JP4511033B2 - Facsを用いて細胞表現型を変化し得る生物活性剤についてスクリーンする方法 - Google Patents
Facsを用いて細胞表現型を変化し得る生物活性剤についてスクリーンする方法 Download PDFInfo
- Publication number
- JP4511033B2 JP4511033B2 JP2000544822A JP2000544822A JP4511033B2 JP 4511033 B2 JP4511033 B2 JP 4511033B2 JP 2000544822 A JP2000544822 A JP 2000544822A JP 2000544822 A JP2000544822 A JP 2000544822A JP 4511033 B2 JP4511033 B2 JP 4511033B2
- Authority
- JP
- Japan
- Prior art keywords
- cell
- cells
- exocytosis
- preferred
- dye
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000000034 method Methods 0.000 title claims abstract description 97
- 238000012216 screening Methods 0.000 title claims abstract description 25
- 239000012867 bioactive agent Substances 0.000 title claims description 105
- 238000001943 fluorescence-activated cell sorting Methods 0.000 title claims description 4
- 230000022983 regulation of cell cycle Effects 0.000 claims abstract description 25
- 210000004027 cell Anatomy 0.000 claims description 640
- 230000028023 exocytosis Effects 0.000 claims description 166
- 108090000623 proteins and genes Proteins 0.000 claims description 91
- 102000004169 proteins and genes Human genes 0.000 claims description 90
- 239000008187 granular material Substances 0.000 claims description 77
- 150000007523 nucleic acids Chemical class 0.000 claims description 67
- 102000039446 nucleic acids Human genes 0.000 claims description 65
- 108020004707 nucleic acids Proteins 0.000 claims description 65
- 230000000694 effects Effects 0.000 claims description 57
- 102000004190 Enzymes Human genes 0.000 claims description 56
- 108090000790 Enzymes Proteins 0.000 claims description 56
- 239000006185 dispersion Substances 0.000 claims description 42
- 230000027455 binding Effects 0.000 claims description 35
- 230000004927 fusion Effects 0.000 claims description 33
- 230000001177 retroviral effect Effects 0.000 claims description 25
- 102000000412 Annexin Human genes 0.000 claims description 24
- 108050008874 Annexin Proteins 0.000 claims description 24
- 239000007850 fluorescent dye Substances 0.000 claims description 22
- 230000001413 cellular effect Effects 0.000 claims description 12
- 238000000926 separation method Methods 0.000 claims description 8
- 230000003833 cell viability Effects 0.000 claims description 6
- 210000004881 tumor cell Anatomy 0.000 claims description 6
- 102000034287 fluorescent proteins Human genes 0.000 claims description 5
- 108091006047 fluorescent proteins Proteins 0.000 claims description 5
- 230000004663 cell proliferation Effects 0.000 claims description 3
- 238000003556 assay Methods 0.000 abstract description 52
- 230000004075 alteration Effects 0.000 abstract 1
- 239000000975 dye Substances 0.000 description 131
- 235000018102 proteins Nutrition 0.000 description 88
- 108090000765 processed proteins & peptides Proteins 0.000 description 74
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 69
- 229940088598 enzyme Drugs 0.000 description 55
- 239000003795 chemical substances by application Substances 0.000 description 54
- 239000005090 green fluorescent protein Substances 0.000 description 45
- 230000008859 change Effects 0.000 description 43
- 230000014509 gene expression Effects 0.000 description 40
- 230000000638 stimulation Effects 0.000 description 33
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 32
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 32
- 239000013598 vector Substances 0.000 description 32
- 235000001014 amino acid Nutrition 0.000 description 30
- 229940024606 amino acid Drugs 0.000 description 30
- 150000001413 amino acids Chemical class 0.000 description 30
- 239000000758 substrate Substances 0.000 description 29
- 239000002609 medium Substances 0.000 description 27
- 238000005259 measurement Methods 0.000 description 26
- 102000004196 processed proteins & peptides Human genes 0.000 description 26
- 239000006228 supernatant Substances 0.000 description 25
- 239000012528 membrane Substances 0.000 description 24
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 23
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 23
- 108091006146 Channels Proteins 0.000 description 22
- 230000022131 cell cycle Effects 0.000 description 20
- 239000000126 substance Substances 0.000 description 20
- 108020004414 DNA Proteins 0.000 description 19
- 238000001514 detection method Methods 0.000 description 18
- 239000003814 drug Substances 0.000 description 18
- 108091005948 blue fluorescent proteins Proteins 0.000 description 17
- 230000001965 increasing effect Effects 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- 229940079593 drug Drugs 0.000 description 16
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 16
- 239000000427 antigen Substances 0.000 description 15
- 102000036639 antigens Human genes 0.000 description 15
- 108091007433 antigens Proteins 0.000 description 15
- 210000000170 cell membrane Anatomy 0.000 description 15
- 238000000684 flow cytometry Methods 0.000 description 15
- 239000003068 molecular probe Substances 0.000 description 15
- 238000010186 staining Methods 0.000 description 15
- 125000005504 styryl group Chemical group 0.000 description 15
- 230000006870 function Effects 0.000 description 14
- 238000001516 cell proliferation assay Methods 0.000 description 13
- 230000028327 secretion Effects 0.000 description 13
- 230000035899 viability Effects 0.000 description 13
- 108090000672 Annexin A5 Proteins 0.000 description 12
- 102000004121 Annexin A5 Human genes 0.000 description 12
- 150000001875 compounds Chemical class 0.000 description 12
- 230000007717 exclusion Effects 0.000 description 12
- 210000003630 histaminocyte Anatomy 0.000 description 12
- 239000003112 inhibitor Substances 0.000 description 12
- 102000002268 Hexosaminidases Human genes 0.000 description 11
- 108010000540 Hexosaminidases Proteins 0.000 description 11
- 210000004899 c-terminal region Anatomy 0.000 description 11
- 230000001419 dependent effect Effects 0.000 description 11
- 238000006471 dimerization reaction Methods 0.000 description 11
- 108020001507 fusion proteins Proteins 0.000 description 11
- 102000037865 fusion proteins Human genes 0.000 description 11
- 230000010190 G1 phase Effects 0.000 description 10
- 102000035195 Peptidases Human genes 0.000 description 10
- 108091005804 Peptidases Proteins 0.000 description 10
- 239000004365 Protease Substances 0.000 description 10
- 108010076504 Protein Sorting Signals Proteins 0.000 description 10
- 230000000340 anti-metabolite Effects 0.000 description 10
- 229940100197 antimetabolite Drugs 0.000 description 10
- 239000002256 antimetabolite Substances 0.000 description 10
- 230000003834 intracellular effect Effects 0.000 description 10
- 150000002500 ions Chemical class 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- 241001430294 unidentified retrovirus Species 0.000 description 10
- 108091026890 Coding region Proteins 0.000 description 9
- 102000053187 Glucuronidase Human genes 0.000 description 9
- 108010060309 Glucuronidase Proteins 0.000 description 9
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 238000003570 cell viability assay Methods 0.000 description 9
- 238000011065 in-situ storage Methods 0.000 description 9
- 238000003780 insertion Methods 0.000 description 9
- 230000037431 insertion Effects 0.000 description 9
- 230000003993 interaction Effects 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 230000003362 replicative effect Effects 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- -1 AraC Chemical compound 0.000 description 8
- 108010040476 FITC-annexin A5 Proteins 0.000 description 8
- 108091034117 Oligonucleotide Proteins 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 239000011575 calcium Substances 0.000 description 8
- 230000015556 catabolic process Effects 0.000 description 8
- 238000010276 construction Methods 0.000 description 8
- 238000006731 degradation reaction Methods 0.000 description 8
- 230000012202 endocytosis Effects 0.000 description 8
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 8
- 125000003729 nucleotide group Chemical group 0.000 description 8
- 230000001105 regulatory effect Effects 0.000 description 8
- 238000011160 research Methods 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 210000004739 secretory vesicle Anatomy 0.000 description 8
- 231100000747 viability assay Toxicity 0.000 description 8
- 238000003026 viability measurement method Methods 0.000 description 8
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 7
- 239000004471 Glycine Substances 0.000 description 7
- 102000001400 Tryptase Human genes 0.000 description 7
- 108060005989 Tryptase Proteins 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 230000003698 anagen phase Effects 0.000 description 7
- 229910052791 calcium Inorganic materials 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 238000003776 cleavage reaction Methods 0.000 description 7
- 230000026374 cyclin catabolic process Effects 0.000 description 7
- 238000001952 enzyme assay Methods 0.000 description 7
- 230000004807 localization Effects 0.000 description 7
- 230000037361 pathway Effects 0.000 description 7
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 7
- 230000007017 scission Effects 0.000 description 7
- 230000003248 secreting effect Effects 0.000 description 7
- 230000008685 targeting Effects 0.000 description 7
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 6
- PRDFBSVERLRRMY-UHFFFAOYSA-N 2'-(4-ethoxyphenyl)-5-(4-methylpiperazin-1-yl)-2,5'-bibenzimidazole Chemical compound C1=CC(OCC)=CC=C1C1=NC2=CC=C(C=3NC4=CC(=CC=C4N=3)N3CCN(C)CC3)C=C2N1 PRDFBSVERLRRMY-UHFFFAOYSA-N 0.000 description 6
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 6
- 101150085390 RPM1 gene Proteins 0.000 description 6
- 238000004873 anchoring Methods 0.000 description 6
- 230000012820 cell cycle checkpoint Effects 0.000 description 6
- 239000000306 component Substances 0.000 description 6
- 230000001086 cytosolic effect Effects 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 238000000799 fluorescence microscopy Methods 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- JGVWCANSWKRBCS-UHFFFAOYSA-N tetramethylrhodamine thiocyanate Chemical compound [Cl-].C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=C(SC#N)C=C1C(O)=O JGVWCANSWKRBCS-UHFFFAOYSA-N 0.000 description 6
- 230000004568 DNA-binding Effects 0.000 description 5
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 5
- 102100037872 Intercellular adhesion molecule 2 Human genes 0.000 description 5
- 101710148794 Intercellular adhesion molecule 2 Proteins 0.000 description 5
- 230000018199 S phase Effects 0.000 description 5
- 150000001720 carbohydrates Chemical class 0.000 description 5
- 235000018417 cysteine Nutrition 0.000 description 5
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 230000001939 inductive effect Effects 0.000 description 5
- 230000000977 initiatory effect Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 235000013930 proline Nutrition 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 239000013603 viral vector Substances 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 5
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 5
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- 108020004705 Codon Proteins 0.000 description 4
- 108050006400 Cyclin Proteins 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 4
- 238000009825 accumulation Methods 0.000 description 4
- 230000006907 apoptotic process Effects 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 229960002685 biotin Drugs 0.000 description 4
- 239000011616 biotin Substances 0.000 description 4
- 235000014633 carbohydrates Nutrition 0.000 description 4
- 125000002091 cationic group Chemical group 0.000 description 4
- 238000003783 cell cycle assay Methods 0.000 description 4
- 230000032823 cell division Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000005194 fractionation Methods 0.000 description 4
- 229930182480 glucuronide Natural products 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000002438 mitochondrial effect Effects 0.000 description 4
- 210000004940 nucleus Anatomy 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 230000002062 proliferating effect Effects 0.000 description 4
- 238000011282 treatment Methods 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- 108091005957 yellow fluorescent proteins Proteins 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- IXZONVAEGFOVSF-UHFFFAOYSA-N 2-(5'-chloro-2'-phosphoryloxyphenyl)-6-chloro-4-(3H)-quinazolinone Chemical compound OP(O)(=O)OC1=CC=C(Cl)C=C1C1=NC(=O)C2=CC(Cl)=CC=C2N1 IXZONVAEGFOVSF-UHFFFAOYSA-N 0.000 description 3
- 102000000844 Cell Surface Receptors Human genes 0.000 description 3
- 108010001857 Cell Surface Receptors Proteins 0.000 description 3
- 208000031879 Chédiak-Higashi syndrome Diseases 0.000 description 3
- 108010014173 Factor X Proteins 0.000 description 3
- 206010020751 Hypersensitivity Diseases 0.000 description 3
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- 102000018697 Membrane Proteins Human genes 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- 108010090804 Streptavidin Proteins 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 230000007815 allergy Effects 0.000 description 3
- 125000003275 alpha amino acid group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000000692 anti-sense effect Effects 0.000 description 3
- YBHILYKTIRIUTE-UHFFFAOYSA-N berberine Chemical compound C1=C2CC[N+]3=CC4=C(OC)C(OC)=CC=C4C=C3C2=CC2=C1OCO2 YBHILYKTIRIUTE-UHFFFAOYSA-N 0.000 description 3
- 229940093265 berberine Drugs 0.000 description 3
- QISXPYZVZJBNDM-UHFFFAOYSA-N berberine Natural products COc1ccc2C=C3N(Cc2c1OC)C=Cc4cc5OCOc5cc34 QISXPYZVZJBNDM-UHFFFAOYSA-N 0.000 description 3
- 102000005936 beta-Galactosidase Human genes 0.000 description 3
- 108010005774 beta-Galactosidase Proteins 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 235000020958 biotin Nutrition 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 210000003855 cell nucleus Anatomy 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 210000001723 extracellular space Anatomy 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 230000002209 hydrophobic effect Effects 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 238000011527 multiparameter analysis Methods 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 229930014626 natural product Natural products 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 238000004806 packaging method and process Methods 0.000 description 3
- 230000004962 physiological condition Effects 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 108010054624 red fluorescent protein Proteins 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- 238000012163 sequencing technique Methods 0.000 description 3
- 210000000130 stem cell Anatomy 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- ATCJTYORYKLVIA-SRXJVYAUSA-N vamp regimen Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1.C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C(C45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 ATCJTYORYKLVIA-SRXJVYAUSA-N 0.000 description 3
- 238000012800 visualization Methods 0.000 description 3
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 2
- BGWLYQZDNFIFRX-UHFFFAOYSA-N 5-[3-[2-[3-(3,8-diamino-6-phenylphenanthridin-5-ium-5-yl)propylamino]ethylamino]propyl]-6-phenylphenanthridin-5-ium-3,8-diamine;dichloride Chemical compound [Cl-].[Cl-].C=1C(N)=CC=C(C2=CC=C(N)C=C2[N+]=2CCCNCCNCCC[N+]=3C4=CC(N)=CC=C4C4=CC=C(N)C=C4C=3C=3C=CC=CC=3)C=1C=2C1=CC=CC=C1 BGWLYQZDNFIFRX-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- 102000016736 Cyclin Human genes 0.000 description 2
- 102000002427 Cyclin B Human genes 0.000 description 2
- 108010060385 Cyclin B1 Proteins 0.000 description 2
- 230000006820 DNA synthesis Effects 0.000 description 2
- OZLGRUXZXMRXGP-UHFFFAOYSA-N Fluo-3 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(Cl)C(=O)C=C3OC3=CC(O)=C(Cl)C=C32)N(CC(O)=O)CC(O)=O)=C1 OZLGRUXZXMRXGP-UHFFFAOYSA-N 0.000 description 2
- 230000010337 G2 phase Effects 0.000 description 2
- 102100032340 G2/mitotic-specific cyclin-B1 Human genes 0.000 description 2
- 101710171049 Gene 21 protein Proteins 0.000 description 2
- 102100031132 Glucose-6-phosphate isomerase Human genes 0.000 description 2
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical compound NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 description 2
- 101000944380 Homo sapiens Cyclin-dependent kinase inhibitor 1 Proteins 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- 229930182816 L-glutamine Natural products 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- 101150117895 LAMP2 gene Proteins 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 108091005461 Nucleic proteins Proteins 0.000 description 2
- 108010038807 Oligopeptides Proteins 0.000 description 2
- 102000015636 Oligopeptides Human genes 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- 108010067902 Peptide Library Proteins 0.000 description 2
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 2
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 2
- 102100036691 Proliferating cell nuclear antigen Human genes 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 241000269370 Xenopus <genus> Species 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- DPKHZNPWBDQZCN-UHFFFAOYSA-N acridine orange free base Chemical compound C1=CC(N(C)C)=CC2=NC3=CC(N(C)C)=CC=C3C=C21 DPKHZNPWBDQZCN-UHFFFAOYSA-N 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 230000001919 adrenal effect Effects 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 208000006673 asthma Diseases 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 238000005452 bending Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N benzoquinolinylidene Natural products C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 238000000339 bright-field microscopy Methods 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 230000033366 cell cycle process Effects 0.000 description 2
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 2
- 229960003677 chloroquine Drugs 0.000 description 2
- 210000003737 chromaffin cell Anatomy 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000004624 confocal microscopy Methods 0.000 description 2
- 238000004163 cytometry Methods 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 210000003890 endocrine cell Anatomy 0.000 description 2
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 210000002907 exocrine cell Anatomy 0.000 description 2
- 239000012894 fetal calf serum Substances 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 239000012737 fresh medium Substances 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 229960001340 histamine Drugs 0.000 description 2
- 102000049765 human CDKN1A Human genes 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000009830 intercalation Methods 0.000 description 2
- 230000002687 intercalation Effects 0.000 description 2
- DRAVOWXCEBXPTN-UHFFFAOYSA-N isoguanine Chemical compound NC1=NC(=O)NC2=C1NC=N2 DRAVOWXCEBXPTN-UHFFFAOYSA-N 0.000 description 2
- 229940043355 kinase inhibitor Drugs 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 210000001700 mitochondrial membrane Anatomy 0.000 description 2
- 230000011278 mitosis Effects 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- 230000000858 peroxisomal effect Effects 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000037452 priming Effects 0.000 description 2
- 150000003148 prolines Chemical group 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 230000004850 protein–protein interaction Effects 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 102000016914 ras Proteins Human genes 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 125000006850 spacer group Chemical group 0.000 description 2
- 238000009987 spinning Methods 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 238000012916 structural analysis Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- 238000010798 ubiquitination Methods 0.000 description 2
- 230000034512 ubiquitination Effects 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- KUHSEZKIEJYEHN-BXRBKJIMSA-N (2s)-2-amino-3-hydroxypropanoic acid;(2s)-2-aminopropanoic acid Chemical compound C[C@H](N)C(O)=O.OC[C@H](N)C(O)=O KUHSEZKIEJYEHN-BXRBKJIMSA-N 0.000 description 1
- CHADEQDQBURGHL-UHFFFAOYSA-N (6'-acetyloxy-3-oxospiro[2-benzofuran-1,9'-xanthene]-3'-yl) acetate Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(OC(C)=O)C=C1OC1=CC(OC(=O)C)=CC=C21 CHADEQDQBURGHL-UHFFFAOYSA-N 0.000 description 1
- NCYCYZXNIZJOKI-IOUUIBBYSA-N 11-cis-retinal Chemical compound O=C/C=C(\C)/C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-IOUUIBBYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- XQCZBXHVTFVIFE-UHFFFAOYSA-N 2-amino-4-hydroxypyrimidine Chemical compound NC1=NC=CC(O)=N1 XQCZBXHVTFVIFE-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- ARQXEQLMMNGFDU-JHZZJYKESA-N 4-methylumbelliferone beta-D-glucuronide Chemical compound C1=CC=2C(C)=CC(=O)OC=2C=C1O[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O ARQXEQLMMNGFDU-JHZZJYKESA-N 0.000 description 1
- YXHLJMWYDTXDHS-IRFLANFNSA-N 7-aminoactinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=C(N)C=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 YXHLJMWYDTXDHS-IRFLANFNSA-N 0.000 description 1
- 108700012813 7-aminoactinomycin D Proteins 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 108010027410 Adenovirus E3 Proteins Proteins 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 102000007698 Alcohol dehydrogenase Human genes 0.000 description 1
- 108010021809 Alcohol dehydrogenase Proteins 0.000 description 1
- 101000879393 Aplysia californica Synaptobrevin Proteins 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 102100022146 Arylsulfatase A Human genes 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101100533230 Caenorhabditis elegans ser-2 gene Proteins 0.000 description 1
- 102100029968 Calreticulin Human genes 0.000 description 1
- 108090000549 Calreticulin Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 102000005367 Carboxypeptidases Human genes 0.000 description 1
- 108010006303 Carboxypeptidases Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108010036867 Cerebroside-Sulfatase Proteins 0.000 description 1
- 108010077544 Chromatin Chemical group 0.000 description 1
- 102000003858 Chymases Human genes 0.000 description 1
- 108090000227 Chymases Proteins 0.000 description 1
- 108010077840 Complement C3a Proteins 0.000 description 1
- 102100031506 Complement C5 Human genes 0.000 description 1
- 108010078546 Complement C5a Proteins 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 108010069514 Cyclic Peptides Proteins 0.000 description 1
- 102000001189 Cyclic Peptides Human genes 0.000 description 1
- 108010068192 Cyclin A Proteins 0.000 description 1
- 102100025191 Cyclin-A2 Human genes 0.000 description 1
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 1
- 102000000634 Cytochrome c oxidase subunit IV Human genes 0.000 description 1
- 108050008072 Cytochrome c oxidase subunit IV Proteins 0.000 description 1
- 102000019265 Cytochrome c1 Human genes 0.000 description 1
- 108010007528 Cytochromes c1 Proteins 0.000 description 1
- 238000012287 DNA Binding Assay Methods 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 241000283155 Delphinidae Species 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 102000018711 Facilitative Glucose Transport Proteins Human genes 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 102000006575 G-Protein-Coupled Receptor Kinases Human genes 0.000 description 1
- 108010008959 G-Protein-Coupled Receptor Kinases Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 241000699694 Gerbillinae Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108091052347 Glucose transporter family Proteins 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 108010051696 Growth Hormone Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 229920000209 Hexadimethrine bromide Polymers 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000745711 Homo sapiens Cytochrome P450 3A4 Proteins 0.000 description 1
- 101000967216 Homo sapiens Eosinophil cationic protein Proteins 0.000 description 1
- 101000712530 Homo sapiens RAF proto-oncogene serine/threonine-protein kinase Proteins 0.000 description 1
- 101001132698 Homo sapiens Retinoic acid receptor beta Proteins 0.000 description 1
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 1
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000003746 Insulin Receptor Human genes 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 102100025390 Integrin beta-2 Human genes 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 1
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102100039064 Interleukin-3 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- JTTHKOPSMAVJFE-VIFPVBQESA-N L-homophenylalanine Chemical compound OC(=O)[C@@H](N)CCC1=CC=CC=C1 JTTHKOPSMAVJFE-VIFPVBQESA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- FGBAVQUHSKYMTC-UHFFFAOYSA-M LDS 751 dye Chemical compound [O-]Cl(=O)(=O)=O.C1=CC2=CC(N(C)C)=CC=C2[N+](CC)=C1C=CC=CC1=CC=C(N(C)C)C=C1 FGBAVQUHSKYMTC-UHFFFAOYSA-M 0.000 description 1
- 101150048357 Lamp1 gene Proteins 0.000 description 1
- 101710128836 Large T antigen Proteins 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 1
- 102000014944 Lysosome-Associated Membrane Glycoproteins Human genes 0.000 description 1
- 108010064171 Lysosome-Associated Membrane Glycoproteins Proteins 0.000 description 1
- 206010064912 Malignant transformation Diseases 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102000014171 Milk Proteins Human genes 0.000 description 1
- 108010011756 Milk Proteins Proteins 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 101100059444 Mus musculus Ccnb1 gene Proteins 0.000 description 1
- 101100005613 Mus musculus Ccnb2 gene Proteins 0.000 description 1
- 101000934300 Mus musculus Cyclin-A2 Proteins 0.000 description 1
- 101000601947 Mus musculus Proliferating cell nuclear antigen Proteins 0.000 description 1
- 241000282339 Mustela Species 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- GKXJWSZPLIKUPS-IUNAMMOKSA-N N-[(2Z,6Z)-2,6-bis(hydroxyimino)cyclohexylidene]hydroxylamine Chemical compound O\N=C1\CCC\C(=N\O)C1=NO GKXJWSZPLIKUPS-IUNAMMOKSA-N 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 102000003729 Neprilysin Human genes 0.000 description 1
- 108090000028 Neprilysin Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 102000002488 Nucleoplasmin Human genes 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 229940116355 PI3 kinase inhibitor Drugs 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108091093037 Peptide nucleic acid Proteins 0.000 description 1
- 241000233805 Phoenix Species 0.000 description 1
- 208000012641 Pigmentation disease Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000004257 Potassium Channel Human genes 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 102100033479 RAF proto-oncogene serine/threonine-protein kinase Human genes 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101001113506 Rattus norvegicus Proliferating cell nuclear antigen Proteins 0.000 description 1
- 101000639133 Rattus norvegicus Vesicle-associated membrane protein 2 Proteins 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 102100040756 Rhodopsin Human genes 0.000 description 1
- 108090000820 Rhodopsin Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000001332 SRC Human genes 0.000 description 1
- 108060006706 SRC Proteins 0.000 description 1
- 102100038803 Somatotropin Human genes 0.000 description 1
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 1
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 1
- 101150052863 THY1 gene Proteins 0.000 description 1
- 206010043189 Telangiectasia Diseases 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- MZZINWWGSYUHGU-UHFFFAOYSA-J ToTo-1 Chemical compound [I-].[I-].[I-].[I-].C12=CC=CC=C2C(C=C2N(C3=CC=CC=C3S2)C)=CC=[N+]1CCC[N+](C)(C)CCC[N+](C)(C)CCC[N+](C1=CC=CC=C11)=CC=C1C=C1N(C)C2=CC=CC=C2S1 MZZINWWGSYUHGU-UHFFFAOYSA-J 0.000 description 1
- 108060008539 Transglutaminase Proteins 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NTECHUXHORNEGZ-UHFFFAOYSA-N acetyloxymethyl 3',6'-bis(acetyloxymethoxy)-2',7'-bis[3-(acetyloxymethoxy)-3-oxopropyl]-3-oxospiro[2-benzofuran-1,9'-xanthene]-5-carboxylate Chemical compound O1C(=O)C2=CC(C(=O)OCOC(C)=O)=CC=C2C21C1=CC(CCC(=O)OCOC(C)=O)=C(OCOC(C)=O)C=C1OC1=C2C=C(CCC(=O)OCOC(=O)C)C(OCOC(C)=O)=C1 NTECHUXHORNEGZ-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 238000004115 adherent culture Methods 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 230000001800 adrenalinergic effect Effects 0.000 description 1
- PPQRONHOSHZGFQ-LMVFSUKVSA-N aldehydo-D-ribose 5-phosphate Chemical group OP(=O)(O)OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PPQRONHOSHZGFQ-LMVFSUKVSA-N 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 208000025341 autosomal recessive disease Diseases 0.000 description 1
- GOOXRYWLNNXLFL-UHFFFAOYSA-H azane oxygen(2-) ruthenium(3+) ruthenium(4+) hexachloride Chemical compound N.N.N.N.N.N.N.N.N.N.N.N.N.N.[O--].[O--].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Ru+3].[Ru+3].[Ru+4] GOOXRYWLNNXLFL-UHFFFAOYSA-H 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 102000007478 beta-N-Acetylhexosaminidases Human genes 0.000 description 1
- 108010085377 beta-N-Acetylhexosaminidases Proteins 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- BQRGNLJZBFXNCZ-UHFFFAOYSA-N calcein am Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O)=C(OC(C)=O)C=C1OC1=C2C=C(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(=O)C)C(OC(C)=O)=C1 BQRGNLJZBFXNCZ-UHFFFAOYSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 210000002309 caveolated cell Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000012578 cell culture reagent Substances 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 230000004635 cellular health Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 210000003763 chloroplast Anatomy 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000013065 commercial product Substances 0.000 description 1
- 229920002055 compound 48/80 Polymers 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000021953 cytokinesis Effects 0.000 description 1
- 210000004395 cytoplasmic granule Anatomy 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000005202 decontamination Methods 0.000 description 1
- 230000003588 decontaminative effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 238000007877 drug screening Methods 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 230000002900 effect on cell Effects 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000000295 emission spectrum Methods 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000009144 enzymatic modification Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 238000003028 enzyme activity measurement method Methods 0.000 description 1
- 238000009585 enzyme analysis Methods 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000000695 excitation spectrum Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 230000006126 farnesylation Effects 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 108010027225 gag-pol Fusion Proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Chemical compound C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 1
- 230000006130 geranylgeranylation Effects 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000008134 glucuronides Chemical class 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 150000002333 glycines Chemical class 0.000 description 1
- 229930004094 glycosylphosphatidylinositol Natural products 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- 210000002288 golgi apparatus Anatomy 0.000 description 1
- 239000001056 green pigment Substances 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 239000003966 growth inhibitor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- PNDZEEPOYCVIIY-UHFFFAOYSA-N indo-1 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C=2N=C3[CH]C(=CC=C3C=2)C(O)=O)N(CC(O)=O)CC(O)=O)=C1 PNDZEEPOYCVIIY-UHFFFAOYSA-N 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229940076264 interleukin-3 Drugs 0.000 description 1
- 102000010681 interleukin-8 receptors Human genes 0.000 description 1
- 108010038415 interleukin-8 receptors Proteins 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 239000002555 ionophore Substances 0.000 description 1
- 230000000236 ionophoric effect Effects 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 101150066555 lacZ gene Proteins 0.000 description 1
- 150000002611 lead compounds Chemical class 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 210000001699 lower leg Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000006674 lysosomal degradation Effects 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 230000036212 malign transformation Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000005171 mammalian brain Anatomy 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000012533 medium component Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- RXMBKOPBFXCPDD-UHFFFAOYSA-N methoxyphosphonamidous acid Chemical compound COP(N)O RXMBKOPBFXCPDD-UHFFFAOYSA-N 0.000 description 1
- CWWARWOPSKGELM-SARDKLJWSA-N methyl (2s)-2-[[(2s)-2-[[2-[[(2s)-2-[[(2s)-2-[[(2s)-5-amino-2-[[(2s)-5-amino-2-[[(2s)-1-[(2s)-6-amino-2-[[(2s)-1-[(2s)-2-amino-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-5 Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)OC)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CCCN=C(N)N)C1=CC=CC=C1 CWWARWOPSKGELM-SARDKLJWSA-N 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 235000021239 milk protein Nutrition 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- QCTHLCFVVACBSA-JVNHZCFISA-N n-[(2s,3r,4r,5s,6r)-4,5-dihydroxy-6-(hydroxymethyl)-2-(4-methyl-2-oxochromen-7-yl)oxyoxan-3-yl]acetamide Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC=C(C(C)=CC(=O)O2)C2=C1 QCTHLCFVVACBSA-JVNHZCFISA-N 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 210000000633 nuclear envelope Anatomy 0.000 description 1
- 108060005597 nucleoplasmin Proteins 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 230000026792 palmitoylation Effects 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 239000002935 phosphatidylinositol 3 kinase inhibitor Substances 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- 150000008039 phosphoramides Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- INAAIJLSXJJHOZ-UHFFFAOYSA-N pibenzimol Chemical compound C1CN(C)CCN1C1=CC=C(N=C(N2)C=3C=C4NC(=NC4=CC=3)C=3C=CC(O)=CC=3)C2=C1 INAAIJLSXJJHOZ-UHFFFAOYSA-N 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 230000019612 pigmentation Effects 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000025726 positive regulation of exocytosis Effects 0.000 description 1
- 230000024715 positive regulation of secretion Effects 0.000 description 1
- 108020001213 potassium channel Proteins 0.000 description 1
- 108010066381 preproinsulin Proteins 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000003087 receptor blocking agent Substances 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000004161 regulation of exocytosis Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000000284 resting effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 230000008684 selective degradation Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 230000004960 subcellular localization Effects 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 108060008004 synaptotagmin Proteins 0.000 description 1
- 102000003137 synaptotagmin Human genes 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 208000009056 telangiectasis Diseases 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 230000036962 time dependent Effects 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 102000003601 transglutaminase Human genes 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 230000009750 upstream signaling Effects 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 230000011215 vesicle docking Effects 0.000 description 1
- 230000001018 virulence Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- 238000001086 yeast two-hybrid system Methods 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images













Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5011—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing antineoplastic activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1079—Screening libraries by altering the phenotype or phenotypic trait of the host
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5014—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing toxicity
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/502—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing non-proliferative effects
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/43504—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from invertebrates
- G01N2333/43595—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from invertebrates from coelenteratae, e.g. medusae
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/46—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans from vertebrates
- G01N2333/47—Assays involving proteins of known structure or function as defined in the subgroups
- G01N2333/4701—Details
- G01N2333/4718—Lipocortins
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Chemical & Material Sciences (AREA)
- Molecular Biology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Analytical Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- Toxicology (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- General Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- Plant Pathology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Biophysics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating Or Analysing Materials By The Use Of Chemical Reactions (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Measuring Pulse, Heart Rate, Blood Pressure Or Blood Flow (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Cephalosporin Compounds (AREA)
Description
(発明の分野)
本発明は、細胞パラメータの変化を検出する新規方法、詳細には、ペプチドなどの化学ライブラリィを含む、有機分子の総合的化学ライブラリィなどの小さな分子のライブラリィを、蛍光活性化セルソーター(FACS)機器を用いて標的分子への結合についてスクリーンする新規方法に関する。
【0002】
(発明の背景)
薬剤の発見および候補薬剤をスクリーンしてリード化合物を同定する分野は急速に発展している。新規および有効な候補薬剤を同定し、特徴化する従来の方法には、天然生成物または合成製造物を単離し、続いて既知のまたは未知の標的について検査することが含まれる。参照、例えば、WO94/24314, Gallop et al., J. Med. Chem. 37(9):1233 (1994); Gallop et al., J. Med. Chem. 37(10):1385(1994); Ellman, Acc. Chem. Res. 29:132(1996); Gordon et al., E. J. Med. Chem. 30:388s (1994); Gordon et al., Acc. Chem. Res. 29:144(1996); WO95/12608、すべて出典明示により本明細書の一部とする。
【0003】
これらのライブラリィのスクリーンは様々な方法で行われる。一つの方法はビーズへの付着と色素による視覚化を伴う;参照、Neslter et al., Bioorg. Med. Chem. Lett. 6(12):1327(1996)。別の方法はビースおよび蛍光活性化セルソーター(FACS)を使用している;参照、Needles et al., PNAS USA 90:10700(1993)およびVetter et al., Bioconjugate Chem. 6:319(1995)。
【0004】
蛍光活性化セルソーター(FACS)はフロー・サイトメトリーとも呼ばれ、蛍光発光を含む光学的特性に基づいて個々の細胞を分別するのに使用される。蛍光活性化セルソーターは一般に速いので、比較的短時間で大きな細胞集団をスクリーンし得る。
【0005】
FACSスクリーンなどの迅速で費用のかからないスクリーンが特に重要である多くの例がある。このような領域は細胞周期アッセイである。細胞はM期に始まり、様々な成長段階を経て循環する。M期において有糸分裂および細胞質分裂(細胞分裂)が発生する。M期の後にG1期が続き、G1期において細胞は高速度の生合成および成長を再び始める。S期はDNA合成で始まり、細胞核のDNAの内容量が倍加すると終わる。次いで、細胞はG2期に入り、凝集された染色体が現れることにより示される有糸分裂が始まると終わる。定期的に分化される細胞はG1期で停止され、それ以上細胞分裂をしない。
【0006】
悪性細胞の顕著な特徴は、制御されない増殖である。この表現型は遺伝子の変異の蓄積から得たものであり、その大部分は細胞周期を通して継代を促進する。癌細胞は成長制御シグナルを無視し、細胞分裂を続ける。rasなどの典型的な癌遺伝子は細胞周期のG1期からS期への不適切な転移を導き、増殖細胞外シグナルを模倣する。細胞周期チェックポイント制御により、確実にゲノムが正確に複製および分離される。細胞周期チェックポイント制御が失われると、ゲノムが不安定になり、悪性形質転換をもたらす変異の蓄積が大いに促進される。よって、p53およびATM(変異性毛細管拡張失調症)などのチェックポイント制御因子はまた、腫瘍抑制因子としても機能する。従って、治療剤で細胞周期チェックポイント経路を調節すると、正常細胞と腫瘍細胞との間の相違を利用して、放射線療法と化学療法の選択性を向上させ、新規な癌治療を導くことができる。
【0007】
従って、本発明の目的は、細胞周期チェックポイント制御の調節因子についてのスクリーンに有効な組成物および方法を提供することである。
【0008】
迅速なスクリーン方法が特別な使用を見出した別の領域は、エキソサイトーシスのアッセイの領域である。エキソサイトーシスは、分泌小胞の細胞原形質膜との融合であり、2つの主な機能を有する。一つは細胞外空間に小胞内容物を放出することであり、もう1つは原形質膜自体に新しいタンパク質および脂質を組み込むことである。
【0009】
エキソサイトーシスは2つの種類:構造性および制御性に分けられる。すべての真核生物細胞は構造性エキソサイトーシスを示し、形成後の分泌小胞の連続融合により特徴づけられる。制御性エキソサイトーシスは、外分泌性細胞、内分泌性細胞および神経細胞を含む特定の細胞に制限される。制御性エキソサイトーシスは、適切なシグナルを受け取るときのみ、通常は(常にではないが)細胞基質遊離Ca2+濃度が増加すると、原形質膜と融合する分泌小胞の蓄積をもたらす。
【0010】
制御性エキソサイトーシスは多くの分化した細胞に重要であり、しばしば特定の細胞は同じエキソサイトーシス顆粒から多数の媒介物質を放出し得る。エキソサイトーシス顆粒は標的細胞において調整された生理的反応を産生するために協力して機能する。これらの制御性エキソサイトーシス細胞には、神経細胞(神経伝達物質放出)、副腎性クロマフィン細胞(アドレナリン分泌)、膵臓腺房細胞(消化酵素分泌)、膵臓β細胞(インシュリン分泌)、肥満細胞(ヒスタミン分泌)、乳房細胞(乳タンパク質分泌)、精子(酵素分泌)、卵細胞(受精外被の発生)および含脂肪細胞(グルコース輸送体の原形質膜への挿入)が含まれる。さらに現在の理論において、小胞のドッキングおよび融合の基本的メカニズムは酵母菌から哺乳動物の脳へと保存されることが示唆されている。
【0011】
さらに、エキソサイトーシスに関する疾患が知られている。例えば、肥満細胞からの炎症性媒介物質の放出は、喘息を含む様々な疾患を引き起こす。米国だけで5000万以上の人々が喘息、鼻炎またはアレルギーによるいくつかの他の疾患にかかっている。アレルギーに対する治療は、肥満細胞により放出される媒介物質を遮断すること(抗ヒスタミン)、ステロイドなどの非特異的抗炎症剤および肥満細胞安定剤に制限され、肥満細胞安定剤はアレルギーの症状を制限するのにわずかに有効であるにすぎない。同様に、Chediak-Higashi症候群(CHS)は、稀な常染色体の劣性疾患であり、その疾患において好中球、単核細胞およびリンパ球などのほとんどの細胞は大きな細胞質顆粒を含有する。同様の疾患がマウス、ミンク、ウシ、ネコおよびシャチについて報告されており、CHS(beigeまたはbgと呼ばれる)のマウスの相同器官が最もよく特徴づけられている。参照、Perou et al., J. Biol. Chem. 272(47):29790(1997)およびBarbosa et al., Nature 382:262(1996)、両方とも出典明示により本明細書の一部とする。
【0012】
さらに、広範な一連の精神医学的疾患は神経伝達物質エキソサイトーシスと媒介物質の再取り込みとの間の不均衡の結果であると広く考えられている。
【0013】
非常に多くの医薬品は、主にエキソサイトーシス媒介物質の受容体を遮断することによりエキソサイトーシス媒介物質を特に阻害するように設計されてきた。しかしながら、この方法は、単一の受容体遮断剤が多くの異なる媒介物質の作用を克服し得ないことから限度がある。
【0014】
従って、本発明の目的は、エキソサイトーシスにおける変化をスクリーンする方法、特にこのようなエキソサイトーシスを媒介し得る薬剤をスクリーンする方法を提供することである。また、アッセイの共通事項を減少し、特異性を増加するようなスクリーン方法を提供することも目的である。
【0015】
(発明の要旨)
上に概記した目的に一致して、本発明は、細胞表現型を変化させる能力、または変化を調節する能力について生物活性剤をスクリーンする方法を提供する。この方法は、少なくとも1つの候補生物活性剤と細胞集団とを合わせ、そしてFACS機器における該細胞の分別を、少なくとも3、4または5の細胞パラメーターに基づく該細胞の分離により行うことを一般的に含む。候補剤は、候補生物活性剤をコードする融合核酸を含有する分子ライブラリィの一部である。
【0016】
更なる態様で、本発明は、細胞集団での、種々の状態での単独細胞での、または種々の生物活性剤との組み合せでの、エキソサイトーシスにおける変化の検定によってFACS機器での細胞を分別することを含む。本発明は、該細胞パラメーターが光分散、蛍光色素取り込み、蛍光色素放出、アネキシン顆粒結合、表面顆粒酵素活性、顆粒特異的タンパク質の量よりなる群から選ばれる少なくとも3の特性でもって変化の検定をなして、FACS機器での細胞を分別することを含む。
【0017】
本発明が提供するのは、細胞でのエキソサイトーシスを調節し得る生物活性剤についてスクリーンする方法である。この方法は、候補生物活性剤と細胞集団を合わせ、該細胞をエキソサイトーシスを正常に起こすような状態にすることを含む。細胞のFACS機器での分別は、光分散、蛍光色素取り込み、蛍光色素放出、アネキシン顆粒結合、表面顆粒酵素活性、顆粒特異的タンパク質の量よりなる群から選ばれる少なくとも3の特性についての変化の検定による。該特性の少なくとも1つにおける変化を、候補生物活性剤に接触していない細胞と比較すると、該生物活性剤がエキソサイトーシスを調節するのが分かる。
【0018】
生物活性剤をスクリーンする方法の好ましい実施態様において、選択される特性は、蛍光色素放出、アネキシン顆粒結合、表面顆粒酵素活性、顆粒特異的タンパク質の量よりなる群から選ばれる少なくとも1つの特性を含む。
【0019】
蛍光色素取り込みが検出されるとき、色素はスチリル色素である。蛍光色素放出が検出されるとき、色素は低pH濃度色素またはスチリル色素であり得る。
【0020】
好ましい実施態様において、表面顆粒酵素活性の検出は、インシトゥ酵素学アッセイまたは集団による酵素アッセイでなされる。酵素基質は検出可能基質であり得る。好ましくは、酵素基質はFRET構築体にカップルする。FRET構築体はプロテアーゼ部位で分割された2つの蛍光タンパク質を含む。この場合、プロテアーゼ部位は顆粒タンパク質に特異的である。
【0021】
好ましい実施態様において、顆粒特異的タンパク質が検出される。顆粒特異的タンパク質はいかなる検出可能タンパク質でもあり得る。ある実施態様において、顆粒特異的タンパク質は、顆粒特異的タンパク質およびFRET構築体であり得る検出可能分子を含む融合タンパク質である。
【0022】
別の実施態様において、細胞におけるエキソサイトーシスを調節し得る生物活性剤についてのスクリーン方法が提供され、該方法は、少なくとも1つの候補生物活性剤と、顆粒特異的タンパク質をコードする核酸および標識を含む融合核酸を各含有する細胞集団とを合わすことを含む。細胞はエキソサイトーシスを正常に起こす状態に置かれ、標識量での変化が検出される。標識量での変化は生物活性剤がエキソサイトーシスを調節したことを示す。好ましくは、標識は、エピトープ・タグまたは蛍光分子である。好ましい実施態様において、蛍光分子はFRET構築体である。
【0023】
別の態様において、本発明は、エキソサイトーシスの調節剤をスクリーンする方法を提供する。この方法は、候補生物活性剤、検出可能顆粒特異的タンパク質をコードする核酸を含む細胞、およびこのタンパク質の検出用剤を組み合せることを含む。細胞はエキソサイトーシスを正常に起こす状態に置かれ、タンパク質の存在または不存在が測定される。
【0024】
他の好ましい実施態様において、本発明は、細胞でのエキソサイトーシスを調節し得る生物活性剤をスクリーンする方法を提供する。この方法は、少なくとも1つの候補生物活性剤と細胞集団を合わすことを含む。細胞はエキソサイトーシスを正常に起こす状態に置かれ、蛍光アネキシンが加えられる。細胞表面での蛍光アネキシン量の変化が検査される。
【0025】
他の好ましい実施態様において、本発明は、細胞でのエキソサイトーシスを調節し得る生物活性剤をスクリーンする方法を提供する。この方法は、低pH濃度色素を取り込む細胞の集団を提供することを含む。低pH濃度色素を保持する細胞は、少なくとも1つの候補生物活性剤と合されて、エキソサイトーシスを正常に起こす状態に置かれる。低pH濃度色素の放出が検出される。放出色素量の変化は、生物活性剤がエキソサイトーシスを調節したことを示す。
【0026】
他の好ましい実施態様において、本発明は、細胞でのエキソサイトーシスを調節し得る生物活性剤をスクリーンする方法を提供する。この方法は、少なくとも1つの候補生物活性剤と細胞集団を合すことを含む。細胞はエキソサイトーシスを正常に起こす状態に置かれ、顆粒酵素に特異的な蛍光基質が加えられる。顆粒酵素に特異的な蛍光基質の検出において、蛍光基質量の変化が生物活性剤のエキソサイトーシス調節の指標となる。好ましい実施態様において、基質はFRET構築体を含む。
【0027】
更なる態様において、本発明は、細胞周期を調節し得る生物活性剤についてのスクリーン方法および条件を提供する。本発明は、候補生物活性剤のライブラリィと細胞集団を組合せ、FACS機器での細胞を少なくとも細胞生存能アッセイ、増殖アッセイ、細胞相アッセイに基づく細胞分離によって分別することを含む。
【0028】
更なる態様において、本発明は、細胞ライブラリィにおける融合核酸ライブラリィを発現することを含む。融合核酸は、候補生物活性剤および検出可能部分をコードする核酸を含む。FACS機器における細胞の分別は、細胞の分離による。細胞表現型が細胞周期であるとき、細胞の分別は、細胞の生存能、増殖および細胞相に基づく。
【0029】
(図面の簡単な説明)
図1A、1B、1Cは、実施例1の3つのレトロウイルス構築を模式的に示す。図1Aは、CRU5−GFP−p21構築であって、CRU5プロモーター、ψレトロパッキングシグナル、p21のコード領域に融合したGFPのコード領域、次いでLTRを含む。図1Bは、CRU5−GFP−p21C構築を示し、p21のC末24アミノ酸を含む。図1Cは、CRU5−GFP−pUCmut構築体を示し、これは3アラニン置換を有するCRU5−p21Cp21の変異体である。
【0030】
図2A、2B、2C、2Dは、実施例1の実験の結果を示す。図2Aは、前方および側面分散を用いる生存能アッセイを示す。特性割合を表す細胞が集められる。図2Bは、ベクターのGFPの蛍光を示す。図2Cは、増殖アッセイにおけるPKH26、包含色素の使用を表す;細胞を拘束する既知のタンパク質であるp21を含有する細胞は、輝く蛍光を保持し、一方、対照の細胞は、増殖を続けて色素を希釈し蛍光を無くする。図2Dは、細胞相アッセイにおける Hoechst 33342 の使用を表す。
【0031】
図3は、G1相における細胞拘束剤、p21に感染のJurkat細胞に対するAraC処置の効果を示す。AraCは、分割細胞に毒性のヌクレオチド類似体である。従って、拘束された細胞周期であるこれらの細胞が生存する。下側の線はp21の挿入のないJurkat細胞を示し,上側の線はp21挿入のJurkat細胞を示す。
【0032】
図4Aおよび4Bは、集団についてのエキソサイトーシス酵素活性アッセイの結果を示す。図4Aは、DMSO(−)またはイオノマイシン(+)と組み合わせた細胞の上澄み液におけるグルクロニダーゼまたはヘキソサミニダーゼ活性を示す。図4Bは、種々の量のIgEアンチDNPで感受性化され、増加量の抗原BSA−DNPで刺激された細胞の上澄み液におけるヘキソサミニダーゼ活性を示す。
【0033】
図5A−5Fは、フロー・サイトメーター、側面分散対前方分散に見られたエキソサイトシス・分散変化を示す。RBL-2H3細胞(図5Aおよび5D)およびMC−9細胞(図5B、5C、5E、5F)について2変数ヒストグラムとしてプロットした。イオン透過担体での刺激後、細胞の観察を0分(図5Aおよび5C)、5分(図5E)、10分(5D)、30分(図5Bおよび5F)で行った。
【0034】
図6A−6Eは、FACSによるエキソサイトーシスを検出するスチリル色素アッセイの結果を示す。細胞は、DMSO(左ピーク)またはイオノマイシン(右ピーク)と、FM4−64(図6Aおよび6B)またはF1−43(図6C、6D、6E)のいずれかの存在下にて、組み合わせた。図6Aおよび6Cは蛍光チャネル1で検出された細胞を示す。図6Bおよび6Dは、蛍光チャネル3で検出された細胞を示す。図6Eは、蛍光チャネル1でのフロ−・サイトメトリーで検出された平均チャネルシフトを棒グラフで示す。種々の量のPl−3キナーゼ阻害剤ウォルトマニンとの前インキュベートが、FM1−43の存在下にイオン透過担体(棒1−4)またはDMSO(棒5)の投与前になされた。
【0035】
図7A−7Dは、FACSによるエキソサイトーシスのアネキシン-V検出アッセイの結果を示すグラフである。細胞は、DMSO(図7Aおよび7B)またはイオノマイシン(図7Cおよび7D)のいずれかと組み合わせ、次いでヨウ化プロピジウム(図7Aおよび7C)およびアネキシン-V-FITC(図7Bおよび7D)の両者で着色された。
【0036】
図8A−8Cは、FACSにより可視化されたエキソサイトーシス細胞のインシトゥ酵素学アッセイの結果を表すグラフである。細胞は、DMSO(図8A)またはイオン透過担体(図8Bおよび8C)と組み合わせ、次いでインシトゥグルクロニダーゼ活性について着色した。図8Cは細胞表面酵素活性のpHプロファイルを示し、棒グラフで、フロー・サイトメトリー中の平均チャネルシフトにより測定された最大シグナルの百分率を表す。
【0037】
図9は、チャネル1中に検出された蛍光強度のヒストグラフである。細胞に、LYSOTRACKER GREEN(商標)を負荷し、DMSO(左)またはイオノマイシン(右)のいずれかと組み合わせ、フロー・サイトメトリーで測定した。
【0038】
図10A−10Hは、LYSOTRACKER GREEN(商標)、アネキシン-V-APC、前方および側面分散を含む複数パラメーター分析の結果を示す。図10A−10Dおよび10E−10Hの各々は、イオノマイシンの増加量で処理され、次いで4パラメーターのフロー・サイトメーターで同時に観測された細胞を示す。細胞に、低pH濃度色素を負荷し、刺激し、アネキシン-V-APCで着色した。図10A−10Dは、側面対前方の光分散の2変数ヒストグラムを示し、図10E−10Hは、アネキシン-V-APC対低pH濃度色素シグナルの2変数ヒストグラムを示す。
【0039】
図11は、FM1−43の存在下で刺激され、アネキシン-V-APCで着色された細胞についてのグラフである。イオノマイシン刺激後の種々の時点で、細胞の分析をフロー・サイトメトリーおよび酵素活性について上澄み液(細胞上澄み液)で行った。パラメーター、前方分散、FM−143、アネキシン-V-APC、ヘキソサミニダーゼを、各パラメーターについての最大応答に比較してグラフとした。カルシウム情報伝達について、分離管の細胞にFlou−3を負荷し、同様に処理した。
【0040】
発明の詳細な説明
本発明は、蛍光活性化セルソーター(FACS)機器の使用を介した、種々の細胞性パラメーターの評価またはアッセイによる、細胞周期規制、エキソサイトーシス、小分子毒性、細胞表面受容体発現、酵素発現等のような細胞表現型の変化の検出に関する。下記により詳述するように、種々の細胞表現型の変化の検出を可能にするために測定できる多くのパラメーターが存在する。多数のこれらのパラメーターの連続的、または好ましくは同時のアッセイにより、速く正確なスクリーニングを成し得る。
【0041】
好ましい態様において、本明細書に概説の方法は、細胞表現型の調節剤のスクリーンに使用する。アッセイし得る細胞表現型は、細胞周期規制を含む細胞アポトーシス、エキソサイトーシス、小分子への毒性、受容体を含む多くの部分の発現(特に細胞表面受容体)、接着分子、サイトカイン分泌、タンパク質−タンパク質相互作用等を含むが、これらに限定されない。
【0042】
好ましい態様において、本方法は、細胞周期規制の評価に使用する。この態様において、好ましい細胞性パラメーターまたはアッセイは、細胞生存能アッセイ、細胞が特定の細胞周期段階に拘束されているかのアッセイ(「細胞増殖アッセイ」)およびどの段階に細胞が拘束されているかのアッセイ(「細胞相アッセイ」)である。これらのパラメーターの1以上のアッセイまたは測定により、細胞周期規制の変化だけでなく、細胞周期規制経路の異なる段階の変化の検出も可能である。これは、天然の細胞の評価のために行い得、例えば、腫瘍細胞タイプの病原力の定量、または細胞周期規制に対する作用を試験する候補薬剤の効果の評価のために行い得る。この方法において、候補薬剤の速く正確なスクリーニングを、細胞周期規制を調節する薬剤の同定のために行い得る。
【0043】
このように、本方法は細胞の集団の、一つの成長相から他への移動、または一つの成長相への拘束をもらすことができる生物活性薬剤の解明に有用である。ある態様において、細胞を、特定の成長相に拘束し、その相から出すか、他の相へ移動させることが望ましい。あるいは、細胞を一つの成長相、例えばG1に拘束させることが、むしろ細胞周期を通して移動させるより望ましいことがある。同様に、ある状況下において、非拘束であるが、遅く移動する集団の細胞を、次の相にまたは細胞周期を通して加速させるか、または次の相の発現の遅延が望ましいことがあり得る。例えば、細胞相変化の開始に寄与するある酵素、例えば、キナーゼ、ホスファターゼ、プロテアーゼまたはユビキチン化酵素の活性を変えることが可能であり得る。
【0044】
好ましい態様において、本明細書に概説の方法は、G1相に拘束されていない細胞で行う;即ち、これらの細胞は、腫瘍細胞のように急速にまたは抑制できない成長および複製をしている。この方法において、細胞周期規制を変えることができる、即ち、細胞のG1のような細胞周期チェックポイントへの拘束をもたらす(S、G2およびMのような他の相への拘束もまた望ましいが)薬剤の発見のために候補薬剤を評価する。あるいは、候補薬剤を、細胞の集団の増殖をもたらすことができる薬剤、すなわち、一般にG1に拘束されている細胞;例えば、末梢血細胞、最終分化細胞、培養中の幹細胞等の再増殖を開始させる薬剤の評価に使用する。
【0045】
したがって、好ましい態様において、本発明は細胞の集団における細胞周期規制の変化のスクリーニングのための方法を提供する。「変化」および「修飾」(本明細書では交換可能に使用する)なる用語は、本明細書での使用に関して、測定するパラメーターまたは表現型の増加および減少の両方を含むことができる。細胞周期規制の内容に関する「変化」または「調節」は、一般に、二つのうちの一つを意味する。好ましい態様において、他の細胞または異なる条件下での同じ細胞と比較して、変化は細胞の細胞周期に変化、即ち、一つの相に拘束された細胞の増殖、または拘束細胞のその拘束相からの移動および細胞周期の開始をもたらす。あるいは、細胞の特定の相を通った進行を変え得る。即ち、細胞が特定の成長相を通って移動するのにかかる時間の長さを加速または遅延し得る。例えば、細胞は通常G1相を数時間経過する。薬剤の添加はG1相を延長し得る。
【0046】
測定は、各測定において全ての条件が同じであるか、種々の条件下、生物活性薬剤有りまたは無しで、または細胞周期過程の異なる段階で測定できる。例えば、細胞周期規制の測定は、候補生物活性薬剤が存在するおよび候補生物活性薬剤が不存在である細胞集団で測定できる。他の例において、細胞周期規制の測定は、細胞集団の条件または環境が他のものと異なって測定する。例えば、細胞を生理学的シグナル、例えば、ホルモン、抗体、ペプチド、抗原、サイトカイン、成長因子、活動電位、薬理学的試薬(即ち、化学療法剤等)、または他の細胞(即ち、細胞−細胞接触)の存在下または非存在下評価し得る。他の例において、細胞周期規制の測定は、細胞周期過程の異なる段階で測定する。更に別の例において、細胞周期規制の測定は、条件が同じであり、変化が一つの細胞または細胞集団と他の細胞または細胞集団の間であるもので行う。
【0047】
「細胞の集団」または「細胞のライブラリー」または「複数の細胞」は、本明細書では少なくとも2個の細胞を意味し、少なくとも約103が好ましく、少なくとも約105が特に好ましく、少なくとも約108から109が特に好ましい。集団またはサンプルは、一次または二次培養のいずれかからの異なる細胞タイプの混合物を含むことができるが、一つの細胞タイプのみを含むサンプルが好ましく、例えば、サンプルが一つの細胞系、特に腫瘍細胞系由来であり得る(特に、下に概説のとき)。細胞は、同時にまたはそうでなく、M、G1、SおよびG2を含む任意の細胞相である。好ましい態様において、複製または増幅している細胞を使用する。これは、候補生物活性薬剤の導入のためのレトロウイルスベクターの使用を可能にし得る。あるいは、非複製細胞を使用し得、他のベクター(アデノウイルスおよびレンチウイルスベクターのような)を使用できる。加えて、必要ではないが、細胞は色素および抗体と適合性である。
【0048】
本発明で使用するのに好ましい細胞タイプは、調節する細胞表現型により変化する。適当な細胞は、特に胸部、皮膚、肺、頸部、結直腸、白血病、脳等を含む全てのタイプの腫瘍細胞を含む、動物(マウス、ラット、ハムスターおよびアレチネズミを含む齧歯類)、霊長類、およびヒト細胞を含む哺乳類細胞を含むが、これらに限定されない。下に概説のように、更なる細胞タイプをエキソサイトーシスのスクリーニングに使用し得る。
【0049】
好ましい態様において、細胞周期規制法は、細胞生存能、細胞増殖および細胞相を含む数個の異なる細胞パラメーターのアッセイにより、細胞をFACS機器に分類することを含む。
【0050】
好ましい態様において、細胞性変化の欠乏が実験条件(すなわち、候補生物活性薬剤の導入)のためであり、細胞死のためでないことを確認するために、細胞生存能をアッセイする。使用できる多くの適当な細胞生存能アッセイがある。限定でないが、光散乱、生存能色素染色および排除色素染色の使用ができる。
【0051】
好ましい態様において、光分散アッセイを、当分野で周知されているように、生存能アッセイとして使用する。FACSで見たとき、細胞はその前方および90度(側面)光分散特性で測定して、特定の特徴を有する。これらの分散特性は、細胞のサイズ、形および顆粒含量を示す。これらの特性は、生存能の読み出しとして測定する二つのパラメーターを説明する。簡単に、死ぬまたは死んだ細胞は一般に凝縮しており、90°分散を変える;同様に、膜小泡形成が前方分散を変え得る。光分散の強度または細胞屈折指数の変化は生存能の変化を示す。
【0052】
このように、一般に、光分散アッセイに関して、特定の細胞タイプの生存細胞集団を評価して、その前方および側面分散特性を測定する。これは続いて使用できる分散の標準化を設定する。
【0053】
好ましい態様において、生存能アッセイは生存能染色を利用する。死んだまたは死ぬ細胞を染色するが、生育細胞を染色しない多くの既知の生存能色素がある。例えば、アネキシンVは、2価イオン依存的方法でリン脂質(ホスホチジルセリン)に特異的結合を示すタンパク質ファミリーのメンバーである。このタンパク質は、アポトーシス(計画細胞死)の測定に、細胞表面へのホスファチジルセリンの曝露がこの過程の顕著な初期シグナルであるため広く使用されている。適当な生存能色素は、アネキシン、エチジウムホモダイマー−1、DEADレッド、ヨウ化プロピジウム、SYTOXグリーン等および当分野で既知の他のものである;出典明示により本明細書の一部とするMolecular Probes Handbook of Fluorescent Probes and Research Chemicals, Haugland, Sixth Edition参照;特に、285頁のApoptosis Assayおよび16章参照。
【0054】
細胞生存能のための生存能色素染色のプロトコールは既知であり、前記Molecular Probes参照。この態様において、アネキシンのような生存能色素を、直接または間接的に標識し、細胞集団と合わせる。アネキシンは、例えば、PharMingen, San Diego, CaliforniaまたはCaltag Laboratories, Millbrae, Californiaから商品として入手可能である。好ましくは、生存能色素は、色素の濃度が約100ng/mlから約500ng/ml、より好ましくは約500ngから約1μg/ml、および最も好ましくは約1μg/mlから約5μg/mlである溶液として提供する。好ましい態様において、生存能色素は直接標識する。例えば、アネキシンはフルオレッセインイソチオシアネート(FITC)、Alexa色素、TRITC、AMCA、APC、tri-color、Cy−5および、当分野で既知であるか、商品として利用可能な他のもののような螢光色素で標識し得る。別の好ましい態様において、生存能色素は、ビオチンのようなハプテンのような第1標識で標識し、第2蛍光標識を使用する。他の第1および第2標識ペアは、当業者によく知られているように使用できる。
【0055】
添加したら、生存能色素染色を細胞と一定時間インキュベートし、必要であれば洗浄する。細胞を、非生存可能細胞を除くために、下に概説のように分別する。
【0056】
好ましい態様において、排除色素染色を生存能アッセイに使用する。排除色素は、生存細胞から排除されるもの、即ち、受動的に取りこまれないものである(それらは生存細胞の細胞膜を浸透しない)。しかし、死んだまたは死ぬ細胞の透過性により、それらは死亡細胞により取りこまれる。一般にはあるが、常にではなく、排除色素はDNAに、例えば、挿入(インターカレーション)を介して結合する。好ましくは排除色素は、DNAの非存在下で蛍光でないか、蛍光が乏しい;これで洗浄段階の必要性がなくなる。あるいは、第2標識の使用を必要とする排除色素も使用し得る。好ましい排除色素は、臭化エチジウム;エチジウムホモダイマー−1;ヨウ化プロピジウム;SYTOXグリーン核酸染色;Calcein AM、BCECF AM;フルオレッセイン二酢酸;TOTO(登録商標)およびTO-PRO(登録商標)(Molecular Probesから;前掲、16章参照)および当分野で既知の他のものである。
【0057】
細胞生存能のための排除色素染色のプロトコールは既知であり、Moelcular Probesカタログ、前掲参照。一般に、排除色素を、約100ng/mlから約500μg/ml、より好ましくは約500ng/mlから約1μg/ml、および最も好ましくは約0.1μg/mlから約5μg/mlの濃度で細胞に添加し、約0.5μg/mlが特に好ましい。細胞および排除色素を一定時間インキュベートし、必要に応じて洗浄し、次いで細胞を下に概略のように分別し、集団から非生存可能細胞を除去する。
【0058】
加えて、例えば、生存細胞(即ち、分泌されたプロテアーゼ等)または死亡細胞(即ち、媒体中の細胞内酵素の存在;例えば、細胞内プロテアーゼ、ミトコンドリア酵素等)の細胞外酵素活性の測定ができる、酵素アッセイを含んで行い得る他の細胞生存能アッセイがある。出典明示により本明細書の一部とするMolecular Probes Handbook of Fluorescent Probes and Research Chemicals, Haugland, Sixth Edition参照;特に16章参照。
【0059】
好ましい態様において、少なくとも一つの細胞生存能アッセイを行い、蛍光が同等な場合、少なくとも二つの異なる細胞生存能アッセイが好ましい。一つの生存能アッセイのみを行う場合、好ましい態様は光分散アッセイ(前方および側面分散両方)を使用する。二つの生存能アッセイを行うとき、好ましい態様は光分散および色素排除を使用し、光分散および生存能色素染色も可能であり、全3つを、ある場合、同様に行う。このように、生存能アッセイは、非生存可能または死亡細胞から生存能細胞の分離を可能にする。
【0060】
細胞生存能アッセイに加えて、好ましい態様は細胞増殖アッセイを使用する。本明細書の「増殖アッセイ」は、細胞集団が増殖、即ち複製しているかまたは複製していないかの測定を可能にするアッセイを意味する。
【0061】
好ましい態様において、増殖アッセイは色素包含アッセイである。色素包含アッセイは、細胞相を分けるための希釈効果に基づく。簡単に、色素(一般に、下に概説のような蛍光色素)を細胞に導入し、細胞により取りこませる。一度取りこまれたら、色素を細胞に捕獲し、外に分散させない。細胞集団が分割するために色素は比例して希釈される。即ち、包含色素の導入後、細胞を一定時間インキュベートさせる;経時的に蛍光を失う細胞は分割し、蛍光を残す細胞は非成長相に拘束される。
【0062】
一般に、包含色素の導入は、二つの方法の一つで行い得る。色素が細胞に受動的に入ることができないか(例えば、荷電されている)、細胞が色素を取りこむように処理しなければならない;例えば、電気パルスの使用を介して。あるいは、色素は、受動的に細胞に入ることができるが、一度取りこまれたら、細胞の外に分散しないように修飾する。例えば、包含色素の酵素的修飾は、包含色素を荷電し得、したがって細胞の外に拡散できなくする。例えば、Molecular Probes CellTracker(登録商標)色素は、蛍光クロロメチル誘導体であり、細胞内に自由に拡散し、次いでグルタチオンS−トランスフェラーゼ介在反応が膜不浸透性色素を作る。
【0063】
適当な包含色素は、CellTracker(登録商標)色素のMolecular Probesライン、PKH26(Sigma)、および当分野で既知の他のものを含むが、限定されない。これらはCellTracker(登録商標)Blue、CellTracker(登録商標)Yellow-Green、CellTracker(登録商標)Green、CellTracker(登録商標)Orangeを含むが、限定されない。Molecular Probes Handbook、前掲参照;特に15章。
【0064】
一般に、包含色素は約100ng/mlから約5μg/mlの範囲の濃度で細胞に提供する。約500ng/mlから約1μg/mlが好ましい。洗浄段階を使用し得、またはし得ない。好ましい態様において、候補生物活性剤を本明細書に記載の細胞と合わせる。細胞および包含色素を一定時間インキュベートし、細胞を分割させ、そして色素希釈をさせる。時間の長さは特定の細胞の細胞周期に依存する。一般に、少なくとも約2細胞分割が好ましく、少なくとも約3が特に好ましく、少なくとも約4が特別に好ましい。細胞を次いで下に概略のように分別し、複製する細胞およびしない細胞の集団を作る。当業者には認められるように、ある場合、例えば、抗増殖剤のスクリーニングの時に、明るい(即ち蛍光)細胞を集める;他の態様において、例えば、増殖剤のスクリーニングのために、低い蛍光の細胞を集める。変化は、異なる時点かまたは異なる細胞集団で蛍光の測定により決定し、互いにまたは標準と測定結果を比較する。
【0065】
好ましい態様において、増殖アッセイは代謝拮抗物質アッセイである。一般に、代謝拮抗物質アッセイは、G1またはG2静止相への細胞拘束をもたらす薬剤が望ましいとき、最も有用である。代謝拮抗物質増殖アッセイにおいて、分割細胞を殺す毒性代謝拮抗物質の使用は分割しない細胞のみの生存をもたらす。適当な代謝拮抗物質は、メトトレキサート、シスプラチン、タキソール、AraCのようなヌクレオチドアナログ等のような標準化学療法剤を含むが、これらに限定されない。加えて、代謝拮抗物質アッセイは発現により細胞死をもたらす遺伝子の使用を含む。
【0066】
代謝拮抗物質を添加する濃度は、特定の代謝拮抗物質の毒性に依存し、当分野で既知のように決定する。代謝拮抗物質を添加し、細胞を一般に一定時間インキュベートする;再び、正確な時間は代謝拮抗物質の特性および同一性、および特定の細胞集団の細胞周期時間に依存する。一般に、少なくとも1回の細胞分割が起こるのに十分な時間である。
【0067】
好ましい態様において、少なくとも一回の増殖アッセイを行い、1回以上が好ましい。このように、増殖アッセイは増殖細胞の集団および拘束細胞の集団をもたらす。
【0068】
好ましい態様において、上記に概略の1回またはそれ以上の増殖アッセイの後または同時に、少なくとも1回の細胞相アッセイを行う。「細胞相」アッセイは、どの細胞相、M、G1、SまたはG2に細胞が拘束されているかを決定する。
【0069】
好ましい態様において、細胞相アッセイはDNA結合アッセイである。簡単に、DNA結合色素を細胞に挿入し、受動的に取りこませる。細胞内に入ったら、DNA結合色素はDNAに、一般に、挿入(インターカレーション)により結合するが、ある場合、色素は主溝または副溝結合化合物であり得る。色素の量は、このように直接細胞中のDNAの量に依存し、これは細胞相で変化する。G2およびM相細胞は、G1相細胞の2倍のDNA含量を有し、S相細胞は、S相のどの点に細胞があるかに依存して中間の量を有する。適当なDNA結合色素は浸透性であり、Hoechst 33342および33258、アクリジンオレンジ、7−AAD、LDS 751、DAPIおよびSYTO 16を含むが、これらに限定されない;Molecular Probes Handbook、前掲;特に8および16章。
【0070】
一般にDNA結合染料は約1μg/mlないし約5μg/mlの範囲の濃度で添加する。該染料を細胞に加えて一定時間インキュベートするが、この時間の長さは選択される染料にいく分か依存する。1つの態様では、染料の添加直後に測定値をとる。次ぎに以下に概説されるように細胞を分別し、種々の量の染料を含む、従って種々の量のDNAを含む細胞集団を作製し、このようにして複製している細胞から複製していない細胞を分離する。当業者には理解されるであろうが、ある場合、例えば増殖阻害剤に関してスクリーンする場合には、最小の蛍光(従って、1コピーのゲノム)しか持たない細胞を、複製している、従って1コピーを超えるゲノムDNAを含む細胞から分離することができる。変化は、種々の時点で、または種々の細胞集団で蛍光を測定し、その測定値を互いに、または標準と比較することにより決定される。
【0071】
好ましい態様では、細胞相アッセイとはサイクリン分解アッセイである。この態様では、スクリーンに先立ち(および一般に以下に概説されるような候補生物活性剤の導入に先立ち)、細胞に融合核酸を導入する。該融合核酸はサイクリン分解ボックスをコードする核酸と検出可能な分子をコードずる核酸とを含んでなる。「サイクリン分解ボックス」は、当技術分野で公知であり、特定の細胞相の間に該ボックスを含むタンパク質のユビキチン化経路によって分解を引き起こす配列である。すなわち、例えば、G1サイクリンはG1では安定であるが、G1サイクリン分解ボックスが存在するためにS期では分解され得る。従って、サイクリン分解ボックスと検出可能な分子、例えば緑色蛍光タンパク質とを連結することで、検出可能な分子の有無を利用して、その細胞集団の細胞相を同定することができる。好ましい態様では、複数のボックス、好ましくは各々異なる4種のボックスを用い、細胞相の検出が可能となる。
【0072】
いくつかのサイクリン分解ボックスが当術分野で知られており、例えば、サイクリンAは配列
【化1】
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
【0073】
該サイクリン分解ボックスをコードする核酸は、検出可能な分子をコードする核酸に機能し得る形で連結する。該融合タンパク質は当技術分野で公知の方法によって構築される。例えば、該分解ボックスをコードする核酸を、検出可能な分子をコードする核酸に連結する。本明細書において「検出可能な分子」とは、検出可能な分子を含む細胞または化合物が、それを含まない細胞から識別可能となる分子、すなわち、抗原TAGと呼ばれることがあるエピトープ、特異的酵素、または蛍光分子を意味する。好ましい蛍光分子としては、限定されるものではないが、緑色蛍光タンパク質(GFP)、青色蛍光タンパク質(BFP)、黄色蛍光タンパク質(YFP)、赤色蛍光タンパク質(RFP)、ならびにルシフェラーゼおよびβ−ガラクトシダーゼをはじめとする酵素が挙げられる。抗原TAGを用いる場合、好ましい態様では細胞表面抗原を利用する。エピトープとしては細胞膜には通常見られない検出可能なペプチドのいずれかであることが好ましく、エピトープが細胞に通常見られるものであれば増強が検出され得る場合があるが、これは一般に好ましくない。同様に、例えば新規な産物または発色産物を生成する酵素など検出可能な酵素分子を用いてもよい。
【0074】
従って、細胞相アッセイ後の分別の結果、種々の細胞相にある少なくとも2つの細胞集団が得られる。
【0075】
好ましい態様では、本方法を用い、細胞周期チェックポイント経路の活性化または抑制、およびチェックポイント欠陥の改善をはじめとする、その細胞周期調節を変調する能力に関して候補生物活性剤をスクリーンする。候補生物活性剤は、細胞集団に外生的に加えることもできるし、あるいは本明細書にさらに記載されるように細胞中へ導入することもできる。
【0076】
本明細書において「候補生物活性剤」または「外生化合物」とは、例えば、タンパク質、小型の有機分子、炭水化物(多糖類を含む)、ポリヌクレオチド、脂質などのいずれかの分子をいう。一般に、複数のアッセイ混合物を種々の薬剤濃度を用いてパネルに流し、種々の濃度に対しての種々の示差応答を得る。典型的には、これらの濃度のうち1つ、すなわち0または検出レベルに満たない濃度が負の対照として用いられる。さらに、正の対照も使用できる。例えば、細胞周期アッセイでは、細胞周期を変化させることが知られている薬剤を使用してもよい。例えば、p21は、G1サイクリン−CDK複合体と結合することにより、G1細胞相にある細胞を拘束させることが知られている分子である。同様に、エキソサイトーシスに対してはエキソサイトーシスを誘導することが知られている化合物を以下にさらに十分概説されるように使用できる。
【0077】
候補薬剤は多くの化学クラスを包含するが、典型的にはそれらは有機分子、好ましくは分子量が100ダルトンより大きく約2,500ダルトン未満の小型の有機分子である。候補薬剤はタンパク質との構造相互作用、特に水素結合に必要な官能基を含んでなり、典型的には少なくとも1つのアミン、カルボニル、ヒドロキシルまたはカルボキシル基、好ましくは少なくとも2つの官能化学基を含む。該候補薬剤は環式炭素または複素環式構造および/または上記の1以上の官能基で置換された芳香族もしくはポリ芳香族構造を含んでなる。候補薬剤はまた、ペプチド、糖類、脂肪酸、ステロイド、プリン、ピリミジン、誘導体、構造類似体またはそれらの組合せを含む生体分子間に見出せる。特に好ましいのはペプチドである。
【0078】
候補薬剤は合成または天然化合物のライブラリーを含む多様な供給源から得られる。例えば、ランダム化オリゴヌクレオチドの発現をはじめ、多様な有機化合物および生体分子のランダムな、また所定の合成には多くの手段が利用できる。あるいは、細菌、真菌、植物および動物の抽出物形態における天然化合物のライブラリーが入手可能であるか、または容易に作製できる。さらに、天然の、または合成により作製したライブラリーまたは化合物は従来の化学的、物理的、生化学的手段により容易に修飾される。公知の薬理活性物質は、アシル化、アルキル化、エステル化、アミド化などの所定のまたはランダムな化学修飾を受けて構造類似体を形成し得る。
【0079】
好ましい態様では、該候補生物活性剤はタンパク質である。本明細書において「タンパク質」とは共有結合した少なくとも2つのアミノ酸を意味し、タンパク質、ポリペプチド、オリゴペプチドおよびペプチドが含まれる。該タンパク質は天然に存在するアミノ酸およびペプチド結合または合成ペプチド模倣構造からなってもよい。従って本明細書において「アミノ酸」または「ペプチド残基」とは、天然に存在するアミノ酸と合成アミノ酸の双方を意味する。例えば、ホモフェニルアラニン、シトルリンおよびノルロイシンは、本発明の目的のためのアミノ酸と考えられる。また「アミノ酸」には、プロリンおよびヒドロキシプロリンなどのイミノ酸残基が含まれる。その側鎖は(R)配置または(S)配置のいずれであってもよい。好ましい態様では、該アミノ酸は(S)またはL配置である。天然に存在しない側鎖を用いる場合は、例えばインビボ分解を妨げるまたは遅延させるために非アミノ酸置換基を用いてもよい。また、化学ブロッキング基またはその他の化学置換基を付加してもよい。
【0080】
好ましい態様では、候補生物活性剤は天然に存在するタンパク質または天然に存在するタンパク質の断片である。従って、例えば、タンパク質またはタンパク質性細胞抽出物のランダムなもしくは所定の消化物を含む細胞抽出物を用いてもよい。このように、本明細書に記載の系のスクリーンのためには、原核生物および真核生物のタンパク質ライブラリーを作製すればよい。本態様において特に好ましいのは、細菌、真菌、ウイルスおよび哺乳類タンパク質のライブラリーであり、後者が好ましく、ヒトタンパク質が特に好ましい。
【0081】
好ましい態様では、該候補生物活性剤は、約5ないし約30のアミノ酸からなるペプチドであり、約5ないし約20のアミノ酸からなるものが好ましく、約7ないし約15のアミノ酸からなるものが特に好ましい。該ペプチドは上記に概説したような天然に存在するタンパク質の消化物、ランダムペプチド、または「偏りのある」ランダムペプチドであってもよい。本明細書において「ランダム化された」または文法上の同義語は、核酸およびペプチドがそれぞれ実質的にランダムなヌクレオチドおよびアミノ酸からなることを意味する。一般にこれらのランダムペプチド(または以下に論じる核酸)は化学的に合成されるので、いずれの位置にいずれのヌクレオチドまたはアミノ酸でも組み込める。この合成法は、ランダム化されたペプチドまたは核酸を作製し、その長さおよび配列においてあり得る組合せのすべてまたは大部分の形成を可能にし、ランダム化されたタンパク質性生理活性剤候補のライブラリーを形成するよう設計することができる。
【0082】
1つの態様では、該ライブラリーは十分にランダム化され、いずれの位置でも優先のまたは一定の配列は存在しない。好ましい態様では、該ライブラリーには偏りがある。すなわち、配列内のある位置では一定であるか、または限られた数の可能性から選択される。例えば、好ましい態様では、ヌクレオチドまたはアミノ酸残基は、例えば、疎水性アミノ酸、親水性残基、立体的に偏りのある(小さいまたは大きい)残基、架橋のためのシステイン、SH−3ドメインのためのプロリン、リン酸化部位のためのセリン、トレオニン、チロシンもしくはヒスチジンの作出などに対して、またはプリンに対してといった所定のクラス内でランダム化される。
【0083】
好ましい態様では、該候補生物活性剤は核酸である。本明細書において「核酸」もしくは「オリゴヌクレオチド」または文法上の同義語は、共有結合した少なくとも2つのヌクレオチドを意味する。本発明の核酸は一般にホスホジエステル結合を含むが、以下に概説されるいくつかの場合では、例えば、ホスホルアミド(Beaucage, et al., Tetrahedron, 49(10):1925 (1993)およびその参照文献; Letsinger, J. Org. Chem., 35:3800 (1970); Sprinzl, et al., Eur. J. Biochem., 81:579 (1977); Letsinger, et al., Nucl. Acids Res., 14:3487 (1986); Sawai, et al., Chem. Lett., 805 (1984); Letsinger , et al., J. Am. Chem. Soc., 110:4470 (1988); およびPauwels, et al., Chemica Scripta, 26:141 (1986))、ホスホロチオエート(Mag, et al., Nucleic Acids Res., 19:1437 (1991);および米国特許第 5,644,048号)、ホスホロジチオエート(Briu, et al., J. Am. Chem. Soc., 111:2321 (1989))、O−メチルホスホロアミダイト結合(Eckstein , Oligonucleotides and Analogues: A Practical Approach, Oxford University Press参照)、ならびにペプチド核酸主鎖および結合(Egholm, J. Am. Chem. Sci., 114:1895 (1992); Meier, et al., Chem. Int. Ed. Engl., 31:1008 (1992); Nielsen, Nature, 365:566 (1993); Carisson, et al., Nature, 380:207 (1996)参照。なお、これらは出典明示により本明細書の一部とする)を含んでなる選択的主鎖を有し得る核酸類似体が含まれる。
【0084】
その他の核酸類似体としては正電荷主鎖(Denpcy, et al., Proc. Natl. Acad. Sci. USA, 92:6097 (1995));非イオン性主鎖(米国特許第5,386,023号、同第5,637,684号;同第5,602,240号;同5,216,141号;および同第4,469,863号; Kiedroeshi, et al., Angew. Chem. Intl. Ed. English, 30;423 (1991); Letsinger, et al., J. Am. Chem. Sci., 110:4470 (1988); Letsinger, et al., Nucleoside & Nucleotide, 13:1597 (1994); Chapters 2 and 3, ASC Symposium Series 580, "Carbohydrate modifications in Antisense Research", Ed. Y.S. Sanghui and P. Dan Cook; Mesmaeker, et al., Bioorganic & Medicinal Chem. Lett., 4:395 (1994); Jeffs, et al., J. Biomolecular NMR, 34:17 (1994); Tetrahedron Lett., 37:743 (1996))、ならびに米国特許第5,235,033号、同5,034,506号、およびChapters 6 and 7, ASC Symposium Series 580, "Carbohydrate Modifications in Antisense Research", Ed. Y.S. Sanghui and P.Dan Cookに記載のものをはじめとする非リボース主鎖を有するものが挙げられる。
【0085】
1以上の炭素環式糖を含む核酸もまた本核酸定義内に含まれる(Jenkins, et al., Chem. Soc. Rev., (1995) pp. 169-176参照)。いくつかの核酸類似体がRawls, C & E News, June 2, 1997, page 35に記載されている。これらの参照文献はすべて出典明示して本明細書の一部とする。リボース−リン酸主鎖のこれらの修飾を行い、標識などの付加部分の付加を助けたり、あるいは生理学的環境下でかかる分子の安定性を高め、半減期を延ばしてもよい。さらに、天然に存在する核酸と類似体の混合物を作製することもできる。あるいは、種々の核酸類似体の混合物や天然に存在する核酸と類似体の混合物を作製してもよい。該核酸は明示したように一本鎖であっても二本鎖であってもよく、あるいは二本鎖または一本鎖配列の双方の部分を含んでもよい。該核酸は、デオキシリボヌクレオチドとリボヌクレオチドのいずれかの組合せ、およびウラシル、アデニン、チミン、シトシン、グアニン、イノシン、キサンチン、ヒポキサンチン、イソシトシン、イソグアニンなどを含む塩基のいずれかの組合せを含む限り、DNA(ゲノムDNAおよびcDNAの双方)、RNAまたはハイブリッドであってもよい。
【0086】
一般にタンパク質に関して上記したように、核酸候補生物活性剤は天然に存在する核酸、ランダム核酸、または「偏りのある」ランダム核酸であってよい。例えば、タンパク質に関して先に概説したように原核生物ゲノムまたは真核生物ゲノムの消化物を用いてもよい。
【0087】
好ましい態様では、該候補生物活性剤は有機化学部分であり、多様なものが文献から得られる。
【0088】
好ましい態様では、種々の候補生物活性剤のライブラリーを用いる。好ましくは、該ライブラリーはランダム化された薬剤の十分構造的に多様な集団を提供して、特定の標的との結合を可能とするに見込みとして十分な範囲の多様性を達成する。従って、相互作用ライブラリーは、メンバーの少なくとも1つがそれに標的に対する親和性を与える構造を持つよう十分大きくなければならない。相互作用ライブラリーの必要とされる絶対的なサイズを測るのは難しいが、自然が免疫応答でヒントを与えてくれる。すなわち、多様な107ないし108個の異なる抗体は、生物が直面する大部分のあり得る抗原と相互作用するに十分な親和性を有する少なくとも1つの組合せを与える。また、公表されているインビトロ選択技術も、107ないし108のライブラリーサイズが、標的親和性を有する構造を見出すに十分であることを示している。本明細書で一般に提案されるものなど7ないし20アミノ酸長のペプチドのすべての組合せに関するライブラリーは、207(109)ないし2020をコードする可能性を有する。従って、107ないし108の異なる分子のライブラリーを用い、本方法では理論的に完全な相互作用ライブラリーの「実働」サブセットをアミノ酸7個とし、形態サブセットを2020とする。このように好ましい態様では、主題の方法において少なくとも106、好ましくは107、より好ましくは少なくとも108、最も好ましくは少なくとも109個の異なる配列が同時に解析される。好ましい方法はライブラリーのサイズおよび多様性を最大にする。
【0089】
該候補生物活性剤は細胞または細胞集団に結合または付加される。種々の態様に好適な細胞種は先に概説されている。該候補生物活性剤と細胞は結合させる。当業者には理解されるであろうが、これは候補薬剤を細胞表面へ、細胞を含む培地へ、または細胞が増殖している、もしくは接触している表面へ該候補薬剤を添加する。例えば、薬剤を細胞に導入するベクターを用いることにより該薬剤を細胞に付加する(ずなわち、薬剤が核酸またはタンパク質である場合)ことをはじめ、多くの方法のいずれで達成してもよい。
【0090】
好ましい態様では、該候補生物活性剤は、ウイルスベクターをはじめとするベクターを用いて宿主細胞へ導入される核酸またはタンパク質(この場合タンパク質とはタンパク質、オリゴペプチドおよびペプチドを含む)のいずれかである。ベクター、好ましくはウイルスベクターの選択は細胞種による。細胞が複製する場合、以下にさらに十分記載されるようにレトロウイルスベクターを用いる。細胞が複製しない場合(すなわちある増殖期で拘束されている場合)には、レンチウイルスおよびアデノウイルスベクターをはじめとするその他のウイルスベクターを使用してもよい。
【0091】
好ましい態様では、細胞は複製するかまたは複製を誘導できるかのいずれかであり、PCT US87/01019およびPCT US87/01048に一般に概説されているように、レトロウイルスベクターを使用して候補生物活性剤を細胞に導入する。なおこの双方は出典明示により本明細書の一部とする。一般に、レトロウイルスベクターのライブラリーはヘルパーは欠くが、gag、polおよびenvをはじめとする必要なすべてのトランスタンパク質、ならびにシスにψパッケージングシグナルを有するRNA分子を産生できるレトロウイルスパッケージング細胞系統を用いて作製される。便宜には、レトロウイルスDNA構築主鎖にライブラリーを作製し、当技術分野において十分に公知である技術を用いて標準的なオリゴヌクレオチド合成を行い、候補薬剤またはタンパク質、例えばランダムペプチドをコードする核酸のいずれかを作製する。DNAライブラリーを作製した後、該ライブラリーを第1のプライマーにクローン化する。第1のプライマーはレトロウイルス構築物に挿入される「カセット」として機能する。第1のプライマーは一般に、例えば、必要とされる調節配列(例えば、翻訳、転写、プロモーターなど)、融合パートナー、制限エンドヌクレアーゼ(クローニングおよびサブクローニング)部位、拘束コドン(好ましくは3フレームすべての)、第2鎖プライミングのための相補性領域(好ましくは停止コドン領域の末端に小さな欠失または挿入としてランダムな領域に起こり得る)などをはじめとするいくつかのエレメントを含む。
【0092】
次いで、第2のプライマーを加えるが、これは一般に相補性領域のいくつかまたはすべてからなり、第1のプライマーおよびサブクローニングのための第2の唯一の制限部位に必要な任意の配列をプライミングする。DNAポリメラーゼを加えて二本鎖オリゴヌクレオチドを作製する。この二本鎖オリゴヌクレオチドを適当なサブクローニング制限エンドヌクレアーゼで切断して以下に記載する標的レトロウイルスベクターにサブクローン化する。
【0093】
いずれの数の好適なレトロウイルスベクターを使用してもよい。一般に、レトロウイルスベクターは、以下でさらに十分に記載される選択マーカー遺伝子、5'LTRに対してセンスまたはアンチセンスに置かれた、第2の遺伝子を発現させるプロモーター、SINにおけるCRUS(合成LTR)テトラサイクリン調節エレメント、細胞特異的プロモーターなどを含み得る。
【0094】
好ましいレトロウイルスベクターとしては、ネズミ幹細胞ウイルス(MSCV)(Hawley et al., Gene Therapy 1:136(1994)参照)および改変MFGウイルス(Rivere et al., Genetics 92:8733(1995))およびPCT US97/01019に概説されたpBABEを基にしたベクターが挙げられる。
【0095】
レトロウイルスは候補薬剤の発現のための誘導プロモーターおよび構成プロモーターを含み得る。例えば選択過程のある相の間でのみ、またはある細胞相においてのみペプチド発現を誘導する必要がある場合(すなわち、E2F応答プロモーター、p53応答プロモーター、サイクリンプロモーターなどの相特異的プロモーターを用いる場合)がある。誘導プロモーターおよび構成プロモーター双方の多数のもが知られている。
【0096】
さらに、標的細胞における単一ベクターの組み込み後にレトロウイルス挿入部の誘導発現が可能となるようにレトロウイルスベクターを配置することができるが、完全な系が単一のレトロウイルスに含まれていることが重要である。3’LTRエンハンサー/プロモーターレトロウイルス欠失変異体の自己不活化(SIN)特性を組み込んだTet誘導性のレトロウイルスが設計されている(Hoffman et al., PNAS USA 93:5185(1996))。細胞におけるこのベクターの発現はテトラサイクリンまたは他の有効な類似体の存在下では実質的には検出できない。しかしながら、Tetの不在下では誘導後48時間以内に発現が最大を示し、誘導可能なレトロウイルスを含む細胞の全集団の発現が一様に増加するが、このことは、感染細胞集団内では発現が一様に調節されるということを示す。同様に、関連する系は、Tetの存在下でDNAに結合するが、Tetの不在下では除去されるような変異TetDNA結合ドメインを使用する。これらの系はいずれも好適である。
【0097】
好ましい態様では、候補生物活性剤は融合パートナーに結合している。本明細書において「融合パートナー」または「機能的グループ」とは、候補生物活性剤と会合し、そのクラスのライブラリーのすべてのメンバーに共通の機能または能力を付与する配列を意味する。融合パートナーは異種(すなわち、宿主にとり天然でない)、または合成のもの(いずれの細胞にとっても天然でない)であり得る。好適な融合パートナーとしては、限定されるものではないが、a)候補生物活性剤を立体配位上制限されまたは安定な形で提供される以下に定義の提示構造、b)候補生物活性剤を細胞下または細胞外区画へ局在化させる、以下に定義される標的配列、c)候補生物活性剤かまたはそれらをコードする核酸のいずれかの精製または単離を可能にする以下に定義のレスキュー配列、d)候補生物活性剤またはそれをコードする核酸に安定性または分解からの保護、例えばタンパク質分解耐性を付与する安定配列、e)ペプチド二量化を可能にする二量化配列、あるいはf)a)、b)、c)、d)およびe)ならびに要すればリンカー配列のいずれかの組合せが挙げられる。
【0098】
好ましい態様では、融合パートナーは提示構造である。本明細書みにおいて「提示構造」または文法上の同義語は、候補生物活性剤と融合した場合に候補薬剤に立体配位的上制限された形をとらせる配列を意味する。タンパク質は立体配位上拘束されたドメインを介して互いに強く相互作用する。当技術分野で公知のように、自由に回転するアミノおよびカルボキシル末端を有する小さなペプチドは強い機能を有していても、かかるペプチド構造の薬理活性剤への変換は、ペプチド模倣合成に関する側鎖の位置を予測できないために困難である。従って、立体配位上拘束された構造でのペプチドの提示は、その後の医薬の製造にも有益であるし、またおそらくペプチドと標的タンパク質とのより高い親和性の相互作用をもたらす。この事実は、細菌ファージ系において生物学的に作製した短いペプチドを用いる組合せライブラリー作製系において認められている。何人かの研究者が、ランダム化されたペプチド構造を提示する小ドメイン分子を構築している。
【0099】
候補生物活性剤は核酸またはペプチドのいずれであってもよいが、提示構造はペプチド候補薬剤とともに用いるのが好ましい。従って合成提示構造、すなわち人工ポリペプチドはランダム化されたペプチドを立体配位上制限されたドメインとして提示できる。一般に、かかる提示構造は、ランダム化されたペプチドのN末端に結合した第1の部分と、そのペプチドのC末端に結合した第2の部分とを含んでなり、すなわち該ペプチドは、以下に概説されるように、変異体を作製できるが提示構造に挿入されている。ランダム化された発現産物の機能的単離を高めるには、標的細胞において発現された場合に最小の生物学的活性しか持たないように提示構造を選択または設計する。
【0100】
好ましい提示構造は、外側のループにそれを提示することにより、ペプチドへの接近性が最大となる。従って、好適な提示構造としては、限定されるものではないが、ミニボディ構造、βシートターン上のループ、構造に重要でない残基がランダム化されている超らせん幹構造、ジンクフィンガードメイン、システイン結合(ジスルフィド)構造、トランスグルタミナーゼ結合構造、環状ペプチド、Bループ構造、らせんバレルまたはらせん束、ロイシンジッパーモチーフなどが挙げられる。
【0101】
好ましい態様では、該提示構造は超らせん構造であり、外側のループでランダム化ペプチドの提示を可能にする。例えば、出典明示により本明細書の一部とする、Myazka et al., Biochem. 33:2362-2373(1994)参照。この系を用いて、研究者らは適当な標的と高い親和性の相互作用が可能なペプチドを単離している。一般に、超らせん構造は6ないし20個の間のランダムな位置にある。
【0102】
好ましい超らせん提示構造は以下のとおりである。
【化8】
【0103】
好ましい態様では、該提示構造はミニボディ構造である。「ミニボディ」は実質的に最小の抗体相補性領域からなる。該ミニボディ提示構造は一般に折り畳まれたタンパク質において三次構造の単一面に沿って提示される2個のランダム化領域を提供する。例えば、Bianchi et al., J. Mol. Biol. 236(2):648-59(1994)およびそこで引用されている文献参照、なお、すべて出典明示により本明細書の一部とする。研究者らはこの最小ドメインが溶液中で安定であることを示し、組合せライブラリーにおいて相選択系を使用して炎症性サイトカインIL−8にKd=10-7といった高い親和性を示すペプチド領域を用いてミニボディを選択している。
【0104】
好ましいミニボディ提示構造は以下のとおりである。
【化9】
【0105】
好ましい態様では、該提示構造は一般に2個のシステイン残基を含む配列であり、その結果ジスルフィド結合が形成され、立体配位上拘束された配列が得られる。この態様は分泌標的配列を用いる場合には特に好ましい。当業者には理解されるであろうが、いずれの数のランダム配列でも、スペーサーまたは連結配列を伴い伴わなくて、システイン残基でフランクされ得る。他の態様では、効果的な提示構造はランダム領域自体によって作出され得る。例えば、ランダム領域は、システイン残基で「処理」するが、適当な酸化還元条件下では提示構造に類似した高度に架橋した構造の立体配位をもたらし得る。同様に、ランダム化領域は制御されてβシートまたはαへリックス構造を与える一定数の残基を含み得る。
【0106】
好ましい態様では、該融合パートナーは標的配列である。当業者には理解されるであろうが、細胞内でのタンパク質の局在化は有効濃度を高め、かつ機能を決定するための単純な方法である。例えば、RAF1はミトコンドリア膜に局在した場合にはBCL−2の抗アポトーシス作用を阻害し得る。同様に、膜に結合したSosはTリンパ球においてRas媒介性のシグナル伝達を誘導する。これらの機構はリガンドの探索領域を限定する原理によるものと考えられ、すなわち、タンパク質の原形質膜への局在化はそのリガンドの探索を細胞質の三次元空間に対して膜付近の二次元空間に限定することとなる。あるいはまた、タンパク質の濃度も局在化という性質により簡単に高められる。核へタンパク質を往復させることによりそれらをより小さい空間に制限し、それにより濃度が高まる。最後に、リガンドまたは標的は特定のコンパートメントに簡単に局在化され得るし、阻害剤も適当に局在化するはずである。
【0107】
このように好適な標的配列としては、限定されるものではないが、発現産物の生物活性を維持しつつ発現産物を所定の分子またはクラスの分子と結合させることができる結合配列(例えば、酵素阻害剤または基質配列を用いてある関連する酵素クラスを標的化する);それ自体またはともに結合しているタンパク質の選択的分解のシグナルを伝達する配列;ならびに、候補発現産物を、a)ゴルジ体、小胞体、核、仁、核膜、ミトコンドリア、葉緑体、分泌小胞、リソソーム、および細胞膜などの細胞下の位置、およびb)分泌シグナルを介する細胞外の位置を含む、細胞の所定の場所へ構成的に局在化させ得るシグナル配列が挙げられる。細胞下への局在化または分泌を介する細胞外への局在化のいずれかが特に好ましい。
【0108】
好ましい態様では、標的化配列は核局在化シグナル(NLS)である。NLSは一般に、NLSが生じる完全なタンパク質を細胞核へ向かわせるように働く、正電荷を有する(塩基性)短いドメインである。SV40(サルのウイルス)ラージT抗原(
【化10】
Kalderon(1984), et al., Cell, 39:499-509;ヒトレチノイン酸受容体−β核局在化シグナル (
【化11】
;NFkB p50(
【化12】
【化13】
【化14】
Dingwall, et al., Cell, 30:449-458 1982 and Dingwall et al., J. Cell Biol., 107:841-849;1988)などの二塩基性NLSをはじめとする多数のNLSアミノ酸配列が報告されている。多数の局在化研究では、合成ペプチドに組み込まれたNLSまたは通常は細胞核に向かわないリレポータータンパク質につぎ足されたNLSは、これらのペプチドおよびリポータータンパク質を核内に集中させることが証明されている。例えば、Dingwall, and Laskey, Ann. Rev. Cell Biol., 2:367-390, 1986; Bonnerot, et al., Proc. Natl. Acad. Sci. USA, 84:6795-6799, 1987; Galileo, et al., Proc. Natl. Acad. Sci. USA, 87:458-462, 1990参照。
【0109】
好ましい態様では、該標的化配列は膜固定シグナル配列である。多くの細胞内事象が原形質で起こるということの他、多くの寄生虫および病原体が膜に結合するので、これは特に有用である。このように、膜結合ペプチドライブラリーは、これらの過程における重要な要素の同定、ならびに有効な阻害剤の発見の双方に有用である。本発明は、ランダムな発現産物を細胞外または細胞質空間で提示する方法を提供する;図3参照。細胞外提示に関しては、ペプチド提示構造のカルボキシル末端に膜固定領域が提供される。ランダムな発現産物領域は細胞表面で発現され、細胞外空間に提示され、他の表面分子(それらの機能に影響する)または細胞外媒体中に存在する分子と結合できるようになる。かかる分子の結合により、その分子と結合するペプチドを発現する細胞に機能を付与できる。細胞質領域は、きわだった特徴がなくてもよいし、または細胞外のランダムな発現産物領域が結合する場合、細胞に機能(キナーゼ、ホスファターゼの活性化、機能を遂行するための他の細胞成分との結合)を付与するドメインを包含することもできる。同様に、ランダムな発現産物を含む領域は、細胞質領域に包含されることが可能で、かつ、貫通膜領域および細胞外領域は不変のままであるか、または明らかな機能を有する。
【0110】
膜固定配列は、当技術分野で十分公知であり、哺乳類の貫通膜分子の遺伝的幾何学に基づく。ペプチドは、シグナル配列(本明細書ではssTMと表す)に基づいて膜に挿入され、疎水性貫通膜ドメイン(本明細書ではTM)を必要とする。この貫通膜タンパク質は、貫通膜ドメインの5'側にコードされる領域は細胞外に、3'部分をコードする領域は細胞内になるように膜に挿入する。もちろん、これらの貫通膜ドメインが可変領域の5'側に置かれた場合には、それらは細胞内ドメインとしてそれを固定する働きをするであろうし、これはいくつかの態様においては望ましいものであり得る。ssTMおよびTMは、多様な膜結合タンパク質として公知であり、従ってこれらの配列は、特定のタンパク質由来の対として、または各成分を異なるタンパク質から取って使用してもよく、あるいは該配列は人工送達ドメインとして合成してもよいし、また全体が共通配列に由来してもよい。
【0111】
当業者には理解されるであろうが、ssTMおよびTM双方を含む膜固定配列は多様なタンパク質について公知であり、これらのうちいずれを用いてもよい。特に好ましい膜固定配列としては、限定されるものではないが、CD8、ICAM−2、IL−8R、CD4およびLFA−1が挙げられる。
【0112】
有用な配列としては、1)IL−2受容体β鎖(残基1−26はシグナル配列、241−265は貫通膜残基;Hatakeyama et al., Science 244:551(1989) and von Heijne et al., Eur. J. Biochem. 174:871(1988)参照)およびインスリン受容体β鎖(残基1−27はシグナル配列、957−959は貫通膜ドメイン、および960−1382は細胞質ドメイン;Hatakeyama, supra, and Ebina et al., Cell 40:747(1983)参照)などのクラスI内在性膜タンパク質;2)中性エンドペプチダーゼ(残基29−51は貫通膜ドメイン、2−28は細胞質ドメイン;Malfroy et al., Biochem. Biophys. Res. Commun. 144:59(1987)参照)などのクラスII内在性膜タンパク質;3)ヒトシトクロムP450 NF25(Hatakeyama, supra)などのタイプIIIタンパク質;ならびに4)ヒトP−糖タンパク質(Hatakeyama, supra)などのタイプIVタンパク質由来の配列が挙げられる。特に好ましいのはCD8およびICAM−2である。例えば、CD8およびICAM−2由来のシグナル配列は、転写物の最も5'末端に位置する。これらは、CD−8の場合はアミノ酸1−32(
【化15】
【化16】
【化17】
【化18】
【0113】
あるいは、膜固定配列はGPIアンカーを含み、その結果として例えばDAF(
【化19】
アンカー部位に太字のセリンを有する、Homans et al., Nature 333(6170):269-72(1968),およびMoran et al., J. Biol. Chem. 266:1250(1991)参照)においては、グリコシル−ホスファチジルイノシトール結合を介して該分子と脂質二重層の間の共有結合を形成する。これを達成するには、Thy−1由来のGPI配列を貫通膜配列の代わりに可変領域の3'にカセットとして挿入できる。
【0114】
同様に、ミリスチル化配列は膜固定配列として働き得る。c−srcのミリスチル化によりそれが原形質膜に補充されることが知られている。該タンパク質の最初の14個のアミノ酸:
【化20】
【化21】
【化22】
【化23】
【0115】
好ましい態様では、該標的化配列は、例えばLamp−2のようなリソソーム分解配列(KFERQ;Dice, Ann. N.Y. Acad. Sci. 674:58(1992);またはLamp−1由来のリソソーム膜配列(
【化24】
【化25】
Koneckl et al., Biochem. Biophys. Res. Comm. 205:1-5(1994)、双方ともイタリック体は貫通膜ドメイン、下線は細胞質標的シグナルを示す)をはじめとするリソソーム標的化配列である。
【0116】
あるいは、該標的化配列は、ミトコンドリア・マトリックス配列(例えば、酵母アルコールデヒドロゲナーゼIII;
【化26】
【化27】
【化28】
【化29】
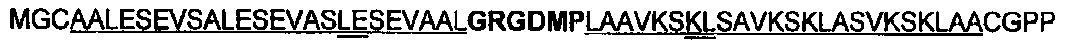
該標的配列はまた、カルレチクリン由来配列(KDEL;Pelham, Royal SocietyLondon Transactions B; 1-10(1992))またはアデノウイルスE3/19Kタンパク質由来の配列(
【化30】
【0118】
さらに標的配列はまた、ペルオキシソーム配列(例えば、ルシフェラーゼ由来のペルオキシソームマトリックス配列;SKL;Ketler et al., PNAS USA 4:3264(1987));ファルネシル化配列(例えば、P21 H-ras1;
【化31】
【化32】
【化33】
【0119】
好ましい態様では、該標的化配列は候補翻訳産物の分泌に作用できる分泌シグナル配列である。可変ペプチド領域の5'側に置かれた公知の分泌シグナル配列は多数あり、ペプチド領域から切断されて細胞外空間への分泌を果たす。分泌シグナル配列とそれらの関連のないタンパク質への転移能力は十分公知である。例えば、Sllhavy, et al., (1985) Microbiol. Rev. 49, 398-418。これは、宿主細胞以外の、例えば該ペプチドを発現する細胞のような標的細胞の表面に結合できる、またはその生理学に作用できるペプチドを作製するのに特に有用である。好ましい試みにおいて、融合産物は一連の分泌シグナルペプチド−提示構造−ランダムな発現産物領域−提示構造を含むように配置する。この方法において、ペプチドライブラリーを発現した細胞の近傍で増殖した標的細胞は、分泌したペプチドに浸される。標的細胞はペプチドの存在に応答して、例えばペプチドが表面受容体に結合するか、または取り込まれて細胞内の標的に結合することにより生理学的変化を示し、種々の分泌スキームのいずれかによって分泌細胞が局在化し、該ペプチドが所定の作用を引き起こす。作用の例としては、デザイナーサイトカイン(すなわち、造血幹細胞を分裂させ、それらの全能性を維持できる幹細胞因子)、癌細胞を自発的アポトーシスに導く因子、標的細胞の細胞表面に結合してそれらを特異的に標識する因子など様々なものが挙げられる。
【0120】
IL−2(
【化34】
【化35】
【化36】
【化37】
【化38】
好ましい態様では、該融合パートナーはレスキュー配列である。レスキュー配列は、候補薬剤またはそれをコードする核酸のいずれかを精製または単離するために使用できる配列である。このようにペプチドレスキュー配列としては、例えば、Ni親和性カラムと併用するHis6タグ、および検出、免疫沈降またはFACS(蛍光活性化細胞分別)のためのエピトープ標識などの精製配列が挙げられる。好適なエピトープタグとしては、myc(市販の9E10抗体と併用)、細菌酵素BirAのBSPビオチン化標的配列、fluタグ、lacZおよびGSTが挙げられる。
【0122】
あるいは、レスキュー配列は、PCR、関連技術またはハイブリダイゼーションによるレトロウイルス構築体の迅速かつ容易な単離を可能にするプローブ標的部位として働く特異なオリゴヌクレオチド配列であってもよい。
【0123】
好ましい態様では、該融合するパートナーは、候補生物活性剤またはそれをコードする核酸に安定性を付与する安定配列である。このように、例えば、VarahavskyのN−End則に従い、ペプチドのユビキチン化を保護するため、開始メチオニン(MGまたはMGGO)の後にグリシンを組み込むことによりペプチドは安定化され、このようにして細胞質中での長い半減期が付与され得る。同様に、C末端の2個のプロリンは、ペプチドにカルボキシペプチダ−ゼの作用に対する強い耐性を付与する。プロリンの前に2個のグリシンが存在すると、可変性が付与されるとともに、2個のプロリンで一連の事象が起こる構造候補ペプチド構造へ伝わるのを防げる。このような好ましい安定配列を以下に示す:MG(X)nGGPP、ここでXはいずれかのアミノ酸であり、nは少なくとも4の整数である。
【0124】
1つの態様では、該融合パートナーは二量化配列である。二量化配列により、通常の生理学的条件下で結合を維持するに十分な親和性で、あるランダムなペプチドと別のランダムなペプチドとの非共有結合が可能となる。このことにより、細胞あたり2個のペプチドが生じ、次いで二量化して108(104×104)の有効なライブラリーが形成されるとすると、ランダムペプチドの小さなライブラリー(例えば、104)が効果的に大きなライブラリーとなる。それはまた、要すればより長いランダムペプチドの形成も可能にし、あるいはより構造的に複雑なランダムペプチド分子の形成も可能にする。この二量体は、ホモ二量体であってもヘテロ二量体であってもよい。
【0125】
二量化の配列は、自己凝集する単一の配列、または2つの配列であってもよく、各配列は異なるレトロウイルス構築体内で生成する。すなわち、二量化配列1を持つ第一のランダムペプチドおよび二量化配列2をもつ第二のランダムペプチド双方をコードする核酸は、細胞への導入と該核酸の発現に基づき、二量化配列1が二量化配列2と会合して新らしいランダムペプチド構造を形成するようにしたものである。
【0126】
適切な二量化配列は広範な配列を包含する。相当数のタンパク質−タンパク質相互作用部位が知られている。さらに、二量化配列はまた酵母の2ハイブリッドシステム、伝統的な生化学的親和性結合研究法などの標準的方法、あるいは本発明方法を用いても解明することができる。
【0127】
融合パートナーは生物学と活性が許す限り、構造中のいずれの部分(すなわち、N−末端、C−末端、内部)にあってもよい。
好ましい態様において、融合パートナーは、PCT US97/01019に一般的に記載されているように、リンカーまたはつなぎ鎖配列を包含して、妨害されていない潜在的標的と候補薬剤が相互作用するのを可能とする。例えば、候補生物活性剤がペプチドである場合、有用なリンカーはグリシン−セリン・ポリマー(例えば、(GS)n、(GSGGS)nおよび(GGGS)nを包含し、nは少なくとも1の整数である)、グリシン−アラニン・ポリマー、アラニン−セリン・ポリマー、およびシェーカー型カリウムチャンネルのつなぎ鎖などの他の柔軟性リンカー、さらに当業者が認める多様な他の柔軟性リンカーを包含する。グリシン−セリン・ポリマーは、このアミノ酸の両方が比較的非構造的であり、それ故に成分間の中性的つなぎ鎖として働くことができるので、好適である。第二に、セリンは親水性であり、したがって、球状のグリシン鎖であり得るものを可溶化することが可能である。第三に、同様の鎖は、例えば、一本鎖抗体などの組換えタンパク質のサブユニットを連接するのに有効であることが示されている。
【0128】
さらに、提示構造を含む融合パートナーは、修飾、ランダム化、および/または成熟化してランダム化発現産物の提示方向を変えることもできる。例えば、ループ塩基の決定因子を修飾し、ランダム化アミノ酸配列を保持した内部ループペプチドの三級構造を僅かに修飾することが可能である。
【0129】
好ましい態様においては、融合パートナーを合わせて用いる。このように、例えば、提示構造、標的配列、レスキュー配列および安定配列の多様な合わせをリンカー配列の存在下または不存在下に使用することができる。
【0130】
このように、候補薬剤はこれらの成分を含むことが可能であり、したがって、各断片が異なるペプチドをコードし得る異なるランダムヌクレオチド配列を含む断片ライブラリーの生成に使用することができる。連結反応産物は次いで大腸菌などのバクテリアに形質転換し、一般手法として概括されている(Kitamura, PNAS USA 92: 9146-9150 (1996); 出典明示により本明細書の一部とする)ように、得られるライブラリーからDNAを調製する。
【0131】
レトロウイルスパッケージシステムにライブラリーDNAを導入すると、感染性ウイルスに転換する。適切なレトロウイルスパッケージシステムは、これらに限定されるものではないが、ビング(Bing)およびBOSC23細胞株(WO94/19478;Soneoka et al., Nucleic Acid Res. 23 (4): 628 (1995); Finer et al., Blood 83: 43 (1994)に記載)、フェオニックス(Pheonix)パッケージ株、例えば、PhiNX−ecoおよびPhiNX−ampho(下記)、292T+gag−polおよびレトロウイルスエンベロープ、PA317、および細胞株(Markowitz et al., Virology 167: 400 (1988), Markowitz et al., J. Virol. 62: 1120 (1988); Li et al., PNAS USA 93: 11658 (1996); Kinsella et al., Human Gene Therapy 7: 1405 (1996)に概説)を包含する(これらの文献すべてを出典明示により本明細書の一部とする)。好ましいシステムはPCT/US97/01019に開示されたPhiNX−ecoおよびPhiNX−amphoまたは類似の細胞株である。
【0132】
細胞が複製しない場合は、他のウイルスベクター、例えば、アデノウイルスベクター、ネコ免疫ウイルス(FIV)ベクターなどを使用してもよい。
好ましい態様において、候補薬剤をウイルスベクターにより細胞に導入する場合には、候補ペプチド薬剤を検出可能な分子に連結するが、本発明方法は少なくとも1つの発現アッセイ法を包含する。発現アッセイ法は、候補生物活性剤が発現されたかどうか、すなわち、候補ペプチド薬剤が細胞内に存在するかどうかの決定を可能とするアッセイ法である。このように、標識などの検出可能な分子の発現に候補薬剤の発現を連結することにより、候補ペプチド薬剤の存在または不存在を決定し得る。したがって、この態様において、候補薬剤は検出可能な分子に操作可能に連結される。一般に、これは融合核酸を創製することにより実施される。融合核酸は候補生物活性剤(上に概説したように融合パートナーを含んでもよい)をコードする第一核酸と検出可能な分子をコードする第二核酸を含んでなる。「第一」および「第二」という用語は、融合核酸の5’−3’方向に関して配列の方向を付与することを意味するものではない。例えば、融合配列の5’−3’方向を仮定すると、第二核酸に対し5’、または第二核酸に対し3’に位置してもよい。本態様において好ましい検出可能な分子は、これらに限定されるものではないが、蛍光タンパク質、例えば、GFP、YFP、BFPおよびRFPなどを包含し、取分け前者が好ましい。
【0133】
一般に、候補薬剤は、薬剤−標的相互作用を好む反応条件下に細胞に加えられる(上に概説したように、細胞外または細胞内のいずれか)。一般に、これは生理的条件である。インキュベーションは最適活性を容易とする温度で実施し得るが、典型的には4℃および40℃の間である。インキュベーション時間は最適活性のために選択されるが、迅速な高処理能力スクリーンを容易にするように最適化してもよい。典型的には、0.1と1時間の間で十分である。過剰の試薬は一般に除去または洗い流す。
【0134】
様々な他の試薬類を本アッセイ法に含めることができる。これらは塩、中性タンパク質、例えば、アルブミン、洗剤などを包含し、これらは最適なタンパク質−タンパク質結合を容易にし、および/または非特異的もしくは共通の相互作用を低減させるために用いる。また、その他アッセイの効率を改善する試薬、例えば、プロテアーゼ阻害剤、ヌクレアーゼ阻害剤、抗微生物剤などを用いてもよい。成分の混合物は検出に必要な順番で添加する。細胞の洗浄またはすすぎは異なる時点で当業者が認めるように実施するが、濾過および遠心分離の使用も包含する。第二標識部位(本明細書では「第二標識体」ともいう)を用いる場合、それらの部位は、好ましくは、非特異的結合を減らすために、過剰の非結合標的分子を除去した後に添加する。しかし、一部の条件下では、成分のすべてを同時に加えてもよい。
【0135】
好ましい態様において、細胞は蛍光細胞活性化セルソーター(FACS)により分別する。本発明において、細胞周期規制は、共通事項を低下させ、特異的性を高めるような複数のパラメーターにより評価する。それに対し、FACSは、2つの異なるまたは無関係の特性を同時に評価するためにこれまで用いられてきたが、これら2つの特性を有する細胞を同定はするが、合わされた特性の共通事項を低下させるものではない。
【0136】
このように、細胞は1種以上のアッセイ法、例えば、細胞生存能アッセイ、増殖アッセイ、細胞相アッセイ、および(候補薬剤が検出可能な部分と共に発現される場合の)発現アッセイなどに基づきFACSにおいて分別または濃厚化される。1種以上のこれらアッセイ法からの結果は、候補生物活性剤に暴露しなかった細胞と、または候補薬剤での誘導前の同一細胞と比較する。これらの結果の変化は、当該薬剤が細胞周期規制を調節することを示している。
【0137】
本発明の利点は、候補薬剤ライブラリーが細胞のライブラリーで試験し得るということであり、その理由は本方法が、非常に稀な事象を検出し得るような非常に高い特異的性によって単一細胞の分別を可能とするからである。複数のレーザー経路を使用することにより、70%を超える精度で10 6 中1の分別精度を可能とする。
【0138】
さらに、本発明は、複数の細胞周期規制の同定法に加えて、他の細胞特性の同定法と合わせることができる。例えば、全般的な細胞健常性のパラメーターは、例えば、カルシウム応答を示す色素インド−1を用いることにより測定および選択することができる。当業者が常套的に同定する他の細胞性パラメーターは、これらに限定されるものではないが、細胞の大きさ、細胞の形、レドックス状態、DNA含量、核酸配列、染色質構造、RNA含量、総タンパク質、抗原、脂質、表面タンパク質、細胞内受容体、酸化性代謝、DNAの合成と分解、および細胞内pHなどである。
【0139】
好ましい態様において、各測定値は細胞が複数レーザーのビーム経路を通過する際に、個々の細胞から同時に決定される。あるいは、測定は連続的になされる。細胞周期規制または細胞周期規制における変化を検出するための1種以上のパラメーターを使用することにより、共通事項を減少させ、特異的性を高める。所望性質のパラメーターに合致する細胞を、所望パラメーターに合致しない細胞から物理的に分別することができるか、またはそれらの細胞は細胞集団中の百分比により同定することができる。
【0140】
一般に、Koは≦1μMであることが好ましく、FACS分別に存在する剪断力の存在下に結合の保持を可能とする。好ましい態様において、細胞は非常な高速で分別される、例えば、その速度は毎秒約5,000を超える分別事象、好ましくは毎秒約10,000を超える分別事象、特に好ましくは毎秒約25,000を超える分別事象、取分け好ましいのは、約50,000〜100,000を超える速度である。
【0141】
刺激および染色用に処理加工した細胞は、一般に緩衝液に取り込み、サイトメトリーに先立ち濾過する。細胞はFACSCAN(ベオトン・ディキンソン・インク、レーザー線488nm)またはMo−Flo(サイトメーション、インク、レーザー線350nM幅広いバンド(UV)、488nmおよび647nm)サイトメーターにより分析することができる。細胞は、所望により、Mo−Floにより分別する。
【0142】
細胞を顕微鏡使用により分析する場合、刺激または染色後の細胞は通常スライドガラスに載せ、ドーバースリップさせる。これらは倒立顕微鏡(すなわち、TE300、ニコン)上、標準のBFP、FITC、またはTRITC(例えば)フィルターセットを用いる明視野および蛍光顕微鏡検査により直接可視化する。また、画像は倒立共焦点走査顕微鏡(ツアイス、インク、バイオ−ラッド、インク)により、標準のFITCおよびTRITC(例えば、)フィルターセットを用いることができる。
【0143】
分別により所望の性質を有する細胞の集団を得る。好ましい態様において、該パラメーターは細胞周期規制を調節する少なくとも1つの候補生物活性剤を同定するために設定する。
【0144】
好ましい態様においては、該生物活性剤を特徴づける。これは当業者の認める通りに進め、通常、活性剤の構造分析、同定、結合親和性および機能を包含する。一般に、同定すると、該生物活性剤は再合成され、標的細胞と合わせて、種々の条件下、また、他の種々の生物活性剤の存在下または不存在下に細胞周期規制変調を確認する。該生物活性は細胞周期規制を調節するために治療有効量を調製し、適切な医薬担体と合わせる。
【0145】
好ましい態様において、細胞集団は候補薬剤の存在下または不存在下に種々の実験条件に付すことができる。条件の変化はpH、温度、緩衝液または塩濃度などであるが、これらに限定されるものではない。好ましい態様において、pHは一般にpHの増減により、通常、約0.5ないし約3のpH単位で変化させる。あるいは、温度の変更は、好ましくは、約5℃ないし約30℃の上昇または下降により行う。同様に、塩濃度は、好ましくは、約0.1Mないし約2Mの増減により変更する。
【0146】
当業者は理解するところであるが、本明細書に提供されるアッセイの段階は順序を変えることができる。しかし、さらに理解し得ることは、種々の選択肢(選択された化合物、性質、または段階の順序について)が本明細書において提供される一方、その選択肢は、それぞれが個々に提供されるものであり、それぞれが本明細書に提供された他の選択肢から個々に分離されるものである。さらに、アッセイの感度を上げる技術上明瞭な既知の工程は、本発明の範囲内にある。例えば、さらには、洗浄工程、または分離、単離工程があってもよい。さらに理解されることは、ある場合には、検出が細胞内でのものであるが、培地中でも起こり得るものであり、その逆もある。
【0147】
好ましい態様において、細胞の表現型はエキソサイトーシスであり、本発明の方法と組成物はエキソサイトーシスの変化の検出を指向するものであって、再びFACS機器を使用する。そこには多くのパラメーターがあり、それを評価または検定してエキソサイトーシス経路での変化を検出し得るが、その検出は光分散、蛍光色素取込み、蛍光色素放出、顆粒暴露、表面顆粒酵素活性、および顆粒特異的タンパク質の量などを包含するが、これらに限定されるものではない。これらパラメーターの一つ以上を検定または測定することにより、エキソサイトーシスの変化のみならず、エキソサイトーシス経路の異なる段階での変化をも検出することが可能である。さらに、複数パラメーター分析は、共通事項、または検出される「偽陽性」を低下させる。この様式において、候補薬剤の迅速で正確なスクリーンが、エキソサイトーシスを調節する薬剤を同定するために実施し得る。
【0148】
好ましい態様において、本発明は細胞集団のエキソサイトーシスにおける変化をスクリーンする方法を提供する。エキソサイトーシスと関連する「変化」または「変調」が意味するのは、1個の細胞を他のもう1つの細胞に比較したときの、または同一細胞において異なる条件下に置いたときの、エキソサイトーシスの量の増減である。測定値はすべての条件が各測定値について同一である場合について、または様々な条件下に、生物活性剤の存在下または不存在下、またはエキソサイトーシス過程の異なる段階で決定することができる。例えば、エキソサイトーシスの測定値は細胞集団において、候補生物活性剤が存在する場合について、また、候補生物活性剤が存在しない場合について決定することができる。もう一つの例において、エキソサイトーシスの測定値は細胞集団の条件または環境が他のもう一つのものと異なる場合について決定される。例えば、細胞は、生理的シグナル、例えば、エキソサイトーシス誘発物質(すなわち、Ca++、イオノマイシンなど)、ホルモン、抗体、ペプチド、抗原、サイトカイン、成長因子、作用タンパク質、または他の細胞(すなわち、細胞−細胞接触)などの存在下または不存在下に評価してもよい。他の例において、エキソサイトーシスの測定値はエキソサイトーシス工程の異なる段階で定量する。さらに他の例において、エキソサイトーシスの測定値は、条件が同じで、その変化が1個の細胞または細胞集団、およびもう一つの細胞または細胞集団の間にある場合について測定する。
【0149】
本明細書において「細胞集団」とは、上記定義の細胞のサンプルを意味する。この態様において、細胞は、好ましくは(要件ではない)、急速に増殖し、レトロウイルスに感染性であり、かつ、色素および抗体と適合し得るものである。この態様において使用する好ましい細胞型は、これらに限定されるものではないが、肥満細胞、神経細胞、副腎クロム親和性細胞、好塩基球、膵臓β細胞を含む内分泌細胞、外分泌細胞を含む膵臓腺房細胞、好中球、単球、リンパ球、乳房細胞、精子、卵子細胞とPMN白血球、内皮細胞、脂肪細胞、および筋肉細胞などを包含する。
【0150】
エキソサイトーシス法は細胞の少なくとも3種の性質における変化を検定することにより、FACS機器において細胞を分別することからなるが、その性質は光分散、蛍光色素取込み、蛍光色素放出、顆粒暴露、表面顆粒酵素活性および顆粒特異的タンパク質の量からなる群から選択される。好ましい態様において、各測定値は細胞が複数レーザーのビーム経路を通過する際に、個々の細胞から同時に決定される。あるいは、測定は連続的になされる。エキソサイトーシスまたはエキソサイトーシスにおける変化を検出するたの1種以上のパラメーターを使用することにより、共通事項を減少させ、特異的性を高める。所望性質のパラメーターに合致する細胞を、所望パラメーターに合致しない細胞から物理的に分別することができるか、またはそれらの細胞は細胞集団中の百分比により同定することができる。
【0151】
好ましい態様において、光分散の変化を検定し、細胞集団中のエキソサイトーシス変化を定量する。FACSで検視するとき、細胞はその前方および90度(側面)への光分散性により測定される特別な性質をもつ。これらの分散性は細胞の大きさ、形および顆粒含量を示している。プロ−エキソサイトーシス刺激により細胞を活性化すると、細胞の前方および側面光分散性双方が相当に変化する。これらの性質はエキソサイトーシス事象に対する読み出し値として測定されるべき2つのパラメーターとみなされる。これらの性質は細胞のエキソサイトーシスの度合いに比例して変化し、同様に経時的エキソサイトーシス事象に依存する。光分散強度の変化、または細胞屈折率の変化は、異なる時点で同じ細胞において、または異なる条件下または候補生物活性剤の存在下もしくは不存在下に同じ細胞に比較して、あるいは異なる細胞または細胞集団に比較して、エキソサイトーシスの変化を示すものである。
【0152】
本明細書に提供した一態様において、細胞集団はエキソサイトーシスを刺激することが知られている薬剤と合わせ、光分散性を定量する。望ましいエキソサイトーシス活性を示す光分散性の細胞を同定および/または分別し得る。本明細書にて使用するエキソサイトーシス活性とは活性を欠く場合も包含する。好ましい態様において、候補生物活性剤はエキソサイトーシス刺激に先立ってまたは同時に細胞集団と併合する。より詳しくは下記に概説する。この態様において、光分散性が、a)既知のエキソサイトーシス刺激剤および候補生物活性剤と合わせた細胞集団と、b)候補生物活性剤が存在しない場合の既知エキソサイトーシス刺激剤と合わせた細胞集団と、で相違する場合、候補生物活性剤はエキソサイトーシスを調節すると決定することができる。ある場合には、エキソサイトーシスを誘導する生物活性剤を同定するために、エキソサイトーシスの阻害剤を含ませるか、またはエキソサイトーシス刺激剤を排除することが望ましいこともある。好ましくは、光分散性は、エキソサイトーシス活性を示す少なくとも1つ、好ましくは他の2つの性質と合わせて測定する。一般的な光分散測定についての方法は、さらに以下の文献に記載されている:Perretti, et al., J. Pharmacol. Methods, 23 (3) 187-194 (1990)およびHide et al., J. Cell Biol., 123 (3) 585-593 (1993)(出典明示により両文献を本明細書の一部とする)。一般に、ベースラインから少なくとも約5%の変化が好ましく、より好ましくは少なくとも約25%であり、特に好ましくは少なくとも約50%であり、そして取分け好ましいのは少なくとも約75%ないし100%である。この場合のベースラインとは、エキソサイトーシス刺激前の細胞の光分散性を一般に意味する。本明細書に提供した各事例において、ベースラインは対照のパラメーターについて設定してもよい。例えば、ベースラインは特定細胞、異なる条件下での類似細胞のエキソサイトーシス測定にて、またはエキソサイトーシスの際の特定時点において設定してもよい。
【0153】
もう一つの好ましい態様においては、蛍光色素取込みの変化を評価する。好ましい蛍光色素はスチリル色素であり、これはエンドサイトーシスに関連してエキソサイトーシス活性を示す色素であり、場合により共役エンドサイトーシスという。共役エンドサイトーシスを支持する理論は、エキソサイトーシスを受ける細胞は細胞容積と膜の完全性を維持するために、エンドサイトーシスをも受けなければならないということである。このように、エキソサイトーシスの刺激に基づき、エンドサイトーシスも増大し、スチリル色素取込み量を定量することにより間接的なエキソサイトーシス測定値を得る。
【0154】
本明細書にて提供される態様においては、細胞はスチリル色素溶液に漬け、プロ−エキソサイトーシス刺激剤により刺激し、色素を定量する。好ましくは、エキソサイトーシス刺激の後、細胞を遠心し、吸引し、新たな緩衝液に再懸濁する。好ましい態様において、候補生物活性剤は本明細書に記載のように細胞と合わす。ある事例において、候補生物活性剤はエキソサイトーシスの阻害剤をもつ細胞と、またはプロ−エキソサイトーシス刺激剤をもたない細胞と合わせることができる。好ましくは、プロ−エキソサイトーシス刺激剤は細胞集団に加えるが、この集団では色素の蛍光シグナルが劇的に増大するに至る。細胞関連のシグナル増大はスチリル色素の共役エンドサイトーシスによるものであり、時間と強度においてエキソサイトーシス応答に比例する。逆に、このシグナルはエキソサイトーシスが阻害されるか、または誘導されなければ増大しない。変化は、異なる時点で、または異なる細胞集団において蛍光を測定し、その測定値を互いにまたは標準値に比較して定量する。一般に、ベースラインから少なくとも約50%の変化が好ましく、より好ましくは少なくとも約75%〜100%であり、特に好ましくは少なくとも約250%であり、そして取分け好ましいのは少なくとも約1000%〜2000%である。この場合のベースラインとは、エキソサイトーシス刺激前の細胞のスチリル色素取込みを意味する。
【0155】
好ましいスチリル色素は、FM1−43、FM4−64、FM14−68、FM2−10、FM4−84、FM1−84、FM14−27、FM14−29、FM3−25、FM3−14、FM5−55、RH414、FM6−55、FM10−75、FM1−18、FM9−49、FM4−95、FM4−59、FM9−40、およびその合わせなどを包含するが、これらに限定されるものではない。FM1−43などの好ましい色素は水中で僅かに蛍光性であるが、膜と会合した場合には非常に蛍光性であり、その結果、色素の取込みが容易に識別できる。適切な色素は市販入手可能である。すなわち、モレキュラー・プローブ・インク、ユージン、オレゴン、「蛍光性プローブと研究試薬の便覧」第6版、1996、特に第17章、およびより詳しくは、第17章、第2項(参照関連の章を含む)(出典明示により本明細書の一部とする)。好ましくは、色素は溶液として提供されるが、色素濃度は約25ないし1000〜5000nMであり、好ましくは約50ないし約1000nMであり、特に好ましくは約50ないし250である。スチリル色素の使用はさらに、Betz, et al., Current Opinion in Neurobiology, 6: 365-371 (1995) (出典明示により本明細書の一部とする)に記載されている。好ましくは、蛍光色素取込みは、少なくとも1種のエキソサイトーシス活性の指示薬、好ましくは2種の他の指示薬と合わせて測定する。
【0156】
もう一つの好ましい態様においては、蛍光色素放出の変化を評価する。本発明は、通常リソゾームの染色に使用される低pH濃度色素が、低pHでエキソサイトーシス顆粒を染色するという発見に一部向けられる。一般に、これらの色素は受動的に細胞により取込まれ、顆粒中に濃縮される;しかし、細胞は色素を取込むように、すなわち、共役エンドサイトーシスにより誘導される。好ましい態様においては、細胞集団を低pH濃度色素に浸け、細胞が色素を取込むようにする。好ましくは、細胞を洗浄する。細胞はプロ−エキソサイトーシス刺激剤および/または阻害剤に暴露することができる。好ましい態様において、候補生物活性剤は細胞集団および、好ましくは、プロ−エキソサイトーシス刺激剤と合わせる。蛍光を評価する。細胞間のまたは同一細胞内での異なる時点での蛍光色素放出の変化はエキソサイトーシスの変化を示す。好ましくは、この変化は、細胞間、最も好ましくは添加した異なる生物活性剤をもつ細胞間にある。一般に、ベースラインから少なくとも約5%の変化が好ましく、より好ましくは少なくとも約25%であり、特に好ましくは少なくとも約50%であり、そして取分け好ましいのは少なくとも約100%である。この場合のベースラインとは、刺激前の細胞中の色素量を一般に意味する。
【0157】
この態様において、低pH濃度色素が好ましい。かかる低pH濃縮色素は、アクリジン・オレンジ、リソトラッカー(LYSOTRACKER)(商標)レッド、リソトラッカー(商標)グリーン、およびリソトラッカー(商標)ブルーなどであるが、これらに限定されるものではない。かかる色素は市販入手可能である。すなわち、上記モレキュラー・プローブからであり、特に、第17章、第17章の第4項、および引用された「関連の章」、すなわち、第23章である。好ましい態様において、該色素は溶液で投与するが、その場合の色素濃度は約50nMないし約25μMであり、好ましくは約5μMないし約25μMであり、そして特に好ましくは約1ないし5μMである。低pH濃度色素の使用については、Haller, et al., Cell Calcium, 19 (2): 157-165 (1996)(出典明示により本明細書の一部とする)に一般的に記載されている(リソゾームの研究に関して)。
【0158】
蛍光色素放出の変化を評価する場合の替わり得る態様においては、上清に放出された蛍光を評価する。この態様においては、エンドサイトーシスした膜を可逆的に標識するスチリル色素、または低pH濃度色素のいずれかを使用する。この態様においては、色素が細胞中に受動的にまたは誘導により取込まれるように、細胞集団を色素に浸ける。次いで、細胞を好適に洗浄する。細胞はプロ−エキソサイトーシス刺激剤および/または阻害剤に、また任意には候補生物活性剤に暴露することができる。プロ−エキソサイトーシス刺激剤に暴露する細胞は細胞外培地に色素を放出する。培地中の蛍光は測定または検出することができる。この過程はある場合に細胞の脱染という。任意には、培地中色素の検出を改善および容易にする薬剤が添加されてもよい。例えば、ミセル形成洗剤、例えば、3−[(3−クロラミドプロピル)ジメチルアンモニオ]−1−プロパン硫酸塩(CHAPS)は蛍光を増加させて、少量のエキソサイトーシス活性の検出を可能とする。色素放出の変化は、同一細胞内での、細胞間での、また、最も好ましくは、添加された異なる生物活性剤をもつ細胞間でのエキソサイトーシスの変化を示すものである。一般に、ベースラインから少なくとも約5%の変化が好ましく、より好ましくは少なくとも約25%であり、特に好ましくは少なくとも約50%であり、そして取分け好ましいのは少なくとも約100%である。この場合のベースラインとは、エキソサイトーシス刺激前の色素の放出を意味する。好ましくは、培地中で測定する場合の色素放出は、少なくとも1種の他のエキソサイトーシス指示薬の評価と合わせる。
【0159】
好ましい態様においては、顆粒暴露での変化を定量する。顆粒はエキソサイトーシスの間、培地に暴露する、すなわち、顆粒は細胞膜と融合し、その内容物を暴露し/放出する。したがって、顆粒暴露はエキソサイトーシス活性を示すものであり、それが欠如することはエキソサイトーシスが誘導されなかったか、または阻害されたことを示す。好ましくは、顆粒暴露は顆粒に特異的的に結合する検出可能な薬剤により検出される。本明細書で使用される検出可能な薬剤の例は、アネキシンVであるが、これは二価イオン依存性様式でリン脂質(ホスホチジルセリン)に特異的的に結合するタンパク質ファミリーの一員である。このタンパク質は、ホスホチジルセリンの細胞表面露出がこの過程の折り紙付き初期シグナルであるため、アポトーシス(プログラムされた細胞死)の測定に広く使用されている。驚くべきことに、アネキシンVは、分泌過程でエキソサイトーシス顆粒が細胞表面に暴露されるとエキソサイトーシス顆粒に特異的的に結合するということが決定されている。細胞の内部にある顆粒は標識されない。アネキシンVのこの性質は、エキソサイトーシス依存性結合に基づく単一のエキソサイトーシスアッセイを創製するために、使用される。細胞のエキソサイトーシス刺激により、細胞は時間と強度においてエキソサイトーシス応答に比例してアネキシン結合および蛍光シグナルの増加を示す。
【0160】
この態様において、アネキシンは直接または間接的に標識し、細胞集団と合わせる。アネキシンは市販品として入手できる。すなわち、ファーミンゲン、サンジエゴ、カリフォルニア、またはカルタック・ラボラトリー、ミルブラエ、カリフォルニアから入手できる。好ましくは、アネキシンは溶液として提供し、その場合のアネキシン濃度は約100ng/mlないし約500ng/mlであり、より好ましくは約500ng/mlないし約1μg/mlであり、最も好ましくは約1μg/mlないし約5μg/mlである。好ましい態様において、アネキシンを直接標識する。例えば、アネキシンは蛍光発色団、例えば、フルオレセインイソチオシアネート(FITC)、アレキサ(Alexa)色素、TRITC、AMCA、APC、トリカラー、Cy−5、および技術上既知または市販入手可能な他の発色団などで標識することができる。替わり得る好ましい態様において、アネキシンは第一標識、例えば、ビオチンなどのハプテンで標識し、また、第二蛍光標識には蛍光ストレプトアビジンなどを使用する。他の第一および第二標識対は当業者が認めるように使用することができる。
【0161】
好ましい態様において、細胞は正常にエキソサイトーシスを引起す条件に付す。任意に、候補生物活性剤が細胞に加えられる。ある事例では、候補薬剤が阻害を逆転し得るかどうかを決めるためにエキソサイトーシスの阻害剤を包含させること、または該薬剤がエキソサイトーシスを誘導するどうかを決めるためにエキソサイトーシス刺激剤なしに候補生物活性剤を加えることが望ましい。細胞は、好ましくは洗浄し、蛍光を顕微鏡で、またはフローサイトメーター上で検出する。アネキシン結合の検出における変化は、同一条件下で、および/またはそれと併用した薬剤の存在下、同一細胞内での、または異なる細胞間でのエキソサイトーシスの変化を示している。一般に、ベースラインから少なくとも約25%の変化が好ましく、より好ましくは少なくとも約50%であり、特に好ましくは少なくとも約100%であり、そして取分け好ましいのは少なくとも約500%である。この場合のベースラインとは、エキソサイトーシス刺激前のアネキシン結合量を意味する。
【0162】
もう一つの好ましい態様において、顆粒暴露はベルベリンまたはルテニウムレッドなどのカチオン性色素により検出する。かかるカチオン性色素は分泌顆粒を特異的的に染色する。このように、エキソサイトーシスが生じ、分泌顆粒が細胞表面で暴露されるとき、蛍光の増大が検出される。好ましい態様において、カチオン性色素はエキソサイトーシス刺激剤および/または阻害剤の存在下または不存在下に、任意には、候補生物活性剤の存在下または不存在下に細胞集団と合わせる。特に好ましい態様においては、候補生物活性剤がエキソサイトーシス活性を調節し得るかどうかを決めるために、ベルベリンを細胞とエキソサイトーシス刺激剤と候補生物活性剤と合わせる。好ましくは、細胞を洗浄し、次いで、蛍光を定量する。好ましい態様において、カチオン性色素の評価は少なくとも1種の他のエキソサイトーシス指示薬の評価と合わせる。色素は技術上知られているとおりに細胞と合わせる。ベルベリンについて記載する一般的な方法論は、Berlin and Enerback, Int. Arch. Allergy Appl. Immunol., 73 (3): 256-262 (1984)(出典明示により本明細書の一部とする)に記載されている。一般に、ベースラインから少なくとも約5%の変化が好ましく、より好ましくは少なくとも約25%であり、特に好ましくは少なくとも約50%であり、そして取分け好ましいのは少なくとも約100%である。この場合のベースラインとは、刺激前の色素結合量を意味する。
【0163】
同様に、ConA−FITCを使用することができるが、その理由は本明細書に概括したと同様の様式で、それが顆粒タンパク質上の炭水化物に結合するからである。
【0164】
もう一つの好ましい態様においては、表面顆粒酵素活性の変化を定量する。分泌顆粒はエキソサイトーシス過程の一部として放出されるプロテアーゼおよびグリコシダーゼなどの酵素を含んでいる。多くの場合、これらの酵素は、低pHのために顆粒内で不活性であるが、生理的pHの細胞外培地に曝されると活性化する。これらの酵素活性は細胞外培地成分としての色原体または発蛍光体基質を用い測定することができる。これは様々な方法でエキソサイトーシス細胞の検出を可能とする。
【0165】
一態様において、ある場合に本明細書では集団に基づく酵素アッセイと呼ぶが、色原体または発蛍光体基質の切断を経てのシグナル生成を培地中で定量することができる。すなわち、培地中検出可能な反応生成物の量は、存在する酵素量、すなわち、エキソサイトーシスの量に関係する。この態様において、検出可能となるのは培地であって、細胞ではない。
【0166】
好ましい態様において、細胞はエキソサイトーシス刺激剤、任意には候補生物活性剤に付される。色原体または発蛍光体基質を培地に加え、酵素が細胞外基質を切断する際のシグナルの変化を評価する。
【0167】
替り得る好ましい態様において、ある場合に本明細書では「インシトゥ酵素学的アッセイ」と呼ぶが、切断により沈殿する発蛍光体基質を使用する。すなわち、エキソサイトーシスにより相当量の酵素活性が会合した細胞/顆粒に残存し、活性部位で沈殿する蛍光基質を用い可視化することができる。例えば、ELF−97グルクロニドなどのグルクロニダーゼの基質はエキソサイトーシス細胞上に沈殿するが、休止細胞には沈殿せず、その結果、細胞は蛍光の増大を示すことができる。蛍光はエキソサイトーシスの直接の測定値であり、開口分泌した酵素のpH最適化を反映して、pH依存性である。この方法はまたその酵素プロフィールに基づいて顆粒の異なるサブタイプを識別する方法を提供する。
【0168】
好ましい態様において、細胞集団はエキソサイトーシス刺激剤に付され、次いで検出可能な基質とインキュベートする。候補生物活性剤を任意に加える。細胞を洗浄し、次いで、顕微鏡またはフロー・サイトメーターで観察する。
【0169】
好ましい顆粒酵素は、キマーゼ、トリプターゼ、アリールスルファターゼA、ベータ−ヘキソサミニダーゼ、ベータ−グルクロニダーゼ、およびベータ−D−ガラクトシダーゼなどを包含するが、これらに限定されるものではない。基質はELF−97グルクロニド、N−アセチル−ベータ−D−グルクロニド、ペプチドに共役したELF−97などであり、その多くが市販購入可能である。すなわち、上記モレキュラー・プローブからであり、特に、第10章、より詳しくは第10章の第2項、および引用された「関連の章」である。
【0170】
検出可能な基質とは、基質が本明細書にさらに記載しているような蛍光分子を含んでなるか、あるいは基質または切断された基質に特異的的な蛍光分子、すなわち、蛍光抗体により検出し得るということを意味する。好ましい態様において、基質は2種類の蛍光タンパク質、すなわち、青色および緑色蛍光タンパク質(BFPおよびGFP)と他の同様の分子から形成された検出可能な分子を含んでなる。技術上知られるように、近接位置にあるこれら2種類のタンパク質を有するGFPとBFGの構築体は蛍光共鳴エネルギー転移(FRET)を可能とする。すなわち、GFPの励起スペクトルはBFPの発光スペクトルに重なる。したがって、BFPを励起するとGFPが発光する。もしプロテアーゼ切断部位をGFPとBFPの間に組込み、「FRET構築体」を形成するなら、該構築体を切断する活性プロテアーゼにFRET構築体を暴露すると、GFPとBFP分子が別々に離れる。その結果、GFPの励起はBFP発光とBFP発光喪失に至る。
【0171】
好ましくは、FRET構築体の2つの蛍光タンパク質間に挿入されたプロテアーゼ依存性切断部位は顆粒特異的酵素に特異的的である。このように、FRET構築体は、FRET構築体の切断部位に特異的的な顆粒特異的酵素の検出に使用し得る。この態様において、細胞または培地と合わされるプロテアーゼ基質はFRET構築体を包含する。FRETシステムは切断状態および未切断状態の検出可能分子の検出を可能とし、二者間を識別する。このシステムについては以下に記載されている:Xu et al., Nucleic Acid Res. 26 (8): 2034 (1998); and Miyawaki et al., Nature 388 (6645): 882-887 (1997) (出典明示により両文献を本明細書の一部とする)。
【0172】
細胞または培地に添加される基質の量は、酵素特異的活性および基質それ自体に部分的に依存するが、一般的には約250nMないし約1mMであり、好ましくは約1μMないし約100μMであり、特に好ましくは約1μMないし約10μMである。一般に、ベースラインから少なくとも約5%の変化が好ましく、好ましくは少なくとも約25%であり、特に好ましくは少なくとも約100%であり、そして取分け好ましいのは少なくとも約1000%である。この場合のベースラインとは、エキソサイトーシス誘導前の基質切断量を意味する。
【0173】
好ましい態様において、顆粒特異的タンパク質量の変化が定量される。分泌顆粒は、これらタンパク質の特異的性により顆粒区画に特異的的に標的化されるタンパク質を含んでいる。エキソサイトーシス誘導により、顆粒特異的タンパク質は、その表面に露呈され、検出される。
【0174】
好ましい態様において、検出可能な顆粒特異的タンパク質は細胞集団と合わせて、エキソサイトーシスを誘導することの知られている条件に付される。任意には、候補生物活性剤は細胞集団および検出可能な顆粒特異的タンパク質と合わせ、顆粒特異的タンパク質を検出する。顆粒特異的タンパク質とは、VAMPおよびシナプトタグミンなどであるが、限定されない。また、顆粒特異的タンパク質の定義に含まれるものとして、エキソサイトーシスに際し放出される調節物質、例えば、セロトニン、ヒスタミン、ヘパリン、ホルモンなどがあるが、これらに限定されない。
【0175】
顆粒タンパク質の定量は数種の方法で実施し得る。一態様においては、顆粒特異的タンパク質に対する標識抗体(蛍光性抗体など)を使用する。他の態様においては、細胞は顆粒タンパク質と検出可能な分子からなる融合タンパク質を含むように加工調製する。好ましい態様において、検出可能な分子を検出のために細胞に加える。例えば、直接または間接に標識した抗体のいずれかを使用することができる。好ましい態様では第一標識抗体、好ましくは蛍光標識抗体を用いる。もう一つの態様では第一および第二標識体、例えば、標識した第二抗体を用いる。一般にこの態様では、顆粒タンパク質に特異的的に結合する薬剤または直接もしくは間接に標識し得る化合物を使用することができる。
【0176】
好ましい態様において、標識体は細胞中に組入れるように加工する。例えば、組換えタンパク質は、顆粒特異的タンパク質と検出可能な分子との融合タンパク質であり、細胞集団に導入する。これは一般に顆粒特異的タンパク質と検出可能な分子とからなる融合タンパク質をコードする融合核酸で細胞を形質転換することにより実施される。これは一般に技術的に知られたとおりに実施するが、細胞型に依存する。一般に、哺乳動物細胞については、レトロウイルスのベクターおよび方法が好ましい。
【0177】
融合タンパク質は技術上既知の方法により構築される。例えば、顆粒特異的タンパク質をコードする核酸が検出可能分子と共に結合される。本明細書において検出可能な分子とは、検出可能な分子含む細胞または化合物を、この分子を含まない細胞または化合物と識別し得る分子を意味する。、すなわち、ある場合に抗原TAGと称されるエピトープまたは蛍光分子である。好ましい蛍光分子は、GFP、BFP、YFP、およびルシフェラーゼ、β−ガラクトシダーゼを含む酵素などを包含するが、これらに限定されない。これらの構築体は、エキソサイトーシスに際し、顆粒に対し内部にあるエピトープが細胞表面に露呈され、そのため検出可能となるような様式で調製することができる。エピトープは、好ましくは検出可能なペプチドであるが、一般には細胞質膜上に見出されない。しかし、ある場合には、そのエピトープが細胞上に正常に見出されるならば、増加が検出され得るが、これは一般的に好ましいことではない。
【0178】
好ましい態様において、融合タンパク質または検出可能な顆粒特異的タンパク質を含む細胞集団は、エキソサイトーシス条件に付される。任意に、候補生物活性剤および/またはエキソサイトーシス阻害剤が含まれる。好ましくは、細胞を洗浄する。細胞上の蛍光を検出する。一般に、ベースラインから少なくとも約5%の変化が好ましく、より好ましくは少なくとも約25%であり、特に好ましくは少なくとも約50%であり、そして取分け好ましいのは少なくとも約100%である。一般に、この場合のベースラインとは、エキソサイトーシス刺激前の蛍光量を意味する。
【0179】
本発明においては、エキソサイトーシスの同じ特性が複数のパラメーターにより評価され、その結果、共通事項が低下し、特異的性が上昇する。それに対し、FACSは、従来2つの異なるまたは無関係の特性を同時に評価するために使用されていたが、これら2つの特性をもつ細胞を同定するが、合わされた特性に対する共通事項を低減させない。しかし、本発明は複数のエキソサイトーシス性の同定に加えて、上に概説したように、他の細胞性パラメーターの同定と合わせることができる。
【0180】
好ましい態様において、細胞は正常にエキソサイトーシスを引起す条件に付される。プロ−エキソサイトーシス薬剤は、イオノマイシン、Ca++、イオノホア(イオノマイシン、AZ3187)、化合物48/80、サブスタンスP、補体C3a/C5a、トリプシン、トリプターゼ、インシュリン、インターロイキン−3、特異的IgE、アレルゲン、抗IgE、または抗IgG受容体抗体などを包含する。これらは技術上知られているように、一般に1ピコモルないし10μMの範囲で、化合物に依存する濃度で提供される。ある事例では、エキソサイトーシスを阻害する薬剤と細胞を合わせることが望ましい。エキソサイトーシス阻害剤はウオートマンニン(Wortmannin)、ジェネスタイン(Genestein)およびその他技術上既知のものであるが、これらに限定されるものではない。
【0181】
好ましい態様において、本方法はエキソサイトーシスを調節する能力に対し候補生物活性剤を選抜するために使用する。候補生物活性剤はエキソサイトーシス刺激の前、途上または以後に細胞集団と合わせるが、刺激前が好ましい。ある場合には、エキソサイトーシスが誘導されない場合またはエキソサイトーシスが阻害される場合の細胞について、本明細書では「候補薬剤」とも称する候補生物活性剤の効果を定量することが望ましい。候補生物活性剤は外因性に細胞集団に加えてもよいし、またはさらに本明細書に記載するように細胞に導入してもよい。
【0182】
好ましい態様においては、細胞周期アッセイについての上記のように、異なる候補生物活性剤のライブラリーを使用する。
上記のように、候補生物活性剤は細胞または細胞集団に合わせるか添加する。さらに、上に概説したように、好ましい態様では核酸候補薬剤および融合パートナーを利用する。好ましくはレトロウイルス構築体である。
【0183】
候補薬剤が核酸である場合、技術上既知の方法、例えば、リン酸カルシウム、エレクトロポレーション、およびインジェクションがこれらを細胞に導入するために使用される。エキソサイトーシス刺激剤は一般に生理的条件下に細胞と合わせる。インキュベーションは最適活性を容易にする温度、一般には4℃と40℃の間の温度で実施する。インキュベーション時間は最適活性のために選定するが、迅速で高処理量のスクリーンを容易にするために最適化してもよい。
【0184】
上記のように、様々な他の試薬がアッセイに含まれてもよく、細胞は上記のように分別される。分別の結果として所望のエキソサイトーシス細胞集団を得る。好ましい態様においては、エキソサイトーシスを調節する少なくとも1種の候補生物活性剤を同定するために、パラメーターを設定する。
【0185】
好ましい態様において、生物活性剤が特性化される。これは当業者の認識どおりに進めるが、通常、薬剤の構造分析、同定、結合親和性および機能を包含する。一般に、同定すると、該生物活性剤は再合成され、標的細胞と合わせて、種々の条件下、また、他の種々の薬剤の存在下または不存在下にエキソサイトーシスの変調を確認する。該生物活性剤は、エキソサイトーシスを調節するために治療有効量で調製され、適切な医薬担体と組み合わされる。
【0186】
好ましい態様において、細胞集団は、候補薬剤の存在下または不存在下に、また、エキソサイトーシス刺激または阻害の存在下または不存在下に、様々の実験条件に付される。条件の変化はpH、温度、緩衝液または塩濃度などであるが、これらに限定されない。好ましい態様において、pHは、一般にpHの増減により、通常、約0.5ないし約3のpH単位で変化させる。あるいは、温度の変更は、好ましくは、約5℃ないし約30℃の上昇または下降により行う。同様に、塩濃度は、好ましくは、約0.1Mないし約2Mの増減により変更する。
【0187】
好ましい態様において、調節すべき細胞の表現型は小分子(または他の候補薬剤)の毒性である。これらは一般に上に概説したように、細胞生存能アッセイに対するものである。小分子の用量応答は、最大機能応答保有の細胞を比較し、次いで、そこに多少ともこれらの細胞と会合する毒性があるか否かを見直すことで比較することも可能である。
【0188】
好ましい態様において、細胞の表現型は細胞表面受容体の発現または活性に関与する。16種までの細胞表面マーカーを同時に追跡することが可能であるが、好ましいのは8種までである。特定の細胞表面マーカーの存在または不存在は、どの細胞表面発現が研究対象細胞と会合した重要な機能パラメーターを反映しているか、いずれかの細胞表面タンパク質に対する、直接またな間接に会合した抗体により検出することができる。小分子などの候補薬剤の効果は、次いで個々のまたは複数のマーカーについて試験することができる。
【0189】
好ましい態様において、細胞の表現型は、一次測定と同時に起こる、または一次測定の結果である、生物事象を伝え得る蛍光ベースのリポーターシステムなど、酵素の発現または活性に関与する。このリポーターシステムは、細胞質における活性な上流シグナル伝達経路の、または核内転写もしくは翻訳事象並びに核もしくは細胞からの輸出事象の読み出しである。
【0190】
好ましい態様において、細胞の表現型は、二量化などのタンパク質−タンパク質相互作用(または他の結合リガンド間の相互作用)に関与し、それが候補薬剤により中断または刺激される。これらの事象は2つの標識化した結合リガンド間のFRETの出現または消失により測定し得る。
【0191】
以下の実施例は、上記本発明の使用方法につきさらに詳細に説明し、同様に本発明の様々な局面を実施するために期待される最良の形態を明らかにするものである。これらの実施例は、如何なる意味でも本発明の真の範囲を制限するものではなく、むしろ説明を目的として提供されるものである。本明細書に引用した文献はすべて出典明示により本明細書の一部とする。
【0192】
(発明を実施するための最良の形態)
【実施例】
実施例1:
正対照としてp21を用いる細胞周期アッセイ
材料および方法:ベクター構築
p21遺伝子のコード領域を、開始メチオニンを含む上流領域のプライマー
【化39】
【化40】
【0193】
シングルPCR産物を、CRU5−GFPレトロウイルスベクター(Rigel,IInc.)中で、プライマー内のフランキングBstXI部位から直接クローンとして発生させた。この得られた構築体、CRU−GFP−p21Fは、C末端位置でFLAG−エピトープと2位の位置でGly挿入を伴うヒトp21タンパク質と融合した(フレーム内)GFPをコードする(図1)。p21のC-末端の24個のアミノ酸を、PCRプライマー:
【化41】
【化42】
【化43】
【化44】
【0194】
レトロウイルス形質転換;1.5ml DMEM(DMEM+10%FBS+Pen/Strep)において106細胞で、フェニックス(Phoenix)E細胞を、6ウェルプレート中でまき、16時間37℃でインキュベートした。CaCl2沈殿トランスフェクションを、50μMクロロキンの存在下、8時間37℃で、CRU5−IRES−GFPベクターまたはCRU5−p21F−IRES−GFPクローンで行った(2μlDNA(1μg/μl)、30.5μl、2M CaCl2、217.5μlH2O、0.5ml)。トランスフェクション培地を、除去し、2mlの完全なDMEMで置換し、該細胞をさらに16時間、37℃でインキュベートした。該培地を、1.5mlの完全なRPM1(RPM1+10%FBS+Pen/Strep)にかえて、48時間、37℃でインキュベートした。トランスフェクションしたプレートからのウイルス上清を、濾過し(0.45μm)、6ウェルプレートに移した。異所性受容体を発現するジャカットT細胞(JurkatE)の100μl数(5×106細胞)を各ウエルに添加した。ポリブレンを終濃度0.5μg/mlで添加した。該プレートをパラフィルムで密封し、2500rpmで、90分間、32℃で遠心分離した。パラフィルムを除去し、該プレートを37℃でインキュベートした。培地を、16時間後に、4mlの完全RPM1に替え、72時間、37℃でインキュベートした。
【0195】
細胞周期のFACS−アッセイ
レトロウイルスベクター形質転換細胞を集め、完全RPM1中に105細胞/mlで再懸濁した。4μMPKH26細胞のトラッキング色素(Sigma)のある容積(1ml)を細胞に添加し、5分、25℃でインキュベートした。該懸濁液を、5倍希釈し、該細胞を400×g、10分、25℃で集めた。該細胞を、さらに6mlの完全RPM1で2回洗浄し、6ウェルプレート中で、3×105細胞/mlで、72時間インキュベートした。標識した細胞を集め、5μg/mlのヘキスト(Hoechst)33342(分子プローブ)を含む完全RPM1中、106細胞/mlで再懸濁し、2時間37℃でインキュベートした。染色細胞を集め、FACS緩衝液(PBS/0.5%FCS/5μg/ml ヘキスト33342)中で、>106細胞/mlで再懸濁した。該細胞を、3つのレンズを有するMoFloフロー・サイトメトリー分析に供し、前方、側面の分散を480nm系のアルゴンレーザーによって誘因し、分散光を分散前方の検出器および480nmバンドパスフィルターで集めた。GFPを480nm系のアルゴンレーザーによって励起させ、530nmバンドパスフィルターから分散光を集めた。PKH26細胞のトラッキング色素を533nm系のHeNe−レーザーによって励起させ、570nmバンドパスフィルターから分散光を集めた。ヘキスト33342色素を、UVレーザーによって励起し、分散光を450nmバンドパスフィルターから集めた。
【0196】
結果
ジャカットT細胞を、GFPに融合したヒトp21(Gp21)をコードするレトロウイルスベクター、またはPCNA結合C末端の24アミノ酸(Gp21C)を用いて形質導入した(図1)。p21C末端の24個のアミノ酸の非PCNA結合変異体型[GP21Cmut、Cyrol et al.,Oncogene 16:311(1998)]は、負の対照として働く。形質導入したp21の発現は、ウエスタンブロッティングによるFLAG−エピトープにより内生タンパク質と区別することができる(示さない)。融合タンパク質の発現は、FACSにおいてGFP蛍光によって示された(図2B)。
形質導入細胞を細胞追跡化合物pkh26で標識した。この化合物は赤色蛍光脂肪族分子を細胞膜中に導入し、選択的な分割よって細胞周期と蛍光強度との相関関係を明かにする:拘束細胞は細胞追跡色素の明るさを保つ:細胞周期中の細胞はシグナルが薄くぼんやりいる。図2Cで示したように、GFP上に捕捉された生存GFP−p21発現細胞は、同一GFPレベルを発現するベクター形質導入細胞よりも強い赤色蛍光を示し、細胞周期の拘束を表す。同様の効果が、Gp21C発現細胞で見られる。しかし、Gp21Cmutは、非発現細胞と同一であった。同一のGFP捕捉細胞のDNA含量を、図2Dに示した。Gp21発現細胞は、細胞周期のG1期で拘束され、Gp21C発現細胞は、G1およびG2チェックポイント蓄積を示し、前記の結果と一致する(Wade Harper, et al., 1993;Cayrol etal., 1998)。Gp21mut発現細胞は、通常の細胞周期分布を示す。存続可能の拘束発現細胞(3つの開始パラメーターを満たす)は、DNA含量を基に別個のチャンバーに分別した:左屈曲(deflection)、G1;右屈曲、G2。
【0197】
実施例2
集団を基にしたエキソサイティック酵素活性測定
材料:全ての化学物質はSigma Chemical Co.から得た。色素およびグルクロニドは、Molecular Probes, Inc.から得た。細胞系MC−9およびRBL−2H3はAmerican Type Culture Collection(ATCC)から得た。細胞培養試薬はFisher Scientificから、そして分子生物学的試薬はClontech Inc.から得た。
【0198】
細胞培養:MC−9細胞を、フラスコ中に、L−アルギニン(116mg/ml)、L−アスパラギン(36mg/ml)、ピルビン酸ナトリウム(1mM)、非必須アミノ酸(0.1mM)、葉酸(6mg/ml)、2−メルカプトエタノール(0.05mM)、L−グルタミン(2mM)、熱不活性化ウシ胎児血清(10%)および10%T-stim条件培地(Collaborative Research, Inc.)を添加したDMEMを含む培地中の懸濁培養として維持した。細胞を0.25から2×106/mlの間の密度に保った。実験はトリパンブルー除外で測定して95%より大きい生存可能の細胞でのみ行った。RBL−2H3細胞を、2mM L−グルタミンおよびEarl’s BSS、15%熱不活性化ウシ胎児血清を添加されたEagles MEMを含む培地中の非被覆(組織培養処理)フラスコ上の接着培養として維持した。細胞を継代(0.05%トリプシン)し、1日以上コンフルエントにならないようにした。
【0199】
エキソサイトーシス刺激プロトコール:実験を、NaCl(137mM)、KCl(2.7mM)、CaCl2(1.8mM)、MgCl2(1mM)、グルコース(5.6mM)、Hepes(20mM、pH7.4)およびウシ血清アルブミン(0.1%)から成る修飾タイロード緩衝液(MT中)で行った。MC−9細胞を400×gで回転させ、培地を吸引した。次いで、細胞をMTで洗浄し、再回転/吸引し、5×106細胞/mlの濃度でMTに取りこませた。次いで、細胞をDMSOまたはイオン透過担体のいずれかで30分(または時間経過の場合、時間を変える)処理した。次いで、酵素分析のために、細胞を回収した上清とペレット化した;ある場合、細胞はフローサイトメトリーのために処理した。全ての刺激は37℃で行った。RBL−2H34細胞の刺激は、接着細胞を1回MTで洗浄し、次いで刺激剤を含む温かいMT(1ml/106細胞)の添加により行った。細胞を37℃で30分インキュベートし、上清を更なる分析のために回収した。実施例のいつくかにおいて(実施例4−6)、プレートに結合した細胞をアネキシンで染色し、次いで、フローサイトメトリーのための更なる処理のためにNo-Zyme(Collaborative Research, Inc.)を使用してフラスコから回収した。RBL−2H3細胞の抗原架橋による刺激のために、細胞を、完全培地中で50ng/mlの濃度のIgE抗-DNP(Sigma Chemical Co.)と一晩インキュベートした。翌日、それらをMTで一回洗浄し、DNPに結合したウシ血清アルブミンをを刺激剤として100ng/mlの濃度で使用した以外、上記のように刺激した。
【0200】
集団をベースにした酵素アッセイ:酵素アッセイを、エキソサイティック刺激に続いて細胞上清およびペレットで行った。細胞上清を刺激後に回収し、氷上で冷やし、5000×g回転後上清を回収して酵素活性分析をした。同様に、細胞ペレットを0.1%トリトンX−100含有MT中に回収/融解させ、5000×g回転後上清を回収して酵素活性分析をした。各分析に関して、100μlの融解物および上清を、2mM基質(4−メチルウンベリフェリルβ−Dグルクロニド[グルクロニダーゼ基質]または4−メチルウンベリフェリルN−アセチルβ−Dグルコサミニド[ヘキソサミニダーゼ基質])を含む100μlの反応緩衝液(40mMクエン酸、pH4.5)と、固体黒色96ウェルプレート(Costar, Inc.)中で混合し、37℃で15分インキュベートした。プレートを蛍光プレートリーダー(Wallac, Inc.)で、励起380nm/放出440nmフィルターを3分毎に5回使用して読取り、酵素の速度を得た;分析は3回行った。
【0201】
フローサイトメトリー:刺激および染色のために処理された細胞を、血球計算前に氷上のMTに取りこみ、100μmフィルターを通して濾過した。細胞をFACSCAN(Becton Dickinson Inc., レーザーライン458nm)またはMo−Flo(Cytomation, Inc., レーザーライン広いバンドで350nM(UV)、488nmおよび647nm)Cytometerを使用して分析した。望ましい場合、Mo−Floを使用して細胞を分類した。
【0202】
結果:結果を図4に示す。細胞上清中の酵素活性をMC9(A)およびRBL−2H3(B)細胞について、種々の条件下で測定した。A)MC−9細胞をDMSO(−)または2μmロノマイシン(+)存在下で30分刺激した。上清を回収し、グルクロニダーゼまたはヘキソサミニダーゼ活性についで分析した。顆粒酵素活性の刺激による放出は明白である。B)RBL−2H3細胞を16時間、種々の量のIgE抗−DNPで感作し、増加した量の抗原BSA−DNPに曝すことによりエキソサイトーシスによる分泌を刺激した。両方の抗体および抗原の用量応答が、上清ヘキソサミニダーゼ活性の測定において明白である。
【0203】
実施例3:肥満細胞エクソサイトーシス性光分散変化
実施例2に記載のように細胞を調製し、光分散性を測定した。
【0204】
結果:結果を図5に示す。フローサイトメーター上で観察された光分散の変化(側面分散対前方分散)を、RBL−2H3細胞(A、D)およびMC−9細胞(B、C、E、F)についての二変数ヒストグラムとしてプロットする。細胞はイオン透過担体A23187(0.5ug/mL)で刺激し、様々な時点[0分(A、C)、5分(E)、10分(D)および30分(B、F)]で観察した。時間に依存した分散の変化は、最初の10分間で有意な変化が起こっている両方のセルラインで明らかであり、これらの細胞における大量のエクソサイトーシスを示すものである。
【0205】
実施例4:FACSによるスチリル色素が肥満細胞エクソサイトーシスを検出する
スチリル色素着色:細胞は上記のように調製した。スチリル色素(FM1−43またはFM4−64;Molecular Probes,Inc.)をMT中250nMの最終濃度に希釈し、刺激緩衝液に加えた(実施例2、参照)。刺激プロトコルの後、細胞を遠沈し、吸引し、新しい氷冷MTに再懸濁した。この細胞について、フローサイトメーターでの分析の準備が完了した(実施例2、参照)。
【0206】
結果:結果を図6に示す。MC−9細胞は、FM4−64(A、B)またはFM1−43(C、D、E)のいずれかの存在下で刺激した(青=DMSO、赤=2μMイオノマイシン)。A)蛍光チャンネル1のフローサイトメーターで検出されたFM4−64標識化細胞。B)蛍光チャンネル3のフローサイトメーターで検出されたFM4−64標識化細胞。C)蛍光チャンネル1のフローサイトメーターで検出されたFM1−43標識化細胞。D)蛍光チャンネル3のフローサイトメトリーで検出されたFM1−43標識化細胞。いずれの色素を用いても、明白な刺激依存性の蛍光強度の増強がある;FM4−64は最も赤色偏位であり、且つチャンネル3で専ら検出され、一方FM1−43はチャンネル1および3の両方でより広域の蛍光が検出されている。E)MC−9細胞を、種々の用量のPI−3キナーゼ阻害剤ウォートマニン(1μM−棒グラフ1、100nM−棒グラフ2、10nM−棒グラフ3、および0nM−棒グラフ1および2)と共に予備インキュベートした後、FM1−43の存在下でA23187(0.5ug/mL、棒グラフ1−4)またはDMSO(棒グラフ5)で刺激した。蛍光チャンネル1のフローサイトメーターで検出された平均のチャンネルシフトを棒グラフとしてプロットする。肥満細胞のエクソサイトーシスの阻害剤として知られるウォートマニンは、FM1−43シグナルの用量依存的減少を惹起し、FM1−43シグナルがMC−9肥満細胞セルラインの分解の程度を反映していることを示す。
【0207】
実施例5:FACSによりアネキシン−V着色が肥満細胞エキソサイトーシスを検出する
材料:アネキシン−Vビオチン、アネキシン−V FITCおよびストレプトアビジンAPCはCaltag Laboratoriesより取得した。本実施例で使用する他の材料および方法は、他の実施例、特に実施例2から採用することができる。
【0208】
アネキシン−V着色:エキソサイトーシス刺激後の細胞を、MT中1/100の希釈で室温下に10分間アネキシン−PITCで着色した。次いでこの細胞をMTで1回洗浄し、MTに溶解し、フローサイトメーターまたは顕微鏡で調べた。間接的標識化については、アネキシン−ビオチンを1/200の希釈で刺激過程の間にMTに加えた。次に細胞を氷冷MT中でペレット化し、遠沈し、吸引し、ストレプトアビジン−APCを添加した氷冷MTに1/200の希釈で溶解し、氷上に10分間保持した。細胞をペレット化し、ストレプトアビジン−APCを吸引した後、細胞をMTに再懸濁し、フローサイトメーターで観察した。幾つかの実験では、顕微鏡で視覚化するためにストレプトアビジン・アレキサ488または594(Molecular Probes,Inc.)といったような異なる二次物質を適用した。
【0209】
結果:結果を図7に示す。MC−9細胞は、DMSO(図AおよびB)または2μmのイオノマイシン(図CおよびD)のいずれかで刺激し、次いでヨウ化プロピジウム[PI](図AおよびC)およびアネキシン−V−FITC(図BおよびD)の両方で着色した。この用量のイオノマイシンによる刺激は、エキソサイトーシスを行っている細胞で有意なPI着色の増加がないことにより証明されるように、原形質膜を傷つけない。図DおよびBの比較により認められるように、脱顆粒は、アネキシン結合の有意な増加をもたらす。
【0210】
実施例6:アネキシン−V−FITCは共焦点顕微鏡検査により視覚化されたMC−9細胞中のエキソサイトーシス性顆粒を着色する
顕微鏡検査:刺激または着色後の細胞を、スライドガラス上に載せ、カバーガラスをかぶせ;標準的BFP、FITCまたはTRITCフィルターセットを使用する倒立顕微鏡(TE300、Nicon)上で明視野および蛍光顕微鏡検査によって直接視覚化した。幾つかの像は、標準的FITCおよびTRITCフィルターセットを使用する倒立共焦点顕微鏡(Zeiss,Inc.、Bio-Rad,Inc.)を使用して得られた。
【0211】
結果:MC−9細胞は、2μmイオノマイシンまたはDMSOで刺激し、アネキシン−V−FITCで着色し、共焦点顕微鏡用のスライドガラスに載せた(結果は示されていない)。刺激されなかった生存細胞は、全て低値のアネキシン結合を示し、細胞表面顆粒の着色を示さない。
【0212】
実施例7:アネキシン−V−FITCは、抗原架橋により刺激したRBL−2H3細胞中のエキソサイトーシス顆粒を着色する
別途下記する場合を除き、本実施例のための方法は先の実施例に記載されている。
【0213】
結果:RBL−2H3細胞をIgEで感作し、MT緩衝液のみ、またはBSA−DNP抗原(100ng/mL)のいずれかによりエキソサイトーシスが起こるよう刺激し、次いでアネキシン−V−FITCで着色した。MC−9細胞に見られるパターンと同様、抗原刺激された細胞中に、多数の細胞表面顆粒が着色される。刺激されなかった生存細胞のアネキシン−V着色は無視できるものである(データは示されていない)。
【0214】
実施例8:FACSで視覚化される細胞のエキソサイトーシスについてのインシトゥ酵素法
インシトゥ酵素法:MC−9細胞を上記のようにエキソサイトーシスを目的として刺激し、次いで、基質ELF−97グルクロニド(250μM)を含有する酵素基質緩衝液(BSAを含有しないMT、pH4.3−7.4の範囲)中、37℃で15分間でインキュベートした。この細胞をMT中で1回洗浄し、次いで顕微鏡またはフローサイトメーター上で観察した。方法の他の詳細は前実施例に記載されている。
【0215】
結果:結果を図8に示す。MC−9細胞をDMSO(図A)またはA23187(0.5μg/mL - 図BおよびC)で刺激し、次いでインシトゥ・グルクロニダーゼ活性を調べるために着色した。A)細胞表面酵素沈降物を示すELF−97検出のフローサイトメーター・ヒストグラム。DMSO処理した細胞では極めて低いシグナルが見られる。B)細胞表面酵素沈降物を示すELF−97検出のフローサイトメーター・ヒストグラム。イオン透過担体による分泌刺激時にシグナルの有意な増強が見られる。C)細胞表面酵素活性のpHプロフィール。異なるpHで作成したMT緩衝液を使用して、フローサイトメーター中に見られるシグナルのpHプロフィールを作成した。棒グラフは、観察された最大シグナルのパーセントを示す(フローサイトメーターでの平均チャンネルシフトにより測定)。酵素活性は、pH依存性であり、pH6未満でピークとなる;このことは、酸性分泌性顆粒から誘導された酵素活性と合致する。
【0216】
実施例9:Lysotracker緑は、エキソサイトーシス時に肥満細胞顆粒から放出され、FACSにより検出できる。
Lysotracker色素着色:Lysotracker色素(青、緑、および赤)を完全培地中1μMの最終濃度に希釈し、これらの存在下で細胞を37℃で60分間インキュベートすることにより、Lysotracker色素を細胞中に負荷した。負荷後、細胞をMTで2回洗浄すると、さらなる分析または刺激への準備が完了した。方法の詳細は前実施例に記載されている。
【0217】
結果:結果を図9に示す。MC−9細胞にLysotracker緑を1時間負荷し、次いでDMSOまたはイオノマイシン(2μM)のいずれかで刺激し、フローサイトメーターで観察した。チャンネル1で検出された蛍光強度のヒストグラムを示す;DMSO対照と比較してイオン透過担体刺激された試料には、シグナルの有意な喪失が見られ、これは、分泌顆粒からのLysotracker緑色素の放出を反映するものである。
【0218】
実施例10:複数パラメータ分析 − Lysotracker緑、アネキシン−V−APC、前方および側面分散
別途に下記しない限り、前実施例に記載の方法を使用した。
【0219】
結果:結果を図10に示す。MC−9細胞を異なる用量のイオノマイシン(0μM−A、E;1μM−B、F;2μM−C、G;および3μM−D、H)で処理し、4パラメータのフローサイトメーターで同時に観察した。細胞にlysotracker緑を1時間負荷し、次いで刺激し、アネキシン−VAPCを目的として着色した。図A−D:側面対前方光分散の二変数ヒストグラム。前方分散が増加し側面分散が減少するにつれて、両パラメータが左から右に用量依存的に変化することに注目されたい。図E−H:アネキシン−V−APC対Lysotracker緑シグナルの二変数ヒストグラム。エキソサイトーシスが増加(左から右へ)するにつれてアネキシンシグナルが大きくなり、lysotrackerシグナルが減少する。これは、細胞表面顆粒へのアネキシン−Vの結合、および顆粒が細胞外環境に暴露されるために顆粒からlysotrackerが失われることを反映するものである。
【0220】
実施例11:複数パラメータ分析 − FM1−43、アネキシン−V−APC、前方および側面分散
別途下記しない限り、上記の方法を使用した。
【0221】
結果:MC−9細胞をDMSOまたはイオノマイシン(2μM)のいずれかで処理し、4パラメータのフローサイトメーターで同時に観察した。細胞はFM1−43の存在下で刺激し、アネキシン−V−APCを目的として着色した(データは示されていない)。30分の時点で両方のパラメータに刺激依存的変化があった。両シグナルに刺激依存的増強があった。この変化は、細胞表面顆粒へのアネキシン−Vの結合、およびMC9細胞中へのFM1−43色素の同時結合エンドサイトーシスを反映するものである。
【0222】
実施例12:FACSでの同時複数パラメータ測定が集団に基づく酵素読み出しと相関する
カルシウムシグナル検定:MC−9またはRBL−2H3細胞をMT中で1回洗浄し、MT中のCa++感受性プローブFluo-3(1μM、Molecular Probes,Inc.)を37℃で20分間負荷した。細胞を温MTで1回洗浄し、次いで上記プロトコルにしたがって刺激した。細胞内Ca++濃度上昇によるシグナルを、フローサイトメーター(下記参照)、蛍光顕微鏡検査、または蛍光板読み取り機による読み取り(Wallac,Inc.)のいずれかを用いて視覚化した。細胞の負荷は、0.1%トリトンX−100を含有するMTで細胞内色素を放出させることにより測定した。
別途下記しない限り、上記の方法を使用した。
【0223】
結果:結果を図11に示す。MC−9細胞をFM1−43の存在下で刺激し、上の方法に記載のようにアネキシン−V−APC着色した。イオノマイシン刺激後の様々な時点で、細胞を氷上に置き、フローサイトメトリーにより分析するか、または酵素活性について分析した(細胞上清)。前方分散、FM1−43、アネキシン−V−APCおよびヘキソサミニダーゼのパラメータを、各パラメータについての最大反応に対してグラフ上にプロットする。カルシウムシグナルについては、別の試験管の細胞にFluo-3を負荷し、同一の操作を行った。サイトメトリーに基づくパラメータの時間経過によると、このパラメータがヘキソサミニダーゼの放出により測定されるエキソサイトーシスとかなりよく相関する。この実施例において、前方分散は、時間と共に正および負の両方に変化する効果を示している。
【0224】
実施例12:VAMP−GFPおよびVAMP−FRET構築体の発現
cDNA構築体:
VAMP−GFP構築体:ラットVAMP−2 cDNA(R.Scheller(スタンフォード大学)より取得)をPCR修飾して、(1) 発現およびインビボ安定性をそれぞれ促進するための、共通Kozakおよびグリシンの挿入(a.a.2)をコードする5'BstXI部位;(2) 3'末端にBamHI部位を有するセリン−グリシンリンカー、を導入した。CdimGFP(Clontech,Inc.)由来のGFPコード化配列をPCR修飾して、停止コドンをコードする3'BstXI部位を導入した。この修飾したrVAMPおよびGFP PCR断片を、セリン−グリシンリンカー中の共通のBamHI部位を介してライゲーションすることにより、VAMP−GFP融合物を構築し、以下の配列を有するフレーム内融合タンパク質を製造した:
【0225】
【化45】
【0226】
このVAMP−GFP融合配列を、直接的BstXI部位を有する96.7レトロウイルス・ベクター中にクローニングしてpVGを作成した。配列は、両方向に配列決定することにより確認した。ウエスタン分析および蛍光顕微鏡検査により、トランスフェクトされ感染した細胞での適正な発現を確認した。
【0227】
Trp−FRET構築体:cGFP(Clontech,Inc.)由来のGFPコード化配列をPCR修飾して、(1) 発現およびインビボ安定性をそれぞれ促進するための、共通Kozakおよびグリシンの挿入(a.a.2)をコードする5'BstXI部位;(2) C末端にAla228をコードする3'末端SacII部位、を作り出した。cBFP(Clontech,Inc.)由来のBFPコード化配列をPCR修飾して、(1)Ser2をコードする5'BamHI部位;(2) 停止コドンをコードする3'末端BstXIを作り出した。GSGSスペーサーが側面に位置し因子Xおよびトリプターゼプロテアーゼ開裂部位をコードするSacII-BamHI変換リンカー(GSGSIEGRLRKQGSCS)を用いて、GFPとBFPとを融合させ、以下の配列を有するフレーム内融合タンパク質を作成した:
【0228】
【化46】
【0229】
このVAMP−GFP融合配列を、レトロウイルスベクター96.7のBamHIおよびBstXI中にクローニングしてpGX/TBを作成した。配列は、両方向に配列決定することにより確認した。ウエスタン分析および蛍光顕微鏡検査により、トランスフェクトされ感染した細胞での適正な発現を確認した。
【0230】
VAMP−GFPコード化配列をPCR修飾して、C末端にAla228をコードする3'末端SacII部位を作り出した。この断片をXhoIおよびSacIIで開裂し、pGX/TBのXhoI/SacII部位中にクローニングして、rVAMP−2−BFP−因子X/トリプターゼ部位−GFP融合タンパク質をコードする、以下の配列のpVGX/TB(Trp−FRET)を作成した。
【化47】
【0231】
このrVAMP−2−BFP−因子X/トリプターゼ部位−GFP融合配列を両方向に配列決定することにより確認した。ウエスタン分析および蛍光顕微鏡検査およびFACS分析により、トランスフェクトされ感染した細胞での適正な発現を確認した。
【0232】
トランスフェクションおよび感染:MC−9およびRBL−2H3細胞にVamp構築体を発現する組換えレトロウイルスを感染させるために、以下の方法を実施した。第一日目、Phoenix EまたはA細胞(G.Nolan(スタンフォード大学)より入手)を、培地(DMEM、10%FBS)1.5mLを入れた6ウェルプレートに8x10E5細胞で接種した。二日目、この細胞中にクロロキン50μMの存在下でCaPO4沈澱法を用いてDNA 5μgをトランスフェクトした。沈澱を細胞と共に37℃で8時間インキュベートし、この時点で培地を除去し、新たな培地で1回洗浄し、新しいMC−9またはRBL−2H3培地のいずれかと交換した。次いでこの細胞を32℃で48−72時間インキュベートした。Phoenix細胞からの上清(ウイルス上清)を1000xgで10分間遠沈し、硫酸プロタミンを最終濃度5μg/mLとなるまで加えた。この上清を、6ウェルプレート(ウェル当たり5x10E5細胞)に入れたMC−9またはRBL−2HS(新たにトリプシン処理)細胞に加え、混合物を室温下に1000xgで90分間遠沈した。次にこの細胞を32℃で16時間インキュベートした。ウイルス上清を除去し、新しい培地を加えた。感染の24時間後に標的遺伝子の発現が認められた。
【図面の簡単な説明】
【図1A−1C】 図1A、1B、1Cは、実施例1の3つのレトロウイルス構築を模式的に示す。図1Aは、CRU5−GFP−p21構築であって、CRU5プロモーター、ψレトロパッキングシグナル、p21のコード領域に融合したGFPのコード領域、次いでLTRを含む。図1Bは、CRU5−GFP−p21C構築を示し、p21のC末24アミノ酸を含む。図1Cは、CRU5−GFP−pUCmut構築体を示し、これは3アラニン置換を有するCRU5−p21Cp21の変異体である。
【図2A−2D】 図2A、2B、2C、2Dは、実施例1の実験の結果を示す。図2Aは、前方および側面分散を用いる生存能アッセイを示す。特性割合を表す細胞が集められる。図2Bは、ベクターのGFPの蛍光を示す。図2Cは、増殖アッセイにおけるPKH26、包含色素の使用を表す;細胞を拘束する既知のタンパク質であるp21を含有する細胞は、輝く蛍光を保持し、一方、対照の細胞は、増殖を続けて色素を希釈し蛍光を無くする。図2Dは、細胞相アッセイにおける Hoechst 33342 の使用を表す。
【図3】 図3は、G1相における細胞拘束剤、p21に感染のJurkat細胞に対するAraC処置の効果を示す。AraCは、分割細胞に毒性のヌクレオチド類似体である。従って、拘束された細胞周期であるこれらの細胞が生存する。下側の線はp21の挿入のないJurkat細胞を示し,上側の線はp21挿入のJurkat細胞を示す。
【図4Aおよび4B】 図4Aおよび4Bは、集団についてのエキソサイトーシス酵素活性アッセイの結果を示す。図4Aは、DMSO(−)またはイオノマイシン(+)と組み合わせた細胞の上澄み液におけるグルクロニダーゼまたはヘキソサミニダーゼ活性を示す。図4Bは、種々の量のIgEアンチDNPで感受性化され、増加量の抗原BSA−DNPで刺激された細胞の上澄み液におけるヘキソサミニダーゼ活性を示す。
【図5A−5F】 図5A−5Fは、フロー・サイトメーター、側面分散対前方分散に見られたエキソサイトシス・分散変化を示す。RBL-2H3細胞(図5Aおよび5D)およびMC−9細胞(図5B、5C、5E、5F)について2変数ヒストグラムとしてプロットした。イオン透過担体での刺激後、細胞の観察を0分(図5Aおよび5C)、5分(図5E)、10分(5D)、30分(図5Bおよび5F)で行った。
【図6A−6D】 図6A−6Eは、FACSによるエキソサイトーシスを検出するスチリル色素アッセイの結果を示す。細胞は、DMSO(左ピーク)またはイオノマイシン(右ピーク)と、FM4−64(図6Aおよび6B)またはF1−43(図6C、6D、6E)のいずれかの存在下にて、組み合わせた。図6Aおよび6Cは蛍光チャネル1で検出された細胞を示す。図6Bおよび6Dは、蛍光チャネル3で検出された細胞を示す。
【図6E】 図6Eは、蛍光チャネル1でのフロ−・サイトメトリーで検出された平均チャネルシフトを棒グラフで示す。種々の量のPl−3キナーゼ阻害剤ウォルトマニンとの前インキュベートが、FM1−43の存在下にイオン透過担体(棒1−4)またはDMSO(棒5)の投与前になされた。
【図7A−7D】 図7A−7Dは、FACSによるエキソサイトーシスのアネキシン-V検出アッセイの結果を示すグラフである。細胞は、DMSO(図7Aおよび7B)またはイオノマイシン(図7Cおよび7D)のいずれかと組み合わせ、次いでヨウ化プロピジウム(図7Aおよび7C)およびアネキシン-V-FITC(図7Bおよび7D)の両者で着色された。
【図8A−8C】 図8A−8Cは、FACSにより可視化されたエキソサイトーシス細胞のインシトゥ酵素学アッセイの結果を表すグラフである。細胞は、DMSO(図8A)またはイオン透過担体(図8Bおよび8C)と組み合わせ、次いでインシトゥグルクロニダーゼ活性について着色した。図8Cは細胞表面酵素活性のpHプロファイルを示し、棒グラフで、フロー・サイトメトリー中の平均チャネルシフトにより測定された最大シグナルの百分率を表す。
【図9】 図9は、チャネル1中に検出された蛍光強度のヒストグラフである。細胞に、LYSOTRACKER GREEN(商標)を負荷し、DMSO(左)またはイオノマイシン(右)のいずれかと組み合わせ、フロー・サイトメトリーで測定した。
【図10A−10H】 図10A−10Hは、LYSOTRACKER GREEN(商標)、アネキシン-V-APC、前方および側面分散を含む複数パラメーター分析の結果を示す。図10A−10Dおよび10E−10Hの各々は、イオノマイシンの増加量で処理され、次いで4パラメーターのフロー・サイトメーターで同時に観測された細胞を示す。細胞に、低pH濃度色素を負荷し、刺激し、アネキシン-V-APCで着色した。図10A−10Dは、側面対前方の光分散の2変数ヒストグラムを示し、図10E−10Hは、アネキシン-V-APC対低pH濃度色素シグナルの2変数ヒストグラムを示す。
【図11】 図11は、FM1−43の存在下で刺激され、アネキシン-V-APCで着色された細胞についてのグラフである。イオノマイシン刺激後の種々の時点で、細胞の分析をフロー・サイトメトリーおよび酵素活性について上澄み液(細胞上澄み液)で行った。パラメーター、前方分散、FM−143、アネキシン-V-APC、ヘキソサミニダーゼを、各パラメーターについての最大応答に比較してグラフとした。カルシウム情報伝達について、分離管の細胞にFlou−3を負荷し、同様に処理した。
Claims (10)
- 細胞表現型を変化し得る生物活性剤についてスクリーンする方法であって、
a)少なくとも1つの候補生物活性剤と細胞集団とを組み合せて;
b)FACS機器における該細胞の分別を、該細胞表現型を測定することができる少なくとも5の細胞パラメーターに基づく該細胞の分離により行う;
ことを含む方法。 - 候補生物活性剤のライブラリィを該集団と組み合す、請求項1の方法。
- 細胞表現型を変化し得る生物活性剤についてスクリーンする方法であって、
a)候補生物活性剤を各コードする核酸のライブラリィを細胞集団に導入し;
b)FACS機器における該細胞の分別を、該細胞表現型を測定することができる少なくとも3の細胞パラメーターに基づく該細胞の分離により行う;
ことを含む方法。 - 該ライブラリィがレトロウイルス・ライブラリィである、請求項3の方法。
- 該細胞表現型がエキソサイトーシスであり、該細胞パラメーターが光分散、蛍光色素取り込み、蛍光色素放出、アネキシン顆粒結合、表面顆粒酵素活性、顆粒特異的タンパク質の量よりなる群から選ばれる、請求項3または4の方法。
- 該細胞を、エキソサイトーシスを正常に起こすような状態にすることをさらに含む、請求項5の方法。
- 該表現型が細胞周期調節であり、該細胞パラメーターが細胞の生存能、増殖および細胞相を含む、請求項3または4の方法。
- 該核酸が下記を含有する融合核酸を含む、請求項3、4、5、6または7の方法:
a)該候補生物活性剤をコードする該核酸;
b)検出可能部分。 - 該細胞が腫瘍細胞である、請求項1、2、3、4、5、6、7または8の方法。
- 該検出可能部分が蛍光タンパク質である、請求項8の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/062,330 US6897031B1 (en) | 1998-04-17 | 1998-04-17 | Multiparameter FACS assays to detect alterations in exocytosis |
| US09/062,330 | 1998-04-17 | ||
| US09/157,748 | 1998-09-21 | ||
| US09/157,748 US6461813B2 (en) | 1998-09-21 | 1998-09-21 | Multiparameter FACS assays to detect alterations in cell cycle regulation |
| PCT/US1999/008345 WO1999054494A2 (en) | 1998-04-17 | 1999-04-16 | Multiparameter facs assays to detect alterations in cellular parameters |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2002512362A JP2002512362A (ja) | 2002-04-23 |
| JP2002512362A5 JP2002512362A5 (ja) | 2006-06-01 |
| JP4511033B2 true JP4511033B2 (ja) | 2010-07-28 |
Family
ID=26742137
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000544822A Expired - Lifetime JP4511033B2 (ja) | 1998-04-17 | 1999-04-16 | Facsを用いて細胞表現型を変化し得る生物活性剤についてスクリーンする方法 |
Country Status (12)
| Country | Link |
|---|---|
| EP (2) | EP1071809B1 (ja) |
| JP (1) | JP4511033B2 (ja) |
| CN (1) | CN100334227C (ja) |
| AT (1) | ATE248226T1 (ja) |
| AU (1) | AU3565499A (ja) |
| CA (1) | CA2325597C (ja) |
| DE (1) | DE69910756T2 (ja) |
| DK (1) | DK1071809T3 (ja) |
| ES (1) | ES2207203T3 (ja) |
| NZ (1) | NZ508120A (ja) |
| PT (1) | PT1071809E (ja) |
| WO (1) | WO1999054494A2 (ja) |
Families Citing this family (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030215798A1 (en) | 1997-06-16 | 2003-11-20 | Diversa Corporation | High throughput fluorescence-based screening for novel enzymes |
| US7090976B2 (en) | 1999-11-10 | 2006-08-15 | Rigel Pharmaceuticals, Inc. | Methods and compositions comprising Renilla GFP |
| US6651008B1 (en) | 1999-05-14 | 2003-11-18 | Cytokinetics, Inc. | Database system including computer code for predictive cellular bioinformatics |
| US6743576B1 (en) | 1999-05-14 | 2004-06-01 | Cytokinetics, Inc. | Database system for predictive cellular bioinformatics |
| US7151847B2 (en) | 2001-02-20 | 2006-12-19 | Cytokinetics, Inc. | Image analysis of the golgi complex |
| JP5389307B2 (ja) * | 2000-06-01 | 2014-01-15 | インスティチュート・パスツール | 単細胞レベルでの生物発光性Ca++リポーターとしてのキメラ性GFP−エクオリン |
| US7218764B2 (en) | 2000-12-04 | 2007-05-15 | Cytokinetics, Inc. | Ploidy classification method |
| US6956961B2 (en) | 2001-02-20 | 2005-10-18 | Cytokinetics, Inc. | Extracting shape information contained in cell images |
| US7016787B2 (en) | 2001-02-20 | 2006-03-21 | Cytokinetics, Inc. | Characterizing biological stimuli by response curves |
| EP1415156B1 (en) | 2001-07-10 | 2009-09-02 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for detecting the activation state of multiple proteins in single cells |
| US7393656B2 (en) | 2001-07-10 | 2008-07-01 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and compositions for risk stratification |
| US7381535B2 (en) | 2002-07-10 | 2008-06-03 | The Board Of Trustees Of The Leland Stanford Junior | Methods and compositions for detecting receptor-ligand interactions in single cells |
| US7235353B2 (en) | 2003-07-18 | 2007-06-26 | Cytokinetics, Inc. | Predicting hepatotoxicity using cell based assays |
| GB2423988A (en) | 2003-07-18 | 2006-09-13 | Cytokinetics Inc | Characterizing biological stimuli by response curves |
| US20050014217A1 (en) | 2003-07-18 | 2005-01-20 | Cytokinetics, Inc. | Predicting hepatotoxicity using cell based assays |
| EP1668355A4 (en) * | 2003-08-28 | 2011-11-09 | Celula Inc | METHODS AND APPARATUS FOR SORTING CELLS USING AN OPTICAL SWITCH IN A MICROFLUIDIC CHANNEL NETWORK |
| JP4454998B2 (ja) | 2003-09-29 | 2010-04-21 | 株式会社ニコン | 細胞観察装置および細胞観察方法 |
| US7323318B2 (en) | 2004-07-15 | 2008-01-29 | Cytokinetics, Inc. | Assay for distinguishing live and dead cells |
| JP4557685B2 (ja) | 2004-11-15 | 2010-10-06 | 独立行政法人理化学研究所 | 蛍光蛋白質 |
| KR20080073293A (ko) | 2005-10-14 | 2008-08-08 | 메디뮨 엘엘씨 | 항체 라이브러리의 세포 디스플레이 |
| FR2902440B1 (fr) * | 2006-06-19 | 2008-08-29 | Univ Paris Curie | Methode de detection et de denombrement des cellules en cytodierese |
| CN101802182A (zh) | 2007-08-21 | 2010-08-11 | 诺达利蒂公司 | 用于诊断、预后和治疗方法的方法 |
| US8227202B2 (en) | 2008-07-10 | 2012-07-24 | Nodality, Inc. | Methods for diagnosis, prognosis and methods of treatment |
| US8309306B2 (en) | 2008-11-12 | 2012-11-13 | Nodality, Inc. | Detection composition |
| US9459246B2 (en) | 2009-09-08 | 2016-10-04 | Nodality, Inc. | Induced intercellular communication |
| US20110193950A1 (en) * | 2010-02-09 | 2011-08-11 | Stephen Liye Chen | Application of microscopic linear array scanner for measuring rate of change of live organisms |
| WO2012013204A1 (en) * | 2010-07-28 | 2012-02-02 | Canvax Biotech Sl | Novel ultrasensitive cell based sensors and uses thereof |
| US9005909B2 (en) | 2011-01-06 | 2015-04-14 | Rigel Pharmaceuticals, Inc. | Whole blood assay for measuring AMPK activation |
| WO2016153819A1 (en) | 2015-03-25 | 2016-09-29 | The Board Of Trustees Of The Leland Stanford Junior University | Single cell analysis using secondary ion mass spectrometry |
| EP3443123B1 (en) | 2016-04-11 | 2021-08-11 | Board of Regents, The University of Texas System | Methods and compositions for detecting single t cell receptor affinity and sequence |
| CN110412179A (zh) * | 2018-04-26 | 2019-11-05 | 缪荣明 | 一种液相色谱检测颗粒酶a的方法 |
| US12352765B2 (en) | 2018-11-15 | 2025-07-08 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods of prognosis and classification for preeclampsia |
| JP7577340B2 (ja) * | 2019-10-24 | 2024-11-05 | 国立大学法人千葉大学 | 細胞内脂質代謝酵素活性定量測定法、細胞内脂質代謝酵素活性定量測定用キット及び脂質代謝酵素活性制御剤のスクリーニング方法 |
| MX2022008801A (es) * | 2020-01-15 | 2022-11-07 | Absci Corp | Enriquecimiento de células específico de la actividad. |
| US20230296622A1 (en) | 2020-08-17 | 2023-09-21 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods of predicting time to onset of labor |
| EP4343775A1 (en) | 2022-09-22 | 2024-03-27 | SurgeCare | A method for determining a medical outcome for an individual, related electronic system and computer program |
| CN120539027B (zh) * | 2025-05-26 | 2025-12-02 | 古贝春集团有限公司 | 窖泥老化快速检测方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4304720A (en) * | 1980-04-30 | 1981-12-08 | Merck & Co., Inc. | Fluorescein esters and ethers and the preparation thereof |
| US5266464A (en) * | 1988-02-10 | 1993-11-30 | Ict Pharmaceuticals, Inc. | Method of screening for protein inhibitors and activators |
| AU6179690A (en) * | 1989-08-09 | 1991-03-11 | Michael Reese Hospital And Medical Center | Method for detection and treatment of multiple drug-resistant tumor cells and useful colchicine derivative probes |
| US5264341A (en) * | 1989-08-30 | 1993-11-23 | Eli Lilly And Company | Selective cloning for high monoclonal antibody secreting hybridomas |
| JPH04252128A (ja) * | 1990-08-03 | 1992-09-08 | Systemix Inc | 免疫化された宿主における変性された異種移植器官系 |
| US5849600A (en) * | 1993-11-10 | 1998-12-15 | The Mclean Hospital Corporation | Diagnostic assays for Alzheimer's disease |
| US5723288A (en) * | 1994-05-06 | 1998-03-03 | The University Of North Carolina At Chapel Hill | Method of fluorescent detection of nucleic acids and cytoskeleton elements using bis-dicationic aryl furans, and kits useful therefor |
| GB9506466D0 (en) * | 1994-08-26 | 1995-05-17 | Prolifix Ltd | Cell cycle regulated repressor and dna element |
| GB9516138D0 (en) * | 1995-08-05 | 1995-10-04 | Medical Res Council | Improvements in or relating to methods of screening substances |
| JPH09100289A (ja) * | 1995-10-02 | 1997-04-15 | Fujisawa Pharmaceut Co Ltd | 新規化合物fr190895、fr190872及びfr190873、その製造方法およびその用途 |
| US20030215798A1 (en) * | 1997-06-16 | 2003-11-20 | Diversa Corporation | High throughput fluorescence-based screening for novel enzymes |
| KR20060079258A (ko) * | 1996-10-18 | 2006-07-05 | 제넨테크, 인크. | 항-ErbB2 항체 |
| JPH11164694A (ja) * | 1997-08-15 | 1999-06-22 | Chugai Pharmaceut Co Ltd | 細胞周期調節タンパク質 |
-
1999
- 1999-04-16 ES ES99917563T patent/ES2207203T3/es not_active Expired - Lifetime
- 1999-04-16 CN CNB998070904A patent/CN100334227C/zh not_active Expired - Lifetime
- 1999-04-16 NZ NZ508120A patent/NZ508120A/en not_active IP Right Cessation
- 1999-04-16 EP EP99917563A patent/EP1071809B1/en not_active Expired - Lifetime
- 1999-04-16 JP JP2000544822A patent/JP4511033B2/ja not_active Expired - Lifetime
- 1999-04-16 AU AU35654/99A patent/AU3565499A/en not_active Abandoned
- 1999-04-16 DK DK99917563T patent/DK1071809T3/da active
- 1999-04-16 DE DE69910756T patent/DE69910756T2/de not_active Expired - Lifetime
- 1999-04-16 EP EP03019052A patent/EP1363127A3/en not_active Withdrawn
- 1999-04-16 AT AT99917563T patent/ATE248226T1/de active
- 1999-04-16 CA CA2325597A patent/CA2325597C/en not_active Expired - Lifetime
- 1999-04-16 PT PT99917563T patent/PT1071809E/pt unknown
- 1999-04-16 WO PCT/US1999/008345 patent/WO1999054494A2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| EP1071809B1 (en) | 2003-08-27 |
| WO1999054494A2 (en) | 1999-10-28 |
| ES2207203T3 (es) | 2004-05-16 |
| CN1304489A (zh) | 2001-07-18 |
| WO1999054494A3 (en) | 2000-02-10 |
| EP1363127A3 (en) | 2004-03-24 |
| CA2325597C (en) | 2010-09-21 |
| DK1071809T3 (da) | 2003-12-01 |
| DE69910756T2 (de) | 2004-07-08 |
| EP1363127A2 (en) | 2003-11-19 |
| JP2002512362A (ja) | 2002-04-23 |
| AU3565499A (en) | 1999-11-08 |
| ATE248226T1 (de) | 2003-09-15 |
| DE69910756D1 (de) | 2003-10-02 |
| CA2325597A1 (en) | 1999-10-28 |
| CN100334227C (zh) | 2007-08-29 |
| PT1071809E (pt) | 2004-01-30 |
| EP1071809A2 (en) | 2001-01-31 |
| NZ508120A (en) | 2003-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4511033B2 (ja) | Facsを用いて細胞表現型を変化し得る生物活性剤についてスクリーンする方法 | |
| CN106574292B (zh) | 利用作为指示剂的miRNA的表达区分期望的细胞类型的方法 | |
| KR20020059370A (ko) | 융합 라이브러리의 제작 및 사용을 위한 방법 및 조성물 | |
| WO2007035553A2 (en) | Methods and compositions for detecting neoplastic cells | |
| EP1228233B1 (en) | Methods and compositions comprising renilla gfp | |
| US6461813B2 (en) | Multiparameter FACS assays to detect alterations in cell cycle regulation | |
| US20130310278A1 (en) | Multiparameter FACS Assays to Detect Alterations in Cellular Parameters and to Screen Small Molecule Libraries | |
| US20030099932A1 (en) | Retroviral vectors with separation sequences | |
| US20050277116A1 (en) | Compositions and methods for the identification of protein interactions in vertebrate cells | |
| JP4388372B2 (ja) | 高スループット小分子薬物発見のための培養ヒト肥満細胞および好塩基球の産生 | |
| US6897031B1 (en) | Multiparameter FACS assays to detect alterations in exocytosis | |
| JP2004503206A (ja) | 致死表現系を引き起こす物質を同定するための方法およびその物質 | |
| AU2004202242B2 (en) | Multiparameter FACS assays to detect alterations in cellular parameters and to screen small molecule libraries | |
| Setu et al. | Calcyphosine is a microtubule-associated protein required for spindle formation and function | |
| CELLULAR | Fisher et al. | |
| Setu | Functional Study of a Drosophila Gene CG10126, and Its Mammalian Ortholog Calcyphosine in Cultured Cells | |
| US7435546B2 (en) | System to detect inducible translocation | |
| WO2010064142A1 (en) | Screening method for the identification of inhibitors of viral entry | |
| EP2123770A1 (en) | Method for determining the DNA repair capacity of a cell | |
| Micklem et al. | A new live-cell reporter strategy to simultaneously monitor mitochondrial biogenesis and morphology | |
| KR20070048386A (ko) | p53 종양 억제단백질과 형광단백질의 융합단백질을발현하는 형질전환 세포주 및 이를 이용한 p53 종양억제단백질의 활성과 관련된 물질의 검색방법 | |
| Wong | The interaction between FHL1B and PP2A (Cbeta) and their functions in regulating cell cycle progression |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060405 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060405 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090402 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090428 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090724 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090827 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20100315 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100406 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100506 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130514 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140514 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |