JP2004346182A - 水性組成物、発泡シートおよび壁紙 - Google Patents
水性組成物、発泡シートおよび壁紙 Download PDFInfo
- Publication number
- JP2004346182A JP2004346182A JP2003144626A JP2003144626A JP2004346182A JP 2004346182 A JP2004346182 A JP 2004346182A JP 2003144626 A JP2003144626 A JP 2003144626A JP 2003144626 A JP2003144626 A JP 2003144626A JP 2004346182 A JP2004346182 A JP 2004346182A
- Authority
- JP
- Japan
- Prior art keywords
- polymerization
- emulsion
- pva
- ethylene
- wallpaper
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Paper (AREA)
Abstract
【課題】ホルムアルデヒド揮散などの環境負荷が少なく、かつ、着色が少なく、発泡性および耐ブロッキング性に優れ、さらには耐風化性に優れる水性組成物、発泡シートおよび壁紙を提供すること。
【解決手段】1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体を分散剤とし、ホルムアルデヒド濃度が1ppm以下であるエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)、発泡剤(B)および無機フィラー(C)からなる水性組成物、上記組成物から得られる発泡シートおよび壁紙。
【選択図】 なし
【解決手段】1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体を分散剤とし、ホルムアルデヒド濃度が1ppm以下であるエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)、発泡剤(B)および無機フィラー(C)からなる水性組成物、上記組成物から得られる発泡シートおよび壁紙。
【選択図】 なし
Description
【0001】
【発明の属する技術分野】
本発明は、水性組成物、発泡シートおよび壁紙に関する。さらに詳しくは、ホルムアルデヒドを、ほとんどまたは全く含有しない環境負荷の少ない水性組成物、および前記組成物を用いて得られる、着色がなく、発泡性および耐ブロッキング性に優れ、さらには耐風化性に優れる発泡シートおよび壁紙に関する。
【0002】
【従来の技術】
従来、発泡シートや壁紙の用途には、難燃性に優れるなどの利点を有していることから、塩化ビニルを含有する共重合体からなるエマルジョンが使用されている。ところが、塩化ビニルを含有する共重合体からなるエマルジョンを使用した発泡シートや壁紙は、焼却処分時に有害なダイオキシンを発生することから、その代替品が強く求められている。
【0003】
この問題を解決する方法として、アクリル樹脂系の装飾シート材に係る発明が開示されている(特許文献1)。しかしながらこのアクリル樹脂系の装飾シート材は、発泡性および耐折性の点において不満足なものであった。そこで、エチレンにバーサチック酸ビニルなどのビニルエステルを共重合して得られるエマルジョンを使用することにより、耐折性などが改善されたが(特許文献2)、その発泡性は未だ十分なものとはいえなかった。また、エチレン−酢酸ビニル共重合体系樹脂エマルジョンは、その調製時に重合開始剤としてソジウムホルムアルデヒドスルホキシレート(通称ロンガリット、以下ロンガリットと記述する)が頻用されており、ロンガリットは分解時にホルムアルデヒドを発生することから、該エマルジョンを用いた壁紙は、室内に用いた場合にシックハウス症候群を引き起こす恐れがある。このように、発泡シートや壁紙用途における塩化ビニルを含有する共重合体からなるエマルジョンに対して、充分な性能を有する代替品は未だに得られていないというのが実状である。さらに、従来の発泡シートや壁紙には年月の経過とともに風合が変化するなどの問題点があった。
【0004】
【特許文献1】
特開平7−108640号公報(特許請求の範囲)
【特許文献2】
特開2000−95915号公報(特許請求の範囲)
【0005】
【発明が解決しようとする課題】
本発明は、上記従来技術の欠点を解消するためになされたものであり、ホルムアルデヒド揮散などの環境負荷が少なく、かつ、着色が少なく、発泡性および耐ブロッキング性に優れ、さらには耐風化性に優れる水性組成物、発泡シートおよび壁紙を提供することを目的とするものである。
【0006】
【課題を解決するための手段】
本発明の上記目的は、1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体を分散剤とし、ホルムアルデヒド濃度が1ppm以下であるエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)、発泡剤(B)および無機フィラー(C)からなる水性組成物によって達成される。
【0007】
【発明の実施の形態】
本発明において、エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に分散剤として用いられる、1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体(以下、高1,2−グリコール結合含有PVAと略記する場合がある)の製造方法としては特に制限はなく、公知の方法が使用可能である。具体的には、1,2−グリコール結合量が上記の値になるようにビニレンカーボネートをビニルエステル系単量体と共重合した後、得られた重合体をけん化する方法、ビニルエステル系単量体の重合温度を通常の条件より高い温度、例えば75〜200℃として加圧下にビニルエステル系単量体を重合した後、得られた重合体をけん化する方法などが挙げられる。後者の方法においては、重合温度は95〜190℃であることが好ましく、100〜180℃であることが特に好ましい。また加圧条件としては、重合系が沸点以下になるように選択することが重要であり、好適には0.2MPa以上、さらに好適には0.3MPa以上である。また上限は5MPa以下が好適であり、さらに3MPa以下がより好適である。上記の重合はラジカル重合開始剤の存在下、塊状重合法、溶液重合法、懸濁重合法、乳化重合法などいずれの方法でも行うことができるが、溶液重合、特にメタノールを溶媒とする溶液重合法が好適である。このようにして得られたビニルエステル重合体を、メタノール系溶媒中でアルカリ触媒を用いてけん化するなど、従来公知のけん化方法でけん化することにより、目的とする高1,2−グリコール結合含有PVAが得られる。
上記の重合に用いられるビニルエステル系単量体としては、蟻酸ビニル、酢酸ビニル、プロピオン酸ビニル、ピバリン酸ビニルなどが挙げられるが、酢酸ビニルが経済的にみて好ましい。
【0008】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に分散剤として用いられるビニルアルコール系重合体の1,2−グリコール結合の含有量は、1.9モル%以上であることが必要であり、より好ましくは1.95モル%以上、さらに好ましくは2.0モル%以上、最適には2.1モル%以上である。1,2−グリコール結合の含有量が1.9モル%未満の場合、得られる発泡シートや壁紙の耐ブロッキング性が低下する懸念が生じる。また、1,2−グリコール結合の含有量は4モル%以下であることが好ましく、さらに好ましくは3.5モル%以下、最適には3.2モル%以下である。ここで、ビニルアルコール系重合体の1,2−グリコール結合の含有量はNMRスペクトルの解析から求められる。
【0009】
上記の高1,2−グリコール結合含有PVAの粘度平均重合度(以下重合度と略す)は、各種の状況に応じて選定すればよく、特に制限はないが、粉末化時の作業性の観点から100〜3000が好適であり、より好ましくは150〜2000、さらに好ましくは200〜1800である。一方、高1,2−グリコール結合含有PVAのけん化度も特に制限されないが、70〜99モル%であることが好ましく、80〜98モル%がより好ましく、83〜95モル%がさらに好ましい。
【0010】
また、上記の高1,2−グリコール結合含有PVAは、本発明の効果を損なわない範囲で共重合可能なエチレン性不飽和単量体を共重合したものでも良い。このようなエチレン性不飽和単量体としては、例えば、炭素数4以下のα−オレフィン(エチレン、プロピレン、ブチレン、イソブチレン)、アクリル酸、メタクリル酸、フマル酸、(無水)マレイン酸、イタコン酸、アクリロニトリル、メタクリロニトリル、アクリルアミド、メタクリルアミド、トリメチル−(3−アクリルアミド−3−ジメチルプロピル)−アンモニウムクロリド、アクリルアミド−2−メチルプロパンスルホン酸およびそのナトリウム塩、エチルビニルエーテル、ブチルビニルエーテル、塩化ビニル、臭化ビニル、フッ化ビニル、塩化ビニリデン、フッ化ビニリデン、テトラフルオロエチレン、ビニルスルホン酸ナトリウム、アリルスルホン酸ナトリウム、N−ビニルピロリドン、N−ビニルホルムアミド、N−ビニルアセトアミドなどのN−ビニルアミド類が挙げられる。
これらのエチレン性不飽和単量体の含有量は20モル%以下が好ましく、1〜20モル%の範囲内であることがより好ましい。これらのエチレン性不飽和単量体の中では、炭素数4以下のα−オレフィンが好適に用いられ、エチレンがより好適に用いられる。
また、チオール酢酸、メルカプトプロピオン酸などのチオール化合物の存在下でビニルエステル系単量体を重合するか、またはビニルエステル系単量体と上記エチレン性不飽和単量体とを共重合し、得られた(共)重合体をけん化することによって得られる末端変性物を用いることもできる。
【0011】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に、分散剤として用いられる高1,2−グリコール結合含有PVAの使用量については特に制限はないが、本発明の目的をより好適に達成するためには、分散質(エチレン−酢酸ビニル共重合体系樹脂)100重量部に対して好ましくは2〜15重量部、より好ましくは3〜10重量部の範囲である。高1,2−グリコール結合含有PVAの使用量が上記範囲内にあるとき、得られる発泡シートや壁紙の発泡性および耐ブロッキング性が向上する。
【0012】
本発明において、エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)は、主にエチレンおよび酢酸ビニルを乳化重合して得られるエマルジョンであるが、本発明の目的を損なわない範囲で、エチレンおよび酢酸ビニルと共重合可能なエチレン性不飽和単量体および/またはジエン系単量体を共重合したエマルジョンでもよい。エチレンおよび酢酸ビニルと共重合可能な単量体としては、プロピレン、イソブチレンなどのα−オレフィン、塩化ビニル、フッ化ビニル、ビニリデンクロリド、ビニリデンフルオリドなどのハロゲン化オレフィン、アクリル酸、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、アクリル酸2−エチルヘキシル、アクリル酸ドデシル、アクリル酸2−ヒドロキシエチルなどのアクリル酸およびそのエステル、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸2−エチルヘキシル、メタクリル酸2−エチルヘキシル、メタクリル酸ドデシル、メタクリル酸2−ヒドロキシエチルなどのメタクリル酸およびそのエステル、アクリル酸ジメチルアミノエチル、メタクリル酸ジメチルアミノエチルおよびこれらの四級化物、さらには、アクリルアミド、メタクリルアミド、N−メチロールアクリルアミド、N,N−ジメチルアクリルアミド、アクリルアミド−2−メチルプロパンスルホン酸およびそのナトリウム塩などのアクリルアミド系単量体、スチレン、α−メチルスチレン、p−メチルスチレンスルホン酸およびそのナトリウム塩またはカリウム塩などのスチレン系単量体、その他N−ビニルピロリドン、バーサチック酸ビニルなど、また、ブタジエン、イソプレン、クロロプレンなどのジエン系単量体、さらにはトリアリルシアヌレート、トリアリルイソシアヌレート、ジアリルフタレートなどの多官能性単量体が挙げられる。得られる発泡シートや壁紙に難燃性を付与する観点で、エチレンおよび酢酸ビニルに対してバーサチック酸ビニル、アクリル酸2−エチルヘキシル、メタクリル酸2−エチルヘキシルなどを共重合することがある。
【0013】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)の分散質中のエチレン含有量は、5〜50重量%が好ましく、さらに好ましくは7〜40重量%、最適には10〜30重量%である。エチレンの含有量が上記範囲内にあるとき、得られる発泡シートや壁紙の発泡性および耐ブロッキング性が向上する。
【0014】
本発明に使用される上記エマルジョン(A)を製造する際、重合開始剤としては過酸化水素と酒石酸および/またはその金属塩からなるレドックス系重合開始剤を用いる。酒石酸には右旋性のL(+)酒石酸、左旋性のD(−)酒石酸、これら対掌体のラセミ化合物であるDL酒石酸があり、特に制限されないが、これらの中でもL(+)酒石酸を用いた場合、得られるエマルジョンの着色が少なく、さらに乳化重合コントロール性も良好である。また、酒石酸の金属塩を用いることも可能であり、金属の種類は特に制限されないが、通常、酒石酸ナトリウムが用いられる。中でもL(+)酒石酸ナトリウムが好ましく用いられる。L(+)酒石酸ナトリウムを用いた場合、上記利点に加えて、乳化重合後に通常行われるアンモニア、苛性ソーダなどのアルカリによるpH調整も不要となる長所がある。
過酸化水素の使用量は、全単量体100重量部に対して0.01〜0.2重量部であることが好適であり、さらに好適には0.02〜0.15重量部である。
【0015】
過酸化水素と酒石酸および/またはその金属塩の使用割合は特に制限されないが、通常過酸化水素100重量部に対して、酒石酸および/またはその金属塩が50〜300重量部、好ましくは70〜250重量部、より好ましくは80〜200重量部である。酒石酸および/またはその金属塩をこの範囲内で使用することにより、得られるエマルジョンの着色が少なく、また重合コントロール性も良好となる。
【0016】
本発明に使用される上記エマルジョン(A)を製造する際、乳化重合系のpHを3〜7、好ましくは4〜6に調整する。pHをこの範囲内に調整することにより、得られる発泡シートや壁紙の着色が少なく、その発泡性および耐ブロッキング性が良好となり、また重合コントロール性も良好となる。乳化重合系のpHの調整方法は特に制限されず、任意の緩衝剤を用いることが可能であるが、通常、酢酸ナトリウム、酢酸/酢酸ナトリウム系、水酸化ナトリウム、炭酸ナトリウムなどが好ましく用いられる。
本発明に使用される上記エマルジョン(A)を製造する際、乳化重合系のpHとは重合初期から重合終了までのpHを言い、重合のどの時点においてもpHが3〜7にあることが好適である。
【0017】
また、本発明に使用される上記エマルジョン(A)を製造する際には、乳化重合系に鉄化合物を添加することが好適である。鉄化合物としては特に制限されないが、塩化第一鉄、硫酸第一鉄、塩化第二鉄、硝酸第二鉄、硫酸第二鉄から選ばれる少なくとも1種の鉄化合物が好ましく用いられ、中でも塩化第一鉄、硫酸第一鉄が特に好ましく用いられる。
鉄化合物の使用量は特に制限されないが、全単量体に対して好ましくは1〜100ppm、より好ましくは5〜50ppmである。鉄化合物をこの範囲内で使用することにより、得られるエマルジョンの着色が少なく、また重合コントロール性も良好となる。
【0018】
本発明に使用される上記エマルジョン(A)を製造する際、上記のレドックス系重合開始剤の添加方法は特に制限されない。過酸化水素の添加方法としては、通常の乳化重合で行われる方法、すなわち、重合開始初期にショットで添加する方法、重合中に逐次的に添加する方法などが挙げられる。酒石酸および/またはその金属塩は、乳化重合初期にその全量を添加して用いてもよく、乳化重合中に逐次的に添加してもよいが、通常はその全量を乳化重合初期に添加して用いる。鉄化合物も、通常は乳化重合初期にその全量を添加して用いる。
【0019】
上記エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)の製造は、加圧下、好適には2〜7MPaの加圧下に行われるが、乳化重合途中で、例えば残存酢酸ビニル濃度が10重量%となったところでエチレンの一部を放出して、重合初期より圧力を0.5〜3.5MPa低下させた後、好適には1〜3MPa低下させた後に、重合を完結させることが好適である。
【0020】
また、本発明に使用される上記エマルジョン(A)を製造する際、乳化重合後に、0.004〜0.03MPa、好適には0.006〜0.025MPaの減圧下において、10〜70℃、好適には15〜60℃で、0.5〜5時間、好適には1〜4時間かけて脱エチレンを行うことで、より確実に未反応酢酸ビニルモノマー量を低減することが可能である。
【0021】
本発明に用いられる上記エマルジョン(A)に含まれるホルムアルデヒド濃度は1ppm以下であることが必須である。このようなエマルジョン(A)は上記の製造方法により好適に得られる。ここでエマルジョン(A)中のホルムアルデヒド濃度は、ガス検知管を用いて測定された数値をいう。エマルジョン(A)中に含まれるホルムアルデヒド濃度が1ppmを超える場合、いわゆるノンホルムアルデヒドエマルジョンということは出来ず、本発明の目的を達成することができない。
【0022】
本発明において、上記エマルジョン(A)はそのままで使用可能であるが、必要に応じて、本発明の効果を損なわない範囲で、従来公知の各種エマルジョンを添加して用いることができる。
また、本発明において上記エマルジョン(A)に用いられる分散剤は、上記した高1,2−グリコール結合含有PVAであるが、必要に応じて、従来公知のアニオン性、ノニオン性またはカチオン性の界面活性剤や、ヒドロキシエチルセルロースなどを併用することもできる。
【0023】
本発明において使用される発泡剤(B)としては、加熱、減圧などの操作により発泡が起こるものであれば特に制限はないが、熱分解型の発泡剤、熱膨張性中空微小球体などが挙げられ、特に熱膨張性中空微小球体が好ましく用いられる。熱分解型の発泡剤としては、ジニトロソペンタメチレンテトラミン、N,N’−ジメチル−N,N’−ジニトロソテレフタルアミド、ベンゼンスルホニルヒドラジド、p−トルエンスルホニルヒドラジド、p,p’−オキシビス(ベンゼンスルホニルヒドラジド)、3,3’−ジスルホニルヒドラジドジフェニルスルホン、1,3−ベンゼンジスルホニルヒドラジド、p−トルエンスルホニルセミカルバジド、アゾビスイソブチロニトリル、アゾジカルボンアミド、ジエチルアゾジカルボキシレートなどが挙げられる。
【0024】
熱膨張性中空微小球体とは、加熱により膨張・発泡させることができる微小球体からなる発泡剤であり、たとえばポリ塩化ビニリデン、塩化ビニリデンとアクリロニトリルの共重合体、ポリアクリロニトリル、アクリロニトリルとアクリル酸メチルの共重合体などからなる殻部分の内部にエタン、プロパン、ブタン、ペンタン、ヘキサン、ヘプタンなどの低沸点炭化水素を含有する粒径1〜50μmの球体である。市販品としては、たとえば松本油脂社製の商品名「マイクロスフェアF−85D」を用いることができる。
【0025】
本発明において使用される無機フィラー(C)としては、水酸化アルミニウム、水酸化マグネシウム、水酸化バリウム、炭酸カルシウム、炭酸マグネシウム、硫酸カルシウム、硫酸バリウム、水酸化第一鉄、塩基性炭酸亜鉛、塩基性炭酸鉛、珪砂、クレー、タルク、シリカ類、二酸化チタン、ケイ酸マグネシウムなどが挙げられる。好ましくは水酸化アルミニウム、水酸化マグネシウム、炭酸カルシウム、炭酸マグネシウム、水酸化第一鉄、塩基性炭酸亜鉛、二酸化チタンである。
【0026】
上記エマルジョン(A)に対する発泡剤(B)の含有量は特に限定されないが、上記エマルジョン(A)100重量部(固形分基準)に対して好ましくは5〜50重量部、より好ましくは5〜40重量部である。発泡剤(B)が少なすぎる場合には得られる発泡シートや壁紙の発泡性が劣る懸念が生じ、一方、発泡剤(B)が多過ぎる場合には得られる発泡シートや壁紙の耐ブロッキング性が低下する懸念が生じる。
【0027】
また、上記エマルジョン(A)に対する無機フィラー(C)の含有量も特に限定されないが、上記エマルジョン(A)100重量部(固形分基準)に対して好ましくは50〜350重量部であり、さらに好ましくは80〜300重量部である。無機フィラー(C)が少なすぎる場合には、得られる発泡シートや壁紙の耐ブロッキング性が劣る懸念が生じ、一方、無機フィラー(C)が多すぎる場合には、得られる発泡シートや壁紙の発泡性が劣る懸念が生じる。
本発明の水性組成物は、エマルジョン(A)、発泡剤(B)および無機フィラー(C)の所定量を混合することにより得られる。
【0028】
本発明の発泡シートおよび壁紙を得る方法は特に限定されないが、たとえば、上記水性組成物をロールコーター、リバースロールコーター、ドクターコーターなどのコーティング方式やスクリーン印刷、グラビア印刷、彫刻ロール印刷、フレキソ印刷などの凹凸印刷方式を用いて紙に塗布または印刷し、乾燥後、発泡処理、エンボス加工を施すことなどにより得られる。
【0029】
本発明の水性組成物、発泡シートおよび壁紙は、ホルムアルデヒドをほとんどまたは全く含有しないので環境負荷が少なく、さらに着色が少なく、発泡性、耐ブロッキング性および耐風化性に優れる。そのために、本発明の壁紙は、環境配慮型の内装用壁紙などとして広範に好適に用いられる。
【0030】
【実施例】
次に、実施例および比較例により本発明をさらに詳細に説明する。なお、以下の実施例および比較例において「部」および「%」は、特に断らない限り重量基準を意味する。また、得られた組成物中の未反応酢酸ビニルモノマー量、ホルムアルデヒド含有量などの物性を下記の要領で評価した。
【0031】
(評価方法)
(1)ホルムアルデヒド含有量
10mLガラスバイアルにエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を0.1g採取し、40℃で1時間加温した後、ガス検知管(No.171SB:光明理化学工業製、100mL吸引)で測定した。
(2)発泡性
水性組成物を、乾燥後の厚さが0.15mmとなるように壁紙用基紙(防湿加工未処理紙)に塗布し、100℃で10分間乾燥させ、さらに180℃で1分間加熱することにより発泡させて、その発泡倍率を下式により評価した。
発泡倍率=(発泡体の厚さ)/(乾燥塗膜の厚さ:0.15mm)
(3)耐ブロッキング性
水性組成物を発泡させた、2つの発泡シートの塗膜面同士を重ねて合わせ、45℃、90%RH(相対湿度)の条件下、5kg/cm2の荷重をかけて24時間放置した。その後、発泡シート同士を剥がし、以下の基準により評価した。
○(抵抗なし)
△(抵抗あり)
×(材料破壊)
(4)発泡シートからのホルムアルデヒド放散量
JIS R3503(化学分析用ガラス器具)で規定された大きさ240mmのデシケータの底部に300mLの蒸留水を入れた容器をおき、その上に2.5cm×7.5cmに切り出した上記発泡シート10枚を支持金具に固定し、それぞれが接触しない様に20℃で24時間放置し、放散するホルムアルデヒドを蒸留水に吸収させて試料溶液とした。この試料溶液を用い、JAS木材編「普通合板」第19頁に示されるアセチルアセトン法によりホルムアルデヒド量を測定した。
(5)着色性
上記の発泡シートを空気雰囲気下、100℃で10分間放置し、その後発泡シートを目視により観察し、着色状態を以下の基準により判断した。
○(着色なし)
△(微黄色に着色)
×(黄色に着色)
(6)耐風化性
上記の発泡シートを40℃において3ケ月放置し、発泡状態の変化を以下の基準により評価した。
変化率(%)=[1−{(3ケ月後の発泡体の厚さ)/(発泡直後の発泡体の厚さ)}]×100
【0032】
製造例1
攪拌機、窒素導入口、開始剤導入口を備えた5L(リットル)加圧反応槽に酢酸ビニル2940g、メタノール60gおよび酒石酸0.088gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(シクロヘキサン−1−カルボニトリル)(V−40)をメタノールに溶解した濃度0.2g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を120℃に昇温した。このときの反応槽圧力は0.5MPaであった。上記の開始剤溶液2.5mLを注入し重合を開始した。重合中は重合温度を120℃に維持し、上記の開始剤溶液を10.0mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は0.5MPaであった。重合開始から3時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は24%であった。
【0033】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が25%となるように調整した。このポリ酢酸ビニルのメタノール溶液400g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)3.8gを添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約20分で系がゲル化した。ゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−1)を得た。得られたPVA(PVA−1)のけん化度は88モル%であった。
【0034】
また、重合後に未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を添加し、ポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ1700であった。
【0035】
上記の精製PVAを、そのけん化度が99.9モル%以上になるまでけん化した後、十分にメタノール洗浄を行い、次いで90℃減圧乾燥を2日間行った。得られたPVAをDMSO−D6に溶解し、トリフルオロ酢酸を数滴加えてプロトンNMR測定用の試料を調製した。500MHzのプロトンNMR測定装置(JEOL製;GX−500)を用い、80℃で該測定試料のプロトンNMRスペクトルを測定した。
ビニルアルコール単位のメチン由来のピークは3.2〜4.0ppm(積分値A)、1,2−グリコール結合の1つのメチン由来のピークは3.25ppm(積分値B)に帰属され、次式によりPVAの1,2−グリコール結合含有量を算出できる。
1,2−グリコール結合含有量(モル%)=B/A×100
上記の精製PVAの1,2−グリコール結合量は2.2モル%であった。
【0036】
製造例2
攪拌機、窒素導入口、開始剤導入口を備えた5L加圧反応槽に酢酸ビニル2850g、メタノール150gおよび酒石酸0.086gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(N−ブチル−2−メチルプロピオンアミド)をメタノールに溶解した濃度0.1g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を150℃に昇温した。このときの反応槽圧力は1.0MPaであった。上記の開始剤溶液15.0mLを注入し重合を開始した。重合中は重合温度を150℃に維持し、上記の開始剤溶液を15.8mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は1.0MPaであった。重合開始から4時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は35%であった。
【0037】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が25%となるように調整した。このポリ酢酸ビニルのメタノール溶液400g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)3.8gを添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約3分で反応系がゲル化した。このゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−2)を得た。得られたPVA(PVA−2)のけん化度は88モル%であった。
【0038】
また、重合後に未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を加えて、ポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ、1000であった。該精製PVAの1,2−グリコール結合量を500MHzプロトンNMR測定装置を用いて前述のとおり求めたところ、2.5モル%であった。
【0039】
製造例3
攪拌機、窒素導入口、開始剤導入口を備えた5L加圧反応槽に酢酸ビニル2700g、メタノール300gおよび酒石酸0.081gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(N−ブチル−2−メチルプロピオンアミド)をメタノールに溶解した濃度0.05g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を180℃に昇温した。このときの反応槽圧力は1.6MPaであった。上記の開始剤溶液0.4mLを注入し重合を開始した。重合中は重合温度を180℃に維持し、上記の開始剤溶液を10.6mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は1.6MPaであった。重合開始から4時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は27%であった。
【0040】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が30%となるように調整した。このポリ酢酸ビニルのメタノール溶液333g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)を3.8g添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約3分で反応系がゲル化した。このゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−4)を得た。得られたPVA(PVA−4)のけん化度は88モル%であった。
【0041】
また、重合後、未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を加えて、40℃でポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ、500であった。該精製PVAの1,2−グリコール結合量を500MHzプロトンNMR測定装置を用いて前述のとおり求めたところ、2.9モル%であった。
【0042】
製造例4
製造例1において、ポリ酢酸ビニルをけん化する際のアルカリモル比を0.013とした他は製造例1と同様にして、けん化度95モル%、重合度1700、1,2−グリコール結合量2.2モル%のPVA−5を得た。
【0043】
実施例1
窒素吹き込み口、温度計、撹拌機を備えた耐圧50LオートクレーブにPVA−1(1,2−グリコール結合量2.2モル%、重合度1700、けん化度88モル%)1061g、イオン交換水19440g、L(+)酒石酸8.3g、酢酸ナトリウム10g、塩化第一鉄0.4gを仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して4.4MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を2MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.8であった。10%水酸化ナトリウム水溶液を120g添加してエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度54.4%、エチレン含量20重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。
【0044】
このエマルジョン184部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。表1中、Etはエチレン−酢酸ビニル共重合体系樹脂中のエチレン含有量を示す。また、分散剤の量(phm)は、エチレンと酢酸ビニルの合計量(エチレン−酢酸ビニル共重合体系樹脂の重量)100重量部に対する分散剤の重量部を示す。
【0045】
実施例2
実施例1においてPVA−1の代わりにPVA−2(1,2−グリコール結合量2.5モル%、重合度1000、けん化度88モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0046】
比較例1
実施例1においてL(+)酒石酸の代わりにロンガリットを、PVA−1の代わりにPVA−3{重合度1700、けん化度88モル%:(株)クラレ製PVA−217}を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0047】
実施例3
実施例1においてPVA−1の代わりにPVA−4(1,2−グリコール結合量2.9モル%、重合度500、けん化度88モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0048】
実施例4
実施例1においてPVA−1の代わりにPVA−5(1,2−グリコール結合量2.2モル%、重合度1700、けん化度95モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0049】
実施例5
窒素吹き込み口、温度計、撹拌機を備えた耐圧60LオートクレーブにPVA−1を4063g、イオン交換水を25064g、L(+)酒石酸を8.3g、酢酸ナトリウムを10g、塩化第一鉄を0.4g仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して4.4MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を2MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.8であった。10%水酸化ナトリウム水溶液を120g添加しエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度50.3%、エチレン含量19重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。このエマルジョン199部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。
【0050】
実施例6
窒素吹き込み口、温度計、撹拌機を備えた耐圧50LオートクレーブにPVA−1を1061g、イオン交換水を19440g、L(+)酒石酸を8.3g、酢酸ナトリウムを10g、塩化第一鉄を0.4g仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して2.5MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を1MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.7であった。10%水酸化ナトリウム水溶液を120g添加しエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度54%、エチレン含量7重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。このエマルジョン185部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。
【0051】
比較例2
実施例1において熱膨張性中空微小球体を用いなかった他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0052】
比較例3
実施例1において炭酸カルシウムを用いなかった他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0053】
実施例7
実施例1において熱膨張性中空微小球体を45部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0054】
実施例8
実施例1において炭酸カルシウムを60部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0055】
実施例9
実施例1において炭酸カルシウムを300部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0056】
【表1】
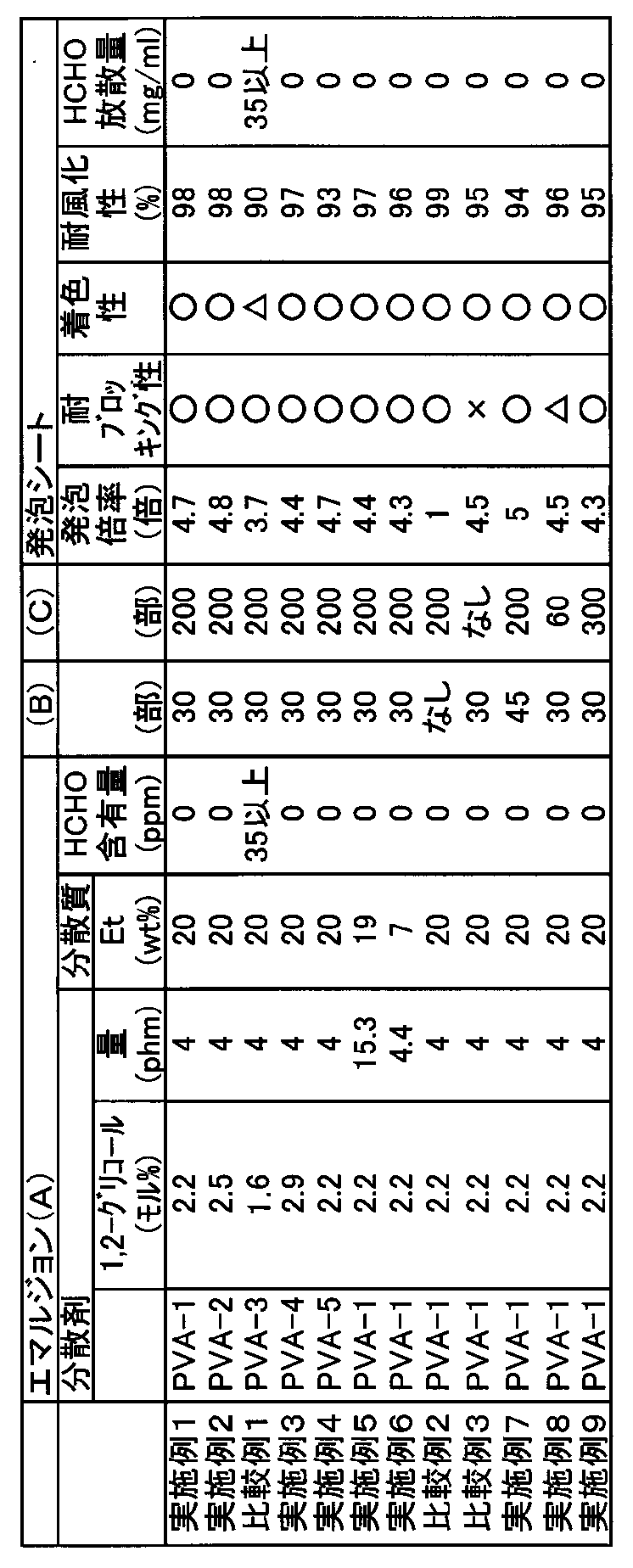 【0057】
【0057】
【発明の効果】
本発明の水性組成物、発泡シートおよび壁紙は、ホルムアルデヒドをほとんどまたは全く含有しないので、環境負荷が少なく、さらに着色が少なく、発泡性、耐ブロッキング性および耐風化性に優れる。そのため、環境配慮型の内装用壁紙などとして広範に好適に用いられる。
【発明の属する技術分野】
本発明は、水性組成物、発泡シートおよび壁紙に関する。さらに詳しくは、ホルムアルデヒドを、ほとんどまたは全く含有しない環境負荷の少ない水性組成物、および前記組成物を用いて得られる、着色がなく、発泡性および耐ブロッキング性に優れ、さらには耐風化性に優れる発泡シートおよび壁紙に関する。
【0002】
【従来の技術】
従来、発泡シートや壁紙の用途には、難燃性に優れるなどの利点を有していることから、塩化ビニルを含有する共重合体からなるエマルジョンが使用されている。ところが、塩化ビニルを含有する共重合体からなるエマルジョンを使用した発泡シートや壁紙は、焼却処分時に有害なダイオキシンを発生することから、その代替品が強く求められている。
【0003】
この問題を解決する方法として、アクリル樹脂系の装飾シート材に係る発明が開示されている(特許文献1)。しかしながらこのアクリル樹脂系の装飾シート材は、発泡性および耐折性の点において不満足なものであった。そこで、エチレンにバーサチック酸ビニルなどのビニルエステルを共重合して得られるエマルジョンを使用することにより、耐折性などが改善されたが(特許文献2)、その発泡性は未だ十分なものとはいえなかった。また、エチレン−酢酸ビニル共重合体系樹脂エマルジョンは、その調製時に重合開始剤としてソジウムホルムアルデヒドスルホキシレート(通称ロンガリット、以下ロンガリットと記述する)が頻用されており、ロンガリットは分解時にホルムアルデヒドを発生することから、該エマルジョンを用いた壁紙は、室内に用いた場合にシックハウス症候群を引き起こす恐れがある。このように、発泡シートや壁紙用途における塩化ビニルを含有する共重合体からなるエマルジョンに対して、充分な性能を有する代替品は未だに得られていないというのが実状である。さらに、従来の発泡シートや壁紙には年月の経過とともに風合が変化するなどの問題点があった。
【0004】
【特許文献1】
特開平7−108640号公報(特許請求の範囲)
【特許文献2】
特開2000−95915号公報(特許請求の範囲)
【0005】
【発明が解決しようとする課題】
本発明は、上記従来技術の欠点を解消するためになされたものであり、ホルムアルデヒド揮散などの環境負荷が少なく、かつ、着色が少なく、発泡性および耐ブロッキング性に優れ、さらには耐風化性に優れる水性組成物、発泡シートおよび壁紙を提供することを目的とするものである。
【0006】
【課題を解決するための手段】
本発明の上記目的は、1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体を分散剤とし、ホルムアルデヒド濃度が1ppm以下であるエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)、発泡剤(B)および無機フィラー(C)からなる水性組成物によって達成される。
【0007】
【発明の実施の形態】
本発明において、エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に分散剤として用いられる、1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体(以下、高1,2−グリコール結合含有PVAと略記する場合がある)の製造方法としては特に制限はなく、公知の方法が使用可能である。具体的には、1,2−グリコール結合量が上記の値になるようにビニレンカーボネートをビニルエステル系単量体と共重合した後、得られた重合体をけん化する方法、ビニルエステル系単量体の重合温度を通常の条件より高い温度、例えば75〜200℃として加圧下にビニルエステル系単量体を重合した後、得られた重合体をけん化する方法などが挙げられる。後者の方法においては、重合温度は95〜190℃であることが好ましく、100〜180℃であることが特に好ましい。また加圧条件としては、重合系が沸点以下になるように選択することが重要であり、好適には0.2MPa以上、さらに好適には0.3MPa以上である。また上限は5MPa以下が好適であり、さらに3MPa以下がより好適である。上記の重合はラジカル重合開始剤の存在下、塊状重合法、溶液重合法、懸濁重合法、乳化重合法などいずれの方法でも行うことができるが、溶液重合、特にメタノールを溶媒とする溶液重合法が好適である。このようにして得られたビニルエステル重合体を、メタノール系溶媒中でアルカリ触媒を用いてけん化するなど、従来公知のけん化方法でけん化することにより、目的とする高1,2−グリコール結合含有PVAが得られる。
上記の重合に用いられるビニルエステル系単量体としては、蟻酸ビニル、酢酸ビニル、プロピオン酸ビニル、ピバリン酸ビニルなどが挙げられるが、酢酸ビニルが経済的にみて好ましい。
【0008】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に分散剤として用いられるビニルアルコール系重合体の1,2−グリコール結合の含有量は、1.9モル%以上であることが必要であり、より好ましくは1.95モル%以上、さらに好ましくは2.0モル%以上、最適には2.1モル%以上である。1,2−グリコール結合の含有量が1.9モル%未満の場合、得られる発泡シートや壁紙の耐ブロッキング性が低下する懸念が生じる。また、1,2−グリコール結合の含有量は4モル%以下であることが好ましく、さらに好ましくは3.5モル%以下、最適には3.2モル%以下である。ここで、ビニルアルコール系重合体の1,2−グリコール結合の含有量はNMRスペクトルの解析から求められる。
【0009】
上記の高1,2−グリコール結合含有PVAの粘度平均重合度(以下重合度と略す)は、各種の状況に応じて選定すればよく、特に制限はないが、粉末化時の作業性の観点から100〜3000が好適であり、より好ましくは150〜2000、さらに好ましくは200〜1800である。一方、高1,2−グリコール結合含有PVAのけん化度も特に制限されないが、70〜99モル%であることが好ましく、80〜98モル%がより好ましく、83〜95モル%がさらに好ましい。
【0010】
また、上記の高1,2−グリコール結合含有PVAは、本発明の効果を損なわない範囲で共重合可能なエチレン性不飽和単量体を共重合したものでも良い。このようなエチレン性不飽和単量体としては、例えば、炭素数4以下のα−オレフィン(エチレン、プロピレン、ブチレン、イソブチレン)、アクリル酸、メタクリル酸、フマル酸、(無水)マレイン酸、イタコン酸、アクリロニトリル、メタクリロニトリル、アクリルアミド、メタクリルアミド、トリメチル−(3−アクリルアミド−3−ジメチルプロピル)−アンモニウムクロリド、アクリルアミド−2−メチルプロパンスルホン酸およびそのナトリウム塩、エチルビニルエーテル、ブチルビニルエーテル、塩化ビニル、臭化ビニル、フッ化ビニル、塩化ビニリデン、フッ化ビニリデン、テトラフルオロエチレン、ビニルスルホン酸ナトリウム、アリルスルホン酸ナトリウム、N−ビニルピロリドン、N−ビニルホルムアミド、N−ビニルアセトアミドなどのN−ビニルアミド類が挙げられる。
これらのエチレン性不飽和単量体の含有量は20モル%以下が好ましく、1〜20モル%の範囲内であることがより好ましい。これらのエチレン性不飽和単量体の中では、炭素数4以下のα−オレフィンが好適に用いられ、エチレンがより好適に用いられる。
また、チオール酢酸、メルカプトプロピオン酸などのチオール化合物の存在下でビニルエステル系単量体を重合するか、またはビニルエステル系単量体と上記エチレン性不飽和単量体とを共重合し、得られた(共)重合体をけん化することによって得られる末端変性物を用いることもできる。
【0011】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を製造する際に、分散剤として用いられる高1,2−グリコール結合含有PVAの使用量については特に制限はないが、本発明の目的をより好適に達成するためには、分散質(エチレン−酢酸ビニル共重合体系樹脂)100重量部に対して好ましくは2〜15重量部、より好ましくは3〜10重量部の範囲である。高1,2−グリコール結合含有PVAの使用量が上記範囲内にあるとき、得られる発泡シートや壁紙の発泡性および耐ブロッキング性が向上する。
【0012】
本発明において、エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)は、主にエチレンおよび酢酸ビニルを乳化重合して得られるエマルジョンであるが、本発明の目的を損なわない範囲で、エチレンおよび酢酸ビニルと共重合可能なエチレン性不飽和単量体および/またはジエン系単量体を共重合したエマルジョンでもよい。エチレンおよび酢酸ビニルと共重合可能な単量体としては、プロピレン、イソブチレンなどのα−オレフィン、塩化ビニル、フッ化ビニル、ビニリデンクロリド、ビニリデンフルオリドなどのハロゲン化オレフィン、アクリル酸、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、アクリル酸2−エチルヘキシル、アクリル酸ドデシル、アクリル酸2−ヒドロキシエチルなどのアクリル酸およびそのエステル、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸2−エチルヘキシル、メタクリル酸2−エチルヘキシル、メタクリル酸ドデシル、メタクリル酸2−ヒドロキシエチルなどのメタクリル酸およびそのエステル、アクリル酸ジメチルアミノエチル、メタクリル酸ジメチルアミノエチルおよびこれらの四級化物、さらには、アクリルアミド、メタクリルアミド、N−メチロールアクリルアミド、N,N−ジメチルアクリルアミド、アクリルアミド−2−メチルプロパンスルホン酸およびそのナトリウム塩などのアクリルアミド系単量体、スチレン、α−メチルスチレン、p−メチルスチレンスルホン酸およびそのナトリウム塩またはカリウム塩などのスチレン系単量体、その他N−ビニルピロリドン、バーサチック酸ビニルなど、また、ブタジエン、イソプレン、クロロプレンなどのジエン系単量体、さらにはトリアリルシアヌレート、トリアリルイソシアヌレート、ジアリルフタレートなどの多官能性単量体が挙げられる。得られる発泡シートや壁紙に難燃性を付与する観点で、エチレンおよび酢酸ビニルに対してバーサチック酸ビニル、アクリル酸2−エチルヘキシル、メタクリル酸2−エチルヘキシルなどを共重合することがある。
【0013】
エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)の分散質中のエチレン含有量は、5〜50重量%が好ましく、さらに好ましくは7〜40重量%、最適には10〜30重量%である。エチレンの含有量が上記範囲内にあるとき、得られる発泡シートや壁紙の発泡性および耐ブロッキング性が向上する。
【0014】
本発明に使用される上記エマルジョン(A)を製造する際、重合開始剤としては過酸化水素と酒石酸および/またはその金属塩からなるレドックス系重合開始剤を用いる。酒石酸には右旋性のL(+)酒石酸、左旋性のD(−)酒石酸、これら対掌体のラセミ化合物であるDL酒石酸があり、特に制限されないが、これらの中でもL(+)酒石酸を用いた場合、得られるエマルジョンの着色が少なく、さらに乳化重合コントロール性も良好である。また、酒石酸の金属塩を用いることも可能であり、金属の種類は特に制限されないが、通常、酒石酸ナトリウムが用いられる。中でもL(+)酒石酸ナトリウムが好ましく用いられる。L(+)酒石酸ナトリウムを用いた場合、上記利点に加えて、乳化重合後に通常行われるアンモニア、苛性ソーダなどのアルカリによるpH調整も不要となる長所がある。
過酸化水素の使用量は、全単量体100重量部に対して0.01〜0.2重量部であることが好適であり、さらに好適には0.02〜0.15重量部である。
【0015】
過酸化水素と酒石酸および/またはその金属塩の使用割合は特に制限されないが、通常過酸化水素100重量部に対して、酒石酸および/またはその金属塩が50〜300重量部、好ましくは70〜250重量部、より好ましくは80〜200重量部である。酒石酸および/またはその金属塩をこの範囲内で使用することにより、得られるエマルジョンの着色が少なく、また重合コントロール性も良好となる。
【0016】
本発明に使用される上記エマルジョン(A)を製造する際、乳化重合系のpHを3〜7、好ましくは4〜6に調整する。pHをこの範囲内に調整することにより、得られる発泡シートや壁紙の着色が少なく、その発泡性および耐ブロッキング性が良好となり、また重合コントロール性も良好となる。乳化重合系のpHの調整方法は特に制限されず、任意の緩衝剤を用いることが可能であるが、通常、酢酸ナトリウム、酢酸/酢酸ナトリウム系、水酸化ナトリウム、炭酸ナトリウムなどが好ましく用いられる。
本発明に使用される上記エマルジョン(A)を製造する際、乳化重合系のpHとは重合初期から重合終了までのpHを言い、重合のどの時点においてもpHが3〜7にあることが好適である。
【0017】
また、本発明に使用される上記エマルジョン(A)を製造する際には、乳化重合系に鉄化合物を添加することが好適である。鉄化合物としては特に制限されないが、塩化第一鉄、硫酸第一鉄、塩化第二鉄、硝酸第二鉄、硫酸第二鉄から選ばれる少なくとも1種の鉄化合物が好ましく用いられ、中でも塩化第一鉄、硫酸第一鉄が特に好ましく用いられる。
鉄化合物の使用量は特に制限されないが、全単量体に対して好ましくは1〜100ppm、より好ましくは5〜50ppmである。鉄化合物をこの範囲内で使用することにより、得られるエマルジョンの着色が少なく、また重合コントロール性も良好となる。
【0018】
本発明に使用される上記エマルジョン(A)を製造する際、上記のレドックス系重合開始剤の添加方法は特に制限されない。過酸化水素の添加方法としては、通常の乳化重合で行われる方法、すなわち、重合開始初期にショットで添加する方法、重合中に逐次的に添加する方法などが挙げられる。酒石酸および/またはその金属塩は、乳化重合初期にその全量を添加して用いてもよく、乳化重合中に逐次的に添加してもよいが、通常はその全量を乳化重合初期に添加して用いる。鉄化合物も、通常は乳化重合初期にその全量を添加して用いる。
【0019】
上記エチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)の製造は、加圧下、好適には2〜7MPaの加圧下に行われるが、乳化重合途中で、例えば残存酢酸ビニル濃度が10重量%となったところでエチレンの一部を放出して、重合初期より圧力を0.5〜3.5MPa低下させた後、好適には1〜3MPa低下させた後に、重合を完結させることが好適である。
【0020】
また、本発明に使用される上記エマルジョン(A)を製造する際、乳化重合後に、0.004〜0.03MPa、好適には0.006〜0.025MPaの減圧下において、10〜70℃、好適には15〜60℃で、0.5〜5時間、好適には1〜4時間かけて脱エチレンを行うことで、より確実に未反応酢酸ビニルモノマー量を低減することが可能である。
【0021】
本発明に用いられる上記エマルジョン(A)に含まれるホルムアルデヒド濃度は1ppm以下であることが必須である。このようなエマルジョン(A)は上記の製造方法により好適に得られる。ここでエマルジョン(A)中のホルムアルデヒド濃度は、ガス検知管を用いて測定された数値をいう。エマルジョン(A)中に含まれるホルムアルデヒド濃度が1ppmを超える場合、いわゆるノンホルムアルデヒドエマルジョンということは出来ず、本発明の目的を達成することができない。
【0022】
本発明において、上記エマルジョン(A)はそのままで使用可能であるが、必要に応じて、本発明の効果を損なわない範囲で、従来公知の各種エマルジョンを添加して用いることができる。
また、本発明において上記エマルジョン(A)に用いられる分散剤は、上記した高1,2−グリコール結合含有PVAであるが、必要に応じて、従来公知のアニオン性、ノニオン性またはカチオン性の界面活性剤や、ヒドロキシエチルセルロースなどを併用することもできる。
【0023】
本発明において使用される発泡剤(B)としては、加熱、減圧などの操作により発泡が起こるものであれば特に制限はないが、熱分解型の発泡剤、熱膨張性中空微小球体などが挙げられ、特に熱膨張性中空微小球体が好ましく用いられる。熱分解型の発泡剤としては、ジニトロソペンタメチレンテトラミン、N,N’−ジメチル−N,N’−ジニトロソテレフタルアミド、ベンゼンスルホニルヒドラジド、p−トルエンスルホニルヒドラジド、p,p’−オキシビス(ベンゼンスルホニルヒドラジド)、3,3’−ジスルホニルヒドラジドジフェニルスルホン、1,3−ベンゼンジスルホニルヒドラジド、p−トルエンスルホニルセミカルバジド、アゾビスイソブチロニトリル、アゾジカルボンアミド、ジエチルアゾジカルボキシレートなどが挙げられる。
【0024】
熱膨張性中空微小球体とは、加熱により膨張・発泡させることができる微小球体からなる発泡剤であり、たとえばポリ塩化ビニリデン、塩化ビニリデンとアクリロニトリルの共重合体、ポリアクリロニトリル、アクリロニトリルとアクリル酸メチルの共重合体などからなる殻部分の内部にエタン、プロパン、ブタン、ペンタン、ヘキサン、ヘプタンなどの低沸点炭化水素を含有する粒径1〜50μmの球体である。市販品としては、たとえば松本油脂社製の商品名「マイクロスフェアF−85D」を用いることができる。
【0025】
本発明において使用される無機フィラー(C)としては、水酸化アルミニウム、水酸化マグネシウム、水酸化バリウム、炭酸カルシウム、炭酸マグネシウム、硫酸カルシウム、硫酸バリウム、水酸化第一鉄、塩基性炭酸亜鉛、塩基性炭酸鉛、珪砂、クレー、タルク、シリカ類、二酸化チタン、ケイ酸マグネシウムなどが挙げられる。好ましくは水酸化アルミニウム、水酸化マグネシウム、炭酸カルシウム、炭酸マグネシウム、水酸化第一鉄、塩基性炭酸亜鉛、二酸化チタンである。
【0026】
上記エマルジョン(A)に対する発泡剤(B)の含有量は特に限定されないが、上記エマルジョン(A)100重量部(固形分基準)に対して好ましくは5〜50重量部、より好ましくは5〜40重量部である。発泡剤(B)が少なすぎる場合には得られる発泡シートや壁紙の発泡性が劣る懸念が生じ、一方、発泡剤(B)が多過ぎる場合には得られる発泡シートや壁紙の耐ブロッキング性が低下する懸念が生じる。
【0027】
また、上記エマルジョン(A)に対する無機フィラー(C)の含有量も特に限定されないが、上記エマルジョン(A)100重量部(固形分基準)に対して好ましくは50〜350重量部であり、さらに好ましくは80〜300重量部である。無機フィラー(C)が少なすぎる場合には、得られる発泡シートや壁紙の耐ブロッキング性が劣る懸念が生じ、一方、無機フィラー(C)が多すぎる場合には、得られる発泡シートや壁紙の発泡性が劣る懸念が生じる。
本発明の水性組成物は、エマルジョン(A)、発泡剤(B)および無機フィラー(C)の所定量を混合することにより得られる。
【0028】
本発明の発泡シートおよび壁紙を得る方法は特に限定されないが、たとえば、上記水性組成物をロールコーター、リバースロールコーター、ドクターコーターなどのコーティング方式やスクリーン印刷、グラビア印刷、彫刻ロール印刷、フレキソ印刷などの凹凸印刷方式を用いて紙に塗布または印刷し、乾燥後、発泡処理、エンボス加工を施すことなどにより得られる。
【0029】
本発明の水性組成物、発泡シートおよび壁紙は、ホルムアルデヒドをほとんどまたは全く含有しないので環境負荷が少なく、さらに着色が少なく、発泡性、耐ブロッキング性および耐風化性に優れる。そのために、本発明の壁紙は、環境配慮型の内装用壁紙などとして広範に好適に用いられる。
【0030】
【実施例】
次に、実施例および比較例により本発明をさらに詳細に説明する。なお、以下の実施例および比較例において「部」および「%」は、特に断らない限り重量基準を意味する。また、得られた組成物中の未反応酢酸ビニルモノマー量、ホルムアルデヒド含有量などの物性を下記の要領で評価した。
【0031】
(評価方法)
(1)ホルムアルデヒド含有量
10mLガラスバイアルにエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)を0.1g採取し、40℃で1時間加温した後、ガス検知管(No.171SB:光明理化学工業製、100mL吸引)で測定した。
(2)発泡性
水性組成物を、乾燥後の厚さが0.15mmとなるように壁紙用基紙(防湿加工未処理紙)に塗布し、100℃で10分間乾燥させ、さらに180℃で1分間加熱することにより発泡させて、その発泡倍率を下式により評価した。
発泡倍率=(発泡体の厚さ)/(乾燥塗膜の厚さ:0.15mm)
(3)耐ブロッキング性
水性組成物を発泡させた、2つの発泡シートの塗膜面同士を重ねて合わせ、45℃、90%RH(相対湿度)の条件下、5kg/cm2の荷重をかけて24時間放置した。その後、発泡シート同士を剥がし、以下の基準により評価した。
○(抵抗なし)
△(抵抗あり)
×(材料破壊)
(4)発泡シートからのホルムアルデヒド放散量
JIS R3503(化学分析用ガラス器具)で規定された大きさ240mmのデシケータの底部に300mLの蒸留水を入れた容器をおき、その上に2.5cm×7.5cmに切り出した上記発泡シート10枚を支持金具に固定し、それぞれが接触しない様に20℃で24時間放置し、放散するホルムアルデヒドを蒸留水に吸収させて試料溶液とした。この試料溶液を用い、JAS木材編「普通合板」第19頁に示されるアセチルアセトン法によりホルムアルデヒド量を測定した。
(5)着色性
上記の発泡シートを空気雰囲気下、100℃で10分間放置し、その後発泡シートを目視により観察し、着色状態を以下の基準により判断した。
○(着色なし)
△(微黄色に着色)
×(黄色に着色)
(6)耐風化性
上記の発泡シートを40℃において3ケ月放置し、発泡状態の変化を以下の基準により評価した。
変化率(%)=[1−{(3ケ月後の発泡体の厚さ)/(発泡直後の発泡体の厚さ)}]×100
【0032】
製造例1
攪拌機、窒素導入口、開始剤導入口を備えた5L(リットル)加圧反応槽に酢酸ビニル2940g、メタノール60gおよび酒石酸0.088gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(シクロヘキサン−1−カルボニトリル)(V−40)をメタノールに溶解した濃度0.2g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を120℃に昇温した。このときの反応槽圧力は0.5MPaであった。上記の開始剤溶液2.5mLを注入し重合を開始した。重合中は重合温度を120℃に維持し、上記の開始剤溶液を10.0mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は0.5MPaであった。重合開始から3時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は24%であった。
【0033】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が25%となるように調整した。このポリ酢酸ビニルのメタノール溶液400g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)3.8gを添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約20分で系がゲル化した。ゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−1)を得た。得られたPVA(PVA−1)のけん化度は88モル%であった。
【0034】
また、重合後に未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を添加し、ポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ1700であった。
【0035】
上記の精製PVAを、そのけん化度が99.9モル%以上になるまでけん化した後、十分にメタノール洗浄を行い、次いで90℃減圧乾燥を2日間行った。得られたPVAをDMSO−D6に溶解し、トリフルオロ酢酸を数滴加えてプロトンNMR測定用の試料を調製した。500MHzのプロトンNMR測定装置(JEOL製;GX−500)を用い、80℃で該測定試料のプロトンNMRスペクトルを測定した。
ビニルアルコール単位のメチン由来のピークは3.2〜4.0ppm(積分値A)、1,2−グリコール結合の1つのメチン由来のピークは3.25ppm(積分値B)に帰属され、次式によりPVAの1,2−グリコール結合含有量を算出できる。
1,2−グリコール結合含有量(モル%)=B/A×100
上記の精製PVAの1,2−グリコール結合量は2.2モル%であった。
【0036】
製造例2
攪拌機、窒素導入口、開始剤導入口を備えた5L加圧反応槽に酢酸ビニル2850g、メタノール150gおよび酒石酸0.086gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(N−ブチル−2−メチルプロピオンアミド)をメタノールに溶解した濃度0.1g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を150℃に昇温した。このときの反応槽圧力は1.0MPaであった。上記の開始剤溶液15.0mLを注入し重合を開始した。重合中は重合温度を150℃に維持し、上記の開始剤溶液を15.8mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は1.0MPaであった。重合開始から4時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は35%であった。
【0037】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が25%となるように調整した。このポリ酢酸ビニルのメタノール溶液400g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)3.8gを添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約3分で反応系がゲル化した。このゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−2)を得た。得られたPVA(PVA−2)のけん化度は88モル%であった。
【0038】
また、重合後に未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を加えて、ポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ、1000であった。該精製PVAの1,2−グリコール結合量を500MHzプロトンNMR測定装置を用いて前述のとおり求めたところ、2.5モル%であった。
【0039】
製造例3
攪拌機、窒素導入口、開始剤導入口を備えた5L加圧反応槽に酢酸ビニル2700g、メタノール300gおよび酒石酸0.081gを仕込み、室温下に窒素ガスによるバブリングをしながら反応槽圧力を2.0MPaまで昇圧して10分間放置した後、放圧するという操作を3回繰り返して系中を窒素置換した。開始剤溶液として2,2’−アゾビス(N−ブチル−2−メチルプロピオンアミド)をメタノールに溶解した濃度0.05g/L溶液を調製し、窒素ガスによるバブリングを行って窒素置換した。次いで上記の反応槽内温を180℃に昇温した。このときの反応槽圧力は1.6MPaであった。上記の開始剤溶液0.4mLを注入し重合を開始した。重合中は重合温度を180℃に維持し、上記の開始剤溶液を10.6mL/hrの割合で連続添加して重合を実施した。重合中の反応槽圧力は1.6MPaであった。重合開始から4時間後に反応槽を冷却して重合を停止した。このときの反応系の固形分濃度は27%であった。
【0040】
次いで、30℃減圧下にメタノールを時々添加しながら未反応酢酸ビニルモノマーの除去を行い、ポリ酢酸ビニルのメタノール溶液(濃度33%)を得た。得られたポリ酢酸ビニル溶液にメタノールを加えて濃度が30%となるように調整した。このポリ酢酸ビニルのメタノール溶液333g(溶液中のポリ酢酸ビニル100g)に、アルカリ溶液(NaOHの10%メタノール溶液)を3.8g添加して{ポリ酢酸ビニル中の酢酸ビニル単位に対するアルカリのモル比(MR)0.008}、40℃でポリ酢酸ビニルのけん化を行った。アルカリ添加後、約3分で反応系がゲル化した。このゲル化物を粉砕器にて粉砕し、1時間放置してけん化を進行させた後、酢酸メチル1000gを加えて残存するアルカリを中和した。フェノールフタレイン指示薬を用いて中和の終了を確認後、濾別して得られた白色固体状のPVAにメタノール1000gを加えて室温で3時間放置洗浄した。上記洗浄操作を3回繰り返した後、遠心脱液して得られたPVAを70℃の乾燥機中に2日間放置して乾燥PVA(PVA−4)を得た。得られたPVA(PVA−4)のけん化度は88モル%であった。
【0041】
また、重合後、未反応酢酸ビニルモノマーを除去して得られたポリ酢酸ビニルのメタノール溶液に、アルカリモル比が0.5となるようにアルカリ溶液を加えて、40℃でポリ酢酸ビニルのけん化を行った。ゲル化した反応物を粉砕し、60℃で5時間放置してけん化を進行させた後、メタノールによるソックスレー洗浄を3日間実施した。次いで80℃で3日間減圧乾燥を行って精製PVAを得た。該PVAの平均重合度を常法のJIS K6726に準じて測定したところ、500であった。該精製PVAの1,2−グリコール結合量を500MHzプロトンNMR測定装置を用いて前述のとおり求めたところ、2.9モル%であった。
【0042】
製造例4
製造例1において、ポリ酢酸ビニルをけん化する際のアルカリモル比を0.013とした他は製造例1と同様にして、けん化度95モル%、重合度1700、1,2−グリコール結合量2.2モル%のPVA−5を得た。
【0043】
実施例1
窒素吹き込み口、温度計、撹拌機を備えた耐圧50LオートクレーブにPVA−1(1,2−グリコール結合量2.2モル%、重合度1700、けん化度88モル%)1061g、イオン交換水19440g、L(+)酒石酸8.3g、酢酸ナトリウム10g、塩化第一鉄0.4gを仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して4.4MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を2MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.8であった。10%水酸化ナトリウム水溶液を120g添加してエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度54.4%、エチレン含量20重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。
【0044】
このエマルジョン184部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。表1中、Etはエチレン−酢酸ビニル共重合体系樹脂中のエチレン含有量を示す。また、分散剤の量(phm)は、エチレンと酢酸ビニルの合計量(エチレン−酢酸ビニル共重合体系樹脂の重量)100重量部に対する分散剤の重量部を示す。
【0045】
実施例2
実施例1においてPVA−1の代わりにPVA−2(1,2−グリコール結合量2.5モル%、重合度1000、けん化度88モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0046】
比較例1
実施例1においてL(+)酒石酸の代わりにロンガリットを、PVA−1の代わりにPVA−3{重合度1700、けん化度88モル%:(株)クラレ製PVA−217}を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0047】
実施例3
実施例1においてPVA−1の代わりにPVA−4(1,2−グリコール結合量2.9モル%、重合度500、けん化度88モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0048】
実施例4
実施例1においてPVA−1の代わりにPVA−5(1,2−グリコール結合量2.2モル%、重合度1700、けん化度95モル%)を用いた他は、実施例1と同様にして乳化重合を行い、エチレン−酢酸ビニル共重合体系樹脂エマルジョンを得た。得られたエマルジョンを用い、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0049】
実施例5
窒素吹き込み口、温度計、撹拌機を備えた耐圧60LオートクレーブにPVA−1を4063g、イオン交換水を25064g、L(+)酒石酸を8.3g、酢酸ナトリウムを10g、塩化第一鉄を0.4g仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して4.4MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を2MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.8であった。10%水酸化ナトリウム水溶液を120g添加しエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度50.3%、エチレン含量19重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。このエマルジョン199部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。
【0050】
実施例6
窒素吹き込み口、温度計、撹拌機を備えた耐圧50LオートクレーブにPVA−1を1061g、イオン交換水を19440g、L(+)酒石酸を8.3g、酢酸ナトリウムを10g、塩化第一鉄を0.4g仕込み、95℃で完全に溶解し、その後60℃に冷却し、窒素置換を行った。次に酢酸ビニル22360gを仕込んだ後、エチレンを導入して2.5MPaまで加圧し、0.4%過酸化水素水溶液1000gを5時間かけて圧入し、60℃で乳化重合を行った。重合初期の重合系のpHは5.2であった。残存酢酸ビニル濃度が10%となったところで、エチレンを放出してエチレン圧力を1MPaとし、3%過酸化水素水溶液50gを圧入して重合を完結させた。冷却後、重合系のpHを確認したところ、pH=4.7であった。10%水酸化ナトリウム水溶液を120g添加しエマルジョンのpHを5.5に調整し、60メッシュのステンレス製金網を用い、ろ過した。その結果、固形分濃度54%、エチレン含量7重量%のエチレン−酢酸ビニル共重合体系樹脂エマルジョンが得られた。このエマルジョン185部(固形分で100部)に熱膨張性中空微小球体(松本油脂社製マイクロスフェアF−85D)30部、炭酸カルシウム200部、水30部を加えてホモミキサーで分散させ、水性組成物を調製した。この組成物の評価を前述の方法により行った。結果を表1に示す。
【0051】
比較例2
実施例1において熱膨張性中空微小球体を用いなかった他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0052】
比較例3
実施例1において炭酸カルシウムを用いなかった他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0053】
実施例7
実施例1において熱膨張性中空微小球体を45部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0054】
実施例8
実施例1において炭酸カルシウムを60部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0055】
実施例9
実施例1において炭酸カルシウムを300部とした他は、実施例1と同様にして水性組成物を調製し、評価を実施例1と同様に行った。結果を併せて表1に示す。
【0056】
【表1】
【発明の効果】
本発明の水性組成物、発泡シートおよび壁紙は、ホルムアルデヒドをほとんどまたは全く含有しないので、環境負荷が少なく、さらに着色が少なく、発泡性、耐ブロッキング性および耐風化性に優れる。そのため、環境配慮型の内装用壁紙などとして広範に好適に用いられる。
Claims (4)
- 1,2−グリコール結合を1.9モル%以上有するビニルアルコール系重合体を分散剤とし、ホルムアルデヒド濃度が1ppm以下であるエチレン−酢酸ビニル共重合体系樹脂エマルジョン(A)、発泡剤(B)および無機フィラー(C)からなる水性組成物。
- 発泡剤が、熱膨張性中空微小球体である請求項1記載の水性組成物。
- 請求項1または2のいずれかに記載の水性組成物を用いて得られる発泡シート。
- 請求項3記載の発泡シートを用いて得られる壁紙。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003144626A JP2004346182A (ja) | 2003-05-22 | 2003-05-22 | 水性組成物、発泡シートおよび壁紙 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003144626A JP2004346182A (ja) | 2003-05-22 | 2003-05-22 | 水性組成物、発泡シートおよび壁紙 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004346182A true JP2004346182A (ja) | 2004-12-09 |
Family
ID=33532034
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003144626A Pending JP2004346182A (ja) | 2003-05-22 | 2003-05-22 | 水性組成物、発泡シートおよび壁紙 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004346182A (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008525569A (ja) * | 2004-12-22 | 2008-07-17 | アクゾ ノーベル エヌ.ブイ. | 化学組成物および方法 |
| JP2008525570A (ja) * | 2004-12-22 | 2008-07-17 | アクゾ ノーベル エヌ.ブイ. | 化学組成物および方法 |
| KR101223613B1 (ko) | 2009-10-16 | 2013-01-17 | 황규성 | 수성발포용액, 이를 이용하는 기재시트 발포코팅방법 및 그로 인해 제조되는 발포코팅 시트제품 |
| KR101243586B1 (ko) * | 2005-03-09 | 2013-03-20 | 가부시키가이샤 구라레 | 수성 에멀젼 및 코팅 |
| JP2018069714A (ja) * | 2016-11-04 | 2018-05-10 | 凸版印刷株式会社 | 発泡壁紙原反 |
| JP2019099933A (ja) * | 2017-11-30 | 2019-06-24 | 凸版印刷株式会社 | 発泡壁紙 |
| CN114907814A (zh) * | 2022-06-10 | 2022-08-16 | 浙江中天东方氟硅材料股份有限公司 | 发泡型陶瓷化隔热防火硅酮密封胶及其制备方法 |
-
2003
- 2003-05-22 JP JP2003144626A patent/JP2004346182A/ja active Pending
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008525569A (ja) * | 2004-12-22 | 2008-07-17 | アクゾ ノーベル エヌ.ブイ. | 化学組成物および方法 |
| JP2008525570A (ja) * | 2004-12-22 | 2008-07-17 | アクゾ ノーベル エヌ.ブイ. | 化学組成物および方法 |
| KR101243586B1 (ko) * | 2005-03-09 | 2013-03-20 | 가부시키가이샤 구라레 | 수성 에멀젼 및 코팅 |
| KR101223613B1 (ko) | 2009-10-16 | 2013-01-17 | 황규성 | 수성발포용액, 이를 이용하는 기재시트 발포코팅방법 및 그로 인해 제조되는 발포코팅 시트제품 |
| JP2018069714A (ja) * | 2016-11-04 | 2018-05-10 | 凸版印刷株式会社 | 発泡壁紙原反 |
| JP2019099933A (ja) * | 2017-11-30 | 2019-06-24 | 凸版印刷株式会社 | 発泡壁紙 |
| JP7183537B2 (ja) | 2017-11-30 | 2022-12-06 | 凸版印刷株式会社 | 発泡壁紙 |
| CN114907814A (zh) * | 2022-06-10 | 2022-08-16 | 浙江中天东方氟硅材料股份有限公司 | 发泡型陶瓷化隔热防火硅酮密封胶及其制备方法 |
| CN114907814B (zh) * | 2022-06-10 | 2024-05-17 | 浙江中天东方氟硅材料股份有限公司 | 发泡型陶瓷化隔热防火硅酮密封胶及其制备方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2432202C2 (ru) | Микросферы | |
| US11208538B2 (en) | Thermally expandable microspheres prepared from bio-based monomers | |
| JP2001502366A (ja) | 自己架橋性水性分散体 | |
| US20140087088A1 (en) | Process for producing foamed wallpapers | |
| CN101133117B (zh) | 水性乳液和涂料 | |
| JP2004346182A (ja) | 水性組成物、発泡シートおよび壁紙 | |
| JP5388424B2 (ja) | 水性エマルジョン及び接着剤 | |
| JP4128287B2 (ja) | 難燃性発泡シート用エマルジョン組成物および難燃性発泡シート | |
| US5446072A (en) | Emulsion compositions for flameproof foam sheet | |
| JPH09104798A (ja) | 壁装飾用熱膨張性被覆組成物および凹凸模様を有する壁紙 | |
| JP2003171493A (ja) | 水性組成物、発泡シートおよび壁紙 | |
| JP2004189890A (ja) | 水性組成物、発泡シートおよび壁紙 | |
| JPH02140271A (ja) | 中空ポリマー顔料とこれを用いた塗料組成物 | |
| JP4810819B2 (ja) | 水性エマルジョン組成物及び発泡壁紙 | |
| JP3629369B2 (ja) | 水性エマルジョン組成物及び発泡シート | |
| JP4416898B2 (ja) | 木工用接着剤 | |
| JP3933528B2 (ja) | 発泡材料用エマルジョン、及び発泡材料用エマルジョン組成物 | |
| JP4416888B2 (ja) | 紙工用接着剤 | |
| JP3970101B2 (ja) | 発泡材料用エマルジョン、及び発泡材料用エマルジョン組成物 | |
| JPH111597A (ja) | 水性エマルジョン組成物 | |
| JP3774400B2 (ja) | エチレン−酢酸ビニル共重合体系樹脂エマルジョンの製造方法 | |
| JPH11166097A (ja) | 水性エマルジョン組成物 | |
| JPS6139354B2 (ja) | ||
| JPH09241466A (ja) | 乳化重合用分散安定剤 | |
| JP4578659B2 (ja) | 水性エマルジョン組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050809 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20071015 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071023 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20080219 |