;
4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato;
4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato;
4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato;
4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato;
4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato; y 4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato.
5. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
6. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es (R)-4-(3-(3-(ciclopropilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
7. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es (R)-4-(3-(3-(ciclopentilamino)pirrolidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
8. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona.
9. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
10. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona.
11. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-((1-(trifluorometil)ciclopropil)amino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona.
12. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
13. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona.
14. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
15. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona.
16. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-(ciclopentilamino)azetidin-1-carbonil)-4-fluorobencil)1:talazin-1(2H)-ona clorhidrato.
17. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-(isopropilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato.
18. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclopropilmetil)amino)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato.
19. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-(isobutilamino)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato.
20. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclopropilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato.
21. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(4-fluoro-3-(3-((isopropilamino)metil)azetidin-1-carbonil)bencil)ftalazin-1(2H)-ona clorhidrato.
22. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-(((ciclopropilmetil)amino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato.
23. El compuesto de acuerdo con la reivindicación 4, en donde el compuesto es 4-(3-(3-((ciclobutilamino)metil)azetidin-1-carbonil)-4-fluorobencil)ftalazin-1(2H)-ona clorhidrato.
24. Una composición farmacéutica para su uso en el tratamiento de cánceres que comprende el compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 23.
25. La composición farmacéutica para su uso de acuerdo con la reivindicación 24,
en donde los cánceres pueden deberse al defecto genérico del gen de fusión de BRCA1, BRCA2 y ERG.
26. La composición farmacéutica para su uso de acuerdo con la reivindicación 24,
en donde los cánceres se seleccionan del grupo que consiste en cáncer de mama, cáncer de ovario, cáncer de páncreas, cáncer gástrico, cáncer de pulmón, cáncer colorrectal, tumor cerebral, cáncer de próstata y sarcoma de Ewing.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20130110170 | 2013-09-13 | ||
| KR1020140120152A KR101670126B1 (ko) | 2013-09-13 | 2014-09-11 | 신규 프탈라지논 유도체 및 그 제조방법 |
| PCT/KR2014/008523 WO2015037939A1 (en) | 2013-09-13 | 2014-09-12 | A novel phtalazinone derivatives and manufacturing process thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2953834T3 true ES2953834T3 (es) | 2023-11-16 |
Family
ID=53024930
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14843941T Active ES2953834T3 (es) | 2013-09-13 | 2014-09-12 | Derivados novedosos de ftalazinona y su procedimiento de fabricación |
Country Status (25)
| Country | Link |
|---|---|
| US (3) | USRE49338E1 (es) |
| EP (1) | EP3030557B1 (es) |
| JP (1) | JP6180643B2 (es) |
| KR (1) | KR101670126B1 (es) |
| CN (1) | CN105793248B (es) |
| AU (1) | AU2014319132C1 (es) |
| BR (1) | BR112016004979B1 (es) |
| CA (1) | CA2923614C (es) |
| DK (1) | DK3030557T3 (es) |
| ES (1) | ES2953834T3 (es) |
| FI (1) | FI3030557T3 (es) |
| HR (1) | HRP20230998T1 (es) |
| HU (1) | HUE062770T2 (es) |
| IL (1) | IL244548B (es) |
| LT (1) | LT3030557T (es) |
| MX (1) | MX379467B (es) |
| MY (1) | MY195343A (es) |
| PL (1) | PL3030557T3 (es) |
| PT (1) | PT3030557T (es) |
| RS (1) | RS64477B1 (es) |
| RU (1) | RU2636585C2 (es) |
| SG (1) | SG11201600549TA (es) |
| SI (1) | SI3030557T1 (es) |
| SM (1) | SMT202300268T1 (es) |
| WO (1) | WO2015037939A1 (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101670126B1 (ko) * | 2013-09-13 | 2016-10-27 | 일동제약(주) | 신규 프탈라지논 유도체 및 그 제조방법 |
| JP2021516229A (ja) * | 2018-02-28 | 2021-07-01 | ザ トラスティーズ オブ ザ ユニバーシティ オブ ペンシルバニア | 低親和性ポリ(ad−リボース)ポリメラーゼ1依存性細胞毒性剤 |
| US11390608B2 (en) * | 2020-04-21 | 2022-07-19 | Idience Co., Ltd. | Crystalline forms of phthalazinone compound |
| US12060345B2 (en) | 2020-04-21 | 2024-08-13 | Idience Co., Ltd. | Process for preparing a phthalazinone derivative and intermediates thereof |
| WO2021225407A1 (ko) * | 2020-05-08 | 2021-11-11 | 주식회사 티씨노바이오사이언스 | 엑토뉴클레오티드 피로포스파타아제-포스포디에스터라아제의 저해 활성을 갖는 신규한 프탈라진 유도체 및 이들의 용도 |
| US12059419B2 (en) | 2020-10-16 | 2024-08-13 | Idience Co., Ltd. | Pharmaceutical composition comprising phthalazinone derivatives |
| US20230391752A1 (en) * | 2020-10-22 | 2023-12-07 | Chulalongkorn University | Pyrrolidine-3-carboxamide derivatives and related uses |
| TWI883310B (zh) * | 2021-03-04 | 2025-05-11 | 南韓商愛迪恩斯股份有限公司 | 酞嗪酮化合物在製備用於治療人類奧拉帕尼抗藥性癌症之藥物的用途 |
| KR20220125183A (ko) | 2021-03-04 | 2022-09-14 | 아이디언스 주식회사 | Parp 억제제에 내성을 가진 환자군의 암 치료제로서의 프탈라지논 유도체의 항암제 용도 |
| US20250312342A1 (en) * | 2021-06-02 | 2025-10-09 | Idience Co., Ltd. | Methods of treating disorders with phthalazinone derivatives |
| US20250120970A1 (en) * | 2022-01-25 | 2025-04-17 | Idience Co., Ltd. | Pharmaceutical composition including phthalazinone derivative for co-administration with anticancer drug |
| WO2024025389A1 (ko) * | 2022-07-29 | 2024-02-01 | 아이디언스 주식회사 | 프탈라지논 유도체의 중간체의 제조방법 |
| CN117229260B (zh) * | 2023-11-13 | 2024-02-27 | 中国药科大学 | DNA聚合酶θ与聚ADP核糖聚合酶1双靶点抑制剂及其制备方法和医药用途 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL223343B1 (pl) | 2000-10-30 | 2016-10-31 | Kudos Pharm Ltd | Zastosowanie związku, kompozycja farmaceutyczna zawierająca związek oraz pochodne ftalazynonowe |
| DE60335359D1 (de) | 2002-04-30 | 2011-01-27 | Kudos Pharm Ltd | Phthalazinonderivate |
| US7449464B2 (en) | 2003-03-12 | 2008-11-11 | Kudos Pharmaceuticals Limited | Phthalazinone derivatives |
| BRPI0408284B8 (pt) | 2003-03-12 | 2021-05-25 | Kudos Pharm Ltd | compostos derivados de ftalazinona, seu uso e composição farmacêutica compreendendo os mesmos |
| WO2007138355A1 (en) | 2006-05-31 | 2007-12-06 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Pyrrolo[1,2-a]pyrazin-1(2h)-one and pyrrolo[1,2-d][1,2,4]triazin-1(2h)-one derivatives as inhibitors of poly(adp-ribose)polymerase(parp) |
| GB0610680D0 (en) | 2006-05-31 | 2006-07-12 | Istituto Di Ricerche D Biolog | Therapeutic compounds |
| DK2120579T3 (da) * | 2006-12-28 | 2014-02-03 | Abbvie Inc | Inhibitorer af poly(ADP-ripose)polymerase |
| CN101030723A (zh) | 2007-04-04 | 2007-09-05 | 钟志华 | 电磁变相发动机 |
| ES2524787T3 (es) | 2007-11-15 | 2014-12-12 | Msd Italia S.R.L. | Derivados de piridazinona como inhibidores de PARP |
| AR070221A1 (es) * | 2008-01-23 | 2010-03-25 | Astrazeneca Ab | Derivados de ftalazinona inhibidores de polimerasas, composiciones farmaceuticas que los contienen y usos de los mismos para prevenir y/o tratar tumores cancerigenos,lesiones isquemicas y otras enfermedades asociadas. |
| GB0804755D0 (en) | 2008-03-14 | 2008-04-16 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| US8871765B2 (en) | 2010-07-27 | 2014-10-28 | Cadila Healthcare Limited | Substituted 4-(4-fluoro-3-(piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one derivatives as poly (ADP-ribose) polymerase-1 inhibitors |
| CN102372716A (zh) | 2010-08-09 | 2012-03-14 | 江苏恒瑞医药股份有限公司 | 酞嗪酮类衍生物、其制备方法及其在医药上的应用 |
| CN102372706A (zh) | 2010-08-09 | 2012-03-14 | 江苏恒瑞医药股份有限公司 | 酞嗪酮类衍生物、其制备方法及其在医药上的应用 |
| CN102372698A (zh) | 2010-08-10 | 2012-03-14 | 江苏恒瑞医药股份有限公司 | 酞嗪酮类衍生物、其制备方法及其在医药上的应用 |
| KR101528688B1 (ko) | 2010-12-02 | 2015-06-12 | 상하이 드 노보 파마테크 컴퍼니 리미티드 | 헤테로사이클릭 유도체, 이의 제조 방법 및 이의 의학적 용도 |
| CN102485721B (zh) | 2010-12-03 | 2015-12-09 | 曹亚 | 取代的2,3-二氮杂萘酮化合物及其用途 |
| CN103130723B (zh) * | 2011-11-30 | 2015-01-14 | 成都地奥制药集团有限公司 | 一种多聚(adp-核糖)聚合酶抑制剂 |
| KR101670126B1 (ko) * | 2013-09-13 | 2016-10-27 | 일동제약(주) | 신규 프탈라지논 유도체 및 그 제조방법 |
-
2014
- 2014-09-11 KR KR1020140120152A patent/KR101670126B1/ko active Active
- 2014-09-12 PL PL14843941.7T patent/PL3030557T3/pl unknown
- 2014-09-12 MX MX2016002998A patent/MX379467B/es unknown
- 2014-09-12 RS RS20230721A patent/RS64477B1/sr unknown
- 2014-09-12 RU RU2016108038A patent/RU2636585C2/ru active
- 2014-09-12 US US16/447,174 patent/USRE49338E1/en active Active
- 2014-09-12 WO PCT/KR2014/008523 patent/WO2015037939A1/en not_active Ceased
- 2014-09-12 PT PT148439417T patent/PT3030557T/pt unknown
- 2014-09-12 DK DK14843941.7T patent/DK3030557T3/da active
- 2014-09-12 AU AU2014319132A patent/AU2014319132C1/en active Active
- 2014-09-12 CN CN201480049174.5A patent/CN105793248B/zh active Active
- 2014-09-12 SI SI201432038T patent/SI3030557T1/sl unknown
- 2014-09-12 EP EP14843941.7A patent/EP3030557B1/en active Active
- 2014-09-12 BR BR112016004979-9A patent/BR112016004979B1/pt active IP Right Grant
- 2014-09-12 FI FIEP14843941.7T patent/FI3030557T3/fi active
- 2014-09-12 ES ES14843941T patent/ES2953834T3/es active Active
- 2014-09-12 MY MYPI2016700730A patent/MY195343A/en unknown
- 2014-09-12 SM SM20230268T patent/SMT202300268T1/it unknown
- 2014-09-12 LT LTEPPCT/KR2014/008523T patent/LT3030557T/lt unknown
- 2014-09-12 HU HUE14843941A patent/HUE062770T2/hu unknown
- 2014-09-12 JP JP2016542643A patent/JP6180643B2/ja active Active
- 2014-09-12 US US15/021,336 patent/US9682973B2/en active Active
- 2014-09-12 CA CA2923614A patent/CA2923614C/en active Active
- 2014-09-12 SG SG11201600549TA patent/SG11201600549TA/en unknown
- 2014-09-12 HR HRP20230998TT patent/HRP20230998T1/hr unknown
-
2016
- 2016-03-11 IL IL244548A patent/IL244548B/en active IP Right Grant
-
2017
- 2017-03-10 US US15/455,631 patent/US9844550B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2953834T3 (es) | Derivados novedosos de ftalazinona y su procedimiento de fabricación | |
| US20260042762A1 (en) | Mertk degraders and uses thereof | |
| US20210363139A1 (en) | Cxcr4 inhibitors and uses thereof | |
| US20240016942A1 (en) | Stat degraders and uses thereof | |
| CN106029659A (zh) | 谷氨酰胺酶抑制剂 | |
| HK1209632A1 (en) | Irak inhibitors and uses thereof | |
| CN113582974A (zh) | 一类作为蛋白降解剂的化合物及其制备方法和医药用途 | |
| ES2919572T3 (es) | Compuestos antiproliferación y usos de los mismos | |
| TW202024096A (zh) | 高活性sting蛋白激動劑 | |
| TW202110848A (zh) | 取代的稠合雙環類衍生物、其製備方法及其在醫藥上的應用 | |
| CN120981448A (zh) | 含氮大环类化合物及其制备方法和应用 | |
| WO2020169058A1 (zh) | Pd-l1拮抗剂化合物 | |
| TWI901793B (zh) | 多功能環二核苷酸及其用途 | |
| HK1224280B (zh) | 酞嗪酮衍生物及其制备方法 | |
| WO2024264070A2 (en) | Akt inhibitors | |
| HK40078355A (en) | Antiproliferation compounds and uses thereof | |
| HK40047065B (en) | Antiproliferation compounds and uses thereof | |
| HK40047065A (en) | Antiproliferation compounds and uses thereof |