FR2736055A1 - Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant - Google Patents
Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant Download PDFInfo
- Publication number
- FR2736055A1 FR2736055A1 FR9507817A FR9507817A FR2736055A1 FR 2736055 A1 FR2736055 A1 FR 2736055A1 FR 9507817 A FR9507817 A FR 9507817A FR 9507817 A FR9507817 A FR 9507817A FR 2736055 A1 FR2736055 A1 FR 2736055A1
- Authority
- FR
- France
- Prior art keywords
- ile
- leu
- formula
- sep
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 35
- 102000004196 processed proteins & peptides Human genes 0.000 title claims abstract description 23
- 238000000034 method Methods 0.000 title claims description 10
- 239000008194 pharmaceutical composition Substances 0.000 title claims description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 25
- -1 alkyl radical Chemical class 0.000 claims abstract description 10
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 7
- 150000001413 amino acids Chemical class 0.000 claims abstract description 5
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 4
- 150000002367 halogens Chemical class 0.000 claims abstract description 4
- 125000000539 amino acid group Chemical group 0.000 claims abstract description 3
- 125000005518 carboxamido group Chemical group 0.000 claims abstract description 3
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 3
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 3
- 230000015572 biosynthetic process Effects 0.000 claims description 12
- 238000003776 cleavage reaction Methods 0.000 claims description 8
- 230000007017 scission Effects 0.000 claims description 8
- 238000003786 synthesis reaction Methods 0.000 claims description 7
- CIUQDSCDWFSTQR-UHFFFAOYSA-N [C]1=CC=CC=C1 Chemical group [C]1=CC=CC=C1 CIUQDSCDWFSTQR-UHFFFAOYSA-N 0.000 claims description 6
- 239000011347 resin Substances 0.000 claims description 6
- 229920005989 resin Polymers 0.000 claims description 6
- MDXGYYOJGPFFJL-QMMMGPOBSA-N N(alpha)-t-butoxycarbonyl-L-leucine Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)OC(C)(C)C MDXGYYOJGPFFJL-QMMMGPOBSA-N 0.000 claims description 5
- 239000002253 acid Substances 0.000 claims description 5
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 claims description 4
- 238000010511 deprotection reaction Methods 0.000 claims description 4
- MOTOSAGBNXXRRE-UHFFFAOYSA-N 2-phenylsulfanylacetic acid Chemical compound OC(=O)CSC1=CC=CC=C1 MOTOSAGBNXXRRE-UHFFFAOYSA-N 0.000 claims description 3
- 125000000217 alkyl group Chemical group 0.000 claims description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 2
- 239000004480 active ingredient Substances 0.000 claims description 2
- 229910052739 hydrogen Inorganic materials 0.000 claims description 2
- 239000001257 hydrogen Substances 0.000 claims description 2
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 claims description 2
- 231100000252 nontoxic Toxicity 0.000 claims description 2
- 230000003000 nontoxic effect Effects 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 239000007790 solid phase Substances 0.000 claims description 2
- 125000001424 substituent group Chemical group 0.000 claims description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims 2
- 150000001412 amines Chemical group 0.000 claims 1
- 125000002431 aminoalkoxy group Chemical group 0.000 claims 1
- BLNWTAHYTCHDJH-UHFFFAOYSA-O hydroxy(oxo)azanium Chemical compound O[NH+]=O BLNWTAHYTCHDJH-UHFFFAOYSA-O 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 abstract description 14
- 230000010076 replication Effects 0.000 abstract description 8
- 102000004190 Enzymes Human genes 0.000 abstract description 5
- 108090000790 Enzymes Proteins 0.000 abstract description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 abstract description 5
- 108010017640 Aspartic Acid Proteases Proteins 0.000 abstract description 4
- 102000004580 Aspartic Acid Proteases Human genes 0.000 abstract description 4
- 108010076039 Polyproteins Proteins 0.000 abstract description 4
- 230000001225 therapeutic effect Effects 0.000 abstract description 4
- 101710172711 Structural protein Proteins 0.000 abstract description 3
- 239000002243 precursor Substances 0.000 abstract description 3
- 125000003545 alkoxy group Chemical group 0.000 abstract description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 abstract description 2
- 239000000539 dimer Substances 0.000 abstract description 2
- 229910052760 oxygen Inorganic materials 0.000 abstract description 2
- 239000001301 oxygen Substances 0.000 abstract description 2
- ATTZFSUZZUNHBP-UHFFFAOYSA-N Piperonyl sulfoxide Chemical compound CCCCCCCCS(=O)C(C)CC1=CC=C2OCOC2=C1 ATTZFSUZZUNHBP-UHFFFAOYSA-N 0.000 abstract 1
- 239000005864 Sulphur Substances 0.000 abstract 1
- 125000003282 alkyl amino group Chemical group 0.000 abstract 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 16
- 208000015181 infectious disease Diseases 0.000 description 10
- 230000000840 anti-viral effect Effects 0.000 description 9
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 7
- 239000000758 substrate Substances 0.000 description 7
- 229910052717 sulfur Inorganic materials 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 230000003612 virological effect Effects 0.000 description 6
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 150000002333 glycines Chemical class 0.000 description 5
- KPYXMALABCDPGN-HYOZMBHHSA-N (4s)-5-[[(2s)-6-amino-1-[[(2s,3s)-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2r)-1-[[2-[[2-[[(1s)-3-amino-1-carboxy-3-oxopropyl]amino]-2-oxoethyl]amino]-2-oxoethyl]amino]-1-oxo-3-sulfanylpropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]a Chemical compound NC(=O)C[C@@H](C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@H](CS)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN)CC1=CC=C(O)C=C1 KPYXMALABCDPGN-HYOZMBHHSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 108010016183 Human immunodeficiency virus 1 p16 protease Proteins 0.000 description 4
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 4
- 230000002458 infectious effect Effects 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 3
- 230000036436 anti-hiv Effects 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 230000000120 cytopathologic effect Effects 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 230000010534 mechanism of action Effects 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 235000019419 proteases Nutrition 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 230000007704 transition Effects 0.000 description 3
- 230000009385 viral infection Effects 0.000 description 3
- 210000002845 virion Anatomy 0.000 description 3
- DNGHLMOUASDWRU-CCKIYBGTSA-N (2s,3s)-2-[[2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s,3s)-2-[[(2s)-6-amino-2-[[(2s)-2-[[(2s,3s)-2-amino-3-methylpentanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]hexanoyl]amino]-3-methylpentanoyl]amino]-4-methylpentanoyl]amino]-3-phenylpropano Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(O)=O)CC1=CC=CC=C1 DNGHLMOUASDWRU-CCKIYBGTSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- 108010010369 HIV Protease Proteins 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000012678 infectious agent Substances 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 230000002797 proteolythic effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 238000010532 solid phase synthesis reaction Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 230000029812 viral genome replication Effects 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- OBMZMSLWNNWEJA-XNCRXQDQSA-N C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 Chemical compound C1=CC=2C(C[C@@H]3NC(=O)[C@@H](NC(=O)[C@H](NC(=O)N(CC#CCN(CCCC[C@H](NC(=O)[C@@H](CC4=CC=CC=C4)NC3=O)C(=O)N)CC=C)NC(=O)[C@@H](N)C)CC3=CNC4=C3C=CC=C4)C)=CNC=2C=C1 OBMZMSLWNNWEJA-XNCRXQDQSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 108700010908 HIV-1 proteins Proteins 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- FORGMRSGVSYZQR-YFKPBYRVSA-N L-leucinamide Chemical compound CC(C)C[C@H](N)C(N)=O FORGMRSGVSYZQR-YFKPBYRVSA-N 0.000 description 1
- KFKWRHQBZQICHA-STQMWFEESA-N Leu-Phe Chemical group CC(C)C[C@H](N)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 KFKWRHQBZQICHA-STQMWFEESA-N 0.000 description 1
- 241001430197 Mollicutes Species 0.000 description 1
- 241000204031 Mycoplasma Species 0.000 description 1
- 230000006181 N-acylation Effects 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 101710176384 Peptide 1 Proteins 0.000 description 1
- 101710149951 Protein Tat Proteins 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- LJPDJTPZNJKXPW-QMMMGPOBSA-N Tert-butyl n-[1-(aminocarbonyl)-3-methylbutyl]carbamate Chemical compound CC(C)C[C@@H](C(N)=O)NC(=O)OC(C)(C)C LJPDJTPZNJKXPW-QMMMGPOBSA-N 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 229940124522 antiretrovirals Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- IYRWEQXVUNLMAY-UHFFFAOYSA-N carbonyl fluoride Chemical class FC(F)=O IYRWEQXVUNLMAY-UHFFFAOYSA-N 0.000 description 1
- 125000005392 carboxamide group Chemical group NC(=O)* 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 239000000039 congener Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- LONYOMRPNGXPGP-UHFFFAOYSA-N ethene-1,1-diol Chemical group [CH2][C](O)O LONYOMRPNGXPGP-UHFFFAOYSA-N 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 230000000799 fusogenic effect Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000004030 hiv protease inhibitor Substances 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- SHDMMLFAFLZUEV-UHFFFAOYSA-N n-methyl-1,1-diphenylmethanamine Chemical compound C=1C=CC=CC=1C(NC)C1=CC=CC=C1 SHDMMLFAFLZUEV-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical compound O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 230000020978 protein processing Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000002976 reverse transcriptase assay Methods 0.000 description 1
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000012882 sequential analysis Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 230000017613 viral reproduction Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
- C07K14/08—RNA viruses
- C07K14/15—Retroviridae, e.g. bovine leukaemia virus, feline leukaemia virus human T-cell leukaemia-lymphoma virus
- C07K14/155—Lentiviridae, e.g. human immunodeficiency virus [HIV], visna-maedi virus or equine infectious anaemia virus
- C07K14/16—HIV-1 ; HIV-2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/55—Protease inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06043—Leu-amino acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16211—Human Immunodeficiency Virus, HIV concerning HIV gagpol
- C12N2740/16222—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Gastroenterology & Hepatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Communicable Diseases (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Engineering & Computer Science (AREA)
- AIDS & HIV (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
La présente invention se rapporte au domaine de la chimie organique et plus particulièrement à celui de la chimie thérapeutique. Elle a plus particulièrement pour objet un nonapeptide synthétique de formule (CF DESSIN DANS BOPI) dans laquelle Ar est un radical phényle non substitué ou substitué. Les composés de formule I sont des inhibiteurs de replication du HIV en agissant comme inhibiteur d'une petite aspartyl protéase dimère qui clive spécifiquement les précurseurs d'une polyprotéine codant pour les protéines de structure et les enzymes constitutives du virus HIV. Utilisation comme médicaments.
Description
NOUVEAUX THIOPHENOXY PEPTIDES,
LEUR PROCEDE DE PREPARATION
ET LES COMPOSITIONS PHARMACEUTIQUES QUI EN RENFERMENT
La présente invention se rapporte au domaine de la chimie organique et plus particulièrement à celui de la chimie thérapeutique.
LEUR PROCEDE DE PREPARATION
ET LES COMPOSITIONS PHARMACEUTIQUES QUI EN RENFERMENT
La présente invention se rapporte au domaine de la chimie organique et plus particulièrement à celui de la chimie thérapeutique.
Elle a plus particulièrement pour objet de nouveaux peptides contenant de la phenylalanine dans lequel le groupe méthylène a été remplacé par un isostère portant un groupe thiophénoxy.
Spécifiquement, l'invention se rapporte à un dérivé de nonapeptide synthétique de formule Ile-Arg-Lys-Ile-Phe-Leu-Asp
Gly-Ile dont le groupe méthylène porté par la molécule de phénylalanine a été remplacé par un atome de soufre, c'est-àdire que le changement de structure selon l'invention peut s'écrire de la façon suivante
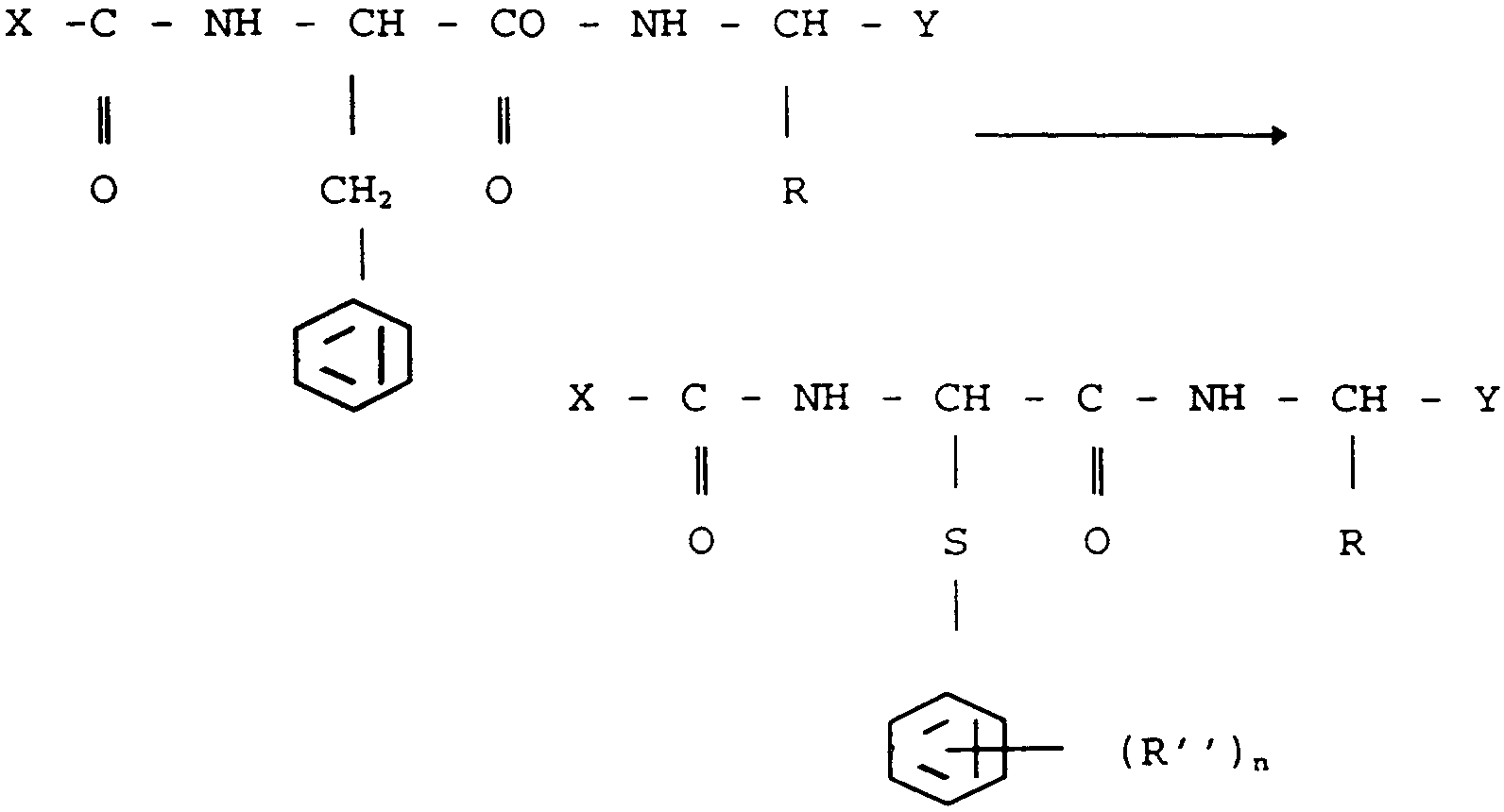
dans lequel X et Y sont des résidus d'acides aminés, protégés ou non ou des peptides, R est un radical alcoyle, linéaire ou ramifié.
Gly-Ile dont le groupe méthylène porté par la molécule de phénylalanine a été remplacé par un atome de soufre, c'est-àdire que le changement de structure selon l'invention peut s'écrire de la façon suivante
dans lequel X et Y sont des résidus d'acides aminés, protégés ou non ou des peptides, R est un radical alcoyle, linéaire ou ramifié.
et R2 est un radical alcoyle, phényle, un halogène, un nitro ou un amino et n est égal à 0, 1,2 ou 3.
Les composés selon l'invention de formule I peuvent donc s'écrire plus précisément de la manière simplifiée suivante

dans laquelle Ar est un radical phényle non substitué ou substitué.
dans laquelle Ar est un radical phényle non substitué ou substitué.
Les composés de formule I sont des inhibiteurs de replication du HIV en agissant comme inhibiteur d'une petite aspartyl protéase dimère qui clive spécifiquement les précurseurs d'une polyprotéine codant pour les protéines de structure et les enzymes constitutives du virus (Martin S.A, Recent Advances in the Design of HIV proteinase inhibitors, Antiviral Res. 17 (1992) 265-278).
Les composés de formule générale I sont préparés en utilisant un synthon de formule Acide [(Boc-Leu) - amino] phényl sulfanyl acétique de formule II
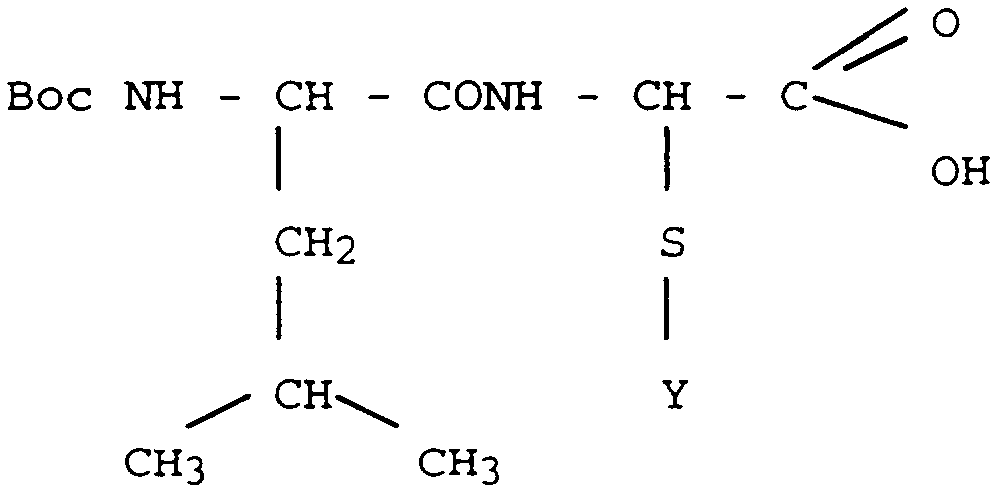
Boc = t.butoxycarbonyl dans laquelle Y est un radical phényle, non substitué ou substitué par un, deux ou trois substituants R qui est employé au cours de la synthèse peptidique sur matrice pour introduire le motif fondamental sur lequel repose la présente invention.
Ce synthon est préparé par la méthode suivante

<tb> Boc <SEP> - <SEP> Leucinamide <SEP> 7 <SEP> [(Boc <SEP> Leu)amino]
<tb> + <SEP> glyoxylate <SEP> d'allyle <SEP> hydroxyacétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> (Boc <SEP> Leu) <SEP> amino]acetoxy
<tb> <SEP> acétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> [(Boc <SEP> Leu) <SEP> amino]phényl <SEP> sulfanyl
<tb> <SEP> acétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> Acide <SEP> [(Boc <SEP> Leu) <SEP> amino]
<tb> <SEP> phénylsulfanylacétique <SEP> (2)
<tb>
En utilisant la méthode de synthèse peptidique en phase solide, suivant le mode opératoire décrit par Nguyen et al (J.Chem.Soc,
Perkin Transact. 1 (1987) 1915-1919), on accroche au départ de ce synthon les différents aminoacides qui constituent la chaîne de ce nonapeptide.On part d'une résine MBHA (p.méthyl benzhydrylamine) ou d'une résine CM (résine chlorométhylée réticulée à 1%) contenant 0,40 mmol d'Ile par gramme. Le couplage a été effectué en utilisant deux équivalents de BOC amino-acide et deux équivalents de [benzotriazolyloxy trisdimethyl aminophosphonium] hexafluoro phosphate (BOP) (fourni par Novabiochem). Après couplage, on effectue la déprotection avec 50 % d'acide trifluoroacétique < TFA) dans le chlorure de méthylène (DCM). On peut également réaliser en une seule étape la déprotection de la chaîne et le clivage de la résine, en employant de l'acide fluorhydrique en présence d'anisole. Le peptide résultant de la condensation est purifié dans une solution aqueuse d'acétonitrile.Les peptides suivants ont été préparés successivement
Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Gly-Ile OH
FmOC-Ile-Arg-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-NH2
Ile-Arg-Lys-Ile-Leu (S) -Phe-Leu-Asp-Gly-NH2
FmOC-Ile-Ary-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile OH
Ile-Ar-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile (OH) dans cet énoncé et dans ce qui suit, le symbole FmOC signifie
Fmoc = 9-fluorenylmethoxycarbonyl
Le schéma, ci-joint, explicite les différentes étapes de la synthèse du synthon (2).
<tb> + <SEP> glyoxylate <SEP> d'allyle <SEP> hydroxyacétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> (Boc <SEP> Leu) <SEP> amino]acetoxy
<tb> <SEP> acétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> [(Boc <SEP> Leu) <SEP> amino]phényl <SEP> sulfanyl
<tb> <SEP> acétate <SEP> d'allyle
<tb> <SEP> 1
<tb> <SEP> Acide <SEP> [(Boc <SEP> Leu) <SEP> amino]
<tb> <SEP> phénylsulfanylacétique <SEP> (2)
<tb>
En utilisant la méthode de synthèse peptidique en phase solide, suivant le mode opératoire décrit par Nguyen et al (J.Chem.Soc,
Perkin Transact. 1 (1987) 1915-1919), on accroche au départ de ce synthon les différents aminoacides qui constituent la chaîne de ce nonapeptide.On part d'une résine MBHA (p.méthyl benzhydrylamine) ou d'une résine CM (résine chlorométhylée réticulée à 1%) contenant 0,40 mmol d'Ile par gramme. Le couplage a été effectué en utilisant deux équivalents de BOC amino-acide et deux équivalents de [benzotriazolyloxy trisdimethyl aminophosphonium] hexafluoro phosphate (BOP) (fourni par Novabiochem). Après couplage, on effectue la déprotection avec 50 % d'acide trifluoroacétique < TFA) dans le chlorure de méthylène (DCM). On peut également réaliser en une seule étape la déprotection de la chaîne et le clivage de la résine, en employant de l'acide fluorhydrique en présence d'anisole. Le peptide résultant de la condensation est purifié dans une solution aqueuse d'acétonitrile.Les peptides suivants ont été préparés successivement
Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Gly-Ile OH
FmOC-Ile-Arg-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-NH2
Ile-Arg-Lys-Ile-Leu (S) -Phe-Leu-Asp-Gly-NH2
FmOC-Ile-Ary-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile OH
Ile-Ar-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile (OH) dans cet énoncé et dans ce qui suit, le symbole FmOC signifie
Fmoc = 9-fluorenylmethoxycarbonyl
Le schéma, ci-joint, explicite les différentes étapes de la synthèse du synthon (2).
L'invention a encore pour objet les compositions pharmaceutiques, notamment destinées au traitement des infections virales dues au virus HIV, qui contiennent, à titre de principe actif, au moins un composé de formule générale I
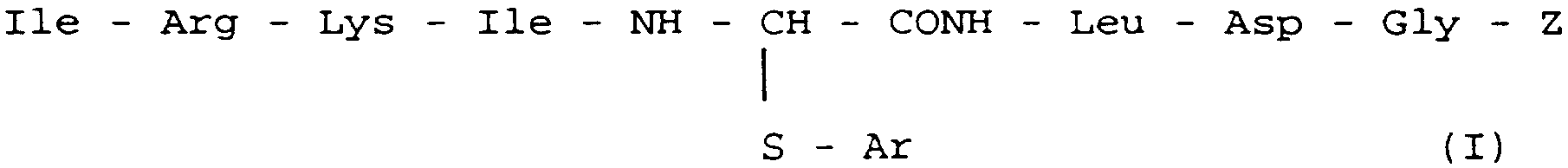
dans laquelle Z est de l'hydrogène ou un reste Ile
Ar est un radical phényle, non substitué ou substitué en association ou en mélange avec un excipient ou un véhicule inerte, non toxique, pharmaceutiquement acceptable.
dans laquelle Z est de l'hydrogène ou un reste Ile
Ar est un radical phényle, non substitué ou substitué en association ou en mélange avec un excipient ou un véhicule inerte, non toxique, pharmaceutiquement acceptable.
Parmi les composés de formule générale I, on utilisera de préférence comme principe actif, celui pour lequel Ar est un radical phényle. On pourra également utiliser des composés pour lesquels Ar est un phényle substitué par un, deux ou trois radicaux choisis dans le groupe formé par un alcoyle inférieur, un alcoxy inférieur, un trifluorométhyle, un trifluorométhoxy, un nitro, un carboxamido, un cyano, les halogènes et un phényle.
Le virus du SIDA produit une aspartyl-protéase dimère qui coupe spécifiquement les précurseurs de polyprotéine qui code pour les protéines structurelles et les enzymes constitutifs du virus.
Cette activité protéolytique est nécessaire pour la production de virions infectieux matures et est, par conséquent, un objectif intéressant pour une intervention sur le plan thérapeutique.
Les chimistes de chimie thérapeutique ont essayé de concevoir et de synthétiser des inhibiteurs de cet enzyme aspartyl protéase qui joue un rôle décisif.
La plupart des laboratoires ont employé le concept d'analogue d'état de transition. Ce concept consiste à synthétiser le substrat de peptide le plus court possible, dans lequel la liaison amide, normalement clivée, est remplacée par une fonction non hydrolysable mimant un motif de l'état de transition tétrahédrique.
Jusqu'à présent, un grand nombre de motifs mimant 1état de transition tétrahédrique de différents états ont été mis en présence de la protéase de HIV1. Il s'agit d'isostères aminoéthyléniques (RICH D.H et al. J.Med.Chem. 33 (1990) 12851288), d'analogues de statine (HUY K.Y et al. FASEB J. 5 (1991) 2606-2610 - VENAUD S et al. Res. Virol. 143 (1992) 311-319), d'isostères d'acide phosphinique (Grobeiny D et al.
Biochem.Biophys.Res.Commun. 169 (1990) 1111-1116), de difluorocétones (SHAM H.L et ai. Biochem.Biophys.Res.Commun.
175 (1991) 914-919), d'isostères dihydroxyéthylène et hydroxyéthylamine (THAISRIVONGS S et al. J.Med.Chem. 34 (1991) 2344-2356 - RICH D.H et al. J.Med.Chem. 34 (1991) 1222-1225).
Les inhibiteurs de protéase de HIV1 sont également conçus en tenant compte de la structure tertiaire de l'enzyme. Ces composés peuvent être classifiés comme inhibiteurs symétriques et comme inhibiteurs de dimérisation. Dans un souci d'augmenter l'étendue générale et l'approche du peptide anti-HIV, les demandeurs ont fait porter leur étude sur le nouveau concept d'inhibiteur de HIV2 basé sur les observations expérimentales suivantes 1. Si après analyse séquentielle des séquences de protéines
structurales et des enzymes des virions matures infectieux
on ne peut mettre en évidence un substrat spécifique de la
protéase HIV, on remarque cependant certaines spécificités
dans les sites de clivage effectués par ces virions
infectieux.
structurales et des enzymes des virions matures infectieux
on ne peut mettre en évidence un substrat spécifique de la
protéase HIV, on remarque cependant certaines spécificités
dans les sites de clivage effectués par ces virions
infectieux.
2. Beaucoup de peptides qui représentent des modèles connus de
site de traitement protéolytique à l'intérieur des
polyprotéines de HIV1 ont été considérés comme étant clivés
avec précision par une protéase synthétique ou recombinante
de HIVî.
site de traitement protéolytique à l'intérieur des
polyprotéines de HIV1 ont été considérés comme étant clivés
avec précision par une protéase synthétique ou recombinante
de HIVî.
3. Plusieurs peptides ont été conçus à partir de la déduction
du séquençage des fonctions amide ou carboxyle terminale des
protéines des HIV1 matures. Parmi celles-ci, le peptide
synthétique Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Glu-Leu a été
trouvé comme étant clivé entre le résidu LEU-PHE, cette
coupure correspondant au site de clivage 727/728 pol normal.
du séquençage des fonctions amide ou carboxyle terminale des
protéines des HIV1 matures. Parmi celles-ci, le peptide
synthétique Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Glu-Leu a été
trouvé comme étant clivé entre le résidu LEU-PHE, cette
coupure correspondant au site de clivage 727/728 pol normal.
En supposant que le remplacement du groupe méthylène du
résidu phénylalanine dans le peptide I par un hétéroatome
tel que le soufre, ne va pas changer le site de clivage
entre LEU et PHE, il est possible de mentionner ici, la
synthèse et les propriétés inhibitrices surprenante vis-à
vis de HIV1, de nouveaux thiophénoxy isostères du peptide I.
résidu phénylalanine dans le peptide I par un hétéroatome
tel que le soufre, ne va pas changer le site de clivage
entre LEU et PHE, il est possible de mentionner ici, la
synthèse et les propriétés inhibitrices surprenante vis-à
vis de HIV1, de nouveaux thiophénoxy isostères du peptide I.
Le modèle de base de ce nouveau peptide contenant une
glycine substituée en a est représentée par la formule I.
glycine substituée en a est représentée par la formule I.
D'une manière habituelle, une glycine a-substituée dans
laquelle le carbone en a est relié à un atome d'azote
d'oxygène ou de soufre est instable. Cependant, différentes
glycines a-substituées N-acétylées de ce type, ont été
décrites dans la littérature. L'acylation du groupe amino
mène à la stabilisation de la molécule en délocalisant les
électrons de l'azote sur la liaison peptidique. Au lieu
d'utiliser une simple N-acylation pour fournir la stabilité
chimique de telles glycines a-substituées, les demandeurs
ont utilisé la liaison peptidique LEU-PHE de façon à mimer
le peptide I.
laquelle le carbone en a est relié à un atome d'azote
d'oxygène ou de soufre est instable. Cependant, différentes
glycines a-substituées N-acétylées de ce type, ont été
décrites dans la littérature. L'acylation du groupe amino
mène à la stabilisation de la molécule en délocalisant les
électrons de l'azote sur la liaison peptidique. Au lieu
d'utiliser une simple N-acylation pour fournir la stabilité
chimique de telles glycines a-substituées, les demandeurs
ont utilisé la liaison peptidique LEU-PHE de façon à mimer
le peptide I.
On a synthétisé les peptides énumérés dans le Tableau 1 par la technique de la synthèse en phase solide. Cette synthèse nécessite l'emploi d'un synthon clé(2). Celui-ci a été préparé selon la procédure suivante 0 l'amide de BOC-LEUCINE (3) a été condensé avec l'hydrate de
glyoxalate d'allyle (4) pour fournir le dérivé a-hydroxylé
correspondant donc l'ester allylique de BOC-LEU-GLY (5).
glyoxalate d'allyle (4) pour fournir le dérivé a-hydroxylé
correspondant donc l'ester allylique de BOC-LEU-GLY (5).
Après acétylation de la fonction hydroxylée, l'ester
résultant (6) a été déplacé par un thiophénol nucléophile.
résultant (6) a été déplacé par un thiophénol nucléophile.
L'élimination du groupe ester allylique en utilisant le
complexe (bis palladium triphénylphosphine) en présence de
triphénylphosphine a conduit au synthon désiré (2) sous la
forme d'un mélange de diastéréoisomères. On doit noter que
l'emploi d'autres esters d'acide glyoxylique a été trouvé
non approprié pour la condensation sur l'amide BOC-LEUCINE.
complexe (bis palladium triphénylphosphine) en présence de
triphénylphosphine a conduit au synthon désiré (2) sous la
forme d'un mélange de diastéréoisomères. On doit noter que
l'emploi d'autres esters d'acide glyoxylique a été trouvé
non approprié pour la condensation sur l'amide BOC-LEUCINE.
Vraisemblablement, les conditions de déprotection finale de
la fonction ester ont conduits au clivage de la liaison
peptidique. En outre, la condensation directe entre l'acide
glyoxylique et l'acide BOC-LEUCINE s'est avérée être
inopérante dans les conditions expérimentales de l'essai
dans la mesure où la synthèse en phase solide des peptides
énumérés au Tableau 1, exige des grammes de synthon (2).
la fonction ester ont conduits au clivage de la liaison
peptidique. En outre, la condensation directe entre l'acide
glyoxylique et l'acide BOC-LEUCINE s'est avérée être
inopérante dans les conditions expérimentales de l'essai
dans la mesure où la synthèse en phase solide des peptides
énumérés au Tableau 1, exige des grammes de synthon (2).
Cette étude a été effectuée en employant le composé (2) sous la forme du mélange de diastéréoisomères. En tenant compte des résultats anti-HIV inattendus, la séparation par HPLC en phase inverse du mélange de (2) et ou la synthèse énantiomérique du peptide correspondant a été effectuée dans une deuxième étape de recherche.
TEST D'ACTIVITE DE LA PROTEASE HIV1
Les peptides modèles énumérés au Tableau 1 ont été mis en incubation avec la protéase de HIV1 partiellement purifiée en employant une procédure standard et les produits de clivage ont été analysés par HPLC en phase inverse. Comme on l'avait supposé, les résultats ont été conformes, à l'hypothèse seul le modèle de peptide 1 a été clivé d'une manière surprenante, les peptides contenant du soufre (2,7,8,9,10 et 11) ont été résistants à tout clivage protéolytique dans les conditions de l'essai. En liaison, les peptides contenants du soufre ont été ajoutés à un essai en employant le peptide I comme substrat de modèle. On a constaté qu'ils n'étaient pas des inhibiteurs à des concentrations de molarité égale aux substrats (environ 2mM).
Les peptides modèles énumérés au Tableau 1 ont été mis en incubation avec la protéase de HIV1 partiellement purifiée en employant une procédure standard et les produits de clivage ont été analysés par HPLC en phase inverse. Comme on l'avait supposé, les résultats ont été conformes, à l'hypothèse seul le modèle de peptide 1 a été clivé d'une manière surprenante, les peptides contenant du soufre (2,7,8,9,10 et 11) ont été résistants à tout clivage protéolytique dans les conditions de l'essai. En liaison, les peptides contenants du soufre ont été ajoutés à un essai en employant le peptide I comme substrat de modèle. On a constaté qu'ils n'étaient pas des inhibiteurs à des concentrations de molarité égale aux substrats (environ 2mM).
ACTIVITE ANTI-VIRALE
Les composés représentatifs énumérés au Tableau 1 ont été également testés pour leur aptitude à inhiber l'infection par
HIV1 en culture de cellules L'effet fusogénique de HIV1 dans la lignée cellulaire MT4 a été déterminé comme décrit par REY et coll. comme montré dans le Tableau 1. Certains des nouveaux thiophénoxypeptides de l'invention ont été trouvés actifs comme inhibiteurs de la replication de HIV.
Les composés représentatifs énumérés au Tableau 1 ont été également testés pour leur aptitude à inhiber l'infection par
HIV1 en culture de cellules L'effet fusogénique de HIV1 dans la lignée cellulaire MT4 a été déterminé comme décrit par REY et coll. comme montré dans le Tableau 1. Certains des nouveaux thiophénoxypeptides de l'invention ont été trouvés actifs comme inhibiteurs de la replication de HIV.
Les composés les plus actifs ont été (9) et (11). Ces résultats montrent que la protection du groupement N-terminal par un groupe FMOC dans les composés (8) ou (10) a entraîné une perte importante de l'aptitude à l'inhibition de la replication virale en comparaison avec les congénères dont le groupement Nterminal est libre (9) et (11). En outre, si on prend en considération les résultats précédents, publiés par BILLICH et al (J. Biol. Chem. 263 (1988) 17905-17908), qui ont montré que les peptides synthétiques à partir de 7 à 18 amino acide de long, peuvent être employés comme substrats modèles et inhibiteurs, pour l'investigation de la protéase, on a trouvé que la longueur minimale pour les peptides contenant de la glycine substituée par un S-phényl en a, était de 9 ou 10 amino acides.Dans cette perspective, à la fois les dipeptides (2) et (7) qui contiennent la fraction S définie, se sont avérés n'être pas actifs en tant qu'inhibiteur de la replication virale. En ce qui concerne le résidu terminal carboné, ce résultat préliminaire semble indiquer que le groupe carboxyle (11) ou le groupe carboxamide (9) pourrait être approprié en tant qu'inhibiteurs de la replication du virus HIV.
Cette étude a montré que des inhibiteurs modérement puissants de la replication de HIV1, incorporant un isostère de phénylalanine pourraient être identifiés. A la connaissance des demandeurs, c'est la première fois que des peptides synthétiques qui ne sont pas des substrats ou des inhibiteurs de protéase de HIV1 peuvent être actifs sur l'infection par
HIV1 dans les cultures de cellule de MT4. Cette nouvelle classe de peptides synthétiques de la protéase de HIV qui est basée sur le remplacement isostérique d'un groupe méthylène par un atome de soufre dans un résidu phénylalanine positionné sur le site de clivage d'un substrat peptidique synthétique de protéase de HIV, pourrait être intéressant.
HIV1 dans les cultures de cellule de MT4. Cette nouvelle classe de peptides synthétiques de la protéase de HIV qui est basée sur le remplacement isostérique d'un groupe méthylène par un atome de soufre dans un résidu phénylalanine positionné sur le site de clivage d'un substrat peptidique synthétique de protéase de HIV, pourrait être intéressant.
Assurément, la question du mécanisme d'action pour cette nouvelle classe de composés semble être cruciale. Il n'y a pas de doute que la réponse à cette question pourrait nécessiter encore des investigations. Les essais destinés à identifier la cible dans le cycle de replication de HIV déclanchée par cette nouvelle classe de composés, devront être poursuivis.
Cependant, en analysant les essais effectués pour déterminer le mécanisme par lequel ces composés interfèrent avec le cycle de replication virale à l'intérieur de la cellule, les résultats rapportés ici laissent supposer que si certains peptides incorporent un motif thiophénoxy, ils ne sont pas cependant actifs au niveau de l'inhibition du virus HIV. Ce manque d'activité peut être associé à une faible perméabilité membranaire de ces composés vis-à-vis de cellules MT4, pour ces nouveaux peptides synthétiques. En effet, pour autant que les composés comme (9) et (11) qui ont une fonction ester terminale libre, ont montré le plus grand effet anti-HIV, les peptides (8) et (10) ont été trouvés inactifs dans des cultures infectées. Il apparait donc qu'à l'intérieur de cette nouvelle classe de peptides synthétiques qui incorpore la fraction thiophénoxy, les composés qui entrent dans la cellule infectée, avec le plus grand effet, pourraient être de bons candidats pour des études de mécanisme d'action.
En résumé, une nouvelle série d'inhibiteurs de la replication du virus HIV en culture cellulaire qui favorise le remplacement par un atome de soufre dans le résidu phénylalanine a été développée. Si le mécanisme d'action de ces nouveaux composés synthétiques demeure peu connu, une fois que l'optimisation de ces résultats annexés aura été achevée, ils pourraient représenter une nouvelle approche dans la recherche de nouveaux médicaments anti-HIV.
MODE OPERATOIRE POUR L'EVALUATION DES PROPRIETES
ANTIRETROVIRALES DE NOUVEAUX ANALOGUES PEPTIDIQUES INCLUANT UN
RESIDU GLYCINE a- SUBSTITUE
METHODE
L'évaluation de l'effet antiviral est basée sur l'étude de l'effet cytopathogène du virus HIV 1 sur la lignée cellulaire MT4.
ANTIRETROVIRALES DE NOUVEAUX ANALOGUES PEPTIDIQUES INCLUANT UN
RESIDU GLYCINE a- SUBSTITUE
METHODE
L'évaluation de l'effet antiviral est basée sur l'étude de l'effet cytopathogène du virus HIV 1 sur la lignée cellulaire MT4.
La lignée MT4 a pour origine des cellules T isolées à partir d'un patient, transformées par le virus HTLV 1. Cette lignée est mycoplasmée. Les mycoplasmes sont des agents infectieux ubiquitaires, bactéries vivant à la surface des MT4 en tant qu'hôtes naturels. Cette bactérie de l'ordre de 300 à 700 nm est responsable du grand effet cytopathogène du HIV par la formation de cellules géantes (fusion par la gp 120)
SYNCITIA. Cette infection par HIV est observée 4 à 5 jours après l'infection, suivie par la mort des cellules.
SYNCITIA. Cette infection par HIV est observée 4 à 5 jours après l'infection, suivie par la mort des cellules.
Cet effet cytopathogène est directement corrélé à l'infection des cellules par le virus, à sa réplication intracellulaire et à l'expression des antigènes viraux par les cellules. Une inhibition de cet effet correspond donc à une inhibition de la multiplication du virus HIV 1. Cette lignée lymphoblastoïde infectée par HIV 1 peut être utilisée pour la production virale.
L'action du traitement par agents infectieux est permanent. En effet, il est présent avant, pendant et après l'infection virale.
Les perspectives antivirales portent essentiellement sur les inhibiteurs de la protéase qui contrôle la maturation des protéines et donc la production de particules infectieuses, mais aussi sur les inhibiteurs de la protéine TAT qui participe au réveil et à la dissémination du virus régulateur positif de la transcription virale et enfin sur les inhibiteurs de la transcriptase inversé (reverse transcriptase) qui transforme 1'ARN viral en ADN double brin, renfermant le message viral et qui s'intègre à l'état de provirus dans 1'ADN de la cellule hôte.
Des dilutions successives sont effectuées dans du milieu à 10% afin de pouvoir cultiver les MT4 pendant 8 jours et de pouvoir lire la formation des syncitia.
TEST MT4 - avant infection 3.106 cellules/100 ul sont distribuées dans une microplaque à 96 puits, centrifugées 3 fois à 2000 tpm et le culot est préincubé avec 100 ul de concentrations successives de l'antiviral à tester, 1 heure à 37 CO2.
- infection
Elle est réalisée en micropuits en rajoutant une dilution 10-3 du virus HIV (cette dilution du virus HIV 1 est déterminée pour induire la formation de syncitia en 4 à 5 jours). L'antiviral est toujours présent lors de 1 l'infection, la concentration finale du virus est alors de 5.10-4.
Elle est réalisée en micropuits en rajoutant une dilution 10-3 du virus HIV (cette dilution du virus HIV 1 est déterminée pour induire la formation de syncitia en 4 à 5 jours). L'antiviral est toujours présent lors de 1 l'infection, la concentration finale du virus est alors de 5.10-4.
- après infection
Après une incubation de 1 heure à 37 sous CO2, les MT4 sont lavées 3 fois avec du RPMI 1640 et mises en culture à raison de 3.105 cellules pour 1 ml de chacune des concentrations des composés à tester en plaques 24 puits. Ce jour est considéré comme JO.
Après une incubation de 1 heure à 37 sous CO2, les MT4 sont lavées 3 fois avec du RPMI 1640 et mises en culture à raison de 3.105 cellules pour 1 ml de chacune des concentrations des composés à tester en plaques 24 puits. Ce jour est considéré comme JO.
A J3 ou J4, les cellules MT4 sont diluées au 1/3, de nouveau dans les différentes concentrations de 1'antiviral.
Chaque jour, l'apparition de syncitia est observée au microscope pour voir s'il y a un peu de retard par rapport au témoin HIV 1.
A J8, le dosage de la transcriptase inverse est effectué. Si les cellules ne sont pas infectées, c'est donc qu'il y a une protection par l'antiviral testé.
La dose IC50, concentration de l'antiviral qui inhibe 50% de la valeur de la transcriptase inverse du témoin HIV 1 sera déterminée.
- Numéro des composés
1 Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Gly-Ile-OH
8 Fmoc-Ile-Arg-Lys-Ile-Leu < S) -Phe-Leu-Asp-Gly-NH2
9 Ile-Arg-Lys-Ile-Leu(S) -Phe-Leu-Asp-Gly-NH2 10 Fmoc-Ile-Arg-Lys-Leu(S)-Phe-Leu-Asp-Gly-Ile-OH 11 Ile-Arg-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile-OH
2 BocNH--Leu(S)-PheOH
7 BocNH-Leu(S)-PheOCH2-CH-=CH2
Leu-(S)-Phe

1 Ile-Arg-Lys-Ile-Leu-Phe-Leu-Asp-Gly-Ile-OH
8 Fmoc-Ile-Arg-Lys-Ile-Leu < S) -Phe-Leu-Asp-Gly-NH2
9 Ile-Arg-Lys-Ile-Leu(S) -Phe-Leu-Asp-Gly-NH2 10 Fmoc-Ile-Arg-Lys-Leu(S)-Phe-Leu-Asp-Gly-Ile-OH 11 Ile-Arg-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Ile-OH
2 BocNH--Leu(S)-PheOH
7 BocNH-Leu(S)-PheOCH2-CH-=CH2
Leu-(S)-Phe

<tb> Numéro <SEP> composés <SEP> IC50 <SEP> M <SEP> AM <SEP> TI(ID50/IC50) <SEP>
<tb> <SEP> 1 <SEP> inactif
<tb> <SEP> 8 <SEP> inactif/toxique
<tb> <SEP> 9 <SEP> 5 <SEP> + <SEP> 2 <SEP> 50
<tb> <SEP> 10 <SEP> 100 <SEP> + <SEP> 50 <SEP> 10
<tb> <SEP> 11 <SEP> 10 <SEP> + <SEP> 5 <SEP> 10
<tb> <SEP> 2 <SEP> inactif
<tb> <SEP> 7 <SEP> inactif
<tb>
IC50 : concentration requise pour inhiber la formation de
syncitia de 50% par rapport à l'essai témoin
TI : index thérapeutique concentration requise pour
provoquer la mort de 50% de cellules MT4 non infectées
(IDso) par rapport à la concentration requise pour
inhiber la formation de syncitia de 50% (IC50)
Essais effectués sur cellules MT4. Souche virale HIV-1 Bru
<tb> <SEP> 1 <SEP> inactif
<tb> <SEP> 8 <SEP> inactif/toxique
<tb> <SEP> 9 <SEP> 5 <SEP> + <SEP> 2 <SEP> 50
<tb> <SEP> 10 <SEP> 100 <SEP> + <SEP> 50 <SEP> 10
<tb> <SEP> 11 <SEP> 10 <SEP> + <SEP> 5 <SEP> 10
<tb> <SEP> 2 <SEP> inactif
<tb> <SEP> 7 <SEP> inactif
<tb>
IC50 : concentration requise pour inhiber la formation de
syncitia de 50% par rapport à l'essai témoin
TI : index thérapeutique concentration requise pour
provoquer la mort de 50% de cellules MT4 non infectées
(IDso) par rapport à la concentration requise pour
inhiber la formation de syncitia de 50% (IC50)
Essais effectués sur cellules MT4. Souche virale HIV-1 Bru
Claims (9)
-
 REVENDICATIONS 1. Un nonapeptide synthétique dont la structure s'écrit
REVENDICATIONS 1. Un nonapeptide synthétique dont la structure s'écrit simplifée estet n est égal à O, 1, 2 ou 3
simplifée estet n est égal à O, 1, 2 ou 3 - 2. Un nonapeptide selon la revendication 1, dont la formulecarboxamido ou un cyanoun amino alcoxy, trifluorométhyle, trifluorométhoxy,et R'' est un radical alcoyle, phényle, halogène, un nitro,R est un radical alcoyle linéaire ou ramifiéprotégés ou non, ou des peptidesdans laquelle X et Y sont les résidus d'acides aminés,2, à savoir .dans laquelle Ar est un radical phényle, non substitué ou substitué -
- 3.Un nonapeptide selon la revendication 1 ou la revendication
 formule IIformule acide[(BOC Leu)-amino]phényl sulfanyl acétique derevendications 1 à 4, dans lequel on utilise un synthon deIle-Arg-Lys-Ile-Leu(s)-phe-Leu-Asp-Gly-NH2
formule IIformule acide[(BOC Leu)-amino]phényl sulfanyl acétique derevendications 1 à 4, dans lequel on utilise un synthon deIle-Arg-Lys-Ile-Leu(s)-phe-Leu-Asp-Gly-NH2 - 4. Un peptide selon la revendication 1 ou 2, à savoir Ile-Arg-Lys-Ile-Leu(S) -Phe-Leu-Asp-Gly-Ile-OH
- 5. Un procédé d'obtention des peptides selon l'une desrésines en phase solide.constituent la chaîne de ce nonapeptide par la technique desauquel on accroche les différents amino acides quisubstitué par un, deux ou trois substituants Rdans laquelle Y est un radical phényle, non substitué oufluorhydrique en présence d'anisole.chaîne et le clivage de la résine à l'aide d'acidetrifluoroacétique ou bien on réalise la déprotection de lapuis on déprotège la fonction amine terminale par l'acidevéhicule inerte, non toxique, pharmaceutiquement-acceptable.en association ou en mélange avec un excipient ou unet Ar est un radical phényle, non substitué ou substituédans laquelle Z est de l'hydrogène ou un reste Ile
 principe actif au moins un composé de formule générale I
principe actif au moins un composé de formule générale I - 6, Les compositions pharmaceutiques qui contiennent à titre de
- 7. A titre d'intermédiaire pour la synthèse des composés deformule I FmOC-Ile-Arg-Lys-Ile-Leu(S)-Phe-Leu-Asp-Gly-Z
- 8. A titre d'intermédiaire pour la synthèse des composés deformule IFmOC-Ile-Arg-Lys-Ile-Leu(S) -Phe-Leu-Asp-Gly-Ile OH
- 9. A titre d'intermédiaire pour la synthèse des composés deformule I, l'acide[(Boc Leu)amino] phénylsulfanylacétique
Priority Applications (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9507817A FR2736055B1 (fr) | 1995-06-29 | 1995-06-29 | Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant |
| AU64624/96A AU706799B2 (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides, method for preparing same and pharmaceutical compositions containing said peptides |
| US08/793,647 US5844078A (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides, method for preparing same, and pharmaceutical compositions containing said peptides |
| APAP/P/1997/000964A AP683A (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides, method for preparing same, and pharmaceutical compositions containing said peptides. |
| CA002197861A CA2197861A1 (fr) | 1995-06-29 | 1996-06-28 | Peptides phenyles, leur procede de preparation et les compositions pharmaceutiques qui en renferment |
| PL96318880A PL318880A1 (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides, method of obtaining them and pharmaceutic preparations containing such peptides |
| IL12034196A IL120341A (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides their preparation and pharmaceutical compositions containing them |
| KR1019970701333A KR970705576A (ko) | 1995-06-29 | 1996-06-28 | 페닐 펩티드, 이를 제조하는 방법, 및 이 펩티드를 함유하는 약제학적 조성물(phenyl peptides, method for preparing same, and pharmaceutical compositions containing said peptides) |
| PCT/FR1996/001008 WO1997001576A2 (fr) | 1995-06-29 | 1996-06-28 | Peptides phenyles, leur procede de preparation et les compositions pharmaceutiques qui en renferment |
| EP96924035A EP0787143A2 (fr) | 1995-06-29 | 1996-06-28 | Peptides phenyles, leur procede de preparation et les compositions pharmaceutiques qui en renferment |
| NZ312726A NZ312726A (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides; preparation; medicaments |
| JP9504212A JPH10505613A (ja) | 1995-06-29 | 1996-06-28 | フェニルペプチドと、その製造方法と、このペプチドを含む医薬品組成物 |
| BR9606455A BR9606455A (pt) | 1995-06-29 | 1996-06-28 | Peptídeo sintético processo de obtenção de peptídeos composições farmacêuticas e intermediário |
| CN96190943A CN1165521A (zh) | 1995-06-29 | 1996-06-28 | 苯基肽,该肽及含有所述肽的药物组合物的制备方法 |
| HU9701940A HUP9701940A3 (en) | 1995-06-29 | 1996-06-28 | Phenyl peptides, method for preparing same, and pharmaceutical compositions containing said peptides |
| NO970895A NO970895D0 (no) | 1995-06-29 | 1997-02-27 | Fenylpeptider, fremgangsmåte for fremstilling av disse, og farmasöytiske preparater inneholdende disse peptider |
| OA60971A OA10403A (fr) | 1995-06-29 | 1997-02-28 | Nouveaux peptides phényles leur procédé de préparation et les compositions pharmaceutiques qui en referment |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9507817A FR2736055B1 (fr) | 1995-06-29 | 1995-06-29 | Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2736055A1 true FR2736055A1 (fr) | 1997-01-03 |
| FR2736055B1 FR2736055B1 (fr) | 1997-09-12 |
Family
ID=9480502
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9507817A Expired - Lifetime FR2736055B1 (fr) | 1995-06-29 | 1995-06-29 | Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US5844078A (fr) |
| EP (1) | EP0787143A2 (fr) |
| JP (1) | JPH10505613A (fr) |
| KR (1) | KR970705576A (fr) |
| CN (1) | CN1165521A (fr) |
| AP (1) | AP683A (fr) |
| AU (1) | AU706799B2 (fr) |
| BR (1) | BR9606455A (fr) |
| CA (1) | CA2197861A1 (fr) |
| FR (1) | FR2736055B1 (fr) |
| HU (1) | HUP9701940A3 (fr) |
| IL (1) | IL120341A (fr) |
| NO (1) | NO970895D0 (fr) |
| NZ (1) | NZ312726A (fr) |
| OA (1) | OA10403A (fr) |
| PL (1) | PL318880A1 (fr) |
| WO (1) | WO1997001576A2 (fr) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1292438B1 (it) * | 1997-06-30 | 1999-02-08 | Zambon Spa | Derivati peptidomimetici inibitori suicidi della proliferazione del hiv |
| US6118226A (en) | 1998-07-31 | 2000-09-12 | Federal-Mogul World Wide, Inc. | Electrodeless neon light module for vehicle lighting systems |
| US8886480B2 (en) | 2011-06-27 | 2014-11-11 | Synaptics Incorporated | System and method for signaling in gradient sensor devices |
| US9188675B2 (en) | 2012-03-23 | 2015-11-17 | Synaptics Incorporated | System and method for sensing multiple input objects with gradient sensor devices |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0094815A2 (fr) * | 1982-05-18 | 1983-11-23 | Smithkline Beckman Corporation | Oligopeptides prémédicamenteux |
| US4454065A (en) * | 1982-05-18 | 1984-06-12 | Smithkline Beckman Corporation | Oligopeptide prodrugs |
| EP0331921A1 (fr) * | 1988-02-11 | 1989-09-13 | Warner-Lambert Company | Inhibiteurs de la rénine, contenant des alpha-hétéroatome-acides aminés |
| WO1991010679A2 (fr) * | 1990-01-22 | 1991-07-25 | Warner Lambert Co | PEPTIDES INHIBANT LA RENINE, PRESENTANT UN ACIDE AMINE A 'alpha'-HETEROATOMES EN POSITION P3 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1193598A (fr) * | 1982-05-06 | 1985-09-17 | Rafael Foguet | Derives d'acide 2-amino-benzoique et methode de preparation |
-
1995
- 1995-06-29 FR FR9507817A patent/FR2736055B1/fr not_active Expired - Lifetime
-
1996
- 1996-06-28 KR KR1019970701333A patent/KR970705576A/ko not_active Withdrawn
- 1996-06-28 AP APAP/P/1997/000964A patent/AP683A/en active
- 1996-06-28 PL PL96318880A patent/PL318880A1/xx unknown
- 1996-06-28 WO PCT/FR1996/001008 patent/WO1997001576A2/fr not_active Ceased
- 1996-06-28 EP EP96924035A patent/EP0787143A2/fr not_active Withdrawn
- 1996-06-28 JP JP9504212A patent/JPH10505613A/ja active Pending
- 1996-06-28 BR BR9606455A patent/BR9606455A/pt not_active Application Discontinuation
- 1996-06-28 US US08/793,647 patent/US5844078A/en not_active Expired - Fee Related
- 1996-06-28 AU AU64624/96A patent/AU706799B2/en not_active Ceased
- 1996-06-28 CA CA002197861A patent/CA2197861A1/fr not_active Abandoned
- 1996-06-28 IL IL12034196A patent/IL120341A/xx active IP Right Grant
- 1996-06-28 CN CN96190943A patent/CN1165521A/zh active Pending
- 1996-06-28 NZ NZ312726A patent/NZ312726A/en unknown
- 1996-06-28 HU HU9701940A patent/HUP9701940A3/hu unknown
-
1997
- 1997-02-27 NO NO970895A patent/NO970895D0/no not_active Application Discontinuation
- 1997-02-28 OA OA60971A patent/OA10403A/fr unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0094815A2 (fr) * | 1982-05-18 | 1983-11-23 | Smithkline Beckman Corporation | Oligopeptides prémédicamenteux |
| US4454065A (en) * | 1982-05-18 | 1984-06-12 | Smithkline Beckman Corporation | Oligopeptide prodrugs |
| EP0331921A1 (fr) * | 1988-02-11 | 1989-09-13 | Warner-Lambert Company | Inhibiteurs de la rénine, contenant des alpha-hétéroatome-acides aminés |
| WO1991010679A2 (fr) * | 1990-01-22 | 1991-07-25 | Warner Lambert Co | PEPTIDES INHIBANT LA RENINE, PRESENTANT UN ACIDE AMINE A 'alpha'-HETEROATOMES EN POSITION P3 |
Non-Patent Citations (3)
| Title |
|---|
| KINGSBURY, WILLIAM D. ET AL: "Synthesis of.alpha.-thiophenylglycine peptides. Novel peptide substrates useful in the study of microbial peptide transport", INT. J. PEPT. PROTEIN RES. (1986), 27(6), 659-65 CODEN: IJPPC3;ISSN: 0367-8377 * |
| KRAUS, J. L. ET AL: "Synthesis and biological activities of new N-formylated methionyl peptides containing an.alpha.-substituted glycine residue", EUR. J. MED. CHEM. (1992), 27(1), 19-26 CODEN: EJMCA5;ISSN: 0223-5234 * |
| REPINE, JOSEPH T. ET AL: "Renin inhibitors containing.alpha.-heteroatom amino acids as P2 residues", J. MED. CHEM. (1992), 35(6), 1032-42 CODEN: JMCMAR;ISSN: 0022-2623 * |
Also Published As
| Publication number | Publication date |
|---|---|
| AU706799B2 (en) | 1999-06-24 |
| AU6462496A (en) | 1997-01-30 |
| BR9606455A (pt) | 1998-07-14 |
| CA2197861A1 (fr) | 1997-01-16 |
| PL318880A1 (en) | 1997-07-07 |
| EP0787143A2 (fr) | 1997-08-06 |
| US5844078A (en) | 1998-12-01 |
| NZ312726A (en) | 1998-07-28 |
| WO1997001576A2 (fr) | 1997-01-16 |
| AP9700964A0 (en) | 1997-04-30 |
| WO1997001576A3 (fr) | 1997-05-15 |
| HUP9701940A2 (hu) | 1998-03-02 |
| NO970895L (no) | 1997-02-27 |
| JPH10505613A (ja) | 1998-06-02 |
| NO970895D0 (no) | 1997-02-27 |
| FR2736055B1 (fr) | 1997-09-12 |
| KR970705576A (ko) | 1997-10-09 |
| CN1165521A (zh) | 1997-11-19 |
| OA10403A (fr) | 2001-12-05 |
| IL120341A0 (en) | 1997-06-10 |
| HUP9701940A3 (en) | 2001-07-30 |
| AP683A (en) | 1998-09-30 |
| IL120341A (en) | 2001-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Meek | Inhibitors of HIV-1 protease | |
| AU633017B2 (en) | Therapeutics for aids based on inhibitors of hiv protease | |
| AU757783B2 (en) | Hepatitis C inhibitor peptides | |
| WO2000052033A1 (fr) | Nouveaux derives de tetrapeptides cycliques et leur utilisation comme medicaments | |
| EP0623144A1 (fr) | Peptides synthetiques pour la detoxication des endotoxines bacteriennes et pour le traitement des chocs septiques | |
| EP0756603A1 (fr) | Multirepresentation d'un analogue peptidique du substrat de la dppiv, notamment de type kpr, afin d'inhiber l'entree du hiv dans les cellules | |
| JPH06501938A (ja) | ペプチドよりなるhiv複製阻害剤 | |
| JPH0832720B2 (ja) | タフトシン類似体、その製法及び医薬組成物 | |
| WO1987003601A1 (fr) | Peptides capables d'inhiber les interactions entre des antigenes et les lymphocytes t4, produits qui en sont derives et leurs applications | |
| FR2736055A1 (fr) | Nouveaux thiophenoxy peptides, leur procede de preparation et les compositions pharmaceutiques en renfermant | |
| JP2001525315A (ja) | 新規メタロプロテアーゼ阻害剤、その治療的利用およびその合成における開始化合物の製造方法 | |
| EP0443573A2 (fr) | Inhibiteurs d'HIV-protéase contenant aminoacides dérivés | |
| RU2238950C2 (ru) | Производные гемина и их фармацевтически приемлемые соли, способ получения, применение и фармацевтическая композиция | |
| BE1003996A3 (fr) | Peptides sdk, leur procede de preparation ainsi que des compositions therapeutiques en contenant. | |
| EP0832110A1 (fr) | Composes antiviraux de type peptidique | |
| Qasmi et al. | Synthesis of N-glyoxylyl peptides and their in vitro evaluation as HIV-1 protease inhibitors | |
| EP0722460B1 (fr) | Derives de peptides therapeutiquement actifs dans la cascade de coagulation sanguine, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| CA2091151A1 (fr) | Inhibiteurs de la protease retrovirale | |
| JPH0952900A (ja) | 亜鉛エンドペプチダーゼ24−15阻害剤として使用可能な新規ペプチド誘導体 | |
| Niddam et al. | Thiophenoxy peptides: a new class of HIV replication inhibitors | |
| MXPA97001482A (es) | Peptidos fenilos, su procedimiento de preparaciony las composiciones farmaceuticas que loscontienen | |
| Frogier et al. | Fluorinated analogues of the p17/p24 sequence incorporating 3-fluoro and 3, 3-difluoro phenylalanines as potential inhibitors of HIV protease | |
| Camp | Design and synthesis of inhibitors for the HIV-1 protease | |
| FR2646353A1 (fr) | Utili sation de peptides contenant de la statine pour la preparation de medicaments utiles pour le traitement des maladies virales | |
| KR0178575B1 (ko) | 항바이러스 활성을 갖는 펩티드 모방체 화합물 및 이를 포함하는 조성물 |