ES2954774T3 - Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1H)-cetona y su método de preparación - Google Patents
Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1H)-cetona y su método de preparación Download PDFInfo
- Publication number
- ES2954774T3 ES2954774T3 ES20849905T ES20849905T ES2954774T3 ES 2954774 T3 ES2954774 T3 ES 2954774T3 ES 20849905 T ES20849905 T ES 20849905T ES 20849905 T ES20849905 T ES 20849905T ES 2954774 T3 ES2954774 T3 ES 2954774T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- crystalline form
- formula
- measured
- weight loss
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Se proporcionan formas cristalinas de compuesto de pirazina-2(1H)-cetona y un método de preparación del mismo; se describen específicamente un método para preparar el compuesto de fórmula (II) y sus formas cristalinas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1 H)-cetona y su método de preparación
La presente solicitud reivindica las siguientes prioridades:
El documento CN201910731663.3, de 8 de agosto de 2019;
El documento CN201911059460.0, de 1 de noviembre de 2019.
Campo de la técnica
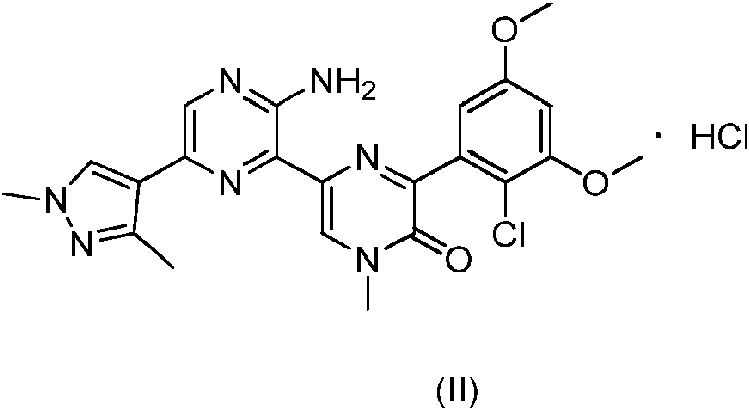
La presente descripción se refiere a una forma cristalina de un compuesto de pirazín-2(1H)-ona y a su método de preparación, se refiere en concreto a un método de preparación de un compuesto de fórmula (II) y a una forma cristalina del mismo.
Antecedentes
El receptor del factor de crecimiento de fibroblastos (FGFR) es un receptor para la transducción de la señal del factor de crecimiento de fibroblastos (FGF); la familia de FGFR consiste en cuatro miembros (FGFR1, FGFR2, FGFR3, FGFR4) y FGFR es una glicoproteína que consiste en un dominio extracelular de tipo inmunoglobulina (Ig), una región transmembranaria hidrófoba y una porción intracelular que incluye una región de tipo tirosina cinasa. El factor de crecimiento de fibroblastos (FGF) desempeña una función importante en muchos procesos reguladores fisiológicos, tales como la proliferación celular, la diferenciación celular, la migración celular y la angiogénesis, a través de estos receptores (FGFR). Existen muchas pruebas que vinculan las anomalías de la vía de señalización del FGF (alta expresión, amplificación génica, mutaciones de genes, reorganización cromosómica, etc.) directamente con muchos procesos patológicos, tales como proliferación, migración, invasión y angiogénesis de células tumorales. Por lo tanto, el FGFR ha surgido como una clase importante de dianas terapéuticas que ha atraído mucho interés de investigación y desarrollo.
La solicitud de patente internacional WO2017/070708A1 describe compuestos de 2-piridazín-3(2H)-ona sustituidos con 2-arilo y 2-heteroarilo como inhibidores de la tirosina cinasa del FGFR. La solicitud de patente internacional WO2013/061081A1 describe benzopirazinas anticancerígenas a través de la inhibición de la cinasa de los FGFR. La solicitud de patente internacional WO2016/134320A1 describe heterociclos bicíclicos inhibidores del FGFR.
Contenido de la presente invención
La presente descripción proporciona una forma cristalina A de un compuesto de fórmula (II), y el patrón de difracción de rayos X en polvo del mismo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,99 ± 0,20°, 9,54 ± 0,20° y 10,45 ± 0,20°.
En algunas realizaciones de la presente descripción, el patrón de difracción de rayos X en polvo de la forma cristalina A del compuesto de fórmula (II) comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,99 ± 0,20°, 5,85 ± 0,20°, 9,54 ± 0,20°, 10,45 ± 0,20°, 11,45 ± 0,20°, 13,19 ± 0,20°, 15,93 ± 0,20° y 19,48 ± 0,20°.
En algunas realizaciones de la presente descripción, el patrón de difracción de rayos X en polvo de la forma cristalina A del compuesto de fórmula (II) comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,99°, 5,85°, 9,54°, 9,94°, 10,45°, 11,45°, 13,19°, 13,92°, 14,51°, 15,11°, 15,93°, 16,86°, 17,94°, 19,48°, 19,90°, 20,80°, 21,60°, 21,97°, 23,28°, 23,66°, 25,52°, 26,19°, 27,47°, 28,66°, 29,03° y 29,42°. En algunas realizaciones de la presente descripción, el patrón de XRPD de la forma cristalina A del compuesto de fórmula (II) es como se muestra en la figura 1.
En algunas realizaciones de la presente descripción, los datos analíticos del patrón de XRPD de la forma cristalina A del compuesto de fórmula (II) son como los mostrados en la tabla 1.
Tabla 1: Datos analíticos del patrón de XRPD de la forma cristalina A del compuesto de fórmula (II)

En algunas realizaciones de la presente descripción, la curva de calorimetría diferencial de barrido de la forma cristalina A del compuesto de fórmula (II), medida a una velocidad de calentamiento de 10 °C/min, tiene un pico endotérmico con un inicio a 201,7 °C ± 2,0 °C, y un pico endotérmico con un inicio a 254,8 °C ± 2,0 °C.
En algunas realizaciones de la presente descripción, el patrón de DSC de la forma cristalina A del compuesto de fórmula (II) es como se muestra en la figura 2.
En algunas realizaciones de la presente descripción, la curva de análisis termogravimétrico de la forma cristalina A del compuesto de fórmula (II), medida a una velocidad de calentamiento de 10 °C/min, muestra una pérdida de peso del 0,5876 % a 63,0 °C ± 3,0 °C, se produjo una pérdida de peso del 2,4156 % a 127,4 °C ± 3,0 °C, se produjo una pérdida de peso del 8,8666 % a 209,4 °C ± 3,0 °C y se produjo una pérdida de peso del 11,8416 % a 263,2 °C ± 3,0 °C.
En algunas realizaciones de la presente descripción, el patrón de TGA de la forma cristalina A del compuesto de fórmula (II) es como se muestra en la figura 3.
La presente descripción da a conocer una forma cristalina B del compuesto de fórmula (II), y su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 10,57 ± 0,20°, 14,59 ± 0,20° y 16,05 ± 0,20°.
En algunas realizaciones de la presente descripción, el patrón de difracción de rayos X en polvo de la forma cristalina B del compuesto de fórmula (II) comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,60 ± 0,20°, 10,57 ± 0,20°, 13,82 ± 0,20°, 14,59 ± 0,20°, 16,05 ± 0,20°, 19,52 ± 0,20°, 21,45 ± 0,20° y 27,16 ± 0,20°.
En algunas realizaciones de la presente descripción, el patrón de difracción de rayos X en polvo de la forma cristalina B del compuesto de fórmula (II) comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,60°, 5,87°, 9,06°, 9,54°, 10,57°, 11,42°, 13,21°, 13,82°, 14,59°, 16,05°, 17,23°, 18,16°, 19,52°, 20,12°, 20,22°, 21,45°, 22,00°, 22,95°, 23,61°, 24,06°, 24,61°, 25,85°, 26,29°, 27,16°, 28,44°, 28,68°, 30,94°, 32,07°, 32,40°, 33,01°, 33,94°, 34,82°, 35,64°, 36,95° y 38,88°.
En algunas realizaciones de la presente descripción, el patrón de XRPD de la forma cristalina B del compuesto de fórmula (II) es como se muestra en la figura 4.
En algunas realizaciones de la presente descripción, los datos analíticos del patrón de XRPD de la forma cristalina B del compuesto de fórmula (II) son como los mostrados en la tabla 2.
Tabla 2: Datos analíticos del patrón de XRPD de la forma cristalina B del compuesto de fórmula (II)

En algunas realizaciones de la presente descripción, la curva de calorimetría diferencial de barrido de la forma cristalina B del compuesto de fórmula (II), medida a una velocidad de calentamiento de 10 °C/min, tiene un pico endotérmico con un inicio a 224,4 °C ± 2,0 °C, y un pico endotérmico con un inicio a 257,4 °C ± 2,0 °C.
En algunas realizaciones de la presente descripción, el patrón de DSC de la forma cristalina B del compuesto de fórmula (II) es como se muestra en la figura 5.
En algunas realizaciones de la presente descripción, la curva de análisis termogravimétrico de la forma cristalina B del compuesto de fórmula (II), medida a una velocidad de calentamiento de 10 °C/min, muestra una pérdida de peso del 0,1558 % a 90,9 °C ± 3,0 °C, se produjo una pérdida de peso del 6,6758 % a 206,5 °C ± 3,0 °C, y se produjo una pérdida de peso del 9,4648 % a 255,9 °C ± 3,0 °C.
En algunas realizaciones de la presente descripción, el patrón de TGA de la forma cristalina B del compuesto de fórmula (II) es como se muestra en la figura 6.
La presente descripción también da a conocer un uso de la forma cristalina A y de la forma cristalina B en la fabricación de un medicamento para tratar enfermedades relacionadas con el FGFR.
Efecto técnico
La forma cristalina A y la forma cristalina B del compuesto de fórmula (II) son fármacos estables y fáciles de formular. De acuerdo con las realizaciones experimentales de la sal de trifluoroacetato del compuesto de fórmula (I), se puede observar que la forma cristalina A y la forma cristalina B del compuesto de fórmula (II) exhiben una mejor actividad inhibidora contra el FGFR de tipo salvaje, y la selectividad por FGFR2 y 3 frente a FGFR1 y 4 es mayor. El índice farmacocinético de la forma cristalina A y de la forma cristalina B del compuesto de fórmula (II) en los ratones es bueno.
La sal de trifluoroacetato del compuesto de fórmula (I) muestra una mejor actividad inhibidora contra el FGFR de tipo salvaje, y la selectividad por FGFR2 y 3 frente a FGFR1 y 4 es mayor. El índice farmacocinético de la sal de trifluoroacetato del compuesto de fórmula (I) en los ratones es bueno.
Definición y descripción
A menos que se indique lo contrario, los siguientes términos y frases utilizados en este documento tienen los siguientes significados. Un término o frase específicos no deben considerarse indefinidos o poco claros en ausencia de una definición concreta, sino que deben entenderse en el sentido habitual. Cuando aparece un nombre comercial en este documento, se pretende hacer referencia a su correspondiente producto o ingrediente activo.
Los compuestos de la presente descripción se pueden preparar mediante varios métodos sintéticos conocidos por los expertos en la técnica, incluidas las realizaciones descritas a continuación, las realizaciones formadas mediante la combinación de las realizaciones descritas a continuación con otros métodos de síntesis química, y las alternativas equivalentes bien conocidas por los expertos en la técnica, en donde las realizaciones preferidas incluyen, pero no se limitan a ellas, las realizaciones de la presente descripción.
Las reacciones químicas de las realizaciones de la presente descripción se llevan a cabo en un solvente adecuado, y el solvente debe ser adecuado para el cambio químico de la presente descripción y los reactivos y materiales requeridos. Para obtener los compuestos de la presente descripción, a veces es necesario que los expertos en la materia modifiquen o seleccionen las etapas de síntesis o los esquemas de reacción en función de las realizaciones existentes.
La presente descripción se describirá específicamente a continuación por medio de realizaciones, pero el alcance de la presente descripción no se limita a las mismas.
Todos los solventes usados en la presente descripción están disponibles comercialmente y pueden usarse directamente sin purificación adicional.
En la presente descripción se utilizan las siguientes abreviaturas: eq se refiere a equivalente, equivalentes; PE se refiere a éter de petróleo; DMSO se refiere a sulfóxido de dimetilo; MeOH se refiere a metanol; y TFA se refiere a ácido trifluoroacético.
Los solventes usados en la presente descripción están disponibles comercialmente y los compuestos disponibles comercialmente se nombran según el repertorio del proveedor. Cuando los solventes mixtos se agregan a la solución de reacción, cada solvente puede mezclarse primero y luego agregarse a la solución de reacción; o cada solvente puede añadirse a la solución de reacción cuando le toque y mezclarse en el sistema de reacción.
Los compuestos se nombran de acuerdo con los principios de nomenclatura convencionales en el campo o por el programa informático ChemDraw®, y los compuestos disponibles comercialmente se nombran según el repertorio del proveedor.
Método del difractómetro de rayos X en polvo (XRPD) en la presente descripción
Aproximadamente de 10 a 20 mg de la muestra se sometieron a detección por XRPD.
Los parámetros detallados de la XRPD son los siguientes:
Tubo de rayos X: Cu, k2, (A = 1,54056 Á)
Voltaje del tubo de rayos X: 40 kV, corriente del tubo de rayos X: 40 mA
Ranura de divergencia: 0,60 mm
Rendija del detector: 10,50 mm
Rendija antidispersión: 7,10 mm
Intervalo de escaneo: 4-40°
Tamaño del paso: 0,02°
Duración del paso: 0,12 s
Velocidad de rotación de la bandeja de muestras: 15 rpm
Método del calorímetro diferencial de barrido (DSC) en la presente descripción
Las muestras (0,5 a 1 mg) se colocaron en un crisol de aluminio de DSC para la prueba y el método fue: de 25 °C a 300 o 350 °C, 10 °C/min.
Método del analizador termogravimétrico (TGA) en la presente descripción
Las muestras (2 a 5 mg) se colocaron en un crisol de platino de TGA para la prueba, en las condiciones de 25 ml/min de N2 a una velocidad de calentamiento de 10 °C/min, en donde la muestra se calentó desde la temperatura ambiente hasta 300 °C o hasta una pérdida de peso del 20 %.
Breve descripción de los dibujos
La figura 1 es el patrón de XRPD de la forma cristalina A del compuesto de fórmula (II) medido por radiación Cu-Ka.
La figura 2 es el patrón de DSC de la forma cristalina A del compuesto de fórmula (II).
La figura 3 es el patrón de TGA de la forma cristalina A del compuesto de fórmula (II).
La figura 4 es el patrón de XRPD de la forma cristalina B del compuesto de fórmula (II) medido por radiación Cu-Ka.
La figura 5 es el patrón de DSC de la forma cristalina B del compuesto de fórmula (II).
La figura 6 es el patrón de TGA de la forma cristalina B del compuesto de fórmula (II).
La invención se define en las reivindicaciones adjuntas. Las realizaciones no abarcadas por las reivindicaciones se proporcionan con fines de referencia.
Descripción detallada de la realización
La presente descripción se describe en detalle a continuación por medio de realizaciones, pero esto no implica ninguna limitación adversa de la descripción.
Realización 1: Preparación del compuesto de fórmula (I) y la sal de trifluoroacetato del mismo

Etapa 1: Síntesis del compuesto BB-1-3
El compuesto BB-1 -2 (2,0 g, 11,49 mmol, 1 eq) y el compuesto BB-1 -1 (2,6 g, 11,49 mmol, 1 eq) se disolvieron en agua (6,0 m) y dioxano (25,0 ml), seguido de la adición de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (841 mg, 1,15 mmol, 0,1 eq) y carbonato de potasio (4,8 g, 34,48 mmol, 3 eq), y la mezcla se calentó a 100 °C y se hizo reaccionar durante 16 horas bajo la protección de nitrógeno. La mezcla de reacción obtenida se filtró a presión reducida y se evaporó a sequedad por evaporación rotatoria, y el producto bruto se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:0 a 0:1) para obtener el compuesto BB-1 -3. MS (ESI) m/z: 190,0 [M+H]+ .
Etapa 2: Síntesis del compuesto BB-1
El compuesto BB-1-3 (0,5 g, 2,64 mmol, 1 eq) y la piridina (209 mg, 2,64 mmol, 213,28 μl, 1 eq) se añadieron al cloroformo (20,0 ml) y la mezcla se enfrió a 0 °C, y luego se le añadió bromo (422 mg, 2,64 mmol, 136,22 μl, 1 eq). La reacción se llevó a cabo a la temperatura ambiente de 28 °C durante 18 horas. La reacción se inactivó con tiosulfato de sodio (1,0 ml), luego se filtró a presión reducida, el filtrado se concentró y el producto bruto se purificó mediante cromatografía en columna de gel de sílice ultrarrápida (éter de petróleo:acetato de etilo = 1:0 a 1:1). Se obtuvo el compuesto BB-1. MS (ESI) m/z: 267,9 [M+H]+ .
RMN 1H (400 MHz, CD3DO) 5: 8,12 (s, 1 H), 7,90 (s, 1 H), 3,86 (s, 3 H), 2,43 (s, 3 H).
Etapa 3: Síntesis del compuesto 2
Bajo la protección de nitrógeno, el compuesto BB-2-1 (2,0 g, 18,77 mmol, 2,17 ml, 1 eq, HCl) se disolvió en clorobenceno (15,0 ml), y luego se le añadió el compuesto BB-2-2 (8,3 g, 65,69 mmol, 5,8 ml, 3,5 eq) gota a gota a 25 °C; la mezcla se calentó lentamente a 90 °C y se agitó durante 16 horas. Se le agregaron agua (30,0 ml) y acetato de etilo (30,0 ml) al sistema de reacción, la mezcla se dejó reposar, luego se separaron las capas y la fase acuosa se extrajo tres veces con acetato de etilo (20,0 ml, 20,0 ml, 20,0 ml). Las fases orgánicas se combinaron, se lavaron una vez con solución saturada de cloruro de sodio (30,0 ml) y finalmente se secaron con sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida. Se separó el producto bruto y se purificó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:0 a 2:1) para obtener el compuesto 2. MS (ESI) m/z: 178,7 [M+1]+ .
RMN 1H (400 MHz, CDCla ) 5: 7,26 (s, 1H), 3,61 (s, 3H).
Etapa 4: Síntesis del compuesto 4
El compuesto 2 (0,2 g, 1,12 mmol, 1 eq) y el compuesto 3 (213 mg, 1,17 mmol, 1,05 eq) se disolvieron en una solución mixta de dioxano (1,5 ml) y agua (1,5 ml) en un tubo de microondas bajo la protección de nitrógeno, se le añadieron tetrakis(trifenilfosfina)paladio (65 mg, 55,86 pmol, 0,05 eq) y carbonato de sodio (130 mg, 1,23 mmol, 1,1 eq), y la mezcla se agitó a 120 °C durante 30 minutos con radiación de microondas. La mezcla de reacción se concentró directamente. El producto bruto se separó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:0 a 0:1) (detección por TLC con éter de petróleo:acetato de etilo = 1:1) para obtener el compuesto 4. MS (ESI) m/z: 281,0 [M+1]+ .
RMN 1H (400 MHz, CDCla ) 5: 7,64 (d, 2H), 7,28 (s, 1H), 6,59 (t, 1H), 3,86 (s, 6H), 3,61 (s, 3H).
Etapa 5: Síntesis del compuesto 0027-1
El compuesto 4 (250 mg, 890,61 pmol, 1 eq) se disolvió en una solución mixta de acetonitrilo (20,0 ml) y diclorometano (5,0 ml) bajo la protección de nitrógeno; se le añadió una solución de cloruro de sulfonilo (84 mg, 623,43 pmol, 62,33 μl, 0,7 eq) en acetonitrilo (2,5 ml) lentamente, gota a gota, a 0 °C y la mezcla se agitó a 0 °C durante 10 minutos. La reacción se inactivó añadiendo metanol (5,0 ml) a la mezcla de reacción y luego la mezcla de reacción se concentró hasta sequedad bajo presión reducida. El producto bruto se separó por cromatografía en columna (éter de petróleo:acetato de etilo = 1:0 a 1:1) (detección por TLC en éter de petróleo:acetato de etilo = 1:1) para obtener el compuesto 0027-1. MS (ESI) m/z: 314,9 [M+H]+ .
Etapa 6: Síntesis del compuesto de fórmula (I)
En un matraz de tres bocas, el compuesto 0027-1 (59 mg, 186,49 pmol, 1 eq), bis(pinacolato)diboro (52 mg, 205,14 μmol, 1,1 eq), acetato de paladio (5 mg, 20,51 pmol, 0,11 eq), 2-diciclohexilfosfino-2,4,6-triisopropilbifenilo (20 mg, 41,03 pmol, 0,22 eq) y acetato de potasio (60 mg, 615,42 pmol, 3,3 eq) se añadieron a una solución de dioxano (4,0 ml); el aire del sistema de reacción se reemplazó por nitrógeno; bajo saturación de nitrógeno, el sistema de reacción se elevó a 100 °C y se agitó con reflujo durante 30 min, se enfrió a 25 °C, luego se le añadieron el compuesto BB-1 (50 mg, 186,49 pmol, 1 eq), el complejo de diclorometano de dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio (15 mg, 18,65 pmol, 0,1 eq), carbonato de potasio (77 mg, 559,47 pmol, 3 eq), dioxano (4,0 ml) y agua (2,0 ml); el aire del sistema de reacción se reemplazó por nitrógeno; bajo saturación de nitrógeno, la temperatura se elevó a 100 °C y la mezcla se agitó con reflujo durante 8 horas. La mezcla de reacción se concentró directamente. Se separó el producto bruto obtenido y se purificó por cromatografía líquida de alta resolución (columna cromatográfica: Boston Green ODS 150 x 30 mm 5 μm; fase móvil: [agua (0,1 % TFA)-ACN]; B %: 30 % a 60 %, 8 min), y se obtuvo la sal de trifluoroacetato
del compuesto de fórmula (I). MS (ESI) m/z: 468,2 [M+H]+ . RMN 1 H (400 MHz, CD3OD) 5: 8,79 (s, 1H), 8,09 (m, 2H), 6,76 (m, 2H), 3,93 (s, 3H), 3,89 (s, 3H), 3,84 (s, 3H), 3,80 (s, 3H), 2,54 (s, 3H). La sal se disolvió en diclorometano y la mezcla se lavó con carbonato de sodio saturado; la fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y el filtrado se evaporó a sequedad mediante evaporación rotatoria para obtener el compuesto de fórmula (I). RMN 1 H (400 MHz, CDCl3) 5: 8,51 (s, 1 H), 8,15 (s, 1 H), 6,71 (d, J = 2,8 Hz, 1 H), 6,63 (d, J = 2,8 Hz, 1H), 6,43 (brs, 2H), 3,94 (s, 3H), 3,92 (s, 3H), 3,84 (s, 3H), 3,79 (s, 3H), 2,55 (s, 3H).
Realización 2: Preparación del compuesto de fórmula (II)

El compuesto de fórmula (I) (44,4 g, 94,89 mmol, 1 eq) se disolvió en tetrahidrofurano (450 ml), seguido de la adición gota a gota de una solución de cloruro de hidrógeno (4 M, 94,89 ml, 4 eq) en acetato de etilo y la mezcla se agitó a 25 °C durante 3 horas. La mezcla de reacción se filtró para obtener un sólido amarillo, que se secó con una bomba de aceite. Se obtuvo el compuesto de fórmula (II).
RMN 1 H (400 MHz, DMSO-ote) 5: 8,71 (s, 1H), 8,18 (s, 2H), 6,82 (d, J = 2,8 Hz, 1H), 6,75 (d, J = 2,8 Hz, 1H) 3,91 (s, 3H), 3,81 (s, 3H), 3,80 (s, 3H), 3,71 (s, 3H), 2,44 (s, 3H).
Realización 3: Preparación de la forma cristalina A del compuesto de fórmula (II)
Se pesaron 400 mg del compuesto de fórmula (II) y se colocaron en un matraz de vidrio de 40 ml, se le añadieron 20 ml de etanol, y luego la mezcla se agitó para formar una suspensión. La muestra se colocó en un agitador magnético (50 °C) y se agitó durante 60 horas (protegido de la luz). La muestra se centrifugó rápidamente y el sólido residual se colocó en un horno de secado al vacío, y se secó al vacío a 45 °C durante la noche para eliminar el solvente residual y obtener la forma cristalina A.
Realización 4: Preparación de la forma cristalina B del compuesto de fórmula (II)
Se pesaron 400 mg del compuesto de fórmula (II) y se colocaron en un matraz de vidrio de 40 ml, se le añadieron 20 ml de acetato de etilo, y luego la mezcla se agitó para formar una suspensión. La muestra se colocó en un agitador magnético (50 °C) y se agitó durante 60 horas (protegido de la luz). La muestra se centrifugó rápidamente y el sólido residual se colocó en un horno de secado al vacío, y se secó al vacío a 45 °C durante la noche para eliminar el solvente residual y obtener la forma cristalina B del compuesto de fórmula (II).
Realización experimental 1: evaluación in vitro de la actividad inhibidora de la cinasa de tipo salvaje Los valores de CI50 se midieron con la prueba de actividad cinasa marcada con el isótopo 33 P (Reaction Biology Corp) para evaluar la capacidad inhibidora de los compuestos analizados sobre los FGFR1 y FGFR4 humanos.
Condiciones del tampón: ácido 4-(2-hidroxietil)-1 -piperazinaetanosulfónico a 20 mM (Hepes) (pH 7,5), MgCl2 a 10 mM, ácido etilenglicol-bis-(éter 2-aminoetílico)tetraacético (EGTA) a 1 mM, 0,02 % de polioxietilenlauriléter (Brij35), 0,02 mg/ml de albúmina de suero bovino (BSA), 0,1 mM de vanadato de sodio (Na3 VO4 ), ditiotreitol (DTT) a 2 mM, DMSO al 1%.
Etapas experimentales: a la temperatura ambiente, los compuestos analizados se disolvieron en DMSO para preparar una solución a 10 mM para su uso posterior. El sustrato se disolvió en el tampón recién preparado, se le añadió la cinasa problema y se mezcló bien la mezcla. La solución de DMSO disuelta con el compuesto problema se añadió a la mezcla de reacción anterior utilizando una técnica acústica (Echo 550). La concentración de los compuestos en la mezcla de reacción fue 10 μM, 3,33 μM, 1,11 μM, 0,370 μM, 0,123 μM, 41,2 nM, 13,7 nM, 4,57 nM, 1,52 nM, 0,508 nM o 10 μM, 2,50 μM, 0,62 μM, 0,156 μM, 39,1 nM, 9,8 nM, 2,4 nM, 0,61 nM, 0,15 nM, 0,038 nM. Después de 15 minutos de incubación, se le añadió 3 3 P-ATP (actividad de 0,01 μCi/μl, las concentraciones correspondientes se recogen en la tabla 3) para iniciar la reacción. El número del proveedor, el número de lote y la información sobre la concentración en la mezcla de reacción de FGFR1, FGFR4 y sus sustratos se recogen en la tabla 3. Después de incubar la reacción de cinasa durante 120 minutos a temperatura ambiente, la mezcla de reacción se depositó en papel de filtro de intercambio iónico P81 (Whatman # 3698-915). Después de lavar repetidamente el papel de filtro con una solución de ácido fosfórico al 0,75 %, se midió la radiactividad del sustrato fosforilado que quedaba en el papel de filtro. Los datos de actividad cinasa se expresaron mediante la comparación de la actividad cinasa del compuesto problema con la actividad cinasa del grupo de control (que solo contenía DMSO). El valor de CI50 se obtuvo ajustando la curva con el programa informático Prism4 (GraphPad). Los resultados experimentales se muestran en la tabla 4.
Tabla 3: Información relativa a la cinasa, el sustrato y el ATP en la prueba in vitro.

Tabla 4: Resultados de las pruebas de cribado in vitro para los compuestos de la presente descripción

Conclusión: La sal de trifluoroacetato del compuesto de fórmula (I) presenta una mejor actividad inhibidora contra el FGFR de tipo salvaje, y la selectividad por FGFR2 y 3 frente a FGFR1 y 4 fue mayor.
Realización experimental 2: evaluación farmacocinética de compuestos
Propósitos experimentales: analizar la farmacocinética del compuesto en los ratones
Materiales experimentales:
Ratones CD-1 (macho), vehículo (solución acuosa con metilcelulosa al 0,5 % (p/v) y Tween 80 al 0,5 % (v/v)), sal de trifluoroacetato del compuesto 0027.
1. Preparación de la formulación para la administración:
El vehículo era una solución acuosa de Tween 80 al 0,5 % (v/v) y metilcelulosa al 0,5 % (p/v), y se preparó de acuerdo con el siguiente procedimiento:
a. Se añadió alrededor del 50 % del volumen de agua pura en un recipiente adecuado y se calentó a aproximadamente 60 °C a 70 °C.
b. Cuando la temperatura del agua alcanzó el margen de valores especificado, se apagó el calentador. La cantidad necesaria de metilcelulosa se añadió lentamente al recipiente y la mezcla se agitó continuamente.
c. La mezcla se agitó continuamente a 4 °C hasta que la mezcla se volvió transparente a simple vista. d. Se añadió el volumen necesario de Tween 80 a la solución anterior. La mezcla se agitó continuamente hasta que el Tween 80 se dispersó y se volvió transparente a simple vista.
e. La solución anterior se ajustó al volumen final con una cantidad apropiada de agua pura.
f. La mezcla se agitó continuamente hasta que se formó una solución homogénea.
Preparación de formulación para la administración intragástrica:
a. se pesó una cantidad apropiada del producto problema en un matraz de vidrio;
b. se le añadió un 70 % del volumen de vehículo (solución acuosa de metilcelulosa al 0,5 % (p/v) y Tween 80 al 0,5 % (v/v));
c. la formulación se agitó hasta que quedó homogénea a simple vista, con sonicación en baño de agua cuando fue necesario;
d. se complementó el volumen restante de metilcelulosa al 0,5 % y Tween 80 al 0,5 %, y la mezcla se agitó uniformemente.
2. Administración
A los animales de los grupos 1 y 2 se les administraron 5 mg/ml y 30 mg/ml del compuesto mediante una única administración intragástrica, respectivamente, con un volumen de dosis de 10 ml/kg.
Los animales se pesaron antes de la administración y el volumen de administración se calculó de acuerdo con el peso corporal.
3. Recolección y procesamiento de las muestras
Se recogieron muestras de sangre completa (30 gl) en tiempos concretos (0,25, 0,5, 1, 2, 4, 6, 8, 24 horas) por extracción desde la vena safena y se anotó la hora de recolección en el registro de la prueba. El error aceptable para los puntos de tiempo de recolección fue de ±1 minuto para los puntos temporales en el plazo de 1 hora desde la administración y de ±5 % del tiempo teórico para otros puntos temporales.
Todas las muestras de sangre se transfirieron inmediatamente a tubos de centrífuga comerciales etiquetados que contenían K2-EDTA. Después de recolectar las muestras de sangre, estas se centrifugaron a 4 °C y 3200 rpm durante 10 minutos, y se aspiró el plasma sobrenadante y luego se colocó rápidamente en hielo seco a una temperatura de -20 °C o menos para el análisis por LC-MS. Y se calcularon los parámetros farmacocinéticos y resultado experimental: véase la tabla 5.
Tabla 5 Resultados de la prueba farmacocinética

Conclusión: El índice farmacocinético de la sal de trifluoroacetato del compuesto de fórmula (I) en los ratones es bueno.
Claims (10)
1. Una forma cristalina A de un compuesto de fórmula (II), en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1,54056 Á: 4,99 ± 0,20°, 9,54 ± 0,20°, y 10,45 ± 0,20°,

2. La forma cristalina A según se define en la reivindicación 1, en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1.54056 Á: 4,99 ± 0,20°, 5,85 ± 0,20°, 9,54 ± 0,20°, 10,45 ± 0,20°, 11,45 ± 0,20°, 13,19 ± 0,20°, 15,93 ± 0,20° y 19,48 ± 0,20°.
3. La forma cristalina A según se define en la reivindicación 2, en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1.54056 Á: 4,99°, 5,85°, 9,54°, 9,94°, 10,45°, 11,45°, 13,19°, 13,92°, 14,51°, 15,11°, 15,93°, 16,86°, 17,94°, 19,48°, 19,90°, 20,80°, 21,60°, 21,97°, 23,28°, 23,66°, 25,52°, 26,19°, 27,47°, 28,66°, 29,03° y 29,42°.
4. La forma cristalina A según se define en cualquiera de las reivindicaciones 1 a 3, en la que su curva de calorimetría diferencial de barrido, medida a una velocidad de calentamiento de 10 °C/min, tiene un pico endotérmico con un inicio a 201,7 °C ± 2,0 °C y tiene un pico endotérmico con un inicio a 254,8 °C ± 2,0 °C.
5. La forma cristalina A según se define en una cualquiera de las reivindicaciones 1 a 3, en la que su curva de análisis termogravimétrico, medida a una velocidad de calentamiento de 10 °C/min, muestra una pérdida de peso del 0,5876 % a 63,0 °C ± 3,0 °C, se produjo una pérdida de peso del 2,4156 % a 127,4 °C ± 3,0 °C, se produjo una pérdida de peso del 8,8666 % a 209,4 °C ± 3,0 °C y se produjo una pérdida de peso del 11,8416 % a 263,2 °C ± 3,0 °C.
6. Una forma cristalina B del compuesto de fórmula (II), en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1.54056 Á: 10,57 ± 0,20°, 14,59 ± 0,20°, y 16,05 ± 0,20°,

7. La forma cristalina B según se define en la reivindicación 6, en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1.54056 Á: 4,60 ± 0,20°, 10,57 ± 0,20°, 13,82 ± 0,20°, 14,59 ± 0,20°, 16,05 ± 0,20°, 19,52 ± 0,20°, 21,45 ± 0,20° y 27,16 ± 0,20°.
8. La forma cristalina B según se define en la reivindicación 7, en la que su patrón de difracción de rayos X en polvo comprende picos de difracción característicos en los siguientes ángulos 20, medidos a una longitud de onda A = 1.54056 Á: 4,60°, 5,87°, 9,06°, 9,54°, 10,57°, 11,42°, 13,21°, 13,82°, 14,59°, 16,05°, 17,23°, 18,16°, 19,52°, 20,12°, 20,22°, 21,45°, 22,00°, 22,95°, 23,61°, 24,06°, 24,61°, 25,85°, 26,29°, 27,16°, 28,44°, 28,68°, 30,94°, 32,07°, 32,40°, 33,01 °, 33,94°, 34,82°, 35,64°, 36,95° y 38,88°.
9. La forma cristalina B según se define en cualquiera de las reivindicaciones 6 a 8, en la que su curva de calorimetría diferencial de barrido, medida a una velocidad de calentamiento de 10 °C/min, tiene un pico endotérmico con un inicio a 224,4 °C ± 2,0 °C y tiene un pico endotérmico con un inicio a 257,4 °C ± 2,0 °C.
10. La forma cristalina B según se define en cualquiera de las reivindicaciones 6 a 8, en la que su curva de análisis termogravimétrico, medida a una velocidad de calentamiento de 10 °C/min, muestra una pérdida de peso del 0,1558 % a 90,9 °C ± 3,0 °C, se produjo una pérdida de peso del 6,6758 % a 206,5 °C ± 3,0 °C y se produjo una pérdida de peso del 9,4648 % a 255,9 °C ± 3,0 °C.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910731663 | 2019-08-08 | ||
| CN201911059460 | 2019-11-01 | ||
| PCT/CN2020/106885 WO2021023192A1 (zh) | 2019-08-08 | 2020-08-04 | 吡嗪-2(1h)-酮类化合物的a晶型和b晶型及其制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2954774T3 true ES2954774T3 (es) | 2023-11-24 |
Family
ID=74503846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20849905T Active ES2954774T3 (es) | 2019-08-08 | 2020-08-04 | Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1H)-cetona y su método de preparación |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11603366B2 (es) |
| EP (1) | EP4011866B1 (es) |
| JP (1) | JP7198388B2 (es) |
| CN (1) | CN114174270B (es) |
| ES (1) | ES2954774T3 (es) |
| WO (1) | WO2021023192A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11680061B2 (en) | 2019-08-08 | 2023-06-20 | Zhangzhou Pien Tze Huang Pharmaceutical Co., Ltd. | Crystal forms C and E of pyrazin-2(1H)-one compound and preparation method therefor |
| CN114269723B (zh) * | 2019-08-08 | 2023-09-12 | 漳州片仔癀药业股份有限公司 | 吡嗪-2(1h)-酮类化合物的d晶型及其制备方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201118654D0 (en) | 2011-10-28 | 2011-12-07 | Astex Therapeutics Ltd | New compounds |
| RS58514B1 (sr) * | 2012-06-13 | 2019-04-30 | Incyte Holdings Corp | Supstituisana triciklična jedinjenja kao inhibitori fgfr |
| SG11201500125QA (en) | 2012-07-11 | 2015-02-27 | Blueprint Medicines Corp | Inhibitors of the fibroblast growth factor receptor |
| RS63405B1 (sr) * | 2013-10-25 | 2022-08-31 | Blueprint Medicines Corp | Inhibitori receptora faktora rasta fibroblasta |
| TWI712601B (zh) * | 2015-02-20 | 2020-12-11 | 美商英塞特公司 | 作為fgfr抑制劑之雙環雜環 |
| US10208024B2 (en) | 2015-10-23 | 2019-02-19 | Array Biopharma Inc. | 2-aryl- and 2-heteroaryl-substituted 2-pyridazin-3(2H)-one compounds as inhibitors of FGFR tyrosine kinases |
| KR102583737B1 (ko) * | 2017-03-03 | 2023-09-26 | 오클랜드 유니서비시즈 리미티드 | Fgfr 키나제 저해제 및 약학적 용도 |
| JP7168149B2 (ja) | 2018-02-08 | 2022-11-09 | ▲ザン▼州片仔▲ファン▼薬業股▲フン▼有限公司 | Fgfr阻害剤としてのピラジン-2(1h)-オン系化合物 |
| US11680061B2 (en) | 2019-08-08 | 2023-06-20 | Zhangzhou Pien Tze Huang Pharmaceutical Co., Ltd. | Crystal forms C and E of pyrazin-2(1H)-one compound and preparation method therefor |
| CN114174272B (zh) | 2019-08-08 | 2023-05-12 | 漳州片仔癀药业股份有限公司 | 吡嗪-2(1h)-酮类化合物的制备方法 |
| CN114269723B (zh) | 2019-08-08 | 2023-09-12 | 漳州片仔癀药业股份有限公司 | 吡嗪-2(1h)-酮类化合物的d晶型及其制备方法 |
-
2020
- 2020-08-04 ES ES20849905T patent/ES2954774T3/es active Active
- 2020-08-04 CN CN202080054655.0A patent/CN114174270B/zh active Active
- 2020-08-04 WO PCT/CN2020/106885 patent/WO2021023192A1/zh not_active Ceased
- 2020-08-04 US US17/633,336 patent/US11603366B2/en active Active
- 2020-08-04 EP EP20849905.3A patent/EP4011866B1/en active Active
- 2020-08-04 JP JP2022507753A patent/JP7198388B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP4011866B1 (en) | 2023-07-26 |
| CN114174270A (zh) | 2022-03-11 |
| US11603366B2 (en) | 2023-03-14 |
| JP7198388B2 (ja) | 2022-12-28 |
| EP4011866A1 (en) | 2022-06-15 |
| JP2022535624A (ja) | 2022-08-09 |
| EP4011866A4 (en) | 2022-08-24 |
| CN114174270B (zh) | 2023-06-06 |
| WO2021023192A1 (zh) | 2021-02-11 |
| US20220315559A1 (en) | 2022-10-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114981268B (zh) | 嘧啶-4(3h)-酮类杂环化合物、其制备方法及其在医药学上的应用 | |
| EP4592296A1 (en) | Novel tetraheterocycle compound | |
| WO2019223766A1 (zh) | 一种fgfr抑制剂、其制备方法和在药学上的应用 | |
| CN107459508A (zh) | 作为p97复合物的抑制剂的稠合嘧啶 | |
| ES2960702T3 (es) | Formas cristalinas C y E del compuesto de pirazin-2(1H)-ona y método de preparación de las mismas | |
| EP3191475B1 (en) | Crystalline forms of 6-((6, 7-dimethoxyquinazolin-4-yl)oxy) -n,2-dimethylbenzofuran-3-carboxamide | |
| WO2020182062A1 (zh) | Fgfr4激酶抑制剂及其制备方法和用途 | |
| ES2954774T3 (es) | Forma cristalina A y forma cristalina B del compuesto de pirazina-2(1H)-cetona y su método de preparación | |
| ES3007814T3 (en) | Pyrazine-2(1h)-ketone compound preparation method | |
| ES2968388T3 (es) | Forma cristalina D del compuesto pirazina-2(1H)-cetona y método para su preparación | |
| KR20240004646A (ko) | Sting를 활성화할 수 있는 헤테로사이클릭 화합물 | |
| ES2989088T3 (es) | Forma de sal y forma de cristal que sirven como compuestos inhibidores de FGFR y VEGFR, y método de preparación de las mismas | |
| US10766862B2 (en) | Crystal form and salt form of and preparation method for tyrosine kinase inhibitor | |
| TWI732249B (zh) | Fgfr抑制劑、其製備方法和應用 | |
| CN112480109B (zh) | 吡啶并[2,3-b]吡嗪-3(4H)-酮类衍生物及其用途 | |
| WO2020224585A1 (zh) | 一种mTORC1/2双激酶活性抑制剂的盐型、晶型及其制备方法 | |
| CN114075135B (zh) | 含邻氨基吡啶炔基的化合物的盐及其制备方法和应用 | |
| WO2021143875A1 (zh) | 一种氮杂吲哚衍生物的晶型及其应用 | |
| BR112019009448B1 (pt) | Cristal de um composto, seu uso e composição farmacêutica | |
| BR112019009448A2 (pt) | cristal de um composto, seu uso e composição farmacêutica |