JP7146016B2 - Shp2の活性を阻害するための化合物および組成物 - Google Patents
Shp2の活性を阻害するための化合物および組成物 Download PDFInfo
- Publication number
- JP7146016B2 JP7146016B2 JP2021076566A JP2021076566A JP7146016B2 JP 7146016 B2 JP7146016 B2 JP 7146016B2 JP 2021076566 A JP2021076566 A JP 2021076566A JP 2021076566 A JP2021076566 A JP 2021076566A JP 7146016 B2 JP7146016 B2 JP 7146016B2
- Authority
- JP
- Japan
- Prior art keywords
- mmol
- methyl
- tert
- azaspiro
- carboxylate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Peptides Or Proteins (AREA)
Description
本発明の化合物を調製する方法、このような化合物を含む医薬調製物、ならびにSHP2
の異常な活性と関連する疾患または障害の管理においてこのような化合物および組成物を
使用する方法を提供する。
維持および遊走を含めた複数の細胞機能に寄与するPTPN11遺伝子によってコードさ
れた非受容体タンパク質チロシンホスファターゼである。SHP2は、Ras-マイトジ
ェン活性化タンパク質キナーゼ、JAK-STATまたはホスホイノシトール3-キナー
ゼ-AKT経路を介するシグナル伝達に関与する。
H2)、触媒ドメイン(PTP)、およびC-末端尾部を有する。2つのSH2ドメイン
は、SHP2の細胞内局在および機能的制御を調節する。その分子は、N-SH2ドメイ
ンとPTPドメインの両方に由来する残基を含む結合網によって安定化された、不活性な
自己阻害型立体構造で存在する。例えば、サイトカインまたは成長因子による刺激によっ
て触媒部位が曝露されて、SHP2が酵素的に活性化される。
の疾患、例えばヌーナン症候群、レオパード症候群、若年性骨髄単球性白血病、神経芽細
胞腫、黒色腫、急性骨髄性白血病、ならびに乳房、肺および結腸のがんにおいて同定され
ている。したがって、SHP2は、様々な疾患を処置する新規な治療を開発するための非
常に魅力的な標的となっている。本発明の化合物は、SHP2の活性を阻害する小分子の
必要を満たすものである。

[式中、
X1は、NおよびCHから選択され、X2は、CR3bであり、X3は、Sおよび結合か
ら選択され、Y1は、NおよびCR7から選択され、R7は、水素、アミノ、ハロ、C1
~3アルキル、C1~3アルコキシおよびヒドロキシから選択され、Y2は、NおよびC
R8から選択され、R8は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C3~6
シクロアルキル、C1~4アルキル、ハロ置換C1~3アルキル、ハロ置換C1~3アル
キル-スルファニル、C1~3アルコキシ、ハロ置換C1~3アルコキシ、C1~3アル
コキシ-C1~3アルコキシ、C6~10アリールおよびC6~10アリール-C0~1
アルコキシから選択され、Y3は、NおよびCR9から選択され、R9は、水素、アミノ
、ハロ、C1~3アルキル、-NH(C3~5シクロアルキル)、C1~3アルコキシお
よびヒドロキシから選択され、R1は、水素、ハロ、ハロ置換C1~2アルキル、ハロ置
換C1~2アルコキシ、C1~2アルキル-ヒドロキシおよびシアノから選択され、また
はR1およびR8は、R1およびR8が付着している炭素原子と一緒になって、1,3-
ジオキソール、フェニル、ピリジン、シクロペンテン、ジヒドロフラン、ジヒドロピラン
から選択される環を形成し、前記1,3-ジオキソール、フェニル、ピリジン、シクロペ
ンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールまたはジヒドロピランは、
非置換であってもよく、または1~2個のハロ基で置換されていてもよく、R2は、水素
およびハロから選択され、R3aは、水素、メチルおよびハロ置換C1~2アルキルから
選択され、R3bは、水素、メチルおよびアミノから選択され、R4aおよびR4bは、
それぞれ独立に、水素、ヒドロキシおよびフルオロから選択され、ただし、R4aおよび
R4bは、両方共はOHではあり得ず、ただし、R4aおよびR4bは、同時にOHおよ
びFではあり得ず、R5aは、アミノおよびアミノ-メチルから選択され、R5bは、O
H、アミノ、フルオロ、C1~6アルキル、メトキシ-カルボニル、C3~6シクロアル
キル-C1~3アルキル、ヒドロキシ置換C1~3アルキル、C1~2アルコキシ置換C
1~3アルキル、ならびにO、SおよびNから選択される1~4個のヘテロ原子を含有す
る5~6員のヘテロアリール環から選択され、R5bの前記C1~6アルキルまたはC1
~2アルコキシ置換C1~3アルキルは、非置換であるか、または1~3個のフッ素で置
換されており、ただし、R5aがアミノである場合、R5bは、OH、アミノまたはフル
オロではあり得ず、またはR5aおよびR5bは、R5aおよびR5bが付着している炭
素原子と一緒になって、

[式中、*Cは、R5aおよびR5bが付着している炭素原子を表し、R10は、アミノ
であり、R11aは、水素、ヒドロキシ、フルオロ、C1~3アルキルおよびヒドロキシ
-メチルから選択され、R11bは、フルオロ、メチルおよび水素から選択され、ただし
、R11aおよびR11bは、両方が同時にOHおよびフルオロではあり得ず、R11c
は、水素、C1~3アルキルおよびヒドロキシ-メチルから選択され、R12は、水素、
ハロ、ヒドロキシ、C1~3アルキル、ハロ置換C1~3アルキル、ハロ置換C1~3ア
ルコキシおよびC1~3アルコキシから選択され、R13は、水素、ハロおよびC1~3
アルキルから選択され、R14は、水素およびフルオロから選択され、ただし、R12お
よびR13は、両方が同時にOHおよびフルオロではあり得ず、R15は、水素およびフ
ルオロから選択される]
から選択される基を形成し、R6aおよびR6bは、それぞれ独立に、水素、ヒドロキシ
およびフルオロから選択され、ただし、R6aおよびR6bは、両方共はOHではあり得
ず、ただし、R6aおよびR6bは、両方が同時にOHおよびフルオロではあり得ない]
または薬学的に許容されるその塩を提供し、ただし、式Iの化合物は、

から選択される化合物を含まない。
体、個々の異性体および異性体混合物、または薬学的に許容されるその塩を、1つまたは
複数の適切な添加剤との混合物として含有する医薬組成物を提供する。
理および/または総体症状を防止する、阻害する、または寛解させることができる、動物
の疾患を処置する方法を提供し、この方法は、動物に、治療有効量の式Iの化合物もしく
はそのN-オキシド誘導体、個々の異性体および異性体混合物、または薬学的に許容され
るその塩を投与するステップを含む。
理および/または総体症状を防止する、阻害する、または寛解させることができる、動物
の疾患を処置する方法を提供し、この方法は、動物に、治療有効量の式Iの化合物もしく
はそのN-オキシド誘導体、個々の異性体および異性体混合物、または薬学的に許容され
るその塩を、抗がん治療剤と同時にまたは逐次的に組み合わせて投与するステップを含む
。
する動物の疾患を処置するための医薬の製造における、式Iの化合物の使用を提供する。
ッグ誘導体、保護された誘導体、個々の異性体および異性体混合物、ならびに薬学的に許
容されるその塩を調製する方法を提供する。
本明細書を通して使用される一般用語は、好ましくは本開示の文脈において、別段指定
されない限り以下の意味を有し、ここでより一般的な用語は、それがどこで使用されてい
ても、互いに独立により具体的な定義によって置き換えることができ、またはそのままで
あってもよく、したがって本発明のより詳細な実施形態を定義することができる。
化水素部分を指す。別段提示されない限り、アルキルは、1~7個の炭素原子を有する炭
化水素部分(C1~7アルキル)、または1~4個の炭素原子を有する炭化水素部分(C
1~4アルキル)を指す。アルキルの代表例として、メチル、エチル、n-プロピル、イ
ソ-プロピル、n-ブチル、sec-ブチル、イソ-ブチル、tert-ブチル、n-ペ
ンチル、イソペンチル、ネオペンチル、n-ヘキシル、3-メチルヘキシル、2,2-ジ
メチルペンチル、2,3-ジメチルペンチル、n-ヘプチル、n-オクチル、n-ノニル
、n-デシル等が挙げられるが、それらに限定されない。置換アルキルは、ハロゲン、ヒ
ドロキシまたはアルコキシ基から選択される1つまたは複数、例えば1個、2個または3
個の置換基を含有するアルキル基である。ハロ置換アルキルおよびハロ置換アルコキシは
、直鎖または分岐のいずれかであってよく、それには、メトキシ、エトキシ、ジフルオロ
メチル、トリフルオロメチル、ペンタフルオロエチル、ジフルオロメトキシ、トリフルオ
ロメトキシ等が含まれる。
の集合体を意味する。例えば、アリールは、フェニルまたはナフチル、好ましくはフェニ
ルであり得る。「アリーレン」は、アリール基から導出された二価の基を意味する。
1つまたは複数は、ヘテロ原子である。例えば、C5~10ヘテロアリールは、炭素原子
によって示される通り最小5員であるが、これらの炭素原子は、ヘテロ原子によって置き
換えることができる。結果的に、C5~10ヘテロアリールには、ピリジル、インドリル
、インダゾリル、キノキサリニル、キノリニル、ベンゾフラニル、ベンゾピラニル、ベン
ゾチオピラニル、ベンゾ[1,3]ジオキソール、イミダゾリル、ベンゾ-イミダゾリル
、ピリミジニル、フラニル、オキサゾリル、イソオキサゾリル、トリアゾリル、テトラゾ
リル、ピラゾリル、チエニル等が含まれる。
不飽和の単環式、縮合二環式または架橋多環式環の集合体を意味する。例えば、C3~1
0シクロアルキルには、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシ
ル、シクロヘキセニル等が含まれる。
示されている環炭素の1つまたは複数は、-O-、-N=、-NR-、-C(O)-、-
S-、-S(O)-または-S(O)2-から選択される部分によって置き換えられてお
り、Rは、水素、C1~4アルキルまたは窒素保護基である。例えば、本発明の化合物を
説明するために本願で使用されるC3~8ヘテロシクロアルキルには、モルホリノ、ピロ
リジニル、ピロリジニル-2-オン、ピペラジニル、ピペリジニル、ピペリジニルオン(
piperidinylone)、1,4-ジオキサ-8-アザ-スピロ[4.5]デカ-8-イル、チ
オモルホリノ、スルファノモルホリノ、スルホノモルホリノ等が含まれる。
またはヨードであってもよい。
H-PTP3、Syp、PTP1D、PTP2C、SAP-2またはPTPN11として
も公知である。
72V、T、D;E76G、Q、K(ALL);G60A;D61Y;E69V;F71
K;A72V;T73I;E76G、K;R289G;G503V(AML);G60R
、D61Y、V、N;Y62D;E69K;A72T、V;T73I;E76K、V、G
、A、Q;E139D;G503A、R;Q506P(JMML);G60V;D61V
;E69K;F71L;A72V;E76A(MDS);Y63C(CMML);Y62
C;E69K;T507K(神経芽細胞腫);V46L;N58S;E76V(肺がん)
;R138Q(黒色腫);E76G(結腸がん)が含まれるが、それらに限定されない。
原子も、(R)-、(S)-または(R,S)-立体構造、好ましくは(R)-または(
S)-立体構造で存在することができる。二重結合、または特に環における置換基は、シ
ス-(=Z-)またはトランス(=E-)形態で存在することができる。したがって、化
合物は、異性体混合物として、または好ましくは純粋な異性体として、好ましくは純粋な
ジアステレオマーまたは純粋なエナンチオマーとして存在することができる。
(例えば、単一の化合物、単一の塩)を含む。「化合物」は、(例えば、医薬製剤におい
て)式Iの2つ以上の化合物(またはその塩)が存在することを除外せず、「a」は、単
に不定冠詞を表す。したがって「a」は、好ましくは「1つまたは複数」と読み取ること
ができ、あるいは、あまり好ましくはないが「1つ」と読み取ることができる。
-オキシドおよび/またはそれらの互変異性体もさらに含むことを企図する。
薬学的に許容される)その塩」は、特に、式Iの化合物が、そのまま存在するか、あるい
はそのN-オキシドとの混合物において、互変異性体として存在するか(例えば、ケト-
エノール、ラクタム-ラクチム、アミド-イミド酸またはエナミン-イミン互変異性に起
因して)、もしくはその互変異性体との(例えば、引き起こされた等価反応)混合物にお
いて、または式Iの化合物の塩、ならびに/またはこれらの形態のいずれかもしくはこの
ような形態の2つもしくはそれを超える混合物として存在し得ることを意味する。
体変動を含む。本発明の化合物または薬学的に許容されるその塩の同位体変動は、少なく
とも1つの原子が、同じ原子番号を有するが、通常自然に見出される原子質量とは異なる
原子質量を有する原子によって置き換えられているものと定義される。本発明の化合物お
よび薬学的に許容されるその塩に取り込むことができる同位体の例として、水素、炭素、
窒素および酸素の同位体、例えば2H、3H、11C、13C、14C、15N、17O
、18O、35S、18F、36Clおよび123Iが挙げられるが、それらに限定され
ない。本発明の化合物および薬学的に許容されるその塩の、ある特定の同位体変動、例え
ば放射性同位体、例えば3Hまたは14Cが取り込まれるものは、薬物および/または基
質の組織分布研究において有用である。特定の例では、調製および検出を容易にするため
に、3Hおよび14C同位体を使用することができる。他の例では、2Hなどの同位体に
よる置換は、より高い代謝安定性から得られる特定の治療上の利点、例えばインビボ半減
期の増大または必要投与量の低減をもたらすことができる。本発明の化合物または薬学的
に許容されるその塩の同位体変動は、一般に、適切な試薬の適切な同位体変動を使用して
、従来の手順によって調製することができる。
する本発明の一態様では、式Ia

[式中、X3は、Sから選択され、Y1は、NおよびCR7から選択され、R7は、水素
、アミノ、ハロ、C1~3アルキル、C1~3アルコキシから選択され、Y2は、Nおよ
びCR8から選択され、R8は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C3
~6シクロアルキル、C1~4アルキル、ハロ置換C1~3アルキル、ハロ置換C1~3
アルキル-スルファニル、C1~3アルコキシ、ハロ置換C1~3アルコキシ、C1~3
アルコキシ-C1~3アルコキシ、C6~10アリールおよびC6~10アリール-C0
~1アルコキシから選択され、またはR1およびR8は、R1およびR8が付着している
炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロ
ピロールから選択される環を形成し、Y3は、NおよびCR9から選択され、R9は、水
素、アミノ、ハロ、C1~3アルキル、C1~3アルコキシおよびヒドロキシから選択さ
れ、R1は、水素、ハロ、ハロ置換C1~2アルキルから選択され、R2は、水素および
クロロから選択され、R4aは、水素、ヒドロキシおよびフルオロから選択され、R6b
は、水素、ヒドロキシおよびフルオロから選択され、R10は、アミノであり、R11c
は、水素およびC1~3アルキルから選択される]
の化合物または薬学的に許容されるその塩が提供される。
ハロおよびアミノから選択され、Y2が、NおよびCR8から選択され、R8が、水素、
ハロ、アミノ、ジメチル-アミノ、シアノ、ハロ置換C1~2アルキル、C1~2アルコ
キシ、シクロプロピル、シクロペンチル、シクロペンチル-メトキシ、ハロ置換C1~2
アルコキシ、フェニル、メトキシ-エトキシ、テトラヒドロ-2H-ピラン-4-イル、
フェノキシおよびベンゾキシから選択され、Y3が、NおよびCR9から選択され、R9
が、水素、アミノ、ハロ、C1~2アルコキシ、シクロプロピル、トリフルオロメチル、
トリフルオロメチル-スルファニル、イソプロピルおよびヒドロキシから選択され、R1
が、水素、ハロ、トリフルオロメチル、トリフルオロメトキシ、C1~2アルキルおよび
シアノから選択され、R2が、水素、フルオロおよびクロロから選択され、R4aが、水
素であり、R6bが、水素であり、R10が、アミノであり、R11cが、水素、メチル
およびエチルから選択される化合物または薬学的に許容されるその塩が提供される。




から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X3は、結合から選択され、Y1は、CR7であり、R7は、水素、クロロおよ
びフルオロから選択され、Y2は、CR8であり、R8は、水素、ハロ、アミノ、ジメチ
ル-アミノ、シアノ、C3~6シクロアルキル、C1~4アルキル、ハロ置換C1~3ア
ルキル、ハロ置換C1~3アルキル-スルファニル、C1~3アルコキシ、ハロ置換C1
~3アルコキシ、C1~3アルコキシ-C1~3アルコキシ、C6アリールおよびC6ア
リール-C0~1アルコキシから選択され、Y3は、CR9から選択され、R9は、水素
、クロロ、フルオロおよびメチルから選択され、R1は、水素、クロロ、フルオロから選
択され、R2は、水素から選択され、R4aは、水素、ヒドロキシおよびフルオロから選
択され、R6bは、水素、ヒドロキシおよびフルオロから選択され、R10は、アミノで
あり、R11cは、水素、C1~3アルキルおよびヒドロキシ-メチルから選択される]
の化合物または薬学的に許容されるその塩が提供される。





から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X3は、Sから選択され、Y1は、NおよびCR7から選択され、R7は、水素
、アミノ、ハロ、C1~3アルキル、C1~3アルコキシから選択され、Y2は、Nおよ
びCR8から選択され、R8は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C3
~6シクロアルキル、C1~4アルキル、ハロ置換C1~3アルキル、ハロ置換C1~3
アルキル-スルファニル、C1~3アルコキシ、ハロ置換C1~3アルコキシ、C1~3
アルコキシ-C1~3アルコキシ、C6~10アリールおよびC6~10アリール-C0
~1アルコキシから選択され、またはR1およびR8は、R1およびR8が付着している
炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロ
ピロールから選択される環を形成し、Y3は、NおよびCR9から選択され、R9は、水
素、アミノ、ハロ、C1~3アルキル、C1~3アルコキシおよびヒドロキシから選択さ
れ、R1は、水素、ハロ、ハロ置換C1~2アルキルおよびハロ置換C1~2アルコキシ
から選択され、R2は、水素およびハロから選択され、R3aは、水素およびメチルから
選択され、R4aは、水素、ヒドロキシおよびフルオロから選択され、R5bは、C1~
6アルキルから選択され、R6bは、水素、ヒドロキシおよびフルオロから選択される]
の化合物または薬学的に許容されるその塩が提供される。
ハロおよびアミノから選択され、Y2が、NおよびCR8から選択され、R8が、水素、
ハロ、アミノ、シアノ、ハロ置換C1~2アルキル、C1~2アルコキシおよびハロ置換
C1~2アルコキシから選択され、Y3が、NおよびCR9から選択され、R9が、水素
、アミノ、ハロ、C1~2アルコキシおよびヒドロキシから選択され、R1が、ハロ、ト
リフルオロメチル、トリフルオロメトキシ、C1~2アルキル、ニトロ、ヒドロキシおよ
びシアノから選択され、またはR1およびR8が、R1およびR8が付着している炭素原
子と一緒になって、1,3-ジオキソランおよびピリジンから選択される環を形成し、前
記1,3-ジオキソランまたはピリジンが、非置換であってもよく、または1~2個のハ
ロ基で置換されていてもよく、R2が、水素、フルオロおよびクロロから選択され、R3
aが、水素およびメチルから選択され、R4aが、水素であり、R6bが、水素である化
合物または薬学的に許容されるその塩が提供される。



から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X3は、結合から選択され、Y1は、CR7であり、R7は、水素、クロロおよ
びフルオロから選択され、Y2は、CR8であり、R8は、水素、ハロ、アミノ、ジメチ
ル-アミノ、シアノ、C3~6シクロアルキル、C1~4アルキル、ハロ置換C1~3ア
ルキル、ハロ置換C1~3アルキル-スルファニル、C1~3アルコキシ、ハロ置換C1
~3アルコキシ、C1~3アルコキシ-C1~3アルコキシ、C6アリールおよびC6ア
リール-C0~1アルコキシから選択され、Y3は、CR9から選択され、R9は、水素
、クロロ、フルオロおよびメチルから選択され、R1は、水素、クロロ、フルオロから選
択され、R2は、水素から選択され、R4aは、水素、ヒドロキシおよびフルオロから選
択され、R5bは、C1~6アルキルから選択され、R6bは、水素、ヒドロキシおよび
フルオロから選択される]
の化合物または薬学的に許容されるその塩が提供される。

から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X1は、NおよびCHから選択され、X3は、Sから選択され、Y1は、Nおよ
びCR7から選択され、R7は、水素、アミノ、ハロ、C1~3アルキル、C1~3アル
コキシから選択され、Y2は、NおよびCR8から選択され、R8は、水素、ハロ、アミ
ノ、ジメチル-アミノ、シアノ、C3~6シクロアルキル、C1~4アルキル、ハロ置換
C1~3アルキル、ハロ置換C1~3アルキル-スルファニル、C1~3アルコキシ、ハ
ロ置換C1~3アルコキシ、C1~3アルコキシ-C1~3アルコキシ、C6~10アリ
ールおよびC6~10アリール-C0~1アルコキシから選択され、またはR1およびR
8は、R1およびR8が付着している炭素原子と一緒になって、シクロペンテン、2,3
-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、Y3は、Nお
よびCR9から選択され、R9は、水素、アミノ、ハロ、C1~3アルキル、C1~3ア
ルコキシおよびヒドロキシから選択され、R1は、水素、ハロ、ハロ置換C1~2アルキ
ルから選択され、R2は、水素およびハロから選択され、R3aは、水素およびメチルか
ら選択され、R3bは、水素およびメチルから選択され、R4aは、水素、ヒドロキシお
よびフルオロから選択され、R5bは、C1~6アルキルから選択され、R6bは、水素
、ヒドロキシおよびフルオロから選択される]
の化合物が提供される。
ハロおよびアミノから選択され、Y2が、NおよびCR8から選択され、R8が、水素、
ハロ、アミノ、シアノ、ハロ置換C1~2アルキル、C1~2アルコキシおよびハロ置換
C1~2アルコキシから選択され、Y3が、NおよびCR9から選択され、R9が、水素
、アミノ、ハロ、C1~2アルコキシおよびヒドロキシから選択され、R1が、ハロ、ト
リフルオロメチル、C1~2アルキルおよびシアノから選択され、R2が、水素、フルオ
ロおよびクロロから選択され、R3が、水素およびメチルから選択され、R4aが、水素
であり、R6bが、水素である化合物または薬学的に許容されるその塩が提供される。


から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X1は、NおよびCHから選択され、X3は、Sから選択され、Y1は、Nおよ
びCR7から選択され、R7は、水素、アミノ、ハロ、C1~3アルキル、C1~3アル
コキシから選択され、Y2は、NおよびCR8から選択され、R8は、水素、ハロ、アミ
ノ、ジメチル-アミノ、シアノ、C3~6シクロアルキル、C1~4アルキル、ハロ置換
C1~3アルキル、ハロ置換C1~3アルキル-スルファニル、C1~3アルコキシ、ハ
ロ置換C1~3アルコキシ、C1~3アルコキシ-C1~3アルコキシ、C6~10アリ
ールおよびC6~10アリール-C0~1アルコキシから選択され、またはR1およびR
8は、R1およびR8が付着している炭素原子と一緒になって、シクロペンテン、2,3
-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、Y3は、Nお
よびCR9から選択され、R9は、水素、アミノ、ハロ、C1~3アルキル、C1~3ア
ルコキシおよびヒドロキシから選択され、R1は、水素、ハロ、ハロ置換C1~2アルキ
ル、ハロ置換C1~2アルコキシおよびシアノから選択され、R2は、水素およびハロか
ら選択され、R3aは、水素、メチルおよびハロ置換C1~2アルキルから選択され、R
3bは、水素、メチルおよびアミノから選択され、R4aは、水素、ヒドロキシおよびフ
ルオロから選択され、R6bは、水素、ヒドロキシおよびフルオロから選択され、R10
は、アミノであり、R11aは、水素、ヒドロキシ、フルオロ、C1~3アルキルおよび
ヒドロキシ-メチルから選択され、R11bは、フルオロ、メチルおよび水素から選択さ
れ、ただし、R11aおよびR11bは、両方が同時にOHおよびフルオロではあり得ず
、R12は、水素、ハロ、ヒドロキシ、C1~3アルキル、ハロ置換C1~3アルキル、
ハロ置換C1~3アルコキシおよびC1~3アルコキシから選択され、R13は、水素、
ハロおよびC1~3アルキルから選択され、ただし、R12およびR13は、両方が同時
にOHおよびフルオロではあり得ず、R14は、水素およびフルオロから選択され、R1
5は、水素およびフルオロから選択される]
の化合物または薬学的に許容されるその塩が提供される。





から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X1は、NおよびCHから選択され、X3は、結合から選択され、Y1は、CR
7であり、R7は、水素、クロロおよびフルオロから選択され、Y2は、CR8であり、
R8は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C3~6シクロアルキル、C
1~4アルキル、ハロ置換C1~3アルキル、ハロ置換C1~3アルキル-スルファニル
、C1~3アルコキシ、ハロ置換C1~3アルコキシ、C1~3アルコキシ-C1~3ア
ルコキシ、C6アリールおよびC6アリール-C0~1アルコキシから選択され、Y3は
、CR9から選択され、R9は、水素、クロロ、フルオロおよびメチルから選択され、R
1は、水素、クロロ、フルオロから選択され、R2は、水素から選択され、R3aは、メ
チルから選択され、R3bは、アミノから選択され、R4aは、水素、ヒドロキシおよび
フルオロから選択され、R6bは、水素、ヒドロキシおよびフルオロから選択され、R1
0は、アミノであり、R11aは、水素、ヒドロキシ、フルオロ、C1~3アルキルおよ
びヒドロキシ-メチルから選択され、R11bは、フルオロ、メチルおよび水素から選択
され、ただし、R11aおよびR11bは、両方が同時にOHおよびフルオロではあり得
ず、R12は、水素、ハロ、ヒドロキシ、C1~3アルキル、ハロ置換C1~3アルキル
、ハロ置換C1~3アルコキシおよびC1~3アルコキシから選択され、R13は、水素
、ハロおよびC1~3アルキルから選択され、ただし、R12およびR13は、両方が同
時にOHおよびフルオロではあり得ず、R14は、水素およびフルオロから選択され、R
15は、水素およびフルオロから選択される]
の化合物または薬学的に許容されるその塩が提供される。


から選択される化合物または薬学的に許容されるその塩が提供される。

[式中、X1は、NおよびCHから選択され、Y1は、NおよびCR7から選択され、R
7は、水素、ハロおよびアミノから選択され、Y2は、NおよびCR8から選択され、R
8は、水素、ハロ、アミノ、シアノ、ハロ置換C1~3アルキル、C1~3アルコキシお
よびハロ置換C1~3アルコキシから選択され、Y3は、NおよびCR9から選択され、
R9は、水素、アミノ、ハロ、C1~3アルコキシおよびヒドロキシから選択され、R1
は、ハロ、ハロ置換C1~2アルキル、ハロ置換C1~2アルコキシ、C1~2アルキル
およびシアノから選択され、R2は、水素およびハロから選択され、R3aは、水素およ
びメチルから選択され、R3bは、水素およびメチルから選択され、R4aは、水素、ヒ
ドロキシおよびフルオロから選択され、R6bは、水素、ヒドロキシおよびフルオロから
選択され、R10は、アミノであり、R11aは、水素、ヒドロキシ、フルオロ、C1~
3アルキルおよびヒドロキシ-メチルから選択され、R11bは、フルオロ、メチルおよ
び水素から選択され、R11cは、水素、C1~3アルキルおよびヒドロキシ-メチルか
ら選択され、R12は、水素、ハロ、ヒドロキシ、C1~3アルキル、ハロ置換C1~3
アルキル、ハロ置換C1~3アルコキシおよびC1~3アルコキシから選択され、R13
は、水素、ハロおよびC1~3アルキルから選択され、ただし、R12およびR13は、
両方が同時にOHおよびフルオロではあり得ない]
の化合物または薬学的に許容されるその塩が提供される。


から選択される化合物または薬学的に許容されるその塩が提供される。
Src相同性-2ホスファターゼ(SHP2)は、増殖、分化、細胞周期維持および遊
走を含めた複数の細胞機能に寄与するPTPN11遺伝子によってコードされたタンパク
質チロシンホスファターゼである。SHP2は、Ras-マイトジェン活性化タンパク質
キナーゼ、JAK-STATまたはホスホイノシトール3-キナーゼ-AKT経路を介す
るシグナル伝達に関与する。SHP2は、受容体チロシンキナーゼ、例えばErbB1、
ErbB2およびc-Metによって、Erk1およびErk2(Erk1/2、Erk
)MAPキナーゼの活性化を媒介する。
H2)、触媒ドメイン(PTP)、およびC-末端尾部を有する。2つのSH2ドメイン
は、SHP2の細胞内局在および機能的制御を調節する。その分子は、N-SH2ドメイ
ンとPTPドメインの両方に由来する残基を含む結合網によってそれ自体の活性を阻害す
る、不活性な立体構造で存在する。成長因子刺激への応答において、SHP2は、ドッキ
ングタンパク質、例えばGab1およびGab2上の特定のチロシン-リン酸化部位に、
そのSH2ドメインを介して結合する。これによって立体構造変化が誘発され、それによ
りSHP2が活性化される。
ド症候群、若年性骨髄単球性白血病、神経芽細胞腫、黒色腫、急性骨髄性白血病、ならび
に乳房、肺および結腸のがんにおいて同定されている。SHP2は、血小板由来成長因子
の受容体(PDGF-R)、線維芽細胞成長因子(FGF-R)および上皮成長因子(E
GF-R)を含めた様々な受容体チロシンキナーゼにとって重要な下流シグナル伝達分子
である。またSHP2は、がん発生の必須条件である細胞形質転換をもたらすおそれがあ
る、マイトジェン活性化タンパク質(MAP)キナーゼ経路を活性化するために重要な下
流シグナル伝達分子である。SHP2のノックダウンは、SHP2突然変異またはEML
4/ALK転位を有する肺がん細胞株の細胞成長、ならびにEGFR増幅した乳がんおよ
び食道がんを著しく阻害した。またSHP2は、胃癌、未分化大細胞リンパ腫および膠芽
腫の癌遺伝子の下流で活性化される。
LS(複数の黒子(lentigene)、心伝導異常、両眼隔離症、肺動脈弁狭窄症、生殖器異
常、成長遅滞、感音難聴)およびNS(心臓欠陥、頭蓋顔面奇形および低身長を含めた先
天性異常)を引き起こす。両方の障害は、正常な細胞の成長および分化に必要な、RAS
/RAF/MEK/ERKマイトジェン活性化タンパク質キナーゼ経路の構成成分の生殖
系列突然変異によって引き起こされる常染色体優性症候群ファミリーの一部である。この
経路の異常な制御は、特に心臓の発達に対して深刻な作用をもたらして、弁膜中隔(valv
uloseptal)の欠損および/または肥大型心筋症(HCM)を含めた様々な異常をもたら
す。MAPKシグナル伝達経路の撹乱は、これらの障害の中心となるものとして確立され
ており、KRAS、NRAS、SOS1、RAF1、BRAF、MEK1、MEK2、S
HOC2およびCBLの突然変異を含めた、この経路に沿ったいくつかの候補遺伝子が、
ヒトにおいて同定されてきた。NSおよびLSにおいて最も一般的に変異した遺伝子は、
PTPN11である。PTPN11の生殖系列突然変異(SHP2)は、NSの症例、お
よびある特定の特色をNSと共有しているLSを有するほとんどすべての患者の症例の約
50%において見出されている。NSについて、タンパク質におけるY62DおよびY6
3C置換は、ほとんどインバリアントであり、最も一般的な突然変異の1つである。これ
らの両方の突然変異は、ホスファターゼとそのリン酸化シグナル伝達パートナーとの結合
を撹乱することなく、SHP2の触媒作用的に不活性な立体構造に影響を及ぼす。
は、JMML、小児期骨髄増殖性障害(MPD)を有する患者の約35%において生じる
。これらの機能獲得型の突然変異は、典型的に、N-SH2ドメインまたはホスファター
ゼドメインにおける点突然変異であり、それによって触媒ドメインとN-SH2ドメイン
の間の自己阻害を防止して、SHP2活性をもたらす。
群(MDS)の約10%、B細胞急性リンパ芽球性白血病(B-ALL)の約7%、およ
び急性骨髄性白血病(AML)の約4%において同定されている。
ドメインによって形成された界面に位置するアミノ酸の変化を引き起こし、分子内の阻害
性相互作用を撹乱して、触媒ドメインの活動亢進をもたらす。
して作用する。RTK変化(EGFRamp、Her2amp、FGFRamp、Met
amp、転位/活性化RTK、すなわちALK、BCR/ABL)を含有するがんには、
食道、乳房、肺、結腸、胃、神経膠腫、頭部および頸部のがんが含まれる。
る。様々なサブタイプ、主に扁平上皮がん(<50%)および腺癌が存在する。食道腺癌
および扁平上皮がんでは、RTKの発現率が高い。したがって、本発明のSHP2阻害剤
は、革新的な処置戦略のために用いることができる。
薬物に対して抵抗性を生じる。管腔A、管腔B、Her2様、およびトリプルネガティブ
/基底様を含めた4つの主なサブタイプの乳がんがある。トリプルネガティブ乳房がん(
TNBC)は、特定の標的治療が存在しない侵襲性の乳房がんである。上皮成長因子受容
体I(EGFR)は、TNBCにおける有望な標的として出現してきた。SHP2を介す
るHer2ならびにEGFRの阻害は、乳房がんにおいて有望な治療となり得る。
(主に腺癌および扁平上皮癌)の約85%を占める。細胞傷害性の化学療法は、まだ処置
の重要な一部ではあるが、腫瘍におけるEGFRおよびALKなどの遺伝的変化に基付く
標的治療は、標的治療から利益を得られる可能性がより高い。
ることが知られており、BRAF突然変異は、結腸直腸がんの10~15%において生じ
る。結腸直腸腫瘍が、EGFRを過剰発現することが実証されている患者のサブセットに
ついて、これらの患者は、抗EGFR治療に対して、好ましい臨床応答を呈する。
シンリン酸化によって反映されるチロシンキナーゼの異常発現は、当技術分野で知られて
いる。c-met(HGF受容体)、FGF受容体2、およびerbB2/neuの3つ
の受容体-チロシンキナーゼは、しばしば胃癌において増幅される。したがって、異なる
シグナル経路の破壊は、異なるタイプの胃がんの進行に寄与し得る。
。未分化リンパ腫キナーゼ(ALK)遺伝子のゲノム変化は、神経芽細胞腫の病変形成に
寄与すると想定されている。
、主に頭部および頸部の扁平上皮癌(SCCHN)における予後不良および放射線治療に
対する抵抗性と相関性がある。EGFRシグナル伝達の妨害は、受容体刺激、細胞増殖を
阻害し、侵襲性および転移を低減する。したがって、EGFRは、SCCHNにおける新
しい抗がん治療の主要標的である。
本発明の化合物およびこのような化合物を含む医薬調製物を調製する方法を提供する。本
発明の別の態様は、治療有効量の本発明の概要に定義されている式Iの化合物を、それを
必要としている患者に投与するステップを含む、SHP2媒介性障害を処置する方法に関
する。
のではないが、JMML、AML、MDS、B-ALL、神経芽細胞腫、食道がん、乳房
がん、肺がん、結腸がん、胃がん、頭部および頸部がんから選択されるがんである、前述
の方法に関する。
用となり得る。したがってさらなる一態様として、本発明は、NS、LS、JMML、A
ML、MDS、B-ALL、神経芽細胞腫、食道がん、乳房がん、肺がん、結腸がん、胃
がん、頭部および頸部がんから選択される障害を処置する方法に関する。
合物または2つもしくはそれを超える他の薬理学的に活性な化合物と、有用に組み合わせ
ることができる。例えば、式(I)の化合物または薬学的に許容されるその塩は、先に定
義した通り、化学療法剤、例えば有糸分裂阻害剤、例えばタキサン、ビンカアルカロイド
、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビンまた
はビンフルニン、および他の抗がん剤、例えばシスプラチン、5-フルオロウラシルまた
は5-フルオロ-2-4(1H,3H)-ピリミジンジオン(5FU)、フルタミドまた
はゲムシタビンから選択される1つまたは複数の薬剤と組み合わせて、同時、順次または
別個に投与することができる。
ることができる。
る。
髄腔内、局所または鼻腔内投与される前述の方法に関する。
。
義されている式Iの化合物と組み合わせて、それを必要としている患者に投与するステッ
プを含む、SHP2媒介性障害を処置する方法に関する。
別の態様では、本発明は、1つまたは複数の薬学的に許容される担体(添加物)および
/または賦形剤と一緒に製剤化された、治療有効量の前述の化合物の1つまたは複数を含
む薬学的に許容される組成物を提供する。以下に詳説する通り、本発明の医薬組成物は、
(1)経口投与、例えば水薬(水性または非水性溶液剤または懸濁液剤)、錠剤、例えば
頬側、舌下および全身吸収を標的にしたもの、ボーラス剤、散剤、顆粒剤、舌に適用する
ためのペースト剤、(2)非経口投与、例えば皮下、筋肉内、静脈内もしくは硬膜外注射
によるもの、例えば滅菌溶液剤もしくは懸濁液剤、または持続放出製剤として、(3)局
所適用、例えば皮膚に適用されるクリーム剤、軟膏剤、または徐放パッチ剤もしくはスプ
レー剤、(4)腟内もしくは直腸内、例えばペッサリー剤、クリーム剤もしくは発泡剤と
して、(5)舌下、(6)眼、(7)経皮、(8)経鼻、(9)肺、あるいは(10)髄
腔内に合わせて適合されたものを含めた固体または液体形態で投与するために、特別に製
剤化され得る。
において、任意の医学的処置に適用できる妥当な損益比でいくつかの所望の治療効果をも
たらすのに有効な、本発明の化合物、材料または化合物を含む組成物の量を意味する。
度の毒性、刺激、アレルギー応答、または他の問題もしくは合併症なしに、ヒトおよび動
物の組織と接触させて使用するのに適しており、妥当な損益比に見合う化合物、材料、組
成物および/または剤形を指すために用いられる。
れる材料、組成物またはビヒクル、例えば液体または固体充填剤、賦形剤、添加剤、製造
助剤(例えば、滑沢剤、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウムも
しくはステアリン酸亜鉛、またはステアリン(steric)酸)、または身体のある臓器もし
くは一部から身体の別の臓器もしくは一部に対象化合物を運搬または輸送する被包性溶媒
材料を意味する。各担体は、製剤の他の成分と適合性があり、患者にとって有害でないと
いう意味で「許容され」なければならない。薬学的に許容される担体として働くことがで
きる材料のいくつかの例として、(1)糖、例えばラクトース、グルコースおよびスクロ
ース、(2)デンプン、例えばトウモロコシデンプンおよびバレイショデンプン、(3)
セルロースおよびその誘導体、例えばカルボキシメチルセルロースナトリウム、エチルセ
ルロースおよび酢酸セルロース、(4)粉末化トラガント、(5)麦芽、(6)ゼラチン
、(7)タルク、(8)添加剤、例えばカカオバターおよび坐剤ワックス、(9)油、例
えばピーナッツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、トウモロコシ油および大
豆油、(10)グリコール、例えばプロピレングリコール、(11)ポリオール、例えば
グリセリン、ソルビトール、マンニトールおよびポリエチレングリコール、(12)エス
テル、例えばオレイン酸エチルおよびラウリン酸エチル、(13)寒天、(14)緩衝剤
、例えば水酸化マグネシウムおよび水酸化アルミニウム、(15)アルギン酸、(16)
発熱物質を含まない水、(17)等張食塩水、(18)リンガー溶液、(19)エチルア
ルコール、(20)pH緩衝液、(21)ポリエステル、ポリカーボネートおよび/また
はポリ酸無水物、ならびに(22)医薬製剤に用いられる他の非毒性の適合性のある物質
が挙げられる。
ルキルアミノを含有することができ、したがって、薬学的に許容される酸と共に薬学的に
許容される塩を形成することができる。これに関して、用語「薬学的に許容される塩」は
、本発明の化合物の相対的に非毒性の、無機および有機酸付加塩を指す。これらの塩は、
投与ビヒクルもしくは剤形の製造方法において現場で調製することができ、またはその遊
離塩基形態の本発明の精製化合物を、適切な有機または無機酸と別個に反応させ、こうし
て形成された塩を、その後の精製中に単離することによって調製することができる。代表
的な塩には、臭化水素酸塩、塩酸塩、硫酸塩、重硫酸塩、リン酸塩、硝酸塩、酢酸塩、吉
草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、
乳酸塩、リン酸塩、トシル酸塩、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、
酒石酸塩、ナプシル酸(napthylate)塩、メシル酸塩、グルコヘプトン酸塩、ラクトビオ
ン酸塩、およびラウリルスルホン酸塩等が含まれる。(例えば、Berge et al. (1977) “
Pharmaceutical Salts” J. Pharm. Sci. 66:1-19参照)。
物の従来の非毒性の塩または第四級アンモニウム塩が含まれる。例えば、このような従来
の非毒性の塩には、無機酸、例えば塩酸塩、臭化水素酸、硫酸、スルファミン酸、リン酸
、硝酸等に由来する塩、および有機酸、例えば酢酸、プロピオン酸、コハク酸、グリコー
ル酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パルミチン
酸、マレイン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、安息香酸、サリ
チル酸(salicyclic)、スルファニル酸、2-アセトキシ安息香酸、フマル酸、トルエン
スルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、イセチオン酸(isothi
onic)等から調製された塩が含まれる。
、したがって、薬学的に許容される塩基と共に薬学的に許容される塩を形成することがで
きる。こうした場合、用語「薬学的に許容される塩」は、本発明の化合物の相対的に非毒
性の、無機および有機塩基付加塩を指す。これらの塩は、投与ビヒクルもしくは剤形の製
造方法において現場で同様に調製することができ、あるいはその遊離酸形態の精製化合物
を、適切な塩基、例えば薬学的に許容される金属カチオンの水酸化物、炭酸塩もしくは重
炭酸塩、アンモニア、または薬学的に許容される第一級、第二級もしくは第三級有機アミ
ンと別個に反応させることによって調製することができる。代表的なアルカリまたはアル
カリ土類塩には、リチウム、ナトリウム、カリウム、カルシウム、マグネシウム、および
アルミニウム塩等が含まれる。塩基付加塩を形成するのに有用な代表的な有機アミンには
、エチルアミン、ジエチルアミン、エチレンジアミン、エタノールアミン、ジエタノール
アミン、ピペラジン等が含まれる。(例えば、上記Berge et al.を参照されたい)。
ネシウム、ならびに着色剤、放出剤、コーティング剤、甘味剤、香味剤および賦香剤、保
存剤、ならびに抗酸化剤が、組成物中に存在することもできる。
酸、システイン塩酸塩、硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウ
ム等、(2)油溶性抗酸化剤、例えばパルミチン酸アスコルビル、ブチル化ヒドロキシア
ニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロ
ピル、アルファ-トコフェロール等、および(3)金属キレート剤、例えばクエン酸、エ
チレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸等が挙げられる。
または非経口投与に適した製剤が含まれる。製剤は、好都合には単位剤形で提示すること
ができ、調剤分野で周知の任意の方法によって調製することができる。単一の剤形を生成
するために担体材料と組み合わせることができる活性成分の量は、処置を受ける宿主、特
定の投与方法に応じて変わる。単一の剤形を生成するために担体材料と組み合わせること
ができる活性成分の量は、一般に、治療効果をもたらす化合物の量である。一般にこの量
は、100パーセントのうち、活性成分が約0.1パーセント~約99パーセント、好ま
しくは約5パーセント~約70パーセント、最も好ましくは約10パーセント~約30パ
ーセントの範囲である。
ーム、ミセル形成剤、例えば胆汁酸およびポリマー性担体、例えばポリエステルおよびポ
リ酸無水物からなる群から選択される添加剤、ならびに本発明の化合物を含む。ある特定
の実施形態では、前述の製剤は、本発明の化合物を経口で生体利用可能にする。
択で1つまたは複数の補助成分と会合させるステップを含む。一般に、製剤は、本発明の
化合物を液体担体、または微粉砕した固体担体、またはその両方と、均一かつ十分に会合
させ、次に必要に応じて生成物を成形することによって調製される。
含有する、カプセル剤、カシェ剤、丸剤、錠剤、ロゼンジ剤(フレーバーベース、通常ス
クロースおよびアカシアまたはトラガントを使用する)、散剤、顆粒剤、または水性もし
くは非水性液体中の溶液剤もしくは懸濁液剤として、または水中油もしくは油中水液体エ
マルジョンとして、またはエリキシル剤もしくはシロップ剤として、またはトローチ剤(
不活性なベース、例えばゼラチンおよびグリセリン、またはスクロースおよびアカシアを
使用する)として、および/または洗口剤等の形態であってもよい。また本発明の化合物
は、ボーラス剤、舐剤またはペースト剤として投与することができる。
、トローチ剤(trouche)等)では、活性成分は、1つまたは複数の薬学的に許容される
担体、例えばクエン酸ナトリウムまたはリン酸二カルシウム、ならびに/または(1)充
填剤もしくは増量剤、例えばデンプン、ラクトース、スクロース、グルコース、マンニト
ール、および/もしくはケイ酸、(2)結合剤、例えばカルボキシメチルセルロース、ア
ルギネート、ゼラチン、ポリビニルピロリドン、スクロースおよび/もしくはアカシアな
ど、(3)保湿剤、例えばグリセロール、(4)崩壊剤、例えば寒天、炭酸カルシウム、
バレイショもしくはタピオカデンプン、アルギン酸、ある特定のシリケート、および炭酸
ナトリウム、(5)溶解遅延剤、例えばパラフィン、(6)吸収促進物質、例えば第四級
アンモニウム化合物および界面活性剤、例えばポロキサマーおよびラウリル硫酸ナトリウ
ム、(7)湿潤剤、例えばセチルアルコール、グリセロールモノステアレート、および非
イオン性界面活性剤など、(8)吸収剤、例えばカオリンおよびベントナイト粘土、(9
)滑沢剤、例えばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポ
リエチレングリコール、ラウリル硫酸ナトリウム、ステアリン酸亜鉛、ステアリン酸ナト
リウム、ステアリン酸、およびこれらの混合物、(10)着色剤、ならびに(11)徐放
剤、例えばクロスポビドンもしくはエチルセルロースのいずれかと混合される。カプセル
剤、錠剤および丸剤の場合、医薬組成物は、緩衝剤を含むこともできる。類似のタイプの
固体組成物も、ラクトースまたは乳糖、ならびに高分子量ポリエチレングリコールなどの
添加剤を使用する軟質および硬質シェル型ゼラチンカプセル中の充填剤として用いること
ができる。
作成され得る。圧縮錠剤は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチル
セルロース)、滑沢剤、不活性賦形剤、保存剤、崩壊剤(例えば、デンプングリコール酸
ナトリウムまたは架橋カルボキシメチルセルロースナトリウム)、表面活性剤または分散
化剤を使用して調製することができる。成型錠剤は、不活性な液体賦形剤で水分を与えた
粉末化化合物の混合物を、適切な機械で成型することによって作成することができる。
び顆粒剤は、任意選択で刻み目をつけることができ、またはコーティングおよびシェル、
例えば腸溶コーティングおよび医薬製剤分野で周知の他のコーティングを用いて調製する
ことができる。またこれらの製剤は、それに含まれる活性成分を、例えばヒドロキシプロ
ピルメチルセルロース、他のポリマーマトリックス、リポソームおよび/またはミクロス
フェアを、所望の放出プロファイルをもたらす異なる割合で使用して、緩徐放出または徐
放するために製剤化することができる。これらの製剤は、急速放出に合わせて製剤化する
ことができ、例えば凍結乾燥させることができる。これらの製剤は、例えば細菌保持フィ
ルタを介して濾過することによって、または滅菌水もしくはいくつかの他の注入可能な滅
菌媒体に、使用直前に溶解させることができる滅菌固体組成物の形態に滅菌剤を取り込む
ことによって、滅菌することができる。これらの組成物は、任意選択で乳白剤を含有する
こともでき、活性成分(複数可)だけを、または活性成分(複数可)を優先的に、胃腸管
のある特定の部分に任意選択で遅延方式により放出させる組成物であり得る。使用できる
包埋組成物の例として、ポリマー物質およびワックスが挙げられる。また活性成分は、適
切な場合、前述の添加剤の1つまたは複数を伴うマイクロ被包形態であってもよい。
、マイクロエマルジョン剤、溶液剤、懸濁液剤、シロップ剤およびエリキシル剤が含まれ
る。液体剤形は、活性成分に加えて、当技術分野で一般に使用される不活性賦形剤、例え
ば水または他の溶媒、可溶化剤および乳化剤、例えばエチルアルコール、イソプロピルア
ルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレ
ングリコール、1,3-ブチレングリコール、油(特に、綿実、落花生、トウモロコシ、
胚芽、オリーブ、ヒマシおよびゴマ油)、グリセロール、テトラヒドロフリルアルコール
、ポリエチレングリコールおよびソルビタンの脂肪酸エステル、ならびにこれらの混合物
などを含有することができる。
濁化剤、甘味剤、香味剤、着色剤、賦香剤、ならびに保存剤を含むこともできる。
コール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶性セルロー
ス、アルミニウムメタヒドロキシド、ベントナイト、寒天およびトラガント、ならびにこ
れらの混合物として含有することができる。
でき、この坐剤は、本発明の1つまたは複数の化合物を、例えばカカオバター、ポリエチ
レングリコール、坐剤ワックスまたはサリチレートを含み、室温で固体であるが体温で液
体になり、したがって直腸または膣腔内で溶融し、活性化合物を放出させる、1つまたは
複数の適切な非刺激性の添加剤または担体と混合することによって調製することができる
。
るような担体を含有する、ペッサリー、タンポン、クリーム、ゲル、ペースト、発泡剤ま
たはスプレーの製剤が含まれる。
ペースト剤、クリーム剤、ローション剤、ゲル剤、溶液剤、パッチ剤および吸入剤が含ま
れる。活性化合物は、滅菌条件下で、薬学的に許容される担体、および必要とされ得る任
意の保存剤、緩衝液または噴霧剤と混合することができる。
剤、例えば動物性および植物性脂肪、油、ワックス、パラフィン、デンプン、トラガント
、セルロース誘導体、ポリエチレングリコール、シリコーン、ベントナイト、ケイ酸、タ
ルクならびに酸化亜鉛、またはこれらの混合物を含有することができる。
ク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウムおよびポリアミド粉末、またはこれ
らの物質の混合物を含有することができる。スプレー剤は、さらに通例の噴霧剤、例えば
クロロフルオロ炭化水素および揮発性の非置換炭化水素、例えばブタンおよびプロパンを
含有することができる。
る。このような剤形は、化合物を適切な媒体に溶解または分散させることによって作成す
ることができる。また、皮膚を介する化合物の流動を増大するために、吸収促進剤を使用
することができる。このような流動の速度は、律速膜を提供するか、または化合物をポリ
マーマトリックスもしくはゲルに分散させることによって調節することができる。
る。
または複数の薬学的に許容される滅菌の等張水性もしくは非水性溶液剤、分散剤、懸濁液
剤もしくはエマルジョン剤、または使用直前に注入可能な滅菌溶液剤もしくは分散剤に再
構成することができる滅菌散剤と組み合わせて含み、これらは、糖、アルコール、抗酸化
剤、緩衝液、静菌剤、製剤を所期のレシピエントの血液と等張にする溶質、または懸濁化
剤もしくは増粘剤を含有することができる。
として、水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール、ポ
リエチレングリコール等)、ならびに適切なこれらの混合物、植物油、例えばオリーブ油
、ならびに注射可能な有機エステル、例えばオレイン酸エチルが挙げられる。適切な流動
性は、例えばコーティング材料、例えばレシチンを使用することによって、分散剤の場合
には必要な粒径を維持することによって、および界面活性剤を使用することによって維持
することができる。
有することもできる。対象化合物に対する微生物作用の防止は、様々な抗菌剤および抗真
菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸等を含めることによ
って確保することができる。等張剤、例えば糖、塩化ナトリウム等を、組成物に含めるこ
とが望ましい場合もある。さらに、注射可能な医薬形態の延長吸収は、遅延吸収させる薬
剤、例えばモノステアリン酸アルミニウムおよびゼラチンを含めることによってもたらす
ことができる。
収を緩徐させることが望ましい。これは、水溶性が低い結晶性または非晶質性材料の懸濁
液を使用することによって達成することができる。次に、薬物の吸収速度は、その溶解速
度に応じて決まり、溶解速度は、結晶の大きさおよび結晶形に応じて決まり得る。あるい
は、非経口投与される薬物形態の遅延吸収は、油ビヒクルに薬物を溶解または懸濁させる
ことによって達成される。
の対象化合物のマイクロ被包マトリックスを形成することによって作成される。薬物とポ
リマーの比、および用いられる特定のポリマーの性質に応じて、薬物放出速度を調節する
ことができる。他の生分解性ポリマーの例として、ポリ(オルトエステル)およびポリ(
無水物)が含まれる。また、注射可能なデポー製剤は、身体組織と適合性があるリポソー
ムまたはマイクロエマルジョンに薬物を捕捉することによって調製される。
は例えば0.1~99%(より好ましくは10~30%)の活性成分を薬学的に許容され
る担体と組み合わせて含有する医薬組成物として与えることができる。
は、当然のことながら、各投与経路に適した形態で与えられる。例えば、錠剤またはカプ
セル剤の形態で、注射剤、吸入剤、目のローション剤、軟膏剤、坐剤等、注射、注入また
は吸入による投与により、局所にはローションまたは軟膏によって、および直腸には坐剤
によって投与される。経口投与が好ましい。
、経腸および局所投与以外の、通常は注射による投与方法を意味し、それには、静脈内、
筋肉内、動脈内、髄腔内、嚢内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下
、関節内、嚢下、くも膜下、脊髄内および胸骨内の注射および注入が含まれるが、それら
に限定されない。
いう句は、本明細書で使用される場合、患者の系に入り、したがって代謝および他の類似
のプロセスを受けるように、中枢神経系に直接的に投与する以外の、化合物、薬物または
他の材料の投与、例えば皮下投与を意味する。
内および局所として、散剤、軟膏剤または液滴剤によって、頬側および舌下を含めた、任
意の適切な投与経路によって、治療のためにヒトおよび他の動物に投与することができる
。
/または本発明の医薬組成物は、当業者に公知の従来の方法によって、薬学的に許容され
る剤形に製剤化される。
よび投与方法に合った望ましい治療応答を、患者への毒性なしに達成するのに有効な活性
成分の量を得るために、変わり得る。
もしくはアミドの活性、投与経路、投与時間、用いられる特定の化合物の排出速度または
代謝、吸収の速度および程度、処置期間、用いられる特定の化合物と組み合わせて使用さ
れる他の薬物、化合物および/または材料、処置を受ける患者の年齢、性別、体重、状態
、全体的な健康状態および過去の既往歴、ならびに医術で周知の同様の因子を含めた様々
な因子に応じて決まる。
。例えば医師または獣医は、医薬組成物において用いられる本発明の化合物の用量を、所
望の治療効果を達成するために必要なレベルよりも低いレベルで開始し、所望の効果が達
成されるまで投与量を徐々に増大することができる。
である化合物の量となる。このような有効用量は、一般に、前述の因子に応じて変わるこ
とになる。一般に、患者のための本発明の化合物の経口、静脈内、脳室内および皮下用量
は、示された鎮痛効果のために使用される場合、1日に体重1キログラム当たり約0.0
001~約100mgの範囲である。
される2つ、3つ、4つ、5つ、6つまたはそれを超える下位用量として、任意選択で単
位剤形により投与することができる。好ましい投与は、1日1回の投与である。
として投与することが好ましい。
のに好都合な任意の方法で投与するために製剤化することができる。
/または賦形剤と一緒に製剤化された、治療有効量の前述の対象化合物の1つまたは複数
を含む薬学的に許容される組成物を提供する。以下に詳説する通り、本発明の医薬組成物
は、(1)経口投与、例えば水薬(水性または非水性溶液剤または懸濁液剤)、錠剤、ボ
ーラス剤、散剤、顆粒剤、舌に適用するためのペースト剤、(2)非経口投与、例えば皮
下、筋肉内もしくは静脈内注射によるもの、例えば滅菌溶液剤もしくは懸濁液剤として、
(3)局所適用、例えば皮膚、肺もしくは粘膜に適用されるクリーム剤、軟膏剤もしくは
スプレー剤として、または(4)腟内もしくは直腸内、例えばペッサリー剤、クリーム剤
もしくは発泡剤として、(5)舌下もしくは頬側、(6)眼、(7)経皮、あるいは(8
)経鼻に合わせて適合されたものを含めた固体または液体形態で投与するために、特別に
製剤化され得る。
、ブタおよびヒツジ、ならびに一般に家禽およびペットを含めた、処置を必要としている
任意の動物である。
ことができ、抗菌剤、例えばペニシリン、セファロスポリン、アミノグリコシドおよびグ
リコペプチドと併用して投与することもできる。したがって、併用治療は、最初に投与さ
れた治療剤の治療効果が、その後の治療剤が投与されるまでに完全に消失してしまわない
ように、活性化合物を逐次的、同時および別個に投与することを含む。
ィを改善することができる。その例として、トリメトリン(Trimetrine)(Dordunoo, S.
K., et al., Drug Development and Industrial Pharmacy, 17(12), 1685-1713, 1991
およびREV5901(Sheen, P. C., et al., J Pharm Sci 80(7), 712-714, 1991)
が挙げられる。数ある中でも、マイクロ乳化は、吸収を循環系の代わりにリンパ系に優先
的に直接方向付けることによって肝臓を迂回し、肝胆道循環における化合物の破壊を防止
することによって、バイオアベイラビリティを促進する。
一般に安全と認められる(GRAS)状態のものであり、本発明の化合物を可溶化するこ
とができ、溶液が複合水相(ヒトの胃腸管において見出されるものなど)と接触する後の
段階において、本発明の化合物をマイクロ乳化することができるものである。通常、これ
らの要件を満たす両親媒性成分は、HLB(親水性と親油性のバランス)値が2~20で
あり、それらの構造は、C-6~C-20の範囲の直鎖脂肪族基を含有している。それら
の例は、ポリエチレン-グリコール化脂肪グリセリドおよびポリエチレングリコールであ
る。
glycol(すべて、Gattefosse Corporation、Saint
Priest、フランスによって製造され、流通されている)、PEG-モノオレエート
、PEG-ジオレエート、PEG-モノラウレートおよびジラウレート、レシチン、ポリ
ソルベート80等(米国および世界中のいくつかの企業によって生成され、流通されてい
る)を含めた、市販の両親媒性担体が、特に企図される。
脂質に共有結合により付着することができ、毒性作用をもたらさずにインビボで耐容性を
示す(すなわち生体適合性がある)ものである。適切なポリマーには、ポリエチレングリ
コール(PEG)、ポリ乳酸(ポリラクチドとも呼ばれる)、ポリグリコール酸(ポリグ
リコリドとも呼ばれる)、ポリ乳酸-ポリグリコール酸コポリマー、およびポリビニルア
ルコールが含まれる。好ましいポリマーは、分子量が約100または120ダルトンから
約5,000または10,000ダルトンまで、より好ましくは約300ダルトン~約5
,000ダルトンのものである。特に好ましい一実施形態では、ポリマーは、分子量が約
100~約5,000ダルトン、より好ましくは分子量が約300~約5,000ダルト
ンのポリエチレングリコールである。特に好ましい一実施形態では、ポリマーは、750
ダルトンのポリエチレングリコール(PEG(750))である。またポリマーは、それ
に含まれるモノマーの数によって定義することができる。本発明の好ましい一実施形態は
、少なくとも約3つのモノマーからなるポリマー、例えば3つのモノマーからなるPEG
ポリマー(およそ150ダルトン)を利用する。
メトキサゾリン(polymethoxazoline)、ポリエチルオキサゾリン、ポリヒドロキシプロ
ピルメタクリルアミド、ポリメタクリルアミド、ポリジメチルアクリルアミド、および誘
導体化セルロース、例えばヒドロキシメチルセルロースまたはヒドロキシエチルセルロー
スが含まれる。
キレン、アクリル酸エステルとメタクリル酸エステルのポリマー、ポリビニルポリマー、
ポリグリコリド、ポリシロキサン、ポリウレタンおよびそれらのコポリマー、セルロース
、ポリプロピレン、ポリエチレン、ポリスチレン、乳酸とグリコール酸のポリマー、ポリ
酸無水物、ポリ(オルト)エステル、ポリ(酪(butic)酸)、ポリ(吉草酸)、ポリ(
ラクチド-co-カプロラクトン)、ポリサッカライド、タンパク質、ポリヒアルロン酸
、ポリシアノアクリレート、ならびにそれらのブレンド、混合物またはコポリマーからな
る群から選択される生体適合性ポリマーを含む。
リシア文字のアルファ、ベータまたはガンマによって指定される環式オリゴ糖である。6
個未満のグルコース単位を有するシクロデキストリンは、存在するかどうか未知である。
グルコース単位は、アルファ-1,4-グルコシド結合によって連結する。糖単位のいす
形配座の結果、すべての第二級ヒドロキシル基(C-2、C-3における)は、環の一方
の側に位置し、C-6におけるすべての第一級ヒドロキシル基は、他方の側に位置する。
その結果、外側面は親水性となり、シクロデキストリンは水溶性になる。それとは対照的
に、シクロデキストリンの空洞は、原子C-3およびC-5の水素、およびエーテル様の
酸素によって覆われているので、疎水性である。これらのマトリックスは、例えばステロ
イド化合物、例えば17ベータ-エストラジオールを含めた相対的に疎水性の様々な化合
物と複合体を形成する(例えば、van Uden et al. Plant Cell Tiss. Org. Cult. 38:1-3
-113(1994)参照)。複合体形成は、ファンデルワールス相互作用および水素結合形成に
よって行われる。シクロデキストリンの化学的性質の総説については、Wenz, Agnew. Che
m. Int. Ed. Engl., 33:803-822 (1994)を参照されたい。
て決まる。例えば、それらの水溶性は、不溶性(例えば、トリアセチル-ベータ-シクロ
デキストリン)から、147%の可溶性(w/v)(G-2-ベータ-シクロデキストリ
ン)の範囲にある。さらに、それらは多くの有機溶媒に可溶性を示す。シクロデキストリ
ンの特性は、様々な製剤構成成分の可溶性を増大または低減することによって、それらの
可溶性を調節することができる。
er(I)、ら(米国特許第3,453,259号明細書)およびGrameraら(米国特許第
3,459,731号明細書)は、電気的に中性のシクロデキストリンを記載している。
他の誘導体には、カチオン特性を有するシクロデキストリン[Parmeter(II)、米国特
許第3,453,257号明細書]、不溶性架橋シクロデキストリン(Solms、米国特許
第3,420,788号明細書)、およびアニオン特性を有するシクロデキストリン[Pa
rmeter(III)、米国特許第3,426,011号明細書]が含まれる。アニオン特性
を有するシクロデキストリン誘導体の中でも、カルボン酸、亜リン酸、亜ホスフィン酸、
ホスホン酸、リン酸、チオホスホン酸、チオスルフィン酸、およびスルホン酸は、親シク
ロデキストリンに付加されている[上記Parmeter(III)参照]。さらに、スルホアル
キルエーテルシクロデキストリン誘導体は、Stellaら(米国特許第5,134,127号
明細書)によって記載されている。
ームは、膜のタイプおよびサイズによって特徴付けることができる。小型単層小胞(SU
V)は、単一の膜を有し、典型的に直径が0.02~0.05μmの範囲である。大型単
層小胞(LUV)は、典型的に0.05μmよりも大きい。少数層の大型小胞および多層
小胞は、複数の、通常は同心円状の膜層を有し、典型的に0.1μmよりも大きい。同心
円状ではないいくつかの膜、すなわちより大きい小胞内に含有されているいくつかのより
小さい小胞を有するリポソームは、多胞体小胞と呼ばれる。
ポソーム膜は、担持能力が高いリポソームを提供するように製剤化される。あるいはまた
はさらに、本発明の化合物は、リポソームのリポソーム二層内に含有されるか、またはリ
ポソームのリポソーム二層上に吸着され得る。本発明の化合物は、脂質界面活性剤と凝集
体を形成し、リポソームの内部空間内に担持されていてもよい。こうした場合、リポソー
ム膜は、活性薬剤と界面活性剤の凝集体による破壊作用に抵抗するように製剤化される。
EG)によって誘導体化された脂質を含有し、したがってPEG鎖は、脂質二層の内表面
からリポソームによって囲い込まれた内部空間に延び、脂質二層の外部から取り囲む環境
に延びる。
性剤と活性薬剤の凝集体(例えば、対象となる活性薬剤を含有するエマルジョンまたはミ
セル)は、本発明によるリポソームの内部空間内に捕捉され得る。界面活性剤は、活性薬
剤を分散させ、可溶化するように作用し、それに限定されるものではないが、様々な鎖長
(例えば約C14~約C20)の生体適合性のあるリゾホスファチジルコリン(LPC)
を含めた、任意の適切な脂肪族、脂環式または芳香族の界面活性剤から選択することがで
きる。またPEG-脂質などのポリマー誘導体化脂質は、ミセル/膜の融合を阻害するよ
うに作用し、ポリマーを界面活性剤分子に添加すると、界面活性剤のCMCを低減し、ミ
セルの形成を助けるので、ミセルの形成に利用することができる。マイクロモル範囲のC
MCを有する界面活性剤が好ましい。より高いCMCの界面活性剤を利用して、本発明の
リポソーム内に捕捉されたミセルを調製することができるが、ミセル界面活性剤モノマー
は、リポソームの二層安定性に影響を及ぼすことができ、所望の安定性を有するリポソー
ムを設計する際の因子となる。
れ得る。例えば米国特許第4,235,871号明細書、PCT出願国際公開第96/1
4057号パンフレット;New RRC, Liposomes:A practical approach, IRL Press, Oxf
ord (1990), pages 33-104;Lasic DD, Liposomes from physics to applications, Else
vier Science Publishers BV, Amsterdam, 1993参照。
形成しておいたリポソーム内に拡散させることによって、例えば予め形成しておいたリポ
ソームを、脂質グラフト化ポリマーから構成されたミセルに曝露することによって、リポ
ソームにおいて望ましい誘導体化脂質の最終的なモルパーセントに相当する脂質濃度で調
製することができる。親水性ポリマーを含有するリポソームは、当技術分野で公知の通り
、均質化、脂質-電場による水和(lipid-field hydration)または押出技術によっても
形成され得る。
有するように調製される。サイズ分類する1つの有効な方法では、選択された均一な細孔
径を有する一連のポリカーボネート膜を介して、リポソームの水性懸濁液を押し出す。膜
の細孔径は、その膜を介して押し出すことによって生成したほぼ最大サイズのリポソーム
に相当する。例えば米国特許第4,737,323号明細書(1988年4月12日)参
照。
在に依存して変わる。例えば、放出は、例えば胃内のような低pHでのみ放出するか、ま
たは腸内のようなより高いpHでのみ放出するpH感受性コーティングを使用して、pH
に依存するように操作することができる。胃を通過するまで放出しないようにするために
、腸溶コーティングを使用することができる。複数のコーティングまたは異なる材料に被
包されたシアンアミドの混合物を使用して、胃内で初めて放出させ、その後腸内で放出さ
せることができる。放出はまた、カプセルからの拡散によって水分の取込みまたは薬物の
放出を増大することができる塩または細孔形成剤を含めることによって操作することがで
きる。薬物の可溶性を改変する添加剤を使用して、放出速度を調節することもできる。マ
トリックスの分解またはマトリックスからの放出を促進する薬剤を取り込むこともできる
。それらの薬剤は、薬物に添加し、別個の相として(すなわち微粒子として)添加し、ま
たは化合物に応じてポリマー相に共に溶解させることができる。あらゆる場合において、
その量は、0.1~30パーセント(w/wポリマー)とすべきである。分解促進剤のタ
イプには、無機塩、例えば硫酸アンモニウムおよび塩化アンモニウム、有機酸、例えばク
エン酸、安息香酸およびアスコルビン酸、無機塩基、例えば炭酸ナトリウム、炭酸カリウ
ム、炭酸カルシウム、炭酸亜鉛および水酸化亜鉛、ならびに有機塩基、例えば硫酸プロタ
ミン、スペルミン、コリン、エタノールアミン、ジエタノールアミンおよびトリエタノー
ルアミン、ならびに界面活性剤、例えばTween(登録商標)およびPluronic
(登録商標)が含まれる。マトリックスに微細構造を加える細孔形成剤(すなわち水溶性
化合物、例えば無機塩および糖)は、微粒子として添加される。その範囲は、1~30パ
ーセント(w/wポリマー)とすべきである。
れは、例えば粒子を粘膜接着性ポリマーでコーティングすることによって、または被包材
料として粘膜接着性ポリマーを選択することによって達成することができる。その例とし
て、遊離カルボキシル基を有するほとんどのポリマー、例えばキトサン、セルロース、特
にポリアクリレートが挙げられる(本明細書で使用される場合、ポリアクリレートは、ア
クリレート基および修飾アクリレート基、例えばシアノアクリレートおよびメタクリレー
トを含めたポリマーを指す)。
本発明は、特に、式Iの化合物(または式Iの化合物を含む医薬組成物)を、本明細書
に列挙する疾患の1つまたは複数の処置に使用することに関する。処置に対する応答は、
例えば、疾患の症状の1つまたは複数を、完全に治癒または寛解するまで部分的または完
全に除去することによって実証される場合に有益となる。
使用することができる。
ブ(Inilotinib);ニロチニブ(Tasigna(登録商標));ダサチニブ(BMS-
345825);ボスチニブ(SKI-606);ポナチニブ(AP24534);バフ
ェチニブ(INNO406);ダヌセルチブ(PHA-739358)、AT9283(
CAS1133385-83-7);サラカチニブ(AZD0530);ならびにN-[
2-[(1S,4R)-6-[[4-(シクロブチルアミノ)-5-(トリフルオロメチ
ル)-2-ピリミジニル]アミノ]-1,2,3,4-テトラヒドロナフタレン-1,4
-イミン-9-イル]-2-オキソエチル]-アセトアミド(PF-03814735、
CAS942487-16-3)。
;5-クロロ-N4-(2-(イソプロピルスルホニル)フェニル)-N2-(2-メト
キシ-4-(4-(4-メチルピペラジン-1-イル)ピペリジン-1-イル)フェニル
)ピリミジン-2,4-ジアミン;GSK1838705A;およびCH5424802
。
フェニブ。
)で販売されている);PKC412(ミドスタウリン);タヌチニブ(tanutinib)、
ソラフェニブ、スニチニブ、ミドスタウリン、レスタウルチニブ、KW-2449、キザ
ルチニブ(AC220)およびクレノラニブ(crenolanib)。
ocheから商標アバスチン(登録商標)で販売されている)、アキシチニブ、(N-メ
チル-2-[[3-[(E)-2-ピリジン-2-イルエテニル]-1H-インダゾール
-6-イル]スルファニル]ベンズアミド、AG013736としても公知であり、PC
T国際公開第01/002369号パンフレットに記載されている)、ブリバニブアラニ
ネート((S)-((R)-1-(4-(4-フルオロ-2-メチル-1H-インドール
-5-イルオキシ)-5-メチルピロロ[2,1-f][1,2,4]トリアジン-6-
イルオキシ)プロパン-2-イル)2-アミノプロパノエート、BMS-582664と
しても公知)、モテサニブ(N-(2,3-ジヒドロ-3,3-ジメチル-1H-インド
ール-6-イル)-2-[(4-ピリジニルメチル)アミノ]-3-ピリジンカルボキサ
ミド、PCT国際公開第02/066470号パンフレットに記載されている)、パシレ
オチド(SOM230としても公知であり、PCT国際公開第02/010192号パン
フレットに記載されている)、ソラフェニブ(商標Nexavar(登録商標)で販売さ
れている);
erceptin(登録商標)で販売されている)、ネラチニブ(HKI-272、(2
E)-N-[4-[[3-クロロ-4-[(ピリジン-2-イル)メトキシ]フェニル]
アミノ]-3-シアノ-7-エトキシキノリン-6-イル]-4-(ジメチルアミノ)ブ
タ-2-エナミドとしても公知であり、PCT国際公開第05/028443号パンフレ
ットに記載されている)、ラパチニブまたはジトシル酸ラパチニブ(GlaxoSmit
hKlineから商標Tykerb(登録商標)で販売されている);トラスツズマブエ
ムタンシン(米国では、アド-トラスツズマブエムタンシン、商標Kadcyla)-細
胞傷害剤メルタンシン(DM1)に連結しているモノクローナル抗体トラスツズマブ(ハ
ーセプチン)からなる抗体-薬物コンジュゲート;
(登録商標)およびMabThera(登録商標)で販売されている)、トシツモマブ(
GlaxoSmithKlineから商標Bexxar(登録商標)で販売されている)
、オファツムマブ(GlaxoSmithKlineから商標Arzerra(登録商標
)で販売されている);
標Tarceva(登録商標)で販売されている)、リニファニブ(N-[4-(3-ア
ミノ-1H-インダゾール-4-イル)フェニル]-N’-(2-フルオロ-5-メチル
フェニル)尿素、ABT869としても公知であり、Genentechから利用可能で
ある)、リンゴ酸スニチニブ(Pfizerから商標Sutent(登録商標)で販売さ
れている)、ボスチニブ(4-[(2,4-ジクロロ-5-メトキシフェニル)アミノ]
-6-メトキシ-7-[3-(4-メチルピペラジン-1-イル)プロポキシ]キノリン
-3-カルボニトリル、SKI-606としても公知であり、米国特許第6,780,9
96号明細書に記載されている)、ダサチニブ(Bristol-Myers Squi
bbから商標Sprycel(登録商標)で販売されている)、アルマラ(armala)(パ
ゾパニブとしても公知であり、GlaxoSmithKlineから商標Votrien
t(登録商標)で販売されている)、イマチニブおよびメシル酸イマチニブ(Novar
tisから商標Gilvec(登録商標)およびGleevec(登録商標)で販売され
ている);
売されている)、塩酸ゲムシタビン(Eli Lilly and Companyから
商標Gemzar(登録商標)で販売されている)、ネララビン((2R,3S,4R,
5R)-2-(2-アミノ-6-メトキシ-プリン-9-イル)-5-(ヒドロキシメチ
ル)オキソラン-3,4-ジオール、GlaxoSmithKlineから商標Arra
non(登録商標)およびAtriance(登録商標)で販売されている);
atin(登録商標)で販売されており、米国特許第4,169,846号明細書に記載
されている);
a(登録商標)で販売されている)、N-[4-[(3-クロロ-4-フルオロフェニル
)アミノ]-7-[[(3”S”)-テトラヒドロ-3-フラニル]オキシ]-6-キナ
ゾリニル]-4(ジメチルアミノ)-2-ブテンアミド、Boehringer Ing
elheimから商標Tovok(登録商標)で販売されている)、セツキシマブ(Br
istol-Myers Squibbから商標Erbitux(登録商標)で販売され
ている)、パニツムマブ(Amgenから商標Vectibix(登録商標)で販売され
ている);
(登録商標)で販売されている);
genから商標Neupogen(登録商標)で販売されている);
グフィルグラスチム(Amgenから商標Neulasta(登録商標)で販売されてい
る)、レナリドミド(CC-5013としても公知であり、商標Revlimid(登録
商標)で販売されている)、サリドマイド(商標Thalomid(登録商標)で販売さ
れている);
、Seattle Genetics,Incから利用可能である);
AMG-951としても公知であり、Amgen/Genentechから利用可能であ
る);
)フェニル]-4-(メチルスルホニル)-ベンズアミド(GDC-0449としても公
知であり、PCT国際公開第06/028958号パンフレットに記載されている);
ルスルホニル)ピペラジン-1-イル]メチル]チエノ[3,2-d]ピリミジン-4-
イル]モルホリン(GDC0941としても公知であり、PCT国際公開第09/036
082号パンフレットおよび国際公開第09/055730号パンフレットに記載されて
いる)、2-メチル-2-[4-[3-メチル-2-オキソ-8-(キノリン-3-イル
)-2,3-ジヒドロイミダゾ[4,5-c]キノリン-1-イル]フェニル]プロピオ
ニトリル(BEZ235またはNVP-BEZ235としても公知であり、PCT国際公
開第06/122806号パンフレットに記載されている);
れている);
1-シクロヘキサエン-1-イル]メチル]-1-ピペラジニル]-N-[[4-[[(
1R)-3-(4-モルホリニル)-1-[(フェニルチオ)メチル]プロピル]アミノ
]-3-[(トリフルオロメチル)スルホニル]フェニル]スルホニル]ベンズアミド(
ABT-263としても公知であり、PCT国際公開第09/155386号パンフレッ
トに記載されている);
1029872-29-4、ACC Corp.から利用可能である);
商標)で販売されている)、レトロゾール(Novartisから商標Femara(登
録商標)で販売されている)、アナストロゾール(商標Arimidex(登録商標)で
販売されている);
(登録商標)で販売されている)、塩酸トポテカン(GlaxoSmithKlineか
ら商標Hycamtin(登録商標)で販売されている);
も公知であり、商標Toposar(登録商標)、VePesid(登録商標)およびE
topophos(登録商標)で販売されている)、テニポシド(VM-26としても公
知であり、商標Vumon(登録商標)で販売されている);
で販売されている)、リダフォロリムス(以前はデフォロリムス(deferolimus)として
公知であり、(1R,2R,4S)-4-[(2R)-2[(1R,9S,12S,15
R,16E,18R,19R,21R、23S,24E,26E,28Z,30S,32
S,35R)-1,18-ジヒドロキシ-19,30-ジメトキシ-15,17,21,
23、29,35-ヘキサメチル-2,3,10,14,20-ペンタオキソ-11,3
6-ジオキサ-4-アザトリシクロ[30.3.1.04,9]ヘキサトリアコンタ-1
6,24,26,28-テトラエン-12-イル]プロピル]-2-メトキシシクロヘキ
シルジメチルホスフィネート、AP23573およびMK8669としても公知であり、
PCT国際公開第03/064383号パンフレットに記載されている)、エベロリムス
(Novartisから商標Afinitor(登録商標)で販売されている);
チル)ホスホン酸一水和物(Novartisから商標Zometa(登録商標)で販売
されている);
ethから商標Mylotarg(登録商標)で販売されている);
びWAY-207294とも呼ばれ、Hangzhou Sage Chemical
Co.,Ltd.から利用可能である);
(登録商標)で販売されている);
も公知であり、商標Sandostatin(登録商標)およびSandostatin
LAR(登録商標)で販売されている);
thから商標Neumega(登録商標)で販売されている);
(登録商標)で販売されている);
標Prolia(登録商標)で販売されている);
ら商標Nplate(登録商標)で販売されている;
標)で販売されている);
51,871としても公知であり、ACC Corpから利用可能である)、ロバツムマ
ブ(robatumumab)(CAS番号934235-44-6);
;
;
675,206として公知である、Pfizerから利用可能なIgG2モノクローナル
抗体)、イピリムマブ(CTLA-4抗体、MDX-010としても公知である、CAS
番号477202-00-9);
kから商標Zolinza(登録商標)で販売されている);
Temodar(登録商標)およびTemodal(登録商標)で販売されている)、ダ
クチノマイシン(アクチノマイシン-Dとしても公知であり、商標Cosmegen(登
録商標)で販売されている)、メルファラン(L-PAM、L-サルコリシン、およびフ
ェニルアラニンマスタードとしても公知であり、商標Alkeran(登録商標)で販売
されている)、アルトレタミン(ヘキサメチルメラミン(HMM)としても公知であり、
商標Hexalen(登録商標)で販売されている)、カルムスチン(商標BiCNU(
登録商標)で販売されている)、ベンダムスチン(商標Treanda(登録商標)で販
売されている)、ブスルファン(商標Busulfex(登録商標)およびMylera
n(登録商標)で販売されている)、カルボプラチン(商標Paraplatin(登録
商標)で販売されている)、ロムスチン(CCNUとしても公知であり、商標CeeNU
(登録商標)で販売されている)、シスプラチン(CDDPとしても公知であり、商標P
latinol(登録商標)およびPlatinol(登録商標)-AQで販売されてい
る)、クロラムブシル(商標Leukeran(登録商標)で販売されている)、シクロ
ホスファミド(商標Cytoxan(登録商標)およびNeosar(登録商標)で販売
されている)、ダカルバジン(DTIC、DICおよびイミダゾールカルボキサミドとし
ても公知であり、商標DTIC-Dome(登録商標)で販売されている)、アルトレタ
ミン(ヘキサメチルメラミン(HMM)としても公知であり、商標Hexalen(登録
商標)で販売されている)、イホスファミド(商標Ifex(登録商標)で販売されてい
る)、プロカルバジン(商標Matulane(登録商標)で販売されている)、メクロ
レタミン(ナイトロジェンマスタード、ムスチンおよび塩酸メクロレタミン(mechloroet
hamine)としても公知であり、商標Mustargen(登録商標)で販売されている)
、ストレプトゾシン(商標Zanosar(登録商標)で販売されている)、チオテパ(
チオホスホアミド、TESPAおよびTSPAとしても公知であり、商標Thiople
x(登録商標)で販売されている;
びTICE(登録商標)BCGで販売されている)、デニロイキンジフチトクス(商標O
ntak(登録商標)で販売されている);
bex(登録商標)で販売されている)、ブレオマイシン(商標lenoxane(登録
商標)で販売されている)、ダウノルビシン(塩酸ダウノルビシン(dauorubicin)、ダ
ウノマイシン、および塩酸ルビドマイシンとしても公知であり、商標Cerubidin
e(登録商標)で販売されている)、ダウノルビシンリポソーム(ダウノルビシンクエン
酸塩リポソーム、商標DaunoXome(登録商標)で販売されている)、ミトキサン
トロン(DHADとしても公知であり、商標Novantrone(登録商標)で販売さ
れている)、エピルビシン(商標Ellence(商標)で販売されている)、イダルビ
シン(商標Idamycin(登録商標)、Idamycin PFS(登録商標)で販
売されている)、マイトマイシンC(商標Mutamycin(登録商標)で販売されて
いる);
ル)-4-フルオロ-N2-{(1S)-2,2,2-トリフルオロ-1-[4’-(メ
チルスルホニル)ビフェニル-4-イル]エチル}-L-ロイシンアミドとしても公知で
あり、Lanzhou Chon Chemicals、ACC Corp.、およびC
hemieTekから利用可能であり、PCT国際公開第03/075836号パンフレ
ットに記載されている);
ら商標Lxempra(登録商標)で販売されている);
7-デメトキシゲルダナマイシン、KOS-953および17-AAGとしても公知であ
り、SIGMAから利用可能であり、米国特許第4,261,989号明細書に記載され
ている);
romacta(登録商標)およびRevolade(登録商標)で販売されている);
e(登録商標)で販売されている);
売されている);
dron(登録商標)で販売されている)、ビカルタミド(商標Casodex(登録商
標)で販売されている)、フルタミド(商標Fulexin(商標)で販売されている)
;
されている);
いる);
275、2-(2-クロロフェニル)-5,7-ジヒドロキシ-8-[(3S,4R)-
3-ヒドロキシ-1-メチル-4-ピペリジニル]-4-クロメノンとしても公知であり
、米国特許第5,621,002号明細書に記載されている);
ロイプロリド(Bayer AGから商標Viadure(登録商標)で販売されており
、Sanofi-AventisからEligard(登録商標)で販売されており、A
bbott LabからLupron(登録商標)で販売されている);
-9-オキソ-5β,20-エポキシタキサ-11-エン-2α,4,13α-トリイル
-4-アセテート-2-ベンゾエート-13-[(2R,3S)-3-{[(tert-
ブトキシ)カルボニル]アミノ}-2-ヒドロキシ-3-フェニルプロパノエート)、ラ
ロタキセル((2α,3ξ,4α,5β,7α,10β,13α)-4,10-ビス(ア
セチルオキシ)-13-({(2R,3S)-3-[(tert-ブトキシカルボニル)
アミノ]-2-ヒドロキシ-3-フェニルプロパノイル}オキシ)-1-ヒドロキシ-9
-オキソ-5,20-エポキシ-7,19-シクロタキサ-11-エン-2-イルベンゾ
エート);
フチル)エチル]-4-[3-(トリフルオロメチル)フェニル]-1,2,3,6-テ
トラヒドロピリジンとしても公知であり、米国特許第5,266,573号明細書に記載
されている);
で販売されており、MerckからGardasil(登録商標)で販売されている;
ade(登録商標)で販売されている);
eustatin(登録商標)で販売されている)、5-フルオロウラシル(商標Adr
ucil(登録商標)で販売されている)、6-チオグアニン(商標Purinetho
l(登録商標)で販売されている)、ペメトレキセド(商標Alimta(登録商標)で
販売されている)、シタラビン(アラビノシルシトシン(Ara-C)としても公知であ
り、商標Cytosar-U(登録商標)で販売されている)、シタラビンリポソーム(
リポソームAra-Cとしても公知であり、商標DepoCyt(商標)で販売されてい
る)、デシタビン(商標Dacogen(登録商標)で販売されている)、ヒドロキシ尿
素(商標Hydrea(登録商標)、Droxia(商標)およびMylocel(商標
)で販売されている)、フルダラビン(商標Fludara(登録商標)で販売されてい
る)、フロクスウリジン(商標FUDR(登録商標)で販売されている)、クラドリビン
(2-クロロデオキシアデノシン(2-CdA)としても公知であり、商標Leusta
tin(商標)で販売されている)、メトトレキセート(アメトプテリン、メトトレキサ
ートナトリウム(sodim)(MTX)としても公知であり、商標Rheumatrex(
登録商標)およびTrexall(商標)で販売されている)、ペントスタチン(商標N
ipent(登録商標)で販売されている);
る)、ゾレドロン酸(商標Zometa(登録商標)で販売されている);
、デシタビン(商標Dacogen(登録商標)で販売されている);
登録商標)で販売されている)、ビンブラスチン(硫酸ビンブラスチン、ビンカロイコブ
ラスチンおよびVLBとしても公知であり、商標Alkaban-AQ(登録商標)およ
びVelban(登録商標)で販売されている)、ビンクリスチン(硫酸ビンクリスチン
、LCR、およびVCRとしても公知であり、商標Oncovin(登録商標)およびV
incasar Pfs(登録商標)で販売されている)、ビノレルビン(商標Nave
lbine(登録商標)で販売されている)、パクリタキセル(商標TaxolおよびO
nxal(商標)で販売されている);
)、トレチノイン(全トランスレチノイン酸、ATRAとしても公知であり、商標Ves
anoid(登録商標)で販売されている)、イソトレチノイン(13-cis-レチノ
イン酸、商標Accutane(登録商標)、Amnesteem(登録商標)、Cla
ravis(登録商標)、Clarus(登録商標)、Decutan(登録商標)、I
sotane(登録商標)、Izotech(登録商標)、Oratane(登録商標)
、Isotret(登録商標)、およびSotret(登録商標)で販売されている)、
ベキサロテン(商標Targretin(登録商標)で販売されている);
ナトリウム、リン酸ヒドロコルチゾンナトリウムとしても公知であり、商標Ala-Co
rt(登録商標)、リン酸ヒドロコルチゾン、Solu-Cortef(登録商標)、H
ydrocort Acetate(登録商標)およびLanacort(登録商標)で
販売されている)、デキサメタゾン(dexamethazone)((8S,9R,10S,11S
,13S,14S,16R,17R)-9-フルオロ-11,17-ジヒドロキシ-17
-(2-ヒドロキシアセチル)-10,13,16-トリメチル-6,7,8,9,10
,11,12,13,14,15,16,17-ドデカヒドロ-3H-シクロペンタ[a
]フェナントレン-3-オン)、プレドニゾロン(商標Delta-Cortel(登録
商標)、Orapred(登録商標)、Pediapred(登録商標)およびPrel
one(登録商標)で販売されている)、プレドニゾン(商標Deltasone(登録
商標)、Liquid Red(登録商標)、Meticorten(登録商標)および
Orasone(登録商標)で販売されている)、メチルプレドニゾロン(6-メチルプ
レドニゾロン、酢酸メチルプレドニゾロン、コハク酸メチルプレドニゾロンナトリウムと
しても公知であり、商標Duralone(登録商標)、Medralone(登録商標
)、Medrol(登録商標)、M-Prednisol(登録商標)およびSolu-
Medrol(登録商標)で販売されている);
であり、商標Proleukin(登録商標)で販売されている)、インターロイキン-
11(オプレルベキン(oprevelkin)としても公知であり、商標Neumega(登録商
標)で販売されている)、アルファインターフェロンアルファ(IFN-アルファとして
も公知であり、商標Intron(登録商標)A、およびRoferon-A(登録商標
)で販売されている);
標)で販売されている);
いる);
sta(登録商標)で販売されている);
(登録商標)で販売されている);
gace(登録商標)で販売されている);
)、アスパラギナーゼ(L-アスパラギナーゼ、エルウィニアL-アスパラギナーゼとし
ても公知であり、商標Elspar(登録商標)およびKidrolase(登録商標)
で販売されている);
neから商標Rezonic(登録商標)およびZunrisa(登録商標)で販売され
ている);および
コボリン(ロイコボリンカルシウム、シトロボラム因子およびフォリン酸としても公知で
ある)。
性に悪影響を及ぼし得る先行技術であることを承認するものと理解されるべきではない。
本発明はまた、本発明の化合物を調製する方法を含む。記載の反応では、反応性官能基
、例えばヒドロキシ、アミノ、イミノ、チオまたはカルボキシ基が、最終生成物において
望ましい場合、それらを保護して、反応における望ましくない関与を回避する必要があり
得る。従来の保護基は、標準技法に従って使用することができ、例えばT.W. Greene and
P. G. M. Wuts in “Protective Groups in Organic Chemistry”, John Wiley and Sons
, 1991を参照されたい。
調製することができる。
反応スキームI:

式中、X1、X2、Y1、Y2、Y3、R1、R2、R3a、R3b、R4a、R4b、
R5a、R5b、R6aおよびR6bは、本発明の概要によって定義されている通りであ
り、Qは、脱離基、例えばヨウ化物等である。式Iの化合物は、式2の化合物を式3の化
合物と、適切な溶媒(例えばジオキサン等)、適切な金属リガンド(例えばTMEDA等
)、適切な金属ハロゲン化物(例えばCu(I)I等)および適切な塩(例えばK3PO
4等)が存在する状態で反応させることによって、調製することができる。反応は約80
℃~約140℃の温度範囲で進行し、完了するのに約1時間~約24時間かかり得る。
F等)、適切なカップリング剤(例えばCuTC等)および適切な塩(例えば炭酸カリウ
ム等)が存在する状態で反応させることによって、調製することができる。反応は約80
℃~約140℃の温度範囲で進行し、完了するのに約1時間~約24時間かかり得る。
媒(例えばジオキサン等)、適切な金属ハロゲン化物(例えばCu(I)I等)、適切な
塩基(例えば炭酸セシウム等)および適切なリガンド(例えば1,10-フェナントロリ
ン等)が存在する状態で反応させることによって、調製することができる。反応は約80
℃~約140℃の温度範囲で進行し、完了するのに約1時間~約24時間かかり得る。

式中、X1、X2、Y1、Y2、Y3、R1、R2、R3a、R3b、R4a、R4b、
R5a、R5b、R6aおよびR6bは、本発明の概要によって定義されている通りであ
り、Qは、脱離基、例えばヨウ化物等である。式Iの化合物は、式4の化合物を式5の化
合物と、適切な溶媒(例えばMeCN、DMF等)、適切なカップリング剤(例えばBO
P-Cl、BOP等)および適切な触媒(例えばDBU等)が存在する状態で反応させる
ことによって、調製することができる。反応は約80℃~約140℃の温度範囲で進行し
、完了するのに約1時間~約24時間かかり得る。
本発明の化合物は、化合物の遊離塩基形態を、薬学的に許容される無機または有機酸と
反応させることによって、薬学的に許容される酸付加塩として調製することができる。あ
るいは、本発明の化合物の薬学的に許容される塩基付加塩は、化合物の遊離酸形態を、薬
学的に許容される無機または有機塩基と反応させることによって調製することができる。
することができる。この種類の修飾は、当技術分野で公知であり、それには、所与の生物
系(例えば、血液、リンパ系、中枢神経系、精巣)への浸透を増大し、バイオアベイラビ
リティを増大し、可溶性を増大して非経口投与(例えば、注射、注入)を可能にし、代謝
を変化させ、かつ/または分泌速度を変化させる修飾が含まれる。このタイプの修飾の例
として、例えばポリエチレングリコールを用いるエステル化、ピバロイルオキシまたは脂
肪酸置換基を用いる誘導体化、カルバメートへの変換、芳香環のヒドロキシル化、および
芳香族環におけるヘテロ原子の置換が挙げられるが、それらに限定されない。式Iの化合
物、ならびに/またはそのN-オキシド、互変異性体および/または(好ましくは薬学的
に許容される)塩がどこで言及されようと、これは、このような修飾された式を含むと同
時に、好ましくは式Iの分子、それらのN-オキシド、それらの互変異性体および/また
はそれらの塩を意味する。
ことができる。遊離形態の新規な式Iの化合物と、例えば新規な化合物の精製または同定
において中間体として使用できる塩を含めた、それらの塩の形態の式Iの化合物の間の密
接な関係を考慮すると、式Iの1つまたは複数の化合物に関するいかなる言及も、本明細
書を通して、遊離形態の化合物、および/または適切で好都合な場合、それらの1つもし
くは複数の塩、ならびに1つまたは複数の溶媒和物、例えば水和物に言及すると理解され
るべきである。
化合物から酸付加塩として、特に薬学的に許容される塩として形成される。適切な無機酸
は、例えばハロゲン酸、例えば塩酸、硫酸またはリン酸である。適切な有機酸は、例えば
カルボン酸、ホスホン酸、スルホン酸もしくはスルファミン酸、例えば酢酸、プロピオン
酸、オクタン酸、デカン酸、ドデカン酸、グリコール酸、乳酸、フマル酸、コハク酸、マ
ロン酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、リンゴ酸、酒石酸、クエ
ン酸、アミノ酸、例えばグルタミン酸もしくはアスパラギン酸、マレイン酸、ヒドロキシ
マレイン酸、メチルマレイン酸、シクロヘキサンカルボン酸、アダマンタンカルボン酸、
安息香酸、サリチル酸、4-アミノサリチル酸、フタル酸、フェニル酢酸、マンデル酸、
ケイ皮酸、メタン-もしくはエタン-スルホン酸、2-ヒドロキシエタンスルホン酸、エ
タン-1,2-ジスルホン酸、ベンゼンスルホン酸、4-トルエンスルホン酸、2-ナフ
タレンスルホン酸、1,5-ナフタレン-ジスルホン酸、2-もしくは3-メチルベンゼ
ンスルホン酸、メチル硫酸、エチル硫酸、ドデシル硫酸、N-シクロヘキシルスルファミ
ン酸、N-メチル-、N-エチル-もしくはN-プロピル-スルファミン酸、または他の
有機プロトン酸、例えばアスコルビン酸である。
素酸塩を使用することも可能である。治療上の使用では、薬学的に許容される塩または遊
離化合物だけが用いられ(医薬調製物の形態で適用できる場合)、したがってこれらが好
ましい。
付加塩形態から調製することができる。例えば、酸付加塩形態の本発明の化合物は、適切
な塩基(例えば、水酸化アンモニウム溶液、水酸化ナトリウム等)を用いて処理すること
によって、対応する遊離塩基に変換することができる。塩基付加塩形態の本発明の化合物
は、適切な酸(例えば、塩酸等)を用いて処理することによって、対応する遊離酸に変換
することができる。
、硫黄、二酸化硫黄、トリフェニルホスフィン、水素化ホウ素リチウム、水素化ホウ素ナ
トリウム、三塩化リン、三臭化物等)を用いて、適切な不活性有機溶媒(例えば、MeC
N、エタノール、ジオキサン水溶液等)中で0~80℃において処理することによって調
製することができる。
できる(例えば、さらなる詳細については、Saulnier et al., (1994), Bioorganic and
Medicinal Chemistry Letters, Vol. 4, p. 1985を参照されたい)。例えば、適切なプロ
ドラッグは、本発明の非誘導体化化合物を、適切なカルバミル化剤(例えば、1,1-ア
シルオキシアルキルカルバノクロリデート(acyloxyalkylcarbanochloridate)、炭酸パ
ラ-ニトロフェニル等)と反応させることによって調製することができる。
きる。保護基の創作およびそれらの除去に適用できる技術の詳細な説明は、T. W. Greene
, “Protecting Groups in Organic Chemistry”, 3rd edition, John Wiley and Sons,
Inc., 1999に見出すことができる。
して調製または形成することができる。本発明の化合物の水和物は、好都合には、水性/
有機溶媒の混合物から再結晶化させることによって、有機溶媒、例えばダイオキシン、テ
トラヒドロフランまたはメタノールを使用して調製することができる。
のジアステレオマー化合物を形成し、ジアステレオマーを分離し、光学的に純粋なエナン
チオマーを回収することによって、それらの個々の立体異性体として調製することができ
る。エナンチオマーの分割は、本発明の化合物の共有結合性のジアステレオマー誘導体を
使用して実施することができるが、解離できる複合体が好ましい(例えば、結晶性ジアス
テレオマー塩)。ジアステレオマーは、明確な物理的特性(例えば、融点、沸点、可溶性
、反応性等)を有し、これらの相違点を利用することによって容易に分離することができ
る。ジアステレオマーは、クロマトグラフィーによって、または好ましくは可溶性の差異
に基づく分離/分割技術によって分離することができる。次に、ラセミ化を生じるおそれ
がない任意の実用的な手段によって、分割剤と共に光学的に純粋なエナンチオマーを回収
する。化合物の立体異性体をそれらのラセミ混合物から分割するのに適用できる技術のよ
り詳細な説明は、Jean Jacques, Andre Collet, Samuel H. Wilen, “Enantiomers, Race
mates and Resolutions”, John Wiley And Sons, Inc., 1981に見出すことができる。
(a)反応スキームIおよびIIの方法、ならびに
(b)任意選択で、本発明の化合物を、薬学的に許容される塩に変換すること、
(c)任意選択で、本発明の化合物の塩形態を、塩ではない形態に変換すること、
(d)任意選択で、本発明の化合物の未酸化形態を、薬学的に許容されるN-オキシドに
変換すること、
(e)任意選択で、本発明の化合物のN-オキシド形態を、その未酸化形態に変換するこ
と、
(f)任意選択で、本発明の化合物の個々の異性体を、異性体混合物から分割すること、
(g)任意選択で、本発明の非誘導体化化合物を、薬学的に許容されるプロドラッグ誘導
体に変換すること、ならびに
(h)任意選択で、本発明の化合物のプロドラッグ誘導体を、その非誘導体化形態に変換
すること
を含む方法によって作成することができる。
当技術分野で公知の方法と同様にもしくは以下の実施例に開示されている通りに調製する
ことができる。
の周知の方法を同様に使用できることを理解されよう。
に役立つものである。実施例で使用するいくつかの略語は以下の通りである:酢酸(Ac
OH);MeCN(MeCN);トリエチルアミン(TEA);テトラヒドロフラン(T
HF);水性(aq);飽和(sat.);気圧(atm.);2,2’-ビス-ジフェ
ニルホスファニル-[1,1’]ビナフタレニル(BINAP);4-ジメチルアミノピ
リジン(DMAP);tert-ブトキシカルボニル(Boc);1,1-カルボニルジ
イミダゾール(CDI);二炭酸ジ-tert-ブチル(Boc2O);ベンゾトリアゾ
ール-1-イル-オキシ-トリス-(ジメチルアミノ)-ホスホニウムヘキサフルオロホ
スフェート(BOP);ジクロロメタン(DCM);ジエチルエーテル(Et2O);p
-トルエンスルホン酸(PTSA);酢酸エチル(EtOAc);エタノール(EtOH
);リチウムビス(トリメチルシリル)アミド(LHMDS);アゾジカルボン酸ジイソ
プロピル(DIAD);N,N-ジイソプロピル-エチルアミン(DIEAまたはDIP
EA);N,N-ジメチルホルムアミド(DMF);ジメチルスルホキシド(DMSO)
;ジフェニルホスホリルアジド(DPPA);時間(h);2-(1H-7-アザベンゾ
トリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホ
スフェート(HATU);高速液体クロマトグラフィー(HPLC);イソプロピルアル
コール(IPA);水素化アルミニウムリチウム(LAH);質量分析と組み合わせた液
体クロマトグラフィー(LCMS);リチウムジイソプロピルアミド(LDA);メタノ
ール(MeOH);ミリリットル(mL);分(min);マイクロ波(MW);ナトリ
ウムビス(トリメチルシリル)アミド(NHMDS);n-ブチルリチウム(n-BuL
i);1,1-ビス(ジフェニルホスフィノ)-フェロセンジクロロパラジウム(II)
(PdCl2(dppf));トリス(ジベンジリデンアセトン)ジパラジウム(0)(
Pd2(dba)3);ジクロロビス(トリフェニルホスフィン)パラジウム(II)(
PdCl2(PPh3)2);室温(RT);テトラ-n-ブチルアンモニウムフルオリ
ド(TBAF);tert-ブチルジメチルシリルクロリド(TBSCl);トリフルオ
ロ酢酸(TFA);テトラヒドロフラン(THF);薄層クロマトグラフィー(TLC)
;保持時間(tR);(S)-(-)-2,2’-ビス(ジ-p-トリルホスフィノ)-
1,1’-ビナフチル((S)-TolBINAP);および4,5-ビス(ジフェニル
ホスフィノ)-9,9-ジメチルキサンテン(キサントホス)。
ナトリウム2-アミノ-3-クロロピリジン-4-チオレート

ジオキサン(13mL)中の3-クロロ-4-ヨードピリジン-2-アミン(1.0g
、3.93mmol)、キサントホス(136mg、0.236mmol)およびPd(
OAc)2(44mg、0.196mmol)の溶液に、室温でおよびN2雰囲気下で、
メチル3-メルカプトプロパノエート(479μL、4.32mmol)を添加し、続い
てDIPEA(1.37mL、7.86mmol)を添加した。得られた溶液を100℃
で2時間撹拌した。室温に冷却した後、反応混合物をEtOAc(20mL)で希釈し、
セライトのパッドに通して濾過し、続いてEtOAcで洗浄(25mL)した。合わせた
濾液を減圧下で濃縮し、残留物をシリカクロマトグラフィー(0から10%の勾配のMe
OH/DCM)により精製して、メチル3-((2-アミノ-3-クロロピリジン-4-
イル)チオ)プロパノエート(970mg、3.93mmol)を得た。MSm/z24
7.1(M+H)+。
THF(14mL)中のメチル3-((2-アミノ-3-クロロピリジン-4-イル)
チオ)プロパノエート(1.04g、4.22mmol)の溶液に、室温でおよびN2下
で、ナトリウムエトキシド(EtOH中21重量%、1.65mL、4.43mmol)
を室温でおよびN2雰囲気下で添加した。室温で40分間激しく撹拌した後、反応混合物
をDCM(30mL)で希釈し、これを5分間超音波処理した。形成されて得られた固体
を濾別し、続いてDCMで洗浄(5mL)し、減圧下で乾燥させて、ナトリウム2-アミ
ノ-3-クロロピリジン-4-チオレート(770mg、4.22mmol)を得た。1H
NMR (400 MHz, メタノール-d4) δ ppm 7.23 (d, J=5.56 Hz, 1 H), 6.82 (d, J=5.56 H
z, 1 H).
化アリールまたは臭化アリールを使用して作製した。

2-(トリフルオロメトキシ)ピリジン-3-チオール

DCM(15mL)中の2-(トリフルオロメトキシ)ピリジン-3-オール(0.7
5g、4.19mmol)およびEt3N(1.17mL、8.38mmol)の-78
℃溶液に、トリフルオロメタンスルホン酸無水物(DCM中1M、6.28mL、6.2
8mmol)を添加した。得られた溶液を-78℃で30分間撹拌した。反応混合物を飽
和NaHCO3水溶液(25mL)で慎重に希釈し、得られた混合物をDCM(2×15
mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除
去した。残留物をシリカクロマトグラフィー(0から40%の勾配のEtOAc/ヘプタ
ン)により精製して、2-(トリフルオロメトキシ)ピリジン-3-イルトリフルオロメ
タンスルホネート(1.25g、4.02mmol)を得た。MSm/z312.0(M
+H)+。
ジオキサン(10mL)中の2-(トリフルオロメトキシ)ピリジン-3-イルトリフ
ルオロメタンスルホネート(1.25g、4.02mmol)、キサントホス(139m
g、0.241mmol)およびPd(OAc)2(45mg、0.201mmol)の
溶液に、室温でおよびN2雰囲気下で、メチル3-メルカプトプロパノエート(489μ
L、4.42mmol)を添加し、続いてDIPEA(1.4mL、8.03mmol)
を添加した。得られた溶液を100℃で2時間撹拌した。室温に冷却した後、反応混合物
をEtOAc(20mL)で希釈し、セライトのパッドに通して濾過し、続いてEtOA
cで洗浄(25mL)した。合わせた濾液を減圧下で濃縮し、残留物をカラムクロマトグ
ラフィー(シリカゲル、0から25%の勾配のEtOAc/ヘプタン)により精製して、
メチル3-((2-(トリフルオロメトキシ)ピリジン-3-イル)チオ)プロパノエー
ト(1.025g、3.64mmol)を得た。MSm/z282.1(M+H)+。
THF(12mL)中のメチル3-((2-(トリフルオロメトキシ)ピリジン-3-
イル)チオ)プロパノエート(1.025g、3.64mmol)の溶液に、室温でおよ
びN2下で、ナトリウムエトキシド(EtOH中21重量%、1.43mL、3.83m
mol)を添加した。室温で40分間激しく撹拌した後、反応混合物をDCM(40mL
)で希釈し、5分間超音波処理した。揮発物を減圧下で除去し、残留物をDCM中に懸濁
させ、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れた。有機相を分離し、水相を
DCM(2×15mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮
発物を減圧下で除去した。水相を1N HCl水溶液で酸性化し、DCM(3×10mL
)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去し
て、粗製の2-(トリフルオロメトキシ)ピリジン-3-チオール(711mg、3.6
4mmol)を得た。MSm/z194.1(M-H)-。
ナトリウム3-クロロ-2-(ピロリジン-1-イル)ピリジン-4-チオレート

DMSO(10mL)中の3-クロロ-2-フルオロ-4-ヨードピリジン(2.0g
、7.77mmol)およびピロリジン(1.93mL、23.31mmol)の溶液を
、70℃で30分間撹拌した。室温に冷却した後、得られた混合物を、飽和NH4Cl水
溶液を含有する分液漏斗に注ぎ入れ、Et2O(5×10mL)で抽出した。合わせた有
機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去して、3-クロロ-4-ヨー
ド-2-(ピロリジン-1-イル)ピリジン(1.66g、5.38mmol)を得た。
MSm/z309.0(M+H)+。
ジオキサン(11mL)中の3-クロロ-4-ヨード-2-(ピロリジン-1-イル)
ピリジン(1.66g、5.38mmol)、キサントホス(187mg、0.323m
mol)およびPd(OAc)2(60mg、0.269mmol)の溶液に、室温でお
よびN2雰囲気下で、メチル3-メルカプトプロパノエート(655μL、5.92mm
ol)を添加し、続いてDIPEA(1.88mL、10.76mmol)を添加した。
得られた溶液を100℃で2時間撹拌した。室温に冷却した後、反応混合物をEtOAc
(20mL)で希釈し、セライトのパッドに通して濾過し、続いてEtOAcで洗浄(2
5mL)した。合わせた濾液を減圧下で濃縮し、残留物をカラムクロマトグラフィー(シ
リカゲル、0から30%の勾配のEtOAc/ヘプタン)により精製して、メチル3-(
(3-クロロ-2-(ピロリジン-1-イル)ピリジン-4-イル)チオ)プロパノエー
ト(1.62g、5.38mmol)を得た。MSm/z301.2(M+H)+。
THF(20mL)中のメチル3-((3-クロロ-2-(ピロリジン-1-イル)ピ
リジン-4-イル)チオ)プロパノエート(1.62g、5.38mmol)の溶液に、
ナトリウムエトキシド(EtOH中21重量%、2.39mL、6.39mmol)を室
温でおよびN2雰囲気下で添加した。室温で40分間激しく撹拌した後、反応物をDCM
(40mL)で希釈し、これを5分間超音波処理した。揮発物を減圧下で除去し、残留物
をさらに精製することなく使用した。MSm/z215.1(M-H)-。
化アリールを使用して作製した。

3-アミノ-2-(トリフルオロメチル)ベンゼンチオール

DMF(25mL)中の3-フルオロ-2-(トリフルオロメチル)アニリン(2.2
1g、12.35mmol)、Cs2CO3(12.08g、37.1mmol)および
2-メチルプロパン-2-チオール(4.18mL、37.1mmol)の混合物を、1
30℃で18時間撹拌した。室温に冷却した後、反応混合物を、H2O(50mL)を含
有する分液漏斗に注ぎ入れ、EtOAc(100mL)で抽出した。有機相をH2O(2
×25mL)、ブライン(2×25mL)で洗浄し、MgSO4で脱水し、濾過し、揮発
物を減圧下で除去して、3-(tert-ブチルチオ)-2-(トリフルオロメチル)ア
ニリン(3.08mg、12.35mmol)を得た。MSm/z250.1(M+H)
+。
濃HCl(308mL)中の3-(tert-ブチルチオ)-2-(トリフルオロメチ
ル)アニリン(7.19g、31.3mmol)の溶液を、85℃で2時間撹拌した。室
温に冷却した後、N2流を溶液に16時間通した。揮発物を減圧下で除去し、得られた固
体を濾別し、ヘプタンで洗浄し、真空下で乾燥させて、3-アミノ-2-(トリフルオロ
メチル)ベンゼンチオール(7.19g、31.3mmol)を得た。MSm/z194
.0(M+H)+。
ナトリウム3-クロロ-2-シクロプロピルピリジン-4-チオレート

ジオキサン(7mL)中の2,3-ジクロロ-4-ヨードピリジン(1.0g、3.6
5mmol)、キサントホス(127mg、0.219mmol)およびPd(OAc)
2(41mg、0.183mmol)の混合物に、室温でおよびN2雰囲気下で、メチル
3-メルカプトプロパノエート(445μL、4.02mmol)を添加し、続いてDI
PEA(1.28mL、7.3mmol)を添加した。得られた溶液を100℃で4.5
時間撹拌した。室温に冷却した後、反応混合物をEtOAc(20mL)で希釈し、セラ
イトのパッドに通して濾過し、続いてEtOAcで洗浄(25mL)した。合わせた濾液
を減圧下で濃縮し、残留物をシリカクロマトグラフィー(10から50%の勾配のEtO
Ac/ヘプタン)により精製して、メチル3-((2,3-ジクロロピリジン-4-イル
)チオ)プロパノエート(965mg、5.38mmol)を得た。MSm/z266.
1(M+H)+。
トルエン:H2O(10:1;13mL)中のメチル3-((2,3-ジクロロピリジ
ン-4-イル)チオ)プロパノエート(800mg、3.19mmol)、n-BuPA
d2(86mg、0.240mmol)、Pd(OAc)2(36mg、0.160mm
ol)、Cs2CO3(3.12g、9.58mmol)およびシクロプロピルトリフル
オロホウ酸カリウム(709mg、4.79mmol)の混合物を、100℃で4.5時
間撹拌した。室温に冷却した後、反応混合物を、飽和NH4Cl水溶液を含有する分液漏
斗に注ぎ入れ、EtOAc(3×15mL)で抽出した。合わせた有機相をMgSO4で
脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー(10
から40%の勾配のEtOAc/ヘプタン)により精製して、メチル3-((3-クロロ
-2-シクロプロピルピリジン-4-イル)チオ)プロパノエート(380mg、1.3
98mmol)を得た。MSm/z272.1(M+H)+。
THF(5mL)中のメチル3-((3-クロロ-2-シクロプロピルピリジン-4-
イル)チオ)プロパノエート(380mg、1.398mmol)の溶液に、ナトリウム
エトキシド(EtOH中21重量%、0.548mL、1.468mmol)を室温でお
よびN2雰囲気下で添加した。室温で30分間激しく撹拌した後、揮発物を減圧下で除去
して、ナトリウム3-クロロ-2-シクロプロピルピリジン-4-チオレート(290m
g、1.398mmol)を得、これをさらに精製することなく使用した。MSm/z1
86.1(M+H)+。
6-アミノ-2,3-ジクロロピリジン-4-チオール
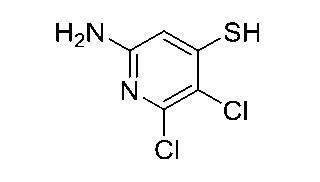
THF(60mL)中の5,6-ジクロロピリジン-2-アミン(2.445g、15
mmol)の0℃溶液に、LiHMDS(THF中1M、33.0mL、33.0mmo
l)を滴下添加し、反応混合物を0℃で10分間撹拌した。THF(20mL)中のBo
c2O(3.60g、16.5mmol)を添加し、得られた混合物をこの温度で15分
間撹拌した。反応混合物を室温に加温し、1N HCl水溶液を用いてpH4にした。水
層を分離し、EtOAc(2×20mL)で抽出した。合わせた有機相を飽和NaHCO
3水溶液で洗浄し、MgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物を
シリカクロマトグラフィー(0から40%の勾配のEtOAc/ヘプタン)により精製し
て、tert-ブチル(5,6-ジクロロピリジン-2-イル)カルバメート(3.12
g、11.86mmol)を得た。MSm/z207.8(M+H-tBu)+。
THF(20mL)中のジイソプロピルアミン(3.25mL、22.80mmol)
の-78℃溶液に、n-BuLi(ヘキサン中2.5M、9.12mL、22.80mm
ol)を滴下添加し、反応混合物を-78℃で1時間撹拌した。THF(20mL)中の
tert-ブチル(5,6-ジクロロピリジン-2-イル)カルバメート(3.0g、1
1.40mmol)を添加し、得られた混合物を-78℃で2時間撹拌した。THF(2
0mL)中のI2(3.04g、11.97mmol)を添加し、混合物を-78℃で3
0分間撹拌した。室温まで加温した後、反応混合物をH2Oで慎重に希釈し、EtOAc
(2×50mL)で抽出した。合わせた有機相を飽和Na2S2O3水溶液、ブラインで
洗浄し、MgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロ
マトグラフィー(0から40%の勾配のEtOAc/ヘプタン)により精製して、ter
t-ブチル(5,6-ジクロロ-4-ヨードピリジン-2-イル)カルバメート(3.3
3g、4.792mmol)を得た。MSm/z332.8(M+H-tBu)+。
ジオキサン(10mL)中のtert-ブチル(5,6-ジクロロ-4-ヨードピリジ
ン-2-イル)カルバメート(1.0g、2.57mmol)、キサントホス(89mg
、0.154mmol)およびPd(OAc)2(29mg、0.129mmol)の溶
液に、室温でおよびN2雰囲気下で、メチル3-メルカプトプロパノエート(313μL
、2.83mmol)を添加し、続いてDIPEA(0.9mL、5.14mmol)を
添加した。得られた溶液を100℃で2時間撹拌した。室温に冷却した後、反応混合物を
EtOAc(20mL)で希釈し、セライトのパッドに通して濾過し、続いてEtOAc
で洗浄(25mL)した。合わせた濾液を減圧下で濃縮し、残留物をシリカクロマトグラ
フィー(0から25%の勾配のEtOAc/ヘプタン)により精製して、メチル3-((
6-((tert-ブトキシカルボニル)アミノ)-2,3-ジクロロピリジン-4-イ
ル)チオ)プロパノエート(668mg、1.752mmol)を得た。MSm/z32
5.1(M+H-tBu)+。
DCM(10mL)中のメチル3-((6-((tert-ブトキシカルボニル)アミ
ノ)-2,3-ジクロロピリジン-4-イル)チオ)プロパノエート(668mg、1.
75mmol)およびTFA(1.35mL)の溶液を、室温で1時間撹拌した。この後
、揮発物を減圧下で除去して、6-アミノ-2,3-ジクロロピリジン-4-チオール(
342mg、1.75mmol)を得、これをさらに精製することなく次のステップにお
いて使用した。MSm/z194.6(M+H)+。
ナトリウム3-(トリフルオロメチル)ピリジン-4-チオレート

DMF(8mL)中の4-クロロ-3-(トリフルオロメチル)ピリジン(535mg
、2.95mmol)、炭酸カリウム(407mg、2.95mmol)およびメチル3
-メルカプトプロパノエート(0.343mL、3.09mmol)の溶液を、室温で1
時間撹拌した。反応混合物をEtOAc(60mL)で希釈し、H2O(3×60mL)
で洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮して、メチル3-((3-(トリ
フルオロメチル)ピリジン-4-イル)チオ)プロパノエート(710mg、2.68m
mol)を透明油状物として得た。MSm/z266.1(M+H)+。
THF(5.4mL)中のメチル3-((3-(トリフルオロメチル)ピリジン-4-
イル)チオ)プロパノエート(710mg、2.68mmol)の溶液に、ナトリウムエ
トキシド(EtOH中21重量%、1.01mL、2.94mmol)を室温でおよびN
2雰囲気下で添加した。室温で1時間激しく撹拌した後、追加のナトリウムエトキシド(
EtOH中21重量%、0.25mL、0.44mmol)を添加し、反応混合物を室温
で30分間撹拌した。揮発物を減圧下で除去し、残留物をDCM(3mL)中に懸濁させ
た。懸濁液を濾過し、減圧下で乾燥させて、ナトリウム3-(トリフルオロメチル)ピリ
ジン-4-チオレート(216mg、1.074mmol)を黄褐色固体として得た。M
Sm/z180.1(M+2H-Na)+。
ナトリウム3-クロロ-2-メチルピリジン-4-チオレート

DMF(25mL)中の3,4-ジクロロ-2-メチルピリジン(3.05g、18.
83mmol)、炭酸カリウム(2.73g、19.77mmol)およびメチル3-メ
ルカプトプロパノエート(2.19mL、19.8mmol)の溶液を、室温で4時間撹
拌した。反応混合物をEtOAc(125mL)で希釈し、H2O(3×100mL)で
洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮した。残留物をシリカゲルでのカラ
ムクロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)により精製して、
メチル3-((3-クロロ-2-メチルピリジン-4-イル)チオ)プロパノエート(1
.07g)を得た。MSm/z246.0(M+H)+。1H NMR (400 MHz, クロロホル
ム-d) δ ppm 8.27 (d, J=5.27 Hz, 1 H), 6.97 (d, J=5.27 Hz, 1 H), 3.71-3.82 (m, 3
H), 3.26 (t, J=7.53 Hz, 2 H), 2.78 (t, J=7.53 Hz, 2 H), 2.63 (s, 3 H).
THF(9mL)中のメチル3-((3-クロロ-2-メチルピリジン-4-イル)チ
オ)プロパノエート(1.07g、4.35mmol)の溶液に、ナトリウムエトキシド
(EtOH中21重量%、1.8mL、4.82mmol)を室温でおよびN2雰囲気下
で添加した。1時間激しく撹拌した後、揮発物を減圧下で除去し、残留物をDCM(20
mL)中に懸濁させた。沈殿物を濾別し、減圧下で乾燥させて、ナトリウム3-クロロ-
2-メチルピリジン-4-チオレートを白色固体としての白色粉末(850mg)として
得た。MSm/z160.0(M+H-Na)+。1H NMR (400 MHz, DMSO-d6) δ ppm 7
.36 (d, J=5.31 Hz, 1 H), 6.97 (d, J=5.31 Hz, 1 H), 2.30 (s, 3 H).
ナトリウム2-メトキシ-3-(トリフルオロメチル)ピリジン-4-チオレート

THF(20mL)中のジイソプロピルアミン(0.966mL、6.77mmol)
の-78℃溶液に、n-BuLi(ヘキサン中1.6M、4.23mL、6.77mmo
l)を滴下添加し、反応混合物を-78℃で5分間撹拌した。THF(10mL)中の2
-メトキシ-3-(トリフルオロメチル)ピリジン(1.2g、6.77mmol)の溶
液を添加し、得られた混合物を-78℃で2時間撹拌した。THF(5mL)中のI2(
1.72g、6.77mmol)を-78℃で添加し、得られた混合物を30分以内で室
温に加温し、この温度で30分間さらに撹拌した。揮発物を減圧下で除去し、残留物をE
t2O(200mL)に溶解した。有機層を飽和Na2S2O3水溶液(200mL)、
飽和NH4Cl水溶液(200mL)および飽和NaHCO3水溶液(200mL)で順
次洗浄し、MgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカク
ロマトグラフィー(0から25%の勾配のEtOAc/ヘプタン)により精製して、4-
ヨード-2-メトキシ-3-(トリフルオロメチル)ピリジン(540mg、1.354
mmol)を得た。MSm/z304.0(M+H)+。
ジオキサン(1.5mL)中の4-ヨード-2-メトキシ-3-(トリフルオロメチル
)ピリジン(540mg、1.354mmol)、キサントホス(63mg、0.108
mmol)およびPd(OAc)2(12mg、0.054mmol)の溶液に、室温で
およびN2雰囲気下で、メチル3-メルカプトプロパノエート(158μL、1.422
mmol)を添加し、続いてDIPEA(0.47mL、2.71mmol)を添加した
。得られた溶液を105℃で30分間撹拌した。室温に冷却した後、反応混合物をEtO
Ac(10mL)で希釈し、セライトのパッドに通して濾過し、続いてEtOAcで洗浄
(15mL)した。合わせた濾液を濃縮し、残留物をシリカクロマトグラフィー(0から
50%の勾配のEtOAc/ヘプタン)により精製して、メチル3-((2-メトキシ-
3-(トリフルオロメチル)ピリジン-4-イル)チオ)プロパノエート(344mg、
1.165mmol)を得た。MSm/z296.1(M+H)+。
THF(2.3mL)中のメチル3-((2-メトキシ-3-(トリフルオロメチル)
ピリジン-4-イル)チオ)プロパノエート(340mg、1.151mmol)の溶液
に、ナトリウムエトキシド(EtOH中21重量%、0.52mL、1.382mmol
)を室温でおよびN2雰囲気下で添加した。室温で30分間激しく撹拌した後、揮発物を
減圧下で除去し、残留物をDCM(10mL)中に懸濁させた。得られた懸濁液を濾過し
、減圧下で乾燥させて、3-クロロ-2-メチルピリジン-4-チオレート(850mg
、4.31mmol)を白色固体として得た。MSm/z210.0(M+H)+。
2-(トリフルオロメチル)ピリジン-3-チオール

窒素雰囲気下、ジオキサン(12mL)中の3-ブロモ-2-(トリフルオロメチル)
ピリジン(1.0g、4.42mmol)、キサントホス(256mg、0.442mm
ol)、Pd2(dba)3(203mg、0.221mmol)の溶液に、2-エチル
ヘキシル-3-メルカプトプロパノエート(1.1mL、4.87mmol)を室温で添
加し、続いてDIPEA(1.55mL、8.85mmol)を添加した。得られた混合
物にMW反応器内110℃で1時間照射を行った。室温に冷却した後、反応混合物をセラ
イトのパッドに通して濾過し、続いてEtOAc(25mL)で洗浄した。合わせた濾液
を減圧下で濃縮し、得られた残留物をシリカクロマトグラフィー(0から30%の勾配の
EtOAc/ヘプタン)により精製して、2-エチルヘキシル3-((2-(トリフルオ
ロメチル)ピリジン-3-イル)チオ)プロパノエート(1.41g、3.88mmol
)を得た。MSm/z364.0(M+H)+。
THF(8mL)中の2-エチルヘキシル3-((2-(トリフルオロメチル)ピリジ
ン-3-イル)チオ)プロパノエート(1.0g、2.75mmol)の溶液に、-78
℃でおよびN2雰囲気下で、カリウムtert-ブトキシド(THF中1M、8.25m
L、8.25mmol)を添加した。-78℃で20分間激しく撹拌した後、反応物をK
2CO3(H2O中2M、0.5mL)でクエンチし、揮発物を減圧下で除去した。残留
物を、K2CO3(H2O中2M、30mL)を含有する分液漏斗に注ぎ入れた。混合物
をEt2O(2×20mL)で抽出し、水相をpH4になるまで6N HClで酸性化し
、得られた濁った懸濁液をCHCl3/IPA(9/1;3×20mL)で抽出して、2
-(トリフルオロメチル)ピリジン-3-チオール(380mg、2.12mmol)を
得た。MSm/z180.0(M+H)+。
3-アミノ-2-クロロベンゼンチオール

DMF(650mL)中の2-メチルプロパン-2-チオール(137mL、1216
mmol)、2-クロロ-3-フルオロアニリン(63.2g、437mmol)および
炭酸セシウム(283g、868mmol)の懸濁液を、120℃で16時間撹拌した。
室温に冷却した後、反応混合物をEtOAc(500mL)で希釈し、H2O、ブライン
で洗浄し、Na2SO4で脱水し、濾過し、減圧下で濃縮して、3-(tert-ブチル
チオ)-2-クロロアニリン(111.2g、423mmol)を得た。MSm/z21
6.1(M+H)+。
3-(tert-ブチルチオ)-2-クロロアニリン(53g、246mmol)およ
び濃HCl(700mL)の懸濁液を、45℃で8時間および室温で16時間激しく撹拌
した。0℃に冷却した後、懸濁液を濾過し、固体を濃HCl(100mL)およびヘキサ
ン(3×100mL)で洗浄し、減圧下で乾燥させて、3-アミノ-2-クロロベンゼン
チオール塩酸塩(42g、214mmol)を得た。MSm/z159.6(M+H)+
。
4-フェニルピペリジン-4-アミン

MeOH中のN-(1-ベンジル-4-フェニルピペリジン-4-イル)アセトアミド
(400mg、1.3mmol)およびPd/C(10重量%、138mg)の懸濁液を
、水素雰囲気下で16時間激しく撹拌した。反応混合物をセライトのパッドに通して濾過
し、揮発物を減圧下で除去した。得られた残留物をEtOAcに溶解し、これを飽和Na
HCO3水溶液、ブラインで洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮して、
N-(4-フェニルピペリジン-4-イル)アセトアミドを得、これをさらに精製するこ
となく次のステップに持ち込んだ。
MeOH/ジオキサン(1/1、4mL)中のN-(4-フェニルピペリジン-4-イ
ル)アセトアミド(150mg、0.69mmol)および4N LiOH(2.1mL
、8.40mmol)の懸濁液を、100℃で16時間撹拌した。室温に冷却した後、揮
発物を減圧下で除去し、残った水相をEtOAc(3×5mL)で抽出した。合わせた有
機相をブラインで洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮して、4-フェニ
ルピペリジン-4-アミンを無色油状物として得、これをさらに精製することなく使用し
た。
tert-ブチル((4-(ピラジン-2-イル)ピペリジン-4-イル)メチル)カル
バメート

DMF(30mL)中の水素化ナトリウム(鉱油中60%、1.90g、47.7mm
ol)の懸濁液に、DMF(5mL)中の2-(ピラジン-2-イル)アセトニトリル(
1.90g、15.90mmol)を0℃で10分以内に滴下添加した。得られた混合物
を0℃で30分間撹拌した。DMF(5mL)中のN-ベンジル-2-クロロ-N-(2
-クロロエチル)エタンアミン(4.7g、17.5mmol)を0℃で添加し、得られ
た混合物を0℃で15分間および90℃で16時間撹拌した。室温に冷却した後、反応混
合物を飽和NaHCO3水溶液で希釈し、EtOAc(3×25mL)で抽出した。合わ
せた有機相をNa2SO4で脱水し、濾過し、減圧下で濃縮し、得られた残留物を、ヘキ
サンで摩砕することにより精製して、1-ベンジル-4-(ピラジン-2-イル)ピペリ
ジン-4-カルボニトリル(1.60g、5.76mmol)を得た。
NH3(MeOH中7N、50mL)中の1-ベンジル-4-(ピラジン-2-イル)
ピペリジン-4-カルボニトリル(1.50g、5.39mmol)の溶液に、ラネーニ
ッケル(水中50%、750mg)を室温で添加した。得られた懸濁液を、出発物質が消
費されるまで(約16時間)水素雰囲気(60psi)下室温で激しく撹拌した。反応混
合物をセライトのパッドに通して濾過し、続いてMeOH(50mL)で洗浄した。揮発
物を減圧下で除去して、(1-ベンジル-4-(ピラジン-2-イル)ピペリジン-4-
イル)メタンアミン(1.20g、4.25mmol)を得、これをさらに精製すること
なく次のステップにおいて使用した。MSm/z319(M+H)+。
DCM(50mL)中の(1-ベンジル-4-(ピラジン-2-イル)ピペリジン-4
-イル)メタンアミン(1.20g、4.25mmol)、Et3N(1.17mL、8
.51mmol)およびBoc2O(1.95mL、8.51mmol)の溶液を、室温
で2時間撹拌した。反応物をH2Oで希釈し、これをDCM(3×25mL)で抽出した
。合わせた有機相をブラインで洗浄し、Na2SO4で脱水し、濾過し、揮発物を減圧下
で除去した。得られた残留物をシリカクロマトグラフィー(0から100%の勾配のEt
OAc/ヘプタン)により精製して、tert-ブチル((1-ベンジル-4-(ピラジ
ン-2-イル)ピペリジン-4-イル)メチル)カルバメート(1.30g、3.40m
mol)を得た。MSm/z383(M+H)+。
MeOH(20mL)中のtert-ブチル((1-ベンジル-4-(ピラジン-2-
イル)ピペリジン-4-イル)メチル)カルバメート(1.50g、3.93mmol)
およびPd(OH)2(炭素上20%、600mg、水分50%)の懸濁液を、水素雰囲
気(50psi)下室温で3時間激しく撹拌した。反応混合物をセライトのパッドに通し
て濾過し、続いてMeOH(50mL)で洗浄した。揮発物を減圧下で除去して、ter
t-ブチル((4-(ピラジン-2-イル)ピペリジン-4-イル)メチル)カルバメー
ト(1.10g、3.76mmol)を得、これをさらに精製することなく使用した。M
Sm/z283(M+H)+。
のヘテロ芳香族アセトニトリルを使用して作製した。

tert-ブチル((4-イソブチルピペリジン-4-イル)メチル)カルバメート

LHMDSの溶液(THF中1M、16.45mL、16.45mmol)に、THF
(37.4mL)中の1-ベンジルピペリジン-4-カルボニトリル(1.50g、7.
49mmol)の溶液を-78℃で添加した。得られた黄色溶液を-78℃で1時間撹拌
した。1-ヨード-2-メチルプロパン(5.60mL、48.7mmol)を添加し、
反応混合物を室温まで加温し、撹拌を3日間続けた。飽和NH4Cl水溶液(約30mL
)を0℃で添加し、混合物をEtOAcで抽出した。有機相を水(50mL)およびブラ
イン(50mL)で洗浄した。各水層をEtOAcで抽出し、合わせた有機相をNa2S
O4で脱水し、濾過し、減圧下で濃縮して、粗製の1-ベンジル-4-イソブチルピペリ
ジン-4-カルボニトリル(2.54g)を黄色油状物として得、これをさらに精製する
ことなく直接使用した。MSm/z257.3(M+H)+。
MeOH(38.7mL)中の粗製の1-ベンジル-4-イソブチルピペリジン-4-
カルボニトリル(2.48g)、Boc2O(6.33g、29.0mmol)および塩
化ニッケル(II)水和物(1.15g、4.84mmol)の溶液を、室温で15分間
撹拌した。水素化ホウ素ナトリウム(2.56g、67.7mmol)を0℃で少量ずつ
添加し、撹拌を室温で18時間続けた。追加の水素化ホウ素ナトリウム(2.56g、6
7.7mmol)を0℃で添加し、得られた混合物を35℃で18時間撹拌した。室温に
冷却した後、揮発物を減圧下で除去し、得られた残留物をDCM(100mL)中に懸濁
させ、セライトのパッドに通して濾過した。濾液を減圧下で濃縮し、得られた残留物をシ
リカクロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)により精製して
、tert-ブチル((1-ベンジル-4-イソブチルピペリジン-4-イル)メチル)
カルバメート(482mg、1.34mmol)を無色油状物として得た。MSm/z3
61.4(M+H)+。
MeOH(6.7mL)中のtert-ブチル((1-ベンジル-4-イソブチルピペ
リジン-4-イル)メチル)カルバメート(482mg、1.34mmol)およびPd
/C(10重量%、142mg)の懸濁液を、水素雰囲気下で18時間激しく撹拌した。
混合物をセライトのパッドに通して濾過し、続いてMeOHで洗浄し、揮発物を減圧下で
除去して、tert-ブチル((4-イソブチルピペリジン-4-イル)メチル)カルバ
メート(338mg、1.25mmol)を得、これをさらに精製することなく直接使用
した。MSm/z271.3(M+H)+。
カンを使用して合成した。

ラセミtert-ブチルtrans-((3-ヒドロキシ-4-メチルピペリジン-4-
イル)メチル)カルバメート

THF(20mL)中の水素化リチウム(0.118g、14.8mmol)の溶液に
、アセトンシアノヒドリン(1.4mL、14.8mmol)を0℃で添加した。得られ
た反応混合物を室温で2時間撹拌した。揮発物を減圧下で除去して、白色固体を得た。T
HF(60mL)中のこの固体の溶液に、3-ベンジル-6-メチル-7-オキサ-3-
アザビシクロ[4.1.0]ヘプタン(2.0g、9.85mmol)を室温で滴下添加
した。溶液を14時間加熱還流した。室温に冷却した後、水(10mL)を添加し、得ら
れた混合物をEtOAc(3×100mL)で抽出した。合わせた有機相をブラインで洗
浄し、Na2SO4で脱水し、濾過し、減圧下で濃縮した。得られた残留物をシリカクロ
マトグラフィー(0から20%の勾配のEtOAc/ヘプタン)により精製して、ラセミ
trans-1-ベンジル-3-ヒドロキシ-4-メチルピペリジン-4-カルボニトリ
ル(0.70g、3.0mmol)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 7.36-7.
22 (m, 5 H), 5.25 (d, J=6.0 Hz, 1 H), 3.70-3.67 (m, 1 H), 3.49 (dd, J=13.2, 10.4
Hz, 2 H), 2.37 (m, 3 H), 1.88-1.74 (m, 2 H), 1.25 (s, 3 H).MSm/z231.2
(M+H)+。
アンモニア(EtOH中7N;80mL)中のラセミtrans-1-ベンジル-3-
ヒドロキシ-4-メチルピペリジン-4-カルボニトリル(1.3g、5.6mmol)
およびラネーニッケル(水中50%、600mg)の懸濁液を、水素雰囲気(バルーン)
下室温で6時間激しく撹拌した。混合物をN2下でセライトに通して濾過し、MeOHで
洗浄した。揮発物を減圧下で除去して、trans-4-(アミノメチル)-1-ベンジ
ル-4-メチルピペリジン-3-オール(1.6g、4.79mmol)を得、これをさ
らに精製することなく次のステップにおいて使用した。MSm/z235.2(M+H)
+。
CHCl3(70mL)中のtrans-4-(アミノメチル)-1-ベンジル-4-
メチルピペリジン-3-オール(1.6g、4.79mmol)、Boc2O(2.84
mL、12.4mmol)およびNaHCO3(0.935g、11.1mmol)の溶
液を、室温で14時間撹拌した。混合物をDCMで希釈し、氷水およびブラインで洗浄し
、Na2SO4で脱水し、濾過し、減圧下で濃縮した。得られた残留物をシリカクロマト
グラフィー(0から5%の勾配のMeOH/DCM)により精製して、ラセミtert-
ブチルtrans-(1-ベンジル-3-ヒドロキシ-4-メチルピペリジン-4-イル
)メチル)カルバメート(1.1g、3.3mmol)を得た。MSm/z335.3(
M+H)+。
MeOH(60mL)中のラセミtert-ブチルtrans-((1-ベンジル-3
-ヒドロキシ-4-メチルピペリジン-4-イル)メチル)カルバメート(1.1g、3
.3mmol)およびPd(OH)2(木炭上20%;0.250g)の懸濁液を、水素
雰囲気(バルーン)下室温で6時間激しく撹拌した。得られた混合物をセライトに通して
濾過し、MeOHで洗浄し、減圧下で濃縮した。残留物をヘキサン(10mL)およびジ
エチルエーテル(2mL)で摩砕して、ラセミtert-ブチルtrans-((3-ヒ
ドロキシ-4-メチルピペリジン-4-イル)メチル)カルバメート(0.70g、2.
87mmol)を白色粉末として得た。1H NMR (400 MHz, メタノール-d4) δ ppm 3.42
(dd, J=9.9, 4.4 Hz, 1 H), 3.12 (d, J=13.9 Hz, 1 H), 2.94-2.84 (m, 2 H), 2.82-2.6
8 (m, 2 H), 2.62 (dd, J=12.5, 10.0 Hz, 1 H), 1.44 (s, 9 H), 1.41-1.30 (m, 2 H),
0.91 (s, 3 H).MSm/z245.1(M+H)+。
ラセミtert-ブチルcis-((3-ヒドロキシ-4-メチルピペリジン-4-イル
)メチル)カルバメート

THF(30mL)中のラセミtrans-1-ベンジル-3-ヒドロキシ-4-メチ
ルピペリジン-4-カルボニトリル(2.0g、8.70mmol)、トリフェニルホス
フィン(3.41g、13.0mmol)およびDIAD(2.63g、13.0mmo
l)の溶液を、0℃で10分間撹拌した。4-ニトロ安息香酸(2.18g、13.0m
mol)を少量ずつ添加し、得られた混合物を室温で16時間撹拌した。混合物を水で希
釈し、EtOAcで抽出した。合わせた有機相をブラインで洗浄し、Na2SO4で脱水
し、濾過し、減圧下で濃縮した。得られた残留物をMeOHで摩砕して、ラセミcis-
1-ベンジル-4-シアノ-4-メチルピペリジン-3-イル4-ニトロベンゾエート(
1.5g、3.96mmol)を得、これをさらに精製することなく使用した。MSm/
z380(M+H)+。
MeOH(20mL)中のラセミcis-1-ベンジル-4-シアノ-4-メチルピペ
リジン-3-イル4-ニトロベンゾエート(1.5g、3.96mmol)および炭酸カ
リウム(1.07g、7.92mmol)の溶液を、0℃で10分間および室温で1時間
激しく撹拌した。揮発物を減圧下で除去した。得られた残留物を水で希釈し、EtOAc
(3×)で抽出した。合わせた有機相をブラインで洗浄し、Na2SO4で脱水し、濾過
し、減圧下で濃縮した。得られた残留物をシリカクロマトグラフィー(0から15%の勾
配のEtOAc/ヘプタン)により精製して、ラセミcis-1-ベンジル-3-ヒドロ
キシ-4-メチルピペリジン-4-カルボニトリル(0.8g、3.5mmol)を得た
。1H NMR (400 MHz, CDCl3) δ ppm 7.36-7.26 (m, 5 H), 3.99 (d, J=12.4 Hz, 1 H), 3
.67 (d, J=12.8, 1 H), 3.60-3.51 (m, 2 H), 3.11-3.07 (m, 2 H), 2.76-2.69 (m, 2 H)
, 2.24 (dd, J=12.8, 6.0 Hz, 1 H), 1.87-1.80 (m, 1 H), 1.54 (s, 3 H).MSm/z2
31(M+H)+。
アンモニア(EtOH中7N;20mL)中のcis-1-ベンジル-3-ヒドロキシ
-4-メチルピペリジン-4-カルボニトリル(800mg、3.5mmol)およびラ
ネーニッケル(水中50%、700mg)の懸濁液を、水素雰囲気(バルーン)下室温で
16時間激しく撹拌した。混合物をN2雰囲気下でセライトに通して濾過し、MeOHで
すすいだ。揮発物を減圧下で除去して、ラセミcis-4-(アミノメチル)-1-ベン
ジル-4-メチルピペリジン-3-オール(700mg、3.0mmol)を得、これを
さらに精製することなく次のステップにおいて使用した。MSm/z235.2(M+H
)+。
DCM(10mL)中のcis-4-(アミノメチル)-1-ベンジル-4-メチルピ
ペリジン-3-オール(700mg、3.0mmol)、Boc2O(1.1mL、2.
99mmol)およびEt3N(860μL、5.98mmol)の溶液を、室温で2時
間撹拌した。混合物をDCMで希釈し、氷水およびブラインで洗浄し、Na2SO4で脱
水し、濾過し、減圧下で濃縮した。得られた残留物をシリカクロマトグラフィー(0から
50%の勾配のEtOAc/ヘプタン)により精製して、ラセミtert-ブチルcis
-(1-ベンジル-3-ヒドロキシ-4-メチルピペリジン-4-イル)メチル)カルバ
メート(700mg、2.10mmol)を得、これをさらに精製することなく次のステ
ップにおいて使用した。MSm/z335(M+H)+。
MeOH(20mL)中のラセミtert-ブチルcis-(1-ベンジル-3-ヒド
ロキシ-4-メチルピペリジン-4-イル)メチル)カルバメート(700mg、2.1
mmol)およびPd(木炭上10%;300mg)の懸濁液を、水素雰囲気(バルーン
)下室温で5時間激しく撹拌した。得られた混合物をセライトに通して濾過し、MeOH
で洗浄し、減圧下で濃縮した。得られた残留物をシリカクロマトグラフィー(0から10
%の勾配のMeOH/DCM)により精製して、ラセミtert-ブチルcis-((3
-ヒドロキシ-4-メチルピペリジン-4-イル)メチル)カルバメート(200mg、
0.8mmol)を白色粉末として得た。1H NMR (400 MHz, メタノール-d4) δ ppm 3.7
3-3.67 (m, 1 H), 3.59 (dd, J=11.1, 7.7 Hz, 1 H), 3.15-2.99 (m, 4 H), 1.90 (m, 1
H), 1.62 (m, 1 H), 1.47 (m, 1 H), 1.44 (s, 9 H), 0.96 (s, 3 H).MSm/z245
(M+H)+。
ラセミtert-ブチル1-(1,1-ジメチルエチルスルフィンアミノ(dimethylethy
lsulfinamino))-2,2-ジフルオロ-8-アザスピロ[4.5]デカン-8-カルボ
キシレート

NHMDSの溶液(THF中1M、8.68mL、8.68mmol)に、THF(5
mL)中のtert-ブチル1-オキソ-8-アザスピロ[4.5]デカン-8-カルボ
キシレート(2.0g、7.89mmol)の溶液を-78℃で添加した。この温度で3
0分間撹拌した後、THF(10mL)中のN-フルオロベンゼンスルホンアミド(2.
49g、7.89mmol)の溶液を添加した。-78℃で3時間撹拌した後、混合物を
飽和NaHCO3水溶液(100mL)で希釈し、DCM(3×100mL)で抽出した
。合わせた有機相をブラインで洗浄し、Na2SO4で脱水し、濾過し、減圧下で濃縮し
た。得られた残留物をシリカクロマトグラフィー(0から25%の勾配のEtOAc/ヘ
プタン)により精製して、ラセミtert-ブチル2-フルオロ-1-オキソ-8-アザ
スピロ[4.5]デカン-8-カルボキシレート(351mg、1.29mmol)、M
Sm/z272.1(M+H)+、および出発物質と共溶出したtert-ブチル2,2
-ジフルオロ-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレートを
得た。合わせたジフルオロケトン含有画分をシリカクロマトグラフィー(0から5%の勾
配のMeOH/DCM)により精製して、tert-ブチル2,2-ジフルオロ-1-オ
キソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(573mg、1.98
mmol)を得た。MSm/z290.1(M+H)+。
THF(4mL)中のtert-ブチル2,2-ジフルオロ-1-オキソ-8-アザス
ピロ[4.5]デカン-8-カルボキシレート(220mg、0.76mmol)、ラセ
ミ2-メチルプロパン-2-スルフィンアミド(184mg、1.52mmol)および
チタン(IV)エトキシド(0.640mL、3.0mmol)の溶液を、90℃で30
分間撹拌した。0℃に冷却した後、水素化ホウ素リチウム(33mg、1.5mmol)
を一度に添加した。30分間撹拌した後、反応混合物をMeOHの添加によりクエンチし
た。揮発物を減圧下で除去した。得られた残留物をブラインで希釈し、EtOAc(4×
10mL)で抽出した。合わせた有機相をNa2SO4で脱水し、濾過し、減圧下で濃縮
した。得られた残留物をシリカクロマトグラフィー(10から50%の勾配のEtOAc
/ヘプタン)により精製して、ラセミtert-ブチル1-(1,1-ジメチルエチルス
ルフィンアミノ)-2,2-ジフルオロ-8-アザスピロ[4.5]デカン-8-カルボ
キシレートを白色粉末(190mg、0.48mmol)として得た。MSm/z395
.2(M+H)+。
tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンアミノ)-2-フル
オロ-8-アザスピロ[4.5]デカン-8-カルボキシレート

ザスピロ[4.5]デカン-8-カルボキシレート(78mg、0.28mmol)、チ
タン(IV)エトキシド(235μL、1.1mmol)および(R)-2-メチルプロ
パン-2-スルフィンアミド(68mg、0.56mmol)の溶液を、90℃で1時間
撹拌した。0℃に冷却した後、水素化ホウ素リチウム(12mg、0.56mmol)を
一度に添加した。30分間撹拌した後、反応混合物をMeOHの添加によりクエンチした
。揮発物を減圧下で除去した。得られた残留物をブラインで希釈し、EtOAc(4×1
0mL)で抽出し、合わせた有機相をNa2SO4で脱水し、濾過し、減圧下で濃縮した
。得られた残留物をシリカクロマトグラフィー(0から50%の勾配のEtOAc/ヘプ
タン)により精製して、tert-ブチル1-((R)-1,1-ジメチルエチルスルフ
ィンアミノ)-2-フルオロ-8-アザスピロ[4.5]デカン-8-カルボキシレート
(異性体混合物、64mg、0.17mmol)を得た。MSm/z377.3(M+H
)+。
tert-ブチル1-オキソ-8-アザスピロ[4.5]デカ-2-エン-8-カルボキ
シレート

DMF(328mL)中のtert-ブチル4-ホルミルピペリジン-1-カルボキシ
レート(35.0g、164mmol)、リチウムtert-ブトキシド(15.77g
、197mmol)およびアリルブロミド(11.54mL、189mmol)の混合物
を、0℃で1時間撹拌した。混合物を、飽和NH4Cl水溶液/H2O(1/1、500
mL)を含有する分液漏斗に注ぎ入れ、これをEt2O(5×50mL)で抽出した。合
わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留
物をシリカクロマトグラフィー(0から25%の勾配のEtOAc/ヘプタン)により精
製して、tert-ブチル4-アリル-4-ホルミルピペリジン-1-カルボキシレート
(24g、95mmol)を無色油状物として得た。1H NMR (400 MHz, クロロホルム-d)
δ ppm 9.52 (s, 1 H), 5.53-5.76 (m, 1 H), 4.96-5.19 (m, 2 H), 3.80 (br. s, 2 H)
, 2.97 (t, J=11.49 Hz, 2 H), 2.26 (d, J=7.33 Hz, 2 H), 1.95 (dt, J=13.71, 3.13 H
z, 2 H), 1.38-1.58 (m, 11 H).
N2雰囲気下、THF(300mL)中のtert-ブチル4-アリル-4-ホルミル
ピペリジン-1-カルボキシレート(24g、95mmol)の溶液に、臭化ビニルマグ
ネシウム(THF中1M、118mL、118mmol)を-78℃で添加した。得られ
た混合物を1時間以内で室温まで加温した。混合物を、飽和NH4Cl水溶液(250m
L)を含有する分液漏斗に注ぎ入れ、これをEtOAc(4×50mL)で抽出した。合
わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去して、tert-ブ
チル4-アリル-4-(1-ヒドロキシアリル)ピペリジン-1-カルボキシレート(2
6.7g、95mmol)を無色油状物として得、これをさらに精製することなく次の反
応において使用した。1H NMR (400 MHz, クロロホルム-d) δ ppm 9.52 (s, 1 H), 5.56-
5.75 (m, 1 H), 5.05-5.18 (m, 2 H), 3.80 (br. s., 2 H), 2.97 (t, J=11.49 Hz, 2 H)
, 2.26 (d, J=7.33 Hz, 2 H), 1.96 (dt, J=13.83, 3.06 Hz, 2 H), 1.49-1.60 (m, 2 H)
, 1.41-1.49 (m, 9 H).
DCM(380mL)中のtert-ブチル4-アリル-4-(1-ヒドロキシアリル
)ピペリジン-1-カルボキシレート(26.7g、95mmol)およびデス-マーチ
ンペルヨージナン(44.3g、105mmol)の混合物を、室温で1時間撹拌した。
混合物を、NaHCO3/Na2SO3飽和水溶液(1/1、300mL)を含有する分
液漏斗に注ぎ入れ、これをDCM(4×50mL)で抽出した。合わせた有機相をMgS
O4で脱水し、濾過し、揮発物を減圧下で除去して、白色固体を得た。この固体をヘプタ
ン(250mL)中に懸濁させ、5分間超音波処理した。白色懸濁液をセライトのパッド
に通して濾過し、揮発物を減圧下で除去して、tert-ブチル4-アクリロイル-4-
アリルピペリジン-1-カルボキシレート(26.5g、95mmol)を黄色油状物と
して得、これをさらに精製することなく次の反応において使用した。1H NMR (400 MHz,
クロロホルム-d) δ ppm 6.81 (dd, J=16.93, 10.36 Hz, 1 H), 6.40 (dd, J=16.80, 1.8
9 Hz, 1 H), 5.71 (dd, J=10.36, 2.02 Hz, 1 H), 5.46-5.66 (m, 1 H), 4.91-5.14 (m,
2 H), 3.78 (br. s., 2 H), 2.96 (br. s, 2 H), 2.25-2.39 (m, 2 H), 1.97-2.15 (m, 2
H), 1.37-1.57 (m, 11 H).
トルエン(脱気、850mL)中のtert-ブチル4-アクリロイル-4-アリルピ
ペリジン-1-カルボキシレート(26.5g、95mmol)の溶液に、トルエン(脱
気、100mL)中のGrubbs II触媒(2.02g、2.38mmol)を添加
した。得られた混合物を85℃で45分間撹拌した。溶媒を減圧下で除去し、得られた残
留物をシリカクロマトグラフィー(0から40%の勾配のEtOAc/ヘプタン)により
精製して、tert-ブチル1-オキソ-8-アザスピロ[4.5]デカ-2-エン-8
-カルボキシレート(20.76g、83mmol)を茶色固体として得た。トルエン(
540mL)中のこの化合物およびDDQ(565mg、2.49mmol)の溶液を、
室温で15分間撹拌した。得られた明赤色溶液をセライトのパッドに通して濾過した。木
炭(200g)を濾液に添加し、得られた懸濁液を室温で2時間撹拌した。混合物をセラ
イトのパッドに通して濾過し、濾液を減圧下で濃縮した。得られた残留物をシリカクロマ
トグラフィー(0から40%の勾配のEtOAc/ヘプタン)により精製して、tert
-ブチル1-オキソ-8-アザスピロ[4.5]デカ-2-エン-8-カルボキシレート
(15.6g、62.3mmol)を白色固体として得た。1H NMR (400 MHz, クロロホ
ルム-d) δ ppm 7.63-7.74 (m, 1 H), 6.20 (dt, J=5.81, 2.15 Hz, 1 H), 3.99-4.25 (m
, 2 H), 2.92 (t, J=11.62 Hz, 2 H), 2.63 (s, 2 H), 1.72-1.86 (m, 2 H), 1.49 (s, 9
H), 1.29 (d, J=12.88 Hz, 2 H).
(1R,3R)-ベンジル1-((R)-1,1-ジメチルエチルスルフィンアミノ)-
3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート

N2雰囲気下、Et2O(100mL)中のtert-ブチル1-オキソ-8-アザス
ピロ[4.5]デカ-2-エン-8-カルボキシレート(4.2g、16.71mmol
)およびCuI(6.37g、33.4mmol)の懸濁液に、MeLi(THF中1.
6M、31.3mL、50.1mmol)を0℃で添加した。0℃で90分間撹拌した後
、混合物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ、これをEtOAc(
3×15mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減
圧下で除去した。得られた残留物をシリカクロマトグラフィー(0から50%の勾配のE
tOAc/ヘプタン)により精製して、tert-ブチル3-メチル-1-オキソ-8-
アザスピロ[4.5]デカ-2-エン-8-カルボキシレート(4.23g、15.82
mmol)を無色油状物として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 3.89-4
.00 (m, 1 H), 3.83 (d, J=13.39 Hz, 1 H), 3.11 (ddd, J=13.64, 10.36, 3.28 Hz, 1 H
), 2.99 (ddd, J=13.58, 10.42, 3.54 Hz, 1 H), 2.47-2.59 (m, 1 H), 2.19-2.36 (m, 2
H), 1.74-1.97 (m, 2 H), 1.50-1.65 (m, 2 H), 1.48 (s, 9 H), 1.33-1.44 (m, 2 H),
1.17 (d, J=6.32 Hz, 3 H).
DCM(80mL)中のtert-ブチル3-メチル-1-オキソ-8-アザスピロ[
4.5]デカ-2-エン-8-カルボキシレート(4.23g、15.82mmol)お
よびTFA(17mL)の混合物を、室温で30分間撹拌した。揮発物を減圧下で除去し
た。得られた残留物、DIPEA(13.82mL、79mmol)およびクロロギ酸ベ
ンジル(3.39mL、23.73mmol)の混合物を、室温で16時間撹拌した。混
合物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ、これをDCM(3×25
mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除
去した。得られた残留物をシリカクロマトグラフィー(0から40%の勾配のEtOAc
/ヘプタン)により精製して、ベンジル3-メチル-1-オキソ-8-アザスピロ[4.
5]デカン-8-カルボキシレート(4.58g、15.20mmol)を薄黄色油状物
として得た。MSm/z302.2(M+H)+。
ベンジル3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシ
レート(4.58g、15.20mmol)を、キラルSFCにより、以下の通り、すな
わち:カラム:IA 21×250mm、流速:70g/分、移動相:CO2中45%(
9/1 EtOH/MeCN)、検出:220nm UVでさらに精製して、(R)-ベ
ンジル3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレー
ト(2.02g、6.70mmol)、TR:2.0分;および(S)-ベンジル3-メ
チル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(2.11
g、7.0mmol)、TR:3.6分を得た。
THF(67mL)中の(R)-ベンジル3-メチル-1-オキソ-8-アザスピロ[
4.5]デカン-8-カルボキシレート(2.02g、6.70mmol)、チタン(I
V)エトキシド(5.62mL、26.8mmol)および(R)-2-メチルプロパン
-2-スルフィンアミド(1.625g、13.4mmol)の溶液を、65℃で16時
間撹拌した。混合物を-78℃に冷却し、MeOH(12mL)、続いて水素化ホウ素リ
チウム(0.438g、20.11mmol)を添加した。得られた混合物を-78℃か
ら室温で16時間撹拌した。飽和NH4Cl水溶液をゆっくりと添加して過剰のホウ水素
化物をクエンチし、続いてEtOAc(100mL)を添加した。得られた混合物を15
分間激しく撹拌し、次いでセライトのパッドに通して濾過した。揮発物を減圧下で除去し
、得られた残留物をシリカクロマトグラフィー(5から90%の勾配のEtOAc/ヘプ
タン)により精製して、(1R,3R)-ベンジル1-((R)-1,1-ジメチルエチ
ルスルフィンアミノ)-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシ
レート(1.94g、4.77mmol)を白色固体として得た。MSm/z407.3
(M+H)+。
-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレートは、上記手順ま
たは上記手順を変更したものを用い、(S)-ベンジル3-メチル-1-オキソ-8-ア
ザスピロ[4.5]デカン-8-カルボキシレートを出発物質として使用して合成した。
(1R,3R)-tert-ブチル3-((tert-ブチルジメチルシリル)オキシ)
-1-((R)-1,1-ジメチルエチルスルフィンアミノ)-8-アザスピロ[4.5
]デカン-8-カルボキシレート)

THF(60mL)中のCuCl(142mg、1.432mmol)、(S)-To
lBINAP(972mg、1.432mmol)およびナトリウムtert-ブトキシ
ド(138mg、1.432mmol)の混合物を、室温で30分間撹拌した。THF(
20mL)中のビス(ピナコラト)ジボラン(13.34g、52.5mmol)を添加
し、得られた混合物を室温で10分間撹拌した。THF(50mL)中のtert-ブチ
ル1-オキソ-8-アザスピロ[4.5]デカ-2-エン-8-カルボキシレート(12
.0g、47.7mmol)を、続いてMeOH(3.9mL、95mmol)を添加し
た。得られた混合物を室温で16時間撹拌した。H2O(150mL)、続いて過ホウ酸
ナトリウム(36.7g、239mmol)を添加し、得られた混合物を室温で1時間激
しく撹拌した。得られた緑色懸濁液をセライトのパッドに通して濾過し、飽和NaHCO
3水溶液/飽和Na2SO3水溶液(1/1、300mL)を含有する分液漏斗に注ぎ入
れ、EtOAc(4×40mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾
過し、揮発物を減圧下で除去して、粗製の(R)-tert-ブチル3-ヒドロキシ-1
-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレートを得た。この混合物
のエナンチオマーの定量は、90%eeを示す(Rt(S):1.59分、Rt(R):
1.80分;キラルSFC;カラム:IA 4.6×100mm、流速:70g/分、移
動相:CO2中5~55%MeOH、検出:220nm UV)。DMF(120mL)
中の粗製の(R)-tert-ブチル3-ヒドロキシ-1-オキソ-8-アザスピロ[4
.5]デカン-8-カルボキシレート(47.7mmol)、イミダゾール(4.87g
、71.6mmol)およびTBSCl(8.99g、59.6mmol)の混合物を、
室温で16時間撹拌した。反応混合物を、飽和NH4Cl水溶液/H2O(1/1、25
0mL)を含有する分液漏斗に注ぎ入れ、これをEt2O(5×50mL)で抽出した。
合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残
留物をシリカクロマトグラフィー(0から30%の勾配のEtOAc/ヘプタン)により
精製して、(R)-tert-ブチル3-((tert-ブチルジメチルシリル)オキシ
)-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(13.11
5g、34.2mmol)を無色油状物として得、これは静置すると固化した。
THF(100mL)中の(R)-tert-ブチル3-((tert-ブチルジメチ
ルシリル)オキシ)-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレ
ート(8.0g、20.86mmol)、チタン(IV)エトキシド(17.49mL、
83.0mmol)および(R)-2-メチルプロパン-2-スルフィンアミド(5.0
6g、41.7mmol)の溶液を、65℃で16時間撹拌した。-78℃に冷却した後
、MeOH(15mL)、続いて水素化ホウ素リチウム(1.363g、62.6mmo
l)を添加した。得られた混合物を-78℃で16時間撹拌した。飽和NH4Cl水溶液
をゆっくりと添加して過剰のホウ水素化物をクエンチし、続いてEtOAc(100mL
)を添加した。得られた混合物を15分間激しく撹拌し、次いでセライトのパッドに通し
て濾過した。揮発物を減圧下で除去し、得られた残留物をシリカクロマトグラフィー(0
から50%の勾配のEtOAc/ヘプタン)により精製して、(1R,3R)-tert
-ブチル3-((tert-ブチルジメチルシリル)オキシ)-1-((R)-1,1-
ジメチルエチルスルフィンアミノ)-8-アザスピロ[4.5]デカン-8-カルボキシ
レート(5.3g、10.84mmol)を白色固体として得た。MSm/z489.3
(M+H)+および389.3(M+H-Boc)+。
(1R,3R)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート

ジメチルシリル)オキシ)-1-((R)-1,1-ジメチルエチルスルフィンアミノ)
-8-アザスピロ[4.5]デカン-8-カルボキシレート(3.84g、7.86mm
ol)およびTBAF(THF中1M;8.64mL、8.64mmol)の混合物を、
室温で30分間撹拌した。揮発物を減圧下で除去し、得られた残留物をシリカクロマトグ
ラフィー(0から10%の勾配のMeOH/DCM)により精製して、(1R、3R)-
tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-ヒド
ロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート(2.94g、7.8
6mmol)を得た。MSm/z375.3(M+H)+。
(1R,3S)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート

THF(80mL)中の(1R,3R)-tert-ブチル1-((R)-1,1-ジ
メチルエチルスルフィンアミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-
8-カルボキシレート(3.0g、8.01mmol)、トリフェニルホスフィン(4.
2g、16.02mmol)およびイソキノリン-1-カルボン酸(4.16g、24.
03mmol)の溶液に、DIAD(3.1mL、16.02mmol)を添加した。得
られた混合物を室温で1時間撹拌した。反応物をEtOAc(50mL)で希釈し、セラ
イトのパッドに通して濾過し、飽和NaHCO3水溶液を含有する分液漏斗に注ぎ入れ、
EtOAc(3×25mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し
、揮発物を減圧下で除去した。得られた残留物をシリカクロマトグラフィー(0から4%
の勾配のMeOH/DCM)により精製して、(2S,4R)-8-(tert-ブトキ
シカルボニル)-4-((R)-1,1-ジメチルエチルスルフィンアミノ)-8-アザ
スピロ[4.5]デカン-2-イルイソキノリン-1-カルボキシレート(3.65g、
6.89mmol)を橙色固体として得た。MSm/z530.3(M+H)+。
THF/H2O(1/1、70mL)中の(2S,4R)-8-(tert-ブトキシ
カルボニル)-4-((R)-1,1-ジメチルエチルスルフィンアミノ)-8-アザス
ピロ[4.5]デカン-2-イルイソキノリン-1-カルボキシレート(3.65g、6
.89mmol)および水酸化リチウム(2.95g、68.9mmol)の混合物を、
室温で2時間撹拌した。混合物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ
、これをEtOAc(3×15mL)で抽出した。合わせた有機相をMgSO4で脱水し
、濾過し、揮発物を減圧下で除去した。得られた残留物をシリカクロマトグラフィー(0
から10%の勾配のMeOH/DCM)により精製して、(1R,3S)-tert-ブ
チル1-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-ヒドロキシ-8-
アザスピロ[4.5]デカン-8-カルボキシレート(2.35g、6.27mmol)
を白色固体として得た。MSm/z275.2(M+H-Boc)+。
(1R,3S)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-メトキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート

チルエチルスルフィンアミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8
-カルボキシレート(500mg、1.335mmol)、酸化銀(I)(340mg、
1.468mmol)およびヨードメタン(0.25mL、4.0mmol)の混合物を
、室温で24時間および45℃で24時間撹拌した(光から保護した)。室温に冷却した
後、混合物をセライトのパッドに通して濾過し、揮発物を減圧下で除去し、得られた残留
物をシリカクロマトグラフィー(0から5%の勾配のMeOH/DCM)により精製して
、(1R,3S)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィン
アミノ)-3-メトキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート(2
48mg、0.638mmol)を得た。MSm/z289.2(M+H-Boc)+。
アミノ)-3-メトキシ-8-アザスピロ[4.5]デカン-8-カルボキシレートは、
上記手順または上記手順を変更したものを用い、(1R,3R)-tert-ブチル1-
((R)-1,1-ジメチルエチルスルフィンアミノ)-3-ヒドロキシ-8-アザスピ
ロ[4.5]デカン-8-カルボキシレートを出発物質として使用して合成した。
ラセミtert-ブチル1-((tert-ブトキシカルボニル)アミノ)-3,3-ジ
フルオロ-8-アザスピロ[4.5]デカン-8-カルボキシレート

MeOH(4mL)中のtert-ブチル3-((tert-ブチルジメチルシリル)
オキシ)-1-(1,1-ジメチルエチルスルフィンアミノ)-8-アザスピロ[4.5
]デカン-8-カルボキシレート(365mg、0.746mmol)およびHCl(ジ
オキサン中4M、1.86mL、7.46mmol)の混合物を、40℃で1時間撹拌し
た。室温に冷却した後、揮発物を減圧下で除去して、粗製の4-アミノ-8-アザスピロ
[4.5]デカン-2-オールを白色固体として得た。MSm/z171.1(M+H)
+。
THF(15mL)中の粗製の4-アミノ-8-アザスピロ[4.5]デカン-2-オ
ール、DIPEA(2.6mL、14.92mmol)およびBoc2O(407mg、
1.865mmol)の混合物を、室温で16時間撹拌した。混合物を、飽和NH4Cl
水溶液を含有する分液漏斗に注ぎ入れ、これをEt2O(5×10mL)で抽出した。合
わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留
物をシリカクロマトグラフィー(10から80%の勾配のEtOAc/ヘプタン)により
精製して、tert-ブチル1-((tert-ブトキシカルボニル)アミノ)-3-ヒ
ドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート(275mg、0.
742mmol)を得た。MSm/z271.3(M+H-Boc)+。
DCM(7.5mL)中のtert-ブチル1-((tert-ブトキシカルボニル)
アミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレート(
275mg、0.742mmol)およびデス-マーチンペルヨージナン(472mg、
1.113mmol)の混合物を、0℃で2時間撹拌した。混合物を、飽和NaHCO3
水溶液を含有する分液漏斗に注ぎ入れ、これをDCM(3×10mL)で抽出した。合わ
せた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留物
をシリカクロマトグラフィー(5から75%の勾配のEtOAc/ヘプタン)により精製
して、tert-ブチル1-((tert-ブトキシカルボニル)アミノ)-3-オキソ
-8-アザスピロ[4.5]デカン-8-カルボキシレート(135mg、0.366m
mol)を得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.57 (d, J=9.09 Hz, 1 H)
, 4.16 (d, J=8.08 Hz, 1 H), 3.89-4.08 (m, 2 H), 2.77-2.93 (m, 2 H), 2.71 (dd, J=
18.95, 8.08 Hz, 1 H), 2.50 (d, J=18.19 Hz, 1 H), 2.07-2.24 (m, 2 H), 1.76 (td, J
=12.82, 4.67 Hz, 1 H), 1.58-1.70 (m, 1 H), 1.42-1.53 (m, 18 H), 1.25-1.38 (m, 1
H).
DCM(1mL)中のtert-ブチル1-((tert-ブトキシカルボニル)アミ
ノ)-3-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(95mg
、0.258mmol)およびDeoxoFluor(190μL、1.031mmol
)の混合物を、50℃で48時間撹拌した。混合物を、飽和NaHCO3水溶液/氷を含
有する分液漏斗に注ぎ入れ、これをEtOAc(3×5mL)で抽出した。合わせた有機
相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留物をシリカ
クロマトグラフィー(0から30%の勾配のEtOAc/ヘプタン)により精製して、t
ert-ブチル1-((tert-ブトキシカルボニル)アミノ)-3,3-ジフルオロ
-8-アザスピロ[4.5]デカン-8-カルボキシレート(52mg、0.133mm
ol)を得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.55 (d, J=9.35 Hz, 1 H),
3.78-4.02 (m, 3 H), 2.64-2.86 (m, 2 H), 2.38-2.59 (m, 1 H), 2.10-2.32 (m, 1 H),
1.79-2.10 (m, 2 H), 1.58 (qd, J=12.72, 3.79 Hz, 1 H), 1.27-1.52 (m, 21 H).
-ジフルオロ-8-アザスピロ[4.5]デカン-8-カルボキシレートは、上記手順ま
たは上記手順を変更したものを用い、キラル的に純粋な(1R,3R)-tert-ブチ
ル3-((tert-ブチルジメチルシリル)オキシ)-1-((R)-1,1-ジメチ
ルエチルスルフィンアミノ)-8-アザスピロ[4.5]デカン-8-カルボキシレート
を出発物質として使用して合成した。
(1R,3S)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-フルオロ-8-アザスピロ[4.5]デカン-8-カルボキシレート

ジメチルエチルスルフィンアミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン
-8-カルボキシレート(400mg、1.068mmol)およびDAST(DCM中
1M、1.87mL、1.87mmol)の混合物を、0℃で90分間撹拌した。反応混
合物を飽和NaHCO3水溶液(5mL)の添加によりクエンチした。0℃で10分間撹
拌した後、相を分離し、水層をDCM(2×5mL)で抽出した。合わせた有機相をMg
SO4で脱水し、濾過し、揮発物を減圧下で除去して、(1R,3S)-tert-ブチ
ル1-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-フルオロ-8-アザ
スピロ[4.5]デカン-8-カルボキシレートを得、これをさらに精製することなく次
のステップにおいて使用した。MSm/z277.2(M+H-Boc)+。
(1R,3R)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-フルオロ-8-アザスピロ[4.5]デカン-8-カルボキシレート

チルエチルスルフィンアミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8
-カルボキシレート(200mg、0.534mmol)およびDAST(DCM中1M
、934μL、0.934mmol)の混合物を、0℃で90分間撹拌した。反応混合物
を飽和NaHCO3水溶液(5mL)の添加によりクエンチした。室温で10分間撹拌し
た後、相を分離し、水層をDCM(2×5mL)で抽出した。合わせた有機相をMgSO
4で脱水し、濾過し、揮発物を減圧下で除去して、(1R,3R)-tert-ブチル1
-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-フルオロ-8-アザスピ
ロ[4.5]デカン-8-カルボキシレートを得、これをさらに精製することなく次のス
テップにおいて使用した。MSm/z277.2(M+H-Boc)+。
tert-ブチル4-オキソ-2-オキサ-8-アザスピロ[4.5]デカン-8-カル
ボキシレート

6日の手順に従って、tert-ブチル4-ヒドロキシ-2-オキサ-8-アザスピロ[
4.5]デカン-8-カルボキシレートを、1-tert-ブチル4-エチルピペリジン
-1,4-ジカルボキシレートから4ステップで調製した。1H NMR (400 MHz, クロロホ
ルム-d) δ ppm 4.13 (dd, J=10.1, 4.6 Hz, 1 H), 4.03 (dd, J=4.6, 2.0 Hz, 1 H), 3.
78-3.71 (m, 2 H), 3.69 (d, J=8.6 Hz, 1 H), 3.67-3.58 (m, 2 H), 3.29 (m, 1 H), 3.
16 (m, 1 H), 1.78 (m, 2 H), 1.58 (m, 1 H), 1.50 (m, 2 H), 1.47 (s, 9 H).MSm/
z258.1(M-H)+。
ロ[4.5]デカン-8-カルボキシレート(544mg、2.11mmol)およびデ
ス-マーチンペルヨージナン(1.39g、3.17mmol)の溶液を、0℃で2時間
撹拌した。飽和NaHCO3水溶液/飽和Na2S2O3水溶液(1/1、10mL)を
添加し、有機相を分離し、水相をDCM(3×10mL)で抽出した。合わせた有機相を
Na2SO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留物をシリカク
ロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)により精製して、te
rt-ブチル4-オキソ-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキ
シレート(470mg、1.84mmol)を無色油状物として得、これは静置すると結
晶化した。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.08 (s, 2 H), 4.05 (s, 2 H),
3.88 (dt, J = 13.7, 4.9 Hz, 2 H), 3.12 (ddd, J = 13.6, 9.8, 3.6 Hz, 2 H), 1.75 (
ddd, J = 13.9, 9.7, 4.2 Hz, 2 H), 1.58-1.51 (m, 2 H), 1.48 (s, 9 H).MSm/z2
56.2(M+H)+。
(S)-tert-ブチル4-((R)-1,1-ジメチルエチルスルフィンアミノ)-
2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレート

.5]デカン-8-カルボキシレート(220mg、0.86mmol)、チタン(IV
)エトキシド(725μL、3.45mmol)および(R)-2-メチルプロパン-2
-スルフィンアミド(209mg、1.72mmol)の溶液を、90℃で1時間撹拌し
た。0℃に冷却した後、水素化ホウ素リチウム(23mg、1.06mmol)を添加し
た。30分間撹拌した後、反応混合物をMeOHの添加によりクエンチした。揮発物を減
圧下で除去した。得られた残留物をブラインで希釈し、これをEtOAc(4×10mL
)で抽出した。合わせた有機相をNa2SO4で脱水し、濾過し、揮発物を減圧下で除去
し、得られた残留物をシリカクロマトグラフィー(0から100%の勾配のEtOAc/
ヘプタン)により精製して、(S)-tert-ブチル4-((R)-1,1-ジメチル
エチルスルフィンアミノ)-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボ
キシレート(170mg、0.47mmol)を得た。MSm/z361.1(M+H)
+。
ンおよびスルホンアミドを使用して合成した。

(3S,4S)-tert-ブチル4-((R)-1,1-ジメチルエチルスルフィンア
ミノ)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレ
ートおよび(3R,4S)-tert-ブチル4-((R)-1,1-ジメチルエチルス
ルフィンアミノ)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-カ
ルボキシレート

THF(24mL)中のtert-ブチル4-オキソ-2-オキサ-8-アザスピロ[
4.5]デカン-8-(2.47g、9.67mmol)の溶液に、LHMDS(THF
中1M、9.67mL、9.67mmol)を-78℃で添加した。混合物をこの温度で
30分間撹拌した後、THF(10mL)中のヨードメタン(0.605mL、9.67
mmol)を添加した。得られた混合物を室温に加温し、1時間撹拌した。反応混合物を
EtOAcおよび飽和NaHCO3水溶液で希釈した。有機層をブラインで洗浄し、Na
2SO4で脱水し、濾過し、減圧下で濃縮した。得られた茶色油状物をシリカクロマトグ
ラフィー(0から20%の勾配のEtOAc/ヘプタン)により精製して、tert-ブ
チル3-メチル-4-オキソ-2-オキサ-8-アザスピロ[4.5]デカン-8-カル
ボキシレート(318mg、1.18mmol)を得た。MSm/z270.2(M+H
)+。
THF(4mL)中のtert-ブチル3-メチル-4-オキソ-2-オキサ-8-ア
ザスピロ[4.5]デカン-8-カルボキシレート(318mg、1.18mmol)、
チタン(IV)エトキシド(990μL、4.72mmol)および(R)-2-メチル
プロパン-2-スルフィンアミド(286mg、2.361mmol)の溶液を、90℃
で90分間撹拌した。0℃に冷却した後、水素化ホウ素リチウム(65.3mg、3.0
0mmol)を一度に添加し、得られた混合物を室温で16時間撹拌した。飽和NH4C
l水溶液をゆっくりと添加して過剰のホウ水素化物をクエンチし、続いてEtOAc(2
5mL)を添加した。得られた混合物を15分間激しく撹拌し、次いでセライトのパッド
に通して濾過した。有機相を飽和NaHCO3水溶液およびブラインで洗浄し、Na2S
O4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留物をシリカクロマトグ
ラフィー(0から100%の勾配のEtOAc/ヘプタン)により精製して、(4S)-
tert-ブチル4-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-メチ
ル-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレート(88mg、
0.235mmol)を得た。MSm/z375.2(M+H)+。
ジアステレオマーを、キラルSFC.カラム:LUXC4 30×250mm、流速:
80g/分、移動相:CO2中20%MeOH、検出:210nmにより分離して、(3
R,4S)-tert-ブチル4-((R)-1,1-ジメチルエチルスルフィンアミノ
)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレート
、Rt=4.0分;および(3S,4S)-tert-ブチル4-((R)-1,1-ジ
メチルエチルスルフィンアミノ)-3-メチル-2-オキサ-8-アザスピロ[4.5]
デカン-8-カルボキシレート、TR=4.55分を得た。
アミノ)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシ
レートの代替調製:
THF(220mL)中のジイソプロピルアミン(23.4mL、166mmol)の
-10℃溶液に、n-BuLi(ヘキサン中2.5M、64.1mL、160mmol)
を滴下添加した。この温度で30分間撹拌した後、THF(50mL)中の1-tert
-ブチル4-エチルピペリジン-1,4-ジカルボキシレート(27.5g、107mm
ol)を滴下添加し、得られた混合物を0℃で30分間撹拌した。(S)-2-((te
rt-ブチルジメチルシリル)オキシ)プロパナール(20.47mL、102mmol
)を添加し、混合物を0℃で1時間および室温で1時間撹拌した。反応物を飽和NaHC
O3水溶液/H2O(1:4、125mL)で希釈し、EtOAc(50mL)を添加し
、相を分離した。水相をEtOAc(3×100mL)でさらに抽出した。合わせた有機
相をNa2SO4で脱水し、濾過し、溶媒を減圧下で除去した。得られた残留物をさらに
精製することなく次のステップにおいて使用した。MSm/z346.4(M+H-Bo
c)+。
THF(600mL)中の粗製の1-tert-ブチル4-エチル4-((2S)-2
-((tert-ブチルジメチルシリル)オキシ)-1-ヒドロキシプロピル)ピペリジ
ン-1,4-ジカルボキシレート(95g、214mmol)の溶液に、水素化ホウ素リ
チウム(7.0g、321mmol)を少量ずつ添加し、得られた混合物を室温で16時
間撹拌した。0℃に冷却した後、飽和NaHCO3水溶液/H2O(1/2、150mL
)を添加し、得られた混合物を、ガス発生が観察されなくなるまで激しく撹拌した。Et
OAc(100mL)を添加し、混合物を濾過し、相を分離し、水相をEtOAc(3×
50mL)でさらに抽出した。合わせた有機相をブラインで洗浄し、Na2SO4で脱水
し、濾過し、揮発物を減圧下で除去して、tert-ブチル4-((2S)-2-((t
ert-ブチルジメチルシリル)オキシ)-1-ヒドロキシプロピル)-4-(2-ヒド
ロキシエチル)ピペリジン-1-カルボキシレート(64.8g、161mmol)を得
、これをさらに精製することなく次のステップにおいて使用した。
THF(500mL)中のtert-ブチル4-((2S)-2-((tert-ブチ
ルジメチルシリル)オキシ)-1-ヒドロキシプロピル)-4-(2-ヒドロキシエチル
)ピペリジン-1-カルボキシレート(64.8g、161mmol)およびTBAF(
THF中1M、242mL、242mmol)の溶液を、室温で2時間撹拌した。飽和N
aHCO3水溶液/H2O(1:2、150mL)を添加し、相を分離し、水相をEtO
Ac(3×100mL)でさらに抽出した。合わせた有機相をブラインで洗浄し、Na2
SO4で脱水し、濾過し、揮発物を減圧下で除去した。得られた残留物をシリカクロマト
グラフィー(20から100%の勾配のEtOAc/ヘプタン)により精製して、ter
t-ブチル4-((2S)-1,2-ジヒドロキシプロピル)-4-(2-ヒドロキシエ
チル)ピペリジン-1-カルボキシレート(39.25g、136mmol)を半固体の
無色油状物として得た。
THF(600mL)中のNaH(10.60g、424mmol)の0℃懸濁液に、
THF(200mL)中のtert-ブチル4-((2S)-1,2-ジヒドロキシプロ
ピル)-4-(2-ヒドロキシエチル)ピペリジン-1-カルボキシレート(35.06
g、121mmol)および4-トルエンスルホニルクロリド(23.1g、121mm
ol)の溶液を滴下添加した。得られた混合物を0℃で1時間撹拌した。飽和NH4Cl
水溶液(約5mL)を-20℃でゆっくりと添加し、反応物を、ガス発生が観察されなく
なるまで激しく撹拌した。この時点で、飽和NH4Cl水溶液(100mL)、続いてブ
ライン(100mL)を添加し、混合物をEtOAc(3×100mL)で抽出した。合
わせた有機相をNa2SO4で脱水し、濾過し、溶媒を減圧下で除去して、(3S)-t
ert-ブチル4-ヒドロキシ-3-メチル-2-オキサ-8-アザスピロ[4.5]デ
カン-8-カルボキシレート(32.19g、119mmol)を得、これをさらに精製
することなく次のステップにおいて使用した。MSm/z171.1(M-Boc)-。
DCM(300mL)中の(3S)-tert-ブチル4-ヒドロキシ-3-メチル-
2-オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレート(32.19g、
119mmol)およびデス-マーチンペルヨージナン(67.4g、154mmol)
の溶液を、0℃で2時間撹拌した。混合物を室温まで加温し、揮発物を減圧下で除去した
。得られた残留物をシリカクロマトグラフィー(0から40%の勾配のEtOAc/ヘプ
タン)により精製して、(3S)-tert-ブチル3-メチル-4-オキソ-2-オキ
サ-8-アザスピロ[4.5]デカン-8-カルボキシレート(27.68g、92mm
ol)を淡黄色油状物として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.09 (d,
J=9.60 Hz, 1 H), 3.66-3.86 (m, 4 H), 3.03 (ddd, J=13.77, 9.73, 3.79 Hz, 1 H), 2
.90 (ddd, J=13.64, 10.23, 3.41 Hz, 1 H), 1.68 (ddd, J=13.83, 9.92, 4.29 Hz, 1 H)
, 1.41-1.59 (m, 2 H), 1.30-1.40 (m, 10 H), 1.20-1.25 (m, 3 H).
THF(300mL)中の(3S)-tert-ブチル3-メチル-4-オキソ-2-
オキサ-8-アザスピロ[4.5]デカン-8-カルボキシレート(22.52g mg
、84mmol)、チタン(IV)エトキシド(70.1mL、334mmol)および
(R)-2-メチルプロパン-2-スルフィンアミド(21g、173mmol)の溶液
を、90℃で21時間撹拌した。-4℃に冷却した後、MeOH(30mL)を添加し、
続いて水素化ホウ素リチウム(1.82g、84mmol)を滴下添加(反応温度を2℃
未満に維持しながら)し、得られた混合物を-4℃で1時間撹拌した。飽和NH4Cl水
溶液をゆっくりと添加して過剰のホウ水素化物(半固体)をクエンチし、続いてEtOA
c(500mL)を添加した。得られた混合物を室温で15分間激しく撹拌し、次いでセ
ライトのパッドに通して濾過し、続いてEtOAc(500mL)で洗浄した。揮発物を
減圧下で除去し、得られた残留物をシリカクロマトグラフィー(0から100%の勾配の
EtOAc/ヘプタン)により精製して、(3S,4S)-tert-ブチル4-((R
)-1,1-ジメチルエチルスルフィンアミノ)-3-メチル-2-オキサ-8-アザス
ピロ[4.5]デカン-8-カルボキシレートを95:5のジアステレオマー混合物(副
ジアステレオマーは(3R,4S)-tert-ブチル4-((R)-1,1-ジメチル
エチルスルフィンアミノ)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン
-8-カルボキシレート)として得た。
ジアステレオマーを、キラルSFC.カラム:LC-4 30×250mm、流速:1
00g/分、移動相:CO2中30%MeOH、検出:225nm、TR:0.95分(
副ジアステレオマーTR:0.55分)により分離して、(3S,4S)-tert-ブ
チル4-((R)-1,1-ジメチルエチルスルフィンアミノ)-3-メチル-2-オキ
サ-8-アザスピロ[4.5]デカン-8-カルボキシレート(19g、50.68mm
ol)を得た。MSm/z375.2。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.24
-4.16 (m, 1 H), 4.03-3.94 (m, 1 H), 3.91-3.85 (m, 1 H), 3.84-3.78 (m, 1 H), 3.64
(d, J=9.1 Hz, 1 H), 3.50-3.42 (m, 1 H), 3.32 (d, J=10.1 Hz, 1 H), 2.99-2.85 (m,
2 H), 1.82 (td, J=18.1, 15.1, 7.7 Hz, 2 H), 1.62-1.53 (m, 1 H), 1.53-1.47 (m, 1
H), 1.46 (s, 9 H), 1.22 (d, J=6.4 Hz, 3 H).
ドアルカンを使用して合成した。

(1R)-tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンアミノ)
-2-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート

THF(24mL)中のtert-ブチル1-オキソ-8-アザスピロ[4.5]デカ
ン-8-カルボキシレート(2.2g、8.68mmol)の溶液に、LHMDS(TH
F中1M、8.68mL、8.68mmol)を0~5℃で添加した。混合物をこの温度
で30分間撹拌した後、ヨードメタン(0.543mL、8.68mmol)を添加した
。得られた混合物を室温に加温し、さらに2時間撹拌した。反応混合物をEtOAcと飽
和NaHCO3水溶液で希釈した。有機相をブラインで洗浄し、Na2SO4で脱水し、
濾過し、減圧下で濃縮した。得られた茶色油状物をシリカクロマトグラフィー(0から2
5%の勾配のEtOAc/ヘプタン)により精製して、ラセミtert-ブチル2-メチ
ル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.3g、
4.86mmol)を得た。MSm/z268.1。(M+H)+。
THF(10mL)中のラセミtert-ブチル2-メチル-1-オキソ-8-アザス
ピロ[4.5]デカン-8-カルボキシレート(267mg、0.999mmol)、チ
タン(IV)エトキシド(837μL、3.99mmol)および(R)-2-メチルプ
ロパン-2-スルフィンアミド(242mg、1.997mmol)の溶液を、85℃で
24時間撹拌した。-78℃に冷却した後、MeOH(12mL)、続いて水素化ホウ素
リチウム(65.3mg、3.00mmol)を添加した。得られた混合物を-78℃か
ら室温で16時間撹拌した。飽和NH4Cl水溶液をゆっくりと添加して過剰のホウ水素
化物をクエンチし、続いてEtOAc(100mL)を添加した。得られた混合物を15
分間激しく撹拌し、次いでセライトのパッドに通して濾過した。揮発物を減圧下で除去し
、得られた残留物をシリカクロマトグラフィー(0から60%の勾配のEtOAc/ヘプ
タン(0.25%のEt3Nを含有))により精製して、(1R)-tert-ブチル1
-((R)-1,1-ジメチルエチルスルフィンアミノ)-2-メチル-8-アザスピロ
[4.5]デカン-8-カルボキシレート(92mg、0.247mmol)を得た。M
Sm/z373.1(M+H)+。
ラセミ(1S,2S,3S)-tert-ブチル1-((tert-ブトキシカルボニル
)アミノ)-2,3-ジヒドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシ
レート

tert-ブチル1-オキソ-8-アザスピロ[4.5]デカ-2-エン-8-カルボ
キシレート(2g、7.96mmol)および塩化セリウム(III)七水和物(3.2
6g、8.75mmol)の混合物に、MeOH(60mL)およびTHF(20mL)
をN2雰囲気下で添加した。得られた混合物を室温で1時間撹拌し、0℃に冷却し、水素
化ホウ素ナトリウム(0.60g、15.9mmol)を添加した。反応混合物を0℃で
1時間撹拌し、次いで1N NaOH(75mL)に注ぎ入れた。混合物をEt2O(3
×50mL)で抽出し、合わせた抽出物をNa2SO4で脱水し、濾過し、減圧下で濃縮
した。残留物をシリカクロマトグラフィー(10から60%の勾配のEtOAc/ヘプタ
ン、0.25%のNEt3を含有)により精製して、ラセミtert-ブチル1-ヒドロ
キシ-8-アザスピロ[4.5]デカ-2-エン-8-カルボキシレート(2.01g、
7.93mmol)を橙色油状物として得た。1H NMR (400 MHz, クロロホルム-d) δ pp
m 5.99-5.94 (m, 1 H), 5.89-5.83 (m, 1 H), 4.32 (s, 1 H), 3.79-3.65 (m, 2 H), 3.2
9-3.12 (m, 2 H), 2.36-2.27 (m, 1 H), 2.26-2.15 (m, 1 H), 1.81-1.71 (m, 1 H), 1.5
5-1.50 (m, 2 H), 1.49 (s, 9 H), 1.43-1.36 (m, 1 H).MSm/z276.2(M+N
a)+。
DCM(64mL)中のラセミtert-ブチル1-ヒドロキシ-8-アザスピロ[4
.5]デカ-2-エン-8-カルボキシレート(1.63g、6.43mmol)の溶液
に、tert-ブチルヒドロペルオキシド(デカン中5.5M溶液、1.4mL、7.7
2mmol)およびバナジルアセチルアセトネート(156mg、0.643mmol)
をN2雰囲気下0℃で順次添加した。反応混合物を0℃で20分間および室温で15時間
撹拌した。反応混合物を飽和Na2SO3水溶液(50mL)に注ぎ入れ、DCM(3×
25mL)で抽出した。合わせた抽出物をNa2SO4で脱水し、濾過し、減圧下で濃縮
した。残留物をシリカクロマトグラフィー(10から60%の勾配のEtOAc/ヘプタ
ン、0.25%のNEt3を含有)により精製して、ラセミ(1R,2R,5S)-te
rt-ブチル2-ヒドロキシ-6-オキサスピロ[ビシクロ[3.1.0]ヘキサン-3
,4’-ピペリジン]-1’-カルボキシレート(805mg、単一ジアステレオマー)
を得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 3.92-3.78 (m, 3 H), 3.65 (t, J=2
.27 Hz, 1 H), 3.56 (t, J=2.27 Hz, 1 H), 2.99-2.82 (m, 2 H), 2.22 (d, J=14.65 Hz,
1 H), 1.78 (br. s., 1 H), 1.70-1.58 (m, 3 H), 1.47-1.40 (m, 10 H).MSm/z1
70.1(M+H-Boc)+。
窒素雰囲気下、THF(15mL)中のラセミ(1R,2R,5S)-tert-ブチ
ル2-ヒドロキシ-6-オキサスピロ[ビシクロ[3.1.0]ヘキサン-3,4’-ピ
ペリジン]-1’-カルボキシレート(805mg、2.99mmol)、トリフェニル
ホスフィン(1.57g、5.98mmol)およびイミノジカルボン酸ジ-tert-
ブチル(1.30g、5.98mmol)の溶液に、DIAD(1.16mL、5.98
mmol)を0℃でゆっくりと添加した。得られた混合物を0℃で5分間撹拌し、室温で
10分間撹拌し、次いで40℃に15時間加熱した。反応混合物をEtOAc(25mL
)で希釈し、飽和NH4Cl水溶液に注ぎ入れ、EtOAc(3×10mL)で抽出した
。合わせた抽出物をNa2SO4で脱水し、濾過し、減圧下で濃縮した。残留物をシリカ
クロマトグラフィー(0から40%の勾配の、0.25%のNEt3を含有するEtOA
c/0.25%のNEt3を含有するヘプタン)により精製して、ラセミ(1R,2S,
5S)-tert-ブチル2-((ジ-tert-ブトキシカルボニル)アミノ)-6-
オキサスピロ[ビシクロ[3.1.0]ヘキサン-3,4’-ピペリジン]-1’-カル
ボキシレート(480mg;単一ジアステレオマー)を得た。MSm/z491.3(M
+Na)+。
CHCl3(4mL)中のラセミ(1R,2S,5S)-tert-ブチル2-((ジ
-tert-ブトキシカルボニル)アミノ)-6-オキサスピロ[ビシクロ[3.1.0
]ヘキサン-3,4’-ピペリジン]-1’-カルボキシレート(346mg)の溶液に
、HOAc(0.2mL)を添加した。反応混合物を室温で5時間撹拌し、減圧下で濃縮
して、粗製のラセミ(3aS,6S,6aS)-N,N’-ビス-(tert-ブチルカ
ルボニル)-6-ヒドロキシテトラヒドロスピロ[シクロペンタ[d]オキサゾール-4
,4’-ピペリジン]-2(5H)-オン(295mg;単一ジアステレオマー)を透明
油状物として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.74 (dd, J=7.45, 1.39
Hz, 1 H), 4.48 (br. s, 1 H), 4.42 (d, J=7.33 Hz, 1 H), 4.04 (br. s., 2 H), 2.78
(br. s., 2 H), 2.03-1.99 (m, 2 H), 1.93-1.79 (m, 3 H), 1.66-1.58 (m, 1 H), 1.56
(s, 9 H), 1.46 (s, 9 H).MSm/z435.2(M+Na)+。
MeOH(1.5mL)中の粗製のラセミ(3aS,6S,6aS)-N,N’-ビス
-(tert-ブチルカルボニル)-6-ヒドロキシテトラヒドロスピロ[シクロペンタ
[d]オキサゾール-4,4’-ピペリジン]-2(5H)-オン(125mg)の溶液
に、Cs2CO3(20mg、0.06mmol)を添加し、反応混合物を室温で5時間
撹拌した。混合物をEtOAc(10mL)および飽和NH4Cl水溶液/水(1:1、
10mL)で希釈した。分離した水層をEtOAc(2×10mL)で抽出した。合わせ
た抽出物をMgSO4で脱水し、濾過し、減圧下で濃縮した。残留物をSFC(Prin
cetone 2-EP 20×150mm 5μm、CO2/MeOH 80g/分
120bar)により精製して、ラセミ(1S,2S,3S)-tert-ブチル1-(
(tert-ブトキシカルボニル)アミノ)-2,3-ジヒドロキシ-8-アザスピロ[
4.5]デカン-8-カルボキシレート(44mg、単一ジアステレオマー)を透明油状
物として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 5.02 (d, J=9.35 Hz, 1 H),
4.17-4.10 (m, 1 H), 4.04 (br. s, 1 H), 3.96 (br. s, 3 H), 2.83 (d, J=12.13 Hz, 2
H), 2.22-2.10 (m, 1 H), 1.98 (br. s, 2 H), 1.76 (td, J=12.88, 4.55 Hz, 1 H), 1.
64-1.52 (m, 1 H), 1.46 (s, 9 H), 1.45 (s, 9 H).MSm/z409.3(M+Na)
+。
tert-ブチル((1R,3R)-3-(トリフルオロメチル)-8-アザスピロ[4
.5]デカン-1-イル)カルバメート

THF(40mL)中のベンジル1-オキソ-8-アザスピロ[4.5]デカ-2-エ
ン-8-カルボキシレート(3.05g、10.7mmol)の溶液に、トリメチル(ト
リフルオロメチル)シラン(THF中2M、6.41mL)およびTBAF(THF中1
M、0.214mL)を0℃で添加し、得られた混合物を0℃で1.5時間撹拌した。反
応混合物を2M HCl水溶液(10mL)で慎重に希釈し、0℃で1時間撹拌した。溶
液を飽和NH4Cl水溶液(50mL)でさらに希釈し、EtOAc(3×50mL)で
抽出した。合わせた有機層をNa2SO4で脱水し、濾過し、揮発物を減圧下で除去した
。残留物をシリカクロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)に
より精製して、ベンジル1-オキソ-3-(トリフルオロメチル)-8-アザスピロ[4
.5]デカン-8-カルボキシレートを無色油状物(2.22g、6.25mmol)と
して得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.45-7.31 (m, 5 H), 5.16 (s, 2
H), 3.84 (dd, J=8.9, 5.1 Hz, 1 H), 3.30 (ddd, J=13.5, 9.5, 3.4 Hz, 1 H), 3.21 (
ddd, J=13.5, 9.8, 3.6 Hz, 1 H), 3.03-2.87 (m, 1 H), 2.66 (ddd, J=18.8, 8.4, 1.5
Hz, 1 H), 2.46 (dd, J=18.9, 10.7 Hz, 1 H), 2.38-2.25 (m, 1 H), 1.97-1.79 (m, 2 H
), 1.70-1.58 (m, 1 H), 1.54 (m, 3 H). 19F NMR (376 MHz, クロロホルム-d) δ ppm
-72.08 (d, J=8.0 Hz). 13C NMR (101 MHz, クロロホルム-d) δ ppm 215.93, 155.18,
136.67, 128.53, 128.07, 127.93, 125.74 (q, J=263 Hz), 67.24, 47.96, 40.35, 39.86
, 37.30, 32.77 (q, J=29 Hz), 33.77 (q, J=3 Hz), 31.89, 31.10.
THF(50mL)中のベンジル1-オキソ-3-(トリフルオロメチル)-8-アザ
スピロ[4.5]デカン-8-カルボキシレート(2.22g、6.25mmol)、(
R)-tert-ブタンスルフィンアミド(1.514g、12.50mmol)および
テトラエトキシチタン(5.70g、5.24mL、25mmol)の混合物を、80℃
に16時間加熱した。反応混合物を-78℃に冷却し、次いでMeOH(10mL)およ
び水素化ホウ素リチウム(0.408g、18.74mmol)を添加した。反応混合物
を3時間かけて室温に加温した。反応混合物を飽和NH4Cl水溶液(50mL)で慎重
に希釈した。得られた不均一混合物をセライトに通して濾過し、EtOAcで洗浄した。
濾液の層を分離し、水層をEtOAc(2×25mL)で抽出した。合わせた有機層をN
a2SO4で脱水し、濾過し、揮発物を減圧下で除去して、粗製のベンジル(1R)-1
-(((R)-tert-ブチルスルフィニル)アミノ)-3-(トリフルオロメチル)
-8-アザスピロ[4.5]デカン-8-カルボキシレートを白色固体として得、これを
さらに精製することなく次のステップにおいて使用した。MS:m/z461.3(M+
H)+。
MeOH(25mL)中の粗製のベンジル(1R)-1-(((R)-tert-ブチ
ルスルフィニル)アミノ)-3-(トリフルオロメチル)-8-アザスピロ[4.5]デ
カン-8-カルボキシレート(2.88g、6.25mmol)の溶液に、HCl(ジオ
キサン中4M、3.13mL)を添加した。得られた溶液を周囲温度で1時間撹拌した。
揮発物を減圧下で除去し、得られた残留物を減圧下で2時間乾燥させた。残留物をCH2
Cl2に溶解し、DIPEA(5.57mL、31.3mmol)および二炭酸ジ-te
rt-ブチル(2.05g、9.4mmol)を添加した。得られた混合物を周囲温度で
72時間撹拌した。反応混合物を飽和NH4Cl水溶液(50mL)で希釈し、CH2C
l2(3×25mL)で抽出した。合わせた有機層をNa2SO4で脱水し、濾過し、揮
発物を減圧下で除去した。得られた残留物をシリカクロマトグラフィー(0から100%
の勾配のEtOAc/ヘプタン)により精製して、ベンジル(1R,3R)-1-((t
ert-ブトキシカルボニル)アミノ)-3-(トリフルオロメチル)-8-アザスピロ
[4.5]デカン-8-カルボキシレート(0.90g、1.97mmol)を白色固体
として、ベンジル(1R,3R)-1-((tert-ブトキシカルボニル)アミノ)-
3-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート
およびベンジル(1R,3S)-1-((tert-ブトキシカルボニル)アミノ)-3
-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレートの
1.47gの混合物と共に得た。混合物を分取SFC(カラム:IB 21×250mm
、10%IPA共溶媒)によりさらに精製して、ベンジル(1R,3R)-1-((te
rt-ブトキシカルボニル)アミノ)-3-(トリフルオロメチル)-8-アザスピロ[
4.5]デカン-8-カルボキシレート(0.60g、1.32mmol)[1H NMR (400
MHz, クロロホルム-d) δ ppm 7.34-7.20 (m, 5 H), 5.06 (s, 2 H), 4.39 (d, J=9.6 H
z, 1 H), 3.96-3.75 (m, 3 H), 2.99 (t, J=11.0 Hz, 2 H), 2.65 (dq, J=18.3, 9.1 Hz,
1 H), 2.24 (dt, J=15.3, 8.3 Hz, 1 H), 1.77 (dd, J=13.9, 9.7 Hz, 1 H), 1.66 (dd,
J=13.9, 8.4 Hz, 1 H), 1.60-1.41 (m, 3 H), 1.39 (s, 9 H), 1.31-1.16 (m, 2 H)]お
よびベンジル(1R,3S)-1-((tert-ブトキシカルボニル)アミノ)-3-
(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート(0
.50g、1.09mmol)[1H NMR (400 MHz, クロロホルム-d) δ ppm 7.44-7.30 (
m, 5 H), 5.15 (s, 2 H), 4.36 (d, J=9.1 Hz, 1 H), 4.24-4.01 (m, 2 H), 3.88 (q, J=
8.7 Hz, 1 H), 2.91 (dd, J=29.7, 16.1 Hz, 2 H), 2.72 (ddt, J=14.6, 9.7, 5.0 Hz, 1
H), 2.30-2.11 (m, 2 H), 1.77-1.60 (m, 2 H), 1.46 (m, 12 H), 1.27-1.15 (m, 1 H)]
を得た。
EtOH(40mL)中のベンジル(1R,3R)-1-((tert-ブトキシカル
ボニル)アミノ)-3-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8
-カルボキシレート(0.90g、1.97mmol)およびPd/C(10重量%、2
00mg)の混合物を、H2雰囲気(バルーン)下で2時間水素化した。混合物を窒素で
5分間スパージし、次いでセライトに通して濾過し、EtOHですすいだ。濾液を減圧下
で濃縮し、減圧下で乾燥させて、粗製のtert-ブチル((1R,3R)-3-(トリ
フルオロメチル)-8-アザスピロ[4.5]デカン-1-イル)カルバメート(625
mg、1.94mmol)を白色泡状物として得、これを精製することなく直接使用した
。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.54 (d, J=9.7 Hz, 1 H), 3.84 (q, J=8.
8 Hz, 1 H), 3.00 (tt, J=12.1, 4.0 Hz, 2 H), 2.79-2.63 (m, 3 H), 2.28 (ddd, J=13.
5, 8.8, 6.8 Hz, 1 H), 2.19 (d, J=8.5 Hz, 1 H), 1.80 (qd, J=14.0, 9.1 Hz, 2 H), 1
.63 (qd, J=9.0, 3.4 Hz, 2 H), 1.47 (m, 12 H). 19F NMR (376 MHz, クロロホルム-d)
δ ppm -71.42 (d, J=9.6 Hz).
4.5]デカン-1-イル)カルバメート(355mg、1.09mmol)を、上記手
順に従って、ベンジル(1R,3S)-1-((tert-ブトキシカルボニル)アミノ
)-3-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレ
ート(0.50g、1.09mmol)から調製した。1H NMR (400 MHz, クロロホルム-
d) δ ppm 4.45 (d, J=9.3 Hz, 1 H), 3.80 (q, J=9.2 Hz, 1 H), 3.15-2.96 (m, 2 H),
2.79 (t, J=11.9 Hz, 1 H), 2.67 (tt, J=13.8, 6.9 Hz, 2 H), 2.17 (dd, J=13.7, 9.1
Hz, 2 H), 1.73 (td, J=13.2, 4.3 Hz, 1 H), 1.68-1.56 (m, 1 H), 1.54-1.31 (m, 12 H
), 1.26-1.13 (m, 1 H), 0.84 (d, J=4.6 Hz, 1 H).
tert-ブチル((1S,3R)-2,2-ジフルオロ-3-メチル-8-アザスピロ
[4.5]デカン-1-イル)カルバメート

THF(15mL)中のLiHMDS(THF中1M、3.7mL、3.7mmol)
の-78℃溶液に、THF(5mL)中のベンジル(R)-3-メチル-1-オキソ-8
-アザスピロ[4.5]デカン-8-カルボキシレート(1.0g、3.32mmol)
を滴下添加した。-78℃で30分間撹拌した後、THF(5mL)中のN-フルオロ-
N-(フェニルスルホニル)ベンゼンスルホンアミド(1.15g、3.65mmol)
を添加し、得られた混合物をこの温度で30分間撹拌した。室温に加温した後、反応混合
物を、飽和NaHCO3水溶液(25mL)を含有する分液漏斗に注ぎ入れ、これをEt
OAc(3×30mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮
発物を減圧下で除去した。残留物をシリカクロマトグラフィー(0から40%の勾配のE
tOAc/ヘプタン)により精製して、ベンジル(3R)-2-フルオロ-3-メチル-
1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.05g、3
.32mmol)を得た。MSm/z320.2(M+H)+。
THF(15mL)中のLiHMDS(THF中1M、3.45mL、3.45mmo
l)の-78℃溶液に、THF(5mL)中のベンジル(3R)-2-フルオロ-3-メ
チル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.0g
、3.13mmol)を添加した。-78℃で30分間撹拌した後、THF(5mL)中
のN-フルオロ-N-(フェニルスルホニル)ベンゼンスルホンアミド(1.086g、
3.44mmol)を添加し、得られた混合物をこの温度で1時間撹拌した。室温に加温
した後、反応混合物を、飽和NaHCO3水溶液(25mL)を含有する分液漏斗に注ぎ
入れ、EtOAc(3×30mL)で抽出した。合わせた有機相をMgSO4で脱水し、
濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー(0から40%
の勾配のEtOAc/ヘプタン)により精製して、ベンジル(R)-2,2-ジフルオロ
-3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(
1.17g、3.32mmol)を得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.
30-7.48 (m, 5 H), 5.16 (s, 2 H), 3.75-3.99 (m, 2 H), 3.32-3.44 (m, 1 H), 3.28 (d
dd, J=13.52, 9.47, 3.54 Hz, 1 H), 2.25-2.47 (m, 1 H), 2.17 (ddd, J=13.20, 7.52,
2.78 Hz, 1 H), 1.72-1.92 (m, 2 H), 1.42-1.62 (m, 2 H), 1.24 (dd, J=7.33, 0.76 Hz
, 3 H). 19F NMR (376 MHz, クロロホルム-d) δ ppm -119.79 (br. d, J=277.64 Hz, 1
F), -123.89 (d, J=271.90 Hz, 1 F).
THF(35mL)中のベンジル(R)-2,2-ジフルオロ-3-メチル-1-オキ
ソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.17g、3.32m
mol)、チタン(IV)エトキシド(2.91mL、13.87mmol)および(R
)-2-メチルプロパン-2-スルフィンアミド(841mg、6.94mmol)の溶
液を、60℃で4時間撹拌した。-78℃に冷却した後、MeOH(3.5mL)、続い
て水素化ホウ素リチウム(0.227g、10.40mmol)を添加した。得られた混
合物を-78℃から室温で2時間撹拌した。飽和NH4Cl水溶液をゆっくりと添加し、
続いてEtOAc(100mL)を添加した。得られた混合物を5分間激しく撹拌し、次
いでセライトのパッドに通して濾過した。揮発物を減圧下で除去し、残留物をシリカクロ
マトグラフィー(0から50%の勾配のEtOAc/ヘプタン)により精製して、ベンジ
ル(1S,3R)-1-(((R)-tert-ブチルスルフィニル)アミノ)-2,2
-ジフルオロ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート(
700mg、1.582mmol)を単一ジアステレオマーとして得た。MSm/z44
3.3(M+H)+。
MeOH(10mL)中のベンジル(1S,3R)-1-(((R)-tert-ブチ
ルスルフィニル)アミノ)-2,2-ジフルオロ-3-メチル-8-アザスピロ[4.5
]デカン-8-カルボキシレート(700mg、1.582mmol)の溶液に、HCl
(ジオキサン中4M、3.95mL、15.82mmol)を添加した。得られた混合物
を45℃で30分間撹拌した。室温に冷却した後、揮発物を減圧下で除去した。THF(
20mL)中のこの残留物、Boc2O(432mg、1.977mmol)およびDI
PEA(2.76mL、15.82mmol)の溶液を、室温で16時間撹拌した。混合
物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ、EtOAc(3×10mL
)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去し
、残留物をシリカクロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)に
より精製して、ベンジル(1S,3R)-1-((tert-ブトキシカルボニル)アミ
ノ)-2,2-ジフルオロ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボ
キシレート(645mg、1.471mmol)を得た。MSm/z383.3(M+H
-tBu)+。
MeOH(10mL)中のPd/C(10重量%、78mg)の懸濁液を、H2雰囲気
(バルーン)下で5分間激しく撹拌した。MeOH(10mL)中のベンジル(1S,3
R)-1-((tert-ブトキシカルボニル)アミノ)-2,2-ジフルオロ-3-メ
チル-8-アザスピロ[4.5]デカン-8-カルボキシレート(645mg、1.47
1mmol)を添加し、得られた懸濁液をH2雰囲気下で16時間激しく撹拌した。反応
混合物をセライトのパッドに通して濾過し、パッドをDCMで洗浄し、揮発物を減圧下で
除去して、tert-ブチル((1S,3R)-2,2-ジフルオロ-3-メチル-8-
アザスピロ[4.5]デカン-1-イル)カルバメート(448mg、1.471mmo
l)を得、これをさらに精製することなく使用した。MSm/z305.3(M+H)+
。
tert-ブチル((1R,2S,3R)-2-フルオロ-3-メチル-8-アザスピロ
[4.5]デカン-1-イル)カルバメート(A)およびtert-ブチル((1S,2
R,3R)-2-フルオロ-3-メチル-8-アザスピロ[4.5]デカン-1-イル)
カルバメート(B);
(A:B=4:1の混合物)

THF(30mL)中のLiHMDS(THF中1M、7.31mL、7.31mmo
l)の-78℃溶液に、THF(10mL)中のベンジル(R)-3-メチル-1-オキ
ソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(2.0g、6.64mm
ol)を滴下添加した。-78℃で30分間撹拌した後、THF(10mL)中のN-フ
ルオロ-N-(フェニルスルホニル)ベンゼンスルホンアミド(2.3g、7.30mm
ol)を添加し、得られた混合物をこの温度で30分間撹拌した。室温に加温した後、反
応混合物を、飽和NaHCO3水溶液(25mL)を含有する分液漏斗に注ぎ入れ、Et
OAc(3×30mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮
発物を減圧下で除去した。残留物をシリカクロマトグラフィー(0から40%の勾配のE
tOAc/ヘプタン)により精製して、ジアステレオマーであるベンジル(2S,3R)
-2-フルオロ-3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カル
ボキシレート(主)およびベンジル(2R,3R)-2-フルオロ-3-メチル-1-オ
キソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(副)の4:1混合物(
1.81g、5.67mmol)を得た。このジアステレオマー混合物をさらに精製する
ことなく次のステップにおいて使用した。MSm/z320.2(M+H)+。
THF(35mL)中の、ジアステレオマーであるベンジル(2S,3R)-2-フル
オロ-3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレー
ト(主)およびベンジル(2R,3R)-2-フルオロ-3-メチル-1-オキソ-8-
アザスピロ[4.5]デカン-8-カルボキシレート(副)の4:1混合物(1.95g
、6.11mmol)、チタン(IV)エトキシド(5.12mL、24.42mmol
)および(R)-2-メチルプロパン-2-スルフィンアミド(1.48g、12.21
mmol)の混合物を、60℃で4時間撹拌した。-78℃に冷却した後、MeOH(3
.5mL)、続いて水素化ホウ素リチウム(0.399g、18.32mmol)を添加
した。得られた混合物を-78℃から室温で2時間撹拌した。飽和NH4Cl水溶液をゆ
っくりと添加して過剰のホウ水素化物をクエンチし、続いてEtOAc(100mL)を
添加した。得られた混合物を5分間激しく撹拌し、次いでセライトのパッドに通して濾過
した。揮発物を減圧下で除去し、残留物をシリカクロマトグラフィー(0から50%の勾
配のEtOAc/ヘプタン)により精製して、ベンジル(1R,2S,3R)-1-((
(R)-tert-ブチルスルフィニル)アミノ)-2-フルオロ-3-メチル-8-ア
ザスピロ[4.5]デカン-8-カルボキシレートおよびベンジル(1S,2R,3R)
-1-(((R)-tert-ブチルスルフィニル)アミノ)-2-フルオロ-3-メチ
ル-8-アザスピロ[4.5]デカン-8-カルボキシレートの4:1混合物(2.05
g、4.83mmol)を得た。このジアステレオマー混合物をさらに精製することなく
次のステップにおいて使用した。MSm/z443.3(M+H)+。
MeOH(20mL)中のベンジル(1R,2S,3R)-1-(((R)-tert
-ブチルスルフィニル)アミノ)-2-フルオロ-3-メチル-8-アザスピロ[4.5
]デカン-8-カルボキシレートおよびベンジル(1S,2R,3R)-1-(((R)
-tert-ブチルスルフィニル)アミノ)-2-フルオロ-3-メチル-8-アザスピ
ロ[4.5]デカン-8-カルボキシレートの4:1混合物(2.05mg、4.83m
mol)の溶液に、HCl(ジオキサン中4M、12mL、48.0mmol)を添加し
た。得られた混合物を45℃で30分間撹拌した。室温に冷却した後、揮発物を減圧下で
除去した。THF(40mL)中のこの残留物、Boc2O(1.32g、6.04mm
ol)およびDIPEA(8.43mL、48.3mmol)の混合物を、室温で1時間
撹拌した。反応混合物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ、EtO
Ac(3×25mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発
物を減圧下で除去し、残留物をシリカクロマトグラフィー(0から50%の勾配のEtO
Ac/ヘプタン)により精製して、ベンジル(1R,2S,3R)-1-((tert-
ブトキシカルボニル)アミノ)-2-フルオロ-3-メチル-8-アザスピロ[4.5]
デカン-8-カルボキシレートおよびベンジル(1S,2R,3R)-1-((tert
-ブトキシカルボニル)アミノ)-2-フルオロ-3-メチル-8-アザスピロ[4.5
]デカン-8-カルボキシレートの4:1混合物(1.51g、3.59mmol)を得
た。このジアステレオマー混合物をさらに精製することなく次のステップにおいて使用し
た。MSm/z383.3(M+H-tBu)+。
MeOH(10mL)中のPd/C(10重量%、78mg)の懸濁液を、H2雰囲気
(バルーン)下で5分間激しく撹拌した。MeOH(10mL)中のベンジル(1R,2
S,3R)-1-((tert-ブトキシカルボニル)アミノ)-2-フルオロ-3-メ
チル-8-アザスピロ[4.5]デカン-8-カルボキシレートおよびベンジル(1S,
2R,3R)-1-((tert-ブトキシカルボニル)アミノ)-2-フルオロ-3-
メチル-8-アザスピロ[4.5]デカン-8-カルボキシレートの4:1混合物(1.
50g、3.57mmol)を添加し、得られた懸濁液をH2雰囲気下で16時間激しく
撹拌した。反応混合物をセライトのパッドに通して濾過し、パッドをDCMで洗浄し、揮
発物を減圧下で除去して、tert-ブチル((1R,2S,3R)-2-フルオロ-3
-メチル-8-アザスピロ[4.5]デカン-1-イル)カルバメートおよびtert-
ブチル((1S,2R,3R)-2-フルオロ-3-メチル-8-アザスピロ[4.5]
デカン-1-イル)カルバメートの4:1混合物(1.07g、3.74mmol)を得
た。このジアステレオマー混合物をさらに精製することなく次のステップにおいて使用し
た。MSm/z305.3(M+H)+。
2-((1S,2S,3R)-2-フルオロ-3-メチル-8-アザスピロ[4.5]デ
カン-1-イル)イソインドリン-1,3-ジオン

THF/MeOH(9:1 20mL)中の(2S,3R)-ベンジル2-フルオロ-
3-メチル-1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレート(1
.89g、5.92mmol;40%の(2R,3R)-2-フルオロ-3-メチル-1
-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレートを含有)の溶液に、
LiBH4(THF中2M、11.84mL、23.67mmol)を-78℃で添加し
た。得られた混合物を-78℃で30分間撹拌した。溶液に、飽和NH4Cl水溶液をゆ
っくりと添加し、混合物を室温まで加温した。混合物をEtOAc(3×)で抽出し、合
わせた有機相をブラインで洗浄し、MgSO4で脱水し、濾過し、揮発物を減圧下で除去
した。得られた残留物をシリカクロマトグラフィー(0から50%の勾配のEtOAc/
ヘプタン)により精製して、(1R,2S,3R)-ベンジル2-フルオロ-1-ヒドロ
キシ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート(970m
g、3.02mmol)を得た。MSm/z322.2(M+H)+。1H NMR (400 MHz,
メタノール-d4) δ 7.37-7.28 (m, 5 H), 5.10 (s, 2 H), 4.47 (dt, J=54.4, 4.7 Hz,
1 H), 3.86 (d, J=12.9 Hz, 2 H), 3.65 (dd, J=18.0, 4.7 Hz, 1 H), 3.20-3.03 (m, 2
H), 2.39-2.21 (m, 1 H), 2.20-2.10 (m, 1 H), 1.75-1.60 (m, 2 H), 1.45 (d, J=13.4
Hz, 1 H), 1.29 (d, J=13.1 Hz,1 H), 1.08 (d, J=7.1 Hz, 3 H), 0.96 (dd, J=13.3, 8.
5 Hz, 1 H).
THF(20mL)中のベンジル(1R,2S,3R)-2-フルオロ-1-ヒドロキ
シ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.03g
、3.20mmol)、トリフェニルホスフィン(1.68g、6.41mmol)およ
びフタルイミド(0.943g、6.41mmol)の溶液に、DIAD(1.25mL
、6.41mmol)を滴下添加した。得られた混合物を55℃で16時間撹拌した。室
温に冷却した後、反応混合物を、飽和NH4Cl水溶液を含有する分液漏斗に注ぎ入れ、
EtOAc(5×10mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し
、揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー(0から50%の勾配
のEtOAc/ヘプタン)により精製して、ベンジル(1S,2S,3R)-1-(1,
3-ジオキソイソインドリン-2-イル)-2-フルオロ-3-メチル-8-アザスピロ
[4.5]デカン-8-カルボキシレート(2.3g、UV吸収に基づく純度約50%)
を得た。この物質をさらに精製することなく次のステップにおいて使用した。MSm/z
451.2(M+H)+。
MeOH(15mL)中のPd/C(10重量%、170mg)の懸濁液を、H2雰囲
気(バルーン)下で5分間激しく撹拌した。次いで、MeOH(15mL)中のベンジル
(1S,2S,3R)-1-(1,3-ジオキソイソインドリン-2-イル)-2-フル
オロ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート(UV吸収
に基づく純度50%、3.20mmol)を添加し、得られた懸濁液をH2雰囲気下で2
.5時間激しく撹拌した。反応混合物をセライトのパッドに通して濾過し、パッドをDC
Mで洗浄し、揮発物を減圧下で除去して、2-((1S,2S,3R)-2-フルオロ-
3-メチル-8-アザスピロ[4.5]デカン-1-イル)イソインドリン-1,3-ジ
オン(294mg、0.929mmol)を得た。MSm/z317.2(M+H)+。
tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンアミド)-2-(ヒ
ドロキシメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート

“Suna, E. et al., (J. Org. Chem. 2014, 79, 3715-3724)における手順に従って、T
HF(8mL)、MeOH(0.811mL、20mmol)およびパラホルムアルデヒ
ド(660mg、22mmol)を厚肉圧力容器に添加し、反応混合物を100℃で70
分間加熱した。反応混合物を室温に冷却した。室温に冷却すると、容器の底に沈殿物が形
成された。追加のTHF(1.2mL)を添加して、メトキシメタノールの濃度をTHF
中2M溶液に調整した。MeOHが限定試薬であることに基づいて、定量的収率と推定さ
れた。
tert-ブチル1-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレー
ト(238g、939mmol)を含有するフラスコに、(R)-(+)-2-メチル-
2-プロパンスルフィンアミド(171g、1409mmol)をN2雰囲気下で入れた
。チタン(IV)エトキシド(985mL、4.70mol)を添加し、次いで混合物を
105℃に4時間加熱した。加熱マントルを取り外し、混合物を、機械式オーバーヘッド
撹拌機とN2導入口アダプターを有する250mLの添加漏斗とを備え、冷水浴中で冷却
された10Lの4口フラスコに、N2流下でFEPチューブカニューレを介してEtOA
c(6L)により真空移送した。水(288mL)を、添加漏斗を介して30~45分間
かけて滴下添加した結果、大量の薄黄色塩が沈殿した。懸濁液を、浴を取り外した状態で
15分間エージングした後、混合物全体をセライトに通して濾過し、EtOAc(2×1
L)で洗浄した。次いで、濾液を水(3×1L)で洗浄し、減圧下で濃縮した。濃縮およ
びヘプタン(2L)の逆添加時に、水を共沸除去し、それによりフラスコの内壁に塩の濁
った白色膜が析出した。薄茶色混合物を中間焼結ガラス漏斗に通して濾過した(EtOA
cおよびヘプタンですすいだ)。濾液を、EtOAcの大部分が除去されるまでさらに濃
縮し、追加のヘプタンを添加した(1L)。混合物を減圧下でさらに濃縮して沈殿物を生
成し、追加のヘプタン(500mL)を添加して混合物の流動性を保った。混合物を室温
で撹拌し、次いで氷浴で冷却した後、固体を真空濾過により単離した。固体を氷冷ヘプタ
ンで3回洗浄した。固体を減圧下で乾燥させて、(R,E)-tert-ブチル1-((
tert-ブチルスルフィニル)イミノ)-8-アザスピロ[4.5]デカン-8-カル
ボキシレートをクリーム色固体(408.9g、収率66.7%)として得た。1H NMR (
400 MHz, クロロホルム-d) δ ppm 3.93 (m, 2 H), 3.05 (m, 3 H), 2.71 (dt, J=19.6,
7.2 Hz, 1 H), 1.96-1.71 (m, 6 H), 1.49 (s, 9 H), 1.42 (m, 2 H), 1.27 (s, 9 H).
THF(27.3mL)中の(R,E)-tert-ブチル1-((tert-ブチル
スルフィニル)イミノ)-8-アザスピロ[4.5]デカン-8-カルボキシレート(1
.95g、5.47mmol)の溶液に、LiHMDS(THF中1M、6.02mL)
をN2雰囲気下-78℃でゆっくりと添加し、反応混合物を-78℃で10分間撹拌した
。メトキシメタノール(THF中2M、9.57mL)を約10分間かけて滴下添加し、
反応混合物を0℃まで加温し、撹拌をこの温度で1時間続けた。反応混合物を飽和NH4
Cl水溶液(20mL)で慎重に希釈し、EtOAcで抽出した。合わせた有機層をブラ
インで洗浄し、MgSO4で脱水し、減圧下で濃縮した。残留物をカラムクロマトグラフ
ィー(シリカゲル、0から100%のEtOAc/ヘプタン)により精製して、(E)-
tert-ブチル1-(((R)-tert-ブチルスルフィニル)イミノ)-2-(ヒ
ドロキシメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート(1.14
g)を白色固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 4.90 (t, J=5.1 Hz, 1
H), 3.83 (dd, J=14.1, 8.5 Hz, 2 H), 3.67 (dt, J=9.5, 4.5 Hz, 1 H), 3.39 (td, J=9
.7, 4.7 Hz, 1 H), 3.16 (dt, J=9.0, 5.8 Hz, 1 H), 2.84 (d, J=60.4 Hz, 2 H), 2.08-
1.85 (m, 2 H), 1.84-1.62 (m, 2 H), 1.62-1.49 (m, 1 H), 1.39 (s, 12 H), 1.13 (s,
9 H).
THF(12mL)およびMeOH(1.2mL)中の(E)-tert-ブチル1-
(((R)-tert-ブチルスルフィニル)イミノ)-2-(ヒドロキシメチル)-8
-アザスピロ[4.5]デカン-8-カルボキシレート(1.025g、2.65mmo
l)の溶液に、LiBH4(87mg、3.98mmol)をN2雰囲気下でおよび-7
8℃で添加した。反応混合物を-78℃で5分間撹拌し、次いで室温まで加温した。反応
混合物を飽和NaHCO3水溶液(5mL)で慎重に希釈し、EtOAcで抽出した。合
わせた有機層をブラインで洗浄し、MgSO4で脱水し、減圧下で濃縮した。残留物をカ
ラムクロマトグラフィー(シリカゲル、0から20%の勾配のMeOH/DCM)により
精製して、tert-ブチル1-((R)-1,1-ジメチルエチルスルフィンアミド)
-2-(ヒドロキシメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート
(778mg)を白色固体として得た。1H NMR(400MHz、DMSO-d6)
は、4.98ppmでアミンの診断NHシグナルの存在を示した(d、J=10.4Hz
、1H)。
(2R,3S)-3-メチル-2-(トリフルオロメチル)-8-アザスピロ[4.5]
デカン-1-アミン

THF(30mL)中の(S)-ベンジル3-メチル-1-オキソ-8-アザスピロ[
4.5]デカン-8-カルボキシレート(2.1g、6.97mmol)の-78℃溶液
に、LiHMDS(THF中1M、8.36mL、8.36mmol)を添加し、反応混
合物を-78℃で30分間撹拌した。クロロトリエチルシラン(1.23mL、7.32
mmol)を添加し、反応混合物を室温に加温し、この温度で18時間撹拌した。反応混
合物を、飽和NH4Cl水溶液(125mL)を含有する分液漏斗に注ぎ入れ、ヘプタン
(3×125mL)で抽出した。合わせた有機抽出物をMgSO4で脱水し、濾過し、減
圧下で濃縮した。残留物をシリカクロマトグラフィー(0から30%の勾配のEtOAc
/ヘプタン)により精製して、(S)-ベンジル3-メチル-1-((トリエチルシリル
)オキシ)-8-アザスピロ[4.5]デカ-1-エン-8-カルボキシレート(2.7
8g、6.69mmol)を透明油状物として得た。MSm/z416.3(M+H)+
。
DMF(43mL)中の(S)-ベンジル3-メチル-1-((トリエチルシリル)オ
キシ)-8-アザスピロ[4.5]デカ-1-エン-8-カルボキシレート(2.14g
、5.15mmol)、1-トリフルオロメチル-1,2-ベンゾヨードキソール-3-
(1H)-オン 60重量%、添加剤として40重量%のCelatom(登録商標)F
W-80含有(Togni’s II、4.07g、7.72mmol)およびCu(I
)SCN(63mg、0.515mmol)の混合物を、50℃で3日間撹拌した。室温
に冷却した後、反応混合物をEt2Oで希釈し、濾過し、フィルターパッドをEt2O(
150mL)で洗浄した。有機層を飽和NaHCO3水溶液(2×150mL)で洗浄し
、MgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグ
ラフィー(0から30%の勾配のEtOAc/ヘプタン)により精製して、(2R,3S
)-ベンジル3-メチル-1-オキソ-2-(トリフルオロメチル)-8-アザスピロ[
4.5]デカン-8-カルボキシレート(1.52g、4.12mmol)を透明油状物
として得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.30-7.45 (m, 5 H), 5.15 (s,
2 H),4.00 (dt, J=13.36, 4.99 Hz, 1 H), 3.79 (dt, J=13.49, 4.93 Hz, 1 H), 3.26-3
.39 (m, 1 H), 3.20 (ddd, J=13.49, 9.72, 3.64 Hz, 1 H), 2.55-2.70 (m, 1 H), 2.42-
2.55 (m, 1 H), 2.29 (dd, J=13.30, 6.53 Hz, 1 H), 1.81-1.92 (m, 1 H), 1.65 (ddd,
J=13.49, 9.47, 3.89 Hz, 1 H), 1.45-1.58 (m, 1 H), 1.38 (br. d, J=12.30 Hz, 2 H),
1.32 (d, J=6.27 Hz, 3 H). 19F NMR (376 MHz) δ ppm -66.32 (br. d, J=7.76 Hz).
MSm/z370.2(M+H)+。
EtOH(7.5mL)中のO-メチルヒドロキシルアミン塩酸塩(2.896g、3
4.7mmol)、(2R,3S)-ベンジル3-メチル-1-オキソ-2-(トリフル
オロメチル)-8-アザスピロ[4.5]デカン-8-カルボキシレート(854mg、
2.312mmol)およびピリジン(3.74mL、46.2mmol)の混合物を、
90℃で18時間撹拌した。室温に冷却した後、反応混合物を、H2O(60mL)およ
び飽和CuSO4水溶液(60mL)を含有する分液漏斗に注ぎ入れ、これをEt2O(
3×120mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を
減圧下で除去した。残留物をシリカクロマトグラフィー(0から30%の勾配のEtOA
c/ヘプタン)により精製して、(2R,3S)-ベンジル1-(メトキシイミノ)-3
-メチル-2-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-8-カルボ
キシレートの2つのE/Z立体異性体の混合物(750mg、1.882mmol)を透
明油状物として得た。MSm/z399.1(M+H)+。
MeOH(2mL)中のPd/C(10重量%、68.8mg)の懸濁液を、H2雰囲
気(バルーン)下室温で5分間激しく撹拌した。この懸濁液に、MeOH(4mL)中の
(2R,3S)-ベンジル1-(メトキシイミノ)-3-メチル-2-(トリフルオロメ
チル)-8-アザスピロ[4.5]デカン-8-カルボキシレートのE/Z異性体(51
5mg、1.293mmol)の溶液を添加し、得られた混合物をH2雰囲気下で30分
間激しく撹拌した。反応混合物をセライトのパッドに通して濾過し、DCM(70mL)
で洗浄した。濾液を減圧下で濃縮して、(2R,3S)-3-メチル-2-(トリフルオ
ロメチル)-8-アザスピロ[4.5]デカン-1-オンO-メチルオキシムのE/Z異
性体の混合物(351mg、1.275mmol)を透明油状物として得た。MSm/z
265.1(M+H)+。
MeCN(1.5mL)中の(2R,3S)-3-メチル-2-(トリフルオロメチル
)-8-アザスピロ[4.5]デカン-1-オンO-メチルオキシムのE/Z異性体の混
合物(210mg、0.795mmol)、Et3N(0.886mL、6.36mmo
l)およびBnBr(0.378mL、3.18mmol)の溶液を、室温で1.5時間
撹拌した。反応混合物を、H2O(25mL)および飽和NH4Cl水溶液(25mL)
を含有する分液漏斗に注ぎ入れ、DCM(3×50mL)で抽出した。合わせた有機抽出
物をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマト
グラフィー(0から50%の勾配のEtOAc/ヘプタン)により精製して、(2R,3
S)-8-ベンジル-3-メチル-2-(トリフルオロメチル)-8-アザスピロ[4.
5]デカン-1-オンO-メチルオキシムのE/Z異性体の混合物(219mg、0.6
18mmol)を透明油状物として得た。MSm/z355.2(M+H)+。
THF(1mL)中の(2R,3S)-8-ベンジル-3-メチル-2-(トリフルオ
ロメチル)-8-アザスピロ[4.5]デカン-1-オンO-メチルオキシムのE/Z異
性体(150mg、0.423mmol)の0℃溶液に、BH3-THF錯体(THF中
1M、6.35mL、6.35mmol)を滴下添加し、反応混合物を10分間かけて室
温に加温し、80℃に加熱し、この温度で24時間撹拌した。0℃に冷却した後、反応混
合物をH2O(5mL)で慎重に希釈した。ガスの発生が停止した後(5分)、反応混合
物を室温に加温し、NaOH水溶液(2M、3mL、6mmol)を添加し、反応混合物
を80℃で2時間撹拌した。反応混合物を、NaOH水溶液(1M、20mL)を含有す
る分液漏斗に注ぎ入れ、DCM(3×20mL)で抽出した。合わせた有機抽出物をMg
SO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をHPLC(勾配溶出、水
中45~70%アセトニトリル、5mM NH4OH修飾剤)により精製して、(2R,
3S)-8-ベンジル-3-メチル-2-(トリフルオロメチル)-8-アザスピロ[4
.5]デカン-1-アミンのエピマーの混合物(81mg、0.248mmol)を橙色
油状物として得た。MSm/z327.1(M+H)+。
窒素雰囲気下、MeOH(2mL)中の(2R,3S)-8-ベンジル-3-メチル-
2-(トリフルオロメチル)-8-アザスピロ[4.5]デカン-1-アミンのエピマー
(90.6mg、0.278mmol)の溶液に、Pd/C(10重量%、59.1mg
、0.056mmol)を添加した。反応混合物をH2雰囲気(バルーン)下で26時間
激しく撹拌した。反応混合物をDCMで希釈し、セライトで濾過し、パッドをDCM(1
00mL)で洗浄した。揮発物を減圧下で除去して、(2R,3S)-3-メチル-2-
(トリフルオロメチル)-8-アザスピロ[4.5]デカン-1-アミンのエピマーの混
合物(68mg、0.276mmol)を茶色油状物として得た。MSm/z237.2
(M+H)+。
2,8-ジアザスピロ[4.5]デカ-1-エン-1-アミン塩酸塩

室温のDCM(3mL)中のtert-ブチル2-オキソ-1,8-ジアザスピロ[4
.5]デカン-8-カルボキシレート(300mg、1.180mmol)の溶液に、五
硫化リン(110mg、0.495mmol)、続いてヘキサメチルジシロキサン(2.
26mL、10.6mmol)を添加した。反応物を室温で3時間撹拌し、次いでEtO
Acで希釈し、セライトに通して濾過した。濾液を減圧下で濃縮した。粗生成物をシリカ
クロマトグラフィー(0から80%の勾配のEtOAc/ヘプタン)により精製して、t
ert-ブチル1-チオキソ-2,8-ジアザスピロ[4.5]デカン-8-カルボキシ
レート(0.290g、1.07mmol)を白色固体として得た。1H NMR (400 MHz, D
MSO-d6) δ ppm 10.39 (s, 1 H), 3.66 (dt, J=13.6, 4.9 Hz, 2 H), 3.09 (s, 2 H), 2.
78 (t, J=7.8 Hz, 2 H), 1.95 (t, J=7.8 Hz, 2 H), 1.57 (dd, J=6.6, 4.8 Hz, 4 H), 1
.39 (s, 9 H).MSm/z271(M+H)+。
THF(3mL)中の1-チオキソ-2,8-ジアザスピロ[4.5]デカン-8-カ
ルボキシレート(100mg、0.370mmol)の溶液に、ヨードメタン(0.23
1mL、3.70mmol)を滴下添加した。得られた溶液を室温で16時間撹拌した。
反応混合物の色はゆっくりと黄色が強くなり、割り当てられた反応時間撹拌した後、結果
として薄黄色沈殿物を得た。反応混合物を濃縮し、真空下で乾燥させて、黄色固体を得た
。黄色固体をMeOH(2mL)に溶かし、アンモニア(MeOH中7M溶液、3mL)
で処理し、密封管内で100℃に8時間加熱した。反応物を室温に冷却し、減圧下で濃縮
して、固体を得、これをMeCNと共に超音波処理し、濾過した。濾液を減圧下で濃縮し
、残留物をシリカクロマトグラフィー(0から30%の勾配のMeOH/DCM)により
精製して、tert-ブチル1-アミノ-2,8-ジアザスピロ[4.5]デカ-1-エ
ン-8-カルボキシレート(87mg、0.343mmol)を得た。1H NMR (400 MHz,
DMSO-d6) δ ppm 9.38 (s, 1 H), 8.81 (d, J=25.2 Hz, 2 H), 3.98 (s, 2 H), 3.55 (t
, J=7.0 Hz, 2 H), 2.82 (s, 2 H), 2.12 (t, J=7.1 Hz, 2 H), 1.74 (td, J=12.9, 4.7
Hz, 2 H), 1.57 (d, J=12.7 Hz, 2H), 1.41 (s, 9 H).MSm/z254(M+H)+。
DCM(3mL)中のtert-ブチル1-アミノ-2,8-ジアザスピロ[4.5]
デカ-1-エン-8-カルボキシレート(86mg、0.339mmol)の溶液に、H
Cl(ジオキサン中4M溶液、0.5mL、2.0mmol)を室温で添加し、反応物を
16時間撹拌した。反応混合物を濃縮し、残留物をMeCNで摩砕し、濾過して、2,8
-ジアザスピロ[4.5]デカ-1-エン-1-アミン(57.7mg、0.254mm
ol)を黄褐色固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 9.64 (s, 1 H), 9.
39-9.23 (m, 1 H), 9.15 (s, 1 H), 9.07 (s, 1 H), 8.70 (d, J=12.5 Hz, 1 H), 3.54 (
t, J=6.9 Hz, 2 H), 3.32 (d, J=13.3 Hz, 2 H), 3.05-2.88 (m, 2 H), 2.18 (t, J=6.9
Hz, 2 H), 2.01 (td, J=13.7, 4.3 Hz, 2 H), 1.80 (d, J=13.8 Hz, 2 H).MSm/z1
54(M+H)+。
(4R)-4-アミノ-2-メチル-8-アザスピロ[4.5]デカン-2-オール

H2O(5mL)中の(2R,4R)-4-アミノ-8-アザスピロ[4.5]デカン
-2-オール二塩酸塩(623mg、2.56mmol)、Na2CO3(1.36g、
12.80mmol)およびクロロギ酸ベンジル(1.05g、6.14mmol)の混
合物を、室温で30分間激しく撹拌した。THF(0.5mL)を添加し、得られた混合
物を室温で18時間撹拌した。混合物を水およびDCMで希釈した。分離した水層をDC
M(2×10mL)で抽出した。合わせた有機相をNa2SO4で脱水し、濾過し、減圧
下で濃縮した。得られた残留物をシリカクロマトグラフィー(0から100%の勾配のE
tOAc/ヘプタン)により精製して、(1R,3R)-ベンジル1-(((ベンジルオ
キシ)カルボニル)アミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8-
カルボキシレート(940mg、2.14mmol)を白色泡状物として得た。MSm/
z439.3(M+H)+。
DCM(6mL)中の(1R,3R)-ベンジル1-(((ベンジルオキシ)カルボニ
ル)アミノ)-3-ヒドロキシ-8-アザスピロ[4.5]デカン-8-カルボキシレー
ト(440mg、1.003mmol)およびデス-マーチンペルヨージナン(638m
g、1.505mmol)の混合物を、0℃で1時間および室温で18時間撹拌した。反
応混合物を飽和NaHCO3水溶液/飽和Na2S2O3水溶液(1:1、25mL)で
希釈した。分離した水相をDCM(3×15mL)で抽出した。合わせた有機相をブライ
ンで洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮した。得られた残留物をシリカ
クロマトグラフィー(0から70%の勾配のEtOAc/ヘプタン)により精製して、(
R)-ベンジル1-(((ベンジルオキシ)カルボニル)アミノ)-3-オキソ-8-ア
ザスピロ[4.5]デカン-8-カルボキシレート(415mg、0.951mmol)
を白色泡状物として得た。MSm/z437.2(M+H)+。
THF(15mL)中のMeLi(THF中1.2M、2.61mL、3.13mmo
l)の溶液に、THF(5mL)中の(R)-ベンジル1-(((ベンジルオキシ)カル
ボニル)アミノ)-3-オキソ-8-アザスピロ[4.5]デカン-8-カルボキシレー
ト(415mg、0.951mmol)を-30から-40℃で滴下添加した。得られた
混合物を-30から-40℃で20分間撹拌した。混合物をNaHSO4(H2O中10
%溶液)およびEtOAcで希釈し、激しく撹拌しながら室温まで加温した。混合物を、
飽和NaHCO3水溶液を含有する分液漏斗に注ぎ入れ、相を分離した。水相をEtOA
c(15mL)でさらに抽出し、合わせた有機相をNa2SO4で脱水し、濾過し、減圧
下で濃縮した。水(10mL)およびTHF(1mL)中の得られた残留物(313mg
)、Na2CO3(498mg、4.70mmol)およびクロロギ酸ベンジル(295
mg、1.729mmol)の溶液を、室温で3日間激しく撹拌した。混合物をEtOA
cで希釈し、分離した水相をEtOAc(3×15mL)で抽出した。合わせた有機相を
減圧下で濃縮した。得られた残留物をシリカクロマトグラフィー(0から50%の勾配の
EtOAc/ヘプタン)により精製して、2つのジアステレオマー:ジアステレオマーA
(112mg、0.25mmol)を無色半固体、MSm/z453.3(M+H)+と
して、ジアステレオマーB(45mg、0.010mmol)を白色泡状物、MSm/z
453.3(M+H)+として得た。
MeOH(8mL)中のジアステレオマーA(50mg、0.11mmol)およびP
d/C(10重量%;12mg、0.011mmol)の混合物を、水素雰囲気下で2時
間激しく撹拌した。セライトを添加し、混合物をセライトのパッドに通して濾過し、続い
てDCMで洗浄した。濾液を減圧下で濃縮して、(4R)-4-アミノ-2-メチル-8
-アザスピロ[4.5]デカン-2-オールを無色固体として得、これをさらに精製する
ことなく使用した。MSm/z185.2(M+H)+。
マーBを出発物質として使用して合成した。
2-((1S,2S,3R)-2-フルオロ-3-メチル-8-アザスピロ[4.5]デ
カン-1-イル)イソインドリン-1,3-ジオン

THF(16.5mL)中のベンジル(1R,2S,3R)-2-フルオロ-1-ヒド
ロキシ-3-メチル-8-アザスピロ[4.5]デカン-8-カルボキシレート(760
mg、2.365mmol)の溶液に、トリフェニルホスフィン(744mg、2.85
mmol)およびDIAD(0.557mL、2.84mmol)を添加した。得られた
混合物を0℃で20分間撹拌し、ジフェニルホスホルアジデート(0.787mL、3.
55mmol)を添加した。反応混合物を室温まで加温し、この温度で18時間撹拌した
。反応混合物を、EtOAc(30mL)を含有する分液漏斗に注ぎ入れ、有機相を飽和
NH4Cl水溶液およびブラインで洗浄した。合わせた有機相をMgSO4で脱水し、濾
過し、揮発物を減圧下で除去した。得られた残留物をシリカクロマトグラフィー(0から
50%の勾配のEtOAc/ヘプタン)により精製して、ベンジル(1S,2S,3R)
-1-アジド-2-フルオロ-3-メチル-8-アザスピロ[4.5]デカン-8-カル
ボキシレート(432mg、1.247mmol)を得た。MSm/z347.2(M+
H)+。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.43-7.31 (m, 5 H), 5.15 (s, 2 H
), 4.48 (dt, J=54.4, 7.5 Hz, 1 H), 3.93 (s, 2 H), 3.61 (dd, J=16.1, 6.9 Hz, 1 H)
, 3.13-2.95 (m, 2 H), 2.31-2.13 (m, 1 H), 1.96 (dd, J=13.1, 9.3 Hz, 1 H), 1.81-1
.64 (m, 2 H), 1.47 (s, 1 H), 1.32-1.19 (m, 2 H), 1.16 (d, J=6.7 Hz, 3 H).
EtOH(12.2mL)中のPd/C(10重量%、65mg)およびベンジル(1
S,2S,3R)-1-アジド-2-フルオロ-3-メチル-8-アザスピロ[4.5]
デカン-8-カルボキシレート(423mg、1.221mmol)の懸濁液を、H2雰
囲気(バルーン)下で16時間激しく撹拌した。反応混合物をセライトのパッドに通して
濾過し、揮発物を減圧下で除去して、粗製の(1S,2S,3R)-2-フルオロ-3-
メチル-8-アザスピロ[4.5]デカン-1-アミン(235mg、0.966mmo
l)を得、これをさらに精製することなく使用した。MSm/z317.2(M+H)+
。1H NMR (400 MHz, クロロホルム-d) δ ppm 4.15 (dt, J=55.5, 8.1 Hz, 1 H), 2.95 (
dt, J=12.5, 3.7 Hz, 2 H), 2.87 (dd, J=16.6, 8.0 Hz, 1 H), 2.74 (tdd, J=12.4, 7.3
, 2.8 Hz, 2 H), 2.19-2.02 (m, 1 H), 1.95 (dd, J=13.4, 8.4 Hz, 1 H), 1.71-1.48 (m
, 4 H), 1.34-1.23 (m, 3 H), 1.18-1.09 (m, 4 H).
ラセミ(1S,2S,3S)-1-アミノ-8-アザスピロ[4.5]デカン-2,3-
ジオールトリフルオロ酢酸塩

t-ブトキシカルボニル)アミノ)-2,3-ジヒドロキシ-8-アザスピロ[4.5]
デカン-8-カルボキシレート(21mg、0.054mmol)の溶液に、トリフルオ
ロ酢酸(0.1mL、1.298mmol)を添加し、混合物を30℃で30分間撹拌し
た。揮発物を減圧下で除去して、粗製のラセミ(1S,2S,3S)-1-アミノ-8-
アザスピロ[4.5]デカン-2,3-ジオールトリフルオロ酢酸塩(単一ジアステレオ
マー)を透明油状物として得、これをさらに精製することなく直接使用した。MSm/z
187.1(M+H)+。
(3S,4S)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-アミ
ン

(R)-1,1-ジメチルエチルスルフィンアミノ)-3-メチル-2-オキサ-8-ア
ザスピロ[4.5]デカン-8-カルボキシレート(9.4g、25.1mmol)に、
HCl(ジオキサン中4M溶液、35mL、140mmol)をゆっくりと添加した。反
応混合物を50℃で40分間加熱し、撹拌をN2流下で18時間続けた。反応混合物を減
圧下で濃縮した。残留物をMeOHおよびMeCNで希釈し、減圧下で濃縮した。残留物
を超音波処理下MeCNで摩砕した。固体を濾過し、高真空中で乾燥させて、(3S,4
S)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-アミン塩酸塩を
白色粉末として得た。
ミン塩酸塩は、(3S,4S)-3-メチル-2-オキサ-8-アザスピロ[4.5]デ
カン-4-アミン塩酸塩について記載した通りの手順に従って、(R)-2-((ter
t-ブチルジメチルシリル)オキシ)プロパナールから出発し、(S)-tert-ブチ
ルスルフィンアミドを使用して調製した。
(R)-2-メチル-N-((R)-8-アザスピロ[4.5]デカン-1-イル)プロ
パン-2-スルフィンアミド

,1-ジメチルエチルスルフィンアミノ)-8-アザスピロ[4.5]デカン-8-カル
ボキシレート(1g、2.79mmol)の撹拌溶液に、硫酸(0.623mL、11.
2mmol)をゆっくりと添加した。反応混合物を室温で1時間撹拌し、pH=12にな
るまでNaOH水溶液で希釈した。混合物をDCM(3×30mL)で抽出した。合わせ
た有機層を残留水の除去のために相分離器に通過させ、減圧下で濃縮して、粗製の(R)
-2-メチル-N-((R)-8-アザスピロ[4.5]デカン-1-イル)プロパン-
2-スルフィンアミド(642mg)を得、これをさらに精製することなく直接使用した
。
(1R,3R)-3-メチル-8-アザスピロ[4.5]デカン-1-アミン

1,1-ジメチルエチルスルフィンアミノ)-3-メチル-8-アザスピロ[4.5]デ
カン-8-カルボキシレート(182mg、0.448mmol)およびHCl(ジオキ
サン中4M溶液、6.7mL)の混合物に、MW中140℃で23時間照射を行った。混
合物を減圧下で濃縮した。残留物をEt2O(10mL)中に懸濁させた。透明溶液を除
去し、残った固体を減圧下で乾燥させて、粗製の(1R,3R)-3-メチル-8-アザ
スピロ[4.5]デカン-1-アミン塩酸塩(126mg)を灰クリーム色固体として得
た。
は、上記手順または上記手順を変更したものを用い、(1R,3S)-ベンジル1-((
R)-1,1-ジメチルエチルスルフィンアミノ)-3-メチル-8-アザスピロ[4.
5]デカン-8-カルボキシレートを出発物質として使用して合成した。
(R)-8-アザスピロ[4.5]デカン-1-アミン

スルフィンアミノ)-8-アザスピロ[4.5]デカン-8-カルボキシレート(4.6
6g、13mmol)の溶液に、HCl(ジオキサン中4M、32.5mL、130mm
ol)を添加した。混合物を50℃で1時間撹拌した。揮発物を減圧下で除去し、残留物
をトルエン(5mL)およびEt2O(10mL)中に懸濁させ、混合物を減圧下で濃縮
して、粗製の(R)-8-アザスピロ[4.5]デカン-1-アミン塩酸塩を得、これを
さらに精製することなく直接使用した。MSm/z155.1(M+H)+。
されたアミンを使用して合成した。


tert-ブチル((1-(4-アミノ-5-ヨード-1-メチル-6-オキソ-1,6
-ジヒドロピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバ
メート

DMF(15mL)中の6-アミノ-3-メチルピリミジン-2,4(1H,3H)-
ジオン(1.0g、7.09mmol)、tert-ブチル((4-メチルピペリジン-
4-イル)メチル)カルバメート(1.78g、7.79mmol)、BOP(6.27
g、14.17mmol)およびDBU(5.34mL、35.4mmol)の混合物を
、室温で2時間撹拌した。得られた混合物を、飽和NH4Cl水溶液(25mL)、水(
25mL)を含有する分液漏斗に注ぎ入れ、Et2O(3×15mL)およびDCM(3
×15mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧
下で除去し、残留物をシリカクロマトグラフィー(0から10%の勾配のMeOH/DC
M)により精製して、tert-ブチル((1-(4-アミノ-1-メチル-6-オキソ
-1,6-ジヒドロピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル
)カルバメート(3.6g;不純)を薄黄色固体として得た。MSm/z351.9(M
+H)+。この化合物をさらに精製することなく次のステップにおいて使用した。
DMF(14mL)中の粗製のtert-ブチル((1-(4-アミノ-1-メチル-
6-オキソ-1,6-ジヒドロピリミジン-2-イル)-4-メチルピペリジン-4-イ
ル)メチル)カルバメート(理論上7.09mmol)およびNIS(1.76g、7.
80mmol)の混合物を、室温で1時間撹拌した。得られた混合物を、飽和Na2S2
O3水溶液(25mL)、飽和NH4Cl水溶液(25mL)および水(25mL)を含
有する分液漏斗に注ぎ入れ、EtOAc(3×15mL)で抽出した。合わせた有機相を
MgSO4で脱水し、濾過し、揮発物を減圧下で除去し、残留物をシリカクロマトグラフ
ィー(0から10%の勾配のMeOH/DCM)により精製して、不純なtert-ブチ
ル((1-(4-アミノ-5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピリ
ミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(2.5
4g)を得た。MSm/z478.2(M+H)+。この化合物をさらに精製することな
く次のステップにおいて使用した。
tert-ブチル((3S,4S)-8-(4-アミノ-5-ヨード-1-メチル-6-
オキソ-1,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オキサ-8-アザ
スピロ[4.5]デカン-4-イル)カルバメート
DMF(15mL)中の6-アミノ-3-メチルピリミジン-2,4(1H,3H)-
ジオン(1g、7.09mmol)、(3S,4S)-3-メチル-2-オキサ-8-ア
ザスピロ[4.5]デカン-4-アミン(1.81g、7.44mmol)、BOP(6
.27g、14.17mmol)およびDBU(7.48mL、49.6mmol)の混
合物を、室温で60時間撹拌した。得られた混合物をHPLC(勾配溶出、水中2~12
%MeCN、5mM NH4OH修飾剤)により精製して、6-アミノ-2-((3S,
4S)-4-アミノ-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-
イル)-3-メチルピリミジン-4(3H)-オン(2.08g、7.09mmol)を
得た。MSm/z294.3(M+H)+。
DMF(14mL)中の6-アミノ-2-((3S,4S)-4-アミノ-3-メチル
-2-オキサ-8-アザスピロ[4.5]デカン-8-イル)-3-メチルピリミジン-
4(3H)-オン(2.08g、7.09mmol)、Boc2O(1.55g、7.0
9mmol)およびDIPEA(2.5mL、14.18mmol)の混合物を、室温で
1時間撹拌した。得られた混合物を、飽和NH4Cl水溶液(75mL)を含有する分液
漏斗に注ぎ入れ、これをDCM(3×15mL)で抽出した。合わせた有機相をMgSO
4で脱水し、濾過し、揮発物を減圧下で除去して、tert-ブチル((3S,4S)-
8-(4-アミノ-1-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)
-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カルバメート
(2.79g、7.09mmol)を得た。MSm/z394.4(M+H)+。この化
合物をさらに精製することなく次のステップにおいて使用した。
DMF(14mL)中のtert-ブチル((3S,4S)-8-(4-アミノ-1-
メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オキ
サ-8-アザスピロ[4.5]デカン-4-イル)カルバメート(2.79g、7.09
mmol)およびNIS(1.76g、7.80mmol)の混合物を、室温で1時間撹
拌した。得られた混合物を、飽和Na2S2O3水溶液(25mL)、飽和NH4Cl水
溶液(25mL)および水(25mL)を含有する分液漏斗に注ぎ入れ、DCM(3×2
0mL)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で
除去し、残留物をシリカクロマトグラフィー(0から5%の勾配のMeOH/DCM)に
より精製して、tert-ブチル((3S,4S)-8-(4-アミノ-5-ヨード-1
-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オ
キサ-8-アザスピロ[4.5]デカン-4-イル)カルバメート(1.51g、2.9
1mmol)を得た。MSm/z521.0(M+H)+。
物質および中間体を使用して合成した。


tert-ブチル((1-(5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピ
リミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート

THF(4mL)中の2,4-ジクロロ-5-ヨードピリミジン(1g、3.64mm
ol)およびNaOH水溶液(2M、2.73mL、5.46mmol)の混合物を、室
温で90時間撹拌した。混合物を、HCl水溶液(1M)を用いてpH1に酸性化した。
水層をEtOAc(2×)で抽出した。合わせた有機層をMgSO4で処理し、濾過し、
揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー(0%から10%の勾配
のMeOH/DCM)により精製して、2-クロロ-5-ヨードピリミジン-4(3H)
-オン(195mg、0.760mmol)を薄黄色固体として得た。1H NMR (400 MHz,
メタノール-d4) δ ppm 8.46 (s, 1 H).MSm/z256.7(M+H)+。
DMF(39mL)中の2-クロロ-5-ヨードピリミジン-4(3H)-オン(1g
、3.90mmol)の溶液に、LDA(THF/ヘプタン/エチルベンゼン中2.5M
、2.92mL、5.85mmol)を0℃で滴下添加した。混合物を0℃で5分間撹拌
した。ヨウ化メチル(364μL、5.85mmol)を添加し、混合物を室温まで加温
し、この温度で18時間撹拌した。混合物を水(20mL)で慎重に希釈した。水層をE
tOAc(2×)で抽出した。合わせた有機層を飽和NH4Cl水溶液(2×)、続いて
ブラインで洗浄した。有機層をMgSO4で処理し、濾過し、揮発物を減圧下で除去した
。残留物質をシリカクロマトグラフィー(20から70%の勾配のEtOAc/ヘプタン
)により精製して、2-クロロ-5-ヨード-3-メチルピリミジン-4(3H)-オン
(663mg)を薄茶色固体として得た。1H NMR (400 MHz, DMSO-d6) δ ppm 8.33 (s,
1 H), 3.58 (s, 3 H).MSm/z270.8(M+H)+。
DMF(1mL)中の2-クロロ-5-ヨード-3-メチルピリミジン-4(3H)-
オン(77.1mg、0.285mmol)、tert-ブチル((4-メチルピペリジ
ン-4-イル)メチル)カルバメート(78mg、0.342mmol)およびDIPE
A(0.149mL、0.855mmol)の混合物に、マイクロ波反応器内120℃で
2時間照射を行った。室温に冷却した後、反応混合物をEtOAcで希釈し、これを飽和
NH4Cl水溶液(2×)、続いてブラインで洗浄した。有機層をMgSO4で脱水し、
濾過し、減圧下で除去した。残留物をシリカクロマトグラフィー(0から100%の勾配
のEtOAc/ヘプタン)により精製して、tert-ブチル((1-(5-ヨード-1
-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)-4-メチルピペリジ
ン-4-イル)メチル)カルバメート(76.9mg)を白色固体として得た。1H NMR (
400 MHz, クロロホルム-d) δ ppm 8.17 (s, 1 H), 4.68 (s, 1 H), 3.54 (s, 3 H), 3.4
8-3.39 (m, 2 H), 3.35-3.22 (m, 2 H), 1.71-1.57 (m, 2 H), 1.57-1.39 (m, 11 H), 1.
34-1.21 (m, 2 H), 1.03 (s, 3 H).MSm/z463.0(M+H)+。
tert-ブチル((1-(5-ブロモ-4-メトキシピリミジン-2-イル)-4-メ
チルピペリジン-4-イル)メチル)カルバメート

-2-クロロ-4-メトキシピリミジン(200mg、0.895mmol)およびte
rt-ブチル((4-メチルピペリジン-4-イル)メチル)カルバメート(225mg
、0.985mmol)の混合物を、120℃に2時間加熱した。反応混合物を室温に冷
却し、EtOAc(50mL)で希釈し、ブライン(50mL)で洗浄した。分離した水
層をEtOAc(50mL)で抽出した。合わせた有機層をNa2SO4で脱水し、濾過
し、減圧下で濃縮して、粗製のtert-ブチル((1-(5-ブロモ-4-メトキシピ
リミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(42
4mg)を茶橙色固体として得、これをさらに精製することなく直接使用した。MSm/
z417.2(M+H)+。
tert-ブチル((1-(5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピ
リジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート

EtOH(11.6mL)中の6-クロロピリジン-2(1H)-オン(500mg、
3.86mmol)、K2CO3(800mg、5.79mmol)およびヨウ化メチル
(0.360mL、5.79mmol)の混合物を、70℃で18時間撹拌した。反応混
合物を室温に冷却し、揮発物を減圧下で除去し、残留物を水中に懸濁させた。水層をEt
OAc(2×)で抽出した。合わせた有機層をMgSO4で脱水し、濾過し、揮発物を減
圧下で除去した。残留物をシリカクロマトグラフィー(0から10%の勾配のMeOH/
DCM)により精製して、6-クロロ-1-メチルピリジン-2(1H)-オン(472
mg、3.29mmol)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 7.39 (dd, J=9.2
, 7.3 Hz, 1 H), 6.49 (dd, J=7.3, 1.2 Hz, 1 H), 6.41 (dd, J=9.2, 1.1 Hz, 1 H), 3.
55 (s, 3 H).MSm/z144.0(M+H)+。
DMF(1mL)中の6-クロロ-1-メチルピリジン-2(1H)-オン(55mg
、0.383mmol)、DIPEA(200μL、1.15mmol)およびtert
-ブチル((4-メチルピペリジン-4-イル)メチル)カルバメート(96mg、0.
421mmol)の溶液に、マイクロ波反応器内140℃で2時間照射を行った。室温に
冷却した後、反応混合物をEtOAcで希釈した。有機層を飽和NH4Cl水溶液(2×
)、続いてブラインで洗浄した。有機層をMgSO4で脱水し、濾過し、揮発物を減圧下
で除去した。残留物をシリカクロマトグラフィー(0から100%の勾配のEtOAc/
ヘプタン)により精製して、tert-ブチル((4-メチル-1-(1-メチル-6-
オキソ-1,6-ジヒドロピリジン-2-イル)ピペリジン-4-イル)メチル)カルバ
メート(53mg、0.158mmol)を得た。1H NMR (400 MHz, DMSO-d6) δ ppm 7
.33 (dd, J=8.9, 7.4 Hz, 1 H), 6.94 (t, J=6.3 Hz, 1 H), 6.10-6.04 (m, 1 H), 3.35
(s, 3 H), 2.95-2.85 (m, 4 H), 2.77 (s, 2 H), 1.56-1.47 (m, 2 H), 1.42-1.30 (m, 1
1 H), 0.89 (s, 3 H).MSm/z336.6(M+H)+
THF(2mL)中のtert-ブチル((4-メチル-1-(1-メチル-6-オキ
ソ-1,6-ジヒドロピリジン-2-イル)ピペリジン-4-イル)メチル)カルバメー
ト(53mg、0.158mmol)およびNIS(41.2mg、0.174mmol
)の溶液を、室温で18時間撹拌した。混合物をEtOAcで希釈した。有機層を飽和N
a2S2O3水溶液:飽和NH4Cl水溶液(1:1)、続いてブラインで2回洗浄した
。有機層をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカク
ロマトグラフィー(0から100%の勾配のEtOAc/ヘプタン)により精製して、t
ert-ブチル((1-(5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピリ
ジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(34.4
mg、0.075mmol)を得た。1H NMR (400 MHz, クロロホルム-d) δ ppm 7.79 (
d, J=7.8 Hz, 1 H), 5.57 (d, J=7.9 Hz, 1 H), 4.64 (s, 1 H), 3.50 (s, 3 H), 3.42 (
s, 1 H), 3.09-2.97 (m, 2 H), 2.96-2.85 (m, 2 H), 2.84-2.70 (m, 2 H), 1.62-1.50 (
m, 2 H), 1.46-1.30 (m, 11 H), 0.92 (s, 3 H).MSm/z462.0(M+H)+。
tert-ブチル((1-(5-ヨード-4-((4-メトキシベンジル)オキシ)ピリ
ミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート

DMF(50mL)中の、国際公開第2011022440号パンフレットにおける方
法に従って調製した2-クロロ-4-((4-メトキシベンジル)オキシ)ピリミジン(
118mg、0.519mmol)の溶液に、tert-ブチル((4-メチルピペリジ
ン-4-イル)メチル)カルバメート(130mg、0.519mmol)を添加した。
反応物を48時間撹拌し、EtOAc:水(1:1、100mL)中に希釈した。混合物
をEtOAc(3×)で抽出し、合わせた有機抽出物をブライン(3×)で洗浄し、Mg
SO4で脱水し、濾過し、減圧下で濃縮して、粗製のtert-ブチル((1-(4-(
(4-メトキシベンジル)オキシ)ピリミジン-2-イル)-4-メチルピペリジン-4
-イル)メチル)カルバメートを黄色油状物(202mg)として得、これをさらに精製
することなく使用した。MSm/z443(M+H)+。
MeCN(50mL)中の粗製のtert-ブチル((1-(4-((4-メトキシベ
ンジル)オキシ)ピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)
カルバメート(718mg)の溶液に、NIS(365mg、1.62mmol)を室温
で一度に添加した。反応物は色が橙色であるように見え、飽和Na2S2O3水溶液(5
mL)を添加し、反応物を濃縮して透明溶液を得た。混合物をDCM(100mL)およ
び飽和Na2S2O3水溶液(100mL)で希釈した。分離した水層をDCM(3×)
で抽出した。合わせた有機抽出物をブラインで洗浄し、MgSO4で脱水し、濾過し、減
圧下で濃縮して、tert-ブチル((1-(5-ヨード-4-((4-メトキシベンジ
ル)オキシ)ピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カル
バメート(800mg、1.41mmol)を白色泡状物として得た。MSm/z569
(M+H)+。
tert-ブチル((3S,4S)-8-(5-ブロモ-1,4-ジメチル-6-オキソ
-1,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オキサ-8-アザスピロ
[4.5]デカン-4-イル)カルバメート

AcOH(15mL)中の3,6-ジメチルピリミジン-2,4(1H,3H)-ジオ
ン(0.5g、3.57mmol)の混合物に、臭素(0.230mL、4.46mmo
l)を滴下添加した。混合物を室温で1時間撹拌した。反応混合物を飽和NaS2O3水
溶液(5mL)で希釈し、5分間激しく撹拌し、0.1M NaOH水溶液(10mL)
でさらに希釈し、15分間撹拌した。混合物をDCM(4×)で抽出し、合わせた有機層
をNa2SO4で脱水し、濾別し、減圧下で濃縮した。残留物をトルエン(3mL)中に
懸濁させ、混合物を濃縮し、減圧下で乾燥させて、粗製の5-ブロモ-3,6-ジメチル
ピリミジン-2,4(1H,3H)-ジオン(720mg)を白色固体として得、これを
さらに精製することなく直接使用した。MSm/z221.0(M+H)+。
DMF(3mL)中の5-ブロモ-3,6-ジメチルピリミジン-2,4(1H,3H
)-ジオン(0.3g、1.37mmol)、(3S,4S)-3-メチル-2-オキサ
-8-アザスピロ[4.5]デカン-4-アミンビス塩酸塩(0.350g、1.438
mmol)およびBOP(1.212g、2.74mmol)の混合物を、N2雰囲気下
で約10分間撹拌した。DBU(1.445mL、9.59mmol)を添加し、反応混
合物を終夜撹拌した。反応混合物を水(3mL)で希釈し、揮発性成分の約3/4を減圧
下で除去した。残留物をブラインおよびEtOAcで希釈し、分離した水層をEtOAc
で抽出した。合わせた有機層を水およびブラインで洗浄し、Na2SO4で脱水し、濾別
し、減圧下で濃縮して、粗製の2-((3S,4S)-4-アミノ-3-メチル-2-オ
キサ-8-アザスピロ[4.5]デカン-8-イル)-5-ブロモ-3,6-ジメチルピ
リミジン-4(3H)-オン(1.0g)を得、これをさらに精製することなく直接使用
した。MSm/z373.2(M+H)+。
DMF(5mL)中の粗製の2-((3S,4S)-4-アミノ-3-メチル-2-オ
キサ-8-アザスピロ[4.5]デカン-8-イル)-5-ブロモ-3,6-ジメチルピ
リミジン-4(3H)-オン(509mg、1.37mmol)の溶液に、Boc2O(
0.35mL、1.51mmol)およびDIPEA(0.526mL、3.01mmo
l)を添加した。混合物を室温でおよびN2雰囲気下で終夜撹拌した。反応混合物を飽和
NaHCO3水溶液およびEtOAcで希釈した。分離した水層をEtOAcで抽出した
。合わせた有機層を水およびブラインで洗浄し、Na2SO4で脱水し、濾過し、減圧下
で濃縮した。残留物をカラムクロマトグラフィー(シリカゲル、10から100%の勾配
のEtOAc/ヘプタン)により精製して、tert-ブチル((3S,4S)-8-(
5-ブロモ-1,4-ジメチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)
-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カルバメート
(200mg)を白色綿毛状固体として得た。MSm/z473.2(M+H)+。
6-アミノ-5-((2,3-ジクロロフェニル)チオ)-3-メチルピリミジン-2,
4(1H,3H)-ジオン

DMF(5mL)中の6-アミノ-3-メチルピリミジン-2,4(1H,3H)-ジ
オン(1.0g、7.09mmol)およびNBS(1.58g、8.86mmol)の
混合物を、室温で16時間撹拌した。得られた混合物を水(20mL)で希釈し、形成さ
れた固体を濾過し、続いて水(2×5mL)で洗浄して、6-アミノ-5-ブロモ-3-
メチルピリミジン-2,4(1H,3H)-ジオン(1.1g、5.0mmol)をオフ
ホワイトの固体として得た。MSm/z222.1(M+H)+。
ジオキサン(2mL)中の6-アミノ-5-ブロモ-3-メチルピリミジン-2,4(
1H,3H)-ジオン(200mg、0.909mmol)、2,3-ジクロロベンゼン
チオール(326mg、1.818mmol)、Cu(I)I(34.6mg、0.18
2mmol)、TMEDA(55μL、0.364mmol)およびK3PO4(579
mg、2.73mmol)の混合物を、100℃で20時間撹拌した。室温に冷却した後
、反応混合物をHPLC(勾配溶出、水中5~20%MeCN、5mM NH4OH修飾
剤)により精製して、6-アミノ-5-((2,3-ジクロロフェニル)チオ)-3-メ
チルピリミジン-2,4(1H,3H)-ジオン(120.0mg、0.377mmol
)を白色固体として得た。MSm/z318.2(M+H)+。
ールを使用して作製した。

6-アミノ-5-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)-3-(
(2-(トリメチルシリル)エトキシ)メチル)ピリミジン-2,4(1H,3H)-ジ
オン

ヘキサメチルジシラザン(5mL)中の6-アミノウラシル(1.0g、7.87mm
ol)および硫酸アンモニウム(52mg、0.393mmol)の混合物を、130℃
で16時間撹拌した。反応混合物を室温に冷却し、沈殿物を濾別し、揮発物を減圧下で除
去した。残留物をトルエン(10mL)に溶解し、SEMCl(2.1mL、11.80
mmol)を添加した。得られた混合物を室温で90分間撹拌した。揮発物を減圧下で除
去し、残留物をシリカクロマトグラフィー(0から15%の勾配のMeOH/DCM)に
より精製して、6-アミノ-3-((2-(トリメチルシリル)エトキシ)メチル)ピリ
ミジン-2,4(1H,3H)-ジオン(720mg、2.80mmol)を得た。1H N
MR (400 MHz, DMSO-d6) δ ppm 10.44 (s, 1 H), 6.32 (br. s, 2 H), 5.10 (s, 2 H), 4
.57 (d, J=2.02 Hz, 1 H), 3.42-3.60 (m, 2 H), 0.79-0.91 (m, 2 H), 0.00 (m, 9 H).
DMF(5mL)中の6-アミノ-3-((2-(トリメチルシリル)エトキシ)メチ
ル)ピリミジン-2,4(1H,3H)-ジオン(720mg、2.80mmol)およ
びNBS(747mg、4.20mmol)の混合物を、室温で16時間撹拌した。得ら
れた混合物を水(20mL)で希釈し、形成された固体を濾別し、続いて水(2×10m
L)で洗浄して、6-アミノ-5-ブロモ-3-((2-(トリメチルシリル)エトキシ
)メチル)ピリミジン-2,4(1H,3H)-ジオン(941mg、2.80mmol
)を得た。MSm/z336.1(M+H)+。
ジオキサン(5mL)中の6-アミノ-5-ブロモ-3-((2-(トリメチルシリル
)エトキシ)メチル)-ピリミジン-2,4(1H,3H)-ジオン(941mg、2.
80mmol)、Cu(I)I(57mg、0.297mmol)、TMEDA(90μ
L、0.595mmol)およびK3PO4(947mg、4.46mmol)の混合物
を、100℃で14時間撹拌した。室温に冷却した後、反応混合物をシリカクロマトグラ
フィー(0から10%の勾配のMeOH/DCM)により精製して、6-アミノ-5-(
(2-(トリフルオロメチル)ピリジン-3-イル)チオ)-3-((2-(トリメチル
シリル)エトキシ)メチル)ピリミジン-2,4(1H,3H)-ジオン(400mg、
0.921mmol)を得た。1H NMR (400 MHz, DMSO-d6) δppm 10.98 (s, 1 H), 8.47
(dd, J=4.29, 1.01 Hz, 1 H), 7.45-7.65 (m, 2 H), 6.91 (br. s, 2 H), 5.18 (s, 2 H
), 3.42-3.71 (m, 2 H), 0.75-1.01 (m, 2 H), -0.02-0.02 (m, 9 H). 19F NMR (376 MH
z, DMSO-d6) δppm -63.48.
2-クロロ-5-((2,3-ジクロロフェニル)チオ)-4-メトキシピリミジン

ジオキサン(3mL)中の6-クロロ-3-ヨード-2-メトキシピリジン(100m
g、0.371mmol)、2,3-ジクロロベンゼンチオール(100mg、0.55
7mmol)、1,10-フェナントロリン(26.7mg、0.148mmol)、C
u(I)I(14.1mg、0.074mmol)およびCs2CO3(242mg、0
.742mmol)の混合物を、100℃で1時間撹拌した。反応混合物を室温に冷却し
、EtOAcで希釈し、セライトのパッドに通して濾過した。有機層を飽和NH4Cl水
溶液で洗浄した。有機層をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残
留物をシリカクロマトグラフィー(0から100%の勾配のEtOAc/ヘプタン)によ
り精製して、2-クロロ-5-((2,3-ジクロロフェニル)チオ)-4-メトキシピ
リミジン(81.6mg、0.255mmol)を得た。1H NMR (400 MHz, DMSO-d6) δ
ppm 7.75 (d, J=7.9 Hz, 1 H), 7.56 (dd, J=8.0, 1.4 Hz, 1 H), 7.30 (t, J=8.0 Hz,
1 H), 7.22 (s, 1 H), 6.94 (dd, J=8.0, 1.2 Hz, 1 H), 3.89 (s, 3 H).MSm/z32
2.0(M+H)+。
4-アミノ-6-フルオロ-3-((2-(トリフルオロメチル)ピリジン-3-イル)
チオ)ピリジン-2(1H)-オン

AcOH(19mL)中の4-アミノ-6-フルオロピリジン-2(1H)-オン(4
90mg、3.83mmol)の0℃溶液に、NIS(818mg、3.63mmol)
を添加した。固化した反応混合物を室温に加温し、1時間撹拌した。揮発物を減圧下で除
去した。残留物を、飽和Na2S2O3水溶液(15mL)、飽和NH4Cl水溶液(1
5mL)および水(15mL)を含有する分液漏斗に移した。混合物をEt2O(4×5
0mL)で抽出し、合わせた有機抽出物をMgSO4で脱水し、濾過し、揮発物を減圧下
で除去した。残留物をシリカクロマトグラフィー(0から5%の勾配のMeOH/DCM
)により精製して、4-アミノ-6-フルオロ-3-ヨードピリジン-2(1H)-オン
(54mg、0.213mmol)を白色固体として得た。MSm/z255.0(M+
H)+。
ジオキサン(0.5mL)中の4-アミノ-6-フルオロ-3-ヨードピリジン-2(
1H)-オン(40mg、0.157mmol)、2-(トリフルオロメチル)ピリジン
-3-チオール(33.9mg、0.189mmol)、TMEDA(9.51μL、0
.063mmol)、K3PO4(66.9mg、0.315mmol)およびCu(I
)I(6.00mg、0.031mmol)の混合物を、100℃で90分間撹拌した。
室温に冷却した後、混合物をEtOAc(2mL)で希釈し、5分間撹拌し、セライトの
パッドに通して濾過した。揮発物を減圧下で除去し、残留物をシリカクロマトグラフィー
(0から70%の勾配のEtOAc/ヘプタン、続いて0から10%の勾配のMeOH/
DCM)により精製して、4-アミノ-6-フルオロ-3-((2-(トリフルオロメチ
ル)ピリジン-3-イル)チオ)ピリジン-2(1H)-オン(22mg、0.072m
mol)を黄色固体として得た。MSm/z306.0(M+H)+。
4-アミノ-6-フルオロ-1-メチル-3-((2-(トリフルオロメチル)ピリジン
-3-イル)チオ)ピリジン-2(1H)-オン

EtOH(15mL)中の4-アミノ-6-フルオロピリジン-2(1H)-オン(5
70mg、4.45mmol)、K2CO3(922mg、6.67mmol)およびM
eI(278μL、4.45mmol)の懸濁液を、70℃で15時間撹拌した。室温に
冷却した後、反応混合物を濾過し、EtOHですすいだ。濾液を水(40mL)中に懸濁
させ、EtOAc(2×40mL)およびトリフルオロエタノール(DCM中10%、8
×40mL)で抽出した。合わせた有機抽出物をMgSO4で脱水し、濾過し、揮発物を
減圧下で除去した。残留物をシリカクロマトグラフィー(0から10%の勾配のMeOH
/DCM)により精製して、4-アミノ-6-フルオロ-1-メチルピリジン-2(1H
)-オン(356mg、2.51mmol)を白色固体として得た。MSm/z142.
8(M+H)+。
AcOH(8mL)中の4-アミノ-6-フルオロ-1-メチルピリジン-2(1H)
-オン(356mg、2.505mmol)の溶液に、DMF(2mL)中のNIS(5
52mg、2.455)の溶液をシリンジポンプにより30分間かけて添加し、得られた
混合物を室温でさらに20分間撹拌した。揮発物を減圧下で除去し、残留物をEt2Oに
溶解し、飽和Na2S2O3水溶液(15mL)、飽和NH4Cl水溶液(15mL)お
よび水(15mL)を含有する分液漏斗に注ぎ入れ、これをEt2O(8×50mL)で
抽出した。合わせた有機抽出物をMgSO4で脱水し、濾過し、減圧下で濃縮した。残留
物をシリカクロマトグラフィー(20から70%の勾配のEtOAc/ヘプタン)により
精製して、4-アミノ-6-フルオロ-3-ヨード-1-メチルピリジン-2(1H)-
オン(純度80%、345mg)を得た。MSm/z268.8(M+H)+。
ジオキサン(3.4mL)中の4-アミノ-6-フルオロ-3-ヨード-1-メチルピ
リジン-2(1H)-オン(340mg、上記参照)、2-(トリフルオロメチル)ピリ
ジン-3-チオール(227mg、1.269mmol)、TMEDA(0.061mL
、0.406mmol)、K3PO4(431mg、2.03mmol)およびCu(I
)I(38.7mg、0.203mmol)の混合物を、100℃で1.5時間撹拌した
。追加のCu(I)I(38.7mg、0.203mmol)およびTMEDA(61μ
L、0.406mmol)を添加し、反応混合物を100℃で2.5時間撹拌した。室温
に冷却した後、反応混合物をEtOAc(10mL)で希釈し、5分間撹拌し、セライト
のパッドに通して濾過し、EtOAcで洗浄した。揮発物を減圧下で除去し、残留物をシ
リカクロマトグラフィー(0から80%のEtOAc/ヘプタン)により精製して、4-
アミノ-6-フルオロ-1-メチル-3-((2-(トリフルオロメチル)ピリジン-3
-イル)チオ)ピリジン-2(1H)-オン(81mg、0.254mmol)を白色固
体として得た。MSm/z320.1(M+H)+。
2-クロロ-5-(2,3-ジクロロフェニル)-4-メトキシピリミジン

メトキシピリミジン(200mg、0.895mmol)、(2,3-ジクロロフェニル
)ボロン酸(171mg、0.895mmol)、PdCl2(dppf)CH2Cl2
付加物(73.1mg、0.090mmol)およびK2CO3(495mg、3.58
mmol)の懸濁液を、N2流で5分間脱気し、50℃に1.5時間加熱した。反応混合
物をEtOAc(100mL)と水(50mL)との間で分配した。分離した有機層をブ
ライン(50mL)で洗浄し、Na2SO4で脱水し、濾過し、減圧下で濃縮した。残留
物をカラムクロマトグラフィー(シリカゲル、0から30%の勾配のEtOAc/ヘプタ
ン)により精製して、2-クロロ-5-(2,3-ジクロロフェニル)-4-メトキシピ
リミジン(17mg)を白色固体として得た。MSm/z289.1(M+H)+。
ピロ[4.5]デカン-8-イル)-3-メチル-5-((2-(トリフルオロメチル)
ピリジン-3-イル)チオ)ピリミジン-4(3H)-オン

-5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)-3
-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カルバメート(4
2mg、0.081mmol)、2-(トリフルオロメチル)ピリジン-3-チオール(
22mg、0.121mmol)、Cu(I)I(3.1mg、0.016mmol)、
TMEDA(5μL、0.032mmol)およびK3PO4(51mg、0.243m
mol)の混合物を、100℃で90分間撹拌した。室温に冷却した後、反応混合物を、
K2CO3水溶液(2M、2mL)を含有する分液漏斗に注ぎ入れ、DCM(3×5mL
)で抽出した。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去し
た。残留物をDCM(5mL)に溶解し、TFA(1mL)を添加した。室温で20分間
撹拌した後、揮発物を減圧下で除去し、残留物をHPLC(勾配溶出、水中15~40%
MeCN、5mM NH4OH修飾剤)により精製して、6-アミノ-2-((3S,4
S)-4-アミノ-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-イ
ル)-3-メチル-5-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピ
リミジン-4(3H)-オン(20.0mg)を白色固体として得た。
(2,3-ジクロロフェニル)チオ)-3-メチルピリミジン-4(3H)-オン

メチルピリミジン-2,4(1H,3H)-ジオン(60mg、0.189mmol)、
tert-ブチル((4-メチルピペリジン-4-イル)メチル)カルバメート(64.
6mg、0.283mmol)、BOP(250mg、0.566mmol)およびDB
U(142μL、0.943mmol)の混合物を、室温で2時間撹拌した。得られた混
合物を、水を含有する分液漏斗に注ぎ入れ、これをEtOAc(3×5mL)で抽出した
。合わせた有機相をMgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物を
DCM(5mL)に溶解し、TFA(1mL)を添加した。室温で10分間撹拌した後、
揮発物を減圧下で除去し、残留物をHPLC(勾配溶出、水中35~60%MeCN、5
mM NH4OH修飾剤)により精製して、6-アミノ-2-(4-(アミノメチル)-
4-メチルピペリジン-1-イル)-5-((2,3-ジクロロフェニル)チオ)-3-
メチルピリミジン-4(3H)-オン(20.0mg)を白色固体として得た。
オールおよびヨード-ピリミジノン中間体を使用して合成した。
















[実施例67]
ピロ[4.5]デカン-8-イル)-5-((3-クロロ-2-メチルピリジン-4-イ
ル)チオ)-3-メチルピリミジン-4(3H)-オン

-8-(4-アミノ-5-ヨード-1-メチル-6-オキソ-1,6-ジヒドロピリミジ
ン-2-イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル
)カルバメート(145mg、0.279mmol)、ナトリウム3-クロロ-2-メチ
ルピリジン-4-チオレート(101mg、0.558mmol)、Cu(I)I(10
.6mg、0.056mmol)、TMEDA(17μL、0.112mmol)および
K3PO4(178mg、0.838mmol)の混合物を、100℃で90分間撹拌し
た。室温に冷却した後、反応混合物を、K2CO3水溶液(2M、30mL)を含有する
分液漏斗に注ぎ入れ、DCM(3×30mL)で抽出した。合わせた有機相をMgSO4
で脱水し、濾過し、揮発物を減圧下で除去した。残留物をDCM(5mL)に溶解し、T
FA(1mL)を添加した。室温で90分間撹拌した後、揮発物を減圧下で除去し、残留
物をHPLC(勾配溶出、水中10~30%MeCN、5mM NH4OH修飾剤)によ
り精製して、6-アミノ-2-((3S,4S)-4-アミノ-3-メチル-2-オキサ
-8-アザスピロ[4.5]デカン-8-イル)-5-((3-クロロ-2-メチルピリ
ジン-4-イル)チオ)-3-メチルピリミジン-4(3H)-オン(36.1mg)を
白色固体として得た。
オールおよびヨード-ピリミジノン中間体を使用して合成した。





[実施例88]
ピロ[4.5]デカン-8-イル)-3-((2-(トリフルオロメチル)ピリジン-3
-イル)チオ)ピリジン-2(1H)-オン

ロ-3-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピリジン-2(1
H)-オン(22mg、0.072mmol)および(3S,4S)-3-メチル-2-
オキサ-8-アザスピロ[4.5]デカン-4-アミン(21mg、0.086mmol
)の溶液を、100℃で5時間撹拌した。室温に冷却した後、揮発物を減圧下で除去し、
残留物をHPLC(勾配溶出、水中45~70%MeCN、5mM NH4OH修飾剤)
により精製して、4-アミノ-6-((3S,4S)-4-アミノ-3-メチル-2-オ
キサ-8-アザスピロ[4.5]デカン-8-イル)-3-((2-(トリフルオロメチ
ル)ピリジン-3-イル)チオ)ピリジン-2(1H)-オン(6.1mg)を白色固体
として得た。1H NMR (400 MHz, メタノール-d4) δ ppm 8.27-8.37 (m, 1 H), 7.48 (d,
J=7.58 Hz, 1 H), 7.41 (dd, J=8.21, 4.42 Hz, 1 H), 5.40 (s, 1 H), 4.22 (dd, J=6.4
4, 4.93 Hz, 1 H), 3.83 (d, J=8.84 Hz, 1 H), 3.68 (d, J=8.84 Hz, 1 H), 3.53 (td,
J=11.68, 5.94 Hz, 2 H), 3.03-3.20 (m, 2 H), 3.01 (d, J=4.80 Hz, 1 H), 1.77-1.92
(m, 2 H), 1.59-1.74 (m, 2 H), 1.22 (d, J=6.32 Hz, 3 H).C20H25F3N5O2
S(M+H)+のHRMSm/z計算値456.1676、実測値456.1623。I
C50=0.042μM。
[実施例89]
ピロ[4.5]デカン-8-イル)-1-メチル-3-((2-(トリフルオロメチル)
ピリジン-3-イル)チオ)ピリジン-2(1H)-オン

ルオロ-1-メチル-3-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)
ピリジン-2(1H)-オン(80mg、0.251mmol)および(3S,4S)-
3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-アミン(73.1mg
、0.086mmol)の溶液を、100℃で20時間撹拌した。室温に冷却した後、揮
発物を減圧下で除去し、残留物をHPLC(勾配溶出、水中45~70%MeCN、0.
1%TFA修飾剤)により精製して、4-アミノ-6-((3S,4S)-4-アミノ-
3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-イル)-1-メチル-
3-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピリジン-2(1H)
-オン(84.8mg)をそのTFA塩として橙色油状物として得た。1H NMR (400 MHz,
メタノール-d4) δ ppm 8.33 (dd, J=4.04, 2.02 Hz, 1 H), 7.33-7.43 (m, 2 H), 5.71
(s, 1 H), 4.23-4.36 (m, 1 H), 3.95 (d, J=9.09 Hz, 1 H), 3.84 (d, J=9.09 Hz, 1 H
), 3.47 (br.d, J=2.78 Hz, 1 H), 3.46 (s, 3 H), 3.15-3.26 (m, 2 H), 2.60-2.86 (m,
2 H), 1.88-2.02 (m, 3 H), 1.76 (br.d, J=12.13 Hz, 1 H), 1.32 (d, J=6.57 Hz, 3 H
).C21H27F3N5O2S(M+H)+のHRMSm/z計算値470.1832、
実測値470.1826。IC50=0.429μM。
[実施例90]
(2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピリミジン-4(3H)-オ
ン

MeCN(5mL)中の6-アミノ-5-((2-(トリフルオロメチル)ピリジン-
3-イル)チオ)-3-((2-(トリメチルシリル)エトキシ)メチル)ピリミジン-
2,4(1H,3H)-ジオン(200mg、0.460mmol)、BOP-Cl(1
52mg、0.598mmol)およびtert-ブチル((4-メチルピペリジン-4
-イル)メチル)カルバメート(158mg、0.690mmol)の混合物に、DBU
(210μL)を添加した。得られた混合物を100℃で2時間撹拌した。室温に冷却し
た後、揮発物を減圧下で除去し、残留物をシリカクロマトグラフィー(0から50%の勾
配のEtOAc/ヘプタン)により精製して、tert-ブチル((1-(4-アミノ-
6-オキソ-5-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)-1-(
(2-(トリメチルシリル)エトキシ)メチル)-1,6-ジヒドロピリミジン-2-イ
ル)-4-メチルピペリジン-4-イル)メチル)カルバメート(32mg)を得た。1H
NMR (400 MHz, クロロホルム-d) δppm 8.39 (d, J=4.04 Hz, 1 H), 7.58 (d, J=8.08 H
z, 1 H), 7.29 (dd, J=8.08, 4.55 Hz, 1 H), 5.22 (s, 2 H), 4.67 (t, J=6.06 Hz, 1 H
), 4.12 (q, J=7.16 Hz, 1 H), 3.87 (t, J=8.08 Hz, 2 H), 3.62-3.77 (m, 2 H), 3.37
(t, J=9.98 Hz, 2 H), 3.09 (d, J=6.57 Hz, 2 H), 2.05 (s, 2 H), 1.57 (ddd, J=13.33
, 9.54, 3.41 Hz, 2 H), 1.37-1.51 (m, 12 H), 1.26 (t, J=7.07 Hz, 3 H), 1.00 (s, 3
H), 0.84-0.97 (m, 3 H), 0.02 (br. s, 9 H).
DCM(2mL)中のtert-ブチル((1-(4-アミノ-6-オキソ-5-((
2-(トリフルオロメチル)ピリジン-3-イル)チオ)-1-((2-(トリメチルシ
リル)エトキシ)メチル)-1,6-ジヒドロピリミジン-2-イル)-4-メチルピペ
リジン-4-イル)メチル)カルバメート(32mg、0.050mmol)の溶液に、
TFA(0.5mL)を添加し、得られた混合物を50℃で30分間撹拌した。室温に冷
却した後、揮発物を減圧下で除去し、残留物をMeOH(2mL)に溶解し、エチレンジ
アミン(50mg、0.75mmol)を添加した。得られた混合物を室温で16時間撹
拌した。揮発物を減圧下で除去し、残留物をHPLC(勾配溶出、水中15~40%Me
CN、5mM NH4OH修飾剤)により精製して、6-アミノ-2-(4-(アミノメ
チル)-4-メチルピペリジン-1-イル)-5-((2-(トリフルオロメチル)ピリ
ジン-3-イル)チオ)ピリミジン-4(3H)-オン(4.1mg、9.69μmol
)を白色固体として得た。1H NMR (400 MHz, メタノール-d4) δppm 8.33 (d, J=3.79 Hz
, 1 H), 7.56 (d, J=8.08 Hz, 1 H), 7.43 (dd, J=8.08, 4.55 Hz, 1 H), 3.96 (dt, J=1
3.77, 4.74 Hz, 2 H), 3.39-3.52 (m, 2 H), 2.52-2.62 (m, 2 H), 1.54 (ddd, J=13.71,
9.92, 4.17 Hz, 2 H), 1.39-1.49 (m, 3 H), 1.04 - 1.13 (m, 2 H). 19F NMR (376 MH
z, メタノール-d4) δppm -66.39.C17H22F3N6OS(M+H)+のHRMS計
算値415.1519、実測値415.1528。IC50=0.066μM。
[実施例91]
イル)-4-メチルピペリジン-4-イル)メチル)カルバメート

DMF(2mL)中のtert-ブチル((1-(5-ヨード-4-((4-メトキシ
ベンジル)オキシ)ピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル
)カルバメート(198mg、0.348mmol)の溶液に、K2CO3(96mg、
0.697mmol)、銅-1-チオフェン-2-カルボキシレート(27mg、0.1
39mmol)および2-(トリフルオロメチル)ピリジン-3-チオール(90mg、
491mmol)を添加した。混合物にマイクロ波反応器内120℃で30分間照射を行
った。反応物を水(100mL)に注ぎ入れ、混合物をDCM(4×)で抽出した。合わ
せた有機抽出物をブラインで洗浄し、MgSO4で脱水し、濾過し、減圧下で濃縮した。
残留物をシリカクロマトグラフィー(0から50%の勾配のEtOAc/ヘプタン)によ
り精製して、tert-ブチル((1-(4-((4-メトキシベンジル)オキシ)-5
-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピリミジン-2-イル)
-4-メチルピペリジン-4-イル)メチル)カルバメート(110mg)を透明油状物
として得た。MSm/z620(M+H)+。
DCM(2mL)中のtert-ブチル((1-(4-((4-メトキシベンジル)オ
キシ)-5-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピリミジン-
2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(60mg、0.
097mmol)の溶液に、TFA(2mL)を添加した。反応物を室温で30分間撹拌
し、減圧下で濃縮した。残留物質をHPLC(勾配溶出、水中10~30%MeCN、0
.1%TFA修飾剤)により精製して、2-(4-(アミノメチル)-4-メチルピペリ
ジン-1-イル)-5-((2-(トリフルオロメチル)ピリジン-3-イル)チオ)ピ
リミジン-4(3H)-オン(50.0mg、0.125mmol)を得た。
護されたアミンおよびチオール中間体を使用して合成した。


[実施例98]
クロロフェニル)チオ)-3-メチルピリミジン-4(3H)-オン

ジオキサン(3mL)中のtert-ブチル((1-(5-ヨード-1-メチル-6-
オキソ-1,6-ジヒドロピリミジン-2-イル)-4-メチルピペリジン-4-イル)
メチル)カルバメート(76.9mg、0.166mmol)、2,3-ジクロロベンゼ
ンチオール(44.7mg、0.249mmol)、1,10-フェナントロリン(12
.0mg、0.067mmol)、Cu(I)I(6.3mg、0.033mmol)お
よびCs2CO3(108mg、0.333mmol)の混合物を、100℃で1時間撹
拌した。室温に冷却した後、反応物をEtOAcで希釈し、セライトのパッドに通して濾
過した。有機層を飽和NH4Cl水溶液(2×)、続いてブラインで洗浄した。有機層を
MgSO4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグラ
フィー(0から100%の勾配のEtOAc/ヘプタン)により精製して、tert-ブ
チル((1-(5-((2,3-ジクロロフェニル)チオ)-1-メチル-6-オキソ-
1,6-ジヒドロピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)
カルバメートを得た。MSm/z513.0(M+H)+
DCM(3mL)およびTFA(1mL)中のtert-ブチル((1-(5-((2
,3-ジクロロフェニル)チオ)-1-メチル-6-オキソ-1,6-ジヒドロピリミジ
ン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメートの混合物を、
室温で1時間撹拌した。揮発物を減圧下で除去し、残留物をHPLC(勾配溶出、水中1
5~40%MeCN、0.1%TFA修飾剤)により精製して、2-(4-(アミノメチ
ル)-4-メチルピペリジン-1-イル)-5-((2,3-ジクロロフェニル)チオ)
-3-メチルピリミジン-4(3H)-オン(TFA塩、36mg)を得た。
オールおよびヨード-ピリミジノン中間体を使用して合成した。

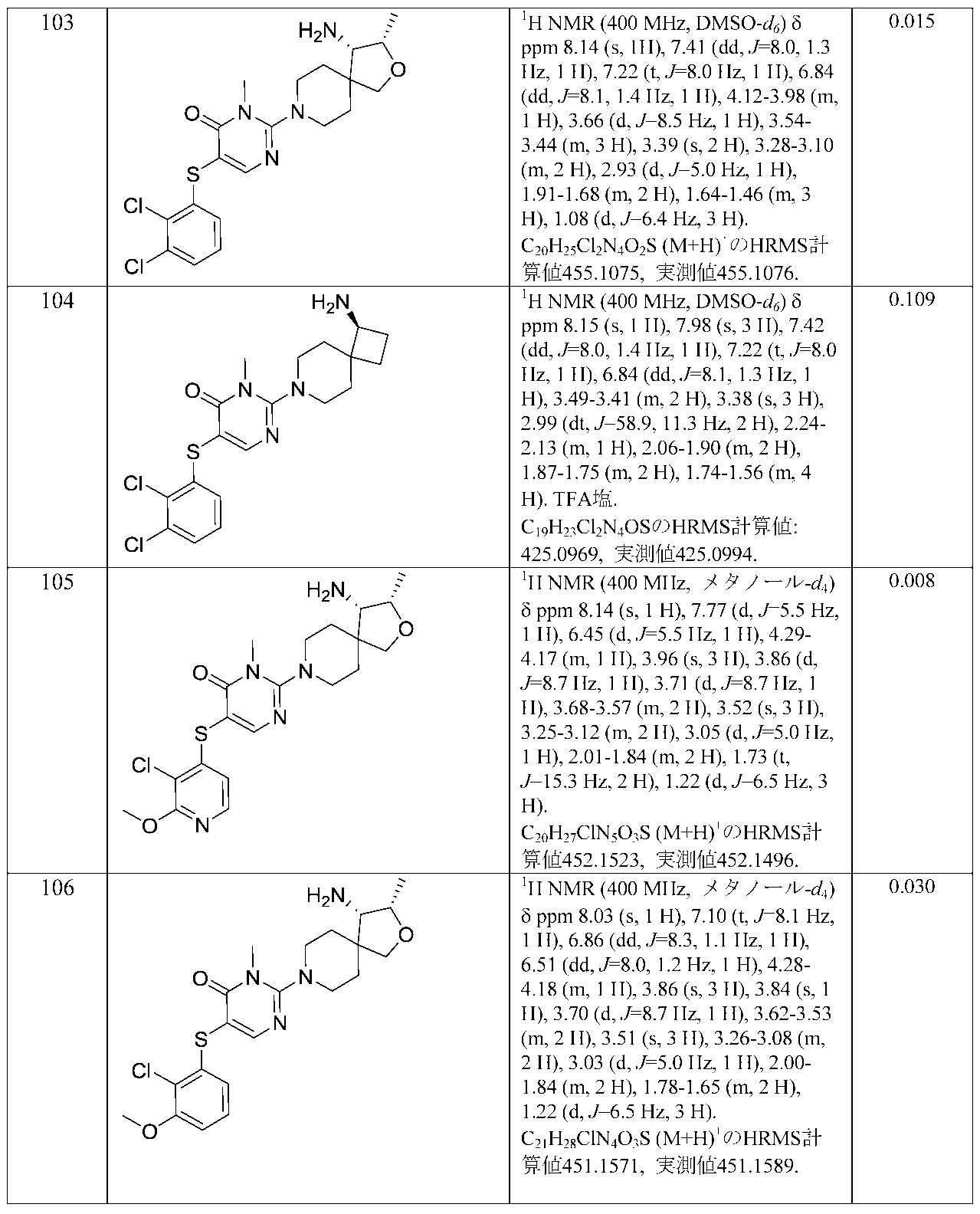


[実施例116]
クロロフェニル)チオ)-1-メチルピリジン-2(1H)-オン

ジオキサン(3mL)中のtert-ブチル((1-(5-ヨード-1-メチル-6-
オキソ-1,6-ジヒドロピリジン-2-イル)-4-メチルピペリジン-4-イル)メ
チル)カルバメート(34.4mg、0.075mmol)、2,3-ジクロロベンゼン
チオール(20mg、0.112mmol)、1,10-フェナントロリン(5.4mg
、0.030mmol)、Cu(I)I(2.8mg、0.015mmol)およびCs
2CO3(48.6mg、0.149mmol)の混合物を、100℃で1時間撹拌した
。室温に冷却した後、反応物をEtOAcで希釈し、セライトのパッドに通して濾過した
。有機層を飽和NH4Cl水溶液(2×)、続いてブラインで洗浄した。有機層をMgS
O4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー
(0から100%の勾配のEtOAc/ヘプタン)により精製して、tert-ブチル(
(1-(5-((2,3-ジクロロフェニル)チオ)-1-メチル-6-オキソ-1,6
-ジヒドロピリジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメ
ートを得た。MSm/z512.0(M+H)+
DCM(3mL)およびTFA(1mL)中のtert-ブチル((1-(5-((2
,3-ジクロロフェニル)チオ)-1-メチル-6-オキソ-1,6-ジヒドロピリジン
-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(上記参照)の
混合物を、室温で1時間撹拌した。揮発物を減圧下で除去し、残留物をHPLC(勾配溶
出、水中15~40%MeCN、0.1%TFA修飾剤)により精製して、6-(4-(
アミノメチル)-4-メチルピペリジン-1-イル)-3-((2,3-ジクロロフェニ
ル)チオ)-1-メチルピリジン-2(1H)-オン(TFA塩、5mg)を得た。
発物質および中間体を使用して合成した。

[実施例118]
クロロフェニル)チオ)ピリジン-2(1H)-オン

DMF(1mL)中の2-クロロ-5-((2,3-ジクロロフェニル)チオ)-4-
メトキシピリミジン(81.6mg、0.255mmol)、DIPEA(89μL、0
.509mmol)およびtert-ブチル((4-メチルピペリジン-4-イル)メチ
ル)カルバメート(69.7mg、0.305mmol)の混合物に、マイクロ波反応器
内140℃で2時間照射を行った。室温に冷却した後、混合物をEtOAcで希釈した。
有機層を飽和NH4Cl水溶液(2×)、続いてブラインで洗浄した。有機層をMgSO
4で脱水し、濾過し、揮発物を減圧下で除去した。残留物をシリカクロマトグラフィー(
0から100%の勾配のEtOAc/ヘプタン)により精製して、tert-ブチル((
1-(5-((2,3-ジクロロフェニル)チオ)-6-メトキシピリジン-2-イル)
-4-メチルピペリジン-4-イル)メチル)カルバメート(26.1mg)を白色固体
として得た。1H NMR (400 MHz, メタノール-d4) δ ppm 7.53 (d, J=8.4 Hz, 1 H), 7.21
(dd, J=8.0, 1.4 Hz, 1 H), 7.04 (t, J=8.0 Hz, 1 H), 6.51 (dd, J=8.1, 1.3 Hz, 1 H
), 6.38 (d, J=8.5 Hz, 1 H), 3.96-3.87 (m, 2 H), 3.82 (s, 3 H), 3.49-3.39 (m, 4 H
), 3.01 (d, J=5.3 Hz, 2 H), 1.59-1.47 (m, 3 H), 1.44 (s, 9 H), 1.42-1.33 (m, 2 H
), 1.00 (s, 3 H).MSm/z512.1(M+H)+。
DCM(2mL)中のtert-ブチル((1-(5-((2,3-ジクロロフェニル
)チオ)-6-メトキシピリジン-2-イル)-4-メチルピペリジン-4-イル)メチ
ル)カルバメート(26.1mg、0.051mmol)およびBBr3(DCM中1M
、0.153mL、0.153mmol)の溶液を、0℃で1時間撹拌した。揮発物を減
圧下で除去して、tert-ブチル((1-(5-((2,3-ジクロロフェニル)チオ
)-4-メトキシピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)
カルバメート(26.1mg、0.051mmol)を得た。MSm/z412.1(M
+H)+。
tert-ブチル((1-(5-((2,3-ジクロロフェニル)チオ)-4-メトキ
シピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(
26.1mg、0.051mmol)およびHCl(ジオキサン中4M、2mL、8mm
ol)の混合物を、90℃で20時間撹拌した。室温に冷却した後、揮発物を減圧下で除
去し、残留物をHPLC(勾配溶出、水中15~40%MeCN、0.1%TFA修飾剤
)により精製して、2-(4-(アミノメチル)-4-メチルピペリジン-1-イル)-
5-((2,3-ジクロロフェニル)チオ)ピリミジン-4(3H)-オン(TFA塩、
13.0mg)を得た。
発物質および中間体を使用して合成した。

[実施例121]
4-アミノ-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-8-イル)-
3,6-ジメチルピリミジン-4(3H)-オン
N2雰囲気下、マイクロ波バイアル内のDMF中のtert-ブチル((3S,4S)
-8-(5-ブロモ-1,4-ジメチル-6-オキソ-1,6-ジヒドロピリミジン-2
-イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カル
バメート(130mg、0.276mmol)、K3PO4(176mg、0.827m
mol)、ナトリウム2-アミノ-3-クロロピリジン-4-チオレート(76mg、0
.414mmol)およびCu(I)I(10.50mg、0.055mmol)の混合
物に、TMEDA(0.017mL、0.110mmol)を添加した。反応混合物にマ
イクロ波中150℃で1時間照射を行った。反応混合物を水/EtOAcで希釈し、Et
OAc(2×)およびDCM(2×)で抽出した。合わせた有機層をNa2SO4で脱水
し、濾別し、減圧下で濃縮した。残留物を分取TLC(シリカゲル、1mm、DCM/M
eOH=95/5)により精製して、tert-ブチル((3S,4S)-8-(5-(
(2-アミノ-3-クロロピリジン-4-イル)チオ)-1,4-ジメチル-6-オキソ
-1,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オキサ-8-アザスピロ
[4.5]デカン-4-イル)カルバメートを得た。MSm/z551.3(M+H)+
。
tert-ブチル((3S,4S)-8-(5-((2-アミノ-3-クロロピリジン
-4-イル)チオ)-1,4-ジメチル-6-オキソ-1,6-ジヒドロピリミジン-2
-イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カル
バメートをDCM(3mL)中に溶解し/懸濁させ、TFA(0.5mL)を添加した。
混合物を約20分間撹拌し、減圧下で濃縮した。残留物に、MeCN(5%の水を含有)
および固体NaHCO3を添加した。混合物を5分間激しく撹拌し、シリンジフィルター
(2μm)に通して濾過した。濾液を減圧下で濃縮し、分取TLC(シリカゲル、1mm
、DCM/MeOH=90:10)により精製した。シリカバンドをDCM/MeOHで
洗浄し、濾液を減圧下で濃縮した。残留物をMeCNに溶解し、シリンジフィルター(2
μm)に通して濾過し、凍結乾燥して、5-((2-アミノ-3-クロロピリジン-4-
イル)チオ)-2-((3S,4S)-4-アミノ-3-メチル-2-オキサ-8-アザ
スピロ[4.5]デカン-8-イル)-3,6-ジメチルピリミジン-4(3H)-オン
(3.6mg)を得た。
発物質および中間体を使用して合成した。

[実施例123]
ピロ[4.5]デカン-8-イル)-5-(3-クロロ-4-メチルフェニル)-3-メ
チルピリミジン-4(3H)-オン

トルエン(1mL)中の(3-クロロ-4-メトキシフェニル)ボロン酸(41.0m
g、0.241mmol)、Cs2CO3(182mg、0.558mmol)およびP
d(PPh3)4(16.69mg、0.014mmol)の混合物に、N2雰囲気下で
、EtOH(1mL)中のtert-ブチル((3S,4S)-8-(4-アミノ-5-
ヨード-1-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-イル)-3-メチ
ル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カルバメート(50mg
、0.096mmol)の溶液を添加した。混合物にマイクロ波反応器内100℃で30
分間照射を行った。反応混合物を水およびEtOAcで希釈した。分離した水層をEtO
Ac(2×)で抽出し、合わせた有機層をNa2SO4で脱水し、濾別し、減圧下で濃縮
して、粗製のtert-ブチル((3S,4S)-8-(4-アミノ-5-(3-クロロ
-4-メチルフェニル)-1-メチル-6-オキソ-1,6-ジヒドロピリミジン-2-
イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-イル)カルバ
メート(83mg)を茶色固体として得、これをさらに精製することなく次の反応におい
て直接使用した。MSm/z518.3(M+H)+。
N2雰囲気下、DCM(1mL)中の粗製のtert-ブチル((3S,4S)-8-
(4-アミノ-5-(3-クロロ-4-メチルフェニル)-1-メチル-6-オキソ-1
,6-ジヒドロピリミジン-2-イル)-3-メチル-2-オキサ-8-アザスピロ[4
.5]デカン-4-イル)カルバメート(83mg)に、TFA(1mL)を添加した。
混合物を1時間撹拌し、トルエン(1mL)で希釈し、減圧下で濃縮した。残留物をMe
OHに溶解し、NH3(MeOH中7M)で塩基性化し、混合物を減圧下で濃縮した。残
留物をMeCN/水(2/1)に溶解し、シリンジフィルター(0.2μm)に通して濾
過し、HPLC(勾配溶出、水中25~50%MeCN、5mM NH4OH修飾剤)に
より精製して、6-アミノ-2-((3S,4S)-4-アミノ-3-メチル-2-オキ
サ-8-アザスピロ[4.5]デカン-8-イル)-5-(3-クロロ-4-メチルフェ
ニル)-3-メチルピリミジン-4(3H)-オン(19mg)を白色固体として得た。
発物質および中間体を使用して合成した。


















[実施例198]
ロロフェニル)ピリミジン-4(3H)-オン

THF(1.88mL)および水(0.375mL)中のtert-ブチル((1-(
5-ブロモ-4-メトキシピリミジン-2-イル)-4-メチルピペリジン-4-イル)
メチル)カルバメート(105mg、0.225mmol)、(2,3-ジクロロフェニ
ル)ボロン酸(42.9mg、0.225mmol)、PdCl2(dppf)CH2C
l2付加物(18.4mg、0.023mmol)およびK2CO3(124mg、0.
900mmol)の懸濁液を、N2流で5分間脱気し、50℃に4時間加熱した。反応混
合物をEtOAc(100mL)と水(50mL)との間で分配した。分離した有機層を
ブライン(50mL)で洗浄し、Na2SO4で脱水し、濾過し、減圧下で濃縮した。残
留物をカラムクロマトグラフィー(シリカゲル、0から30%の勾配のEtOAc/ヘプ
タン)により精製して、tert-ブチル((1-(5-(2,3-ジクロロフェニル)
-4-メトキシピリミジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カ
ルバメート(17mg)を無色固体として得た。MSm/z481.3(M+H)。
tert-ブチル((1-(5-(2,3-ジクロロフェニル)-4-メトキシピリミ
ジン-2-イル)-4-メチルピペリジン-4-イル)メチル)カルバメート(17mg
、0.028mmol)をHBr(AcOH中33%、0.4mL)に溶解し、混合物を
90℃で1時間撹拌した。反応混合物を減圧下で濃縮した。残留物をMeOHに溶解し、
NH3(MeOH中7M)で塩基性化し、混合物を減圧下で濃縮した。残留物をMeCN
/水(2:1)に溶解し、シリンジフィルター(0.2μm)に通して濾過し、HPLC
(勾配溶出、水中15~40%MeCN、5mM NH4OH修飾剤)により精製して、
2-(4-(アミノメチル)-4-メチルピペリジン-1-イル)-5-(2,3-ジク
ロロフェニル)ピリミジン-4(3H)-オン(6mg)を白色固体として得た。
[実施例199]
]デカン-8-イル)-5-(2,3-ジクロロフェニル)ピリミジン-4(3H)-オ
ン

N2雰囲気下、DMSO(1.18mL)およびDIPEA(0.59mL)中の2-
クロロ-5-(2,3-ジクロロフェニル)-4-メトキシピリミジン(55mg、0.
177mmol)および(3S,4S)-3-メチル-2-オキサ-8-アザスピロ[4
.5]デカン-4-アミンビス塩酸塩(47.3mg、0.194mmol)の混合物を
、120℃に2時間加熱した。反応混合物を室温に冷却し、EtOAc(50mL)で希
釈し、ブライン(25mL)で洗浄した。分離した水層をEtOAc(25mL)で抽出
した。合わせた有機層をNa2SO4で脱水し、濾過し、減圧下で濃縮して、粗製の(3
S,4S)-8-(5-(2,3-ジクロロフェニル)-4-メトキシピリミジン-2-
イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-アミン(67
mg)を茶橙色固体として得、これをさらに精製することなく直接使用した。MSm/z
423.2(M+H)+。
(3S,4S)-8-(5-(2,3-ジクロロフェニル)-4-メトキシピリミジン
-2-イル)-3-メチル-2-オキサ-8-アザスピロ[4.5]デカン-4-アミン
(67mg、0.158mmol)をHBr(AcOH中33%、1mL)に溶解し、混
合物を90℃で1時間撹拌した。反応混合物を減圧下で濃縮した。残留物をMeOHに溶
解し、NH3(MeOH中7M)で塩基性化し、混合物を減圧下で濃縮した。残留物をM
eCN/水(2:1)に溶解し、シリンジフィルター(0.2μm)に通して濾過し、H
PLC(勾配溶出、水中10~30%MeCN、5mM NH4OH修飾剤)により精製
して、2-((3S,4S)-4-アミノ-3-メチル-2-オキサ-8-アザスピロ[
4.5]デカン-8-イル)-5-(2,3-ジクロロフェニル)ピリミジン-4(3H
)-オン(20mg)を白色固体として得た。
発物質および中間体を使用して合成した。

本発明の化合物を、SHP2活性を選択的に阻害するそれらの能力について評価した。
本明細書に記載の本発明の化合物の阻害特性は、以下のアッセイのいずれか1つを試験す
ることによって明らかになり得る。
SHP2は、ビス-チロシル-リン酸化(phorphorylated)ペプチドとそのSrc相同
性2(SH2)ドメインの結合を介して、アロステリックに活性化される。後の活性化ス
テップによって、SHP2の自己阻害性界面が放出され、それによってSHP2タンパク
質チロシンホスファターゼ(PTP)は活性になり、基質認識および反応触媒作用に利用
可能になる。SHP2の触媒活性は、迅速な蛍光アッセイ型式において代替基質DiFM
UPを使用してモニターした。
レンプレートの平底低フランジの非結合表面(Corning、カタログ番号3575)
内で、最終的な反応体積25μLおよび以下のアッセイバッファー条件:60mMのHE
PES、pH7.2、75mMのNaCl、75mMのKCl、1mMのEDTA、0.
05%P-20、5mMのDTTを使用して実施した。
ッセイを使用してモニターし、このアッセイでは、0.5nMのSHP2を、0.5μM
のペプチドIRS1_pY1172(dPEG8)pY1222(配列:H2N-LN(
pY)IDLDLV(dPEG8)LST(pY)ASINFQK-アミド)(配列番号
1)と共にインキュベートした。25℃で30~60分インキュベートした後、代替基質
DiFMUP(Invitrogen、カタログ番号D6567)を反応物に添加し、2
5℃で30分間インキュベートした。次に、bpV(Phen)(Enzo Life
Sciencesカタログ番号ALX-270-204)の160μM溶液5μlを添加
することによって、反応を慎重に希釈した。蛍光シグナルを、マイクロプレートリーダー
(Envision、Perki-Elmer)を使用して、それぞれ励起波長および発
光波長340nmおよび450nmを使用してモニターした。阻害剤の用量応答曲線を、
正規化IC50回帰曲線フィッティングを使用し、対照をベースとする正規化を用いて分
析した。本発明の化合物のIC50の結果は、先の実施例および表1~7に示されている
。
AlphaScreen(登録商標)SureFire(商標)ホスホ-ERK1/2
キット(PerkinElmer)を使用するp-ERK細胞アッセイ:KYSE-52
0細胞(30,000個の細胞/ウェル)を、96ウェルプレート培養物中で終夜成長さ
せ、20、6.6、2.2、0.74、0.24、0.08、0.027μMの濃度のS
hp2阻害剤で、37℃において2時間処理した。インキュベーションを、SureFi
reホスホ-細胞外シグナル制御キナーゼ(pERK)アッセイキット(PerkinE
lmer)を用いて供給した溶解バッファー(PerkinElmer)30μLを添加
することによって終了させた。試料を、製造者の指示に従って処理した。pERKからの
蛍光シグナルを、2101マルチラベルリーダー(Perkin Elmer Envi
sion)を使用して二重に測定した。阻害百分率を、全ERKシグナルによって正規化
し、DMSOビヒクル対照と比較した。
KYSE-520細胞(1500個の細胞/ウェル)を、24ウェルプレートの培地3
00μL(10%FBSを含有するRPMI-1640、Lonza)に蒔いた。薬物処
置では、様々な濃度(20、10、5、2.5、1.25μM)の本発明の化合物を、細
胞を蒔いてから24時間後および5日後に添加した。11日目に、コロニーを、0.2%
クリスタルバイオレット(MP Biomedicals)で染色し、その後、Spec
tramaxリーダー(Thermo Scientific)を使用して定量化するた
めに、20%AcOHに溶解させた。細胞増殖アッセイにおいて、細胞(1500個の細
胞/ウェル)を、96ウェルプレートの培地100μL(10%FBSを含有するRPM
I-1640、Lonza)に蒔いた。6日目に、Celltiter-Glo試薬(P
romega)50μLを添加し、発光シグナルを、供給者の指示(Promega)に
従って決定した。
本発明は、以下の態様を含む。
[1]
式I

X 1 は、NおよびCHから選択され、
X 2 は、CR 3b であり、
X 3 は、Sおよび結合から選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシおよびヒドロキシから選択され、
Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6~10 アリールおよびC 6~10 アリール-C 0~1 アルコキシから選択され、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルキル、-NH(C 3~5 シクロアルキル)、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、水素、ハロ、ハロ置換C 1~2 アルキル、ハロ置換C 1~2 アルコキシ、C 1~2 アルキル-ヒドロキシおよびシアノから選択され、またはR 1 およびR 8 は、R 1 およびR 8 が付着している炭素原子と一緒になって、1,3-ジオキソール、フェニル、ピリジン、シクロペンテン、ジヒドロフラン、ジヒドロピランから選択される環を形成し、前記1,3-ジオキソール、フェニル、ピリジン、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールまたはジヒドロピランは、非置換であってもよく、または1~2個のハロ基で置換されていてもよく、
R 2 は、水素およびハロから選択され、
R 3a は、水素、メチルおよびハロ置換C 1~2 アルキルから選択され、
R 3b は、水素、メチルおよびアミノから選択され、
R 4a およびR 4b は、それぞれ独立に、水素、ヒドロキシおよびフルオロから選択され、ただし、R 4a およびR 4b は、両方共はOHではあり得ず、ただし、R 4a およびR 4b は、同時にOHおよびFではあり得ず、
R 5a は、アミノおよびアミノ-メチルから選択され、
R 5b は、OH、アミノ、フルオロ、C 1~6 アルキル、メトキシ-カルボニル、C 3~6 シクロアルキル-C 1~3 アルキル、ヒドロキシ置換C 1~3 アルキル、C 1~2 アルコキシ置換C 1~3 アルキル、ならびにO、SおよびNから選択される1~4個のヘテロ原子を含有する5~6員のヘテロアリール環から選択され、R 5b の前記C 1~6 アルキルまたはC 1~2 アルコキシ置換C 1~3 アルキルは、非置換であるか、または1~3個のフッ素で置換されており、ただし、R 5a がアミノである場合、R 5b は、OH、アミノまたはフルオロではあり得ず、またはR 5a およびR 5b は、R 5a およびR 5b が付着している炭素原子と一緒になって、

から選択される基を形成し、
R 6a およびR 6b は、それぞれ独立に、水素、ヒドロキシおよびフルオロから選択され、ただし、R 6a およびR 6b は、両方共はOHではあり得ず、ただし、R 6a およびR 6b は、両方が同時にOHおよびフルオロではあり得ない]
の化合物または薬学的に許容されるその塩であって、ただし、式Iの化合物が、

[2]
式Ia

X 3 は、Sから選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシから選択され、
Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6~10 アリールおよびC 6~10 アリール-C 0~1 アルコキシから選択され、またはR 1 およびR 8 は、R 1 およびR 8 が付着している炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、水素、ハロ、ハロ置換C 1~2 アルキルから選択され、
R 2 は、水素およびクロロから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択され、
R 10 は、アミノであり、
R 11c は、水素およびC 1~3 アルキルから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[3]
Y 1 が、NおよびCR 7 から選択され、R 7 が、水素、ハロおよびアミノから選択され、
Y 2 が、NおよびCR 8 から選択され、R 8 が、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、ハロ置換C 1~2 アルキル、C 1~2 アルコキシ、シクロプロピル、シクロペンチル、シクロペンチル-メトキシ、ハロ置換C 1~2 アルコキシ、フェニル、メトキシ-エトキシ、テトラヒドロ-2H-ピラン-4-イル、モルホリノ、フェノキシおよびベンゾキシから選択され、
Y 3 が、NおよびCR 9 から選択され、R 9 が、水素、アミノ、ハロ、C 1~2 アルコキシ、シクロプロピル、トリフルオロメチル、トリフルオロメチル-スルファニル、イソプロピルおよびヒドロキシから選択され、
R 1 が、水素、ハロ、トリフルオロメチル、トリフルオロメトキシ、C 1~2 アルキルおよびシアノから選択され、
R 2 が、水素、フルオロおよびクロロから選択され、
R 4a が、水素であり、
R 6b が、水素であり、
R 10 が、アミノであり、
R 11c が、水素、メチルおよびエチルから選択される、[2]に記載の化合物または薬学的に許容されるその塩。
[4]




から選択される、[3]に記載の化合物または薬学的に許容されるその塩。
[5]
式Ia

[式中、
X 3 は、結合から選択され、
Y 1 は、CR 7 であり、R 7 は、水素、クロロおよびフルオロから選択され、
Y 2 は、CR 8 であり、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6 アリールおよびC 6 アリール-C 0~1 アルコキシから選択され、
Y 3 は、CR 9 から選択され、R 9 は、水素、クロロ、フルオロおよびメチルから選択され、
R 1 は、水素、クロロ、フルオロから選択され、
R 2 は、水素から選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択され、
R 10 は、アミノであり、
R 11c は、水素、C 1~3 アルキルおよびヒドロキシ-メチルから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[6]





[7]
式Ib

X 3 は、Sから選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシから選択され、
Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6~10 アリールおよびC 6~10 アリール-C 0~1 アルコキシから選択され、またはR 1 およびR 8 は、R 1 およびR 8 が付着している炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、水素、ハロ、ハロ置換C 1~2 アルキルから選択され、
R 2 は、水素およびハロから選択され、
R 3a は、水素およびメチルから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 5b は、C 1~6 アルキルから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[8]
Y 1 が、NおよびCR 7 から選択され、R 7 が、水素、ハロおよびアミノから選択され、
Y 2 が、NおよびCR 8 から選択され、R 8 が、水素、ハロ、アミノ、シアノ、ハロ置換C 1~2 アルキル、C 1~2 アルコキシおよびハロ置換C 1~2 アルコキシから選択され、
Y 3 が、NおよびCR 9 から選択され、R 9 が、水素、アミノ、ハロ、C 1~2 アルコキシおよびヒドロキシから選択され、
R 1 が、ハロ、トリフルオロメチル、トリフルオロメトキシ、C 1~2 アルキル、ニトロ、ヒドロキシおよびシアノから選択され、またはR 1 およびR 8 が、R 1 およびR 8 が付着している炭素原子と一緒になって、1,3-ジオキソランおよびピリジンから選択される環を形成し、前記1,3-ジオキソランまたはピリジンが、非置換であってもよく、または1~2個のハロ基で置換されていてもよく、
R 2 が、水素、フルオロおよびクロロから選択され、
R 3 が、水素およびメチルから選択され、
R 4a が、水素であり、
R 5b が、メチルであり、
R 6b が、水素である、[7]に記載の化合物または薬学的に許容されるその塩。
[9]



[10]
式Ib

X 3 は、結合から選択され、
Y 1 は、CR 7 であり、R 7 は、水素、クロロおよびフルオロから選択され、
Y 2 は、CR 8 であり、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6 アリールおよびC 6 アリール-C 0~1 アルコキシから選択され、
Y 3 は、CR 9 から選択され、R 9 は、水素、クロロ、フルオロおよびメチルから選択され、
R 1 は、水素、クロロ、フルオロから選択され、
R 2 は、水素から選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 5b は、C 1~6 アルキルであり、
R 6b は、水素、ヒドロキシおよびフルオロから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[11]

[12]
式Ic

X 1 は、NおよびCHから選択され、
X 3 は、Sから選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシから選択され、
Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6~10 アリールおよびC 6~10 アリール-C 0~1 アルコキシから選択され、またはR 1 およびR 8 は、R 1 およびR 8 が付着している炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、水素、ハロ、ハロ置換C 1~2 アルキルおよびハロ置換C 1~2 アルコキシから選択され、
R 2 は、水素およびハロから選択され、
R 3a は、水素およびメチルから選択され、
R 3b は、水素およびメチルから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 5b は、C 1~6 アルキルから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択される]
の[1]に記載の化合物。
[13]
Y 1 が、NおよびCR 7 から選択され、R 7 が、水素、ハロおよびアミノから選択され、
Y 2 が、NおよびCR 8 から選択され、R 8 が、水素、ハロ、アミノ、シアノ、ハロ置換C 1~2 アルキル、C 1~2 アルコキシおよびハロ置換C 1~2 アルコキシから選択され、
Y 3 が、NおよびCR 9 から選択され、R 9 が、水素、アミノ、ハロ、C 1~2 アルコキシおよびヒドロキシから選択され、
R 1 が、ハロ、トリフルオロメチル、トリフルオロメトキシ、C 1~2 アルキルおよびシアノから選択され、
R 2 が、水素、フルオロおよびクロロから選択され、
R 3 が、水素およびメチルから選択され、
R 4a が、水素であり、
R 6b が、水素である、[12]に記載の化合物または薬学的に許容されるその塩。
[14]


[15]
式Id

X 1 は、NおよびCHから選択され、
X 3 は、Sから選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシから選択され、
Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6~10 アリールおよびC 6~10 アリール-C 0~1 アルコキシから選択され、またはR 1 およびR 8 は、R 1 およびR 8 が付着している炭素原子と一緒になって、シクロペンテン、2,3-ジヒドロフラン、2,3-ジヒドロピロールから選択される環を形成し、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルキル、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、水素、ハロ、ハロ置換C 1~2 アルキルおよびシアノから選択され、
R 2 は、水素およびハロから選択され、
R 3a は、水素、メチルおよびハロ置換C 1~2 アルキルから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択され、
R 10 は、アミノであり、
R 11a は、水素、ヒドロキシ、フルオロ、C 1~3 アルキルおよびヒドロキシ-メチルから選択され、
R 11b は、フルオロ、メチルおよび水素から選択され、ただし、R 11a およびR 11b は、両方が同時にOHおよびフルオロではあり得ず、
R 12 は、水素、ハロ、ヒドロキシ、C 1~3 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルコキシおよびC 1~3 アルコキシから選択され、
R 13 は、水素、ハロおよびC 1~3 アルキルから選択され、ただし、R 12 およびR 13 は、両方が同時にOHおよびフルオロではあり得ず、
R 14 は、水素およびフルオロから選択され、
R 15 は、水素およびフルオロから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[16]





[17]
式Id

X 1 は、NおよびCHから選択され、
X 3 は、結合から選択され、
Y 1 は、CR 7 であり、R 7 は、水素、クロロおよびフルオロから選択され、
Y 2 は、CR 8 であり、R 8 は、水素、ハロ、アミノ、ジメチル-アミノ、シアノ、C 3~6 シクロアルキル、C 1~4 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルキル-スルファニル、C 1~3 アルコキシ、ハロ置換C 1~3 アルコキシ、C 1~3 アルコキシ-C 1~3 アルコキシ、C 6 アリールおよびC 6 アリール-C 0~1 アルコキシから選択され、
Y 3 は、CR 9 から選択され、R 9 は、水素、クロロ、フルオロおよびメチルから選択され、
R 1 は、水素、クロロ、フルオロから選択され、
R 2 は、水素から選択され、
R 3a は、メチルから選択され、
R 3b は、アミノから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、
R 6b は、水素、ヒドロキシおよびフルオロから選択され、
R 10 は、アミノであり、
R 11a は、水素、ヒドロキシ、フルオロ、C 1~3 アルキルおよびヒドロキシ-メチルから選択され、
R 11b は、フルオロ、メチルおよび水素から選択され、ただし、R 11a およびR 11b は、両方が同時にOHおよびフルオロではあり得ず、
R 12 は、水素、ハロ、ヒドロキシ、C 1~3 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルコキシおよびC 1~3 アルコキシから選択され、
R 13 は、水素、ハロおよびC 1~3 アルキルから選択され、ただし、R 12 およびR 13 は、両方が同時にOHおよびフルオロではあり得ず、
R 14 は、水素およびフルオロから選択され、
R 15 は、水素およびフルオロから選択される]
の[1]に記載の化合物または薬学的に許容されるその塩。
[18]


[19]
式Ie

X 1 は、NおよびCHから選択され、
Y 1 は、NおよびCR 7 から選択され、R 7 は、水素、ハロおよびアミノから選択され、Y 2 は、NおよびCR 8 から選択され、R 8 は、水素、ハロ、アミノ、シアノ、ハロ置換C 1~3 アルキル、C 1~3 アルコキシおよびハロ置換C 1~3 アルコキシから選択され、
Y 3 は、NおよびCR 9 から選択され、R 9 は、水素、アミノ、ハロ、C 1~3 アルコキシおよびヒドロキシから選択され、
R 1 は、ハロ、ハロ置換C 1~2 アルキル、ハロ置換C 1~2 アルコキシ、C 1~2 アルキルおよびシアノから選択され、
R 2 は、水素およびハロから選択され、
R 3a は、水素およびメチルから選択され、
R 3b は、水素およびメチルから選択され、
R 4a は、水素、ヒドロキシおよびフルオロから選択され、R 6b は、水素、ヒドロキシおよびフルオロから選択され、
R 10 は、アミノであり、
R 11a は、水素、ヒドロキシ、フルオロ、C 1~3 アルキルおよびヒドロキシ-メチルから選択され、
R 11b は、フルオロ、メチルおよび水素から選択され、
R 11c は、水素、C 1~3 アルキルおよびヒドロキシ-メチルから選択され、
R 12 は、水素、ハロ、ヒドロキシ、C 1~3 アルキル、ハロ置換C 1~3 アルキル、ハロ置換C 1~3 アルコキシおよびC 1~3 アルコキシから選択され、
R 13 は、水素、ハロおよびC 1~3 アルキルから選択され、ただし、R 12 およびR 13 は、両方が同時にOHおよびフルオロではあり得ない]
の[1]に記載の化合物または薬学的に許容されるその塩。
[20]


[21]
[1]に記載の化合物または薬学的に許容されるその塩を、SHP2の活性によって媒介される疾患または障害を予防的または治療的に処置するのに有効な量で、このような処置を必要としているヒトに投与するステップを含む、処置の方法。
[22]
SHP2の活性によって媒介される前記疾患または障害が、ヌーナン症候群、レオパード症候群、若年性骨髄単球性白血病、神経芽細胞腫、黒色腫、急性骨髄性白血病、乳房がん、食道がん、肺がん、結腸がん、頭部がん、神経芽細胞腫、頭部および頸部の扁平上皮癌、胃癌、未分化大細胞リンパ腫、ならびに膠芽腫から選択される、[21]に記載の方法。
Claims (3)
- 式(5a):

R4aは、水素、ヒドロキシおよびフルオロから選択され、
R6bは、水素、ヒドロキシおよびフルオロから選択され、
R10は、アミノであり、
R11cは、水素およびC1~3アルキルから選択される]
の中間体、または薬学的に許容されるその塩。 - R4aは水素であり、
R6bは水素であり、
R10はアミノであり、
R11cは、水素、メチルおよびエチルから選択される、
請求項1に記載の中間体、または薬学的に許容されるその塩。 -

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562181871P | 2015-06-19 | 2015-06-19 | |
| US62/181,871 | 2015-06-19 | ||
| JP2017565688A JP6878316B2 (ja) | 2015-06-19 | 2016-06-15 | Shp2の活性を阻害するための化合物および組成物 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017565688A Division JP6878316B2 (ja) | 2015-06-19 | 2016-06-15 | Shp2の活性を阻害するための化合物および組成物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021130662A JP2021130662A (ja) | 2021-09-09 |
| JP7146016B2 true JP7146016B2 (ja) | 2022-10-03 |
Family
ID=56194529
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017565688A Active JP6878316B2 (ja) | 2015-06-19 | 2016-06-15 | Shp2の活性を阻害するための化合物および組成物 |
| JP2021076566A Active JP7146016B2 (ja) | 2015-06-19 | 2021-04-28 | Shp2の活性を阻害するための化合物および組成物 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017565688A Active JP6878316B2 (ja) | 2015-06-19 | 2016-06-15 | Shp2の活性を阻害するための化合物および組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10308660B2 (ja) |
| EP (1) | EP3310771B1 (ja) |
| JP (2) | JP6878316B2 (ja) |
| CN (1) | CN107787323B (ja) |
| ES (1) | ES2824576T3 (ja) |
| WO (1) | WO2016203405A1 (ja) |
Families Citing this family (109)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ713361A (en) | 2009-08-17 | 2017-06-30 | Memorial Sloan Kettering Cancer Center | Heat shock protein binding compounds, compositions, and methods for making and using same |
| US11466017B2 (en) | 2011-03-10 | 2022-10-11 | Board Of Regents, The University Of Texas System | Heterocyclic inhibitors of PTPN11 |
| ES2699351T3 (es) | 2014-01-17 | 2019-02-08 | Novartis Ag | Derivados de 1-piridazin/triazin-3-il-piper(-azina)/idina/pirolidina y composiciones de las mismas para inhibir la actividad de SHP2 |
| JO3517B1 (ar) | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| WO2015107494A1 (en) | 2014-01-17 | 2015-07-23 | Novartis Ag | 1 -(triazin-3-yi_/pyridazin-3-yl)-piper(-azine)idine derivatives and compositions thereof for inhibiting the activity of shp2 |
| JP6607870B2 (ja) * | 2014-05-13 | 2019-11-20 | メモリアル スローン ケタリング キャンサー センター | Hsp70モジュレーターならびにその作製および使用方法 |
| WO2016203405A1 (en) * | 2015-06-19 | 2016-12-22 | Novartis Ag | Compounds and compositions for inhibiting the activity of shp2 |
| EP3310779B1 (en) | 2015-06-19 | 2019-05-08 | Novartis AG | Compounds and compositions for inhibiting the activity of shp2 |
| US10287266B2 (en) | 2015-06-19 | 2019-05-14 | Novartis Ag | Compounds and compositions for inhibiting the activity of SHP2 |
| WO2017210134A1 (en) | 2016-05-31 | 2017-12-07 | Board Of Regents, University Of Texas System | Heterocyclic inhibitors of ptpn11 |
| MX388576B (es) | 2016-06-07 | 2025-03-20 | Jacobio Pharmaceuticals Co Ltd | Derivados heterociclicos novedosos utiles como inhibidores de shp2. |
| WO2017216706A1 (en) | 2016-06-14 | 2017-12-21 | Novartis Ag | Compounds and compositions for inhibiting the activity of shp2 |
| EP4302834A3 (en) | 2016-07-12 | 2024-07-17 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric shp2 inhibitors |
| WO2018057884A1 (en) | 2016-09-22 | 2018-03-29 | Relay Therapeutics, Inc. | Shp2 phosphatase inhibitors and methods of use thereof |
| TW202500565A (zh) | 2016-10-24 | 2025-01-01 | 美商傳達治療有限公司 | Shp2磷酸酶抑制劑及其使用方法 |
| CA3048340A1 (en) * | 2017-01-10 | 2018-07-19 | Novartis Ag | Pharmaceutical combination comprising an alk inhibitor and a shp2 inhibitor |
| KR20190110588A (ko) | 2017-01-23 | 2019-09-30 | 레볼루션 메디슨즈, 인크. | 알로스테릭 shp2 억제제로서의 피리딘 화합물 |
| JP7240319B2 (ja) | 2017-01-23 | 2023-03-15 | レヴォリューション・メディスンズ,インコーポレイテッド | アロステリックshp2阻害剤としての二環式化合物 |
| EA202190196A1 (ru) | 2017-03-23 | 2021-08-31 | Джакобио Фармасьютикалс Ко., Лтд. | Новые гетероциклические производные, применимые в качестве ингибиторов shp2 |
| EP3630770B1 (en) | 2017-05-26 | 2024-08-28 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine derivatives as shp2 phosphatase inhibitors |
| CA3074690A1 (en) | 2017-09-07 | 2019-03-14 | Revolution Medicines, Inc. | Shp2 inhibitor compositions and methods for treating cancer |
| JP7447002B2 (ja) | 2017-09-11 | 2024-03-11 | クルーゾン・ファーマシューティカルズ・インコーポレイテッド | SHP2のオクタヒドロシクロペンタ[c]ピロールのアロステリック阻害剤 |
| EP3687997A1 (en) | 2017-09-29 | 2020-08-05 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine derivatives as shp2 phosphatase inhibitors |
| TW201930292A (zh) | 2017-10-12 | 2019-08-01 | 美商銳新醫藥公司 | 作為變構shp2抑制劑的吡啶、吡嗪和三嗪化合物 |
| JP7361693B2 (ja) * | 2017-12-15 | 2023-10-16 | レヴォリューション・メディスンズ,インコーポレイテッド | アロステリックshp2阻害剤としての多環式化合物 |
| JP7335882B2 (ja) | 2018-02-13 | 2023-08-30 | ブルーレイ セラピューティクス (シャンハイ) カンパニー,リミティド | ピリミジン縮合環式化合物及びその製造方法、並びに使用 |
| IL276232B2 (en) | 2018-03-02 | 2024-04-01 | Otsuka Pharma Co Ltd | Pyrazine derivatives, pharmaceutical compositions comprising them and their use for treating diseases |
| US12138263B2 (en) | 2018-03-21 | 2024-11-12 | Relay Therapeutics, Inc. | Pyrazolo[3,4-b]pyrazine SHP2 phosphatase inhibitors and methods of use thereof |
| RU2020133727A (ru) * | 2018-03-21 | 2022-04-21 | Сучжоу Пухе Биофарма Ко., Лтд. | Ингибиторы shp2 и их применение |
| AU2019240299B2 (en) | 2018-03-21 | 2023-06-22 | D.E. Shaw Research, Llc | SHP2 phosphatase inhibitors and methods of use thereof |
| MX2020010719A (es) | 2018-04-10 | 2020-11-06 | Revolution Medicines Inc | Composiciones de inhibidores de shp2, metodos para tratar el cancer y metodos para identificar a un sujeto con mutaciones en shp2. |
| MX2020011528A (es) | 2018-05-02 | 2021-02-09 | Navire Pharma Inc | Inhibidores heterociclicos sustituidos de ptpn11. |
| MX2021000795A (es) | 2018-07-24 | 2021-04-12 | Taiho Pharmaceutical Co Ltd | Compuestos heterociclicos para inhibir la actividad de shp2. |
| CN112601750B (zh) * | 2018-08-10 | 2023-10-31 | 纳维尔制药有限公司 | Ptpn11(shp2)抑制剂 |
| CR20210175A (es) | 2018-09-18 | 2021-06-01 | Nikang Therapeutics Inc | Derivados de anillo tricíclico condensado como inhibidores de la fosfatasa de homología a src 2 (shp2) |
| US20210393623A1 (en) | 2018-09-26 | 2021-12-23 | Jacobio Pharmaceuticals Co., Ltd. | Novel Heterocyclic Derivatives Useful as SHP2 Inhibitors |
| CN112867718B (zh) * | 2018-09-29 | 2025-03-04 | 诺华股份有限公司 | 用于抑制shp2活性的化合物和组合物的制造 |
| IL305106B2 (en) | 2018-09-29 | 2025-08-01 | Novartis Ag | Process of manufacture of a compound for inhibiting the activity of shp2 |
| EP3860717A1 (en) | 2018-10-03 | 2021-08-11 | Gilead Sciences, Inc. | Imidozopyrimidine derivatives |
| JP2022508651A (ja) | 2018-10-08 | 2022-01-19 | レヴォリューション・メディスンズ,インコーポレイテッド | 癌を処置するためのshp2阻害剤組成物および方法 |
| BR112021005733A2 (pt) | 2018-10-17 | 2021-07-27 | Array Biopharma Inc. | inibidores de proteína tirosina fosfatase |
| CN117143079A (zh) * | 2018-11-06 | 2023-12-01 | 上海奕拓医药科技有限责任公司 | 一种螺芳环化合物及其应用 |
| EP3889153A4 (en) | 2018-11-30 | 2022-09-07 | Tuojie Biotech (Shanghai) Co., Ltd. | PYRIMIDINE AND FIVE-MEMBERED HETEROCYCLIC NITROGEN DERIVATIVE, METHOD OF MANUFACTURE THEREOF AND MEDICAL USES THEREOF |
| MX2021002804A (es) | 2018-12-05 | 2021-07-15 | Mirati Therapeutics Inc | Terapias de combinacion. |
| WO2020156242A1 (zh) * | 2019-01-31 | 2020-08-06 | 贝达药业股份有限公司 | Shp2抑制剂及其应用 |
| MX2021010319A (es) | 2019-03-01 | 2021-12-10 | Revolution Medicines Inc | Compuestos biciclicos de heteroarilo y usos de estos. |
| MX2021010323A (es) | 2019-03-01 | 2021-12-10 | Revolution Medicines Inc | Compuestos bicíclicos de heterociclilo y usos de este. |
| US11033547B2 (en) | 2019-03-07 | 2021-06-15 | Merck Patent Gmbh | Carboxamide-pyrimidine derivatives as SHP2 antagonists |
| CU20210080A7 (es) | 2019-04-02 | 2022-05-11 | Array Biopharma Inc | Triazinas sustituidas como inhibidores de proteína tirosina fosfatasa |
| JP7586834B2 (ja) | 2019-04-08 | 2024-11-19 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Shp2拮抗薬としてのピリミジノン誘導体 |
| KR20220054285A (ko) | 2019-06-28 | 2022-05-02 | 투오지에 바이오텍 (상하이) 컴퍼니 리미티드 | 피리미딘 5 원 질소 헤테로고리형 유도체, 이의 제조 방법 및 이의 약학적 용도 |
| CN111704611B (zh) * | 2019-07-25 | 2022-01-14 | 上海凌达生物医药有限公司 | 一类芳基螺环类shp2抑制剂化合物、制备方法和用途 |
| EP3772513A1 (en) * | 2019-08-09 | 2021-02-10 | C.N.C.C.S. S.c.a.r.l. Collezione Nazionale Dei Composti Chimici e Centro Screening | Shp2 inhibitors |
| GB201911928D0 (en) | 2019-08-20 | 2019-10-02 | Otsuka Pharma Co Ltd | Pharmaceutical compounds |
| CN114502165A (zh) * | 2019-09-23 | 2022-05-13 | 苏州浦合医药科技有限公司 | Shp2抑制剂及其用途 |
| MX2022003454A (es) | 2019-09-24 | 2022-04-19 | Relay Therapeutics Inc | Inhibidores de fosfatasa shp2 y metodos para su fabricacion y uso. |
| TW202132316A (zh) | 2019-11-04 | 2021-09-01 | 美商銳新醫藥公司 | Ras抑制劑 |
| AU2020377925A1 (en) | 2019-11-04 | 2022-05-05 | Revolution Medicines, Inc. | Ras inhibitors |
| IL322454A (en) | 2019-11-04 | 2025-09-01 | Revolution Medicines Inc | ras inhibitors |
| CA3156359A1 (en) | 2019-11-08 | 2021-05-14 | Adrian Liam Gill | Bicyclic heteroaryl compounds and uses thereof |
| EP4065231A1 (en) | 2019-11-27 | 2022-10-05 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| CA3164995A1 (en) | 2019-12-20 | 2021-06-24 | Erasca, Inc. | Tricyclic pyridones and pyrimidones |
| WO2021142026A1 (en) | 2020-01-07 | 2021-07-15 | Revolution Medicines, Inc. | Shp2 inhibitor dosing and methods of treating cancer |
| CN114761394B (zh) * | 2020-01-16 | 2024-03-29 | 浙江海正药业股份有限公司 | 吡啶或嘧啶类衍生物及其制备方法和用途 |
| CN113135910A (zh) * | 2020-01-19 | 2021-07-20 | 北京诺诚健华医药科技有限公司 | 嘧啶-4(3h)-酮类杂环化合物、其制备方法及其在医药学上的应用 |
| CN114846005B (zh) * | 2020-01-21 | 2024-04-02 | 贝达药业股份有限公司 | Shp2抑制剂及其应用 |
| EP4110338A1 (en) | 2020-02-28 | 2023-01-04 | Novartis AG | A triple pharmaceutical combination comprising dabrafenib, an erk inhibitor and a shp2 inhibitor |
| IL299131A (en) | 2020-06-18 | 2023-02-01 | Revolution Medicines Inc | Methods for delaying, preventing and treating acquired resistance to RAS inhibitors |
| US12441737B2 (en) | 2020-07-08 | 2025-10-14 | Novartis Ag | Manufacture of compounds and compositions for inhibiting the activity of SHP2 |
| CN115916765B (zh) * | 2020-07-10 | 2025-04-18 | 浙江海正药业股份有限公司 | 吡啶或嘧啶类衍生物及其制备方法和用途 |
| US20250195521A1 (en) | 2020-09-03 | 2025-06-19 | Revolution Medicines, Inc. | Use of sos1 inhibitors to treat malignancies with shp2 mutations |
| CA3194067A1 (en) | 2020-09-15 | 2022-03-24 | Revolution Medicines, Inc. | Ras inhibitors |
| JP2023544450A (ja) | 2020-09-23 | 2023-10-23 | エラスカ・インコーポレイテッド | 三環式ピリドン及びピリミドン |
| WO2022133345A1 (en) | 2020-12-18 | 2022-06-23 | Erasca, Inc. | Tricyclic pyridones and pyrimidones |
| JP7849366B2 (ja) | 2020-12-22 | 2026-04-21 | キル・レガー・セラピューティクス・インコーポレーテッド | Sos1阻害剤およびその使用 |
| EP4039685A1 (en) | 2021-02-08 | 2022-08-10 | Irbm S.P.A. | Azabicyclic shp2 inhibitors |
| EP4067358A1 (en) | 2021-04-02 | 2022-10-05 | C.N.C.C.S. S.c.a.r.l. Collezione Nazionale Dei Composti Chimici e Centro Screening | (s)-1-(5-((pyridin-3-yl)thio)pyrazin-2-yl)-4'h,6'h-spiro[piperidine-4,5'-pyrrolo[1,2-b]pyrazol]-4'-amine derivatives and similar compounds as shp2 inhibitors for the treatment of e.g. cancer |
| KR20240017811A (ko) | 2021-05-05 | 2024-02-08 | 레볼루션 메디슨즈, 인크. | 암의 치료를 위한 ras 억제제 |
| JP2024516450A (ja) | 2021-05-05 | 2024-04-15 | レボリューション メディシンズ インコーポレイテッド | 共有結合性ras阻害剤及びその使用 |
| TW202309052A (zh) | 2021-05-05 | 2023-03-01 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2022259157A1 (en) | 2021-06-09 | 2022-12-15 | Novartis Ag | A triple pharmaceutical combination comprising dabrafenib, trametinib and a shp2 inhibitor |
| TW202317100A (zh) | 2021-06-23 | 2023-05-01 | 瑞士商諾華公司 | 包含kras g12c抑制劑的藥物組合及其用於治療癌症之用途 |
| KR20240055778A (ko) | 2021-09-01 | 2024-04-29 | 노파르티스 아게 | Tead 억제제를 포함하는 제약 조합물 및 암의 치료를 위한 이의 용도 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| JP2025510572A (ja) | 2022-03-08 | 2025-04-15 | レボリューション メディシンズ インコーポレイテッド | 免疫不応性肺癌を治療するための方法 |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| EP4345101A1 (en) | 2022-09-29 | 2024-04-03 | Irbm S.P.A. | Azole derivatives as shp2 inhibitors |
| AU2024241633A1 (en) | 2023-03-30 | 2025-11-06 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
| CR20250420A (es) | 2023-04-07 | 2025-11-20 | Revolution Medicines Inc | Inhibidores macrocíclicos de ras |
| AR132338A1 (es) | 2023-04-07 | 2025-06-18 | Revolution Medicines Inc | Inhibidores de ras |
| CN121464140A (zh) | 2023-04-14 | 2026-02-03 | 锐新医药公司 | Ras抑制剂的结晶形式、含有其的组合物及其使用方法 |
| CN121100123A (zh) | 2023-04-14 | 2025-12-09 | 锐新医药公司 | Ras抑制剂的结晶形式 |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| AU2024321817A1 (en) | 2023-08-08 | 2026-03-05 | Quanta Therapeutics, Inc. | Combination therapies with kras modulators |
| AU2024360465A1 (en) | 2023-10-12 | 2026-04-09 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2025106901A1 (en) | 2023-11-17 | 2025-05-22 | Quanta Therapeutics, Inc. | Combination cancer therapies with a kras modulator and an rtk-mapk pathway inhibitor |
| TW202542151A (zh) | 2023-12-22 | 2025-11-01 | 美商銳格醫藥有限公司 | Sos1抑制劑及其用途 |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| TW202547461A (zh) | 2024-05-17 | 2025-12-16 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026060400A1 (en) | 2024-09-16 | 2026-03-19 | Quanta Therapeutics, Inc. | Combination therapies with kras modulators |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004002368A (ja) | 2002-04-04 | 2004-01-08 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JP2006511622A (ja) | 2002-12-18 | 2006-04-06 | グラクソ グループ リミテッド | 抗菌剤 |
| JP2009532452A (ja) | 2006-04-03 | 2009-09-10 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | ポリ(adp−リボース)ポリメラーゼ(parp)阻害剤としてのアミド置換インダゾール及びベンゾトリアゾール誘導体 |
| JP2011507849A (ja) | 2007-12-19 | 2011-03-10 | アムジエン・インコーポレーテツド | 細胞周期阻害剤としての縮合ピリジン、ピリミジンおよびトリアジン化合物 |
| US20110152243A1 (en) | 2009-12-23 | 2011-06-23 | Abbott Laboratories | Novel thienopyrrole compounds |
| JP2011529066A (ja) | 2008-07-23 | 2011-12-01 | バーテックス ファーマシューティカルズ インコーポレイテッド | 三環式ピラゾロピリジンキナーゼ阻害剤 |
| JP2014534974A (ja) | 2011-11-11 | 2014-12-25 | アッヴィ・インコーポレイテッド | Nampt阻害剤 |
| JP6473457B2 (ja) | 2014-01-17 | 2019-02-20 | ノバルティス アーゲー | Shp2の活性を阻害するための1−(トリアジン−3−イル/ピリダジン−3−イル)−ピペリジン/ピペラジン誘導体およびその組成物 |
| JP6534389B2 (ja) | 2014-01-17 | 2019-06-26 | ノバルティス アーゲー | Shp2の活性を阻害するためのn−アザスピロシクロアルカン置換n−ヘテロアリール化合物および組成物 |
| JP6718889B2 (ja) | 2015-06-19 | 2020-07-08 | ノバルティス アーゲー | Shp2の活性を阻害するための化合物および組成物 |
| JP6878316B2 (ja) | 2015-06-19 | 2021-05-26 | ノバルティス アーゲー | Shp2の活性を阻害するための化合物および組成物 |
Family Cites Families (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH445129A (fr) | 1964-04-29 | 1967-10-15 | Nestle Sa | Procédé pour la préparation de composés d'inclusion à poids moléculaire élevé |
| US3459731A (en) | 1966-12-16 | 1969-08-05 | Corn Products Co | Cyclodextrin polyethers and their production |
| US3453257A (en) | 1967-02-13 | 1969-07-01 | Corn Products Co | Cyclodextrin with cationic properties |
| US3426011A (en) | 1967-02-13 | 1969-02-04 | Corn Products Co | Cyclodextrins with anionic properties |
| US3453259A (en) | 1967-03-22 | 1969-07-01 | Corn Products Co | Cyclodextrin polyol ethers and their oxidation products |
| JPS6041077B2 (ja) | 1976-09-06 | 1985-09-13 | 喜徳 喜谷 | 1,2‐ジアミノシクロヘキサン異性体のシス白金(2)錯体 |
| US4235871A (en) | 1978-02-24 | 1980-11-25 | Papahadjopoulos Demetrios P | Method of encapsulating biologically active materials in lipid vesicles |
| US4261989A (en) | 1979-02-19 | 1981-04-14 | Kaken Chemical Co. Ltd. | Geldanamycin derivatives and antitumor drug |
| FR2567518B1 (fr) | 1984-07-11 | 1987-11-13 | Sanofi Sa | Nouveaux composes a noyau heterocyclique azote, leur preparation et les medicaments qui en contiennent |
| US4737323A (en) | 1986-02-13 | 1988-04-12 | Liposome Technology, Inc. | Liposome extrusion method |
| WO1987004928A1 (fr) | 1986-02-24 | 1987-08-27 | Mitsui Petrochemical Industries, Ltd. | Agents therapeutiques de la neuropathie |
| US5266573A (en) | 1989-08-07 | 1993-11-30 | Elf Sanofi | Trifluoromethylphenyltetrahydropyridines for the treatment and/or prophylaxis of intestinal motility disorders |
| KR0166088B1 (ko) | 1990-01-23 | 1999-01-15 | . | 수용해도가 증가된 시클로덱스트린 유도체 및 이의 용도 |
| DK169008B1 (da) | 1990-06-01 | 1994-07-25 | Holec Lk A S | Fremgangsmåde og skærm til afskærmning af en strømtransformer samt strømtransformer med en sådan afskærmning |
| WO1994014794A1 (fr) * | 1992-12-28 | 1994-07-07 | Yoshitomi Pharmaceutical Industries, Ltd. | Derive d'acide 8-methoxyquinolonecarboxylique |
| US5455258A (en) * | 1993-01-06 | 1995-10-03 | Ciba-Geigy Corporation | Arylsulfonamido-substituted hydroxamic acids |
| EP0647450A1 (en) | 1993-09-09 | 1995-04-12 | BEHRINGWERKE Aktiengesellschaft | Improved prodrugs for enzyme mediated activation |
| IL115849A0 (en) | 1994-11-03 | 1996-01-31 | Merz & Co Gmbh & Co | Tangential filtration preparation of liposomal drugs and liposome product thereof |
| AU4198400A (en) | 1999-04-06 | 2000-10-23 | Krenitsky Pharmaceuticals Inc. | Neurotrophic thio substituted pyrimidines |
| PE20010306A1 (es) | 1999-07-02 | 2001-03-29 | Agouron Pharma | Compuestos de indazol y composiciones farmaceuticas que los contienen utiles para la inhibicion de proteina kinasa |
| GB0018891D0 (en) | 2000-08-01 | 2000-09-20 | Novartis Ag | Organic compounds |
| JP4272338B2 (ja) | 2000-09-22 | 2009-06-03 | バイエル アクチェンゲゼルシャフト | ピリジン誘導体 |
| US6995162B2 (en) | 2001-01-12 | 2006-02-07 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| ATE407678T1 (de) | 2001-10-17 | 2008-09-15 | Boehringer Ingelheim Pharma | Pyrimidinderivate, arzneimittel enthaltend diese verbindungen, deren verwendung und verfahren zu ihrer herstellung |
| PT1478648E (pt) | 2002-02-01 | 2014-07-15 | Ariad Pharma Inc | Compostos contendo fósforo e suas utilizações |
| CN100430052C (zh) | 2002-03-05 | 2008-11-05 | 默克弗罗斯特加拿大有限公司 | 组织蛋白酶半胱氨酸蛋白酶抑制剂 |
| TWI275390B (en) | 2002-04-30 | 2007-03-11 | Wyeth Corp | Process for the preparation of 7-substituted-3- quinolinecarbonitriles |
| AU2004218228A1 (en) | 2003-03-07 | 2004-09-16 | Merck Sharp & Dohme Limited | Tetrahydropyran compounds as tachykinin antagonists |
| US7399865B2 (en) | 2003-09-15 | 2008-07-15 | Wyeth | Protein tyrosine kinase enzyme inhibitors |
| WO2005095384A1 (en) | 2004-04-01 | 2005-10-13 | Astellas Pharma Inc. | Pyrazine derivatives and pharmaceutical use thereof as adenosine antagonists |
| WO2005117909A2 (en) | 2004-04-23 | 2005-12-15 | Exelixis, Inc. | Kinase modulators and methods of use |
| ES2377430T3 (es) | 2004-09-02 | 2012-03-27 | Genentech, Inc. | Inhibidores piridílicos de la señalización de hedgehog |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| CA2622372A1 (en) | 2005-09-13 | 2007-03-22 | Palau Pharma, S.A. | 2-aminopyrimidine derivatives as modulators of the histamine h4 receptor activity |
| WO2007112368A1 (en) * | 2006-03-28 | 2007-10-04 | Takeda Pharmaceutical Company Limited | Preparation of (r)-3-aminopiperidine dihydrochloride |
| US7515405B2 (en) | 2006-07-25 | 2009-04-07 | Hewlett-Packard Development Company, L.P. | Anti-rotation mechanism for an electronic device |
| US20090137549A1 (en) | 2006-11-09 | 2009-05-28 | Paul John Edward | Novel compounds useful for the treatment of degenerative & inflamatory diseases |
| US8252803B2 (en) | 2007-02-12 | 2012-08-28 | Merck Sharp & Dohme Corp. | Piperidine derivatives |
| CA2681162C (en) | 2007-03-15 | 2015-11-24 | Novartis Ag | Benzyl and pyridine derivatives as modulators of hedgehog pathway |
| EP2205242B1 (en) | 2007-09-12 | 2015-04-15 | Genentech, Inc. | Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents, and methods of use |
| JP5348725B2 (ja) | 2007-10-25 | 2013-11-20 | ジェネンテック, インコーポレイテッド | チエノピリミジン化合物の製造方法 |
| AU2009238590A1 (en) | 2008-04-22 | 2009-10-29 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| JP2011522865A (ja) | 2008-06-13 | 2011-08-04 | ノバルティス アーゲー | Ia心不全および癌の治療に有用なプロテインキナーゼd阻害剤としての2,4’−ビピリジニル化合物 |
| CA2727174A1 (en) | 2008-06-20 | 2010-01-21 | Jiangao Song | Aryl gpr119 agonists and uses thereof |
| US8168784B2 (en) | 2008-06-20 | 2012-05-01 | Abbott Laboratories | Processes to make apoptosis promoters |
| NZ603525A (en) | 2008-06-27 | 2015-02-27 | Celgene Avilomics Res Inc | Pyrimidine based compound and uses thereof |
| WO2010048149A2 (en) | 2008-10-20 | 2010-04-29 | Kalypsys, Inc. | Heterocyclic modulators of gpr119 for treatment of disease |
| CN102356066A (zh) | 2008-12-10 | 2012-02-15 | 同和药品株式会社 | 新型2,6-取代的-3-硝基吡啶衍生物、其制备方法及包含有其的药物组合物 |
| FR2941696B1 (fr) | 2009-02-05 | 2011-04-15 | Sanofi Aventis | Derives d'azaspiranyl-alkylcarbamates d'heterocycles a 5 chainons, leur preparation et leur application en therapeutique |
| WO2010121212A2 (en) | 2009-04-17 | 2010-10-21 | H. Lee Moffit Cancer Center And Research Institute, Inc. | Indoline scaffold shp-2 inhibitors and method of treating cancer |
| NZ713361A (en) | 2009-08-17 | 2017-06-30 | Memorial Sloan Kettering Cancer Center | Heat shock protein binding compounds, compositions, and methods for making and using same |
| WO2011078143A1 (ja) | 2009-12-22 | 2011-06-30 | 塩野義製薬株式会社 | ピリミジン誘導体およびそれらを含有する医薬組成物 |
| ES2689103T3 (es) | 2010-06-30 | 2018-11-08 | Fujifilm Corporation | Nuevo derivado de nicotinamida o sal del mismo |
| JP2013531037A (ja) | 2010-07-13 | 2013-08-01 | メルク・シャープ・エンド・ドーム・コーポレイション | スピロ環式化合物 |
| PE20130774A1 (es) | 2010-07-29 | 2013-06-26 | Rigel Pharmaceuticals Inc | Compuestos heterociclicos activadores de ampk y metodos para emplearlos |
| US8609672B2 (en) | 2010-08-27 | 2013-12-17 | University Of The Pacific | Piperazinylpyrimidine analogues as protein kinase inhibitors |
| CA2813162C (en) | 2010-10-20 | 2015-06-16 | Pfizer Inc. | Pyridine-2- derivatives as smoothened receptor modulators |
| US9321760B2 (en) | 2011-09-12 | 2016-04-26 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013096093A1 (en) | 2011-12-21 | 2013-06-27 | Merck Sharp & Dohme Corp. | Compounds as dgat-1 inhibitors |
| EA026353B1 (ru) * | 2012-01-17 | 2017-03-31 | Астеллас Фарма Инк. | Соединение пиразинкарбоксамида |
| CN104540830A (zh) | 2012-06-07 | 2015-04-22 | 霍夫曼-拉罗奇有限公司 | 端锚聚合酶的吡唑并嘧啶酮和吡唑并吡啶酮抑制剂 |
| JP6099753B2 (ja) | 2012-10-03 | 2017-03-22 | アドビナス セラピューティクス リミテッド | スピロ環化合物、その組成物及びその医薬応用 |
| WO2014160521A1 (en) | 2013-03-15 | 2014-10-02 | Blueprint Medicines Corporation | Piperazine derivatives and their use as kit modulators |
| ES2699351T3 (es) | 2014-01-17 | 2019-02-08 | Novartis Ag | Derivados de 1-piridazin/triazin-3-il-piper(-azina)/idina/pirolidina y composiciones de las mismas para inhibir la actividad de SHP2 |
| KR102400920B1 (ko) * | 2014-05-01 | 2022-05-20 | 셀젠 콴티셀 리서치, 인크. | 리신 특이적 데메틸라제-1의 억제제 |
| US10174032B2 (en) | 2014-05-05 | 2019-01-08 | Signalrx Pharmaceuticals, Inc. | Heterocyclic compound classes for signaling modulation |
| EP3310779B1 (en) | 2015-06-19 | 2019-05-08 | Novartis AG | Compounds and compositions for inhibiting the activity of shp2 |
-
2016
- 2016-06-15 WO PCT/IB2016/053549 patent/WO2016203405A1/en not_active Ceased
- 2016-06-15 EP EP16731665.2A patent/EP3310771B1/en active Active
- 2016-06-15 JP JP2017565688A patent/JP6878316B2/ja active Active
- 2016-06-15 US US15/736,989 patent/US10308660B2/en active Active
- 2016-06-15 ES ES16731665T patent/ES2824576T3/es active Active
- 2016-06-15 CN CN201680035706.9A patent/CN107787323B/zh active Active
-
2021
- 2021-04-28 JP JP2021076566A patent/JP7146016B2/ja active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004002368A (ja) | 2002-04-04 | 2004-01-08 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JP2006511622A (ja) | 2002-12-18 | 2006-04-06 | グラクソ グループ リミテッド | 抗菌剤 |
| JP2009532452A (ja) | 2006-04-03 | 2009-09-10 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | ポリ(adp−リボース)ポリメラーゼ(parp)阻害剤としてのアミド置換インダゾール及びベンゾトリアゾール誘導体 |
| JP2011507849A (ja) | 2007-12-19 | 2011-03-10 | アムジエン・インコーポレーテツド | 細胞周期阻害剤としての縮合ピリジン、ピリミジンおよびトリアジン化合物 |
| JP2011529066A (ja) | 2008-07-23 | 2011-12-01 | バーテックス ファーマシューティカルズ インコーポレイテッド | 三環式ピラゾロピリジンキナーゼ阻害剤 |
| US20110152243A1 (en) | 2009-12-23 | 2011-06-23 | Abbott Laboratories | Novel thienopyrrole compounds |
| JP2014534974A (ja) | 2011-11-11 | 2014-12-25 | アッヴィ・インコーポレイテッド | Nampt阻害剤 |
| JP6473457B2 (ja) | 2014-01-17 | 2019-02-20 | ノバルティス アーゲー | Shp2の活性を阻害するための1−(トリアジン−3−イル/ピリダジン−3−イル)−ピペリジン/ピペラジン誘導体およびその組成物 |
| JP6534389B2 (ja) | 2014-01-17 | 2019-06-26 | ノバルティス アーゲー | Shp2の活性を阻害するためのn−アザスピロシクロアルカン置換n−ヘテロアリール化合物および組成物 |
| JP6718889B2 (ja) | 2015-06-19 | 2020-07-08 | ノバルティス アーゲー | Shp2の活性を阻害するための化合物および組成物 |
| JP6878316B2 (ja) | 2015-06-19 | 2021-05-26 | ノバルティス アーゲー | Shp2の活性を阻害するための化合物および組成物 |
Non-Patent Citations (4)
| Title |
|---|
| FORNS,P. et al.,Pyrazine-based Syk kinase inhibitors,Bioorganic & Medicinal Chemistry Letters,2012年,Vol.22, No.8,p.2784-2788 |
| STN International,file CAPLUS [online],[令和4年5月18日検索],CAS AN= 1973:515409 & TOSUNYAN,A.O. et al, Synthesis of tetrahydropyrans, tetrahydrothiopyrans, and 4-piperidinones from vinylacetylenic alcohols, Dokl. Vses. Konf. Khim. Atsetilena, 4th, 1972, vol.1, p.293-5,CAS Registry No. 50289-09-3の化合物参照 |
| STN International,file REGISTRY [online],[令和4年5月13日検索],CAS Registry No. 1468803-33-9 (Entered STN: 03 Nov 2013 ), 1439897-97-8 (Entered STN: 20 Jun 2013 ), 1416374-13-4 (Entered STN: 10 Jan 2013 )参照 |
| STN International,file REGISTRY [online],[令和4年5月13日検索],CAS Registry No. 1781072-03-4 (Entered STN: 15 Jun 2015 ), Registry No. 1779573-14-6 (Entered STN: 12 Jun 2015 ), Registry No. 1779573-00-0 (Entered STN: 12 Jun 2015 ), Registry No. 1779130-03-8 (Entered STN: 12 Jun 2015 ), Registry No. 1779124-66-1 (Entered STN: 12 Jun 2015 ), Registry No. 1774896-82-0 (Entered STN: 07 Jun 2015 ), Registry No. 1707602-31-0 (Entered STN: 19 May 2015 )等参照 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180251471A1 (en) | 2018-09-06 |
| JP6878316B2 (ja) | 2021-05-26 |
| EP3310771A1 (en) | 2018-04-25 |
| CN107787323B (zh) | 2020-09-01 |
| CN107787323A (zh) | 2018-03-09 |
| JP2021130662A (ja) | 2021-09-09 |
| WO2016203405A1 (en) | 2016-12-22 |
| US10308660B2 (en) | 2019-06-04 |
| EP3310771B1 (en) | 2020-07-22 |
| JP2018517746A (ja) | 2018-07-05 |
| ES2824576T3 (es) | 2021-05-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7146016B2 (ja) | Shp2の活性を阻害するための化合物および組成物 | |
| JP7278359B2 (ja) | Shp2の活性を阻害するための化合物および組成物 | |
| JP6718889B2 (ja) | Shp2の活性を阻害するための化合物および組成物 | |
| JP6523303B2 (ja) | Shp2の活性を阻害するための1−ピリダジン/トリアジン−3−イル−ピペラジン/ピペリジン/ピロリジン誘導体およびその組成物 | |
| JP6534389B2 (ja) | Shp2の活性を阻害するためのn−アザスピロシクロアルカン置換n−ヘテロアリール化合物および組成物 | |
| JP6473457B2 (ja) | Shp2の活性を阻害するための1−(トリアジン−3−イル/ピリダジン−3−イル)−ピペリジン/ピペラジン誘導体およびその組成物 | |
| EP3310779B1 (en) | Compounds and compositions for inhibiting the activity of shp2 | |
| BR112018075663B1 (pt) | Compostos para inibição da atividade de shp2, seu uso, e composição farmacêutica | |
| HK1228375A1 (en) | N-azaspirocycloalkane substituted n-heteroaryl compounds and compositions for inhibiting the activity of shp2 | |
| HK1228375B (en) | N-azaspirocycloalkane substituted n-heteroaryl compounds and compositions for inhibiting the activity of shp2 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210528 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210528 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220531 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220830 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220906 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220920 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7146016 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |