JP7266896B2 - β修飾リン酸化合物前駆体、β修飾リン酸化合物、反応阻害剤及びこれを含む医薬並びに反応阻害方法 - Google Patents
β修飾リン酸化合物前駆体、β修飾リン酸化合物、反応阻害剤及びこれを含む医薬並びに反応阻害方法 Download PDFInfo
- Publication number
- JP7266896B2 JP7266896B2 JP2020505121A JP2020505121A JP7266896B2 JP 7266896 B2 JP7266896 B2 JP 7266896B2 JP 2020505121 A JP2020505121 A JP 2020505121A JP 2020505121 A JP2020505121 A JP 2020505121A JP 7266896 B2 JP7266896 B2 JP 7266896B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- reaction
- carbon atoms
- compound
- modified phosphate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon or a metal, e.g. chelates or vitamin B12
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/02—Phosphorylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H11/00—Compounds containing saccharide radicals esterified by inorganic acids; Metal salts thereof
- C07H11/04—Phosphates; Phosphites; Polyphosphates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/19—Purine radicals with arabinosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
- C07H19/207—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids the phosphoric or polyphosphoric acids being esterified by a further hydroxylic compound, e.g. flavine adenine dinucleotide or nicotinamide-adenine dinucleotide
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Description

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。*はリン酸化を受けてリン酸基が結合する結合手であり、リン酸化前においては水素又はリン酸基以外の置換基が結合していることを意味する。)

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、又は下記式2Dで示されるモノリン酸基、ジリン酸基、又はトリリン酸基である。

(ここで、nは1~3の整数を示す。Z1は、ヒドロキシル基、又はグリシン、アラニン、バリン、ロイシン、フェニルアラニン、トリプトファン、メチオニン若しくはプロリンのメチルエステル、エチルエステル、イソプロピルエステル、n-ブチルエステル、ベンジルエステル若しくはフェニルエステルである。Z2は、水素、炭素数1~4のアルキル基、ハロゲン、又はフェニル基である。nが2以上の場合においてそれぞれのZ1は同一又は異なっていてもよい。)
Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。*はリン酸化を受けてリン酸基が結合する結合手であり、リン酸化前においては水素又はリン酸基以外の置換基が結合していることを意味する。)

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。)
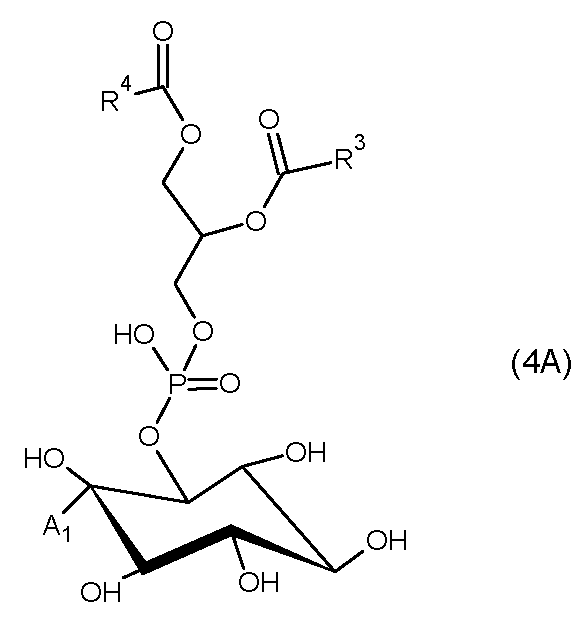
(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。R3はアラキドン酸、リノール酸、及びリノレン酸から選択される不飽和脂肪酸であり、R4はステアリン酸、パルミチン酸から選択される飽和脂肪酸である。)

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、炭素数1~20のアルケニル基から選択される。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。)

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、又は下記式2Dで示されるモノリン酸基、ジリン酸基、又はトリリン酸基である。

(ここで、nは1~3の整数を示す。Z1は、ヒドロキシル基、又はグリシン、アラニン、バリン、ロイシン、フェニルアラニン、トリプトファン、メチオニン若しくはプロリンのメチルエステル、エチルエステル、イソプロピルエステル、n-ブチルエステル、ベンジルエステル若しくはフェニルエステルである。Z2は、水素、炭素数1~4のアルキル基、ハロゲン、又はフェニル基である。nが2以上の場合においてそれぞれのZ1は同一又は異なっていてもよい。)
Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。)

(ここで、A2は-S-、-S+(R1)-、-S+(S-R1)-、-Se+(R1)-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。)

(ここで、A2は-S-、-S+(R1)-、-S+(S-R1)-、-Se+(R1)-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、又は下記式2Dで示されるモノリン酸基、ジリン酸基、又はトリリン酸基である。

(ここで、nは1~3の整数を示す。Z1は、ヒドロキシル基、又はグリシン、アラニン、バリン、ロイシン、フェニルアラニン、トリプトファン、メチオニン若しくはプロリンのメチルエステル、エチルエステル、イソプロピルエステル、n-ブチルエステル、ベンジルエステル若しくはフェニルエステルである。Z2は、水素、炭素数1~4のアルキル基、ハロゲン、又はフェニル基である。nが2以上の場合においてそれぞれのZ1は同一又は異なっていてもよい。)
Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。)
(1)β修飾リン酸化合物前駆体
以下、本発明のβ修飾リン酸化合物前駆体について説明する。本発明のβ修飾リン酸化合物前駆体は、リン酸化反応によりリン酸化されるβ修飾リン酸化合物前駆体であって、下記式1Aの部分構造を分子内に有する。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。*はリン酸化を受けてリン酸基が結合する結合手であり、リン酸化前においては水素又はリン酸基以外の置換基が結合していることを意味する。)
次に、β修飾リン酸化合物について説明する。本発明のβ修飾リン酸化合物は、式1Aのβ修飾リン酸化合物前駆体において、α位の炭素に結合する酸素がリン酸化された化合物である。具体的には、下記式1Bで示される部分構造を有するβ修飾リン酸化合物である。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、炭素数1~20のアルケニル基から選択される。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。)
次に、反応阻害剤及び反応阻害方法について説明する。本発明の反応阻害剤は、リン酸化を伴う反応におけるリン酸化後の反応の進行を阻害する反応阻害剤であって、上記式1Aで示される部分構造を分子内に有するβ修飾リン酸化合物前駆体を含有する。そして、上記式1Aのβ修飾リン酸化化合物前駆体がリン酸化されて上記式1Bのβ修飾リン酸化化合物が生成する。このβ修飾リン酸化化合物は、以下の反応メカニズムにより、リン酸化後の反応を阻害する。

(ここで、A2は-S-、-S+R1-、-S+-S-R1-、-Se+R1-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示し、L2は炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基を示す。L1とL2は互いに連結して4~6員の環構造を形成してもよく、環構造は炭素、窒素、酸素、及び硫黄から選択される1種類以上の元素で構成される。L1及びL2は、それぞれヒドロキシル基、カルボキシル基、アミノ基、アルキル基、アリール基、炭素数15~30の飽和脂肪酸及び/又は不飽和脂肪酸を1以上含むリン脂質、モノリン酸基、ジリン酸基、又はトリリン酸基、及び塩基(ここで、塩基はアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルを意味する)から選択される1又は2以上の置換基を有していてもよい。)
本発明の反応阻害剤は、上記のようにリン酸化後の反応を阻害することから、医薬、農薬などの薬剤における有効成分として有用であり、特に、医薬用途が好ましい。医薬の種類としては、錠剤、カプセル、丸剤、散剤、顆粒剤、細粒剤、ゼリー剤、液剤などを挙げることができる。
次に、β修飾リン酸化合物前駆体の製造方法について説明する。式1Aのβ修飾リン酸化合物前駆体は、下記式のスキームで合成することができる。具体的には、β位の炭素にヒドロキシル基などが結合した化合物を出発物質とし、このヒドロキシル基以外のヒドロキシル基に保護基を結合して保護する。次に、β炭素に結合したヒドロキシル基をA1で置換し、最後に保護基を脱離させることで式1Aの化合物を合成することができる。出発物質や保護基の種類、反応条件(濃度、温度等)などは、合成するβ修飾リン酸化合物前駆体によって異なる。

(1)β修飾リン酸化合物前駆体
β修飾リン酸化合物前駆体としては、下記式2Aで示されるヌクレオシド誘導体を挙げることができる。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、又は下記式2Dで示されるモノリン酸基、ジリン酸基、又はトリリン酸基である。

(ここで、nは1~3の整数を示す。Z1は、ヒドロキシル基、又はグリシン、アラニン、バリン、ロイシン、フェニルアラニン、トリプトファン、メチオニン若しくはプロリンのメチルエステル、エチルエステル、イソプロピルエステル、n-ブチルエステル、ベンジルエステル若しくはフェニルエステルである。Z2は、水素、炭素数1~4のアルキル基、ハロゲン、又はフェニル基である。nが2以上の場合においてそれぞれのZ1は同一又は異なっていてもよい。)
Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。*はリン酸化を受けてリン酸基が結合する結合手であり、リン酸化前においては水素又はリン酸基以外の置換基が結合していることを意味する。)





上記式2Aで示されるヌクレオシド誘導体がリン酸化を受けたあとのβ修飾リン酸化合物としては、下記式2Bで示される部分構造を有するβ修飾リン酸化合物を挙げることができる。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、又は上記式2Dで示されるモノリン酸基、ジリン酸基、又はトリリン酸基である。Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。)
β修飾リン酸化合物前駆体が式2Aで示されるヌクレオシド誘導体であり、β修飾リン酸化合物が上記式2Bで示されるヌクレオシド誘導体である場合、中間体である活性種として下記式2Cで示される化合物が生成する。

(ここで、A2は-S-、-S+(R1)-、-S+(S-R1)-、-Se+(R1)-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L3は水素、モノリン酸基、ジリン酸基、又はトリリン酸基である。Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。)
ヌクレオシド誘導体は、ポリメラーゼ阻害剤であり、DNAの複製やRNAの転写反応を阻害するため、細胞やウイルスの増殖などを抑制する。このため、式2Aで示されるヌクレオシド誘導体は、増殖阻害剤であり、ウイルス性疾患や癌などの治療薬として有用である。
以下、ヌクレオシド誘導体の製造方法について説明する。具体的な反応条件などは、後述する実施例において詳細に説明するため、ここでは、いくつかのヌクレオシド誘導体について、合成方法(製造方法)の概略を説明する。
下記の合成スキームに沿って説明する。以下で説明する合成スキームにおいて、数字は化合物の番号を表す。まず、ヌクレオシド(下記スキームではアデノシン)を出発物質とし、1,3-ジクロロ-1,1,3,3-テトライソプロピルジシロキサン(TPDSCl2)をピリジンなどの溶媒中で反応させる。これにより、リボースの3’ヒドロキシル基と5’ヒドロキシル基との間でシロキサン結合の環状構造を形成させ、3’位と5’位のヒドロキシル基を保護する(化合物1)。次に、N-フェニルトリフルオロメタンスルホンイミドを添加して、N,N-ジメチル-4-アミノピリジン(DMAP)などの求核剤をジクロロメタン(DCM)などの溶媒中で反応させて、リボースの2’位をトリフルオロスルホン酸基とする(化合物2)。次に、N,N-ジメチルホルムアミド(DMF)などの存在下でチオ酢酸カリウムなどのチオール基を有する化合物を反応させて、リボースの2’位にチオエステルを形成させる(化合物3)。さらにアンモニア/メタノール溶液中で反応させることでチオエステル基をチオール基に変換する(化合物4)。続いて、トリエチルアミン三フッ化水素酸塩(3HF-Et3N)とテトラヒドロフラン(THF)とを添加してリボースの3’位と5’位の保護基を脱離させてヒドロキシル基にする。

化合物4までの合成スキームは上記の式(2A-1A)の化合物の合成の場合と同じである。続いて、テトラヒドロフラン(THF)とアゾジカルボン酸ジイソプロピル(DIAD)の混合溶媒などの溶媒中で1-プロパンチオールなどのチオアルキル化合物と反応させてリボースの2’位をジスルフィドアルキル基に変換する(化合物5)。次に、溶媒中で塩化ベンゾイル(BzCl)と反応させて、塩基のアミノ基をベンゾイル基で保護する。続いて、トリエチルアミン三フッ化水素酸塩(3HF-Et3N)とTHFとを添加してリボースの3’位と5’位の保護基を脱離させてヒドロキシル基にする(化合物11)。

化合物2までの合成スキームは上記の式(2A-1A)の化合物の合成の場合と同じである。次に、ジメチルジセレニド、水素化ホウ素ナトリウム、さらにはTHF等の溶媒中で反応させる(化合物12)。さらに、フッ化テトラ-n-ブチルアンモニウム(TBAF)、THFなどを添加してリボースの3’位と5’位の保護基を脱離させてヒドロキシル基にする(化合物13)。

次に、ハロゲン化合物の合成スキームについて説明する。化合物2までの合成スキームは上記の式(2A-1A)の化合物の合成の場合と同じである。次に、ハロゲン化リチウムを添加して加熱してハロゲンを導入する。ハロゲン化リチウムとしては、フッ化リチウム、塩化リチウム、臭化リチウム、ヨウ化リチウムを挙げることができる。続いて、トリエチルアミン三フッ化水素酸塩(3HF-Et3N)とTHFとを添加してリボースの3’位と5’位の保護基を脱離させてヒドロキシル基にする。

(1)β修飾リン酸化合物前駆体
β修飾リン酸化合物前駆体としては、下記式3Aで示されるメバロン酸誘導体を挙げることができる。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。)
また、上記式3Aで示されるメバロン酸誘導体がリン酸化を受けたあとのβ修飾リン酸化合物としては、下記式3Bで示される部分構造を有するβ修飾リン酸化合物を挙げることができる。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。)
特に、β修飾リン酸化合物前駆体が式3Aで示されるメバロン酸誘導体であり、β修飾リン酸化合物が上記式3Bで示されるメバロン酸誘導体である場合、中間体である活性種として下記式3Cで示される化合物が生成する。

(ここで、A2は-S-、-S+(R1)-、-S+(S-R1)-、-Se+(R1)-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。)
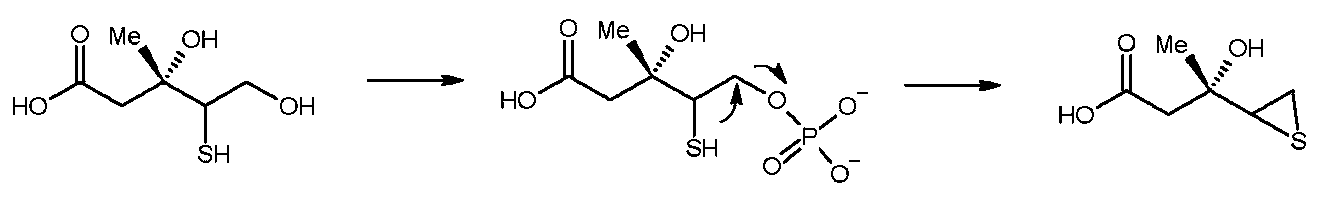
式3Aで示されるメバロン酸誘導体は、メバロン酸経路を阻害するため、コレステロールの生成を抑制する。このため、式3Aで示されるメバロン酸誘導体は、コレステロール生成阻害剤であり、高脂血症や高コレステロール血症などの治療薬として有用である。
(1)β修飾リン酸化合物前駆体
β修飾リン酸化合物前駆体としては、下記式4Aで示されるホスファチジルイノシトール誘導体を挙げることができる。

(ここで、A1は-SR1、-S-S-R1、-SeR1、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。R3はアラキドン酸、リノール酸、及びリノレン酸から選択される不飽和脂肪酸であり、R4はステアリン酸、パルミチン酸から選択される飽和脂肪酸である。)
特に、β修飾リン酸化合物前駆体が式4Aで示されるホスファチジルイノシトール誘導体であり、β修飾リン酸化合物が上記式4Bで示されるホスファチジルイノシトール誘導体である場合、中間体である活性種として下記式4Cで示される化合物が生成する。

(ここで、A2は-S-、-S+(R1)-、-S+(S-R1)-、-Se+(R1)-、又は-X+-(ここで、Xはフッ素、塩素、臭素、及びヨウ素から選択されるハロゲンを意味する)を示し、R1は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。R3はアラキドン酸、リノール酸、及びリノレン酸から選択される不飽和脂肪酸であり、R4はステアリン酸、パルミチン酸から選択される飽和脂肪酸である。)

式4Aで示されるホスファチジルイノシトールは、PI3K活性阻害剤であり、癌、悪性リンパ腫、白血病、リウマチなどの治療薬として有用である。
式4Aで示されるホスファチジルイノシトールの製造方法は、式2Aで示されるヌクレオシド誘導体と同様の方法で製造することができる。すなわち、式4Aのホスファチジルイノシトールは、以下の手順で合成することができる。まず、シロキサン類を保護基としてイノシトールの2’以外のヒドロキシル基を保護する。次に、2’位にトリフルオロスルホン酸基を導入したのち、チオエステルからチオール基に変換する。最後に保護基をイノシトールから脱離させる。


(1)化合物1の合成
アルゴン(Ar)雰囲気下で、pyridine(65mL)にadenosine(3.01g,10.1mmol,1.0eq.)を溶かし、TPDSCl2(3.2mL,10.1mmol,1.0eq.)を加え、室温で46時間撹拌した。減圧下で溶媒を留去し、酢酸エチルと水(0.1M HCl aq.,H2O,sat.NaHCO3 aq.,brine)で分液した。無水硫酸ナトリウムを加えて乾燥した後、綿栓ろ過し、減圧下で溶媒を留去した。中性フラッシュカラムクロマトグラフィー(CHCl3(MeOH 0-4%))にて精製し、化合物1(4.14g,80%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ1.06-1.13(m,28H),3.31(s,1H),4.01-4.16(m,3H),4.56(d,J=5.6Hz,1H),5.07-5.11(m,1H),5.79(s,2H),5.97(d,J=1.2Hz,1H),7.97(s,1H),8.28(s,1H).
Ar雰囲気下、氷冷下で化合物1(1.00g,1.83mmol,1.0eq.)とDMAP(650mg,5.5mmol,3eq.)を脱水DCM(17mL)に溶かし、N-Phenyltrifluoromethanesulfonimide(803mg,2.2mmol,1.2eq.)を加えて2時間撹拌した。氷冷したジクロロメタンと水(0.1M AcOH aq.,sat. NaHCO3 aq.,brine)で分液した。無水硫酸ナトリウムを加えて乾燥した後、綿栓ろ過し、減圧下で溶媒を留去した。中性フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=1/1)で精製し、化合物2(946mg,81%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ1.06-1.11(m,28H,),4.02-4.20(m,3H),5.26-5.29(m,1H),5.72(s,2H),5.78(d,J=4.8 Hz,1H),6.10(s,1H),7.96(s,1H),8.26(s,1H).
Ar雰囲気下で化合物2(2.80g,4.36mmol,1.0eq.)及び、脱水アセトニトリルで共沸したチオ酢酸カリウム(1.06g,9.28mmol,2.1eq.)を脱水DMF(9mL)に溶かし、14.5時間撹拌した。減圧下で溶媒を留去し、ヘキサン/酢酸エチル=1/5混合溶媒と水(sat. NaHCO3 aq.,brine)で分液した。無水硫酸ナトリウムを加えて乾燥した後、綿栓ろ過し、減圧下で溶媒を留去した。中性フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=2/3)で精製し、化合物3(1.72g,67%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ1.01-1.18(m,28H),2.15(s,3H),3.97-4.03(m,2H),4.23-4.27(m,1H),4.54-4.58(m,1H),5.01-5.06(m,1H),6.12(s,1H),6.40(d,J=7.6Hz,1H),7.90(s,1H),8.23(s,1H).
Ar雰囲気下、氷冷下で化合物3(1.42g,2.50mmol)を7M NH3-MeOH(25mL)に溶かし1時間撹拌した。減圧下で溶媒を留去し、酢酸エチルと水(sat. NaHCO3 aq.,brine)で分液した。無水硫酸ナトリウムを加えて乾燥した後、綿栓ろ過し、減圧下で溶媒を留去し、化合物4(1.75g,quant.)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ1.00-1.18(m,28H),1.44(d,J=8.4Hz,1H),3.81-3.91(m,2H),4.03-4.07(m,1H),4.20-4.24(m,1H),4.58-4.63(m,1H),5.76(s,1H),6.40(d,J=7.6Hz,1H),8.11(s,1H),8.33(s,1H).
化合物4(304mg,0.571mmol,1.0eq.)をTHF(1.9mL,0.3M to 化合物4)に溶かし、DIAD(124μL,0.628mmol,1.1eq.)を加えて18時間撹拌した。1-propanethiol(3.1mL,33.7mmol,59eq.)を加え、80℃に加熱して29時間撹拌し、室温まで放冷した。減圧下で溶媒を留去し、中性フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=1/1)で精製し、化合物5(230mg,67%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ0.88-0.92(m,3H),1.06-1.17(m,28H),1.53-1.58(m,2H),2.53(t,J=14.4Hz,2H),3.88-3.94(m,2H),4.01-4.05(m,1H),4.15-4.20(m,1H),4.74(t,J=9.2Hz,1H),5.68(s,1H),6.49(d,J=7.2Hz,1H),7.97(s,1H),8.32(s,1H).
Ar雰囲気下、氷冷下で化合物5(314mg,0.523mmol,1.0eq.)をpyridine(2.1mL,0.25M to 化合物5)に溶かし、そこにBzCl(91μL,0.785mmol,1.5eq.)を加えて3時間30分撹拌した。BzCl(60μL,0.523mmol 1.0eq.)を加え、さらに1時間撹拌した。BzCl(30μL,0.262mmol 0.5eq.)を加え、さらに40分撹拌した。BzCl(60μL,0.523mmol 1.0eq.)を加え、さらに40分撹拌した。4mLの水を加え、5分撹拌した後、28%アンモニア水溶液を8mL加えて15分撹拌した。酢酸エチルと水(sat. NaHCO3 aq.,brine)で分液した。無水硫酸ナトリウムを加えて乾燥した後、綿栓ろ過し、減圧下で溶媒を留去した。中性フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=1/1)で精製し、化合物6(183mg,50%) を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ0.88-0.92(m,3H),1.01-1.18(m,28H),1.55-1.61(m,2H),2.57(t,J=15.2Hz,2H),3.91-3.97(m,2H),4.03-4.07(m,1H),4.18-4.22(m,1H),4.74(t,J=9.2Hz,1H),6.57(d,J=7.2Hz,1H),7.52(t,J=8.0Hz,2H),7.61(t,J=7.2Hz,2H),8.02(d,J=7.2Hz,1H),8.16(s,1H),8.80(s,1H),9.08(s,1H);13C-NMR(100MHz,CDCl3):δ12.5,12.9,13.0,13.1,13.6,17.1,17.4,17.5,22.1,41.3,61.7,63.3,73.9,83.9,84.8,123.0,127.9,128.9,132.8,133.9,142.0,149.6,151.6,152.6,164.7;HRMS(ESI+)calc.m/z704.28(M+H+),726.26(M+Na+),found m/z 704.2821(M+H+),726.2623(M+Na+).
Ar雰囲気下で、化合物6(241mg,0.342mmol,1.0eq.)をTHF(3.4mL)に溶かし、3HF-Et3N(139μL,0.855mmol,2.5eq.)を加えて2時間20分撹拌した。減圧下で溶媒を留去し、中性フラッシュカラムクロマトグラフィー(CHCl3(MeOH 0-5%))で分離し、化合物7(154mg,98%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CD3SOCD3):δ0.78-0.81(m,3H),1.40-1.45(m,2H),2.49-2.55(m,2H),3.66-3.77(m,2H),3.82-3.84(m,1H),3.96-3.99(m,1H),4.41-4.45(m,1H),5.14(t,J=3.6Hz,1H),5.92(d,J=4.4Hz,1H),6.66(d,J=4.8Hz,1H),7.55(t,J=8.0Hz,1H),7.64(t,J=7.6Hz,1H),8.03(d,J=7.2Hz,1H),8.64(s,1H),8.74(s,1H),11.21(br,1H);13C-NMR(100MHz,CD3OD):δ13.1,22.9,41.9,61.2,63.2,72.8,86.0,86.4,124.6,129.4,129.7,133.8,134.9,144.8,151.0,153.1,153.3,168.0;HRMS(ESI+)calc.m/z462.13(M+H+),484.11(M+Na+),found m/z 462.1288(M+H+),484.1121(M+Na+).
アルゴン雰囲気下、0℃にてSM(154mg,0.344mmol,1.0eq.)をpyridine(3.4ml,0.1M to 化合物7)に溶かし、撹拌しながら、BzCl(46μl,0.401 mmol,1.2eq.)を加えた。TLCにて反応を追跡しながら、撹拌開始から2h,4h,7h,10h後にそれぞれBzClを0.6eq.,0.6eq.,1.2eq.,1.2eq.ずつ加えて、反応開始から11h後にMeOH(5ml)を加えて10min撹拌した。溶媒を減圧留去した後、EtOAc-水(sat.NaHCO3 aq.,brine)にて分液を行った。芒硝乾燥したのち溶媒留去を行い、中性フラッシュカラム(H/A=1/1)にて精製して化合物8(133mg,68%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ0.738(t,J=7.2Hz,3H),1.38(m,2H),2.42(m,2H),3.96(t,J=7.6Hz,1H),4.30(m,1H),4.70-4.72(m,2H),4.82(t,J=7.6Hz,1H),5.44(brs,1H),6.59(d,J=7.2Hz,1H),7.37-7.50(m,6H),7.97-8.00(m,4H),8.15(s,1H),8.71(s,1H),9.51(brs,1H);13C-NMR(100MHz,CDCl3):δ12.8,14.2,21.1,21.9,41.1,60.5,61.3,63.8,74.4,82.3,85.5,122.7,128.0,128.5,128.8,129.4,129.7,129.8,132.8,133.4,133.6,142.2,149.5,151.3,152.6,165.0,166.5,171.3;HRMS(ESI+)calc.m/z566.15(M+H+),588.14(M+Na+),604.11(M+K+),found m/z 544.1461(M+H+),588.1327(M+Na+),604.1017(M+K+).
Ar雰囲気下、0℃にてphenylphosphorodichloride(9μl,0.06mmol,1.5eq. in 150μl THF)、脱水TEA(28μl,0.2mmol,5.0eq. in 150μl THF)、化合物8(23mg,0.04mmol,1.0eq. in 200μl THF and 28μl TEA)を加え、1.5h撹拌した後、反応系を室温に戻し、2.5h撹拌した。その後MQを150μl加え、overnight撹拌した後、HPLCにて精製した。反応収率はHPLCで計算し62%であった。HRMS(ESI‐) calc. m/z 720.14(M‐), found m/z 719.7807(M‐), HRMS(ESI+) calc. m/z 722.15(M+H+),found m/z 722.1443(M + H+).
Ar雰囲気下で、化合物4(549mg,1.04mmol,1.0eq.)をTHF(10mL)に溶かし、3HF-Et3N(424μL,2.6mmol,2.5eq.)を加えて2時間40分撹拌した。減圧下で溶媒を留去し、DCMで吸引ろ過、洗浄し、化合物10(266mg,89%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CD3SOCD3):δ3.31-3.39(m,2H),3.73-3.76(m,4H),4.24-4.29(m,1H),5.78(d,J=6.0Hz,1H),6.36(d,J=7.6Hz,1H),7.29(s,2H),8.12(s,1H),8.30(s,1H).
Ar雰囲気下で、化合物5(553mg,0.920mmol,1.0eq.)をTHF(9.2mL)に溶かし、3HF-Et3N(375μL,2.30mmol,2.5eq.)を加えて2時間撹拌した。減圧下で溶媒を留去し、中性フラッシュカラムクロマトグラフィー(CHCl3 (MeOH 0-5%))で分離し、化合物11(337mg,quant.)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CD3OD):δ0.74-0.78(m,3H),1.36-1.43(m,2H),2.44-2.47(m,2H),3.84-3.91(m,4H),4.51(t,J=8.8Hz,1H),6.54(d,J=7.2Hz,1H),8.16(s,1H),8.32(s,1H).
?78℃で、無水DCM(4.2ml)中の塩化ホスホリル(130μl、1.4mmol、1.0eq.)の溶液に、フェノール(132mg、1.4mmol、1.0eq.)及びトリエチルアミン(132mg)の溶液を加えた。無水DCM(1.4ml)中の195μl、1.4mmol、1.0eq.)を滴下した。同じ温度で3時間撹拌した後、反応混合物をL-アラニンイソプロピルエステル塩酸塩(235mg、1.4mmol、1.0eq.)で一度に処理し、続いてトリエチルアミン(390μl、2.8mmol, 2.0eq.)を滴下した。それを?78℃でさらに1時間撹拌し、次いで1時間かけて室温まで温めた。0℃に冷却した後、上記混合物を、無水DCM(1.4ml)中の化合物11(94.2mg、0.280mmol、0.2当量)及びNMI(111μl、1.4mmol、1.0当量)の溶液でさらに処理し、0℃で16時間撹拌した。反応混合物をH2O(7.0ml)で処理し、DCMで抽出した。有機層を0.5Mの希HCl及び食塩水で順次洗浄した。無水Na2SO4で乾燥した後、それを濃縮し、次いでシリカゲルカラムクロマトグラフィー(DCM/MeOH=-10/1)及びHPLCで精製して所望の生成物Pro Aを得た(48mg,38%)。得られた化合物のNMR情報は以下のとおりである。
1H-NMR(400MHz,DMSO):δ-0.06 (t,J=7.6Hz,3H),0.18(d,J=6.4Hz,2H),0.23-0.27(m,4H),0.30(d,J=6.8Hz,3H),0.49-0.59(m,2H),1.64-1.66(m,2H),2.78-2.91(m,1H),3.06-3.13(m,2H),3.36-3.50(m,2H),3.64-3.70(m,1H),3.83-3.89(m,1H),5.08-5.21(m,2H),5.68(d,J=8.0Hz,1H),6.29-6.33(m,3H),6.45-6.49(m,4H),7.28(s,1H),7.35(s,1H);13C-NMR(100MHz,DMSO):δ12.5,19.6,21.2,21.3,49.7,62.4,67.8,72.6,83.6,118.8,120.0,120.1,124.4,129.5,140.0,149.0,150.7,152.5,156.0,172.5;31P-NMR(162MHz,CDCl3):δ4.27(P(R)),4.67(P(S));HRMS(ESI+)calc.m/z 627.18(M+H+),649.16(M+Na+),665.14(M+K+),found m/z 627.1786(M+H+),649.1611(M+Na+),665.1328(M+K+).
氷冷下でジメチルジセレニド(300μL,3.17mmol)を溶解したエタノール(5mL)に水素化ホウ素ナトリウム(289mg,7.64mmol)を加えた。化合物2(3.36g,5.30mmol)をTHF(20mL)に溶解し、反応溶液に加え、70℃で3.5時間撹拌した。ジメチルジセレニド(100μL,1.06mmol)と水素化ホウ素ナトリウム(62.3mg,1.64mmol)を加え、60℃で2.5時間撹拌した。1M塩酸で中和した後、溶媒を留去した。残渣を酢酸エチル(50mL)に溶解し水で3回、飽和食塩水1回で洗浄し、有機層を芒硝乾燥した。溶媒を留去した後、中性フラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル=1/1→9/1→酢酸エチル/メタノール=9/1)により精製し、化合物12(1.90g,61%)を得た。本反応には耐圧容器を用いた。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,CDCl3):δ0.89-1.22(m,28H),1.95(s,3H),3.72-3.90(m,2H),4.04-4.23(m,2H),4.73(dd,J=8.4Hz,10.4Hz,1H),5.64(br,s,2H),6.49(d,J=7.6Hz,1H),8.26(s,1H);13C-NMR(100MHz,DMSO):δ5.801,12.70,13.15,13.16,13.86,17.13,17.21,17.25,17.27,17.48,17.51,17.57,17.64,49.87,61.23,74.94,84.23,84.58,119.56,139.32,149.93,152.754;HRMS(ESI+)calc.m/z588.19(M+H+),610.18(M+Na+),found m/z 588.1945(M+H+),610.1768(M+Na+).
氷冷下で化合物12(1.83g,3.12mmol)をTHF(18mL)に溶解し、TBAF(1M in THF,7.8mL,7.80mmol)を加え、17.5時間撹拌した。溶媒を留去し、中性フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール=95/5→85/15)で精製し、化合物13(1.04g,97%)を得た。得られた化合物の1H-NMR情報は以下のとおりである。
1H-NMR(400MHz,DMSO-d6):δ1.85(s,3H),3.64-3.83(m,4H),4.43(m,1H),5.73(dd,J=4.0Hz,6.0Hz,1H),6.45(dd,J=3.6Hz,7.6Hz,1H),7.28(s,2H),8.28(s,1H);13C-NMR(100MHz,DMSO):δ4.17,49.1,59.8,73.3,84.1,85.0,118.3,139.3,149.2,152.5,156.0;HRMS(ESI+)calc.m/z346.04(M+H+),368.02(M+Na+),found m/z 346.0402(M+H+),368.0214(M+Na+).
POCl3(134μL,1.50mmol)をジクロロメタン(4.5mL)に溶解し、-78℃で撹拌した。フェノール(132.6mg,1.46mmol)とトリエチルアミン(201uL,1.50mmol)をジクロロメタン(1.5mL)に溶解し、滴下し、3時間撹拌した。L-イソプロピルアラニン塩酸塩(248.4mg,1.54mmol)とトリエチルアミン(201uL,3.00mmol)を加え、更に1時間撹拌し、室温まで上昇させた。化合物13(94.2mg,0.27mmol)とNMI(114uL,1.50mmol)をジクロロメタン(1.5mL)に溶解し滴下した。0℃で20.5時間撹拌した後、水でクエンチした。RP-HPLC(MiliQ/ACN=20/80→50/50)により精製を行い、Pro SelenoA(33.5mg,19%)を得た。得られた化合物のNMR情報は以下のとおりである。
1H-NMR(400MHz,CD2Cl2):δ1.01-1.25(m,9H),1.88(s,3H),3.64-3.89(m,3H),4.20-4.83(4m,H),5.82-6.04(m,2H),6.45(d,1H),7.05-7.43(m,7H),8.13(d,2H);13C-NMR(100MHz,CD2Cl2):δ4.95,20.24,21.84,48.93,50.26,66.50,79.71,83.39,84.81,119.2,120.7,125.0,130.1,140.1,149.5,151.2,153.0,156.6,173.1;HRMS(ESI+)calc.m/z615.11(M+H+),found m/z 615.1244(M+H+);
リン酸トリメチル(84.1mg)にセレノアデノシン(化合物13:101mg,0.30mmol)とトリブチルアミン(643μL,2.70mmol)を加え、-30℃で撹拌しながら、塩化ホスホリル(84μL,0.90mmol)を滴下し、16時間撹拌した。アセトニトリル(1.6mL)にビス(トリブチルアンモニウム)ピロリン酸(993.5mg,1.51mmol)とトリブチルアミン(600μL)を加え、24時間撹拌した。1M TEAB(5mL)を加え、凍結乾燥をした。HPLC(DEAE-2SW,1Mギ酸アンモニウム/MQ=0/100→50/50)で反応の進行を分析し、HPLC(C18 Hydrosphere,50mM TEAB/ACN=100/0→70/30)で精製をし、三リン酸体のトリエチルアンモニウム塩を得た。0.1M過塩素酸ナトリウムのアセトン溶液を加え、吸引ろ過により三リン酸体のナトリウム塩(93.9mg,43%)を得た。得られた化合物のNMR情報は以下のとおりである。
1H-NMR(400MHz,D2O):δ8.28(s,1H),8.03(s,1H),6.40(d,J=8MHz,1H),4.42(t,J=10MHz,1H),4.23(d,J=1.6MHz,2H),3.98(m,1H),3,74(q,J=7.2MHz,1H),1.64(s,3H);13C-NMR(100MHz,D2O):δ4.26,48.19,63.43,71.78,82.69,85.85,118.1,140.6,148.6,152.7,155.6;31P-NMR(160MHz,D2O):δ?5.39(br,1P),?10.28(d,J=17MHz,1P),?20.81(br,1P);HRMS(ESI-)calc.m/z583.92(M?H)?,found m/z 583.9202(M?H)?;
化合物9の溶液(MeCN:H2O=2:1,35mM,100uL)、HEPES Buffer(50uL,pH=9.0,500mM)、H2O(332.5uL)、DTT水溶液(1M,17.5uL)を加え、混合した後、25℃で20時間インキュベートした。HPLC(条件は下記に記載)で分析し、ピークの同定はHRMS(ESI)で行った。その結果のグラフを図2に示す。
<HPLC condition>
Column: Hydrosphere C18 250×4.6mm S-5μm 12nm
Eluent: A) 50mM TEAA buffer, 5% ACN
B) ACN
Gradient: B conc. 0-10%(0-10min),10-100%(10-22.5min),100%(22.5-30min)
Flow rate: 1mL/min Detection:260nm

ヒト肝がん細胞HuH-7由来でHBVゲノム(遺伝子型C)が恒常的に複製する細胞株EB-HBCeを24ウェルプレートに播種し、化合物A-1(化合物10)及びA-2(化合物11)を終濃度10又は100μMで添加し9日間培養した。その間、3日毎に培地を交換するとともに同濃度の化合物を添加した。培養した細胞からTRI Reagent(Molecular Research Center社)によってtotal RNAを抽出し、DNaseI及びRNase阻害剤の処理を行った。SuperScript VILO cDNA synthesis kit(Invitrogen社)によりcDNAを合成し、SYBR qPCR Mix Kit(東洋紡社)を用いた定量PCRによってHBV複製中間体であるウイルスRNA(pgRNA)を定量した。その結果を図3に示す。左側のグラフから順に、コントロール、化合物A-1を10μM、化合物A-1を100μM、化合物A-2を10μM、化合物A-2を100μM添加した結果である。また、最も右側のグラフは、肝炎の治療薬であるETV(エンテカビル)を10μM添加した結果である。
下記の鋳型RNA(0.2μM,最終濃度,以下同様),下記のプライマーDNA(0.2μM),dCTP,dGTP,dTTP(各100μM),Tris-HCl(pH8.3,5mM),KCl(5mM),DTT(0.2mM),MgCl2(0.5mM)を含む溶液に、AMV逆転写酵素(20U/μL)を0.25μLと、dATPあるいはddATPあるいはdASeTP(100μM)を加え、42℃で3時間インキュベートした。Microcon 100Kを用いて、限外ろ過を3回行った後、Amicon 3Kを用いてタンパク溶液を濃縮した。タンパク溶液を用いて、再度鎖伸長反応を行った(鋳型RNA 0.2μM,プライマーDNA 0.2μM,100μM each dNTP,Tris-HCl(pH8.3) 2.5mM,KCl 5mM,DTT 0.2mM,0.5mM MgCl2,AMV逆転写酵素 0.1U/μL)。
反応時間5分、30分、60分において10μLのサンプルを取り、2×変性バッファーに加えて、反応を停止した。各サンプルを20% dPAGE(7.5M Urea, 1×TBE,7.5% formamide,20mA const.)で電気泳動を行い、蛍光発光で伸長核酸を定量検出した。その結果を図4に示す。
プライマーDNA:5’-(FAM)-GGTGGACTTTCGC-3’
鋳型RNA:5’-ACGACGUGCGAAAGUCCACC-3’
ヒト肝がん細胞HuH-7由来でHBVゲノム(遺伝子型C)が恒常的に複製する細胞株EB-HBCeを24ウェルプレートに播種し、各化合物を種々の終濃度で添加し9日間培養した。その間、3日毎に培地を交換するとともに同濃度の化合物を添加した。
培養上清を回収しPNE溶液(8.45% PEG6000, 0.445M NaCl,13mM EDTA)添加によってウイルス粒子を沈殿させた後、DNase I(タカラバイオ社)及びRNase A(タカラバイオ社)で37℃、1時間処理することによって粒子外の核酸を除いた。さらに、Proteinase Kで一晩処理した後、DNAをフェノール/クロロホルム抽出しエタノール沈殿を行った。沈殿を可溶化した後、SYBR qPCR Mix Kit(東洋紡社)を用いてHBV DNAの定量測定を行った。その結果を図5に示す。図中の「SelenoA」は化合物13を、「entecavir」はエンテカビルを、それぞれ添加した結果を表す。
(1)抗HIV活性測定
Dulbecco’s modified Eagle’s Medium(DMEM)(SIGMA/Cat.No.D5796)に、最終濃度10%のfetal bovine serum(Japan Bio Serum社)を添加した培地で、37℃、5% CO2の条件でTZM-bl細胞を培養した。TZM-bl細胞を96well microplateに播種した(1.3×104cells/100μL DMEM+10%FBS)。翌日、薬剤溶液とHIV-1(NL4-3, 10ng)をこの順に培養液に添加した。2日後、培養上清200μLを除去し、1×Steady Glo(Promega/Cat.No.E2510)を100μL加え、かきとった80μLの細胞破砕液を専用のプレート(Coster/Cat.No3912) IESEL, VERITAS Microplate Luminometer(Promega)でLuciferaseを定量し、薬剤の抗HIV-1活性(EC50)を算出した。異なるタイプのウイルスを用い、複数の化合物について実験を行った。その結果を下記表(A)に示す。表中、「AZT」はアジドチミジン、「Lamivudine」はラミブジン、「Didanosine」はジダノシンを意味する。
MTT assayは、Celltiter 96 Non-radioactive Cell Proliferation Assay(Promega)を使用した。 MT-4細胞にHIV-1 NL4-3をMOI=0.001で感染させた。(37℃, 1-1.5時間)HIV-1感染又は非感染MT-4細胞(2.5×105/ml,100μL)を96ウェルマイクロプレートに分注し(最終DMSO濃度;0.5%)、37℃,5%CO2で培養を開始した。培養5日目に培養上清(100μL)を除去し、dye solution(MTT試薬)15μLを各ウェルに添加し、CO2インキュベーター内で1時間培養した。続いてsolubilization solution/stop mixを100μL 各ウェルに添加し、よく混和し4℃で一晩静置した。プレートを室温に戻してからDD570/690を分光光度計(BIO-TEK ELx808)で測定した(CC50)。その結果を下記表(B)に示す。

Claims (4)
- リン酸化を伴うDNAポリメラーゼによる核酸伸長反応におけるリン酸化後の核酸伸長反応の進行を阻害する反応阻害剤であって、前記式2Aで示されるヌクレオシド誘導体又は該ヌクレオシド誘導体を3’末端に有する核酸であるβ修飾リン酸化合物前駆体を含有し、DNAポリメラーゼの反応を阻害することを特徴とする反応阻害剤。

(ここで、A 1 は-SR 1 、-S-S-R 1 、-SeR 1 、又は-X(ここで、Xはフッ素、塩素、臭素、ヨウ素から選択されるハロゲンを意味する)を示し、R 1 は水素、炭素数1~20のアルキル基、炭素数1~20のアリール基、又は炭素数1~20のアルケニル基である。L 3 は水素、又は下記式2D―1で示されるモノリン酸基、若しくは下記式2Eで示されるトリリン酸基である。

(ここで、Z 1 は、ヒドロキシル基、又はグリシン、アラニン、バリン、ロイシン、フェニルアラニン、トリプトファン、メチオニン若しくはプロリンのメチルエステル、エチルエステル、イソプロピルエステル、n-ブチルエステル、ベンジルエステル若しくはフェニルエステルである。Z 2 は、水素、炭素数1~4のアルキル基、ハロゲン、又はフェニル基である。)

Bはアデニン、グアニン、シトシン、チミン、ウラシル、N-メチルアデニン、N-ベンゾイルアデニン、2-メチルチオアデニン、2-アミノアデニン、7-メチルグアニン、N-イソブチリルグアニン、5-フルオロシトシン、5-ブロモシトシン、5-メチルシトシン、4-N-メチルシトシン、4-N,N-ジメチルシトシン、5-フルオロウラシル、5-ブロモウラシル、5-クロロウラシル、又は5,6-ジヒドロウラシルから選択される塩基である。*はリン酸化を受けてリン酸基が結合する結合手であり、リン酸化前においては水素が結合していることを意味する。) - 前記A 1 は、-SH,-S-S-C 3 H 5 、及び-SeCH 3 から選択され、
前記Z 1 は、ヒドロキシル基及びアラニンのイソプロピルエステルから選択され、
前記Z 2 は、水素及びフェニル基から選択され、
前記Bは、アデニン、グアニン、シトシン、チミン及びウラシルから選択されることを特徴とする請求項1に記載の反応阻害剤。 - 請求項1又は2に記載の反応阻害剤によってリン酸化を伴うDNAポリメラーゼによる核酸伸長反応におけるリン酸化後の核酸伸長反応の進行を阻害する反応阻害方法(ただし人間を治療する方法を除く)であって、
前記式2Aで示されるβ修飾リン酸化合物前駆体を用意する工程と、
前記β修飾リン酸化合物前駆体をリン酸化して下記式2Bで示されるβ修飾リン酸化合物を生成させるとともに、前記β修飾リン酸化合物を部分開裂させて活性種を生成させる工程と、

(ここで、A 1 、L 3 、Bは前記式2Aで定義したとおりである。)
を備えることを特徴とする反応阻害方法。 - 前記A 1 は、-SH,-S-S-C 3 H 5 、及び-SeCH 3 から選択され、
前記Z 1 は、ヒドロキシル基及びアラニンのイソプロピルエステルから選択され、
前記Z 2 は、水素及びフェニル基から選択され、
前記Bは、アデニン、グアニン、シトシン、チミン及びウラシルから選択されることを特徴とする請求項3に記載の反応阻害方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018043329 | 2018-03-09 | ||
| JP2018043329 | 2018-03-09 | ||
| PCT/JP2019/009213 WO2019172394A1 (ja) | 2018-03-09 | 2019-03-08 | β修飾リン酸化合物前駆体、β修飾リン酸化合物、反応阻害剤及びこれを含む医薬並びに反応阻害方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2019172394A1 JPWO2019172394A1 (ja) | 2021-02-18 |
| JP7266896B2 true JP7266896B2 (ja) | 2023-05-01 |
Family
ID=67846033
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020505121A Active JP7266896B2 (ja) | 2018-03-09 | 2019-03-08 | β修飾リン酸化合物前駆体、β修飾リン酸化合物、反応阻害剤及びこれを含む医薬並びに反応阻害方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11597745B2 (ja) |
| EP (1) | EP3763724A4 (ja) |
| JP (1) | JP7266896B2 (ja) |
| CN (1) | CN111836823B (ja) |
| WO (1) | WO2019172394A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12359254B2 (en) | 2019-07-31 | 2025-07-15 | Japan Science And Technology Agency | Primer, device for producing double-stranded DNA using primer, and method for producing double-stranded DNA using primer |
| JP7551134B2 (ja) | 2019-07-31 | 2024-09-17 | 国立研究開発法人科学技術振興機構 | プライマー及びこれを用いた二本鎖dnaの製造装置並びに二本鎖dnaの製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012117246A1 (en) | 2011-03-01 | 2012-09-07 | Nucana Biomed Limited | Phosphoramidate derivatives of 5 - fluoro - 2 ' - deoxyuridine for use in the treatment of cancer |
| WO2014160484A1 (en) | 2013-03-13 | 2014-10-02 | Idenix Pharmaceuticals, Inc. | Amino acid phosphoramidate pronucleotides of 2'-cyano, azido and amino nucleosides for the treatment of hcv |
| WO2015038596A1 (en) | 2013-09-11 | 2015-03-19 | Emory University | Nucleotide and nucleoside compositions and uses related thereto |
| CN105646629A (zh) | 2014-11-25 | 2016-06-08 | 广州市恒诺康医药科技有限公司 | L-核苷类化合物及其应用 |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0365556B1 (en) * | 1987-06-05 | 1995-07-12 | Genentech, Inc. | Nucleoside analogues |
| JPH09136842A (ja) | 1995-11-14 | 1997-05-27 | Shigemasa Sawada | 抗慢性関節リウマチ剤 |
| WO2002077002A2 (en) * | 2001-03-22 | 2002-10-03 | Research Foundation Of The City University Of New York | Synthesis of selenium-derivatized nucleosides, nucleotides, phosphoramidites, triphosphates and nucleic acids |
| WO2013177188A1 (en) * | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| JP6165848B2 (ja) | 2012-05-22 | 2017-07-19 | イデニク ファーマシューティカルズ エルエルシー | 肝疾患のためのd−アミノ酸化合物 |
| CN104540843A (zh) | 2012-06-29 | 2015-04-22 | 塞纳研究股份有限公司 | 用作营养补剂的含硒核苷 |
| EP3071590A4 (en) * | 2013-11-21 | 2017-07-19 | SeNA Research, Inc. | Methods for structural determination of selenium derivatized nucleic acid complexes |
-
2019
- 2019-03-08 EP EP19763431.4A patent/EP3763724A4/en active Pending
- 2019-03-08 JP JP2020505121A patent/JP7266896B2/ja active Active
- 2019-03-08 WO PCT/JP2019/009213 patent/WO2019172394A1/ja not_active Ceased
- 2019-03-08 CN CN201980017573.6A patent/CN111836823B/zh active Active
- 2019-03-08 US US16/977,525 patent/US11597745B2/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012117246A1 (en) | 2011-03-01 | 2012-09-07 | Nucana Biomed Limited | Phosphoramidate derivatives of 5 - fluoro - 2 ' - deoxyuridine for use in the treatment of cancer |
| WO2014160484A1 (en) | 2013-03-13 | 2014-10-02 | Idenix Pharmaceuticals, Inc. | Amino acid phosphoramidate pronucleotides of 2'-cyano, azido and amino nucleosides for the treatment of hcv |
| WO2015038596A1 (en) | 2013-09-11 | 2015-03-19 | Emory University | Nucleotide and nucleoside compositions and uses related thereto |
| CN105646629A (zh) | 2014-11-25 | 2016-06-08 | 广州市恒诺康医药科技有限公司 | L-核苷类化合物及其应用 |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
Non-Patent Citations (13)
| Title |
|---|
| Biochemistry,1981年,20,3056-3062 |
| Biochemistry,1982年,21,5870-5877 |
| Biochimica et Biophysica Acta,1979年,561,223-231 |
| Bioorganic & Medicinal Chemistry Letters,2012年,22,4203-4205 |
| Bioorganic & Medicinal Chemistry,2012年,20,2416-2418 |
| Chemical & Pharmaceutical Bulletin,1981年,29,2408-2412 |
| European Journal of Biochemistry,1978年,87,45-54 |
| Journal of Organic Chemistry,1980年,45,4830-4834 |
| Journal of Organic Chemistry,2010年,75,637-641 |
| Journal of the American Chemical Society,1996年,118,11715-11719 |
| Nucleic Acids Research,2008年,36,970-983 |
| Organic & Biomolecular Chemistry,2004年,2,120-126 |
| Tetrahedron Letters,1960年,n.8,20-22 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3763724A4 (en) | 2022-01-12 |
| US11597745B2 (en) | 2023-03-07 |
| CN111836823A (zh) | 2020-10-27 |
| JPWO2019172394A1 (ja) | 2021-02-18 |
| EP3763724A1 (en) | 2021-01-13 |
| US20210284679A1 (en) | 2021-09-16 |
| CN111836823B (zh) | 2023-06-30 |
| WO2019172394A1 (ja) | 2019-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7053754B2 (ja) | HCV治療に対するβ-D-2’-デオキシ-2’-α-フルオロ-2’-β-C-置換-2-修飾-N6-置換プリンヌクレオチド | |
| Slusarczyk et al. | Phosphoramidates and phosphonamidates (ProTides) with antiviral activity | |
| Mahmoud et al. | Antiviral nucleoside and nucleotide analogs: a review | |
| AU2012223012B2 (en) | Phosphoramidate derivatives of 5 - fluoro - 2 ' - deoxyuridine for use in the treatment of cancer | |
| CN106188192B (zh) | 含d-氨基酸酯的核苷氨基磷酸/膦酸酯衍生物及其医药用途 | |
| WO2006121820A1 (en) | Phosphoramidate prodrugs for treatment of viral infection | |
| KR20060008297A (ko) | 2'-데옥시-β-L-뉴클레오시드의 제조방법 | |
| JP2005503358A5 (ja) | ||
| CN103435672A (zh) | 含有取代苄基的新型核苷磷酸酯前药的结构与合成 | |
| WO2010068708A2 (en) | 3'-azido purine nucleotide prodrugs for treatment of viral infections | |
| CN106661075A (zh) | 二磷酸和三磷酸前体药物 | |
| JP7266896B2 (ja) | β修飾リン酸化合物前駆体、β修飾リン酸化合物、反応阻害剤及びこれを含む医薬並びに反応阻害方法 | |
| US11021497B2 (en) | Compositions and methods for synthesis of phosphorylated molecules | |
| Slusarczyk et al. | Synthesis and biological evaluation of 6-substituted-5-fluorouridine ProTides | |
| CN1812995A (zh) | 工业化规模的核苷合成 | |
| US12539332B2 (en) | Anti-viral and hepatic-targeted drugs | |
| Pomeisl et al. | Synthesis of fluorinated acyclic nucleoside phosphonates with 5-azacytosine base moiety | |
| Congiatu | Design, synthesis and biological evaluation of some novel nucleotide prodrugs as potential anticancer agents | |
| Alanazi | Design, synthesis, and biological evaluation of novel nucleotide prodrugs as potential therapeutics | |
| CN118978559A (zh) | 一种磷酰胺酯核苷类药物前药的合成方法及药物应用 | |
| Pileggi | Novel synthetic pathways for the preparation of ProTides as potential therapeutic agents | |
| US20040158054A1 (en) | Di-ribonucleotides as specific viral RNA-polymerase inhibitors for the treatment or prevention of viral infections | |
| Guga et al. | Nucleotides and nucleic acids: mononucleotides | |
| CN120025389A (zh) | 一种磷酰胺酯核苷类药物化合物及其合成方法与药物应用 | |
| Suzol | Novel nucleoside analogues with bases modified with (β-halo) vinyl sulfone or β-keto sulfone as probes to study RNA/DNA-Proteins interactions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211112 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20221108 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230105 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230411 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230412 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7266896 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |