FR2745568A1 - Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides - Google Patents
Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides Download PDFInfo
- Publication number
- FR2745568A1 FR2745568A1 FR9702461A FR9702461A FR2745568A1 FR 2745568 A1 FR2745568 A1 FR 2745568A1 FR 9702461 A FR9702461 A FR 9702461A FR 9702461 A FR9702461 A FR 9702461A FR 2745568 A1 FR2745568 A1 FR 2745568A1
- Authority
- FR
- France
- Prior art keywords
- sep
- acid
- radicals
- phosgene
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 239000002253 acid Substances 0.000 title claims abstract description 55
- 238000004519 manufacturing process Methods 0.000 title description 4
- 150000004820 halides Chemical class 0.000 title 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 claims abstract description 44
- 238000000034 method Methods 0.000 claims abstract description 33
- 239000003054 catalyst Substances 0.000 claims abstract description 31
- 150000008064 anhydrides Chemical class 0.000 claims abstract description 24
- 239000002904 solvent Substances 0.000 claims abstract description 7
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 claims abstract description 4
- -1 anhydride acids Chemical class 0.000 claims description 27
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 claims description 15
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 11
- 125000003118 aryl group Chemical group 0.000 claims description 10
- 238000007872 degassing Methods 0.000 claims description 10
- 125000005110 aryl thio group Chemical group 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- 125000000217 alkyl group Chemical group 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 125000001424 substituent group Chemical group 0.000 claims description 7
- 125000004104 aryloxy group Chemical group 0.000 claims description 6
- 150000007513 acids Chemical class 0.000 claims description 5
- 125000003545 alkoxy group Chemical group 0.000 claims description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 claims description 5
- 150000003254 radicals Chemical class 0.000 claims description 5
- 239000001569 carbon dioxide Substances 0.000 claims description 4
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 4
- 238000000926 separation method Methods 0.000 claims description 4
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 3
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 3
- 125000004414 alkyl thio group Chemical group 0.000 claims description 3
- 150000005840 aryl radicals Chemical class 0.000 claims description 3
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 3
- 125000001188 haloalkyl group Chemical group 0.000 claims description 3
- 239000000203 mixture Substances 0.000 claims description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 2
- 125000005842 heteroatom Chemical group 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- 229910052760 oxygen Inorganic materials 0.000 claims description 2
- 239000001301 oxygen Substances 0.000 claims description 2
- 229920006395 saturated elastomer Polymers 0.000 claims description 2
- 229910052717 sulfur Inorganic materials 0.000 claims description 2
- 239000011593 sulfur Substances 0.000 claims description 2
- 150000002763 monocarboxylic acids Chemical class 0.000 abstract description 2
- 238000006243 chemical reaction Methods 0.000 description 31
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 19
- 239000012429 reaction media Substances 0.000 description 15
- 238000012360 testing method Methods 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 8
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 8
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 8
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 235000021355 Stearic acid Nutrition 0.000 description 7
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 7
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 7
- 239000008117 stearic acid Substances 0.000 description 7
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 150000001805 chlorine compounds Chemical class 0.000 description 5
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 5
- PGZVFRAEAAXREB-UHFFFAOYSA-N 2,2-dimethylpropanoyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OC(=O)C(C)(C)C PGZVFRAEAAXREB-UHFFFAOYSA-N 0.000 description 4
- SMNDYUVBFMFKNZ-UHFFFAOYSA-N 2-furoic acid Chemical compound OC(=O)C1=CC=CO1 SMNDYUVBFMFKNZ-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 239000005711 Benzoic acid Substances 0.000 description 4
- UHOVQNZJYSORNB-MZWXYZOWSA-N benzene-d6 Chemical compound [2H]C1=C([2H])C([2H])=C([2H])C([2H])=C1[2H] UHOVQNZJYSORNB-MZWXYZOWSA-N 0.000 description 4
- 235000010233 benzoic acid Nutrition 0.000 description 4
- LPNBBFKOUUSUDB-UHFFFAOYSA-N p-toluic acid Chemical compound CC1=CC=C(C(O)=O)C=C1 LPNBBFKOUUSUDB-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-OUBTZVSYSA-N Carbon-13 Chemical compound [13C] OKTJSMMVPCPJKN-OUBTZVSYSA-N 0.000 description 3
- 238000005660 chlorination reaction Methods 0.000 description 3
- 238000005755 formation reaction Methods 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- OBETXYAYXDNJHR-SSDOTTSWSA-M (2r)-2-ethylhexanoate Chemical compound CCCC[C@@H](CC)C([O-])=O OBETXYAYXDNJHR-SSDOTTSWSA-M 0.000 description 2
- RPAJSBKBKSSMLJ-DFWYDOINSA-N (2s)-2-aminopentanedioic acid;hydrochloride Chemical class Cl.OC(=O)[C@@H](N)CCC(O)=O RPAJSBKBKSSMLJ-DFWYDOINSA-N 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- JVSFQJZRHXAUGT-UHFFFAOYSA-N 2,2-dimethylpropanoyl chloride Chemical compound CC(C)(C)C(Cl)=O JVSFQJZRHXAUGT-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- WTBAHSZERDXKKZ-UHFFFAOYSA-N octadecanoyl chloride Chemical compound CCCCCCCCCCCCCCCCCC(Cl)=O WTBAHSZERDXKKZ-UHFFFAOYSA-N 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- JUJWROOIHBZHMG-RALIUCGRSA-N pyridine-d5 Chemical compound [2H]C1=NC([2H])=C([2H])C([2H])=C1[2H] JUJWROOIHBZHMG-RALIUCGRSA-N 0.000 description 2
- 229910052594 sapphire Inorganic materials 0.000 description 2
- 239000010980 sapphire Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- MLQBTMWHIOYKKC-KTKRTIGZSA-N (z)-octadec-9-enoyl chloride Chemical compound CCCCCCCC\C=C/CCCCCCCC(Cl)=O MLQBTMWHIOYKKC-KTKRTIGZSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- WFSGQBNCVASPMW-UHFFFAOYSA-N 2-ethylhexanoyl chloride Chemical compound CCCCC(CC)C(Cl)=O WFSGQBNCVASPMW-UHFFFAOYSA-N 0.000 description 1
- OFTKFKYVSBNYEC-UHFFFAOYSA-N 2-furoyl chloride Chemical compound ClC(=O)C1=CC=CO1 OFTKFKYVSBNYEC-UHFFFAOYSA-N 0.000 description 1
- GPZXFICWCMCQPF-UHFFFAOYSA-N 2-methylbenzoyl chloride Chemical compound CC1=CC=CC=C1C(Cl)=O GPZXFICWCMCQPF-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 150000001721 carbon Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- MVPPADPHJFYWMZ-IDEBNGHGSA-N chlorobenzene Chemical group Cl[13C]1=[13CH][13CH]=[13CH][13CH]=[13CH]1 MVPPADPHJFYWMZ-IDEBNGHGSA-N 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- CCGKOQOJPYTBIH-UHFFFAOYSA-N ethenone Chemical compound C=C=O CCGKOQOJPYTBIH-UHFFFAOYSA-N 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 125000004440 haloalkylsulfinyl group Chemical group 0.000 description 1
- 125000004441 haloalkylsulfonyl group Chemical group 0.000 description 1
- 125000004995 haloalkylthio group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- REEZZSHJLXOIHL-UHFFFAOYSA-N octanoyl chloride Chemical compound CCCCCCCC(Cl)=O REEZZSHJLXOIHL-UHFFFAOYSA-N 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- PNQBEPDZQUOCNY-UHFFFAOYSA-N trifluoroacetyl chloride Chemical compound FC(F)(F)C(Cl)=O PNQBEPDZQUOCNY-UHFFFAOYSA-N 0.000 description 1
- 238000013022 venting Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/58—Preparation of carboxylic acid halides
- C07C51/60—Preparation of carboxylic acid halides by conversion of carboxylic acids or their anhydrides or esters, lactones, salts into halides with the same carboxylic acid part
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
Abstract
Procédé de phosgénation d'acides monocarboxyliques et/ou anhydrides caractérisé en ce que l'on traite l'acide et/ou l'anhydride, en présence ou non de solvant, par un excès molaire de phosgène, de préférence 2 à 15 fois plus de phosgène que d'acide, à des températures comprises entre 80 et 200 deg.C et à des pressions comprises entre 2 et 60 bar, avec ou sans catalyseur, de préférence en absence de tout catalyseur. Procédé de plus caractérisé en ce que la pression est en outre utilisée pour faciliter la séparation de l'acide chlorhydrique, du gaz carbonique et du phosgène, dans une colonne extérieure au réacteur.
Description
Phosgénation sous pression des acides et/ou anhydrides pour la
production des chlorures d'acides.
production des chlorures d'acides.
La présente invention a pour objet un procédé original pour accéder aux chlorures d'acides par phosgénation des acides monocarboxyliques et/ou anhydrides correspondants sous pression avec ou sans catalyseur, de préférence en absence de catalyseur.
Les procédés classiques avec catalyseur consistent à injecter du phosgène dans l'acide seul ou en solution à pression ordinaire et à des températures comprises entre 80 et 1500C. On utilise généralement un excès de phosgène. Les évents constitués d'un mélange de phosgène, de gaz carbonique et d'acide chlorhydrique sont non séparables à pression ordinaire, sauf à utiliser des condenseurs à très basse température, ce qui occasionne toujours une perte de phosgène.
La chimie est régie par les équations réactionnelles suivantes:
(1) RCOOH + COCU2 < RCOCl + HCI + CO2 (ki)
(2) RCOOH + RCOC1 H (RCO)20 + HCl (k-2/k2)
(3) (RC0)20 + 2COC12 o 2RCOCl + 2C02 (k3)
A pression ordinaire la réaction (1) est limitée par la concentration en phosgène, fonction de la température. La disparition de l'acide est donc relativement rapide mais le chlorure d'acide formé réagit sur l'acide présent pour conduire à l'anhydride selon la réaction (2) dont la transformation ultérieure en chlorure d'acide est lente (réaction (3)).
(1) RCOOH + COCU2 < RCOCl + HCI + CO2 (ki)
(2) RCOOH + RCOC1 H (RCO)20 + HCl (k-2/k2)
(3) (RC0)20 + 2COC12 o 2RCOCl + 2C02 (k3)
A pression ordinaire la réaction (1) est limitée par la concentration en phosgène, fonction de la température. La disparition de l'acide est donc relativement rapide mais le chlorure d'acide formé réagit sur l'acide présent pour conduire à l'anhydride selon la réaction (2) dont la transformation ultérieure en chlorure d'acide est lente (réaction (3)).
Ainsi, pour activer la réaction (3), il est nécessaire d'avoir recours à un ou plusieurs catalyseurs, d'où une littérature abondante pour ces dérivés. L'utilisation d'un catalyseur présente néanmoins plusieurs inconvénients. Leur coût tout d'abord puis leur influence sur le choix des matériaux car les catalyseurs rendent souvent le système réactionnel très corrosif. Ensuite, il favorise la formation de produits secondaires (tels que par ex. cétène) et le développement de coloration.
Enfin, il implique une purification du chlorure d'acide par distillation ou cristallisation.
Comme exemple de tels procédés, on citera par exemple la demande de brevet français FR 2585351 (EP 213976), dont le contenu est incorporé ici par référence, qui décrit la préparation de chlorures d'acides par phosgénation de l'acide carboxylique correspondant. Ce document présente comme une nécessité l'emploi d'un catalyseur afin d'obtenir des chlorures d'acides dans des conditions économiquement acceptables. Un des objets de cette demande concerne notamment le catalyseur utilisé pour faire la réaction de phosgénation.
Par ailleurs, le demandeur de la demande EP 213976 cite comme document de l'état de la technique un brevet américain (USP 2657233) qui, selon le demandeur, divulguerait l'utilisation d'une forte pression associée à une forte température pour produire les chlorures d'acides. Toutefois, la lecture de ce document montre que l'invention concerne ici un procédé de production de chlorures d'acides dicarboxyliques par phosgénation, sous fortes pression et température, de l'acide dicarboxylique correspondant. Ce document enseigne bien le fait que pour la production de monochlorures d'acides, la méthode classique telle que décrite plus haut est tout à fait satisfaisante et que finalement une amélioration ne peut être attendue que par une optimisation des catalyseurs utilisés, ce que confirment les documents FR 2585351 précité et FR 2254547 ou
EP 545774 par exemple.
EP 545774 par exemple.
L'invention cherche à éviter les inconvénients précités, notamment liés à l'utilisation de catalyseurs.
L'invention concerne un procédé de phosgénation d'acides monocarboxyliques et/ou anhydrides caractérisé en ce que l'on traite l'acide et/ou l'anhydride, en présence ou non de solvant, par un excès molaire de phosgène, de préférence 2 à 15 fois plus de phosgène que d'acide, à une température comprise entre 80 et 200 C et à une pression comprise entre 2 et 60 bar (1 bar = 105 Pa), avec ou sans catalyseur, de préférence en absence de tout catalyseur. On opère généralement en système fermé (pression autogène) ou en système ouvert (pression régulée par un dégazage partiel par ex.). Le procédé est généralement mis en oeuvre en continu ou en semi continu. De préférence, on opère en système ouvert en faisant un dégazage partiel. Le dégazage doit se faire en veillant à ce que reste un excès de phosgène. Cela passe soit par l'élimination sélective de l'acide chlorhydrique et du gaz carbonique, tout en gardant l'excès de phosgène et un peu de HC1 (pour ne pas faire d'anhydride mais pas trop pour ne pas trop ralentir la réaction finale) soit par un dégazage incluant du phosgène, ce dernier étant réalimenté en même temps. La température est avantageusement choisie entre 100 et 1500C, de préférence entre 110 et 130"C, alors que la pression est choisie de préférence entre 6 et 40 bar. Les conditions de température et pression sont déterminées par la nature de l'acide monocarboxylique et/ou de l'anhydride et du chlorure correspondant, notamment le point critique et/ou le point de décomposition.
Les avantages de la phosgénation sous pression selon l'invention sont de pouvoir a) permettre d'éliminer les condenseurs à basse température et b) de se dispenser de solvant et/ou de catalyseur. Cela permet d'éviter la purification finale du chlorure d'acide obtenu, d'avoir une séparation simple en fin de réaction, de réduire le coût des utilités, et d'une façon générale les avantages sont ceux déjà discutés plus haut liés à l'absence de catalyseur. On verra dans 1' exemple 1 que l'effet de l'ajout d'un catalyseur est quasiment gommé, c'est à dire que le gain de productivité obtenu par l'utilisation du procédé selon l'invention avec catalyseur par rapport au même procédé sans catalyseur est très faible en regard des inconvénients apportés par l'emploi d'un tel catalyseur. On verra aussi que le procédé selon l'invention sans catalyseur permet de convertir totalement l'acide engagé en chlorure en moins de temps que ne le ferait un procédé classique avec catalyseur. Enfin, le procédé selon l'invention permet un accroissement de productivité par rapport au procédé Batch ("par paquets") semi continu à pression ordinaire.
Le procédé selon l'invention est avantageusement mis en oeuvre pour la chloration des acides de formule RCOOH en chlorure d'acide RCOCI, R étant défini comme:
- un radical aliphatique ayant de 1 à 22 atomes de carbone, linéaire ou ramifié, saturé ou non, éventuellement substitué a) par un ou plusieurs atomes d'halogène identiques ou différents, b) par un ou plusieurs group nitro ou c) par au moins un groupe aryle (de préférence phényl), aryloxy ou arylthlo, chacun de ces groupes étant non substitué ou substitué;
- un radical cycloaliphatique ayant de 3 à 8 atomes de carbone, portant ou non un ou plusieurs substituants choisis parmi a) les atomes d'halogène, b) les radicaux alkyles ou haloalkyles, c) les groupes nitro et d) les radicaux aryles (de préférence phényl), aryloxy ou arylthio ceux-ci pouvant eux-mêmes être non substitués ou substitués;
- un radical carbocyclique aromatique, non substitué ou substitué par un ou plusieurs substituants choisis dans le groupe comprenant les atomes d'halogène, les radicaux alkyles ou haloalkyles (de préférence CF3) ayant de 1 à 12 atomes de carbone, les radicaux alkylthio ou haloalkylthio ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfinyl ou haloalkylsulfinyl ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfonyl ou haloalkylsulfonyl ayant de 1 à 6 atomes de carbone, les radicaux alkyloxy ou haloalkyloxy ayant de 1 à 6 atomes de carbone, les radicaux aryles, arylthio ou aryloxy, le groupe nitro;
- un radical hétérocyclique, aromatique ou non, à 5 ou 6 sommets, ayant un ou plusieurs hétéroatomes, identiques ou différents, choisis parmi les atomes d'oxygène, de soufre et d'azote et pouvant être non substitué ou substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène, les groupes nitro, les radicaux alkyles, halogénoalkyles, alkyloxy, haloalkyloxy, aryles, arylthio, aryloxy, et/ou pouvant être éventuellement condensé avec un carbocyle aromatique, lui même non substitué ou substitué.
- un radical aliphatique ayant de 1 à 22 atomes de carbone, linéaire ou ramifié, saturé ou non, éventuellement substitué a) par un ou plusieurs atomes d'halogène identiques ou différents, b) par un ou plusieurs group nitro ou c) par au moins un groupe aryle (de préférence phényl), aryloxy ou arylthlo, chacun de ces groupes étant non substitué ou substitué;
- un radical cycloaliphatique ayant de 3 à 8 atomes de carbone, portant ou non un ou plusieurs substituants choisis parmi a) les atomes d'halogène, b) les radicaux alkyles ou haloalkyles, c) les groupes nitro et d) les radicaux aryles (de préférence phényl), aryloxy ou arylthio ceux-ci pouvant eux-mêmes être non substitués ou substitués;
- un radical carbocyclique aromatique, non substitué ou substitué par un ou plusieurs substituants choisis dans le groupe comprenant les atomes d'halogène, les radicaux alkyles ou haloalkyles (de préférence CF3) ayant de 1 à 12 atomes de carbone, les radicaux alkylthio ou haloalkylthio ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfinyl ou haloalkylsulfinyl ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfonyl ou haloalkylsulfonyl ayant de 1 à 6 atomes de carbone, les radicaux alkyloxy ou haloalkyloxy ayant de 1 à 6 atomes de carbone, les radicaux aryles, arylthio ou aryloxy, le groupe nitro;
- un radical hétérocyclique, aromatique ou non, à 5 ou 6 sommets, ayant un ou plusieurs hétéroatomes, identiques ou différents, choisis parmi les atomes d'oxygène, de soufre et d'azote et pouvant être non substitué ou substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène, les groupes nitro, les radicaux alkyles, halogénoalkyles, alkyloxy, haloalkyloxy, aryles, arylthio, aryloxy, et/ou pouvant être éventuellement condensé avec un carbocyle aromatique, lui même non substitué ou substitué.
D'une manière générale, lorsqu'un groupe aryle (ou un de ses dérivés tels que aryloxy, arylthio) ou un carbocyle aromatique est mentionné, il faut considérer, même si ceci n'est pas dit au moment ou un tel radical apparaît afin d'alléger le présent exposé, que celui-ci peut porter des substituants choisis dans le groupe comprenant les atomes d'halogène, les radicaux alkyles, halogénoalkyles, alkyloxy, haloalkyloxy, alkylthio, haloalkylthio, alkylsulfinyl, haloalkylsulfinyl, alkylsulfonyl, haloalkylsulfonyl, aryles, aryloxy, arylthio, nitro.
Le procédé selon l'invention est également avantageusement mis en oeuvre pour la chloration des anhydrides de formule (RCO)20 ou des anhydrides mixtes (RCO)O(OCR') en chlorure d'acide RCOC1 et R'COCl, R et R' étant définis comme R ci-dessus et R et R' ne représentant pas le même radical en même temps.
Le procédé selon l'invention convient également pour la chloration de mélanges d'acides et d'anhydrides.
Ce procédé selon l'invention est également caractérisé en ce que la pression est en outre utilisée pour faciliter la séparation de l'acide chlorhydrique éventuel, du gaz carbonique et du phosgène, dans une colonne extérieure au réacteur, et ceci sans avoir recours à des condenseurs à basse température, source on l'a déjà vu de perte de COC12. La séparation devient dès lors plus simple, donc plus économique, qu'avec les procédés connus, et conduit à du phosgène facilement recyclable et à de l'acide chlorhydrique pur.
Les exemples suivant sont donnés à titre purement illustratifs de l'invention, qu'ils ne limitent en aucune façon. Ils montrent les avantages liés au procédé selon l'invention.
Exemple 1. Production de chlorure de stéaroyle par action du phosgène sur l'acide stéarique (Essai type n 413).
Dans un tube en saphir monocristallin (1) de diamètres externe et interne de 10 et 8 mm respectivement, conçu pour résister à de fortes pressions, sont pesés 0,175g (0,615 mmol-concentration 0,41M) d'acide stéarique et 0,920g (8,171 mmol) de chlorobenzène. Un tube scellé (2) de diamètre 5mm contenant du benzène deutérié est également introduit dans le tube (1) pour assurer la présence d'un lock externe, nécessaire à l'analyse RMN qui suivra. Le tube (1) est alors fermé, plongé dans un bain d'acétone/carboglace (-78"C) puis relié à une bouteille de phosgène. On condense alors dans le tube (1) 0,606g (6,127 mmol) de phosgène (concentration 4,08M). Après retour à température ambiante, le milieu réactionnel se présente sous la forme d'une suspension d'acide stéarique dans un liquide incolore et homogène. Le tube est alors introduit dans le cryo-aimant d'un spectromètre de RMN préchauffé à 117"C. Un premier spectre est enregistré 11 mn après introduction dans le cryo-aimant, soit une durée correspondant au réglage du spectromètre et à la stabilisation du réacteur à la température de consigne. Un programme automatique permet ensuite d'enregistrer des spectres à intervalles réguliers (typiquement toutes les 5 mn).
Le pourcentage molaire de chaque composé est déterminé par intégration des triplets correspondant aux protons méthylèniques en alpha de la fonction carbonyle.
On obtient alors les résultats suivants.

<tb>
N <SEP> essai <SEP> Temp. <SEP> [Acide] <SEP> [COCl2] <SEP> Vol. <SEP> k <SEP> observé* <SEP> t1/2 <SEP> P**
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb>

<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb>
<tb> 413 <SEP> 117 <SEP> 0,41 <SEP> 4.08 <SEP> 1.5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb>
* la constante cinétique k de pseudo premier ordre est calculée par linéarisation selon la formule : -Ln(1-TT)= kt avec TT = taux de transformation de l'acide et t = temps.
<tb>
* la constante cinétique k de pseudo premier ordre est calculée par linéarisation selon la formule : -Ln(1-TT)= kt avec TT = taux de transformation de l'acide et t = temps.
** P = Productivité calculée à TT = 50%.
En opérant comme à l'exemple ci-dessus en faisant varier différents paramètres {température, pression, etc.), on obtient les résultats donnés dans les tableaux ci-dessous.
a) influence de la température:

<tb> N <SEP> essai <SEP> Temp. <SEP> [Acide] <SEP> [COCl2] <SEP> Vol. <SEP> k <SEP> observé <SEP> t1/2 <SEP> P
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-' <SEP> min.
<tb>
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-' <SEP> min.
<tb>
<SEP> 415 <SEP> 83 <SEP> 0,42 <SEP> 4,3 <SEP> 1,5 <SEP> 0,44 <SEP> 94 <SEP> 40
<tb> <SEP> 421 <SEP> 101 <SEP> 0,39 <SEP> 4,94 <SEP> 1,6 <SEP> 1,18 <SEP> 35 <SEP> 100
<tb> <SEP> 413 <SEP> 117 <SEP> 0,41 <SEP> 4,08 <SEP> 1,5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb> <SEP> 416 <SEP> 121 <SEP> 0,39 <SEP> 4,69 <SEP> 1,6 <SEP> 3,2 <SEP> 13 <SEP> 270
<tb>
b) présence du catalyseur décrit dans l'exemple 1 de la demande FR 2585351:
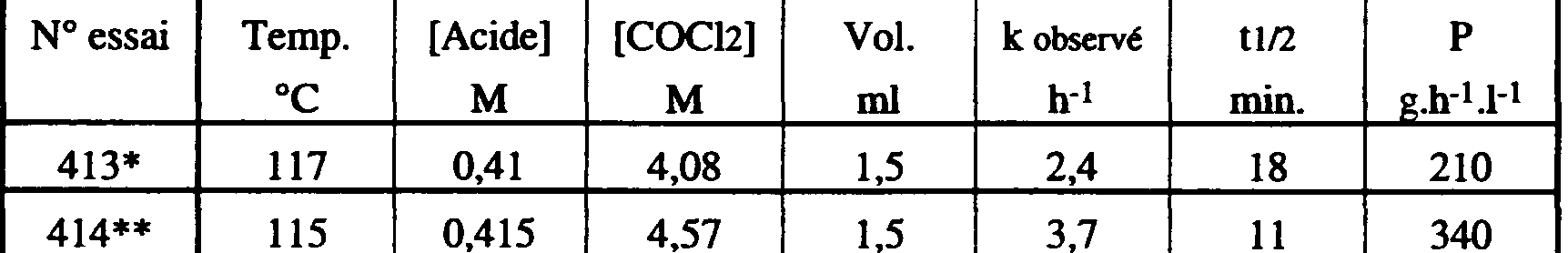
<tb> <SEP> 421 <SEP> 101 <SEP> 0,39 <SEP> 4,94 <SEP> 1,6 <SEP> 1,18 <SEP> 35 <SEP> 100
<tb> <SEP> 413 <SEP> 117 <SEP> 0,41 <SEP> 4,08 <SEP> 1,5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb> <SEP> 416 <SEP> 121 <SEP> 0,39 <SEP> 4,69 <SEP> 1,6 <SEP> 3,2 <SEP> 13 <SEP> 270
<tb>
b) présence du catalyseur décrit dans l'exemple 1 de la demande FR 2585351:
<tb> N <SEP> essai <SEP> Temp. <SEP> [Acide] <SEP> [COCl2j <SEP> Vol. <SEP> k <SEP> observ <SEP> tl,2 <SEP> P
<tb> <SEP> C <SEP> M <SEP> M <SEP> mi <SEP> h-1 <SEP> min.
<tb>
<tb> <SEP> C <SEP> M <SEP> M <SEP> mi <SEP> h-1 <SEP> min.
<tb>
<SEP> 413* <SEP> 117 <SEP> 0,41 <SEP> ~ <SEP> 4,08 <SEP> 1,5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb> <SEP> 414** <SEP> 115 <SEP> 0,415 <SEP> 4,57 <SEP> 1,5 <SEP> 3,7 <SEP> 11 <SEP> 340
<tb>
* sans catalyseur.
<tb> <SEP> 414** <SEP> 115 <SEP> 0,415 <SEP> 4,57 <SEP> 1,5 <SEP> 3,7 <SEP> 11 <SEP> 340
<tb>
* sans catalyseur.
** avec catalyseur (chlorure d'hexa n-butylguanidinium)(0,02% mol).
Comme on l'a déjà dit plus haut, L'effet de l'ajout d'un catalyseur est quasiment gommé, c'est à dire que le gain de productivité obtenu par l'utilisation du procédé selon l'invention avec catalyseur par rapport au même procédé sans catalyseur est très faible.
c) influence de la concentration en phosgène

<tb> N0 <SEP> essai <SEP> Temp. <SEP> [Acide] <SEP> [COCl2] <SEP> Vol. <SEP> observé <SEP> tlN <SEP> P
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb> <SEP> 413 <SEP> 117 <SEP> 0,41 <SEP> 4,08 <SEP> 1,5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb> <SEP> 420 <SEP> 114 <SEP> 0,47 <SEP> 3,15 <SEP> 1,3 <SEP> 2,1 <SEP> 20 <SEP> 185
<tb> d) influence du solvant:

<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb> <SEP> 413 <SEP> 117 <SEP> 0,41 <SEP> 4,08 <SEP> 1,5 <SEP> 2,4 <SEP> 18 <SEP> 210
<tb> <SEP> 420 <SEP> 114 <SEP> 0,47 <SEP> 3,15 <SEP> 1,3 <SEP> 2,1 <SEP> 20 <SEP> 185
<tb> d) influence du solvant:
<tb> N <SEP> essai <SEP> Temp. <SEP> [Acide] <SEP> [COCl2] <SEP> Vol. <SEP> K <SEP> observé <SEP> t1/2 <SEP> P
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb> <SEP> 415* <SEP> 83 <SEP> 0,42 <SEP> 4,3 <SEP> 1,5 <SEP> 0,44 <SEP> 94 <SEP> 40
<tb> <SEP> 425** <SEP> 80 <SEP> 0,885 <SEP> 10 <SEP> 1,4 <SEP> 0,63 <SEP> 66 <SEP> 50
<tb>
* solvant = chlorobenzène.
<tb> <SEP> C <SEP> M <SEP> M <SEP> ml <SEP> h-1 <SEP> min. <SEP> g.h-1.1-1
<tb> <SEP> 415* <SEP> 83 <SEP> 0,42 <SEP> 4,3 <SEP> 1,5 <SEP> 0,44 <SEP> 94 <SEP> 40
<tb> <SEP> 425** <SEP> 80 <SEP> 0,885 <SEP> 10 <SEP> 1,4 <SEP> 0,63 <SEP> 66 <SEP> 50
<tb>
* solvant = chlorobenzène.
** solvant = COC12.
e) influence du dégazage:
L'influence de travailler avec (essai 417) ou sans (essai 413) dégazage est montrée à la figure I ci-jointe.
L'influence de travailler avec (essai 417) ou sans (essai 413) dégazage est montrée à la figure I ci-jointe.
Le test 413 est déjà décrit et le test 417 est sensiblement le même mais en utilisant 0,16 g (0,56 mmol) d'acide, 0,875 g de chlorobenzène et 0,88 g (8,9 mmol) de phosgène. Pour le test 417, lorsque 90% de l'acide est transformé (soit après environ 45 mn), le tube est refroidi de façon à réaliser le dégazage. Après ce
dégazage, le tube est à nouveau chauffé jusqu'à 117 C après y avoir réintroduit
0,99 g de phosgène.
dégazage, le tube est à nouveau chauffé jusqu'à 117 C après y avoir réintroduit
0,99 g de phosgène.
Par ailleurs, d'après cette figure 1 on constate que l'on obtient un taux de
transformation de 100% de l'acide stéarique en moins de 2 heures (environ 75 mn
lorsque l'on opère avec un dégazage à 117 C (essai type n 417)). Cela permet de
comparer notre invention aux résultats donnés dans FR 2585351. En effet, dans ce
document l'acide stéarique est transformé totalement en chlorure lorsque l'on
opère en présence de 0,02% molaire de catalyseur à une température de 120
125 C mais ceci en 4 heures. On constate donc bien comme on l'a déjà dit que le
procédé selon l'invention sans catalyseur permet de convertir totalement l'acide engagé en chlorure en moins de temps que ne le ferait un procédé classique avec catalyseur.
transformation de 100% de l'acide stéarique en moins de 2 heures (environ 75 mn
lorsque l'on opère avec un dégazage à 117 C (essai type n 417)). Cela permet de
comparer notre invention aux résultats donnés dans FR 2585351. En effet, dans ce
document l'acide stéarique est transformé totalement en chlorure lorsque l'on
opère en présence de 0,02% molaire de catalyseur à une température de 120
125 C mais ceci en 4 heures. On constate donc bien comme on l'a déjà dit que le
procédé selon l'invention sans catalyseur permet de convertir totalement l'acide engagé en chlorure en moins de temps que ne le ferait un procédé classique avec catalyseur.
Exemple 2. Phosgénation de l'acide pivalique:
a) Une première approche a été faite en réalisant une réaction de phosgénation sous pression de cet acide en masse. En opérant comme à l'exemple 1 avec une sonde multinoyaux 10mm mais en utilisant la pyridine deutériée pour le lock à Ja place de benzène deutérié, on constate que les réactions de phosgénation sous pression de l'acide pivalique en chlorure d'acide sont d'ordre 1.
a) Une première approche a été faite en réalisant une réaction de phosgénation sous pression de cet acide en masse. En opérant comme à l'exemple 1 avec une sonde multinoyaux 10mm mais en utilisant la pyridine deutériée pour le lock à Ja place de benzène deutérié, on constate que les réactions de phosgénation sous pression de l'acide pivalique en chlorure d'acide sont d'ordre 1.
Les résultats sont les suivants
al) à 81"C, on a trouvé une constante de vitesse égale à 0,28 h- (conditions : 0,75 g d'acide (7,3 mmol) et 1,5 g de phosgène (15,2 mmol)).
al) à 81"C, on a trouvé une constante de vitesse égale à 0,28 h- (conditions : 0,75 g d'acide (7,3 mmol) et 1,5 g de phosgène (15,2 mmol)).
a2) à 115"C, on a trouvé une constante de vitesse égale à 3,00 h- (conditions: 0,692 g d'acide (6,78 mmol) et 1,25 g de phosgène (12,7 mmol)).
b) Une deuxième étude est réalisée pour suivre le cinétique de la réaction de phosgénation de l'acide pivalique dans le chlorobenzène et non plus en masse.
Contrairement à l'étude en masse, il n'est plus possible de distinguer les protons
CH3 de l'acide et du chlorure et, par conséquent, il n'est pas possible de suivre la réaction d'avancement de la réaction dans le chlorobenzène.
CH3 de l'acide et du chlorure et, par conséquent, il n'est pas possible de suivre la réaction d'avancement de la réaction dans le chlorobenzène.
Nous avons toutefois tenté de distinguer l'acide et le chlorure par RMN du carbone puisque les déplacements chimiques (par rapport au tétraméthylsilane
TMS) des carbones des groupes carbonyle et des carbones quaternaires des groupes tertiobutyle sont très différents. On a des déplacements chimiques de 185,8 ppm pour COOH, 180,8 pour COC1, 38,9 ppm pour le carbone quaternaire de l'acide et 49,5 ppm pour le carbone quaternaire du chlorure d'acide. On a utilisé un spectromètre AMX 300 opérant à 75 MHz pour le carbone 13 et équipé d'une sonde multinoyaux lOmm. Les déplacements chimiques (o) des raies de résonance carbone sont exprimés par rapport au tétraméthylsilane (TMS). Comme sous a), on utilise la pyridine deutériée (lock externe).
TMS) des carbones des groupes carbonyle et des carbones quaternaires des groupes tertiobutyle sont très différents. On a des déplacements chimiques de 185,8 ppm pour COOH, 180,8 pour COC1, 38,9 ppm pour le carbone quaternaire de l'acide et 49,5 ppm pour le carbone quaternaire du chlorure d'acide. On a utilisé un spectromètre AMX 300 opérant à 75 MHz pour le carbone 13 et équipé d'une sonde multinoyaux lOmm. Les déplacements chimiques (o) des raies de résonance carbone sont exprimés par rapport au tétraméthylsilane (TMS). Comme sous a), on utilise la pyridine deutériée (lock externe).
Dès lors, on a fait un suivi qui montre que, à 80"C, au bout de 1 heure 45 minutes le chlorure d'acide est largement majoritaire même s'il reste un peu d'acide (conditions : 0,06 g d'acide (0,6 mmol), 0,943 g de chlorobenzène et 0,735 g de phosgène (7,43 mmol)).
Exemple 3. Phosgénation de l'anhydride pivalique
Les analyses RMN du proton sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 300 MHz pour le proton et équipé d'une sonde
QNP íH/l3C/l9F/3lP gradient-z 5mm. Les déplacements chimiques (o) des raies de résonances du proton sont exprimés par rapport au tétraméthylsilane (TMS).
Les analyses RMN du proton sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 300 MHz pour le proton et équipé d'une sonde
QNP íH/l3C/l9F/3lP gradient-z 5mm. Les déplacements chimiques (o) des raies de résonances du proton sont exprimés par rapport au tétraméthylsilane (TMS).
Pour cet exemple, et pour les exemples 4 à 6 qui suivent, le tube en saphir monocristaijin (1) a des diamètres externe et interne de 5 et 4 mm respectivement.
Comme pour l'analyse RMN 1H sous pression de la réaction de phosgénation en masse de l'acide (cf. 2a), il a été possible de distinguer les protons
CH3 de l'anhydride pivalique (o=1,24 ppm) et du chlorure d'acide (8=1,31 ppm).
CH3 de l'anhydride pivalique (o=1,24 ppm) et du chlorure d'acide (8=1,31 ppm).
La réaction a été faite à 80 C avec un excès molaire de phosgène de 3 par rapport à l'anhydride.
Quand on trace l'opposé du logarithme népérien de la proportion molaire relative en anhydride pivalique en fonction du temps, on obtient une droite.
L'ordre apparent de la réaction de formation du chlorure de pivaloyle à partir d'anhydride pivalique est donc de 1. La pente de cette droite est égale à la constante de vitesse de la réaction : k = 1,6.10-2 min-l. Le temps de demi-vie tl/2, représentant le temps nécessaire pour que la concentration en anhydride soit diminuée de moitié, est égal à ln2/k (t1/2 = 40 minutes environ).
Exemple 4. Phosgénation de l'acide octanoique:
En opérant comme à l'exemple 2a), on constate que les réactions en masse de phosgénation sous pression de l'acide octanoïque en chlorure d'acide sont d'ordre 1. Les résultats sont les suivants:
1) à 79"C, on a trouvé une constante de vitesse égale à 0,25 h- (conditions : 0,79 g d'acide (6,9 mmol) et 1,68 g de phosgène (17 mmol)).
En opérant comme à l'exemple 2a), on constate que les réactions en masse de phosgénation sous pression de l'acide octanoïque en chlorure d'acide sont d'ordre 1. Les résultats sont les suivants:
1) à 79"C, on a trouvé une constante de vitesse égale à 0,25 h- (conditions : 0,79 g d'acide (6,9 mmol) et 1,68 g de phosgène (17 mmol)).
2) à 124"C, on a trouvé une constante de vitesse égale à 1,68 h-1 (conditions : 0,78 g d'acide (6,85 mmol) et 1,35 g de phosgène (13,7 mmol)).
Exemple 5. Phosgénation de l'acide trifluoroacétique:
Les analyses RMN du fluor sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 300 MHz pour le proton et équipé d'une sonde
QNP IH/t3C/19F/31P gradient-z 5mm. Les déplacements chimiques des raies de résonances du fluor sont exprimés par rapport à l'acide trifluoroacétique (TFA).
Les analyses RMN du fluor sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 300 MHz pour le proton et équipé d'une sonde
QNP IH/t3C/19F/31P gradient-z 5mm. Les déplacements chimiques des raies de résonances du fluor sont exprimés par rapport à l'acide trifluoroacétique (TFA).
La réaction est suivie par l'apparition d'une raie de résonance à 0,3 ppm qui correspond au chlorure de trifluoroacétyl, l'acide étant à 0 ppm puisque c'est la référence.
La réaction est lente puisque le taux de transformation en chlorure est voisin de 20% après un temps de chauffage à 107"C durant 4 heures. Toutefois, cette réaction de phosgénation sous pression en masse se fait (conditions : 0,22 g d'acide (1,93 mmol) et 0,464 g de phosgène (4,7 mmol)).
Exemple 6. Phosgénation de l'acide benzoïque:
Les analyses RMN du 13C sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 75 MHz pour le carbone 13 et équipé d'une sonde QNP 1H/13C119F/31P gradient-z 5mm. Les déplacements chimiques (o) des raies de résonance carbone sont exprimés par rapport au tétraméthylsilane (TMS).
Les analyses RMN du 13C sous pression ont été effectuées sur un spectromètre AMX 300 opérant à 75 MHz pour le carbone 13 et équipé d'une sonde QNP 1H/13C119F/31P gradient-z 5mm. Les déplacements chimiques (o) des raies de résonance carbone sont exprimés par rapport au tétraméthylsilane (TMS).
Par RMN 1H il est très difficile de distinguer l'acide benzoïque du chlorure d'acide. C'est pourquoi l'essai a été fait avec de l'acide benzoïque enrichi en carbone 13 (carbone du groupe carbonyle) pour effectuer un suivi cinétique à 90"C de la réaction sous pression en masse par RMN sur ce carbone 13. En effet, les raies de résonance carbone de l'acide et du chlorure d'acide sont situées respectivement à 8=170 ppm et 8=167 ppm.
Quand on trace l'opposé du logarithme népérien de la proportion molaire relative en acide benzoïque en fonction du temps, on obtient une droite (conditions initiales : 0,077 g d'acide (0,63 mmol) et 0,422 g de phosgène (4,27 mmol) - Température : 90"C). L'ordre apparent de la réaction de formation du chlorure de benzoyle à partir de l'acide benzoïque est donc de 1. La pente de cette droite est égale à la constante de vitesse de la réaction : k = 0,28 h-l. Le temps de demi-vie tel/2, représentant le temps nécessaire pour que la concentration en acide soit diminuée de moitié, est égal à ln2/k (tel/2 = 2h30 environ).
Mode opératoire général pour des essais à plus grande échelle que les précédents:
Les essais suivants ont été réalisés dans un réacteur autoclave de deux litres équipé d'un condenseur et d'un système de régulation de pression. Le volume total de l'autoclave et des accessoires est de 2,25 litres. Dans le réacteur parfaitement anhydre et purgé à l'argon, on introduit le monochlorobenzène et l'acide organique puis on ajoute le phosgène aux environ s de 20"C. On ferme la vanne mise à l'air puis on règle la consigne d'ouverture de la vanne de régulation à la pression choisie. On porte alors le milieu réactionnel à 1200C le plus rapidement possible.
Les essais suivants ont été réalisés dans un réacteur autoclave de deux litres équipé d'un condenseur et d'un système de régulation de pression. Le volume total de l'autoclave et des accessoires est de 2,25 litres. Dans le réacteur parfaitement anhydre et purgé à l'argon, on introduit le monochlorobenzène et l'acide organique puis on ajoute le phosgène aux environ s de 20"C. On ferme la vanne mise à l'air puis on règle la consigne d'ouverture de la vanne de régulation à la pression choisie. On porte alors le milieu réactionnel à 1200C le plus rapidement possible.
Les pourcentages d'acide, d'anhydride et de chlorure dans le milieu réactionnel sont déterminés par un suivi RMN du proton.
Exemple 7. Phosgénation de l'acide pivalique:
Dans le réacteur, on introduit 61,3 g (0,6 mole) d'acide pivalique et 890 g de monochlorobenzène puis on ajoute 597 g (6 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. Il se forme environ 0,3% d'anhydride pendant les premières 30 minutes. L'anhydride formé se phosgène. La réaction est terminée au bout de deux heures. Le taux d'acide résiduel est inférieur à 0,5% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure de pivaloyle obtenu est supérieur à 99,5% molaire.
Dans le réacteur, on introduit 61,3 g (0,6 mole) d'acide pivalique et 890 g de monochlorobenzène puis on ajoute 597 g (6 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. Il se forme environ 0,3% d'anhydride pendant les premières 30 minutes. L'anhydride formé se phosgène. La réaction est terminée au bout de deux heures. Le taux d'acide résiduel est inférieur à 0,5% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure de pivaloyle obtenu est supérieur à 99,5% molaire.
Exemple 8. Phosgénation de l'acide 2-éthylhexanoïque:
Dans le réacteur, on introduit 87 g (0,6 mole) d'acide 2-éthylhexanoïque et 890 g de monochlorobenzène puis on ajoute 607 g (6,14 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de une heure 30 minutes. Les taux d'acide et d'anhydride résiduels sont nuls et le taux de chlorure de 2éthylhexanoyle obtenu est supérieur à 99,8% molaire.
Dans le réacteur, on introduit 87 g (0,6 mole) d'acide 2-éthylhexanoïque et 890 g de monochlorobenzène puis on ajoute 607 g (6,14 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de une heure 30 minutes. Les taux d'acide et d'anhydride résiduels sont nuls et le taux de chlorure de 2éthylhexanoyle obtenu est supérieur à 99,8% molaire.
Exemple 9. Phosgénation de l'acide octanoïque:
Dans le réacteur, on introduit 86,6 g (0,6 mole) d'acide octanoïque et 890 g de monochlorobenzène puis on ajoute 600 g (6,07 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. Au bout de deux heures, le taux d'acide résiduel est d'environ 1% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure d'octanoyle obtenu est de 99% molaire.
Dans le réacteur, on introduit 86,6 g (0,6 mole) d'acide octanoïque et 890 g de monochlorobenzène puis on ajoute 600 g (6,07 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs et on chauffe le milieu réactionnel. Au bout de deux heures, le taux d'acide résiduel est d'environ 1% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure d'octanoyle obtenu est de 99% molaire.
Exemple 10. Phosgénation de l'acide stéarique:
Dans le réacteur, on introduit 170,4 g (0,6 mole) d'acide stéarique et 890 g de monochlorobenzène puis on ajoute 594 g (6 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs. On chauffe le milieu réactionnel à 1200C et on le maintient pendant 1 heure 30 minutes à cette température puis on le chauffe à 150"C et le maintient 1 heure à cette nouvelle température. Le taux d'acide résiduel est alors d'environ 1,4% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure de stéaroyle obtenu est de 98,6% molaire. La pression maximale atteinte a été de 9 bars relatifs.
Dans le réacteur, on introduit 170,4 g (0,6 mole) d'acide stéarique et 890 g de monochlorobenzène puis on ajoute 594 g (6 moles) de phosgène en 30 minutes environ en maintenant le température du milieu réactionnel à 25"C maximum. La consigne de pression est réglée à 10,5 bars relatifs. On chauffe le milieu réactionnel à 1200C et on le maintient pendant 1 heure 30 minutes à cette température puis on le chauffe à 150"C et le maintient 1 heure à cette nouvelle température. Le taux d'acide résiduel est alors d'environ 1,4% molaire, le taux d'anhydride résiduel est nul et le taux de chlorure de stéaroyle obtenu est de 98,6% molaire. La pression maximale atteinte a été de 9 bars relatifs.
Exemple 11. Phosgénation de l'acide oléique:
Dans le réacteur, on introduit 200 g (0,7 mole) d'acide oléique et 725 g de monochlorobenzène puis on ajoute 574 g (5,8 moles) de phosgène. La consigne de pression est réglée à 11,2 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de 1 heure 30 min. Le taux d'acide résiduel est de 0,5% molaire et le taux de chlorure d'oléoyle obtenu est de 99,5% molaire.
Dans le réacteur, on introduit 200 g (0,7 mole) d'acide oléique et 725 g de monochlorobenzène puis on ajoute 574 g (5,8 moles) de phosgène. La consigne de pression est réglée à 11,2 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de 1 heure 30 min. Le taux d'acide résiduel est de 0,5% molaire et le taux de chlorure d'oléoyle obtenu est de 99,5% molaire.
Exemple 12. Phosgénation de l'acide p-toluique:
Dans le réacteur, on introduit 81,4 g (0,6 mole) d'acide p-toluique et 892 g de monochlorobenzène puis on ajoute 582 g (5,9 moles) de phosgène. La consigne de pression est réglée à 11.2 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de 3 heures. L'analyse du milieu réactionnel final est effectuée par chromatographie en phase gazeuse. Le taux d'acide résiduel est de 0,3% molaire et le taux de chlorure de toluoyle obtenu est de 99,7% molaire.
Dans le réacteur, on introduit 81,4 g (0,6 mole) d'acide p-toluique et 892 g de monochlorobenzène puis on ajoute 582 g (5,9 moles) de phosgène. La consigne de pression est réglée à 11.2 bars relatifs et on chauffe le milieu réactionnel. La réaction est terminée au bout de 3 heures. L'analyse du milieu réactionnel final est effectuée par chromatographie en phase gazeuse. Le taux d'acide résiduel est de 0,3% molaire et le taux de chlorure de toluoyle obtenu est de 99,7% molaire.
Exemple 13. Phosgénation de l'acide 2-furoique:
Dans le réacteur, on introduit 110 g (0,98 mole) d'acide 2-furoique et 900 g de monochlorobenzène puis on ajoute 615 g (6,2 moles) de phosgène. La consigne de pression est réglée à 11,2 bars relatifs et on chauffe le milieu réactionnel. La pression est régulée par ajout d'argon. La réaction est terminée au bout de 3 heures. Le taux d'acide résiduel est de 5,5% molaire et le taux de chlorure de 2-furoyle obtenu est de 94,5% molaire.
Dans le réacteur, on introduit 110 g (0,98 mole) d'acide 2-furoique et 900 g de monochlorobenzène puis on ajoute 615 g (6,2 moles) de phosgène. La consigne de pression est réglée à 11,2 bars relatifs et on chauffe le milieu réactionnel. La pression est régulée par ajout d'argon. La réaction est terminée au bout de 3 heures. Le taux d'acide résiduel est de 5,5% molaire et le taux de chlorure de 2-furoyle obtenu est de 94,5% molaire.
Claims (10)
1. Procédé de phosgénation d'acides monocarboxyliques et/ou anhydrides caractérisé en ce que l'on traite l'acide et/ou l'anhydride, en présence ou non de solvant, par un excès molaire de phosgène à une température comprise entre 80 et 200"C et à une pression comprise entre 2 et 60 bar, avec ou sans catalyseur.
2. Procédé selon la revendication 1 caractérisé en ce que l'on opère avec un excès molaire de phosgène par rapport à l'acide de 2 à 15 fois plus de phosgène.
3. Procédé selon la revendication 1 ou 2 caractérisé en ce que l'on opère en absence de catalyseur.
4. Procédé selon les revendications 1, 2 ou 3 caractérisé en ce que l'on opère en système ouvert en faisant un dégazage partiel.
5. Procédé selon l'une des revendications 1 à 4 caractérisé en ce que la température est comprise entre 100 et 1500C, de préférence entre 110 et 130"C.
6. Procédé selon l'une des revendications précédentes caractérisé en ce que la pression est comprise entre 6 et 40 bar.
7. Procédé selon l'une quelconque des revendications précédentes caractérisé en ce quun acide monocarboxylique de formule RCOOH est converti en chlorure d'acide RCOCl, R étant défini comme:
- un radical aliphatique ayant de 1 à 22 atomes de carbone, linéaire ou ramifié, saturé ou non, éventuellement substitué a) par un ou plusieurs atomes d'halogène identiques ou différents, b) par un ou plusieurs group nitro ou c) par au moins un groupe aryle (de préférence phényl), aryloxy ou arylthio, chacun de ces groupes étant non substitué ou substitué;
- un radical cycloaliphatique ayant de 3 à 8 atomes de carbone, portant ou non un ou plusieurs substituants choisis parmi a) les atomes d'halogène, b) les radicaux alkyles ou haloalkyles, c) les groupes nitro et d) les radicaux aryles (de préférence phényl), aryloxy ou arylthio ceux-ci pouvant eux-mêmes être non substitués ou substitués;
- un radical carbocyclique aromatique, non substitué ou substitué par un ou plusieurs substituants choisis dans le groupe comprenant les atomes d'halogène, les radicaux alkyles ou haloalkyles (de préférence CF3) ayant de 1 à 12 atomes de carbone, les radicaux alkylthio ou haloalkylthio ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfinyl ou haloalkylsulfinyl ayant de 1 à 6 atomes de carbone, les radicaux alkylsulfonyl ou haloalkylsulfonyl ayant de 1 à 6 atomes de carbone, les radicaux alkyloxy ou haloalkyloxy ayant de 1 à 6 atomes de carbone, les radicaux aryles, arylthio ou aryloxy, le groupe nitro;
- un radical hétérocyclique, aromatique ou non, à 5 ou 6 sommets, ayant un ou plusieurs hétéroatomes, identiques ou différents, choisis parmi les atomes d'oxygène, de soufre et d'azote et pouvant être non substitué ou substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène, les groupes nitro, les radicaux alkyles, halogénoalkyles, alkyloxy, haloalkyloxy, aryles, arylthio, aryloxy, et/ou pouvant être éventuellement condensé avec un carbocyle aromatique, lui même non substitué ou substitué.
8. Procédé selon l'une quelconque des revendications 1 à 6 caractérisé en ce qu'un anhydride de formule (RCO)20 ou un anhydride mixte (RCO)O(OCR') est converti en chlorure d'acide RCOC1 et R'COCI, R et R' étant définis comme R dans la revendication 7 et R et R' ne représentant pas le même radical en même temps.
9 Procédé selon l'une quelconque des revendications 1 à 6 caractérisé en ce qu'un mélange d'acides et d'anhydrides est converti en chlorure d'acide monocarboxylique.
10. Procédé selon l'une des revendications précédente caractérisé en ce que la pression est en outre utilisée pour faciliter la séparation de l'acide chlorhydrique éventuel, du gaz carbonique et du phosgène, dans une colonne extérieure au réacteur.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9702461A FR2745568B1 (fr) | 1996-02-29 | 1997-02-25 | Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9602795A FR2745567B1 (fr) | 1996-02-29 | 1996-02-29 | Phosgenation sous pression des acides pour la prodution des chlorures d'acides |
| FR9702461A FR2745568B1 (fr) | 1996-02-29 | 1997-02-25 | Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2745568A1 true FR2745568A1 (fr) | 1997-09-05 |
| FR2745568B1 FR2745568B1 (fr) | 1998-08-21 |
Family
ID=9489904
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9602795A Expired - Fee Related FR2745567B1 (fr) | 1996-02-29 | 1996-02-29 | Phosgenation sous pression des acides pour la prodution des chlorures d'acides |
| FR9702461A Expired - Fee Related FR2745568B1 (fr) | 1996-02-29 | 1997-02-25 | Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9602795A Expired - Fee Related FR2745567B1 (fr) | 1996-02-29 | 1996-02-29 | Phosgenation sous pression des acides pour la prodution des chlorures d'acides |
Country Status (9)
| Country | Link |
|---|---|
| JP (1) | JPH09323953A (fr) |
| KR (1) | KR100599065B1 (fr) |
| CN (1) | CN1071303C (fr) |
| BR (1) | BR9700326A (fr) |
| DE (1) | DE19707285A1 (fr) |
| FR (2) | FR2745567B1 (fr) |
| GB (1) | GB2310661B (fr) |
| HU (1) | HU222273B1 (fr) |
| IL (1) | IL120272A0 (fr) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007108046A (ja) * | 2005-10-14 | 2007-04-26 | Idemitsu Kosan Co Ltd | 二層分離温度測定装置およびその測定方法 |
| BRPI0908991A2 (pt) * | 2008-03-18 | 2015-10-13 | Mitsui Chemicals Agro Inc | método para produzir derivado de ácido acilacético contendo flúor, método para produzir derivado de éster de ácido pirazolcarboxílico contendo flúor e método para produzir derivado de ácido pirazolcarboxílixo contendo flúor |
| CN104072347B (zh) * | 2014-06-30 | 2016-07-13 | 湖南海利化工股份有限公司 | 4-烷氧基-1,1,1-三氟-3-丁烯-2-酮的制备方法 |
| CN105384641A (zh) * | 2015-10-26 | 2016-03-09 | 安徽广信农化股份有限公司 | 一种对硝基苯甲酰氯的废水处理工艺 |
| CN105384640B (zh) * | 2015-10-26 | 2017-07-04 | 安徽广信农化股份有限公司 | 一种对硝基苯甲酰氯的尾气处理工艺 |
| CN105254505A (zh) * | 2015-10-26 | 2016-01-20 | 安徽广信农化股份有限公司 | 一种对硝基苯甲酰氯的精制工艺 |
| CN105524017B (zh) * | 2015-12-24 | 2017-06-30 | 江苏瀚联生物科技有限公司 | 2‑甲基氨基‑5‑叔丁基‑1,3,4‑噻二唑的制备方法 |
| CN105585478A (zh) * | 2016-03-08 | 2016-05-18 | 天津市敬业精细化工有限公司 | 一种酰氯的制备方法 |
| CN106674166B (zh) * | 2016-12-20 | 2018-09-25 | 重庆市化工研究院 | 呋喃甲酰氯的制备方法 |
| CN113402383A (zh) * | 2020-03-17 | 2021-09-17 | 上海祖玥新材料科技有限公司 | 一种2-乙基己酸与光气反应合成2-乙基己酰氯的方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB402328A (en) * | 1932-04-29 | 1933-11-30 | Ig Farbenindustrie Ag | Improvements in the manufacture of acetyl-chloride |
| FR2232532A1 (en) * | 1973-06-05 | 1975-01-03 | Poudres & Explosifs Ste Nale | Gas phase acid chloride prodn. - from acid and phosgene in presence of active carbon catalyst |
| EP0220516A1 (fr) * | 1985-10-09 | 1987-05-06 | Bayer Ag | Procédé de fabrication de chlorures d'acides carboxyliques aromatiques |
| EP0531826A2 (fr) * | 1991-09-07 | 1993-03-17 | BASF Aktiengesellschaft | Procédé de préparation d'halogénures d'acide carboxylique |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL175521C (nl) * | 1973-04-26 | 1984-11-16 | Hoechst Ag | Werkwijze voor het bereiden van carbonzuurchloriden. |
-
1996
- 1996-02-29 FR FR9602795A patent/FR2745567B1/fr not_active Expired - Fee Related
-
1997
- 1997-02-20 IL IL12027297A patent/IL120272A0/xx unknown
- 1997-02-24 DE DE19707285A patent/DE19707285A1/de not_active Withdrawn
- 1997-02-25 FR FR9702461A patent/FR2745568B1/fr not_active Expired - Fee Related
- 1997-02-25 KR KR1019970005815A patent/KR100599065B1/ko not_active Expired - Fee Related
- 1997-02-27 JP JP9044230A patent/JPH09323953A/ja active Pending
- 1997-02-27 GB GB9704047A patent/GB2310661B/en not_active Expired - Fee Related
- 1997-02-28 BR BR9700326A patent/BR9700326A/pt not_active IP Right Cessation
- 1997-02-28 CN CN97104714A patent/CN1071303C/zh not_active Expired - Fee Related
- 1997-02-28 HU HU9700535A patent/HU222273B1/hu not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB402328A (en) * | 1932-04-29 | 1933-11-30 | Ig Farbenindustrie Ag | Improvements in the manufacture of acetyl-chloride |
| FR2232532A1 (en) * | 1973-06-05 | 1975-01-03 | Poudres & Explosifs Ste Nale | Gas phase acid chloride prodn. - from acid and phosgene in presence of active carbon catalyst |
| EP0220516A1 (fr) * | 1985-10-09 | 1987-05-06 | Bayer Ag | Procédé de fabrication de chlorures d'acides carboxyliques aromatiques |
| EP0531826A2 (fr) * | 1991-09-07 | 1993-03-17 | BASF Aktiengesellschaft | Procédé de préparation d'halogénures d'acide carboxylique |
Also Published As
| Publication number | Publication date |
|---|---|
| HU222273B1 (hu) | 2003-05-28 |
| FR2745568B1 (fr) | 1998-08-21 |
| GB9704047D0 (en) | 1997-04-16 |
| FR2745567B1 (fr) | 1998-04-10 |
| GB2310661B (en) | 1998-12-02 |
| HUP9700535A3 (en) | 1999-06-28 |
| FR2745567A1 (fr) | 1997-09-05 |
| CN1163881A (zh) | 1997-11-05 |
| DE19707285A1 (de) | 1997-09-04 |
| IL120272A0 (en) | 1997-06-10 |
| KR100599065B1 (ko) | 2006-12-05 |
| BR9700326A (pt) | 1998-10-27 |
| GB2310661A (en) | 1997-09-03 |
| JPH09323953A (ja) | 1997-12-16 |
| KR970061845A (ko) | 1997-09-12 |
| HUP9700535A2 (hu) | 1998-06-29 |
| HU9700535D0 (en) | 1997-04-28 |
| CN1071303C (zh) | 2001-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FR2745568A1 (fr) | Phosgenation sous pression des acides et/ou anhydrides pour la production des chlorures d'acides | |
| FR2846651A1 (fr) | Procede de fabrication d'acides carboxyliques | |
| EP0648731B1 (fr) | Procédé d'hydroxycarbonylation du butadiène | |
| WO2008006977A1 (fr) | Procédé de préparation du 2-hydroxy-4-(méthylthio)butyronitrile et de la méthionine | |
| EP0021525B1 (fr) | Procédé pour la fabrication de composés carboxylés | |
| FR2887248A1 (fr) | Procede de fabrication d'acides carboxyliques | |
| US4131742A (en) | Cobalt-catalyzed oxidation of hydrocarbons | |
| EP1165481A1 (fr) | Procede d'oxydation d'hydrocarbures, d'alcools et/ou de cetones | |
| EP1345880B1 (fr) | Procede de preparation d'acides carboxyliques par carbonylation au palladium. | |
| EP0637579A1 (fr) | Procédé pour la préparation de 1-chloro-1-fluoroéthane et/ou de 1,1-difluoroéthane | |
| FR2767821A1 (fr) | Phosgenation sous pression des alcools pour la production des chloroformiates | |
| US5258530A (en) | Process for producing biphenyltetracarboxylic dianhydride | |
| FR2510101A1 (fr) | Procede d'hydrolyse catalytique en phase vapeur de chlorure de benzal ou son substitut avec un halogene ou un trifluoromethyle, pour former du benzaldehyde ou son substitut | |
| FR2470761A1 (fr) | Procede de preparation de l'acide trimellitique | |
| US4131741A (en) | Cobalt-catalyzed oxidation of C3 to C7 saturated aliphatic hydrocarbons to oxygenated products | |
| EP0326455B1 (fr) | Procédé de préparation de chloranil | |
| US4086267A (en) | Cobalt-catalyzed oxidation of C3 to C7 saturated aliphatic hydrocarbons to oxygenated products including acetic acid | |
| EP0612711B1 (fr) | Procédé d'hydroxycarbonylation de lactones | |
| EP1268384A1 (fr) | Procede d'oxydation d'hydrocarbures en acides | |
| FR2731218A1 (fr) | Procede de preparation de l'acide 3-chloropropionique | |
| EP0690836B1 (fr) | Procede d'isomerisation d'acides carboxyliques | |
| KR820002063B1 (ko) | 클로로 안식향산의 제조방법 | |
| JP2003342227A (ja) | ビフェニルテトラカルボン酸の製造法 | |
| FR2541993A1 (fr) | Procede de preparation d'acide adipique | |
| FR2471361A1 (fr) | Procede de production d'acide isophtalique |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ST | Notification of lapse |
Effective date: 20081031 |