JP6055817B2 - ナフチリジンの調製方法 - Google Patents
ナフチリジンの調製方法 Download PDFInfo
- Publication number
- JP6055817B2 JP6055817B2 JP2014509568A JP2014509568A JP6055817B2 JP 6055817 B2 JP6055817 B2 JP 6055817B2 JP 2014509568 A JP2014509568 A JP 2014509568A JP 2014509568 A JP2014509568 A JP 2014509568A JP 6055817 B2 JP6055817 B2 JP 6055817B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- reaction
- alkyl
- ethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[*-]C=C(C(*)=O)C(c1cc([N+]([O-])=O)cnc1Cl)=O Chemical compound C[*-]C=C(C(*)=O)C(c1cc([N+]([O-])=O)cnc1Cl)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/76—Nitrogen atoms to which a second hetero atom is attached
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description
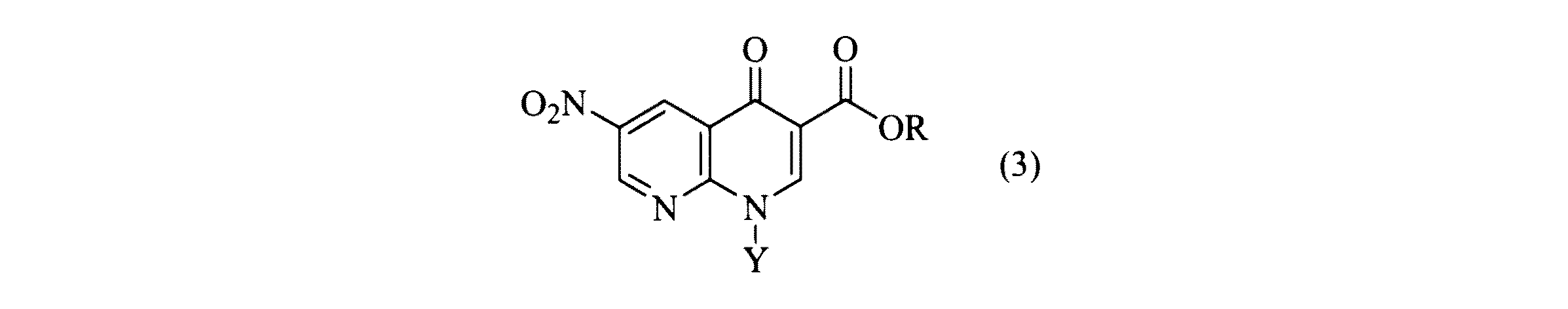
Qは、所望により置換された窒素含有ヘテロシクリルまたは所望により置換された窒素含有ヘテロアリールであり、Yは、低級アルキル基である)
前記方法が:
a)式(2)の化合物を、
b)工程(a)からの式(3)の化合物のニトロ基を還元し、式(4)の化合物を得ること、
d)工程(c)からの式(5)の化合物を加水分解し、式(6)の化合物を得ること、および
前記方法が、
a)式(2)の化合物を、
b)工程a)からの式(2a)の化合物を、非求核塩基および低級アルキルN,N−低級ジアルキルアミノアクリレートで処理し、式(2b)の化合物を得ること(式中、RおよびR’は独立して低級アルキルである)、および
前記方法が
a)式(6)の化合物を、
b)工程a)から得られた式(1)の化合物を弱塩基溶液で洗浄し、精製することを含む、方法を提供する。
(前記、
pは0〜6、qは0〜2であり、Ra、Rb、Rcはそれぞれ独立して、H、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜7シクロアルキル、C4〜7シクロアルケニル、アリール、ヘテロシクリル、ヘテロアリール、C1〜6アルキルアリール、C1〜6アルキルヘテロアリールおよびC1〜6アルキルヘテロシクリルから選択され、前記アルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクリル、ヘテロアリール、C1〜6アルキルアリール、C1〜6アルキルヘテロアリールまたはC1〜6アルキルヘテロシクリルは、所望により、ハロゲン、ヒドロキシ、低級アルキル、低級アルコキシ、−CO2H、CF3、CN、フェニル、NH2およびNO2から選択される1〜6の同一または異なる基により置換されうる
またはRbおよびRcが同一の窒素原子に結合している場合、それらは該原子と共に結合し、5〜7員窒素含有ヘテロ環を形成する)
により置換されうる。
a)式(2a)の化合物を得るのに十分な条件下、
(i)50℃において、鉄(3.0eqs)および塩酸(4.0eqs)
(ii)55〜60℃において、鉄(3.0eqs)および酢酸(13〜15vol)
(iii)ラネーニッケル、水素、メタノールもしくはDCM+メタノール(1:1)もしくは水メタノールまたは酢酸エチル
(iv)ラネーニッケル、水素、TFAまたはメタノール中の酢酸(15%)
(v)55〜60℃において、3:1のアセトン:水またはアセトン:塩酸(1Nまたは2N)中の亜ジチオン酸ナトリウム(4.0eqs)
(vi)炭素上のパラジウム、水素、(メタノールまたはメタノール+1N塩酸触媒)および
(vii)炭素上の10%パラジウム(湿潤)、水素(2kg/cm2の圧力)、メタノール、25〜30℃
a)式(4)の化合物を
N,N−ジメチルホルムアミド(500g)および塩化チオニル(6.0kg)を、窒素雰囲気下、25〜30℃において、フラスコに連続的に加え、反応混合物を加熱還流した。
反応を、10.0mLのメタノールで1.0mLの反応混合物をクエンチさせ(quenching)、2−クロロ−5−ニトロニコチン酸(10%以上とすべきではない)に対するHPLCを記録することにより観察した。
反応完了後(〜3時間)、反応マス(reaction mass)を40〜45℃まで冷却し、有機溶媒を、減圧条件下、45℃以下で蒸留した。残査を25〜30℃まで冷却し、25〜30℃において、攪拌しながらアセトニトリル(10.0L)を添加した。
工程1で調製した酸性塩化物溶液を、25〜30℃において、反応フラスコへ加え、混合物を2.5時間攪拌した。
反応混合物は、0.5mLのメタノールで0.5mLの反応混合物をクエンチさせ(quenching)、2−クロロ−5−ニトロニコチン酸のメチルエステル(5.0%以上とすべきではない)に対するHPLCを記録することにより観察した。
反応完了後(〜4時間)、4.0Lのエチルアミン水溶液(70%)を、反応温度を25〜30℃において維持したまま、ゆっくり添加した(〜2.5時間)。
反応混合物を2.5時間、加熱還流し、アクリル酸エステルを含有する中間体(2.0%以上とすべきではない)に対するHPLCにより観察した。反応は8時間以内に完了することがわかった。反応混合物を、次いで25〜30℃まで冷却し、この温度において3時間維持した。
反応混合物を、〜3時間0〜5℃に静置し、固体はヌッチェフィルターを通して濾過した。溶媒は真空条件下における濾過により除去した。
ウェットケーキをジエチルエーテル(25.0L)中に分散させ、十分に混合し、ヌッチェフィルターを通して濾過した。
溶媒は真空条件下における濾過により除去し、次いで原料を乾燥用トレーに入れた。
50〜55℃において、熱風乾燥機を用いて固体を水分含量が2.0%以下となるまで乾燥させ、次いで、25〜30℃まで冷却し、固体状の望ましいエステルを収率68%で得た。
1H NMR (DMSO-d6, 500MHz): 1.31(3H, t, J =7.0 Hz), 1.40(3H, t, J =7.0 Hz), 4.27(2H, q, J =7.0 Hz), 4.54(2H, q, J =7.0 Hz), 8.96(1H, s), 9.07(1H, d, J =2.5 Hz), 9.59 (1H, d, J =2.5 Hz). 13C NMR (DMSO-d6, 125MHz): 14.16, 14.53, 46.63, 60.27, 113.00, 121.54, 131.45, 140.97, 147.70, 150.61, 151.22, 163.49, 172.57.
反応容器に窒素ガスを連続的に2〜3分間流した。メタノール(255.0L)を窒素雰囲気下、25〜30℃において、反応溶器に加えた。
306gのパラジウム炭素およびエチル1−エチル−6ニトロ−4−オキソ−1,8−ナフチリジン−3−カルボキシレート(5.1kg)を窒素雰囲気下、25〜30℃において、連続的に加えた。
容器の圧力を窒素により1.0kg/cm2まで高め、次いで、徐々に開放した。この作業を三度繰り返した。反応容器を水素ガスで満し、圧力を1.0kg/cm2まで高め、次いで、徐々に開放した。この作業を三度繰り返し、結果、25〜30℃における水素ガスの圧力は2.0kg/cm2まで高まり、その後60〜72時間維持された。
反応の進行は、出発物質(2.0%以上とすべきではない)の消費量およびヒドロキシルアミン中間体(10.0%以上とすべきではない)に対するHPLCにより観察した。
反応の進行が非常に遅かった場合、水素の圧力を徐々に開放し、更に10%のパラジウム炭素(50g)およびメタノール(50.0L)を加え、水素の圧力(2.0kg/cm2)を再設した。
反応完了後(64〜75時間)、水素の圧力を徐々に開放し、反応容器を、窒素ガスで1.0kg/cm2となるまで充填した。窒素の圧力を徐々に開放し、ジクロロメタン(128.0L)を加えた。
ヌッチェフィルターにおいて、ハイフローベッド(Hyflow bed)を2.60kgのハイフローおよび29.1Lのジクロロメタンを用いて調製した。濾過水は廃棄した。
反応混合物を、次いで、真空条件下、ハイフローベッドを通して濾過した。ハイフローベッドはメタノール(38.3L)およびジクロロメタン(38.3L)の混合物を用いて洗浄した。
濾過器を再度真空条件下にさらした。濾過水を蒸溜フラスコへ移し、溶媒を真空条件下、50℃以下で蒸発させた。
フラスコを25〜30℃まで冷却し、25〜30℃においてアセトン(25.5L)を加えた。反応混合物を25〜30℃において攪拌し、真空条件下、ヌッチェフィルターを通して濾過した。
この原料を乾燥用トレーに均等に移し、40〜45℃において、熱風乾燥機を用いて、水分含量が0.5%以下となるまで乾燥させ、次いで25〜30℃まで冷却し固体状の望ましい化合物を65%の収率で得た。
1H NMR (DMSO-d6, 500MHz): 1.29(3H, t, J = 8.4Hz), 1.35(3H, t, J =7.0Hz), 4.23(2H, q, J =7.0Hz), 4.43(2H, q, J =7.0Hz), 5.80(2H, s), 7.67(1H, s), 8.26(1H, d, J = 2.0 Hz), 8.67 (1H, s). 13C NMR (DMSO-d6, 125MHz): 14.32, 15.08, 45.81, 59.51, 108.77, 115.12, 123.87, 140.08, 140.67, 143.57, 146.82, 164.69, 173.30.
工程1:18.0Lの1,2−ジクロロエタン(水分含量0.1%以下)およびホウ化水素(785g)を、窒素雰囲気下、25〜30℃において連続的にフラスコへ加えた。
反応溶液を0〜5℃まで冷却し、4.30Lの酢酸(水分含量0.1%以下)を、0〜10℃に維持したまま反応フラスコへ徐々に加えた。
この反応混合物を25〜30℃まで温め、この温度を12時間維持した。
2−インダノン(1.80kg)を攪拌下、0〜5℃において反応混合物に加え、これに次いで酢酸(136.8L)を0〜5℃の温度に保ちながら徐々に加えた。
反応混合物を0〜5℃において10〜15分攪拌した。13.8Lのトリアセトキシ水素化ホウ素ナトリウム溶液(工程1で調製した)を0〜5℃において、それぞれ4.6Lの3つの受け入れ容器(instalments)に徐々に加え、0〜5℃において2時間攪拌した。
次いで、硫酸ナトリウム(10.1kg)および2−インダノン(878g)を0〜5℃において反応混合物へ連続的に加えた。8.0Lのトリアセトキシ水素化ホウ素ナトリウム溶液(工程1で調製した)を0〜5℃において反応混合物に徐々に加え、反応混合物を0〜5℃で10〜12時間維持した。
反応の進行は、出発物質の消失に対するTLCにより観察した。反応完了後(15時間)、真空条件下、60℃以下において溶媒を除去し、反応マスを25〜30℃まで冷却した。
ジクロロメタン(72.0L)を攪拌下、25〜30℃において、反応混合物に添加し、攪拌を20〜30分の間継続した。
次いで、反応混合物のpHを、炭酸水素ナトリウム水溶液(36kgの炭酸水素ナトリウムを324Lの水に溶解することにより調製)により、7〜8に調整し、反応混合物を25〜30℃において、10〜15分攪拌した。
二層は15〜20分間を超えて分離していることができ、次いで有機層が分離した。
水層を36Lのジクロロメタンで二回洗浄し、全ての有機層を溜め、水(2×36L)および塩化ナトリウム溶液(7.2kgの塩化ナトリウムを28.8Lの水に溶解することにより調製)で二回洗浄した。最終的に無水硫酸ナトリウム上において25〜30℃で15〜20分間攪拌することにより乾燥させた。
溶液はヌッチェフィルターを通して濾過した。濾過水を反応容器へ移し、溶媒を真空条件下、40℃以下で蒸発させた。
シリカゲル(25.2kg)をプラグカラム(plugged column)へ加え、粗製反応マスをカラム上へ担持させた。カラムをジクロロメタン(1728.0L)、ジクロロメタン(864L)中の5%酢酸エチル、ジクロロメタン(865L)中の7.5%酢酸エチルおよびジクロロメタン(1500L)中の10%酢酸エチルで溶出した。
画分をTLCにより分析し、同時に純物質を含有する画分を反応容器中へ貯留した。
溶媒を、減圧条件下、45℃以下において蒸留し、残存マスを25〜30℃まで冷却した。
ジエチルエーテル(14.4L)を25〜30℃において反応容器へ加え、2時間攪拌した。反応マスはヌッチェフィルターを通して濾過した。
固体を乾燥用トレーに広げ、35〜40℃において、熱風乾燥機を用いて乾燥させ、2.2kgの望ましい固体状の生成物を得た。
1H NMR (DMSO-d6, 500MHz): 1.29(3H, t, J =7.0Hz), 1.36(3H, t, J = 7.0Hz), 2.84-2.88(2H, m), 3.5-3.39(2H, m), 4.22(2H, q, J = 7.0Hz), 4.31-4.33(1H,m), 4.44(2H, q, J = 7.0Hz), 6.73(1H, d, J = 6.5Hz), 7.16-7.17(2H. m), 7.25-7.27(2H, m), 7.59(1H, d, J = 3.0 Hz), 8.35(1H, d, J =3.0 Hz), 8.68 (1H, s). 13C NMR (DMSO-d6, 125MHz): 14.29, 15.10, 45.85, 53.07, 59.52, 109.00, 112.15, 123.78, 124.60, 126.36, 140.04, 140.69, 141.21, 142.78, 146.65, 164.72, 173.17.
エタノール(42.0L)を25〜30℃において反応用器に加え、次いで攪拌しながらエチル1−エチル−6−(インダン−2−イルアミノ)−4−オキソ−1,8−ナフチリジン−3−カルボキシレート(4.20kg)を加えた。
水酸化ナトリウム水溶液(3.4kgの水素ナトリウムを42.0Lの水に溶解させることにより調製)を25〜30℃において反応混合物に加え、反応温度を50〜55℃に上昇させた。
反応混合物を50〜55℃において2時間攪拌し、反応の進行をTLCにより観察した。加水分解の完了後(〜3時間)、反応マスを25〜30℃まで冷却し、クエン酸水溶液(5.2kgのクエン酸を47.0Lの水に溶解することにより調製)を添加することによりpHを5〜6に調整した。
反応マスを25〜30℃において、20〜25分の間攪拌し、濾過し、固体マスを水(42.0L)及びアセトン(21.0L)で洗浄した。
原料を乾燥用トレーに移し、70〜75℃において、熱風乾燥機で水分含量が1.0%に減少するまで乾燥し、望ましい固体状の化合物(90%)を得た。
1H NMR (DMSO-d6, 500MHz): 1.40(3H, t, J = 7.0Hz), 2.86-2.90(2H, m), 3.37-3.41(2H, m), 4.38(1H, d, J = 5.5 Hz), 4.62(2H, q, J = 7.0Hz), 7.06(1H, d, J = 6.0Hz), 7.17-7.18(2H. m), 7.26-7.27(2H, m), 7.60(1H, d, J = 2.5 Hz), 8.52(1H, d, J =2.0 Hz), 9.00 (1H, s), 15.30(1H, s). 13C NMR (DMSO-d6, 125MHz): 15.19, 46.92, 52.92, 107.12, 109.42, 121.58, 124.63, 126.41, 140.11, 141.12, 143.33, 143.43, 145.93, 166.12, 177.53.
工程1:160.0Lのジクロロメタン(水分含量を0.1%以上とすべきではない)、1−エチル−6−(インダン−2−イルアミノ)−4−オキソ−1,8−ナフチリジン−3−カルボン酸(4.0kg)およびトリエチルアミン(3.5kg)を窒素雰囲気下、25〜30℃において、反応容器に連続的に加え、反応混合物を10〜15℃まで冷却した。
塩化ピバノイル(4.1kg)を、反応温度を10〜15℃に保ちながら、反応混合物へ徐々に加えた。
次いで、反応温度25〜30℃まで上昇させ、攪拌した。
反応の進行は、出発物質の消失に対するTLCにより観察した。
反応完了後(3〜4時間)、反応混合物を15〜20℃にまで再度冷却し、攪拌し、かつ反応混合物を15〜20℃に保ちながら、モルフォリン(6.0kg)を加えた。
N,N−ジメチル−4−アミノピリジン(194g)およびDMF(2.0L)を15〜20℃において反応混合物へ加え、加熱還流した。
反応の進行は、ピバノイルエステル中間体の消失に対するTLCにより観察し、12〜13時間以内に完了することが判明した。
反応混合物を15〜20℃まで冷却し、次いで、攪拌しながら重炭酸ナトリウム水溶液(5.6kgの重炭酸ナトリウムを56.0Lの水に溶解することにより調製)を添加することによりクエンチさせた。
有機層を分離し、塩化ナトリウム水溶液(23.0kgの塩化ナトリウムを57.0Lの水に溶解することにより調製)で洗浄した。有機層を分離し、無水硫酸ナトリウム(4.0kg)を用いて攪拌することにより乾燥した。
有機層を、ヌッチェフィルターを通して濾過し、硫酸ナトリウムをジクロロメタンで洗浄した。濾過液はフラスコへ移し、真空条件下、40℃以下において蒸発させた。
フラスコ中の結果得られた材料を25〜30℃まで冷却し、ジエチルエーテル(40.0L)に懸濁させた。
分離した固体を、ヌッチェフィルターを用いて濾過し、ジエチルエーテル(8.0L)で洗浄し、単離した湿式固体をジクロロメタン(20.0L)へ溶解した。
次いで、その溶液(10.0L)を、ジクロロメタン(36.0L)次いでジクロロメタン中10Lの10%メタノールを用いてシリカゲルプラグ(10.0kg)を通して濾過した。シリカゲルフィルターは真空条件下、乾燥させた。
同様に、溶液(10.0L)の残部を別のシリカゲルプラグ(10.0kg)を通して濾過した。複合濾過液を真空条件下、40℃以下において蒸発させ、次いで、残留固体を25〜30℃において、攪拌しながら、酢酸エチル(20.0L)に懸濁させた。
分離した固体を、ヌッチェフィルターを通して濾過し、酢酸エチル(4.0L)で洗浄した。フィルターを減圧条件下、乾燥し、次いで、材料を乾燥用トレーに移し、40〜45℃において乾燥させ、2.36kg得た。
1H NMR (DMSO-d6, 500MHz): 1.49 (3H, t, J = 7.2Hz), 2.91(2H, dd, J = 3.5Hz, 16.0 Hz), 3.42-3.47(4H, m), 3.80(6H, s), 4.25-4.26(1H, bd), 4.40-4.48(3H, m), 7.20-7.25(4H, m), 7.82(1H, d, J = 3.0Hz), 8.09 (1H, s), 8.18(1H, d, J = 3.0Hz). 13C NMR (CDCl3, 150MHz): 15.27, 39.87, 43.05, 46.66, 48.09, 53.93, 66.80, 67.40, 113.27, 116.71, 123.22, 124.92, 126.78, 140.87, 141.46, 141.83, 141.90, 143.84, 166.27, 173.38.
エチル1−エチル−6−(インダン−2−イルアミノ)−4−オキソ−1,8−ナフチリジン−3−カルボキシレート
2−インダノン(210mg、1.6mmol)を反応混合物に添加し、イミン形成が完了(LCMSによる出発物質の消失により示される)(〜30分から1時間)するまで攪拌した。
2−ピコリンボラン錯体(140mg、1.3mmol)を反応混合物へ加え、激しく攪拌し、イミン還元を完了(LCMSによるイミンの消失)させる。
反応混合物をビーカーへ移し、酢酸エチル(20mL)で希釈し、氷冷の水酸化ナトリウム水溶液(1.0M)(〜90mL、酢酸のほぼ90%をクエンチするのに十分な)を加え、次いで、固体重炭酸ナトリウムで塩基性化した。
有機層を分離し、塩水で洗浄し、硫酸マグネシウム上で乾燥させて、減圧下、蒸発乾固した。粗物をジクロロメタン(20mL)中に溶解し、ヘキサン(〜20mL)を滴下し懸濁させた。
分離した固体をジエチルエーテルおよびヘキサンの1/1混合物で洗浄し、黄色の固体物として望ましい化合物を得た(収率:70%、HPLC純度:95%)。
2−インダノン(180mg、1.4mmol)を反応混合物に加え、1時間攪拌した。
反応混合物を無水ジエチルエーテル(10mL)で希釈し、分離した固体を濾過し、冷ジエチルエーテルで洗浄し、真空条件下乾燥させ、暗黄色固体状のイミン(275mg、74%)を得た。
反応混合物をビーカーへ移し、酢酸エチル(40mL)で希釈し、氷冷の水酸化ナトリウム水溶液(35mL,2.0M)(酢酸のほぼ90%をクエンチするのに十分な)を添加し、次いで、反応混合物を、固体重炭酸ナトリウムでpH7〜8まで塩基性化した。
有機層を分離し、塩水で洗浄し、硫酸マグネシウム上で乾燥し、減圧下、蒸発乾固し、暗黄色の固体物を得た(260mg、95%、HPLC純度:87%)
2−インダノンを反応混合物に加え、イミン形成が完了するまで攪拌した(LCMSによる出発物質の消失により示される)(〜30分から1時間)。
反応混合物にボロン還元剤(表1に記載したように)を添加し、激しく攪拌し、イミン還元を完了(LCMS中のイミンの消失)させた。
反応混合物をビーカーへ移し、酢酸エチルで希釈し、氷冷の水酸化ナトリウム水溶液(1.0M)(酢酸のほぼ90%をクエンチするのに十分な)を加え、次いで、更に反応混合物を、固体重炭酸ナトリウムでpH7〜8まで塩基性化した。
有機層を分離し、塩水で洗浄し、硫酸マグネシウム上で乾燥し、減圧下、蒸発乾固した。
表1に規定される、粗物はシリカゲルのプラグをDCM中の酢酸エチルの勾配を増大させながら通過し、暗黄色固体状の望ましい生成物を提供した。
6−(2,3−ジヒドロ−1H−インデン−2−イルアミノ)−1−エチル−3−(モルフォリン−4−イルカルボニル)−1,8−ナフチリジン−4(1H)−オン
エチルクロロギ酸エステル(143μL、1.5mmol)を、シリンジを介して反応混合物へ添加し、反応混合物を10分攪拌した。
冷浴を除去し、反応温度を室温まで上昇できるようにし、反応が完了するよう攪拌した(〜20分)。
反応混合物を再度冷浴中で冷却し、モルフォリン(261mg、3mmol)を、シリンジを介して加えた。
反応混合物を室温で、アミド形成が完了するまで攪拌する(〜30分)。
反応混合物を再度、冷浴中で冷却し、メタノールを数滴添加することによりクエンチし、次いで、酢酸エチルで希釈した。
有機層を炭酸水素ナトリウム(飽和水溶液)、塩水で洗浄し、分離し、硫酸マグネシウム上で乾燥し、蒸発乾固した。
粗物をメタノール(5mL)に溶解し、熱水(7mL、〜70℃)を加えた。
分離した固体を濾過し、高真空条件下、乾燥し、望ましい淡黄色固体状の原料(350mg、84%、HPLC純度:99%)を得た。
反応混合物を、反応が完了するまで(〜1時間)50℃に加熱した。
反応を炭酸水素ナトリウム(飽和水溶液)でクエンチし、DMCにより抽出した。
抽出物を混合し、水および塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、減圧下で濃縮した。
粗物は、DMC中のメタノールの勾配(1%〜5%)を増加させながらシリカの小さなプラグを通過し、望ましい淡黄色固体物を得た。
Claims (18)
- 式(1)の化合物の調製方法であって、
Qは、所望により置換された窒素含有ヘテロシクリルまたは所望により置換された窒素含有ヘテロアリールであり、Yは、C1〜C6アルキル基である)
前記方法が:
a)式(2)の化合物を、
b)工程(a)からの式(3)の化合物のニトロ基を還元し、式(4)の化合物を得ること、
d)工程(c)からの式(5)の化合物を加水分解し、式(6)の化合物を得ること、および
- 工程a)における前記酸性活性化剤が塩化チオニルであり、前記反応が塩素系溶媒中で起こる、請求項1に記載の方法。
- 前記反応を、触媒量のDMF存在下で行う、請求項2に記載の方法。
- 前記非求核塩基がトリエチルアミン(TEA)である、請求項1〜3のいずれか一項に記載の方法。
- 前記C1〜C6アルキル−N,N−C1〜C6ジアルキルアミノアクリレートがエチルN,N−ジメチルアミノアクリレートである、請求項1〜4のいずれか一項に記載の方法。
- 前記C1〜C6アルキルアミンがエチルアミンである、請求項1〜5のいずれか一項に記載の方法。
- 前記ニトロ基の還元工程が、式(3)の化合物を鉄または亜ジチオン酸ナトリウムから選択される適切な還元剤にさらすこと、または式(3)の化合物を、触媒としてパラジウム(Pd)若しくはラネーニッケルを用いて加水分解することを含む、請求項1〜6のいずれか一項に記載の方法。
- 工程(c)における前記還元剤が、水素化ホウ素ナトリウム、水素化ホウ素リチウム、水素化ホウ素カリウム、トリアセトキシ水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウム、トリエチル水素化ホウ素リチウム、ピリジンボラン、ピコリンボラン、5−エチル−2−メチルピリジンボランおよびtert−ブチルアミン−ボランから選択される、ホウ素を含有する還元剤である、請求項1〜7のいずれか一項に記載の方法。
- 工程(c)を、助溶媒として用いられるC1〜C6カルボン酸とともに、有機溶媒の存在下行う、請求項1〜8のいずれか一項に記載の方法。
- 工程(c)をイミンの前記初期生成を伴う2工程の処理として行う、請求項1〜9のいずれか一項に記載の方法。
- 工程(b)において、前記式(4)の化合物を1〜2.5モル当量の2−インダノンで処理する、請求項1〜10のいずれか一項に記載の方法。
- 工程e)における、前記酸性活性化剤がクロロギ酸アルキルまたは塩酸である、請求項1〜11のいずれか一項に記載の方法。
- 反応工程(e)を、塩素系溶媒中、TEAの存在下で行う、請求項12に記載の方法。
- 工程(e)が、ジクロロメタン中、0〜20℃の温度において、式(6)の化合物の溶液を1〜2モル当量の塩化チオニルまたは塩化オキサリルを用いて処理することを含む、請求項1〜13のいずれか一項に記載の方法。
- 前記溶液がジクロロメタンに対して、5〜10%の濃度のDMFを含む、請求項14に記載の方法。
- 前記QHがモルフォリンである、請求項1〜15のいずれか一項に記載の方法。
- 工程(a)が、式(2a)の化合物を得るのに十分な条件下、
RおよびYは独立してC1〜C6アルキル基である) - 工程(c)が、式(4)の化合物を、式(5)の化合物を得るのに十分な条件下、2−インダノンおよび適切な還元剤で処理することを含み、
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2011901791A AU2011901791A0 (en) | 2011-05-12 | Chemical Processes | |
| AU2011901791 | 2011-05-12 | ||
| PCT/AU2012/000533 WO2012151640A1 (en) | 2011-05-12 | 2012-05-11 | Methods for preparing naphthyridines |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2014513142A JP2014513142A (ja) | 2014-05-29 |
| JP6055817B2 true JP6055817B2 (ja) | 2016-12-27 |
Family
ID=47138578
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014509568A Active JP6055817B2 (ja) | 2011-05-12 | 2012-05-11 | ナフチリジンの調製方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9133188B2 (ja) |
| EP (1) | EP2707367B1 (ja) |
| JP (1) | JP6055817B2 (ja) |
| CN (1) | CN103649086A (ja) |
| AU (1) | AU2012253237B2 (ja) |
| CA (1) | CA2835450C (ja) |
| NZ (1) | NZ615466A (ja) |
| WO (1) | WO2012151640A1 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5373613B2 (ja) | 2006-10-16 | 2013-12-18 | バイオノミクス リミテッド | 新規な抗不安薬化合物 |
| US10954231B2 (en) | 2006-10-16 | 2021-03-23 | Bionomics Limited | Anxiolytic compounds |
| CA2828780A1 (en) | 2011-03-02 | 2012-09-07 | Bionomics Limited | Novel small-molecules as therapeutics |
| EP2707367B1 (en) | 2011-05-12 | 2019-10-09 | Bionomics Limited | Methods for preparing naphthyridines |
| AU2013204159B2 (en) * | 2013-03-15 | 2015-05-07 | Bionomics Limited | A Crystalline Form of an Anxiolytic Compound |
| CN105492440A (zh) * | 2013-03-15 | 2016-04-13 | 生态学有限公司 | 抗焦虑化合物的盐、共晶体和多晶型物 |
| WO2016196017A1 (en) * | 2015-06-04 | 2016-12-08 | Antriabio, Inc. | Amine pegylation methods for the preparation of site-specific protein conjugates |
| WO2018102885A1 (en) * | 2016-12-09 | 2018-06-14 | Bionomics Limited | Modulators of nicotinic acetylcholine receptors and uses thereof |
| US12358901B2 (en) | 2018-10-16 | 2025-07-15 | Autifony Therapeutics Limited | KV3 modulators |
Family Cites Families (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3429887A (en) | 1965-01-26 | 1969-02-25 | Sterling Drug Inc | 1,7-naphthyridine-3-carboxylic acid derivatives and their preparation |
| JPS5023036B1 (ja) | 1970-12-29 | 1975-08-05 | ||
| JPS5023037B1 (ja) | 1970-12-29 | 1975-08-05 | ||
| JPS5132594A (en) | 1974-09-11 | 1976-03-19 | Kanebo Ltd | 1 88 nafuchirijinjudotai no seiho |
| JPS5331690A (en) | 1976-09-01 | 1978-03-25 | Shionogi & Co Ltd | Oxadithiacephalosporins |
| JPS54163596A (en) | 1978-06-09 | 1979-12-26 | Daiichi Seiyaku Co | Flonaphlizine derivative |
| JPS55111486A (en) | 1979-02-19 | 1980-08-28 | Dai Ichi Seiyaku Co Ltd | Preparation of pro 3,2,-b -1,8-naphthyridine derivative |
| JPS55151584A (en) | 1979-05-11 | 1980-11-26 | Dai Ichi Seiyaku Co Ltd | Preparation of 1,8-naphthyridine derivative |
| JPS56115787A (en) | 1980-02-19 | 1981-09-11 | Dai Ichi Seiyaku Co Ltd | Preparation of furo (3,2-b)(1,8)naphthyridine derivative |
| JPS56118081A (en) | 1980-02-22 | 1981-09-16 | Dai Ichi Seiyaku Co Ltd | Preparation of 6-methoxy-1,8-naphthyridine |
| JPS56118083A (en) | 1980-02-22 | 1981-09-16 | Dai Ichi Seiyaku Co Ltd | Furo(3,2-b)(1,8)naphthyridine derivative |
| JPS5726688A (en) | 1980-07-24 | 1982-02-12 | Dai Ichi Seiyaku Co Ltd | Furo 3,2-b 1,8-naphthyridine-7-carboxylic acid derivative |
| JPS57109790A (en) | 1980-12-26 | 1982-07-08 | Dai Ichi Seiyaku Co Ltd | Furo(3,2-b)1,8-naphthyridine-7-carboxylic acid derivative |
| US4404201A (en) | 1981-11-13 | 1983-09-13 | Warner-Lambert Company | Cephalosporins |
| JPS5993080A (ja) | 1982-11-18 | 1984-05-29 | Sumitomo Chem Co Ltd | 1,5−ナフチリジンカルボン酸誘導体 |
| ATE113953T1 (de) | 1988-05-10 | 1994-11-15 | Ici Plc | Cephalosporine, verfahren zu ihrer herstellung und pharmazeutische präparate. |
| US5095015A (en) | 1990-07-24 | 1992-03-10 | Neurogen Corporation | Certain azacycloalkyl imidazopyrimidines; a new class of gaba brain receptor ligands |
| US5182386A (en) | 1991-08-27 | 1993-01-26 | Neurogen Corporation | Certain imidazoquinoxalinones; a new class of gaba brain receptor ligands |
| US5182290A (en) | 1991-08-27 | 1993-01-26 | Neurogen Corporation | Certain oxazoloquinolinones; a new class of GABA brain receptor ligands |
| IT1251202B (it) | 1991-09-13 | 1995-05-04 | Mediolanum Farmaceutici Spa | 6-amminochinoloni, sintesi ed impiego come antibatterici |
| US5212310A (en) | 1991-12-19 | 1993-05-18 | Neurogen Corporation | Certain aryl fused imidazopyrimidines; a new class of GABA brain receptor ligands |
| US5306819A (en) | 1992-08-27 | 1994-04-26 | Neurogen Corporation | Certain aryl a cycloalkyl fused imidazopyrazinols; and new class of GABA brain receptor ligands |
| US5750702A (en) | 1993-10-27 | 1998-05-12 | Neurogen Corporation | Certain pyrrolo pyridine-3-carboxamides; a new class of GABA brain receptor ligands |
| US5484944A (en) | 1993-10-27 | 1996-01-16 | Neurogen Corporation | Certain fused pyrrolecarboxanilides and their use as GABA brain receptor ligands |
| US6211365B1 (en) | 1996-01-19 | 2001-04-03 | Neurogen Corporation | Fused pyrrolecarboxamides; a new class of GABA brain receptor ligands |
| US5804686A (en) | 1996-01-19 | 1998-09-08 | Neurogen Corporation | fused pyrrolecarboxamides; a new class of GABA brain receptor ligands |
| US6013650A (en) | 1996-03-14 | 2000-01-11 | Neurogen Corporation | Certain aryl fused imidazopyrimidines; a new class of GABA brain receptor ligands |
| WO1997034870A1 (en) | 1996-03-22 | 1997-09-25 | Neurogen Corporation | Certain fused pyrrolecarboxamides as gaba brain receptor ligands |
| US5723462A (en) | 1996-04-26 | 1998-03-03 | Neurogen Corporation | Certain fused pyrrolecarboxamides a new class of GABA brain receptor ligands |
| US6143760A (en) | 1997-08-25 | 2000-11-07 | Neurogen Corporation | Substituted 4-oxo-napthyridine-3-carboxamides: GABA brain receptor ligands |
| WO1999043682A1 (en) | 1998-02-26 | 1999-09-02 | Neurogen Corporation | 2-(het-)aryl-4-(cyclic amino substituted) heteroaryl fused pyridine derivatives, their preparation and their use as (ant-)agonists for gaba (a) brain receptors |
| DE19826050A1 (de) * | 1998-06-12 | 1999-12-16 | Bayer Ag | Verfahren zur Herstellung von Chinolon- und Naphthyridoncarbonsäuren und deren Ester |
| US6177569B1 (en) | 1998-08-25 | 2001-01-23 | Neurogen Corporation | Oxo-pyridoimidazole-carboxamides: GABA brain receptor ligands |
| WO2000037471A1 (en) | 1998-12-23 | 2000-06-29 | Neurogen Corporation | 2-amino-9-alkylpurines: gaba brain receptor ligands |
| BR0010308A (pt) | 1999-05-06 | 2002-01-08 | Neurogen Corp | Composto, composição farmacêutica, uso de um composto, e, métodos para o tratamento de uma doença ou distúrbio associados com o agonismo patogênico, agonismo inverso ou antagonismo do receptor de gabaa, para localizar receptores de gabaa em uma amostra de tecido, para inibir a ligação de um composto de benzodiazepina a um receptor de gabaa e para alterar a atividade da transdução de sinal dos receptores de gabaa, e, composição farmacêutica embalada |
| US6297256B1 (en) | 1999-06-15 | 2001-10-02 | Neurogen Corporation | Aryl and heteroaryl substituted pyridino derivatives GABA brain receptor ligands |
| CN1390217A (zh) | 1999-11-12 | 2003-01-08 | 神经原公司 | 二环和三环杂芳族化合物 |
| AU2001264932A1 (en) | 2000-05-26 | 2001-12-11 | Neurogen Corporation | Oxo-imidazopyrimidine-carboxamides and their use as gaba brain receptor ligands |
| WO2002000623A2 (en) | 2000-06-26 | 2002-01-03 | Neurogen Corporation | Aryl fused substituted 4-oxy-pyridines |
| US6423956B1 (en) | 2000-07-28 | 2002-07-23 | Optical Biopsy Technologies | Fiber-coupled, high-speed, integrated, angled-dual-axis confocal scanning microscopes employing vertical cross-section scanning |
| ES2250464T3 (es) | 2000-08-16 | 2006-04-16 | Neurogen Corporation | Derivados de piridina 2,4- sustituidos. |
| BR0113697A (pt) | 2000-09-06 | 2003-07-22 | Neurogen Corp | Composto ou um sal farmaceuticamente aceitável do mesmo, composição farmacêutica, uso de um composto ou sal, embalagem e métodos para, para alterar a atividade transdutora de sinal dos receptores de gabaa, para tratar ansiedade, depressão, um distúrbio do sono, ou demência de alzheimer, para demonstrar a presença dos receptores de gabaa em células ou amostras de tecido e para preparar um composto |
| US20020151591A1 (en) | 2000-10-17 | 2002-10-17 | Anabella Villalobos | Combination use of acetylcholinesterase inhibitors and GABAa inverse agonists for the treatment of cognitive disorders |
| AUPR283801A0 (en) | 2001-02-01 | 2001-03-01 | Australian National University, The | Chemical compounds and methods |
| WO2002069948A1 (en) | 2001-03-01 | 2002-09-12 | Pfizer Products Inc. | Use of gabaa inverse agonists in combination with nicotine receptor partial agonists, estrogen, selective estrogen modulators, or vitamin e for the treatment of cognitive disorders |
| EP1392692B1 (en) | 2001-03-27 | 2006-11-15 | Neurogen Corporation | (oxo-pyrazolo¬1,5a|pyrimidin-2-yl)alkyl-carboxamides |
| AU2002352868A1 (en) | 2001-11-27 | 2003-06-10 | Merck & Co., Inc. | 4-aminoquinoline compounds |
| CA2468015A1 (en) | 2001-11-27 | 2003-06-05 | Merck & Co., Inc. | 2-aminoquinoline compounds |
| US8163769B2 (en) * | 2002-03-12 | 2012-04-24 | Abbott Laboratories | Antibacterial compounds |
| NZ536061A (en) | 2002-05-14 | 2008-09-26 | Univ California | Substituted quinolone carboxylic acids, their derivatives, site of action, and uses thereof |
| IS7839A (is) | 2002-11-22 | 2004-05-23 | Merck Frosst Canada Ltd. | 4-oxó-1-(3-setið fenýl-1,4-díhýdró-1,8-naftýridín-3-karboxamíð fosfódíesterasa-4 hindrar |
| US7829694B2 (en) | 2002-11-26 | 2010-11-09 | Medtronic, Inc. | Treatment of neurodegenerative disease through intracranial delivery of siRNA |
| TW200427688A (en) | 2002-12-18 | 2004-12-16 | Glaxo Group Ltd | Antibacterial agents |
| JP2005162726A (ja) | 2003-01-24 | 2005-06-23 | Tanabe Seiyaku Co Ltd | ピラゾロピリミジン化合物およびその製法 |
| TW200418835A (en) | 2003-01-24 | 2004-10-01 | Tanabe Seiyaku Co | A pyrazolopyrimidine compound and a process for preparing the same |
| US20050031652A1 (en) | 2003-02-25 | 2005-02-10 | Allergan, Inc. | Compositions and methods comprising memantine and polyanionic polymers |
| MXPA05009688A (es) | 2003-03-12 | 2005-12-05 | Abbott Lab | Derivados de naftiridina como agentes antibacterianos. |
| DE502005007840D1 (de) | 2004-01-31 | 2009-09-17 | Sanofi Aventis Deutschland | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US7402674B2 (en) | 2004-01-31 | 2008-07-22 | Sanofi-Aventis Deutschland Gmbh, | 7-Phenylamino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments |
| US7498341B2 (en) | 2004-01-31 | 2009-03-03 | Sanofi Aventis Deutschland Gmbh | Heterocyclically substituted 7-amino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments |
| DE102004004971B3 (de) | 2004-01-31 | 2005-09-15 | Aventis Pharma Deutschland Gmbh | Cycloalkyl substituierte 7-Amino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arnzeimittel |
| DE102004004972B3 (de) | 2004-01-31 | 2005-09-15 | Aventis Pharma Deutschland Gmbh | Heterocyclisch substituierte 7-Amino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| US7470706B2 (en) | 2004-01-31 | 2008-12-30 | Sanofi-Aventis Deutschland Gmbh | Cycloalkyl-substituted 7-amino-4-quinolone-3-carboxylic acid derivatives, process for their preparation and their use as medicaments |
| PE20060285A1 (es) | 2004-03-30 | 2006-05-08 | Aventis Pharma Inc | Piridonas sustituidas como inhibidores de pol(adp-ribosa)-polimerasa (parp) |
| MX2007005112A (es) | 2004-11-01 | 2007-06-26 | Hoffmann La Roche | Quinolinas como intensificadores alostericos de receptores de acido gamma-aminobutirico-b. |
| JP2008521905A (ja) | 2004-12-03 | 2008-06-26 | メルク エンド カムパニー インコーポレーテッド | キノリンタキキニン受容体拮抗薬 |
| KR100978954B1 (ko) | 2005-06-02 | 2010-08-30 | 에프. 호프만-라 로슈 아게 | Gaba-b 증강인자로서 3-메테인설폰일퀴놀린 |
| AU2006299092A1 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl-(1,2,4)-oxadiazol-5-one derivatives with phenyl group, processes for their preparation and their use as pharmaceuticals |
| TW200824678A (en) | 2006-08-11 | 2008-06-16 | Combinatorx Inc | Methods and compositions for the treatment of neurodegenerative disorders |
| ES2341166B1 (es) | 2006-10-16 | 2011-05-17 | Technological Resources Pty. Limited | Procedimiento de clasificacion de material extraido de minas. |
| JP5373613B2 (ja) * | 2006-10-16 | 2013-12-18 | バイオノミクス リミテッド | 新規な抗不安薬化合物 |
| CA2687931C (en) * | 2007-05-31 | 2016-05-24 | Boehringer Ingelheim International Gmbh | Ccr2 receptor antagonists and uses thereof |
| WO2009089263A2 (en) * | 2008-01-07 | 2009-07-16 | Ardea Biosciences Inc. | Novel compositions and methods of use |
| EP2431362A4 (en) * | 2009-05-14 | 2012-10-10 | Dainippon Sumitomo Pharma Co | 3- (4-AMINOPHENYL) -2-FURANCARIC ACID DERIVATIVE AND PHARMACEUTICAL ACCEPTABLE SALT THEREOF |
| AR076687A1 (es) | 2009-05-18 | 2011-06-29 | Infinity Pharmaceuticals Inc | Isoxazolinas como inhibidores de la amidahidrolasa de acidos grasos y com-posiciones farmaceuticas que los contienen |
| WO2010143733A1 (en) * | 2009-06-09 | 2010-12-16 | Takeda Pharmaceutical Company Limited | Novel fused cyclic compound and use thereof |
| CA2828780A1 (en) | 2011-03-02 | 2012-09-07 | Bionomics Limited | Novel small-molecules as therapeutics |
| US20140051701A1 (en) * | 2011-03-02 | 2014-02-20 | Bionomics Limited | Methods of treating a disease or condition of the central nervous system |
| EP2707367B1 (en) | 2011-05-12 | 2019-10-09 | Bionomics Limited | Methods for preparing naphthyridines |
-
2012
- 2012-05-11 EP EP12782057.9A patent/EP2707367B1/en active Active
- 2012-05-11 US US14/116,196 patent/US9133188B2/en active Active
- 2012-05-11 NZ NZ615466A patent/NZ615466A/en not_active IP Right Cessation
- 2012-05-11 WO PCT/AU2012/000533 patent/WO2012151640A1/en not_active Ceased
- 2012-05-11 AU AU2012253237A patent/AU2012253237B2/en active Active
- 2012-05-11 CN CN201280019994.0A patent/CN103649086A/zh active Pending
- 2012-05-11 JP JP2014509568A patent/JP6055817B2/ja active Active
- 2012-05-11 CA CA2835450A patent/CA2835450C/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2707367A4 (en) | 2014-12-03 |
| EP2707367B1 (en) | 2019-10-09 |
| EP2707367A1 (en) | 2014-03-19 |
| CA2835450C (en) | 2020-05-12 |
| US20140128600A1 (en) | 2014-05-08 |
| CN103649086A (zh) | 2014-03-19 |
| NZ615466A (en) | 2016-02-26 |
| CA2835450A1 (en) | 2012-11-15 |
| JP2014513142A (ja) | 2014-05-29 |
| AU2012253237A1 (en) | 2013-03-14 |
| AU2012253237B2 (en) | 2015-09-24 |
| WO2012151640A1 (en) | 2012-11-15 |
| US9133188B2 (en) | 2015-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6055817B2 (ja) | ナフチリジンの調製方法 | |
| JP6926329B2 (ja) | 6−(2−ヒドロキシ−2−メチルプロポキシ)−4−(6−(6−((6−メトキシピリジン−3−イル)メチル)−3,6−ジアザビシクロ[3.1.1]ヘプタン−3−イル)ピリジン−3−イル)ピラゾロ[1,5−a]ピリジン−3−カルボニトリルの調製のためのプロセス | |
| CN113784963B (zh) | 用作ret激酶抑制剂的化合物及其应用 | |
| RU2728829C1 (ru) | Новое изоиндолиновое производное, включающая его фармацевтическая композиция и его применение | |
| JP7548814B2 (ja) | Ido阻害剤および/またはido-hdac二重阻害剤としての多環式化合物 | |
| AU2017250302A1 (en) | Inhibitors of activin receptor-like kinase | |
| CN118660887A (zh) | 三环稠杂环类pde3/4双重抑制及其用途 | |
| CN113454081A (zh) | 咪唑并吡啶基化合物及其用于治疗增生性疾病的用途 | |
| CN113164475A (zh) | Dyrk1a的大环抑制剂 | |
| CN116113416B (zh) | 四环类衍生物、其制备方法和其医药上的用途 | |
| TW201536766A (zh) | 3-(5-胺基-2-甲基-4-氧代喹唑啉-3(4h)-基)六氫吡啶-2,6-二酮之合成 | |
| JP4977465B2 (ja) | ピロロ[2,3−c]ピリジン化合物、その製造方法および用途 | |
| JP6033788B2 (ja) | 置換されたメチルアミン、セロトニン5−ht6受容体アンタゴニスト、製造のための方法及びその使用 | |
| JP2026053339A (ja) | Ahrアゴニスト | |
| EP4249077A1 (en) | Novel acridinium salt and method for producing same | |
| KR20230026411A (ko) | 방향족 에터 화합물의 제조 방법 | |
| CN115605460A (zh) | 杂环化合物的合成 | |
| EA008304B1 (ru) | 8-замещённые имидазопиридины | |
| AU2003249262B2 (en) | Process for the preparation of imidazo(1,2-a)pyridine-3-acetamides | |
| CN114249723A (zh) | 一种唑吡坦及其关键中间体的制备方法 | |
| JP7716147B2 (ja) | 置換されたイソインドリン-1,3-ジオン類pde4阻害剤及びその薬物応用 | |
| CN121293164A (zh) | 一种小分子化合物及其制备方法与在抗癌药物中的应用 | |
| CN116082352A (zh) | 一种加兰他敏衍生物及其制备方法和用途 | |
| HK40079222B (zh) | 芳香醚类化合物的制备方法 | |
| HK40058867A (en) | Imidazopyridinyl compounds and use thereof for treatment of proliferative disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20150218 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20150910 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20150925 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20151224 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160520 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160729 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160930 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161011 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20161104 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20161205 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6055817 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |