JP2017196544A - 分離材及びカラム - Google Patents
分離材及びカラム Download PDFInfo
- Publication number
- JP2017196544A JP2017196544A JP2016086980A JP2016086980A JP2017196544A JP 2017196544 A JP2017196544 A JP 2017196544A JP 2016086980 A JP2016086980 A JP 2016086980A JP 2016086980 A JP2016086980 A JP 2016086980A JP 2017196544 A JP2017196544 A JP 2017196544A
- Authority
- JP
- Japan
- Prior art keywords
- group
- functional group
- separation material
- hydrophobic polymer
- polymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Landscapes
- Treatment Of Liquids With Adsorbents In General (AREA)
- Coating Of Shaped Articles Made Of Macromolecular Substances (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Abstract
Description
[1] 疎水性高分子粒子と、該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、上記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む、分離材。
[2] 上記水酸基以外の官能基が、カルボキシ基、エポキシ基、又はアミノ基である、[1]に記載の分離材。
[3] 水中での5%圧縮変形弾性率が70MPa以上である、[1]又は[2]に記載の分離材。
[4] カラムに充填した場合、カラム圧0.3MPaのときに通液速度が500cm/h以上である、[1]〜[3]のいずれかに記載の分離材。
[5] 上記疎水性高分子粒子が、スチレン系モノマに由来する構造単位を有する疎水性高分子を含む、[1]〜[4]のいずれかに記載の分離材。
[6] 上記官能基含有高分子が、多糖類又はその変性体に由来する構造を含む、[1]〜[5]のいずれかに記載の分離材。
[7] 上記官能基含有高分子が、アガロース又はその変性体に由来する構造を含む、[1]〜[6]のいずれかに記載の分離材。
[8] 上記官能基含有高分子が、架橋されている、[1]〜[7]のいずれかに記載の分離材。
[9] 上記疎水性高分子粒子の平均粒径が10〜500μmである、[1]〜[8]のいずれかに記載の分離材。
[10] 上記疎水性高分子粒子が多孔構造を有する、[1]〜[9]のいずれかに記載の分離材。
[11] [1]〜[10]のいずれかに記載の分離材を備えるカラム。
本実施形態の分離材は、疎水性高分子粒子と、該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、上記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む。なお、例えば、疎水性高分子粒子が多孔構造を有するような態様である場合において、「疎水性高分子粒子の表面」とは、疎水性高分子粒子の外側の表面のみでなく、疎水性高分子粒子の内部における細孔の表面を含むものとする。本実施形態の分離材によれば、タンパク質の非特異吸着が低減されると共に、カラムに充填したときに優れた通液性を有する。また、本実施形態の分離材は、耐久性、耐アルカリ性及び耐圧性にも優れ、カラムに充填したときの吸着量(動的吸着量)も実用上充分に高いと考えられる。
本実施形態に係る疎水性高分子粒子は、疎水性を有する高分子(以下、場合により「疎水性高分子」という)を含む粒子である。疎水性高分子粒子の製造方法に特に制限はないが、例えば、疎水性を有する高分子を形成可能なモノマを重合させる方法が挙げられる。モノマとしては、疎水性を有する高分子を形成可能なものであれば、特に限定されないが、例えば、スチレン系モノマが挙げられる。すなわち、上記疎水性高分子粒子は、例えば、スチレン系モノマに由来する構造単位を有する疎水性高分子を含んでいてもよい。
1)粒子又は分離材を、超音波分散装置を使用して水(界面活性剤等の分散剤を含む)に分散させ、1質量%の粒子又は分離材を含む分散液を調製する。
2)粒度分布計(シスメックスフロー、シスメックス株式会社製)を用いて、上記分散液中の粒子又は分離材約1万個の画像により平均粒径及び粒径のC.V.(変動係数)を測定する。
本実施形態に係る被覆層は、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む。なお、上記官能基含有高分子において、上記官能基は化学結合により高分子に結合している。被覆層がこのようなものであると、イオン交換性又はリガンドとの結合性が付与される。
本実施形態に係る被覆層は、例えば、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体を吸着させる工程(吸着処理工程)と、上記水酸基を有する高分子又はその変性体を架橋する工程(架橋処理工程)と、架橋された水酸基を有する高分子又はその変性体に官能基を導入する工程(官能基導入工程)と、を備える方法により形成できる。以下、これらの工程について、具体的に説明する。
吸着処理工程においては、例えば、水酸基を有する高分子又はその変性体の溶液に、疎水性高分子粒子を含浸させることにより、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体を吸着させる。これにより、疎水性高分子粒子表面に、水酸基を有する高分子又はその変性体が固定化される。水酸基を有する高分子又はその変性体の溶液の溶媒としては、水酸基を有する高分子又はその変性体を溶解することのできるものであれば、特に限定されないが、水が最も一般的である。溶媒に溶解させる高分子の濃度は、5〜20(mg/mL)が好ましい。
架橋処理工程においては、例えば、架橋剤を用いて、疎水性高分子粒子表面に吸着された水酸基を有する高分子又はその変性体を架橋反応させて、これらの架橋体を形成する。当該架橋体は、例えば、水酸基を有する3次元架橋網目構造を有する。
官能基導入工程は、上記架橋体に官能基を導入する工程である。上記架橋体に官能基を導入する方法に特に制限はなく、任意の方法を選択できる。上記架橋体に官能基を導入する方法としては、例えば、上記架橋体に官能基を直接導入する方法、及び上記架橋体が有する水酸基を介して官能基を導入する方法が挙げられる。官能基の導入方法に特に制限はなく、官能基の種類等により適宜選択できる。水酸基を介して官能基を導入する方法としては、例えば、ハロゲン化アルキル化合物を用いる方法が挙げられる。なお、水酸基を介して官能基を導入する場合、官能基は、架橋体が有する水酸基のうちの少なくとも一部に導入されていればよい。
微小圧縮試験機(Fisher製)を用いて、室温(20〜25℃)条件にて荷重負荷速度1mN/秒で、四角柱の平滑な端面(50μm×50μm)により、予め水中に浸漬させた分離材を50mNまで圧縮したときの荷重及び圧縮変位を測定する。得られた測定値から、分離材が5%圧縮変形したときの圧縮弾性率(5%K値)を下記式により求める。また、上記測定中の変位量が最も大きく変化する点の荷重を破壊強度(mN)とする。
5%K値(MPa)=(3/21/2)・F・S−3/2・R−1/2
F:分離材が5%圧縮変形したときの荷重(mN)
S:分離材が5%圧縮変形したときの圧縮変位(mm)
R:分離材の半径(mm)
(疎水性の多孔質高分子粒子の合成)
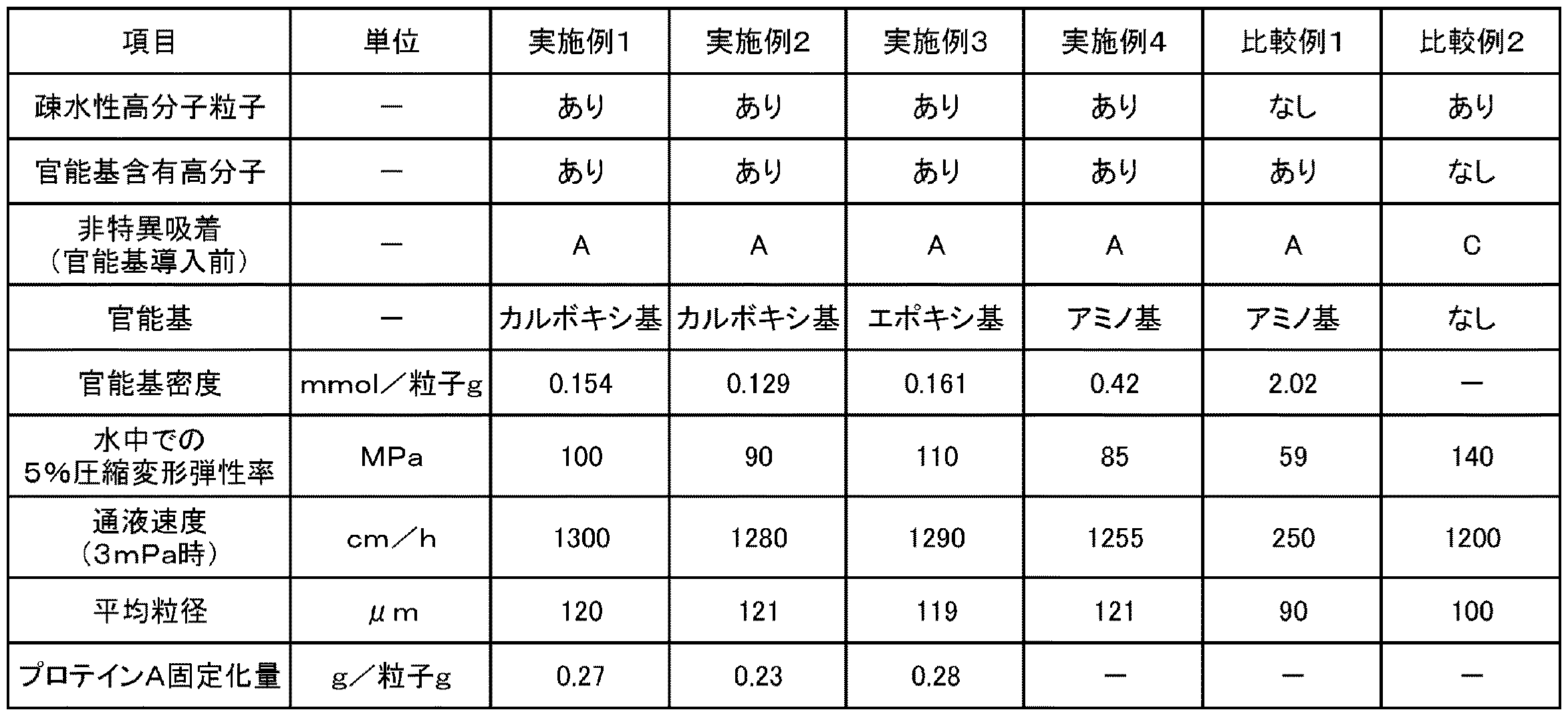
500mLの三口フラスコに、モノマとしての純度96%のジビニルベンゼン(新日鉄住金株式会社製、商品名:DVB960)16g、多孔質化剤としてのヘキサノール16g及びジエチルベンゼン16g、並びに開始剤としての過酸化ベンゾイル0.64gをポリビニルアルコール(0.5質量%)分散剤水溶液に混合して、混合液を得た。得られた混合液を、マイクロプロセスサーバーを使用して乳化した後、得られた乳化液をフラスコに移し、80℃のウォーターバスで加熱しながら、撹拌機を用いて約8時間撹拌して粒子を得た。得られた粒子をろ別して、更にアセトンで洗浄し、疎水性の多孔質高分子粒子1を得た。得られた多孔質高分子粒子1の粒径をフロー型粒径測定装置で測定し、平均粒径を算出した。結果を表1に示す。
アガロース水溶液(濃度2質量%)480mLに水酸化ナトリウム0.98g、及びグリシジルフェニルエーテル4.90gを投入して60℃で6時間反応させ、アガロースにフェニル基を導入した。得られた変性アガロースをイソプロピルアルコールで再沈殿させ、洗浄した。変性アガロースの疎水基含有量を下記方法により算出したところ、14.2%であった。
乾燥状態の粉末アガロース(変性されていないアガロース)と、揮発分0.1質量%未満まで乾燥させた変性アガロースとをそれぞれ70℃の純水に溶解させ、0.05質量%の水溶液サンプルを調製した。
・疎水基含有量(%)=(CAG/(CHAG+CAG))×100
・CAG:変性されているアガロース構成単位の濃度(mmol/L)
=A/εGPE×1000
・CHAG:変性されていないアガロース構成単位の濃度(mmol/L)
=(変性されてないアガロース構成単位の濃度(g/L)/アガロース構成単位(306g/mol))×1000
・A:疎水基導入アガロースの真の吸光度
=疎水基を導入したアガロースの吸光度−変性されていないアガロースの吸収
・εGPE:グリシジルフェニルエーテルの吸光係数
=1372(L/(mol・cm))
・変性されていないアガロースの吸収=変性されてないアガロースの吸光度×(変性アガロースのサンプル濃度(mmol/L)/変性されてないアガロースのサンプル濃度(mmol/L))
・変性されてないアガロース構成単位の濃度(g/L)=変性アガロースのサンプル濃度(質量%)×10−変性されているアガロース構成単位の濃度(g/L)
・変性されているアガロース構成単位の濃度(g/L)=(CAG×変性されているアガロース構成単位(456g/mol))/1000
20mg/mLの変性アガロース水溶液に多孔質高分子粒子1を70mL/粒子gの濃度で投入し、55℃で24h撹拌して、多孔質高分子粒子1に変性アガロースを吸着させた。次いで、変性アガロースを吸着させた多孔質高分子粒子1をろ別して、更に熱水で洗浄した。
多孔質高分子粒子1の表面及び細孔内部に吸着した変性アガロースを次のようにして架橋した。変性アガロースが吸着した粒子10gを0.4M水酸化ナトリウム水溶液に分散させ、0.02Mエピクロロヒドリンを添加し、24時間室温にて撹拌した。その後、2質量%の熱ドデシル硫酸ナトリウム水溶液で洗浄後、純水で更に洗浄し、乾燥させることで、変性アガロースの架橋体を被覆層として有する架橋体被覆粒子を得た。
得られた架橋体被覆粒子0.2gを、BSA(Bovine Serum Albumin)濃度24mg/mLのTris−塩酸緩衝液(pH8.0)20mLに加え、24時間室温で撹拌した。その後、遠心分離で上澄み液を採取し、分光光度計で上澄み液の280nmの吸光度を測定することによって求めた上澄み液のBSA濃度より、粒子に吸着したBSA量を算出した。粒子1mLあたりのBSA量吸着量が、1mg以下である場合を「A」、1mg超5mg未満である場合を「B」、5mg以上である場合を「C」として評価した。結果を表1に示す。
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄して、架橋体にエポキシ基を導入した。次いで、粒子に、水10mL、及び1Mのメルカプト酢酸ナトリウム水溶液10mLを加え、室温で24時間撹拌し、反応させた。反応終了後、粒子をろ別し、1000mLの水で洗浄して、架橋体にカルボキシ基を導入した。これにより本実施形態の分離材を得た。なお、本実施例における官能基導入剤は、メルカプト酢酸ナトリウムである。
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gに、0.1Nの塩酸水溶液20gを加え、室温で30分撹拌した。撹拌後の分離材を吸引してろ別し、さらに、水500mLで洗浄し、洗浄液のpHが5以上であることを確認した。洗浄後の分離材に0.5Mの塩化ナトリウム水溶液10gを加え、室温で30分撹拌した。撹拌後の分離材の懸濁液を0.01N水酸化ナトリウム水溶液でpH7を終点として滴定することにより、乾燥状態の分離材1g当たりのカルボキシ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
得られた分離材0.09gに、プロテインAを0.02g、WSC(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩)を0.02g、及びpH5.8に調整した50mMのPBSを2ml加えたのち、室温で24時間撹拌した。その後、粒子をろ別及び洗浄することによりリガンド固定化粒子を得た。
24時間撹拌後の反応溶液から粒子をろ別した後、所定濃度の粒子を含む試料を調製した。当該試料の吸光度(280nm)を測定し、予め作成した検量線により、リガンド固定化量を算出した。結果を表1に示す。
得られた分離材の水中での5%圧縮変形弾性率を以下のようにして算出した。結果を表1に示す。
F:分離材が5%圧縮変形したときの荷重(mN)
S:分離材が5%圧縮変形したときの圧縮変位(mm)
R:分離材の半径(mm)
得られた分離材を、濃度30質量%のスラリー(溶媒:メタノール)として、φ7.8×300mmのステンレスカラムに、15分充填した。その後、カラムに流速を変えながら水を通し、流速とカラム圧との関係を測定し、0.3MPa時の通液速度(線流速)を測定した。結果を表1に示す。
実施例1と同様にして、架橋体被覆粒子を作製した。
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄して、架橋体にエポキシ基を導入した。次いで、粒子に、水酸化ナトリウム5gを水15mLに溶解させた水酸化ナトリウム水溶液15mLと、メルカプトプロピオン酸5gとを加え、室温で24時間撹拌し、反応させた。反応終了後、粒子をろ別し、1000mLの水で洗浄して、架橋体にカルボキシ基を導入した。これにより本実施形態の分離材を得た。なお、本実施例における官能基導入剤は、メルカプトプロピオン酸である。
実施例1と同様にして、架橋体被覆粒子を作製した。
得られた架橋体被覆粒子(乾燥質量1g)を、0.4Mの水酸化ナトリウム水溶液10mLに加えた後、エピクロロヒドリン0.6gを更に加え、室温で撹拌しながら3時間反応させた。反応終了後、粒子をろ別し、500mLの水で洗浄し、架橋体にエポキシ基を導入した。これにより本実施形態の分離材を得た。
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gに水10g,1Nの塩酸水溶液1g加え、70℃で1時間撹拌し、反応させた。反応後、0.1N水酸化ナトリウムで中和滴定することにより、乾燥状態の分離材1g当たりのエポキシ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
得られた分離材0.09gに、プロテインAを0.02g、及びpH10の0.5M炭酸バッファー(和光純薬工業社製の炭酸水素ナトリウム及び炭酸ナトリウムと、RO水とで調製)を2mL加え、30℃で24時間撹拌した。その後、粒子をろ別及び洗浄することによりリガンド固定化粒子を得た。
24時間撹拌後の反応溶液から粒子をろ別した後、所定濃度の粒子を含む試料を調製した。当該試料の吸光度(280nm)を測定し、予め作成した検量線により、リガンド固定化量を算出した。結果を表1に示す。
実施例1と同様にして、架橋体被覆粒子を作製した。
得られた架橋体被覆粒子(乾燥質量20g)を、5Mの水酸化ナトリウム水溶液200mLに加え、室温で1時間放置した。別途、ジエチルアミノエチルクロライド塩酸塩の所定量(10g)を200mL添加し、水溶液の温度を70℃まで上げ、撹拌しながら2時間反応させた。反応終了後、ろ過、水/エタノール(体積比5/1)で3回洗浄し、架橋体にジエチルアミノエチル(DEAE)基を導入した。これにより本実施形態の分離材を得た。
得られた分離材の官能基量を以下のように測定した。湿潤状態の分離材0.5gを0.1N水酸化ナトリウム20mLに1時間浸漬し、室温で撹拌した。その後、溶液のpHが7以下となるように水で洗浄を行った。洗浄後の分離材を0.1N塩酸20mLに浸漬し、1時間撹拌させた。撹拌後の分離材をろ別後、塩酸水溶液を中和滴定することにより、乾燥状態の分離材1g当たりのアミノ基のモル量(mmol)を測定した。この結果を官能基密度(mmol/粒子g)として、表1に示す。
市販のアガロース粒子(Capto DEAE(GEヘルスケア製、商品名))をそのまま分離材として用い、実施例1と同様に、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。
多孔質高分子粒子1をそのまま分離材として用い、実施例1と同様に、平均粒径、タンパク質の非特異吸着能、水中での5%圧縮変形弾性率、及びカラム特性(通液速度)を評価した。結果を表1に示す。
Claims (11)
- 疎水性高分子粒子と、
該疎水性高分子粒子の表面の少なくとも一部を被覆する被覆層と、を備え、
前記被覆層が、水酸基及び水酸基以外の官能基を有する官能基含有高分子を含む、分離材。 - 前記水酸基以外の官能基が、カルボキシ基、エポキシ基、又はアミノ基である、請求項1に記載の分離材。
- 水中での5%圧縮変形弾性率が70MPa以上である、請求項1又は2に記載の分離材。
- カラムに充填した場合、カラム圧0.3MPaのときに通液速度が500cm/h以上である、請求項1〜3のいずれか一項に記載の分離材。
- 前記疎水性高分子粒子が、スチレン系モノマに由来する構造単位を有する疎水性高分子を含む、請求項1〜4のいずれか一項に記載の分離材。
- 前記官能基含有高分子が、多糖類又はその変性体に由来する構造を含む、請求項1〜5のいずれか一項に記載の分離材。
- 前記官能基含有高分子が、アガロース又はその変性体に由来する構造を含む、請求項1〜6のいずれか一項に記載の分離材。
- 前記官能基含有高分子が、架橋されている、請求項1〜7のいずれか一項に記載の分離材。
- 前記疎水性高分子粒子の平均粒径が10〜500μmである、請求項1〜8のいずれか一項に記載の分離材。
- 前記疎水性高分子粒子が多孔構造を有する、請求項1〜9のいずれか一項に記載の分離材。
- 請求項1〜10のいずれか一項に記載の分離材を備えるカラム。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016086980A JP6759679B2 (ja) | 2016-04-25 | 2016-04-25 | 分離材及びカラム |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016086980A JP6759679B2 (ja) | 2016-04-25 | 2016-04-25 | 分離材及びカラム |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017196544A true JP2017196544A (ja) | 2017-11-02 |
| JP6759679B2 JP6759679B2 (ja) | 2020-09-23 |
Family
ID=60237002
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016086980A Expired - Fee Related JP6759679B2 (ja) | 2016-04-25 | 2016-04-25 | 分離材及びカラム |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6759679B2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020036151A1 (ja) * | 2018-08-14 | 2020-02-20 | 日立化成株式会社 | 分離材、分離材の製造方法及びカラム |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6463858A (en) * | 1987-04-24 | 1989-03-09 | Unilever Nv | Substrate and manufacture thereof |
| JPH01254247A (ja) * | 1988-04-01 | 1989-10-11 | Mitsubishi Kasei Corp | 複合化分離剤及びその製造法 |
| US5030352A (en) * | 1990-01-25 | 1991-07-09 | Purdue Research Foundation | Coated media for chromatography |
| JPH055731A (ja) * | 1991-06-28 | 1993-01-14 | Sekisui Chem Co Ltd | アフイニテイクロマトグラフイー用担体 |
| JP2002507934A (ja) * | 1997-06-25 | 2002-03-12 | アメルシャム・ファルマシア・バイオテック・アクチボラグ | コートされたポリマー物品およびその使用 |
| JP2008232764A (ja) * | 2007-03-19 | 2008-10-02 | Tosoh Corp | 充填床用新規充填剤及びその用途 |
| WO2011125673A1 (ja) * | 2010-03-31 | 2011-10-13 | Jsr株式会社 | アフィニティークロマトグラフィー用充填剤 |
| JP2013512078A (ja) * | 2009-12-01 | 2013-04-11 | エクステラ・メディカル・エルエルシー | 表面固定した多糖類を用いた、血液からのサイトカイン除去法 |
-
2016
- 2016-04-25 JP JP2016086980A patent/JP6759679B2/ja not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6463858A (en) * | 1987-04-24 | 1989-03-09 | Unilever Nv | Substrate and manufacture thereof |
| JPH01254247A (ja) * | 1988-04-01 | 1989-10-11 | Mitsubishi Kasei Corp | 複合化分離剤及びその製造法 |
| US5030352A (en) * | 1990-01-25 | 1991-07-09 | Purdue Research Foundation | Coated media for chromatography |
| JPH04505968A (ja) * | 1990-01-25 | 1992-10-15 | パーデュー・リサーチ・ファウンデーション | クロマトグラフィー用被覆媒体 |
| JPH055731A (ja) * | 1991-06-28 | 1993-01-14 | Sekisui Chem Co Ltd | アフイニテイクロマトグラフイー用担体 |
| JP2002507934A (ja) * | 1997-06-25 | 2002-03-12 | アメルシャム・ファルマシア・バイオテック・アクチボラグ | コートされたポリマー物品およびその使用 |
| JP2008232764A (ja) * | 2007-03-19 | 2008-10-02 | Tosoh Corp | 充填床用新規充填剤及びその用途 |
| JP2013512078A (ja) * | 2009-12-01 | 2013-04-11 | エクステラ・メディカル・エルエルシー | 表面固定した多糖類を用いた、血液からのサイトカイン除去法 |
| WO2011125673A1 (ja) * | 2010-03-31 | 2011-10-13 | Jsr株式会社 | アフィニティークロマトグラフィー用充填剤 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020036151A1 (ja) * | 2018-08-14 | 2020-02-20 | 日立化成株式会社 | 分離材、分離材の製造方法及びカラム |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6759679B2 (ja) | 2020-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6848203B2 (ja) | 分離材及びカラム | |
| WO2016117574A1 (ja) | 分離材 | |
| JP6790834B2 (ja) | 分離材 | |
| JPWO2018181738A1 (ja) | 分離材 | |
| JP6911464B2 (ja) | 分離材及びカラム | |
| JP6778381B2 (ja) | 分離材 | |
| JP6972606B2 (ja) | 分離材、カラム、及び分離材の製造方法 | |
| JP6759679B2 (ja) | 分離材及びカラム | |
| JP2021007915A (ja) | 被覆粒子の製造方法及びカラムの製造方法 | |
| WO2018181925A1 (ja) | ゲルろ過用分離材及び水溶性高分子物質の精製方法 | |
| JP2020044496A (ja) | 分離材及び充填カラム | |
| JP6939021B2 (ja) | 分離材及びカラム | |
| JP2017125815A (ja) | 分離材及びカラム | |
| WO2020036151A1 (ja) | 分離材、分離材の製造方法及びカラム | |
| JP6746919B2 (ja) | 分離材及びカラム | |
| JP6676975B2 (ja) | 分離材及びカラム | |
| JP6874276B2 (ja) | 分離材及びカラム | |
| JP2020203266A (ja) | 分離材及びカラム | |
| JP6926573B2 (ja) | 分離材及びカラム | |
| JP6852259B2 (ja) | 分離材及びカラム | |
| JP6834132B2 (ja) | 分離材及びカラム | |
| WO2018174022A1 (ja) | 分離材 | |
| JP6676976B2 (ja) | 分離材及びカラム | |
| JP6778378B2 (ja) | 分離材及びカラム | |
| JP2020015809A (ja) | 多孔質ポリマー粒子、分離材、カラム及び多孔質ポリマー粒子の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190325 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200115 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200121 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200319 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200804 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200817 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 6759679 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| LAPS | Cancellation because of no payment of annual fees |