RU2400577C2 - Способ получения высокомодульного волокна из среднепрочных углеродных волокон - Google Patents
Способ получения высокомодульного волокна из среднепрочных углеродных волокон Download PDFInfo
- Publication number
- RU2400577C2 RU2400577C2 RU2008134392/04A RU2008134392A RU2400577C2 RU 2400577 C2 RU2400577 C2 RU 2400577C2 RU 2008134392/04 A RU2008134392/04 A RU 2008134392/04A RU 2008134392 A RU2008134392 A RU 2008134392A RU 2400577 C2 RU2400577 C2 RU 2400577C2
- Authority
- RU
- Russia
- Prior art keywords
- pcco
- carbon
- modulus
- carb
- bundle
- Prior art date
Links
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 title claims abstract description 11
- 239000000835 fiber Substances 0.000 title claims abstract description 11
- 229910052799 carbon Inorganic materials 0.000 title claims abstract description 10
- 238000000034 method Methods 0.000 title claims description 19
- 229920000049 Carbon (fiber) Polymers 0.000 claims description 9
- 239000004917 carbon fiber Substances 0.000 claims description 9
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims description 5
- 238000010438 heat treatment Methods 0.000 claims 4
- 238000004513 sizing Methods 0.000 claims 3
- 239000002994 raw material Substances 0.000 claims 1
- 239000002131 composite material Substances 0.000 abstract description 5
- 230000000694 effects Effects 0.000 abstract description 4
- 239000003795 chemical substances by application Substances 0.000 abstract 2
- 238000007669 thermal treatment Methods 0.000 abstract 2
- 239000007858 starting material Substances 0.000 abstract 1
- 239000000126 substance Substances 0.000 abstract 1
- 239000003054 catalyst Substances 0.000 description 21
- 239000000203 mixture Substances 0.000 description 15
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 10
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 229910052979 sodium sulfide Inorganic materials 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 8
- 239000011734 sodium Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 229910052742 iron Inorganic materials 0.000 description 7
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 238000006243 chemical reaction Methods 0.000 description 5
- MPMSMUBQXQALQI-UHFFFAOYSA-N cobalt phthalocyanine Chemical compound [Co+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 MPMSMUBQXQALQI-UHFFFAOYSA-N 0.000 description 5
- 238000001663 electronic absorption spectrum Methods 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 159000000000 sodium salts Chemical class 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 229910017052 cobalt Inorganic materials 0.000 description 4
- 239000010941 cobalt Substances 0.000 description 4
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- KMHSUNDEGHRBNV-UHFFFAOYSA-N 2,4-dichloropyrimidine-5-carbonitrile Chemical compound ClC1=NC=C(C#N)C(Cl)=N1 KMHSUNDEGHRBNV-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- -1 N- (4′-hydroxyphenyl) -N- (carboxymethyl) sulfamoyl phthalocyanine cobalt Chemical compound 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- KGCBHUMVJDXKJF-UHFFFAOYSA-N [H]C([H])(Cl)[Co] Chemical compound [H]C([H])(Cl)[Co] KGCBHUMVJDXKJF-UHFFFAOYSA-N 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 229920002239 polyacrylonitrile Polymers 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 3
- MIINHRNQLVVCEW-UHFFFAOYSA-N 132-16-1 Chemical compound [Fe+2].C12=CC=CC=C2C(N=C2[N-]C(C3=CC=CC=C32)=N2)=NC1=NC([C]1C=CC=CC1=1)=NC=1N=C1[C]3C=CC=CC3=C2[N-]1 MIINHRNQLVVCEW-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 238000007265 chloromethylation reaction Methods 0.000 description 2
- 229910002804 graphite Inorganic materials 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- IEQIEDJGQAUEQZ-UHFFFAOYSA-N phthalocyanine Chemical class N1C(N=C2C3=CC=CC=C3C(N=C3C4=CC=CC=C4C(=N4)N3)=N2)=C(C=CC=C2)C2=C1N=C1C2=CC=CC=C2C4=N1 IEQIEDJGQAUEQZ-UHFFFAOYSA-N 0.000 description 2
- 150000004032 porphyrins Chemical class 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- VLDPXPPHXDGHEW-UHFFFAOYSA-N 1-chloro-2-dichlorophosphoryloxybenzene Chemical compound ClC1=CC=CC=C1OP(Cl)(Cl)=O VLDPXPPHXDGHEW-UHFFFAOYSA-N 0.000 description 1
- ZWLKKKRIYPQRQD-UHFFFAOYSA-N 14285-59-7 Chemical compound [H+].[H+].[H+].[H+].[Co+2].C12=CC(S(=O)(=O)[O-])=CC=C2C(N=C2[N-]C(C3=CC=C(C=C32)S([O-])(=O)=O)=N2)=NC1=NC([C]1C=CC(=CC1=1)S([O-])(=O)=O)=NC=1N=C1[C]3C=CC(S([O-])(=O)=O)=CC3=C2[N-]1 ZWLKKKRIYPQRQD-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 206010019345 Heat stroke Diseases 0.000 description 1
- 229910021575 Iron(II) bromide Inorganic materials 0.000 description 1
- JMXFKIBMOCWDSZ-UHFFFAOYSA-N P(=O)(O)(O)C[Co] Chemical compound P(=O)(O)(O)C[Co] JMXFKIBMOCWDSZ-UHFFFAOYSA-N 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 229910052946 acanthite Inorganic materials 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 1
- 239000011609 ammonium molybdate Substances 0.000 description 1
- 235000018660 ammonium molybdate Nutrition 0.000 description 1
- 229940010552 ammonium molybdate Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- SLUNEGLMXGHOLY-UHFFFAOYSA-N benzene;hexane Chemical compound CCCCCC.C1=CC=CC=C1 SLUNEGLMXGHOLY-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000003495 flagella Anatomy 0.000 description 1
- 238000005755 formation reaction Methods 0.000 description 1
- ANSXAPJVJOKRDJ-UHFFFAOYSA-N furo[3,4-f][2]benzofuran-1,3,5,7-tetrone Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1C(=O)OC2=O ANSXAPJVJOKRDJ-UHFFFAOYSA-N 0.000 description 1
- 239000010439 graphite Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- GYCHYNMREWYSKH-UHFFFAOYSA-L iron(ii) bromide Chemical compound [Fe+2].[Br-].[Br-] GYCHYNMREWYSKH-UHFFFAOYSA-L 0.000 description 1
- 150000002678 macrocyclic compounds Chemical class 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005272 metallurgy Methods 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 238000003918 potentiometric titration Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- OGFYIDCVDSATDC-UHFFFAOYSA-N silver silver Chemical group [Ag].[Ag] OGFYIDCVDSATDC-UHFFFAOYSA-N 0.000 description 1
- XUARKZBEFFVFRG-UHFFFAOYSA-N silver sulfide Chemical group [S-2].[Ag+].[Ag+] XUARKZBEFFVFRG-UHFFFAOYSA-N 0.000 description 1
- 229940056910 silver sulfide Drugs 0.000 description 1
- XNRABACJWNCNEQ-UHFFFAOYSA-N silver;azane;nitrate Chemical compound N.[Ag+].[O-][N+]([O-])=O XNRABACJWNCNEQ-UHFFFAOYSA-N 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- CYTQBVOFDCPGCX-UHFFFAOYSA-N trimethyl phosphite Chemical compound COP(OC)OC CYTQBVOFDCPGCX-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Yarns And Mechanical Finishing Of Yarns Or Ropes (AREA)
- Inorganic Fibers (AREA)
- Catalysts (AREA)
Abstract
Изобретение относится к технологии получения высокомодульных углеродных волокон из среднепрочных волокон на основе полиакрилонитрильных жгутиков и может быть использовано для производства высококачественных композитов. В качестве исходного сырья используют среднепрочное углеродное волокно с линейной плотностью 200-1600 текс и модулем упругости 200-250 ГПа. Это волокно подвергают крутке до величины 30-60 круток/м при содержании в нем аппрета более 1%. Дополнительно аппретируют волокно при содержании аппрета менее 1%. Затем подкрученный жгут подвергают первичной термообработке при 2300-2500°С в течение 1-10 мин до мод значения модуля упругости углеродного жгута не менее 300 ГПа. Затем проводят вторую термообработку при температуре не ниже 3000°С в течение 1-20 сек при вытяжке жгута до 10% до возрастания модуля упругости углеродного жгута до величины не менее 450 ГПа. Высокое качество достигается за счет компактной формы получаемых углеродных жгутов, обеспечивающей высокое содержание углеродных волокон в композите и максимальную реализацию механических свойств композиционного материала. 2 табл.
Description
Изобретение относится к металлургии, в частности, к способам получения из среднепрочных волокон на основе полиакрилонитрильных жгутиков высокомодульных углеродных волокон, которые могут затем использоваться для производства высококачественных композитов.
Высокое качество достигается за счет компактной формы получаемых углеродных жгутов, обеспечивающей высокое содержание углеродных волокон в композите и максимальную реализацию механических свойств композиционного материала. Получение таких углеродных волокон традиционным способом невозможно.
Известен способ получения углеродного волокна с повышенным модулем упругости, описанный в Трудах НИИГрафита "Директивный технологический процесс производства жгута углеродного марки ВПР-19С", инв. №256, 1987.
Известный способ заключается в том, что процесс проводят в статических условиях с использованием высокотемпературной обработки. Окисленное полиакрилонитрильное волокно в виде пасм длиной до 1800 мм загружают в графитовые тигли, которые помещают в электрические печи сопротивления и подвергают волокно высокотемпературной обработке в диапазоне температур 2800-3000°С.
Недостатком известного способа является то, что волокно по этому способу получают только в дискретном виде.
Известен способ получения высокомодульного углеродного волокна, описанный в патенте ВБР №1295289 по кл. С1А, опубл. 1972 г.
Известный способ заключается в том, что окисляют в среде воздуха полиакрилонитрильное волокно до плотности 1,0-1,4 г/ куб. см, затем окисленное волокно карбонизируют при температуре 675-725°С в течение 60-150 с в нейтральной среде, пропитывают 10-15%-ным раствором борной кислоты при температуре пропитки 40-70°С, затем сушат тепловым ударом при 200-300°С и подвергают высокотемпературной обработке в интервале температур 2000-2100°С не менее 20 с и с вытяжкой 0,-1,5%.
В результате получают углеродное волокно с плотностью 1,85-2,05 г/куб.см и модулем упругости 435-470 ГПа.
Недостатком известного способа является нестабильность процесса и соответственно недостаточная воспроизводимость результатов.
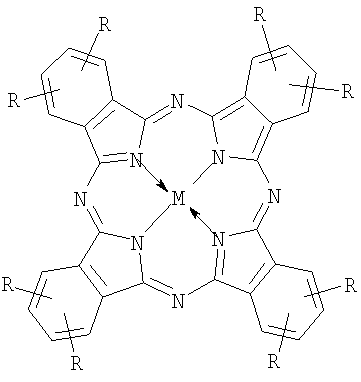 |
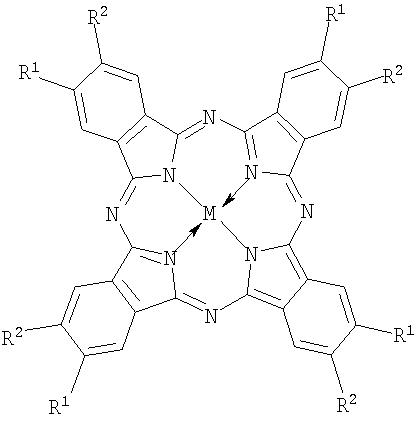 |
| I: (R+)nPcM, где | II: (R-)nPcM, где |
| R=-CH2N+(CH3)2CH2CH2OH Cl- (choln-PcM), n=2÷8, M=Co, Fe; | R=R1=R2=-C(O)O- Na+ (carb8-PcM), n=8, M=Co, Fe; |
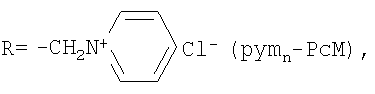 n=2÷8, M=Co, Fe; |
R=R1=-C(O)O- Na+ R2=H (carb4-PcM), n=4, M=Co; |
| (R-)nPcM, где | R-R1=-S(O)2O- Na+, R2=H (sul4-PcM), n=4, M=Co |
| R=-CH2P(=O)(O- Na+)2 (phosn-PcM), n=8, M=Co |
Поставленная задача также решается тем, что мольное соотношение компонентов в ассоциате 1:1÷2.
Известен способ окисления сульфида натрия [1] с использованием катализатора, представляющего собой N-(4′-гидроксифенил)-N-(карбоксиметил)сульфамоил фталоцианин кобальта, как описано выше. Он отличается невысокой активностью.
Задача настоящего изобретения состоит в разработке способа каталитического окисления сульфида натрия, который бы обеспечивал высокую конверсию.
Поставленная задача решается тем, что процесс окисления сульфида натрия проводят в водном растворе с использованием кислорода воздуха с указанным выше катализатором при комнатной температуре.
Около 20 лет назад на примере порфиринов, а затем - смесей порфиринов и фталоцианинов было установлено, что при смешивании растворов комплексов с четырьмя противоположными по знаку заряда заместителями (R+ или R-) образуются прочные ассоциаты (в литературе используется и другие названия - супрамолекулярные агрегаты, комплексы, ионные пары с переносом заряда и проч.) строго определенного состава - димеры и триммеры [Т.Shimidzu and Т.Iyoda, Chem. Phys. Lett. 1981. p.853.; H.Segawa, H.Nishino, Т.Kamikawa, K.Honda and Т.Shimidzu, Chem. Lett. 1989. p.1917; S.Gaspard, C.R., Acad. Sci. Paris. 1984. 298. p.379]. Использование нами термина «ассоциаты» обусловлено следующими причинами. Тетрапиррольные макроциклы в растворах склонны к взаимодействию друг с другом посредством нековалентного связывания. В результате образуются агрегаты, состоящие из нескольких одинаковых молекул. Для того чтобы отличать такие агрегаты от изучаемых нами, мы используем термин «ассоциаты», так как наши образования состоят из разных молекул. Использовать термин «комплексы» также неудобно, так как исходные молекулы являются фталоцианиновыми комплексами. Таким образом, выражение «надмолекулярный ионный ассоциат» обозначает систему, состоящую из нескольких молекул (надмолекулярный), различных по своим свойствам, связанных между собой преимущественно ионными связями, но не только.
Фталоцианины с катионными аммониометильными заместителями choln-PcM и pymn-PcM получали хлорметилированием фталоцианина кобальта или фталоцианина железа α,α′-дихлорметиловым эфиром и последующим взаимодействием хлорметилзамещенного производного с 2-(диметиламино)этанолом или пиридином соответственно. Среднюю степень замещения регулировали, изменяя время реакции хлорметилирования.
Аналогично получали и анионный фосфонатометилзамещенный фталоцианин кобальта phosn-PcCo, однако при взаимодействии с хлорметилзамещенным производным использовали триметилфосфит или триэтилфосфит с последующим гидролизом диалкилфосфонатных групп.
Натриевую соль тетракарбоксифталоцианина кобальта carb4-PcCo получали нейтрализацией известного 2,9,16,23-тетракарбоксифталоцианина кобальта [С.А.Михаленко, Л.И.Соловьева, Е.А.Лукьянец // ЖОХ. 2004. Т.74. Вып.3. С.496-505].
Натриевую соль октакарбоксифталоцианина кобальта carb8-PcCo получали по способу, описанному в работе [Патент РФ 2304582, 2007, БИ №23].
Натриевую соль октакарбоксифталоцианина железа carb8-PcFe получали по методике, аналогичной carb8-PcCo.
Натриевую соль тетрасульфофталоцианина кобальта sul4-PcCo получают как в работе [Rollman L.D. Ivamoto R.T. J. Amer. Chem. Soc. 1968. V.90. №5. P.1455].
Ассоциаты получали путем смешивания водных растворов фталоцианинов с разноименно заряженными заместителями. Состав ассоциатов регулировали, изменяя стехиометрическое соотношение компонентов (R-)nPcM:(R+)nPcM. Состав катализаторов в соответствии с изобретением приведен в таблице 1.
Каталитическая активность катализаторов, выраженная в молях окисленного сульфида натрия на моль катализатора в минуту (моль(Na2S)ок/моль(катализатора)мин), приведена в таблице 2.
Как видно из таблицы 2, некоторые катализаторы показали каталитическую активность на один - три порядка большую, в сравнении с прототипом в таких же условиях: при окислении кислородом воздуха при комнатной температуре.
Таким образом, получен активный катализатор, который позволяет эффективно проводить процесс окисления сульфида натрия при комнатной температуре в атмосфере воздуха.
Нижеприведенные примеры иллюстрируют предлагаемое изобретение.
Пример 1. Получение октакис(N-(2-гидроксиэтил)-N,N-диметиламмо- ниометил)фталоцианина кобальта (chol8-PcCo).
К 11 г (0,082 моль) хлористого алюминия добавляют при перемешивании 3 мл триэтиламина. После охлаждения массы до температуры 70-80°С к смеси приливают 6 мл (0, 0075 моль) α,α′-дихлорметилового эфира, а затем загружают 3 г (0,0052 моль) фталоцианина кобальта. Смесь нагревают в течение 3 часов при перемешивании и температуре 90-93°С, после чего выгружают на лед. Осадок отфильтровывают, промывают водой, метанолом и сушат. Выход октакис(хлорметил)фталоцианина кобальта 5,65 г (78,6%).). Электронный спектр поглощения, λmax=673 нм (ДМФА).
Найдено, %: Cl 29,11.
Вычислено % Cl 29,56.
К 0,7 г (0,00073 моль) окстакис(хлорметил)фталоцианина кобальта добавляют 5 мл диметилформамида и 1,5 мл 2-(диметиламино)этанола, после чего смесь нагревают при перемешивании на кипящей водяной бане в течение 2 часов. Осадок отфильтровывают, промывают ацетоном, переосаждают из метанола с ацетоном и сушат. Выход 1,0 г (83,3%) комплекса (I). Электронный спектр поглощения, λmax=672 нм (Н2О).
Найдено, %: Cl 16,51; N 13,02.
Вычислено для C80H134N18O10C18Co, %: Cl 16,95; N 13,4.
Пример 2. Получение октакис(пиридиниометил)фталоцианина кобальта октахлорид (pym8-PcCo).
К 0,49 г (0,00051 моль) окстакис(хлорметил)фталоцианина кобальта (примеры 1-2) добавляют 5,0 мл пиридина, после чего смесь нагревают при перемешивании на кипящей водяной бане в течение 2 часов. Осадок отфильтровывают, промывают ацетоном, переосаждают из метанола с ацетоном и сушат. Выход 0,43 г (83,3%) комплекса (II). Электронный спектр поглощения, λmax=672 нм (Н2О).
Найдено, %: Cl 16,51; N 13,02.
Вычислено для C90H74N18C18Co, %: Cl 16,95; N 13,4.
Пример 3. Получение октакис(фосфонометил)фталоцианина кобальта (phos8-PcCo).
К 2,0 г (0,00208 моль) окстакис(хлорметил)фталоцианина кобальта, полученного как в примере 1, добавляют 5 мл триэтилфосфита и смесь нагревают при 150°С в течение 2 ч. Избыток триэтилфосфита удаляют в вакууме, продукт переосаждают из бензола гексаном. Выход октакис[(диэтоксифосфонил)метил]фталоцианина кобальта 3,0 г (81,3%) комплекса (I). Электронный спектр поглощения, λmax=685 нм (H2O).
Найдено, %: Р 13,31; Со 3,51.
Вычислено для C72H104CoN8O24P8, %: P 13.98; Со 3.33.
Смесь 0,50 г (0,000282 моль) выше полученного эфира и 1 мл концентрированной бромистоводородной кислоты нагревают при 110°С в течение 2 ч. Избыток бромистоводородной кислоты отгоняют в вакууме, остаток промывают водой, спиртом и сушат. Выход продукта 0,27 г (72,3%). Электронный спектр поглощения, λmax=684 нм (водный раствор NaOH, рН 10).
Найдено, %: Р 18,1; Со 4,2.
Вычислено для C40H40CoN8O24P8, %: P 18,72; Со 4,45.
Пример 4. Получение окта-4,5-карбоксифталоцианина железа (carb8-PcFe).
Данный фталоцианин железа получали взаимодействием пиромеллитового диангидрида с безводным бромидом железа (II) в присутствии мочевины, сульфата натрия и молибдата аммония (в молярном соотношении 4:1:10:10:0,1) при 205-210°С в течение трех часов с последующим омылением полученного технического тетраимида октакарбоксифталоцианина железа 25% раствором гидроокиси калия (кипячением в течение 25 часов) до калиевой соли октакарбоксифталоцианина железа. Затем подкисляли ее 10% водным раствором соляной кислоты до свободной кислоты с последующей обработкой последней водным раствором гидроокиси натрия. Выход натриевой соли технического октакарбонатного фталоцианина железа составил 35%. Полученную соль очищают от примесей, в том числе и от олигомерных продуктов методом колоночной хроматографии на оксиде алюминия. Элюент - фосфатный буфер рН 8. Из фракции с Rf 0,9 (на пластинке silufol) выделяют с выходом 10% (в расчете на диангидрид пиромеллитовой кислоты) carb8-PcFe. Готовый продукт сушат при 105-110°С в вакууме над P2O5 до постоянной массы.
Найдено, %: С 42,98; Н 1,10; N 9,81. C40H8FeN8Na8O16.
Вычислено, %: С 43,82; Н 0,74; N 10,22.
Пример 5. Получение катализатора - ассоциата pym8-PcCo:carb8-PcCo=2:1.
К 1 мл 3,30·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 6,60·10-5 М водного раствора pym8-PcCo, перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Пример 6. Получение катализатора - ассоциата pym8-PcCo:carb8-РсСо=1:1.
К 1 мл 3,30·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 3,30·10-5 М водного раствора pym8-PcCo и перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Пример 7. Получение катализатора, включающего ассоциат pym8-РсСо:carb8-PcCo=1:2.
К 1 мл 6,60·10-5 М водного раствора carb8-PcCo прибавляют 1 мл 3,30·10-5 М водного раствора pym8-PcCo и перемешивают 15 мин. Концентрация раствора полученного ассоциата составляет 1,65·10-5 М.
Примеры 8-24.
Остальные катализаторы согласно таблице 1 получали аналогичным образом.
Пример 25. Каталитическое окисление сульфида натрия.
Смешивают в реакторе 10 мл (0,024 М) сульфида натрия и 0,37 мл водного раствора катализатора, содержащего (1,65·10-5 М) pym8PcCo:carb8PcCo=2:1 (пример 5). Конечная концентрация катализатора составила 5,0·10-7 М. Реакцию проводили при комнатной температуре в атмосфере воздуха при интенсивном перемешивании в течение 15 минут. Анализы на остаточный сульфид натрия после опыта проводили методом потенциометрического титрования на универсальном иономере И-500 по стандартной методике раствором азотнокислого аммиаката серебра. Измерительный электрод - сульфид-серебряный, электрод сравнения - хлор-серебряный.
По результатам анализа конверсия сульфида натрия составила 100% или 3,1·103 моль(Na2S)/моль(катализатора)мин.
| Таблица 1 | |||
| № примера | Состав катализатора | ||
| (R+)nPcM | (R-)nPcM | (R+)nPcM:(R-)nPcM | |
| 5 | pym8-PcCo | carb8-PcCo | 2:1 |
| 6 | 1:1 | ||
| 7 | 1:2 | ||
| 8 | chol8-PcCo | carb8-PcFe | 1:1 |
| 10 | chol8-PcCo | carb4-PcCo | 2:1 |
| 11 | chol8-PcCo | carb8-PcCo | 2:1 |
| 12 | 1:1 | ||
| 13 | 1:2 | ||
| 14 | pym8-PcCo | carb8-PcFe | 2:1 |
| 15 | 1:1 | ||
| 16 | 1:2 | ||
| 17 | pym4-PcCo | carb4-PcCo | 2:1 |
| 18 | 1:1 | ||
| 19 | chol8-PcFe | carb8-PcCo | 1:1 |
| 20 | chol8-PcFe | carb8-PcFe | 1:1 |
| 21 | pym8-PcFe | carb8-PcFe | 1:1 |
| 22 | pym8-PcFe | carb8-PcCo | 1:1 |
| 23 | pym8-PcCo | phos8-PcCo | 1:1 |
| 24 | pym4-PcCo | sul4-PcCo | 1:1 |
Примеры 26-44.
Процесс проводили по примеру 25, но с использованием катализаторов по примерам 6-24. Результаты приведены в таблице 2.
Таким образом, как видно из таблицы 2, предложенный катализатор обладает активностью, большей, чем у прототипа в 4 и более раз: самый активный катализатор показал активность в 62 раза выше, чем у прототипа.
| Таблица 2 | ||||
| примера | Состав катализатора | (R+)nPcM:(R-)nPcM | моль(Na2S)/моль (катализатора)мин, 103 | |
| (R+)nPcM | (R-)nPcM | |||
| 25 | pym8-PcCo | carb8-PcCo | 2:1 | 3,1 |
| 26 | 1:1 | 1,1 | ||
| 27 | 1:2 | 2,1 | ||
| 28 | chol8-PcCo | carb8-PcFe | 1:1 | 0,4 |
| 29 | chol4-PcCo | carb4-PcCo | 1:2 | 1,0 |
| 30 | 2:1 | 2,1 | ||
| 31 | chol8-PcCo | carb8-PcCo | 2:1 | 1,8 |
| 32 | 1:1 | 0,4 | ||
| 33 | 1:2 | 1,5 | ||
| 34 | pym8-PcCo | carb8-PcFe | 2:1 | 1,1 |
| 35 | 1:1 | 0,5 | ||
| 36 | 1:2 | 0,8 | ||
| 37 | pym4-РсСо | carb4-PcCo | 1:2 | 2,1 |
| 38 | 2:1 | 1,3 | ||
| 39 | chol8-PcFe | carb8-PcCo | 1:1 | 0,8 |
| 40 | chol8-PcFe | carb8-PcFe | 1:1 | 0,5 |
| 41 | pym8-PcFe | carb8-PcCo | 1:1 | 1,1 |
| 42 | pym8-PcFe | carb8-PcFe | 1:1 | 0,7 |
| 43 | pym8-PcCo | phos8-PcCo | 1:2 | 0,3 |
| 44 | pym4-PcCo | sul4-PcCo | 1:1 | 0,2 |
Claims (1)
- Способ получения высокомодульного волокна из среднепрочных низкомодульных углеродных волокон, включающем использование углеродного волокна на основе ПАН-сырья, изготовление из него жгута, его первую термообработку при температуре не ниже 2200°С, и вторую высокоскоростную термообработку при температуре не ниже 3000°С, отличающийся тем, что в качестве исходного сырья используют среднепрочное углеродное волокно с линейной плотностью 200-1600 текс и модулем упругости 200-250 ГПа, которое подвергают крутке до величины 30-60 круток/м при содержании в нем аппрета более 1% и дополнительно аппретируют при содержании аппрета менее 1%, затем подкрученный жгут подвергают первичной термообработке при температуре 2300-2500°С в течение 1-10 мин до возрастания модуля упругости углеродного жгута до значения не менее 300 ГПа, затем проводят вторую термообработку в течение нескольких - 1-20 с при вытяжке жгута до 10% до возрастания модуля упругости углеродного жгута до величины не менее 450 ГПа.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2008134392/04A RU2400577C9 (ru) | 2008-08-21 | 2008-08-21 | Способ получения высокомодульного волокна из среднепрочных углеродных волокон |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2008134392/04A RU2400577C9 (ru) | 2008-08-21 | 2008-08-21 | Способ получения высокомодульного волокна из среднепрочных углеродных волокон |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| RU2008134392A RU2008134392A (ru) | 2010-02-27 |
| RU2400577C2 true RU2400577C2 (ru) | 2010-09-27 |
| RU2400577C9 RU2400577C9 (ru) | 2010-12-27 |
Family
ID=42127595
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2008134392/04A RU2400577C9 (ru) | 2008-08-21 | 2008-08-21 | Способ получения высокомодульного волокна из среднепрочных углеродных волокон |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2400577C9 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2555468C2 (ru) * | 2013-11-13 | 2015-07-10 | Акционерное общество "Научно-исследовательский институт конструкционных материалов на основе графита "НИИграфит" | Способ термической обработки углеродосодержащих волокнистых материалов |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4869856A (en) * | 1986-08-07 | 1989-09-26 | Toho Rayon Co., Ltd. | Method for producing carbon fibers from acrylonitrile fiber strands |
| EP0100410B1 (en) * | 1982-06-09 | 1990-01-17 | Toray Industries, Inc. | High strength and high elongation carbon fiber bundle and process for producing the same |
| US5256344A (en) * | 1989-02-23 | 1993-10-26 | Hercules Incorporated | Process of thermally stabilizing pan fibers |
| RU2016147C1 (ru) * | 1991-10-17 | 1994-07-15 | Государственный научно-исследовательский институт конструкционных материалов на основе графита | Способ получения высокомодульного углеродного волокна |
| RU2130516C1 (ru) * | 1996-09-10 | 1999-05-20 | Акционерное общество "Балаковские волокна" | Способ получения углеродного волокна |
| RU2220235C2 (ru) * | 2001-04-23 | 2003-12-27 | Государственное предприятие Всероссийский научно-исследовательский институт полимерных волокон с опытным заводом | Углеродная крученая нить для композиционных материалов и способ ее получения |
| JP2006307407A (ja) * | 2005-03-29 | 2006-11-09 | Toray Ind Inc | 炭素繊維および、炭素繊維の製造方法 |
-
2008
- 2008-08-21 RU RU2008134392/04A patent/RU2400577C9/ru active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0100410B1 (en) * | 1982-06-09 | 1990-01-17 | Toray Industries, Inc. | High strength and high elongation carbon fiber bundle and process for producing the same |
| US4869856A (en) * | 1986-08-07 | 1989-09-26 | Toho Rayon Co., Ltd. | Method for producing carbon fibers from acrylonitrile fiber strands |
| US5256344A (en) * | 1989-02-23 | 1993-10-26 | Hercules Incorporated | Process of thermally stabilizing pan fibers |
| RU2016147C1 (ru) * | 1991-10-17 | 1994-07-15 | Государственный научно-исследовательский институт конструкционных материалов на основе графита | Способ получения высокомодульного углеродного волокна |
| RU2130516C1 (ru) * | 1996-09-10 | 1999-05-20 | Акционерное общество "Балаковские волокна" | Способ получения углеродного волокна |
| RU2220235C2 (ru) * | 2001-04-23 | 2003-12-27 | Государственное предприятие Всероссийский научно-исследовательский институт полимерных волокон с опытным заводом | Углеродная крученая нить для композиционных материалов и способ ее получения |
| JP2006307407A (ja) * | 2005-03-29 | 2006-11-09 | Toray Ind Inc | 炭素繊維および、炭素繊維の製造方法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2555468C2 (ru) * | 2013-11-13 | 2015-07-10 | Акционерное общество "Научно-исследовательский институт конструкционных материалов на основе графита "НИИграфит" | Способ термической обработки углеродосодержащих волокнистых материалов |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2008134392A (ru) | 2010-02-27 |
| RU2400577C9 (ru) | 2010-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Eggerding et al. | Synthesis of dihydroxycyclopropenone (deltic acid) | |
| KR20160048378A (ko) | 초음파를 이용한 그래핀옥사이드의 제조방법 및 제조장치 | |
| CN109942821A (zh) | 羟基氨基改性硅油阳离子乳液的制备方法及其产品和应用 | |
| RU2400577C2 (ru) | Способ получения высокомодульного волокна из среднепрочных углеродных волокон | |
| DE69408919T2 (de) | Verfahren zur Herstellung von Polydimethylsiloxanen | |
| CN113403435B (zh) | 一种聚乙二醇均三嗪衍生物鞣剂的制备方法及其应用 | |
| SU1189327A3 (ru) | Катализатор дл получени 3-цианпиридина | |
| EP1207135B1 (en) | Phosphation reagent, process and use | |
| CN102977138A (zh) | 一种清洁工艺制备毒死蜱的方法 | |
| RU2381066C1 (ru) | Катализатор и способ окисления сульфида натрия | |
| DE3734469A1 (de) | Verfahren zur herstellung von pyromellitsaeure | |
| JPS585897B2 (ja) | 芳香族カルボン酸クロライドの製造方法 | |
| US2993010A (en) | Activation of dehydration catalysts | |
| CN112915993B (zh) | 一种联产草酰胺和氨基甲酸甲酯的催化剂及制备方法 | |
| DE1443187A1 (de) | Verfahren zur Herstellung eines Fluorierungskatalysators | |
| IT8223828A1 (it) | Catalizzatore particolarmente per la sintesi del metanolo | |
| DE3688817T2 (de) | 5-azido-deoxyuridinverbindungen und deren herstellung. | |
| Wu et al. | Two novel molybdenum complexes containing [Mo2O2S2] 2+ fragment: synthesis, crystal structures and catalytic studies | |
| CN109180476B (zh) | 一种高碳醇酯及其制备方法 | |
| CN115215806B (zh) | 一种磺胺-6-甲氧基嘧啶的合成方法 | |
| JPS63248724A (ja) | 繊維状酸化アンチモンの製造方法 | |
| CN119259066B (zh) | 一种乙苯脱氢催化剂及其制备方法和应用 | |
| JPH02300233A (ja) | 窒化炭素の酸処理誘導体およびその製造法 | |
| CN105597836B (zh) | 丝光Beta沸石及其制备方法和应用以及载体及其应用 | |
| Moosath et al. | Studies on hexachloroceric acid: I. Isolation of hexachloroceric acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TH4A | Reissue of patent specification |