JP2010143944A - イミダゾ[1,2−b]ピリダジン類の新規な製造法 - Google Patents
イミダゾ[1,2−b]ピリダジン類の新規な製造法 Download PDFInfo
- Publication number
- JP2010143944A JP2010143944A JP2010025306A JP2010025306A JP2010143944A JP 2010143944 A JP2010143944 A JP 2010143944A JP 2010025306 A JP2010025306 A JP 2010025306A JP 2010025306 A JP2010025306 A JP 2010025306A JP 2010143944 A JP2010143944 A JP 2010143944A
- Authority
- JP
- Japan
- Prior art keywords
- group
- lower alkyl
- alkyl group
- metal
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 CCCCN([*+])C=NS(c([n](c(cc1)n2)nc1Cl)c2Cl)(=O)=O Chemical compound CCCCN([*+])C=NS(c([n](c(cc1)n2)nc1Cl)c2Cl)(=O)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N47/00—Biocides, pest repellants or attractants, or plant growth regulators containing organic compounds containing a carbon atom not being member of a ring and having no bond to a carbon or hydrogen atom, e.g. derivatives of carbonic acid
- A01N47/08—Biocides, pest repellants or attractants, or plant growth regulators containing organic compounds containing a carbon atom not being member of a ring and having no bond to a carbon or hydrogen atom, e.g. derivatives of carbonic acid the carbon atom having one or more single bonds to nitrogen atoms
- A01N47/28—Ureas or thioureas containing the groups >N—CO—N< or >N—CS—N<
- A01N47/36—Ureas or thioureas containing the groups >N—CO—N< or >N—CS—N< containing the group >N—CO—N< directly attached to at least one heterocyclic ring; Thio analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Environmental Sciences (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Plant Pathology (AREA)
- Pest Control & Pesticides (AREA)
- Agronomy & Crop Science (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
【解決手段】下記一般式(I)で表されるイミダゾ[1,2−b]ピリダジン化合物と有機金属化合物とを遷移金属触媒の存在下で反応させ、下記一般式(II)で表される6位に炭素原子を介する置換基を有するイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミド誘導体の製造法。
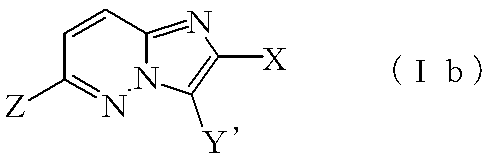
〔式(I)中、Xはハロゲン原子を、Yは水素原子を、Zはハロゲン原子またはOSO2R3を示す。〕
〔式(II)中、Rは低級アルキル基、低級アルキル基で置換されていてもよい低級シクロアルキル基、低級アルケニル基または低級アルキニル基を示す。〕
【選択図】なし
Description
即ち、本発明は、
(1)式(I):
式:
式(II):
(2)遷移金属触媒の金属がパラジウム、ニッケルまたは鉄である上記(1)記載の製造法、
(3)遷移金属触媒の金属がニッケルである上記(1)記載の製造法、
(4)有機金属化合物の金属がマグネシウムまたは亜鉛である上記(1)記載の製造法、
(5)Rが、低級アルキル基または低級アルキル基で置換されていてもよい低級シクロアルキル基である上記(1)記載の製造法、
(6)XおよびZが塩素原子である上記(1)記載の製造法、
(7)Yが水素原子で、且つRが低級アルキル基である上記(1)記載の製造法、
(8)有機金属化合物の金属がマグネシウムまたは亜鉛である上記(3)記載の製造法、
(9)有機金属化合物がハロゲン化低級アルキルマグネシウムまたはハロゲン化低級アルキル亜鉛である上記(8)記載の製造法、
(10)有機金属化合物がハロゲン化プロピルマグネシウムまたはハロゲン化プロピル亜鉛で、且つニッケル触媒が[1,3−ビス(ジフェニルホスフィノ)プロパン]ニッケル(II)ジクロリドまたはビス(トリフェニルホスフィン)ニッケル(II)ジクロリドである上記(9)記載の製造法、
(11)式(Ia):
式:
(12)式(Ib):
式:
本明細書において、低級アルキル基、低級アルケニル基および低級アルキニル基等における「低級」とは、これらのアルキル基等が1または2〜6個の炭素原子、好ましくは、1または2〜4個の炭素原子によって構成されていることをいう。例えば、直鎖状または分枝鎖状のC1-6アルキル基、C2-6アルケニル基およびC2−6アルキニル基が挙げられる。また、「低級シクロアルキル基」とは、炭素数が3〜7のC3−7シクロアルキル基をいう。
6−エチル−2−メチルイミダゾ[1,2−b]ピリダジンの合成
1H NMR (CDCl3,δ): 1.33 (3H,t,J=7.5 Hz), 2.48 (3H,s), 2.82 (2H,q,J=7.5 Hz), 6.87 (1H,d,J=9.2 Hz), 7.65 (1H,s), 7.72 (1H,d,J=9.2 Hz)
IR (Neat, cm-1): 2973, 2934, 2876, 1543, 1460, 1382, 1333, 1300, 1263, 1155, 1125, 1057, 1000, 820 ,726, 699
6−エチル−2−メチルイミダゾ[1,2−b]ピリダジン−3−スルホンアミドの合成
mp 215.0-215.5℃
1H NMR (DMSO-d6,δ): 1.30 (3H,t,J=7.5 Hz), 2.57 (3H,s), 2.93 (2H,q,J=7.5 Hz), 7.39 (1H,d,J=9.3 Hz), 7.47 (2H,brs), 8.08 (1H,d,J=9.3 Hz).
IR (Nujol, cm-1) : 3304, 3177, 3090, 1546, 1540, 1507, 1463, 1389, 1362, 1341, 1309, 1201, 1166, 1127, 1086, 1057, 959, 900, 9864, 824, 772, 686, 670, 652, 591, 525.
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
mp 73.9-80.0℃
1H NMR(CDCl3,δ): 1.01(3H, t, J=7.4 Hz), 1.78(2H, m), 2.79(2H, t, J=7.6 Hz), 6.96 (1H, d, J=9.3 Hz), 7.75(1H, d, J=9.3 Hz), 7.80(1H, s).
IR(Nujol, cm-1): 3122, 1466, 1377, 1314, 1302.
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジン−3−スルホンアミドの合成
mp 174-5℃
1H NMR (DMSO-d6,δ): 0.96 (3H,t,J=7.4 Hz), 1.7-1.9 (2H,m), 2.8-3.0 (2H,m), 7.53 (1H,d,J=9.5 Hz), 7.82 (2H,brs), 8.19 (1H,d,J=9.4 Hz)
IR (Nujol, cm-1): 3377, 3324, 3189, 1545, 1364, 1322, 1187, 1166, 821, 680, 597
6−n−ブチル−2−クロロイミダゾ[1,2−b]ピリダジンの合成
mp 61.0-63.0℃
1H NMR (CDCl3,δ): 0.96(3H, t, J=7.3 Hz),1.41(2H, tq, J=7.5, 7.3 Hz),1.73(2H, tt, J=7.8, 7.5 Hz), 2.81(2H, t, J=7.8 Hz), 6.96(1H, d, J=9.4 Hz), 7.74(1H, d, J=9.4 Hz), 7.79(1H, s).
IR(Nujol, cm-1): 3115, 3061, 1545, 1466, 1378, 1326, 1276, 817.
6−n−ブチル−2−クロロイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 165.5-166.5℃
1H NMR(DMSO-d6,δ): 0.93(3H, t, J=7.3 Hz), 1.37(2H, tq, J=7.5, 7.3 Hz), 1.72(2H, tt, J=7.9, 7.5 Hz), 2.93(2H, t, J=7.9 Hz), 7.53(1H, d, J=9.4 Hz), 7.80(2H, s), 8.18(1H, d, J=9.4 Hz).
IR(Nujol, cm-1): 3412, 3360, 3287, 3197, 1546, 1464, 1376, 1321, 1172.
N’−(2−クロロ−6−シクロプロピルイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジイソブチルホルムアミジンの合成
mp 154.0-160.0℃
1H NMR(CDCl3,δ): 0.74(6H, d, J=6.7 Hz), 0.95(6H, d, J=6.7 Hz), 1.00-1.10(2H, m), 1.10-1.25(2H, m), 1.85-2.10(2H, m), 2.10-2.20(1H, m), 3.19(2H, d, J=7.5 Hz), 3.28(2H, d, J=7.5 Hz), 6.98(1H, d, J=9.4 Hz), 7.78(1H, d, J=9.4 Hz), 8.45(1H, s).
IR(Nujol)ν(cm-1): 1613, 1464, 1334, 1318, 1143, 909, 859, 661.
2−クロロ−6−シクロプロピルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 194.0-196.0℃
NMR(DMSO-d6,δ): 1.10-1.25(4H, m), 2.30-2.45(1H, m), 7.36(1H, d, J=9.4 Hz), 7.78(2H, brs), 8.12(1H, d, J=9.4Hz).
IR(Nujol, cm-1): 3348, 3247, 1553, 1468, 1455, 1358, 1316, 1170, 908, 825, 662.
N’−(2−クロロ−6−エテニルイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジイソブチルホルムアミジンの合成
mp 194.0-198.0℃
1H NMR(CDCl3,δ): 0.71(6H, d, J=6.7 Hz), 0.94(6H, d, J=6.6 Hz), 1.85-2.10(2H, m), 3.17(2H, d, J=7.5 Hz), 3.26(2H, d, J=7.7 Hz), 5.77(1H, d, J=11.1 Hz), 6.16(1H, d, J=17.8 Hz), 6.82(1H, dd, J=17.8, 11.1 Hz), 7.46(1H, d, J=9.5 Hz), 7.89(1H, d, J=9.5 Hz), 8.50(1H, s).
IR(Nujol, cm-1): 1614, 1456, 1350, 1319, 1145, 913, 859, 664, 612.
2−クロロ−6−エテニルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 229.0-233.0℃
1H NMR(DMSO-d6,δ): 5.87(1H, d, J=11.2 Hz), 6.50(1H, d, J=17.9 Hz), 6.86(1H, dd, J=17.9, 11.2 Hz), 7.89(2H, s), 7.96(1H, d, J=9.6 Hz), 8.26(1H, d, J=9.6 Hz).
IR(Nujol, cm-1): 3316, 3183, 1466, 1368, 1321, 1167.
N’−(2−クロロ−6−(1−プロペニル)イミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジイソブチルホルムアミジンの合成
mp E,Z混合物の為未測定。
1H NMR(CDCl3,δ):[E体]0.72(6H, d, J=6.6 Hz), 0.94(6H, d, J=6.6 Hz), 1.85-2.10(2H, m), 2.00(3H, dd, J=6.9, 1.5 Hz), 3.17(2H, d, J=7.6 Hz), 3.26(2H, d, J=7.7 Hz), 6.51(1H, dq, J=16.0, 1.5 Hz), 6.71(1H, dq, J=16.0, 6.9 Hz), 7.35(1H, d, J=9.5 Hz), 7.82(1H, d, J=9.5 Hz), 8.50(1H, s).
1H NMR(CDCl3,δ):[Z体]0.72(6H, d, J=6.6 Hz), 0.92(6H, d, J=6.6 Hz), 1.85-2.10(2H, m), 2.21(3H, dd, J=7.3, 1.8Hz), 3.12(2H, d, J=7.5 Hz), 3.25(2H, d, J=7.7 Hz), 6.23(1H, dq, J=11.9, 7.3 Hz), 6.40(1H, dq, J=11.9, 1.8 Hz), 7.19(1H, d, J=9.5 Hz), 7.85(1H, d, J=9.5 Hz), 8.43(1H, s).
IR(Nujol, cm-1): 1609, 1456, 1351, 1319, 1144, 911.
(E)−2−クロロ−6−(1−プロペニル)イミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 225.0-229.0℃
1H NMR(DMSO-d6,δ): 1.98(3H, dd, J=6.8, 1.7 Hz), 6.71(1H, dq, J=16.0, 1.7 Hz), 7.01(1H, dq, J=16.0, 6.8 Hz), 7.83(2H, s), 7.84(1H, d, J=9.5 Hz), 8.19(1H, d, J=9.6 Hz).
IR(Nujol, cm-1): 3323, 3179, 1662, 1550, 1466, 1360, 1325, 1173.
2−クロロ−6−イソブチルイミダゾ[1,2−b]ピリダジンの合成
mp 71.0-72.5℃
1H NMR(CDCl3,δ): 0.98(6H, d, J=6.6 Hz), 2.09(1H, m), 2.68(2H, d, J=7.3 Hz), 6.94(1H, d, J=9.3 Hz), 7.75(1H, d, J=9.3 Hz), 7.81(1H,s).
IR(Nujol, cm-1): 3126, 3059, 1545, 1466, 1369, 1331, 1320, 1279, 803.
2−クロロ−6−イソブチルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 168.0-169.5℃
1H NMR(DMSO-d6,δ): 0.93(6H, d, J=6.6 Hz), 2.14(1H, m), 2.82(2H, d, J=7.4 Hz), 7.51(1H, d, J=9.4 Hz), 7.80(2H, s), 8.19(1H, d, J=9.4Hz).
IR(Nujol, cm-1): 3316, 3180, 3117, 1548, 1469, 1362, 1336, 1321, 1200, 1173, 849, 678.
2−クロロ−6−エチルイミダゾ[1,2−b]ピリダジンの合成
1H NMR(CDCl3,δ): 1.35(3H, t, J=7.6 Hz), 2.85(2H, q, J=7.6 Hz), 6.97(1H, d, J=9.3 Hz), 7.75(1H, d, J=9.3 Hz), 7.80(1H, s).
IR(Nujol, cm-1): 3121, 3058, 1544, 1471, 1318, 1280, 1262, 1189, 1142, 1121, 1059, 983, 953, 822.
2−クロロ−6−エチルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 204-205℃
1H NMR(DMSO-d6,δ): 1.31(3H, t, J=7.6 Hz), 2.95(2H, q, J=7.6 Hz), 7.54(1H, d, J=9.4 Hz), 7.82(2H, brs), 8.19(1H, d, J=9.4 Hz).
IR(Nujol, cm-1): 3317, 3211, 1365, 1356, 1325, 1172, 829, 668.
2−メチル−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
1H NMR(CDCl3,δ): 1.00(3H, t, J=7.4 Hz), 1.7-1.9(2H, m), 2.48(3H, d, J=0.7 Hz), 2.77(2H, t, J=7.5 Hz), 6.85(1H, d, J=9.2 Hz), 7.66(1H, d, J=0.7 Hz), 7.72(1H, d, J=9.2 Hz).
IR(Nujol, cm-1): 2961, 1541, 1464, 1326, 1296, 1153, 1124, 989, 816, 726.
2−メチル−6−n−プロピルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 178-179℃(dec.)
1H NMR(DMSO-d6,δ): 0.96(3H, t, J=7.3 Hz), 1.7-1.9(2H, m), 2.56(3H, s), 2.8-2.9(2H, m), 7.39(1H, d, J=9.3 Hz), 7.46(2H, brs), 8.08(1H, d, J=9.3 Hz).
IR(Nujol, cm-1): 3384, 3327, 1543, 1508, 1420, 1348, 1327, 1309, 1162, 827.
N,N−ジメチル−N’−(6−n−プロピル−2−トリフルオロメチルイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)ホルムアミジンの合成
mp 219.3-220.4℃
1H NMR(DMSO-d6,δ): 0.95(3H, t, J=7.3 Hz), 1.71(2H, m), 2.88(2H, t, J=7.7 Hz), 2.92(3H, s), 3.28(3H, s), 7.59(1H, d, J=9.5 Hz), 8.33(1H, d, J=9.5 Hz), 8.54(1H, s).
IR(Nujol, cm-1): 1635, 1334, 1318, 1169, 1153, 920, 619.
6−n−プロピル−2−トリフルオロメチルイミダゾ[1,2−b]ピリダジン−3−イルスルホンアミドの合成
mp 151.0-151.7℃
1H NMR(DMSO-d6,δ): 0.97(3H, t, J=7.3 Hz), 1.78(2H, m), 2.96(2H, t, J=7.7 Hz), 7.62(1H, d, J=9.5 Hz), 7.97(2H, brs), 8.36(1H, d, J=9.5 Hz).
IR(Nujol, cm-1): 3356, 1550, 1465, 1373, 1362, 1322, 1199, 1179, 1151, 608.
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
2−クロロ−6−n−プロピルイミダゾ[1,2−b]ピリダジンの合成
N’−(2,6−ジクロロイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジメチルホルムアミジンの合成
1H NMR(DMSO-d6,δ): 2.94(3H, s), 3.26(3H, s), 7.71(1H, d, J=9.5 Hz), 8.34(1H, d, J=9.5 Hz), 8.43(1H, s).
N’−(2,6−ジクロロイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジイソブチルホルムアミジンの合成
mp 151.0-154.0℃
1H NMR(CDCl3,δ): 0.76(6H, d, J=6.7 Hz), 0.97 (6H, d, J=6.7 Hz), 1.90-2.10(2H, m), 3.23(2H, d, J=7.6 Hz), 3.28(2H, d, J=7.7 Hz), 7.26(1H, d, J=9.5 Hz), 7.90(1H, d, J=9.5 Hz), 8.51(1H, s).
IR(Nujol, cm-1): 1615, 1456, 1324, 1311, 1146, 910, 858, 654.
N’−(6−クロロ−2−トリフルオロメチルイミダゾ[1,2−b]ピリダジン−3−イルスルホニル)−N,N−ジメチルホルムアミジンの合成
mp 203.7-205.0℃
1H NMR(DMSO-d6,δ): 2.95(3H, s), 3.29(3H, s), 7.79(1H, d, J=9.6 Hz), 8.47(1H, s), 8.52(1H, d, J=9.6 Hz).
IR(Nujol, cm-1): 1636, 1526, 1456, 1321, 1201, 1159, 1130, 1115, 922, 816, 628, 616.
Claims (11)
- 式(I):
式:
式(II):
- 遷移金属触媒の金属がパラジウム、ニッケルまたは鉄である請求項1記載の製造法。
- 遷移金属触媒の金属がニッケルである請求項1記載の製造法。
- 有機金属化合物の金属がマグネシウムまたは亜鉛である請求項1記載の製造法。
- Rが、低級アルキル基または低級アルキル基で置換されていてもよい低級シクロアルキル基である請求項1記載の製造法。
- XおよびZが塩素原子である請求項1記載の製造法。
- Rが低級アルキル基である請求項1記載の製造法。
- 有機金属化合物の金属がマグネシウムまたは亜鉛である請求項3記載の製造法。
- 有機金属化合物がハロゲン化低級アルキルマグネシウムまたはハロゲン化低級アルキル亜鉛である請求項8記載の製造法。
- 有機金属化合物がハロゲン化プロピルマグネシウムまたはハロゲン化プロピル亜鉛で、且つニッケル触媒が[1,3−ビス(ジフェニルホスフィノ)プロパン]ニッケル(II)ジクロリドまたはビス(トリフェニルホスフィン)ニッケル(II)ジクロリドである請求項9記載の製造法。
- 式(Ia):
式:
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010025306A JP5197645B2 (ja) | 2002-07-29 | 2010-02-08 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002219786 | 2002-07-29 | ||
| JP2002219786 | 2002-07-29 | ||
| JPPCT/JP03/00244 | 2003-01-15 | ||
| PCT/JP2003/000244 WO2003061388A1 (en) | 2002-01-18 | 2003-01-15 | Fused heterocyclic sulfonylurea compound, herbicide containing the same, and method of controlling weed with the same |
| JP2003085617 | 2003-03-26 | ||
| JP2003085617 | 2003-03-26 | ||
| JP2010025306A JP5197645B2 (ja) | 2002-07-29 | 2010-02-08 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004524110A Division JP4481818B2 (ja) | 2002-07-29 | 2003-07-16 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010143944A true JP2010143944A (ja) | 2010-07-01 |
| JP5197645B2 JP5197645B2 (ja) | 2013-05-15 |
Family
ID=34810057
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004524110A Expired - Lifetime JP4481818B2 (ja) | 2002-07-29 | 2003-07-16 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
| JP2010025306A Expired - Lifetime JP5197645B2 (ja) | 2002-07-29 | 2010-02-08 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004524110A Expired - Lifetime JP4481818B2 (ja) | 2002-07-29 | 2003-07-16 | イミダゾ[1,2−b]ピリダジン類の新規な製造法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US7307165B2 (ja) |
| JP (2) | JP4481818B2 (ja) |
| KR (1) | KR101027360B1 (ja) |
| CN (1) | CN100341875C (ja) |
| AT (1) | ATE459626T1 (ja) |
| DE (1) | DE60331558D1 (ja) |
| DK (1) | DK1541575T3 (ja) |
| ES (1) | ES2340582T3 (ja) |
| PT (1) | PT1541575E (ja) |
| TW (1) | TWI315309B (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8148524B2 (en) * | 2005-08-30 | 2012-04-03 | Sumitomo Chemical Company, Limited | Process for producing sulfonyl chloride compound |
| US7750000B2 (en) * | 2005-09-02 | 2010-07-06 | Bayer Schering Pharma Ag | Substituted imidazo[1,2b]pyridazines as kinase inhibitors, their preparation and use as medicaments |
| ES2315166B1 (es) * | 2006-04-28 | 2009-12-11 | Sumitono Chemical Company, Limited | Composicion herbicida. |
| GR20090100384A (el) * | 2009-07-08 | 2011-02-18 | Αχιλλεας Τσουκαλης | Αντλια ινσουλινης |
| WO2011114280A2 (en) | 2010-03-18 | 2011-09-22 | Basf Se | Fungicidal compositions comprising comprising a phosphate solubilizing microorganism and a fungicidally active compound |
| US8516150B2 (en) | 2011-04-11 | 2013-08-20 | Honeywell International Inc. | Systems and methods for multiple computer dataloading using a standard dataloader |
| CN106489973A (zh) * | 2015-09-07 | 2017-03-15 | 江苏龙灯化学有限公司 | 一种除草组合物 |
| CN106889098A (zh) * | 2017-03-10 | 2017-06-27 | 南京华洲药业有限公司 | 一种含丙嗪嘧磺隆与西草净的除草组合物及其应用 |
| CN119524806B (zh) * | 2024-10-29 | 2025-09-26 | 兰州城市学院 | 一种咪唑柱芳烃浸渍硅二氧化碳吸附材料的制备和应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6438091A (en) * | 1986-03-20 | 1989-02-08 | Takeda Chemical Industries Ltd | Condensed heterocyclic sulfonylurea |
| JPH01139582A (ja) * | 1987-08-31 | 1989-06-01 | Takeda Chem Ind Ltd | スルホニル尿素誘導体の製造方法 |
| JPH01316379A (ja) * | 1988-03-30 | 1989-12-21 | Takeda Chem Ind Ltd | 縮合複素環類の製造方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1329614A (zh) * | 1998-10-21 | 2002-01-02 | 武田药品工业株式会社 | 稠合的哒嗪衍生物、它们的制备方法和用途 |
| TWI327462B (en) | 2002-01-18 | 2010-07-21 | Sumitomo Chemical Co | Condensed heterocyclic sulfonyl urea compound, a herbicide containing the same, and a method for weed control using the same |
-
2003
- 2003-07-16 AT AT03771268T patent/ATE459626T1/de active
- 2003-07-16 CN CNB038179768A patent/CN100341875C/zh not_active Expired - Lifetime
- 2003-07-16 JP JP2004524110A patent/JP4481818B2/ja not_active Expired - Lifetime
- 2003-07-16 KR KR1020057001687A patent/KR101027360B1/ko not_active Expired - Lifetime
- 2003-07-16 ES ES03771268T patent/ES2340582T3/es not_active Expired - Lifetime
- 2003-07-16 DE DE60331558T patent/DE60331558D1/de not_active Expired - Lifetime
- 2003-07-16 US US10/522,798 patent/US7307165B2/en not_active Expired - Lifetime
- 2003-07-16 DK DK03771268.4T patent/DK1541575T3/da active
- 2003-07-16 PT PT03771268T patent/PT1541575E/pt unknown
- 2003-07-17 TW TW092119486A patent/TWI315309B/zh not_active IP Right Cessation
-
2010
- 2010-02-08 JP JP2010025306A patent/JP5197645B2/ja not_active Expired - Lifetime
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6438091A (en) * | 1986-03-20 | 1989-02-08 | Takeda Chemical Industries Ltd | Condensed heterocyclic sulfonylurea |
| JPH01139582A (ja) * | 1987-08-31 | 1989-06-01 | Takeda Chem Ind Ltd | スルホニル尿素誘導体の製造方法 |
| JPH01316379A (ja) * | 1988-03-30 | 1989-12-21 | Takeda Chem Ind Ltd | 縮合複素環類の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| JPN6009063274; 第4版 実験化学講座19 有機合成I-炭化水素・ハロゲン化合物- , 1992, 第104-110頁, 丸善株式会社 * |
| JPN6009063275; Tamao,K et al.,: Journal of the American Chemical Society Vol.94, No.12, 1972, pages 4374-4376 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4481818B2 (ja) | 2010-06-16 |
| KR20050025191A (ko) | 2005-03-11 |
| TW200404072A (en) | 2004-03-16 |
| ES2340582T3 (es) | 2010-06-07 |
| PT1541575E (pt) | 2010-05-05 |
| JPWO2004011466A1 (ja) | 2005-11-24 |
| US7307165B2 (en) | 2007-12-11 |
| CN1671707A (zh) | 2005-09-21 |
| DE60331558D1 (de) | 2010-04-15 |
| ATE459626T1 (de) | 2010-03-15 |
| TWI315309B (en) | 2009-10-01 |
| KR101027360B1 (ko) | 2011-04-11 |
| CN100341875C (zh) | 2007-10-10 |
| JP5197645B2 (ja) | 2013-05-15 |
| DK1541575T3 (da) | 2010-05-03 |
| US20050171108A1 (en) | 2005-08-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5197645B2 (ja) | イミダゾ[1,2−b]ピリダジン類の新規な製造法 | |
| TWI703132B (zh) | 用於製備二芳基硫乙內醯脲化合物之方法(一) | |
| US6114580A (en) | N-arylsulfilimine compounds and their use as catalysts in the preparation of N-arylarylsulfonamide compounds | |
| CN103772297B (zh) | 手性六元氮杂环卡宾前体化合物及其制备方法和应用 | |
| CA2940614A1 (en) | Naphthylamide compound, preparation method and use thereof | |
| CA3096963A1 (en) | A new method for synthesis of deuterated amide and deuterated sulfonamide | |
| BR112020025580A2 (pt) | processo de preparação de compostos tricíclicos | |
| JP2023171735A (ja) | 3,6-ジ置換イミダゾ[1,2-b]ピリダジン誘導体の製造方法 | |
| CN101479283A (zh) | 作为复分解反应催化剂的新型钌络合物 | |
| CN107973779A (zh) | 一种n-(2-吡啶/嘧啶基)吲哚衍生物的制备方法 | |
| CN108314658B (zh) | 一种多取代噁唑衍生物的制备方法 | |
| CN103380127B (zh) | 制备n-磺酰基取代的羟吲哚的方法 | |
| CN109180682B (zh) | 一种具有双环骨架的手性氮杂环卡宾前体化合物及其制备方法 | |
| EP1541575B1 (en) | Novel process for producing imidazo(1,2-b)pyridazine derivative | |
| CN102549002A (zh) | 金属配合物、吡啶基膦化合物及甲基丙烯酸烷基酯的制备方法 | |
| WO2015135438A1 (zh) | 一种卤代硫化合物及其制备方法和应用 | |
| CN102746327A (zh) | 肼化合物的制造方法、以及肼化合物的制造中间体及其制造方法 | |
| Jakobsen et al. | N-sulphonylformamidines; preparation and characterisation | |
| CN113024604B (zh) | 一种c3-烯基化的2-吡啶酮类衍生物的制备方法 | |
| CN101781260A (zh) | 一种含氮杂环胺类化合物及其制备方法 | |
| CN103204818A (zh) | 一种多取代喹唑啉衍生物的制备方法 | |
| CN119930511B (zh) | 温和条件下锰配合物催化合成n-杂环化合物的方法 | |
| CN115710286B (zh) | 一种顺式苯乙烯基硼衍生物的制备方法 | |
| WO2018023493A1 (zh) | 芳基腈类化合物的制备方法 | |
| CN115385948A (zh) | 螺双二氢苯并噻咯双噁唑啉类化合物、其制备方法及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120309 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120724 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120919 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20120919 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120919 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130122 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130205 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20160215 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5197645 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| EXPY | Cancellation because of completion of term |