JP7311642B2 - オリゴヌクレオチドを脱保護するための方法 - Google Patents
オリゴヌクレオチドを脱保護するための方法 Download PDFInfo
- Publication number
- JP7311642B2 JP7311642B2 JP2021574989A JP2021574989A JP7311642B2 JP 7311642 B2 JP7311642 B2 JP 7311642B2 JP 2021574989 A JP2021574989 A JP 2021574989A JP 2021574989 A JP2021574989 A JP 2021574989A JP 7311642 B2 JP7311642 B2 JP 7311642B2
- Authority
- JP
- Japan
- Prior art keywords
- column
- buffer solution
- phosphate
- oligonucleotide
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/36—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving ionic interaction, e.g. ion-exchange, ion-pair, ion-suppression or ion-exclusion
- B01D15/361—Ion-exchange
- B01D15/363—Anion-exchange
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/06—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/10—Selective adsorption, e.g. chromatography characterised by constructional or operational features
- B01D15/20—Selective adsorption, e.g. chromatography characterised by constructional or operational features relating to the conditioning of the sorbent material
- B01D15/203—Equilibration or regeneration
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Saccharide Compounds (AREA)
- Peptides Or Proteins (AREA)
Description
a)逆相クロマトグラフィー
b)濃縮および脱塩
c)溶液中での酸不安定な5’ヒドロキシ保護基の除去、ならびに
d)さらなる濃縮および脱塩。
[本発明1001]
オリゴヌクレオチドの5’-O-オリゴヌクレオチド末端にある酸不安定な5’ヒドロキシ保護基の、酸を用いるオンカラム脱保護による除去を含む、オリゴヌクレオチドを精製するための方法。
[本発明1002]
カラムがイオン交換クロマトグラフィーカラム、好ましくは陰イオン交換クロマトグラフィーカラムである、本発明1001の方法。
[本発明1003]
酸不安定な5’ヒドロキシ保護基が、4,4’-ジメトキシトリチル、4-メトキシトリチル、トリチル、9-フェニル-キサンテン-9-、9-(p-トリル)-キサンテン-9-イルから、またはtert-ブチルジメチルシリルから、好ましくは4,4’-ジメトキシトリチルから選択される、本発明1001または1002の方法。
[本発明1004]
a リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの1回目の平衡化を行う工程、
b 粗オリゴヌクレオチドの希アンモニア水溶液をカラムに投入する工程、
c リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの2回目の平衡化を行う工程、
d リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いてカラムを洗浄する工程、
e リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの3回目の平衡化を行う工程、
f 酸を用いるオンカラム脱保護を行う工程、
g リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの4回目の平衡化を行う工程、
h リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いて、脱保護されたオリゴヌクレオチドを溶出する工程、ならびに、それに続いて、リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いてカラムを無勾配で洗浄する工程
を含む、本発明1001から1003のいずれかの方法。
[本発明1005]
バッファー溶液中のリン酸塩が、アルカリリン酸塩またはその混合物から選択されるリン酸塩であるが、好ましくはリン酸一ナトリウムもしくはリン酸二ナトリウムまたはそれらの混合物である、本発明1004の方法。
[本発明1006]
バッファー溶液中のリン酸塩含有量が、10mMから40mMの間、好ましくは20mMから30mMの間で選択される、本発明1004または1005の方法。
[本発明1007]
カラムに投入されるアンモニア水溶液が、カラム容積1L当たり8~20g、好ましくはカラム容積1L当たり10~15gのオリゴヌクレオチド総含有量を有する、本発明1004から1006のいずれかの方法。
[本発明1008]
バッファー溶液中の有機溶媒が、極性のプロトン性溶媒または極性の非プロトン性溶媒から、好ましくは極性の非プロトン性溶媒から、より好ましくはアセトニトリルから選択される、本発明1004または1007のいずれかの方法。
[本発明1009]
ハロゲン化アルカリが塩化ナトリウムである、本発明1004から1008のいずれかの方法。
[本発明1010]
洗浄する工程(d)におけるバッファー溶液が、0.2M~1.0M、好ましくは0.4M~0.7Mの塩化ナトリウムを含む、本発明1004から1009のいずれかの方法。
[本発明1011]
溶出する工程(h)におけるバッファー溶液が、1.5.M~3.0M、好ましくは1.8M~2.5Mの塩化ナトリウムを含む、本発明1004から1010のいずれかの方法。
[本発明1012]
酸がプロトン酸である、本発明1001から1011のいずれかの方法。
[本発明1013]
酸が、酢酸、好ましくは、水中の酢酸の濃度が50~95重量%、より好ましくは70~90重量%、さらにより好ましくは75~85重量%である酢酸水溶液である、本発明1001から1012のいずれかの方法。
[本発明1014]
オンカラム脱保護を行う工程(f)における酸溶液の流速が、1.5L/分から2.5L/分の間で選択される、本発明1001から1013のいずれかの方法。
[本発明1015]
工程(a)~(e)および(g)~(h)におけるバッファー溶液の流速が、2.0L/分から3.0L/分の間で選択される、本発明1001から1014のいずれかの方法。
[本発明1016]
精製工程(a)~(h)の後に行われる、以下の工程
(i)タンジェンシャルフロー濾過によって濾液を精製水で洗浄することを含む、脱塩および濃縮する工程、ならびに
(j)脱塩および濃縮する工程から得られた濾液を凍結乾燥する工程
をさらに含む、本発明1001から1014のいずれかの方法。
[本発明1017]
オリゴヌクレオチドが、修飾されていてもよいDNA、RNA、もしくはLNAヌクレオシドモノマー、またはそれらの組合せからなり、かつ、10~40、好ましくは10~25ヌクレオチド長である、本発明1001から1015のいずれかの方法。
a1)固体支持体上の保護された5’ヒドロキシル基をデブロッキングすること、
a2)活性化ホスホラミダイトとしての第1のヌクレオシドと固体支持体上の遊離5’ヒドロキシル基とを結合させること、
a3)P結合した各々のヌクレオシドを酸化または硫化して、各々のホスホジエステル(P=O)または各々のホスホロチオエート(P=S)を形成させること、
a4)任意で、固体支持体上の任意の未反応5’ヒドロキシル基をキャッピングすること、
a5)固体支持体に結合された第1のヌクレオシドの5’ヒドロキシル基をデブロッキングすること、
a6)活性化されたホスホラミダイトとしての第2のヌクレオシドを結合させて、各々のP-O結合したダイマーを形成させること、
a7)P-O結合した各々のジヌクレオシドを酸化または硫化して、各々のホスホジエステル(P=O)または各々のホスホロチオエート(P=S)を形成させること、
a8)任意で、任意の未反応5’ヒドロキシル基をキャッピングすること、
a9)所望の配列が組み立てられるまで、前述の工程a5~a8を繰り返すこと。
a リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの1回目の平衡化を行う工程、
b 粗オリゴヌクレオチドの希アンモニア水溶液を陰イオン交換カラムに投入する工程
c リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの2回目の平衡化を行う工程、
d リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いてカラムを洗浄する工程、
e リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの3回目の平衡化を行う工程、
f 酸を用いるオンカラム脱保護を行う工程、
g リン酸塩および有機溶媒を含むバッファー溶液を用いて陰イオン交換カラムの4回目の平衡化を行う工程、
h リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いて、脱保護されたオリゴヌクレオチドを溶出する工程、ならびに、それに続いて、リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いて陰イオン交換カラムを無勾配で洗浄する工程
を含む。
(i)タンジェンシャルフロー濾過によって濾液を精製水で洗浄することを含む、脱塩および濃縮する工程、ならびに
(j)脱塩および濃縮の工程から得られた濾液を凍結乾燥する工程
をさらに含む。
SEQ ID No.1:cucagtaacattgacaccac’
Ac2O=酢酸無水物
(d)A=(デオキシ)アデノシン
(d)C=(デオキシ)シチジン
(d)G=(デオキシ)グアノシン
DCA=ジクロロ酢酸
DCI=4,5-ジシアノイミダゾール
DMT=4,4’-ジメトキシトリチル
CV=カラム容積
Et3N=トリエチルアミン
EtOH=エタノール
MeCN=アセトニトリル
MOE=2-メトキシエチル
NA=該当なし
NaOAc=酢酸ナトリウム
NMI=N-メチルイミダゾール
PADS=フェニルアセチルジスルフィド
PhMe=トルエン
T=チミジン
U=ウリジン
下線を引いたヌクレオシドは、2’-MOEヌクレオシドである
a)
標題化合物をAKTA oligopilot-100を用いて72mmolスケールで「DMTオン型」として合成した。




実施例1a)で得られた粗物質(338.87g)の溶液を、いくつかの平衡工程、カラム洗浄工程、オンカラム脱トリチル工程、および最終生成物溶出工程からなる精製工程に供した。
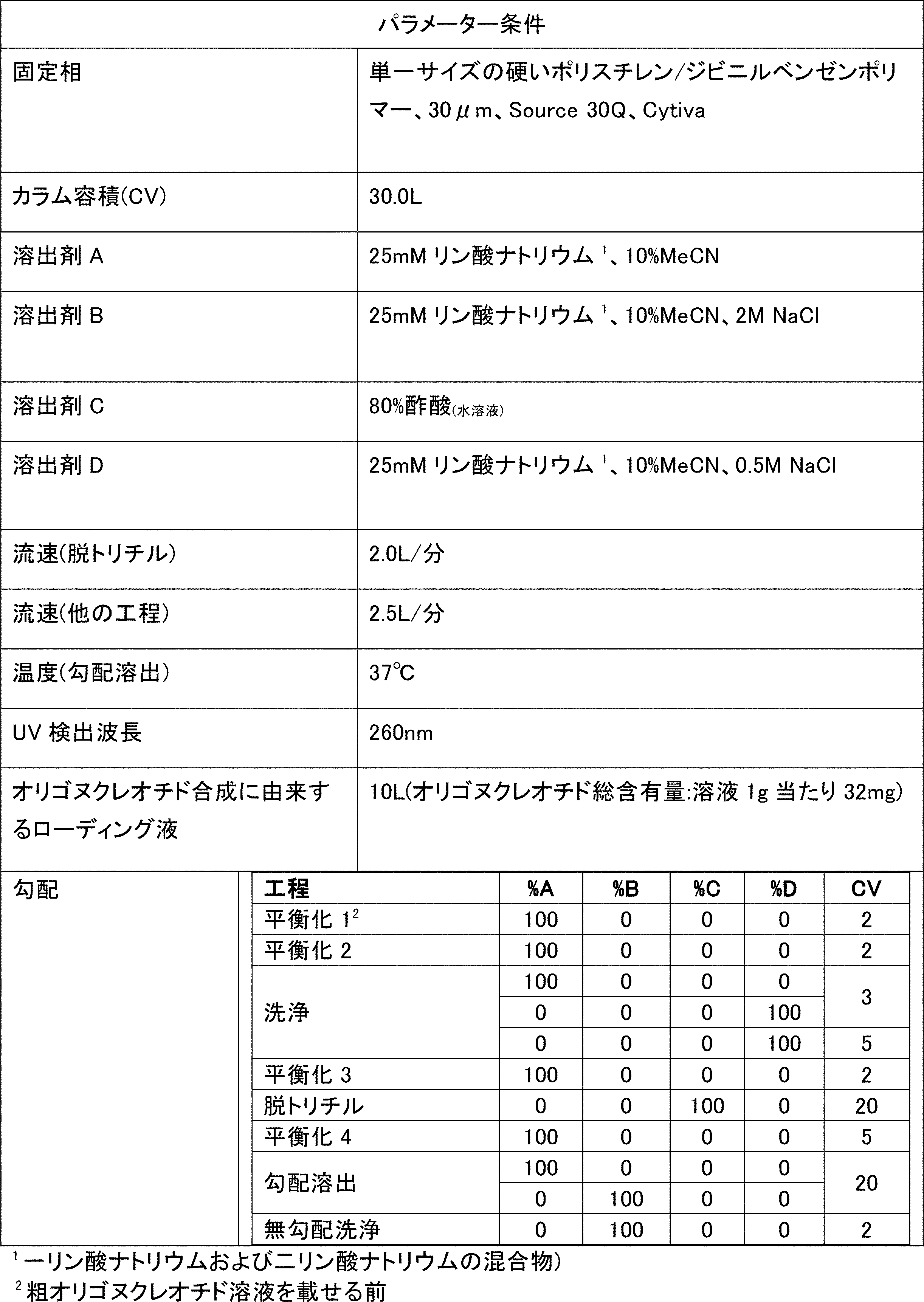
2つの精製バッチ(例1b)を、タンジェンシャルフロー濾過/凍結乾燥工程のために混合した。

a)
実施例1aに従って標題化合物を調製した。
粗物質の一部を、下記の表のパラメーターを用いたHPLC(Amberchrom XT20樹脂を充填したカラム)によって精製した。

実施例2bから得られた画分の混合物のpHを、氷酢酸を脱トリチル剤として用いてpH3に調整し、続いて、10M NaOHを用いてpHを5に再調整した。pH調整した溶液をエタノールに添加することにより、標題化合物を沈殿させた。得られた収率は66.0%であった。
Claims (13)
- オリゴヌクレオチドの5’-O-オリゴヌクレオチド末端にある酸不安定な5’ヒドロキシ保護基の、酢酸を用いるオンカラム脱保護による除去を含む、オリゴヌクレオチドを精製するための方法であって、以下の工程:
a リン酸塩および極性非プロトン性溶媒を含むバッファー溶液を用いて陰イオン交換カラムの1回目の平衡化を行う工程、
b 粗オリゴヌクレオチドの希アンモニア水溶液をカラムに投入する工程、
c リン酸塩および極性非プロトン性溶媒を含むバッファー溶液を用いて陰イオン交換カラムの2回目の平衡化を行う工程、
d リン酸塩、極性非プロトン性溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いてカラムを洗浄する工程、
e リン酸塩および極性非プロトン性溶媒を含むバッファー溶液を用いて陰イオン交換カラムの3回目の平衡化を行う工程、
f 酢酸を用いるオンカラム脱保護を行う工程、
g リン酸塩および極性非プロトン性溶媒を含むバッファー溶液を用いて陰イオン交換カラムの4回目の平衡化を行う工程、
h リン酸塩、極性非プロトン性溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いて、脱保護されたオリゴヌクレオチドを溶出する工程、ならびに、それに続いて、リン酸塩、有機溶媒、およびハロゲン化アルカリを含むバッファー溶液を用いてカラムを無勾配で洗浄する工程
を含み、
バッファー溶液中のリン酸塩含有量が、10mMから40mMの間で選択され、
カラムに投入されるアンモニア水溶液が、カラム容積1L当たり8~20gのオリゴヌクレオチド総含有量を有し、
ハロゲン化アルカリが塩化ナトリウムである、方法。 - 酸不安定な5’ヒドロキシ保護基が、4,4’-ジメトキシトリチル、4-メトキシトリチル、トリチル、9-フェニル-キサンテン-9-イル、9-(p-トリル)-キサンテン-9-イルから、またはtert-ブチルジメチルシリルから選択される、請求項1に記載の方法。
- バッファー溶液中のリン酸塩が、アルカリリン酸塩またはその混合物から選択されるリン酸塩である、請求項1または2に記載の方法。
- バッファー溶液中のリン酸塩含有量が、20mMから30mMの間で選択される、請求項1~3のいずれか一項に記載の方法。
- カラムに投入されるアンモニア水溶液が、カラム容積1L当たり10~15gのオリゴヌクレオチド総含有量を有する、請求項1~4のいずれか一項に記載の方法。
- バッファー溶液中の有機溶媒が、極性非プロトン性溶媒がアセトニトリルである、請求項1~5のいずれか一項に記載の方法。
- 洗浄する工程(d)におけるバッファー溶液が、0.2M~1.0Mを含む、請求項1~6のいずれか一項に記載の方法。
- 溶出する工程(h)におけるバッファー溶液が、1.5M~3.0Mを含む、請求項1~7のいずれか一項に記載の方法。
- 酢酸が、水中の酢酸の濃度が50~95重量%である酢酸水溶液である、請求項1~8のいずれか一項に記載の方法。
- オンカラム脱保護を行う工程(f)における酸溶液の流速が、1.5L/分から2.5L/分の間で選択される、請求項1~9のいずれか一項に記載の方法。
- 工程(a)~(e)および(g)~(h)におけるバッファー溶液の流速が、2.0L/分から3.0L/分の間で選択される、請求項1~10のいずれか一項に記載の方法。
- 精製工程(a)~(h)の後に行われる、以下の工程
(i)タンジェンシャルフロー濾過によって濾液を精製水で洗浄することを含む、脱塩および濃縮する工程、ならびに
(j)脱塩および濃縮する工程から得られた濾液を凍結乾燥する工程
をさらに含む、請求項1~11のいずれか一項に記載の方法。 - オリゴヌクレオチドが、修飾されていてもよいDNA、RNA、もしくはLNAヌクレオシドモノマー、またはそれらの組合せからなり、かつ、10~40ヌクレオチド長である、請求項1~12のいずれか一項に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19185225.0 | 2019-07-09 | ||
| EP19185225 | 2019-07-09 | ||
| PCT/EP2020/068922 WO2021004977A1 (en) | 2019-07-09 | 2020-07-06 | Process for the deprotection of oligonucleotides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022537182A JP2022537182A (ja) | 2022-08-24 |
| JP7311642B2 true JP7311642B2 (ja) | 2023-07-19 |
Family
ID=67226011
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021574989A Active JP7311642B2 (ja) | 2019-07-09 | 2020-07-06 | オリゴヌクレオチドを脱保護するための方法 |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP3997100A1 (ja) |
| JP (1) | JP7311642B2 (ja) |
| KR (1) | KR102746639B1 (ja) |
| CN (1) | CN114051499A (ja) |
| AU (1) | AU2020309307B2 (ja) |
| IL (1) | IL289440B2 (ja) |
| MX (1) | MX2022000113A (ja) |
| TW (1) | TWI872090B (ja) |
| WO (1) | WO2021004977A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117143165B (zh) * | 2023-08-30 | 2026-02-03 | 上海奥锐特生物科技有限公司 | 一种寡核苷酸的纯化方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007063940A1 (ja) | 2005-12-02 | 2007-06-07 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素標識化合物の製造方法 |
| WO2007066567A1 (ja) | 2005-12-06 | 2007-06-14 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素標識有機化合物の製造方法 |
| WO2019023439A1 (en) | 2017-07-28 | 2019-01-31 | Celgene Corporation | PROCESS FOR THE PREPARATION OF OLIGONUCLEOTIDE COMPOUNDS |
| WO2020191252A1 (en) | 2019-03-20 | 2020-09-24 | Wave Life Sciences Ltd. | Technologies useful for oligonucleotide preparation |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7273933B1 (en) * | 1998-02-26 | 2007-09-25 | Isis Pharmaceuticals, Inc. | Methods for synthesis of oligonucleotides |
| US20050136458A1 (en) * | 1998-12-31 | 2005-06-23 | Oligos, Etc, Inc. | Method for nucleic acid preparation |
| AU2003216830A1 (en) * | 2002-03-21 | 2003-10-08 | Avecia Biotechnology Inc. | Purification methods for oligonucleotides and their analogs |
| US7655790B2 (en) * | 2002-07-12 | 2010-02-02 | Sirna Therapeutics, Inc. | Deprotection and purification of oligonucleotides and their derivatives |
| US6989442B2 (en) * | 2002-07-12 | 2006-01-24 | Sirna Therapeutics, Inc. | Deprotection and purification of oligonucleotides and their derivatives |
| US7345163B2 (en) * | 2002-08-28 | 2008-03-18 | Quiatech Ab | Process for separating and deprotecting oligonucleotides |
| DK3719128T3 (da) * | 2017-12-01 | 2025-03-03 | Suzhou Ribo Life Science Co Ltd | Dobbelt-strenget oligonukleotid, sammensætning og konjugat bestaende dobbelt-strenget oligonukleotid, fremgangsmåde herfor og anvendelse heraf |
-
2020
- 2020-07-06 JP JP2021574989A patent/JP7311642B2/ja active Active
- 2020-07-06 MX MX2022000113A patent/MX2022000113A/es unknown
- 2020-07-06 WO PCT/EP2020/068922 patent/WO2021004977A1/en not_active Ceased
- 2020-07-06 AU AU2020309307A patent/AU2020309307B2/en active Active
- 2020-07-06 KR KR1020217041103A patent/KR102746639B1/ko active Active
- 2020-07-06 CN CN202080047541.3A patent/CN114051499A/zh active Pending
- 2020-07-06 IL IL289440A patent/IL289440B2/en unknown
- 2020-07-06 EP EP20735609.8A patent/EP3997100A1/en active Pending
- 2020-07-08 TW TW109123049A patent/TWI872090B/zh active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007063940A1 (ja) | 2005-12-02 | 2007-06-07 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素標識化合物の製造方法 |
| WO2007066567A1 (ja) | 2005-12-06 | 2007-06-14 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素標識有機化合物の製造方法 |
| WO2019023439A1 (en) | 2017-07-28 | 2019-01-31 | Celgene Corporation | PROCESS FOR THE PREPARATION OF OLIGONUCLEOTIDE COMPOUNDS |
| WO2020191252A1 (en) | 2019-03-20 | 2020-09-24 | Wave Life Sciences Ltd. | Technologies useful for oligonucleotide preparation |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2021004977A1 (en) | 2021-01-14 |
| US20220410035A1 (en) | 2022-12-29 |
| IL289440A (en) | 2022-02-01 |
| BR112021026636A2 (pt) | 2022-03-22 |
| TWI872090B (zh) | 2025-02-11 |
| MX2022000113A (es) | 2022-02-10 |
| AU2020309307A1 (en) | 2021-11-04 |
| JP2022537182A (ja) | 2022-08-24 |
| IL289440B2 (en) | 2025-09-01 |
| CA3139610A1 (en) | 2021-01-14 |
| EP3997100A1 (en) | 2022-05-18 |
| IL289440B1 (en) | 2025-05-01 |
| AU2020309307B2 (en) | 2023-03-30 |
| KR20220009437A (ko) | 2022-01-24 |
| KR102746639B1 (ko) | 2024-12-24 |
| CN114051499A (zh) | 2022-02-15 |
| TW202117013A (zh) | 2021-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12043642B2 (en) | Process for the purification of oligonucleotides | |
| US20240092823A1 (en) | Process for the de-tritylation of oligonucleotides | |
| JP7311642B2 (ja) | オリゴヌクレオチドを脱保護するための方法 | |
| US12616921B2 (en) | Process for the deprotection of oligonucleotides | |
| CA3139610C (en) | Process for the deprotection of oligonucleotides | |
| WO2021001329A1 (en) | Process for the preparation of high water affinity type products with controlled humidity | |
| HK40060848A (en) | Process for the deprotection of oligonucleotides | |
| BR112021026636B1 (pt) | Processo para a purificação de oligonucleotídeos | |
| HK40051957B (zh) | 用於纯化寡核苷酸的方法 | |
| HK40051957A (en) | Process for the purification of oligonucleotides | |
| WO2023067038A1 (en) | Process for oligonucleotide purification |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211216 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20221226 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230314 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230607 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230706 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7311642 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |