JP3816593B2 - キラータンパク質 - Google Patents
キラータンパク質 Download PDFInfo
- Publication number
- JP3816593B2 JP3816593B2 JP23439396A JP23439396A JP3816593B2 JP 3816593 B2 JP3816593 B2 JP 3816593B2 JP 23439396 A JP23439396 A JP 23439396A JP 23439396 A JP23439396 A JP 23439396A JP 3816593 B2 JP3816593 B2 JP 3816593B2
- Authority
- JP
- Japan
- Prior art keywords
- gene
- dna
- protein
- killer
- mutant
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images



Landscapes
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Detergent Compositions (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Saccharide Compounds (AREA)
Description
【発明の属する技術分野】
本発明は、キラータンパク質、該タンパク質をコードする遺伝子、該遺伝子によって形質転換された形質転換体及び該キラータンパク質の用途に関する。
【0002】
【従来の技術】
酵母又はその近縁菌類がキラートキシンと呼ばれる抗菌活性を示すキラータンパク質を産生することが知られて以来、いくつかの分泌タンパク質性のキラートキシンが発見されており、これらの酵母等によって生産されるキラータンパク質の種々の性質又は作用機序が知られている。
【0003】
例えば、ハンゼヌラ・アノマラ(Hansenula anomala)が産生するKh-IIキラータンパク質は、作用pHを3〜6にもち、パパインによって分解されず、麹菌プロテアーゼによって徐々に分解されることが報告されている(特公平5-70639 号公報、特公平5-70640 号公報)。
【0004】
また、サッカロミセス・セレビシエ(Saccharomyces cerevisiae) が産生するK1キラータンパク質(二本鎖RNAプラスミド上にコードされている。)は、標的となる酵母のプロトンポンプを阻害することにより感受性酵母を殺すことが知られており、クルイベロミセス・ラクティス(Kluyveromyces lacits)菌が産生するキラータンパク質(直鎖状の二本鎖DNAプラスミド上にコードされている。)は、本来キチナーゼ活性を有するが、感受性酵母のアデニレートサイクラーゼを阻害することが知られており、ピチア・クルイベリ(Pchia kluyveri)菌が産生するキラータンパク質は、標的酵母の脂質二重層膜に穴をあけることが知られている。
【0005】
自然界には、酵母(広くは有核系微生物)が抗生物質的作用を有するタンパク質性毒素を生産・分泌し、互いに相克するという現象がある。この現象は、かかるタンパク質性毒素は標的菌に対し特異的で毒性を示すが自己に対しては無害であるというタンパク質工学上興味ある事象である。従って、昨今のいわゆる「State of Arts」を考慮すると、遺伝子工学的手法を用いてタンパク質性毒素を改変することにより、カビ等の病原性真菌に対するタンパク質性抗生物質を創製し、真菌症の治療、洗剤等に利用できるタンパク質を開発する必要があることは明らかである。また、前記キラータンパク質のように小分子量のタンパク質の立体構造を決定し、その活性部位領域の立体構造から非タンパク質性抗菌化合物をドラッグデザインすることも可能である。
【0006】
一方、エイズに感染したり、免疫力が低下したり、老化が進行すると、真菌に感染しやすくなるため、全身に感染症状を呈する感染症が発症するようになる。このため、感染症を治療目的とする抗生剤(抗生物質)の開発が試みられ、すでに、有機化合物であるアクレアシンAやパプラカンデンBといった細胞壁成分のグルカンを合成阻害する抗カビ性の抗生物質が知られている。
【0007】
しかし、これら抗生物質は肝毒性が高く、薬剤耐性を誘発するなどの問題点がある。従って、薬剤耐性を誘発せず、しかも安全性に優れた抗生物質が要求されている。
そこで、キラータンパク質を抗生物質や洗剤等に利用できるようにするため、病原性真菌類に有効であり、アルカリ耐性かつプロテアーゼ耐性を示すキラータンパク質の作製が必要となってきた。
【0008】
従来より、ハンゼヌラ・ムラキ(Hansenula mrakii) が産生するキラータンパク質が、抗カビ剤や洗剤等に有用であることが知られている。
しかし、ハンゼヌラ・ムラキが産生するキラータンパク質は微量であるため大量生産が困難であり、抗菌剤等として実際に適用することは困難である。また、該キラータンパク質はプロテアーゼに感受性であるため失活しやすい。さらに、動物細胞などを培養する際にキラータンパク質の代わりに種々の抗菌剤(例えばファンギゾン等)を用いることもできるが、これらの抗菌剤は熱に不安定であるため、培養液の滅菌処理にしばしば利用される高温滅菌(例えばオートクレーブ)をすることができない。従って、培養液の滅菌とは別途に滅菌しなければならず、操作が煩雑である。
【0009】
一方、遺伝子工学的手法によりタンパク質を大量生産しようとする場合、その有用形質転換宿主として酵母サッカロミセス・セレビシエを用いることが適当であるが、サッカロミセス・セレビシエは、ハンゼヌラ・ムラキが産生するキラータンパク質に感受性でその標的となるため、ハンゼヌラ・ムラキ由来のキラータンパク質の遺伝子を導入することができない。
【0010】
【発明が解決しようとする課題】
本発明は、キラータンパク質、該タンパク質をコードする遺伝子、該遺伝子によって形質転換された形質転換体、並びに抗菌石鹸及び抗カビ剤を提供することを目的とする。
【0011】
【課題を解決するための手段】
本発明者は、上記課題に基づいて鋭意研究を行った結果、ハンゼヌラ・ムラキ(Hansenula mrakii)が産生する微量なHM-1キラータンパク質をサッカロミセス・セレビシエを用いた遺伝子工学的手法により大量生産し、しかも、プロテアーゼ耐性を有し、かつ病原性真菌に一層有効であるキラータンパク質を創製することに成功し、本発明を完成するに至った。
【0012】
すなわち、本発明は、配列番号1で表されるアミノ酸配列のうち第98番目及び/又は第122番目のアルギニンがアラニンに変異したアミノ酸配列を実質的に含み、抗菌活性を有するキラータンパク質である。
さらに、本発明は、配列番号1で表されるアミノ酸配列のうち第98番目及び/又は第122番目のアルギニンがアラニンに変異したアミノ酸配列を実質的にコードするDNAを含むキラータンパク質遺伝子である。該遺伝子としては、配列番号2又は3で表される塩基配列を実質的に含むものが挙げられる。
【0013】
ここで、「実質的に」とは、本発明のキラータンパク質が抗菌活性を有する限り、また、本発明の遺伝子がキラータンパク質の活性を発現させる機能を有する限り、当該タンパク質(ポリペプチド)に含まれるアミノ酸配列、又は当該遺伝子の塩基配列に欠失、置換、挿入等の変異が生じてもよいことを意味する。
【0014】
従って、例えば本発明のキラータンパク質に含まれる第1番目のメチオニン(Met)が欠失しているものなども、このアミノ酸配列の変化によるタンパク質に含まれる。また、本発明のキラータンパク質に含まれるアミノ酸をコードする塩基配列のほか、縮重コドンにおいてのみ異なる同一のポリペプチドをコードする縮重異性体も本発明の遺伝子に含まれる。
【0015】
但し、配列番号1で表されるアミノ酸配列のうち、変異箇所(第98番目のアルギニンに代わるアラニン及び/又は122番目のアルギニンに代わるアラニン)には、上記欠失、置換、挿入等の変異は生じないものとし、また、アラニンをコードする塩基配列には、アラニンの縮重異性体による変異以外は上記欠失、置換、挿入等の変異は生じないものとする。
【0016】
さらに、本発明は、前記キラータンパク質遺伝子を含む組換え体DNAである。
さらに、本発明は、前記組換え体DNA、及び前記キラータンパク質耐性遺伝子を含む組換え体DNAによって形質転換された形質転換体である。
さらに、本発明は、前記形質転換体を培地に培養し、得られる培養物からキラータンパク質を採取することを特徴とするキラータンパク質の製造方法である。
【0017】
さらに、本発明は、前記キラータンパク質を有効成分として含む抗菌剤である。
さらに、本発明は、前記キラータンパク質を抗菌成分として含む抗菌石鹸である。
以下、本発明を詳細に説明する。
【0018】
【発明の実施の形態】
本発明のキラータンパク質(以下、キラータンパク質を「HM-1」という) は、遺伝子工学的手法により変異を導入してプロテアーゼ耐性を付与したHM-1遺伝子を、あらかじめHM-1耐性遺伝子(HM-1感受性遺伝子を破壊して得られる遺伝子)を導入してHM-1耐性を付与した酵母に導入し、発現させることにより得られるものである。本発明のHM-1は抗菌活性が上昇しており、かつプロテアーゼ耐性で作用pHをアルカリ側にもつものである。また、本発明のHM-1は、酵母サッカロミセス・セレビシエを宿主として産生させることが可能となったため、効率よく得ることができる。
【0019】
本発明のHM-1を得るための手法を説明する。
まず、ハンゼヌラ・ムラキが産生するHM-1遺伝子をクローニングし、得られる遺伝子の一部を変異することによりHM-1変異遺伝子を作製する。一方、HM-1耐性遺伝子を酵母サッカロミセス・セレビシエに導入し、HM-1耐性の形質転換体(形質転換酵母)を作製する。得られる形質転換酵母に前記変異遺伝子を導入し、変異遺伝子及びHM-1耐性遺伝子両者を含む形質転換体を作製する。そして、得られる形質転換体を培養することにより、本発明のHM-1を得ることができる。
【0020】
(1) HM-1遺伝子のクローニング
▲1▼ ハンゼヌラ・ムラキからのゲノムDNAの調製
まず、ハンゼヌラ・ムラキを培養し、得られる培養物からゲノムDNAを調製する。ハンゼヌラ・ムラキとしては、IFO 0895又はIFO 8075などが挙げられる。
【0021】
ハンゼヌラ・ムラキの培養は、通常の方法により行うことができる(実験医学別冊:酵母による遺伝子実験法、14〜15頁、羊土社、1994年)。例えば、YPD培地などの栄養培地又は最小培地に菌株を接種し、30℃で対数増殖期まで培養する。次に、培養菌株からゲノムDNAを調製する。ゲノムDNAの調製についても、公知手法を用いることができる(Methods in Enzymology,194,218-219 )。
【0022】
▲2▼ 公知アミノ酸配列からのオリゴヌクレオチドの設計
既に公知であるHM-1のアミノ酸配列(FEBS LETT.,195,253-257,1986) に基づいて、ゲノムDNAとのハイブリダイゼーションを行うためのヌクレオチドを設計する。例えば、HM-1の公知アミノ酸配列のうち、第20番目のトリプトファン(Trp)から第26番目のスレオニン(Thr) までのアミノ酸配列をコードするヌクレオチド20マー(配列番号4)、及び第33番目のアスパラギン(Asn) から第38番目のバリン(Val) までのアミノ酸配列をコードするヌクレオチドのアンチコドンの17マー(配列番号5)の2種のオリゴヌクレオチドプローブを合成する。ヌクレオチドの合成は、例えば、ABI 社(Applied Biosystems Inc.) 製の自動DNA合成装置等を用いて行うことができる。得られるヌクレオチドは、ゲノムDNAとのハイブリダイゼーションに使用する。
【0023】
▲3▼ ハイブリダイゼーション
前記▲1▼で調製されたゲノムDNAを適当な制限酵素(例えばHindIII、EcoRI又はBamHI等)で切断し、得られるDNA断片と前記▲2▼で合成された2種のオリゴヌクレオチドプローブとを用いたサザンブロットハイブリダイゼーション(サザンブロッティング)を行う。サザンブロッティングについては、公知の方法により行うことができる(FEBS LETT.,195,253-257,1986)。
【0024】
サザンブロッティングの結果目的とされるDNA断片を得た場合は、該DNA断片の物理地図を作製し、適当な制限酵素、例えばEcoRI 、Hind III等で処理することにより適当な長さの断片にする。なお、得られる断片をもとに、目的とされるDNA断片に不要のDNA領域が含まれていることがわかった場合は、さらに制限酵素EcoRI 、Hind III等で消化して不要のDNA領域を除き、前記と同様に、2種の合成オリゴヌクレオチドプローブ(配列番号4及び5)を用いてゲノムDNAライブラリーから目的とするHM-1遺伝子をスクリーニングする。
【0025】
▲4▼ 塩基配列の決定
前記▲3▼で得られたDNA断片の塩基配列を決定する。塩基配列の決定は、公知の手法、例えばジデオキシ法、マキサム−ギルバート法等により行うことができ、ABI 社製のオートシークエンサー等を用いてもよい。
【0026】
(2) HM-1耐性形質転換体の作製
サッカロミセス・セレビシエがHM-1に対し感受性であるのは、HM-1と相互作用し、HM-1感受性を表現する遺伝子がサッカロミセス属菌に存在しているためであると予想することができる。従って、本発明においては、まずHM-1感受性遺伝子をクローニングした後、HM-1感受性遺伝子を破壊することによりHM-1耐性遺伝子を作製する。そして、得られるHM-1耐性遺伝子を酵母に形質転換することによりHM-1耐性形質転換体を作製する。
【0027】
▲1▼ サッカロミセス・セレビシエのゲノムDNAライブラリーの調製
サッカロミセス・セレビシエのHM-1感受性遺伝子をクローニングするため、まず、サッカロミセス酵母ベクターYCp50に連結されたサッカロミセス・セレビシエのゲノムDNAライブラリーを作製する。ライブラリーは、サッカロミセス・セレビシエのゲノムDNAを制限酵素BamHIで消化し、得られる断片をYCp50と連結することにより作製することができる。
【0028】
▲2▼ HM-1耐性変異株の調製
次に、サッカロミセス・セレビシエのゲノムDNAライブラリーを導入するためのHM-1耐性変異株(ウラシル要求性)を分離・調製する。ウラシル要求性の変異株を宿主として用いるのは、ウラシル非要求性遺伝子をもつライブラリーのみをスクリーニングするためである。
【0029】
HM-1耐性変異株の分離法としては、EMS(エチルメタンスルホン酸)若しくはニトロソグアニジンで誘発する方法、紫外線照射による方法又はトランスポゾンTyを利用する方法が挙げられる。
【0030】
例えば、EMSを用いたHM-1耐性変異株の調製の場合は、以下のように行う。すなわち、サッカロミセス・セレビシエBJ1824(a, ura3,leu2,trp1,pep4)をYPD培地に接種し、増殖させた後集菌し、リン酸カリウムバッファーで洗浄後、同じバッファーに懸濁する。これにEMSを加え、激しくボルテックスし、30℃で30分振盪後、等量のチオ硫酸ナトリウムを加え、よく混合する。集菌し滅菌水で2回洗い、滅菌水に懸濁、希釈する。
【0031】
このようにして調製された前記細胞懸濁液を、HM-1を含むYPD寒天培地にプレーテイングし、30℃で36時間のインキュベートにより、HM-1耐性変異株を取得する。
【0032】
▲3▼ ゲノムDNAライブラリーのHM-1耐性変異株への導入
前記▲1▼で得られたDNAライブラリーを、前記▲2▼で作製されたHM-1耐性変異株(以下「rhk1-1」という) に導入することにより、rhk1-1の形質転換を行う。DNAライブラリーの導入は、通常の方法(例えば酢酸リチウム法等)を用いることができる。
【0033】
▲4▼ ゲノムDNAライブラリー導入株の選別
rhk1-1を形質転換した段階では、ゲノムDNAを含まない酵母も存在するため、ゲノムDNAが導入された酵母のみを選別する必要がある。
そこで、YCp50 がURA3遺伝子を含むことを利用して、▲3▼により得られた形質転換体を、CMドロップアウト培地からウラシルを除いた培地を用いて培養することにより、ゲノムDNAが導入された形質転換体のみを選別する。
【0034】
ここで、CMドロップアウト培地とは、アミノ酸フリーの酵母ナイトローゲンベース0.67%、グルコース2%、並びにアミノ酸及び核酸の混合物であるドロップアウトパウダー0.13%を含む培地である。
【0035】
宿主rhk1-1はウラシル要求性であるためウラシルを含まない培地では生育できないが、宿主中に導入されたプラスミドYCp50 がウラシル非要求性遺伝子であるURA3遺伝子を保有するため、rhk1-1はウラシル無添加培地でも生育することができる。この性質を利用して、ウラシル非要求性のコロニー(Ura+ 形質転換体)を得ることができる。
【0036】
▲5▼ HM-1感受性遺伝子を含むコロニーの選別
得られたウラシル非要求性の形質転換体の中には、ゲノムDNAのうちHM-1感受性遺伝子を含むYCp50プラスミドを保有するもののほか、HM-1感受性遺伝子とは異なる遺伝子を含むYCp50プラスミドを保有するもの、及びYCp50しか保有しないものが混在している。そこで、HM-1感受性遺伝子を含むYCp50プラスミドを保有する形質転換体のみを選択するため、レプリカ法(J.Bacteriol.,63,399,1952) によりUra+形質転換体をHM-1を含むCMドロップアウト寒天培地へレプリカする。これにより、HM-1感受性遺伝子を含むコロニーのみを取得することができる。
得られたコロニーからのプラスミドDNAの調製は、ゲノムDNAの調製法の記載(前記(1) の▲1▼)に準じて行う。
【0037】
▲6▼ インサートDNAの確認及びクローニング
インサートDNA(HM-1感受性遺伝子が含まれているDNA断片) を確認するために、前記▲5▼により得られたプラスミドを制限酵素BamHIで消化し、電気泳動する。得られるDNAをさらに切りとって縮めるために、該DNAの物理地図を作成し、種々の制限酵素で消化する。得られたDNA断片をベクターYCp50にサブクローニングし、rhk1-1に導入し、選択培地(HM-1を含む合成寒天培地)上でHM-1に対する感受性を試験する。そして、感受性を有することが確認されたDNAの塩基配列を決定し、HM-1感受性遺伝子をクローニングする。
【0038】
▲7▼ 遺伝子破壊によるHM-1耐性遺伝子の構築及びHM-1耐性株の作出
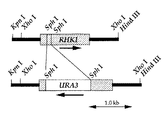
前記▲6▼のようにしてクローニングされたHM-1感受性遺伝子(以下「RHK1遺伝子」という) の一部を制限酵素(例えばSphI)で切断し、別に同じ制限酵素で消化したURA3 DNA断片(ニューイングランド・バイオラボ社)をRHK1遺伝子断片の間に挿入することにより、RHK1遺伝子はURA3遺伝子により破壊され、RHK1遺伝子の制限酵素切断部位をURA3遺伝子で置き換えたキメラ遺伝子(RHK-1-URA3キメラDNA)が構築される。なお、RHK1遺伝子のURA3遺伝子による破壊は、四分子分析により確認することができる。
このキメラDNAをrhk1-1に導入し、HM-1耐性の形質転換体であるサッカロミセス・セレビシエBJ1824rhk1△::URA3を作製する。
【0039】
(3) 変異型HM-1遺伝子の構築
公知のHM-1にプロテアーゼ耐性を付与するため、HM-1のアミノ酸配列の一部に変異(置換)を導入する。
本発明では、例えば配列番号1で表されるアミノ酸配列のうち、第1番目のメチオニン(Met) から数えて98番目及び/又は122番目のアルギニン(Arg) をアラニン(Ala) に置換することにより変異型HM-1を作製する。但し、変異は第98番目又は第122番目の一方に導入してもよく、両方に導入してもよい。
【0040】
なお、配列番号1で表されるアミノ酸配列には、第119番目及び第123番目にもアルギニンが存在するため、これらの部位についても変異を導入して得られる変異型HM-1遺伝子の構築も試みる。
【0041】
変異の導入は、公知の方法を用いることができる。例えば、点突然変異導入法、ポリメラーゼ連鎖反応(PCR)を利用した突然変異の導入法等が挙げられる。
変異型HM-1遺伝子は、例えばLA PCR in vitroミュタージェネシスキット(宝酒造社製)を用いて構築することができる。
【0042】
すなわち、前記(1) でクローニングされたHM-1遺伝子領域を含むDNA断片の5'側をキロシーケンス用デレーションキット(宝酒造)で短くした後、BamHIリンカーを連結する。この5'側にBamHI部位をもつBamHI-HindIII断片をpUC118へ挿入し、鋳型のDNAとする。
一方、HM-1の第98、119、122又は123番目のアルギニン(Arg) をアラニン(Ala)に変えるために、予想される相補鎖の配列に従ってオリゴヌクレオチドを化学合成し、プライマーを作製する。
【0043】
次に、前記鋳型及びプライマーを用いて、キットの説明書に従ってPCRを行い、変異型遺伝子を作製する。得られた変異型遺伝子をpUC18のEcoRI部位とHindIII部位との間に挿入し、大腸菌 DH5αへ導入する。得られた形質転換体をLB培地で培養することによりプラスミドを調製し、次いでHidIIIとBamHIでこのプラスミドを消化後、アガロース電気泳動で精製する。この精製された変異型遺伝子断片を、酵母ベクターYEp51のHindIIIとBamHI部位に挿入してYEp51のGal10プロモーターの下流につないだ後、大腸菌DH5αに形質転換し、プラスミドを抽出精製する。
【0044】
(4) 変異型HM-1の精製及び活性の測定
前記(2) で作成された形質転換酵母サッカロミセス・セレビシエBJ1824rhk1△::URA3に変異型HM-1遺伝子を導入した後、形質転換体を培地に培養する。得られる培養物を通常のタンパク質精製手法に従って精製する。
【0045】
精製は、塩析、セファデックスG-50などを用いたゲル濾過、CMセルロース、DEAE等を用いたイオン交換クロマトグラフィー、抗HM-1モノクローナル抗体を利用したアフィニティクロマトグラフィー、その他逆相クロマトグラフィー等を単独で、又は適宜組み合わせることにより行うことができる。
【0046】
精製されたタンパク質が目的のものであるか否かの確認は、SDSポリアクリルアミドゲル電気泳動、ウエスタンブロッティング等により行うことができる。ここで、HM-1を精製する際に、培養物中に変異型HM-1の生産がされているか否かの検定を行う。検定は、抗HM-1モノクローナル抗体又はポリクローナル抗体を用いたウエスタンブロッティング等により行うことができる。
【0047】
また、生産された変異型HM-1のキラー活性の検定は、ハンゼヌラ・アノマラ(Hansenula anomala) IFO 0569株を含む寒天培地上に、前記のようにして精製された変異型HM-1を浸したペーパーディスクを置き、生じる阻止円のサイズを測定することにより行うことができる。
【0048】
(5) 抗菌石鹸及び抗菌剤
本発明のHM-1を抗菌石鹸として使用する場合は、使用する対象を特に限定しない。例えば、手洗い用洗剤、衣類や寝具(シーツ、枕カバー等)の洗剤、又は衣類の洗濯後のコーティング等に使用することができる。
また、前記キラータンパク質のように小分子量のタンパク質の立体構造を決定し、その活性部位領域の立体構造から非タンパク質性抗菌化合物をドラッグデザインすることも可能である。
【0049】
【実施例】
以下、実施例により本発明をさらに具体的に説明する。但し、本発明は、これら実施例にその技術的範囲を限定するものではない。
〔実施例1〕HM-1遺伝子のクローニング
(1) ゲノムDNAライブラリーの調製
▲1▼ ゲノムDNAの調製
ハンゼヌラ・ムラキIFO 0895を酵母エキス1%、ペプトン2%、ソルビトール2%からなるYPD培地に接種し、30℃で対数増殖期まで培養した後、染色体DNAを調製した。
【0050】
すなわち、HM-1感受性コロニーをYPD 液体培地1.5 mlに植えて一晩培養し、得られた培養液をエッペンドルフチューブに移し、遠心後水で1回洗った。
これを0.5 mlのソルビトール溶液(1M ソルビトール, 100mM EDTA(pH7.5))に懸濁後、ザイモリエース酵素溶液(10mg/ml zymolyase-100T;生化学工業社製)0.02ml を加え、37℃で攪拌しながら60分間インキュベートした。3,000rpmで30秒遠心後、細胞をトリス溶液(50mM Tris-HCl(pH7.5), 20mM EDTA)0.5mlに懸濁し、さらに10%SDS 0.05mlを加え、よく混ぜた。これを65℃で30分間インキュベートし、4M酢酸カリウム溶液を0.25ml加え、よく混ぜた後0℃で60分間静置した。
【0051】
次に、12,000rpm、4℃で5分間遠心した。上清を別のエッペンドルフチューブに移し、等量のイソプロパノールを加えよく混ぜた後、室温で5分間静置した。12,000rpm、室温で10秒間遠心した後、上清を捨て、沈殿を70%エタノールで洗浄した。沈殿をTE溶液(10mM Tris-HCl(pH8), 1mM EDTA)0.2ml に溶解後、RNase 酵素溶液(1mg/ml RNaseA) 5μLを加え、37℃で30分間インキュベートした。フェノール抽出後、3M 酢酸ナトリウム溶液20μLとともにイソプロパノール0.2ml を加え、よく混ぜ合わせ、室温で5分間置いた後、12,000rpmで5分間遠心し、沈殿を70%エタノールで洗った。乾燥後の100 μLのTE溶液に溶解した。
このようにして得られた染色体DNAは約10μgであった。
【0052】
▲2▼ ゲノムDNAライブラリーの作製
まず、λファージのクローニングベクターEMBL3をBamHIで切断し、BamHI切断面を有するEMBL3 DNA を得た。次に、上記ハンゼヌラ・ムラキ IFO 0895 株から得られた染色体DNAを制限酵素Sau3AIで部分消化した。
【0053】
このようにして得られた処理液を、in vitroパッケージングキット(STRATAGENE 社製) に添加して、in vitroパッケージング法(Horn, B.,Methods in Enzymology,68,299(1979)) により当該DNA をファージ粒子に導入し、ハンゼヌラ・ムラキIFO 0895のゲノムDNAライブラリーを作製した(Weiss,B. et al.,J.Biol.Chem.,243,4543(1968))。
【0054】
別に、山本らにより報告されているHM-1のアミノ酸配列(FEBS LETT.,195,253-257,1986) のうち、第20番目のトリプトファンから第26番目のスレオニンに相当するアミノ酸配列をコードするヌクレオチド20マー(以下「プローブ1」という;配列番号4)、及び第33番目のアスパラギンから第38番目のバリンに相当するヌクレオチドのアンチコドンの17マー(以下「プローブ2」という;配列番号5)の2種の合成オリゴヌクレオチドプローブを作製した。
【0055】
(2) ゲノムDNAのクローニング
制限酵素HindIII、EcoRI又はBamHIで切断した染色体DNAと、プローブ1及びプローブ2とを、サザンブロットハイブリダイゼーションさせた(Gene 137, 265-270,1993)。
【0056】
その結果、1本のバンドが検出されたので、次に前記プローブ1及びプローブ2を用いて、同様にゲノムDNAライブラリーからスクリーニングし、3.6kb のHindIII DNA 断片を含むDNA(λHMK08)を釣り上げた。得られた3.6kb の HindIII DNA断片の物理地図を作成し、プローブ1及びプローブ2とハイブリダイズする断片をEcoRI 及びHind IIIで消化し、DNA断片を縮小した。
その結果、DNAの両端にEcoRI 及びHind III部位を有する2.4kb の断片(2.4kb EcoRI-HindIII DNA) を得た。
【0057】
〔実施例2〕HM-1耐性酵母の作製
(1) ゲノムDNAライブラリーの作製
シャトルベクターYCp50に連結されたサッカロミセス・セレビシエのDNAライブラリーは、サッカロミセス・セレビシエのゲノムDNAを制限酵素Sau3AIで部分消化し、得られる断片とBamHIで切断したYCp50とをT4 DNAリガーゼで連結することにより作製した。
【0058】
(2) HM-1耐性変異株(rhk1-1) の作製
サッカロミセス・セレビシエBJ1824(a, ura3,leu2,trp1,pep4)を5mlのYPD培地に接種し、30℃で細胞濃度2×108 細胞/mlまで増殖させ、2.5mlを遠心集菌した。50mMリン酸カリウムバッファー(pH7.0)で2回洗い、10mlの同じバッファーに懸濁した。これに300μlのEMS(エチルメタンスルホン酸)を加え、激しくボルテックスした。30℃で30分振盪後、等量のチオ硫酸ナトリウムを加え、よく混合した。集菌し滅菌水で2回洗い、滅菌水に懸濁、希釈した。
このようにして調製された上記細胞懸濁液を3μg/mlのHM-1を含むYPD寒天培地にプレーテイングし、30℃で36時間のインキュベートにより、HM-1耐性変異株rhk1-1を取得した。
【0059】
(3) DNAライブラリーのrhk1-1への導入
こうして得られたrhk1-1に上記のYCp50-ゲノムDNAライブラリーを導入する形質転換を、酢酸リチウム法に従って行った。
【0060】
すなわち、100 mlのYPD 培地にrhk1-1を植え、OD600 が1.0 〜2.0 になるまで30℃で培養した。培養後、3,000rpm、5分間の遠心により集菌し、5mlのTE(10mM Tris-HCl(pH7.5), 1mM EDTA)に懸濁した。同様の遠心により集菌して5mlの酢酸リチウム溶液に懸濁した。再度遠心により集菌し、1mlの酢酸リチウム溶液(100mM酢酸リチウム, 10mM Tris-HCl(pH7.5), 1mM EDTA) に懸濁した。これを30℃で60分間振盪した後、400 μgのキャリアDNA(仔ウシ胸腺DNA)を加え、エッペンドルフチューブに100 μLずつ分注した。これに1μgのDNAを加え、30℃で30分間保温した。これに700 μLのPEG溶液(35 %(w/v) ポリエチレングリコール4000、100mM 酢酸リチウム、10mM Tris-HCl(pH7.5)、1mM EDTA) を加え、30℃で50分間保温した後、42℃で5分間熱ショックを与えた。次いで、4秒間遠心し、上清をアスピレーターにより除去し、沈殿を500 μLのTEで洗浄した後、100 μLのTEに懸濁した。
その結果、YCp50-ゲノムDNAライブラリーが導入された形質転換体を得た。
【0061】
(4) HM-1感受性コロニーの選別
CMドロップアウト培地(アミノ酸フリーの酵母ナイトローゲンベース0.67%、グルコース2%、並びにアミノ酸及び核酸の混合物であるドロップアウトパウダー0.13%からなる)よりウラシルを除いた培地の上に細胞懸濁液を広げて30℃で培養した。宿主rhk1-1はウラシル要求性であるが、YCp50 を有するrhk1-1の形質転換体であれば、YCp50 のもつURA3遺伝子の存在によりウラシル無添加培地でも生育することができることを利用して、ウラシル非要求性のコロニー(Ura+ 形質転換体)20,000 個を得た。
【0062】
得られたウラシル非要求性の形質転換体の中には、ゲノムDNAのうちHM-1感受性遺伝子を含むYCp50プラスミドを保有するもののほか、HM-1感受性遺伝子とは異なる遺伝子を含むYCp50プラスミドを保有するもの、及びYCp50しか保有しないものが混在している。そこで、HM-1感受性遺伝子を含むYCp50プラスミドを持つ形質転換体のみを選別するために、Ura+形質転換体を3μg/mlのHM-1を含むCMドロップアウト寒天培地へレプリカした。
その結果、HM-1感受性のコロニーを取得し、実施例1(1) のゲノムDNAの調製の記載に準じてコロニーよりプラスミドを調製した。
【0063】
このプラスミドが有するインサートDNAを確認するために、プラスミドを含む10μlのTE溶液を制限酵素BamHIで消化し、電気泳動したところ、13.4kbのインサートDNAをもつプラスミドであることがわかった。このインサートDNAをさらに切りとって縮めるために、種々の制限酵素で消化した(図1(1) 〜(5) )。得られたそれぞれのDNA断片をベクターYCp50にサブクローニングし、rhk1-1に導入し、それぞれのDNA断片について、選択培地(HM-1を含む合成寒天培地)上でHM-1に対する感受性を試験した。
【0064】
その結果、HM-1存在下でrhk1-1の生育が阻止されたことから(図1Aの(3) )、HM-1感受性を表現する遺伝子領域は2.5kb のKpnI-ScaI断片に局在することが示された(図1(3) )。
【0065】
(5) DNA配列の決定
次に、この2.5kb断片の塩基配列を決定した結果、配列番号6で表される配列が得られ、この配列中に、458アミノ酸からなるオープンリーディングフレーム(ORF) が含まれていた。このORFをコードする遺伝子をRHK1と命名した。
【0066】
(6) 遺伝子破壊によるHM-1耐性株の作出
YCp50 にRHK1が連結された遺伝子(YCp50-RHK1)を制限酵素KpnI及びHind IIIで消化後、RHK1断片を切り出してpUC118に連結した。得られたpUC118-RHK1 を制限酵素SphIで切断し、別にSphIで消化したURA3 DNA断片と連結することにより、RHK1遺伝子のSphI-SphI領域をURA3遺伝子で置き換えた(図2)。RHK1-URA3 キメラDNAをもつpUC118を制限酵素XhoIで切断後、RHK1-URA3 キメラDNA 断片をウラシル要求性株BJ1824(ura3, leu2, trp1, pep4)へ酢酸リチウム法により導入し、選択培地(ウラシルを含まないCMドロップアウト培地)上でウラシル非要求性株を選別した。RHK1遺伝子がURA3遺伝子により破壊されているか否か、すなわちURA3遺伝子がRHK1遺伝子の内部に組み込まれているか否かは、RHK1遺伝子をプローブとしてゲノムサザンブロッティングにより確認した。
【0067】
得られたウラシル非要求性株がHM-1耐性であるか否かを試験した結果、該ウラシル非要求性株は、HM-1を含むYPD培地で生育することからHM-1耐性株と判断された(サッカロミセス・セレビシエ(BJ1824rhk1 △::URA3) )を作製した。
【0068】
〔実施例3〕変異型HM-1の構築と発現
HM-1は、プロテアーゼの一種であるトリプシンと長時間反応させると分解されることが知られている。そこで、トリプシンによる作用を受けにくい変異型HM-1を作製するために、配列番号1で表されるアミノ酸配列のうち、第1番目のメチオニン(Met) から数えて98番目又は122番目のアルギニン(Arg) を、アラニン(Ala) に変換した。
また、119 番目及び123番目のアルギニンについても同様にアラニンに変換して試験を行った。
【0069】
(1) 発現プラスミドの構築条件
HM-1領域を含む2.4kb EcoRI-HindIII DNA断片の5'側をキロシーケンス用デレーションキット(宝酒造)で短くした後、BamHIリンカーを連結した。この5'側にBamHI部位をもつBamHI-HindIII断片をpUC118へ挿入し、鋳型のDNAとした。
【0070】
別にHM-1タンパク質の98番目、122番目、第119番目又は第123番目のArgをAlaに変えるために、予想される相補鎖の配列に従ってオリゴヌクレオチド23マーを化学合成し、プライマーとした(それぞれ、R98Aプライマー(配列番号7)、R122A プライマー(配列番号8)、R119Aプライマー(配列番号9)、R123Aプライマー(配列番号10))。
【0071】
変異型HM-1遺伝子は、LA PCR in vitroミュタージェネシスキット(宝酒造)を用いて構築した。すなわち、BamHI及びHind III制限部位を両端に持つプラスミド(pUC118-BamHI-HindIII)を鋳型DNAとして用い、前記各プライマーとミーュタージェネシスキットに含まれるM13RVプライマーとの間でPCRを行った。別に、同じキットにあるMUT1プライマーとM13M4プライマーのと間でもPCRを行った。以下、同じキットの手順に従って変異型遺伝子(HM-1R98A、HM-1R122A、HM-1R122A及びHM-1R122A)を作製した後、それぞれをpUC18のEcoRIとHindIII部位に挿入し、大腸菌 DH5αへ導入した。得られた形質転換体をLB培地で培養することによりプラスミドを調製し、次いでHidIIIとBamHIでこのプラスミドを消化後、アガロース電気泳動で精製した。この精製された変異型遺伝子断片を、酵母ベクターYEp51のHindIIIとBamHI部位へ挿入してYEp51のGal10プロモーターの下流につないだ後、大腸菌DH5αに形質転換し、プラスミドを抽出精製した。得られたプラスミドをサッカロミセス・セレビシエBJ1824rhk1△::URA3へ導入し、形質転換体を得た(図3)。形質転換体の選択は、CMドロップアウト培地からロイシンを除いたものにより行った。
【0072】
次いで、形質転換体を、HM-1感受性菌106細胞をまいたYPD 寒天培地上にストリークすることにより、キラー活性を測定した(J.Fermentation & Bioengineering, 80,423-428,1995)。
【0073】
その結果、変異型遺伝子HM-1R98A(配列番号2)及びHM-1R122A(配列番号3)に活性が認められた(図4)。
なお、変異型遺伝子HM-1R122A を保有する形質転換体(Saccharomyces cerevisiae BJ1824rhk1△::URA3/YEp51-HM-1R122A)は、工業技術院生命工学工業技術研究所に、FERM P- 15811 として寄託されている。
【0074】
(2) 変異型HM-1の精製
サッカロミセス・セレビシエBJ1824rhk1△::URA3/YEp51-HM-1R98A(またはYEp51-HM-1R122A)をショ糖0.5%を含むYPG培地(酵母エキス1%、ペプトン2%、ガラクトース2%)にて30℃でOD600nmが50になるまで(約72時間)培養し、その上清液を得た。
【0075】
まず、培地中に変異型HM-1が産生されているかどうかは、HM-1に対するポリクローナル抗体でウエスタンブロッテイングすることにより、HM-1と同じ位置に新規な変異型HM-1のバンドが検出されるか否かを調べることにより判断した。
次に、上記産生された変異型HM-1のうちどのHM-1がキラー活性を有するかを検定した。新規HM-1タンパク質がキラー活性をもつことは、YPD寒天培地上でハンゼヌラ・アノマラIFO0569株を検定菌とすることにより判定した。
【0076】
すなわち、YPD寒天培地上にステンレスカップを置き、次いでハンゼヌラ・アノマラIFO 0569株1×106細胞/mlを含むYPD軟寒天培地を重層後、ステンレスカップを除いて得られた穴へ、変異型HM-1を含む培養液を入れ、30℃にて一夜放置後、生じる阻止円を測定することにより検定を行った。
【0077】
次に、活性の認められた変異型HM-1を精製するために、サッカロミセス・セレビシエBJ1824rhk1△::URA3/YEp51-HM-1R98A(またはYEp51-HM-1R122A)を、500 mlの三角フラスコ中、200 mlのYPG + 0.5% Sucrose培地で、5日培養した(30℃,120rpm) 。
【0078】
培養物を7000rpmで10分遠心し、培養上清を0.45μm のフィルターに通し、遠心式濃縮機で濃縮した。得られた濃縮液を、分子量3500カットの透析チューブを用いて多量の水に対して透析した。透析後遠心式濃縮機で試料を濃縮し、Sephadex G-50(Fine 2.5×96cm) を用いてゲル濾過を行った。溶出は20mMトリメチルアミン- 酢酸(pH5.0) バッファーを用いた。
【0079】
活性画分を集め、遠心式濃縮機で濃縮後、70mlの20mM水酸化アンモニウム- 酢酸(pH8.5) バッファーに溶解し、2M の同バッファーでpHを8.0 に調整した。この試料をSuperQトヨパール650S(1.9×7cm)のカラムにかけ、20mM水酸化アンモニウム- 酢酸(pH8.5) バッファーで溶出し、溶出液を10mMトリメチルアミン- 酢酸(pH3.0) バッファーで透析した。
【0080】
得られた試料を、10mMトリメチルアミン- ギ酸(pH3.0) バッファーで平衡化したSPトヨパール650S(1.5×7cm)にかけ、1M の同バッファーで溶出した。活性画分を集め、10mMトリメチルアミン- ギ酸(pH3.0) バッファーで透析した後、HPLC(SP-5PW(7.5mm ×7.5cm); トーソー) を行った。HPLCは、10mMトリメチルアミン- ギ酸(pH3.0) バッファーと2M トリメチルアミン- ギ酸(pH3.0) バッファーとのリニアグラジエントにより行い、最終的に変異型HM-1を精製した。
こうして得られた標品は、20%アクリルアミドゲルを含むSDS-ポリアクリルアミドゲル電気泳動で均一のバンドを示した。
【0081】
〔実施例4〕本発明の変異型HM-1の性質
(1) プロテアーゼに対する分解性(プロテアーゼ耐性)
配列番号1で表されるアミノ酸配列のうち第98番目をアラニンに変異させた変異型HM-1における第98番目のアラニンと第99番目のセリンとの間の結合、及び配列番号1で表されるアミノ酸配列のうち第122番目をアラニンに変異させた変異型HM-1における第122番目のアラニンと第123番目のアルギニンとの間の結合が、トリプシンにより切断されるか否かを検討した。
【0082】
精製した変異型HM-1タンパク質にトリプシンを加えて作用させたところ、トリプシンによる消化は認められなかったことから、変異型HM-1タンパク質は強いトリプシン耐性を示した。
【0083】
(2) キラー活性
本発明の精製変異型HM-1についてキラー活性を調べた結果、HM-1と同等のキラー活性が認められた。
【0084】
【発明の効果】
本発明により、変異型キラータンパク質、該タンパク質をコードする遺伝子、及び該遺伝子によって形質転換された形質転換体が提供される。
本発明のタンパク質は強いプロテアーゼ耐性を示し、しかも大量生産することができるため、例えば抗菌剤又は抗菌性石鹸等に利用することができる。
【0085】
【配列表】

【図面の簡単な説明】
【図1】 HM-1感受性の試験結果を示す写真である(生物の形態)。
【図2】 HM-1耐性遺伝子の構築図である。
【図3】本発明の形質転換体の構築図である。
【図4】変異型HM-1のキラー活性を示す写真である(生物の形態)。
Claims (2)
- 配列番号6で表されるキラータンパク質感受性遺伝子を破壊することにより得られたキラータンパク質耐性遺伝子をサッカロミセス・セレビシエに導入することにより得られたキラータンパク質耐性サッカロミセス・セレビシエ株を、
( 1 )配列番号1で表されるアミノ酸配列のうち第 98 番目及び/若しくは第 122 番目のアルギニンがアラニンに変異したアミノ酸配列を実質的にコードするDNAを含むキラータンパク質遺伝子を含む組み換え体 DNA 、又は( 2 )配列番号2若しくは3で表される塩基配列を実質的に含むキラータンパク質遺伝子を含む組み換え体 DNA で、
形質転換したサッカロミセス・セレビシエ形質転換体。 - 請求項1記載の形質転換体を培地に培養し、得られる培養物からキラータンパク質を採取することを特徴とするキラータンパク質の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP23439396A JP3816593B2 (ja) | 1996-09-04 | 1996-09-04 | キラータンパク質 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP23439396A JP3816593B2 (ja) | 1996-09-04 | 1996-09-04 | キラータンパク質 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH1075789A JPH1075789A (ja) | 1998-03-24 |
| JP3816593B2 true JP3816593B2 (ja) | 2006-08-30 |
Family
ID=16970302
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP23439396A Expired - Fee Related JP3816593B2 (ja) | 1996-09-04 | 1996-09-04 | キラータンパク質 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP3816593B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2006030492A1 (ja) | 2004-09-14 | 2008-07-31 | 日本製紙株式会社 | Glp−1誘導体が集積された植物及び植物貯蔵器官とその生産方法 |
| JP2023553341A (ja) * | 2020-11-25 | 2023-12-21 | エスペロバックス インコーポレイティド | in vivoでの治療タンパク質の酵母ベースの発現 |
-
1996
- 1996-09-04 JP JP23439396A patent/JP3816593B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JPH1075789A (ja) | 1998-03-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Valdivieso et al. | CAL1, a gene required for activity of chitin synthase 3 in Saccharomyces cerevisiae. | |
| Van Dyck et al. | A single‐stranded DNA binding protein required for mitochondrial DNA replication in S. cerevisiae is homologous to E. coli SSB. | |
| Nakano et al. | The small GTP‐binding protein Rho1 is a multifunctional protein that regulates actin localization, cell polarity, and septum formation in the fission yeast Schizosaccharomyces pombe | |
| HU204097B (en) | Process for producing cloning system relating to kluyveromyces species | |
| RU2105812C1 (ru) | Полипептид, обладающий уратоксидазной активностью, фрагмент днк, обеспечивающий экспрессию полипептида с уратоксидазной активностью (варианты), вектор экспрессии, содержащий фрагмент днк, обеспечивающий экспрессию полипептида с уратоксидазной активностью (варианты), штамм, экспрессирующий полипептид, обладающий уратоксидазной активностью (варианты), способ получения полипептида, обладающего уратоксидазной активностью | |
| KR0159107B1 (ko) | 우레이트 산화효소 활성 단백질, 이 단백질을 암호화하는 재조합 유전자, 발현 벡터, 미생물 및 형질전환세포 | |
| JPH10500024A (ja) | 無毒、非毒素原性、非病原性の発現系、並びにその中で利用するためのプロモーター及びターミネーター | |
| KR100359563B1 (ko) | 아우레오바시딘 민감성 조절 단백질을 코딩하는 유전자 | |
| US5827706A (en) | Cyclosporin synthetase | |
| JP3468523B2 (ja) | ガゼインキナーゼ▲i▼と相互作用するタンパク質に関連する方法および物質 | |
| Barki et al. | Isolation of a Candida albicans DNA sequence conferring adhesion and aggregation on Saccharomyces cerevisiae | |
| KR0131063B1 (ko) | 디-아미노산 옥시다제 생산성 형질 전환체 | |
| US5484724A (en) | DNA encoding GLSI | |
| JP3816593B2 (ja) | キラータンパク質 | |
| Zimmermann et al. | Cloning and expression of the complement fixation antigen-chitinase of Coccidioides immitis | |
| US6326477B1 (en) | Process for modifying glucose repression | |
| Perfect et al. | Identification of a Cryptococcus neoformans gene that directs expression of the cryptic Saccharomyces cerevisiae mannitol dehydrogenase gene | |
| WO1986003774A1 (en) | A method of transforming fungi with a vector | |
| JPH03503843A (ja) | ホスホリパーゼa2の精製方法およびホスホリパーゼa2様ポリペプチドの製造方法 | |
| Santhanam et al. | A reassessment of flocculosin-mediated biocontrol activity of Pseudozyma flocculosa through CRISPR/Cas9 gene editing | |
| Schweizer et al. | Genetic control of Yarrowia lipolytica fatty acid synthetase biosynthesis and function | |
| KR100452393B1 (ko) | 오레오바시딘감수성조절유전자 | |
| JPH10504452A (ja) | 新規トポイソメラーゼiv、対応するヌクレオチド配列およびその使用 | |
| JP2748347B2 (ja) | アスペルギルス・ソーヤ形質転換体 | |
| JP3535535B2 (ja) | 糸状菌の形質転換用プラスミドおよびそれを用いた糸状菌の育種法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20050830 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051028 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060207 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060315 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060516 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060608 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100616 Year of fee payment: 4 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100616 Year of fee payment: 4 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100616 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110616 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120616 Year of fee payment: 6 |
|
| LAPS | Cancellation because of no payment of annual fees |