FR2717802A1 - Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. - Google Patents
Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. Download PDFInfo
- Publication number
- FR2717802A1 FR2717802A1 FR9403560A FR9403560A FR2717802A1 FR 2717802 A1 FR2717802 A1 FR 2717802A1 FR 9403560 A FR9403560 A FR 9403560A FR 9403560 A FR9403560 A FR 9403560A FR 2717802 A1 FR2717802 A1 FR 2717802A1
- Authority
- FR
- France
- Prior art keywords
- group
- formula
- alkylene
- compound
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000000034 method Methods 0.000 title claims abstract description 21
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 8
- 238000002360 preparation method Methods 0.000 title claims description 17
- 230000008569 process Effects 0.000 title claims description 9
- 150000001491 aromatic compounds Chemical class 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 139
- -1 hydroxymethylene group Chemical group 0.000 claims description 79
- 125000002947 alkylene group Chemical group 0.000 claims description 58
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 40
- 150000003839 salts Chemical class 0.000 claims description 31
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 19
- 150000001450 anions Chemical class 0.000 claims description 18
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 16
- 239000002253 acid Substances 0.000 claims description 16
- 125000001424 substituent group Chemical group 0.000 claims description 16
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 14
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 claims description 14
- 125000003545 alkoxy group Chemical group 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 14
- 229910052739 hydrogen Inorganic materials 0.000 claims description 14
- 239000001257 hydrogen Substances 0.000 claims description 14
- 239000004480 active ingredient Substances 0.000 claims description 13
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 12
- 238000010511 deprotection reaction Methods 0.000 claims description 12
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 12
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims description 12
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 10
- 125000005843 halogen group Chemical group 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 9
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 8
- 125000003277 amino group Chemical group 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- 125000004076 pyridyl group Chemical group 0.000 claims description 8
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 7
- 125000004414 alkyl thio group Chemical group 0.000 claims description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 7
- 239000011707 mineral Substances 0.000 claims description 7
- 150000007522 mineralic acids Chemical class 0.000 claims description 7
- 150000007524 organic acids Chemical class 0.000 claims description 7
- 125000001544 thienyl group Chemical group 0.000 claims description 7
- 239000012359 Methanesulfonyl chloride Substances 0.000 claims description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 6
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 claims description 6
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 claims description 6
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 claims description 6
- 235000005985 organic acids Nutrition 0.000 claims description 6
- RASGVORPWAYILQ-UHFFFAOYSA-N 1-azabicyclo[3.2.2]nonane Chemical compound C1CC2CCN1CCC2 RASGVORPWAYILQ-UHFFFAOYSA-N 0.000 claims description 5
- FMEHIMDNLRASDU-UHFFFAOYSA-N 1-azabicyclo[3.3.1]nonane Chemical compound C1CCN2CCCC1C2 FMEHIMDNLRASDU-UHFFFAOYSA-N 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000002883 imidazolyl group Chemical class 0.000 claims description 5
- 125000001041 indolyl group Chemical group 0.000 claims description 5
- 125000001624 naphthyl group Chemical group 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- 125000004187 tetrahydropyran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 5
- 238000005917 acylation reaction Methods 0.000 claims description 4
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 4
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 claims description 4
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 claims description 3
- 125000004846 (C1-C4) allyl group Chemical group 0.000 claims description 3
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 3
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 3
- CSKNWIPXDBBWRW-UHFFFAOYSA-N 4-phenyl-1-azabicyclo[2.2.2]octane Chemical compound C1CN(CC2)CCC21C1=CC=CC=C1 CSKNWIPXDBBWRW-UHFFFAOYSA-N 0.000 claims description 3
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 claims description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 3
- 230000006179 O-acylation Effects 0.000 claims description 3
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 3
- 125000001231 benzoyloxy group Chemical group C(C1=CC=CC=C1)(=O)O* 0.000 claims description 3
- 150000001721 carbon Chemical group 0.000 claims description 3
- 125000004432 carbon atom Chemical group C* 0.000 claims description 3
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 claims description 3
- 125000005170 cycloalkyloxycarbonyl group Chemical group 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 239000012948 isocyanate Substances 0.000 claims description 3
- 150000002513 isocyanates Chemical class 0.000 claims description 3
- 125000005493 quinolyl group Chemical group 0.000 claims description 3
- 150000003335 secondary amines Chemical class 0.000 claims description 3
- 150000003512 tertiary amines Chemical class 0.000 claims description 3
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- 125000006701 (C1-C7) alkyl group Chemical group 0.000 claims description 2
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 claims description 2
- 125000004429 atom Chemical group 0.000 claims description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 2
- 125000000043 benzamido group Chemical group [H]N([*])C(=O)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 2
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 claims description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 2
- 125000000623 heterocyclic group Chemical group 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 229910052760 oxygen Inorganic materials 0.000 claims description 2
- 239000001301 oxygen Substances 0.000 claims description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 2
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 claims description 2
- 150000003457 sulfones Chemical class 0.000 claims description 2
- 150000003462 sulfoxides Chemical class 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims 2
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 claims 1
- 150000003863 ammonium salts Chemical class 0.000 claims 1
- 150000002431 hydrogen Chemical group 0.000 claims 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 claims 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 claims 1
- 102000009493 Neurokinin receptors Human genes 0.000 abstract description 2
- 108050000302 Neurokinin receptors Proteins 0.000 abstract description 2
- 239000002464 receptor antagonist Substances 0.000 abstract description 2
- 229940044551 receptor antagonist Drugs 0.000 abstract description 2
- 150000001408 amides Chemical class 0.000 abstract 1
- 125000005936 piperidyl group Chemical group 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 101
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 69
- 239000000203 mixture Substances 0.000 description 56
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 51
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 45
- 239000000047 product Substances 0.000 description 44
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 40
- 239000000243 solution Substances 0.000 description 40
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 36
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 27
- 229910052938 sodium sulfate Inorganic materials 0.000 description 23
- 235000011152 sodium sulphate Nutrition 0.000 description 23
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- 239000000377 silicon dioxide Substances 0.000 description 19
- 239000002904 solvent Substances 0.000 description 17
- 239000011541 reaction mixture Substances 0.000 description 16
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 15
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 13
- 150000002825 nitriles Chemical class 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 150000001412 amines Chemical class 0.000 description 10
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 10
- 238000006243 chemical reaction Methods 0.000 description 10
- 239000012047 saturated solution Substances 0.000 description 10
- 125000004189 3,4-dichlorophenyl group Chemical group [H]C1=C([H])C(Cl)=C(Cl)C([H])=C1* 0.000 description 9
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 8
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 8
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 6
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 6
- 238000007796 conventional method Methods 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- QDZOEBFLNHCSSF-PFFBOGFISA-N (2S)-2-[[(2R)-2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-1-[(2R)-2-amino-5-carbamimidamidopentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-N-[(2R)-1-[[(2S)-1-[[(2R)-1-[[(2S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]pentanediamide Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CCCNC(N)=N)C1=CC=CC=C1 QDZOEBFLNHCSSF-PFFBOGFISA-N 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 5
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 5
- 208000002193 Pain Diseases 0.000 description 5
- 102100024304 Protachykinin-1 Human genes 0.000 description 5
- 101800003906 Substance P Proteins 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- JVCBVWTTXCNJBJ-UHFFFAOYSA-N 1-azabicyclo[2.2.1]heptane Chemical compound C1CC2CCN1C2 JVCBVWTTXCNJBJ-UHFFFAOYSA-N 0.000 description 4
- ZRQHMGUMVCESKV-UHFFFAOYSA-N 2-(3-propan-2-yloxyphenyl)acetic acid Chemical compound CC(C)OC1=CC=CC(CC(O)=O)=C1 ZRQHMGUMVCESKV-UHFFFAOYSA-N 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- HEAUFJZALFKPBA-YRVBCFNBSA-N Neurokinin A Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)C(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCCCN)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)O)C1=CC=CC=C1 HEAUFJZALFKPBA-YRVBCFNBSA-N 0.000 description 4
- 101800000399 Neurokinin A Proteins 0.000 description 4
- 102400000097 Neurokinin A Human genes 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 102000003141 Tachykinin Human genes 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 125000004494 ethyl ester group Chemical group 0.000 description 4
- 239000012458 free base Substances 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 150000003254 radicals Chemical class 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 108060008037 tachykinin Proteins 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- STHHLVCQSLRQNI-UHFFFAOYSA-N 1-azabicyclo[3.2.1]octane Chemical compound C1C2CCN1CCC2 STHHLVCQSLRQNI-UHFFFAOYSA-N 0.000 description 3
- GUGQQGROXHPINL-UHFFFAOYSA-N 2-oxobutanoyl chloride Chemical compound CCC(=O)C(Cl)=O GUGQQGROXHPINL-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- 208000019695 Migraine disease Diseases 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 150000001414 amino alcohols Chemical class 0.000 description 3
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 3
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 3
- 238000010876 biochemical test Methods 0.000 description 3
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 3
- 230000004054 inflammatory process Effects 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 3
- 206010027599 migraine Diseases 0.000 description 3
- 150000003141 primary amines Chemical class 0.000 description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 3
- 125000006833 (C1-C5) alkylene group Chemical group 0.000 description 2
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 2
- TYJQMJYIXSXHBO-UHFFFAOYSA-N 1-azabicyclo[2.2.2]octane Chemical compound C1CC2CCN1CC2.C1CC2CCN1CC2 TYJQMJYIXSXHBO-UHFFFAOYSA-N 0.000 description 2
- FVMDYYGIDFPZAX-UHFFFAOYSA-N 3-hydroxyphenylacetic acid Chemical compound OC(=O)CC1=CC=CC(O)=C1 FVMDYYGIDFPZAX-UHFFFAOYSA-N 0.000 description 2
- QRDSDKAGXMWBID-UHFFFAOYSA-N 5-azabicyclo[3.1.0]hexane Chemical compound C1CCN2CC21 QRDSDKAGXMWBID-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 101001125071 Homo sapiens Neuromedin-K receptor Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 108010040718 Neurokinin-1 Receptors Proteins 0.000 description 2
- 108010040722 Neurokinin-2 Receptors Proteins 0.000 description 2
- 102100029409 Neuromedin-K receptor Human genes 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 102100037342 Substance-K receptor Human genes 0.000 description 2
- 108010072901 Tachykinin Receptors Proteins 0.000 description 2
- 102000007124 Tachykinin Receptors Human genes 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 230000003042 antagnostic effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 210000000748 cardiovascular system Anatomy 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 229920001429 chelating resin Polymers 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 150000004683 dihydrates Chemical class 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 230000010355 oscillation Effects 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- FLNYLINBEZROPL-NSOVKSMOSA-N (2s,3s)-2-benzhydryl-n-[(2-methoxyphenyl)methyl]-1-azabicyclo[2.2.2]octan-3-amine Chemical compound COC1=CC=CC=C1CN[C@@H]1[C@H](C(C=2C=CC=CC=2)C=2C=CC=CC=2)N2CCC1CC2 FLNYLINBEZROPL-NSOVKSMOSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- NQHFSECYQAQZBN-LSYPWIJNSA-M 1-[(3s)-3-(3,4-dichlorophenyl)-3-[2-(4-phenyl-1-azoniabicyclo[2.2.2]octan-1-yl)ethyl]piperidin-1-yl]-2-(3-propan-2-yloxyphenyl)ethanone;chloride Chemical compound [Cl-].CC(C)OC1=CC=CC(CC(=O)N2C[C@](CC[N+]34CCC(CC3)(CC4)C=3C=CC=CC=3)(CCC2)C=2C=C(Cl)C(Cl)=CC=2)=C1 NQHFSECYQAQZBN-LSYPWIJNSA-M 0.000 description 1
- YEOSPWRTBXRGIM-UHFFFAOYSA-N 1-azabicyclo[2.2.0]hexane Chemical compound C1CC2CCN21 YEOSPWRTBXRGIM-UHFFFAOYSA-N 0.000 description 1
- SJZKULRDWHPHGG-UHFFFAOYSA-N 1-benzylpiperidin-4-one Chemical compound C1CC(=O)CCN1CC1=CC=CC=C1 SJZKULRDWHPHGG-UHFFFAOYSA-N 0.000 description 1
- MYFKLQFBFSHBPA-UHFFFAOYSA-N 1-chloro-2-methylsulfanylethane Chemical compound CSCCCl MYFKLQFBFSHBPA-UHFFFAOYSA-N 0.000 description 1
- GCUOLJOTJRUDIZ-UHFFFAOYSA-N 2-(2-bromoethoxy)oxane Chemical compound BrCCOC1CCCCO1 GCUOLJOTJRUDIZ-UHFFFAOYSA-N 0.000 description 1
- SGVWGQRMFAWWKV-UHFFFAOYSA-N 2-(3,4-dichlorophenyl)-4-(oxan-2-yloxy)butan-1-amine Chemical compound C=1C=C(Cl)C(Cl)=CC=1C(CN)CCOC1CCCCO1 SGVWGQRMFAWWKV-UHFFFAOYSA-N 0.000 description 1
- GQFIZSUSVABAPV-UHFFFAOYSA-N 2-(3,4-difluorophenyl)-4-(oxan-2-yloxy)butan-1-amine Chemical compound C=1C=C(F)C(F)=CC=1C(CN)CCOC1CCCCO1 GQFIZSUSVABAPV-UHFFFAOYSA-N 0.000 description 1
- GNPYERUNJMDEFQ-UHFFFAOYSA-N 2-(3,4-difluorophenyl)acetonitrile Chemical compound FC1=CC=C(CC#N)C=C1F GNPYERUNJMDEFQ-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- FSQSNFFVEJCHQQ-UHFFFAOYSA-N 2-benzamidoethyl acetate Chemical compound CC(=O)OCCNC(=O)C1=CC=CC=C1 FSQSNFFVEJCHQQ-UHFFFAOYSA-N 0.000 description 1
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical group BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 1
- MTEZLAATISORQK-UHFFFAOYSA-N 2-methoxyacetamide Chemical compound COCC(N)=O MTEZLAATISORQK-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 241000238876 Acari Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- NWPVUNFVASHTBK-UHFFFAOYSA-N C(CN(CC1)CC2)C12C1=CC=CC=C1.C(CN(CC1)CC2)C12C1=CC=CC=C1 Chemical compound C(CN(CC1)CC2)C12C1=CC=CC=C1.C(CN(CC1)CC2)C12C1=CC=CC=C1 NWPVUNFVASHTBK-UHFFFAOYSA-N 0.000 description 1
- XTDKZSUYCXHXJM-UHFFFAOYSA-N COC1OCCCC1 Chemical compound COC1OCCCC1 XTDKZSUYCXHXJM-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 206010065929 Cardiovascular insufficiency Diseases 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 208000014085 Chronic respiratory disease Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 1
- 201000010374 Down Syndrome Diseases 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101000655188 Homo sapiens Tachykinin-3 Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 206010026749 Mania Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- NHXYSAFTNPANFK-HDMCBQFHSA-N Neurokinin B Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCSC)NC(=O)[C@@H](N)CC(O)=O)C1=CC=CC=C1 NHXYSAFTNPANFK-HDMCBQFHSA-N 0.000 description 1
- 102000046798 Neurokinin B Human genes 0.000 description 1
- 102000002002 Neurokinin-1 Receptors Human genes 0.000 description 1
- 101800002813 Neurokinin-B Proteins 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 229940083963 Peptide antagonist Drugs 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000007107 Stomach Ulcer Diseases 0.000 description 1
- 102100037346 Substance-P receptor Human genes 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102100033009 Tachykinin-3 Human genes 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010047163 Vasospasm Diseases 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 150000007960 acetonitrile Chemical class 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- AXJDEHNQPMZKOS-UHFFFAOYSA-N acetylazanium;chloride Chemical compound [Cl-].CC([NH3+])=O AXJDEHNQPMZKOS-UHFFFAOYSA-N 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000005298 acute pain Diseases 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical compound [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000002421 anti-septic effect Effects 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 230000002567 autonomic effect Effects 0.000 description 1
- IUKQLMGVFMDQDP-UHFFFAOYSA-N azane;piperidine Chemical compound N.C1CCNCC1 IUKQLMGVFMDQDP-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 229910010277 boron hydride Inorganic materials 0.000 description 1
- KVNRLNFWIYMESJ-UHFFFAOYSA-N butyronitrile Chemical compound CCCC#N KVNRLNFWIYMESJ-UHFFFAOYSA-N 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001352 cyclobutyloxy group Chemical group C1(CCC1)O* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003113 cycloheptyloxy group Chemical group C1(CCCCCC1)O* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002933 cyclohexyloxy group Chemical group C1(CCCCC1)O* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004410 cyclooctyloxy group Chemical group C1(CCCCCCC1)O* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001887 cyclopentyloxy group Chemical group C1(CCCC1)O* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 1
- 201000003146 cystitis Diseases 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000006612 decyloxy group Chemical group 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- UMNKXPULIDJLSU-UHFFFAOYSA-N dichlorofluoromethane Chemical compound FC(Cl)Cl UMNKXPULIDJLSU-UHFFFAOYSA-N 0.000 description 1
- 229940099364 dichlorofluoromethane Drugs 0.000 description 1
- WBKFWQBXFREOFH-UHFFFAOYSA-N dichloromethane;ethyl acetate Chemical compound ClCCl.CCOC(C)=O WBKFWQBXFREOFH-UHFFFAOYSA-N 0.000 description 1
- SYZWSSNHPZXGML-UHFFFAOYSA-N dichloromethane;oxolane Chemical compound ClCCl.C1CCOC1 SYZWSSNHPZXGML-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 208000000718 duodenal ulcer Diseases 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- ZYBWTEQKHIADDQ-UHFFFAOYSA-N ethanol;methanol Chemical compound OC.CCO ZYBWTEQKHIADDQ-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- MDKXBBPLEGPIRI-UHFFFAOYSA-N ethoxyethane;methanol Chemical compound OC.CCOCC MDKXBBPLEGPIRI-UHFFFAOYSA-N 0.000 description 1
- FQTIYMRSUOADDK-UHFFFAOYSA-N ethyl 3-bromopropanoate Chemical compound CCOC(=O)CCBr FQTIYMRSUOADDK-UHFFFAOYSA-N 0.000 description 1
- XBPOBCXHALHJFP-UHFFFAOYSA-N ethyl 4-bromobutanoate Chemical compound CCOC(=O)CCCBr XBPOBCXHALHJFP-UHFFFAOYSA-N 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000005095 gastrointestinal system Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- RMIODHQZRUFFFF-UHFFFAOYSA-N methoxyacetic acid Chemical compound COCC(O)=O RMIODHQZRUFFFF-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- CWWARWOPSKGELM-SARDKLJWSA-N methyl (2s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-2-[[(2s)-5-amino-2-[[(2s)-5-amino-2-[[(2s)-1-[(2s)-6-amino-2-[[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-5 Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)OC)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 CWWARWOPSKGELM-SARDKLJWSA-N 0.000 description 1
- RAVVJKCSZXAIQP-UHFFFAOYSA-N methyl 5-bromopentanoate Chemical compound COC(=O)CCCCBr RAVVJKCSZXAIQP-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- HRQOQSWMKUGORD-UHFFFAOYSA-N n-(4-piperidin-1-ylphenyl)acetamide Chemical compound C1=CC(NC(=O)C)=CC=C1N1CCCCC1 HRQOQSWMKUGORD-UHFFFAOYSA-N 0.000 description 1
- HWXVFCFSOLPLJX-UHFFFAOYSA-N n-[4-(4-benzylpiperidin-1-yl)-2-(3,4-dichlorophenyl)butyl]-2-(oxan-2-yloxy)acetamide Chemical compound C1=C(Cl)C(Cl)=CC=C1C(CNC(=O)COC1OCCCC1)CCN1CCC(CC=2C=CC=CC=2)CC1 HWXVFCFSOLPLJX-UHFFFAOYSA-N 0.000 description 1
- AHPIWLXEAYEKID-UHFFFAOYSA-N n-[4-(4-benzylpiperidin-1-yl)-2-(3,4-dichlorophenyl)butyl]-2-hydroxyacetamide Chemical compound C=1C=C(Cl)C(Cl)=CC=1C(CNC(=O)CO)CCN(CC1)CCC1CC1=CC=CC=C1 AHPIWLXEAYEKID-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 125000006501 nitrophenyl group Chemical group 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000000414 obstructive effect Effects 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- 150000004885 piperazines Chemical class 0.000 description 1
- 150000003053 piperidines Chemical class 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 201000007094 prostatitis Diseases 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- PGKXDIMONUAMFR-AREMUKBSSA-N saredutant Chemical compound C([C@H](CN(C)C(=O)C=1C=CC=CC=1)C=1C=C(Cl)C(Cl)=CC=1)CN(CC1)CCC1(NC(C)=O)C1=CC=CC=C1 PGKXDIMONUAMFR-AREMUKBSSA-N 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005930 sec-butyloxycarbonyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 208000000143 urethritis Diseases 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/52—Oxygen atoms attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
L'invention a pour objet des composés de formule: (CF DESSIN DANS BOPI) dans laquelle: - Am représente un reste de pipéridine ou de pipérazine substitué; - Ar représente un phényle éventuellement substitué; un thiényle; un benzothiényle; un naphtyle; un indolyle éventuellement N-substitué - R1 représente un omega-(C1 -C4 )alcoxy(C2 -C4 )alkylène; un omega-(C1 -C4 )-alkylcarbonyloxy(C2 -C4 )alkylène; un omega-benzoyloxy(C2 -C4 )alkylène; un omega-hydroxy(C2 -C4 )-alkylène; un omega-(C1 -C4 )alkylthio(C2 -C4 )alkylène; un omega-(C1 -C4 )alkylcarbonyl(C2 -C4 )alkylène; un omega-carboxy(C2 -C4 )alkylène; un omega-(C1 -C4 -alcoxycarbonyl(C2 -C4 ) alkylène; - T représente une liaison directe; un groupe hydroxyméthylène; un groupe alcoxyméthylène dans lequel l'alcoxy est en C1 -C4 ; un groupe alkylène en C1 -C5 ; un vinylène; un atome d'oxygène; un groupe -NR7 -; - R7 représente un hydrogène ou un alkyle en C1 -C4 ; - Z représente un phényle éventuellement substitué; un naphtyle éventuellement substitué; un pyridyle; un thiényle; un indolyle; un quinoléyle; un benzothiényle; un imidazolyle; APPLICATION: antagonistes des récepteurs des neurokinines.
Description
La présente invention a pour objet de nouveaux composés aromatiques, un procédé pour leur préparation et les compositions pharmaceutiques en contenant cn tant que principe actif.
Plus particulièrement, la présente invention concerne une nouvelle classe de composés aromatiques à usage thérapeutique, dans les phénomènes pathologiques qui impliquent le système des tachykinines comme par exemple de manière non limitative: la douleur (D. Regoli et al., Life Sciences, 1987, 40, 109-117), l'allergie et l'inflammation (J.E. Morlay et al., Life Sciences, 1987, 41, 527-544), l'insuffisance circulatoire (J. Losay et al., 1977, Substance P, Von Euler, U.S. and Pernow cd., 287293, Raven Press, New York), les troubles gastr(intestinaux (D. Regoli et al., Trcnds
Pharmacol. Sci., 1985, 6, 481-484), les troubles respiratoires (J. Mizrahi et al.,
Pharmacology, 1982,25,39-50).
Pharmacol. Sci., 1985, 6, 481-484), les troubles respiratoires (J. Mizrahi et al.,
Pharmacology, 1982,25,39-50).
Dans les années récentes de nombreux travaux de recherche ont été effectués sur les tachykinines et leurs récepteurs. Les tachykinines sont distribuées à la fois dans le système nerveux central et dans le système nerveux périphérique. Les récepteurs aux tachykinines ont été reconnus et sont classés en trois types : NK1, NK2,NK3. La substance P (SP) est le ligand endogène des récepteurs NK1, la neurokinine A (NKA) celui des récepteurs NK2 et la neurokinine B (NK, celui des récepteurs NK3.
Les récepteurs NK1, NK2, NK3 ont été mis CII évidence chez différentes espèces.
Une revue récente de CA. Maggi et al. fait le point sur les récepteurs aux tachykinines et leurs antagonistes (J. Autonomic Pharmacol., 1993, 13, 23-93).
Parmi les antagonistes spécifiques du récepteur NK1 on peut citer les composés non peptidiques suivants : CP-96345 (J. Med. Chem., 1992, 35, 2591-2600), RP68651 (Proc. Natl. Acad. Sci. USA, 1991, 88, 10208-10212), SR 140333 (Curr. J.
Pharmacol., 1993, 250, 403-413).
Pour le récepteur NK2, un antagoniste sélectif non peptidique, le SR 48968 a été décrit en détail (Life Sci., 1992, 50 PL101-PL106).
On a maintenant trouvé que certains composés aromatiques possèdent des propriétés pharmacologiques intéressantes, en tant qu'antagonistes des récepteurs des neurokinines et sont notamment utiles pour le traitement de toute pathologie substance
P et neurokinine dépendante.
P et neurokinine dépendante.
Ainsi selon un de ses aspects, la présente invention concerne des composés de formule:
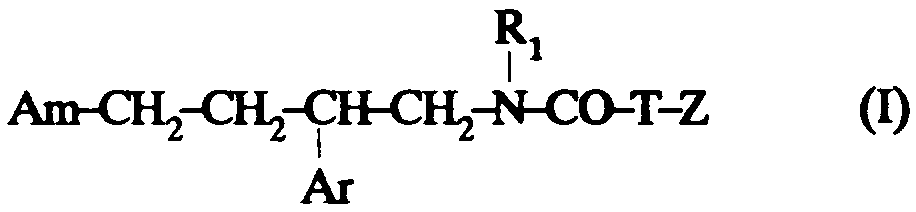
dans laquelle:
- Am représente:
i- soit un groupe Am1 de formule:

dans laquelle J1 représente: il soit un groupe

dans lequel R2 représente un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy cn
C1-C4, un alkyle en C1-C4, un trifluorométhyle, lesdits substituants étant identiques ou différents; un pyridyle ; un thiényle ; un pyrimidyle ; un imidazolyle non substitué ou substitué par un alkyle en C1 -C4 ; i2- soit un groupe

dans lequel R2 est tel que défini ci-dessus; i3- soit un groupe

dans lequel:
- R2 est tel que défini ci-dessus;
- x est zéro ou un;
- X1 représente un hydrogène; un hydroxyle ; un alcoxy en C1 -C4; un alkyl-
carbonyloxy dans lequel l'alkyle est en C1-C6 ; un benzoyloxy ; un carboxy ; un
alcoxycarbonyle dans lequel l'alcoxy est en C1-C4 ; un amino ; un groupe
-NR3COR4 ; un cyano ; un groupe -CH2NH2; un groupe -CH2NR3COR4; un
groupe -CH2OH; un groupe -CH2-GAlk dans lequel Alk représente un alkyle en
C1-C4 ; un groupe -CH2-O-COR4;
- ou bien X1 forme avec l'atome de carbone auquel il est lié et avec l'atome de
carbone voisin dans la pipéridine une liaison supplémentaire;
- R3 représente un hydrogène ou un alkyle en C1 -C4;
- R4 représente un alkyle en C1 -C7; un cycloalkyle en C3-C7 non substitué ou
substitué par un ou plusieurs méthyles ; un phényle ; un pyridyle;
i-soit un groupe dans lequel:
- R2 est tel que défini cidessus;
- X2 représente un atome d'oxygène ; un atome de soufre ; un sulfoxyde ; une
sulfone ; un groupe ZR3-; un groupe NCGA1k dans lequel Alk représente
un alkyle en C1-C4 ; un group-N-(CH2)p-NR5R6 ;
- R3 est tel que défini cidessus;
-pest un, deux ou trois;
- R5 et R6 représentent chacun indépendamment un hydrogène ou un alkyle en
C1-C4
- ou bien R5 et R6 ensemble avec l'atome d'azote auquel ils sont liés constituent un
hétérocycle choisi parmi la pyrrolidine, la pipéridine ou la morphotine,
ii- soit un groupe Am2 de formule:

dans laquelle J2 représente:
/
r soit un groupe R2-N\
/
H2- soit un groupe R2-CH2-N\ dans lesquels R2 est tel que défini ci-dessus;
iii- soit un groupe Am3 de formule:

dans laquelle X3, X4, X5 ensemble et avec l'atome d'azote auquel ils sont liés forment un système azabicyclique ou azatricyclique contenant de 5 à 9 atomes de carbone, non substitué ou substitué par un phényle ou un benzyle;
- A- représente un anion;
- Ar représente un phényle non substitué ou substitué une ou plusieurs fois par un
substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy en C1-C4,
un alkyle en C1-C4, un trifluorométhyle, lesdits substituants étant identiques ou
différents ; un thiényle ; un benzothiényle ; un naphtyle ; un indolyle non substitué
ou N-substitué par un alkyle en C1-C4 ou un benzoyle;
- R1 représente un #-(C1-C4)alcoxy(C2-C4)alkylène ; un *(C1-C4
alkylcarbonyloxy(C2-Cq)alkylène ; un o-benzoyloxy(C2-Cq)aIkylène ; un o >
hydroxy(C2-C4)-alkylène; un #-(C1-C4)alkylthio(C2-C4)alkylène ; un o > (C1-
C4)alkylcarbonyl(C2-C4)alkylène ; un #-carboxy(C2-C4)alkylène ; un *(C1-
C4)-alcoxycarbonyl(C2-C4)alkylène ;
- T représente une liaison directe ; un groupe hydroxyméthylène ; un groupe
alcoxyméthylène dans lequel l'alcoxy est en C1-C4 ; un groupe alkylène en C1 -C5;
un vinylène ; un atome d'oxygène ; un groupe -NR7-;
- R7 représente un hydrogène ou un alkyle en C1-C4;
- Z représente:
- un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène ; un trifluorométhyle ; un cyano ; un hydroxyle ; un nitro ; un amino non substitué ou substitué une ou deux fois par un alkyle en C1 -C4; un benzylamino ; un carboxy ; un alkyle en C1-C10 ; un cycloalkyle en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle; un alcoxy en C1-C10 ; un cycloalkyloxy en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle ; un mercapto; un alkylthio en C1-C10 ; un alkylcarbonyloxy dans lequel l'allyle est en C1-C6 ; un alkylcarbonylamino dans lequel l'alkyle est en C1-C6; un benzoylamino ; un alcoxycarbonyle dans lequel l'alcoxy est en C1 -C4 ; un cycloalkyloxycarbonyle dans lequel le cycloalkyle est en C3-C7 ; un carbamoyle non substitué ou substitué une ou deux fois par un allyle en C1-C4 ; un uréido non substitué ou substitué une ou deux fois en position 3 par un alkyle en C1-C4 ou un cycloalkyle en C3-C7 ; un (1-pyrrolidino)carbonylamino, lesdits substituants étant identiques ou différents;
- un naphtyle non substitué ou substitué une ou plusieurs fois par un halogène, un trifluorométhyle, un alkyle en C1-C4, un hydroxyle, un alcoxy en C1-C4;
- un pyridyle ; un thiényle ; un indolyle ; un quinoléyle ; un benzothiényle ; un imidazolyle; avec la limitation que R1 ne peut pas représenter un *(Cl-C4)alcoxy (C2-C4)alkylène ou un o-(C1 Cq)alkylcarbonyloxy(C2-Cq)allrylène lorsque Am représente un groupe Am; ainsi que leurs sels éventuels avec des acides minéraux ou organiques ou leurs sels d'ammonium quaternaire éventuels.
dans laquelle:
- Am représente:
i- soit un groupe Am1 de formule:
dans laquelle J1 représente: il soit un groupe
dans lequel R2 représente un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy cn
C1-C4, un alkyle en C1-C4, un trifluorométhyle, lesdits substituants étant identiques ou différents; un pyridyle ; un thiényle ; un pyrimidyle ; un imidazolyle non substitué ou substitué par un alkyle en C1 -C4 ; i2- soit un groupe
dans lequel R2 est tel que défini ci-dessus; i3- soit un groupe
dans lequel:
- R2 est tel que défini ci-dessus;
- x est zéro ou un;
- X1 représente un hydrogène; un hydroxyle ; un alcoxy en C1 -C4; un alkyl-
carbonyloxy dans lequel l'alkyle est en C1-C6 ; un benzoyloxy ; un carboxy ; un
alcoxycarbonyle dans lequel l'alcoxy est en C1-C4 ; un amino ; un groupe
-NR3COR4 ; un cyano ; un groupe -CH2NH2; un groupe -CH2NR3COR4; un
groupe -CH2OH; un groupe -CH2-GAlk dans lequel Alk représente un alkyle en
C1-C4 ; un groupe -CH2-O-COR4;
- ou bien X1 forme avec l'atome de carbone auquel il est lié et avec l'atome de
carbone voisin dans la pipéridine une liaison supplémentaire;
- R3 représente un hydrogène ou un alkyle en C1 -C4;
- R4 représente un alkyle en C1 -C7; un cycloalkyle en C3-C7 non substitué ou
substitué par un ou plusieurs méthyles ; un phényle ; un pyridyle;
i-soit un groupe dans lequel:
- R2 est tel que défini cidessus;
- X2 représente un atome d'oxygène ; un atome de soufre ; un sulfoxyde ; une
sulfone ; un groupe ZR3-; un groupe NCGA1k dans lequel Alk représente
un alkyle en C1-C4 ; un group-N-(CH2)p-NR5R6 ;
- R3 est tel que défini cidessus;
-pest un, deux ou trois;
- R5 et R6 représentent chacun indépendamment un hydrogène ou un alkyle en
C1-C4
- ou bien R5 et R6 ensemble avec l'atome d'azote auquel ils sont liés constituent un
hétérocycle choisi parmi la pyrrolidine, la pipéridine ou la morphotine,
ii- soit un groupe Am2 de formule:
dans laquelle J2 représente:
/
r soit un groupe R2-N\
/
H2- soit un groupe R2-CH2-N\ dans lesquels R2 est tel que défini ci-dessus;
iii- soit un groupe Am3 de formule:
dans laquelle X3, X4, X5 ensemble et avec l'atome d'azote auquel ils sont liés forment un système azabicyclique ou azatricyclique contenant de 5 à 9 atomes de carbone, non substitué ou substitué par un phényle ou un benzyle;
- A- représente un anion;
- Ar représente un phényle non substitué ou substitué une ou plusieurs fois par un
substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy en C1-C4,
un alkyle en C1-C4, un trifluorométhyle, lesdits substituants étant identiques ou
différents ; un thiényle ; un benzothiényle ; un naphtyle ; un indolyle non substitué
ou N-substitué par un alkyle en C1-C4 ou un benzoyle;
- R1 représente un #-(C1-C4)alcoxy(C2-C4)alkylène ; un *(C1-C4
alkylcarbonyloxy(C2-Cq)alkylène ; un o-benzoyloxy(C2-Cq)aIkylène ; un o >
hydroxy(C2-C4)-alkylène; un #-(C1-C4)alkylthio(C2-C4)alkylène ; un o > (C1-
C4)alkylcarbonyl(C2-C4)alkylène ; un #-carboxy(C2-C4)alkylène ; un *(C1-
C4)-alcoxycarbonyl(C2-C4)alkylène ;
- T représente une liaison directe ; un groupe hydroxyméthylène ; un groupe
alcoxyméthylène dans lequel l'alcoxy est en C1-C4 ; un groupe alkylène en C1 -C5;
un vinylène ; un atome d'oxygène ; un groupe -NR7-;
- R7 représente un hydrogène ou un alkyle en C1-C4;
- Z représente:
- un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène ; un trifluorométhyle ; un cyano ; un hydroxyle ; un nitro ; un amino non substitué ou substitué une ou deux fois par un alkyle en C1 -C4; un benzylamino ; un carboxy ; un alkyle en C1-C10 ; un cycloalkyle en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle; un alcoxy en C1-C10 ; un cycloalkyloxy en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle ; un mercapto; un alkylthio en C1-C10 ; un alkylcarbonyloxy dans lequel l'allyle est en C1-C6 ; un alkylcarbonylamino dans lequel l'alkyle est en C1-C6; un benzoylamino ; un alcoxycarbonyle dans lequel l'alcoxy est en C1 -C4 ; un cycloalkyloxycarbonyle dans lequel le cycloalkyle est en C3-C7 ; un carbamoyle non substitué ou substitué une ou deux fois par un allyle en C1-C4 ; un uréido non substitué ou substitué une ou deux fois en position 3 par un alkyle en C1-C4 ou un cycloalkyle en C3-C7 ; un (1-pyrrolidino)carbonylamino, lesdits substituants étant identiques ou différents;
- un naphtyle non substitué ou substitué une ou plusieurs fois par un halogène, un trifluorométhyle, un alkyle en C1-C4, un hydroxyle, un alcoxy en C1-C4;
- un pyridyle ; un thiényle ; un indolyle ; un quinoléyle ; un benzothiényle ; un imidazolyle; avec la limitation que R1 ne peut pas représenter un *(Cl-C4)alcoxy (C2-C4)alkylène ou un o-(C1 Cq)alkylcarbonyloxy(C2-Cq)allrylène lorsque Am représente un groupe Am; ainsi que leurs sels éventuels avec des acides minéraux ou organiques ou leurs sels d'ammonium quaternaire éventuels.
Lorsque Am représente Aml ou Am2 on peut former des sels des composés de formule (T). Ces sels comprennent aussi bien ceux avec des acides minéraux ou organiques qui permettent une séparation ou une cristallisation convenable des composés de formule (I), tels que l'acide picrique ou l'acide oxalique ou un acide optiquement actif, par exemple un acide mandélique ou camphosulfonique, que ceux qui forment des sels pharmaceutiquement acceptables tels que le chlorhydrate, le bromhydrate, le sulfate, l'hydrogénosulfate, le dihydrogénophosphate, le méthanesulfonate, le méthylsulfate, le maléate, le fumarate, le 2-naphtalènesulfonate, le glycolate, le gluconate, le citrate, l'iséthionatc.
Lorsque Am représente Aml on peut former les sels d'ammonium quaternaire des composés de formule (I). Le groupe Am est alors représenté par le groupe Amq de formule:

dans laquelle:
- J1 est tel que défini ci-dessus pour (1);
- Q représente un alkyle en C1-C6 ou un benzyle ; ledit substituant étant soit en
position axiale soit en position équatoriale;
- A(9 est un anion.
dans laquelle:
- J1 est tel que défini ci-dessus pour (1);
- Q représente un alkyle en C1-C6 ou un benzyle ; ledit substituant étant soit en
position axiale soit en position équatoriale;
- A(9 est un anion.
les composés de formule (1) selon l'invention comprennent aussi bien les racémiques, les isomères optiquement purs, ainsi que les isomères axiaux et équatoriaux lorsque dans le composé de formule (1), Am représente Am4.
Les anions A sont ceux normalement utilisés pour salifier les ions d'ammonium quaternaire, de préférence les ions chlorure, bromure, iodure, hydrogénosulfate, méthanesulfonate, paratoluènesulfonate, acétate, benzènesulfonate.
Préférentiellement, on utilise les anions pharmaceutiquement acceptables par exemple le chlorure, le méthanesulfonate ou le benzènesulfonate.
Dans la présente description les groupes alkyles ou les groupes alcoxy sont droits ou ramifiés ; par atome d'halogène on entend un atome de chlore, de brome, de fluor ou d'iode.
Lorsque Z est un groupe phénylc, celui Ci pcut erre mono ou disubstitué notamment en position 2,4 mais aussi par exemple en position 2, 3 ou 4, 5 ou 3, 4, ou 3, 5 ; il peut aussi être trisubstitué, notamment en position 2, 4, 6 mais aussi par exemple en 2, 3, 4 ou 2, 3, 5 ou 2, 4, 5 ou 3, 4, 5; tétrasubstitué, par ezcemple en 2, 3, 4, 5 ; ou pentasubstitué. Dans les substituants du groupe phényle, par alkyle en C1- C10 on entend par exemple un méthyle, un éthyle, un n-propyle, un isopropyle, un nbutyle, un isobutyle, un sec-butyle, un tert-butyle, un pentyle ou n-pcntylc, un hcxyle ou n-hexyle, un heptyle ou un n-heptyle, un octyle ou n-octyle, un nonyle ou nnonyle, un décyle ou un n-décyle; par cycloalkyle en C3-Cg éventuellement substitué par un méthyle on entend par exemple un cyclopropyle, un cyclobutyle, un cyclopentyle, un 1-, 2- ou 3-méthylcyclopentyle, un cyclohexyle, un 1-, 2-, 3- ou 4-méthylcyclohexyle, un cycloheptyle ou un cyclooctyle ; par alcoxy en C1-C10 on entend par exemple un méthoxy, un éthoxy, un n-propoxy, un isopropoxy, un nbutoxy, un isobutoxy, un sec-butoxy, un tert-butoxy, un pentyloxy, un hexyloxy, un heptyloxy, un octyloxy, un nonyloxy ou un décyloxy ; par cycloalkyloxy en C3 C8 éventuellement substitué par un méthyle on entend par exemple un cyclopropyloxy, un cyclobutyloxy, un cyclopentyloxy, un 1-, 2- ou 3-méthylcyclopentyloxy, un cyclohexyloxy, un 1-, 2-, 3- ou 4-méthylcydobexyloxy, un cycloheptyloxy ou un cyclooctyloxy; par alkylthio en C1-C10 on entend par exemple un méthylthio, un éthylthio, un n-propylthio, un isopropylthio, un n-butylthio, un isobutylthio, un secbutylthio, un tert-butylthio, un pentylthio, un htxylthio, un heptylthio, un octylthio, un nonylthio ou un décylthio ; par aurylcarbonyloxy dans lequel l'allyle est en C1 C6 on entend par exemple un formyloxy, un acétyloxy, un propionyloxy, un butyryloxy, un valéryloxy, un caproyloxy, un heptanoyloxy ; par un alkylcarbonylamino dans lequel l'alkyle est en C1-C6 on entend par exemple un formylamino, un acétylamino, un propionylamino, un butyrylamino, un isobutyrylamino, un valérylamino, un caproylamino ou un heptanoylamino ; par alcoxycarbonyle dans lequel l'alcoxy est en
C1-C4 on entend par exemple un méthoxycarbonyle, un éthoxycarbonyle, un npropoxycarbonyle, un isopropoxycarbonyle, n-butoxycarbonyle, un isobutoxycarbonyle, un sec-butoxycarbonyle ou un tert-butoxycarbonyle ; par cycloatkyloxycarbonyle dans lequel le cycloallcyle est en C3-C7 on entend par exemple un cyclopropyloxycarbonyle, un cyclobutyloxycarbonyle, un cyclopentyloxycarbonyle, un cyclohexyloxycarbonyle ou un cycloheptyloxycarbonyle.
C1-C4 on entend par exemple un méthoxycarbonyle, un éthoxycarbonyle, un npropoxycarbonyle, un isopropoxycarbonyle, n-butoxycarbonyle, un isobutoxycarbonyle, un sec-butoxycarbonyle ou un tert-butoxycarbonyle ; par cycloatkyloxycarbonyle dans lequel le cycloallcyle est en C3-C7 on entend par exemple un cyclopropyloxycarbonyle, un cyclobutyloxycarbonyle, un cyclopentyloxycarbonyle, un cyclohexyloxycarbonyle ou un cycloheptyloxycarbonyle.
De manière avantageuse, le radical Z représente un phényle non substitué ou substitué une ou plusieurs fois par un atome d'halogène, plus particulièrement un atome de chlore, de fluor ou d'iode, un trifluorométhyle, un alkyle cn C1 -C4, un hydroxyle, un alcoxy en C1-C4 ; un naphtyle non substitué ou substitué une ou plusieurs fois par un halogène, un trifluorométhyle, un alkyle en C1 -C4, un hydroxyle, un alcoxy en C1-C4 ; un pyridyle ; un thiényle ; un indolyle ; un quinoléyle ; un benzothiényle ; un imidazolyle.
Un groupe de composés préférés de l'invention est constitué par les composés de formule (I) dans laquelle Ar, R1, T et Z sont tels que définis précédemment et Am est le groupe de formule:

dans lequel Rû x et X1 sont tels que définis précdemment.
dans lequel Rû x et X1 sont tels que définis précdemment.
Les composés particulièrement préférés sont ceux de formule (dans laquelle à la fois:
- Am représente un groupe de formule:

dans laquelle X1 est tel que défini précédemment ;
- R1 représente un groupe 2-hydroxyéthyle ou un groupe 2-acetoxysthylc ou un
groupe 2-méthoxyéthyle;
- T représente une liaison;
- Z représente un phényle;
- Ar est tel que défini précédemment.
- Am représente un groupe de formule:
dans laquelle X1 est tel que défini précédemment ;
- R1 représente un groupe 2-hydroxyéthyle ou un groupe 2-acetoxysthylc ou un
groupe 2-méthoxyéthyle;
- T représente une liaison;
- Z représente un phényle;
- Ar est tel que défini précédemment.
Un autre groupe de composés particulièrement préférés sont ceux de formule (I) dans laquelle à la fois:
- Am représente un groupe de formule:

- Am représente un groupe de formule:
- x est zéro ou un;
- T représente un méthylène;
- Z représente un 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (1);
les sels d'ammonium quaternaire particulièrement préférés selon la présente invention sont ceux de formule (1) dans laquelle à la fois:
- Am représente un groupe Am4 dans lequel J1 représente: * soit un groupe

* soit un groupe

- T représente un méthylène;
- Z représente un 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (1);
les sels d'ammonium quaternaire particulièrement préférés selon la présente invention sont ceux de formule (1) dans laquelle à la fois:
- Am représente un groupe Am4 dans lequel J1 représente: * soit un groupe
* soit un groupe
- Q est tel que défini précédemment et est en position axiale;
- T représente un méthylène;
- Z représente un groupe 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (1);
- A représente un anion, préférentiellement un anion pharmaceutiquement
acceptable.
- T représente un méthylène;
- Z représente un groupe 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (1);
- A représente un anion, préférentiellement un anion pharmaceutiquement
acceptable.
Un autre groupe de composés préférés de l'invention est constitué par les composés de formule (f) dans laquelle Ar, R1, T et Z sont tels que définis précédemment et Am est le groupe Am3.
Le radical

représenté par le substituant Am3O+ dans la formule (I) est de préférence le résidu d'un système azabicyclique ou azatricydique choisi parmi: (a) 1 -azoniabicyclo[22.0]hexane (b) 1-azoniabicyclo[3.1 .0]hexane (c) 1-azoniabicyclo[2.2.1]heptane
(d) 1-azoniabicyclo[2.2.2]octane (quinuclidine) (e) 1-azoniabicyclo(3.2. 1]octane (f) 1-azoniabicydo[3.2.2]nonane (g) 1-azoniabicyclo[3.3.1]nonane (h) hexahydro 1H-pyrrolizinium-4 (i) octahydroindolizinium-4 (.j) octahydro 2H-quinolizinium-5 (k) 1-azoniatricyclo[3.3.1.1.3.7]décane (1) 4-phényl-l-azoniabicyclo[2.2.2]octane (phényl-4 quinuclidine).
représenté par le substituant Am3O+ dans la formule (I) est de préférence le résidu d'un système azabicyclique ou azatricydique choisi parmi: (a) 1 -azoniabicyclo[22.0]hexane (b) 1-azoniabicyclo[3.1 .0]hexane (c) 1-azoniabicyclo[2.2.1]heptane
(d) 1-azoniabicyclo[2.2.2]octane (quinuclidine) (e) 1-azoniabicyclo(3.2. 1]octane (f) 1-azoniabicydo[3.2.2]nonane (g) 1-azoniabicyclo[3.3.1]nonane (h) hexahydro 1H-pyrrolizinium-4 (i) octahydroindolizinium-4 (.j) octahydro 2H-quinolizinium-5 (k) 1-azoniatricyclo[3.3.1.1.3.7]décane (1) 4-phényl-l-azoniabicyclo[2.2.2]octane (phényl-4 quinuclidine).
Les composés particulièrement préférés sont ceux de formule (I) dans laquelle à la fois:
- Am représente un radical (d) ou (I) tels que définis ci-dessus pour le radical
Am3+, avec A(9 représentant un anion, préférentiellement un anion
pharmaceutiquement acceptable;
- T représente un méthylène;
- Z représente un groupe 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (I).
- Am représente un radical (d) ou (I) tels que définis ci-dessus pour le radical
Am3+, avec A(9 représentant un anion, préférentiellement un anion
pharmaceutiquement acceptable;
- T représente un méthylène;
- Z représente un groupe 3-isopropoxyphényle;
- Ar et R1 sont tels que définis pour les composés de formule (I).
Selon un autre de ses aspects, la présente invention concerne un procédé pour la préparation des composés de formule (I) et de leurs sels, caractérisé en ce que:
1) on traite un composé de formule:

dans laquelle Ar est tel que défini précédemment, R'1 représente un o (Cl- C4)alcoxy(C2-C4)alkylène, un Fhydroxy(C2-C4)alkylène, un > (C1-
C4)alkylthio(C2-C4)alkylène, un #-(C1-C4)alkylcarbonyl(C2-C4)alkylène, un o- carboxy(C2-C4)alkylène, un #(-(C1-C4)alcoxycarbonyl(C2-C4)alkylène, étant entendu que lorsque R'1 représente un #-hydroaxy(C2-C4)alkylène, l'hydroxyle peut être protégé, et E représente l'hydrogène ou un groupe Oprotecteur tel que par exemple le tétrahydropyran-2-yle,
- soit avec un dérivé fonctionnel d'un acide de formule:
HOCO-T'-Z (n') dans laquelle Z est tel que défini précédemment et T' représente une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'alcoxy est en C1-
C4, un groupe aikylène en C1-Cs ou un vinylène, lorsqu'on doit préparer un composé de formule (I) où T est une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'alcoxy est en C1 -C4, un groupe aikylène en C1 -C5 ou un vinylène,
- soit avec un chloroformiate de formule:
CICOOZ R dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (l)où T est un oxygène,
- soit avec un isocyanate de formule:
O=C=N-Z (V) dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (1) où T est un groupe -NR7- dans lequel R7 est un hydrogène,
- soit avec un chlorure de carbamoyle de formule:

1) on traite un composé de formule:
dans laquelle Ar est tel que défini précédemment, R'1 représente un o (Cl- C4)alcoxy(C2-C4)alkylène, un Fhydroxy(C2-C4)alkylène, un > (C1-
C4)alkylthio(C2-C4)alkylène, un #-(C1-C4)alkylcarbonyl(C2-C4)alkylène, un o- carboxy(C2-C4)alkylène, un #(-(C1-C4)alcoxycarbonyl(C2-C4)alkylène, étant entendu que lorsque R'1 représente un #-hydroaxy(C2-C4)alkylène, l'hydroxyle peut être protégé, et E représente l'hydrogène ou un groupe Oprotecteur tel que par exemple le tétrahydropyran-2-yle,
- soit avec un dérivé fonctionnel d'un acide de formule:
HOCO-T'-Z (n') dans laquelle Z est tel que défini précédemment et T' représente une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'alcoxy est en C1-
C4, un groupe aikylène en C1-Cs ou un vinylène, lorsqu'on doit préparer un composé de formule (I) où T est une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'alcoxy est en C1 -C4, un groupe aikylène en C1 -C5 ou un vinylène,
- soit avec un chloroformiate de formule:
CICOOZ R dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (l)où T est un oxygène,
- soit avec un isocyanate de formule:
O=C=N-Z (V) dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (1) où T est un groupe -NR7- dans lequel R7 est un hydrogène,
- soit avec un chlorure de carbamoyle de formule:
dans laquelle Z est tel que défini précédemment, et R'7 représente un alkylc cn
C1-C4, lorsqu'on doit préparer un composé de formule (1) où T est un groupe -NR7dans lequel R7 est un aikyle en C1-C4, pour obtenir un composé de formule:

C1-C4, lorsqu'on doit préparer un composé de formule (1) où T est un groupe -NR7dans lequel R7 est un aikyle en C1-C4, pour obtenir un composé de formule:
2) éventuellement lorsque R'1 représente un oe-hydroxy(C2-C4)aIkylène, on effectue une réaction d'O-acylation pour obtenir un composé de formule:

dans laquelle E, Ar, T et Z sont tels que définis précédemment et R"1 représente un J (C1-C4)alkylcarbonyloxy(C2-C4)alkylène ou un #-benzoyloxy(C2-C4)alkylène, ou on protège l'hydroxyle;
3) puis on hydrolyse le composé ainsi obtenu à l'étape 1) ou à l'étape 2), pour obtenir l'alcool de formule:

dans laquelle E, Ar, T et Z sont tels que définis précédemment et R"1 représente un J (C1-C4)alkylcarbonyloxy(C2-C4)alkylène ou un #-benzoyloxy(C2-C4)alkylène, ou on protège l'hydroxyle;
3) puis on hydrolyse le composé ainsi obtenu à l'étape 1) ou à l'étape 2), pour obtenir l'alcool de formule:
4) on traite l'alcool (VIII) avec le chlorure de méthanesulfonyle pour obtenir le mésylate de formule:

S) on fait réagir le mésylate (
- soit avec une amine secondaire cyclique de formule:

dans laquelle J1 est tel que défini précédemment pour (I), étant entendu que lorsque J1 représente le groupe:
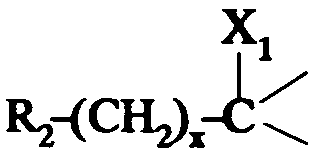
où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés;
- soit avec une amine secondaire cyclique de formule:

dans laquelle J2 est tel que défini précédemment pour (1),
- soit avec une amine tertiaire cyclique de formule:

dans laquelle X3, X4 et X5 sont tels que définis préodemment pour (I) ; et
6) - soit, lorsqu'on utilise une amine secondaire de formule (X) ou (xI), et après déprotection éventuelle des groupes hydroxyles ou du groupe amino, on transforme éventuellement, le produit ainsi obtenu en un dc ses sels avec un acide minéral ou organique ou un de ses sels d'ammonium quaternaire,
- soit, lorsqu'on utilise une amine tertiaire dc formule (XII) et après déprotectiou
éventuelle du groupe hydroxyle, on isole le produit ainsi obtenu ou bien,
éventuellement on échange l'anion méthanesulfonate du sel quaternaire ainsi
obtenu avec un autre anion, par exemple un anion pharmaccutiquement
acceptable.
- soit avec une amine secondaire cyclique de formule:
dans laquelle J1 est tel que défini précédemment pour (I), étant entendu que lorsque J1 représente le groupe:
où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés;
- soit avec une amine secondaire cyclique de formule:
dans laquelle J2 est tel que défini précédemment pour (1),
- soit avec une amine tertiaire cyclique de formule:
dans laquelle X3, X4 et X5 sont tels que définis préodemment pour (I) ; et
6) - soit, lorsqu'on utilise une amine secondaire de formule (X) ou (xI), et après déprotection éventuelle des groupes hydroxyles ou du groupe amino, on transforme éventuellement, le produit ainsi obtenu en un dc ses sels avec un acide minéral ou organique ou un de ses sels d'ammonium quaternaire,
- soit, lorsqu'on utilise une amine tertiaire dc formule (XII) et après déprotectiou
éventuelle du groupe hydroxyle, on isole le produit ainsi obtenu ou bien,
éventuellement on échange l'anion méthanesulfonate du sel quaternaire ainsi
obtenu avec un autre anion, par exemple un anion pharmaccutiquement
acceptable.
Selon une variante du procédé, lorsque Am représente Am1 ou Am2
1') on traite un composé de formule:

dans laquelle Ar et R1 sont tels que définis précédemment et J représente un groupe J1 ou un groupe J2 tel que définis précédemment, étant entendu que lorsque J1 représente le groupe:

où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés, et/ou lorsque
R1 représente un groupe -hydroxy(C2-C4)alkylène, l'hydroxyle peut être protégé, avec l'un des composés (E), (il), (V) ou (VI) tels que définis précédemment, et
2') après déprotection éventuelle des groupes hydroxyles ou du groupe amino, on transforme éventuellement le produit obtenu en un de ses sels avec un acide minéral ou organique ou en un de ses sels d'ammonium quatcrnaire.
1') on traite un composé de formule:
dans laquelle Ar et R1 sont tels que définis précédemment et J représente un groupe J1 ou un groupe J2 tel que définis précédemment, étant entendu que lorsque J1 représente le groupe:
où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés, et/ou lorsque
R1 représente un groupe -hydroxy(C2-C4)alkylène, l'hydroxyle peut être protégé, avec l'un des composés (E), (il), (V) ou (VI) tels que définis précédemment, et
2') après déprotection éventuelle des groupes hydroxyles ou du groupe amino, on transforme éventuellement le produit obtenu en un de ses sels avec un acide minéral ou organique ou en un de ses sels d'ammonium quatcrnaire.
Les composés de formule:

à la condition que R'1 soit différent d'un #-(C1-C4)alcoxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
à la condition que R'1 soit différent d'un #-(C1-C4)alcoxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
Les composés de formule:

à la condition que R1 soit différent d'un {Cl-C4)alcoxy(C2-C4)alkylène ou d'un #-alkylcarbonyloxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
à la condition que R1 soit différent d'un {Cl-C4)alcoxy(C2-C4)alkylène ou d'un #-alkylcarbonyloxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
Les composés de formule:

à la condition que R1 soit différent d'un o > -(C1-C4)alcoxy(C2-C4)aIkylène ou d'un #-(C1-C4)alkylcarbonyloxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
à la condition que R1 soit différent d'un o > -(C1-C4)alcoxy(C2-C4)aIkylène ou d'un #-(C1-C4)alkylcarbonyloxy(C2-C4)alkylène, sont nouveaux et font partie de l'invention.
Les composés de formule:

sont nouveaux et font partie de l'invention.
sont nouveaux et font partie de l'invention.
Les groupes O-protecteurs éventuellement utilisés pour obtenir un composé de formule (f) dans laquelle R1 représente un w-hydroxy(C2-C4)aaaylène et/ou X1 représente un hydroxy sont les groupes O-protecteurs classiques bien connus de l'homme de l'art tels que, par exemple, le tétrahydropyran-2-yle, I'acétyle ou le benzoyle.
Les groupes N-protecteurs éventuellement utilisés pour obtenir un composé de formule (I) dans laquelle X1 représente un amino sont les groupes N-protecteurs classiques bien connus de l'homme de l'art tels que, par exemple, le groupe trityle, méthoxytrityle, ten-butoxycarbonyle ou benzyloxycarbonyle.
De façon particulière, lorsqu'on utilise comme groupe O-protecteur un groupe acétyle ou un groupe benzoyle, le composé de formule (1) obtenu représente le produit final dans lequel R1 représente un -acétoxy(C2-C4)alkylène et/ou X1 représente un acétoxy ou R1 représente un -benzoyloxy(C2-C4)alkylène et/ou X1 représente un benzoyloxy.
Selon que t'on veut obtenir un composé de formule (I) totalement ou partiellement protégé, on choisit les groupes Oprotecteurs ct/ou N-protecteurs appropriés.
Dans l'étape 1) ou dans l'étape 1'), comme dénvé fonctionnel de l'acide (III), on utilise l'acide lui-même, convenablement activé par exemple par le 1,3dicylohexylcarbodiimide ou par l'hexafluorophosphate de benzotriazol-1- yloxytris(diméthylamino)phosphonium (BOP), ou bien un des dérivés fonctionnels qui réagissent avec les amines, par exemple un anhydride, un anhydride mixte, le chlorure d'acide, ou un ester activé, comme l'ester de paranitrophényle.
Lorsqu'on utilise un chloroformiate de formule (W) la réaction s'effectue dans un solvant tel que le dichlorométhane, à une température comprise entre OeC et la température ambiante et en présence d'une base telle que la triéthylamine.
Lorsqu'on utilise un isocyanate de formule (V) la réaction s'effectue dans un solvant inerte tel que le dichlorométhane ou le benzène pendant une nuit à tempature ambiante.
Lorsqu'on utilise un chlorure de carbamoyle de formule (VI), la réaction s'effectue dans un solvant tel que le toluène ou le 1,2-dichloroéthane, à une température comprise entre 80 et 1100C et en présence d'une base telle que la triéthylamine.
Lorsque comme produit de départ on utilise un composé de formule (II) où E représente un tétrahydropyran-2-yle (top), le procédé de la présente invention peut être représenté et illustré par le Schéma 1 ci-après:
SCHEMA 1
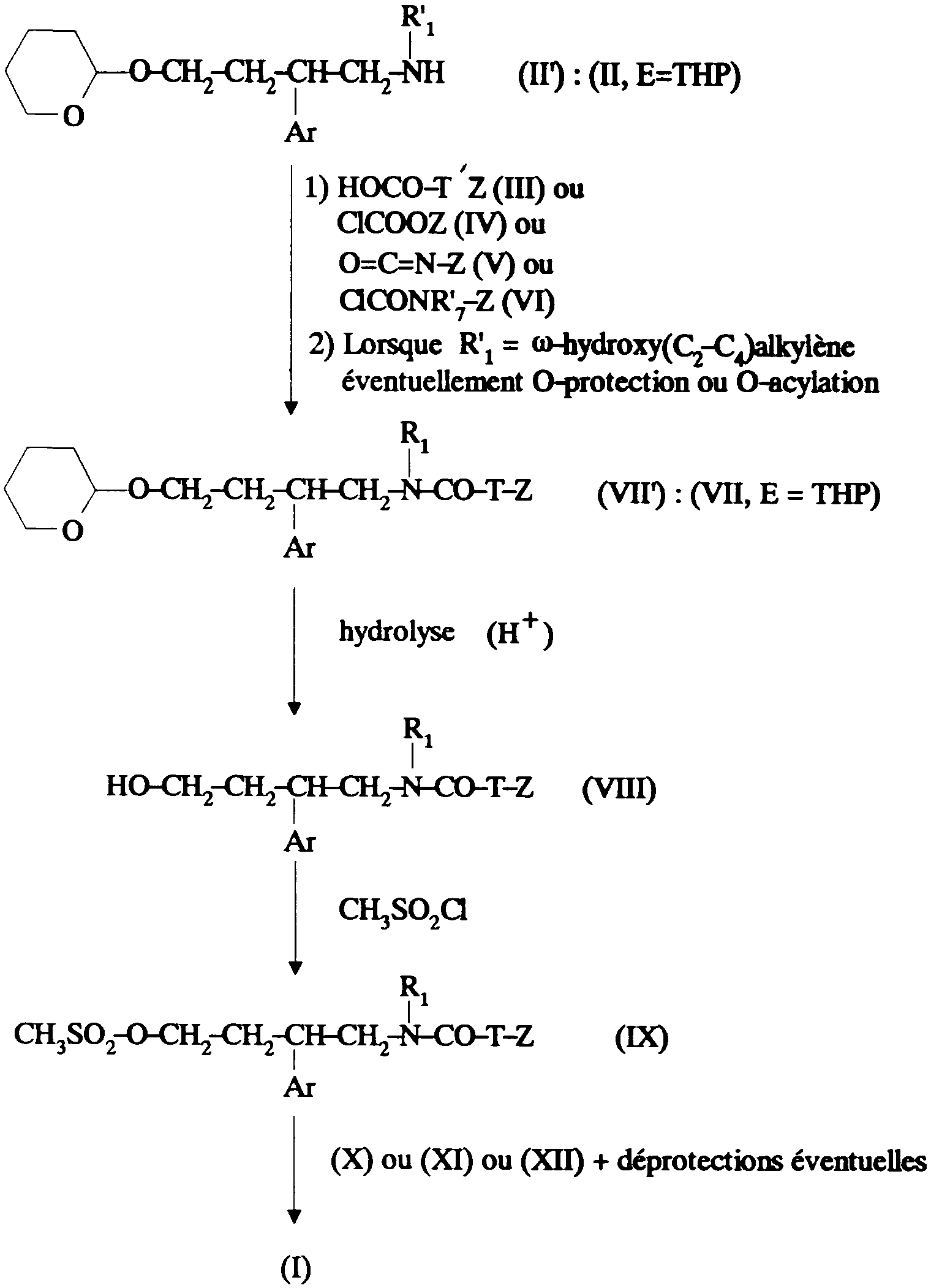
SCHEMA 1
<tb> <SEP> R'
<tb> <SEP> O <SEP> : <SEP> (11, <SEP> E='flIP)
<tb> <SEP> o <SEP> Ar
<tb> <SEP> 1) <SEP> HOCO-T <SEP> Z <SEP> (III) <SEP> ou
<tb> <SEP> CICOOZ <SEP> (IV) <SEP> ou
<tb> <SEP> O=C=N(V)ou
<tb> <SEP> dCONR'-Z <SEP> (VI)
<tb> <SEP> 2) <SEP> Lorsque <SEP> R'1 <SEP> = <SEP> (shydrozcy(C2-C4)al41ène
<tb> <SEP> éventuellement <SEP> Osrotection <SEP> ou <SEP> Osculation
<tb> R1
<tb> GCH2-CH2tH2 <SEP> lRl <SEP> T-Z <SEP> (VII): <SEP> (VII, <SEP> E <SEP> = <SEP> THP)
<tb> <SEP> Ar
<tb> <SEP> hydrolyse <SEP> (H <SEP> +)
<tb> <SEP> R1
<tb> <SEP> HO < H2 <SEP> CH2vf T-Z <SEP> (VII <SEP> I)
<tb> <SEP> Ar
<tb> <SEP> i <SEP> CH3SO2C1
<tb> <SEP> R1
<tb> <SEP> CH,SO <SEP> ICH-CH2AC <SEP> T <SEP> Z <SEP> (IX)
<tb> <SEP> I
<tb> <SEP> (X) <SEP> ou <SEP> (XI) <SEP> ou <SEP> (XII) <SEP> + <SEP> déprotections <SEP> éventuelles
<tb> <SEP> (I)
<tb>
Les réactions du composé (II') avec les réactifs (III), (IV), (V) ou (VI) se déroulent comme décrit ci-dessus.
<tb> <SEP> O <SEP> : <SEP> (11, <SEP> E='flIP)
<tb> <SEP> o <SEP> Ar
<tb> <SEP> 1) <SEP> HOCO-T <SEP> Z <SEP> (III) <SEP> ou
<tb> <SEP> CICOOZ <SEP> (IV) <SEP> ou
<tb> <SEP> O=C=N(V)ou
<tb> <SEP> dCONR'-Z <SEP> (VI)
<tb> <SEP> 2) <SEP> Lorsque <SEP> R'1 <SEP> = <SEP> (shydrozcy(C2-C4)al41ène
<tb> <SEP> éventuellement <SEP> Osrotection <SEP> ou <SEP> Osculation
<tb> R1
<tb> GCH2-CH2tH2 <SEP> lRl <SEP> T-Z <SEP> (VII): <SEP> (VII, <SEP> E <SEP> = <SEP> THP)
<tb> <SEP> Ar
<tb> <SEP> hydrolyse <SEP> (H <SEP> +)
<tb> <SEP> R1
<tb> <SEP> HO < H2 <SEP> CH2vf T-Z <SEP> (VII <SEP> I)
<tb> <SEP> Ar
<tb> <SEP> i <SEP> CH3SO2C1
<tb> <SEP> R1
<tb> <SEP> CH,SO <SEP> ICH-CH2AC <SEP> T <SEP> Z <SEP> (IX)
<tb> <SEP> I
<tb> <SEP> (X) <SEP> ou <SEP> (XI) <SEP> ou <SEP> (XII) <SEP> + <SEP> déprotections <SEP> éventuelles
<tb> <SEP> (I)
<tb>
Les réactions du composé (II') avec les réactifs (III), (IV), (V) ou (VI) se déroulent comme décrit ci-dessus.
Dans le cas particulier où R'1 représente un ù)-hydroxy(C2-C4)aa:ylène, éventuellement on protège l'hydroxyle, ou éventuellement on effectue une réaction d'O-acylation selon les méthodes habituelles.
L'intermédiaire (VII') ainsi obtenu est déprotégé par hydrolyse acide pour conduire au composé (vus), étant entendu que lorsque R1 représente un ehydroxy(C2- C4)alkylène et que l'hydroxyle est protégé, ce groupe protecteur est conservé dans ces conditions opératoires.
On prépare ensuite le mésylate (IN pour le substituer par une amine secondaire cyclique de formule (X) ou (XI) ou par une amine tertiaire cyclique de formule (XII).
On obtient finalement après déprotections éventuelles des groupes hydroxyles ou du groupe amino, les composés (I) selon l'invention.
Lorsqu'on fait réagir, selon une variante du procédé, le composé (Xm) avec l'un des composés (III), (il), (V) ou (vu), le produit obtenu peut soit représenter le produit final, soit posséder un ou plusieurs groupes protecteurs. Dans ce dernier cas, les groupes O-protecteurs et/ou les groupes N-protecteurs sont éventuellement hydrolysés selon les méthodes habituelles.
Les produits de formule (I) ainsi obtenus sont:
- soit isolés sous forme de base libre ou de sel, selon les techniques classiques, lorsque Am représente Aml ou Am2,
- soit, lorsque Am représente Am3, on isole le produit ainsi obtenu ou bien, éventuellement on échange l'anion méthanesulfonate du sel quaternaire ainsi obtenu avec un autre anion pharmaceutiquement acceptable.
- soit isolés sous forme de base libre ou de sel, selon les techniques classiques, lorsque Am représente Aml ou Am2,
- soit, lorsque Am représente Am3, on isole le produit ainsi obtenu ou bien, éventuellement on échange l'anion méthanesulfonate du sel quaternaire ainsi obtenu avec un autre anion pharmaceutiquement acceptable.
Lorsque le composé de formule (I) dans laquelle Am représente Aml ou Am2 est obtenu sous forme de base libre, la salification est effectuée par traitement avec l'acide choisi dans un solvant organique. Par traitement de la base libre, dissoute par exemple dans un alcool tel que l'isopropanol, avec une solution de l'acide choisi dans le même solvant, on obtient le sel correspondant qui est isolé selon les techniques classiques.
Ainsi, on prépare par exemple le chlorhydrate, le bromhydrate, le sulfate, l'hydrogénosulfate, le dihydrogénophosphate, le méthanesulfonate, l'oxalate, le maléate, le fumarate, le naphtalène-2 sulfonate.
A la fin de la réaction, les composés de formule (I) dans laquelle Am représente
Am1 ou Art2 peuvent être isolés sous forme d'un de leurs sels, par exemple le chlorhydrate, ou l'oxalate ; dans ce cas, s'il est nécessaire, la base libre peut être préparée par neutralisation dudit sel avec une base minérale ou organique, telle que l'hydroxyde de sodium ou la triéthylamine ou avec un carbonate ou bicarbonate alcalin, tel que le carbonate ou bicarbonate de sodium ou de potassium.
Am1 ou Art2 peuvent être isolés sous forme d'un de leurs sels, par exemple le chlorhydrate, ou l'oxalate ; dans ce cas, s'il est nécessaire, la base libre peut être préparée par neutralisation dudit sel avec une base minérale ou organique, telle que l'hydroxyde de sodium ou la triéthylamine ou avec un carbonate ou bicarbonate alcalin, tel que le carbonate ou bicarbonate de sodium ou de potassium.
L'anion méthanesulfonate issu de la réaction entre l'amine tertiaire de formule (XII) et le dérivé méthanesulfonyloxy de formule (1 peut être échangé, in situ ou après isolement du composé de formule (I) dans laquelle Am représente un groupe
Am3 dans lequel A(t est l'ion méthanesulfonate, par un autre anion A, selon les méthodes conventionnelles, par exemple par échange en solution avec une solution saturée de chlorure de sodium ou avec une solution d'acide chlorhydrique lorsque
A représente un anion chlorure ou par échange de l'anion par élution du composé (I) sur une résine échangeuse d'ion, par exemple l'Amberlite IRA68 ou
Duolite A375 (8).
Am3 dans lequel A(t est l'ion méthanesulfonate, par un autre anion A, selon les méthodes conventionnelles, par exemple par échange en solution avec une solution saturée de chlorure de sodium ou avec une solution d'acide chlorhydrique lorsque
A représente un anion chlorure ou par échange de l'anion par élution du composé (I) sur une résine échangeuse d'ion, par exemple l'Amberlite IRA68 ou
Duolite A375 (8).
Les sels d'ammonium quaternaires formés avec l'azote de la pipéridine sont préparés par réaction des bases libres des composés de formule (1) dans laquelle Am représente Aml, pour lesquelles les fonctions aminées autres, éventuellement présentes sont N-protégées par un groupe N-protecteur habituel, tel que par exemple le tertbutoxycarbonyle, avec un excès d'agent alkylant de formule:
A-Q dans lequel A est tel que défini précédemment pour (I), de préférence un chlorure ou un iodure et Q est tel que défini précédemment pour (I) et on chauffe le mélange réactionnel dans un solvant, par exemple le dichlorométhane, le chloroforme, l'acétone ou l'acétonitrile, à une température comprise entre la température ambiante et le reflux pendant une à plusieurs heures pour obtenir après traitement selon les méthodes habituelles et après déprotection éventuelle, un mélange des isomères axiaux et équatoriaux des sels d'ammonium quaternaires.
A-Q dans lequel A est tel que défini précédemment pour (I), de préférence un chlorure ou un iodure et Q est tel que défini précédemment pour (I) et on chauffe le mélange réactionnel dans un solvant, par exemple le dichlorométhane, le chloroforme, l'acétone ou l'acétonitrile, à une température comprise entre la température ambiante et le reflux pendant une à plusieurs heures pour obtenir après traitement selon les méthodes habituelles et après déprotection éventuelle, un mélange des isomères axiaux et équatoriaux des sels d'ammonium quaternaires.
De préférence, A représente un iodure qui peut être échangé par un autre anion ou par un anion pharmacologiquement acceptable, par exemple un chlorure, par élution du composé (I) sur une résine échangeuse d'ion, par exemple l'Amberlite IRA68 t) ou
Duolite A375(!).
Duolite A375(!).
Les isomères sont séparés selon les méthodes habituelles, par exemple par chromatographie ou par recristallisation.
La résolution des mélanges racémiques (I) permet d'isoler les énantiomères.
Les composés de départ de formule (Il) sont préparés à partir de nitriles de formule

dans laquelle E et Ar sont tels que définis ci-dessus, par réduction puis substitution de l'amine obtenue.
dans laquelle E et Ar sont tels que définis ci-dessus, par réduction puis substitution de l'amine obtenue.
La réduction des nitriles de formule (XW)s'effectue par hydrogénation dans un alcanol tel que l'éthanol, en présence d'un catalyseur tel que par exemple le nickel de
Raney 8 et on obtient, après isolation selon les méthodes classiques, l'amine primaire de formule:
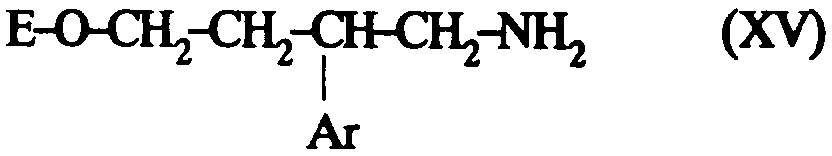
Raney 8 et on obtient, après isolation selon les méthodes classiques, l'amine primaire de formule:
Pour préparer un composé de formule (11) dans laquelle R'1 représente un groupe o-hydIoxy(C2-C4)alkylène, on traite l'amine (XV) par le chloure d'éthyloxalyle, par l'hémimalonate d'éthyle ou par l'hémisuccinate d'éthyle par exemple et on obtient les dérivés N-acylés correspondants. Les groupes carbonyles sont alors réduits selon les méthodes habituelles, telles que l'action d'un agent réducteur comme par exemple un hydrure métallique, tel que l'hydrure de lithium ct d'aluminium ou par un hydrure du bore, tel que le diméthylsulfure de borane. La réduction est réalisée dans un solvant, tel que le tétrahydrofurane ou le toluène à une température comprise entre la température ambiante et 60-C. L'amine ainsi obtenue de formule:

(II, R'= #-hydroxy(C2-C4)alkylène) est isolée selon les méthodes habituelles.
(II, R'= #-hydroxy(C2-C4)alkylène) est isolée selon les méthodes habituelles.
De façon particulière on peut utiliser la réaction de l'acide glycolique avec l'amine (XV), en présence de BOP et de triéthylamine, pour obtenir le dérivé N-acylé correspondant qui, après réduction du groupe carbonyle, permet d'obtenir un composé de formule (11) dans laquelle R'1 représente un groupe 2-hydroxyéthyle.
Lorsque le groupe w-hydroxy(C2-C4)alkylène constitue le groupe R1 du composé final de formule (I), il est nécessaire de protéger, la fonction hydroxyle. Cette étape de protection peut s'effectuer de manière classique soit au niveau du composé (II), soit au niveau du composé (VII), en utilisant un groupe Oprotecteur qui ne sera pas hydrolysé lors de la déprotection du groupe E (étape 3).
Pour préparer un composé de formule (II) dans laquelle R'1 représente un groupe -(Cl-C4)alcoxy(C2-C4)alkylène, on fait réagir l'amine (XV) avec un acide * (Cî-C4)alcoxy(C2-C4)alcanoïque, en présence de BOP et d'une base telle que la triéthylamine, puis on réduit le groupe carbonyle du dérivé N-acylé obtenu intermédiairement.
Pour préparer un composé de formule (11) dans laquelle R'1 représente un #- alcoxycarbonyl(C2-C4)alkylène, on fait réagir l'amine (XV) avec un ehalogéno(C2- C4 > atkylènecarboxylate de (C1-C4) alkyle tel que par exemple le 3-bromopropionate d'éthyle, le 4-bromobutyrate d'éthyle ou le 5-bromovalérate méthyle
L'hydrolyse des composés de formule (II) dans laquelle R'1 représente un o-(C1- C4)alcoxycaibonyl(C2-C4)alkylène conduit, selon les méthodes classiques, aux composés de formule (11) dans laquelle R'1 représente un oe-carboxy(C2-C4)alkylène.
L'hydrolyse des composés de formule (II) dans laquelle R'1 représente un o-(C1- C4)alcoxycaibonyl(C2-C4)alkylène conduit, selon les méthodes classiques, aux composés de formule (11) dans laquelle R'1 représente un oe-carboxy(C2-C4)alkylène.
Pour préparer un composé de formule (11) dans laquelle R'1 représente un alkylthlo(C2-C4)alkylène, on fait réagir l'amine (XV) avec un #-halogéno(C2- C4)alkylènethioaLkyle(C1-C4) tel que par exemple le 2-chloro-1-méthylthioéthane.
Les nitriles de formule (XW) sont préparés à partir de nitriles commerciaux ou connus de formule:
Ar-CH2-CN (XVI) par alkylation avec un composé de formule: E-OCH2-CH2-G (XVII) dans laquelle E est tel que défini ci-dessus et G est un atome d'halogène, par exemple le brome.
Ar-CH2-CN (XVI) par alkylation avec un composé de formule: E-OCH2-CH2-G (XVII) dans laquelle E est tel que défini ci-dessus et G est un atome d'halogène, par exemple le brome.
De préférence, la synthèse des nitriles de formule (XIV) où E est un groupe tétrahydropyran-2-yle est réalisé à partir d'un dérivé tétrahydropyranyloxy obtenu par réaction entre un alcanol de formule Br CH2)2-OH et le 3,4-dihydro-2H-pyrane pour conduire au composé:

qui est ensuite mis en réaction, en présence d'hydrure alcalin avec le dérivé acétonitrile (XVI) pour préparer l'intermédiaire,

qui est ensuite mis en réaction, en présence d'hydrure alcalin avec le dérivé acétonitrile (XVI) pour préparer l'intermédiaire,
Les composés de départ de formule (Xm) sont préparés à partir des nitriles de formule:

dans laquelle J et Ar sont tels que définis précédemment, par réduction puis substitution de l'amine obtenue.
La réduction des nitriles (XVIII) s'effectue selon le procédé décrit précdemment pour la préparation des composés de formule (II). On obtient l'amine primaire de formule:

La substitution de l'amine primaire (XIX) par le groupe R1 s'effectue en utilisant les mêmes procédés que ceux décrits précédemment pour la préparation des composés de formule (II). Dans le cas particulier où R1 est un ehydroxy(C2-C4)alkylène, éventuellement après protection de l'amine secondaire obtenue, on effectue une O acylation, puis après déprotection de l'amine, on obtient un composé de formule tXE) dans laquelle R1 est un #-(C1-C4)alkylcarbonyloxy(C2-C4)alkylène ou un o > - benzoyloxy(C2-C4)alkylène.
La synthèse des nitriles de formule (XVIII) est effectuée selon des méthodes connues en mettant en réaction sur des dérivés chlorés de formule:
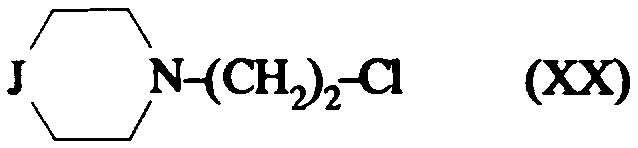
un dérivé nitrile de formule:
Ar-CH2-CN (XVI) en présence d'amidure de sodium dans un solvant tel que le toluène à des températures comprises entre 30 et 80-C.
un dérivé nitrile de formule:
Ar-CH2-CN (XVI) en présence d'amidure de sodium dans un solvant tel que le toluène à des températures comprises entre 30 et 80-C.
Le dérivé chloré (XX) est préparé par action d'un réactif chlorant tel que par exemple le chlorure de thionyle sur le dérivé hydroxylé de formule:

lui-même préparé à partir de l'amine de formule:

sur laquelle on fait réagir, l'oxyde d'éthylène étant entendu que lorsque J1 représente le groupe:

où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés.
lui-même préparé à partir de l'amine de formule:
sur laquelle on fait réagir, l'oxyde d'éthylène étant entendu que lorsque J1 représente le groupe:
où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés.
Les pipéridines de formule (X) sont connues ou prépares par des méthodes connues, telles que celles décrites dans EP-A-0428434, EP-A-0474561, EP-A0515240 et EP-A-559438.
Par exemple, lorsque dans une pipéridine de formule (X), J1 représente un groupe
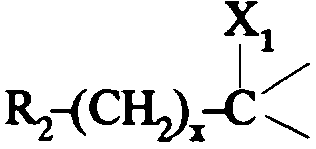
dans lequel R2 est un pyrid2yle, x est zéro et X1 est l'hydroxyle, on fait réagir la 2-bromopyridine sur la N-benzyl-4-pipéridone dans un solvant en présence de butyllithium pour préparer la N-benzyl-4-hydrxy-4tpyrid-2-ylcF pipéridine.
dans lequel R2 est un pyrid2yle, x est zéro et X1 est l'hydroxyle, on fait réagir la 2-bromopyridine sur la N-benzyl-4-pipéridone dans un solvant en présence de butyllithium pour préparer la N-benzyl-4-hydrxy-4tpyrid-2-ylcF pipéridine.
Les composés de formule (X) dans laquelle X1 représente un hydroxyle ct qui portent un groupe protecteur sur l'azote de la pipéridine, peuvent subir une réaction de
Ritter par action de l'acétonitrile pour préparer les composés de formule (X) dans laquelle X1 est un acétamido.
Ritter par action de l'acétonitrile pour préparer les composés de formule (X) dans laquelle X1 est un acétamido.
Par hydrolyse en milieu acide, on prépare ensuite les composés de formule (X) dans laquelle X1 est un amino. Eventuellement, on peut effectuer la substitution du groupe amino par un groupe R'3 = alkyle en C1 -C4. Par action d'un dérivé fonctionnel d'un acide R4COOH, on prépare les composés de formule (X) dans laquelle X1 est le groupe R4CONR3.
Les pipérazines de formule (Xl) sont connues ou prépayées par des méthodes connues, telles que celles décrites dans EP-A-0474561.
Les amines de formule (XII) sont celles décrites dans la littérature, par exemple celles citées ci-après:
(a') 1-azabicyclo[2.2.0]hexane préparé selon CA. Grob et al., Helv. Chim. Acta,
1964; (47), 8, 2145-55.

(a') 1-azabicyclo[2.2.0]hexane préparé selon CA. Grob et al., Helv. Chim. Acta,
1964; (47), 8, 2145-55.
(b') 1-azabicyclo[3.1.0]hexane préparé selon A.L Logothetis, J. Am. Chem. Soc., 1965, (87), 4, 749-754.

(c') 1-azabicyclo[2.2.1]heptane préparé selon Gassman et al., J. Am. Chem. Soc., 1968, (90), 5, 1355-6.

(d') 1-azabicyclo[2.2.2]octane ou quinuclidine.

(e') 1-azabicyclo[3.2.1]octane préparé selon B. Thill et al., J. Org. Chem., 1968, (33), 12,4376-80.

(P) 1-azabicyclo[3.2.2]nonane préparé selon C. Ruggles et al., J. Am. Chez.
Soc., 1988, (110), 17, 5692-8.

(g') 1-azabicyclo[3.3.1]nonane préparé selon S. Miyano et al., J. Chem. Soc.,
Perkin Trans 1, 1988,5, 1057-63.

Perkin Trans 1, 1988,5, 1057-63.
(h') hexahydro 1H-pyrrolizine-4 préparée selon P. Edwards et al., Tetrahedron
Letters, 1984, (25), 9, 939-42.

(i') octahydro indolizine-4 préparée selon J. Chastanet et al., J. Org. Chem., 1985, (50), 16, 2910-14.

Letters, 1984, (25), 9, 939-42.
(i') octahydro indolizine-4 préparée selon J. Chastanet et al., J. Org. Chem., 1985, (50), 16, 2910-14.
(.i') octahydro 2H-quinolizine-5 préparée selon P. Edwards et al., Tetrahedron
Letters, 1984, (25), 9, 939-42.

Letters, 1984, (25), 9, 939-42.
(k') 1-azatricyclo[3.3.1.1.3.7]décane, ou 1-azadamantane, préparée selon Y.
Bubnov et al., J. Organomet. Chem., 1991, 412 (1-2), 1-8.

(1') 4-phényl-1-azabicydo[2.2.2joctane, ou phényl-4 quinuclidine, préparé selon T. Perrine, J. Org. Chem., 1957, 22, 1484-1489.

(1') 4-phényl-1-azabicydo[2.2.2joctane, ou phényl-4 quinuclidine, préparé selon T. Perrine, J. Org. Chem., 1957, 22, 1484-1489.
La résolution des mélanges racémiques (')permet d'isoler les énantiomères (1*).
fi est cependant préférable d'effectuer le dédoublement des racémiques sur les aminoalcools intermédiaires de formule:

susceptibles de donner des sels avec des acides optiquement actifs.
susceptibles de donner des sels avec des acides optiquement actifs.
Les aminoalcools (XV, E = H) s'obtiennent à partir des composés (XV*, E = THP) après déprotection par hydrolyse en milieu acide.
Les énantiomères (XV*, E = H) sont alors séparés par des méthodes classiques telles que celles décrites dans EP-A-0428434 et EP-AX474561.
La préparation des composés de formule (VffI*) optiquement purs est illustrée dans le SCHEMA 2 ci-après où "*" signifie que l'atome de carbone ainsi marqué est de configuration (+) ou (-) déterminée.
SCHEMA 2

I;)CHMiLCYNH <SEP> (XV):(E <SEP> T
<tb> <SEP> Ar
<tb> <SEP> Ar
<tb> <SEP> HO{: <SEP> 1FCHz <SEP> NH2 <SEP> (XV) <SEP> :(E=H)
<tb> <SEP> Ar
<tb> <SEP> séparation <SEP> des <SEP> énantiomères
<tb> <SEP> *
<tb> <SEP> HO{H24I2wl2 <SEP> NH2 <SEP> (XV*) <SEP> : <SEP> (E <SEP> = <SEP> H)
<tb> <SEP> I
<tb> <SEP> Ar
<tb> <SEP> *
<tb> 3L.
<tb>
<tb> <SEP> Ar
<tb> <SEP> Ar
<tb> <SEP> HO{: <SEP> 1FCHz <SEP> NH2 <SEP> (XV) <SEP> :(E=H)
<tb> <SEP> Ar
<tb> <SEP> séparation <SEP> des <SEP> énantiomères
<tb> <SEP> *
<tb> <SEP> HO{H24I2wl2 <SEP> NH2 <SEP> (XV*) <SEP> : <SEP> (E <SEP> = <SEP> H)
<tb> <SEP> I
<tb> <SEP> Ar
<tb> <SEP> *
<tb> 3L.
<tb>
<SEP> 0 <SEP> (XV*) <SEP> :(E <SEP> TIP)
<tb> <SEP> l <SEP> R'
<tb> R', <SEP> (Il'
<tb> <SEP> <SEP> Ar <SEP> H
<tb> <SEP> 1) <SEP> (II <SEP> ou(N)ou <SEP> 03 <SEP> ou <SEP> (vI) <SEP> ,
<tb> <SEP> 2) <SEP> Osculation <SEP> éventuelle <SEP> lorsque
<tb> <SEP> R'1 <SEP> = <SEP> (shydroxy(C2{:4)alkylène
<tb> <SEP> R1
<tb> <SEP> CZCICT-Z <SEP> (VII'*)
<tb> <SEP> O <SEP> Ar
<tb> <SEP> H+
<tb> <SEP> (VIII*)
<tb>
Le composé ainsi obtenu de formule:

est alors soumis à l'action du chlorure de méthanesulfonyle pour conduire au dérivé (IX*) optiquement pur de formule:

<tb> <SEP> l <SEP> R'
<tb> R', <SEP> (Il'
<tb> <SEP> <SEP> Ar <SEP> H
<tb> <SEP> 1) <SEP> (II <SEP> ou(N)ou <SEP> 03 <SEP> ou <SEP> (vI) <SEP> ,
<tb> <SEP> 2) <SEP> Osculation <SEP> éventuelle <SEP> lorsque
<tb> <SEP> R'1 <SEP> = <SEP> (shydroxy(C2{:4)alkylène
<tb> <SEP> R1
<tb> <SEP> CZCICT-Z <SEP> (VII'*)
<tb> <SEP> O <SEP> Ar
<tb> <SEP> H+
<tb> <SEP> (VIII*)
<tb>
Le composé ainsi obtenu de formule:
est alors soumis à l'action du chlorure de méthanesulfonyle pour conduire au dérivé (IX*) optiquement pur de formule:
Les produits de formule (I) dans laquelle T représente un groupe hydroxyméthylène, alcoxyméthylène en C1 -C4, possèdent deux centres d'asymétrie.
Dans ce cas, les diastéréoisomères et les isomères purs peuvent être préparés par réaction de l'aminoalcool optiquement pur (in*, E = H) et de l'acide HOCOT"-Ar soit optiquement pur, soit racémique, dans lequel r représente un groupe hydroxyméthylène ou un groupe alcoxyméthylène en C1-C4.
La réaction du mésylate (six*) avec un composé de formule (X) ou un composé de formule (XI) ou un composé de formule (XII), selon les conditions décrites pour l'obtention de (I) ci-dessus, permet la préparation des composés (1*), après déprotections éventuelles, et transformation éventuelle des produits ainsi obtenus cn un de leurs sels
Les composés de formule (I) ci-dessus comprennent également ceux dans lesquels un ou plusieurs atomes d'hydrogène ou de carbone ont été remplacés par leur isotope radioactif par exemple le tritium ou le carbone-14. De tels composés marqués sont utiles dans des travaux de recherche, de métabolisme ou de pharmacocinétique, dans des essais biochimiques en tant que ligand de récepteurs.
Les composés de formule (I) ci-dessus comprennent également ceux dans lesquels un ou plusieurs atomes d'hydrogène ou de carbone ont été remplacés par leur isotope radioactif par exemple le tritium ou le carbone-14. De tels composés marqués sont utiles dans des travaux de recherche, de métabolisme ou de pharmacocinétique, dans des essais biochimiques en tant que ligand de récepteurs.
Les composés selon l'invention ont fait l'objet d'essais biochimiques.
Certains composés (I) et (1*) et leurs sels ont montré des propriétés antagonistes de la liaison de la Substance P dans des essais réalisés sur des membranes de cortex de rat et de cellules lymphoblastiques IM9, selon MA. Cascieri et al., J. Biol. Chem., 1983, 258 5158-5164 et D.D. Paya et al., J. Immunol., 1984, 133.3260-3265.
Certains composés et leurs sels ont montré des propriétés antagonistes de la NKA dans des essais réalisés sur des membranes de duodénum de rat, selon L Bergstom et al., J. Immunol., 1987, 2, 764-771.
Les composés qui se sont révélés être de puissants antagonistes de la Substance P inhibent la fixation de celle-ci à son récepteur avec une constante d'inhibition (Ki) comprise entre 10-8 M et 10-10 M dans les différents essais biochimiques réalisés.
Les composés qui se sont révélés être de puissants antagonistes de la Neurokinine
A inhibent la fixation de celle-ci à son récepteur avec une constante d'inhibition (Ki) comprise entre 10-8 M et 10-10 M dans les différents essais biochimiques réalisés.
A inhibent la fixation de celle-ci à son récepteur avec une constante d'inhibition (Ki) comprise entre 10-8 M et 10-10 M dans les différents essais biochimiques réalisés.
Les composés de la présente invention sont notamment des principes actifs dc compositions pharmaceutiques, dont la toxicité est compatible avec leur utilisation en tant que médicaments.
Les composés de formule (I) ci-dessus peuvent être utilisés à des doses journalières de 0,01 à 100 mg par kilo de poids corporel du mammifère à traiter, de préférence à des doses journalières de 0,1 à 50 mg/kg. chez l'être humain, la dose peut varier de préférence de 0,5 à 4000 mg par jour, plus particulièrement de 2,5 à 1000 mg par jour selon l'âge du sujet à traiter ou le type de traitement : prophylactique ou curatif.
Pour leur utilisation comme médicaments, les composés de formule (I) sont généralement administrés en unité de dosage. Lesdites unités de dosage sont dc préférence formulées dans des compositions pharmaceutiques dans lesquellcs le principe actif est mélangé avec un excipient pharmaceutique.
Ainsi, selon un autre de ses aspects, la présente invention concerne des compositions pharmaceutiques renfermant, en tant que principe actif, un composé de formule (I).
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, sublinguale, sous-cutanée, intramusculairc, intraveineuse, transdermique, locale ou rectale, les principes actifs peuvent être administrés sous formes unitaires d'administration, en mélange avec des supports pharmaceutiques classiques, aux animaux et aux êtres humains. Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, les gélules, les poudres, les granules et les solutions ou suspensions orales, les formes d'administration sublinguale et buccale, les formes d'administration sous cutanée, intramusculaire, intraveineuse, intranasale ou intraoculaire et les formes d'administration rectale.
Lorsque l'on prépare une composition solide sous forme de comprimés, on mélange le principe actif principal avec un véhicule pharmaceutique tel que la gélatine, l'amidon, le lactose, le stéarate de magnésium, le talc, la gomme arabique ou analogues. On peut enrober les comprimés dc saccharose ou d'autres matires appropriées ou encore les traiter de telle sorte qu'ils aient une activité prolongée ou retardée et qu'ils libèrent d'une facon continue une quantité prédéterminée de principe actif.
On obtient une préparation en gélules en mélangeant le principe actif avec un diluant et en versant le mélange obtenu dans des gélules molles ou dures.
Une préparation sous forme de sirop ou d'élixir peut contenir le principe actif conjointement avec un édulcorant, acalorique de préférence, du méthylparaben et du propylparaben comme antiseptique, ainsi qu'un agent donnant du goût et un colorant approprié.
Les poudres ou les granules dispersibles dans l'eau peuvent contenir le principe actif en mélange avec des agents de dispersion ou des agents mouillants, ou des agents de mise en suspension, comme la polyvinylpyrrolidone, de même qu'avec des édulcorants ou des correcteurs du goût.
Pour une administration rectale, on recourt à des suppositoires qui sont préparés avec des liants fondant à la température rectale, par exemple du beurre de cacao ou des polyéthylèneglycols.
Pour une administration parentérale, intranasale ou intraoculalre, on utilise des suspensions aqueuses, des solutions salines isotoniques ou des solutions stériles et injectables qui contiennent des agents de dispersion et/ou des agents mouillants pharmacologiquement compatibles, par exemple le propylèneglycol ou le butylèneglycol.
Pour une administration par inhalation on utilise un aérosol contenant par exemple du trioléate de sorbitane ou de l'acide oléique ainsi que du trichlorofluorométhane, du dichlorofluorométhane, du dichlorotétrafluorométhane ou tout autre gaz propulseur biologiquement compatible.
Le principe actif peut être formulé également sous forme de microcapsules, éventuellement avec un ou plusieurs supports ou additifs.
Dans chaque unité de dosage le principe actif de formule (')est présent dans les quantités adaptées aux doses journalières envisagées. En général chaque unité de dosage est convenablement ajustée selon le dosage et le type d'administration prévu, par exemple comprimés, gélules et similaires, sachets, ampoules, sirops et similaires, gouttes de façon à ce qu'une telle unité de dosage contienne de 0,5 à 1000 mg de principe actif, de préférence de 2,5 à 250 mg devant être administrés une à quatre fois par jour.
Selon un autre de ses aspects, la présente invention concerne l'utilisation des produits de formule (I) pour la préparation de médicaments destinés à traiter des troubles physiologiques associés à un excès de tachykinines, notamment de Substance
P et toutes les pathologies neurokinine-dépendantes du système respiratoire, gastrointestinal, urinaire, immunitaire, cardiovasculaire et du système nerveux central ainsi que la douleur et la migraine.
P et toutes les pathologies neurokinine-dépendantes du système respiratoire, gastrointestinal, urinaire, immunitaire, cardiovasculaire et du système nerveux central ainsi que la douleur et la migraine.
Par exemple et de manière non limitative:
- douleurs aigues et chroniques liées par exemple à la migraine, aux douleurs du
cancéreux et de l'angineux, aux processus inflammatoires chroniques tels que
l'ostéoarthrite et l'arthrite rhumatoïde,
- les inflammations telles que les maladies respiratoires chroniques obstructives,
I'asthme, les allergies, les rhinites, l'hypersensibilité par exemple pollens et
acariens, arthrites rhumatoïdes, ostéoarthrites, psoriasis, colites ulcératives,
maladie de Crohn, inflammation des intestins (colon irritable), prostatite, vessie
neurologique, cystite, urétrite, néphrite,
- les maladies du système immunitaire liées à la suppression ou à la stimulation
des fonctions des cellules immunes par exemple l'arthrite rhumatoïde, le psoriasis,
la maladie de Crohn, la diabète, le lupus,
- les maladies du système nerveux central telles que l'anxiété, la dépression, la
psychose, la schizophrénie, la manie, la démence, l'épilepsie, la maladie de
Parkinson, la maladie d'Alzheimer, la drogue-dépendance, le syndrome de Down
et la chorée d'Huntington ainsi que les maladies neurodégénératives,
- les maladies du système gastro-intestinal telles que nausées, colon irritable,
ulcères gastriques et duodénaux, diarrhées, hypersécrétions,
- les maladies du système cardiovasculaire telles que l'hypertension, les aspects
vasculaires de la migraine, les oedèmes, la thrombose, l'angine de poitrine et les
spasmes vasculaires.
- douleurs aigues et chroniques liées par exemple à la migraine, aux douleurs du
cancéreux et de l'angineux, aux processus inflammatoires chroniques tels que
l'ostéoarthrite et l'arthrite rhumatoïde,
- les inflammations telles que les maladies respiratoires chroniques obstructives,
I'asthme, les allergies, les rhinites, l'hypersensibilité par exemple pollens et
acariens, arthrites rhumatoïdes, ostéoarthrites, psoriasis, colites ulcératives,
maladie de Crohn, inflammation des intestins (colon irritable), prostatite, vessie
neurologique, cystite, urétrite, néphrite,
- les maladies du système immunitaire liées à la suppression ou à la stimulation
des fonctions des cellules immunes par exemple l'arthrite rhumatoïde, le psoriasis,
la maladie de Crohn, la diabète, le lupus,
- les maladies du système nerveux central telles que l'anxiété, la dépression, la
psychose, la schizophrénie, la manie, la démence, l'épilepsie, la maladie de
Parkinson, la maladie d'Alzheimer, la drogue-dépendance, le syndrome de Down
et la chorée d'Huntington ainsi que les maladies neurodégénératives,
- les maladies du système gastro-intestinal telles que nausées, colon irritable,
ulcères gastriques et duodénaux, diarrhées, hypersécrétions,
- les maladies du système cardiovasculaire telles que l'hypertension, les aspects
vasculaires de la migraine, les oedèmes, la thrombose, l'angine de poitrine et les
spasmes vasculaires.
La présente invention inclut aussi une méthode pour traiter lesdites affections aux doses indiquées ci-dessus.
Dans les Préparations et dans les exemples on utilise les abréviations suivantes:
EtOH : éthanol
MeOH: méthanol
Ether : éther diéthylique
Ether iso : éther diisopropylique
DMF: diméthylformamide
AcOEt : acétate d'éthyle
DCM : dichlorométhane
THF: tétrahydrofürane
NaOH : soude
iPr: isopropyle
TA: température ambiante
F: point de fusion
RMN : résonnance magnétique nucléaire.
EtOH : éthanol
MeOH: méthanol
Ether : éther diéthylique
Ether iso : éther diisopropylique
DMF: diméthylformamide
AcOEt : acétate d'éthyle
DCM : dichlorométhane
THF: tétrahydrofürane
NaOH : soude
iPr: isopropyle
TA: température ambiante
F: point de fusion
RMN : résonnance magnétique nucléaire.
s : singulet
se : singulet élargi d : doublet
td : triplet dédoublé
sept : septuplet
m: massif
PREPARATIONS.
se : singulet élargi d : doublet
td : triplet dédoublé
sept : septuplet
m: massif
PREPARATIONS.
Préparation 1
Acide 3-isopropoxyphénylacétique.
Acide 3-isopropoxyphénylacétique.
a) Ester éthylique de l'acide 3-hydroxyphénylacétique.
On chauffe à reflux pendant une nuit un mélange de 55 g d'acide 3hydroxyphénylacétique dans 400 ml d'EtOH absolu et quelques gouttes d'acide sulfurique concentré. On évapore sous vide le mélange réactionnel, reprend le résidu à l'éther, lave à l'eau, par une solution saturée d'hydrogénocarbonate de sodium, sèche sur sulfate de magnésium et évapore sous vide le solvant. On obtient 58 g du produit attendu, sous forme d'huile, qui est utilisé tel quel à l'étape suivante.
b) Ester éthylique de l'acide 3-isopropoxyphénylacétique.
On chauffe à 80-100'C pendant 8 heures un mélange de 58 g du composé obtenu à l'étape précédente, 88 g de carbonate de potassium et 108 g de 2-iodopropame dans 300 ml de DMF. On évapore sous vide le mélange réactionnel, reprend le résidu à l'AcOEt, lave par une solution à 10 % de carbonate de potassium, sèche sur sulfate de magnésium et évapore sous vide le solvant. On chromatographie le résidu sur silice, en éluant par du DCM. On obtient 61 g du produit attendu, sous forme d'huile, qui est utilisé tel quel à l'étape suivante.
c) Acide 3-isopropoxyphénylacétique.
On chauffe à reflux pendant 2 heures un mélange de 31 g du composé obtenu à l'étape précédente, 20 g de NaOH dans 400 ml d'EtOH. On évapore sous vide le mélange réactionnel, reprend le résidu à l'eau, acidifie par ajout d'acide chlorhydrique concentré jusqu'à pH = 1, extrait à l'éther, lave à l'eau, sèche sur sulfate de magnésium et évapore sous vide le solvant. On obtient 27 g du produit attendu, F = 33-35-C
Préparation 2
2-(3,4-Difluorophényl)-4-(tétrahydropyran-2-yloxy)butylamine.
Préparation 2
2-(3,4-Difluorophényl)-4-(tétrahydropyran-2-yloxy)butylamine.
a) 2d3,4-difluorophényl)-4 tétrahydropyran-2-yloxy)butanenitrile.
A une solution de 2,5 g de 3,4-difluorophénylacétonitrile dans 250 ml de THF, on ajoute, par portions, 6,53 g d'hydrure de sodium à 60 % dans l'huile et laisse 3 heures sous agitation à TA. On refroidit à -20'C, ajoute, goutte à goutte, une solution de 34,1 g de 1-bromo-2-(tétrahydropyran-2-yloxy)éthane dans 70 ml de IMF et laisse 18 heures sous agitation en laissant remonter la température à TA. On évapore sous vide le mélange réactionnel, reprend le résidu dans l'éther, lave à l'eau, par une solution tampon pH = 4, à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice en éluant par du toluène. On obtient 15 g du produit attendu qui est utilisé tel quel à l'étape suivante.
b) 2d3,4-difluorophényl)-4 tétrahydropyran-2-yloxy)butylamine.
On hydrogène à TA et à pression atmosphérique, un mélange de 15 g du composé obtenu à l'étape précédente, du nickel de Raney qD (10 % de la quantité de nitrile de départ) dans 200 ml d'EtOH absolu et 30 ml d'ammoniaque concentré. On filtre le catalyseur sur Célite Q et évapore sous vide le filtrat. On reprend le résidu à l'éther, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On obtient 14 g du produit attendu qui est utilisé tel quel à L'EXEMPLE 4, étape A.
EXEMPLE 1
Chlorhydrate de N-[4d4-benzylpiprid-1-yl 2 3,4-dichlorophenyl)butyl]- N-(2-hydroxyéthyl)3-isopropoxyphe'nyl)acétamide, hémihydrate.

Chlorhydrate de N-[4d4-benzylpiprid-1-yl 2 3,4-dichlorophenyl)butyl]- N-(2-hydroxyéthyl)3-isopropoxyphe'nyl)acétamide, hémihydrate.
A) N-[4-(4-Benzylpipérid-1-yl)-2-(3,4-dichlorophényl)butyl]glycolamide.
A une solution de 14,3 g de dichlorohydrate de 4 < 4-benzylpipérid-1-yl 2- (3,4-dichlorophényl)butylamine (préparée dans EP-A-428434), 2,52 g d'acide glycolique, 9 g de triéthylamine dans 150 ml dc DCM, on ajoute 13,26 g de BOP et laisse 1 heure sous agitation à TA. On évapore sous vide le mélange réactionnel, extrait le résidu à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, seche sur sulfate de sodium et évapore sous vide le solvant. On obtient 13,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
B) N-[4-(4-Benzylpipérid-1-yl)-2-(3,4-dichlorophényl)butyl]tétrahydropyran-2-
yloxy-acétamide.
yloxy-acétamide.
On acidifie à pH = 1 une solution de 14 g du composé obtenu à l'étape précédente dans 100 ml de DCM, par ajout d'une solution saturée d'acide chlorhydrique dans l'éther. Puis on ajoute une solution de 2,8 g de 3,4-dihydro-2H-pyrane dans 20 ml de
DCM et laisse 1 heure sous agitation à TA. On concentre de moitié le mélange réactionnel, lave par une solution à 10 % de NaOH, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/2; v/v). On obtient 6 g du produit attendu qui est utilisé tel quel à l'étape suivante.
DCM et laisse 1 heure sous agitation à TA. On concentre de moitié le mélange réactionnel, lave par une solution à 10 % de NaOH, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/2; v/v). On obtient 6 g du produit attendu qui est utilisé tel quel à l'étape suivante.
C) N-[2-(tétrahydropyran-2-yloxy)éthyl]-4 4-benzylpipérid-1-ylF2d3,4- dichloro-phényl)butylamine.
On chauffe à 6oeC une suspension de 1,3 g d'hydrure d'aluminium et de lithium dans 50 ml de 'IMF et ajoute, goutte à goutte, une solution de 6 g du composé obtenu à l'étape précédente dans 50 ml de THF. On chauffe 3 heures à reflux puis après refroidissement, on hydrolyse le mélange réactionnel par ajout de 1 ml d'eau, puis 1 ml de NaOH 3N et 3 ml d'eau. On filtre les sels minéraux et évapore sous vide le filtrat.
On obtient 4,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
D) N- [4-(4-Benzylpipérid- 1 -yl23,4dichlorophénybutyl]-N-[2- tétrahydropyran-2-yloxy)éthyl] -(3-isopropoxyphényl)acétamide.
A un mélange de 4,5 g du composé obtenu à l'étape précédente, 1 g de triéthylamine, 1,94 g d'acide 3-isopropoxyphénylacétique dans 100 ml de DCM, on ajoute 4,42 g de BOP. Puis on laisse 1 heure sous agitation à TA et évapore sous vide.
On reprend le résidu à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/2,5 ; v/v). On obtient 3,2 g du produit attendu.
E) Chlorhydrate de N- (4-(4-benzylpipérid- 1-yl)-2-(3,4-dichlorophényl)butyl]-
N-(2-hydroxyéthyl)-(3-isopropoxyphényl)acétamide, hémihydrate.
N-(2-hydroxyéthyl)-(3-isopropoxyphényl)acétamide, hémihydrate.
On acidifie à pH = 1 une solution de 3,2 g du composé obtenu à l'étape précédente dans 50 ml de MeOH par ajout d'une solution saturée d'acide chlorhydrique dans l'éther. On laisse 1 heure sous agitation à TA et évapore sous vide. On reprend le résidu à l'eau, neutralise par ajout d'une solution à 10 % de NaOH, extrait à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de magnésium et évapore sous vide. On chromatographie le résidu sur silice en éluant par le mélange
DCM/MeOH (100/3 ; v/v). On reprend le produit obtenu dans du DCM, acidifie jusqu'à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 2,5 g du produit attendu après cristallisation dans le mélange DCM/Pentane.
DCM/MeOH (100/3 ; v/v). On reprend le produit obtenu dans du DCM, acidifie jusqu'à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 2,5 g du produit attendu après cristallisation dans le mélange DCM/Pentane.
Spectre de RMN à 200 Mhz dans DMSO.
1,3ppm:d:6H
1,4 ppm : m : 7 H 2,5à3,8ppm :m : 17H
4,6 ppm: sept: 1 H 4,7 à 5,05 ppm: td: 1 H
6,5 à 7,9 : m : 12 H 10,4ppm:se:1H
EXEMPLE 2 Chlorhydrate de N-[4-(4-acétamido-4-phénylpipérid-1-yl)-2-(3,4- dichlorophényl)butyl]-N-(2-méthoxyéthyl)benzamide, dihydrate.

1,4 ppm : m : 7 H 2,5à3,8ppm :m : 17H
4,6 ppm: sept: 1 H 4,7 à 5,05 ppm: td: 1 H
6,5 à 7,9 : m : 12 H 10,4ppm:se:1H
EXEMPLE 2 Chlorhydrate de N-[4-(4-acétamido-4-phénylpipérid-1-yl)-2-(3,4- dichlorophényl)butyl]-N-(2-méthoxyéthyl)benzamide, dihydrate.
A) N-[2-(3,4-dichlorophényl)-4 tétrahydropyran-2-
yloxy)butyl]méthoxyacétamide .
yloxy)butyl]méthoxyacétamide .
On refroidit à 0'C un mélange de 8,6 g de 23,4dichlorophényl- (tétrahydropyran-2-yloxy)butylamine (décrite dans EP-A-0474561), 2,46 g d'acide méthoxyacétique, 5,5 g de triéthylamine dans 50 ml de DCM, on ajoute 14,5 g de BOP et on laisse 1 heure sous agitation. Après évaporation sous vide du mélange réactionnel, on reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/1 ; v/v). On obtient 7,5 g du produit attendu qui est utilisé tel quel à l'étape Suivante.
B) Nd2-méthoxyéthylF2-(3,4-dichlorophényl 4{tétrahydropyran-2-ylozcyk
butylamine.
butylamine.
On chauffe à reflux une suspension de 1,5 g d'hydrure d'aluminium et de lithium dans 30 ml de flIF et ajoute goutte à goutte une solution de 7,5 g du composé obtenu à l'étape précédente dans 40 ml de THF. On laisse 3 heures au reflux sous agitation puis, après refroidissement, on hydrolyse le mélange réactionnel par ajout de 1 ml d'eau. On filtre les sels minéraux et évapore sous vide le filtrat. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/1,5 ; v/v). On obtient 4,1 g du produit attendu qui est utilisé tel quel à l'étape suivante.
C) N-[2-(3,4-dichlorophénylt4 tétrahydropyran-2-yloxy)butyl]-N{2- méthoxyéthylFbenzamide.
A une solution de 2 g du composé obtenu à l'étape précédente, 1,7 g de triéthylamine dans 50 ml de DCM, on ajoute 0,745 g de chlorure dc benzoyle et on laisse 1 heure sous agitation à TA. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On chromatographie le résidu sur silice H en éluant par le gradient du mélange
DCM/MeOH de (100/0,5 ; v/v) jusqu'à (100/1 ; v/v). On obtient 2,2 g du produit attendu qui est utilisé tel quel à l'étape suivante.
DCM/MeOH de (100/0,5 ; v/v) jusqu'à (100/1 ; v/v). On obtient 2,2 g du produit attendu qui est utilisé tel quel à l'étape suivante.
D) N- [2-(3,4-dichlorophényl4-hydroxybutyl]-N2-métboxyéthyl)bermmide.
On acidifie à pH = 1 une solution de 2,2 g du composé obtenu à l'étape proEdente dans 50 ml de MeOH par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et laisse 1 heure sous agitation à TA. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On obtient 1,6 g du produit attendu qui est utilisé tel quel à l'étape suivante.
E) N-[2-(3,4-dichlorophényl)-4-méthanesulfonyloxybubl]-N{2-méthoxyéthylF
benzamide.
benzamide.
A une solution de 1,6 g du composé obtenu à l'étape précédente, 0,5 g de triéthylamine dans 50 ml de DCM on ajoute 0,555 g de chlorure de méthanesulfonylc et on laisse 30 minutes sous agitation à TA. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide le solvant. On obtient 1 g du produit attendu qui est utilisé tel quel à l'étape suivante.
F) Chlorhydrate de N-14-(4-acétamido-4-phénylpipérid-1-ylt2{3,4-dichloF
phényl)butyl]-N-(2-méthoxyéthyl)benzamide, dihydratc.
phényl)butyl]-N-(2-méthoxyéthyl)benzamide, dihydratc.
On chauffe au reflux pendant 5 heures un mélange de 1,9 g du composé obtenu à l'étape précédente, 1,7 g de paratoluènesulfonate de 4-acitamido-4-phényIpipCndint, 2,8 g de carbonate de potassium dans 20 ml d'acétonitrile ct 20 ml dc DMF. Après refroidissement, on verse le mélange réactionnel dans l'eau, extrait à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice H cn éluant par le mélange DCM/MeOH (100/3 ; v/v). On reprend le produit obtenu dans du DCM, acidifie jusqu'à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 1,4 g du produit attendu après crstalisation dans le mélange DCM/éther, F = 139-142-C.
EXEMPLE 3
Chlorhydrate de N-[4-(4-acétamide4-phénylpipérid-1-ylt2 3,4- dichlorophényl)butyl] -N-(2-acétoxyéthyl)benzamide, hémihydrate.

Chlorhydrate de N-[4-(4-acétamide4-phénylpipérid-1-ylt2 3,4- dichlorophényl)butyl] -N-(2-acétoxyéthyl)benzamide, hémihydrate.
A) Ester éthylique de l'acide N-[3,4-dichlorophényl)-4-(tétrahydropyran-2-
yloxy)butyl]oxamique .
yloxy)butyl]oxamique .
On refroidit à 0 C un mélange de 19 g de 2-(3,4-dichlorophény1)-4- (tétrahydropyran-2-yloxy)butylamine, 7 g de triéthylamine dans 100 ml de DCM, puis on ajoute goutte à goutte, 8,2 g de chlorure d'éthyloxalyle et laisse 30 minutes sous agitation en laissant remonter la température à TA. Après évaporation sous vide, on reprend le résidu à l'éther, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/1 ; v/v). On obtient 16 g du produit attendu qui est utilisé tel quel à l'étape suivante.
B) N-(2-hydroxyéthyl 2-(3,4-dichlorophényl 4 tétrahydropyran-2-
yloxy)butylamine.
yloxy)butylamine.
On chauffe à 60'C une suspension de 1,7 g d'hydrure d'aluminium ct dc lithium dans 60 ml de '111F et ajoute, goutte à goutte, une solution de 16 g du composé obtenu à l'étape précédente dans 40 ml de THF. On laisse 4 heures au reflux sous agitation puis, après refroidissement, on hydrolyse le mélange réactionnel par ajout de 2 ml d'eau, puis 2 ml de NaOH 3N et 6 ml d'eau. On filtre les sels minéraux et évapore sous vide le filtrat. On chromatographie le résidu sur silice en éluant par le mélange
DCM/MeOH (100/5 ; v/v). On obtient 7,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
DCM/MeOH (100/5 ; v/v). On obtient 7,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
C) N-[2d3,4-dichlorophényl 4 tétrahydropyran-2-yloxy)butyl]-Nd2-
hydroxyéthyl)benzamide .
hydroxyéthyl)benzamide .
On refroidit à o.C un mélange de 2,5 g du composé obtenu à l'étape précédente, 0,843 g d'acide benzoique, 1,4 g de triéthylamine dans 50 ml de DCM et ajoute 3,7 g de BOP. On laisse 1 heure sous agitation et évapore sous vide. On reprend le résidu à l'éther, lave à l'eau, par une solution tampon pH = 4, à l'eau, puis sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange DCM/MeOH (100/1,5 ; v/v). On obtient 1,2 g du produit attendu qui est utilisé tel quel à l'étape suivante.
D) N-(2-acétoxyéthylN- [23,4dichlorop1tnyl4-hydroxybutyl]bcnzide.
A une solution de 1,2 g du composé obtenu à l'étape précédente, 0,3 g dc triéthylamine dans 30 ml de DCM, on ajoute 0,255 g de chlorure d'acétyle et laisse 30 minutes sous agitation à TA. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On reprend le résidu dans 20 ml de MeOH, ajoute 0,06 g de paratoluènesulfonate de pyridinium, laisse une nuit sous agitation à TA puis 30 minutes à reflux. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium ct évapore sous vide. On obtient 0,7 g du produit attendu qui est utilisé tel quel à l'étape suivante.
E) N-(2-acétoxyéthyl}N-[2d3,4-dichlorophényl 4-méthanesulfonyloxybutyl]-
benzamide.
benzamide.
A une solution de 0,7 g du composé obtenu à l'étape précédente, 0,180 g de triéthylamine dans 30 ml de DCM, on ajoute 0,206 g de chlorure de méthanesulfonyle et on laisse 30 minutes sous agitation à TA. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, par une solution saturée d'hydrogénocarbonate de sodium, à l'eau, sèche sur sulfate de sodium et évapore sous vide. On obtient 0,72 g du produit attendu qui est utilisé tel quel à l'étape suivante.
F) Chlorhydrate de N-[4-(4-acétamido-4-phénylpipérid-1-yl)-2-(3,4-A)
dichlorophénylFbutyl]-N{2-acétoxyéthyl)benzamide, hémihydrate.
dichlorophénylFbutyl]-N{2-acétoxyéthyl)benzamide, hémihydrate.
On chauffe à 80 C pendant 3 heures un mélange de 0,72 g du composé obtenu à l'étape précédente, 0,76 g de 4-acétamide4-phénylpipéridine dans 2 ml de DMF.
Après refroidissement, on verse le mélange réactionnel dans l'eau, filtre le précipité formé et lave à l'eau. On reprend le solide dans l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le gradient du mélange DCM/MeOH de (100/1,5 ; v/v) jusqu'à (10/5 ; v/v). On reprend le produit obtenu dans du DCM, acidifie jusqu'à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 0,4 g du produit attendu après cristallisation dans le mélange DCM/éther, F = 136-138C.
EXEMPLE 4
Chlorhydrate de N-[4q4-acétamido-4-phenylpipérid-1-yl 2 3,4 dicOloro- phényl)butyl]-N-(2-hydroxyéthyl)benzamide, sesquihydrate.

Chlorhydrate de N-[4q4-acétamido-4-phenylpipérid-1-yl 2 3,4 dicOloro- phényl)butyl]-N-(2-hydroxyéthyl)benzamide, sesquihydrate.
On agite à TA pendant 1 heure un mélange de 0,2 g du composé obtenu à l'EXEMPLE 3, et 0,02 g d'hydroxyde de lithium monohydrate dans 10 ml de MeOH.
On évapore sous vide, reprend le résidu au DCM, lave à l'eau, sèche sur sulfate dc sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange DCM/MeOH (100/5 ; v/v) puis (100/10 ; v/v). On reprend le produit obtenu dans du DCM, acidifie à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 0,07 g du produit attendu, F = 177-186 C.
EXEMPLES
Chlorhydrate de N-[4q4-acétamido-4-phénylpipérid-1-yl}2 3,4 difluoro phényl)butyl] -N{2-hydroxyéthyl)benzamide.
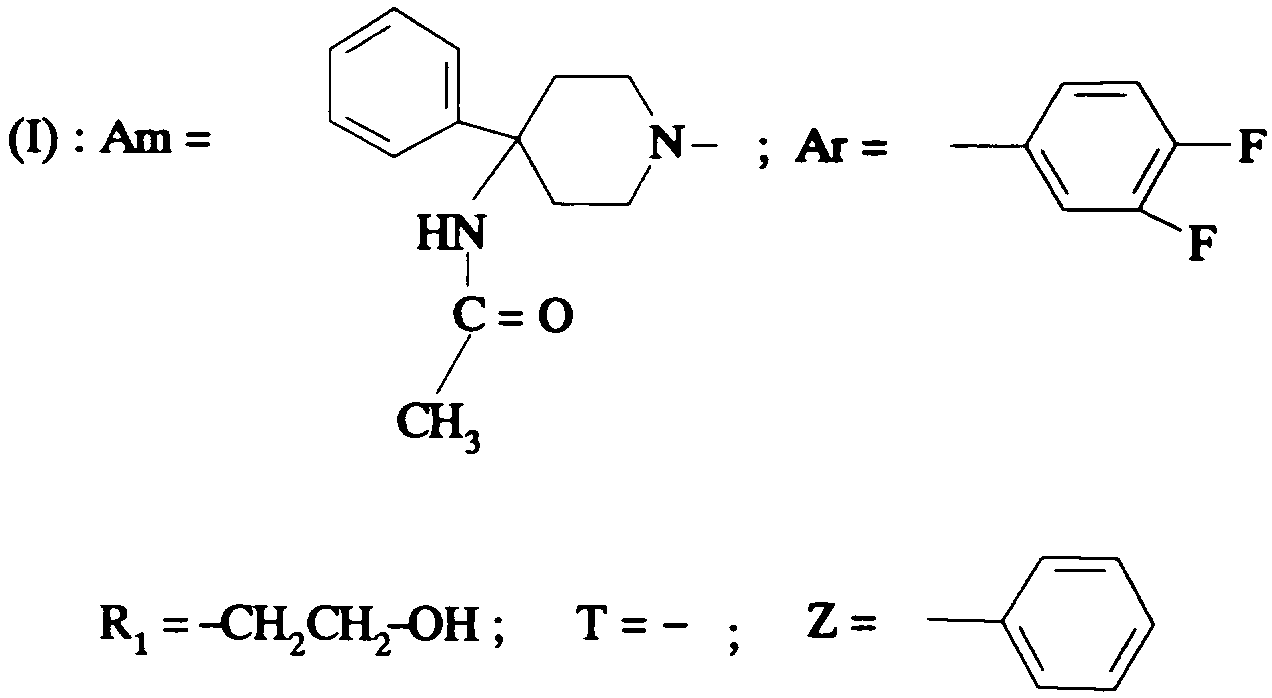
Chlorhydrate de N-[4q4-acétamido-4-phénylpipérid-1-yl}2 3,4 difluoro phényl)butyl] -N{2-hydroxyéthyl)benzamide.
A) Ester éthylique de l'acide N-[2{3,4 difluorophényl 4{tétrahydropyran-2-
yloxy)-butyl]oxamique .
yloxy)-butyl]oxamique .
On refroidit à OeC un mélange de 14 g du composé obtenu à la Préparation 2,5,94 g de triéthylamine dans 100 ml de DCM et ajoute 6,69 g du chlorure d'éthyloxalyle. On laisse 30 minutes sous agitation en laissant remonter la température à TA puis évapore sous vide. On reprend le résidu à l'éther, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice en éluant par le mélange
DCM/MeOH (100/1 ; v/v). On obtient 16,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
DCM/MeOH (100/1 ; v/v). On obtient 16,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
B) N-(2-hydroxyéthyl)-2-(3,4-difluorophényl)-4-tétrahydropyran-2-yloxy)-
butylamine.
butylamine.
On chauffe à reflux une suspension de 6,3 g d'hydrure d'aluminium et de lithium dans 180 ml de 'IMF et ajoute, goutte à goutte, une solution de 16,5 g du composé obtenu à l'étape précédente dans 100 ml de THF. On laisse 18 heures à reflux sous agitation puis après refroidissement on hydrolyse le mélange réactionnel par ajout de 6 ml d'eau, puis 6 ml de NaOH 4N et 18 ml d'eau. On filtre les sels minéraux sur Célite et et évapore sous vide le filtrat. On reprend le résidu au DCM, sèche sur sulfate de sodium et évapore sous vide le solvant. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (95/5 ; v/v). On obtient 5,5 g du produit attendu qui est utilisé tel quel à l'étape suivante.
C) N-(2-benzoyloxyéthyl)-N-[2-(3,4-difluorophényle4-(tétrahydropyran-2-
yloxy)butyl]-benzamide.
yloxy)butyl]-benzamide.
On refroidie à 0 C un mélange de 3,2 g du composé obtenu à l'étape précédente, 2,56 g de triéthylamine dans 50 ml de DCM et ajoute goutte à goutte une solution de 2,73 g de chlorure de benzoyle dans 50 ml de DCM. On laisse 1 heure sous agitation en laissant remonter la température à TA et évapore sous vide. On reprend le résidu à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore vous vide. On chromatographie le résidu sur silice en éluant par le mélange DCM/MeOH (100/1 ; v/v). On obtient S g du produit attendu qui est utilisé tel quel à l'étape suivante.
D) N{2-benzoyloncyéthyl N-[2 3,4-difluorophényl 4- hydroxybutyl]benzamide.
A une solution de 5 g du composé obtenu à l'étape précédente dans 60 ml de
MeOH, on ajoute 0,230 g de paratoluènesulfonate de pyridinium et chauffe 30 minutes à reflux. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On obtient 4 g du produit attendu qui est utilise tel quel à l'étape suivante.
MeOH, on ajoute 0,230 g de paratoluènesulfonate de pyridinium et chauffe 30 minutes à reflux. On évapore sous vide, reprend le résidu à l'AcOEt, lave à l'eau, sèche sur sulfate de sodium et évapore sous vide. On obtient 4 g du produit attendu qui est utilise tel quel à l'étape suivante.
E) N-(2-benzoyloxyéthyl)-N- (2-(3,4-difluorophény1)-4-
méthanesulfonyloxybutyl]-benzamide.
méthanesulfonyloxybutyl]-benzamide.
A un mélange de 4 g du composé obtenu à l'étape précédente, 1,16 g de triéthylamine dans 40 ml de DCM, on ajoute une solution de 1,11 g de chlorure de méthanesulfonyle dans 40 ml de DCM. On laisse 30 minutes sous agitation à TA et évapore sous vide. On reprend le résidu à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On obtient 4 g du produit attendu qui est utilisé tel quel à l'étape suivante.
F) N- [4-(4-acétamido-4-phénylpipénd- 1 -yl)-2-(3,4-difluorophinyl)butyl]-N-
(2-benzoyloxyéthyl)benaamide.
On chauffe à 80-C pendant 4 heures un mélange de 2,2 g du composé obtenu à l'étape précédente, 1,77 g de paratoluènesulfonate de 4-acétamidoX phénylpipéridine, 2,29 g de carbonate de potassium dans 15 ml d'acétonitrile et 2 ml de
DMF. Après refroidissement, on verse le mélange réactionnel dans de l'eau glacée, extrait à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange
DCM/MeOH (100/2 ; v/v). On obtient 1,7 g du produit attendu qui est utilisé tel quel à l'étape suivante.
DMF. Après refroidissement, on verse le mélange réactionnel dans de l'eau glacée, extrait à l'AcOEt, lave à l'eau, par une solution saturée de chlorure de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange
DCM/MeOH (100/2 ; v/v). On obtient 1,7 g du produit attendu qui est utilisé tel quel à l'étape suivante.
G) Chlorhydrate de N- [4-(4-acétamido-4-phénylpipérid- 1 -yl23,4-difluoro
phényl)butyl]-N-(2-hydroxyéthyl)benzamide.
phényl)butyl]-N-(2-hydroxyéthyl)benzamide.
A une solution de 1 g du composé obtenu à l'étape précédente dans 10 ml de
MeOH, on ajoute 0,067 g de NaOH et laisse 1 heure 30 minutes sous agitation à TA
On évapore sous vide, reprend le résidu au DCM, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange DCM/MeOH (95/5; v/v). On reprend le résidu dans du DCM, acidifie à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 0,52 g du produit attendu après cristallisation dans le mélange pentane/éther, F = 184-1860C.
MeOH, on ajoute 0,067 g de NaOH et laisse 1 heure 30 minutes sous agitation à TA
On évapore sous vide, reprend le résidu au DCM, lave à l'eau, par une solution saturée de chlorure de sodium, sèche sur sulfate de sodium et évapore sous vide. On chromatographie le résidu sur silice H en éluant par le mélange DCM/MeOH (95/5; v/v). On reprend le résidu dans du DCM, acidifie à pH = 1 par ajout d'une solution saturée d'acide chlorhydrique dans l'éther et évapore sous vide. On obtient 0,52 g du produit attendu après cristallisation dans le mélange pentane/éther, F = 184-1860C.
EXEMPLE 6
Chlorhydrate de N- [44-acétamido-4-phénylpipérid- I -yl23,4- difluorophényl)butyl]-N-(2-benzoyloxyéthyl)benzamide.

Chlorhydrate de N- [44-acétamido-4-phénylpipérid- I -yl23,4- difluorophényl)butyl]-N-(2-benzoyloxyéthyl)benzamide.
A une solution de 0,7 g du composé obtenu à EXEMPLE 5, étape F dans du
DCM, on ajoute jusqu'à pH = 1, une solution saturée d'acide chlorhydrique dans l'éther et on évapore sous vide. On obtient 0,24 g du produit attendu après cristallisation dans le mélange pentane/éther, F = 148-150'C.
DCM, on ajoute jusqu'à pH = 1, une solution saturée d'acide chlorhydrique dans l'éther et on évapore sous vide. On obtient 0,24 g du produit attendu après cristallisation dans le mélange pentane/éther, F = 148-150'C.
Claims (16)
1. Composé de formule:

dans laquelle: - Am représente: i- soit un groupe Amî de formule:
dans laquelle J1 représente:
il soit un groupe R2CH = dans lequel R2 représente un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy en C1-C4, un aLkyle en C1-C4, un trifluorométhyle, lesdits substituants étant identiques ou différents ; un pyridyle ; un thiényle ; un pyrimidyle ; un imidazolyle non substitué ou substitué par un alkyle en C1 -C4; i2-soit un groupe
dans lequel R2 est tel que défini cideusus; i3- soit un groupe

dans lequel: - R2 est tel que défini ci-dessus; - x est zéro ou un; - X1 représente un hydrogène ; un hydroxyle ; un alcoxy en C1 -C4; un alkyl- carbonyloxy dans lequel l'allyle est en C1-C6; un benzoyloxy ; un carboxy ; un alcoxycarbonyle dans lequel l'alcoxy est en C1 -C4; un amino ; un groupe -NR3COR4; un cyano ; un groupe -CH2NH2; un groupe -CH2NR3COR4 ; un
groupe -CH2OH; un groupe -CH2-O-Alk dans lequel Alk représente un alkyle
en C1-C4 ; un groupe -CH2-O-COR4;
- ou bien X1 forme avec l'atome de carbone auquel il est lié et avec l'atome de
carbone voisin dans la pipéridine une liaison supplémentaire;
- R3 représente un hydrogène ou un alkyle en C1 -C4;
- R4 représente un alkyle en C1-C7 ; un cycloalkyle en C3-C7 non substitué ou
substitué par un ou plusieurs méthyles; un phényle ; un pyridyle;
i4- soit un groupe R2-X2-CH-
|
dans lequel:
R2 est tel que défini ci-dessus;
X2 représente un atome d'oxygène ; un atome de soufre ; un sulfoxyde ; une
sulfone ; un groupe N-; un groupe -NCO-Alk dans lequel Alk représente
|
un alkyle en C1-C4; ; un group-N-(CH2)p-NR5R6 ;
|
- R3 est tel que défini cidessus;
-pest un, deux ou trois;
- R5 et R6 représentent chacun indépendamment un hydrogène ou un aikyle en
C1-C4
- ou bien R5 et R6 ensemble avec l'atome d'azote auquel ils sont liés constituent
un hétérocycle choisi panni la pyrrolidine, la pipéridine ou la morpholine;
ii- soit un groupe Am2 de formule:
dans laquelle J2 représente:
/
ii1-soit un groupe R2-N\
/ soit soit un groupe R2{:H2-N\ dans lesquels R2 est tel que défini ci dessus; iii- soit un groupe Am3 de formule:
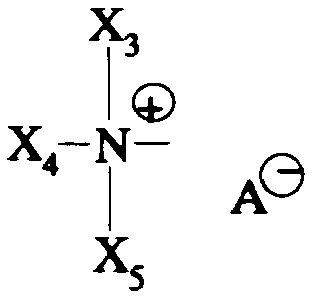
d'ammonium quaternaire éventuels.
ainsi que ses sels éventuels avec des acides minéraux ou organiques ou ses sels
Am représente un groupe Am3;
(C2-C4)alkylène ou un (o-(Cl-C4)alkylcarbonyloxy(C2-C4)alkylène lorsque
avec la limitation que R1 ne peut pas représenter un w-(C1-C4)alcoxy
- un pyridyle ; un thiényle ; un indolyle ; un quinoléyle ; un benzothiényle ; un imidazolyle;
- un naphtyle non substitué ou substitué une ou plusieurs fois par un halogène, un trifluorométhyle, un allyle en C1-C4, un hydroxyle, un alcoxy en C1-C4;
- un phényle non substitué ou substitué une ou plusieurs fois par un substituant choisi parmi : un atome d'halogène ; un trifluorométhyle ; un cyano; un hydroxyle ; un nitro ; un amino non substitué ou substitué une ou deux fois par un alkyle en C1 -C4; un benzylamino ; un carboxy ; un alkyle en C1-C10 ; un cycloalkyle en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle ; un alcoxy en C1 -C10; un cycloalkyloxy en C3-Cg non substitué ou substitué une ou plusieurs fois par un méthyle ; un mercapto; un alkylthio en C1-C10 ; un alkylcarbonyloxy dans lequel l'alkyle est en C1-C6 ; un alkylcarbonylamino dans lequel l'alkyle est en C1-C6; un benzoylamino ; un alcoxycarbonyle dans lequel l'alcoxy est en C1 -C4; un cycloalkyloxycarbonyle dans lequel le cycloalkyle est en C3-C7 ; un carbamoyle non substitué ou substitué une ou deux fois par un allyle en C1-C4 ; un uréido non substitué ou substitué une ou deux fois en position 3 par un alkyle en C1 -C4 ou un cycloalkyle en C3-C7; un (1-pyrrolidino)carbonylamino, lesdits substituants étant identiques ou différents;
- Z représente:
- R7 représente un hydrogène ou un alkyle en C1 -C4;
C1-Cs; un vinylène ; un atome d'oxygène ; un groupe -NR7-;
alcoxyméthylène dans lequel l'alcoxy est en C1 -C4; un groupe alkylène en
- T représente une liaison directe ; un groupe hydroxyméthylène ; un groupe
(C1-C4)-alcoxycarbonyl(C2-C4)alkylène ;
(C1-C4)alkylcarbonyl(C2-C4)alkylène ; un ecarboxy(C2-C4)alkylène; un #-
hydroxy(C2-C4)-alkylène ; un #-(C1-C4)alkylthio(C2-C4)alkylène ; un o-
alkylcarbonyloxy(C2-C4)alkylène ; un o-benzoyloxy(C2-Cq)aIkylène ; un w-
- R1 représente un #-(C1-C4)alcoxy(C2-C4)alkylène ; un (C1-C4
non substitué ou N-substitué par un alkyle en C1 -C4 ou un benzyle;
identiques ou différents ; un thiényle ; un benzothiényle; un naphtyle ; un indolyle
C1-C4, un alkyle en C1-C4, un trifluorométhyle, lesdits substituants étant
substituant choisi parmi : un atome d'halogène, un hydroxyle, un alcoxy en
- Ar représente un phényle non substitué ou substitué une ou plusieurs fois par un
- A représente un anion;
carbone, non substitué ou substitué par un phényle ou un benzyle;
forment un système azabicyclique ou azatricyclique contenant de 5 à 9 atomes de
dans laquelle X3, X4, X5 ensemble et avec l'atome d'azote auquel ils sont liés
2. Composé selon la revendication 1 dans lequel Am représente le groupe de formule:

0 est un anion.
position axiale soit en position équatoriale;
- Q représente un alkyle en C1 -C6 ou un benzyle ; ledit substituant étant soit en
~ J1 est tel que défini ci-dessus pour (1) dans la revendication 1;
dans laquelle:
3. Composé selon la revendication 1 de formule (1) dans laquelle Am est le groupe de formule:
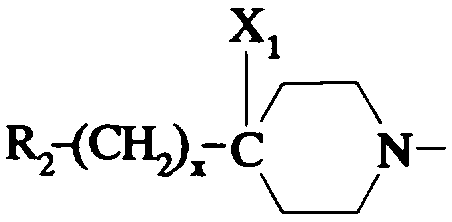
quaternaire.
ses sels avec des acides minéraux ou organiques ou ses sels d'ammonium
dans laquelle R2, x et X1 sont tels que définis pour (1) dans la revendication 1 et
4. Composés selon l'une des revendications 1 ou 3 de formule:
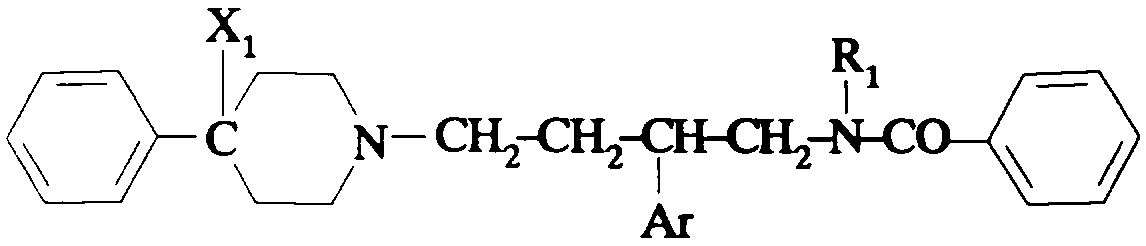
ses sels d'ammonium quaternaire.
groupe 2-méthoxyéthyle et ses sels avec des acides minéraux ou organiques ou
et R1 représente un groupe 2-hydroxyéthylc ou un groupe 2-acétoxyéthyle ou un
dans laquelle X1 et Ar sont tels que définis dans la revendication 1
5. Composé selon l'une des revendications 1 ou 3 de formule:

acides minéraux ou organiques ou ses sels d'ammonium quaternaire.
- Ar, R1 et x sont tels que définis dans la revendication 1 et ses sels avec des
dans laquelle:
6. Sel d'ammonium quaternaire d'un composé selon l'une des revendications 1 ou 2
de formule:
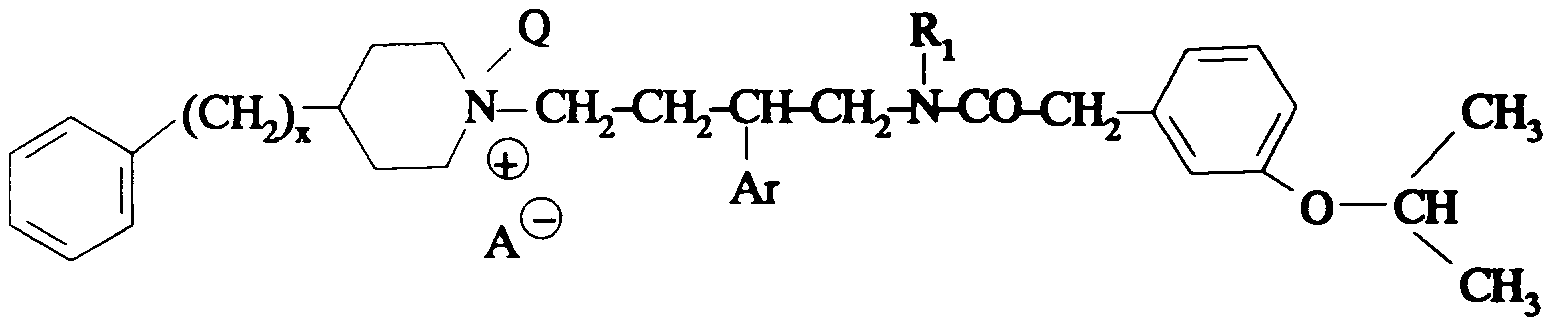
et Q est en position axiale.
dans laquelle x, Ar, R1, Q, A sont tels que définis dans les revendications 1 et 2
7. Composé selon la revendication 1 caractérisé en ce que Am3 est le résidu d'un
système azabicyclique ou azatricyclique choisi parmi:
(a) 1-azoniabicyclo[2.2.0]hexane (b) 1-azoniabicyclot3.1.0]hexane
(c) 1-azoniabicyclo[2.2.1]heptane (d) 1 -azoniabicyclo[2.2.2]octane
(e) 1-azoniabicyclot3.2.1]octane
(f) 1-azoniabicyclo[3.2.2]nonane (g) 1-azoniabicyclo[3.3.1]nonane
(h) hexahydro 1H-pyrrolizinium-4
(i) octahydroindolizinium-4
(j) octahydro 2H-quinolizinium-5
(k) 1-azoniatricyclo[3.3.1.1.3.7]décane
(1) 4-phényl-1 -azoniabicyclo[2.2.2]octane.
8. Composé selon la revendication 7 de formule choisie parmi:

dans lesquelles A , Ar, R1 sont tels que définis dans le revendication 1.
9. Composé selon l'une des revendications 1 à 8 sous forme d'un sel
pharmaceutiquement acceptable.
10. Procédé pour la préparation d'un composé selon la revendication 1 et de ses sels
éventuels, caractérisé en ce que:
1) on traite un composé de formule:
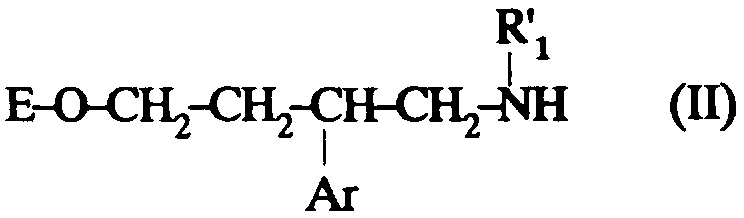
dans laquelle Ar est tel que défini précédemment, R'1 représente un *(C1- C4)alcoxy(C2-C4)alkylène, un -hydroxy(C2-C4)alkylène, un *(C1-
C4)alkylthio(C2-C4)alkylène, un #-(C1-C4)alkylcarbonyl(C2-C4)alkylène, un -carboxy(C2-C4)alkylène, un #(-(C1-C4)alcoxycarbonyl(C2-C4)alkylène, étant entendu que lorsque R'1 représente un ehydroxy(C2-C4)alkylène, l'hydroxyle peut être protégé, et E représente l'hydrogène ou un groupe O- protecteur tel que par exemple le tétrahydropyran-2-yle, - soit avec un dérivé fonctionnel d'un acide de formule: HOCO-T-Z (ici) dans laquelle Z est tel que défini précédemment et T représente une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'aicoxy est en C1-C4, un groupe alkylène en C1-Cs ou un vinylène, lorsqu'on doit préparer un composé de formule (')où T est une liaison directe, un groupe hydroxyméthylène, un groupe alcoxyméthylène dans lequel l'alcoxy est en C1
C4, un groupe alkylène en C1-Cs ou un vinylène,
- soit avec un chloroformiate de formule: ClCOOZ m dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (I) où T est un oxygène,
- soit avec un isocyanate de formule:
O=C=N-Z (V) dans laquelle Z est tel que défini précédemment, lorsqu'on doit préparer un composé de formule (I) où T est un groupe -NR7- dans lequel R7 est un hydrogène,
- soit avec un chlorure de carbamoyle de formule:
dans laquelle Z est tel que défini précédemment, et R'7 représente un alkyle en
C1-C4, lorsqu'on doit préparer un composé de formule ( où T est un groupe
NR7- dans lequel R7 est un alkyle en C1 -C4, pour obtenir un composé de formule:

2) éventuellement lorsque R'1 représente un Fhydroxy(C2-C4)alkylène, on effectue une réaction d'O-acylation pour obtenir un composé de formule:

dans laquelle E, Ar, T et Z sont tels que définis précédemment et R"1 représente un #-(C1-C4)alkylcarbonyloxy(C2-C4)alkylène ou un #-benzoyloxy(C2-
C4)alkylène, ou on protège l'hydroxyle; 3) puis on hydrolyse le composé ainsi obtenu à l'étape 1) ou à l'étape 2), pour obtenir l'alcool de formule:

4) on traite l'alcool (Vin) avec le chlorure de méthanesulfonyle pour obtenir le mésylate de formule:

5) on fait réagir le mésylate (IX)
- soit avec une amine secondaire cyclique de formule:
dans laquelle J1 est tel que défini à la revendication 1 pour (1), étant entendu que lorsque J1 représente le groupe:

où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés;
- soit avec une amine secondaire cyclique de formule:
dans laquelle J2 est tel que défini à la revendication 1 pour (I), - soit avec une amine tertiaire cyclique de formule:

acceptable.
obtenu avec un autre anion, par exemple un anion pharmaceutiquement
éventuellement on échange l'anion méthanesulfonate du sel quaternaire ainsi
éventuelle du groupe hydroxyle, on isole le produit ainsi obtenu ou bien,
- soit, lorsqu'on utilise une amine tertiaire de formule (XII) et après déprotection
minéral ou organique ou un de ses sels d'ammonium quaternaire,
transforme éventuellement, le produit ainsi obtenu en un de ses sels avec un acide
déprotection éventuelle des groupes hydroxyles ou du groupe amino, on
6) - soit, lorsqu'on utilise une amine secondaire de formule (x)ou (cl), et après
dans laquelle X3, X4 et X5 sont tels que définis précédemment pour (I) ; et
11. Procédé pour la préparation d'un composé selon la revendication 1 dans lequel Am
représente Am1 ou Am2, caractérisé en ce que:
1') on traite un composé de formule:

dans laquelle Ar et R1 sont tels que définis dans la revendication 1 et J représente un groupe J1 ou un groupe J2 tel que définis dans la revendication 1, étant entendu que lorsque J représente le groupe:

où X1 est un hydroxyle ou un amino, ces groupes peuvent être protégés, et/ou lorsque R1 représente un groupe -hydroxy(C2-C4)alkylène, l'hydroxyle peut être protégé, avec l'un des composés (m), (il), (V) ou (VI) tels que définis à la revendication 10, et
2') après déprotection éventuelle des groupes hydroxyles ou du groupe amino, on
transforme éventuellement le produit obtenu en un de ses sels avec un acide
minéral ou organique ou en un de ses sels d'ammonium quaternaire.
12. Composé de formule:

que R'1 soit différent d'un -(C1-C4)alcoxy(C2-C4)alkylène.
dans laquelle Ar, R'1 et E sont tels que définis à la revendication 10, à la condition
13. Composé de formule:

condition que R1 soit différent d'un (ss C1-C4)alcoxy(C2-C4)alkylène ou d'un o > -alkylcarbonyloxy(C2-C4)alkylène.
dans laquelle Ar, R1, T et Z sont tels que définis à la revendication 1, à la
14. Composé de formule:

que R1 soit différent d'un o-(Cl-C4)alcoxy(C2-C4)alkylène ou d'un *(C1- C)all:ylcarbonyloxy(C2-C4)alkylène.
dans laquelle Ar, R1, T et Z sont tels que définis à la revendication 1 à la condition
15. Composé de formule:

l'invention.
et Ar et R1 sont définis à la revendication 1 sont nouveaux et font partie de
dans laquelle J représente un groupe J1 ou J2 tels que définis à la revendication 1
16. Composition pharmaceutique contenant à titre de principe actif un composé selon
l'une quelconque des revendications 1 à 8.
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9403560A FR2717802B1 (fr) | 1994-03-25 | 1994-03-25 | Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. |
| EP95914415A EP0700382A1 (fr) | 1994-03-25 | 1995-03-24 | Nouveaux amides n-arylaliphatiques-n-alkyl-fonctionnalises, procede pour leur preparation et compositions pharmaceutiques en contenant |
| AU21422/95A AU2142295A (en) | 1994-03-25 | 1995-03-24 | Novel n-arylaliphatic-n-alkyl-functionalised amides, method for their prepation and pharmaceutical compositions containing same |
| JP7525003A JPH08511277A (ja) | 1994-03-25 | 1995-03-24 | n−アリール脂肪族−n−アルキルで機能化された新規なアミド、これらを製造する方法及びこれらが存在する薬学的組成物 |
| PCT/FR1995/000369 WO1995026335A1 (fr) | 1994-03-25 | 1995-03-24 | Nouveaux amides n-arylaliphatiques-n-alkyl-fonctionnalises, procede pour leur preparation et compositions pharmaceutiques en contenant |
| HU9503366A HUT73226A (en) | 1994-03-25 | 1995-03-24 | With arylaliphatic and alkyl-groups n-substituted novel amides, method for their preparation and pharmaceutical compositions containing same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9403560A FR2717802B1 (fr) | 1994-03-25 | 1994-03-25 | Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2717802A1 true FR2717802A1 (fr) | 1995-09-29 |
| FR2717802B1 FR2717802B1 (fr) | 1996-06-21 |
Family
ID=9461451
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9403560A Expired - Fee Related FR2717802B1 (fr) | 1994-03-25 | 1994-03-25 | Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP0700382A1 (fr) |
| JP (1) | JPH08511277A (fr) |
| AU (1) | AU2142295A (fr) |
| FR (1) | FR2717802B1 (fr) |
| HU (1) | HUT73226A (fr) |
| WO (1) | WO1995026335A1 (fr) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5688960A (en) * | 1995-05-02 | 1997-11-18 | Schering Corporation | Substituted oximes, hydrazones and olefins useful as neurokinin antagonists |
| US5696267A (en) * | 1995-05-02 | 1997-12-09 | Schering Corporation | Substituted oximes, hydrazones and olefins as neurokinin antagonists |
| US5789422A (en) * | 1996-10-28 | 1998-08-04 | Schering Corporation | Substituted arylalkylamines as neurokinin antagonists |
| US6063926A (en) * | 1998-11-18 | 2000-05-16 | Schering Corporation | Substituted oximes as neurokinin antagonists |
| US6440192B2 (en) | 1997-04-10 | 2002-08-27 | Valeo | Filtration device and process for its manufacture |
| WO2002083134A1 (fr) | 2001-04-12 | 2002-10-24 | Pharmacopeia, Inc. | Aryl et biaryl piperidines utilisees en tant qu'antagonistes de la mch |
| US7034056B2 (en) | 2001-03-21 | 2006-04-25 | Pharmacopeia Drug Discovery, Inc. | Aryl and biaryl compounds having MCH modulatory activity |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW449590B (en) * | 1995-04-14 | 2001-08-11 | Boehringer Ingelheim Kg | New arylglycinamide derivatives, processes for the manufacture thereof and pharmaceutical compositions containing these compounds |
| DE19519245C2 (de) * | 1995-04-14 | 2003-04-30 | Boehringer Ingelheim Kg | Neue Arylglycinamidderivate, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende pharmazeutische Zusammensetzungen |
| US6413959B1 (en) | 1995-04-14 | 2002-07-02 | Boehringer Ingelheim Kg | Method of treating depression with arylglycinamide derivatives |
| US5654316A (en) * | 1995-06-06 | 1997-08-05 | Schering Corporation | Piperidine derivatives as neurokinin antagonists |
| FR2759585B1 (fr) * | 1997-02-17 | 1999-06-11 | Sanofi Sa | Formulations pharmaceutiques presentees sous forme seche pour l'administration orale d'un compose ammonium quaternaire cyclique |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0428434A2 (fr) * | 1989-11-06 | 1991-05-22 | Sanofi | Composés aromatiques aminés et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0474561A1 (fr) * | 1990-09-05 | 1992-03-11 | Sanofi | Arylalkylamines, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0515240A1 (fr) * | 1991-05-03 | 1992-11-25 | Sanofi | Nouveaux composés N-alkylènepipéridino et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0559538A1 (fr) * | 1992-03-03 | 1993-09-08 | Sanofi | Sels quaternaires de pipéridines 4-substitués, leur préparation et compositions pharmaceutiques les contenant |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2696178B1 (fr) * | 1992-09-30 | 1994-12-30 | Sanofi Elf | Amides basiques quaternaires, procédé pour leur préparation et compositions pharmaceutiques en contenant. |
| GB9321557D0 (en) * | 1992-11-03 | 1993-12-08 | Zeneca Ltd | Carboxamide derivatives |
| ATE158580T1 (de) * | 1993-05-06 | 1997-10-15 | Merrell Pharma Inc | Substituierte pyrrolidin-3-alkyl-piperidine verwendbar als tachykinin-antagonisten |
| GB9310713D0 (en) * | 1993-05-24 | 1993-07-07 | Zeneca Ltd | Aryl substituted heterocycles |
-
1994
- 1994-03-25 FR FR9403560A patent/FR2717802B1/fr not_active Expired - Fee Related
-
1995
- 1995-03-24 AU AU21422/95A patent/AU2142295A/en not_active Abandoned
- 1995-03-24 WO PCT/FR1995/000369 patent/WO1995026335A1/fr not_active Ceased
- 1995-03-24 HU HU9503366A patent/HUT73226A/hu unknown
- 1995-03-24 EP EP95914415A patent/EP0700382A1/fr not_active Withdrawn
- 1995-03-24 JP JP7525003A patent/JPH08511277A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0428434A2 (fr) * | 1989-11-06 | 1991-05-22 | Sanofi | Composés aromatiques aminés et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0474561A1 (fr) * | 1990-09-05 | 1992-03-11 | Sanofi | Arylalkylamines, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0515240A1 (fr) * | 1991-05-03 | 1992-11-25 | Sanofi | Nouveaux composés N-alkylènepipéridino et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques les contenant |
| EP0559538A1 (fr) * | 1992-03-03 | 1993-09-08 | Sanofi | Sels quaternaires de pipéridines 4-substitués, leur préparation et compositions pharmaceutiques les contenant |
Non-Patent Citations (1)
| Title |
|---|
| CARLO ALBERTO MAGGI ET AL.: "Tachykinin receptors and tachykinin receptors antagonists", J. AUTON. PHARMACOL., vol. 13, 1993, pages 13 - 23 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5688960A (en) * | 1995-05-02 | 1997-11-18 | Schering Corporation | Substituted oximes, hydrazones and olefins useful as neurokinin antagonists |
| US5696267A (en) * | 1995-05-02 | 1997-12-09 | Schering Corporation | Substituted oximes, hydrazones and olefins as neurokinin antagonists |
| US5840725A (en) * | 1995-05-02 | 1998-11-24 | Schering Corporation | Substituted oximes, hydrazones and olefins as neurokinin antagonists |
| US5789422A (en) * | 1996-10-28 | 1998-08-04 | Schering Corporation | Substituted arylalkylamines as neurokinin antagonists |
| US6440192B2 (en) | 1997-04-10 | 2002-08-27 | Valeo | Filtration device and process for its manufacture |
| US6063926A (en) * | 1998-11-18 | 2000-05-16 | Schering Corporation | Substituted oximes as neurokinin antagonists |
| US7034056B2 (en) | 2001-03-21 | 2006-04-25 | Pharmacopeia Drug Discovery, Inc. | Aryl and biaryl compounds having MCH modulatory activity |
| WO2002083134A1 (fr) | 2001-04-12 | 2002-10-24 | Pharmacopeia, Inc. | Aryl et biaryl piperidines utilisees en tant qu'antagonistes de la mch |
| US6887889B2 (en) | 2001-04-12 | 2005-05-03 | Pharmacopeia Drug Discovery, Inc. | Aryl and biaryl piperdines with MCH modulatory activity |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0700382A1 (fr) | 1996-03-13 |
| HU9503366D0 (en) | 1996-02-28 |
| JPH08511277A (ja) | 1996-11-26 |
| AU2142295A (en) | 1995-10-17 |
| FR2717802B1 (fr) | 1996-06-21 |
| WO1995026335A1 (fr) | 1995-10-05 |
| HUT73226A (en) | 1996-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0591040B1 (fr) | Amides basiques quaternaires comme antagonistes des tachykinines | |
| EP0807111B1 (fr) | Composes heterocycliques substitues, procede pour leur preparation et compositions pharmaceutiques les contenant | |
| EP0512901B1 (fr) | Composés polycycliques aminés et leurs énantiomères, procédé pour leur préparation et compositions pharmaceutiques en contenant | |
| EP0559538B1 (fr) | Sels quaternaires de pipéridines 4-substitués, leur préparation et compositions pharmaceutiques les contenant | |
| EP0673928B1 (fr) | Nouveaux dérivés de la N-(3,4-dichlorophényl-propyl)-pipéridine comme antagonistes sélectifs du récepteur NK3 humain | |
| EP0515240B1 (fr) | Composés N-(aminoalkyl)pipéridine et leurs énantiomères comme antagonistes des récepteurs des neurokinines, procédés pour leur préparation et compositions pharmaceutiques les contenant | |
| FR2718136A1 (fr) | Composés aromatiques aminés, procédé pour leur obtention et compositions pharmaceutiques les contenant. | |
| EP1101757A1 (fr) | Composés hétérocycliques comme antagonistes de récepteurs de la tachykinine | |
| EP1241168A1 (fr) | Composés antagonistes sélectifs du récepteur NK3 humain, procédé pour leur obtention et compositions pharmaceutiques les contenant | |
| US5965580A (en) | Piperidine derivatives, process for obtaining them and pharmaceutical compositions containing them | |
| FR2717802A1 (fr) | Nouveaux composés aromatiques, procédé pour leur préparation et compositions pharmaceutiques en contenant. | |
| EP1150970A1 (fr) | Derives de (1-phenacy-3-phenyl-3-piperidylethyl)piperidine, procede pour leur obtention et compositions pharmaceutiques les contenant | |
| FR2717804A1 (fr) | Sels de composés hétéroaromatiques azotés substitués, procédé pour leur préparation et compositions pharmaceutiques en contenant. | |
| EP0915882A1 (fr) | Derives de 1-azoniabicyclo[2.2.1]heptane et compositions pharmaceutiques les contenant | |
| FR2729952A1 (fr) | Composes heterocycliques substitues, procede pour leur preparation et compositions pharmaceutiques les contenant | |
| FR2688218A1 (fr) | Sels d'ammonium quaternaires de composes aromatiques amines, leur preparation et compositions pharmaceutiques les contenant. | |
| FR2676227A1 (fr) | Nouveaux intermediaires pour la synthese de composes dialkylenepiperidino. | |
| FR2676226A1 (fr) | Nouveaux intermediaires pour la synthese de composes dialkylenepiperidino. | |
| FR2676225A1 (fr) | Nouveaux intermediaires pour la synthese de composes dialkylenepiperidino. | |
| FR2717477A1 (fr) | Composés antagonistes sélectifs du récepteur NK3 humain et leur utilisation comme médicaments et outils de diagnostic. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |