KR20140059236A - Hcv 프로테아제 억제제의 결정 형태 - Google Patents
Hcv 프로테아제 억제제의 결정 형태 Download PDFInfo
- Publication number
- KR20140059236A KR20140059236A KR1020147006888A KR20147006888A KR20140059236A KR 20140059236 A KR20140059236 A KR 20140059236A KR 1020147006888 A KR1020147006888 A KR 1020147006888A KR 20147006888 A KR20147006888 A KR 20147006888A KR 20140059236 A KR20140059236 A KR 20140059236A
- Authority
- KR
- South Korea
- Prior art keywords
- compound
- hydrate
- diffraction pattern
- rti
- powder diffraction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 CC1C*CC1 Chemical compound CC1C*CC1 0.000 description 3
- AWECGHXJUXCNBT-SCSAIBSYSA-M CC(C)(C)[C@@H](C(O)=O)NC([O-])=O Chemical compound CC(C)(C)[C@@H](C(O)=O)NC([O-])=O AWECGHXJUXCNBT-SCSAIBSYSA-M 0.000 description 2
- AWECGHXJUXCNBT-SCSAIBSYSA-N CC(C)(C)[C@@H](C(O)=O)NC(O)=O Chemical compound CC(C)(C)[C@@H](C(O)=O)NC(O)=O AWECGHXJUXCNBT-SCSAIBSYSA-N 0.000 description 1
- WEPJLNOQGOQHBW-UHFFFAOYSA-N CC1(C)OB(C2C(CCCCl)C2)OC1(C)C Chemical compound CC1(C)OB(C2C(CCCCl)C2)OC1(C)C WEPJLNOQGOQHBW-UHFFFAOYSA-N 0.000 description 1
- GYPYUCZGOXVHSG-UHFFFAOYSA-N CCNc1cc(OC)ccc1NC Chemical compound CCNc1cc(OC)ccc1NC GYPYUCZGOXVHSG-UHFFFAOYSA-N 0.000 description 1
- ZORHSASAYVIBLY-UHNVWZDZSA-N COC([C@H](C1)NC[C@@H]1O)=O Chemical compound COC([C@H](C1)NC[C@@H]1O)=O ZORHSASAYVIBLY-UHNVWZDZSA-N 0.000 description 1
- UOLCFTFGJFPJOK-UTKZUKDTSA-N COC([C@H](C1)NC[C@@H]1Oc1nc(cc(cc2)OC)c2nc1CCCCCC1CC1)=O Chemical compound COC([C@H](C1)NC[C@@H]1Oc1nc(cc(cc2)OC)c2nc1CCCCCC1CC1)=O UOLCFTFGJFPJOK-UTKZUKDTSA-N 0.000 description 1
- AGAHETWGCFCMDK-UHFFFAOYSA-N COc(cc1)cc(N)c1N Chemical compound COc(cc1)cc(N)c1N AGAHETWGCFCMDK-UHFFFAOYSA-N 0.000 description 1
- CHTYMWBYHAIEOF-UHFFFAOYSA-N COc(cc1)cc(nc2O)c1nc2O Chemical compound COc(cc1)cc(nc2O)c1nc2O CHTYMWBYHAIEOF-UHFFFAOYSA-N 0.000 description 1
- LWDSJSSAQHMGHA-PRJDIBJQSA-N O[C@H]1C(CCCCl)C1 Chemical compound O[C@H]1C(CCCCl)C1 LWDSJSSAQHMGHA-PRJDIBJQSA-N 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C35/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring
- C07C35/02—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring monocyclic
- C07C35/04—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a ring other than a six-membered aromatic ring monocyclic containing a three or four-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C269/00—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C269/04—Preparation of derivatives of carbamic acid, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups from amines with formation of carbamate groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/32—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C271/34—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of rings other than six-membered aromatic rings with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/14—Preparation of carboxylic acid esters from carboxylic acid halides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/013—Esters of alcohols having the esterified hydroxy group bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/02—Esters of acyclic saturated monocarboxylic acids having the carboxyl group bound to an acyclic carbon atom or to hydrogen
- C07C69/12—Acetic acid esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/16—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0808—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/12—Cyclic peptides with only normal peptide bonds in the ring
- C07K5/126—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Communicable Diseases (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Veterinary Medicine (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Cephalosporin Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
본 발명은 HCV 억제 화합물의 상이한 형태에 관한 것이다.
Description
본 발명은 HCV 억제 화합물의 상이한 형태에 관한 것이다. HCV 억제 화합물은 치료 및 연구 응용을 갖는다.
C형 간염 바이러스(HCV) 감염은 주요 건강 문제점이다. HCV 감염은 상당수의 감염된 개체들에 있어서, 만성 간 질환, 예컨대 간경화 및 간세포 암종을 초래한다. 몇몇 바이러스적으로-코딩된 효소는 메탈로프로테아제(NS2-3), 세린 프로테아제(NS3), 헬리카제(NS3), 및 RNA-의존성 RNA 폴리머라제(NS5B)를 비롯한, 치료적 개입을 위해 추정되는 표적들이다.
HCV 감염의 가능한 치료법은 문헌 [Balsano, Mini Rev . Med . Chem . 8(4):307-318, 2008], [Roenn et al ., Current Topics in Medicinal Chemistry 8:533-562, 2008], [Sheldon et al ., Expert Opin . Investig . Drugs 16(8):1171-1181, 2007], 및 [De Francesco et al ., Antiviral Research 58:1-16, 2003]을 비롯한 다양한 문헌에서 논의되어 있다.
HCV 프로테아제 활성을 억제할 수 있는 마크로락탐 화합물을 기재하는 공개문헌의 예로는 [McCauley et al ., WO2011014487]; [Harper et al ., WO2010011566]; [Liverton et al ., WO2009134624]; [McCauley et al ., WO2009108507]; [Liverton et al ., WO2009010804]; [Liverton et al ., WO2008057209]; [Liverton et al ., WO2008051477]; [Liverton et al ., WO2008051514]; [Liverton et al ., WO2008057208]; [Crescenzi et al ., WO2007148135]; [Di Francesco et al ., WO2007131966]; [Holloway et al ., WO2007015855]; [Holloway et al ., WO2007015787]; [Holloway et al ., WO2007016441]; [Holloway et al ., WO2006119061]; [Liverton et al ., J. Am . Chem. Soc ., 130:4607-4609, 2008]; 및 [Liverton et al ., Antimicrobial Agents and Chemotherapy 54:305-311, 2010]이 포함된다.
발명의 개요
본 발명은 상이한 형태의 하기 화합물 A 또는 그의 제약 염을 포함한다:
<화합물 A>

결정질 화합물 A에 관한 본 발명의 측면은, 치료 유효량의 상기 화합물 및 제약상 허용되는 담체를 포함하는 제약 조성물; HCV 감염 환자를 상기 화합물로 치료하는 것; 의학에서의 상기 화합물의 용도; 환자에서 HCV를 치료하는데 사용하기 위한 의약의 제조; 및 아세톤 및 물을 사용하여 화합물 A로부터 수화물 III을 제조하는 방법을 포함한다.
본 발명의 다른 실시양태, 측면 및 특징은 본원에 추가로 기재되거나 다음의 설명, 실시예 및 첨부된 특허청구범위로부터 명백할 것이다.
도 1은 화합물 A 결정질 수화물 II의 X-선 회절 패턴을 도시한다.
도 2는 화합물 A 수화물 III의 X-선 회절 패턴을 도시한다.
도 3은 화합물 A 수화물 III의 열중량 분석 곡선을 도시한다.
도 4는 화합물 A 수화물 III의 시차 주사 열량측정 곡선을 도시한다.
도 5는 화합물 A 수화물 III에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 6은 결정질 화합물 A Na-염의 X-선 회절 패턴을 도시한다.
도 7은 결정질 화합물 A K-염의 X-선 회절 패턴을 도시한다.
도 8은 화합물 A 결정질 수화물 I의 X-선 회절 패턴을 도시한다.
도 9는 화합물 A 수화물 I에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 10은 화합물 A 결정질 수화물 IV의 X-선 회절 패턴을 도시한다.
도 11은 화합물 A 수화물 IV에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 12는 화합물 A 결정질 수화물 V의 X-선 회절 패턴을 도시한다.
도 13은 화합물 A 수화물 V에 관한 고체 상태 C-13 CPMAS NMR을 도시한다,
도 14는 화합물 A 결정질 수화물 VI의 X-선 회절 패턴을 도시한다.
도 15는 화합물 A 수화물 VI에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 2는 화합물 A 수화물 III의 X-선 회절 패턴을 도시한다.
도 3은 화합물 A 수화물 III의 열중량 분석 곡선을 도시한다.
도 4는 화합물 A 수화물 III의 시차 주사 열량측정 곡선을 도시한다.
도 5는 화합물 A 수화물 III에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 6은 결정질 화합물 A Na-염의 X-선 회절 패턴을 도시한다.
도 7은 결정질 화합물 A K-염의 X-선 회절 패턴을 도시한다.
도 8은 화합물 A 결정질 수화물 I의 X-선 회절 패턴을 도시한다.
도 9는 화합물 A 수화물 I에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 10은 화합물 A 결정질 수화물 IV의 X-선 회절 패턴을 도시한다.
도 11은 화합물 A 수화물 IV에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
도 12는 화합물 A 결정질 수화물 V의 X-선 회절 패턴을 도시한다.
도 13은 화합물 A 수화물 V에 관한 고체 상태 C-13 CPMAS NMR을 도시한다,
도 14는 화합물 A 결정질 수화물 VI의 X-선 회절 패턴을 도시한다.
도 15는 화합물 A 수화물 VI에 관한 고체 상태 C-13 CPMAS NMR을 도시한다.
HCV 활성을 억제할 수 있는 마크로락탐 화합물은 생체내 HCV 활성 억제, 시험관내 HCV 활성 억제, 및 HCV NS3 효소 활성 억제를 비롯한 상이한 용도를 갖는다. HCV 활성의 생체내 억제는 치료 응용에 사용될 수 있다. 시험관내 HCV 활성 억제는 HCV 내성 돌연변이체를 수득하는데 사용되는 것, HCV 레플리콘 또는 효소 활성을 억제하는 관능기의 능력을 추가로 특성화하는 것, 및 HCV 복제 또는 프로테아제 활성을 연구하는 것을 비롯한 상이한 응용을 갖는다.
화합물 A는 문헌 [Harper et al ., WO2010011566]에 기재되어 있다. 문헌 [Harper et al ., WO2010011566]은 HCV 레플리콘 활성 및 NS3/4A를 억제하는 화합물 A의 능력을 나타내는 데이터를 포함한다.
화합물 A 형태
6개의 상이한 화합물 A 수화물을 확인하였다 (형태 I, II, III, IV, 및 V). 수화물 III은 확인된 가장 안정한 수화물 형태였다.
수화물 II에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선 (즉, 방사선 공급원은 Cu Kα1 및 Kα2 방사선의 조합임)을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 1에 도시되어 있다.
제2 실시양태에서, 수화물은 약 11.7, 16.6, 및 11.2 도(degree)의 약 2θ 값 (즉, 2θ 값에서 반사)을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
본원에서 제공된 2θ 값에 관해서 "약"에 대한 언급은 ± 0.1을 나타낸다.
본 실시양태 및 이하에 유사 실시양태에서 용어 "약"은 2θ 값 각각을 변형시키는 것으로 이해되며; 예를 들어, 표현 "약 11.7, 16.6, 및 11.2"는 "약 11.7, 약 16.6, 및 약 11.2"에 대한 약칭이다.
수화물 II에 관한 제3 실시양태에서, 수화물은 약 11.7, 16.6, 11.2, 15.1, 및 16.1 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 II에 관한 제4 실시양태에서, 수화물은 약 11.7, 16.6, 11.2, 15.1, 16.1, 23.0, 20.9, 8.0, 23.9, 25.0, 16.8, 17.8, 19.8, 22.5, 및 8.3 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
또 다른 실시양태에서, 수화물 II는 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 II는 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
수화물 III에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 2에 도시되어 있다.
수화물 III에 관한 제2 실시양태에서, 수화물은 약 20.5, 5.0, 및 18.2 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 III에 관한 제3 실시양태에서, 수화물은 약 20.5, 5.0, 18.2, 20.1, 및 20.7 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 III에 관한 제4 실시양태에서, 수화물은 약 20.5, 5.0, 18.2, 20.1, 20.7, 14.1, 23.7, 13.1, 18.9, 15.3, 20.9, 11.4, 18.4, 14.7, 및 13.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 III에 관한 제5 실시양태에서, 수화물은 도 3에 제공된 바와 같은 열중량 분석을 특징으로 한다.
수화물 III에 관한 제6 실시양태에서, 수화물은 도 4에 제공된 바와 같은 시차 주사 열량측정 곡선을 특징으로 한다.
제7 실시양태는 수화물 III에 관한 것이고, 여기서 수화물은 도 5에 제공된 고체 상태 탄소-13 CPMAS NMR 스펙트럼을 특징으로 한다.
제8 실시양태는 수화물 III에 관한 것이고, 여기서 수화물은 약 5.14, 6.31, 12.49, 18.35, 26.81, 28.03, 30.33, 31.27, 34.95, 35.99, 38.68, 42.01, 54.93, 56.39, 60.14, 74.20, 107.02, 120.11, 121.60, 129.73, 134.35, 135.95, 142.89, 148.47, 155.37, 157.32, 160.90, 168.32, 172.17, 및 175.53 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 한다.
본원에서 제공된 고체 상태 탄소-13 CPMAS NMR 2θ 값에 관해서 "약"에 대한 언급은 ± 0.1을 나타낸다.
또 다른 실시양태에서, 수화물 III은 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 III은 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
추가 측면에서 화합물 A는 K 또는 Na 결정질 염으로서 제공된다. 이들 염은 수화물이다.
화합물 A 결정질 Na 염에 관한 제1 실시양태에서, 화합물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 6에 도시되어 있다.
화합물 A 결정질 Na 염에 관한 제2 실시양태에서, 화합물은 약 18.4, 9.1, 및 9.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
화합물 A 결정질 Na 염에 관한 제3 실시양태에서, 화합물은 약 18.4, 9.1, 9.8, 9.6, 19.3, 15.3 및 16.5 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
화합물 A 결정질 Na 염에 관한 제4 실시양태에서, 화합물은 약 18.4, 9.1, 9.8, 9.6, 19.3, 15.3, 16.5, 22.5, 17.4, 20.2, 8.4, 21.3, 26.9, 4.8, 및 26.2 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
또 다른 실시양태에서, 화합물 A 결정질 Na 염은 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 화합물 A 결정질 Na 염은 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
화합물 A 결정질 K 염에 관한 제1 실시양태에서, 화합물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 7에 도시되어 있다.
화합물 A 결정질 K 염에 관한 제2 실시양태에서, 화합물은 약 18.2, 8.9, 및 20.3 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
화합물 A 결정질 K 염에 관한 제3 실시양태에서, 화합물은 약 18.2, 8.9, 20.3, 18.7 및 22.5 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
화합물 A 결정질 K 염에 관한 제4 실시양태에서, 화합물은 약 18.2, 8.9, 20.3, 18.7, 22.5, 8.4, 19.6, 16.7, 27.1, 10.3, 21.9, 9.4, 21.2, 25.9, 및 12.5 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
또 다른 실시양태에서, 화합물 A 결정질 K 염은 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 화합물 A 결정질 K 염은 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
수화물 I에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 8에 도시되어 있다.
수화물 I에 관한 제2 실시양태에서, 수화물은 약 8.6, 20.6, 및 26.6 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 I에 관한 제3 실시양태에서, 수화물은 약 8.6, 20.6, 26.6, 17.4, 및 16.6 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 I에 관한 제4 실시양태에서, 수화물은 약 8.6, 20.6, 26.6, 17.4, 16.6, 12.2, 21.2, 18.8, 15.0, 23.0, 14.1 및 16.9 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
제7 실시양태는 수화물 I에 관한 것이고, 여기서 수화물은 도 9에 제공된 고체 상태 탄소-13 CPMAS NMR 스펙트럼을 특징으로 한다.
제8 실시양태는 수화물 I에 관한 것이고, 여기서 수화물은 약 4.22, 7.23, 11.45, 17.79, 24.04, 26.95, 28.29, 31.15, 32.47, 32.47, 33.46, 34.03, 35.74, 42.32, 53.50, 56.05, 56.96, 77.49, 108.95, 119.65, 122.55, 131.05, 133.13, 135.38, 142.28, 150.78, 156.03, 157.99, 161.36, 171.40, 173.42, 174.30 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 한다.
또 다른 실시양태에서, 수화물 I은 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 I은 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
수화물 IV에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 10에 도시되어 있다.
수화물 IV에 관한 제2 실시양태에서, 수화물은 약 14.7, 11.5, 및 7.1 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 IV에 관한 제3 실시양태에서, 수화물은 약 14.7, 11.5, 7.1, 9.3, 및 15.6 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 IV에 관한 제4 실시양태에서, 수화물은 약 14.7, 11.5, 7.1, 9.3, 15.6, 7.7, 및 8.0 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
제7 실시양태는 수화물 IV에 관한 것이고, 여기서 수화물은 도 11에 제공된 고체 상태 탄소-13 CPMAS NMR 스펙트럼을 특징으로 한다.
제8 실시양태는 수화물 IV에 관한 것이고, 여기서 수화물은 약 3.90, 5.30, 6.99, 10.49, 13.13, 17.81, 24.73, 27.52, 28.14, 29.42, 31.02, 32.80, 36.08, 39.22, 42.45, 53.62, 55.93, 59.14, 60.76, 74.77, 109.22, 111.19, 11.38, 120.24, 122.50, 133.96, 139.74, 147.2, 148.90, 154.65, 158.25, 159.53, 160.12, 170.14, 171.05, 172.08, 173.47, 및 174.46 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 한다.
또 다른 실시양태에서, 수화물 IV는 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 IV는 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
수화물 V에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 12에 도시되어 있다.
수화물 V에 관한 제2 실시양태에서, 수화물은 약 9.1, 18.3, 및 19.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 V에 관한 제3 실시양태에서, 수화물은 약 9.1, 18.3, 19.8, 15.2, 및 23.2 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 V에 관한 제4 실시양태에서, 수화물은 약 9.1, 18.3, 19.8, 15.2, 23.2, 10.9, 17.6 및 23.9 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
제7 실시양태는 수화물 V에 관한 것이고, 여기서 수화물은 도 13에 제공된 고체 상태 탄소-13 CPMAS NMR 스펙트럼을 특징으로 한다.
제8 실시양태는 수화물 V에 관한 것이고, 여기서 수화물은 약 7.86, 8.92, 13.10, 18.31, 23.72, 27.44, 28.47, 30.77, 35.79, 36.25, 37.15, 37.15, 42.95, 53.13, 55.67, 57.31, 60.47, 62.06, 75.09, 110.59, 112.24, 118.32, 132.18, 134.05, 135.83, 139.88, 148.30, 155.19, 157.97, 159.41, 170.31 및 175.20 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 한다.
또 다른 실시양태에서, 수화물 V는 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 V는 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
수화물 VI에 관한 제1 실시양태에서, 수화물은 3개 이상의 특징적 피크를 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다. 특징적 피크는 도 14에 도시되어 있다.
수화물 VI에 관한 제2 실시양태에서, 수화물은 약 20.5, 12.8, 및 19.4 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 VI에 관한 제3 실시양태에서, 수화물은 약 20.5, 12.8, 19.4, 21.2, 및 16.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
수화물 VI에 관한 제4 실시양태에서, 수화물은 약 20.5, 12.8, 19.4, 21.2, 16.8, 13.9, 5.0, 18.5, 23.7, 및 26.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 한다.
제7 실시양태는 수화물 VI에 관한 것이고, 여기서 수화물은 도 15에 제공된 고체 상태 탄소-13 CPMAS NMR 스펙트럼을 특징으로 한다.
제8 실시양태는 수화물 VI에 관한 것이고, 여기서 수화물은 약 4.87, 6.24, 11.70, 12.85, 18.36, 26.55, 28.31m 31.51, 34.98, 38.47, 42.09, 54.27, 56.12, 60.10, 73.49, 73.97, 105.91, 108.04, 118.39, 119.75, 121.33, 129.96, 133.87, 136.13, 142.26, 142.97, 146.85, 148.36, 154.97, 157.32, 160.71, 168.23, 172.21 및 175.34 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 한다.
또 다른 실시양태에서, 수화물 VI은 실질적으로 순수하다. "실질적으로 순수"에 대한 언급은 특정의 형태가 존재하는 화합물의 적어도 50%를 구성함을 의미한다. 상이한 실시양태에서, 수화물 VI은 존재하는 화합물 A의 적어도 75%, 적어도 85%, 적어도 90%, 적어도 95%, 또는 약 94%-98%를 구성한다.
또 다른 측면은 아세톤/물 및 건조의 사용을 포함하는, 화합물 A로부터 수화물 III의 제조 방법에 관한 것이다. 아세톤/물의 상이한 비를 이용할 수 있다. 상이한 실시양태에서, 아세톤 대 물 비는 80:20 v/v 아세톤 대 물 내지 0:100 v/v 아세톤 대 물이다. 추가 실시양태에서, 아세톤 대 물 비는 65:35, 50:50, 약 65:35, 또는 약 50:50이고/거나; 화합물 A의 수화물 II 또는 수화물 IV는 수화물 III의 제조를 위한 출발점으로서 사용되고/거나; 화합물은 아세톤에 초기에 용해된다.
아세톤:물 비에 관해서 "약"에 대한 언급은 각각의 성분에 대해 ± 10의 범위를 나타낸다. 예를 들어, 비 65:35에 관한 "약"은, 아세톤이 55-75로 변할 수 있음을 나타낸다.
한 실시양태에서, 조물질 (수화물 II)을 아세톤에 용해시킨다. 물을 공정에 첨가하여 용매 조성물을 85:15 아세톤:물이 되도록 한다. 그 다음 공정을 시딩하여 결정화를 개시한 다음, 물을 첨가하여 수율을 향상시키고, 최종 용매 조성은 50:50 아세톤:물이다.
동위원소 농축(
enrichment
)
본원에 기재된 화합물에서의 원자는 그의 본질적 동위 원소 존재율을 나타낼 수 있거나, 원자 중 1개 이상은 동일 원자수를 갖지만 주로 자연 상태에서 발견되는 원자 질량 또는 질량수와 상이한 원자 질량 또는 질량수를 갖는 특정의 동위 원소로 인위적으로 농축될 수 있다. 본 발명은 본원에 기재된 화합물의 모든 적합한 동위 원소 변형을 포함하는 것으로 여겨진다. 예를 들어, 수소 (H)의 상이한 동위원소 형태로는 프로튬 (1H) 및 듀테륨 (2H)이 포함된다. 프로튬은 자연에서 발견되는 지배적인 수소 동위 원소이다. 듀테륨의 농축은 특정 치료상 이점, 예컨대 생체내 반감기 증가 또는 투여 요건 감소를 제공해 줄 수 있거나, 생체 시료의 특성화를 위한 표준으로서 유용한 화합물을 제공할 수 있다.
본원에 기재된 동위 원소-농축된 화합물은 과도한 실험 없이 당업자에게 주지된 통상적인 기술에 의해 또는 본원에 제공된 반응식 및 실시예에 기재된 것과 유사한 공정에 의해 적절한 동위 원소-농축된 시약 및/또는 중간체를 사용하여 제조될 수 있다.
투여 및 조성물
제약상 허용되는 염은 환자, 바람직하게는 인간에게 투여하기에 적합하다. 적합한 염으로는, 예를 들어, 화합물의 용액을 제약상 허용되는 산, 예컨대 염산, 황산, 아세트산, 트리플루오로아세트산, 또는 벤조산의 용액과 혼합함으로써 형성될 수 있는 산 부가염이 포함된다. 산성 모이어티를 갖는 화합물을 적합한 제약상 허용되는 염과 혼합하여, 예를 들어, 알칼리 금속염 (예를 들어, 나트륨 또는 칼륨 염), 알칼리 토금속염 (예를 들어, 칼슘 또는 마그네슘 염), 및 적합한 유기 리간드와 함께 형성된 염, 예컨대 4급 암모늄 염을 제공할 수 있다. 또한, 산 (-COOH) 또는 알콜 기가 존재하는 경우에, 화합물의 용해도 또는 가수분해 특성을 변형시키기 위하여 제약상 허용되는 에스테르를 사용할 수 있다.
화합물 A를 HCV로 감염된 환자에게 투여할 수 있다. 용어 "투여" 및 그의 변형 (예를 들어, 화합물을 "투여함")은 치료를 필요로 하는 개체에게 화합물을 제공함을 의미한다. 화합물이 1종 이상의 다른 활성제 (예를 들어, HCV 감염을 치료하는데 유용한 항바이러스제)와 조합하여 제공되는 경우에, "투여" 및 그의 변형은 각각 화합물 또는 염 및 다른 작용제의 동시 및 순차적 제공을 포함하는 것으로 이해된다.
본원에 사용된 바와 같이, 용어 "조성물"은 특정된 성분을 포함하는 생성물뿐만 아니라, 특정된 성분을 배합함으로써 직접 또는 간접적으로 생성되는 임의의 생성물을 포함하는 것으로 의도된다.
"제약상 허용되는"이란 제약 조성물의 성분이 서로 상용성이고 그의 수용자에게 적합함을 의미한다.
본원에 사용된 바와 같은 용어 "대상체" (대안으로 본원에서 "환자"로 언급됨)는 치료, 관찰 또는 실험의 대상인 동물, 바람직하게는 포유동물, 가장 바람직하게는 인간을 지칭한다.
용어 "유효량"은 치료적 또는 예방적 효과를 발휘하기에 충분한 양을 나타낸다. HCV로 감염된 환자의 경우, 유효량은 다음 효과 중 하나 이상을 달성하기에 충분하다: 복제하는 HCV의 능력을 감소시키고, HCV 로드(load)를 감소시키고, 바이러스 제거(clearance)를 증가시킨다. HCV로 감염되지 않은 환자의 경우, 유효량은 다음 중 하나 이상을 달성하기에 충분하다: HCV 감염에 대한 감수성(susceptibility) 감소, 및 만성 질환에 관한 지속적인 감염을 확립하는 감염 바이러스의 능력의 감소.
HCV NS3 프로테아제를 억제하고 HCV 감염을 치료하고/하거나 HCV 감염의 가능성 또는 증상의 중증도를 저하시키기 위하여, 화합물은, 임의로 염의 형태로, 활성제를 활성제의 작용 부위와 접촉하게 하는 수단에 의해 투여될 수 있다. 이들은 개별적인 치료제로서 또는 치료제의 조합물로서 제약과 함께 사용하는데 이용할 수 있는 통상적인 수단에 의해 투여될 수 있다. 이들은 단독으로 투여될 수 있지만, 전형적으로는 선택된 투여 경로 및 표준 제약 실무에 근거하여 선택된 제약 담체와 함께 투여된다.
화합물은, 예를 들어, 다음의 경로들 중 하나 이상에 의해 투여될 수 있다: 유효량의 화합물 및 제약상 허용되는 담체 (예를 들어, 인간 환자에게 투여하기에 적합한 담체), 아주반트 및 비히클을 함유하는 제약 조성물의 단위 투여의 형태로, 경구로, 비경구로 (피하 주사, 정맥내, 근육내, 흉골내 주사 또는 주입 기술을 포함함), 흡입에 의해 (예컨대 분무 형태로), 또는 직장내로. 경구 투여에 적합한 액체 제제 (예를 들어, 현탁액, 시럽, 엘릭시르 등)는 매질, 예컨대 물, 글리콜, 오일, 알콜 등을 이용할 수 있다. 경구 투여에 적합한 고체 제제 (예를 들어, 분말, 환제, 캡슐 및 정제)는 고체 부형제, 예컨대 전분, 당, 카올린, 윤활제, 결합제, 붕해제 등을 이용할 수 있다. 비경구 조성물은 전형적으로는 담체로서 멸균수 및 임의로 다른 성분, 예컨대 용해도 보조제를 이용한다. 주사용 액제는, 예를 들어, 생리 식염수 용액, 글루코스 용액 또는 생리 식염수와 글루코스의 혼합물을 함유하는 용액을 포함하는 담체를 사용하여 제조할 수 있다. 제약 조성물의 제조시 사용하기에 적합한 방법에 관한 추가 지침이 문헌 [Remington : The Science and Practice of Pharmacy, 21th edition (Lippincott Williams & Wilkins, 2006)]에 제공되어 있다.
치료 화합물은 단일 용량으로 또는 분할 용량으로 하루에 포유동물 (예: 인간) 체중 1 ㎏당 0.001 내지 1000 ㎎의 투여량 범위로 경구 투여할 수 있다. 한 투여량 범위는 단일 용량으로 또는 분할 용량으로 경구로 하루에 체중 1 ㎏당 0.01 내지 500 ㎎이다. 또 다른 투여량 범위는 단일 또는 분할 용량으로 경구로 하루에 체중 1 ㎏당 0.1 내지 100 ㎎이다. 경구 투여용으로, 조성물은 치료할 환자에 대한 투여량의 증상적 조정을 위해 활성 성분 1.0 내지 500 ㎎, 특히 활성 성분 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 및 750 ㎎을 함유하는 정제 또는 캡슐의 형태로 제공될 수 있다. 임의의 특정 환자의 경우 투여의 구체적 용량 수준 및 빈도를 달리할 수 있고, 이용된 구체적 화합물의 활성, 화합물의 대사적 안정성 및 작용 기간, 연령, 체중, 전체적인 건강, 성별, 식이, 투여 방식과 시간, 배설률, 약물 병용, 특정 병태의 중증도, 및 치료를 겪는 숙주를 비롯한 다양한 요인에 따라 좌우될 것이다.
본 발명의 다른 실시양태는 다음을 포함한다:
(a) 유효량의 화합물 A 및 제약상 허용되는 담체를 포함하는 제약 조성물.
(b) (a)에 있어서, HCV 항바이러스제, 면역조절제, 및 항감염제로 이루어진 군으로부터 선택된 제2 치료제를 추가로 포함하는 제약 조성물.
(c) (b)에 있어서, HCV 항바이러스제가 HCV 프로테아제 억제제, NS5A 억제제, 및 HCV NS5B 폴리머라제 억제제로 이루어진 군으로부터 선택된 항바이러스제인 제약 조성물.
(d) (i) 화합물 A 및 (ii) HCV 항바이러스제, 면역조절제, 및 항감염제로 이루어진 군으로부터 선택된 제2 치료제인 제약 조합물로서; 여기서 화합물 A 및 제2 치료제가 각각 조합물을 HCV 복제의 억제, HCV 감염의 치료 및/또는 HCV 감염의 가능성 또는 증상의 중증도의 감소에 효과적이 되도록 하는 양으로 이용되는 것인 제약 조합물.
(e) (d)에 있어서, HCV 항바이러스제가 HCV 프로테아제 억제제, NS5A 억제제, 및 HCV NS5B 폴리머라제 억제제로 이루어진 군으로부터 선택된 항바이러스제인 조합물.
(f) HCV 복제의 억제를 필요로 하는 대상체에게 유효량의 화합물 A를 투여하는 것을 포함하는, 상기 대상체에서 HCV 복제를 억제하는 방법.
(g) HCV 감염의 치료 및/또는 HCV 감염의 가능성 또는 증상의 중증도의 감소를 필요로 하는 대상체에게 유효량의 화합물 A를 투여하는 것을 포함하는, 상기 대상체에서 HCV 감염을 치료하고/거나 HCV 감염의 가능성 또는 증상의 중증도를 감소시키는 방법.
(h) (g)에 있어서, 화합물을 HCV 항바이러스제, 면역조절제, 및 항감염제로 이루어진 군으로부터 선택된 유효량의 1종 이상의 제2 치료제와 조합하여 투여하는 것인 방법.
(i) (h)에 있어서, HCV 항바이러스제가 HCV 프로테아제 억제제, NS5A 억제제 및 HCV NS5B 폴리머라제 억제제로 이루어진 군으로부터 선택된 항바이러스제인 방법.
(j) HCV 복제의 억제를 필요로 하는 대상체에게 (a), (b) 또는 (c)의 제약 조성물 또는 (d) 또는 (e)의 조합물을 투여하는 것을 포함하는, 상기 대상체에서 HCV 복제를 억제하는 방법.
(k) HCV 감염의 치료 및/또는 HCV 감염의 가능성 또는 증상의 중증도의 감소를 필요로 하는 대상체에게 (a), (b) 또는 (c)의 제약 조성물 또는 (d) 또는 (e)의 조합물을 투여하는 것을 포함하는, 상기 대상체에서 HCV 감염을 치료하고/거나 HCV 감염의 가능성 또는 증상의 중증도를 감소시키는 방법.
본 발명은 또한 의학에서 사용하기 위한 본 발명의 화합물; 또는 (a) HCV 복제의 억제 또는 (b) HCV 감염의 치료 및/또는 HCV 감염의 가능성 또는 증상의 중증도의 감소를 위한, (i) 의약에서 사용하거나 (ii) 의약의 제조에서 사용하기 위한 본 발명의 화합물을 포함한다. 이들 용도에서, 본 발명의 화합물은 임의로 HCV 항바이러스제, 항감염제, 및 면역조절제로부터 선택된 1종 이상의 제2 치료제와 조합하여 이용될 수 있다.
HCV
억제 활성
HCV NS3 활성, HCV 레플리콘 활성, 및 HCV 복제 활성을 억제하는 화합물의 능력은 당업계에 주지된 기술을 사용하여 평가될 수 있다. (예를 들어, 문헌 [Carroll et al., J. Biol . Chem . 278: 11979-11984, 2003] 참조.)
한 그러한 검정은 하기 및 문헌 [Mao et al., Anal . Biochem . 373:1-8, 2008] 및 [Mao et al., WO2006/102087]에 기재된 바와 같은 HCV NS3 프로테아제 시간-분해 형광 (TRF) 검정이다. NS3 프로테아제 검정을, 예를 들어, 50 mM HEPES, pH 7.5, 150 mM NaCl, 15% 글리세롤, 0.15% 트리톤(TRITON) X-100, 10 mM DTT, 및 0.1% PEG 8000을 함유하는 100 ㎕ 검정 완충제의 최종 부피 중에서 수행할 수 있다. NS3 및 NS4A를 30분 동안 DMSO 중에서 다양한 농도의 억제제와 함께 사전-배양시킨다. 반응은 TRF 펩티드 기질 (최종 농도 100 nM)을 첨가함으로써 개시된다. 기질의 NS3 매개 가수분해를 100 ㎕의 500 mM MES, pH 5.5로 실온에서 1시간 후 켄칭한다. 생성물 형광을 400 ㎲ 지연으로 340 nm에서 여기 및 615 nm에서 발광으로 빅토르(VICTOR) V2 또는 퓨전(FUSION) 형광광도계 (퍼킨 엘머 라이프 앤드 어낼리티컬 사이언시즈(Perkin Elmer Life and Analytical Sciences))를 사용하여 검출한다. 상이한 효소 형태의 시험 농도를 선택하여 10-30의 신호 대 배경비 (S/B)를 초래한다. IC50 값은 데이터에 대한 표준 4-파라미터 피트(fit)를 사용하여 얻는다. Ki 값은 하기 수학식 1을 사용하여 IC50 값으로부터 얻는다.
<수학식 1>
상기 식에서, [S]는 반응 중 기질 펩티드의 농도이고 KM은 미하엘리스(Michaelis) 상수이다. 문헌 [P. Gallinari et al., 38 BIOCHEM. 5620-32(1999)]; [P. Gallinari et al., 72 J. VIROL. 6758-69 (1998)]; [M. Taliani et al., 240 ANAL. BIOCHEM. 60-67 (1996)]; 및 [Mao et al ., Analytical Biochemistry 373: 1-8, (2008)]을 참조한다.
약어
BOC: t-부톡시카르보닐
Cbz: 벤질옥시카르보닐
CDI: 1,1'-카르보닐디이미다졸
CIP: 2-클로로-1-메틸피리디늄 아이오다이드
CPME: 시클로펜틸 메틸 에테르
DABO: 1,4-디아자비시클로[2.2.2.]옥탄
DBA: 염 디벤질아민
DBU: 1,8-디아조비시클로[5.4.0]운데스-7-엔
DCC: N,N'-디시클로헥실카르보디이미드
DIC: N, N'-디이소프로필카르보디이미드
DIPEA: 디이소프로필에틸아민
DMAc: N,N-디메틸아세트아미드
DMF: N,N-디메틸포름아미드
DMPU: N,N-디메틸프로필렌우레아
DMSO: 디메틸술폭시드
EDC: 1-에틸-3-(3-디메틸아미노프로필) 카르보디이미드
EtOAc: 에틸 아세테이트
Fmoc: 9-플루오레닐메틸옥시카르보닐
HATU: 2-(1H-7-아자벤조트리아졸-1-일)-1,1,3,3-테트라메틸우로늄 헥사플루오로포스페이트
HOAT: 1-히드록시-7-아자벤조트리아졸
HOBT: 1-히드록시벤조트리아졸
HOPO: 2-히드록시피리딘-N-옥시드
HOSu: N-히드록시숙신이미드
IPA: 이소프로판올
IPAc: 이소프로필 아세테이트
MTBE: t-부틸 메틸 에테르
MsOH 및 MSA: CH3SO3H 또는 메탄술폰산
Moz: p-메톡시벤질옥시카르보닐
Msz: 4-메틸술피닐벤질옥시카르보닐
NMP: N-메틸피롤리돈
PFP: 펜타플루오로페놀
T3P: 프로필포스폰산 무수물
TBA: t-부틸 아민
TEA: 트리에틸아민
THF: 테트라히드로푸란
pTSA 및 TsOH는 각각 p-톨루엔술폰산에 관한 약어이다.
실시예
하기 제공된 실시예는 본 발명 및 그의 실시를 설명하고자 하는 것이다. 특허청구범위에 달리 제공되지 않는 한, 실시예는 발명의 범위 또는 취지에 대한 제한으로서 해석되지 않는다.
실시예
1: 2-[2-(3-
클로로
-프로필)-
시클로프로필
]-4,4,5,5-
테트라메틸[1,3,2]디옥사보롤란
(화합물 3)의 제조
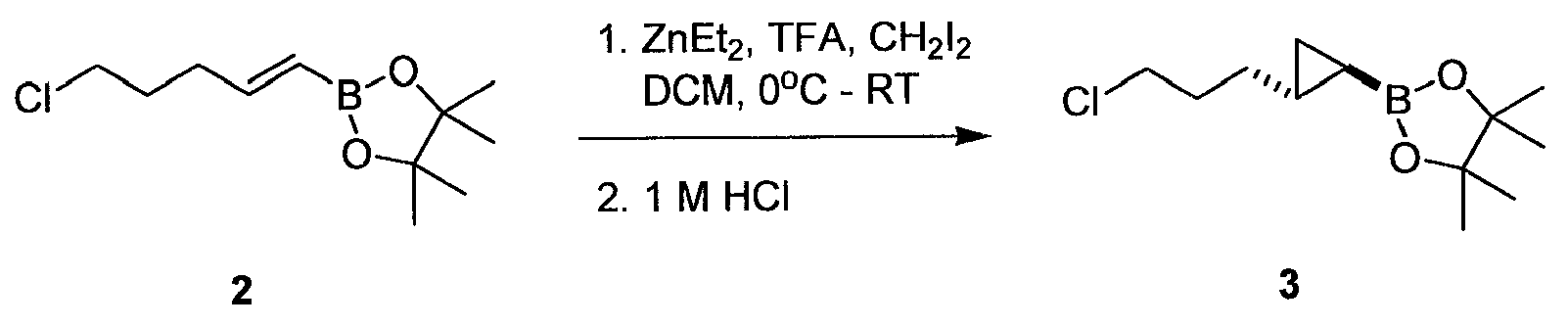
화합물 2는 문헌 [Shirakawa et al . Synthesis 11:1814-1820, 2004]에 기재된 바와 같이 제조할 수 있다.
화합물 3을 다음과 같이 제조하였다: N2 하에 질소 유입구, 기계 교반기, 적하 깔대기 및 열전쌍이 장착된 5 L 플라스크에 800 mL 디클로로메탄 및 800 mL의, 헵탄 중 1 M 디에틸아연 용액 (0.8 mol, 1.07 당량)을 첨가하였다. 용액을 빙조를 사용하여 내부 온도 3℃로 냉각하였다. 그 다음 플라스크에 200 mL 디클로로메탄 중 57.6 mL 트리플루오로아세트산 (0.748 mol, 1.0 당량)의 용액을, 내부 온도를 10℃ 미만으로 유지하면서 1시간에 걸쳐 적하 깔대기로부터 첨가하였다. 생성된 현탁액을 3℃에서 30분 동안 교반하였다. 그 다음 플라스크에 72.4 mL 디아이오도메탄 (0.897 mol, 1.2 당량)을 한 번에 첨가하였다. 30분 동안 3℃에서 교반 후, 172 mL의 2 (0.748 mol, 1.0 당량)를 용액에 한 번에 첨가하였다. 그 다음 플라스크를 실온으로 가온하고 백색 침전물이 형성되기 시작하였다. 3시간 후, GC 분석은 반응이 90% 전환되었음을 나타냈다. 현탁액을 추가의 17시간 또는 2의 완전한 소모가 관찰될 때까지 숙성시켰다. 그 시점에서, 800 mL의 1 M HCl (0.8 mol, 1.07 당량)을 첨가하고 +5℃ 발열이 관찰되었다. 2상의 혼합물을 30분 동안 교반하여 침전 고체를 용해시키고 유기층을 분리하였다. 200 mL 디클로로메탄을 사용한 수성층의 추출, 500 mL 염수를 사용한 합해진 유기층의 세척, 및 진공 중 농축으로 194 g의 3을 황색 오일 (DCM 중 74 wt%)로서 수득하였다.

실시예
2: 2-(3-
클로로
-프로필)-
시클로프로판올
(화합물 4)의 제조

질소 유입구, 기계 교반기, 적하 깔대기 및 열전쌍이 장착된 3 L 플라스크에1 L 메탄올 중 143 g의 3 (0.585 mol, 1.0 당량)을 첨가하였다. 그 다음 플라스크에 적하 깔대기로부터 58.5 mL의 10 M 수산화나트륨 (0.585 mol, 1.0 당량)을 30분에 걸쳐 첨가하고, 그 동안 외부 냉각과 함께 내부 온도를 10℃ 미만으로 유지하였다. 30분 동안 교반 후, 120 mL의 30 wt% 과산화수소 용액 (1.17 mol, 2 당량)을, 내부 온도를 10℃ 미만으로 유지하면서 1시간에 걸쳐 적하 깔대기로부터 서서히 첨가하였다. 첨가의 완료시, 그 다음 생성된 무색 슬러리를 30분 동안 또는 3의 완전한 소모가 GC에 의해 관찰될 때까지 주위 온도에서 교반하였다. 2 M HCl (375 mL)을, 내부 온도를 10℃ 미만으로 유지하면서 30분에 걸쳐 적하 깔대기로부터 첨가하였다. 그 다음 이러한 투명한 황색 용액에 500 mL의, Na2SO3의 1M 용액을 내부 온도를 10℃ 미만으로 유지하면서 적하 깔대기로부터 서서히 첨가하였다. 그 다음 생성된 현탁액을 여과하고 3 x 200 mL MTBE로 추출하였다. 농축 후 실리카겔 칼럼 크로마토그래피 (6:4 헥산:에틸 아세테이트)하여, 피나콜을 제거하여, 60.6 g의 생성물 4를 투명 오일 (90 wt %)로서 수득하였다.

실시예
3: 2-
펜트
-4-이닐-
시클로프로판올
(
rac
-화합물 5)의 제조

온도 프로브, N2 유입구, 및 격막(septum)이 장착된 2구 15 mL 플라스크에 1 g의 4 (7.28 mmol, 1.0 당량) 및 3.0 mL THF를 첨가하였다. 용액을 빙조를 사용하여 내부 온도 0℃로 냉각하였다. 이 용액에 2.95 mL의 33 wt% n-헥실리튬 (7.28 mmol, 1.0 당량)을 1시간에 걸쳐 시린지 펌프를 통해 서서히 첨가하였다. 내부 온도는 6.8℃로 상승하였고 용액은 황색이 되었다. 온도 프로브, N2 유입구, 및 격막이 장착된 분리 3구 100 mL 플라스크에서 0.82 g의 리튬 아세틸리드-에틸렌디아민 착물 (8.01 mmol, 1.1 당량)을 실온에서 5.0 mL의 DMPU 중에서 슬러리화하였다. 이러한 실온 슬러리에, 탈양성자화된 시클로프로판올의 냉 용액을 5분에 걸쳐 캐뉼라를 통해 옮겼다. 첨가 후, 갈색 혼합물을 3시간 동안 또는 98% 초과의 전환이 GC에 의해 관찰될 때까지 가열 맨틀을 사용하여 내부 온도 52℃로 가열하였다. 갈색 혼합물을 빙조를 사용하여 3℃로 냉각한 다음 빙조를 제거하여 동결을 방지하였다. 여기에 17.5 mL의 0.5 N HCl을 서서히 첨가하고 빙조를 적용하여 내부 온도를 21℃ 미만으로 유지하였다. 그 다음 혼합물을 10 mL MTBE 및 5 mL의 물로 희석한 후에 분별 깔대기에 옮기고 수성층을 제거하였다. 수성층을 15 mL MTBE로 1회 추출한 다음 합해진 유기층을 20 mL 물 이어서 20 mL 염수로 세척하였다. 그 다음 유기층을 진공중에 농축하여 1.27 g의 rac-5를 황색 오일 (72 wt%)로서 수득하였다.

실시예
4: 아세트산
라세미
트랜스-2-
펜트
-4-이닐-
시클로프로필
에스테르 (
rac
-화합물 6)의 제조

N2 하에 질소 유입구, 기계 교반기, 적하 깔대기 및 열전쌍이 장착된 5 L 플라스크에 31.2 g의 rac-5 (251 mmol, 1.0 당량), 350 mL의 MTBE 및 45.5 mL의 트리에틸아민 (327 mmol, 1.3 당량)을 첨가한 후에 아세톤/빙조 중에 용액을 내부 온도 < 5℃로 냉각하였다. 용액에 23.7 mL 아세틸 클로라이드 (301 mmol, 1.1 당량)를 내부 온도를 <10℃로 유지하면서 30분에 걸쳐 적하 깔대기로부터 첨가하였다. 그 다음 생성된 슬러리를 실온으로 가온하고 2시간 동안 숙성시켰다. 그 다음 반응 혼합물을 200 mL의 물로 희석하였다. 유기층을 200 mL의 2 N HCl 및 이어서 300 mL의 포화 NaHCO3로 세척한 후에 MgSO4 상에서 건조시켰다. 용매를 진공중에 제거하여 41.8 g의 rac-6을 수득하였다.

실시예
5: (1
R
, 2
R
)-2-
펜트
-4-이닐-
시클로프로판올
(
ent
-화합물-5)의 제조

오버헤드 교반기 및 온도 프로브가 장착된 1 L 플라스크에 MTBE (44.8 g, 0.27 mol) 중 rac-6의 60 wt% 용액 및 수성 0.1 M pH 7 인산염 완충제로 포화된 추가의 730 mL의 MTBE를 첨가하여, 60 g/l의 rac-6의 최종 용액 농도를 얻었다. 플라스크를 빙조에 위치시켜 가수분해 반응 전반에 걸쳐 내부 온도를 대략 10℃로 유지하였고, 이는 730 mg 노보짐 435를 첨가함으로써 개시되었다. 반응물을 전환이 41%에 도달할 때까지 대략 4시간 동안 10℃에서 숙성시키고, 이 시점에서 ent-5의 ee는 96%이었다. 그 다음 반응 혼합물을 150-ml 중간-세공 유리 필터 펀넬을 통해 여과하고 고체 부동화 효소를 80 ml MTBE로 3회 세척하였다. 그 다음 생성된 MTBE 용액을 헵탄으로 용매 교환하였다. 헵탄 중 혼합물 (39.2 kg, 대략 50 L)을 바이오타지 플래시(Biotage Flash) 400 L 카트리지 (40 x 60 cm, 40 kg 실리카겔, 60 옹스트롬, 40-63 um)에 적용하고 165 L의 2.5:97.5, 75 L의 10:90, 및 330 L의 25:75 EtOAc/헵탄 (v/v)으로 순차적으로 용리시켰다. 혼합물을 칼럼에 적용한 후, 18 L 분획을 취하였다. 알콜 ent-5의 풍부(rich) 절단 분획을 TLC (실리카, 20% EtOAc/헵탄)에 의해 위치시킨 다음 GC (HP-1, 30 m x 320 um x 0.25 um 필름, 9.14 psi 일정 He 압력, 15:1 분할, 5분 동안 50℃ 그 다음 275℃로 25도/분 및 5분 유지, 알콜의 RT 8.8분)에 의해 분석하였다. 분획 15 내지 21개를 농축하여 3.48 kg (80 wt%, 92% ee)의 목적 ent-5 (화합물 7)를 수득하였다.
GC: 레스텍 RT-Bdex SA (30 m x 0.25 mm x 0.25 ㎛), 60 cm/s 선 속도, 20:1 분할, 120℃ 등온선, tr(5) = 25.0, 29.6분, tr(6) = 17.1, 17.5분.
실시예
6: (S)-3,3-디메틸-2-((1R,2R)-2-
펜트
-4-이닐-
시클로프로폭시카르보닐아미노
)-부티르산 (화합물 8)의 제조

기계 교반기, 열전쌍 및 환류 냉각기가 장착된 50 L 환저 플라스크에 화합물 7 (3.477 kg, NMR에 의해 81 wt%, 92 % ee) 및 14.1 L (5 L/kg)의 후니그 염기를 첨가하였다. 생성된 균질 용액에 내부 온도를 21-25℃로 유지하면서 CDI를 고체로서 나누어 첨가하였다. 생성된 슬러리를 1시간 동안 실온에서 숙성시켰다. 슬러리에 L-tert-류신을 고체로서 첨가하고 반응 혼합물을 2.5시간 동안 내부 온도 95℃로 가열하였다. 반응 혼합물을 실온으로 냉각하고 17 L의 물로 희석하였다. 혼합물을 30분 동안 숙성시켜 모든 고체를 용해시킨 다음 100 L 원통형 추출기에 옮겼다. 그 다음 수성층을 12 L의 MTBE로 세척하였다. 수성층을 8 L의 MTBE로 세척하였다. 생성된 수성층을 진한 HCl을 사용하여 pH를 최종 pH 1.5-2.0으로 조정하였다. 2상 혼합물을 MTBE (2 X 12 L)로 추출하고 합해진 유기상을 6 L의 물 이어서 5 L의 염수로 세척하였다.
그 다음 MTBE 층을 진공을 통해 기계 교반기, 열전쌍, 및 배치 농축기(batch concentrator)가 장착된 50 L 환저 플라스크에 옮기고 증류 동안 배치의 내부 온도를 < 20℃로 유지하면서 용매를 감압하에 제거하였다. 그 다음 용매를 ~ 5 L의 CPME로 플러싱함으로써 시클로펜틸 메틸 에테르 (CPME)로 교환한 다음 최종 부피 ~ 20 L로 희석하였다. 이 물질을 추가 정제 없이 다음 반응에서 사용하였다.
분석 샘플을 무색 오일로서 실리카겔 크로마토그래피에 의해 수득하였다:

실시예
7: 6-
메톡시
-
퀴녹살린
-2,3-
디올
(화합물 10)의 제조

기계 교반기, 열전쌍 및 냉각기가 장착된 50 L 플라스크에 질소하에 4-메톡시-1,2-페닐렌디아민 디히드로클로라이드 염 (화합물 9) (2.65 kg, 98 wt%, 12.30 mol), 옥살산 (1.582 kg, 98 wt.%, 17.22 mol) 및 3 N HCl (수성) (17.8 L)을 첨가하였다. 회색 불균질 슬러리를 7.25 시간 동안 스팀을 사용하여 90℃로 가열하였다. 반응을 HPLC에 의해 모니터링하였다. 그 다음 생성된 회색 슬러리를 밤새 내부 온도 20℃로 냉각하였다. 슬러리를 여과하고, 물 (1.0-1.5 L/Kg)을 사용하여 옮겼다. 담회색 고체를 2 케이크(cake) 부피 물 (5.0-5.5 L/Kg)로 세척하였다. 고체를 24시간 동안 진공/N2 스윕(sweep)하에 건조시키고, 이때 고체는 여전히 매우 습윤 상태이다. 그 다음 생성물을 메탄올로 슬러리 세척하고, 진공 오븐 중 40-45℃에서 48시간에 걸쳐 건조시켜 화합물 10을 HPLC 검정에 의한 99.95% 순도의 회백색 생성물로서 수득하였다. NMR에 의하면 메탄올은 없었고 KF= 0.05 wt.% 물이었다.
HPLC 조건: 조르박스 이클립스 플러스(Zorbax Eclipse Plus) C18 50 x 4.6 mm, 1.8 um, 1.5 mL/분, 210 nm, 25℃, 용리액: 물 0.1% H3PO4 (A), 아세토니트릴 (B). 90% A 0분, 5% A 5분, 5% A 6분.
화합물 9 (디아민 HCl 염) 0.394분
화합물 10 1.55분 (때때로 2개의 피크)
실시예
8: 2,3-
디클로로
-6-
메톡시퀴녹살린
(화합물 11)의 제조

기계 교반기, 열전쌍 및 냉각기가 장착된 22 L 환저 플라스크에, 2,3-디클로로-6-메톡시퀴녹살론 화합물 10 (3.8 kg)을 첨가하였다. POCl3 (5.92 L, 99%)를 실온에서 서서히 충전하였다. 회색 슬러리를 20시간 동안 98℃로 가열하였다. 2-3 시간 후 슬러리는 회색에서 녹색, 그 다음 황색으로 변했고 최종적으로 균질한 적색으로 변했다. 슬러리가 POCl3 중에서 균질해짐에 따라, 상당한 양의 HCl 가스 배출(off-gassing)이 초래되었다. 암적색의 균질한 용액을 80℃ 미만으로 서서히 냉각하였다. 이 시점에서, 19 L의 아세토니트릴 (5.0 L/Kg)을 충전하고 이로써 담갈색 슬러리가 생성되었다. 반응물을 빙조에서 10-15℃로 냉각하고 100 L 원통형 용기 중 45.6 L의 냉수 (12.0 L/Kg)로 역(reverse) 켄칭하였다. 이러한 발열 켄칭을 27℃ 미만으로 유지하였다. MeCN (~ 4L)을 사용하여 슬러리를 옮겼다. 갈색 슬러리를 여과하고 5 L의 물을 사용하여 플라스크를 세척하였다. 고체를 1 케이크 부피의 물 (~5 L)로 세척하였다. 여액의 pH는 산성이었다. 그 다음 고체를 2 케이크 부피의 5% 탄산수소나트륨 (~20.00 L)으로 대체 세척하였다. pH는 8-9이었다. 슬러리 세척을 2 케이크 부피의 물 (총 20 L)로 수행하였다. pH는 변화하지 않았다. 고체를 감압 및 질소류(nitrogen flow)하에 72시간 동안 건조시켜 HPLC 검정에 의한 99.5% 순도의 황갈색(tan) 생성물 화합물 11을 수득하였고 KF= 0.5 wt.% 물이었다.
HPLC 조건: 조르박스 이클립스 플러스 C18 50 x 4.6 mm, 1.8 um, 1.5 mL/분, 210 nm, 25℃; 용리액: 물 0.1% H3PO4 (A), 아세토니트릴 (B). 90% A 0분, 5% A 5분, 5% A 6분.
화합물 10 1.55분 (때때로 2개의 피크)
화합물 11 4.55분
분석 샘플을 실리카겔 크로마토그래피에 의해 그리고 무색 발포체로서 수득하였다:

실시예
9: (2S,4R)-4-(3-
클로로
-7-
메톡시퀴녹살린
-2-
일옥시
)-2-(
메톡시카르보닐
)
피롤리디늄
메탄술포네이트
(14)의 제조

주위 온도에서 DMAc (500 ml, KF < 150) 중 2,3-디클로로퀴녹살린 11 (100 g, 0.437 mol) 및 N-Boc-4-트랜스-히드록시-L-프롤린 메틸 에스테르 (12, 118 g, 0.48 mol)의 슬러리에 DBU (86 g, 0.568 mol)를 첨가하였다. 슬러리를 ~35시간 동안 40-45℃에서 교반하였다. 그 다음 배치를 15℃로 냉각하였다. 에틸 아세테이트 (1.2 L) 이어서 시트르산 (10%, 504 mL, 162 mmol)을 내부 온도가 < 25℃로 유지되는 동안 첨가하였다. 유기상을 10% 시트르산 (200 mL) 및 물 (200 mL)의 용액 이어서 물 (400 mL x 2)로 세척하였다. 유기상을 공비 건조시키고 최종 부피 ~880 mL에서 MeCN으로 용매 교환하였다. MeSO3H (36 mL, 0.555 mol)를 첨가하고 반응 혼합물을 ~16시간 동안 40℃에서 숙성시켰다. 반응 슬러리에 35℃에서 2시간에 걸쳐 MTBE (1.05 L)를 적가하였다. 그 다음, 배치를 0-5℃로 추가로 냉각하고 2-3시간 동안 숙성시킨 후 여과하였다. 습윤 케이크를 MTBE 중 30% MeCN으로 대체 세척하고 (600 mL x 2), 40℃에서 진공 오븐 건조시켜 생성물 14를 수득하였다.

HPLC 조건: 하이퍼실 골드(Hypersil Gold) PFP 칼럼, 150 x 4.6 mm, 3.0 um; 칼럼 온도 40℃; 유량 1.8 mL/분; 및 파장 215 nm.

실시예
10: (S)-2-(((1R,2R)-2-(5-(6-
메톡시
-3-((3R,5S)-5-(
메톡시카르보닐
)
피롤리딘
-3-
일옥시
)
퀴녹살린
-2-일)
펜트
-4-이닐)
시클로프로폭시
)
카르보닐아미노
)-3,3-
디메틸부탄산
(16) 및
알킨
마크로시클릭
에스테르 (17)의 제조
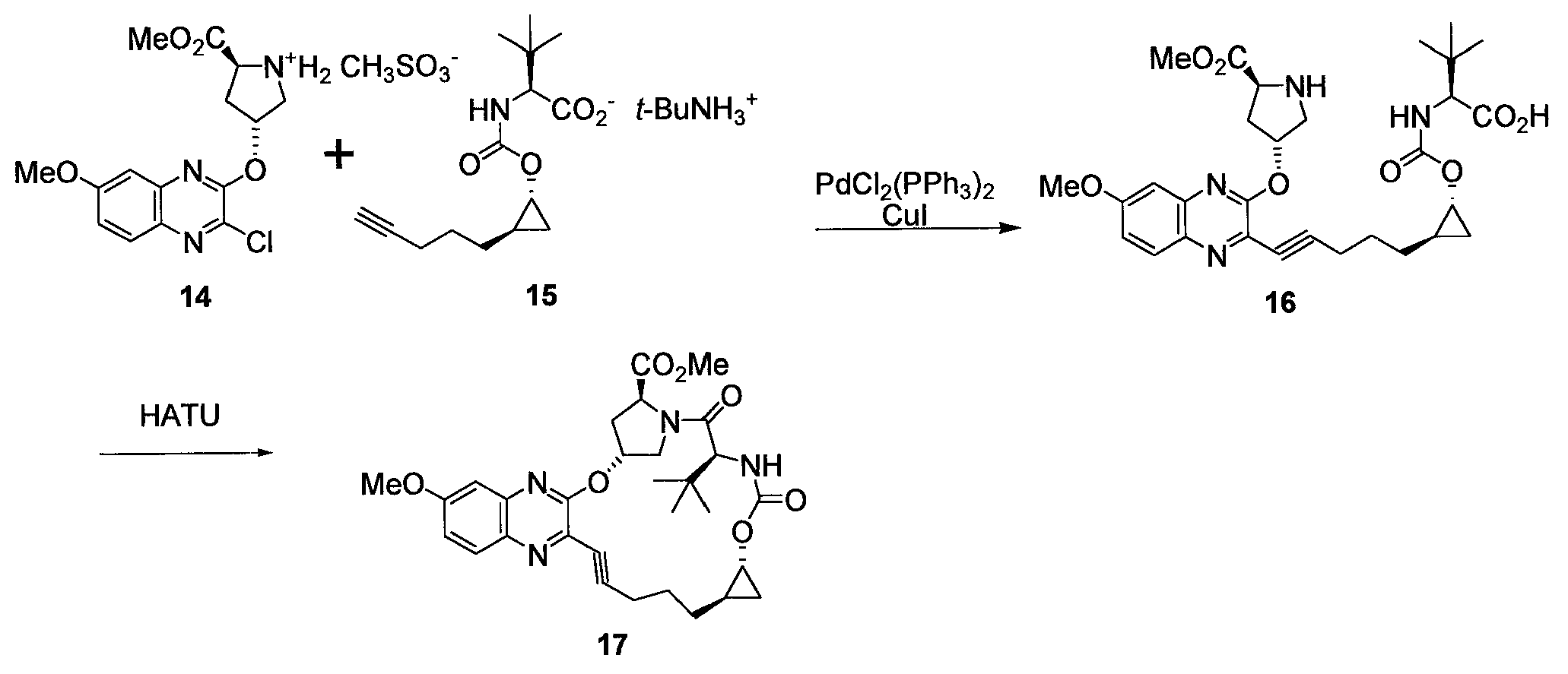
3구 플라스크에 아이오딘화구리(I) (0.219 g, 1.152 mmol), 클로로퀴녹살린 MsOH 염 14 (50 g, 115 mmol), 알킨산 TBA 염 15 (49.3 g, 121 mmol), 및 비스(트리페닐포스핀)팔라듐(II) 디클로라이드 (0.404 g, 0.573 mmol)를 첨가하였다. 플라스크를 N2로 진공 탈기하였다. MeOH (500 ml)를 첨가하고 반응 혼합물을 N2로 다시 진공 탈기하였다. 트리에틸아민 (32.1 ml, 230 mmol)을 첨가하였다. 반응 용액을 3-5 시간 동안 35℃에서 숙성시켰다. 그 다음 배치를 진공중에 ~100 mL의 부피로 농축하였다. THF (250 mL) 및 EtOAc (250 mL)를 첨가하였다. 반응 혼합물을 5℃ 미만으로 냉각하였다. 반응 용액이 pH ~2로 조정될 때까지 HCl 용액 (1 N, ~180 mL)을 5℃ 미만에서 서서히 첨가하였다. NaCl 수용액 (10%, 350 mL)을 첨가하였다. 분리된 수성상을 THF (250 mL) 및 EtOAc (250 mL)의 용액으로 역추출(back-extract)하였다. 합해진 유기상을 10% NaCl 수용액 (500 mL)으로 세척하였다. 용액의 KF가 500 ppm 미만이 될 때까지 유기상을 20℃ 미만에서 THF와 진공중에 공비 농축하였다. 그 다음, 반응 용매를 20℃ 미만에서 진공중에 DMAc (650 mL)로 교환하였다.
주위 온도에서 DMAc (650 mL) 중 HATU (55.1 g, 145 mmol)의 용액을 N2로 진공 탈기하였다. 그 다음 용액을 0℃로 냉각하고 DIPEA (58.5 mL, 335 mmol)를 0-5℃ 미만에서 적가하였다. 그 다음, DMAc 중 알킨 퀴녹살린산 16 (65 g 검정(assay), 112 mmol)의 상기 용액을 내부 온도를 0℃로 유지하면서 10시간에 걸쳐 적가하였다. 첨가 후, 배치를 추가의 2시간 동안 0℃에서 교반하였다. EtOAc (750 mL)를 5℃ 미만에서 첨가하였다. 10% NaCl 수용액 (400 mL), 물 (125 mL) 및 1 N HCl 용액 (100 mL)의 용액을 배치 온도를 5℃ 미만으로 유지하면서 서서히 첨가하였다. 그 다음 용액을 1 N HCl (~25 mL)을 사용하여 pH = 2로 조정하였다. 분리된 수성상을 EtOAc (500 mL)로 역추출하였다. 합해진 유기상을 10% NaCl 수용액 (500 mL)으로 세척하였다. 10% NaCl 수용액 (500 mL)을 합해진 유기상에 첨가한 후, 혼합 용액을 0-5℃로 냉각하였다. 1 N NaOH 수용액 (~25 mL)을 첨가하여 pH = ~7로 조정하였다. 분리된 유기상을 셀라이트를 통해 여과하고 최종 부피 300 mL에서 IPA로 용매 교환하였다. 아세트산 (5.0 mL)을 첨가한 다음, 배치를 30분 동안 환류로 가열하였다. 슬러리를 60℃로 냉각하고 물 (250 mL)을 1시간에 걸쳐 적가하였다. 첨가 후, 배치를 추가의 30분 동안 숙성시킨 후에 약 2시간 내에 주위 온도로 서서히 냉각하였다. 적어도 1시간 숙성시킨 후, 배치를 여과하였다. 습윤 케이크를 50% 수성 IPA (100 mL)로 대체 세척하였다. 주위 온도에서 흡입 건조시켜 56 g의 마크로시클릭 알킨 에스테르 17을 수득하였다.

IPC HPLC 조건: 아센티스 익스프레스(Ascentis Express) C18 칼럼, 100 x 4.6 mm, 2.7 마이크로미터; 칼럼 온도 40℃; 유량 1.8 mL/분; 및 파장 215 nm.

실시예
11:
마크로시클릭
에스테르 18의 제조

THF (100 mL) 중 알킨 마크로시클릭 에스테르 17 (10.0 g, 17.71 mmol) 및 5% Pd/C 50% 습윤 (3.5 g, 0.822 mmol)의 혼합물을 적어도 10시간 동안 40 psig의 수소하에 주위 온도에서 수소화시켰다. 반응 완료시, 배치를 셀라이트를 통해 여과하고 여과된 촉매를 THF (100 mL)로 세척하였다. 합해진 여액을 최종 부피 ~50 mL에서 진공중 IPA로 용매 교환하였다. 슬러리를 약 1시간 동안 환류로 가열하였다. 그 다음 배치를 50℃로 냉각하고 물 (30 mL)을 1시간에 걸쳐 적가하였다. 배치를 2시간에 걸쳐 0℃ 미만으로 서서히 냉각하고 추가의 1시간 동안 0℃에서 교반한 후에 여과하였다. 습윤 케이크를 물 (17.5 mL) 중 57% IPA의 냉 용액 (0-5℃)으로 세척하였다. 주위 온도에서 흡입 건조시켜 8.5 g의 목적 마크로시클릭 에스테르 18을 수득하였다.

IPC HPLC 조건: 아센티스 익스프레스 C18 칼럼, 100 x 4.6 mm, 2.7 마이크로미터; 칼럼 온도 40℃; 유량 1.8 mL/분; 및 파장 215 nm.

실시예
12:
마크로시클릭산
(19)의 제조

주위 온도에서 MeOH (720 mL) 중 마크로시클릭 에스테르 18 (90 g, 158.3 mmol)의 슬러리에 2 M NaOH (237.4 mL, 475 mmol)를 적가하였다. 반응 혼합물을 2-3 시간 동안 50℃에서 숙성시켰다. 반응 용액을 35-40℃로 냉각하고 50% 수성 MeOH (70 mL) 중 5 N HCl을 적가하였다. 배치를 유리 산 반수화물 19 (~100 mg)로 시딩하고 40℃에서 30분 내지 1시간 동안 숙성시켰다. 50% 수성 MeOH (30 mL) 중 추가의 5 N HCl을 40℃에서 2-4 시간에 걸쳐 적가하였다. 슬러리를 추가의 1시간 동안 숙성시킨 후에 주위 온도로 냉각하였다. 슬러리를 추가의 1시간 동안 숙성시킨 후에 여과하였다. 습윤 케이크를 물 중 65% MeOH (3x 270 mL, 대체 세척, 슬러리 세척 및 대체 세척)로 세척하였다. 주위 온도에서 흡입 건조 또는 60-80℃에서 건조 N2 스윕으로 진공 오븐 건조시켜 85.6 g의 마크로시클릭산 반수화물 19를 회백색 고체로서 수득하였다.

IPC HPLC 조건: 하이퍼실 골드 PFP 칼럼, 150 x 4.6 mm, 3.0 ㎛, 칼럼 온도 40℃; 유량 1.8 mL/분; 및 파장 215 nm

실시예
13: 화합물 A의 제조
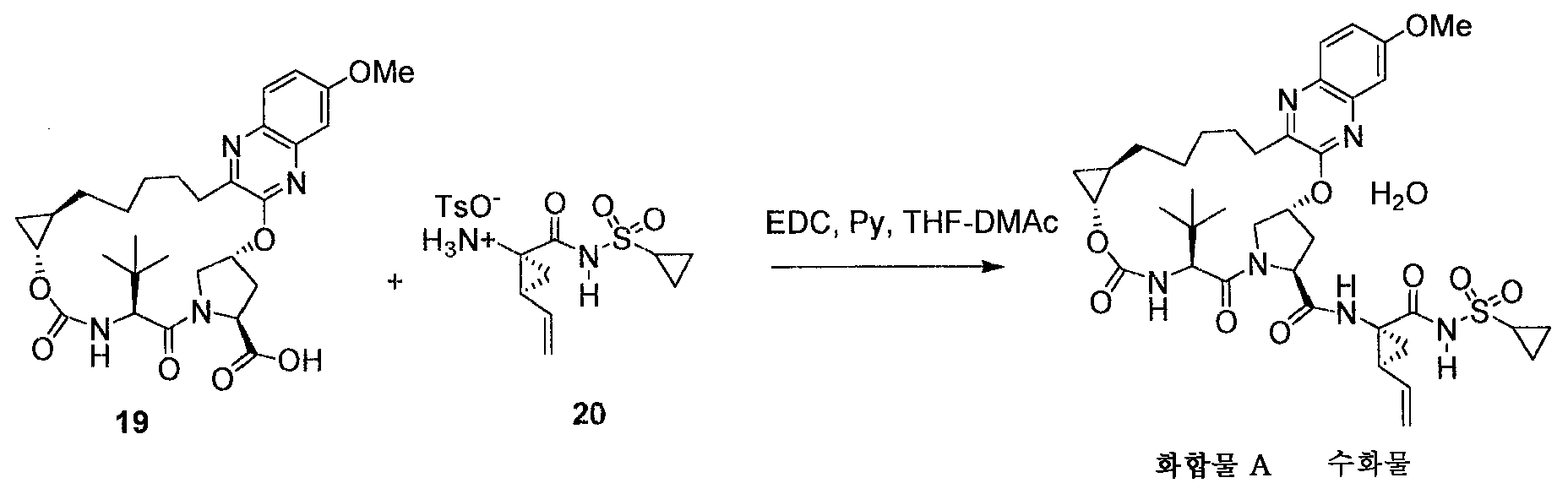
마크로시클릭산 반수화물 19 (10.16 g, 18.03 mmol)를 THF (50 - 90 mL)에 용해시켰다. 용액을 최종 부피 100 mL에서 공비 건조시켰다. 술폰아미드 pTSA 염 20 (7.98 g, 1.983 mmol) 이어서 DMAc (15 mL)를 주위 온도에서 첨가하였다. 배치를 0-10℃로 냉각하고 피리딘 (10 mL)을 적가하였다. 그 다음, EDC HCl (4.49 g, 23.44 mmol)을 0-10℃에서 나누어 또는 한 번에 첨가하였다. 반응 혼합물을 1시간 동안 0-10℃에서 숙성시킨 다음, 2-4시간 동안 15-20℃로 가온하였다. MeOAc (100 mL) 이어서 물 (50 mL) 중 5% NaCl 중 15wt% 시트르산을, 외부 냉각과 함께 내부 온도를 < 25℃로 유지하면서 첨가하였다. 분리된 유기상을 물 (50 mL) 중 5% NaCl 중 15wt% 시트르산 이어서 5% NaCl (50 mL)로 세척하였다. 유기상을 최종 부피 ~80 mL에서 아세톤으로 용매 교환하였다. 물 (10 mL)을 35-40℃에서 적가하였다. 배치를 화합물 A 일수화물 형태 III (~10 mg)으로 시딩하고 35-40℃에서 0.5-1 시간 동안 숙성시켰다. 추가의 물 (22 mL)을 35-40℃에서 2-4 시간에 걸쳐 적가하였다. 슬러리를 2-4시간 동안 20℃에서 숙성시킨 후에 여과하였다. 습윤 케이크를 물 중 60% 아세톤 (40 mL x 2)으로 대체 세척하였다. 주위 온도에서 흡입 건조시켜 화합물 A 일수화물 형태 III을 백색 고체로서 수득하였다.

HPLC 조건: 아센티스 익스프레스 칼럼, 10 cm x 4.6 mm x 2.7 ㎛; 칼럼 온도 40℃; 유량 1.8 mL/분; 및 파장 215 nm

실시예
14: 화합물 A 제조를 위한 대안적인 절차

오버헤드 교반기가 장착된 50 L 플라스크에 마크로시클릭산 19 (1.06 kg 조, 1.00 당량), 아민-pTSA (862 g 조, 1.12 당량) 및 MeCN 7.42 L를 19℃에서 첨가하였다. 슬러리를 수조에서 냉각하고, 피리딘 (2.12 L, 13.8 당량)을 첨가하고, 15분 숙성시킨 다음, EDC (586 g, 1.60 당량)를 한 번에 첨가하고, 1.5시간 숙성시키고 그 동안 이는 투명한 균질 용액으로 변했다.
용액을 수조에서 냉각한 다음, 2 N HCl (1.7 L)로 켄칭하고, 시딩하고 (9.2 g), 15분 동안 숙성시키고, 수성 HCl의 나머지를 2.5시간에 걸쳐 첨가하였다. 황색 슬러리가 형성되었다. 슬러리를 실온에서 밤새 숙성시키고, 여과하고, MeCN/물 (1:1 v/v) 8 L로 세척하여, 화합물 A (수화물 II)를 수득하였다.
화합물 A를 실온에서 아세톤 4 L에 용해시키고, 여과하고, 오버헤드 교반기를 갖춘 12 L RBF에 옮겼고, 추가의 아세톤 1 L로 세정하고, 50℃로 가열하고, 물 0.9 L를 첨가하고, 10 g 시딩하고, 15분 숙성시킨 다음, 2.5시간에 걸쳐 물 0.8 L를 첨가하고, 2.5시간에 걸쳐 추가의 물 3.3v를 첨가하고, 가열을 중지하고, 실온으로 냉각하고, 실온에서 밤새 숙성시키고, 여과하고, 물/아세톤 (1:1 v/v) 4 L로 세척하고, 진공하에 공기 건조시켰다. 화합물 A 수화물 III, 670 g을 회백색 고체로서 수득하였다.
실시예
15:
마크로시클릭
에스테르 (18)의 대안적인 제조

3구 플라스크에 아이오딘화구리(I) (0.020 g, 0.104 mmol), 클로로퀴녹살린 MsOH 염 14 (4.5 g, 10.5 mmol), 알킨산 TBA 염 15 (4.4 g, 10.9 mmol), 및 비스(트리페닐포스핀)팔라듐(II) 디클로라이드 (0.036 g, 0.052 mmol)를 첨가하였다. 플라스크를 N2로 진공 탈기하였다. MeOH (45 ml)를 첨가하고 반응 혼합물을 N2로 다시 진공 탈기하였다. 트리에틸아민 (2.89 ml, 20.7 mmol)을 첨가하였다. 반응 용액을 3-5 시간 동안 35℃에서 숙성시켰다. 그 다음 배치를 진공중에 ~9 mL의 부피로 농축하였다. THF (23 mL) 및 EtOAc (23 mL)를 첨가하였다. 반응 혼합물을 5℃ 미만으로 냉각하였다. 반응 용액이 pH ~2로 조정될 때까지 HCl 용액 (1 N, ~16 mL)을 5℃ 미만으로 서서히 첨가하였다. NaCl 수용액 (10%, 32 mL)을 첨가하였다. 분리된 수성상을 THF (23 mL) 및 EtOAc (23 mL)의 용액으로 역추출하였다. 합해진 유기상을 10% NaCl 수용액 (45 mL)으로 세척하였다. 용매를 20℃ 미만에서 진공중에 MeOH (75 mL)로 교환하였다.
반응 혼합물에 DARCO KB-B (1.0 g)를 첨가하고, 생성된 현탁액을 1시간 동안 20℃에서 교반한 후 셀라이트를 통해 여과하였다. 습윤 케이크를 MeOH (25 mL)로 세척하였다. 합해진 여액을 적어도 5시간 동안 주위 온도에서 수소 1 기압 하에 펄먼 촉매(Pearlman's catalyst) (1.2 g, 탄소상 20% Pd(OH)2, 50% 습윤)의 존재하에 수소화시켰다. 반응 완료후, 현탁액을 셀라이트를 통해 여과하고 산 21 함유 여액을 DMAc (65 mL)로 용매 교환하였다.
주위 온도에서 DMAc (65 mL) 중 HATU (5.05 g, 13.3 mmol)의 용액을 N2로 진공 탈기하였다. 용액을 0℃로 냉각하고 DIPEA (5.4 mL, 30.9 mmol)를 0-5℃에서 적가하였다. 그 다음, DMAc 중 산 21 (5.98 g 검정, 10.2 mmol)의 상기 용액을 내부 온도를 0℃로 유지하면서 10시간에 걸쳐 적가하였다. 첨가 후, 배치를 추가의 2시간 동안 0℃에서 교반하여 마크로시클릭 에스테르 18을 수득하였다. 마크로시클릭 에스테르 18의 후처리 절차 및 단리는 실시예 11에 기재된 바와 동일하였다.
실시예
16: 화합물 A 수화물
II
수화물 II를, 화합물 A 유리 염기를 75% RH (상대 습도) 이상의 물 활성을 갖는 아세토니트릴:물 비를 함유하는 용액에 첨가하여 아세토니트릴 용매화물을 형성시킨 다음 승온에서 건조시킴으로써 제조하였다.
수화물 II를 X-선 분말 회절, 열중량 분석, 및 시차 주사 열량측정 곡선 TG를 비롯한, 각양각색의 방법에 의해 특성화하였다. X-선 분말 회절 패턴을 PW3040/60 콘솔을 갖춘 필립스 어낼리티컬 엑스퍼트(Philips Analytical X'Pert) PRO X-선 회절 시스템 상에서 발생시켰다. PW3373/00 세라믹 Cu LEF X-선 관 K-알파 방사선을 공급원으로서 사용하였다.
DSC 데이터를 TA 인스트루먼츠(Instruments) DSC 2910 또는 등가물을 사용하여 획득하였다. 2 내지 6 mg 샘플을 팬(pan)에 측량 도입하고 뚜껑을 닫았다. 그 다음 이러한 팬을 크림핑(crimping)하고 열량계 셀(cell)에 샘플 위치에 위치시켰다. 공(empty) 팬을 참조 위치에 위치시켰다. 열량계 셀을 폐쇄하고 질소류를 셀에 통과시켰다. 샘플을 10℃/분의 가열 속도로 대략 250℃의 온도로 가열하는 가열 프로그램을 설정하였다. 가열 프로그램을 시작하였다. 시행이 완료된 때, 데이터를 시스템 소프트웨어에 함유된 DSC 분석 프로그램을 사용하여 분석하였다. 열 사상(thermal event)이 관찰되는 온도 범위에 걸쳐 그 상하에 있는 기준선 온도점 사이에 열 사상을 통합하였다. 보고된 데이터는 개시 온도, 피크 온도 및 엔탈피였다.
TG 데이터를 퍼킨 엘머 모델 TGA 7을 사용하여 획득하였다. 질소류하에 및 대략 250℃의 최대 온도로 10℃/분의 가열 속도를 사용하여 실험을 수행하였다. 자동으로 중량 산정법에 의해 측정(taring the balance) 후, 5 내지 20 mg의 샘플을 백금 팬에 첨가하고, 퍼니스(furnace)를 올리고, 가열 프로그램을 시작하였다. 중량/온도 데이터를 기기에 의해 자동으로 수집하였다. 기기 소프트웨어 내에 델타(Delta) Y 함수를 선택하고 온도 (이들 온도 간에 중량 손실이 산출됨)를 선택함으로써 결과의 분석을 수행하였다. 중량 손실을 분해/증발의 개시까지 보고하였다.
도 1은 화합물 A의 결정질 수화물 II의 특징적 X-선 회절 패턴을 도시한다. 수화물 II는 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 1>

실시예
17: 수화물
III
수화물 III을 X-선 분말 회절, 열중량 분석, 시차 주사 열량측정 곡선 TG, 및 고체 상태 탄소-13 핵 자기 공명 (NMR) 스펙트럼을 비롯한, 각양각색의 방법에 의해 특성화하였다. X-선 분말 회절 패턴을 PW3040/60 콘솔을 갖춘 필립스 어낼리티컬 엑스퍼트 PRO X-선 회절 시스템 상에서 발생시켰다. PW3373/00 세라믹 Cu LEF X-선 관 K-알파 방사선을 공급원으로서 사용하였다.
DSC 데이터를 TA 인스트루먼츠 DSC 2910 또는 등가물을 사용하여 획득하였다. 2 내지 6 mg 샘플을 팬에 측량 도입하고 뚜껑을 닫았다. 그 다음 이러한 팬을 크림핑하고 열량계 셀에 샘플 위치에 위치시켰다. 공 팬을 참조 위치에 위치시켰다. 열량계 셀을 폐쇄하고 질소류를 셀에 통과시켰다. 샘플을 10℃/분의 가열 속도로 대략 250℃의 온도로 가열하는 가열 프로그램을 설정하였다. 가열 프로그램을 시작하였다. 시행이 완료된 때, 데이터를 시스템 소프트웨어에 함유된 DSC 분석 프로그램을 사용하여 분석하였다. 열 사상이 관찰되는 온도 범위에 걸쳐 그 상하에 있는 기준선 온도점 사이에 열 사상을 통합하였다. 보고된 데이터는 개시 온도, 피크 온도 및 엔탈피였다.
TG 데이터를 퍼킨 엘머 모델 TGA 7을 사용하여 획득하였다. 질소류하에 및 대략 250℃의 최대 온도로 10℃/분의 가열 속도를 사용하여 실험을 수행하였다. 자동으로 중량 산정법에 의해 측정 후, 5 내지 20 mg의 샘플을 백금 팬에 첨가하고, 퍼니스를 올리고, 가열 프로그램을 시작하였다. 중량/온도 데이터를 기기에 의해 자동으로 수집하였다. 기기 소프트웨어 내에 델타 Y 함수를 선택하고 온도 (이들 온도 간에 중량 손실이 산출됨)를 선택함으로써 결과의 분석을 수행하였다. 중량 손실을 분해/증발의 개시까지 보고하였다.
탄소-13 스펙트럼을 브루커(Bruker) 4 mm H/F/X BB 이중 공명 CPMAS 프로브를 사용하여 브루커 AV400 NMR 분광계 상에서 기록하였다. 스펙트럼을 80 kHz에서 양성자/탄소-13 가변-증폭 간섭 편파(variable-amplitude cross-polarization: VACP)를 이용하여 접촉 시간 3 ms로 수집하였다. 데이터 획득에 사용된 다른 실험 파라미터는 100 kHz의 양성자 90-도 펄스, 100 kHz에서의 스피날(SPINAL)64 데커플링(decoupling), 2 s의 펄스 지연, 및 26824 스캔에 관한 신호가산평균(signal averaging)이었다. 매직-앵글 스피닝(magic-angle spinning: MAS) 속도를 13 kHz로 설정하였다. 10 Hz의 로렌치안(Lorentzian) 선 폭확대를 스펙트럼에 적용한 후에 푸리에 변환(Fourier Transformation)시켰다. 화학 이동을 제2 참조 물질로서 글리신의 카르보닐 탄소 (176.70 ppm)를 사용하여 TMS 스케일 상에서 보고하였다.
도 2는 결정질 수화물 III의 특징적 X-선 회절 패턴이다. 수화물 III은 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 2>

도 3은 결정질 수화물 III의 전형적인 열중량 분석 곡선을 도시한다.
도 4는 결정질 수화물 III의 시차 주사 열량측정 곡선을 도시한다.
도 5는 화합물 A 수화물 III에 관한 고체 상태 C-13 CPMAS NMR을 도시한다. 수화물 III에 관한 특징적 피크는 5.14, 6.31, 12.49, 18.35, 26.81, 28.03, 30.33, 31.27, 34.95, 35.99, 38.68, 42.01, 54.93, 56.39, 60.14, 74.20, 107.02, 120.11, 121.60, 129.73, 134.35, 135.95, 142.89, 148.47, 155.37, 157.32, 160.90, 168.32, 172.17, 및 175.53 ppm에서 관찰되었다.
실시예
18: 추가의 수화물
수화물 I, IV, V, 및 VI을 X-선 분말 회절 및 탄소-13 NMR에 의해 특성화하였다. X-선 분말 회절 패턴을 PW3040/60 콘솔을 갖춘 필립스 어낼리티컬 엑스퍼트 PRO X-선 회절 시스템 상에서 발생시켰다. PW3373/00 세라믹 Cu LEF X-선 관 K-알파 방사선을 공급원으로서 사용하였다. 탄소-13 스펙트럼을 브루커 4 mm H/F/X BB 이중 공명 CPMAS 프로브를 사용하여 브루커 AV400 NMR 분광계 상에서 기록하였다. 스펙트럼을 80 kHz에서 양성자/탄소-13 가변-증폭 간섭 편파(VACP)를 이용하여 100 kHz의 양성자 90-도 펄스, 100 kHz에서의 스피날64 데커플링으로 수집하였다. 30 Hz의 로렌치안 선 폭확대를 스펙트럼에 적용한 후에 푸리에 변환시켰다. 화학 이동을 제2 참조 물질로서 글리신의 카르보닐 탄소 (176.70 ppm)를 사용하여 TMS 스케일 상에서 보고하였다. 다른 실험 파라미터는 하기 각각의 섹션에 개요가 서술되어 있다.
수화물 I
수화물 I을, 유리 형태를 순(neat) 메탄올에 첨가하고, 실온에서 ~24 h 동안 평형화시키고, 공기 건조시킴으로써 제조하였다.
도 8은 본 발명의 화합물 I의 결정질 수화물 형태 I의 특징적 X-선 회절 패턴이다. 수화물 형태 I은 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 3>
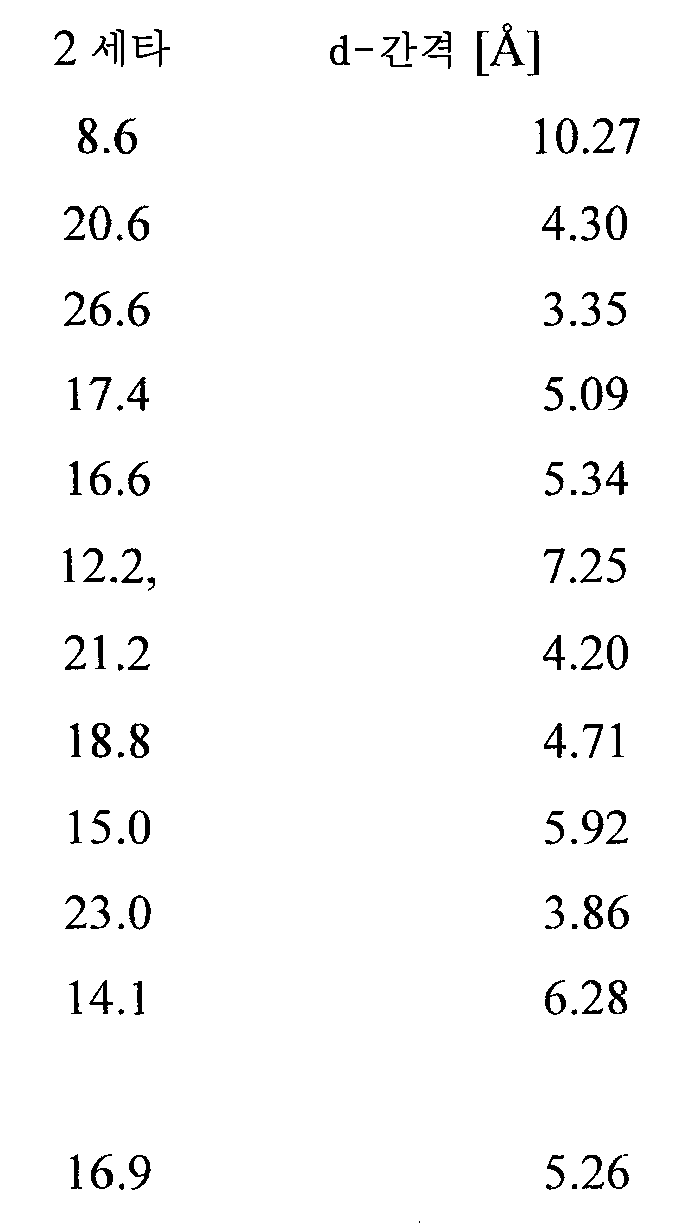
수화물 I을 그의 고체 상태 탄소-13 핵 자기 공명 (NMR) 스펙트럼을 기반으로 특성화하였다. 데이터 획득에 사용된 다른 실험 파라미터는 2 s의 펄스 지연, 및 20480 스캔에 관한 신호가산평균이었다. 매직-앵글 스피닝 (MAS) 속도를 13 kHz로 설정하고 온도를 270 K로 설정하였다. (도 9.) 수화물 I에 관한 특징적 피크는 4.22, 7.23, 11.45, 17.79, 24.04, 26.95, 28.29, 31.15, 32.47, 32.47, 33.46, 34.03, 35.74, 42.32, 53.50, 56.05, 56.96, 77.49, 108.95, 119.65, 122.55, 131.05, 133.13, 135.38, 142.28, 150.78, 156.03, 157.99, 161.36, 171.40, 173.42, 174.30 ppm에서 관찰되었다.
수화물
IV
수화물 IV를, 칼륨 염을 1 당량의 HCl과 함께 1:1 아세톤:물의 용액에 첨가하고 실온에서 건조시킴으로써 제조하였다.
도 10은 결정질 수화물 IV의 특징적 X-선 회절 패턴이다. 수화물 IV는 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 4>

수화물 IV를 그의 고체 상태 탄소-13 핵 자기 공명 (NMR) 스펙트럼을 기반으로 특성화하였다. 데이터 획득에 사용된 다른 실험 파라미터는 2 s의 펄스 지연, 및 1245 스캔에 관한 신호가산평균이었다. 매직-앵글 스피닝 (MAS) 속도를 13 kHz로 설정하고 온도를 275 K로 설정하였다. (도 11.) 수화물 IV에 관한 특징적 피크는 3.90, 5.30, 6.99, 10.49, 13.13, 17.81, 24.73, 27.52, 28.14, 29.42, 31.02, 32.80, 36.08, 39.22, 42.45, 53.62, 55.93, 59.14, 60.76, 74.77, 109.22, 111.19, 11.38, 120.24, 122.50, 133.96, 139.74, 147.2, 148.90, 154.65, 158.25, 159.53, 160.12, 170.14, 171.05, 172.08, 173.47, 및 174.46 ppm에서 관찰되었다.
수화물 V
수화물 V를, 수화물 IV를 81% 초과의 상대 습도에 노출시킴으로써 제조하였다.
도 12는 결정질 수화물 V의 특징적 X-선 회절 패턴이다. 수화물 V는 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 5>

수화물 V를 그의 고체 상태 탄소-13 핵 자기 공명 (NMR) 스펙트럼을 기반으로 특성화하였다. (도 13.) 스펙트럼을 80 kHz에서 양성자/탄소-13 가변-증폭 간섭 편파(VACP)를 이용하여 접촉 시간 2 ms로 수집하였다. 데이터 획득에 사용된 다른 실험 파라미터는 3 s의 펄스 지연, 및 3425 스캔에 관한 신호가산평균이었다. 매직-앵글 스피닝(MAS) 속도를 13 kHz로 설정하였다. 수화물 V에 관한 특징적 피크는 7.86, 8.92, 13.10, 18.31, 23.72, 27.44, 28.47, 30.77, 35.79, 36.25, 37.15, 37.15, 42.95, 53.13, 55.67, 57.31, 60.47, 62.06, 75.09, 110.59, 112.24, 118.32, 132.18, 134.05, 135.83, 139.88, 148.30, 155.19, 157.97, 159.41, 170.31 및 175.20 ppm에서 관찰되었다.
수화물
VI
수화물 VI을, 유리 염기를 메탄올/아세톤의 50/50 혼합물에 첨가하고 실온에서 건조시킴으로써 제조하였다.
도 14는 결정질 수화물 VI의 특징적 X-선 회절 패턴이다. 수화물 VI은 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 6>

수화물 VI을 그의 고체 상태 탄소-13 핵 자기 공명 (NMR) 스펙트럼을 기반으로 특성화하였다. (도 15.) 스펙트럼을 80 kHz에서 양성자/탄소-13 가변-증폭 간섭 편파(VACP)를 이용하여 접촉 시간 3 ms로 수집하였다. 데이터 획득에 사용된 다른 실험 파라미터는 2 s의 펄스 지연, 및 3425 스캔에 관한 신호가산평균이었다.
수화물 VI에 관한 특징적 피크는 4.87, 6.24, 11.70, 12.85, 18.36, 26.55, 28.31m 31.51, 34.98, 38.47, 42.09, 54.27, 56.12, 60.10, 73.49, 73.97, 105.91, 108.04, 118.39, 119.75, 121.33, 129.96, 133.87, 136.13, 142.26, 142.97, 146.85, 148.36, 154.97, 157.32, 160.71, 168.23, 172.21 및 175.34 ppm에서 관찰되었다.
수화물 I, II, 및 III의 물에서의 상대 안정성은 12시간 후 주로 수화물 II 및 5일 후 주로 수화물 III을 나타냈다. 수화물 II, III 및 IV의 안정성은 25℃ 및 50℃에서 아세톤, 물 및 이들의 혼합물 중 주로 수화물 III을 나타냈다.
수화물 형태의 상대 안정성은, 실온에서 0.072 내지 1의 범위에 이르는 물 활성에서 아세톤:물 중 경쟁적 슬러리 전환(turnover) 실험에 의해 측정하였다. 모든 경우에서, 수화물 형태 III은 이들 실험으로부터 생성된 고체였고, 이는 조사된 용매 중의 수화물 형태 중에서 가장 안정함을 나타내는 것이다.
실시예
19: 화합물 A K
+
및
Na
+
염 제조
화합물 A K+ 및 Na+ 염을 하기와 같이 제조하였다:
화합물 K
Na
-염의 제조


30 L 2:1 MeCN:EtOH를 함유하는, 기계 교반기, 열전쌍 및 질소 유입구가 장착된 50 L 재킷(jacketed) 원통형 용기에 3.3 kg 화합물 A 유리 산을 첨가하였다. 그 다음 이를 인-라인(in-line) 필터를 통해 기계 교반기, 열전쌍 및 질소 유입구가 장착된 72 L RBF에 옮겼다. 이 용액에 EtOH 중 KOEt를 1시간에 걸쳐 첨가하였다. EtOH 중 20% KOEt의 첨가 후 용액을 형태 II로 시딩하였다. 생성된 슬러리를 주위 온도에서 3시간 동안 교반한 다음 여과하였다.
그 다음 현탁액을 여과하고, 2 x 10 L의 MeCN으로 세척하고 N2/진공 스윕하에 건조시켜 3.40 kg의 화합물 A 칼륨 염 (98.3 wt%, 98.1 LCAP, 99% 수율)을 수득하였다.
HPLC 조건: 조르박스 이클립스 플러스 C18 50 x 4.6 mm, 1.8 ㎛, A=0.1% 인산, C=아세토니트릴: 10% 내지 95% C, 5분; 95% C, 6분; 10% C, 6.1분; 2분 후, 1.5 mL/분, 230 nm, 25℃.
화합물 A 5.41 분
화합물 A
Na
-염의 제조


질소 하에 100 mL 2구 환저 플라스크에서, 화합물 A 유리 산을 40 mL EtOH에 용해시키고 0.2 mL 물을 50℃에서 첨가하였다. 여기에 1.30 mL 2 M 수산화나트륨 용액을 30분에 걸쳐 서서히 첨가하고, 고체가 형성될 때까지 50℃에서 유지한 다음 실온으로 서서히 냉각하고 3시간 동안 숙성시켰다. 그 다음 슬러리를 빙조에서 냉각한 다음 현탁액을 여과시키고, 2 x 10 mL의 MeCN으로 세척하고 N2/진공 스윕하에 건조시켜 2.01 g의 화합물 A 나트륨 염 (98.3 LCAP, 99% 수율)을 수득하였다.
HPLC 조건: 조르박스 이클립스 플러스 C18 50 x 4.6 mm, 1.8 ㎛, A=0.1% 인산, C=아세토니트릴: 10% 내지 95% C, 5분; 95% C, 6분; 10% C, 6.1분; 2분 후, 1.5 mL/분, 230 nm, 25℃.
화합물 A 5.41 분
실시예
20: 화합물 A K
+
및
Na
+
염 특성화
X-선 분말 회절 패턴을 PW3040/60 콘솔을 갖춘 필립스 어낼리티컬 엑스퍼트 PRO X-선 회절 시스템 상에서 발생시켰다. PW3373/00 세라믹 Cu LEF X-선 관 K-알파 방사선을 공급원으로서 사용하였다.
DSC 데이터를 TA 인스트루먼츠 DSC 2910 또는 등가물을 사용하여 획득하였다. 2 내지 6 mg 샘플을 팬에 측량 도입하고 뚜껑을 닫았다. 그 다음 이러한 팬을 크림핑하고 열량계 셀에 샘플 위치에 위치시켰다. 공 팬을 참조 위치에 위치시켰다. 열량계 셀을 폐쇄하고 질소류를 셀에 통과시켰다. 샘플을 10℃/분의 가열 속도로 300℃ 초과의 온도로 가열하는 가열 프로그램을 설정하였다. 가열 프로그램을 시작하였다. 시행이 완료된 때, 데이터를 시스템 소프트웨어에 함유된 DSC 분석 프로그램을 사용하여 분석하였다. 열 사상이 관찰되는 온도 범위에 걸쳐 그 상하에 있는 기준선 온도점 사이에 열 사상을 통합하였다. 보고된 데이터는 개시 온도, 피크 온도 및 엔탈피였다.
TG 데이터를 퍼킨 엘머 모델 TGA 7을 사용하여 획득하였다. 질소류하에 및 300℃ 초과의 최대 온도로 10℃/분의 가열 속도를 사용하여 실험을 수행하였다. 자동으로 중량 산정법에 의해 측정 후, 5 내지 20 mg의 샘플을 백금 팬에 첨가하고, 퍼니스를 올리고, 가열 프로그램을 시작하였다. 중량/온도 데이터를 기기에 의해 자동으로 수집하였다. 기기 소프트웨어 내에 델타 Y 함수를 선택하고 온도 (이들 온도 간에 중량 손실이 산출됨)를 선택함으로써 결과의 분석을 수행하였다. 중량 손실을 분해/증발의 개시까지 보고하였다.
화합물 A
Na
-염
도 6은 화합물 A의 결정질 Na-염의 특징적 X-선 회절 패턴을 도시한다. Na-염은 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 7>

화합물 A K-염
도 11은 화합물 A의 결정질 K-염의 특징적 X-선 회절 패턴을 도시한다. K-염은 하기 d-간격에 상응하는 특징적 반사를 나타냈다:
<표 8>

본 출원을 통해 기재된 참조 문헌 중 어떤 것도 청구된 발명의 선행 기술로서 인정되는 것은 아니다.
Claims (6)
- a) (i) 약 20.5, 5.0, 및 18.2 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴; 또는 (ii) 약 5.14, 6.31, 12.49, 18.35, 26.81, 28.03, 30.33, 31.27, 34.95, 35.99, 38.68, 42.01, 54.93, 56.39, 60.14, 74.20, 107.02, 120.11, 121.60, 129.73, 134.35, 135.95, 142.89, 148.47, 155.37, 157.32, 160.90, 168.32, 172.17, 및 175.53 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 하는 수화물 III;
b) 약 11.7, 16.6, 및 11.2 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 하는 수화물 II;
c) 약 18.4, 9.1, 및 9.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 하는 결정질 Na 염;
d) 약 18.2, 8.9, 및 20.3 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴을 특징으로 하는 결정질 K 염;
e) (i) 약 8.6, 20.6, 및 26.6 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴; 또는 (ii) 약 4.22, 7.23, 11.45, 17.79, 24.04, 26.95, 28.29, 31.15, 32.47, 32.47, 33.46, 34.03, 35.74, 42.32, 53.50, 56.05, 56.96, 77.49, 108.95, 119.65, 122.55, 131.05, 133.13, 135.38, 142.28, 150.78, 156.03, 157.99, 161.36, 171.40, 173.42, 174.30 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 하는 수화물 I;
f) (i) 약 14.7, 11.5, 및 7.1 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴; 또는 (ii) 약 3.90, 5.30, 6.99, 10.49, 13.13, 17.81, 24.73, 27.52, 28.14, 29.42, 31.02, 32.80, 36.08, 39.22, 42.45, 53.62, 55.93, 59.14, 60.76, 74.77, 109.22, 111.19, 11.38, 120.24, 122.50, 133.96, 139.74, 147.2, 148.90, 154.65, 158.25, 159.53, 160.12, 170.14, 171.05, 172.08, 173.47, 및 174.46 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 하는 수화물 IV;
g) (i) 약 9.1, 18.3, 및 19.8 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴; 또는 (ii) 약 7.86, 8.92, 13.10, 18.31, 23.72, 27.44, 28.47, 30.77, 35.79, 36.25, 37.15, 37.15, 42.95, 53.13, 55.67, 57.31, 60.47, 62.06, 75.09, 110.59, 112.24, 118.32, 132.18, 134.05, 135.83, 139.88, 148.30, 155.19, 157.97, 159.41, 170.31 및 175.20에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 하는 수화물 V;
h) (i) 약 20.5, 12.8, 및 19.4 도의 2θ 값을 포함하는, 구리 Kα 방사선을 사용하여 수득된 X-선 분말 회절 패턴; 또는 (ii) 약 4.87, 6.24, 11.70, 12.85, 18.36, 26.55, 28.31m 31.51, 34.98, 38.47, 42.09, 54.27, 56.12, 60.10, 73.49, 73.97, 105.91, 108.04, 118.39, 119.75, 121.33, 129.96, 133.87, 136.13, 142.26, 142.97, 146.85, 148.36, 154.97, 157.32, 160.71, 168.23, 172.21 및 175.34 ppm에서의 피크를 포함하는 고체 상태 탄소-13 CPMAS NMR을 특징으로 하는 수화물 VI
으로 이루어진 군으로부터 선택된 형태를 갖는 화합물 A. - 치료 유효량의 제1항의 화합물 및 제약상 허용되는 담체를 포함하는 제약 조성물.
- 치료 유효량의 제1항의 화합물을 환자에게 투여하는 단계를 포함하는, 환자에서 HCV를 치료하는 방법.
- 유효량의 제1항의 화합물을 포함하는, 환자에서 HCV를 치료하는데 사용되는 의약의 제조.
- 제1항에 있어서, HCV의 치료에 사용하기 위한 화합물.
- 제1항의 화합물인 상기 화합물 A가 80:20 v/v 아세톤 대 물 내지 0:100 v/v 아세톤 대 물의 비를 갖는 아세톤/물을 사용하여 화합물 A를 결정화시킨 다음 건조시키는 것을 포함하는 공정에 의해 제조된 수화물 III인, 제1항의 화합물의 제조 방법.
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161525462P | 2011-08-19 | 2011-08-19 | |
| US61/525,462 | 2011-08-19 | ||
| US201161533439P | 2011-09-12 | 2011-09-12 | |
| US61/533,439 | 2011-09-12 | ||
| US201161533915P | 2011-09-13 | 2011-09-13 | |
| US61/533,915 | 2011-09-13 | ||
| US201161539540P | 2011-09-27 | 2011-09-27 | |
| US61/539,540 | 2011-09-27 | ||
| PCT/US2012/051168 WO2013028465A1 (en) | 2011-08-19 | 2012-08-16 | Crystal forms of a hcv protease inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20140059236A true KR20140059236A (ko) | 2014-05-15 |
Family
ID=47746773
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020147006858A Withdrawn KR20140053330A (ko) | 2011-08-19 | 2012-08-16 | 마크로락탐의 제조를 위한 방법 및 중간체 |
| KR1020147006888A Withdrawn KR20140059236A (ko) | 2011-08-19 | 2012-08-16 | Hcv 프로테아제 억제제의 결정 형태 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020147006858A Withdrawn KR20140053330A (ko) | 2011-08-19 | 2012-08-16 | 마크로락탐의 제조를 위한 방법 및 중간체 |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US9238604B2 (ko) |
| EP (3) | EP2744336B1 (ko) |
| JP (2) | JP2014521750A (ko) |
| KR (2) | KR20140053330A (ko) |
| CN (2) | CN103874414A (ko) |
| AU (2) | AU2012299218A1 (ko) |
| BR (2) | BR112014003802A2 (ko) |
| CA (2) | CA2844386A1 (ko) |
| MX (2) | MX2014001944A (ko) |
| RU (2) | RU2014110399A (ko) |
| WO (3) | WO2013028470A1 (ko) |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| BR112014003802A2 (pt) | 2011-08-19 | 2017-06-13 | Merck Sharp & Dohme | composto, composto composição farmacêutica, uso de um composto, método para fabricar um composto, e, preparação de um medicamento |
| UA119315C2 (uk) | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | Інгібітори вірусу гепатиту с |
| EA025560B1 (ru) | 2012-10-19 | 2017-01-30 | Бристол-Майерс Сквибб Компани | Ингибиторы вируса гепатита с |
| EP2914613B1 (en) | 2012-11-02 | 2017-11-22 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2764866A1 (en) | 2013-02-07 | 2014-08-13 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| JP6342922B2 (ja) | 2013-03-07 | 2018-06-13 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | C型肝炎ウイルス阻害剤 |
| WO2014145095A1 (en) | 2013-03-15 | 2014-09-18 | Gilead Sciences, Inc. | Macrocyclic and bicyclic inhibitors of hepatitis c virus |
| SG11201602693QA (en) | 2013-10-18 | 2016-05-30 | Merck Sharp & Dohme | Methods and intermediates for preparing macrolactams |
| WO2015095437A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Methods and intermediates for the preparation of macrolactams |
| ES2933350T3 (es) * | 2014-01-24 | 2023-02-06 | Turning Point Therapeutics Inc | Macrociclos de diarilo como moduladores de proteína quinasas |
| MX373056B (es) | 2014-06-06 | 2020-05-20 | Abbvie Inc | Formas cristalinas de glecaprevir. |
| AU2016287568B2 (en) | 2015-07-02 | 2020-08-20 | Turning Point Therapeutics, Inc. | Chiral diaryl macrocycles as modulators of protein kinases |
| RU2765181C2 (ru) | 2015-07-06 | 2022-01-26 | Тёрнинг Поинт Терапьютикс, Инк. | Полиморфная форма диарильного макроцикла |
| PT3733187T (pt) | 2015-07-21 | 2024-11-08 | Turning Point Therapeutics Inc | Macrociclo de diarilo quiral e utilização do mesmo no tratamento do cancro |
| CN109641874A (zh) | 2016-05-10 | 2019-04-16 | C4医药公司 | 用于靶蛋白降解的c3-碳连接的戊二酰亚胺降解决定子体 |
| EP3454856B1 (en) | 2016-05-10 | 2024-09-11 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| ES2990061T3 (es) | 2016-05-10 | 2024-11-28 | C4 Therapeutics Inc | Degronímeros espirocíclicos para la degradación de proteínas diana |
| CN109790143A (zh) | 2016-05-10 | 2019-05-21 | C4医药公司 | 用于靶蛋白降解的胺连接的c3-戊二酰亚胺降解决定子体 |
| US10689400B2 (en) | 2016-07-28 | 2020-06-23 | Turning Point Therapeutics, Inc. | Macrocycle kinase inhibitors |
| TWI808958B (zh) | 2017-01-25 | 2023-07-21 | 美商特普醫葯公司 | 涉及二芳基巨環化合物之組合療法 |
| CN110769822A (zh) | 2017-06-20 | 2020-02-07 | C4医药公司 | 用于蛋白降解的n/o-连接的降解决定子和降解决定子体 |
| CN111182903A (zh) | 2017-07-28 | 2020-05-19 | 特普医药公司 | 巨环化合物及其用途 |
| LT3728271T (lt) | 2017-12-19 | 2022-12-12 | Turning Point Therapeutics, Inc. | Makrocikliniai junginiai, skirti ligų gydymui |
| CN111057045A (zh) * | 2019-12-18 | 2020-04-24 | 安徽红杉生物医药科技有限公司 | Hcv ns3/4a蛋白酶抑制剂中间体及其合成方法、应用 |
| CN112174982A (zh) * | 2020-09-10 | 2021-01-05 | 上海希迈医药科技有限公司 | 一种洛普替尼晶型及其制备方法 |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4871868A (en) | 1987-03-11 | 1989-10-03 | Takeda Chemical Industries, Ltd. | Production of substituted acetylenic compounds |
| GB9207987D0 (en) * | 1992-04-10 | 1992-05-27 | Smithkline Beecham Plc | Novel container and closure |
| US5716960A (en) * | 1995-01-13 | 1998-02-10 | U.S. Bioscience Inc. And Individuals | Crystalline trimetrexate salts and the process for making the same |
| NO317155B1 (no) | 1997-02-04 | 2004-08-30 | Ono Pharmaceutical Co | <omega>-cykloalkyl-prostagladin-E<N>2</N>-derivater |
| ZA98879B (en) * | 1997-02-04 | 1998-08-03 | Ono Pharmaceutical Co | Omega-cycloalkyl-prostaglandin e2 derivatives |
| AU2006215041B2 (en) * | 2005-02-18 | 2012-05-31 | Mitsubishi Tanabe Pharma Corporation | Salt of proline derivative, solvate thereof, and production method thereof |
| US7834145B2 (en) | 2005-03-22 | 2010-11-16 | Merck Sharp & Dohme Corp. | HCV protease substrates |
| AU2006242475B2 (en) | 2005-05-02 | 2011-07-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| UA90909C2 (en) * | 2005-07-20 | 2010-06-10 | Мерк Шарп Энд Домэ Корп. | Hcv ns3 protease inhibitors |
| TWI387603B (zh) | 2005-07-20 | 2013-03-01 | Merck Sharp & Dohme | Hcv ns3蛋白酶抑制劑 |
| BRPI0614205A2 (pt) | 2005-08-01 | 2016-11-22 | Merck & Co Inc | composto, composição farmacêutica, e, uso de composto |
| CN101394829A (zh) | 2006-03-07 | 2009-03-25 | 宝洁公司 | 用于氧化性染色角蛋白纤维的组合物和使用此类组合物的方法 |
| GB0609492D0 (en) | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| JP2010507656A (ja) | 2006-10-24 | 2010-03-11 | メルク エンド カムパニー インコーポレーテッド | Hcvns3プロテアーゼ阻害剤 |
| CN101583372A (zh) | 2006-10-24 | 2009-11-18 | 默克公司 | Hcv ns3蛋白酶抑制剂 |
| WO2008057209A1 (en) | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| AU2007318164B2 (en) | 2006-10-27 | 2013-02-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| EP2160392A2 (en) | 2007-05-03 | 2010-03-10 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| AU2008277377B2 (en) | 2007-07-19 | 2013-08-01 | Msd Italia S.R.L. | Macrocyclic compounds as antiviral agents |
| AU2009217551B2 (en) | 2008-02-25 | 2014-07-31 | Msd Italia S.R.L. | Therapeutic compounds |
| WO2009131196A1 (ja) * | 2008-04-24 | 2009-10-29 | 武田薬品工業株式会社 | 置換ピロリジン誘導体およびその用途 |
| EP2274288A2 (en) | 2008-04-24 | 2011-01-19 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| AU2009241445A1 (en) | 2008-04-28 | 2009-11-05 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| PT2540350E (pt) * | 2008-07-22 | 2014-08-27 | Merck Sharp & Dohme | Combinações de um composto de quinoxalina macrocílico o qual é um inibidor da protease ns3 do hcv com outros agentes do hcv |
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| EP2470189A4 (en) * | 2009-08-27 | 2013-01-23 | Merck Sharp & Dohme | METHOD FOR PRODUCING PROTEASE INHIBITORS OF HEPATITIS C VIRUS |
| BR112014003802A2 (pt) | 2011-08-19 | 2017-06-13 | Merck Sharp & Dohme | composto, composto composição farmacêutica, uso de um composto, método para fabricar um composto, e, preparação de um medicamento |
-
2012
- 2012-08-16 BR BR112014003802A patent/BR112014003802A2/pt not_active IP Right Cessation
- 2012-08-16 KR KR1020147006858A patent/KR20140053330A/ko not_active Withdrawn
- 2012-08-16 WO PCT/US2012/051177 patent/WO2013028470A1/en not_active Ceased
- 2012-08-16 CN CN201280050361.6A patent/CN103874414A/zh active Pending
- 2012-08-16 JP JP2014526217A patent/JP2014521750A/ja active Pending
- 2012-08-16 US US14/239,393 patent/US9238604B2/en active Active
- 2012-08-16 MX MX2014001944A patent/MX2014001944A/es unknown
- 2012-08-16 MX MX2014001945A patent/MX2014001945A/es unknown
- 2012-08-16 WO PCT/US2012/051168 patent/WO2013028465A1/en not_active Ceased
- 2012-08-16 WO PCT/US2012/051182 patent/WO2013028471A1/en not_active Ceased
- 2012-08-16 RU RU2014110399/04A patent/RU2014110399A/ru not_active Application Discontinuation
- 2012-08-16 EP EP12826404.1A patent/EP2744336B1/en not_active Not-in-force
- 2012-08-16 AU AU2012299218A patent/AU2012299218A1/en not_active Abandoned
- 2012-08-16 EP EP12825540.3A patent/EP2744507A4/en not_active Withdrawn
- 2012-08-16 EP EP12825726.8A patent/EP2744331A4/en not_active Withdrawn
- 2012-08-16 CN CN201280050382.8A patent/CN103889439A/zh active Pending
- 2012-08-16 BR BR112014003798A patent/BR112014003798A2/pt not_active IP Right Cessation
- 2012-08-16 RU RU2014110400/04A patent/RU2014110400A/ru not_active Application Discontinuation
- 2012-08-16 CA CA2844386A patent/CA2844386A1/en not_active Abandoned
- 2012-08-16 CA CA2844388A patent/CA2844388A1/en not_active Abandoned
- 2012-08-16 KR KR1020147006888A patent/KR20140059236A/ko not_active Withdrawn
- 2012-08-16 JP JP2014526215A patent/JP2014524442A/ja active Pending
- 2012-08-16 AU AU2012299223A patent/AU2012299223A1/en not_active Abandoned
- 2012-08-16 US US14/239,391 patent/US9073825B2/en active Active
- 2012-08-16 US US14/239,389 patent/US9242917B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2744336A4 (en) | 2014-12-31 |
| US20140243519A1 (en) | 2014-08-28 |
| BR112014003802A2 (pt) | 2017-06-13 |
| EP2744507A1 (en) | 2014-06-25 |
| WO2013028465A1 (en) | 2013-02-28 |
| US9242917B2 (en) | 2016-01-26 |
| EP2744331A1 (en) | 2014-06-25 |
| RU2014110400A (ru) | 2015-09-27 |
| CA2844388A1 (en) | 2013-02-28 |
| AU2012299223A1 (en) | 2014-02-27 |
| JP2014521750A (ja) | 2014-08-28 |
| KR20140053330A (ko) | 2014-05-07 |
| BR112014003798A2 (pt) | 2017-03-01 |
| US9073825B2 (en) | 2015-07-07 |
| MX2014001945A (es) | 2014-03-27 |
| EP2744507A4 (en) | 2015-01-28 |
| US20140206605A1 (en) | 2014-07-24 |
| US20140200343A1 (en) | 2014-07-17 |
| MX2014001944A (es) | 2014-03-27 |
| AU2012299218A1 (en) | 2014-02-20 |
| CN103889439A (zh) | 2014-06-25 |
| EP2744336A1 (en) | 2014-06-25 |
| US9238604B2 (en) | 2016-01-19 |
| CN103874414A (zh) | 2014-06-18 |
| WO2013028470A1 (en) | 2013-02-28 |
| CA2844386A1 (en) | 2013-02-28 |
| EP2744336B1 (en) | 2017-07-05 |
| EP2744331A4 (en) | 2015-01-21 |
| WO2013028471A1 (en) | 2013-02-28 |
| RU2014110399A (ru) | 2015-09-27 |
| JP2014524442A (ja) | 2014-09-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20140059236A (ko) | Hcv 프로테아제 억제제의 결정 형태 | |
| JP6524355B2 (ja) | Tlr7アゴニストとしての7−(チアゾール−5−イル)ピロロピリミジン化合物 | |
| KR100652535B1 (ko) | Impdh 효소의 억제제 | |
| ES2470568T3 (es) | Inhibidores macroc�clicos del virus de la hepatitis C | |
| CN102414199B (zh) | 二芳基醚 | |
| JP6034802B2 (ja) | 大環状ラクタムの調製のための方法および中間体 | |
| CA2899763C (en) | Nucleoside phosphoramidate compound and use thereof | |
| TWI542585B (zh) | C型肝炎病毒抑制劑 | |
| EP3816163A1 (en) | Cell necrosis inhibitor, preparation method therefor and use thereof | |
| WO2016127916A1 (zh) | 作为长效dpp-iv抑制剂的取代的氨基六元饱和杂脂环类 | |
| CN114555607A (zh) | 一类靶向蛋白质水解通路的功能分子及其制备和应用 | |
| WO2022171072A1 (zh) | 一种二氢嘧啶类化合物、其制备方法及其应用 | |
| WO2025016441A1 (zh) | 新型GalNAc的靶向递送片段及其制备和应用 | |
| WO2015095430A1 (en) | Methods and intermediates for the preparation of macrolactams | |
| EP3498712B1 (en) | Spirocyclic indolone polyethylene glycol carbonate compound, composition, preparation method and use thereof | |
| RU2793918C2 (ru) | Ингибитор некроза клеток, способ его получения и применения | |
| CN106967141A (zh) | 核苷氨基磷酸酯化合物及其医药组合物和用途 | |
| HK1136824B (en) | Macrocyclic inhibitors of hepatitis c virus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20140314 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |