JP3623802B2 - 金属配位子含有漂白組成物 - Google Patents
金属配位子含有漂白組成物 Download PDFInfo
- Publication number
- JP3623802B2 JP3623802B2 JP50704898A JP50704898A JP3623802B2 JP 3623802 B2 JP3623802 B2 JP 3623802B2 JP 50704898 A JP50704898 A JP 50704898A JP 50704898 A JP50704898 A JP 50704898A JP 3623802 B2 JP3623802 B2 JP 3623802B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- bleaching composition
- cycloalkyl
- amide
- ligand
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21C—PRODUCTION OF CELLULOSE BY REMOVING NON-CELLULOSE SUBSTANCES FROM CELLULOSE-CONTAINING MATERIALS; REGENERATION OF PULPING LIQUORS; APPARATUS THEREFOR
- D21C9/00—After-treatment of cellulose pulp, e.g. of wood pulp, or cotton linters ; Treatment of dilute or dewatered pulp or process improvement taking place after obtaining the raw cellulosic material and not provided for elsewhere
- D21C9/10—Bleaching ; Apparatus therefor
- D21C9/1026—Other features in bleaching processes
- D21C9/1036—Use of compounds accelerating or improving the efficiency of the processes
-
- C—CHEMISTRY; METALLURGY
- C11—ANIMAL OR VEGETABLE OILS, FATS, FATTY SUBSTANCES OR WAXES; FATTY ACIDS THEREFROM; DETERGENTS; CANDLES
- C11D—DETERGENT COMPOSITIONS; USE OF SINGLE SUBSTANCES AS DETERGENTS; SOAP OR SOAP-MAKING; RESIN SOAPS; RECOVERY OF GLYCEROL
- C11D3/00—Other compounding ingredients of detergent compositions covered in group C11D1/00
- C11D3/39—Organic or inorganic per-compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/006—Catalysts comprising hydrides, coordination complexes or organic compounds comprising organic radicals, e.g. TEMPO
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/10—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C11—ANIMAL OR VEGETABLE OILS, FATS, FATTY SUBSTANCES OR WAXES; FATTY ACIDS THEREFROM; DETERGENTS; CANDLES
- C11D—DETERGENT COMPOSITIONS; USE OF SINGLE SUBSTANCES AS DETERGENTS; SOAP OR SOAP-MAKING; RESIN SOAPS; RECOVERY OF GLYCEROL
- C11D3/00—Other compounding ingredients of detergent compositions covered in group C11D1/00
- C11D3/39—Organic or inorganic per-compounds
- C11D3/3902—Organic or inorganic per-compounds combined with specific additives
- C11D3/3905—Bleach activators or bleach catalysts
- C11D3/3932—Inorganic compounds or complexes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/70—Oxidation reactions, e.g. epoxidation, (di)hydroxylation, dehydrogenation and analogues
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/70—Complexes comprising metals of Group VII (VIIB) as the central metal
- B01J2531/72—Manganese
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/84—Metals of the iron group
- B01J2531/842—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
- B01J31/181—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine
- B01J31/184—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine mixed aromatic/aliphatic ring systems, e.g. indoline
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Inorganic Chemistry (AREA)
- Materials Engineering (AREA)
- Detergent Compositions (AREA)
- Paper (AREA)
- Silver Salt Photography Or Processing Solution Therefor (AREA)
- Silicon Polymers (AREA)
- Cosmetics (AREA)
Description
発明の分野
本発明は漂白触媒としての大環状金属配位子錯体の使用に関し、特には酸化性漂白反応を増強するための触媒としての大環状テトラアミド配位子の遷移金属錯体に関する。
関連技術の簡単な記載
過酸化水素、及び水溶液中で過酸化水素を生じる他のペルオキシ化合物が布及び表面の漂白における使用について長い間知られている。しかしながら、過ホウ酸ナトリウムのようなペルオキシ化合物(一水和物もしくは四水和物、過炭酸ナトリウム等)は、低温(例えば、38.8℃/100゜F以下)では、漂白性能が比較的穏やかである。過安息香酸のような有機過酸は強力な酸化体であるが、しばしば、高価で厄介な方法によって安定化しなければ不安定である。加えて、予め調製された過酸には、しばしば、製造の対費用効果がない。漂白剤活性剤、又はエステル、ケトン、ニトリル等の過酸前駆体は、しばしば、ペルオキシ化合物の効力の強化に有効である。しかしながら、これらの漂白剤活性剤は通常化学量論量又はそれより大きな量で存在していなければならず、同様に製造するのに費用がかさむ。
遷移金属キレート、特にマンガン及び鉄を用いるものがペルオキシ化合物のための漂白触媒として知られている。これらは、例えば、Favreら,米国特許第5,246,621号、Braggら,米国特許第5,002,682号、Postlethwaite,米国特許第4,119,557号、及びEllis,Jr.ら,米国特許第4,900,871号によって説明されている。これらの遷移金属キレートは、例えば、適切なペルオキシ化合物、例えば、過ホウ酸ナトリウム一水和物を用いる布の洗濯において用いることができる。
これらの遷移金属キレートはペルオキシ化合物の酸化力を改善することが立証されてはいるが、漂白剤活性剤として用いた場合には布への染料の移行を媒介し、それに損傷を与えることさえある。
一定の遷移金属キレートは無関係の目的で研究されている。例えば、高酸化状態の遷移金属の錯体はタンパク質マトリックスの影響の下で多くの生物学的反応において酸化体として機能することが知られており、近年、特定のモノオキシゲナーゼ触媒の作用機構及び反応性の理解について幅広い関心が生じている。
代表的なプログラムがCollins,T.J.,“Designing Ligands for Oxidizing Complexes",Accounts of Chemical Research,279,Vol.27,No.9(1994)に記述されている。この論文では、高度に酸化性の金属中心に配位させる場合に酸化性分解に対して抵抗性である配位子を得るための設計指向のアプローチが説明されている。幾つかのジアミノ−N−ジフェノキシド及びジアミノ−N−アルコキシド非環式キレート化合物並びに大環状テトラアミド−Nキレート化合物がこのCollinsのAccounts of Chemical Researchの論文に記述されている。
大環状テトラアミド配位子のアジドベースの合成ルートがカリフォルニア工科大学のUffelman,E.S.,Ph.D.の学位論文(1992)に記述されている。加えて、アジドベースのルートによるアリール架橋テトラアミド配位子の合成は芳香族ジアミンを出発原料として用いることにより進行させることができる。
しかしながら、当該技術分野では、一定の大環状テトラアミド配位子がペルオキシ化合物のための新規かつ著しく有効な漂白剤活性剤を提供することは認識されていない。加えて、これらの型の化合物が染料移行阻害、抗汚れ沈着及び染み抜きの領域において非常に有利であることは、教示も、開示も、示唆もされていない。
発明の要旨
本発明は、
(a)構造:

を有する酸化的に安定な漂白剤活性剤、及び
(b)有効量の酸化性化合物の供給源
を含む漂白組成物を包含する。
表面活性剤、充填剤、ビルダー、金属イオン封鎖剤、酸化防止剤、酵素、蛍光白化剤、染料、着色料、顔料、及び他の標準的なクリーニング及び/又は洗濯補助剤を添加することができる。
好ましい漂白剤活性剤は大環状テトラアミド化合物である。これらのうち、この配位子の環状構造に直接融合する置換芳香族置換基を有するものが特に好ましい。
例えば、好ましい化合物は、構造:
改良された染料移行阻害特性を有するペルオキシ漂白剤活性剤が必要とされている。改良された抗汚れ沈着特性を有する漂白剤活性剤が必要とされている。ユニークな染み抜き性能を有する漂白剤活性剤を提供するさらに別の必要性が存在する。緩衝溶液中で長期触媒安定性を有する新規な漂白剤活性剤を提供するさらなる必要性が存在する。酸化性化合物に比較して化学量論量で用いることができる新規な漂白剤活性剤を提供するさらなる必要性が存在する。
【図面の簡単な説明】
図1は、本発明の大環状テトラアミド配位子をアジドルートで調製するための合成ルートを示す。
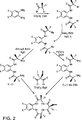
図2は、本発明の大環状テトラアミド配位子を、芳香族ジアミンを出発原料として用いてアジドルートで調製するための合成ルートを示す。
図3は、本発明の好ましい態様の長期触媒安定性を対照と比較するグラフである。
好ましい態様の詳細な説明
本発明は、
(a)構造:
を有する酸化的に安定な漂白剤活性剤、及び
(b)有効量の酸化性化合物の供給源
を含む漂白組成物を包含する。
これらのうち、好ましい本発明の大環状テトラアミド配位子は、漂白剤活性剤の性能特性の様々な群において驚くほど有効であることが立証されている。
これらの配位子は、Gordon−WylieらのSYNTHESIS OF MACROCYCLIC TETRAAMIDO−N LIGANDSと題する現在出願されている米国特許出願の第08/681,187号及びCollinsらのLONG−VIVED HOMOGENOUS OXIDATION CATALYSTSと題するWO98/03263に説明される手順に従って調製され、これらの特許の両者は参照することによりここに組み込まれる。
これらの本発明の化合物の特に好ましい態様は、構造:
X及びZ基はH、又は電子供与基もしくは電子吸引基であり得る。電子吸引基には、Br、I、最も好ましくはClのようなハロゲンが含まれる。さらに、SO3 -、OSO3 -、OSO3R(Rは、限定されることなく、H、アルキル、アリール、アルキルアリールと定義される)及びNO2 -が適切な基である。電子供与基には、アルコキシ(限定されることなく、メトキシ、エトキシ、プロポキシ及びブトキシ)、アルキル(限定されることなく、メチル、エチル、プロピル、n−ブチル及びt−ブチル)及び水素が含まれる。これらの基は、金属配位子錯体の電子密度を変化させ、その反応性に強い影響を与える。
R'及びR"は本発明の大環状テトラアミド配位子の長期触媒安定性に強い影響を及ぼすものと思われる。各々個別にH、アルキル、アルケニル、アリール、アルキニル、ハロゲン、アルコキシ、又はフェノキシ置換基から選択することができるが、短鎖アルキルが好ましい。特に好ましいのは、R'及びR"が同じであって、エチル及びメチルから選択される場合、又はR'及びR"が結合してシクロアルキル環もしくはシクロアルケニル環、特にはシクロペンチルもしくはシクロヘキシルを形成する場合である。このシクロアルキル環は少なくとも1つの炭素以外の他の原子、例えば、これらに限定されることなく、N、O、もしくはSを含んでいてもよい。
金属MはI、II、III、IV、VもしくはVIの酸化状態を有する遷移金属であるか;又は元素周期表の6、7、8、9、10及び11族から選択される。これは、好ましくは、Fe、Mn、Cr、Cu、Co、Ni、Mo、Zn、及びWからなる群より選択される。それらの混合物も可能である。
Qは化学量論基準でその化合物の電荷(一般には負;好ましくは−1)と釣り合うあらゆる対イオンである。したがって、一般に正に荷電した対イオンが好ましく選択され:限定されるものではないが、アルカリ金属対イオン(例えば、K、Li、Na)、NR* 4及びPR* 4(式中、R*個別にH、アルキル、アリール、アルキルアリール、アルケニルから選択されるか、又は一緒に融合して炭素以外の原子を少なくとも1つ含んでいてもよいシクロアルキルもしくはシクロアルケニルもしくはアリール環を形成することができる)から選択される。
LはMに結合することができるあらゆる不安定性配位子である。これらには、好ましくは、H2O、Cl、及びC=Nが含まれるが、これらに限定されるものではない。
これらの化合物の錯体性のため、本明細書においてはそれらを命名することはないが、便宜上、それらに存在する置換基によって言及する。例えば、上に表される構造は5,6(4,5−ジ−X−ベンゾ)−3,8,11,13−テトラオキソ−2,2,9,9−テトラメチル−12,12−ジエチル−1,4,7,10−テトラアザシクロトリデカン(又は、テトラメチルジエチルジ−X−ベンゼン(TMDE−DXB、式中、X=Cl、H、Me、OMe))と呼ぶことができる。したがって、便宜上、上記構造において、2つのメチル基が各々配位子のアミン構成要素上に存在し、かつR'及びR"として作用する2つのエチル基が存在する場合、その化合物はTMDEと呼ばれる。X及びZ基が両者ともクロロである場合、その化合物はDCBと呼ばれる。配位子の好ましい遷移金属は鉄であり、それによりその化合物はFeDCBと呼ばれる。
本発明の大環状テトラアミド配位子は真の触媒であるため、漂白組成物に添加されるそれらの量は一般に化学量論的である。しかしながら、本発明の組成物に約0.0001〜約999,999ppmで添加することが好ましく、より好ましくは0.001〜100,000ppmであるが、これに限定されるものではない。
下記の実験の部においては、好ましい大環状テトラアミド化合物の選択された合成が説明される。加えて、これらの本発明の大環状配位子の染料移行阻害特性、長期触媒活性及び染み抜き性能を示す試験を行った。
酸化性化合物
酸化性化合物は有機化合物であっても無機化合物であってもよい。−O−O−過酸化物結合を有するペルオキシ化合物が好ましい。典型的な化合物には過酸化水素、過酸化水素付加物、水溶液中で過酸化水素を生成することが可能な化合物、有機過酸化物、過硫酸塩、過リン酸塩、及び過ケイ酸塩が含まれる。過酸化水素付加物にはアルカリ金属(例えば、ナトリウム、リチウム、カリウム)炭酸塩ペルオキシ水和物及び過酸化尿素が含まれる。水溶液中で過酸化水素を生成することが可能な化合物にはアルカリ金属(ナトリウム、カリウム、リチウム)過ホウ酸塩(一及び四水和物)が含まれる。この過ホウ酸塩はAkzo N.V.及びFMC Corporationのような供給業者から購入することができる。その代わりに、アルコールオキシダーゼ酵素及びその適切なアルコール基質を過酸化水素源として用いることができる。有機過酸化物には、限定されることなく、ベンゾイルヒドロペルオキシド及びクメンヒドロペルオキシドが含まれる。過硫酸塩にはペルオキシモノ硫酸カリウム(Oxone▲R▼として販売、E.I.du Pont de Nemours)及びカロ酸(Caro's acid)が含まれる。
ペルオキシ化合物の有効量は少なくとも0.001ppmの活性酸素(A.O.)を生成するのに十分な量である。これに限定されるものではないが、0.001ないし1,000ppmのA.O.を生成することが好ましい。布の漂白には、0.01ないし50ppmのA.O.が好ましい。A.O.測定の記述及び説明はSheldon N.Lewisの論文,“Peracid and Peroxide Oxidation",Oxidation(1969),pp.213−258に見出され、これは参照することによりここに組み込まれる。
クリーニング及び/又は洗濯補助剤
本発明の大環状テトラアミド配位子は酸化体漂白剤又は洗浄剤基剤と組み合わせることができ、この基剤はビルダー並びに、場合により、アニオン性、非イオン性、カチオン性、両性、双性イオン性界面活性剤、及びそれらの混合物から選択される界面活性剤を含む。他の補助物質が存在していてもよい。また、これらの化合物は、硬質表面、染み抜き剤、又は他の表面洗浄/漂白の遂行のために液体基剤中に存在させてもよい。また、これらの化合物はパルプ及び生地の漂白処理にも有用であり得る。これらの化合物の各々及びここでの使用に適する補助物質を以下でさらに論じる。
a.ビルダー
ビルダーは典型的にはアルカリ性ビルダー、すなわち、水溶液中で7〜14、好ましくは9〜12のpHを達成するものである。無機ビルダーの例にはアルカリ金属及びアンモニウム炭酸塩(セスキ炭酸塩及び重炭酸塩を含む)、リン酸塩(オルトリン酸塩、トリポリリン酸塩及びテトラピロリン酸塩を含む)、アルミノケイ酸塩(天然及び合成ゼオライトの両者)及びそれらの混合物が含まれる。炭酸塩が本発明における使用に特に望ましく、これは、それらが低コストであることに加えて、それらの高いアルカリ度及び硬水中に存在し得る硬質イオン(hardness ions)の除去における有効性によるものである。炭酸塩を優勢ビルダーとして用いることができる。ケイ酸塩(Na2O:SiO2、4:1ないし1:1、最も好ましくは3:1ないし1:1のモジュラス)も用いることができる。ケイ酸塩も、それらの水中での溶解度及びガラス状マトリックスを形成する能力により、洗浄剤用のビルダーとして有利に用いることができる。
有機ビルダーも使用に適しており、アルキル金属及びアンモニウムスルホコハク酸塩、ポリアクリレート、ポリマレイン酸塩、アクリル酸及びマレイン酸もしくはマレイン酸もしくはマレイン酸無水物の共重合体、クエン酸びにそれらの混合物からなる群より選択される。
b.充填剤/希釈剤
漂白剤組成物又は洗浄剤用の充填剤は、洗浄又はクリーニングでの使用毎に正しい量又は用量の洗浄又はクリーニング活性剤が送り出されることを保証するために用いられる。NaCl、Na2SO4、及びホウ砂のような塩が好ましい。糖のような有機希釈剤も可能である。液体での遂行においては、溶媒(例えば、これに限定されることなく、アルカノール、グリコール、グリコールエステル、炭化水素、ケトン、及びカルボン酸)、液体界面活性剤及び水を希釈剤として用いることができる。
c.界面活性剤
一般に、特定の目的とする汚れを除去するために界面活性剤が、例えば、油性汚れに対しては非イオン性界面活性剤が、かつ粒状汚れに対してはアニオン性界面活性剤が漂白剤又は洗浄剤配合物に添加される。しかしながら、一般的に述べると、酸化体漂白組成物は界面活性剤をほとんど、もしくは全く含まないことがある。
特に有効な界面活性剤はアニオン性界面活性剤であるものと思われる。このようなアニオン性界面活性剤の例には、アンモニウム、置換アンモニウム(例えば、モノ−、ジ−、及びトリ−エタノールアンモニウム)、C6〜C20脂肪酸及びロジン酸のアルカリ金属及びアルカリ土類金属塩、直鎖及び分岐アルキルベンゼンスルホン酸塩、アルキル硫酸塩、アルキルエーテル硫酸塩、アルカンスルホン酸塩、オレフィンスルホン酸塩、ヒドロキシアルカンスルホン酸塩、脂肪酸モノグリセリド硫酸塩、アルキルグリセリルエーテル硫酸塩、アシルサルコシネート並びにアシルN−メチルタウリドが含まれる。アルキルベンゼンスルホン酸塩のようなアルキルアリールスルホン化界面活性剤が好ましい。
他の使用に好ましい界面活性剤には、Neodolの商品名でShell Chemical Companyにより販売されるもののような直鎖エトキシル化アルコールが含まれる。他の適切な非イオン性界面活性剤には、6ないし16炭素原子の平均長及び平均でアルコールのモル当たり2ないし20モルのエチレンオキシドを有する直鎖エトキシル化アルコール;6ないし16炭素原子の平均長並びに平均でアルコールのモル当たり0〜10モルのエチレンオキシド及び1ないし10モルのプロピレンオキシドを有する、直鎖及び分岐鎖の第1及び第2エトキシル化プロポキシル化アルコール;8ないし16炭素原子の平均鎖長及び平均でアルコールのモル当たり1.5ないし30モルのエチレンオキシドを有する、エトキシル化アルキルフェノールとしても知られる、直鎖及び分岐アルキルフェノキシ(ポリエトキシ)アルコール;並びにそれらの混合物が含まれ得る。
さらに適切な非イオン性界面活性剤には、ポリオキシエチレンカルボン酸エステル、脂肪酸グリセロールエステル、脂肪酸及びエトキシル化脂肪酸アルカノールアミド、プロピレンオキシド及びエチレンオキシドの特定のブロック共重合体、並びにプロピレンオキシド及びエチレンオキシドとプロポキシル化エチレンジアミンとのブロック共重合体が含まれ得る。アミン酸化物、ホスフィン酸化物、スルホキシド、及びそれらのエトキシル化誘導体のような半極性非イオン性界面活性剤も含まれる。
適切なカチオン性界面活性剤には、典型的には窒素原子に連結する基の1つがC12〜C18アルキル基であり、かつ他の3つの基がフェニル基のような置換基を有していてもよい短鎖アルキル基である四級アンモニウム化合物が含まれ得る。
さらに、アニオン性水溶性基、カチオン性基及び疎水性有機基を含む適切な両性及び双性イオン性界面活性剤には、アミノカルボン酸及びそれらの塩、アミノジカルボン酸及びそれらの塩、アルキルベタイン、アルキルアミノプロピルベタイン、スルホベタイン、アルキルイミダゾリニウム誘導体、特定の四級アンモニウム化合物、特定の四級ホスホニウム化合物及び特定の三級スルホニウム化合物が含まれ得る。潜在的に適切な双性イオン性界面活性剤の他の例がJonesの米国特許第4,005,029号の11〜15欄に記述されていることを見出すことができ、これは参照することによりここに組み込まれる。
本発明における使用に適し得るアニオン性、非イオン性、カチオン性及び両性界面活性剤のさらなる例はKirk−Othmer,Encyclopedia of Chemical Technology,第3版、Vol.22,pp.347−387及び“McCutcheon's Detergents and Emulsifiers",北米版、1983に記載されており、これらは参照することによりここに組み込まれる。
上述のように、漂白剤もしくは洗浄剤漂白製品が所望される場合には、他の一般的な洗浄剤補助剤を添加することができる。例えば、洗浄剤組成物が所望される場合、以下の範囲(重量%)が実用的であるように思われる。
本明細書中の組成物の幾つかにおいては、キレート剤を含めることが特に好ましく、最も好ましくはアミノポリホスホン酸塩を含める。これらのキレート剤は、最適性能を達成するため、酸化体の溶液安定性の維持を補助する。このようにして、これらは遊離重金属イオンをキレート化するように作用する。キレート剤は、遊離重金属イオンのキレート化の点で有効である多くの既知薬剤から選択される。キレート剤は加水分解及び酸化体による急速な酸化に対して耐性であるべきである。好ましくは、1〜9の酸解離定数(pKa)を有するべきであり、これはそれが低pHで解離して金属カチオンへの結合を増強することを示す。最も好ましいキレート剤はアミノポリホスホン酸塩であり、これはDequestの商品名でMonsanto Companyから購入することができる。それらの例はDequest2000、2041、2060及び2066である(Bossu,米国特許第4,473,507号、第12欄第63行ないし第13欄第22行も参照、この特許は参照することによりここに組み込まれる)。Dequest2010のようなポリホスホン酸塩も使用に適する。エチレンジアミン四酢酸(EDTA)及びニトリロ三酢酸(NTA)のような他のキレート剤も使用に適し得る。さらに別の新たな好ましいキレート剤は、W.R.GraceからのHampshire 1,3PDTA及びCiba−Geigy A.G.からのChel DTPA100#Fのような新規プロピレンジアミン四酢酸塩である。前述のものの混合物も適し得る。キレート剤の有効量は、洗浄溶液中に1〜1,000、より好ましくは5〜500、最も好ましくは10〜100ppmのキレート剤の範囲である。
e.他の補助剤:
標準的な洗浄剤又は酸化漂白剤補助剤を本発明に含めることができる。
これらには酵素が含まれ、これはこれらの洗浄剤又は酸化漂白剤製品において特に望ましい補助物質である。しかしながら、酵素安定化剤を含めることが好ましいことがある。
プロテアーゼが特に好ましいクラスの酵素の1つである。これらは酸性、中性及びアルカリ性プロテアーゼから選択される。この“酸性”、“中性”及び“アルカリ性”という用語はその酵素の活性が最適であるpHを指す。中性プロテアーゼの例にはミレザイム(Milezyme)(Miles Laboratoryから入手可能)及びトリプシン、天然プロテアーゼが含まれる。アルカリ性プロテアーゼは広く様々な源から入手可能であり、典型的には様々な微生物(例えば、枯草菌(Bacillis subtilisis)から産生される。アルカリ性プロテアーゼの典型的な例には、International BioSyntheticsからのマキサターゼ(Maxatase)及びマキサカル(Maxacal)、Novo Industri A/Sから入手可能なアルカレース(Alcalase)、サビネース(Savinase)及びエスペレース(Esperase)が含まれる。Stanislowskiらの米国特許第4,511,490号(参照することによりここに組み込まれる)も参照のこと。
さらなる適切な酵素は炭水化物加水分解酵素であるアミラーゼである。アミラーゼ及びプロテアーゼの混合物を含めることも好ましい。適切なアミラーゼにはソサイイット・ラピデース(Societe Rapidase)からのラピデース(Rapidase)、ミレス・ラボラトリーからのミレザイム(Milezyme)、及びInternational BioSyntheticsからのマキサミル(Maxamyl)が含まれる。
さらに別の適切な酵素は、Taiの米国特許第4,479,881号、Murataらの米国特許第4,443,355号、Barbesgaardらの米国特許4,435,307号、及びOhyaらの米国特許第3,983,082号に記述されるもののようなセルラーゼであり、これらの特許は参照することによりここに組み込まれる。
さらに別の適切な酵素は、Silverの米国特許第3,950,277号、及びThomらの米国特許第4,707,291号に記述されるもののようなリパーゼであり、これらの特許は参照することによりここに組み込まれる。
ここで関心のあるさらなる酵素はセイヨウワサビペルオキシダーゼ及びWO93/24628号に開示されるもののようなペルオキシダーゼであり、この特許は参照することによりここに組み込まれる。
酵素は、洗浄剤/漂白剤/クリーニング剤基剤の0〜5重量%、より好ましくは0.01〜3重量%、最も好ましくは0.1〜2重量%の量が存在し得る。前述の加水分解酵素のいずれかの混合物が望ましく、特にはプロテアーゼ/アミラーゼ配合物が望ましい。
加えて、任意の補助剤にはモナストラル(Monastral)ブルー及びアントラキノン染料(例えば、Zielskeの米国特許第4,661,293号及び第4,746,461号に記述されるもの)のような染料が含まれる。
適切な着色料でもある顔料を、限定することなく、二酸化チタン、ウルトラマリーンブルー(Changらの米国特許第4,708,816号も参照)及び着色アルミノケイ酸塩から選択することができる。
蛍光白化剤がさらに別の望ましい補助剤である。これらにはスチルベン、スチレン、及びナフタレン誘導体が含まれ、紫外線の入射により蛍光を可視波長で放出する。これらのFWAもしくは光沢剤は、汚れ及び洗浄の繰り返しにより薄黒くなっている布の外観を改良するのに有用である。好ましいFWAは、Ciba−Geigy A.G.からのTinopal 5 BMX−C及びTinopal RBS、並びにMobay ChemicalsからのPhorwite RKHである。適切なFWAの例は米国特許第1,298,577号、第2,076,011号、第2,026,054号、第2,026,566号、第1,393,042号;並びに米国特許第3,951,960号、第4,298,290号、第3,993,659号、第3,980,713号及び第3,627,758号に見出すことができ、これらは参照することによりここに組み込まれる。
カルボキシメチルセルロースのような抗再沈着剤(anti−redeposition agents)が潜在的に望ましい。次に、適切なアニオン性界面活性剤のようなフォームブースター(foam boosters)が本明細書に含めるのに適切なものであり得る。また、特定の界面活性剤の使用の結果過剰の泡立ちが生じる場合には、アルキル化ポリシクロキサン、例えばジメチルポリシロキサンのような消泡剤が望ましい。芳香剤もこれらの組成物において望ましい補助剤である。
さらなる有機漂白剤活性剤を添加することが可能であり、これにはエステル(Fongらの米国特許第4,778,618号及びRowlandらの米国特許第5,182,045号を参照)、ケトン、イミド(Kaaretの米国特許第5,478,569号を参照)及びニトリルが含まれるがこれらに限定されるものではない。
これらの添加物は0〜50%、より好ましくは0〜30%、最も好ましくは0〜10%の範囲の量で存在し得る。特定の場合には、個々の補助剤の幾つかが他の範疇と重複してもよい。しかしながら、本発明では、これらの補助剤の各々がそれらの様々な範疇で別々の性能上の利益を提供することが意図されている。
実験の部
酸化的に抵抗性のテトラアミド配位子の合成
材料 全ての溶媒及び試薬は試薬級であり(Aldrich,Aldrich Sure−Seal,Fisher)、受け取ったまま用いた。微量分析は米国インディアナ州インディアナポリスのMidwest Microlabsにより行なわれた。
質量分析 エレクトロスプレーイオン化質量スペクトルは、アナリティカ・オブ・ブランドフォード(Analytica of Brandford)エレクトロスプレーインターフェースを装着したFinnigan−MAT SSQ700(米国カリフォルニア州サンノゼ)質量分析計で得た。2400〜3400Vのエレクトロスプレー電圧を用いた。試料をアセトニトリル又はジクロロメタンのいずれかに約10ピコモル/リットルの濃度で溶解し、データ取得の前に1リットル/分の流速で直接注入することによりESIインターフェースに導入し、かつデータ取得の前に導入した。陽イオン電子衝撃イオン化(770ev)MS実験は、INCOSデータシステムと協働するFinnigan−MA T4615四極子質量分析計で行った。イオン源の温度は150℃であり、マニホールドチャンバーの温度は100℃であった。試料はガスクロマトグラフ又は直接挿入探索子により導入した。陽イオン高速原子衝撃質量スペクトルは、INCOSデータシステムと組み合わせたFinnigan−MAT212磁気セクター装置で取得した。加速電圧は3kVであり、イオン源の温度は約70℃であった。イオン・テック(Ion Tech)サドルフィールド(saddle field)高速原子ガンをキセノンを用いて8keVで用いた。チオグリセロールをFABマトリックスとして用いた。陽イオン電子衝撃イオン化(70cV)MS/MS実験はFinnigan−MAT TSQ/700タンデム四極子質量分析計で行った。試料は直接挿入探索子によって導入した。イオン源は150℃に維持し、マニホールドチャンバーは70℃で保持した。衝突誘発解離(Collision−induced dissociation)(CID)は、マニホールド内の圧力が0.9〜2.5×10-6トール(120〜333μPa)に到達するまで中心rf単独衝突八極子(center rf−only collision octapole)にアルゴンを導入することにより達成した。CID生成物イオンの名目上のイオン動力学的エネルギーは<35Ev(実験参考値)であった。高解像度データはEB形状のJEOL JMS AX−505H二重焦点質量分析計で7500の解像度を用いて得た。試料はガスクロマトグラフ又は直接挿入探索子によって導入した。質量スペクトルの取得の間、ペルフルオロケロセンを加熱導入口を用いてイオン源に導入した。正確な質量の割り当てはペルフルオロケロセンの質量からコンピュータ支援内挿により得た。GC/MS条件:カラム、20m×0.25mmDB−1701(J & W Sientific);キャリヤガス、直線速度40cm/秒のヘリウム;注入口、125℃;カラム温度、35℃で3分間、次いで100℃まで10℃/分で昇温;注入、分割モード、約50:1の比。
分光学的方法 300MHz1H NMRスペクトル及び75MHz10C NMRスペクトルはIBM AF300装置でオックスフォード超電導磁石システムを用いて得、データ取得はブルーカー(Bruker)ソフトウェアにより制御した。赤外線スペクトルはマッキントッシュII(Macintosh II)コンピュータで制御されたマットソン・ギャラクシー(Mattson Galaxy)シリーズ5000FTIR分光計で得た。UV/可視スペクトルはゼニス(Zenith)Z−425/SXコンピュータによって駆動されるヒューレット・パッカード(Hewlett Packard)8452A分光光度計で得た。通常のX−バンドEPRスペクトルは、オックスフォードESR−900ヘリウム流クリオスタットを備えるブルーカーER300分光計で記録した。メスバウアースペクトルは定加速装置で得、異性シフトは298Kの金属鉄標準に対して報告した。印加した磁場による多結晶試料の配向を回避するため、試料を凍結ヌジョールに懸濁させた。
大環状テトラアミド−N供与体配位子の合成
一般反応スキーム
以下に示されるのは、本発明の大環状テトラアミド配位子を合成するための好ましい反応例である。
以下の実施例1〜25において、この反応工程の様々な部分を説明する。実施例26〜32では本発明の性能特性及び利点を示す。
実施例1
α−メチルアラニン及びジエチルマロニルクロリドからのマクロリンカー中間体(A−L−A)(テトラメチルジエチル置換中間体)合成
均圧滴下ロート(250mL)及び隔壁を付した2頸フラスコ(1L)をN2気流下に置いて、α−アミノ酪酸(即ち、α−メチルアラニン)(20.62g,0.2モル)及び乾燥ピリジン(250mL,4Åモレキュラーシーブで乾燥したもの)をそのフラスコに添加して攪拌しながら60〜70℃に加熱してから、乾燥ピリジン(100mL,4Åモレキュラーシーブで乾燥したもの)中に溶解させたジエチルマロニルクロリド(23.23mL,0.135モル)を滴下ロートに添加する。滴下ロート中の内容物を反応液に添加(滴下,1h)して、N2気流下又は乾燥管を付してアシル化を進行させる(60〜70℃,30〜36h)。アシル化が完了したら、H2O(30mL)を添加して攪拌(60〜70℃,24h)することにより反応を停止させる。溶媒の容量をロータリーエバポレーターで少なくしてオイルを得てから、濃HCl(約25mL)を加えて最終pHを2〜3にする。この熱い溶液を冷蔵庫内(4℃,15h)に入れ、得られた淡褐色生成物を濾取して、アセトニトリル(2×100mL)で徹底的に洗浄する。その風乾後の白色生成物(16.5〜19.8g,50〜60%収率)は、デシケーター内に貯蔵しなければならない。この生成物は、通常、閉環反応のためには十分に純粋であるが、場合により再結晶を行ってもよい。物性測定:1H NMRスペクトル(d2−ピリジン)〔ppm〕:8.9(s,2H,NH,アミド);2.2(q,4H),1.8(s,12H);1.2(t,6H)。IR(ヌジョール混合):〔cm-1〕=3310(アミドNH);1721(カルボキシルCO),1623(アミドCO)。元素分析:C19H21N2O9の計算値;C,54.53;H,7.93;N,8.48。実測値;C,54.48;H,7.88;N,8.47%。
実施例2
α−メチルアラニン及びジエチルマロニルクロリドからの大規模マクロリンカー中間体(A−L−A)(TMDE置換中間体)合成
均圧滴下ロート(250mL)及び隔壁を付した2頸フラスコ(1L)をN2気流下に置いて、α−アミノ酪酸(即ち、α−メチルアラニン)(20.62g,0.2モル)及び乾燥ピリジン(250mL,0.4nm(4Å)モレキュラーシーブで乾燥したもの)をそのフラスコに添加して攪拌しながら60〜70℃に加熱してから、乾燥ピリジン(100mL,0.4nm(4Å)モレキュラーシーブで乾燥したもの)中に溶解させたジエチルマロニルクロリド(23.23mL,0.135モル)を滴下ロートに添加する。滴下ロート中の内容物を反応液に添加(滴下,1h)して、N2気流下又は乾燥管を付してアシル化を進行させる(60〜70℃,30〜36h)。アシル化が完了したら、H2O(30mL)を添加して攪拌(60〜70℃,24h)することにより反応を停止させる。溶媒の容量をロータリーエバポレーターで少なくしてオイルを得てから、濃HCl(約25mL)を加えて最終pHを2〜3にする。この熱い溶液を冷蔵庫内(4℃,15h)に入れ、得られた淡褐色生成物を濾取して、アセトニトリル(2×100mL)で徹底的に洗浄する。その風乾後の白色生成物(16.5〜19.8g,50〜60%収率)は、デシケーター内に貯蔵しなければならない。この生成物は、通常、閉環反応のためには十分に純粋であるが、場合により再結晶を行ってもよい。物性測定:1H NMRスペクトル(d2−ピリジン)〔ppm〕:8.9(s,2H,NH,アミド);2.2(q,4H),1.8(s,12H);1.2(t,6H)。IR(ヌジョール混合):〔cm-1〕=3310(アミドNH);1721(カルボキシルCO),1623(アミドCO)。元素分析:C19H21N2O9の計算値;C,54.53;H,7.93;N,8.48。実測値;C,54.48;H,7.88;N,8.47%。
実施例2
α−メチルアラニン及びジエチルマロニルクロリドからの大規模マクロリンカー中間体(A−L−A)(TMDE置換中間体)合成
均圧滴下ロート(250mL)及び隔壁を付した2頸フラスコ(2L,RB+クライゼン)をN2気流下に置いて、α−アミノ酪酸(即ち、α−メチルアラニン)(90.3g,0.9モル)を添加し、無水ピリジン(1.4L,確実密閉)をそのフラスコにカニューレで添加し、反応混合物を45〜55℃に加熱して攪拌する。ピリジン(100mL,確実密閉)の後にジエチルマロニルクロリド(104.4mL,0.61モル)を滴下ロートにカニューレで添加する。滴下ロート中の内容物を反応液に添加(滴下,3〜4h)してから滴下ロートを外し、N2気流下でアシル化を進行させる(55〜65℃,120〜130h)。アシル化が完了したら、H2O(100mL)を添加して攪拌(60〜70℃,24〜36h)することにより反応を停止させる。溶媒の容量をロータリーエバポレーターで少なくしてオイルを得てから、濃HCl(約110mL)を加えて最終pHを2〜3にする。この熱い溶液を冷蔵庫内(4℃,15h)に入れ、得られた淡褐色生成物を濾取して、三角フラスコ中で攪拌することによりアセトニトリル(700mL,150mL)で徹底的に洗浄する。その風乾後の白色生成物(87.9g,60%収率)を乳鉢と乳棒で粉砕してデシケーター内に貯蔵する。この大規模反応アミド中間生成物は、閉環反応に使用する前に再結晶を要するようである。
実施例3
上記からのTMDE置換中間体の再結晶
実施例2からの粗製TMDE中間体(50.4g,0.153モル)を、NA2CO3(16.2g,0.153モル)を3回に分けてゆっくり添加して良好な攪拌と穏和な加熱で過剰な発泡を避けることにより、H2O(500mL,脱イオン水)中に溶解させる。この溶液を沸騰させ、濾過し、濃HCl(30mL,0.36モル)で酸性にする。この溶液を冷やして(一晩,4℃)白色析出物を濾取してアセトニトリル(250mL)で洗浄する。その風乾後の生成物(38.8g,再結晶収率77〜90%)は、デシケーター内に貯蔵しなければならない。
実施例4
ヘキサメチル(HM)中間体(A−L−A)
HM中間体の合成は、ジエチルマロニルクロリドをジメチルマロニルクロリド(17.8mL,0.135モル)に代え、そのアシル化剤の低沸点のために反応温度を55〜65℃に低下させなければならなかったことを除けば、実施例2におけるTMDE中間体の合成と同一である。ヘキサメチル中間体の収率は45〜60%である。物性測定:1H NMR(d5−ピリジン,〔ppm〕):9.2〜9.8(br s,2H(カルボキシOH)),8.23(s,2H(アミド)),1.87(s 12H(CH3)),1.74(s 6H(CH3))。IR(ヌジョール/NaCl):〔cm-1〕=3317.0(アミドNH);1717.9(カルボキシルCO),1625.7(アミドCO)。元素分析(100℃で乾燥したもの):C13H22N2O6の計算値;C,51.63;H,7.34;N,9.27。実測値;C,51.64;H,7.35;N,9.33%。
実施例5
HM中間体の再結晶
TMDEアミド中間体と同じやり方で粗製ヘキサメチル(HM)中間体を再結晶した。HMアミド中間体の僅かに高い水溶性のために、少ないH2Oを用いなければならない。
実施例6
ジシクロヘキシルジエチル中間体
丸底フラスコ(500mL)に1−アミノ−1−シクロヘキサンカルボン酸(15g,0.1モル)を仕込んでから、均圧滴下ロート(40mL)を付し、隔壁でキャップをし、そして窒素でパージする。無水ピリジン(300mL)をその反応フラスコにカニューレで滴下ロートを通して添加し、20mLを滴下ロートに添加する。その系を加熱し始めて温度を60℃に安定化させる。60℃に到達したら、その反応に使用される全ジエチルマロニルクロリドの3分の1(即ち、6mL,0.033モル)をシリンジを介して滴下ロートに添加する。このピリジンとジエチルマロニルクロリドの混合液を反応液に滴下してアシル化を12時間進行させる。第2量(6mL,0.033モル)及び第3量(6mL,0.033モル)を12時間の間隔をあけて添加する。全てのアシル化剤を添加して反応させた後(全反応時間48〜56h)、20mLの水を反応液に滴下する。反応液を更に24時間加熱してモノ及びビスオキサゾロン中間体を開環させてジアミドジカルボン酸を生成させる。ロータリーエバポレーターによりピリジンを除去すると、淡黄色性淡褐色スラッジが生成するので、これを濃HClでpH2にする。こ粗生成物を濾取し、アセトニトリルで洗浄し、乾燥して白色のジシクロヘキシルジエチルアミド中間体(16g,74%)を得る。物性測定:1H NMR(d5−ピリジン)〔ppm〕:8.30(s,2H,NHアミド),2.60(m,4H,シクロヘキシル),2.25(q,4H,エチルCH2),2.15(m,4H,シクロヘキシル),1.8〜1.5(m,10H,シクロヘキシル),1.25(m,2H,シクロヘキシル),1.20(t,6H,エチルCH3)。13C NMR広幅吸収(broadband decoupled)(d5−ピリジン)〔ppm〕:178.0(カルボキシCO),174.3(アミドCO),60.5(シクロヘキシルquat),59.4(マロニルquat),33.0(シクロヘキシルCH2),30.3(エチルCH2),26.0(シクロヘキシルCH2),22.3(シクロヘキシルCH2),9.9(エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕:3307(アミドNH);3150(sh,br,m,アミドNH/カルボキシOH),3057(s,str,水素結合アミドNY/カルボキシOH),1717(s,str,カルボキシルCO),1621(s,str,アミドCO)。元素分析:C21H24N:O6の計算値;C,61.44;H,8.35;N,6.82。実測値;C,61.41;H,8.38;N,6.90%。
実施例7
ジシクロヘキシルジエチルモノオキサゾロン
1.35ジエチルマロニルクロリドの理論量でのジシクロヘキシルジエチル中間体反応(熱及び水,上記を参照のこと)を停止させるのに失敗すると、2シクロヘキシルアミノ酸は、ジシクロヘキシルジエチルアミド中間体及びモノオキサゾロン生成物の混合物をもたらす。このジシクロヘキシルジエチルモノオキサゾロン生成物は、沸騰シクロヘキサン中で中程度に溶解性であるが、ジシクロヘキシルジエチルアミド中間体はそうではないので、この生成物混合物は簡単に分離して、少量の残留CH2Cl2を含有する約10gの混合されたアミド中間体とモノオキサゾロンは、400〜500mLのシクロヘキサン中での激しい攪拌で沸騰した。不溶性のジシクロヘキシルジエチルアミド中間体生成物は、熱自然濾過により濾取されたが、モノオキサゾロン生成物は、そのシクロヘキサン溶液を冷却して溶媒を留去すると、徐々に結晶化した。アミド中間体の収量は約6gでモノオキサゾロンの収量は約4gであった。物性測定:1H NMR(d5−ピリジン)〔ppm〕:9.7(s,1H,アミドNH),2.7〜1.6(未分離シクロヘキシル基),1.05(t,6H,エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕:3309(sh,w,アミドNH),3229(s,str,水素結合アミドNH/カルボキシOH),3166(s,str,水素結合アミドNH/カルボキシOH),3083(s,str,水素結合アミドNH/カルボキシOH),1834(s,str,オキサゾロンC=O),1809(s,m,水素結合オキサゾロンC=O),1743(s,str,カルボキシルCO),1663(s,str,オキサゾロンC=N),1639(s,br,str,アミドCO)。元素分析:C22H22N2O5(C6H12)0.25の計算値;C,65.35;H,8.53;N,6.77。実測値;C,65.07;H,8.67;N,6.68%。溶媒和シクロヘキサンの存在は13C NMRにより確認した。
大環化反応
大環状テトラアミド配位子の調製のための幾つかの合成ルートの例は次の通りである。
三塩化リンカップリング
アミド含有中間体(A−L−A)の芳香族1,2−ジアミンへの三塩化リンカップリングで、大環状テトラアミドが安全、安価かつ高収率で生成する。2つの明確に異なるPCl3カップリング法の変法が有用なものであり、その相違は、添加の順序及び用いられる試薬の選択に関連する。これら方法は、主として全ての合成へのマクロリンカー型のアミド中間体の並行的組み込むの故に、橋架けジアミン上に存在する異なる電子置換基又はアミド中間体上に存在する立体的置換基を有する多種多様の異なる大環状化合物の調製に応用可能である。
実施例8
A.三塩化カップリングによる大環合成
長頸フラスコ(250mL)に実施例2〜7のアミド中間体(10ミリモル)を仕込んで攪拌バーを入れてから、オーブン中で焼く(80〜100℃,30〜45分)。その熱いフラスコをN2気流下に置いて、アリールジアミン(10ミリモル)を添加して無水ピリジン(50mL,確実密閉)をカニューレで添加する。そのフラスコを加熱(50〜60℃)してPCl3(d=1.574g/mL,1.72mL,20ミリモル)を過剰に還流させることなくできるだけ速やかにシリンジで添加する。これは発熱反応なので、注意を要する。次いで、温度を還流まで又は還流直前(100〜115℃)まで上げて、N2気流下で反応を進行させる(48h)。アシル化が完了したら、フラスコの内容物をHCl(1規定,約60mL)で最終pH2まで酸性にする。この混合液を三角フラスコに移して(フラスコを濯ぎ洗うのに水を使用する)CH2Cl2と一緒に攪拌(300mL,2〜3h)してから、追加のCH2Cl2(2×150mL)で抽出する。合わせた有機層を希HCl(0.1M,2×100mL)で洗浄してからNa2CO3の薄い水溶液(2×5g/100mL)で洗浄する。有機溶媒をロータリーエバポレーターで除去して粗生成物を得る(30%)。この粗生成物の重量は、通常、ジアミンの当初重量に等しい。
B.三塩化リンカップリングによる大環合成
長頸フラスコ(250mL)にMgSO4(5g)、攪拌バー、アリールジアミン(10ミリモル)及びピリジン(50mL,0.4nm(4Å)モレキュラーシーブで乾燥したもの)を仕込んでからN2気流下に置いて、PCl3(d=1.754g/mL,1.72mL,20ミリモル)をシリンジで添加し、その混合液を30分間還流させると、橙/黄色析出物が生成する。この混合液を幾分冷却して、アミド中間体(10ミリモル)を添加してから、その混合液をN2気流下で還流させる(115℃,48h)。アシル化が完了したら、フラスコの内容物をHCl(1規定,約60mL)で最終pH2まで酸性にする。この混合液を三角フラスコに移してCH2Cl2と一緒に攪拌(2×150mL)する。合わせた有機層を希HCl(0.1M,2×100mL)で洗浄してからNa2CO3の薄い水溶液(2×5g/100mL)で洗浄する。有機溶媒をロータリーエバポレーターで除去して粗生成物を得る(30%)。この粗生成物の重量は、通常、ジアミンの当初重量に等しい。
注意:大規模大環化反応では、閉環時間は還流下で4〜5日まで長引くので、反応の終盤に存在するピリジンの殆どは、酸性化の前にロータリーエバポレーターにより除去されていまう。
実施例9
TMDE中間体+DCBジアミンからのTMDE−DCB
1,2−ジアミノ−4,5−ジクロロベンゼン(1.77g,10ミリモル)をアリールジアミンとして、TMDEアミド中間体(3.3g,10ミリモル)と共に、A法又はB法のPCl3大環化反応に用いた。粗製大環状生成物(2.7g)を最少量の熱95%EtOHから留去により再結晶して、純粋なTMDE−DCB(1.5g,32%)を得た。物性測定:1H NMR(CH2Cl2)〔ppm〕:7.65(s,1H,ArH),7.35(s,2H,アミドNH),6.45(s,1H,アミドNH),1.90(q,4H,エチルCH2),1.57(s,12H、RCH3),0.85(t,6H,エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕=3454(痕跡ROH),3346(br,アミドNH),1706&1688&1645(アミドCO)。元素分析:C22H28Cl2N4O4の計算値;C,53.51;H,5.99;N,11.89。実測値;C,53.58;H,6.09;N,11.89。
実施例10
TMDE中間体+BジアミンからのTMDE−B
1,2−ジアミノベンゼン(即ち、o−フェニレンジアミン)(1.08g,10ミリモル)をアリールジアミンとして、TMDEアミド中間体(3.3g,10ミリモル)と共に、A法又はB法の大環化反応に用いた。粗製大環状生成物(1.5g)を最少量の熱95%EtOHから留去により再結晶して、純粋なTMDE−B(ジアミンから25%)を得た。物性測定:1H NMR(CDCl3)〔ppm〕:7.55(m,2H,ArH),7.48(s,br,2H,アリールアミドNH),7.17(m,2H,ArH),6.46(s,br,2H,アルキルアミドNH),2.07(m,br,4H,エチルCH2),1.60(s,12H,RCH3),0.89(t,6H,エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕=3395&3363(アミドNH),1702&1680&1652&1635(アミドCO)。元素分析:C21H10N4O4・H2Oの計算値;C,59.98;H,7.67;N,13.32。実測値;C,60.18;H,7.20;N,13.18。
実施例11
TMDE中間体+DCBジアミンからのTMDE−DMB
1,2−ジアミノ−4,5−ジメチルベンゼン(1.36g,10ミリモル)をアリールジアミンとして、テトラメチルジエチルアミド中間体(3.3g,10ミリモル)と共に、A法又はB法のPCl3大環化反応に用いた。粗製大環状生成物(1.6g)を最少量の熱95%EtOHから留去により再結晶して、純粋なTMDE−DMB(ジアミンから25%)を得た。物性測定:1H NMR(DMSO d6)〔ppm〕:8.00(s,2H,アミドNH),7.67(s,2H,アミドNH),7.28(s,2H,ArH),2.17(s,6H,アリールCH3),1.99(q,4H,エチルCH1),1.46(s,12H,RCH3),0.75(t,6H,エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕:3446(s,m,痕跡ROH),3362(s,str,アミドNH),3348(sh,m,アミドNH),3332(s,str,HアミドNH),1696(アミドCO),1679(アミドCO),1641(アミドCO),1584(s,m/w,アリール環/アミド)。元素分析:C23H14N4O4の計算値;C,74.16;H,7.96;N,13.01。実測値;C,64.09,64.28;H,8.04,7.92;N,12.86,13.04。
実施例12
TMDEアミド中間体+DMOBジアミンからのTMDE−DMOB
上記のようにして調製した1,2−ジアミノ−4,5−ジメチルベンゼン・2HBr(5.0g,15ミリモル)をアリールジアミンとして、直接、テトラメチルジエチルアミド中間体(5.0g,15ミリモル)と共に、A法又はB法の1.5倍規模PCl3大環化反応に用いた。粗製大環状生成物(3.57g)を最少量の熱80〜85%EtOH(1g/40mL)から留去により再結晶して、純粋なTMDE−DMOB(ジアミンから30%)を得た。物性測定:1H NMR(CH2Cl2)〔ppm〕:7.26(s,2H,アミドNH),7.01(s,2H,ArH),6.41(s,2H,アミドNH),3.80(s,6H,アリールOCH3),2.07(q,br,4H,エチルCH3),1.54(s,12H,RCH3),0.90(t,6H,エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕:3451(s,m,水素結合H2O),3391&3347(アミドNH),1695&1670&1655(アミドCO)。元素分析:C23H34N4O6(H2O)0.22の計算値(ESU);C,58.96;H,7.46;N,11.96。実測値;C,58.90;H,7.26;N,11.76。溶媒和H2Oの存在は1H NMR及びIRにより確認した。
実施例13
TMDE中間体+NapジアミンからのTMDE−Nap
4,5−ジアミノナフタレン(1.68g,10ミリモル)をアリールジアミンとして、テトラメチルジエチルアミド中間体(3.3g,10ミリモル)と共に、A法又はB法のPCl3大環化反応に用いた。未最高化収率はジアミンから15〜20%であった。1H NMR(CDCl3)〔ppm〕:8.05(s,2H,ArH環),7.75(m,2H,ArH環),7.55(s,2H,ArアミドNH),7.35(m,2H,ArH1環),6.45(s,2H,アルキルアミドNH),2.15(m,br,4H,エチルCH2),1.65(s,12H,RCH3),0.90(t,6H,エチルCH3)。
実施例14
HM中間体+DCBジアミンからのHM−DCB
1,2−ジアミノ−4,5−ジクロロベンゼン(1.77g,10ミリモル)をアリールジアミンとして、ヘキサメチルアミド中間体(3.02g,10ミリモル)と共に、A法又はB法のPCl3大環化反応に用いた。粗製大環状生成物(1.33g,30%)を最少量のn−プロパノールから留去により再結晶して、第1晶収率が60%であった。物性測定:1H NMR〔ppm〕:7.69(s,2H,ArH),7.39(s,2H,アミドNH),6.44(s,2H,アミドNH),1.58(s,12H,アームメチル),1.53(s,6H,マロネートメチル),小さなn−プロパノールのピークが認められた。IR(ヌジョール/NaCl)〔cm-1〕:3503(s,br,m−w,n−プロパノールOH),3381(sh,m,アミドNH),3338(s,str,アミドNH),1689(s,str,アミドCO),1643(s,str,アミドCO)。元素分析:C17H24N4O4Cl2(C3H8O)0.2の計算値;C,51.70;H,5.57;N,12.30。実測値;C,51.69;H,5.63;N,12.33。
実施例15
HM中間体+DMOB又はBジアミンからのHM−DMOB及びHM−B
実施例14と同じ方法に従って、HM中間体を用いてHM−B及びHM−DMOBも合成し、ジクロロ誘導体について実施例14で得られた結果と類似の結果を得た。CDCl3中でのHM−DMOBについての1H NMRデータ〔ppm〕:7.65(s,2H,アミドNH),7.21(s,2H,アリールCH),6.72(s,2H,アミドNH),4.00(s,6H,メトキシCH3),1.76(s,12H,アームメチル),1.58(s,6H,マロネートメチル)。d5ピリジン中でのHM−Bについての1H NMRデータ〔ppm〕:8.55(s,2H,アミドNH),8.40(s,2H,アミドNH),7.81(m,2Hh,ArHaa1bb1),7.10(m,2H,ArHaa1bb1),1.77(s,12H,アームメチル),1.73(s,6H,マロネートメチル)。これらのアミドピークは、水、酸等の不純物の存在で0.1のオーダーでシフトする傾向がある。
実施例16
ジシクロヘキシルDE中間体+DCBジアミンからのジシクロヘキシルDE−DCB
1,2−ジアミノ−4,5−ジクロロベンゼン(1.77g,10ミリモル)をアリールジアミンとして、ジシクロヘキシルジエチルアミド中間体(3.3g,10ミリモル)と共に、A法又はB法のPCl3大環化反応に用いた。立体障害が増すので、閉環時間を増やすことが推奨される(通常の48hに対して3〜4日)この反応の間の副生成物としてのシクロヘキシルオキサゾロンは、酸塩基処理によっては除去できないので、最初に単離したCH2Cl2可溶性生成物をペンタンで磨き潰し洗浄してこのオキサゾロンを除去することが必要である。ペンタン洗浄液を濃縮すれば、このオキサゾロンの再利用が可能になる。粗製ペンタン不溶性生成物をCH2Cl2又はCHCl3中に溶解させ、僅かに曇るまでシクロヘキサンを添加してから、空気中で蒸発させることにより再結晶し、濾取して、白色微結晶性ジシクロヘキシルDE−DCB生成物を得た(1.38g,ジアミンから25%)。ニートの熱トルエンからの留去を伴う再結晶も有望である。物性測定:1H NMR(CDCl3)〔ppm〕:7.70(s,2H,ArH),7.45(s,2H,アミドNH),6.45(s,2H,アミドNH),2.35(m,br,4H,シクロヘキシル),2.00(m,br,8H,シクロヘキシル/エチルCH3),1.70(m,br,8H,シクロヘキシル),1.30(m,br,4H,シクロヘキシル),0.90(t,6H,エチルCH3)。元素分析(100℃で乾燥したもの):C27H34Cl2N4O42(C6H12)0.2の計算値;C,59.60;H,6.81;N,9.86。実測値;C,59.60;H,6.77;N,9.77。溶媒シクロヘキサンの存在は1H及び13C NMRにより確認した。
実施例17
ジシクロヘキシルDE中間体+BジアミンからのジシクロヘキシルDE−B
1,2−ジアミノベンゼン(o−フェニレンジアミン,1.08g,10ミリモル)をアリールジアミンとして、ジシクロヘキシルDE−DCBの調製と類似の調製に用い、ジシクロヘキシルDE−Bを得た(1.25g,ジアミンから26%)物性測定:1H NMR(DC3CN)〔ppm〕:7.62(s,2H,アリールアミドNH),7.51(m,2H,ArH),7.18(m,2H,ArH),6.71(s,2H,アルキルアミドNH),2.12(m,6H,シクロヘキシル),1.85(q&m,エチルCH2&シクロヘキシル),1.62(m,シクロヘキシル),1.37(m,シクロヘキシル),0.90(t,6H,エチルCH3),0.85(m,シクロヘキシル)。IR(ヌジョール/NaCl)〔cm-1〕:3750(s,m,H2O),3385(s,str,アミドNH),314(s,str,アミドNH),3258(s,m,br,水素結合アミドNH),1694(s,str,アミドCO),1651(s,str,アミドCO),1594(s,m,アリール環/アミド)。
実施例18
ジシクロヘキシルジエチルビスオキサゾロン
この生成物は、ジシクロヘキシルジエチルアミド中間体とo−フェニレンジアミンとのPCl3大環化反応の副生成物として得られた。このビスオキサゾロンは、酸塩基処理によっては除去されない(中性分子であり非常に有機溶媒可溶性である)。この粗製大環/オキサゾロン生成物をペンタンで洗浄すると、殆どのビスオキサゾロンがペンタン中に抽出される。ペンタン層を空気中で蒸発させると、純粋なビスオキサゾロンが大きな透明プリズム晶(1cm×1cm×0.5cm)として得られる。嵩高な疎水性シクロヘキシル基のために、このオキサゾロンは、対応するメチル誘導体よりも加水分解に対してずっと抵抗性である。ビスオキサゾロンの物性測定:1H NMR(CD3CN)〔ppm〕:2.05(q,4H,エチルCH2),1.8〜1.4(未分離シクロヘキシル基),0.88(t,tH,エチルCH3)。13C NMR広幅吸収(CD3CN)〔ppm〕:181.0(オキサゾロンC=O),162.7(オキサゾロンC=N),69.0(オキサゾロンシクロヘキシルquat),49.0(マロネートquat),34.3(シクロヘキシルメチレン),25.5(シクロヘキシルメチレン),24.9(マロネートメチレン),21.8(シクロヘキシルメチレン),8.3(エチルCH3)。IR(ヌジョール/NaCl)〔cm-1〕:1822(s,str,br,オキサゾロンC=O),1662(s,str,オキサゾロンC=N)。元素分析(50℃で乾燥したもの):C22H10N2O4の計算値;C,67.36;H,8.07;N,7.48。実測値;C,67.26;H,8.15;N,7.64。
キレート錯体の合成
実施例19
[Et1N]2及び[Et1N]3[それぞれ鉄(III)クロロTMDE−DCBモノアニオン及び鉄(III)aquo TMDE−DCBモノアニオンのテトラエチルアンモニウム塩]
上記の実施例10〜18のいずれかのペアレント大環状テトラアミド(525mg,1.1ミリモル)をテトラヒドロフラン(40mL,Aldrich)中にN2気流下で溶解させる。schlenk法を用いて、tert−ブチルリチウム(2.6mL,4.4ミリモル,2,4−ジメチルペンタン中1.7M,Aldrich)をその溶液にN2気流下で−108℃で添加した。次いで、塩化第一鉄(無水、155mg,1.2ミリモル,Alfa)を添加してその溶液を攪拌しながら室温まで温め(16h)、空気に感受性のFeII錯体を海松色析出物として得た。乾燥管から空気を通して(2h)橙色固体を集め、CH2Cl2(2×10mL)で洗浄した。得られた橙色粉末を減圧乾燥した。収量:595mg(93%)。溶媒和が変動すること及び溶解性に限界があるので、更なる使用のために、このリチウム塩をテトラエチルアンモニウム塩に転化させた。CH3OH(50mL)中のリチウム塩(595mg)を[Et4N]+カチオンで予め飽和されたイオン交換カラム(Dowex▲R▼50X2−100)に充填して、橙色バンドをCH3OH(100mL)で溶出させた。溶媒を減圧下で留去した。残渣をCH2Cl2(20mL)中に懸濁させ、その混合液を濾過した。溶媒を減圧下で母液から留去させて、得られた[Et4N]22の橙色吸湿性ガラス状残渣を更に精製することなく使用した。IR(ヌジョール/NaCl)〔cm-1〕:1619((CO)アミド),1575((CO)アミド),1534((CO)アミド)。鉄(III)出発原料の注意深い精製は、このアクシャルのクロロジアニオン性錯体ではなくてアクシャルのaquaモノアニオン性錯体を処理することにより、より便利に行えた。[Et4N]22(550mg,約0.7ミリモル)をCH3CN(50mL)中に溶解させた。テトラフルオロホウ酸銀(140mL,0.7ミリモル)をCH3CN(2mL)中に溶解させてこの溶液に添加して攪拌した(1h)。AgCl析出物を濾去して溶媒を減圧留去させた。得られた[Et4N]3をシリカゲルカラム(CH2Cl2中8%MeOH)から溶出させることにより更に精製した。溶媒を減圧留去して生成物をH2Oから再結晶した。収量:360mg(77%,異なる微結晶性サンプルにおいて水との種々の溶媒和が見られた)。IR(ヌジョール/NaCl)〔cm-1〕:1590((CO)アミド),1565((CO)アミド),1535((CO)アミド)。元素分析:C29H46N5FeO5Cl2(H2O)の計算値;C,50.52;H,7.02;N,10.16;Cl,10.28。実測値;C,50.24;H,6.84;N,9.82;Cl,10.32。ESIMS(陰イオン):m/z522.2,[3−H2O]1-(100%);m/z269.7,[3−H-]2-(18%)。
実施例20
[Et4N]4[鉄(IV)クロロTMDE−DCBモノアニオンのテトラエチルアンモニウム塩]の合成
[Et4N]2(500mg,約0.6ミリモル)をCH2Cl2(30mL)中に溶解させた。硝酸アンモニウムセリウム(IV)(10.3g,18.3ミリモル)をその溶液に添加してその混合液を攪拌した(2h)。固体セリウム塩を濾去した。減圧下で溶媒を留去して減圧乾燥することにより紫色生成物を得た。収量:400mg(95%)。CH2Cl2/Et2Oからの再結晶により紫色結晶を得た。IR(ヌジョール/NaCl)〔cm-1〕:1688((CO)アミド),1611((CO)アミド),1582((CO)アミド)。ESIMS(陰イオン):m/z557,[4]-1(100%);m/z522,[4−Cl]1-(65%)。
実施例21
[Et4N]4[鉄(IV)クロロTMDE−DCBモノアニオンのテトラエチルアンモニウム塩]とNaCNからの[Ph4P]5[鉄(IV)シアノTMDE−DCBモノアニオンのテトラフェニルホスホニウム塩]の合成
[Et4N]4[鉄(IV)クロロTMDE−DCBモノアニオンのテトラエチルアンモニウム塩](225mg,0.33ミリモル)をH2O(10mL)中に懸濁させた。シアン化ナトリウム(140mg,2.85ミリモル)をH2O(10mL)中に溶解させてその懸濁液に添加してその混合液を超音波処理した(Branson1200,0.5h)。この紫色の懸濁液は濃い青色溶液に変化し殆ど全ての固体物質は溶解した。この混合液を濾過して、水に溶かしたPPh4Cl[テトラフェニルホスホニウムクロリド](600mg,1.6ミリモル,10mL,Aldrich)を添加することにより、青色生成物を析出させた。この青色生成物を濾取してH2O(2×10mL)で洗浄した。収量:250mg(0.28ミリモル,85%)。この物質(120mg)を薄層クロマトグラフィー(TLC)(シリカゲルプレート,GF,20cm×20cm×1000m,10:1 CH2Cl2:CH3CN)により更に精製した。この青色物質を、CH3CN:CH2Cl2(1:1,60mL)でシリカゲルから抽出した。溶媒を減圧留去して残渣をCH2Cl2(3mL)中に溶かして濾過した。ペンタン(150mL)を加えると、青色粉末(90mg,0.10ミリモル)が得られた。精製収率:75%。IR(ヌジョール/NaCl)〔cm-1〕:2129(CN),1659((CO)アミド),1598((CO)アミド),1571((CO)アミド)。元素分析:C46H44N5FeOCl2Pの計算値;C,62.18;H,4.99;N,7.88;Cl,7.98。実測値;C,61.96;H,5.04;N,7.84;Cl,8.06。ESIMS(陰イオン):m/z548.2,[5]1-(100%);m/z522.1,[5−CN]1-(20%)。13C−標識シアナイド;m/z549.2,[5]1-(100%);m/z522.1,[5−13CN]1-(8%)。
実施例22
ニトリルシアナイド供給源からの[Ph4P]5[鉄(IV)シアノTMDE−DCBモノアニオンのテトラフェニルホスホニウム塩]の合成
[Ph4P]5[鉄(IV)シアノTMDE−DCBモノアニオンのテトラフェニルホスホニウム塩]は、塩基の存在下でも不存在下でも生成させることができる。塩基の不存在下では、単離処理で溶媒が除去されるにつれて、青色が黄橙色に色あせる。従って、青色固体を得るための生成物単離は、9〜10のpH域の塩基の存在下で最も良好に行われる。次の反応では、溶媒基質としてのCH3CN、CD3CN、CH3CH2CN及び(CH3)2CHCNのそれぞれで5が生成する。記載した触媒反応には塩基を添加しなかった。単離された[Ph4P]5をTBHP(tert−ブチルハイドロペルオキシド)のアセトニトリル溶液に添加することにより、青色化合物が有効な触媒前躯体となることが確認された。この溶媒及び酸化体の両方共が消費されたことは、[Ph4P]5がこの触媒的酸化過程の最終生成物として形成されるとはいえ、それは触媒の失活型ではないことを示している。
実施例23
塩基の存在下での[Ph4P]5の合成
160mg,0.23ミリモル)を選択したニトリル溶媒(6中に溶解された。水酸化テトラエチルアンモニウム塩基を添加(20wt%,0.370,Aldrich)してから、tert−ブチルハイドロペルオキシド(90%,0.605mL,5.4ミリモル)を攪拌しながら滴下(20分)すると青色溶液になった。溶媒を減圧留去すると、オイル状青色残渣(15mL)が残ったので濾過した。この青色物質をPPh4Cl(800mg,2.1ミリモル,Aldrich)の水溶液から析出させ、10を濾取してH2O(2×10mL)で洗浄した。収率:130%。[Ph4P]5に記載した通りに更なる精製を行った。
実施例24
[Et1N]3・H2OについてのX線結晶構造データ及び精密化
C29H48Cl2FeN5O6,M=689.47,三斜晶系、空間群P−1,a=9.899(2);b=11.771(2);c=1.4991(4)nm(14.991(4)Å),=95.33(2);=100.09(2);=92.31(2)゜,V=1.7096(6)nm3(1709.6(6)Å3),Dobs=1.33gcm3,Dcalcd(Z=2)=1.339gcm3,T=293K,=0.071069nm(0.71069Å),=0.64mm-1,トランス係数0.87〜1.00。回折データは、グラファイト単色化Mo−K照射を用いてEnraff−Nonius CAD−4回折計で室温で収集した。データ収集全体で3個の反射データを追跡し、強度においてランダムなふらつきが観察されただけであった。構造は直接法により解析した。炭素に結合した水素原子を0.096nm(0.96Å)のC/H結合距離を有する計算位置に含ませて、ペアレント炭素よりも20%大きな温度因子(thermal parameter)を有するライディングモデル(riding model)を用いて精密化(refine)した。水分子の水素原子は、電子密度差マップ(electron density defference map)から位置決定し、それらの位相(coordinate)を酸素のものよりも20%大きく固定した温度因子で精密化した。精密化は、International Tablesからの走査因子でF2に関する全マトリックス最小二乗法によった。全ての非水素原子は、異方性温度因子で精密化した。最終的な電子密度差マップは特色のないものであった。精密化は、2262の観察した反射データについて重み(weight)1.0/[2(F2)+{0.0652(F2+2F2)/3}2]でR=0.053,wR2=0.112に収斂した。
実施例25
[Et4N]4についてのX線結晶構造データ及び精密化
20±1Cでの[Et4N]4の単結晶は、単斜晶系であり、a=0.9958(2)nm(9.958(2)Å)、b=1.4956(3)nm(14.956(3)Å)、c=2.2688(5)nm(22.688(5)Å),=90.00、=93.83(2)、=90.00、V=3.372(1)nm3(3372(1)Å3)、及びZ=4(dcalcd=1.357gcm3:1(CuK)=6.17mm-1を有する空間群P21/c−C5 2(No.14)である。2(CuK)、115.0を有する4626個の独立吸収補正反射データを−2スキャン及びNiでフィルター処理したCuK照射を用いて補正した。構造は、Crystalytics Companyで修正されたNicolet SHELXTLソフトウェアーでの直接法を用いて解析された。得られた構造因子は、2(CuK)<115.0及びI>3(I)を有する2680の独立反射データについてR1(重みなし、Fに基づく)=0.037に収斂した。10個のメチル基は、sp3混成配置及び0.096nm(0.96Å)の結合長を有する硬質ローター(rigid rotors)として精密化された。各メチル基の初期方向は、それらの水素原子についての異なるフーリエ位置から決定した。各メチル基の最終方向は、3個の回転因子により決定した。それらの水素原子についての異なるフーリエ位置から決定した。硬質ローターメチル基についての精密化位置は、103〜118のC−C−H角度を有する。残りの水素原子は、それぞれの炭素原子上にある理想化された原子(炭素原子のsp2又はsp3混成及び0.096nm(0.96Å)のC−H結合長を想定)としての構造因子計算に含めた。各水素原子の異方性温度因子は、それが共有結合している炭素の相当異方性温度因子の1.2倍に固定された。
実施例26
長期触媒安定性
図3を参照すると、本発明の2つの態様の触媒寿命が比較されている。化合物1は、それぞれが−CH2CH3である置換基R'及びR"を有した。対照には触媒は添加されなかった。
条件は、pH9、室温(21.1℃)でNaHCO3/Na2CO3の緩衝系であった。酸化体は4mM(30%)H2O2であった。それぞれのアステリスクにおいて、12μMのピアシアノール染料を添加した。
このグラフから分かるように、化合物1が存在する場合の染料のそれぞれの添加は、殆ど即時の脱色をもたらした。化合物2、つまりジエチル化合物は、より緩やかな脱色であった。対照だけが非常に緩やかな脱色速度を示した。
前述の事柄から、本発明化合物、特に化合物1は、洗浄液中で洗浄される着色した布から放出される付着性の又は自由に流れる染料を酸化又は脱色するのに有効である。自由に流れる染料種は、別の布のような吸着剤又は吸着性基質上に吸着され得る。かくして、本発明の大環状テトラアミド化合物は、酸化体系、即ち、染料掃去にユニークな恩恵を提供するので、付着性染料の移行を阻止し、かくして洗浄液中での一方の布から他方の布への不要な染料の移行を阻止する。
実施例27〜30は、染料のような吸収性又は吸着性種が溶液中で布のような基質上に吸収又は吸着するのを阻害するのに十分な速度でそれら種を酸化する、本発明の漂白活性剤のユニークな能力の更なる例である。溶液中で速やかに酸化する能力は、本発明の大環状テトラアミド配位子の染料移行阻害特性を提供する。実施例27〜28では、スペクトル及び吸光度−時間曲線を島津分光光度計で記録した。過酸化物又は触媒を添加する前にサンプルを350〜700nmの波長(I)域で走査して、染料の最大吸光度のための波長を確認した。次いで、その分光光度計を極大波長に設定して過酸化物及び/又は触媒を添加した。2分後の極大吸光度の変化を記録した。
アシッドブルー25を600nmで追跡した。サンプルは、2mLの溶液を含有できる1cmのキュベット中で25℃で予め形成した。
実施例27
溶液中でのアシッドブルー25の漂白
アシッドブルー25の溶液〔120mg/l(染料含有量45%),600nmでの初期吸光度は1.2であった〕に:(a)20ppmA.O.H2O2;(b)20ppmA.O.H2O2+1ppmのZ及びYがそれぞれ水素である本発明化合物(以下、FeB);及び(c)20ppmA.O.H2O2と1ppmのZ及びYがそれぞれクロロである本発明化合物(以下、FeDCB)を添加した。以下に示すように、触媒を含有する系だけが、染料の何らかの漂白効果を示した(2分後の600nmにおける吸光度の変化を観察して追跡した)。更なる比較として、次亜塩素酸ナトリウムにより生ずる吸光度の低下を示した(5.25%溶液,20ppmAv.Cl2で添加した)。これら結果を以下の表に纏めた。
実施例28
溶液中でのアシッドオレンジ8の漂白
アシッドオレンジ8の溶液(210mg/l(染料含有量65%),490nmでの初期吸光度は1.2であった)ことを除いて、上記の実施例27の通りに実験を行った。漂白は、490nmにおける吸光度の変化として測定した。
以下の実施例29及び30では次の条件を用いた:0.95gのUltra Tide▲R▼洗濯洗剤(Procter & Gamble)を1.5リッターの温水、2枚の20.32×20.32cm(8×8インチ)綿製標的布(大片)、及び溶液に染料を放出する布と共にTerg−O−Tometerバケツに加えた。標的布の目的は、脱色又は酸化されなかった付着性染料についての染料受容体としての役目を果たす。染料掃去系(H2O2及び触媒)の添加後、Terg−O−Tometerを用いてサンプルを12分間掻き混ぜたてから、周囲温度で水で2分間濯ぎ、そして自動乾燥機内で20分間乾燥させた。
実施例29
ダイレクトレッド79を用いる生地から生地への染料移行
上記の溶液実験(実施例27〜28)で見られた効果をそのような溶液中に存在する生地で確認するために、溶液に0.1gのダイレクトレッド79を放出する布を含有するモデル洗浄液中に清浄な綿製小片を浸漬する実験を行った。標的布により吸収された染料の量は、ΔEの計算値により出した。染料移行阻害性能を標準的染料移行阻害剤であるポリビニルピロリドン(PVP)と比較した。これらデータでは、より小さな評点がより良好である。
実施例30
アシッドレッド151を用いる生地から生地への染料移行
0.1gのアシッドレッド151を放出する放出布を用いることだけを除き、実施例29に従って実験を行った。
実施例31
マスタード及び土の汚れ除去
この実施例は、家庭での洗濯を模した条件下での汚れの除去を実証するものである。マスタード又は天然に存在する粘土で汚れた布を2gのA11▲R▼液体洗濯洗剤及び酸化体系(H2O2又はH2O2と本発明触媒FeDCBのいずれか)で洗浄した。洗浄条件は、Terg−O−Tometerを用いる中水位の温水、冷水濯ぎであった。汚れ除去率を測定し、汚れ除去率(%SRE)を用いて計算した。従って、より高い評点が好ましい。
実施例32
Claims (17)
- 漂白組成物であって、
(a)構造:
(式中、X及びZはH、電子供与基又は電子吸引基であり得、R、R'及びR"はH、アルキル、シクロアルキル、シクロアルケニル、アルケニル、アリール、アルキニル、アルキルアリール、ハロゲン、アルコキシ、フェノキシ、CH2CF3若しくはCF3置換基のあらゆる組み合わせであるか、又は結合して炭素ではない原子を少なくとも1つ含んでいてもよいシクロアルキル環もしくはシクロアルケニル環を形成することができ;MはI、II、III、IV、VもしくはVIの酸化状態を有する遷移金属であるか、又は元素周期表の6、7、8、9、10及び11族から選択され;Qは化学量論基準でこの化合物の電荷と釣り合うあらゆる対イオンであり;Lは不安定性配位子である)を有する大環状テトラアミド配位子である酸化的に安定な漂白剤活性剤、及び
(b)有効量の酸化性化合物の供給源
を含む漂白組成物。 - 漂白組成物であって、
(a)構造:
(式中、X及びZはH、電子供与基又は電子吸引基であり得、R'及びR"はH、アルキル、シクロアルキル、シクロアルケニル、アルケニル、アリール、アルキニル、アルキルアリール、ハロゲン、アルコキシ、もしくはフェノキシ置換基のあらゆる組み合わせであるか、又は結合して炭素ではない原子を少なくとも1つ含んでいてもよいシクロアルキル環もしくはシクロアルケニル環を形成することができ;MはI、II、III、IV、VもしくはVIの酸化状態を有する遷移金属であるか、又は元素周期表の6、7、8、9、10及び11族から選択され;Qは化学量論基準でこの化合物の電荷と釣り合うあらゆる対イオンであり;Lは不安定性配位子である)を有する大環状テトラアミド配位子である酸化的に安定な漂白剤活性剤、及び
(b)有効量の酸化性化合物の供給源
を含む漂白組成物。 - 前記酸化性化合物が、過酸化水素、過酸化水素付加物、水溶液中で過酸化水素を生成することが可能な化合物、有機過酸化物、過硫酸塩、過リン酸塩、及び過ケイ酸塩からなる群から選択されるペルオキシ化合物である、請求項1又は2に記載の漂白組成物。
- X及びZが、H、ハロゲン、SO3、OSO3、OSO3R'''(式中R'''は、H、アルキル、アリール、又はアルキルアリールである)及びNO2からなる群から独立に選択される、請求項1又は2に記載の漂白組成物。
- R'及びR"がH及びC1-6アルキルから選択される、請求項1又は2に記載の漂白組成物。
- 表面活性剤、充填剤、ビルダー、金属イオン封鎖剤、酸化防止剤、酵素、蛍光白化剤、染料、着色料、顔料、及び他の慣用のクリーニング及び/又は洗濯補助剤からなる群から選択される更なる補助剤を含む、請求項1又は2に記載の漂白組成物。
- MがFe又はMnである、請求項1又は2に記載の漂白組成物。
- R'及びR"が、C1-6アルキルから選択されるか、又は一緒に結合して炭素ではない少なくとも1つの元素を有していてもよいシクロアルキルもしくはシクロアルケニルを形成している、請求項1又は2に記載の漂白組成物。
- X及びZがHである、請求項1又は2に記載の漂白組成物。
- 表面又は布を請求項1又は2に記載の組成物と接触させることを含む、表面又は布を漂白する方法。
- 吸収性又は吸着性基質上への吸収性又は吸着性種の、該種及び該基質を漂白剤に曝すことによる吸収及び吸着を阻害する方法において、該種及び該基質を
(1)構造:
(式中、X及びZはH、電子供与基又は電子吸引基であり得、R、R'及びR"はH、アルキル、シクロアルキル、シクロアルケニル、アルケニル、アリール、アルキニル、アルキルアリール、ハロゲン、アルコキシ、フェノキシ、CH2CF3若しくはCF3置換基のあらゆる組み合わせであるか、又は結合して炭素ではない原子を少なくとも1つ含んでいてもよいシクロアルキル環もしくはシクロアルケニル環を形成することができ;MはI、II、III、IV、VもしくはVIの酸化状態を有する遷移金属であるか、又は元素周期表の6、7、8、9、10及び11族から選択され;Qは化学量論基準でこの化合物の電荷と釣り合うあらゆる対イオンであり;Lは不安定性配位子である)を有する大環状テトラアミド配位子である酸化的に安定な漂白剤活性剤、及び
(2)有効量の酸化性化合物の供給源
を含んでなる水溶液に曝すこと含み、
該吸収性又は吸着性種が該溶液中で該基質上に吸収又は吸着するのを阻害するのに十分な速度で酸化されることを特徴とする方法。 - 吸収性又は吸着性基質上への吸収性又は吸着性種の、該種及び該基質を漂白剤に曝すことによる吸収及び吸着を阻害する方法において、該種及び該基質を
(1)構造:
(式中、X及びZはH、電子供与基又は電子吸引基であり得、R'及びR"はH、アルキル、シクロアルキル、シクロアルケニル、アルケニル、アリール、アルキニル、アルキルアリール、ハロゲン、アルコキシ、もしくはフェノキシ置換基のあらゆる組み合わせであるか、又は結合して炭素ではない原子を少なくとも1つ含んでいてもよいシクロアルキル環もしくはシクロアルケニル環を形成することができ;MはI、II、III、IV、VもしくはVIの酸化状態を有する遷移金属であるか、又は元素周期表の6、7、8、9、10及び11族から選択され;Qは化学量論基準でこの化合物の電荷と釣り合うあらゆる対イオンであり;Lは不安定性配位子である)を有する大環状テトラアミド配位子である酸化的に安定な漂白剤活性剤、及び
(2)有効量の酸化性化合物の供給源
を含んでなる水溶液に曝すこと含み、
該吸収性又は吸着性種が該溶液中で該基質上に吸収又は吸着するのを阻害するのに十分な速度で酸化されることを特徴とする方法。 - 酸化性化合物がペルオキシ化合物である、請求項11又は12に記載の方法。
- 前記ペルオキシ化合物が、過酸化水素、過酸化水素付加物、水溶液中で過酸化水素を生成することが可能な化合物、有機過酸化物、過硫酸塩、過リン酸塩、及び過ケイ酸塩からなる群から選択される、請求項13に記載の方法。
- 溶液が更に溶剤を含む、請求項11又は12に記載の方法。
- 溶剤が水である、請求項15記載の方法。
- MがFe又はMnである、請求項11又は12に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/684,670 US5876625A (en) | 1996-07-22 | 1996-07-22 | Metal ligand containing bleaching compositions |
| US08/684,670 | 1996-07-22 | ||
| PCT/US1997/012358 WO1998003625A1 (en) | 1996-07-22 | 1997-07-21 | Metal ligand containing bleaching compositions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2001503073A JP2001503073A (ja) | 2001-03-06 |
| JP3623802B2 true JP3623802B2 (ja) | 2005-02-23 |
Family
ID=24749062
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50704898A Expired - Fee Related JP3623802B2 (ja) | 1996-07-22 | 1997-07-21 | 金属配位子含有漂白組成物 |
| JP50716598A Expired - Fee Related JP3623803B2 (ja) | 1996-07-22 | 1997-07-22 | 金属配位子を含有する漂白組成物 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50716598A Expired - Fee Related JP3623803B2 (ja) | 1996-07-22 | 1997-07-22 | 金属配位子を含有する漂白組成物 |
Country Status (21)
| Country | Link |
|---|---|
| US (3) | US5876625A (ja) |
| EP (2) | EP0918840B1 (ja) |
| JP (2) | JP3623802B2 (ja) |
| KR (2) | KR20000067992A (ja) |
| CN (2) | CN100352902C (ja) |
| AP (2) | AP1013A (ja) |
| AT (2) | ATE225844T1 (ja) |
| AU (2) | AU730906B2 (ja) |
| BR (2) | BR9710538A (ja) |
| CA (2) | CA2261228C (ja) |
| DE (2) | DE69716275T2 (ja) |
| DK (1) | DK0923635T3 (ja) |
| ES (2) | ES2186906T3 (ja) |
| IL (1) | IL128147A0 (ja) |
| NO (2) | NO326521B1 (ja) |
| NZ (2) | NZ333796A (ja) |
| OA (2) | OA10961A (ja) |
| PL (2) | PL188333B1 (ja) |
| PT (1) | PT923635E (ja) |
| RU (2) | RU2193050C2 (ja) |
| WO (2) | WO1998003625A1 (ja) |
Families Citing this family (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6054580A (en) * | 1996-07-22 | 2000-04-25 | Carnegie Mellon University | Long-lived homogenous amide containing macrocyclic compounds |
| US5876625A (en) * | 1996-07-22 | 1999-03-02 | Carnegie Mellon University | Metal ligand containing bleaching compositions |
| US6136223A (en) * | 1996-07-22 | 2000-10-24 | Carnegie Mellon University | Metal ligand containing bleaching compositions |
| US6204234B1 (en) * | 1997-07-09 | 2001-03-20 | The Proctor & Gamble Company | Cleaning compositions comprising a specific oxygenase |
| US6569286B1 (en) * | 1998-09-30 | 2003-05-27 | Warwick International Group Limited | Method for the alkaline bleaching of pulp with a peroxyacid based oxygen bleaching species using an agglomerated bleach activator |
| US6127536A (en) * | 1999-05-25 | 2000-10-03 | The Clorox Company | Synthesis of a tetraamido macrocycle ligand |
| US6297400B1 (en) | 1999-07-02 | 2001-10-02 | The Clorox Company | Synthesis of a tetraamido macrocycle ligand from a novel diamidodiol |
| US6294047B1 (en) * | 1999-07-30 | 2001-09-25 | Institute Of Paper | Methods for reducing fluorescence in paper-containing samples |
| WO2001031110A1 (en) * | 1999-10-25 | 2001-05-03 | The Clorox Company | Low odor, hard surface abrasive cleaner with enhanced soil removal |
| KR20010081172A (ko) * | 2000-02-10 | 2001-08-29 | 성재갑 | 과산화물 표백제를 활성화시키는 전이 금속 착화합물을포함하는 표백 세제 조성물 |
| GB0005090D0 (en) | 2000-03-01 | 2000-04-26 | Unilever Plc | Bleaching and dye transfer inhibiting composition and method for laundry fabrics |
| US6969752B1 (en) | 2000-07-21 | 2005-11-29 | Georgia Tech Research Corporation | Water soluble/dispersible and easy removable cationic adhesives and coating for paper recycling |
| GB0020846D0 (en) * | 2000-08-23 | 2000-10-11 | Unilever Plc | Ligands for bleaching compositions and synthesis thereof |
| US20030050211A1 (en) * | 2000-12-14 | 2003-03-13 | Unilever Home & Personal Care Usa, Division Of Conopco, Inc. | Enzymatic detergent compositions |
| GB0030877D0 (en) | 2000-12-18 | 2001-01-31 | Unilever Plc | Enhancement of air bleaching catalysts |
| US6797196B2 (en) * | 2001-01-10 | 2004-09-28 | Kao Corporation | Bleaching formulation |
| DE10109502A1 (de) * | 2001-02-28 | 2002-09-12 | Rhodia Acetow Gmbh | Verfahren zum Abtrennen von Hemicellulosen aus hemicellulosehaltiger Biomasse sowie die mit dem Verfahren erhältliche Biomasse und Hemicellulose |
| JP4694020B2 (ja) * | 2001-03-30 | 2011-06-01 | 花王株式会社 | 漂白洗浄剤組成物 |
| JP4647126B2 (ja) * | 2001-03-30 | 2011-03-09 | 花王株式会社 | 漂白洗浄剤組成物 |
| JP2002294290A (ja) * | 2001-03-30 | 2002-10-09 | Kao Corp | 漂白洗浄剤組成物 |
| US6833343B2 (en) * | 2001-03-30 | 2004-12-21 | Kao Corporation | Bleaching detergent formulation |
| JP4694021B2 (ja) * | 2001-03-30 | 2011-06-01 | 花王株式会社 | 繊維製品の漂白方法 |
| GB0118934D0 (en) * | 2001-08-02 | 2001-09-26 | Unilever Plc | Improvements relating to bleaching compositions |
| GB0118936D0 (en) * | 2001-08-02 | 2001-09-26 | Unilever Plc | Improvements relating to colour-safe fabric treatment compositions |
| US7115549B2 (en) * | 2001-08-02 | 2006-10-03 | Carnegie Mellon University | Composition comprising macrocyclic tetra-amido metal complex as bleaching catalyst |
| TW591089B (en) * | 2001-08-09 | 2004-06-11 | Cheil Ind Inc | Slurry composition for use in chemical mechanical polishing of metal wiring |
| GB0202783D0 (en) * | 2002-02-06 | 2002-03-27 | Unilever Plc | Granule and composition containing same |
| GB0204505D0 (en) * | 2002-02-26 | 2002-04-10 | Unilever Plc | Detergent composition |
| JP4104966B2 (ja) * | 2002-03-06 | 2008-06-18 | 花王株式会社 | 漂白触媒 |
| GB0205278D0 (en) * | 2002-03-06 | 2002-04-17 | Unilever Plc | Composition comprising macrocyclic tetra-amido N-donor metal-ligand complex as bleaching catalyst |
| GB0205276D0 (en) * | 2002-03-06 | 2002-04-17 | Unilever Plc | Bleaching composition |
| JP2006504809A (ja) | 2002-05-02 | 2006-02-09 | ザ プロクター アンド ギャンブル カンパニー | 洗剤組成物及びその成分 |
| US7060818B2 (en) * | 2003-02-21 | 2006-06-13 | Carnegie Mellon University | Synthesis of macrocyclic tetraamido compounds and new metal insertion process |
| DE10311765A1 (de) * | 2003-03-18 | 2004-09-30 | Bayer Chemicals Ag | Verwendung eines Oxidationssystems enthaltend einen makrocyclischen Metallkomplex und ein Oxidationsmittel zur Entfernung überschüssigen, nichtgebundenen Farbstoffs von gefärbten, textilen Materialien |
| US20040183220A1 (en) * | 2003-03-18 | 2004-09-23 | Avinash Dalmia | Ultra thin layer coating using self-assembled molecules as a separating layer for diffraction grating application |
| DE10311766A1 (de) * | 2003-03-18 | 2004-09-30 | Bayer Chemicals Ag | Oxidationssystem enthaltend einen makrocyclischen Metallkomplex, dessen Herstellung und Verwendung |
| DE10345273A1 (de) * | 2003-09-30 | 2005-04-21 | Clariant Gmbh | Verwendung von Übergangsmetallkomplexen mit Lactamliganden als Bleichkatalysatoren |
| GB0323275D0 (en) * | 2003-10-04 | 2003-11-05 | Unilever Plc | Bleaching composition |
| WO2005035708A1 (en) * | 2003-10-08 | 2005-04-21 | Johnsondiversey, Inc. | Method of use of chlorine dioxide as an effective bleaching agent |
| CN1929921A (zh) * | 2004-01-12 | 2007-03-14 | 西巴特殊化学制品控股公司 | 包含金属络合物和聚磷酸酯的组合物作为过氧化合物用催化剂的用途 |
| US20080000032A1 (en) * | 2004-04-29 | 2008-01-03 | Torsten Wieprecht | Use of Metal Complexes Having Bispyridylpyrimidine or Bispyridyltriazine Ligands as Catalysts for Reactions With Peroxy Compounds for Bleaching Coloured Stains on Hard Surfaces |
| RU2280028C1 (ru) * | 2004-12-20 | 2006-07-20 | Государственное образовательное учреждение высшего профессионального образования "Ивановский государственный химико-технологический университет" | 4, 4'-N, N'-БИС[5-АМИНО-6-ЦИКЛОГЕКСЕНИЛИДЕН-7-(3', 4', 5'-f, g)АЦЕНАФТЕН-2, 2'-ДИСУЛЬФОБИФЕНИЛЕН] КАК ПРЯМОЙ И КИСЛОТНЫЙ КРАСИТЕЛЬ И КАК ИСХОДНОЕ ВЕЩЕСТВО ДЛЯ СИНТЕЗА МАКРОГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ |
| RU2280029C1 (ru) * | 2005-03-29 | 2006-07-20 | Государственное образовательное учреждение высшего профессионального образования "Ивановский государственный химико-технологический университет" | 4,4'-бис(1-инденон-3-ил)-2,2'-дисульфостильбен, проявляющий свойства кислотного красителя |
| WO2007025244A2 (en) | 2005-08-25 | 2007-03-01 | Houle Philip R | Treatment systems for delivery of sensitizer solutions |
| JP2007126776A (ja) * | 2005-11-02 | 2007-05-24 | Nisshin Kagaku Kenkyusho:Kk | 古紙パルプの処理方法および脱墨助剤 |
| RU2386670C2 (ru) * | 2006-01-23 | 2010-04-20 | Дзе Проктер Энд Гэмбл Компани | Композиции, содержащие фермент и оттеночный агент для ткани |
| ES2386650T3 (es) * | 2006-04-04 | 2012-08-24 | Basf Se | Sistemas de blanqueo revestidos en forma de capa con polímeros |
| CN100393734C (zh) * | 2006-05-10 | 2008-06-11 | 吾国强 | 一种四氨基大环金属配合物的合成方法 |
| US7967948B2 (en) * | 2006-06-02 | 2011-06-28 | International Paper Company | Process for non-chlorine oxidative bleaching of mechanical pulp in the presence of optical brightening agents |
| US20080025960A1 (en) * | 2006-07-06 | 2008-01-31 | Manoj Kumar | Detergents with stabilized enzyme systems |
| DE202008001532U1 (de) | 2008-02-01 | 2008-04-10 | Weber, Lothar Ernst Wilhelm | Vorrichtung zum Waschen oder Reinigen von Gegenständen |
| EP2103735A1 (en) | 2008-03-18 | 2009-09-23 | Unilever PLC | Catalytic bleaching of substrates |
| JP5795254B2 (ja) | 2008-04-09 | 2015-10-14 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 金属ヒドラジド錯体化合物の酸化触媒としての使用 |
| US20100010285A1 (en) * | 2008-06-26 | 2010-01-14 | Lumimove, Inc., D/B/A Crosslink | Decontamination system |
| WO2010138941A2 (en) | 2009-05-28 | 2010-12-02 | Gp Cellulose Gmbh | Modified cellulose from chemical kraft fiber and methods of making and using the same |
| US9512237B2 (en) | 2009-05-28 | 2016-12-06 | Gp Cellulose Gmbh | Method for inhibiting the growth of microbes with a modified cellulose fiber |
| US9512563B2 (en) | 2009-05-28 | 2016-12-06 | Gp Cellulose Gmbh | Surface treated modified cellulose from chemical kraft fiber and methods of making and using same |
| US9511167B2 (en) | 2009-05-28 | 2016-12-06 | Gp Cellulose Gmbh | Modified cellulose from chemical kraft fiber and methods of making and using the same |
| US8722881B2 (en) * | 2009-10-13 | 2014-05-13 | Board Of Trustees Of The University Of Arkansas | Method of synthesis of tetradentate amide macrocycle ligand and its metal-complex |
| US20110091283A1 (en) * | 2009-10-14 | 2011-04-21 | University Of Connecticut | Oxidation of environmental contaminants with mixed valent manganese oxides |
| US8584757B2 (en) | 2010-04-21 | 2013-11-19 | Halliburton Energy Services, Inc. | Method and composition for treating fluids before injection into subterranean zones |
| JP2013542171A (ja) | 2010-04-21 | 2013-11-21 | ディスカス デンタル,エルエルシー | 同時に歯をクリーニング及びホワイトニングする方法 |
| US20150342839A1 (en) | 2010-04-21 | 2015-12-03 | Oraceutica LLC | Compositions and methods for whitening teeth |
| EP2395147A1 (en) * | 2010-05-10 | 2011-12-14 | Unilever Plc, A Company Registered In England And Wales under company no. 41424 of Unilever House | Freeness of paper products |
| KR101831472B1 (ko) | 2010-06-28 | 2018-02-22 | 바스프 에스이 | 금속 비함유 표백 조성물 |
| JP6254078B2 (ja) | 2011-05-23 | 2017-12-27 | ゲーペー ツェルローゼ ゲーエムベーハー | 白色度および明度が改善された針葉樹クラフト繊維、ならびにそれを作製する方法および使用する方法 |
| CN104145012A (zh) | 2011-10-25 | 2014-11-12 | 巴斯夫欧洲公司 | 丙烯酸酯共聚物作为抗污垢再沉积剂和去污剂在洗衣过程中的用途 |
| CA2853318A1 (en) | 2011-10-25 | 2013-05-02 | Basf Se | Use of comb or block copolymers as soil antiredeposition agents and soil release agents in laundry processes |
| CN103174007B (zh) * | 2011-11-24 | 2015-05-13 | 东华大学 | 三吡啶基四氮金属配合物在纺织品低温练漂助剂中的应用 |
| CN102493186B (zh) * | 2011-11-24 | 2013-06-26 | 东华大学 | 一种纺织品低温练漂助剂及其制备方法和应用 |
| CN103174010A (zh) * | 2011-11-24 | 2013-06-26 | 东华大学 | 大环多胺桥连金属配合物在纺织品低温练漂助剂中的应用 |
| BR112014017164A8 (pt) | 2012-01-12 | 2017-07-04 | Gp Cellulose Gmbh | método para produzir polpa kraft oxidada e fibras kraft de madeira macia |
| EP3495550A1 (en) | 2012-04-18 | 2019-06-12 | GP Cellulose GmbH | The use of surfactant to treat pulp and improve the incorporation of kraft pulp into fiber for the production of viscose and other secondary fiber products |
| US8888489B2 (en) | 2012-10-23 | 2014-11-18 | Oraceutical Llc | Method of simultaneously cleaning and whitening teeth |
| PL2954115T3 (pl) | 2013-02-08 | 2022-05-02 | Gp Cellulose Gmbh | Włókno siarczanowe z drewna iglastego o polepszonej zawartości a-celulozy i jego zastosowanie w wytwarzaniu chemicznych produktów celulozowych |
| US10138598B2 (en) | 2013-03-14 | 2018-11-27 | Gp Cellulose Gmbh | Method of making a highly functional, low viscosity kraft fiber using an acidic bleaching sequence and a fiber made by the process |
| BR112015020000A2 (pt) | 2013-03-15 | 2017-07-18 | Gp Cellulose Gmbh | fibra kraft quimicamente modificada e métodos de fabricação da mesma |
| US9790452B2 (en) | 2013-03-27 | 2017-10-17 | Basf Se | Block copolymers as soil release agents in laundry processes |
| US9320580B2 (en) | 2013-04-21 | 2016-04-26 | Oraceutical Llc | Hand-held tooth whitening instrument with applicator reservoir for whitening composition and methods of using same |
| ES2650924T3 (es) | 2013-11-27 | 2018-01-23 | Basf Se | Copolímeros aleatorios como agentes de liberación de la suciedad en procedimientos de lavado de ropa |
| AR104940A1 (es) | 2015-06-10 | 2017-08-23 | Chemsenti Ltd | Método para generar dióxido de cloro |
| AR104939A1 (es) | 2015-06-10 | 2017-08-23 | Chemsenti Ltd | Método oxidativo para generar dióxido de cloro |
| ES2907129T3 (es) | 2015-09-25 | 2022-04-22 | Univ Carnegie Mellon | Catalizadores de oxidación basados en compuestos macrocíclicos |
| WO2017186480A1 (en) | 2016-04-26 | 2017-11-02 | Basf Se | Metal free bleaching composition |
| KR102466079B1 (ko) * | 2016-09-09 | 2022-11-10 | 리서치 파운데이션 오브 더 시티 유니버시티 오브 뉴욕 | 자기-조립 펩타이드 중합체 |
| CA3040734A1 (en) | 2016-11-16 | 2018-05-24 | Gp Cellulose Gmbh | Modified cellulose from chemical fiber and methods of making and using the same |
| CN108751717B (zh) * | 2018-06-29 | 2021-05-11 | 河源市东源鹰牌陶瓷有限公司 | 一种防滑釉、防滑砖及其制备方法 |
| RU2713356C1 (ru) * | 2019-05-15 | 2020-02-04 | Сергей Борисович Врублевский | Шихта для композиционного отбеливателя и способ его получения (варианты) |
| CN110359296A (zh) * | 2019-07-02 | 2019-10-22 | 福建凤竹纺织科技股份有限公司 | 一种中小学生校服面料的绿色染色工艺 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1075504A (en) * | 1913-08-14 | 1913-10-14 | Western Electric Co | Telegraph-transmitter. |
| US3736224A (en) * | 1971-06-16 | 1973-05-29 | American Cyanamid Co | Catalyzed oxygen bleaching |
| US3950277A (en) * | 1973-07-25 | 1976-04-13 | The Procter & Gamble Company | Laundry pre-soak compositions |
| US4005029A (en) * | 1973-08-01 | 1977-01-25 | The Procter & Gamble Company | Laundering adjunct |
| US3983082A (en) * | 1975-02-19 | 1976-09-28 | General Electric Company | Intumescent fire retardant material and article |
| GB1565807A (en) * | 1975-12-18 | 1980-04-23 | Uilever Ltd | Process and compositions for cleaning fabrics |
| IL58084A (en) * | 1979-08-22 | 1985-04-30 | Yeda Res & Dev | Process for the preparation of heterocyclic macrocyclic compounds |
| DK187280A (da) * | 1980-04-30 | 1981-10-31 | Novo Industri As | Ruhedsreducerende middel til et fuldvaskemiddel fuldvaskemiddel og fuldvaskemetode |
| US4473507A (en) * | 1981-10-21 | 1984-09-25 | The Procter & Gamble Company | Controlled release laundry bleach product |
| JPS591598A (ja) * | 1982-06-25 | 1984-01-06 | 花王株式会社 | 洗浄剤組成物 |
| GB8306645D0 (en) * | 1983-03-10 | 1983-04-13 | Unilever Plc | Detergent compositions |
| US4758682A (en) * | 1983-03-17 | 1988-07-19 | California Institute Of Technology | Homogeneous coordination compounds as oxidation catalysts |
| US4577042A (en) * | 1983-03-17 | 1986-03-18 | California Institute Of Technology | Homogeneous coordination compounds as oxidation catalysts |
| GB8311865D0 (en) * | 1983-04-29 | 1983-06-02 | Procter & Gamble Ltd | Bleach compositions |
| US4511490A (en) * | 1983-06-27 | 1985-04-16 | The Clorox Company | Cooperative enzymes comprising alkaline or mixtures of alkaline and neutral proteases without stabilizers |
| US4661293A (en) * | 1983-12-01 | 1987-04-28 | The Clorox Company | Method for preparing 1,4-diaminoanthraquinones and intermediates thereof |
| US4708816A (en) * | 1984-01-27 | 1987-11-24 | The Clorox Company | Bleach composition containing controlled density capsules |
| GB8514707D0 (en) * | 1985-06-11 | 1985-07-10 | Unilever Plc | Enzymatic detergent composition |
| US4746461A (en) * | 1986-05-23 | 1988-05-24 | The Clorox Company | Method for preparing 1,4-diaminoanthraquinones and intermediates thereof |
| US4773966A (en) * | 1986-09-29 | 1988-09-27 | Regents Of The University Of Minnesota | Oxidative degradation of lignin with inorganic metal complexes |
| US4778618A (en) * | 1986-11-06 | 1988-10-18 | The Clorox Company | Glycolate ester peracid precursors |
| US4900871A (en) * | 1987-01-02 | 1990-02-13 | Sun Refining And Marketing Company | Hydrocarbon oxidations catalyzed by iron coordination complexes containing a halogenated ligand |
| SU1689477A1 (ru) * | 1988-06-20 | 1991-11-07 | Le I Textilnoi | Komпoзиция для hизkotemпepatуphoгo otбeлиbahия xлoпчatoбуmaжhыx tkaheй |
| MC2034A1 (fr) * | 1988-06-23 | 1990-05-30 | Hoffmann La Roche | Preparations |
| US5032286A (en) * | 1988-10-18 | 1991-07-16 | Boise Cascade Corporation | Pulp mill effluent color removal process |
| US5182045A (en) * | 1989-03-29 | 1993-01-26 | The Clorox Company | Late peracid precursors |
| ES2100925T3 (es) * | 1990-05-21 | 1997-07-01 | Unilever Nv | Activacion de blanqueador. |
| CA2085642A1 (en) * | 1991-12-20 | 1993-06-21 | Ronald Hage | Bleach activation |
| GB9127060D0 (en) * | 1991-12-20 | 1992-02-19 | Unilever Plc | Bleach activation |
| EP0594893B1 (en) * | 1992-10-27 | 1998-02-04 | The Procter & Gamble Company | Detergent compositions inhibiting dye transfer |
| US5288746A (en) * | 1992-12-21 | 1994-02-22 | The Procter & Gamble Company | Liquid laundry detergents containing stabilized glucose/glucose oxidase as H2 O2 generation system |
| CN1146165A (zh) * | 1994-03-08 | 1997-03-26 | 普罗格特-甘布尔公司 | 金属大环催化剂组合物 |
| WO1995031526A1 (en) * | 1994-05-11 | 1995-11-23 | The Procter & Gamble Company | Dye transfer inhibiting compositions with specifically selected metallo catalysts |
| US5451337A (en) * | 1994-05-31 | 1995-09-19 | The Procter & Gamble Co. | Dye transfer inhibition system containing a peroxidase/accelerator system |
| US5560748A (en) * | 1994-06-10 | 1996-10-01 | The Procter & Gamble Company | Detergent compositions comprising large pore size redox catalysts |
| ES2135068T3 (es) * | 1994-06-13 | 1999-10-16 | Unilever Nv | Activacion de blanqueantes. |
| GB9425296D0 (en) * | 1994-12-15 | 1995-02-15 | Ciba Geigy Ag | Inhibition of dye migration |
| US5698476A (en) * | 1995-03-01 | 1997-12-16 | The Clorox Company | Laundry article for preventing dye carry-over and indicator therefor |
| US5876625A (en) * | 1996-07-22 | 1999-03-02 | Carnegie Mellon University | Metal ligand containing bleaching compositions |
| US5847120A (en) * | 1996-07-22 | 1998-12-08 | Carnegie Mellon University | Long-lived homogenous oxidation catalysts |
-
1996
- 1996-07-22 US US08/684,670 patent/US5876625A/en not_active Expired - Lifetime
-
1997
- 1997-02-24 US US08/804,776 patent/US5853428A/en not_active Expired - Fee Related
- 1997-07-21 KR KR1019997000526A patent/KR20000067992A/ko not_active Withdrawn
- 1997-07-21 AU AU36655/97A patent/AU730906B2/en not_active Ceased
- 1997-07-21 ES ES97933485T patent/ES2186906T3/es not_active Expired - Lifetime
- 1997-07-21 CA CA002261228A patent/CA2261228C/en not_active Expired - Fee Related
- 1997-07-21 AT AT97933485T patent/ATE225844T1/de not_active IP Right Cessation
- 1997-07-21 CN CNB971979782A patent/CN100352902C/zh not_active Expired - Fee Related
- 1997-07-21 DE DE69716275T patent/DE69716275T2/de not_active Expired - Fee Related
- 1997-07-21 AP APAP/P/1999/001456A patent/AP1013A/en active
- 1997-07-21 EP EP97933485A patent/EP0918840B1/en not_active Expired - Lifetime
- 1997-07-21 IL IL12814797A patent/IL128147A0/xx unknown
- 1997-07-21 WO PCT/US1997/012358 patent/WO1998003625A1/en not_active Ceased
- 1997-07-21 PL PL97331316A patent/PL188333B1/pl not_active IP Right Cessation
- 1997-07-21 JP JP50704898A patent/JP3623802B2/ja not_active Expired - Fee Related
- 1997-07-21 NZ NZ333796A patent/NZ333796A/xx unknown
- 1997-07-21 RU RU99103697/04A patent/RU2193050C2/ru not_active IP Right Cessation
- 1997-07-21 BR BR9710538A patent/BR9710538A/pt not_active IP Right Cessation
- 1997-07-22 AT AT97934248T patent/ATE233312T1/de not_active IP Right Cessation
- 1997-07-22 BR BR9710514-7A patent/BR9710514A/pt not_active IP Right Cessation
- 1997-07-22 EP EP97934248A patent/EP0923635B1/en not_active Expired - Lifetime
- 1997-07-22 CN CNB971980942A patent/CN100390259C/zh not_active Expired - Fee Related
- 1997-07-22 AP APAP/P/1999/001449A patent/AP905A/en active
- 1997-07-22 AU AU37352/97A patent/AU720042B2/en not_active Ceased
- 1997-07-22 PT PT97934248T patent/PT923635E/pt unknown
- 1997-07-22 DK DK97934248T patent/DK0923635T3/da active
- 1997-07-22 ES ES97934248T patent/ES2197356T3/es not_active Expired - Lifetime
- 1997-07-22 WO PCT/US1997/012744 patent/WO1998003626A2/en not_active Ceased
- 1997-07-22 JP JP50716598A patent/JP3623803B2/ja not_active Expired - Fee Related
- 1997-07-22 NZ NZ333795A patent/NZ333795A/en unknown
- 1997-07-22 CA CA002261229A patent/CA2261229C/en not_active Expired - Fee Related
- 1997-07-22 KR KR1019997000531A patent/KR20000067996A/ko not_active Withdrawn
- 1997-07-22 DE DE69719337T patent/DE69719337T2/de not_active Expired - Fee Related
- 1997-07-22 PL PL97331352A patent/PL331352A1/xx unknown
- 1997-07-22 RU RU99103691/04A patent/RU2193049C2/ru not_active IP Right Cessation
-
1998
- 1998-07-20 US US09/119,459 patent/US6099586A/en not_active Expired - Lifetime
-
1999
- 1999-01-20 OA OA9900012A patent/OA10961A/en unknown
- 1999-01-20 OA OA9900013A patent/OA10962A/en unknown
- 1999-01-21 NO NO19990267A patent/NO326521B1/no not_active IP Right Cessation
- 1999-01-21 NO NO990266A patent/NO990266L/no not_active Application Discontinuation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3623802B2 (ja) | 金属配位子含有漂白組成物 | |
| US6241779B1 (en) | Metal ligand containing bleaching compositions | |
| AU2614895A (en) | Bleach activation | |
| HK1020350B (en) | Metal ligand containing bleaching compositions | |
| MXPA99000790A (es) | Composiciones de blanqueo que contienen ligando metalico | |
| HK1020750B (en) | Metal ligand containing bleaching compositions | |
| MXPA00011039A (en) | Metal ligand containing bleaching compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20031201 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20040119 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040227 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20041102 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20041126 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20081203 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20091203 Year of fee payment: 5 |
|
| LAPS | Cancellation because of no payment of annual fees |