JP2004244366A - リグニン誘導体を含有する抗hiv剤 - Google Patents
リグニン誘導体を含有する抗hiv剤 Download PDFInfo
- Publication number
- JP2004244366A JP2004244366A JP2003035642A JP2003035642A JP2004244366A JP 2004244366 A JP2004244366 A JP 2004244366A JP 2003035642 A JP2003035642 A JP 2003035642A JP 2003035642 A JP2003035642 A JP 2003035642A JP 2004244366 A JP2004244366 A JP 2004244366A
- Authority
- JP
- Japan
- Prior art keywords
- lignin
- derivative
- hiv
- phenol compound
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images







Landscapes
- Medicines Containing Plant Substances (AREA)
Abstract
【課題】リグニン誘導体の一種であるリグノフェノール系誘導体を用いた抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤を提供すること。
【解決手段】(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する抗HIV剤とする。
【選択図】図4
【解決手段】(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する抗HIV剤とする。
【選択図】図4
Description
【0001】
【発明の属する技術分野】
本発明は、リグニンにフェノール化合物を導入して得られるリグニン誘導体の利用に関し、特に、抗ヒトエイズウイルスプロテアーゼ1型(Human immunodeficiency virus type I (HIV−1) protease)剤及び抗エイズウイルス(Human immunodeficiency virus(HIV))剤に関する。
【0002】
【従来の技術】
リグニンは、植物体を構築するリグニン−セルロース複合体の構成成分である。近年、植物細胞壁を維持する作用のみならず、その生理的作用についても着目されている。
例えば、木質化植物材料を有機溶媒で加温洗浄した後、アルカリ水溶液で抽出して得られるリグニン抽出物がウイルス感染防止作用を有することが知られている(特許文献1)。また、木材から得られるクラフトリグニンをカルボキシメチル化して得られる水溶性クラフトリグニンが抗エイズウイルス活性を有していることも知られている(特許文献2)。さらに、ある種のきのこから抽出された水溶性リグニン様物質や、小麦から抽出された水溶性リグニンがHIV−1プロテアーゼ活性を阻害することが知られている(キノコから抽出したリグニンについて非特許文献1、小麦抽出リグニンについて非特許文献2)。
また、合成により得られる水溶性リグニンが抗HIV活性を有していることも報告されている(特許文献1、非特許文献2、非特許文献3)。
【0003】
【特許文献1】
特開平7−206601号公報
【特許文献2】
特開平8−143587号公報
【非特許文献1】
Biosci. Biotechnol. Biochem., 62(3), 575−577, 1998
【非特許文献2】
Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999
【非特許文献3】
Chem. Pharm. Bull. 40(8)2102−2105(1992)
【0004】
【発明が解決しようとする課題】
ここに、リグニン誘導体としては、クラフトリグニン、蒸煮爆砕リグニン、酢酸リグニンなど多種類のリグニン誘導体がある他、リグノセルロース系材料にフェノール化合物を添加し溶媒和させた後、酸を添加することにより得られるリグニンのフェノール誘導体化合物(以下、リグノフェノール系誘導体ともいう。)が知られている(リグノフェノール系誘導体について、特開2001−64494号公報、特開2001−261839号公報、特開2001−131201号公報、特開2001−342353号公報、特開2002−105240号公報等)。
一方、各種リグニン誘導体の構造は、その抽出分離方法によって大きくことなることが知られている。
しかしながら、既に説明したように、抗HIV−1プロテアーゼ活性は限定的な抽出方法かあるいは合成によるリグニン様物質において検出されたと報告されているに過ぎず、全てのリグニン誘導体がHIV−1プロテアーゼ阻害活性を有していると確認されてはいない。また、当該阻害活性のための構造も未だ不明である。
そこで、本発明では、リグニン誘導体の一種であるリグノフェノール系誘導体を用いた抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤を提供することを目的とする。
【0005】
【課題を解決するための手段】
本発明者らは、各種のリグノフェノール系誘導体を調製し、これらの誘導体について抗HIV−1プロテアーゼ活性とHIV感受性細胞におけるHIV感染による細胞変性試験により、有用な抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤について探索した。その結果、ある種のリグノフェノール系誘導体が抗HIV−1プロテアーゼ活性及び抗HIV活性を有することを見出した。
また、本発明者らは、リグノフェノール系誘導体を調製するのに適した方法、すなわち、使用するフェノール化合物の種類、リグノフェノール系誘導体の抽出分離操作、及び修飾手段を見出した。
本発明によれば、以下の手段が提供される。
【0006】
(1) 抗HIV剤であって、
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体、及び
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。
(2)前記フェノール化合物は、1価のフェノール化合物である、(1)記載の抗HIV剤。
(3)前記カルボキシアルキル基におけるアルキル基はメチル基である、(1)又は(2)記載の抗HIV剤。
(4)前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、(1)〜(3)のいずれかに記載の抗HIV剤。
(5)抗HIV剤であって、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体、
(e)前記リグニン一次誘導体に対してアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。
(6)前記2価以上のフェノール化合物は、カテコール、レゾルシノール、及びピロガロールからなる群から選択される1種あるいは2種以上である、(5)記載の抗HIV剤。
(7)前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、(5)又は(6)に記載の抗HIV剤。
(8)前記リグノセルロース系材料は、針葉樹由来である、(7)記載の抗HIV剤。
(9)(1)〜(8)のいずれかに記載のリグニン誘導体を含有する抗HIV−1プロテアーゼ剤。
(10)抗HIV剤を製造する方法であって、以下の(a)〜(g);
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体、
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンの一次誘導体
(e)(d)のリグニン一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
からなる群から選択される1種あるいは2種以上のリグニン誘導体を調製する工程を有し、
これらのリグニン誘導体について以下の工程:
抗HIVプロテアーゼ活性を測定する工程、
HIV感染感受性の非感染細胞株を用いた急性感染系における抗HIV活性を測定する工程、
HIV持続感染細胞株とHIV感染感受性の非感染細胞株とを用いた巨細胞形成系において抗HIV活性を測定する工程、
から選択される1種あるいは2種以上の工程を行い、抗HIV活性を評価することを含む、方法。
【0007】
【発明の実施の形態】
本発明の抗HIV剤及び抗HIV−1プロテアーゼ剤は、リグニン含有材料から所定の手法により分離して得られるリグノフェノール系誘導体を有効成分としている。これらのリグノフェノール系誘導体としては、以下の(a)〜(g)を挙げることができる。
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンの一次誘導体
(e)(d)のリグニン一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体。
【0008】
ここで、上記(a)〜(c)は、フェノール化合物を用いたリグニン一次誘導体に由来し、(d)及び(e)は2価以上のフェノール化合物を用いたリグニン一次誘導体に由来している。
そこで、以下の説明においては、(a)に記載のリグニン一次誘導体を第1のリグニン一次誘導体といい、(a)〜(c)に記載のリグニン誘導体を、(b)及び(c)に記載の二次及び三次誘導体を含めて第1の誘導体というものとし、(d)記載のリグニン一次誘導体を第2のリグニン一次誘導体といい、(d)〜(g)に記載のリグニン誘導体を含めて第2の誘導体というものとする。
第1及び第2の誘導体が由来するリグニン含有材料は入手が容易で再生可能な天然資源であり、この資源を由来材料とすることにより、有効性と安全性のバランスが取れた抗HIV−1プロテアーゼ剤及び抗HIV剤を提供することができる。また、基本骨格を合成手法でなく、大量かつ再生可能に存在する植物資源から得るために、抗HIV剤製造にかかる原料コスト及び工程コストを含む製造コストを低減することができる。
また、リグニン含有材料からリグノフェノール系誘導体は、構造制御や修飾が容易であり、薬物送達性や有効性を確保するような構造を探索し、発見することも容易に達成することができる。
以下、本発明の実施の形態について詳細に説明する。
【0009】
(第1及び第2のリグニン一次誘導体)
本発明において用いるリグニン一次誘導体は、リグニン含有材料をフェノール化合物で溶媒和後、酸を添加し混合して得られるリグニンのフェノール化合物であるリグニン誘導体である。この反応過程によりリグニンのアリールプロパンユニットのベンジル位(側鎖C1位、以下、単にC1位という。)にフェノール化合物がグラフト(導入)されたリグニン誘導体を得ることができる。フェノール化合物は、そのフェノール性水酸基に対してオルト位あるいはパラ位にて前記C1位の炭素原子に結合する。この結果、1,1−ビス(アリール)プロパンユニットがリグニン中に形成される。この反応において、フェノール化合物は、前記C1位に対して選択的に導入されるため、出発原料であるリグニン含有材料におけるC1位における様々な結合を開放し、リグニンマトリックスの多様性を低減し、また、低分子量化することができる。さらに、この結果、従来のリグニンにはなかった各種溶媒への溶解性、熱流動性、熱可塑性など各種の特性を発現することが既に知られている。
なお、ここで、フェノール化合物で溶媒和するとは、液体のフェノール化合物にリグニン含有材料を浸漬する等して溶媒和する他、液体あるいは固体のフェノール化合物を当該フェノール化合物が溶解する溶媒に溶解させたものをリグニン含有材料に適用後、溶媒を留去することでリグニン含有材料にフェノール化合物を収着することによっても達成することができる。
【0010】
濃酸による炭水化物の膨潤に基づく組織構造の破壊と、フェノール化合物によるリグニンの溶媒和とを組み合わせてリグニンの不活性化を抑制しつつ、リグノセルロース系材料を炭水化物とリグノフェノール誘導体とに分離する方法(特開平2−233701号)が知られている。この方法で得られたリグノフェノール誘導体の活用法としては、例えば、セルロース系ファイバー等の成形材料に適用し成形体を作製することが報告されている(特開平9−278904号)。かかるリグノフェノール誘導体は、1,1−ビス(アリール)プロパンを高頻度構成単位として有するリグニン系ポリマーであって、高粘結性を潜在的に有していることがわかっている(特開平9−278904号)。
さらに、かかるリグノフェノール誘導体は、メチロール化することにより架橋性を付与でき、リニアあるいはネットワーク状の架橋構造を構築できると同時に、アルカリ処理によって、再び低分子化して溶媒中に溶解されることも、見出されている(特開2001−261839号公報)。
また、これらの他、リグノフェノール誘導体に関するより一般的な記載及びその製造プロセスについては、国際公開WO99/14223号公報、特開2001−64494号公報、特開2001−261839号公報、特開2001−131201号公報、特開2001−342353号公報、特開2002−105240号公報において記載されている(これらの特許文献に記載の内容は、全て引用により本明細書中に取り込まれるものとする)。
【0011】
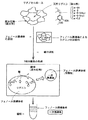
このプロセスによりリグノセルロース系材料からリグノフェノール誘導体を得るシステムにおける構造変換プロセスの一例を図1に示す。
この構造変換プロセスは、図1に示すように、リグノセルロース系材料を予めフェノール化合物で溶媒和しておいた上で、当該リグノセルロース系材料を酸と接触させることにより、リグニンのアリールプロパンユニットのリグニンの複合状態を緩和させ、同時に、天然リグニンのアリールプロパンユニットのC1位(ベンジル位)に選択的に前記フェノール誘導体をグラフティングさせ、リグノフェノール誘導体を生成させ、同時にセルロースとリグノフェノール誘導体とに分離できる。このプロセスにおける構造変換の一例を、図2に示す。
【0012】
リグノフェノール誘導体は、それ自体、リグノセルロース系材料などのリグニン含有材料から反応、分離して得られるリグニン由来のポリマーの混合物である。このため、得られるポリマーにおける導入フェノール誘導体の量や分子量は、原料となるリグニン含有材料のリグニン構造および反応条件により変動する。
【0013】
(リグニン含有材料)
本発明におけるリグニン含有材料には、天然リグニンを含有するリグノセルロース系材料を含む。リグノセルロース系材料は、木質化した材料、主として木材である各種材料、例えば、木粉、チップの他、廃材、端材、古紙などの木本類植物資源に付随する農産廃棄物や工業廃棄物を挙げることができる。また用いる木本類の種類としては、スギ、ヒノキなどの針葉樹、ブナなどの広葉樹等、任意の種類のものを使用することができる。さらに、ケナフ、ジュート、イネ、タケなどの各種草本類植物、それに関連するイネワラ、モミガラなどの農産廃棄物や工業廃棄物なども使用できる。
本発明の第2の誘導体においては、木本類植物や草本類植物のいずれのリグノセルロース系材料も使用できるが、第2の誘導体においては、由来資源の影響が第1の誘導体よりも大きい場合がある。第2の誘導体においては好ましくは、木本類植物資源をリグノセルロース系材料として用い、スギ、マツ、ヒノキなどの針葉樹植物資源由来のリグノセルロース系材料を用いることもできるが、一次誘導体の回収率を考慮すれば、広葉樹や草本類資源を用いることが好ましい。
【0014】
(フェノール化合物)
フェノール化合物としては、1価のフェノール化合物、2価のフェノール化合物、または3価のフェノール化合物などを用いることができる。
1価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいフェノール、1以上の置換基を有していてもよいナフトール、1以上の置換基を有していてもよいアントロール、1以上の置換基を有していてもよいアントロキノンオールなどが挙げられる。
2価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいカテコール、1以上の置換基を有していてもよいレゾルシノール、1以上の置換基を有していてもよいヒドロキノンなどが挙げられる。
3価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいピロガロールなどが挙げられる。
本発明の第1の一次誘導体にあっては、1価のフェノール化合物から選択される1種あるいは2種以上を用いることができる。また、第2の一次誘導体にあっては、2価のフェノール化合物及び3価のフェノール化合物のうち、1種あるいは2種以上を用いることができる。
【0015】
なお、1価から3価のフェノール化合物が有していてもよい置換基の種類は特に限定されず、任意の置換基を有していてもよいが、好ましくは、電子吸引性の基(ハロゲン原子など)以外の基であり、例えば、炭素数が1〜4、好ましくは炭素数が1〜3の低級アルキル基含有置換基である。低級アルキル基含有置換基としては、例えば、低級アルキル基(メチル基、エチル基、プロピル基など)、低級アルコキシ基(メトキシ基、エトキシ基、プロポキシ基など)である。また、アリール基(フェニル基など)の芳香族系の置換基を有していてもよい。また、水酸基含有置換基であってもよい。
【0016】
第1の一次誘導体を得るには、1価のフェノール化合物及び2価以上のフェノール化合物を用いることができる。一次誘導体としての収率を高めるには、好ましくは1価のフェノール化合物を用いる。また、好ましくはp−クレゾール、m−クレゾール、o−クレゾール、2,6−ジメチルフェノール、2,4−ジメチルフェノール、2−メトキシフェノール(Guaiacol)、2,6−ジメトキシフェノール等を用いることができ、より好ましくは、p−クレゾール、フェノール、p−エチルフェノールである。また、第2の一次誘導体を得るには、2価以上のフェノール化合物を用いることができる。2価のフェノール化合物としては、レゾルシノール、ホモカテコール、ハイドロキノン、1,3−ナフタレンジオール等を用いることが好ましく、より好ましくは、レゾルシノール、カテコールである。また、3価のフェノール化合物としては、好ましくは、ピロガロール、フロログルシノール、2−ヒドロキシ−ナフトキノン等を用いることができ、より好ましくは、ピロガロール、フロログルシノールである。第2の一次誘導体を得るには、好ましくは、3価のフェノール化合物を用い、より好ましくは、ピロガロールである。
【0017】
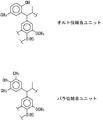
これらのフェノール化合物は、そのフェノール性水酸基に対してオルト位あるいはパラ位の炭素原子がリグニンのアリールプロパンユニットのC1位の炭素に結合することにより、1,1−ビス(アリール)プロパンユニットが形成されることになる。したがって、少なくとも1つの導入サイトを確保するには、オルト位及びパラ位のうち、少なくともひとつの位置に置換基を有していないことが好ましい。
フェノール化合物のフェノール性水酸基のオルト位炭素原子が前記C1位に結合して形成されたユニットをオルト位結合ユニットといい、フェノール化合物のフェノール性水酸基のパラ位炭素原子が前記C1位に結合して形成されたユニットをパラ位結合ユニットという。図3に、オルト位結合ユニット及びパラ位結合ユニットの一例として、フェノール化合物として、それぞれp−クレゾールと、2,6−ジメチルフェノールを用いて形成されるユニットを示す。
【0018】
以上のことから、本発明では、各一次誘導体について、無置換フェノール誘導体の他、少なくとも一つの無置換のオルト位あるいはパラ位を有する各種置換形態のフェノール誘導体の1種あるいは2種以上を適宜選択して用いることができる。
【0019】
オルト位結合ユニットとパラ位結合ユニットとは、例えば、後述するアルカリ処理工程において異なる機能を発現する。オルト位結合ユニットは、緩和なアルカリ処理により導入されたフェノール化合物におけるフェノール性水酸基を消失させるとともにアリールクマラン構造を当該ユニットにおいて生成し、強いアルカリ処理によりアリール基移動に伴って分子形態が変動される。いずれにおいても、オルト位結合ユニットは、アルカリ処理による効率的なリグノフェノール誘導体の低分子化に寄与する。
一方、パラ位結合ユニットは、アルカリ処理によりアリールクマラン構造やその後の分子形態変動を生じず、当該ユニット部位における低分子化には寄与しない。したがって、アルカリ処理耐性を付与する機能を有するといえる。
【0020】
また、使用するフェノール化合物の種類を選択することにより、得られる一次誘導体への後段の二次誘導体化工程での官能基の導入位置や導入頻度等を調節し、第1及び第2の誘導体の構造制御が可能となる。
【0021】
なお、オルト位結合ユニットを有するリグノフェノール誘導体を得るには、少なくとも一つのオルト位(好ましくは全てのオルト位)に置換基を有していないフェノール化合物を用いる。また、少なくとも一つのオルト位(2位あるいは6位)が置換基を有さず、パラ位(4位)に置換基を有するフェノール化合物(典型的には、2,4位置換1価フェノール誘導体)が好ましい。最も好ましくは、全てのオルト位が置換基を有さず、パラ位に置換基を有するフェノール化合物(典型的には、4位置換1価フェノール化合物)である。したがって、4位置換フェノール化合物及び2,4位置換フェノール化合物を1種あるいは2種以上組み合わせて用いることができる。
【0022】
パラ位結合ユニットを有するリグノフェノール誘導体を得るには、パラ位に置換基を有していないフェノール化合物(典型的には、2位(あるいは6位)置換1価フェノール化合物)が好ましく、より好ましくは、同時に、オルト位(好ましくは、全てのオルト位)に置換基を有するフェノール化合物(典型的には2,6位置換1価フェノール化合物)を用いる。すなわち、2位(あるいは6位)置換フェノール化合物及び2、6位置換フェノールのうち1種あるいは2種以上を組み合わせて用いることが好ましい。
【0023】
(酸)
リグニン含有材料と接触させる酸としては、特に限定しないで、リグノフェノール誘導体を生成しうる範囲で各種無機酸や有機酸を使用することができる。したがって、硫酸、リン酸、塩酸などの無機酸の他、p−トルエンスルホン酸、トリフルオロ酢酸、トリクロロ酢酸、ギ酸などを使用することができる。リグニン含有材料としてリグノセルロース系材料を使用する場合には、セルロースを膨潤させる作用を有していることが好ましい。例えば、65重量%以上の硫酸(好ましくは、72重量%の硫酸)、85重量%以上のリン酸、38重量%以上の塩酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリクロロ酢酸、ギ酸などを挙げることができる。好ましい酸は、65重量%以上の硫酸(好ましくは、72重量%の硫酸)、85重量%以上(好ましくは95重量%以上)のリン酸、トリフルオロ酢酸又はギ酸であるが、リグノフェノール誘導体を高収率で得るには、65重量%以上の硫酸(好ましくは、72重量%の硫酸)を好ましく用いることができる。
【0024】
リグニン含有材料中のリグニンを、リグノフェノール誘導体に変換し、分離する方法としては各種方法が採用できる。
第1及び第2の一次誘導体を得る反応工程としては、例えば、第1の図1に示すように、リグニン含有材料に、液体状のフェノール化合物(上記で説明したもの、例えば、p−クレゾール)を浸透させ、リグニンをフェノール誘導体により溶媒和させ、次に、リグノセルロース系材料に酸(上記で説明したもの、例えば、72%硫酸)を添加し混合して、セルロース成分を溶解する(以下、一段法ともいう。)。この方法によると、リグニンが低分子化され、同時にその基本構成単位のC1位にフェノール化合物が導入されたリグノフェノール誘導体がフェノール化合物相に生成される。このフェノール化合物相から、リグノフェノール誘導体が抽出される。リグノフェノール誘導体は、リグニン中のベンジルアリールエーテル結合が開裂して低分子化されたリグニンの低分子化体の集合体として得られる。
図2は、アリールプロパンユニットを有する天然リグニンに対して相分離処理を行うことにより、本発明におけるリグノフェノール誘導体が得られることを示している。
また、他の反応工程としては、リグニン含有材料に、固体状あるいは液体状のフェノール化合物を溶解した溶媒(例えば、エタノールあるいはアセトン)を浸透させた後、溶媒を留去(フェノール誘導体の収着)した場合も、先の方法と同様、リグノフェノール誘導体が生成される(以下、二段法ともいう)。
【0025】
第1のリグニン誘導体は、後段でカルボキシメチル化及び/又はアルカリ処理により、リグニン一誘導体に水溶性を付与する。したがって、一次誘導体としては、有機溶媒区分のリグニン一次誘導体を分離抽出することが有効である。
有機溶媒区分のリグニン一次誘導体を分離回収するには、一段法にあっては、液体のフェノール化合物相を、大過剰のエチルエーテルに加えて得た沈殿物を集めて、アセトンに溶解する。アセトン不溶部を遠心分離により除去し、アセトン可溶部を濃縮する。このアセトン可溶部を、大過剰のエチルエーテルに滴下し、沈殿区分を集める。この沈殿区分から溶媒留去し、第1のリグニン一次誘導体を得る。なお、粗第1の一次誘導体は、フェノール化合物相やアセトン可溶区分を単に減圧蒸留により除去することによって得ることができる。
【0026】
また、二段法にあっては、生成したリグノフェノール誘導体は、液体フェノール化合物にて抽出分離することができる。あるいは、全反応液を過剰の水中に投入し、不溶区分を遠心分離にて集め、脱酸後、乾燥する。この乾燥物にアセトンあるいはアルコールを加えてリグノフェノール誘導体を抽出する。さらに、この可溶区分を1段法における場合と同様に、過剰のエチルエーテル等に滴下して、リグノフェノール誘導体を不溶区分として得ることもできる。
以上、第1の一次誘導体の調製方法の具体例を説明したが、これらに限定されるわけではなく、これらに適宜改良を加えた方法で調製することもできる。
【0027】
また、第2のリグニン一次誘導体は、2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体である。したがって、上記一段法及び二段法における反応液の酸性水溶液相を採取し、この酸性相に対して、フェノール化合物、あるいはベンゼン、キシレン、トルエン、ヘキサン等の有機溶媒、あるいはフェノール化合物とこれらの有機溶媒との混合液を添加して有機溶媒相に可溶な一次誘導体を当該有機溶媒相に分離除去して、得られた酸性水溶液相に水溶性のリグニン一次誘導体を分離採取する。フェノール化合物としては、反応工程に用いたものを利用することが好ましい。リグニン一次誘導体の精製には、この酸性水溶液相を希釈し、透析等により脱酸、乾燥し、さらにこの乾燥物をメタノール等の有機溶媒で抽出した溶液をエーテルに滴下し、沈殿区分として採取する等の方法を採用することができる。有機溶媒相を用いた有機溶媒可溶性リグニン一次誘導体の分離除去は、十分に有機溶媒可溶性誘導体を除去する回数繰り返すことが好ましい。
以上、第2の一次誘導体の調製方法の具体例を説明したが、これらに限定されるわけではなく、これらに適宜改良を加えた方法で調製することもできる。
【0028】
(第1の誘導体)
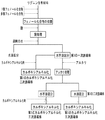
次に、第1の一次誘導体をさらに化工して得られる第1の誘導体について説明する。図4には、第1のリグニン誘導体を得るスキームの例を示す。
アルカリ処理による水溶性化
アルカリ処理は、第1のリグニン一次誘導体をアルカリと接触させることにより行う。好ましくは加熱する。アルカリ処理においては、オルト位結合ユニットにおいて、導入されたフェノール化合物のフェノキシドイオンによるC2位炭素の攻撃が生じる。すなわち、一旦この反応が生じれば、C2アリールエーテル結合が開裂する。
例えば、緩和なアルカリ処理では、一次誘導体が第一のユニットを有する場合、当該導入フェノール誘導体の当該フェノール性水酸基が開裂し、生じたフェノキシドイオンが、C2アリールエーテル結合を構成するC2位を分子内求核反応的にアタックして、当該エーテル結合を開裂させて低分子化することができる。
C2アリールエーテル結合の開裂により、リグニンの母核にフェノール性水酸基が生成されることになり、当該分子内求核反応により、導入フェノール核が、それが導入されたフェニルプロパン単位とクマラン骨格を形成した構造(アリールクマラン単位)が発現される。
これらの結果、フェノール誘導体側にあったフェノール性水酸基が、リグニン母核側に移動されたことになる。
このため、オルト位結合ユニットを有するリグノフェノール誘導体においては、このユニットの存在部位において(1)C2アリールエーテル結の開裂による低分子化、(2)アリールクマラン構造の発現、(3)C2アリールエーテル結合で結合されていたリグニン母核側におけるフェノール性水酸基が発現する。
【0029】
当該アルカリ処理は、具体的には、リグノフェノール誘導体をアルカリ溶液に溶解し、一定時間反応させ、必要であれば、加熱することにより行う。この処理に用いることのできるアルカリ溶液は、リグノフェノール誘導体中の導入フェノール誘導体のフェノール性水酸基を解離させることができるものであればよく、特に、アルカリの種類及び濃度、溶媒の種類等は限定されない。アルカリ下において前記フェノール性水酸基の解離が生じれば、隣接基関与効果により、クマラン構造が形成されるからである。例えば、p−クレゾールを導入したリグノフェノール誘導体では、水酸化ナトリウム溶液を用いることができる。例えば、アルカリ溶液のアルカリ濃度範囲は0.5〜2Nとし、処理時間は1〜5時間程度とすることができる。また、アルカリ溶液中のリグノフェノール誘導体は、加熱されることにより、容易にクマラン構造を発現する。加熱に際しての、温度、圧力等の条件は、特に限定することなく設定することができる。例えば、アルカリ溶液を100℃以上(例えば、140℃程度)に加熱することによりリグノフェノール誘導体の低分子化を達成することができる。さらに、アルカリ溶液を加圧下においてその沸点以上に加熱して一次誘導体の低分子化を行ってもよい。
【0030】
なお、同じアルカリ溶液で同濃度においては、加熱温度が120℃〜140℃の範囲では、加熱温度が高い程、C2−アリールエーテル結合の開裂による低分子化が促進されることがわかっている。また、該温度範囲で、加熱温度が高い程、リグニン母体由来の芳香核由来のフェノール性水酸基が増加し、導入されたフェノール誘導体由来のフェノール性水酸基が減少することがわかっている。したがって、低分子化の程度及びフェノール性水酸基部位のC1位導入フェノール誘導体側からリグニン母体のフェノール核への変換の程度を、反応温度により調整することができる。すなわち、低分子化が促進され、あるいは、より多くのフェノール性水酸基部位がC1位導入フェノール誘導体側からリグニン母体へ変換されたアリールクマラン体を得るには80〜140℃程度の反応温度が好ましい。
【0031】
C1フェノール核の隣接基関与によるC2−アリールエーテルの開裂は、上述したようにアリールクマラン構造の形成を伴うが、リグノフェノール誘導体の低分子化は、必ずしもアリールクラマンが効率よく生成する条件下(140℃付近)で行う必要はなく、より高い温度(例えば170℃付近)で行うこともできる。この場合、一旦生成したクラマン環は開裂し、導入フェノール誘導体側にフェノール性水酸基が再生される結果、140℃処理物とは特性の異なるよりフェノール活性が高い素材を誘導することができる。
【0032】
以上のことから、アルカリ処理における加熱温度は、特に限定されないが好ましくは80℃以上200℃以下である。80℃を大きく下回ると、反応が十分に進行せず、200℃を大きく越えると好ましくない副反応が派生しやすくなるからである。
【0033】
クラマン構造の形成とそれに伴う低分子化のための処理の好ましい一例としては、0.5Nの水酸化ナトリウム水溶液をアルカリ溶液として用い、140℃で加熱時間60分という条件を挙げることができる。また、0.5Nの水酸化ナトリウム水溶液をアルカリ溶液として用い、170℃で加熱時間60分という条件を挙げることができる。
【0034】
アルカリ処理して水溶性のリグニン二次誘導体を得る方法としては、アルカリ処理反応液を中和後、その水溶性区分を遠心分離等により分離採取し、これを透析等により脱塩し、凍結乾燥等する方法を採用することができる。脱塩後の液をそのまま水溶性リグニン二次誘導体含有組成物として用いることもできる。なお、アルカリ処理反応液中和後の水不溶性区分は、水不溶性リグニン二次誘導体として後述するカルボキシアルキル化三次誘導体の前駆体となる。
第1の誘導体としての水溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。
また、水不溶性のリグニン二次誘導体の重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは700以上6000以下である。
【0035】
カルボキシアルキル化による二次誘導体化及び三次誘導体化
第1の一次誘導体及び、水溶性及び/又は水不溶性のリグニン二次誘導体に対して、カルボキシアルキル基を導入することによりカルボキシアルキル化二次誘導体及び三次誘導体を得ることができる。カルボキシルアルキル基におけるアルキル基は、炭素数1〜5の直鎖あるいは分枝アルキル基であることが好ましく、より好ましくは、炭素数1〜3の直鎖アルキル基である。たとえば、メチル基、エチル基、プロピル基を用いることができる。
カルボキシルアルキル基は、リグニン一次誘導体中のアルコール性あるいはフェノール性水酸基に導入される。カルボキシアルキル化は、一般に、アルカリ存在下に、モノハロゲノアルキルカルボン酸と反応させることにより達成することができる。リグニン誘導体のカルボキシアルキル化法は従来公知の各種方法を採用することができる。
例えば、リグニン一次誘導体を、この誘導体を分散できるイソプロパノールなどの有機溶媒で分散し、その後、アルカリ水溶液を加え、必要に応じてさらにイソプロパノールなどの有機溶媒を加えて得られた不均一混合溶液をモノクロロ酢酸などのモノハロゲノ酢酸(カルボキシアルキル化剤)を添加し、攪拌等しながら反応させることができる。また、リグニン一次誘導体を予めアルカリ溶液に浸漬した後、この浸漬物を有機溶媒に分散して、その後反応させることもできる。
なお、アルカリとしては、一般に用いられるアルカリ試薬でよいが、水酸化ナトリウム、水酸化カリウム等のアルカリ金属化合物のような強アルカリが好適に用いられる。また、リグニン一次誘導体等を分散する有機溶媒としては、メタノール、エタノール、イソプロパノール、t−ブタノール等のアルコール類、アセトン、メチルエチルケトン等のケトン類、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン等のエーテル類、ジクロロメタン、クロロホルム等のハロゲン化物、ヘキサン、シクロヘキサン、ベンゼン、トルエン等の炭化水素類等が挙げられ、これらは単独であるいは2種以上混合して使用される。これらの中でも、アルコール類、ケトン類、エーテル類等の極性有機溶媒の1種又は2種以上の組合せが良く、さらに好ましくは、イソプロパノール、アセトン、1,4−ジオキサンが用いられる。
【0036】
モノハロゲノアルキルカルボン酸は、次の一般式〔I〕で表わされるものが好ましく用いられる。
XRCOOH 〔I〕
(上式中、X及びRはそれぞれ以下のものを表わす。
X:F、Cl、Br、I等のハロゲン原子、R:炭素数1〜5の直鎖及び分枝を有するアルキル基。)
上記化合物中、特にXがCl又はBr原子で、炭素数が1〜3の直鎖アルキル基を有するものが好ましい。
【0037】
上記モノハロゲノアルキルカルボン酸の使用量は、リグニンの水酸基量以上あれば良く、水酸基あたり、約1〜3モルであることが好ましい。モノハロゲノアルキルカルボン酸の添加方法としては、固体のまま及び/又はこの有機溶媒の溶液として、一括あるいは連続滴下で添加すれば良い。好ましいのは、有機溶媒の溶液として0.5〜3時間かけた連続滴下の方法であり、特に1〜2時間で連続滴下することが好ましい。また、モノハロゲノアルキルカルボン酸の添加終了後、その温度で0.5〜5時間(より好ましくは1〜4時間)反応を続けることが好適である。なお、モノハロゲノアルキルカルボン酸による反応にあたり、反応液を40℃〜60℃程度の範囲で加熱することが好ましい。
【0038】
カルボキシアルキル化二次及び三次誘導体は、固形生成物の他一部溶解した状態とで得られる。固形生成物は、反応終了後、ろ過等により固形の生成物を分離採取し、この固形生成物を水に溶解させ、希鉱酸、例えば希塩酸、希硫酸等の酸で中性に中和した後、電気透析で脱塩し、凍結乾燥等して得ることができる。また、溶解生成物は、上記ろ液中の有機溶媒を留去し、溶解物を乾固させ、この乾固物を中和前の固形生成物に添加することにより、両者を回収することができる。
【0039】
(第2の誘導体)
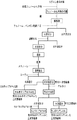
第2の誘導体は、既に説明した第2の一次誘導体とこの一次誘導体をさらにアルカリ処理して得られる水溶性のリグニン二次誘導体及びカルボキシアルキル化した二次及び三次誘導体を含んでいる。以下、第2のリグニン二次誘導体及びカルボキシアルキル化二次及び三次誘導体について説明する。図5には、第2のリグニン誘導体を得るためのスキームの例を示す。
アルカリ処理による水溶性化
アルカリ処理は、第2のリグニン一次誘導体をアルカリと接触させることにより行う。当該アルカリ処理は、第1の誘導体において説明したのと同様の操作及び条件等を採用することができ、好ましいものとして挙げた操作及び条件もそのまま採用できる。第2の水溶性の二次誘導体においては、既に一次誘導体において水溶性であるため、アルカリ処理により、より低分子化されさらに水溶性が向上すると考えられる。なお、アルカリ処理時に得られる水溶性及び/又は水不溶性の二次誘導体は、第1の誘導体におけるのと同様に、カルボキシアルキル化処理してカルボキシアルキル化三次誘導体を得ることができる。
なお、第2の誘導体としての水溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。第2の誘導体としての水不溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。
カルボキシアルキル化による二次誘導体化及び三次誘導体化
第1の誘導体におけるのと同様に、第2の一次誘導体及び、この一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して、カルボキシアルキル基を導入することによりカルボキシアルキル化二次誘導体及び三次誘導体を得ることができる。当該カルボキシアルキル化は、第1の誘導体において説明したのと同様の操作及び条件等を採用することができ、好ましいものとして挙げた操作及び条件もそのまま採用できる。
【0040】
(抗HIV−1プロテアーゼ剤及び抗HIV剤の有効成分としてのリグニン誘導体)以上説明した第1及び第2の誘導体は、本発明者らの研究によれば、好ましい抗HIV−1プロテアーゼ活性及び/又は抗HIV活性を有していることが認められている。なお、これらの誘導体における抗HIV活性は、抗HIV−1プロテアーゼ活性以外の何らかの抗HIV作用に起因する可能性があり、必ずしも抗HIV−1プロテアーゼ活性に基づく抗HIV活性に限定するものでない。したがって、本発明の抗HIV剤も抗HIV−1プロテアーゼ剤を有効成分とするものに限定されないし、本抗HIV剤の有効成分たるリグニン誘導体に抗HIV−1プロテアーゼ活性を必要条件とするものではない。しかしながら、抗HIV剤を探索するにあたり、抗HIV−1プロテアーゼ活性はその指標とすることができる。
【0041】
これらの各種誘導体に対して、HIV−1プロテアーゼ又は抗HIV活性を測定することにより、高い抗HIV−1プロテアーゼ活性又は抗HIV活性を有する誘導体を選択できる。抗HIV−1プロテアーゼ活性を有する場合、抗HIV−1プロテアーゼ剤として使用できる。抗HIV活性を有している場合には、抗HIV剤として使用できる。抗HIV剤を探索する場合、少なくとも1つの抗HIV活性を測定する必要があるが、本発明の誘導体については、抗HIV−1プロテアーゼ活性の測定もよい指標とすることができる。したがって、本発明の一つの態様として、これらの各種誘導体について、抗HIV−1プロテアーゼ活性の測定及び抗HIV活性の測定のうちいずれか1つを行うことにより、抗HIV剤を探索する方法を開示できる。また、当該探索工程を有することにより、結果として、抗HIV剤を生産できるため、当該探索工程を有する、抗HIV剤の生産方法も開示できる。
【0042】
これらの誘導体について、好ましい抗HIV−1プロテアーゼ活性測定方法及び抗HIV活性測定方法としては、例えば、以下の方法が挙げられる。
【0043】
抗HIV−1プロテアーゼ活性測定方法
抗HIV−1プロテアーゼ活性は、HIV−1プロテアーゼ活性測定に際し、阻害剤として各誘導体を添加し、それによるHIV−1プロテアーゼ活性の阻害程度を測定することで測定できる。抗HIV−1プロテアーゼ活性の測定方法としては、前記非特許文献1(Biosci. Biotechnol. Biochem., 62(3), 575−577, 1998)、非特許文献2(Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999)の他、従来公知の各種方法を採用することができる。
当該抗HIV−1プロテアーゼ活性は、従来公知の各種方法を採用できる。例えば、酢酸緩衝液とHIV−1プロテアーゼ溶液と各誘導体の各種濃度溶液とを合わせ、適当時間インキュベート後、HIV−1プロテアーゼの基質を加え、所定時間経過後に反応を停止し、基質分解物をHPLC等により測定し、誘導体を添加しない反応系をコントロールとしてHIV−1プロテアーゼ活性を求める方法を採用できる。
【0044】
抗HIV−1プロテアーゼ活性として、IC50(50%細胞変性抑制濃度)が高くとも100μg/ml以下のとき、有効な抗HIV−1プロテアーゼ剤といえる。IC50は、より好ましくは、50μg/ml以下であることが好ましく、より好ましくは、10μg/ml以下であり、さらに好ましくは5μg/ml以下である。
【0045】
抗HIV活性測定方法
また、抗HIV活性の測定方法としては、特開平7−206601号公報に記載の方法、前記非特許文献2(Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999)の他、従来公知の各種方法を採用できる。例えば、HIV感染感受性であって非感染細胞株にHIVを感染させた場合の細胞変性を利用する急性感性系による測定、HIV持続感染細胞株と非感染細胞株とを共存させたとき生じる細胞融合による巨細胞の形成を利用する巨細胞形成系における測定のいずれかあるいは双方を採用できる。これらの方法によれば、抗HIV−1プロテアーゼ活性による、あるいは当該抗HIV−1プロテアーゼ活性によらない抗HIV活性を測定できる。
なお、急性感性系による測定方法は、例えば、HIV感染について高い感受性を有するT細胞株であるMT−4細胞におけるHIV−1感染による細胞変性に対する抑制効果を指標とする。同時に、細胞毒性も指標とすることができる。また、巨細胞形成系による測定方法は、例えば、HIV−1が持続感染したMOLT/LAI細胞と非感染のMOLT−4細胞の混合培養を行い、非感染細胞の表面に存在するHIV−1レセプターが結合し、細胞融合が起こり、巨細胞が形成されることに対する抑制効果を指標とできる。
【0046】
抗HIV活性として、急性感染系における細胞変性を指標としたIC50は、高くとも100μg/ml以下であるとき、有効な抗HIV剤とすることができる。より好ましくは、50μg/ml以下であり、さらに好ましくは10μg/ml以下である。
また、抗HIV活性として、巨細胞系における巨細胞の形成抑制を指標としたIC50は、高くとも100μg/ml以下であるとき、有効な抗HIV剤とすることができる。より好ましくは、50μg/ml以下であり、さらに好ましくは10μg/ml以下である。
一方、これらの各系において同時に測定される50%細胞毒性濃度(CC50)は、10μg/ml以上であることが好ましく、50μg/ml以上であるとより好ましい。あるいは、SI(CC50÷IC50)が10以上であることが好ましく、より好ましくは30以上である。
【0047】
リグニン誘導体が抗HIV−1プロテアーゼ活性を有する場合には、例えば、そのまま、あるいは適当な細胞内取り込み化合物とともに細胞に付与することにより、細胞膜を通過させ、細胞内にて抗HIV−1プロテアーゼ活性を発揮させることができる。また、リグニン誘導体が、抗HIV活性を有する場合には、そのまま、あるいは、細胞内取り込みを容易化する化合物や細胞表面に親和性を付与する化合物とともに適用することができる。
抗HIV−1プロテアーゼ剤や抗HIV剤は、例えば、HIV感染防止剤、HIV増殖抑制剤として用いることができる。
HIV感染防止剤としては、例えば、医療用具や手術用具などを含む各種医療用具、避妊具などの衛生用品、及び治療室や手術室などの医療用領域に適用する感染防止剤としての用途がある。適用方法としては、散布、塗布、噴霧、浸漬等、必要に応じて選択することができる。これらに本剤を適用することで、HIV感染可能性のある用具や領域においてウイルス感染力を低下させ、他者への感染を防止できる。この場合、塗布、散布、浸漬用の溶液としては、5μg/ml〜500μg/mlの濃度とすることができる。より好ましくは、20μg/ml〜200μg/mlの濃度である。
【0048】
また、移植用臓器あるいは移植用組織の感染防止剤として使用できる。移植前の臓器あるいは組織に適用することで、用具や領域に付着した血液や体液などに由来して潜在可能性のあるHIVがこれらの用具等を介して他人に感染するのを防止できる。また、臓器等の提供者に由来してHIV感染可能性のある臓器あるいは組織におけるHIVを殺傷し、あるいは感染力を弱めて、移植による感染を予防することができる。この場合、塗布、散布、浸漬用の溶液としては、5μg/ml〜500μg/mlの濃度とすることができる。より好ましくは、20μg/ml〜200μg/mlの濃度である。
なお、輸血用血液や全血製剤や各種の血液成分製剤の感染防止剤としても使用できる。
【0049】
また、本抗HIV−1プロテアーゼ剤及び本抗HIV剤は、製剤学上許容される賦形剤等と組み合わされて、うがい剤、ローション剤、クリーム剤、ゲル剤、フィルム剤、坐剤や膣坐剤としての各種局所外用剤での感染防止剤としても使用することができる。感染予防剤としては、局所外用の場合、適時、局所に10〜1000μg適用することが好ましい。
【0050】
本リグニン誘導体は、水溶性であり、特にアルカリ処理により低分子化されている場合もある。したがって、経口あるいは腸管経由での投与形態であっても、感染可能性部位に到達させることも可能であり、各種経口剤あるいは腸管経由剤としての感染防止剤及び増殖抑制剤(治療剤)としても有用である。感染防止を目的とする場合には、好ましい用量としては、例えば、0.01〜10mg/kg/日とすることができる。また、HIV増殖抑制を目的とする場合には、好ましい用量としては、例えば、0.01〜100mg/kg/日とすることができる。
さらに、本剤は、経口あるいは腸管経由の栄養補助食品としても有用である。
【0051】
また、体内の局所へ注入する使用形態での抗HIV増殖抑制剤(治療剤)としても使用することができる。HIVはリンパ節等の組織に潜伏し、増殖を開始する。したがって、潜伏期間中、あるいは増殖開始後であっても、HIVの潜伏部位あるいは増殖部位に本剤を注入することにより、HIVの増殖を抑制することができる。この場合、適時、局所に1〜500μg/mlを適量注入することが好ましい。
また、本剤は、吸引治療用剤にも用いることができる。特に、肺におけるHIV増殖抑制に有用である。この場合の用量は、例えば、1〜500μg/kg/日とすることができる。
【0052】
【実施例】
以下、本発明を具体化して実施例について説明する。なお、これらの実施例は本発明を具体的に説明するものであって、本願発明を拘束するものではない。
(実施例1)
第2の各種一次誘導体(試料1〜3)の調製
スギ(Japanese cedar)脱脂木粉に含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、それぞれカテコール、レゾルシノール及びピロガロール3モルをアセトンに溶解させた。そのアセトン溶液にスギ脱脂木粉を一昼夜浸漬させ、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を木粉に収着させた。この収着木粉に72wt%硫酸(20ml/g脱脂木粉)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(10ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を取り除いた後、さらに、硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相を取り除いた。有機相に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相からフェノール:ベンゼン混液で抽出されなくなるまでこの洗浄を数回繰り返した。その後、硫酸相は大過剰の水で希釈(硫酸濃度約10wt%まで)され、透析(透析膜、分子量カットオフ;1000)にて未反応フェノール化合物と酸を取り除いた。透析後に再度生じた沈殿は、遠心分離にて取り除かれ、水溶性リグノフェノールを含む粗水溶液は凍結乾燥によって乾燥された。この乾燥物からメタノール抽出し、抽出液をジエチルエーテルに滴下、沈殿区分をそれぞれリグノカテコールIV、リグノレゾルシノールIV及びリグノピロガロールIV(試料1〜3)とした。
【0053】
(実施例2)
第2の各種水溶性リグニン二次誘導体の調製(試料4〜6)
実施例1で調製した各試料1〜3各約500mgを耐圧管(直径12mm×長さ60mm)に入れ、これに0.5N水酸化ナトリウム10ml、攪拌球(直径3mm)2個を入れ、オイルバス中で170℃、1時間処理した。処理後、冷却して内容物を0.5N塩酸にて中和し、遠心分離(20℃、10,000rpm、5分)で沈殿物と上澄み液とに分離させた。上澄み液を透析(透析膜、分子量カットオフ;1000)して脱塩した後、この時生じた沈殿物は再度遠心分離により取り除き、上澄み液を凍結乾燥、さらに五酸化二リン上で減圧乾燥し、リグノカテコールIV−b、リグノレゾルシノールIV−b及びリグノピロガロールIV−b(試料4〜6)とした。
【0054】
(実施例3)
第2の各種一次誘導体(試料7〜9)の調製
モミガラ、ケナフ(葉を除いた靭皮含む茎部)、タケ(葉を除いた茎部)の脱脂粉末試料を用い、それぞれに含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、ピロガロール3モルをアセトンに溶解させた。そのアセトン溶液に各試料を一昼夜浸漬させた後、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を試料に収着させた。この収着試料に65wt%硫酸(20ml/g脱脂試料)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(10ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を取り除いた後、さらに、硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相を取り除いた。有機相に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相からフェノール:ベンゼン混液で抽出されなくなるまでこの洗浄を数回繰り返した。その後、硫酸相は大過剰の水で希釈(硫酸濃度約10wt%まで)され、透析(透析膜、分子量カットオフ;1000)にて未反応フェノール化合物と酸を取り除いた。透析後に再度生じた沈殿は、遠心分離にて取り除かれ、水溶性リグノフェノールを含む粗水溶液は凍結乾燥によって乾燥された。この乾燥物を、メタノール抽出した液をジエチルエーテルに滴下し、沈殿区分をそれぞれリグノピロガロールIV−RH、リグノピロガロールIV−K及びリグノピロガロールIV−B(試料7〜9)とした。
【0055】
(比較例1)
リグノカテコールV II −b、リグノレゾルシノールV II −b及びリグノピロガロールV II −b(比較試料1〜3)の調製
スギ脱脂木粉に含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、それぞれカテコール、レゾルシノール及びピロガロール3モルをアセトンに溶解させた。そのアセトン溶液にスギ脱脂木粉を一昼夜浸漬させ、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を木粉に収着させた。この収着木粉に95wt%リン酸(20ml/g脱脂木粉)を加えて50℃、1時間、攪拌した。攪拌後、リン酸濃度を10%まで水で希釈して、さらに1時間ホモジナイザーにより攪拌した。これを遠心分離(25℃、10,000rpm、5分)し、沈殿物を脱イオン水で繰り返し洗い、未反応フェノール化合物を除去、脱酸し、凍結乾燥した。この乾燥物をアセトン抽出し、その抽出残渣を乾燥させた。さらにアセトン残渣をメタノール抽出し、抽出液をジエチルエーテルに滴下して沈殿した区分を、それぞれリグノカテコールVII、リグノレゾルシノールVII及びリグノピロガロールVIIとした。
次いで、これら各約500mgを耐圧管(直径12mm×長さ60mm)に入れ、これに0.5N水酸化ナトリウム10ml、攪拌球(直径3mm)2個を入れ、オイルバス中で170℃、1時間処理した。処理後、冷却して内容物を0.5N塩酸にて中和し、遠心分離(20℃、10,000rpm、5分)で沈殿物と上澄み液とに分離させた。上澄み液を透析(透析膜、分子量カットオフ;1000)して脱塩した後、この時生じた沈殿物は再度遠心分離により取り除き、上澄み液を凍結乾燥、さらに五酸化二リン上で減圧乾燥し、リグノカテコールVII−b、リグノレゾルシノールVII−b及びリグノピロガロールVII−b(比較試料1〜3)とした。
【0056】
(実施例4)
第1のカルボキシメチル化二次誘導体(試料10〜13)の調製
モミガラ、ケナフ(葉を除いた靭皮含む茎部)、タケ(葉を除いた茎部)、及びイネワラの脱脂粉末試料を用い、それぞれに含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、p−クレゾール3モルをアセトンに溶解させた。そのアセトン溶液に各試料を一昼夜浸漬させた後、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を試料に収着させた。この収着試料に68wt%硫酸(4ml/g脱脂試料)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(2ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を大過剰のジエチルエーテルに滴下し、さらに、再度分離された硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相をジエチルエーテルに滴下した。フェノール:ベンゼン混液に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相から抽出されなくなるまで同じ操作を数回繰り返した。フェノール:ベンゼン相をジエチルエーテルに滴下し生じた沈殿物を遠心分離によって回収後、これをアセトンに溶解させ、不溶解区分を遠心分離によって除去した。このアセトン抽出溶液をエバポレーターで濃縮後、大過剰のジエチルエーテルに滴下し、その不溶解沈殿物を遠心分離にて回収、さらに沈殿物を少量のジエチルエーテルにて遠心分離により洗浄し、五酸化リン上で減圧乾燥後、第1の一次誘導体として、それぞれリグノクレゾール−RH、リグノクレゾール−K、リグノクレゾール−B、リグノクレゾール−RSを得た。
これら第1の一次誘導体である各リグノクレゾール1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化二次誘導体として、それぞれリグノクレゾール−CM−RH、−CM−K、−CM−B、−CM−RS(試料10〜13)を得た。
【0057】
(実施例5)
第1のカルボキシメチル化二次及び三次誘導体(試料14〜16)の調製
ブナ(Beech)脱脂木粉からリグニン一次誘導体(リグノクレゾール)の調製アセトン脱脂したブナ木粉(Beech)にリグニンあたり3mol倍量のp−クレゾールを収着させた。この収着木粉に72%硫酸(4ml/g 脱脂木粉)を加え、1時間攪拌しながら30℃で反応させた後、反応系を大過剰の水に投入し、硫酸濃度を10%程度まで希釈した。その時、生じた不溶解沈殿物を遠心分離にて分離し、硫酸と未反応のクレゾールを取除くまで完全に洗浄し、五酸化リン上で減圧乾燥させた。これにアセトンを加え、一昼夜攪拌後、不溶物を遠心分離にて取除き、エバポレーターで濃縮後、大過剰のジエチルエーテルに滴下し、その不溶解沈殿物を遠心分離、洗浄し、第1の一次誘導体として、それぞれリグノクレゾールを得た。
これの第1の一次誘導体であるリグノクレゾール1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化二次誘導体として、それぞれリグノクレゾール−CM(試料14)を得た。
さらに、先の第1の一次誘導体であるリグノクレゾール20gを、0.5N水酸化ナトリウム水溶液400mlに完全溶解させ、これをオートクレーブに投入し、140℃(0.25MPa)および170℃(0.68MPa)にて1時間処理した。反応終了後、冷却し、1N塩酸にて中和した。沈殿物を遠心分離にて回収、水にて洗浄し、凍結乾燥後、アルカリ処理物を得た。
これら2種類のアルカリ処理物それぞれ1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化三次誘導体として、それぞれリグノクレゾール−CM−140及びリグノクレゾール−CM−170(試料15、16)を得た。
【0058】
(比較例2)
第1の一次誘導体のアルカリ水溶化体(リグノクレゾール−140及びリグノクレゾール−170(比較試料4、5)の調製
実施例5で調製した第1の一次誘導体であるリグノクレゾール、各20gを、0.5N水酸化ナトリウム水溶液400mlに完全溶解させ、これをオートクレーブに投入し、140℃(0.25MPa)および170℃(0.68MPa)にて1時間処理した。反応終了後、冷却し、1N塩酸にて中和した。上澄み液を遠心分離にて回収し、透析(膜の種類を教えて下さい)して脱塩後、凍結乾燥してアルカリ処理水溶化物である(リグノクレゾール−140及びリグノクレゾール−170(比較試料4、5))を得た。
【0059】
(実施例7)
1.抗HIV−1プロテアーゼ活性の測定
実施例1〜5及び比較例1〜2で調製した試料1〜16及び比較試料1〜5について、以下の条件で抗HIV−1プロテアーゼ活性を測定した。
0.5ml容のマイクロチューブに100mM酢酸緩衝液(200mM塩化ナトリウム、5mMジチオトレイトール(DTT)、10vol%グリセロール、pH4.9)100μL、25μg/mlのrecHIV−1プロテアーゼ(Escherichia coli)溶液100mM酢酸緩衝液(200mM塩化ナトリウム、5mMジチオトレイトール、10vol%グリセロール、5vol%エチレングリコール、pH5.5)20μL、及び各種濃度(5、10、12.5、50、μg/ml)の試料溶液30μLを加え、37℃で5分間インキュベートした後、1mg/ml基質溶液(HIV−1プロテアーゼ基質III;His−Lys−Ala−Val−Leu−p−nitro−Phe−Glu−Ala−Nle−Ser−NH2/H2O)10μLを加えた。反応液中の各濃度は以下の通りであった。
recHIV−1プロテアーゼ3.1μg/ml
基質62.5μg/ml
試料0.9、2.3、4.7、9.4、18.8μg/ml
63mM 酢酸緩衝液(125mM 塩化ナトリウム、3mM DTT、6vol%グリセロール)
【0060】
この反応液を37℃に保持し15分後に10%TFA10μLを加えて反応を終了させた。RecHIV−1プロテアーゼによって分解された基質分解物(His−Lys−Ala−Val−Leu−NH2又はp−nitro−Phe−Glu−Ala−Nle−Ser−NH2)の遊離量からrecHIV−1プロテアーゼの活性を換算するため、反応液を0.45μmのメンブランフィルターでろ過し、ろ液をHPLCに供した。PDA検出器により基質分解物の最大吸収272nmにおけるピーク強度を測定し、試料無添加系をコントロールとして各濃度でのrecHIV−1プロテアーゼ活性を求めた。なお、対照として、クラフトリグニン(アルドリッチ社製、製品番号;8068−05−1)についても試料と同様に試験した。また、公知のHIV−1プロテアーゼインヒビター(アスパラギン酸プロテアーゼインヒビター)であるペプスタチンA(Pepstain A)(シグマ社製、製品番号;P−5318)についても同様に試験した。結果を図6〜12に示す。
【0061】
2.抗HIV活性の測定
ヒトT細胞性白血病ウイルスI画他(HTLV−I)に持続感染しているT細胞株であるMT−4細胞に、ヒト免疫不全ウイルス1型(HIV−1、LAI株)を0.001TCID50/cellの割合で1時間感染させた後、正確に段階希釈(0.49μg/ml、0.98μg/ml、1.96μg/ml、3.91μg/ml、7.81μg/ml、15.62μg/ml、31.25μg/ml、62.5μg/ml、125μg/ml、250μg/ml、500μg/ml、1000μg/ml)した試料を含むRPMI−1640培養液(10%牛胎児血清、ペニシリンG100U/mlとストレプトマイシン100μg/mlを含む)に1.5×105cells/mlの濃度で浮遊させ、96穴の平底培養プレートに1ウェルあたり200μLで培養した。培養5日目に、鏡検によりHIV−1増殖による細胞変性効果(CPE)及び細胞の生育状態(細胞毒性)を観察し、細胞変性が認められる最低希釈濃度(IC)と細胞毒性が認められた最低希釈濃度(CC)とを求めた。なお、対照として、DS8000(デキストラン硫酸8000)とAZT(アジドチミジン)についても同様にICとCCとを求めた。結果を図13に示す。
【0062】
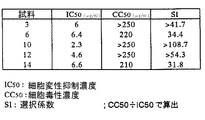
さらに、一部の試料(3、6、10、12、14)について、段階希釈の濃度を0.25〜250μg/mlとして、48穴の平底培養プレートにて1ウェルあたり600μLで培養する以外は上記条件と同様の条件で試験を行い、培養5日目に、生細胞数をトリパンブルー染色で計測し、50%細胞変性抑制濃度(IC50)と50%細胞毒性濃度(CC50)を算出し、加えてSIも算出した。結果を図14に示す。
【0063】
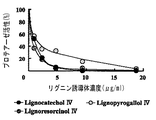
抗HIV−1プロテアーゼ活性については、図6〜12に示すように、試料1〜16においては、HIV−1プロテアーゼ活性を50%阻止濃度がおおよそ5μg/ml以下のものが、試料1〜3(図6)、4〜6(図7)、10〜13(図9)、14〜16(図10)及び対照のペプスタチンA(図12)であった。試料7〜9においては50%阻止濃度は20μg/mlを超えていた(図8)。
これに対し、比較試料1〜3(図11)においては、50%阻止濃度は、10μg/ml以下であった。また、比較試料4は約10μg/mlであり、比較試料5は20μg/mlを超えていた(図12)。対照のクラフトリグニンも20μg/mlを超えていた(図12)。
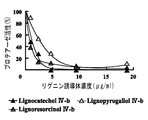
【0064】
抗HIV活性については、図13に示すように、試料1〜9(第2の誘導体群)においては、抗HIV活性は明瞭でなく、毒性との区別が付きにくかった。また、抗HIV−1プロテアーゼ活性において良好な結果を示した試料には細胞毒性が高いものがあることがわかった。例えば、試料1〜6については、レゾルシノール及びカテコール誘導のリグニン誘導体(1,2,4,5)は細胞毒性が高い傾向にあった。これに対し、試料10〜16(第1の誘導体群)については、抗HIV活性は明瞭であった。また、比較試料1〜5は、いずれも、抗HIV活性は不明瞭であった。
【0065】
さらに、図14に示すように、試料1〜6のうちピロガロール誘導のリグニン誘導体(試料2,4)と試料10〜16のうちモミガラ由来のp−クレゾール誘導のカルボキシメチル化誘導体(試料10)、タケ由来のp−クレゾール誘導体カルボキシメチル化誘導体(試料12)、ブナ由来のp−クレゾール誘導アルカリ処理−カルボキシメチル化誘導体(試料14)については、いずれも良好なIC50を示すとともに200μg/ml以上のCC50を示した。結果として高い選択性、すなわち、少なくとも30倍以上のSIを示した。
また、試料1〜16は、これら試験に供した試料と同程度の抗HIV活性を有し、構造類似であることから、試料1〜16も高い抗HIV活性と選択性を有し、硫酸化デキストランに匹敵する活性を示したと思われる。
【0066】
以上のことから、本発明の第1の誘導体及び第2の誘導体は、いずれも、有効な抗HIV剤として使用できることがわかった。
また、ピロガロールなどの3価のフェノール化合物により誘導された第2の誘導体、クレゾールなどの1価のフェノール化合物に誘導されカルボキシアルキル(メチル)化された第1の誘導体は、及び1価のフェノール化合物に誘導されアルカリ処理されカルボキシアルキル(メチル)化された第1の誘導体は、特に、有効な抗HIV剤として使用できることがわかった。
【0067】
【発明の効果】
本発明によれば、リグニン誘導体の一種であるリグノフェノール系誘導体を用いた抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤を提供できる。
【図面の簡単な説明】
【図1】リグノフェノール系誘導体を得る構造変換プロセスの一例を示す図である。
【図2】図1に示す構造変換プロセスにおける構造変換の一例を示す図である。
【図3】オルト位結合ユニットとパラ位結合ユニットとを示す図である。
【図4】リグニン誘導体の相分離及び各種誘導体化のスキームを示す図である。
【図5】リグニン誘導体の相分離及び各種誘導体化のスキームを示す図である。
【図6】試料1〜3の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図7】試料4〜6の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図8】試料7〜9の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図9】試料10〜13の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図10】試料14〜16の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図11】比較試料1〜3の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図12】比較試料4〜5の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図13】試料1〜16及び比較試料1〜5の抗HIV活性の測定結果と使用したリグニン誘導体の由来、フェノール化合物の種類、用いた誘導体化手法等を併せて記載した表を示す図である。
【図14】試料3、6、10、12、14の抗HIV活性測定結果を示す図である。
【発明の属する技術分野】
本発明は、リグニンにフェノール化合物を導入して得られるリグニン誘導体の利用に関し、特に、抗ヒトエイズウイルスプロテアーゼ1型(Human immunodeficiency virus type I (HIV−1) protease)剤及び抗エイズウイルス(Human immunodeficiency virus(HIV))剤に関する。
【0002】
【従来の技術】
リグニンは、植物体を構築するリグニン−セルロース複合体の構成成分である。近年、植物細胞壁を維持する作用のみならず、その生理的作用についても着目されている。
例えば、木質化植物材料を有機溶媒で加温洗浄した後、アルカリ水溶液で抽出して得られるリグニン抽出物がウイルス感染防止作用を有することが知られている(特許文献1)。また、木材から得られるクラフトリグニンをカルボキシメチル化して得られる水溶性クラフトリグニンが抗エイズウイルス活性を有していることも知られている(特許文献2)。さらに、ある種のきのこから抽出された水溶性リグニン様物質や、小麦から抽出された水溶性リグニンがHIV−1プロテアーゼ活性を阻害することが知られている(キノコから抽出したリグニンについて非特許文献1、小麦抽出リグニンについて非特許文献2)。
また、合成により得られる水溶性リグニンが抗HIV活性を有していることも報告されている(特許文献1、非特許文献2、非特許文献3)。
【0003】
【特許文献1】
特開平7−206601号公報
【特許文献2】
特開平8−143587号公報
【非特許文献1】
Biosci. Biotechnol. Biochem., 62(3), 575−577, 1998
【非特許文献2】
Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999
【非特許文献3】
Chem. Pharm. Bull. 40(8)2102−2105(1992)
【0004】
【発明が解決しようとする課題】
ここに、リグニン誘導体としては、クラフトリグニン、蒸煮爆砕リグニン、酢酸リグニンなど多種類のリグニン誘導体がある他、リグノセルロース系材料にフェノール化合物を添加し溶媒和させた後、酸を添加することにより得られるリグニンのフェノール誘導体化合物(以下、リグノフェノール系誘導体ともいう。)が知られている(リグノフェノール系誘導体について、特開2001−64494号公報、特開2001−261839号公報、特開2001−131201号公報、特開2001−342353号公報、特開2002−105240号公報等)。
一方、各種リグニン誘導体の構造は、その抽出分離方法によって大きくことなることが知られている。
しかしながら、既に説明したように、抗HIV−1プロテアーゼ活性は限定的な抽出方法かあるいは合成によるリグニン様物質において検出されたと報告されているに過ぎず、全てのリグニン誘導体がHIV−1プロテアーゼ阻害活性を有していると確認されてはいない。また、当該阻害活性のための構造も未だ不明である。
そこで、本発明では、リグニン誘導体の一種であるリグノフェノール系誘導体を用いた抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤を提供することを目的とする。
【0005】
【課題を解決するための手段】
本発明者らは、各種のリグノフェノール系誘導体を調製し、これらの誘導体について抗HIV−1プロテアーゼ活性とHIV感受性細胞におけるHIV感染による細胞変性試験により、有用な抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤について探索した。その結果、ある種のリグノフェノール系誘導体が抗HIV−1プロテアーゼ活性及び抗HIV活性を有することを見出した。
また、本発明者らは、リグノフェノール系誘導体を調製するのに適した方法、すなわち、使用するフェノール化合物の種類、リグノフェノール系誘導体の抽出分離操作、及び修飾手段を見出した。
本発明によれば、以下の手段が提供される。
【0006】
(1) 抗HIV剤であって、
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体、及び
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。
(2)前記フェノール化合物は、1価のフェノール化合物である、(1)記載の抗HIV剤。
(3)前記カルボキシアルキル基におけるアルキル基はメチル基である、(1)又は(2)記載の抗HIV剤。
(4)前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、(1)〜(3)のいずれかに記載の抗HIV剤。
(5)抗HIV剤であって、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体、
(e)前記リグニン一次誘導体に対してアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。
(6)前記2価以上のフェノール化合物は、カテコール、レゾルシノール、及びピロガロールからなる群から選択される1種あるいは2種以上である、(5)記載の抗HIV剤。
(7)前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、(5)又は(6)に記載の抗HIV剤。
(8)前記リグノセルロース系材料は、針葉樹由来である、(7)記載の抗HIV剤。
(9)(1)〜(8)のいずれかに記載のリグニン誘導体を含有する抗HIV−1プロテアーゼ剤。
(10)抗HIV剤を製造する方法であって、以下の(a)〜(g);
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体、
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンの一次誘導体
(e)(d)のリグニン一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
からなる群から選択される1種あるいは2種以上のリグニン誘導体を調製する工程を有し、
これらのリグニン誘導体について以下の工程:
抗HIVプロテアーゼ活性を測定する工程、
HIV感染感受性の非感染細胞株を用いた急性感染系における抗HIV活性を測定する工程、
HIV持続感染細胞株とHIV感染感受性の非感染細胞株とを用いた巨細胞形成系において抗HIV活性を測定する工程、
から選択される1種あるいは2種以上の工程を行い、抗HIV活性を評価することを含む、方法。
【0007】
【発明の実施の形態】
本発明の抗HIV剤及び抗HIV−1プロテアーゼ剤は、リグニン含有材料から所定の手法により分離して得られるリグノフェノール系誘導体を有効成分としている。これらのリグノフェノール系誘導体としては、以下の(a)〜(g)を挙げることができる。
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンの一次誘導体
(e)(d)のリグニン一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体。
【0008】
ここで、上記(a)〜(c)は、フェノール化合物を用いたリグニン一次誘導体に由来し、(d)及び(e)は2価以上のフェノール化合物を用いたリグニン一次誘導体に由来している。
そこで、以下の説明においては、(a)に記載のリグニン一次誘導体を第1のリグニン一次誘導体といい、(a)〜(c)に記載のリグニン誘導体を、(b)及び(c)に記載の二次及び三次誘導体を含めて第1の誘導体というものとし、(d)記載のリグニン一次誘導体を第2のリグニン一次誘導体といい、(d)〜(g)に記載のリグニン誘導体を含めて第2の誘導体というものとする。
第1及び第2の誘導体が由来するリグニン含有材料は入手が容易で再生可能な天然資源であり、この資源を由来材料とすることにより、有効性と安全性のバランスが取れた抗HIV−1プロテアーゼ剤及び抗HIV剤を提供することができる。また、基本骨格を合成手法でなく、大量かつ再生可能に存在する植物資源から得るために、抗HIV剤製造にかかる原料コスト及び工程コストを含む製造コストを低減することができる。
また、リグニン含有材料からリグノフェノール系誘導体は、構造制御や修飾が容易であり、薬物送達性や有効性を確保するような構造を探索し、発見することも容易に達成することができる。
以下、本発明の実施の形態について詳細に説明する。
【0009】
(第1及び第2のリグニン一次誘導体)
本発明において用いるリグニン一次誘導体は、リグニン含有材料をフェノール化合物で溶媒和後、酸を添加し混合して得られるリグニンのフェノール化合物であるリグニン誘導体である。この反応過程によりリグニンのアリールプロパンユニットのベンジル位(側鎖C1位、以下、単にC1位という。)にフェノール化合物がグラフト(導入)されたリグニン誘導体を得ることができる。フェノール化合物は、そのフェノール性水酸基に対してオルト位あるいはパラ位にて前記C1位の炭素原子に結合する。この結果、1,1−ビス(アリール)プロパンユニットがリグニン中に形成される。この反応において、フェノール化合物は、前記C1位に対して選択的に導入されるため、出発原料であるリグニン含有材料におけるC1位における様々な結合を開放し、リグニンマトリックスの多様性を低減し、また、低分子量化することができる。さらに、この結果、従来のリグニンにはなかった各種溶媒への溶解性、熱流動性、熱可塑性など各種の特性を発現することが既に知られている。
なお、ここで、フェノール化合物で溶媒和するとは、液体のフェノール化合物にリグニン含有材料を浸漬する等して溶媒和する他、液体あるいは固体のフェノール化合物を当該フェノール化合物が溶解する溶媒に溶解させたものをリグニン含有材料に適用後、溶媒を留去することでリグニン含有材料にフェノール化合物を収着することによっても達成することができる。
【0010】
濃酸による炭水化物の膨潤に基づく組織構造の破壊と、フェノール化合物によるリグニンの溶媒和とを組み合わせてリグニンの不活性化を抑制しつつ、リグノセルロース系材料を炭水化物とリグノフェノール誘導体とに分離する方法(特開平2−233701号)が知られている。この方法で得られたリグノフェノール誘導体の活用法としては、例えば、セルロース系ファイバー等の成形材料に適用し成形体を作製することが報告されている(特開平9−278904号)。かかるリグノフェノール誘導体は、1,1−ビス(アリール)プロパンを高頻度構成単位として有するリグニン系ポリマーであって、高粘結性を潜在的に有していることがわかっている(特開平9−278904号)。
さらに、かかるリグノフェノール誘導体は、メチロール化することにより架橋性を付与でき、リニアあるいはネットワーク状の架橋構造を構築できると同時に、アルカリ処理によって、再び低分子化して溶媒中に溶解されることも、見出されている(特開2001−261839号公報)。
また、これらの他、リグノフェノール誘導体に関するより一般的な記載及びその製造プロセスについては、国際公開WO99/14223号公報、特開2001−64494号公報、特開2001−261839号公報、特開2001−131201号公報、特開2001−342353号公報、特開2002−105240号公報において記載されている(これらの特許文献に記載の内容は、全て引用により本明細書中に取り込まれるものとする)。
【0011】
このプロセスによりリグノセルロース系材料からリグノフェノール誘導体を得るシステムにおける構造変換プロセスの一例を図1に示す。
この構造変換プロセスは、図1に示すように、リグノセルロース系材料を予めフェノール化合物で溶媒和しておいた上で、当該リグノセルロース系材料を酸と接触させることにより、リグニンのアリールプロパンユニットのリグニンの複合状態を緩和させ、同時に、天然リグニンのアリールプロパンユニットのC1位(ベンジル位)に選択的に前記フェノール誘導体をグラフティングさせ、リグノフェノール誘導体を生成させ、同時にセルロースとリグノフェノール誘導体とに分離できる。このプロセスにおける構造変換の一例を、図2に示す。
【0012】
リグノフェノール誘導体は、それ自体、リグノセルロース系材料などのリグニン含有材料から反応、分離して得られるリグニン由来のポリマーの混合物である。このため、得られるポリマーにおける導入フェノール誘導体の量や分子量は、原料となるリグニン含有材料のリグニン構造および反応条件により変動する。
【0013】
(リグニン含有材料)
本発明におけるリグニン含有材料には、天然リグニンを含有するリグノセルロース系材料を含む。リグノセルロース系材料は、木質化した材料、主として木材である各種材料、例えば、木粉、チップの他、廃材、端材、古紙などの木本類植物資源に付随する農産廃棄物や工業廃棄物を挙げることができる。また用いる木本類の種類としては、スギ、ヒノキなどの針葉樹、ブナなどの広葉樹等、任意の種類のものを使用することができる。さらに、ケナフ、ジュート、イネ、タケなどの各種草本類植物、それに関連するイネワラ、モミガラなどの農産廃棄物や工業廃棄物なども使用できる。
本発明の第2の誘導体においては、木本類植物や草本類植物のいずれのリグノセルロース系材料も使用できるが、第2の誘導体においては、由来資源の影響が第1の誘導体よりも大きい場合がある。第2の誘導体においては好ましくは、木本類植物資源をリグノセルロース系材料として用い、スギ、マツ、ヒノキなどの針葉樹植物資源由来のリグノセルロース系材料を用いることもできるが、一次誘導体の回収率を考慮すれば、広葉樹や草本類資源を用いることが好ましい。
【0014】
(フェノール化合物)
フェノール化合物としては、1価のフェノール化合物、2価のフェノール化合物、または3価のフェノール化合物などを用いることができる。
1価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいフェノール、1以上の置換基を有していてもよいナフトール、1以上の置換基を有していてもよいアントロール、1以上の置換基を有していてもよいアントロキノンオールなどが挙げられる。
2価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいカテコール、1以上の置換基を有していてもよいレゾルシノール、1以上の置換基を有していてもよいヒドロキノンなどが挙げられる。
3価のフェノール化合物の具体例としては、1以上の置換基を有していてもよいピロガロールなどが挙げられる。
本発明の第1の一次誘導体にあっては、1価のフェノール化合物から選択される1種あるいは2種以上を用いることができる。また、第2の一次誘導体にあっては、2価のフェノール化合物及び3価のフェノール化合物のうち、1種あるいは2種以上を用いることができる。
【0015】
なお、1価から3価のフェノール化合物が有していてもよい置換基の種類は特に限定されず、任意の置換基を有していてもよいが、好ましくは、電子吸引性の基(ハロゲン原子など)以外の基であり、例えば、炭素数が1〜4、好ましくは炭素数が1〜3の低級アルキル基含有置換基である。低級アルキル基含有置換基としては、例えば、低級アルキル基(メチル基、エチル基、プロピル基など)、低級アルコキシ基(メトキシ基、エトキシ基、プロポキシ基など)である。また、アリール基(フェニル基など)の芳香族系の置換基を有していてもよい。また、水酸基含有置換基であってもよい。
【0016】
第1の一次誘導体を得るには、1価のフェノール化合物及び2価以上のフェノール化合物を用いることができる。一次誘導体としての収率を高めるには、好ましくは1価のフェノール化合物を用いる。また、好ましくはp−クレゾール、m−クレゾール、o−クレゾール、2,6−ジメチルフェノール、2,4−ジメチルフェノール、2−メトキシフェノール(Guaiacol)、2,6−ジメトキシフェノール等を用いることができ、より好ましくは、p−クレゾール、フェノール、p−エチルフェノールである。また、第2の一次誘導体を得るには、2価以上のフェノール化合物を用いることができる。2価のフェノール化合物としては、レゾルシノール、ホモカテコール、ハイドロキノン、1,3−ナフタレンジオール等を用いることが好ましく、より好ましくは、レゾルシノール、カテコールである。また、3価のフェノール化合物としては、好ましくは、ピロガロール、フロログルシノール、2−ヒドロキシ−ナフトキノン等を用いることができ、より好ましくは、ピロガロール、フロログルシノールである。第2の一次誘導体を得るには、好ましくは、3価のフェノール化合物を用い、より好ましくは、ピロガロールである。
【0017】
これらのフェノール化合物は、そのフェノール性水酸基に対してオルト位あるいはパラ位の炭素原子がリグニンのアリールプロパンユニットのC1位の炭素に結合することにより、1,1−ビス(アリール)プロパンユニットが形成されることになる。したがって、少なくとも1つの導入サイトを確保するには、オルト位及びパラ位のうち、少なくともひとつの位置に置換基を有していないことが好ましい。
フェノール化合物のフェノール性水酸基のオルト位炭素原子が前記C1位に結合して形成されたユニットをオルト位結合ユニットといい、フェノール化合物のフェノール性水酸基のパラ位炭素原子が前記C1位に結合して形成されたユニットをパラ位結合ユニットという。図3に、オルト位結合ユニット及びパラ位結合ユニットの一例として、フェノール化合物として、それぞれp−クレゾールと、2,6−ジメチルフェノールを用いて形成されるユニットを示す。
【0018】
以上のことから、本発明では、各一次誘導体について、無置換フェノール誘導体の他、少なくとも一つの無置換のオルト位あるいはパラ位を有する各種置換形態のフェノール誘導体の1種あるいは2種以上を適宜選択して用いることができる。
【0019】
オルト位結合ユニットとパラ位結合ユニットとは、例えば、後述するアルカリ処理工程において異なる機能を発現する。オルト位結合ユニットは、緩和なアルカリ処理により導入されたフェノール化合物におけるフェノール性水酸基を消失させるとともにアリールクマラン構造を当該ユニットにおいて生成し、強いアルカリ処理によりアリール基移動に伴って分子形態が変動される。いずれにおいても、オルト位結合ユニットは、アルカリ処理による効率的なリグノフェノール誘導体の低分子化に寄与する。
一方、パラ位結合ユニットは、アルカリ処理によりアリールクマラン構造やその後の分子形態変動を生じず、当該ユニット部位における低分子化には寄与しない。したがって、アルカリ処理耐性を付与する機能を有するといえる。
【0020】
また、使用するフェノール化合物の種類を選択することにより、得られる一次誘導体への後段の二次誘導体化工程での官能基の導入位置や導入頻度等を調節し、第1及び第2の誘導体の構造制御が可能となる。
【0021】
なお、オルト位結合ユニットを有するリグノフェノール誘導体を得るには、少なくとも一つのオルト位(好ましくは全てのオルト位)に置換基を有していないフェノール化合物を用いる。また、少なくとも一つのオルト位(2位あるいは6位)が置換基を有さず、パラ位(4位)に置換基を有するフェノール化合物(典型的には、2,4位置換1価フェノール誘導体)が好ましい。最も好ましくは、全てのオルト位が置換基を有さず、パラ位に置換基を有するフェノール化合物(典型的には、4位置換1価フェノール化合物)である。したがって、4位置換フェノール化合物及び2,4位置換フェノール化合物を1種あるいは2種以上組み合わせて用いることができる。
【0022】
パラ位結合ユニットを有するリグノフェノール誘導体を得るには、パラ位に置換基を有していないフェノール化合物(典型的には、2位(あるいは6位)置換1価フェノール化合物)が好ましく、より好ましくは、同時に、オルト位(好ましくは、全てのオルト位)に置換基を有するフェノール化合物(典型的には2,6位置換1価フェノール化合物)を用いる。すなわち、2位(あるいは6位)置換フェノール化合物及び2、6位置換フェノールのうち1種あるいは2種以上を組み合わせて用いることが好ましい。
【0023】
(酸)
リグニン含有材料と接触させる酸としては、特に限定しないで、リグノフェノール誘導体を生成しうる範囲で各種無機酸や有機酸を使用することができる。したがって、硫酸、リン酸、塩酸などの無機酸の他、p−トルエンスルホン酸、トリフルオロ酢酸、トリクロロ酢酸、ギ酸などを使用することができる。リグニン含有材料としてリグノセルロース系材料を使用する場合には、セルロースを膨潤させる作用を有していることが好ましい。例えば、65重量%以上の硫酸(好ましくは、72重量%の硫酸)、85重量%以上のリン酸、38重量%以上の塩酸、p−トルエンスルホン酸、トリフルオロ酢酸、トリクロロ酢酸、ギ酸などを挙げることができる。好ましい酸は、65重量%以上の硫酸(好ましくは、72重量%の硫酸)、85重量%以上(好ましくは95重量%以上)のリン酸、トリフルオロ酢酸又はギ酸であるが、リグノフェノール誘導体を高収率で得るには、65重量%以上の硫酸(好ましくは、72重量%の硫酸)を好ましく用いることができる。
【0024】
リグニン含有材料中のリグニンを、リグノフェノール誘導体に変換し、分離する方法としては各種方法が採用できる。
第1及び第2の一次誘導体を得る反応工程としては、例えば、第1の図1に示すように、リグニン含有材料に、液体状のフェノール化合物(上記で説明したもの、例えば、p−クレゾール)を浸透させ、リグニンをフェノール誘導体により溶媒和させ、次に、リグノセルロース系材料に酸(上記で説明したもの、例えば、72%硫酸)を添加し混合して、セルロース成分を溶解する(以下、一段法ともいう。)。この方法によると、リグニンが低分子化され、同時にその基本構成単位のC1位にフェノール化合物が導入されたリグノフェノール誘導体がフェノール化合物相に生成される。このフェノール化合物相から、リグノフェノール誘導体が抽出される。リグノフェノール誘導体は、リグニン中のベンジルアリールエーテル結合が開裂して低分子化されたリグニンの低分子化体の集合体として得られる。
図2は、アリールプロパンユニットを有する天然リグニンに対して相分離処理を行うことにより、本発明におけるリグノフェノール誘導体が得られることを示している。
また、他の反応工程としては、リグニン含有材料に、固体状あるいは液体状のフェノール化合物を溶解した溶媒(例えば、エタノールあるいはアセトン)を浸透させた後、溶媒を留去(フェノール誘導体の収着)した場合も、先の方法と同様、リグノフェノール誘導体が生成される(以下、二段法ともいう)。
【0025】
第1のリグニン誘導体は、後段でカルボキシメチル化及び/又はアルカリ処理により、リグニン一誘導体に水溶性を付与する。したがって、一次誘導体としては、有機溶媒区分のリグニン一次誘導体を分離抽出することが有効である。
有機溶媒区分のリグニン一次誘導体を分離回収するには、一段法にあっては、液体のフェノール化合物相を、大過剰のエチルエーテルに加えて得た沈殿物を集めて、アセトンに溶解する。アセトン不溶部を遠心分離により除去し、アセトン可溶部を濃縮する。このアセトン可溶部を、大過剰のエチルエーテルに滴下し、沈殿区分を集める。この沈殿区分から溶媒留去し、第1のリグニン一次誘導体を得る。なお、粗第1の一次誘導体は、フェノール化合物相やアセトン可溶区分を単に減圧蒸留により除去することによって得ることができる。
【0026】
また、二段法にあっては、生成したリグノフェノール誘導体は、液体フェノール化合物にて抽出分離することができる。あるいは、全反応液を過剰の水中に投入し、不溶区分を遠心分離にて集め、脱酸後、乾燥する。この乾燥物にアセトンあるいはアルコールを加えてリグノフェノール誘導体を抽出する。さらに、この可溶区分を1段法における場合と同様に、過剰のエチルエーテル等に滴下して、リグノフェノール誘導体を不溶区分として得ることもできる。
以上、第1の一次誘導体の調製方法の具体例を説明したが、これらに限定されるわけではなく、これらに適宜改良を加えた方法で調製することもできる。
【0027】
また、第2のリグニン一次誘導体は、2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体である。したがって、上記一段法及び二段法における反応液の酸性水溶液相を採取し、この酸性相に対して、フェノール化合物、あるいはベンゼン、キシレン、トルエン、ヘキサン等の有機溶媒、あるいはフェノール化合物とこれらの有機溶媒との混合液を添加して有機溶媒相に可溶な一次誘導体を当該有機溶媒相に分離除去して、得られた酸性水溶液相に水溶性のリグニン一次誘導体を分離採取する。フェノール化合物としては、反応工程に用いたものを利用することが好ましい。リグニン一次誘導体の精製には、この酸性水溶液相を希釈し、透析等により脱酸、乾燥し、さらにこの乾燥物をメタノール等の有機溶媒で抽出した溶液をエーテルに滴下し、沈殿区分として採取する等の方法を採用することができる。有機溶媒相を用いた有機溶媒可溶性リグニン一次誘導体の分離除去は、十分に有機溶媒可溶性誘導体を除去する回数繰り返すことが好ましい。
以上、第2の一次誘導体の調製方法の具体例を説明したが、これらに限定されるわけではなく、これらに適宜改良を加えた方法で調製することもできる。
【0028】
(第1の誘導体)
次に、第1の一次誘導体をさらに化工して得られる第1の誘導体について説明する。図4には、第1のリグニン誘導体を得るスキームの例を示す。
アルカリ処理による水溶性化
アルカリ処理は、第1のリグニン一次誘導体をアルカリと接触させることにより行う。好ましくは加熱する。アルカリ処理においては、オルト位結合ユニットにおいて、導入されたフェノール化合物のフェノキシドイオンによるC2位炭素の攻撃が生じる。すなわち、一旦この反応が生じれば、C2アリールエーテル結合が開裂する。
例えば、緩和なアルカリ処理では、一次誘導体が第一のユニットを有する場合、当該導入フェノール誘導体の当該フェノール性水酸基が開裂し、生じたフェノキシドイオンが、C2アリールエーテル結合を構成するC2位を分子内求核反応的にアタックして、当該エーテル結合を開裂させて低分子化することができる。
C2アリールエーテル結合の開裂により、リグニンの母核にフェノール性水酸基が生成されることになり、当該分子内求核反応により、導入フェノール核が、それが導入されたフェニルプロパン単位とクマラン骨格を形成した構造(アリールクマラン単位)が発現される。
これらの結果、フェノール誘導体側にあったフェノール性水酸基が、リグニン母核側に移動されたことになる。
このため、オルト位結合ユニットを有するリグノフェノール誘導体においては、このユニットの存在部位において(1)C2アリールエーテル結の開裂による低分子化、(2)アリールクマラン構造の発現、(3)C2アリールエーテル結合で結合されていたリグニン母核側におけるフェノール性水酸基が発現する。
【0029】
当該アルカリ処理は、具体的には、リグノフェノール誘導体をアルカリ溶液に溶解し、一定時間反応させ、必要であれば、加熱することにより行う。この処理に用いることのできるアルカリ溶液は、リグノフェノール誘導体中の導入フェノール誘導体のフェノール性水酸基を解離させることができるものであればよく、特に、アルカリの種類及び濃度、溶媒の種類等は限定されない。アルカリ下において前記フェノール性水酸基の解離が生じれば、隣接基関与効果により、クマラン構造が形成されるからである。例えば、p−クレゾールを導入したリグノフェノール誘導体では、水酸化ナトリウム溶液を用いることができる。例えば、アルカリ溶液のアルカリ濃度範囲は0.5〜2Nとし、処理時間は1〜5時間程度とすることができる。また、アルカリ溶液中のリグノフェノール誘導体は、加熱されることにより、容易にクマラン構造を発現する。加熱に際しての、温度、圧力等の条件は、特に限定することなく設定することができる。例えば、アルカリ溶液を100℃以上(例えば、140℃程度)に加熱することによりリグノフェノール誘導体の低分子化を達成することができる。さらに、アルカリ溶液を加圧下においてその沸点以上に加熱して一次誘導体の低分子化を行ってもよい。
【0030】
なお、同じアルカリ溶液で同濃度においては、加熱温度が120℃〜140℃の範囲では、加熱温度が高い程、C2−アリールエーテル結合の開裂による低分子化が促進されることがわかっている。また、該温度範囲で、加熱温度が高い程、リグニン母体由来の芳香核由来のフェノール性水酸基が増加し、導入されたフェノール誘導体由来のフェノール性水酸基が減少することがわかっている。したがって、低分子化の程度及びフェノール性水酸基部位のC1位導入フェノール誘導体側からリグニン母体のフェノール核への変換の程度を、反応温度により調整することができる。すなわち、低分子化が促進され、あるいは、より多くのフェノール性水酸基部位がC1位導入フェノール誘導体側からリグニン母体へ変換されたアリールクマラン体を得るには80〜140℃程度の反応温度が好ましい。
【0031】
C1フェノール核の隣接基関与によるC2−アリールエーテルの開裂は、上述したようにアリールクマラン構造の形成を伴うが、リグノフェノール誘導体の低分子化は、必ずしもアリールクラマンが効率よく生成する条件下(140℃付近)で行う必要はなく、より高い温度(例えば170℃付近)で行うこともできる。この場合、一旦生成したクラマン環は開裂し、導入フェノール誘導体側にフェノール性水酸基が再生される結果、140℃処理物とは特性の異なるよりフェノール活性が高い素材を誘導することができる。
【0032】
以上のことから、アルカリ処理における加熱温度は、特に限定されないが好ましくは80℃以上200℃以下である。80℃を大きく下回ると、反応が十分に進行せず、200℃を大きく越えると好ましくない副反応が派生しやすくなるからである。
【0033】
クラマン構造の形成とそれに伴う低分子化のための処理の好ましい一例としては、0.5Nの水酸化ナトリウム水溶液をアルカリ溶液として用い、140℃で加熱時間60分という条件を挙げることができる。また、0.5Nの水酸化ナトリウム水溶液をアルカリ溶液として用い、170℃で加熱時間60分という条件を挙げることができる。
【0034】
アルカリ処理して水溶性のリグニン二次誘導体を得る方法としては、アルカリ処理反応液を中和後、その水溶性区分を遠心分離等により分離採取し、これを透析等により脱塩し、凍結乾燥等する方法を採用することができる。脱塩後の液をそのまま水溶性リグニン二次誘導体含有組成物として用いることもできる。なお、アルカリ処理反応液中和後の水不溶性区分は、水不溶性リグニン二次誘導体として後述するカルボキシアルキル化三次誘導体の前駆体となる。
第1の誘導体としての水溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。
また、水不溶性のリグニン二次誘導体の重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは700以上6000以下である。
【0035】
カルボキシアルキル化による二次誘導体化及び三次誘導体化
第1の一次誘導体及び、水溶性及び/又は水不溶性のリグニン二次誘導体に対して、カルボキシアルキル基を導入することによりカルボキシアルキル化二次誘導体及び三次誘導体を得ることができる。カルボキシルアルキル基におけるアルキル基は、炭素数1〜5の直鎖あるいは分枝アルキル基であることが好ましく、より好ましくは、炭素数1〜3の直鎖アルキル基である。たとえば、メチル基、エチル基、プロピル基を用いることができる。
カルボキシルアルキル基は、リグニン一次誘導体中のアルコール性あるいはフェノール性水酸基に導入される。カルボキシアルキル化は、一般に、アルカリ存在下に、モノハロゲノアルキルカルボン酸と反応させることにより達成することができる。リグニン誘導体のカルボキシアルキル化法は従来公知の各種方法を採用することができる。
例えば、リグニン一次誘導体を、この誘導体を分散できるイソプロパノールなどの有機溶媒で分散し、その後、アルカリ水溶液を加え、必要に応じてさらにイソプロパノールなどの有機溶媒を加えて得られた不均一混合溶液をモノクロロ酢酸などのモノハロゲノ酢酸(カルボキシアルキル化剤)を添加し、攪拌等しながら反応させることができる。また、リグニン一次誘導体を予めアルカリ溶液に浸漬した後、この浸漬物を有機溶媒に分散して、その後反応させることもできる。
なお、アルカリとしては、一般に用いられるアルカリ試薬でよいが、水酸化ナトリウム、水酸化カリウム等のアルカリ金属化合物のような強アルカリが好適に用いられる。また、リグニン一次誘導体等を分散する有機溶媒としては、メタノール、エタノール、イソプロパノール、t−ブタノール等のアルコール類、アセトン、メチルエチルケトン等のケトン類、ジエチルエーテル、テトラヒドロフラン、1,4−ジオキサン等のエーテル類、ジクロロメタン、クロロホルム等のハロゲン化物、ヘキサン、シクロヘキサン、ベンゼン、トルエン等の炭化水素類等が挙げられ、これらは単独であるいは2種以上混合して使用される。これらの中でも、アルコール類、ケトン類、エーテル類等の極性有機溶媒の1種又は2種以上の組合せが良く、さらに好ましくは、イソプロパノール、アセトン、1,4−ジオキサンが用いられる。
【0036】
モノハロゲノアルキルカルボン酸は、次の一般式〔I〕で表わされるものが好ましく用いられる。
XRCOOH 〔I〕
(上式中、X及びRはそれぞれ以下のものを表わす。
X:F、Cl、Br、I等のハロゲン原子、R:炭素数1〜5の直鎖及び分枝を有するアルキル基。)
上記化合物中、特にXがCl又はBr原子で、炭素数が1〜3の直鎖アルキル基を有するものが好ましい。
【0037】
上記モノハロゲノアルキルカルボン酸の使用量は、リグニンの水酸基量以上あれば良く、水酸基あたり、約1〜3モルであることが好ましい。モノハロゲノアルキルカルボン酸の添加方法としては、固体のまま及び/又はこの有機溶媒の溶液として、一括あるいは連続滴下で添加すれば良い。好ましいのは、有機溶媒の溶液として0.5〜3時間かけた連続滴下の方法であり、特に1〜2時間で連続滴下することが好ましい。また、モノハロゲノアルキルカルボン酸の添加終了後、その温度で0.5〜5時間(より好ましくは1〜4時間)反応を続けることが好適である。なお、モノハロゲノアルキルカルボン酸による反応にあたり、反応液を40℃〜60℃程度の範囲で加熱することが好ましい。
【0038】
カルボキシアルキル化二次及び三次誘導体は、固形生成物の他一部溶解した状態とで得られる。固形生成物は、反応終了後、ろ過等により固形の生成物を分離採取し、この固形生成物を水に溶解させ、希鉱酸、例えば希塩酸、希硫酸等の酸で中性に中和した後、電気透析で脱塩し、凍結乾燥等して得ることができる。また、溶解生成物は、上記ろ液中の有機溶媒を留去し、溶解物を乾固させ、この乾固物を中和前の固形生成物に添加することにより、両者を回収することができる。
【0039】
(第2の誘導体)
第2の誘導体は、既に説明した第2の一次誘導体とこの一次誘導体をさらにアルカリ処理して得られる水溶性のリグニン二次誘導体及びカルボキシアルキル化した二次及び三次誘導体を含んでいる。以下、第2のリグニン二次誘導体及びカルボキシアルキル化二次及び三次誘導体について説明する。図5には、第2のリグニン誘導体を得るためのスキームの例を示す。
アルカリ処理による水溶性化
アルカリ処理は、第2のリグニン一次誘導体をアルカリと接触させることにより行う。当該アルカリ処理は、第1の誘導体において説明したのと同様の操作及び条件等を採用することができ、好ましいものとして挙げた操作及び条件もそのまま採用できる。第2の水溶性の二次誘導体においては、既に一次誘導体において水溶性であるため、アルカリ処理により、より低分子化されさらに水溶性が向上すると考えられる。なお、アルカリ処理時に得られる水溶性及び/又は水不溶性の二次誘導体は、第1の誘導体におけるのと同様に、カルボキシアルキル化処理してカルボキシアルキル化三次誘導体を得ることができる。
なお、第2の誘導体としての水溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。第2の誘導体としての水不溶性リグニン二次誘導体は、重量平均分子量は700以上10000以下とすることが好ましく、より好ましくは、700以上6000以下である。
カルボキシアルキル化による二次誘導体化及び三次誘導体化
第1の誘導体におけるのと同様に、第2の一次誘導体及び、この一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して、カルボキシアルキル基を導入することによりカルボキシアルキル化二次誘導体及び三次誘導体を得ることができる。当該カルボキシアルキル化は、第1の誘導体において説明したのと同様の操作及び条件等を採用することができ、好ましいものとして挙げた操作及び条件もそのまま採用できる。
【0040】
(抗HIV−1プロテアーゼ剤及び抗HIV剤の有効成分としてのリグニン誘導体)以上説明した第1及び第2の誘導体は、本発明者らの研究によれば、好ましい抗HIV−1プロテアーゼ活性及び/又は抗HIV活性を有していることが認められている。なお、これらの誘導体における抗HIV活性は、抗HIV−1プロテアーゼ活性以外の何らかの抗HIV作用に起因する可能性があり、必ずしも抗HIV−1プロテアーゼ活性に基づく抗HIV活性に限定するものでない。したがって、本発明の抗HIV剤も抗HIV−1プロテアーゼ剤を有効成分とするものに限定されないし、本抗HIV剤の有効成分たるリグニン誘導体に抗HIV−1プロテアーゼ活性を必要条件とするものではない。しかしながら、抗HIV剤を探索するにあたり、抗HIV−1プロテアーゼ活性はその指標とすることができる。
【0041】
これらの各種誘導体に対して、HIV−1プロテアーゼ又は抗HIV活性を測定することにより、高い抗HIV−1プロテアーゼ活性又は抗HIV活性を有する誘導体を選択できる。抗HIV−1プロテアーゼ活性を有する場合、抗HIV−1プロテアーゼ剤として使用できる。抗HIV活性を有している場合には、抗HIV剤として使用できる。抗HIV剤を探索する場合、少なくとも1つの抗HIV活性を測定する必要があるが、本発明の誘導体については、抗HIV−1プロテアーゼ活性の測定もよい指標とすることができる。したがって、本発明の一つの態様として、これらの各種誘導体について、抗HIV−1プロテアーゼ活性の測定及び抗HIV活性の測定のうちいずれか1つを行うことにより、抗HIV剤を探索する方法を開示できる。また、当該探索工程を有することにより、結果として、抗HIV剤を生産できるため、当該探索工程を有する、抗HIV剤の生産方法も開示できる。
【0042】
これらの誘導体について、好ましい抗HIV−1プロテアーゼ活性測定方法及び抗HIV活性測定方法としては、例えば、以下の方法が挙げられる。
【0043】
抗HIV−1プロテアーゼ活性測定方法
抗HIV−1プロテアーゼ活性は、HIV−1プロテアーゼ活性測定に際し、阻害剤として各誘導体を添加し、それによるHIV−1プロテアーゼ活性の阻害程度を測定することで測定できる。抗HIV−1プロテアーゼ活性の測定方法としては、前記非特許文献1(Biosci. Biotechnol. Biochem., 62(3), 575−577, 1998)、非特許文献2(Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999)の他、従来公知の各種方法を採用することができる。
当該抗HIV−1プロテアーゼ活性は、従来公知の各種方法を採用できる。例えば、酢酸緩衝液とHIV−1プロテアーゼ溶液と各誘導体の各種濃度溶液とを合わせ、適当時間インキュベート後、HIV−1プロテアーゼの基質を加え、所定時間経過後に反応を停止し、基質分解物をHPLC等により測定し、誘導体を添加しない反応系をコントロールとしてHIV−1プロテアーゼ活性を求める方法を採用できる。
【0044】
抗HIV−1プロテアーゼ活性として、IC50(50%細胞変性抑制濃度)が高くとも100μg/ml以下のとき、有効な抗HIV−1プロテアーゼ剤といえる。IC50は、より好ましくは、50μg/ml以下であることが好ましく、より好ましくは、10μg/ml以下であり、さらに好ましくは5μg/ml以下である。
【0045】
抗HIV活性測定方法
また、抗HIV活性の測定方法としては、特開平7−206601号公報に記載の方法、前記非特許文献2(Biosci. Biotechnol. Biochem., 63(12), 2202−22−4, 1999)の他、従来公知の各種方法を採用できる。例えば、HIV感染感受性であって非感染細胞株にHIVを感染させた場合の細胞変性を利用する急性感性系による測定、HIV持続感染細胞株と非感染細胞株とを共存させたとき生じる細胞融合による巨細胞の形成を利用する巨細胞形成系における測定のいずれかあるいは双方を採用できる。これらの方法によれば、抗HIV−1プロテアーゼ活性による、あるいは当該抗HIV−1プロテアーゼ活性によらない抗HIV活性を測定できる。
なお、急性感性系による測定方法は、例えば、HIV感染について高い感受性を有するT細胞株であるMT−4細胞におけるHIV−1感染による細胞変性に対する抑制効果を指標とする。同時に、細胞毒性も指標とすることができる。また、巨細胞形成系による測定方法は、例えば、HIV−1が持続感染したMOLT/LAI細胞と非感染のMOLT−4細胞の混合培養を行い、非感染細胞の表面に存在するHIV−1レセプターが結合し、細胞融合が起こり、巨細胞が形成されることに対する抑制効果を指標とできる。
【0046】
抗HIV活性として、急性感染系における細胞変性を指標としたIC50は、高くとも100μg/ml以下であるとき、有効な抗HIV剤とすることができる。より好ましくは、50μg/ml以下であり、さらに好ましくは10μg/ml以下である。
また、抗HIV活性として、巨細胞系における巨細胞の形成抑制を指標としたIC50は、高くとも100μg/ml以下であるとき、有効な抗HIV剤とすることができる。より好ましくは、50μg/ml以下であり、さらに好ましくは10μg/ml以下である。
一方、これらの各系において同時に測定される50%細胞毒性濃度(CC50)は、10μg/ml以上であることが好ましく、50μg/ml以上であるとより好ましい。あるいは、SI(CC50÷IC50)が10以上であることが好ましく、より好ましくは30以上である。
【0047】
リグニン誘導体が抗HIV−1プロテアーゼ活性を有する場合には、例えば、そのまま、あるいは適当な細胞内取り込み化合物とともに細胞に付与することにより、細胞膜を通過させ、細胞内にて抗HIV−1プロテアーゼ活性を発揮させることができる。また、リグニン誘導体が、抗HIV活性を有する場合には、そのまま、あるいは、細胞内取り込みを容易化する化合物や細胞表面に親和性を付与する化合物とともに適用することができる。
抗HIV−1プロテアーゼ剤や抗HIV剤は、例えば、HIV感染防止剤、HIV増殖抑制剤として用いることができる。
HIV感染防止剤としては、例えば、医療用具や手術用具などを含む各種医療用具、避妊具などの衛生用品、及び治療室や手術室などの医療用領域に適用する感染防止剤としての用途がある。適用方法としては、散布、塗布、噴霧、浸漬等、必要に応じて選択することができる。これらに本剤を適用することで、HIV感染可能性のある用具や領域においてウイルス感染力を低下させ、他者への感染を防止できる。この場合、塗布、散布、浸漬用の溶液としては、5μg/ml〜500μg/mlの濃度とすることができる。より好ましくは、20μg/ml〜200μg/mlの濃度である。
【0048】
また、移植用臓器あるいは移植用組織の感染防止剤として使用できる。移植前の臓器あるいは組織に適用することで、用具や領域に付着した血液や体液などに由来して潜在可能性のあるHIVがこれらの用具等を介して他人に感染するのを防止できる。また、臓器等の提供者に由来してHIV感染可能性のある臓器あるいは組織におけるHIVを殺傷し、あるいは感染力を弱めて、移植による感染を予防することができる。この場合、塗布、散布、浸漬用の溶液としては、5μg/ml〜500μg/mlの濃度とすることができる。より好ましくは、20μg/ml〜200μg/mlの濃度である。
なお、輸血用血液や全血製剤や各種の血液成分製剤の感染防止剤としても使用できる。
【0049】
また、本抗HIV−1プロテアーゼ剤及び本抗HIV剤は、製剤学上許容される賦形剤等と組み合わされて、うがい剤、ローション剤、クリーム剤、ゲル剤、フィルム剤、坐剤や膣坐剤としての各種局所外用剤での感染防止剤としても使用することができる。感染予防剤としては、局所外用の場合、適時、局所に10〜1000μg適用することが好ましい。
【0050】
本リグニン誘導体は、水溶性であり、特にアルカリ処理により低分子化されている場合もある。したがって、経口あるいは腸管経由での投与形態であっても、感染可能性部位に到達させることも可能であり、各種経口剤あるいは腸管経由剤としての感染防止剤及び増殖抑制剤(治療剤)としても有用である。感染防止を目的とする場合には、好ましい用量としては、例えば、0.01〜10mg/kg/日とすることができる。また、HIV増殖抑制を目的とする場合には、好ましい用量としては、例えば、0.01〜100mg/kg/日とすることができる。
さらに、本剤は、経口あるいは腸管経由の栄養補助食品としても有用である。
【0051】
また、体内の局所へ注入する使用形態での抗HIV増殖抑制剤(治療剤)としても使用することができる。HIVはリンパ節等の組織に潜伏し、増殖を開始する。したがって、潜伏期間中、あるいは増殖開始後であっても、HIVの潜伏部位あるいは増殖部位に本剤を注入することにより、HIVの増殖を抑制することができる。この場合、適時、局所に1〜500μg/mlを適量注入することが好ましい。
また、本剤は、吸引治療用剤にも用いることができる。特に、肺におけるHIV増殖抑制に有用である。この場合の用量は、例えば、1〜500μg/kg/日とすることができる。
【0052】
【実施例】
以下、本発明を具体化して実施例について説明する。なお、これらの実施例は本発明を具体的に説明するものであって、本願発明を拘束するものではない。
(実施例1)
第2の各種一次誘導体(試料1〜3)の調製
スギ(Japanese cedar)脱脂木粉に含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、それぞれカテコール、レゾルシノール及びピロガロール3モルをアセトンに溶解させた。そのアセトン溶液にスギ脱脂木粉を一昼夜浸漬させ、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を木粉に収着させた。この収着木粉に72wt%硫酸(20ml/g脱脂木粉)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(10ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を取り除いた後、さらに、硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相を取り除いた。有機相に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相からフェノール:ベンゼン混液で抽出されなくなるまでこの洗浄を数回繰り返した。その後、硫酸相は大過剰の水で希釈(硫酸濃度約10wt%まで)され、透析(透析膜、分子量カットオフ;1000)にて未反応フェノール化合物と酸を取り除いた。透析後に再度生じた沈殿は、遠心分離にて取り除かれ、水溶性リグノフェノールを含む粗水溶液は凍結乾燥によって乾燥された。この乾燥物からメタノール抽出し、抽出液をジエチルエーテルに滴下、沈殿区分をそれぞれリグノカテコールIV、リグノレゾルシノールIV及びリグノピロガロールIV(試料1〜3)とした。
【0053】
(実施例2)
第2の各種水溶性リグニン二次誘導体の調製(試料4〜6)
実施例1で調製した各試料1〜3各約500mgを耐圧管(直径12mm×長さ60mm)に入れ、これに0.5N水酸化ナトリウム10ml、攪拌球(直径3mm)2個を入れ、オイルバス中で170℃、1時間処理した。処理後、冷却して内容物を0.5N塩酸にて中和し、遠心分離(20℃、10,000rpm、5分)で沈殿物と上澄み液とに分離させた。上澄み液を透析(透析膜、分子量カットオフ;1000)して脱塩した後、この時生じた沈殿物は再度遠心分離により取り除き、上澄み液を凍結乾燥、さらに五酸化二リン上で減圧乾燥し、リグノカテコールIV−b、リグノレゾルシノールIV−b及びリグノピロガロールIV−b(試料4〜6)とした。
【0054】
(実施例3)
第2の各種一次誘導体(試料7〜9)の調製
モミガラ、ケナフ(葉を除いた靭皮含む茎部)、タケ(葉を除いた茎部)の脱脂粉末試料を用い、それぞれに含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、ピロガロール3モルをアセトンに溶解させた。そのアセトン溶液に各試料を一昼夜浸漬させた後、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を試料に収着させた。この収着試料に65wt%硫酸(20ml/g脱脂試料)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(10ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を取り除いた後、さらに、硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相を取り除いた。有機相に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相からフェノール:ベンゼン混液で抽出されなくなるまでこの洗浄を数回繰り返した。その後、硫酸相は大過剰の水で希釈(硫酸濃度約10wt%まで)され、透析(透析膜、分子量カットオフ;1000)にて未反応フェノール化合物と酸を取り除いた。透析後に再度生じた沈殿は、遠心分離にて取り除かれ、水溶性リグノフェノールを含む粗水溶液は凍結乾燥によって乾燥された。この乾燥物を、メタノール抽出した液をジエチルエーテルに滴下し、沈殿区分をそれぞれリグノピロガロールIV−RH、リグノピロガロールIV−K及びリグノピロガロールIV−B(試料7〜9)とした。
【0055】
(比較例1)
リグノカテコールV II −b、リグノレゾルシノールV II −b及びリグノピロガロールV II −b(比較試料1〜3)の調製
スギ脱脂木粉に含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、それぞれカテコール、レゾルシノール及びピロガロール3モルをアセトンに溶解させた。そのアセトン溶液にスギ脱脂木粉を一昼夜浸漬させ、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を木粉に収着させた。この収着木粉に95wt%リン酸(20ml/g脱脂木粉)を加えて50℃、1時間、攪拌した。攪拌後、リン酸濃度を10%まで水で希釈して、さらに1時間ホモジナイザーにより攪拌した。これを遠心分離(25℃、10,000rpm、5分)し、沈殿物を脱イオン水で繰り返し洗い、未反応フェノール化合物を除去、脱酸し、凍結乾燥した。この乾燥物をアセトン抽出し、その抽出残渣を乾燥させた。さらにアセトン残渣をメタノール抽出し、抽出液をジエチルエーテルに滴下して沈殿した区分を、それぞれリグノカテコールVII、リグノレゾルシノールVII及びリグノピロガロールVIIとした。
次いで、これら各約500mgを耐圧管(直径12mm×長さ60mm)に入れ、これに0.5N水酸化ナトリウム10ml、攪拌球(直径3mm)2個を入れ、オイルバス中で170℃、1時間処理した。処理後、冷却して内容物を0.5N塩酸にて中和し、遠心分離(20℃、10,000rpm、5分)で沈殿物と上澄み液とに分離させた。上澄み液を透析(透析膜、分子量カットオフ;1000)して脱塩した後、この時生じた沈殿物は再度遠心分離により取り除き、上澄み液を凍結乾燥、さらに五酸化二リン上で減圧乾燥し、リグノカテコールVII−b、リグノレゾルシノールVII−b及びリグノピロガロールVII−b(比較試料1〜3)とした。
【0056】
(実施例4)
第1のカルボキシメチル化二次誘導体(試料10〜13)の調製
モミガラ、ケナフ(葉を除いた靭皮含む茎部)、タケ(葉を除いた茎部)、及びイネワラの脱脂粉末試料を用い、それぞれに含まれるリグニン基本単位(フェニルプロパン単位、C9)あたり、p−クレゾール3モルをアセトンに溶解させた。そのアセトン溶液に各試料を一昼夜浸漬させた後、アセトンを完全に留去、風乾することにより、それぞれのフェノール化合物を試料に収着させた。この収着試料に68wt%硫酸(4ml/g脱脂試料)を加えて30℃、1時間、攪拌した。攪拌後、有機相に可溶(水に不溶)なリグノフェノール及び過剰な未反応フェノール化合物を抽出するため、フェノール:ベンゼン(7:3,v/v)混液(2ml/g脱脂試料)を加え、再度、15分間攪拌し、遠心分離(30℃、3500rpm、10分)により2相に分離した。フェノール:ベンゼン相を大過剰のジエチルエーテルに滴下し、さらに、再度分離された硫酸相に少量のフェノール:ベンゼン混液を加え攪拌、遠心分離し、フェノール:ベンゼン相をジエチルエーテルに滴下した。フェノール:ベンゼン混液に可溶なリグノフェノール及び未反応フェノール化合物が硫酸相から抽出されなくなるまで同じ操作を数回繰り返した。フェノール:ベンゼン相をジエチルエーテルに滴下し生じた沈殿物を遠心分離によって回収後、これをアセトンに溶解させ、不溶解区分を遠心分離によって除去した。このアセトン抽出溶液をエバポレーターで濃縮後、大過剰のジエチルエーテルに滴下し、その不溶解沈殿物を遠心分離にて回収、さらに沈殿物を少量のジエチルエーテルにて遠心分離により洗浄し、五酸化リン上で減圧乾燥後、第1の一次誘導体として、それぞれリグノクレゾール−RH、リグノクレゾール−K、リグノクレゾール−B、リグノクレゾール−RSを得た。
これら第1の一次誘導体である各リグノクレゾール1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化二次誘導体として、それぞれリグノクレゾール−CM−RH、−CM−K、−CM−B、−CM−RS(試料10〜13)を得た。
【0057】
(実施例5)
第1のカルボキシメチル化二次及び三次誘導体(試料14〜16)の調製
ブナ(Beech)脱脂木粉からリグニン一次誘導体(リグノクレゾール)の調製アセトン脱脂したブナ木粉(Beech)にリグニンあたり3mol倍量のp−クレゾールを収着させた。この収着木粉に72%硫酸(4ml/g 脱脂木粉)を加え、1時間攪拌しながら30℃で反応させた後、反応系を大過剰の水に投入し、硫酸濃度を10%程度まで希釈した。その時、生じた不溶解沈殿物を遠心分離にて分離し、硫酸と未反応のクレゾールを取除くまで完全に洗浄し、五酸化リン上で減圧乾燥させた。これにアセトンを加え、一昼夜攪拌後、不溶物を遠心分離にて取除き、エバポレーターで濃縮後、大過剰のジエチルエーテルに滴下し、その不溶解沈殿物を遠心分離、洗浄し、第1の一次誘導体として、それぞれリグノクレゾールを得た。
これの第1の一次誘導体であるリグノクレゾール1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化二次誘導体として、それぞれリグノクレゾール−CM(試料14)を得た。
さらに、先の第1の一次誘導体であるリグノクレゾール20gを、0.5N水酸化ナトリウム水溶液400mlに完全溶解させ、これをオートクレーブに投入し、140℃(0.25MPa)および170℃(0.68MPa)にて1時間処理した。反応終了後、冷却し、1N塩酸にて中和した。沈殿物を遠心分離にて回収、水にて洗浄し、凍結乾燥後、アルカリ処理物を得た。
これら2種類のアルカリ処理物それぞれ1gを容器に取り、予めイソプロピルアルコール4gで分散させ、40%水酸化ナトリウム水溶液6.25gを加え、十分に静置した。これにイソプロピルアルコール12gを加え、この不均一混合溶液を攪拌下、50℃に維持し、モノクロロ酢酸(リグノクレゾール中の水酸基あたり約1.6mol)をイソプロピルアルコール4gに溶解させた溶液を1時間かけて添加し、その後さらに、50℃で2時間攪拌した。反応終了後、反応系は水酸化ナトリウムを含むリグニン二次誘導体の沈殿物と一部溶解した二次誘導体とを含むアルコール溶液に分離され、この沈殿物とアルコール溶液とをろ別し、アルコール溶液をエバポレータにて留去し、溶解物を乾固させた。次に、沈殿物を水に溶解し、これに少量の水で溶解させた乾固物を加え、1N塩酸にて中和した。このとき生じた沈殿物を遠心分離にて分離洗浄し、取り除き、上澄み液及び洗浄液を回収し、透析にて脱塩、凍結乾燥し、第1のカルボキシメチル化三次誘導体として、それぞれリグノクレゾール−CM−140及びリグノクレゾール−CM−170(試料15、16)を得た。
【0058】
(比較例2)
第1の一次誘導体のアルカリ水溶化体(リグノクレゾール−140及びリグノクレゾール−170(比較試料4、5)の調製
実施例5で調製した第1の一次誘導体であるリグノクレゾール、各20gを、0.5N水酸化ナトリウム水溶液400mlに完全溶解させ、これをオートクレーブに投入し、140℃(0.25MPa)および170℃(0.68MPa)にて1時間処理した。反応終了後、冷却し、1N塩酸にて中和した。上澄み液を遠心分離にて回収し、透析(膜の種類を教えて下さい)して脱塩後、凍結乾燥してアルカリ処理水溶化物である(リグノクレゾール−140及びリグノクレゾール−170(比較試料4、5))を得た。
【0059】
(実施例7)
1.抗HIV−1プロテアーゼ活性の測定
実施例1〜5及び比較例1〜2で調製した試料1〜16及び比較試料1〜5について、以下の条件で抗HIV−1プロテアーゼ活性を測定した。
0.5ml容のマイクロチューブに100mM酢酸緩衝液(200mM塩化ナトリウム、5mMジチオトレイトール(DTT)、10vol%グリセロール、pH4.9)100μL、25μg/mlのrecHIV−1プロテアーゼ(Escherichia coli)溶液100mM酢酸緩衝液(200mM塩化ナトリウム、5mMジチオトレイトール、10vol%グリセロール、5vol%エチレングリコール、pH5.5)20μL、及び各種濃度(5、10、12.5、50、μg/ml)の試料溶液30μLを加え、37℃で5分間インキュベートした後、1mg/ml基質溶液(HIV−1プロテアーゼ基質III;His−Lys−Ala−Val−Leu−p−nitro−Phe−Glu−Ala−Nle−Ser−NH2/H2O)10μLを加えた。反応液中の各濃度は以下の通りであった。
recHIV−1プロテアーゼ3.1μg/ml
基質62.5μg/ml
試料0.9、2.3、4.7、9.4、18.8μg/ml
63mM 酢酸緩衝液(125mM 塩化ナトリウム、3mM DTT、6vol%グリセロール)
【0060】
この反応液を37℃に保持し15分後に10%TFA10μLを加えて反応を終了させた。RecHIV−1プロテアーゼによって分解された基質分解物(His−Lys−Ala−Val−Leu−NH2又はp−nitro−Phe−Glu−Ala−Nle−Ser−NH2)の遊離量からrecHIV−1プロテアーゼの活性を換算するため、反応液を0.45μmのメンブランフィルターでろ過し、ろ液をHPLCに供した。PDA検出器により基質分解物の最大吸収272nmにおけるピーク強度を測定し、試料無添加系をコントロールとして各濃度でのrecHIV−1プロテアーゼ活性を求めた。なお、対照として、クラフトリグニン(アルドリッチ社製、製品番号;8068−05−1)についても試料と同様に試験した。また、公知のHIV−1プロテアーゼインヒビター(アスパラギン酸プロテアーゼインヒビター)であるペプスタチンA(Pepstain A)(シグマ社製、製品番号;P−5318)についても同様に試験した。結果を図6〜12に示す。
【0061】
2.抗HIV活性の測定
ヒトT細胞性白血病ウイルスI画他(HTLV−I)に持続感染しているT細胞株であるMT−4細胞に、ヒト免疫不全ウイルス1型(HIV−1、LAI株)を0.001TCID50/cellの割合で1時間感染させた後、正確に段階希釈(0.49μg/ml、0.98μg/ml、1.96μg/ml、3.91μg/ml、7.81μg/ml、15.62μg/ml、31.25μg/ml、62.5μg/ml、125μg/ml、250μg/ml、500μg/ml、1000μg/ml)した試料を含むRPMI−1640培養液(10%牛胎児血清、ペニシリンG100U/mlとストレプトマイシン100μg/mlを含む)に1.5×105cells/mlの濃度で浮遊させ、96穴の平底培養プレートに1ウェルあたり200μLで培養した。培養5日目に、鏡検によりHIV−1増殖による細胞変性効果(CPE)及び細胞の生育状態(細胞毒性)を観察し、細胞変性が認められる最低希釈濃度(IC)と細胞毒性が認められた最低希釈濃度(CC)とを求めた。なお、対照として、DS8000(デキストラン硫酸8000)とAZT(アジドチミジン)についても同様にICとCCとを求めた。結果を図13に示す。
【0062】
さらに、一部の試料(3、6、10、12、14)について、段階希釈の濃度を0.25〜250μg/mlとして、48穴の平底培養プレートにて1ウェルあたり600μLで培養する以外は上記条件と同様の条件で試験を行い、培養5日目に、生細胞数をトリパンブルー染色で計測し、50%細胞変性抑制濃度(IC50)と50%細胞毒性濃度(CC50)を算出し、加えてSIも算出した。結果を図14に示す。
【0063】
抗HIV−1プロテアーゼ活性については、図6〜12に示すように、試料1〜16においては、HIV−1プロテアーゼ活性を50%阻止濃度がおおよそ5μg/ml以下のものが、試料1〜3(図6)、4〜6(図7)、10〜13(図9)、14〜16(図10)及び対照のペプスタチンA(図12)であった。試料7〜9においては50%阻止濃度は20μg/mlを超えていた(図8)。
これに対し、比較試料1〜3(図11)においては、50%阻止濃度は、10μg/ml以下であった。また、比較試料4は約10μg/mlであり、比較試料5は20μg/mlを超えていた(図12)。対照のクラフトリグニンも20μg/mlを超えていた(図12)。
【0064】
抗HIV活性については、図13に示すように、試料1〜9(第2の誘導体群)においては、抗HIV活性は明瞭でなく、毒性との区別が付きにくかった。また、抗HIV−1プロテアーゼ活性において良好な結果を示した試料には細胞毒性が高いものがあることがわかった。例えば、試料1〜6については、レゾルシノール及びカテコール誘導のリグニン誘導体(1,2,4,5)は細胞毒性が高い傾向にあった。これに対し、試料10〜16(第1の誘導体群)については、抗HIV活性は明瞭であった。また、比較試料1〜5は、いずれも、抗HIV活性は不明瞭であった。
【0065】
さらに、図14に示すように、試料1〜6のうちピロガロール誘導のリグニン誘導体(試料2,4)と試料10〜16のうちモミガラ由来のp−クレゾール誘導のカルボキシメチル化誘導体(試料10)、タケ由来のp−クレゾール誘導体カルボキシメチル化誘導体(試料12)、ブナ由来のp−クレゾール誘導アルカリ処理−カルボキシメチル化誘導体(試料14)については、いずれも良好なIC50を示すとともに200μg/ml以上のCC50を示した。結果として高い選択性、すなわち、少なくとも30倍以上のSIを示した。
また、試料1〜16は、これら試験に供した試料と同程度の抗HIV活性を有し、構造類似であることから、試料1〜16も高い抗HIV活性と選択性を有し、硫酸化デキストランに匹敵する活性を示したと思われる。
【0066】
以上のことから、本発明の第1の誘導体及び第2の誘導体は、いずれも、有効な抗HIV剤として使用できることがわかった。
また、ピロガロールなどの3価のフェノール化合物により誘導された第2の誘導体、クレゾールなどの1価のフェノール化合物に誘導されカルボキシアルキル(メチル)化された第1の誘導体は、及び1価のフェノール化合物に誘導されアルカリ処理されカルボキシアルキル(メチル)化された第1の誘導体は、特に、有効な抗HIV剤として使用できることがわかった。
【0067】
【発明の効果】
本発明によれば、リグニン誘導体の一種であるリグノフェノール系誘導体を用いた抗HIV−1プロテアーゼ剤及び抗エイズウイルス剤を提供できる。
【図面の簡単な説明】
【図1】リグノフェノール系誘導体を得る構造変換プロセスの一例を示す図である。
【図2】図1に示す構造変換プロセスにおける構造変換の一例を示す図である。
【図3】オルト位結合ユニットとパラ位結合ユニットとを示す図である。
【図4】リグニン誘導体の相分離及び各種誘導体化のスキームを示す図である。
【図5】リグニン誘導体の相分離及び各種誘導体化のスキームを示す図である。
【図6】試料1〜3の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図7】試料4〜6の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図8】試料7〜9の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図9】試料10〜13の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図10】試料14〜16の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図11】比較試料1〜3の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図12】比較試料4〜5の抗HIV−1プロテアーゼ活性測定結果を示すグラフ図である。
【図13】試料1〜16及び比較試料1〜5の抗HIV活性の測定結果と使用したリグニン誘導体の由来、フェノール化合物の種類、用いた誘導体化手法等を併せて記載した表を示す図である。
【図14】試料3、6、10、12、14の抗HIV活性測定結果を示す図である。
Claims (10)
- 抗HIV剤であって、
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。 - 前記フェノール化合物は、1価のフェノール化合物である、請求項1記載の抗HIV剤。
- 前記カルボキシアルキル基におけるアルキル基はメチル基である、請求項1又は2記載の抗HIV剤。
- 前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、請求項1〜3のいずれかに記載の抗HIV剤。
- 抗HIV剤であって、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体、
(e)前記リグニン一次誘導体に対してアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
からなる群から選択される1種あるいは2種以上のリグニン誘導体を含有する、抗HIV剤。 - 前記2価以上のフェノール化合物は、カテコール、レゾルシノール、及びピロガロールからなる群から選択される1種あるいは2種以上である、請求項5記載の抗HIV剤。
- 前記リグニン含有材料は、木本類植物及び草本類植物から選択される1種あるいは2種以上のリグノセルロース系材料である、請求項5又は6に記載の抗HIV剤。
- 前記リグノセルロース系材料は、針葉樹由来である、請求項7記載の抗HIV剤。
- 請求項1〜8のいずれかに記載のリグニン誘導体を含有する抗HIV−プロテアーゼ剤。
- 抗HIV剤を製造する方法であって、以下の(a)〜(g);
(a)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体、
(b)(a)記載のリグニン一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体、
(c)フェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合して得られるリグニンの一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、
(d)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンの一次誘導体、及び
(e)(d)のリグニン一次誘導体をアルカリ処理して得られる水溶性のリグニン二次誘導体
(f)(d)記載の一次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化されたリグニン二次誘導体及び、
(g)2価以上のフェノール化合物により溶媒和されたリグニン含有材料に酸を添加し混合した反応液の酸性水溶性区分から分離して得られるリグニンのフェノール化合物の一次誘導体をアルカリ処理して得られる水溶性及び/又は水不溶性のリグニン二次誘導体に対して炭素数1〜5の低級アルキル基を備えるカルボキシアルキル基によりカルボキシアルキル化して得られるリグニン三次誘導体、からなる群から選択される1種あるいは2種以上のリグニン誘導体を調製する工程を有し、
これらのリグニン誘導体について以下の工程:
抗HIVプロテアーゼ活性を測定する工程、
HIV感染感受性の非感染細胞株を用いた急性感染系における抗HIV活性を測定する工程、
HIV持続感染細胞株とHIV感染感受性の非感染細胞株とを用いた巨細胞形成系において抗HIV活性を測定する工程、
から選択される1種あるいは2種以上の工程を行い、抗HIV活性を評価することを含む、方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003035642A JP2004244366A (ja) | 2003-02-13 | 2003-02-13 | リグニン誘導体を含有する抗hiv剤 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003035642A JP2004244366A (ja) | 2003-02-13 | 2003-02-13 | リグニン誘導体を含有する抗hiv剤 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004244366A true JP2004244366A (ja) | 2004-09-02 |
Family
ID=33021009
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003035642A Pending JP2004244366A (ja) | 2003-02-13 | 2003-02-13 | リグニン誘導体を含有する抗hiv剤 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004244366A (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007291287A (ja) * | 2006-04-27 | 2007-11-08 | Osaka Univ | アレルゲン抑制化合物 |
| JP2011256380A (ja) * | 2010-05-14 | 2011-12-22 | Mie Univ | リグニン系材料、その製造方法及びその利用 |
| JP2025036276A (ja) * | 2023-09-01 | 2025-03-14 | 広島県公立大学法人 | コンクリート組成物及びコンクリート硬化体並びにコンクリート補強材 |
-
2003
- 2003-02-13 JP JP2003035642A patent/JP2004244366A/ja active Pending
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007291287A (ja) * | 2006-04-27 | 2007-11-08 | Osaka Univ | アレルゲン抑制化合物 |
| JP2011256380A (ja) * | 2010-05-14 | 2011-12-22 | Mie Univ | リグニン系材料、その製造方法及びその利用 |
| JP2025036276A (ja) * | 2023-09-01 | 2025-03-14 | 広島県公立大学法人 | コンクリート組成物及びコンクリート硬化体並びにコンクリート補強材 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5795444B2 (ja) | マイクロ波化学処理に基づく高等植物及び真菌の多糖類抽出方法 | |
| JP2905289B2 (ja) | ウィルス感染の逆進行性治療の為の混合分子系 | |
| JPH08500589A (ja) | アストラガルス ポリサッカライド免疫調節剤 | |
| DK145505B (da) | Fremgangsmaade til fremstilling af et nitrogenholdigt polysaccharid | |
| CN101678057A (zh) | 从木材提取开环异落叶松树酯酚和二氢栎精的方法 | |
| JP2004238539A (ja) | リグニン系架橋体とその製造方法 | |
| FI84065C (fi) | Foerfarande foer framstaellning av isosilybinfri silibinin. | |
| EP3209300A1 (en) | Extract from a plant of the genus boswellia and related products and uses | |
| CN108272831A (zh) | 红松松针多酚类化合物的提取方法及其应用 | |
| JP2004244366A (ja) | リグニン誘導体を含有する抗hiv剤 | |
| JP2004244367A (ja) | リグノフェノール系誘導体を含有する哺乳動物細胞保護剤 | |
| SU1436875A3 (ru) | Способ получени производных силибинина | |
| JPH06503305A (ja) | 抗hiv−1剤としてのデキストリン硫酸とその組成体 | |
| CA1339111C (en) | Antiviral agent | |
| JPH03206043A (ja) | ウイルス感染防止剤 | |
| CN1111541C (zh) | 一种聚甘露糖醛酸硫酸盐 | |
| Salleh et al. | Radical scavenging activity of lignin extracted from oil palm empty fruit bunch and its effect on glutathione-S-transferase enzymes activity | |
| CA1335259C (en) | Composition of spent liquor from pulping process for antiviral medicine | |
| JPH03120223A (ja) | 抗ウイルス性医薬用組成物 | |
| US5632980A (en) | AIDS therapeutic agents comprising polymers formed from cinnamic acid derivatives | |
| KR101079042B1 (ko) | 항-hiv-1 저해 활성을 갖는 감태 유래 플로로글루시놀 유도체 | |
| JP2602715B2 (ja) | 抗ウィルス性医薬用組成物 | |
| JP4540133B2 (ja) | 抗AIDSウイルス活性を有する新規リン酸化β−グルカン及びそれを含むレトロウイルス感染症治療用薬剤 | |
| RU2499002C1 (ru) | Конъюгаты госсипола и натрийкарбоксиметилцеллюлозы, способы их получения и противовирусные средства на их основе | |
| KR20000070851A (ko) | Hiv 복제에 대한 억제력을 갖는 멜라닌 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20051227 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20061127 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090804 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20091222 |