JP2017510595A - (5,6−ジヒドロ)ピリミド[4,5−e]インドリジン - Google Patents
(5,6−ジヒドロ)ピリミド[4,5−e]インドリジン Download PDFInfo
- Publication number
- JP2017510595A JP2017510595A JP2016560952A JP2016560952A JP2017510595A JP 2017510595 A JP2017510595 A JP 2017510595A JP 2016560952 A JP2016560952 A JP 2016560952A JP 2016560952 A JP2016560952 A JP 2016560952A JP 2017510595 A JP2017510595 A JP 2017510595A
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- alkoxy
- compound
- amino
- give
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/22—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed systems contains four or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
ここで、
R1およびR2は、以下からなる群から独立して選択される:
a)(6−10C)アリール、
b)(1−5C)ヘテロアリール、
ここで、両基は任意的に置換されることができる。
R11はH、ハロゲン、(1−2C)アルキル、(2−3C)アルケニル、(2−3C)アルキニル、(1−2C)アルコキシまたはOC2H3であり、アルキルおよびアルコキシ基のすべてが一つ以上のハロゲンによって任意的に置換されていても良く;
R12は、H、ハロゲン、(1−2C)アルキルまたは(1−2C)アルコキシであり;
R13は、R131CH2、R132O、R133R134N、R135C(O)、R136S、R136S(O)、R136S(O)(NH)、R137SO2、(2−7C)ヘテロシクロアルキル、又は(1−5C)ヘテロアリールであり、ここでヘテロシクロアルキルまたはヘテロアリールが各々(1−2C)アルキル、フルオロ、ヒドロキシル、オキソ、(1−2C)アルコキシ、(1−6C)アルキルカルボニル、(1−6C)アルキルスルホニル、(1−5C)アルコキシカルボニル、(1−6C)アルキルアミノカルボニル、(3−6C)シクロアルキルカルボニル、(2−7C)ヘテロシクロアルキルカルボニルまたはジ[(1−2C)アルキル]アミノで任意的に置換されていても良く、ここでアルキルカルボニル、アルキルスルホニル、アルコキシカルボニル、アルキルアミノカルボニル、シクロアルキルカルボニルまたはヘテロシクロアルキルカルボニルが各々(1−2C)アルキル、フルオロ、ヒドロキシル、シアノ、オキソまたは(1−2C)アルコキシによって任意的に置換されていても良く;
R131は、各々が(1−2C)アルキル、フルオロ、ヒドロキシル、または(1−2C)アルコキシから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキルカルボニルアミノ、(3−6C)シクロアルキルカルボニルアミノ、または(2−7C)ヘテロシクロアルキルカルボニルアミノであり、
R132は、各々(1−2C)アルキル、ハロゲン、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、または(2−7C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(6−10C)アリール、または(1−5C)ヘテロアリールであり;
R133は、各々(1−2C)アルキル、ハロゲン、ヒドロキシル又は(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、または(2−7C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルカルボニル、(1−5C)アルコキシカルボニル、(3−6C)シクロアルキルカルボニル、または(2−7C)ヘテロシクロアルキルカルボニルであり;
R134は、水素または(1−2C)アルキルであり;
R135は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、(2−7C)ヘテロシクロアルキル、オキソ、シアノ、又はアミノから選択された一つ以上の基によって任意的に置換されていても良い、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルアミノ、ジ[(1−6C)アルキル]アミノ、(2−7C)ヘテロシクロアルキルアミノ、または(3−6C)シクロアルキルアミノであり;
R136は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、または(1−2C)アルコキシから選択された一つ以上の基によっ任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキルであり;
R137は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、又は(1−2C)アルコキシから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルアミノ、ジ[(1−6C)アルキル]アミノ、(2−7C)ヘテロシクロアルキルアミノ、または(3−6C)シクロアルキルアミノであり;
R14は、H、ハロゲン、(1−2C)アルキルまたは(1−2C)アルコキシであり;及び
R15は、H、ハロゲンである。
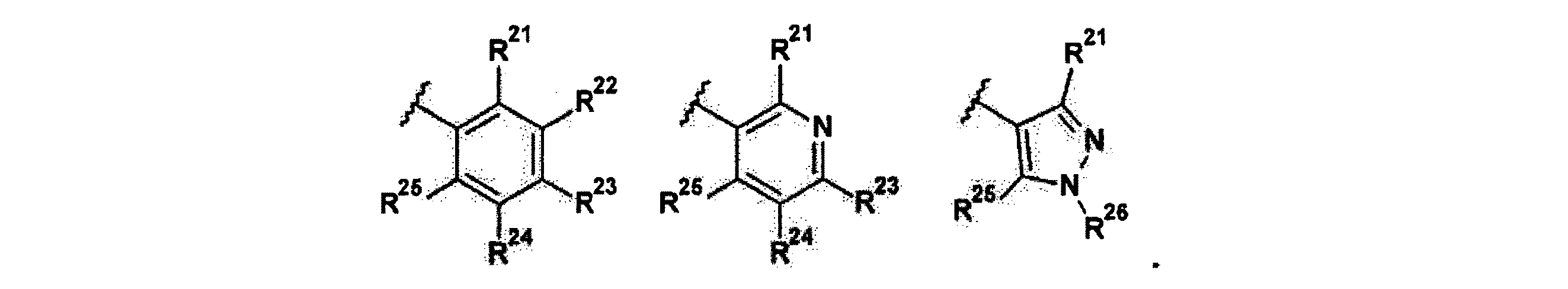
R21は、H、ハロゲン、(1−3C)アルキル、(1−2C)アルコキシ、ヒドロキシ(1−2C)アルキル、(3−4C)シクロアルキル、(2−3C)アルケニル、またはシアノであり;
R22は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり;
R23は、H、ハロゲン、(1−2C)アルキル、(1−2C)アルコキシ、シアノ、又はヒドロキシであり;
R24は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり;
R25は、H、ハロゲン、(1−3C)アルキル、(1−2C)アルコキシ、ヒドロキシ(1−2C)アルキル、(3−4C)シクロアルキル、(2−3C)アルケニル、またはシアノであり;
R26はH、(1−6C)アルキル、(3−6C)シクロアルキル、(2−5C)ヘテロシクロアルキル、(1−2C)アルコキシ[(2−4C)アルコキシ]n(1−6C)アルキルであり、ここでnは1,2,3又は4の整数を表し、アルキル、ヘテロシクロアルキル、および(1−2C)アルコキシ[(2−4C)アルコキシ]n(1−6C)アルキル基のすべては、(1−2C)アルキル、(1−2C)アルコキシ、ヒドロキシル、オキソ、アミノ、(3−6C)シクロアルキル、ジ[(1−2C)アルキル]アミノ、または(2−5C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い。
薬理学的に許容される液体によって、活性な剤は、溶液、懸濁物、エマルジョンの形の例えば注射薬のような流体組成物として、またはスプレー、例えば鼻スプレーとして適用されることができる。
TFA トリフルオロ酢酸
HATU O−(7−アザベンゾトリアゾル−1−イル)1,1,3,3−O-テトラメチルウロニウムヘキサフルオロホスフェート
DMF N,N−ジメチルホルムアミド
THF テトラヒドロフラン
MeOH メタノール
EtOAc 酢酸エチル
DCM ジクロロメタン
Na2SO4 硫酸ナトリウム
TMS−Cl クロロトリメチルシラン
DiPEA N,N−ジイソプロピルエチルアミン
EtOH エタノール
10%Pd/C 炭素上の10%パラジウム
HPLC 高速液体クロマトグラフィ
LCMS 液体クロマトグラフィー質量分析計
NaOH 水酸化ナトリウム
HCl 塩化水素
KOH 水酸化カリウム
NaHCO3 重炭酸ナトリウム
4−DMAP 4−ジメチルアミノピリジン
Boc ブチルオキシカルボニル
Cbz ベンジルオキシカルボニル
HNO3 硝酸
LiHMDS リチウムビス(トリメチルシリル)アミド
DDQ 2,3−ジクロロ−5,6−ジシアノ−p−ベンゾキノン
DBU 1,8−ジアザビシクロ〔5.4.0〕ウンデカ−7−エン
DEAD アゾジカルボン酸ジエチル
エチル2−クロロ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート
(a)5−ブロモ−2−クロロ−ピリミジン−4−アミン
THF(445mL)中の5−ブロモ−2,4−ジクロロピリミジン(150g、658mmol)の溶液に、水酸化アンモニウム(25%水溶液、250mL)が加えられて、得られた反応混合物は、室温で90分間撹拌された。混合物は、その後小容量に蒸発されて、酢酸エチルと水の間で分配された。有機相は分離されて、水および塩水で洗浄され、硫酸ナトリウムで乾燥され、濾過され、濃縮されて、5−ブロモ−2−クロロ−ピリミジン−4−アミン137.3g(定量的収量)を与えた。
メタノール(1L)中の5−ブロモ−2−クロロ−ピリミジン−4−アミン(137.3g、658mmol)の懸濁物に、ナトリウムメトキシド(83.5g、1.54mol)が加えられた。反応混合物は、還流で2時間撹拌された。反応混合物は小容量(〜400mL)に濃縮され、水中の塩化アンモニウム飽和溶液(1.2L)中へと注がれた。この混合物は15分間攪拌され、その後水層は酢酸エチルにより抽出された。一緒にされた酢酸エチル層は塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、濃縮されて、5−ブロモ−2−メトキシピリミジン−4−アミン(133.7g、99.4%)を生成した。
酢酸パラジウム(II)(1.21g、5.5mmol)およびトリフェニルホスフィン(3.40g、13.0mmol)は、無水かつ無酸素のDMF(53mL)に溶かされ、30°Cで5分間撹拌されて、オレンジの懸濁物を与えた。この懸濁物に、DMF(270mL)中の5−ブロモ−2−メトキシピリミジン−4−アミン(44.1g、216mmol、トリエチルアミン(60.2mL、432mmol)、およびDMF(50mL)中のアクリル酸エチル(23.5mL、216mmol)溶液が加えられた。反応混合物は、窒素雰囲気下で100℃に一晩撹拌された。反応混合物は、小容量に蒸発された。水(300mL)および塩水(300mL)が該混合物に加えられ、続いて酢酸エチル(300mL、二回)で抽出された。一緒にされた有機層は、水、塩水により洗浄され、その後硫酸ナトリウムで乾燥され、そして減圧下で濃縮された。粗生成物はシリカカラムクロマトグラフィ(酢酸エチル:ヘプタン=2:1v/v%)により精製されて、標記の化合物を生成した。(38.2g、77%)
メタノール(250mL)中のエチル(E)−3−(4−アミノ−2−メトキシ−ピリミジン−5−イル)プロプ−2−エノアート(12.52g、56.1mmol)の撹拌された溶液に、メタノール/エタノール=3/1v/v%(30mL)中の炭素上の10%パラジウム(1.19g)の懸濁物が加えられた。反応混合物は、窒素雰囲気下、室温で15分間撹拌された。続いて、蟻酸アンモニウム(35.3g、561mmol)が加えられ、得られた反応混合物は一晩還流された。反応混合物の冷却後、追加の蟻酸アンモニウム(20g、317mmol)が加えられ、さらに一晩還流で撹拌が続けられた。反応混合物はDecalite(商標)を通して濾過され、パラジウムカーボン/Decalite(商標)残渣はジクロロメタン/メタノール=8/2v/v%により洗浄され、濾過液は減圧下で濃縮された。残渣はジクロロメタン中に溶かされそして水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、2−メトキシ−6,8−ジヒドロ−5H−ピリド[2,3−d]ピリミジン−7−オン9.4g(94%)を得た。
機械式撹拌装置、温度計、および還流コンデンサが装備されている三つ口フラスコ(500mL)中において、THF(200mL)中で、2−メトキシ−6,8−ジヒドロ−5H−ピリド[2,3−d]ピリミジン−7−オン(4.79g、26.8mmol)が懸濁された。混合物は0°Cへと冷やされ、水素化ナトリウム(オイル中60%分散物、1.18g、29.4mmol)が2回に分けて加えられた。混合物は、0°Cで30分間撹拌された。(1−エトキシカルボニルシクロプロピル)トリフェニルホスホニウムテトラフルオロボレート(13.6g、29.4mmol)が加えられ、得られた懸濁物は還流するまで加熱され、3日間還流温度に保たれた。反応混合物は室温へと冷やされ、塩水/水/EtOAcの1/1/1混合物(450mL)中に注がれた。水層は、酢酸エチルにより抽出された(2回)。一緒にされた有機層は、水、塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、18.05gのオレンジ色のオイルを与えた。粗生成物が、精製することなく直接に次の工程で使われた。
ジクロロメタン(100mL)中のエチル2−メトキシ−5,6,8,9−テトラヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(18.05g、26.2mmol)の撹拌された溶液に、酢酸(3.15g、3mL)および酢酸鉛(IV)(13.9g、31.4mmol)が加えられた。反応混合物は室温で2時間撹拌され、続いてPE濾過器で濾過されて、Pb−塩が除去され、Pb−残渣は2×30mLのDCMにより洗浄された。濾過液は真空で濃縮され、得られた残渣は酢酸エチル(300mL)中に溶かされた。重炭酸ナトリウム溶液(5%)は、pH〜8.5まで加えられた。有機層および水層の両方とも、Decalite(商標)を通して濾過されて、なんらかの残留塩を除去した。その後、水層はEtOAc(2×50mL)により抽出された。一緒にされた有機層は、5%の重炭酸ナトリウム溶液(100mL)、水(100mL)、塩水(50mL)により洗浄され、乾燥され(Na2SO4)、濾過され、そして減圧で濃縮された。粗生成物は、シリカ上のカラムクロマトグラフィ(ヘプタン:酢酸エチル=1/0〜1/1v/v%)により精製されて、標記の化合物を生成した(4.74g、2段階で66%)。
ヨウ化ナトリウム(7.83g、52.2mmol)が、アセトニトリル(150mL)中のエチル2−メトキシ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(4.74g、17.3mmol)の撹拌された溶液に加えられた。アセトニトリル(30mL)中に溶かされたトリメチルシリルクロリド(5.64g、6.59mL)は反応混合物に滴下により加えられ、該混合物は室温で一晩撹拌された。NaI(1当量)が加えられ、そしてアセトニトリル(6mL)中に追加のTMS−Cl(0.94g、1.1mmol)が滴下により加えられ、そして反応系は室温で3日間撹拌された。混合物は濃縮され、そして残渣は200mLのDCM/MeOH(4/1)中で懸濁され、チオ硫酸ナトリウムの飽和溶液(200mL)および水(200mL)の混合物により抽出された。水層はDCM/MeOH(4/1)3x150mLにより抽出された。一緒にされた有機層は硫酸ナトリウムで乾燥され、濾過され、及び溶媒は減圧下で除去されて、黄色の固体を与えた。残渣が減圧下に40℃で18時間乾燥されて、3.89gのエチル2−ヒドロキシ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレートを与えた(86%)。
N、N−ジメチルアニリン(182mg、191uL、1.50mmol)が、アセトニトリル(100mL)中のエチル2−ヒドロキシ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(3.89g、15.0mmol)の溶液に加えられた。アセトニトリル(15mL)中のオキシ塩化リン(V)(11.5g、7.00mL、75.0mmol)の溶液は、反応混合物に滴下により加えられた。黄色の懸濁物は65°Cで4時間加熱され、その間に懸濁物が透明な溶液に変わった。冷却後に、該混合物は25%水性アンモニア(200mL、86.7当量)および氷水(250mL)の撹拌された混合物中に、10°C未満の温度を保ちながら15−20分間で、ゆっくり注がれた。さらに15分間撹拌した後に、固体は濾過された。該固体は、EtOAc200mL中に溶解され、塩水(20mL)により洗浄された。有機層は、硫酸ナトリウムで乾燥され、そして減圧下で濃縮されてオフホワイトの固体を与えた。粗生成物は、シリカ上のカラムクロマトグラフィ(ヘプタン/酢酸エチル=1/0〜1/1v/v%)により精製されて、標記の化合物を生成した(3.05g、73%)。
4−アミノ−N−(2−ヒドロキシ−2−メチル−プロピル)−3−メトキシ−ベンズアミド
(a)N−(2−ヒドロキシ−2−メチル−プロピル)−3−メトキシ−4−ニトロ−ベンズアミド
ジクロロメタン(20mL)中の3−メトキシ−4−ニトロ安息香酸(1g、5.07mmol)、HATU(2.31g、6.1mmol)、および1−アミノ−2−メチル−プロパン−2−オ−ル(1.13g、12.7mmol)の混合物は、氷冷水浴上で撹拌され、そしてDiPEA(2.2mL、12.7mmol)が加えられた。10分間後、この混合物は放置して室温へと温められ、そして1時間撹拌された。該混合物は酢酸エチル(100mL)で希釈され、その後5%の重炭酸ナトリウム溶液(3×75mL)、水、および塩水で洗浄され、硫酸ナトリウムで乾燥され、そして減圧で濃縮されて、1.47gの粗N−(2−ヒドロキシ−2−メチル−プロピル)−3−メトキシ−4−ニトロ−ベンズアミドを生成した(定量的)。
EtOH(70mL)中のN−(2−ヒドロキシ−2−メチル−プロピル)−3−メトキシ−4−ニトロベンズアミド(1.46g、5.44mmol)の溶液は、30°C、1気圧、フルH2モード,1 mL/minでH−Cube連続フローリアクター、10%Pd/Cを使用して水素化された。得られた溶液は減圧下で濃縮され、そして粗生成物はシリカ上のカラムクロマトグラフィ(DCM/MeOH/25%NH3=10/0/0〜9/1/0〜9/0.9/0.1v/v%)により精製されて、標記の化合物を生成した(775mg、60%)。
2−メチル−4−(4−メチルピペラジン−1−イル)アニリン
(a)1−メチル−4−(3−メチル−4−ニトロ−フェニル)ピペラジン
N−メチルピペラジン(752μL、6.79mmol)は4−フルオロ−2−メチル−1−ニトロ−ベンゼン(527mg、3.39mmol)に加えられ、そして得られた混合物は室温で18時間撹拌された。水が反応混合物に加えられ、そして抽出が酢酸エチルにより行われた。一緒にされた有機層は、塩水により洗浄され、その後硫酸ナトリウムで乾燥され、そして減圧下で濃縮された。粗生成物はシリカカラムクロマトグラフィ(DCM/MeOH = 1/0 〜 9/1 v/v%)により精製されて、標記の化合物を生成した(786mg、98%)。
エタノール(10mL)中の1−メチル−4−(3−メチル−4−ニトロ−フェニル)ピペラジン(393mg、1.67mmol)の撹拌された溶液に、エタノール(6mL)中の炭素上の10%パラジウム(35mg)の懸濁物が加えられた。反応混合物は、窒素雰囲気下、室温で15分間撹拌された。続いて、蟻酸アンモニウム(1.05g、16.7mmol)が加えられ、反応混合物は還流温度まで15分間加熱された。反応混合物は冷却され、Decalite(商標)を通して濾過され、減圧下で濃縮された。残渣はジクロロメタン中に溶かされ、重炭酸ナトリウムの飽和溶液で洗浄された。有機層は硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、2−メチル−4−(4−メチルピペラジン−1−イル)アニリン268mg(78%)を得た。
2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボニルクロライド
(a)エチル2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート
n−ブタノール(11mL)中のエチル2−クロロ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(中間体1、381mg、1.37mmol)の懸濁物に、2−メチル−4−(4−メチルピペラジン−1−イル)アニリン(中間体B、268mg、1.3mmol)及びトリフルオロ酢酸(200μL、2.6mmol)が加えられた。反応混合物は、マイクロ波照射下で120°Cで12時間加熱された。反応混合物は減圧下で濃縮され、その残渣は酢酸エチル中に溶かされた。有機層は重炭酸ナトリウムの飽和溶液により洗浄され、硫酸ナトリウムで乾燥して、濾過され、そして減圧下で濃縮された。残渣は、分取HPLCにより精製された。生成物を含有している分画は集められ、蒸発されて、エチル2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレートを与えた(211mg、収率34%)。
無水エタノール8mL中のエチル2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(211mg、0.47mmol)の溶液に、2Mの水酸化ナトリウム溶液(591μL(2.5当量)、1.18mmol)が加えられた。反応混合物は、65°Cで一晩加熱された。反応混合物は蒸発乾固され、高減圧下で乾燥された。得られた残渣が水に溶かされ、室温で一晩撹拌され、凍結乾燥されて標記の粗化合物が産生された。
2−メトキシ−4−モルホリノ−アニリン
モルホリンおよび4−フルオロ−2−メトキシ−1−ニトロ−ベンゼンから出発し、中間体B−aで記載されたのと類似の方法で、この化合物は調製された(1.3g、95%)。このようにして得られた4−(3−メトキシ−4−ニトロ−フェニル)モルホリン(1.3g、5.46mmol)は、THF(45mL)中に溶かされ、酢酸(5mL)が加えられた。
混合物は0°Cまで冷やされ、亜鉛(7.09g、109mmol)が少量づつ加えられ、20°C未満の温度に保たれた。反応混合物は、室温で一晩撹拌された。TLC分析が出発物質の完全転化を示した後、該混合物はDecalite(商標)およびZn−Decalite(商標)を通して濾過され、残渣はEtOAc(20mL)で洗浄された。一緒にされた濾液は1NNaOH溶液(25mL)、続いて水(25mL)および塩水(25mL)で洗浄された。有機層は乾燥され(Na2SO4)、濾過され、且つ減圧下で濃縮されて、2−メトキシ−4−モルホリノ−アニリンを与えた(1.05g、92%)。
2−メチル−6−モルホリノ−ピリジン−3−アミン
モルホリンおよび6−クロロ−3−ニトロ−2−ピコリンから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(156.9mg、82%)。
2−クロロ−4−(4−メチルピペラジン−1−イル)アニリン
(a)3級ブチルN−(4−ブロモ−2−クロロ−フェニル)−N−3級ブトキシカルボニル−カルバメート
DMF(100mL)中の4−ブロモ−2−クロロアニリン(3.0g、14.52mmol)および炭酸カリウム(6g、43.6mmol)の溶液に、ジ−3級ブチルジカーボネート(1.8g、28.3mmol)が加えられた。反応混合物は、室温で48時間撹拌され、その後混合物は水/塩水に注がれ、酢酸エチル(3×50mL)により抽出された。一緒にされた有機層は、塩水(3×50mL)により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(EtOAc/ヘプタン=1/9v/v%)により精製されて、黄色/オレンジ色のオイルとして3級ブチル−(4−ブロモ−2−クロロ−フェニル)−N−3級ブトキシカルボニル−カルバメート(2.5g、42.3%)を得た。
トルエン(20mL)中の3級ブチルN−(4−ブロモ−2−クロロ−フェニル)−N−3級ブトキシカルボニル−カルバメート(500mg、1.23mmol)、1−メチルピペラジン(166μL、1.48mmol)、酢酸パラジウム(II)(27.6mg、0.12mmol)、(+/−)−2,2'−ビス(ジフェニルホスフィノ)−1,1'−ビナフチル(119mg、0.20mmol)、および炭酸セシウム(1.2g、3.69mmol)の混合物は、窒素雰囲気下に100°Cで16時間加熱された。環境温度に冷却後、混合物は濃縮され、そして残渣はジクロロメタンで希釈され、水および塩水で洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。
残渣はカラムクロマトグラフィ(ジクロロメタン/メタノール=100/0〜95/5v/v%)により精製されて、3級ブチルN−3級ブトキシカルボニル−N−[2−クロロ−4−メチルピペラジン−1−イル)フェニル]カルバメートを与えた(480mg、91.6%)。
3級ブチルN−3級ブトキシカルボニル−N−[2−クロロ−4−メチルピペラジン−1−イル)フェニル]カルバメート(260mg、0.61mmol)はDCM(4mL)中に溶解された。TFA(4mL)が加えられ、そして反応混合物は室温で1時間撹拌された。混合物は濃縮され、そして残渣はDCM(10mL)に溶かされ、5%の重炭酸ナトリウム溶液(10mL)に注がれた。水層はDCM(2 x 10 mL)により抽出された。一緒にされた有機層は、PE−フィルターを通して濾過され、減圧で濃縮されて、茶色のオイル(105mg、76%)が与えられ、それは更に精製されることなく使われた。
2−メトキシ−4−(4−メチルピペラジン−1−イル)アニリン
N−メチルピペラジンおよび2−メトキシ−4−フルオロニトロベンゼンから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(1.38g、94%)。
2−エトキシ−4−(4−メチルピペラジン−1−イル)アニリン
(a)2−エトキシ−4−フルオロ−1−ニトロ−ベンゼン
THF(15mL)中のエタノール(0.735mL、12.6mmol)の冷却混合物(0°C)に、水素化ナトリウム(鉱油中60%分散物、553mg、13.83mmol)が加えられた。反応混合物は15分間0°Cで撹拌され、その後THF(25mL)中の2,4−ジフルオロ−1−ニトロ−ベンゼン(1.38mL、12.6mmol)の溶液が、滴下で加えられた。室温でさらに90分間撹拌後、反応系は水によってクエンチされ、混合物がEtOAcで抽出された。一緒にされた有機層は、水および塩水により洗浄され、その後硫酸ナトリウムで乾燥され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=100/0〜85/15v/v%)により精製されて、2−エトキシ−4−フルオロ−1−ニトロ−ベンゼンを得た(2.15g、92%)。
N−メチルピペラジン(603μL、5.44mmol)は2−エトキシ−4−フルオロ−1−ニトロ−ベンゼン(500mg、2.7mmol)に加えられ、そして得られた混合物は室温で18時間撹拌された。水が反応混合物に加えられ、続いて酢酸エチルにより抽出された。一緒にされた有機層は、塩水により洗浄され、その後硫酸ナトリウムで乾燥され、そして減圧下で濃縮された。粗生成物はシリカ上のカラムクロマトグラフィ(DCM/MeOH = 10/0 〜 9/1 v/v%)により精製されて、標記の化合物を生成した(646 mg, 90%)。
エタノール(5mL)中の1−(3−エトキシ−4−ニトロ−フェニル)−4−メチル−ピペラジン(265mg、1.0mmol)の撹拌された溶液に、エタノール(6mL)中の炭素上の10%パラジウム(22mg)の懸濁物が加えられた。反応混合物は、窒素雰囲気下、室温で15分間撹拌された。蟻酸アンモニウム(630mg、10.0mmol)が加えられ、得られた反応混合物は還流温度で15分間加熱された。反応混合物は冷却され、Decalite(商標)を通して濾過された。濾過液は減圧下で濃縮され、そして残渣はその後ジクロロメタンに溶かされ、重炭酸ナトリウムの飽和溶液で洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、2−エトキシ−4−(4−メチルピペラジン−1−イル)アニリン231mg(98%)を得た。
3級ブチル4−(4−アミノ−3−メトキシフェニル)ピペラジン−1−カルボキシレート
3級ブチルピペラジン−1−カルボキシレートおよび2−メトキシ−4−フルオロニトロベンゼンから出発し、中間体B中に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(245mg、91%)。
ベンジル4−(4−アミノ−3−メチル−フェニル)ピペラジン−1−カルボキシレート
ベンジルピペラジン−1−カルボキシレートおよび4−フルオロ−2−メチル−1−ニトロ−ベンゼンから出発し、中間体Cのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(327mg、定量的)。
2−(ジフルオロメトキシ)−4−(4−メチルピペラジン−1−イル)アニリン
DMF(6ml)中の5−フルオロ−2−ニトロ−フェノール(500mg、3.18mmol)の溶液に、2−クロロ−2,2−ジフルオロ酢酸ナトリウム(970mg、6.36mmol)および炭酸二ナトリウム(405mg、3.82mmol)は添加された。反応混合物は、100°Cで3.5時間、その後3日間室温で撹拌された。透明な溶液が得られるまで4MのHCl溶液は加えられ、そして混合物は室温で2時間撹拌された。反応混合物は水で希釈され、EtOAcにより抽出された。一緒にされた有機層は1MのNaOH溶液,塩水により洗浄され、その後硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=100/0〜8/2v/v%)により精製されて、2−(ジフルオロメトキシ)−4−フルオロ−1−ニトロ−ベンゼンを得た(493mg、75%)。
4−メチル−6−モルホリノ−ピリジン−3−アミン
モルホリンおよび2−クロロ−5−ニトロ−4−ピコリンから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(122.9mg、81.5%)。
4−アミノ−3−メトキシ−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび3−メトキシ−4−ニトロ安息香酸から出発し、中間体Aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(980mg、82%)。
4−アミノ−3−メチル−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび4−アミノ−3‐メチル安息香酸から出発し、中間体A-aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(700mg、70%)。
4−アミノ−3−クロロ−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび4−アミノ−3−クロロ安息香酸から出発し、中間体A−aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(1.64g、定量的)。
4−アミノ−3−フルオロ−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび4−アミノ−3−フルオロ安息香酸から出発し、中間体A−aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(170mg、16%)。
4−アミノ−3−エトキシ−N−(1−メチル−4−ピペリジル)ベンズアミド
(a)3−エトキシ−4−ニトロ安息香酸
エタノール(25mL)中の3−フルオロ−4−ニトロ安息香酸(1.50g、8.11mmol)および水酸化カリウム(1.05g、18.6mmol)は、室温で撹拌された。得られた懸濁物は還流へとゆっくり加熱され(10分間)、この間に反応混合物が深紅の溶液になった。還流での10分の加熱の後、激しい撹拌下で粘稠な固体が沈澱した。反応混合物は室温まで冷やされ、そして水(10mL)が加えられた。次に、2MのHCl溶液(9.3mL)がpH<2まで加えられた。得られた沈殿物は激しく撹拌され、濾過されて、そして残渣は水(2×10mL)で洗浄された。残渣は減圧下40°Cで乾燥され、3−エトキシ−4−ニトロ安息香酸1.53gを与えた(89%)。
4−アミノ−1−メチルピペリジンおよび3−エトキシ−4−ニトロ安息香酸から出発し、中間体Aの為に記載されているのと類似の方法で、この化合物は調製されて、4−アミノ−3−エトキシ−N−(1−メチル−4−ピペリジル)ベンズアミド190mgを得た(70%)。
4−アミノ−3−(ジフルオロメトキシ)−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび4−アミノ−3−(ジフルオロメトキシ)ベンゾエートから出発し、中間体A−aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(130mg、29.5%)。
2−メトキシ−4−(1,3,5−トリメチルピラゾール−4−イル)アニリン
(a)3級ブチルN−[2−メトキシ−4−(1,3,5−トリメチルピラゾール−4−イル)フェニル]カルバメート
ジオキサン(4mL)中の3級ブチルN−(4−ブロモ−2−メトキシ−フェニル)カルバメート(150mg、0.5mmol)、1,3,5−トリメチル−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピラゾール(118mg、0.5mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(58mg、0.05mmol)、および炭酸カリウム(207mg、1.5mmol)の混合物は、封管中でマイクロ波の照射下に100°Cで20分間加熱された。環境温度に冷却後、混合物は濃縮され、そして残渣は酢酸エチルで希釈され、水および塩水で洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=100/0〜25/75v/v%)により精製されて、3級ブチルN−[2−メトキシ−4−(1,3,5−トリメチルピラゾール−4−イル)フェニル]カルバメートを与えた(126.8mg、77%)。
3級ブチルN−[2−メトキシ−4−(1,3,5−トリメチルピラゾール−4−イル)フェニル]カルバメート(127mg、0.38mmol)がDCM(2mL)中に溶解された。TFA(3mL)が加えられ、そして反応混合物は室温で1時間撹拌された。混合物は減圧下で濃縮されて、茶色のオイル(313mg)が得られ、更に精製することなく使われた。
2−メトキシ−4−[(1−メチル−4−ピペリジル)オキシ]アニリン
(a)4−(3−メトキシ−4−ニトロ−フェノキシ)−1−メチル−ピペリジン
トルエン(10mL)中の4−フルオロ−2−メトキシ−1−ニトロ−ベンゼン(750mg、4.38mmol)の溶液に、10mLの25%KOH溶液、4−ヒドロキシ−N−メチルピペリジン(1009mg、8.76mmol)、およびテトラ−n−ブチル臭化アンモニウム(282mg、0.876mmol)が加えられた。混合物は、60°Cで一晩加熱された。その後反応混合物は酢酸エチルで希釈され、そして水層は酢酸エチルにより抽出された。一緒にされた有機層は、塩水により洗浄され、その後硫酸ナトリウムで乾燥され、そして蒸発された。残渣はシリカゲル上のフラッシュクロマトグラフィ(ジクロロメタン/メタノール=99/1〜9/1v/v%)により精製されて、標記化合物を得た。(650mg、55.7%)
10%Pd/C(20mg)がエタノール中の懸濁物として、エタノール(5mL)中の4−(3−メトキシ−4−ニトロ−フェノキシ)−1−メチル−ピペリジン(200mg、0.75mmol)の溶液に加えられた。得られた混合物は、室温で15分間撹拌された。蟻酸アンモニウム(473mg、7.5mmol)が加えられ、そして反応混合物は窒素雰囲気下に還流で1時間撹拌された。反応混合物は室温まで冷やされ、そしてDecalite(商標)を通して濾過された。濾過液は減圧下で濃縮され、その後ジクロロメタンが加えられ、そして有機相はNaHCO3の5%溶液により洗浄された。有機相は、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、2−メトキシ−4−[(1−メチル−4−ピペリジル)オキシ]アニリンを得た(169.5mg、95.6%)。
2−メチル−4−[(1−メチル−4−ピペリジル)オキシ]アニリン
1−メチルピペリジン−4−オ−ルおよび4−フルオロ−2−メチル−1−ニトロ−ベンゼンから出発し、中間体Sのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(151.7mg、81%)。
4−(2−ジメチルアミノエチルオキシ)2−メトキシ−アニリン
(a)2−(3−メトキシ−4−ニトロ−フェノキシ)−N,N−ジメチル−エタンアミン
THF(5mL)中のN,N−ジメチルエタノールアミン(651μL、6.43mmol)の冷却溶液(0°C)溶液に、NaH(鉱油中60%分散物、385mg)が徐々に加えられた。懸濁物はさらに30分間かきまぜられ、その後乾燥THF5mL中の4−フルオロ−2−メトキシ1−ニトロ−ベンゼン(1g、5.84mmol)が滴下により加えられた。該溶液は、一晩還流された。溶媒は減圧下で除去され、そして残渣は酢酸エチルと水(それぞれ50mL)の間で分配された。有機層は集められ、そして水層は酢酸エチルによってその後抽出された。一緒にされた有機層は、1MのHCl溶液、水、5%重炭酸ナトリウム溶液、および塩水により洗浄された。有機層は硫酸ナトリウムで乾燥され、蒸発された。残渣はシリカゲル上のフラッシュクロマトグラフィ(ヘプタン/酢酸エチル=9/1〜1/1v/v%)によって溶出液として精製されて、標記化合物を得た(720mg、51.3%)。
中間体Sbの為に記載されているのと類似の方法でこの化合物は調製されて、標記化合物を得た(144.8mg、68.9%)。
4−アミノ−N−(1−メチル−4−ピペリジル)ベンズアミド
4−アミノ−1−メチルピペリジンおよび4−アミノ安息香酸から出発し、中間体A-aの為に記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(170mg、16%)。
2−メチル−4−モルホリノ−アニリン
モルホリンおよび5−フルオロ−2−ニトロトルエンから出発し、中間体Cのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(8.77g、92%)。
2−イソプロポキシ−4−(4−メチルピペラジン−1−イル)アニリン
1−メチルピペラジンおよび4−フルオロ−2−イソプロポキシ−1−ニトロ−ベンゼンから出発し、中間体Gのために記載されているのと類似の方法で、標記化合物は調製されて、2−イソプロポキシ−4−(4−メチルピペラジン−1−イル)アニリンを与えた(337mg、50%)。
2−メトキシ−4−(1−メチルピロリジン−3−イル)オキシ−アニリン
1−メチルピロリジン−3−オールおよび4−フルオロ−2−メトキシ−1−ニトロ−ベンゼンから出発し、中間体Uのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(199mg、84%)。
ベンジル3−(4−アミノ−3−メチル−フェノキシ)アゼチジン−1−カルボキシレート
(a)3級ブチル3−(3−メチル−4−ニトロ−フェノキシ)アゼチジン−1−カルボキシレート
THF(10mL)中のN−Boc−3−ヒドロキシアゼチジン(1.84g、10.6mmol)の冷却溶液(0°C)に、NaH(鉱油中60%分散物、382mg)が徐々に加えられた。該懸濁物はさらに30分間撹拌され、その後乾燥THF10mL中の2−ニトロ−5−フルオロトルエン(1.5g、9.79mmol)が滴下により加えられた。該溶液は、室温で一晩撹拌された。溶媒は減圧下で除去され、そして残渣は酢酸エチルと水(それぞれ50mL)の間で分配された。有機層は集められ、そして水層は酢酸エチルによってその後抽出された。一緒にされた有機層は、1MのHCl溶液、水、5%重炭酸ナトリウム溶液、および塩水により洗浄された。有機層は硫酸ナトリウムで乾燥され、減圧下で濃縮された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=9/1〜1/1v/v%)により精製されて、標記化合物2.24g(75.1%)を得た。
DCM(20mL)中の3級ブチル3−(3−メチル−4−ニトロ−フェノキシ)アゼチジン−1−カルボキシレート(2.24g、7.3mmol)の溶液に、ジオキサン(9.1mL)中の4NのHCl溶液が加えられ、そして反応混合物は室温で3時間撹拌された。
該混合物は減圧下で濃縮され、そして残渣はエタノールによって二回共蒸発されて、脱保護された化合物(1.7g、95%)がを与えられ、更に精製することなく用いられた。DCM(25mL)中の粗3−(3−メチル−4−ニトロ−フェノキシ)アゼチジン塩酸塩(1.7g、7.97mmol)の撹拌された懸濁物に、トリエチルアミン(2.8mL、20mmol)が加えられた。N−(ベンジルオキシカルボニルオキシ)−スクシンイミド(2.19g、8.77mmol)が反応混合物に加えられ、そして該混合物は30分間室温で撹拌された。5%の重炭酸ナトリウム溶液は、反応混合物に加えられた。有機相は集められ、そして水相はジクロロメタンにより抽出された。一緒にされた有機層は、乾燥され(Na2SO4)、濾過され、そして溶媒は蒸発された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=100/0〜50/50v/v%)により精製されて、ベンジル3−(3−メチル−4−ニトロフェノキシ)アゼチジン−1−カルボキシレート2.17g(80%)を得た。
ベンジル3−(3−メチル−4−ニトロフェノキシ)アゼチジン−1−カルボキシレートから出発し、中間体Cのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(145.3mg、39%)。
4−(3,5−ジメチルイソキサゾール−4−イル)−2−メトキシ−アニリン
3級ブチルN−(4−ブロモ−2−メトキシ−フェニル)カルバメートおよび3,5−ジメチル−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)イソオキサゾールから出発し、中間体Rのために記載されているのと類似の方法で、この化合物は調製されて、そして標記化合物を得た(96mg)。
ベンジル3−(4−アミノ−3−メトキシ−フェノキシ)アゼチジン−1−カルボキシレート
4−フルオロ−2−メトキシ−1−ニトロ−ベンゼンおよびN−Boc−3−ヒドロキシアゼチジンから出発し、中間体Zのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(215.1mg、55%)。
4−(4−メチルピペラジン−1−イル)−2−(トリ重水素メトキシ)アニリン
(a)4−フルオロ−1−ニトロ−2−(トリ重水素メトキシ)ベンゼン
アセトン(20mL)中の5−フルオロ−2−ニトロフェノール(1.5g、9.55mmol)の溶液に、K2CO3(2.31g、16.7mmol)が室温で加えられた。得られた懸濁物に、重水素化ヨードメタン(0.71ml、11.46mmol)が加えられ、そして反応混合物は還流で一晩撹拌された。反応混合物の濃縮後、残渣は水と酢酸エチルの間で分配された。酢酸エチル層は、水、塩水により洗浄され、硫酸ナトリウムで乾燥され、そして減圧下で蒸発されて4−フルオロ−1−ニトロ−2−(トリ重水素メトキシ)ベンゼンを得た(1.67g、100%)。
4−フルオロ−1−ニトロ−2−(トリ重水素メトキシ)ベンゼン(1.67g、9.55mmol)およびN−メチルピペラジン(2.12mL、19.1mmol)は、THF(1mL)中で混合され、室温で18時間撹拌された。水の付加後に形成された黄色の沈殿は濾過され、水で洗浄され、減圧下に乾燥されて、1−メチル−4−[4−ニトロ−3−(トリ重水素メトキシ)フェニル]ピペラジンを得た(1.67g、80.7%)。
1−メチル−4−[4−ニトロ−3−(トリ重水素メトキシ)フェニル]ピペラジンから出発し、中間体Bステップbのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(177.8mg)。
2−メチル−6−(4−メチルピペラジン−1−イル)ピリジン−3−アミン
N−メチルピペラジンおよび6−クロロ−3−ニトロ−2−ピコリンから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(188.9mg、92%)。
ベンジル4−(4−アミノ−3−メトキシ−フェニル)ピペラジン−1−カルボキシレート
ベンジルピペラジン−1−カルボキシレートおよび2−メトキシ−4−フルオロニトロベンゼンから出発し、中間体B−aの為に記載されているのと類似の方法で、この化合物は調製された。中間体Cのために記載されている方法を使用した還元後、標記化合物を得た(1.2g、95%)。
4−メトキシ−2−(4−メチルピペラジン−1−イル)ピリミジン−5−アミン
(a)2−クロロ−4−メトキシ−5−ニトロピリミジン
メタノール(30mL)中の2,4−ジクロロ−5−ニトロピリミジン(1.00g、5.2mmol)の溶液に、メタノール(5mL)中のナトリウムメトキシド(278mg、5.2mmol)の溶液が−10°Cで滴下により加えられた。反応混合物は−10°Cで10分間撹拌された。酢酸(5mL)が加えられ、そして反応混合物は、放置して室温まで温められた。混合物の蒸発後、残渣は、5%のNaHCO3溶液と酢酸エチルの間の間で分配された。酢酸エチル層は、水、塩水により洗浄され、硫酸ナトリウムで乾燥され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(ヘプタン/酢酸エチル=4/1v/v%)により精製されて、2−クロロ−4−メトキシ−5−ニトロピリミジン281.9mg(29%)を得た。
2−クロロ−4−メトキシ−5−ニトロピリミジンおよびN−メチルピペラジンから出発し、中間体BステップBのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(212.8mg、89%)。
2−メトキシ−5−メチル−4−(4−メチルピペラジン−1−イル)アニリン
N−メチルピペラジンおよび5−フルオロ−4−メチル−2−ニトロアニソールから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(94.4mg、定量的)。
ベンジル4−(4−アミノ−3−メトキシ−フェニル)−2−メチル−ピペラジン−1−カルボキシレート
ベンジル2−メチルベンジルピペラジン−1−カルボキシレートおよび2−メトキシ−4−フルオロニトロベンゼンから出発し、中間体B−aの為に記載されているのと類似の方法で、この化合物は調製された。中間体Cのために記載されている方法を使用した還元後、標記化合物を得た(295.4mg、97%)。
N4−[3−(ジメチルアミノ)プロピル]−2−メトキシ−N4−メチル−ベンゼン−1,4−ジアミン
N,N',N'−トリメチルプロパン−1,3−ジアミンおよび4−フルオロ−2−メトキシ−1−ニトロ−ベンゼンから出発し、中間体Bのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(148.8mg、94%)。
2−メトキシ−4−(2−メトキシエトキシ)アニリン
2−メトキシエタノールおよび4−フルオロ−2−メトキシ−1−ニトロ−ベンゼンから出発し、中間体Sのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(113.4mg、定量的)。
1−(2−メトキシエチル)−3,5−ジメチル−ピラゾール−4−アミン
(a)1−(2−メトキシエチル)−3,5−ジメチル−4−ニトロ−ピラゾール
DMF(50mL)中の3,5−ジメチル−4−ニトロ−1H−ピラゾール(2.5g、17.7mmol)および炭酸セシウム(6.06g、18.6mmol)の溶液に、2−ブロモエチルメチルエーテル(2.59g、1.75mL、18.6mmol)が加えられた。混合物は100°Cで3.5時間加熱された。室温に冷却後、混合物は水中に注がれ、酢酸エチル(3x50mL)により抽出された。一緒にされた有機層は、塩水(50mL)により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣はカラムクロマトグラフィ(EtOAc/ヘプタン=1/4v/v%)により精製されて、白い結晶質固体として1−(2−メトキシエチル)3,5−ジメチル−4−ニトロ−ピラゾール(2.66g、75.4%)を得た。
1−(2−メトキシエチル)−3,5−ジメチル−4−ニトロ−ピラゾール(245mg、1.22mmol)がメタノール(25mL)中に溶解された。得られた溶液は、30°C、8−10気圧、1mL/分、フルH2モードにおいて、H−Cube連続フロー水素化反応器、10%Pd/Cを使用して水素化された。得られた溶液は減圧下で濃縮され、明るい茶色のオイルとして標記化合物208mg(定量的収量)を生成した。
3,5−ジメチル−1H−ピラゾール−4−アミン
3,5−ジメチル−4−ニトロ−1H−ピラゾールから出発し、中間体Aa−bのために記載されているのと類似の方法で、標記化合物は調製され、そして3,5−ジメチル−1H−ピラゾール−4−アミン110mgを与えた(定量的)。
3,5−ジエチル−1H−ピラゾール−4−アミン
(a)3,5−ジエチル−1H−ピラゾール
水(10mL)中の3,5−ヘプタンジオン(2g、15.6mmol)およびヒドラジン水和物(0.77g、15.8mmol)の溶液に、酢酸(1滴)は加えられ、そして反応混合物は還流まで1時間加熱された。その後反応混合物は冷却され、減圧下で濃縮されて、標記化合物1.8gが提供された。この化合物が、精製することなく直接に次の工程で使われた。
3,5−ジエチル−1H−ピラゾール(1.8g、14.5mmol)および濃硫酸(1.5ml)の冷却混合物(0 °C)に、発煙HNO3(4.35ml)が激しい撹拌の下でゆっくり加えられた。反応混合物は、60°Cで一晩撹拌された。該混合物はその後室温に冷やされ、重炭酸ナトリウムの氷冷飽和溶液に慎重に加えられ、そして15分間攪拌された。その後混合物はEtOAcによって3回抽出され、そして一緒にされた有機層は塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、減圧下で蒸発されて、3,5−ジエチル−4−ニトロ−1H−ピラゾール2.52gを得た。
3,5−ジエチル−4−ニトロ−1H−ピラゾールから出発し、中間体Aa−bのために記載されているのと類似の方法で、標記化合物は調製され、そして3,5−ジエチル−1H−ピラゾール−4−アミンを与えた(174mg、71%)。
5−クロロ−1,3−ジメチル−ピラゾール−4−アミン
(a)5−クロロ−1,3−ジメチル−4−ニトロ−ピラゾール
5−クロロ−1,3−ジメチルピラゾール(1g、7.66mmol)および濃硫酸(750μl)の冷却混合物(0 °C)に、発煙HNO3(2.1mL)が激しい撹拌の下でゆっくり加えられた。反応混合物は、60°Cで一晩撹拌された。該混合物はその後室温に冷やされ、重炭酸ナトリウムの氷冷飽和溶液に慎重に加えられ、そして15分間攪拌された。
その後混合物はEtOAcによって3回抽出され、そして一緒にされた有機層は塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、減圧下で蒸発されて、5−クロロ−1,3−ジメチル−4−ニトロピラゾールを得た(1.1g、82%)。
THF(5mL)中の5−クロロ−1,3−ジメチル−4−ニトロ−ピラゾール(146mg、1mmol)の撹拌された溶液に、酢酸(1.1mL、16mmol)が加えられた。混合物は0°Cまで冷やされ、20°C未満の温度を保ちながら亜鉛(1.31g、20mmol)が少量づつ加えられた。反応混合物は、室温で一晩撹拌された。TLC分析が出発物質の完全転化を示した後、該混合物はDecalite(商標)およびZn−Decalite(商標)を通して濾過され、残渣はDCM/MeOH 9:1で洗浄された。一緒にされた濾過液は、5%NaHCO3溶液、続いて水および塩水により洗浄された。有機層は乾燥され(Na2SO4)、濾過され、且つ減圧下で濃縮されて、標記化合物を与えた(78mg、53%)。
3,5−ジエチル−1−(2−メトキシエチル)ピラゾール−4−アミン
2−ブロモエチルメチルエーテルおよび3,5−ジエチル−4−ニトロ−1H−ピラゾールから出発し、中間体Aaのために記載されているのと類似の方法で、標記化合物は調製され、そして3,5−ジエチル−1H−ピラゾール−4−アミンを与えた(143mg、36%)。
1−(4−アミノ−3,5−ジメチル−ピラゾール−1−イル)−2−メチル−プロパン−2−オ−ル
(a)1−(3,5−ジメチル−4−ニトロ−ピラゾール−1−イル)−2−メチル−プロパン−2−オ−ル
アセトニトリル(10ml)中の3,5−ジメチル−4−ニトロ−1H−ピラゾール(706mg、5mmol)およびDBU(1.49ml、10mmol)の溶液に、イソブチレンオキサイド(669μL、7.5mmol)が加えられた。該混合物は65°Cで72時間加熱された。室温に冷却後、混合物は水中に注がれ、酢酸エチル(3x 50mL)により抽出された。一緒にされた有機層は、1MHCl溶液、水、および塩水(50ml)により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。
残渣はカラムクロマトグラフィ(ヘプタン/EtOAc=100/0〜60/40v/v%)により精製されて、1−(3,5−ジメチル−4−ニトロ−ピラゾール−1−イル)−2−メチル−プロパン−2−オ−ルを得た(799mg、75%)。
1−(3,5−ジメチル−4−ニトロ−ピラゾール−1−イル)−2−メチル−プロパン−2−オ−ルから出発し、中間体Bステップbのために記載されているのと類似の方法で、この化合物は調製されて、標記化合物を得た(229mg、39%)。
3−エチル−5−メチル−イソキサゾール−4−アミン
(a)3級ブチルN−(3−エチル−5−メチル−イソキサゾール−4−イル)カルバメート
ジフェニルホスホリルアジド(2.51mL、11.6mmol)は、トルエン(50mL)中の3−エチル−5−メチル−イソオキサゾール−4−カルボン酸(1.5g、9.67mmol)、トリエチルアミン(2.7mL、19.3mmol)、および3級ブチルアルコール(0.92mL、9.67mmol)の溶液に加えられ、そして100°Cで4時間撹拌された。溶媒は蒸発により除去され、残渣がEtOAc(50mL)中に回収された。有機層は水および塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣は、ヘプタン/酢酸エチル=10/0〜7/3v/v%中のシリカゲル上のフラッシュクロマトグラフィによって溶出液として精製された。
標記化合物を含有する分画はプールされ、そして蒸発されて、標記化合物を得た。(1.67g、76.3%)
3級ブチルN−(3−エチル−5−メチル−イソキサゾール−4−イル)カルバメート(500mg、2.2mmol)は、TFA/ジクロロメタン=1/1v/v%(5mL)に溶解され、室温で1時間撹拌された。得られた混合物は蒸発され、残渣はメタノールに溶かされ、そしてSCX−2カラムを通して濾過された。カラムをメタノールですすいだ後、所望の生成物が0.7Nアンモニア/メタノール溶液により溶出された。得られた溶出液は、減圧で濃縮され、標記化合物を与えた(302.7mg、92%)。
1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]−3,5−ジメチル−ピラゾール−4−アミン
(a)1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]−3,5−ジメチル−4−ニトロ−ピラゾール
THF(10mL)中の3,5−ジメチル−4−ニトロ−1H−ピラゾール(250mg、1.77mmol)、トリエチレングリコールモノメチルエーテル(482μL、3.01mmol)、およびトリフェニルホスフィン(789mg、3.01mmol)の冷却溶液(0°C)に、トルエン中の40%DEAD溶液(1.31mL、3.01mmol)が滴下により加えられた。反応混合物は放置して室温まで温められ、そして3時間撹拌された。酢酸エチルが加えられ、10%NaCl溶液よって洗浄された。有機層は乾燥され(Na2SO4)、濾過され、そして濃縮された。残渣はカラムクロマトグラフィ(DCM/MeOH=99/1〜95/5v/v%)により精製され、1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]−3,5−ジメチル−4−ニトロピラゾール(1.7g、粗)が得られ、次のステップで直接使われた。
1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]−3,5−ジメチル−4−ニトロ−ピラゾール(1.5g、1.77mmol理論値)はTHF(15mL)中に溶解され、そして酢酸(1.6mL)が加えられた。混合物は0°Cまで冷やされ、20°C未満の温度を保ちながら亜鉛(2.3g、35.4mmol)が少量づつ加えられた。反応混合物は、室温で一晩撹拌された。TLC分析が出発物質の完全転化を示した後、該混合物はDecaliteおよびZn−Decaliteを通して濾過され、残渣はEtOAcで洗浄された。一緒にされた濾過液は、1NNaOH溶液、続いて水および塩水により洗浄された。有機層は乾燥され(Na2SO4)、濾過され、そして減圧下で濃縮された。残渣はメタノールに溶かされ、そしてSCX−2カラムを通して濾過された。カラムをメタノールですすいだ後に、所望の生成物は0.7Nアンモニア/メタノール溶液により溶出されて、標記化合物を与えた(340.1mg、74.7%)。
3級ブチル3−(4−アミノ−3,5−ジメチル−ピラゾール−1−イル)アゼチジン−1−カルボキシレート
3,5−ジメチル−4−ニトロ−1H−ピラゾールおよび1−Boc−3−ヒドロキシアゼチジンから出発して、中間体Ahのために記載されているのと類似の方法で、標記化合物は調製され、そして261.1mgの3級ブチル3−(4−アミノ−3,5−ジメチル−ピラゾール−1−イル)アゼチジン−1−カルボキシレートを与えた(定量的)。
3,5−ジメチル−1−(オキセタン−2−イルメチル)ピラゾール−4−アミン
3,5−ジメチル−4−ニトロ−1H−ピラゾールおよび2−ヒドロキシメチルオキセタンから出発し、中間体Ahのために記載されているのと類似の方法で、標記化合物は調製され、そして3,5−ジメチル−1−(オキセタン−2−イルメチル)ピラゾール−4−アミンを与えた(36.2%)。
3,5−ジエチル−1−[2−(2−メトキシエトキシ)エチル]ピラゾール−4−アミン
3,5−ジエチル−4−ニトロ−1H−ピラゾール(中間体Ac−b)および1−ブロモ−2−(2−メトキシエトキシ)−エタンから出発して、中間体Aaのために記載されているのと類似の方法で、標記化合物は調製されて、3,5−ジエチル−1−[2−(2−メトキシエトキシ)エチル]ピラゾール−4−アミン290mgが与えらた(72.2%)。
3,5−ジエチル−1−(オキセタン−3−イル)ピラゾール−4−アミン
3,5−ジエチル−4−ニトロ−1H−ピラゾール(中間体Ac−b)およびトルエン−4−スルホン酸オキセタン−3−イルエステルから出発して、中間体Aaのために記載されているのと類似の方法で、標記化合物は調製されて、3,5−ジエチル−1−(オキセタン−3−イル)ピラゾール−4−アミン165mgを与えた(47.7%。)。
1−(2−ジメチルアミノエチル)−3,5−ジメチル−ピラゾール−4−アミン
3,5−ジメチル−4−ニトロ−1H−ピラゾールおよびN,N−ジメチルエタノールアミンから出発し、中間体Ahのために記載されているのと類似の方法で、標記化合物は調製され、1−(2−ジメチルアミノエチル)−3,5−ジメチル−ピラゾール−4−アミン380.4mgを与えた(定量的)。
(a)エチル2−[4−[(2−ヒドロキシ−2−メチル−プロピル)カルバモイル]−2−メトキシ−アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート
エチル2−クロロ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(中間体1、200mg、0.72mmol)、4−アミノ−N−(2−ヒドロキシ−2−メチル−プロピル)−3−メトキシ−ベンズアミド(中間体A、172mg、0.72mmol)、そして炭酸セシウム、(937mg、2.89mmol)がジオキサン(20mL)中に懸濁された。窒素が混合物を通して30°Cで5分間泡立せられ、続いて9,9−ビス−ジメチル−4,5−ビス(ジフェニルホスフィノ)キサンテン(41.7mg、72μmol)およびトリス(ジベンジリデンアセトン)ジパラジウム(0)(33.0mg、36μモル)が加えられた。反応混合物は、窒素気体のフローの下で80℃で20時間撹拌された。酢酸エチル/水/塩水=1/1/1v/v%(50mL)が反応混合物に加えられ、そして撹拌が15分間続けられた。Decalite(商標)を通した濾過後、水層は分離され、酢酸エチル(2×20mL)で抽出された。一緒にされた有機層はその後水(40mL)、塩水(20mL)により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。粗生成物はシリカ上のカラムクロマトグラフィ(DCM/MeOH = 10/0 〜 9/1 v/v%)により精製されて、標記の化合物を与えた(190mg、55.3%)。
LiHDMS(THF/エチルベンゼン中1M、0.61mL、0.61mmol)は、THF(1mL)中の2,6−ジメチルアニリン(38.5μL、0.31mmol)の冷却溶液(0°C)に加えられた。0°Cで15分間撹拌後に、THF(3mL)中のエチル2−[4−[(2−ヒドロキシ−2−メチル−プロピル)カルバモイル]−2−メトキシ−アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(50mg、0.10mmol)が反応混合物に滴下により加えられ、そして撹拌が0°Cで90分間続けられた。追加のLiHMDS(100μL)が室温で滴下により加えられ、そして撹拌が室温で2時間続けられた。反応混合物は塩化アンモニウム飽和溶液20mLのによってクエンチされ、そして酢酸エチルにより抽出された。
一緒にされた有機層は、水、塩水により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。残渣は、分取HPLCにより精製された。生成物を含有している分画が集められ、そして凍結乾燥されて、N−(2,6−ジメチルフェニル)−2−[4−[(2−ヒドロキシ−2−メチル−プロピル)カルバモイル]−2−メトキシ−アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミドを得た(10mg、18%)。データ:LCMS(B)Rt:14.432分、m/z 555.3(M+H)+。
アセトニトリル(2mL)中の2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボニルクロライド(中間体2、52mg、0.07mmol理論値)の懸濁物に、2−クロロ−6−メチルアニリン(15μL、0.12mmol)および触媒量の4−DMAPが加えられた。反応混合物は50°Cで一晩撹拌された。溶媒の蒸発後、粗生成物は分取HPLCにより精製された。生成物を含有している分画は集められ、減圧下で濃縮された。残渣は、ジクロロメタンと5%NaHCO3溶液の間で分配された。有機相は、PE−フィルターを通して分離され、蒸発されて標記化合物28mgを得た(収率74%)。データ:LCMS(B)Rt:9.852分、m/z 542.2/544.2(M+H)+(塩素イオンパターン)。
中間体Cを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、その対応する酸クロリドから、標記化合物は調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.8mg、17%)。データ:LCMS(A)Rt:8.031分、m/z 587,1/589,1(M+H)+(臭素イオンパターン)。
商業的に入手可能な4−(4−メチルピペラジノ)アニリンを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−o−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18mg、58%)。データ:LCMS(B)Rt:10.456分、m/z 522.3(M+H)+。
中間体Dを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18mg、58%)。データ:LCMS(B)Rt:10.991分、m/z 530.2/532.2(M+H)+(塩素イオンパターン)。
中間体Eを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、11%)。データ:LCMS(B)Rt:10.886分、m/z 562.2(M+H)+。
中間体Oを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、20%)。データ:LCMS(B)Rt:10.469分、m/z 568.3(M+H)+。
中間体Nを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.7mg、10%)。データ:LCMS(B)Rt:10.222分、m/z 612.2/614.3(M+H)+。
中間体Bを使用して中間体2のために記載されているのと類似の方法で、2−[2−メチル−4−(4−メチルピペラジン−1−イル)アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボン酸は調製された。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−メトキシ−6−メチル−アニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(16.1mg、29.1%)。データ:LCMS(B)Rt:9.925分、m/z 554.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジイソプロピルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(15.9mg、35.7%)。データ:LCMS(B)Rt:12.686分、m/z 594.4(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後3,5−ジメチルイソキサゾール−4−アミンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(24.4mg、46.1%)。データ:LCMS(B)Rt:8.408分、m/z 529.3(M+H)+。
中間体Tを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aeと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14mg、27%)。データ:LCMS(B)Rt:9.531分、m/z 613.3(M+H)+。
中間体Pを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−o−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.3mg、14%)。データ:LCMS(B)Rt:12.313分、m/z 608.3(M+H)+。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Abと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14.1mg、37%)。データ:LCMS(B)Rt:7.902分、m/z 543.2(M+H)+。
中間体Hを出発原料として使用し、実施例9のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Aaとその後反応された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(5.2mg、28%)。データ:LCMS(B)Rt:8.140分、m/z 572.3(M+H)+。
中間体Hを出発原料として使用し、実施例9のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸エステルから調製された。実施例1のために記載されているのと類似の方法で、該カルボン酸エステルは2,6−ジメチルアニリンとその後反応された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(16mg、52%)。データ:LCMS(B)Rt:10.268分、m/z 524.3(M+H)+。
中間体Hを出発原料として使用し、実施例9のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Acとその後反応された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(7mg、32%)。データ:LCMS(B)Rt:8.065分、m/z 542.3(M+H)+。
実施例9のために記載されているのと同じ反応系列を用いかつ中間体Hを出発原料として使用して、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Aeとその後反応された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、32%)。データ:LCMS(B)Rt:9.146分、m/z 600.3(M+H)+。
中間体Fを出発原料として使用し、実施例1のために記載されているのと同じ反応系列を用いて、この化合物はその対応するエステルから調製された。実施例1に記載されている手順に従い、該エステルはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.5mg、23.2%)。データ:LCMS(C)Rt:12.686分、m/z 566.4(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(22mg、36%)。データ:LCMS(B)Rt:8.319分、m/z 557.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,4,6−トリメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(16mg、26%)。データ:LCMS(B)Rt:11.240分、m/z 552.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後(2−アミノ−3−メチル−フェニル)メタノールと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6mg、10%)。データ:LCMS(B)Rt:8.876分、m/z 554.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−フルオロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(21.4mg、39.6%)。データ:LCMS(B)Rt:9.878分、m/z 542.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後3−アミノ−2−クロロ−4−メチルピリジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.9mg、28.4%)。データ:LCMS(B)Rt:8.790分、m/z 559.2/561.2(M+H)+(塩素イオンパターン)。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(25.0mg、42.7%)。データ:LCMS(B)Rt:8.128分、m/z 586.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Abと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14.0mg、26.5%)。データ:LCMS(B)Rt:7.146分、m/z 528.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Adと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18mg、29%)。データ:LCMS(B)Rt:8.563分、m/z 562.2/564.2(M+H)+(塩素イオンパターン)。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−エチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14mg、26%)。データ:LCMS(B)Rt:10.708分、m/z 538.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジフルオロアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(20.5mg、37.5%)。データ:LCMS(B)Rt:9.607分、m/z 546.2(M+H)+。
中間体Gを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(10mg、37%)。データ:LCMS(B)Rt:8.939分、m/z 570.3(M+H)+。
中間体Gを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−O−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5mg、9%)。データ:LCMS(B)Rt:11.636分、m/z 566.3(M+H)+。
中間体Jを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Acとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(9.6mg、23%)。データ:LCMS(B)Rt:8.864分、m/z 592.3(M+H)+。
中間体Kを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.2mg、17.4%)。データ:LCMS(B)Rt:10.575分、m/z 510.3(M+H)+。
中間体Lを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(9.8mg、21%)。データ:LCMS(B)Rt:11.000分、m/z 600.2/602.2(M+H)+(塩素イオンパターン)。
中間体Mを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(2.5mg、11%)。データ:LCMS(B)Rt:11.332分、m/z 592.3(M+H)+。
中間体Qを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、12%)。データ:LCMS(B)Rt:11.282分、m/z 616.3(M+H)+。
(a)2−(4−ブロモ2メトキシ・アニリノ)−N−(2,6−ジエチルフェニル)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド
商業的に入手可能な4−ブロモ−2−メトキシアニリンを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(50mg)。
ジオキサン/水(1.5mL/0.3mL)中の2−(4−ブロモ−2−メトキシ−アニリノ)−N−(2,6−ジエチルフェニル)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(22.6mg、0.041mmol)、1−メチル−4−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1H−ピラゾール(17.2mg、0.083mmol)、1,1'−ビス(ジフェニルホスフィノ)フェロセン 塩化パラジウム(II)ジクロロメタン(3.4mg)付加物、及び炭酸カリウム(28.3mg、0.205mmol)の混合物は、封管中で140°Cで60分間、マイクロ波中で加熱された。環境温度に冷却した後、混合物は減圧下で濃縮された。得られた残渣は、酢酸エチルで希釈され、水および塩水で洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮された。分取HPLCを使用して精製は実施されて、標記化合物を得た(15.2mg、38%)。データ:LCMS(B)Rt:17.769分、m/z 548.3(M+H)+。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.8mg、37.7%)。データ:LCMS(B)Rt:11.129分、m/z 553.3(M+H)+。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−O−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8.7mg、21.9%)。データ:LCMS(B)Rt:11.778分、m/z 567.3(M+H)+。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.4mg、28.4%)。データ:LCMS(B)Rt:11.395分、m/z 573.2/575.2(M+H)+(塩素イオンパターン)。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.8mg、27.3%)。データ:LCMS(B)Rt:11.527分、m/z 617.2/619.2(M+H)+(臭素イオンパターン)。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.6mg、33.6%)。データ:LCMS(B)Rt:12.578分、m/z 581.3(M+H)+。
中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(16.9mg、40.1%)。データ:LCMS(B)Rt:8.940分、m/z 601.3(M+H)+。
中間体Tを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14.2mg、40%)。データ:LCMS(B)Rt:10.765分、m/z 537.3(M+H)+。
中間体Tを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(10.5mg、26%)。データ:LCMS(B)Rt:10.077分、m/z 557.2/559.2(M+H)+(塩素イオンパターン)。
中間体Tを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(7.6mg、19%)。データ:LCMS(B)Rt:8.307分、m/z 555.3(M+H)+。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(16.7mg、52.9%)。データ:LCMS(B)Rt:10.556分、m/z 527.3(M+H)+。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(15.6mg、52.9%)。データ:LCMS(B)Rt:10.566分、m/z 547.2/549.2(M+H)+(塩素イオンパターン)。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−O−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.2mg、53.1%)。データ:LCMS(B)Rt:11.075分、m/z 541.3(M+H)+。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(9.38mg、26.4%)。データ:LCMS(B)Rt:10.760分、m/z 590.2/592.2(M+H)+(臭素イオンパターン)。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.54mg、16.6%)。データ:LCMS(B)Rt:11.832分、m/z 555.3(M+H)+。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.4mg、41.0%)。データ:LCMS(B)Rt:8.189分、m/z 545.3(M+H)+。
中間体Uを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18.3mg、53.2%)。データ:LCMS(B)Rt:8.234分、m/z 575.3(M+H)+。
中間体Vを出発原料として使用し、実施例1−aおよび中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(3.7mg、17%)。データ:LCMS(B)Rt:11.254分、m/z 578.3(M+H)+。
中間体Wを使用し、実施例2aのために記載されているのと同じ反応系列を用いて、この化合物はその対応するエチルエステルから調製された。実施例1bに記載されている手順に従い、該エステルはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.5mg、22%)。データ:LCMS(B)Rt:13.950分、m/z 509.3(M+H)+。
中間体Xを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後6−エチル−O−トルイジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13mg、28%)。データ:LCMS(B)Rt:12.031分、m/z 580.3(M+H)+。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。カルボベンゾキシ基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(20mg、64%)。データ:LCMS(B)Rt:9.706分、m/z 508.3(M+H)+。
中間体Yを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8.1mg、25%)。データ:LCMS(B)Rt:12.158分、m/z 567.3(M+H)+。
(a)4−[(7−エトキシカルボニル−5,6−ジヒドロピリミド[4,5−e]インドリジン−2−イル)アミノ]−3−メトキシ−安息香酸
4−アミノ−3−メトキシ安息香酸を出発原料として使用し、中間体1aのために記載されているのと類似の方法で、この化合物は調製された。収量:160mg(43.5%)
DMF(2mL)中の4−[(7−エトキシカルボニル−5,6−ジヒドロピリミド[4,5−e]インドリジン−2−イル)アミノ]−3−メトキシ安息香酸(80mg、0.20mmol)および3−アミノ−1−メチルピペリジン二塩酸塩(36.6mg、0.20mmol)の撹拌された懸濁物に、DiPEA(139μL、0.84mmol)が加えられた。得られた溶液は0°Cへと冷却され、HATU(83.6mg、0.22mmol)が加えられた。冷却は取り除かれ、そして反応混合物は室温で一晩撹拌された。該混合物は、EtOAc/水/塩水1/1/1(30mL)の激しく撹拌された混合液に滴下により加えられた。その後、水層はEtOAc(2×10mL)により抽出された。一緒にされた有機層は水(20mL)、塩水(20mL)により洗浄され、Na2SO4で乾燥され、濾過され、そして減圧下で濃縮されて標記化合物を与えた(70mg、70%)。
粗エチル2−[2−メトキシ−4−[(1−メチル−3−ピペリジル)カルバモイル]アニリノ]−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレートの鹸化およびその後の中間体2bのために記載されているチオニルクロリドとの反応は、対応する酸クロリドを生成する。実施例2に記載されている方法に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8.4mg、19%)。データ:LCMS(B)Rt:11.406分、m/z 580.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジクロロアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(9mg、20.7%)。データ:LCMS(B)Rt:10.404分、m/z 578.2(M+H)+(塩素イオンパターン)。
エチル2−クロロピリミド[4,5−e]インドリジン−7−カルボキシレート
(a)エチル2−メトキシピリミド[4,5−e]インドリジン−7−カルボキシレート
DDQ(1.53g、6.76mmol)が、DCM(50mL)中のエチル2−メトキシ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレート(1.54g、5.63mmol)の撹拌された溶液に加えられた。反応混合物は、室温で3日間攪拌された。追加として200mgのDDQが加えられ、そして反応混合物は室温でさらに7日間撹拌された。該混合物は濾過され、小容量へと減圧下で濃縮された。粗生成物は、シリカ上のカラムクロマトグラフィ(ヘプタン/酢酸エチル=1/0〜1/1v/v%)により精製されて、標記の化合物を生成した(750mg、50%)。
ヨウ化ナトリウム(1.24g、8.29mmol)が、アセトニトリル(19mL)中のエチル2−メトキシ−ピリミド[4,5−e]インドリジン−7−カルボキシレート(750mg、2.76mmol)の撹拌された溶液に加えられた。アセトニトリル(3mL)中のトリメチルシリルクロリド(896mg、1.05mL)の溶液は、反応混合物に滴下により加えられた。該混合物は、室温で一晩撹拌された。 追加のアセトニトリル(6mL)中のヨウ化ナトリウム(3.33g)TMS−Cl(2.4g、2.8mL)が滴下により加えられ、そして該反応系は室温で3日間撹拌された。該混合物は、減圧下で濃縮された。残渣は200mLのDCM/MeOH(4/1)中で懸濁され、チオ硫酸ナトリウム(50mL)および水(100mL)の飽和溶液の混合物により抽出された。水層はDCM/MeOH(4/1、2×150mL)により抽出された。一緒にされた有機層は硫酸ナトリウムで乾燥され、濾過され、及び溶媒は減圧下で除去されて、固体を与えた。該固体は、沸騰酢酸エチル(50mL)中で砕かれた。冷却後、該固体は室温で1時間撹拌され、そして濾過された。残渣が減圧下に40℃で乾燥されて、1.0gの粗エチル2−ヒドロキシ−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキシレートを与えた(定量的収率)。
N、N−ジメチルアニリン(47mg、50μL、1.50mmol)が、アセトニトリル(30mL)中のエチル2−ヒドロキシピリミド[4,5−e]インドリジン−7−カルボキシレート(1.0g、3.89mmol)の溶液に加えられた。アセトニトリル(4mL)中のオキシ塩化リン(V)(2.99g、1.81mL、19.5mmol)の溶液は、反応混合物に滴下により加えられた。茶色/赤の該懸濁物は、65°Cまで4時間加熱された。冷却後、25%水性アンモニア(50mL)の撹拌された混合物および氷水(100mL)中に、10°C未満の温度を保ちながら、混合物はゆっくり注がれた。さらに15分間撹拌した後に、該混合物は酢酸エチルにより抽出された。一緒にされた有機層は、その後、水(50mL)、0.2N HCl(50mL)、塩水(25mL)により洗浄され、乾燥され(Na2SO4)、濾過され、そして減圧下で濃縮された。粗生成物は、シリカ上のカラムクロマトグラフィ(ヘプタン/酢酸エチル=1/0〜1/1v/v%)により精製されて、標記の化合物200mgを生成した。
中間体3及び中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(30mg、45%)。データ:LCMS(B)Rt:12.491分、m/z 551.3(M+H)+。
中間体Bを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17mg、47%)。データ:LCMS(B)Rt:9.602分、m/z 522.3(M+H)+。
中間体Bを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−エチル−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14mg、65%)。データ:LCMS(B)Rt:10.303分、m/z 536.3(M+H)+。
中間体Bを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(30mg、73%)。データ:LCMS(B)Rt:9.914分、m/z 586.2/588.2(M+H)+(臭素イオンパターン)。
中間体Bを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジクロロアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(10mg、36%)。データ:LCMS(B)Rt:9.770分、m/z 562.2(M+H)+(塩素イオンパターン)。
中間体Bを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13mg、34%)。データ:LCMS(B)Rt:10.954分、m/z 550.3(M+H)+。
中間体Dを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(9.6mg、27%)。データ:LCMS(B)Rt:10.844分、m/z 510.2(M+H)+。
中間体Dを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−エチル−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.2mg、30.4%)。データ:LCMS(B)Rt:11.599分、m/z 524.3(M+H)+。
中間体Dを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(12mg、30%)。データ:LCMS(B)Rt:11.151分、m/z 574.2/576.2(M+H)+(臭素イオンパターン)。
中間体3及び中間体Fから出発し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(0.7mg)。データ:LCMS(A)Rt:6.446分、m/z 564.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−エチル−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.8mg、32.2%)。データ:LCMS(B)Rt:10.836分、m/z 552.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18.1mg、30%)。データ:LCMS(B)Rt:10.427分、m/z 602.2/604.2(M+H)+(臭素イオンパターン)。
出発原料として中間体3及び中間体Hから出発し、実施例1のために記載されているのと同じ反応系列を用いて、この化合物はその対応するエチルエステルから調製された。実施例1に記載されている手順に従い、該エチルエステルはその後2,6−ジメチルアニリンと反応された。50%TFA/ジクロロメタンを用いたBoc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(1.4mg、5%)。データ:LCMS(B)Rt:10.283分、m/z 524.3(M+H)+。
出発原料として中間体1及び中間体Hから出発し、実施例1のために記載されているのと同じ反応系列を用いて、この化合物はその対応するエチルエステルから調製された。実施例1に記載されている手順に従い、該エチルエステルはその後2−クロロ−6−メチルアニリンと反応された。50%TFA/ジクロロメタンを用いたBoc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(0.9mg、4%)。データ:LCMS(A)Rt:5.794分、m/z 544.2/546.2(M+H)+(塩素イオンパターン)。
出発原料として中間体1及び中間体Hから出発し、実施例1のために記載されているのと同じ反応系列を用いて、この化合物はその対応するエチルエステルから調製された。実施例1に記載されている手順に従い、該エチルエステルはその後2,6−ジエチルアニリンと反応された。50%TFA/ジクロロメタンを用いたBoc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(20.7mg、72%)。データ:LCMS(A)Rt:6.213分、m/z 552.3(M+H)+。
実施例76の分取HPLC精製の間の副生成物としてこの化合物は単離されて、標記化合物を得た(2mg、7%)。データ:LCMS(A)Rt:6.512分、m/z 550.3(M+H)+。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。カルボベンゾキシ基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(24mg、81%)。データ:LCMS(B)Rt:9.831分、m/z 528.2/530.2(M+H)+(塩素イオンパターン)。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジクロロアニリンと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(27mg、96%)。データ:LCMS(B)Rt:9.853分、m/z 548.2(M+H)+(塩素イオンパターン)。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(18mg、40%)。データ:LCMS(B)Rt:10.916分、m/z 536.3(M+H)+。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−ブロモ−6−メチルアニリンと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(16mg、51%)。データ:LCMS(B)Rt:9.922分、m/z 572.2/574.2(M+H)+(臭素イオンパターン)。
中間体Iを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−エチル−6−メチルアニリンと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(22mg、77%)。データ:LCMS(B)Rt:10.361分、m/z 522.3(M+H)+。
中間体Wを使用し、中間体2aのために記載されているのと同じ反応系列を用いて、この化合物はその対応するエチルエステルから調製された。実施例1bに記載されている手順に従い、該エステルはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.2mg、13%)。データ:LCMS(B)Rt:15.695分、m/z 537.3(M+H)+。
中間体Yを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、12%)。データ:LCMS(B)Rt:10.832分、m/z 539.2(M+H)+。
商業的に入手可能な4−(1−メチル−piperidin−4−イル)−アニリンを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(18.3mg、44%)。データ:LCMS(B)Rt:8.405分、m/z 555.3(M+H)+。
中間体Jを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−クロロ−6−メチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.2mg、41%)。データ:LCMS(B)Rt:11.569分、m/z 594.2/596.2(M+H)+(塩素イオンパターン)。
4−ブロモ−2−エチルアニリンを出発原料として使用し、実施例38−aのために記載されているのと同じ反応系列を用いて、この化合物はその対応するブロミドから調製された。実施例38−bに記載されている手順に従い、該ブロミドはその後2N−メチルピペラジンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.5mg、15%)。データ:LCMS(B)Rt:12.136分、m/z 564.4(M+H)+。
N,N,N−トリメチル−1,3−プロパンジアミンを出発原料として使用し、実施例60のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aeと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.8mg、20%)。データ:LCMS(B)Rt:10.220分、m/z 658.4(M+H)+。
中間体Zを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(6.6mg、17%)。データ:LCMS(B)Rt:7.975分、m/z 543.3(M+H)+。
中間体ZAを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(16.2mg、46%)。データ:LCMS(B)Rt:14.477分、m/z 583.3(M+H)+。
中間体ZBを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(12.8mg、25%)。データ:LCMS(B)Rt:8.441分、m/z 559.3(M+H)+。
中間体ZBを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(8.9mg、19%)。データ:LCMS(B)Rt:8.324分、m/z 529.3(M+H)+。
中間体ZBを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aeと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(14.9mg、34%)。データ:LCMS(B)Rt:9.618分、m/z 587.4(M+H)+。
中間体Rを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8.7mg、21%)。データ:LCMS(B)Rt:12.704分、m/z 596.3(M+H)+。
中間体3及び中間体Sを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(23mg、32%)。データ:LCMS(B)Rt:9.796分、m/z 599.3(M+H)+。
中間体3及び中間体ZEから出発し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(14mg、61%)。データ:LCMS(B)Rt:9.058分、m/z 570.3(M+H)+。
中間体3及び中間体ZEから出発し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Acと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(10.6mg、39%)。データ:LCMS(B)Rt:8.908分、m/z 540.3(M+H)+。
中間体ZEを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Afと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(11.4mg、28%)。データ:LCMS(B)Rt:7.882分、m/z 586.3(M+H)+。
中間体ZCを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(14.6mg、23%)。データ:LCMS(B)Rt:12.878分、m/z 569.4(M+H)+。
中間体ZCを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aeと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.2mg、25%)。データ:LCMS(B)Rt:9.443分、m/z 617.4(M+H)+。
中間体ZDを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.8mg、27%)。データ:LCMS(B)Rt:7.187分、m/z 571.4(M+H)+。
分取HPLCを使用して、実施例20中に記載された化合物からの副生成物としてこの化合物は単離されて、標記化合物を得た(9.1mg)。データ:LCMS(B)Rt:14.555分、m/z 601.3/603.3(M+H)+(塩素イオンパターン)。
(a)2−(4−ブロモ2メトキシ・アニリノ)−N−(2,6−ジメチルフェニル)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド
商業的に入手可能な4−ブロモ−2−メトキシアニリンを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応され、標記化合物を得た(1.35g、84%)。
DMF(4mL)中の2−(4−ブロモ−2−メトキシ−アニリノ)−N−(2,6−ジメチルフェニル)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(1.35g、2.6mmol)およびシアン化亜鉛(321mg、2.73mmol)の溶液に、テトラキス(トリフェニルホスフィン)パラジウム(0)(300mg、0.26mmol)が加えられた。反応混合物は、マイクロ波照射下で170°Cで30分間加熱された。環境温度に冷却した後、該混合物は濃縮され、そして残渣は酢酸エチルで希釈され、水および塩水で洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、標記粗化合物を得た(1.05g、87%)。
MeOH(25mL)中の2−(4−シアノ−2メトキシ・アニリノ)−N−(2,6−ジメチルフェニル)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(750mg、1.61mmol)の撹拌された懸濁物に、水(12.5mL)中の水酸化カリウム(453mg、8.07mmol)溶液が加えられた。反応混合物は、マイクロ波照射下で120°Cで2時間加熱された。メタノール分画の蒸発後、得られた水層は2N HCl溶液の添加によってpH〜2まで酸性化された。ジクロロメタンでの抽出後、一緒にされた有機層はPE−フィルターを通して濾過されて、標記化合物330mgを与えた(収率:42%)。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および4−アミノテトラヒドロピラン塩酸塩から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5mg、18%)。データ:LCMS(B)Rt:14.407分、m/z 567.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および3−アミノ−1−N−Boc−アゼチジンから、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(6.8mg、20%)。データ:LCMS(B)Rt:11.597分、m/z 538.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例17)およびメトキシ酢酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(10mg、49%)。データ:LCMS(B)Rt:12.973分、m/z 596.3(M+H)+。
中間体ZFを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.9mg、20%)。データ:LCMS(B)Rt:7.658分、m/z 588.4(M+H)+。
中間体ZFを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(3.1mg、11%)。データ:LCMS(B)Rt:12.119分、m/z 568.4(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Agとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.3mg、45%)。データ:LCMS(B)Rt:9.102分、m/z 543.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応するカルボン酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Aeとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(21.5mg、76%)。データ:LCMS(B)Rt:9.108分、m/z 614.4(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例17)および3−フルオロシクロ−ブタンカルボン酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(26mg、99%)。データ:LCMS(B)Rt:15.532分、m/z 624.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例17)および3−メチル−オキセタン−3−カルボン酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4mg、17%)。データ:LCMS(B)Rt:13.824分、m/z 622.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例17)およびシクロプロパンカルボン酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11mg、49%)。データ:LCMS(B)Rt:14.744分、m/z 592.3(M+H)+。
DCM(2mL)中のN−(2,6−ジメチルフェニル)−2−(2−メトキシ−4−ピペラジン−1−イル−アニリノ)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(実施例17、20mg、0.038mmol)の撹拌された溶液に、トリエチルアミン(18μL、0.13mmol)およびブタン−1−スルホニルクロリド(5μL、0.042mmol)が加えられた。反応混合物は、一晩室温で撹拌された。該混合物は濃縮され、そして残渣が分取HPLCを使用して精製されて、標記化合物を得た(3mg、13%)。データ:LCMS(B)Rt:17.533分、m/z 644.3(M+H)+。
DCM(2mL)中のN−(2,6−ジメチルフェニル)−2−(2−メトキシ−4−ピペラジン−1−イル−アニリノ)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(実施例17、20mg、0.038mmol)の撹拌された溶液に、イソシアン酸エチル(4μL、0.042mmol)が加えられた。反応混合物は、一晩室温で撹拌された。該混合物は濃縮され、そして残渣が分取HPLCを使用して精製されて、標記化合物を得た(24mg、99%)。データ:LCMS(B)Rt:13.414分、m/z 595.3(M+H)+。
中間体ZGを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aaと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(19.2mg、29%)。データ:LCMS(B)Rt:9.296分、m/z 600.4(M+H)+。
中間体ZHを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(12.2mg、17%)。データ:LCMS(B)Rt:12.915分、m/z 566.4(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および(3S,4R)−4−アミノ−1−Boc−3−メトキシ−ピペリジンから、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(2.7mg、7%)。データ:LCMS(B)Rt:11.799分、m/z 596.4(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および3級ブチル(3S,4R)−4−アミノ−3−フルオロ−ピペリジン−1−カルボキシレートから、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(2.2mg、5%)。データ:LCMS(B)Rt:11.671分、m/z 584.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および1−N−Boc−シス−1,4−シクロヘキシルジアミン塩酸塩から、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(4.6mg、15%)。データ:LCMS(B)Rt:11.581分、m/z 580.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)およびイソプロピルアミン塩酸塩から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(2.9mg、8%)。データ:LCMS(B)Rt:15.663分、m/z 525.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および(1R,5S)−3−オキサビシクロ[3.1.0]ヘキサン−6−アミン塩酸塩から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.5mg、15%)。データ:LCMS(B)Rt:14.186分、m/z 565.2(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)およびモルホリンから、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8.7mg、15%)。データ:LCMS(B)Rt:14.262分、m/z 553.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)およびチオモルホリン1,1−ジオキシドから、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(10.5mg、14%)。データ:LCMS(B)Rt:13.936分、m/z 601.2(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および1−エチル−1,4−ジアゼパン二塩酸塩から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.4mg、7%)。データ:LCMS(B)Rt:11.510分、m/z 594.4(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および3,3−ジフルオロシクロブタンアミン塩酸塩から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.1mg、7%)。データ:LCMS(B)Rt:16.415分、m/z 573.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)およびシクロプロピルアミンから、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.7mg、10%)。データ:LCMS(B)Rt:14.833分、m/z 523.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および2−メトキシエチルアミンから、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.9mg、8%)。データ:LCMS(B)Rt:14.283分、m/z 541.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および2−(1−ピペリジル)エタンアミンから、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(4.2mg、5%)。データ:LCMS(B)Rt:12.340分、m/z 594.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するカルボン酸(実施例103−c)および4−メチルピペリジン−4−カルボニトリル塩酸塩から、この化合物は調製された。精製はシリカゲル上のフラッシュクロマトグラフィ(ジクロロメタン/メタノール=99/1〜9/1v/v%)により実施されて、標記化合物を得た(3.9mg、5%)。データ:LCMS(B)Rt:5.853分、m/z 590.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例17)および1−3級ブトキシカルボニル−3−メチル−アゼチジン−3−カルボン酸から、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(10mg、39%)。データ:LCMS(B)Rt:11.186分、m/z 621.4(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例16)および酢酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17mg、45%)。データ:LCMS(B)Rt:10.096分、m/z 614.3(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例16)および3−フルオロシクロブタンカルボン酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(8mg、20%)。データ:LCMS(B)Rt:12.238分、m/z 672.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2−アミノ−3−メチルベンゾニトリルと反応された。精製はシリカゲル上のフラッシュクロマトグラフィ(ジクロロメタン/メタノール=99/1〜9/1v/v%)により実施されて、標記化合物を得た(8.3mg、10%)。データ:LCMS(B)Rt:10.090分、m/z 549.3(M+H)+。
中間体ZIを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.6mg、14%)。データ:LCMS(B)Rt:12.601分、m/z 582.4(M+H)+。
中間体ZIを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Ahと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(5.7mg、11%)。データ:LCMS(B)Rt:8.660分、m/z 690.4(M+H)+。
中間体ZJを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジエチルアニリンと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(13.4mg、30%)。データ:LCMS(B)Rt:17.314分、m/z 542.3(M+H)+。
中間体ZJを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Ahと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(6.7mg、13%)。データ:LCMS(B)Rt:12.186分、m/z 650.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Ahと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.6mg、28.6%)。データ:LCMS(B)Rt:6.985分、m/z 674.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Aiと反応された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(6.9mg、19.7%)。データ:LCMS(B)Rt:4.784分、m/z 583.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Ajと反応された。精製はシリカゲル上のフラッシュクロマトグラフィ(ジクロロメタン/メタノール=99/1〜9/1v/v%)により実施されて、標記化合物を得た(22.1mg、61.6%)。データ:LCMS(B)Rt:6.495分、m/z 598.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Akと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(22mg、54.4%)。データ:LCMS(B)Rt:7.845分、m/z 658.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Alと反応された。精製はシリカゲル上のフラッシュクロマトグラフィ(ジクロロメタン/メタノール=99/1〜9/1v/v%)により実施されて、標記化合物を得た(23.8mg、64.8%)。データ:LCMS(B)Rt:7.412分、m/z 612.3(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後中間体Amと反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(11.5mg、32.0%)。データ:LCMS(B)Rt:4.944分、m/z 599.3(M+H)+。
中間体Jを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸クロリドから調製された。実施例2に記載されている手順に従い、該酸クロリドはその後2,6−ジメチルアニリンと反応された。データ:LCMS(B)Rt:11.461分、m/z 574.3(M+H)+。
DCM(2mL)中のN−(2,6−ジメチルフェニル)−2−(2−メトキシ−4−ピペラジン−1−イル−アニリノ)−5,6−ジヒドロピリミド[4,5−e]インドリジン−7−カルボキサミド(実施例17、37mg、0.063mmol)の撹拌された溶液に、2−イソシアン酸プロピル(8μL、0.07mmol)が加えられた。反応混合物は、一晩室温で撹拌された。該混合物は濃縮され、そして残渣が分取HPLCを使用して精製されて、標記化合物を得た(6mg、16%)。データ:CMS(B)Rt:14.299分、m/z 609.4(M+H)+。
TTK酵素活性に対する化合物の阻害活性を測定するために、IMAP(商標)アッセイ(Molecular Devices社)が使用された。化合物はジメチルスルホキシド(DMSO)中で、続いてその後IMAP反応緩衝液(10mMトリス−HCl、pH 7.5、10mMMgCl2、0.01%Tween−20、0.1%NaN3、および新たに調製された1mMジチオトレイトール(DTT)から成る)で希釈された。該化合物の溶液は、IMAP反応緩衝液中の完全長のTTK酵素(Life Technologies, Cat. no. PV 3792)と等用量で混ぜ合わせられた。暗所に室温で1時間プレインキュベーションの後、フルオレセイン標識MBP由来の基質ペプチド(Molecular Devices社、cat. no. RP 7123)、続いてATPが加えられ、反応が始められた。最終酵素濃度は50nM、最終基質濃度は3.9nM、そして最終ATP濃度は5μMだった。反応は、暗所に室温で2時間進行されることができた。製造業者(MolecularDevices社)のプロトコ−ルに従って、IMAP progressive binding solutionでのクエンチングによって反応は止められた。フルオレセインの分極は、Envisionマルチモードリーダー(Perkin Elmer, Waltham, MA, U.S.A.)上で測定された。IC50は、XLfit(商標)5ソフトウェアを使用して算出された(ID Business Solutions,Ltd., Surrey, U.K.)。すべての例証された化合物のIC50値が、100nMより小さいとわかった。
MOLT−4癌細胞系は米国菌培養収集所(ATCC、Manassas、VA、米国)から購入され、そして10%ウシ成体血清で補充されたRPMI 1640培地(LifeTechnologies、Bleiswijk、Netherlands)で培養された。化合物は100%DMSO中での段階的に3.16倍に連続して希釈され、続いて水性緩衝液中で更に希釈された。培地中の細胞は384ウェルのウェル中でウェル当たり45μLで播種され、37°Cで5%CO2の湿性雰囲気中で24時間インキュベートされた。化合物溶液5μLが加えられ、そして該プレートはさらに72時間インキュベートされ、その後ATPlite 1Step(商標)(PerkinElmer、Groningen、Netherlands)溶液25μLが各々のウェルに加えられた。発光は、Envisionマルチモードリーダーに記録された。細胞増殖と細胞死を区別するために、インキュベーションの開始時に細胞シグナルが記録された。加えて、0.4%DMSOの存在下で、最大増殖は化合物がない複製のインキュベーションで測定された。成長パーセントが、メインのy軸シグナルとして使われた。最大シグナル、最小シグナル、ヒルパラメータおよびIC50を示す4−パラメータロジスティック曲線を使用し、IDBS XLfit(商標)5を使用した非線形回帰によってIC50は合わせられた。
1−[2−(2−エトキシエトキシ)エチル]−3,5−ジメチル−ピラゾール−4−アミン
(a)2−(2−エトキシエトキシ)エチル4−メチルベンゼンスルホナート
0°Cに冷やされたTHF15mL中のジ(エチレングリコール)エチルエーテル(4.92ml、36.2mmol)の溶液に、水15mL中に溶かされた水酸化ナトリウム(2.46g、61.5mmol)が激しく撹拌しながら加えられた。この混合物に、THF15mL中のトシルクロリド(8.28g、43.4mmol)の溶液が0°Cで10分間にわたり滴下により加えられた。冷却はその後取り除かれ、そして反応混合物は窒素下で1時間撹拌された。TLC分析が出発原料の完全転化を示したあと、該混合物はジエチルエーテル(2×50mL)二回抽出され、そして有機層は1N NaOH溶液(25mL)および水(25mL)により洗浄された。有機層は乾燥され(Na2SO4)、濾過され、そして溶媒は減圧下で取り除かれて、無色の液体として粗2−(2−エトキシエトキシ)エチル4−メチルベンゼンスルホナート10gを生成した(95.8%)。
DMF(10mL)中の3,5−ジメチル−4−ニトロ−1H−ピラゾール(1g、7.08mmol)および炭酸セシウム(2.31g、7.08mmol)の溶液に、2−(2−エトキシエトキシ)エチル4−メチルベンゼンスルホナート(2.04g、7.08mmol)が加えられた。混合物は100°Cで1時間加熱された。該混合物は室温に冷やされ、水/塩水に注がれ、酢酸エチル(100mL)により抽出された。一緒にされた有機層は、塩水(50mL)により洗浄され、硫酸ナトリウムで乾燥され、濾過され、そして減圧下で濃縮されて、標記化合物1.69gを生成した(92.8%)。
メタノール(25mL)中の1−[2−(2−エトキシエトキシ)エチル]−3,5−ジメチル−4−ニトロピラゾール(1.69g、6.57mmol)の撹拌された溶液に、エタノール(1mL)中の炭素上の10%パラジウム(200mg)の懸濁物が加えられた。反応混合物は、窒素雰囲気下、室温で15分間撹拌された。続いて、蟻酸アンモニウム(4.14g、65.7mmol)が加えられ、反応混合物は還流温度まで15分間加熱された。反応混合物は冷却され、Decalite(商標)を通して濾過され、減圧下で濃縮された。残渣はメタノールに溶かされ、そしてSCX−2カラムを通して濾過された。カラムをメタノールで洗い落した後、所望の生成物が0.7Nアンモニア/メタノール溶液により溶出された。得られた溶出液は、減圧で濃縮され、標記化合物を与えた(520mg、34.8%)。
1−[2−(2−メトキシエトキシ)エチル]−3,5−ジメチル−ピラゾール−4−アミンジエチレングリコールメチルエーテルおよび3,5−ジメチル−4−ニトロ−1H−ピラゾールから出発し、中間体Ahのために記載されているのと類似の方法で、標記化合物は調製され、そして1−[2−(2−メトキシエトキシ)エチル]−3,5−ジメチル−ピラゾール−4−アミン500mgを与えた(34.8%)。
1−[2−(2−エトキシエトキシ)エチル]−3,5−ジエチル−ピラゾール−4−アミン
3,5−ジエチル−4−ニトロ−1H−ピラゾール(中間体Ac−b)およびジ(エチレングリコール)エチルエーテルから出発して、中間体Anのために記載されているのと類似の方法で、標記化合物は調製されて、1−[2−(2−エトキシエトキシ)エチル]−3,5−ジエチル−ピラゾール−4−アミン550mgが与えらた(79.8%)。
3,5−ジエチル−1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]ピラゾール−4−アミン
3,5−ジエチル−4−ニトロ−1H−ピラゾール(中間体Ac−b)およびトリエチレングリコールモノメチルエーテルから出発して、中間体Anのために記載されているのと類似の方法で、標記化合物は調製されて、3,5−ジエチル−1−[2−[2−(2−メトキシエトキシ)エトキシ]エチル]ピラゾール−4−アミン660mgが与えらた(41.7%)。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Anとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(32.3mg、53.9%)。データ:LCMS(B)Rt:7.432分、m/z 644.6(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体青とその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(31.5mg、53.7%)。データ:LCMS(B)Rt:6.806分、m/z 630.7(M+H)+。
中間体Fを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Apとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(34.2mg、54.8%)。データ:LCMS(B)Rt:8.430分、m/z 672.7(M+H)+。
中間体ZEを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Apとその後反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(147.4mg、93.4%)。データ:LCMS(A)Rt:4.376分、m/z 658.7(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例151)およびメトキシ酢酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.0mg、61.4%)。データ:LCMS(B)Rt:10.554分、m/z 730.7(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例151)およびBoc−N−エチル−グリシンから、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(15.0mg、53.1%)。データ:LCMS(B)Rt:8.619分、m/z 743.8(M+H)+。
実施例9に記載されている標準HATU−カップリング方法を使用して、中間体2、中間体ZEおよび中間体Aq、およびメトキシ酢酸を使用している実施例151の為に記載されたのと類似の方法で、この化合物はその対応するアミンから調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.8mg、57.1%)。データ:LCMS(B)Rt:9.908分、m/z 760.8(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例16)および1−メチル−3−アゼチジンカルボン酸から、この化合物は調製された。分取HPLCを使用して精製は実施されて、標記化合物を得た(0.5mg、1%)。データ:LCMS(A)Rt:3.892分、m/z 669.7(M+H)+。
中間体ZEを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Ahとその後反応された。Cbz基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(189.4mg、95.7%)。データ:LCMS(A)Rt:3.811分、m/z 660.7(M+H)+。
実施例9に記載されている標準HATUカップリング方法を用いて、その対応するアミン(実施例156)およびBoc−N−エチル−グリシンから、この化合物は調製された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(18.9mg、61.8%)。データ:LCMS(B)Rt:7.255分、m/z 745.8(M+H)+。
中間体ZEを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Akとその後反応された。Cbz基の脱保護の後、実施例9に記載されている標準HATUカップリング方法を用いてBoc−N−エチル−グリシンは導入された。Boc基の脱保護の後、分取HPLCを使用して精製は実施されて、標記化合物を得た(15.8mg、54.2%)。データ:LCMS(B)Rt:8.002分、m/z 729.8(M+H)+。
中間体ZEを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Akとその後反応された。Cbz基の脱保護の後、実施例9に記載されている標準HATUカップリング方法を用いてメトキシ酢酸は導入された。分取HPLCを使用して精製は実施されて、標記化合物を得た(17.3mg、60.4%)。データ:LCMS(B)Rt:9.848分、m/z 716.7(M+H)+。
中間体Rを出発原料として使用し、中間体2のために記載されているのと同じ反応系列を用いて、この化合物はその対応する酸から調製された。実施例9のために記載されているのと類似の方法で、該カルボン酸は中間体Ahとその後反応された。分取HPLCを使用して精製は実施されて、標記化合物を得た(19.5mg、42.6%)。データ:LCMS(B)Rt:10.946分、m/z 684.7(M+H)+。
オーロラ(Aurora)Aに対する化合物の阻害活性を決定するために、LANCE(商標)Ultra TR-FRETアッセイ(Perkin Elmer)が使用された。化合物はジメチルスルホキシド(DMSO)中で、続いてその後LANCE(商標)キナーゼ緩衝液(50mMHepes、pH 7.5、10mMMgCl2、1mMEGTA、0.01%Tween−20、および2mMジチオトレイトール(DTT)から成る)中の4%DMSO中で希釈された。化合物溶液2.5μlは、LANCE(商標)キナーゼ緩衝液中の完全長オーロラ(Aurora)A酵素(Carna Biosciences社、cat. no. 05-101)と等容量で混合された。暗所に室温で1時間プレインキュベーションの後、ULight(商標)標識PLK(Ser137)基質ペプチド(Perkin Elmer社, cat. no. TRF-0110)及びATPが加えられ、反応が始められた。最終酵素濃度は2.5nMであり;最終基質濃度は、25nMであり;最終ATP濃度は、2μMであり;各々のウェルの最終DMSO濃度は、1%であった。2時間後、100mM EDTA5μlの添加によって反応は止められた。室温での5分間のインキュベーションの後、ユウロピウム標識抗ホスホPLK(Ser137)基質抗体(Perkin Elmer社, cat. no. TRF-0203)5μlが加えられ、そしてインキュベーションは暗所に室温で1時間続けられた。時間分解蛍光はEnvisionマルチラベルリーダー(Perkin Elmer社、Waltham、MA、米国)上で測られた。IC50は、XLfit(商標)5ソフトウェアを使用して算出された(ID Business Solutions,Ltd., Surrey, U.K.)。実施例33、138、141、148、149、150、151、152、153、154、155、156、157、158、159、および160の化合物は、100nM以上のIC50値を示した。
オーロラ(Aurora)Cに対する化合物の阻害活性を決定するために、LANCE UltraTR-FRETアッセイ(Perkin Elmer)が使用された。化合物はジメチルスルホキシド(DMSO)中で、続いてその後LANCE(商標)キナーゼ緩衝液(50mMHepes、pH 7.5、10mMMgCl2、1mMEGTA、0.01%Tween−20、および2 mMジチオトレイトール(DTT)から成る)中の4%DMSO中で希釈された。化合物溶液2.5μlは、LANCE(商標)キナーゼ緩衝液中の完全長オーロラ(Aurora)C酵素(Carna社、cat. no. 05-103)と等容量で混合された。暗所に室温で1時間プレインキュベーションの後、ULight(商標)標識PLK(Ser137)基質ペプチド(Perkin Elmer社, cat. no. TRF-0110)及びATPが加えられ、反応が始められた。最終酵素濃度は10nMであり;最終基質濃度は25nMであり;最終ATP濃度は5μMであり;各々のウェルの最終DMSO濃度は1%であった。3時間後、100mM EDTA5μlの添加によって反応は止められた。室温での5分間のインキュベーションの後、ユウロピウム標識抗ホスホPLK(Ser137)基質抗体(Perkin Elmer社, cat. no. TRF-0203)5μlが加えられ、そしてインキュベーションは暗所に室温で1時間続けられた。時間分解蛍光はEnvisionマルチラベルリーダー(Perkin Elmer社、Waltham、MA、米国)上で測られた。IC50は、XLfit(商標)5ソフトウェア(ID Business Solutions, Ltd.,Surrey, U.K.)を使用して算出された。
ポロ様キナーゼ1(PLK1)に対する化合物の阻害活性を決定するために、LANCE(商標)Ultra TR-FRETアッセイ(Perkin Elmer)が使用された。化合物はジメチルスルホキシド(DMSO)中で、続いてその後LANCE(商標)キナーゼ緩衝液(50mMHepes、pH 7.5、10mMMgCl2、1mMEGTA、0.01%Tween−20、および2 mMジチオトレイトール(DTT)から成る)中の4%DMSO中で希釈された。化合物溶液2.5μlは、LANCE(商標)キナーゼ緩衝液中の完全長PLK1酵素(Carna社、cat. no. 05-157)と等容量で混合された。
暗所に室温で1時間プレインキュベーションの後、ULight(商標)標識p70S6K(Thr389)基質ペプチド(PerkinElmer社, cat. no. TRF-0126)及びATPが加えられ、反応が始められた。最終酵素濃度は7.5nMであり;最終基質濃度は50nMであり;最終ATP濃度は5μMであり;各々のウェルの最終DMSO濃度は1%であった。4時間後、100mM EDTA5μlの添加によって反応は止められた。室温での5分間のインキュベーションの後、ユウロピウム標識抗ホスホp70S6(Thr389)基質抗体(Perkin Elmer社, cat. no. TRF-0214)5μlが加えられ、そしてインキュベーションは暗所に室温で1時間続けられた。時間分解蛍光はEnvisionマルチラベルリーダー(Perkin Elmer社、Waltham、MA、米国)上で測られた。IC50は、XLfit(商標)5ソフトウェア(ID Business Solutions, Ltd.,Surrey, U.K.)を使用して算出された。
Claims (15)
- 式Iの化合物:
またはそれの薬学的に許容される塩、ここで、
R1は、以下からなる群から選択される:
ここで、
R11は、H、ハロゲン、(1−2C)アルキル、(2−3C)アルケニル、(2−3C)アルキニル、(1−2C)アルコキシ、またはOC2H3であり、アルキルおよびアルコキシ基のすべてが一つ以上のハロゲンによって任意的に置換されていても良く;
R12は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり;
R13は、R131CH2、R132O、R133R134N、R135C(O)、R136S、R136S(O)、R136S(O)(NH)、R137SO2、(2−7C)ヘテロシクロアルキル、又は(1−5C)ヘテロアリールであり、ここでヘテロシクロアルキルまたはヘテロアリールが各々(1−2C)アルキル、フルオロ、ヒドロキシル、オキソ、(1−2C)アルコキシ、(1−6C)アルキルカルボニル、(1−6C)アルキルスルホニル、(1−5C)アルコキシカルボニル、(1−6C)アルキルアミノカルボニル、(3−6C)シクロアルキルカルボニル、(2−7C)ヘテロシクロアルキルカルボニル、またはジ[(1−6C)アルキル]アミノで任意的に置換されていても良く、ここでアルキルカルボニル、アルキルスルホニル、アルコキシカルボニル、アルキルアミノカルボニル、シクロアルキルカルボニル、またはヘテロシクロアルキルカルボニルが、各々(1−2C)アルキル、フルオロ、ヒドロキシル、シアノ、オキソ、または(1−2C)アルコキシによって任意的に置換されていても良く;
R131は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、または(1−2C)アルコキシから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキルカルボニルアミノ、(3−6C)シクロアルキルカルボニルアミノ、または(2−7C)ヘテロシクロアルキルカルボニルアミノであり;
R132は、各々(1−2C)アルキル、ハロゲン、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、または(2−7C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(6−10C)アリール、または(1−5C)ヘテロアリールであり;
R133は、各々(1−2C)アルキル、ハロゲン、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、または(2−7C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル(1−6C)アルキルカルボニル、(1−5C)アルコキシカルボニル、(3−6C)シクロアルキルカルボニル、(2−7C)ヘテロシクロアルキルカルボニルであり;
R134は、水素または(1−2C)アルキルであり;
R135は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、(2−7C)ヘテロシクロアルキル、オキソ、シアノ、又はアミノから選択された一つ以上の基によって任意的に置換されていても良い、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルアミノ、ジ[(1−6C)アルキル]アミノ、(2−7C)ヘテロシクロアルキルアミノ、または(3−6C)シクロアルキルアミノであり;
R136は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、または(1−2C)アルコキシから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキルであり;
R137は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、又は(1−2C)アルコキシから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルアミノ、ジ[(1−6C)アルキル]アミノ、(2−7C)ヘテロシクロアルキルアミノ、または(3−6C)シクロアルキルアミノであり;
R14は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり;
R15は、H、ハロゲンであり;
R2は、以下からなる群から選択される:
ここで、
R21は、H、ハロゲン、(1−3C)アルキル、(1−2C)アルコキシ、ヒドロキシ(1−2C)アルキル、(3−4C)シクロアルキル、(2−3C)アルケニル、またはシアノであり;
R22は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり、
R23は、H、ハロゲン、(1−2C)アルキル、(1−2C)アルコキシ、シアノ、又はヒドロキシであり;
R24は、H、ハロゲン、(1−2C)アルキル、または(1−2C)アルコキシであり;
R25は、H、ハロゲン、(1−3C)アルキル、(1−2C)アルコキシ、ヒドロキシ(1−2C)アルキル、(3−4C)シクロアルキル、(2−3C)アルケニル、またはシアノであり;
R26はH、(1−6C)アルキル、(3−6C)シクロアルキル、(2−5C)ヘテロシクロアルキル、(1−2C)アルコキシ[(2−4C)アルコキシ]n(1−6C)アルキルであり、ここでnは1,2,3又は4の整数を表し、アルキル、ヘテロシクロアルキル、および(1−2C)アルコキシ[(2−4C)アルコキシ]n(1−6C)アルキル基のすべては、ハロゲン、(1−2C)アルキル、(1−2C)アルコキシ、ヒドロキシル、オキソ、アミノ、(3−6C)シクロアルキル、ジ[(1−2C)アルキル]アミノ、または(2−5C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良く;
ただし、R2の中のR21およびR25のうち1つだけがHであり得る。 - 請求項1に記載の化合物であって、ここで
R13がR132O、R135C(O)、(2−7C)ヘテロシクロアルキル、または(1−5C)ヘテロアリールであり、ヘテロシクロアルキルまたはヘテロアリールが各々(1−2C)アルキル、(1−6C)アルキルカルボニル、(1−6C)アルキルスルホニル、(1−5C)アルコキシカルボニル、(1−6C)アルキルアミノカルボニル、(3−6C)シクロアルキルカルボニル、又は(2−7C)ヘテロシクロアルキルカルボニルで任意的に置換されていても良く、アルキルカルボニル、アルキルスルホニル、アルコキシカルボニル、アルキルアミノカルボニル、シクロアルキルカルボニル、またはヘテロシクロアルキルカルボニルが各々(1−2C)アルキル、フルオロ、(1−2C)アルコキシによって任意的に置換されていても良く;
R132は、各々(1−2C)アルキル、ハロゲン、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、または(2−7C)ヘテロシクロアルキルから選択された一つ以上の基によって任意的に置換されていても良い、(1−6C)アルキル、(3−6C)シクロアルキル、(2−7C)ヘテロシクロアルキル、(6−10C)アリール、または(1−5C)ヘテロアリールからなる群から選択され;
R135は、各々(1−2C)アルキル、フルオロ、ヒドロキシル、(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、(2−7C)ヘテロシクロアルキル、オキソ、シアノ、又はアミノから選択された一つ以上の基によって任意的に置換されていても良い、(2−7C)ヘテロシクロアルキル、(1−6C)アルキルアミノ、ジ[(1−6C)アルキル]アミノ、(2−7C)ヘテロシクロアルキルアミノ、または(3−6C)シクロアルキルアミノからなる群から選択される。 - 請求項1又は2に記載の化合物であって、ここで
R13は、R132O、又はR135C(O)であり;または、R13はピペリジニル、ピペラジニル、モルホリニル、ピラゾリル、またはイソオキサゾリルであり、ここで各々が(1−2C)アルキル、(1−6C)アルキルカルボニル、(1−6C)アルキルスルホニル、(1−5C)アルコキシカルボニル、(1−6C)アルキルアミノカルボニル、(3−6C)シクロアルキルカルボニル、または(2−7C)ヘテロシクロアルキルカルボニルによって任意的に置換されていても良く、ここでアルキルカルボニル、アルキルスルホニル、アルコキシカルボニル、アルキルアミノカルボニル、シクロアルキルカルボニル、またはヘテロシクロアルキルカルボニルが、(1−2C)アルキル、フルオロ、または(1−2C)アルコキシによって任意的に置換されていても良く;
R132は(1−6C)アルキル、ピペリジニル、ピロリジニル、またはアゼチジニルからなる群から選択され、ここで各々が(1−2C)アルキル(1−2C)アルコキシ、またはジ[(1−2C)アルキル]アミノ)から選択される一つ以上の基によって任意的に置換されていても良く;
R135はピペリジニル、チオモルホリニル、モルホリニル、ホモピペラジニル、(1−6C)アルキルアミノ、(3−6C)シクロアルキルアミノ、またはピペジリノアミノ、アゼチジニルアミノ、テトラヒドロピラニルアミノ、または3−オキサビシクロ[3.1.0]ヘキサン−6−アミノからなる群から選択され、ここで各々が(1−2C)アルキル、フルオロ、ヒドロキシル又は(1−2C)アルコキシ、ジ[(1−2C)アルキル]アミノ、(2−7C)ヘテロシクロアルキル、オキソ、シアノ、またはアミノから選択された一つ以上の基によって任意的に置換されていても良い。 - 請求項1〜3のいずれか一つに記載の化合物であって、ここでR1は、以下からなる群から選択される:
- 請求項1〜4のいずれか一つに記載の化合物であって、ここでR12およびR15各々がHであり且つR14がH、フルオロ、クロロ、または(1−2C)アルキルである。
- 請求項1〜5のいずれが一つに記載の化合物であって、ここでR11がH、(1−2C)アルキル、または(1−2C)アルコキシであり、且つアルコキシ基は一つ以上のフッ素によって任意的に置換されていても良い。
- 請求項1〜6のいずれか一つに記載の化合物であって、ここでR2は、以下からなる群から選択される:
- 請求項1〜6のいずれか一つに記載の化合物であって、ここでR2は、以下である:
- 請求項1〜8のいずれか一つに記載の化合物であって、ここでR23がHまたは(1−2C)アルキルであり、かつR22およびR24は各々がHであり、かつR21およびR25はハロゲン、(1−3C)アルキル、メトキシ、ヒドロキシメチル、またはシアノからなる群から独立して選択される。
- 請求項1〜9のいずれか一つに記載の化合物であって、ここでR26がH、(1−6C)アルキル、オキセタニル、アゼチジニル、または(1−2C)アルコキシ[(2−4C)アルコキシ]n(1−6C)アルキルであり、ここでnは1又は2の整数を表し、アルキル、オキセタニル、およびアゼチジニル基のすべてが、(1−2C)アルキル、(1−2C)アルコキシ、ヒドロキシル、ジ[(1−2C)アルキル]アミノ、またはオキセタニルから選択された一つ以上の基によって任意的に置換されていても良い。
- 治療に用いる為の請求項1〜10のいずれか一つに記載の化合物またはその薬学的に許容される塩。
- TTK媒介疾患または症状の治療の為の請求項1〜10のいずれか一つに記載の化合物またはその薬学的に許容される塩。
- TTK媒介疾患または症状の治療の為の薬物の調製の為に、請求項1〜10のいずれか一つに記載の化合物またはその薬学的に許容される塩を用いる方法。
- 請求項1〜10のいずれか一つに従う式Iの化合物またはその薬学的に許容される塩、および一つ以上の薬学的に許容される賦形剤を有する医薬組成物。
- 請求項14に記載の医薬組成物であって、少なくとも一つの任意的な治療的に活性な剤をさらに含有する該組成物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14163734.8 | 2014-04-07 | ||
| EP14163734 | 2014-04-07 | ||
| EP15153207 | 2015-01-30 | ||
| EP15153207.4 | 2015-01-30 | ||
| PCT/EP2015/056839 WO2015155042A1 (en) | 2014-04-07 | 2015-03-30 | (5,6-dihydro)pyrimido[4,5-e]indolizines |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017510595A true JP2017510595A (ja) | 2017-04-13 |
| JP2017510595A5 JP2017510595A5 (ja) | 2018-05-17 |
| JP6518690B2 JP6518690B2 (ja) | 2019-05-22 |
Family
ID=52774240
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016560952A Active JP6518690B2 (ja) | 2014-04-07 | 2015-03-30 | (5,6−ジヒドロ)ピリミド[4,5−e]インドリジン |
Country Status (16)
| Country | Link |
|---|---|
| US (3) | US9856258B2 (ja) |
| EP (1) | EP3129374B1 (ja) |
| JP (1) | JP6518690B2 (ja) |
| KR (1) | KR102432420B1 (ja) |
| CN (1) | CN106132963B (ja) |
| AU (1) | AU2015243694B2 (ja) |
| BR (1) | BR112016022342B1 (ja) |
| CA (1) | CA2944610C (ja) |
| DK (1) | DK3129374T3 (ja) |
| ES (1) | ES2716165T3 (ja) |
| HU (1) | HUE043108T2 (ja) |
| MX (1) | MX368767B (ja) |
| PL (1) | PL3129374T3 (ja) |
| PT (1) | PT3129374T (ja) |
| RU (1) | RU2692479C2 (ja) |
| WO (1) | WO2015155042A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2024544083A (ja) * | 2021-12-15 | 2024-11-27 | シルラジェン,インコーポレイテッド | 修飾ベータ-シクロデキストリンを含む医薬組成物 |
| JP2024546977A (ja) * | 2021-12-15 | 2024-12-26 | シルラジェン,インコーポレイテッド | 新生物疾患の処置方法 |
| JP2025500906A (ja) * | 2021-12-15 | 2025-01-15 | シルラジェン,インコーポレイテッド | 新生物性疾患の治療の使用のための医薬組み合わせ |
| JP2025508012A (ja) * | 2022-03-04 | 2025-03-21 | シルラジェン,インコーポレイテッド | 新生物性疾患の処置において使用するための医薬の組合せ |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9856258B2 (en) * | 2014-04-07 | 2018-01-02 | Netherlands Translational Research Center B.V. | (5,6-dihydro)pyrimido[4,5-E]indolizines |
| FI3283642T3 (fi) | 2015-04-17 | 2024-01-11 | Crossfire Oncology Holding B V | Prognostisia biomarkkereita ttk-inhibiittorikemoterapiaan |
| DK3416964T3 (da) * | 2016-02-19 | 2021-02-22 | Phoenix Molecular Designs | 6-Oxo-N-(1-(benzyl)-1H-pyrazol-4-YL)-6,7,8,9-tetrahydropyrido[3',2':4,5]pyrrolo[1,2-A]pyrazin-2-carboxamid-derivater som P90 ribosomal S6 kinase- (RSK-) hæmmere til behandling af cancer |
| EA202092441A1 (ru) | 2016-06-07 | 2021-05-21 | Джакобио Фармасьютикалс Ко., Лтд. | Новые гетероциклические производные, применимые в качестве ингибиторов shp2 |
| CN106551938B (zh) * | 2016-11-26 | 2017-11-24 | 广东安诺药业股份有限公司 | 一种治疗非酒精性脂肪肝的药物及其应用 |
| KR20240026521A (ko) | 2017-03-23 | 2024-02-28 | 자코바이오 파마슈티칼스 컴퍼니 리미티드 | Shp2 억제제로서 유용한 신규한 헤테로환형 유도체 |
| CN112839935A (zh) | 2018-09-26 | 2021-05-25 | 北京加科思新药研发有限公司 | 可用作shp2抑制剂的新型杂环衍生物 |
| US12378245B2 (en) | 2019-02-11 | 2025-08-05 | Phoenix Molecular Designs | Crystalline forms of an RSK inhibitor |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011525516A (ja) * | 2008-06-26 | 2011-09-22 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | ピラゾロ−キナゾリン |
| JP2014503567A (ja) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | 三環系ピロロ誘導体、その調製方法およびそのキナーゼ阻害剤としての使用 |
| JP2014503521A (ja) * | 2010-12-17 | 2014-02-13 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 過増殖性障害の処置におけるmps−1およびtkk阻害剤としての使用のための、イミダゾピラジン類 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL160915A0 (en) * | 2001-09-19 | 2004-08-31 | Aventis Pharma Sa | Indolizines inhibiting kinase proteins |
| PT1636236E (pt) | 2003-05-22 | 2013-12-16 | Nerviano Medical Sciences Srl | Derivados de pirazol-quinazolina, processo para sua preparação e sua utilização como inibidores de cinase |
| EP2118102B1 (en) | 2006-11-28 | 2013-06-12 | Nerviano Medical Sciences S.r.l. | Tricyclic indoles and (4,5-dihydro) indoles |
| WO2010111406A2 (en) | 2009-03-24 | 2010-09-30 | Myriad Pharmaceuticals, Inc. | Compounds and therapeutic uses thereof |
| EP2424868B1 (en) | 2009-04-29 | 2018-01-10 | Nerviano Medical Sciences S.r.l. | Cdk inhibitor salts |
| JP5851990B2 (ja) | 2009-07-29 | 2016-02-03 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Plk阻害剤の塩 |
| TW201107329A (en) | 2009-07-30 | 2011-03-01 | Oncotherapy Science Inc | Fused imidazole derivative having ttk inhibitory action |
| EP2343297A1 (en) | 2009-11-30 | 2011-07-13 | Bayer Schering Pharma AG | Triazolopyridines |
| JP6523267B2 (ja) | 2013-06-24 | 2019-05-29 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | Fshrの調節剤としてのイミダゾール化合物及びその使用 |
| US9856258B2 (en) * | 2014-04-07 | 2018-01-02 | Netherlands Translational Research Center B.V. | (5,6-dihydro)pyrimido[4,5-E]indolizines |
-
2015
- 2015-03-30 US US15/128,874 patent/US9856258B2/en not_active Ceased
- 2015-03-30 CN CN201580017365.8A patent/CN106132963B/zh active Active
- 2015-03-30 PT PT15712912T patent/PT3129374T/pt unknown
- 2015-03-30 US US17/579,990 patent/USRE50082E1/en active Active
- 2015-03-30 HU HUE15712912A patent/HUE043108T2/hu unknown
- 2015-03-30 CA CA2944610A patent/CA2944610C/en active Active
- 2015-03-30 PL PL15712912T patent/PL3129374T3/pl unknown
- 2015-03-30 EP EP15712912.3A patent/EP3129374B1/en active Active
- 2015-03-30 BR BR112016022342-0A patent/BR112016022342B1/pt active IP Right Grant
- 2015-03-30 KR KR1020167031029A patent/KR102432420B1/ko active Active
- 2015-03-30 US US16/728,900 patent/USRE48974E1/en active Active
- 2015-03-30 JP JP2016560952A patent/JP6518690B2/ja active Active
- 2015-03-30 WO PCT/EP2015/056839 patent/WO2015155042A1/en not_active Ceased
- 2015-03-30 DK DK15712912.3T patent/DK3129374T3/en active
- 2015-03-30 MX MX2016012997A patent/MX368767B/es active IP Right Grant
- 2015-03-30 ES ES15712912T patent/ES2716165T3/es active Active
- 2015-03-30 AU AU2015243694A patent/AU2015243694B2/en active Active
- 2015-03-30 RU RU2016141405A patent/RU2692479C2/ru active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011525516A (ja) * | 2008-06-26 | 2011-09-22 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | ピラゾロ−キナゾリン |
| JP2014503521A (ja) * | 2010-12-17 | 2014-02-13 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング | 過増殖性障害の処置におけるmps−1およびtkk阻害剤としての使用のための、イミダゾピラジン類 |
| JP2014503567A (ja) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | 三環系ピロロ誘導体、その調製方法およびそのキナーゼ阻害剤としての使用 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2024544083A (ja) * | 2021-12-15 | 2024-11-27 | シルラジェン,インコーポレイテッド | 修飾ベータ-シクロデキストリンを含む医薬組成物 |
| JP2024546977A (ja) * | 2021-12-15 | 2024-12-26 | シルラジェン,インコーポレイテッド | 新生物疾患の処置方法 |
| JP2025500906A (ja) * | 2021-12-15 | 2025-01-15 | シルラジェン,インコーポレイテッド | 新生物性疾患の治療の使用のための医薬組み合わせ |
| JP2025508012A (ja) * | 2022-03-04 | 2025-03-21 | シルラジェン,インコーポレイテッド | 新生物性疾患の処置において使用するための医薬の組合せ |
| JP7844660B2 (ja) | 2022-03-04 | 2026-04-13 | シルラジェン,インコーポレイテッド | 新生物性疾患の処置において使用するための医薬の組合せ |
Also Published As
| Publication number | Publication date |
|---|---|
| US9856258B2 (en) | 2018-01-02 |
| EP3129374B1 (en) | 2018-12-19 |
| RU2016141405A (ru) | 2018-05-07 |
| BR112016022342B1 (pt) | 2022-10-04 |
| CN106132963B (zh) | 2019-08-06 |
| US20170096432A1 (en) | 2017-04-06 |
| WO2015155042A1 (en) | 2015-10-15 |
| RU2692479C2 (ru) | 2019-06-25 |
| EP3129374A1 (en) | 2017-02-15 |
| BR112016022342A8 (pt) | 2021-07-20 |
| JP6518690B2 (ja) | 2019-05-22 |
| MX368767B (es) | 2019-10-15 |
| PL3129374T3 (pl) | 2019-07-31 |
| HUE043108T2 (hu) | 2019-07-29 |
| CA2944610C (en) | 2024-01-09 |
| KR102432420B1 (ko) | 2022-08-17 |
| PT3129374T (pt) | 2019-03-25 |
| CA2944610A1 (en) | 2015-10-15 |
| AU2015243694B2 (en) | 2019-01-17 |
| CN106132963A (zh) | 2016-11-16 |
| BR112016022342A2 (pt) | 2017-08-15 |
| USRE48974E1 (en) | 2022-03-15 |
| ES2716165T3 (es) | 2019-06-10 |
| MX2016012997A (es) | 2016-12-07 |
| USRE50082E1 (en) | 2024-08-20 |
| RU2016141405A3 (ja) | 2018-10-02 |
| DK3129374T3 (en) | 2019-04-08 |
| KR20170013866A (ko) | 2017-02-07 |
| AU2015243694A1 (en) | 2016-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6518690B2 (ja) | (5,6−ジヒドロ)ピリミド[4,5−e]インドリジン | |
| AU2016352813B2 (en) | Novel pyrazolo pyrimidine derivatives | |
| ES2904544T3 (es) | Compuestos de indazol como inhibidores de la cinasa FGFR, preparación y uso de los mismos | |
| ES2873515T3 (es) | Derivado de quinolona, sal o estereoisómero del mismo farmacéuticamente aceptable, como inhibidor de AXL | |
| AU2017286380B2 (en) | Azabenzimidazole derivatives as PI3K beta inhibitors | |
| CA2716079C (en) | 4-aryl-2-anilino-pyrimidines as plk kinase inhibitors | |
| ES2620119T3 (es) | Derivados heterocíclicos novedosos como moduladores de la actividad de quinasa | |
| JP2022516469A (ja) | ユビキチン特異的プロテアーゼ1を阻害するための組成物 | |
| CN103012399B (zh) | 7-氧代吡啶并嘧啶类化合物及其药用组合物和应用 | |
| KR20170101908A (ko) | 피리미딘 또는 피리딘계 화합물, 이의 제조방법 및 약학적 용도 | |
| AU2020255100A1 (en) | N-heteroaromatic amide derivatives for treatment of cancer | |
| WO2005063709A1 (ja) | アミド誘導体及び医薬 | |
| TW201219383A (en) | Chemical compounds | |
| JP2025526419A (ja) | グルタミニル-ペプチドシクロトランスフェラーゼ及びグルタミニル-ペプチドシクロトランスフェラーゼ様タンパク質の阻害剤としてのピペリジニルピリジニルカルボニトリル誘導体 | |
| CN112457306A (zh) | 3,5-二取代吡唑化合物作为激酶抑制剂及其应用 | |
| KR20160007348A (ko) | 신규한 트리아졸로피리미디논 또는 트리아졸로피리디논 유도체, 및 이들의 용도 | |
| US20230348462A1 (en) | Imidazo[4,5-c]quinoline compounds and their use as atm kinase inhibitors | |
| CA2967551A1 (en) | Imidazopyridazine derivatives as pi3k.beta. inhibitors | |
| CN114539263B (zh) | 一类含氮并杂环化合物及其药用组合物和应用 | |
| ES2760507T3 (es) | Derivados de imidazopiridazina enlazados a heterociclilo como inhibidores de PI3Kß | |
| WO2022116943A1 (zh) | 取代的稠合双环化合物作为激酶抑制剂及其应用 | |
| JP7053654B2 (ja) | PI3Kβ阻害剤としてのキノキサリン及びピリドピラジン誘導体 | |
| CN103570731A (zh) | 嘧啶并三环或嘧啶并四环类化合物及其药用组合物和应用 | |
| HK1231072A1 (en) | (5,6-dihydro)pyrimido[4,5-e]indolizines | |
| HK1231072B (zh) | (5,6-二氢)嘧啶并[4,5-e]吲嗪 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180330 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180330 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20181213 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181218 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190314 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190409 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190422 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6518690 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |