JP4677684B2 - 高分子−フィラー複合材料の製造方法 - Google Patents
高分子−フィラー複合材料の製造方法 Download PDFInfo
- Publication number
- JP4677684B2 JP4677684B2 JP2001163509A JP2001163509A JP4677684B2 JP 4677684 B2 JP4677684 B2 JP 4677684B2 JP 2001163509 A JP2001163509 A JP 2001163509A JP 2001163509 A JP2001163509 A JP 2001163509A JP 4677684 B2 JP4677684 B2 JP 4677684B2
- Authority
- JP
- Japan
- Prior art keywords
- filler
- plate
- polymer
- mixture
- composite material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0066—Use of inorganic compounding ingredients
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Inorganic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Casting Or Compression Moulding Of Plastics Or The Like (AREA)
- Manufacture Of Porous Articles, And Recovery And Treatment Of Waste Products (AREA)
- Injection Moulding Of Plastics Or The Like (AREA)
- Shaping By String And By Release Of Stress In Plastics And The Like (AREA)
- Molding Of Porous Articles (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
Description
【発明の属する技術分野】
本発明は、高分子−フィラー複合材料の製造方法に関し、より詳しくは、板状フィラーを含有し、弾性率、強度、耐衝撃性等の特性に優れたフィルム材料や発泡体を得ることが可能な高分子−フィラー複合材料の製造方法に関する。
【0002】
【従来の技術】
従来より、高分子材料にフィラーを添加して高分子材料の物性を向上させたり成形性を向上させる試みがなされてきた。高分子材料に添加するフィラーとしては、近年、層状粘土鉱物等に由来した板状フィラーが注目されており、例えば、米国特許5,717,000号公報には、有機物で処理された層状粘土鉱物をポリオレフィン等に添加した後、発泡剤で発泡させて得られた高分子発泡体が開示されている。
【0003】
【発明が解決しようとする課題】
しかしながら、上記公報に開示の方法やその他公知の従来技術に基づいて作製された高分子−フィラー複合材料の、弾性率、強度、衝撃エネルギー等の特性は必ずしも充分とは言えるものではなかった。かかる従来技術の問題点は、高分子材料中に分散されるフィラーの分散状態が充分に制御されていないことに起因するものであると本発明者らは考察するが、従来、層状粘土鉱物等の板状フィラーを高分子材料中で一定の規則性をもって分散させる手法は知られていなかった。
【0004】
本発明はかかる問題点に鑑みてなされたものであり、高分子材料中に分散される板状フィラーの分散状態を制御することができ、弾性率、強度、耐衝撃性等の特性に優れた高分子−フィラー複合材料を生じせしめることの可能な高分子−フィラー複合材料の製造方法を提供することを目的とする。
【0005】
【課題を解決するための手段】
本発明者らは、上記の目的を達成すべく鋭意研究を重ねた結果、特定サイズの板状フィラーが特定濃度で分散された高分子材料を特定の歪み速度で変形させることにより、弾性率、強度、耐衝撃性等の特性に優れた高分子−フィラー複合材料を得ることができることを見出し、本発明を完成させた。
【0006】
すなわち、本発明の高分子−フィラー複合材料の製造方法は、有機化処理された板状フィラーと高分子材料の混合物であって、前記混合物中における前記板状フィラーの平均面積(L2)、平均厚み(d)及び体積分率(c)が、それぞれ下記式(1)、(2)及び(3)を満たす混合物を得る混合物作製工程と、
500nm2≦L2≦100μm2 …(1)
d≦L/20 …(2)
0.2d/L≦c≦4d/L …(3)
前記混合物に対して、歪み速度10s−1以下で変形を与えることにより前記板状フィラーに3次元的なクラスターを形成させる変形工程と、を含み、前記板状フィラーが、スメクタイト、バーミキュライト、ハロイサイト、膨潤性マイカ及びカオリナイトからなる群より選ばれる少なくとも一種であることを特徴とするものである。
【0007】
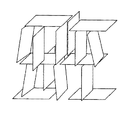
本発明の製造方法においては、上記一般式(1)及び(2)の条件を満たしたサイズの板状フィラーを上記一般式(3)の濃度で高分子材料中で分散させるために、混合物はレオペクシー性及びストレインハードニングを生じうるものとなる。また、混合物を歪み速度10s-1以下でさせるため、高分子材料中で板状フィラーが3次元的なクラスターを形成する。ここで、板状フィラーの3次元的なクラスターとは、高分子材料中で板状フィラーが「カードハウス構造」を形成していることを意味し、そのモデル構造は図1に示される。本発明においては、高分子材料中で板状フィラーがカードハウス構造を形成することにより、高分子−フィラー複合材料の弾性率、強度、耐衝撃性等の特性の向上が達成されるものと考えられる。
【0008】
カードハウス構造は、理想的にはそれぞれの板状フィラーの面の法線が、互いに直交するx軸、y軸又はz軸のいずれかの方向に向いた状態で(すなわち板状フィラーの法線同士は互いに直交した状態で)、板状フィラーが存在した構造をいうが、図1に示されるようにx軸、y軸及びz軸のうち2軸方向においてのみ、上記関係が成立っているものもカードハウス構造に含まれる。また、板状フィラーの濃度(数)はx軸、y軸及びz軸の各方向でそれぞれ異なっていてもよい。更には、図2に示すように板状フィラーの法線同士がほぼ直交するものであれば、全てが直交しないものであってもよい。すなわち、板状フィラーの面の法線と、x軸、y軸及びz軸のいずれかの軸とが平行関係にあることが理想的であるが、法線と軸とが平行関係から多少ずれ、法線と軸のなす角度が平均で45°以下(好ましくは30°以下、更に好ましくは15°以下)であれば、本発明においてカードハウス構造が形成されたものと考えることができる。
【0009】
【発明の実施の形態】
以下、本発明の好適な実施形態について詳細に説明する。
本発明の高分子−フィラー複合材料製造方法は、上述のように混合物作製工程と変形工程とを含むものである。
【0010】
先ず、混合物作製工程について説明する。混合物作製工程においては、板状フィラーと高分子材料の混合物であって、前記混合物中における前記板状フィラーの平均面積(L2)、平均厚み(d)及び体積分率(c)が、それぞれ下記式(1)、(2)及び(3)を満たす混合物を得る。
500nm2≦L2≦100μm2 …(1)
d≦L/20 …(2)
0.2d/L≦c≦4d/L …(3)
【0011】
板状フィラーの平均面積がL2<500nm2又はL2>100μm2の場合では、高分子−フィラー複合材料はレオペクシー性を発現せず、変形を与えてもストレインハードニングの現象は認められない。したがって、カードハウス構造が形成されないと考えられ、得られる高分子−フィラー複合材料の特性が不充分となる。また、L2<500nm2では板状フィラーが細かすぎて充分な補強効果が発現しない。一方、L2>100μm2の場合には高分子−フィラー複合材料を用いて作製される成形体の表面が荒れるため実用性に劣る。板状フィラーの平均面積(L2)は、1000nm2≦L2≦25μm2がより好ましく、2000nm2≦L2≦5μm2が更に好ましい。
【0012】
本発明においては、板状フィラーの平均厚み(d)はL≫dでなければならず、具体的にはd≦L/20でなければならない。d>L/20ではレオペクシー性が発現せず、変形を与えてもストレインハードニングの現象は認められない。したがって、カードハウス構造が形成されないと考えられ、得られる高分子−フィラー複合材料の特性が不充分となる。
【0013】
板状フィラーの面の形状は特に限定されないが、面の長軸及び短軸をそれぞれn及びmとした場合に、n/m≦100、m/d≧5が好ましい。n/m>100、m/d<5ではフィラーが棒状に近くなり、高分子−フィラー複合材料はレオペクシー性を発現し難くなり、変形によるカードハウス構造の形成が困難になる傾向にある。
【0014】
本発明において板状フィラーの体積分率(c)は、0.2d/L≦c≦4d/Lでなければならない。c<0.2d/L又はc>4d/Lでは、高分子−フィラー複合材料はレオペクシー性を発現せず、変形を与えてもストレインハードニングの現象は認められない。したがって、カードハウス構造が形成されないと考えられ、得られる高分子−フィラー複合材料の特性が不充分となる。また、c<0.2d/Lでは高分子−フィラー複合材料を用いて作製される成形体の補強効果が少なく、c>4d/Lでは成形体が硬く脆くなりすぎ実用性に劣る。板状フィラーの体積分率(c)は、0.4d/L≦c≦3.5d/Lがより好ましく、0.5d/L≦c≦3d/Lが更に好ましい。体積分率がこのような範囲内にある場合においては、レオペクシー性の発現がより顕著となり、明確なストレインハードニングが認められてカードハウス構造が形成されるため、得られる高分子−フィラー複合材料の特性が特に優れるようになる。なお、本発明において板状フィラーの体積分率とは、板状フィラーの体積を板状フィラーと高分子材料の合計体積で除した値(小数)であり、体積百分率(%)の1/100の値である。
【0015】
ここで、板状フィラーの体積分率につき図1に示すモデル構造を用いて検討を加える。図1は、一辺の長さがLである正方形の面を有し、厚みd(L≫d)の板状フィラーからなるカードハウス構造を示すものである。この構造が体積pqr(p,q,r≫L)のマトリックス全体に渡って均一に形成されているとすると、マトリックス中に存在する板状フィラーの総数は、約2pqr/L3個である。したがって、この構造が形成されたときの板状フィラーの体積分率は約2d/L(200d/L体積%)となる。図1では板状フィラーが正方形である場合を仮定したが、板状フィラーが極端に細長くならない限り板状フィラーの体積分率は上記値となると考えられ、この値は板状フィラーが四角形以外の形状であっても成立つと考えられる。
【0016】
この構造から板状フィラーがいくつか欠落したとしても、ある体積分率まではマトリックス全体に渡って巨大なクラスターが存在すると考えられる。この体積分率の下限はパーコレーションの臨界点Pcを考えればよく、これは0.2<Pc<0.6の範囲にあると考えられる。したがって、板状フィラーがマトリックス全体に渡って巨大なクラスターを形成するためには、フィラーの体積分率を0.4d/L以上とすることが好ましい。また、マトリックス全体に渡るよりも幾分小さなクラスターを形成しても、粘度の急上昇が起こり得るため、板状フィラーの体積分率の下限は0.2d/Lとなる。実験的にも板状フィラーの濃度がこの値を下回るとどのような変形を与えてもストレインハードニング現象が認められない。これは、カードハウス構造が形成されていないか、形成されていたとしてもクラスターのサイズが小さすぎることによるものと考察される。また、クラスターサイズが小さい場合はマトリックスの粘度を高める効果が発現しない。
【0017】
なお、本発明者らの知見によれば、本発明の製造方法により図2に示すように幾分乱れたカードハウス構造が形成されることがあり、また、一方向で板状フィラーの濃度が高くなることが生じうる。この傾向は、板状フィラーとして後述する層状粘土鉱物由来のものを用いたときに顕著である。このような場合は、板状フィラーの体積分率がある程度まで高くなってもカードハウス構造は形成されるが、体積分率が高くなりすぎると構造形成が不可能になる。これは板状フィラー同士の立体障害が大きいことによるものと考えられ、この点から板状フィラーの体積分率の上限は4d/Lとなる。板状フィラーの体積分率がこの値よりも高い場合は、どのような変形を与えてもストレインハードニング現象は認められず、カードハウス構造が形成されない。
【0018】
本発明において、変形によるカードハウス構造の形成容易性の観点から、板状フィラーは正又は負に帯電しているか、分極していることが好ましい。帯電及び分極の程度は任意であり、板状フィラー間でわずかでも電気的な相互作用が生じれば、レオペクシー性が向上しカードハウス構造の形成が容易になる。この場合において、板状フィラーには電荷対となるような有機イオンが付加されていてもよく、また、板状フィラーの分極を促進するような物質が付加されていてもよい。
【0019】
本発明において用いられる板状フィラーは、上述した条件を満たすものであればよく、その種類は特に制限されない。板状フィラーは高分子材料に添加する前において上述の条件を満たしていなくても、高分子材料と混合物を形成した状態で上述の条件を満たしていればよい。このような板状フィラーとしては、層状粘土鉱物に由来するものが好ましく、中でも高分子材料中で一層ごとに微細分散するように表面を有機化処理した有機化層状粘土鉱物が特に好ましい。板状フィラーとして層状粘土鉱物を用いる場合においては、層状粘土鉱物は一層ごとに完全に分散されていなくてもよく、数層が重なっていてもよい。この場合において、板状フィラーの厚みdとしては、重なり全体の厚みを考慮して平均値を用いる。
【0020】
ここで、層状粘土鉱物とは層状フィロ珪酸塩を意味し、かかる層状粘土鉱物としては、例えば、モンモリロナイト、サポナイト、ヘクトライト、バイデライト、スティブンサイト、ノントロナイト等のスメクタイト系の層状粘土鉱物や、バーミキュライト、ハロイサイト、膨潤性マイカ、カオリナイト等を例示できる。これらは天然のものでも、合成されたものでもよい。また、有機化層状粘土鉱物とは、有機オニウムイオンによって有機化された層状粘土鉱物を意味する。有機オニウムイオンとしては、炭素数が6以上のアルキルオニウムイオンが代表的である。炭素数が6未満であると、有機オニウムイオンの疎水性が低くなるために、ポリオレフィン系オリゴマーやポリオレフィン樹脂を高分子材料として用い、これに層状粘土鉱物を分散させたときの相溶性が低下する恐れがある。
【0021】
層状粘土鉱物の選択に当たり、これを添加する高分子材料との接触面積が大きいものを用いると、層状粘土鉱物の層間距離を広げて分散が可能になることから好ましい。また、層状粘土鉱物の陽イオン交換容量は、50〜200ミリモル当量/100gであることが好ましい。陽イオン交換容量が50ミリモル当量/100g未満の場合には有機オニウムイオンのイオン交換による有機化が不充分となり易いために、結果的に層状粘土鉱物の分散が困難になる場合がある。一方、陽イオン交換容量が200ミリモル当量/100gを超える場合には、層状粘土鉱物の層間結合力が強固であるために有機オニウムイオンのイオン交換による層間への介入が困難になり、結果的に層状粘土鉱物の分散が不充分になる。
【0022】
有機オニウムイオンとしては、第1〜4級アンモニウムイオンやホスフェニウムイオンを用いることができる。アンモニウムイオンとしては、ヘキシルアンモニウムイオン、オクチルアンモニウムイオン、2−エチルヘキシルアンモニウムイオン、ドデシルアンモニウムイオン、ラウリルアンモニウムイオン、オクタデシルアンモニウムイオン、ジオクチルジメチルアンモニウムイオン、トリオクチルアンモニウムイオン、ジオクタデシルジメチルアンモニウムイオン、トリオクチルアンモニウムイオン、トリオクタデシルアンモニウムイオン、ジメチルベンジルオクタデシルアンモニウムイオン、ジメチルオクタデシルアンモニウムイオンが挙げられ、ホスフェニウムイオンとしては、テトラエチルホスフェニウムイオン、トリエチルベンジルホスフェニウムイオン、テトラ−n−ブチルホスフェニウムイオン、トリ−n−ブチルヘキサデシルホスフェニウムイオン、トリ−n−ブチルベンジルホスフェニウムイオンが挙げられる。
【0023】
本発明において上述した板状フィラーと混合物を形成する高分子材料は特に限定されないが、高分子材料の少なくとも一部が、極性基を有する高分子材料であることが好ましい。極性基としては、分極を有した基、酸性基及び塩基性基が挙げられる。高分子材料が極性基を有する高分子材料を含む場合は、変形にともなって板状フィラーがカードハウス構造を形成し易くなる。
【0024】
本発明において用いることのできる高分子材料としては、ポリエステル(ポリエチレンナフタレート、ポリエチレンテレフタレート、ポリブチレンテレフタレート等)、ポリカーボネート、ポリアセタール、ポリアリレート、ポリアミド、ポリアミドイミド、ポリエーテルイミド、ポリフェニレンエーテル、ポリフェニレンサルファイド、ポリエーテルサルホン、ポリエーテルケトン、ポリフタルアミド、ポリエーテルニトリル、ポリエーテルサルホン、ポリベンズイミダゾール、ポリカルボジイミド、ポリシロキサン、アクリル樹脂(ポリメチル(メタ)アクリレート、ポリ(メタ)アクリルアミド等)、アクリルゴム、ポリ4フッ化エチレン、フッ素樹脂、フッ素ゴム、液晶ポリマー、エポキシ樹脂、メラミン樹脂、ユリア樹脂、ジアリルフタレート樹脂、フェノール樹脂、ポリシラン、シリコーン樹脂、ウレタン樹脂、ポリエチレン、ポリプロピレン、ポリブテン、ポリペンテン、エチレン−プロピレン共重合体、エチレン−ブテン共重合体、ポリブタジエン、ポリイソプレン、水添ポリブタジエン、水添ポリイソプレン、エチレン−プロピレン−ジエン共重合体、エチレン−ブテン−ジエン共重合体、ブチルゴム、ポリメチルペンテン、ポリスチレン、スチレン−ブタジエン共重合体、スチレン−水添ブタジエン共重合体、ポリ塩化ビニル、ポリ塩化ビニリデン、ポリフッ化ビニル、ポリフッ化ビニリデン、ポリビニルアルコール、ポリ(エチレン−ビニルアルコール)共重合体(EVOH)、ポリビニルブチラール、酢酸セルロース、ポリ乳酸、ポリヒドロキシブチレート、ポリカプロラクトン、ポリブチレンサクシネート、ポリエチレンサクシネート、アクリロニトリル−ブタジエン−スチレン共重合体(ABS)、アクリロニトリル−ブタジエンゴム(NBR)が挙げられる。これらの高分子材料は単独又は2種以上を組み合わせて(ブレンド又はアロイ)用いることができる。
【0025】
上述した板状フィラーと高分子材料との混合物の製造方法としては、板状フィラーとして層状粘土鉱物を用いる場合は、層状粘土鉱物の有機化を行った後に、有機化層状粘土鉱物を高分子材料と混合する方法が採用可能である。
【0026】
層状粘土鉱物の有機化は、例えば以下のように実施することができる。すなわち、層状粘土鉱物が塊状の場合は、まずこれをボールミル等により粉砕し粉体化する。次いで、ミキサー等を用いてこの粉体を水中に分散させ層状粘土鉱物の水分散物を得る。これとは別に、有機化剤である有機オニウムイオンを含む水溶液を調製する。この水溶液を上記層状粘土鉱物の水分散物に加え混合することにより、層状粘土鉱物中の無機イオンが有機オニウムイオンによりイオン交換され、この混合物から水を除去することにより有機化層状粘土鉱物が得られる。ここで分散媒体としては、水以外にもメタノール、エタノール、プロパノール、イソプロパノール、エチレングリコール、これらの混合物及びこれらと水の混合物等も使用することが可能である。
【0027】
次いで、有機化層状粘土鉱物と高分子材料を混合する。用いる高分子材料が水や有機溶剤等の溶媒に溶解性又は分散性を有する場合は、有機化層状粘土鉱物と高分子材料を、水や有機溶剤等の溶媒に分散若しくは溶解させた後、溶媒を除去することにより混合が可能である。
【0028】
また、高分子材料と有機化層状粘土鉱物とを、高分子材料の融点若しくは軟化点以上に加熱して混合する方法も採用できる。加熱時には、せん断力を加え有機化された層状粘土鉱物を均一に分散させることが好ましく、加熱しつつせん断力を加える手段としては押出機を用いることが好ましい。この際、有機溶媒、オイル等を添加することができ、層状粘土鉱物の分散後あるいは分散過程において、高分子の架橋及び/又は加硫を行ってもよい。
【0029】
上記の方法以外にも、例えば、高分子材料となるべきモノマーに有機化層状粘土鉱物を加え、有機化層状粘土鉱物の存在下で当該モノマーの重合を行い、混合物を得ることも可能である。また、ポリウレタンやポリエステル等のように2以上の成分を混合し反応させることにより高分子材料が生成するような場合は、反応前の各成分の少なくとも1つに予め有機化層状粘土鉱物を添加しておくことにより、混合物を得ることも可能である。
【0030】
次に、本発明の高分子−フィラー複合材料の製造方法における変形工程について説明する。変形工程においては、上述した板状フィラーと高分子材料の混合物に対して、歪み速度10s-1以下で変形を与える。
【0031】
混合物を変形させる歪み速度が10s-1を超す場合は、歪み速度が速すぎて板状フィラーがカードハウス構造を形成することができない。したがって、ストレインハードニング現象も認められず、得られる高分子−フィラー複合材料の特性が不充分となる。本発明において歪み速度(γ’)は、0.0001s-1≦γ’≦5s-1が好ましく、0.0001s-1≦γ’≦5s-1が更に好ましい。歪み速度が0.0001s-1未満である場合は、変形に時間がかかりすぎる傾向にあり、実用性に問題が生じる場合がある。
【0032】
変形工程において、変形を与える温度(T)は、マトリックスとなる高分子材料が融点を有する高分子である場合、その融点をTmとすると、T≧Tm−30℃であることが好ましい。マトリックスとなる高分子材料が非晶性、又は結晶化度が低くガラス転移温度(Tg)付近以上で流動性を示す場合は、T≧Tg−30℃であることが好ましい。高分子材料が可塑剤を含む等してTm、Tgが本来の値よりも低下している場合には、その低下の程度に応じてTを適宜選択することができる。上記の温度範囲よりも低温では、変形流動性が乏しく、板状フィラーがカードハウス構造を形成し難くなる傾向にある。
【0033】
本発明においては、歪み速度10s-1以下の変形を、延伸又は発泡により実施することが好ましい。ここで、延伸には、一軸延伸、二軸延伸の他、ブロー成形による延伸も含まれる。発泡は、樹脂(板状フィラーと高分子材料の混合物)にブタン、ペンタンのような炭化水素、炭酸ガス等を加熱加圧下で含浸せしめ、発泡性樹脂としたものを型内で加熱して発泡させる方法(ガス含浸、型内加熱発泡法)、押出機バレル内で樹脂、発泡剤及び発泡核生成剤をよく混練し、金型を通して大気圧下に放出しながら発泡させる方法(押出し、大気圧下物理発泡法)、物理発泡剤を直接射出成形機に注入して樹脂と混合したり、化学発泡剤を樹脂にブレンドしたものを射出成形機に投入し、熱分解して発生したガスの発泡圧を射出金型に導入して発泡成形を行う方法(射出発泡成形法)、加熱プレス中で樹脂中に混合された分解型発泡剤をガス化し、除圧と同時に発泡する方法(プレス発泡法)、反応しながら重合する樹脂において反応時に発生する炭酸ガスや水を用いて発泡させる方法(化学反応法)等、公知の発泡方法がいずれも採用可能である。発泡剤としては、熱分解により発生するガスを利用する化学発泡剤を用いてもよく、炭酸ガスや不活性ガスを注入、含浸させて放出させる物理発泡剤を用いてもよい。特に、物理発泡剤を用いる場合には、発泡剤が超臨界状態となる加圧、加熱下で、発泡剤を樹脂に含浸、混入させ、後に減圧し、必要に応じて再加熱することにより発泡させることが望ましい。
【0034】
板状フィラーとして層状粘土鉱物由来のものを用いた場合の混合物を、一軸延伸や二軸延伸等により歪み速度10s-1以下で延伸した場合は、伸長(延伸)方向に垂直な方向、及び伸長方向に対して平行であるが板状成形体の厚み方向に対しては垂直な方向の2方向に、層状粘土鉱物が配向してカードハウス構造が形成される傾向にあり、これにより得られる高分子−フィラー複合材料の引張弾性率や衝撃エネルギーが向上する。一方、同様の組成の混合物を発泡により歪み速度10s-1以下で変形させた場合は、空隙部を仕切る隔壁において、隔壁の厚み方向、及び厚み方向と垂直方向に層状粘土鉱物が配向してカードハウス構造を形成する傾向にあり、得られる発泡体の曲げ弾性率、曲げ強さ等の特性が向上する。したがって、本発明の製造方法により得られる高分子−フィラー複合材料は、高い曲げ弾性率、曲げ強さ等の特性が求められる分野に好適に用いることができる。
【0035】
【実施例】
以下、本発明の好適な実施例についてさらに詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0036】
(実施例1)
板状フィラーであるナトリウム型モンモリロナイト(クニミネ工業社製、クニピアF、陽イオン交換容量:119ミリモル当量/100g)80gを80℃の水5000mLに分散させた。一方、有機化剤であるオクタデシルアミン28.5gと濃塩酸11mLを80℃で2000mLの水に溶解させた。この溶液を先のナトリウム型モンモリロナイトの水分散液に加え、これにより有機化モンモリロナイト(以下「C18−Mt」という)を得た。得られたC18−Mtを80℃の水で3回洗浄した後、凍結乾燥し粉砕した。灼残法により求めたC18−Mt中の無機分量は68重量%であった。
【0037】
次いで、マレイン酸変性ポリプロピレン樹脂(エクソン化学社製、PO1015、融点:145℃)にC18−Mtを無機分に換算した量で4重量%添加した。これを二軸押出機(日本製鋼所製、TEX30α)を用いて溶融混練し、C18−Mtがマレイン酸変性ポリプロピレン樹脂中に分散した混合物を得た。この混合物の透過型電子顕微鏡(TEM)写真から、得られた混合物中において層状粘土鉱物は1層毎に剥離して存在していることがわかった。
【0038】
なお、クニピアFの1層の平均面積(L2)は10000nm2であり、平均厚み(d)は1nmであるため式(2)はd≦5nmとなる。すなわち、実施例1においては式(1)及び(2)の要件が満たされている。また、かかる条件においては式(3)は0.002≦c≦0.04となるが、PO1015の密度は0.86g/cm3、クニピアFの密度は2.5g/cm3であるため、板状フィラーの体積分率は0.014となり、実施例1においては式(3)の要件も満たされている。
【0039】
次に、得られた混合物を射出成形機(日精樹脂工業社製、PS40E2ASE)を用いて直方体状の試験片を作製した。そして、伸長粘度試験機(豊田工業大学製)を用いてこの試験片の伸長粘度の時間変化を測定した。得られた結果を図3に示す。なお、この測定において、測定温度は150℃であり、歪み速度(ここでは真歪み速度を意味するため、変形速度は伸度が進むにしたがって速くなる)は0.001〜1s-1であった。
【0040】
また、伸長粘度の時間変化の測定後の試験片から図4に示すようなTEM観察用試験片を切り出し、当該試験片の上面及び前面から板状フィラーの配向状態をTEMにより観察した。上面から観察されたTEM写真を図5に、前面から観察されたTEM写真を図6に示す。
【0041】
(比較例1)
板状フィラーを含有しないマレイン酸変性ポリプロピレン樹脂(エクソン化学社製、PO1015、融点:145℃)を用いて、実施例1と同様にして伸長粘度の時間変化を測定した。得られた結果を図7に示す。
【0042】
(比較例2〜3)
クニピアFの体積分率をそれぞれ0.0018及び0.042とした他は実施例1と同様にして、伸長粘度の時間変化を測定した。
【0043】
以上の実験結果から、実施例1で得られた混合物は0.001〜1s-1の伸長歪み速度範囲でストレインハードニングの現象が生じることがわかった(図3)。一方、板状フィラーを含有しないマレイン酸変性ポリプロピレン樹脂は、0.001〜1s-1の伸長歪み速度範囲でストレインハードニングの現象を全く示さず(図7)、板状フィラーの体積分率が式(3)を満足しない比較例2〜3においても、同様の伸長歪み速度範囲でストレインハードニングの現象を全く示さないことがわかった。
【0044】
また、図5及び図6に示されるように、伸長粘度の時間変化の測定後の試験片においては、伸長方向に垂直な方向、及び伸長方向に対して平行であるが試験片の厚み方向に対しては垂直な方向の2方向に、板状フィラーが配向してカードハウス構造が形成されていることがわかった(かかるカードハウス構造の模式図を図8に示す)。なお、図6における実線の丸で囲んだ板状フィラーは紙面に垂直方向に配向したものを示し、破線の丸で囲んだ板状フィラーは紙面に平行方向に配向したものを示す。
【0045】
(実施例2)
内容積100mLのオートクレーブ(豊田中央研究所社製)に、実施例1で得られた混合物とドライアイス40gを入れ、バルブの付いた蓋を閉めて140℃に加熱したオイルバスに浸した。加熱により超臨界状態になった二酸化炭素を混合物に充分に吸収させるため、1時間放置した後、オイルバスからオートクレーブを取り出し、バルブを急激に開いてオートクレーブ内を急減圧させて、歪み速度10s-1以下で発泡させた。得られた発泡体から凍結破面を作製し、発泡状態を走査型電子顕微鏡(SEM)で観察した。得られたSEM写真を図9に示す。また、発泡体の空隙部(発泡セル)を仕切る隔壁から超薄切片を切り出し、TEMで板状フィラーの配向状態を観察した。得られたTEM写真を図10に示す。
【0046】
(比較例4)
板状フィラーを含有しないマレイン酸変性ポリプロピレン樹脂(エクソン化学社製、PO1015、融点:145℃)を用いて、実施例2と同様にして発泡体を作製し、SEM写真を得た。得られたSEM写真を図11に示す。
【0047】
以上の電子顕微鏡観察の結果、実施例2の発泡体は図9に示すように良好な空隙部(発泡セル)を多数有するものであったのに対し、比較例4の発泡体は図11に示すように、空隙部(発泡セル)は認められるものの形が崩れていたり、潰れているものが多数見受けられた。また、実施例2の発泡体の隔壁には、隔壁の厚み方向に対して水平な面を持つよう配向した板状フィラーと、これに垂直な面を持つように配向した板状フィラーが観察され、隔壁部分で板状フィラーがカードハウス構造を形成していることが示された(図10)。なお、図10における実線の丸で囲んだ板状フィラーは紙面に垂直方向に配向したものを示し、破線の丸で囲んだ板状フィラーは紙面に平行方向に配向したものを示す。
【0048】
(実施例1〜2及び比較例1及び4の特性試験)
実施例1及び比較例1で得られた直方体状の試験片と、実施例2及び比較例4で得られた発泡体からクラックが入らないように注意深く切り出した試験片を用いて、JIS K7171に準拠して曲げ試験を行い、曲げ弾性率及び曲げ強さを求めた。また、各試験片の密度を測定し、曲げ弾性率及び曲げ強さを密度で除して、単位密度あたりの曲げ弾性率及び曲げ強さを算出し、これらをそれぞれ比弾性率及び比強度とした。この結果をまとめて以下の表1に示す。
【0049】
【表1】

非発泡体である実施例1及び比較例1、発泡体である実施例2及び比較例4を比較することにより、曲げ強度及び曲げ強さの実測値はいずれも実施例の方が優れており、単位密度あたりの補正値である比弾性率及び比強度も実施例の方が優れていることがわかった。なお、実施例2は発泡体であるにもかかわらず、比弾性率及び比強度の値が他の実施例や比較例に比べて非常に大きくなっており、良好なカードハウス構造の形成が推測された。
【0051】
(実施例3)
小型卓上プレス(高千穂精機製、TW−07型)を用い、実施例1で得られた混合物を200℃でプレス成形することにより、厚さ1mmのフィルムを作製した。これをフィルム延伸装置(柴山科学器械製作所製、延伸装置 SS−60特型)を用い、初期歪み速度0.5s-1(工学的歪み速度を意味し、延伸が進むにつれて真歪み速度は低下する。なお、変形速度は一定である)、温度130℃で一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。
【0052】
(比較例5)
実施例3と同様の小型卓上プレスを用いて、実施例1で得られた混合物を200℃でプレス成形することにより、厚さ0.5mmのフィルムを作製した。なお、得られたフィルムは未延伸フィルムである。
【0053】
(比較例6)
実施例1で得られた混合物に代えて、板状フィラーを含有しないマレイン酸変性ポリプロピレン樹脂(エクソン化学社製、PO1015、融点:145℃)を用いた他は、実施例3と同様にして厚さ1mmのフィルムを作製した後、更に実施例3と同様にして、一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。
【0054】
(実施例4)
マレイン酸変性された直鎖状低密度ポリエチレン(デュポン社製、Fusabond MB−226D)とC18−Mtを用い、実施例1と同様にして混合物を作製した。なお、C18−Mtは無機分換算で4重量%添加しており、Fusabond MB−226Dの密度は0.92g/cm3であるので、板状フィラーの体積分率は0.015となり、実施例4においては式(1)〜(3)の要件を全て満たしていた。また、得られた混合物のTEM観察を行ったところ板状フィラー(層状粘土鉱物)のほぼ全てが一層毎に分散していた。次いで、得られた混合物を用いて、実施例3と同様にして厚さ1mmのフィルムを作製した後、更に実施例3と同様にして、一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。但し、一軸延伸時の温度は110℃とした。
【0055】
(比較例7)
実施例3と同様の小型卓上プレスを用いて、実施例4で得られた混合物を200℃でプレス成形することにより、厚さ0.5mmのフィルムを作製した。なお、得られたフィルムは未延伸フィルムである。
【0056】
(比較例8)
Fusabond MB−226DとC18−Mtとの混合物に代えて、板状フィラーを含有しないFusabond MB−226Dを用いた他は、実施例4と同様にして厚さ1mmのフィルムを作製した後、更に実施例4と同様にして、一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。
【0057】
(実施例5)
ナイロン6(宇部興産社製、1022B、融点:220℃)とC18−Mtを用い、実施例1と同様にして混合物を作製した。なお、C18−Mtは無機分換算で4重量%添加しており、1022Bの密度は1.13g/cm3であるので、板状フィラーの体積分率は0.018となり、実施例5においては式(1)〜(3)の要件を全て満たしていた。また、得られた混合物のTEM観察を行ったところ板状フィラー(層状粘土鉱物)のほぼ全てが一層毎に分散していた。次いで、得られた混合物を用いて、実施例3と同様にして厚さ1mmのフィルムを作製した後、更に実施例3と同様にして、一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。但し、一軸延伸時の温度は110℃とした。
【0058】
(比較例9)
実施例3と同様の小型卓上プレスを用いて、実施例5で得られた混合物を200℃でプレス成形することにより、厚さ0.5mmのフィルムを作製した。なお、得られたフィルムは未延伸フィルムである。
【0059】
(比較例10)
1022BとC18−Mtとの混合物に代えて、板状フィラーを含有しない1022Bを用いた他は、実施例5と同様にして厚さ1mmのフィルムを作製した後、更に実施例5と同様にして、一軸延伸することにより、厚さ0.5mmの延伸フィルムを得た。
【0060】
(実施例3〜5及び比較例5〜10の特性試験)
実施例3〜5、比較例6、8及び10の延伸フィルムと、比較例5、7及び8の未延伸フィルムとを用いて、JIS K7142及びJIS K7127に準拠して、衝撃エネルギー及び引張弾性率を求めた。得られた結果をまとめて以下の表2に示す。
【0061】
【表2】
表2より、実施例3〜5の延伸フィルムは、同様の組成であるにもかかわらず延伸を行っていないため板状フィラーがカードハウス構造を形成していない未延伸フィルム(比較例5、7及び9)や、同様の延伸を行ったにもかかわらず板状フィラーを含有しない延伸フィルム(比較例6、8及び10)に比べて、より優れた引張弾性率及び衝撃エネルギーを示すことがわかった。
【0063】
【発明の効果】
以上説明したように、本発明によれば、高分子材料中に分散される板状フィラーの分散状態を制御することができ、弾性率、強度、耐衝撃性等の特性に優れた高分子−フィラー複合材料を生じせしめることの可能な高分子−フィラー複合材料の製造方法を提供することが可能になる。
【図面の簡単な説明】
【図1】板状フィラーのカードハウス構造のモデルを示す図である。
【図2】板状フィラーのカードハウス構造のモデルを示す図である。
【図3】実施例1で得られた試験片の伸長粘度の時間変化を示す図である。
【図4】実施例1における伸長粘度の時間変化の測定後の試験片から切り出されたTEM観察用試験片の模式図である。
【図5】図4の模式図における上面から撮影された、実施例1のTEM観察用試験片のTEM写真である。
【図6】図4の模式図における前面から撮影された、実施例1のTEM観察用試験片のTEM写真である。
【図7】比較例1における伸長粘度の時間変化を示す図である。
【図8】実施例1のTEM観察用試験片における板状フィラーの配向状態の模式図である。
【図9】実施例2で得られた発泡体のSEM写真である。
【図10】実施例2で得られた発泡体の隔壁のTEM写真である。
【図11】比較例4で得られた発泡体のSEM写真である。
Claims (5)
- 有機化処理された板状フィラーと高分子材料の混合物であって、
前記混合物中における前記板状フィラーの平均面積(L2)、平均厚み(d)及び体積分率(c)が、それぞれ下記式(1)、(2)及び(3)を満たす混合物を得る混合物作製工程と、
500nm2≦L2≦100μm2 …(1)
d≦L/20 …(2)
0.2d/L≦c≦4d/L …(3)
前記混合物に対して、歪み速度10s−1以下で変形を与えることにより前記板状フィラーに3次元的なクラスターを形成させる変形工程と、を含み、
前記板状フィラーが、スメクタイト、バーミキュライト、ハロイサイト、膨潤性マイカ及びカオリナイトからなる群より選ばれる少なくとも一種であることを特徴とする高分子−フィラー複合材料の製造方法。 - 前記変形を、延伸により実施することを特徴とする請求項1記載の高分子−フィラー複合材料の製造方法。
- 前記変形を、発泡により実施することを特徴とする請求項1記載の高分子−フィラー複合材料の製造方法。
- 前記板状フィラーが、層状粘土鉱物に由来するものであることを特徴とする請求項1〜3のいずれか一項に記載の高分子−フィラー複合材料の製造方法。
- 前記高分子材料の少なくとも一部が、極性基を有する高分子材料であることを特徴とする請求項1〜4のいずれか一項に記載の高分子−フィラー複合材料の製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001163509A JP4677684B2 (ja) | 2001-05-30 | 2001-05-30 | 高分子−フィラー複合材料の製造方法 |
| US10/155,999 US6821464B2 (en) | 2001-05-30 | 2002-05-29 | Process for producing polymer/filler composite material |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001163509A JP4677684B2 (ja) | 2001-05-30 | 2001-05-30 | 高分子−フィラー複合材料の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2002356560A JP2002356560A (ja) | 2002-12-13 |
| JP4677684B2 true JP4677684B2 (ja) | 2011-04-27 |
Family
ID=19006463
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001163509A Expired - Fee Related JP4677684B2 (ja) | 2001-05-30 | 2001-05-30 | 高分子−フィラー複合材料の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US6821464B2 (ja) |
| JP (1) | JP4677684B2 (ja) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002363393A (ja) * | 2001-06-07 | 2002-12-18 | Unitika Ltd | 生分解性ポリエステル樹脂組成物、その製造方法、及びそれより得られる発泡体 |
| JP4116373B2 (ja) * | 2002-09-17 | 2008-07-09 | 帝人化成株式会社 | ポリカーボネート樹脂組成物から形成された発泡体 |
| US20050131126A1 (en) * | 2003-02-27 | 2005-06-16 | Kumin Yang | Production of polymer nanocomposites using supercritical fluids |
| JP4473517B2 (ja) * | 2003-03-25 | 2010-06-02 | ユニチカ株式会社 | 微細な気泡を有する熱可塑性樹脂発泡体およびその製造方法 |
| JP4713063B2 (ja) * | 2003-05-08 | 2011-06-29 | 日本合成化学工業株式会社 | エチレン−酢酸ビニル共重合体ケン化物発泡体およびエチレン−酢酸ビニル共重合体ケン化物発泡体の製造方法 |
| EP1484357A1 (fr) * | 2003-06-06 | 2004-12-08 | Université de Liège | Procédé de préparation de mousses de polyester biodégradables, mousses de polyester telles qu'ainsi obtenues et leur utilisation, et procédé de modification de nanocharges |
| JP4337976B2 (ja) * | 2004-02-20 | 2009-09-30 | 積水化成品工業株式会社 | 熱可塑性芳香族ポリエステル系樹脂発泡体の製造方法 |
| JP4498783B2 (ja) | 2004-03-17 | 2010-07-07 | トヨタ紡織株式会社 | 木質成形体の製造方法 |
| JP2005307192A (ja) * | 2004-03-26 | 2005-11-04 | Toyota Central Res & Dev Lab Inc | ポリオレフィン複合材料、その製造方法及び成形体 |
| EP1787918B1 (en) * | 2004-06-10 | 2012-06-13 | Unitika, Ltd. | Biodegradable gas barrier vessel and process for producing the same |
| WO2006084163A2 (en) * | 2005-02-02 | 2006-08-10 | E.I. Dupont De Nemours And Company | Composite comprising cellulose and thermoplastic polymer |
| JP5196544B2 (ja) * | 2008-03-24 | 2013-05-15 | 曙ブレーキ工業株式会社 | 無機複合化樹脂材料およびその製造方法 |
| CN112639013A (zh) * | 2018-08-29 | 2021-04-09 | 住友化学株式会社 | 水性分散体 |
| JP7380852B2 (ja) * | 2020-04-14 | 2023-11-15 | 三菱電機株式会社 | 樹脂成形用金型及び貯湯タンクユニットの製造方法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4990688A (ja) * | 1972-12-29 | 1974-08-29 | ||
| US4739007A (en) * | 1985-09-30 | 1988-04-19 | Kabushiki Kaisha Toyota Chou Kenkyusho | Composite material and process for manufacturing same |
| US4894411A (en) * | 1987-03-18 | 1990-01-16 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Composite material and process for producing the same |
| JPH0778089B2 (ja) * | 1987-03-26 | 1995-08-23 | 株式会社豊田中央研究所 | 複合材料の製造方法 |
| US5328650A (en) * | 1991-10-08 | 1994-07-12 | Owens Illinois Closure Inc. | Method of distributing pigments in compression molded products |
| JPH07331013A (ja) * | 1994-06-07 | 1995-12-19 | Tosoh Corp | ポリビニルアルコール複合材料およびその溶融成形物 |
| US5773121A (en) * | 1994-07-29 | 1998-06-30 | Isorca Inc. | Syntactic foam core incorporating honeycomb structure for composites |
| US5973053A (en) * | 1995-06-05 | 1999-10-26 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Composite clay material and method for producing the same, blend material and composite clay rubber using the same and production method thereof |
| DE69708605T2 (de) * | 1996-02-23 | 2002-08-01 | The Dow Chemical Co., Midland | Dispersionen aus schichtgespaltenen partikeln in kunststoffschäumen |
| JP3377159B2 (ja) * | 1996-09-04 | 2003-02-17 | 株式会社豊田中央研究所 | 粘土複合ゴム材料の製造方法 |
| JPH10279705A (ja) * | 1997-04-09 | 1998-10-20 | Mitsubishi Chem Corp | 難燃性ポリエステルエラストマーシート及びその用途 |
| DE19882437B4 (de) * | 1998-03-30 | 2004-02-12 | Kabushiki Kaisha Toyota Chuo Kenkyusho, Nagakute | Verfahren zur Herstellung eines ein Polymer umfassenden Verbundmaterials |
| NL1013520C2 (nl) | 1999-07-19 | 2001-01-22 | Dsm Nv | GeÙxtrudeerd polyolefine vormdeel. |
| JP2001123000A (ja) | 1999-10-27 | 2001-05-08 | Sekisui Chem Co Ltd | ポリオレフィン系樹脂発泡体 |
-
2001
- 2001-05-30 JP JP2001163509A patent/JP4677684B2/ja not_active Expired - Fee Related
-
2002
- 2002-05-29 US US10/155,999 patent/US6821464B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002356560A (ja) | 2002-12-13 |
| US20030149156A1 (en) | 2003-08-07 |
| US6821464B2 (en) | 2004-11-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4677684B2 (ja) | 高分子−フィラー複合材料の製造方法 | |
| Adak et al. | Effect of organoclay-type and clay-polyurethane interaction chemistry for tuning the morphology, gas barrier and mechanical properties of clay/polyurethane nanocomposites | |
| Wibowo et al. | Biodegradable nanocomposites from cellulose acetate: Mechanical, morphological, and thermal properties | |
| Sur et al. | Synthesis, structure, mechanical properties, and thermal stability of some polysulfone/organoclay nanocomposites | |
| Chang et al. | Preparation of polyethylene‐octene elastomer/clay nanocomposite and microcellular foam processed in supercritical carbon dioxide | |
| US10538640B2 (en) | Polymer foam and method for preparing the same | |
| Wu et al. | BaTiO3-assisted exfoliation of boron nitride nanosheets for high-temperature energy storage dielectrics and thermal management | |
| Wang et al. | Fabrication of polyamide 6 nanocomposite with improved thermal conductivity and mechanical properties via incorporation of low graphene content | |
| Shi et al. | Reinforcement of mechanical properties of silicone rubber foam by functionalized graphene using supercritical CO2 foaming technology | |
| Li et al. | Polypropylene/hydroxyl-multiwall carbon nanotubes composites: crystallization behavior, mechanical properties, and foaming performance | |
| Jo et al. | Constitutive modeling of HDPE polymer/clay nanocomposite foams | |
| Shao et al. | Morphology and interaction of nanocomposite foams formed with organo-palygorskite and ethylene-vinyl acetate copolymers | |
| JP2007046019A (ja) | 熱可塑性樹脂発泡体およびその製造方法 | |
| KR101042894B1 (ko) | 내열성과 내유성이 우수한 방진 고무 및 이를 위한 나노 콤포지트 조성물 | |
| Zhang et al. | The effect of controlled intercalation on the mechanical performances and dimensional accuracy of material extrusion additive manufactured poly (lactic acid)/organo-montmorillonite nanocomposites | |
| Khorasani et al. | Foaming behavior and cellular structure of microcellular HDPE nanocomposites prepared by a high temperature process | |
| KR101554650B1 (ko) | 나노 복합체 및 나노 복합체 제조 방법 | |
| Zhao et al. | Effect of inorganic nanoparticles on polypropylene in‐mold decoration and microcellular foaming injection molding composites | |
| Zuo et al. | Microcellular foaming and mechanical properties of iPP-iPPF using supercritical CO2 | |
| US20080281011A1 (en) | High temperature resistant, structural polymer foam | |
| Balamurugan et al. | Effects of nanotalc inclusion on mechanical, microstructural, melt shear rheological, and crystallization behavior of polyamide 6‐based binary and ternary nanocomposites | |
| CN110760137A (zh) | 一种高性能阻燃发泡材料及其制备方法 | |
| CN110978366A (zh) | 一种用于提高发泡材料中的功能组分添加量的方法 | |
| Rizvi et al. | Microcellular PP vs. microcellular PP/MMT nanocomposites: A comparative study of their mechanical behavior | |
| Altan et al. | Foam injection molding of polypropylene/zinc oxide nanocomposite with chemical foaming agent: mechanical properties and morphology |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20071211 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100610 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100615 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100831 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101101 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101101 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110104 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110117 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140210 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140210 Year of fee payment: 3 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313532 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140210 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| LAPS | Cancellation because of no payment of annual fees |