FR2765221A1 - Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique - Google Patents
Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique Download PDFInfo
- Publication number
- FR2765221A1 FR2765221A1 FR9707900A FR9707900A FR2765221A1 FR 2765221 A1 FR2765221 A1 FR 2765221A1 FR 9707900 A FR9707900 A FR 9707900A FR 9707900 A FR9707900 A FR 9707900A FR 2765221 A1 FR2765221 A1 FR 2765221A1
- Authority
- FR
- France
- Prior art keywords
- alkyl group
- group
- alkyl
- branched
- straight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- -1 1H-IMIDAZOL-4-YL Chemical class 0.000 title claims description 17
- 238000002360 preparation method Methods 0.000 title claims description 5
- 230000001225 therapeutic effect Effects 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 61
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 24
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 20
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 17
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 9
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims abstract description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 5
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 5
- 239000001301 oxygen Substances 0.000 claims abstract description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 4
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 3
- 125000006527 (C1-C5) alkyl group Chemical group 0.000 claims abstract 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 14
- 238000000034 method Methods 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 6
- 150000002460 imidazoles Chemical class 0.000 claims description 6
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims description 4
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 4
- 125000004434 sulfur atom Chemical group 0.000 claims description 4
- 125000005842 heteroatom Chemical group 0.000 claims description 3
- 239000001257 hydrogen Substances 0.000 claims description 3
- 150000003839 salts Chemical class 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 239000011593 sulfur Substances 0.000 claims description 3
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 2
- 150000007513 acids Chemical class 0.000 claims description 2
- 150000002148 esters Chemical group 0.000 claims description 2
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000006701 (C1-C7) alkyl group Chemical group 0.000 claims 1
- 230000007062 hydrolysis Effects 0.000 claims 1
- 238000006460 hydrolysis reaction Methods 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- 229940126601 medicinal product Drugs 0.000 claims 1
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 abstract 1
- 238000002560 therapeutic procedure Methods 0.000 abstract 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 66
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- 239000000203 mixture Substances 0.000 description 35
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 16
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 14
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- 238000002844 melting Methods 0.000 description 12
- 230000008018 melting Effects 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000012429 reaction media Substances 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 7
- 210000001772 blood platelet Anatomy 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 6
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 6
- 235000019341 magnesium sulphate Nutrition 0.000 description 6
- 125000006255 cyclopropyl carbonyl group Chemical group [H]C1([H])C([H])([H])C1([H])C(*)=O 0.000 description 5
- 230000008961 swelling Effects 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 229940095102 methyl benzoate Drugs 0.000 description 4
- 210000004623 platelet-rich plasma Anatomy 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 0 C*1*CCN(C)C1 Chemical compound C*1*CCN(C)C1 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 239000000010 aprotic solvent Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 3
- 125000002140 imidazol-4-yl group Chemical group [H]N1C([H])=NC([*])=C1[H] 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 3
- 235000019260 propionic acid Nutrition 0.000 description 3
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- ORZUIQRIUPOWOU-UHFFFAOYSA-N 3-(butanoylamino)-4-[4-(5-methyl-1-tritylimidazol-4-yl)piperidin-1-yl]benzoic acid Chemical compound CCCC(=O)NC1=CC(C(O)=O)=CC=C1N1CCC(C2=C(N(C=N2)C(C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)C)CC1 ORZUIQRIUPOWOU-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- CNJJSTPBUHAEFH-UHFFFAOYSA-N methyl 4-fluoro-3-nitrobenzoate Chemical compound COC(=O)C1=CC=C(F)C([N+]([O-])=O)=C1 CNJJSTPBUHAEFH-UHFFFAOYSA-N 0.000 description 2
- RBEDIMSKRPGFEF-UHFFFAOYSA-N n-[2-[4-(5-methyl-1h-imidazol-4-yl)piperidin-1-yl]-5-(4-methylpiperazine-1-carbonyl)phenyl]cyclopropanecarboxamide Chemical compound C1CN(C)CCN1C(=O)C(C=C1NC(=O)C2CC2)=CC=C1N1CCC(C2=C(NC=N2)C)CC1 RBEDIMSKRPGFEF-UHFFFAOYSA-N 0.000 description 2
- IWZKEYLKBHDOFH-UHFFFAOYSA-N n-[2-[4-(5-methyl-1h-imidazol-4-yl)piperidin-1-yl]-5-(piperidine-1-carbonyl)phenyl]butanamide Chemical compound CCCC(=O)NC1=CC(C(=O)N2CCCCC2)=CC=C1N(CC1)CCC1C=1N=CNC=1C IWZKEYLKBHDOFH-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- HHBAOQCGVQTAAT-UHFFFAOYSA-N 4-(5-methyl-1h-imidazol-4-yl)piperidine;dihydrochloride Chemical compound Cl.Cl.N1=CNC(C2CCNCC2)=C1C HHBAOQCGVQTAAT-UHFFFAOYSA-N 0.000 description 1
- BOJWTAQWPVBIPG-UHFFFAOYSA-N 4-fluoro-3-nitrobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C([N+]([O-])=O)=C1 BOJWTAQWPVBIPG-UHFFFAOYSA-N 0.000 description 1
- 206010002383 Angina Pectoris Diseases 0.000 description 1
- JWEVRQIEYIKNDZ-UHFFFAOYSA-N CC1=C(N=CN1)C2CCN(CC2)C3=C(C=C(C=C3)C(=O)OC)[N+](=O)[O-] Chemical compound CC1=C(N=CN1)C2CCN(CC2)C3=C(C=C(C=C3)C(=O)OC)[N+](=O)[O-] JWEVRQIEYIKNDZ-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- OLBVUFHMDRJKTK-UHFFFAOYSA-N [N].[O] Chemical group [N].[O] OLBVUFHMDRJKTK-UHFFFAOYSA-N 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000002337 anti-port Effects 0.000 description 1
- 230000002785 anti-thrombosis Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 229960004676 antithrombotic agent Drugs 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 208000037849 arterial hypertension Diseases 0.000 description 1
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- DNSISZSEWVHGLH-UHFFFAOYSA-N butanamide Chemical compound CCCC(N)=O DNSISZSEWVHGLH-UHFFFAOYSA-N 0.000 description 1
- DVECBJCOGJRVPX-UHFFFAOYSA-N butyryl chloride Chemical compound CCCC(Cl)=O DVECBJCOGJRVPX-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 230000012105 intracellular pH reduction Effects 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- AOXPHVNMBPFOFS-UHFFFAOYSA-N methyl 2-nitrobenzoate Chemical compound COC(=O)C1=CC=CC=C1[N+]([O-])=O AOXPHVNMBPFOFS-UHFFFAOYSA-N 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- JVLFLMAMNYWHGP-UHFFFAOYSA-N n-[2-[4-(5-methyl-1-tritylimidazol-4-yl)piperidin-1-yl]-5-(piperidine-1-carbonyl)phenyl]butanamide Chemical compound CCCC(=O)NC1=CC(C(=O)N2CCCCC2)=CC=C1N(CC1)CCC1C(=C1C)N=CN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 JVLFLMAMNYWHGP-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000010414 supernatant solution Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Composés de formule (I) (CF DESSIN DANS BOPI) dans laquelle R1 représente un atome d'hydrogène ou un groupe (C1 -C4 ) alkyle droit ou ramifié, R2 représente un groupe (C1 -C6 ) alkyle droit ou ramifié, cyclo (C3 -C7 ) alkyle ou cyclo (C3 -C7 ) alkyle (C1 -C5 ) alkyle et R3 représente un groupe OR4 , O (CH2 ) n NR5 R6 , -NHC (NH) NH2 , -NHC (NH) N (CH3 ) 2 -NR5 R6 , - NR5 (CH2 ) n NR6 R7 , un groupe, (CF DESSIN DANS BOPI) , X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8 , -NR8 , -SO et -SO2 -, R4 , R5 , R6 , R7 et R8 étant un atome d'hydrogène, un groupe (C1 -C6 ) alkyle droit ou ramifié, cyclo (C3 -C7 ) alkyle, cyclo (C3 -C7 ) alkyle (C1 -C5 ) alkyle, phényle, phényl (C1 -C6 ) alkyle, hétéroaryle, m est égal à 1 ou 2 et n est égal à 2, 3 ou 4.Application en thérapeutique.
Description
La présente invention a pour objet des dérivés de 4- [(1H-imidazol-4-yl)pipéridin-1-yl]anilide, leur préparation et leur application en thérapeutique.
Les composés de l'invention répondent à la formule (I)
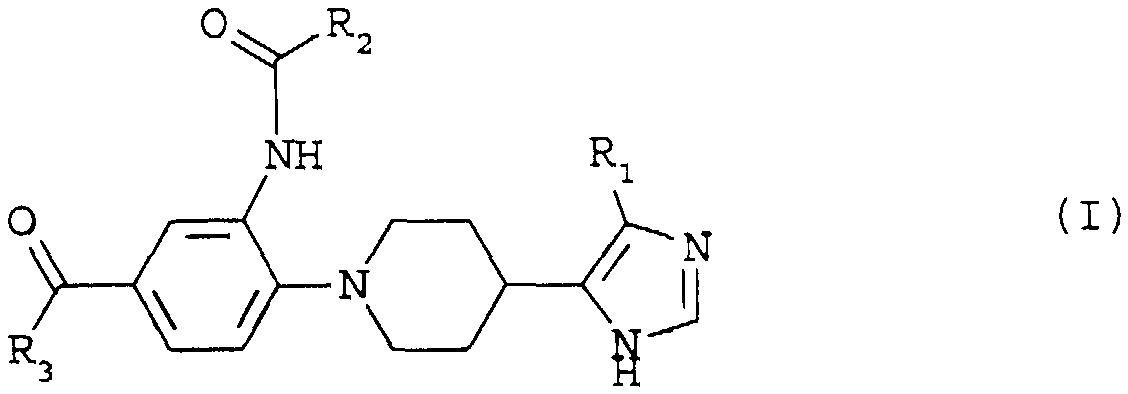
dans laquelle
R1 représente soit un atome d'hydrogène, soit un groupe (C1-C4)alkyle droit ou ramifié,
R2 représente soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs)alkyle et
R3 représente soit un groupe -OR4, soit un groupe -O(CH2)nNR5R6, soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2, soit un groupe -NR5R6, soit un groupe -MR5 (CH2) NR6R7, soit un groupe

dans laquelle
R1 représente soit un atome d'hydrogène, soit un groupe (C1-C4)alkyle droit ou ramifié,
R2 représente soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs)alkyle et
R3 représente soit un groupe -OR4, soit un groupe -O(CH2)nNR5R6, soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2, soit un groupe -NR5R6, soit un groupe -MR5 (CH2) NR6R7, soit un groupe
X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NR8, -SO- et -SO2-, R4, R51 R6, R7 et R8 étant indépendamment l'un de l'autre soit un atome d'hydrogène, soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs) alkyle, soit un groupe phényle, soit un groupe phényl(C1-C6) alkyle, soit un groupe hétéroaryle de 5 à 6 côtés, les hétéroatomes étant choisis parmi les atomes d'oxygène de soufre et d'azote, m est égal à 1 ou 2 et n est égal à 2, 3 ou 4, à l'état de bases libres ou de sels d'addition à des acides pharmaceutiquement acceptables.
Conformément à l'invention, on peut préparer les composés de formule (I) selon les procédés illustrés dans les schémas 1 et 2 ; dans ces schémas le groupe -C(C6H5)3 représente un groupe protecteur triphénylméthyle (groupe trityle).
Pour préparer les composés de formule (Ia) et (Ib), on fait réagir un composé de formule (II) dans laquelle Hal représente un atome d'halogène avec un alcool de formule ROH dans laquelle R représente un groupe (C1-C6)alkyle droit ou ramifié et on obtient un composé de formule (III) que l'on fait réagir avec un composé de formule (Iv) dans laquelle R1 est tel que défini précédemment, dans un solvant aprotique comme le diméthylformamide en présence d'une base comme la
N,N-diisopropyléthylamine, pour obtenir un composé de formule (V) que l'on traite par du chlorure de triphénylméthyle dans un solvant tel que le dichlorométhane en présence d'une base comme la N-méthylmorpholine pour préparer un composé de formule (VI) que l'on soumet à une hydrogénation catalytique et on obtient un composé de formule (VII) que l'on fait réagir avec un composé de formule R2COX dans laquelle X représente un atome d'halogène et R2 est tel que défini précédemment et on obtient un composé de formule (VIII).
N,N-diisopropyléthylamine, pour obtenir un composé de formule (V) que l'on traite par du chlorure de triphénylméthyle dans un solvant tel que le dichlorométhane en présence d'une base comme la N-méthylmorpholine pour préparer un composé de formule (VI) que l'on soumet à une hydrogénation catalytique et on obtient un composé de formule (VII) que l'on fait réagir avec un composé de formule R2COX dans laquelle X représente un atome d'halogène et R2 est tel que défini précédemment et on obtient un composé de formule (VIII).
Ensuite si on veut préparer un composé de formule (Ia) qui correspond à un composé de formule (I) dans laquelle R3 représente un groupe -OR4, R4 étant un atome d'hydrogène ou un groupe (C1-C6)alkyle droit ou ramifié alors on déprotège le noyau imidazole du composé de formule (VIII) correspondant dans des conditions classiques connues de l'homme du métier puis on réalise éventuellement une hydrolyse de la fonction ester.
Si on veut préparer un composé de formule (Ib) qui correspond à un composé de formule (I) dans laquelle R3 représente soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2, alors on fait réagir le composé de formule (VIII) correspondant avec un composé de formule H2NC(NH)N(Rg) 2 dans laquelle Rg représente soit un atome d'hydrogène, soit un groupe méthyle pour obtenir un composé de formule (XI) dont on déprotège le
Schéma 1

noyau imidazole dans des conditions classiques connues de l'homme du métier.
Schéma 1
noyau imidazole dans des conditions classiques connues de l'homme du métier.
Lorsqu'on veut obtenir un composé de formule (Ic) qui correspond à un composé de formule (I) dans laquelle R3 représente soit un groupe -OR4 (R4 étant un groupe cyclo(C3-C7)alkyle, cyclo(C3-C7)alkyle(Cl-Cs)alkyle, phényle, phényl(Cl-C6)alkyle ou hétéroaryle de 5 à 6 côtés les hétéroatomes étant choisis parmi les atomes d'oxygène de soufre et d'azote), soit un groupe -O(CH2)nNR5R6, soit un groupe -NR5R6, soit un groupe -NR5(CH2)nNR6R7 (R5, R6, R7 et n étant tels que définis précédemment), soit un groupe

X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NR8, -So- et -SO2- et R8 et m étant tels que définis précédemment, alors on procède selon le schéma 2.
On traite le composé de formule (VIII) correspondant en milieu basique et on obtient un composé de formule (IX) que l'on fait réagir avec un composé de formule R3H (R3 étant tel que défini ci-dessus) dans un solvant aprotique comme le diméthylformamide en présence de l,l'-carbonyldiimidazole pour préparer un composé de formule (X) dont on déprotège le noyau imidazole dans des conditions classiques connues de l'homme du métier.
Dans une variante selon l'invention, on peut préparer les composés de formule (Ic) en faisant réagir les composés de formule (la) correspondants dans laquelle R3 représente un groupe -OR4, R4 étant un atome d'hydrogène, avec un compose de formule R3H dans un solvant aprotique comme le diméthylformamide en présence d'un base comme la N,N-diisopropyléthylamine et de l,l'-carbonyldiimidazole puis en réalisant une déprotection du noyau imidazole dans des conditions classiques connues de l'homme du métier.
Schéma 2

Les composés de départ sont décrits dans la littérature ou peuvent être préparés selon des méthodes qui y sont décrites ou qui sont connues de l'homme de métier.
Ainsi la préparation des composés de formule (IV) est décrite dans la demande de brevet européen EP 0507650.
Les exemples qui suivent illustrent l'invention sans la limiter. Les micro-analyses et les spectres IR, RMN et de masse confirment la structure des composés obtenus. Les numéros des composés exemplifiés renvoient à ceux du tableau donné plus loin qui illustre les structures chimiques et les propriétés physiques de quelques composés selon l'invention.
Les rapports (x:y) correspondent au rapport (acide:base).
Exemple 1 (composé n0 11)
N-[2-[4-(5-méthyl- 1H- imidazol-4-yl) pipéridin-l-yl]-5-[ (4- méthylpipérazin- l-yl) carbonyl] phényl] cyclopropanecarboxamide 1.1. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-f5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]
benzoate de méthyle
1.1.1 3-amino-4-[4-(5-méthyl-l-(triphénylméthyl)-lH-
imidazol-4-yl)pipéridin-l-yl]benzoate de
méthyle
a) 4-fluoro-3-nitrobenzoate de méthyle
On met 5 g (27 mmoles) d'acide 4-fluoro-3-nitrobenzoïque en solution dans 50 ml de méthanol, on ajoute à la température ambiante 7,88 ml (108 mmoles) de chlorure de thionyle et on chauffe le mélange pendant 2 heures à la température de reflux. On évapore le milieu réactionnel à sec et on reprend le résidu par 150 ml de dichlorométhane. On lave successivement par 2 fois 10 ml d'une solution aqueuse saturée d'hydrogénocarbonate de sodium puis par 50 ml d'eau et on sèche sur sulfate de magnésium.
N-[2-[4-(5-méthyl- 1H- imidazol-4-yl) pipéridin-l-yl]-5-[ (4- méthylpipérazin- l-yl) carbonyl] phényl] cyclopropanecarboxamide 1.1. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-f5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]
benzoate de méthyle
1.1.1 3-amino-4-[4-(5-méthyl-l-(triphénylméthyl)-lH-
imidazol-4-yl)pipéridin-l-yl]benzoate de
méthyle
a) 4-fluoro-3-nitrobenzoate de méthyle
On met 5 g (27 mmoles) d'acide 4-fluoro-3-nitrobenzoïque en solution dans 50 ml de méthanol, on ajoute à la température ambiante 7,88 ml (108 mmoles) de chlorure de thionyle et on chauffe le mélange pendant 2 heures à la température de reflux. On évapore le milieu réactionnel à sec et on reprend le résidu par 150 ml de dichlorométhane. On lave successivement par 2 fois 10 ml d'une solution aqueuse saturée d'hydrogénocarbonate de sodium puis par 50 ml d'eau et on sèche sur sulfate de magnésium.
On obtient 5,37 g de produit.
Rendement = 91 %
Point de fusion = 70 OC
b) 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-3-
nitrobenzoate de méthyle
On met en suspension dans un mélange de 10 ml de dichlorométhane et de 2 ml de diméthylformamide, 2,38 g (10 mmoles) de dichlorhydrate de 4-(5-méthyl-lH-imidazol-4yl)pipéridine auxquels on ajoute 6,89 ml (40 mmoles) de
N,N-diisopropyléthylamine. On refroidit le mélange à - 5 OC et on ajoute goutte à goutte 1,9 g (10 mmoles) de 4-fluoro-3nitrobenzoate de méthyle en solution dans 5 ml de dichlorométhane. On laisse la température du mélange revenir à 0 "C puis on agite pendant 3 heures à cette température. On lave le milieu réactionnel avec 3 fois 10 ml d'eau, on sèche sur sulfate de magnésium et on évapore les solvants.
Point de fusion = 70 OC
b) 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-3-
nitrobenzoate de méthyle
On met en suspension dans un mélange de 10 ml de dichlorométhane et de 2 ml de diméthylformamide, 2,38 g (10 mmoles) de dichlorhydrate de 4-(5-méthyl-lH-imidazol-4yl)pipéridine auxquels on ajoute 6,89 ml (40 mmoles) de
N,N-diisopropyléthylamine. On refroidit le mélange à - 5 OC et on ajoute goutte à goutte 1,9 g (10 mmoles) de 4-fluoro-3nitrobenzoate de méthyle en solution dans 5 ml de dichlorométhane. On laisse la température du mélange revenir à 0 "C puis on agite pendant 3 heures à cette température. On lave le milieu réactionnel avec 3 fois 10 ml d'eau, on sèche sur sulfate de magnésium et on évapore les solvants.
Après trituration dans l'éther, on obtient 2,2 g de produit sous forme d'une poudre beige.
Rendement = 67 8
Point de fusion = 108 OC
c) 4- [4- [5-méthyl-l- (trîphénylméthyl) -îH-imidazol-4-
yl]pipéridin-l-yl]-3-nitrobenzoate de méthyle
On met en suspension dans 190 ml de dichlorométhane à la température ambiante 20 g (58 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl] -3-nitrobenzoate de méthyle, on ajoute 9,6 ml (87 mmoles) de N-méthylmorpholine puis 16,44 g (64 mmoles) de chlorure de trityle. On agite le mélange pendant 48 heures à la température ambiante et on ajoute 200 ml de dichlorométhane. On lave par 2 fois 150 ml d'eau.
Point de fusion = 108 OC
c) 4- [4- [5-méthyl-l- (trîphénylméthyl) -îH-imidazol-4-
yl]pipéridin-l-yl]-3-nitrobenzoate de méthyle
On met en suspension dans 190 ml de dichlorométhane à la température ambiante 20 g (58 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl] -3-nitrobenzoate de méthyle, on ajoute 9,6 ml (87 mmoles) de N-méthylmorpholine puis 16,44 g (64 mmoles) de chlorure de trityle. On agite le mélange pendant 48 heures à la température ambiante et on ajoute 200 ml de dichlorométhane. On lave par 2 fois 150 ml d'eau.
On sèche sur sulfate de magnésium, on filtre et on évapore à sec. On cristallise le résidu obtenu dans l'éther.
On obtient 27 g de produit.
Rendement = 79 %
Point de fusion = 228 OC
d) 3-amino-4- [4- [5-méthyl-l- (triphénylméthyl) -1H-
imidazol-4-yl]pipéridin-l-yl]benzoate de méthyle
Dans 100 ml de tétrahydrofurane, on ajoute 10 g (17,02 mmoles) de 4- [4- [5-méthyl-1- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-l-yl] -3-nitrobenzoate de méthyle et une suspension de Nickel de Raney. On agite le mélange sous atmosphère d'hydrogène jusqu'à décoloration de la solution surnageante, on filtre et on évapore le solvant. On triture le résidu dans l'éther glacé et on filtre.
Point de fusion = 228 OC
d) 3-amino-4- [4- [5-méthyl-l- (triphénylméthyl) -1H-
imidazol-4-yl]pipéridin-l-yl]benzoate de méthyle
Dans 100 ml de tétrahydrofurane, on ajoute 10 g (17,02 mmoles) de 4- [4- [5-méthyl-1- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-l-yl] -3-nitrobenzoate de méthyle et une suspension de Nickel de Raney. On agite le mélange sous atmosphère d'hydrogène jusqu'à décoloration de la solution surnageante, on filtre et on évapore le solvant. On triture le résidu dans l'éther glacé et on filtre.
On obtient 9,74 g de produit.
Rendement = 95 W
Point de fusion = 260 OC
1.1.2. 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-méthyl-1-
(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-
yl]benzoate de méthyle
On met en suspension dans 255 ml de dichlorométhane 25 g (45 mmoles) de 3-amino-4-[4- [5-méthyl-l- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-1-yl]benzoate de méthyle et on refroidit le mélange à 0 OC sous azote. On ajoute 3,74 g (47 mmoles) de pyridine puis goutte à goutte 4,94 g (47 mmoles) de chlorure de cyclopropanecarbonyle et on agite le milieu réactionnel pendant 1 heure à cette température. On laisse le mélange revenir à la température ambiante, on ajoute 200 ml de dichlorométhane puis on lave par 2 fois 150 ml d'eau. On sèche sur sulfate de sodium, on filtre et on évapore à sec. On reprend le résidu dans l'éther, on filtre et on sèche.
Point de fusion = 260 OC
1.1.2. 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-méthyl-1-
(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-
yl]benzoate de méthyle
On met en suspension dans 255 ml de dichlorométhane 25 g (45 mmoles) de 3-amino-4-[4- [5-méthyl-l- (triphénylméthyl) -1H- imidazol-4-yl]pipéridin-1-yl]benzoate de méthyle et on refroidit le mélange à 0 OC sous azote. On ajoute 3,74 g (47 mmoles) de pyridine puis goutte à goutte 4,94 g (47 mmoles) de chlorure de cyclopropanecarbonyle et on agite le milieu réactionnel pendant 1 heure à cette température. On laisse le mélange revenir à la température ambiante, on ajoute 200 ml de dichlorométhane puis on lave par 2 fois 150 ml d'eau. On sèche sur sulfate de sodium, on filtre et on évapore à sec. On reprend le résidu dans l'éther, on filtre et on sèche.
On obtient 25,8 g de produit.
Rendement = 91,9 %
1.1.3. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-
méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]
pipéridin-l-yl]benzoïque
On met en suspension dans 150 ml de méthanol 13 g (20,8 mmoles) de 3-[(cyclopropylcarbonyl)amino]-4-[4-[5- méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]pipéridin-l- yl]benzoate de méthyle, on ajoute 62,42 ml d'une solution aqueuse de soude 1 M (62,42 mmoles) et on chauffe à la température de reflux pendant 3,5 heures. On laisse le mélange pendant une nuit à la température ambiante, on ajoute 400 ml d'eau et on évapore le méthanol. On acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 M, on filtre et on sèche le composé obtenu à 50 OC.
1.1.3. acide 3-[(cyclopropylcarbonyl)amino]-4-[4-[5-
méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]
pipéridin-l-yl]benzoïque
On met en suspension dans 150 ml de méthanol 13 g (20,8 mmoles) de 3-[(cyclopropylcarbonyl)amino]-4-[4-[5- méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl]pipéridin-l- yl]benzoate de méthyle, on ajoute 62,42 ml d'une solution aqueuse de soude 1 M (62,42 mmoles) et on chauffe à la température de reflux pendant 3,5 heures. On laisse le mélange pendant une nuit à la température ambiante, on ajoute 400 ml d'eau et on évapore le méthanol. On acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 M, on filtre et on sèche le composé obtenu à 50 OC.
On obtient 12,1 g de produit.
Rendement = 95 8
Point de fusion = 237 OC 1.2. N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-5- [(4-méthylpipérazin-1-yl)carbonyl]phényl]cyclopropane-
carboxamide
On met en suspension dans 100 ml de dichlorométhane 2,2 g (3,6 mmoles) d'acide 3-[( cyclopropylcarbonyl ) amino]-4- [4- [5- méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoïque, on ajoute 1,4 ml (8 mmoles) de
N,N-diisopropyléthylamine puis on refroidit le mélange à 0 OC. On ajoute alors 0,52 ml (4,6 mmoles) de N-méthylpipérazine en solution dans 30 ml de dichlorométhane, on poursuit l'agitation à cette température pendant 15 minutes puis on ajoute 2,4 g (4,6 mmoles) de benzotriazol-l-yl-oxytripyrrolidinophosphonium hexafluorophosphate. On laisse la température du mélange remonter à la température ambiante pendant une nuit, on ajoute 100 ml de dichlorométhane et on lave la phase organique avec successivement 50 ml d'une solution aqueuse d'hydrogénocarbonate de sodium à 8 %, 100 ml d'une solution aqueuse de chlorure de sodium à 15 % puis 50 ml d'une solution aqueuse de chlorure de sodium à 30 %. On filtre et on sèche sur sulfate de magnésium. ON filtre et on évapore à sec. On reprend le résidu dans 100 ml d'éther et on l'essorez
On obtient 2,1 g de dérivé tritylé que l'on reprend dans 57,6 ml de tétrahydrofurane, on ajoute 60 ml d'eau et 60 ml d'acide acétique puis on chauffe pendant 3 heures à 80 "C. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (90:12:1).
Point de fusion = 237 OC 1.2. N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-5- [(4-méthylpipérazin-1-yl)carbonyl]phényl]cyclopropane-
carboxamide
On met en suspension dans 100 ml de dichlorométhane 2,2 g (3,6 mmoles) d'acide 3-[( cyclopropylcarbonyl ) amino]-4- [4- [5- méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoïque, on ajoute 1,4 ml (8 mmoles) de
N,N-diisopropyléthylamine puis on refroidit le mélange à 0 OC. On ajoute alors 0,52 ml (4,6 mmoles) de N-méthylpipérazine en solution dans 30 ml de dichlorométhane, on poursuit l'agitation à cette température pendant 15 minutes puis on ajoute 2,4 g (4,6 mmoles) de benzotriazol-l-yl-oxytripyrrolidinophosphonium hexafluorophosphate. On laisse la température du mélange remonter à la température ambiante pendant une nuit, on ajoute 100 ml de dichlorométhane et on lave la phase organique avec successivement 50 ml d'une solution aqueuse d'hydrogénocarbonate de sodium à 8 %, 100 ml d'une solution aqueuse de chlorure de sodium à 15 % puis 50 ml d'une solution aqueuse de chlorure de sodium à 30 %. On filtre et on sèche sur sulfate de magnésium. ON filtre et on évapore à sec. On reprend le résidu dans 100 ml d'éther et on l'essorez
On obtient 2,1 g de dérivé tritylé que l'on reprend dans 57,6 ml de tétrahydrofurane, on ajoute 60 ml d'eau et 60 ml d'acide acétique puis on chauffe pendant 3 heures à 80 "C. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (90:12:1).
On obtient 1,12 g de produit après cristallisation dans l'acétate d'éthyle.
Rendement = 69 %
Point de fusion = 188 OC
Exemple 2 (composé n" 2) 4-[4-(5-méthyl-lH-imldazol-4-yl)pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoate de méthyle 2.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-
yl]pipéridin-l-yl]-3-[(1-oxobutyl)amino]benzoate de
méthyle
Sous azote, on met 6,3 g (10,62 mmoles) de 3-amino-4-[4-[5 méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoate de méthyle en suspension dans 55 ml de dichlorométhane et on refroidit le mélange à 0-5 OC. On ajoute alors 0,95 ml (11,74 mmoles) de pyridine et 1,16 ml (11,15 mmoles) de chlorure de butyryle puis on poursuit l'agitation pendant 1 heure à 0-5 OC. On laisse revenir la température du mélange à la température ambiante et on agite pendant une nuit à cette température. On ajoute 90 ml de dichlorométhane, on lave avec 2 fois 80 ml d'eau et on sèche sur sulfate de magnésium. On filtre et on évapore à sec.
Point de fusion = 188 OC
Exemple 2 (composé n" 2) 4-[4-(5-méthyl-lH-imldazol-4-yl)pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoate de méthyle 2.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-
yl]pipéridin-l-yl]-3-[(1-oxobutyl)amino]benzoate de
méthyle
Sous azote, on met 6,3 g (10,62 mmoles) de 3-amino-4-[4-[5 méthyl-l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1- yl]benzoate de méthyle en suspension dans 55 ml de dichlorométhane et on refroidit le mélange à 0-5 OC. On ajoute alors 0,95 ml (11,74 mmoles) de pyridine et 1,16 ml (11,15 mmoles) de chlorure de butyryle puis on poursuit l'agitation pendant 1 heure à 0-5 OC. On laisse revenir la température du mélange à la température ambiante et on agite pendant une nuit à cette température. On ajoute 90 ml de dichlorométhane, on lave avec 2 fois 80 ml d'eau et on sèche sur sulfate de magnésium. On filtre et on évapore à sec.
On obtient 6,5 g de produit sous forme d'un composé amorphe.
Rendement = 98 % 2.2. 4- [4- (5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl] -3- [(I-
oxobutyl)amino]benzoate de méthyle
On solubilise 6 g (10,5 mmoles) de 4-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-3-[(1oxobutyl)amino]benzoate de méthyle dans 170 ml de tétrahydrofurane, on ajoute 170 ml d'eau et 340 ml d'acide acétique puis on chauffe àla température de reflux pendant 5 heures. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (92:8:0,5).
oxobutyl)amino]benzoate de méthyle
On solubilise 6 g (10,5 mmoles) de 4-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-3-[(1oxobutyl)amino]benzoate de méthyle dans 170 ml de tétrahydrofurane, on ajoute 170 ml d'eau et 340 ml d'acide acétique puis on chauffe àla température de reflux pendant 5 heures. On évapore le milieu réactionnel, on reprend le résidu dans l'éther et on essore. On purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol:ammoniaque (92:8:0,5).
On obtient 3,3 g de produit après cristallisation dans l'éther.
Rendement = 82 %
Point de fusion = 166-170 OC
Exemple 3 (composé nO 1) acide 4- [4- (5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl] -3- [(1- oxobutyl)amino]benzoïque
On solubilise 2,9 g (7,87 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl]-3-[(1-oxobutyl)amino]benzoate de méthyle dans 50 ml de méthanol, on ajoute 0,630 g (15,74 mmoles) de soude et on chauffe le mélange à la température de reflux pendant 2 heures. On évapore le méthanol, on reprend le résidu par 70 ml d'eau et on ajuste le pH à 6,5 avec une solution aqueuse d'acide chlorhydrique 3 N. On filtre et on sèche sous vide à 60 OC.
Point de fusion = 166-170 OC
Exemple 3 (composé nO 1) acide 4- [4- (5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl] -3- [(1- oxobutyl)amino]benzoïque
On solubilise 2,9 g (7,87 mmoles) de 4-[4-(5-méthyl-1H- imidazol-4-yl)pipéridin-1-yl]-3-[(1-oxobutyl)amino]benzoate de méthyle dans 50 ml de méthanol, on ajoute 0,630 g (15,74 mmoles) de soude et on chauffe le mélange à la température de reflux pendant 2 heures. On évapore le méthanol, on reprend le résidu par 70 ml d'eau et on ajuste le pH à 6,5 avec une solution aqueuse d'acide chlorhydrique 3 N. On filtre et on sèche sous vide à 60 OC.
On obtient 2,68 g de produit.
Rendement = 92 8
Point de fusion = 291 OC (fusion avec décomposition)
Exemple 4 (composé nO 6)
N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide
Méthode A A.4.1. acide 4- [4- [5-méthyl-l-(triphénylméthyl)-îH-
imidazol-4-yl]pipéridin-l-yl]-3-[(l-oxobutyl)
amino] benzoique
On met 12 g (19,46 mmoles) de 4-[4-[5-méthyl-l-(triphénylmé- thyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(1-oXobutyl)amino] benzoate de méthyle en suspension dans 120 ml de méthanol, on ajoute 38,93 ml (38,93 mmoles) d'une solution aqueuse de soude 0,1 N et on porte le milieu réactionnel à la température de reflux pendant 2 heures. On évapore le méthanol, on refroidit le mélange à 0-5 OC, on l'acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 N et on laisse le milieu réactionnel pendant une nuit à cette température. On filtre, on essore et on sèche sur pentoxyde de phosphore sous vide à 70 OC.
Point de fusion = 291 OC (fusion avec décomposition)
Exemple 4 (composé nO 6)
N-[2-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-l-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide
Méthode A A.4.1. acide 4- [4- [5-méthyl-l-(triphénylméthyl)-îH-
imidazol-4-yl]pipéridin-l-yl]-3-[(l-oxobutyl)
amino] benzoique
On met 12 g (19,46 mmoles) de 4-[4-[5-méthyl-l-(triphénylmé- thyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(1-oXobutyl)amino] benzoate de méthyle en suspension dans 120 ml de méthanol, on ajoute 38,93 ml (38,93 mmoles) d'une solution aqueuse de soude 0,1 N et on porte le milieu réactionnel à la température de reflux pendant 2 heures. On évapore le méthanol, on refroidit le mélange à 0-5 OC, on l'acidifie à pH 4 avec une solution aqueuse d'acide chlorhydrique 3 N et on laisse le milieu réactionnel pendant une nuit à cette température. On filtre, on essore et on sèche sur pentoxyde de phosphore sous vide à 70 OC.
On obtient 10,75 g de produit.
Rendement = 90 %
A.4.2. N-[2-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4- yl]pipéridin-l-yl]-5-(pipéridin-1-ylcarbonyl)phényl]
butanamide
Sous azote on met 1,5 g (2,45 mmoles) d'acide 4-[4-[5-méthyl l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoïque en suspension dans 10 ml de diméthylformamide, on ajoute 0,44 mg (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 40 C pendant 2 heures. On ajoute ensuite 0,208 g (2,45 mmoles) de pipéridine et on laisse le mélange pendant une nuit à 80 OC.
A.4.2. N-[2-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4- yl]pipéridin-l-yl]-5-(pipéridin-1-ylcarbonyl)phényl]
butanamide
Sous azote on met 1,5 g (2,45 mmoles) d'acide 4-[4-[5-méthyl l-(triphénylméthyl)-lH-imidazol-4-yl]pipéridin-l-yl]-3-[(l- oxobutyl)amino]benzoïque en suspension dans 10 ml de diméthylformamide, on ajoute 0,44 mg (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 40 C pendant 2 heures. On ajoute ensuite 0,208 g (2,45 mmoles) de pipéridine et on laisse le mélange pendant une nuit à 80 OC.
On évapore le diméthylformamide, on reprend le résidu par 100 ml de dichlorométhane puis on lave par 2 fois 35 ml d'eau. On sèche sur sulfate de sodium, on filtre et on évapore à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (95:5).
On obtient 1,0 g de produit.
Rendement = 60 %
A.4.3. N-[2-[4-(5-méthyl-1H-imidazol-4-yl)pipéridin-1-yl] 5- (pipéridin- l-ylcarbonyl) phényl] butanamide
On solubilise 1 g (1,47 mmoles) de N-[2-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide dans 25 ml de tétrahydrofurane, on ajoute 25 ml d'eau et 50 ml d'acide acétique puis on chauffe à la température de reflux pendant 2 heures. On évapore le milieu réactionnel et on purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10).
A.4.3. N-[2-[4-(5-méthyl-1H-imidazol-4-yl)pipéridin-1-yl] 5- (pipéridin- l-ylcarbonyl) phényl] butanamide
On solubilise 1 g (1,47 mmoles) de N-[2-[4-[5-méthyl-1- (triphénylméthyl)-lH-imidazol-4-yl]pipéridin-1-yl]-5- (pipéridin-l-ylcarbonyl)phényl]butanamide dans 25 ml de tétrahydrofurane, on ajoute 25 ml d'eau et 50 ml d'acide acétique puis on chauffe à la température de reflux pendant 2 heures. On évapore le milieu réactionnel et on purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10).
On obtient 0,3 g de produit après cristallisation dans l'éther.
Rendement = 54 %
Point de fusion = 139-141,5 OC
Méthode B
Sous azote, on met en suspension 0,7 g (1,89 mmoles) d'acide 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-3-[(1- oxobutyl)amino]benzoïque en suspension dans 7 ml de diméthylformamide, on ajoute 0,34 g (2,08 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures. On laisse la température du milieu réactionnel redescendre à 40 OC, on ajoute 0,16 g (1,89 mmoles) de pipéridine et on chauffe le mélange à 85 OC pendant 2 heures. On évapore le diméthylformamide, on reprend le résidu dans l'éther et on le purifie par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions adéquates, on évapore à sec et on recristallise dans un mélange eau:méthanol (1:1). On filtre et on sèche sous vide sur pentoxyde de phosphore.
Point de fusion = 139-141,5 OC
Méthode B
Sous azote, on met en suspension 0,7 g (1,89 mmoles) d'acide 4-[4-(5-méthyl-lH-imidazol-4-yl)pipéridin-1-yl]-3-[(1- oxobutyl)amino]benzoïque en suspension dans 7 ml de diméthylformamide, on ajoute 0,34 g (2,08 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures. On laisse la température du milieu réactionnel redescendre à 40 OC, on ajoute 0,16 g (1,89 mmoles) de pipéridine et on chauffe le mélange à 85 OC pendant 2 heures. On évapore le diméthylformamide, on reprend le résidu dans l'éther et on le purifie par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions adéquates, on évapore à sec et on recristallise dans un mélange eau:méthanol (1:1). On filtre et on sèche sous vide sur pentoxyde de phosphore.
On obtient 0,215 g de produit.
Rendement = 26 %
Point de fusion = 139-141,5 OC
Exemple 5 (composé nO 4) 4-[4-(5-methyl-1H- imidazol-4-yl)p ipéridin-l-yl]-3-[( I- oxobutyl) amino] -N- (phénylméthyl)benzamide 5.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-yl]
pipéridin-l-yl]-3-[(1-oxobutyl)amino]-N-(phénylméthyl)
benzamide
Sous azote on met 1,5 g (2,44 mmoles) d'acide 4-[4-[5-méthyl 1- (triphénylméthyl) -lH-imidazol-4-yl]pipérîdin-l-yl] -3- [(1- oxobutyl)amino]benzoïque en suspension dans 9 ml de diméthylformamide, on ajoute 0,44 g (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures.
Point de fusion = 139-141,5 OC
Exemple 5 (composé nO 4) 4-[4-(5-methyl-1H- imidazol-4-yl)p ipéridin-l-yl]-3-[( I- oxobutyl) amino] -N- (phénylméthyl)benzamide 5.1. 4-[4-[5-méthyl-1-(triphénylméthyl)-lH-imidazol-4-yl]
pipéridin-l-yl]-3-[(1-oxobutyl)amino]-N-(phénylméthyl)
benzamide
Sous azote on met 1,5 g (2,44 mmoles) d'acide 4-[4-[5-méthyl 1- (triphénylméthyl) -lH-imidazol-4-yl]pipérîdin-l-yl] -3- [(1- oxobutyl)amino]benzoïque en suspension dans 9 ml de diméthylformamide, on ajoute 0,44 g (2,69 mmoles) de l,l'-carbonyldiimidazole et on chauffe le mélange à 60 OC pendant 2 heures.
On laisse la température du milieu réactionnel revenir à 50 OC et on ajoute 0,26 g (2,44 mmoles) de phénylméthylamine puis on chauffe à 80 OC pendant une nuit. On évapore à sec, on reprend le résidu par 100 ml de dichlorométhane et on lave successivement par 40 ml d'une solution aqueuse d'acide acétique 0,1 M puis par 40 ml d'eau. On sèche sur sulfate de magnésium, on filtre et on évapore à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (95:5).
On obtient 1,55 g de produit.
Rendement = 90 % 5.2. 4-[4-(5-méthyl-1H-imidazol-4-yl)pipéridin-1-yl]-3-[(1-
oxobutyl)amino]-N-(phénylméthyl)benzamide
On solubilise 1,55 g (2,21 mmoles) de 4-[4-[5-méthyl-1 (triphénylméthyl)-lH-imidazol-4-yl]pipéridln-l-yl]-3-[(loxobutyl)amino]-N-(phénylméthyl)benzamide dans 35 ml de tétrahydrofurane, on ajoute 35 ml d'eau et 70 ml d'acide acétique puis on chauffe à la température de reflux pendant 3 heures. On évapore le milieu réactionnel et on purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions, on évapore à sec et on laissse cristalliser dans un mélange méthanol:eau. On filtre et on sèche sous vide sur pentoxyde de phosphore à 60 OC.
oxobutyl)amino]-N-(phénylméthyl)benzamide
On solubilise 1,55 g (2,21 mmoles) de 4-[4-[5-méthyl-1 (triphénylméthyl)-lH-imidazol-4-yl]pipéridln-l-yl]-3-[(loxobutyl)amino]-N-(phénylméthyl)benzamide dans 35 ml de tétrahydrofurane, on ajoute 35 ml d'eau et 70 ml d'acide acétique puis on chauffe à la température de reflux pendant 3 heures. On évapore le milieu réactionnel et on purifie le produit par chromatographie sur colonne de gel de silice en éluant par un mélange dichlorométhane:méthanol (90:10). On rassemble les fractions, on évapore à sec et on laissse cristalliser dans un mélange méthanol:eau. On filtre et on sèche sous vide sur pentoxyde de phosphore à 60 OC.
On obtient 0,455 g de produit.
Rendement = 45 %
Point de fusion = 233-235 OC
Légende du tableau
dans la colonne "R2", -c(C3H7) représente un groupe cyclopropyle,
dans la colonne "Sel", "fum" représente un fumarate ; les rapports entre parenthèses représentent le rapport (acide:base) ; l'absence de toute mention signifie que le composé est sous forme de base,
dans la colonne "Point de fusion", (d) coreespond à une fusion avec décomposition.
Point de fusion = 233-235 OC
Légende du tableau
dans la colonne "R2", -c(C3H7) représente un groupe cyclopropyle,
dans la colonne "Sel", "fum" représente un fumarate ; les rapports entre parenthèses représentent le rapport (acide:base) ; l'absence de toute mention signifie que le composé est sous forme de base,
dans la colonne "Point de fusion", (d) coreespond à une fusion avec décomposition.
Tableau


<tb> <SEP> Point <SEP> de
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 1 <SEP> -CH3 <SEP> -C3H7 <SEP> -OH <SEP> - <SEP> 291 <SEP> (d)
<tb> 2 <SEP> -CH3 <SEP> -C3H7 <SEP> -OCH3 <SEP> - <SEP> 166-170
<tb> 3 <SEP> -CH3 <SEP> -C3H7 <SEP> -NHCH3 <SEP> 138-144
<tb> <SEP> \NH
<tb> 4 <SEP> -CH, <SEP> -C,H, <SEP> 233-235
<tb> 6 <SEP> -CH3 <SEP> -C3H7 <SEP> - <SEP> NS <SEP> - <SEP> 139-141,5
<tb> 7 <SEP> /m <SEP> - <SEP> 123-129
<tb> 7 <SEP> -CH3 <SEP> -C3H7 <SEP> -N <SEP> N-CH3
<tb> <SEP> \ff
<tb> 8 <SEP> -CH3 <SEP> -CH2CH(CH3)2 <SEP> - <SEP> N3 <SEP> - <SEP> 177
<tb> 9 <SEP> -CH3 <SEP> ; <SEP> -CH2CH(CH3) <SEP> 2 <SEP> -N <SEP> N <SEP> - <SEP> - <SEP> 120 <SEP> (d)
<tb> <SEP> M
<tb>

<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 1 <SEP> -CH3 <SEP> -C3H7 <SEP> -OH <SEP> - <SEP> 291 <SEP> (d)
<tb> 2 <SEP> -CH3 <SEP> -C3H7 <SEP> -OCH3 <SEP> - <SEP> 166-170
<tb> 3 <SEP> -CH3 <SEP> -C3H7 <SEP> -NHCH3 <SEP> 138-144
<tb> <SEP> \NH
<tb> 4 <SEP> -CH, <SEP> -C,H, <SEP> 233-235
<tb> 6 <SEP> -CH3 <SEP> -C3H7 <SEP> - <SEP> NS <SEP> - <SEP> 139-141,5
<tb> 7 <SEP> /m <SEP> - <SEP> 123-129
<tb> 7 <SEP> -CH3 <SEP> -C3H7 <SEP> -N <SEP> N-CH3
<tb> <SEP> \ff
<tb> 8 <SEP> -CH3 <SEP> -CH2CH(CH3)2 <SEP> - <SEP> N3 <SEP> - <SEP> 177
<tb> 9 <SEP> -CH3 <SEP> ; <SEP> -CH2CH(CH3) <SEP> 2 <SEP> -N <SEP> N <SEP> - <SEP> - <SEP> 120 <SEP> (d)
<tb> <SEP> M
<tb>
<tb> <SEP> Point <SEP> de
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 10 <SEP> -CH3 <SEP> -c <SEP> (C3H7) <SEP> -É <SEP> - <SEP> 240 <SEP> (d)
<tb> 11 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -N <SEP> N-cH <SEP> - <SEP> 188
<tb> <SEP> M
<tb> <SEP> fum
<tb> 12 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> Ne <SEP> < 1:1) <SEP> 130 <SEP> (d)
<tb> 13 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> N43 <SEP> ~ <SEP> 233-237
<tb> 14 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -NHCH(NH)NH2 <SEP> - <SEP> 236-241,5
<tb>
Les composés de l'invention ont fait l'objet d'études pharmacologiques qui ont mis en évidence leurs propriétés inhibitrices de l'échangeur sodium/proton et leur intérêt comme substances à activité thérapeutique.
<tb> NO <SEP> R1 <SEP> R2 <SEP> R3 <SEP> Sel
<tb> <SEP> fusion <SEP> (0C)
<tb> 10 <SEP> -CH3 <SEP> -c <SEP> (C3H7) <SEP> -É <SEP> - <SEP> 240 <SEP> (d)
<tb> 11 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -N <SEP> N-cH <SEP> - <SEP> 188
<tb> <SEP> M
<tb> <SEP> fum
<tb> 12 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> Ne <SEP> < 1:1) <SEP> 130 <SEP> (d)
<tb> 13 <SEP> -CH3 <SEP> -c(C3H7) <SEP> N <SEP> N43 <SEP> ~ <SEP> 233-237
<tb> 14 <SEP> -CH3 <SEP> -c(C3H7) <SEP> -NHCH(NH)NH2 <SEP> - <SEP> 236-241,5
<tb>
Les composés de l'invention ont fait l'objet d'études pharmacologiques qui ont mis en évidence leurs propriétés inhibitrices de l'échangeur sodium/proton et leur intérêt comme substances à activité thérapeutique.
Ainsi, les composés de l'invention ont été soumis à un test d'inhibition du gonflement des plaquettes sanguines de lapin en milieu acide selon la méthode de Grinstein et al. (In
Methods in Enzymology, Fleisher S. And Flusher B., Vol 173, pp 777-790, Academic Press Inc., 1984).
Methods in Enzymology, Fleisher S. And Flusher B., Vol 173, pp 777-790, Academic Press Inc., 1984).
On prélève par ponction cardiaque du sang sur des lapins
Néo-Zélandais, en utilisant un anticoagulant citratedextrose. On obtient le plasma riche en plaquettes (PRP) par centrifugation à 1200 rpm pendant 20 minutes à la température ambiante. Après mesure du volume moyen plaquettaire initial on incube une fraction aliquote de PRP pendant 20 minutes dans un milieu propionate de sodium/acide propionique (140 mM) contenant du chlorure de potassium (1 mM), du chlorure de magnésium (1 mM), du glucose (10 mM) le tout tamponné par de l'Hepes (20 mM) à pH 6,7 et dont l'osmolarité est d'environ 300 mosm/l. L'acide propionique diffuse dans les plaquettes où il se dissocie, provoquant une acidification intra-cellulaire et une activation de l'antiport sodium/proton. L'influx d'ions sodium s'accompagne d'une capture d'eau qui provoque le gonflement des plaquettes. La mesure du volume moyen plaquettaire à la fin de l'incubation, diminuée du volume moyen plaquettaire initial, permet d'estimer le gonflement maximal des plaquettes. On ajoute les produits à tester au milieu d'incubation d' acide propionique aux concentrations voulues, avant l'addition de PRP. Les résultats sont exprimés en pourcentage d'inhibition du gonflement maximum permettant de calculer la CIso ou concentration inhibant de 50% le gonflement maximum.
Néo-Zélandais, en utilisant un anticoagulant citratedextrose. On obtient le plasma riche en plaquettes (PRP) par centrifugation à 1200 rpm pendant 20 minutes à la température ambiante. Après mesure du volume moyen plaquettaire initial on incube une fraction aliquote de PRP pendant 20 minutes dans un milieu propionate de sodium/acide propionique (140 mM) contenant du chlorure de potassium (1 mM), du chlorure de magnésium (1 mM), du glucose (10 mM) le tout tamponné par de l'Hepes (20 mM) à pH 6,7 et dont l'osmolarité est d'environ 300 mosm/l. L'acide propionique diffuse dans les plaquettes où il se dissocie, provoquant une acidification intra-cellulaire et une activation de l'antiport sodium/proton. L'influx d'ions sodium s'accompagne d'une capture d'eau qui provoque le gonflement des plaquettes. La mesure du volume moyen plaquettaire à la fin de l'incubation, diminuée du volume moyen plaquettaire initial, permet d'estimer le gonflement maximal des plaquettes. On ajoute les produits à tester au milieu d'incubation d' acide propionique aux concentrations voulues, avant l'addition de PRP. Les résultats sont exprimés en pourcentage d'inhibition du gonflement maximum permettant de calculer la CIso ou concentration inhibant de 50% le gonflement maximum.
Dans ce test, les CIso des composés les plus intéressants de l'invention sont inférieures à 10 pM.
A ce titre ils peuvent être utilisés dans le traitement et la prévention de différentes formes de pathologies telles que l'hypertension artérielle et pulmonaire, l'arythmie cardiaque, l'ischémie cardiaque, l'infarctus cardiaque, l'insuffisance cardiaque et l'angine de poitrine, les ischémies des organes périphériques, des membres inférieurs et du système nerveux central, les néphropathies, les oedèmes, les fibroses et les cancers, ainsi que les maladies caractérisées par des hyperplasies et hypertrophies des vaisseaux ou du coeur.
Ils peuvent aussi être utilisés pour la protection des organes dans les opérations de chirurgie ou de transplantation d'organe.
Les composés de l'invention peuvent être utilisés seuls ou en association avec d'autres substances telles que les nitrates, les antagonistes du calcium, les bêta-bloquants, les antithrombotiques, les thrombolytiques, les salicylates.
A cet effet ils peuvent être présentés sous toutes formes appropriées à l'administration orale ou parentérale, telles que comprimés, dragées, gélules, capsules, suspensions ou solutions buvables ou injectables, etc. en association avec des excipients convenables. Ces formes sont dosées pour permettre une administration de 1 à 1000 mg/kg de 1 à 4 fois par jour.
Ils peuvent également être présentés sous toutes formes appropriées à l'administration transdermique ou sublinguale.
Claims (6)
1. Composés de formule (I)

dans laquelle
R1 représente soit un atome d'hydrogène, soit un groupe (Cl-C4)alkyle droit ou ramifié,
R2 représente soit un groupe (Cl-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-C5)alkyle et
R3 représente soit un groupe -OR4, soit un groupe -O(CH2)nNRsR6, soit un groupe -NHC(NH)NH2, soit un groupe -NHC(NH)N(CH3)2 soit un groupe -NR5R6, soit un groupe -NRS(CH2)nNR6R7, soit un groupe

X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NRs, -SO- et -SO2-, R4, R5, R6, R7 et Re étant indépendamment l'un de l'autre soit un atome d'hydrogène, soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-.C7)alkyle, soit un groupe cyclo(C3-C7)alkyle(Cl-Cs) alkyle, soit un groupe phényle, soit un groupe phényl(C1-C6) alkyle, soit un groupe hétéroaryle de 5 à 6 côtés les hétéroatomes étant choisis parmi les atomes d'oxygène de soufre et d'azote, m est égal à 1 ou 2 et n est égal à 2, 3 ou 4, ainsi que leurs sels d'addition à des acides pharmaceutiquement acceptables.
2. Procédé de préparation des composés de formule (Ia) selon la revendication 1

dans laquelle R1 représente soit un atome d'hydrogène, soit un groupe (Cl-C4)alkyle droit ou ramifié, R2 représente soit un groupe (C1-C6)alkyie droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle (C1-Cs)alkyle et R4 représente soit un atome d'hydrogène, soit un groupe (Cl-C6)alkyle droit ou ramifié, procédé caractérisé en ce que l'on déprotège le noyau imidazole d'un composé de formule (VIII)

dans laquelle R représente un groupe (C1-C6)alkyle droit ou ramifié pour obtenir un composé de de formule (Ia) dans laquelle R4 représente un groupe (C1-C6)alkyle droit ou ramifié puis que l'on réalise si nécessaire une hydrolyse de la fonction ester pour obtenir un composé de formule (Ia) dans laquelle R4 représente un atome d'hydrogène.
3. Procédé de préparation des composés de formule (Ib) selon la revendication 1

dans laquelle R1 représente soit un atome d'hydrogène, soit un groupe (C1-C4)alkyle droit ou ramifié, R2 représente soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(Q-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle (Cl-C5)alkyle et Rg représente soit un atome d'hydrogène, soit un groupe méthyle, procédé caractérisé en ce que l'on fait réagir un composé de formule (VIII)

dans laquelle R représente un groupe (C1-C6)alkyle droit ou ramifié, R1 représente soit un atome d'hydrogène, soit un groupe (C1-C4)alkyle droit ou ramifié et R2 représente soit un groupe (C1-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle (C1-Cs)alkyle, avec un composé de formule H2NC(NH)N(Rg)2 dans laquelle Rg représente soit un atome d'hydrogène, soit un groupe méthyle pour obtenir un composé de formule (XI)

dont on déprotège le noyau imidazole.
4. Procédé de préparation des composés de formule (Ic)

dans laquelle R1 représente soit un atome d'hydrogène, soit un groupe (Cl-C4)alkyle droit ou ramifié, R2 représente soit un groupe (Cl-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle (C1-C5)alkyle et R3 représente soit un groupe -OR4 (R4 étant un groupe cyclo(C3-C7)alkyle, cyclo(C3-C7)alkyle(Cl-Cs)alkyle, phényle, phényl(C1-C6)alkyle ou hétéroaryle), soit un groupe -O(CH2)nNR5R6, soit un groupe -NRSR6, soit un groupe NR5(CH2)NR6R7 (R5, R6, R7 et n étant tels que définis dans la revendication 1), soit un groupe

dont on déprotège le noyau imidazole.

que l'on fait réagir avec un composé de formule R3H où R3 est tel que défini précédemment et on obtient un composé de formule (X)

dans laquelle R représente un groupe (C1-C6)alkyle droit ou ramifié, R1 représente soit un atome d'hydrogène, soit un groupe (Cl-C4)alkyle droit ou ramifié et R2 représente soit un groupe (Cl-C6)alkyle droit ou ramifié, soit un groupe cyclo(C3-C7)alkyle, soit un groupe cyclo(C3-C7)alkyle (C1-Cs)alkyle, en milieu basique et on obtient un composé de formule (IX)

X étant choisi parmi les atomes d'oxygène et de soufre et les groupes -CHR8, -NR8, -SO- et -SO2- et R8 et m étant tels que définis dans la revendication 1, procédé caractérisé en ce que l'on traite un composé de formule (VIII)
5. Médicament caractérisé en ce qu'il contient un composé selon la revendication 1.
6. Composition pharmaceutique caractérisée en ce qu'elle contient un composé selon la revendication 1 en association avec un excipient pharmaceutiquement acceptable.
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9707900A FR2765221B1 (fr) | 1997-06-25 | 1997-06-25 | Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique |
| EP98932237A EP0991639A1 (fr) | 1997-06-25 | 1998-06-19 | DERIVES DE 4- (1$i(H)-IMIDAZOL-4-YL)PIPERIDIN-1-YL]ANILIDE, LEUR PREPARATION ET LEUR APPLICATION EN THERAPEUTIQUE |
| AU82206/98A AU8220698A (en) | 1997-06-25 | 1998-06-19 | 4-{(1h-imidazol-4-yl)piperidin-1-yl}anilide derivatives, their preparation and application in therapy |
| JP50531399A JP2002506458A (ja) | 1997-06-25 | 1998-06-19 | 4−[(1h−イミダゾール−4−イル)ピペリジン−1−イル]アニリド誘導体、それらの製造及び治療におけるそれらの応用 |
| PCT/FR1998/001288 WO1999000379A1 (fr) | 1997-06-25 | 1998-06-19 | Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique |
| ARP980103032A AR013127A1 (es) | 1997-06-25 | 1998-06-24 | Derivados de 4-[(1h-imidazol-4-il)piperidin-1-il]anilida, su preparacion y su aplicacion en terapeutica |
| ZA985518A ZA985518B (en) | 1997-06-25 | 1998-06-24 | 4-[(1H-imidazol-4-YL) piperidin-1-YL] anilide derivatives their preparation and their application in therapeutics |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR9707900A FR2765221B1 (fr) | 1997-06-25 | 1997-06-25 | Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2765221A1 true FR2765221A1 (fr) | 1998-12-31 |
| FR2765221B1 FR2765221B1 (fr) | 1999-07-30 |
Family
ID=9508386
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR9707900A Expired - Lifetime FR2765221B1 (fr) | 1997-06-25 | 1997-06-25 | Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0991639A1 (fr) |
| JP (1) | JP2002506458A (fr) |
| AR (1) | AR013127A1 (fr) |
| AU (1) | AU8220698A (fr) |
| FR (1) | FR2765221B1 (fr) |
| WO (1) | WO1999000379A1 (fr) |
| ZA (1) | ZA985518B (fr) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027107A3 (fr) * | 1999-10-12 | 2002-01-24 | Bristol Myers Squibb Co | Inhibiteurs heterocycliques d'echange sodium / protons et procedes a cet effet |
| EP4291191A4 (fr) * | 2021-02-12 | 2025-01-15 | The Trustees Of Columbia University In The City Of New York | Nouveaux composés comprenant une nouvelle classe de ligands de transthyrétine pour le traitement de comorbidités communes associées à l'âge |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7501889B2 (en) | 2004-04-26 | 2009-03-10 | Rgb Systems, Inc. | Method and apparatus for implementing soft switching in a class D amplifier |
| EP2734517B1 (fr) | 2011-07-18 | 2017-08-30 | Merck Patent GmbH | Benzamides |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0197840A1 (fr) * | 1985-03-26 | 1986-10-15 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | (Imidazolyl-4) piperidines, leur préparation et leur application en thérapeutique |
| EP0494010A1 (fr) * | 1990-12-31 | 1992-07-08 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Nouvelles 4-(4-imidazolyl) pipéridines substituées en 1, leur préparation ainsi que leurs applications thérapeutiques |
| EP0507650A1 (fr) * | 1991-04-03 | 1992-10-07 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique |
| EP0591027A1 (fr) * | 1992-09-28 | 1994-04-06 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4325822A1 (de) * | 1993-07-31 | 1995-02-02 | Hoechst Ag | Substituierte Benzoylguanidine, Verfahren zu ihrer Herstellung, ihre Verwendung als Medikament oder Diagnostikum sowie sie enthaltendes Medikament |
-
1997
- 1997-06-25 FR FR9707900A patent/FR2765221B1/fr not_active Expired - Lifetime
-
1998
- 1998-06-19 JP JP50531399A patent/JP2002506458A/ja active Pending
- 1998-06-19 EP EP98932237A patent/EP0991639A1/fr not_active Withdrawn
- 1998-06-19 AU AU82206/98A patent/AU8220698A/en not_active Abandoned
- 1998-06-19 WO PCT/FR1998/001288 patent/WO1999000379A1/fr not_active Ceased
- 1998-06-24 AR ARP980103032A patent/AR013127A1/es unknown
- 1998-06-24 ZA ZA985518A patent/ZA985518B/xx unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0197840A1 (fr) * | 1985-03-26 | 1986-10-15 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | (Imidazolyl-4) piperidines, leur préparation et leur application en thérapeutique |
| EP0494010A1 (fr) * | 1990-12-31 | 1992-07-08 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Nouvelles 4-(4-imidazolyl) pipéridines substituées en 1, leur préparation ainsi que leurs applications thérapeutiques |
| EP0507650A1 (fr) * | 1991-04-03 | 1992-10-07 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique |
| EP0591027A1 (fr) * | 1992-09-28 | 1994-04-06 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001027107A3 (fr) * | 1999-10-12 | 2002-01-24 | Bristol Myers Squibb Co | Inhibiteurs heterocycliques d'echange sodium / protons et procedes a cet effet |
| US6887870B1 (en) | 1999-10-12 | 2005-05-03 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| US7326705B2 (en) | 1999-10-12 | 2008-02-05 | Bristol-Myers Squibb Company | Heterocyclic sodium/proton exchange inhibitors and method |
| EP4291191A4 (fr) * | 2021-02-12 | 2025-01-15 | The Trustees Of Columbia University In The City Of New York | Nouveaux composés comprenant une nouvelle classe de ligands de transthyrétine pour le traitement de comorbidités communes associées à l'âge |
Also Published As
| Publication number | Publication date |
|---|---|
| WO1999000379A1 (fr) | 1999-01-07 |
| JP2002506458A (ja) | 2002-02-26 |
| AR013127A1 (es) | 2000-12-13 |
| EP0991639A1 (fr) | 2000-04-12 |
| FR2765221B1 (fr) | 1999-07-30 |
| AU8220698A (en) | 1999-01-19 |
| ZA985518B (en) | 1999-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1499589B1 (fr) | Derives de n-¬phenyl(piperidin-2-yl)methyl|benzamide, leur preparation et leur application en therapeutique | |
| WO2005037781A2 (fr) | Derives de n-[heteroaryl(piperidin-2-yl)methyl]benzamide, leur preparation et leur application en therapeutique | |
| FR2673427A1 (fr) | Derives heterocycliques diazotes n-substitues par un groupement biphenylmethyle, leur preparation, les compositions pharmaceutiques en contenant. | |
| CZ281763B6 (cs) | Indolové deriváty představující 5-HT1 agonisty, způsob jejich přípravy, farmaceutické prostředky obsahující tyto látky, jejich použití a meziprodukty postupu přípravy | |
| EP0306375A1 (fr) | Dérivés de[(pipéridinyl-4)méthyl]-2 tétrahydro-1,2,3,4 isoquinoléine, leur préparation et leur application en thérapeutique | |
| EP1641787B1 (fr) | Dicetopiperazines substituees et leur utilisation comme antagonistes de l'ocytocine | |
| FR2595352A1 (fr) | Derives de l'indole utiles comme medicaments et procede et intermediaires pour leur preparation | |
| CA1095047A (fr) | Procede de preparation de nouveaux derives du 1,4- benzodioxane | |
| FR2765221A1 (fr) | Derives de 4-[(1h-imidazol-4-yl)piperidin-1-yl]anilide, leur preparation et leur application en therapeutique | |
| FR2696177A1 (fr) | Dérivés de pipéridine, leur préparation et leur application en thérapeutique. | |
| CA2489723C (fr) | Nouveaux derives d'aryl-{4-halogeno-4-[(heteroaryl-methylamino)-methyl]-piperidin-1-yl}-methanone, leur procede de preparation et leur utilisation a titre de medicaments | |
| FR2758329A1 (fr) | Derives d'imidazole-4-butane boronique, leur preparation et leur utilisation en therapeutique | |
| FR2687146A1 (fr) | Nouveaux derives de pyrrolidine, leur procede de preparation et les compositions pharmaceutiques qui les contiennent. | |
| CA2166032A1 (fr) | Derives de 1-oxo-2-(phenylsulfonylamino) pentylpiperidine, leur preparation et leur application en therapeutique | |
| EP2917204A1 (fr) | Derives de 1 h-indole-3-carboxamide et leurs utilisation comme antagonistes du p2y12 | |
| FR2765580A1 (fr) | Derives de (1h-imidazol-4-yl)piperidine, leur preparation et leur application en therapeutique | |
| FR2601951A1 (fr) | Nouvelles tetrahydrocarbazolones, composition pharmaceutique les contenant et leur procede de preparation | |
| CA2017044A1 (fr) | Derives de la pyrrolidone, leur procede de preparation et les compositions pharmaceutiques les renfermant | |
| EP0000306A1 (fr) | Hexahydro benzopyrano (3,2-c) pyridines substituées, leur procédé de préparation et médicament les contenant | |
| WO1998022443A1 (fr) | Derives de n-(imidazolylbutyle) benzenesulfonamide ayant une activite antithrombotique | |
| EP0254527A2 (fr) | Dérivés de l'indole tétracycliques | |
| FR2816619A1 (fr) | Derives de benzimidazole, leur preparation et leur application en therapeutique | |
| MC1278A1 (fr) | Derives d'oxadiazolotriazines | |
| WO2000051973A1 (fr) | Derives de cyclobutene-3,4-dione leur preparation et leur application en therapeutique | |
| EP0200583A1 (fr) | Dérivés du 7-hydroxy indole, leurs sels, procédé de préparation, application à titre de médicaments, compositions les renfermant et intermédiaires |