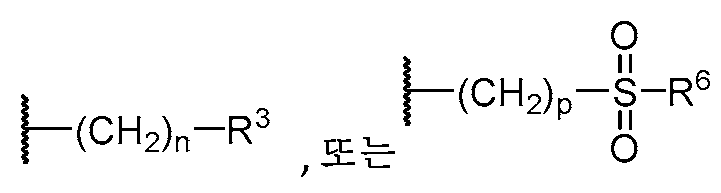개시내용의 상세한 설명
화합물
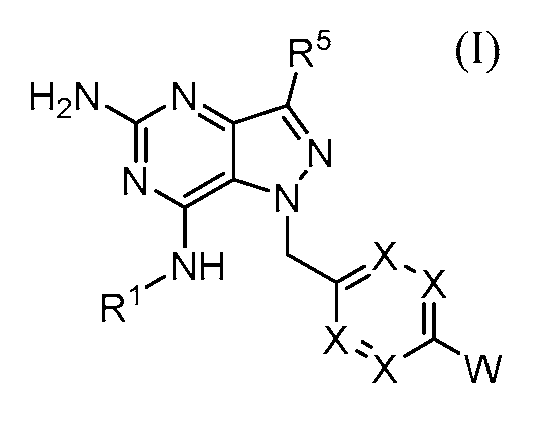
한 측면에서, 본 개시내용의 화합물은 R1 및 R3이 화학식 (I)에 대해 정의된 바와 같은 화학식 (Ia)에 따른 것이다:
한 측면에서, 본 개시내용은
R3은 OH,
인 화학식 (Ia)에 따른 구조를 갖는 화합물을 제공한다.
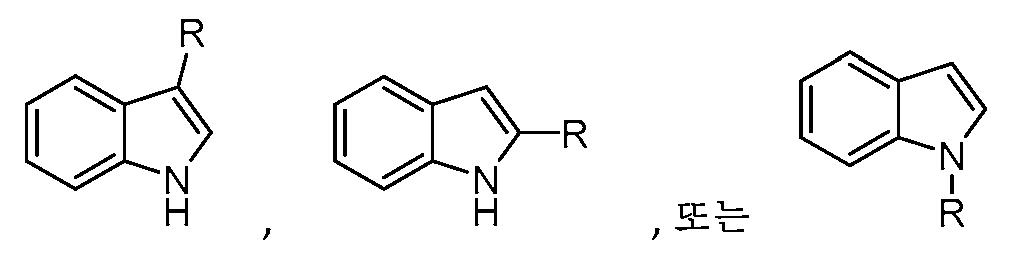
기 R1의 예는
를 포함한다.
R2는 바람직하게는 OMe, O(시클로프로필), 또는 OCHF2이고, 보다 바람직하게는 OMe이다.
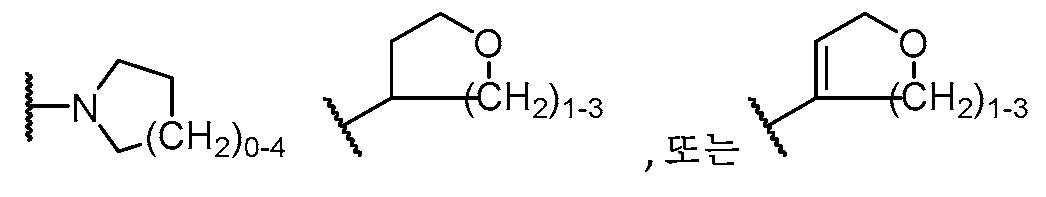
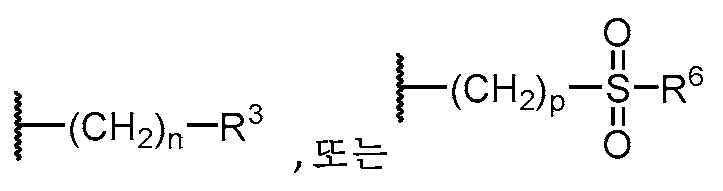
기 R3의 예는 OH,
를 포함한다.
한 측면에서, R5는 H이다.
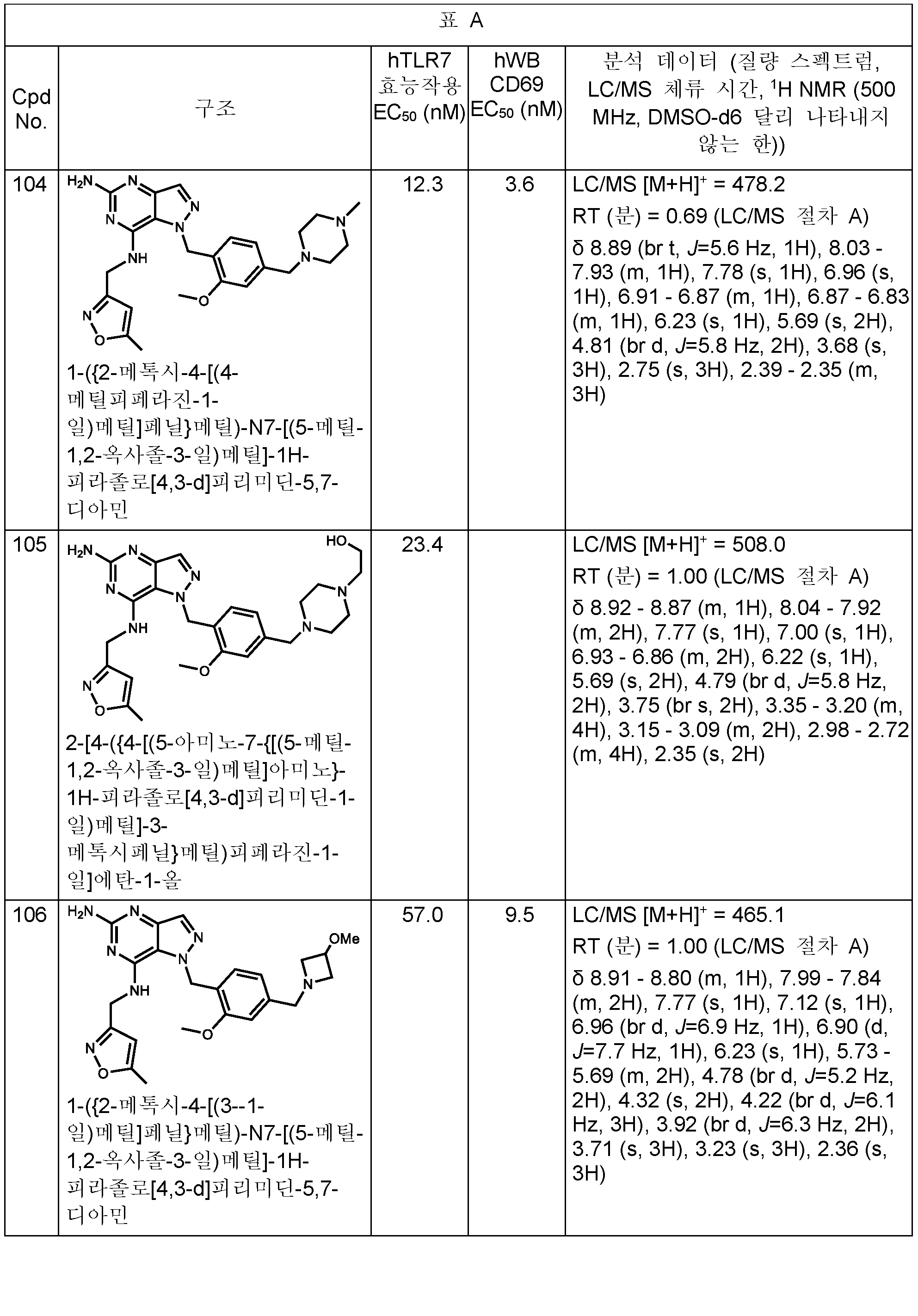
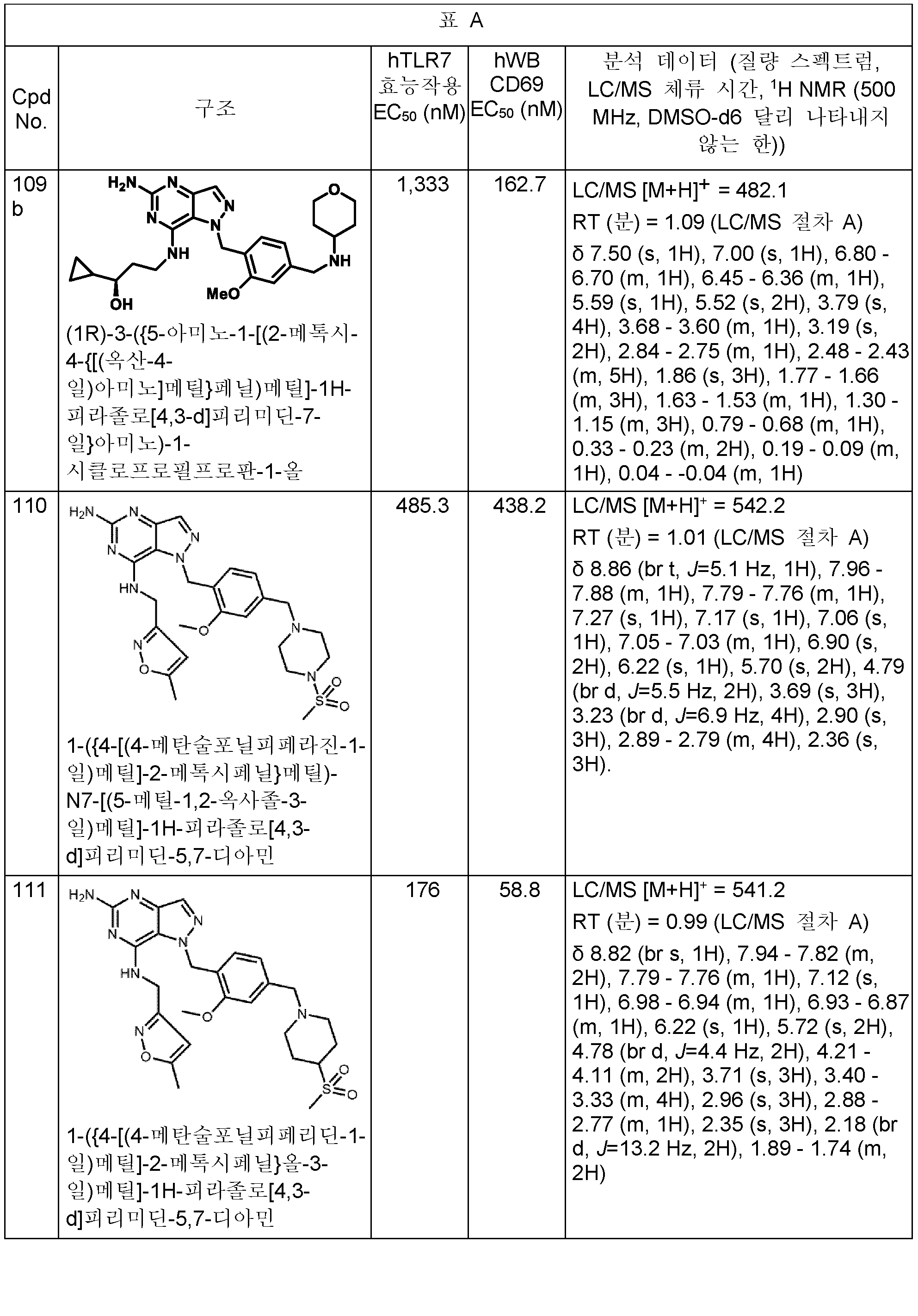
본원에 개시된 화합물의 구체적 예는 하기 표 A에 제시된다. 표는 또한 하기 생물학적 활성과 관련된 데이터를 제공한다: 하기 제공된 절차에 따라 결정된, 인간 TLR7 리포터 검정 및/또는 인간 전혈에서의 CD69 유전자의 유도. 가장 우측 칼럼은 분석 데이터 (질량 스펙트럼, HPLC 체류 시간, 및 NMR)를 포함한다. 한 실시양태에서, 본 개시내용의 화합물은 (a) 1,000 nM 미만의 인간 TLR7 (hTLR7) 효능제 (리포터) 검정 EC50 값 및 (b) 1,000 nM 미만의 인간 전혈 (hWB) CD69 유도 EC50 값을 갖는다. (검정이 다수회 수행되는 경우에, 보고된 값은 평균임).
제약 조성물 및 투여
또 다른 측면에서, 제약상 허용되는 담체 또는 부형제와 함께 제제화되는, 본원에 개시된 화합물 또는 그의 접합체를 포함하는 제약 조성물이 제공된다. 이는 1종 이상의 추가의 제약 활성 성분, 예컨대 생물학적 또는 소분자 약물을 임의로 함유할 수 있다. 제약 조성물은 또 다른 치료제, 특히 항암제와의 조합 요법으로 투여될 수 있다.
제약 조성물은 1종 이상의 부형제를 포함할 수 있다. 사용될 수 있는 부형제는 담체, 표면 활성제, 증점제 또는 유화제, 고체 결합제, 분산 또는 현탁 보조제, 가용화제, 착색제, 향미제, 코팅, 붕해제, 윤활제, 감미제, 보존제, 등장화제, 및 그의 조합을 포함한다. 적합한 부형제의 선택 및 용도는 문헌 [Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003)]에 교시되어 있다.
바람직하게는, 제약 조성물은 (예를 들어, 주사 또는 주입에 의한) 정맥내, 근육내, 피하, 비경구, 척수 또는 표피 투여에 적합하다. 투여 경로에 따라, 활성 화합물은 이를 불활성화시킬 수 있는 산 및 다른 천연 조건의 작용으로부터 이를 보호하기 위한 물질로 코팅될 수 있다. 어구 "비경구 투여"는 통상적으로 주사에 의한, 경장 및 국소 투여 이외의 다른 투여 방식을 의미하며, 비제한적으로 정맥내, 근육내, 동맥내, 척수강내, 피막내, 안와내, 심장내, 피내, 복강내, 경기관, 피하, 각피하, 관절내, 피막하, 지주막하, 척수내, 경막외 및 흉골내 주사 및 주입을 포함한다. 대안적으로, 제약 조성물은 비-비경구 경로, 예컨대 국소, 표피 또는 점막 투여 경로를 통해, 예를 들어 비강내로, 경구로, 질로, 직장으로, 설하로 또는 국소로 투여될 수 있다.
제약 조성물은 멸균 수용액 또는 분산액 형태일 수 있다. 그들은 또한 마이크로에멀젼, 리포솜, 또는 높은 약물 농도를 달성하기에 적합한 다른 정렬된 구조로 제제화될 수 있다. 조성물은 또한 투여 전에 물 중 재구성을 위해, 동결건조물 형태로 제공될 수 있다.
단일 투여 형태를 제조하기 위해 담체 물질과 조합될 수 있는 활성 성분의 양은 치료될 대상체 및 특정한 투여 방식에 따라 달라질 것이며, 일반적으로 치료 효과를 생성시키는 조성물의 양일 것이다. 일반적으로 100%를 기준으로, 이 양은 제약상 허용되는 담체와 조합된 활성 성분의 약 0.01% 내지 약 99%, 바람직하게는 약 0.1% 내지 약 70%, 가장 바람직하게는 활성 성분의 약 1% 내지 약 30% 범위일 것이다.
투여 요법은 치료 반응을 제공하도록 조정된다. 예를 들어, 단일 볼루스가 투여될 수 있거나, 여러 분할 용량이 시간 경과에 따라 투여될 수 있거나, 또는 용량이 상황의 위급성에 의해 지시된 바와 같이 비례적으로 감소 또는 증가될 수 있다. 투여의 용이성 및 투여량의 균일성을 위해 비경구 조성물을 투여 단위 형태로 제제화하는 것이 특히 유리하다. "투여 단위 형태"는 치료될 대상체에 대한 단일 투여량으로서 적합화된 물리적 이산 단위를 지칭하며; 각각의 단위는 목적하는 치료 반응을 생성시키도록 계산된 미리 결정된 양의 활성 화합물을, 필요한 제약 담체와 함께 함유한다.
투여량은 숙주 체중의 약 0.0001 내지 100 mg/kg, 보다 통상적으로 0.01 내지 5 mg/kg 범위이다. 예를 들어 투여량은 0.3 mg/kg 체중, 1 mg/kg 체중, 3 mg/kg 체중, 5 mg/kg 체중 또는 10 mg/kg 체중 또는 1-10 mg/kg, 또는 대안적으로 0.1 내지 5 mg/kg 범위 내일 수 있다. 예시적인 치료 요법은 1주에 1회, 2주마다 1회, 3주마다 1회, 4주마다 1회, 1개월에 1회, 3개월마다 1회, 또는 3 내지 6개월마다 1회 투여이다. 바람직한 투여 요법은 하기 투여 스케줄 중 하나를 사용하여, 정맥내 투여를 통한 1 mg/kg 체중 또는 3 mg/kg 체중을 포함한다: (i) 6회 투여량에 대해 4주마다, 이어서 3개월마다; (ii) 3주마다; (iii) 3 mg/kg 체중 1회에 이은 3주마다 1 mg/kg 체중. 일부 방법에서, 투여량은 약 1-1000 μg/mL, 및 일부 방법에서는 약 25-300 μg/mL의 혈장 항체 농도가 달성되도록 조정된다.
"치료 유효량"의 본 발명의 화합물은 바람직하게는 질환 증상의 중증도에서의 감소, 질환 무증상 기간의 빈도 및 지속기간에서의 증가, 또는 질환 고통으로 인한 손상 또는 장애의 예방을 생성시킨다. 예를 들어, 종양-보유 대상체의 치료를 위해, "치료 유효량"은 바람직하게는 종양 성장을 비치료 대상체에 비해 적어도 약 20%, 보다 바람직하게는 적어도 약 40%, 보다 더 바람직하게는 적어도 약 60%, 더욱 더 바람직하게는 적어도 약 80% 억제한다. 치료 유효량의 치료 화합물은 전형적으로 인간이지만 또 다른 포유동물일 수 있는 대상체에서 종양 크기를 감소시키거나 또는 증상을 달리 호전시킬 수 있다. 2종 이상의 치료제가 조합 치료로 투여되는 경우에, "치료 유효량"은 개별적으로 각 작용제의 효능이 아닌 조합의 전체로서의 효능을 지칭한다.
제약 조성물은 임플란트, 경피 패치, 및 마이크로캡슐화 전달 시스템을 포함한 제어 또는 지속 방출 제제일 수 있다. 생분해성, 생체적합성 중합체, 예컨대 에틸렌 비닐 아세테이트, 폴리무수물, 폴리글리콜산, 콜라겐, 폴리오르토에스테르, 및 폴리락트산이 사용될 수 있다. 예를 들어, 문헌 [Sustained and Controlled Release Drug Delivery Systems, J.R. Robinson, ed., Marcel Dekker, Inc., New York, 1978]을 참조한다.
치료 조성물은 의료 장치 예컨대 (1) 무바늘 피하 주사 장치; (2) 마이크로-주입 펌프; (3) 경피 장치; (4) 주입 장치; 및 (5) 삼투 장치를 통해 투여될 수 있다.
특정 실시양태에서, 제약 조성물은 생체내에서 적절한 분포가 보장되도록 제제화될 수 있다. 예를 들어, 본 발명의 치료 화합물이 혈액-뇌 장벽을 가로지르는 것을 보장하기 위해, 이들은 리포솜 중에 제제화될 수 있으며, 이는 특이적 세포 또는 기관에 대한 선택적 수송을 증진하기 위한 표적화 모이어티를 추가적으로 포함할 수 있다.
산업상 적용성 및 용도
본원에 개시된 TLR7 효능제 화합물은 TLR7의 활성화에 의해 개선될 수 있는 질환 또는 상태의 치료에 사용될 수 있다.
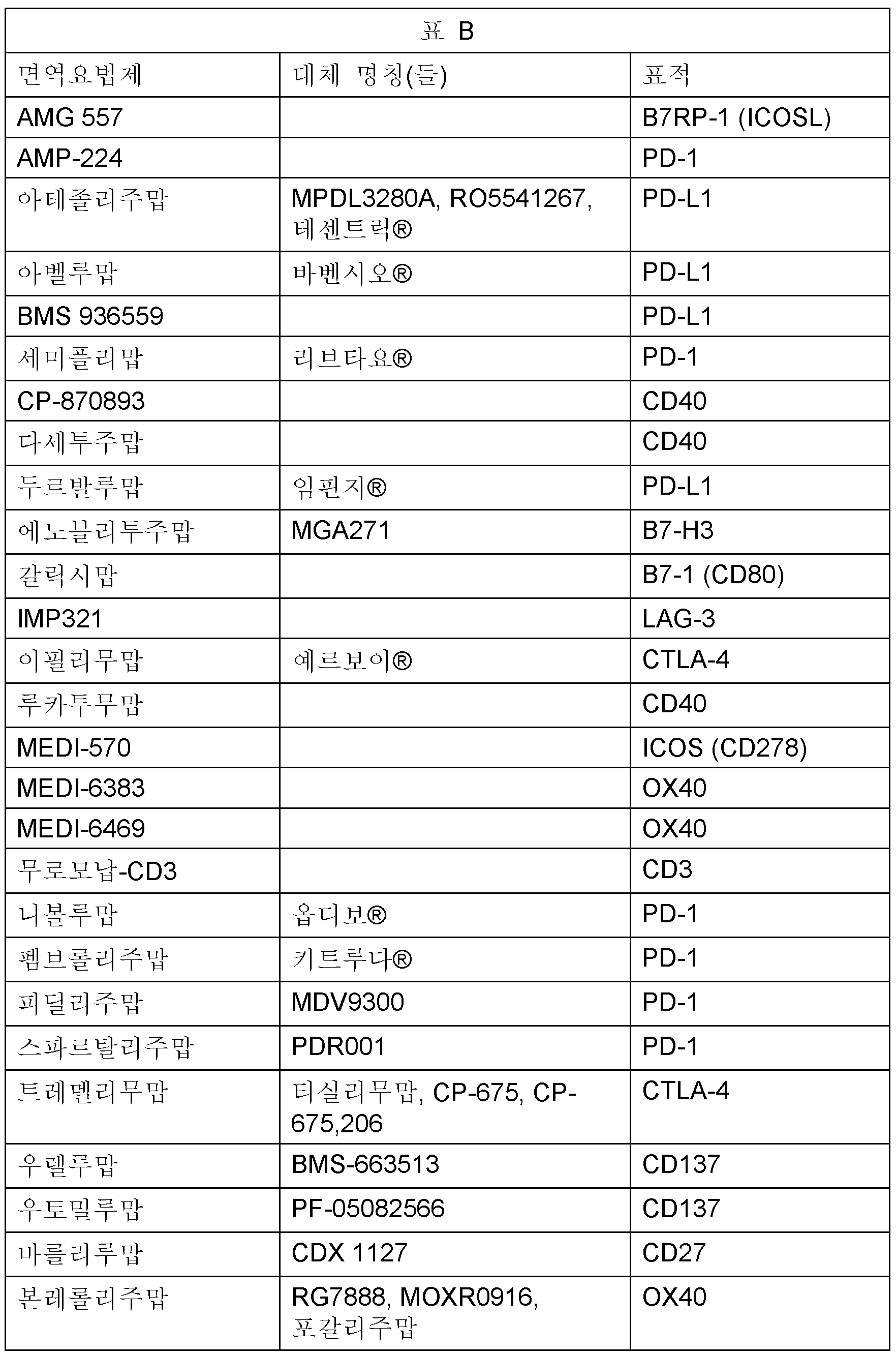
한 실시양태에서, TLR7 효능제는 면역-종양학 작용제로도 알려져 있는 항암 면역요법제와 조합으로 사용된다. 항암 면역요법제는 신체의 면역계를 자극하여, 특히 T 세포의 활성화를 통해 암 세포를 공격하고 파괴함으로써 작용한다. 면역계는 수많은 체크포인트 (조절) 분자를 가져, 면역계가 적당한 표적 세포를 공격하는 것과 면역계가 건강한 정상 세포를 공격하는 것을 방지하는 것 사이의 균형을 유지하도록 돕는다. 일부는 그의 결속이 T 세포 활성화를 촉진하고, 면역 반응을 증진시키는 것을 의미하는 자극제 (상향-조절자)이다. 다른 것은 그의 결속이 T 세포 활성화를 억제하고, 면역 반응을 감소시키는 것을 의미하는 억제제 (하향-조절자 또는 브레이크)이다. 효능작용 면역요법제의 자극성 체크포인트 분자로의 결합은 후자의 활성화 및 암 세포에 대한 증진된 면역 반응으로 이어질 수 있다. 반대로, 길항작용 면역요법제의 억제 체크포인트 분자로의 결합은 후자에 의한 면역계의 하향-조절을 방지하고, 암 세포에 대한 격렬한 반응을 유지하는 것을 보조할 수 있다. 자극성 체크포인트 분자의 예는 B7-1, B7-2, CD28, 4-1BB (CD137), 4-1BBL, ICOS, CD40, ICOS-L, OX40, OX40L, GITR, GITRL, CD70, CD27, CD40, DR3 및 CD28H이다. 억제 체크포인트 분자의 예는 CTLA-4, PD-1, PD-L1, PD-L2, LAG-3, TIM-3, 갈렉틴 9, CEACAM-1, BTLA, CD69, 갈렉틴-1, CD113, GPR56, VISTA, 2B4, CD48, GARP, PD1H, LAIR1, TIM-1, CD96 및 TIM-4이다.
어떠한 항암 면역요법제의 작용 방식이든지, 그의 유효성은 면역계의 일반적 상향조절 예컨대 TLR7의 활성화에 의해 증가될 수 있다. 따라서, 한 실시양태에서, 본 명세서는 암을 앓는 환자에게 항암 면역요법제와 본원에 개시된 TLR7 효능제의 치료상 유효한 조합을 투여하는 것을 포함하는, 이러한 암을 치료하는 방법을 제공한다. 투여 시기는 동시, 순차적, 또는 교대일 수 있다. 투여 방식은 전신 또는 국부일 수 있다. TLR7 효능제는 접합체를 통해, 표적화 방식으로 전달될 수 있다.
상기 기재된 바와 같이 조합 치료로 치료될 수 있는 암의 비제한적 예는 급성 골수성 백혈병, 부신피질 암종, 카포시 육종, 림프종, 항문암, 충수암, 기형양/횡문근양 종양, 기저 세포 암종, 담관암, 방광암, 골암, 뇌암, 유방암, 기관지 종양, 카르시노이드 종양, 심장 종양, 자궁경부암, 척삭종, 만성 림프구성 백혈병, 만성 골수증식성 신생물, 결장암, 결장직장암, 두개인두종, 담관암, 자궁내막암, 상의세포종, 식도암, 감각신경모세포종, 유잉 육종, 안암, 난관암, 담낭암, 위장 카르시노이드 종양, 위장 기질 종양, 배세포 종양, 모발상 세포 백혈병, 두경부암, 심장암, 간암, 하인두암, 췌장암, 신장암, 후두암, 만성 골수 백혈병, 구순암 및 구강암, 폐암, 흑색종, 메르켈 세포 암종, 중피종, 구내암, 구강암, 골육종, 난소암, 음경암, 인두암, 전립선암, 직장암, 타액선암, 피부암, 소장암, 연부 조직 육종, 고환암, 인후암, 갑상선암, 요도암, 자궁암, 질암, 및 외음부암을 포함한다.
본원에 기재된 바와 같이 조합 요법에 사용될 수 있는 항암 면역요법제는 하기를 포함한다: AMG 557, AMP-224, 아테졸리주맙, 아벨루맙, BMS 936559, 세미플리맙, CP-870893, 다세투주맙, 두르발루맙, 에노블리투주맙, 갈릭시맙, IMP321, 이필리무맙, 루카투무맙, MEDI-570, MEDI-6383, MEDI-6469, 무로모납-CD3, 니볼루맙, 펨브롤리주맙, 피딜리주맙, 스파르탈리주맙, 트레멜리무맙, 우렐루맙, 우토밀루맙, 바를리루맙, 본레롤리주맙. 하기 표 B는 그의 대체 명칭 (상표명, 이전 명칭, 연구 코드 또는 동의어) 및 각 표적 체크포인트 분자를 열거한다.
TLR7 효능제를 사용하는 조합 치료의 한 실시양태에서, 항암 면역요법제는 길항작용 항-CTLA-4, 항-PD-1 또는 항-PD-L1 항체이다. 암은 폐암 (비소세포 폐암 포함), 췌장암, 신장암, 두경부암, 림프종 (호지킨 림프종 포함), 피부암 (흑색종 및 메르켈 피부암 포함), 요로상피암 (방광암 포함), 위암, 간세포성암 또는 결장직장암일 수 있다.
TLR7 효능제를 사용하는 조합 치료의 또 다른 실시양태에서, 항암 면역요법제는 길항작용 항-CTLA-4 항체, 바람직하게는 이필리무맙이다.
TLR7 효능제를 사용하는 조합 치료의 또 다른 실시양태에서, 항암 면역요법제는 길항작용 항-PD-1 항체, 바람직하게는 니볼루맙 또는 펨브롤리주맙이다.
본원에 개시된 TLR7 효능제는 백신 보조제로서 유용하다.
본 발명의 실시는 추가로 하기 실시예를 참조하여 이해될 수 있고, 이는 제한이 아니라 예시로 제공된다.
분석 절차
NMR
양성자 핵 자기 공명 (NMR) 스펙트럼을 수득하기 위해 하기 조건을 사용하였다: 용매 및 내부 표준으로서 DMSO-d6 또는 CDCl3을 사용하여 400 Mz 또는 500 Mhz 브루커 기기에서 NMR 스펙트럼을 수득하였다. 조 NMR 데이터를 ADC 랩스(ADC Labs)에 의한 ACD 스펙트러스 버전 2015-01 또는 메스트레노바 소프트웨어를 사용하여 분석하였다.
화학적 이동은 내부 테트라메틸실란 (TMS)으로부터 또는 중수소화 NMR 용매에 의해 추론된 TMS의 위치로부터 백만분율 (ppm) 다운필드로 보고된다. 겉보기 다중도는 단일선-s, 이중선-d, 삼중선-t, 사중선-q 또는 다중선-m으로 보고된다. 광폭화를 나타내는 피크는 br로 또한 나타낸다. 적분은 근사치이다. 적분 강도, 피크 형상, 화학적 이동 및 커플링 상수는 용매, 농도, 온도, pH 및 다른 인자에 따라 달라질 수 있음을 주목해야 한다. 또한, NMR 스펙트럼에서 물 또는 용매 피크와 중첩되거나 교환되는 피크는 신뢰가능한 적분 강도를 제공하지 않을 수 있다. 일부 경우에, NMR 스펙트럼은 물 피크 억제를 사용하여 수득될 수 있으며, 가시적이지 않거나 또는 변경된 형상 및/또는 적분을 갖는 중첩 피크를 생성할 수 있다.
액체 크로마토그래피
하기 정제용 및 분석용 (LC/MS) 액체 크로마토그래피 방법을 사용하였다:
LC/MS 방법 A: 칼럼: BEH C18 2.1 x 50mm; 이동상 A: 물, 0.05% TFA 포함; 이동상 B: 아세토니트릴, 0.05% TFA 포함; 온도: 50℃; 구배: 1.7분에 걸쳐 2-98% B; 유량: 0.8 mL/분.
LC/MS 방법 B: 칼럼: BEH C18 2.1 x 50mm; 이동상 A: 95:5 H2O:아세토니트릴, 0.01M NH4OAc 포함; 이동상 B: 5:95 H2O:아세토니트릴, 0.01M NH4OAc 포함; 온도: 50℃; 구배: 1분에 걸쳐 5-95% B; 유량: 0.8 mL/분.
LC/MS 방법 C: 칼럼: 워터스 엑스브리지 C18, 2.1 mm x 50 mm, 1.7 μm 입자; 이동상 A: 5:95 아세토니트릴:물, 0.1% TFA 포함; 이동상 B: 95:5 아세토니트릴:물, 0.1% TFA 포함; 온도: 50℃; 구배: 3분에 걸쳐 0%B에서 100%B, 이어서 100%B에서 0.50분 유지; 유량: 1 mL/분; 검출: MS 및 UV (220 nm).
LC/MS 방법 D. 칼럼: BEH C18 2.1 x 50mm; 이동상 A: 물, 0.05% TFA 포함; 이동상 B: 아세토니트릴, 0.05% TFA 포함; 온도: 50℃; 구배: 1.0분에 걸쳐 2-98% B, 이어서 98% B에서 0.50분 유지; 유량: 0.8 mL/분. 검출: MS 및 UV (220 nm).
LCMS 방법 E. 칼럼: 엑스브리지 BEH C18 XP (50 x 2.1 mm), 2.5 μm; 이동상 A: 5:95 CH3CN: H2O, 10 mM NH4OAc 포함; 이동상 B: 95:5 CH3CN: H2O, 10 mM NH4OAc 포함; 온도: 50℃; 구배: 3분에 걸쳐 0-100% B; 유량: 1.1 mL/분).
합성 - 일반적 절차
일반적으로, 본원에 개시된 절차는 피라졸로피리미딘 고리계의 1H 또는 2H 위치에서 알킬화된 위치이성질체의 혼합물을 생성한다 (이는 또한 알킬화된 질소를 지칭하는 N1 및 N2 위치이성질체로 각각 지칭됨). 간결하게 하기 위해, N2 위치이성질체는 편의상 나타내지 않았지만, 이들은 초기 생성물 혼합물 중에 존재하고, 예를 들어 정제용 HPLC에 의해 나중에 분리되는 것으로 이해되어야 한다.
위치이성질체의 혼합물은 합성의 초기 단계에서 분리되고, 나머지 합성 단계는 1H 위치이성질체를 사용하여 수행될 수 있거나, 또는 대안적으로, 합성은 위치이성질체의 혼합물을 보유하여 진행되고, 분리는 목적하는 바에 따라 후속 단계에서 실시될 수 있다.
본 발명의 화합물은 유기 합성 기술분야의 통상의 기술자에게 널리 공지된 다수의 방식으로 제조될 수 있다. 본 발명의 화합물은 합성 유기 화학 기술분야에 공지된 합성 방법, 또는 관련 기술분야의 통상의 기술자에 의해 인식되는 바와 같은 그에 대한 변형과 함께 하기 기재된 방법을 사용하여 합성될 수 있다. 바람직한 방법은 하기 기재된 것들을 포함하나, 이에 제한되지는 않는다. 본원에 인용된 모든 참고문헌은 그 전문이 본원에 참조로 포함된다.
본 발명의 화합물은 본 섹션에 기재된 반응 및 기술을 사용하여 제조될 수 있다. 반응은 사용된 시약 및 물질에 적절한 용매 중에서 수행되고, 변환이 영향을 미치기에 적합하다. 또한, 하기 기재된 합성 방법의 기재에서, 용매, 반응 분위기, 반응 온도, 실험 지속기간 및 후처리 절차의 선택을 포함한 모든 제안된 반응 조건은 그 반응에 대한 표준 조건이 되도록 선택되며, 이는 관련 기술분야의 통상의 기술자에 의해 용이하게 인식되어야 하는 것으로 이해되어야 한다. 분자의 다양한 부분에 존재하는 관능기가 제안된 시약 및 반응과 상용성이어야 한다는 것이 유기 합성 분야의 통상의 기술자에 의해 이해된다. 반응 조건과 상용성인 치환기에 대한 이러한 제한은 관련 기술분야의 통상의 기술자에게 용이하게 명백할 것이고, 이어서 대안적 방법이 사용되어야 한다. 이는 때때로 본 발명의 목적 화합물을 수득하기 위해 합성 단계의 순서를 변형하기 위한 또는 또 다른 것에 비해 하나의 특정한 공정 반응식을 선택하기 위한 판단을 필요로 할 것이다. 또한, 이 분야의 임의의 합성 경로의 계획에서 또 다른 주요 고려사항은 본 발명에 기재된 화합물에 존재하는 반응성 관능기의 보호에 사용되는 보호기의 신중한 선택임이 인식될 것이다. 숙련된 진료의에게 많은 대안을 기재하는 권위있는 설명은 문헌 [Greene and Wuts (Protective Groups In Organic Synthesis, Third Edition, Wiley and Sons, 1999)]이다.
화학식 (I)의 화합물은 하기 반응식에 예시된 방법을 참조하여 제조될 수 있다. 여기에 나타낸 바와 같이, 최종 생성물은 화학식 (I)과 동일한 구조 화학식을 갖는 화합물이다. 화학식 (I)의 임의의 화합물은 적절한 치환을 갖는 시약의 적합한 선택에 의해 반응식에 의해 제조될 수 있는 것으로 이해될 것이다. 용매, 온도, 압력 및 다른 반응 조건은 통상의 기술자에 의해 용이하게 선택될 수 있다. 출발 물질은 상업적으로 입수가능하거나 또는 관련 기술분야의 통상의 기술자에 의해 용이하게 제조된다. 화합물의 구성성분은 본원 또는 명세서의 다른 곳에 정의된 바와 같다.
반응식 1
본 발명에 기재된 화합물로의 일반적 경로는 반응식에 예시되며, 여기서 R1, R5, L1, L2, L3, Q1, Q2, X 및 W 치환기는 본문에서 이전에 정의된 바와 같거나 또는 목적하는 최종 치환기로 전환될 수 있는 관능기이다. L은 이탈기 예컨대 할라이드, OH이고, 이는 이탈기 예컨대 트리플레이트, 티오테르 또는 헤테로사이클로 용이하게 전환될 수 있다. 반응식 1에 나타낸 바와 같이, 본 발명의 화합물의 제조를 위한 일반적 절차는 치환된 벤질 유도체 1로 출발하는 것을 포함한다. 적합한 시약을 사용하여 1을 적합하게 보호된 히드라진으로 치환하여 관능화된 벤질 유도체 2를 수득할 수 있다. 예를 들어, 2는 적합한 용매, 예컨대 DMF 중에서 다수의 이용가능한 염기 시약, 예컨대 DIPEA 또는 K2CO3 중 하나를 사용하여 벤질 할라이드, 예컨대 메틸 4-(브로모메틸)-3-메톡시벤조에이트와 적합하게 보호된 히드라진, 예컨대 tert-부틸 히드라진카르복실레이트 사이의 치환 반응, 이어서 문헌에 공지된 표준 조건을 사용한 보호기 제거로부터 생성시킬 수 있다. 고리화를 가져오는 것으로 공지된 조건을 사용한 적합하게 치환된 알케노에이트 3과의 2의 후속 반응은 적절하게 치환된 니트로피라졸 4를 제공할 수 있다. 예를 들어, 벤질 히드라진 2를 적합한 염기를 사용하여 메틸 (Z)-4-(디메틸아미노)-3-니트로-2-옥소부트-3-에노에이트와 고리화 반응시켜 니트로피라졸 4를 제공할 수 있다. 니트로피라졸 4의 아미노피라졸 5로의 환원은 문헌에 공지된 표준 조건, 예컨대 H2 (g)와 Pd-C 또는 Zn (s)과 NH4OAc를 사용하여 달성될 수 있다. 적합하게 치환된 5와 적절하게 관능화된 이미데이트 6의 반응 및 염기성 조건, 예컨대 NaOMe-MeOH 하에 생성된 구안디노 중간체의 고리화에 의해 히드록시피리미딘 7을 제공할 수 있다. 문헌에 공지된 표준 조건을 사용하여 7을 적절하게 치환된 아민 8과 커플링시킨 다음, 필요한 경우에 탈보호하여 화합물 9를 제공한다.
반응식 2
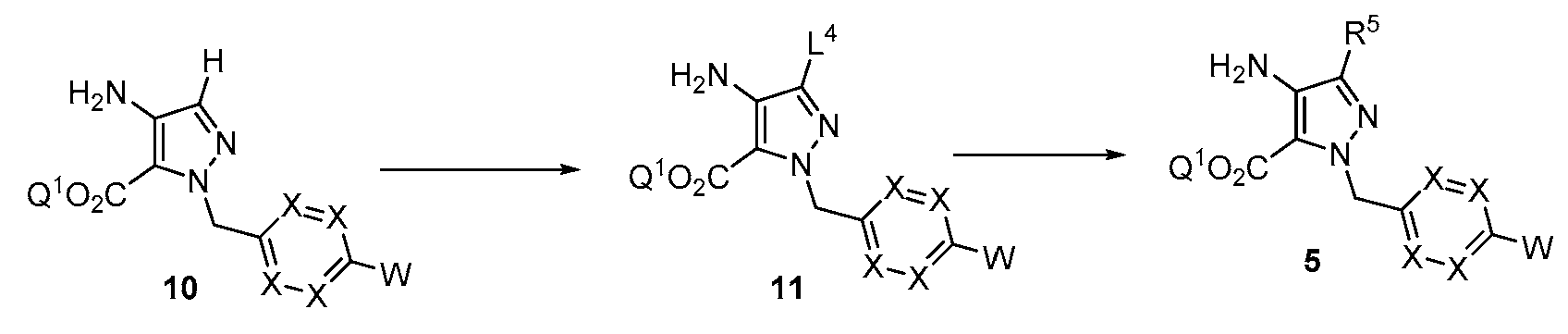
반응식 2에 예시된 바와 같이, R5에서의 기는 피라졸로피리미딘 고리를 형성하기 전에 치환기를 도입하도록 조작될 수 있다. 적합한 이탈기 L4는 후속 화학을 위한 제조에서 아미노피라졸 10에 도입될 수 있다. 예를 들어, 할로겐 기의 도입은 적합한 할로겐화 시약, 예컨대 NBS 또는 NIS를 사용하여 달성될 수 있다. 문헌에 기재된 조건 하에 공지된 탄소-탄소 결합 형성 반응 예컨대 스즈키 반응 또는 공지된 탄소-헤테로원자 반응 예컨대 부흐발트 반응을 사용한 11의 후속 반응을 사용하여 R5에서 알킬, 시클로알킬, 아릴 또는 헤테로아릴 치환기를 도입할 수 있다.
반응식 3
피라졸로피리미딘 9의 대안적 합성은 반응식 3 및 4에 나타낸다. 반응식 1 및 2에 기재된 합성 경로를 사용하여, 화합물 12를 Q4에서 플레이스홀더 관능기를 사용하여 제조할 수 있다. 표준 문헌 조건을 사용하여 아민 8과 커플링시킨 후, Q4를 관련 기술분야의 통상의 기술자에게 이용가능한 다양한 수단을 사용하여 W로 변환시킬 수 있다. 예를 들어, Q4가 에스테르인 경우에, 이를 표준 조건, 예컨대 LiAlH4 또는 LiBH4를 사용하여 1급 알콜로 환원할 수 있고, 적합한 이탈기, 예컨대 -Cl, -Br 또는 -OTs로 변환하고, 이를 다양한 친핵체에 의해 대체할 수 있다. 필요한 경우, 탈보호하여 피라졸로피리딘피리미딘 9를 수득한다. 또 다른 변형에서, 반응식 4, 화합물 12에 나타낸 바와 같은 플레이스홀더 관능기 Q4는 아민 8과의 커플링 전에 화합물 14에서와 같이 W로 변환할 수 있다.
반응식 4
합성 - 구체적 실시예
상기를 추가로 예시하기 위해, 하기 비제한적인 하기 예시적인 합성 반응식이 포함된다. 청구범위의 범주 내의 이들 예의 변형은 관련 기술분야의 통상의 기술자의 이해범위 내에 있으며, 본 개시내용의 범주 내에 속하는 것으로 간주된다. 독자는 본 개시내용 및 관련 기술분야의 통상의 기술에 의해 통상의 기술자가 철저한 실시예 없이도 본원에 개시된 화합물을 제조하고 사용할 수 있을 것임을 인식할 것이다.
100 이상의 번호의 화합물에 대한 분석 데이터는 표 A에서 확인된다.
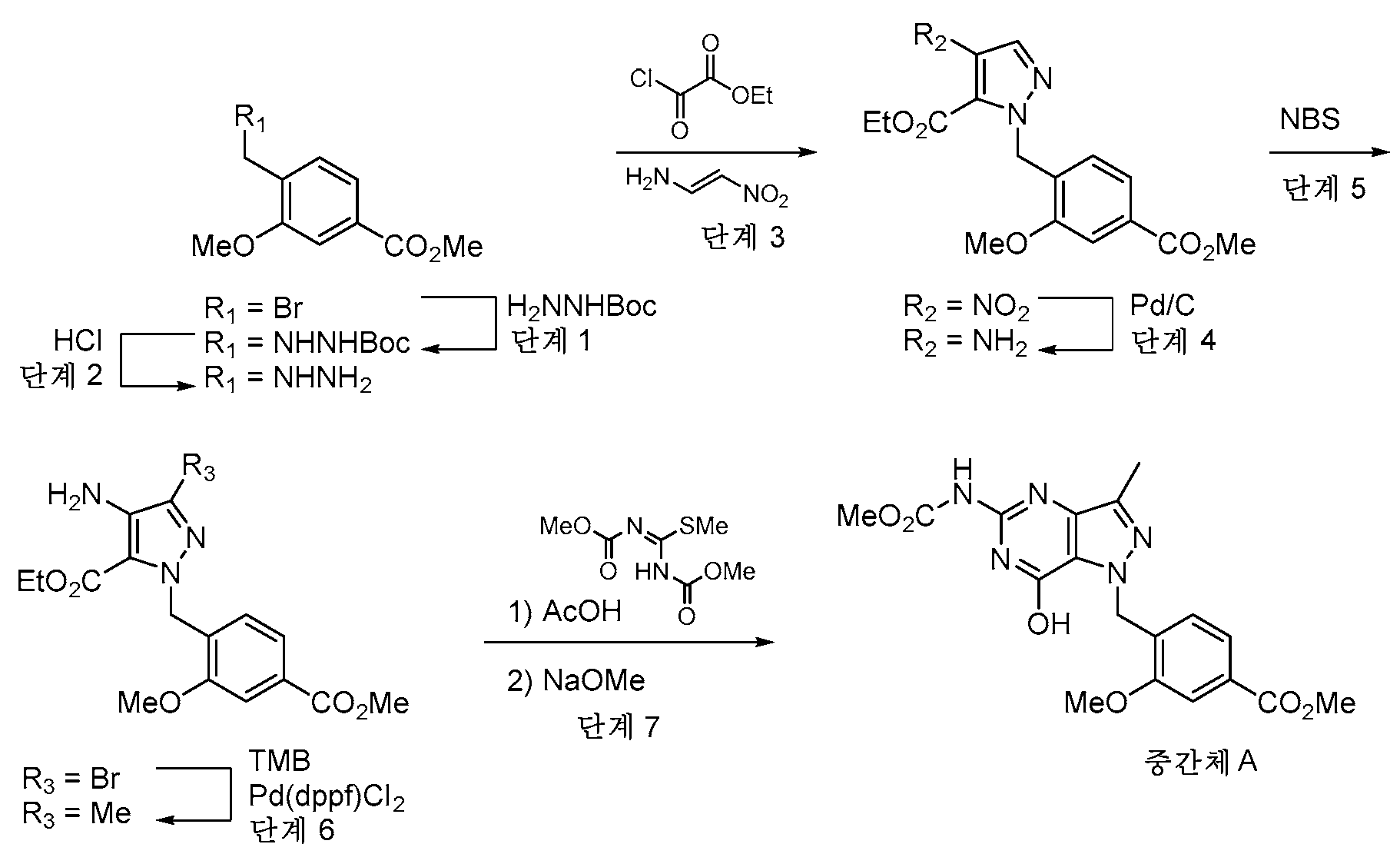
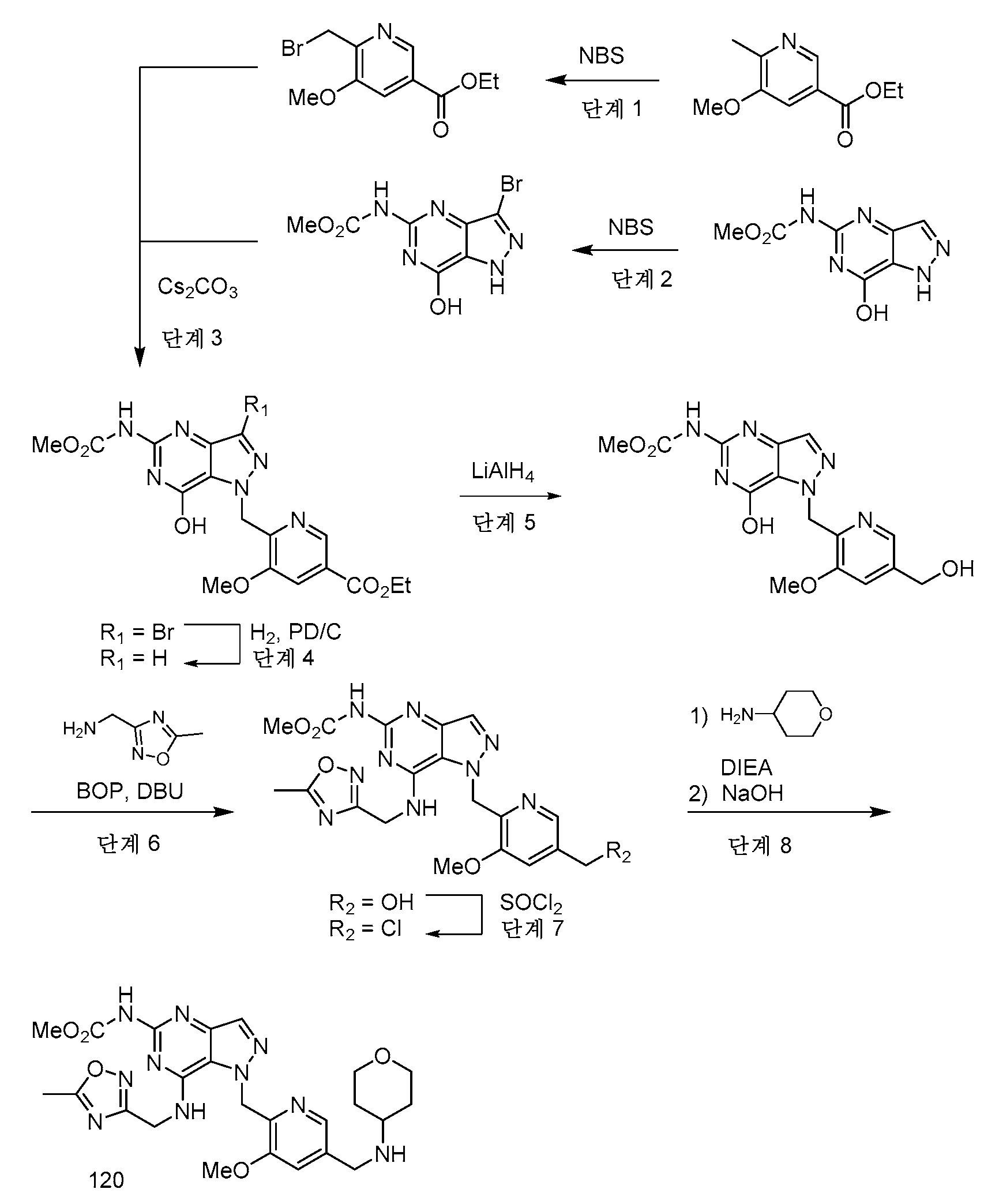
실시예 1 - 중간체 A
중간체 A는 본 개시내용의 화합물의 합성에 유용하다.
단계 1: 실온에서 DMF (24 mL) 중 tert-부틸 히드라진카르복실레이트 (12.75 g, 96 mmol) 및 DIPEA의 용액을 DMF 24 mL 중 메틸 4-(브로모메틸)-3-메톡시벤조에이트 (5 g, 19.30 mmol)의 적가로 첨가 깔때기를 통해 1시간에 걸쳐 처리하였다. 반응 혼합물을 실온에서 밤새 교반하였다. EtOAc (135 mL) 및 H2O (75 mL)를 첨가하고, 2상 혼합물을 30분 동안 교반하였다. 반응 혼합물을 분리 깔때기에 붓고, 수성 층을 제거하였다. 유기 층을 H2O의 2개의 추가의 부분 (75 mL), 10% LiCl 용액의 2개의 부분 (75 mL)으로 세척하고, Na2SO4 상에서 건조시키고, 농축시켰다. 칼럼 크로마토그래피 (이스코, 220 g SiO2, 0% CH2Cl2 (5분)에 이어서 15% EtOAc-CH2Cl2)하여 tert-부틸 2-(2-메톡시-4-(메톡시카르보닐)벤질)히드라진-1-카르복실레이트를 투명한 오일 (3.85 g)로서 수득하였다.
1H NMR (400 MHz, 클로로포름-d) δ 7.64 (dd, J=7.7, 1.5 Hz, 1H), 7.56 (d, J=1.5 Hz, 1H), 7.37 (d, J=7.7 Hz, 1H), 6.08 - 5.87 (m, 1H), 4.07 (s, 2H), 3.94 (d, J=4.6 Hz, 6H), 1.50 - 1.40 (m, 9H).
LC/MS [M+H]+ 311.2; LC RT = 0.80분 (방법 A).
단계 2: tert-부틸 2-(2-메톡시-4-(메톡시카르보닐)벤질)히드라진-1-카르복실레이트 (25.4 g, 82 mmol)를 실온에서 MeOH (164 mL) 중에 용해시켰다. 4 N HCl-디옥산 (123 ml, 59.5 mmol)을 첨가하고, 반응물을 실온에서 밤새 교반하였다. 백색 침전물을 여과에 의해 수집하고, 건조시켜 메틸 4-(히드라지닐메틸)-3-메톡시벤조에이트, 2·HCl (20 g)을 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.12 (br s), 7.62 - 7.55 (m, 1H), 7.53 - 7.47 (m, 2H), 4.10 (s, 2H), 3.88 (s, 3H), 3.87 (s, 3H).
LC/MS [M+H]+ 211.1; LC RT = 0.51분. (방법 A)
단계 3: CH2Cl2 (799 ml) 중 (E)-N,N-디메틸-2-니트로에텐-1-아민 (46.4 g, 400 mmol) 및 피리딘 (420 ml, 5195 mmol)의 용액을 -10℃로 냉각시키고, 에틸 2-클로로-2-옥소아세테이트 (51.4 ml, 460 mmol)로 서서히 처리하였다. 반응 혼합물을 25℃로 2시간에 걸쳐 가온되도록 하고, 밤새 교반하였다. CH2Cl2를 회전 증발에 의해 제거하고, 메틸 4-(히드라지닐메틸)-3-메톡시벤조에이트 디히드로클로라이드 (31.7 g, 112 mmol)를 반응 혼합물에 첨가하였다. 용액을 실온에서 2시간 동안 교반하고, 용매를 진공 하에 제거하였다. 잔류물을 물, 1N 수성 HCl 용액으로 세척하고, EtOAc (3x)로 추출하였다. 유기 층을 Na2SO4 상에서 건조시키고, 농축시켰다. 잔류물을 CH2Cl2 중에 용해시키고, 짧은 실리카 겔 칼럼에 통과시키고, 에탄올로부터 재결정화하여 에틸 1-(2-메톡시-4-(메톡시카르보닐)벤질)-4-니트로-1H-피라졸-5-카르복실레이트 (29.4 g)를 수득하였다.
1H NMR (400 MHz, 클로로포름-d) δ 8.06 (s, 1H), 7.64 (dd, J=7.9, 1.5 Hz, 1H), 7.56 (d, J=1.5 Hz, 1H), 7.13 (d, J=7.8 Hz, 1H), 5.53 (s, 2H), 4.45 (q, J=7.2 Hz, 2H), 3.94 (s, 3H), 3.88 (s, 3H), 1.37 (t, J=7.2 Hz, 3H).
LC/MS [M+Na]+ 386.0; LC RT = 0.98분 (방법 A).
단계 4: 에틸 4-아미노-1-(2-메톡시-4-(메톡시카르보닐)벤질)-1H-피라졸-5-카르복실레이트 (3.04 g, 9.12 mmol, 86% 수율) 및 Pd-C (1.131 g, 0.531 mmol)를 EtOAc/MeOH (1:1) (152 mL) 중에 현탁시켰다. 반응 플라스크를 진공 하에 배기시키고, H2 (3X)로 퍼징한 후, H2 (g)의 풍선 압력 하에 교반하였다. 5시간 후, 반응 혼합물을 셀라이트(CELITE)™을 통해 여과하고, 새로운 Pd-C (1.131 g, 0.531 mmol)를 첨가하였다. 반응 플라스크를 진공 하에 배기시키고, H2 (3X)로 퍼징한 후, H2의 풍선 압력 하에 16시간 동안 교반하였다. 반응 혼합물을 셀라이트™을 통해 여과하고, 농축시키고, 진공 하에 건조시켜 에틸 4-아미노-1-(2-메톡시-4-(메톡시카르보닐)벤질)-1H-피라졸-5-카르복실레이트 (3.04 g)를 크림색 분말로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 7.52 - 7.49 (m, 1H), 7.47 (dd, J=7.9, 1.5 Hz, 1H), 7.19 (s, 1H), 6.40 (d, J=7.8 Hz, 1H), 5.54 (s, 2H), 5.10 (s, 1H), 4.15 (q, J=7.1 Hz, 2H), 3.91 (s, 3H), 3.84 (s, 3H), 1.14 (t, J=7.1 Hz, 3H).
LC/MS [M+H]+ 334.1; LC/RT = 0.85분. (방법 B).
단계 5: 에틸 4-아미노-1-(2-메톡시-4-(메톡시카르보닐)벤질)-1H-피라졸-5-카르복실레이트 (1.65 g, 4.95 mmol)를 CHCl3 (49.5 ml)에 용해시키고, 0℃로 냉각시켰다. NBS (0.925 g, 5.20 mmol)를 첨가하였다. 15분 후, 반응물을 CHCl3 로 희석하고, 10% 수성 티오황산나트륨 용액과 함께 10분 동안 격렬히 교반하였다. 유기 상을 분리하고, H2O로 세척하고, MgSO4 상에서 건조시키고, 농축시켰다. 조 생성물을 칼럼 크로마토그래피 (80g SiO2, 0에서 50% EtOAc-헥산 구배 용리)에 의해 정제하여 에틸 4-아미노-3-브로모-1-(2-메톡시-4-(메톡시카르보닐)벤질)-1H-피라졸-5-카르복실레이트 (1.32 g)를 백색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 7.61 - 7.41 (m, 2H), 6.55 (d, J=8.3 Hz, 1H), 5.56 (s, 2H), 5.02 (s, 2H), 4.20 (q, J=7.1 Hz, 2H), 3.90 (s, 3H), 3.85 (s, 3H), 1.15 (t, J=7.1 Hz, 3H).
LC/MS [M+H]+ 412.2; LC RT = 1.02분 (방법 A).
단계 6: 에틸 4-아미노-3-브로모-1-(2-메톡시-4-(메톡시카르보닐)벤질)-1H-피라졸-5-카르복실레이트 (741.2 mg, 67.1% 수율), K2CO3 (1.098 g, 7.94 mmol) 및 TMB (THF 중 3.5 M) (1.816 ml, 6.36 mmol)를 디옥산 (26.5 ml):물 (5.30 ml) (5:1)에 현탁시켰다. N2의 스트림을 반응 혼합물을 통해 5분 동안 버블링한 후, PdCl2(dppf)-CH2Cl2 부가물 (0.052 g, 0.064 mmol)을 첨가하였다. 교반을 추가로 4분 동안 계속한 후, 반응 플라스크를 밀봉하고, 90℃로 가열하였다. 3시간 후, 추가의 TMB (THF 중 3.5 M; 0.908 mL, 3.18 mmoL) 및 PdCl2(dppf)-CH2Cl2 부가물 (0.052 g, 0.064 mmol)을 첨가하였다. 반응 혼합물을 100℃에서 16시간 동안 교반하였다. 냉각된 반응 혼합물을 EtOAc 100 mL로 희석하고, 추가의 EtOAc로 세척하면서 셀라이트™을 통해 여과하였다. 조 생성물을 4 g 셀라이트™ 상에서 농축시켰다. 칼럼 크로마토그래피 (80g SiO2, 0에서 30% EtOAc-CH2Cl2 구배 용리)로 에틸 4-아미노-1-(2-메톡시-4-(메톡시카르보닐)벤질)-3-메틸-1H-피라졸-5-카르복실레이트 (741 mg)를 크림색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 7.49 (d, J=1.5 Hz, 1H), 7.46 (dd, J=7.9, 1.5 Hz, 1H), 6.40 (d, J=7.8 Hz, 1H), 5.48 (s, 2H), 4.94 - 4.86 (m, 2H), 4.14 (q, J=7.0 Hz, 2H), 3.90 (s, 3H), 3.84 (s, 3H), 2.10 (s, 3H), 1.15 - 1.08 (m, 3H).
LC/MS [M+H]+ 348.2; LC/RT = 0.89분. (방법 A).
단계 7: 에틸 4-아미노-1-(2-메톡시-4-(메톡시카르보닐)벤질)-3-메틸-1H-피라졸-5-카르복실레이트 (742 mg, 2.136 mmol)를 MeOH (10.800 mL) 중에 현탁시키고, 격렬한 교반 하에 서서히 가열하여 물질을 가용화시켰다. 1,3-비스-(메톡시카르보닐)-2-메틸-2-티오슈도우레아 (661 mg, 3.20 mmol)를 첨가하고, 이어서 AcOH (0.611 mL, 10.68 mmol)를 첨가하였다. 반응 혼합물을 실온에서 16시간 동안 교반하였다. AcOH의 추가 부분을 첨가하고 (0.049 mL, 0.854 mmol), 반응물을 실온에서 추가로 72시간 동안 교반한 후, NaOMe (MeOH 중 25 wt%) (5.69 mL, 25.6 mmol)를 첨가하였다. 3시간 동안 교반한 후, 반응 혼합물을 AcOH로 재산성화시켰다. 생성물을 여과에 의해 수집하고, 10분 동안 공기-건조시키고, 화학-건조 오븐에서 완전히 건조시켜 메틸 4-((7-히드록시-5-((메톡시카르보닐)아미노)-3-메틸-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (중간체 A) (722.0 mg)를 크림색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 11.58 - 11.17 (m, 2H), 7.51 (d, J=1.4 Hz, 1H), 7.49 - 7.42 (m, 1H), 6.67 (d, J=7.9 Hz, 1H), 5.67 (s, 2H), 3.90 (s, 3H), 3.84 (s, 3H), 3.71 (s, 3H), 2.31 (s, 3H).
LC/MS [M+H]+ 402.3; LC RT = 0.86분 (방법 A).
실시예 2 - 화합물 112
단계 1: 실온에서 DMF (2491 μl) 중 메틸 4-((7-히드록시-5-((메톡시카르보닐)아미노)-3-메틸-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (중간체 A, 200 mg, 0.498 mmol) 및 BOP (331 mg, 0.747 mmol)의 현탁액을 (5-메틸이속사졸-3-일)메탄아민 (72.6 mg, 0.648 mmol) 및 DBU (3 당량) (225 μl, 1.495 mmol)로 처리하였다. 반응 혼합물을 40℃로 가열하였다. 15분 후, 추가의 DBU (2 당량; 150 μL, 0.997 mmol)를 첨가하였다. 반응 혼합물을 40℃에서 16시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 EtOAc와 반포화 수성 NaHCO3 사이에 분배하였다. 유기 상을 분리하고, 수성 상을 EtOAc (2x)로 추출하였다. 합한 유기 층을 10% 수성 LiCl 용액 및 염수로 순차적으로 세척하고, Na2SO4 상에서 건조시키고, 농축시켰다. 칼럼 크로마토그래피 (12g SiO2, 0에서 10% CH3OH-CH2Cl2 구배 용리)하여 메틸 3-메톡시-4-((5-((메톡시카르보닐)아미노)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)벤조에이트 (201.1 mg)를 수득하였다.
LC/MS [M+H]+ 496.2; LC RT = 0.79분 (방법 A).
단계 2: 메틸 3-메톡시-4-((5-((메톡시카르보닐)아미노)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)벤조에이트 (200 mg, 0.404 mmol)를 실온에서 THF 중에 현탁시키고, 초음파처리하여 용해를 보조하였다. LiAlH4 (THF 중 1M; 807 μL, 0.807 mmol)을 10분에 걸쳐 적가하였다. 20분 후, 반응물을 MeOH로 켄칭하고, EtOAc와 로쉘 염 사이에 분배하였다. 2상 혼합물을 실온에서 2시간 동안 교반하였다. 수성 층을 분리하고, EtOAc (1X)로 재추출하였다. 합한 유기 층을 염수로 세척하고, 농축시켰다. 칼럼 크로마토그래피 (12g SiO2, 0에서 10% CH3OH-CH2Cl2 구배 용리)하여 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (73 mg)를 수득하였다.
LC/MS [M+H]+ 468.4; LC RT = 0.62분. (방법 A).
단계 3: 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (73 mg, 0.156 mmol)를 실온에서 CH2Cl2 (1562 μL) 중에 용해시켰다. SOCl2 (57.0 μl, 0.781 mmol)를 첨가하고, 반응물을 20분 동안 교반하였다. 농축시켜 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (80 mg)를 추가 정제 없이 사용하기에 충분한 순도로 수득하였다.
LC/MS [M+H]+ 486.1; LC RT = 0.83분 (방법 A).
단계 4: 아세토니트릴 (412 μL) 중 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (20 mg, 0.041 mmol)의 원액을 테트라히드로-2H-피란-4-아민 (12.49 mg, 0.123 mmol)으로 처리하였다. 반응물을 40℃에서 밤새 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 농축시키고, 디옥산 (400 μL) 중에 재용해시키고, 10 M NaOH (82 μL, 0.823 mmol)로 처리하였다. 반응 혼합물을 80℃로 5시간 동안 가열하였다. 실온으로 냉각시킨 후, 반응물을 AcOH (42 μL)로 중화시키고, 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, NH4OAc 포함; 구배: 3% B에서 0-분 유지, 20분에 걸쳐 3-43% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 및 UV 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 112 (5.1 mg)를 수득하였다.
화합물 113을 유사하게 제조하였다: 조 생성물을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, NH4OAc 포함; 구배: 2% B에서 0-분 유지, 24분에 걸쳐 2-42% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 113 (8.6 mg)을 수득하였다.
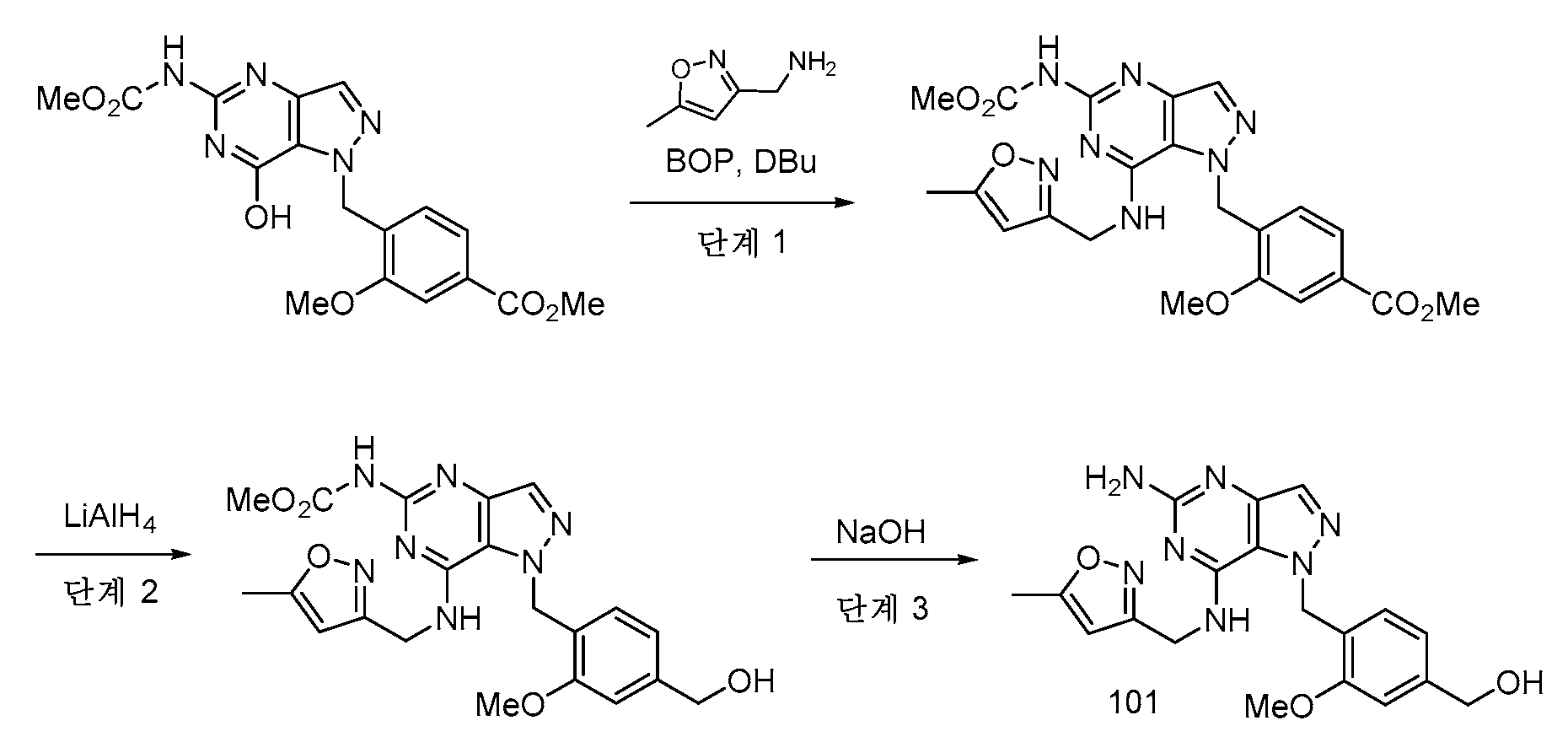
실시예 3 - 화합물 101
단계 1: DMSO (3.9 mL) 중 메틸 4-((7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (US 2020/0038403 A1; 300 mg, 0.774 mmol)의 용액을 (5-메틸이속사졸-3-일)메탄아민 (174 mg, 1.55 mmol), BOP (411 mg, 0.929 mmol) 및 DBU (233 μl, 1.549 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, EtOAc로 희석하고, H2O (3x)로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 메틸 3-메톡시-4-((5-((메톡시카르보닐)아미노)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)벤조에이트 (353 mg, 95% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1H), 7.99 - 7.93 (m, 1H), 7.77 (t, J=5.9 Hz, 1H), 7.49 (d, J=1.5 Hz, 1H), 7.45 (dd, J=7.8, 1.5 Hz, 1H), 6.62 (d, J=7.9 Hz, 1H), 6.10 (d, J=0.9 Hz, 1H), 5.80 (s, 2H), 4.73 (d, J=5.9 Hz, 2H), 3.84 (s, 3H), 3.82 (s, 3H), 3.64 (s, 3H), 2.31 (s, 3H).
LC RT: 0.67분. LC/MS [M+H]+ 482.3 (방법 A)
단계 2: THF (10 mL) 중 메틸 3-메톡시-4-((5-((메톡시카르보닐)아미노)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)벤조에이트 (190 mg, 0.395 mmol)의 용액을 0℃로 냉각시키고, LiAlH4 (THF 중 1M, 691 μL, 0.691 mmol)로 처리하였다. 반응 혼합물을 0℃에서 15분 동안 교반하고, MeOH 및 로쉘 염 (포화 수용액)으로 켄칭하고, 실온에서 1시간 동안 교반하였다. 혼합물을 EtOAc (3x)로 추출하였다. 합한 유기 층을 H2O로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (160 mg, 89% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.77 - 9.75 (m, 1H), 7.90 - 7.88 (m, 1H), 7.72 (br t, J=5.7 Hz, 1H), 6.94 (s, 1H), 6.76 (d, J=7.5 Hz, 1H), 6.61 - 6.57 (m, 1H), 6.15 (d, J=0.8 Hz, 1H), 5.68 (s, 2H), 5.16 (t, J=5.7 Hz, 1H), 4.73 (br d, J=5.8 Hz, 2H), 4.44 (d, J=5.6 Hz, 2H), 3.70 (s, 3H), 3.62 (s, 3H), 2.33 (s, 3H).
LC RT: 0.58분. LCMS [M+H]+ = 454.3 (방법 A)
단계 3: 디옥산 (500 μL) 중 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (22 mg, 0.048 mmol)의 용액을 NaOH (10 M 수용액, 200 μL, 2.0 mmol)로 처리하고, 75℃로 가열하였다. 5시간 후, 반응 혼합물을 실온으로 냉각시키고, HOAc (114 μL, 2.0 mmol)로 중화시키고, 질소의 스트림 하에 농축시켰다. 잔류물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하였다. 조 물질을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴:물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 9% B에서 0-분 유지, 20분에 걸쳐 9-49% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 101 (3.5 mg, 8% 수율)을 수득하였다.
실시예 4 - 화합물 102
SOCl2 (24 μL, 0.33 mmol)을 THF (0.7 mL) 중 (4-((5-아미노-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시페닐)메탄올 (26.3 mg, 0.067 mmol)의 실온 용액에 첨가하였다. 30분 동안 교반한 후, 반응 혼합물을 진공 하에 농축시켰다. 잔류물을 DCM 중에 재용해시키고, 진공 하에 농축시켰다. 잔류물을 DMF (0.7 mL) 중에 용해시키고, 시클로부탄아민 (25.3 mg, 0.355 mmol)으로 처리하고, 실온에서 3시간 동안 교반하였다. 온도를 70℃로 상승시켰다. 반응 혼합물을 추가로 2시간 동안 교반하고, 진공 하에 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하였다. 조 물질을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴:물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 2% B에서 0-분 유지, 20분에 걸쳐 2-42% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 잔류물을 수득하였으며, 이를 추가로 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 0% B에서 0-분 유지, 22분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 102를 비스 TFA 염 (4.0 mg, 11%)으로서 수득하였다.
실시예 5 - 화합물 103
단계 1: DCM (3.5 mL) 중 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (159 mg, 0.35 mmol)의 용액을 SOCl2 (128 μL, 1.76 mmol)로 처리하였다. 반응 혼합물을 실온에서 15분 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 DCM 중에 재용해시키고, 진공 하에 농축시켜 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (182 mg, 100%)를 수득하였다.
LC RT: 0.80분. LCMS [M+H]+ = 472.3 (방법 A)
단계 2: DMF (1.1 mL) 중 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸이속사졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (25 mg, 0.053 mmol)의 용액을 테트라히드로-2H-피란-4-아민 (26.8 mg, 0.265 mmol)으로 처리하였다. 반응 혼합물을 70℃에서 2시간 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 실온에서 디옥산 (0.5 mL) 중에 재용해시키고, NaOH (10M 수용액, 27 μl, 0.27 mmol)로 처리하고, 80℃로 4.5시간 동안 가열하였다. 반응 혼합물을 실온에서 HOAc (15 μl, 0.27 mmol)로 중화시키고, 진공 하에 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 0% B에서 0-분 유지, 20분에 걸쳐 0-30% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 103을 비스 TFA 염 (20.2 mg, 54%)으로서 수득하였다.
하기 화합물을 유사하게 제조하였다: 화합물 104, 화합물 105, 화합물 106, 화합물 110, 및 화합물 111.
실시예 6 - 화합물 107
DMF (0.7 mL) 중 메틸 (1-(4-((시클로부틸아미노)메틸)-2-메톡시벤질)-7-히드록시-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (US 2020/0038403 A1; 30 mg, 0.073 mmol)의 용액을 BOP (57.9 mg, 0.131 mmol), (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민·HCl (54.4 mg, 0.364 mmol) 및 DBU (164 μL, 1.091 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, EtOAc로 희석하고, 포화 NaHCO3 용액 및 H2O로 세척하였다. 유기 층을 진공 하에 농축시켰다. 잔류물을 디옥산 (0.7 mL) 중에 용해시키고, NaOH (10 M 수용액, 0.20 mL, 2.0 mmol)로 처리하고, 75℃로 가열하였다. 4시간 후, 반응 혼합물을 실온으로 냉각시키고, HOAc (0.12 mL, 2.0 mmol)로 중화시키고, 진공 하에 농축시켰다. 조 생성물을 DMF 및 H2O 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 0% B에서 0-분 유지, 20분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 107 (8.6 mg, 26% 수율)을 수득하였다.
실시예 7 - 화합물 114
단계 1: DMSO (9.7 mL) 중 메틸 (7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (US 2020/0038403 A1, 도 7, 화합물 64; 700 mg, 1.95 mmol)의 용액을 (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민·HCl (379 mg, 2.53 mmol), BOP (129 mg, 2.92 mmol) 및 DBU (1.0 mL, 6.8 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, DCM으로 희석하고, H2O로 세척하였다. 유기 층을 H2O (6x)로 세척하고, Na2SO4상에서 건조시키고, 여과하고, 진공 하에 농축시켰다. 잔류물을 DCM/MeOH 중에 용해시키고, 셀라이트™ 상에 흡수시키고, 칼럼 크로마토그래피 (100g C18 골드 칼럼; 이동상 A: 5:95 아세토니트릴:물, 0.05% TFA 함유; 이동상 B: 95:5 아세토니트릴:물, 0.05% TFA 함유; 유량: 60 mL/분, 10-50% 구배)에 의해 정제하였다. 정제된 생성물을 DCM에 용해시키고, 포화 NaHCO3 수용액으로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (372 mg, 42% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.69 - 9.66 (m, 1H), 7.89 (s, 1H), 7.76 (t, J=5.8 Hz, 1H), 6.95 (s, 1H), 6.81 - 6.77 (m, 1H), 6.76 - 6.70 (m, 1H), 5.69 (s, 2H), 5.17 (t, J=5.7 Hz, 1H), 4.89 (d, J=5.7 Hz, 2H), 4.45 (d, J=5.8 Hz, 2H), 3.77 (s, 3H), 3.60 (s, 3H), 2.56 (s, 3H).
LC RT: 0.56분. LC/MS [M+H]+ 455.3 (방법 A)
단계 2: DCM (8.2 mL) 중 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (372 mg, 0.818 mmol)의 용액을 SOCl2 (179 μL, 2.46 mmol)로 처리하였다. 반응 혼합물을 실온에서 10분 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 DCM 중에 재용해시키고, 진공 하에 농축시켜 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (387 mg, 100%)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 11.82 - 11.60 (m, 1H), 9.40 - 9.21 (m, 1H), 8.12 - 8.08 (m, 1H), 7.10 (s, 1H), 7.04 - 6.95 (m, 2H), 5.81 (s, 2H), 5.02 (br d, J=5.3 Hz, 2H), 4.74 (s, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 2.60 (s, 3H).
LC RT: 0.70분. LCMS [M+H]+ = 473.3 (방법 A)
단계 3: DMF (1.5 mL) 중 메틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (34.7 mg, 0.073 mmol)의 용액을 테트라히드로-2H-피란-4-아민 (37.1 mg, 0.367 mmol)으로 처리하였다. 반응물을 75℃에서 1시간 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 디옥산 (1.0 mL) 및 MeOH (0.2 mL) 중에 용해시키고, NaOH (10M 수용액, 0.2 mL, 2.0 mmol)로 처리하고, 75℃에서 2시간 동안 가열하였다. 실온으로 냉각시킨 후, 반응 혼합물을 HOAc (0.12 mL, 2.0 mmol)로 중화시키고, 진공 하에 농축시켰다. 조 생성물을 DMF 및 H2O 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 0% B에서 0-분 유지, 30분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 114 (7.5 mg, 18%)를 수득하였다.
하기 화합물을 유사하게 제조하였다: 화합물 115, 화합물 117, 화합물 120, 화합물 121, 화합물 122 및 화합물 123.
실시예 8 - 화합물 116
디옥산 (0.4 mL) 및 MeOH (0.2 mL) 중 메틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (19 mg, 0.043 mmol)의 용액을 NaOH (10 M 수성 용액, 50 μL, 0.5 mmol)로 처리하고, 50℃로 가열하였다. 30분 후, 반응 혼합물을 실온으로 냉각시키고, HOAc (30 μL, 0.5 mmol)로 중화시키고, 진공 하에 농축시켰다. 잔류물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하였다. 조 물질을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 2% B에서 0-분 유지, 25분에 걸쳐 2-42% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 116 (3.9 mg, 22% 수율)을 수득하였다.
실시예 9 - 화합물 109a
DMSO (1.5 mL) 중 메틸 (7-히드록시-1-(2-메톡시-4-(((테트라히드로-2H-피란-4-일)아미노)메틸)벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (75 mg, 0.170 mmol, US 2020/0038403 A1)의 용액에 (S)-3-아미노-1-시클로프로필프로판-1-올 (39.0 mg, 0.339 mmol), DBU (0.077 mL, 0.509 mmol), 및 BOP (150 mg, 0.339 mmol)를 첨가하고; 반응 혼합물을 70℃에서 2시간 동안 가열하고, 5M NaOH (0.136 mL, 0.678 mmol)로 처리하고, 70℃에서 2시간 동안 가열하였다. 반응 혼합물을 25℃로 냉각시키고, 조 물질을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, NH4OAc 포함; 구배: 3% B에서 0-분 유지, 30분에 걸쳐 3-43% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켰다. 물질을 추가로 정제용 LC/MS를 통해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 0% B에서 0-분 유지, 25분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켰다. 물질을 추가로 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, NH4OAc 포함; 구배: 1% B에서 0-분 유지, 25분에 걸쳐 1-41% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 109a (2.3 mg, 4.69 μmol, 2.77% 수율)를 수득하였다.
화합물 109b를 유사하게 제조하였다.
실시예 10 - 화합물 108
단계 1. DMF (2034 μl) 중 메틸 (7-히드록시-1-(2-메톡시-4-(((테트라히드로-2H-피란-4-일)아미노)메틸)벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (90 mg, 0.203 mmol, US 2020/0038403 A1), (S)-2-아미노-3-시클로프로필프로판-1-올 히드로클로라이드 (93 mg, 0.610 mmol) 및 BOP (135 mg, 0.305 mmol)의 용액에 DBU (153 μl, 1.017 mmol)를 첨가하였다. 반응 혼합물을 실온에서 밤새 희석하고, 물 (2 mL, 0.2% TFA)로 희석하고, Accq 정제용 20x150 mm 엑스브리지 칼럼 (6회 주입): 20% 아세토니트릴/물 (0.1% TFA)에 의해 정제하였다. 12분에 수집된 분획을 동결건조시켜 메틸 (S)-(7-((1-시클로프로필-3-히드록시프로판-2-일)아미노)-1-(2-메톡시-4-(((테트라히드로-2H-피란-4-일)아미노)메틸)벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (65 mg, 59.2% 수율)를 백색 고체로서 수득하였다.
LCMS [M+H]+ = 539.3.
단계 2. 메틸 (S)-(7-((1-시클로프로필-3-히드록시프로판-2-일)아미노)-1-(2-메톡시-4-(((테트라히드로-2H-피란-4-일)아미노)메틸)벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (167 mg, 0.309 mmol)를 디옥산 (5158 μl) 중에 용해시키고, NaOH (619 μl, 3.09 mmol)로 처리하고, 80℃에서 밤새 가열하였다. 반응 혼합물을 HCl로 중화시키고, 농축시켰다. 잔류물을 DMF (4 mL) 중에 용해시키고, 여과하였다. 조 물질을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, NH4OAc 포함; 구배: 0% B에서 0-분 유지, 20분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 108 (60 mg, 40% 수율)을 수득하였다.
화합물 125를 유사하게 제조하였다.
실시예 11 - 화합물 126
단계 1. DMF (1 mL) 중 메틸 4-((7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (50 mg, 0.129 mmol)에 NBS (76 mg, 0.427 mmol)를 첨가하였다. 반응 혼합물을 40℃에서 밤새 교반하고, 25℃로 냉각시키고, MeOH로 희석하고, 여과하여 메틸 4-((3-브로모-7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (40 mg, 0.082 mmol, 63.1% 수율)를 수득하였다.
LC-MS m/z 468.2 [M+2H]+.
1H NMR (400 MHz, DMSO-d6) δ 11.86 - 11.17 (m, 2H), 7.51 (s, 2H), 7.02 - 6.74 (m, 1H), 5.74 (s, 2H), 3.86 (d, J=9.7 Hz, 6H), 3.76 (s, 3H)
단계 2. LiAlH4 (THF 중 1M; 6 mL, 6.00 mmol)를 0℃ (빙조)에서 THF (20 mL) 중 메틸 4-((3-브로모-7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (1 g, 2.145 mmol)의 용액에 천천히 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응물을 0℃ (빙조)에서 포화 Na2SO4 (5.0 ml)의 느린 첨가에 의해 켄칭하였다. 혼합물을 실온에서 30분 동안 교반하였다. 유기 용매를 회전 증발기 상에서 제거하고, 수성 상을 동결건조시켰다. 동결건조된 물질을 MeOH (100ml)로 희석하고, 여과하였다 (3x 10 mL MeOH로 세척). 용매를 제거하고, 물질을 실리카 겔 (DCM-MeOH 0-30%) 상에서 정제하여 메틸 (3-브로모-7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (330 mg, 0.753 mmol, 30% 수율)를 수득하였다.
LC-MS m/z 440.2[M+2H]+.
1H NMR (400 MHz, DMSO-d6) δ 7.05 - 6.95 (m, 1H), 6.87 - 6.76 (m, 2H), 5.66 (s, 2H), 5.23 - 5.14 (m, 1H), 4.52 - 4.43 (m, 2H), 3.82 - 3.72 (m, 6H)
단계 3. 마이크로웨이브 바이알에 메틸 (3-브로모-7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (200 mg, 0.456 mmol) (N2-위치이성질체로 오염된 약 80% 순도), TMB (0.255 ml, 1.825 mmol), [1,1'-비스(디페닐포스피노)페로센]디클로로팔라듐(II) (100 mg, 0.137 mmol), K2CO3 (442 mg, 3.19 mmol), 디옥산 (8 mL) 및 물 (2 mL)을 충전하였다. 반응 혼합물을 마이크로웨이브 오븐에서 120℃에서 1시간 동안 가열하고, EtOAc로 희석하고, 물로 세척하고, Na2SO4 상에서 건조시켰다. 용매를 제거하고, 물질을 실리카 겔 (건조 로딩) 상에서 DCM-MeOH 0-50%로 정제하여 5-아미노-1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (49 mg, 0.093 mmol, 20.43% 수율)을 수득하였다.
LC-MS m/z 316.3[M+H]+.
단계 4. 20 mL 바이알에 5-아미노-1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (50 mg, 0.159 mmol) 및 DCM (2 mL)을 첨가하고, 이어서 SOCl2 (.1 mL, 1.370 mmol)를 실온에서 첨가하였다. 반응 혼합물을 25℃에서 교반하고, 진공 하에 농축시켜 5-아미노-1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (52.9 mg, 0.158 mmol, 100% 수율)을 수득하였으며, 이를 정제 없이 사용하였다.
LC-MS m/z 335.7[M+2H]+.
단계 5. DMF (2 mL) 중 5-아미노-1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (52 mg, 0.156 mmol)에 2-(피페라진-1-일)에탄-1-올 (.1 mL, 0.815 mmol)을 첨가하였다. 반응 혼합물을 25℃에서 밤새 교반하고, 용매를 제거하였다. 물질을 실리카 겔 (건조 로딩) 상에서 DCM-MeOH 0-30%로 정제하여 5-아미노-1-(4-((4-(2-히드록시에틸)피페라진-1-일)메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (53 mg, 0.095 mmol, 61.3% 수율)을 수득하였다.
LC-MS m/z 428.3[M+H]+.
단계 6. DMSO (1.5 mL) 중 5-아미노-1-(4-((4-(2-히드록시에틸)피페라진-1-일)메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (53 mg, 0.124 mmol) 및 (S)-3-아미노-1-시클로프로필프로판-1-올 (30 mg, 0.260 mmol)의 용액에 DBU (0.075 mL, 0.496 mmol) 및 BOP (110 mg, 0.248 mmol)를 첨가하였다. 반응 혼합물을 70℃에서 1시간 동안 가열하였다. 생성물을 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.1% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.1% TFA 포함; 구배: 0% B에서 0-분 유지, 20분에 걸쳐 0-40% B, 이어서 30 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 및 UV 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 126을 수득하였다.
실시예 12 - 화합물 118
단계 1. THF (16 mL) 중 메틸 4-((5-((tert-부톡시카르보닐)아미노)-7-히드록시-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (685 mg, 1.59 mmol; US 2020/0038403; 도 8, 화합물 71)의 용액을 0℃로 냉각시키고, LiAlH4 (THF 중 1 M, 2.8 mL, 2.8 mmol)로 처리하였다. 반응 혼합물을 0℃에서 15분 동안 교반하고, H2O 및 로쉘 염 (포화 수용액)으로 켄칭하고, 실온에서 3시간 동안 교반하였다. 유기 층을 셀라이트™ 상에 흡수시키고, 칼럼 크로마토그래피 (24g SiO2; 0에서 20% MeOH-DCM 구배 용리)에 의해 정제하여 tert-부틸 (7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (460 mg, 72% 수율)를 수득하였다.
1H (400 MHz, DMSO-d6) δ 11.69 - 11.43 (m, 1H), 10.95 - 10.62 (m, 1H), 7.87 - 7.79 (m, 1H), 6.97 (s, 1H), 6.77 (d, J=7.7 Hz, 1H), 6.59 (d, J=7.8 Hz, 1H), 5.66 (s, 2H), 5.16 (t, J=5.8 Hz, 1H), 4.45 (d, J=5.8 Hz, 2H), 3.79 (s, 3H), 1.49 (s, 9H).
LC RT: 0.77분. LC/MS [M+H]+ = 402.2 (방법 D)
단계 2. DMSO (5.7 mL) 중 tert-부틸 (7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (460 mg, 1.15 mmol)의 용액을 (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민·HCl (223 mg, 1.49 mmol), BOP (760 mg, 1.72 mmol) 및 DBU (0.69 mL, 4.6 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, EtOAc로 희석하고, H2O (2x)로 세척하였다. 유기 층을 셀라이트™ 상에 흡수시키고, 칼럼 크로마토그래피 (100g C18 골드 칼럼; 이동상 A: 5:95 아세토니트릴:물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴:물, 0.05% TFA 포함; 유량: 60 mL/분, 30-50% 구배)에 의해 정제하였다. 정제된 생성물을 DCM에 용해시키고, 포화 NaHCO3 수용액으로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 tert-부틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (190 mg, 33% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.24 - 9.15 (m, 1H), 7.87 (s, 1H), 7.72 (t, J=5.8 Hz, 1H), 6.95 (s, 1H), 6.82 - 6.75 (m, 1H), 6.73 - 6.68 (m, 1H), 5.68 (s, 2H), 5.17 (t, J=5.7 Hz, 1H), 4.87 (d, J=5.7 Hz, 2H), 4.44 (d, J=5.7 Hz, 2H), 3.76 (s, 3H), 2.55 (s, 3H), 1.43 (s, 9H).
LC RT: 0.75분. LC/MS [M+H]+ = 497.2 (방법 D)
단계 3. DCM (0.65 mL) 중 tert-부틸 (1-(4-(히드록시메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (161 mg, 0.320 mmol)의 용액을 SOCl2 (71 μL, 0.97 mmol)로 처리하였다. 반응 혼합물을 실온에서 15분 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 DCM 중에 용해시키고, 진공 하에 농축시켜 tert-부틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (166 mg, 100%)를 수득하였다.
LC RT: 0.89분. LC/MS [M+H]+ = 515.2 (방법 D)
단계 4. DMF (1.3 mL) 중 tert-부틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (33 mg, 0.064 mmol)의 용액을 DIEA (113 μL, 0.645 mmol) 및 3-메톡시아제티딘·HCl (23.9 mg, 0.193 mmol)로 처리하였다. 반응 혼합물을 70℃에서 1시간 동안 교반하고, N2 스트림 하에 건조시키고, 이어서 진공 하에 추가로 건조시켰다. 잔류물을 디옥산 (0.6 mL) 중에 용해시키고, HCl (디옥산 중 4 M, 0.82 mL, 3.3 mmol)로 처리하고, 40℃에서 30분 동안 교반하고, 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS를 통해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴:물, 10 mM NH4OAc 포함; 구배: 2% B에서 0-분 유지, 30분에 걸쳐 2-42% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켰다. 단리된 생성물을 추가로 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 0% B에서 0-분 유지, 25분에 걸쳐 0-30% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 118 (9.4 mg, 21%)을 수득하였다.
화합물 119를 유사하게 제조하였다.
실시예 13 - 화합물 127
단계 1. DMSO (2.5 mL) 중 tert-부틸 (7-히드록시-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (200 mg, 0.498 mmol)의 용액을 (5-시클로프로필-1,2,4-옥사디아졸-3-일)메탄아민·HCl (175 mg, 0.996 mmol), BOP (331 mg, 0.747 mmol) 및 DBU (0.30 mL, 2.0 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, EtOAc로 희석하고, H2O (2x)로 세척하였다. 유기 층을 진공 하에 농축시켰다. 조 생성물을 MeOH 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 HPLC에 의해 하기 조건으로 정제하였다: 칼럼: 악시아 C18 100 mm x 30 mm, 5-μm 입자; 이동상 A: 10:90 메탄올: 물, 0.1% TFA 포함; 이동상 B: 90:10 MeOH: 물, 0.1% TFA 포함; 구배: 40% B에서 0-분 유지, 10분에 걸쳐 40-55% B, 이어서 55% B에서 5-분 유지; 유량: 40 mL/분; 220 nm에서 UV 검출; 칼럼 온도: 25℃. 정제된 생성물을 포화 수성 NaHCO3 용액으로 중화시키고, DCM으로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 tert-부틸 (7-(((5-시클로프로필-1,2,4-옥사디아졸-3-일)메틸)아미노)-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (93.2 mg, 36% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.25 - 9.17 (m, 1H), 7.88 (s, 1H), 7.71 (t, J=5.7 Hz, 1H), 6.96 (s, 1H), 6.84 - 6.76 (m, 1H), 6.75 - 6.67 (m, 1H), 5.70 - 5.67 (m, 2H), 5.17 (t, J=5.7 Hz, 1H), 4.84 (d, J=4.6 Hz, 2H), 4.45 (d, J=5.8 Hz, 2H), 3.77 (s, 3H), 2.35 - 2.27 (m, 1H), 1.44 (s, 9H), 1.25 - 1.20 (m, 2H), 1.08 - 1.03 (m, 2H).
LC RT: 0.77분. LC/MS [M+H]+ = 523.4 (방법 D)
단계 2. DCM (3.6 mL) 중 tert-부틸 (7-(((5-시클로프로필-1,2,4-옥사디아졸-3-일)메틸)아미노)-1-(4-(히드록시메틸)-2-메톡시벤질)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (93.2 mg, 0.178 mmol)의 용액을 SOCl2 (39 μL, 0.54 mmol)로 처리하였다. 반응 혼합물을 실온에서 10분 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 DCM 중에 용해시키고, 진공 하에 농축시켜 tert-부틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-시클로프로필-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (95.4 mg, 99% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 11.70 - 11.19 (m, 1H), 9.46 - 9.20 (m, 1H), 8.10 - 8.06 (m, 1H), 7.10 (s, 1H), 6.97 (s, 2H), 5.79 (s, 2H), 4.97 (br d, J=5.2 Hz, 2H), 4.73 (s, 2H), 3.74 (s, 3H), 2.40 - 2.30 (m, 1H), 1.53 (s, 9H), 1.30 - 1.22 (m, 2H), 1.10 - 1.04 (m, 2H).
LC RT: 0.89분. LC/MS [M+H]+ = 541.3 (방법 D).
단계 3. DMF (1.1 mL) 중 tert-부틸 (1-(4-(클로로메틸)-2-메톡시벤질)-7-(((5-시클로프로필-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (30 mg, 0.055 mmol)의 용액을 DIEA (77 μL, 0.44 mmol) 및 테트라히드로-2H-피란-4-아민 (22.4 mg, 0.222 mmol)으로 처리하였다. 반응 혼합물을 60℃에서 1시간 동안 교반한 후, 온도를 65℃로 상승시키고, 교반을 1시간 동안 계속하였다. 반응 혼합물을 N2 스트림 하에 건조시키고, 이어서 진공 하에 추가로 건조시켰다. 잔류물을 디옥산 (1.1 mL) 중에 용해시키고, HCl (디옥산 중 4 M, 0.75 mL, 3 mmol)로 처리하고, 40℃에서 90분 동안 교반하고, 진공 하에 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 2% B에서 0-분 유지, 30분에 걸쳐 2-42% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켰다. 단리된 생성물을 추가로 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 5% B에서 0-분 유지, 20분에 걸쳐 5-70% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 127 (13.6 mg, 47%)을 수득하였다. 분석 데이터에 대해서는 표 A를 참조한다.
화합물 128 및 화합물 129를 유사하게 제조하였다.
실시예 14 - 화합물 130
단계 1. CCl4 (19 mL) 중 에틸 5-메톡시-6-메틸니코티네이트 (1.32 g, 6.77 mmol)의 용액을 NBS (1.44 g, 8.12 mmol) 및 AIBN (0.22 g, 1.4 mmol)으로 처리하였다. 반응 혼합물을 60℃에서 40시간 동안 교반하고, 포화 수성 Na2S2O3 용액으로 세척하였다. 유기 층을 진공 하에 농축시키고, 조 생성물을 칼럼 크로마토그래피 (40g SiO2; 0에서 25% EtOAc-헥산 구배 용리)에 의해 정제하여 에틸 6-(브로모메틸)-5-메톡시니코티네이트 (1.20 g, 4.38 mmol, 65% 수율)를 수득하였다.
1H NMR (400 MHz, 클로로포름-d) δ 8.83 - 8.75 (m, 1H), 7.78 (d, J=1.6 Hz, 1H), 4.65 (s, 2H), 4.43 (q, J=7.1 Hz, 2H), 3.99 (s, 3H), 1.43 (t, J=7.2 Hz, 3H). LC RT: 0.89분.
LC/MS [M+H]+ = 274.1 (방법 D).
단계 2. DMF (50 mL) 중 메틸 (7-히드록시-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (2.51 g, 12.0 mmol)의 용액을 NBS (2.14 g, 12.0 mmol)로 처리하였다. 반응 혼합물을 실온에서 15분 동안 교반하고, 여과하였다. 수집된 고체를 H2O 및 디에틸 에테르로 세척하여 메틸 (3-브로모-7-히드록시-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (3.28 g, 95% 수율)를 수득하였다.
LC RT: 0.57분. LC/MS [M+H]+ = 288.1 (방법 D).
단계 3. DMF (22.5 mL) 중 메틸 (3-브로모-7-히드록시-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (648 mg, 2.25 mmol)의 용액을 에틸 6-(브로모메틸)-5-메톡시니코티네이트 (617 mg, 2.25 mmol) 및 Cs2CO3 (2199 mg, 6.75 mmol)로 처리하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, EtOAc로 희석하고, 포화 NaHCO3 용액 및 H2O로 세척하였다. 유기 층을 진공 하에 농축시켰다. 조 생성물을 칼럼 크로마토그래피 (40g SiO2; 0에서 100% EtOAc-헥산 구배 용리)에 의해 정제하여 에틸 6-((3-브로모-7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-5-메톡시니코티네이트 (653.1 mg, 60% 수율)를 수득하였다.
1H NMR (500 MHz, DMSO-d6) δ 11.61 - 11.41 (m, 1H), 8.49 - 8.47 (m, 1H), 7.81 (d, J=1.6 Hz, 1H), 5.85 (s, 2H), 4.34 (q, J=7.1 Hz, 2H), 3.96 (s, 3H), 3.74 (s, 3H), 1.31 (t, J=7.1 Hz, 3H).
LC RT: 0.86분. LC/MS [M+H]+ =481.2 (방법 D).
단계 4. MeOH (54 mL) 중 에틸 6-((3-브로모-7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-5-메톡시니코티네이트 (542 mg, 1.13 mmol)의 현탁액을 Pd/C (24 mg, 0.23 mmol)로 처리하였다. 반응 플라스크를 진공 하에 배기시키고, H2 (3x)로 퍼징하였다. 반응 혼합물을 H2 분위기 (풍선) 하에 16시간 동안 교반하였다. 반응 플라스크를 진공 하에 배기시키고, N2 (3x)로 퍼징하였다. 반응 혼합물을 DCM으로 희석하고, 셀라이트™을 통해 여과하고, 진공 하에 농축시켜 에틸 6-((7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-5-메톡시니코티네이트 (450 mg, 99% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 8.49 - 8.44 (m, 1H), 7.85 (s, 1H), 7.79 (d, J=1.6 Hz, 1H), 5.86 (s, 2H), 4.33 (q, J=7.1 Hz, 2H), 3.95 (s, 3H), 3.75 (s, 3H), 1.31 (t, J=7.1 Hz, 3H).
LC RT: 0.78분. LC/MS [M+H]+ = 403.0 (방법 D)
단계 5. THF (28 mL) 중 에틸 6-((7-히드록시-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-5-메톡시니코티네이트 (543 mg, 1.35 mmol)의 용액을 0℃로 냉각시키고, LiAlH4 (THF 중 1 M, 2.4 mL, 2.4 mmol)로 처리하였다. 반응 혼합물을 0℃에서 15분 동안 교반하고, H2O 및 로쉘 염 (포화 수용액)으로 켄칭하고, 실온에서 2시간 동안 교반하였다. 유기 층을 셀라이트™ 상에 흡수시키고, 칼럼 크로마토그래피 (40g SiO2; 0에서 10% MeOH-DCM 구배 용리)에 의해 정제하여 메틸 (7-히드록시-1-((5-(히드록시메틸)-3-메톡시피리딘-2-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (191 mg, 39% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 7.89 - 7.84 (m, 1H), 7.80 (s, 1H), 7.37 (d, J=1.5 Hz, 1H), 5.80 - 5.72 (m, 2H), 5.28 (t, J=5.7 Hz, 1H), 4.48 (d, J=5.4 Hz, 2H), 3.87 - 3.81 (m, 3H), 3.74 (s, 3H).
LC RT: 0.56분. LC/MS [M+H]+ = 361.0 (방법 D).
단계 6. DMSO (2.6 mL) 중 메틸 (7-히드록시-1-((5-(히드록시메틸)-3-메톡시피리딘-2-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (190 mg, 0.527 mmol)의 용액을 (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민·HCl (103 mg, 0.685 mmol), BOP (303 mg, 0.685 mmol) 및 DBU (0.28 mL, 1.8 mmol)로 처리하였다. 반응 혼합물을 실온에서 1시간 동안 교반하고, DCM으로 희석하고, H2O (6x)로 세척하였다. 유기 층을 진공 하에 농축시켰다. 조 생성물을 MeOH 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 HPLC에 의해 하기 조건으로 정제하였다: 칼럼: 악시아 C18 100 mm x 30 mm, 5-μm 입자; 이동상 A: 10:90 메탄올: 물, 0.1% TFA 포함; 이동상 B: 90:10 메탄올: 물, 0.1% TFA 포함; 구배: 5% B에서 0-분 유지, 10분에 걸쳐 5-30% B, 이어서 30% B에서 2-분 유지; 유량: 40 mL/분; 220 nm에서 UV 검출; 칼럼 온도: 25℃. 정제된 생성물을 포화 수성 NaHCO3 용액으로 중화시키고, DCM으로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 진공 하에 농축시켜 메틸 (1-((5-(히드록시메틸)-3-메톡시피리딘-2-일)메틸)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (102.4 mg, 43% 수율)를 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ 9.68 (s, 1H), 8.99 (br s, 1H), 7.98 - 7.92 (m, 1H), 7.84 (s, 1H), 7.45 (d, J=1.1 Hz, 1H), 5.77 (s, 2H), 5.35 (br s, 1H), 4.92 (br s, 2H), 4.51 (br s, 2H), 3.88 (s, 3H), 3.61 (s, 3H), 2.57 (s, 3H).
LC RT: 0.61분. LC/MS [M+H]+ = 456.1 (방법 D).
단계 7. DCM (4.5 mL) 중 메틸 (1-((5-(히드록시메틸)-3-메톡시피리딘-2-일)메틸)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (102 mg, 0.225 mmol)의 용액을 SOCl2 (49 μL, 0.68 mmol)로 처리하였다. 반응 혼합물을 실온에서 30분 동안 교반하고, 진공 하에 농축시켰다. 잔류물을 DCM 중에 용해시키고, 진공 하에 농축시켜 메틸 (1-((5-(클로로메틸)-3-메톡시피리딘-2-일)메틸)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (107 mg, 100% 수율)를 수득하였다.
LC RT: 0.67분. LC/MS [M+H]+ = 474.3 (방법 D).
단계 8. DMF (0.7 mL) 중 메틸 (1-((5-(클로로메틸)-3-메톡시피리딘-2-일)메틸)-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (35 mg, 0.074 mmol)의 용액을 DIEA (103 μL, 0.591 mmol) 및 테트라히드로-2H-피란-4-아민 (29.9 mg, 0.295 mmol)으로 처리하였다. 반응 혼합물을 70℃에서 2시간 동안 교반하고, N2 스트림 하에 건조시키고, 이어서 진공 하에 추가로 건조시켰다. 잔류물을 디옥산 (0.8 mL) 중에 용해시키고, NaOH (10M 수용액, 37 μL, 0.37 mmol)로 처리하였다. 반응 혼합물을 60℃로 가열하였다. 추가의 NaOH (10M 수용액, 120 μL, 1.2 mmol)를 반응 혼합물에 8시간의 기간에 걸쳐 첨가하였다. 반응 혼합물을 실온에서 HOAc로 중화시키고, 진공 하에 농축시켰다. 조 생성물을 DMF 중에 용해시키고, PTFE 프릿을 통해 여과하고, 정제용 LC/MS에 의해 하기 조건으로 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 10 mM NH4OAc 포함; 이동상 B: 95:5 아세토니트릴: 물, 10 mM NH4OAc 포함; 구배: 1% B에서 0-분 유지, 20분에 걸쳐 1-41% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 촉발하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켰다. 단리된 생성물을 추가로 정제용 LC/MS를 통해 하기 조건을 사용하여 정제하였다: 칼럼: 엑스브리지 C18, 200 mm x 19 mm, 5-μm 입자; 이동상 A: 5:95 아세토니트릴: 물, 0.05% TFA 포함; 이동상 B: 95:5 아세토니트릴: 물, 0.05% TFA 포함; 구배: 0% B에서 0-분 유지, 25분에 걸쳐 0-40% B, 이어서 100% B에서 0-분 유지; 유량: 20 mL/분; 칼럼 온도: 25℃. 분획 수집을 MS 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 원심 증발을 통해 건조시켜 화합물 130을 비스 TFA 염 (11 mg, 20%)으로서 수득하였다.
화합물 131을 유사하게 제조하였다.
실시예 15 - 화합물 134
단계 1. 0℃에서 DMF (50.0 mL) 중 메틸 (7-히드록시-3-아이오도-1H-피라졸로[4,3-d]피리미딘-5-일)카르바메이트 (5.0 g, 14.92 mmol)의 교반 용액에 Cs2CO3 (9.72 g, 29.8 mmol) 및 메틸 4-(브로모메틸)-3-메톡시벤조에이트 (3.87 g, 14.92 mmol)를 첨가하였다. 반응 혼합물을 0℃에서 1시간 동안 교반하고, 물을 첨가하였다. 침전된 고체를 여과하고, 과량의 물에 이어서 석유 에테르로 세척하였다. 고체를 진공 하에 건조시켰다. 조 화합물을 이스코 콤비플래쉬 크로마토그래피에 의해 클로로포름 중 0-100% 에틸 아세테이트로 용리시켜 정제하여 메틸 4-((7-히드록시-3-아이오도-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (3.88 g, 6.20 mmol, 41.5% 수율)를 회백색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ ppm: 11.69 (br s, 1H), 11.38 (s, 1H), 7.56 - 7.45 (m, 2H), 6.87 - 6.78 (m, 1H), 5.75 (s, 2H), 3.88 (s, 3H), 3.85 (s, 3H), 3.75 (s, 3H).
LC-MS m/z 514.0 [M+H]+.
단계 2. 1,4-디옥산 (35.0 mL) 중 메틸 4-((7-히드록시-3-아이오도-5-((메톡시카르보닐)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (3.5 g, 6.82 mmol)의 교반 용액에 K2CO3 (1.885 g, 13.64 mmol), TMB (1.907 mL, 13.64 mmol) 및 PdCl2(dppf).CH2Cl2 부가물 (0.557 g, 0.682 mmol)을 N2 퍼징 하에 첨가하였다. 반응 혼합물을 100℃에서 6시간 동안 교반하였다. 반응 혼합물을 셀라이트™ 층을 통해 여과하고, 과량의 에틸 아세테이트로 세척하였다. 여과물을 감압 하에 농축시켜 잔류물을 수득하였다. 조 화합물을 이스코 콤비플래쉬 크로마토그래피 (클로로포름 중 0-20% 메탄올)에 의해 정제하여 메틸 4-((5-아미노-7-히드록시-3-메틸-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (2.1 g, 4.10 mmol, 60.1% 수율)를 갈색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ = 10.90 (s, 1H), 7.51 (s, 1H), 7.46 (d, J = 8.0 Hz, 1H), 6.63 - 6.50 (m, 1H), 6.18 - 6.01 (m, 2H), 5.71 - 5.54 (m, 2H), 3.91 (s, 3H), 3.87 - 3.78 (s, 3H), 2.23 (s, 3H).
LC-MS m/z 344.0 [M+H]+.
단계 3. 0℃에서 THF (5.0 mL) 중 메틸 4-((5-아미노-7-히드록시-3-메틸-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시벤조에이트 (0.5 g, 1.456 mmol)의 교반 용액에 LiAlH4 (1.214 mL, 2.91 mmol)를 첨가하였다. 반응 혼합물을 실온으로 가온하고, 1시간 동안 교반하고, 빙냉수로 켄칭하고, 셀라이트™ 층을 통해 여과하고, 이를 과량의 에틸 아세테이트로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 5-아미노-1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (0.31 g, 0.551 mmol, 37.8% 수율)을 갈색 반고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ = 6.99 - 6.95 (m, 1H), 6.73 (br d, J = 7.5 Hz, 1H), 6.44 - 6.38 (m, 1H), 5.75 - 5.49 (m, 2H), 5.26 - 4.99 (m, 1H), 4.44 (s, 2H), 3.87 - 3.80 (m, 3H), 2.23 (s, 3H).
LC-MS m/z 316.3 [M+H]+.
단계 4. DMSO (10.0 mL) 중 5-아미노-1-(4-(히드록시메틸)-2-메톡시벤질)-3-메틸-1H-피라졸로[4,3-d]피리미딘-7-올 (1.1 g, 3.49 mmol)의 교반 용액에 DBU (1.577 mL, 10.47 mmol), BOP (2.314 g, 5.23 mmol) 및 (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민 히드로클로라이드 (0.522 g, 3.49 mmol)를 첨가하였다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. (5-메틸-1,2,4-옥사디아졸-3-일)메탄아민, HCl (0.3 g, 2.0 mmol)을 첨가하였다. 반응 혼합물을 실온에서 16시간 동안 교반하고, EtOAc와 물 사이에 분배하였다. 유기 층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 잔류물을 수득하였다. 조 화합물을 이스코 콤비플래쉬 크로마토그래피에 의해 클로로포름 중 0-20% 메탄올로 용리시켜 정제하여 (4-((5-아미노-3-메틸-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시페닐)메탄올 (0.81 g, 1.243 mmol, 35.6% 수율)을 갈색 고체로서 수득하였다.
1H NMR (400 MHz, DMSO-d6) δ = 7.60 - 7.55 (m, 1H), 7.26 (br t, J = 5.8 Hz, 1H), 6.98 - 6.93 (m, 1H), 6.77 (br d, J = 7.5 Hz, 1H), 6.68 - 6.60 (m, 1H), 5.68 (s, 2H), 5.55 - 5.48 (m, 1H), 5.20 - 5.13 (m, 1H), 4.78 (br d, J = 5.5 Hz, 2H), 4.49 - 4.42 (m, 2H), 3.82 - 3.77 (m, 3H), 2.56 (d, J = 2.0 Hz, 4H), 2.55 - 2.50 (m, 6H).
LC-MS m/z 411.2 [M+H]+.
단계 5. 0℃에서 THF (10.0 mL) 중 (4-((5-아미노-3-메틸-7-(((5-메틸-1,2,4-옥사디아졸-3-일)메틸)아미노)-1H-피라졸로[4,3-d]피리미딘-1-일)메틸)-3-메톡시페닐)메탄올 (0.45 g, 1.096 mmol)의 교반 용액에 SOCl2 (1.0 ml, 13.70 mmol)를 첨가하였다. 반응 혼합물을 0℃에서 1시간 동안 교반하고, 실온으로 가온하고, 감압 하에 농축시켜 조 1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-N7-((5-메틸-1,2,4-옥사디아졸-3-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5,7-디아민 (0.51 g, 100% 수율로 추정됨)을 갈색 고체로서 수득하였으며, 이를 후속 단계에 그대로 사용하였다.
LC-MS m/z 429.4 [M+H]+.
단계 6. DMF (3.0 mL) 중 1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-N7-((5-메틸-1,2,4-옥사디아졸-3-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5,7-디아민 (0.15 g, 0.350 mmol)의 교반 용액에 1-메틸피페라진 (0.053 g, 0.525 mmol) 및 K2CO3 (0.145 g, 1.049 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 90분 동안 교반하고, 셀라이트™ 층을 통해 여과하고, 이를 과량의 에틸 아세테이트로 세척하였다. 여과물을 감압 하에 농축시켜 잔류물을 수득하였다. 조 화합물을 역상 정제용 LC/MS (칼럼: 트리아트-YMC-EXRS (250 mm x 19 mm); 이동상 A: 물 중 10 mM NH4OAc pH-4.5, 이동상 B: CH3CN; 유량: 20 mL/분; 구배: 0/0, 10/15, 20/15, 22/100, 24/0)에 의해 정제하였다. 분획 수집을 MS 및 UV 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 진백 장치를 사용하여 원심 증발을 통해 건조시켜 화합물 134 (12.6 mg, 0.025 mmol, 7.15% 수율)를 수득하였다.
실시예 16 - 화합물 132
DMF (3.0 mL) 중 1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-N7-((5-메틸-1,2,4-옥사디아졸-3-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5,7-디아민 (0.15 g, 0.350 mmol)의 교반 용액에 2-(피페라진-1-일)에탄-1-올 (0.068 g, 0.525 mmol), 2-(피페라진-1-일)에탄-1-올 (0.068 g, 0.525 mmol) 및 K2CO3 (0.097 g, 0.699 mmol)을 첨가하였다. 반응 혼합물을 50℃에서 90분 동안 교반하고, 셀라이트™ 층을 통해 여과하고, 이를 과량의 에틸 아세테이트로 세척하였다. 여과물을 감압 하에 농축시켜 잔류물을 수득하였으며, 이를 역상 정제용 LC/MS (칼럼: 제미니 NX (250 x 21.2 mm) 5 μm, 이동상 A: 물 중 10 mM 중탄산암모늄 9.5 pH, 이동상 B: CH3CN, 유량: 20 mL/분, 구배 T/%B: 0/10, 7/35, 12/35, 12.01/100)에 의해 정제하였다. 분획 수집을 MS 및 UV 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 진백 장치를 사용하여 원심 증발을 통해 건조시켜 화합물 132 (51.2 mg, 0.095 mmol, 27.2% 수율)를 수득하였다.
실시예 17 - 화합물 133
아세토니트릴 (3.0 mL) 중 1-(4-(클로로메틸)-2-메톡시벤질)-3-메틸-N7-((5-메틸-1,2,4-옥사디아졸-3-일)메틸)-1H-피라졸로[4,3-d]피리미딘-5,7-디아민 (0.15 g, 0.350 mmol)의 교반 용액에 테트라히드로-2H-피란-4-아민 히드로클로라이드 (0.072 g, 0.525 mmol), Na2CO3 (0.111 g, 1.049 mmol) 및 KI (0.058 g, 0.350 mmol)를 첨가하였다. 반응 혼합물을 50℃에서 3시간 동안 교반하였다. 반응 혼합물을 셀라이트™ 층을 통해 여과하고, 이를 과량의 에틸 아세테이트로 세척하였다. 여과물을 감압 하에 농축시켜 잔류물을 수득하였다. 조 화합물을 역상 정제용 LC/MS (칼럼: 제미니 NX (250 x 21 mm) x 5 마이크로미터; 이동상 A: 물 중 10 mM NH4OAc, 이동상 B: CH3CN:MeOH (1:1), 유량: 19 mL/분, 구배: 0/35, 12/45)에 의해 정제하였다. 분획 수집을 MS 및 UV 신호에 의해 개시하였다. 목적 생성물을 함유하는 분획을 합하고, 진백을 사용하여 원심 증발을 통해 건조시켜 화합물 133 (17.4 mg, 0.035 mmol, 10.08% 수율)을 수득하였다.
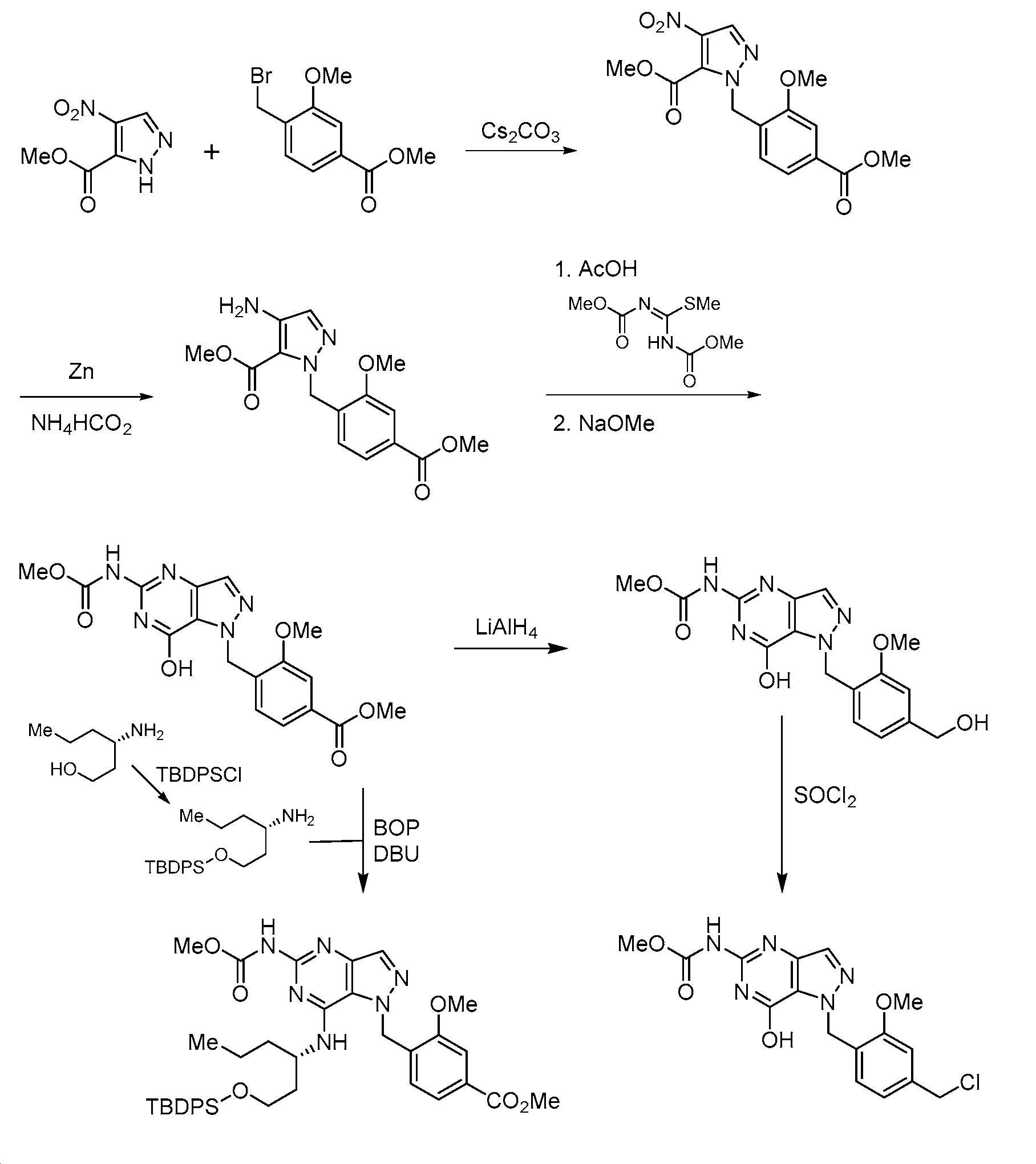
실시예 18 - 출발 물질 및 중간체
하기 차트는 본원에 개시된 TLR7 효능제의 제조를 위한 출발 물질 또는 중간체로서 유용할 수 있는 화합물을 제조하기 위한 반응식을 나타낸다. 반응식은 출발 물질 또는 중간체로서 사용될 수 있는 다른 유사한 화합물을 제조하기 위해 적합화될 수 있다. 사용된 시약은 관련 기술분야에 널리 공지되어 있고, 많은 경우에 그의 사용은 상기 실시예에서 입증되었다.
차트 1
차트 2
차트 3
생물학적 활성
TLR7 효능제로서 본원에 개시된 화합물의 생물학적 활성은 하기 절차에 의해 검정될 수 있다.
인간 TLR7 효능제 활성 검정
이 절차는 본 명세서에 개시된 화합물의 인간 TLR7 (hTLR7) 효능제 활성을 검정하는 방법을 기재한다.
인간 TLR7-분비 배아 알칼리성 포스파타제 (SEAP) 리포터 트랜스진을 보유하는 조작된 인간 배아 신장 블루 세포 (HEK-블루™ TLR 세포; 인비보젠(Invivogen))를 비-선택적인 배양 배지 (10% 소 태아 혈청 (시그마(Sigma))이 보충된 DMEM 고-글루코스 (인비트로겐(Invitrogen))) 중에 현탁시켰다. HEK-블루™ TLR7 세포를 384-웰 조직-배양 플레이트의 각 웰에 첨가하고 (웰 당 15,000개 세포), 16-18시간 동안 37℃, 5% CO2에서 인큐베이션하였다. 화합물 (100 nl)을 HEK-블루™ TLR 세포를 함유하는 웰에 분배하고, 처리된 세포를 37℃, 5% CO2에서 인큐베이션하였다. 18시간 처리 후, 10 마이크로리터의 새로 제조된 퀀티-블루™ 시약 (인비보젠)을 각각의 웰에 첨가하고, 30분 동안 인큐베이션하고 (37℃, 5% CO2), SEAP 수준을 엔비전 플레이트 판독기 (OD = 620 nm)를 사용하여 측정하였다. 반수 최대 유효 농도 값 (EC50; 검정 기준선과 최대값 사이의 반응 중간값을 유도하는 화합물 농도)을 계산하였다.
인간 혈액에서의 제I형 인터페론 유전자 (MX-1) 및 CD69의 유도
제I형 인터페론 (IFN) MX-1 유전자 및 B-세포 활성화 마커 CD69의 유도는 TLR7 경로의 활성화시 발생하는 하류 이벤트이다. 하기는 TLR7 효능제에 반응하는 그의 유도를 측정하는 인간 전혈 검정이다.
헤파린첨가 인간 전혈을 인간 대상체로부터 수거하고, 1mM의 시험 TLR7 효능제 화합물로 처리하였다. 혈액을 RPMI 1640 배지로 희석하고, 에코(Echo)를 사용하여 웰당 10 nL을 적가하여 최종 농도 1uM (혈액 10uL 중 10nL)를 수득하였다. 30초 동안 진탕기에서 혼합한 후, 플레이트를 덮고, 챔버에 37℃에서 o/n=17시간 동안 두었다. 고정/용해 완충제를 제조하고 (H2O 중 5x->1x, 37℃에서 가온; Cat# BD 558049), 나중에 사용하기 위해 투과화 완충제 (얼음 상)를 유지하였다.
표면 마커 염색 (CD69)의 경우: 표면 Abs: 0.045ul hCD14-FITC (써모피셔(ThermoFisher) Cat # MHCD1401) + 0.6ul hCD19-ef450 (써모피셔 Cat # 48-0198-42) + 1.5ul hCD69-PE (cat# BD555531) + 0.855ul FACS 완충제를 제조했다. 3ul/웰을 첨가하고, 1분 동안 1000 rpm 회전시키고, 진탕기에서 30초 동안 혼합하고, 얼음 상에 30분 동안 두었다. 30분 후에 70uL의 미리가온된 1x 고정/용해 완충제를 사용하여 자극을 중지시키고, 펠릭스 메이트를 사용하여 재현탁시키고 (15회, 각 플레이트에 대해 팁을 바꿈), 37℃에서 10분 동안 인큐베이션하였다.
2000 rpm에서 5분 동안 원심분리하고, HCS 플레이트 세척기로 흡인하고, 진탕기에서 30초 동안 혼합한 다음, 이어서 dPBS 70uL로 세척하고, 2xs (2000rpm, 5분 동안) 펠릿화하고, FACS 완충제 50ul로 세척하고, 1xs (2000rpm, 5분 동안) 펠릿화하였다. 진탕기에서 30초 동안 혼합하였다. 세포내 마커 염색 (MX-1)의 경우: 50ul의 BD 투과화 완충제 III을 첨가하고, 진탕기에서 30초 동안 혼합하였다. 얼음 상에서 30분 동안 (암소에서) 인큐베이션하였다. 50uL의 FACS 완충제 2X (투과화 후에 회전 @2300rpm x 5분)로 세척한 다음, 진탕기에서 30초 동안 혼합하였다. MX1 항체()(4812)-알렉사 647: 노부스 바이올로지칼스(Novus Biologicals) #NBP2-43704AF647) 20ul FACS bf + 0.8ul hIgG + 0.04ul MX-1을 함유하는 20ul의 FACS 완충제에 재현탁시켰다. 1000rpm로 1분 동안 회전시키고, 진탕기에서 30초 동안 혼합하고, 샘플을 암소에서 실온에서 45분 동안 인큐베이션한 다음, 이어서 2x FACS 완충제로 세척하였다 (투과 후에 회전 @2300rpm x 5분). 20ul (35uL, 웰 당 총계)의 FACS 완충제를 재현탁시키고, 호일로 덮고, 4℃에서 두어 다음날 판독하였다. 플레이트를 i큐플러스(iQuePlus)에서 판독하였다. 결과를 툴세트에 기록하고, 커브 마스터에서 IC50 곡선을 생성한다. y-축 100%를 1uM의 레시퀴모드로 설정한다.
마우스 혈액에서의 TNF-알파 및 제I형 IFN 반응 유전자의 유도
TNF-알파 및 제I형 IFN 반응 유전자의 유도는 TLR7 경로의 활성화시 발생하는 하류 이벤트이다. 하기는 TLR7 효능제에 반응하는 마우스 전혈에서의 그의 유도를 측정하는 검정이다.
헤파린첨가 마우스 전혈을 Pen-Strep을 함유한 RPMI 1640에 의해 5:4 (50 uL 전혈 및 배지 40 uL)의 비로 희석하였다. 희석된 혈액 90 uL의 부피를 팔콘(Falcon) 편평 바닥 96-웰 조직 배양 플레이트의 웰로 옮기고, 플레이트를 4℃에서 1시간 동안 인큐베이션하였다. 100% DMSO 스톡 중 시험 화합물을 농도 반응 검정 동안 동일한 배지에서 20배 희석한 다음, 이어서 희석된 시험 화합물 10 uL을 웰에 첨가하여 최종 DMSO 농도가 0.5%가 되었다. 대조군 웰은 5% DMSO를 함유하는 10 uL 배지를 수용하였다. 이어서, 플레이트를 5% CO2 인큐베이터에서 37℃에서 17시간 동안 인큐베이션하였다. 인큐베이션 후, 배양 배지 100 uL을 각 웰에 첨가하였다. 플레이트를 원심분리하고, 상청액 130 uL을 ELISA (인비트로젠, 카탈로그 번호 88-7324, 써모-피셔 사이언티픽(Thermo-Fisher Scientific)에 의함)에 의한 TNFa 생산의 검정에 사용하기 위해 제거하였다. 인비트로젠 mRNA 캐처 플러스 키트 (Cat#K1570-02)로부터의 DTT를 함유한 mRNA 캐처 용해 완충제 (1x) 70 uL 부피를 웰 내의 나머지 70 uL 샘플에 첨가하고, 5회 상하로 피펫팅하여 혼합하였다. 이어서, 플레이트를 실온에서 5 - 10분 동안 진탕한 다음, 이어서 프로테이나제 K 2 uL을 각 웰에 첨가하였다 (20 mg/mL). 이어서, 플레이트를 실온에서 15 - 20분 동안 진탕하였다. 이어서, 플레이트를 추가로 가공할 때까지 -80℃에서 저장하였다.
동결된 샘플을 해동시키고, mRNA를 인비트로젠 mRNA 캐처 플러스 키트 (Cat# K1570-02)를 사용하여 제조업체의 지침서에 따라 추출하였다. RNA 추출로부터의 절반 수율의 mRNA를 사용하여 인비트로젠 슈퍼스크립트 IV VILO 마스터 믹스 (Cat# 11756500)를 사용하여 20 μL 리버스 트랜스크립타제 반응물 중에 cDNA를 합성하였다. 택맨(TaqMan)® 실시간 PCR을 써모피셔 (어플라이드 바이오시스템즈(Applied Biosystems))로부터의 퀀트스튜디오 실시간 PCR 시스템을 사용하여 수행하였다. 모든 실시간 PCR 반응을 마우스 IFIT1, IFIT3, MX1 및 PPIA 유전자 발현에 대해 상업적으로 예비설계된 택맨 검정 및 택맨 마스터 믹스를 사용하여 이중으로 실행하였다. PPIA를 하우스키핑 유전자로서 사용하였다. 제조업체의 권장사항을 따랐다. 모든 미가공 데이터 (Ct)를 평균 하우스키핑 유전자 (Ct)에 의거해 정규화하고, 이어서 비교 Ct (ΔΔCt) 방법을 사용하여 실험적 분석에 대한 상대 유전자 발현을 정량화 (RQ)하였다.
정의
"지방족"은 명시된 수의 탄소 원자를 갖거나 (예를 들어, "C3 지방족", "C1-5 지방족", "C1-C5 지방족" 또는 "C1 내지 C5 지방족"에서와 같으며, 후자 3개의 어구는 1 내지 5개의 탄소 원자를 갖는 지방족 모이어티에 대한 동의어임) 또는 탄소 원자의 수가 명확하게 명시되지 않은 경우에는 1 내지 4개의 탄소 원자 (불포화 지방족 모이어티의 경우에는 2 내지 4개의 탄소)를 갖는 직쇄 또는 분지쇄, 포화 또는 불포화, 비-방향족 탄화수소 모이어티를 의미한다. 유사한 이해가 C2-4 알켄, C4-C7 시클로지방족 등에서와 같은 다른 유형에서의 탄소의 수에 적용된다. 유사한 맥락에서, "(CH2)1-3"과 같은 용어는 이러한 용어가 CH2, CH2CH2, 및 CH2CH2CH2를 나타내도록, 아래첨자가 1, 2, 또는 3인 것에 대한 약칭으로서 이해되어야 한다.
"알킬"은 포화 지방족 모이어티를 의미하며, 여기서 탄소 원자의 수의 지정에 대한 동일한 규정이 적용가능하다. 예시로서, C1-C4 알킬 모이어티는 메틸, 에틸, 프로필, 이소프로필, 이소부틸, t-부틸, 1-부틸, 2-부틸 등을 포함하나 이에 제한되지는 않는다. "알칸디일" (때때로 "알킬렌"으로도 지칭됨)은 알킬 기의 2가 대응물, 예컨대
를 의미한다.
"알케닐"은 적어도 1개의 탄소-탄소 이중 결합을 갖는 지방족 모이어티를 의미하며, 여기서 탄소 원자의 수의 지정에 대한 동일한 규정이 적용가능하다. 예시로서, C2-C4 알케닐 모이어티는 에테닐 (비닐), 2-프로페닐 (알릴 또는 프로프-2-에닐), 시스-1-프로페닐, 트랜스-1-프로페닐, E- (또는 Z-) 2-부테닐, 3-부테닐, 1,3-부타디에닐 (부트-1,3-디에닐) 등을 포함하나 이에 제한되지는 않는다.
"알키닐"은 적어도 1개의 탄소-탄소 삼중 결합을 갖는 지방족 모이어티를 의미하며, 여기서 탄소 원자의 수의 지정에 대한 동일한 규정이 적용가능하다. 예시로서, C2-C4 알키닐 기는 에티닐 (아세틸레닐), 프로파르길 (프로프-2-이닐), 1-프로피닐, 부트-2-이닐 등을 포함한다.
"시클로지방족"은 1 내지 3개의 고리를 가지며, 각각의 고리가 3 내지 8개 (바람직하게는 3 내지 6개)의 탄소 원자를 갖는, 포화 또는 불포화, 비-방향족 탄화수소 모이어티를 의미한다. "시클로알킬"은 각각의 고리가 포화된 시클로지방족 모이어티를 의미한다. "시클로알케닐"은 적어도 1개의 고리가 적어도 1개의 탄소-탄소 이중 결합을 갖는 시클로지방족 모이어티를 의미한다. "시클로알키닐"은 적어도 1개의 고리가 적어도 1개의 탄소-탄소 삼중 결합을 갖는 시클로지방족 모이어티를 의미한다. 예시로서, 시클로지방족 모이어티는 시클로프로필, 시클로부틸, 시클로펜틸, 시클로펜테닐, 시클로헥실, 시클로헥세닐, 시클로헵틸, 시클로옥틸 및 아다만틸을 포함하나 이에 제한되지는 않는다. 바람직한 시클로지방족 모이어티는 시클로알킬 모이어티, 특히 시클로프로필, 시클로부틸, 시클로펜틸 및 시클로헥실이다. "시클로알칸디일" (때때로 "시클로알킬렌"으로도 지칭됨)은 시클로알킬 기의 2가 대응물을 의미한다. 유사하게, "비시클로알칸디일" (또는 "비시클로알킬렌") 및 "스피로시클로알칸디일" (또는 "스피로알킬렌")은 비시클로알킬 및 스피로알킬 (또는 "스피로시클로알킬") 기의 2가 대응물을 지칭한다.
"헤테로시클로지방족"은 그의 적어도 1개의 고리 내에서, 3개 이하 (바람직하게는 1 내지 2개)의 탄소가 N, O, 또는 S로부터 독립적으로 선택된 헤테로원자로 대체된 것인 시클로지방족 모이어티를 의미하며, 여기서 N 및 S는 임의로 산화될 수 있고, N은 임의로 4급화될 수 있다. 바람직한 시클로지방족 모이어티는 5- 내지 6-원 크기인 1개의 고리로 이루어진다. 유사하게, "헤테로시클로알킬", "헤테로시클로알케닐", 및 "헤테로시클로알키닐"은 그의 적어도 1개의 고리가 이렇게 하여 변형된, 각각 시클로알킬, 시클로알케닐, 또는 시클로알키닐 모이어티를 의미한다. 예시적인 헤테로시클로지방족 모이어티는 아지리디닐, 아제티디닐, 1,3-디옥사닐, 옥세타닐, 테트라히드로푸릴, 피롤리디닐, 피페리디닐, 피페라지닐, 테트라히드로피라닐, 테트라히드로티오피라닐, 테트라히드로티오피라닐 술폰, 모르폴리닐, 티오모르폴리닐, 티오모르폴리닐 술폭시드, 티오모르폴리닐 술폰, 1,3-디옥솔라닐, 테트라히드로-1,1-디옥소티에닐, 1,4-디옥사닐, 티에타닐 등을 포함한다. "헤테로시클로알킬렌"은 헤테로시클로알킬 기의 2가 대응물을 의미한다.
"알콕시", "아릴옥시", "알킬티오", 및 "아릴티오"는 각각 -O(알킬), -O(아릴), -S(알킬), 및 -S(아릴)을 의미한다. 예는 각각 메톡시, 페녹시, 메틸티오 및 페닐티오이다.
보다 좁은 의미가 지정되지 않는 한, "할로겐" 또는 "할로"는 플루오린, 염소, 브로민 또는 아이오딘을 의미한다.
"아릴"은 각각의 고리가 3 내지 7개의 탄소 원자를 갖고 적어도 1개의 고리가 방향족인 모노-, 비-, 또는 트리시클릭 고리계 (바람직하게는 모노시클릭)를 갖는 탄화수소 모이어티를 의미한다. 고리계 내의 고리는 서로 융합될 수 있거나 (나프틸에서와 같음) 또는 서로 결합될 수 있으며 (비페닐에서와 같음), 비-방향족 고리에 융합 또는 결합될 수 있다 (인다닐 또는 시클로헥실페닐에서와 같음). 추가 예시로서, 아릴 모이어티는 페닐, 나프틸, 테트라히드로나프틸, 인다닐, 비페닐, 페난트릴, 안트라세닐 및 아세나프틸을 포함하나 이에 제한되지는 않는다. "아릴렌"은 아릴 기의 2가 대응물, 예를 들어 1,2-페닐렌, 1,3-페닐렌, 또는 1,4-페닐렌을 의미한다.
"헤테로아릴"은 각각의 고리가 3 내지 7개의 탄소 원자를 갖고 적어도 1개의 고리가 N, O, 또는 S로부터 독립적으로 선택된 1 내지 4개의 헤테로원자를 함유하는 방향족 고리인 모노-, 비-, 또는 트리시클릭 고리계 (바람직하게는 5 내지 7-원 모노시클릭)를 갖는 모이어티를 의미하며, 여기서 N 및 S는 임의로 산화될 수 있고, N은 임의로 4급화될 수 있다. 이러한 적어도 1개의 헤테로원자 함유 방향족 고리는 다른 유형의 고리에 융합될 수 있거나 (벤조푸라닐 또는 테트라히드로이소퀴놀릴에서와 같음) 또는 다른 유형의 고리에 직접 결합될 수 있다 (페닐피리딜 또는 2-시클로펜틸피리딜에서와 같음). 추가 예시로서, 헤테로아릴 모이어티는 피롤릴, 푸라닐, 티오페닐 (티에닐), 이미다졸릴, 피라졸릴, 옥사졸릴, 이속사졸릴, 티아졸릴, 이소티아졸릴, 트리아졸릴, 테트라졸릴, 피리딜, N-옥소피리딜, 피리다지닐, 피리미디닐, 피라지닐, 퀴놀리닐, 이소퀴놀리닐, 퀴나졸리닐, 신놀리닐, 퀴노잘리닐, 나프티리디닐, 벤조푸라닐, 인돌릴, 벤조티오페닐, 옥사디아졸릴, 티아디아졸릴, 페노티아졸릴, 벤즈이미다졸릴, 벤조트리아졸릴, 디벤조푸라닐, 카르바졸릴, 디벤조티오페닐, 아크리디닐 등을 포함한다. "헤테로아릴렌"은 헤테로아릴 기의 2가 대응물을 의미한다.
"비치환되거나 또는 치환된 C1-C5 알킬" 또는 "임의로 치환된 헤테로아릴"에서와 같은 "비치환되거나 또는 치환된" 또는 "임의로 치환된" 어구의 사용에 의해서와 같이 모이어티가 치환될 수 있는 것으로 나타낸 경우에, 이러한 모이어티는 1개 이상의 독립적으로 선택된 치환기, 바람직하게는 수에 있어서 1 내지 5개, 보다 바람직하게는 수에 있어서 1 또는 2개를 가질 수 있다. 치환기 및 치환 패턴은 관련 기술분야의 통상의 기술자에 의해, 치환기가 부착되는 모이어티를 고려하여, 화학적으로 안정하고 관련 기술분야에 공지된 기술뿐만 아니라 본원에 제시된 방법에 의해 합성될 수 있는 화합물을 제공하도록 선택될 수 있다. 모이어티가 "비치환되거나 또는 치환된" 또는 "임의로 치환된" 것으로 확인되는 경우에, 바람직한 실시양태에서 이러한 모이어티는 비치환된다.
"아릴알킬", "(헤테로시클로지방족)알킬", "아릴알케닐", "아릴알키닐", "비아릴알킬" 등은, 예를 들어 벤질, 페네틸, N-이미다조일에틸, N-모르폴리노에틸 등에서와 같이, 경우에 따라 알킬, 알케닐, 또는 알키닐 모이어티에서 개방 (비충족) 원자가를 갖는, 경우에 따라 아릴, 헤테로시클로지방족, 비아릴 등의 모이어티로 치환된 알킬, 알케닐, 또는 알키닐 모이어티를 의미한다. 반대로, "알킬아릴", "알케닐시클로알킬" 등은, 경우에 따라 예를 들어 메틸페닐 (톨릴) 또는 알릴시클로헥실에서와 같이, 경우에 따라 알킬, 알케닐 등의 모이어티로 치환된 아릴, 시클로알킬 등의 모이어티를 의미한다. "히드록시알킬", "할로알킬", "알킬아릴", "시아노아릴" 등은, 경우에 따라 확인된 치환기 (경우에 따라 히드록실, 할로 등) 중 1개 이상으로 치환된 알킬, 아릴 등의 모이어티를 의미한다.
예를 들어, 허용되는 치환기는 알킬 (특히 메틸 또는 에틸), 알케닐 (특히 알릴), 알키닐, 아릴, 헤테로아릴, 시클로지방족, 헤테로시클로지방족, 할로 (특히 플루오로), 할로알킬 (특히 트리플루오로메틸), 히드록실, 히드록시알킬 (특히 히드록시에틸), 시아노, 니트로, 알콕시, -O(히드록시알킬), -O(할로알킬) (특히 -OCF3), -O(시클로알킬), -O(헤테로시클로알킬), -O(아릴), 알킬티오, 아릴티오, =O, =NH, =N(알킬), =NOH, =NO(알킬), -C(=O)(알킬), -C(=O)H, -CO2H, -C(=O)NHOH, -C(=O)O(알킬), -C(=O)O(히드록시알킬), -C(=O)NH2, -C(=O)NH(알킬), -C(=O)N(알킬)2, -OC(=O)(알킬), -OC(=O)(히드록시알킬), -OC(=O)O(알킬), -OC(=O)O(히드록시알킬), -OC(=O)NH2, -OC(=O)NH(알킬), -OC(=O)N(알킬)2, 아지도, -NH2, -NH(알킬), -N(알킬)2, -NH(아릴), -NH(히드록시알킬), -NHC(=O)(알킬), -NHC(=O)H, -NHC(=O)NH2, -NHC(=O)NH(알킬), -NHC(=O)N(알킬)2, -NHC(=NH)NH2, -OSO2(알킬), -SH, -S(알킬), -S(아릴), -S(시클로알킬), -S(=O)알킬, -SO2(알킬), -SO2NH2, -SO2NH(알킬), -SO2N(알킬)2 등을 포함하나 이에 제한되지는 않는다.
치환될 모이어티가 지방족 모이어티인 경우에, 바람직한 치환기는 아릴, 헤테로아릴, 시클로지방족, 헤테로시클로지방족, 할로, 히드록실, 시아노, 니트로, 알콕시, -O(히드록시알킬), -O(할로알킬), -O(시클로알킬), -O(헤테로시클로알킬), -O(아릴), 알킬티오, 아릴티오, =O, =NH, =N(알킬), =NOH, =NO(알킬), -CO2H, -C(=O)NHOH, -C(=O)O(알킬), -C(=O)O(히드록시알킬), -C(=O)NH2, -C(=O)NH(알킬), -C(=O)N(알킬)2, -OC(=O)(알킬), -OC(=O)(히드록시알킬), -OC(=O)O(알킬), -OC(=O)O(히드록시알킬), -OC(=O)NH2, -OC(=O)NH(알킬), -OC(=O)N(알킬)2, 아지도, -NH2, -NH(알킬), -N(알킬)2, -NH(아릴), -NH(히드록시알킬), -NHC(=O)(알킬), -NHC(=O)H, -NHC(=O)NH2, -NHC(=O)NH(알킬), -NHC(=O)N(알킬)2, -NHC(=NH)NH2, -OSO2(알킬), -SH, -S(알킬), -S(아릴), -S(=O)알킬, -S(시클로알킬), -SO2(알킬), -SO2NH2, -SO2NH(알킬), 및 -SO2N(알킬)2이다. 보다 바람직한 치환기는 할로, 히드록실, 시아노, 니트로, 알콕시, -O(아릴), =O, =NOH, =NO(알킬), -OC(=O)(알킬), -OC(=O)O(알킬), -OC(=O)NH2, -OC(=O)NH(알킬), -OC(=O)N(알킬)2, 아지도, -NH2, -NH(알킬), -N(알킬)2, -NH(아릴), -NHC(=O)(알킬), -NHC(=O)H, -NHC(=O)NH2, -NHC(=O)NH(알킬), -NHC(=O)N(알킬)2, 및 -NHC(=NH)NH2이다. 페닐, 시아노, 할로, 히드록실, 니트로, C1-C4 알콕시, O(C2-C4 알칸디일)OH, 및 O(C2-C4 알칸디일)할로가 특히 바람직하다.
치환될 모이어티가 시클로지방족, 헤테로시클로지방족, 아릴 또는 헤테로아릴 모이어티인 경우에, 바람직한 치환기는 알킬, 알케닐, 알키닐, 할로, 할로알킬, 히드록실, 히드록시알킬, 시아노, 니트로, 알콕시, -O(히드록시알킬), -O(할로알킬), -O(아릴), -O(시클로알킬), -O(헤테로시클로알킬), 알킬티오, 아릴티오, -C(=O)(알킬), -C(=O)H, -CO2H, -C(=O)NHOH, -C(=O)O(알킬), -C(=O)O(히드록시알킬), -C(=O)NH2, -C(=O)NH(알킬), -C(=O)N(알킬)2, -OC(=O)(알킬), -OC(=O)(히드록시알킬), -OC(=O)O(알킬), -OC(=O)O(히드록시알킬), -OC(=O)NH2, -OC(=O)NH(알킬), -OC(=O)N(알킬)2, 아지도, -NH2, -NH(알킬), -N(알킬)2, -NH(아릴), -NH(히드록시알킬), -NHC(=O)(알킬), -NHC(=O)H, -NHC(=O)NH2, -NHC(=O)NH(알킬), -NHC(=O)N(알킬)2, -NHC(=NH)NH2, -OSO2(알킬), -SH, -S(알킬), -S(아릴), -S(시클로알킬), -S(=O)알킬, -SO2(알킬), -SO2NH2, -SO2NH(알킬), 및 -SO2N(알킬)2이다. 보다 바람직한 치환기는 알킬, 알케닐, 할로, 할로알킬, 히드록실, 히드록시알킬, 시아노, 니트로, 알콕시, -O(히드록시알킬), -C(=O)(알킬), -C(=O)H, -CO2H, -C(=O)NHOH, -C(=O)O(알킬), -C(=O)O(히드록시알킬), -C(=O)NH2, -C(=O)NH(알킬), -C(=O)N(알킬)2, -OC(=O)(알킬), -OC(=O)(히드록시알킬), -OC(=O)O(알킬), -OC(=O)O(히드록시알킬), -OC(=O)NH2, -OC(=O)NH(알킬), -OC(=O)N(알킬)2, -NH2, -NH(알킬), -N(알킬)2, -NH(아릴), -NHC(=O)(알킬), -NHC(=O)H, -NHC(=O)NH2, -NHC(=O)NH(알킬), -NHC(=O)N(알킬)2, 및 -NHC(=NH)NH2이다. C1-C4 알킬, 시아노, 니트로, 할로, 및 C1-C4알콕시가 특히 바람직하다.
"C1-C5 알킬" 또는 "5 내지 10%"에서와 같이 범위가 언급된 경우에, 이러한 범위는 첫 번째 경우에서는 C1 및 C5 및 두 번째 경우에서는 5% 및 10%에서와 같은 범위의 종점을 포함한다.
특정한 입체이성질체가 구체적으로 (예를 들어, 구조 화학식에서의 관련 입체중심에서 굵은선 또는 파선 결합에 의해, 구조 화학식에서 E 또는 Z 배위를 갖는 것으로서의 이중 결합의 도시에 의해, 또는 입체화학-지정 명명법 또는 기호의 사용에 의해) 나타나 있지 않은 한, 모든 입체이성질체는 순수한 화합물뿐만 아니라 그의 혼합물로서 본 발명의 범주 내에 포함된다. 달리 나타내지 않는 한, 라세미체, 개별 거울상이성질체 (광학적으로 순수하거나 또는 부분적으로 분해됨), 부분입체이성질체, 기하 이성질체, 및 그의 조합 및 혼합물은 모두 본 발명에 의해 포괄된다.
관련 기술분야의 통상의 기술자는 화합물이 본원에 사용된 구조 화학식에 도시된 것들과 등가인 호변이성질체 형태 (예를 들어, 케토 및 엔올 형태), 공명 형태 및 쯔비터이온 형태를 가질 수 있다는 것 및 구조 화학식이 이러한 호변이성질체, 공명 또는 쯔비터이온 형태를 포괄한다는 것을 인지할 것이다.
"제약상 허용되는 에스테르"는 생체내에서 (예를 들어 인간 신체에서) 가수분해되어 모 화합물 또는 그의 염을 생산하거나, 또는 그 자체로 모 화합물의 것과 유사한 활성을 갖는 에스테르를 의미한다. 적합한 에스테르는 C1-C5 알킬, C2-C5 알케닐 또는 C2-C5 알키닐 에스테르, 특히 메틸, 에틸 또는 n-프로필을 포함한다.
"제약상 허용되는 염"은 제약 제제에 적합한 화합물의 염을 의미한다. 화합물이 1개 이상의 염기성 기를 갖는 경우에, 염은 산 부가염, 예컨대 술페이트, 히드로브로마이드, 타르트레이트, 메실레이트, 말레에이트, 시트레이트, 포스페이트, 아세테이트, 파모에이트 (엠보네이트), 히드로아이오다이드, 니트레이트, 히드로클로라이드, 락테이트, 메틸술페이트, 푸마레이트, 벤조에이트, 숙시네이트, 메실레이트, 락토비오네이트, 수베레이트, 토실레이트 등일 수 있다. 화합물이 1개 이상의 산성 기를 갖는 경우에, 염은 칼슘 염, 칼륨 염, 마그네슘 염, 메글루민 염, 암모늄 염, 아연 염, 피페라진 염, 트로메타민 염, 리튬 염, 콜린 염, 디에틸아민 염, 4-페닐시클로헥실아민 염, 벤자틴 염, 나트륨 염, 테트라메틸암모늄 염 등과 같은 염일 수 있다. 다형성 결정질 형태 및 용매화물은 본 발명의 범주 내에 또한 포괄된다.
용어 "대상체"는 영장류 (예를 들어, 인간), 원숭이, 소, 돼지, 양, 염소, 말, 개, 고양이, 토끼, 래트, 또는 마우스를 포함하나, 이에 제한되지는 않는 동물을 지칭한다. 용어 "대상체" 및 "환자"는, 예를 들어 포유동물 대상체, 예컨대 인간과 관련하여 본원에서 참조로 상호교환가능하게 사용된다.
질환 또는 장애를 치료하는 것과 관련하여, 용어 "치료하다", "치료하는" 및 "치료"는 장애, 질환 또는 상태, 또는 장애, 질환 또는 상태와 연관된 증상 중 1종 이상의 완화 또는 제거; 또는 질환, 장애 또는 상태 또는 그의 1종 이상의 증상의 진행, 확산 또는 악화의 저속화를 포함하도록 의도된다. "암의 치료"는 하기 효과: (1) 종양 성장의 (i) 저속화 및 (ii) 완전 성장 정지를 포함하는 어느 정도까지의 억제; (2) 종양 세포 수의 감소; (3) 종양 크기의 유지; (4) 종양 크기의 감소; (5) 말초 기관으로의 종양 세포 침윤의 (i) 감소, (ii) 저속화 또는 (iii) 완전 예방을 포함하는 억제; (6) 전이의 (i) 감소, (ii) 저속화 또는 (iii) 완전 예방을 포함하는 억제; (7) (i) 종양 크기의 유지, (ii) 종양 크기의 감소, (iii) 종양 성장의 저속화, (iv) 침습의 감소, 저속화 또는 예방을 유발할 수 있는 항종양 면역 반응의 증진, 및/또는 (8) 장애와 연관된 1종 이상의 증상의 중증도 또는 수의 어느 정도까지의 경감 중 1종 이상을 지칭한다.
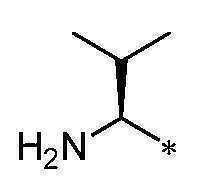
본 명세서의 화학식에서, 결합을 가로지르는 파상선 (
) 또는 결합의 말단에서의 별표 (*)는 공유 부착 부위를 나타낸다. 예를 들어, 화학식
에서 R이
이거나 또는 R이
이다라는 언급은
임을 의미한다.
본 명세서의 화학식에서, 방향족 고리를 그의 2개의 탄소 사이를 가로지르는 결합은 결합에 부착된 기가 암시적으로 그곳에 존재하는 (또는 그려진 경우, 그곳에 명백하게 존재하는) 수소의 제거에 의해 이용가능하게 되는 방향족 고리의 임의의 위치에 위치할 수 있음을 의미한다. 예시로서:
본 개시내용은 본원에 기재된 화합물에서 발생하는 원자의 모든 동위원소를 포함한다. 동위원소는 동일한 원자 번호를 갖지만 상이한 질량수를 갖는 원자를 포함한다. 일반적 예로서 및 비제한적으로, 수소의 동위원소는 중수소 및 삼중수소를 포함한다. 탄소의 동위원소는 13C 및 14C를 포함한다. 본 발명의 동위원소-표지된 화합물은 일반적으로 관련 기술분야의 통상의 기술자에게 공지된 통상의 기술에 의해 또는 본원에 기재된 것과 유사한 방법에 의해, 달리 사용되는 비-표지된 시약 대신에 적절한 동위원소-표지된 시약을 사용하여 제조될 수 있다. 예로서, C1-C3 알킬 기는 중수소화되지 않을 수 있거나, 부분적으로 중수소화되거나, 또는 완전히 중수소화되고, "CH3"은 CH3, 13CH3, 14CH3, CH2T, CH2D, CHD2, CD3 등을 포함한다. 한 실시양태에서, 화합물의 다양한 성분은 그의 천연 동위원소 존재비로 존재한다.
관련 기술분야의 통상의 기술자는 특정 구조가 1종의 호변이성질체 형태 또는 또 다른 것 - 예를 들어, 케토 대 엔올 -로 그려질 수 있고, 2종의 형태는 동등한 것으로 인식될 것이다.
두문자어들 및 약어들
표 C는 본 명세서에 사용된 두문자어 및 약어의 목록을 그의 의미와 함께 제공한다.
참고문헌
본 명세서에 앞에서 제1 저자 (또는 발명자) 및 연도에 따른 약기 방식으로 인용된 하기 참고문헌에 대한 전체 인용이 하기에 제공된다. 각각의 이들 참고문헌은 모든 목적을 위해 본원에 참조로 포함된다.
Akinbobuyi et al., Tetrahedron Lett. 2015, 56, 458, “Facile syntheses of functionalized toll-like receptor 7 agonists”.
Akinbobuyi et al., Bioorg. Med. Chem. Lett. 2016, 26, 4246, “Synthesis and immunostimulatory activity of substituted TLR7 agonists.”
Barberis et al., US 2012/0003298 A1 (2012).
Beesu et al., J. Med. Chem. 2017, 60, 2084, “Identification of High-Potency Human TLR8 and Dual TLR7/TLR8 Agonists in Pyrimidine-2,4-diamines.”
Berghoefer et al., J. Immunol. 2007, 178, 4072, “Natural and Synthetic TLR7 Ligands Inhibit CpG-A- and CpG-C-Oligodeoxynucleotide-Induced IFN-α Production.”
Bonfanti et al., US 2014/0323441 A1 (2015) [2015a].
Bonfanti et al., US 2015/0299221 A1 (2015) [2015b].
Bonfanti et al., US 2016/0304531 A1 (2016).
Carson et al., US 2013/0202629 A1 (2013).
Carson et al., US 8,729,088 B2 (2014).
Carson et al., US 9,050,376 B2 (2015).
Carson et al., US 2016/0199499 A1 (2016).
Chan et al., Bioconjugate Chem. 2009, 20, 1194, “Synthesis and Immunological Characterization of Toll-Like Receptor 7 Agonistic Conjugates.”
Chan et al., Bioconjugate Chem. 2011, 22, 445, “Synthesis and Characterization of PEGylated Toll Like Receptor 7 Ligands.”
Chen et al., US 7,919,498 B2 (2011).
Coe et al., US 9,662,336 B2 (2017).
Cortez and Va, Medicinal Chem. Rev. 2018, 53, 481, “Recent Advances in Small-Molecule TLR7 Agonists for Drug Discovery”.
Cortez et al., US 2017/0121421 A1 (2017).
Cortez et al., US 9,944,649 B2 (2018).
Dellaria et al., WO 2007/028129 A1 (2007).
Desai et al., US 9,127,006 B2 (2015).
Ding et al., WO 2016/107536 A1 (2016).
Ding et al., US 2017/0273983 A1 (2017) [2017a].
Ding et al., WO 2017/076346 A1 (2017) [2017b].
Gadd et al., Bioconjugate Chem. 2015, 26, 1743, “Targeted Activation of Toll-Like Receptors: Conjugation of a Toll-Like Receptor 7 Agonist to a Monoclonal Antibody Maintains Antigen Binding and Specificity.”
Graupe et al., US 8,993,755 B2 (2015).
Embrechts et al., J. Med. Chem. 2018, 61, 6236, “2,4-Diaminoquinazolines as Dual Toll Like Receptor (TLR) 7/8 Modulators for the Treatment of Hepatitis B Virus.”
Halcomb et al., US 9,161,934 B2 (2015).
Hashimoto et al., US 2009/0118263 A1 (2009).
He et al., US 10,487,084 B2 (2019) [2019a].
He et al., US 10,508,115 B2 A1 (2019) [2019b].
Hirota et al., US 6,028,076 (2000).
Holldack et al., US 2012/0083473 A1 (2012).
Isobe et al., US 6,376,501 B1 (2002).
Isobe et al., JP 2004137157 (2004).
Isobe et al., J. Med. Chem. 2006, 49 (6), 2088, “Synthesis and Biological Evaluation of Novel 9-Substituted-8-Hydroxyadenine Derivatives as Potent Interferon Inducers.”
Isobe et al., US 7,521,454 B2 (2009) [2009a].
Isobe et al., US 2009/0105212 A1 (2009) [2009b].
Isobe et al., US 2011/0028715 A1 (2011).
Isobe et al., US 8,148,371 B2 (2012).
Jensen et al., WO 2015/036044 A1 (2015).
Jones et al., US 7,691,877 B2 (2010).
Jones et al., US 2012/0302598 A1 (2012).
Kasibhatla et al., US 7,241,890 B2 (2007).
Koga-Yamakawa et al., Int. J. Cancer 2013, 132 (3), 580, “Intratracheal and oral administration of SM-276001: A selective TLR7 agonist, leads to antitumor efficacy in primary and metastatic models of cancer.”
Li et al., US 9,902,730 B2 (2018).
Lioux et al., US 9,295,732 B2 (2016).
Lund et al., Proc. Nat’l Acad. Sci (USA) 2004, 101 (15), 5598, “Recognition of single-stranded RNA viruses by Toll-like receptor 7.”
Maj et al., US 9,173,935 B2 (2015).
McGowan et al., US 2016/0168150 A1 (2016) [2016a].
McGowan et al., US 9,499,549 B2 (2016) [2016b].
McGowan et al., J. Med. Chem. 2017, 60, 6137, “Identification and Optimization of Pyrrolo[3,2-d]pyrimidine Toll-like Receptor 7 (TLR7) Selective Agonists for the Treatment of Hepatitis B.”
Musmuca et al., J. Chem. Information & Modeling 2009, 49 (7), 1777, “Small-Molecule Interferon Inducers. Toward the Comprehension of the Molecular Determinants through Ligand-Based Approaches.”
Nakamura et al., Bioorg. Med. Chem. Lett. 2013, 13, 669, “Synthesis and evaluation of 8-oxoadenine derivatives as potent Toll-like receptor agonists with high water solubility.”
Ogita et al., US 2007/0225303 A1 (2007).
Ota et al., WO 2019/124500 A1 (2019).
Pilatte et al., WO 2017/216293 A1 (2017).
Poudel et al., US 10,472,361 B2 (2019) [2019a].
Poudel et al., US 10,494,370 B2 (2019) [2019b].
Poudel et al., US 2020/0038403 A1 (2020) [2020a].
Poudel et al., US 2020/0039986 A1 (2020) [2020b].
Purandare et al., WO 2019/209811 A1 (2019).
Pryde, US 7,642,350 B2 (2010).
Sato-Kaneko et al., JCI Insight 2017, 2, e93397, “Combination Immunotherapy with TLR Agonists and Checkpoint Inhibitors Suppresses Head and Neck Cancer”.
Smits et al., The Oncologist 2008, 13, 859, “The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy”.
Vasilakos and Tomai, Expert Rev. Vaccines 2013, 12, 809, “The Use of Toll-like Receptor 7/8 Agonists as Vaccine Adjuvants”.
Vernejoul et al., US 2014/0141033 A1 (2014).
Young et al., US 10,457,681 B2 (2019).
Yu et al., PLoS One 2013, 8 (3), e56514, “Toll-Like Receptor 7 Agonists: Chemical Feature Based Pharmacophore Identification and Molecular Docking Studies.”
Zhang et al., Immunity 2016, 45, 737, “Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA.”
Zhang et al., WO 2018/095426 A1 (2018)>
Zurawski et al., US 2012/0231023 A1 (2012).
상기 본 발명의 상세한 설명은 본 발명의 특정한 부분 또는 측면과 주로 또는 독점적으로 관련된 구절을 포함한다. 이는 명확성 및 편의성을 위한 것이고, 특정한 특색은 그것이 개시된 구절을 넘어 관련될 수 있으며, 본원의 개시내용은 상이한 구절에서 발견된 정보의 모든 적절한 조합을 포함하는 것으로 이해되어야 한다. 유사하게, 본원의 다양한 도면 및 설명은 본 발명의 구체적 실시양태와 관련되어 있지만, 구체적 특색이 특정한 도면 또는 실시양태의 문맥에서 개시되는 경우에, 이러한 특색은 또한 적절한 정도로, 또 다른 도면 또는 실시양태의 문맥에서, 또 다른 특색과 조합되어, 또는 본 발명에서 일반적으로 사용될 수 있는 것으로 이해되어야 한다.
추가로, 본 발명이 특정의 바람직한 실시양태의 면에서 구체적으로 기재되기는 하였지만, 본 발명이 이러한 바람직한 실시양태로 제한되는 것은 아니다. 오히려, 본 발명의 범주는 첨부된 청구범위에 의해 정의된다.